Embed Size (px)
Citation preview

Cytotoxic T Lymphocytes and Vaccine Development 2013
Guest Editors: Zhengguo Xiao, Kim Klonowski, Hanchun Yang, and Julie Curtsinger
BioMed Research International

Cytotoxic T Lymphocytes andVaccine Development 2013

BioMed Research International
Cytotoxic T Lymphocytes andVaccine Development 2013
Guest Editors: Zhengguo Xiao, Kim Klonowski,Hanchun Yang, and Julie Curtsinger

Copyright © 2013 Hindawi Publishing Corporation. All rights reserved.
This is a special issue published in “BioMed Research International.” All articles are open access articles distributed under the CreativeCommons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the originalwork is properly cited.

Editorial BoardThe editorial board of the journal is organized into sections that correspond to
the subject areas covered by the journal.
Agricultural Biotechnology
Ahmad Zuhairi Abdullah, MalaysiaGuihua H. Bai, USAChristopher P. Chanway, CanadaRavindra N. Chibbar, CanadaAdriana S. Franca, BrazilIan Godwin, Australia
Hari B. Krishnan, USACarol A. Mallory-Smith, USAXiaoling Miao, ChinaDennis P. Murr, CanadaRodomiro Ortiz, SwedenEncarnacion Ruiz, Spain
B. C. Saha, USAAbdurrahman Saydut, TurkeyMariam B. Sticklen, USAKok Tat Tan, MalaysiaChiu-Chung Young, Taiwan
Animal Biotechnology
E. S. Chang, USABhanu P. Chowdhary, USANoelle E. Cockett, USAPeter Dovc, SloveniaScott C. Fahrenkrug, USADorian J. Garrick, USAThomas A. Hoagland, USA
Tosso Leeb, SwitzerlandJames D. Murray, USAAnita M. Oberbauer, USAJorge A. Piedrahita, USADaniel Pomp, USAKent M. Reed, USALawrence Reynolds, USA
Lawrence B. Schook, USAMari A. Smits, The NetherlandsLeon Spicer, USAJ. Verstegen, USAMatthew B. Wheeler, USAKenneth L. White, USA
Biochemistry
David Ronald Brown, UKSaulius Butenas, USAVittorio Calabrese, ItalyMiguel Castanho, PortugalFrancis J. Castellino, USARoberta Chiaraluce, ItalyD. M. Clarke, CanadaFrancesca Cutruzzola, ItalyPaul W. Doetsch, USA
Hicham Fenniri, CanadaNick V. Grishin, USAJ. Guy Guillemette, CanadaPaul W. Huber, USAChen-Hsiung Hung, TaiwanMaria Jerzykiewicz, PolandMichael Kalafatis, USAB. E. Kemp, AustraliaPhillip E. Klebba, USA
Wen-Hwa Lee, USAGeorge Makhatadze, USALeonid Medved, USASusan A. Rotenberg, USAJason Shearer, USAAndrei Surguchov, USAJohn B. Vincent, USAY. George Zheng, USA
Bioinformatics
T. Akutsu, JapanMiguel A. Andrade, GermanyMark Y. Borodovsky, USARita Casadio, ItalyDavid Corne, UKSorin Draghici, USA
Eugenio Ferreira, PortugalStavros J. Hamodrakas, GreecePaul Harrison, USAGeorge Karypis, USAGuohui Lin, CanadaSatoru Miyano, Japan
Zoran Obradovic, USAFlorencio Pazos, SpainZhirong Sun, ChinaYing Xu, USAAlexander Zelikovsky, USAAlbert Zomaya, Australia

Biophysics
Miguel Castanho, PortugalP. Bryant Chase, USAKuo-Chen Chou, USARizwan Khan, India
Ali A. Khraibi, Saudi ArabiaRumiana Koynova, USASerdar Kuyucak, AustraliaJianjie Ma, USA
S. B. Petersen, DenmarkPeter Schuck, USAClaudio M. Soares, Portugal
Cell Biology
Omar Benzakour, FranceSanford I. Bernstein, USAPhillip I. Bird, AustraliaEric Bouhassira, USAMohamed Boutjdir, USAChung-Liang Chien, TaiwanRichard Gomer, USAPaul J. Higgins, USAPavel Hozak, Czech Republic
Xudong Huang, USAAnton M. Jetten, USASeamus J. Martin, IrelandManuela Martins-Green, USAShoichiro Ono, USAGeorge Perry, USAM. Piacentini, ItalyGeorge E. Plopper, USALawrence Rothblum, USA
Michael Sheetz, USAJames L. Sherley, USAG. S. Stein, USARichard Tucker, USAThomas van Groen, USAAndre Van Wijnen, USASteve Winder, UKChuanyue Wu, USABin-Xian Zhang, USA
Genetics
Adewale Adeyinka, USAClaude Bagnis, FranceJ. Birchler, USASusan Blanton, USABarry J. Byrne, USAR. Chakraborty, USADomenico Coviello, ItalySarah H. Elsea, USACelina Janion, Poland
J. Spencer Johnston, USAM. Ilyas Kamboh, USAFeige Kaplan, CanadaManfred Kayser, The NetherlandsBrynn Levy, USAXiao Jiang Li, USAThomas Liehr, GermanyJames M. Mason, USAMohammed Rachidi, France
Raj S. Ramesar, South AfricaElliot D. Rosen, USADharambir K. Sanghera, USAMichael Schmid, GermanyMarkus Schuelke, GermanyWolfgang Arthur Schulz, GermanyJorge Sequeiros, PortugalMouldy Sioud, NorwayRongjia Zhou, China
Genomics
Vladimir Bajic, Saudi ArabiaMargit Burmeister, USASettara Chandrasekharappa, USAYataro Daigo, Japan
J. Spencer Johnston, USAVladimir Larionov, USAThomas Lufkin, SingaporeJohn L. McGregor, France
John V. Moran, USAYasushi Okazaki, JapanGopi K. Podila, USAMomiao Xiong, USA

Editorial BoardThe editorial board of the journal is organized into sections that correspond to
the subject areas covered by the journal.
Immunology
Hassan Alizadeh, USAPeter Bretscher, CanadaRobert E. Cone, USATerry L. Delovitch, CanadaAnthony L. DeVico, USANick Di Girolamo, AustraliaDon Mark Estes, USASoldano Ferrone, USAJeffrey A. Frelinger, USAJohn Robert Gordon, Canada
James D. Gorham, USASilvia Gregori, ItalyThomas Griffith, USAYoung S. Hahn, USADorothy E. Lewis, USABradley W. McIntyre, USAR. Lee Mosley, USAMarija Mostarica-Stojkovic, SerbiaHans Konrad Muller, AustraliaAli Ouaissi, France
Kanury V. S. Rao, IndiaYair Reisner, IsraelHarry W. Schroeder, USAWilhelm Schwaeble, UKNilabh Shastri, USAYufang Shi, ChinaPiet Stinissen, BelgiumHannes Stockinger, AustriaGraham R. Wallace, UK
Microbial Biotechnology
Suraini Abd-Aziz, MalaysiaJozef Anne, BelgiumNuri Azbar, TurkeyYoav Bashan, MexicoMarco Bazzicalupo, ItalyHakan Bermek, TurkeyNico Boon, BelgiumJose Luis Campos, SpainYinguang Chen, ChinaLuca Simone Cocolin, Italy
Peter Coloe, AustraliaDaniele Daffonchio, ItalyHan de Winde, The NetherlandsRaf Dewil, BelgiumJose Domingos Fontana, BrazilPetros Gikas, GreeceTom Granstrom, FinlandIsmail Kiran, TurkeyHongjuan Liu, ChinaYanhe Ma, China
Paula Loureiro Paulo, BrazilBernd H. A. Rehm, New ZealandAlberto Reis, PortugalMuthuswamy Sathishkumar, SingaporeRamkrishna Sen, IndiaAngela Sessitsch, AustriaYa-Jie Tang, ChinaOrhan Yenigun, TurkeyEileen Hao Yu, UK
Microbiology
D. Beighton, UKSteven R. Blanke, USAStanley Brul, The NetherlandsIsaac K. O. Cann, USAStephen K. Farrand, USAAlain Filloux, UK
Gad Frankel, UKRoy Gross, GermanyHans-Peter Klenk, GermanyTanya Parish, UKGopi K. Podila, USAFrederick D. Quinn, USA
Didier A. Raoult, FranceIsabel Sa-Correia, PortugalP. L. C. Small, USAMichael Thomm, GermanyH. C. van der Mei, The NetherlandsSchwan William, USA
Molecular Biology
Rudi Beyaert, BelgiumMichael Bustin, USADouglas Cyr, USAK. Iatrou, GreeceLokesh Joshi, Ireland
David W. Litchfield, CanadaWuyuan Lu, USAPatrick Matthias, SwitzerlandJohn L. McGregor, FranceS. L. Mowbray, Sweden
Elena Orlova, UKYeon-Kyun Shin, USAWilliam S. Trimble, CanadaLisa Wiesmuller, GermanyMasamitsu Yamaguchi, Japan

Oncology
Colin Cooper, UKF. M. J. Debruyne, The NetherlandsNathan Ames Ellis, USADominic Fan, USAGary E. Gallick, USADaila S. Gridley, USAXin-yuan Guan, Hong KongAnne Hamburger, USAManoor Prakash Hande, SingaporeBeric Henderson, Australia
Daehee Kang, Republic of KoreaAbdul R. Khokhar, USARakesh Kumar, USAMacus Tien Kuo, USAEric W. Lam, UKSue-Hwa Lin, USAKapil Mehta, USAOrhan Nalcioglu, USAP. J. Oefner, GermanyAllal Ouhtit, Oman
Frank Pajonk, USAWaldemar Priebe, USAF. C. Schmitt, PortugalSonshin Takao, JapanAna Maria Tari, USAHenk G. Van Der Poel, The NetherlandsHaodong Xu, USADavid J. Yang, USA
Pharmacology
Abdel A. Abdel-Rahman, USAM. Badr, USAStelvio M. Bandiera, CanadaRonald E. Baynes, USAR. Keith Campbell, USAHak-Kim Chan, AustraliaMichael D. Coleman, UKJ. Descotes, FranceDobromir Dobrev, Germany
Ayman El-Kadi, CanadaJeffrey Hughes, USAKazim Husain, USAFarhad Kamali, UKMichael Kassiou, AustraliaJoseph J. McArdle, USAMark J. McKeage, New ZealandDaniel T. Monaghan, USAT. Narahashi, USA
Kennerly S. Patrick, USAVickram Ramkumar, USAMichael J. Spinella, USAQuadiri Timour, FranceTodd W. Vanderah, USAVal J. Watts, USADavid J. Waxman, USA
Plant Biotechnology
Prem L. Bhalla, AustraliaJ. R. Botella, AustraliaElvira Gonzalez De Mejia, USAShi-You Ding, USA
Metin Guru, TurkeyH. M. Haggman, FinlandLiwen Jiang, Hong KongP. B. Kirti, India
Yong Pyo Lim, Republic of KoreaGopi K. Podila, USARalf Reski, GermanySudhir Sopory, India
Toxicology
Michael Aschner, USAJuergen Buenger, GermanyMichael L. Cunningham, USALaurence D. Fechter, USA
Hartmut Jaeschke, USAY. James Kang, USAM. Firoze Khan, USAPascal Kintz, France
Qaisar Mahmood, PakistanR. S. Tjeerdema, USAKenneth Turteltaub, USABrad Upham, USA

Editorial BoardThe editorial board of the journal is organized into sections that correspond to
the subject areas covered by the journal.
Virology
Nafees Ahmad, USAEdouard Cantin, USAEllen Collisson, USAKevin M. Coombs, CanadaNorbert K. Herzog, USATom Hobman, CanadaShahid Jameel, India
Fred Kibenge, CanadaFenyong Liu, USAEric Rassart, CanadaGerald G. Schumann, GermanyY.-C. Sung, Republic of KoreaGregory Tannock, Australia
Ralf Wagner, GermanyJianguo Wu, ChinaDecheng Yang, CanadaJiing-Kuan Yee, USAXueping Zhou, ChinaWen-Quan Zou, USA

Contents
Cytotoxic T Lymphocytes and Vaccine Development 2013, Zhengguo Xiao, Kim Klonowski,Hanchun Yang, and Julie CurtsingerVolume 2013, Article ID 865314, 1 page
Characterization of CD8+ T-Cell Responses in the Peripheral Blood and Skin Injection Sites ofMelanoma Patients Treated with mRNA Electroporated Autologous Dendritic Cells (TriMixDC-MEL),Daphne Benteyn, An M. T. Van Nuffel, Sofie Wilgenhof, Jurgen Corthals, Carlo Heirman, Bart Neyns,Kris Thielemans, and Aude BonehillVolume 2013, Article ID 976383, 8 pages
IL-6 Production by Dendritic Cells Is Dispensable for CD8+ Memory T-Cell Generation, Jean-FrancoisDaudelin, Melissa Mathieu, Salix Boulet,and Nathalie LabrecqueVolume 2013, Article ID 126189, 12 pages
Increased Toll-Like Receptor Signaling Pathways Characterize CD8+ Cells in Rapidly Progressive SIVInfection, Maria Cecilia Garibaldi Marcondes, Celsa Spina, Eduardo Bustamante, and Howard FoxVolume 2013, Article ID 796014, 7 pages
What Is Recent in Pancreatic Cancer Immunotherapy?, Elena Niccolai, Domenico Prisco,Mario Milco D’Elios, and Amedeo AmedeiVolume 2013, Article ID 492372, 14 pages
MUC1-Specific Cytotoxic T Lymphocytes in Cancer Therapy: Induction and Challenge, David Roulois,Marc Gregoire, and Jean-Francois FonteneauVolume 2013, Article ID 871936, 10 pages
CpG and Interleukin-15 Synergize to Enhance IFN-γ Production by Activated CD8+ T Cells,Dustin Cobb, Siqi Guo, and Ronald B. SmeltzVolume 2013, Article ID 924023, 12 pages

Hindawi Publishing CorporationBioMed Research InternationalVolume 2013, Article ID 865314, 1 pagehttp://dx.doi.org/10.1155/2013/865314
EditorialCytotoxic T Lymphocytes and Vaccine Development 2013
Zhengguo Xiao,1 Kim Klonowski,2 Hanchun Yang,3 and Julie Curtsinger4
1 Department of Animal and Avian Sciences, University of Maryland, College Park, MD 20742, USA2 Department of Cellular Biology, University of Georgia, Athens, GA 30602, USA3 Department of Veterinary Microbiology and Immunology, College of Veterinary Medicine, China Agricultural University,Beijing 100083, China
4 Center for Immunology, University of Minnesota, Minneapolis, MN 55455, USA
Correspondence should be addressed to Zhengguo Xiao; [email protected]
Received 1 January 2013; Accepted 1 January 2013
Copyright © 2013 Zhengguo Xiao et al.is is an open access article distributed under the Creative CommonsAttribution License,which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
CTLs play a critical role in �ghting chronic virus infectionand cancers. Despite the tremendous efforts spent so far,generating potent and highly effective CTLs remain a majorroadblock for disease control (prevention and therapies).Activation of CTLs requires different stimuli and is in�u-enced by different disease conditions. In this special issue,several papers investigated functions of cytokines, such asIL-15 and IL-6, in activation of CTLs. Several papers arefocusing on the therapeutic effects of CTLs in different can-cers using novel approaches targeting tumor-speci�c CTLs.In addition, potential markers and mechanisms for rapidprogression of AIDS are suggested using SIV model.
In the �rst paper entitled “IL-6 production by dendriticcells is dispensable for 𝐶𝐶𝐶𝐶𝐶+ memory T-cell generation,” Dau-delin et al. describe a new observation that IL-6 is notrequired in APC vaccination.
In the second paper entitled “What is recent in pancreaticcancer immunotherapy?” Niccolai et al. summarize the cur-rent immunotherapy of pancreatic cancer targeting cancer-associated antigens.
In the third paper entitled “MUC1-speci�c cytotoxic Tlymphocytes in cancer therapy: induction and challenge,”Roulois et al. review current knowledge regarding MUC1 asa potential target tumor therapeutic vaccines.
In the fourth paper entitled “Characterization of 𝐶𝐶𝐶𝐶𝐶+T cell responses in the peripheral blood and skin injectionsites of melanoma patients treated with mRNA electroporatedautologous dendritic cells (TriMixDC-MEL),” Benteyn et al.report that functional TAA-speci�c CD𝐶+ T cells are detected
in both the skin and the peripheral blood aer TriMixDC-MEL therapy.
In the �h paper entitled “CpG and interleukin-15 syn-ergize to enhance IFN-production by activated 𝐶𝐶𝐶𝐶𝐶+ T cells,”Cobb et al. present evidence that IL-15 synergizes withCpG in the induction of IFN-𝛾𝛾 in activated CD𝐶+ T cells,expanding our understanding about the function of IL-15.
In the sixth paper entitled “Increased Toll-like receptorsignaling pathways characterize 𝐶𝐶𝐶𝐶𝐶+ cells in rapidly pro-gressive SIV infection,” Marcondes et al. suggest that TLRoverexpression may indicate ineffective T-cell response inrapid progression in HIV infection.
We thank all of the authors for their great contributions tothis special issue and appreciate their patience in processingtheir manuscripts. We also want to thank all of the reviewerswho went through the manuscripts multiple times, providinginsightful suggestions. We really hope this special issue willcontinue to thrive as a timely communication platform forbasic and translational research on CTLs.
Zhengguo XiaoKim KlonowskiHanchun YangJulie Curtsinger






Fold
incr
ease
IFN
-γse
cret
ion
40
30
20
10
0
Neg
ativ
e co
ntr
ol
Tyro
sin
ase
MA
GE
-A3
MA
GE
-C2
gp10
0
PretreatmentPosttreatment
22
7
5
2.5
0
181–
195
185–
199
253–
267
257–
271
Pept
ide
41
Pept
ide
42
Pept
ide
43
Pept
ide
44
Pept
ide
45
Pept
ide
46
Pept
ide
47
Pept
ide
48
Pept
ide
49
Pept
ide
50
Pept
ide
61
Pept
ide
62
Pept
ide
63
Pept
ide
64
Pept
ide
65
Pept
ide
66
Pept
ide
67
Pept
ide
68
Pept
ide
69
Pept
ide
70
Fold
incr
ease
IFN
-γse
cret
ion
PretreatmentPosttreatment



Hindawi Publishing CorporationBioMed Research InternationalVolume 2013, Article ID 126189, 12 pageshttp://dx.doi.org/10.1155/2013/126189
Research ArticleIL-6 Production by Dendritic Cells Is Dispensable forCD8+Memory T-Cell Generation
Jean-François Daudelin,1 Mélissa Mathieu,1, 2 Salix Boulet,1 and Nathalie Labrecque1, 2, 3
1 Maisonneuve-Rosemont Hospital Research Center, University of Montreal, 5415 Boulevard de l’Assomption,Montréal, QC, Canada H1T 2M4
2 Department of Microbiology and Immunology, University of Montreal, Montréal, QC, Canada H3C 3J73 Department of Medicine, University of Montreal, Montréal, QC, Canada H3C 3J7
Correspondence should be addressed to Nathalie Labrecque; [email protected]
Received 18 May 2012; Accepted 19 June 2012
Academic Editor: Zhengguo Xiao
Copyright © 2013 Jean-François Daudelin et al.is is an open access article distributed under the Creative Commons AttributionLicense, which permits unrestricted use, distribution, and reproduction in anymedium, provided the originalwork is properly cited.
Following activation, naïve CD8+ T cells will differentiate into effectors that differ in their ability to survive: some will persist asmemory cells while the majority will die by apoptosis. Signals given by antigen-presenting cells (APCs) at the time of primingmodulate this differential outcome. We have recently shown that, in opposition to dendritic cell (DC), CD40-activated B-(CD40-B) cell vaccination fails to efficiently produce CD8+ memory T cells. Understanding why CD40-B-cell vaccination does not leadto the generation of functional long-lived memory cells is essential to de�ne the signals that should be provided to naïve T cellsby APCs. Here we show that CD40-B cells produce very low amount of IL-6 when compared to DCs. However, supplementationwith IL-6 during CD40-B-cell vaccination did not improve memory generation. Furthermore, IL-6-de�cient DCs maintained thecapacity to promote the formation of functional CD8+ effectors and memory cells. Our results suggest that in APC vaccinationmodels, IL-6 provided by the APCs is dispensable for proper CD8+ T-cell memory generation.
1. Introduction
e recognition of a foreign antigen (Ag) presented byspecialized Ag-presenting cells (APCs) in lymphoid organsby naïve CD8+ T cells leads to their activation, differentiation,and proliferation. is is accompanied by changes in migra-tion properties and gain of effector functions to control theinfection. Aer elimination of the pathogen, most (90–95%)of the activated CD8+ effector T cells (Te) die during thecontraction phase to reset the system for the next challenge.Importantly, a fraction of the Ag-speci�c Te cells will surviveas resting memory T cells (Tm) able to respond quickly to asecond Ag encounter.
During acute infection, two subsets of CD8+ effectors,short-lived effector cells (SLECs; CD127lo and KLRG-1hi),and memory precursor effector cells (MPECs; CD127hi andKLRG-1lo) can be identi�ed at the peak of the response [1–6]. Only MPECs, which represent about 10% of the Ag-speci�c population at the peak of the response, survive and
further differentiate into Tm cells [1–5]. However, a differentpicture emerged in vaccination strategies using Ag-pulsedAPCs [2, 7–11] or Ag plus adjuvant [8, 12]. We and othershave shown that CD8+ T-cell response to immunization withTLR-stimulated DCs follows a different course than responseto infection [2, 7–10]. Due to low in�ammation, the majorityof CD8+ Te cells acquire an MPEC phenotype at the peak ofthe response [2, 7–10]. ese MPECs are very good effectorsendowed with the ability to produce cytokines and kill targetcells [10, 11]. Unlike the MPECs that are generated followinginfection, MPECs obtained following DC vaccination willstill undergo a normal contraction phase [7, 8] and thusonly a fraction of them will become long-lived Tm cells.Similarly, vaccination with Ag plus adjuvant generates a highproportion of CD127hi cells (MPECs) at the peak of theresponse and only a fraction of them will survive as long-lived CD8+ Tm cells [8, 12]. Following vaccination with Agplus adjuvant, it was shown that high level of expression ofIL-6 receptor (R) 𝛼𝛼 chain in combination with high level of

2 BioMed Research International
expression of IL-7R𝛼𝛼 (CD127) better identi�es the MPECsthat will further differentiate into Tm cells [12]. is suggeststhat IL-6 signal might contribute to Tm-cell development.
Until recently, little was known about the potential ofother APCs, such as B cells, to induce a CD8+ T-cell response[11, 13–15]. We and others have shown that CD40-activatedB (CD40-B) cells can prime a functional CD8+ T cell responsein vivo [11, 13–15]. We have shown that as for DC vacci-nation, all effectors acquire a MPEC phenotype followingCD40-B-cell immunization [11]. Furthermore, these MPECshave excellent effector functions as measured by their abilityto secrete cytokines, kill target cells in vivo and clear a bac-terial infection [11]. Although MPECs were generated withCD40-B-cell vaccination, Tm-cell generation was inefficient[11]. erefore, understanding why CD40-B cell vaccinationdoes not lead to the formation of functional long-livedTm cells is essential to de�ne the signals that should beprovided to naïve T cells by APCs to promote efficient Tm-cell differentiation. e reported high level of expression ofIL-6R𝛼𝛼 by prememory CD8+ T cells [12] suggests that IL-6may be one of the missing signal.
IL-6was �rst identi�ed as a B-cell proliferation and differ-entiation factor [16]. Its high affinity receptor is composed ofthe IL-6R𝛼𝛼 chain and the common gp30 chain [16]. As manycytokines, IL-6 has pleiotropic action on different cell typesof the immune system [16]. Speci�cally, on CD8+ T cells, IL-6 was reported to promote the survival of naïve T cells [17–20], to enhance the proliferation of CD8+ T cells followingTCR triggering [14, 20–23] and to synergize with IL-7 or IL-15 to induce Ag-independent proliferation of CD8+ T cells[24]. IL-6 was also shown to contribute to in vivoCD8+ T-cellresponse. Indeed, maximal in vivo CD8+ T cell proliferationfollowing vaccination with CD40-B cells stimulated via the Bcell receptor and TLR7 was dependent on IL-6 production byB cells [14]. Moreover, cytotoxic CD8+ T-cell differentiationwas dependent on IL-6 induction by adjuvant in vaccinationprotocol [25]. Finally, the transfer of CD8+ MPECs into IL-6-de�cient hosts severely impaired the generation of long-lived CD8+ Tm cells [12]. ese studies suggest that IL-6 isessential for optimal and complete in vivo response of CD8+T cells.
e reported in�uences of IL-6 on CD8+ T-cell responselead us to investigate whether IL-6 signal from APCs duringprimingwas necessary to promote the formation of CD8+ Tmcells following APC vaccination. In this paper, we show thatCD40-B cells stimulated with LPS produce very low amountof IL-6 when compared to DCs and that supplementationwith IL-6 during CD40-B-cell vaccination did not improvetheir ability to generate CD8+ Tm cells. Furthermore, vacci-nation with IL-6-de�cient DCs did not impede their abilityto promote the formation of functional CD8+ effectors andmemory T cells.
2. Materials and Methods
2.1. Mice. B6.SJL and OT-I [26] mice were bred at theMaisonneuve-Rosemont Hospital Research Center facility.IL-6 knock-out (KO) (B6.129S2-Il6 tm1Kopf/J) mice [27] werepurchased from e Jackson Laboratory. Mice were housed
in a pathogen-free environment and treated in accordanceto the Canadian Council on Animal Care guidelines. Ouranimal protocol (number: 2007-36) was approved by theMaisonneuve-Rosemont Hospital Research Center AnimalCare Committee.
2.2. B-Cell and DC Cultures. For B-cell culture, lymphocyteswere isolated on a FICOLL gradient from male B6.SJL spleenfollowed by a 4 days culture on irradiated �broblasts stablytransfected with the CD40L cDNA (3T3-CD40L) to generateCD40-B cells [28]. Bone-marrow-derived DCs were gener-ated as previously described [8]. e day before harvesting,lipopolysaccharide (LPS) (1 𝜇𝜇g/mL) was added to DC andCD40-B-cell cultures. e ovalbumin (OVA257–264) peptide(SIINFEKL) (Midwest biotech) was loaded overnight onDCs(2 𝜇𝜇g/mL) and B cells (4𝜇𝜇g/mL).
2.3. Immunization and Analysis of T-Cell Responses. Twodays aer adoptive transfer of 106 OT-I T cells (CD45.2+;from female mice) into female B6.SJL mice (CD45.1+),recipients were immunized intravenously (i.v.) with 0.5 × 106
DCs or 2 × 106 DCs (as indicated in the Figure legend) or2 × 106 CD40-B cells from male mice to induce a CD4+T-cell response against the male minor histocompatibilityantigen HY [29]. Some mice were injected intraperitoneally(i.p) with 500 ng of recombinant mouse IL-6 (R&D Systems).e presence of Te (d4 post-immunization) and Tm (d45postimmunization) cells was evaluated in the same mouse bysequential removal of super�cial lymph nodes as describedpreviously [8]. Functions of Te (d4) and Tm (d60) wereanalyzed as previously described with minor modi�cations[8]. Splenocytes were restimulated with 2 𝜇𝜇g/mL OVA257–264peptide in complete RPMI 1640 for 6 h at 37∘C. For the last3 h, 10 𝜇𝜇g/mL of brefeldin A (Sigma Aldrich) was added. TeandTmcells were identi�ed by �ow cytometry as beingCD8+and CD45.2+.
2.4. Mouse Surgery. Lymph node removal by surgery wasdone as described [30]. Brie�y, mice were anesthetised byinhalation of iso�urane (2%, 1L oxygen). Before the surgery,eye ointment was applied to avoid eye dryness and buprenor-phine was administered subcutaneously (0.05–0.1mg/Kg) asan analgesic. To harvest the brachial and the inguinal lymphnodes, a small incision (5mm) of the skin was made andthe lymph nodes were removed using forceps. e incisionwas closed with one clip (Michel suture clips, 7.5 × 1.75mm,Harvard Apparatus).
2.5. Antibodies, Cytometry, and ELISA. Anti-CD86 (GL-1),-TNF-𝛼𝛼 (MP6-XT22), and -Bcl-2 (3F11) antibodies werepurchased from BD Biosciences. Anti-H-2Kb (AF6-88.5),-CD45.2 (104), -CD44 (1M7), -CD8 (53-6.7), -CD19 (6D5),-CD11c (N418), -CD80 (16-10A1), -IL-6R𝛼𝛼 (D7715A7),-CD43 (1B11), -CD62L (MEL-14), and -IL-2 (JES6-5H4)antibodies were purchased from Biolegend. Anti-I-Ab
(28-16-8S) was purchased from Cedarlane. Anti-CD127(A7R34), -Eomes (Dan11mag), -KLRG1 (2F1), and -granzyme B (16G6) antibodies were purchased from

BioMed Research International 3
eBioscience. Anti-Bcl-6 (7D1) antibody was purchasedfrom Santa Cruz Biotechnology. Anti-CXCR3 (220803)antibody was purchased from R&D Systems. Anti-IFN-𝛾𝛾(XMG1.2) antibody was purchased from Life technologies.OVA peptide loading on Kb MHC was measured by stainingwith the 25-D1.16 Ab [31] followed by staining with a ratanti-mouse IgG1 (A85-1) antibody from BD Biosciences.Cell surface and intracellular stainings for cytokines wereperformed as previously described [8, 32]. Bcl-6 and Eomesintracellular stainings were performed with the FoxP3 kitfrom eBioscience. For Bcl-2 staining, cells were stained for 30minutes in 0.1% saponin (Sigma-Aldrich) and washed twicewithout saponin before cell surface staining. All stainingswere analyzed on a BD FACSCanto I system.
For ELISA, B cells and DCs were cultured as describedabove. Before harvesting, supernatants were collected andELISA was performed against IL-6 (Biolegend), according tothe manufacturer’s protocol.
2.6. Statistical Analysis. Statistical analyses for differencesbetween groups were performed using Mann Whitney test(two experimental groups) or one-way ANOVA followed byGames-Howell posttest (3 experimental groups or more).Data are presented as mean ± standard error of the mean(SEM). All tests were two-sided and 𝑃𝑃 𝑃 𝑃𝑃𝑃𝑃 was consideredstatistically signi�cant. ∗𝑃𝑃 𝑃 𝑃𝑃𝑃𝑃, ∗∗𝑃𝑃 𝑃 𝑃𝑃𝑃𝑃, ∗∗∗𝑃𝑃 𝑃 𝑃𝑃𝑃𝑃𝑃and NS: non-signi�cant.
3. Results and Discussion
3.1. Expression of IL-6R𝛼𝛼 by CD8+ T Cells following Vac-cination with APCs. Our previous work has shown thatvaccination with CD40-B cells matured with LPS and loadedwith the OVA peptide leads to the formation of functionalCD8+ Te cells but not Tm cells [11]. Although the CD8+Te cells generated following CD40-B cell vaccination wereenriched for MPECs (CD127hi and KLRG1lo), they did notsurvive the contraction phase [11]. Since high level of IL-6R𝛼𝛼expression was shown to better identify at the peak of theT-cell response the MPECs that will differentiate into CD8+Tm cells [12], we have evaluated if the MPECs generatedfollowing CD40-B-cell vaccination express high level of IL-6R𝛼𝛼. As shown in Figures 1(a) and 1(b), at the peak of the T-cell response (day 4 in this model) most of the OVA-speci�cCD8+ Te cells express high level of IL-6R𝛼𝛼. Furthermore, theCD8+ Te cells generated following CD40-B-cell vaccinationexpress similar level of IL-6R𝛼𝛼 than those obtained withDC vaccination (Figures 1(a) and 1(b)), which efficientlygenerates CD8+ Tm cells. ese results indicate that MPECsgenerated following CD40-B cell vaccination should be ableto respond to IL-6 during the contraction phase of theresponse. e fact that CD40-B-cell vaccination generatesMPECs expressing high levels of both IL-7R𝛼𝛼 (SupplementalFigure 1 and ref [11] see Supplementary Materials availableonline at doi:10.1155/2012/126189.) and IL-6R𝛼𝛼 suggests thatthese MPECs should received the proper survival signalsallowing them to persist during the contraction phase andfurther differentiate into Tm cells. However, our previous
work has shown that the MPECs obtained with CD40-Bcell vaccination rapidly contract during the T cell responseand do not differentiate into CD8+ Tm cells [11]. issuggests that other survival and differentiation factors mightbe implicated for the differentiation ofMPECs into CD8+ Tmcells.
3.2. IL-6 Supplementation Does Not Enhance CD8+ Tm-CellGeneration following CD40-B-Cell Vaccination. e reportedrole of IL-6 in CD8+ T-cell proliferation and differentiation[12, 14, 20–23, 25] leads us to evaluate if CD40-B cellswere providing IL-6 during the priming of naïve CD8+T cells. IL-6 was quanti�ed in the supernatants obtainedat the end of CD40-B-cell and DC cultures. As shown inFigure 1(c), CD40-B cells produce around 5-fold less IL-6 than DCs. is reduced production of IL-6 might beresponsible for the lack of CD8+ Tm-cell generation withCD40-B-cell vaccination.
To test whether the decreased IL-6 production by CD40-B cells was responsible for their inability to induce CD8+Tm-cell development, we injected IL-6 at the time of CD40-B-cell immunization. e dose of IL-6 was chosen basedon previous publications where IL-6 injection had an effecton T-cell response [33, 34]. As shown in Figure 2, theadministration of IL-6 (500 ng) i.p. at the time of OT-Inaïve CD8+ T-cell priming by CD40-B cells did not enhancethe generation of CD8+ Te and Tm cells. Furthermore, theeffectors generated with or without IL-6 supplementationhad a similar phenotype as determined by the expression ofCD44, CD127, and Bcl-2 (Supplemental Figure 1).
3.3. IL-6 Is Dispensable for the Generation of CD8+ Tm Cellsfollowing Vaccination with DCs. Since it was possible thatthe amount administered and the route of injection did notlead to a sufficient IL-6 signals in naïve OT-I T cells, wetested whether IL-6 production by DCs was necessary forthe generation of long-lived CD8+ Tm cells. To do so, wegenerated DCs from the bone marrow of IL-6-de�cient mice.Before using these IL-6-de�cient DCs in our vaccinationprotocol, we con�rmed that they had a similar phenotypethanwild-typeDCs following LPSmaturation (SupplementalFigure 2). Furthermore, IL-6-de�cient DCs were equallyloaded with the OVA peptide as WT DCs (SupplementalFigure 2). We then compared the OVA-speci�c CD8+ T-cell response following vaccination with IL-6-de�cient or -sufficent DCs. As shown in Figure 3, a similar frequency andnumber (not shown) of CD8+ Te and Tm cells were gen-erated following vaccination with WT or IL-6 KO DCs.Furthermore, the yield of CD8+ Tm cells (% of Te cells thatdeveloped into Tm cells) was similar in both groups (Figure3(b)). ese results show that IL-6 production by APCs atthe priming of naïve CD8+ T cells is not necessary for thegeneration of CD8+ Te and Tm cells. Several reports haveshown that IL-6 can enhance CD8+ T-cell proliferation invitro [14, 20–24] and in vivo [14]. However, the use of IL-6-de�cient DCs did not reduce the number of CD8+ Te cellsgenerated. us, it is possible that the basal level of IL-6present in the host is sufficient for optimal T-cell proliferationor that IL-6 production by DCs is not necessary for maximal

