Embed Size (px)
Citation preview

651.06 - 1 -
1
Chemical Bonding (A-B) I. Valence Bond Approach
II. Molecular Orbital Approach
I. Valence Bonds In the chemical bond between A & B, the two electrons involved in the bond are
indistinguishable and are permitted to interact equally with both nuclei.
A. Hybridization
The four electron orbitals are replaced by equivalent hybrid orbitals where the
number of equivalent hybrid orbitals is determined by the number of ligands to
carbon.
CH4 4 ligands sp3 hybridization
CH2CH2 3 ligands sp2 hybridization with the
nonhybridized p providing additional
overlap
HCCH 2 ligands sp hybridization with two non-
hybridized p orbitals providing
additional overlap
Hybridization, in turn predicts bond length, bond strength and bond angle. This
approach to viewing chemical bonding is useful because of the relative constancy
of the bond properties from molecule to molecule.
ex
is a regular hexagon with all C-C-C bond angles = 120o
O
HH has an H-C-H angle of 118o
651.06 - 2 -
2
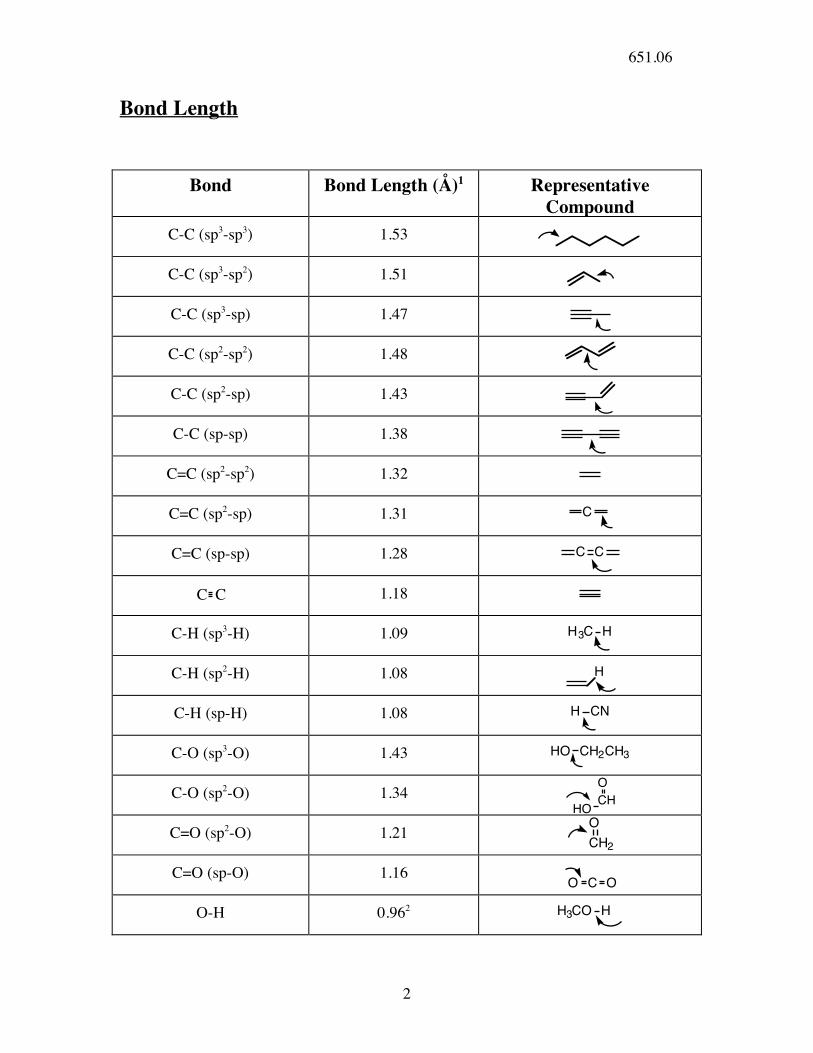
Bond Length
Bond Bond Length (Å)1 Representative Compound
C-C (sp3-sp3) 1.53
C-C (sp3-sp2) 1.51
C-C (sp3-sp) 1.47
C-C (sp2-sp2) 1.48
C-C (sp2-sp) 1.43
C-C (sp-sp) 1.38
C=C (sp2-sp2) 1.32
C=C (sp2-sp) 1.31 C
C=C (sp-sp) 1.28 C C
C C 1.18
C-H (sp3-H) 1.09 H3C H
C-H (sp2-H) 1.08 H
C-H (sp-H) 1.08 H CN
C-O (sp3-O) 1.43 HO CH2CH3
C-O (sp2-O) 1.34
HOCH
O
C=O (sp2-O) 1.21
CH2
O
C=O (sp-O) 1.16
O C O
O-H 0.962 H3CO H

