Embed Size (px)
Citation preview

Chapter I
Introduction
And
Review of literature
Chapter 1 Introduction and Review of Literature
1
INTRODUCTION
11 Coffee
Coffee is an important plantation crop belonging to the family Rubiaceae subfamily
Cinchonoideae and tribe Coffeae (Clifford et al 1989) Rubiaceae is the largest
flowering plant family comprising of 650 genera and 13000 species which are largely
tropical or subtropical (Rova et al 2002)
The word coffee connects with the town Kaffa in Ethiopia reported to be the
place of origin of coffee plants The word coffee derives from the Ottoman Turkish
word kahve via Italian word caffeacute Turkish word is in turn borrowed from the Arabic
word quhwah referring to a type of wine- quaqat-al-bunn (wine of bean)This found
its way into the European languages and became cafeacute(French) caffe (Italian) Kaffee
(German) koffie (Dutch) coffee (English) and Latin Coffea for the botanical genus
The stimulatory effects of roasted coffee beans were well known to the natives of
Africa when the Arabs brought C arabica seeds from Ethiopia to Yemen (Arabian
Peninsula) during the 13th
century and established the first plantations (Monaco et al
1997)
12 Taxonomy of Coffee
The genus Coffea L comprises 103 species (Davis et al 2006) and occurs naturally
in tropical Africa Madagascar the Comoros and Mascarenes (Mauritius and
Reunion) Coffea species are mostly restricted to humid evergreen forest although
some species are found in seasonally dry deciduous forest andor bushland The most
recent classifications of Coffea (Bridson 1988 Bridson 2003 Davis et al 2005
Davis et al 2006) divide the genus into two subgenera subgenus Coffea (95 spp)
and subgenus Baracoffea JF Leroy (eight spp) Coffea subgenus has a wide range of
distribution whereas Coffea subgenus Baracoffea is restricted to the seasonally dry
forest and scrubland of western Madagascar (Davis et al 2005) and they are also
found in NE Kenya and SE Somalia according to (Leroy 1982)
Coffea subgenus Coffea includes the species used in the production of coffee
ie C arabica (Arabica coffee) C canephora (Robusta coffee) and C liberica
(Liberian coffee) C arabica is by far the most important traded species and accounts
for at least 65 of total commercial production C arabica is the only allotetraploid
Coffea species (2n= 4x =44) (Grassias amp Kammacher 1975) all other Coffea species
are diploid (2n = 2x =22) C arabica is also self-compatible (Carvalho et al 1991)
Chapter 1 Introduction and Review of Literature
2
thus far only reported in two other species C heterocalyx (Coulibaly et al 2002) and
C anthonyi (Maurin et al 2007)
121 Classification
Kingdom Plantae
Subkingdom Tracheobionta
Superdivision Spermatophyta
Division Magnoliophyta
Class Magnoliopsida
Subclass Asteridae
Order Rubiales
Family Rubiaceae
Genus Coffea
122 Morphological features
The genus Coffea contains about 25 species These are small trees or shrubs that
originally grew in African tropical forests Some varieties of coffee plant typically
grow over 30 feet But in cultivation for ease of picking of the coffee berry the
coffee tree is seldom allowed over 15 feet The C arabica coffee plant is typically
smaller than the C canephora plants C canephora is shrub type and C arabica is
tree type They have extensive tap root systems with well-developed main vertical
roots and lateral roots that grow parallel to the ground Leaves have a bipolar structure
where leaf pairs are at 90 degree rotation to each other on the stem Leaves are oblong
to ovate in shape with characteristic interpetiolar stipule
Flowers are white are white produced in dense clusters at leaf axils Flowers
are pentamerous Calyx is toothed and corolla is tubular There are five stamens and a
single bifid style Sexually C arabica is autogamous whereas Ccanephora is self-
incompatible The fruits are drupes
123 Classification of coffee
Chevalier (1947) has grouped the valid coffee species into the following sections
(1) Argocoffea
(2) Paracoffea
(3) Mascarocoffea
Chapter 1 Introduction and Review of Literature
3
(4) Eucoffea
The Coffee species belonging to Mascarocoffea section have one common
characteristic the absence of caffeine The Eucoffea (now named Coffea) has been
again divided into five subsections according to diverse criteria tree height
(Nanocoffea) leaf thickness (Pachycoffea) fruit colour (Erythrocoffea Melanocoff-
ea) and geographical distribution (Mozambicoffea) (Table 11) The recent
classification has widely accepted that Coffea sect Paracoffea and Agrocoffea mainly
consist of species from other genera
Table 11The grouping of the species in the subsection Eucoffea (Chevalier 1947)
Section Subsection Species
Eucoffea
Erythrocoffea
C arabica
C canephora
C congensis
Pachycoffea
C abeokutae
C liberica
C klainii
C oyemensis
C dewerei
Melanocoffea
C stenophylla
C carissoi
C mayombensis
Nanocoffea
C humilis
C brevipes
C togoensis
Mozambicoffea
C schumanniana
C eugenioides
C kivuensis
C mufindiensis
C zanguebariae
C racemosa
C ligustroides
C salvatrix
The current sub generic classification comprises of two genera Coffea and Baracoffea
(Table 12) Most species of Coffea belong to Coffea subgenera includes those used
for producing the beverage coffee Coffea subgenera occur throughout the natural
range of the genus in Africa Madagascar and The Mascarenes Subgenera Baracoffea
contains only three accepted species (although five remain undescribed) (Table 12)
and is restricted to the dry forests of western Madagascar Leroy (Leroy 1980) placed
C rhamnifolia a species from Africa (Somalia and Kenya) in Coffea subgen
Chapter 1 Introduction and Review of Literature
4
Baracoffea but this was contested by Bridson (1988) Davis amp Rakotonasolo (2003)
and Davis et al (2005)
Table 12Outline classification of Coffea and Psilanthus with synonyms (Davis amp Rakotonasolo
2003)
Coffea L Sp Pl 172 (1753)
Coffea sub gen Coffea L Type C arabica L 95 species Africa Madagascar Mascarenes
Coffea sub gen Psilanthopsis (AChev)
Psilanthopsis AChev Type Coffea kapakata
Type Paolia jasminoides Chiov (= C rhamnifolia (Chiov) Bridson)
Coffea sub gen Baracoffea
Coffea sect Baracoffea Type Coffea humbertii J-FLeroy
Eight species (including five -yet to be published) West Madagascar
[Paracoffea subgen Insulanoparacoffea J-FLeroy J Agric Trop Bot Appl 14 276 (1967)
nom nud]
Psilanthus Hookf Gen Pl 115 (1873)
Psilanthus subgen Psilanthus Hookf Type Psilanthus mannii Hookf
Two species Africa (central and western)
Psilanthus subgen Afrocoffea (Moens)
Coffea subgenAfrocoffea
Type Psilanthus lebrunianus (Germain amp Kesler) Bridsonc 18 species Africa Asia
Australasia
Coffea sectParacoffeaMiq Fl Ind Batavae 308 (1856)
Psilanthus subgen Paracoffea Type Coffea horsfieldiana Miq
Paracoffea
[Paracoffea subgenAfroparacoffea J-FLeroy J Agric Trop Bot Appl 14 276 (1967)
nom nud]
[Paracoffea subgenMelanoparacoffea J-FLeroy J Agric Trop Bot Appl 14 276 (1967)
nom nud]
13 Economics of Coffee
Coffee is the fourth most valuable traded agricultural commodity (FAO 2004) but is
the second most valuable commodity exported by the developing countries and more
than 75 million people depend on coffee for most of their livelihood (Pendergrast
2009)
India produces both Arabica coffee from C arabica and Robusta coffee from
C canephora As per the economic survey 2012-13 Coffee is planted in India in
around 388195 hectares Out of this area 52 is Ccanephora and 48 is Carabica
Chapter 1 Introduction and Review of Literature
5
Coffee is mostly grown in south India parts of Orissa W Bengal and North east
India Karnataka is the largest producer with 569 of total area under production
which accounts for 71 of the total produce The total crop harvest for 2012-2013 is
placed at 325300 tonnes (httpwww Indicoffeeorgindiacoffeephpp-
age=CoffeeData)
14 Composition of coffee beans
Navellier (1961) gave a mean composition of green coffee as Carbohydratesglucides
(58) lignin (2) lipids (13) proteins (13) ash (4) non-volatile acids (8)
trigonelline (1) and caffeine (1) The caffeine content of green coffee is relatively
limited (10-25) of dry matter and changes little with seed development (Clifford
and Kazi 1987) In coffee seeds the concentration of trigonelline is approximately
2 of dry weight (Clifford 1985 Mazzafera 1991) Mazzafera (1999) found a higher
protein content in the mature beans than in the immature beans but a lower content of
free amino acids with asparagine as the main component Flament ( 2000) estimated
the composition of coffee beans as follows
Proteins 100 (dry weight basis of green coffee)
Carbohydrates 500 -do-
Lipids 117-140 (Arabicas)
76- 95 (Robustas)
Chlorogenic acids 65 (Arabicas)
90 (Robustas)
15 Purine alkaloids structure and role
Purine alkaloids are derived from purine nucleotides (Zulak et al 2007) and found in
over hundred plant species in 13 orders in plant kingdom (Ashihara amp Crozier 1999)
Methylxanthines like caffeine (137-trimethlyxanthine) theobromine (37-methylxan-
thine) and methyluric acids are classified into purine alkaloids (Figure 11) (Table
13)(Ashihara et al 2008) These are known to occur in tea coffee and a number of
non-alcoholic beverages Caffeine was isolated from coffee (Coffea arabica) and tea
(Camellia sinensis) in 1820 (Ashihara amp Crozier 2001)
Chapter 1 Introduction and Review of Literature
6
Figure 11 Purine alkaloid xanthine and uric acid skeleton
Plants that accumulate purine alkaloid are classified into three groups based on
the type of alkaloids they produce caffeine-producing plants which include coffee
tea and mateacute (Ilex paraguariensis) theobromine ndash producing plants are represented
by cacao cocoa tea (Camellia ptilophylla) and Camellia irrawadiensis and
methyluric acid ndash producing plants consist of Coffea dewevrei Coffea liberica C
excelsa and Kucha tea (Camellia assamica)
Table 13 Purine alkaloid structures based on xanthine and uric acid skeleton
Compound Trivial name R1 R2 R3 R4 O-2 Δ2-3
Methylxanthines
Xanthine H H H
1-methylxanthine CH3 H H
3-methyl xanthine H CH3 H
7-methyl xanthine H H CH3
1-3methylxanthine Theophylline CH3 CH3 H
17-methylxanthine Paraxanthine CH3 H CH3
37-methylxanthine Theobromine H CH3 CH3
137-methylxanthine Caffeine CH3 CH3 CH3
Uric acid and methyl uric acids
Uric acid H H H H
137-trimethyluric acid CH3 CH3 CH3 H
1379-tetramethyluric
acid Theacrine CH3 CH3 CH3 CH3
O19-trimethyluric acid Liberine CH3 H H CH3 CH3 Δ2-3
O179-trimethyluric acid Methylliberine CH3 H CH3 CH3 CH3 Δ2-3
16 Role of caffeine in plants
The physiological role of endogenous purine alkaloids and related compounds in
higher plants remains undetermined There are two hypotheses about the role of the
Chapter 1 Introduction and Review of Literature
7
high concentrations of caffeine that accumulate in tea coffee and a few other plant
species The lsquoallelopathic theory or the auto toxic function theoryrsquo proposes that
caffeine in seed coats is released into the soil and inhibits the germination of seeds
around the parent plants (Anaya et al 2006)
The lsquochemical defense theoryrsquo proposes that caffeine in young leaves fruits
and flower buds acts to protect soft tissues from insect pests (Ashihara amp Crozier
2001 Ashihara et al 2008) It has been shown that spraying tomato leaves with a 1
solution of caffeine deters feeding by tobacco horn worms while treatment of
cabbage leaves and orchids with 001ndash01 solutions of caffeine acts as a neurotoxin
and kills or repels slugs and snails (Hollingsworth et al 2002) This work has now
been extended with convincing evidence for chemical defense theory has recently
been obtained with transgenic caffeine ndash producing tobacco plants (Uefuji et al 2005
Kim et al 2006)
17 Distribution of caffeine in plants
Caffeine has been found in 13 orders of the plant kingdom (Ashihara and Suzuki
2004 Anaya et al 2006) In some species the main purine alkaloid is theobromine or
methyl uric acids such as theacrine liberine and methylliberine rather than caffeine
(Ashihara amp Crozier 1999 Ashihara amp Crozier 2001)(Table 14)
Table 14 Distribution of purine alkaloids in plants
SNo Plant species Common
name Major alkaloid Plant parts
containing
alkaloids 1 Coffea arabica Arabica Caffeine Leaves Seeds
2 Coffea canephora Robusta Caffeine Leaves Seeds
3 Coffea liberica Caffeine Theacrine Liberine
Seeds Mature leaves
4 Coffea dewevrei Caffeine Theacrine Liberine
Seeds Mature leaves
5 Camellia sinensis Tea Caffeine Leaves
6 Camellia assamica Assam tea Caffeine Leaves
7 Camellia assamica var
kucha Kucha Theacrine Caffeine Leaves
8 Camellia irrawadensis Theobromine Leaves
9 Camellia ptilophylla Cocoa tea Theobromine Leaves
10 Theobroma cacao Cocoa Theobromine Seeds
11 Paullinia cupuna Guarana Caffeine Seeds
Chapter 1 Introduction and Review of Literature
8
12 Cola nitida Caffeine Seeds
13 Citrus sp Caffeine Pollen
14 Ilex paraguariensis Mate Caffeine Leaves
Adapted from (Ashihara amp Crozier 1999 Ashihara amp Crozier 2001)
18 Caffeine biosynthesis
The major biosynthetic pathway is a four step sequence consisting of three sequential
methylation and one nucleosidase reactions (Figure 12) The xanthine skeleton of
caffeine is derived from purine nucleotides The initial step in caffeine biosynthesis is
the methylation of xanthosine by a SAM- dependent N-methyltransferase In addition
to experiments with radiolabeled precursors substrate specificities of native (Kato et
al 1999) and recombinant N-methyltransferases (Kato et al 2000 Ogawa et al
2001 Mizuno et al 2003a Mizuno et al 2003b Uefuji et al 2003) strongly
suggest that the major route to caffeine is a xanthosine rarr7-methylxanthosinrarr 7-
methylxanthine rarr theobromine rarr caffeine pathway (Figure 12) Although the
information has been obtained mainly from coffee (C arabica) and tea (Camellia
sinensis) the available evidence indicates that the pathway is essentially the same in
other purine alkaloid-forming plants such as mateacute (Ashihara 1993) and cacao
(Koyama et al 2003 Yoneyama et al 2006)
The first step in the caffeine biosynthetic pathway from xanthosine is
conversion of xanthosine to 7-methylxanthosine (Figure 12) This reaction is
catalyzed by the 7-methylxanthosine synthase (xanthosine 7 N-methyltransferase EC
211158) The genes encoding for 7-methylxanthosine synthase CmXRS1 (AB
034699) and CaXMT1 (AB 048793) were isolated from the C arabica (Mizuno et
al 2003a Uefuji et al 2003) The recombinant proteins obtained from these genes
exhibit 7-methylxanthosine synthase activity in vitro Xanthosine monophosphate
(XMP) was not converted to 7-methylxanthosine by the recombinant enzymes thus
inclusion of 7-methy-XMP was proposed (Schulthess et al 1996)
The second step of caffeine biosynthesis involves a nucleosidase which
catalyses the hydrolysis of 7-methylxanthosine Although N-methylnucleosidase (EC
32225) was partially purified from tea leaves (Negishi et al 1988) recent detailed
structural studies on coffee 7-methylxanthosine synthase suggested that the methyl
transfer and nucleoside cleavage may be coupled and catalyzed by a single enzyme
(McCarthy amp McCarthy 2007)
Chapter 1 Introduction and Review of Literature
9
The last two steps of the caffeine synthesis are also catalyzed by a SAM-
dependent N-methyltransferase(s) which is different from the N-methyltransferase
enzyme that catalyzes the first step in the pathway Native N-methyltransferase
activities have been detected in crude and partially purified extracts from tea and
coffee plants (Ashihara amp Suzuki 2004) and a highly purified preparation has been
obtained from young tea leaves (Kato et al 1999) The enzyme was assigned the
name caffeine synthase (EC 211160) that catalyses the last two steps of caffeine
biosynthesis viz the conversion of 7-methylxanthine to caffeine via theobromine
(Figure 12) The gene encoding caffeine synthase was first cloned from young tea
leaves (Kato et al 2000) Since then several genes encoding N-methyltransferases
with different substrate specificity have been reported
Activity of the recombinant theobromine synthase (CTS1 and CaMXMT EC
211159) is specific for the conversion of 7-methylxanthine to theobromine In
contrast the recombinant caffeine synthases (CCS1 and CaDMXMT1) can utilize
paraxanthine theobromine and 7-methylxanthine as shown with tea caffeine synthase
(TCS1) (Mohanpuria et al 2011) Although paraxanthine is the most active substrate
of this recombinant enzyme only limited amounts of paraxanthine are synthesized in
coffee cells hence in vivo caffeine synthase is principally involved in the conversion
of 7-methylxanthine to caffeine via theobromine
Radiolabelled tracer experiments with theobromine accumulating cacao and
Chinese tea (Camellia ptilophylla) showed a limited conversion of theobromine to
caffeine N-methyltransferase which catalyzed the conversion of 7-methylxanthine to
theobromine was detected in crude extracts from C ptilophylla but conversion of
theobromine to caffeine was not observed (Ashihara et al 1998) Genes homologous
to caffeine synthase have been isolated from several theobromine accumulating plants
including cacao (Yoneyama et al 2006) The recombinant enzymes derived from
these genes have 3 N-methyltransferase activity suggesting that these theobromine
accumulating plants contain specific theobromine synthase Caffeine does not occur
in C ptilophylla (Ashihara et al 1998) although it is present in leaves and fruits of
cacao along with theobromine (Koyama et al 2003 Zheng amp Ashihara 2004)
Chapter 1 Introduction and Review of Literature
10
Figure 12 Pathways for the biosynthesis of caffeine
The major pathway is indicated by solid lines (Mizuno et al 2003b Uefuji et al 2003) Alternative pathways in coffee and tea plants are indicated
by dashed lines(Kato et al 1996 Schulthess et al 1996) Enzymes (1) 7-methylxanthosine synthase (2) N-methylnucleosidase (3) Theobromine
synthase (4) Caffeine synthase
N
N
N
N7
H3C
3
1
O
O
Caffeine
N
NH
N
N7
3
O
O
Theobromine
N
N
N
N
H
H
7
O
O
N
N
N
N
H
H
O
O
N
N
N
N
H
H
O
O
Xanthosine
N
N
N
N
H
H
O
O
N
N
N
N
H
H
O
ON
N
N
N
H
H3C
71
O
O
Paraxanthine
CH3
CH3
CH3
CH3CH3
7-methylxanthine
Rib P
Rib
(1)
(4)
Rib
+
CH3
7-methyl
xanthosine
(2)
(3)
SAM SAH
SAM SAH SAM SAH
Xanthosine mono
phosphate
Rib P
7-methyl xanthosine
monophosphate
+
CH3 CH3
Inosine 5-mono
phosphate
Adenosine 5-mono
phosphate
SAM SAH
Guanosine
Guanosine 5-mono
phosphate
Chapter 1 Introduction and Review of Literature
11
181 Source of Xanthosine for caffeine biosynthesis
Xanthosine the initial substrate of purine alkaloid synthesis is supplied by at least four
different pathways de novo purine biosynthesis (de novo route) the degradation
pathways of adenine nucleotides (AMP route) and guanine nucleotides (GMP route)
and the S-adenosyl-L-methionine (SAM) cycle (SAM route) (Figure 13) Purine
nucleotides are synthesized by de novo and salvage pathways (Ashihara amp Crozier
1999 Stasolla et al 2003 Zrenner et al 2006) The de novo synthesis of non purine
precursors like CO2 10-formyltetrahydrofolate 5-phosphoribosyl-1-pyrophosphate and
the amino acids Glycine glutamine and aspartate results in the production of purine
nucleotides
Figure 13 Xanthosine biosynthesis Xanthosine for purine alkaloid biosynthesis is supplied by at
least four routes from adenosine released from the SAM cycle (SAM route) from IMP
originating from de novo purine synthesis (de novo route) from the cellular adenine
nucleotide pool (AMP route) and from the guanine nucleotide pool (GMP route)
Adapted from (Ashihara et al 2008)
The utilization of IMP for caffeine biosynthetic pathway which is formed by the
de novo purine biosynthetic pathway for caffeine biosynthetic pathways was
demonstrated in young tea leaves using 15
N-glycine and 14
C-labelled precursors and
inhibitors of de novo purine biosynthesis (Ito amp Ashihara 1999) Xanthosine is formed
by an IMP rarr XMP rarr xanthosine pathway IMP dehydrogenase (EC 111205) and 5prime-
nucleotidase (EC 3135) catalyze these reactions The inhibition of IMP dehydrogen-
ase by ribavirin inhibits both caffeine and gunanine nucleotide biosynthesis in tea and
coffee leaves (Keya et al 2003)
Chapter 1 Introduction and Review of Literature
12
The portion of xanthosine used for caffeine biosynthesis is derived from adenine
and guanine nucleotide pools which are produce de novo by salvage pathways there are
several potential pathways for xanthosine synthesis from AMP but the AMP rarr IMP
rarr XMPrarr xanthosine route is predominant All three enzymes involved in the
conversion AMP deaminase (EC 3546) IMP dehydrogenase and 5prime-nucleosidase
have been detected in tea leaves (Koshiishi et al 2001) Xanthosine for caffeine
biosynthesis can also be produced from guanine nucleotides by GMPrarrguanosine
rarrxanthosine pathway Guanosine deaminase (EC 35415) activity is found in cell free
extracts from young tea leaves (Negishi et al 1994) but GMP deaminase activity was
not detected in plants (Stasolla et al 2003 Zrenner et al 2006) The absence of GMP
deaminase activity suggests that a GMP rarr IMP rarr XMP rarr xanthosine route is not
functional in plants
The use of radiolabelled purine bases and nucleosides for the study of caffeine
biosynthetic pathway was facilitated by the fact that exogenous purine nucleotides are
rapidly hydrolyzed by the plant tissues forming purine nucleosides andor bases which
are then utilized in pathways which in most instances indirectly lead to xanthosine
Most of these purine compounds including adenine adenosine inosine hypoxanthine
and guanine are converted to their respective nucleotides by salvage enzymes
including adenine phosphoribosyl transferase (EC 2427) hypoxanthineguanine
phosphoribosyl transferase (EC 2428) adenosine kinase (EC 27120) and
inosineguanosine kinase (EC 2421) which usually have high activities in plant cells
The resultant nucleotides then enter the purine alkaloid biosynthetic pathway In the
case of guanosine it may be directly deaminated to xanthosine and utilized for caffeine
biosynthesis (Figure 13) (Ashihara et al 1997)
182 SAM cycle in plants
SAM is the methyl donor for the three methylation steps in the caffeine biosynthetic
pathway In this process SAM is converted to S-adenosyl-L-homocysteine (SAH)
which in turn is hydrolyzed to homocysteine and adenosine Homocysteine is recycled
via the SAM cycle to replenish SAM levels and adenosine is released from the cycle
Adenosine released from the cycle is converted to AMP directly andor indirectly via
adenine AMP is converted to xanthosine and used for caffeine biosynthesis Since
three moles of SAH are produced via the SAM cycle for each mole of caffeine that is
synthesized in theory this pathway has the capacity to be the sole source of both the
Chapter 1 Introduction and Review of Literature
13
purine skeleton and the methyl groups required for caffeine biosynthesis in young tea
leaves (Koshiishi et al 2001)
Methionine
S-Adenosyl-L-methionineHomocysteine
ATP
PPi+ Pi
Tetrahydrofolate
5-Methyl-
tetrahydrofolate
CH3+Adenosine
S-Adenosyl-L-
homocysteine
(xanthine
structure)(Methyl groups)
Caffeine
(1)
(2)(3)
(4)
Figure 14 The SAM cycle (activated methyl cycle) in plants (Ashihara amp Suzuki 2004)
Enzymes SAM synthetase SAM-dependent N-methyltransferases S-adenosyl-
homocysteine (SAH) hydrolase Methionine synthase Adenosine released from the cycle
is salvaged to adenine nucleotides and utilized both for purine structure of caffeine via
xanthosine and for re-synthesis of SAM via ATP
183 N-methyltransferase in caffeine biosynthesis Molecular cloning
The genes encoding for the enzymes involved in the caffeine biosynthesis were
successfully cloned from the coffee family (Ogawa et al 2001 Mizuno et al 2003a
Mizuno et al 2003b Uefuji et al 2003) The conserved amino acid regions of tea CS
(AB31280) the sequences of O-methyltransferases derived from plant origin and
Arabidopsis hypothetical proteins were made use of in designing primers for RT-PCR
and RACE technique Independent groups have isolated N-methyltransferase (NMT)
genes from coffee which have been classified based on the substrate specificity of the
recombinant proteins expressed in Ecoli (Table 15) PCR products thus obtained were
used as the probe to isolate full length cDNA from the library resulting in the
identification of all three N-methyltransferases involved in caffeine biosynthesis
(Figure 12) Several cDNA have been characterized from the coffee plants one for
xanthosine methyltransferase (XMTXRS) three for 7-methylxanthine methyltransferase
or theobromine synthase (MXMTCTS) and three for 37-dimethylxanthine
methyltransferase or caffeine synthase (DXMTCCS) (Table 15) (Kato amp Mizuno
2004)
A single gene encoding for xanthosine methyltransferase (XMTCmXRS1) was
identified which encoded for a polypeptide consisting of 372 amino acids with an
apparent molecular mass of 418kDa It is expressed almost uniformly in aerial tissues
Chapter 1 Introduction and Review of Literature
14
of C arabica including leaves floral buds and immature but not in mature beans
Three genes encoding 7-methylxanthine methyltransferase (theobromine synthase) have
been identified The number of amino acids in the putative polypeptides are 378 for
MXMT1 (427kDa) and 384 for MXMT2 and CTS2 (434kDa) They differ by insertion
or deletion of blocks of several residues in the C-terminal region Their catalytic
properties based on kinetic parameters such as Km values were different They are
expressed in young leaves floral buds and immature but not mature beans Three genes
were identified for 3 7-dimethylxanthine methyltransferase (caffeine synthase) DXMT
CCS1 and CtCS7 each encoding a 43 kDa polypeptide consisting of 384 amino acids
However their kinetic properties differ from each other such as DXMT and CCS1
showed Km values of 1200 and 157microM for theobromine respectively Expressions
profiles are also distinct DXMT being expressed exclusively in immature beans while
CCS1 expression is ubiquitous occurring in all tissues The presence of isoforms of
these enzymes with different properties suggests that caffeine is synthesized through
multiple pathways depending on availability and concentration of the substrate (Mizuno
et al 2001 Ogawa et al 2001 Mizuno et al 2003a Mizuno et al 2003b Uefuji et
al 2003) Genomic clones of theobromine synthase were obtained by PCR- RFLP
from C canephora (Satyanarayana et al 2005 Satyanarayana 2006) The genomic
clones are reported to have highly conserved exons and a variable third intron The
promoter for the theobromine synthase was cloned and characterized (Satyanarayana et
al 2005)
There is high degree of similarity in the coding region of coffee NMT genes
isolated so far though differences exist in the 3prime untranslated region (UTR) for these
genes The deduced amino acid sequence of these enzymes shows more than 85
homology between these genes Phylogenetic analysis indicates that they are more
closely related to C-methyltransferases including those for jasmonic acid salicylic acid
and benzoic acid than to other N-methyltransferases This suggests that coffee N-methyl
transferases belong to a distinct sub-group within plant methyltransferases Their
cellular localization determined by green fluorescent protein fusion method and β-
glucourodinase (GUS) showed that the all the three genes are localized in the cytoplasm
(Ogawa et al 2001 Vinod et al 2007) It was also shown that these were present
along with chlorogenic acid(Vinod et al 2007)
Chapter 1 Introduction and Review of Literature
15
Table 15 N-methyltransferases and their encoding genes involved in caffeine biosynthesis
Gene (Enzyme) Gene name Accession No Description Reference
Xanthosine methyltransferase
( 7-methylxanthosine synthase) (EC 211158)
CaXMT AB 048793 Immature fruit cDNA Screening (Uefuji et al 2003)
CmXRS1 AB 034699 Leaf cDNA library RACE (Mizuno et al 2003a)
CaMXMT1 AB 048794 Leaf cDNA library RACE (Ogawa et al 2001)
7-methylxanthine synthase transferase
(theobromine synthase) ( EC 211159)
CTS1 AB 034700 Immature fruit cDNA Screening (Uefuji et al 2003)
CaMXMT2 AB 984125 Leaf cDNA library Screening (Mizuno et al 2001)
CTS2 AB 054841 Leaf cDNA library Screening (Mizuno et al 2001)
37-methylxanthinemethyltransferase
(Caffeine synthase) (EC 211160)
CaDXMT1 AB 086414 Immature fruit cDNA Screening (Uefuji et al 2003)
CCS1 AB 0861414 Immature fruit cDNA Screening (Uefuji et al 2003)
CtCS7 AB 0864415 Immature fruit cDNA
NMT genomic clones
Theobromine synthase-1
CX10 Complete coding region PCR Satyanarayan 2006
CX8 Complete coding region PCR -do-
Theobromine synthase-1 PG-5 Promoter + coding region RAGE -do-
Theobromine synthase-1 PG-1 Promoter + coding region RAGE Satyanarayan 2006
PG-4 Promoter + coding region RAGE -do-
Theobromine synthase-1 NMT
(AY273814) Complete coding region PCR Kochko A et al 2010
Theobromine synthase-2 NMT
(AY362825) Complete coding region PCR Kochko A et al 2010
Chapter 1 Introduction and Review of Literature
16
19 Catabolism of caffeine
Caffeine is produced in young leaves and immature fruits and continues to accumulate
gradually during the maturation of these organs At the same time caffeine is slowly
degraded with the removal of the three methyl groups resulting in the formation of
xanthine Catabolism of caffeine was first reported in coffee leaves (Kalberer 1965)
Since then a number of tracer experiments using 14
C labelled purine alkaloids have
been reported (Suzuki amp Waller 1984 Ashihara et al 1996 Vitoria amp Mazzafera
1999 Mazzafera 2004) They demonstrated the major catabolic pathway is caffeine rarr
theophylline rarr 3-methylxanthine rarr xanthine Xanthine is further degraded by the
removal by the conventional purine catabolic pathway to CO2 and NH3 via uric acid
allatonin and allantoate (Figure 15) (Ashihara amp Crozier 1999 Stasolla et al 2003
Zrenner et al 2006) Caffeine catabolism usually begins with its conversion to
theophylline catalyzed by N7-demethylase The involvement of P450-dependent mono-
oxygenase activity for this reaction was suggested (Huber amp Baumann 1998
Mazzafera 2004) However the activity of this enzyme has not yet been determined in
cell free extracts [8-14
C] Theophylline degraded to CO2 at much faster rate than [8-14
C]
caffeine indicating that conversion of caffeine to theophylline is a major rate limiting
step of caffeine catabolism and a possible reason why caffeine accumulates in high
concentrations in tissues of C sinensis and C arabica It is widely believed that the
control of caffeine levels in plants is a function of the balance between rates of
synthesis and degradation and this balance seems to vary depending on the plant
species and the tissue developmental stage (Mazzafera 2004)
Chapter 1 Introduction and Review of Literature
17
Figure 15 Caffeine catabolic pathways Caffeine is mainly catabolised via theophylline and 3-
methylxanthine Xanthine is further degraded to CO2 and NH3 by the conventional
oxidative pathway Adapted from (Ashihara et al 2008)
Chapter 1 Introduction and Review of Literature
18
110 Genetic engineering of Coffee
Genetic transformation has potential applications in coffee agriculture for incorporating
genes which could provide desirable traits such as disease and insect resistance
drought and frost tolerance and herbicide resistance Transgenic technology can also be
used to increase nutritional value and improve cup quality produce varieties with
caffeine-free beans and for production of hybrid crops for molecular farming
Identification of target specific genes is one of the pre-requisites for developing
transgenic crops The availability of a large number of EST sequences in coffee and
initiation of coffee genome sequencing may speed up the gene discovery and accelerate
transgenic research efforts in coffee
1101 Insect Resistance
Production of insect resistant coffee plants is one of the major objectives of the
breeding programs The major pests attacking coffee include coffee berry borer (CBB
Hypothenemus hampei) white stem borer (WSB Xylotrechus quadripes) leaf miner
(Perileucoptera coffeela) and root nematodes (Meloidogyne spp and Pratylenchus
spp) The CBB is present in almost all the coffee growing countries and considered to
the most devastating pest in coffee To date there is no reported source of resistance to
CBB in the coffee gene pool Like CBB WSB is another serious pest in C arabica in
India and several other East Asian countries Both CBB and WSB belong to the order
Coleoptera For India controlling WSB is the biggest challenge and has therefore
become the highest research priority Robusta is generally resistant to WSB but the
interspecific Robusta Arabica hybrids are susceptible to WSB Although leaf miner is
not yet a serious pest in India and other East Asian countries it is an economically
important pest in East Africa and Brazil Effective chemical control of CBB and WSB
is difficult due to the nature of their life cycle inside the berry and stem respectively as
well as environmental concern regarding the use of pesticides Biological control
measures are adapted to combat these insect pests with varying degrees of success
Developing coffee plants resistant to these pests using genetic transformation
technology could be one of the alternative strategies to counter pest damage For insect
resistance several different classes of proteins from bacterial plant and animal sources
have been isolated and their insecticidal properties tested against many important pests
Amongst these proteins Bt toxins are most important and several transgenic crops
expressing Bt genes have been commercialized Coffee transgenic plants carrying a
Chapter 1 Introduction and Review of Literature
19
synthetic version of the cry1Ac gene have been produced (Leroy et al 2000 Mishra et
al 2002) Indeed this was the first report of an important agronomic trait being
introduced into a coffee plant The transgenic plants presented similar features in
growth and development compared to normal plants Transgenic plants highly resistant
to leaf miner under greenhouse conditions were tested under field conditions in French
Guyana for 4 years for pest resistance (Perthius et al 2005 Perthius et al 2006)
From 54 independent transformation events 70 of the events were resistant to leaf
miner Unfortunately the field trial was vandalized which led to the termination of the
experiment (Montagnon et al 2005)The effectiveness of Bt genes in controlling
coleopteran pests is well documented in corn and potato which indicates that Bt genes
might be effective against CBB The high toxicity of B thuringiensis sero var
israelensis against first in star larvae of CBB has been demonstrated (Mendez et al
2003) In another experiment an α-amylase inhibitor from Phaseolus vulgaris was
tested against CBB and found to have an inhibitory effect on its growth and
development (Grossi et al 2004)In addition to the CBB and WSB C arabica
varieties are also susceptible to endoparasitic root-knot nematode (Meloidogyne spp)
(Campos et al 1990 Mishra et al 2012)
So far 15 species have been reported to be parasites of coffee Controlling
nematodes is extremely difficult and currently seedling grafting with robusta rootstock
is followed as one of the control measure Sources of resistance specific to root-knot
nematodes have been identified in coffee trees (Bertrand et al 2001) and the Mex-1
gene conferring resistance to M exigua in C arabica is in the process of isolation (Noir
et al 2003)
1102 Tolerance to Abiotic Stress
In many coffee growing countries abiotic stress such as drought and frost are the major
climatic factors that limit coffee production Changes in climatic patterns due to global
climate change are considered increasingly important for coffee cultivation Drought
induces water stress in plants which affects vegetative growth and vigor and triggers
floral abnormalities and poor fruit set It also indirectly increases the incidence of pests
and diseases in the plants C arabica is generally more tolerant to water stress than C
canephora partly due to its extensive deep root system C racemosa is known to be a
good source of drought tolerance In India hybrids between C canephora and C
racemosa have been obtained and are currently under evaluation for drought tolerance
Chapter 1 Introduction and Review of Literature
20
In many coffee growing countries coffee is propagated in marginal areas where the
annual rainfall is below 1000thinspmm with prolonged dry spells of over 4-5 months In
those areas water shortage and unfavorable temperatures constitute major constraints
and the growth and productivity of C canephora are badly affected As with drought
periodic frost also affects coffee production in parts of Brazil The introduction of
drought and frost tolerant genes through genetic transformation would be of great
importance for alleviating these problems Research is now being carried out by several
groups to identify genes involved in biotic as well as abiotic stress The most promising
genetic engineering approach for drought tolerance includes the use of functional or
regulatory genes as well as the transfer of transcription factors In recent years plants
tolerant to high temperature and water stress have been the subject of intense research
(Chaves et al 2003 Chaves et al 2004 Coraggio et al 2004) For achieving drought
tolerance genes that have been targeted include those encoding enzymes involved in
detoxification or osmotic response metabolism enzymes active in signaling proteins
involved in the transport of metabolites and regulating the plant energy status (Chaves
et al 2003 Chaves et al 2004 Coraggio et al 2004) The dissection of molecular
mechanisms related to signal transduction and transcriptional regulation might help in
engineering drought tolerance in coffee
1103 Disease Resistance
Coffee leaf rust (CLR) caused by the fungus Hemileia vastatrix is the most important
disease in coffee with substantial loss to coffee production and productivity in all the
coffee growing countries In addition to leaf rust coffee berry disease (CBD) caused by
fungus Colletotrichum kahawae can be a devastating anthracnose leading to substantial
crop loss in Africa Several other fungal and bacterial diseases may also affect coffee
to a extent C arabica is more susceptible to many diseases than C canephora Though
most of the disease control measures rely upon chemical control they are more
expensive and labour intensive The long-term solution is the breeding of resistant
varieties which is the focus of many breeding programs However breeding for disease
resistant varieties is time consuming due to the perennial nature of coffee with its long
gestation period India has a long history of C arabica breeding especially for leaf rust
resistance and is the first country to demonstrate the existence of multiple races of leaf
rust Resistance to CLR is conditioned primarily by a number of major (SH) genes and
coffee genotypes are classified into different resistance groups based on their
Chapter 1 Introduction and Review of Literature
21
interaction with different rust races pathogen (van der Vossen 2001) Currently C
canephora provides the main source of resistance to pests and diseases including CLR
(H vastatrix) and CBD (C kahawae) and therefore used in breeding programs Other
diploid species like C liberica and C racemosa are being used as a source of resistance
to coffee leaf rust and coffee leaf miner respectively (Guerreiro et al 1999
Sreenivasan et al 1989) The development of coffee varieties resistant to major fungal
diseases such as CLR and CBD using transgenic technology will benefit the coffee
industry immensely During the last 15 years significant progress has been made in the
area of host-pathogen interactions (Staskawicz et al 1995 Feys et al 2000
Shivaprasad et al 2012) and many resistance genes involved in recognizing invading
pathogens have been identified and cloned (Takken et al 2000) A number of
signalling pathways induced because of pathogen infection have been dissected (Dong
et al 1998 Navarro et al 2006) Many antifungal compounds synthesized by plants to
combat fungal infection have been identified (Does et al 1998) Understanding the
specific induction of targeted pathways and identification of specific pathways
responsible for particular fungal resistance is important in order to employ this strategy
in transgenic technology The recent investigation of gene expression during coffee leaf
rust infection could provide an insight into the defence pathways operating in coffee
(Fernandez et al 2004 Nardi et al 2006) Efforts have been made to identify and
clone resistance genes from coffee for achieving durable resistance The genetic and
physical map of two resistance genes SH3gene conferring resistance to rust (Prakash et
al 2004) and the Ck1 gene conferring resistance to C kahawae CBD (Gichuru et al
2006) have been established These genes could be used for molecular marker assisted
breeding programs (Mishra et al 2012)
1104 Production of low-caffeine coffee
A widespread belief that the ingestion of caffeine can have adverse effects on
health resulted in the increased demand for decaffeinated coffee (Ashihara amp Crozier
2001) Several method were used to obtain decaffeinated plants by conventional
breeding techniques using low yielding varieties and mutants that accumulated
relatively low amounts of caffeine when compared to the commercially available ones
Low-caffeine and decaffeinated coffee represent around 10 of the coffee sales
around the world (Ribas et al 2006b) The industrial process for coffee decaffeination
can be expensive and affects the original flavour and aroma in coffee (David 2002)
Chapter 1 Introduction and Review of Literature
22
Transgenic coffee plants with suppressed caffeine synthesis using RNA interference
(RNAi) technology have been obtained (Ogita et al 2003 Ogita et al 2004) Specific
sequences in the 3prime untranslated region of the theobromine synthase gene (CaMXMT1)
were selected for construction of RNAi short and long fragments The caffeine and
theobromine content of the transgenic plants were reduced by ~ 70 when compared to
the untransformed plants In C canephora RNAi technology has also been employed
to silence the N-methyl transferase gene involved in caffeine biosynthesis (Vinod et al
2007) Promoter of an N-methyltransferase (NMT) gene involved in caffeine
biosynthesis was cloned (Satyanarayana et al 2005 Satyanarayana 2006) which will
be very useful for studying the regulation of caffeine biosynthesis
1105 Improvement in cup quality
Improvement in coffee cup quality requires elaborate knowledge of the chemical
constituents as well as the metabolic pathways involved in the elaboration of quality
The constituents of coffee beans include minerals proteins carbohydrates caffeine
chlorogenic acids (CGA) glycosides lipids and many volatile compounds that give
flavour to coffee by roasting Among these the role of three major constituents
sucrose CGA and trigonelline have been studied in coffee The sucrose content of
coffee bean is associated with coffee flavour the higher the sucrose content in green
beans the more intense will be the cup flavour (Clifford et al 1985 de Maria et al
1996) The sucrose content of C arabica (82-83) is higher than C canephora (33ndash
40) The sucrose amino acids ratio in green beans determines the profile of volatile
compounds Manipulating sucrose content in coffee bean is therefore important in
improving cup quality The sucrose synthase gene (CcSUS2) from C canephora has
been cloned and sequenced (Leroy et al 2005) This provides an opportunity to
manipulate the sucrose content in coffee Chlorogenic acids are products of
phenylpropanoid metabolism Chlorogenic acids are a group of hydroxycinnamoyl
quinic acids (HQA) formed by esterification between caffeic acids coumaric acids and
quinic acids (Clifford et al 1999) They are present in relatively large quantities in the
coffee bean and are the precursor of phenolic compounds in roasted beans C
canephora beans contain higher CGA (10) compared to C arabica beans (6-7)
CGA are known to have antioxidant properties as well as being associated with disease
resistance (Campa et al 2003) Genetic manipulations of genes involved in CGA
synthesis can serve either of these purpose by up- or down regulating the pathway
Chapter 1 Introduction and Review of Literature
23
Phenylalanine ammonia-lyase (PAL) catalyzes the first step of the phenylpropanoid
pathway leading to the synthesis of a wide range of chemical compounds including
flavonoids coumarins hydroxycinnamoyl esters and lignins (Hahlbrock amp Scheel
1989) Recently the full length cDNA and corresponding genomic sequences of PAL
from C canephora was isolated characterized and functionally validated (Mahesh et
al 2006 Parvatam et al 2006) This has opened up new possibilities for manipulating
the level of the PAL enzyme in coffee which in turn can be useful for improvement of
cup quality and manipulating antioxidant properties in coffee
1106 Fruit Ripening
Uniformity during fruit ripening is decisively related to cup quality in coffee and
consequently to the value of the product Fruits at the ideal ripening stage produce the
best organoleptic characteristics for coffee The presence of over ripened or green fruits
changes the acidity the bitterness and consequently the cup quality In order to
maximize uniform ripening of coffee fruits it is essential to control the action of genes
involved in the last step of maturation process Ethylene is known to trigger ripening
and increasing ethylene biosynthesis is associated with various stages of ripening
process (Protasio et al 2005) To control coffee fruit maturation two of the major
genes involved in ethylene biosynthesis namely ACC synthase and ACC oxidase
have been cloned (Protasio et al 2005 Neupane et al 1999) Introduction of the ACC
oxidase gene in antisense orientation have been achieved in both C arabica and C
canephora The effect of the transgene on ethylene production and fruit maturation has
yet to be reported The inhibition of genes downstream to the initial ethylene burst is
also an option to control coffee fruit maturation (Ribas et al 2006)
111 RNA interferenceRNAi
The regulation of gene expression at post transcriptional level has recently attracted
much attention because of the discovery of the phenomenon called RNA interference
(RNAi) RNAi based silencing techniques are more efficient than antisense mediated
gene silencing (Wesley et al 2001) The effect was first observed in plants wherein
silencing was observed when some copies of a transgene was in the forward orientation
with respect to the promoter and some in the reverse orientation This resulted in the
formation of double stranded RNA in the system The phenomenon in which
experimentally introduced double stranded RNA (dsRNA) leads to the loss of
expression of the corresponding cellular gene by sequence specific RNA degradation
Chapter 1 Introduction and Review of Literature
24
was termed as RNA interference or RNAi The protective effect of RNA silencing in
reducing virus infectivity supports the view that PTGS has evolved as a mechanism to
defend plants against virus infection and also to moderate the possible deleterious
genome-restructuring (insertional) activity of virus-like mobile genetic elements eg
retrotransposons A growing body of biochemical and genetic data has further
demonstrated that plants animals and yeasts share related mechanisms of specific
degradation of RNAs in which double-stranded forms of RNA are initiator molecules
(Brenstein et al 2001)
The key insight into the process of PGTS was provided by the experiments
conducted by Hamilton amp Baulcombe (1999) who identified the product of RNA
degradation as small RNA (siRNA) species of ~25nt of both sense and antisense
polarity SiRNA are formed and accumulate as double stranded RNA molecules of
defined chemical structures Both genetic and biochemical approaches were undertaken
to understand the basis of silencing Genetic screens were carried out in fungus
Neurospora carassa the alga Chalmydomonas reinhardtii and plant Arabidopsis
thaliana to search for detective mutants in PTGS
A combination of results obtained from several in vivo and in vitro experiments
have gelled into a two step mechanistic model for RNAiPTGS The first step referred
to as the RNAi initiating step involves binding of the RNA nucleases to a larges
dsRNA and its cleavage into discrete ~21-25nt RNA fragments This is ATP-
dependent reaction which involved RNase III - type endonucleases which make
staggered cuts in both strands of the dsRNA leaving 3prime overhang of 2 nucleotides with
5prime- phosphate 3prime-hydroxyl and no modification in the sugar phosphate backbone
(Hamilton amp Baulcombe 1999 Elbashir et al 2001) SiRNAs are the direct products
of dsRNA cleavage by the multi-domain RNase III enzyme DICER (Figure 16)
(Brenstein et al 2001 Karthikeyan et l 2013)
In the second step or commonly known as the effector step the double stranded
siRNAs produced in the first step bind to an RNAi specific protein complex to form a
RISC The complex undergoes activation in the presence of ATP so that the antisense
component of the unwound siRNA becomes exposed and allows the RISC to perform
the downstream RNAi reaction
Chapter 1 Introduction and Review of Literature
25
Several enzymes are involved in RNAi based on their role genes that encode for them
are classified into RNAi initiators RNAi effectors RNA dependent RNA polymerases
and silencing genes Two C elegans genes rdeI and rde4 (rde stands for lsquo RNAi
deficientrsquo) are believed to be involved in the initiation step of RNAi The C elegans
rde1 gene is a member of a large family of genes and is homologous to the Neurospora
qde2 (qde stands for ldquoquelling deficientrdquo) and Arabidopsis AGO1 (AGO stands for
agronaute AGO1 was previously identified to be involved in Arabidopsis
development) Although the functions of these genes are not clear a mammalian
member of RDE1 family has been identified as a translation initiation factor (Thakur
2005) Important genes for the effector step of PTGS in C elegans are rde2 and mut7
genes These genes were identified from heterozygous mutant worms that were unable
to transmit RNAi to their homozygous offsprings Worms with mutated rde2 or mut 7
genes show defective RNAi mut-7 gene encodes a protein with homology to the
nuclease domains of RNAase D and a protein implicated in Werner syndrome (a rapid
ageing disease in humans) (Grishok et al 2001 Kamath-Loeb et al 2012s)
Neurospora qde-1 Arabidopsis SDE-1SGS-2 and C elegans ego-1 appear to encode
RNA dependent RNA polymerase (RdRPs) It assumed that this is a proof that an RdRp
activity is required for RNAi Certainly the existence of an RdRp might explain the
remarkable efficiency of dsRNA induced silencing if it amplifies either the dsRNA
prior to cleavage or the siRNAs directly (Muers 2013) In C elegans ego-1 mutants
(ego stands for lsquoenhancer of glp-1) RNAi functions normally in somatic cells but is
defective in germline cells where ego-1 is primarily expressed In Arabidopsis SDE-
1SGS-2 mutants (SGS stands for suppressor of gene silencing) siRNA are produced
when dsRNA is introduced via an endogenously replicating RNA virus but not
introduced by a transgene It has been proposed that perhaps the viral RdRP is
substituting for the Arabidopsis enzyme in these mutants Random degradative PCR
model suggests that an RdRP uses the guide strand of an siRNA as a primer for the
target mRNA generating a dsRNA substrate for Dicer and thus more siRNAs
Several genes controlling RNA silencing in plants have been identified through
genetic screens of Arabidopsis mutants impaired in transgene induced RNA silencing
They encode a putative RNA-dependent RNA polymerase (SGS2SDE1) a coiled
protein (SGS3) a protein containing PAZ and Piwi domains (AGO1) and an RNA
helicase (SDE3) The putative SGS2SDE1 of Neurospora is related to QDE-1 and
Chapter 1 Introduction and Review of Literature
26
EGO-1 of C elegans whereas PAZPiwi protein AGO is related to QDE-2 of
Neurospora RDE-1of C elegans RNA helicase SDE3 is related to SMG-2 of C
elegans and Mut-6 of Chlamydomonas MUT-7 gene of C elegans encodes a protein
similar to Rnase D whereas the Drosophila DICER gene encode a protein similar to
RNase III An Arabidopsis ortholog of DICER gene has been identified called CAF
SIN1 SUS1(Thakur 2005 Poethig 2009)
Since the RNAi machinery is present constitutively within the eukaryotic cell it
is important to explore the metabolic advantages that are accorded by the RNAi related
proteins during the intrinsic normal growth of cells and development of organisms The
natural RNAi machinery not only keeps the mobile transposable elements from
disrupting the integrity of the genomes as suggested by the analysis of model organisms
like A thaliana C elegans D melanogaster (Tabara et al 1999 Wu-Scharf et al
2000 Aravin et al 2001 Hamilton et al 2002 Lisch 2009) but also participates in
development of organisms Genetic defects in Celegans RNAi genes ego1 and dicer
cause known specific developmental errors (Grishok et al 2000 Grishok et al 2001
Knight amp Bass 2001 Hall et al 2013) Similarly the Argonaute family of genes of A
thaliana (especially the ZWILLE proteins) is also responsible for plant architecture and
meristem development (Carmell et al 2002 Hamant and Pautot 2010) and the Dicer
homologue of A thaliana CAF1 is required for embryo development (Golden et al
2002 Nakano et al 2011) Thus genetic evidence illustrates the role of the RNAi
machinery as a controller of development related genes The mechanistic details of
these developmental processes are beginning to emerge
Chapter 1 Introduction and Review of Literature
27
Figure 16 Mechanism of gene silencing induced double stranded RNA The dicer enzyme to
produce siRNAs cleaves the RNA The siRNA generated bind to the nuclease complex
RISC The antisense component in the siRNA in the RISC guides the complex towards
the cognate mRNA resulting in endonucleolytic cleavage of the mRNA
Chapter 1 Introduction and Review of Literature
28
112 MicroRNA or MiRNA
In 1991 Ambros and co-workers first isolated a lin-4 mutant of Celegans which was
arrested at the first larval stage (Lee et al 1993) Later on the let-7 mutation was
isolated in the same system which was responsible for development through the fourth
larval stage Both lin-4 and let-7 encode short 22-nucleotide mature RNAs and were
called short temporal RNA because they control the temporal development program of
C elegans The mature lin-4 RNA defines (negatively regulates) the mRNA expression
of the lin-14 and lin-28 hetrochronic genes with the antisense mediated repression
mechanism of translation initiation and thus specifies the fate of cells during the first
three larval stages Recent studies have revealed that the short temporal RNAs are
actually members of a group of tiny RNAs (21to 28 nucleotides) called the micro-
RNAs (miRNA) (Bartel 2009) isolated members of which could easily run to a few
thousand which includes both those were identified computationally and by deep
sequencing Some of the components of the RNAi machinery have also been clearly
established as the effector proteins for the maturation of miRNAs
113 MicroRNAs in plants
1131 Discovery
MicroRNAs have been discovered by three basic approaches Direct cloning forward
genetic direct cloning and bioinformatic prediction followed by experimental
validation The most direct method of miRNA discovery is to isolate and clone small
RNAs from biological samples and several groups have used this approach to identify
plant miRNAs (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002 Xie et al 2003 Sunkar et al 2005 Williams et al 2013) The cloning methods
adapted were similar to those used to identify large numbers of animal miRNAs
(Lagos-Quintana et al 2001 Lau et al 2001 Aravin amp Tuschl 2005) which involve
isolating small RNAs followed by oligonucleotide adapter ligation reverse
transcription amplification and sequencing Some protocols incorporate methods to
enrich for Dicer cleavage products (ie molecules with 5prime-phosphates and 3prime-
hydroxyls) and to concatemerize the short cDNAs so that several may be identified in a
single sequencing read (Lau et al 2001 Llave et al 2002a Jagadeeswaran amp Sunkar
2013)The initial cloning experiments in Arabidopsis identified 19 miRNAs belonging
to 15 families (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002) although hundreds of other small RNAs such as siRNAs and RNA degradation
Chapter 1 Introduction and Review of Literature
29
fragments were also cloned through the direct cloning methods To select miRNAs
from the pool of small RNAs secondary structures of the RNA molecules were
examined in compliance with the acknowledged structural features of miRNAs
(Ambros et al 2003) The secondary structures were validated using several ad-hoc
software tools such as the Mfold or RNAfold programs (Reeder et al 2006) Finally
the existence of mature miRNAs was determined by RNA gel blot hybridization These
direct cloning methods were successful in isolating many miRNAs in Arabidopsis
(Reinhart et al 2002 Sunkar amp Zhu 2004) Rice (Sunkar et al 2005) Poplar (Lu et
al 2005) moss (Arazi et al 2005) and Tobacco (Billoud et al 2005)
Although miRNAs were first discovered through forward genetic screens in
worms (Lee et al 1993 Reinhart et al 2000)only few A thaliana miRNA gene
families have been discovered by this method (Chen 2008) in plants MiRNA
involvement in plant mutant phenotypes were not properly inferred till the cloning
experiments established that plant genomes contain numerous miRNAs (Park et al
2002 Reinhart et al 2002 Rhoades et al 2002) MiRNA loss-of-function allele has
been identified by forward genetic screens early extra petala1 is caused by a
transposon insertion ~160bp upstream of the predicted miR164c stem loop resulting in
flowers with extra petals (Baker et al 2005 Irish 2008) The fact that loss-of-function
miRNA mutants have been rarely discovered using forward genetics reflects small
target size for mutagenesis coupled with redundancy in nearly all evolutionarily
conserved plant miRNAs which are encoded by gene families (Table16) Family
members are likely to have overlapping functions buffering against loss at any single
miRNA locus Over expression screens can circumvent redundancy limitations At least
four plant miRNAs miR319 (also known as miR-JAW) miR172 (also known as EAT)
and miR166 were isolated in over expression screens for dominant mutants with
developmental abnormalities (Aukerman amp Sakai 2003 Palatnik et al 2003 Kim et
al 2005 Williams et al 2005b Schommer et al 2012) Mutations in miRNA target
sites which can prevent the entire family of miRNAs from repressing a target gene can
also circumvent redundant functions of miRNA family members
The dominant mutations in the HD-ZIP genes PHB PHV and REVOLUTA
(REV) in Arabidopsis and ROLLEDLEAF1 (RLD1) in Maize result in adaxialization of
leaves andor Vasculature (McConnell amp Barton 1998 McConnell et al 2001 Nelson
et al 2002 Zhong amp Ye 2004 Allen amp Millar 2012) and are all caused by mutations
Chapter 1 Introduction and Review of Literature
30
in miR166 complementary sites (Rhoades et al 2002 Emery et al 2003 Jones-
Rhoades amp Bartel 2004 Mallory et al 2004 Zhong amp Ye 2004)
In both plants and animals cloning was the initial means of large-scale
miRNA discovery (Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Reinhart et al 2002 Mendes et al 2012) However cloning is biased toward
RNAs that are expressed highly and broadly MiRNAs expressed at low levels or
only in specific cell types or in response to certain environmental stimuli were more
difficult to clone Sequence based biases in cloning procedures might also cause
certain miRNAs to be missed Because of these limitations bioinformatics approaches
to identify miRNAs have provided a useful complement to cloning
A straightforward use of bioinformatics has been carried out to find homologs
of known miRNAs both within the same genome and in the genomes of other species
(Pasquinelli et al 2000 Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Mendes et al 2009) A more difficult challenge is to identify miRNAs
unrelated to previously known miRNAs This was first accomplished for
vertebrate nematode and fly miRNAs using algorithms that search for conservation
of sequence and secondary structure (ie miRNA stem-loop precursors) between
species searching for patterns that are characteristic of miRNAs (Lai et al 2003 Lim
et al 2003 Lim et al 2005)