4 BioMed Research International
Isotype
Max
(%
)
CD8+CD45.2+
CD8+CD45.2−
Isotype
CD8+CD45.2+
CD8+CD45.2−
Isotype
CD8+CD45.2+
CD8+CD45.2−
CD40-B CD40-B OVA DC OVA
974
660
422
628
381
613
(a)
DC OVA
0
1
2
MF
I ra
tio
0.5
1.5
CD40-B CD40-B OVA
∗∗∗
∗∗∗
(b)
DC
0
10
20
30
40
pg/
mL
(×
103)
CD40-B
∗
(c)
F 1: CD40-B cell andDC immunizations generate effectors expressing similar levels of IL-6R𝛼𝛼. (a) Expression of IL-6R𝛼𝛼 byOVA-speci�cTe cells at the peak of the T cell response (day 4). 106 female OT-I T cells (CD8+CD45.2+) were adoptively transferred into congenic B6.SJLfemale mice (CD45.1+) followed by immunization two days later with 2 × 106 LPS-matured unloaded CD40-B cells (CD40-B), LPS-maturedCD40-B cells loaded with the OVA peptide (CD40-BOVA) or LPS-matured DCs loaded with the OVA peptide (DCOVA).e representativeoverlay histograms show expression of IL-6R𝛼𝛼 by OVA-speci�c Te cells (CD8+CD45.2+) and endogenous T cells (CD8+CD45.2−). e upperbold number indicates the mean �uorescence intensity (MFI) of OVA-speci�c Te cells while the lower number is for the endogenous CD8+T cells. (b) �uanti�cation of IL-6R𝛼𝛼 expression by effectors. e MFI of IL-6R𝛼𝛼 expression by effector CD8+ T cells (CD8+CD45.2+) wasnormalized to the MFI of the recipient CD8+ T cells (CD8+CD45.2−). Each dot represents one mouse. (c) CD40-B cells produce less IL-6than DCs. Supernatants from CD40-B LPS or DC LPS culture were used to measure IL-6 secretion by ELISA. Mean ± SEM is shown for 3independent experiments. ∗𝑃𝑃 𝑃 0𝑃0𝑃 and ∗∗∗𝑃𝑃 𝑃 0𝑃001.
proliferation of CD8+ T cells. Moreover, IL-6 production byCD40-B cells stimulated via the BCR and TLR7 was reportedto be necessary for the maximal expansion of Ag-speci�cCD8+ T cells following vaccination [14]. us, our resultswith IL-6 KO DCs suggest that different APC types mightproduce different cytokines to promote the full expansionof CD8+ T cells. However, in our hands supplementation ofIL-6 during CD40-B-cell vaccination did not increase T-cellexpansion (Figure 2). is might be explained by the use ofdifferent stimuli (BCR + TLR7 ligand versus LPS) to mature
the CD40B cells that may lead to production of differentcytokines.
�.�. �a���na���n���� �����De���en� DCs�ene�a�es ��n����nalCD8+ Te and Tm Cells. Since IL-6 was shown to in�u-ence cytotoxic T-cell differentiation [25], we have carefullyevaluated the phenotype and functions of the OVA-speci�cCD8+ Te and Tm cells generated following vaccination withWT or IL-6 KO DCs. As shown in Figure 4, both types ofeffectors produce similar amounts of IFN-𝛾𝛾, TNF-𝛼𝛼, IL-2, and
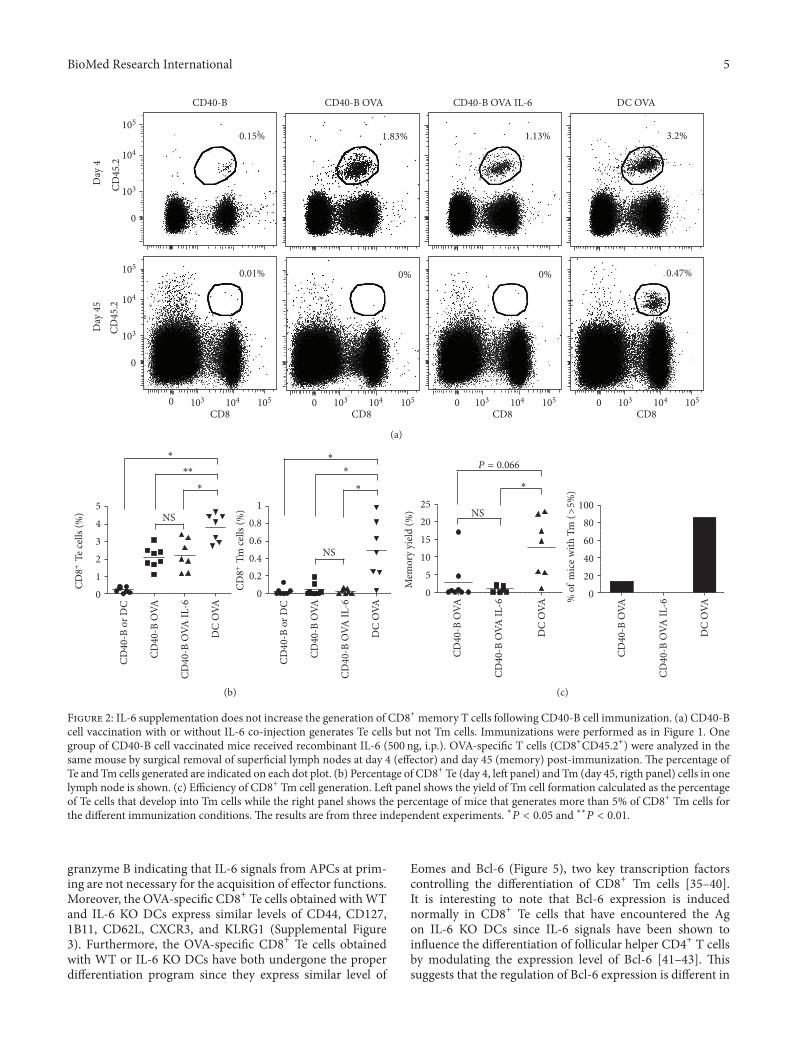
BioMed Research International 5
Day
4D
ay 4
5
CD8 CD8 CD8 CD8
CD40-B CD40-B OVA CD40-B OVA IL-6
0.15% 1.83% 1.13% 3.2%
0.01% 0% 0% 0.47%
0 103 104 105 0 103 104 105 0 103 104 105 0 103 104 105
DC OVAC
D45
.2C
D45
.2
0
103
104
105
0
103
104
105
(a)
CD
40-B
or
DC
CD
40-B
OV
A
CD
40-B
OV
A I
L-6
DC
OV
A
0
1
2
3
4
5
CD
40-B
or
DC
CD
40-B
OV
A
CD
40-B
OV
A I
L-6
DC
OV
A
0
0.2
0.4
0.6
0.8
1NS
NS
CD
8+T
e ce
lls
(%)
CD
8+T
m c
ells
(%
)
∗∗∗∗∗
∗
∗
(b)
CD
40-B
OV
A
CD
40-B
OV
A I
L-6
DC
OV
A
0
5
10
15
20
25
CD
40-B
OV
A
CD
40-B
OV
A I
L-6
DC
OV
A
0
20
40
60
80
100
Mem
ory
yie
ld (
%) NS
% o
f m
ice
wit
h T
m (
>5%
)
∗
(c)
F 2: IL-6 supplementation does not increase the generation of CD8+ memory T cells following CD40-B cell immunization. (a) CD40-Bcell vaccination with or without IL-6 co-injection generates Te cells but not Tm cells. Immunizations were performed as in Figure 1. Onegroup of CD40-B cell vaccinated mice received recombinant IL-6 (500 ng, i.p.). O�A-speci�c T cells (CD8+CD45.2+) were analyzed in thesame mouse by surgical removal of super�cial lymph nodes at day 4 (effector) and day 45 (memory) post-immunization. e percentage ofTe and Tm cells generated are indicated on each dot plot. (b) Percentage of CD8+ Te (day 4, le panel) and Tm (day 45, rigth panel) cells in onelymph node is shown. (c) Efficiency of CD8+ Tm cell generation. Le panel shows the yield of Tm cell formation calculated as the percentageof Te cells that develop into Tm cells while the right panel shows the percentage of mice that generates more than 5% of CD8+ Tm cells forthe different immunization conditions. e results are from three independent experiments. ∗𝑃𝑃 𝑃 𝑃𝑃𝑃𝑃 and ∗∗𝑃𝑃 𝑃 𝑃𝑃𝑃𝑃.
granzyme B indicating that IL-6 signals from APCs at prim-ing are not necessary for the acquisition of effector functions.Moreover, the O�A-speci�c CD8+ Te cells obtained with WTand IL-6 KO DCs express similar levels of CD44, CD127,1B11, CD62L, CXCR3, and KLRG1 (Supplemental Figure3). Furthermore, the O�A-speci�c CD8+ Te cells obtainedwith WT or IL-6 KO DCs have both undergone the properdifferentiation program since they express similar level of
Eomes and Bcl-6 (Figure 5), two key transcription factorscontrolling the differentiation of CD8+ Tm cells [35–40].It is interesting to note that Bcl-6 expression is inducednormally in CD8+ Te cells that have encountered the Agon IL-6 KO DCs since IL-6 signals have been shown toin�uence the differentiation of follicular helper CD4+ T cellsby modulating the expression level of Bcl-6 [41–43]. issuggests that the regulation of Bcl-6 expression is different in

6 BioMed Research International
0 103 104 105 0 103 104 105 0 103 104 105
0
103
104
105
0
103
104
105
Day
4D
ay 4
5
CD
45.2
CD
45.2
CD8 CD8 CD8
DC DC OVA DC OVA IL-6 KO
0.33% 3.61% 2.49%
0.02% 0.41% 0.28%
(a)
CD
8+
Te
cell
s (%
)
CD
8+
Tm
cel
ls (
%)
NS NS NS
Mem
ory
yie
ld (
%)
10
8
6
4
2
0 0 0
0.5
1
1.5 20
15
10
5
DC OVA DC OVA IL-6 KO DC OVA DC OVA IL-6 KO DC OVA DC OVA IL-6 KO
(b)
F 3: Normal generation of CD8+ e�ector and memory T cells following vaccination with IL-6-de�cient DCs. (a) �accination with WTor IL-6 KO DCs generates O��-speci�c CD8+ Te cells and Tm cells. 106 female OT-I T cells (CD8+CD45.2+) were adoptively transferredinto congenic B6.SJL female mice (CD45.1+) followed by immunization two days later with 0.5 × 106WT or IL-6 KO DCs, matured with LPSand loaded or not with O�� peptide. Te and Tm cells were identi�ed as CD8+CD45.2+ by �ow cytometry. e percentage of Te and Tm cellsgenerated are indicated on each dot plot. (b) �uanti�cation of CD8+ T cell response. Percentage of CD8+ Te (day 4, le panel) and Tm (day45, middle panel) cells in one lymph node is shown. e yield of Tm cell formation was calculated as the percentage of Te cells that developinto Tm cells (right panel). e results are from two independent e�periments with at least three mice per group. NS, non-signi�cant.

BioMed Research International 7
100
80
60
40
20
0
IL-2
Gra
nzy
me
B
DC OVA
6932
627
1361
242
718
234
1209
362
100
80
60
40
20
0
100
80
60
40
20
0
100
80
60
40
20
0
CD8+CD45.2+
CD8+CD45.2−
0 103 104 105
(a)
DC OVA IL-6 KO
6521
695
1195
262
721
233
1066
363
CD8+CD45.2+
CD8+CD45.2−
0 103 104 105
100
80
60
40
20
0
IL-2
Gra
nzy
me
B
100
80
60
40
20
0
100
80
60
40
20
0
100
80
60
40
20
0
(b)
F 4: �eneration of functional OVA�speci�c CD8+ effector T cells following immunization with I����de�cient DCs.Mice wereimmunized as in Figure 3 and effector molecules production was analyzed following a short in vitro stimulation with the OVA peptide.�e overlays show production of the different effector molecules by OVA�speci�c Te cells (CD8+CD45.2+) compared to endogenous T cells(CD8+CD45.2−) at day 4 post�immunization with�T (le�) or I��� �O (right) DCs.�eMFI of effectormolecule e�pression byOVA�speci�cCD8+ effectors (upper bold number) and endogenous CD8+ T cells (lower number) are indicated on each overlay.

8 BioMed Research International
0 103 104 105 0 103102 104 105
100
80
60
40
20
0
CD8+CD45.2+
CD8+CD45.2−
CD8+CD45.2+
CD8+CD45.2−
100
80
60
40
20
0
DC
OV
AD
C O
VA
IL
-6 K
O
1369
655
772
412
1652
698
681
435
Eomes Bcl-6
(a)
NS NSEomes
2.5
2
1.5
1
0.5
0
2
1.5
1
0.5
0
MF
I ra
tio
DC OVA DC OVA IL-6 KO
DC OVA DC OVA IL-6 KO
Bcl-6
(b)
F 5: WT or IL-6 KO DC immunization generates effectors expressing similar levels of the transcription factors Eomes and Bcl-6. (a)�e representative overlay histogram shows expression of Eomes and Bcl-6 by O�A-speci�c Te cells (CD8+CD45.2+) and endogenous T cells(CD8+CD45.2−). �e MFI of Bcl-6 or Eomes expression by O�A-speci�c CD8+ effectors (upper bold number) and endogenous CD8+ T cells(lower number) are indicated on each overlay. Mice were immunized as in Figure 3. (b) �uanti�cation of the level of expression of Eomes andBcl-6. �e bar charts show the MFI of expression for Eomes or Bcl-6 by O�A-speci�c CD8+ Te cells normalized to the MFI of endogenousCD8+ T cells. Results are presented as mean ± SEM. At least two mice per group.

BioMed Research International 9
DC
OV
AD
C O
VA
IL
-6 K
O
100
80
60
40
20
0
100
80
60
40
20
0
CD8+CD45.2+
CD8+CD45.2−
CD8+CD45.2+
CD8+CD45.2−
CD8+CD45.2+
CD8+CD45.2−CD8+CD45.2+
CD8+CD45.2−
0 103 104 105 0 103 104 105 0 103 104 105 0 103 104 105
IL-2 Granzyme B
76.5% 25.3% 11.1% 6.7%
80.3% 30.9% 9.2% 3.9%
(a)
NS NS NS
NS NS NS NS
100
80
60
40
20
0
30
20
10
0
8
6
4
2
0
8
6
4
2
0
0
MF
I ra
tio
MF
I ra
tio
MF
I ra
tio
MF
I ra
tio
IL-2
IL-2
+(%
)
(%)50
40
30
30
20
20
1010
0 0
15
10
5
2.5
2
1.5
1
Granzyme B
Gra
nzy
me
B+
DC OVA DC OVA IL-6 KO
DC OVA DC OVA IL-6 KO
DC OVA DC OVA IL-6 KO
DC OVA DC OVA IL-6 KO
(b)
F 6: �eneration of functional O�A-speci�c CD8+memory T cells following immunization with IL-6-de�cient DCs. (a) Functionality ofO�A-speci�c CD8+ Tm cells at day 60 post-immunization. e overlays show production of the different effector molecules by O�A-speci�cT cells (CD8+CD45.2+) compared to endogenous T cells (CD8+CD45.2−) following immunization with WT (top) or IL-6 KO (bottom) DCs.e percentage of cells producing the different effectormolecules is indicated on each histogram. (b)�uanti�cation of cytokine and granzymeB production by O�A-speci�c CD8+ Tm cells. e percentage of cytokines and granzyme B producing O�A-speci�c Tm cells (top) and theamount produced (bottom) are shown at day 60 post-immunization. e MFI of cytokine and granzyme B production by CD8+ Tm cells wasnormalized to the MFI of the recipient CD8+ T cells (MFI ratio). e results are from two independent experiments.
CD4+ versus CD8+ T cells or the endogenous source of IL-6 issufficient to promote Bcl-6 expression in CD8+ Te cells. eproper differentiation of effectors following vaccination withIL-6 KO DCs contrasts with the results obtained by others
where IL-6 induction by adjuvant was critical for cytotoxicT-cell differentiation [25]. One possible explanation is thatvaccination with fully matured DCs bypassed the needs forIL-6. Altogether our results suggest that IL-6 production by

10 BioMed Research International
the DCs involved in the priming of naïve CD8+ T cells isdispensable for the proper differentiation of CD8+ Te cells.
Although CD8+ Tm cells were generated following vac-cination with IL-6-de�cient DCs, it was important to inves-tigate if the Tm cells generated were fully functional. Asshown in Figure 6, OVA-speci�c CD8+ Tm cells obtainedwith both WT and IL-6 KO DCs were similarly functional.ey both produced similar amounts of IFN-𝛾𝛾, IL-2, TNF-𝛼𝛼 and granzyme B (Figure 6). ese results show that IL-6production by APCs during priming of naïve CD8+ T cells isalso dispensable for the generation of fully functional CD8+Tm cells.
Our results show that IL-6 production by DCs is dispens-able for the generation of fully functional CD8+ Tm cells.Furthermore, they also suggest that the lack of productionof IL-6 by CD40-B cells is probably not the explanation fortheir inability to induce the formation of CD8+ Tm cells.Further studies are required to understand why CD40-B-cellvaccination does not promote the generation of CD8+ Tmcells. Possible explanations include differences in the site ofpriming, the level of costimulation, the interaction time withT cells, and the production of other soluble mediators suchas IL-12 or type I IFNs. e ability of IL-6-de�cient DCs topromote the generation of functional CD8+ Tmcells indicatesthat other soluble factors (IL-12 and IL-23) produced byDCs are sufficient to induce the generation of CD8+ Tmcells. Indeed, it was shown by others that vaccination withIL-12 and IL-23 de�cient DCs abrogated CD8+ Tm-celldevelopment [44]. It is also possible that IL-6 plays a roleduring CD8+ Tm-cell differentiation but that it does not haveto be produced by the APCs involved in the T cell priming.
In conclusion, we show that the inability of CD40-B-cell vaccination to induce the formation of CD8+ Tm cellsis not due to their reduced production of IL-6. Similarly,vaccination with IL-6-de�cient DCs did not impede theirability to promote the formation of functional CD8+ Tmcells.us, IL-6 production by theAPCs involved in the priming ofnaïve CD8+ T cells is dispensable for the formation of CD8+Tm cells. Furthermore, our results also highlight the variousrole of IL-6 in different immunization protocol. Vaccinationwith DC does not rely on IL-6 for the full expansion anddifferentiation of CD8+ Te cells while IL-6 is necessary whenadjuvant is used.
Abbreviations Used in This Paper
Ag: AntigenAPC: Antigen-presenting cellCD40-B cell: CD40-activated B cellDC: Dendritic cellIFN: InterferonIL: InterleukinKO: Knock-outMPEC: Memory precursor effector cellNS: Non-signi�cantOVA: OvalbuminR: ReceptorSEM: Standard error of the meanSLEC: Short lived effector cell
Te cell: Effector T cellTm cell: Memory T cellTNF: Tumor necrosis factorWT: Wild-type.
Acknowledgments
e authors thank all the members of the laboratory forhelpful discussion. e authors acknowledge J. Yewdell forkindly providing the anti-Kb-OVA antibody. e authorsthank J. Dubeau and the animal care staff formice husbandry.is work was supported by the Canadian Institutes ofHealth Research (CIHR, MOP-77545 and MOP-115139). M.Mathieu was supported by a studentship from the NaturalSciences and Engineering Research Council of Canada andthe Fonds de la Recherche du Québec-Santé. S. Boulet wassupported by a CIHR postdoctoral fellowship.
References
[1] S. M. Kaech, J. T. Tan, E. J. Wherry, B. T. Konieczny, C. D.Surh, and R. Ahmed, “Selective expression of the interleukin 7receptor identi�es effector CD8 T cells that give rise to long-lived memory cells,” Nature Immunology, vol. 4, no. 12, pp.1191–1198, 2003.
[2] N. S. Joshi, W. Cui, A. Chandele et al., “In�ammation directsmemory precursor and short-lived effector CD8+ T cell fates viathe graded expression of T-bet transcription factor,” Immunity,vol. 27, no. 2, pp. 281–295, 2007.
[3] N. S. Joshi and S. M. Kaech, “Effector CD8 T cell development:a balancing act between memory cell potential and terminaldifferentiation,” Journal of Immunology, vol. 180, no. 3, pp.1309–1315, 2008.
[4] S. Sarkar, V. Kalia, W. N. Haining, B. T. Konieczny, S. Subra-maniam, and R. Ahmed, “Functional and genomic pro�ling ofeffector CD8 T cell subsets with distinct memory fates,” Journalof Experimental Medicine, vol. 205, no. 3, pp. 625–640, 2008.
[5] K. M. Huster, V. Busch, M. Schiemann et al., “Selectiveexpression of IL-7 receptor on memory T cells identi�es earlyCD40L-dependent generation of distinct CD8+ memory T cellsubsets,” Proceedings of the National Academy of Sciences of theUnited States of America, vol. 101, no. 15, pp. 5610–5615, 2004.
[6] V. Kalia, S. Sarkar, S. Subramaniam, W. N. Haining, K. A.Smith, and R. Ahmed, “Prolonged interleukin-2R𝛼𝛼 expressionon virus-speci�c CD8+ T cells favors terminal-effector differen-tiation in vivo,” Immunity, vol. 32, no. 1, pp. 91–103, 2010.
[7] V. P. Badovinac, K. A. Messingham, A. Jabbari, J. S. Haring,and J. T. Harty, “Accelerated CD8+ T-cell memory and prime-boost response aer dendritic-cell vaccination,” Nature Medi-cine, vol. 11, no. 7, pp. 748–756, 2005.
[8] M. H. Lacombe, M. P. Hardy, J. Rooney, and N. Labrecque,“IL-7 receptor expression levels do not identify CD8+ memoryT lymphocyte precursors following peptide immunization,”Journal of Immunology, vol. 175, no. 7, pp. 4400–4407, 2005.
[9] N. L. Pham, V. P. Badovinac, and J. T. Harty, “A default pathwayofmemory CD8 T cell differentiation aer dendritic cell immu-nization is de�ected by encounter with in�ammatory cytokinesduring antigen-driven proliferation,” Journal of Immunology,vol. 183, no. 4, pp. 2337–2348, 2009.

BioMed Research International 11
[10] J. Leignadier and N. Labrecque, “Epitope density in�uencesCD8+ memory T cell differentiation,” PLoS ONE, vol. 5, no. 10,Article ID e13740, 2010.
[11] M. Mathieu, N. Cotta-Grand, J. F. Daudelin, S. Boulet, R.Lapointe, and N. Labrecque, “CD40-activated B cells cane�ciently prime antigen-speci�c na�ve CD8 T cells to generateeffector but notmemory T cells,” PLoSONE, vol. 7, no. 1, ArticleID e30139, 2012.
[12] F. Castellino and R. N. Germain, “Chemokine-guided CD4+T cell help enhances generation of IL-6R𝛼𝛼highIL-7R𝛼𝛼highprememory CD8+ T cells,” Journal of Immunology, vol. 178, no.2, pp. 778–787, 2007.
[13] D. S. Ritchie, J. Yang, I. F. Hermans, and F. Ronchese, “B-lymphocytes activated by CD40 ligand induce an antigen-speci�c anti-tumour immune response by direct and indirectactivation of CD8+ T-cells,” Scandinavian Journal of Immunol-ogy, vol. 60, no. 6, pp. 543–551, 2004.
[14] T. J. Vanden Bush, C. M. Buchta, J. Claudio, and G. A. Bishop,“Cutting edge: importance of IL-6 and cooperation betweeninnate and adaptive immune receptors in cellular vaccinationwith B lymphocytes,” Journal of Immunology, vol. 183, no. 8, pp.4833–4837, 2009.
[15] S. Guo, J. Xu, W. Denning, and Z. Hel, “Induction of protectivecytotoxic T-cell responses by a B-cell-based cellular vaccinerequires stable expression of antigen,” Gene erapy, vol. 16, no.11, pp. 1300–1313, 2009.
[16] T. Kishimoto, “IL-6: from its discovery to clinical applications,”International Immunology, vol. 22, no. 5, pp. 347–352, 2010.
[17] T. K. Teague, P. Marrack, J. W. Kappler, and A. T. Vella, “IL-6 rescues resting mouse T cells from apoptosis,” Journal ofImmunology, vol. 158, no. 12, pp. 5791–5796, 1997.
[18] M. Narimatsu, H. Maeda, S. Itoh et al., “Tissue-speci�c autoreg-ulation of the stat3 gene and its role in interleukin-6-inducedsurvival signals in t cells,” Molecular and Cellular Biology, vol.21, no. 19, pp. 6615–6625, 2001.
[19] T. K. Teague, B. C. Schaefer, D. Hildeman et al., “Activation-induced inhibition of interleukin 6-mediated T cell survival andsignal transducer and activator of transcription 1 signaling,”Journal of Experimental Medicine, vol. 191, no. 6, pp. 915–926,2000.
[20] K. Takeda, T. Kaisho, N. Yoshida, J. Takeda, T. Kishimoto, andS. Akira, “Stat3 activation is responsible for IL-6-dependent Tcell proliferation through preventing apoptosis: generation andcharacterization of T cell- speci�c stat3-de�cient mice,” Journalof Immunology, vol. 161, no. 9, pp. 4652–4660, 1998.
[21] R. D. Garman, K. A. Jacobs, S. C. Clark, and D. H. Raulet, “B-cell-stimulatory factor 2 (𝛽𝛽2 interferon) functions as a secondsignal for interleukin 2 production by mature murine T cells,”Proceedings of the National Academy of Sciences of the UnitedStates of America, vol. 84, no. 21, pp. 7629–7633, 1987.
[22] R. D. Garman and D. H. Raulet, “Characterization of a novelmurine T cell-activating factor,” Journal of Immunology, vol.138, no. 4, pp. 1121–1129, 1987.
[23] H. Sepulveda, A. Cerwenka, T. Morgan, and R. W. Dutton,“CD28, IL-2-independent costimulatory pathways for CD8 Tlymphocyte activation,” Journal of Immunology, vol. 163, no. 3,pp. 1133–1142, 1999.
[24] J. Gagnon, S. Ramanathan, C. Leblanc, A. Cloutier, P. P.McDonald, and S. Ilangumaran, “IL-6, in synergy with IL-7 or
IL-15, stimulates TCR-independent proliferation and func-tional differentiation of CD8+ t lymphocytes,” Journal ofImmunology, vol. 180, no. 12, pp. 7958–7968, 2008.
[25] M. K. MacLeod, A. S. McKee, A. David et al., “Vaccineadjuvants aluminum and monophosphoryl lipid A providedistinct signals to generate protective cytotoxic memory CD8T cells,” Proceedings of the National Academy of Sciences of theUnited States of America, vol. 108, no. 19, pp. 7914–7919, 2011.
[26] K. A. Hogquist, S. C. Jameson, W. R. Heath, J. L. Howard, M. J.Bevan, and F. R. Carbone, “T cell receptor antagonist peptidesinduce positive selection,” Cell, vol. 76, no. 1, pp. 17–27, 1994.
[27] M. Kopf, H. Baumann, G. Freer et al., “Impaired immune andacute-phase responses in interleukin-6-de�cient mice,” Nature,vol. 368, no. 6469, pp. 339–342, 1994.
[28] M. Mathieu, N. Cotta-Grand, J. F. Daudelin, S. Boulet, R.Lapointe, and N. Labrecque, “CD40-activated B cells cane�ciently prime antigen-speci�c na�ve CD8+ T cells to generateeffector but not memory T cells,” PLoS ONE, vol. 7, no. 1,Article ID e30139, 2012.
[29] A. M. Livingstone and M. Kühn, “Dendritic cells need T cellhelp to prime cytotoxic T cell responses to strong antigens,”European Journal of Immunology, vol. 29, no. 9, pp. 2826–2834,1999.
[30] M. Mathieu and N. Labrecque, “Murine super�cial lymph nodesurgery,” Journal of Visualized Experiments, vol. 21, no. 63,Article ID e3444, 2012.
[31] A. Porgador, J. W. Yewdell, Y. Deng, J. R. Bennink, and R. N.Germain, “Localization, quantitation, and in situ detection ofspeci�c peptide- MHC class I complexes using a monoclonalantibody,” Immunity, vol. 6, no. 6, pp. 715–726, 1997.
[32] V. Ostiguy, E. L. Allard, M. Marquis, J. Leignadier, and N.Labrecque, “IL-21 promotes T lymphocyte survival by acti-vating the phosphatidylinositol-3 kinase signaling cascade,”Journal of Leukocyte Biology, vol. 82, no. 3, pp. 645–656, 2007.
[33] L. Haynes, S.M. Eaton, E.M. Burns,M. Rincon, and S. L. Swain,“In�ammatory cytokines overcome age-related defects in CD4T cell responses in vivo,” Journal of Immunology, vol. 172, no. 9,pp. 5194–5199, 2004.
[34] V. Singh, S. Jain, U. Gowthaman et al., “Co-administration ofIL-1+IL-6+TNF-𝛼𝛼 with Mycobacterium tuberculosis infectedmacrophages vaccine induces better protective T cell memorythan BCG,” PLoS ONE, vol. 6, no. 1, Article ID e16097, 2011.
[35] A. Banerjee, S.M.Gordon, A.M. Intlekofer et al., “Cutting edge:the transcription factor eomesodermin enables CD8+ T cells tocompete for thememory cell niche,” Journal of Immunology, vol.185, no. 9, pp. 4988–4992, 2010.
[36] H. Ichii, A. Sakamoto, M. Hatano et al., “Role for BcL-6 in thegeneration and maintenance of memory CD8+ T cells,” NatureImmunology, vol. 3, no. 6, pp. 558–563, 2002.
[37] H. Ichii, A. Sakamoto, Y. Kuroda, and T. Tokuhisa, “Bc16 actsas an ampli�er for the generation and proliferative capacity ofcentral memory CD8+ T cells,” Journal of Immunology, vol. 173,no. 2, pp. 883–891, 2004.
[38] A. M. Intlekofer, N. Takemoto, C. Kao et al., “Requirement forT-bet in the aberrant differentiation of unhelpedmemory CD8+T cells,” Journal of Experimental Medicine, vol. 204, no. 9, pp.2015–2021, 2007.
[39] A. M. Intlekofer, N. Takemoto, E. J. Wherry et al., “Effector andmemory CD8+ T cell fate coupled by T-bet and eomesodermin,”Nature Immunology, vol. 6, pp. 1236–1244, 2005.

12 BioMed Research International
[40] N. Takemoto, A. M. Intlekofer, J. T. Northrup, E. J. Wherry,and S. L. Reiner, “Cutting edge: IL-12 inversely regulates T-betand eomesodermin expression during pathogen-induced CD8+T cell differentiation,” Journal of Immunology, vol. 177, no. 11,pp. 7515–7519, 2006.
[41] D. Eto, C. Lao, D. DiToro et al., “IL-21 and IL-6 are critical fordifferent aspects of B cell immunity and redundantly induceoptimal follicular helper CD4 T cell (T) differentiation,” PLoSONE, vol. 6, no. 3, Article ID e17739, 2011.
[42] J. A. Harker, G. M. Lewis, L. Mack, and E. I. Zuniga, “Lateinterleukin-6 escalates T follicular helper cell responses andcontrols a chronic viral infection,” Science, vol. 334, no. 6057,pp. 825–829, 2011.
[43] R. I. Nurieva, Y. Chung, G. J. Martinez et al., “Bcl6 mediates thedevelopment of T follicular helper cells,” Science, vol. 325, no.5943, pp. 1001–1005, 2009.
[44] C. J. Henry, J. M. Grayson, K. L. Brzoza-Lewis et al., “eroles of IL-12 and IL-23 in CD8+ T cell-mediated immunityagainst Listeriamonocytogenes: insights from aDCvaccinationmodel,” Cellular Immunology, vol. 264, no. 1, pp. 23–31, 2010.

Hindawi Publishing CorporationBioMed Research InternationalVolume 2013, Article ID 796014, 7 pageshttp://dx.doi.org/10.1155/2013/796014
Research ArticleIncreased Toll-Like Receptor Signaling Pathways CharacterizeCD8+ Cells in Rapidly Progressive SIV Infection
Maria Cecilia Garibaldi Marcondes,1 Celsa Spina,2 Eduardo Bustamante,1 and Howard Fox3
1 Molecular and Integrative Neurosciences Department, e Scripps Research Institute, La Jolla, CA 92037, USA2 Department of Pathology, UCSD School of Medicine, AIDS Research Center, La Jolla, CA 92037, USA3 Department of Pharmacology and Experimental Neuroscience, University of Nebraska Medical Center, Omaha, NE 68198, USA
Correspondence should be addressed to Maria Cecilia Garibaldi Marcondes; [email protected]
Received 18 May 2012; Accepted 9 November 2012
Academic Editor: Zhengguo Xiao
Copyright © 2013 Maria Cecilia Garibaldi Marcondes et al.is is an open access article distributed under the Creative CommonsAttribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work isproperly cited.
Similar to HIV infection in humans, SIV infection in macaques induces progressive loss of immune cell components and function,resulting in immune de�ciency in nearly all untreated infected sub�ects. In SIV-infectedmacaques, 25% of animals develop terminalAIDS within 6 months of infection. e factors responsible for the development of such rapid progression are unknown. We havepreviously found that defects in CD8+ T cells detectable from early infection correlate to rapid progression to simian AIDS. etranscriptional screening of molecular �ngerprints on di�erent steps along the activation/e�ector process of splenic CD8+ cells attermination revealed a distinction in rapid compared to regular progressors, which was characterized by a decrease in classic Tcell receptor (TCR) components, and an increase in Toll-like receptor (TLR) and apoptotic pathways. A TLR pathway screeningin lymphoid and myeloid cells from both the spleen and from the central nervous system of infected macaques revealed that theupregulation of TLR is not in the innate immune compartment, but rather in lymphoid cells that contain adaptive immune cells.�ur �ndings suggest that opposing e�ects of TCR speci�c signaling and TLR engagement may drive the CD8 phenotypic failurethat determines a rapid disease course in HIV infection.
1. Introduction
SIV infection in macaques and HIV infection in humansfollow a similar pattern. e SIV-infected rhesus macaquemodel has been useful for studying many aspects of HIVpathogenesis. �ne such �nding was a crucial role for CD8+cells, where their acute depletion in the early infection periodleads to high viremia and rapid progression [1–3]. Even inthe absence of this manipulation, rapid progression (≤200days) occurs in 25% of SIV infected animals [4–6]. Hallmarksof rapid progressing (RAP) animals include a low antibodyresponse to the virus [7], fatal immunode�ciency linked tode�cits in tissue-homing memory CD4 cells [8, 9], and asevere central nervous system (CNS) disease characterized byencephalitis [4, 10].
Little is known on how the CD8 performance in�uencesthe CD4 decline in spontaneous rapid progression. Poor
antivirus CD8+ cytotoxic T lymphocyte (CTL) response wasshown in RAPs [11–14], both in SIV and HIV. CTLs becomedefective regardless of epitope escape [10, 15–17]. In RAPs,known peptide-speci�c CTL populations collapse along witha decrease of activation/memory markers in all CD8+ cells,and a loss of memory CD4 cells [10]. Likewise, experimentalinduction of CD8 collapse using depleting antibodies (asabove) leads to loss of memory/activated CD4 cells [10].ese observations suggest that dysfunctional CD8+ cellscould contribute to disease progression in HIV-infectedindividuals.
De�cient CD8+ cell responsiveness is a potential cause ofuncontrolled virus replication, driving disease progression,and failure to maintain the CD4 memory pool. To testthe basis of CD8+ cell collapse, we compared molecularchanges triggered by the virus in splenic CD8+ cells isolatedfrom RAPs and those that did not (regular progressors,

2 BioMed Research International
REGs), with CD8+ cells from uninfected controls. Along keysteps of the CD8 stimulation process (activation, regulation,and effector function), we have identi�ed transcriptionalalterations that may help understand CD8 functional de�citsobserved in correlation to rapid progression andAIDS.esealterations were mainly related to initial steps of the CD8stimulation, which in REGs followed a typical T-cell receptor(TCR) speci�c engagement pattern, but in RAPs, a TLR-triggered pathway was rather activated.
Given that Toll-like receptors (TLRs) have been exten-sively proposed as adjuvants for HIV vaccination approaches,we determined that it was important to perform an in-depthinvestigation of TLR expression pattern and associated coad-aptors in correlation to rapid progression. We also examinedresulting activation pathways at the transcriptional level bothin the innate and in the adaptive immune compartments ofSIV-infected macaques exhibiting the accelerated develop-ment of AIDS, in comparison to animals that follow a morechronic course. We further con�rmed the TLR activation tooccur predominantly in the adaptive T-cells compartment,by comparing the expression of TLR pathway components inlymphoid cells in comparison to themyeloid from the spleen.Our results suggest that TLR engagement and inefficientvirus-speci�c TCR signaling are linked to CD8 phenotypiccharacteristics of rapid progression in HIV infection. eoverexpression of TLRsmay be predictive of disease outcomeas a marker of hyperactivation in the absence of effectivespeci�c T-cell response, and potentially constitute a markerof immune senescence.
2. Material and Methods
2.1. Animals. Rhesus macaques were infected with a stockderived from SIVmac251, containing 1.25 ng of p27 (gag) Ag.Animals 298, 332, and 357 were uninfected and sacri�cedas controls. Animals 350, 353, and 354 were sacri�ced at73, 77, and 80 days aer infection (p.i.), respectively, andcomprised the 11 wk p.i. group (REG 11 weeks pi). Animals417, 418, 523, and 529 developed signs of simian AIDS andwere sacri�ced at days 56, 82, 140, and 34 p.i., respectively(RAP). Some of these animals have been characterized inprevious studies [10, 17, 18].
2.2. Cells. At necropsy, splenic and brain cells were obtainedand either were used for �ow cytometry or culture, or cry-opreserved as previously described [19]. Cells were thawedand washed in fetal bovine serum and were counted andincubated with anti-CD8 or anti-CD11b magnetic beads formagnetic separation and enrichment of CD8+ T cells usingMiltenyi Biotech, Inc., separation system (Miltenyi Biotec,Auburn, CA, USA) following the manufacturer’s protocol.e purity of cells was con�rmed by �ow cytometry asdescribed [19], using non-overlapping anti-human CD8-PE(clone DK25; DAKO, Carpinteria, CA, USA) and anti-Mac-1-PE (clone M1/70, Roche, Indianapolis, IN, USA). Biotiny-lated anti-rhesus macaque CD3 (Biosource) and anti-humanCD4 (L200 clone, BD Bioscience) were also used. Cellswere acquired in a FACSCalibur using CellQuest soware
(BD Immunocytometry Systems, San Jose, CA, USA) andanalyzed using FlowJo 6.2.1 soware (Tree Star, Ashland, OR,USA).
2.3. RNA and qRT-PCR. RNA from CD8+ and CD11b+ cellpellets were extracted using Ambion Totally RNA kit (LifeTechnologies, Carlsbad, CA, USA). First strand kit (SABio-sciences, Qiagen, Valencia, CA, USA) for cDNA synthesiswas performed. Isolated CD8+-enriched cells were screenedusing a custom made PCR array (SABiosciences). CD11b-enriched and CD11b-depleted cells were analyzed using apathway array analysis of rhesus macaque TLR pathways(RT2 Pro�ler PCR array, PAQQ-018C; SABiosciences). SyBrGreen real-time PCRswere performed inABI Prism 7900HTinstrument (Applied Biosystems) using ABI Prism 7900 SDS2.1. For gene expression, raw data threshold was normalizedusing GAPDH or the average of 5 housekeeping genes, asindicated at �gures legends, yielding dCt values.
2.4. Statistical Analysis. e screening of CD8 functionalperformance was made on 48 genes and analyzed usingone-way ANOVA and post hoc Tukey’s tests, using Prism(GraphPad Soware). e TLR pathway analysis on sortedcells was performed on 86 genes and comparison betweengroups was performed using Student’s 𝑡𝑡𝑡𝑡-test, using the web-based analysis tool PCR Array Data Analysis Web Portal(http://www.sabioscience.com/).
3. Results
3.1. RAPs Have TCR-Mediated Signaling and Several Func-tional Molecules at Low Levels but Highly UpregulatedTLR Coadaptor MyD88 in CD8+ Cells. SIV-infected RAPmacaques have poor antiviral CD8+ response and fail tomaintain activation [10]. To understand the nature of theCD8 collapse, we have analyzed changes in splenicCD8+ cellsfrom rhesus macaques infected with a SIVmac251-derivedstock.ree groups were examined: (1) REGs at 11 weeks p.i.,(2) RAPs at an average of 11 weeks p.i., and (3) uninfectedcontrols.We �rst performed a screening for expression of keyactivation and functional molecules (Figure 1). For that, pureCD8+ cells were isolated from the spleen, using magneticbead-labeled antibodies [19]. e purity of the CD8+ cellisolates by FACSwas ≥95%; mRNAwas extracted and reversetranscribed. Using quantitative PCRwe compared the controland infected animals, grouping the infected monkeys bydifferent progression patterns.
Interestingly, of the 48 gene transcripts examined, 12showed signi�cant (𝑃𝑃𝑃𝑃 𝑃𝑃 𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃) differences between the groupsby ANOVA.ese fell in twomain categories. Post hoc Tukeytesting revealed that four transcripts were downregulated inthe RAP animals when compared to REG animals and/orthe uninfected controls (Figure 1(a)). ese comprised keycomponents of CD8+ T cells and were strongly down-regulated relative to the uninfected controls. TCR𝛽𝛽𝛽𝛽 andCD3𝛾𝛾𝛾𝛾 are critical components of the antigen recognition andsignaling process in CD8+ T cells, whereas granzyme A ispart of the cytotoxic arsenal. While their downregulation

