Embed Size (px)
Citation preview

JOURNAL OF INTERFERON AND CYTOKINE RESEARCH 16:217-224 (1996)Mary Ann Liebert, Inc.
Effect of Interferon Therapy on Bone Marrow Morphology inChronic Myeloid Leukemia: A Cytochemical andImmunohistochemical Study of Trephine Biopsies
JUERGEN THIELE,1 THOMAS KARL ZIRBES,1 HANS MICHAEL KVASNICKA,1 JOHANN LORENZEN,1NORBERT NIEDERLE,2 LUTZ-DIETRICH LEDER,3 and ROBERT FISCHER1
ABSTRACT
The effect of interferon (IFN) therapy on bone marrow features in chronic myeloid leukemia (CML) has beenstudied on successive trephine biopsies (mean interval 13 ± 8 months) by cytochemical and immunohistochemi¬cal methods in combination with morphometry and in comparison with a control group of patients who receivedmonotherapy by busulfan (BU). Following IFN administration (IFN- frequently in combination with IFN-y),there was a decrease in neutrophil granulopoiesis accompanied by a significant expansion of erythroid precur¬sors and increased numbers of hemosiderin-laden macrophages. These changes corresponded with the hémato¬logie response in 21 of the 25 patients investigated. Numbers of megakaryocytes and reticulin/collagen fiber den¬sity increased during treatment. Most conspicuously, in responding patients atypical micromegakaryocytes,usually characterizing CML, were partially replaced by normal-sized cells of this lineage. These features are inkeeping with the assumption of a reappearance of the normal hematopoietic cell clone as the result of IFN ther¬apy, which was not found in the BU-treated control group. On the other hand, a relevant subpopulation of mi¬cromegakaryocytes (about 30%) was still maintained. This result probably relates to the failure to improvemyelofibrosis more effectively. Analysis of cell proliferation (proliferating cell nuclear antigen-PCNA) andapoptosis (in situ end labeling) revealed a reduction in PCNA labeling and increased numbers of cells undergoingprogrammed death. Ident ideation of the activated subset of macrophages ( -D-galactosyl residues expression)by appropriate lectin histochemistry disclosed an increase in the number of GSA-I binding cells. These d ndingswere exclusively limited to IFN administration and reflect an inhibitory effect of IFN on cell proliferation andstimulation of programmed cell death. The latter phenomenon probably results in increased phagocytosis ofclonally transformed myeloid cells by GSA-I-positive (activated) macrophages.
INTRODUCTION tion that IFN exerts its pleiotropic effect on the bone marrow byinfluencing cell proliferation ' and various differentiation
Generally, therapeutic regimens in Philadelphia chromo- processes.'91 Furthermore, reactivity of this agent is character-some-positive chronic myeloid leukemia (Ph1+ CML) are ized by modulating complex immune responses involving the
designed to inhibit the evolution from the stable phase into bias- mesenchymal compartment (microenvironment) and itstic crisis, in which patients die of bone marow insufficiency labyrinth-like cytokine-mediated functional network."0"18' A(bleeding and infection). In this context, the application of inter- wealth of data has been accumulated that, particularly in CML,feron (IFN) has been proven a powerful novel approach for its the dose-dependent and inhibitory efficacy of IFN is directedantineoplastic potency and has been the subject of several re- against the proliferation of the Ph1+ clone, presumably by down-cently published clinical trials including larger series of pa- regulation of oncogen expression.0318'For this reason, a persis¬tiente.(l~8) Experimental studies are in keeping with the assump- tence of at least some elements of the nontransformed (i.e., Ph1 ")
'Institute of Pathology, University of Cologne, Cologne, Germany.department of Oncology and Hematology, Klinikum Leverkusen, Leverkusen, Germany.'Institute of Pathology, University of Essen, Essen, Germany.
217

218 THIELE ET AL.
hematopoiesis is necessary to restore the normal diploid clone(hématologie and cytogenetic remission) following appropriateIFN treatment.0' In consideration of these findings, however,there is a significant lack of information concerning the morpho¬logic changes induced by IFN in the course of CML therapy thatmay reflect its biologic properties. For this purpose, the presentstudy was focused on the evaluation of bone marrow features as¬sociated with IFN administration in Ph1 + CML. In pursuit of thisaim, cytochemical and immunohistochemical methods in com¬
bination with morphometry were applied for an unequivocalidentification of the various cell elements and bone marrow
structures, including maikers for proliferation and apoptosis(programmed cell death).
