Embed Size (px)
Citation preview

Proc. Nat. Acad. Sci. USAVol. 71, No. 4, pp. 1118-1122, April 1974
Electron Transfer to Clostridial Rubredoxin: Kinetics of the Reductionby Hexaammineruthenium(II), Vanadous and Chromous Ions
(iron-sulfur proteins/activation enthalpies/activation entropies/rubredoxin"""11 entropy difference/ionic strength effects)
CHARLES A. JACKS, LARRY E. BENNETT*, WAYNE N. RAYMOND, AND WALTER LOVENBERGtDepartment of Chemistry, San Diego State University, San Diego, California 92115; and t Experimental Therapeutics Branch,National Heart Institute, Bethesda, Maryland
Communicated by Henry Taube, December 3, 1973
ABSTRACT The rate constants (25°, M-1 see-'), activa-tion enthalpies (keal/mol), and activation entropies (e.u.)for the second-order reduction of oxidized clostridialrubredoxin by Ru(NH3)s0+, V(H20),?+, and Cr(H20)e' + atI = 0.10 have been determined or estimated to be 9.5 X104, '-1.4, -31 (pH 6.3-7.0); 1.6 X 104, 0.1, -40 (pH3.5-4.5); and 1.2 X 108, -0, - -44 (pH 3.5-4.0), respec-tively. Ionic strength dependencies for the vanadium re-action are suggestive of a direct interaction of the re-ductants with the Fe(SR)4-1 site of oxidized rubredoxin.The results are consistent with outer-sphere mechanismsfor all three reductants and an especially high inherentouter-sphere reactivity of rubredoxin. This high reactivitylevel is attributed to the low activation enthalpy demandsof the iron-sulfur site of rubredoxin. Thus, the possibilityis raised that the rate of reaction of rubredoxin with itsphysiological counterparts may be determined largely bythe activation entropy demands imposed by the physiolog-ical reactant. Evidence is presented in support of an ab-solute entropy decrease of about 7.5 e.u. on going fromoxidized to reduced rubredoxin, which is presumablyattributable to the charge increase from Fe(SR)41- toFe(SR)42- at the redox site.
Over the past decade iron-sulfur proteins have emerged as animportant class of redox metalloproteins, functioning in suchdiverse processes as respiration, photosynthesis, nitrogen fixa-tion, biosynthesis, and degradative metabolism (1-5). Thesimpler proteins of this class appear to function physiologicallyas electron carriers; e.g., ferredoxins, adrenodoxin, and puti-daredoxin (1-5). The structural characteristics of their metalsites have been reasonably well defined by extensive studiesof their static properties (6), including refined x-ray deter-minations for the structures of crystalline clostridial rubre-doxin (1 Fe) (7), high potential iron protein (4 Fe) (8), and aferredoxin (8 Fe) (8). In constrast, little is known of theirkinetic properties.We have begun a systematic investigation of the reactivity
patterns of these proteins with a kinetic study of the reactionof the oxidized form of clostridial rubredoxin, Rd",', withsimple inorganic reductants which have been well character-ized in their mechanistic behavior (1).
Rubredoxin was chosen because it is the least complex ofthe iron-sulfur proteins (molecular weight -6,000) and offersthe advantage of an established structure for Rd"I (7). The
Abbreviations: RdIII, oxidized rubredoxin; RdII, reducedrubredoxin.* To whom correspondence should be addressed at San DiegoState University.
single iron center is coordinated close to the surface of thecrystalline protein by four cysteinyl sulfurs at the apices of adistorted tetrahedron (7). Tetrahedral coordination is re-tained in solution (9). From a preliminary difference map itappears that the dimensions of the immediate metal environ-ment in the crystal change by no more than about 0.1 A in thereduced form, RdV, from those in the oxidized form (L. H.Jensen, personal communication).
Tetrahedral coordination of iron by divalent sulfur is foundin all the structures of iron-sulfur proteins which have beensolved by x-ray diffraction (7, 8) or proposed on the basis ofsubstantial evidence (6). For those proteins containing acluster of two or more iron centers, bridging positions betweenthe metal centers are occupied by acid-labile sulfur. Theclusters are bound to their proteins via terminal (nonbridging)coordination of the iron centers by cysteinyl sulfurs. Eachcluster appears characteristically to undergo reduction by oneelectron, which for several two-iron species seems to be local-ized on one metal center (6). To the extent that redox changesare localized on one center and the reactivity of that center isnot greatly different for coordination by bridging labile sulfurversus terminal cysteinyl sulfur, the reactions of rubredoxinshould provide a representative indication of the redox re-activity of the metal sites in iron-sulfur proteins.
