Embed Size (px)
Citation preview

Pharmacokinetic Interaction between JBP485 and Cephalexinin Rats
Jian Zhang, Changyuan Wang, Qi Liu, Qiang Meng, Jian Cang, Huijun Sun, Ying Gao,Taiichi Kaku, and Kexin Liu
Department of Clinical Pharmacology, College of Pharmacy (J.Z., C.W., Q.L., Q.M., J.C., H.S., K.L.), and Provincial KeyLaboratory (Y.G.), Dalian, Liaoning, Dalian Medical University, China; and Japan Bioproducts Industry Co. Ltd., Tomigaya,
Shibuya-ku, Tokyo, Japan (T.K.)
Received January 5, 2010; accepted March 10, 2010
ABSTRACT:
The purpose of this study was to investigate the pharmacokineticmechanism of interaction between JBP485 (cyclo-trans-4-L-hy-droxyprolyl-L-serine, a dipeptide) and cephalexin when they werecoadministered in rats. The plasma concentrations of JBP485 andcephalexin were both decreased significantly after oral combina-tion, but little difference was observed after simultaneous intrave-nous administration of the two agents, suggesting that the inter-action target localized in the intestine during the absorptionprocess. The uptake in everted intestinal sacs and absorption injejunal perfusions of JBP485 and cephalexin were dramaticallyreduced after drug combination. When JBP485 and cephalexinwere coadministered, both the decrease in accumulative renalexcretion (81.9–68.1% of JBP485 and 91.8–74.5% of cephalexin)and in renal clearance (2.89–1.87 ml/min/kg JBP485 and 2.23–1.58ml/min/kg cephalexin) indicated that transporter(s) other than H�/
peptide transporter (PEPT) 2 are involved in the process of excre-tion. Probenecid could reduce renal excretion of JBP485 andcephalexin. Moreover, the decreased uptake of JBP485 with pro-benecid, p-aminohippuate, or benzylpenicillin in kidney slicescould be explained by an inhibition in the kidney via organic aniontransporters (OATs), at least in part. The accumulation of JBP485 inhuman (h) OAT1- or hOAT3-human embryonic kidney (HEK) 293cells was greater than that in vector-HEK293 cells, and the uptakecould be inhibited by probenecid. These findings further confirmedthat the pharmacokinetic mechanism of the drug-drug interactionbetween JBP485 and cephalexin could be explained by their inhi-bition of the same transporters in the intestinal mucosa (PEPT1)and kidneys (PEPT2 and OATs). We provide the first evidence thatJBP485 is not only a substrate of PEPTs but also is excretedthrough OATs.
Drug-drug interactions arising from inhibition of the same trans-porters may lead to adverse effects. Some drugs with harmful drug-drug interactions have been withdrawn from the market. Numerousamino acid transporter systems are responsible for the uptake of freeamino acids, whereas the uptake of di- and tripeptides is achieved bythe intestinal low-affinity/high-capacity peptide transporter (PEPT1).PEPT1 is primarily expressed in the brush-border membrane of thesmall intestine (Leibach and Ganapathy, 1996). PEPT2 is expressedspecifically on the apical (luminal) membrane of epithelial cells of theproximal tubules of the kidney. They are responsible not only fornutrient transport across absorptive cell membranes but also for theabsorption (in the small intestines) and reabsorption (in the kidneys)of peptides or peptide-like compounds, such as angiotensin-convert-
ing enzyme inhibitors, cefadroxil, and valacyclovir (Ganapathy et al.,1994).
Cyclo-trans-4-L-hydroxyprolyl-L-serine (JBP485) is a dipeptide(Liu et al., 2000) (Fig. 1A) first isolated from Laennec (Yang et al.,2009), which is a proprietary product made from the hydrolysate ofhuman placenta (Annaert and Brouwer, 2005) in Japan, and has beensynthesized by chemical means. Animal experiments have indicatedthat JBP485 exhibits obvious liver protective effects after oral admin-istration (Liu et al., 1998; Wu et al., 2008; Yang et al., 2009). JBP485was well absorbed through the gastrointestinal tract. Our previousstudies have shown that its absorption can be inhibited by glycylsar-cosine (Gly-sar), which is a dipeptide model drug and the substrate ofPEPTs. These studies have suggested that JBP485 is recognized bythe peptide transporter system in the gastrointestinal tract. Cephalexin,as a first-generation oral cephalosporin (Fig. 1B), has a broad spec-trum of antibacterial activity (Ogawa et al., 1994). It has already beendemonstrated that dipeptides and cephalexin share certain structuralfeatures, such as a peptide bond with an �-amino group and a terminalcarboxylic acid group (Bretschneider et al., 1999). This structuralsimilarity is apparently the basis for the molecular mimicry, enabling
This work was supported by the National Natural Science Foundation of China[Grants 30672498 and 30873118]; and the Liaoning Provincial Key Laboratory[Grant 2008S078].
Article, publication date, and citation information can be found athttp://dmd.aspetjournals.org.
doi:10.1124/dmd.110.032060.
ABBREVIATIONS: PEPT, peptide transporter; JBP485, cyclo-trans-4-L-hydroxyprolyl-L-serine; Gly-sar, glycylsarcosine; OAT, organic aniontransporter; LC-MS/MS, liquid chromatography-tandem mass spectrometry; PAH, p-aminohippuate; PCG, benzylpenicillin; h, human; HEK,human embryonic kidney; KRB, Krebs-Ringer bicarbonate; HBSS, Hanks’ balanced salt solution; AUC, area under the plasma concentration-timecurve.
0090-9556/10/3806-930–938$20.00DRUG METABOLISM AND DISPOSITION Vol. 38, No. 6Copyright © 2010 by The American Society for Pharmacology and Experimental Therapeutics 32060/3588628DMD 38:930–938, 2010 Printed in U.S.A.
930
at ASPE
T Journals on Septem
ber 15, 2018dm
d.aspetjournals.orgD
ownloaded from

the peptide transporters to accept cephalexin as a substrate (Amidonand Lee, 1994). The oral absorption of cephalexin is rapid and safe.Moreover, the renal secretion of most cephalosporins is believed to bemediated by organic anion transporters (OATs) located on the baso-lateral membrane of proximal tubule epithelia (Takeda et al., 2002;Shitara et al., 2003).