MATERIALS AND METHODS
Patients
A group of 25 patients (15 males and 10 females: median age36 years) with Ph1+ CML was recruited from a clinical trial onIFN(4) in a random fashion and enrolled into this study. Criteriafor eligibility included representative bone marrow biopsies be¬fore randomization and in the course of treatment (mostly two tothree biopsies, mean interval 13 ± 8 months, range 1-64 monthsbetween first and last examination). The diagnosis of CML was
strictly confirmed by relevant clinical, morphologic, and cytoge¬netic findings and included only patients in a stable phase withless than 15% myeloblasts and promyelocytes in the peripheralblood and thorough workup with complete hospital and officerecords. At closure of this study (deadline July 1994), 8 patientswere still alive and 17 dead, with an overall survival time of 62months. Therapeutic regimens consisted of IFN-a2b (4 X 106U/m2/day as initial dosage) combined with IFN-y(supplied in 11patients at a constant daily dose of 50 ^g).<24) Following induc¬tion therapy, an appropriate maintenance treatment was appliedin accordance with leukocyte counts and toxicity. Because addi¬tion of low-dose IFN-y has failed to show any therapeutic bene-fit(2) and, additionally, no differences in marrow changes were
observable, patients were grouped together for the purpose ofthis study. Following standard criteria,'4'5' there was a completehématologie response in 21 and a major cytogenetic remission in3 of the 25 patients comprising our study group. For assessmentof the IFN-specific marrow changes, data on a control group of23 patients with Phl+ CML (11 males and 12 females; medianage 50 years) who received monotherapy with busulfan (BU)were also recorded. These parameters were partly derived from a
previously published study.09'Bone marrow biopsies
After informed consent, representative trephine biopsies ofthe bone marrow (mean size 20 X 1.3 mm) were performed andprocessed by low-concentrated (2-3%) formalin fixation, fol¬lowed by décalcification in EDTA and paraffin wax embedding.Routine staining procedure included Giemsa (overview), peri¬odic acid-Schiff reagent, naphthol-AS-D-chloroacetate esterase(neutrophil granulopoiesis), Gomori's silver impregnation (reti-culin-collagen fibers), and Perl's reaction for iron (hemosiderin-laden macrophages). For a selective staining of marrow cellsseveral monoclonal antibodies were used: CD61 (megakaryo-
poiesis). Ret40f (nucleated erythroid precursors), PG-M1(macrophages), and a lectin derived from Griffonia simplici¬folia I (GSA-I), specific for -galactose binding sites to identifythe so-called activated subpopulation of macrophages.<20_22) Toassess cell proliferation, the monoclonal antibody PC 10 was em¬
ployed, which is directed against proliferating cell nuclear anti¬gen (PCNA).'23·24' The rate of apoptosis (programmed or physio¬logic cell death) was determined by in situ end labeling of DNAstrand breaks.'25-27' A double-immunostaining procedure was
applied for the simultaneous demonstration of PG-M1- andGSA-I-positive macrophages engulfing apoptotic cells. Allmonoclonal antibodies and further reagents were purchasedfrom Dako-Diagnostica GmbH (Hamburg, Germany). GSA-Iwas purchased from E-Y Laboratories (San Mateo, CA). Detailsof immunohistochemical staining procedures involving the alka¬line phosphatase-antialkaline phosphatase technique have beendescribed in previous communications.'28·29'
MorphometryMorphometric analysis was performed by a manual optic
planimeter (VIDAS-Zeiss-Kontron) with a standard program set
(Videoplan software) on large biopsy specimens with an artifact-free mean marrow area of 9.6 ± 5.0 mm2. Frequencies of neu-
trophils, granulocytes, megakaryocytes, and nucleated erythroidprecursors, as well as macrophages and their subpopulations,were determined at X500 magnification by calculation of fatcell- and edema-free hematopoietic tissue and the number of cor¬
responding cells. An identical method was applied for the assess¬
ment of PCNA-positive as well as apoptotic elements, whichwere calculated as relative values (percentages of nucleatedhematopoietic cells). Sizes of megakaryocytes were evaluated ata magnification of X1250 and more than 100 cells of this lineagerandomly selected from each bone marrow sample. In addition tosize measurements involving the total megakaryopoiesis, twoclasses of this cell lineage were distinguished according to previ¬ous studies'3*-33': micromegakaryocytes (size s 200 /um2) andnormal-sized megakaryocytes displaying a sectioned area a
250 µ, )2. Reticulin and collagen (argyrophilic) fiber densitywas measured in specimens following silver impregnation(Gomori's stain) by counting the number of intersections (1)with the lines of a grid ocular at a magnification of X500 in atleast 20 randomly selected fields free from osseous trabeculaeand (2) also expressed per square millimeter hematopoietic tis¬sue. In this context it should be emphasized that all measure¬
ments were not calculated per marrow area but refer to
hematopoietic tissue (i.e., cellularity), in consideration of thetherapy-related changes (reduction in hematopoiesis followedby expansion of adipose tissue and interstitial edema) that other¬wise preclude a direct comparison of variables. Statistical evalu¬ation included Wilcoxon's matched-pair test to determine in-traindividual changes in morphometric parameters betweensuccessive trephine biopsies.