651.06 - 3 -
3
Bond Bond Length (Å) Representative Compound
O-H 1.052 O
CHOH
C-N (sp3-N) 1.47 H2N CH3
C-N (sp2-N) 1.38 H2N CH
O
C=N (sp2-N) 1.28 HN CHCH3
C N 1.14 HC N
N-H 1.032 R3N H
N-H 1.012
H3CN H
H
C-S (sp3-S) 1.82 HS CH3
C-S (sp2-S) 1.75 PhS SPh
C-S (sp-S) 1.68 H3CS CN
C=S (sp-S) 1.67
S-H 1.332 H3CS H
C-F (sp3-F) 1.40
C-F (sp2-F) 1.34
C-F (sp-F) 1.27
C-Cl (sp3-Cl) 1.79
C-Cl (sp2-Cl) 1.73
C-Cl (sp-Cl) 1.63
C-Br (sp3-Br) 1.97
C-Br (sp2-Br) 1.88
651.06 - 4 -
4
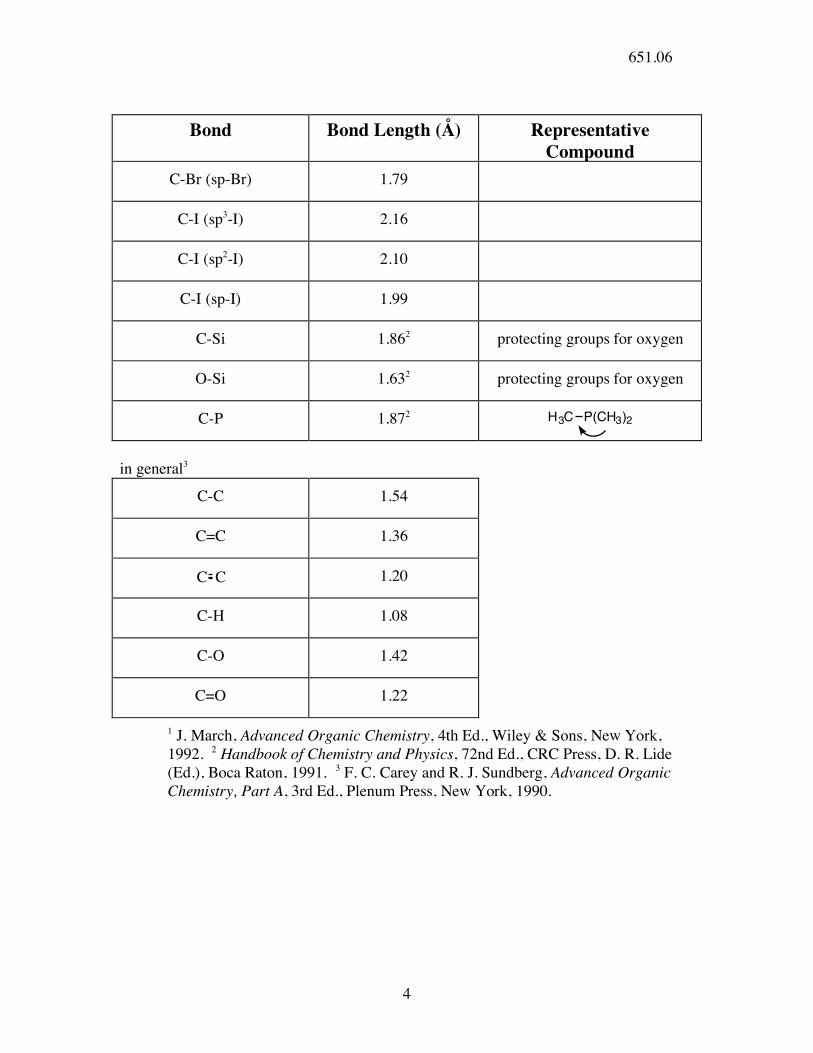
Bond Bond Length (Å) Representative
Compound C-Br (sp-Br) 1.79
C-I (sp3-I) 2.16
C-I (sp2-I) 2.10
C-I (sp-I) 1.99
C-Si 1.862 protecting groups for oxygen
O-Si 1.632 protecting groups for oxygen
C-P 1.872 H3C P(CH3)2
in general3
C-C 1.54
C=C 1.36
C C 1.20
C-H 1.08
C-O 1.42
C=O 1.22
1 J. March, Advanced Organic Chemistry, 4th Ed., Wiley & Sons, New York, 1992. 2 Handbook of Chemistry and Physics, 72nd Ed., CRC Press, D. R. Lide (Ed.), Boca Raton, 1991. 3 F. C. Carey and R. J. Sundberg, Advanced Organic Chemistry, Part A, 3rd Ed., Plenum Press, New York, 1990.

651.06 - 5 -
5
Bond Strength
Bond Bond Strength (kcal/mol)
Bond Bond Strength (kcal/mol)
O-O 343 Br-H3 87
I-I 363 C-O1 88
Br-Br 453 C-Si2 90
C-I 521 N-H1 93
Cl-Cl 573 C-H1 98
C-S 611 Cl-H3 102
C-Sn 632 H-H3 103
C-Br 661 C-F3 108
I-H 713 O-H1 110
C-N 721 C=N1 143
C-Cl 791 C=C1 148
S-H 821 C=O1 177
C-C 841 C C 1 200
H-CH2Ph 853 H-CH2CH3 1003
F-CH3 1083 H-CHCH2 1063
H-OCH3 1023 H-CCH 1323
H-NH2 1033 H-C6H5 1113
H-NHCH3 923 CH3-CH3 903
H-CN 1243 H2C=CH2 1723
H-CHCH3OH 933 HC CH 2303 1 J. March, Advanced Organic Chemistry, 4th Ed., Wiley & Sons, New York, 1992. 2 Handbook of Chemistry and Physics, 72nd Ed., CRC Press, D. R. Lide (Ed.), Boca Raton, 1991. 3 F. C. Carey and R. J. Sundberg, Advanced Organic Chemistry, Part A, 3rd Ed., Plenum Press, New York, 1990.

