Embed Size (px)
Citation preview

Emerging Sequencing Technologies
TAC Presentation
Jay Shendure
February 27, 2004

Genome Resequencing
• Sanger Sequencing (state-of-the-art):
– ~ 0.1 cents per unfinished base
– ~ $40,000,000 per 6.5x diploid human genome coverage
– ~ 2 million bases per machine per day (24 bp/s)
(~20,000 instrument-days)
• But the goals of a ULCS genome requires…
– ~ 0.0000025 cents per unfinished base
– ~ $1,000 per 6.5x diploid human genome coverage
– ~ 40 billion bases per instrument-day (~450,000 bp/s)
(~1 instrument-day)

Emerging Alternative Technologies
• Cyclic Array Sequencing
– Amplified
• Pyrosequencing
• Sequencing By Synthesis with Fluor-dNTPs
• Sequencing by Cleavage / Ligation (MPSS)
– Single Molecule
• Sequencing By Synthesis with Fluor-dNTPs
• Nanopore Sequencing (Single Molecule)
• Sequencing By Hybridization
• Microelectrophoretic Sequencing

The 24 Hour Genome
• Throughput: 450,000 bp/s
• Coverage: 6.5-fold x 6e9 bases
• Raw Accuracy: 99.7%
• Read Lengths: 20 bp – 60 bp (at least)
• Final Accuracy: >99.999%
• Template Prep: ~ 1 billion
• Assumption: cost to own & operate a single instrument will be
roughly similar to conventional sequencing (100% uptime -> $1000
/ day)
• Just one of many possible scenarios.
• Tradeoffs between error and throughput.

Microelectrophoretic Sequencing
• Throughput: 28 bp/s
• Read Lengths: 400 - 800 bp
• Accuracy: ~99.9%
Advanatages:
• Based on an extraordinarily well-tested technology.
• Long read-lengths.
Disadvantages:
• Difficult to imagine how 4 orders of magnitude will be achieved without a more radical departure in terms of parallelization.
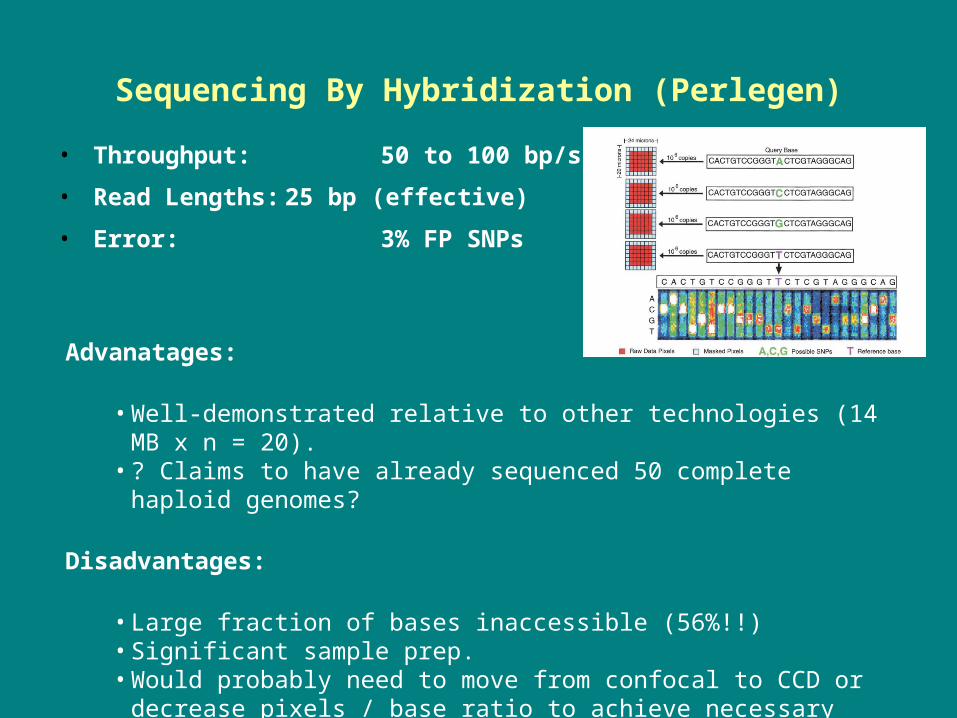
Sequencing By Hybridization (Perlegen)
• Throughput: 50 to 100 bp/s
• Read Lengths: 25 bp (effective)
• Error: 3% FP SNPs
Advanatages:
• Well-demonstrated relative to other technologies (14 MB x n = 20).• ? Claims to have already sequenced 50 complete haploid genomes?
Disadvantages:
• Large fraction of bases inaccessible (56%!!)• Significant sample prep.• Would probably need to move from confocal to CCD or decrease pixels
/ base ratio to achieve necessary speeds.