RESULTS
Changes in bone marrow features of IFN-treated patients were
most conspicuously expressed in those 21 cases with a hemato-

IFN EFFECT ON BONE MARROW MORPHOLOGY 219
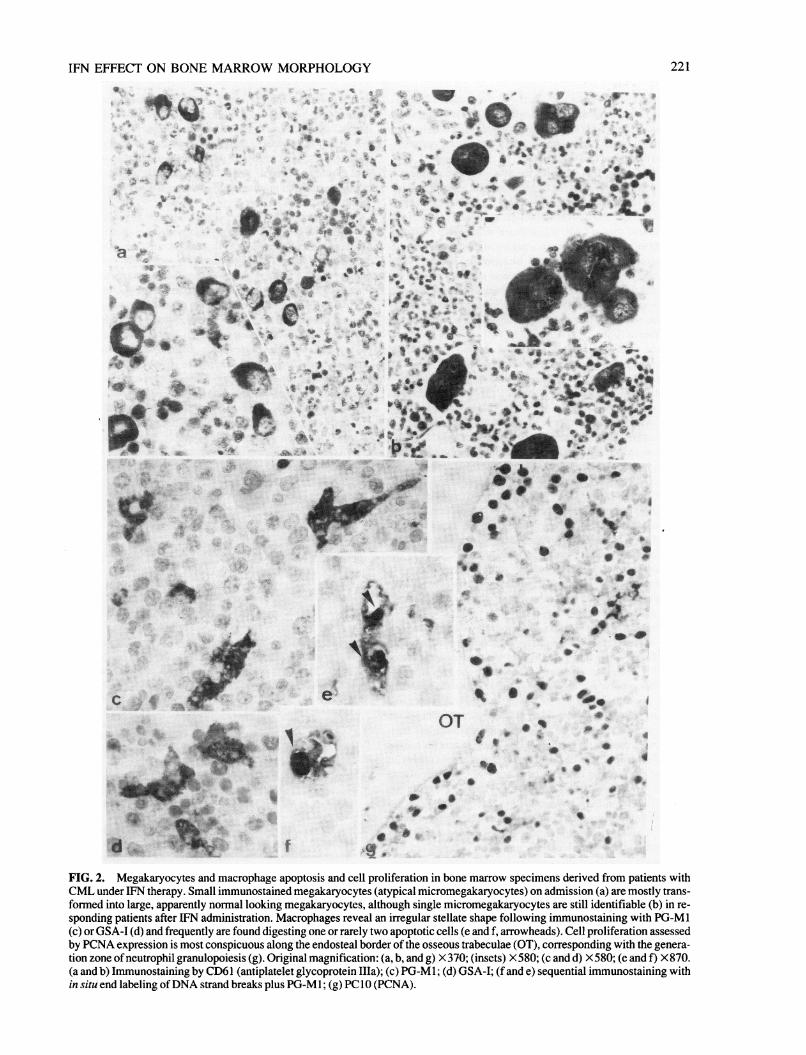
logic remission and partially reflected the improvement in labo¬ratory data. An overall reduction in cellularity (hematopoietictissue) was observed in sequential biopsies amounting to ap¬proximately 15% (range 0-34%). As may be derived from Table1 in comparison with the BU-treated control group, IFN admin¬istration resulted in a decreased growth of neutrophil granu-lopoiesis and a significant expansion of nucleated erythroid pre¬cursors showing large islets (Fig. la and b). This major effect isfurther demonstrated by the corresponding granulocyte/erythro-cyte (G/E) ratio (10.4:1 on admission versus 5.2:1 after treat¬ment) and by the dramatic drop in the white blood cell count(135 ± 120 versus 25 ± 63 X" 109/liter). Similarly, ameliora¬tion of anemia was obvious, considering the hemoglobin leveland the erythrocyte count in association with the increase in ery¬throid precursors and the prevalence of patients showing hemo-siderin-laden macrophages. On the other hand, number ofmegakaryocytes and content of argyrophilic (reticulin and colla¬gen) fibers increased during the course of disease (Fig. 1 c and d).Moreover, not only the frequency of megakaryocytes was fre¬quently increased, but most remarkably their average size (Fig.2a and b). A more refined analysis of this IFN-specific effect on
megakaryocyte sizes disclosed that so-called micromegakary¬ocytes (size S 200 µ 2) were predominant (about 90%) beforetherapy (Fig. 2a). IFN administration was able to reduce this ab¬normal subpopulation to about a half of the absolute frequencyand further generated a prevalence of normal-sized megakary¬ocytes (Fig. 2b and Table 1). In addition to these alterations,macrophages also revealed an increase in number, including the
subset of so-called activated (i.e., GSA-I-positive) cells (Fig. 2cand d). Simultaneous assessment of programmed cell death or
apoptosis (Fig. 2e and f) and cell proliferation by PCNA expres¬sion (Fig. 2g) disclosed that, in contrast to BU, IFN exerted a sig¬nificant stimulation on the first and an inhibitory effect on thelatter phenomenon (Table 1).
DISCUSSION
Our findings on sequentially performed bone marrow biopsiesimplicate a partial reappearance of the normal diploid Ph1" cellclone in CML following IFN treatment. Although at first sightreduction in neutrophil granulopoiesis and expansion of ery¬throid precursors could be regarded as a nonspecific or toxic ef¬fect comparable with the BU-treated control group, alterations ofmegakaryopoiesis were not in support of this hypothesis. Agrowing body of evidence has been provided that in CML thiscell lineage is characterized by small hypolobulated dwarf or mi¬croforms. <28·30~33> These micromegakaryocytes measure approxi¬mately 180 /im2 in size,'33' which is not different from the valuegiven in Table 1. Following IFN administration in respondingpatients of our trial with minimal residual disease,'34' there is a
significant decrease in the fraction of CML-specific mi¬cromegakaryocytes. This atypical cell population is partially(about 70%) replaced by larger elements exhibiting normal mea¬
surements.'33' However, because of the overall increase in fre¬quency of megakaryocytes following treatment, a significant
Table 1. Histologic Characteristics of Bone Marrow Biopsies Evaluated Before and During IFNAdministration in 25 Patients with CML in Comparison with BU Treatment"
ParameterBeforeIFN
AfterIFN BU treatment
Cellularity, % hematopoiesisNeutrophil granulopoiesis, mm2Erythroid precursors, mm2G/EMegakaryocytes
Frequency, mm2Size, total, µ 2
Micromegakaryocytes(size < 200 µ 2)frequency, mm2
Normal-sized megakaryocytes(size a 250 µ 2)frequency, mm2
Fibers, Xl02i/mm2Macrophages
Total, mm2GSA-I positive, mm2Hemosiderin-laden/ratio
PCNA, %Apoptosis, %
983885 ±1183
375 ± 28410.4:1
50 ±29178 ± 25
44 ±26
5 ±432 ± 18
35 ±521 ± 3
4/256.4 ± 2.7
0.32 ± 0.15
833527 ± 1059
685 ±5125.2:1
67 ± 35277 ± 79
22 ± 12
4165
2729
49 ± 1526 ± 11
19/253.3 ± 0.9
0.65 ± 0.07
793340 ± 883
413 ± 1238.0:1
82 ± 58179 ± 53*
62 ± 49*
16 ± 12*45 ± 23*
53 ± 1214 + 4*
11/256.2 ± 1.8*
0.33 ± 0.07*
aMean values ± standard deviation were calculated per area of hematopoiesis; levels of significance between successive biopsiesare/7 s 0.05-0.0001, with the exception of granulopoiesis in the IFN group andp < 0.01-0.001 in the BU group for those parametersindicated by asterisks.

