Embed Size (px)
Citation preview

Molecular and Cellular Pathobiology
EGF Receptor Activates MET through MAPK to EnhanceNon–Small Cell Lung Carcinoma Invasion and BrainMetastasis
Jerrica L. Breindel1, Jonathan W. Haskins1, Elizabeth P. Cowell2, Minghui Zhao1, Don X. Nguyen1, andDavid F. Stern1
AbstractMET amplification as a mechanism of acquired resistance to EGF receptor (EGFR)-targeted therapies in non–
small cell lung carcinoma (NSCLC) led to investigation of novel combinations of EGFR andMETkinase inhibitors.However, promiscuous interactions between MET and ERBB family members have made it difficult to evaluatethe effects of MET on EGFR signaling, both independent of drug treatment and in the context of drug resistance.We addressed this issue by establishing a 32Dmodel cell system wherein ERBBs or MET are expressed alone andin combination. Using this model, we determined that EGFR signaling is sufficient to induce MET phosphor-ylation, although MET activation is enhanced by coexpression of ERBB3. EGFR–MET cross-talk was not direct,but occurred by a combined regulation of MET levels and intermediary signaling through mitogen-activatedprotein kinases (MAPK). In NSCLCs harboring either wild-type or mutant EGFR, inhibiting EGFR or MAPKreduced MET activation and protein levels. Furthermore, MET signaling promoted EGFR-driven migration andinvasion. Finally, EGFR–MET signaling was enhanced in a highly metastatic EGFR-mutant cell subpopulation,compared with the indolent parental line, and MET attenuation decreased the incidence of brain metastasis.Overall, our results establish that EGFR–MET signaling is critical for aggressive behavior of NSCLCs andrationalize its continued investigation as a therapeutic target for tumors harboring both wild-type and mutantEGFR at early stages of progression. Cancer Res; 73(16); 5053–65. �2013 AACR.
IntroductionActivatingmutations in EGF receptor (EGFR) are commonly
found in non–small cell lung carcinoma (NSCLC), and cellsexpressing mutant alleles depend on EGFR signaling for sur-vival. Drugs targeting the EGFR kinase domain are effective forNSCLCs; unfortunately, responding tumors eventually prog-ress owing to acquired resistance. Mechanisms of resistance toEGFR tyrosine kinase inhibitors (TKI) include secondarymuta-tions in EGFR and activation of compensatory receptor tyro-sine kinases (RTK), such as MET. In 5% to 20% of recurrentpatients, MET activation sustains tumor cell survival and isassociated with relapse (1–4). Accordingly, EGFR and METcombination therapies are in clinical trials for patients har-boring MET amplification and resistance to EGFR TKIs (5).Bidirectional signaling between EGFR and MET occurs in
both EGFR TKI-resistant and drug-na€�ve NSCLC cells. Notably,in NSCLC cells addicted to EGFR mutations, EGFR inhibition
reduces basal MET phosphorylation (6–8). Conversely, METinhibition in cells with high basal MET activity orMET ampli-fication reduces basal phosphorylation of EGFR and ERBBfamily members, ERBB2 and ERBB3 (1, 2, 9, 10). In MET-amplified cells, MET signaling through ERBB3 maintainsPI3K/Akt cell survival signaling despite EGFR inhibition (1).In addition, in NSCLC with wild-type EGFR or EGFR TKIresistancemutations, ligand stimulation of EGFR inducesMETactivation, and vice versa (8, 11, 12).
Mechanisms and outcomes of these biochemical events aredifficult to interpret owing to the complexity of inter-ERBBinteractions, which may involve promiscuous ERBB hetero-dimerization or indirect signaling cascades. Proposedmechan-isms of ERBB–MET cross-talk include direct interaction, acti-vation through autocrine regulation, and signaling throughintermediary proteins (12, 13). Moreover, receptor cross-talk insome cells may not be bidirectional, but rather unilateral fromEGFR to MET (14). In cancer cells encoding wild-type EGFR,MET regulates invasion and cell motility in an EGF-dependentmanner (8, 12). Significantly, it is unknown if EGFR–METcross-talk in lung cancers with EGFR mutations modulatessimilar phenotypes, particularly in patients with advanceddisease and metastatic relapse. Defining the specific biologiccontexts of ERBB–MET cross-talk, identifying their mechan-isms of action, and characterizing their function may helpexpand clinical settings where combinatorial EGFR-MET ther-apies would benefit patients with NSCLC.
Authors' Affiliations: 1Department of Pathology, YaleUniversity School ofMedicine; and 2Yale University; New Haven, Connecticut
Note: Supplementary data for this article are available at Cancer ResearchOnline (http://cancerres.aacrjournals.org/).
Corresponding Author: David F. Stern, Yale University School of Medi-cine, BML 348, 310 Cedar Street, NewHaven, CT 06510. Phone: 203-785-4832; Fax: 203-785-7467; E-mail: [email protected]
doi: 10.1158/0008-5472.CAN-12-3775
�2013 American Association for Cancer Research.
CancerResearch
www.aacrjournals.org 5053
on June 6, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 21, 2013; DOI: 10.1158/0008-5472.CAN-12-3775

To systematically examine ERBB–MET cross-talk, we used acellular system enabling analysis of functional interactionsbetween individual ERBBs and MET in isolation or combina-tion. We found that EGFR is sufficient to stabilize and cross-activate MET in the absence of other ERBBs. Activation isindirect, involving MEK/p38MAPK signaling, and is enhancedby ERBB3. This EGFR–MET axis is also active in NSCLC cells,where MET facilitates EGF-induced migration and invasionirrespective of EGFR mutational status. Finally, EGFR–METactivation is enhanced in an experimental model of metastaticNSCLC cells and potentiates brain metastasis.
Materials and MethodsCell culture
32Dmousemyeloid cells [American Type Culture Collection(ATCC)] were grown in RPMI with 10% heat-inactivated FBSand 10% WEHI-3B conditioned medium (CM). WEHI-3B(ATCC) cells were grown in RPMI with 10% heat-inactivatedFBS. For WEHI-CM, WEHI-3B cells were grown in RPMI with1%FBS for 4 days,mediumwas collected, and passed through a0.22 mm filter. A549, H441, H2030, H1650, HCC827, H1975 cells(ATCC), PC9, and PC9-BrM3 cells (15) were grown in RPMIwith 10% FBS. SYF�/� mouse embryonic fibroblasts (MEF;ref. 16) were grown in Dulbecco's Modified Eagle Mediumwith10% FBS. All media contained 1% penicillin/streptomycin,L-glutamine, and sodium pyruvate.
CloningSite-directed mutagenesis was conducted with QuikChange
Multi Site-Directed Mutagenesis Kit (Agilent Technologies)following the manufacturer's protocol. Primers were designedusing the manufacturer's guidelines and obtained from Inte-grated DNA Technologies. The pRK5TKneo-MET-V5/His(Genentech) plasmid was used to make pRK5TKneo-MET-K1110A-V5/His, pRK5TKneo-MET-Y1234/1235F-V5/His, andpRK5TKneo-MET-Y1349/1354F-V5/His.
Plasmids, electroporation, and virus production32D cells were electroporated at 240 V, 1 pulse, 35 milli-
seconds, with pBABE-EGFR (Addgene), pBABE-ERBB3 (NancyHynes, Friedrich-Miescher Institute, Basel, Switzerland),pBABE-EGFR-L858R (Don Nguyen, Yale University Schoolof Medicine, New Haven, CT), pCDNA3(-)-EGFR-L858R/T790M (Katerina Politi, Yale University, New Haven, CT),pRTK-V5/His-Met, pRTK-V5/His-Met-K1110A, pRTK-V5/His-Met-Y1234F/Y1235F, or pRTK-V5/His-Met-Y1349F/Y1354F.Cells were selected and then maintained at 2 and 1 mg/mLpuromycin (R&D Systems) or 100 and 50 mg/mL G418, respec-tively (GIBCO).
Control and MET knockdown lentivirus was producedby cotransfecting HEK 293T cells with pLKO-shRNA con-structs (Sigma-Aldrich), pMD2.G, and psPAX2 (Addgene)using lipofectamine (Invitrogen). Supernatants were col-lected daily for 3 days, combined, and concentrated withCentricon plus-20 filters (Millipore Corporation). Cells wereinfected overnight in 4 mg/mL polybrene, selected andmaintained at 2 and 1 mg/mL puromycin, respectively.