Nanopore Sequencing
• Throughput: 10 bp/s (–> 30e6
bp/s?)
• Read Lengths: 1 bp
• Accuracy: ~60%Advanatages:
• Potential for extraordinarily rapid sequencing
• Potential for zero-sample prep
Disadvantages:
• No demonstration of single-base-pair resolution at internal base-positions
• Will likely require significant pore engineering

Cyclic Array Methods
• Molecules: Single vs. Amplified
• Acquisition: Real-time vs. Off-line Acquisition
• Plone Amplification
– In Situ Polonies
– Emulsion Amplified Beads
– Capture Beads
– Picowell PCR
• Enzymatic: Polymerase vs. RE/Ligase

Cyclic Array Methods
• (1) Homopolymers!
• (2) Removal of labels (for FL-dNTP methods)
• (3) Maintainance of synchrony (for amplified methods)
• (4) High sensitivity of optics (for single molecule methods)
• (5) Spatial separation of random or ordered array features
• (6) Method of clonal amplification
• (7) Feature miniaturization
• (8) Rate of data acquisition

Multiplex Pyrosequencing (454)
• Throughput: <100 bp/s ??
• Read Lengths: 40 – 150 bp
• Accuracy: >99% ??
Advanatages:
• Functional technology.• Reasonable homopolymer solution.• Based almost purely on unmodified nucleotides
Disadvantages:
• Requirement for real-time imaging of sequencing by extension may be a significant bottleneck for improving per-instrument throughput.
• Diffusion and light scattering creates demand for spatial separation between features; ordered arrays

Polony FISSEQ
• Throughput: ~1 bp/s
• Read Lengths: 5 - 8 bp
• Accuracy: ~94%
Advanatages:
• Integrates plone generation and sequencing to a single platform.
• Beat Poisson with polony exclusion principle.
Disadvantages:
• Miniaturization definitely possible, but difficulties in minaturizing sequencable polonies.
• Polonies visible to confocal scanner but invisible to CCD
• Observed errors are systematic rather than random.

Massively Parallel Signature Sequencing (Lynx)
• Throughput: 86 bp/s
• Read Lengths: 12 – 16 bp
• Accuracy: 91%?
Advanatages:
• Demonstrated method for tag sequencing
• CCD imaging
• No homopolymer problem.
Disadvantages:
• Observed enzyme efficiencies are poor -> limited read-lengths
• Long cycle times, long scan times (low signal?).

Single Molecule Sequencing (Webb, Quake, Genovoxx)
• Throughput: ~100 bp/s (333 bp/s?)
• Read Lengths: ~4 bp (45 bp?)
• Accuracy: 96% (82%?)
Advanatages:
• Can proceed asynchronously.• Misincorporation events terminate (maybe).• No need for amplification step in sample prep.
Disadvantages:
• Blinking, photobleaching -> missed events.• Misincorporation events terminate (maybe).• Succesful experiments have involved sequencing non-consecutive bases.

Key Points
(1) Very, very high accuracy is critical.
(2) Throughput as a function of method of data acquisition is a major bottleneck (CCD!)
(3) Real-time methods have much larger constraints in terms of parallelization relative to “off-line” methods.

Key Points
(4) Throughput of sequencing must be matched by throughput of library generation and throughput of plone generation.
(5) The primary advantages of single-molecule methods are asynchrony and template preparation, not data-density.
(6) Combinations of methods for clonal amplification and sequencing give rise to many alternatives.

Key Points
(7) A partial homopolymer solution will suffice for many initial goals.
(8) For SBS methods, substitution error-rate may be << than indel error-rate.
(9) Extending past the point of high accuracy is still informative for matching back to a reference genome.

Cyclic Array vs Nanopore vs Microelectrophoretic vs SBH
Single vs Amplifed
Real-time vs Off-line
Polonies vs Emulsion Beads vs Lynx Beads vs Nanowell PCR vs Rolling Circle Beads
Linear Quantitation vs Steric Reversible Terminators vs 3’ Reversible Terminators vs FRET
Polymerase vs RE/Ligase
Modified vs Unmodified Nucleotides vs Mixtures vs Multistep
Direct vs Indirect Labeling
Klenow vs BST vs Sequenase
CCD vs Confocal
Random Arrays vs Ordered Arrays
Polymerase Trapping vs Enzyme Reloading
TCEP vs BME vs MESNA
Technology Agnostism

(1) Generate flanked library with purely in vitro methods.
(2) Generate 1-micron “clonal beads” via emulsion PCR (or other method?).
(3) Enrich amplified beads from “empty” beads.
(4) Sequence acrylamide-immoblized beads in parallel via FISSEQ protocol.
Strategy Overview

Clonal PCR Amplification with Emulsions
(a) Protocol adopted from Dressman et al. (PNAS 2003)
(b) Sub-picoliter aqueous compartments => isolated reaction chambers
(c) Compartments also contain paramagnetic beads to which PCR products end up immoblized (via biotin-streptavidin interaction).
(d) All copies of amplified DNA on same bead are the same, but amplified DNA on different beads is different (AMPLIFIED CLONALITY, just like polonies!)
(e) ~100 million beads per PCR tube!!!