220 THIELE ET AL.
fÜ* !Qb
:3 ß-'
".,;··$"*
FIG. 1. Erythropoiesis and reticulin fibers in the bone marrow of patients with CML under IFN therapy. On admission, only tinyassemblies of immunostained erythroid precursors are recognizable (a), whereas following IFN administration a significant increasein the extent of erythroid islets is evident (b). Scanty reticulin fibers on admission (c) are transformed into gross myelofibrosis ac¬
companied by megakaryocyte growth about 14 months following IFN treatment (d). Original magnification: (a and b) X580; (c andd) X370. (a and b) Immunostaining by Ret40f (antiglycophorin C); (c and d) silver impregnation (Gomori's stain).
IFN EFFECT ON BONE MARROW MORPHOLOGY 221
<§*· .>·. t*
9 »-jr*
X· %-
I*4 h
îOTîV FfÇ|
"a * S·*
if.
, '/«:.*.· ;_wW^fe
* i
/·' *jL
\ ·
teOT
f.M§
• ·
,··
*.« #
FIG. 2. Megakaryocytes and macrophage apoptosis and cell proliferation in bone marrow specimens derived from patients withCML under IFN therapy. Small immunostained megakaryocytes (atypical micromegakaryocytes) on admission (a) are mostly trans¬formed into large, apparently normal looking megakaryocytes, although single micromegakaryocytes are still identifiable (b) in re¬
sponding patients after IFN administration. Macrophages reveal an irregular stellate shape following immunostaining with PG-M 1(c) or GSA-I (d) and frequently are found digesting one or rarely two apoptotic cells (e and f, arrowheads). Cell proliferation assessedby PCNA expression is most conspicuous along the endosteal border of the osseous trabeculae (OT), corresponding with the genera¬tion zone ofneutrophil granulopoiesis (g). Original magnification: (a, b, and g) X370; (insets) X580;(candd) X580;(eandf) X870.(a and b) Immunostaining by CD61 (antiplatelet glycoprotein Ilia); (c) PG-M 1 ; (d) GSA-I; (f and e) sequential immunostaining within situ end labeling of DNA strand breaks plus PG-M 1 ; (g) PC 10 (PCNA).

222 THIELE ET AL.
amount of micromegakaryocytes is still maintained (Table 1).For this reason, from the morphologic point of view, alterationsin megakaryocyte sizes in addition to changes in the G/E ratio are
in keeping with the assumption that the leukemic cell mass is re¬
duced in the course of IFN therapy.The inability of IFN to inhibit the evolution of bone marrow
fibrosis may be related to the fact that atypical micromegakary¬ocytes are not replaced more thoroughly by normal elements ofthis cell lineage. Previous morphologic studies have repeatedlydemonstrated that in CML a significant relationship exists be¬tween fiber content and micromegakaryocyte growth and thatmegakaryocyte-rich subtypes may be addressed as premyelofi-brotic stages.'28·35·36' These findings are in congruence with ex¬
perimental data that provide convincing arguments for complexcytokine-mediated interactions between megakaryocytes and fi¬broblasts, which are held to be responsible for the generation ofmyelofibrosis.'37^1 ' '
Culture studies on various subsets of macrophages indicatethat activation is induced by IFN administration'42"14' and thatthis state is characterized by an expression of terminal a-D-galactosyl residues on their surface.'20-22' This carbohydratemoiety is recognized by GSA-I.'20-22·45"48' Major functions of ac¬
tivated macrophages consist of phagocytosis of cell debris, in¬volvement in iron turnover, and killing of tumor cells.'49' In thiscontext, activated macrophages were found to produce tumornecrosis factor, which belongs to a mediator superfamily capableof inducing various forms of cell death.'5"' Increase in the totalnumber of PG-M 1-positive macrophages, in particular theirGSA-I-reactive subpopulation, is most likely related to thesefunctions. Considering iron metabolism and bone marrow
macrophages, it is noteworthy that sequential examinations re¬
vealed no hemosiderin deposits in CML in the majority of pa¬tients on admission.'5 ' ' Following IFN administration, the preva¬lence of patients displaying small numbers of hemosiderin-ladenmacrophages was significantly increased, which points to a stim¬ulation of iron turnover.