MATERIALS AND METHODS
Clostridial rubredoxin was isolated and purified by the methodof Lovenberg and Williams (10). Samples for kinetic experi-ments had Am0/A490 = 2.6 with several separately isolatedsamples yielding indistinguishable results.
Electrolytic reduction at a platinum electrode of reagentgrade V205 in excess HCl yielded solutions of vanadyl ionwhich was reduced, under nitrogen, to V(H20)62+ at a mercuryelectrode or by amalgamated zinc with either method yieldingindistinguishable kinetic results. Solutions of Cr(H20)62+ weresimilarly prepared by reduction of acidic chromic chloride(twice recrystallized) solutions with amalgamated zinc. TheRu(NH3)6CI3 salt obtained from Matthey Bishop was re-crystallized according to the Armor procedure (11) and testedspectrophotometrically for purity. Amalgamated zinc wasused to generate Ru(NH3)62+ under nitrogen in solution con-taining Tris HCl adjusted to the desired pH (usually 7.0).Reductant concentrations were determined by spectrallymonitoring the product solutions following reaction with ex-cess Co(NH3)5Cl2+ in 0.1 M HCl04. The vanadium resultagreed with that determined by monitoring the excesschromium(VI) remaining after oxidation to VO2+ by HCrOc4.
1118
Electron Transfer to Clostridial Rubredoxin 1119
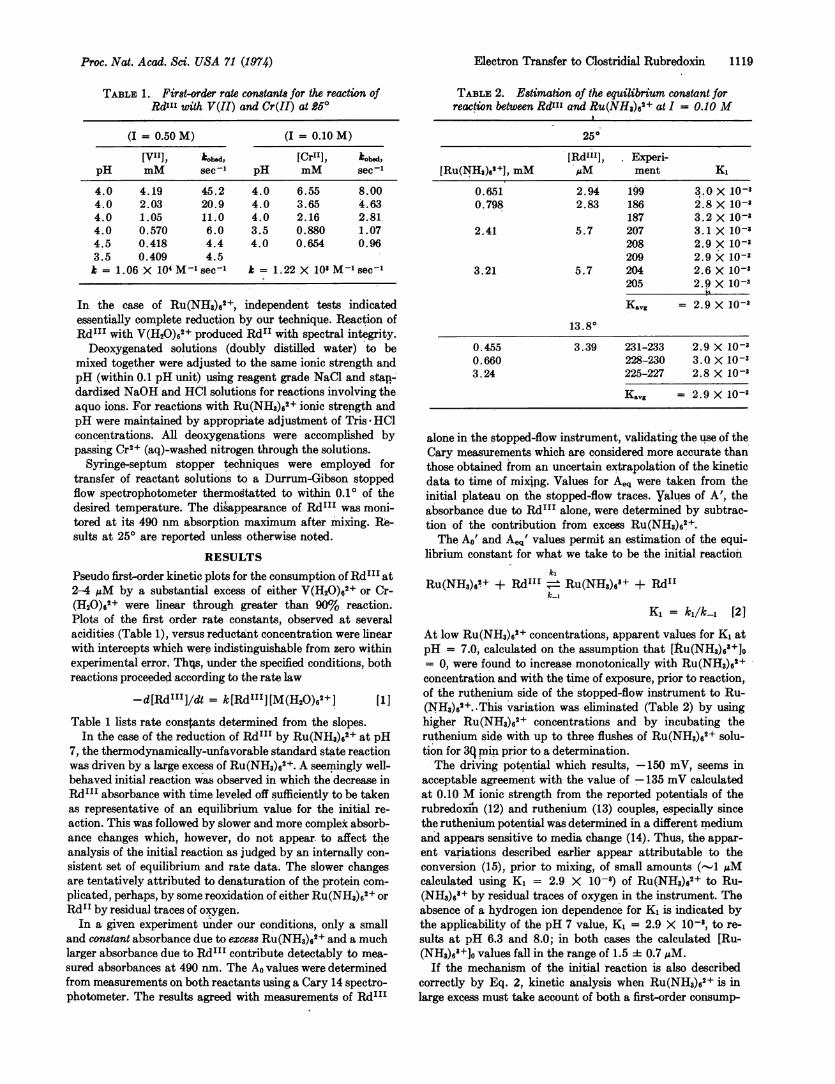
TABLE 1. First-order rate constants for the reaction ofRdI"' with V(II) and Cr(II) at 250
(I = 0.50 M) (I = 0.10 M)
[VII]X, kbsd, [Cri"], kobd,pH mM sec pH mM sec'
4.0 4.19 45.2 4.0 6.55 8.004.0 2.03 20.9 4.0 3.65 4.634.0 1.05 11.0 4.0 2.16 2.814.0 0.570 6.0 3.5 0.880 1.074.5 0.418 4.4 4.0 0.654 0.963.5 0.409 4.5k = 1.06 X 10' M-1 sec'1 k 1.22 X 101 M-1 sect
In the case of Ru(NH3)62+, independent tests indicatedessentially complete reduction by our technique. Reaction ofRdIII with V(H20)62+ produced Rd" with spectral integrity.Deoxygenated solutions (doubly distilled water) to be
mixed together were adjusted to the same ionic strength andpH (within 0.1 pH unit) using reagent grade NaCl and sta.L-dardized NaOH and HCl solutions for reactions involving theaquo ions. For reactions with Ru(NH3)62+ ionic strength andpH were maintained by appropriate adjustment of Tris * HClconcentrations. All deoxygenations were accomplished bypassing Cr2+ (aq)-washed nitrogen through the solutions.