651.06 - 6 -
6
Bond Angle
Atoms Compound Carbon Hybridization Angle (o)
H-C-H ethane2 sp3 109
H-C-H ethylene3 sp2 117
H-C-C acetylene3 sp 180
O-C=O formic acid2 sp2 124
H-C-O formic acid2 sp2 118
C-C=O propynal2 sp2 123
C-O-C dimethyl ether2 - 110
C-O-C diphenyl ether1 - 124 1 J. March, Advanced Organic Chemistry, 4th Ed., Wiley & Sons, New York, 1992. 2 Handbook of Chemistry and Physics, 72nd Ed., CRC Press, D. R. Lide (Ed.), Boca Raton, 1991. 3 G. M. Loudon, Organic Chemistry, 2rd Ed., Benjamin/Cummings, Menlo Park, California, 1988.

651.06 - 7 -
7
B. Resonance
1. When one Lewis structure is inadequate for describing a molecular structure:
The “true” molecular structure is derived from a quantum mechanical
summing of each contributing structure.
2. The amount a particular Lewis acid structure contributes to the ground state of
a hybrid structure is dependent on its stability. The greater contribution is
made by the more stable structure.
3. Enhanced stability is associated with electron delocalization.
Resonance concept indicates that the valence bond approach to chemical bonding
has difficulty in treating certain types of chemical structures.
H
H
H
HH
H
cyclopropane
Each carbon has to bond to two other hydrogens and two other carbons. This
requires an sp3 hybridization.
However, to account for angle strain, each C-C bond is given more p character
while the C-H bond is given more s character
% S character
C-C 17
C-H 33
Reactivity: Br2
Br Br
BrNaCN
no reaction
H
O
H
O
H
O

651.06 - 8 -
8
Other deficiencies of Valence Bond theory:
• Why does the SN2 mechanism proceed with stereochemical inversion?
• Why is benzene so much more stable than other conjugated polyenes?
• The stereoselectivity of the Diels-Alder reaction:
+
CO2Et
CO2Et CO2Et
CO2Et
H
H
However, another model exists that can explain these phenomena…
II. Molecular Orbital Theory MO Theory - Bonding electron pairs are not localized between specific atoms.
Orbitals are extended over entire molecule.
Ψ - is the wave function which describes the interaction of the electron with other
electrons and nuclei of the molecule
Molecular orbitals are the linear combination of atomic orbitals
Ψmc = c1φ1 + c2φ2 + .... cnφn
φ - basis functions (usually atomic orbitals)
s p c1 - n - weighting coefficients which indicate the contribution of each atomic
orbital to the molecular orbital.
this is referred to as the LCAO-MO approximation

651.06 - 9 -
9
A. Hydrogen HA - HB
ΨMO = ϕ1SA - ϕ1SB
ΨMO = ϕ1SA + ϕ1SB
The number of molecular orbitals formed always equals the number of atomic
orbitals used.
!1SA !1SB
"E
"E
H H
H H
HHEnergy

651.06
10
B. Hybrid orbitals - Methane
1) orient methane with the carbon at the origin of a coordinate system
H1 is in the +x, +y, +z quadrant
H2 is in the -x, -y, +z quadrant
H3 is in the +x, -y, -z quadrant
H4 is in the -x, +y, -z quadrant
2) linearly combine the atomic orbitals
χsp31 = ϕ2s + ϕ2px
+ ϕ2py + ϕ2pz
χsp32 = ϕ2s - ϕ2px
- ϕ2py + ϕ2pz
χsp33 = ϕ2s + ϕ2px
- ϕ2py - ϕ2pz
χsp34 = ϕ2s - ϕ2px
+ ϕ2py - ϕ2pz
ϕ2s + ϕ2px + ϕ2py
+ ϕ2pz
+z
-z
-x
+x
-y+y
+z
-z
-x
+x
-y+y
+z
-z
-x
+x
-y+y
+z
-z
-x
+x
-y+y
= χsp3
+z
-z
-x
+x
-y+y

651.06
11
C. Bond Orbitals
ΨMOσ1 = χsp3
1 - ϕ1S1
C
H
C H
C H
ΨMOσ1 = χsp3
1 + ϕ1S1
others
ΨMOσ2 = χsp3
2 + ϕ1S2
ΨMOσ3 = χsp3
3 + ϕ1S3
ΨMOσ4 = χsp3
4 + ϕ1S4