The clear-cut identification of apoptosis (programmed celldeath) in bone marrow tissue is a controversial issue inhistopathology and has given rise to discussion and contro¬versy.'52"57' Problems are mostly generated by the very rapid na¬ture of this process and the instant degradation of the senescentcells by macrophages.'52·57·58' In vitro studies have demonstratedthat aging cells are recognized and disposed by macrophages viathe vitronectin receptor.'54·58' This pathomechanism has beensuggested to present the major pathway that prevents the releaseof cellular content from disintegrated cells that would otherwisehave the potential to cause inflammatory reactions similar tonecrosis.'52-57' A promising approach to identify cell death hasbeen the ability of DNA polymerase I to incorporate labeled nu-cleotides into the fragmented DNA characteristic for apopto¬sis.'25-27' Although much has yet to be learned about pro¬grammed cell death, it has been repeatedly emphasized thatapoptosis is a regulated phenomenon, capable of being inhibitedand activated.'52·55-57' Apoptosis occurs in a wide range of physi¬ologic conditions and may be induced by cytotoxic treat¬ment.'55·58-60' rjsjng myeloid cell lines as a model system to studyregulation of proliferation and differentiation, it has been foundthat some hematopoietic cytokines were able to inhibit trans¬forming growth factor /3,-mediated apoptosis.'59·61-63' In the con¬
text ofCML, it is noteworthy that the characteristic translation of
the abl gene from chromosome 9-22 (Philadephia chromosome)results in the synthesis of a chimeric bcr-abl protein that gener¬ates an increased activity of abl-tyrosine kinase.'64' One effect ofthis alteration is a marked increase in resistance to drug-inducedapoptosis.' ' Our finding of a significant stimulation of apop¬tosis (Table 1 ) following IFN therapy is a further argument for a
(partial) regeneration of normal hematopoiesis or regression ofthe Ph1 + (bcr/abl) cell lineages.
In most tissues, PCNA reactivity is limited to the proliferatingcompartment, although in some tumors the proportion of posi¬tively stained cells exceeds the fraction detected by other mark¬ers. PCNA is not only detected during the replicative cell cyclebut also in DNA repair. Therefore, caution must be applied wheninterpreting staining results.'24' In comparison with normal bonemarrow in CML, a relatively low rate of proliferation has beenfound in cell culture studies'68·69' that is also demonstrable by thelow index of PCNA labeling (Table 1 ). This feature documentsthe prolongation of granulocyte life span in CML and has beenassumed to be the result of suppression of apoptosis. Probablythis mechanism presents the major contributory factor for the ex¬
pansion of the leukemic cell mass.'70' In consideration of our
findings, particularly in comparison with the BU-treated controlgroup, it is tempting to speculate that IFN has a significant andspecific impact on at least two conversely acting pathomech-anisms, that is, inhibition of proliferation of precursor cells as
well as enhancement of apoptosis-inducing effects, involvingparticularly the Ph1 + cell clone.
ACKNOWLEDGMENT
Supported by Grant No. DFG Th 390/1-4 from the DeutscheForschungsgemeinschaft.
REFERENCES
1. MORRA, E., LAZZARINO, M., ALÌMENA, G, LIBERATI,A.M., GRIGNANI, F., MANDELLI, F., and BERNASCONI, C.(1992). The role of interferon in the treatment of chronic myeloge¬nous leukemia: results and prospects. Leuk Lymph 6,305-315.
2. KLOKE, O., WANDL, U., OPALKA, B., MORITZ, T., NAGEL-HIEMKE, M., FRANZ, T„ HIRCHE, H., SEEBER, S., andNIEDERLE, N. (1992). A prospective randomized comparison ofsingle-agent interferon (IFN)-alpha with the combination of IFN-al-pha and low-dose IFN-gamma in chronic myelogenous leukaemia.Eur. J. Haematol. 58,93-98.
3. KANTARJIAN, H.M., and TALPAZ, M. (1993). Long-term fol¬low-up results of alpha interferon therapy in chronic myelogenousleukemia at M.D. Anderson Cancer Center. Leuk. Lymph. 11,169-174.
4. NIEDERLE, N., KLOKE, O., WANDL, U.B„ BECHER, R.,MORITZ, T., and OPALKA, B. (1993). Long-term treatment ofchronic myelogenous leukemia with different interferons: resultsfrom three studies. Leuk. Lymph. 9,111-119.
5. OZER, H., GEORGE, S.L., SCHIFFER, CA., RAO, ., RAO, . ., WURSTER-HILL, D.H., ARTHUR, D.D., POWELL, B.,GOTTLIEB, ., PETERSON, B.A./ RAI, ., TESTA, J.R,.LEBEAU, M., TANTRAVAHI, R., and BLOOMFIELD, CD.( 1993). Prolonged subcutaneous administration of recombinant a2binterferon in patients with previously untreated Philadelphia chro¬mosome-positive chronic-phase chronic myelogenous leukemia: ef-

IFN EFFECT ON BONE MARROW MORPHOLOGY 223
feet on remission duration and survival. Cancer and LeukemiaGroup Study 8583. Blood 82,2975-2984.
6. ITALIAN COOPERATIVE STUDY GROUP ON CHRONICMYELOID LEUKEMIA (1994). Interferon alfa-2a as comparedwith conventional chemotherapy for the treatment of chronicmyeloid leukemia. N. Engl. J. Med. 330,820-825.
7. HEHLMANN, R., HEIMPEL, ., HASFORD, J., KOLB, H.J.,PRALLE, ., HOSSFELD, D.K., QUEISSER, W., LOEFFLER,H., HOCHHAUS, ., HEINZE, B., GEORGII, ., et al. (1994).Randomized comparison of interferon- with busulfan and hydrox-yurea in chronic myelogenous leukemia. Blood 84,4064-4077.
8. AULITZKY, W.E., PESCHEL, C, SCHNELLER, F., and HUBER,C. (1995). Biotherapy of chronic myelogenous leukemia. Ann.Hematol. 70,113-120.
9. STELLA, C.C., and CAZZOLA, M. (1988). Interferons as biologicmodulators of hematopoietic cell proliferation and differentiation.Haematologica (Pavia) 73,225-237.