BioMed Research International 3
TCRβ∗ ∗
∗
dCt
dCt
dCt
dCt
CD3γ PD-1Granzyme A
Control REG RAP Control REG RAP Control REG RAP Control REG RAP
20
18
16
14
12
10
22
20
18
16
14
12
∗∗
∗∗ ∗∗18
16
14
12
10
8
201816141210
86420
(a)
∗∗
dCt
dCt
Fas Bax Caspase 3 TNFα
Control REG RAP Control REG RAP Control REG RAP Control REG RAP
dCt
dCt
20
18
16
14
12
10
∗∗∗∗
∗∗
∗∗
∗∗
23
21
19
17
15
13
22
20
18
16
14
∗∗∗
19
17
15
13
9
11
∗
∗
dCt
Tim3 MyD88 PP1β Cyclin B1
Control REG RAP Control REG RAP Control REG RAP
dCt
20
18
16
14
12
10
∗∗
dCt
18
16
14
12
10
8
∗∗∗
∗∗∗∗
21
19
17
15
13
11
dCt
Control REG RAP
21
19
17
15
13
11
(b)
F 1: Gene expression in CD8+ cells in uninfected and SIV infected REG and RAP monkeys. e expression of activation moleculeswas normalized against GAPDH. (a) Genes decreased in RAPs. Multiple group signi�cances demonstrated by one-way ANOVA (TCR𝛽𝛽𝛽𝛽 =0.0483, CD3𝛾𝛾𝛾𝛾 = 0.014�, PD-1 = 0.0044, granzyme A = 0.0033). (b) Genes increased in RAPS. Multiple group signi�cances demonstrated byone-way ANOVA (Fas = 0.0015, Bax = 0.0002, Caspase 3 = 0.0034, TNF𝛼𝛼𝛼𝛼 = 0.0056, Tim3 = 0.0180, MyD88 = 0.0001, PP1𝛽𝛽𝛽𝛽 = 0.0114, CyclinB1 = 0.00��). For both (a) and (b) signi�cance between the groups was determined by post hoc Tukey testing, and indicated by ∗𝑃𝑃𝑃𝑃 𝑃𝑃 𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃,∗∗𝑃𝑃𝑃𝑃 𝑃𝑃 𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃, ∗∗∗𝑃𝑃𝑃𝑃 𝑃𝑃 𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃.
can certainly compromise function, the downregulation ofPD-1 (programmed cell death-1) may result in a reductionof CD8+ inhibitory functions. On the other hand, PD-1downmodulation could also be a compensatory mechanismin an attempt to prevent CD8 exhaustion.
Eight of the transcripts were signi�cantly upregulated inRAP animals (Figure 1(b)). Six of these were signi�cantly andstrongly increased relative to both the uninfected controlsand the REG animals, and three can be directly linked toapoptosis: Fas, Bax, and Caspase 3. Another Tim3 representsan inhibitory molecule, although in contrast to the PD-1inhibitory molecule found downregulated above it is highlyupregulated. TNF-𝛼𝛼𝛼𝛼 expression is also markedly increased,but neither other cytokines were examined (IFN-𝛾𝛾𝛾𝛾, IL2, IL17,IL21) nor cytokine receptors (IL2R𝛼𝛼𝛼𝛼, IL7R, IL15R𝛼𝛼𝛼𝛼, IL21R)were altered. e sixth molecule upregulated relative to bothREGs and uninfected controls was MyD88. is increase wasstriking and intriguing given the key role ofMyD88 in linkingTLR recognition to NF-𝜅𝜅𝜅𝜅B activation. In fact, the inductionof TNF-𝛼𝛼𝛼𝛼 is one of the major effects of such activation.Two additional transcripts were found increased in the
RAPs relative to the REGs, the serine/threonine phosphatasePP1𝛽𝛽𝛽𝛽 and Cyclin B1, both of which are involved in cellularproliferation.
3.2. TLR Activation Is Concentrated in the Adaptive ImmuneCompartment. e decrease in TCR𝛽𝛽𝛽𝛽, CD3𝛾𝛾𝛾𝛾, and granzymeAand increase inTim3 are consistentwith the lack of adaptiveimmune function in RAP CD8+ cells that we found in aprevious study [10]. Tim3, in particular, has been shownto reduce cytotoxicity in exhausted CD8 cells during HIVinfection [20], suggesting that the elevation of Tim3 in rapidprogressors is consistent with a phenotype of exhaustionwithin the CD8+ compartment.
e strong increase in MyD88 along with the increasedTNF𝛼𝛼𝛼𝛼 was consistent with potential activation of the TLRpathway. Since the differences between RAP and REG CD8+cells were identi�ed among initial activation molecules, wehypothesized that antigen presenting cells could be involvedin the TLR activation. We thus examined the innate myeloidmononuclear cells, which include APC, from the spleniccells by selecting for CD11b+, as well as the CD11b− cells

4 BioMed Research International
6
5
4
3
2
1
0
Cel
ls (
%)
CD3+CD4+ CD3+CD8+
ControlREGRAP
∗
F 2: Percentage of T cells in the brain of controls andSIV-infected rhesus macaques exhibiting REG or RAP diseasecourse. Brain cell suspensions were stainedwith �uorescent-labelledantibodies against CD3, CD4, and CD8 surface markers to identifyCD3+CD4+ T helper cells and CD3+CD8+ cytotoxic T cells, using�ow cytometry. ∗𝑃𝑃𝑃𝑃 𝑃𝑃 𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃 in comparison to uninfected controlanimals.
mainly consisting of T and B adaptive immune cells, for theexpression of 84 genes related to the TLR signaling pathwayand innate immunity. Strikingly the differences between RAPand REG were almost exclusive in the CD11b− lymphoidadaptive immune compartment (Figure 2), with signi�cantincreases in RAP animals in three of the TLRs (TLR3, 6, and9) and multiple TLR adapter and interacting proteins, as wellas the downstream NF𝜅𝜅𝜅𝜅B and JNK/p38 signaling pathways(Table 1). ese include, as identi�ed above, increases inMyD88 and TNF𝛼𝛼𝛼𝛼. In contrast, in the CD11b+ myeloid cells,only one gene was signi�cantly different between the RAPsand REGs, TLR10 (2.61-fold, 𝑃𝑃𝑃𝑃 𝑃𝑃 𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃).
3.3. Isolated Brain Immune Cells fromRAPAnimals Also Showa Predominance on TLR-Mediated Response. RAP animalshave a high incidence of SIV encephalitis (SIVE). WhileSIV-speci�c CD8+ T cells are present in the brains of theseanimals, they are ineffective in controlling the high brainviral load characterizing SIVE [10]. Similar proportions ofCD8+ T cells are indeed present in brains of both REGsand RAPs (Figure 2). In order to ascertain whether the TLRpathways are also elevated in theCNS, we isolatedCD8+CNSin�ltrating cells and examined them for expression of theTLR components using quantitative real-time PCR. Indeed�ve TLRs (TLR 1, 2, 3, 8, and 9) aswell as, similar to the spleenlymphocytes, multiple TLR adapter and interacting proteinswere upregulated in RAP animals. e downstream NF𝜅𝜅𝜅𝜅Band JNK/p38 signaling pathways are increased; however, wenote that some were decreased, including TLR4 (Table 2).
Overall, most examined molecules were increased, and, asfound in the spleen, signi�cant increases in MyD88 andTNF𝛼𝛼𝛼𝛼were observed in the CD8+ cells in the brains of RAPs.
Overall, we observed that rapid progression is character-ized by a strong MyD88-dependent TLR activation in theabsence of an effective anti-SIV speci�c response, causingan unbalance that can lead to exacerbated in�ammatoryresponse and lack of control over viral load. is wasobserved both in the spleen and in the CNS.
4. Discussion
Rapid progressors have low or absent virus-speci�c CD8+pool in the spleen and brain, as accessed by the number ofGag and Tat-speci�c cells, con�rming previous �ndings [10].is reduction may be on the base of a low TCR signalingand activation through nonspeci�c pathways such as TLR,generating rather bystander CD8+ cells, activated to becomeproin�ammatory but not e�cient against the infection. Herewe show that the expression of TLRs is increased on splenicCD8+ cells from RAP animals in comparison to controls.Downstream adaptor molecule MyD88 was increased inRAPs, suggesting that in rapid progression upregulation ofTLRs is followed by its engagement. e increase of TLR9in splenic CD8+ cells from RAPs compared to REGs wasvalidated by detection of intracellular levels by FACS (notshown).
ese results put forward a potential mechanismfor previous results from rapid progressors expressingMamuA∗01 class I haplotype. When compared to regularMamuA∗01+ progressors, not only they showed a reducedTat and Gag-speci�c pool [10], but their CD8+ T cells failedto proliferate speci�cally in response to Gag CM9 and TatSL8 peptides in vitro.
e initial failure of the TCR signaling originated fromthe absence of a speci�c pool and predominance of TLRpathways results in differences on performance and reg-ulation of survival and cell cycle molecules, favoring theupregulation of molecules related to apoptosis. Indeed, directTLR stimulation on T cells may result in increased apoptosis[21]. TLR2, in particular, which was upregulated both inthe spleen and in the brain of RAP animals in comparisonto REGs, can trigger MyD88-mediated apoptosis, involvingFas and Caspase 8 [22], depending on levels of Bcl-2 familymolecules of pro- and antiapoptotic proteins [23]. We foundupregulation of Fas and Caspases as well as decrease in Bcl-2in splenic CD8+ cells from RAP animals.
Several studies suggest that the action of TLR in CD8activation is indirect and may require the participation ofinnate immune cells [24–26]. In the context of infection,CD8+ cells express TLR, but HIV does not infect these cells.Nevertheless, TLR agonists can directly stimulate CD8+ Tcells [21, 27, 28]. We analyzed the performance of isolatedCD11b cells from REGs and RAPs regarding the expressionof TLRs and its adaptors. However, only TLR10 showeda signi�cant upregulation in correlation with poor diseaseoutcome in the innate compartment. is is interesting sincethe role of TLR10, which is mostly expressed by B cells andDCs [29], is not clear.

BioMed Research International 5
T 1: Comparison of expression levels of TLR signaling pathway and innate immunity genes in SIV-infected REG and RAP CD11b-depleted splenic cells. Expression levels were normalized using the average of 5 housekeeping genes and the ratio between RAP and REGanimals� dCT values was calculated issuing fold change. Shown are the genes whose expression is signi�cantly changed between the groups,organized by functional categories of the TLRs and their adapter as well as interacting proteins, the downstreamNF𝜅𝜅𝜅𝜅B and JNK/p38 signalingpathways, as well as other related molecules (MAPK8IP3 is present in both the adapter/interacting and the JNK/p38 categories).
TLRs and adapter/interacting Fold RAP/REG 𝑃𝑃𝑃𝑃 value NF𝜅𝜅𝜅𝜅B & JNK/p38 Fold RAP/REG 𝑃𝑃𝑃𝑃 value Other Fold RAP/REG 𝑃𝑃𝑃𝑃 valueGPC1 9.43 0.0168 ESCIT 10.05 0.0235 CCL2 13.42 0.0193HRAS 5.72 0.0061 IL10 12.73 0.0497 CD86 8.47 0.0216LY86 9.96 0.0455 IRAK2 8.07 0.0121 CSF3 12.03 0.0161MAL 10.28 0.0028 IRAK4 11.15 0.0310 IL6 11.17 0.0239MAPK8IP3 12.26 0.0031 IRF3 9.74 0.0077 IRF1 9.23 0.0006MYD88 12.39 0.0450 JUN 11.35 0.0387 IRF7 14.37 0.0412PGLYRP3 12.08 0.0388 MAP2K3 11.65 0.0229 LTA 10.12 0.0196SARM1 7.38 0.0335 MAP2K4 8.80 0.0282 PRKRA 12.03 0.0258TIRAP 7.53 0.0275 MAPK12 10.05 0.0077 TBK1 10.79 0.0476TLR3 13.90 0.0142 MAPK8IP3 12.26 0.0031TLR6 4.10 0.0472 NFKB2 8.70 0.0317TLR9 5.60 0.0489 NFKBIB 12.67 0.0242TOLLIP 4.70 0.0082 REL 6.45 0.0270
RELB 9.45 0.0401TNF 9.51 0.0034
TNFRSF1A 8.12 0.0050
T 2: Comparison of expression levels of TLR signaling pathway and innate immunity genes in SIV-infected REG and RAP CD8+ cellsisolated from the brain. Expression levels were normalized using the average of 5 housekeeping genes and the ratio between RAP and REGanimals� dCT values was calculated issuing fold change. Shown are the genes whose expression is signi�cantly changed between the groups,organized by functional categories of the TLRs and their adapter as well as interacting proteins, the downstreamNF𝜅𝜅𝜅𝜅B and JNK/p38 signalingpathways, as well as other related molecules.
TLRs and adapter/interacting Fold RAP/REG 𝑃𝑃𝑃𝑃 value NF𝜅𝜅𝜅𝜅B & JNK/p38 Fold RAP/REG 𝑃𝑃𝑃𝑃 value Other Fold RAP/REG 𝑃𝑃𝑃𝑃 valueHRAS 1.43 0.0436 IL10 2.56 0.0373 CCL2 27.49 0.0338MYD88 2.55 0.0183 IRF3 0.26 0.0123 IL12B 0.17 0.0387PELI2 0.41 0.0011 MAP2K3 1.62 0.0400 IL2 3.38 0.0075TLR1 1.95 0.0437 MAP4K4 0.46 0.0040 IRF1 3.08 0.0016TLR2 4.60 0.0429 MAPK10 1.38 0.0374 IRF7 4.91 0.0009TLR3 1.11 0.0417 NFKB1 1.65 0.0151 LTA 1.27 0.0245TLR4 0.51 0.0483 NFKBIB 2.03 0.0300 PTGS2 2.57 0.0371TLR8 2.64 0.0238 NFKBIE 1.37 0.0496 TBK1 1.20 0.0276TLR9 1.71 0.0242 PPARA 0.44 0.0489TOLLIP 1.78 0.0376 REL 1.88 0.0032
TNF 4.47 0.0482TNFRSF1A 2.72 0.0090
Even though TLRs have been proposed as adjuvantsin HIV vaccination approaches. For example, stimulationof TLR4, TLR2, TLR6, TLR7, and TLR8 has been usedto enhance HIV-1-speci�c cellular, humoral, and mucosalimmunity [30]. CpG ODNs, TLR9 ligands, are also effectiveadjuvants for peptide-based HIV-1 vaccines [31]. e mag-nitude and quality of the CD8+ response to HIV is improvedwhen antigens are conjugated with TLR7/8 agonists [32,33]. However, even in the absence of high affinity TCRengagement, TLR7 agonists can induce activation of CD8+ Tcells [21], suggesting that virus- speci�c CTLs in the presence
of synthetic agonists may be improved if the development ofan antigen-speci�c pool precedes the TLR stimulation.
Importantly, the response triggered by TLR engagementis not necessarily protective. Other studies have revealed acontroversial role for TLRs in vaccination approaches. Forinstance, in a mouse model of HIV-1 infection, sustainedTLR7 stimulation causes activation and disruption of thelymphoid system, leading to pathology and poor outcome[34]. Interestingly, data from SIV/macaque model provideadditional support for a role of TLR pathways in HIV-1 pathogenesis. SIV infection in rhesus macaques induces

6 BioMed Research International
strong immune activation and pathogenesis while in sootymangabeys, the natural host of SIV, immune activation issigni�cantly lower while disease is not observed. Conversely,stimulation with SIV, TLR7, or TLR9 ligands induced muchstronger IFN production by pDCs derived from rhesusmacaques compared with pDCs from sooty mangabeys. ishappens in close correlation with genetic differences in IRF-7 between these 2 species, which could offer a potentialexplanation for differences in pDC responses to ligands [35].TLR polymorphisms have been also found in humans inassociation with both HIV progression and protection [36].
In another study, monkeys intravaginally treated withTLR7 and TLR9 agonists prior to SIV challenge had sig-ni�cantly higher set point viremia than control animalsthat were PBS-treated [37], suggesting that the activationinduced by direct TLR engagement on CD8+ cells is notenough to produce efficient antiviral response. Furthermore,on CD4 cells, it may enhance vaginal SIV transmissionor posttransmission-SIV replication. us, TLR engagementahead of the development of a virus-speci�c pool can gener-ate poorCD8performance characteristic of rapid progression[10].
Our results show that a failure in controlling viral load,both peripherally and in the brain, is strongly associatedto a poor TCR-mediated response, lack of development ofantiviral response, and characteristics of exhaustion in theCD8 compartment. In addition, immune cells from rapidprogressors show instead a robust activation of MyD88-mediated TLR pathways, including downstream NF𝜅𝜅𝜅𝜅b andJNK/p38 pathways, and lead to upregulation of cell deathmarkers. Our results suggest that there is a high risk instimulating TLR response ahead of the development of astrong antiviral response.e use of TLR as vaccine adjuvantscan be positive but it should be used with caution, inindividuals where signs of speci�c response are detectable. Inaddition, hyperactivation of TLR pathways can be a strongpredictive of rapid progression.
Acknowledgments
e authors thank Dr. Bruce Torbett (e Scripps ResearchInstitute) for interesting discussions. e work was devel-oped with funding from CFAR/NIH 2 P30 AI36214,1R03DA027936-01, and 1R21DA029491-01A1. is is thepaper number 21770 of e Scripps Research Institute.
References
[1] E. S. Roberts, M. A. Zandonatti, D. D. Watry et al., “Inductionof pathogenic sets of genes in macrophages and neurons inneuroAIDS,” American Journal of Pathology, vol. 162, no. 6, pp.2041–2057, 2003.
[2] L. J. Madden, M. A. Zandonatti, C. T. Flynn et al., “CD8+ celldepletion ampli�es the acute retroviral syndrome,” Journal ofNeuroVirology, vol. 10, no. 1, pp. 58–66, 2004.
[3] S. J. Bissel, G. Wang, A. M. Trichel, M. Murphey-Corb, andC. A. Wiley, “Longitudinal analysis of monocyte/macrophageinfection in simian immunode�ciency virus-infected, CD8+T-cell-depleted macaques that develop lentiviral encephalitis,”
American Journal of Pathology, vol. 168, no. 5, pp. 1553–1569,2006.
[4] S. V. Westmoreland, E. Halpern, and A. A. Lackner, “Simianimmunode�ciency virus encephalitis in rhesus macaques isassociated with rapid disease progression,” Journal of NeuroVi-rology, vol. 4, no. 3, pp. 260–268, 1998.
[5] S. M. Smith, B. Holland, C. Russo, P. J. Dailey, P. A. Marx, andR. I. Connor, “Retrospective analysis of viral load and SIV anti-body responses in rhesus macaques infected with pathogenicSIV: predictive value for disease progression,” AIDS Researchand Human Retroviruses, vol. 15, no. 18, pp. 1691–1701, 1999.
[6] S. I. Staprans, P. J. Dailey, A. Rosenthal et al., “Simian immun-ode�ciency virus disease course is predicted by the extent ofvirus replication during primary infection,” Journal of Virology,vol. 73, no. 6, pp. 4829–4839, 1999.
[7] M. Dykhuizen, J. L. Mitchen, D. C. Monte�ori et al., “Determi-nants of disease in the simian immunode�ciency virus-infectedrhesus macaque: characterizing animals with low antibodyresponses and rapid progression,” Journal of General Virology,vol. 79, part 10, pp. 2461–2467, 1998.
[8] L. J. Picker, S. I. Hagen, R. Lum et al., “Insufficient productionand tissue delivery of CD4+ memory T cells in rapidly pro-gressive simian immunode�ciency virus infection,” Journal ofExperimental Medicine, vol. 200, no. 10, pp. 1299–1314, 2004.
[9] A. Okoye, M. Meier-Schellersheim, J. M. Brenchley et al., “Pro-gressive CD4+ central-memory T cell decline results in CD4+effector-memory insufficiency and overt disease in chronic SIVinfection,” Journal of Experimental Medicine, vol. 204, no. 9, pp.2171–2185, 2007.
[10] M. C. G. Marcondes, S. Sopper, U. Sauermann et al., “CD4de�cits and disease course acceleration can be driven by acollapse of the CD8 response in rhesus macaques infected withsimian immunode�ciency virus,” AIDS, vol. 22, no. 12, pp.1441–1452, 2008.
[11] D. T. Evans, D. H. O�Connor, P. Jing et al., “Virus-speci�c cyto-toxic T-lymphocyte responses select for amino-acid variation insimian immunode�ciency virus Env andNef,”Nature Medicine,vol. 5, no. 11, pp. 1270–1276, 1999.
[12] V. M. Hirsch, S. Santra, S. Goldstein et al., “Immune failurein the absence of profound CD4+ T-lymphocyte depletionin simian immunode�ciency virus-infected rapid progressormacaques,” Journal of Virology, vol. 78, no. 1, pp. 275–284, 2004.
[13] D. T. Evans, P. Jing, T. M. Allen et al., “De�nition of �venew simian immunode�ciency virus cytotoxic t-lymphocyteepitopes and their restricting major histocompatibility complexclass I molecules: evidence for an in�uence on disease progres-sion,” Journal of Virology, vol. 74, no. 16, pp. 7400–7410, 2000.
[14] R. A. Koup, J. T. Safrit, Y. Cao et al., “Temporal association ofcellular immune responses with the initial control of viremiain primary human immunode�ciency virus type 1 syndrome,”Journal of Virology, vol. 68, no. 7, pp. 4650–4655, 1994.
[15] C. M. Hay, D. J. Ruhl, N. O. Basgoz et al., “Lack of viralescape and defective in vivo activation of human immunode�-ciency virus type 1-speci�c cytotoxic T lymphocytes in rapidlyprogressive infection,” Journal of Virology, vol. 73, no. 7, pp.5509–5519, 1999.
[16] S. A. Islam, C. M. Hay, K. E. Hartman et al., “Persistence ofhuman immunode�ciency virus type 1-speci�c cytotoxic T-lymphocyte clones in a subject with rapid disease progression,”Journal of Virology, vol. 75, no. 10, pp. 4907–4911, 2001.
[17] M. C. G. Marcondes, T. H. Burdo, S. Sopper et al., “Enrichmentand persistence of virus-speci�c CTL in the brain of simian

BioMed Research International 7
immunode�ciency virus-infected monkeys is associated with aunique cytokine environment,” Journal of Immunology, vol. 178,no. 9, pp. 5812–5819, 2007.
[18] M. C. G. Marcondes, C. M. S. Lanigan, T. H. Burdo, D.D. Watry, and H. S. Fox, “Increased expression of monocyteCD44v6 correlates with the development of encephalitis in rhe-sus macaques infected with simian immunode�ciency virus,”Journal of Infectious Diseases, vol. 197, no. 11, pp. 1567–1576,2008.
[19] M. C. G. Marcondes, E. M. E. Burudi, S. Huitron-Resendiz etal., “Highly activated CD8+ T cells in the brain correlate withearly central nervous system dysfunction in simian immunod-e�ciency virus infection,” Journal of Immunology, vol. 167, no.9, pp. 5429–5438, 2001.
[20] A. Sakhdari, S. Mujib, B. Vali et al., “Tim-3 negatively regulatescytotoxicity in exhausted CD8 + T cells in HIV infection,” PLoSOne, vol. 7, no. 7, Article ID 40146, 2012.
[21] N. Funderburg, A. A. Luciano,W. Jiang, B. Rodriguez, S. F. Sieg,andM.M. Lederman, “Toll-like receptor ligands induce humanT cell activation anddeath, amodel forHIVpathogenesis,”PLoSOne, vol. 3, no. 4, Article ID e1915, 2008.
[22] A. O. Aliprantis, R. B. Yang, D. S. Weiss, P. Godowski, and A.Zychlinsky, “e apoptotic signaling pathway activated by Toll-like receptor-2,” EMBO Journal, vol. 19, no. 13, pp. 3325–3336,2000.
[23] M. Raisova, A. M. Hossini, J. Eberle et al., “e Bax/Bcl-2 ratio determines the susceptibility of human melanomacells to CD95/Fas-mediated apoptosis,” Journal of InvestigativeDermatology, vol. 117, no. 2, pp. 333–340, 2001.
[24] A.Meier, G. Alter, N. Frahm et al., “MyD88-dependent immuneactivation mediated by human immunode�ciency virus type 1-encoded toll-like receptor ligands,” Journal of Virology, vol. 81,no. 15, pp. 8180–8191, 2007.
[25] G. Mancuso, M. Gambuzza, A. Midiri et al., “Bacterial recogni-tion by TLR7 in the lysosomes of conventional dendritic cells,”Nature Immunology, vol. 10, no. 6, pp. 587–594, 2009.
[26] A. S. Beignon, K. McKenna, M. Skoberne et al., “Endocytosisof HIV-1 activates plasmacytoid dendritic cells via Toll-likereceptor-viral RNA interactions,” Journal of Clinical Investiga-tion, vol. 115, no. 11, pp. 3265–3275, 2005.
[27] C. L. Ahonen, C. L. Doxsee, S. M. McGurran et al., “CombinedTLRandCD40 triggering induces potentCD8+ Tcell expansionwith variable dependence on type I IFN,” Journal of Experimen-tal Medicine, vol. 199, no. 6, pp. 775–784, 2004.
[28] B. Brichacek, C. Vanpouille, Y. Kiselyeva et al., “Contrastingroles for TLR ligands in HIV-1 pathogenesis,” PLoS One, vol.5, no. 9, Article ID e12831, pp. 1–12, 2010.
[29] U. Hasan, C. Chaffois, C. Gaillard et al., “Human TLR10 isa functional receptor, expressed by B cells and plasmacytoiddendritic cells, which activates gene transcription throughMyD88,” Journal of Immunology, vol. 174, no. 5, pp. 2942–2950,2005.
[30] A. A. C. Lemckert, J. Goudsmit, andD.H. Barouch, “Challengesin the search for an HIV vaccine,” European Journal of Epidemi-ology, vol. 19, no. 6, pp. 513–516, 2004.
[31] M. Tritel, A. M. Stoddard, B. J. Flynn et al., “Prime-boost vacci-nation with HIV-1 gag protein and cytosine phosphate guano-sine oligodeoxynucleotide, followed by adenovirus, inducessustained and robust humoral and cellular immune responses,”Journal of Immunology, vol. 171, no. 5, pp. 2538–2547, 2003.
[32] U. Wille-Reece, B. J. Flynn, K. Loré et al., “HIV Gag proteinconjugated to a Toll-like receptor 7/8 agonist improves themagnitude and quality of 1 and CD8+ T cell responses innonhuman primates,” Proceedings of the National Academy ofSciences of the United States of America, vol. 102, no. 42, pp.15190–15194, 2005.
[33] U. Wille-Reece, B. J. Flynn, K. Loré et al., “Toll-like receptoragonists in�uence the magnitude and quality of memory Tcell responses aer prime-boost immunization in nonhumanprimates,” Journal of Experimental Medicine, vol. 203, no. 5, pp.1249–1258, 2006.
[34] S. Baenziger, M. Heikenwalder, P. Johansen et al., “TriggeringTLR7 inmice induces immune activation and lymphoid systemdisruption, resembling HIV-mediated pathology,” Blood, vol.113, no. 2, pp. 377–388, 2009.
[35] J. N. Mandl, A. P. Barry, T. H. Vanderford et al., “DivergentTLR7 and TLR9 signaling and type I interferon production dis-tinguish pathogenic and nonpathogenic AIDS virus infections,”Nature Medicine, vol. 14, no. 10, pp. 1077–1087, 2008.
[36] D. Y. Oh, K. Baumann, O. Hamouda et al., “A frequentfunctional toll-like receptor 7 polymorphism is associated withaccelerated HIV-1 disease progression,” AIDS, vol. 23, no. 3, pp.297–307, 2009.
[37] E. Chung, S. B. Amrute, K. Abel et al., “Characterization ofvirus-responsive plasmacytoid dendritic cells in the rhesusmacaque,” Clinical and Diagnostic Laboratory Immunology, vol.12, no. 3, pp. 426–435, 2005.

Hindawi Publishing CorporationBioMed Research InternationalVolume 2013, Article ID 492372, 14 pageshttp://dx.doi.org/10.1155/2013/492372
Review ArticleWhat Is Recent in Pancreatic Cancer Immunotherapy?
Elena Niccolai,1 Domenico Prisco,2 Mario Milco D’Elios,1, 3 and Amedeo Amedei1, 3, 4
1 Department of Internal Medicine, University of Florence and Patologia Medica Unit Department of Biomedicine,Azienda Ospedaliero-Universitaria Careggi, 50134 Florence, Italy
2 Department of Medical and Surgical Critical Care, University of Florence and Patologia Medica Unit Department of Biomedicine,Azienda Ospedaliero Universitaria Careggi, 50134 Florence, Italy
3 Center of Oncologic Minimally Invasive Surgery, University of Florence, Largo Brambilla 3, 50134 Florence, Italy4 Division of Immunology, Department of Internal Medicine, University of Florence, Viale Pieraccini, 6, 50134 Florence, Italy
Correspondence should be addressed to Amedeo Amedei; aamedei�uni�.it
Received 21 May 2012; Accepted 6 July 2012
Academic Editor: Julie Curtsinger
Copyright © 2013 Elena Niccolai et al. is is an open access article distributed under the Creative Commons Attribution License,which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Pancreatic cancer (PC) represents an unresolved therapeutic challenge, due to the poor prognosis and the reduced responseto currently available treatments. Pancreatic cancer is the most lethal type of digestive cancers, with a median survival of 4–6months. Only a small proportion of PC patients is curative by surgical resection, whilst standard chemotherapy for patients inadvanced disease generates only modest effects with considerable toxic damages. us, new therapeutic approaches, speciallyspeci�c treatments such as immunotherapy, are needed. In this paper we analyze recent preclinical and clinical efforts towardsimmunotherapy of pancreatic cancer, including passive and active immunotherapy approaches, designed to target pancreatic-cancer-associated antigens and to elicit an antitumor response in vivo.
1. Introduction
Pancreatic cancer (PC) represents an unresolved therapeuticchallenge, due to the poor prognosis and the reduced re-sponse to currently available treatments. Pancreatic canceris the most lethal type of digestive cancers, with a mediansurvival (MS) of 4–6 months [1]. ere are three principalPC types: ductal adenocarcinoma, neuroendocrine tumors(rare), and cystic neoplasm (less than 1% of pancreaticcancers) [1]. Pancreatic ductal adenocarcinoma accounts for90% of cancers of the pancreas and has the poorest outcome,representing the 4th most common cause of cancer-relateddeath among men and women [2].
e only potentially curative therapy for pancreatic can-cer is surgical resection. Unfortunately, only 20% PC patientsare resectable at the time of diagnosis, and among thosepatients who undergo resection and have tumor-free mar-gins, the 5-year survival rate aer surgery is 10% to 25%[3]. Gemcitabine, with or without erlotinib, represents thestandard chemotherapy but the bene�t is only modest, andmost patients do not survive longer than 6 months [4, 5].
Development of novel agents and approaches is urgent-ly needed in conjunction with improvement in access toclinical trials for patients. Since there are different evidencesthat pancreatic adenocarcinomas elicit antitumor immuneresponses [6–9] speci�c immunotherapy could be of greatimportance in the PC treatment. In support of the PC-speci�c immunotherapy approaches there are numerous datashowing how PC patients generate B and T cells speci�cto antigens expressed on autologous pancreatic tumor cells[10–12], such as Wilms’ tumor gene 1 (WT1) (75%) [13],mucin 1 (MUC1) (over 85%) [14], human telomerase reversetranscriptase (hTERT) (88%) [15], mutated K-RAS (73%)[16], survivin (77%) [17], carcinoembryonic antigen (CEA)(over 90%) [18], HER-2/neu (61.2%) [19], p53 (67%) [20],and 𝛼𝛼-enolase [21]. Furthermore, the analysis of immunein�ltrates in human tumors has demonstrated a positivecorrelation between prognosis and presence of humoralresponse to pancreatic antigens (MUC-1 and mesothelin)[8, 9, 22] or of tumor-in�ltrating T cells [23].
In this paper we analyze recent preclinical and clin-ical efforts towards immunotherapy of pancreatic cancer,

2 BioMed Research International
including passive immunotherapy approaches, such as theuse of antibodies or effector cells generated in vitro, andactive immunotherapic strategies, whose goal is to stimulatean antitumor response in vivo, by means of vaccination.
2. Passive Immunotherapy
2.1. Humoral Immunity: e Role of Monoclonal Antibod-ies. Speci�c recognition and elimination of pathologicalorganisms or malignant cells by antibodies were proposedover a century ago by Paul Ehrlich, who is credited forconceptualizing the “magic bullet” theory of targeted therapy.Over the past 30 years, antibody cancer therapeutics havebeen developed and used clinically in an effort to realize thepotential of targeted therapy. Antibodies can target antigensdifferentially expressed in tumor cells (tumor-associatedantigens (TAAs)) or can be used to block molecules involvedin cancer progression or angiogenesis. e immunoglobulinscan invoke tumor cell death by blocking ligand-receptorgrowth and survival pathways. In addition, innate immuneeffector mechanisms: antibody-dependent cellular cytotox-icity (ADCC), complement-mediated cytotoxicity (CMC),and antibody-dependent cellular phagocytosis (ADCP), areemerging as equally important [24].
Although unconjugated antibodies have had efficacy,molecular genetics and chemical modi�cations to mono-clonal antibodies (mAbs) have advanced their clinical utility.For example, modi�cation of immune effector engagementhas improved pharmacokinetic pro�les, and conjugatingcytotoxic agents to mAbs has enhanced targeted therapeu-tic delivery to tumors. e increasing facility of antibodymodi�cations has made it possible to construct diverse andefficacious mAb-based therapeutics.
e humoral immune response to mesothelin has beenfound to be a favorable prognostic factor for pancreatic cancer[8, 22, 25, 26]. Mesothelin is a 40 kDa protein present innormalmesothelial cells of the pericardium, pleura, and peri-toneum, but overexpressed inmesotheliomas ovarian cancers[27] and detected in 90–100% of pancreatic adenocarcinomas[28, 29]. Different antibodies tomesothelin have been studiedand in particular SS1P, a murine single-chain Fv, speci�cfor human mesothelin, which has been fused to PE38, a38 kDa portion of Pseudomonas exotoxin A (PE-A). Aerbinding to mesothelin and subsequent internalization intocells, it inhibits protein synthesis and results in apoptosis[30]. In phase I clinical studies SS1P was found to be welltolerated, with self-limiting pleuritis as the dose-limitingtoxicity. Also, the administration of a version of SS1P withreleasable PEGylation resulted in complete regression of amesothelin-expressing human carcinoma in mice with onlya single dose [30–32]. MORAb-009, a monoclonal antibodyagainst mesothelin, is being tested in a phase I trial of 11patients (threewith pancreatic cancer) [33]. One of themwhohad previously progressed on gemcitabine showed diseasestabilization on computed tomography (CT) and a dropin CA19-9 (carbohydrate antigen 19-9). Two fully human,antihumanmesothelin antibodies,M912 andHN1, have beendeveloped, which bind mesothelin-positive cells and result in
their lysis via ADCC [34, 35]. Similar to SS1P, HN1 has beenfused to truncated PE-A immunotoxin, although its bindingsite on mesothelin probably binds a distinct but overlappingepitope to that of SS1P [35].
MUC1 (mucin-1, CD227) is a polymorphic, glycosylatedtype I transmembrane protein present in glandularepithelium of different tissues (pancreas, breast, lung) andoverexpressed (aberrantly glycosylated) in 90% of pancreaticcancers [36, 37]. It inhibits cell-cell and cell-stromainteractions and functions as a signal transducer in the cancerprogression, including tumor invasion and metastasis [38].Evidences suggest that circulating anti-MUC1-IgG is afavorable prognostic factor for pancreatic cancer [22].Downregulation of MUC1 expression in human PC cell lineS2-013 by RNAi signi�cantly decreased proliferation in vitroand in nude mice [39]. In a murine model, the use of MUC1-speci�c 90�ttrium-labelledmoAbPAM4 in combinationwithgemcitabine as a radiosensitiser [40] increased inhibition oftumor growth and prolonged animal survival. To date, it isundergoing phase I trial for stage III or IV PC patients.
In vitro study showed that 213Bi-C595 was speci�callycytotoxic to MUC1-expressing PC cells in a concentration-dependent manner compared to controls. 213Bi-C595 is amoAb targeting the protein core of MUC1, conjugated withthe 𝛼𝛼-particle-emitting 213bismuth [37].
PankoMab (Glycotope, Germany) is a murine anti-human MUC-1 antibody that binds to a carbohydrate-induced conformational tumor epitope of MUC-1, greatlyincreasing its tumor speci�city [41]. PankoMab can induceADCC of MUC-1 positive cells and can also induce deathfollowing internalization by inhibition of RNA polymerasewhen linked to 𝛽𝛽-amanitin. e humanized version ofPankoMab has been shown to react to the tumor expressedMUC-1 in multiple human carcinomas, although no clinicaltrials have been published [42].
e epidermal growth factor receptor 2 (HER2), atransmembrane receptor tyrosine kinase, is overexpressedin up to 45% of pancreatic cancer. An anti-Her-2/neuantibody, known as Herceptin (Genentech Inc., CA, USA)or trastuzumab, has been used with some success to treatPC murine models. Treatments with trastuzumab prolongedsurvival and reduced liver metastasis in nude mice ortho-topically challenged with human pancreatic tumor cell linesthat expressed Her-2/neu at low levels. e pancreatic lineswere sensitive to ADCC lysis by trastuzumab in vitro [43].Similar results were found when nude mice (challenged withHer-2/neu high expressing human PC cell lines) were treatedwith both trastuzumab and 5-�uorouracil [44]. e com-bination of treatments signi�cantly inhibited tumor growthcompared with either treatment alone. When combined withmatuzumab, an anti-EGFR antibody, trastuzumab treatment,resulted in inhibited PC growth in a nude mouse [45]. Also,this combined treatment was more effective than treatmentwith either antibody alone or combined with gemcitabine[46].
Carcinoembryonic antigen (CEA), a member of a familyof cell surface glycoproteins involved in cell adhesion, isfrequently overexpressed in various types of human cancers.