Key Advances with Plone FISSEQ
(1) In situ Polonies -> Beads => Major reduction in physical size (& greater consistency). Comes at a price.
(2) Confocal Scanner - > CCD imaging => ~25x fold increase in data acquisition rate.
(3) Operating near the limit of optical resolution.
(4) Integration of software for stage operation, data collection, image alignment, bead identification, and base-calling.
(5) Iron cores provide sharp resolution of bead centroids in at high bead-densities.
(6) Revision to cycling protocols (no trapping or alkylation, TCEP cleavage, unlabeled follow-up)
(7) Improvements to emulsion PCR technology to generate sufficient signal for bead-sequencing.
(8) Disappearance of systematic errors (unexplained!)
(9) 6% gels are stable over 30+ cycles -> read-lengths of 14-15 bases.

FISSEQ (G1 Slide)
(C..A..G..T) x 7.5 = 30 quarter-cycles
T1 CACACACACACACACTCCACCA
T2 GTGTGTGTGTGTGTGTCCACCA
T3 AGTGCTCACACACGTGATCCAC
T4 CAGCCGAACGACCGATCCACCA
T5 ATGTGAGAGCTGTCGTCCACCA

0.5% of full gel area, 10X, 3 uM beads

Current Protocol
1. Extend with labeled base.
2. Wash
3. Image
4. Cleave label with TCEP
5. Wash
6. Extend with unlabeled base
7. Wash
~30 minute non-imaging cycle time.

Current Analysis Algorithm
1. Align images with allowances for gel warping.
2. Define white-light (WL) pixels as connected objects.
3. Extract data from each Cy5 image and a single Cy3 image at WL pixels only.
4. Calculate Cy5 / Cy3 ratios for each bead (avg value over WL pixels)
5. Rank-order ratios choose cutoff based on a priori knowledge of fraction of
beads expected to add. Sub-classify as “Class 1” or “Class 2”.
6. Repeat till “signature” is obtained for each bead in all images.
7. Determine base sequence from signature and knowledge of base-cycle-order.
8. Match signatures to database of known templates (Hamming distance)
9. Classify sequences as perfect matches, matches with one substitution, one
single-base insertion, one single-base deletion, or “complex” deviations.
10. Discard “complex” deviations as low-quality reads.
11. Determine error rates for each class as # of errors / total # of bases sequenced

~0.005% of gel area, 3 uM beads, 10x, cycle 1

~0.005% of gel area, 3 uM beads, 10x, cycle 1

cy3
cy5

• Throughput: ~6000 bp/s
• Read Lengths: 14-15 bp
• Beads analyzed (0 – 1 error) 1,627,646
• Beads discarded (2+ errors) 520,689
• # of bases sequenced (total) 23,703,953
• # bases sequenced (unique) 73
• Avg fold coverage 324,711
• Homopolymer solution None
• Pixels used per bead (analysis) ~3.6
G1 Slide

cy3
cy5
Class I (blue, 83%) vs. Class II (red, 17%) base-calls

Total Class I (83%) Class II
(17%)
Bases 23,703,953 19,729,897 3,974,056
Insertions 331,110 93,909 237,201
Deletions 339,242 131,340 207,902
Substitutions 10,840 757 10,083
Total Class I Class II
Insertions 0.986 0.995 0.940
Deletions 0.986 0.993 0.948
Substitutions 0.9995 0.99996 0.997
Error Analysis
Not included: PCR error, homopolymer error, difficult contexts
Class of substitution error determined by inserted base

6 bp/s 6000 bp/s 60,000 bp/s 450,000 bp/s
5-8 bp 14-15 bp 27 bp 60 bp
92% ~99% >99.9% >99.99%
5 5 1 million 1 billion
0 v 1+ 0 v 1+ 0 v 1 v 2+ Full
Past Present Future 1 Future 2
Still a long way to go…

Short-Term Challenges
(1) 5 templates -> 1 million templates
(2) Implementation of FRET homopolymer solution.
(3) 30 cycles -> 40 cycles
(4) 6000 bp/s -> 60,000 bp/s.
(5) Better base-calling algorithms (source of errors?)
(6) Boosting clonal bead-generation throughput.
(7) Need method for rapid generation of template libraries.
(8) Automation of Cycling & Integration of Cycling / Imaging.
(9) How to generate paired reads in this system?