10. MURPHY, M. PERUSSIA, B„ and TRINCHIERI, G. (1988).Effects of recombinant tumor necrosis factor, lymphotoxin, and im¬mune interferon on proliferation and differentiation of enrichedhematopoietic precursors cells. Exp. Hematol. 16, 131-138.
11. PELUS, L.M., OTTMANN, O.G., and NOCKA, K.H. (1988).Synergistic inhibition of human marrow granulocyte-macrophageprogenitors cells by prostaglandin E and recombinant interferon- ,-ß, and gamma and an effect mediated by tumor necrosis factor. J.Immunol. 140,479-484.
12. GALVANI, D.W., and CAWLEY, J.C. (1989). Mechanism of ac¬
tion of alpha interferon in chronic granulocytic leukaemia: evidencefor preferential inhibition of late progenitors. Br. J. Haematol. 73,475-479.
13. SHAN, B., VAZQUEZ, E., and LEWIS, J.A. (1990). Interferon se¬
lectively inhibits the expression of mitochondrial genes: a novelpathway for interferon-mediated responses. EMBO J. 9,4307^1314.
14. GALVANI, D.W., and CAWLEY, J.C. (1990). The effects of inter¬feron on human long-term bone marrow culture. Leuk. Res. 14,525-531.
15. OSTERHOLZ,J., DOWDING, C, GUO, A.P., SICZKOWSKI, M„and GOLDMAN, J.M. ( 1991 ). Interferon- alters the distribution ofCFU-GM between the adherent and nonadherent compartments inlong-term cultures of chronic myeloid leukemia marrow. Exp.Hematol. 19,326-331.
16. DOWDING, C, GOU, A.P., OSTERHOLZ, J., SICZKOWSKI, M.,GOLDMAN, J., and GORDON, M. (1991). Interferon- overridesthe deficient adhesion of chronic myeloid leukemia primitive pro¬genitor cells to bone marrow stromal cells. Blood 78,499-505.
17. DEISSEROTH, A.B., ZAHNG, W., CHA, Y., YUAN, T., CHEN,H., SIMS, S., WEDRYCHOWSKI, ., GAO, P.-Q., HUSTON, L.,FILACCIO, M., CLAXTON, D., KORNBLAU, S„ JOHNSON, E.,HOWARD, O.M.Z., ANDERSSON, B., DEL GIGLIO, ., GRES-SOT, L„ KANTARJIAN, H., TALPAZ, M., KHOURI, I., CHAM-PLIN, R., ANDREEFF, M., GAOZZA, E., SEONG, D., SUH, S.-P.,ELLERSON, D., HU, G., and CHOU, M. (1992). New directions inthe biology and therapy of chronic myeloid leukemia. Leuk. Lymph.6,89-95.
18. DOWDING, C, GORDON, M., GUO, A.P., MAISON, D., OS¬TERHOLZ, J., SICZKOWSKI, M., and GOLDMAN, J. (1993).Potential mechanisms of action of interferon-alpha in CML. Leuk.Lymph. 11,185-191.
19. THIELE, J., KVASNICKA, H.M., NIEDERLE, N., ZIRBES, T.K.,SCHMIDT, M., DAMMASCH, J., MEUTER, B.R., LEDER, L.D.,KLOKE, O., DIEHL, V., and FISCHER, R. (1995). The impact ofinterferon versus busulfan therapy on the reticulin stain-measuredfibrosis in CML—a comparative morphometric study on sequentialtrephine biopsies. Ann. Hematol. 70, 121-128.
20. MADDOX, D.E., SHIBATA, S., and GOLDSTEIN, I.J. (1982).
Stimulated macrophages express a new glycoprotein receptor reac¬
tive with Grijfonia simplicifolia I-B4 isolectin. Proc. Nati. Acad. Sci.USA 79,166-170.
21. WARFEL, A.H., ZUCKER-FRANKLIN, D.. and ZHENG, Z.Y.(1991). Macrophage membrane glycoproteins that bind Grijfoniasimplicifolia I-B4: effect on cytotoxicity and protein secretion. J.Cell. Physiol. 147,265-273.
22. TABOR, D.R., LARRY, C.H., and JACOBS, R.F. (1989).Differential induction of macrophage GSIB4-binding-activity. J.Leukoc. Biol. 45,452^157.
23. HALL, P.A., LEVISON, D.A., WOODS, A.L., YU, C.C.W., KEL-LOCK, DB., WATKINS, J.A., BARNES, D.M., GILLET, CE.,CAMPLEJOHN, R., DOVER, R„ WASEEM, N.H., and LANE,D.P. (1990). Proliferating cell nuclear antigen (PCNA) immunolo-calization in paraffin sections: an index of cell proliferation with ev¬
idence of deregulated expression in some neoplasms. J. Pathol. 162,285-294.
24. MCCORMICK, D., and HALL, P.A. (1992). The complexities ofproliferating cell nuclear antigen. Histopathology 21,591-594.
25. ANSARI, B., COATES, P.J., GREENSTEIN, B.D., and HALL,P.A. (1993). In situ end-labelling detects DNA strand breaks inapoptosis and other physiological and pathological states. J. Pathol.170, 1-8.
26. FEHSEL, K„ KRÖNCKE, K.D., KOLB, H., and KOLB-BACH-OFEN, V. (1994). In situ nick-translation detects focal apoptosis inthymuses of glucocorticoid- and lipopolysaccharide-treated mice. J.Histochem. Cytochem. 42,613-619.