Strategies have been used to modify drugs that are absorbed poorlyby targeting them toward receptors/transporters for improved bio-availability (Sakaeda et al., 2001; Anand et al., 2002; Manfredini etal., 2002). To observe whether the combination of JBP485 and cepha-lexin affects either of the components, to identify the pharmacokineticmechanism(s) of interaction, and to provide a rationale for the clinicaluse of the drug combination, we have established an LC-MS/MSmethod for the determination of JBP485 and cephalexin. We used invivo oral administration, in situ intestinal perfusions, in vitro evertedsmall intestinal sac preparations, in vivo urinary excretion, in vitrokidney slices, and transfected cell uptake studies to investigate thechange in pharmacokinetics when JBP485 and cephalexin were usedtogether. Our results for renal excretion first indicated that JBP485might be a substrate of OATs. These findings demonstrate the impor-tance of recognizing that, when JBP485 and cephalexin are coadmin-istered, the target transporters are not only PEPTs but also OATs.
Materials and Methods
Chemicals. JBP485 was provided by Japan Bioproducts Industry Co. Ltd.(Tokyo, Japan). Reference standards of cephalexin and paracetamol (internalstandard), with purities of �99.0%, were purchased from the National Institutefor the Control of Pharmaceutical and Biological Products (Beijing, China).Probenecid, p-aminohippurate (PAH), benzylpenicillin (PCG), and Gly-sarwere purchased from Sigma-Aldrich (St. Louis, MO). Methanol was of high-performance liquid chromatography grade (Tedia, Carson City, NV). Thestable transfectants expressing hOAT1- or hOAT3-HEK293 and vector cells(mock) were a generous gift from Professor Yuichi Sugiyama, GraduateSchool of Pharmaceutical Sciences, University of Tokyo (Tokyo, Japan) andLi-kun Gong (Shanghai Institute of Materia Medicable, Chinese Academy ofScience, Shanghai, China). All other reagents and solvents were of analyticalgrade, and were commercially available.
Animals. Male Wistar rats that weighed 220 to 250 g were obtained fromthe Experimental Animal Center of Dalian Medical University (Dalian, China;permit number SCXK 2008-0002). They were allowed free access to water andchow diet but were fasted for 12 h with water ad libitum before the pharma-cokinetic experiments. All of the animal experiments were performed accord-ing to local institutional guidelines for the care and use of laboratory animals.
Pharmacokinetic Interaction Studies in Rats. In all cases, rats were fastedovernight and were anesthetized with ether before the onset of each experi-ment. JBP485 and cephalexin were dissolved in normal saline or buffersolution and administered to the rats in aqueous solution.
In vivo absorption experiment in rats. Rats were divided randomly intothree groups: 1) JBP485 alone (25 mg/kg) as control; 2) cephalexin alone (50mg/kg) as control; and 3) JBP485 (25 mg/kg) � cephalexin (50 mg/kg) as the
experimental group. The drugs were administered orally by gavage. Bloodsamples were collected at 1, 5, 10, 20, 30, 60, 120, 180, 240, 360, 480, and 600min for JBP485 and cephalexin determination, as described below.
In vitro everted intestinal sac preparation. The abdomen was opened by amidline incision, and the jejunum was removed by cutting across the upper endof the duodenum (i.e., �2 cm distal to the ligament of Treitz) and the lowerend of the ileum and manually stripping the mesentery. The small intestine waswashed out carefully with cold normal oxygenated saline, using a syringeequipped with a blunt end. Intestinal segments (10 � 1 cm) were evertedaccording to the conventional technique described by Wilson and Wiseman(1954) with modifications. The everted intestine was placed in glucose-salineat room temperature in a flat dish. A thread ligature was tied around one endto facilitate subsequent identification and to check for perforation. The emptysac was filled with 1 ml of Krebs-Ringer buffer (KRB) (pH 7.4) containing 0.5mM MgCl2, 4.5 mM KCl, 120 mM NaCl, 0.7 mM Na2HPO4, 1.5 mMNaH2PO4, 1.2 mM CaCl2, 15 mM NaHCO3, 10 mM glucose, and 20 mg/l ofphenolsulfonphthalein as a nonabsorbable marker. The distended sac wasplaced in incubation medium (mucosal solution) containing 1) 0.5 mMJBP485, 2) 1 mM cephalexin, and 3) 0.5 mM JBP485 � 1 mM cephalexin. Theincubation medium was surrounded by a water jacket maintained at 37°C. Agas mixture of 95% O2 and 5% CO2 was bubbled through the externalincubation medium during the incubation period. At the end of the incubationperiod, the serosal fluid was drained through a small incision into a test tube.Samples were collected for JBP485 and cephalexin determination as describedbelow.
In situ jejunal perfusion technique. A laparotomy was performed after etheranesthesia, and an inflow cannula made of Silastic tubing was inserted in thejejunum approximately 1 cm below the ligament of Treitz (Zhang et al., 2009).An outflow cannula was set up at a distance of 10 cm. The bile duct was ligatedto prevent possible enterohepatic circulation. The jejunal segment was thenflushed with saline solution (prewarmed to 37°C) to remove residual intestinalcontents. Oxygenated perfusion solution was delivered with a peristaltic pumpat a flow rate of 4 ml/15 min through an inlet tube water-jacketed at 37°C,before its entry into the jejunal segment. The solution for jejunal perfusion wasthe same as the KRB above. After a 30-min equilibration period, 0.5 mMJBP485 alone, 1 mM cephalexin alone, or 0.5 mM JBP485 � 1 mM cephalexindissolved in KRB was administered. Portal vein blood was collected using avein-detained needle, purchased from a clinic, at certain times for JBP485 andcephalexin determination, as described below.
In vivo plasma concentration and renal excretion. Rats were anesthetizedwith ether and grouped according to the following method and with intrave-nous administration via the jugular vein: 1) JBP485 (25 mg/kg), 2) cephalexin(50 mg/kg), or 3) JBP485 (25 mg/kg) � cephalexin (50 mg/kg). Blood sampleswere collected at 1, 5, 10, 30, 60, 120, 240, 360, 480, and 600 min. The bladderwas cannulated with polyethylene tubing, the distal end of which flowed intoan Eppendorf tube resting on a small pad of ice (Zhang et al., 2009). Urine wascollected directly from the bladder at 0.5, 1, 2, 4, 6, 8, 10, 12, and 24 h. Inaddition, the two drugs were administered together with probenecid (100mg/kg). The concentrations of JBP485 and cephalexin were measured. Thecumulative urinary excretion and renal clearance were calculated.