27-mer Library (Tengs & Meyerson)
• Cut Site = 5’-NNNNNNNNNACNNNNNCTCCNNNNNNN-3’
• Enzyme = BsaXI
• Large jump in complexity (5 -> 1.2 million)
• Defined sizes and internal fixed bases are a good sanity-checks
• Human.
• Sanger traces available for error estimation.
• Succesfully amplified and proven clonality, but haven’t actually sequenced more the 2 cycles.
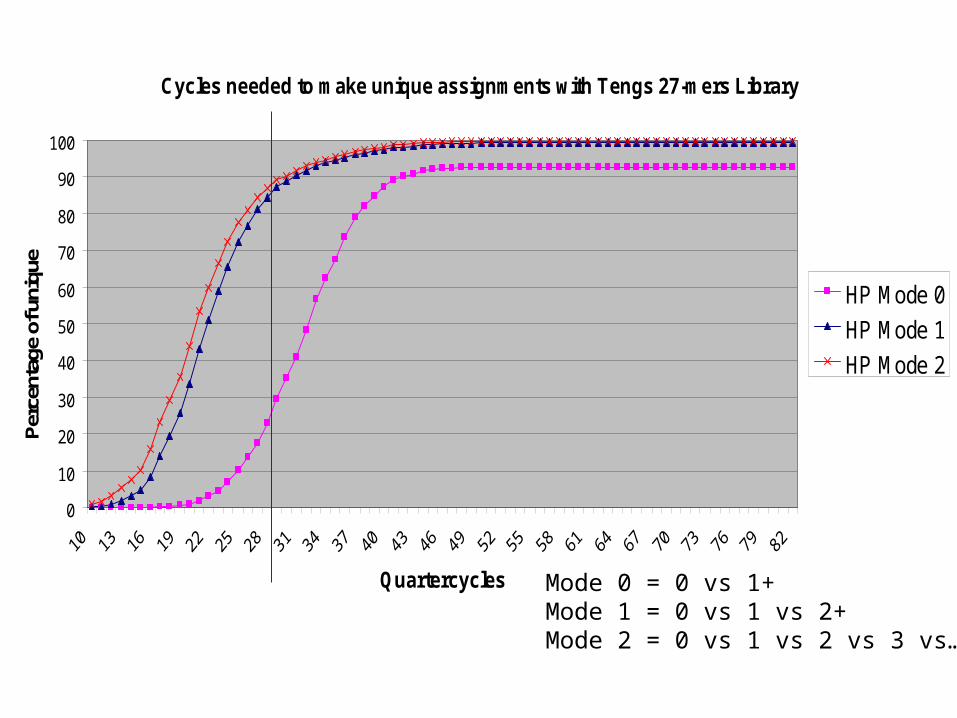
Cycles needed to make unique assignments with Tengs 27-mers Library
0
10
20
30
40
50
60
70
80
90
100
Quartercycles
Per
cent
age
of u
niqu
e
HP Mode 0
HP Mode 1
HP Mode 2
Mode 0 = 0 vs 1+Mode 1 = 0 vs 1 vs 2+Mode 2 = 0 vs 1 vs 2 vs 3 vs…

0
2
4
6
8
10
12
14
1
Modes
% o
f u
nd
etec
tab
le S
NP
s
Avg Mode 0
Avg Mode 1
Avg Mode 2
Mode 0 = 0 vs 1+Mode 1 = 0 vs 1 vs 2+Mode 2 = 0 vs 1 vs 2 vs 3 vs…

FRET as a solution to the resolving homopolymers
2+ populations of reversibly labeled dNTPs
Multiple-label strands will FRET, but single-labeled strands will not.
Get around quenching. Positive signal on different channel.
Internally controlled. Futher multiplexing with more fluors possible.
C C G A AC C G T T A C G C
Cy3 Cy5
C C G A C C G T A C G C
Cy3
C C G A AC C G T T A C G C
Cy3 Cy5
C C G A C C G T A C G C
Cy5

Acknowledgements
GeorgeGreg Porreca
Rob MitraAaron Lee
Church Lab
Paul McEwanKevin McKernan

• Summary of altered strategy• 5-mer library description• Image of current bead-density (SBE)• Sample Cy3 vs. Cy5 plot• Summary statistics
• 27-mer library description• Aaron’s simulation graphs• GP: proof-of-clonality
• Description of FRET concept• Initial FRET data
• Slide on plethora of sequencing technologies





