Syringe-septum stopper techniques were employed fortransfer of reactant solutions to a Durrum-Gibson stoppedflow spectrophotometer thermostatted to within 0.1° of thedesired temperature. The disappearance of Rd"' was moni-tored at its 490 nm absorption maximum after mixing. Re-sults at 250 are reported unless otherwise noted.
RESULTSPseudo first-order kinetic plots for the consumption of RdIII at2-4 AM by a substantial excess of either V(H20)62+ or Cr-(H20) 2+ rwere linear through greater than 90% reaction.Plots of the first order rate constants, observed at severalacidities (Table 1), versus reductant concentration were linearwith intercepts which were indistinguishable from zero withinexperimental error. Thqs, under the specified conditions, bothreactions proceeded according to the rate law
-d[Rd"I]/dt = k[RdII'][M(H2O)62] [11]Table 1 lists rate constants determined from the slopes.In the case of the reduction of RdIII by Ru(NH3)62+ at pH
7, the thermodynamically-unfavorable standard state reactionwas driven by a large excess of Ru(NHa)62+. A seemingly well-behaved initial reaction was observed in which the decrease inRd"I' absorbance with time leveled off sufficiently to be takenas representative of an equilibrium value for the initial re-action. This was followed by slower and more complex absorb-ance changes which, however, do not appear. to affect theanalysis of the initial reaction as judged by an internally con-sistent set of equilibrium and rate data. The slower changesare tentatively attributed to denaturation of the protein com-plicated, perhaps, by some reoxidation of either Ru(NH3)62+ orRdII by residual traces of oxygen.
In a given experiment under our conditions, only a smalland constant absorbance due to excess Ru(NH3)62+ and a muchlarger absorbance due to RdIII contribute detectably to mea-sured absorbances at 490 nm. The Ao values were determinedfrom measurements on both reactants using a Cary 14 spectro-photometer. The results agreed with measurements of RdIII
TABLE 2. Estimation of the equilibrium constant forreaction between Rd", and Ru(NH3)62+ at I = 0.10 M
250
[Rd"'], Experi-[Ru(NHs),'2], mMIM ment K,
0.651 2.94 199 3.0 X 10-30.798 2.83 186 2.8 X 10-i
187 3.2 X 10-82.41 5.7 207 3.1 X 10-3
208 2.9 X 10-3209 2.9 X 10-3
3.21 5.7 204 2.6 X 10-3205 2.9 X 10-3
i'Kavg = 2.9 X 10-3
13.80
0.455 3.39 231-233 2.9 X 10-30.660 228-230 3.0X 10-33.24 225-227 2.8 X 10-3
Kavg = 2.9 X 10-3
alone in the stopped-flow instrument, validating the use of theCary measurements which are considered more accurate thanthose obtained from an uncertain extrapolation of the kineticdata to time of mixjpg. Values for Aeq were taken from theinitial plateau on the stopped-flow traces. values of A', theabsorbance due to Rd"' alone, were determined by subtrac-tion of the contribution from excess Ru(NH3)62+.The Ao' and A.' values permit an estimation of the equi-
librium constant for what we take to be the initial reactionki
Ru(NHa)Y2+ + RdI'I = Ru(NH3)63+ + Rd"
K, = k,/1kc [2]
At low Ru(NH3)6g+ concentrations, apparent values for K, atpH = 7.0, calculated on the assumption that [Itu(NH3)6'+]o= 0, were found to increase monotonically with Ru(NH3)62+concentration and with the time of exposure, prior to reaction,of the ruthenium side of the stopped-flow instrument to Ru-(NH3)62. -This variation was eliminated (Table 2) by usinghigher Ru(NH3)62, concentrations and by incubating theruthenium side with up to three flushes of Ru(NH3)62+ solu-tion for 3q mun prior to a determination.The driving potential which results, -150 mV, seems in