BioMed Research International 3
Many anti-CEA antibodies have been used for immunother-apy, such as hMN-14 (labetuzumab), which has been shownto induce ADCC in vitro with CEA+ colon tumor cellsand inhibited growth of lung metastases in nude mice [47].A phase I/II trial with hMN-14 in PC patients has beencompleted but the results have not been published [48].
EGFR is a transmembrane glycoprotein receptor, over-expressed in 90% of pancreatic tumors [49], which inducestumor cell proliferation and neovascularization; also thisexpression is associated with worse prognosis [50, 51]. Block-ing EGFR signaling decreases growth and metastasis of pan-creatic tumor in animal models and enhances the effects ofgemcitabine [52, 53].
Cetuximab (Erbitux or IMC-C225) is a chimeric mono-clonal antibody generated from fusion of the variable regionof themurine anti-EGFRmonoclonal antibodyM225 and thehuman IgG1 constant region. Promising laboratory resultshave led cetuximab to be tested in clinical trials. A phaseIII randomized study by Southwestern Oncology Group(SWOG) tested the efficacy of cetuximab and gemcitabinecombination in patients with advanced PC. e mediansurvival was 6months in the gemcitabine arm and 6.5monthsin the combination arm for an overall hazard ratio (HR) of1.09 (𝑃𝑃 𝑃 𝑃𝑃𝑃𝑃). e corresponding progression free survivalwas 3months and 3.5months, respectively.e study failed todemonstrate a clinically signi�cant advantage of the additionof cetuximab to gemcitabine [54]. In an ongoing phase IItrial with trimodal therapy of cetuximab, gemcitabine andintensity modulated radiotherapy (IMRT) for patients withadvanced PC; there was no increase in toxicity pro�le [55].One-year survival was 57% while median survival has notbeen reached.
Matuzumab (EMD72000) is a humanized IgG1 mon-oclonal antibody to the human EGFR. Laboratory studieshave shown promising inhibitory effects on tumor growthand angiogenesis, including L3.6pl in an orthotopic ratmodel [56]. In a phase I study of combined treatment withmatuzumab and gemcitabine, eight out of 12 patients withadvanced pancreatic adenocarcinoma showed partial re-sponse or stable disease [57].
Vascular endothelial growth factor (VEGF) plays a pivotalrole in the control of angiogenesis, tumor growth, andmetastasis [58]. VEGF and its receptors are overexpressedin PC and have been demonstrated to be a poor prognosticfactor. ere is suggestion that elevated serum VEGF levelscorrelate with tumor stage, disease recurrence, and survival[59]. Development of therapeutic strategies directed towardsthe VEGFmediated signaling axis has been extensively testedin patients with advanced PC.
Bevacizumab (Avastin) is a recombinant humanized anti-VEGF monoclonal antibody. A pilot study demonstratedthat bevacizumab, when added to gemcitabine in patientswith metastatic PC, resulted in a signi�cant improvement inresponse, survival, and progression-free survival [60]. iswas immediately followed by a phase III trial by CALGBcomparing gemcitabine plus bevacizumab to gemcitabineplus placebo and showing no bene�t for bevacizumab addi-tion [61]. e AviTa phase III trial that examined treatmentwith gemcitabine plus erlotinib with either bevacizumab or
placebo has been closed. Bevacizumab, however, may havea role in palliative treatment of chemotherapy-resistant PC.In a case report, a patient with stage IV disease initiallyunresponsive to gemcitabine, 5-FU, irinotecan, and cisplatinsubsequently responded with the addition of bevacizumab[62].
2.2. Cellular Mediated Immunity: Adoptive T Cell Transfer.Adoptive T cell transfer is a form of immunotherapy in whichpatient’s own T cells are expanded and reinfused into thepatient. In particular, this method involves harvesting thepatient’s peripheral blood T lymphocytes, stimulating andexpanding the autologous tumor-reactive T cells using IL-2and CD3-speci�c antibody, before subsequently transferringthem back into the patient. Adoptive T cell therapy dependson the ability to optimally select or genetically engineercells with targeted antigen speci�city and then to inducethe cell proliferation preserving their effector function andengrament and homing abilities. Currently, there are noFDA-approved adoptive T cell therapy protocols for cancer,but T cell therapies have shown activity inmicemodels and inselected clinical applications. For example, adoptive transferof telomerase-speci�c T cells was studied in a syngeneicPC murine model [63]. T cells were produced in vitro bycoculturing human lymphocytes with telomerase peptide-pulsed dendritic cells (DCs) or in vivo by injection of peptidewith adjuvant into C57BL/6 mice. Telomerase is a reversetranscriptase that contains an RNA template used to synthe-size telomeric repeats onto chromosomal ends. Activation oftelomerase and its maintenance of telomeres play a role inimmortalization of human cancer cells, as telomeres shrinkaer each cell division [64]. Telomerase activity is foundin 92–95% of pancreatic cancers [65, 66] and is associatedwith increased potential of invasion and metastasis and poorprognosis [67, 68]. Upregulation of telomerase may also beresponsible for the development of chemotherapy resistance[69]. Animals treated with these T cells showed signi�cantlydelayed disease progression [63].
Adoptive transfer of MUC1-speci�c cytotoxic T-lymphocytes (CTLs) was able to completely eradicateMUC1-expressing tumors inmice [70]. In this perspective, ina clinical study, MUC-1-speci�c autologous T cells, isolatedfrom patient PBMCs (peripheral blood mononuclear cells),were expanded by incubation with an MUC-1-presentingcell line prior to administration in PC patients. e meansurvival time for unresectable patients in this study was 5months [71]. However, patients with resectable pancreaticcancer had 1-, 2-, and 3-year survival rates of 83.3, 32.4, and19.4%, respectively, and amean survival time of 17.8 months.In a similar study, Kondo et al. isolated adherent cells frompatient PBMCs to generatematureDCs that were then pulsedwith MUC-1 peptide. e pulsed DCs were administered,along with autologous expanded MUC-1-speci�c T cells, topatients with unresectable or recurrent pancreatic cancer.Remarkably, a complete response was observed in onepatient with lung metastases, and the MS time of the wholegroup was 9.8 months, suggesting that the addition of pulsedDCs may have improved the outcome [72].

4 BioMed Research International
T 1: Pancreatic cancer-associated antigens for immune targeting.
PC-associatedantigens Characteristics and functions Location Tumor expression References
CEA Glycoprotein, normally expressed only in oncofetal tissues. Functionsas cell-adhesion molecule. First tumor antigen to be described.
Cell surface(GPI-linked) Overexpressed [148, 152]
Her2-neu A receptor tyrosine kinase, member of the EGF-receptor family,involved in cell growth and differentiation. Transmembrane Over-expressed [149]
MUC-1
Type I transmembrane glycoprotein, expressed on apical surface ofductal and glandular epithelial cells at low levels. Extracellular
domain has a polypeptide core with multiple tandem repeats of 20aminoacids.
Transmembrane Over-expressed,hypo-glycosylation [37, 153]
P53Tumor suppressor that regulates cell cycle. Normally inhibits survivalat the transcription level and can initiate apoptosis if DNA damage is
irreparable.Intracellular Mutated self [154, 155]
SurvivinMember of IAP family. Inhibits caspase activation, is found in mosthuman tumors and fetal tissue, but is completely absent in terminally
differentiated cells.Intracellular Over-expressed [150]
K-ras Mutated form of ras, a GTPase important for cell proliferation,differentiation, and survival. Intracellular Mutated self [156]
Telomerase Ribonucleoprotein that is responsible for RNA-dependent synthesisof telomeric DNA. TERT is its catalytic subunit. Intracellular Over-expressed [65]
VEGFR2A tyrosine kinase and member of platelet-derived growth factor
family. Receptor for VEGF with functions in blood vesseldevelopment.
Transmembrane Over-expressed [157]
MesothelinGPI-linked glycoprotein, expressed on the surface of mesothelialcells lining the pleura, peritoneum, and pericardium at low levels.
Binding partner of CA125/MUC16.
Cell surface(GPI-linked) Over-expressed [27, 158]
Alfa-enolaseGlycolytic enzyme that also acts as a surface plasminogen receptor. Isfound in a variety of tissue, on the cell surface as well as within the
nucleus and cytosol.
Cell surface,Intracellular
Over-expressed,post-translationalmodi�ed (i.e.,acetylated)
[21, 151, 159]
3. Active Immunotherapy: Vaccine Strategies
Vaccination involves administering a tumor antigen withthe aim of stimulating tumor-speci�c immunity. Antigenscould be delivered in the form of DNA or peptides, as wellas tumor cells or antigen-pulsed DCs. To be considered anideal tumor vaccine candidate, expression of the antigenmust be restricted to the tumor or only minimally expressedelsewhere in the body. Table 1 summaries a list of majorcandidate pancreas tumor-associated antigens for immunetargeting. Additional synergistic help is added to elicit amore vigorous and effective immune response, such ascytokines and immunostimulating compounds. Vaccinationagainst tumor antigens is an attractive approach to adjuvanttreatment aersurgery, when tumor-induced immune sup-pression is minimal [73–75].
3.1. Vaccines Using Whole Cells. e simplest vaccine ap-proach that has been applied to cancer is the inoculation ofpatients with irradiated tumor cells. is approach remainsa potent vehicle for generating antitumor immunity becausetumor cells express all relevant candidate TAAs, includingboth known and unidenti�ed. In the clinical setting, theuse of autologous tumor cell depends on the availabilityof an adequate number of them. As only 10–15% of PC
patients diagnosed are eligible for surgical, autologous pan-creatic cancer cells may not be provided in most of thepatients. Moreover, even if the patients are treated by surgicalresection, it is difficult to prepare sufficient numbers oftumor cells due to the length of culture time and risk ofcontamination [76, 77]. To elude this problem, allogeneictumor cell linesmay be used instead of autologous tumor cells[78]. is strategy has many advantages: (1) speci�c TAAsdo not need to be identi�ed for vaccination, (2) allogeneictumor cell lines are well characterized as TAAs source, (3)allogeneic tumor cell lines can grow well in vitro; thus, thereis no limiting factor for preparation of tumor cells, (4) itis not necessary to determine HLA typing of patients andallogeneic tumor cells, because autologous DCs can processand presentmultiple TAAs from allogeneic tumor cells owingto cross-presentation in the context of appropriate MHCclass I and II alleles [75, 79], (5) polyclonal antigen-speci�cT cells (CD4+/CD8+) can be generated, which may protectagainst tumor escape variants, and (6) the tumor cell vaccineplatform can be easily modi�ed. For example, tumor cellscan be transduced to express immunomodulatory cytokinessuch as granulocyte macrophage colony-stimulating factor(GM-CSF), which has shown signi�cant antitumor effect invivo [80]. GM-CSF is an important growth factor for granu-locytes and monocytes and has a crucial role in the growth

BioMed Research International 5
and differentiation of DCs. In a phase I clinical trial,Jaffee et al. [80] used allogeneic GM-CSF-secreting whole-cell tumor vaccine for pancreatic cancer, based on theconcept that the GM-CSF localization in the implantedtumor environment together with the shared tumor antigenexpressed by the primary cancer would effectively inducean antitumor immune response. In this study two PC celllines (PANC 10.05 and PANC 6.03) were used as the vaccine,both genetically modi�ed to express GM-CSF and thenirradiated to prevent further cell division. 14 PC patientswho had undergone pancreatic duodenectomy eight weeksbefore were given variable doses of the vaccine intradermally.ree of the eight patients who received ≥10 × 107 vaccinecells developed postvaccination delayed-type hypersensitiv-ity (DTH) responses associated with increased disease-freesurvival time and remained disease free for longer than 25months aer diagnosis. Side effects were mainly limited tolocal skin reactions at the site of vaccination.
In a recently completed phase II study 60 patients withresected pancreatic adenocarcinoma received �ve treatmentsof 2.5 × 108 vaccine cells, together with 5-FU and radio-therapy [81]. e reported MS was 26 months, with a one-and two-year survival of 88% and 76%, respectively. Inthese two studies, a PC cell vaccine induced a CD8+ T cellresponse, speci�c to mesothelin, regardless of HLA matchbetween the tumor vaccine and recipient—demonstratingthat cross-priming had occurred [80, 82]. Mesothelin is aparticularly promising cancer vaccine target owing to itslow level of expression in nontumor tissues and high levelsof expression in pancreatic as well as other cancers (i.e.,ovarian) [83]. Laheru et al. [84] administrated GMCSF-secreting allogeneic PC cells in sequence with cyclophos-phamide in patients with advanced pancreatic cancer. eapproach showed minimal treatment-related toxicity andmesothelin-speci�c T cell responses. Moreover, combinationof the vaccine and cyclophosphamide resulted in MS in agemcitabine-resistant population similar to chemotherapyalone. It was also reported that combination of the vaccinesand chemoradiation demonstrated an overall survival thatcompares favorably with published data for resected pancreascancer [85].
Tumor cell vaccines have also been modi�ed to expressepitopes, which increase antibody-mediated uptake by DCs.Normally, MUC-1 expressed on tumors is immunogenicowing to overexpression and tumor-restricted hypoglycosy-lation [86]. e NewLink Genetics Corporation (IA, USA)has developed a whole-cell vaccine expressing MUC-1 mod-i�ed to express 𝛼𝛼-gal epitopes, which is the focus of multipleclinical trials [87–90].is vaccine takes advantage of anti-𝛼𝛼-gal antibodies that are found in most people due to exposureto gastrointestinal �ora, resulting in increased uptake of thevaccine in an antibody-dependent manner [91]. In murinemodels, the addition of such 𝛼𝛼-gal epitopes to a Muc-1+ PCwhole-cell vaccine resulted in increased production of anti-Muc-1 antibodies, enhanced tumor-speci�c T cell responses,and increased survival aer challenge with Muc-1+ B16 cellsin 𝛼𝛼-gal knockout mice, previously sensitized to 𝛼𝛼-gal [92].
3.2. Peptide Vaccines. Peptide-based cancer vaccines arepreparations made from antigenic protein fragments (calledepitopes), that represent theminimal immunogenic region ofantigens [93, 94], designed to enhance the T cell response,especially the CD8+. Induction of CTLs needs peptidesderived from TAAs to be presented on the surface of APCs(antigens presenting cells), such as DCs, in the context ofHLA molecules. e major advantages of peptide vaccinesare that they are simple, stable, safe, economical, and do notrequire manipulation of patient tissues, whose availabilitymay be limited. However, there are also several obstacles thatlimit the widespread usefulness of peptide vaccines: (1) alimited number of known synthesized short peptides cannotbe available in many HLA molecules [95–97], (2) impairedfunction ofAPCs in patients with advanced pancreatic cancer[76, 98], (3) CTLs may be ineffective in reacting with PC cellsdownregulated by certain tumor antigens and MHC class Imolecules, which may appear during the course of tumorprogression [99], (4) regulatory T cells (Tregs) or MDSCs(myeloid-derived suppressor cells) in tumor environmentproduce immunosuppressive cytokines such as IL-10 andTGF-𝛽𝛽 [100].
Anyway, a number of peptide vaccines have under-gone phase I/II clinical trials [12, 101], showing encourag-ing results, due to their ability to produce cancer-speci�cresponses in PC patients (Table 2). In a phase I study, vac-cination with a 100 mer peptide of the MUC-1 extracellulartandem repeat generated a MUC-1-speci�c T cell responsein some PC patients with two of the 15 patients alive at61 months [102]. Moreover, in a separate phase I clinicaltrial using the same peptide vaccine, the production of anti-MUC-1 circulating antibodies was detected in patients withinoperable PC, although no signi�cant impact on survivalwas discovered [103].
In a phase I trial, Miyazawa et al. administered a peptidevaccine for human VEGF receptor, (VEGFR)2-169 epitope,in patients with advanced PC, in combination with gemc-itabine, observing an antigen-speci�c DTH and VEGFR2-speci�c CD8+ cells in 61% patients, with an overall MStime of 8.7 months [101]. A randomized, placebo-controlled,multicenter, phase II/III study of this VEGFR2–169 peptidevaccine therapy, combined with gemcitabine, is currentlyunderway in patients with unresectable advanced or recur-rent PC [104]. In similar studies, a telomerase-based vaccine,consisting of the human telomerase reverse transcriptase(GV1001) peptide, was found to induce a telomerase-speci�cimmune response in 63% of evaluable patients, as measuredby DTH in unresectable PC. ose with a positive DTH werefound to live longer than those that did not have a positiveDTH [105]. In addition, augmented immune responses andprolonged survival were observed following vaccination ofadvanced PC patients with telomerase peptide and GM-CSF[105]. More recently, a phase III clinical trial was performedin which the effect of gemcitabine treatment on survival wascompared with gemcitabine treatment in combination withGV1001 therapy in unresectable and metastatic PC patients[106]. However, the trial was terminated when no survivalbene�t was found.

6 BioMed Research International
T 2: Peptide vaccines-based clinical trial.
Peptide(Adjuvant) Combination Patients enrolled Phase of
the study Clinical results References
100 mer MUC1(SB-AS2 adjuvant)
16 with resected orlocally advanced PC I Detectable MUC1-speci�c humoral and T-cell
responses were detected in some patients. [102]
100 mer MUC1(incompleteFreund’s adjuvant)
6 with advanced PC I One patient showed a tendency for increasedcirculating anti-MUC1 IgG antibody. [103]
VEGFR2-169 Gemcitabine 21 with unresectable PC I
Speci�c cytoto�ic T lymphocytes (CTL) reacting tothe VEGFR2-169 peptide were induced in 11 (61%)of the 18 evaluable patients. e disease control ratewas 67%, and the median overall survival time was
8.7 months.
[101]
Telomerase(GM-CSF) 48 with advanced PC I/II
Immune responses were observed in 24 of 38evaluable patients. One-year survival for the
evaluable patients in the intermediate dose groupwas 25%.
[105]
Mutant K-ras(GM-CSF)
10 with resected and 38with advanced PC I/II
Immune response to the peptide vaccine showedprolonged survival compared to nonresponders.
K-ras-speci�c T cells were selectively accumulatedin the tumor.
[77]
Mutant K-ras(GM-CSF) 24 with resected PC Pilot study
Vaccination proved to be safe and tolerable withhowever no elicitable immunogenicity and
unproven efficacy.[107]
13 mer mutant ras 5 with PC and 7 withcolorectal cancer II
is vaccine is safe, can induce speci�c immuneresponses, and it appears to have a positive outcome
in overall survival. e �ve pancreatic cancerpatients have shown a mean disease-free survival
(DFS) of 35.2+ months and a mean overall survival(OS) of 44.4+ months.
[108]
Mutant ras longpeptide 23 with resected PC I
17 of 20 evaluable patients (85%) respondedimmunologically to the vaccine. Ten-year survival
was 20% (four patients out of 20 evaluable).[111]
Surviving1 with liver metastasisof PC refractory togemcitabine
Case reporte patient initially underwent partial remission ofliver metastasis which proceeded aer 6 months intoa complete remission with duration of 8 months.
[17]
Personalizedpeptide vaccine Gemcitabine 11 with advanced PC I
e 6- and 12-month survival rates for patients whoreceived >3 vaccinations (𝑛𝑛 𝑛 𝑛𝑛) were 80% and
20%, respectively.[12]
e most interesting results have come from studiesof K-Ras-targeted peptide vaccines. Gjertsen et al. [77]�rst reported mutant K-ras peptide vaccines for PC. In aphase I/II trial involving 48 PC patients, they studied raspeptide in combination with GM-CSF, since native epitopeshave relatively low immunogenicity [77]. Peptide-speci�cimmunity was induced in 58% of patients. Of patients withadvanced disease, those who responded to treatment showedincreased survival compared to nonresponders. Recently,another group reported that vaccination of 24 PC patientswith K-ras peptide in combination with GM-CSF proved tobe safe without tumor regression [107]. In another pilot vac-cine study, pancreatic and colorectal patients were vaccinatedwith K-Ras peptides containing patient-speci�c mutations.ree of the �ve PC patients displayed an antigen-speci�cimmune response to a K-Ras [108]. Disease progressionwas observed in the two PC patients that did not respond
to the vaccine, with the responders having no evidence ofdisease. Of the PC patients, a mean disease-free survival of35.2 months and a mean overall survival of 44.4 monthswere observed. Such results with peptide vaccines are highlyencouraging.
e more attractive peptide-based vaccines may be syn-thetic long peptides to induce antigen-speci�c polyclonalCD8+ andCD4+ Tcells [109]. Long synthetic peptides cannotbind directly on MHC class I or II molecules, but they needto be processed and presented by DCs. So, the long peptidevaccines can present MHC class I- and II-restricted epitopesfor long time, thus eliciting both CD4+- and CD8+-mediatedimmune recognition [110] and probably inducing a robusttherapeutic T cell response. In a phase I study using longsynthetic mutant ras peptides, Wedén et al. [111] treated23 patients who were vaccinated aer surgical PC resection.Long-term immunologicalmemory responses to the vaccines

BioMed Research International 7
were observed. Strikingly, 10-year survival was 20% (fourpatients out of 20 evaluable) versus zero (0/87) in a cohortof nonvaccinated patient treated in the same period.
To increase the immunogenicity of peptide vaccines,some groups have mutated key anchor residues in thepeptides such that binding to MHC-I molecules, and conse-quently the presentation to CD8+ T cells, is increased. isis particularly important when vaccinating against TAAs, asthey are oen weak or only intermediate binders to HLAmolecules [112–116]. An MUC-1 peptide vaccine modi�edin this way was shown to enhance production of IFN-𝛾𝛾 frompatient and normal donor T cells. MUC-1-speci�c T cellclones, generated by stimulation with this peptide, could lysetargets pulsed with native Muc-1 epitope as well as HLA-A2+ MUC-1+ human tumor cells in vitro [117]. Notably, onecase has been reported in which vaccination with a modi�edHLA-A2-restricted survivin peptide resulted in remission ofliver metastasis in one PC patient [17].
Another approach in cancer peptide-vaccination consistsin using personalized peptide vaccines based on the tumor-antigen epitopes that are most immunogenic for a particularpatient. In a phase I clinical trial, Yanagimoto et al. appliedthis strategy, in combination with gemcitabine therapy, topancreatic cancer. Prior to vaccination, T cells from patientPBMCs were screened against a panel of tumor antigen-derived peptides. Patients were vaccinated only with the pep-tides to which they had a response [12]. An increase in tumorantigen-speci�c T cell responses was observed from the 13evaluable patients with no correlation to clinical responsesor humoral responses following vaccination, although 11patients experienced either reduction in tumor size. Mediansurvival time was 7.6 months. A similar phase II study waspublished in 2010 by the same group, showing an MS time of9 months and a 1-year survival of 38% [118].
3.3. DNA Vaccination. Vaccination with DNA representsa simple vehicle for in vivo transfection and antigen pro-duction. A DNA vaccine is composed of a plasmid DNAthat encodes for a TAA under the control of a mammalianpromoter and can be easily produced in the bacteria [119]. Itcan be administered to humans via intramuscular injectionwith or without electroporation. Compared with cell-basedvaccines, this vaccination strategy offers more advantages;in fact, while cell-based vaccines become less effective overtime because the induced immune system recognizes them asforeign and quickly destroys them,DNAvaccines can provideprolonged antigen expression, leading to ampli�cation ofimmune responses and inducing memory responses againstweakly immunogenic TAAs. Moreover, as DNA might betaken up by cells and the encoded antigen is processedthrough both endogenous and exogenous pathways, DNAvaccines administered via intramuscular injection allow foran immune response to multiple potential epitopes withinan antigen to be generated regardless of the recipient’s MHCpro�le [120]. Actually DNA vaccines are ongoing trials in dif-ferent tumors [121–123] and being studied inmurinemodelsof pancreatic cancer. In a murine PC study, an MUC-1DNAvaccinewas able to induce a signi�cantMUC-1-speci�c
CTL response and had both prophylactic and therapeuticeffects in tumor-bearing mice [124]. Similarly, in anotherPC murine model, vaccination with either murine or humanfull-length survivin DNA generated an antitumor-speci�cresponse, increased in�ltration of tumor with lymphocytesand increased survival [125]. Furthermore, Gaffney et al.studied the mesothelin DNA vaccine in combination withthe antiglucocorticoid-induced TNF receptor antibody (anti-GITR) in mice with syngeneic mesothelin-expressing pan-creatic cancer [126]. 50% of animals treated with mesothelinwere tumor free 25 days aer tumor injection compared to 0%of nontreatedmice.is increased to 94%with the addition ofanti-GITR. e agonist anti-GITR served to enhance T cell-mediated response of the vaccine [127, 128].
3.4. Antigen-Pulsed DCs. Antigen-speci�c T cell responsesare initiated by DCs. ey capture antigens secreted or shedby tumor cells and present peptides in association with theMHC class I and II molecules. is results in the expressionand upregulation of cytokines and costimulatory moleculeswhich in turn stimulate CD4+ and CD8+ T cells to mountan antitumor response [129]. erefore, a major area ofinvestigation in cancer immunotherapy involves the designof DCs-based cancer vaccines [130]. Autologous DCs can beused in tumor vaccination (1) pulsed with synthetic peptidederived from the known tumor antigens [131], tumor celllysates [132], or apoptotic tumor cells [133], (2) transfectedwith whole-tumor mRNA [134] or with mRNA or cDNA of aspeci�c antigen [135] and (3) fusedwith tumor cells to induceantigen-speci�c polyclonal CTL responses [136].
DC-based vaccines have been used in different PCstudies. Schmidt et al. intratumorally vaccinated with wholetumor mRNA transfected DCs and found an antitumor-speci�c immune response and signi�cantly decreased tumorvolume in a murine PC model [137]. Apoptotic PC lysateshave also been evaluated as a source of antigens and havebeen demonstrated to elicit stronger antitumor lytic activitywhen used to stimulate autologous human CD8+ T cellsin vitro compared with those stimulated with tumor lysate-pulsed DCs [138]. In cases in which an immunogenic tumorantigen is known, autologous DCs have been transfectedwith or virally transduced to express, the mRNA or cDNAof a speci�c tumor antigen (Table 3). A vaccine consist-ing of liposomal MUC1-transfected autologous DCs wasevaluated in a clinical phase I/II trial. In MUC1 peptide-loaded DC vaccines in PC patients following resection oftheir primary tumors, four of the 12 patients followed forover four years were alive, all without evidence of recurrence[139]. Moreover, MUC1-speci�c immune responses werealso observed even in patients with pretreated and advanceddisease, following immunization with DCs transfected withMUC1 cDNA [140]. is technique does not require that theexact immunogenic epitopes of the antigen be identi�ed, asfull-length protein is transfected.
In another study, three patients with resected PC fol-lowing neoadjuvant chemoradiotherapy were given monthlyinjections of autologous, monocyte-derived DCs loaded withthe mRNA of CEA for six months [141]. No toxicities were

8 BioMed Research International
T 3: DC-based vaccines clinical trial.
DC-based vaccines Patients enrolled Phase of thestudy Clinical results References
MUC1 peptide-loadedDC
12 with resected pancreaticand biliary cancer I/II 4 of the 12 patients followed for over four years were
alive [139]
DC transfected withMUC1 cDNA
10 with advanced breast,pancreatic, or papillary cancer I/II
4 patients showed a 2- to 10-fold increase in thefrequency of MUC1-speci�c IFN-gamma-secretingCD8+ T cells.
[140]
mRNA CEA-loadedDC 3 with resected PC Pilot study
e immunizations were well tolerated withoutevidence of adverse events. All patients were alivewithout evidence of disease at more than 30 monthsfrom the original diagnosis.
[141]
Peptides (mutant p53-and k-ras-loaded DC
39 patients with several typesof cancer (lung, breast,pancreatic, ovarian, colon,others)
I
10 (26%) of 38 patients had detectable CTL againstmutant p53 or K-ras, and 2 patients were positive forCTL at baseline. Positive IFN-𝛾𝛾 responses occurred in16 patients (42%) aer vaccination, whereas 4 patientshad positive IFN-𝛾𝛾 reaction before vaccination. Cellularimmunity to mutant p53 and K-ras oncopeptides isassociated with longer survival.
[142]
DC engineered(secreting IL-2)
17 patients with several typesof cancer (3 metastaticpancreatic, 5 colorectal, 9 liver,cancer)
Pilot study
Treatment was well tolerated. DC treatment induced amarked increase of in�ltrating CD8+ T lymphocytes inthree of 11 tumor biopsies analyzed. A partial responsewas observed in one patient with pancreatic carcinoma.
[143]
reported, and all patients remained disease free formore than30 months from diagnosis.
Pulse can also be performed with peptides from multipletumor antigens, as was performed in a Phase I clinical studyby Carbone et al. Patients with various cancers, includingpancreatic cancer, immunized with p53 and K-ras peptide-pulsed PBMCs, saw increased survival [142]. In addition,autologous DCs, virally transduced to express IL-12, havealso been used in cancer treatment. One PC patient receivingthis treatment had a partial response in studies by Mazzoliniet al. [143]. As the treatment DCs were not loaded withtumor antigens, cross-presentation of tumor antigens musthave occurred. Moreover, DCs have been fused with tumorcells to induce antigen-speci�c polyclonal CTL responses[136]. In the DC/tumor cell fusion approach, whole TAAsincluding those known and those yet unidenti�ed are pro-cessed endogenously and presented by MHC class I and IIpathways in the context of costimulatory signals [144–146].In particular, this technique has been used to treat mice in aPC model, resulting in the generation of CD8+ T cells withtumor-speci�c cytolytic activity and tumor re�ection [147].
4. Conclusion
Pancreatic cancer is a dismal disease that has a high mor-bidity and mortality, and at present there are not effectivechemotherapeutic treatments, especially for patients withadvanced and metastatic diseases. For all these reasonsAlphais of prime importance to investigate new pancreaticcancer treatments. In this paper we have analyzed the variousstrategies of the immunotherapeutic approach, some ofwhich are still used in animal models; others are alreadybeing exploited in clinical trials. Immunotherapy is certainly
a promising treatment for pancreatic cancer, because it ishighly speci�c for cancer cells and therefore without the sideeffects associated with traditional chemotherapy. But at themoment there are not antigens expressed only by PC cells;in fact the antigens used as the target of immunotherapictreatments are self-protein or overexpressed [65, 148–150] intumor cells or present in acetylated form [151], with the riskof autoimmune phenomena. However, the data obtained indifferent clinical trials showed an increase in the survival ofpatients treated with PC immunotherapy alone [72, 105, 108,111] or in combination with chemotherapy treatments [12,54, 101], with very minimal autoimmune manifestations. Inconclusion we can say that immunotherapy may be includedamong the future treatments for pancreatic cancer, especiallyfor inoperable patients, but for the effectiveness of thisinnovative treatment is essential to overcome some obstacles:(a) �nding speci�c markers for pancreatic cancer cells, (b)mitigating the immune suppressive effects of tumor cells, (c)early diagnosis of the tumor so as to act in a timely mannerbefore the cancer spreads in other locations.
Acknowledgments
e authors thank Italian Ministry of University and Re-search and Ente Cassa di Risparmio di Firenze for supportingtheir studies.
References
[1] M. W. Saif, “Pancreatic neoplasm in 2011: an update,” Journal ofthe Pancreas, vol. 12, no. 4, pp. 316–321, 2011.
[2] Surveillance Epidemiology and End Results (SEER), U.S.Cancer Statistics: 1999–2007 Incidence and Mortality Report,http://www.seer.cancer.gov/publications/uscs.html.

BioMed Research International 9
[3] M. W. Saif, “Controversies in the adjuvant treatment of pancre-atic adenocarcinoma,” Journal of the Pancreas, vol. 8, no. 5, pp.545–552, 2007.
[4] H. A. Burris III, M. J. Moore, J. Andersen et al., “Improvementsin survival and clinical bene�t with gemcitabine as �rst-line therapy for patients with advanced pancreas cancer: arandomized trial,” Journal of Clinical Oncology, vol. 15, no. 6,pp. 2403–2413, 1997.
[5] M. J. Moore, D. Goldstein, J. Hamm et al., “Erlotinib plusgemcitabine compared with gemcitabine alone in patients withadvanced pancreatic cancer: a phase III trial of the NationalCancer Institute of Canada Clinical Trials Group,” Journal ofClinical Oncology, vol. 25, no. 15, pp. 1960–1966, 2007.
[6] J. Yokokawa, C. Palena, P. Arlen et al., “Identi�cation of novelhuman CTL epitopes and their agonist epitopes of mesothelin,”Clinical Cancer Research, vol. 11, no. 17, pp. 6342–6351, 2005.
[7] M. H. Andersen, L. O. Pedersen, J. C. Becket, and P. T. Straten,“Identi�cation of a cytotoxic T lymphocyte response to theapoptosis inhibitor protein survivin in cancer patients,” CancerResearch, vol. 61, no. 3, pp. 869–872, 2001.
[8] F. M. Johnston, M. C. B. Tan, B. R. Tan Jr. et al., “Circulatingmesothelin protein and cellular antimesothelin immunity inpatients with pancreatic cancer,” Clinical Cancer Research, vol.15, no. 21, pp. 6511–6518, 2009.
[9] Y. Kotera, J. D. Fontenot, G. Pecher, R. S.Metzgar, andO. J. Finn,“Humoral immunity against a tandem repeat epitope of humanmucinMUC-1 in sera from breast, pancreatic, and colon cancerpatients,” Cancer Research, vol. 54, no. 11, pp. 2856–2860, 1994.
[10] B. Kubuschok, F. Neumann, R. Breit et al., “Naturally occur-ring T-cell response against mutated p21 Ras oncoprotein inpancreatic cancer,” Clinical Cancer Research, vol. 12, no. 4, pp.1365–1372, 2006.
[11] L. Wenandy, R. B. Sørensen, L. Sengeløv, I. M. Svane, P. T.Straten, and M. H. Andersen, “e immunogenicity of thehTERT540-548 peptide in cancer,” Clinical Cancer Research,vol. 14, no. 1, pp. 4–7, 2008.
[12] H. Yanagimoto, T. Mine, K. Yamamoto et al., “Immunologicalevaluation of personalized peptide vaccination with gemc-itabine for pancreatic cancer,” Cancer Science, vol. 98, no. 4, pp.605–611, 2007.
[13] Y. Oji, S. Nakamori, M. Fujikawa et al., “Overexpression of theWilms’ tumor geneWT1 in pancreatic ductal adenocarcinoma,”Cancer Science, vol. 95, no. 7, pp. 583–587, 2004.
[14] M. Ueda, Y. Miura, O. Kunihiro et al., “MUC1 overexpressionis the most reliable marker of invasive carcinoma in intraductalpapillary-mucinous tumor (IPMT),” Hepato-Gastroenterology,vol. 52, no. 62, pp. 398–403, 2005.
[15] K. Seki, T. Suda, Y. Aoyagi et al., “Diagnosis of pancreaticadenocarcinoma by detection of human telomerase reversetranscriptasemessenger RNA in pancreatic juice with sample�uali�cation,” Clinical Cancer Research, vol. 7, no. 7, pp.1976–1981, 2001.
[16] M. K. Gjertsen, A. Bakka, J. Breivik et al., “Vaccination withmutant ras peptides and induction of T-cell responsiveness inpancreatic carcinoma patients carrying the corresponding RASmutation,” e Lancet, vol. 346, no. 8987, pp. 1399–1400, 1995.
[17] M. Wobser, P. Keikavoussi, V. Kunzmann, M. Weininger, M.H. Andersen, and J. C. Becker, “Complete remission of livermetastasis of pancreatic cancer under vaccination with a HLA-A2 restricted peptide derived from the universal tumor antigen
survivin,” Cancer Immunology, Immunotherapy, vol. 55, no. 10,pp. 1294–1298, 2006.
[18] K. Yamaguchi, M. Enjoji, and M. Tsuneyoshi, “Pancreatoduo-denal carcinoma: a clinicopathologic study of 304 patientsand immunohistochemical observation for CEA and CA19-9,”Journal of Surgical Oncology, vol. 47, no. 3, pp. 148–154, 1991.
[19] M. Komoto, B. Nakata, R. Amano et al., “HER2 overexpressioncorrelates with survival aer curative resection of pancreaticcancer,” Cancer Science, vol. 100, no. 7, pp. 1243–1247, 2009.
[20] H. Maacke, A. Kessler, W. Schmiegel et al., “Overexpression ofp53 protein during pancreatitis,” British Journal of Cancer, vol.75, no. 10, pp. 1501–1504, 1997.
[21] P. Cappello, B. Tomaino, R. Chiarle et al., “An integratedhumoral and cellular response is elicited in pancreatic can-cer by 𝛼𝛼-enolase, a novel pancreatic ductal adenocarcinoma-associated antigen,” International Journal of Cancer, vol. 125, no.3, pp. 639–648, 2009.
[22] Y. Hamanakai, Y. Suehiro, M. Fukui, K. Shikichi, K. Imai, and Y.Hinoda, “Circulating anti-MUC1 IGG antibodies as a favorableprognostic factor for pancreatic cancer,” International Journal ofCancer, vol. 103, no. 1, pp. 97–100, 2003.
[23] F. Pagès, J. Galon, M. C. Dieu-Nosjean, E. Tartour, C. Sautès-Fridman, and W. H. Fridman, “Immune in�ltration in humantumors: a prognostic factor that should not be ignored,” Onco-gene, vol. 29, no. 8, pp. 1093–1102, 2010.
[24] X. R. Jiang, A. Song, S. Bergelson et al., “Advances in theassessment and control of the effector functions of therapeuticantibodies,” Nature Reviews Drug Discovery, vol. 10, no. 2, pp.101–111, 2011.
[25] I. Hellstrom, E. Friedman, T. Verch et al., “Anti-mesothelinantibodies and circulating mesothelin relate to the clinical statein ovarian cancer patients,” Cancer Epidemiology Biomarkersand Prevention, vol. 17, no. 6, pp. 1520–1526, 2008.
[26] M. Ho, R. Hassan, J. Zhang et al., “Humoral immune responseto mesothelin in mesothelioma and ovarian cancer patients,”Clinical Cancer Research, vol. 11, no. 10, pp. 3814–3820, 2005.
[27] R. Hassan, T. Bera, and I. Pastan, “Mesothelin: a new target forimmunotherapy,” Clinical Cancer Research, vol. 10, no. 12 I, pp.3937–3942, 2004.
[28] P. Argani, C. Iacobuzio-Donahue, B. Ryu et al., “Mesothelin isoverexpressed in the vastmajority of ductal adenocarcinomas ofthe pancreas: identi�cation of a new pancreatic cancer markerby serial analysis of gene expression (SAGE),” Clinical CancerResearch, vol. 7, no. 12, pp. 3862–3868, 2001.
[29] R. Hassan, Z. G. Laszik, M. Lerner, M. Raffeld, R. Postier, andD. Brackett, “Mesothelin is overexpressed in pancreaticobiliaryadenocarcinomas but not in normal pancreas and chronicpancreatitis,” American Journal of Clinical Pathology, vol. 124,no. 6, pp. 838–845, 2005.
[30] R. Hassan, S. Bullock, A. Premkumar et al., “Phase I studyof SS1P, a recombinant anti-mesothelin immunotoxin givenas a bolus I.V. infusion to patients with mesothelin-expressingmesothelioma, ovarian, and pancreatic cancers,”Clinical CancerResearch, vol. 13, no. 17, pp. 5144–5149, 2007.
[31] D. Filpula andH. Zhao, “Releasable PEGylation of proteins withcustomized linkers,” Advanced Drug Delivery Reviews, vol. 60,no. 1, pp. 29–49, 2008.
[32] R. J. Kreitman, R. Hassan, D. J. FitzGerald, and I. Pastan, “PhaseI trial of continuous infusion anti-mesothelin recombinantimmunotoxin SS1P,” Clinical Cancer Research, vol. 15, no. 16,pp. 5274–5279, 2009.