Most of the methods identified numerous miRNAs but are necessarily more
relaxed in terms of allowed structures Some approaches take advantage of the high
complementarity of plant miRNAs to target messages implementing the requirement
that the candidate has conserved complementarity to mRNAs (Jones-Rhoades amp Bartel
2004 Adai et al 2005) This additional filter has been useful for distinguishing
authentic plant miRNAs from false positives which later extended to mammalian
miRNA gene prediction (Xie et al 2005 Mendes et al 2012)
Another criteria used by most algorithms in the filters is evolutionary
conservation In plants mature miRNA sequences are conserved to a higher degree
than their precursor sequences For example Arabidopsis and rice diverged more than
130 million years ago (Friis et al 2004) but some miRNAs are highly conserved
in these plant species (Reinhart et al 2002) Furthermore various parameters for the
secondary structures such as free energy the number of paired residues within a
Chapter 1 Introduction and Review of Literature
31
miRNA or the number and size of bulges are also used to validate the predictions
(Jones-Rhoades amp Bartel 2004 Wang et al 2004 Adai et al 2005 Mendes et al
2012)
1132 Conserved microRNAs in Plants
Till date cloning genetics and bioinformatics screens have resulted in annotation of
approximately 184 potential Arabidopsis miRNAs (miRBase release 13) These
miRNAs seem to arise through gene duplication and subsequent diversification and
represent approximately 100 families (Maher et al 2006 Axtell 2013) Twenty one
families represented by 92 genes are clearly conserved in species beyond Arabidopsis
(Table 15) The presence of these miRNA families in other sequenced plant genomes
indicates that different plant species have an evolutionarily fluid set of miRNAs
(Willmann amp Poethig 2007) Recent studies on mosses one of the most ancient land
plants have shown that at least 14 miRNA families are conserved in eudicots and
mosses and therefore it appears that plant miRNAs share a common ancient origin
(Arazi et al 2005 Axtell amp Bartel 2005 Talmor-Neiman et al 2006 Zhang et al
2006 Axtell 2013) The number of members per family in a genome ranges from 1-32
with the exception of miR-430 which is represented by a cluster of ~80 loci in zebra
fish (Giraldez et al 2005)
In plants the number of members in each family correlates among examined
species certain families contain numerous members in all three species (eg miR156
miR166 miR169) whereas others consistently contain fewer genes (eg miR162
mir168 miR394) (Table 16) Although it is still unclear why certain miRNA are
encoded by more than one gene in plants (eg 12 genes encoding miR156) this
correlation might suggest functional significance of various miRNA family sizes Most
annotated plant miRNAs are conserved throughout flowering plants while a few
miRNAs are found only in a single sequenced genome and thus they could be
evolutionarily lsquoyoungrsquo miRNAs For example Arabidopsis has several miRNA
families that are not found in poplar or rice suggesting that miRNAs could be
generated continuously during evolution (Allen et al 2004a Rajagopalan et al
2006 Fahlgren et al 2007 Axtell 2012 de Alba et al 2013) Consistent with the
notion that non conserved miRNAs are of a more recent evolutionary origin these
miRNAs are mainly encoded by a single locus in the genome Within each family the
mature miRNA is always located in the same arm
Chapter 1 Introduction and Review of Literature
32
Table 16 MicroRNA families conserved in plants
miRNA
family
Arabidopsis Oryza Populus
miR156 12 12 11 miR159319 6 8 15 miR160 3 6 8 miR162 2 2 3 miR164 3 5 6 miR166 9 12 17 miR167 4 9 8 miR168 2 2 2 miR169 14 17 32 miR171 4 7 10 miR172 5 3 9 miR390 3 1 4 miR393 2 2 4 miR394 2 1 2 miR395 6 19 10 miR396 2 5 7 miR397 2 2 3 miR398 3 2 3 miR399 6 11 12 miR408 1 1 1 miR403 1 0 2 miR437 0 1 0 miR444 0 1 0 miR445 0 9 0 Total 92 127 169
All known miRNA families that are conserved between more than one plant species are listed
together with the number of genes identified in the sequenced genomes Rice miRNA families that have orthologs in maize but do not appear to have orthologs in the eudicots (Arabidopsis and Populus) are marked with an asterisk The following families contain miRNA genes annotated with
more than one number miR156 (miR156 and miR157) miR159319 (miR159 and miR319) miR166 (miR165 and miR166) miR171 (miR170 and miR171) and miR390 (miR390 and miR391)
of the stem loop (5prime- or 3prime) as it would be expected if the genes carry common ancestry
(Figure 17) The sequence of the mature miRNA and to some extent the segment on
the opposite arm of the hairpin to which it pairs is highly conserved between the
members of the same family (both within and between the species) while the sequence
secondary structure and length of the intervening ldquolooprdquo region can be highly divergent
within the same family
The patterns of paring and non pairing nucleotides are often conserved between
homologous miRNA stem loops from different species (Figure 17) The significance of
conserved mismatches is opined to guide DCL1 to cleave at the appropriate positions
along the stem loop
Chapter 1 Introduction and Review of Literature
33
Figure 17 Representative stem loop of miR164 Segments corresponding to the mature miRNAs
are shown in red (Adapted from Rajagopalan et al 2006)
Although many miRNAs are highly conserved in plants the degree of conservation in
most cases is quite low between plants and animals one exception is miR854 A recent
study has revealed that miR854 is conserved between Arabidopsis and animals and that
the Arabidopsis miR854 differs from the human miR854 only by a single nucleotide
(Arteaga-Vazquez et al 2006 Axtell 2012 de Alba et al 2013) The Arabidopsis
miR854 has multiple binding sites in the 3prime-untranslated region (UTR) of
OLIGOURIDYLATE-binding-PROTEIN mRNA 1bA (UBP1b) and represses the
translation of the UBP1b mRNA The animal miR854 is also complementary to a site in
the 3prime-UTR of the UBP1b homolog in animals However it is currently unclear how the
animal miR854 regulates its target mRNA Another distinction between plant and
animal miRNAs is the genomic organization of the miRNA genes While plant miRNA
genes are located predominantly in the intergenic regions animal miRNA genes tend to
localize in the intragenic regions both in the introns or exons of protein coding genes
Moreover miRNA genes are often clustered within a single precursor transcript
whereas individual plant miRNAs are derived from individual precursor RNAs (Bartel
Chapter 1 Introduction and Review of Literature
34
2004 Bonnet et al 2004 Kim 2005 Vaucheret et al 2006 Marco et al 2013)
1133 Non-conserved miRNA and challenges in annotation
Most miRNAs are conserved throughout flowering plants (Table 15) but many more
were found only in a single sequenced genome thus considered to be of a more recent
evolutionary origin The extended homology between few non conserved miRNAs
precursors and their target genes provides strong evidence that these relatively ldquoYoungrdquo
miRNAs arose from the duplication of the target gene segments (Allen et al 2004)
Several non-conserved miRNAs including miR161 miR163 miR173 miR447
miR475 and miR476 are known to direct cleavage of target transcripts (Allen et al
2004a Adai et al 2005 Sunkar et al 2005 de Alba et al 2013) It is difficult to
confidently predict targets for many because it is not possible to use complementary
site as a filter against false-positive target predictions It is also difficult to
confidently annotate non-conserved miRNAs as miRNA rather than siRNAs The
established minimal standard for miRNA annotation is a small RNA with detectable
expression and the potential to form a stem-loop when joined to flanking genomic
sequence (Kasschau et al 2003) In practice these requirements are too loose to
definitively categorize many small RNAs cloned from plants Many plant siRNAs are
detectable on blots (Vazquez et al 2004b) and hundreds of thousands of non-
miRNA genomic sequences can be predicted to fold into secondary structures that
resemble structures of plant miRNA precursors (Jones-Rhoades amp Bartel 2004)
Therefore without conservation of both sequence and secondary structure it is
difficult to be confident that a given cloned RNA originated from a stem-loop (ie is a
miRNA) rather than from a double-stranded RNA (ie is a siRNA) In fact many of
the thousands o f small RNAs cloned from Arabidopsis (Gustafson et al 2005) would
probably meet the literal requirements for annotation as miRNAs A few of these
sequences might be miRNAs but others that meet the literal criteria probably are not
so The challenges of annotating non conserved small RNAs were evidenced by
three related small silencing RNAs that were originally annotated as miRNAs
that turned out to be trans-acting siRNAs (tasiRNAs)(Kanno et al2013)
Although most miRNAs that are broadly conserved among flowering plants are
now being identified and experimentally validated (Table 16)(Jones-Rhoades amp
Bartel 2004) the challenges and ambiguities for classifying non-conserved miRNAs
prevent meaningful estimates of the total number of miRNA genes in Arabidopsis and
Chapter 1 Introduction and Review of Literature
35
other plant genomes It is possible that miRNAs might have escaped detection
because they are non-conserved and expressed in specific tissues or conditions and can
only be identified by using high- throughput sequencing
1134 MicroRNA biogenesis
11341 Transcription of microRNA precursor
Plant miRNAs are primarily found in the genomic regions that are not associated with
the protein coding genes (Reinhart et al 2002) and it appears that most if not all plant
miRNAs are produced from their own transcriptional units Plant miRNA genes are
occasionally clustered near each other in the genome suggesting transcription of
multiple miRNAs from a single primary transcript [eg the miR395 cluster (Grishok
et al 2001 Jones-Rhoades amp Bartel 2004b)] but this polycistronic arrangement of
miRNA genes appears far less frequently in plants than in animals (Bartel 2004)
Northern blot EST and mapping evidence indicate that plant miRNA primary
transcripts (also known as pri-miRNAs) as in animals are longer than needed to
encompass the miRNA stem-loops (Palatnik et al 2003 Jones-Rhoades amp Bartel
2004b) At least some of these pri-miRNA transcripts appear to be spliced
polyadenylated (Kurihara amp Watanabe 2004) and capped (Xie Z et al 2005) Two
rice miRNAs are contained within transcripts that contain exon junctions within the
presumptive stem-loop precursor implying that in these cases splicing is a prerequisite
for Dicer recognition (Sunkar et al 2005) Plant pri-miRNAs are over 1kb in length
and they are usually preceded by typical TATA box motifs They can also undergo
canonical splicing polyadenylation and capping All these observations indicates RNA
polymerase II is probably responsible for transcribing most plant miRNAs (Xie Z et
al 2005) as appears to be the case for many animal miRNAs Relatively little is
known about the regulation of miRNA transcription in plants but there is no reason
to suspect that this regulation would differ from that of protein-coding transcripts as
the bioinformatics analysis of the sequence upstream of the transcriptional start
sites of the miRNA genes showed the presence of putative binding motifs of
number of known transcription factors (Megraw et al 2006 Sethupathy 2013)
11342 MicroRNA processing and export
In plants maturation of miRNA is a step wise process A miRNA gene is transcribed to
pri-miRNA which is much longer than the pre-miRNA possessing the characteristic
stem-loop structure Like transcription of most protein-coding genes the miRNA
Chapter 1 Introduction and Review of Literature
36
transcription is processed by RNA polymerase II (Pol II) enzymes and the pri-
miRNAs are 5prime-capped and 3prime-polyadenylated (Lee et al 2004 Xie Z et al 2005)
Mature miRNAs are produced from pri-miRNAs through at least two sequential
processing steps by RNase III-type endonucleases In animals pri-miRNAs are first
processed at the bottom portion of the stem by Drosha a nuclear-localized RNase III
enzyme to liberate pre-miRNAs The pre-miRNAs are then exported to the cytoplasm
with the help of exportin-5The pre-miRNAs are further processed by Dicer another
RNaseIII enzyme to a duplex consisting of the miRNA and the antisense strands
(miRNA) Since plants do not have any Drosha homologs the two sequential
processing steps seem to be carried out by DCL1 a Dicer homolog in the nucleus
(Kurihara amp Watanabe 2004 de Alba et al 2013 Rogers amp Chen 2013)
The processing of pri-miRNAs to pre-miRNAs by DCL1 is assisted by two
other proteins HYPONASTY LEAVES1 (HYL1) and SERRATE (SE) HYL1 belongs to a
family of double-stranded RNA (dsRNA) binding protein and interacts with DCL1 in
vitro and in vivo (Lu amp Fedoroff 2000 Hiraguri et al 2005 de Alba et al 2013
Rogers amp Chen 2013) Loss-of-function mutations in the HYL1 gene result in
decreased accumulation of miRNAs and concomitant accumulation of pri-miRNAs
(Han et al 2004 Vazquez et al 2004a Kurihara et al 2006) SE a C2H2 zinc finger
protein interacts with DCL1 and HYL1 and plays a role in the processing of pri-
miRNAs to pre-miRNAs (Lobbes et al 2006 Yang L et al 2006) In addition the
nuclear cap-binding complex proteins CBP20 and CBP80 that are known as ABA
HYPER SENSITIVE 1(ABH1) have been shown to be required for proper miRNA
processing (Gregory et al 2008 Laubinger et al 2008) Recently it has been
demonstrated that DCL1 can release 21-nt RNAs from dsRNA as well as from
synthetic miR167 precursor RNAs in vitro However this cleavage is error prone and
inefficient in the absence of HYL1 and SE which accelerate the rate of DCL1-mediated
cleavage and promote accurate processing of miRNAs (Dong et al 2008 Rogers amp
Chen 2013)
11343 Methylation of the miRNAmiRNAduplex
After the miRNAmiRNAduplexes are released from the pre-miRNAs by the DCL1
activities the duplexes are methylated at the 2primeOH of the3prime-ends by HUA ENHANCER1
(HEN1) a small RNA methyltransferase (Yu et al 2005 Yang Z et al 2006 Rogers
amp Chen 2013 ) This step which is distinct from the RNase III-mediated processing
Chapter 1 Introduction and Review of Literature
37
step distinguishes the biogenesis of plant miRNAs from that of animal miRNAs
Mutations in the HEN1 gene were first isolated in a morphological screening for floral
development although the HEN1 mutants display pleiotropic phenotypes similar to the
developmental defects observed in the DCL1 mutants (Chen et al 2002 Park et al
2002 Rogers amp Chen 2013)
Figure 18 Biogenesis of microRNA
A Biogenesis of plant MicroRNA B Biogenesis of metazoananimal MicroRNA
The miRNA (red) is incorporated into RISC and the miRNA(blue) in degraded
This enzyme has two dsRNA-binding domains and nuclear localization signal It
is also conserved in fungi and is required for the processing of siRNAs as well as of
miRNAs (Park et al 2002 Boutet et al 2003 Xie et al 2003) Although the HEN1
protein is localized to the nucleus its methylation activity in the cytoplasm cannot be
ignored (Xie et al 2004 Rogers amp Chen 2013)
Chapter 1 Introduction and Review of Literature
38
The methylation of the miRNAmiRNA duplex is required to protect miRNAs
against the 3prime-end uridylation activity and subsequent degradation (Li et al 2005) In
the hen1 mutants accumulation of miRNAs is reduced to a lower level Also the
miRNAs appear to be heterogeneous in their sizes such that a ladder of bands rather
than a single species is observed for most miRNAs in the mutants (Li et al 2005)
MiRNA cloning and sequencing analyses revealed the presence of a number of U
residues at the 3prime-end of miRNAs in the hen1 mutants Moreover these nucleotides do
not correspond to the miRNAs in the pre-miRNAs indicating that these nucleotides are
added after the processing of the miRNAs from pre-miRNAs (Li et al 2005)
Uridylation of RNAs has also been proven to be correlated with RNA decay (Shen amp
Goodman 2004 Mullen amp Marzluff 2008) suggesting that uridylation attracts an
exonuclease which degrades miRNAs in plants
11344 MiRNA incorporation in to the RISC and its nuclear export
The methylated miRNAmiRNA duplexes undergo RISC assembly In this process
the miRNA of the duplexes is selectively incorporated into the RISC and the miRNA
appears to be removed and subsequently degraded (Hammond et al 2000 Hutvagner
amp Zamore 2002 Schwarz et al 2003 de Alba et al 2013 Rogers amp Chen 2013)
The ARGONAUTE 1(AGO1) proteins are core components of the RISC complex
They contain two structural domains the N-terminal PAZ domain that binds to the 3prime-
end of single-stranded RNAs and the C-terminal piwi domains that has a structure
similar to that of RNase H (Cerutti et al 2000 Parker et al 2004) Several AGO
proteins possess endonucleolytic activities within the PIWI domain and cleave target
mRNAs in the middle of the site complementary to miRNA (Liu et al 2004 Meister et
al 2004 Miyoshi et al 2005) Mutations in the AGO1 gene cause miRNA target
genes to be ectopically expressed and the levels of some miRNAs are reduced in the
mutants (Kidner amp Martienssen 2004 Vaucheret et al 2004 Kidner amp Martienssen
2005 Ronemus et al 2006) Moreover the phenotype of the AGO1 hypomorphic
alleles display defects in lateral organ polarity and leaf and flower morphology as
observed in the dcl1 mutants and null AGO1 alleles are embryonically lethal
supporting the close relationship between the AGO proteins and miRNA processing
(Kidner amp Martienssen 2004 Vaucheret et al 2004 de Alba et al 2013 Rogers amp
Chen 2013)
The production of miRNA miRNA duplexes occur in the nucleus while
Chapter 1 Introduction and Review of Literature
39
miRNA-RISC complexes are present in the cytoplasm where target mRNAs are
located This discordance of functional sites requires an export step during miRNA
biogenesis HASTY the Arabidopsis homolog of exportin-5 is thought to be involved
in miRNA nuclear export (Bollman et al 2003 Park et al 2005)The hasty mutant
exhibits pleiotropic defects in plant development and reduced accumulation of most
miRNAs as in the DCL1 or AGO1 mutants Howeverit is unclear whether the HASTY
proteins export the miRNA miRNA duplexes or the miRNA-RISC complexes in
plants
11345 Feedback regulation of miRNA biogenesis
MiRNA biogenesis is under feedback regulation such that the DCL1 and AGO1 genes
are themselves regulated by miRNAs DCL1 is itself a target of miR162 which leads to
the cleavage of the DCL1 mRNA (Xie et al 2003 de Alba et al 2013) Consistent
with this the levels of DCL1 mRNA are elevated in the mutants of miRNA processing
genes in which the miR162 abundance is reduced In addition there is a predicted
hairpin structure of miR838 in the 14th
intron of the DCL1 primary transcript
(Rajagopalan et al 2006) This prediction implies that DCL1-mediated processing
on this site can result in the cleavage of the DCL1 primary transcript into two truncated
fragments the N-terminal capped fragment and the C-terminal polyadenylated
fragment If this is the case it is possible that miRNA biogenesis competes with the
splicing event of the DCL1 pre-mRNA
The AGO1 is also regulated by miRNA There is a complementary site for
miR168 binding in the AGO1 mRNA and the transcript level of the AGO1 mRNA is
reduced through an mRNA cleavage mechanism (Vaucheret et al 2006 de Alba et al
2013 Rogers amp Chen 2013) indicating that the AGO1 mRNA levels are tightly
controlled by the levels of the AGO1 protein
1135 Mechanism of miRNA action
11351 MiRNA-directed mRNA cleavage
By forming the miRNA-RISC assembly miRNAs can direct the RISC to regulate gene
expression by two post transcriptional mechanisms mRNA cleavage or translational
repression (Bartel 2004 Jones-Rhoades et al 2006 Dalmay 2013) The degree
of miRNA-mRNA complementarity plays an important role for the
determination of the mechanism to be used A general rule is that while perfect or
Chapter 1 Introduction and Review of Literature
40
near-perfect complementarity induces mRNA cleavage central mismatches trigger
translational repression (Carrington amp Ambros 2003 Jones-Rhoades amp Bartel 2004)
Unlike most animal miRNAs plant miRNAs have near-perfect or perfect
complementarity to their targets and mRNA cleavage is assumed to be a predominant
mechanism by which miRNAs regulate gene expression in plants
In RNA cleavage mechanism miRNAs guide the AGO component of RISC to
cleave a single phosphodiester bond of the target mRNA within the miRNA-binding
site (Llave et al 2002b Tang et al 2003) The truncated fragments are then released
and degraded freeing the RISC to recognize and cleave another target mRNA This has
been validated by the detection of 3prime cleavage products that have 5prime ends that start at the
middle of the complementary regions The mRNA cleavage products are then further
degraded by other mechanisms The 3prime cleavage products of some miRNA targets are
degraded by the 5prime-3prime exonuclease XRN4 an Arabidopsis homolog of the major Yeast
mRNA degrading exonuclease Xrn1p It has been observed that the 3prime cleavage
products of some target mRNAs were accumulated in the xrn4 mutants (Souret et al
2004 Dalmay 2013) However the 3prime cleavage products of other miRNA targets do
not accumulate in the xrn4 mutants indicating that there are also XRN4 independent
mechanisms The 5prime cleavage products are typically untraceable by RNA gel blot
hybridization but can be detected by sensitive PCR based methods It has been found
that the 5prime cleavage products tend to obtain an oligo U tail at the 3prime ends This 3prime U tail
is correlated with shortening of the 5prime cleavage products from the 5prime end leading to the
conclusion that the 3prime uridylation causes 5 prime- 3 prime exonucleolytic decay of the 5prime cleavage
products (Shen amp Goodman 2004)
DCL1 HYL1 and SE interact with each other and are co-localized in the
nuclear bodies termed Dicing bodies or D-bodies in which some pri-miRNA shave
been found suggesting that D-bodies serve as a center for active miRNA processing
(Fang amp Spector 2007 Fujioka et al 2007 Song et al 2007) The miRNA
processing site appears to be distinct from the Cajal bodies that have previously been
found as a siRNA processing site (Pontes amp Pikaard 2008) The D-bodies include
SmD3 and SmB proteins that are found in the Cajal bodies However At collin an
additional marker for the Cajal bodies is not found in the D-bodies Moreover the D-
bodies are present in an At collin mutant lacking the Cajal bodies The pri-miRNAs of
miR171A and miR159A genes have been found to be co-localized with HYL1 and
Chapter 1 Introduction and Review of Literature
41
DCL1 in the D- bodies that are distinct from the Cajal bodies (Song et al 2007)
11352 MiRNA-directed translational repression
Translation repression is another mechanism exerted by plant miRNAs The regulatory
mechanism of miR172 which regulates flowering time and floral organ identity is
consistent with translation repression MiR172 over production does not affect the
transcript levels of APETALA2 (AP2) and AP2- like target mRNAs Instead the AP2
protein level is reduced in the miR172 over expressing mutant (Aukerman amp Sakai
2003 Chen 2004 Dalmay 2013) It was later shown that miR172 can also direct the
cleavage of the target mRNAs although the steady-state mRNA level is unchanged due
to feedback regulation (Schwab et al 2005 Jung et al 2007) It is clear that miR172
regulates its target genes primarily by translational repression rather than mRNA
cleavage Recently it has been reported that miR156157 and miR854 also regulate
their target genes through translational repression (Arteaga-Vazquez et al 2006
Gandikota et al 2007 Dalmay 2013) It was once thought that translational repression
is an exception to the rule However experimental evidence suggest a widespread
coexistence of translational repression and mRNA cleavage (Brodersen et al 2008)
1136 Regulatory roles of miRNAs in plant development
The miRNA target prediction has been a difficult task in order to obtain regulatory
targets without bringing in too many false predictions Plant growth and developmental
processes regulated by miRNAs are quite diverse and enormous Furthermore plant
miRNAs work cooperatively or antagonistically to establish a balanced regulation This
notion is well consistent with the findings that mutations in the regulators of miRNA
biogenesis transport and processing such as AGO1 DCL1 HYL1 HEN1 ABH1
and HASTY cause pleiotropic phenotypes (Mallory amp Vaucheret 2006 de Alba et al
2013) To date the roles of miRNAs have been mostly deduced from obtaining miRNA
over expressing transgenic plants or gain-of-function mutants in which miRNA-
resistant target genes are ectopically expressed
11361 Phase transition
Plants pass through a series of developmental phase transitions during their life span
beginning with seed germination continuing through vegetative phase change
reproductive phase change flowering initiation and finally seed production for the next
generation Because of their importance in understanding plant developmental
Chapter 1 Introduction and Review of Literature
42
processes genes regulating developmental transitions have been extensively studied
and underlying molecular mechanisms have been explored in various plant species
Majority of researches were mainly focused on vegetative phase change and
reproductive transition in Arabidopsis and Maize (Poethig 2003 Baurle amp Dean
2006) Particularly the mutations of the genes encoding various regulators of miRNA
biogenesis and transport such as HST SE and AGO exhibit altered juvenile to adult
vegetative phase change and vegetative to reproductive change (Hunter et al 2003
Peragine et al 2004 Vazquez et al 2004a Jones-Rhoades et al 2006 de Alba et al
2013)
Over-expression of the miR156a gene causes late flowering and delays
vegetative phase change (Wu amp Poethig 2006 Gandikota et al 2007 Wang et al
2008) It has been shown that miR156 negatively regulates a group of genes encoding
SQUAMOSA PROMOTER-BINDING PROTEIN-LIKE (Lewis et al 2007 de Alba et
al 2013 Rogers amp Chen 2013) transcription factors such as SPL3 SPL4 and SPL5
that promote flowering initiation In contrast transgenic plants over expressing
miR156-resistant SPL345 genes are early flowering Interestingly the endogenous
level of miR156 is temporally regulated In the early juvenile vegetative phase the
level of miR156 is very high but decreases rapidly before the onset of the adult juvenile
phase (Wu amp Poethig 2006)
The Arabidopsis early activation tagged (eat-D) mutant exhibits early flowering
with disrupted floral structure The mutant phenotypes are caused by over expression of
the miR172a-2gene (Aukerman amp Sakai 2003) MiR172 regulates a small group of
genes encoding APETALLA2 (AP2)-like transcription factors such as TARGET OF
EAT1 (TOE1) TOE2 SCHLAFMUTZE (SMZ) and SCHNARCHZAPFEN(SNZ)
TOE1 TOE2 SMZ and SNZ have been shown to participate in their productive phase
change by repressing a floral integrator FLOWERING LOCUS T (FT)(Jung et al
2007) Consistent with this toe1-2D 35STOE2 35SSMZ and 35SSNZ plants
exhibit late flowering (Jung et al 2007) MiR172 is also regulated temporally and
highly expressed in the adult vegetative phase both in Arabidopsis and maize (Chuck et
al 2007) Because the temporal expression patterns of miR156 and miR172 are
complementary to each other it has been suggested that the two miRNAs interact with
each other in regulating phase change (Chuck et al 2009 de Alba et al 2013) In
support of this view it has been shown that miR172 is down-regulated in the
Chapter 1 Introduction and Review of Literature
43
Corngrass1(Cg1) mutant in which miR156 is overproduced (Chuck et al 2007 de
Alba et al 2013) However there is no direct evidence for the direct linkage between
miR156 and miR172 It is also envisioned that miR156 and miR172 would not be
directly related For example miR156 regulates plastochron index that is the inverse of
leaf initiation rate and thus frequently used to analyze temporal patterns of plant
development In contrast miR172 does not affect plastochron (Wang et al 2008) The
down-regulation of miR172 in the maize miR156-over producing mutants would be
due to the prolonged juvenile vegetative phase in the mutants
Another miRNA that regulates flowering initiation is miR159 It regulates
MYB33 and MYB65 which are closely related to the barley GAMY B transcription
factor that responds to gibberellic acid (GA) The level of miR159 is elevated in GA-
treated seedlings but reduced in the GA biosynthetic ga1 mutant Transgenic plants
over producing miR159 exhibit delayed floral induction under short days (Achard et
al 2004) It is been suggested that MYB33 mediates GA signal to regulate LEAFY
(LFY) These observations indicate that miR159 plays an important role in the GA
signaling pathways that regulate proper floral induction under short days (Achard et al
2004)
11362 Floral development
Arabidopsis flowers consist of four distinct organs They arise in a characteristic
pattern within concentric rings called whorls from outside to inside four sepals four
petals six stamens and the central pistil consisting of two carpels Identities of the
floral organs are determined by three classes of regulatory genes classA classB and
class C genes (Krizek amp Fletcher 2005) The A and C class genes act antagonistically
to restrict their expression domains (Bowman et al 1991)
It is notable that miR172 also regulates floral architecture in addition to its role
in the timing of flowering initiation MiR172 inhibits the translation of the AP2 mRNA
(Chen 2004) Transgenic plants overproducing miR172 not only exhibit early
flowering but also show abnormal floral development which is quite similar to those
of the class A gene mutants such as the ap2 mutants (Aukerman amp Sakai 2003 de
Alba et al 2013) When the activity of the class A genes is inactivated that of the class
C genes such as AGAMOUS (AG) is elevated As a result the miR172- overproducing
transgenic plants exhibit no petals along with homeotic transformation of the sepals to
Chapter 1 Introduction and Review of Literature
44
the carpels (Aukerman amp Sakai 2003 Chen 2004)
Interestingly miR166 also regulates floral architecture The role of miR165166
in the regulation of meristem activity is closely linked to floral meristem formation and
organization (Zhao et al 2007 Miyashima et al 2013) Floral structure is severely
disrupted in the miR165166-overproducing mutants such as men1 and jba-1D in
which miR166 is overproduced (Williams et al 2005b Jung amp Park 2007) The men1
and jba-1D mutants have smaller gynoecium and reduced carpel number In an extreme
case the jba-1D homozygous plants lack visible carpels (Williams et al 2005b)
Similar phenotypes have been observed in the miR165 overproducers (Zhao et al
2007) It has been suggested that the phenotypes are possibly caused by altered AG
expression in the floral meristem (Jung amp Park 2007 Turchi et al 2013)
Other miRNAs also affect floral development Overproduction of miRNAs
involved in auxin signaling also affects floral architecture Transgenic plants
overproducing miR167 have defects in the formation of ovule and anther with reduced
fertility (Ru et al 2006) It has been shown that miR167 targets ARF6 and ARF8 that
play a role in gynoecium and stamen development and in regulating fertility (Ru et al
2006 Wu et al 2006) These observations suggest that auxin signals are also closely
related to floral organogenesis In addition transgenic plants over-producing miR164
and the cuc1cuc2 double mutants show fused sepals and reduced petals (Laufs et al
2004 Turchi et al 2013) suggesting that miR164 is also related to floral meristem
activity and boundary specification of the meristematic region
11363 Shoot apical meristem development
Shoot apical meristem comprises self-maintaining totipotent cells The shoot apical
meristem activity affects diverse aspects of plant developmental processes including
leaf development vascular development and lateral organ polarity (Bowman et al
2002) Because miR165166 plays a primary role in meristem formation
overproduction of miR165166 and multiple knockout mutants of its target genes
exhibit pleiotropic pheonotypes (McConnell et al 2001 Emery et al 2003 Kim et al
2005 Williams et al 2005b)
The targets of miR165166 include genes encoding the homeo domain-leucine
zipper (HD-ZIP) class III transcription factors such as PHABULOSA(PHB)
PHAVOLUTA (PHV) REVOLUTA(REV) ATHB15CORONA (CNA) and ATHB8
Chapter 1 Introduction and Review of Literature
45
PHB PHV and ATHB15 have overlapping functions in controlling meristem
development (Turchi et al 2013) Indeed the phb phv athb15 triple mutants have an
enlarged shoot apical meristem Consistent with this the miR166- overproducing men1
and jba-1D mutants exhibit enlarged shoot apical meristems (Kim et al 2005
Williams et al 2005b Turchi et al 2013) In the mutants the shoot apical meristems
are multiplied and large
The development of shoot apical meristems is also regulated by the WUSCHEL
(WUS)ndashCLAVATA (CLV) signaling pathway WUS positively regulates cell
proliferation In contrast CLV3 which is expressed in stem cells limits the WUS
expression and thus inhibits cell proliferation in the SAM (Sharma et al 2003)
Consistent with this notion WUS is expressed to a higher level in jba-1D
explaining the ectopic enlargement of the SAM in the mutant (Williams et al 2005)
While WUS is closely related to miR165166 in the shoot apical meristem
development the underlying molecular mechanisms are currently unclear It has been
observed that the miR166-over producing men1 plants have an arrested meristem
development (Jung amp Park 2007) In addition the heterozygous men1+ and the
homozygous men1 plants show different expression patterns of WUS and CLV3 It
appears that miR166 regulates WUS indirectly and influences the expressional balance
between WUS and CLV3 through a negative feedback loop (Jung amp Park 2007 de
Alba et al 2013)
The phv phb athb15 triple mutant has an enlarged shoot apical meristems the
rev phb phv mutant does not have functional shoot apical meristems (Prigge et al
2005) This distinction may be explained by the fact that while miR166 targets
primarily PHB PHV and ATHB15 miR165 targets all the five HD-ZIP III genes (Zhou
et al 2007) Consistent with this transgenic plants overproducing miR166 are distinct
from those overproducing miR165 The miR165-overproducing transgenic plants lack
functional shoot apical meristem and a pin-like structure is formed at the apical region
of the mutant These phenotypes are quite similar to rev phb phv triple mutant (Zhou et
al 2007 Liu et al 2013) The phenotypic distinction may because by the difference
of the cleavage efficiencies of the target mRNAs In accordance with this notion a few
35S miR166a transgenic lines exhibit no shoot apical meristems The men1
homozygous plants also show arrested meristem development (Jung amp Park 2007)
Chapter 1 Introduction and Review of Literature
46
MiR164 cleaves the CUC1 and CUC2 mRNAs as well as the NAC1 mRNA
CUC1 and CUC2 determines the organ boundary in the meristem Phenotypes of
transgenic plants that overproduce miR164 are similar to those of the cuc1cuc2 double
mutant that exhibits embryonic developmental defects such as cup-shaped cotyledons
and fused cotyledons (Laufs et al 2004) When a miR164-resistant CUC2 is
expressed the boundary domain is enlarged CUC also plays a role in embryonic shoot
meristem formation by regulating the SHOOT MERISTEMLESS gene It was therefore
concluded that miR164-mediated cleavage of CUC1 and CUC2 mRNAs determines the
sizes of the boundary domains in the shoot apical meristem and regulates separation of
the organs by restricting cell proliferation (Laufs et al 2004 de Alba et al 2013)
11364 Leaf development
The role of miR319 has been extensively studied in leaf development A dominant
jaw-D mutant overproducing miR319 shows uneven curved leaves with enlarged size
and five TCP genes which are closely related to the CINCINNATA (CIN) gene in
Antirrhinum majus are down regulated in the mutant suggesting that miR319 mediated
regulation of the TCP transcription factor genes are important for the final step of
leaf shaping (Palatnik et al 2003 Naz et al 2013) In addition to leaf shape and size
leaf polarity is also specified by miRNA MiR166-mediated cleavage of the HD-ZIPIII
mRNAs is a well to establish leaf polarity This discovery originates from the gain-of-
function mutants such as phb-1d and phv-1d wherein point mutations in the
complementary sites to miRNA helps the mRNA to evade or resist from miRNA-
mediated cleavage (McConnell et al 2001 Emery et al 2003 Naz et al 2013)
Plant leaves have a dorso-ventral polarity consisting of the adaxial (upper
surface) and abaxial (lower surface) sides The adaxial side is closer to the shoot
apical meristem Adaxial identity is specified by functionally redundant class III HD-
ZIP family genes such as PHB PHV and REV Ectopic expression of each genes
result in adaxialized leaves Dominant phb-1d and phv-1d mutants exhibit
transformation of the abaxial to adaxial fate and have rod-shaped leaves In contrast
miR165166-overproducing plants show pin-like cotyledons radicalized leaves and
abaxialized leaves (Chitwood et al 2007 Pattanayak et al 2013)
Leaf asymmetry is determined by miR166-mediated restriction of the
expression domains of the HD-ZIPIII genes The HD-ZIPIII genes are expressed in the
Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
1
INTRODUCTION
11 Coffee
Coffee is an important plantation crop belonging to the family Rubiaceae subfamily
Cinchonoideae and tribe Coffeae (Clifford et al 1989) Rubiaceae is the largest
flowering plant family comprising of 650 genera and 13000 species which are largely
tropical or subtropical (Rova et al 2002)
The word coffee connects with the town Kaffa in Ethiopia reported to be the
place of origin of coffee plants The word coffee derives from the Ottoman Turkish
word kahve via Italian word caffeacute Turkish word is in turn borrowed from the Arabic
word quhwah referring to a type of wine- quaqat-al-bunn (wine of bean)This found
its way into the European languages and became cafeacute(French) caffe (Italian) Kaffee
(German) koffie (Dutch) coffee (English) and Latin Coffea for the botanical genus
The stimulatory effects of roasted coffee beans were well known to the natives of
Africa when the Arabs brought C arabica seeds from Ethiopia to Yemen (Arabian
Peninsula) during the 13th
century and established the first plantations (Monaco et al
1997)
12 Taxonomy of Coffee
The genus Coffea L comprises 103 species (Davis et al 2006) and occurs naturally
in tropical Africa Madagascar the Comoros and Mascarenes (Mauritius and
Reunion) Coffea species are mostly restricted to humid evergreen forest although
some species are found in seasonally dry deciduous forest andor bushland The most
recent classifications of Coffea (Bridson 1988 Bridson 2003 Davis et al 2005
Davis et al 2006) divide the genus into two subgenera subgenus Coffea (95 spp)
and subgenus Baracoffea JF Leroy (eight spp) Coffea subgenus has a wide range of
distribution whereas Coffea subgenus Baracoffea is restricted to the seasonally dry
forest and scrubland of western Madagascar (Davis et al 2005) and they are also
found in NE Kenya and SE Somalia according to (Leroy 1982)
Coffea subgenus Coffea includes the species used in the production of coffee
ie C arabica (Arabica coffee) C canephora (Robusta coffee) and C liberica
(Liberian coffee) C arabica is by far the most important traded species and accounts
for at least 65 of total commercial production C arabica is the only allotetraploid
Coffea species (2n= 4x =44) (Grassias amp Kammacher 1975) all other Coffea species
are diploid (2n = 2x =22) C arabica is also self-compatible (Carvalho et al 1991)
Chapter 1 Introduction and Review of Literature
2
thus far only reported in two other species C heterocalyx (Coulibaly et al 2002) and
C anthonyi (Maurin et al 2007)
121 Classification
Kingdom Plantae
Subkingdom Tracheobionta
Superdivision Spermatophyta
Division Magnoliophyta
Class Magnoliopsida
Subclass Asteridae
Order Rubiales
Family Rubiaceae
Genus Coffea
122 Morphological features
The genus Coffea contains about 25 species These are small trees or shrubs that
originally grew in African tropical forests Some varieties of coffee plant typically
grow over 30 feet But in cultivation for ease of picking of the coffee berry the
coffee tree is seldom allowed over 15 feet The C arabica coffee plant is typically
smaller than the C canephora plants C canephora is shrub type and C arabica is
tree type They have extensive tap root systems with well-developed main vertical
roots and lateral roots that grow parallel to the ground Leaves have a bipolar structure
where leaf pairs are at 90 degree rotation to each other on the stem Leaves are oblong
to ovate in shape with characteristic interpetiolar stipule
Flowers are white are white produced in dense clusters at leaf axils Flowers
are pentamerous Calyx is toothed and corolla is tubular There are five stamens and a
single bifid style Sexually C arabica is autogamous whereas Ccanephora is self-
incompatible The fruits are drupes
123 Classification of coffee
Chevalier (1947) has grouped the valid coffee species into the following sections
(1) Argocoffea
(2) Paracoffea
(3) Mascarocoffea
Chapter 1 Introduction and Review of Literature
3
(4) Eucoffea
The Coffee species belonging to Mascarocoffea section have one common
characteristic the absence of caffeine The Eucoffea (now named Coffea) has been
again divided into five subsections according to diverse criteria tree height
(Nanocoffea) leaf thickness (Pachycoffea) fruit colour (Erythrocoffea Melanocoff-
ea) and geographical distribution (Mozambicoffea) (Table 11) The recent
classification has widely accepted that Coffea sect Paracoffea and Agrocoffea mainly
consist of species from other genera
Table 11The grouping of the species in the subsection Eucoffea (Chevalier 1947)
Section Subsection Species
Eucoffea
Erythrocoffea
C arabica
C canephora
C congensis
Pachycoffea
C abeokutae
C liberica
C klainii
C oyemensis
C dewerei
Melanocoffea
C stenophylla
C carissoi
C mayombensis
Nanocoffea
C humilis
C brevipes
C togoensis
Mozambicoffea
C schumanniana
C eugenioides
C kivuensis
C mufindiensis
C zanguebariae
C racemosa
C ligustroides
C salvatrix
The current sub generic classification comprises of two genera Coffea and Baracoffea
(Table 12) Most species of Coffea belong to Coffea subgenera includes those used
for producing the beverage coffee Coffea subgenera occur throughout the natural
range of the genus in Africa Madagascar and The Mascarenes Subgenera Baracoffea
contains only three accepted species (although five remain undescribed) (Table 12)
and is restricted to the dry forests of western Madagascar Leroy (Leroy 1980) placed
C rhamnifolia a species from Africa (Somalia and Kenya) in Coffea subgen
Chapter 1 Introduction and Review of Literature
4
Baracoffea but this was contested by Bridson (1988) Davis amp Rakotonasolo (2003)
and Davis et al (2005)
Table 12Outline classification of Coffea and Psilanthus with synonyms (Davis amp Rakotonasolo
2003)
Coffea L Sp Pl 172 (1753)
Coffea sub gen Coffea L Type C arabica L 95 species Africa Madagascar Mascarenes
Coffea sub gen Psilanthopsis (AChev)
Psilanthopsis AChev Type Coffea kapakata
Type Paolia jasminoides Chiov (= C rhamnifolia (Chiov) Bridson)
Coffea sub gen Baracoffea
Coffea sect Baracoffea Type Coffea humbertii J-FLeroy
Eight species (including five -yet to be published) West Madagascar
[Paracoffea subgen Insulanoparacoffea J-FLeroy J Agric Trop Bot Appl 14 276 (1967)
nom nud]
Psilanthus Hookf Gen Pl 115 (1873)
Psilanthus subgen Psilanthus Hookf Type Psilanthus mannii Hookf
Two species Africa (central and western)
Psilanthus subgen Afrocoffea (Moens)
Coffea subgenAfrocoffea
Type Psilanthus lebrunianus (Germain amp Kesler) Bridsonc 18 species Africa Asia
Australasia
Coffea sectParacoffeaMiq Fl Ind Batavae 308 (1856)
Psilanthus subgen Paracoffea Type Coffea horsfieldiana Miq
Paracoffea
[Paracoffea subgenAfroparacoffea J-FLeroy J Agric Trop Bot Appl 14 276 (1967)
nom nud]
[Paracoffea subgenMelanoparacoffea J-FLeroy J Agric Trop Bot Appl 14 276 (1967)
nom nud]
13 Economics of Coffee
Coffee is the fourth most valuable traded agricultural commodity (FAO 2004) but is
the second most valuable commodity exported by the developing countries and more
than 75 million people depend on coffee for most of their livelihood (Pendergrast
2009)
India produces both Arabica coffee from C arabica and Robusta coffee from
C canephora As per the economic survey 2012-13 Coffee is planted in India in
around 388195 hectares Out of this area 52 is Ccanephora and 48 is Carabica
Chapter 1 Introduction and Review of Literature
5
Coffee is mostly grown in south India parts of Orissa W Bengal and North east
India Karnataka is the largest producer with 569 of total area under production
which accounts for 71 of the total produce The total crop harvest for 2012-2013 is
placed at 325300 tonnes (httpwww Indicoffeeorgindiacoffeephpp-
age=CoffeeData)
14 Composition of coffee beans
Navellier (1961) gave a mean composition of green coffee as Carbohydratesglucides
(58) lignin (2) lipids (13) proteins (13) ash (4) non-volatile acids (8)
trigonelline (1) and caffeine (1) The caffeine content of green coffee is relatively
limited (10-25) of dry matter and changes little with seed development (Clifford
and Kazi 1987) In coffee seeds the concentration of trigonelline is approximately
2 of dry weight (Clifford 1985 Mazzafera 1991) Mazzafera (1999) found a higher
protein content in the mature beans than in the immature beans but a lower content of
free amino acids with asparagine as the main component Flament ( 2000) estimated
the composition of coffee beans as follows
Proteins 100 (dry weight basis of green coffee)
Carbohydrates 500 -do-
Lipids 117-140 (Arabicas)
76- 95 (Robustas)
Chlorogenic acids 65 (Arabicas)
90 (Robustas)
15 Purine alkaloids structure and role
Purine alkaloids are derived from purine nucleotides (Zulak et al 2007) and found in
over hundred plant species in 13 orders in plant kingdom (Ashihara amp Crozier 1999)
Methylxanthines like caffeine (137-trimethlyxanthine) theobromine (37-methylxan-
thine) and methyluric acids are classified into purine alkaloids (Figure 11) (Table
13)(Ashihara et al 2008) These are known to occur in tea coffee and a number of
non-alcoholic beverages Caffeine was isolated from coffee (Coffea arabica) and tea
(Camellia sinensis) in 1820 (Ashihara amp Crozier 2001)
Chapter 1 Introduction and Review of Literature
6
Figure 11 Purine alkaloid xanthine and uric acid skeleton
Plants that accumulate purine alkaloid are classified into three groups based on
the type of alkaloids they produce caffeine-producing plants which include coffee
tea and mateacute (Ilex paraguariensis) theobromine ndash producing plants are represented
by cacao cocoa tea (Camellia ptilophylla) and Camellia irrawadiensis and
methyluric acid ndash producing plants consist of Coffea dewevrei Coffea liberica C
excelsa and Kucha tea (Camellia assamica)
Table 13 Purine alkaloid structures based on xanthine and uric acid skeleton
Compound Trivial name R1 R2 R3 R4 O-2 Δ2-3
Methylxanthines
Xanthine H H H
1-methylxanthine CH3 H H
3-methyl xanthine H CH3 H
7-methyl xanthine H H CH3
1-3methylxanthine Theophylline CH3 CH3 H
17-methylxanthine Paraxanthine CH3 H CH3
37-methylxanthine Theobromine H CH3 CH3
137-methylxanthine Caffeine CH3 CH3 CH3
Uric acid and methyl uric acids
Uric acid H H H H
137-trimethyluric acid CH3 CH3 CH3 H
1379-tetramethyluric
acid Theacrine CH3 CH3 CH3 CH3
O19-trimethyluric acid Liberine CH3 H H CH3 CH3 Δ2-3
O179-trimethyluric acid Methylliberine CH3 H CH3 CH3 CH3 Δ2-3
16 Role of caffeine in plants
The physiological role of endogenous purine alkaloids and related compounds in
higher plants remains undetermined There are two hypotheses about the role of the
Chapter 1 Introduction and Review of Literature
7
high concentrations of caffeine that accumulate in tea coffee and a few other plant
species The lsquoallelopathic theory or the auto toxic function theoryrsquo proposes that
caffeine in seed coats is released into the soil and inhibits the germination of seeds
around the parent plants (Anaya et al 2006)
The lsquochemical defense theoryrsquo proposes that caffeine in young leaves fruits
and flower buds acts to protect soft tissues from insect pests (Ashihara amp Crozier
2001 Ashihara et al 2008) It has been shown that spraying tomato leaves with a 1
solution of caffeine deters feeding by tobacco horn worms while treatment of
cabbage leaves and orchids with 001ndash01 solutions of caffeine acts as a neurotoxin
and kills or repels slugs and snails (Hollingsworth et al 2002) This work has now
been extended with convincing evidence for chemical defense theory has recently
been obtained with transgenic caffeine ndash producing tobacco plants (Uefuji et al 2005
Kim et al 2006)
17 Distribution of caffeine in plants
Caffeine has been found in 13 orders of the plant kingdom (Ashihara and Suzuki
2004 Anaya et al 2006) In some species the main purine alkaloid is theobromine or
methyl uric acids such as theacrine liberine and methylliberine rather than caffeine
(Ashihara amp Crozier 1999 Ashihara amp Crozier 2001)(Table 14)
Table 14 Distribution of purine alkaloids in plants
SNo Plant species Common
name Major alkaloid Plant parts
containing
alkaloids 1 Coffea arabica Arabica Caffeine Leaves Seeds
2 Coffea canephora Robusta Caffeine Leaves Seeds
3 Coffea liberica Caffeine Theacrine Liberine
Seeds Mature leaves
4 Coffea dewevrei Caffeine Theacrine Liberine
Seeds Mature leaves
5 Camellia sinensis Tea Caffeine Leaves
6 Camellia assamica Assam tea Caffeine Leaves
7 Camellia assamica var
kucha Kucha Theacrine Caffeine Leaves
8 Camellia irrawadensis Theobromine Leaves
9 Camellia ptilophylla Cocoa tea Theobromine Leaves
10 Theobroma cacao Cocoa Theobromine Seeds
11 Paullinia cupuna Guarana Caffeine Seeds
Chapter 1 Introduction and Review of Literature
8
12 Cola nitida Caffeine Seeds
13 Citrus sp Caffeine Pollen
14 Ilex paraguariensis Mate Caffeine Leaves
Adapted from (Ashihara amp Crozier 1999 Ashihara amp Crozier 2001)
18 Caffeine biosynthesis
The major biosynthetic pathway is a four step sequence consisting of three sequential
methylation and one nucleosidase reactions (Figure 12) The xanthine skeleton of
caffeine is derived from purine nucleotides The initial step in caffeine biosynthesis is
the methylation of xanthosine by a SAM- dependent N-methyltransferase In addition
to experiments with radiolabeled precursors substrate specificities of native (Kato et
al 1999) and recombinant N-methyltransferases (Kato et al 2000 Ogawa et al
2001 Mizuno et al 2003a Mizuno et al 2003b Uefuji et al 2003) strongly
suggest that the major route to caffeine is a xanthosine rarr7-methylxanthosinrarr 7-
methylxanthine rarr theobromine rarr caffeine pathway (Figure 12) Although the
information has been obtained mainly from coffee (C arabica) and tea (Camellia
sinensis) the available evidence indicates that the pathway is essentially the same in
other purine alkaloid-forming plants such as mateacute (Ashihara 1993) and cacao
(Koyama et al 2003 Yoneyama et al 2006)
The first step in the caffeine biosynthetic pathway from xanthosine is
conversion of xanthosine to 7-methylxanthosine (Figure 12) This reaction is
catalyzed by the 7-methylxanthosine synthase (xanthosine 7 N-methyltransferase EC
211158) The genes encoding for 7-methylxanthosine synthase CmXRS1 (AB
034699) and CaXMT1 (AB 048793) were isolated from the C arabica (Mizuno et
al 2003a Uefuji et al 2003) The recombinant proteins obtained from these genes
exhibit 7-methylxanthosine synthase activity in vitro Xanthosine monophosphate
(XMP) was not converted to 7-methylxanthosine by the recombinant enzymes thus
inclusion of 7-methy-XMP was proposed (Schulthess et al 1996)
The second step of caffeine biosynthesis involves a nucleosidase which
catalyses the hydrolysis of 7-methylxanthosine Although N-methylnucleosidase (EC
32225) was partially purified from tea leaves (Negishi et al 1988) recent detailed
structural studies on coffee 7-methylxanthosine synthase suggested that the methyl
transfer and nucleoside cleavage may be coupled and catalyzed by a single enzyme
(McCarthy amp McCarthy 2007)
Chapter 1 Introduction and Review of Literature
9
The last two steps of the caffeine synthesis are also catalyzed by a SAM-
dependent N-methyltransferase(s) which is different from the N-methyltransferase
enzyme that catalyzes the first step in the pathway Native N-methyltransferase
activities have been detected in crude and partially purified extracts from tea and
coffee plants (Ashihara amp Suzuki 2004) and a highly purified preparation has been
obtained from young tea leaves (Kato et al 1999) The enzyme was assigned the
name caffeine synthase (EC 211160) that catalyses the last two steps of caffeine
biosynthesis viz the conversion of 7-methylxanthine to caffeine via theobromine
(Figure 12) The gene encoding caffeine synthase was first cloned from young tea
leaves (Kato et al 2000) Since then several genes encoding N-methyltransferases
with different substrate specificity have been reported
Activity of the recombinant theobromine synthase (CTS1 and CaMXMT EC
211159) is specific for the conversion of 7-methylxanthine to theobromine In
contrast the recombinant caffeine synthases (CCS1 and CaDMXMT1) can utilize
paraxanthine theobromine and 7-methylxanthine as shown with tea caffeine synthase
(TCS1) (Mohanpuria et al 2011) Although paraxanthine is the most active substrate
of this recombinant enzyme only limited amounts of paraxanthine are synthesized in
coffee cells hence in vivo caffeine synthase is principally involved in the conversion
of 7-methylxanthine to caffeine via theobromine
Radiolabelled tracer experiments with theobromine accumulating cacao and
Chinese tea (Camellia ptilophylla) showed a limited conversion of theobromine to
caffeine N-methyltransferase which catalyzed the conversion of 7-methylxanthine to
theobromine was detected in crude extracts from C ptilophylla but conversion of
theobromine to caffeine was not observed (Ashihara et al 1998) Genes homologous
to caffeine synthase have been isolated from several theobromine accumulating plants
including cacao (Yoneyama et al 2006) The recombinant enzymes derived from
these genes have 3 N-methyltransferase activity suggesting that these theobromine
accumulating plants contain specific theobromine synthase Caffeine does not occur
in C ptilophylla (Ashihara et al 1998) although it is present in leaves and fruits of
cacao along with theobromine (Koyama et al 2003 Zheng amp Ashihara 2004)
Chapter 1 Introduction and Review of Literature
10
Figure 12 Pathways for the biosynthesis of caffeine
The major pathway is indicated by solid lines (Mizuno et al 2003b Uefuji et al 2003) Alternative pathways in coffee and tea plants are indicated
by dashed lines(Kato et al 1996 Schulthess et al 1996) Enzymes (1) 7-methylxanthosine synthase (2) N-methylnucleosidase (3) Theobromine
synthase (4) Caffeine synthase
N
N
N
N7
H3C
3
1
O
O
Caffeine
N
NH
N
N7
3
O
O
Theobromine
N
N
N
N
H
H
7
O
O
N
N
N
N
H
H
O
O
N
N
N
N
H
H
O
O
Xanthosine
N
N
N
N
H
H
O
O
N
N
N
N
H
H
O
ON
N
N
N
H
H3C
71
O
O
Paraxanthine
CH3
CH3
CH3
CH3CH3
7-methylxanthine
Rib P
Rib
(1)
(4)
Rib
+
CH3
7-methyl
xanthosine
(2)
(3)
SAM SAH
SAM SAH SAM SAH
Xanthosine mono
phosphate
Rib P
7-methyl xanthosine
monophosphate
+
CH3 CH3
Inosine 5-mono
phosphate
Adenosine 5-mono
phosphate
SAM SAH
Guanosine
Guanosine 5-mono
phosphate
Chapter 1 Introduction and Review of Literature
11
181 Source of Xanthosine for caffeine biosynthesis
Xanthosine the initial substrate of purine alkaloid synthesis is supplied by at least four
different pathways de novo purine biosynthesis (de novo route) the degradation
pathways of adenine nucleotides (AMP route) and guanine nucleotides (GMP route)
and the S-adenosyl-L-methionine (SAM) cycle (SAM route) (Figure 13) Purine
nucleotides are synthesized by de novo and salvage pathways (Ashihara amp Crozier
1999 Stasolla et al 2003 Zrenner et al 2006) The de novo synthesis of non purine
precursors like CO2 10-formyltetrahydrofolate 5-phosphoribosyl-1-pyrophosphate and
the amino acids Glycine glutamine and aspartate results in the production of purine
nucleotides
Figure 13 Xanthosine biosynthesis Xanthosine for purine alkaloid biosynthesis is supplied by at
least four routes from adenosine released from the SAM cycle (SAM route) from IMP
originating from de novo purine synthesis (de novo route) from the cellular adenine
nucleotide pool (AMP route) and from the guanine nucleotide pool (GMP route)
Adapted from (Ashihara et al 2008)
The utilization of IMP for caffeine biosynthetic pathway which is formed by the
de novo purine biosynthetic pathway for caffeine biosynthetic pathways was
demonstrated in young tea leaves using 15
N-glycine and 14
C-labelled precursors and
inhibitors of de novo purine biosynthesis (Ito amp Ashihara 1999) Xanthosine is formed
by an IMP rarr XMP rarr xanthosine pathway IMP dehydrogenase (EC 111205) and 5prime-
nucleotidase (EC 3135) catalyze these reactions The inhibition of IMP dehydrogen-
ase by ribavirin inhibits both caffeine and gunanine nucleotide biosynthesis in tea and
coffee leaves (Keya et al 2003)
Chapter 1 Introduction and Review of Literature
12
The portion of xanthosine used for caffeine biosynthesis is derived from adenine
and guanine nucleotide pools which are produce de novo by salvage pathways there are
several potential pathways for xanthosine synthesis from AMP but the AMP rarr IMP
rarr XMPrarr xanthosine route is predominant All three enzymes involved in the
conversion AMP deaminase (EC 3546) IMP dehydrogenase and 5prime-nucleosidase
have been detected in tea leaves (Koshiishi et al 2001) Xanthosine for caffeine
biosynthesis can also be produced from guanine nucleotides by GMPrarrguanosine
rarrxanthosine pathway Guanosine deaminase (EC 35415) activity is found in cell free
extracts from young tea leaves (Negishi et al 1994) but GMP deaminase activity was
not detected in plants (Stasolla et al 2003 Zrenner et al 2006) The absence of GMP
deaminase activity suggests that a GMP rarr IMP rarr XMP rarr xanthosine route is not
functional in plants
The use of radiolabelled purine bases and nucleosides for the study of caffeine
biosynthetic pathway was facilitated by the fact that exogenous purine nucleotides are
rapidly hydrolyzed by the plant tissues forming purine nucleosides andor bases which
are then utilized in pathways which in most instances indirectly lead to xanthosine
Most of these purine compounds including adenine adenosine inosine hypoxanthine
and guanine are converted to their respective nucleotides by salvage enzymes
including adenine phosphoribosyl transferase (EC 2427) hypoxanthineguanine
phosphoribosyl transferase (EC 2428) adenosine kinase (EC 27120) and
inosineguanosine kinase (EC 2421) which usually have high activities in plant cells
The resultant nucleotides then enter the purine alkaloid biosynthetic pathway In the
case of guanosine it may be directly deaminated to xanthosine and utilized for caffeine
biosynthesis (Figure 13) (Ashihara et al 1997)
182 SAM cycle in plants
SAM is the methyl donor for the three methylation steps in the caffeine biosynthetic
pathway In this process SAM is converted to S-adenosyl-L-homocysteine (SAH)
which in turn is hydrolyzed to homocysteine and adenosine Homocysteine is recycled
via the SAM cycle to replenish SAM levels and adenosine is released from the cycle
Adenosine released from the cycle is converted to AMP directly andor indirectly via
adenine AMP is converted to xanthosine and used for caffeine biosynthesis Since
three moles of SAH are produced via the SAM cycle for each mole of caffeine that is
synthesized in theory this pathway has the capacity to be the sole source of both the
Chapter 1 Introduction and Review of Literature
13
purine skeleton and the methyl groups required for caffeine biosynthesis in young tea
leaves (Koshiishi et al 2001)
Methionine
S-Adenosyl-L-methionineHomocysteine
ATP
PPi+ Pi
Tetrahydrofolate
5-Methyl-
tetrahydrofolate
CH3+Adenosine
S-Adenosyl-L-
homocysteine
(xanthine