In vitro uptake in kidney slices. Rats were anesthetized with ether and fixedin the supine position on the operating table; kidneys were incised, decapsu-
FIG. 1. Chemical structures of JBP485 (A) and cephalexin (B).MW, molecular weight.
931INTERACTION OF JBP485 AND CEPHALEXIN WITH PEPTs AND OATs
at ASPE
T Journals on Septem
ber 15, 2018dm
d.aspetjournals.orgD
ownloaded from

lated, and immediately placed into oxygenated buffer at 4°C as described byNozaki et al. (2007). We investigated the viability of kidney slices by theuptake of PAH (a classic substrate of OAT1) in a concentration-dependentmanner. In brief, kidneys were cut into slices accurately using a ZQP-86 tissueslicer (thickness 300 �m, surface area approximately 0.15 cm2; Zhixin Co.,Ltd., Shanghai, China) and prepared in buffer. After preincubation for 3 minunder a carbogen atmosphere at 37°C in 6-well culture plates with gentleshaking, kidney slices were transferred to 24-well culture plates containingfresh carbogen-saturated JBP485 and/or cephalexin for further incubation. Inaddition, JBP485 in the presence or absence of Gly-sar (20 mM), probenecid(2 mM), PAH (0.5 mM), or PCG (0.5 mM) was used. The uptake wasmeasured at 0, 1, 5, 10, and 15 min. At the end of the incubation period, kidneyslices were washed with ice-cold Hanks’ balanced salt solution (HBSS) (pH7.5). Accumulated concentrations of JBP485 and cephalexin were determinedby LC-MS/MS after the kidney slices were homogenized. Krebs-bicarbonateslicing buffer consisted of 120 mM NaCl, 16.2 mM KCl, 1 mM CaCl2, 1.2 mMMgSO4, and 10 mM NaH2PO4/Na2HPO4, adjusted to pH 7.4.
Uptake Experiments Using Transporter Expression Systems. Uptakeexperiments with hOAT1- or hOAT3-HEK293 and mock cells were performedat 37°C in HBSS adjusted to pH 7.4. Cultured cells were washed and prein-cubated in the transport buffer for 15 min at 37°C. The uptake was initiated byadding transport buffer (1 ml) containing drugs including JBP485 (0.5 mM),with or without cephalexin (1 mM). After incubation for the designated timesat 37°C, the experiment was terminated by removal of the medium, followedby washing three times with 1 ml of ice-cold HBSS. The inhibition ofprobenecid (2 mM), PAH (0.5 mM), and PCG (0.5 mM) was also investigated.Samples were then analyzed by LC-MS/MS after homogenization.
Determination of JBP485 and Cephalexin by LC-MS/MS. Plasma sam-ples (50 �l) were added to 50 �l of the internal standard solution (200 ng/mlparacetamol) and 400 �l of methyl alcohol for deproteinization. After centrif-ugation at 12,000g for 10 min, the upper organic layer was transferred into apolythene tube and dried with nitrogen at 37°C. The dried residue wasdissolved in 200 �l of the mobile phase. Urine samples were diluted 20 timeswith the mobile phase. The other preparations were the same as the plasmasamples. The kidney slices were mixed with 300 �l of normal saline afterweighing and then homogenized (IKA-T10 homogenizer; IKA, Staufen, Ger-many) in an ice-bath environment. The other preparations were handled thesame as the plasma samples. To KRB and cell lysate samples (25 �l), 25 �l ofinternal standard (200 ng/ml paracetamol), 50 �l of methyl cyanide was added,followed by centrifugation for 10 min at 12,000g. Ten microliters of eachsample were injected for LC-MS/MS (API 3200; Applied Biosystems, FosterCity, CA) for analysis.
Data Analysis. Pharmacokinetics of JBP485 and cephalexin plasma con-centrations. The main pharmacokinetic parameters were calculated accordingto the 3P97 program. The quality of the fit was judged by evaluating the S.E.of parameter estimates and the coefficient of determination (r2) and by visualinspection of the residual plots. The main pharmacokinetic parameters werecalculated using eqs. 1 to 5. The plasma clearance (CLp) was calculated by thefollowing:
CLp � Dose/AUCi.v. (1)
where AUCi.v. is the area under the plasma concentration-time profile afterintravenous injection.
AUCi.v. � A/� � B/� (2)
The oral availability (F) was calculated as follows:
F � AUCp.o./AUCi.v. (3)
where AUCp.o. is the AUC after oral administration. It was calculated by thetrapezoidal rule. The renal clearance of JBP485 or cephalexin (CLr) wascalculated as follows:
CLR � Atotal/AUCi.v. (4)
where Atotal is the cumulative amount of JBP485 or cephalexin excreted inurine over 24 h. The uptake clearance of JBP485 or cephalexin (CLuptake) wascalculated as follows:
CLuptake � Atotal/AUC�0360� (5)
where Atotal is the cumulative uptake amount of JBP485 or cephalexin inkidney slices over 60 min. AUC(0360) is the area under the JBP485 orcephalexin plasma concentration-time curve from 0 to 60 min, as determinedby the trapezoidal rule.
Statistical analysis. Statistical analysis was performed using the SPSS11.5package. Test results are expressed as means � S.D. To test for statisticallysignificant differences among multiple treatments for a given parameter, one-way analysis of variance was performed. If p � 0.05 or p � 0.01, differenceswere considered statistically significant.
Results
Pharmacokinetic Interaction between JBP485 and Cephalexinin Intravenous and Oral Administration In Vivo. To understandthe target of interaction between JBP485 and cephalexin, the drugswere administered simultaneously by the intravenous or oral routes.As shown in Fig. 2, A and B, when the two drugs were administeredorally in combination, their plasma concentrations and AUCs weredecreased significantly compared with those for the control groups.The AUCs of JBP485 and cephalexin were only 35.2 and 23.4% ofthose of the respective control groups, and the Ka values of JBP485and cephalexin were approximately 70 and 50% of those of theircontrol groups, respectively. In addition, the other kinetic data forJBP485 such as t1/2� (2-fold), Tmax (delayed), Cmax (reduced to nearly10%), mean retention time (increased approximately 2.4-fold), and F(dropped to 35%) were all changed after drug combination comparedwith single administration. These parameters for cephalexin showedsimilar changes when it was coadministered with JBP485 (Table 1).However, when cephalexin and JBP485 were coadministered intrave-nously, their plasma concentrations and pharmacokinetic parameterswere almost unchanged compared with those of the correspondingcontrol groups (Fig. 2, C and D; Table 1).