acceptable agreement with the value of -135 mV calculatedat 0.10 M ionic strength from the reported potentials of therubredoxiii (12) and ruthenium (13) couples, especially sincethe ruthenium potential was determined in a different mediumand appears sensitive to media change (14). Thus, the appar-ent variations described earlier appear attributable to theconversion (15), prior to mixing, of small amounts (-1 uMcalculated using K, = 2.9 X 10-8) of Ru(NH3)62+ to Ru-(NH3)63+ by residual traces of oxygen in the instrument. Theabsence of a hydrogen ion dependence for K, is indicated bythe applicability of the pH 7 value, K, = 2.9 X 10-8, to re-sults at pH 6.3 and 8.0; in both cases the calculated [Ru-(NH3)6+]O values fall in the range of 1.5 0.7 MM.
If the mechanism of the initial reaction is also describedcorrectly by Eq. 2, kinetic analysis when Ru(NH3)2+ is inlarge excess must take account of both a first-order consump-
Proc. Nat. Acad. Sci. USA 71 (1974)

Proc. Nat. Acad. Sci. USA 71 (1974)
TABLE 3. First order rate constants for the reaction ofRd"'I with Ru(NH3)'2+ at 25°, I = 0.10 M and
pH = 7.0 (Tris.HCl)
Experi- [Ru(NH3)62 +], Cobbdyment mM %.obd sec' kavg, sec'
194 0.110 19-76 12.2195 19-78 14.0 13.1191 0.303 33-85 30.8192 30-84 31.7 29.9193 28-91 27.2188 0.537 38-90 53.6189 38-90 56.6197 0.651 42-95 66.3198 52-96 59.6 64.9199 55-96 68.8185 0.798 39-87 75.7186 41-88 78.6 77.1187 38-93 77.1
207-209 2.41 70-96 222 it 10%204-205 3.21 73-96 273-- 10%213-216 0.654 31-93 58.0 (pH = 8.0)218-220 0.673 54-90 67.3 (pH = 6.3)
ki = 9.5 it 0.3 X 104 M-' sec' (see text).
tion of Rd"I and its regeneration via a second-order backreaction between Rd" and Ru(NH3)63+, the latter beingpresent in small concentration prior to mixing. The resultantrate law was integrated (16) to give
where the rate coefficient is the slope of the least square plot ofkobsd versus [Ru(NH3)62+] at pH 7. An analogous plot arrivedat under the incorrect assumption, [Ru(NH3)68+]o = 0, yieldeda second order rate constant of 9.0 X 104 M-1 sec-1 indicatinglittle kinetic sensitivity to the levels of [Ru(NH3)63+]o en-countered under our conditions.The rate constant given in Eq. [SI is considered to reflect the
true [Ru(NH3)62+] dependence more accurately than 10.4 4±0.4 X 104M'sec-, the average result ofkob8d/[Ru(NH3)62+]calculated independently for each experiment. The discrep-ancy between these numbers and a y-intercept (2.5 sec') ofthe pH 7 plot which seems slightly outside experimentalerror is suggestive of a small uniform consumption of Rd"'which is independent of [Ru(NH3)62+]. This may be due to thepresence of H202 (from the Ru(NHB)62+-02 reaction) whichotherwise did not appear to introduce kinetic complications.The first-order rate constants obtained at the higher Ru-
(NH3)62+ concentrations of the equilibrium studies were notincluded in the second-order rate constant determination be-cause they carry with them greater uncertainties. These un-certainties result from data being accessible only over the last30% of reaction and, relatedly, from a high sensitivity of theseparticular kob8d's to small uncertainties in Aeq' as the result ofthe secondary changes. The results are included in Table 3 toindicate that, within the constraints imposed by the system,there is no evidence for rate saturation as the concentrationof Ru(NH3)62+ is increased.