Growth factor stimulation and drug treatmentCells were stimulated with 10 ng/mL EGF, 50 ng/mL neur-
egulin (NRG), or 50 ng/mL hepatocyte growth factor (HGF;R&D Systems) for the indicated times. Kinase inhibitors suni-tinib, imatinib, nilotinib, tozasertib, gefitinib, U0126, SB203580,dasatinib (LC Laboratories); BMS-754807, AZD-4547, BI2536(ChemieTek); BMS-536924, MK2206, PLX4032, PD0332991(Sellek Chemicals); PP2, AR-A014118 (Sigma-Aldrich); Jnkinhibitor II, Syk inhibitor II (EMD Chemicals); PF-573208,CMPD1, BRD7389, SB41542, PHA665752 (Tocris); AZD-7762,NSC625982 (Axon Medchem); CIP-1374 (AlloStem Therapeu-tics); and PP2 (Calbiochem) were used at 1 mmol/L for theindicated times. Actinomycin D (Sigma) was used at 10 mg/mL,cycloheximide (Sigma) at 10 ng/mL, and bortezomib (LCLaboratories) at 100 nmol/L.
ImmunoprecipitationCells were lysed in TX100 lysis buffer (20 mmol/L Tris–HCl
pH 7.5, 150 mmol/L NaCl, 1 mmol/L EGTA, 1 mmol/L EDTA,and 1% Triton X-100). Immunoprecipitations were conductedovernight at 4�Cwith 2mg protein lysate, 4mg anti-Met or anti-Cbl antibody (Santa Cruz Biotechnology), and Protein A/GUltralink Resin (Invitrogen), washed three times in TX100buffer, and resuspended in 2� Laemmli sample buffer.
ImmunoblottingCell lysates were prepared in 2� sample buffer or NETN lysis
buffer [150 mmol/L NaCl, 1 mmol/L EDTA, 50 mmol/L Tris–HCl pH 7.8, 1% NP40 substitute (Fluka)]. Immunoblots onpolyvinylidene difluoride (PVDF) were blocked in 5% nonfatmilk/PBST (Dulbecco's PBS, 0.1% Tween-20). Antibodiesagainst phospho-EGFR-Y1068, phospho-ERBB3-Y1197, phos-pho-MET-Y1003, phospho-MET-Y1234/1235, phospho-Akt-S473, Akt, phospho-p44/42MAPK(Erk1/2)-T202/Y204, p44/42MAPK(Erk1/2), phospho-p38MAPK-T180/Y182, p38MAPK(Cell Signaling Technology); EGFR, ERBB3, MET, ubiquitin,cCBL, GAPDH (SantaCruz Biotechnology); andV5 (Invitrogen)were incubated overnight at 4�C in 5% milk/PBST. Anti-phos-pho-tyrosine (Cell Signaling Technology) antibody was incu-bated 3 hours at room temperature. Membranes were washedin PBST and incubated 1 hour in horseradish peroxidase–conjugated secondary antibodies in 5% milk/PBST.
RNA isolation and real-time PCRRNA was isolated using the RNeasy Mini Plus Kit with
QIAshredder columns (Qiagen) and cDNA was synthesizedusing the iScript Kit (Bio-Rad). Real-time PCR (RT-PCR) wasconducted on a Bio-Rad iCycler RT-PCR machine by combin-ing cDNA, TaqMan Universal Master Mix, and premixed FAM-labeled probes (Applied Biosystems). mRNA quantity wascalculated relative to glyceraldehyde-3-phosphate dehydroge-nase (GAPDH) using the 2�DDCt method.
Migration, invasion, and growth assaysFor migration assays, cells were preincubated 3 hours with
10 ng/mL EGF, 1 mmol/L PHA665752, or 1 mmol/L PF-04217903, then plated at 5� 104 cells per well in 24-well plateswith 8-mm filter inserts (BD Biosciences). Conditions were
Breindel et al.
Cancer Res; 73(16) August 15, 2013 Cancer Research5054
on June 6, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 21, 2013; DOI: 10.1158/0008-5472.CAN-12-3775

maintained and cells migrated overnight from 0.1% FBStoward 10% FBS. Cells were stained and cell number wasaveraged from threefields of viewper chamber. Invasion assayswere conducted similarly but with 1.4 � 105 cells per well inMatrigel invasion chambers (BD Biosciences). For growthassays, cells were plated at 1 � 104 cells per well in a 12-welldish and counted daily for 5 days. Results are the average of atleast three replicates and P values were calculated by t test.
Colony formation assayCells were plated at 2,000 cells per well in a 6-well dish and
grown for 7 days, then washed in PBS, fixed 10 minutes on icewith coldmethanol, and stained 2 hourswith 0.1% crystal violet/PBS. Cells were rinsed 30minutes in PBS and air-dried. Colonieswere counted using scanned images with ImageJ software.
In vivo metastasis assayAnimal procedures were carried out in accordance with the
Yale Institutional Animal Care and Use Committee. A total of2.5� 104 cells were suspended in 0.1 mL PBS and injected intothe right ventricle of 6-week-old nude athymic mice. Metas-tasis was detected by bioluminescence with an IVIS Spectrum(Perkin Elmer) as previously described (17). Tumor incidencewas quantified by luminescent signal in limbs, spine, or brainin whole body images at given time points and presentedas Kaplan–Meier curves. P values were calculated by log-ranktest. When possible, animals were sacrificed for harvest oftissues to confirm metastasis by quantifying organ-specificluminescence.
ResultsSignaling through EGFR leads to delayed activation ofMETDiscrepancies in ERBB–MET interactions among NSCLC
cell lines arise from underlying differences in ERBBmutationalstatus and relative expression levels. Because ERBBs cross-activate, it was uncertain which individual ERBB(s) interactwith MET or if intermediary signaling pathways indirectlyintegrate MET with ERBB signaling. To distinguish thesepossibilities, we used 32D cells that lack ERBBs and MET.Because epithelial cell lines express one or more ERBBs, use ofmurine hematopoietic 32D cells has been invaluable for elu-cidating ERBB interactions in reconstruction experiments (18,19). RT-PCR verified that 32D cells are devoid of EGFR, ERBB2,ERBB3, ERBB4, MET, and agonists EGF, TGF-a, AREG, BTC,NRG1, and HGF, consistent with previous studies (data notshown; refs. 18, 19). We then ectopically expressed EGFR andMET alone or in combination (Fig. 1A).In 32D cells that are engineered to express human EGFR or
MET singly, EGF stimulated EGFR phosphorylation and HGFstimulated MET phosphorylation, as expected (Supplemen-tary Fig. S1A). EGF did not induce MET phosphorylation andHGF did not induce EGFR phosphorylation, verifying spec-ificity of ligands for their receptors (Supplementary Fig. S1A).In cells expressing both EGFR and MET (EGFR/MET), HGFfailed to induce phosphorylation of EGFR at Y1068 (Supple-mentary Fig. S1B), however, EGF increased MET phosphor-
ylation and protein levels (Fig. 1B). Although EGF inducesrapid phosphorylation of EGFR, EGF-induced phosphoryla-tion of MET was delayed, occurring after 2 hours and increas-ing through 10 hours (Fig. 1B). EGF stimulation inducedan increase in total MET levels without affecting the abun-dance of MET mRNA (Fig. 1C). Also, quantification of phos-pho-MET to total MET revealed increased stoichiometry ofMET phosphorylation induced by EGF (Fig. 1D). Overall,EGFR upregulates MET abundance and relative Tyr phos-phorylation over an extended time frame, and does notrequire other ERBB family receptors.
EGFR and MET kinases are required for EGF-dependentactivation of MET
EGF-induced MET phosphorylation could occur throughdirect phosphorylation of MET by EGFR, EGF-dependentactivation of MET autophosphorylation, or another interme-diary kinase. To investigate the importance of kinase activity inthis receptor interaction, small-molecule TKIs gefitinib andPHA665752 were used to inhibit EGFR and MET, respectively.Specificity of inhibitors for the cognate receptors was verified(Supplementary Fig. S2A and S2B). Although slight changes inphospho-EGFR were seen with single inhibitors, these changesreflected similar differences in total protein level and were notseen consistently (Fig. 1E). Treatment of EGFR/MET 32D cellswith gefitinib prevents EGF-induced MET phosphorylation,while PHA665752 completely abolishes both EGF-induced andbasal phosphorylation of MET (Fig. 1E). Similarly, EGF wasunable to induce robust phosphorylation of kinase-inactiveMET mutants K1110A and Y1234F/Y1235F, while still activat-ing wild-type MET receptor and docking site-mutant Y1349F/Y1354F (Supplementary Fig. S2C). Hence, EGF-induced METphosphorylation in 32D cells requires activity of both EGFRand MET kinases.