structure)(Methyl groups)
Caffeine
(1)
(2)(3)
(4)
Figure 14 The SAM cycle (activated methyl cycle) in plants (Ashihara amp Suzuki 2004)
Enzymes SAM synthetase SAM-dependent N-methyltransferases S-adenosyl-
homocysteine (SAH) hydrolase Methionine synthase Adenosine released from the cycle
is salvaged to adenine nucleotides and utilized both for purine structure of caffeine via
xanthosine and for re-synthesis of SAM via ATP
183 N-methyltransferase in caffeine biosynthesis Molecular cloning
The genes encoding for the enzymes involved in the caffeine biosynthesis were
successfully cloned from the coffee family (Ogawa et al 2001 Mizuno et al 2003a
Mizuno et al 2003b Uefuji et al 2003) The conserved amino acid regions of tea CS
(AB31280) the sequences of O-methyltransferases derived from plant origin and
Arabidopsis hypothetical proteins were made use of in designing primers for RT-PCR
and RACE technique Independent groups have isolated N-methyltransferase (NMT)
genes from coffee which have been classified based on the substrate specificity of the
recombinant proteins expressed in Ecoli (Table 15) PCR products thus obtained were
used as the probe to isolate full length cDNA from the library resulting in the
identification of all three N-methyltransferases involved in caffeine biosynthesis
(Figure 12) Several cDNA have been characterized from the coffee plants one for
xanthosine methyltransferase (XMTXRS) three for 7-methylxanthine methyltransferase
or theobromine synthase (MXMTCTS) and three for 37-dimethylxanthine
methyltransferase or caffeine synthase (DXMTCCS) (Table 15) (Kato amp Mizuno
2004)
A single gene encoding for xanthosine methyltransferase (XMTCmXRS1) was
identified which encoded for a polypeptide consisting of 372 amino acids with an
apparent molecular mass of 418kDa It is expressed almost uniformly in aerial tissues
Chapter 1 Introduction and Review of Literature
14
of C arabica including leaves floral buds and immature but not in mature beans
Three genes encoding 7-methylxanthine methyltransferase (theobromine synthase) have
been identified The number of amino acids in the putative polypeptides are 378 for
MXMT1 (427kDa) and 384 for MXMT2 and CTS2 (434kDa) They differ by insertion
or deletion of blocks of several residues in the C-terminal region Their catalytic
properties based on kinetic parameters such as Km values were different They are
expressed in young leaves floral buds and immature but not mature beans Three genes
were identified for 3 7-dimethylxanthine methyltransferase (caffeine synthase) DXMT
CCS1 and CtCS7 each encoding a 43 kDa polypeptide consisting of 384 amino acids
However their kinetic properties differ from each other such as DXMT and CCS1
showed Km values of 1200 and 157microM for theobromine respectively Expressions
profiles are also distinct DXMT being expressed exclusively in immature beans while
CCS1 expression is ubiquitous occurring in all tissues The presence of isoforms of
these enzymes with different properties suggests that caffeine is synthesized through
multiple pathways depending on availability and concentration of the substrate (Mizuno
et al 2001 Ogawa et al 2001 Mizuno et al 2003a Mizuno et al 2003b Uefuji et
al 2003) Genomic clones of theobromine synthase were obtained by PCR- RFLP
from C canephora (Satyanarayana et al 2005 Satyanarayana 2006) The genomic
clones are reported to have highly conserved exons and a variable third intron The
promoter for the theobromine synthase was cloned and characterized (Satyanarayana et
al 2005)
There is high degree of similarity in the coding region of coffee NMT genes
isolated so far though differences exist in the 3prime untranslated region (UTR) for these
genes The deduced amino acid sequence of these enzymes shows more than 85
homology between these genes Phylogenetic analysis indicates that they are more
closely related to C-methyltransferases including those for jasmonic acid salicylic acid
and benzoic acid than to other N-methyltransferases This suggests that coffee N-methyl
transferases belong to a distinct sub-group within plant methyltransferases Their
cellular localization determined by green fluorescent protein fusion method and β-
glucourodinase (GUS) showed that the all the three genes are localized in the cytoplasm
(Ogawa et al 2001 Vinod et al 2007) It was also shown that these were present
along with chlorogenic acid(Vinod et al 2007)
Chapter 1 Introduction and Review of Literature
15
Table 15 N-methyltransferases and their encoding genes involved in caffeine biosynthesis
Gene (Enzyme) Gene name Accession No Description Reference
Xanthosine methyltransferase
( 7-methylxanthosine synthase) (EC 211158)
CaXMT AB 048793 Immature fruit cDNA Screening (Uefuji et al 2003)
CmXRS1 AB 034699 Leaf cDNA library RACE (Mizuno et al 2003a)
CaMXMT1 AB 048794 Leaf cDNA library RACE (Ogawa et al 2001)
7-methylxanthine synthase transferase
(theobromine synthase) ( EC 211159)
CTS1 AB 034700 Immature fruit cDNA Screening (Uefuji et al 2003)
CaMXMT2 AB 984125 Leaf cDNA library Screening (Mizuno et al 2001)
CTS2 AB 054841 Leaf cDNA library Screening (Mizuno et al 2001)
37-methylxanthinemethyltransferase
(Caffeine synthase) (EC 211160)
CaDXMT1 AB 086414 Immature fruit cDNA Screening (Uefuji et al 2003)
CCS1 AB 0861414 Immature fruit cDNA Screening (Uefuji et al 2003)
CtCS7 AB 0864415 Immature fruit cDNA
NMT genomic clones
Theobromine synthase-1
CX10 Complete coding region PCR Satyanarayan 2006
CX8 Complete coding region PCR -do-
Theobromine synthase-1 PG-5 Promoter + coding region RAGE -do-
Theobromine synthase-1 PG-1 Promoter + coding region RAGE Satyanarayan 2006
PG-4 Promoter + coding region RAGE -do-
Theobromine synthase-1 NMT
(AY273814) Complete coding region PCR Kochko A et al 2010
Theobromine synthase-2 NMT
(AY362825) Complete coding region PCR Kochko A et al 2010
Chapter 1 Introduction and Review of Literature
16
19 Catabolism of caffeine
Caffeine is produced in young leaves and immature fruits and continues to accumulate
gradually during the maturation of these organs At the same time caffeine is slowly
degraded with the removal of the three methyl groups resulting in the formation of
xanthine Catabolism of caffeine was first reported in coffee leaves (Kalberer 1965)
Since then a number of tracer experiments using 14
C labelled purine alkaloids have
been reported (Suzuki amp Waller 1984 Ashihara et al 1996 Vitoria amp Mazzafera
1999 Mazzafera 2004) They demonstrated the major catabolic pathway is caffeine rarr
theophylline rarr 3-methylxanthine rarr xanthine Xanthine is further degraded by the
removal by the conventional purine catabolic pathway to CO2 and NH3 via uric acid
allatonin and allantoate (Figure 15) (Ashihara amp Crozier 1999 Stasolla et al 2003
Zrenner et al 2006) Caffeine catabolism usually begins with its conversion to
theophylline catalyzed by N7-demethylase The involvement of P450-dependent mono-
oxygenase activity for this reaction was suggested (Huber amp Baumann 1998
Mazzafera 2004) However the activity of this enzyme has not yet been determined in
cell free extracts [8-14
C] Theophylline degraded to CO2 at much faster rate than [8-14
C]
caffeine indicating that conversion of caffeine to theophylline is a major rate limiting
step of caffeine catabolism and a possible reason why caffeine accumulates in high
concentrations in tissues of C sinensis and C arabica It is widely believed that the
control of caffeine levels in plants is a function of the balance between rates of
synthesis and degradation and this balance seems to vary depending on the plant
species and the tissue developmental stage (Mazzafera 2004)
Chapter 1 Introduction and Review of Literature
17
Figure 15 Caffeine catabolic pathways Caffeine is mainly catabolised via theophylline and 3-
methylxanthine Xanthine is further degraded to CO2 and NH3 by the conventional
oxidative pathway Adapted from (Ashihara et al 2008)
Chapter 1 Introduction and Review of Literature
18
110 Genetic engineering of Coffee
Genetic transformation has potential applications in coffee agriculture for incorporating
genes which could provide desirable traits such as disease and insect resistance
drought and frost tolerance and herbicide resistance Transgenic technology can also be
used to increase nutritional value and improve cup quality produce varieties with
caffeine-free beans and for production of hybrid crops for molecular farming
Identification of target specific genes is one of the pre-requisites for developing
transgenic crops The availability of a large number of EST sequences in coffee and
initiation of coffee genome sequencing may speed up the gene discovery and accelerate
transgenic research efforts in coffee
1101 Insect Resistance
Production of insect resistant coffee plants is one of the major objectives of the
breeding programs The major pests attacking coffee include coffee berry borer (CBB
Hypothenemus hampei) white stem borer (WSB Xylotrechus quadripes) leaf miner
(Perileucoptera coffeela) and root nematodes (Meloidogyne spp and Pratylenchus
spp) The CBB is present in almost all the coffee growing countries and considered to
the most devastating pest in coffee To date there is no reported source of resistance to
CBB in the coffee gene pool Like CBB WSB is another serious pest in C arabica in
India and several other East Asian countries Both CBB and WSB belong to the order
Coleoptera For India controlling WSB is the biggest challenge and has therefore
become the highest research priority Robusta is generally resistant to WSB but the
interspecific Robusta Arabica hybrids are susceptible to WSB Although leaf miner is
not yet a serious pest in India and other East Asian countries it is an economically
important pest in East Africa and Brazil Effective chemical control of CBB and WSB
is difficult due to the nature of their life cycle inside the berry and stem respectively as
well as environmental concern regarding the use of pesticides Biological control
measures are adapted to combat these insect pests with varying degrees of success
Developing coffee plants resistant to these pests using genetic transformation
technology could be one of the alternative strategies to counter pest damage For insect
resistance several different classes of proteins from bacterial plant and animal sources
have been isolated and their insecticidal properties tested against many important pests
Amongst these proteins Bt toxins are most important and several transgenic crops
expressing Bt genes have been commercialized Coffee transgenic plants carrying a
Chapter 1 Introduction and Review of Literature
19
synthetic version of the cry1Ac gene have been produced (Leroy et al 2000 Mishra et
al 2002) Indeed this was the first report of an important agronomic trait being
introduced into a coffee plant The transgenic plants presented similar features in
growth and development compared to normal plants Transgenic plants highly resistant
to leaf miner under greenhouse conditions were tested under field conditions in French
Guyana for 4 years for pest resistance (Perthius et al 2005 Perthius et al 2006)
From 54 independent transformation events 70 of the events were resistant to leaf
miner Unfortunately the field trial was vandalized which led to the termination of the
experiment (Montagnon et al 2005)The effectiveness of Bt genes in controlling
coleopteran pests is well documented in corn and potato which indicates that Bt genes
might be effective against CBB The high toxicity of B thuringiensis sero var
israelensis against first in star larvae of CBB has been demonstrated (Mendez et al
2003) In another experiment an α-amylase inhibitor from Phaseolus vulgaris was
tested against CBB and found to have an inhibitory effect on its growth and
development (Grossi et al 2004)In addition to the CBB and WSB C arabica
varieties are also susceptible to endoparasitic root-knot nematode (Meloidogyne spp)
(Campos et al 1990 Mishra et al 2012)
So far 15 species have been reported to be parasites of coffee Controlling
nematodes is extremely difficult and currently seedling grafting with robusta rootstock
is followed as one of the control measure Sources of resistance specific to root-knot
nematodes have been identified in coffee trees (Bertrand et al 2001) and the Mex-1
gene conferring resistance to M exigua in C arabica is in the process of isolation (Noir
et al 2003)
1102 Tolerance to Abiotic Stress
In many coffee growing countries abiotic stress such as drought and frost are the major
climatic factors that limit coffee production Changes in climatic patterns due to global
climate change are considered increasingly important for coffee cultivation Drought
induces water stress in plants which affects vegetative growth and vigor and triggers
floral abnormalities and poor fruit set It also indirectly increases the incidence of pests
and diseases in the plants C arabica is generally more tolerant to water stress than C
canephora partly due to its extensive deep root system C racemosa is known to be a
good source of drought tolerance In India hybrids between C canephora and C
racemosa have been obtained and are currently under evaluation for drought tolerance
Chapter 1 Introduction and Review of Literature
20
In many coffee growing countries coffee is propagated in marginal areas where the
annual rainfall is below 1000thinspmm with prolonged dry spells of over 4-5 months In
those areas water shortage and unfavorable temperatures constitute major constraints
and the growth and productivity of C canephora are badly affected As with drought
periodic frost also affects coffee production in parts of Brazil The introduction of
drought and frost tolerant genes through genetic transformation would be of great
importance for alleviating these problems Research is now being carried out by several
groups to identify genes involved in biotic as well as abiotic stress The most promising
genetic engineering approach for drought tolerance includes the use of functional or
regulatory genes as well as the transfer of transcription factors In recent years plants
tolerant to high temperature and water stress have been the subject of intense research
(Chaves et al 2003 Chaves et al 2004 Coraggio et al 2004) For achieving drought
tolerance genes that have been targeted include those encoding enzymes involved in
detoxification or osmotic response metabolism enzymes active in signaling proteins
involved in the transport of metabolites and regulating the plant energy status (Chaves
et al 2003 Chaves et al 2004 Coraggio et al 2004) The dissection of molecular
mechanisms related to signal transduction and transcriptional regulation might help in
engineering drought tolerance in coffee
1103 Disease Resistance
Coffee leaf rust (CLR) caused by the fungus Hemileia vastatrix is the most important
disease in coffee with substantial loss to coffee production and productivity in all the
coffee growing countries In addition to leaf rust coffee berry disease (CBD) caused by
fungus Colletotrichum kahawae can be a devastating anthracnose leading to substantial
crop loss in Africa Several other fungal and bacterial diseases may also affect coffee
to a extent C arabica is more susceptible to many diseases than C canephora Though
most of the disease control measures rely upon chemical control they are more
expensive and labour intensive The long-term solution is the breeding of resistant
varieties which is the focus of many breeding programs However breeding for disease
resistant varieties is time consuming due to the perennial nature of coffee with its long
gestation period India has a long history of C arabica breeding especially for leaf rust
resistance and is the first country to demonstrate the existence of multiple races of leaf
rust Resistance to CLR is conditioned primarily by a number of major (SH) genes and
coffee genotypes are classified into different resistance groups based on their
Chapter 1 Introduction and Review of Literature
21
interaction with different rust races pathogen (van der Vossen 2001) Currently C
canephora provides the main source of resistance to pests and diseases including CLR
(H vastatrix) and CBD (C kahawae) and therefore used in breeding programs Other
diploid species like C liberica and C racemosa are being used as a source of resistance
to coffee leaf rust and coffee leaf miner respectively (Guerreiro et al 1999
Sreenivasan et al 1989) The development of coffee varieties resistant to major fungal
diseases such as CLR and CBD using transgenic technology will benefit the coffee
industry immensely During the last 15 years significant progress has been made in the
area of host-pathogen interactions (Staskawicz et al 1995 Feys et al 2000
Shivaprasad et al 2012) and many resistance genes involved in recognizing invading
pathogens have been identified and cloned (Takken et al 2000) A number of
signalling pathways induced because of pathogen infection have been dissected (Dong
et al 1998 Navarro et al 2006) Many antifungal compounds synthesized by plants to
combat fungal infection have been identified (Does et al 1998) Understanding the
specific induction of targeted pathways and identification of specific pathways
responsible for particular fungal resistance is important in order to employ this strategy
in transgenic technology The recent investigation of gene expression during coffee leaf
rust infection could provide an insight into the defence pathways operating in coffee
(Fernandez et al 2004 Nardi et al 2006) Efforts have been made to identify and
clone resistance genes from coffee for achieving durable resistance The genetic and
physical map of two resistance genes SH3gene conferring resistance to rust (Prakash et
al 2004) and the Ck1 gene conferring resistance to C kahawae CBD (Gichuru et al
2006) have been established These genes could be used for molecular marker assisted
breeding programs (Mishra et al 2012)
1104 Production of low-caffeine coffee
A widespread belief that the ingestion of caffeine can have adverse effects on
health resulted in the increased demand for decaffeinated coffee (Ashihara amp Crozier
2001) Several method were used to obtain decaffeinated plants by conventional
breeding techniques using low yielding varieties and mutants that accumulated
relatively low amounts of caffeine when compared to the commercially available ones
Low-caffeine and decaffeinated coffee represent around 10 of the coffee sales
around the world (Ribas et al 2006b) The industrial process for coffee decaffeination
can be expensive and affects the original flavour and aroma in coffee (David 2002)
Chapter 1 Introduction and Review of Literature
22
Transgenic coffee plants with suppressed caffeine synthesis using RNA interference
(RNAi) technology have been obtained (Ogita et al 2003 Ogita et al 2004) Specific
sequences in the 3prime untranslated region of the theobromine synthase gene (CaMXMT1)
were selected for construction of RNAi short and long fragments The caffeine and
theobromine content of the transgenic plants were reduced by ~ 70 when compared to
the untransformed plants In C canephora RNAi technology has also been employed
to silence the N-methyl transferase gene involved in caffeine biosynthesis (Vinod et al
2007) Promoter of an N-methyltransferase (NMT) gene involved in caffeine
biosynthesis was cloned (Satyanarayana et al 2005 Satyanarayana 2006) which will
be very useful for studying the regulation of caffeine biosynthesis
1105 Improvement in cup quality
Improvement in coffee cup quality requires elaborate knowledge of the chemical
constituents as well as the metabolic pathways involved in the elaboration of quality
The constituents of coffee beans include minerals proteins carbohydrates caffeine
chlorogenic acids (CGA) glycosides lipids and many volatile compounds that give
flavour to coffee by roasting Among these the role of three major constituents
sucrose CGA and trigonelline have been studied in coffee The sucrose content of
coffee bean is associated with coffee flavour the higher the sucrose content in green
beans the more intense will be the cup flavour (Clifford et al 1985 de Maria et al
1996) The sucrose content of C arabica (82-83) is higher than C canephora (33ndash
40) The sucrose amino acids ratio in green beans determines the profile of volatile
compounds Manipulating sucrose content in coffee bean is therefore important in
improving cup quality The sucrose synthase gene (CcSUS2) from C canephora has
been cloned and sequenced (Leroy et al 2005) This provides an opportunity to
manipulate the sucrose content in coffee Chlorogenic acids are products of
phenylpropanoid metabolism Chlorogenic acids are a group of hydroxycinnamoyl
quinic acids (HQA) formed by esterification between caffeic acids coumaric acids and
quinic acids (Clifford et al 1999) They are present in relatively large quantities in the
coffee bean and are the precursor of phenolic compounds in roasted beans C
canephora beans contain higher CGA (10) compared to C arabica beans (6-7)
CGA are known to have antioxidant properties as well as being associated with disease
resistance (Campa et al 2003) Genetic manipulations of genes involved in CGA
synthesis can serve either of these purpose by up- or down regulating the pathway
Chapter 1 Introduction and Review of Literature
23
Phenylalanine ammonia-lyase (PAL) catalyzes the first step of the phenylpropanoid
pathway leading to the synthesis of a wide range of chemical compounds including
flavonoids coumarins hydroxycinnamoyl esters and lignins (Hahlbrock amp Scheel
1989) Recently the full length cDNA and corresponding genomic sequences of PAL
from C canephora was isolated characterized and functionally validated (Mahesh et
al 2006 Parvatam et al 2006) This has opened up new possibilities for manipulating
the level of the PAL enzyme in coffee which in turn can be useful for improvement of
cup quality and manipulating antioxidant properties in coffee
1106 Fruit Ripening
Uniformity during fruit ripening is decisively related to cup quality in coffee and
consequently to the value of the product Fruits at the ideal ripening stage produce the
best organoleptic characteristics for coffee The presence of over ripened or green fruits
changes the acidity the bitterness and consequently the cup quality In order to
maximize uniform ripening of coffee fruits it is essential to control the action of genes
involved in the last step of maturation process Ethylene is known to trigger ripening
and increasing ethylene biosynthesis is associated with various stages of ripening
process (Protasio et al 2005) To control coffee fruit maturation two of the major
genes involved in ethylene biosynthesis namely ACC synthase and ACC oxidase
have been cloned (Protasio et al 2005 Neupane et al 1999) Introduction of the ACC
oxidase gene in antisense orientation have been achieved in both C arabica and C
canephora The effect of the transgene on ethylene production and fruit maturation has
yet to be reported The inhibition of genes downstream to the initial ethylene burst is
also an option to control coffee fruit maturation (Ribas et al 2006)
111 RNA interferenceRNAi
The regulation of gene expression at post transcriptional level has recently attracted
much attention because of the discovery of the phenomenon called RNA interference
(RNAi) RNAi based silencing techniques are more efficient than antisense mediated
gene silencing (Wesley et al 2001) The effect was first observed in plants wherein
silencing was observed when some copies of a transgene was in the forward orientation
with respect to the promoter and some in the reverse orientation This resulted in the
formation of double stranded RNA in the system The phenomenon in which
experimentally introduced double stranded RNA (dsRNA) leads to the loss of
expression of the corresponding cellular gene by sequence specific RNA degradation
Chapter 1 Introduction and Review of Literature
24
was termed as RNA interference or RNAi The protective effect of RNA silencing in
reducing virus infectivity supports the view that PTGS has evolved as a mechanism to
defend plants against virus infection and also to moderate the possible deleterious
genome-restructuring (insertional) activity of virus-like mobile genetic elements eg
retrotransposons A growing body of biochemical and genetic data has further
demonstrated that plants animals and yeasts share related mechanisms of specific
degradation of RNAs in which double-stranded forms of RNA are initiator molecules
(Brenstein et al 2001)
The key insight into the process of PGTS was provided by the experiments
conducted by Hamilton amp Baulcombe (1999) who identified the product of RNA
degradation as small RNA (siRNA) species of ~25nt of both sense and antisense
polarity SiRNA are formed and accumulate as double stranded RNA molecules of
defined chemical structures Both genetic and biochemical approaches were undertaken
to understand the basis of silencing Genetic screens were carried out in fungus
Neurospora carassa the alga Chalmydomonas reinhardtii and plant Arabidopsis
thaliana to search for detective mutants in PTGS
A combination of results obtained from several in vivo and in vitro experiments
have gelled into a two step mechanistic model for RNAiPTGS The first step referred
to as the RNAi initiating step involves binding of the RNA nucleases to a larges
dsRNA and its cleavage into discrete ~21-25nt RNA fragments This is ATP-
dependent reaction which involved RNase III - type endonucleases which make
staggered cuts in both strands of the dsRNA leaving 3prime overhang of 2 nucleotides with
5prime- phosphate 3prime-hydroxyl and no modification in the sugar phosphate backbone
(Hamilton amp Baulcombe 1999 Elbashir et al 2001) SiRNAs are the direct products
of dsRNA cleavage by the multi-domain RNase III enzyme DICER (Figure 16)
(Brenstein et al 2001 Karthikeyan et l 2013)
In the second step or commonly known as the effector step the double stranded
siRNAs produced in the first step bind to an RNAi specific protein complex to form a
RISC The complex undergoes activation in the presence of ATP so that the antisense
component of the unwound siRNA becomes exposed and allows the RISC to perform
the downstream RNAi reaction
Chapter 1 Introduction and Review of Literature
25
Several enzymes are involved in RNAi based on their role genes that encode for them
are classified into RNAi initiators RNAi effectors RNA dependent RNA polymerases
and silencing genes Two C elegans genes rdeI and rde4 (rde stands for lsquo RNAi
deficientrsquo) are believed to be involved in the initiation step of RNAi The C elegans
rde1 gene is a member of a large family of genes and is homologous to the Neurospora
qde2 (qde stands for ldquoquelling deficientrdquo) and Arabidopsis AGO1 (AGO stands for
agronaute AGO1 was previously identified to be involved in Arabidopsis
development) Although the functions of these genes are not clear a mammalian
member of RDE1 family has been identified as a translation initiation factor (Thakur
2005) Important genes for the effector step of PTGS in C elegans are rde2 and mut7
genes These genes were identified from heterozygous mutant worms that were unable
to transmit RNAi to their homozygous offsprings Worms with mutated rde2 or mut 7
genes show defective RNAi mut-7 gene encodes a protein with homology to the
nuclease domains of RNAase D and a protein implicated in Werner syndrome (a rapid
ageing disease in humans) (Grishok et al 2001 Kamath-Loeb et al 2012s)
Neurospora qde-1 Arabidopsis SDE-1SGS-2 and C elegans ego-1 appear to encode
RNA dependent RNA polymerase (RdRPs) It assumed that this is a proof that an RdRp
activity is required for RNAi Certainly the existence of an RdRp might explain the
remarkable efficiency of dsRNA induced silencing if it amplifies either the dsRNA
prior to cleavage or the siRNAs directly (Muers 2013) In C elegans ego-1 mutants
(ego stands for lsquoenhancer of glp-1) RNAi functions normally in somatic cells but is
defective in germline cells where ego-1 is primarily expressed In Arabidopsis SDE-
1SGS-2 mutants (SGS stands for suppressor of gene silencing) siRNA are produced
when dsRNA is introduced via an endogenously replicating RNA virus but not
introduced by a transgene It has been proposed that perhaps the viral RdRP is
substituting for the Arabidopsis enzyme in these mutants Random degradative PCR
model suggests that an RdRP uses the guide strand of an siRNA as a primer for the
target mRNA generating a dsRNA substrate for Dicer and thus more siRNAs
Several genes controlling RNA silencing in plants have been identified through
genetic screens of Arabidopsis mutants impaired in transgene induced RNA silencing
They encode a putative RNA-dependent RNA polymerase (SGS2SDE1) a coiled
protein (SGS3) a protein containing PAZ and Piwi domains (AGO1) and an RNA
helicase (SDE3) The putative SGS2SDE1 of Neurospora is related to QDE-1 and
Chapter 1 Introduction and Review of Literature
26
EGO-1 of C elegans whereas PAZPiwi protein AGO is related to QDE-2 of
Neurospora RDE-1of C elegans RNA helicase SDE3 is related to SMG-2 of C
elegans and Mut-6 of Chlamydomonas MUT-7 gene of C elegans encodes a protein
similar to Rnase D whereas the Drosophila DICER gene encode a protein similar to
RNase III An Arabidopsis ortholog of DICER gene has been identified called CAF
SIN1 SUS1(Thakur 2005 Poethig 2009)
Since the RNAi machinery is present constitutively within the eukaryotic cell it
is important to explore the metabolic advantages that are accorded by the RNAi related
proteins during the intrinsic normal growth of cells and development of organisms The
natural RNAi machinery not only keeps the mobile transposable elements from
disrupting the integrity of the genomes as suggested by the analysis of model organisms
like A thaliana C elegans D melanogaster (Tabara et al 1999 Wu-Scharf et al
2000 Aravin et al 2001 Hamilton et al 2002 Lisch 2009) but also participates in
development of organisms Genetic defects in Celegans RNAi genes ego1 and dicer
cause known specific developmental errors (Grishok et al 2000 Grishok et al 2001
Knight amp Bass 2001 Hall et al 2013) Similarly the Argonaute family of genes of A
thaliana (especially the ZWILLE proteins) is also responsible for plant architecture and
meristem development (Carmell et al 2002 Hamant and Pautot 2010) and the Dicer
homologue of A thaliana CAF1 is required for embryo development (Golden et al
2002 Nakano et al 2011) Thus genetic evidence illustrates the role of the RNAi
machinery as a controller of development related genes The mechanistic details of
these developmental processes are beginning to emerge
Chapter 1 Introduction and Review of Literature
27
Figure 16 Mechanism of gene silencing induced double stranded RNA The dicer enzyme to
produce siRNAs cleaves the RNA The siRNA generated bind to the nuclease complex
RISC The antisense component in the siRNA in the RISC guides the complex towards
the cognate mRNA resulting in endonucleolytic cleavage of the mRNA
Chapter 1 Introduction and Review of Literature
28
112 MicroRNA or MiRNA
In 1991 Ambros and co-workers first isolated a lin-4 mutant of Celegans which was
arrested at the first larval stage (Lee et al 1993) Later on the let-7 mutation was
isolated in the same system which was responsible for development through the fourth
larval stage Both lin-4 and let-7 encode short 22-nucleotide mature RNAs and were
called short temporal RNA because they control the temporal development program of
C elegans The mature lin-4 RNA defines (negatively regulates) the mRNA expression
of the lin-14 and lin-28 hetrochronic genes with the antisense mediated repression
mechanism of translation initiation and thus specifies the fate of cells during the first
three larval stages Recent studies have revealed that the short temporal RNAs are
actually members of a group of tiny RNAs (21to 28 nucleotides) called the micro-
RNAs (miRNA) (Bartel 2009) isolated members of which could easily run to a few
thousand which includes both those were identified computationally and by deep
sequencing Some of the components of the RNAi machinery have also been clearly
established as the effector proteins for the maturation of miRNAs
113 MicroRNAs in plants
1131 Discovery
MicroRNAs have been discovered by three basic approaches Direct cloning forward
genetic direct cloning and bioinformatic prediction followed by experimental
validation The most direct method of miRNA discovery is to isolate and clone small
RNAs from biological samples and several groups have used this approach to identify
plant miRNAs (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002 Xie et al 2003 Sunkar et al 2005 Williams et al 2013) The cloning methods
adapted were similar to those used to identify large numbers of animal miRNAs
(Lagos-Quintana et al 2001 Lau et al 2001 Aravin amp Tuschl 2005) which involve
isolating small RNAs followed by oligonucleotide adapter ligation reverse
transcription amplification and sequencing Some protocols incorporate methods to
enrich for Dicer cleavage products (ie molecules with 5prime-phosphates and 3prime-
hydroxyls) and to concatemerize the short cDNAs so that several may be identified in a
single sequencing read (Lau et al 2001 Llave et al 2002a Jagadeeswaran amp Sunkar
2013)The initial cloning experiments in Arabidopsis identified 19 miRNAs belonging
to 15 families (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002) although hundreds of other small RNAs such as siRNAs and RNA degradation
Chapter 1 Introduction and Review of Literature
29
fragments were also cloned through the direct cloning methods To select miRNAs
from the pool of small RNAs secondary structures of the RNA molecules were
examined in compliance with the acknowledged structural features of miRNAs
(Ambros et al 2003) The secondary structures were validated using several ad-hoc
software tools such as the Mfold or RNAfold programs (Reeder et al 2006) Finally
the existence of mature miRNAs was determined by RNA gel blot hybridization These
direct cloning methods were successful in isolating many miRNAs in Arabidopsis
(Reinhart et al 2002 Sunkar amp Zhu 2004) Rice (Sunkar et al 2005) Poplar (Lu et
al 2005) moss (Arazi et al 2005) and Tobacco (Billoud et al 2005)
Although miRNAs were first discovered through forward genetic screens in
worms (Lee et al 1993 Reinhart et al 2000)only few A thaliana miRNA gene
families have been discovered by this method (Chen 2008) in plants MiRNA
involvement in plant mutant phenotypes were not properly inferred till the cloning
experiments established that plant genomes contain numerous miRNAs (Park et al
2002 Reinhart et al 2002 Rhoades et al 2002) MiRNA loss-of-function allele has
been identified by forward genetic screens early extra petala1 is caused by a
transposon insertion ~160bp upstream of the predicted miR164c stem loop resulting in
flowers with extra petals (Baker et al 2005 Irish 2008) The fact that loss-of-function
miRNA mutants have been rarely discovered using forward genetics reflects small
target size for mutagenesis coupled with redundancy in nearly all evolutionarily
conserved plant miRNAs which are encoded by gene families (Table16) Family
members are likely to have overlapping functions buffering against loss at any single
miRNA locus Over expression screens can circumvent redundancy limitations At least
four plant miRNAs miR319 (also known as miR-JAW) miR172 (also known as EAT)
and miR166 were isolated in over expression screens for dominant mutants with
developmental abnormalities (Aukerman amp Sakai 2003 Palatnik et al 2003 Kim et
al 2005 Williams et al 2005b Schommer et al 2012) Mutations in miRNA target
sites which can prevent the entire family of miRNAs from repressing a target gene can
also circumvent redundant functions of miRNA family members
The dominant mutations in the HD-ZIP genes PHB PHV and REVOLUTA
(REV) in Arabidopsis and ROLLEDLEAF1 (RLD1) in Maize result in adaxialization of
leaves andor Vasculature (McConnell amp Barton 1998 McConnell et al 2001 Nelson
et al 2002 Zhong amp Ye 2004 Allen amp Millar 2012) and are all caused by mutations
Chapter 1 Introduction and Review of Literature
30
in miR166 complementary sites (Rhoades et al 2002 Emery et al 2003 Jones-
Rhoades amp Bartel 2004 Mallory et al 2004 Zhong amp Ye 2004)
In both plants and animals cloning was the initial means of large-scale
miRNA discovery (Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Reinhart et al 2002 Mendes et al 2012) However cloning is biased toward
RNAs that are expressed highly and broadly MiRNAs expressed at low levels or
only in specific cell types or in response to certain environmental stimuli were more
difficult to clone Sequence based biases in cloning procedures might also cause
certain miRNAs to be missed Because of these limitations bioinformatics approaches
to identify miRNAs have provided a useful complement to cloning
A straightforward use of bioinformatics has been carried out to find homologs
of known miRNAs both within the same genome and in the genomes of other species
(Pasquinelli et al 2000 Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Mendes et al 2009) A more difficult challenge is to identify miRNAs
unrelated to previously known miRNAs This was first accomplished for
vertebrate nematode and fly miRNAs using algorithms that search for conservation
of sequence and secondary structure (ie miRNA stem-loop precursors) between
species searching for patterns that are characteristic of miRNAs (Lai et al 2003 Lim
et al 2003 Lim et al 2005)
Most of the methods identified numerous miRNAs but are necessarily more
relaxed in terms of allowed structures Some approaches take advantage of the high
complementarity of plant miRNAs to target messages implementing the requirement
that the candidate has conserved complementarity to mRNAs (Jones-Rhoades amp Bartel
2004 Adai et al 2005) This additional filter has been useful for distinguishing
authentic plant miRNAs from false positives which later extended to mammalian
miRNA gene prediction (Xie et al 2005 Mendes et al 2012)
Another criteria used by most algorithms in the filters is evolutionary
conservation In plants mature miRNA sequences are conserved to a higher degree
than their precursor sequences For example Arabidopsis and rice diverged more than
130 million years ago (Friis et al 2004) but some miRNAs are highly conserved
in these plant species (Reinhart et al 2002) Furthermore various parameters for the
secondary structures such as free energy the number of paired residues within a
Chapter 1 Introduction and Review of Literature
31
miRNA or the number and size of bulges are also used to validate the predictions
(Jones-Rhoades amp Bartel 2004 Wang et al 2004 Adai et al 2005 Mendes et al
2012)
1132 Conserved microRNAs in Plants
Till date cloning genetics and bioinformatics screens have resulted in annotation of
approximately 184 potential Arabidopsis miRNAs (miRBase release 13) These
miRNAs seem to arise through gene duplication and subsequent diversification and
represent approximately 100 families (Maher et al 2006 Axtell 2013) Twenty one
families represented by 92 genes are clearly conserved in species beyond Arabidopsis
(Table 15) The presence of these miRNA families in other sequenced plant genomes
indicates that different plant species have an evolutionarily fluid set of miRNAs
(Willmann amp Poethig 2007) Recent studies on mosses one of the most ancient land
plants have shown that at least 14 miRNA families are conserved in eudicots and
mosses and therefore it appears that plant miRNAs share a common ancient origin
(Arazi et al 2005 Axtell amp Bartel 2005 Talmor-Neiman et al 2006 Zhang et al
2006 Axtell 2013) The number of members per family in a genome ranges from 1-32
with the exception of miR-430 which is represented by a cluster of ~80 loci in zebra
fish (Giraldez et al 2005)
In plants the number of members in each family correlates among examined
species certain families contain numerous members in all three species (eg miR156
miR166 miR169) whereas others consistently contain fewer genes (eg miR162
mir168 miR394) (Table 16) Although it is still unclear why certain miRNA are
encoded by more than one gene in plants (eg 12 genes encoding miR156) this
correlation might suggest functional significance of various miRNA family sizes Most
annotated plant miRNAs are conserved throughout flowering plants while a few
miRNAs are found only in a single sequenced genome and thus they could be
evolutionarily lsquoyoungrsquo miRNAs For example Arabidopsis has several miRNA
families that are not found in poplar or rice suggesting that miRNAs could be
generated continuously during evolution (Allen et al 2004a Rajagopalan et al
2006 Fahlgren et al 2007 Axtell 2012 de Alba et al 2013) Consistent with the
notion that non conserved miRNAs are of a more recent evolutionary origin these
miRNAs are mainly encoded by a single locus in the genome Within each family the
mature miRNA is always located in the same arm
Chapter 1 Introduction and Review of Literature
32
Table 16 MicroRNA families conserved in plants
miRNA
family
Arabidopsis Oryza Populus
miR156 12 12 11 miR159319 6 8 15 miR160 3 6 8 miR162 2 2 3 miR164 3 5 6 miR166 9 12 17 miR167 4 9 8 miR168 2 2 2 miR169 14 17 32 miR171 4 7 10 miR172 5 3 9 miR390 3 1 4 miR393 2 2 4 miR394 2 1 2 miR395 6 19 10 miR396 2 5 7 miR397 2 2 3 miR398 3 2 3 miR399 6 11 12 miR408 1 1 1 miR403 1 0 2 miR437 0 1 0 miR444 0 1 0 miR445 0 9 0 Total 92 127 169
All known miRNA families that are conserved between more than one plant species are listed
together with the number of genes identified in the sequenced genomes Rice miRNA families that have orthologs in maize but do not appear to have orthologs in the eudicots (Arabidopsis and Populus) are marked with an asterisk The following families contain miRNA genes annotated with
more than one number miR156 (miR156 and miR157) miR159319 (miR159 and miR319) miR166 (miR165 and miR166) miR171 (miR170 and miR171) and miR390 (miR390 and miR391)
of the stem loop (5prime- or 3prime) as it would be expected if the genes carry common ancestry
(Figure 17) The sequence of the mature miRNA and to some extent the segment on
the opposite arm of the hairpin to which it pairs is highly conserved between the
members of the same family (both within and between the species) while the sequence
secondary structure and length of the intervening ldquolooprdquo region can be highly divergent
within the same family
The patterns of paring and non pairing nucleotides are often conserved between
homologous miRNA stem loops from different species (Figure 17) The significance of
conserved mismatches is opined to guide DCL1 to cleave at the appropriate positions
along the stem loop
Chapter 1 Introduction and Review of Literature
33
Figure 17 Representative stem loop of miR164 Segments corresponding to the mature miRNAs
are shown in red (Adapted from Rajagopalan et al 2006)
Although many miRNAs are highly conserved in plants the degree of conservation in
most cases is quite low between plants and animals one exception is miR854 A recent
study has revealed that miR854 is conserved between Arabidopsis and animals and that
the Arabidopsis miR854 differs from the human miR854 only by a single nucleotide
(Arteaga-Vazquez et al 2006 Axtell 2012 de Alba et al 2013) The Arabidopsis
miR854 has multiple binding sites in the 3prime-untranslated region (UTR) of
OLIGOURIDYLATE-binding-PROTEIN mRNA 1bA (UBP1b) and represses the
translation of the UBP1b mRNA The animal miR854 is also complementary to a site in
the 3prime-UTR of the UBP1b homolog in animals However it is currently unclear how the
animal miR854 regulates its target mRNA Another distinction between plant and
animal miRNAs is the genomic organization of the miRNA genes While plant miRNA
genes are located predominantly in the intergenic regions animal miRNA genes tend to
localize in the intragenic regions both in the introns or exons of protein coding genes
Moreover miRNA genes are often clustered within a single precursor transcript
whereas individual plant miRNAs are derived from individual precursor RNAs (Bartel
Chapter 1 Introduction and Review of Literature
34
2004 Bonnet et al 2004 Kim 2005 Vaucheret et al 2006 Marco et al 2013)
1133 Non-conserved miRNA and challenges in annotation
Most miRNAs are conserved throughout flowering plants (Table 15) but many more
were found only in a single sequenced genome thus considered to be of a more recent
evolutionary origin The extended homology between few non conserved miRNAs
precursors and their target genes provides strong evidence that these relatively ldquoYoungrdquo
miRNAs arose from the duplication of the target gene segments (Allen et al 2004)
Several non-conserved miRNAs including miR161 miR163 miR173 miR447
miR475 and miR476 are known to direct cleavage of target transcripts (Allen et al
2004a Adai et al 2005 Sunkar et al 2005 de Alba et al 2013) It is difficult to
confidently predict targets for many because it is not possible to use complementary
site as a filter against false-positive target predictions It is also difficult to
confidently annotate non-conserved miRNAs as miRNA rather than siRNAs The
established minimal standard for miRNA annotation is a small RNA with detectable
expression and the potential to form a stem-loop when joined to flanking genomic
sequence (Kasschau et al 2003) In practice these requirements are too loose to
definitively categorize many small RNAs cloned from plants Many plant siRNAs are
detectable on blots (Vazquez et al 2004b) and hundreds of thousands of non-
miRNA genomic sequences can be predicted to fold into secondary structures that
resemble structures of plant miRNA precursors (Jones-Rhoades amp Bartel 2004)
Therefore without conservation of both sequence and secondary structure it is
difficult to be confident that a given cloned RNA originated from a stem-loop (ie is a
miRNA) rather than from a double-stranded RNA (ie is a siRNA) In fact many of
the thousands o f small RNAs cloned from Arabidopsis (Gustafson et al 2005) would
probably meet the literal requirements for annotation as miRNAs A few of these
sequences might be miRNAs but others that meet the literal criteria probably are not
so The challenges of annotating non conserved small RNAs were evidenced by
three related small silencing RNAs that were originally annotated as miRNAs
that turned out to be trans-acting siRNAs (tasiRNAs)(Kanno et al2013)
Although most miRNAs that are broadly conserved among flowering plants are
now being identified and experimentally validated (Table 16)(Jones-Rhoades amp
Bartel 2004) the challenges and ambiguities for classifying non-conserved miRNAs
prevent meaningful estimates of the total number of miRNA genes in Arabidopsis and
Chapter 1 Introduction and Review of Literature
35
other plant genomes It is possible that miRNAs might have escaped detection
because they are non-conserved and expressed in specific tissues or conditions and can
only be identified by using high- throughput sequencing
1134 MicroRNA biogenesis
11341 Transcription of microRNA precursor
Plant miRNAs are primarily found in the genomic regions that are not associated with
the protein coding genes (Reinhart et al 2002) and it appears that most if not all plant
miRNAs are produced from their own transcriptional units Plant miRNA genes are
occasionally clustered near each other in the genome suggesting transcription of
multiple miRNAs from a single primary transcript [eg the miR395 cluster (Grishok
et al 2001 Jones-Rhoades amp Bartel 2004b)] but this polycistronic arrangement of
miRNA genes appears far less frequently in plants than in animals (Bartel 2004)
Northern blot EST and mapping evidence indicate that plant miRNA primary
transcripts (also known as pri-miRNAs) as in animals are longer than needed to
encompass the miRNA stem-loops (Palatnik et al 2003 Jones-Rhoades amp Bartel
2004b) At least some of these pri-miRNA transcripts appear to be spliced
polyadenylated (Kurihara amp Watanabe 2004) and capped (Xie Z et al 2005) Two
rice miRNAs are contained within transcripts that contain exon junctions within the
presumptive stem-loop precursor implying that in these cases splicing is a prerequisite
for Dicer recognition (Sunkar et al 2005) Plant pri-miRNAs are over 1kb in length
and they are usually preceded by typical TATA box motifs They can also undergo
canonical splicing polyadenylation and capping All these observations indicates RNA
polymerase II is probably responsible for transcribing most plant miRNAs (Xie Z et
al 2005) as appears to be the case for many animal miRNAs Relatively little is
known about the regulation of miRNA transcription in plants but there is no reason
to suspect that this regulation would differ from that of protein-coding transcripts as
the bioinformatics analysis of the sequence upstream of the transcriptional start
sites of the miRNA genes showed the presence of putative binding motifs of
number of known transcription factors (Megraw et al 2006 Sethupathy 2013)
11342 MicroRNA processing and export
In plants maturation of miRNA is a step wise process A miRNA gene is transcribed to
pri-miRNA which is much longer than the pre-miRNA possessing the characteristic
stem-loop structure Like transcription of most protein-coding genes the miRNA
Chapter 1 Introduction and Review of Literature
36
transcription is processed by RNA polymerase II (Pol II) enzymes and the pri-
miRNAs are 5prime-capped and 3prime-polyadenylated (Lee et al 2004 Xie Z et al 2005)
Mature miRNAs are produced from pri-miRNAs through at least two sequential
processing steps by RNase III-type endonucleases In animals pri-miRNAs are first
processed at the bottom portion of the stem by Drosha a nuclear-localized RNase III
enzyme to liberate pre-miRNAs The pre-miRNAs are then exported to the cytoplasm
with the help of exportin-5The pre-miRNAs are further processed by Dicer another
RNaseIII enzyme to a duplex consisting of the miRNA and the antisense strands
(miRNA) Since plants do not have any Drosha homologs the two sequential
processing steps seem to be carried out by DCL1 a Dicer homolog in the nucleus
(Kurihara amp Watanabe 2004 de Alba et al 2013 Rogers amp Chen 2013)
The processing of pri-miRNAs to pre-miRNAs by DCL1 is assisted by two
other proteins HYPONASTY LEAVES1 (HYL1) and SERRATE (SE) HYL1 belongs to a
family of double-stranded RNA (dsRNA) binding protein and interacts with DCL1 in
vitro and in vivo (Lu amp Fedoroff 2000 Hiraguri et al 2005 de Alba et al 2013
Rogers amp Chen 2013) Loss-of-function mutations in the HYL1 gene result in
decreased accumulation of miRNAs and concomitant accumulation of pri-miRNAs
(Han et al 2004 Vazquez et al 2004a Kurihara et al 2006) SE a C2H2 zinc finger
protein interacts with DCL1 and HYL1 and plays a role in the processing of pri-
miRNAs to pre-miRNAs (Lobbes et al 2006 Yang L et al 2006) In addition the
nuclear cap-binding complex proteins CBP20 and CBP80 that are known as ABA
HYPER SENSITIVE 1(ABH1) have been shown to be required for proper miRNA
processing (Gregory et al 2008 Laubinger et al 2008) Recently it has been
demonstrated that DCL1 can release 21-nt RNAs from dsRNA as well as from
synthetic miR167 precursor RNAs in vitro However this cleavage is error prone and
inefficient in the absence of HYL1 and SE which accelerate the rate of DCL1-mediated
cleavage and promote accurate processing of miRNAs (Dong et al 2008 Rogers amp
Chen 2013)
11343 Methylation of the miRNAmiRNAduplex
After the miRNAmiRNAduplexes are released from the pre-miRNAs by the DCL1
activities the duplexes are methylated at the 2primeOH of the3prime-ends by HUA ENHANCER1
(HEN1) a small RNA methyltransferase (Yu et al 2005 Yang Z et al 2006 Rogers
amp Chen 2013 ) This step which is distinct from the RNase III-mediated processing
Chapter 1 Introduction and Review of Literature
37
step distinguishes the biogenesis of plant miRNAs from that of animal miRNAs
Mutations in the HEN1 gene were first isolated in a morphological screening for floral
development although the HEN1 mutants display pleiotropic phenotypes similar to the
developmental defects observed in the DCL1 mutants (Chen et al 2002 Park et al
2002 Rogers amp Chen 2013)
Figure 18 Biogenesis of microRNA
A Biogenesis of plant MicroRNA B Biogenesis of metazoananimal MicroRNA
The miRNA (red) is incorporated into RISC and the miRNA(blue) in degraded
This enzyme has two dsRNA-binding domains and nuclear localization signal It
is also conserved in fungi and is required for the processing of siRNAs as well as of
miRNAs (Park et al 2002 Boutet et al 2003 Xie et al 2003) Although the HEN1
protein is localized to the nucleus its methylation activity in the cytoplasm cannot be
ignored (Xie et al 2004 Rogers amp Chen 2013)
Chapter 1 Introduction and Review of Literature
38
The methylation of the miRNAmiRNA duplex is required to protect miRNAs
against the 3prime-end uridylation activity and subsequent degradation (Li et al 2005) In
the hen1 mutants accumulation of miRNAs is reduced to a lower level Also the
miRNAs appear to be heterogeneous in their sizes such that a ladder of bands rather
than a single species is observed for most miRNAs in the mutants (Li et al 2005)
MiRNA cloning and sequencing analyses revealed the presence of a number of U
residues at the 3prime-end of miRNAs in the hen1 mutants Moreover these nucleotides do
not correspond to the miRNAs in the pre-miRNAs indicating that these nucleotides are
added after the processing of the miRNAs from pre-miRNAs (Li et al 2005)
Uridylation of RNAs has also been proven to be correlated with RNA decay (Shen amp
Goodman 2004 Mullen amp Marzluff 2008) suggesting that uridylation attracts an
exonuclease which degrades miRNAs in plants
11344 MiRNA incorporation in to the RISC and its nuclear export
The methylated miRNAmiRNA duplexes undergo RISC assembly In this process
the miRNA of the duplexes is selectively incorporated into the RISC and the miRNA
appears to be removed and subsequently degraded (Hammond et al 2000 Hutvagner
amp Zamore 2002 Schwarz et al 2003 de Alba et al 2013 Rogers amp Chen 2013)
The ARGONAUTE 1(AGO1) proteins are core components of the RISC complex
They contain two structural domains the N-terminal PAZ domain that binds to the 3prime-
end of single-stranded RNAs and the C-terminal piwi domains that has a structure
similar to that of RNase H (Cerutti et al 2000 Parker et al 2004) Several AGO
proteins possess endonucleolytic activities within the PIWI domain and cleave target
mRNAs in the middle of the site complementary to miRNA (Liu et al 2004 Meister et
al 2004 Miyoshi et al 2005) Mutations in the AGO1 gene cause miRNA target
genes to be ectopically expressed and the levels of some miRNAs are reduced in the
mutants (Kidner amp Martienssen 2004 Vaucheret et al 2004 Kidner amp Martienssen
2005 Ronemus et al 2006) Moreover the phenotype of the AGO1 hypomorphic
alleles display defects in lateral organ polarity and leaf and flower morphology as
observed in the dcl1 mutants and null AGO1 alleles are embryonically lethal
supporting the close relationship between the AGO proteins and miRNA processing
(Kidner amp Martienssen 2004 Vaucheret et al 2004 de Alba et al 2013 Rogers amp
Chen 2013)
The production of miRNA miRNA duplexes occur in the nucleus while
Chapter 1 Introduction and Review of Literature
39
miRNA-RISC complexes are present in the cytoplasm where target mRNAs are
located This discordance of functional sites requires an export step during miRNA
biogenesis HASTY the Arabidopsis homolog of exportin-5 is thought to be involved
in miRNA nuclear export (Bollman et al 2003 Park et al 2005)The hasty mutant
exhibits pleiotropic defects in plant development and reduced accumulation of most
miRNAs as in the DCL1 or AGO1 mutants Howeverit is unclear whether the HASTY
proteins export the miRNA miRNA duplexes or the miRNA-RISC complexes in
plants
11345 Feedback regulation of miRNA biogenesis
MiRNA biogenesis is under feedback regulation such that the DCL1 and AGO1 genes
are themselves regulated by miRNAs DCL1 is itself a target of miR162 which leads to
the cleavage of the DCL1 mRNA (Xie et al 2003 de Alba et al 2013) Consistent
with this the levels of DCL1 mRNA are elevated in the mutants of miRNA processing
genes in which the miR162 abundance is reduced In addition there is a predicted
hairpin structure of miR838 in the 14th
intron of the DCL1 primary transcript
(Rajagopalan et al 2006) This prediction implies that DCL1-mediated processing
on this site can result in the cleavage of the DCL1 primary transcript into two truncated
fragments the N-terminal capped fragment and the C-terminal polyadenylated
fragment If this is the case it is possible that miRNA biogenesis competes with the
splicing event of the DCL1 pre-mRNA
The AGO1 is also regulated by miRNA There is a complementary site for
miR168 binding in the AGO1 mRNA and the transcript level of the AGO1 mRNA is
reduced through an mRNA cleavage mechanism (Vaucheret et al 2006 de Alba et al
2013 Rogers amp Chen 2013) indicating that the AGO1 mRNA levels are tightly
controlled by the levels of the AGO1 protein
1135 Mechanism of miRNA action
11351 MiRNA-directed mRNA cleavage
By forming the miRNA-RISC assembly miRNAs can direct the RISC to regulate gene
expression by two post transcriptional mechanisms mRNA cleavage or translational
repression (Bartel 2004 Jones-Rhoades et al 2006 Dalmay 2013) The degree
of miRNA-mRNA complementarity plays an important role for the
determination of the mechanism to be used A general rule is that while perfect or
Chapter 1 Introduction and Review of Literature
40
near-perfect complementarity induces mRNA cleavage central mismatches trigger
translational repression (Carrington amp Ambros 2003 Jones-Rhoades amp Bartel 2004)
Unlike most animal miRNAs plant miRNAs have near-perfect or perfect
complementarity to their targets and mRNA cleavage is assumed to be a predominant
mechanism by which miRNAs regulate gene expression in plants
In RNA cleavage mechanism miRNAs guide the AGO component of RISC to
cleave a single phosphodiester bond of the target mRNA within the miRNA-binding
site (Llave et al 2002b Tang et al 2003) The truncated fragments are then released
and degraded freeing the RISC to recognize and cleave another target mRNA This has
been validated by the detection of 3prime cleavage products that have 5prime ends that start at the
middle of the complementary regions The mRNA cleavage products are then further
degraded by other mechanisms The 3prime cleavage products of some miRNA targets are
degraded by the 5prime-3prime exonuclease XRN4 an Arabidopsis homolog of the major Yeast
mRNA degrading exonuclease Xrn1p It has been observed that the 3prime cleavage
products of some target mRNAs were accumulated in the xrn4 mutants (Souret et al
2004 Dalmay 2013) However the 3prime cleavage products of other miRNA targets do
not accumulate in the xrn4 mutants indicating that there are also XRN4 independent
mechanisms The 5prime cleavage products are typically untraceable by RNA gel blot
hybridization but can be detected by sensitive PCR based methods It has been found
that the 5prime cleavage products tend to obtain an oligo U tail at the 3prime ends This 3prime U tail
is correlated with shortening of the 5prime cleavage products from the 5prime end leading to the
conclusion that the 3prime uridylation causes 5 prime- 3 prime exonucleolytic decay of the 5prime cleavage
products (Shen amp Goodman 2004)
DCL1 HYL1 and SE interact with each other and are co-localized in the
nuclear bodies termed Dicing bodies or D-bodies in which some pri-miRNA shave
been found suggesting that D-bodies serve as a center for active miRNA processing
(Fang amp Spector 2007 Fujioka et al 2007 Song et al 2007) The miRNA
processing site appears to be distinct from the Cajal bodies that have previously been
found as a siRNA processing site (Pontes amp Pikaard 2008) The D-bodies include
SmD3 and SmB proteins that are found in the Cajal bodies However At collin an
additional marker for the Cajal bodies is not found in the D-bodies Moreover the D-
bodies are present in an At collin mutant lacking the Cajal bodies The pri-miRNAs of
miR171A and miR159A genes have been found to be co-localized with HYL1 and
Chapter 1 Introduction and Review of Literature
41
DCL1 in the D- bodies that are distinct from the Cajal bodies (Song et al 2007)
11352 MiRNA-directed translational repression
Translation repression is another mechanism exerted by plant miRNAs The regulatory