651.06
12
D. MO Treatment of Cyclopropane H
H
H
HH
H
each carbon bonded to 4 other atoms, valence bond indicates sp3 but, H - C - H bond is ~120o therefore sp2!
The Walsh hybridization model for cyclopropane. All carbons are sp2 hybridized, two of the sp2 orbitals form
σ bonds to hydrogen with the third in the center of the ring. The p orbitals lie in the plane of the ring.
Above are the energy level diagram of the hybrid orbitals of cyclopropane and a representation of it's
molecular orbitals. The "σ type" orbitals arise from the overlap of sp2 hybrid orbitals with σ2 and σ3 being
degenerate and antibonding. The "π type" orbitals are made from p orbitals with π1 and π2 degenerate and
π3 being antibonding. The resulting electron density of cyclopropane is shown on the right.
MO treatment of cyclopropane explains chemistry of molecule much better than
valence bond treatment because chemistry of molecule is more typical of alkenes
than that of alkanes.
Br Br
Br+ HBr
+ Br2
!1
!2 !3
"1 "2
"3
Energy
non-bonding level
H
H
H
H
H
H

651.06
13
E. Construction of Molecular Orbitals
1) Symmetry
Only orbitals of the same symmetry can interact
a. constructed hybrid orbitals (oxygen is sp3) OHH
b. constructed bond orbitals
OHH
OHH
ϕ1 ϕ2
Question: How much bonding which holds a water molecule together is due
to the interaction between ϕ1 and ϕ2?
Problem: Question can not be answered until ϕ1 and ϕ2 are symmetry
adapted.
Molecules posses elements of symmetry. All molecular orbitals which
describe a structure must be symmetric or antisymmetric with respect to
each of the molecule’s elements of symmetry.
OHH
O
HH
OHH
axis of symmetry plane of symmetry plane of symmetry
ϕ1 and ϕ2 are neither symmetrical or asymmetric relative to C2 axis
ϕ1 and ϕ2 are neither symmetrical or asymmetrical relative to σ plane
ϕ1 and ϕ2 are symmetrical relative to σ' plane

651.06
14
To adapt:
ΨA = ϕ1 - ϕ2
O
HH
symmetric to C2 axis symmetric to σ plane symmetric to σ‘ plane
ΨB = ϕ1 + ϕ2
O
HH
antisymmetric to C2 axis antisymmetric to σ plane symmetric to σ‘ plane
Bonding orbitals are now symmetry adapted
Note: Orbitals do not have to be entirely symmetric or asymmetric relative to the symmetry elements of the molecule. ΨB is twice antisymmetric and once symmetric
ΨB = ϕ1 + ϕ2
ΨA = ϕ1 - ϕ2
O
HH
O
HH
O
HH
O
HH
!1 !2

651.06
15
Because ϕ1 and ϕ2 both possess two electrons, both higher and lower energy
levels will be occupied. No stabilization. Overlap between ϕ1 and ϕ2
contributes little bonding which holds the water molecule together.
III. Perturbation
!E
!E
"#
"+
$2
$1
"# = $2 - %$1
"+ = $1 + %$2 % = (0-1)
Working Example of PMO Theory
A. Hyperconjugation [Why are tertiary carbocations more stable?]
Enthalpy (ΔHo) for Ionization of Alkyl Chlorides (Gas Phase)
R Cl R
+Cl-
+
R ΔHo, kcal/mol CH3 229
CH3CH2 190
CH3CH2CH2 193
CH3CH2CH2CH2 193
(CH3)2CH 168
(CH3)3C 153

651.06
16
B. PMO Treatment
H C
H-C bond is symmetric relative to symmetry element of the molecule
P orbital is antisymmetric relative to symmetry of the molecule P
C C
Ha
Hb
Hc
H
H
C CHc
H
H
ϕa ϕb
C
Ha
Hb
Hc
H
HC
C
Hb
Hc
H
H
Ha
C
ΨS = ϕa + ϕb ΨA = ϕa - ϕb
C C
Ha
Hb
Hc
H
H
C C
Ha
Hb
Hc
H
H