Enhanced MET signaling is one mechanism by whichNSCLC patients with EGFR-activating mutations, such asEGFR L858R, become resistant to therapy with the EGFRinhibitor erlotinib (2–4). Recent evidence suggests that METand activated EGFR also interact in drug-na€�ve lung cancercells independent of drug-resistance mechanisms (6–9, 12). Todetermine if activated EGFR alleles commonly found in NSCLCactivate MET, MET was expressed in 32D cells in combinationwith EGFR L858R. Similar to wild-type EGFR, EGFR L858Rinduced MET phosphorylation after 4 hours of EGF stimula-tion, and activation was prevented by gefitinib (Fig. 1F). Theseresults show for the first time that EGFR activates METindependent of other ERBB family members, and that bothwild-type and activated EGFR induce MET phosphorylation.
ERBB3 enhances EGFR-induced MET phosphorylationERBB3 is nearly devoid of intrinsic catalytic activity, and
functions through heteromers with other ERBBs (20, 21).Although ERBB3 phosphorylation can be dependent on METin gefitinib-resistant NSCLC cells, it is unclear whether ERBB3and MET interact in drug-na€�ve cells or if another ERBBfamily member is required (1). To investigate signaling inter-actions between ERBB3 and MET, ERBB3 was coexpressedwithMET alone or with EGFR/MET in 32D cells. Coexpression
EGFR–MET Signaling in NSCLC Metastasis
www.aacrjournals.org Cancer Res; 73(16) August 15, 2013 5055
on June 6, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 21, 2013; DOI: 10.1158/0008-5472.CAN-12-3775

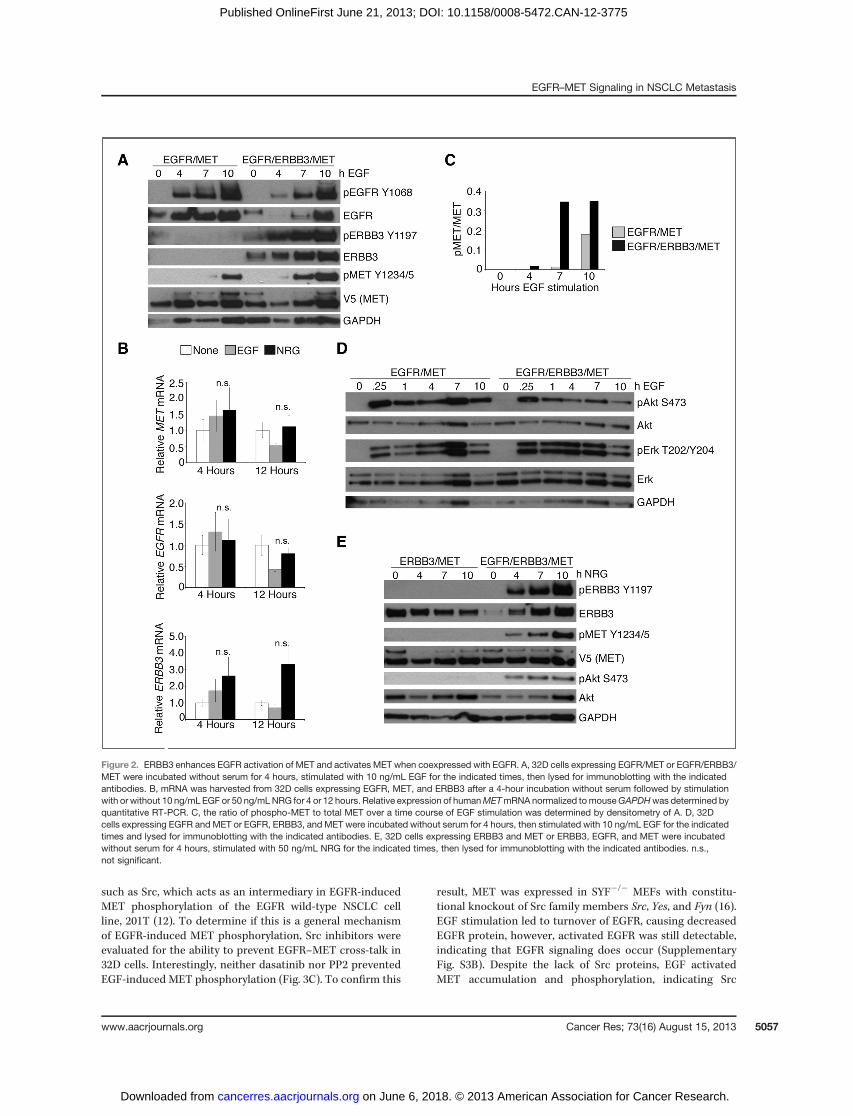
of ERBB3 enhanced EGF-induced MET phosphorylation com-pared with EGFR/MET alone (Fig. 2A). Similar to EGFR/METcells, stimulation of EGFR/ERBB3/MET cells with ERBBligands did not affect MET mRNA levels (Fig. 2B). IncreasedEGF-dependent phospho-MET relative to total MET occurswithin 4 hours in EGFR/ERBB3/MET cells and is maximal by7 hours (Fig. 2C). Hence, ERBB3 enhances EGF-inducedMET phosphorylation, likely through signal amplification andbroadening by EGFR/ERBB3 heteromers. In fact, coexpressionof EGFR and ERBB3 alters EGF-induced downstream signal-ing, leading to decreased phospho-Akt, increased phospho-Erk, and possibly increased total extracellular signal–regulat-ed kinase (Erk; Fig. 2D).
Having determined that ERBB3 alters signaling to MET byEGFR, we investigated whether MET can be activated byERBB3 alone. Stimulation of ERBB3/MET cells with the ERBB3ligand NRG1 for up to 10 hours failed to activate MET or Akt(Fig. 2E). Lack of ERBB3/MET signaling activity suggests thatthese receptors cannot act as direct dimerization partners.However, when EGFR is coexpressed in ERBB3/MET cells, NRGinduces MET phosphorylation (Fig. 2E), indicating that ERBB3can induce MET phosphorylation when it has a catalyticallyactive signaling partner. Therefore, MET cannot act as afunctional dimerization partner for ERBB3, but ligand-activat-ed ERBB3 is able to induce MET phosphorylation when coex-pressed with EGFR.
EGFR activates MET through protein stability andMAPKsignaling in 32D cells
EGFR may activate MET through increased autocrine METsignaling by increasing transcription or stability of MET orHGF. EGF-inducedMET activation is actinomycin D–sensitiveand thus requires transcription (Supplementary Fig. S3A).However, MET activation was not associated with changes inMETmRNA levels (Fig. 1C) and HGFmRNA was not expressed(data not shown). Therefore, transcriptional dependency in32D cells involves another component of the pathway.
The changes observed in MET levels without an increase inMETmRNA suggest instead that EGFR stabilizes MET protein.32D cells expressing MET or EGFR/MET were treated withthe protein synthesis inhibitor, cycloheximide, and the half-lifeof MET protein was evaluated. EGF failed to stabilize MET incells expressing MET alone, as expected (Fig. 3A). However,MET was stabilized by coexpression of EGFR and furtherstabilized when EGFR was activated by EGF (Fig. 3A). Over-expression of EGFR increased the MET half-life from 3.5 to5.5 hours, whereas EGF-stimulation of EGFR further increasedthe half-life to 6.3 hours (Fig. 3B). It is likely that the increasein MET half-life by EGFR expression is caused by basal EGFRactivity from high expression levels. Overall, MET protein isstabilized by EGFR signaling in 32D cells.
In addition to regulating MET protein stability, it ispossible that EGFR activates MET through signaling proteins
Figure 1. EGFR activation increases MET protein and phosphorylation in 32D cells. A, 32D cells were transfected with constructs for expression of humanEGFR or MET, and protein expression was tested by immunoblotting. B, 32D cells expressing EGFR andMET were incubated without serum for 4 hours andthen stimulated with 10 ng/mL EGF for the indicated times. Following stimulation, cells were lysed and analyzed by immunoblotting with the indicatedantibodies. C, mRNA was collected from 32D cells after a 4-hour incubation without serum followed by stimulation with or without 10 ng/mL EGF for 4 or 12hours. Relative expression of human MET mRNA normalized to mouse GAPDH was determined by quantitative RT-PCR. D, the ratio of phospho-METto totalMETagainst duration of EGFstimulationwasdeterminedby densitometry of B. E, EGFR/MET32Dcellswere incubatedwithout serum for 4 hours, thenincubated for 4 hours with 1 mmol/L PHA665752, 1 mmol/L gefitinib, and/or 10 ng/mL EGF as indicated and lysed for immunoblotting with the indicatedantibodies. F, EGFR L858R/MET 32D cells were incubated for 4 hours without serum, then incubated with 1 mmol/L gefitinib and/or 10 ng/mL EGF for4 hours and lysed for immunoblotting with the indicated antibodies. n.s., not significant.
Breindel et al.