mechanism of miR172 which regulates flowering time and floral organ identity is
consistent with translation repression MiR172 over production does not affect the
transcript levels of APETALA2 (AP2) and AP2- like target mRNAs Instead the AP2
protein level is reduced in the miR172 over expressing mutant (Aukerman amp Sakai
2003 Chen 2004 Dalmay 2013) It was later shown that miR172 can also direct the
cleavage of the target mRNAs although the steady-state mRNA level is unchanged due
to feedback regulation (Schwab et al 2005 Jung et al 2007) It is clear that miR172
regulates its target genes primarily by translational repression rather than mRNA
cleavage Recently it has been reported that miR156157 and miR854 also regulate
their target genes through translational repression (Arteaga-Vazquez et al 2006
Gandikota et al 2007 Dalmay 2013) It was once thought that translational repression
is an exception to the rule However experimental evidence suggest a widespread
coexistence of translational repression and mRNA cleavage (Brodersen et al 2008)
1136 Regulatory roles of miRNAs in plant development
The miRNA target prediction has been a difficult task in order to obtain regulatory
targets without bringing in too many false predictions Plant growth and developmental
processes regulated by miRNAs are quite diverse and enormous Furthermore plant
miRNAs work cooperatively or antagonistically to establish a balanced regulation This
notion is well consistent with the findings that mutations in the regulators of miRNA
biogenesis transport and processing such as AGO1 DCL1 HYL1 HEN1 ABH1
and HASTY cause pleiotropic phenotypes (Mallory amp Vaucheret 2006 de Alba et al
2013) To date the roles of miRNAs have been mostly deduced from obtaining miRNA
over expressing transgenic plants or gain-of-function mutants in which miRNA-
resistant target genes are ectopically expressed
11361 Phase transition
Plants pass through a series of developmental phase transitions during their life span
beginning with seed germination continuing through vegetative phase change
reproductive phase change flowering initiation and finally seed production for the next
generation Because of their importance in understanding plant developmental
Chapter 1 Introduction and Review of Literature
42
processes genes regulating developmental transitions have been extensively studied
and underlying molecular mechanisms have been explored in various plant species
Majority of researches were mainly focused on vegetative phase change and
reproductive transition in Arabidopsis and Maize (Poethig 2003 Baurle amp Dean
2006) Particularly the mutations of the genes encoding various regulators of miRNA
biogenesis and transport such as HST SE and AGO exhibit altered juvenile to adult
vegetative phase change and vegetative to reproductive change (Hunter et al 2003
Peragine et al 2004 Vazquez et al 2004a Jones-Rhoades et al 2006 de Alba et al
2013)
Over-expression of the miR156a gene causes late flowering and delays
vegetative phase change (Wu amp Poethig 2006 Gandikota et al 2007 Wang et al
2008) It has been shown that miR156 negatively regulates a group of genes encoding
SQUAMOSA PROMOTER-BINDING PROTEIN-LIKE (Lewis et al 2007 de Alba et
al 2013 Rogers amp Chen 2013) transcription factors such as SPL3 SPL4 and SPL5
that promote flowering initiation In contrast transgenic plants over expressing
miR156-resistant SPL345 genes are early flowering Interestingly the endogenous
level of miR156 is temporally regulated In the early juvenile vegetative phase the
level of miR156 is very high but decreases rapidly before the onset of the adult juvenile
phase (Wu amp Poethig 2006)
The Arabidopsis early activation tagged (eat-D) mutant exhibits early flowering
with disrupted floral structure The mutant phenotypes are caused by over expression of
the miR172a-2gene (Aukerman amp Sakai 2003) MiR172 regulates a small group of
genes encoding APETALLA2 (AP2)-like transcription factors such as TARGET OF
EAT1 (TOE1) TOE2 SCHLAFMUTZE (SMZ) and SCHNARCHZAPFEN(SNZ)
TOE1 TOE2 SMZ and SNZ have been shown to participate in their productive phase
change by repressing a floral integrator FLOWERING LOCUS T (FT)(Jung et al
2007) Consistent with this toe1-2D 35STOE2 35SSMZ and 35SSNZ plants
exhibit late flowering (Jung et al 2007) MiR172 is also regulated temporally and
highly expressed in the adult vegetative phase both in Arabidopsis and maize (Chuck et
al 2007) Because the temporal expression patterns of miR156 and miR172 are
complementary to each other it has been suggested that the two miRNAs interact with
each other in regulating phase change (Chuck et al 2009 de Alba et al 2013) In
support of this view it has been shown that miR172 is down-regulated in the
Chapter 1 Introduction and Review of Literature
43
Corngrass1(Cg1) mutant in which miR156 is overproduced (Chuck et al 2007 de
Alba et al 2013) However there is no direct evidence for the direct linkage between
miR156 and miR172 It is also envisioned that miR156 and miR172 would not be
directly related For example miR156 regulates plastochron index that is the inverse of
leaf initiation rate and thus frequently used to analyze temporal patterns of plant
development In contrast miR172 does not affect plastochron (Wang et al 2008) The
down-regulation of miR172 in the maize miR156-over producing mutants would be
due to the prolonged juvenile vegetative phase in the mutants
Another miRNA that regulates flowering initiation is miR159 It regulates
MYB33 and MYB65 which are closely related to the barley GAMY B transcription
factor that responds to gibberellic acid (GA) The level of miR159 is elevated in GA-
treated seedlings but reduced in the GA biosynthetic ga1 mutant Transgenic plants
over producing miR159 exhibit delayed floral induction under short days (Achard et
al 2004) It is been suggested that MYB33 mediates GA signal to regulate LEAFY
(LFY) These observations indicate that miR159 plays an important role in the GA
signaling pathways that regulate proper floral induction under short days (Achard et al
2004)
11362 Floral development
Arabidopsis flowers consist of four distinct organs They arise in a characteristic
pattern within concentric rings called whorls from outside to inside four sepals four
petals six stamens and the central pistil consisting of two carpels Identities of the
floral organs are determined by three classes of regulatory genes classA classB and
class C genes (Krizek amp Fletcher 2005) The A and C class genes act antagonistically
to restrict their expression domains (Bowman et al 1991)
It is notable that miR172 also regulates floral architecture in addition to its role
in the timing of flowering initiation MiR172 inhibits the translation of the AP2 mRNA
(Chen 2004) Transgenic plants overproducing miR172 not only exhibit early
flowering but also show abnormal floral development which is quite similar to those
of the class A gene mutants such as the ap2 mutants (Aukerman amp Sakai 2003 de
Alba et al 2013) When the activity of the class A genes is inactivated that of the class
C genes such as AGAMOUS (AG) is elevated As a result the miR172- overproducing
transgenic plants exhibit no petals along with homeotic transformation of the sepals to
Chapter 1 Introduction and Review of Literature
44
the carpels (Aukerman amp Sakai 2003 Chen 2004)
Interestingly miR166 also regulates floral architecture The role of miR165166
in the regulation of meristem activity is closely linked to floral meristem formation and
organization (Zhao et al 2007 Miyashima et al 2013) Floral structure is severely
disrupted in the miR165166-overproducing mutants such as men1 and jba-1D in
which miR166 is overproduced (Williams et al 2005b Jung amp Park 2007) The men1
and jba-1D mutants have smaller gynoecium and reduced carpel number In an extreme
case the jba-1D homozygous plants lack visible carpels (Williams et al 2005b)
Similar phenotypes have been observed in the miR165 overproducers (Zhao et al
2007) It has been suggested that the phenotypes are possibly caused by altered AG
expression in the floral meristem (Jung amp Park 2007 Turchi et al 2013)
Other miRNAs also affect floral development Overproduction of miRNAs
involved in auxin signaling also affects floral architecture Transgenic plants
overproducing miR167 have defects in the formation of ovule and anther with reduced
fertility (Ru et al 2006) It has been shown that miR167 targets ARF6 and ARF8 that
play a role in gynoecium and stamen development and in regulating fertility (Ru et al
2006 Wu et al 2006) These observations suggest that auxin signals are also closely
related to floral organogenesis In addition transgenic plants over-producing miR164
and the cuc1cuc2 double mutants show fused sepals and reduced petals (Laufs et al
2004 Turchi et al 2013) suggesting that miR164 is also related to floral meristem
activity and boundary specification of the meristematic region
11363 Shoot apical meristem development
Shoot apical meristem comprises self-maintaining totipotent cells The shoot apical
meristem activity affects diverse aspects of plant developmental processes including
leaf development vascular development and lateral organ polarity (Bowman et al
2002) Because miR165166 plays a primary role in meristem formation
overproduction of miR165166 and multiple knockout mutants of its target genes
exhibit pleiotropic pheonotypes (McConnell et al 2001 Emery et al 2003 Kim et al
2005 Williams et al 2005b)
The targets of miR165166 include genes encoding the homeo domain-leucine
zipper (HD-ZIP) class III transcription factors such as PHABULOSA(PHB)
PHAVOLUTA (PHV) REVOLUTA(REV) ATHB15CORONA (CNA) and ATHB8
Chapter 1 Introduction and Review of Literature
45
PHB PHV and ATHB15 have overlapping functions in controlling meristem
development (Turchi et al 2013) Indeed the phb phv athb15 triple mutants have an
enlarged shoot apical meristem Consistent with this the miR166- overproducing men1
and jba-1D mutants exhibit enlarged shoot apical meristems (Kim et al 2005
Williams et al 2005b Turchi et al 2013) In the mutants the shoot apical meristems
are multiplied and large
The development of shoot apical meristems is also regulated by the WUSCHEL
(WUS)ndashCLAVATA (CLV) signaling pathway WUS positively regulates cell
proliferation In contrast CLV3 which is expressed in stem cells limits the WUS
expression and thus inhibits cell proliferation in the SAM (Sharma et al 2003)
Consistent with this notion WUS is expressed to a higher level in jba-1D
explaining the ectopic enlargement of the SAM in the mutant (Williams et al 2005)
While WUS is closely related to miR165166 in the shoot apical meristem
development the underlying molecular mechanisms are currently unclear It has been
observed that the miR166-over producing men1 plants have an arrested meristem
development (Jung amp Park 2007) In addition the heterozygous men1+ and the
homozygous men1 plants show different expression patterns of WUS and CLV3 It
appears that miR166 regulates WUS indirectly and influences the expressional balance
between WUS and CLV3 through a negative feedback loop (Jung amp Park 2007 de
Alba et al 2013)
The phv phb athb15 triple mutant has an enlarged shoot apical meristems the
rev phb phv mutant does not have functional shoot apical meristems (Prigge et al
2005) This distinction may be explained by the fact that while miR166 targets
primarily PHB PHV and ATHB15 miR165 targets all the five HD-ZIP III genes (Zhou
et al 2007) Consistent with this transgenic plants overproducing miR166 are distinct
from those overproducing miR165 The miR165-overproducing transgenic plants lack
functional shoot apical meristem and a pin-like structure is formed at the apical region
of the mutant These phenotypes are quite similar to rev phb phv triple mutant (Zhou et
al 2007 Liu et al 2013) The phenotypic distinction may because by the difference
of the cleavage efficiencies of the target mRNAs In accordance with this notion a few
35S miR166a transgenic lines exhibit no shoot apical meristems The men1
homozygous plants also show arrested meristem development (Jung amp Park 2007)
Chapter 1 Introduction and Review of Literature
46
MiR164 cleaves the CUC1 and CUC2 mRNAs as well as the NAC1 mRNA
CUC1 and CUC2 determines the organ boundary in the meristem Phenotypes of
transgenic plants that overproduce miR164 are similar to those of the cuc1cuc2 double
mutant that exhibits embryonic developmental defects such as cup-shaped cotyledons
and fused cotyledons (Laufs et al 2004) When a miR164-resistant CUC2 is
expressed the boundary domain is enlarged CUC also plays a role in embryonic shoot
meristem formation by regulating the SHOOT MERISTEMLESS gene It was therefore
concluded that miR164-mediated cleavage of CUC1 and CUC2 mRNAs determines the
sizes of the boundary domains in the shoot apical meristem and regulates separation of
the organs by restricting cell proliferation (Laufs et al 2004 de Alba et al 2013)
11364 Leaf development
The role of miR319 has been extensively studied in leaf development A dominant
jaw-D mutant overproducing miR319 shows uneven curved leaves with enlarged size
and five TCP genes which are closely related to the CINCINNATA (CIN) gene in
Antirrhinum majus are down regulated in the mutant suggesting that miR319 mediated
regulation of the TCP transcription factor genes are important for the final step of
leaf shaping (Palatnik et al 2003 Naz et al 2013) In addition to leaf shape and size
leaf polarity is also specified by miRNA MiR166-mediated cleavage of the HD-ZIPIII
mRNAs is a well to establish leaf polarity This discovery originates from the gain-of-
function mutants such as phb-1d and phv-1d wherein point mutations in the
complementary sites to miRNA helps the mRNA to evade or resist from miRNA-
mediated cleavage (McConnell et al 2001 Emery et al 2003 Naz et al 2013)
Plant leaves have a dorso-ventral polarity consisting of the adaxial (upper
surface) and abaxial (lower surface) sides The adaxial side is closer to the shoot
apical meristem Adaxial identity is specified by functionally redundant class III HD-
ZIP family genes such as PHB PHV and REV Ectopic expression of each genes
result in adaxialized leaves Dominant phb-1d and phv-1d mutants exhibit
transformation of the abaxial to adaxial fate and have rod-shaped leaves In contrast
miR165166-overproducing plants show pin-like cotyledons radicalized leaves and
abaxialized leaves (Chitwood et al 2007 Pattanayak et al 2013)
Leaf asymmetry is determined by miR166-mediated restriction of the
expression domains of the HD-ZIPIII genes The HD-ZIPIII genes are expressed in the
Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
2
thus far only reported in two other species C heterocalyx (Coulibaly et al 2002) and
C anthonyi (Maurin et al 2007)
121 Classification
Kingdom Plantae
Subkingdom Tracheobionta
Superdivision Spermatophyta
Division Magnoliophyta
Class Magnoliopsida
Subclass Asteridae
Order Rubiales
Family Rubiaceae
Genus Coffea
122 Morphological features
The genus Coffea contains about 25 species These are small trees or shrubs that
originally grew in African tropical forests Some varieties of coffee plant typically
grow over 30 feet But in cultivation for ease of picking of the coffee berry the
coffee tree is seldom allowed over 15 feet The C arabica coffee plant is typically
smaller than the C canephora plants C canephora is shrub type and C arabica is
tree type They have extensive tap root systems with well-developed main vertical
roots and lateral roots that grow parallel to the ground Leaves have a bipolar structure
where leaf pairs are at 90 degree rotation to each other on the stem Leaves are oblong
to ovate in shape with characteristic interpetiolar stipule
Flowers are white are white produced in dense clusters at leaf axils Flowers
are pentamerous Calyx is toothed and corolla is tubular There are five stamens and a
single bifid style Sexually C arabica is autogamous whereas Ccanephora is self-
incompatible The fruits are drupes
123 Classification of coffee
Chevalier (1947) has grouped the valid coffee species into the following sections
(1) Argocoffea
(2) Paracoffea
(3) Mascarocoffea
Chapter 1 Introduction and Review of Literature
3
(4) Eucoffea
The Coffee species belonging to Mascarocoffea section have one common
characteristic the absence of caffeine The Eucoffea (now named Coffea) has been
again divided into five subsections according to diverse criteria tree height
(Nanocoffea) leaf thickness (Pachycoffea) fruit colour (Erythrocoffea Melanocoff-
ea) and geographical distribution (Mozambicoffea) (Table 11) The recent
classification has widely accepted that Coffea sect Paracoffea and Agrocoffea mainly
consist of species from other genera
Table 11The grouping of the species in the subsection Eucoffea (Chevalier 1947)
Section Subsection Species
Eucoffea
Erythrocoffea
C arabica
C canephora
C congensis
Pachycoffea
C abeokutae
C liberica
C klainii
C oyemensis
C dewerei
Melanocoffea
C stenophylla
C carissoi
C mayombensis
Nanocoffea
C humilis
C brevipes
C togoensis
Mozambicoffea
C schumanniana
C eugenioides
C kivuensis
C mufindiensis
C zanguebariae
C racemosa
C ligustroides
C salvatrix
The current sub generic classification comprises of two genera Coffea and Baracoffea
(Table 12) Most species of Coffea belong to Coffea subgenera includes those used
for producing the beverage coffee Coffea subgenera occur throughout the natural
range of the genus in Africa Madagascar and The Mascarenes Subgenera Baracoffea
contains only three accepted species (although five remain undescribed) (Table 12)
and is restricted to the dry forests of western Madagascar Leroy (Leroy 1980) placed
C rhamnifolia a species from Africa (Somalia and Kenya) in Coffea subgen
Chapter 1 Introduction and Review of Literature
4
Baracoffea but this was contested by Bridson (1988) Davis amp Rakotonasolo (2003)
and Davis et al (2005)
Table 12Outline classification of Coffea and Psilanthus with synonyms (Davis amp Rakotonasolo
2003)
Coffea L Sp Pl 172 (1753)
Coffea sub gen Coffea L Type C arabica L 95 species Africa Madagascar Mascarenes
Coffea sub gen Psilanthopsis (AChev)
Psilanthopsis AChev Type Coffea kapakata
Type Paolia jasminoides Chiov (= C rhamnifolia (Chiov) Bridson)
Coffea sub gen Baracoffea
Coffea sect Baracoffea Type Coffea humbertii J-FLeroy
Eight species (including five -yet to be published) West Madagascar
[Paracoffea subgen Insulanoparacoffea J-FLeroy J Agric Trop Bot Appl 14 276 (1967)
nom nud]
Psilanthus Hookf Gen Pl 115 (1873)
Psilanthus subgen Psilanthus Hookf Type Psilanthus mannii Hookf
Two species Africa (central and western)
Psilanthus subgen Afrocoffea (Moens)
Coffea subgenAfrocoffea
Type Psilanthus lebrunianus (Germain amp Kesler) Bridsonc 18 species Africa Asia
Australasia
Coffea sectParacoffeaMiq Fl Ind Batavae 308 (1856)
Psilanthus subgen Paracoffea Type Coffea horsfieldiana Miq
Paracoffea
[Paracoffea subgenAfroparacoffea J-FLeroy J Agric Trop Bot Appl 14 276 (1967)
nom nud]
[Paracoffea subgenMelanoparacoffea J-FLeroy J Agric Trop Bot Appl 14 276 (1967)
nom nud]
13 Economics of Coffee
Coffee is the fourth most valuable traded agricultural commodity (FAO 2004) but is
the second most valuable commodity exported by the developing countries and more
than 75 million people depend on coffee for most of their livelihood (Pendergrast
2009)
India produces both Arabica coffee from C arabica and Robusta coffee from
C canephora As per the economic survey 2012-13 Coffee is planted in India in
around 388195 hectares Out of this area 52 is Ccanephora and 48 is Carabica
Chapter 1 Introduction and Review of Literature
5
Coffee is mostly grown in south India parts of Orissa W Bengal and North east
India Karnataka is the largest producer with 569 of total area under production
which accounts for 71 of the total produce The total crop harvest for 2012-2013 is
placed at 325300 tonnes (httpwww Indicoffeeorgindiacoffeephpp-
age=CoffeeData)
14 Composition of coffee beans
Navellier (1961) gave a mean composition of green coffee as Carbohydratesglucides
(58) lignin (2) lipids (13) proteins (13) ash (4) non-volatile acids (8)
trigonelline (1) and caffeine (1) The caffeine content of green coffee is relatively
limited (10-25) of dry matter and changes little with seed development (Clifford
and Kazi 1987) In coffee seeds the concentration of trigonelline is approximately
2 of dry weight (Clifford 1985 Mazzafera 1991) Mazzafera (1999) found a higher
protein content in the mature beans than in the immature beans but a lower content of
free amino acids with asparagine as the main component Flament ( 2000) estimated
the composition of coffee beans as follows
Proteins 100 (dry weight basis of green coffee)
Carbohydrates 500 -do-
Lipids 117-140 (Arabicas)
76- 95 (Robustas)
Chlorogenic acids 65 (Arabicas)
90 (Robustas)
15 Purine alkaloids structure and role
Purine alkaloids are derived from purine nucleotides (Zulak et al 2007) and found in
over hundred plant species in 13 orders in plant kingdom (Ashihara amp Crozier 1999)
Methylxanthines like caffeine (137-trimethlyxanthine) theobromine (37-methylxan-
thine) and methyluric acids are classified into purine alkaloids (Figure 11) (Table
13)(Ashihara et al 2008) These are known to occur in tea coffee and a number of
non-alcoholic beverages Caffeine was isolated from coffee (Coffea arabica) and tea
(Camellia sinensis) in 1820 (Ashihara amp Crozier 2001)
Chapter 1 Introduction and Review of Literature
6
Figure 11 Purine alkaloid xanthine and uric acid skeleton
Plants that accumulate purine alkaloid are classified into three groups based on
the type of alkaloids they produce caffeine-producing plants which include coffee
tea and mateacute (Ilex paraguariensis) theobromine ndash producing plants are represented
by cacao cocoa tea (Camellia ptilophylla) and Camellia irrawadiensis and
methyluric acid ndash producing plants consist of Coffea dewevrei Coffea liberica C
excelsa and Kucha tea (Camellia assamica)
Table 13 Purine alkaloid structures based on xanthine and uric acid skeleton
Compound Trivial name R1 R2 R3 R4 O-2 Δ2-3
Methylxanthines
Xanthine H H H
1-methylxanthine CH3 H H
3-methyl xanthine H CH3 H
7-methyl xanthine H H CH3
1-3methylxanthine Theophylline CH3 CH3 H
17-methylxanthine Paraxanthine CH3 H CH3
37-methylxanthine Theobromine H CH3 CH3
137-methylxanthine Caffeine CH3 CH3 CH3
Uric acid and methyl uric acids
Uric acid H H H H
137-trimethyluric acid CH3 CH3 CH3 H
1379-tetramethyluric
acid Theacrine CH3 CH3 CH3 CH3
O19-trimethyluric acid Liberine CH3 H H CH3 CH3 Δ2-3
O179-trimethyluric acid Methylliberine CH3 H CH3 CH3 CH3 Δ2-3
16 Role of caffeine in plants
The physiological role of endogenous purine alkaloids and related compounds in
higher plants remains undetermined There are two hypotheses about the role of the
Chapter 1 Introduction and Review of Literature
7
high concentrations of caffeine that accumulate in tea coffee and a few other plant
species The lsquoallelopathic theory or the auto toxic function theoryrsquo proposes that
caffeine in seed coats is released into the soil and inhibits the germination of seeds
around the parent plants (Anaya et al 2006)
The lsquochemical defense theoryrsquo proposes that caffeine in young leaves fruits
and flower buds acts to protect soft tissues from insect pests (Ashihara amp Crozier
2001 Ashihara et al 2008) It has been shown that spraying tomato leaves with a 1
solution of caffeine deters feeding by tobacco horn worms while treatment of
cabbage leaves and orchids with 001ndash01 solutions of caffeine acts as a neurotoxin
and kills or repels slugs and snails (Hollingsworth et al 2002) This work has now
been extended with convincing evidence for chemical defense theory has recently
been obtained with transgenic caffeine ndash producing tobacco plants (Uefuji et al 2005
Kim et al 2006)
17 Distribution of caffeine in plants
Caffeine has been found in 13 orders of the plant kingdom (Ashihara and Suzuki
2004 Anaya et al 2006) In some species the main purine alkaloid is theobromine or
methyl uric acids such as theacrine liberine and methylliberine rather than caffeine
(Ashihara amp Crozier 1999 Ashihara amp Crozier 2001)(Table 14)
Table 14 Distribution of purine alkaloids in plants
SNo Plant species Common
name Major alkaloid Plant parts
containing
alkaloids 1 Coffea arabica Arabica Caffeine Leaves Seeds
2 Coffea canephora Robusta Caffeine Leaves Seeds
3 Coffea liberica Caffeine Theacrine Liberine
Seeds Mature leaves
4 Coffea dewevrei Caffeine Theacrine Liberine
Seeds Mature leaves
5 Camellia sinensis Tea Caffeine Leaves
6 Camellia assamica Assam tea Caffeine Leaves
7 Camellia assamica var
kucha Kucha Theacrine Caffeine Leaves
8 Camellia irrawadensis Theobromine Leaves
9 Camellia ptilophylla Cocoa tea Theobromine Leaves
10 Theobroma cacao Cocoa Theobromine Seeds
11 Paullinia cupuna Guarana Caffeine Seeds
Chapter 1 Introduction and Review of Literature
8
12 Cola nitida Caffeine Seeds
13 Citrus sp Caffeine Pollen
14 Ilex paraguariensis Mate Caffeine Leaves
Adapted from (Ashihara amp Crozier 1999 Ashihara amp Crozier 2001)
18 Caffeine biosynthesis
The major biosynthetic pathway is a four step sequence consisting of three sequential
methylation and one nucleosidase reactions (Figure 12) The xanthine skeleton of
caffeine is derived from purine nucleotides The initial step in caffeine biosynthesis is
the methylation of xanthosine by a SAM- dependent N-methyltransferase In addition
to experiments with radiolabeled precursors substrate specificities of native (Kato et
al 1999) and recombinant N-methyltransferases (Kato et al 2000 Ogawa et al
2001 Mizuno et al 2003a Mizuno et al 2003b Uefuji et al 2003) strongly
suggest that the major route to caffeine is a xanthosine rarr7-methylxanthosinrarr 7-
methylxanthine rarr theobromine rarr caffeine pathway (Figure 12) Although the
information has been obtained mainly from coffee (C arabica) and tea (Camellia
sinensis) the available evidence indicates that the pathway is essentially the same in
other purine alkaloid-forming plants such as mateacute (Ashihara 1993) and cacao
(Koyama et al 2003 Yoneyama et al 2006)
The first step in the caffeine biosynthetic pathway from xanthosine is
conversion of xanthosine to 7-methylxanthosine (Figure 12) This reaction is
catalyzed by the 7-methylxanthosine synthase (xanthosine 7 N-methyltransferase EC
211158) The genes encoding for 7-methylxanthosine synthase CmXRS1 (AB
034699) and CaXMT1 (AB 048793) were isolated from the C arabica (Mizuno et
al 2003a Uefuji et al 2003) The recombinant proteins obtained from these genes
exhibit 7-methylxanthosine synthase activity in vitro Xanthosine monophosphate
(XMP) was not converted to 7-methylxanthosine by the recombinant enzymes thus
inclusion of 7-methy-XMP was proposed (Schulthess et al 1996)
The second step of caffeine biosynthesis involves a nucleosidase which
catalyses the hydrolysis of 7-methylxanthosine Although N-methylnucleosidase (EC
32225) was partially purified from tea leaves (Negishi et al 1988) recent detailed
structural studies on coffee 7-methylxanthosine synthase suggested that the methyl
transfer and nucleoside cleavage may be coupled and catalyzed by a single enzyme
(McCarthy amp McCarthy 2007)
Chapter 1 Introduction and Review of Literature
9
The last two steps of the caffeine synthesis are also catalyzed by a SAM-
dependent N-methyltransferase(s) which is different from the N-methyltransferase
enzyme that catalyzes the first step in the pathway Native N-methyltransferase
activities have been detected in crude and partially purified extracts from tea and
coffee plants (Ashihara amp Suzuki 2004) and a highly purified preparation has been
obtained from young tea leaves (Kato et al 1999) The enzyme was assigned the
name caffeine synthase (EC 211160) that catalyses the last two steps of caffeine
biosynthesis viz the conversion of 7-methylxanthine to caffeine via theobromine
(Figure 12) The gene encoding caffeine synthase was first cloned from young tea
leaves (Kato et al 2000) Since then several genes encoding N-methyltransferases
with different substrate specificity have been reported
Activity of the recombinant theobromine synthase (CTS1 and CaMXMT EC
211159) is specific for the conversion of 7-methylxanthine to theobromine In
contrast the recombinant caffeine synthases (CCS1 and CaDMXMT1) can utilize
paraxanthine theobromine and 7-methylxanthine as shown with tea caffeine synthase
(TCS1) (Mohanpuria et al 2011) Although paraxanthine is the most active substrate
of this recombinant enzyme only limited amounts of paraxanthine are synthesized in
coffee cells hence in vivo caffeine synthase is principally involved in the conversion
of 7-methylxanthine to caffeine via theobromine
Radiolabelled tracer experiments with theobromine accumulating cacao and
Chinese tea (Camellia ptilophylla) showed a limited conversion of theobromine to
caffeine N-methyltransferase which catalyzed the conversion of 7-methylxanthine to
theobromine was detected in crude extracts from C ptilophylla but conversion of
theobromine to caffeine was not observed (Ashihara et al 1998) Genes homologous
to caffeine synthase have been isolated from several theobromine accumulating plants
including cacao (Yoneyama et al 2006) The recombinant enzymes derived from
these genes have 3 N-methyltransferase activity suggesting that these theobromine
accumulating plants contain specific theobromine synthase Caffeine does not occur
in C ptilophylla (Ashihara et al 1998) although it is present in leaves and fruits of
cacao along with theobromine (Koyama et al 2003 Zheng amp Ashihara 2004)
Chapter 1 Introduction and Review of Literature
10
Figure 12 Pathways for the biosynthesis of caffeine
The major pathway is indicated by solid lines (Mizuno et al 2003b Uefuji et al 2003) Alternative pathways in coffee and tea plants are indicated
by dashed lines(Kato et al 1996 Schulthess et al 1996) Enzymes (1) 7-methylxanthosine synthase (2) N-methylnucleosidase (3) Theobromine
synthase (4) Caffeine synthase
N
N
N
N7
H3C
3
1
O
O
Caffeine
N
NH
N
N7
3
O
O
Theobromine
N
N
N
N
H
H
7
O
O
N
N
N
N
H
H
O
O
N
N
N
N
H
H
O
O
Xanthosine
N
N
N
N
H
H
O
O
N
N
N
N
H
H
O
ON
N
N
N
H
H3C
71
O
O
Paraxanthine
CH3
CH3
CH3
CH3CH3
7-methylxanthine
Rib P
Rib
(1)
(4)
Rib
+
CH3
7-methyl
xanthosine
(2)
(3)
SAM SAH
SAM SAH SAM SAH
Xanthosine mono
phosphate
Rib P
7-methyl xanthosine
monophosphate
+
CH3 CH3
Inosine 5-mono
phosphate
Adenosine 5-mono
phosphate
SAM SAH
Guanosine
Guanosine 5-mono
phosphate
Chapter 1 Introduction and Review of Literature
11
181 Source of Xanthosine for caffeine biosynthesis
Xanthosine the initial substrate of purine alkaloid synthesis is supplied by at least four
different pathways de novo purine biosynthesis (de novo route) the degradation
pathways of adenine nucleotides (AMP route) and guanine nucleotides (GMP route)
and the S-adenosyl-L-methionine (SAM) cycle (SAM route) (Figure 13) Purine
nucleotides are synthesized by de novo and salvage pathways (Ashihara amp Crozier
1999 Stasolla et al 2003 Zrenner et al 2006) The de novo synthesis of non purine
precursors like CO2 10-formyltetrahydrofolate 5-phosphoribosyl-1-pyrophosphate and
the amino acids Glycine glutamine and aspartate results in the production of purine
nucleotides
Figure 13 Xanthosine biosynthesis Xanthosine for purine alkaloid biosynthesis is supplied by at
least four routes from adenosine released from the SAM cycle (SAM route) from IMP
originating from de novo purine synthesis (de novo route) from the cellular adenine
nucleotide pool (AMP route) and from the guanine nucleotide pool (GMP route)
Adapted from (Ashihara et al 2008)
The utilization of IMP for caffeine biosynthetic pathway which is formed by the
de novo purine biosynthetic pathway for caffeine biosynthetic pathways was
demonstrated in young tea leaves using 15
N-glycine and 14
C-labelled precursors and
inhibitors of de novo purine biosynthesis (Ito amp Ashihara 1999) Xanthosine is formed
by an IMP rarr XMP rarr xanthosine pathway IMP dehydrogenase (EC 111205) and 5prime-
nucleotidase (EC 3135) catalyze these reactions The inhibition of IMP dehydrogen-
ase by ribavirin inhibits both caffeine and gunanine nucleotide biosynthesis in tea and
coffee leaves (Keya et al 2003)
Chapter 1 Introduction and Review of Literature
12
The portion of xanthosine used for caffeine biosynthesis is derived from adenine
and guanine nucleotide pools which are produce de novo by salvage pathways there are
several potential pathways for xanthosine synthesis from AMP but the AMP rarr IMP
rarr XMPrarr xanthosine route is predominant All three enzymes involved in the
conversion AMP deaminase (EC 3546) IMP dehydrogenase and 5prime-nucleosidase
have been detected in tea leaves (Koshiishi et al 2001) Xanthosine for caffeine
biosynthesis can also be produced from guanine nucleotides by GMPrarrguanosine
rarrxanthosine pathway Guanosine deaminase (EC 35415) activity is found in cell free
extracts from young tea leaves (Negishi et al 1994) but GMP deaminase activity was
not detected in plants (Stasolla et al 2003 Zrenner et al 2006) The absence of GMP
deaminase activity suggests that a GMP rarr IMP rarr XMP rarr xanthosine route is not
functional in plants
The use of radiolabelled purine bases and nucleosides for the study of caffeine
biosynthetic pathway was facilitated by the fact that exogenous purine nucleotides are
rapidly hydrolyzed by the plant tissues forming purine nucleosides andor bases which
are then utilized in pathways which in most instances indirectly lead to xanthosine
Most of these purine compounds including adenine adenosine inosine hypoxanthine
and guanine are converted to their respective nucleotides by salvage enzymes
including adenine phosphoribosyl transferase (EC 2427) hypoxanthineguanine
phosphoribosyl transferase (EC 2428) adenosine kinase (EC 27120) and
inosineguanosine kinase (EC 2421) which usually have high activities in plant cells
The resultant nucleotides then enter the purine alkaloid biosynthetic pathway In the
case of guanosine it may be directly deaminated to xanthosine and utilized for caffeine
biosynthesis (Figure 13) (Ashihara et al 1997)
182 SAM cycle in plants
SAM is the methyl donor for the three methylation steps in the caffeine biosynthetic
pathway In this process SAM is converted to S-adenosyl-L-homocysteine (SAH)
which in turn is hydrolyzed to homocysteine and adenosine Homocysteine is recycled
via the SAM cycle to replenish SAM levels and adenosine is released from the cycle
Adenosine released from the cycle is converted to AMP directly andor indirectly via
adenine AMP is converted to xanthosine and used for caffeine biosynthesis Since
three moles of SAH are produced via the SAM cycle for each mole of caffeine that is
synthesized in theory this pathway has the capacity to be the sole source of both the
Chapter 1 Introduction and Review of Literature
13
purine skeleton and the methyl groups required for caffeine biosynthesis in young tea
leaves (Koshiishi et al 2001)
Methionine
S-Adenosyl-L-methionineHomocysteine
ATP
PPi+ Pi
Tetrahydrofolate
5-Methyl-
tetrahydrofolate
CH3+Adenosine
S-Adenosyl-L-
homocysteine
(xanthine
structure)(Methyl groups)
Caffeine
(1)
(2)(3)
(4)
Figure 14 The SAM cycle (activated methyl cycle) in plants (Ashihara amp Suzuki 2004)
Enzymes SAM synthetase SAM-dependent N-methyltransferases S-adenosyl-
homocysteine (SAH) hydrolase Methionine synthase Adenosine released from the cycle
is salvaged to adenine nucleotides and utilized both for purine structure of caffeine via
xanthosine and for re-synthesis of SAM via ATP
183 N-methyltransferase in caffeine biosynthesis Molecular cloning
The genes encoding for the enzymes involved in the caffeine biosynthesis were
successfully cloned from the coffee family (Ogawa et al 2001 Mizuno et al 2003a
Mizuno et al 2003b Uefuji et al 2003) The conserved amino acid regions of tea CS
(AB31280) the sequences of O-methyltransferases derived from plant origin and
Arabidopsis hypothetical proteins were made use of in designing primers for RT-PCR
and RACE technique Independent groups have isolated N-methyltransferase (NMT)
genes from coffee which have been classified based on the substrate specificity of the
recombinant proteins expressed in Ecoli (Table 15) PCR products thus obtained were
used as the probe to isolate full length cDNA from the library resulting in the
identification of all three N-methyltransferases involved in caffeine biosynthesis
(Figure 12) Several cDNA have been characterized from the coffee plants one for
xanthosine methyltransferase (XMTXRS) three for 7-methylxanthine methyltransferase
or theobromine synthase (MXMTCTS) and three for 37-dimethylxanthine
methyltransferase or caffeine synthase (DXMTCCS) (Table 15) (Kato amp Mizuno
2004)
A single gene encoding for xanthosine methyltransferase (XMTCmXRS1) was
identified which encoded for a polypeptide consisting of 372 amino acids with an
apparent molecular mass of 418kDa It is expressed almost uniformly in aerial tissues
Chapter 1 Introduction and Review of Literature
14
of C arabica including leaves floral buds and immature but not in mature beans
Three genes encoding 7-methylxanthine methyltransferase (theobromine synthase) have
been identified The number of amino acids in the putative polypeptides are 378 for
MXMT1 (427kDa) and 384 for MXMT2 and CTS2 (434kDa) They differ by insertion
or deletion of blocks of several residues in the C-terminal region Their catalytic
properties based on kinetic parameters such as Km values were different They are
expressed in young leaves floral buds and immature but not mature beans Three genes
were identified for 3 7-dimethylxanthine methyltransferase (caffeine synthase) DXMT
CCS1 and CtCS7 each encoding a 43 kDa polypeptide consisting of 384 amino acids
However their kinetic properties differ from each other such as DXMT and CCS1
showed Km values of 1200 and 157microM for theobromine respectively Expressions
profiles are also distinct DXMT being expressed exclusively in immature beans while
CCS1 expression is ubiquitous occurring in all tissues The presence of isoforms of
these enzymes with different properties suggests that caffeine is synthesized through
multiple pathways depending on availability and concentration of the substrate (Mizuno
et al 2001 Ogawa et al 2001 Mizuno et al 2003a Mizuno et al 2003b Uefuji et
al 2003) Genomic clones of theobromine synthase were obtained by PCR- RFLP
from C canephora (Satyanarayana et al 2005 Satyanarayana 2006) The genomic
clones are reported to have highly conserved exons and a variable third intron The
promoter for the theobromine synthase was cloned and characterized (Satyanarayana et
al 2005)
There is high degree of similarity in the coding region of coffee NMT genes
isolated so far though differences exist in the 3prime untranslated region (UTR) for these
genes The deduced amino acid sequence of these enzymes shows more than 85
homology between these genes Phylogenetic analysis indicates that they are more
closely related to C-methyltransferases including those for jasmonic acid salicylic acid
and benzoic acid than to other N-methyltransferases This suggests that coffee N-methyl
transferases belong to a distinct sub-group within plant methyltransferases Their
cellular localization determined by green fluorescent protein fusion method and β-
glucourodinase (GUS) showed that the all the three genes are localized in the cytoplasm
(Ogawa et al 2001 Vinod et al 2007) It was also shown that these were present
along with chlorogenic acid(Vinod et al 2007)
Chapter 1 Introduction and Review of Literature
15
Table 15 N-methyltransferases and their encoding genes involved in caffeine biosynthesis
Gene (Enzyme) Gene name Accession No Description Reference
Xanthosine methyltransferase
( 7-methylxanthosine synthase) (EC 211158)
CaXMT AB 048793 Immature fruit cDNA Screening (Uefuji et al 2003)
CmXRS1 AB 034699 Leaf cDNA library RACE (Mizuno et al 2003a)
CaMXMT1 AB 048794 Leaf cDNA library RACE (Ogawa et al 2001)
7-methylxanthine synthase transferase
(theobromine synthase) ( EC 211159)
CTS1 AB 034700 Immature fruit cDNA Screening (Uefuji et al 2003)
CaMXMT2 AB 984125 Leaf cDNA library Screening (Mizuno et al 2001)
CTS2 AB 054841 Leaf cDNA library Screening (Mizuno et al 2001)
37-methylxanthinemethyltransferase
(Caffeine synthase) (EC 211160)
CaDXMT1 AB 086414 Immature fruit cDNA Screening (Uefuji et al 2003)
CCS1 AB 0861414 Immature fruit cDNA Screening (Uefuji et al 2003)
CtCS7 AB 0864415 Immature fruit cDNA
NMT genomic clones
Theobromine synthase-1
CX10 Complete coding region PCR Satyanarayan 2006
CX8 Complete coding region PCR -do-
Theobromine synthase-1 PG-5 Promoter + coding region RAGE -do-
Theobromine synthase-1 PG-1 Promoter + coding region RAGE Satyanarayan 2006
PG-4 Promoter + coding region RAGE -do-
Theobromine synthase-1 NMT
(AY273814) Complete coding region PCR Kochko A et al 2010
Theobromine synthase-2 NMT
(AY362825) Complete coding region PCR Kochko A et al 2010
Chapter 1 Introduction and Review of Literature
16
19 Catabolism of caffeine
Caffeine is produced in young leaves and immature fruits and continues to accumulate
gradually during the maturation of these organs At the same time caffeine is slowly
degraded with the removal of the three methyl groups resulting in the formation of
xanthine Catabolism of caffeine was first reported in coffee leaves (Kalberer 1965)
Since then a number of tracer experiments using 14
C labelled purine alkaloids have
been reported (Suzuki amp Waller 1984 Ashihara et al 1996 Vitoria amp Mazzafera
1999 Mazzafera 2004) They demonstrated the major catabolic pathway is caffeine rarr
theophylline rarr 3-methylxanthine rarr xanthine Xanthine is further degraded by the
removal by the conventional purine catabolic pathway to CO2 and NH3 via uric acid
allatonin and allantoate (Figure 15) (Ashihara amp Crozier 1999 Stasolla et al 2003
Zrenner et al 2006) Caffeine catabolism usually begins with its conversion to
theophylline catalyzed by N7-demethylase The involvement of P450-dependent mono-
oxygenase activity for this reaction was suggested (Huber amp Baumann 1998
Mazzafera 2004) However the activity of this enzyme has not yet been determined in
cell free extracts [8-14
C] Theophylline degraded to CO2 at much faster rate than [8-14
C]
caffeine indicating that conversion of caffeine to theophylline is a major rate limiting
step of caffeine catabolism and a possible reason why caffeine accumulates in high
concentrations in tissues of C sinensis and C arabica It is widely believed that the
control of caffeine levels in plants is a function of the balance between rates of
synthesis and degradation and this balance seems to vary depending on the plant
species and the tissue developmental stage (Mazzafera 2004)
Chapter 1 Introduction and Review of Literature
17
Figure 15 Caffeine catabolic pathways Caffeine is mainly catabolised via theophylline and 3-
methylxanthine Xanthine is further degraded to CO2 and NH3 by the conventional
oxidative pathway Adapted from (Ashihara et al 2008)
Chapter 1 Introduction and Review of Literature
18
110 Genetic engineering of Coffee
Genetic transformation has potential applications in coffee agriculture for incorporating
genes which could provide desirable traits such as disease and insect resistance
drought and frost tolerance and herbicide resistance Transgenic technology can also be
used to increase nutritional value and improve cup quality produce varieties with
caffeine-free beans and for production of hybrid crops for molecular farming
Identification of target specific genes is one of the pre-requisites for developing
transgenic crops The availability of a large number of EST sequences in coffee and
initiation of coffee genome sequencing may speed up the gene discovery and accelerate
transgenic research efforts in coffee
1101 Insect Resistance
Production of insect resistant coffee plants is one of the major objectives of the
breeding programs The major pests attacking coffee include coffee berry borer (CBB
Hypothenemus hampei) white stem borer (WSB Xylotrechus quadripes) leaf miner
(Perileucoptera coffeela) and root nematodes (Meloidogyne spp and Pratylenchus
spp) The CBB is present in almost all the coffee growing countries and considered to
the most devastating pest in coffee To date there is no reported source of resistance to
CBB in the coffee gene pool Like CBB WSB is another serious pest in C arabica in
India and several other East Asian countries Both CBB and WSB belong to the order
Coleoptera For India controlling WSB is the biggest challenge and has therefore
become the highest research priority Robusta is generally resistant to WSB but the
interspecific Robusta Arabica hybrids are susceptible to WSB Although leaf miner is
not yet a serious pest in India and other East Asian countries it is an economically
important pest in East Africa and Brazil Effective chemical control of CBB and WSB
is difficult due to the nature of their life cycle inside the berry and stem respectively as
well as environmental concern regarding the use of pesticides Biological control
measures are adapted to combat these insect pests with varying degrees of success
Developing coffee plants resistant to these pests using genetic transformation
technology could be one of the alternative strategies to counter pest damage For insect
resistance several different classes of proteins from bacterial plant and animal sources
have been isolated and their insecticidal properties tested against many important pests
Amongst these proteins Bt toxins are most important and several transgenic crops
expressing Bt genes have been commercialized Coffee transgenic plants carrying a
Chapter 1 Introduction and Review of Literature
19
synthetic version of the cry1Ac gene have been produced (Leroy et al 2000 Mishra et
al 2002) Indeed this was the first report of an important agronomic trait being
introduced into a coffee plant The transgenic plants presented similar features in
growth and development compared to normal plants Transgenic plants highly resistant
to leaf miner under greenhouse conditions were tested under field conditions in French
Guyana for 4 years for pest resistance (Perthius et al 2005 Perthius et al 2006)
From 54 independent transformation events 70 of the events were resistant to leaf
miner Unfortunately the field trial was vandalized which led to the termination of the
experiment (Montagnon et al 2005)The effectiveness of Bt genes in controlling
coleopteran pests is well documented in corn and potato which indicates that Bt genes
might be effective against CBB The high toxicity of B thuringiensis sero var
israelensis against first in star larvae of CBB has been demonstrated (Mendez et al
2003) In another experiment an α-amylase inhibitor from Phaseolus vulgaris was
tested against CBB and found to have an inhibitory effect on its growth and
development (Grossi et al 2004)In addition to the CBB and WSB C arabica
varieties are also susceptible to endoparasitic root-knot nematode (Meloidogyne spp)
(Campos et al 1990 Mishra et al 2012)
So far 15 species have been reported to be parasites of coffee Controlling
nematodes is extremely difficult and currently seedling grafting with robusta rootstock
is followed as one of the control measure Sources of resistance specific to root-knot
nematodes have been identified in coffee trees (Bertrand et al 2001) and the Mex-1
gene conferring resistance to M exigua in C arabica is in the process of isolation (Noir
et al 2003)
1102 Tolerance to Abiotic Stress
In many coffee growing countries abiotic stress such as drought and frost are the major
climatic factors that limit coffee production Changes in climatic patterns due to global
climate change are considered increasingly important for coffee cultivation Drought
induces water stress in plants which affects vegetative growth and vigor and triggers
floral abnormalities and poor fruit set It also indirectly increases the incidence of pests
and diseases in the plants C arabica is generally more tolerant to water stress than C
canephora partly due to its extensive deep root system C racemosa is known to be a
good source of drought tolerance In India hybrids between C canephora and C
racemosa have been obtained and are currently under evaluation for drought tolerance
Chapter 1 Introduction and Review of Literature
20
In many coffee growing countries coffee is propagated in marginal areas where the
annual rainfall is below 1000thinspmm with prolonged dry spells of over 4-5 months In
those areas water shortage and unfavorable temperatures constitute major constraints
and the growth and productivity of C canephora are badly affected As with drought
periodic frost also affects coffee production in parts of Brazil The introduction of
drought and frost tolerant genes through genetic transformation would be of great
importance for alleviating these problems Research is now being carried out by several
groups to identify genes involved in biotic as well as abiotic stress The most promising
genetic engineering approach for drought tolerance includes the use of functional or
regulatory genes as well as the transfer of transcription factors In recent years plants
tolerant to high temperature and water stress have been the subject of intense research
(Chaves et al 2003 Chaves et al 2004 Coraggio et al 2004) For achieving drought
tolerance genes that have been targeted include those encoding enzymes involved in
detoxification or osmotic response metabolism enzymes active in signaling proteins
involved in the transport of metabolites and regulating the plant energy status (Chaves
et al 2003 Chaves et al 2004 Coraggio et al 2004) The dissection of molecular
mechanisms related to signal transduction and transcriptional regulation might help in
engineering drought tolerance in coffee
1103 Disease Resistance
Coffee leaf rust (CLR) caused by the fungus Hemileia vastatrix is the most important
disease in coffee with substantial loss to coffee production and productivity in all the
coffee growing countries In addition to leaf rust coffee berry disease (CBD) caused by
fungus Colletotrichum kahawae can be a devastating anthracnose leading to substantial
crop loss in Africa Several other fungal and bacterial diseases may also affect coffee
to a extent C arabica is more susceptible to many diseases than C canephora Though
most of the disease control measures rely upon chemical control they are more
expensive and labour intensive The long-term solution is the breeding of resistant
varieties which is the focus of many breeding programs However breeding for disease
resistant varieties is time consuming due to the perennial nature of coffee with its long
gestation period India has a long history of C arabica breeding especially for leaf rust
resistance and is the first country to demonstrate the existence of multiple races of leaf
rust Resistance to CLR is conditioned primarily by a number of major (SH) genes and
coffee genotypes are classified into different resistance groups based on their
Chapter 1 Introduction and Review of Literature
21
interaction with different rust races pathogen (van der Vossen 2001) Currently C
canephora provides the main source of resistance to pests and diseases including CLR
(H vastatrix) and CBD (C kahawae) and therefore used in breeding programs Other
diploid species like C liberica and C racemosa are being used as a source of resistance
to coffee leaf rust and coffee leaf miner respectively (Guerreiro et al 1999
Sreenivasan et al 1989) The development of coffee varieties resistant to major fungal
diseases such as CLR and CBD using transgenic technology will benefit the coffee
industry immensely During the last 15 years significant progress has been made in the
area of host-pathogen interactions (Staskawicz et al 1995 Feys et al 2000
Shivaprasad et al 2012) and many resistance genes involved in recognizing invading
pathogens have been identified and cloned (Takken et al 2000) A number of
signalling pathways induced because of pathogen infection have been dissected (Dong
et al 1998 Navarro et al 2006) Many antifungal compounds synthesized by plants to
combat fungal infection have been identified (Does et al 1998) Understanding the
specific induction of targeted pathways and identification of specific pathways
responsible for particular fungal resistance is important in order to employ this strategy
in transgenic technology The recent investigation of gene expression during coffee leaf
rust infection could provide an insight into the defence pathways operating in coffee
(Fernandez et al 2004 Nardi et al 2006) Efforts have been made to identify and
clone resistance genes from coffee for achieving durable resistance The genetic and
physical map of two resistance genes SH3gene conferring resistance to rust (Prakash et
al 2004) and the Ck1 gene conferring resistance to C kahawae CBD (Gichuru et al
2006) have been established These genes could be used for molecular marker assisted
breeding programs (Mishra et al 2012)
1104 Production of low-caffeine coffee
A widespread belief that the ingestion of caffeine can have adverse effects on
health resulted in the increased demand for decaffeinated coffee (Ashihara amp Crozier
2001) Several method were used to obtain decaffeinated plants by conventional
breeding techniques using low yielding varieties and mutants that accumulated
relatively low amounts of caffeine when compared to the commercially available ones
Low-caffeine and decaffeinated coffee represent around 10 of the coffee sales
around the world (Ribas et al 2006b) The industrial process for coffee decaffeination
can be expensive and affects the original flavour and aroma in coffee (David 2002)
Chapter 1 Introduction and Review of Literature
22
Transgenic coffee plants with suppressed caffeine synthesis using RNA interference
(RNAi) technology have been obtained (Ogita et al 2003 Ogita et al 2004) Specific
sequences in the 3prime untranslated region of the theobromine synthase gene (CaMXMT1)
were selected for construction of RNAi short and long fragments The caffeine and
theobromine content of the transgenic plants were reduced by ~ 70 when compared to
the untransformed plants In C canephora RNAi technology has also been employed
to silence the N-methyl transferase gene involved in caffeine biosynthesis (Vinod et al
2007) Promoter of an N-methyltransferase (NMT) gene involved in caffeine
biosynthesis was cloned (Satyanarayana et al 2005 Satyanarayana 2006) which will
be very useful for studying the regulation of caffeine biosynthesis
1105 Improvement in cup quality
Improvement in coffee cup quality requires elaborate knowledge of the chemical
constituents as well as the metabolic pathways involved in the elaboration of quality
The constituents of coffee beans include minerals proteins carbohydrates caffeine
chlorogenic acids (CGA) glycosides lipids and many volatile compounds that give
flavour to coffee by roasting Among these the role of three major constituents
sucrose CGA and trigonelline have been studied in coffee The sucrose content of
coffee bean is associated with coffee flavour the higher the sucrose content in green
beans the more intense will be the cup flavour (Clifford et al 1985 de Maria et al
1996) The sucrose content of C arabica (82-83) is higher than C canephora (33ndash
40) The sucrose amino acids ratio in green beans determines the profile of volatile
compounds Manipulating sucrose content in coffee bean is therefore important in
improving cup quality The sucrose synthase gene (CcSUS2) from C canephora has
been cloned and sequenced (Leroy et al 2005) This provides an opportunity to
manipulate the sucrose content in coffee Chlorogenic acids are products of
phenylpropanoid metabolism Chlorogenic acids are a group of hydroxycinnamoyl
quinic acids (HQA) formed by esterification between caffeic acids coumaric acids and
quinic acids (Clifford et al 1999) They are present in relatively large quantities in the
coffee bean and are the precursor of phenolic compounds in roasted beans C
canephora beans contain higher CGA (10) compared to C arabica beans (6-7)
CGA are known to have antioxidant properties as well as being associated with disease
resistance (Campa et al 2003) Genetic manipulations of genes involved in CGA
synthesis can serve either of these purpose by up- or down regulating the pathway
Chapter 1 Introduction and Review of Literature
23
Phenylalanine ammonia-lyase (PAL) catalyzes the first step of the phenylpropanoid
pathway leading to the synthesis of a wide range of chemical compounds including
flavonoids coumarins hydroxycinnamoyl esters and lignins (Hahlbrock amp Scheel
1989) Recently the full length cDNA and corresponding genomic sequences of PAL
from C canephora was isolated characterized and functionally validated (Mahesh et
al 2006 Parvatam et al 2006) This has opened up new possibilities for manipulating
the level of the PAL enzyme in coffee which in turn can be useful for improvement of
cup quality and manipulating antioxidant properties in coffee
1106 Fruit Ripening
Uniformity during fruit ripening is decisively related to cup quality in coffee and
consequently to the value of the product Fruits at the ideal ripening stage produce the
best organoleptic characteristics for coffee The presence of over ripened or green fruits
changes the acidity the bitterness and consequently the cup quality In order to
maximize uniform ripening of coffee fruits it is essential to control the action of genes
involved in the last step of maturation process Ethylene is known to trigger ripening
and increasing ethylene biosynthesis is associated with various stages of ripening
process (Protasio et al 2005) To control coffee fruit maturation two of the major
genes involved in ethylene biosynthesis namely ACC synthase and ACC oxidase
have been cloned (Protasio et al 2005 Neupane et al 1999) Introduction of the ACC
oxidase gene in antisense orientation have been achieved in both C arabica and C
canephora The effect of the transgene on ethylene production and fruit maturation has
yet to be reported The inhibition of genes downstream to the initial ethylene burst is
also an option to control coffee fruit maturation (Ribas et al 2006)
111 RNA interferenceRNAi
The regulation of gene expression at post transcriptional level has recently attracted
much attention because of the discovery of the phenomenon called RNA interference
(RNAi) RNAi based silencing techniques are more efficient than antisense mediated
gene silencing (Wesley et al 2001) The effect was first observed in plants wherein
silencing was observed when some copies of a transgene was in the forward orientation
with respect to the promoter and some in the reverse orientation This resulted in the
formation of double stranded RNA in the system The phenomenon in which
experimentally introduced double stranded RNA (dsRNA) leads to the loss of
expression of the corresponding cellular gene by sequence specific RNA degradation
Chapter 1 Introduction and Review of Literature
24
was termed as RNA interference or RNAi The protective effect of RNA silencing in
reducing virus infectivity supports the view that PTGS has evolved as a mechanism to
defend plants against virus infection and also to moderate the possible deleterious
genome-restructuring (insertional) activity of virus-like mobile genetic elements eg
retrotransposons A growing body of biochemical and genetic data has further
demonstrated that plants animals and yeasts share related mechanisms of specific
degradation of RNAs in which double-stranded forms of RNA are initiator molecules
(Brenstein et al 2001)
The key insight into the process of PGTS was provided by the experiments
conducted by Hamilton amp Baulcombe (1999) who identified the product of RNA
degradation as small RNA (siRNA) species of ~25nt of both sense and antisense
polarity SiRNA are formed and accumulate as double stranded RNA molecules of
defined chemical structures Both genetic and biochemical approaches were undertaken
to understand the basis of silencing Genetic screens were carried out in fungus
Neurospora carassa the alga Chalmydomonas reinhardtii and plant Arabidopsis
thaliana to search for detective mutants in PTGS
A combination of results obtained from several in vivo and in vitro experiments
have gelled into a two step mechanistic model for RNAiPTGS The first step referred
to as the RNAi initiating step involves binding of the RNA nucleases to a larges
dsRNA and its cleavage into discrete ~21-25nt RNA fragments This is ATP-