Drug Interaction in Intestinal Uptake Studies In Vitro. Toexclude the impact of changes in physiological conditions and to furtherconfirm that the target of the interaction between JBP485 and cephalexinwas in the small intestine, we applied the rat everted gut sac model(Cummins et al., 2003) to investigate the interaction between JBP485 andcephalexin in vitro (Fig. 3). The serosal side concentrations of JBP485and cephalexin in the experimental groups were both decreased signifi-cantly compared with those of the corresponding control group (Fig. 3, Aand B). The AUCs of the experimental group were 52.9% (for JBP485)and 42.5% (for cephalexin) of that of the respective control groups. Thisresult suggested that JBP485 and cephalexin could inhibit the intestinaluptake of the other in vitro.
In Situ Single-Pass Intestinal Perfusion Studies of Drug Inter-actions. Because the uptake in everted small intestinal sacs in vitro islimited by their lack of an intact blood supply, we also performed insitu jejunal perfusions to clarify the mechanism of the interactionbetween JBP485 and cephalexin. When the two drugs were perfusedsimultaneously, the plasma concentrations of JBP485 and cephalexinin the portal vein were significantly lower than that of control groups(Fig. 4, A and B). AUCs of the experimental group were 43.6 and47.9% of controls, respectively.
Drug Interactions in Renal Excretion In Vivo. The cumulativeurine excretion during 24 h was 81.9 (for JBP485) and 91.8% (forcephalexin) when JBP485 (25 mg/kg) or cephalexin (50 mg/kg) wasadministered intravenously alone (Fig. 5, A and B). This findingindicated that renal excretion is the major route of excretion for thetwo drugs. When JBP485 and cephalexin were administered intrave-nously at the same time, the cumulative urine excretion decreased to68.1 (for JBP485) and to 74.5% (for cephalexin) of the correspondingcontrols, respectively (Fig. 5, A and B). The renal clearance rate of
932 ZHANG ET AL.
at ASPE
T Journals on Septem
ber 15, 2018dm
d.aspetjournals.orgD
ownloaded from

FIG. 2. Mean plasma concentration-timecurves of JBP485 and cephalexin afteroral and intravenous administration inrats. A, plasma concentration of JBP485after oral administration. B, plasma con-centration of cephalexin after oral admin-istration. C, plasma concentration ofJBP485 after intravenous administration.D, plasma concentration of JBP485 afterintravenous administration. Statisticaldifferences between each set of pointswere compared with those for the controlgroups by a two-tailed unpaired t test,with p � 0.05 as the limit of significance(�, p � 0.05; ��, p � 0.01). Data areexpressed as means � S.D. (n � 5).
FIG. 3. Time profile of JBP485 and cephalexin transported ineverted small intestinal sac preparations. A, serosal lateral concen-tration of JBP485. B, serosal lateral concentration of cephalexin.Statistical differences of each points were compared with those forthe control groups by a two-tailed unpaired t test, with p � 0.05 asthe limit of significance (�, p � 0.05; ��, p � 0.01). Data areexpressed as means � S.D. (n � 5).
TABLE 1
Pharmacokinetic parameters of JBP485 and cephalexin after oral or intravenous administration
Values represent means � S.D. (n � 5). Statistical analyses were conducted using a two-sided unpaired Student’s t test.
Parameters JBP485 JBP485 � Cephalexin Cephalexin Cephalexin � JBP485
OralCmax (�g/ml) 12.75 � 0.77 1.75 � 0.52a 53.97 � 4.09 6.41 � 1.43a
Tmax (min) 60 240a 60 120a
Ka (1/min) 0.11 � 0.0042 0.077 � 0.0013 0.0047 � 0.0006 0.0024 � 0.0001Ke (1/min) 0.0064 � 0.0004 0.0029 � 0.0011 0.0044 � 0.0022 0.0021 � 0.0016MRT (min) 194.07 � 1.64 469.784 � 2.47a 208.87 � 1.59 361.52 � 1.94b
AUC03 (min � �g/ml) 2724.63 � 83.81 958.97 � 76.32a 13657.48 � 95.10 3159.78 � 88.23a
t1/2� (min) 135.19 � 4.06 247.96 � 2.06b 86.98 � 3.13 157.42 � 2.69b
Vd/F (l/kg) 0.30 � 0.02 0.67 � 0.04 0.77 � 0.03 1.74 � 0.01CLp/F (ml/kg/min) 1.91 � 0.32 1.93 � 0.13 3.39 � 0.32 3.74 � 0.41
IntravenousF (%) 26.1 � 4.23 9.2 � 0.96a 81.2 � 9.72 18.8 � 1.81a
Cmax (�g/ml) 131.24 � 3.21 114.36 � 2.79 482.84 � 5.08 476.51 � 2.68Ke (1/min) 0.0085 � 0.0017 0.0098 � 0.0004 0.0079 � 0.0008 0.0080 � 0.0009MRT (min) 145.53 � 1.84 169.83 � 1.47 101.22 � 1.09 102.33 � 1.56AUC03 (min � �g/ml) 10455.37 � 130.17 11119.61 � 109.61 16817.26 � 163.34 16716.96 � 128.52t1/2� (min) 134.24 � 1.02 132.86 � 1.31 86.80 � 2.01 87.48 � 1.36Vd (l/kg) 0.28 � 0.04 0.23 � 0.01 0.42 � 0.03 0.42 � 0.05CLp (ml/kg/min) 2.39 � 0.19 2.25 � 0.15 3.32 � 0.32 3.36 � 0.18CLR (ml/kg/min) 2.23 � 0.16 1.58 � 0.09b 2.89 � 0.15 1.87 � 0.15b
MRT, mean retention time.a p � 0.001, compared with single administration.b p � 0.01.
933INTERACTION OF JBP485 AND CEPHALEXIN WITH PEPTs AND OATs
at ASPE
T Journals on Septem
ber 15, 2018dm
d.aspetjournals.orgD
ownloaded from

JBP485 or cephalexin was decreased to 70.9 (Fig. 5C) or 64.7% (Fig.5D) of that of the control group.