651.06
17
Qualitative MO Theory
Effect of electronegativity on MO energy levels:
nC
nN
nO
Bond formation:
Look at reaction of Lewis base with Lewis acid: B: A
How do we show this in MO theory? By mixing their MOs!
C C
Ha
Hb
HcH
H
C C
Ha
Hb
HcH
H
C C
Ha
Hb
HcH
H
! " #$A
!
$A
C C
Ha
Hb
HcH
H
$A + #!
651.06
18
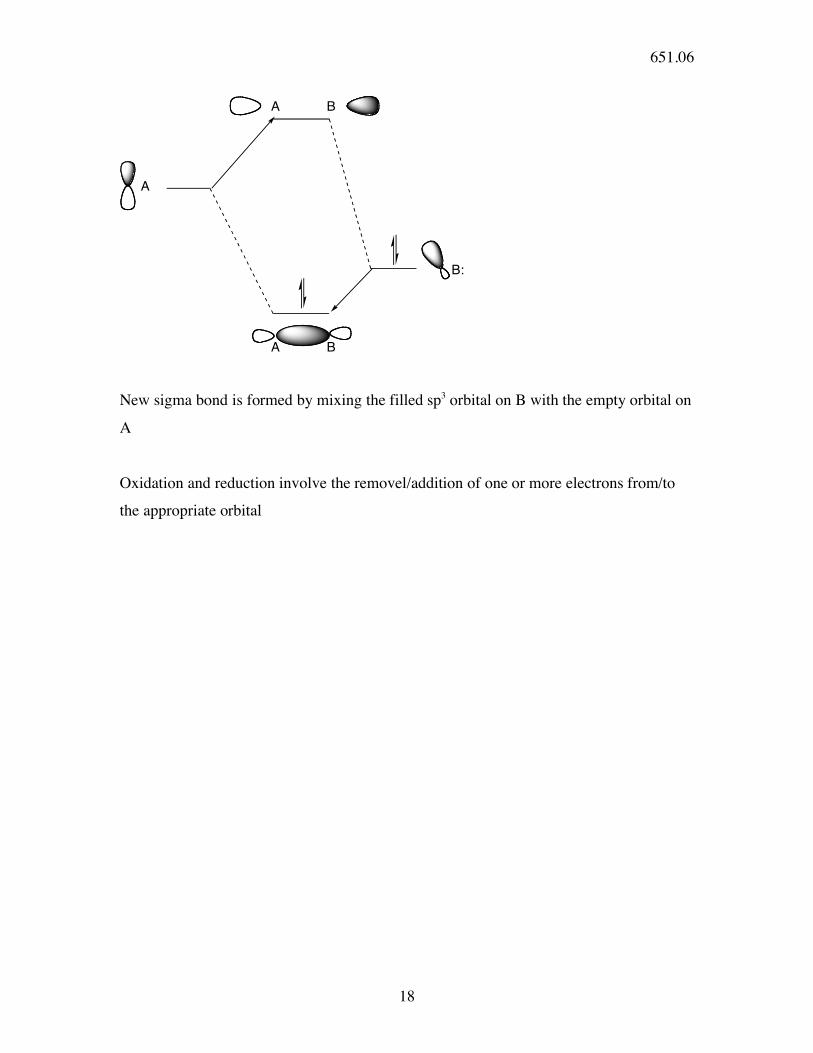
B:
A
A B
A B
New sigma bond is formed by mixing the filled sp3 orbital on B with the empty orbital on
A
Oxidation and reduction involve the removel/addition of one or more electrons from/to
the appropriate orbital

651.06
19
Energy Levels in MO Theory
HΨ = EΨ H = Hamiltonian energy operator Ψ = electron amplitude function (wavefunction) E = the total energy of the system consisting of both kinetic and potential energy HΨ is non-commutable (i.e. ΨHΨ ≠ HΨ2) Ψ2 is the probability of finding an electron
!2
"#
#
$"#
#
$"#
#
$ dxdydz = 1 short form: !2
" d# = 1
HΨ = EΨ ΨHΨ = EΨ2
!"!d# = E !2
$$ d# % E =!"!d#$
! 2d#$
- solve for H2+
LCAO Approximation: Ψ = c1χ1 + c2χ2
E =(c1!1 + c2! 2)H(c1!1 + c2!2 )d"#
(c1!1 + c2!2 )2d"#
E =c1!1Hc
1!1+ c
1!1Hc
2!2+ c
2!2Hc
1!1+ c
2!2Hc
2!2[ ]d"#
c1
2!1
2 + 2c1c2!1!2+ c
2
2!2
2( )d"#
Substitutions:
H11= !
1" H!1d#
H12= H
21= !
1" H!2d# = !
2" H!1d#
H22= !
2" H!2d#
S11= !
1
2d#"
S12= !
1" !2d#
S22= !
2"2d#
E =c1
2H11+ 2c
1c2H12+ c
2
2H22
c1
2S11+ 2c
1c2S12+ c
2
2S22

651.06
20
To find minimum value of the energy (as required by the variation principle), E is differentiated relative to each coefficient to give the secular equations:
!E
!c1
= c1H11" ES
11( ) + c
2H12" ES
12( ) = 0
!E
!c2
= c1H12" ES
12( ) + c
2H22" ES
22( ) = 0
Determining the energy levels then requires solving the secular determinant derived from the secular equations
H11! ES
11H12! ES
12
H12! ES
12H22! ES
22
= 0
Simplifying Assumptions:
Sik is the overlap integral = !i
"#
#
$ !kd%
where i = k there is complete overlap and Sik = 1 where i ≠ k the orbitals are orthogonal and Sik = 0 Hii= !
i" H!id#
Hii = α Hik= !
i" H!kd#
Hik = β

651.06
21
Energy Levels in MO Theory Solutions
1) solve for E: H11! ES
11H12! ES
12
H12! ES
12H22! ES
22
= 0
now becomes
! " E # " 0
# " 0 ! " E= 0
! " E #
# ! " E= 0
! 2 " 2!E + E2 " # 2 = 0
E2" 2!E + !
2" #
2( ) = 0
using quadratic formula
E =2! ± 4! 2 " 4 ! 2 " # 2( )
2
roots: E = α + β E = α − β 2) solve for c1 and c2 plug E back into secular equation to solve for c1 and c2 c1 ( α - E ) + c2 ( β ) = 0 c1 ( β ) + c2 ( α - E ) = 0
c1
c2
=!"
# ! E or
c1
c2
=!" + E
#
when E = α + β c1 = c2 when E = α − β c1 = - c2