Cancer Res; 73(16) August 15, 2013 Cancer Research5056
on June 6, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 21, 2013; DOI: 10.1158/0008-5472.CAN-12-3775
such as Src, which acts as an intermediary in EGFR-inducedMET phosphorylation of the EGFR wild-type NSCLC cellline, 201T (12). To determine if this is a general mechanismof EGFR-induced MET phosphorylation, Src inhibitors wereevaluated for the ability to prevent EGFR–MET cross-talk in32D cells. Interestingly, neither dasatinib nor PP2 preventedEGF-induced MET phosphorylation (Fig. 3C). To confirm this
result, MET was expressed in SYF�/� MEFs with constitu-tional knockout of Src family members Src, Yes, and Fyn (16).EGF stimulation led to turnover of EGFR, causing decreasedEGFR protein, however, activated EGFR was still detectable,indicating that EGFR signaling does occur (SupplementaryFig. S3B). Despite the lack of Src proteins, EGF activatedMET accumulation and phosphorylation, indicating Src
Figure 2. ERBB3 enhances EGFR activation of MET and activates MET when coexpressed with EGFR. A, 32D cells expressing EGFR/MET or EGFR/ERBB3/MET were incubated without serum for 4 hours, stimulated with 10 ng/mL EGF for the indicated times, then lysed for immunoblotting with the indicatedantibodies. B, mRNA was harvested from 32D cells expressing EGFR, MET, and ERBB3 after a 4-hour incubation without serum followed by stimulationwith or without 10 ng/mL EGF or 50 ng/mLNRG for 4 or 12 hours. Relative expression of humanMETmRNA normalized tomouseGAPDHwas determined byquantitative RT-PCR. C, the ratio of phospho-MET to total MET over a time course of EGF stimulation was determined by densitometry of A. D, 32Dcells expressing EGFR and MET or EGFR, ERBB3, and MET were incubated without serum for 4 hours, then stimulated with 10 ng/mL EGF for the indicatedtimes and lysed for immunoblotting with the indicated antibodies. E, 32D cells expressing ERBB3 and MET or ERBB3, EGFR, and MET were incubatedwithout serum for 4 hours, stimulated with 50 ng/mL NRG for the indicated times, then lysed for immunoblotting with the indicated antibodies. n.s.,not significant.
EGFR–MET Signaling in NSCLC Metastasis
www.aacrjournals.org Cancer Res; 73(16) August 15, 2013 5057
on June 6, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 21, 2013; DOI: 10.1158/0008-5472.CAN-12-3775

family proteins are not absolutely required for EGF-inducedMET phosphorylation (Supplementary Fig. S3B). Thus,although Src is involved in EGFR-induced MET activationin some contexts, it is not a unique mediator of and is notrequired for EGFR–MET cross-talk.
To identify intermediaries in EGF-induced MET phosphor-ylation in 32D cells, pools of protein kinase inhibitors weretested for interference with this process (Supplementary Fig.S3C). Of these, inhibitor pools A, C, and D prevented EGF-induced MET phosphorylation. Pool C yielded inconsistentresults among experiments, and only partially inhibited Metphosphorylation. Pool A includes at least one agent,BMS754807, which is now reported to have some inhibitoryactivity on MET. For these reasons, we focused on pool D,composed of mitogen-activated protein kinase (MAPK) sig-naling inhibitors.
To evaluate the role of MAPK in EGFR-MET signal-ing, inhibitors of MEK1/2 and p38MAPK, U0126 andSB203580, were tested for ability to prevent EGFR-METcrosstalk. These inhibitors had moderate or no effect on
EGF-induced MET phosphorylation singly, but nearlycompletely prevent it when combined (Fig. 3D). This sug-gests that EGF-induced MET phosphorylation worksthrough the RAS–ERK–p38MAPK pathway, and compensa-tion occurs through parallel MAPK cascades when only oneis inhibited.
Although gefitinib inhibits EGF-induced MET phosphor-ylation in cells with wild-type EGFR or EGFR L858R (Fig. 1Eand F), it is ineffective against the resistance mutation,T790M. To determine if MEK and p38MAPK inhibitorsprevent EGFR activation of MET in cells with T790M muta-tions, EGFR L858R/T790M (LT) receptors were coexpressedwith MET in 32D cells. Gefitinib did not affect EGFR or METphosphorylation in these cells, but the combination ofU0126/SB203580 substantially decreased MET phosphoryla-tion (Fig. 3E). Hence, MEK/p38MAPK inhibitors can inhibitEGFR–MET signaling in cells where gefitinib is not effective.Overall, EGFR activation of MET in 32D cells occurs throughincreased stability of MET protein and intermediary signal-ing through MAPK.
Figure 3. EGFR signaling stabilizesMET protein and induces METphosphorylation through MAPKsignaling. A, 32D cells expressingMET or EGFR and MET wereincubated without serum for 4hours, pretreated with or without10 ng/mL EGF for 15 minutes,then incubated with 10 ng/mLcycloheximide (CHX) for theindicated times and lysed. TotalMET (V5) and GAPDH levels wereanalyzedby immunoblotting. B, thehalf-life of MET was calculated onthe basis of densitometry of A.Results from 3 experiments wereaveraged and P values werecalculated by t test. C, EGFR/MET32D cells were incubated withoutserum for 4 hours, incubatedwith 1mmol/L gefitinib (Gef), dasatinib(Das), or PP2, incubated with orwithout 10 ng/mL EGF for 4 hours,and lysed for immunoblotting withthe indicated antibodies. D, 32Dcells expressing EGFR and MET orE, EGFR L858R/T790M and METwere incubatedwithout serum for 4hours, incubated with 1 mmol/L ofthe indicated agents, stimulatedwith 10 ng/mL EGF for 4 hours,then lysed for immunoblotting withthe indicated antibodies.
Breindel et al.
Cancer Res; 73(16) August 15, 2013 Cancer Research5058
on June 6, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 21, 2013; DOI: 10.1158/0008-5472.CAN-12-3775

EGFR regulatesMETatmultiple levels inNSCLC cell linesTo confirm the presence of EGFR–MET cross-talk in NSCLC
cell lines, the EGFR-mutant cell line, HCC827, was treated withgefitinib for up to 24 hours. Gefitinib treatment induced rapiddecreases in MET phosphorylation and delayed decreases inMET protein levels (Fig. 4A). These different rates of changesuggest that EGFR can affect MET phosphorylation indepen-dent of MET protein levels. Unlike 32D cells, EGFR signaling inHCC827 cells regulates MET protein and MET mRNA withsimilar kinetics following gefitinib treatment (Fig. 4B and C),emphasizing the complex dynamics of EGFR–MET signaling inNSCLC cell lines. Similar changes in MET phosphorylation,protein, and mRNA were observed following gefitinib treat-ment of another EGFR-mutant cell line, PC9 (SupplementaryFig. S4).Although changes in MET protein levels following gefitinib
treatment may be caused by decreased mRNA abundance,there is evidence for EGFR-dependent modulation of MET atthe protein level through MET ubiquitination. Following stim-ulation by HGF, MET is ubiquitinated by the E3 ligase, c-CBL,and internalized for intracellular trafficking or degradation(22–25). Although three cell lines with wild-type EGFR under-went robust MET ubiquitination following ligand stimulation,three of four cell lines with EGFR-activating mutations did not(Fig. 4D). Importantly,METubiquitinationwas associatedwithphosphorylation of c-CBL (Fig. 4D). This corroborates ourfinding that, in 32D cells, EGFR stabilizes MET protein levels
independent of transcriptional changes and indicates thatEGFR modulates MET at multiple levels in NSCLC cell lines.
To determine if intermediary signaling cross-talk occursthrough MAPK in NSCLC cell lines, HCC827 cells were treatedwith U0126/SB203580. Interestingly, treatment with eithergefitinib or U0126/SB203580 caused a reduction in total METlevels, accompanied by elimination of phospho-MET (Fig. 4E).This confirms that signaling observed in 32D cells resemblesNSCLC cell lines and that MEK/p38MAPK pathways act assignaling intermediaries between EGFR and MET in multiplecell types. Although NSCLC cells display a more complicatedrelationship between EGFR and MET than 32D cells, NSCLCcells similarly show independent regulation of MET proteinand phosphorylation by EGFR signaling. Importantly, theexistence of EGFR–MET cross-talk in multiple NSCLC celllines indicates that EGFR-induced MET activation may havea biologic significance.