To further examine the mechanism for renal excretion of JBP485and cephalexin, the drugs were administered intravenously with 200mg/kg probenecid (a classic substrate of OATs) (Khamdang et al.,2003). Probenecid inhibited further the renal excretion of JBP485 andcephalexin when the three drugs were administered together (Fig. 6, Aand B). Probenecid also reduced the cumulative renal excretion ofJBP485 or cephalexin when it was given in conjunction with thesedrugs (Fig. 6, C and D). These findings indicate that JBP485 andcephalexin are excreted via OATs in the kidneys.
Drug Interaction in Kidney Slices. To further understand thetarget transporter related to renal excretion of JBP485 and cephalexin,we used rat fresh kidney slices to investigate the uptake of the twodrugs. Cephalexin (1 mM) could significantly inhibit JBP485 (0.5mM) uptake in renal slices (Fig. 7A). The rate of uptake (Liu et al.,1992) calculated with eq. 5 for JBP485 was 50.2% of the controlgroup. Conversely, JBP485 (0.5 mM) could also inhibit the uptake ofcephalexin (1 mM) in renal slices (Fig. 7B), and the rate of uptake was74.4% of the control group. To clarify the excretion mechanism ofJBP485, we investigated the effects of Gly-sar, probenecid, PAH (aspecific substrate of OAT1), and PCG (a specific substrate of OAT3) onJBP485 uptake. The uptake of JBP485 was significantly inhibited byGly-sar (Fig. 8A), probenecid (Fig. 8B), PAH (Fig. 8C), and PCG (Fig.8D), suggesting that JBP485 is a substrate for both PEPTs and OATs.
Uptake Interaction of JBP485 and Cephalexin in hOAT1- orhOAT3-HEK293 Cells. To examine whether JBP485 or cephalexin
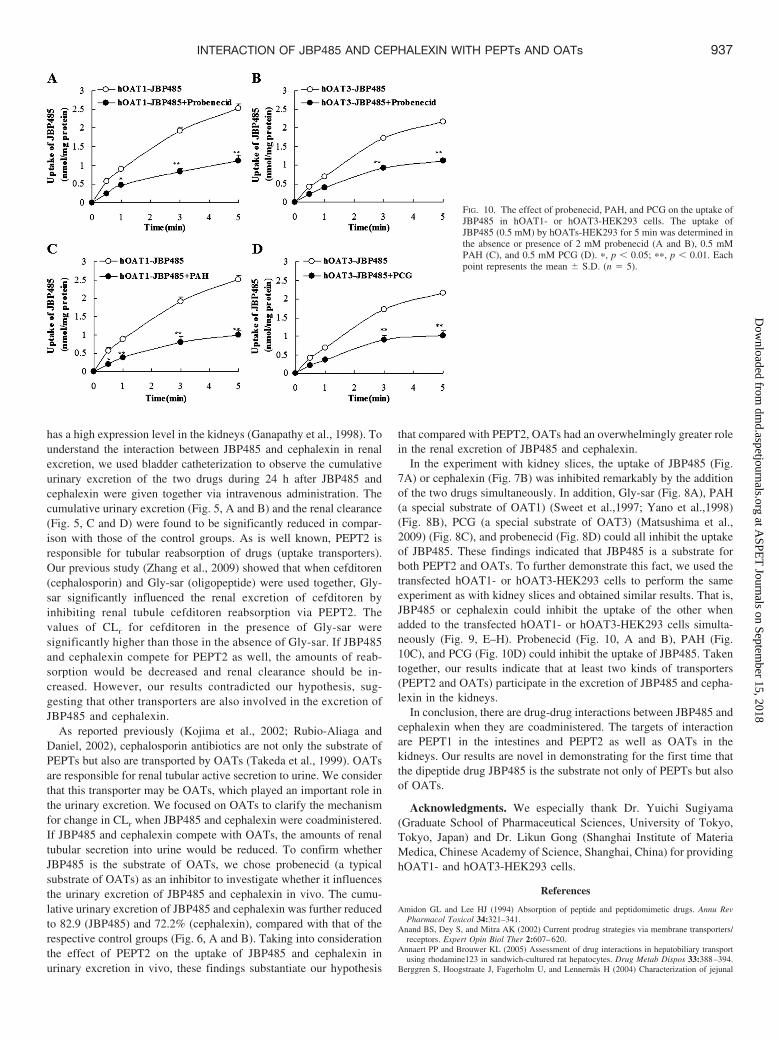
is the substrate of OAT1 or OAT3, the time profiles of JBP485 andcephalexin uptake in hOAT1- or hOAT3- and vector-HEK293 cellswere investigated. The uptake of JBP485 by hOAT1 (Fig. 9A) andhOAT3 (Fig. 9B) was markedly greater than that in vector-HEK293cells at all time points. Similar uptake by hOAT1 (Fig. 9C) andhOAT3 (Fig. 9D) was also found for cephalexin. This observationsuggests that JBP485 and cephalexin are substrates of OAT1 andOAT3, respectively. The uptakes of JBP485 in OAT1 (Fig. 9E) andOAT3 (Fig. 9F) and the uptakes of cephalexin in OAT1 (Fig. 9G) and OAT3(Fig. 9H) were inhibited significantly by addition of JBP485 andcephalexin simultaneously in the transfected cell system. Further-more, the uptake of JBP485 in hOAT1- or hOAT3-HEK293 cellscould be markedly inhibited by the typical OAT substrate probenecid(Fig. 10A for OAT1 and Fig. 10B for OAT3), the OAT1 substratePAH (Fig. 10C), and the OAT3 substrate PCG (Fig. 10D), suggestingthat OAT1 and OAT3 are involved in the uptake of JBP485.
Discussion
Clinical drug-drug interactions are expected to enhance the bene-ficial effect of a particular drug or reduce its toxicity. However, somedrug combinations will enhance the toxic effect or reduce the phar-macological effect; these are undesirable drug-drug interactions. An-tibiotics are used widely, and it is important to pay attention topotentially adverse effects when the antibiotics are combined withother drugs. Because of the broad substrate specificity of drug trans-porters, drug-drug interactions involving these transporters emergefrequently. Quinidine, which is an inhibitor of P-glycoprotein, could
FIG. 4. Mean plasma concentration-time curves of JBP485 andcephalexin during intrajejunal perfusions. A, plasma concentrationof JBP485. B, plasma concentration of cephalexin. Statistical dif-ferences of each of the points were compared with those for thecontrol groups by a two-tailed unpaired t test, with p � 0.05 as thelimit of significance (�, p � 0.05; ��, p � 0.01). Data are expressedas means � S.D. (n � 5).