651.06
22
Solutions (cont’d) 3) normalization remember !
2
" d# = 1 Ψ = χ1 + χ2
!2
" d# = $1+ $
2( )"2
d#
!2
" d# = $1"2d# + $
2"2d# + 2 $
1" $2d#
!
2
" d# = 1+ 1+ 0 = 2 to normalize divide through by 2:
!2
" d# = 12" $
1
2d# + 1
2" $2
2d# + 2 1
2" $1$2d#
!2
" d# = 12+ 12+ 0 = 1
for E = α + β, Ψ1 = c1χ1 + c1χ2
normalized Ψ1 = 1
2c1
c1χ1 + 1
2c1
c1χ2
for E = α − β, Ψ2 = c1χ1 - c1χ2 normalized Ψ2 =
1
2c1
c1χ1 - 1
2c1
c1χ2

651.06
23
IN GENERAL Ψj = cj1χ1 + cj2χ2 + ... + cjiχi The secular equations will be:
cji(H11 - S11Ej) + cj2(H12 - S12Ej) + ... + cji(H1i - S1iEj) = 0
cji(H21 - S21Ej) + cj2(H22 - S22Ej) + ... + cji(H2i - S2iEj) = 0
.
.
.
cji(Hi1 - Si1Ej) + cj2(Hi2 - Si2Ej) + ... + cji(Hii - SiiEj) = 0

651.06
24
Huckel π Electron Method
1) Hik, Sik conditions are same as before
2) Only π electrons are considered
3) Basis functions consist of n p orbitals, one on each of the carbon atoms of the π system.
Ψ = c1χ1 + c2χ2 χ1 χ2 where χ are p orbitals and not atoms
step 1
secular equation: c1(H11 - S11E) + c2(H12 - S12E) = 0
c1(H21 - S21E) + c2(H22 - S22E) = 0
secular determinant:
H11! S
11E H
12! S
12E
H21! S
21E H
22! S
22E= 0
" ! E #
# " ! E= 0
step 2
c1(α - E) + c2(β) = 0 c1 = c2 for E = α + β
c1(β) + c2(α - E) = 0 c1 = -c2 for E = α − β

651.06
25
step 3
Ψ1 = 1
2c1
c1χ1 + 1
2c1
c1χ2 Ψ2 = 1
2c1
c1χ1 - 1
2c1
c1χ2
step 4
draw energy diagram
E2 = α − β antibonding
!2
= 1
2
" #
$ % &1 '
1
2
" #
$ % &2 1
2 12
E1 = α + β bonding
!1
= 1
2
" #
$ % &1 + 1
2
" #
$ % &2 1
2 12
E2
E1
!2
!1
-"
+"
#

651.06
26
Butadiene
!1 !4!2 !3
Ψ = c1χ1 + c2χ2 + c3χ3 + c4χ4
1) set up secular equations and solve the secular determinant
c1(H11 - S11E) + c2(H12 - S12E) + c3(H13 - S13E) + c4(H14 - S14E) =0
c1(H21 - S21E) + c2(H22 - S22E) + c3(H23 - S23E) + c4(H24 - S24E) =0
c1(H31 - S31E) + c2(H32 - S32E) + c3(H33 - S33E) + c4(H34 - S34E) =0
c1(H41 - S41E) + c2(H42 - S42E) + c3(H43 - S43E) + c4(H44 - S44E) =0
H11! S
11E H
12! S
12E H
13! S
13E H
14! S
14E
H21! S
21E H
22! S
22E H
23! S
23E H
24! S
24E
H31! S
31E H
32! S
32E H
33! S
33E H
34! S
34E
H41! S
41E H
42! S
42E H
43! S
43E H
44! S
44E
= 0
! " E # 0 0
# ! " E # 0
0 # ! " E #
0 0 # ! " E
= 0
x 1 0 0
1 x 1 0
0 1 x 1
0 0 1 x
= 0 = x4 - 3x2 + 1
solve for x: x2=3 ± 9 ! 4
2=3 ± 5
2" x = ±
3 ± 5
2
x = ± 0.618, ± 1.618

651.06
27
substitute
E4 = α -1.618β
Ε3 = α - 0.618β
E2 = α + 0.618β
E1 = α + 1.618β
2) Plug E’s back into secular equations and solve for coefficients
Ψ4 = 0.372χ1 - 0.602χ2 + 0.602χ3 - 0.372χ4
Ψ3 = 0.602χ1 - 0.372χ2 - 0.372χ3 +0.602χ4
Ψ2 = 0.602χ1 + 0.372χ2 - 0.372χ3 - 0.602χ4
Ψ1 = 0.372χ1 + 0.602χ2 + 0.602χ3 + 0.372χ4
E4 = ! - 1.618"
#4
E3 = ! - 0.618"
#3
#2
#1
E2 = ! + 0.618"
E1 = ! +1.618"
non-bonding level