MET regulates EGFR-induced migration and invasion inNSCLC cells
MET regulates many cellular phenotypes, including cellgrowth and motility. We compared EGFR-only with EGFR/MET 32D cells and found that EGFR–MET cross-talk had noeffect on cell viability (data not shown). BecauseMET signalingis also important for cell motility, it is possible that EGFRactivation of MET modulates this phenotype. MET is requiredfor EGF-induced cell invasion and motility in EGFR wild-type
Figure 4. EGFR regulates MET phosphorylation, protein, and mRNA levels in NSCLC cells. A, HCC827 cells were incubated with 1 mmol/L gefitinib for theindicated times then lysed for immunoblotting with the indicated antibodies. B, MET protein levels were determined by densitometry of A and normalized toGAPDH. Results were averaged among 3 experiments and P values were calculated by t test. C, HCC827 cells were incubated with 1 mmol/L gefitinibfor the indicated times. mRNA was isolated and the relative expression of humanMETmRNA normalized to human GAPDH was determined by quantitativeRT-PCR. D, NSCLC cell lines with the indicated EGFRmutations were incubated overnight without serum then with 50 ng/mL HGF for 10minutes asmarked.Cells were lysed and MET or Cbl immunoprecipitations (IP) were conducted followed by immunoblotting with the indicated antibodies. E, HCC827cells were incubated overnight with 1 mmol/L of the indicated agents and lysed for immunoblotting with the indicated antibodies. �, P < 0.05.
EGFR–MET Signaling in NSCLC Metastasis
www.aacrjournals.org Cancer Res; 73(16) August 15, 2013 5059
on June 6, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 21, 2013; DOI: 10.1158/0008-5472.CAN-12-3775

NSCLC cells but it was unclear if this is true in cells with EGFR-activating mutations (12). Hence, we confirmed the previouslyreported MET-dependence for EGF-induced migration andinvasion in the EGFR wild-type NSCLC cell line A549 (Supple-mentary Fig. S5A) and tested additional cell lines with variousEGFR mutations. Indeed, PHA665752 prevented EGF-inducedmigration of NSCLC cell lines with both wild-type and acti-vated EGFR (Fig. 5A). Similarly, MET inhibition preventedEGF-induced invasion through Matrigel of both EGFR wild-type and mutant NSCLC cells (Fig. 5B). This was confirmedwith an additional highly specific MET inhibitor, PF-04217903,which has strong activity in NSCLC cells (Supplementary Fig.S5B and S5C). Therefore, EGFR–MET signaling is active inNSCLC cell lines withmultiple EGFR-activating mutations andmediates aggressive phenotypes including migration andinvasion.
EGFR–MET signaling is important for metastaticbehavior
MET promotes EGF-induced NSCLC cell invasion, so it mayhave a role in the biology of metastatic lung cancer cellsaddicted to EGFR. To investigate this, we compared EGFR–MET signaling in an EGFR-mutant NSCLC cell line, PC9, and itsmetastatic subpopulation, PC9-BrM3 (15, 26, 27). Both parentalPC9 and PC9-BrM3 cell lines harbor constitutively active EGFRD746–750 and are dependent on EGFR signaling for survival.However, PC9-BrM3 cells have a marked increase in thecapacity to invade and colonize distant organs, most notablythe brain (15).
EGFR inhibition in both PC9 and PC9-BrM3 cells decreasesMET phosphorylation and protein levels, confirming the pres-ence of EGFR–MET signaling in these cells (Fig. 6A). However,
the mechanism of EGFR–MET signaling differs, as MEK/p38MAPK inhibition reduces MET phosphorylation and pro-tein levels in PC9-BrM3 cells, but has little effect on PC9 cells(Fig. 6A). Different requirements for MAPK in EGFR–METsignaling may correlate with lower Erk phosphorylationobserved in PC9 compared with PC9-BrM3 cells (Fig. 6B). Inaddition,mRNA levels of bothEGFR andMET trended higher inPC9-BrM3 than PC9 cells (Fig. 6C). Although possibly coinci-dental, these differences in EGFR–MET signaling raised thepossibility that this pathway contributes to the highly meta-static behavior of PC9-BrM3 cells.
To directly evaluate the role of MET in EGFR-driven phe-notypes of PC9 and PC9-BrM3 cells, a lentiviral system wasused to induce MET knockdown (Supplementary Fig. S6A).Similar to growth studies in 32D cells, MET knockdown did notaffect growth of PC9 or PC9-BrM3 cells (Fig. 6D). In contrastwith other NCSLC cells, MET was not required for EGF-induced cell invasion of parental PC9 cells and knockdowninconsistently affected cell migration (Fig. 6E). Interestingly,MET knockdown prevented both EGF-induced cell migrationand invasion in PC9-BrM3 cells (Fig. 6E). This was confirmedwith PHA665752 treatment, showing consistent cellularresponses to MET inhibition and knockdown (SupplementaryFig. S6B). The role of MET in clonogenic potential also differedbetween the cell lines. MET knockdown with two differentshort hairpin RNAs (shRNA) yielded inconsistent changes inclonogenic colony formation of PC9 cells, suggesting theseeffects were notMET specific (Fig. 6F). However, knockdown ofMET in PC9-BrM3 cells consistently reduced clonogenic col-ony formation (Fig. 6F). Although EGFR–MET cross-talk existsin parental PC9 cells, the functional consequence of EGFR–MET cross-talk is different in metastatic cells where it
Figure 5. MET promotes EGF-induced cell migration and invasion of NSCLC cell lines. NSCLC cell lines were pretreated for 3 hours with 10 ng/mL EGF (EGF),1 mmol/L PHA665752 (PHA), or both (EGFþPHA), then plated in migration or invasion chambers and allowed to migrate overnight. Results are representedrelative to no treatment controls (none) and are the average of at least 3 experiments. P values were calculated by t test. wt, wild-type.
Breindel et al.
Cancer Res; 73(16) August 15, 2013 Cancer Research5060
on June 6, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 21, 2013; DOI: 10.1158/0008-5472.CAN-12-3775

enhances clonogenicity and invasion, independently of cellsurvival. Thus, although NSCLC cells generally require mutantEGFR signaling for tumorigenesis, cross-talk with MET mayfurther enhance metastatic progression.MET signaling modulates metastasis in many epithelial
cancers (28–31). In patients with NSCLC, MET expression andphosphorylation correlate with the incidence of brain meta-stasis, but the functions of MET in lung cancer metastasisremain uncharacterized (32). A rate-limiting step in metastasisis invasion and colonization of distant organs by cancercells following dissemination into the bloodstream (33). To
assess effects of EGFR–MET signaling on metastatic coloniza-tion, we injected luciferase-marked PC9-BrM3 cells into thearterial circulation of immunocompromised mice. Incidenceand burden of metastasis were compared following intracar-diac injection of PC9-BrM3 cells stably expressing control orMETknockdownvectors. Surprisingly, no significant differencewas seen in overall tumor burden between control and METknockdown (Fig. 7A). Although MET knockdown had no effecton bone metastasis, incidence of brain metastasis was sig-nificantly delayed and/or attenuated (Fig. 7B and C). Whenpossible, animals were sacrificed to confirm metastasis by
Figure 6. EGFR–MET signaling is enhanced in metastatic cells. A, PC9 or PC9-BrM3 cells were incubated for 24 hours with 1 mmol/L of the indicatedagents then lysed for immunoblotting with the indicated antibodies. B, PC9 or PC9-BrM3 cells were lysed and immunoblotting was conducted withthe indicated antibodies. C, RNA was isolated from PC9 and PC9-BrM3 cells and expression levels of EGFR and MET mRNA normalized toGAPDH were determined by quantitative RT-PCR. D, growth of PC9 and PC9-BrM3 cells with control (shScr) or MET knockdown was counted over5 days and is represented as cell number averaged among 3 experiments. E, PC9 and PC9-BrM3 cells with control (shScr) or MET knockdownwere pretreated with or without 10 ng/mL EGF for 3 hours then plated in migration or invasion chambers and allowed to migrate overnight. Results arerepresented as EGF-induced migration or invasion relative to shScr and are the average of at least 3 experiments. F, clonogenic colony formationof PC9 and PC9-BrM3 cells with control (shScr) or MET knockdown was tested by plating cells at low density and staining for colonies after 6days of growth. The number of colonies was quantified using ImageJ software and is averaged among at least 6 wells. Representative stained plates areshown. A–F, P values were calculated by t test; �, P < 0.05; ��, P < 0.01.