10 BioMed Research International
[33] D. K. Armstrong, D. Laheru, W. W. Ma et al., “A phase 1 studyof MORAb-009, a monoclonal antibody against mesothelin inpancreatic cancer,mesothelioma andovarian adenocarcinoma,”Journal of Clinical Oncology, vol. 25, no. 18S, Article ID 14041,2007.
[34] Y. Feng, D. Xiao, Z. Zhu et al., “A novel human monoclonalantibody that binds with high affinity to mesothelin-expressingcells and kills them by antibody-dependent cell-mediatedcytotoxicity,” Molecular Cancer erapeutics, vol. 8, no. 5, pp.1113–1118, 2009.
[35] M. Ho, M. Feng, R. J. Fisher, C. Rader, and I. Pastan, “Anovel high-affinity humanmonoclonal antibody tomesothelin,”International Journal of Cancer, vol. 128, no. 9, pp. 2020–2030,2011.
[36] H. Yamasaki, S. Ikeda, M. Okajima et al., “Expression andlocalization of MUC1, MUC2, MUC5AC and small intestinalmucin antigen in pancreatic tumors,” International Journal ofOncology, vol. 24, no. 1, pp. 107–113, 2004.
[37] C. F. Qu, Y. Li, Y. J. Song et al., “MUC1 expression in primaryand metastaticpancreatic cancer cells for in vitro treatment by213Bi-C595 radioimmunoconjugate,” British Journal of Cancer,vol. 91, no. 12, pp. 2086–2093, 2004.
[38] E. Levi, D. S. Klimstra, A. Andea, O. Basturk, and N. V. Adsay,“MUC1 and MUC2 in pancreatic neoplasia,” Journal of ClinicalPathology, vol. 57, no. 5, pp. 456–462, 2004.
[39] H. Tsutsumida, B. J. Swanson, P. K. Singh et al., “RNAinterference suppression of MUC1 reduces the growth rateand metastatic phenotype of human pancreatic cancer cells,”Clinical Cancer Research, vol. 12, no. 10, pp. 2976–2987, 2006.
[40] D. V. Gold, D. E. Modrak, K. Schutsky, and T. M. Cardillo,“Combined 90Yttrium-Dota-Labeled PAM4 antibody radioim-munotherapy and gemcitabine radiosensitization for the treat-ment of a human pancreatic cancer xenogra,” InternationalJournal of Cancer, vol. 109, no. 4, pp. 618–626, 2004.
[41] A. Danielczyk, R. Stahn, D. Faulstich et al., “PankoMab: apotent new generation anti-tumour MUC1 antibody,” CancerImmunology, Immunotherapy, vol. 55, no. 11, pp. 1337–1347,2006.
[42] X. N. Fan, U. Karsten, S. Goletz, and Y. Cao, “Reactivity ofa humanized antibody (hPankoMab) towards a tumor-relatedMUC1 epitope (TA-MUC1) with various human carcinomas,”Pathology Research and Practice, vol. 206, no. 8, pp. 585–589,2010.
[43] G. Pratesi, G. Petrangolini, M. Tortoreto et al., “Antitumor effi-cacy of trastuzumab in nude mice orthotopically xenograedwith human pancreatic tumor cells expressing low levels ofHER-2/neu,” Journal of Immunotherapy, vol. 31, no. 6, pp.537–544, 2008.
[44] H. Saeki, S. Yanoma, S. Takemiya et al., “Antitumor activity ofa combination of trastuzumab (Herceptin) and oral �uoropy-rimidine S-1 on human epidermal growth factor receptor 2-overexpressing pancreatic cancer,” Oncology Reports, vol. 18,no. 2, pp. 433–439, 2007.
[45] C. Larbouret, B. Robert, I. Navarro-Teulon et al., “In vivotherapeutic synergism of anti-epidermal growth factor receptorand anti-HER2monoclonal antibodies against pancreatic carci-nomas,”Clinical Cancer Research, vol. 13, no. 11, pp. 3356–3362,2007.
[46] C. Larbouret, B. Robert, C. Bascoul-Mollevi et al., “Combinedcetuximab and trastuzumab are superior to gemcitabine in thetreatment of human pancreatic carcinoma xenogras,” Annalsof Oncology, vol. 21, no. 1, pp. 98–103, 2010.
[47] R. D. Blumenthal, L. Osorio, M. K. Hayes, I. D. Horak, H. J.Hansen, and D. M. Goldenberg, “Carcinoembryonic antigenantibody inhibits lung metastasis and augments chemotherapyin a human colonic carcinoma xenogra,” Cancer Immunology,Immunotherapy, vol. 54, no. 4, pp. 315–327, 2005.
[48] A Phase I/II Study of Radioimmunotherapy with 90Y-Humanized MN-14 IgG Administered as a Single Dose toPatients with Refractory Advanced/Metastatic PancreaticCarcinoma (NCT00041639), http://clinicaltrials.gov/.
[49] N. R. Lemoine, C. M. Hughes, C. M. Barton et al., “eepidermal growth factor receptor in human pancreatic cancer,”Journal of Pathology, vol. 166, no. 1, pp. 7–12, 1992.
[50] Y. Yamanaka, H. Friess, M. S. Kobrin, M. Buchler, H. G.Beger, and M. Korc, “Coexpression of epidermal growth factorreceptor and ligands in human pancreatic cancer is associatedwith enhanced tumor aggressiveness,” Anticancer Research, vol.13, no. 3, pp. 565–569, 1993.
[51] C. J. Bruns, C. C. Solorzano, M. T. Harbison et al., “Blockadeof the epidermal growth factor receptor signaling by a noveltyrosine kinase inhibitor leads to apoptosis of endothelial cellsand therapy of human pancreatic carcinoma,” Cancer Research,vol. 60, no. 11, pp. 2926–2935, 2000.
[52] S. S. W. Ng, M. S. Tsao, T. Nicklee, and D. W. Hedley, “Effectsof the epidermal growth factor receptor inhibitor OSI-774,Tarceva, on downstream signaling pathways and apoptosis inhuman pancreatic adenocarcinoma,” Molecular Cancer era-peutics, vol. 1, no. 10, pp. 777–783, 2002.
[53] J. Baselga and C. L. Arteaga, “Critical update and emergingtrends in epidermal growth factor receptor targeting in cancer,”Journal of Clinical Oncology, vol. 23, no. 11, pp. 2445–2459,2005.
[54] P. A. Philip, J. Benedetti, C. L. Corless et al., “Phase IIIstudy comparing gemcitabine plus cetuximab versus gemc-itabine in patients with advanced pancreatic adenocarcinoma:Southwest oncology group-directed intergroup trial S0205,”Journal of Clinical Oncology, vol. 28, no. 22, pp. 3605–3610,2010.
[55] R. Krempien, M. W. Munter, C. Timke et al., “Cetux-imab in combination with intensity modulated radiotherapy(IMRT) and gemcitabine for patients with locally advancedpancreatic cancer: a prospective phase II trial [PARC-StudyISRCTN56652283],” Journal of Clinical Oncology, vol. 25, no.18S, article 4573, 2007.
[56] C. Bangard, A. Gossmann, A. Papyan, S. Tawadros, M.Hellmich, and C. J. Bruns, “Magnetic resonance imaging inan orthotopic rat model: blockade of epidermal growth factorreceptor with EMD72000 inhibits human pancreatic carcinomagrowth,” International Journal of Cancer, vol. 114, no. 1, pp.131–138, 2005.
[57] U. Graeven, B. Kremer, T. Südhoff et al., “Phase I study ofthe humanised anti-EGFR monoclonal antibody matuzumab(EMD 72000) combined with gemcitabine in advanced pan-creatic cancer,” British Journal of Cancer, vol. 94, no. 9, pp.1293–1299, 2006.
[58] M. Korc, “Pathways for aberrant angiogenesis in pancreaticcancer,” Molecular Cancer, vol. 2, article 8, 2003.
[59] A. J. Karayiannakis, H. Bolanaki, K. N. Syrigos et al., “Serumvascular endothelial growth factor levels in pancreatic cancerpatients correlate with advanced and metastatic disease andpoor prognosis,” Cancer Letters, vol. 194, no. 1, pp. 119–124,2003.

BioMed Research International 11
[60] H. L. Kindler, G. Friberg, D. A. Singh et al., “Phase II trialof bevacizumab plus gemcitabine in patients with advancedpancreatic cancer,” Journal of Clinical Oncology, vol. 23, no. 31,pp. 8033–8040, 2005.
[61] H. L. Kindler, D. Niedzwiecki, D. Hollis et al., “Gemcitabineplus bevacizumab compared with gemcitabine plus placebo inpatients with advanced pancreatic cancer: phase III trial of theCancer and Leukemia Group B (CALGB 80303),” Journal ofClinical Oncology, vol. 28, no. 22, pp. 3617–3622, 2010.
[62] H. W. Bruckner, V. R. Hrehorovich, and H. S. Sawhney, “Beva-cizumab as treatment for chemotherapy-resistant pancreaticcancer,”Anticancer Research, vol. 25, no. 5, pp. 3637–3639, 2005.
[63] J. Schmidt, E. Ryschich, E. Sievers, I. G. H. Schmidt-Wolf, M.W. B�chler, and A. M�rten, “Telomerase-speci�c T-cells killpancreatic tumor cells in vitro and in vivo,” Cancer, vol. 106, no.4, pp. 759–764, 2006.
[64] W. C.Hahn, “Role of telomeres and telomerase in the pathogen-esis of human cancer,” Journal of Clinical Oncology, vol. 21, no.10, pp. 2034–2043, 2003.
[65] E.Hiyama, T. Kodama, K. Shinbara et al., “Telomerase activity isdetected in pancreatic cancer but not in benign tumors,” CancerResearch, vol. 57, no. 2, pp. 326–331, 1997.
[66] S. J. Myung, M. H. Kim, Y. S. Kim et al., “Telomerase activityin pure pancreatic juice for the diagnosis of pancreatic cancermay be complementary to K-ras mutation,” GastrointestinalEndoscopy, vol. 51, no. 6, pp. 708–713, 2000.
[67] N. Sato, N.Maehara, K.Mizumoto et al., “Telomerase activity ofcultured human pancreatic carcinoma cell lines correlates withtheir potential for migration and invasion,” Cancer, vol. 91, no.3, pp. 496–504, 2001.
[68] S. J. Tang, J. A. Dumot, L. Wang et al., “Telomerase activity inpancreatic endocrine tumors,” American Journal of Gastroen-terology, vol. 97, no. 4, pp. 1022–1030, 2002.
[69] N. Sato, K. Mizumoto, M. Kusumoto et al., “Up-regulationof telomerase activity in human pancreatic cancer cells aerexposure to etoposide,” British Journal of Cancer, vol. 82, no. 11,pp. 1819–1826, 2000.
[70] P. Mukherjee, A. R. Ginardi, C. S. Madsen et al., “Micewith spontaneous pancreatic cancer naturally develop MUC-1-speci�c CTLs that eradicate tumors when adoptively trans-ferred,” Journal of Immunology, vol. 165, no. 6, pp. 3451–3460,2000.
[71] T. Kawaoka, M. Oka, M. Takashima et al., “Adoptive im-munotherapy for pancreatic cancer: cytotoxic T lymphocytesstimulated by the MUC1-expressing human pancreatic cancercell line YPK-1,” Oncology Reports, vol. 20, no. 1, pp. 155–163,2008.
[72] H. Kondo, S. Hazama, T. Kawaoka et al., “Adoptive immuno-therapy for pancreatic cancer usingMUC1 peptide-pulsed den-dritic cells and activated T lymphocytes,” Anticancer Research,vol. 28, no. 1B, pp. 379–387, 2008.
[73] T. Hensler, H. Hecker, K. Heeg et al., “Distinct mechanismsof immunosuppression as a consequence of major surgery,”Infection and Immunity, vol. 65, no. 6, pp. 2283–2291, 1997.
[74] G. Shakhar and S. Ben-Eliyahu, “Potential prophylactic mea-sures against postoperative immunosuppression: could theyreduce recurrence rates in oncological patients?” Annals ofSurgical Oncology, vol. 10, no. 8, pp. 972–992, 2003.
[75] H. Weighardt, C. D. Heidecke, K. Emmanuilidis et al., “Sepsisaer major visceral surgery is associated with sustained and
interferon-𝛾𝛾-resistant defects of monocyte cytokine produc-tion,” Surgery, vol. 127, no. 3, pp. 309–315, 2000.
[76] S. Koido, E. Hara, S. Homma et al., “Dendritic/pancreatic carci-noma fusions for clinical use: comparative functional analysis ofhealthy-versus patient-derived fusions,” Clinical Immunology,vol. 135, no. 3, pp. 384–400, 2010.
[77] M. K. Gjertsen, T. Buanes, A. R. Rosseland et al., “Intradermalras peptide vaccination with granulocyte-macrophage colony-stimulating factor as adjuvant: clinical and immunologicalresponses in patients with pancreatic adenocarcinoma,” Inter-national Journal of Cancer, vol. 92, no. 3, pp. 441–450, 2001.
[78] S. Koido, E. Hara, S. Homma et al., “Dendritic cells fused withallogeneic colorectal cancer cell line present multiple colorec-tal cancer-speci�c antigens and induce antitumor immunityagainst autologous tumor cells,” Clinical Cancer Research, vol.11, no. 21, pp. 7891–7900, 2005.
[79] H. Saito, P. Dubsky, C. Dantin, O. J. Finn, J. Banchereau,and A. K. Palucka, “Cross-priming of cyclin B1, MUC-1 andsurvivin-speci�c CD8+ T cells by dendritic cells loaded withkilled allogeneic breast cancer cells,”Breast Cancer Research, vol.8, no. 6, article R65, 2006.
[80] E. M. Jaffee, R. H. Hruban, B. Biedrzycki et al., “Novelallogeneic granulocyte-macrophage colony-stimulating factor-secreting tumor vaccine for pancreatic cancer: a phase I trial ofsafety and immune activation,” Journal of Clinical Oncology, vol.19, no. 1, pp. 145–156, 2001.
[81] D. Laheru, C. Yeo, B. Biedrzycki et al., “A safety and efficacy trialof lethally irradiated allogeneic pancreatic tumor cells trans-fected with the GM-CSF gene in combination with adjuvantchemoradiotherapy for the treatment of adenocarcinoma of thepancreas,” Journal of Clinical Oncology, vol. 25, no. 18S, article3010, 2007.
[82] A. M. omas, L. M. Santarsiero, E. R. Lutz et al., “Mesothelin-speci�c CD8+ T cell responses provide evidence of in vivo cross-priming by antigen-presenting cells in vaccinated pancreaticcancer patients,” Journal of Experimental Medicine, vol. 200, no.3, pp. 297–306, 2004.
[83] R. Hassan and M. Ho, “Mesothelin targeted cancer immuno-therapy,” European Journal of Cancer, vol. 44, no. 1, pp. 46–53,2008.
[84] D. Laheru, E. Lutz, J. Burke et al., “Allogeneic granulo-cyte macrophage colony-stimulating factor-secreting tumorimmunotherapy alone or in sequence with cyclophosphamidefor metastatic pancreatic cancer: a pilot study of safety, feasibil-ity, and immune activation,” Clinical Cancer Research, vol. 14,no. 5, pp. 1455–1463, 2008.
[85] K. M. Hege, K. Jooss, and D. Pardoll, “GM-CSF gene-modifedcancer cell immunotherapies: of mice and men,” InternationalReviews of Immunology, vol. 25, no. 5-6, pp. 321–352, 2006.
[86] R. E. Beatson, J. Taylor-Papadimitriou, and J. M. Burchell,“MUC1 immunotherapy,” Immunotherapy, vol. 2, no. 3, pp.305–327, 2010.
[87] A Phase I/II Study of an Antitumor Vaccination UsingAlpha(1, 3) Galactosyltransferase ExpressingAllogeneic TumorCells in Patients with Pancreatic Cancer (NCT00255827),http://clinicaltrials.gov/.
[88] A Phase II Study of Low Dose HyperAcute(R)-PancreaticCancer Vaccine in Subjects with Surgically Resected PancreaticCancer (NCT00614601), http://clinicaltrials.gov/.

12 BioMed Research International
[89] A Phase II Study of HyperAcute(R)-Pancreatic Cancer Vac-cine in Subjects with Surgically Resected Pancreatic Cancer(NCT00569387), http://clinicaltrials.gov/.
[90] R. B. Mandell, R. Flick, W. R. Staplin et al., “e 𝛼𝛼galhyperAcute technology: enhancing immunogenicity of antivi-ral vaccines by exploiting the natural 𝛼𝛼gal-mediated zoonoticblockade,” Zoonoses and Public Health, vol. 56, no. 6-7, pp.391–406, 2009.
[91] B. A. Macher and U. Galili, “e Gal𝛼𝛼1,3Gal𝛽𝛽1,4GlcNAc-R (𝛼𝛼-Gal) epitope: a carbohydrate of unique evolution and clinicalrelevance,” Biochimica et Biophysica Acta, vol. 1780, no. 2, pp.75–88, 2008.
[92] T. Deguchi, M. Tanemura, E. Miyoshi et al., “Increased immu-nogenicity of tumor-associated antigen, mucin 1, engineered toexpress 𝛼𝛼-Gal epitopes: a novel approach to immunotherapyin pancreatic cancer,” Cancer Research, vol. 70, no. 13, pp.5259–5269, 2010.
[93] A. W. Purcell, J. McCluskey, and J. Rossjohn, “More than onereason to rethink the use of peptides in vaccine design,” NatureReviews Drug Discovery, vol. 6, no. 5, pp. 404–414, 2007.
[94] M. S. Bijker, C. J. M. Melief, R. Offringa, and S. H. van der Burg,“Design and development of synthetic peptide vaccines: past,present and future,” Expert Review of Vaccines, vol. 6, no. 4, pp.591–603, 2007.
[95] S. Kanodia and W. M. Kast, “Peptide-based vaccines for cancer:realizing their potential,” Expert Review of Vaccines, vol. 7, no.10, pp. 1533–1545, 2008.
[96] C. J. Voskens, S. E. Strome, and D. A. Sewell, “Syntheticpeptide-based cancer vaccines: lessons learned and hurdlesto overcome,” Current Molecular Medicine, vol. 9, no. 6, pp.683–693, 2009.
[97] S. Mocellin, P. Pilati, and D. Nitti, “Peptide-based anticancervaccines: recent advances and future perspectives,” CurrentMedicinal Chemistry, vol. 16, no. 36, pp. 4779–4796, 2009.
[98] H. Yanagimoto, S. Takai, S. Satoi et al., “Impaired function ofcirculating dendritic cells in patients with pancreatic cancer,”Clinical Immunology, vol. 114, no. 1, pp. 52–60, 2005.
[99] T. A. Waldmann, “Immunotherapy: past, present and future,”Nature Medicine, vol. 9, no. 3, pp. 269–277, 2003.
[100] O. J. Finn, “Molecular origins of cancer: cancer immunology,”e New England Journal of Medicine, vol. 358, no. 25, pp.2704–2715, 2008.
[101] M.Miyazawa, R.Ohsawa, T. Tsunoda et al., “Phase I clinical trialusing peptide vaccine for human vascular endothelial growthfactor receptor 2 in combination with gemcitabine for patientswith advanced pancreatic cancer,” Cancer Science, vol. 101, no.2, pp. 433–439, 2010.
[102] R. K. Ramanathan, K. M. Lee, J. McKolanis et al., “Phase Istudy of aMUC1 vaccine composed of different doses ofMUC1peptide with SB-AS2 adjuvant in resected and locally advancedpancreatic cancer,” Cancer Immunology, Immunotherapy, vol.54, no. 3, pp. 254–264, 2005.
[103] K. Yamamoto, T. Ueno, T. Kawaoka et al., “MUC1 peptidevaccination in patients with advanced pancreas or biliary tractcancer,”Anticancer Research, vol. 25, no. 5, pp. 3575–3579, 2005.
[104] International Clinical Trials Registry Platform (ICTRP),http://apps.who.int/trialsearch/trial.aspx?trialid=JPRN-UMIN000001664.
[105] S. L. Bernhardt, M. K. Gjertsen, S. Trachsel et al., “Telomerasepeptide vaccination of patients with non-resectable pancreaticcancer: a dose escalating phase I/II study,” British Journal ofCancer, vol. 95, no. 11, pp. 1474–1482, 2006.
[106] T. Buanes, J. Maurel, W. Liauw, M. Hebbar, and J. Nemunaitis,“A randomized Phase III study of gemcitabine (G) versusGV1001 in sequential combination with G in patients withunresectable and metastatic pancreatic cancer (PC),” Journal ofClinical Oncology, vol. 27, no. 15S, article 4601, 2009.
[107] G. K. Abou-Alfa, P. B. Chapman, J. Feilchenfeldt et al., “Tar-geting mutated K-ras in pancreatic adenocarcinoma using anadjuvant vaccine,” American Journal of Clinical Oncology, vol.34, no. 3, pp. 321–325, 2011.
[108] A. Toubaji, M. Achtar, M. Provenzano et al., “Pilot study ofmutant ras peptide-based vaccine as an adjuvant treatmentin pancreatic and colorectal cancers,” Cancer Immunology,Immunotherapy, vol. 57, no. 9, pp. 1413–1420, 2008.
[109] C. J. M. Melief and S. H. van der Burg, “Immunotherapy ofestablished (pre)malignant disease by synthetic long peptidevaccines,” Nature Reviews Cancer, vol. 8, no. 5, pp. 351–360,2008.
[110] M. S. Bijker, S. J. F. van den Eeden, K. L. Franken, C. J.M.Melief,S. H. van der Burg, and R. Offringa, “Superior induction of anti-tumor CTL immunity by extended peptide vaccines involvesprolonged, DC-focused antigen presentation,” European Jour-nal of Immunology, vol. 38, no. 4, pp. 1033–1042, 2008.
[111] S. Wedén, M. Klemp, I. P. Gladhaug et al., “Long-term follow-up of patients with resected pancreatic cancer following vacci-nation against mutant K-ras,” International Journal of Cancer,vol. 128, no. 5, pp. 1120–1128, 2011.
[112] J. Begley and A. Ribas, “Targeted therapies to improve tumorimmunotherapy,” Clinical Cancer Research, vol. 14, no. 14, pp.4385–4391, 2008.
[113] L. Chapatte, M. Ayyoub, S. Morel et al., “Processing of tumor-associated antigen by the proteasomes of dendritic cells controlsin vivo T-cell responses,” Cancer Research, vol. 66, no. 10, pp.5461–5468, 2006.
[114] A. N. Houghton and J. A. Guevara-Patiño, “Immune recog-nition of self in immunity against cancer,” Journal of ClinicalInvestigation, vol. 114, no. 4, pp. 468–471, 2004.
[115] L. M. Weiner, R. Surana, and J. Murray, “Vaccine preventionof cancer: can endogenous antigens be targeted?” CancerPrevention Research, vol. 3, no. 4, pp. 410–415, 2010.
[116] Z. Yu, M. R. eoret, C. E. Touloukian et al., “Poor immuno-genicity of a self/tumor antigen derives from peptide-MHC-Iinstability and is independent of tolerance,” Journal of ClinicalInvestigation, vol. 114, no. 4, pp. 551–559, 2004.
[117] K. Y. Tsang, C. Palena, J. Gulley, P. Arlen, and J. Schlom, “Ahuman cytotoxic T-lymphocyte epitope and its agonist epitopefrom the nonvariable number of tandem repeat sequence ofMUC-1,”Clinical Cancer Research, vol. 10, no. 6, pp. 2139–2149,2004.
[118] H. Yanagimoto, H. Shiomi, S. Satoi et al., “A phase II studyof personalized peptide vaccination combined with gemc-itabine for non-resectable pancreatic cancer patients,” OncologyReports, vol. 24, no. 3, pp. 795–801, 2010.
[119] R. G. Webster and H. L. Robinson, “DNA vaccines: a review ofdevelopments,” BioDrugs, vol. 8, no. 4, pp. 273–292, 1997.
[120] G. Eschenburg, A. Stermann, R. Preissner, H. A. Meyer, and H.N. Lode, “DNA vaccination: using the patient’s immune systemto overcome cancer,” Clinical and Developmental Immunology,vol. 2010, Article ID 169484, 14 pages, 2010.
[121] A Phase I Clinical Trial to Evaluate the Safety andImmunogenicity of a Mammaglobin-A DNA Vaccine in Breast

BioMed Research International 13
Cancer Patients with Metastatic Disease (NCT00807781),http://clinicaltrials.gov/.
[122] DNA Vaccine Coding for the Rhesus Prostate Speci�c Antigen(rhPSA) and Electroporation in Patients with Relapsed ProstateCancer, A Phase I/II Study (NCT00859729), http:// clinicaltri-als.gov/.
[123] A Phase Ia/Ib Study of the Safety and Immunogenicityof a Xenogeneic Tyrosinase DNA Vaccine Melanoma(NCT00471133), http://clinicaltrials.gov/.
[124] Y. Rong, D. Jin, W. Wu et al., “Induction of protective and ther-apeutic anti-pancreatic cancer immunity using a reconstructedMUC1 DNA vaccine,” BMC Cancer, vol. 9, article 191, 2009.
[125] K. Zhu, H. Qin, S. C. Cha et al., “Survivin DNA vaccinegenerated speci�c antitumor effects in pancreatic carcinomaand lymphoma mouse models,” Vaccine, vol. 25, no. 46, pp.7955–7961, 2007.
[126] M. C. Gaffney, P. Goedegebuure, H. Kashiwagi et al., “DNAvaccination targeting mesothelin combined with anti-GITRantibody induces rejection of pancreatic adenocarcinoma,”American Association For Cancer Research, vol. 47, article 329a,2006.
[127] A. D. Cohen, A. Diab, M. A. Perales et al., “Agonist anti-GITRantibody enhances vaccine-induced CD8+ T-cell responsesand tumor immunity,” Cancer Research, vol. 66, no. 9, pp.4904–4912, 2006.
[128] E. M. Esparza and R. H. Arch, “Glucocorticoid-induced TNFreceptor functions as a costimulatory receptor that promotessurvival in early phases of T cell activation,” Journal of Immunol-ogy, vol. 174, no. 12, pp. 7869–7874, 2005.
[129] R. M. Steinman, “e dendritic cell system and its role inimmunogenicity,” Annual Review of Immunology, vol. 9, pp.271–296, 1991.
[130] J. Banchereau and A. K. Palucka, “Dendritic cells as therapeuticvaccines against cancer,”Nature Reviews Immunology, vol. 5, no.4, pp. 296–306, 2005.
[131] F. O. Nestle, S. Alijagic, M. Gilliet et al., “Vaccination ofmelanoma patients with peptide- or tumor lysate-pulsed den-dritic cells,” Nature Medicine, vol. 4, no. 3, pp. 328–332, 1998.
[132] A. Mackensen, B. Herbst, J. L. Chen et al., “Phase I study inmelanoma patients of a vaccine with peptide-pulsed dendriticcells generated in vitro from CD34+ hematopoietic progenitorcells,” International Journal of Cancer, vol. 89, no. 2, pp.385–392, 2000.
[133] A. K. Palucka, H. Ueno, J. Connolly et al., “Dendritic cellsloaded with killed allogeneic melanoma cells can induceobjective clinical responses and MART-1 speci�c CD8+ T-cell immunity,” Journal of Immunotherapy, vol. 29, no. 5, pp.545–557, 2006.
[134] E. Gilboa and J. Vieweg, “Cancer immunotherapy with mRNA-transfected dendritic cells,” Immunological Reviews, vol. 199, pp.251–263, 2004.
[135] S. K. Nair, D. Boczkowski, M. Morse, R. I. Cumming, H. K.Lyerly, and E. Gilboa, “Induction of primary carcinoembryonicantigen (CEA)-speci�c cytotoxic T lymphocytes in vitro usinghuman dendritic cells transfected with RNA,” Nature Biotech-nology, vol. 16, no. 4, pp. 364–369, 1998.
[136] J. Gong, D. Chen, M. Kashiwaba, and D. Kufe, “Induction ofantitumor activity by immunization with fusions of dendriticand carcinoma cells,”NatureMedicine, vol. 3, no. 5, pp. 558–561,1997.
[137] T. Schmidt, C. Ziske, A.Märten et al., “Intratumoral immuniza-tion with tumor RNA-pulsed dendritic cells confers antitumorimmunity in a C57BL/6 pancreatic murine tumor model,”Cancer Research, vol. 63, no. 24, pp. 8962–8967, 2003.
[138] M. Schnurr, C. Scholz, S. Rothenfusser et al., “Apoptoticpancreatic tumor cells are superior to cell lysates in promotingcross-priming of cytotoxic T cells and activate NK and 𝛾𝛾𝛾𝛾Tcells,” Cancer Research, vol. 62, no. 8, pp. 2347–2352, 2002.
[139] A. J. Lepisto, A. J. Moser, H. Zeh et al., “A phase I/II studyof a MUC1 peptide pulsed autologous dendritic cell vaccine asadjuvant therapy in patientswith resected pancreatic and biliarytumors,” Cancer erapy, vol. 6, no. B, pp. 955–964, 2008.
[140] G. Pecher, A. Häring, L. Kaiser, and E. iel, “Mucin gene(MUC1) transfected dendritic cells as vaccine: results of a phaseI/II clinical trial,” Cancer Immunology, Immunotherapy, vol. 51,no. 11-12, pp. 669–673, 2002.
[141] M. A. Morse, S. K. Nair, D. Boczkowski et al., “e feasibilityand safety of immunotherapy with dendritic cells loaded withCEA mRNA following neoadjuvant chemoradiotherapy andresection of pancreatic cancer,” International Journal of Gas-trointestinal Cancer, vol. 32, no. 1, pp. 1–6, 2002.
[142] D. P. Carbone, I. F. Ciernik, M. J. Kelley et al., “Immunizationwithmutant p53- andK-ras-derived peptides in cancer patients:immune response and clinical outcome,” Journal of ClinicalOncology, vol. 23, no. 22, pp. 5099–5107, 2005.
[143] G.Mazzolini, C. Alfaro, B. Sangro et al., “Intratumoral injectionof dendritic cells engineered to secrete interleukin-12 by recom-binant adenovirus in patients with metastatic gastrointestinalcarcinomas,” Journal of Clinical Oncology, vol. 23, no. 5, pp.999–1010, 2005.
[144] S. Koido, E. Hara, S. Homma, K. Fujise, J. Gong, and H. Tajiri,“Dendritic/tumor fusion cell-based vaccination against cancer,”Archivum Immunologiae et erapiae Experimentalis, vol. 55,no. 5, pp. 281–287, 2007.
[145] J. Gong, S. Koido, and S. K. Calderwood, “Cell fusion: fromhybridoma to dendritic cell-based vaccine,” Expert Review ofVaccines, vol. 7, no. 7, pp. 1055–1068, 2008.
[146] S. Koido, E. Hara, S. Homma, T. Ohkusa, J. Gong, and H. Tajiri,“Cancer immunotherapy by fusions of dendritic cells and tumorcells,” Immunotherapy, vol. 1, no. 1, pp. 49–62, 2009.
[147] M. Yamamoto, T. Kamigaki, K. Yamashita et al., “Enhancementof anti-tumor immunity by high levels of 1 and 17 with acombination of dendritic cell fusion hybrids and regulatory Tcell depletion in pancreatic cancer,” Oncology Reports, vol. 22,no. 2, pp. 337–343, 2009.
[148] F. V. Ona, N. Zamcheck, P. Dhar, T. Moore, and H. Z.Kupchik, “Carcinoembryonic antigen (CEA) in the diagnosisof pancreatic cancer,” Cancer, vol. 31, no. 2, pp. 324–327, 1973.
[149] S. Lei, H. E. Appert, B. Nakata, D. R. Domenico, K. Kim, andJ. M. Howard, “Overexpression of HER2/neu oncogene in pan-creatic cancer correlates with shortened survival,” InternationalJournal of Pancreatology, vol. 17, no. 1, pp. 15–21, 1995.
[150] B. M. Ryan, N. O’Donovan, and M. J. Duffy, “Survivin: a newtarget for anti-cancer therapy,” Cancer Treatment Reviews, vol.35, no. 7, pp. 553–562, 2009.
[151] W. Zhou, M. Capello, C. Fredolini et al., “Mass spectrometryanalysis of the post-translational modi�cations of r-enolasefrom pancreatic ductal adenocarcinoma cells,” Journal of Pro-teome Research, vol. 9, no. 6, pp. 2929–2936, 2010.
[152] H. H. Emina and H. L. Kaufman, “CEA-based vaccines,” ExpertReview of Vaccines, vol. 1, no. 1, pp. 49–63, 2002.

14 BioMed Research International
[153] C. K. Tang, M. Katsara, and V. Apostolopoulos, “Strategies usedfor MUC1 immunotherapy: human clinical studies,” ExpertReview of Vaccines, vol. 7, no. 7, pp. 963–975, 2008.
[154] A. Scarpa, P. Capelli, K. Mukai et al., “Pancreatic adenocar-cinomas frequently show p53 gene mutations,” e AmericanJournal of Pathology, vol. 142, no. 5, pp. 1534–1543, 1993.
[155] F. Chen, W. Wang, and W. S. El-Deiry, “Current strategies totarget p53 in cancer,” Biochemical Pharmacology, vol. 80, no. 5,pp. 724–730, 2010.
[156] C. Almoguera, D. Shibata, K. Forrester, J. Martin, N. Arnheim,and M. Perucho, “Most human carcinomas of the exocrinepancreas contain mutant c-K-ras genes,” Cell, vol. 53, no. 4, pp.549–554, 1988.
[157] J. Itakura, T. Ishiwata, H. Friess et al., “Enhanced expression ofvascular endothelial growth factor in human pancreatic can-cer correlates with local disease progression,” Clinical CancerResearch, vol. 3, no. 8, pp. 1309–1316, 1997.
[158] M. Li, U. Bharadwaj, R. Zhang et al., “Mesothelin is a malignantfactor and therapeutic vaccine target for pancreatic cancer,”Molecular Cancer erapeutics, vol. 7, no. 2, pp. 286–296, 2008.
[159] H. J. Kang, S. K. Jung, S. J. Kim, and S. J. Chung, “Structureof human 𝛼𝛼-enolase (hENO1), a multifunctional glycolyticenzyme,” Acta Crystallographica D, vol. 64, no. 6, pp. 651–657,2008.