651.06
28
Mobile Bond Order (π bond order)
for butadiene: a qualitative assesment of bonding
Ψ1 c1 c2 c3 c4 c2 - c3 strong bonding
c1 - c2 , c3 - c4 fair bonding
Ψ2 c1 - c2, c3 - c4 fair bonding
c2 - c3 antibonding
Pij = Σncicj Pij = mobile bond order
n = # of electrons in an occuppied orbital
ci , cj = normalized coefficients for atoms i & j
P12 = (n1c1c2)Ψ1 + (n2c1c2)Ψ2
= 2(0.372)(0.602) + 2(0.602)(0.372)
= 0.896
P23 = (n1c2c3)Ψ1 + (n2c2c3)Ψ2
= 2(0.602)(0.602) + 2(0.372)(-0.372)
= 0.448
P34 = (n1c3c4)Ψ1 + (n2c3c4)Ψ2
= 2(0.602)(0.372) + 2(0.372)(0.602)
= 0.896
for the σ bond, add 1.0 to each calculated Pij:
H2C CH
CH
CH2
1.896 1.8961.448

651.06
29
π Bond Energy:
E! = ni
" Ei Eπ = π bond energy
n = # of electrons in each orbital Ei = energy of each orbital
for butadiene
Eπ = 2(α + 1.62β) + 2(α + 0.62β)
= 4α + 4.48β
Resonance Energy:
EHDE = Eπ - Eloc EHDE = Huckel delocalization energy (or resonance energy) Eπ = π bond energy Eloc = π bond energy of the component, isolated bonds
for butadiene:
EHDE = 4α + 4.48β - 4(α + β)
= 0.48β
for benzene:
Eπ = 2(α + 2β) + 4(α + β) = 6α + 8β
EHDE = 6α + 8β - 6α − 6β = 2β
Experimental value for resonance energy of benzene = 36 kcal/mol
β = -18 kcal/mol for carbon systems

651.06
30
Simplify
For linear polyenes of formula CnHn+2:
E = α + mjiβ where mj = 2cos (jπ / n+1)
j = mo #
n = # of carbons
# of molecular orbitals = # of basis orbitals ≈ in Huckel treatment to # of p
orbitals
coefficient crj of atom r in the jth molecular orbital:
crj =2
n +1sin
rj!
n +1
" #
$ %
For cyclic conjugated systems (Frost’s Circle)
Ej = ! + 2" cos2#J
N
$ %
& ' J = 0, 1, 2, ..., n-1 the orbital number
counting counter-clockwise from lowest vertex
N = ring size
J = 0
J = 1
J = 2J = 3
J = 4
! + 2"! + 0.618"
! # 1.618"

651.06
31
Aromaticity
cyclobutadiene benzene
Huckel Molecular Orbital Treatment
cyclobutadiene DE = 2(α + 2β) + 2α − 4(α + β) = 0
benzene DE = 2(α + 2β) + 4(α + β) − 6(α + β) = 2β
Experimental Data:
cyclobutadiene is extremely reactive
35
K
O
medium pressuremercury lamp
7 K, argon matrixguest - host ratio1 : 100 - 500
O
+

651.06
32
4n + 2 rule (Huckel’s Rule)
Species is aromatic if:
1) cyclic array of p orbitals
2) fully conjugated
3) planar
4) contains 4n + 2 π electrons
Physical measurements relevant to aromaticity:
1) bond lengths
aromatic molecules show bond lenths of 1.38 - 1.40Ao and are uniform
around the ring
2) diamagnetic anisotropy
in a magnetic field, π electron circulation leads to a magnetic field which
opposes the externally applied magnetic field
H0 nuclei within cone are shielded, nuclei on periphery of cone are
deshielded
3) thermodynamic characteristics
(a) heats of hydrogenation
49.8 kcal/mol
28.6 kcal/mol
stabilization associated with benzene = 3(28.6 kcal/mol) - 49.8 kcal/mol = 36 kcal/mol

651.06
33
(b) hydride & proton dissociation constants
Apply criterion for aromatic systems:
2e-
6e-
6e-
6e-
N
HN O S
O
6e-
6e-
6e-
6e-
6e-
Terms
aromatic - as discussed
antiaromatic - 4n system which is strongly destabilized relative to conjugated but
noncyclic equivalent
vs.
nonaromatic - 4n system which is isolable but not possessed of any stabilization relative
to the noncyclic equivalent
vs.
homoaromatic - 4n + 2, stabilized, cyclic conjugated system which is formed by
bypassing a saturated carbon atom
Ha
Hb
HaHb
Ha is 5.8 ppm upfield of Hb ⇒ ring current