EGFR–MET Signaling in NSCLC Metastasis
www.aacrjournals.org Cancer Res; 73(16) August 15, 2013 5061
on June 6, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 21, 2013; DOI: 10.1158/0008-5472.CAN-12-3775
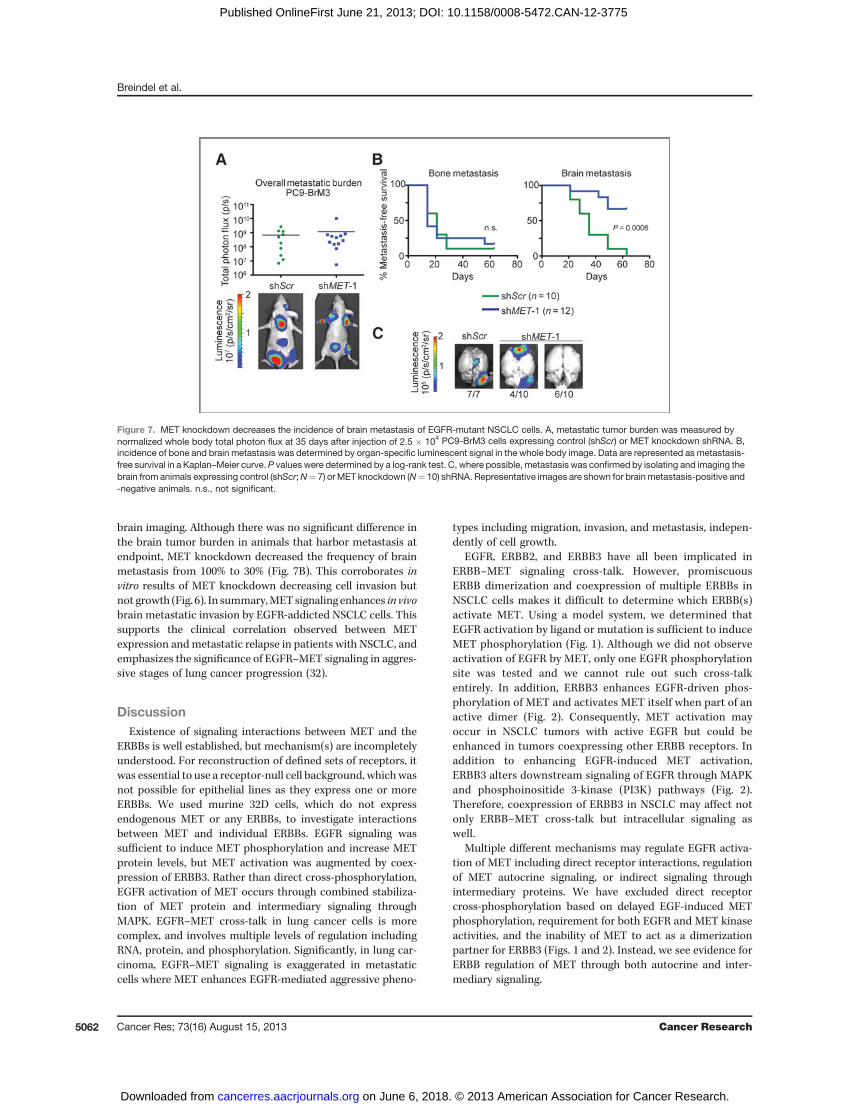
brain imaging. Although there was no significant difference inthe brain tumor burden in animals that harbor metastasis atendpoint, MET knockdown decreased the frequency of brainmetastasis from 100% to 30% (Fig. 7B). This corroborates invitro results of MET knockdown decreasing cell invasion butnot growth (Fig. 6). In summary,METsignaling enhances in vivobrain metastatic invasion by EGFR-addicted NSCLC cells. Thissupports the clinical correlation observed between METexpression andmetastatic relapse in patients with NSCLC, andemphasizes the significance of EGFR–MET signaling in aggres-sive stages of lung cancer progression (32).
DiscussionExistence of signaling interactions between MET and the
ERBBs is well established, but mechanism(s) are incompletelyunderstood. For reconstruction of defined sets of receptors, itwas essential to use a receptor-null cell background, whichwasnot possible for epithelial lines as they express one or moreERBBs. We used murine 32D cells, which do not expressendogenous MET or any ERBBs, to investigate interactionsbetween MET and individual ERBBs. EGFR signaling wassufficient to induce MET phosphorylation and increase METprotein levels, but MET activation was augmented by coex-pression of ERBB3. Rather than direct cross-phosphorylation,EGFR activation of MET occurs through combined stabiliza-tion of MET protein and intermediary signaling throughMAPK. EGFR–MET cross-talk in lung cancer cells is morecomplex, and involves multiple levels of regulation includingRNA, protein, and phosphorylation. Significantly, in lung car-cinoma, EGFR–MET signaling is exaggerated in metastaticcells where MET enhances EGFR-mediated aggressive pheno-
types including migration, invasion, and metastasis, indepen-dently of cell growth.
EGFR, ERBB2, and ERBB3 have all been implicated inERBB–MET signaling cross-talk. However, promiscuousERBB dimerization and coexpression of multiple ERBBs inNSCLC cells makes it difficult to determine which ERBB(s)activate MET. Using a model system, we determined thatEGFR activation by ligand or mutation is sufficient to induceMET phosphorylation (Fig. 1). Although we did not observeactivation of EGFR by MET, only one EGFR phosphorylationsite was tested and we cannot rule out such cross-talkentirely. In addition, ERBB3 enhances EGFR-driven phos-phorylation of MET and activates MET itself when part of anactive dimer (Fig. 2). Consequently, MET activation mayoccur in NSCLC tumors with active EGFR but could beenhanced in tumors coexpressing other ERBB receptors. Inaddition to enhancing EGFR-induced MET activation,ERBB3 alters downstream signaling of EGFR through MAPKand phosphoinositide 3-kinase (PI3K) pathways (Fig. 2).Therefore, coexpression of ERBB3 in NSCLC may affect notonly ERBB–MET cross-talk but intracellular signaling aswell.
Multiple different mechanisms may regulate EGFR activa-tion of MET including direct receptor interactions, regulationof MET autocrine signaling, or indirect signaling throughintermediary proteins. We have excluded direct receptorcross-phosphorylation based on delayed EGF-induced METphosphorylation, requirement for both EGFR and MET kinaseactivities, and the inability of MET to act as a dimerizationpartner for ERBB3 (Figs. 1 and 2). Instead, we see evidence forERBB regulation of MET through both autocrine and inter-mediary signaling.
Figure 7. MET knockdown decreases the incidence of brain metastasis of EGFR-mutant NSCLC cells. A, metastatic tumor burden was measured bynormalized whole body total photon flux at 35 days after injection of 2.5 � 104 PC9-BrM3 cells expressing control (shScr) or MET knockdown shRNA. B,incidence of bone and brain metastasis was determined by organ-specific luminescent signal in the whole body image. Data are represented as metastasis-free survival in a Kaplan–Meier curve. P values were determined by a log-rank test. C, where possible, metastasis was confirmed by isolating and imaging thebrain from animals expressing control (shScr;N¼ 7) orMET knockdown (N¼ 10) shRNA. Representative images are shown for brainmetastasis-positive and-negative animals. n.s., not significant.
Breindel et al.
Cancer Res; 73(16) August 15, 2013 Cancer Research5062
on June 6, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 21, 2013; DOI: 10.1158/0008-5472.CAN-12-3775

In some cell lines, EGFR activates MET by inducing METtranscription, thereby increasing the concentration of MET atthe membrane, leading to more receptor collisions, homodi-mer formation, and MET activation (12, 34, 35). In our modelsystem, EGFR regulates MET at the protein level by extendingthe MET half-life (Fig. 3), which would also increase METavailability and enhance autocrine activation. In papillary renalcarcinoma, MET mutations lead to increased recycling anddecreased degradation, causing increased activation of METthat drives tumorigenesis (36). We see decreased MET ubiqui-tination in EGFR-mutant NSCLC cells, which would similarlyalter MET recycling and degradation (Fig. 4). Therefore, activeEGFR in NSCLC tumors may regulate MET through alteredreceptor trafficking to enhance already robust tumorigenicsignaling.Dulak and colleagues reported that Src is required for EGFR-
induced MET activation independent of MET transcription,indicating MET can also be regulated through intermediarysignaling pathways (12). We found that MAPK signaling cor-related with higher ERBB-driven MET phosphorylation andwas required for EGFR-dependentMET activation in cells withboth wild-type and mutant EGFR (Figs. 3 and 4). Differentrequirements for Src and MAPK signaling may depend on thecell line, and it is possible that both pathways act as inter-mediates in different contexts. In addition, Src can enhanceactivation of MAPK in cancer cells directly or by binding toEGFR, so these pathways may have additive functions (37).Investigation of additional experimental models and tumorsamples could reveal the relative roles of these proteins inEGFR–MET cross-talk in different cell types and contexts.Existence of EGFR–MET signaling in multiple normal and
cancer cell lines suggests that MET modulates biologic out-comes of EGFR signaling. Although both receptors activatePI3K/Akt signaling, MET was not required for cell survival in32D or NSCLC cells. EGFR and MET also commonly regulatecell motility andMET promotes EGF-induced cell motility andinvasion in EGFR wild-type NSCLC cells (8, 12). NSCLC cellswith EGFR mutations have constitutively hyperactive EGFRsignaling and itwas unclear ifMETcould regulate EGFR-drivenphenotypes in these cells. We found, however, that METfacilitates EGF-induced migration and invasion in EGFR-mutant NSCLC cells (Fig. 5), showing that MET signalingenhances other aggressive phenotypes of EGFR-mutant lungcancers.Moreover, we discovered differences in EGFR–MET cross-
talk in NSCLC cell lines of varying metastatic potential. Inmetastatic PC9-BrM3 derivatives, but not parental PC9 cells,EGFR–MET signaling enhanced migration, invasion, and col-ony formation, and was dependent onMAPK signaling (Fig. 6).Differences between PC9 and PC9-BrM3 cells reveal context-dependent requirements for EGFR–MET signaling in meta-static NSCLC cells. Because PC9-BrM3 cells were selectedto be highly metastatic in vivo, enhanced signaling in thesecells suggests EGFR–MET cross-talk could be a mediator ofmetastasis and that the mechanism of cross-talk can changethroughout progression.MET regulates metastasis of gastric cancer, renal papillary
carcinomas, and breast cancer and MET expression and activ-
ity in NSCLC correlate with occurrence of brain metastasis(29, 32, 38, 39). In addition, MET copy number, expression, andphosphorylation are enriched in NSCLC brain metastasescompared with primary tumors (32). Despite association ofMET with metastasis, a direct role for MET in metastasis ofNSCLC has not been previously identified. We observed thatMET knockdown in highly metastatic NSCLC PC9-BrM3 cellscaused significant reduction in metastasis to the brain (Fig. 7).Differences between brain and other organ sites may dependon the brain being a more stringent organ to invade, which issupported by delayed detection of brain metastasis versusother sites in ourmodel. In addition, differences in brain tumorincidence but not burden suggest that an important role ofMET in lung cancer metastasis may be to promote invasion ofthe brain environment. The significance of this result is under-scored by the fact that the brain is themajormetastatic site andsource of morbidity in patients with lung cancer (40). Theinability of mouse HGF to activate human MET makes itdifficult to address the relative contributions of EGFR–METcross-talk versus HGF paracrine MET activation towardmetastasis with this model, but does not detract from theimportance of MET in promoting NSCLC progression. Theseresults support further investigation of the role of MET inEGFR-mutant NSCLC brain metastasis and the use of METinhibitors to prevent metastatic progression of NSCLC inpatients.