Studies at 13.80 yielded (i) an equilibrium constant (Table
[doRdII- Peqlll][Ruo"' + Rdo'I - RdeqIII] [Rdo011 - RdeqIII][(RdOI"Ruo"I') 4 (Rdo11)2- (Rd I"'Rdeq"I)][(Rdo"I'Ruo"I') + (RdoIII)2 - (RdeqIII)2] Rdo"'[Rdt'I - Rdeqlll][RUOIII + RdoII - Rdeq1I]o
[3]
where Rd"' = [Rd"'], Ru"'l = [Ru(NH3)6o+] and kobd =k,[Ru(NH3)62+]. Eq. 3 can be expressed in terms of the Rd"'absorbance, A', as
[Ao' -Aeq'][X + Ao' - Aeq'][(XAo') + (Aot)2- (Aeq t)2
X ln [A' - Aeq'] [(XAt') + (Aot)2 - (At'Aeq')] =kIb8dt [4]Ao'[At' - Aeq'][X + Ao' - Aeq'I
where X = (path length) (RdIII extinction coefficient)[Ru(NH3)6'+]0
= (2.0 cm) (8850 M-'cm-') [Ru(NH$)6+l]oand [Ru(NH3)68+]0 is calculated for each run from Aot, Aeq'and K, = 2.9 X 10-1.
Plots of the left hand side of Eq 4 versus time had zero y-intercepts within experimental error and were linear over theindicated percent of reaction observed, yielding the kobsd valueslisted in Table 3. A plot of the average values at pH 7 versusthe five lowest Ru(NH3)62+ concentrations was linear. Theaverage value obtained at pH 8 fell about 10% below thisplot, suggesting the possibility of an acid-dependence athigher pH. However, the average result at pH 6.3 was coin-cident with the pH 7 plot within experimental error. Thus, atleast over this pH range, the reaction follows the rate law
_ d[Rd"'] = (9.5 i 0.3 X 104M-sec-)dt
X [RdIIII[Ru(NH3)62+ 5
2) which was indistinguishable from that observed at 250 and,(ii)kl = 8.3 X 104 M-I sec- from the least-squares slope of alinear plot of pseudo first-order rate constants versus severalRu(NH3)62+ concentrations. At 37.9° the secondary changesoccurred so early that no acceptable plateau after the initialreaction was reached. In an attempt to surmount this diffi-culty, an estimate was made of the absorbance following thefirst reaction and this was iterated to minimize the ratio,standard error of the slope: slope, of the kinetic plots usingearly data. The equilibrium constant calculated using thisresult did not appear in conflict with the temperature inde-pendence described earlier. However, the rate constant, ki =8.1 X 104 M-l sec', obtained as before from reactions atseveral reductant concentrations is actually lower than thatat 250. We consider it quite possible that this number isartificially low as a result of the limitations imposed by thesecondary changes. Only data for the lower two temperatureswere included in an Eyring plott to determine the activationparameters which are possibly of more qualitative than quanti-tative significance.
In the case of the more tractable V(H2O)62+-RdIII re-action, a linear Eyring plot was obtained over the tempera-ture range 15-35°, and measurements were extended to sev-eral ionic strengths. A summary of the kinetic data is pre-sented in Table 4. (The estimate of AH* - 0 for the chromousreaction arises from a single experiment at 15° which, if ac-curate, would indicate AS* -44 e.u.)
t A plot of log (k/T) versus l/T with a slope of AH*/2.3 R.
1120 Biochemistry: Jacks et al.

Electron Transfer to Clostridial Rubredoxin 1121
TABLE 4. Summary of kinetic data for the reduction of clostridial rubredoxin
k (250) AH* AS*Reductant (M-1 sec-1) I (M) pH (kcal/mol) (e.u.)