FIG. 5. Urine excretion curves and renal clearances after intrave-nous injection of JBP485 and cephalexin. A, urine excretion ofJBP485. B, urine excretion of cephalexin. C, renal clearance ofJBP485. D, renal clearance of cephalexin. Data are expressed asmeans � S.D. (�, p � 0.05 versus control; ��, p � 0.01 versuscontrol; n � 5).
934 ZHANG ET AL.
at ASPE
T Journals on Septem
ber 15, 2018dm
d.aspetjournals.orgD
ownloaded from

increase plasma concentrations of digoxin because quinidine blocksbiliary and urinary excretion of digoxin via P-glycoprotein (Hedmanet al., 1991). Because the therapeutic range of digoxin is narrow,changes in its plasma concentration are potentially very serious.Research and development of peptide drugs is an area of much current
interest. Oligopeptides are known to be absorbed through brush-border membrane PEPT1 into intestinal epithelial cells (Kusuhara andSugiyama, 2002). �-Lactam antibiotics with a unique amide structurecan also be recognized by PEPTs (Berggren et al., 2004; Daniel andKottra, 2004) in the intestines. Thus, when combined with each other,
FIG. 6. Effects of probenecid on urinary excretion of JBP485 andcephalexin in rats in vivo. A, the cumulative urinary excretion ofJBP485 when JBP485, cephalexin, and probenecid were coadminis-tered; B, the cumulative urinary excretion of cephalexin when JBP485,cephalexin, and probenecid were coadministered; C, the cumulativeurinary excretion of JBP485 when JBP485 and probenecid were co-administered; D, the cumulative urinary excretion of cephalexin whencephalexin and probenecid were coadministered. Statistical differencesbetween each set of points were compared with those for the controlgroups by a two-tailed unpaired t test, with p � 0.05 as the limit ofsignificance (�, p � 0.05; ��, p � 0.01). Data are expressed asmeans � S.D. (n � 5).
FIG. 7. Time course of JBP485 (A) and cephalexin (B) uptake intokidney slices (�, p � 0.05; ��, p � 0.01; mean � S.D.; n � 3).
FIG. 8. The inhibition effects of Gly-sar (A), probenecid (B), PAH(C), and PCG (D) on JBP485 uptake in kidney slices. Statisticaldifferences between each set of points were compared with thecontrol groups by a two-tailed unpaired t test, with p � 0.05 as thelimit of significance (�, p � 0.05; ��, p � 0.01). Data are expressedas means � S.D. (n � 5).
935INTERACTION OF JBP485 AND CEPHALEXIN WITH PEPTs AND OATs
at ASPE
T Journals on Septem
ber 15, 2018dm
d.aspetjournals.orgD
ownloaded from

their pharmacological effect will be affected. With this in mind, in thisstudy we set up a sensitive and efficient LC-MS/MS method forsimultaneous detection of JBP485 and cephalexin to interpret themechanism(s) of their interaction, providing a strong basis for clinicaltherapy.
When JBP485 (25 mg/kg) and cephalexin (50 mg/kg) were admin-istered together orally, their plasma concentrations were significantlydecreased in comparison with those of the control groups (Fig. 2, Aand B). The bioavailability of JBP485 was decreased to 35% of thecontrol group and that of cephalexin was decreased to 23% of thecontrol group (Table 1). These results indicate that there is obviousmutual inhibition in intestinal absorption when the drugs are coad-ministered orally. Ka values were only approximately half of those fortheir corresponding control groups after oral combination (Table 1),indicating that the absorption velocity of the two drugs is delayedsignificantly. These results show clearly that when peptide drugs and�-lactam antibiotics are coadministered by the oral route, the extentand velocity of their absorption are both inhibited, obviously causingtheir therapeutic effect to be depressed. Nonetheless, there was noinhibition phenomenon when JBP485 and cephalexin were adminis-
tered intravenously at the same time (Fig. 2, A and B); there were nostatistically significant differences in CLp, AUCs, and Ke (Table 1).The pharmacokinetic contrast between the two routes of administra-tion indicated that the small intestine is the location of the interactionbetween JBP485 and cephalexin.
We focused on the small intestine, using everted gut sacs to observethe interaction between JBP485 and cephalexin. Cephalexin is gen-erally accepted as a substrate of PEPTs (Berggren et al., 2004). WhenJBP485 and cephalexin were used simultaneously, the respectiveconcentrations on the serosa side were both reduced (Fig. 3, A and B),suggesting that JBP485 is absorbed through PEPT1 in the gastroin-testinal tract in vitro. To confirm this speculation, we also appliedjejunal perfusions in situ to investigate the mechanism of the interac-tion between JBP485 and cephalexin. We found that the concentrationsof the two drugs in the portal vein were significantly lower than those in therespective control group, respectively (Fig. 4, A and B). These ob-servations indicate that JBP485 and cephalexin may compete forPEPT1 in the small intestine and that the mechanism of drug-druginteraction involves the inhibition of the absorption of each other.
PEPT1 is expressed extensively in the small intestines, and PEPT2
FIG. 9. Time profiles of the uptake of JBP485 and cephalexin byhOAT1- or hOAT3- and vector-HEK293 cells and the mutualinteraction between JBP485 and cephalexin. The time-dependentuptake of JBP485 (0.5 mM) (A and B) and cephalexin (1 mM) (Cand D) by hOAT1- or hOAT3- and vector-HEK293 cells wereexamined at 37°C. The mutual inhibition between JBP485 andcephalexin (E–H) in hOAT1-/hOAT3-HEK293 cells was alsoexamined. �, p � 0.05; ��, p � 0.01. Each point represents themean � S.D. (n � 5).