Previous clinical trials for MET inhibitors in NSCLC werefocused on patients who were resistant to EGFR TKIs due toMET amplification. The ability of both wild-type and mutantEGFR to activate MET, as well as MET regulation of EGFR-driven migration, invasion, and metastasis, suggests that METinhibitors may be beneficial to patients with varying muta-tional status. In fact, a recent clinical trial of combinationEGFR-MET inhibitors that did not prescreen patients formutation status found increased progression free and overallsurvival in patients with wild-type EGFR NSCLC (41). Inaddition, EGFR–MET signaling becomes more important atlater stages of NSCLC progression, indicating that use of METinhibitors in patients with early-stage cancer may preventoccurrence of invasion andmetastasis and should be evaluatedas a therapeutic option. Further investigation of EGFR–METcross-talk will be useful for determining which patientsmay benefit most from combination therapies and for iden-tifying potential new targets to prevent EGFR-induced METactivation.
Disclosure of Potential Conflicts of InterestNo potential conflicts of interest were disclosed.
Authors' ContributionsConception and design: J.L. Breindel, D.F. SternDevelopment of methodology: J.L. BreindelAcquisition of data (provided animals, acquired and managed patients,provided facilities, etc.): J.W. Haskins, E.P. Cowell, M. Zhao, D.X. NguyenAnalysis and interpretation of data (e.g., statistical analysis, biostatistics,computational analysis): J.L. Breindel, E.P. Cowell, D.X. Nguyen, D.F. SternWriting, review, and/or revision of the manuscript: J.L. Breindel, J.W.Haskins, E.P. Cowell, D.X. Nguyen, D.F. SternAdministrative, technical, or material support (i.e., reporting or orga-nizing data, constructing databases): M. ZhaoStudy supervision: D.F. Stern
EGFR–MET Signaling in NSCLC Metastasis
www.aacrjournals.org Cancer Res; 73(16) August 15, 2013 5063
on June 6, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 21, 2013; DOI: 10.1158/0008-5472.CAN-12-3775

AcknowledgmentsThe authors thank Drs. A. Koleske, D. DiMaio, and S. Agarwal for cell lines, N.
Hynes for plasmids and K. Politi for plasmids and advice about EGFR signaling inlung cancer.
Grant SupportThis work was funded in part by the DOD CDMRP Breast Cancer Research
Program #W81XWH-08-1-0780 (J.L. Breindel), CMB training grant #T32GM007223 (J.W. Haskins), USPHS grant R01CA45708 (D.F. Stern), and UnitingAgainst Lung Cancer (D.X. Nguyen). D.X. Nguyen is a scholar of the V
Foundation for Cancer Research, Yale Center for Clinical Investigation, andYoung Investigator of the International Association for the Study of LungCancer.
The costs of publication of this article were defrayed in part by thepayment of page charges. This article must therefore be hereby markedadvertisement in accordance with 18 U.S.C. Section 1734 solely to indicate thisfact.
Received September 27, 2012; revised May 9, 2013; accepted May 28, 2013;published OnlineFirst June 21, 2013.
References1. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO,
et al. MET amplification leads to gefitinib resistance in lung cancer byactivating ERBB3 signaling. Science 2007;316:1039–43.
2. Bean J, Brennan C, Shih JY, Riely G, Viale A, Wang L, et al. METamplification occurs with or without T790Mmutations in EGFRmutantlung tumors with acquired resistance to gefitinib or erlotinib. Proc NatlAcad Sci U S A 2007;104:20932–7.
3. Chen YT, Chang JW, Liu HP, Yu TF, Chiu YT, Hsieh JJ, et al. Clinicalimplications of high MET gene dosage in non–small cell lung cancerpatients without previous tyrosine kinase inhibitor treatment. J ThoracOncol 2011;6:2027–35.
4. Sequist LV, Bell DW, Lynch TJ, Haber DA. Molecular predictors ofresponse to epidermal growth factor receptor antagonists in non–small-cell lung cancer. J Clin Oncol 2007;25:587–95.
5. BruggerW, ThomasM. EGFR-TKI resistant non–small cell lung cancer(NSCLC): new developments and implications for future treatment.Lung Cancer 2012;77:2–8.
6. Guo A, Villen J, Kornhauser J, Lee KA, Stokes MP, Rikova K, et al.Signaling networks assembled by oncogenic EGFR and c-Met. ProcNatl Acad Sci U S A 2008;105:692–7.
7. Kubo T, Yamamoto H, Lockwood WW, Valencia I, Soh J, Peyton M,et al. MET gene amplification or EGFR mutation activate MET in lungcancers untreated with EGFR tyrosine kinase inhibitors. Int J Cancer2009;124:1778–84.
8. Xu Y, Liu H, Chen J, Zhou Q. Acquired resistance of lung adenocar-cinoma to EGFR-tyrosine kinase inhibitors gefitinib and erlotinib.Cancer Biol Ther 2010;9:572–82.
9. Agarwal S, Zerillo C, Kolmakova J, Christensen JG, Harris LN,Rimm DL, et al. Association of constitutively activated hepatocytegrowth factor receptor (Met) with resistance to a dual EGFR/Her2inhibitor in non–small-cell lung cancer cells. Br J Cancer 2009;100:941–9.
10. Tanizaki J, Okamoto I, Sakai K, NakagawaK. Differential roles of trans-phosphorylated EGFR, HER2, HER3, and RET as heterodimerisationpartners of MET in lung cancer with MET amplification. Br J Cancer2011;105:807–13.
11. Yamada T, Matsumoto K, Wang W, Li Q, Nishioka Y, Sekido Y, et al.Hepatocyte growth factor reduces susceptibility to an irreversibleepidermal growth factor receptor inhibitor in EGFR-T790M mutantlung cancer. Clin Cancer Res 2010;16:174–83.
12. Dulak AM, Gubish CT, Stabile LP, Henry C, Siegfried JM. HGF-independent potentiation of EGFR action by c-Met. Oncogene 2011;30:3625–35.
13. Reznik TE, Sang Y, Ma Y, Abounader R, Rosen EM, Xia S, et al.Transcription-dependent epidermal growth factor receptor activationby hepatocyte growth factor. Mol Cancer Res 2008;6:139–50.
14. Yano S, Wang W, Li Q, Matsumoto K, Sakurama H, Nakamura T, et al.Hepatocyte growth factor induces gefitinib resistance of lung adeno-carcinoma with epidermal growth factor receptor-activating muta-tions. Cancer Res 2008;68:9479–87.