Hindawi Publishing CorporationBioMed Research InternationalVolume 2013, Article ID 871936, 10 pageshttp://dx.doi.org/10.1155/2013/871936
Review ArticleM�C1-��eci�c C�toto�ic � ����hoc�tes in Cancer �hera���Induction and Challenge
David Roulois,1, 2, 3 Marc Grégoire,1, 2, 3 and Jean-François Fonteneau1, 2, 3
1 UMR892, INSERM, Institut de Recherche érapeutique, Université de Nantes, 8 quai Moncousu, BP70721,44007 Nantes Cedex 1, France
2 CNRS, UMR6299, Institut de Recherche érapeutique, Université de Nantes, 8 quai Moncousu, BP70721,44007 Nantes Cedex 1, France
3 Faculté de Médecine, Université de Nantes, 44035 Nantes Cedex 1, France
Correspondence should be addressed to Jean-François Fonteneau; [email protected]
Received 18 May 2012; Accepted 6 July 2012
Academic Editor: Julie Curtsinger
Copyright © 2013 David Roulois et al. is is an open access article distributed under the Creative Commons Attribution License,which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
MUC1 glycoprotein is oen found overexpressed and hypoglycosylated in tumor cells from numerous cancer types. Since itsdiscovery MUC1 has been an attractive target for antitumor immunotherapy. Indeed, in vitro and in vivo experiments have shownT-cell-speci�c responses against MUC1 in an HLA-restricted and HLA-unrestricted manner, although some animal models havehighlighted the possible development of tolerogenic responses against this antigen. ese observations permit the development ofnew T-cell vaccine strategies capable of inducing an MUC1-speci�c cytotoxic T cell response in cancer patients. Some of thesestrategies are now being tested in clinical trials against different types of cancer. To date, encouraging clinical responses havebeen observed with some MUC1 vaccines in phase II/III clinical trials. is paper compiles knowledge regarding MUC1 as apromising tumor antigen for antitumor therapeutic vaccines applicable to numerous cancers. We also summarize the results ofMUC1-vaccine-based clinical trials.
1. Introduction
With the increasing number of cancers, the development ofinnovative cancer therapies is a great challenge. One of theseinnovative strategies is immunotherapy. Since the discoverythat the immune system can control cancer progression,which has been conceptualized in the “three Es” theory[1] for “elimination, equilibrium, and escape,” supportingthe implications of the immune system in the control andselection of tumor cells, scientists and clinicians have triedto exploit this phenomenon to induce an antitumor immuneresponse in cancer patients. Major goals in the �eld ofimmunotherapy are to understand how the immune systemcan be speci�cally activated against cancer cells and toidentify relevant antigenic cancer targets.
e �rst human tumor-associated antigen (TAA) to bediscovered, recognized by cytotoxic CD8+ T lymphocytes(CTL), was MA�E-A1 which was identi�ed from tumor-in�ltrating lymphocytes obtained aer culture of amelanoma
biopsy [2]. Since then, many other TAAs have been identi�ed(for review see [3]). Certain TAAs are restricted to one ora few cancer types, such as the “mutated TAA” (BCR-ABLfusion, B-raf, k-ras, N-ras, p53, etc.), or the “differentiationTAA” (Melan-A/MART1, gp100, CEA, PSA, etc.), whereasothers are shared between a wide range of cancers, suchas the “shared tumor-speci�c TAA” (MA�E, N�ESO-1,SSX, etc.) or the “overexpressed TAA” (HER-2/Neu, p53,Telomerase, MUC1, etc.) (http://www.archive.cancerimmu-nity.org/peptidedatabase/Tcellepitopes.htm).
Mucin 1 (MUC1) belongs to the “overexpressed TAA”category, even if this overexpression is not the only hallmarkof MUC1 in tumor cells, since it is oen accompanied bymodi�cation of MUC1 glycosylation status. In healthy cells,MUC1 is a glycoprotein expressed at the apical surface ofepithelial cells and characterized by a high glycosylationlevel. In cancers, this glycoprotein is oen overexpressedby tumor cells, with a loss of polarity and, interestingly,a modi�cation of its glycosylation pattern [4]. Both the

2 BioMed Research International
overexpression and the modi�cation of its glycosylationstatus make this protein highly immunogenic and, thus, aninteresting target in cancer immunotherapy. In this paper,we focus on the immunogenic properties of the MUC1glycoprotein. However, it should be noted that MUC1 hasalso been described as having an oncogenic role (for reviewsee: [5, 6]). Firstly, we describe the differences betweenMUC1 expression in healthy cells and tumor cells whichrenders MUC1 more immunogenic when it is expressed bytumor cells. Secondly, we discuss the problem of inductionof tolerance against MUC1 which can impair the antitumorimmune response. We list the immunotherapy strategiesfor inducing an antitumor response in patients, which arebeing developed in vitro and in mouse models. Finally, wediscuss MUC1-based immunotherapy clinical trials againstcancers.
2. MUC1: An Overexpressed, Hypoglycosylated,Tumor-Associated Antigen
eMUC1 genewas cloned in the early 1990s [7, 8]. It belongsto themucin family, comprising 21members.MUC1 encodesa highly glycosylated, type I transmembrane glycoprotein,with a variable number of 20-amino-acid repeat sequencesreferred to as “variable number tandem repeat” (VNTR)(Figure 1).e number of VNTR is variable from one allele toanother, varying from 25 to 120 VNTR per MUC1 molecule,with the alleles containing 40 and 66 VNTR being the mostfrequent in the northern European population [7]. EachVNTR contains �ve potential sites of O-glycosylation onserine or threonine.
Towards the end of the 1980s, differences betweenthe MUC1 expressed by healthy mammary cells andby breast cancer cells were observed using the mono-clonal antibody, SM-3, speci�c for the MUC1 core pro-tein stripped of sugars [9, 10]. Indeed, SM-3 mAb usedin histology recognized 91% of breast cancer samples,but showed little or no reactivity with healthy mam-mary cells [9]. e SM-3 mAb was also reactive againstlung, colon, and ovarian carcinoma, but failed to stainthe healthy cell counterparts [10]. ese studies showedthat, in different types of carcinoma, MUC1 is hypo-glycosylated. is hypoglycosylation allows recognition bythe SM-3 mAb. Since then, MUC1 was considered tobe a “tumor-associated antigen” which can be targetedin immunotherapy using monoclonal antibodies. It hassubsequently been shown that the glycosylation structuresof MUC1 expressed by normal breast cells and tumorcells are different. Indeed, in healthy cells MUC1 containsextended, core 2-based glycans that are formed by N-acetylglucosamine attachment to the GalNAc of core 1, whileon the MUC1 expressed by tumor cells the glycans areshorter, core 1-based and richer in ST, Tn, and T glycans[11–13].
ese differences observed between MUC1 expressedby tumor cells and healthy cells prompted research on thecapacity of T lymphocytes to recognize tumoral MUC1
epitopes. T lymphocytes usually recognize, with their T-cell receptors, peptides from endogenous or exogenousantigens presented in association with MHC molecules.Surprisingly, the �rst report of recognition of MUC1by T lymphocytes was shown to be non-MHC-restricted[14]. Indeed the T lymphocytes obtained from a pan-creatic cancer patient did not recognize an MUC1 pep-tide presented by an MHC molecule, but rather, directly,the hypoglycosylated core of MUC1. is unusual anti-gen recognition was con�rmed by other groups [15–18].e reactivity of these T cells was inhibited by SM-3 mAb, which showed that these T cells recognize thehypoglycosylated core of MUC1. Furthermore, these Tcells failed to recognize healthy epithelial cells, whichwere not stained by the SM-3 mAb. Finally, Hinoda andcolleagues described an increased recognition of gastrictumor cells cultured with benzyl-2-acetamido-2-deoxy-𝛼𝛼-D-galactopyranoside (BGN), a competitive inhibitor of O-glycosylation, by an HLA-unrestricted, MUC1-speci�c CTLline [19]. All of these results suggest that a non-MHC-restricted, hypoglycosylated, MUC1-speci�c T-cell responsecan be present spontaneously in cancer patients.
In the mid 1990s, efforts were made to identify HLA-restricted, MUC1-speci�c T-cell responses. Indeed, progressmade in the understanding of the T-lymphocyte responseagainst tumor cells allowed the development of new strate-gies to identify TAA, such as “reverse immunology”. isconsists of inducing in vitro T-lymphocyte responses againstpeptides from a candidate TAA. Peptides are selected fortheir capacity to bind a particular HLA allele. e capacityof peptide-responding T cells to recognize a tumor cellline which expresses the candidate TAA and the particularHLA allele is then tested to validate the epitope. Using thisapproach, Domenech and colleagues identi�ed a peptideencoded by the VNTR (STAPPAHGV) with the abilityto bind to several HLA class I alleles: HLA-A1, -A2.1, -A3, and -A11 [20]. ey were able to generate cytotoxicCD8+ T lymphocytes speci�c for HLA-A11/STAPPAHGV,but did not validate the presentation of this peptide byHLA-A11+ tumor cells. Using HLA-A∗0201/Kb transgenicmice, Apostolopoulos and colleagues identi�ed two peptidesfrom the VNTR able to bind HLA-A∗0201 molecule, whichare the most common HLA class I allele in the Caucasianpopulation: the peptide, STAPPAHGV, previously describedby Domenech’s team, and a new peptide, APDTRPA [21].ese peptides were able to induce CD8+ cytotoxic T-cell responses in HLA-A∗0201/Kb mice. e T cells wereable to lyse the HLA-A∗0201+ MUC1+ breast cancer cellline, MCF-7. Brossart and colleagues then selected twoother peptides fromMUC1:MUC1(20–28) LLLLTVLTVandMUC1(950–958) STAPPVHNV, which exhibit a good affin-ity for the HLA-A∗0201 molecule [22]. e STAPPVHNVpeptide is not encoded by the VNTR, but by the region�anking it, whereas the LLLLTVLTV peptide is encodedby the signal sequence of MUC1. Brossart and colleaguesgenerated two CD8+ T-cell clones speci�c for the HLA-A∗0201/MUC1(20–28) and HLA-A∗0201/MUC1(950–958)complexes. ese clones were able to recognize MUC1+HLA-A∗0201+ tumor cell lines from different types of

BioMed Research International 3
Variable number tandem repeats
(20 to 120 VNTR per MUC1 molecule)
Hypoglycosylated
tumor cells
Core-2glycosylations
Core 1 glycosylations: low leveland aberrant glycosylations
(Tn antigen)
Unglycosylated MUC1 (potentialglycosylation site on serine or
threonine in red)
MUC-1
PDTRPAPGSTAPPAHGVTSA
Hyperglycosylated
normal cellsMUC-1
Unglycosylated
unnaturalMUC-1
F 1: Structure of the MUC1 glycoprotein in normal and tumor cells.
cancer: breast, pancreatic, and renal. In another study,the same researchers showed that these MUC1-speci�c T-cell clones were also able to recognize multiple myelomacells and primary acute myelogenous leukemia blasts [23].More recently, we showed that MUC1(950–958) peptide ispresented to MUC1-speci�c CD8+ T cells by HLA-A∗0201+malignant pleural mesothelioma cell lines [24]. In addition,Ninkovic and colleagues reported that some glycosylatedpeptides from the VNTR of MUC1, notably the decamerSAP10 [SAPDT(GalNAc)RPAPG], can be generated by theimmunoproteasome of dendritic cells [25]. is glycosylatedpeptide can be presented by HLA class I molecules andrecognized by CD8+ T cells. e nonglycosylated peptidewas also recognized by CD8+ T cells, whereas a peptide witha longer sugar chain (Gal-GalNac) did not bind the HLA-A∗0201 molecule. Finally, MUC1 can also be recognizedby CD4+ T lymphocytes. However, in this case, only oneHLA-DR3-restricted epitope encoded by the VNTR has beenidenti�ed by �reverse immunology� [26]. Unfortunately, thecapacity for presentation by tumor cells was not tested sincethey do not express HLA class II molecules.
Regarding the recognition of MUC1 peptides by HLA-restricted T lymphocytes, the importance of the MUC1glycosylation status is not as clear as in the case of HLA-unrestricted recognition of MUC1. We showed recentlythat the MUC1 glycosylation level does not affect therecognition of mesothelioma tumor cells by an HLA-A∗0201�MUC1(950–958)-speci�c CD8+ T-cell clone [24].Indeed, we observed that some tumor cell lines recog-nized by the CD8+ T-cell clone were weakly stainedwith the SM-3 antibody and another mAb speci�c forhypoglycosylated MUC1, VU-3-C6. Furthermore, MPM
cell lines treated with benzyl-2-acetamido-2-deoxy-𝛼𝛼-D-galactopyranoside (BGN), a competitive inhibitor of O-glycosylation, were not better recognized by the T-cell clone,despite an increased staining with the SM-3 and VU-3-C6 mAb. However, other HLA-restricted MUC1 epitopesseem to be dependent on the hypoglycosylation status ofMUC1, at least for the induction of the MUC1-speci�c T-cell response by antigen-presenting cells such as dendriticcells (DC). Indeed, Hiltbold and Colleagues reported thatglycosylation of long peptides, consisting of �ve MUC1tandem-repeat regions, decreased the processing and theHLA-A1 restricted cross-presentation to CD8+ T cells by DCof a nine-amino-acid peptide contained in this long peptide[27]. Furthermore, the modi�cation of MUC1 glycosylationin cancer cells may increase the capacity of DC to acquirethis antigen for cross-presentation. Indeed, in cancer, solubleMUC1 presents an aberrant pattern of glycosylation, notablythe Tn antigen, which can be recognized and internalizedby the C-type lectin receptor macrophage galactose-typelectin (CLR MGL) present on DC and macrophages [28,29]. is internalization of the soluble form of this tumoralform of MUC1 antigen by DC has also been shown tobe mediated by the mannose receptor [30]. However, theretention of internalizedMUC1 in the early endosome by thisreceptor inhibits its presentation to CD4+ T cells, whereasthe nonglycosylated form of the antigen is well presented.Conversely, Vlad and colleagues reported that DC exposedto a long-glycosylated MUC1 peptide were able to processandpresent a glycosylated shorter peptide, in associationwithclass II HLA, to CD4+ T cells suggesting that glycosylatedMUC1 peptide can be processed by dendritic cells. [28].In addition, it has been shown that the glycosylation status

4 BioMed Research International
in�uences, but does not inhibit, the cleavage of MUC1 bythe immunoproteasome expressed by mature DC [31]. All ofthese studies underline the capacity of dendritic cells (DC)to distinguish normal from aberrant MUC1. is shows theimportance of MUC1 glycosylation, which affects differentlythe presentation of abnormal MUC1 in MHC class I and IImolecules according to the MUC1 epitope studied.
3. Human MUC1 Mouse Model and Tolerance
With the goal of studying the immunogenicity of MUC1 invivo and of developingMUC1-based cancer immunotherapy,human MUC1 transgenic mouse (TG mice) tumor modelswere set up by the group of Papadimitriou [32].ese authorsreported that the expression of human MUC1 in TG micewas closely similar to that observed in human tissue. Sincethen, different teams have used these TG mice to studytolerance against MUC1 and how to induce in vivo an MUC1tumor-speci�cT-cell response [33–35]. Rowse and colleaguescompared tumor growth of MUC1+ tumors in TG or Wtmice [35]. ey also compared the induction of an MUC1-speci�c humoral response a�er immunization with MUC1peptide. MUC1+ tumors grew in TG mice, whereas theywere rejected in Wt mice, suggesting that a tolerance toMUC1 is present in TG mice. MUC1 immunization of Wtmice also allowed a switch of immunoglobulin to the IgGsubtype, whilst this switch was not observed in TGmice. Twoother studies have also demonstrated the establishment oftolerance against MUC1 in TG mice [36, 37]. In a �rst study,the authors showed that a CD4+ T-cell response againstMUC1 occurred inWtmice following challenge withMUC1-expressing B16 tumor cells, whereas this response was absentin TG mice [37]. When CD4+ T cells were transferred fromB16-bearing Wt mice to B16-bearing TG mice, an increase insurvivalwas observed. Furthermore,Wtmice challengedwithMUC1-expressing B16 tumors mounted an IgG responseagainst MUC1, whereas this IgG response was absent in TGmice [36]. Human MUC1 TG mice have since been crossedwith conditional-endometriosis transgenicmice, showing thepresence of regulatory Foxp3+ T cells and antibodies againstMUC1 during endometriosis, with potential implicationsfor cancer progression [38]. All of these studies, thus, showthat a certain degree of MUC1 tolerance is established invivo. MUC1-based immunotherapy strategies should takeinto account this phenomenon.
Indeed, these in vivo studies highlight the potentialproblem of MUC1 tolerance in humans. MUC1 is naturallyexpressed, and tolerogenic responses can occur in patientsleading to tumor escape against the immune system. eMUC1 TG mouse tumor model seems to back up the ideaof tolerance against MUC1 in humans, but the modi�cationsof MUC1 glycosylation observed in human cancer cells werenot taken into account in these studies. In vivo investigationsusing vaccination against MUC1 with glycosylated or nong-lycosylated peptide suggest that tolerance is not establishedagainst epitopes of MUC1 carrying a tumor-speci�c patternof glycosylation, such as the Tn antigen [39–41]. us,abnormal glycosylation patterns can be recognized as foreign
rather than selfantigen, which seems to be important in theinduction of a speci�c T-cell response [39, 40]. MUC1 can,thus, be considered as an “abnormal selfantigen” when itsglycosylation status is modi�ed in tumor cells and this is aninteresting property that could be exploited in immunother-apy [42].
�. ��ra�e�� �o �nduce MUC1-�pec��cT-Cell Responses
Different approaches have been developed to induce MUC1-speci�c T-cell responses in cancer patients. e majority ofthese are based on the capacity of DC to activate speci�cT-cell immune responses [53]. We have summarized theseapproaches in Table 1 and focus on the two most-studiedstrategies: MUC1-encoding nucleic acids or peptidic vacci-nation.
One approach consists of the utilization of MUC1 DNAas a vaccine [50, 52, 54]. is strategy has the advantageof activating different populations of lymphocytes, contraryto the use of MHC class I or class II peptides. In thesestudies, injections of MUC1 cDNA vaccine to tumor-bearingmice were able to induce tumor regressions. However, intwo studies, the effector T cells were identi�ed as CD8+cytotoxic T cells [52, 54], whereas in the last one, theantitumor response was attributed to CD4+ T cells [50].Studies are now being performed to improve this approach,notably by adding to the MUC1 cDNA a maturation signalfor DC. Indeed, one approach consists of coupling MUC1DNA with DNA corresponding to the expression of a heatshock protein (HSP70) which has already been reported toimprove the stimulatory properties of DC [55]. In this study,MUC1/HSP70-coupled DNA increased the capacity of DCto induce effective cytotoxic T-cell responses and inhibitedtumor cell growth [45]. Another strategy which has beendeveloped to improve MUC1 cDNA vaccination consists ofcombining the vaccine with a tumor-cell-death inducer, suchas a cDNA interfering with the expression of ANT2, a proteinimplicated in carcinogenesis [51]. CD8+ T-cell responseswere more effective using this combination than with the useof MUC1 cDNA alone. To target MUC1 cDNA expressiononly to DC, in vitro transfection of human DC with MUC1mRNA has also been developed [46]. ese MUC1-mRNA-transfected DC were able to induce MUC1-speci�c CD8+T lymphocytes that can kill pancreatic cancer cells. Finally,to improve the MUC1 cDNA strategies, another interestingstudy suggested the use of a modi�ed MUC1 se�uence, inorder to prevent normal glycosylation of the MUC1 proteinin dendritic and cancer cells [47].
Another vaccination strategy which has been studiedpreclinically, in mice, consists of the use of MUC1 peptideor glycopeptides. For this strategy, the use of glycopeptideswith tumor-speci�c sugar motifs, such as the Tn antigen,seems to be more efficient than the use of normal peptides ineliciting an effective immune response [39, 40]. e MUC1peptide can also be modi�ed to increase its penetration intoantigen-presenting cells, for example, the MUC1-MPA(11)Ppeptide which improved the induction of tumor regression in

BioMed Research International 5
T 1: Strategies for the induction of anti-MUC1 T cell responses that are being studied in vitro and in vivo.
Author Strategy Cancer Model Effect
Deguchi et al. [43] 𝛼𝛼-gal epitope to increaseimmunogenicity of MUC1
Pancreatic cancermouse model
Antibody induction, mouse tumor regression,induction of T cell responses
Kovjazin et al. [44] ImMucin peptide 21mer Mouse/PBMC ofpatients
CD4+ and CD8+ T lymphocyte responses in vitroand in vivo
Choi et al. [45] DNA vaccine (MUC1/HSP70) B16 mice cytotoxic T cell response induction/ tumorgrowth inhibition
Chen et al. [46] MUC1 mRNA, dendritic celltransfection Pancreatic cancer MUC1 mRNA-transfected dendritic cells can
induce MUC1-speci�c CD8+ T cell responses
Wright et al. [47] MUC1 peptide with substitution ofO-glycosylation site
Humanadenocarcinoma
O-glycosylation site substitution improvesimmunogenicity
Kobukai et al. [48] MPA11P vehicle of a 30mer MUC1peptide Mouse Reduction of tumor size, lymphocyte in�ltration
Lakshmiarayananet al. [49]
Tripartite MUC1 vaccine (TLR2,elpher, MUC1 glycopeptides)
Mouse model ofmammary cancer
IgG antibodies, cytotoxic T lymphocytes,activation of innate immune response
Sugiura et al. [50] MUC1 DNA vaccine Mouse/colon Induction of CD4+ T cell responses, not CD8+
Ryan et al. [39, 40] TN MUC1 glycopeptide Mouse T cell responses against glycosylated peptides, butnot unglycosylated peptides
Choi et al. [51] MUC1 DNA vaccination, enhancedby mANT2 shRNA Mouse melanoma Combination enhanced effects of DNA
vaccination, MUC1 CD8+ T cell responsesJeon et al. [52] DNA vaccination Mouse Tumor growth inhibition, CD8+ IFN-𝛾𝛾 increased
an animalmodel [48]. Targeting ofMUC1peptide to antigen-presenting cells can also be improved by adding oxidized (T-cell response, weak antibody level) or reduced mannan (Tcell response, high antibody level) [41]. Another interestingapproach was recently developed using a plant model whichcan produce MUC1 glycopeptides that are able to breaktolerance in MUC1 Tg mice vaccinated with this peptideby the production of MUC1-speci�c antibodies [56]. Somelong MUC1 peptides containing MHC class I and class IIepitopes (21mer) have also been developed to induce CD8+and CD4+ T-cell responses [44]. CD8+ T-cell responsesobtained in mice with the long peptide seem to be strongerthan with an MUC1 9mer peptide. Peptidic MUC1 vaccineshave also been combined with other reagents to improvetheir efficiency. For example, MUC1 glycopeptides have beencovalently associatedwith T-helper peptide andTLR2 agonistin a multimodal vaccine [49]. is vaccine strategy wasreported to induce a high level of MUC1-speci�c antibodiesand MUC1-speci�c cytotoxic T lymphocytes, which showedsuperior capability to prevent tumors growth in mice thanunglycosylated peptides.
5. MUC1-Based Immunotherapy:Clinical Studies
Some of these preclinical strategies are now being assessedfor MUC1-based immunotherapy in clinical trials for dif-ferent types of cancers. More than sixty clinical trialsinterested in MUC1 protein are currently in progress(http://www.clinicaltrials.gov/). e majority of these aredeveloping vaccination strategies against MUC1 to treat
cancer. is large number of trials highlights the clinicalinterest in MUC1 vaccination. Only three clinical studieshave reached the phase IIB/III stage. We have summarizedin Table 2 the strategies which are currently being developedin these clinical trials. Some of these studies have shown apossible clinical effect of this vaccine in inducing an MUC1-speci�c T-cell response. Among them, some major strategieshave emerged and reached phase III.
e BLP25 liposome vaccine (stimuvax or L-BLP25)is a liposomal vaccine containing a 25-amino-acid MUC1peptide corresponding to the core peptide of MUC1 (STAP-PAHGVTSAPDTRPAPGSTAPP) and coupled with a palmi-toyl lysine residue at the C terminus which increases incor-poration of this lipopeptide into the liposome particle [74,75]. is strategy is mostly being used in nonsmall-celllung carcinoma (NSCLC) and is well tolerated [62, 76].Even though no immunological response was observed, thisvaccine seems to have enhanced patient survival for stageIIIB/IV NSCLC in a phase II clinical trial [63]. A phase IIIstudy is in progress to con�rm this result [64].
Another major strategy is TG4010, from Transgene SA,which is a recombinant virus of the Modi�ed VacciniaAnkara (MVA), encoding MUC1 and IL-2 (MVA MUC1-IL2). Unlike the L-BLP25 which is being used on NSCLConly, TG4010 is being used against several types of cancer,including prostate cancer, renal cell carcinoma (RCC), andNSCLC [58–60]. In prostate cancer, evidence of biologicalactivity of the vaccine has been observed, with improvedprostate-speci�c antigen (PSA) level doubling time [59]. InRCC, preliminary results were encouraging showing that thisvaccine is well tolerated and indicating some evidence ofMUC1-speci�c CD4+ and CD8+ T-cell responses [60]. e
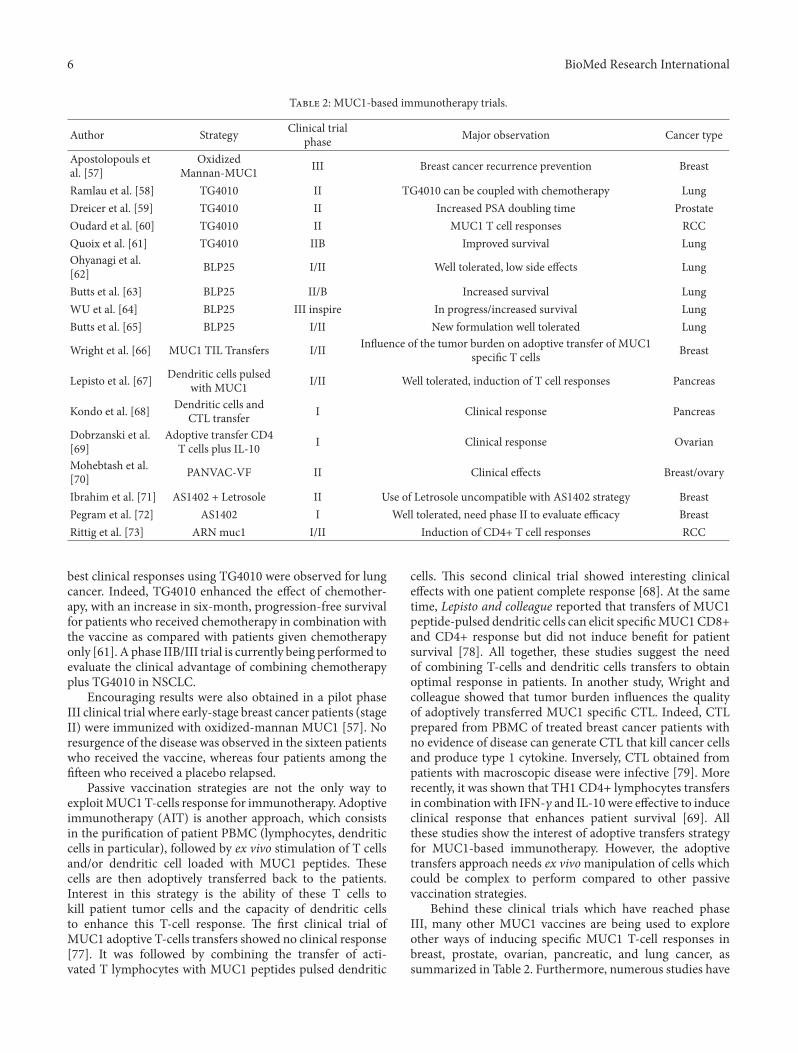
6 BioMed Research International
T 2: MUC1-based immunotherapy trials.
Author Strategy Clinical trialphase Major observation Cancer type
Apostolopouls etal. [57]
OxidizedMannan-MUC1 III Breast cancer recurrence prevention Breast
Ramlau et al. [58] TG4010 II TG4010 can be coupled with chemotherapy LungDreicer et al. [59] TG4010 II Increased PSA doubling time ProstateOudard et al. [60] TG4010 II MUC1 T cell responses RCCQuoix et al. [61] TG4010 IIB Improved survival LungOhyanagi et al.[62] BLP25 I/II Well tolerated, low side effects Lung
Butts et al. [63] BLP25 II/B Increased survival LungWU et al. [64] BLP25 III inspire In progress/increased survival LungButts et al. [65] BLP25 I/II New formulation well tolerated Lung
Wright et al. [66] MUC1 TIL Transfers I/II In�uence of the tumor burden on adoptive transfer of MUC1speci�c T cells Breast
Lepisto et al. [67] Dendritic cells pulsedwith MUC1 I/II Well tolerated, induction of T cell responses Pancreas
Kondo et al. [68] Dendritic cells andCTL transfer I Clinical response Pancreas
Dobrzanski et al.[69]
Adoptive transfer CD4T cells plus IL-10 I Clinical response Ovarian
Mohebtash et al.[70] PANVAC-VF II Clinical effects Breast/ovary
Ibrahim et al. [71] AS1402 + Letrosole II Use of Letrosole uncompatible with AS1402 strategy BreastPegram et al. [72] AS1402 I Well tolerated, need phase II to evaluate efficacy BreastRittig et al. [73] ARN muc1 I/II Induction of CD4+ T cell responses RCC
best clinical responses using TG4010 were observed for lungcancer. Indeed, TG4010 enhanced the effect of chemother-apy, with an increase in six-month, progression-free survivalfor patients who received chemotherapy in combination withthe vaccine as compared with patients given chemotherapyonly [61]. A phase IIB/III trial is currently being performed toevaluate the clinical advantage of combining chemotherapyplus TG4010 in NSCLC.
Encouraging results were also obtained in a pilot phaseIII clinical trial where early-stage breast cancer patients (stageII) were immunized with oxidized-mannan MUC1 [57]. Noresurgence of the disease was observed in the sixteen patientswho received the vaccine, whereas four patients among the��een who received a placebo relapsed.
Passive vaccination strategies are not the only way toexploitMUC1T-cells response for immunotherapy. Adoptiveimmunotherapy (AIT) is another approach, which consistsin the puri�cation of patient PBMC (lymphocytes, dendriticcells in particular), followed by ex vivo stimulation of T cellsand/or dendritic cell loaded with MUC1 peptides. esecells are then adoptively transferred back to the patients.Interest in this strategy is the ability of these T cells tokill patient tumor cells and the capacity of dendritic cellsto enhance this T-cell response. e �rst clinical trial ofMUC1 adoptive T-cells transfers showed no clinical response[77]. It was followed by combining the transfer of acti-vated T lymphocytes with MUC1 peptides pulsed dendritic
cells. is second clinical trial showed interesting clinicaleffects with one patient complete response [68]. At the sametime, Lepisto and colleague reported that transfers of MUC1peptide-pulsed dendritic cells can elicit speci�cMUC1CD8+and CD4+ response but did not induce bene�t for patientsurvival [78]. All together, these studies suggest the needof combining T-cells and dendritic cells transfers to obtainoptimal response in patients. In another study, Wright andcolleague showed that tumor burden in�uences the �ualityof adoptively transferred MUC1 speci�c CTL. Indeed, CTLprepared from PBMC of treated breast cancer patients withno evidence of disease can generate CTL that kill cancer cellsand produce type 1 cytokine. Inversely, CTL obtained frompatients with macroscopic disease were infective [79]. Morerecently, it was shown that TH1 CD4+ lymphocytes transfersin combinationwith IFN-𝛾𝛾 and IL-10 were effective to induceclinical response that enhances patient survival [69]. Allthese studies show the interest of adoptive transfers strategyfor MUC1-based immunotherapy. However, the adoptivetransfers approach needs ex vivo manipulation of cells whichcould be complex to perform compared to other passivevaccination strategies.
Behind these clinical trials which have reached phaseIII, many other MUC1 vaccines are being used to exploreother ways of inducing speci�c MUC1 T-cell responses inbreast, prostate, ovarian, pancreatic, and lung cancer, assummarized in Table 2. Furthermore, numerous studies have

BioMed Research International 7
described MUC1 as an oncogenic protein, advocating thedevelopment of pharmacological studies to counteract thisprotein. is is the case for toxins which target the cytoplas-mic tail (see Figure 1) of MUC1 [80, 81] or molecules thatdownregulate MUC1 protein [82, 83]. Both pharmacologicaland immunotherapeutic strategies based on MUC1 to treatcancer should now be actively pursued.
Abbreviations
MPM: Malignant pleural mesotheliomaMUC1: Mucin1TAA: Tumor associated antigen.
Acknowledgments
isworkwas �nanced by INSERM, La ligue r�gionale contrele Cancer (CSIRGO), theARC (Association pour la recherchecontre le cancer), the Nantes Hospital and the ARSMESO44association.
References
[1] G. P. Dunn, L. J. Old, and R. D. Schreiber, “e three Es ofcancer immunoediting,” Annual Review of Immunology, vol. 22,pp. 329–360, 2004.
[2] C. Traversari, P. van der Bruggen, I. F. Luescher et al., “Anonapeptide encoded by human gene MAGE-1 is recognizedon HLA-A1 by cytolytic T lymphocytes directed against tumorantigen MZ2-E,” Journal of Experimental Medicine, vol. 176, no.5, pp. 1453–1457, 1992.
[3] J. H. Kessler and C. J. M. Melief, “Identi�cation of T-cellepitopes for cancer immunotherapy,” Leukemia, vol. 21, no. 9,pp. 1859–1874, 2007.
[4] S. Müller, K. Alving, J. Peter-Katalinic, N. Zachara, A. A.Gooley, and F. G. Hanisch, “High density O-glycosylation ontandem repeat peptide from secretory MUC1 of T47D breastcancer cells,” e Journal of Biological Chemistry, vol. 274, no.26, pp. 18165–18172, 1999.
[5] D. W. Kufe, “Functional targeting of the MUC1 oncogen inhuman cancers,” Cancer Biology and erapy, vol. 8, no. 13, pp.1197–1203, 2009.
[6] D. Kufe, “Oncogenic function of the MUC1 receptor subunit ingene regulation,”Oncogene, vol. 29, no. 42, pp. 5663–5666, 2010.
[7] S. J. Gendler, C. A. Lancaster, J. Taylor-Papadimitriou et al.,“Molecular cloning and expression of human tumor-associatedpolymorphic epithelial mucin,” e Journal of Biological Chem-istry, vol. 265, no. 25, pp. 15286–15293, 1990.
[8] M. S. Lan, S. K. Batra, W. N. Qi, R. S. Metzgar, and M. A.Hollingsworth, “Cloning and sequencing of a human pancreatictumor mucin cDNA,” e Journal of Biological Chemistry, vol.265, no. 25, pp. 15294–15299, 1990.
[9] J. Burchell, S. Gendler, J. Taylor-Papadimitriou et al., “Develop-ment and characterization of breast cancer reactivemonoclonalantibodies directed to the core protein of the human milkmucin,” Cancer Research, vol. 47, no. 20, pp. 5476–5482, 1987.
[10] A. Girling, J. Bartkova, J. Burchell, S. Gendler, C. Gillett,and J. Taylor-Paradimitriou, “A core protein epitope of thepolymorphic epithelial mucin detected by the monoclonalantibody SM-3 is selectively exposed in a range of primary
carcinomas,” International Journal of Cancer, vol. 43, no. 6, pp.1072–1076, 1989.
[11] H. Takeuchi, K. Kato, K. Denda-Nagai, F. G. Hanisch, H.Clausen, and T. Irimura, “e epitope recognized by the uniqueanti-MUC1 monoclonal antibody MY.1E12 involves sialyl𝛼𝛼2-3galactosyl𝛽𝛽1-3N-acetylgalactosaminide linked to a distinctthreonine residue in the MUC1 tandem repeat,” Journal ofImmunological Methods, vol. 270, no. 2, pp. 199–209, 2002.
[12] K. O. Lloyd, J. Burchell, V. Kudryashov, B. W. T. Yin, and J.Taylor-Papadimitriou, “Comparison of O-linked carbohydratechains in MUC-1 mucin from normal breast epithelial cell linesand breast carcinoma cell lines: demonstration of simpler andfewer glycan chains in tumor cells,” e Journal of BiologicalChemistry, vol. 271, no. 52, pp. 33325–33334, 1996.
[13] G. F. Springer, “T and Tn, general carcinoma autoantigens,”Science, vol. 224, no. 4654, pp. 1198–1206, 1984.
[14] D. L. Barnd, M. S. Lan, R. S. Metzgar, and O. J. Finn, “Speci�c,major histocompatibility complex-unrestricted recognition oftumor-associated mucins by human cytotoxic T cells,” Proceed-ings of the National Academy of Sciences of the United States ofAmerica, vol. 86, no. 18, pp. 7159–7163, 1989.
[15] C. G. Ioannides, B. Fisk, K. R. Jerome, T. Irimura, J. T.Wharton, and O. J. Finn, “Cytotoxic T cells from ovarianmalignant tumors can recognize polymorphic epithelial mucincore peptides,” Journal of Immunology, vol. 151, no. 7, pp.3693–3703, 1993.
[16] T. Takahashi, Y. Makiguchi, Y. Hinoda et al., “Expression ofMUC1 on myeloma cells and induction of HLA-unrestrictedCTL against MUC1 from a multiple myeloma patient,” Journalof Immunology, vol. 153, no. 5, pp. 2102–2109, 1994.
[17] H. Noto, T. Takahashi, Y. Makiguchi, T. Hayashi, Y. Hinoda,and K. Imai, “Cytotoxic T lymphocytes derived from bonemarrowmononuclear cells of multiple myeloma patients recog-nize an underglycosylated form of MUC1 mucin,” InternationalImmunology, vol. 9, no. 5, pp. 791–798, 1997.
[18] K. R. Jerome, D. L. Barnd, K. M. Bendt et al., “Cytotoxic T-lymphocytes derived frompatients with breast adenocarcinomarecognize an epitope present on the protein core of a mucinmolecule preferentially expressed by malignant cells,” CancerResearch, vol. 51, no. 11, pp. 2908–2916, 1991.
[19] Y. Hinoda, T. Takahashi, T. Hayashi et al., “Enhancementof reactivity of anti-MUC1 core protein antibody and killingactivity of anti-MUC1 cytotoxic T cells by deglycosylation oftarget tissues or cells,” Journal of Gastroenterology, vol. 33, no.2, pp. 164–171, 1998.
[20] N. Domenech, R. A. Henderson, and O. J. Finn, “Identi�cationof an HLA-A11-restricted epitope from the tandem repeatdomain of the epithelial tumor antigen mucin,” Journal ofImmunology, vol. 155, no. 10, pp. 4766–4774, 1995.
[21] V. Apostolopoulos, V. Karanikas, J. S. Haurum, and I. F. C.McKenzie, “Induction ofHLA-A2-restrictedCTLs to themucin1 human breast cancer antigen,” Journal of Immunology, vol.159, no. 11, pp. 5211–5218, 1997.
[22] P. Brossart, K. S. Heinrich, G. Stuhler et al., “Identi�cationof HLA-A2-restricted T-cell epitopes derived from the MUC1tumor antigen for broadly applicable vaccine therapies,” Blood,vol. 93, no. 12, pp. 4309–4317, 1999.
[23] P. Brossart, A. Schneider, P. Dill et al., “e epithelial tumorantigen MUC1 is expressed in hematological malignanciesand is recognized by MUC1-speci�c cytotoxic T-lymphocytes,”Cancer Research, vol. 61, no. 18, pp. 6846–6850, 2001.