936 ZHANG ET AL.
at ASPE
T Journals on Septem
ber 15, 2018dm
d.aspetjournals.orgD
ownloaded from
has a high expression level in the kidneys (Ganapathy et al., 1998). Tounderstand the interaction between JBP485 and cephalexin in renalexcretion, we used bladder catheterization to observe the cumulativeurinary excretion of the two drugs during 24 h after JBP485 andcephalexin were given together via intravenous administration. Thecumulative urinary excretion (Fig. 5, A and B) and the renal clearance(Fig. 5, C and D) were found to be significantly reduced in compar-ison with those of the control groups. As is well known, PEPT2 isresponsible for tubular reabsorption of drugs (uptake transporters).Our previous study (Zhang et al., 2009) showed that when cefditoren(cephalosporin) and Gly-sar (oligopeptide) were used together, Gly-sar significantly influenced the renal excretion of cefditoren byinhibiting renal tubule cefditoren reabsorption via PEPT2. Thevalues of CLr for cefditoren in the presence of Gly-sar weresignificantly higher than those in the absence of Gly-sar. If JBP485and cephalexin compete for PEPT2 as well, the amounts of reab-sorption would be decreased and renal clearance should be in-creased. However, our results contradicted our hypothesis, sug-gesting that other transporters are also involved in the excretion ofJBP485 and cephalexin.
As reported previously (Kojima et al., 2002; Rubio-Aliaga andDaniel, 2002), cephalosporin antibiotics are not only the substrate ofPEPTs but also are transported by OATs (Takeda et al., 1999). OATsare responsible for renal tubular active secretion to urine. We considerthat this transporter may be OATs, which played an important role inthe urinary excretion. We focused on OATs to clarify the mechanismfor change in CLr when JBP485 and cephalexin were coadministered.If JBP485 and cephalexin compete with OATs, the amounts of renaltubular secretion into urine would be reduced. To confirm whetherJBP485 is the substrate of OATs, we chose probenecid (a typicalsubstrate of OATs) as an inhibitor to investigate whether it influencesthe urinary excretion of JBP485 and cephalexin in vivo. The cumu-lative urinary excretion of JBP485 and cephalexin was further reducedto 82.9 (JBP485) and 72.2% (cephalexin), compared with that of therespective control groups (Fig. 6, A and B). Taking into considerationthe effect of PEPT2 on the uptake of JBP485 and cephalexin inurinary excretion in vivo, these findings substantiate our hypothesis
that compared with PEPT2, OATs had an overwhelmingly greater rolein the renal excretion of JBP485 and cephalexin.
In the experiment with kidney slices, the uptake of JBP485 (Fig.7A) or cephalexin (Fig. 7B) was inhibited remarkably by the additionof the two drugs simultaneously. In addition, Gly-sar (Fig. 8A), PAH(a special substrate of OAT1) (Sweet et al.,1997; Yano et al.,1998)(Fig. 8B), PCG (a special substrate of OAT3) (Matsushima et al.,2009) (Fig. 8C), and probenecid (Fig. 8D) could all inhibit the uptakeof JBP485. These findings indicated that JBP485 is a substrate forboth PEPT2 and OATs. To further demonstrate this fact, we used thetransfected hOAT1- or hOAT3-HEK293 cells to perform the sameexperiment as with kidney slices and obtained similar results. That is,JBP485 or cephalexin could inhibit the uptake of the other whenadded to the transfected hOAT1- or hOAT3-HEK293 cells simulta-neously (Fig. 9, E–H). Probenecid (Fig. 10, A and B), PAH (Fig.10C), and PCG (Fig. 10D) could inhibit the uptake of JBP485. Takentogether, our results indicate that at least two kinds of transporters(PEPT2 and OATs) participate in the excretion of JBP485 and cepha-lexin in the kidneys.
In conclusion, there are drug-drug interactions between JBP485 andcephalexin when they are coadministered. The targets of interactionare PEPT1 in the intestines and PEPT2 as well as OATs in thekidneys. Our results are novel in demonstrating for the first time thatthe dipeptide drug JBP485 is the substrate not only of PEPTs but alsoof OATs.
Acknowledgments. We especially thank Dr. Yuichi Sugiyama(Graduate School of Pharmaceutical Sciences, University of Tokyo,Tokyo, Japan) and Dr. Likun Gong (Shanghai Institute of MateriaMedica, Chinese Academy of Science, Shanghai, China) for providinghOAT1- and hOAT3-HEK293 cells.
References
Amidon GL and Lee HJ (1994) Absorption of peptide and peptidomimetic drugs. Annu RevPharmacol Toxicol 34:321–341.
Anand BS, Dey S, and Mitra AK (2002) Current prodrug strategies via membrane transporters/receptors. Expert Opin Biol Ther 2:607–620.
Annaert PP and Brouwer KL (2005) Assessment of drug interactions in hepatobiliary transportusing rhodamine123 in sandwich-cultured rat hepatocytes. Drug Metab Dispos 33:388–394.
Berggren S, Hoogstraate J, Fagerholm U, and Lennernas H (2004) Characterization of jejunal
FIG. 10. The effect of probenecid, PAH, and PCG on the uptake ofJBP485 in hOAT1- or hOAT3-HEK293 cells. The uptake ofJBP485 (0.5 mM) by hOATs-HEK293 for 5 min was determined inthe absence or presence of 2 mM probenecid (A and B), 0.5 mMPAH (C), and 0.5 mM PCG (D). �, p � 0.05; ��, p � 0.01. Eachpoint represents the mean � S.D. (n � 5).
937INTERACTION OF JBP485 AND CEPHALEXIN WITH PEPTs AND OATs
at ASPE
T Journals on Septem
ber 15, 2018dm
d.aspetjournals.orgD
ownloaded from

absorption and apical efflux of ropivacaine, lidocaine and bupivacaine in the rat using in situand in vitro absorption models. Eur J Pharmacol 21:553–560.
Bretschneider B, Brandsch M, and Neubert R (1999) Intestinal transport of �-lactam antibiotics:analysis of the affinity at the H�/peptide symporter (PEPT1), and the uptake into Caco-2 cellmonolayers and the transepithelial flux. Pharm Res 16:55–61.
Cummins CL, Salphati L, Reid MJ, and Benet LZ (2003) In vivo modulation of intestinal CYP3Ametabolism by P-glycoprotein: studies using the rat single-pass intestinal perfusion model.J Pharmacol Exp Ther 305:306–314.
Daniel H and Kottra G (2004) The proton oligopeptide cotransporter family SLC15 in physiologyand pharmacology. Pflugers Arch 447:610–618.
Ganapathy ME, Huang W, Wang H, Ganapathy V, and Leibach FH (1998) Valacyclovir: asubstrate for the intestinal and renal peptide transporters PEPT1 and PEPT2. Biochem BiophysRes Commun 246:470–475.