dependent reaction which involved RNase III - type endonucleases which make
staggered cuts in both strands of the dsRNA leaving 3prime overhang of 2 nucleotides with
5prime- phosphate 3prime-hydroxyl and no modification in the sugar phosphate backbone
(Hamilton amp Baulcombe 1999 Elbashir et al 2001) SiRNAs are the direct products
of dsRNA cleavage by the multi-domain RNase III enzyme DICER (Figure 16)
(Brenstein et al 2001 Karthikeyan et l 2013)
In the second step or commonly known as the effector step the double stranded
siRNAs produced in the first step bind to an RNAi specific protein complex to form a
RISC The complex undergoes activation in the presence of ATP so that the antisense
component of the unwound siRNA becomes exposed and allows the RISC to perform
the downstream RNAi reaction
Chapter 1 Introduction and Review of Literature
25
Several enzymes are involved in RNAi based on their role genes that encode for them
are classified into RNAi initiators RNAi effectors RNA dependent RNA polymerases
and silencing genes Two C elegans genes rdeI and rde4 (rde stands for lsquo RNAi
deficientrsquo) are believed to be involved in the initiation step of RNAi The C elegans
rde1 gene is a member of a large family of genes and is homologous to the Neurospora
qde2 (qde stands for ldquoquelling deficientrdquo) and Arabidopsis AGO1 (AGO stands for
agronaute AGO1 was previously identified to be involved in Arabidopsis
development) Although the functions of these genes are not clear a mammalian
member of RDE1 family has been identified as a translation initiation factor (Thakur
2005) Important genes for the effector step of PTGS in C elegans are rde2 and mut7
genes These genes were identified from heterozygous mutant worms that were unable
to transmit RNAi to their homozygous offsprings Worms with mutated rde2 or mut 7
genes show defective RNAi mut-7 gene encodes a protein with homology to the
nuclease domains of RNAase D and a protein implicated in Werner syndrome (a rapid
ageing disease in humans) (Grishok et al 2001 Kamath-Loeb et al 2012s)
Neurospora qde-1 Arabidopsis SDE-1SGS-2 and C elegans ego-1 appear to encode
RNA dependent RNA polymerase (RdRPs) It assumed that this is a proof that an RdRp
activity is required for RNAi Certainly the existence of an RdRp might explain the
remarkable efficiency of dsRNA induced silencing if it amplifies either the dsRNA
prior to cleavage or the siRNAs directly (Muers 2013) In C elegans ego-1 mutants
(ego stands for lsquoenhancer of glp-1) RNAi functions normally in somatic cells but is
defective in germline cells where ego-1 is primarily expressed In Arabidopsis SDE-
1SGS-2 mutants (SGS stands for suppressor of gene silencing) siRNA are produced
when dsRNA is introduced via an endogenously replicating RNA virus but not
introduced by a transgene It has been proposed that perhaps the viral RdRP is
substituting for the Arabidopsis enzyme in these mutants Random degradative PCR
model suggests that an RdRP uses the guide strand of an siRNA as a primer for the
target mRNA generating a dsRNA substrate for Dicer and thus more siRNAs
Several genes controlling RNA silencing in plants have been identified through
genetic screens of Arabidopsis mutants impaired in transgene induced RNA silencing
They encode a putative RNA-dependent RNA polymerase (SGS2SDE1) a coiled
protein (SGS3) a protein containing PAZ and Piwi domains (AGO1) and an RNA
helicase (SDE3) The putative SGS2SDE1 of Neurospora is related to QDE-1 and
Chapter 1 Introduction and Review of Literature
26
EGO-1 of C elegans whereas PAZPiwi protein AGO is related to QDE-2 of
Neurospora RDE-1of C elegans RNA helicase SDE3 is related to SMG-2 of C
elegans and Mut-6 of Chlamydomonas MUT-7 gene of C elegans encodes a protein
similar to Rnase D whereas the Drosophila DICER gene encode a protein similar to
RNase III An Arabidopsis ortholog of DICER gene has been identified called CAF
SIN1 SUS1(Thakur 2005 Poethig 2009)
Since the RNAi machinery is present constitutively within the eukaryotic cell it
is important to explore the metabolic advantages that are accorded by the RNAi related
proteins during the intrinsic normal growth of cells and development of organisms The
natural RNAi machinery not only keeps the mobile transposable elements from
disrupting the integrity of the genomes as suggested by the analysis of model organisms
like A thaliana C elegans D melanogaster (Tabara et al 1999 Wu-Scharf et al
2000 Aravin et al 2001 Hamilton et al 2002 Lisch 2009) but also participates in
development of organisms Genetic defects in Celegans RNAi genes ego1 and dicer
cause known specific developmental errors (Grishok et al 2000 Grishok et al 2001
Knight amp Bass 2001 Hall et al 2013) Similarly the Argonaute family of genes of A
thaliana (especially the ZWILLE proteins) is also responsible for plant architecture and
meristem development (Carmell et al 2002 Hamant and Pautot 2010) and the Dicer
homologue of A thaliana CAF1 is required for embryo development (Golden et al
2002 Nakano et al 2011) Thus genetic evidence illustrates the role of the RNAi
machinery as a controller of development related genes The mechanistic details of
these developmental processes are beginning to emerge
Chapter 1 Introduction and Review of Literature
27
Figure 16 Mechanism of gene silencing induced double stranded RNA The dicer enzyme to
produce siRNAs cleaves the RNA The siRNA generated bind to the nuclease complex
RISC The antisense component in the siRNA in the RISC guides the complex towards
the cognate mRNA resulting in endonucleolytic cleavage of the mRNA
Chapter 1 Introduction and Review of Literature
28
112 MicroRNA or MiRNA
In 1991 Ambros and co-workers first isolated a lin-4 mutant of Celegans which was
arrested at the first larval stage (Lee et al 1993) Later on the let-7 mutation was
isolated in the same system which was responsible for development through the fourth
larval stage Both lin-4 and let-7 encode short 22-nucleotide mature RNAs and were
called short temporal RNA because they control the temporal development program of
C elegans The mature lin-4 RNA defines (negatively regulates) the mRNA expression
of the lin-14 and lin-28 hetrochronic genes with the antisense mediated repression
mechanism of translation initiation and thus specifies the fate of cells during the first
three larval stages Recent studies have revealed that the short temporal RNAs are
actually members of a group of tiny RNAs (21to 28 nucleotides) called the micro-
RNAs (miRNA) (Bartel 2009) isolated members of which could easily run to a few
thousand which includes both those were identified computationally and by deep
sequencing Some of the components of the RNAi machinery have also been clearly
established as the effector proteins for the maturation of miRNAs
113 MicroRNAs in plants
1131 Discovery
MicroRNAs have been discovered by three basic approaches Direct cloning forward
genetic direct cloning and bioinformatic prediction followed by experimental
validation The most direct method of miRNA discovery is to isolate and clone small
RNAs from biological samples and several groups have used this approach to identify
plant miRNAs (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002 Xie et al 2003 Sunkar et al 2005 Williams et al 2013) The cloning methods
adapted were similar to those used to identify large numbers of animal miRNAs
(Lagos-Quintana et al 2001 Lau et al 2001 Aravin amp Tuschl 2005) which involve
isolating small RNAs followed by oligonucleotide adapter ligation reverse
transcription amplification and sequencing Some protocols incorporate methods to
enrich for Dicer cleavage products (ie molecules with 5prime-phosphates and 3prime-
hydroxyls) and to concatemerize the short cDNAs so that several may be identified in a
single sequencing read (Lau et al 2001 Llave et al 2002a Jagadeeswaran amp Sunkar
2013)The initial cloning experiments in Arabidopsis identified 19 miRNAs belonging
to 15 families (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002) although hundreds of other small RNAs such as siRNAs and RNA degradation
Chapter 1 Introduction and Review of Literature
29
fragments were also cloned through the direct cloning methods To select miRNAs
from the pool of small RNAs secondary structures of the RNA molecules were
examined in compliance with the acknowledged structural features of miRNAs
(Ambros et al 2003) The secondary structures were validated using several ad-hoc
software tools such as the Mfold or RNAfold programs (Reeder et al 2006) Finally
the existence of mature miRNAs was determined by RNA gel blot hybridization These
direct cloning methods were successful in isolating many miRNAs in Arabidopsis
(Reinhart et al 2002 Sunkar amp Zhu 2004) Rice (Sunkar et al 2005) Poplar (Lu et
al 2005) moss (Arazi et al 2005) and Tobacco (Billoud et al 2005)
Although miRNAs were first discovered through forward genetic screens in
worms (Lee et al 1993 Reinhart et al 2000)only few A thaliana miRNA gene
families have been discovered by this method (Chen 2008) in plants MiRNA
involvement in plant mutant phenotypes were not properly inferred till the cloning
experiments established that plant genomes contain numerous miRNAs (Park et al
2002 Reinhart et al 2002 Rhoades et al 2002) MiRNA loss-of-function allele has
been identified by forward genetic screens early extra petala1 is caused by a
transposon insertion ~160bp upstream of the predicted miR164c stem loop resulting in
flowers with extra petals (Baker et al 2005 Irish 2008) The fact that loss-of-function
miRNA mutants have been rarely discovered using forward genetics reflects small
target size for mutagenesis coupled with redundancy in nearly all evolutionarily
conserved plant miRNAs which are encoded by gene families (Table16) Family
members are likely to have overlapping functions buffering against loss at any single
miRNA locus Over expression screens can circumvent redundancy limitations At least
four plant miRNAs miR319 (also known as miR-JAW) miR172 (also known as EAT)
and miR166 were isolated in over expression screens for dominant mutants with
developmental abnormalities (Aukerman amp Sakai 2003 Palatnik et al 2003 Kim et
al 2005 Williams et al 2005b Schommer et al 2012) Mutations in miRNA target
sites which can prevent the entire family of miRNAs from repressing a target gene can
also circumvent redundant functions of miRNA family members
The dominant mutations in the HD-ZIP genes PHB PHV and REVOLUTA
(REV) in Arabidopsis and ROLLEDLEAF1 (RLD1) in Maize result in adaxialization of
leaves andor Vasculature (McConnell amp Barton 1998 McConnell et al 2001 Nelson
et al 2002 Zhong amp Ye 2004 Allen amp Millar 2012) and are all caused by mutations
Chapter 1 Introduction and Review of Literature
30
in miR166 complementary sites (Rhoades et al 2002 Emery et al 2003 Jones-
Rhoades amp Bartel 2004 Mallory et al 2004 Zhong amp Ye 2004)
In both plants and animals cloning was the initial means of large-scale
miRNA discovery (Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Reinhart et al 2002 Mendes et al 2012) However cloning is biased toward
RNAs that are expressed highly and broadly MiRNAs expressed at low levels or
only in specific cell types or in response to certain environmental stimuli were more
difficult to clone Sequence based biases in cloning procedures might also cause
certain miRNAs to be missed Because of these limitations bioinformatics approaches
to identify miRNAs have provided a useful complement to cloning
A straightforward use of bioinformatics has been carried out to find homologs
of known miRNAs both within the same genome and in the genomes of other species
(Pasquinelli et al 2000 Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Mendes et al 2009) A more difficult challenge is to identify miRNAs
unrelated to previously known miRNAs This was first accomplished for
vertebrate nematode and fly miRNAs using algorithms that search for conservation
of sequence and secondary structure (ie miRNA stem-loop precursors) between
species searching for patterns that are characteristic of miRNAs (Lai et al 2003 Lim
et al 2003 Lim et al 2005)
Most of the methods identified numerous miRNAs but are necessarily more
relaxed in terms of allowed structures Some approaches take advantage of the high
complementarity of plant miRNAs to target messages implementing the requirement
that the candidate has conserved complementarity to mRNAs (Jones-Rhoades amp Bartel
2004 Adai et al 2005) This additional filter has been useful for distinguishing
authentic plant miRNAs from false positives which later extended to mammalian
miRNA gene prediction (Xie et al 2005 Mendes et al 2012)
Another criteria used by most algorithms in the filters is evolutionary
conservation In plants mature miRNA sequences are conserved to a higher degree
than their precursor sequences For example Arabidopsis and rice diverged more than
130 million years ago (Friis et al 2004) but some miRNAs are highly conserved
in these plant species (Reinhart et al 2002) Furthermore various parameters for the
secondary structures such as free energy the number of paired residues within a
Chapter 1 Introduction and Review of Literature
31
miRNA or the number and size of bulges are also used to validate the predictions
(Jones-Rhoades amp Bartel 2004 Wang et al 2004 Adai et al 2005 Mendes et al
2012)
1132 Conserved microRNAs in Plants
Till date cloning genetics and bioinformatics screens have resulted in annotation of
approximately 184 potential Arabidopsis miRNAs (miRBase release 13) These
miRNAs seem to arise through gene duplication and subsequent diversification and
represent approximately 100 families (Maher et al 2006 Axtell 2013) Twenty one
families represented by 92 genes are clearly conserved in species beyond Arabidopsis
(Table 15) The presence of these miRNA families in other sequenced plant genomes
indicates that different plant species have an evolutionarily fluid set of miRNAs
(Willmann amp Poethig 2007) Recent studies on mosses one of the most ancient land
plants have shown that at least 14 miRNA families are conserved in eudicots and
mosses and therefore it appears that plant miRNAs share a common ancient origin
(Arazi et al 2005 Axtell amp Bartel 2005 Talmor-Neiman et al 2006 Zhang et al
2006 Axtell 2013) The number of members per family in a genome ranges from 1-32
with the exception of miR-430 which is represented by a cluster of ~80 loci in zebra
fish (Giraldez et al 2005)
In plants the number of members in each family correlates among examined
species certain families contain numerous members in all three species (eg miR156
miR166 miR169) whereas others consistently contain fewer genes (eg miR162
mir168 miR394) (Table 16) Although it is still unclear why certain miRNA are
encoded by more than one gene in plants (eg 12 genes encoding miR156) this
correlation might suggest functional significance of various miRNA family sizes Most
annotated plant miRNAs are conserved throughout flowering plants while a few
miRNAs are found only in a single sequenced genome and thus they could be
evolutionarily lsquoyoungrsquo miRNAs For example Arabidopsis has several miRNA
families that are not found in poplar or rice suggesting that miRNAs could be
generated continuously during evolution (Allen et al 2004a Rajagopalan et al
2006 Fahlgren et al 2007 Axtell 2012 de Alba et al 2013) Consistent with the
notion that non conserved miRNAs are of a more recent evolutionary origin these
miRNAs are mainly encoded by a single locus in the genome Within each family the
mature miRNA is always located in the same arm
Chapter 1 Introduction and Review of Literature
32
Table 16 MicroRNA families conserved in plants
miRNA
family
Arabidopsis Oryza Populus
miR156 12 12 11 miR159319 6 8 15 miR160 3 6 8 miR162 2 2 3 miR164 3 5 6 miR166 9 12 17 miR167 4 9 8 miR168 2 2 2 miR169 14 17 32 miR171 4 7 10 miR172 5 3 9 miR390 3 1 4 miR393 2 2 4 miR394 2 1 2 miR395 6 19 10 miR396 2 5 7 miR397 2 2 3 miR398 3 2 3 miR399 6 11 12 miR408 1 1 1 miR403 1 0 2 miR437 0 1 0 miR444 0 1 0 miR445 0 9 0 Total 92 127 169
All known miRNA families that are conserved between more than one plant species are listed
together with the number of genes identified in the sequenced genomes Rice miRNA families that have orthologs in maize but do not appear to have orthologs in the eudicots (Arabidopsis and Populus) are marked with an asterisk The following families contain miRNA genes annotated with
more than one number miR156 (miR156 and miR157) miR159319 (miR159 and miR319) miR166 (miR165 and miR166) miR171 (miR170 and miR171) and miR390 (miR390 and miR391)
of the stem loop (5prime- or 3prime) as it would be expected if the genes carry common ancestry
(Figure 17) The sequence of the mature miRNA and to some extent the segment on
the opposite arm of the hairpin to which it pairs is highly conserved between the
members of the same family (both within and between the species) while the sequence
secondary structure and length of the intervening ldquolooprdquo region can be highly divergent
within the same family
The patterns of paring and non pairing nucleotides are often conserved between
homologous miRNA stem loops from different species (Figure 17) The significance of
conserved mismatches is opined to guide DCL1 to cleave at the appropriate positions
along the stem loop
Chapter 1 Introduction and Review of Literature
33
Figure 17 Representative stem loop of miR164 Segments corresponding to the mature miRNAs
are shown in red (Adapted from Rajagopalan et al 2006)
Although many miRNAs are highly conserved in plants the degree of conservation in
most cases is quite low between plants and animals one exception is miR854 A recent
study has revealed that miR854 is conserved between Arabidopsis and animals and that
the Arabidopsis miR854 differs from the human miR854 only by a single nucleotide
(Arteaga-Vazquez et al 2006 Axtell 2012 de Alba et al 2013) The Arabidopsis
miR854 has multiple binding sites in the 3prime-untranslated region (UTR) of
OLIGOURIDYLATE-binding-PROTEIN mRNA 1bA (UBP1b) and represses the
translation of the UBP1b mRNA The animal miR854 is also complementary to a site in
the 3prime-UTR of the UBP1b homolog in animals However it is currently unclear how the
animal miR854 regulates its target mRNA Another distinction between plant and
animal miRNAs is the genomic organization of the miRNA genes While plant miRNA
genes are located predominantly in the intergenic regions animal miRNA genes tend to
localize in the intragenic regions both in the introns or exons of protein coding genes
Moreover miRNA genes are often clustered within a single precursor transcript
whereas individual plant miRNAs are derived from individual precursor RNAs (Bartel
Chapter 1 Introduction and Review of Literature
34
2004 Bonnet et al 2004 Kim 2005 Vaucheret et al 2006 Marco et al 2013)
1133 Non-conserved miRNA and challenges in annotation
Most miRNAs are conserved throughout flowering plants (Table 15) but many more
were found only in a single sequenced genome thus considered to be of a more recent
evolutionary origin The extended homology between few non conserved miRNAs
precursors and their target genes provides strong evidence that these relatively ldquoYoungrdquo
miRNAs arose from the duplication of the target gene segments (Allen et al 2004)
Several non-conserved miRNAs including miR161 miR163 miR173 miR447
miR475 and miR476 are known to direct cleavage of target transcripts (Allen et al
2004a Adai et al 2005 Sunkar et al 2005 de Alba et al 2013) It is difficult to
confidently predict targets for many because it is not possible to use complementary
site as a filter against false-positive target predictions It is also difficult to
confidently annotate non-conserved miRNAs as miRNA rather than siRNAs The
established minimal standard for miRNA annotation is a small RNA with detectable
expression and the potential to form a stem-loop when joined to flanking genomic
sequence (Kasschau et al 2003) In practice these requirements are too loose to
definitively categorize many small RNAs cloned from plants Many plant siRNAs are
detectable on blots (Vazquez et al 2004b) and hundreds of thousands of non-
miRNA genomic sequences can be predicted to fold into secondary structures that
resemble structures of plant miRNA precursors (Jones-Rhoades amp Bartel 2004)
Therefore without conservation of both sequence and secondary structure it is
difficult to be confident that a given cloned RNA originated from a stem-loop (ie is a
miRNA) rather than from a double-stranded RNA (ie is a siRNA) In fact many of
the thousands o f small RNAs cloned from Arabidopsis (Gustafson et al 2005) would
probably meet the literal requirements for annotation as miRNAs A few of these
sequences might be miRNAs but others that meet the literal criteria probably are not
so The challenges of annotating non conserved small RNAs were evidenced by
three related small silencing RNAs that were originally annotated as miRNAs
that turned out to be trans-acting siRNAs (tasiRNAs)(Kanno et al2013)
Although most miRNAs that are broadly conserved among flowering plants are
now being identified and experimentally validated (Table 16)(Jones-Rhoades amp
Bartel 2004) the challenges and ambiguities for classifying non-conserved miRNAs
prevent meaningful estimates of the total number of miRNA genes in Arabidopsis and
Chapter 1 Introduction and Review of Literature
35
other plant genomes It is possible that miRNAs might have escaped detection
because they are non-conserved and expressed in specific tissues or conditions and can
only be identified by using high- throughput sequencing
1134 MicroRNA biogenesis
11341 Transcription of microRNA precursor
Plant miRNAs are primarily found in the genomic regions that are not associated with
the protein coding genes (Reinhart et al 2002) and it appears that most if not all plant
miRNAs are produced from their own transcriptional units Plant miRNA genes are
occasionally clustered near each other in the genome suggesting transcription of
multiple miRNAs from a single primary transcript [eg the miR395 cluster (Grishok
et al 2001 Jones-Rhoades amp Bartel 2004b)] but this polycistronic arrangement of
miRNA genes appears far less frequently in plants than in animals (Bartel 2004)
Northern blot EST and mapping evidence indicate that plant miRNA primary
transcripts (also known as pri-miRNAs) as in animals are longer than needed to
encompass the miRNA stem-loops (Palatnik et al 2003 Jones-Rhoades amp Bartel
2004b) At least some of these pri-miRNA transcripts appear to be spliced
polyadenylated (Kurihara amp Watanabe 2004) and capped (Xie Z et al 2005) Two
rice miRNAs are contained within transcripts that contain exon junctions within the
presumptive stem-loop precursor implying that in these cases splicing is a prerequisite
for Dicer recognition (Sunkar et al 2005) Plant pri-miRNAs are over 1kb in length
and they are usually preceded by typical TATA box motifs They can also undergo
canonical splicing polyadenylation and capping All these observations indicates RNA
polymerase II is probably responsible for transcribing most plant miRNAs (Xie Z et
al 2005) as appears to be the case for many animal miRNAs Relatively little is
known about the regulation of miRNA transcription in plants but there is no reason
to suspect that this regulation would differ from that of protein-coding transcripts as
the bioinformatics analysis of the sequence upstream of the transcriptional start
sites of the miRNA genes showed the presence of putative binding motifs of
number of known transcription factors (Megraw et al 2006 Sethupathy 2013)
11342 MicroRNA processing and export
In plants maturation of miRNA is a step wise process A miRNA gene is transcribed to
pri-miRNA which is much longer than the pre-miRNA possessing the characteristic
stem-loop structure Like transcription of most protein-coding genes the miRNA
Chapter 1 Introduction and Review of Literature
36
transcription is processed by RNA polymerase II (Pol II) enzymes and the pri-
miRNAs are 5prime-capped and 3prime-polyadenylated (Lee et al 2004 Xie Z et al 2005)
Mature miRNAs are produced from pri-miRNAs through at least two sequential
processing steps by RNase III-type endonucleases In animals pri-miRNAs are first
processed at the bottom portion of the stem by Drosha a nuclear-localized RNase III
enzyme to liberate pre-miRNAs The pre-miRNAs are then exported to the cytoplasm
with the help of exportin-5The pre-miRNAs are further processed by Dicer another
RNaseIII enzyme to a duplex consisting of the miRNA and the antisense strands
(miRNA) Since plants do not have any Drosha homologs the two sequential
processing steps seem to be carried out by DCL1 a Dicer homolog in the nucleus
(Kurihara amp Watanabe 2004 de Alba et al 2013 Rogers amp Chen 2013)
The processing of pri-miRNAs to pre-miRNAs by DCL1 is assisted by two
other proteins HYPONASTY LEAVES1 (HYL1) and SERRATE (SE) HYL1 belongs to a
family of double-stranded RNA (dsRNA) binding protein and interacts with DCL1 in
vitro and in vivo (Lu amp Fedoroff 2000 Hiraguri et al 2005 de Alba et al 2013
Rogers amp Chen 2013) Loss-of-function mutations in the HYL1 gene result in
decreased accumulation of miRNAs and concomitant accumulation of pri-miRNAs
(Han et al 2004 Vazquez et al 2004a Kurihara et al 2006) SE a C2H2 zinc finger
protein interacts with DCL1 and HYL1 and plays a role in the processing of pri-
miRNAs to pre-miRNAs (Lobbes et al 2006 Yang L et al 2006) In addition the
nuclear cap-binding complex proteins CBP20 and CBP80 that are known as ABA
HYPER SENSITIVE 1(ABH1) have been shown to be required for proper miRNA
processing (Gregory et al 2008 Laubinger et al 2008) Recently it has been
demonstrated that DCL1 can release 21-nt RNAs from dsRNA as well as from
synthetic miR167 precursor RNAs in vitro However this cleavage is error prone and
inefficient in the absence of HYL1 and SE which accelerate the rate of DCL1-mediated
cleavage and promote accurate processing of miRNAs (Dong et al 2008 Rogers amp
Chen 2013)
11343 Methylation of the miRNAmiRNAduplex
After the miRNAmiRNAduplexes are released from the pre-miRNAs by the DCL1
activities the duplexes are methylated at the 2primeOH of the3prime-ends by HUA ENHANCER1
(HEN1) a small RNA methyltransferase (Yu et al 2005 Yang Z et al 2006 Rogers
amp Chen 2013 ) This step which is distinct from the RNase III-mediated processing
Chapter 1 Introduction and Review of Literature
37
step distinguishes the biogenesis of plant miRNAs from that of animal miRNAs
Mutations in the HEN1 gene were first isolated in a morphological screening for floral
development although the HEN1 mutants display pleiotropic phenotypes similar to the
developmental defects observed in the DCL1 mutants (Chen et al 2002 Park et al
2002 Rogers amp Chen 2013)
Figure 18 Biogenesis of microRNA
A Biogenesis of plant MicroRNA B Biogenesis of metazoananimal MicroRNA
The miRNA (red) is incorporated into RISC and the miRNA(blue) in degraded
This enzyme has two dsRNA-binding domains and nuclear localization signal It
is also conserved in fungi and is required for the processing of siRNAs as well as of
miRNAs (Park et al 2002 Boutet et al 2003 Xie et al 2003) Although the HEN1
protein is localized to the nucleus its methylation activity in the cytoplasm cannot be
ignored (Xie et al 2004 Rogers amp Chen 2013)
Chapter 1 Introduction and Review of Literature
38
The methylation of the miRNAmiRNA duplex is required to protect miRNAs
against the 3prime-end uridylation activity and subsequent degradation (Li et al 2005) In
the hen1 mutants accumulation of miRNAs is reduced to a lower level Also the
miRNAs appear to be heterogeneous in their sizes such that a ladder of bands rather
than a single species is observed for most miRNAs in the mutants (Li et al 2005)
MiRNA cloning and sequencing analyses revealed the presence of a number of U
residues at the 3prime-end of miRNAs in the hen1 mutants Moreover these nucleotides do
not correspond to the miRNAs in the pre-miRNAs indicating that these nucleotides are
added after the processing of the miRNAs from pre-miRNAs (Li et al 2005)
Uridylation of RNAs has also been proven to be correlated with RNA decay (Shen amp
Goodman 2004 Mullen amp Marzluff 2008) suggesting that uridylation attracts an
exonuclease which degrades miRNAs in plants
11344 MiRNA incorporation in to the RISC and its nuclear export
The methylated miRNAmiRNA duplexes undergo RISC assembly In this process
the miRNA of the duplexes is selectively incorporated into the RISC and the miRNA
appears to be removed and subsequently degraded (Hammond et al 2000 Hutvagner
amp Zamore 2002 Schwarz et al 2003 de Alba et al 2013 Rogers amp Chen 2013)
The ARGONAUTE 1(AGO1) proteins are core components of the RISC complex
They contain two structural domains the N-terminal PAZ domain that binds to the 3prime-
end of single-stranded RNAs and the C-terminal piwi domains that has a structure
similar to that of RNase H (Cerutti et al 2000 Parker et al 2004) Several AGO
proteins possess endonucleolytic activities within the PIWI domain and cleave target
mRNAs in the middle of the site complementary to miRNA (Liu et al 2004 Meister et
al 2004 Miyoshi et al 2005) Mutations in the AGO1 gene cause miRNA target
genes to be ectopically expressed and the levels of some miRNAs are reduced in the
mutants (Kidner amp Martienssen 2004 Vaucheret et al 2004 Kidner amp Martienssen
2005 Ronemus et al 2006) Moreover the phenotype of the AGO1 hypomorphic
alleles display defects in lateral organ polarity and leaf and flower morphology as
observed in the dcl1 mutants and null AGO1 alleles are embryonically lethal
supporting the close relationship between the AGO proteins and miRNA processing
(Kidner amp Martienssen 2004 Vaucheret et al 2004 de Alba et al 2013 Rogers amp
Chen 2013)
The production of miRNA miRNA duplexes occur in the nucleus while
Chapter 1 Introduction and Review of Literature
39
miRNA-RISC complexes are present in the cytoplasm where target mRNAs are
located This discordance of functional sites requires an export step during miRNA
biogenesis HASTY the Arabidopsis homolog of exportin-5 is thought to be involved
in miRNA nuclear export (Bollman et al 2003 Park et al 2005)The hasty mutant
exhibits pleiotropic defects in plant development and reduced accumulation of most
miRNAs as in the DCL1 or AGO1 mutants Howeverit is unclear whether the HASTY
proteins export the miRNA miRNA duplexes or the miRNA-RISC complexes in
plants
11345 Feedback regulation of miRNA biogenesis
MiRNA biogenesis is under feedback regulation such that the DCL1 and AGO1 genes
are themselves regulated by miRNAs DCL1 is itself a target of miR162 which leads to
the cleavage of the DCL1 mRNA (Xie et al 2003 de Alba et al 2013) Consistent
with this the levels of DCL1 mRNA are elevated in the mutants of miRNA processing
genes in which the miR162 abundance is reduced In addition there is a predicted
hairpin structure of miR838 in the 14th
intron of the DCL1 primary transcript
(Rajagopalan et al 2006) This prediction implies that DCL1-mediated processing
on this site can result in the cleavage of the DCL1 primary transcript into two truncated
fragments the N-terminal capped fragment and the C-terminal polyadenylated
fragment If this is the case it is possible that miRNA biogenesis competes with the
splicing event of the DCL1 pre-mRNA
The AGO1 is also regulated by miRNA There is a complementary site for
miR168 binding in the AGO1 mRNA and the transcript level of the AGO1 mRNA is
reduced through an mRNA cleavage mechanism (Vaucheret et al 2006 de Alba et al
2013 Rogers amp Chen 2013) indicating that the AGO1 mRNA levels are tightly
controlled by the levels of the AGO1 protein
1135 Mechanism of miRNA action
11351 MiRNA-directed mRNA cleavage
By forming the miRNA-RISC assembly miRNAs can direct the RISC to regulate gene
expression by two post transcriptional mechanisms mRNA cleavage or translational
repression (Bartel 2004 Jones-Rhoades et al 2006 Dalmay 2013) The degree
of miRNA-mRNA complementarity plays an important role for the
determination of the mechanism to be used A general rule is that while perfect or
Chapter 1 Introduction and Review of Literature
40
near-perfect complementarity induces mRNA cleavage central mismatches trigger
translational repression (Carrington amp Ambros 2003 Jones-Rhoades amp Bartel 2004)
Unlike most animal miRNAs plant miRNAs have near-perfect or perfect
complementarity to their targets and mRNA cleavage is assumed to be a predominant
mechanism by which miRNAs regulate gene expression in plants
In RNA cleavage mechanism miRNAs guide the AGO component of RISC to
cleave a single phosphodiester bond of the target mRNA within the miRNA-binding
site (Llave et al 2002b Tang et al 2003) The truncated fragments are then released
and degraded freeing the RISC to recognize and cleave another target mRNA This has
been validated by the detection of 3prime cleavage products that have 5prime ends that start at the
middle of the complementary regions The mRNA cleavage products are then further
degraded by other mechanisms The 3prime cleavage products of some miRNA targets are
degraded by the 5prime-3prime exonuclease XRN4 an Arabidopsis homolog of the major Yeast
mRNA degrading exonuclease Xrn1p It has been observed that the 3prime cleavage
products of some target mRNAs were accumulated in the xrn4 mutants (Souret et al
2004 Dalmay 2013) However the 3prime cleavage products of other miRNA targets do
not accumulate in the xrn4 mutants indicating that there are also XRN4 independent
mechanisms The 5prime cleavage products are typically untraceable by RNA gel blot
hybridization but can be detected by sensitive PCR based methods It has been found
that the 5prime cleavage products tend to obtain an oligo U tail at the 3prime ends This 3prime U tail
is correlated with shortening of the 5prime cleavage products from the 5prime end leading to the
conclusion that the 3prime uridylation causes 5 prime- 3 prime exonucleolytic decay of the 5prime cleavage
products (Shen amp Goodman 2004)
DCL1 HYL1 and SE interact with each other and are co-localized in the
nuclear bodies termed Dicing bodies or D-bodies in which some pri-miRNA shave
been found suggesting that D-bodies serve as a center for active miRNA processing
(Fang amp Spector 2007 Fujioka et al 2007 Song et al 2007) The miRNA
processing site appears to be distinct from the Cajal bodies that have previously been
found as a siRNA processing site (Pontes amp Pikaard 2008) The D-bodies include
SmD3 and SmB proteins that are found in the Cajal bodies However At collin an
additional marker for the Cajal bodies is not found in the D-bodies Moreover the D-
bodies are present in an At collin mutant lacking the Cajal bodies The pri-miRNAs of
miR171A and miR159A genes have been found to be co-localized with HYL1 and
Chapter 1 Introduction and Review of Literature
41
DCL1 in the D- bodies that are distinct from the Cajal bodies (Song et al 2007)
11352 MiRNA-directed translational repression
Translation repression is another mechanism exerted by plant miRNAs The regulatory
mechanism of miR172 which regulates flowering time and floral organ identity is
consistent with translation repression MiR172 over production does not affect the
transcript levels of APETALA2 (AP2) and AP2- like target mRNAs Instead the AP2
protein level is reduced in the miR172 over expressing mutant (Aukerman amp Sakai
2003 Chen 2004 Dalmay 2013) It was later shown that miR172 can also direct the
cleavage of the target mRNAs although the steady-state mRNA level is unchanged due
to feedback regulation (Schwab et al 2005 Jung et al 2007) It is clear that miR172
regulates its target genes primarily by translational repression rather than mRNA
cleavage Recently it has been reported that miR156157 and miR854 also regulate
their target genes through translational repression (Arteaga-Vazquez et al 2006
Gandikota et al 2007 Dalmay 2013) It was once thought that translational repression
is an exception to the rule However experimental evidence suggest a widespread
coexistence of translational repression and mRNA cleavage (Brodersen et al 2008)
1136 Regulatory roles of miRNAs in plant development
The miRNA target prediction has been a difficult task in order to obtain regulatory
targets without bringing in too many false predictions Plant growth and developmental
processes regulated by miRNAs are quite diverse and enormous Furthermore plant
miRNAs work cooperatively or antagonistically to establish a balanced regulation This
notion is well consistent with the findings that mutations in the regulators of miRNA
biogenesis transport and processing such as AGO1 DCL1 HYL1 HEN1 ABH1
and HASTY cause pleiotropic phenotypes (Mallory amp Vaucheret 2006 de Alba et al
2013) To date the roles of miRNAs have been mostly deduced from obtaining miRNA
over expressing transgenic plants or gain-of-function mutants in which miRNA-
resistant target genes are ectopically expressed
11361 Phase transition
Plants pass through a series of developmental phase transitions during their life span
beginning with seed germination continuing through vegetative phase change
reproductive phase change flowering initiation and finally seed production for the next
generation Because of their importance in understanding plant developmental
Chapter 1 Introduction and Review of Literature
42
processes genes regulating developmental transitions have been extensively studied
and underlying molecular mechanisms have been explored in various plant species
Majority of researches were mainly focused on vegetative phase change and
reproductive transition in Arabidopsis and Maize (Poethig 2003 Baurle amp Dean
2006) Particularly the mutations of the genes encoding various regulators of miRNA
biogenesis and transport such as HST SE and AGO exhibit altered juvenile to adult
vegetative phase change and vegetative to reproductive change (Hunter et al 2003
Peragine et al 2004 Vazquez et al 2004a Jones-Rhoades et al 2006 de Alba et al
2013)
Over-expression of the miR156a gene causes late flowering and delays
vegetative phase change (Wu amp Poethig 2006 Gandikota et al 2007 Wang et al
2008) It has been shown that miR156 negatively regulates a group of genes encoding
SQUAMOSA PROMOTER-BINDING PROTEIN-LIKE (Lewis et al 2007 de Alba et
al 2013 Rogers amp Chen 2013) transcription factors such as SPL3 SPL4 and SPL5
that promote flowering initiation In contrast transgenic plants over expressing
miR156-resistant SPL345 genes are early flowering Interestingly the endogenous
level of miR156 is temporally regulated In the early juvenile vegetative phase the
level of miR156 is very high but decreases rapidly before the onset of the adult juvenile
phase (Wu amp Poethig 2006)
The Arabidopsis early activation tagged (eat-D) mutant exhibits early flowering
with disrupted floral structure The mutant phenotypes are caused by over expression of
the miR172a-2gene (Aukerman amp Sakai 2003) MiR172 regulates a small group of
genes encoding APETALLA2 (AP2)-like transcription factors such as TARGET OF
EAT1 (TOE1) TOE2 SCHLAFMUTZE (SMZ) and SCHNARCHZAPFEN(SNZ)
TOE1 TOE2 SMZ and SNZ have been shown to participate in their productive phase
change by repressing a floral integrator FLOWERING LOCUS T (FT)(Jung et al
2007) Consistent with this toe1-2D 35STOE2 35SSMZ and 35SSNZ plants
exhibit late flowering (Jung et al 2007) MiR172 is also regulated temporally and
highly expressed in the adult vegetative phase both in Arabidopsis and maize (Chuck et
al 2007) Because the temporal expression patterns of miR156 and miR172 are
complementary to each other it has been suggested that the two miRNAs interact with
each other in regulating phase change (Chuck et al 2009 de Alba et al 2013) In
support of this view it has been shown that miR172 is down-regulated in the
Chapter 1 Introduction and Review of Literature
43
Corngrass1(Cg1) mutant in which miR156 is overproduced (Chuck et al 2007 de
Alba et al 2013) However there is no direct evidence for the direct linkage between
miR156 and miR172 It is also envisioned that miR156 and miR172 would not be
directly related For example miR156 regulates plastochron index that is the inverse of
leaf initiation rate and thus frequently used to analyze temporal patterns of plant
development In contrast miR172 does not affect plastochron (Wang et al 2008) The
down-regulation of miR172 in the maize miR156-over producing mutants would be
due to the prolonged juvenile vegetative phase in the mutants
Another miRNA that regulates flowering initiation is miR159 It regulates
MYB33 and MYB65 which are closely related to the barley GAMY B transcription
factor that responds to gibberellic acid (GA) The level of miR159 is elevated in GA-
treated seedlings but reduced in the GA biosynthetic ga1 mutant Transgenic plants
over producing miR159 exhibit delayed floral induction under short days (Achard et
al 2004) It is been suggested that MYB33 mediates GA signal to regulate LEAFY
(LFY) These observations indicate that miR159 plays an important role in the GA
signaling pathways that regulate proper floral induction under short days (Achard et al
2004)
11362 Floral development
Arabidopsis flowers consist of four distinct organs They arise in a characteristic
pattern within concentric rings called whorls from outside to inside four sepals four
petals six stamens and the central pistil consisting of two carpels Identities of the
floral organs are determined by three classes of regulatory genes classA classB and
class C genes (Krizek amp Fletcher 2005) The A and C class genes act antagonistically
to restrict their expression domains (Bowman et al 1991)
It is notable that miR172 also regulates floral architecture in addition to its role
in the timing of flowering initiation MiR172 inhibits the translation of the AP2 mRNA
(Chen 2004) Transgenic plants overproducing miR172 not only exhibit early
flowering but also show abnormal floral development which is quite similar to those
of the class A gene mutants such as the ap2 mutants (Aukerman amp Sakai 2003 de
Alba et al 2013) When the activity of the class A genes is inactivated that of the class
C genes such as AGAMOUS (AG) is elevated As a result the miR172- overproducing
transgenic plants exhibit no petals along with homeotic transformation of the sepals to
Chapter 1 Introduction and Review of Literature
44
the carpels (Aukerman amp Sakai 2003 Chen 2004)
Interestingly miR166 also regulates floral architecture The role of miR165166
in the regulation of meristem activity is closely linked to floral meristem formation and
organization (Zhao et al 2007 Miyashima et al 2013) Floral structure is severely
disrupted in the miR165166-overproducing mutants such as men1 and jba-1D in
which miR166 is overproduced (Williams et al 2005b Jung amp Park 2007) The men1
and jba-1D mutants have smaller gynoecium and reduced carpel number In an extreme
case the jba-1D homozygous plants lack visible carpels (Williams et al 2005b)
Similar phenotypes have been observed in the miR165 overproducers (Zhao et al
2007) It has been suggested that the phenotypes are possibly caused by altered AG
expression in the floral meristem (Jung amp Park 2007 Turchi et al 2013)
Other miRNAs also affect floral development Overproduction of miRNAs
involved in auxin signaling also affects floral architecture Transgenic plants
overproducing miR167 have defects in the formation of ovule and anther with reduced
fertility (Ru et al 2006) It has been shown that miR167 targets ARF6 and ARF8 that
play a role in gynoecium and stamen development and in regulating fertility (Ru et al
2006 Wu et al 2006) These observations suggest that auxin signals are also closely
related to floral organogenesis In addition transgenic plants over-producing miR164
and the cuc1cuc2 double mutants show fused sepals and reduced petals (Laufs et al
2004 Turchi et al 2013) suggesting that miR164 is also related to floral meristem
activity and boundary specification of the meristematic region
11363 Shoot apical meristem development
Shoot apical meristem comprises self-maintaining totipotent cells The shoot apical
meristem activity affects diverse aspects of plant developmental processes including
leaf development vascular development and lateral organ polarity (Bowman et al
2002) Because miR165166 plays a primary role in meristem formation
overproduction of miR165166 and multiple knockout mutants of its target genes
exhibit pleiotropic pheonotypes (McConnell et al 2001 Emery et al 2003 Kim et al
2005 Williams et al 2005b)
The targets of miR165166 include genes encoding the homeo domain-leucine
zipper (HD-ZIP) class III transcription factors such as PHABULOSA(PHB)
PHAVOLUTA (PHV) REVOLUTA(REV) ATHB15CORONA (CNA) and ATHB8
Chapter 1 Introduction and Review of Literature
45
PHB PHV and ATHB15 have overlapping functions in controlling meristem
development (Turchi et al 2013) Indeed the phb phv athb15 triple mutants have an
enlarged shoot apical meristem Consistent with this the miR166- overproducing men1
and jba-1D mutants exhibit enlarged shoot apical meristems (Kim et al 2005
Williams et al 2005b Turchi et al 2013) In the mutants the shoot apical meristems
are multiplied and large
The development of shoot apical meristems is also regulated by the WUSCHEL
(WUS)ndashCLAVATA (CLV) signaling pathway WUS positively regulates cell
proliferation In contrast CLV3 which is expressed in stem cells limits the WUS
expression and thus inhibits cell proliferation in the SAM (Sharma et al 2003)
Consistent with this notion WUS is expressed to a higher level in jba-1D
explaining the ectopic enlargement of the SAM in the mutant (Williams et al 2005)
While WUS is closely related to miR165166 in the shoot apical meristem
development the underlying molecular mechanisms are currently unclear It has been
observed that the miR166-over producing men1 plants have an arrested meristem
development (Jung amp Park 2007) In addition the heterozygous men1+ and the
homozygous men1 plants show different expression patterns of WUS and CLV3 It
appears that miR166 regulates WUS indirectly and influences the expressional balance
between WUS and CLV3 through a negative feedback loop (Jung amp Park 2007 de
Alba et al 2013)
The phv phb athb15 triple mutant has an enlarged shoot apical meristems the
rev phb phv mutant does not have functional shoot apical meristems (Prigge et al
2005) This distinction may be explained by the fact that while miR166 targets
primarily PHB PHV and ATHB15 miR165 targets all the five HD-ZIP III genes (Zhou
et al 2007) Consistent with this transgenic plants overproducing miR166 are distinct
from those overproducing miR165 The miR165-overproducing transgenic plants lack
functional shoot apical meristem and a pin-like structure is formed at the apical region
of the mutant These phenotypes are quite similar to rev phb phv triple mutant (Zhou et
al 2007 Liu et al 2013) The phenotypic distinction may because by the difference
of the cleavage efficiencies of the target mRNAs In accordance with this notion a few
35S miR166a transgenic lines exhibit no shoot apical meristems The men1
homozygous plants also show arrested meristem development (Jung amp Park 2007)
Chapter 1 Introduction and Review of Literature
46
MiR164 cleaves the CUC1 and CUC2 mRNAs as well as the NAC1 mRNA
CUC1 and CUC2 determines the organ boundary in the meristem Phenotypes of
transgenic plants that overproduce miR164 are similar to those of the cuc1cuc2 double
mutant that exhibits embryonic developmental defects such as cup-shaped cotyledons
and fused cotyledons (Laufs et al 2004) When a miR164-resistant CUC2 is
expressed the boundary domain is enlarged CUC also plays a role in embryonic shoot
meristem formation by regulating the SHOOT MERISTEMLESS gene It was therefore
concluded that miR164-mediated cleavage of CUC1 and CUC2 mRNAs determines the
sizes of the boundary domains in the shoot apical meristem and regulates separation of
the organs by restricting cell proliferation (Laufs et al 2004 de Alba et al 2013)
11364 Leaf development
The role of miR319 has been extensively studied in leaf development A dominant
jaw-D mutant overproducing miR319 shows uneven curved leaves with enlarged size
and five TCP genes which are closely related to the CINCINNATA (CIN) gene in
Antirrhinum majus are down regulated in the mutant suggesting that miR319 mediated
regulation of the TCP transcription factor genes are important for the final step of
leaf shaping (Palatnik et al 2003 Naz et al 2013) In addition to leaf shape and size
leaf polarity is also specified by miRNA MiR166-mediated cleavage of the HD-ZIPIII
mRNAs is a well to establish leaf polarity This discovery originates from the gain-of-
function mutants such as phb-1d and phv-1d wherein point mutations in the
complementary sites to miRNA helps the mRNA to evade or resist from miRNA-
mediated cleavage (McConnell et al 2001 Emery et al 2003 Naz et al 2013)
Plant leaves have a dorso-ventral polarity consisting of the adaxial (upper
surface) and abaxial (lower surface) sides The adaxial side is closer to the shoot
apical meristem Adaxial identity is specified by functionally redundant class III HD-
ZIP family genes such as PHB PHV and REV Ectopic expression of each genes
result in adaxialized leaves Dominant phb-1d and phv-1d mutants exhibit
transformation of the abaxial to adaxial fate and have rod-shaped leaves In contrast
miR165166-overproducing plants show pin-like cotyledons radicalized leaves and
abaxialized leaves (Chitwood et al 2007 Pattanayak et al 2013)
Leaf asymmetry is determined by miR166-mediated restriction of the
expression domains of the HD-ZIPIII genes The HD-ZIPIII genes are expressed in the
Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
3
(4) Eucoffea
The Coffee species belonging to Mascarocoffea section have one common
characteristic the absence of caffeine The Eucoffea (now named Coffea) has been
again divided into five subsections according to diverse criteria tree height
(Nanocoffea) leaf thickness (Pachycoffea) fruit colour (Erythrocoffea Melanocoff-
ea) and geographical distribution (Mozambicoffea) (Table 11) The recent
classification has widely accepted that Coffea sect Paracoffea and Agrocoffea mainly
consist of species from other genera
Table 11The grouping of the species in the subsection Eucoffea (Chevalier 1947)
Section Subsection Species
Eucoffea
Erythrocoffea
C arabica
C canephora
C congensis
Pachycoffea
C abeokutae
C liberica
C klainii
C oyemensis
C dewerei
Melanocoffea
C stenophylla
C carissoi
C mayombensis
Nanocoffea
C humilis
C brevipes
C togoensis
Mozambicoffea
C schumanniana
C eugenioides
C kivuensis
C mufindiensis
C zanguebariae
C racemosa
C ligustroides
C salvatrix
The current sub generic classification comprises of two genera Coffea and Baracoffea
(Table 12) Most species of Coffea belong to Coffea subgenera includes those used
for producing the beverage coffee Coffea subgenera occur throughout the natural
range of the genus in Africa Madagascar and The Mascarenes Subgenera Baracoffea
contains only three accepted species (although five remain undescribed) (Table 12)
and is restricted to the dry forests of western Madagascar Leroy (Leroy 1980) placed
C rhamnifolia a species from Africa (Somalia and Kenya) in Coffea subgen
Chapter 1 Introduction and Review of Literature
4
Baracoffea but this was contested by Bridson (1988) Davis amp Rakotonasolo (2003)
and Davis et al (2005)
Table 12Outline classification of Coffea and Psilanthus with synonyms (Davis amp Rakotonasolo
2003)
Coffea L Sp Pl 172 (1753)
Coffea sub gen Coffea L Type C arabica L 95 species Africa Madagascar Mascarenes
Coffea sub gen Psilanthopsis (AChev)
Psilanthopsis AChev Type Coffea kapakata
Type Paolia jasminoides Chiov (= C rhamnifolia (Chiov) Bridson)
Coffea sub gen Baracoffea
Coffea sect Baracoffea Type Coffea humbertii J-FLeroy
Eight species (including five -yet to be published) West Madagascar
[Paracoffea subgen Insulanoparacoffea J-FLeroy J Agric Trop Bot Appl 14 276 (1967)
nom nud]
Psilanthus Hookf Gen Pl 115 (1873)
Psilanthus subgen Psilanthus Hookf Type Psilanthus mannii Hookf
Two species Africa (central and western)
Psilanthus subgen Afrocoffea (Moens)
Coffea subgenAfrocoffea
Type Psilanthus lebrunianus (Germain amp Kesler) Bridsonc 18 species Africa Asia
Australasia
Coffea sectParacoffeaMiq Fl Ind Batavae 308 (1856)
Psilanthus subgen Paracoffea Type Coffea horsfieldiana Miq
Paracoffea
[Paracoffea subgenAfroparacoffea J-FLeroy J Agric Trop Bot Appl 14 276 (1967)
nom nud]
[Paracoffea subgenMelanoparacoffea J-FLeroy J Agric Trop Bot Appl 14 276 (1967)
nom nud]
13 Economics of Coffee
Coffee is the fourth most valuable traded agricultural commodity (FAO 2004) but is
the second most valuable commodity exported by the developing countries and more
than 75 million people depend on coffee for most of their livelihood (Pendergrast
2009)
India produces both Arabica coffee from C arabica and Robusta coffee from
C canephora As per the economic survey 2012-13 Coffee is planted in India in
around 388195 hectares Out of this area 52 is Ccanephora and 48 is Carabica
Chapter 1 Introduction and Review of Literature
5
Coffee is mostly grown in south India parts of Orissa W Bengal and North east
India Karnataka is the largest producer with 569 of total area under production
which accounts for 71 of the total produce The total crop harvest for 2012-2013 is
placed at 325300 tonnes (httpwww Indicoffeeorgindiacoffeephpp-
age=CoffeeData)
14 Composition of coffee beans
Navellier (1961) gave a mean composition of green coffee as Carbohydratesglucides
(58) lignin (2) lipids (13) proteins (13) ash (4) non-volatile acids (8)
trigonelline (1) and caffeine (1) The caffeine content of green coffee is relatively
limited (10-25) of dry matter and changes little with seed development (Clifford
and Kazi 1987) In coffee seeds the concentration of trigonelline is approximately
2 of dry weight (Clifford 1985 Mazzafera 1991) Mazzafera (1999) found a higher
protein content in the mature beans than in the immature beans but a lower content of
free amino acids with asparagine as the main component Flament ( 2000) estimated
the composition of coffee beans as follows
Proteins 100 (dry weight basis of green coffee)
Carbohydrates 500 -do-
Lipids 117-140 (Arabicas)
76- 95 (Robustas)
Chlorogenic acids 65 (Arabicas)
90 (Robustas)
15 Purine alkaloids structure and role
Purine alkaloids are derived from purine nucleotides (Zulak et al 2007) and found in
over hundred plant species in 13 orders in plant kingdom (Ashihara amp Crozier 1999)
Methylxanthines like caffeine (137-trimethlyxanthine) theobromine (37-methylxan-
thine) and methyluric acids are classified into purine alkaloids (Figure 11) (Table
13)(Ashihara et al 2008) These are known to occur in tea coffee and a number of
non-alcoholic beverages Caffeine was isolated from coffee (Coffea arabica) and tea
(Camellia sinensis) in 1820 (Ashihara amp Crozier 2001)
Chapter 1 Introduction and Review of Literature
6
Figure 11 Purine alkaloid xanthine and uric acid skeleton
Plants that accumulate purine alkaloid are classified into three groups based on
the type of alkaloids they produce caffeine-producing plants which include coffee
tea and mateacute (Ilex paraguariensis) theobromine ndash producing plants are represented
by cacao cocoa tea (Camellia ptilophylla) and Camellia irrawadiensis and
methyluric acid ndash producing plants consist of Coffea dewevrei Coffea liberica C
excelsa and Kucha tea (Camellia assamica)
Table 13 Purine alkaloid structures based on xanthine and uric acid skeleton
Compound Trivial name R1 R2 R3 R4 O-2 Δ2-3
Methylxanthines
Xanthine H H H
1-methylxanthine CH3 H H
3-methyl xanthine H CH3 H
7-methyl xanthine H H CH3
1-3methylxanthine Theophylline CH3 CH3 H
17-methylxanthine Paraxanthine CH3 H CH3
37-methylxanthine Theobromine H CH3 CH3
137-methylxanthine Caffeine CH3 CH3 CH3
Uric acid and methyl uric acids
Uric acid H H H H
137-trimethyluric acid CH3 CH3 CH3 H
1379-tetramethyluric
acid Theacrine CH3 CH3 CH3 CH3
O19-trimethyluric acid Liberine CH3 H H CH3 CH3 Δ2-3
O179-trimethyluric acid Methylliberine CH3 H CH3 CH3 CH3 Δ2-3
16 Role of caffeine in plants
The physiological role of endogenous purine alkaloids and related compounds in
higher plants remains undetermined There are two hypotheses about the role of the
Chapter 1 Introduction and Review of Literature
7
high concentrations of caffeine that accumulate in tea coffee and a few other plant
species The lsquoallelopathic theory or the auto toxic function theoryrsquo proposes that
caffeine in seed coats is released into the soil and inhibits the germination of seeds
around the parent plants (Anaya et al 2006)
The lsquochemical defense theoryrsquo proposes that caffeine in young leaves fruits
and flower buds acts to protect soft tissues from insect pests (Ashihara amp Crozier
2001 Ashihara et al 2008) It has been shown that spraying tomato leaves with a 1
solution of caffeine deters feeding by tobacco horn worms while treatment of
cabbage leaves and orchids with 001ndash01 solutions of caffeine acts as a neurotoxin
and kills or repels slugs and snails (Hollingsworth et al 2002) This work has now
been extended with convincing evidence for chemical defense theory has recently
been obtained with transgenic caffeine ndash producing tobacco plants (Uefuji et al 2005
Kim et al 2006)
17 Distribution of caffeine in plants
Caffeine has been found in 13 orders of the plant kingdom (Ashihara and Suzuki
2004 Anaya et al 2006) In some species the main purine alkaloid is theobromine or
methyl uric acids such as theacrine liberine and methylliberine rather than caffeine
(Ashihara amp Crozier 1999 Ashihara amp Crozier 2001)(Table 14)
Table 14 Distribution of purine alkaloids in plants
SNo Plant species Common
name Major alkaloid Plant parts
containing
alkaloids 1 Coffea arabica Arabica Caffeine Leaves Seeds
2 Coffea canephora Robusta Caffeine Leaves Seeds
3 Coffea liberica Caffeine Theacrine Liberine
Seeds Mature leaves
4 Coffea dewevrei Caffeine Theacrine Liberine
Seeds Mature leaves
5 Camellia sinensis Tea Caffeine Leaves
6 Camellia assamica Assam tea Caffeine Leaves
7 Camellia assamica var
kucha Kucha Theacrine Caffeine Leaves
8 Camellia irrawadensis Theobromine Leaves
9 Camellia ptilophylla Cocoa tea Theobromine Leaves
10 Theobroma cacao Cocoa Theobromine Seeds
11 Paullinia cupuna Guarana Caffeine Seeds
Chapter 1 Introduction and Review of Literature
8
12 Cola nitida Caffeine Seeds
13 Citrus sp Caffeine Pollen
14 Ilex paraguariensis Mate Caffeine Leaves
Adapted from (Ashihara amp Crozier 1999 Ashihara amp Crozier 2001)
18 Caffeine biosynthesis
The major biosynthetic pathway is a four step sequence consisting of three sequential
methylation and one nucleosidase reactions (Figure 12) The xanthine skeleton of
caffeine is derived from purine nucleotides The initial step in caffeine biosynthesis is
the methylation of xanthosine by a SAM- dependent N-methyltransferase In addition
to experiments with radiolabeled precursors substrate specificities of native (Kato et
al 1999) and recombinant N-methyltransferases (Kato et al 2000 Ogawa et al
2001 Mizuno et al 2003a Mizuno et al 2003b Uefuji et al 2003) strongly
suggest that the major route to caffeine is a xanthosine rarr7-methylxanthosinrarr 7-
methylxanthine rarr theobromine rarr caffeine pathway (Figure 12) Although the
information has been obtained mainly from coffee (C arabica) and tea (Camellia
sinensis) the available evidence indicates that the pathway is essentially the same in
other purine alkaloid-forming plants such as mateacute (Ashihara 1993) and cacao
(Koyama et al 2003 Yoneyama et al 2006)
The first step in the caffeine biosynthetic pathway from xanthosine is
conversion of xanthosine to 7-methylxanthosine (Figure 12) This reaction is
catalyzed by the 7-methylxanthosine synthase (xanthosine 7 N-methyltransferase EC
211158) The genes encoding for 7-methylxanthosine synthase CmXRS1 (AB
034699) and CaXMT1 (AB 048793) were isolated from the C arabica (Mizuno et
al 2003a Uefuji et al 2003) The recombinant proteins obtained from these genes
exhibit 7-methylxanthosine synthase activity in vitro Xanthosine monophosphate
(XMP) was not converted to 7-methylxanthosine by the recombinant enzymes thus
inclusion of 7-methy-XMP was proposed (Schulthess et al 1996)
The second step of caffeine biosynthesis involves a nucleosidase which
catalyses the hydrolysis of 7-methylxanthosine Although N-methylnucleosidase (EC