651.06
34
[10] annulene
cyclodecapentaenes
10e-
4n + 2 system, cyclic but does not possess aromatic stabilization
all cis bonds are strongly destabilized due to angle strain (ref. 1a-e)
H
H
trans, cis, trans, cis, cis - minimun angle strain but severe
nonbonded repulsion between the two internal hydrogens (ref. 1a-d, f)
h! +
NMR does not indicate ring current, nonaromaticity is due to
nonplanarity (ref 1g, h)
Belted Aromatics (ref 1i-l)
HbHa
placing unsaturated carbon in system relieves the irresolvable
conflict between angle strain and nonbonded steric interactions (ref 1m-
o)
Ha and Hb shifts ⇒ ring current
x-ray indicates no significant alternation in c-c bond lengths around the ring
now: 4n + 2 cyclic fully conjugated planar (ref 1p) ∴ aromatic further examples of belted aromatics
651.06
35
HN
(ref 1q-t)
O
(ref 1q, u)
[14] annulene
HHHH
(ref 1v-y)
[18] annulene
H
H
H
H
H
H
(ref 1z, aa-ac)
C18
(ref 1ad, ae)
651.06
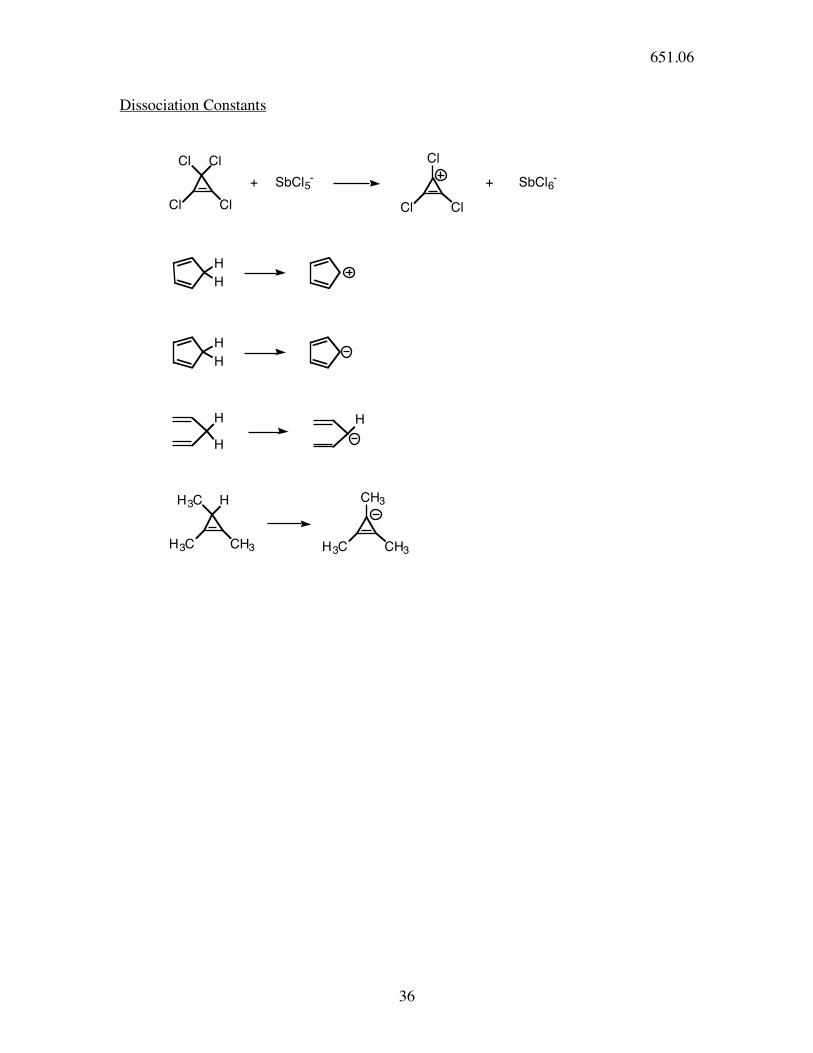
36
Dissociation Constants
Cl Cl
ClCl
+ SbCl5-
ClCl
Cl
+ SbCl6-
H
H
H
H
H
H
H
H3C H
CH3H3C CH3H3C
CH3

651.06
37
References: 1 a) Top. Nonbenzenoid Aromat. Chem 1973, 1, 121 (review) b) Acc. Chem. Res. 1972, 5, 272 (review) c) Snyder, Nonbenzenoid Aromatics, vol. 1; Academic Press: NY, 1969, pp. 63-116 (rev) d) Garratt, Aromaticity; Wiley: NY, 1986, pp.113-147 (rev) e) JACS 1971, 4966 f) J. Chem. Phys. 1952, 1489 g) Croat. Chem. Acta. 1977, 49, 441 h) JACS 1971, 4966 i) Pure Appl. Chem. 1982, 54, 1015 (rev) j) Isr. J. Chem. 1980, 20, 215 k) Chimia 1968, 22, 21 l) Angew. Chem. Int. Ed. Engl. 1967, 6, 385 m)Angew. Chem. Int. Ed. Engl. 1964, 3, 228 n) Angew. Chem. Int. Ed. Engl. 1964, 3, 642 o) Tetrahedron Lett. 1966, 1569 p) Acc. Chem. Res. 1988, 21, 243 q) Angew. Chem. Int. Ed. Engl. 1964, 3, 642 r) JACS 1964, 3168 s) JACS 1967, 6310 t) Chem. Commun. 1967, 1039 u) Tetrahedron Lett. 1965, 3613 v) JACS 1964, 521 w) JACS 1972, 471 x) JACS 1973, 3893 y) Helv. Chim. Acta. 1995, 78, 679 z) JACS 1962, 274 aa) JACS 1972, 8644 ab) Helv. Chim. Acta. 1974, 57, 2276 ac) Helv. Chim. Acta. 1982, 65, 1885 ad) JACS 1990, 4966 ae) Nature 1994, 369, 199





