Ganapathy V, Brandsch M, and Leibach FH (1994) Intestinal transport of amino acids andpeptides, in Physiology of the Gastrointestinal Tract (Johnson LR ed) 3rd ed, pp 1773–1794,Raven Press, New York.
Hedman A, Angelin B, Arvidsson A, Beck O, Dahlqvist R, Nilsson B, Olsson M, and Schenck-Gustafsson K (1991) Digoxin-verapamil interaction: reduction of biliary but not renal digoxinclearance in humans. Clin Pharmacol Ther 49:256–262.
Khamdang S, Teda M, Babu E, Marreli M, and Shafran SD (2003) Interaction of human and ratorganic anion transporter 2 with various cephalosporin antibiotics. Eur J Pharmacol 465:l-7.
Kojima R, Sekine T, Kawachi M, Cha SH, Suzuki Y, and Endou H (2002) Immunolocalizationof multispecific organic anion transporters, OAT1, OAT2, and OAT3, in rat kidney. J Am SocNephrol 13:848–857.
Kusuhara H and Sugiyama Y (2002) Role of transporters in the tissue-selective distribution andelimination of drugs: transporters in the liver, small intestine, brain and kidney. J ControlRelease 78:43–54.
Leibach FH and Ganapathy V (1996) Peptide transporters in the intestine and the kidney. AnnuRev Nutr 16:99–119.
Liu KX, Kato Y, Kaku T, and Sugiyama Y (1998) Human placental extract stimulates liverregeneration in rats. Biol Pharm Bull 21:44–49.
Liu KX, Kato Y, Kaku TI, Santa T, Imai K, Yagi A, Ishizu T, and Sugiyama Y (2000)Hydroxyprolylserine derivatives JBP923 and JBP485 exhibit the antihepatitis activities aftergastrointestinal absorption in rats. J Pharmacol Exp Ther 294:510–515.
Liu KX, Kato Y, Narukawa M, Kim DC, Hanano M, Higuchi O, Nakamura T, and Sugiyama Y(1992) Importance of the liver in plasma clearance of hepatocyte growth factors in rats. Am JPhysiol 263:G642–G649.
Manfredini S, Pavan B, Vertuani S, Scaglianti M, Compagnone D, Biondi C, Scatturin A,Tanganelli S, Ferraro L, Prasad P, et al. (2002) Design, synthesis and activity of ascorbic acidprodrugs of nipecotic, kynurenic and diclophenamic acids, liable to increase neurotropicactivity. J Med Chem 45:559–562.
Matsushima S, Maeda K, Inoue K, Ohta KY, Yuasa H, Kondo T, Nakayama H, Horita S,Kusuhara H, and Sugiyama Y(2009) The inhibition of human multidrug and toxin extrusion
1 is involved in the drug-drug interaction caused by cimetidine. Drug Metab Dispos 37:555–559.
Nozaki Y, Kusuhara H, Kondo T, Hasegawa M, Shiroyanagi Y, Nakazawa H, Okano T, andSugiyama Y (2007) Characterization of the uptake of organic anion transporter (OAT) 1 andOAT3 substrates by human kidney slices. J Pharmacol Exp Ther 321:362–369.
Ogawa M, Suzuki H, Sawada Y, Hanano M, and Sugiyama Y (1994) Kinetics of active effluxvia choroid plexus of �-lactam antibiotics from the CSF into the circulation. Am J Physiol266:R392–R399.
Rubio-Aliaga I and Daniel H (2002) Mammalian peptide transporters as targets for drug delivery.Trends Pharmacol Sci 23:434–440.
Sakaeda T, Tada Y, Sugawara T, Ryu T, Hirose F, Yoshikawa T, Hirano K, Kupczyk-Subotkowska L, Siahaan TJ, Audus KL, et al. (2001) Conjugation with L-glutamate for in vivobrain drug delivery. J Drug Target 9:23–37.
Shitara Y, Li AP, Kato Y, Lu C, Ito K, Itoh T, and Sugiyama Y (2003) Function of uptaketransporters for taurocholate and estradiol 17�-D-glucuronide in cryopreserved human hepa-tocytes. Drug Metab Pharmacokinet 18:33–41.
Sweet DH, Wolff NA, and Pritchard JB (1997) Expression cloning and characterization ofROAT1. The basolateral organic anion transporter in rat kidney. J Biol Chem 272:30088–30095.
Takeda M, Babu E, Narikawa S, and Endou H (2002) Interaction of human organic aniontransporters with various cephalosporin antibiotics. Eur J Pharmacol 438:137–142.
Takeda M, Tojo A, Sekine T, Hosoyamada M, Kanai Y, and Endou H (1999) Role of organicanion transporter 1 (OAT1) in cephaloridine (CER)-induced nephrotoxicity. Kidney Int 56:2128–2136.
Wilson TH and Wiseman G (1954) The use of sacs of everted small intestine for the study of thetransference of substances from the mucosal to the serosal surface. J Physiol 123:116–125.
Wu J, Wang C, Liu Q, Yang T, Zhang Q, Peng J, Gao Y, Sun H, Kaku T, and Liu K (2008)Protective effect of JBP485 on concanavalin A-induced hepatocyte toxicity in primary cul-tured rat hepatocytes. Eur J Pharmacol 589:299–305.
Yang T, Wu J, Wang C, Liu Q, Ma X, Peng J, Kaku T, and Liu K (2009) Protective effect ofJBP485 on concanavalin A-induced liver injury in mice. J Pharm Pharmacol 61:767–774.
Yano I, Takayama A, Takano M, Inatani M, Tanihara H, Ogura Y, Honda Y, and Inui K (1998)Pharmacokinetics and pharmacodynamics of acetazolamide in patients with transient intraoc-ular pressure elevation. Eur J Clin Pharmacol 54:63–68.
Zhang Q, Liu Q, Wu J, Wang C, Peng J, Ma X, and Liu K (2009) PEPT1 involved in the uptakeand transepithelial transport of cefditoren in vivo and in vitro. Eur J Pharmacol 612:9–14.
Address correspondence to: Dr. Kexin Liu, Department of Clinical Pharma-cology, College of Pharmacy, Dalian Medical University, 9 West Section, LvshunSouth Road, Lvshunkou District, Dalian 116044, China. E-mail: [email protected]
938 ZHANG ET AL.
at ASPE
T Journals on Septem
ber 15, 2018dm
d.aspetjournals.orgD
ownloaded from





