Ru(NH3)62+ 9.5 X 104 0.10 6.3-7.0 (1.4) (-31)V(H2O)62+ 1.1 X 104 0.50 3.5-4.5 0.1 -40
1.6 X 104 0.10 4.04.0 X 104 0.010 4.0
Cr(HpO)62+ 1.2 X 103 0.10 3.5-4.0 -(0) (-44)
DISCUSSION
Since Ru(NH3)62+ retains its ligands for times greatly exceed-ing those of the redox experiments and does not present abonding site to the iron center of Rd"', an outer-sphere re-action between these species is virtually assured. Both therate and activation parameters of the V(H2O)62+-RdIII re-action provide evidence against an inner-sphere reactionwhich is rate-limited by substitution on vanadium (1). Sub-stitution into the iron coordination sphere by a water mole-cule which remains coordinated to vanadium (assuming sucha process were sterically feasible) seems incompatible with thehigh rate observed, since water is a very inefficient bridgingligand for electron transfer (17).Support for an outer-sphere mechanism is provided by a
comparison with the rate of reduction of Rd," by Cr(H2O)62 .The kv/kcr ratio of r13 with Rd"I (I = 0.10) falls withinthe theoretically rationalized range of --10-60 observed witha number of other oxidants when both reductants react viaouter-sphere paths (17). §
Further details of the reaction are suggested by the varia-tion in rate of the vanadium reaction with ionic strength.The rates at the two lower ionic strengths suggest thatV(H2O)62+ is reacting at a site of -1 charge. This is consistentwith a direct approach to the uninegative Fe(SR)4- cluster bythe reductants.That no inner-sphere path more facile than --'103 M- sec1
is presented to Cr(H2O)62+ by Rd"I seems significant sincesterically unhindered thiolate sulfur is a highly efficient bridg-ing function, at least on Co(III) (19). The approach of Cr-(H2O)52+ to a potentially bridging thiolate sulfur may besterically restricted by the protein in solution. Alternatively,tetrahedrally-ligated iron(III) is not expected to be as re-sponsive kinetically to an inner-sphere option as octahedrally-ligated cobalt(III).The results with Ru(NH3)62+ and V(H2O)62+ lead to an
important conclusion of this study: the iron center of clostridialrubredoxin can be oxidized or reduced very rapidly by outer-sphere agents. A direct demonstration of this is provided by acalculation, from K1 and ki, of a rate constant, k-1 = 3.3 X10' M-1 sec-1, for the microscopically reversible outer-spherereaction between RdII and Ru(NH3)63+.A less direct estimation of the inherent reactivity of
rubredoxin is obtained from calculations of the apparent self-exchange rate constant, kRdRd, for electron transfer betweenRd,' and Rd"I which involves no net free energy change.
§ Our recently determined rate constant of 1.3 X 103 M-l sec-'(250, I = 0.10) for the V(H2O)62+-Ru(NH3)63+ reaction is largerthan that determined earlier without a stopped-flow instrument(18). The ratio of the new value to k = 1.0 X 102 A-l sec-(230 I = 0.08) for the Cr(H2O)62+-Ru(NH3)63+ reaction (28)falls in the range observed with other oxidants and is in excellentagreement with the rubredoxin results.
Using the relative theory of Marcus (20) which has been dis-cussed extensively elsewhere (21, 22, 1), the expression appro-priate to the Ru(NH3)62+-Rd'I' reaction is
log kRdRd = 2 log kRuRd -log kRuRu - 0.059 [61
where a small log f correction term has been neglected. Weestimate log kRunu = 3.5 at I = 0.10 from the data available(13). With the vanadium data our value for the V(H2O)62+-Ru(NH3)63+ reaction was used to eliminate the need for kvvwhich is not available at I = 0.10. The calculations yield re-
markably similar results for kRdRd, 1.0 X 109 and 1.7 X 108M-1 sec', respectively. There are reasons to believe thatsuch apparent values may not reflect the true rubredoxin self-exchange rate with complete accuracy. Nevertheless, theysupport the qualitative conclusion of a high inherent reac-tivity of rubredoxin. In the context of inorganic chemistry, theresults are interesting since they provide the first measure ofreactivity for tetrahedrally-coordinated iron.The nature of the reactivity of RdIII with our reductants can
be examined further using the activation parameters of Table4. The activation enthalpies are so low for all three reactionsthey would approach being diffusion-controlled were it notfor the substantial entropy barriers. At our ionic strength asmall contribution to the low activation enthalpies may comefrom a favorable electrostatic interaction between reactants.In any event, the process of activating RdIII for reduction doesnot require much of a heat input. Since AH 00 for the Ru-(NH3)62+-RdIII reaction a similar conclusion can be made re-garding the activation of Rd"I for oxidation by Ru(NH3)6,+.In particular, the enthalpic barrier, 4Hi*, for reorganizing theiron coordination sphere of rubredoxin prior to electron trans-fer, appears to be small. It seems likely that this aspect ofrubredoxin's high reactivity will be reflected in its physio-logical reactions as well.