15. Nguyen DX, Chiang AC, Zhang XH, Kim JY, Kris MG, Ladanyi M, et al.WNT/TCF signaling through LEF1 and HOXB9 mediates lung adeno-carcinoma metastasis. Cell 2009;138:51–62.
16. Klinghoffer RA, Sachsenmaier C, Cooper JA, Soriano P. Src familykinases are required for integrin but not PDGFR signal transduction.EMBO J 1999;18:2459–71.
17. Nguyen KS, Kobayashi S, Costa DB. Acquired resistance to epidermalgrowth factor receptor tyrosine kinase inhibitors in non–small-cell lungcancers dependent on the epidermal growth factor receptor pathway.Clin Lung Cancer 2009;10:281–9.
18. Pinkas-Kramarski R, Soussan L, Waterman H, Levkowitz G, Alroy I,Klapper L, et al. Diversification of Neu differentiation factor and epi-dermal growth factor signaling by combinatorial receptor interactions.EMBO J 1996;15:2452–67.
19. Pierce JH, Ruggiero M, Fleming TP, Di Fiore PP, Greenberger JS,Varticovski L, et al. Signal transduction through the EGF receptortransfected in IL-3–dependent hematopoietic cells. Science 1988;239:628–31.
20. Guy PM, Platko JV, Cantley LC, Cerione RA, Carraway KL III. Insectcell-expressed p180erbB3 possesses an impaired tyrosine kinaseactivity. Proc Natl Acad Sci U S A 1994;91:8132–6.
21. Riese DJ II, van Raaij TM, Plowman GD, Andrews GC, Stern DF.The cellular response to neuregulins is governed by complexinteractions of the erbB receptor family. Mol Cell Biol 1995;15:5770–6.
22. HammondDE,UrbeS,VandeWoudeGF,ClagueMJ.Down-regulationof MET, the receptor for hepatocyte growth factor. Oncogene 2001;20:2761–70.
23. Peschard P, Fournier TM, Lamorte L, Naujokas MA, Band H, LangdonWY, et al. Mutation of the c-Cbl TKB domain binding site on the Metreceptor tyrosine kinase converts it into a transforming protein. MolCell 2001;8:995–1004.
24. Carter S, Urbe S, Clague MJ. The met receptor degradation pathway:requirement for Lys48-linked polyubiquitin independent of protea-some activity. J Biol Chem 2004;279:52835–9.
25. Abella JV, Peschard P, Naujokas MA, Lin T, Saucier C, Urbe S, et al.Met/Hepatocyte growth factor receptor ubiquitination suppressestransformation and is required for Hrs phosphorylation. Mol Cell Biol2005;25:9632–45.
26. Koizumi F, Shimoyama T, Taguchi F, Saijo N, Nishio K. Establishmentof a human non–small cell lung cancer cell line resistant to gefitinib. IntJ Cancer 2005;116:36–44.
27. Sasaki Y, Shinkai T, Eguchi K, Tamura T, Ohe Y, Ohmori T, et al.Prediction of the antitumor activity of new platinum analogs based ontheir ex vivo pharmacodynamics as determined by bioassay. CancerChemother Pharmacol 1991;27:263–70.
28. Meiners S, Brinkmann V, Naundorf H, Birchmeier W. Role of morpho-genetic factors in metastasis of mammary carcinoma cells. Oncogene1998;16:9–20.
29. Corso S, Migliore C, Ghiso E, De Rosa G, Comoglio PM, Giordano S.Silencing the MET oncogene leads to regression of experimentaltumors and metastases. Oncogene 2008;27:684–93.
30. Cassinelli G, Lanzi C, Petrangolini G, Tortoreto M, Pratesi G, CuccuruG, et al. Inhibition of c-Met and prevention of spontaneous metastaticspreading by the 2-indolinone RPI-1. Mol Cancer Ther 2006;5:2388–97.
31. Navab R, Liu J, Seiden-Long I, Shih W, Li M, Bandarchi B, et al. Co-overexpression of Met and hepatocyte growth factor promotes sys-temic metastasis in NCI-H460 non–small cell lung carcinoma cells.Neoplasia 2009;11:1292–300.
32. Benedettini E, Sholl LM, Peyton M, Reilly J, Ware C, Davis L, et al. Metactivation in non–small cell lung cancer is associated with de novo
Breindel et al.
Cancer Res; 73(16) August 15, 2013 Cancer Research5064
on June 6, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 21, 2013; DOI: 10.1158/0008-5472.CAN-12-3775

resistance to EGFR inhibitors and the development of brain metasta-sis. Am J Pathol 2010;177:415–23.
33. Nguyen DX, Bos PD, Massague J. Metastasis: from dissemination toorgan-specific colonization. Nat Rev Cancer 2009;9:274–84.
34. Presnell SC, Stolz DB, Mars WM, JoM, Michalopoulos GK, Strom SC.Modifications of the hepatocyte growth factor/c-met pathwayby constitutive expression of transforming growth factor-alpha in ratliver epithelial cells. Mol Carcinog 1997;18:244–55.
35. Accornero P, Miretti S, Cucuzza LS, Martignani E, Baratta M. Epider-mal growth factor and hepatocyte growth factor cooperate to enhancecell proliferation, scatter, and invasion in murine mammary epithelialcells. J Mol Endocrinol 2010;44:115–25.
36. Joffre C, Barrow R, Menard L, Calleja V, Hart IR, Kermorgant S. A directrole forMet endocytosis in tumorigenesis.NatCell Biol 2011;13:827–37.
37. Biscardi JS, Belsches AP, Parsons SJ. Characterization of humanepidermal growth factor receptor and c-Src interactions in humanbreast tumor cells. Mol Carcinog 1998;21:261–72.
38. Schmidt L, Duh FM, Chen F, Kishida T, Glenn G, Choyke P, et al.Germline and somatic mutations in the tyrosine kinase domain of theMET proto-oncogene in papillary renal carcinomas. Nat Genet1997;16:68–73.
39. Ponzo MG, Lesurf R, Petkiewicz S, O'Malley FP, Pinnaduwage D,Andrulis IL, et al. Met induces mammary tumors with diversehistologies and is associated with poor outcome and humanbasal breast cancer. Proc Natl Acad Sci U S A 2009;106:12903–8.
40. Hess KR, Varadhachary GR, Taylor SH, Wei W, Raber MN, Lenzi R,et al. Metastatic patterns in adenocarcinoma. Cancer 2006;106:1624–33.
41. Eathiraj S, Palma R, Volckova E, Hirschi M, France DS, Ashwell MA,et al. Discovery of a novel mode of protein kinase inhibition charac-terized by the mechanism of inhibition of human mesenchymal-epi-thelial transition factor (c-Met) protein autophosphorylation by ARQ197. J Biol Chem 2011;286:20666–76.
EGFR–MET Signaling in NSCLC Metastasis
www.aacrjournals.org Cancer Res; 73(16) August 15, 2013 5065
on June 6, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 21, 2013; DOI: 10.1158/0008-5472.CAN-12-3775

2013;73:5053-5065. Published OnlineFirst June 21, 2013.Cancer Res Jerrica L. Breindel, Jonathan W. Haskins, Elizabeth P. Cowell, et al. Cell Lung Carcinoma Invasion and Brain Metastasis
Small−EGF Receptor Activates MET through MAPK to Enhance Non
Updated version
10.1158/0008-5472.CAN-12-3775doi:
Access the most recent version of this article at:
Material
Supplementary
http://cancerres.aacrjournals.org/content/suppl/2013/06/21/0008-5472.CAN-12-3775.DC1
Access the most recent supplemental material at:
Cited articles
http://cancerres.aacrjournals.org/content/73/16/5053.full#ref-list-1
This article cites 41 articles, 17 of which you can access for free at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerres.aacrjournals.org/content/73/16/5053To request permission to re-use all or part of this article, use this link
on June 6, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 21, 2013; DOI: 10.1158/0008-5472.CAN-12-3775






![Review Article Regulation of the Ras-MAPK and PI3K-mTOR ... · Cytosolic kinase SK Tumor suppressor/oncogenic isoforms, activates/inhibits mTORC. Breast, lung [ , ] Cytosolic kinase](https://img.dokumen.tips/doc/110x75/6080c0d51308b03b786a8817/review-article-regulation-of-the-ras-mapk-and-pi3k-mtor-cytosolic-kinase-sk.jpg)











![Involvement of the EGF Receptor in MAPK Signaling ... · of the field mediated by joint activation of the signal transduction pathways MAPK-ERK1/2 and -p38 [21, 22]. Other authors](https://img.dokumen.tips/doc/110x75/6060111ff0ec692f300940d5/involvement-of-the-egf-receptor-in-mapk-signaling-of-the-field-mediated-by-joint.jpg)