8 BioMed Research International
[24] D. Roulois, V. Vignard, F. Gueugnon et al., “Recognition ofpleural mesothelioma by mucin-1(950–958)/human leukocyteantigen A∗0201-speci�c CD8+ T-cells,” European RespiratoryJournal, vol. 38, no. 5, pp. 1117–1126, 2011.
[25] T. Ninkovic, L. Kinarsky, K. Engelmann et al., “Identi�cation ofO-glycosylated decapeptides within the MUC1 repeat domainas potential MHC class I (A2) binding epitopes,” MolecularImmunology, vol. 47, no. 1, pp. 131–140, 2009.
[26] E. M. Hiltbold, P. Ciborowski, and O. J. Finn, “Naturallyprocessed class II epitope from the tumor antigen MUC1primes human CD4+ T cells,” Cancer Research, vol. 58, no. 22,pp. 5066–5070, 1998.
[27] E. M. Hiltbold, M. D. Alter, P. Ciborowski, and O. J. Finn,“Presentation ofMUC1 tumor antigen by class IMHC andCTLfunction correlate with the glycosylation state of the proteintaken up by dendritic cells,” Cellular Immunology, vol. 194, no.2, pp. 143–149, 1999.
[28] A. M. Vlad, S. Muller, M. Cudic et al., “Complex carbohydratesare not removed during processing of glycoproteins by dendriticcells: processing of tumor antigen MUC1 glycopeptides forpresentation to major histocompatibility complex class II-restricted T cells,” Journal of Experimental Medicine, vol. 196,no. 11, pp. 1435–1446, 2002.
[29] C. Napoletano, A. Rughetti, M. P. Agervig Tarp et al., “Tumor-associated Tn-MUC1 glycoform is internalized through themacrophage galactose-type C-type lectin and delivered to theHLA class I and II compartments in dendritic cells,” CancerResearch, vol. 67, no. 17, pp. 8358–8367, 2007.
[30] E. M. Hiltbold, A. M. Vlad, P. Ciborowski, S. C. Watkins, andO. J. Finn, “e mechanism of unresponsiveness to circulatingtumor antigen MUC1 is a block in intracellular sorting andprocessing by dendritic cells,” Journal of Immunology, vol. 165,no. 7, pp. 3730–3741, 2000.
[31] T. Ninkovic and F. G. Hanisch, “O-glycosylated human MUC1repeats are processed in vitro by immunoproteasomes,” Journalof Immunology, vol. 179, no. 4, pp. 2380–2388, 2007.
[32] N. Peat, S. J. Gendler, E. N. Lalani, T. Duhig, and J. Taylor-Papadimitriou, “Tissue-speci�c expression of a human poly-morphic epithelial mucin (MUC1) in transgenic mice,” CancerResearch, vol. 52, no. 7, pp. 1954–1960, 1992.
[33] M. M. Soares, V. Mehta, and O. J. Finn, “ree differentvaccines based on the 140-amino acid MUC1 peptide withseven tandemly repeated tumor-speci�c epitopes elicit distinctimmune effector mechanisms in wild-type versus MUC1-transgenic mice with different potential for tumor rejection,”Journal of Immunology, vol. 166, no. 11, pp. 6555–6563, 2001.
[34] K. G. Kohlgraf, A. J. Gawron, M. Higashi et al., “Tumor-speci�c immunity inMUC1.Tgmice induced by immunizationwith peptide vaccines from the cytoplasmic tail of CD227(MUC1),” Cancer Immunology, Immunotherapy, vol. 53, no. 12,pp. 1068–1084, 2004.
[35] G. J. Rowse, R. M. Tempero, M. L. VanLith, M. A.Hollingsworth, and S. J. Gendler, “Tolerance and immunity toMUC1 in a human MUC1 transgenic murine model,” CancerResearch, vol. 58, no. 2, pp. 315–321, 1998.
[36] R. M. Tempero, G. J. Rowse, S. J. Gendler, and M. A.Hollingsworth, “Passively transferred anti-MUC1 antibodiescause neither autoimmune disorders nor immunity againsttransplanted tumors in MUC1 transgenic mice,” InternationalJournal of Cancer, vol. 80, no. 4, pp. 595–599, 1999.
[37] R. M. Tempero, M. L. VanLith, K. Morikane, G. J. Rowse, S. J.Gendler, andM.A.Hollingsworth, “CD4+ lymphocytes provide
MUC1-speci�c tumor immunity in vivo that is undetectablein vitro and is absent in MUC1 transgenic mice,” Journal ofImmunology, vol. 161, no. 10, pp. 5500–5506, 1998.
[38] R. A. Budiu, I. Diaconu, R. Chrissluis, A. Dricu, R. P. Edwards,and A. M. Vlad, “A conditional mouse model for humanMUC1-positive endometriosis shows the presence of anti-MUC1 antibodies and Foxp3+ regulatory T cells,” DiseaseModels and Mechanisms, vol. 2, no. 11-12, pp. 593–603, 2009.
[39] S. O. Ryan, A. M. Vlad, K. Islam, J. Gariépy, and O. J. Finn,“Tumor-associated MUC1 glycopeptide epitopes are not sub-ject to self-tolerance and improve responses to MUC1 peptideepitopes in MUC1 transgenic mice,” Biological Chemistry, vol.390, no. 7, pp. 611–618, 2009.
[40] S. O. Ryan, M. S. Turner, J. Gariépy, and O. J. Finn, “Tumorantigen epitopes interpreted by the immune system as self orabnormal-self differentially affect cancer vaccine responses,”Cancer Research, vol. 70, no. 14, pp. 5788–5796, 2010.
[41] C. K. Tang, K. C. Sheng, D. Pouniotis et al., “Oxidized andreduced mannan mediated MUC1 DNA immunization induceeffective anti-tumor responses,” Vaccine, vol. 26, no. 31, pp.3827–3834, 2008.
[42] A. M. Farkas and O. J. Finn, “Vaccines based on abnormal self-antigens as tumor-associated antigens: immune regulation,”Seminars in Immunology, vol. 22, no. 3, pp. 125–131, 2010.
[43] T. Deguchi, M. Tanemura, E. Miyoshi et al., “Increasedimmunogenicity of tumor-associated antigen, mucin 1, engi-neered to express 𝛼𝛼-Gal epitopes: a novel approach toimmunotherapy in pancreatic cancer,” Cancer Research, vol. 70,no. 13, pp. 5259–5269, 2010.
[44] R. Kovjazin, I. Volovitz, Y. Kundel et al., “ImMucin: a noveltherapeutic vaccine with promiscuous MHC binding for thetreatment of MUC1-expressing tumors,” Vaccine, vol. 29, no.29-30, pp. 4676–4686, 2011.
[45] D. H. Choi, J. K. Woo, Y. Choi, H. S. Seo, and C. W. Kim, “Anovel chimeric DNA vaccine: enhancement of preventive andtherapeutic efficacy of DNA vaccine by fusion of Mucin 1 to aheat shock protein 70 gene,” Molecular Medicine Reports, vol. 4,no. 5, pp. 885–890, 2011.
[46] J. Chen, H. Y. Li, D. Wang, J. J. Zhao, and X. Z. Guo, “Humandendritic cells transfected with ampli�ed MUC1 mRNA stimu-late cytotoxic T lymphocyte responses against pancreatic cancerin vitro,” Journal of Gastroenterology and Hepatology, vol. 26, no.10, pp. 1509–1518, 2011.
[47] S. E. Wright, I. S. Quinlin, K. A. Rewers-Felkins, K. E. Dom-browski, and C. A. Phillips, “Retention of immunogenicity pro-duced bymucin1 peptides with glycosylation site substitutions,”Immunopharmacology and Immunotoxicology, vol. 32, no. 4, pp.647–655, 2010.
[48] S. Kobukai, G. J. Kremers, J. G. Cobb et al., “Induction ofantitumor immunity by dendritic cells loaded with membrane-translocating mucin 1 peptide antigen,” Translational Oncology,vol. 4, no. 1, pp. 1–8, 2011.
[49] V. Lakshminarayanan, P. ompson, M. A. Wolfert et al.,“Immune recognition of tumor-associated mucin MUC1 isachieved by a fully synthetic aberrantly glycosylated MUC1tripartite vaccine,” Proceedings of the National Academy ofSciences of the United States of America, vol. 109, no. 1, pp.261–266, 2012.
[50] D. Sugiura, S. Aida, K. Denda-nagai et al., “Differential effectormechanisms induced by vaccination with MUC1 DNA in therejection of colon carcinoma growth at orthotopic sites and

BioMed Research International 9
metastases,” Cancer Science, vol. 99, no. 12, pp. 2477–2484,2008.
[51] Y. Choi, Y. H. Jeon, J. Y. Jang, J. K. Chung, and C. W. Kim,“Treatment with mANT2 shRNA enhances antitumor thera-peutic effects induced by MUC1 DNA vaccination,” Molecularerapy, vol. 19, no. 5, pp. 979–989, 2011.
[52] Y. H. Jeon, Y. Choi, H. J. Kim et al., “In vivo bioluminescencevisualization of antitumor effects by humanMUCI vaccination,”Molecular Imaging, vol. 6, no. 5, pp. 297–303, 2007.
[53] C. Trumpeller, M. P. Longhi, M. Caskey et al., “Dendriticcell-targeted protein vaccines: a novel approach to induce T-cell immunity,” Journal of Internal Medicine, vol. 271, no. 2, pp.183–192, 2012.
[54] Y. Rong, D. Jin, W. Wu et al., “Induction of protective and ther-apeutic anti-pancreatic cancer immunity using a reconstructedMUC1 DNA vaccine,” BMC Cancer, vol. 9, article 191, 2009.
[55] D. Massé, F. Ebstein, G. Bougras, J. Harb, K. Me�ah, andM. Grégoire, “Increased expression of inducible HSP70 inapoptotic cells is correlated with their efficacy for antitumorvaccine therapy,” International Journal of Cancer, vol. 111, no.4, pp. 575–583, 2004.
[56] J. Pinkhasov, M. L. Alvarez, M. M. Rigano et al., “Recombinantplant-expressed tumour-associated MUC1 peptide is immuno-genic and capable of breaking tolerance in MUC1.Tg mice,”Plant Biotechnology Journal, vol. 9, no. 9, pp. 991–1001, 2011.
[57] V. Apostolopoulos, G. A. Pietersz, A. Tsibanis et al., “Pilotphase III immunotherapy study in early-stage breast cancerpatients using oxidized mannan-MUC1 [ISRCTN71711835],”Breast Cancer Research, vol. 8, no. 3, article R27, 2006.
[58] R. Ramlau, E. Quoix, J. Rolski et al., “A phase II study of Tg4010(Mva-Muc1-Il2) in association with chemotherapy in patientswith stage III/IV non-small cell lung cancer,” Journal oforacicOncology, vol. 3, no. 7, pp. 735–744, 2008.
[59] R. Dreicer, W. M. Stadler, F. R. Ahmann et al., “MVA-MUC1-IL2 vaccine immunotherapy (TG4010) improves PSA doublingtime in patients with prostate cancer with biochemical failure,”Investigational New Drugs, vol. 27, no. 4, pp. 379–386, 2009.
[60] S. Oudard, O. Rixe, B. Beuselinck et al., “A phase II studyof the cancer vaccine TG4010 alone and in combination withcytokines in patients withmetastatic renal clear-cell carcinoma:clinical and immunological �ndings,” Cancer Immunology,Immunotherapy, vol. 60, no. 2, pp. 261–271, 2011.
[61] E. Quoix, R. Ramlau, V. Westeel et al., “erapeutic vaccinationwith TG4010 and �rst-line chemotherapy in advanced non-small-cell lung cancer: a controlled phase 2B trial,” e LancetOncology, vol. 12, no. 12, pp. 1125–1133, 2011.
[62] F. Ohyanagi, T. Horai, I. Sekine et al., “Safety of BLP25 liposomevaccine (L-BLP25) in Japanese patients with unresectable stageIII nsclc aer primary chemoradiotherapy: preliminary resultsfrom a phase I/II study,” Japanese Journal of Clinical Oncology,vol. 41, no. 5, Article ID hyr021, pp. 718–722, 2011.
[63] C. Butts, A. Maksymiuk, G. Goss et al., “Updated survivalanalysis in patients with stage IIIB or IV non-small-cell lungcancer receiving BLP25 liposome vaccine (L-BLP25): phase IIBrandomized, multicenter, open-label trial,” Journal of CancerResearch and Clinical Oncology, vol. 137, no. 9, pp. 1337–1342,2011.
[64] Y. L.Wu, K. Park, R. A. Soo et al., “INSPIRE: a phase III study ofthe BLP25 liposome vaccine (L-BLP25) in Asian patients withunresectable stage III non-small cell lung cancer,” BMC Cancer,vol. 11, artcle 430, 2011.
[65] C. Butts, R. N. Murray, C. Smith et al., “A multicenter open-label study to assess the safety of a new formulation of BLP25liposome vaccine in patients with unresectable stage III non-small-cell lung cancer,” Clinical Lung Cancer, vol. 11, no. 6, pp.391–395, 2010.
[66] S. E. Wright, K. A. Rewers-Felkins, I. S. Quinlin et al., “Tumorburden in�uences cytotoxic T cell development in metastaticbreast cancer patients�a phase III study tumor burden in�u-ences CTL development,” Immunological Investigations, vol. 38,no. 8, pp. 820–838, 2009.
[67] A. J. Lepisto, A. J. Moser, H. Zeh et al., “A phase I/II studyof a MUC1 peptide pulsed autologous dendritic cell vaccine asadjuvant therapy in patientswith resected pancreatic and biliarytumors,” Cancer erapy, vol. 6, no. B, pp. 955–964, 2008.
[68] H. Kondo, S. Hazama, T. Kawaoka et al., “Adoptiveimmunotherapy for pancreatic cancer using MUC1 peptide-pulsed dendritic cells and activated T lymphocytes,” AnticancerResearch, vol. 28, no. 1B, pp. 379–387, 2008.
[69] M. J. Dobrzanski, K. A. Rewers-Felkins, K. A. Samad et al.,“Immunotherapy with IL-10- and IFN-gamma-producing CD4effector cells modulate, “Natural” and, “Inducible” CD4 TRegcell subpopulation levels: observations in four cases of patientswith ovarian cancer,” Cancer Immunology, Immunotherapy, vol.61, no. 6, pp. 839–854, 2012.
[70] M. Mohebtash, K. Y. Tsang, R. A. Madan et al., “A pilot studyof MUC-1/CEA/TRICOM poxviral-based vaccine in patientswith metastatic breast and ovarian cancer,” Clinical CancerResearch, vol. 17, no. 22, pp. 7164–7173, 2011.
[71] N. K. Ibrahim, K. O. Yariz, I. Bondarenko et al., “Randomizedphase II trial of letrozole plus anti-MUC1 antibody AS1402in hormone receptor-positive locally advanced or metastaticbreast cancer,” Clinical Cancer Research, vol. 17, no. 21, pp.6822–6830, 2011.
[72] M. D. Pegram, V. F. Borges, N. Ibrahim et al., “Phase I doseescalation pharmacokinetic assessment of intravenous human-ized anti-MUC1 antibody AS1402 in patients with advancedbreast cancer,” Breast Cancer Research, vol. 11, no. 5, article R73,2009.
[73] S. M. Rittig, M. Haentschel, K. J. Weimer et al., “Intradermalvaccinations with RNA coding for TAA generate CD8+ andCD4+ immune responses and induce clinical bene�t in vacci-nated patients,” Molecular erapy, vol. 19, no. 5, pp. 990–999,2011.
[74] S. Sharma, M. K. Srivastava, M. Harris-White, J. M. Lee, and S.Dubinett, “MUC1 peptide vaccine mediated antitumor activityin non-small cell lung cancer,” Expert Opinion on Biologicalerapy, vol. 11, no. 8, pp. 987–990, 2011.
[75] R. Sangha and S. North, “L-BLP25: a MUC1-targeted peptidevaccine therapy in prostate cancer,”Expert Opinion onBiologicalerapy, vol. 7, no. 11, pp. 1723–1730, 2007.
[76] M. Palmer, J. Parker, S. Modi et al., “Phase I study of theBLP25 (MUC1 peptide) liposomal vaccine for active speci�cimmunotherapy in stage IIIB/IV non-small-cell lung cancer,”Clinical Lung Cancer, vol. 3, no. 1, pp. 49–57, 2001.
[77] T. Kawaoka, M. Takashima, K. Yamamoto, T. Ueno, and M.Oka, “Adoptive immunotherapy using MUC1�speci�c CTLsfor unresectable pancreatic cancer,” Nippon Rinsho, vol. 64,supplement 1, pp. 279–282, 2006.
[78] A. J. Lepisto, A. J. Moser, H. Zeh et al., “A phase I/II studyof a MUC1 peptide pulsed autologous dendritic cell vaccine asadjuvant therapy in patientswith resected pancreatic and biliarytumors,” Cancer erapy, vol. 6, no. B, pp. 955–964, 2008.

10 BioMed Research International
[79] S. E. Wright, K. A. Rewers-Felkins, I. S. Quinlin et al., “Tumorburden in�uences cytotoxic T cell development in metastaticbreast cancer patients—a phase I/II study,” ImmunologicalInvestigations, vol. 38, no. 8, pp. 820–838, 2009.
[80] D. Raina, R. Ahmad, H. Rajabi et al., “Targeting cysteine-mediated dimerization of the MUC1-C oncoprotein in humancancer cells,” International Journal of Oncology, vol. 40, no. 5,pp. 1643–1649, 2012.
[81] D. Raina, M. Kosugi, R. Ahmad et al., “Dependence on theMUC1-C oncoprotein in non-small cell lung cancer cells,”Molecular Cancererapeutics, vol. 10, no. 5, pp. 806–816, 2011.
[82] M. J. Brayman, N. Dharmaraj, E. Lagow, and D. D. Carson,“MUC1 expression is repressed by protein inhibitor of activatedsignal transducer and activator of transcription-y,” MolecularEndocrinology, vol. 21, no. 11, pp. 2725–2737, 2007.
[83] D. Roulois, C. Blanquart, C. Panterne et al., “Downregulationof MUC1 expression and its recognition by CD8+ T cells onthe surface ofmalignant pleuralmesothelioma cells treatedwithHDACi,” European Journal of Immunology, vol. 42, no. 3, pp.783–789, 2012.

Hindawi Publishing CorporationBioMed Research InternationalVolume 2013, Article ID 924023, 12 pageshttp://dx.doi.org/10.1155/2013/924023
Research ArticleCpG and Interleukin-15 Synergize to Enhance IFN-𝛾𝛾𝛾𝛾 Productionby Activated CD8+ T Cells
Dustin Cobb,1 Siqi Guo,2 and Ronald B. Smeltz1
1 Department of Microbiology and Immunology, Virginia Commonwealth University, P.O. Box 980678, Richmond, VA 23298, USA2 Department of Microbiology, Old Dominion University, 4211 Monarch Way, Suite 300, Norfolk, VA 23508, USA
Correspondence should be addressed to Ronald B. Smeltz; [email protected]
Received 17 May 2012; Accepted 6 August 2012
Academic Editor: Kim Klonowski
Copyright © 2013 Dustin Cobb et al. is is an open access article distributed under the Creative Commons Attribution License,which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Interleukin-15 (IL-15) regulates the development and maintenance of memory CD8+ T cells. Paradoxically, we previously reportedthat IL-15 could enhance CD8+ T-cell responses to IL-12, a proin�ammatory cytokine required for optimal priming of effectorCD8+ T cells. To expand the physiological relevance of these �ndings, we tested IL-15 for its ability to enhance T-cell responses tobacterial CpG. Expectedly, CpG enhanced the production of IFN-𝛾𝛾𝛾𝛾 by CD8+ T cells polyclonally activated with anti-CD3. However,addition of IL-15 to CpG-stimulated cultures led to a striking increase in IFN-𝛾𝛾𝛾𝛾 production. e effect of CpG and IL-15 was alsoevident with CD8+ T cells recovered from mice infected with the parasite Trypanosoma cruzi (T. cruzi) and restimulated withantigen. e observed synergy between CpG and IL-15 occurred in an IL-12-dependent manner, and this effect could even bedemonstrated in cocultures of activated CD8+ T cells and CD4+CD25+ regulatory T cells. Although IFN-𝛾𝛾𝛾𝛾 was not essential forCpG-induced IL-12, the ability of CpG and IL-15 to act on CD8+ T cells required expression of the IFN-𝛾𝛾𝛾𝛾-inducible transcriptionfactor T-bet. ese data have important implications for development of vaccines and design of therapies to boost CD8+ T-cellresponses to infectious agents and tumors.
1. Introduction
Interleukin-15 (IL-15) is a member of the common 𝛾𝛾𝛾𝛾 chaincytokine family that is important for development andmaintenance of memory CD8+ T cells [1, 2]. IL-15 has shownpromise in its ability to enhance vaccine-mediated immunityagainst pathogens. For example, vaccination against theintracellular protozoan parasiteTrypanosoma cruzi (T. cruzi),the causative agent of human Chagas disease, was improvedwhen combined with a plasmid encoding IL-15 [3]. IL-15can also promote the effector functions of CD8+ T cells[4]. For instance, we previously demonstrated that IL-15enhances IFN-𝛾𝛾𝛾𝛾 production by human CD8+ T cells byincreasing T cell responsiveness to IL-12 [5]. Others havedemonstrated that adoptive immunotherapy of solid tumorswas enhanced by inducing a lymphopenic environment inthe tumor-bearing host prior to adoptive transfer of T cells,and that IL-15-mediated lymphopenia-induced proliferation(as well as proin�ammatory cytokines released in response
to total body irradiation) was an important component ofeffective therapy [6].
CpG motifs are unmethylated dinucleotides that arepresent in bacterial DNA, and they were previously dis-covered to possess powerful immunostimulatory properties[7, 8]. Binding of CpG to Toll-like receptor 9 (TLR-9) onantigen-presenting cells (APCs) induces APC maturationthrough up-regulation of MHC class II, and the costimula-tory molecules CD40 and CD86 [9, 10]. CpG can also stim-ulate production of proin�ammatory cytokines, particularlyIL-12, which promotes T-cell priming and differentiation[11]. us, CpG has the potential to enhance both innateand adaptive immunity and represent a powerful agent thatcan be used as an adjuvant for vaccine-induced immunity.Indeed, the immunostimulatory properties of bacterial CpGhave been recapitulated by the use of synthetic oligodeoxynu-cleotides (ODNs). For example, synthetic CpG have beenshown to increase both natural and vaccine-induced immuneresponses to T. cruzi [12].

2 BioMed Research International
Based on our previous studies with IL-15, and to examinethe efficacy of IL-15 in a clinically relevantmanner, we soughtto determine as a proof-of-concept if IL-15 could synergizewith CpG to enhance CD8+ T-cell responses. We reportthat the combination of IL-15 and CpG led to a signi�cantincrease in IFN-𝛾𝛾𝛾𝛾 production by CD8+ T cells, includingthe enhancement of IFN-𝛾𝛾𝛾𝛾 production by T. cruzi-speci�cCD8+ T cells in an antigen-speci�c manner. Mechanistically,the observed synergy between IL-15 and CpG was criticallydependent uponAPC-derived IL-12.epotency of the com-bined effect of CpG-induced IL-12 and IL-15was also evidentin cocultures of CD8+ T cells and naturally occurring (i.e.,thymus-derived), CD4+CD25+ regulatory T cells (nTreg),which are well known for their potent ability to suppressT-cell activation. Impressively, both proliferation and IFN-𝛾𝛾𝛾𝛾 production were markedly enhanced by these cytokineseven in the presence of high numbers of nTreg. AlthoughIFN-𝛾𝛾𝛾𝛾was not essential for CpG-induced IL-12, the ability ofCpG and IL-15 to act on CD8+ T cells required expressionof the IFN-𝛾𝛾𝛾𝛾-inducible transcription factor T-bet. eseresults have important implications for future developmentof preventative vaccines that combine the potency of TLRagonists such as CpGwith cytokines known to promote long-term memory.
2. Materials and Methods
2.1. Mice and Infections. Age-matched female C57BL/6,Tbx21−/−, B6.129 IL12p35−/−, and Ifng−/−micewere obtainedfrom e Jackson Laboratory and were used between six andeight weeks of age. Mice were housed in an Association forAssessment and Accreditation of Laboratory Animal Care(AAALAC)- accredited facility under pathogen-free condi-tions and used in accordance with an Institutional AnimalCare and Use Committee- (IACUC)- approved protocol. Forinfections, mice were injected intraperitoneally with 1 × 106tissue-culture-derived T. cruzi trypomastigotes (CL strain).
2.2. �eagents and Cell Puri�cations. Recombinant human IL-15 was purchased from R&D Systems and used at 1 ng/mL,10 ng/mL, or 100 ng/mL. CpG ODN 231627 was purchasedfrom TIB MolBiol. For antigen-speci�c ex vivo recall re-sponses to T. cruzi, Tskb20 peptide was used. Tskb20 pep-tide (ANYKFTLV) was synthesized by and purchased fromAbgent (San Diego, CA). For in vitro neutralization of IFN-𝛾𝛾𝛾𝛾and IL-12, LEAF-puri�ed anti-IFN-𝛾𝛾𝛾𝛾 (clone XMG1.2, Biole-gend) and anti-IL-12p40 (clone C17.8, Biolegend) antibodieswere used at a concentration of 5 𝜇𝜇𝜇𝜇g/mL. CD8+ T cells wereisolated from naïve and T. cruzi-infected mice by positiveselection using CD8 Microbeads (MACS, Miltenyi) andpurity was greater than 95%.y1.2− splenocytes were isolat-ed by depleting y1.2+ cells from naïve or T. cruzi-infectedmice using CD90.2 Microbeads and LD columns (MACS,Miltenyi) and purities were greater than 98%. CD8+ T cellswere activated with soluble anti-CD3 (0.1𝜇𝜇𝜇𝜇g/mL, 145-2C11).CD4+CD25+ nTreg were prepared from pooled lymph nodesof naïve mice using Treg isolation kits (Miltenyi Biotec).Purity of nTreg populations (CD4+CD25+) was routinely
checked by FACS and greater than 93% and puri�ed nTregexpressed Foxp3 as determined by intracellular FACS.
2.3. In Vitro and Ex Vivo Cell Cultures. 2 × 105 CD8+ T cellsfrom naïve mice were cocultured with 2 × 106 live y1.2-depleted splenocytes in complete RPMI 1640 (10% FBS,10mM Hepes, 1 IU/mL penicillin and 100𝜇𝜇𝜇𝜇g/mL strepto-mycin, 2mML-glutamine, 1×10−5M2-𝛽𝛽𝛽𝛽-mercaptoethanol).Cells were stimulated with 0.1 𝜇𝜇𝜇𝜇g/mL of LEAF-puri�ed anti-CD3 in the presence of 0.5𝜇𝜇𝜇𝜇M CpG and recombinant humanIL-15 for 72 hours. For antigen-speci�c responses, 2 × 106splenocytes from T. cruzi-infected mice were cultured withTskb20 peptide (1 ng/mL) for 72 hours.
2.4. CFSE Labeling. Subsequent to column puri�cation,CD8+ T cells were labeled with CFSE by incubating T cellsat 100 × 106 cells/mL in 5 𝜇𝜇𝜇𝜇M of CFSE/HBSS for 5 minutes atroom temperature. Labeling was quenched by addition of anequal volume of fetal bovine serum. Cells were washed andresuspended in complete RPMI.
2.5. Coculture Assay with Treg. Cocultures of CFSE-labeledCD8+ T cells, APC, and Treg were established by adding2.5 × 105 CD8+ T cells, 1 × 106 APC, and decreasingnumbers of Treg (starting with a 1 : 1 suppressor : responderratio) per mL of complete RPMI to wells of 48-well plates.Low-endotoxin/azide-free anti-CD3 antibody (LEAF, 2C11,Biolegend) was used at 0.5 𝜇𝜇𝜇𝜇g/mL. In addition, recombinanthuman IL-15 (R&D Systems) and recombinant mouse IL-12(R&D Systems) were used at 1 ng/mL and 0.1 ng/mL, respec-tively. Aer 72 hours, cells were harvested and supernatantsstored frozen at −80∘C until use. Cells were stained with anti-CD8 allophycocyanin (Biolegend), and 30,000 live eventswere analyzed on a Beckman-Coulter FC500 instrument.�uanti�cation of cell division was determined by gating oneither CFSE+ cells or CD8+ T cells and determining thefrequency of cells in each gate/CFSE peak. Supernatants weretested for IFN-𝛾𝛾𝛾𝛾 as described below.
2.6. Measurement of Cytokines. Cell culture supernatantswere collected aer 72 hours, or in some cases aer 24 hours,and analyzed for the presence of IFN-𝛾𝛾𝛾𝛾 and IL-12p40 usingBiolegend’s ELISA MAX Standard Sets according to theirrecommendations.
2.7. Statistical Analysis. Data were analyzed using one-wayANOVA, Tukey multiple comparison procedures (SigmaPlot11.0, Systat Soware, Inc.). A 𝑃𝑃𝑃𝑃 value <0.05 was consideredsigni�cant. Data are represented asmeans±SD of experimen-tal groups.
3. Results
3.1. IL-15 and CpG Synergize to Enhance IFN-𝛾𝛾𝛾𝛾 Productionby CD8+ T Cells. Previous studies have demonstrated theimportance of IL-15 in regulating the development andfunction of memory CD8+ T cells. Additionally, we reported
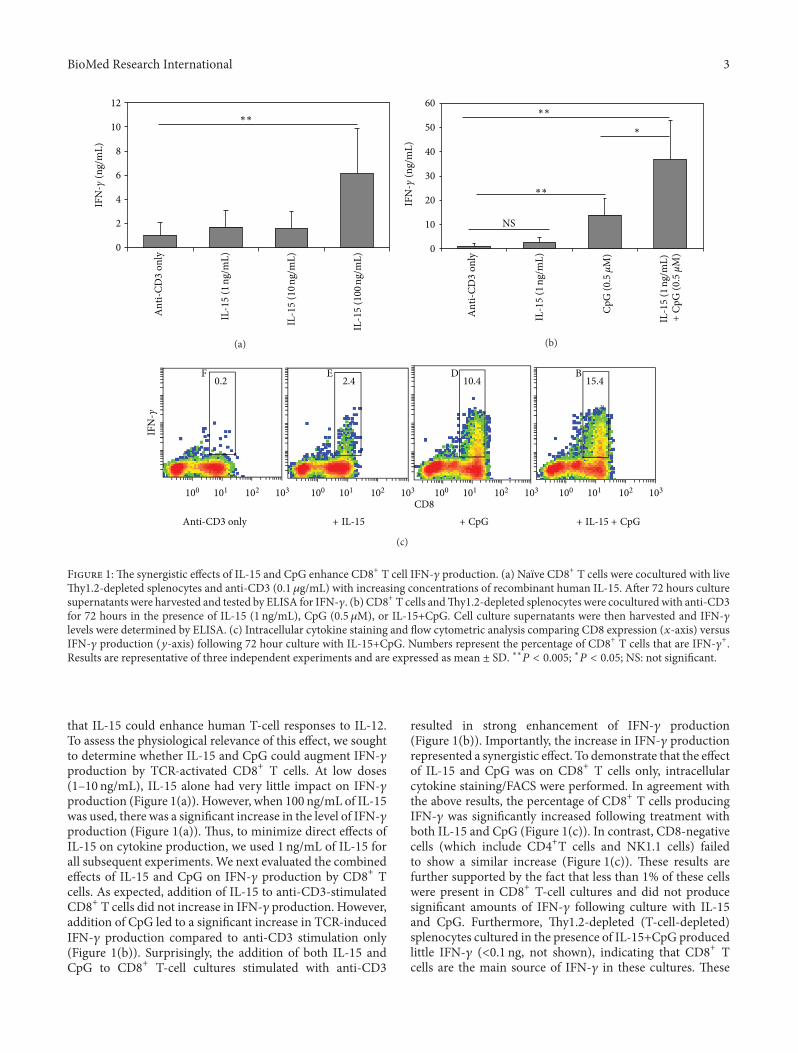
BioMed Research International 3
0
2
4
6
8
10
12IF
N-
(ng/
mL
)
∗∗
An
ti-C
D3
on
ly
IL-1
5 (1
ng/
mL
)
IL-1
5 (1
0 n
g/m
L)
IL-1
5 (1
00 n
g/m
L)
(a)
0
10
20
30
40
50
60
An
ti-C
D3
on
ly
+
IFN
-(n
g/m
L)
∗∗
∗∗
∗
NS
IL-1
5 (1
ng/
mL
)
IL-1
5 (1
ng/
mL
)C
pG
(0.
5 M
)
Cp
G (
0.5
M)
(b)
Anti-CD3 only
IFN
-
0.2F E D
CD8
+ IL-15 + CpG + IL-15 + CpG
2.4 10.4 15.4B
100 101 102 103 100 101 102 103 100 101 102 103 100 101 102 103
(c)
F 1: e synergistic effects of IL-15 and CpG enhance CD8+ T cell IFN-𝛾𝛾𝛾𝛾 production. (a) Naïve CD8+ T cells were cocultured with livey1.2-depleted splenocytes and anti-CD3 (0.1 𝜇𝜇𝜇𝜇g/mL) with increasing concentrations of recombinant human IL-15. Aer 72 hours culturesupernatants were harvested and tested by ELISA for IFN-𝛾𝛾𝛾𝛾. (b) CD8+ T cells andy1.2-depleted splenocytes were coculturedwith anti-CD3for 72 hours in the presence of IL-15 (1 ng/mL), CpG (0.5 𝜇𝜇𝜇𝜇M), or IL-15+CpG. Cell culture supernatants were then harvested and IFN-𝛾𝛾𝛾𝛾levels were determined by ELISA. (c) Intracellular cytokine staining and �ow cytometric analysis comparing CD8 expression (𝑥𝑥𝑥𝑥-axis) versusIFN-𝛾𝛾𝛾𝛾 production (𝑦𝑦𝑦𝑦-axis) following 72 hour culture with IL-15+CpG. Numbers represent the percentage of CD8+ T cells that are IFN-𝛾𝛾𝛾𝛾+.Results are representative of three independent experiments and are expressed as mean ± SD. ∗∗𝑃𝑃𝑃𝑃 𝑃𝑃 𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃; ∗𝑃𝑃𝑃𝑃 𝑃𝑃 𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃; NS: not signi�cant.
that IL-15 could enhance human T-cell responses to IL-12.To assess the physiological relevance of this effect, we soughtto determine whether IL-15 and CpG could augment IFN-𝛾𝛾𝛾𝛾production by TCR-activated CD8+ T cells. At low doses(1–10 ng/mL), IL-15 alone had very little impact on IFN-𝛾𝛾𝛾𝛾production (Figure 1(a)). However, when 100 ng/mL of IL-15was used, there was a signi�cant increase in the level of IFN-𝛾𝛾𝛾𝛾production (Figure 1(a)). us, to minimize direct effects ofIL-15 on cytokine production, we used 1 ng/mL of IL-15 forall subsequent experiments. We next evaluated the combinedeffects of IL-15 and CpG on IFN-𝛾𝛾𝛾𝛾 production by CD8+ Tcells. As expected, addition of IL-15 to anti-CD3-stimulatedCD8+ T cells did not increase in IFN-𝛾𝛾𝛾𝛾 production. However,addition of CpG led to a signi�cant increase in TCR-inducedIFN-𝛾𝛾𝛾𝛾 production compared to anti-CD3 stimulation only(Figure 1(b)). Surprisingly, the addition of both IL-15 andCpG to CD8+ T-cell cultures stimulated with anti-CD3
resulted in strong enhancement of IFN-𝛾𝛾𝛾𝛾 production(Figure 1(b)). Importantly, the increase in IFN-𝛾𝛾𝛾𝛾 productionrepresented a synergistic effect. To demonstrate that the effectof IL-15 and CpG was on CD8+ T cells only, intracellularcytokine staining/FACS were performed. In agreement withthe above results, the percentage of CD8+ T cells producingIFN-𝛾𝛾𝛾𝛾 was signi�cantly increased following treatment withboth IL-15 and CpG (Figure 1(c)). In contrast, CD8-negativecells (which include CD4+T cells and NK1.1 cells) failedto show a similar increase (Figure 1(c)). ese results arefurther supported by the fact that less than 1% of these cellswere present in CD8+ T-cell cultures and did not producesigni�cant amounts of IFN-𝛾𝛾𝛾𝛾 following culture with IL-15and CpG. Furthermore, y1.2-depleted (T-cell-depleted)splenocytes cultured in the presence of IL-15+CpG producedlittle IFN-𝛾𝛾𝛾𝛾 (𝑃𝑃0.1 ng, not shown), indicating that CD8+ Tcells are the main source of IFN-𝛾𝛾𝛾𝛾 in these cultures. ese

4 BioMed Research International
IFN
-(n
g/m
L)
120
100
80
60
40
20
0
No
sti
m.
Tsk
b20
Tsk
b20
+IL
-15
Tsk
b20
+C
pG
∗∗
∗∗
∗
Tsk
b20
+IL
-15
+C
pG
NS
Uninfected
Infected
(a)
IFN
-(n
g/m
L)
No
sti
m.
IL-1
5
Cp
G
IL-1
5+
Cp
G
2
1.5
1
0.5
0
(b)
F 2: e synergistic effects of IL-15 and CpG enhance T. cruzi-speci�c IFN-𝛾𝛾𝛾𝛾 production by CD8+ T cells. C57BL/6 mice were infectedwithTrypanosoma cruzi. On day 9 postinfection,mice were euthanized and spleens harvested. (a) Splenocytes were cultured for 72 hours withTskb20 peptide (1 ng/mL) to induce an antigen-speci�c CD8+ T cell response. Culture supernatants were tested by ELISA to determine thelevels of IFN-𝛾𝛾𝛾𝛾 production. (b) Puri�ed CD8+ T cells were cultured in the presence of IL-15, CpG, or IL-15+CpG for 72 hours. Culturesupernatants were tested by ELISA for IFN-𝛾𝛾𝛾𝛾. ∗∗𝑃𝑃𝑃𝑃 𝑃𝑃 𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃; ∗𝑃𝑃𝑃𝑃 𝑃𝑃 𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃; NS: not signi�cant. Results are representative of at least threeindependent experiments with two mice per group and are expressed as mean ± SD.
results demonstrate that IL-15 can enhance the ability ofCpG to promote IFN-𝛾𝛾𝛾𝛾 production by TCR-activated CD8+T cells, without directly inducing IFN-𝛾𝛾𝛾𝛾. ese data areconsistent with a report in which the combination of IL-15and CpG-enhanced CD69 expression on human T cells [6].
3.2. IL-15 and CpG Can Synergize to Increase the T-Cell IFN-𝛾𝛾𝛾𝛾 Response to the Intracellular Parasite T. cruzi in an Antigen-Speci�c�anner. Because of the potent synergy between CpGand IL-15 in enhancing IFN-𝛾𝛾𝛾𝛾production byT cells polyclon-ally stimulatedwith anti-CD3, we next wanted to determine ifthis effect could be extended to antigen-speci�c CD8+ T cells.To test this, we utilized a murine model of T. cruzi infection.Infection with the intracellular protozoan parasite T. cruzielicits a strong CD8+ T-cell-mediated IFN-𝛾𝛾𝛾𝛾 response thatis necessary for host protection. Previous studies have alsoshown that successful vaccine-induced immunity against T.cruzi induces a strong CD8+ T-cell and IFN-𝛾𝛾𝛾𝛾 response [13–16]. Hence, mice were infected with T. cruzi and splenocytesharvested 9 days postinfection (day 9 p.i.). is time pointwas chosen because we can detect T. cruzi-speci�c CD8+ Tcells in infected mice by FACS on this day [17], and it hasbeen demonstrated to peak at approximately 10–12 d.p.i. [18].Splenocytes from uninfected and T. cruzi-infected mice weresubsequently restimulated with Tskb20, a peptide previouslyidenti�ed as an immunodominant epitope of the CD8+T-cell response to T. cruzi. Addition of IL-15 to Tskb20peptide-stimulated splenocyte cultures did not signi�cantlyincrease IFN-𝛾𝛾𝛾𝛾 production. However, similar to its effects onpolyclonally activated CD8+ T cells from uninfected mice,CpG induced a signi�cant increase in Tskb20 antigen-speci�cIFN-𝛾𝛾𝛾𝛾. Addition of IL-15 to CpG led to a further increase
in IFN-𝛾𝛾𝛾𝛾 production by Tskb20-speci�c CD8+ T cells, albeitnot as signi�cant as that observed with na�ve T cells (Figure2(a)). Since these T cells were derived from infectedmice andhighly activated, we wanted to determine whether IL-15 andCpG could drive IFN-𝛾𝛾𝛾𝛾 production in the absence of TCRstimulation. To test this, CD8+ T cells were puri�ed fromT. cruzi-infected mice and cultured in the presence of IL-15,CpG, or IL-15+CpG.Although the levels of IFN-𝛾𝛾𝛾𝛾productionwere lower without TCR stimulation, the synergistic effectsof IL-15 and CpG were still evident (Figure 2(b)). us,comparable to anti-CD3-stimulated CD8+ T cells, IL-15and CpG can synergize to promote antigen-speci�c IFN-𝛾𝛾𝛾𝛾 production by CD8+ T cells. Of note, IFN-𝛾𝛾𝛾𝛾 levels fromy1.2-depleted (T cell-depleted) splenocytes cultured in thepresence of IL-15/CpGwereminimal (𝑃𝑃1 ng/mL, not shown),indicating aminor contribution of APC to IFN-𝛾𝛾𝛾𝛾 production.
3.3. e Enhancement of T-Cell IFN-𝛾𝛾𝛾𝛾 Production by IL-15and CpG Requires IL-12 Production by Antigen-PresentingCells. Previously it was shown that IL-15 is essential forCpG-induced activation of dendritic cells, including theproduction of IL-12 [19]. erefore, we sought to determinewhether the synergistic effect of IL-15 and CpG was aconsequence of increased IL-12 production by APC. To testthis, APC cultureswere prepared by depletion ofy1.2+ cellsfrom splenocytes obtained from either uninfected orT. cruzi-infected mice, and subsequently cultured with IL-15, CpG,or IL-15 plus CpG. Indeed, stimulation of APC with CpGresulted in a signi�cant increase in IL-12p40 production.Surprisingly, IL-15 alone had no signi�cant effect on IL-12p40 production. Furthermore, the combination of IL-15and CpG did not further increase the levels of IL-12p40 over