In terms of current theories of electron transfer (20-22, 1),a low AHj* can be understood as a result of the evolutionaryselection of tetrahedrally-ligated iron as the redox site. Sincethe redox electron populates an approximately nonbonding,"eg" orbital directed between the ligands, its effect on metal-ligand distances is expected to be small. As a result, theenthalpy demands for reorganization of the first coordinationsphere during activation are small.No completely satisfying account can be offered of the
activational entropy barriers encountered in our reactions.In all three reactions they are comparable to those observedwith tripositive oxidants in simple reactions in spite of thereversal in charge type which, in itself, is expected to have aneffect in the opposite direction. A portion of this entropybarrier, -- -13 e.u., can be attributed to the losses in trans-lational and rotational entropy which occur on forming theouter-sphere collision complex (20-22, 1).
Proc. Nat. Acad. Sci. USA 71 (1974)

Proc. Nat. Acad. Sci. USA 71 (1974)
It is evident in the current-theoretical concepts of electrontransfer reactions that some fraction of the net entropy changecan contribute to the entropy of activation of a cross reaction(20-22). Only for the Ru(NH3)62+-RdIII reaction do we havedata bearing directly on this point; an approximate value of-11.6 e.u. is calculated for the observed net entropy changefrom our limited equilibrium results. An absolute entropychange of -4.1 1.9 e.u. for conversion of Ru(NH3)62+ (aq)to Ru(NHS)68+ (aq) (on a scale where S0H+ = -5.5 e.u. andS0H, = 31.2 e.u.) (23) leaves --' -7.5 e.u. (same scale) forthe net change for conversion of RdIII to RdII. ¶ A contributionof - 11.6/2 e.u. to the activation entropy from the netentropy change seems possible for this reaction.The higher entropy barriers for the vanadium and chro-
mium reactions seem attributable, in part, to their larger half-reaction contributions (above scale) of -47.5 e.u. (24) and-65.5 e.u. (25, 26), respectively, to the net entropy change.
The entropy decreases on going from M(H20)62+ (aq) toM(H20)68" (aq) reflect an increased vibration-orientationpolarization of the medium as the charge increases.The physical nature of a portion of our observed entropy
barriers can be appreciated from the following model wherewe have attempted to adhere to the admonitions of Marcus(20). In proceeding to the activated complex the nuclear co-ordinates of the surrounding media, as well as the first coordi-iation sphere, must adjust to an electronic distribution in thereacting species such that the charge on the ML6 species isintermediate between 2+ and 3+ (>2.5+ for the rutheniumreaction, AG' > 0; <2.5+ for the others, AG0 < 0).
In view of the decreases in absolute entropy recorded abovefor a unit increase in charge on the aquo ions, it appears thatthe increased vibration-orientation polarization of the mediumaround M(H20)6 in the activated complex can make a signifi-cant negative contribution to the entropy of activation. WithRu(NHS)62+ this sort of contribution would appear to be muchless. It should be noted that a partitioning of the entropybarrier between the two reactants can only be a first approxi-mation. Current understanding of this area leaves open thepossibility that their mutual inteaction may give rise to en-tropy effects which are unique to each reaction.One objective of the preceding discussion is to point out that
only a part of the observed entropy barriers reflect the activa-tion requirements of rubredoxin. The net entropy decrease ongoing from RdIII to RdI may reflect a greater polarization ofthe environment around the more highly charged FeII(SR)42-moiety. Also there is evidence for small conformationaldifferences between oxidized and reduced forms (10). Aspectsof these ground state differences may be reflected in rubre-doxin's activation requirements. In any event,'a physiologicalreductant (or oxidant) may well impose entropy demandswhich differ significantly from those imposed by our-reactants.
In conclusion, we present 4 speculative view of the reac-tivity of rubredoxin with its physiological counterparts.Initially an encounter complex is formed in which there maybe specific interactions, 'e.g., electrostatic, between the re-
¶ It is notable that the partial entropy change for the rutheniumcouple (23) is surprisingly less negative than that recordedfor the analogous cobalt couple (27a), perhaps because of anoma-lies with ruthenium, or of substantial media differences, as isalso the case with our reaction, from the I = 0 standard entropychanges normally recorded (27b).
actants. The enthalpic barrier to reaction presented by theiron-sulfur cluster is small. To the extent that the enthalpicbarrier associated with the physiological reactant is also small,the rate of reaction could be determined largely by the specificentropy requirements for activation which have evolved forthis pair of reactants.We wish to acknowledge the technical assistance of Eleanor
Bruckwick and invaluable discussions with Jim Cobble, BobLinck, Peter Rock, and Ed O'Neal regarding this problem.Support for'this research from the National Science Foundation(GP-12223) and the donors of the Petroleum Research Fundadministered by the American Chemical Society (5189-AC3,5) isacknowledged with appreciation.1. Bennett, L. E. (1973) Progr. Inorg. Chem. 18, 1-176.2. Malkin, R. & Rabinowitz, J. C. (1967) Annu. Rev. Biochem.