27. COATES, P.J., SAVE, V., ANSARI, B., and HALL, P.A. (1995).Demonstration of DNA damage/repair in individual cells using insitu end labelling: association of p53 with sites of DNA damage. J.Pathol. 176, 19-26.
28. THIELE, J., KVASNICKA, H.M., TITIUS, B.R., PARPERT, U.,NEBEL, R., ZANKOVICH, R„ DIENEMANN, D., STEIN, H.,DIEHL, V., and FISCHER, R. (1993). Histological features of prog¬nostic significance in CML—an immunohistochemical and mor¬
phometric study (multivariate regression analysis) on trephine biop¬sies of the bone marrow. Ann. Hematol. 66,291-302.
29. THIELE, J., HOEFER, M., KVASNICKA, H.M., BERTSCH, H.P.,ZANKOVICH, R., and FISCHER, R. (1993). Erythropoiesis inCML—immunomorphometric quantification, PCNA-reactivity,and influence on survival. Hematol. Pathol. 7,239-249.
30. BURKHARDT, R., BARTL, R., JAEGER, K„ FRISCH, B„KETTNER, G, MAHL, G, and SUND, M. (1984). Chronic myelo-proliferative disorders (CMPD). Pathol. Res. Pract. 179, 131-186.
31. BURKHARDT, R. (1988). Bone marrow in megakaryocytic disor¬ders. Hematol. Oncol. Clin. North Am. 2,695-733.
32. GEORGII, ., VYKOUPIL, K.F., BUHR, T., CHORITZ, H.,DÖHLER, U., KALOUTSI, V., and WERNER, M. (1990). Chronicmyeloproliferative disorders in bone marrow biopsies. Pathol. Res.Pract. 186,3-27.
33. THIELE, J., and FISCHER, R. (1991). Megakaryocytopoiesis inhaematological disorders: diagnostic features of bone marrow biop¬sies. Virchows Arch. [A] 418,81-97.
34. OPALKA. B., WANDL, U.B., BECHER, R., KLOKE, O.,NAGEL-HIEMKE, M., MORITZ, T., BEER, U., SEEBER, S., andNIEDERLE, R. (1991). Minimal residual disease in patients withchronic myelogenous leukemia undergoing long-term treatmentwith recombinant interferon a-2b alone or in combination with in¬terferon gamma. Blood 78,2188-2193.
35. LAZZARINO, M., MORRA, E., CASTELLO, ., INVERARDI,D., COCÍ, ., PAGNUCCO, G, MAGRINI, U., , G, andBERNASCONI, C. (1986). Myelofibrosis in chronic granulocyticleukaemia: clinicopathologic correlations and prognostic signifi¬cance. Br. J. Haematol. 64,227-240.
36. BUHR, T., CHORITZ, H., and GEORGII, A. (1992). The impact ofmegakaryocyte proliferation for the evolution of myelofibrosis.

±* ¿Jl THIELE ET AL.
Histologie follow-up study in 186 patients with chronic myeloidleukemia. Virchows Arch. [A] 420,473^178.
37. CASTRO-MALASPINA, H. (1984). Pathogenesis of myelofibro¬sis: role of ineffective megakaryopoiesis and megakaryocyte com¬
ponents. In: Myelofibrosis and the Biology ofConnective Tissue. P.Berk, H. Castro-Malaspina, and L. Wasserman (eds.) New York:Liss, pp. 427^154.
38. MACCARTHY, D.M. (1985). Fibrosis of the bone marrow: contentand causes. Br. J. Haematol. 59, 1-7.
39. YONEKURA, S., NAGAO, T., and AR1MORI, S. (1985).Increased growth and collagen synthesis of bone marrow fibroblastsfrom patients with chronic myelocytic leukaemia. Br. J. Haematol.61,93-99.
40. KIMURA. ., , ., and KURAMOTO, A. (1988). Effectsof platelet-derived growth factor, epidermal growth factor and trans¬
forming growth factor-ß on growth of human marrow fibroblasts.Br.J. Haematol. 69,9-12.
41. KATOH, O., KIMURA, ., ITOH, T., and KURAMOTO, A.(1990). Platelet derived growth factor messenger RNA is increasedin bone marrow megakaryocytes in patients with myeloproliferativedisorders. Am. J. Haematol. 35, 145-150.
42. PACE, J.L., RUSSELL, S.W., TORRES, B.A., JOHNSON, H.M.,and GRAY, P.W. ( 1983). Recombinant mouse gamma interferon in¬duces the priming step in macrophage activation for tumor cellkilling. J. Immunol. 130,2011-2013.
43. VARESIO, L., BLASI, E., THURMAN, GB., TALMADGE, J.E.,WILTROUT, R.H., and HERBERMANN, R.B. ( 1984). Potent acti¬vation of mouse macrophages by recombinant interferon-gamma.Cancer Res. 44,4465^t469.
44. HERRIOTT, J.J., and LEU, R.W. (1987). Activation of mouse
macrophages for migration inhibition and for tumor cytotoxicity ismediated by interferon-gamma priming and triggering by variousagents. J. Interferon Res. 7, 165-171.
45. MADDOX, D.E., GOLDSTEIN, I.J., and LOBUGLIO, A.F.(1982). Grijfonia simplicijolia I lectin mediates macrophage-in-duced cytotoxicity against Ehrlich ascites tumor. Cell. Immunol. 71,202-207.
46. IRIMURA, T., NORTH, S.M., and NICOLSON, GL. (1987).Glycoprotein profiles of macrophages at different stages of activa¬tion as revealed by lectin binding after electrophoretic separation.Eur. J. Immunol. 17,73-78.