BioMed Research International 5
4
3
2
1
0
IL-1
2p40
(n
g/m
L)
Uninfected
Infected
No
sti
m.
IL-1
5
Cp
G
Cp
G+
IL-1
5(a)
50
40
30
20
10
0
An
ti-C
D3
on
ly
IFN
-(n
g/m
L)
IL-1
5
Cp
G
IL-1
5+
Cp
G
IL-1
5+
Cp
G+
iso
typ
e
IL-1
5+
Cp
G+
anti
-IL
-12
NS∗
(b)
IFN
-(n
g/m
L)
120
100
80
60
40
20
0
Wild-tybe
No
sti
m.
Tsk
b20
Tsk
b20
+IL
-15
Tsk
b20
+C
pG
∗∗
Tsk
b20
+IL
-15
+C
pG
IL-12p35−/−
(c)
F 3: IL-12 is required for CpG+IL-15-mediated enhancement of IFN-𝛾𝛾𝛾𝛾 production. (a) Splenocytes were harvested from the spleensof uninfected (�lled bars) or T. cruzi-infected wild-type (open bars) mice on day 9 p.i. y1.2-depleted splenocytes were then cultured inthe presence of IL-15, CpG, or both IL-15 and CpG. Aer 72 hours, culture supernatants were harvested and tested for IL-12p40 by ELISA.(b) CD8+ T cells and live y1.2-depleted APC were cocultured with anti-CD3 for 72 hours in the presence of IL-15, CpG, IL-15+CpG,IL-15+CpG+isotype antibody, or IL-15+CpG+anti-IL-12p40 antibody. Cell culture supernatants were then harvested and IFN-𝛾𝛾𝛾𝛾 levels weredetermined by ELISA. (c) Splenocytes from wild-type (�lled bars) or IL-12p35−/− (open bars) mice infected with T. cruzi were cultured for 72hours with Tskb20 peptide. ∗∗𝑃𝑃𝑃𝑃 𝑃𝑃 𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃; ∗𝑃𝑃𝑃𝑃 𝑃𝑃 𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃; NS: not signi�cant. Results are representative of three to four independent experimentswith at least two mice per group and expressed as mean ± SD.
that of CpG alone (Figure 3(a)). us, these data show thatalthough CpG induces IL-12 secretion, synergy between IL-15 and CpG is not the result of an IL-15-mediated increase inIL-12 production.
Although CpG are known to stimulate IL-12 production,it was important from a mechanistic standpoint to deter-mine if CpG-induced IL-12p40 was in fact required for IL-15+CpG-induced IFN-𝛾𝛾𝛾𝛾 production by CD8+ T cells. Toaddress this issue, we �rst neutralized IL-12p40 in CD8+Tcell cultures stimulated with anti-CD3, CpG, and/or IL-15.Clearly, neutralization of IL-12p40 led to a dramatic reduc-tion in CpG+IL-15-induced IFN-𝛾𝛾𝛾𝛾 production (Figure 3(b)).Since both IL-12 and IL-23 share the IL-12p40 subunit, wewanted to con�rm the importance of APC-derived IL-12in CpG+IL-15-mediated effects on T. cruzi-speci�c CD8+ T
cells. To do so, we used APCs puri�ed from IL12p35−/− mice(which speci�cally lack IL-12). Similar to results shown inFigure 3(b), splenocytes from T. cruzi-infected IL12p35−/−mice failed to exhibit an increase in antigen-speci�c IFN-𝛾𝛾𝛾𝛾production following IL-15 and CpG treatment (Figure 3(c)).Furthermore, experiments revealed that addition of recombi-nant IL-12 recapitulated the effect of CpG (data not shown).us, the ability of IL-15 and CpG to synergize and enhanceIFN-𝛾𝛾𝛾𝛾 production by T. cruzi-speci�c CD8+ T cells is drivenby CpG-induced IL-12 production by APC.
3.4. Synergy between IL-12 and IL-15 Overcomes Potent Sup-pression of CD8+ T-Cell Responses by CD4+CD25+ RegulatoryT Cells. Since IL-12 was the primary mediator of the CpGeffect, we next wanted to determine if IL-12 could synergize

6 BioMed Research International
No Treg
PBS12
8
D
100 101 102 103
FL 1 log
100 101 102 103
FL 1 log
100 101 102 103
FL 1 log
100 101 102 103
FL 1 log
100 101 102 103
FL 1 log
100 101 102 103
FL 1 log
100 101 102 103
FL 1 log
100 101 102 103
FL 1 log
100 101 102 103
FL 1 log
100 101 102 103
FL 1 log
100 101 102 103
FL 1 log
100 101 102 103
FL 1 log
100 101 102 103
FL 1 log
No Treg
-CD3
C
220
220
220
220
220
220
220
220
220
220
220
220
81.6%
81.9%
81.9%
89.8%
89.8%
87.6%
87.6%
92.9%
92.9%
96.1%
96.1%
81.6%
23.8%
26.8%
+ Treg 1:1
-CD3
K
+ Treg 1:2
-CD3
O
33.7%
35.8%
35.8%
46.5%
46.5%
63%
63%
43.5%
43.5%
33.7%
∗∗
∗∗
HL
M
N RJ
Q
P
I
∗∗∗∗∗∗
∗∗∗
∗∗∗
+ IL-15
+ IL-12
+ IL-15 + IL-12
(a)
50
40
30
20
10
0
∗∗∗
An
ti-C
D3
con
tro
l
+T
reg
(1:1
)
+IL
-15
+IL
-12
+IL
-15
+IL
-12
+T
reg
(1:2
)
+IL
-15
+IL
-12
+IL
-15
+IL
-12
CD
8+
T c
ells
wit
h 1
–3
div
isio
ns
(%)
(b)
F 4: Continued.
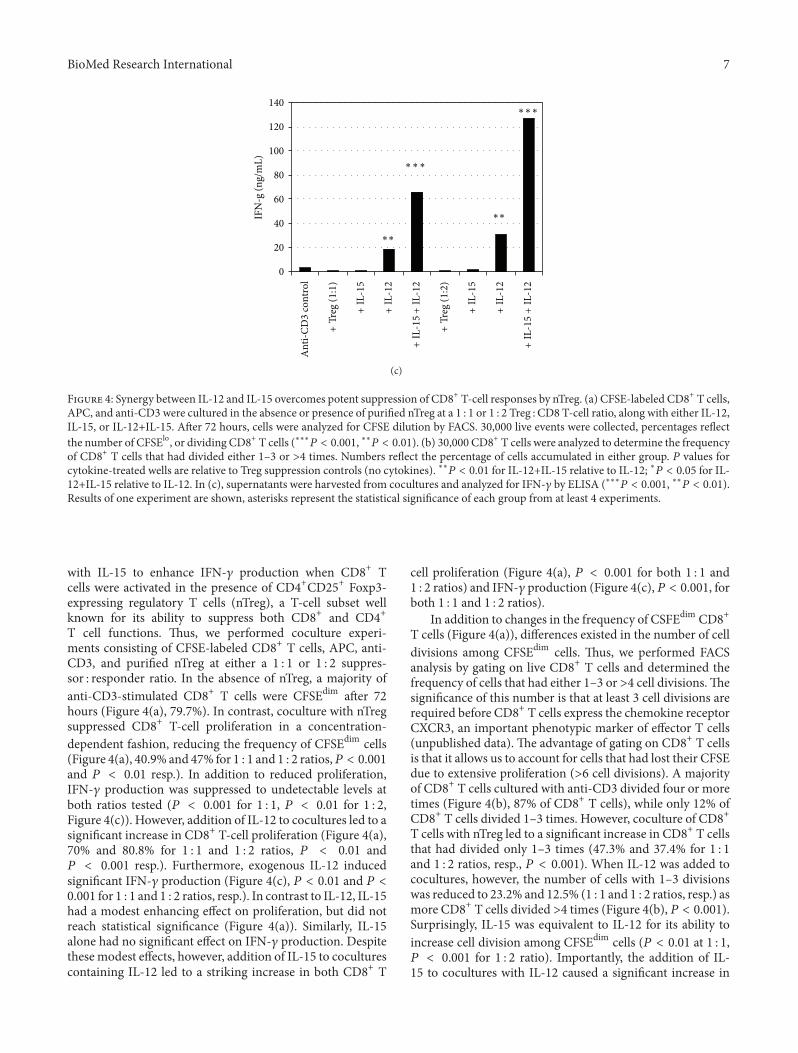
BioMed Research International 7
0
20
40
60
80
100
120
140
An
ti-C
D3
con
tro
l
+T
reg
(1:1
)
+IL
-15
+IL
-12
+IL
-15
+IL
-12
+T
reg
(1:2
)
+IL
-15
+IL
-12
+IL
-15
+IL
-12
∗∗
∗∗
∗∗∗
∗∗∗
IFN
-g (
ng/
mL
)
(c)
F 4: Synergy between IL-12 and IL-15 overcomes potent suppression of CD8+ T-cell responses by nTreg. (a) CFSE-labeled CD8+ T cells,APC, and anti-CD3 were cultured in the absence or presence of puri�ed nTreg at a 1 : 1 or 1 : 2 Treg : CD8 T-cell ratio, along with either IL-12,IL-15, or IL-12+IL-15. Aer 72 hours, cells were analyzed for CFSE dilution by FACS. 30,000 live events were collected, percentages re�ectthe number of CFSElo, or dividing CD8+ T cells (∗∗∗𝑃𝑃𝑃𝑃 𝑃𝑃 𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃, ∗∗𝑃𝑃𝑃𝑃 𝑃𝑃 𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃). (b) 30,000 CD8+ T cells were analyzed to determine the frequencyof CD8+ T cells that had divided either 1–3 or >4 times. Numbers re�ect the percentage of cells accumulated in either group. 𝑃𝑃𝑃𝑃 values forcytokine-treated wells are relative to Treg suppression controls (no cytokines). ∗∗𝑃𝑃𝑃𝑃 𝑃𝑃 𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃 for IL-12+IL-15 relative to IL-12; ∗𝑃𝑃𝑃𝑃 𝑃𝑃 𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃 for IL-12+IL-15 relative to IL-12. In (c), supernatants were harvested from cocultures and analyzed for IFN-𝛾𝛾𝛾𝛾 by ELISA (∗∗∗𝑃𝑃𝑃𝑃 𝑃𝑃 𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃, ∗∗𝑃𝑃𝑃𝑃 𝑃𝑃 𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃).Results of one experiment are shown, asterisks represent the statistical signi�cance of each group from at least 4 experiments.
with IL-15 to enhance IFN-𝛾𝛾𝛾𝛾 production when CD8+ Tcells were activated in the presence of CD4+CD25+ Foxp3-expressing regulatory T cells (nTreg), a T-cell subset wellknown for its ability to suppress both CD8+ and CD4+T cell functions. us, we performed coculture experi-ments consisting of CFSE-labeled CD8+ T cells, APC, anti-CD3, and puri�ed nTreg at either a 1 : 1 or 1 : 2 suppres-sor : responder ratio. In the absence of nTreg, a majority ofanti-CD3-stimulated CD8+ T cells were CFSEdim aer 72hours (Figure 4(a), 79.7%). In contrast, coculture with nTregsuppressed CD8+ T-cell proliferation in a concentration-dependent fashion, reducing the frequency of CFSEdim cells(Figure 4(a), 40.9% and 47% for 1 : 1 and 1 : 2 ratios,𝑃𝑃𝑃𝑃 𝑃𝑃 𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃and 𝑃𝑃𝑃𝑃 𝑃𝑃 𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃 resp.). In addition to reduced proliferation,IFN-𝛾𝛾𝛾𝛾 production was suppressed to undetectable levels atboth ratios tested (𝑃𝑃𝑃𝑃 𝑃𝑃 𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃 for 1 : 1, 𝑃𝑃𝑃𝑃 𝑃𝑃 𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃 for 1 : 2,Figure 4(c)). However, addition of IL-12 to cocultures led to asigni�cant increase in CD8+ T-cell proliferation (Figure 4(a),70% and 80.8% for 1 : 1 and 1 : 2 ratios, 𝑃𝑃𝑃𝑃 𝑃𝑃 𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃 and𝑃𝑃𝑃𝑃 𝑃𝑃 𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃 resp.). Furthermore, exogenous IL-12 inducedsigni�cant IFN-𝛾𝛾𝛾𝛾 production (Figure 4(c), 𝑃𝑃𝑃𝑃 𝑃𝑃 𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃 and 𝑃𝑃𝑃𝑃 𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃 for 1 : 1 and 1 : 2 ratios, resp.). In contrast to IL-12, IL-15had a modest enhancing effect on proliferation, but did notreach statistical signi�cance (Figure 4(a)). Similarly, IL-15alone had no signi�cant effect on IFN-𝛾𝛾𝛾𝛾 production. Despitethese modest effects, however, addition of IL-15 to coculturescontaining IL-12 led to a striking increase in both CD8+ T
cell proliferation (Figure 4(a), 𝑃𝑃𝑃𝑃 𝑃𝑃 𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃 for both 1 : 1 and1 : 2 ratios) and IFN-𝛾𝛾𝛾𝛾 production (Figure 4(c), 𝑃𝑃𝑃𝑃 𝑃𝑃 𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃, forboth 1 : 1 and 1 : 2 ratios).
In addition to changes in the frequency of CSFEdim CD8+T cells (Figure 4(a)), differences existed in the number of celldivisions among CFSEdim cells. us, we performed FACSanalysis by gating on live CD8+ T cells and determined thefrequency of cells that had either 1–3 or >4 cell divisions. esigni�cance of this number is that at least 3 cell divisions arerequired before CD8+ T cells express the chemokine receptorCXCR3, an important phenotypic marker of effector T cells(unpublished data). e advantage of gating on CD8+ T cellsis that it allows us to account for cells that had lost their CFSEdue to extensive proliferation (>6 cell divisions). A majorityof CD8+ T cells cultured with anti-CD3 divided four or moretimes (Figure 4(b), 87% of CD8+ T cells), while only 12% ofCD8+ T cells divided 1–3 times. However, coculture of CD8+T cells with nTreg led to a signi�cant increase in CD8+ T cellsthat had divided only 1–3 times (47.3% and 37.4% for 1 : 1and 1 : 2 ratios, resp., 𝑃𝑃𝑃𝑃 𝑃𝑃 𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃). When IL-12 was added tococultures, however, the number of cells with 1–3 divisionswas reduced to 23.2% and 12.5% (1 : 1 and 1 : 2 ratios, resp.) asmore CD8+ T cells divided >4 times (Figure 4(b), 𝑃𝑃𝑃𝑃 𝑃𝑃 𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃).Surprisingly, IL-15 was equivalent to IL-12 for its ability toincrease cell division among CFSEdim cells (𝑃𝑃𝑃𝑃 𝑃𝑃 𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃 at 1 : 1,𝑃𝑃𝑃𝑃 𝑃𝑃 𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃 for 1 : 2 ratio). Importantly, the addition of IL-15 to cocultures with IL-12 caused a signi�cant increase in

8 BioMed Research International
CD8+ T cell division, with only 5% and 3% of CD8+ T cellswith 1–3 cell divisions (Figure 4(b), 𝑃𝑃𝑃𝑃 𝑃𝑃 𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃, 𝑃𝑃𝑃𝑃 𝑃𝑃 𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃for difference between IL-12 and IL-12+IL-15 at 1 : 1 ratio,𝑃𝑃𝑃𝑃 𝑃𝑃 𝑃𝑃𝑃𝑃𝑃𝑃𝑃𝑃 for difference between IL-12 and IL-12+IL-15 at1 : 2 ratio). ese results demonstrate that synergy betweenIL-12 and IL-15 is effective even in the presence of potentsuppression by nTreg, and that synergy increases not onlythe frequency of CFSEdim cells, but also the number of celldivisions within dividing cells.
3.5. IFN-𝛾𝛾𝛾𝛾 Is Not Required for CpG-Induced IL-12 Produc-tion. Stimulation with CpG, as well as signals provided bymemory CD8+ T cells such as IFN-𝛾𝛾𝛾𝛾, TNF-𝛼𝛼𝛼𝛼, GM-CSF, andCD40L can cooperate in a synergistic fashion to promote IL-12p70 production [20]. For this reason, and because of theimportance of IFN-𝛾𝛾𝛾𝛾 to successful vaccine-induced immuneresponses, we wished to determine if IFN-𝛾𝛾𝛾𝛾 was required forCpG-dependent upregulation of IL-12 by antigen-presentingcells. us, we stimulated y1.2-depleted splenocytes fromnaïve, wild-type, or Ifng−/− mice with CpG for 72 hours andtested supernatants for IL-12p40 protein. Although the levelsof IL-12p40 were decreased in Ifng−/− cultures relative towild-type cultures, there was no statistically signi�cant dif-ference between the groups (Figure 5(a), le). Additionally,y1.2-depleted splenocytes from T. cruzi-infected Ifng−/−mice stimulated with CpG produced as much IL-12p40as wild-type splenocytes (Figure 5(a), right). ese resultssuggest that APC-derived IFN-𝛾𝛾𝛾𝛾 is not absolutely requiredfor CpG-induced IL-12 production but may be necessary foroptimal expression of IL-12. To further address the role ofIFN-𝛾𝛾𝛾𝛾 in CpG-induced IL-12 by APC, we set up coculturesconsisting of y1.2-depleted splenocytes from wild-typeuninfected mice with puri�ed CD8+ T cells derived fromeither wild-type or Ifng−/− mice previously infected withT. cruzi. As expected, the addition of CpG to anti-CD3-stimulated cultures led to a strong induction of IL-12p40.Importantly, CpG-induced IL-12p40 production was notsigni�cantly affectedwhen IFN-𝛾𝛾𝛾𝛾-de�cient CD8+ T cells wereutilized (Figure 5(b)). ese results con�rm that IFN-𝛾𝛾𝛾𝛾 is notrequired for IL-12 production by APC in response to CpGstimulation.
3.6. e Synergistic Effects of IL-15 and CpG Are Dependentupon T-bet Expression. e T-box transcription factor T-bet (Tbx21) is critical for effector and memory CD8+ T-cell function, including IFN-𝛾𝛾𝛾𝛾 production [21]. However,Eomes (Eomesodermin), a related transcription factor, canprovide overlapping and redundant functions with T-bet[22]. Given that Eomes has the potential to compensate forT-bet functions, we wished to determine if the ability of IL-15andCpG to enhance IFN-𝛾𝛾𝛾𝛾 production by CD8+ T cells couldbe maintained in the absence of T-bet. us, CD8+ T cellsfrom T-bet-de�cient (Tbx21−/−) mice were cocultured withAPC in the presence of IL-15+CpG. Surprisingly, however, T-bet-de�ciency in CD8+ T cells had a dramatic consequence,as the ability of IL-15+CpG to synergize and promote IFN-𝛾𝛾𝛾𝛾 production was lost (Figure 6). ese results demonstrate
that synergy between IL-15 and CpG requires the expressionof T-bet and suggests that Eomes cannot compensate for itsabsence.
4. Discussion
Because of their critical role in immunity to tumors, viruses,and intracellular parasites such as T. cruzi [13–16, 23–25], a major goal of vaccine development is to harness thepotency of CD8+ T-cell responses. However, a challenge indeveloping CD8+ T-cell-based vaccines is the generation ofpotent effector T cells as well as long-term memory. Onepromising approach is the incorporation of adjuvants that actas TLR agonists and can augment CD8+ T-cell immunity. Forexample, synthetic CpG ODNs that express unmethylatedCpG motifs have been the focus of much attention fortheir potential use as vaccine adjuvants due to their abilityto promote type I immunity, including antigen-speci�c T-and B-cell responses [26]. Recently, common gamma-chainfamily cytokines such as IL-15 has also been considered ascandidates for improving vaccine responses. While there arenumerous studies showing the bene�cial effects of CpGor IL-15, the potential synergy of these two immune modulatorsremains to be determined.
In this study, our goal was to determine if IL-15 and CpGcould function in a synergistic manner to promote CD8+ T-cell responses. We show that CpG alone had the ability toenhance IFN-𝛾𝛾𝛾𝛾 production by polyclonally activated CD8+ Tcells. However, when CpG and IL-15 were used in combina-tion, there was a profound and synergistic increase in IFN-𝛾𝛾𝛾𝛾. e synergistic effect required only low concentrationsof IL-15 that otherwise could not directly induce IFN-𝛾𝛾𝛾𝛾. Toexpand upon this observation, we wished to determine ifthe synergistic effect could be recapitulated with antigen-speci�c CD8+ T cells. Splenocytes from T. cruzi-infectedmice restimulated with Tskb20 peptide in the presence ofboth CpG and IL-15 showed an increase in IFN-𝛾𝛾𝛾𝛾 productionthat was greater than with CpG alone or IL-15 alone. ese�ndings support the notion that synergy between CpG andIL-15 can enhance antigen-speci�c CD8+ T-cell responses.Although synergy was less dramatic with T cells from T.cruzi-infected mice, we believe the difference is that Tskb20-speci�c T cells, which are generated in vivo in response to anatural infection, have differentiated under highly stimula-tory conditions and may be refractory to further stimulationex vivo. If true, it would suggest that the efficacy of CpGand IL-15 may be better suited for preventative vaccinesas opposed to therapeutic vaccines. Nevertheless, synergybetween CpG and IL-15 was evident when used to stimulateT. cruzi-speci�c CD8+ T cells in the absence of antigen.
Stimulation of APC with CpG promotes their maturationand increases the production of proin�ammatory cytokines,especially IL-12, that favor the development of type I immu-nity [11, 26, 27]. us, we wanted to determine the role ofIL-12 in CpG-mediated synergy with IL-15 on CD8+ T-cellfunction. Not surprisingly, we observed that T-cell-depletedsplenocytes from naïve or T. cruzi-infected mice producedIL-12 in response to stimulation with CpG, whereas IL-15

BioMed Research International 9
5
4
3
2
1
0
IL-1
2p40
(n
g/m
L)
IL-1
2p40
(n
g/m
L)
Wild-type APC Wild-type APC
NS
2
1.6
1.2
0.8
0.4
0IFN-g− /− APCIFN-g− /− APC
(a)
IL-1
2p40
(n
g/m
L)
NS3.5
3
2.5
2
1.5
1
0.5
0
APCs + WT CD8
APCs +
No stim. Anti-CD3 Anti-CD3 + CpG
IFN-g− /− CD8
(b)
F 5: T cell-derived IFN-𝛾𝛾𝛾𝛾 is not required for CpG-induced IL-12 production. (a) y1.2-depleted splenocytes were isolated from thespleens of naïve (le) or T. cruzi-infected (right) wild-type and Ifng −/− mice on day 9 p.i. y1.2-depleted splenocytes were then stimulatedwith CpG. Aer 72 hours, culture supernatants were harvested and tested for IL-12p40 by ELISA. (b) �ild-type (�lled bars) or Ifng −/− (openbars) CD8+ T cells were puri�ed from the spleens of T. cruzi-infected mice on day 9 p.i. and cocultured with wild-type y1.2-depletedAPC for 72 hours in the presence of anti-CD3 or anti-CD3+CpG. Cell culture supernatants were then harvested and IL-12p40 levels weredetermined by ELISA. Results are representative of two independent experiments with two mice per group and are expressed as mean ± SD.NS: not signi�cant.
had no effect on IL-12 production. Importantly, addition ofIL-15 to CpG-stimulated cultures did not lead to a furtherincrease in IL-12, suggesting that the synergy between CpGand IL-15 is not due to IL-15-mediated enhancement of IL-12 production.is observation is in slight contradiction to arecent study in which it was shown that IL-15 is important forCpG-induced IL-12 production by dendritic cells [19]. How-ever, this discrepancy could be explained by the possibilitythat low levels of IL-15 were produced as a result of T. cruziinfection,making it such that additional treatment with IL-15
could not further enhance CpG-induced IL-12 production.To determine the importance of this IL-12, we observed thatneutralization of IL-12 abrogated the synergistic effect onIFN-𝛾𝛾𝛾𝛾 production by CD8+ T cells. Furthermore, antigen-speci�c CD8+ T cells stimulated in the presence of CpG andIL-15 produced signi�cantly less IFN-𝛾𝛾𝛾𝛾 when cultured withIL-12-de�cient APC. us, our results demonstrate that theenhancement of CD8+ T-cell IFN-𝛾𝛾𝛾𝛾 production provided bythe combination of CpG and IL-15 requires CpG-induced IL-12.

10 BioMed Research International
Anti-CD3 only
50
40
30
20
10
0IL-15 CpG IL-15 + CpG
IFN
-(n
g/m
L)
Wild-type
NS
∗
Tbx21− /−
F 6: e combined effects of IL-15 and CpG on CD8+ T cellIFN-𝛾𝛾𝛾𝛾 production require T-bet. Wild-type (�lled bars) or Tbx21−/−(open bars) CD8+ T cells andy1.2-depleted APCwere coculturedwith anti-CD3 for 72 hours in the presence of IL-15, CpG, or IL-15+CpG. Cell culture supernatants were then harvested and IFN-𝛾𝛾𝛾𝛾levels were determined by ELISA. Results are representative of twoindependent experiments and are expressed as means ± SD. ∗𝑃𝑃𝑃𝑃 𝑃𝑃0.05� NS: not signi�cant.
In a previous study, it was reported that memory CD8+T cells are required for APC to produce optimal levels ofIL-12 in response to CpG stimulation [20]. Speci�cally, itwas observed that IFN-𝛾𝛾𝛾𝛾, TNF-𝛼𝛼𝛼𝛼, and GM-CSF produced byCD44hi memory CD8+ T cells synergized with CpG to primedendritic cells for further IL-12 production. For this reason,we examined whether T cell-derived IFN-𝛾𝛾𝛾𝛾 was necessaryfor CpG-induced IL-12 production by APC. However, ourresults indicated that IFN-𝛾𝛾𝛾𝛾, regardless of the source, was notnecessary for APC to produce IL-12 in response to CpG stim-ulation. us, although IFN-𝛾𝛾𝛾𝛾 appeared to be dispensable inour study, it remains possible that TNF-𝛼𝛼𝛼𝛼 and/or GM-CSFfromT. cruzi-speci�cmemoryCD8+ T cells could be primingAPC for CpG-induced IL-12 production. Nevertheless, IL-12is critical for the observed synergistic effects between CpGand IL-15. e �ndings we report here compliment thosereported by Wysocka et al. [6], in which the bene�cial effectsof CpG and IL-15 were observed following the treatment ofhuman NK and CD8+ T cells isolated from patients withcutaneous T-cell lymphoma. Even though analysis of CD8+T cells was limited to upregulation of CD69 expression, CpG,and IL-15-augmented IFN-𝛾𝛾𝛾𝛾 production by peripheral bloodmononuclear cells. Numerous studies have shown that CpGcan increase the immunogenicity of vaccines against cancerand infectious diseases. For instance, CpG can acceleratethe development and enhance the magnitude of vaccine-induced immune responses. In the case of T. cruzi infection,CD8+ T cells are critical for host protection. However,their development is delayed in comparison to other viraland bacterial infections and this delay has been attributedto poor TLR stimulation. Indeed, CpG administration incombination with a TLR-2 agonist signi�cantly acceleratedthe development of CD8+ T-cell responses to T. cruzi [28].us, vaccine approaches against infectious agents will likelybene�t greatly from the immunostimulatory properties ofCpG.
With respect to the contribution of IL-15, we observedthat IL-15 increases cell division among activated CD8+ Tcells, as well as increases cell viability. is is not a trivialpoint. For example, local IL-15 production in the heart ofT. cruzi-infected persons correlates with increased numbersof CD8+ T cells [29]. Furthermore, an important �nding wereport here is that IL-15 can synergize with IL-12, the maineffector of CpG stimulation, to overcome potent suppressionmediated by CD4+CD25+ regulatory T cells (nTreg). ecombination of IL-15 and IL-12 provided a signi�cant boostto both CD8+T-cell proliferation and IFN-𝛾𝛾𝛾𝛾 productiondespite high numbers of Treg. Since Treg function has beenreported to interfere with immune responses to T. cruzi[30, 31], this would be another advantage of using CpG andIL-15 to boost immunity to T. cruzi. We propose that theability of IL-15 to promote CD8+ T-cell survival and celldivision, as well as function in the presence of Treg-mediatedsuppression, contributes to its effectiveness in synergizingwith CpG. It should be noted that although the effectsdescribed above for IL-15 impact CD8+ T cell numbers, itwas recently reported that IL-15 may enhance expression ofthe low-affinity IL-12 receptor 𝛽𝛽𝛽𝛽1 in bothmouse and humans[32, 33].
To identify the T-cell-intrinsic factors that are importantfor the reported effects of CpG and IL-15, we turned ourattention to the transcription factors T-bet and Eomes. T-bet and Eomes are critical for CD8+ T-cell developmentand function [21]. For example, T-bet regulates the gener-ation of antigen-speci�c CD8+ T cells [17] and promoteseffector functions like IFN-𝛾𝛾𝛾𝛾 production [34]. Likewise,Eomes performs similar functions such as promoting theproduction of cytotoxic effector molecules and IFN-𝛾𝛾𝛾𝛾 [22].Here, we demonstrate that T-bet is required for the syner-gistic enhancement of IFN-𝛾𝛾𝛾𝛾 production by CpG and IL-15.Although Eomes can compensate for many of the functionsof T-bet in CD8+ T cells [21], it did not appear to mediatethe synergistic effect of CpG and IL-15 and their ability toenhance IFN-𝛾𝛾𝛾𝛾 production. erefore, T-bet expression inCD8+ T cells is critical for the bene�cial effects of CpG andIL-15 treatment.
In summary, we propose that CpG causes APC to releaseIL-12, a powerful cytokine that serves as “Signal 3” forCD8+ T cells. However, the IL-12 cannot act unless Tcells are activated and express the appropriate receptor torespond to IL-12. IL-15, in addition to its ability to promoteCD8+ T-cell survival and proliferation, may enhance T-cellresponsiveness to IL-12 and thus increase IFN-𝛾𝛾𝛾𝛾. We believethe synergy will enhance both immediate and long-termCD8+ T-cell responses. is has great potential in clinicaltrials, because CpG do not appear to have the same toxicity astreatment with exogenous IL-12, which has been reported incancer patients despite positive therapeutic effects. In termsof IL-15, adoptive transfer of T cells into a lymphopenictumor-bearing host (which typically occurs aer whole bodyirradiation) leads to increased IL-15-dependent proliferation,expansion, and CD8+ T-cell effector function. e resultsof our study provide additional evidence of the potentialefficacy of CpG and IL-15 and how it may improve vaccineapproaches against cancer and infectious diseases. Not only

BioMed Research International 11
do we demonstrate that CpG and IL-15 are more effectivewhen used in combination, but our �ndings also providenew insight into the immunemechanisms responsible for theobserved synergy between CpG and IL-15. Because of theconsiderable interest in TLR agonists as vaccine adjuvants,we believe our study will provide additional evidence thatTLR agonists, when combined with common gamma chaincytokines, have the potential to dramatically enhance vaccineefficacy.
Acknowledgment
is work was supported by funds provided by the omas F.and Kate Miller Jeffress Memorial Trust to R. B. Smeltz.
References
[1] K. S. Schluns, K. Williams, A. Ma, X. X. Zheng, and L.Lefrançois, “Cutting edge: requirement for IL-15 in the gen-eration of primary and memory antigen-speci�c CD8 T cells,”Journal of Immunology, vol. 168, no. 10, pp. 4827–4831, 2002.
[2] M. K. Kennedy, M. Glaccum, S. N. Brown et al., “Reversibledefects in natural killer and memory CD8 T cell lineagesin interleukin 15-de�cient mice,” Journal of ExperimentalMedicine, vol. 191, no. 5, pp. 771–780, 2000.
[3] C. S. Eickhoff, J. R. Vasconcelos, N. L. Sullivan et al., “Co-administration of a plasmid DNA encoding IL-15 improveslong-term protection of a genetic vaccine against Trypanosomacruzi,” PLoS Neglected Tropical Diseases, vol. 5, no. 3, p. e983,2011.
[4] C. A. Klebanoff, S. E. Finkelstein, D. R. Surman et al., “IL-15enhances the in vivo antitumor activity of tumor-reactive CD8+T Cells,” Proceedings of the National Academy of Sciences of theUnited States of America, vol. 101, no. 7, pp. 1969–1974, 2004.
[5] R. B. Smeltz, “Profound enhancement of the IL-12/IL-18pathway of IFN-𝛾𝛾𝛾𝛾 secretion in human CD8+ memory T cellsubsets via IL-15,” Journal of Immunology, vol. 178, no. 8, pp.4786–4792, 2007.
[6] M.Wysocka, B.M. Benoit, S. Newton, L. Azzoni, L. J.Montaner,and A. H. Rook, “Enhancement of the host immune responsesin cutaneous T-cell lymphoma by CpG oligodeoxynucleotidesand IL-15,” Blood, vol. 104, no. 13, pp. 4142–4149, 2004.
[7] H. Wagner, “Bacterial CpG DNA activates immune cells tosignal infectious danger,” Advances in Immunology, no. 73, pp.329–368, 1999.
[8] T. Sparwasser, T. Miethke, G. Lipford et al., “Bacterial DNAcauses septic shock,” Nature, vol. 386, no. 6623, pp. 336–337,1997.
[9] H. Hemmi, O. Takeuchi, T. Kawai et al., “A Toll-like receptorrecognizes bacterial DNA,” Nature, vol. 408, no. 6813, pp.740–745, 2000.
[10] T. Sparwasser, E. S. Koch, R. M. Vabulas et al., “BacterialDNA and immunostimulatory CpG oligonucleotides triggermaturation and activation of murine dendritic cells,” EuropeanJournal of Immunology, vol. 28, no. 6, pp. 2045–2054, 1998.
[11] D. M. Klinman, A. K. Yi, S. L. Beaucage, J. Conover, andA. M. Krieg, “CpG motifs present in bacterial DNA rapidlyinduce lymphocytes to secrete interleukin 6, interleukin 12, andinterferon 𝛾𝛾𝛾𝛾,” Proceedings of the National Academy of Sciences ofthe United States of America, vol. 93, no. 7, pp. 2879–2883, 1996.
[12] D. F. Ho, C. S. Eickhoff, O. K. Giddings, J. R. C. Vas-concelos, and M. M. Rodrigues, “Trans-sialidase recombi-nant protein mixed with CpG motif-containing oligodeoxynu-cleotide induces protective mucosal and systemic Trypanosomacruzi immunity involving CD8+ CTL and B cell-mediatedcross-priming,” Journal of Immunology, vol. 179, no. 10, pp.6889–6900, 2007.
[13] M. E. Rottenberg, A. Riarte, L. Sporrong et al., “Outcome ofinfection with different strains of Trypanosoma cruzi in micelacking CD4 and/or CD8,” Immunology Letters, vol. 45, no. 1-2,pp. 53–60, 1995.
[14] R. L. Tarleton, M. J. Grusby, M. Postan, and L. H. Glimcher,“Trypanosoma cruzi infection in MHC-de�cient mice: furtherevidence for the role of both class I- and class II-restrictedT cellsin immune resistance and disease,” International Immunology,vol. 8, no. 1, pp. 13–22, 1996.
[15] F. Tzelepis, B. C. G. De Alencar, M. L. O. Penido, R. T.Gazzinelli, P. M. Persechini, and M. M. Rodrigues, “Distinctkinetics of effector CD8+ cytotoxic T cells aer infection withTrypanosoma cruzi in naïve or vaccinated mice,” Infection andImmunity, vol. 74, no. 4, pp. 2477–2481, 2006.
[16] A. Y. Limon-Flores, R. Cervera-Cetina, J. L. Tzec-Arjona et al.,“Effect of a combination DNA vaccine for the prevention andtherapy of Trypanosoma cruzi infection in mice: role of CD4+and CD8+ T cells,” Vaccine, vol. 28, no. 46, pp. 7414–7419, 2010.
[17] D. Cobb, S. Guo, A. M. Lara, P. Manque, G. Buck, andR. B. Smeltz, “T-bet-dependent regulation of CD8+ T-cellexpansion during experimental Trypanosoma cruzi infection,”Immunology, vol. 128, no. 4, pp. 589–599, 2009.
[18] D. L. Martin, D. B. Weatherly, S. A. Laucella et al., “CD8+ T-Cellresponses to Trypanosoma cruzi are highly focused on strain-variant trans-sialidase epitopes.,” PLoS pathogens, vol. 2, no. 8,p. e77, 2006.
[19] S. Kuwajima, T. Sato, K. Ishida, H. Tada, H. Tezuka, and T.Ohteki, “Interleukin 15-dependent crosstalk between conven-tional and plasmacytoid dendritic cells is essential for CpG-induced immune activation,” Nature Immunology, vol. 7, no. 7,pp. 740–746, 2006.
[20] K. L. Wong, L. F. M. Tang, F. C. Lew et al., “CD44high memoryCD8 T cells synergize with CpG DNA to activate dendritic cellIL-12p70 production,” Journal of Immunology, vol. 183, no. 1,pp. 41–50, 2009.
[21] L. H. Glimcher, M. J. Townsend, B. M. Sullivan, and G. M.Lord, “Recent developments in the transcriptional regulation ofcytolytic effector cells,” Nature Reviews Immunology, vol. 4, no.11, pp. 900–911, 2004.
[22] E. L. Pearce, A. C. Mullen, G. A. Martins et al., “Control ofeffector CD8+ T cell function by the transcription factor eome-sodermin,” Science, vol. 302, no. 5647, pp. 1041–1043, 2003.
[23] A. E. Albers, L. Strauss, T. Liao, T. K. Hoffmann, and A. M.Kaufmann, “T cell-tumor interaction directs the developmentof immunotherapies in head and neck cancer,” Clinical andDevelopmental Immunology, vol. 2010, Article ID 236378, 2010.
[24] C. Parodi, A. M. Padilla, and M. A. Basombrío, “Protectiveimmunity against Trypanosoma cruzi,” Memorias do InstitutoOswaldo Cruz, vol. 104, no. 1, pp. 288–294, 2009.
[25] S. N. Woolard and U. Kumaraguru, “Viral vaccines and CTLresponse,” Journal of Biomedicine and Biotechnology, vol. 2010,Article ID 141657, 6 pages, 2010.
[26] C. Bode, G. Zhao, F. Steinhagen, T. Kinjo, and D. M. Klinman,“CpG DNA as a vaccine adjuvant,” Expert Review of Vaccines,vol. 10, no. 4, pp. 499–511, 2011.

12 BioMed Research International
[27] A. M. Krieg, “CpG motifs in bacterial DNA and their immuneeffects,” Annual Review of Immunology, vol. 20, pp. 709–760,2002.
[28] A. M. Padilla, L. J. Simpson, and R. L. Tarleton, “InsufficientTLR activation contributes to the slow development of CD8+T cell responses in Trypanosoma cruzi infection,” Journal ofImmunology, vol. 183, no. 2, pp. 1245–1252, 2009.
[29] S. G. Fonseca, M. M. Reis, V. Coelho et al., “Locally producedsurvival cytokines IL-15 and IL-7 may be associated to thepredominance of CD8+ T cells at heart lesions of humanchronic chagas disease cardiomyopathy,” Scandinavian Journalof Immunology, vol. 66, no. 2-3, pp. 362–371, 2007.
[30] F. F. de Araújo, R. Corrêa-Oliveira, M. O. Rocha et al.,“Foxp3+CD25(high) CD4+ regulatory T cells from indetermi-nate patients with Chagas disease can suppress the effectorcells and cytokines and reveal altered correlations with diseaseseverity,” Immunobiology, vol. 217, no. 8, pp. 768–777, 2012.
[31] F. F. de Araújo, D. M. Vitelli-Avelar, A. Teixeira-Carvalho etal., “Regulatory T cells phenotype in different clinical forms ofchagas’ disease,” PLoS Neglected Tropical Diseases, vol. 5, no. 5,p. e992, 2011.
[32] C. Y. Wu, R. R. Warrier, X. Wang, D. H. Presky, and M.K. Gately, “Regulation of interleukin-12 receptor 𝛽𝛽𝛽𝛽1 chainexpression and interleukin-12 binding by human peripheralbloodmononuclear cells,”European Journal of Immunology, vol.27, no. 1, pp. 147–154, 1997.
[33] T.Musikacharoen,A.Oguma, Y. Yoshikai,N.Chiba, A.Masuda,and T. Matsuguchi, “Interleukin-15 induces IL-12 receptor 𝛽𝛽𝛽𝛽1gene expression through PU.1 and IRF 3 by targeting chromatinremodeling,” Blood, vol. 105, no. 2, pp. 711–720, 2005.
[34] B. M. Sullivan, A. Juedes, S. J. Szabo, M. Von Herrath, andL. H. Glimcher, “Antigen-driven effector CD8 T cell functionregulated by T-bet,” Proceedings of the National Academy ofSciences of the United States of America, vol. 100, no. 26, pp.15818–15823, 2003.





