36, 113-148.3. Buchanon, B. B. & Arnon, D. I. (1970) Advan. Enzymol. 33,
119-176.4. Palmer, G. & Brintzinger, H. (1972) in Electrons and Coupled
Energy Transfer in Biological Systems, eds. King, T. E. &Klingen-berg, M. (Marcel Dekker, New York), Vol. 1B,pp. 379-476.
5. Kimura, T. (1968) Structure and Bonding 5, 1-40.6.' (a) Eaton, W. A., Palmer, G., Fee, J. A., Kimura, T. &
Lovenberg, W. (1971) Proc. Nat. Acad. Sci. USA 68, 3015-3025; (b) Dunham, W. R., Palmer, G., Sands, R. H. &Bearden, A. J. (1971) Biochim. Biophys. Acta 253, 373-384.
7. Watenpaugh, K. D., Sieker, L. C., Herriott, J. R. & Jensen,L. H. (1971) Cold Spring Harbor Symp. Quant. Biol. 36,359-367.
8. Carter, Jr., C. W., Kraut, J., Greer, S. T., Alden, R. A.,Sieker, L. C., Adman, E. & Jensen, L. H. (1972) Proc.Nat. Acad. Sci. USA 69, 3526-3529.
9. Eaton, W. A. & Lovenberg, W. (1970) J. Amer. Chem. Soc.92, 7195-7198.
10. Lovenberg, W. & Williams, W. M. (1969) Biochemistry 8,141-148.
11. Armor, J. N. (1970) Ph.D. Thesis, Stanford University,described in ref. 15.
12. Lovenberg, W. & Sobel, B. E. (1965) Proc. Nat. Acad. Sci.USA 54, 193-199.
13. Meyer, T. J. & Taube, H. (1968) Inorg Chem. 7, 2369-2379.14. Lim, H. S., Barclay, D. J. & Anson, F. C. (1972) Inorg.
Chem. 11, 1460-1466.15. Pladziewicz, J. R., Meyer, T. J., Broomhead, J. A. & Taube,
H. (1973) Inorg. Chem. 12, 639-643.16. Frost, A. A. & Pearson, R. G. (1961) Kinetics and Mecha-
nism (Wiley, New York), 2ndjed., p. 186.17. Toppen, D. L. & Linck, R. G. (1971) Inorg. Chem. 10,
2635-2636.18. Endicott, J. F. & Taube, H. (1964) J. Amer. Chem. Soc. 86,
1686-1691.19. Lane, R. H. & Bennett, L. E. (1970) J. Amer. Chem. Soc.
92, 1089-1090.20. Marcus, R. A. (1964) Annu. Rev. Phys. Chem. 15, 155-196
and references therein.21. Sutin, N. (1962) Annu. Rev. Nucl. Sci. 12, 285-328.22. Reynolds, W. L. & Lumry, R. W. (1966) Mechanisms of
Electron Transfer (Ronald Press, New York).23. Lavallee, D. K., Lavellee, C., Sullivan, J. C. & Deutsch, E.
(1973) Inorg. Chem. 12, 570-574.24. (a) Newton, T. W. & Baker, F. B. (1964) J. Phys. Chem. 68,
228-232; (b) Newton, T. W. & Rabideau, S. W. (1959)J. Phys. Chem. 63, 365-370.
25. Linck, R. G. (1971) in Transition Metals in HomogeneousCatalysis, ed. Schrauzer, G. N.' (Marcel Dekker, New York),p. 329.
26. Latimer, W. M. (1952) "Oxidation Potentials (PrenticeHall, Englewood Cliffs, N.J.), 2nd ed., p. 246.
27. (a) Rock, P. A. (1968) Inorg. Chem. 7, 837-840; (b) Kim, J.J. & Rock, P. A. (1969) Inorg. Chem. 8, 563-566.
28. Endicott, J. F. & Taube, H. (1965) Inorg. Chem. 4,437-445.
1122 Biochemistry: Jacks et al.





