47. TAKACS, B., and STAEHLI, C. (1987). Activated macrophagesand antibodies against the plant lectin GSI-B4, recognize the sametumor-associated structure (TAS). J. Immunol. 138, 1999-2007.
48. WARFEL, A.H., and ZUCKER-FRANKLIN, D. (1992). Specificligation of surface a-D-galactosyl epitopes markedly affects thequantity of four major proteins secreted by macrophages. J. Leukoc.Biol. 52,80-84.
49. ADAMS, D.O., and HAMILTON, T.A. (1984). The cell biology ofmacrophage activation. Annu. Rev. Immunol. 2,283-318.
50. SMITH, CA., FARRAH, T., and GOODWIN, R.G. (1994). TheTNF receptor superfamily of cellular and viral proteins: activation,costimulation, and death. Cell 76,959-962.
51. SOKAL, J.E., SHEERIN, K.A. ( 1986). Decreased stainable marrowiron in chronic granulocytic leukemia. Am. J. Med. 81,395-399.
52. ARENDS, M.J.,and WYLLIE,A.H. ( 1991 ). Apoptosis: mechanismand roles in pathology. Int. Rev. Exp. Pathol. 32,223-254.
53. ALLEN, P.D., BUSTIN, S.A., and NEWLAND, A.C. (1993). Therole of apoptosis (programmed cell death) in haemopoiesis and theimmune system. Blood Rev. 7,63-73.
54. WILLIAMS, G.T. (1994). Apoptosis in the immune system. J.Pathol. 173, 1^1.
55. KERR, J.F.R., WINTERFORD, CM., and HARMON, B.V. (1994).
Apoptosis. Its significance in cancer and cancer therapy. Cancer 73,2013-2026.
56. KOURY, M.J. (1992). Programmed cell death (apoptosis) inhematopoiesis. Exp. Hematol. 20,391-394.
57. PAYNE, CM., BERNSTEIN, C, and BERNSTEIN, H. (1995).Apoptosis overview emphasizing the role of oxidative stress, DNAdamage and signal-transduction pathways. Leuk. Lymph. 19,43-93.
58. SAVILL, J., FADOK, V., HENSON, P., and HASLETT, C. (1993).Phagocyte recognition of cells undergoing apoptosis. Immunol.Today 14, 131-136.
59. LOTEM, J., and SACHS, L. (1992). Hematopoietic cytokines in¬hibit apoptosis induced by transforming growth factor ß 1 and cancer
chemotherapy compounds in myeloid leukemic cells. Blood 80,1750-1757.
60. LI, X„ GONG, J„ FELDMAN, E., SEITER, ., TRAGNOS, F., andDARZYNKKIEWICZ, Z. (1994). Apoptotic cell death during treat¬ment of leukemias. Leuk. Lymph. 13,65-70.
61. COLOTTA, F., RE, F., POLENTARUTTI, N., SOZZANI, S„ andMANTOVANI, A. (1992). Modulation of granulocyte survival andprogrammed cell death by cytokines and bacterial products. Blood80,2012-2020.
62. HOFFMAN, B., and LIEBERMANN, D.A. (1994). Molecular con¬
trols of apoptosis: differentiation/growth arrest primary responsegenes, proto-oncogenes, and tumor suppressor genes as positive andnegative modulators. Oncogene 9, 1807-1812.
63. SELVAKUMARAN, M., LIN, H.K.,TJIN THAM SJIN, R., REED,J.C, LIEBERMANN, D.A., and HOFFMAN, B. (1994). The novelprimary response gene MyD 118 and the protooncogenes myb, myc,
" and bcl-2 modulate transforming growth factor ß 1 -induced apopto¬sis of myeloid leukemia cells. Mol. Cell. Biol. 14,2352-2360.
64. MCWHIRTER, J.R., and WANG, J.Y.J. (1991). Activation of ty¬rosine kinase and microfilament-binding functions of c-abl by bersequences in bcr/abl fusion proteins. Mol. Cell' Biol. 11,1553-1565.
65. MARTIN, S.J., LENNON, S.V., BONHAM, A.M., and COTTER,T.G. (1990). Induction of apoptosis (programmed cell death) in hu¬man leukemic HL-60 cells by inhibition of RNA and protein synthe¬sis. J. Immunol. 145, 1859-1867.
66. MCGAHON, ., BISSONNETTE, R., SCHMITT, M., COTTER,K.M., GREEN, D.R., and COTTER, T.C. (1994). Bcr-abl maintainsresistance of chronic myelogenous leukemia cells to apoptotic celldeath. Blood 83,1179-1187.
67. COTTER, T.C. (1995). Bcr-abl: an anti-apoptosis gene in chronicmyelogenous leukemia. Leuk. Lymph. 18,231-236.
68. DOERMER, P., LAU, ., and WILMANNS, W. (1980). Kinetics ofbone marrow cell production in human acute and chronic leukemias.Leuk. Res. 4,231-237.
69. STRIFE, ., and CLARKSON, B. ( 1988). Biology of chronic myel¬ogenous leukaemia: is discordant maturation the primary defect?Semin. Hematol. 25, 1-19.
70. GORDON, M.Y. (1993). Regulation of growth in chronic myeloidleukemia. Leuk. Lymph. 11,75-79.
Address reprint requests to:Dr. Juergen Thiele
Institute of PathologyUniversity of Cologne
Joseph-Stelzmann-Strasse 9D-50924 Köln, Germany
Received 5 May 1995/Accepted 31 October 1995





