32225) was partially purified from tea leaves (Negishi et al 1988) recent detailed
structural studies on coffee 7-methylxanthosine synthase suggested that the methyl
transfer and nucleoside cleavage may be coupled and catalyzed by a single enzyme
(McCarthy amp McCarthy 2007)
Chapter 1 Introduction and Review of Literature
9
The last two steps of the caffeine synthesis are also catalyzed by a SAM-
dependent N-methyltransferase(s) which is different from the N-methyltransferase
enzyme that catalyzes the first step in the pathway Native N-methyltransferase
activities have been detected in crude and partially purified extracts from tea and
coffee plants (Ashihara amp Suzuki 2004) and a highly purified preparation has been
obtained from young tea leaves (Kato et al 1999) The enzyme was assigned the
name caffeine synthase (EC 211160) that catalyses the last two steps of caffeine
biosynthesis viz the conversion of 7-methylxanthine to caffeine via theobromine
(Figure 12) The gene encoding caffeine synthase was first cloned from young tea
leaves (Kato et al 2000) Since then several genes encoding N-methyltransferases
with different substrate specificity have been reported
Activity of the recombinant theobromine synthase (CTS1 and CaMXMT EC
211159) is specific for the conversion of 7-methylxanthine to theobromine In
contrast the recombinant caffeine synthases (CCS1 and CaDMXMT1) can utilize
paraxanthine theobromine and 7-methylxanthine as shown with tea caffeine synthase
(TCS1) (Mohanpuria et al 2011) Although paraxanthine is the most active substrate
of this recombinant enzyme only limited amounts of paraxanthine are synthesized in
coffee cells hence in vivo caffeine synthase is principally involved in the conversion
of 7-methylxanthine to caffeine via theobromine
Radiolabelled tracer experiments with theobromine accumulating cacao and
Chinese tea (Camellia ptilophylla) showed a limited conversion of theobromine to
caffeine N-methyltransferase which catalyzed the conversion of 7-methylxanthine to
theobromine was detected in crude extracts from C ptilophylla but conversion of
theobromine to caffeine was not observed (Ashihara et al 1998) Genes homologous
to caffeine synthase have been isolated from several theobromine accumulating plants
including cacao (Yoneyama et al 2006) The recombinant enzymes derived from
these genes have 3 N-methyltransferase activity suggesting that these theobromine
accumulating plants contain specific theobromine synthase Caffeine does not occur
in C ptilophylla (Ashihara et al 1998) although it is present in leaves and fruits of
cacao along with theobromine (Koyama et al 2003 Zheng amp Ashihara 2004)
Chapter 1 Introduction and Review of Literature
10
Figure 12 Pathways for the biosynthesis of caffeine
The major pathway is indicated by solid lines (Mizuno et al 2003b Uefuji et al 2003) Alternative pathways in coffee and tea plants are indicated
by dashed lines(Kato et al 1996 Schulthess et al 1996) Enzymes (1) 7-methylxanthosine synthase (2) N-methylnucleosidase (3) Theobromine
synthase (4) Caffeine synthase
N
N
N
N7
H3C
3
1
O
O
Caffeine
N
NH
N
N7
3
O
O
Theobromine
N
N
N
N
H
H
7
O
O
N
N
N
N
H
H
O
O
N
N
N
N
H
H
O
O
Xanthosine
N
N
N
N
H
H
O
O
N
N
N
N
H
H
O
ON
N
N
N
H
H3C
71
O
O
Paraxanthine
CH3
CH3
CH3
CH3CH3
7-methylxanthine
Rib P
Rib
(1)
(4)
Rib
+
CH3
7-methyl
xanthosine
(2)
(3)
SAM SAH
SAM SAH SAM SAH
Xanthosine mono
phosphate
Rib P
7-methyl xanthosine
monophosphate
+
CH3 CH3
Inosine 5-mono
phosphate
Adenosine 5-mono
phosphate
SAM SAH
Guanosine
Guanosine 5-mono
phosphate
Chapter 1 Introduction and Review of Literature
11
181 Source of Xanthosine for caffeine biosynthesis
Xanthosine the initial substrate of purine alkaloid synthesis is supplied by at least four
different pathways de novo purine biosynthesis (de novo route) the degradation
pathways of adenine nucleotides (AMP route) and guanine nucleotides (GMP route)
and the S-adenosyl-L-methionine (SAM) cycle (SAM route) (Figure 13) Purine
nucleotides are synthesized by de novo and salvage pathways (Ashihara amp Crozier
1999 Stasolla et al 2003 Zrenner et al 2006) The de novo synthesis of non purine
precursors like CO2 10-formyltetrahydrofolate 5-phosphoribosyl-1-pyrophosphate and
the amino acids Glycine glutamine and aspartate results in the production of purine
nucleotides
Figure 13 Xanthosine biosynthesis Xanthosine for purine alkaloid biosynthesis is supplied by at
least four routes from adenosine released from the SAM cycle (SAM route) from IMP
originating from de novo purine synthesis (de novo route) from the cellular adenine
nucleotide pool (AMP route) and from the guanine nucleotide pool (GMP route)
Adapted from (Ashihara et al 2008)
The utilization of IMP for caffeine biosynthetic pathway which is formed by the
de novo purine biosynthetic pathway for caffeine biosynthetic pathways was
demonstrated in young tea leaves using 15
N-glycine and 14
C-labelled precursors and
inhibitors of de novo purine biosynthesis (Ito amp Ashihara 1999) Xanthosine is formed
by an IMP rarr XMP rarr xanthosine pathway IMP dehydrogenase (EC 111205) and 5prime-
nucleotidase (EC 3135) catalyze these reactions The inhibition of IMP dehydrogen-
ase by ribavirin inhibits both caffeine and gunanine nucleotide biosynthesis in tea and
coffee leaves (Keya et al 2003)
Chapter 1 Introduction and Review of Literature
12
The portion of xanthosine used for caffeine biosynthesis is derived from adenine
and guanine nucleotide pools which are produce de novo by salvage pathways there are
several potential pathways for xanthosine synthesis from AMP but the AMP rarr IMP
rarr XMPrarr xanthosine route is predominant All three enzymes involved in the
conversion AMP deaminase (EC 3546) IMP dehydrogenase and 5prime-nucleosidase
have been detected in tea leaves (Koshiishi et al 2001) Xanthosine for caffeine
biosynthesis can also be produced from guanine nucleotides by GMPrarrguanosine
rarrxanthosine pathway Guanosine deaminase (EC 35415) activity is found in cell free
extracts from young tea leaves (Negishi et al 1994) but GMP deaminase activity was
not detected in plants (Stasolla et al 2003 Zrenner et al 2006) The absence of GMP
deaminase activity suggests that a GMP rarr IMP rarr XMP rarr xanthosine route is not
functional in plants
The use of radiolabelled purine bases and nucleosides for the study of caffeine
biosynthetic pathway was facilitated by the fact that exogenous purine nucleotides are
rapidly hydrolyzed by the plant tissues forming purine nucleosides andor bases which
are then utilized in pathways which in most instances indirectly lead to xanthosine
Most of these purine compounds including adenine adenosine inosine hypoxanthine
and guanine are converted to their respective nucleotides by salvage enzymes
including adenine phosphoribosyl transferase (EC 2427) hypoxanthineguanine
phosphoribosyl transferase (EC 2428) adenosine kinase (EC 27120) and
inosineguanosine kinase (EC 2421) which usually have high activities in plant cells
The resultant nucleotides then enter the purine alkaloid biosynthetic pathway In the
case of guanosine it may be directly deaminated to xanthosine and utilized for caffeine
biosynthesis (Figure 13) (Ashihara et al 1997)
182 SAM cycle in plants
SAM is the methyl donor for the three methylation steps in the caffeine biosynthetic
pathway In this process SAM is converted to S-adenosyl-L-homocysteine (SAH)
which in turn is hydrolyzed to homocysteine and adenosine Homocysteine is recycled
via the SAM cycle to replenish SAM levels and adenosine is released from the cycle
Adenosine released from the cycle is converted to AMP directly andor indirectly via
adenine AMP is converted to xanthosine and used for caffeine biosynthesis Since
three moles of SAH are produced via the SAM cycle for each mole of caffeine that is
synthesized in theory this pathway has the capacity to be the sole source of both the
Chapter 1 Introduction and Review of Literature
13
purine skeleton and the methyl groups required for caffeine biosynthesis in young tea
leaves (Koshiishi et al 2001)
Methionine
S-Adenosyl-L-methionineHomocysteine
ATP
PPi+ Pi
Tetrahydrofolate
5-Methyl-
tetrahydrofolate
CH3+Adenosine
S-Adenosyl-L-
homocysteine
(xanthine
structure)(Methyl groups)
Caffeine
(1)
(2)(3)
(4)
Figure 14 The SAM cycle (activated methyl cycle) in plants (Ashihara amp Suzuki 2004)
Enzymes SAM synthetase SAM-dependent N-methyltransferases S-adenosyl-
homocysteine (SAH) hydrolase Methionine synthase Adenosine released from the cycle
is salvaged to adenine nucleotides and utilized both for purine structure of caffeine via
xanthosine and for re-synthesis of SAM via ATP
183 N-methyltransferase in caffeine biosynthesis Molecular cloning
The genes encoding for the enzymes involved in the caffeine biosynthesis were
successfully cloned from the coffee family (Ogawa et al 2001 Mizuno et al 2003a
Mizuno et al 2003b Uefuji et al 2003) The conserved amino acid regions of tea CS
(AB31280) the sequences of O-methyltransferases derived from plant origin and
Arabidopsis hypothetical proteins were made use of in designing primers for RT-PCR
and RACE technique Independent groups have isolated N-methyltransferase (NMT)
genes from coffee which have been classified based on the substrate specificity of the
recombinant proteins expressed in Ecoli (Table 15) PCR products thus obtained were
used as the probe to isolate full length cDNA from the library resulting in the
identification of all three N-methyltransferases involved in caffeine biosynthesis
(Figure 12) Several cDNA have been characterized from the coffee plants one for
xanthosine methyltransferase (XMTXRS) three for 7-methylxanthine methyltransferase
or theobromine synthase (MXMTCTS) and three for 37-dimethylxanthine
methyltransferase or caffeine synthase (DXMTCCS) (Table 15) (Kato amp Mizuno
2004)
A single gene encoding for xanthosine methyltransferase (XMTCmXRS1) was
identified which encoded for a polypeptide consisting of 372 amino acids with an
apparent molecular mass of 418kDa It is expressed almost uniformly in aerial tissues
Chapter 1 Introduction and Review of Literature
14
of C arabica including leaves floral buds and immature but not in mature beans
Three genes encoding 7-methylxanthine methyltransferase (theobromine synthase) have
been identified The number of amino acids in the putative polypeptides are 378 for
MXMT1 (427kDa) and 384 for MXMT2 and CTS2 (434kDa) They differ by insertion
or deletion of blocks of several residues in the C-terminal region Their catalytic
properties based on kinetic parameters such as Km values were different They are
expressed in young leaves floral buds and immature but not mature beans Three genes
were identified for 3 7-dimethylxanthine methyltransferase (caffeine synthase) DXMT
CCS1 and CtCS7 each encoding a 43 kDa polypeptide consisting of 384 amino acids
However their kinetic properties differ from each other such as DXMT and CCS1
showed Km values of 1200 and 157microM for theobromine respectively Expressions
profiles are also distinct DXMT being expressed exclusively in immature beans while
CCS1 expression is ubiquitous occurring in all tissues The presence of isoforms of
these enzymes with different properties suggests that caffeine is synthesized through
multiple pathways depending on availability and concentration of the substrate (Mizuno
et al 2001 Ogawa et al 2001 Mizuno et al 2003a Mizuno et al 2003b Uefuji et
al 2003) Genomic clones of theobromine synthase were obtained by PCR- RFLP
from C canephora (Satyanarayana et al 2005 Satyanarayana 2006) The genomic
clones are reported to have highly conserved exons and a variable third intron The
promoter for the theobromine synthase was cloned and characterized (Satyanarayana et
al 2005)
There is high degree of similarity in the coding region of coffee NMT genes
isolated so far though differences exist in the 3prime untranslated region (UTR) for these
genes The deduced amino acid sequence of these enzymes shows more than 85
homology between these genes Phylogenetic analysis indicates that they are more
closely related to C-methyltransferases including those for jasmonic acid salicylic acid
and benzoic acid than to other N-methyltransferases This suggests that coffee N-methyl
transferases belong to a distinct sub-group within plant methyltransferases Their
cellular localization determined by green fluorescent protein fusion method and β-
glucourodinase (GUS) showed that the all the three genes are localized in the cytoplasm
(Ogawa et al 2001 Vinod et al 2007) It was also shown that these were present
along with chlorogenic acid(Vinod et al 2007)
Chapter 1 Introduction and Review of Literature
15
Table 15 N-methyltransferases and their encoding genes involved in caffeine biosynthesis
Gene (Enzyme) Gene name Accession No Description Reference
Xanthosine methyltransferase
( 7-methylxanthosine synthase) (EC 211158)
CaXMT AB 048793 Immature fruit cDNA Screening (Uefuji et al 2003)
CmXRS1 AB 034699 Leaf cDNA library RACE (Mizuno et al 2003a)
CaMXMT1 AB 048794 Leaf cDNA library RACE (Ogawa et al 2001)
7-methylxanthine synthase transferase
(theobromine synthase) ( EC 211159)
CTS1 AB 034700 Immature fruit cDNA Screening (Uefuji et al 2003)
CaMXMT2 AB 984125 Leaf cDNA library Screening (Mizuno et al 2001)
CTS2 AB 054841 Leaf cDNA library Screening (Mizuno et al 2001)
37-methylxanthinemethyltransferase
(Caffeine synthase) (EC 211160)
CaDXMT1 AB 086414 Immature fruit cDNA Screening (Uefuji et al 2003)
CCS1 AB 0861414 Immature fruit cDNA Screening (Uefuji et al 2003)
CtCS7 AB 0864415 Immature fruit cDNA
NMT genomic clones
Theobromine synthase-1
CX10 Complete coding region PCR Satyanarayan 2006
CX8 Complete coding region PCR -do-
Theobromine synthase-1 PG-5 Promoter + coding region RAGE -do-
Theobromine synthase-1 PG-1 Promoter + coding region RAGE Satyanarayan 2006
PG-4 Promoter + coding region RAGE -do-
Theobromine synthase-1 NMT
(AY273814) Complete coding region PCR Kochko A et al 2010
Theobromine synthase-2 NMT
(AY362825) Complete coding region PCR Kochko A et al 2010
Chapter 1 Introduction and Review of Literature
16
19 Catabolism of caffeine
Caffeine is produced in young leaves and immature fruits and continues to accumulate
gradually during the maturation of these organs At the same time caffeine is slowly
degraded with the removal of the three methyl groups resulting in the formation of
xanthine Catabolism of caffeine was first reported in coffee leaves (Kalberer 1965)
Since then a number of tracer experiments using 14
C labelled purine alkaloids have
been reported (Suzuki amp Waller 1984 Ashihara et al 1996 Vitoria amp Mazzafera
1999 Mazzafera 2004) They demonstrated the major catabolic pathway is caffeine rarr
theophylline rarr 3-methylxanthine rarr xanthine Xanthine is further degraded by the
removal by the conventional purine catabolic pathway to CO2 and NH3 via uric acid
allatonin and allantoate (Figure 15) (Ashihara amp Crozier 1999 Stasolla et al 2003
Zrenner et al 2006) Caffeine catabolism usually begins with its conversion to
theophylline catalyzed by N7-demethylase The involvement of P450-dependent mono-
oxygenase activity for this reaction was suggested (Huber amp Baumann 1998
Mazzafera 2004) However the activity of this enzyme has not yet been determined in
cell free extracts [8-14
C] Theophylline degraded to CO2 at much faster rate than [8-14
C]
caffeine indicating that conversion of caffeine to theophylline is a major rate limiting
step of caffeine catabolism and a possible reason why caffeine accumulates in high
concentrations in tissues of C sinensis and C arabica It is widely believed that the
control of caffeine levels in plants is a function of the balance between rates of
synthesis and degradation and this balance seems to vary depending on the plant
species and the tissue developmental stage (Mazzafera 2004)
Chapter 1 Introduction and Review of Literature
17
Figure 15 Caffeine catabolic pathways Caffeine is mainly catabolised via theophylline and 3-
methylxanthine Xanthine is further degraded to CO2 and NH3 by the conventional
oxidative pathway Adapted from (Ashihara et al 2008)
Chapter 1 Introduction and Review of Literature
18
110 Genetic engineering of Coffee
Genetic transformation has potential applications in coffee agriculture for incorporating
genes which could provide desirable traits such as disease and insect resistance
drought and frost tolerance and herbicide resistance Transgenic technology can also be
used to increase nutritional value and improve cup quality produce varieties with
caffeine-free beans and for production of hybrid crops for molecular farming
Identification of target specific genes is one of the pre-requisites for developing
transgenic crops The availability of a large number of EST sequences in coffee and
initiation of coffee genome sequencing may speed up the gene discovery and accelerate
transgenic research efforts in coffee
1101 Insect Resistance
Production of insect resistant coffee plants is one of the major objectives of the
breeding programs The major pests attacking coffee include coffee berry borer (CBB
Hypothenemus hampei) white stem borer (WSB Xylotrechus quadripes) leaf miner
(Perileucoptera coffeela) and root nematodes (Meloidogyne spp and Pratylenchus
spp) The CBB is present in almost all the coffee growing countries and considered to
the most devastating pest in coffee To date there is no reported source of resistance to
CBB in the coffee gene pool Like CBB WSB is another serious pest in C arabica in
India and several other East Asian countries Both CBB and WSB belong to the order
Coleoptera For India controlling WSB is the biggest challenge and has therefore
become the highest research priority Robusta is generally resistant to WSB but the
interspecific Robusta Arabica hybrids are susceptible to WSB Although leaf miner is
not yet a serious pest in India and other East Asian countries it is an economically
important pest in East Africa and Brazil Effective chemical control of CBB and WSB
is difficult due to the nature of their life cycle inside the berry and stem respectively as
well as environmental concern regarding the use of pesticides Biological control
measures are adapted to combat these insect pests with varying degrees of success
Developing coffee plants resistant to these pests using genetic transformation
technology could be one of the alternative strategies to counter pest damage For insect
resistance several different classes of proteins from bacterial plant and animal sources
have been isolated and their insecticidal properties tested against many important pests
Amongst these proteins Bt toxins are most important and several transgenic crops
expressing Bt genes have been commercialized Coffee transgenic plants carrying a
Chapter 1 Introduction and Review of Literature
19
synthetic version of the cry1Ac gene have been produced (Leroy et al 2000 Mishra et
al 2002) Indeed this was the first report of an important agronomic trait being
introduced into a coffee plant The transgenic plants presented similar features in
growth and development compared to normal plants Transgenic plants highly resistant
to leaf miner under greenhouse conditions were tested under field conditions in French
Guyana for 4 years for pest resistance (Perthius et al 2005 Perthius et al 2006)
From 54 independent transformation events 70 of the events were resistant to leaf
miner Unfortunately the field trial was vandalized which led to the termination of the
experiment (Montagnon et al 2005)The effectiveness of Bt genes in controlling
coleopteran pests is well documented in corn and potato which indicates that Bt genes
might be effective against CBB The high toxicity of B thuringiensis sero var
israelensis against first in star larvae of CBB has been demonstrated (Mendez et al
2003) In another experiment an α-amylase inhibitor from Phaseolus vulgaris was
tested against CBB and found to have an inhibitory effect on its growth and
development (Grossi et al 2004)In addition to the CBB and WSB C arabica
varieties are also susceptible to endoparasitic root-knot nematode (Meloidogyne spp)
(Campos et al 1990 Mishra et al 2012)
So far 15 species have been reported to be parasites of coffee Controlling
nematodes is extremely difficult and currently seedling grafting with robusta rootstock
is followed as one of the control measure Sources of resistance specific to root-knot
nematodes have been identified in coffee trees (Bertrand et al 2001) and the Mex-1
gene conferring resistance to M exigua in C arabica is in the process of isolation (Noir
et al 2003)
1102 Tolerance to Abiotic Stress
In many coffee growing countries abiotic stress such as drought and frost are the major
climatic factors that limit coffee production Changes in climatic patterns due to global
climate change are considered increasingly important for coffee cultivation Drought
induces water stress in plants which affects vegetative growth and vigor and triggers
floral abnormalities and poor fruit set It also indirectly increases the incidence of pests
and diseases in the plants C arabica is generally more tolerant to water stress than C
canephora partly due to its extensive deep root system C racemosa is known to be a
good source of drought tolerance In India hybrids between C canephora and C
racemosa have been obtained and are currently under evaluation for drought tolerance
Chapter 1 Introduction and Review of Literature
20
In many coffee growing countries coffee is propagated in marginal areas where the
annual rainfall is below 1000thinspmm with prolonged dry spells of over 4-5 months In
those areas water shortage and unfavorable temperatures constitute major constraints
and the growth and productivity of C canephora are badly affected As with drought
periodic frost also affects coffee production in parts of Brazil The introduction of
drought and frost tolerant genes through genetic transformation would be of great
importance for alleviating these problems Research is now being carried out by several
groups to identify genes involved in biotic as well as abiotic stress The most promising
genetic engineering approach for drought tolerance includes the use of functional or
regulatory genes as well as the transfer of transcription factors In recent years plants
tolerant to high temperature and water stress have been the subject of intense research
(Chaves et al 2003 Chaves et al 2004 Coraggio et al 2004) For achieving drought
tolerance genes that have been targeted include those encoding enzymes involved in
detoxification or osmotic response metabolism enzymes active in signaling proteins
involved in the transport of metabolites and regulating the plant energy status (Chaves
et al 2003 Chaves et al 2004 Coraggio et al 2004) The dissection of molecular
mechanisms related to signal transduction and transcriptional regulation might help in
engineering drought tolerance in coffee
1103 Disease Resistance
Coffee leaf rust (CLR) caused by the fungus Hemileia vastatrix is the most important
disease in coffee with substantial loss to coffee production and productivity in all the
coffee growing countries In addition to leaf rust coffee berry disease (CBD) caused by
fungus Colletotrichum kahawae can be a devastating anthracnose leading to substantial
crop loss in Africa Several other fungal and bacterial diseases may also affect coffee
to a extent C arabica is more susceptible to many diseases than C canephora Though
most of the disease control measures rely upon chemical control they are more
expensive and labour intensive The long-term solution is the breeding of resistant
varieties which is the focus of many breeding programs However breeding for disease
resistant varieties is time consuming due to the perennial nature of coffee with its long
gestation period India has a long history of C arabica breeding especially for leaf rust
resistance and is the first country to demonstrate the existence of multiple races of leaf
rust Resistance to CLR is conditioned primarily by a number of major (SH) genes and
coffee genotypes are classified into different resistance groups based on their
Chapter 1 Introduction and Review of Literature
21
interaction with different rust races pathogen (van der Vossen 2001) Currently C
canephora provides the main source of resistance to pests and diseases including CLR
(H vastatrix) and CBD (C kahawae) and therefore used in breeding programs Other
diploid species like C liberica and C racemosa are being used as a source of resistance
to coffee leaf rust and coffee leaf miner respectively (Guerreiro et al 1999
Sreenivasan et al 1989) The development of coffee varieties resistant to major fungal
diseases such as CLR and CBD using transgenic technology will benefit the coffee
industry immensely During the last 15 years significant progress has been made in the
area of host-pathogen interactions (Staskawicz et al 1995 Feys et al 2000
Shivaprasad et al 2012) and many resistance genes involved in recognizing invading
pathogens have been identified and cloned (Takken et al 2000) A number of
signalling pathways induced because of pathogen infection have been dissected (Dong
et al 1998 Navarro et al 2006) Many antifungal compounds synthesized by plants to
combat fungal infection have been identified (Does et al 1998) Understanding the
specific induction of targeted pathways and identification of specific pathways
responsible for particular fungal resistance is important in order to employ this strategy
in transgenic technology The recent investigation of gene expression during coffee leaf
rust infection could provide an insight into the defence pathways operating in coffee
(Fernandez et al 2004 Nardi et al 2006) Efforts have been made to identify and
clone resistance genes from coffee for achieving durable resistance The genetic and
physical map of two resistance genes SH3gene conferring resistance to rust (Prakash et
al 2004) and the Ck1 gene conferring resistance to C kahawae CBD (Gichuru et al
2006) have been established These genes could be used for molecular marker assisted
breeding programs (Mishra et al 2012)
1104 Production of low-caffeine coffee
A widespread belief that the ingestion of caffeine can have adverse effects on
health resulted in the increased demand for decaffeinated coffee (Ashihara amp Crozier
2001) Several method were used to obtain decaffeinated plants by conventional
breeding techniques using low yielding varieties and mutants that accumulated
relatively low amounts of caffeine when compared to the commercially available ones
Low-caffeine and decaffeinated coffee represent around 10 of the coffee sales
around the world (Ribas et al 2006b) The industrial process for coffee decaffeination
can be expensive and affects the original flavour and aroma in coffee (David 2002)
Chapter 1 Introduction and Review of Literature
22
Transgenic coffee plants with suppressed caffeine synthesis using RNA interference
(RNAi) technology have been obtained (Ogita et al 2003 Ogita et al 2004) Specific
sequences in the 3prime untranslated region of the theobromine synthase gene (CaMXMT1)
were selected for construction of RNAi short and long fragments The caffeine and
theobromine content of the transgenic plants were reduced by ~ 70 when compared to
the untransformed plants In C canephora RNAi technology has also been employed
to silence the N-methyl transferase gene involved in caffeine biosynthesis (Vinod et al
2007) Promoter of an N-methyltransferase (NMT) gene involved in caffeine
biosynthesis was cloned (Satyanarayana et al 2005 Satyanarayana 2006) which will
be very useful for studying the regulation of caffeine biosynthesis
1105 Improvement in cup quality
Improvement in coffee cup quality requires elaborate knowledge of the chemical
constituents as well as the metabolic pathways involved in the elaboration of quality
The constituents of coffee beans include minerals proteins carbohydrates caffeine
chlorogenic acids (CGA) glycosides lipids and many volatile compounds that give
flavour to coffee by roasting Among these the role of three major constituents
sucrose CGA and trigonelline have been studied in coffee The sucrose content of
coffee bean is associated with coffee flavour the higher the sucrose content in green
beans the more intense will be the cup flavour (Clifford et al 1985 de Maria et al
1996) The sucrose content of C arabica (82-83) is higher than C canephora (33ndash
40) The sucrose amino acids ratio in green beans determines the profile of volatile
compounds Manipulating sucrose content in coffee bean is therefore important in
improving cup quality The sucrose synthase gene (CcSUS2) from C canephora has
been cloned and sequenced (Leroy et al 2005) This provides an opportunity to
manipulate the sucrose content in coffee Chlorogenic acids are products of
phenylpropanoid metabolism Chlorogenic acids are a group of hydroxycinnamoyl
quinic acids (HQA) formed by esterification between caffeic acids coumaric acids and
quinic acids (Clifford et al 1999) They are present in relatively large quantities in the
coffee bean and are the precursor of phenolic compounds in roasted beans C
canephora beans contain higher CGA (10) compared to C arabica beans (6-7)
CGA are known to have antioxidant properties as well as being associated with disease
resistance (Campa et al 2003) Genetic manipulations of genes involved in CGA
synthesis can serve either of these purpose by up- or down regulating the pathway
Chapter 1 Introduction and Review of Literature
23
Phenylalanine ammonia-lyase (PAL) catalyzes the first step of the phenylpropanoid
pathway leading to the synthesis of a wide range of chemical compounds including
flavonoids coumarins hydroxycinnamoyl esters and lignins (Hahlbrock amp Scheel
1989) Recently the full length cDNA and corresponding genomic sequences of PAL
from C canephora was isolated characterized and functionally validated (Mahesh et
al 2006 Parvatam et al 2006) This has opened up new possibilities for manipulating
the level of the PAL enzyme in coffee which in turn can be useful for improvement of
cup quality and manipulating antioxidant properties in coffee
1106 Fruit Ripening
Uniformity during fruit ripening is decisively related to cup quality in coffee and
consequently to the value of the product Fruits at the ideal ripening stage produce the
best organoleptic characteristics for coffee The presence of over ripened or green fruits
changes the acidity the bitterness and consequently the cup quality In order to
maximize uniform ripening of coffee fruits it is essential to control the action of genes
involved in the last step of maturation process Ethylene is known to trigger ripening
and increasing ethylene biosynthesis is associated with various stages of ripening
process (Protasio et al 2005) To control coffee fruit maturation two of the major
genes involved in ethylene biosynthesis namely ACC synthase and ACC oxidase
have been cloned (Protasio et al 2005 Neupane et al 1999) Introduction of the ACC
oxidase gene in antisense orientation have been achieved in both C arabica and C
canephora The effect of the transgene on ethylene production and fruit maturation has
yet to be reported The inhibition of genes downstream to the initial ethylene burst is
also an option to control coffee fruit maturation (Ribas et al 2006)
111 RNA interferenceRNAi
The regulation of gene expression at post transcriptional level has recently attracted
much attention because of the discovery of the phenomenon called RNA interference
(RNAi) RNAi based silencing techniques are more efficient than antisense mediated
gene silencing (Wesley et al 2001) The effect was first observed in plants wherein
silencing was observed when some copies of a transgene was in the forward orientation
with respect to the promoter and some in the reverse orientation This resulted in the
formation of double stranded RNA in the system The phenomenon in which
experimentally introduced double stranded RNA (dsRNA) leads to the loss of
expression of the corresponding cellular gene by sequence specific RNA degradation
Chapter 1 Introduction and Review of Literature
24
was termed as RNA interference or RNAi The protective effect of RNA silencing in
reducing virus infectivity supports the view that PTGS has evolved as a mechanism to
defend plants against virus infection and also to moderate the possible deleterious
genome-restructuring (insertional) activity of virus-like mobile genetic elements eg
retrotransposons A growing body of biochemical and genetic data has further
demonstrated that plants animals and yeasts share related mechanisms of specific
degradation of RNAs in which double-stranded forms of RNA are initiator molecules
(Brenstein et al 2001)
The key insight into the process of PGTS was provided by the experiments
conducted by Hamilton amp Baulcombe (1999) who identified the product of RNA
degradation as small RNA (siRNA) species of ~25nt of both sense and antisense
polarity SiRNA are formed and accumulate as double stranded RNA molecules of
defined chemical structures Both genetic and biochemical approaches were undertaken
to understand the basis of silencing Genetic screens were carried out in fungus
Neurospora carassa the alga Chalmydomonas reinhardtii and plant Arabidopsis
thaliana to search for detective mutants in PTGS
A combination of results obtained from several in vivo and in vitro experiments
have gelled into a two step mechanistic model for RNAiPTGS The first step referred
to as the RNAi initiating step involves binding of the RNA nucleases to a larges
dsRNA and its cleavage into discrete ~21-25nt RNA fragments This is ATP-
dependent reaction which involved RNase III - type endonucleases which make
staggered cuts in both strands of the dsRNA leaving 3prime overhang of 2 nucleotides with
5prime- phosphate 3prime-hydroxyl and no modification in the sugar phosphate backbone
(Hamilton amp Baulcombe 1999 Elbashir et al 2001) SiRNAs are the direct products
of dsRNA cleavage by the multi-domain RNase III enzyme DICER (Figure 16)
(Brenstein et al 2001 Karthikeyan et l 2013)
In the second step or commonly known as the effector step the double stranded
siRNAs produced in the first step bind to an RNAi specific protein complex to form a
RISC The complex undergoes activation in the presence of ATP so that the antisense
component of the unwound siRNA becomes exposed and allows the RISC to perform
the downstream RNAi reaction
Chapter 1 Introduction and Review of Literature
25
Several enzymes are involved in RNAi based on their role genes that encode for them
are classified into RNAi initiators RNAi effectors RNA dependent RNA polymerases
and silencing genes Two C elegans genes rdeI and rde4 (rde stands for lsquo RNAi
deficientrsquo) are believed to be involved in the initiation step of RNAi The C elegans
rde1 gene is a member of a large family of genes and is homologous to the Neurospora
qde2 (qde stands for ldquoquelling deficientrdquo) and Arabidopsis AGO1 (AGO stands for
agronaute AGO1 was previously identified to be involved in Arabidopsis
development) Although the functions of these genes are not clear a mammalian
member of RDE1 family has been identified as a translation initiation factor (Thakur
2005) Important genes for the effector step of PTGS in C elegans are rde2 and mut7
genes These genes were identified from heterozygous mutant worms that were unable
to transmit RNAi to their homozygous offsprings Worms with mutated rde2 or mut 7
genes show defective RNAi mut-7 gene encodes a protein with homology to the
nuclease domains of RNAase D and a protein implicated in Werner syndrome (a rapid
ageing disease in humans) (Grishok et al 2001 Kamath-Loeb et al 2012s)
Neurospora qde-1 Arabidopsis SDE-1SGS-2 and C elegans ego-1 appear to encode
RNA dependent RNA polymerase (RdRPs) It assumed that this is a proof that an RdRp
activity is required for RNAi Certainly the existence of an RdRp might explain the
remarkable efficiency of dsRNA induced silencing if it amplifies either the dsRNA
prior to cleavage or the siRNAs directly (Muers 2013) In C elegans ego-1 mutants
(ego stands for lsquoenhancer of glp-1) RNAi functions normally in somatic cells but is
defective in germline cells where ego-1 is primarily expressed In Arabidopsis SDE-
1SGS-2 mutants (SGS stands for suppressor of gene silencing) siRNA are produced
when dsRNA is introduced via an endogenously replicating RNA virus but not
introduced by a transgene It has been proposed that perhaps the viral RdRP is
substituting for the Arabidopsis enzyme in these mutants Random degradative PCR
model suggests that an RdRP uses the guide strand of an siRNA as a primer for the
target mRNA generating a dsRNA substrate for Dicer and thus more siRNAs
Several genes controlling RNA silencing in plants have been identified through
genetic screens of Arabidopsis mutants impaired in transgene induced RNA silencing
They encode a putative RNA-dependent RNA polymerase (SGS2SDE1) a coiled
protein (SGS3) a protein containing PAZ and Piwi domains (AGO1) and an RNA
helicase (SDE3) The putative SGS2SDE1 of Neurospora is related to QDE-1 and
Chapter 1 Introduction and Review of Literature
26
EGO-1 of C elegans whereas PAZPiwi protein AGO is related to QDE-2 of
Neurospora RDE-1of C elegans RNA helicase SDE3 is related to SMG-2 of C
elegans and Mut-6 of Chlamydomonas MUT-7 gene of C elegans encodes a protein
similar to Rnase D whereas the Drosophila DICER gene encode a protein similar to
RNase III An Arabidopsis ortholog of DICER gene has been identified called CAF
SIN1 SUS1(Thakur 2005 Poethig 2009)
Since the RNAi machinery is present constitutively within the eukaryotic cell it
is important to explore the metabolic advantages that are accorded by the RNAi related
proteins during the intrinsic normal growth of cells and development of organisms The
natural RNAi machinery not only keeps the mobile transposable elements from
disrupting the integrity of the genomes as suggested by the analysis of model organisms
like A thaliana C elegans D melanogaster (Tabara et al 1999 Wu-Scharf et al
2000 Aravin et al 2001 Hamilton et al 2002 Lisch 2009) but also participates in
development of organisms Genetic defects in Celegans RNAi genes ego1 and dicer
cause known specific developmental errors (Grishok et al 2000 Grishok et al 2001
Knight amp Bass 2001 Hall et al 2013) Similarly the Argonaute family of genes of A
thaliana (especially the ZWILLE proteins) is also responsible for plant architecture and
meristem development (Carmell et al 2002 Hamant and Pautot 2010) and the Dicer
homologue of A thaliana CAF1 is required for embryo development (Golden et al
2002 Nakano et al 2011) Thus genetic evidence illustrates the role of the RNAi
machinery as a controller of development related genes The mechanistic details of
these developmental processes are beginning to emerge
Chapter 1 Introduction and Review of Literature
27
Figure 16 Mechanism of gene silencing induced double stranded RNA The dicer enzyme to
produce siRNAs cleaves the RNA The siRNA generated bind to the nuclease complex
RISC The antisense component in the siRNA in the RISC guides the complex towards
the cognate mRNA resulting in endonucleolytic cleavage of the mRNA
Chapter 1 Introduction and Review of Literature
28
112 MicroRNA or MiRNA
In 1991 Ambros and co-workers first isolated a lin-4 mutant of Celegans which was
arrested at the first larval stage (Lee et al 1993) Later on the let-7 mutation was
isolated in the same system which was responsible for development through the fourth
larval stage Both lin-4 and let-7 encode short 22-nucleotide mature RNAs and were
called short temporal RNA because they control the temporal development program of
C elegans The mature lin-4 RNA defines (negatively regulates) the mRNA expression
of the lin-14 and lin-28 hetrochronic genes with the antisense mediated repression
mechanism of translation initiation and thus specifies the fate of cells during the first
three larval stages Recent studies have revealed that the short temporal RNAs are
actually members of a group of tiny RNAs (21to 28 nucleotides) called the micro-
RNAs (miRNA) (Bartel 2009) isolated members of which could easily run to a few
thousand which includes both those were identified computationally and by deep
sequencing Some of the components of the RNAi machinery have also been clearly
established as the effector proteins for the maturation of miRNAs
113 MicroRNAs in plants
1131 Discovery
MicroRNAs have been discovered by three basic approaches Direct cloning forward
genetic direct cloning and bioinformatic prediction followed by experimental
validation The most direct method of miRNA discovery is to isolate and clone small
RNAs from biological samples and several groups have used this approach to identify
plant miRNAs (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002 Xie et al 2003 Sunkar et al 2005 Williams et al 2013) The cloning methods
adapted were similar to those used to identify large numbers of animal miRNAs
(Lagos-Quintana et al 2001 Lau et al 2001 Aravin amp Tuschl 2005) which involve
isolating small RNAs followed by oligonucleotide adapter ligation reverse
transcription amplification and sequencing Some protocols incorporate methods to
enrich for Dicer cleavage products (ie molecules with 5prime-phosphates and 3prime-
hydroxyls) and to concatemerize the short cDNAs so that several may be identified in a
single sequencing read (Lau et al 2001 Llave et al 2002a Jagadeeswaran amp Sunkar
2013)The initial cloning experiments in Arabidopsis identified 19 miRNAs belonging
to 15 families (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002) although hundreds of other small RNAs such as siRNAs and RNA degradation
Chapter 1 Introduction and Review of Literature
29
fragments were also cloned through the direct cloning methods To select miRNAs
from the pool of small RNAs secondary structures of the RNA molecules were
examined in compliance with the acknowledged structural features of miRNAs
(Ambros et al 2003) The secondary structures were validated using several ad-hoc
software tools such as the Mfold or RNAfold programs (Reeder et al 2006) Finally
the existence of mature miRNAs was determined by RNA gel blot hybridization These
direct cloning methods were successful in isolating many miRNAs in Arabidopsis
(Reinhart et al 2002 Sunkar amp Zhu 2004) Rice (Sunkar et al 2005) Poplar (Lu et
al 2005) moss (Arazi et al 2005) and Tobacco (Billoud et al 2005)
Although miRNAs were first discovered through forward genetic screens in
worms (Lee et al 1993 Reinhart et al 2000)only few A thaliana miRNA gene
families have been discovered by this method (Chen 2008) in plants MiRNA
involvement in plant mutant phenotypes were not properly inferred till the cloning
experiments established that plant genomes contain numerous miRNAs (Park et al
2002 Reinhart et al 2002 Rhoades et al 2002) MiRNA loss-of-function allele has
been identified by forward genetic screens early extra petala1 is caused by a
transposon insertion ~160bp upstream of the predicted miR164c stem loop resulting in
flowers with extra petals (Baker et al 2005 Irish 2008) The fact that loss-of-function
miRNA mutants have been rarely discovered using forward genetics reflects small
target size for mutagenesis coupled with redundancy in nearly all evolutionarily
conserved plant miRNAs which are encoded by gene families (Table16) Family
members are likely to have overlapping functions buffering against loss at any single
miRNA locus Over expression screens can circumvent redundancy limitations At least
four plant miRNAs miR319 (also known as miR-JAW) miR172 (also known as EAT)
and miR166 were isolated in over expression screens for dominant mutants with
developmental abnormalities (Aukerman amp Sakai 2003 Palatnik et al 2003 Kim et
al 2005 Williams et al 2005b Schommer et al 2012) Mutations in miRNA target
sites which can prevent the entire family of miRNAs from repressing a target gene can
also circumvent redundant functions of miRNA family members
The dominant mutations in the HD-ZIP genes PHB PHV and REVOLUTA
(REV) in Arabidopsis and ROLLEDLEAF1 (RLD1) in Maize result in adaxialization of
leaves andor Vasculature (McConnell amp Barton 1998 McConnell et al 2001 Nelson
et al 2002 Zhong amp Ye 2004 Allen amp Millar 2012) and are all caused by mutations
Chapter 1 Introduction and Review of Literature
30
in miR166 complementary sites (Rhoades et al 2002 Emery et al 2003 Jones-
Rhoades amp Bartel 2004 Mallory et al 2004 Zhong amp Ye 2004)
In both plants and animals cloning was the initial means of large-scale
miRNA discovery (Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Reinhart et al 2002 Mendes et al 2012) However cloning is biased toward
RNAs that are expressed highly and broadly MiRNAs expressed at low levels or
only in specific cell types or in response to certain environmental stimuli were more
difficult to clone Sequence based biases in cloning procedures might also cause
certain miRNAs to be missed Because of these limitations bioinformatics approaches
to identify miRNAs have provided a useful complement to cloning
A straightforward use of bioinformatics has been carried out to find homologs
of known miRNAs both within the same genome and in the genomes of other species
(Pasquinelli et al 2000 Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Mendes et al 2009) A more difficult challenge is to identify miRNAs
unrelated to previously known miRNAs This was first accomplished for
vertebrate nematode and fly miRNAs using algorithms that search for conservation
of sequence and secondary structure (ie miRNA stem-loop precursors) between
species searching for patterns that are characteristic of miRNAs (Lai et al 2003 Lim
et al 2003 Lim et al 2005)
Most of the methods identified numerous miRNAs but are necessarily more
relaxed in terms of allowed structures Some approaches take advantage of the high
complementarity of plant miRNAs to target messages implementing the requirement
that the candidate has conserved complementarity to mRNAs (Jones-Rhoades amp Bartel
2004 Adai et al 2005) This additional filter has been useful for distinguishing
authentic plant miRNAs from false positives which later extended to mammalian
miRNA gene prediction (Xie et al 2005 Mendes et al 2012)
Another criteria used by most algorithms in the filters is evolutionary
conservation In plants mature miRNA sequences are conserved to a higher degree
than their precursor sequences For example Arabidopsis and rice diverged more than
130 million years ago (Friis et al 2004) but some miRNAs are highly conserved
in these plant species (Reinhart et al 2002) Furthermore various parameters for the
secondary structures such as free energy the number of paired residues within a
Chapter 1 Introduction and Review of Literature
31
miRNA or the number and size of bulges are also used to validate the predictions
(Jones-Rhoades amp Bartel 2004 Wang et al 2004 Adai et al 2005 Mendes et al
2012)
1132 Conserved microRNAs in Plants
Till date cloning genetics and bioinformatics screens have resulted in annotation of
approximately 184 potential Arabidopsis miRNAs (miRBase release 13) These
miRNAs seem to arise through gene duplication and subsequent diversification and
represent approximately 100 families (Maher et al 2006 Axtell 2013) Twenty one
families represented by 92 genes are clearly conserved in species beyond Arabidopsis
(Table 15) The presence of these miRNA families in other sequenced plant genomes
indicates that different plant species have an evolutionarily fluid set of miRNAs
(Willmann amp Poethig 2007) Recent studies on mosses one of the most ancient land
plants have shown that at least 14 miRNA families are conserved in eudicots and
mosses and therefore it appears that plant miRNAs share a common ancient origin
(Arazi et al 2005 Axtell amp Bartel 2005 Talmor-Neiman et al 2006 Zhang et al
2006 Axtell 2013) The number of members per family in a genome ranges from 1-32
with the exception of miR-430 which is represented by a cluster of ~80 loci in zebra
fish (Giraldez et al 2005)
In plants the number of members in each family correlates among examined
species certain families contain numerous members in all three species (eg miR156
miR166 miR169) whereas others consistently contain fewer genes (eg miR162
mir168 miR394) (Table 16) Although it is still unclear why certain miRNA are
encoded by more than one gene in plants (eg 12 genes encoding miR156) this
correlation might suggest functional significance of various miRNA family sizes Most
annotated plant miRNAs are conserved throughout flowering plants while a few
miRNAs are found only in a single sequenced genome and thus they could be
evolutionarily lsquoyoungrsquo miRNAs For example Arabidopsis has several miRNA
families that are not found in poplar or rice suggesting that miRNAs could be
generated continuously during evolution (Allen et al 2004a Rajagopalan et al
2006 Fahlgren et al 2007 Axtell 2012 de Alba et al 2013) Consistent with the
notion that non conserved miRNAs are of a more recent evolutionary origin these
miRNAs are mainly encoded by a single locus in the genome Within each family the
mature miRNA is always located in the same arm
Chapter 1 Introduction and Review of Literature
32
Table 16 MicroRNA families conserved in plants
miRNA
family
Arabidopsis Oryza Populus
miR156 12 12 11 miR159319 6 8 15 miR160 3 6 8 miR162 2 2 3 miR164 3 5 6 miR166 9 12 17 miR167 4 9 8 miR168 2 2 2 miR169 14 17 32 miR171 4 7 10 miR172 5 3 9 miR390 3 1 4 miR393 2 2 4 miR394 2 1 2 miR395 6 19 10 miR396 2 5 7 miR397 2 2 3 miR398 3 2 3 miR399 6 11 12 miR408 1 1 1 miR403 1 0 2 miR437 0 1 0 miR444 0 1 0 miR445 0 9 0 Total 92 127 169
All known miRNA families that are conserved between more than one plant species are listed
together with the number of genes identified in the sequenced genomes Rice miRNA families that have orthologs in maize but do not appear to have orthologs in the eudicots (Arabidopsis and Populus) are marked with an asterisk The following families contain miRNA genes annotated with
more than one number miR156 (miR156 and miR157) miR159319 (miR159 and miR319) miR166 (miR165 and miR166) miR171 (miR170 and miR171) and miR390 (miR390 and miR391)
of the stem loop (5prime- or 3prime) as it would be expected if the genes carry common ancestry
(Figure 17) The sequence of the mature miRNA and to some extent the segment on
the opposite arm of the hairpin to which it pairs is highly conserved between the
members of the same family (both within and between the species) while the sequence
secondary structure and length of the intervening ldquolooprdquo region can be highly divergent
within the same family
The patterns of paring and non pairing nucleotides are often conserved between
homologous miRNA stem loops from different species (Figure 17) The significance of
conserved mismatches is opined to guide DCL1 to cleave at the appropriate positions
along the stem loop
Chapter 1 Introduction and Review of Literature
33
Figure 17 Representative stem loop of miR164 Segments corresponding to the mature miRNAs
are shown in red (Adapted from Rajagopalan et al 2006)
Although many miRNAs are highly conserved in plants the degree of conservation in
most cases is quite low between plants and animals one exception is miR854 A recent
study has revealed that miR854 is conserved between Arabidopsis and animals and that
the Arabidopsis miR854 differs from the human miR854 only by a single nucleotide
(Arteaga-Vazquez et al 2006 Axtell 2012 de Alba et al 2013) The Arabidopsis
miR854 has multiple binding sites in the 3prime-untranslated region (UTR) of
OLIGOURIDYLATE-binding-PROTEIN mRNA 1bA (UBP1b) and represses the
translation of the UBP1b mRNA The animal miR854 is also complementary to a site in
the 3prime-UTR of the UBP1b homolog in animals However it is currently unclear how the
animal miR854 regulates its target mRNA Another distinction between plant and
animal miRNAs is the genomic organization of the miRNA genes While plant miRNA
genes are located predominantly in the intergenic regions animal miRNA genes tend to
localize in the intragenic regions both in the introns or exons of protein coding genes
Moreover miRNA genes are often clustered within a single precursor transcript
whereas individual plant miRNAs are derived from individual precursor RNAs (Bartel
Chapter 1 Introduction and Review of Literature
34
2004 Bonnet et al 2004 Kim 2005 Vaucheret et al 2006 Marco et al 2013)
1133 Non-conserved miRNA and challenges in annotation
Most miRNAs are conserved throughout flowering plants (Table 15) but many more
were found only in a single sequenced genome thus considered to be of a more recent
evolutionary origin The extended homology between few non conserved miRNAs
precursors and their target genes provides strong evidence that these relatively ldquoYoungrdquo
miRNAs arose from the duplication of the target gene segments (Allen et al 2004)
Several non-conserved miRNAs including miR161 miR163 miR173 miR447
miR475 and miR476 are known to direct cleavage of target transcripts (Allen et al
2004a Adai et al 2005 Sunkar et al 2005 de Alba et al 2013) It is difficult to
confidently predict targets for many because it is not possible to use complementary
site as a filter against false-positive target predictions It is also difficult to
confidently annotate non-conserved miRNAs as miRNA rather than siRNAs The
established minimal standard for miRNA annotation is a small RNA with detectable
expression and the potential to form a stem-loop when joined to flanking genomic
sequence (Kasschau et al 2003) In practice these requirements are too loose to
definitively categorize many small RNAs cloned from plants Many plant siRNAs are
detectable on blots (Vazquez et al 2004b) and hundreds of thousands of non-
miRNA genomic sequences can be predicted to fold into secondary structures that
resemble structures of plant miRNA precursors (Jones-Rhoades amp Bartel 2004)
Therefore without conservation of both sequence and secondary structure it is
difficult to be confident that a given cloned RNA originated from a stem-loop (ie is a
miRNA) rather than from a double-stranded RNA (ie is a siRNA) In fact many of
the thousands o f small RNAs cloned from Arabidopsis (Gustafson et al 2005) would
probably meet the literal requirements for annotation as miRNAs A few of these
sequences might be miRNAs but others that meet the literal criteria probably are not
so The challenges of annotating non conserved small RNAs were evidenced by
three related small silencing RNAs that were originally annotated as miRNAs
that turned out to be trans-acting siRNAs (tasiRNAs)(Kanno et al2013)
Although most miRNAs that are broadly conserved among flowering plants are
now being identified and experimentally validated (Table 16)(Jones-Rhoades amp
Bartel 2004) the challenges and ambiguities for classifying non-conserved miRNAs
prevent meaningful estimates of the total number of miRNA genes in Arabidopsis and
Chapter 1 Introduction and Review of Literature
35
other plant genomes It is possible that miRNAs might have escaped detection
because they are non-conserved and expressed in specific tissues or conditions and can
only be identified by using high- throughput sequencing
1134 MicroRNA biogenesis
11341 Transcription of microRNA precursor
Plant miRNAs are primarily found in the genomic regions that are not associated with
the protein coding genes (Reinhart et al 2002) and it appears that most if not all plant
miRNAs are produced from their own transcriptional units Plant miRNA genes are
occasionally clustered near each other in the genome suggesting transcription of
multiple miRNAs from a single primary transcript [eg the miR395 cluster (Grishok
et al 2001 Jones-Rhoades amp Bartel 2004b)] but this polycistronic arrangement of
miRNA genes appears far less frequently in plants than in animals (Bartel 2004)
Northern blot EST and mapping evidence indicate that plant miRNA primary
transcripts (also known as pri-miRNAs) as in animals are longer than needed to
encompass the miRNA stem-loops (Palatnik et al 2003 Jones-Rhoades amp Bartel
2004b) At least some of these pri-miRNA transcripts appear to be spliced
polyadenylated (Kurihara amp Watanabe 2004) and capped (Xie Z et al 2005) Two
rice miRNAs are contained within transcripts that contain exon junctions within the
presumptive stem-loop precursor implying that in these cases splicing is a prerequisite
for Dicer recognition (Sunkar et al 2005) Plant pri-miRNAs are over 1kb in length
and they are usually preceded by typical TATA box motifs They can also undergo
canonical splicing polyadenylation and capping All these observations indicates RNA
polymerase II is probably responsible for transcribing most plant miRNAs (Xie Z et
al 2005) as appears to be the case for many animal miRNAs Relatively little is
known about the regulation of miRNA transcription in plants but there is no reason
to suspect that this regulation would differ from that of protein-coding transcripts as
the bioinformatics analysis of the sequence upstream of the transcriptional start
sites of the miRNA genes showed the presence of putative binding motifs of
number of known transcription factors (Megraw et al 2006 Sethupathy 2013)
11342 MicroRNA processing and export
In plants maturation of miRNA is a step wise process A miRNA gene is transcribed to
pri-miRNA which is much longer than the pre-miRNA possessing the characteristic
stem-loop structure Like transcription of most protein-coding genes the miRNA
Chapter 1 Introduction and Review of Literature
36
transcription is processed by RNA polymerase II (Pol II) enzymes and the pri-
miRNAs are 5prime-capped and 3prime-polyadenylated (Lee et al 2004 Xie Z et al 2005)
Mature miRNAs are produced from pri-miRNAs through at least two sequential
processing steps by RNase III-type endonucleases In animals pri-miRNAs are first
processed at the bottom portion of the stem by Drosha a nuclear-localized RNase III
enzyme to liberate pre-miRNAs The pre-miRNAs are then exported to the cytoplasm
with the help of exportin-5The pre-miRNAs are further processed by Dicer another
RNaseIII enzyme to a duplex consisting of the miRNA and the antisense strands
(miRNA) Since plants do not have any Drosha homologs the two sequential
processing steps seem to be carried out by DCL1 a Dicer homolog in the nucleus
(Kurihara amp Watanabe 2004 de Alba et al 2013 Rogers amp Chen 2013)
The processing of pri-miRNAs to pre-miRNAs by DCL1 is assisted by two
other proteins HYPONASTY LEAVES1 (HYL1) and SERRATE (SE) HYL1 belongs to a
family of double-stranded RNA (dsRNA) binding protein and interacts with DCL1 in
vitro and in vivo (Lu amp Fedoroff 2000 Hiraguri et al 2005 de Alba et al 2013
Rogers amp Chen 2013) Loss-of-function mutations in the HYL1 gene result in
decreased accumulation of miRNAs and concomitant accumulation of pri-miRNAs
(Han et al 2004 Vazquez et al 2004a Kurihara et al 2006) SE a C2H2 zinc finger
protein interacts with DCL1 and HYL1 and plays a role in the processing of pri-
miRNAs to pre-miRNAs (Lobbes et al 2006 Yang L et al 2006) In addition the
nuclear cap-binding complex proteins CBP20 and CBP80 that are known as ABA
HYPER SENSITIVE 1(ABH1) have been shown to be required for proper miRNA
processing (Gregory et al 2008 Laubinger et al 2008) Recently it has been
demonstrated that DCL1 can release 21-nt RNAs from dsRNA as well as from
synthetic miR167 precursor RNAs in vitro However this cleavage is error prone and
inefficient in the absence of HYL1 and SE which accelerate the rate of DCL1-mediated
cleavage and promote accurate processing of miRNAs (Dong et al 2008 Rogers amp
Chen 2013)
11343 Methylation of the miRNAmiRNAduplex
After the miRNAmiRNAduplexes are released from the pre-miRNAs by the DCL1
activities the duplexes are methylated at the 2primeOH of the3prime-ends by HUA ENHANCER1
(HEN1) a small RNA methyltransferase (Yu et al 2005 Yang Z et al 2006 Rogers
amp Chen 2013 ) This step which is distinct from the RNase III-mediated processing
Chapter 1 Introduction and Review of Literature
37
step distinguishes the biogenesis of plant miRNAs from that of animal miRNAs
Mutations in the HEN1 gene were first isolated in a morphological screening for floral
development although the HEN1 mutants display pleiotropic phenotypes similar to the
developmental defects observed in the DCL1 mutants (Chen et al 2002 Park et al
2002 Rogers amp Chen 2013)
Figure 18 Biogenesis of microRNA
A Biogenesis of plant MicroRNA B Biogenesis of metazoananimal MicroRNA
The miRNA (red) is incorporated into RISC and the miRNA(blue) in degraded
This enzyme has two dsRNA-binding domains and nuclear localization signal It
is also conserved in fungi and is required for the processing of siRNAs as well as of
miRNAs (Park et al 2002 Boutet et al 2003 Xie et al 2003) Although the HEN1
protein is localized to the nucleus its methylation activity in the cytoplasm cannot be
ignored (Xie et al 2004 Rogers amp Chen 2013)
Chapter 1 Introduction and Review of Literature
38
The methylation of the miRNAmiRNA duplex is required to protect miRNAs
against the 3prime-end uridylation activity and subsequent degradation (Li et al 2005) In
the hen1 mutants accumulation of miRNAs is reduced to a lower level Also the
miRNAs appear to be heterogeneous in their sizes such that a ladder of bands rather
than a single species is observed for most miRNAs in the mutants (Li et al 2005)
MiRNA cloning and sequencing analyses revealed the presence of a number of U
residues at the 3prime-end of miRNAs in the hen1 mutants Moreover these nucleotides do
not correspond to the miRNAs in the pre-miRNAs indicating that these nucleotides are
added after the processing of the miRNAs from pre-miRNAs (Li et al 2005)
Uridylation of RNAs has also been proven to be correlated with RNA decay (Shen amp
Goodman 2004 Mullen amp Marzluff 2008) suggesting that uridylation attracts an
exonuclease which degrades miRNAs in plants
11344 MiRNA incorporation in to the RISC and its nuclear export
The methylated miRNAmiRNA duplexes undergo RISC assembly In this process
the miRNA of the duplexes is selectively incorporated into the RISC and the miRNA
appears to be removed and subsequently degraded (Hammond et al 2000 Hutvagner
amp Zamore 2002 Schwarz et al 2003 de Alba et al 2013 Rogers amp Chen 2013)
The ARGONAUTE 1(AGO1) proteins are core components of the RISC complex
They contain two structural domains the N-terminal PAZ domain that binds to the 3prime-
end of single-stranded RNAs and the C-terminal piwi domains that has a structure
similar to that of RNase H (Cerutti et al 2000 Parker et al 2004) Several AGO
proteins possess endonucleolytic activities within the PIWI domain and cleave target
mRNAs in the middle of the site complementary to miRNA (Liu et al 2004 Meister et
al 2004 Miyoshi et al 2005) Mutations in the AGO1 gene cause miRNA target
genes to be ectopically expressed and the levels of some miRNAs are reduced in the
mutants (Kidner amp Martienssen 2004 Vaucheret et al 2004 Kidner amp Martienssen
2005 Ronemus et al 2006) Moreover the phenotype of the AGO1 hypomorphic
alleles display defects in lateral organ polarity and leaf and flower morphology as
observed in the dcl1 mutants and null AGO1 alleles are embryonically lethal
supporting the close relationship between the AGO proteins and miRNA processing
(Kidner amp Martienssen 2004 Vaucheret et al 2004 de Alba et al 2013 Rogers amp
Chen 2013)
The production of miRNA miRNA duplexes occur in the nucleus while
Chapter 1 Introduction and Review of Literature
39
miRNA-RISC complexes are present in the cytoplasm where target mRNAs are
located This discordance of functional sites requires an export step during miRNA
biogenesis HASTY the Arabidopsis homolog of exportin-5 is thought to be involved
in miRNA nuclear export (Bollman et al 2003 Park et al 2005)The hasty mutant
exhibits pleiotropic defects in plant development and reduced accumulation of most
miRNAs as in the DCL1 or AGO1 mutants Howeverit is unclear whether the HASTY
proteins export the miRNA miRNA duplexes or the miRNA-RISC complexes in
plants
11345 Feedback regulation of miRNA biogenesis
MiRNA biogenesis is under feedback regulation such that the DCL1 and AGO1 genes
are themselves regulated by miRNAs DCL1 is itself a target of miR162 which leads to
the cleavage of the DCL1 mRNA (Xie et al 2003 de Alba et al 2013) Consistent
with this the levels of DCL1 mRNA are elevated in the mutants of miRNA processing
genes in which the miR162 abundance is reduced In addition there is a predicted
hairpin structure of miR838 in the 14th
intron of the DCL1 primary transcript
(Rajagopalan et al 2006) This prediction implies that DCL1-mediated processing
on this site can result in the cleavage of the DCL1 primary transcript into two truncated
fragments the N-terminal capped fragment and the C-terminal polyadenylated
fragment If this is the case it is possible that miRNA biogenesis competes with the
splicing event of the DCL1 pre-mRNA
The AGO1 is also regulated by miRNA There is a complementary site for
miR168 binding in the AGO1 mRNA and the transcript level of the AGO1 mRNA is
reduced through an mRNA cleavage mechanism (Vaucheret et al 2006 de Alba et al
2013 Rogers amp Chen 2013) indicating that the AGO1 mRNA levels are tightly
controlled by the levels of the AGO1 protein
1135 Mechanism of miRNA action
11351 MiRNA-directed mRNA cleavage
By forming the miRNA-RISC assembly miRNAs can direct the RISC to regulate gene
expression by two post transcriptional mechanisms mRNA cleavage or translational
repression (Bartel 2004 Jones-Rhoades et al 2006 Dalmay 2013) The degree
of miRNA-mRNA complementarity plays an important role for the
determination of the mechanism to be used A general rule is that while perfect or
Chapter 1 Introduction and Review of Literature
40
near-perfect complementarity induces mRNA cleavage central mismatches trigger
translational repression (Carrington amp Ambros 2003 Jones-Rhoades amp Bartel 2004)
Unlike most animal miRNAs plant miRNAs have near-perfect or perfect
complementarity to their targets and mRNA cleavage is assumed to be a predominant
mechanism by which miRNAs regulate gene expression in plants
In RNA cleavage mechanism miRNAs guide the AGO component of RISC to
cleave a single phosphodiester bond of the target mRNA within the miRNA-binding
site (Llave et al 2002b Tang et al 2003) The truncated fragments are then released
and degraded freeing the RISC to recognize and cleave another target mRNA This has
been validated by the detection of 3prime cleavage products that have 5prime ends that start at the
middle of the complementary regions The mRNA cleavage products are then further
degraded by other mechanisms The 3prime cleavage products of some miRNA targets are
degraded by the 5prime-3prime exonuclease XRN4 an Arabidopsis homolog of the major Yeast
mRNA degrading exonuclease Xrn1p It has been observed that the 3prime cleavage
products of some target mRNAs were accumulated in the xrn4 mutants (Souret et al
2004 Dalmay 2013) However the 3prime cleavage products of other miRNA targets do
not accumulate in the xrn4 mutants indicating that there are also XRN4 independent
mechanisms The 5prime cleavage products are typically untraceable by RNA gel blot
hybridization but can be detected by sensitive PCR based methods It has been found
that the 5prime cleavage products tend to obtain an oligo U tail at the 3prime ends This 3prime U tail
is correlated with shortening of the 5prime cleavage products from the 5prime end leading to the
conclusion that the 3prime uridylation causes 5 prime- 3 prime exonucleolytic decay of the 5prime cleavage
products (Shen amp Goodman 2004)
DCL1 HYL1 and SE interact with each other and are co-localized in the
nuclear bodies termed Dicing bodies or D-bodies in which some pri-miRNA shave
been found suggesting that D-bodies serve as a center for active miRNA processing
(Fang amp Spector 2007 Fujioka et al 2007 Song et al 2007) The miRNA
processing site appears to be distinct from the Cajal bodies that have previously been
found as a siRNA processing site (Pontes amp Pikaard 2008) The D-bodies include
SmD3 and SmB proteins that are found in the Cajal bodies However At collin an
additional marker for the Cajal bodies is not found in the D-bodies Moreover the D-
bodies are present in an At collin mutant lacking the Cajal bodies The pri-miRNAs of
miR171A and miR159A genes have been found to be co-localized with HYL1 and
Chapter 1 Introduction and Review of Literature
41
DCL1 in the D- bodies that are distinct from the Cajal bodies (Song et al 2007)
11352 MiRNA-directed translational repression
Translation repression is another mechanism exerted by plant miRNAs The regulatory
mechanism of miR172 which regulates flowering time and floral organ identity is
consistent with translation repression MiR172 over production does not affect the
transcript levels of APETALA2 (AP2) and AP2- like target mRNAs Instead the AP2
protein level is reduced in the miR172 over expressing mutant (Aukerman amp Sakai
2003 Chen 2004 Dalmay 2013) It was later shown that miR172 can also direct the
cleavage of the target mRNAs although the steady-state mRNA level is unchanged due
to feedback regulation (Schwab et al 2005 Jung et al 2007) It is clear that miR172
regulates its target genes primarily by translational repression rather than mRNA
cleavage Recently it has been reported that miR156157 and miR854 also regulate
their target genes through translational repression (Arteaga-Vazquez et al 2006
Gandikota et al 2007 Dalmay 2013) It was once thought that translational repression
is an exception to the rule However experimental evidence suggest a widespread
coexistence of translational repression and mRNA cleavage (Brodersen et al 2008)
1136 Regulatory roles of miRNAs in plant development
The miRNA target prediction has been a difficult task in order to obtain regulatory
targets without bringing in too many false predictions Plant growth and developmental
processes regulated by miRNAs are quite diverse and enormous Furthermore plant
miRNAs work cooperatively or antagonistically to establish a balanced regulation This
notion is well consistent with the findings that mutations in the regulators of miRNA
biogenesis transport and processing such as AGO1 DCL1 HYL1 HEN1 ABH1
and HASTY cause pleiotropic phenotypes (Mallory amp Vaucheret 2006 de Alba et al
2013) To date the roles of miRNAs have been mostly deduced from obtaining miRNA
over expressing transgenic plants or gain-of-function mutants in which miRNA-
resistant target genes are ectopically expressed
11361 Phase transition
Plants pass through a series of developmental phase transitions during their life span
beginning with seed germination continuing through vegetative phase change
reproductive phase change flowering initiation and finally seed production for the next
generation Because of their importance in understanding plant developmental
Chapter 1 Introduction and Review of Literature
42
processes genes regulating developmental transitions have been extensively studied
and underlying molecular mechanisms have been explored in various plant species
Majority of researches were mainly focused on vegetative phase change and
reproductive transition in Arabidopsis and Maize (Poethig 2003 Baurle amp Dean
2006) Particularly the mutations of the genes encoding various regulators of miRNA
biogenesis and transport such as HST SE and AGO exhibit altered juvenile to adult
vegetative phase change and vegetative to reproductive change (Hunter et al 2003
Peragine et al 2004 Vazquez et al 2004a Jones-Rhoades et al 2006 de Alba et al
2013)
Over-expression of the miR156a gene causes late flowering and delays
vegetative phase change (Wu amp Poethig 2006 Gandikota et al 2007 Wang et al
2008) It has been shown that miR156 negatively regulates a group of genes encoding
SQUAMOSA PROMOTER-BINDING PROTEIN-LIKE (Lewis et al 2007 de Alba et
al 2013 Rogers amp Chen 2013) transcription factors such as SPL3 SPL4 and SPL5
that promote flowering initiation In contrast transgenic plants over expressing
miR156-resistant SPL345 genes are early flowering Interestingly the endogenous
level of miR156 is temporally regulated In the early juvenile vegetative phase the
level of miR156 is very high but decreases rapidly before the onset of the adult juvenile
phase (Wu amp Poethig 2006)
The Arabidopsis early activation tagged (eat-D) mutant exhibits early flowering
with disrupted floral structure The mutant phenotypes are caused by over expression of
the miR172a-2gene (Aukerman amp Sakai 2003) MiR172 regulates a small group of
genes encoding APETALLA2 (AP2)-like transcription factors such as TARGET OF
EAT1 (TOE1) TOE2 SCHLAFMUTZE (SMZ) and SCHNARCHZAPFEN(SNZ)
TOE1 TOE2 SMZ and SNZ have been shown to participate in their productive phase
change by repressing a floral integrator FLOWERING LOCUS T (FT)(Jung et al
2007) Consistent with this toe1-2D 35STOE2 35SSMZ and 35SSNZ plants
exhibit late flowering (Jung et al 2007) MiR172 is also regulated temporally and
highly expressed in the adult vegetative phase both in Arabidopsis and maize (Chuck et
al 2007) Because the temporal expression patterns of miR156 and miR172 are
complementary to each other it has been suggested that the two miRNAs interact with
each other in regulating phase change (Chuck et al 2009 de Alba et al 2013) In
support of this view it has been shown that miR172 is down-regulated in the
Chapter 1 Introduction and Review of Literature
43
Corngrass1(Cg1) mutant in which miR156 is overproduced (Chuck et al 2007 de
Alba et al 2013) However there is no direct evidence for the direct linkage between
miR156 and miR172 It is also envisioned that miR156 and miR172 would not be
directly related For example miR156 regulates plastochron index that is the inverse of
leaf initiation rate and thus frequently used to analyze temporal patterns of plant
development In contrast miR172 does not affect plastochron (Wang et al 2008) The
down-regulation of miR172 in the maize miR156-over producing mutants would be
due to the prolonged juvenile vegetative phase in the mutants
Another miRNA that regulates flowering initiation is miR159 It regulates
MYB33 and MYB65 which are closely related to the barley GAMY B transcription
factor that responds to gibberellic acid (GA) The level of miR159 is elevated in GA-
treated seedlings but reduced in the GA biosynthetic ga1 mutant Transgenic plants
over producing miR159 exhibit delayed floral induction under short days (Achard et
al 2004) It is been suggested that MYB33 mediates GA signal to regulate LEAFY
(LFY) These observations indicate that miR159 plays an important role in the GA
signaling pathways that regulate proper floral induction under short days (Achard et al
2004)
11362 Floral development
Arabidopsis flowers consist of four distinct organs They arise in a characteristic
pattern within concentric rings called whorls from outside to inside four sepals four
petals six stamens and the central pistil consisting of two carpels Identities of the
floral organs are determined by three classes of regulatory genes classA classB and
class C genes (Krizek amp Fletcher 2005) The A and C class genes act antagonistically
to restrict their expression domains (Bowman et al 1991)
It is notable that miR172 also regulates floral architecture in addition to its role
in the timing of flowering initiation MiR172 inhibits the translation of the AP2 mRNA
(Chen 2004) Transgenic plants overproducing miR172 not only exhibit early
flowering but also show abnormal floral development which is quite similar to those
of the class A gene mutants such as the ap2 mutants (Aukerman amp Sakai 2003 de
Alba et al 2013) When the activity of the class A genes is inactivated that of the class
C genes such as AGAMOUS (AG) is elevated As a result the miR172- overproducing
transgenic plants exhibit no petals along with homeotic transformation of the sepals to
Chapter 1 Introduction and Review of Literature
44
the carpels (Aukerman amp Sakai 2003 Chen 2004)
Interestingly miR166 also regulates floral architecture The role of miR165166
in the regulation of meristem activity is closely linked to floral meristem formation and
organization (Zhao et al 2007 Miyashima et al 2013) Floral structure is severely
disrupted in the miR165166-overproducing mutants such as men1 and jba-1D in
which miR166 is overproduced (Williams et al 2005b Jung amp Park 2007) The men1
and jba-1D mutants have smaller gynoecium and reduced carpel number In an extreme
case the jba-1D homozygous plants lack visible carpels (Williams et al 2005b)
Similar phenotypes have been observed in the miR165 overproducers (Zhao et al
2007) It has been suggested that the phenotypes are possibly caused by altered AG
expression in the floral meristem (Jung amp Park 2007 Turchi et al 2013)
Other miRNAs also affect floral development Overproduction of miRNAs
involved in auxin signaling also affects floral architecture Transgenic plants
overproducing miR167 have defects in the formation of ovule and anther with reduced
fertility (Ru et al 2006) It has been shown that miR167 targets ARF6 and ARF8 that
play a role in gynoecium and stamen development and in regulating fertility (Ru et al
2006 Wu et al 2006) These observations suggest that auxin signals are also closely
related to floral organogenesis In addition transgenic plants over-producing miR164
and the cuc1cuc2 double mutants show fused sepals and reduced petals (Laufs et al
2004 Turchi et al 2013) suggesting that miR164 is also related to floral meristem
activity and boundary specification of the meristematic region
11363 Shoot apical meristem development
Shoot apical meristem comprises self-maintaining totipotent cells The shoot apical
meristem activity affects diverse aspects of plant developmental processes including
leaf development vascular development and lateral organ polarity (Bowman et al
2002) Because miR165166 plays a primary role in meristem formation
overproduction of miR165166 and multiple knockout mutants of its target genes
exhibit pleiotropic pheonotypes (McConnell et al 2001 Emery et al 2003 Kim et al
2005 Williams et al 2005b)
The targets of miR165166 include genes encoding the homeo domain-leucine
zipper (HD-ZIP) class III transcription factors such as PHABULOSA(PHB)
PHAVOLUTA (PHV) REVOLUTA(REV) ATHB15CORONA (CNA) and ATHB8
Chapter 1 Introduction and Review of Literature
45
PHB PHV and ATHB15 have overlapping functions in controlling meristem
development (Turchi et al 2013) Indeed the phb phv athb15 triple mutants have an
enlarged shoot apical meristem Consistent with this the miR166- overproducing men1
and jba-1D mutants exhibit enlarged shoot apical meristems (Kim et al 2005
Williams et al 2005b Turchi et al 2013) In the mutants the shoot apical meristems
are multiplied and large
The development of shoot apical meristems is also regulated by the WUSCHEL
(WUS)ndashCLAVATA (CLV) signaling pathway WUS positively regulates cell
proliferation In contrast CLV3 which is expressed in stem cells limits the WUS
expression and thus inhibits cell proliferation in the SAM (Sharma et al 2003)
Consistent with this notion WUS is expressed to a higher level in jba-1D
explaining the ectopic enlargement of the SAM in the mutant (Williams et al 2005)
While WUS is closely related to miR165166 in the shoot apical meristem
development the underlying molecular mechanisms are currently unclear It has been
observed that the miR166-over producing men1 plants have an arrested meristem
development (Jung amp Park 2007) In addition the heterozygous men1+ and the
homozygous men1 plants show different expression patterns of WUS and CLV3 It
appears that miR166 regulates WUS indirectly and influences the expressional balance
between WUS and CLV3 through a negative feedback loop (Jung amp Park 2007 de
Alba et al 2013)
The phv phb athb15 triple mutant has an enlarged shoot apical meristems the
rev phb phv mutant does not have functional shoot apical meristems (Prigge et al
2005) This distinction may be explained by the fact that while miR166 targets
primarily PHB PHV and ATHB15 miR165 targets all the five HD-ZIP III genes (Zhou
et al 2007) Consistent with this transgenic plants overproducing miR166 are distinct
from those overproducing miR165 The miR165-overproducing transgenic plants lack
functional shoot apical meristem and a pin-like structure is formed at the apical region
of the mutant These phenotypes are quite similar to rev phb phv triple mutant (Zhou et
al 2007 Liu et al 2013) The phenotypic distinction may because by the difference
of the cleavage efficiencies of the target mRNAs In accordance with this notion a few
35S miR166a transgenic lines exhibit no shoot apical meristems The men1
homozygous plants also show arrested meristem development (Jung amp Park 2007)
Chapter 1 Introduction and Review of Literature
46
MiR164 cleaves the CUC1 and CUC2 mRNAs as well as the NAC1 mRNA
CUC1 and CUC2 determines the organ boundary in the meristem Phenotypes of
transgenic plants that overproduce miR164 are similar to those of the cuc1cuc2 double
mutant that exhibits embryonic developmental defects such as cup-shaped cotyledons
and fused cotyledons (Laufs et al 2004) When a miR164-resistant CUC2 is
expressed the boundary domain is enlarged CUC also plays a role in embryonic shoot
meristem formation by regulating the SHOOT MERISTEMLESS gene It was therefore
concluded that miR164-mediated cleavage of CUC1 and CUC2 mRNAs determines the
sizes of the boundary domains in the shoot apical meristem and regulates separation of
the organs by restricting cell proliferation (Laufs et al 2004 de Alba et al 2013)
11364 Leaf development
The role of miR319 has been extensively studied in leaf development A dominant
jaw-D mutant overproducing miR319 shows uneven curved leaves with enlarged size
and five TCP genes which are closely related to the CINCINNATA (CIN) gene in
Antirrhinum majus are down regulated in the mutant suggesting that miR319 mediated
regulation of the TCP transcription factor genes are important for the final step of
leaf shaping (Palatnik et al 2003 Naz et al 2013) In addition to leaf shape and size
leaf polarity is also specified by miRNA MiR166-mediated cleavage of the HD-ZIPIII
mRNAs is a well to establish leaf polarity This discovery originates from the gain-of-
function mutants such as phb-1d and phv-1d wherein point mutations in the
complementary sites to miRNA helps the mRNA to evade or resist from miRNA-
mediated cleavage (McConnell et al 2001 Emery et al 2003 Naz et al 2013)
Plant leaves have a dorso-ventral polarity consisting of the adaxial (upper
surface) and abaxial (lower surface) sides The adaxial side is closer to the shoot
apical meristem Adaxial identity is specified by functionally redundant class III HD-
ZIP family genes such as PHB PHV and REV Ectopic expression of each genes
result in adaxialized leaves Dominant phb-1d and phv-1d mutants exhibit
transformation of the abaxial to adaxial fate and have rod-shaped leaves In contrast
miR165166-overproducing plants show pin-like cotyledons radicalized leaves and
abaxialized leaves (Chitwood et al 2007 Pattanayak et al 2013)
Leaf asymmetry is determined by miR166-mediated restriction of the
expression domains of the HD-ZIPIII genes The HD-ZIPIII genes are expressed in the
Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
4
Baracoffea but this was contested by Bridson (1988) Davis amp Rakotonasolo (2003)
and Davis et al (2005)
Table 12Outline classification of Coffea and Psilanthus with synonyms (Davis amp Rakotonasolo
2003)
Coffea L Sp Pl 172 (1753)
Coffea sub gen Coffea L Type C arabica L 95 species Africa Madagascar Mascarenes
Coffea sub gen Psilanthopsis (AChev)
Psilanthopsis AChev Type Coffea kapakata
Type Paolia jasminoides Chiov (= C rhamnifolia (Chiov) Bridson)
Coffea sub gen Baracoffea
Coffea sect Baracoffea Type Coffea humbertii J-FLeroy
Eight species (including five -yet to be published) West Madagascar
[Paracoffea subgen Insulanoparacoffea J-FLeroy J Agric Trop Bot Appl 14 276 (1967)
nom nud]
Psilanthus Hookf Gen Pl 115 (1873)
Psilanthus subgen Psilanthus Hookf Type Psilanthus mannii Hookf
Two species Africa (central and western)
Psilanthus subgen Afrocoffea (Moens)
Coffea subgenAfrocoffea
Type Psilanthus lebrunianus (Germain amp Kesler) Bridsonc 18 species Africa Asia
Australasia
Coffea sectParacoffeaMiq Fl Ind Batavae 308 (1856)
Psilanthus subgen Paracoffea Type Coffea horsfieldiana Miq
Paracoffea
[Paracoffea subgenAfroparacoffea J-FLeroy J Agric Trop Bot Appl 14 276 (1967)
nom nud]
[Paracoffea subgenMelanoparacoffea J-FLeroy J Agric Trop Bot Appl 14 276 (1967)
nom nud]
13 Economics of Coffee
Coffee is the fourth most valuable traded agricultural commodity (FAO 2004) but is
the second most valuable commodity exported by the developing countries and more
than 75 million people depend on coffee for most of their livelihood (Pendergrast
2009)
India produces both Arabica coffee from C arabica and Robusta coffee from
C canephora As per the economic survey 2012-13 Coffee is planted in India in
around 388195 hectares Out of this area 52 is Ccanephora and 48 is Carabica
Chapter 1 Introduction and Review of Literature
5
Coffee is mostly grown in south India parts of Orissa W Bengal and North east
India Karnataka is the largest producer with 569 of total area under production
which accounts for 71 of the total produce The total crop harvest for 2012-2013 is
placed at 325300 tonnes (httpwww Indicoffeeorgindiacoffeephpp-
age=CoffeeData)
14 Composition of coffee beans
Navellier (1961) gave a mean composition of green coffee as Carbohydratesglucides
(58) lignin (2) lipids (13) proteins (13) ash (4) non-volatile acids (8)
trigonelline (1) and caffeine (1) The caffeine content of green coffee is relatively
limited (10-25) of dry matter and changes little with seed development (Clifford
and Kazi 1987) In coffee seeds the concentration of trigonelline is approximately
2 of dry weight (Clifford 1985 Mazzafera 1991) Mazzafera (1999) found a higher
protein content in the mature beans than in the immature beans but a lower content of
free amino acids with asparagine as the main component Flament ( 2000) estimated
the composition of coffee beans as follows
Proteins 100 (dry weight basis of green coffee)
Carbohydrates 500 -do-
Lipids 117-140 (Arabicas)
76- 95 (Robustas)
Chlorogenic acids 65 (Arabicas)
90 (Robustas)
15 Purine alkaloids structure and role
Purine alkaloids are derived from purine nucleotides (Zulak et al 2007) and found in
over hundred plant species in 13 orders in plant kingdom (Ashihara amp Crozier 1999)
Methylxanthines like caffeine (137-trimethlyxanthine) theobromine (37-methylxan-
thine) and methyluric acids are classified into purine alkaloids (Figure 11) (Table
13)(Ashihara et al 2008) These are known to occur in tea coffee and a number of
non-alcoholic beverages Caffeine was isolated from coffee (Coffea arabica) and tea
(Camellia sinensis) in 1820 (Ashihara amp Crozier 2001)
Chapter 1 Introduction and Review of Literature
6
Figure 11 Purine alkaloid xanthine and uric acid skeleton
Plants that accumulate purine alkaloid are classified into three groups based on
the type of alkaloids they produce caffeine-producing plants which include coffee
tea and mateacute (Ilex paraguariensis) theobromine ndash producing plants are represented
by cacao cocoa tea (Camellia ptilophylla) and Camellia irrawadiensis and
methyluric acid ndash producing plants consist of Coffea dewevrei Coffea liberica C
excelsa and Kucha tea (Camellia assamica)
Table 13 Purine alkaloid structures based on xanthine and uric acid skeleton
Compound Trivial name R1 R2 R3 R4 O-2 Δ2-3
Methylxanthines
Xanthine H H H
1-methylxanthine CH3 H H
3-methyl xanthine H CH3 H
7-methyl xanthine H H CH3
1-3methylxanthine Theophylline CH3 CH3 H
17-methylxanthine Paraxanthine CH3 H CH3
37-methylxanthine Theobromine H CH3 CH3
137-methylxanthine Caffeine CH3 CH3 CH3
Uric acid and methyl uric acids
Uric acid H H H H
137-trimethyluric acid CH3 CH3 CH3 H
1379-tetramethyluric
acid Theacrine CH3 CH3 CH3 CH3
O19-trimethyluric acid Liberine CH3 H H CH3 CH3 Δ2-3
O179-trimethyluric acid Methylliberine CH3 H CH3 CH3 CH3 Δ2-3
16 Role of caffeine in plants
The physiological role of endogenous purine alkaloids and related compounds in
higher plants remains undetermined There are two hypotheses about the role of the
Chapter 1 Introduction and Review of Literature
7
high concentrations of caffeine that accumulate in tea coffee and a few other plant
species The lsquoallelopathic theory or the auto toxic function theoryrsquo proposes that
caffeine in seed coats is released into the soil and inhibits the germination of seeds
around the parent plants (Anaya et al 2006)
The lsquochemical defense theoryrsquo proposes that caffeine in young leaves fruits
and flower buds acts to protect soft tissues from insect pests (Ashihara amp Crozier
2001 Ashihara et al 2008) It has been shown that spraying tomato leaves with a 1
solution of caffeine deters feeding by tobacco horn worms while treatment of
cabbage leaves and orchids with 001ndash01 solutions of caffeine acts as a neurotoxin
and kills or repels slugs and snails (Hollingsworth et al 2002) This work has now
been extended with convincing evidence for chemical defense theory has recently
been obtained with transgenic caffeine ndash producing tobacco plants (Uefuji et al 2005
Kim et al 2006)
17 Distribution of caffeine in plants
Caffeine has been found in 13 orders of the plant kingdom (Ashihara and Suzuki
2004 Anaya et al 2006) In some species the main purine alkaloid is theobromine or
methyl uric acids such as theacrine liberine and methylliberine rather than caffeine
(Ashihara amp Crozier 1999 Ashihara amp Crozier 2001)(Table 14)
Table 14 Distribution of purine alkaloids in plants
SNo Plant species Common
name Major alkaloid Plant parts
containing
alkaloids 1 Coffea arabica Arabica Caffeine Leaves Seeds
2 Coffea canephora Robusta Caffeine Leaves Seeds
3 Coffea liberica Caffeine Theacrine Liberine
Seeds Mature leaves
4 Coffea dewevrei Caffeine Theacrine Liberine
Seeds Mature leaves
5 Camellia sinensis Tea Caffeine Leaves
6 Camellia assamica Assam tea Caffeine Leaves
7 Camellia assamica var
kucha Kucha Theacrine Caffeine Leaves
8 Camellia irrawadensis Theobromine Leaves
9 Camellia ptilophylla Cocoa tea Theobromine Leaves
10 Theobroma cacao Cocoa Theobromine Seeds
11 Paullinia cupuna Guarana Caffeine Seeds
Chapter 1 Introduction and Review of Literature
8
12 Cola nitida Caffeine Seeds
13 Citrus sp Caffeine Pollen
14 Ilex paraguariensis Mate Caffeine Leaves
Adapted from (Ashihara amp Crozier 1999 Ashihara amp Crozier 2001)
18 Caffeine biosynthesis
The major biosynthetic pathway is a four step sequence consisting of three sequential
methylation and one nucleosidase reactions (Figure 12) The xanthine skeleton of
caffeine is derived from purine nucleotides The initial step in caffeine biosynthesis is
the methylation of xanthosine by a SAM- dependent N-methyltransferase In addition
to experiments with radiolabeled precursors substrate specificities of native (Kato et
al 1999) and recombinant N-methyltransferases (Kato et al 2000 Ogawa et al
2001 Mizuno et al 2003a Mizuno et al 2003b Uefuji et al 2003) strongly
suggest that the major route to caffeine is a xanthosine rarr7-methylxanthosinrarr 7-
methylxanthine rarr theobromine rarr caffeine pathway (Figure 12) Although the
information has been obtained mainly from coffee (C arabica) and tea (Camellia
sinensis) the available evidence indicates that the pathway is essentially the same in
other purine alkaloid-forming plants such as mateacute (Ashihara 1993) and cacao
(Koyama et al 2003 Yoneyama et al 2006)
The first step in the caffeine biosynthetic pathway from xanthosine is
conversion of xanthosine to 7-methylxanthosine (Figure 12) This reaction is
catalyzed by the 7-methylxanthosine synthase (xanthosine 7 N-methyltransferase EC
211158) The genes encoding for 7-methylxanthosine synthase CmXRS1 (AB
034699) and CaXMT1 (AB 048793) were isolated from the C arabica (Mizuno et
al 2003a Uefuji et al 2003) The recombinant proteins obtained from these genes
exhibit 7-methylxanthosine synthase activity in vitro Xanthosine monophosphate
(XMP) was not converted to 7-methylxanthosine by the recombinant enzymes thus
inclusion of 7-methy-XMP was proposed (Schulthess et al 1996)
The second step of caffeine biosynthesis involves a nucleosidase which
catalyses the hydrolysis of 7-methylxanthosine Although N-methylnucleosidase (EC
32225) was partially purified from tea leaves (Negishi et al 1988) recent detailed
structural studies on coffee 7-methylxanthosine synthase suggested that the methyl
transfer and nucleoside cleavage may be coupled and catalyzed by a single enzyme
(McCarthy amp McCarthy 2007)
Chapter 1 Introduction and Review of Literature
9
The last two steps of the caffeine synthesis are also catalyzed by a SAM-
dependent N-methyltransferase(s) which is different from the N-methyltransferase
enzyme that catalyzes the first step in the pathway Native N-methyltransferase
activities have been detected in crude and partially purified extracts from tea and
coffee plants (Ashihara amp Suzuki 2004) and a highly purified preparation has been
obtained from young tea leaves (Kato et al 1999) The enzyme was assigned the
name caffeine synthase (EC 211160) that catalyses the last two steps of caffeine
biosynthesis viz the conversion of 7-methylxanthine to caffeine via theobromine
(Figure 12) The gene encoding caffeine synthase was first cloned from young tea
leaves (Kato et al 2000) Since then several genes encoding N-methyltransferases
with different substrate specificity have been reported
Activity of the recombinant theobromine synthase (CTS1 and CaMXMT EC
211159) is specific for the conversion of 7-methylxanthine to theobromine In
contrast the recombinant caffeine synthases (CCS1 and CaDMXMT1) can utilize
paraxanthine theobromine and 7-methylxanthine as shown with tea caffeine synthase
(TCS1) (Mohanpuria et al 2011) Although paraxanthine is the most active substrate
of this recombinant enzyme only limited amounts of paraxanthine are synthesized in
coffee cells hence in vivo caffeine synthase is principally involved in the conversion
of 7-methylxanthine to caffeine via theobromine
Radiolabelled tracer experiments with theobromine accumulating cacao and
Chinese tea (Camellia ptilophylla) showed a limited conversion of theobromine to
caffeine N-methyltransferase which catalyzed the conversion of 7-methylxanthine to
theobromine was detected in crude extracts from C ptilophylla but conversion of
theobromine to caffeine was not observed (Ashihara et al 1998) Genes homologous
to caffeine synthase have been isolated from several theobromine accumulating plants
including cacao (Yoneyama et al 2006) The recombinant enzymes derived from
these genes have 3 N-methyltransferase activity suggesting that these theobromine
accumulating plants contain specific theobromine synthase Caffeine does not occur
in C ptilophylla (Ashihara et al 1998) although it is present in leaves and fruits of
cacao along with theobromine (Koyama et al 2003 Zheng amp Ashihara 2004)
Chapter 1 Introduction and Review of Literature
10
Figure 12 Pathways for the biosynthesis of caffeine
The major pathway is indicated by solid lines (Mizuno et al 2003b Uefuji et al 2003) Alternative pathways in coffee and tea plants are indicated
by dashed lines(Kato et al 1996 Schulthess et al 1996) Enzymes (1) 7-methylxanthosine synthase (2) N-methylnucleosidase (3) Theobromine
synthase (4) Caffeine synthase
N
N
N
N7
H3C
3
1
O
O
Caffeine
N
NH
N
N7
3
O
O
Theobromine
N
N
N
N
H
H
7
O
O
N
N
N
N
H
H
O
O
N
N
N
N
H
H
O
O
Xanthosine
N
N
N
N
H
H
O
O
N
N
N
N
H
H
O
ON
N
N
N
H
H3C
71
O
O
Paraxanthine
CH3
CH3
CH3
CH3CH3
7-methylxanthine
Rib P
Rib
(1)
(4)
Rib
+
CH3
7-methyl
xanthosine
(2)
(3)
SAM SAH
SAM SAH SAM SAH
Xanthosine mono
phosphate
Rib P
7-methyl xanthosine
monophosphate
+
CH3 CH3
Inosine 5-mono
phosphate
Adenosine 5-mono
phosphate
SAM SAH
Guanosine
Guanosine 5-mono
phosphate
Chapter 1 Introduction and Review of Literature
11
181 Source of Xanthosine for caffeine biosynthesis
Xanthosine the initial substrate of purine alkaloid synthesis is supplied by at least four
different pathways de novo purine biosynthesis (de novo route) the degradation
pathways of adenine nucleotides (AMP route) and guanine nucleotides (GMP route)
and the S-adenosyl-L-methionine (SAM) cycle (SAM route) (Figure 13) Purine
nucleotides are synthesized by de novo and salvage pathways (Ashihara amp Crozier
1999 Stasolla et al 2003 Zrenner et al 2006) The de novo synthesis of non purine
precursors like CO2 10-formyltetrahydrofolate 5-phosphoribosyl-1-pyrophosphate and
the amino acids Glycine glutamine and aspartate results in the production of purine
nucleotides
Figure 13 Xanthosine biosynthesis Xanthosine for purine alkaloid biosynthesis is supplied by at
least four routes from adenosine released from the SAM cycle (SAM route) from IMP
originating from de novo purine synthesis (de novo route) from the cellular adenine
nucleotide pool (AMP route) and from the guanine nucleotide pool (GMP route)
Adapted from (Ashihara et al 2008)
The utilization of IMP for caffeine biosynthetic pathway which is formed by the
de novo purine biosynthetic pathway for caffeine biosynthetic pathways was
demonstrated in young tea leaves using 15
N-glycine and 14
C-labelled precursors and
inhibitors of de novo purine biosynthesis (Ito amp Ashihara 1999) Xanthosine is formed
by an IMP rarr XMP rarr xanthosine pathway IMP dehydrogenase (EC 111205) and 5prime-
nucleotidase (EC 3135) catalyze these reactions The inhibition of IMP dehydrogen-
ase by ribavirin inhibits both caffeine and gunanine nucleotide biosynthesis in tea and
coffee leaves (Keya et al 2003)
Chapter 1 Introduction and Review of Literature
12
The portion of xanthosine used for caffeine biosynthesis is derived from adenine
and guanine nucleotide pools which are produce de novo by salvage pathways there are
several potential pathways for xanthosine synthesis from AMP but the AMP rarr IMP
rarr XMPrarr xanthosine route is predominant All three enzymes involved in the
conversion AMP deaminase (EC 3546) IMP dehydrogenase and 5prime-nucleosidase
have been detected in tea leaves (Koshiishi et al 2001) Xanthosine for caffeine
biosynthesis can also be produced from guanine nucleotides by GMPrarrguanosine
rarrxanthosine pathway Guanosine deaminase (EC 35415) activity is found in cell free
extracts from young tea leaves (Negishi et al 1994) but GMP deaminase activity was
not detected in plants (Stasolla et al 2003 Zrenner et al 2006) The absence of GMP
deaminase activity suggests that a GMP rarr IMP rarr XMP rarr xanthosine route is not
functional in plants
The use of radiolabelled purine bases and nucleosides for the study of caffeine
biosynthetic pathway was facilitated by the fact that exogenous purine nucleotides are
rapidly hydrolyzed by the plant tissues forming purine nucleosides andor bases which
are then utilized in pathways which in most instances indirectly lead to xanthosine
Most of these purine compounds including adenine adenosine inosine hypoxanthine
and guanine are converted to their respective nucleotides by salvage enzymes
including adenine phosphoribosyl transferase (EC 2427) hypoxanthineguanine
phosphoribosyl transferase (EC 2428) adenosine kinase (EC 27120) and
inosineguanosine kinase (EC 2421) which usually have high activities in plant cells
The resultant nucleotides then enter the purine alkaloid biosynthetic pathway In the
case of guanosine it may be directly deaminated to xanthosine and utilized for caffeine
biosynthesis (Figure 13) (Ashihara et al 1997)
182 SAM cycle in plants
SAM is the methyl donor for the three methylation steps in the caffeine biosynthetic
pathway In this process SAM is converted to S-adenosyl-L-homocysteine (SAH)
which in turn is hydrolyzed to homocysteine and adenosine Homocysteine is recycled
via the SAM cycle to replenish SAM levels and adenosine is released from the cycle
Adenosine released from the cycle is converted to AMP directly andor indirectly via
adenine AMP is converted to xanthosine and used for caffeine biosynthesis Since
three moles of SAH are produced via the SAM cycle for each mole of caffeine that is
synthesized in theory this pathway has the capacity to be the sole source of both the
Chapter 1 Introduction and Review of Literature
13
purine skeleton and the methyl groups required for caffeine biosynthesis in young tea
leaves (Koshiishi et al 2001)
Methionine
S-Adenosyl-L-methionineHomocysteine
ATP
PPi+ Pi
Tetrahydrofolate
5-Methyl-
tetrahydrofolate
CH3+Adenosine
S-Adenosyl-L-
homocysteine
(xanthine
structure)(Methyl groups)
Caffeine
(1)
(2)(3)
(4)
Figure 14 The SAM cycle (activated methyl cycle) in plants (Ashihara amp Suzuki 2004)
Enzymes SAM synthetase SAM-dependent N-methyltransferases S-adenosyl-
homocysteine (SAH) hydrolase Methionine synthase Adenosine released from the cycle
is salvaged to adenine nucleotides and utilized both for purine structure of caffeine via
xanthosine and for re-synthesis of SAM via ATP
183 N-methyltransferase in caffeine biosynthesis Molecular cloning
The genes encoding for the enzymes involved in the caffeine biosynthesis were
successfully cloned from the coffee family (Ogawa et al 2001 Mizuno et al 2003a
Mizuno et al 2003b Uefuji et al 2003) The conserved amino acid regions of tea CS
(AB31280) the sequences of O-methyltransferases derived from plant origin and
Arabidopsis hypothetical proteins were made use of in designing primers for RT-PCR
and RACE technique Independent groups have isolated N-methyltransferase (NMT)
genes from coffee which have been classified based on the substrate specificity of the
recombinant proteins expressed in Ecoli (Table 15) PCR products thus obtained were
used as the probe to isolate full length cDNA from the library resulting in the
identification of all three N-methyltransferases involved in caffeine biosynthesis
(Figure 12) Several cDNA have been characterized from the coffee plants one for
xanthosine methyltransferase (XMTXRS) three for 7-methylxanthine methyltransferase
or theobromine synthase (MXMTCTS) and three for 37-dimethylxanthine
methyltransferase or caffeine synthase (DXMTCCS) (Table 15) (Kato amp Mizuno
2004)
A single gene encoding for xanthosine methyltransferase (XMTCmXRS1) was
identified which encoded for a polypeptide consisting of 372 amino acids with an
apparent molecular mass of 418kDa It is expressed almost uniformly in aerial tissues
Chapter 1 Introduction and Review of Literature
14
of C arabica including leaves floral buds and immature but not in mature beans
Three genes encoding 7-methylxanthine methyltransferase (theobromine synthase) have
been identified The number of amino acids in the putative polypeptides are 378 for
MXMT1 (427kDa) and 384 for MXMT2 and CTS2 (434kDa) They differ by insertion
or deletion of blocks of several residues in the C-terminal region Their catalytic
properties based on kinetic parameters such as Km values were different They are
expressed in young leaves floral buds and immature but not mature beans Three genes
were identified for 3 7-dimethylxanthine methyltransferase (caffeine synthase) DXMT
CCS1 and CtCS7 each encoding a 43 kDa polypeptide consisting of 384 amino acids
However their kinetic properties differ from each other such as DXMT and CCS1
showed Km values of 1200 and 157microM for theobromine respectively Expressions
profiles are also distinct DXMT being expressed exclusively in immature beans while
CCS1 expression is ubiquitous occurring in all tissues The presence of isoforms of
these enzymes with different properties suggests that caffeine is synthesized through
multiple pathways depending on availability and concentration of the substrate (Mizuno
et al 2001 Ogawa et al 2001 Mizuno et al 2003a Mizuno et al 2003b Uefuji et
al 2003) Genomic clones of theobromine synthase were obtained by PCR- RFLP
from C canephora (Satyanarayana et al 2005 Satyanarayana 2006) The genomic
clones are reported to have highly conserved exons and a variable third intron The
promoter for the theobromine synthase was cloned and characterized (Satyanarayana et
al 2005)
There is high degree of similarity in the coding region of coffee NMT genes
isolated so far though differences exist in the 3prime untranslated region (UTR) for these
genes The deduced amino acid sequence of these enzymes shows more than 85
homology between these genes Phylogenetic analysis indicates that they are more
closely related to C-methyltransferases including those for jasmonic acid salicylic acid
and benzoic acid than to other N-methyltransferases This suggests that coffee N-methyl
transferases belong to a distinct sub-group within plant methyltransferases Their
cellular localization determined by green fluorescent protein fusion method and β-
glucourodinase (GUS) showed that the all the three genes are localized in the cytoplasm
(Ogawa et al 2001 Vinod et al 2007) It was also shown that these were present
along with chlorogenic acid(Vinod et al 2007)
Chapter 1 Introduction and Review of Literature
15
Table 15 N-methyltransferases and their encoding genes involved in caffeine biosynthesis
Gene (Enzyme) Gene name Accession No Description Reference
Xanthosine methyltransferase
( 7-methylxanthosine synthase) (EC 211158)
CaXMT AB 048793 Immature fruit cDNA Screening (Uefuji et al 2003)
CmXRS1 AB 034699 Leaf cDNA library RACE (Mizuno et al 2003a)
CaMXMT1 AB 048794 Leaf cDNA library RACE (Ogawa et al 2001)
7-methylxanthine synthase transferase
(theobromine synthase) ( EC 211159)
CTS1 AB 034700 Immature fruit cDNA Screening (Uefuji et al 2003)
CaMXMT2 AB 984125 Leaf cDNA library Screening (Mizuno et al 2001)
CTS2 AB 054841 Leaf cDNA library Screening (Mizuno et al 2001)
37-methylxanthinemethyltransferase
(Caffeine synthase) (EC 211160)
CaDXMT1 AB 086414 Immature fruit cDNA Screening (Uefuji et al 2003)
CCS1 AB 0861414 Immature fruit cDNA Screening (Uefuji et al 2003)
CtCS7 AB 0864415 Immature fruit cDNA
NMT genomic clones
Theobromine synthase-1
CX10 Complete coding region PCR Satyanarayan 2006
CX8 Complete coding region PCR -do-
Theobromine synthase-1 PG-5 Promoter + coding region RAGE -do-
Theobromine synthase-1 PG-1 Promoter + coding region RAGE Satyanarayan 2006
PG-4 Promoter + coding region RAGE -do-
Theobromine synthase-1 NMT
(AY273814) Complete coding region PCR Kochko A et al 2010
Theobromine synthase-2 NMT
(AY362825) Complete coding region PCR Kochko A et al 2010
Chapter 1 Introduction and Review of Literature
16
19 Catabolism of caffeine
Caffeine is produced in young leaves and immature fruits and continues to accumulate
gradually during the maturation of these organs At the same time caffeine is slowly
degraded with the removal of the three methyl groups resulting in the formation of
xanthine Catabolism of caffeine was first reported in coffee leaves (Kalberer 1965)
Since then a number of tracer experiments using 14
C labelled purine alkaloids have
been reported (Suzuki amp Waller 1984 Ashihara et al 1996 Vitoria amp Mazzafera
1999 Mazzafera 2004) They demonstrated the major catabolic pathway is caffeine rarr
theophylline rarr 3-methylxanthine rarr xanthine Xanthine is further degraded by the
removal by the conventional purine catabolic pathway to CO2 and NH3 via uric acid
allatonin and allantoate (Figure 15) (Ashihara amp Crozier 1999 Stasolla et al 2003
Zrenner et al 2006) Caffeine catabolism usually begins with its conversion to
theophylline catalyzed by N7-demethylase The involvement of P450-dependent mono-
oxygenase activity for this reaction was suggested (Huber amp Baumann 1998
Mazzafera 2004) However the activity of this enzyme has not yet been determined in
cell free extracts [8-14
C] Theophylline degraded to CO2 at much faster rate than [8-14
C]
caffeine indicating that conversion of caffeine to theophylline is a major rate limiting
step of caffeine catabolism and a possible reason why caffeine accumulates in high
concentrations in tissues of C sinensis and C arabica It is widely believed that the
control of caffeine levels in plants is a function of the balance between rates of
synthesis and degradation and this balance seems to vary depending on the plant
species and the tissue developmental stage (Mazzafera 2004)
Chapter 1 Introduction and Review of Literature
17
Figure 15 Caffeine catabolic pathways Caffeine is mainly catabolised via theophylline and 3-
methylxanthine Xanthine is further degraded to CO2 and NH3 by the conventional
oxidative pathway Adapted from (Ashihara et al 2008)
Chapter 1 Introduction and Review of Literature
18
110 Genetic engineering of Coffee
Genetic transformation has potential applications in coffee agriculture for incorporating
genes which could provide desirable traits such as disease and insect resistance
drought and frost tolerance and herbicide resistance Transgenic technology can also be
used to increase nutritional value and improve cup quality produce varieties with
caffeine-free beans and for production of hybrid crops for molecular farming
Identification of target specific genes is one of the pre-requisites for developing
transgenic crops The availability of a large number of EST sequences in coffee and
initiation of coffee genome sequencing may speed up the gene discovery and accelerate
transgenic research efforts in coffee
1101 Insect Resistance
Production of insect resistant coffee plants is one of the major objectives of the
breeding programs The major pests attacking coffee include coffee berry borer (CBB
Hypothenemus hampei) white stem borer (WSB Xylotrechus quadripes) leaf miner
(Perileucoptera coffeela) and root nematodes (Meloidogyne spp and Pratylenchus
spp) The CBB is present in almost all the coffee growing countries and considered to
the most devastating pest in coffee To date there is no reported source of resistance to
CBB in the coffee gene pool Like CBB WSB is another serious pest in C arabica in
India and several other East Asian countries Both CBB and WSB belong to the order
Coleoptera For India controlling WSB is the biggest challenge and has therefore
become the highest research priority Robusta is generally resistant to WSB but the
interspecific Robusta Arabica hybrids are susceptible to WSB Although leaf miner is
not yet a serious pest in India and other East Asian countries it is an economically
important pest in East Africa and Brazil Effective chemical control of CBB and WSB
is difficult due to the nature of their life cycle inside the berry and stem respectively as
well as environmental concern regarding the use of pesticides Biological control
measures are adapted to combat these insect pests with varying degrees of success
Developing coffee plants resistant to these pests using genetic transformation
technology could be one of the alternative strategies to counter pest damage For insect
resistance several different classes of proteins from bacterial plant and animal sources
have been isolated and their insecticidal properties tested against many important pests
Amongst these proteins Bt toxins are most important and several transgenic crops
expressing Bt genes have been commercialized Coffee transgenic plants carrying a
Chapter 1 Introduction and Review of Literature
19
synthetic version of the cry1Ac gene have been produced (Leroy et al 2000 Mishra et
al 2002) Indeed this was the first report of an important agronomic trait being
introduced into a coffee plant The transgenic plants presented similar features in
growth and development compared to normal plants Transgenic plants highly resistant
to leaf miner under greenhouse conditions were tested under field conditions in French
Guyana for 4 years for pest resistance (Perthius et al 2005 Perthius et al 2006)
From 54 independent transformation events 70 of the events were resistant to leaf
miner Unfortunately the field trial was vandalized which led to the termination of the
experiment (Montagnon et al 2005)The effectiveness of Bt genes in controlling
coleopteran pests is well documented in corn and potato which indicates that Bt genes
might be effective against CBB The high toxicity of B thuringiensis sero var
israelensis against first in star larvae of CBB has been demonstrated (Mendez et al
2003) In another experiment an α-amylase inhibitor from Phaseolus vulgaris was
tested against CBB and found to have an inhibitory effect on its growth and
development (Grossi et al 2004)In addition to the CBB and WSB C arabica
varieties are also susceptible to endoparasitic root-knot nematode (Meloidogyne spp)
(Campos et al 1990 Mishra et al 2012)
So far 15 species have been reported to be parasites of coffee Controlling
nematodes is extremely difficult and currently seedling grafting with robusta rootstock
is followed as one of the control measure Sources of resistance specific to root-knot
nematodes have been identified in coffee trees (Bertrand et al 2001) and the Mex-1
gene conferring resistance to M exigua in C arabica is in the process of isolation (Noir
et al 2003)
1102 Tolerance to Abiotic Stress
In many coffee growing countries abiotic stress such as drought and frost are the major
climatic factors that limit coffee production Changes in climatic patterns due to global
climate change are considered increasingly important for coffee cultivation Drought
induces water stress in plants which affects vegetative growth and vigor and triggers
floral abnormalities and poor fruit set It also indirectly increases the incidence of pests
and diseases in the plants C arabica is generally more tolerant to water stress than C
canephora partly due to its extensive deep root system C racemosa is known to be a
good source of drought tolerance In India hybrids between C canephora and C
racemosa have been obtained and are currently under evaluation for drought tolerance
Chapter 1 Introduction and Review of Literature
20
In many coffee growing countries coffee is propagated in marginal areas where the
annual rainfall is below 1000thinspmm with prolonged dry spells of over 4-5 months In
those areas water shortage and unfavorable temperatures constitute major constraints
and the growth and productivity of C canephora are badly affected As with drought
periodic frost also affects coffee production in parts of Brazil The introduction of
drought and frost tolerant genes through genetic transformation would be of great
importance for alleviating these problems Research is now being carried out by several
groups to identify genes involved in biotic as well as abiotic stress The most promising
genetic engineering approach for drought tolerance includes the use of functional or
regulatory genes as well as the transfer of transcription factors In recent years plants
tolerant to high temperature and water stress have been the subject of intense research
(Chaves et al 2003 Chaves et al 2004 Coraggio et al 2004) For achieving drought
tolerance genes that have been targeted include those encoding enzymes involved in
detoxification or osmotic response metabolism enzymes active in signaling proteins
involved in the transport of metabolites and regulating the plant energy status (Chaves
et al 2003 Chaves et al 2004 Coraggio et al 2004) The dissection of molecular
mechanisms related to signal transduction and transcriptional regulation might help in
engineering drought tolerance in coffee
1103 Disease Resistance
Coffee leaf rust (CLR) caused by the fungus Hemileia vastatrix is the most important
disease in coffee with substantial loss to coffee production and productivity in all the
coffee growing countries In addition to leaf rust coffee berry disease (CBD) caused by
fungus Colletotrichum kahawae can be a devastating anthracnose leading to substantial
crop loss in Africa Several other fungal and bacterial diseases may also affect coffee
to a extent C arabica is more susceptible to many diseases than C canephora Though
most of the disease control measures rely upon chemical control they are more
expensive and labour intensive The long-term solution is the breeding of resistant
varieties which is the focus of many breeding programs However breeding for disease
resistant varieties is time consuming due to the perennial nature of coffee with its long
gestation period India has a long history of C arabica breeding especially for leaf rust
resistance and is the first country to demonstrate the existence of multiple races of leaf
rust Resistance to CLR is conditioned primarily by a number of major (SH) genes and
coffee genotypes are classified into different resistance groups based on their
Chapter 1 Introduction and Review of Literature
21
interaction with different rust races pathogen (van der Vossen 2001) Currently C
canephora provides the main source of resistance to pests and diseases including CLR
(H vastatrix) and CBD (C kahawae) and therefore used in breeding programs Other
diploid species like C liberica and C racemosa are being used as a source of resistance
to coffee leaf rust and coffee leaf miner respectively (Guerreiro et al 1999
Sreenivasan et al 1989) The development of coffee varieties resistant to major fungal
diseases such as CLR and CBD using transgenic technology will benefit the coffee
industry immensely During the last 15 years significant progress has been made in the
area of host-pathogen interactions (Staskawicz et al 1995 Feys et al 2000
Shivaprasad et al 2012) and many resistance genes involved in recognizing invading
pathogens have been identified and cloned (Takken et al 2000) A number of
signalling pathways induced because of pathogen infection have been dissected (Dong
et al 1998 Navarro et al 2006) Many antifungal compounds synthesized by plants to
combat fungal infection have been identified (Does et al 1998) Understanding the
specific induction of targeted pathways and identification of specific pathways
responsible for particular fungal resistance is important in order to employ this strategy
in transgenic technology The recent investigation of gene expression during coffee leaf
rust infection could provide an insight into the defence pathways operating in coffee
(Fernandez et al 2004 Nardi et al 2006) Efforts have been made to identify and
clone resistance genes from coffee for achieving durable resistance The genetic and
physical map of two resistance genes SH3gene conferring resistance to rust (Prakash et
al 2004) and the Ck1 gene conferring resistance to C kahawae CBD (Gichuru et al
2006) have been established These genes could be used for molecular marker assisted
breeding programs (Mishra et al 2012)
1104 Production of low-caffeine coffee
A widespread belief that the ingestion of caffeine can have adverse effects on
health resulted in the increased demand for decaffeinated coffee (Ashihara amp Crozier
2001) Several method were used to obtain decaffeinated plants by conventional
breeding techniques using low yielding varieties and mutants that accumulated
relatively low amounts of caffeine when compared to the commercially available ones
Low-caffeine and decaffeinated coffee represent around 10 of the coffee sales
around the world (Ribas et al 2006b) The industrial process for coffee decaffeination
can be expensive and affects the original flavour and aroma in coffee (David 2002)
Chapter 1 Introduction and Review of Literature
22
Transgenic coffee plants with suppressed caffeine synthesis using RNA interference
(RNAi) technology have been obtained (Ogita et al 2003 Ogita et al 2004) Specific
sequences in the 3prime untranslated region of the theobromine synthase gene (CaMXMT1)
were selected for construction of RNAi short and long fragments The caffeine and
theobromine content of the transgenic plants were reduced by ~ 70 when compared to
the untransformed plants In C canephora RNAi technology has also been employed
to silence the N-methyl transferase gene involved in caffeine biosynthesis (Vinod et al
2007) Promoter of an N-methyltransferase (NMT) gene involved in caffeine
biosynthesis was cloned (Satyanarayana et al 2005 Satyanarayana 2006) which will
be very useful for studying the regulation of caffeine biosynthesis
1105 Improvement in cup quality
Improvement in coffee cup quality requires elaborate knowledge of the chemical
constituents as well as the metabolic pathways involved in the elaboration of quality
The constituents of coffee beans include minerals proteins carbohydrates caffeine
chlorogenic acids (CGA) glycosides lipids and many volatile compounds that give
flavour to coffee by roasting Among these the role of three major constituents
sucrose CGA and trigonelline have been studied in coffee The sucrose content of
coffee bean is associated with coffee flavour the higher the sucrose content in green
beans the more intense will be the cup flavour (Clifford et al 1985 de Maria et al
1996) The sucrose content of C arabica (82-83) is higher than C canephora (33ndash
40) The sucrose amino acids ratio in green beans determines the profile of volatile
compounds Manipulating sucrose content in coffee bean is therefore important in
improving cup quality The sucrose synthase gene (CcSUS2) from C canephora has
been cloned and sequenced (Leroy et al 2005) This provides an opportunity to
manipulate the sucrose content in coffee Chlorogenic acids are products of
phenylpropanoid metabolism Chlorogenic acids are a group of hydroxycinnamoyl
quinic acids (HQA) formed by esterification between caffeic acids coumaric acids and
quinic acids (Clifford et al 1999) They are present in relatively large quantities in the
coffee bean and are the precursor of phenolic compounds in roasted beans C
canephora beans contain higher CGA (10) compared to C arabica beans (6-7)
CGA are known to have antioxidant properties as well as being associated with disease
resistance (Campa et al 2003) Genetic manipulations of genes involved in CGA
synthesis can serve either of these purpose by up- or down regulating the pathway
Chapter 1 Introduction and Review of Literature
23
Phenylalanine ammonia-lyase (PAL) catalyzes the first step of the phenylpropanoid
pathway leading to the synthesis of a wide range of chemical compounds including
flavonoids coumarins hydroxycinnamoyl esters and lignins (Hahlbrock amp Scheel
1989) Recently the full length cDNA and corresponding genomic sequences of PAL
from C canephora was isolated characterized and functionally validated (Mahesh et
al 2006 Parvatam et al 2006) This has opened up new possibilities for manipulating
the level of the PAL enzyme in coffee which in turn can be useful for improvement of
cup quality and manipulating antioxidant properties in coffee
1106 Fruit Ripening
Uniformity during fruit ripening is decisively related to cup quality in coffee and
consequently to the value of the product Fruits at the ideal ripening stage produce the
best organoleptic characteristics for coffee The presence of over ripened or green fruits
changes the acidity the bitterness and consequently the cup quality In order to
maximize uniform ripening of coffee fruits it is essential to control the action of genes
involved in the last step of maturation process Ethylene is known to trigger ripening
and increasing ethylene biosynthesis is associated with various stages of ripening
process (Protasio et al 2005) To control coffee fruit maturation two of the major
genes involved in ethylene biosynthesis namely ACC synthase and ACC oxidase
have been cloned (Protasio et al 2005 Neupane et al 1999) Introduction of the ACC
oxidase gene in antisense orientation have been achieved in both C arabica and C
canephora The effect of the transgene on ethylene production and fruit maturation has
yet to be reported The inhibition of genes downstream to the initial ethylene burst is
also an option to control coffee fruit maturation (Ribas et al 2006)
111 RNA interferenceRNAi
The regulation of gene expression at post transcriptional level has recently attracted
much attention because of the discovery of the phenomenon called RNA interference
(RNAi) RNAi based silencing techniques are more efficient than antisense mediated
gene silencing (Wesley et al 2001) The effect was first observed in plants wherein
silencing was observed when some copies of a transgene was in the forward orientation
with respect to the promoter and some in the reverse orientation This resulted in the
formation of double stranded RNA in the system The phenomenon in which
experimentally introduced double stranded RNA (dsRNA) leads to the loss of
expression of the corresponding cellular gene by sequence specific RNA degradation
Chapter 1 Introduction and Review of Literature
24
was termed as RNA interference or RNAi The protective effect of RNA silencing in
reducing virus infectivity supports the view that PTGS has evolved as a mechanism to
defend plants against virus infection and also to moderate the possible deleterious
genome-restructuring (insertional) activity of virus-like mobile genetic elements eg
retrotransposons A growing body of biochemical and genetic data has further
demonstrated that plants animals and yeasts share related mechanisms of specific
degradation of RNAs in which double-stranded forms of RNA are initiator molecules
(Brenstein et al 2001)
The key insight into the process of PGTS was provided by the experiments
conducted by Hamilton amp Baulcombe (1999) who identified the product of RNA
degradation as small RNA (siRNA) species of ~25nt of both sense and antisense
polarity SiRNA are formed and accumulate as double stranded RNA molecules of
defined chemical structures Both genetic and biochemical approaches were undertaken
to understand the basis of silencing Genetic screens were carried out in fungus
Neurospora carassa the alga Chalmydomonas reinhardtii and plant Arabidopsis
thaliana to search for detective mutants in PTGS
A combination of results obtained from several in vivo and in vitro experiments
have gelled into a two step mechanistic model for RNAiPTGS The first step referred
to as the RNAi initiating step involves binding of the RNA nucleases to a larges
dsRNA and its cleavage into discrete ~21-25nt RNA fragments This is ATP-
dependent reaction which involved RNase III - type endonucleases which make
staggered cuts in both strands of the dsRNA leaving 3prime overhang of 2 nucleotides with
5prime- phosphate 3prime-hydroxyl and no modification in the sugar phosphate backbone
(Hamilton amp Baulcombe 1999 Elbashir et al 2001) SiRNAs are the direct products
of dsRNA cleavage by the multi-domain RNase III enzyme DICER (Figure 16)
(Brenstein et al 2001 Karthikeyan et l 2013)
In the second step or commonly known as the effector step the double stranded
siRNAs produced in the first step bind to an RNAi specific protein complex to form a
RISC The complex undergoes activation in the presence of ATP so that the antisense
component of the unwound siRNA becomes exposed and allows the RISC to perform
the downstream RNAi reaction
Chapter 1 Introduction and Review of Literature
25
Several enzymes are involved in RNAi based on their role genes that encode for them
are classified into RNAi initiators RNAi effectors RNA dependent RNA polymerases
and silencing genes Two C elegans genes rdeI and rde4 (rde stands for lsquo RNAi
deficientrsquo) are believed to be involved in the initiation step of RNAi The C elegans
rde1 gene is a member of a large family of genes and is homologous to the Neurospora
qde2 (qde stands for ldquoquelling deficientrdquo) and Arabidopsis AGO1 (AGO stands for
agronaute AGO1 was previously identified to be involved in Arabidopsis
development) Although the functions of these genes are not clear a mammalian
member of RDE1 family has been identified as a translation initiation factor (Thakur
2005) Important genes for the effector step of PTGS in C elegans are rde2 and mut7
genes These genes were identified from heterozygous mutant worms that were unable
to transmit RNAi to their homozygous offsprings Worms with mutated rde2 or mut 7
genes show defective RNAi mut-7 gene encodes a protein with homology to the
nuclease domains of RNAase D and a protein implicated in Werner syndrome (a rapid
ageing disease in humans) (Grishok et al 2001 Kamath-Loeb et al 2012s)
Neurospora qde-1 Arabidopsis SDE-1SGS-2 and C elegans ego-1 appear to encode
RNA dependent RNA polymerase (RdRPs) It assumed that this is a proof that an RdRp
activity is required for RNAi Certainly the existence of an RdRp might explain the
remarkable efficiency of dsRNA induced silencing if it amplifies either the dsRNA
prior to cleavage or the siRNAs directly (Muers 2013) In C elegans ego-1 mutants
(ego stands for lsquoenhancer of glp-1) RNAi functions normally in somatic cells but is
defective in germline cells where ego-1 is primarily expressed In Arabidopsis SDE-
1SGS-2 mutants (SGS stands for suppressor of gene silencing) siRNA are produced
when dsRNA is introduced via an endogenously replicating RNA virus but not
introduced by a transgene It has been proposed that perhaps the viral RdRP is
substituting for the Arabidopsis enzyme in these mutants Random degradative PCR
model suggests that an RdRP uses the guide strand of an siRNA as a primer for the
target mRNA generating a dsRNA substrate for Dicer and thus more siRNAs
Several genes controlling RNA silencing in plants have been identified through
genetic screens of Arabidopsis mutants impaired in transgene induced RNA silencing
They encode a putative RNA-dependent RNA polymerase (SGS2SDE1) a coiled
protein (SGS3) a protein containing PAZ and Piwi domains (AGO1) and an RNA
helicase (SDE3) The putative SGS2SDE1 of Neurospora is related to QDE-1 and
Chapter 1 Introduction and Review of Literature
26
EGO-1 of C elegans whereas PAZPiwi protein AGO is related to QDE-2 of
Neurospora RDE-1of C elegans RNA helicase SDE3 is related to SMG-2 of C
elegans and Mut-6 of Chlamydomonas MUT-7 gene of C elegans encodes a protein
similar to Rnase D whereas the Drosophila DICER gene encode a protein similar to
RNase III An Arabidopsis ortholog of DICER gene has been identified called CAF
SIN1 SUS1(Thakur 2005 Poethig 2009)
Since the RNAi machinery is present constitutively within the eukaryotic cell it
is important to explore the metabolic advantages that are accorded by the RNAi related
proteins during the intrinsic normal growth of cells and development of organisms The
natural RNAi machinery not only keeps the mobile transposable elements from
disrupting the integrity of the genomes as suggested by the analysis of model organisms
like A thaliana C elegans D melanogaster (Tabara et al 1999 Wu-Scharf et al
2000 Aravin et al 2001 Hamilton et al 2002 Lisch 2009) but also participates in
development of organisms Genetic defects in Celegans RNAi genes ego1 and dicer
cause known specific developmental errors (Grishok et al 2000 Grishok et al 2001
Knight amp Bass 2001 Hall et al 2013) Similarly the Argonaute family of genes of A
thaliana (especially the ZWILLE proteins) is also responsible for plant architecture and
meristem development (Carmell et al 2002 Hamant and Pautot 2010) and the Dicer
homologue of A thaliana CAF1 is required for embryo development (Golden et al
2002 Nakano et al 2011) Thus genetic evidence illustrates the role of the RNAi
machinery as a controller of development related genes The mechanistic details of
these developmental processes are beginning to emerge
Chapter 1 Introduction and Review of Literature
27
Figure 16 Mechanism of gene silencing induced double stranded RNA The dicer enzyme to
produce siRNAs cleaves the RNA The siRNA generated bind to the nuclease complex
RISC The antisense component in the siRNA in the RISC guides the complex towards
the cognate mRNA resulting in endonucleolytic cleavage of the mRNA
Chapter 1 Introduction and Review of Literature
28
112 MicroRNA or MiRNA
In 1991 Ambros and co-workers first isolated a lin-4 mutant of Celegans which was
arrested at the first larval stage (Lee et al 1993) Later on the let-7 mutation was
isolated in the same system which was responsible for development through the fourth
larval stage Both lin-4 and let-7 encode short 22-nucleotide mature RNAs and were
called short temporal RNA because they control the temporal development program of
C elegans The mature lin-4 RNA defines (negatively regulates) the mRNA expression
of the lin-14 and lin-28 hetrochronic genes with the antisense mediated repression
mechanism of translation initiation and thus specifies the fate of cells during the first
three larval stages Recent studies have revealed that the short temporal RNAs are
actually members of a group of tiny RNAs (21to 28 nucleotides) called the micro-
RNAs (miRNA) (Bartel 2009) isolated members of which could easily run to a few
thousand which includes both those were identified computationally and by deep
sequencing Some of the components of the RNAi machinery have also been clearly
established as the effector proteins for the maturation of miRNAs
113 MicroRNAs in plants
1131 Discovery
MicroRNAs have been discovered by three basic approaches Direct cloning forward
genetic direct cloning and bioinformatic prediction followed by experimental
validation The most direct method of miRNA discovery is to isolate and clone small
RNAs from biological samples and several groups have used this approach to identify
plant miRNAs (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002 Xie et al 2003 Sunkar et al 2005 Williams et al 2013) The cloning methods
adapted were similar to those used to identify large numbers of animal miRNAs
(Lagos-Quintana et al 2001 Lau et al 2001 Aravin amp Tuschl 2005) which involve
isolating small RNAs followed by oligonucleotide adapter ligation reverse
transcription amplification and sequencing Some protocols incorporate methods to
enrich for Dicer cleavage products (ie molecules with 5prime-phosphates and 3prime-
hydroxyls) and to concatemerize the short cDNAs so that several may be identified in a
single sequencing read (Lau et al 2001 Llave et al 2002a Jagadeeswaran amp Sunkar
2013)The initial cloning experiments in Arabidopsis identified 19 miRNAs belonging
to 15 families (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002) although hundreds of other small RNAs such as siRNAs and RNA degradation
Chapter 1 Introduction and Review of Literature
29
fragments were also cloned through the direct cloning methods To select miRNAs
from the pool of small RNAs secondary structures of the RNA molecules were
examined in compliance with the acknowledged structural features of miRNAs
(Ambros et al 2003) The secondary structures were validated using several ad-hoc
software tools such as the Mfold or RNAfold programs (Reeder et al 2006) Finally
the existence of mature miRNAs was determined by RNA gel blot hybridization These
direct cloning methods were successful in isolating many miRNAs in Arabidopsis
(Reinhart et al 2002 Sunkar amp Zhu 2004) Rice (Sunkar et al 2005) Poplar (Lu et
al 2005) moss (Arazi et al 2005) and Tobacco (Billoud et al 2005)
Although miRNAs were first discovered through forward genetic screens in
worms (Lee et al 1993 Reinhart et al 2000)only few A thaliana miRNA gene
families have been discovered by this method (Chen 2008) in plants MiRNA
involvement in plant mutant phenotypes were not properly inferred till the cloning
experiments established that plant genomes contain numerous miRNAs (Park et al
2002 Reinhart et al 2002 Rhoades et al 2002) MiRNA loss-of-function allele has
been identified by forward genetic screens early extra petala1 is caused by a
transposon insertion ~160bp upstream of the predicted miR164c stem loop resulting in
flowers with extra petals (Baker et al 2005 Irish 2008) The fact that loss-of-function
miRNA mutants have been rarely discovered using forward genetics reflects small
target size for mutagenesis coupled with redundancy in nearly all evolutionarily
conserved plant miRNAs which are encoded by gene families (Table16) Family
members are likely to have overlapping functions buffering against loss at any single
miRNA locus Over expression screens can circumvent redundancy limitations At least
four plant miRNAs miR319 (also known as miR-JAW) miR172 (also known as EAT)
and miR166 were isolated in over expression screens for dominant mutants with
developmental abnormalities (Aukerman amp Sakai 2003 Palatnik et al 2003 Kim et
al 2005 Williams et al 2005b Schommer et al 2012) Mutations in miRNA target
sites which can prevent the entire family of miRNAs from repressing a target gene can
also circumvent redundant functions of miRNA family members
The dominant mutations in the HD-ZIP genes PHB PHV and REVOLUTA
(REV) in Arabidopsis and ROLLEDLEAF1 (RLD1) in Maize result in adaxialization of
leaves andor Vasculature (McConnell amp Barton 1998 McConnell et al 2001 Nelson
et al 2002 Zhong amp Ye 2004 Allen amp Millar 2012) and are all caused by mutations
Chapter 1 Introduction and Review of Literature
30
in miR166 complementary sites (Rhoades et al 2002 Emery et al 2003 Jones-
Rhoades amp Bartel 2004 Mallory et al 2004 Zhong amp Ye 2004)
In both plants and animals cloning was the initial means of large-scale
miRNA discovery (Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Reinhart et al 2002 Mendes et al 2012) However cloning is biased toward
RNAs that are expressed highly and broadly MiRNAs expressed at low levels or
only in specific cell types or in response to certain environmental stimuli were more
difficult to clone Sequence based biases in cloning procedures might also cause
certain miRNAs to be missed Because of these limitations bioinformatics approaches
to identify miRNAs have provided a useful complement to cloning
A straightforward use of bioinformatics has been carried out to find homologs
of known miRNAs both within the same genome and in the genomes of other species
(Pasquinelli et al 2000 Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Mendes et al 2009) A more difficult challenge is to identify miRNAs
unrelated to previously known miRNAs This was first accomplished for
vertebrate nematode and fly miRNAs using algorithms that search for conservation
of sequence and secondary structure (ie miRNA stem-loop precursors) between
species searching for patterns that are characteristic of miRNAs (Lai et al 2003 Lim
et al 2003 Lim et al 2005)
Most of the methods identified numerous miRNAs but are necessarily more
relaxed in terms of allowed structures Some approaches take advantage of the high
complementarity of plant miRNAs to target messages implementing the requirement
that the candidate has conserved complementarity to mRNAs (Jones-Rhoades amp Bartel
2004 Adai et al 2005) This additional filter has been useful for distinguishing
authentic plant miRNAs from false positives which later extended to mammalian
miRNA gene prediction (Xie et al 2005 Mendes et al 2012)
Another criteria used by most algorithms in the filters is evolutionary
conservation In plants mature miRNA sequences are conserved to a higher degree
than their precursor sequences For example Arabidopsis and rice diverged more than
130 million years ago (Friis et al 2004) but some miRNAs are highly conserved
in these plant species (Reinhart et al 2002) Furthermore various parameters for the
secondary structures such as free energy the number of paired residues within a
Chapter 1 Introduction and Review of Literature
31
miRNA or the number and size of bulges are also used to validate the predictions
(Jones-Rhoades amp Bartel 2004 Wang et al 2004 Adai et al 2005 Mendes et al
2012)
1132 Conserved microRNAs in Plants
Till date cloning genetics and bioinformatics screens have resulted in annotation of
approximately 184 potential Arabidopsis miRNAs (miRBase release 13) These
miRNAs seem to arise through gene duplication and subsequent diversification and
represent approximately 100 families (Maher et al 2006 Axtell 2013) Twenty one
families represented by 92 genes are clearly conserved in species beyond Arabidopsis
(Table 15) The presence of these miRNA families in other sequenced plant genomes
indicates that different plant species have an evolutionarily fluid set of miRNAs
(Willmann amp Poethig 2007) Recent studies on mosses one of the most ancient land
plants have shown that at least 14 miRNA families are conserved in eudicots and
mosses and therefore it appears that plant miRNAs share a common ancient origin
(Arazi et al 2005 Axtell amp Bartel 2005 Talmor-Neiman et al 2006 Zhang et al
2006 Axtell 2013) The number of members per family in a genome ranges from 1-32
with the exception of miR-430 which is represented by a cluster of ~80 loci in zebra
fish (Giraldez et al 2005)
In plants the number of members in each family correlates among examined
species certain families contain numerous members in all three species (eg miR156
miR166 miR169) whereas others consistently contain fewer genes (eg miR162
mir168 miR394) (Table 16) Although it is still unclear why certain miRNA are
encoded by more than one gene in plants (eg 12 genes encoding miR156) this
correlation might suggest functional significance of various miRNA family sizes Most
annotated plant miRNAs are conserved throughout flowering plants while a few
miRNAs are found only in a single sequenced genome and thus they could be
evolutionarily lsquoyoungrsquo miRNAs For example Arabidopsis has several miRNA
families that are not found in poplar or rice suggesting that miRNAs could be
generated continuously during evolution (Allen et al 2004a Rajagopalan et al
2006 Fahlgren et al 2007 Axtell 2012 de Alba et al 2013) Consistent with the
notion that non conserved miRNAs are of a more recent evolutionary origin these
miRNAs are mainly encoded by a single locus in the genome Within each family the
mature miRNA is always located in the same arm
Chapter 1 Introduction and Review of Literature
32
Table 16 MicroRNA families conserved in plants
miRNA
family
Arabidopsis Oryza Populus
miR156 12 12 11 miR159319 6 8 15 miR160 3 6 8 miR162 2 2 3 miR164 3 5 6 miR166 9 12 17 miR167 4 9 8 miR168 2 2 2 miR169 14 17 32 miR171 4 7 10 miR172 5 3 9 miR390 3 1 4 miR393 2 2 4 miR394 2 1 2 miR395 6 19 10 miR396 2 5 7 miR397 2 2 3 miR398 3 2 3 miR399 6 11 12 miR408 1 1 1 miR403 1 0 2 miR437 0 1 0 miR444 0 1 0 miR445 0 9 0 Total 92 127 169
All known miRNA families that are conserved between more than one plant species are listed
together with the number of genes identified in the sequenced genomes Rice miRNA families that have orthologs in maize but do not appear to have orthologs in the eudicots (Arabidopsis and Populus) are marked with an asterisk The following families contain miRNA genes annotated with
more than one number miR156 (miR156 and miR157) miR159319 (miR159 and miR319) miR166 (miR165 and miR166) miR171 (miR170 and miR171) and miR390 (miR390 and miR391)
of the stem loop (5prime- or 3prime) as it would be expected if the genes carry common ancestry
(Figure 17) The sequence of the mature miRNA and to some extent the segment on
the opposite arm of the hairpin to which it pairs is highly conserved between the
members of the same family (both within and between the species) while the sequence
secondary structure and length of the intervening ldquolooprdquo region can be highly divergent
within the same family
The patterns of paring and non pairing nucleotides are often conserved between
homologous miRNA stem loops from different species (Figure 17) The significance of
conserved mismatches is opined to guide DCL1 to cleave at the appropriate positions
along the stem loop
Chapter 1 Introduction and Review of Literature
33
Figure 17 Representative stem loop of miR164 Segments corresponding to the mature miRNAs
are shown in red (Adapted from Rajagopalan et al 2006)
Although many miRNAs are highly conserved in plants the degree of conservation in
most cases is quite low between plants and animals one exception is miR854 A recent
study has revealed that miR854 is conserved between Arabidopsis and animals and that
the Arabidopsis miR854 differs from the human miR854 only by a single nucleotide
(Arteaga-Vazquez et al 2006 Axtell 2012 de Alba et al 2013) The Arabidopsis
miR854 has multiple binding sites in the 3prime-untranslated region (UTR) of
OLIGOURIDYLATE-binding-PROTEIN mRNA 1bA (UBP1b) and represses the
translation of the UBP1b mRNA The animal miR854 is also complementary to a site in
the 3prime-UTR of the UBP1b homolog in animals However it is currently unclear how the
animal miR854 regulates its target mRNA Another distinction between plant and
animal miRNAs is the genomic organization of the miRNA genes While plant miRNA
genes are located predominantly in the intergenic regions animal miRNA genes tend to
localize in the intragenic regions both in the introns or exons of protein coding genes
Moreover miRNA genes are often clustered within a single precursor transcript
whereas individual plant miRNAs are derived from individual precursor RNAs (Bartel
Chapter 1 Introduction and Review of Literature
34
2004 Bonnet et al 2004 Kim 2005 Vaucheret et al 2006 Marco et al 2013)
1133 Non-conserved miRNA and challenges in annotation
Most miRNAs are conserved throughout flowering plants (Table 15) but many more
were found only in a single sequenced genome thus considered to be of a more recent
evolutionary origin The extended homology between few non conserved miRNAs
precursors and their target genes provides strong evidence that these relatively ldquoYoungrdquo
miRNAs arose from the duplication of the target gene segments (Allen et al 2004)
Several non-conserved miRNAs including miR161 miR163 miR173 miR447
miR475 and miR476 are known to direct cleavage of target transcripts (Allen et al
2004a Adai et al 2005 Sunkar et al 2005 de Alba et al 2013) It is difficult to
confidently predict targets for many because it is not possible to use complementary
site as a filter against false-positive target predictions It is also difficult to
confidently annotate non-conserved miRNAs as miRNA rather than siRNAs The
established minimal standard for miRNA annotation is a small RNA with detectable
expression and the potential to form a stem-loop when joined to flanking genomic
sequence (Kasschau et al 2003) In practice these requirements are too loose to
definitively categorize many small RNAs cloned from plants Many plant siRNAs are
detectable on blots (Vazquez et al 2004b) and hundreds of thousands of non-
miRNA genomic sequences can be predicted to fold into secondary structures that
resemble structures of plant miRNA precursors (Jones-Rhoades amp Bartel 2004)
Therefore without conservation of both sequence and secondary structure it is
difficult to be confident that a given cloned RNA originated from a stem-loop (ie is a
miRNA) rather than from a double-stranded RNA (ie is a siRNA) In fact many of
the thousands o f small RNAs cloned from Arabidopsis (Gustafson et al 2005) would
probably meet the literal requirements for annotation as miRNAs A few of these
sequences might be miRNAs but others that meet the literal criteria probably are not
so The challenges of annotating non conserved small RNAs were evidenced by
three related small silencing RNAs that were originally annotated as miRNAs
that turned out to be trans-acting siRNAs (tasiRNAs)(Kanno et al2013)
Although most miRNAs that are broadly conserved among flowering plants are
now being identified and experimentally validated (Table 16)(Jones-Rhoades amp
Bartel 2004) the challenges and ambiguities for classifying non-conserved miRNAs
prevent meaningful estimates of the total number of miRNA genes in Arabidopsis and
Chapter 1 Introduction and Review of Literature
35
other plant genomes It is possible that miRNAs might have escaped detection
because they are non-conserved and expressed in specific tissues or conditions and can
only be identified by using high- throughput sequencing
1134 MicroRNA biogenesis
11341 Transcription of microRNA precursor
Plant miRNAs are primarily found in the genomic regions that are not associated with
the protein coding genes (Reinhart et al 2002) and it appears that most if not all plant
miRNAs are produced from their own transcriptional units Plant miRNA genes are
occasionally clustered near each other in the genome suggesting transcription of
multiple miRNAs from a single primary transcript [eg the miR395 cluster (Grishok
et al 2001 Jones-Rhoades amp Bartel 2004b)] but this polycistronic arrangement of
miRNA genes appears far less frequently in plants than in animals (Bartel 2004)
Northern blot EST and mapping evidence indicate that plant miRNA primary
transcripts (also known as pri-miRNAs) as in animals are longer than needed to
encompass the miRNA stem-loops (Palatnik et al 2003 Jones-Rhoades amp Bartel
2004b) At least some of these pri-miRNA transcripts appear to be spliced
polyadenylated (Kurihara amp Watanabe 2004) and capped (Xie Z et al 2005) Two
rice miRNAs are contained within transcripts that contain exon junctions within the
presumptive stem-loop precursor implying that in these cases splicing is a prerequisite
for Dicer recognition (Sunkar et al 2005) Plant pri-miRNAs are over 1kb in length
and they are usually preceded by typical TATA box motifs They can also undergo
canonical splicing polyadenylation and capping All these observations indicates RNA
polymerase II is probably responsible for transcribing most plant miRNAs (Xie Z et
al 2005) as appears to be the case for many animal miRNAs Relatively little is
known about the regulation of miRNA transcription in plants but there is no reason
to suspect that this regulation would differ from that of protein-coding transcripts as
the bioinformatics analysis of the sequence upstream of the transcriptional start
sites of the miRNA genes showed the presence of putative binding motifs of
number of known transcription factors (Megraw et al 2006 Sethupathy 2013)
11342 MicroRNA processing and export
In plants maturation of miRNA is a step wise process A miRNA gene is transcribed to
pri-miRNA which is much longer than the pre-miRNA possessing the characteristic
stem-loop structure Like transcription of most protein-coding genes the miRNA
Chapter 1 Introduction and Review of Literature
36
transcription is processed by RNA polymerase II (Pol II) enzymes and the pri-
miRNAs are 5prime-capped and 3prime-polyadenylated (Lee et al 2004 Xie Z et al 2005)
Mature miRNAs are produced from pri-miRNAs through at least two sequential
processing steps by RNase III-type endonucleases In animals pri-miRNAs are first
processed at the bottom portion of the stem by Drosha a nuclear-localized RNase III
enzyme to liberate pre-miRNAs The pre-miRNAs are then exported to the cytoplasm
with the help of exportin-5The pre-miRNAs are further processed by Dicer another
RNaseIII enzyme to a duplex consisting of the miRNA and the antisense strands
(miRNA) Since plants do not have any Drosha homologs the two sequential
processing steps seem to be carried out by DCL1 a Dicer homolog in the nucleus
(Kurihara amp Watanabe 2004 de Alba et al 2013 Rogers amp Chen 2013)
The processing of pri-miRNAs to pre-miRNAs by DCL1 is assisted by two
other proteins HYPONASTY LEAVES1 (HYL1) and SERRATE (SE) HYL1 belongs to a
family of double-stranded RNA (dsRNA) binding protein and interacts with DCL1 in
vitro and in vivo (Lu amp Fedoroff 2000 Hiraguri et al 2005 de Alba et al 2013
Rogers amp Chen 2013) Loss-of-function mutations in the HYL1 gene result in
decreased accumulation of miRNAs and concomitant accumulation of pri-miRNAs
(Han et al 2004 Vazquez et al 2004a Kurihara et al 2006) SE a C2H2 zinc finger
protein interacts with DCL1 and HYL1 and plays a role in the processing of pri-
miRNAs to pre-miRNAs (Lobbes et al 2006 Yang L et al 2006) In addition the
nuclear cap-binding complex proteins CBP20 and CBP80 that are known as ABA
HYPER SENSITIVE 1(ABH1) have been shown to be required for proper miRNA
processing (Gregory et al 2008 Laubinger et al 2008) Recently it has been
demonstrated that DCL1 can release 21-nt RNAs from dsRNA as well as from
synthetic miR167 precursor RNAs in vitro However this cleavage is error prone and
inefficient in the absence of HYL1 and SE which accelerate the rate of DCL1-mediated
cleavage and promote accurate processing of miRNAs (Dong et al 2008 Rogers amp
Chen 2013)
11343 Methylation of the miRNAmiRNAduplex
After the miRNAmiRNAduplexes are released from the pre-miRNAs by the DCL1
activities the duplexes are methylated at the 2primeOH of the3prime-ends by HUA ENHANCER1
(HEN1) a small RNA methyltransferase (Yu et al 2005 Yang Z et al 2006 Rogers
amp Chen 2013 ) This step which is distinct from the RNase III-mediated processing
Chapter 1 Introduction and Review of Literature
37
step distinguishes the biogenesis of plant miRNAs from that of animal miRNAs
Mutations in the HEN1 gene were first isolated in a morphological screening for floral
development although the HEN1 mutants display pleiotropic phenotypes similar to the
developmental defects observed in the DCL1 mutants (Chen et al 2002 Park et al
2002 Rogers amp Chen 2013)
Figure 18 Biogenesis of microRNA
A Biogenesis of plant MicroRNA B Biogenesis of metazoananimal MicroRNA
The miRNA (red) is incorporated into RISC and the miRNA(blue) in degraded
This enzyme has two dsRNA-binding domains and nuclear localization signal It
is also conserved in fungi and is required for the processing of siRNAs as well as of
miRNAs (Park et al 2002 Boutet et al 2003 Xie et al 2003) Although the HEN1
protein is localized to the nucleus its methylation activity in the cytoplasm cannot be
ignored (Xie et al 2004 Rogers amp Chen 2013)
Chapter 1 Introduction and Review of Literature
38
The methylation of the miRNAmiRNA duplex is required to protect miRNAs
against the 3prime-end uridylation activity and subsequent degradation (Li et al 2005) In
the hen1 mutants accumulation of miRNAs is reduced to a lower level Also the
miRNAs appear to be heterogeneous in their sizes such that a ladder of bands rather
than a single species is observed for most miRNAs in the mutants (Li et al 2005)
MiRNA cloning and sequencing analyses revealed the presence of a number of U
residues at the 3prime-end of miRNAs in the hen1 mutants Moreover these nucleotides do
not correspond to the miRNAs in the pre-miRNAs indicating that these nucleotides are
added after the processing of the miRNAs from pre-miRNAs (Li et al 2005)
Uridylation of RNAs has also been proven to be correlated with RNA decay (Shen amp
Goodman 2004 Mullen amp Marzluff 2008) suggesting that uridylation attracts an
exonuclease which degrades miRNAs in plants
11344 MiRNA incorporation in to the RISC and its nuclear export
The methylated miRNAmiRNA duplexes undergo RISC assembly In this process
the miRNA of the duplexes is selectively incorporated into the RISC and the miRNA
appears to be removed and subsequently degraded (Hammond et al 2000 Hutvagner
amp Zamore 2002 Schwarz et al 2003 de Alba et al 2013 Rogers amp Chen 2013)
The ARGONAUTE 1(AGO1) proteins are core components of the RISC complex
They contain two structural domains the N-terminal PAZ domain that binds to the 3prime-
end of single-stranded RNAs and the C-terminal piwi domains that has a structure
similar to that of RNase H (Cerutti et al 2000 Parker et al 2004) Several AGO
proteins possess endonucleolytic activities within the PIWI domain and cleave target
mRNAs in the middle of the site complementary to miRNA (Liu et al 2004 Meister et
al 2004 Miyoshi et al 2005) Mutations in the AGO1 gene cause miRNA target
genes to be ectopically expressed and the levels of some miRNAs are reduced in the
mutants (Kidner amp Martienssen 2004 Vaucheret et al 2004 Kidner amp Martienssen
2005 Ronemus et al 2006) Moreover the phenotype of the AGO1 hypomorphic
alleles display defects in lateral organ polarity and leaf and flower morphology as
observed in the dcl1 mutants and null AGO1 alleles are embryonically lethal
supporting the close relationship between the AGO proteins and miRNA processing
(Kidner amp Martienssen 2004 Vaucheret et al 2004 de Alba et al 2013 Rogers amp
Chen 2013)
The production of miRNA miRNA duplexes occur in the nucleus while
Chapter 1 Introduction and Review of Literature
39
miRNA-RISC complexes are present in the cytoplasm where target mRNAs are
located This discordance of functional sites requires an export step during miRNA
biogenesis HASTY the Arabidopsis homolog of exportin-5 is thought to be involved
in miRNA nuclear export (Bollman et al 2003 Park et al 2005)The hasty mutant
exhibits pleiotropic defects in plant development and reduced accumulation of most
miRNAs as in the DCL1 or AGO1 mutants Howeverit is unclear whether the HASTY
proteins export the miRNA miRNA duplexes or the miRNA-RISC complexes in
plants
11345 Feedback regulation of miRNA biogenesis
MiRNA biogenesis is under feedback regulation such that the DCL1 and AGO1 genes
are themselves regulated by miRNAs DCL1 is itself a target of miR162 which leads to
the cleavage of the DCL1 mRNA (Xie et al 2003 de Alba et al 2013) Consistent
with this the levels of DCL1 mRNA are elevated in the mutants of miRNA processing
genes in which the miR162 abundance is reduced In addition there is a predicted
hairpin structure of miR838 in the 14th
intron of the DCL1 primary transcript
(Rajagopalan et al 2006) This prediction implies that DCL1-mediated processing
on this site can result in the cleavage of the DCL1 primary transcript into two truncated
fragments the N-terminal capped fragment and the C-terminal polyadenylated
fragment If this is the case it is possible that miRNA biogenesis competes with the
splicing event of the DCL1 pre-mRNA
The AGO1 is also regulated by miRNA There is a complementary site for
miR168 binding in the AGO1 mRNA and the transcript level of the AGO1 mRNA is
reduced through an mRNA cleavage mechanism (Vaucheret et al 2006 de Alba et al
2013 Rogers amp Chen 2013) indicating that the AGO1 mRNA levels are tightly
controlled by the levels of the AGO1 protein
1135 Mechanism of miRNA action
11351 MiRNA-directed mRNA cleavage
By forming the miRNA-RISC assembly miRNAs can direct the RISC to regulate gene
expression by two post transcriptional mechanisms mRNA cleavage or translational
repression (Bartel 2004 Jones-Rhoades et al 2006 Dalmay 2013) The degree
of miRNA-mRNA complementarity plays an important role for the
determination of the mechanism to be used A general rule is that while perfect or
Chapter 1 Introduction and Review of Literature
40
near-perfect complementarity induces mRNA cleavage central mismatches trigger
translational repression (Carrington amp Ambros 2003 Jones-Rhoades amp Bartel 2004)
Unlike most animal miRNAs plant miRNAs have near-perfect or perfect
complementarity to their targets and mRNA cleavage is assumed to be a predominant
mechanism by which miRNAs regulate gene expression in plants
In RNA cleavage mechanism miRNAs guide the AGO component of RISC to
cleave a single phosphodiester bond of the target mRNA within the miRNA-binding
site (Llave et al 2002b Tang et al 2003) The truncated fragments are then released
and degraded freeing the RISC to recognize and cleave another target mRNA This has
been validated by the detection of 3prime cleavage products that have 5prime ends that start at the
middle of the complementary regions The mRNA cleavage products are then further
degraded by other mechanisms The 3prime cleavage products of some miRNA targets are
degraded by the 5prime-3prime exonuclease XRN4 an Arabidopsis homolog of the major Yeast
mRNA degrading exonuclease Xrn1p It has been observed that the 3prime cleavage
products of some target mRNAs were accumulated in the xrn4 mutants (Souret et al
2004 Dalmay 2013) However the 3prime cleavage products of other miRNA targets do
not accumulate in the xrn4 mutants indicating that there are also XRN4 independent
mechanisms The 5prime cleavage products are typically untraceable by RNA gel blot
hybridization but can be detected by sensitive PCR based methods It has been found
that the 5prime cleavage products tend to obtain an oligo U tail at the 3prime ends This 3prime U tail
is correlated with shortening of the 5prime cleavage products from the 5prime end leading to the
conclusion that the 3prime uridylation causes 5 prime- 3 prime exonucleolytic decay of the 5prime cleavage
products (Shen amp Goodman 2004)
DCL1 HYL1 and SE interact with each other and are co-localized in the
nuclear bodies termed Dicing bodies or D-bodies in which some pri-miRNA shave
been found suggesting that D-bodies serve as a center for active miRNA processing
(Fang amp Spector 2007 Fujioka et al 2007 Song et al 2007) The miRNA
processing site appears to be distinct from the Cajal bodies that have previously been
found as a siRNA processing site (Pontes amp Pikaard 2008) The D-bodies include
SmD3 and SmB proteins that are found in the Cajal bodies However At collin an
additional marker for the Cajal bodies is not found in the D-bodies Moreover the D-
bodies are present in an At collin mutant lacking the Cajal bodies The pri-miRNAs of
miR171A and miR159A genes have been found to be co-localized with HYL1 and
Chapter 1 Introduction and Review of Literature
41
DCL1 in the D- bodies that are distinct from the Cajal bodies (Song et al 2007)
11352 MiRNA-directed translational repression
Translation repression is another mechanism exerted by plant miRNAs The regulatory
mechanism of miR172 which regulates flowering time and floral organ identity is
consistent with translation repression MiR172 over production does not affect the
transcript levels of APETALA2 (AP2) and AP2- like target mRNAs Instead the AP2
protein level is reduced in the miR172 over expressing mutant (Aukerman amp Sakai
2003 Chen 2004 Dalmay 2013) It was later shown that miR172 can also direct the
cleavage of the target mRNAs although the steady-state mRNA level is unchanged due
to feedback regulation (Schwab et al 2005 Jung et al 2007) It is clear that miR172
regulates its target genes primarily by translational repression rather than mRNA
cleavage Recently it has been reported that miR156157 and miR854 also regulate
their target genes through translational repression (Arteaga-Vazquez et al 2006
Gandikota et al 2007 Dalmay 2013) It was once thought that translational repression
is an exception to the rule However experimental evidence suggest a widespread
coexistence of translational repression and mRNA cleavage (Brodersen et al 2008)
1136 Regulatory roles of miRNAs in plant development
The miRNA target prediction has been a difficult task in order to obtain regulatory
targets without bringing in too many false predictions Plant growth and developmental
processes regulated by miRNAs are quite diverse and enormous Furthermore plant
miRNAs work cooperatively or antagonistically to establish a balanced regulation This
notion is well consistent with the findings that mutations in the regulators of miRNA
biogenesis transport and processing such as AGO1 DCL1 HYL1 HEN1 ABH1
and HASTY cause pleiotropic phenotypes (Mallory amp Vaucheret 2006 de Alba et al
2013) To date the roles of miRNAs have been mostly deduced from obtaining miRNA
over expressing transgenic plants or gain-of-function mutants in which miRNA-
resistant target genes are ectopically expressed
11361 Phase transition
Plants pass through a series of developmental phase transitions during their life span
beginning with seed germination continuing through vegetative phase change
reproductive phase change flowering initiation and finally seed production for the next
generation Because of their importance in understanding plant developmental
Chapter 1 Introduction and Review of Literature
42
processes genes regulating developmental transitions have been extensively studied
and underlying molecular mechanisms have been explored in various plant species
Majority of researches were mainly focused on vegetative phase change and
reproductive transition in Arabidopsis and Maize (Poethig 2003 Baurle amp Dean
2006) Particularly the mutations of the genes encoding various regulators of miRNA
biogenesis and transport such as HST SE and AGO exhibit altered juvenile to adult
vegetative phase change and vegetative to reproductive change (Hunter et al 2003
Peragine et al 2004 Vazquez et al 2004a Jones-Rhoades et al 2006 de Alba et al
2013)
Over-expression of the miR156a gene causes late flowering and delays
vegetative phase change (Wu amp Poethig 2006 Gandikota et al 2007 Wang et al
2008) It has been shown that miR156 negatively regulates a group of genes encoding
SQUAMOSA PROMOTER-BINDING PROTEIN-LIKE (Lewis et al 2007 de Alba et
al 2013 Rogers amp Chen 2013) transcription factors such as SPL3 SPL4 and SPL5
that promote flowering initiation In contrast transgenic plants over expressing
miR156-resistant SPL345 genes are early flowering Interestingly the endogenous
level of miR156 is temporally regulated In the early juvenile vegetative phase the
level of miR156 is very high but decreases rapidly before the onset of the adult juvenile
phase (Wu amp Poethig 2006)
The Arabidopsis early activation tagged (eat-D) mutant exhibits early flowering
with disrupted floral structure The mutant phenotypes are caused by over expression of
the miR172a-2gene (Aukerman amp Sakai 2003) MiR172 regulates a small group of
genes encoding APETALLA2 (AP2)-like transcription factors such as TARGET OF
EAT1 (TOE1) TOE2 SCHLAFMUTZE (SMZ) and SCHNARCHZAPFEN(SNZ)
TOE1 TOE2 SMZ and SNZ have been shown to participate in their productive phase
change by repressing a floral integrator FLOWERING LOCUS T (FT)(Jung et al
2007) Consistent with this toe1-2D 35STOE2 35SSMZ and 35SSNZ plants
exhibit late flowering (Jung et al 2007) MiR172 is also regulated temporally and
highly expressed in the adult vegetative phase both in Arabidopsis and maize (Chuck et
al 2007) Because the temporal expression patterns of miR156 and miR172 are
complementary to each other it has been suggested that the two miRNAs interact with
each other in regulating phase change (Chuck et al 2009 de Alba et al 2013) In
support of this view it has been shown that miR172 is down-regulated in the
Chapter 1 Introduction and Review of Literature
43
Corngrass1(Cg1) mutant in which miR156 is overproduced (Chuck et al 2007 de
Alba et al 2013) However there is no direct evidence for the direct linkage between
miR156 and miR172 It is also envisioned that miR156 and miR172 would not be
directly related For example miR156 regulates plastochron index that is the inverse of
leaf initiation rate and thus frequently used to analyze temporal patterns of plant
development In contrast miR172 does not affect plastochron (Wang et al 2008) The
down-regulation of miR172 in the maize miR156-over producing mutants would be
due to the prolonged juvenile vegetative phase in the mutants
Another miRNA that regulates flowering initiation is miR159 It regulates
MYB33 and MYB65 which are closely related to the barley GAMY B transcription
factor that responds to gibberellic acid (GA) The level of miR159 is elevated in GA-
treated seedlings but reduced in the GA biosynthetic ga1 mutant Transgenic plants
over producing miR159 exhibit delayed floral induction under short days (Achard et
al 2004) It is been suggested that MYB33 mediates GA signal to regulate LEAFY
(LFY) These observations indicate that miR159 plays an important role in the GA
signaling pathways that regulate proper floral induction under short days (Achard et al
2004)
11362 Floral development
Arabidopsis flowers consist of four distinct organs They arise in a characteristic
pattern within concentric rings called whorls from outside to inside four sepals four
petals six stamens and the central pistil consisting of two carpels Identities of the
floral organs are determined by three classes of regulatory genes classA classB and
class C genes (Krizek amp Fletcher 2005) The A and C class genes act antagonistically
to restrict their expression domains (Bowman et al 1991)
It is notable that miR172 also regulates floral architecture in addition to its role
in the timing of flowering initiation MiR172 inhibits the translation of the AP2 mRNA
(Chen 2004) Transgenic plants overproducing miR172 not only exhibit early
flowering but also show abnormal floral development which is quite similar to those
of the class A gene mutants such as the ap2 mutants (Aukerman amp Sakai 2003 de
Alba et al 2013) When the activity of the class A genes is inactivated that of the class
C genes such as AGAMOUS (AG) is elevated As a result the miR172- overproducing
transgenic plants exhibit no petals along with homeotic transformation of the sepals to
Chapter 1 Introduction and Review of Literature
44
the carpels (Aukerman amp Sakai 2003 Chen 2004)
Interestingly miR166 also regulates floral architecture The role of miR165166
in the regulation of meristem activity is closely linked to floral meristem formation and
organization (Zhao et al 2007 Miyashima et al 2013) Floral structure is severely
disrupted in the miR165166-overproducing mutants such as men1 and jba-1D in
which miR166 is overproduced (Williams et al 2005b Jung amp Park 2007) The men1
and jba-1D mutants have smaller gynoecium and reduced carpel number In an extreme
case the jba-1D homozygous plants lack visible carpels (Williams et al 2005b)
Similar phenotypes have been observed in the miR165 overproducers (Zhao et al
2007) It has been suggested that the phenotypes are possibly caused by altered AG
expression in the floral meristem (Jung amp Park 2007 Turchi et al 2013)
Other miRNAs also affect floral development Overproduction of miRNAs
involved in auxin signaling also affects floral architecture Transgenic plants
overproducing miR167 have defects in the formation of ovule and anther with reduced
fertility (Ru et al 2006) It has been shown that miR167 targets ARF6 and ARF8 that
play a role in gynoecium and stamen development and in regulating fertility (Ru et al
2006 Wu et al 2006) These observations suggest that auxin signals are also closely
related to floral organogenesis In addition transgenic plants over-producing miR164
and the cuc1cuc2 double mutants show fused sepals and reduced petals (Laufs et al
2004 Turchi et al 2013) suggesting that miR164 is also related to floral meristem
activity and boundary specification of the meristematic region
11363 Shoot apical meristem development
Shoot apical meristem comprises self-maintaining totipotent cells The shoot apical
meristem activity affects diverse aspects of plant developmental processes including
leaf development vascular development and lateral organ polarity (Bowman et al
2002) Because miR165166 plays a primary role in meristem formation
overproduction of miR165166 and multiple knockout mutants of its target genes
exhibit pleiotropic pheonotypes (McConnell et al 2001 Emery et al 2003 Kim et al
2005 Williams et al 2005b)
The targets of miR165166 include genes encoding the homeo domain-leucine
zipper (HD-ZIP) class III transcription factors such as PHABULOSA(PHB)
PHAVOLUTA (PHV) REVOLUTA(REV) ATHB15CORONA (CNA) and ATHB8
Chapter 1 Introduction and Review of Literature
45
PHB PHV and ATHB15 have overlapping functions in controlling meristem
development (Turchi et al 2013) Indeed the phb phv athb15 triple mutants have an
enlarged shoot apical meristem Consistent with this the miR166- overproducing men1
and jba-1D mutants exhibit enlarged shoot apical meristems (Kim et al 2005
Williams et al 2005b Turchi et al 2013) In the mutants the shoot apical meristems
are multiplied and large
The development of shoot apical meristems is also regulated by the WUSCHEL
(WUS)ndashCLAVATA (CLV) signaling pathway WUS positively regulates cell
proliferation In contrast CLV3 which is expressed in stem cells limits the WUS
expression and thus inhibits cell proliferation in the SAM (Sharma et al 2003)
Consistent with this notion WUS is expressed to a higher level in jba-1D
explaining the ectopic enlargement of the SAM in the mutant (Williams et al 2005)
While WUS is closely related to miR165166 in the shoot apical meristem
development the underlying molecular mechanisms are currently unclear It has been
observed that the miR166-over producing men1 plants have an arrested meristem
development (Jung amp Park 2007) In addition the heterozygous men1+ and the
homozygous men1 plants show different expression patterns of WUS and CLV3 It
appears that miR166 regulates WUS indirectly and influences the expressional balance
between WUS and CLV3 through a negative feedback loop (Jung amp Park 2007 de
Alba et al 2013)
The phv phb athb15 triple mutant has an enlarged shoot apical meristems the
rev phb phv mutant does not have functional shoot apical meristems (Prigge et al
2005) This distinction may be explained by the fact that while miR166 targets
primarily PHB PHV and ATHB15 miR165 targets all the five HD-ZIP III genes (Zhou
et al 2007) Consistent with this transgenic plants overproducing miR166 are distinct
from those overproducing miR165 The miR165-overproducing transgenic plants lack
functional shoot apical meristem and a pin-like structure is formed at the apical region
of the mutant These phenotypes are quite similar to rev phb phv triple mutant (Zhou et
al 2007 Liu et al 2013) The phenotypic distinction may because by the difference
of the cleavage efficiencies of the target mRNAs In accordance with this notion a few
35S miR166a transgenic lines exhibit no shoot apical meristems The men1
homozygous plants also show arrested meristem development (Jung amp Park 2007)
Chapter 1 Introduction and Review of Literature
46
MiR164 cleaves the CUC1 and CUC2 mRNAs as well as the NAC1 mRNA
CUC1 and CUC2 determines the organ boundary in the meristem Phenotypes of
transgenic plants that overproduce miR164 are similar to those of the cuc1cuc2 double
mutant that exhibits embryonic developmental defects such as cup-shaped cotyledons
and fused cotyledons (Laufs et al 2004) When a miR164-resistant CUC2 is
expressed the boundary domain is enlarged CUC also plays a role in embryonic shoot
meristem formation by regulating the SHOOT MERISTEMLESS gene It was therefore
concluded that miR164-mediated cleavage of CUC1 and CUC2 mRNAs determines the
sizes of the boundary domains in the shoot apical meristem and regulates separation of
the organs by restricting cell proliferation (Laufs et al 2004 de Alba et al 2013)
11364 Leaf development
The role of miR319 has been extensively studied in leaf development A dominant
jaw-D mutant overproducing miR319 shows uneven curved leaves with enlarged size
and five TCP genes which are closely related to the CINCINNATA (CIN) gene in
Antirrhinum majus are down regulated in the mutant suggesting that miR319 mediated
regulation of the TCP transcription factor genes are important for the final step of
leaf shaping (Palatnik et al 2003 Naz et al 2013) In addition to leaf shape and size
leaf polarity is also specified by miRNA MiR166-mediated cleavage of the HD-ZIPIII
mRNAs is a well to establish leaf polarity This discovery originates from the gain-of-
function mutants such as phb-1d and phv-1d wherein point mutations in the
complementary sites to miRNA helps the mRNA to evade or resist from miRNA-
mediated cleavage (McConnell et al 2001 Emery et al 2003 Naz et al 2013)
Plant leaves have a dorso-ventral polarity consisting of the adaxial (upper
surface) and abaxial (lower surface) sides The adaxial side is closer to the shoot
apical meristem Adaxial identity is specified by functionally redundant class III HD-
ZIP family genes such as PHB PHV and REV Ectopic expression of each genes
result in adaxialized leaves Dominant phb-1d and phv-1d mutants exhibit
transformation of the abaxial to adaxial fate and have rod-shaped leaves In contrast
miR165166-overproducing plants show pin-like cotyledons radicalized leaves and
abaxialized leaves (Chitwood et al 2007 Pattanayak et al 2013)
Leaf asymmetry is determined by miR166-mediated restriction of the
expression domains of the HD-ZIPIII genes The HD-ZIPIII genes are expressed in the
Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
5
Coffee is mostly grown in south India parts of Orissa W Bengal and North east
India Karnataka is the largest producer with 569 of total area under production
which accounts for 71 of the total produce The total crop harvest for 2012-2013 is
placed at 325300 tonnes (httpwww Indicoffeeorgindiacoffeephpp-
age=CoffeeData)
14 Composition of coffee beans
Navellier (1961) gave a mean composition of green coffee as Carbohydratesglucides
(58) lignin (2) lipids (13) proteins (13) ash (4) non-volatile acids (8)
trigonelline (1) and caffeine (1) The caffeine content of green coffee is relatively
limited (10-25) of dry matter and changes little with seed development (Clifford
and Kazi 1987) In coffee seeds the concentration of trigonelline is approximately
2 of dry weight (Clifford 1985 Mazzafera 1991) Mazzafera (1999) found a higher
protein content in the mature beans than in the immature beans but a lower content of
free amino acids with asparagine as the main component Flament ( 2000) estimated
the composition of coffee beans as follows
Proteins 100 (dry weight basis of green coffee)
Carbohydrates 500 -do-
Lipids 117-140 (Arabicas)
76- 95 (Robustas)
Chlorogenic acids 65 (Arabicas)
90 (Robustas)
15 Purine alkaloids structure and role
Purine alkaloids are derived from purine nucleotides (Zulak et al 2007) and found in
over hundred plant species in 13 orders in plant kingdom (Ashihara amp Crozier 1999)
Methylxanthines like caffeine (137-trimethlyxanthine) theobromine (37-methylxan-
thine) and methyluric acids are classified into purine alkaloids (Figure 11) (Table
13)(Ashihara et al 2008) These are known to occur in tea coffee and a number of
non-alcoholic beverages Caffeine was isolated from coffee (Coffea arabica) and tea
(Camellia sinensis) in 1820 (Ashihara amp Crozier 2001)
Chapter 1 Introduction and Review of Literature
6
Figure 11 Purine alkaloid xanthine and uric acid skeleton
Plants that accumulate purine alkaloid are classified into three groups based on
the type of alkaloids they produce caffeine-producing plants which include coffee
tea and mateacute (Ilex paraguariensis) theobromine ndash producing plants are represented
by cacao cocoa tea (Camellia ptilophylla) and Camellia irrawadiensis and
methyluric acid ndash producing plants consist of Coffea dewevrei Coffea liberica C
excelsa and Kucha tea (Camellia assamica)
Table 13 Purine alkaloid structures based on xanthine and uric acid skeleton
Compound Trivial name R1 R2 R3 R4 O-2 Δ2-3
Methylxanthines
Xanthine H H H
1-methylxanthine CH3 H H
3-methyl xanthine H CH3 H
7-methyl xanthine H H CH3
1-3methylxanthine Theophylline CH3 CH3 H
17-methylxanthine Paraxanthine CH3 H CH3
37-methylxanthine Theobromine H CH3 CH3
137-methylxanthine Caffeine CH3 CH3 CH3
Uric acid and methyl uric acids
Uric acid H H H H
137-trimethyluric acid CH3 CH3 CH3 H
1379-tetramethyluric
acid Theacrine CH3 CH3 CH3 CH3
O19-trimethyluric acid Liberine CH3 H H CH3 CH3 Δ2-3
O179-trimethyluric acid Methylliberine CH3 H CH3 CH3 CH3 Δ2-3
16 Role of caffeine in plants
The physiological role of endogenous purine alkaloids and related compounds in
higher plants remains undetermined There are two hypotheses about the role of the
Chapter 1 Introduction and Review of Literature
7
high concentrations of caffeine that accumulate in tea coffee and a few other plant
species The lsquoallelopathic theory or the auto toxic function theoryrsquo proposes that
caffeine in seed coats is released into the soil and inhibits the germination of seeds
around the parent plants (Anaya et al 2006)
The lsquochemical defense theoryrsquo proposes that caffeine in young leaves fruits
and flower buds acts to protect soft tissues from insect pests (Ashihara amp Crozier
2001 Ashihara et al 2008) It has been shown that spraying tomato leaves with a 1
solution of caffeine deters feeding by tobacco horn worms while treatment of
cabbage leaves and orchids with 001ndash01 solutions of caffeine acts as a neurotoxin
and kills or repels slugs and snails (Hollingsworth et al 2002) This work has now
been extended with convincing evidence for chemical defense theory has recently
been obtained with transgenic caffeine ndash producing tobacco plants (Uefuji et al 2005
Kim et al 2006)
17 Distribution of caffeine in plants
Caffeine has been found in 13 orders of the plant kingdom (Ashihara and Suzuki
2004 Anaya et al 2006) In some species the main purine alkaloid is theobromine or
methyl uric acids such as theacrine liberine and methylliberine rather than caffeine
(Ashihara amp Crozier 1999 Ashihara amp Crozier 2001)(Table 14)
Table 14 Distribution of purine alkaloids in plants
SNo Plant species Common
name Major alkaloid Plant parts
containing
alkaloids 1 Coffea arabica Arabica Caffeine Leaves Seeds
2 Coffea canephora Robusta Caffeine Leaves Seeds
3 Coffea liberica Caffeine Theacrine Liberine
Seeds Mature leaves
4 Coffea dewevrei Caffeine Theacrine Liberine
Seeds Mature leaves
5 Camellia sinensis Tea Caffeine Leaves
6 Camellia assamica Assam tea Caffeine Leaves
7 Camellia assamica var
kucha Kucha Theacrine Caffeine Leaves
8 Camellia irrawadensis Theobromine Leaves
9 Camellia ptilophylla Cocoa tea Theobromine Leaves
10 Theobroma cacao Cocoa Theobromine Seeds
11 Paullinia cupuna Guarana Caffeine Seeds
Chapter 1 Introduction and Review of Literature
8
12 Cola nitida Caffeine Seeds
13 Citrus sp Caffeine Pollen
14 Ilex paraguariensis Mate Caffeine Leaves
Adapted from (Ashihara amp Crozier 1999 Ashihara amp Crozier 2001)
18 Caffeine biosynthesis
The major biosynthetic pathway is a four step sequence consisting of three sequential
methylation and one nucleosidase reactions (Figure 12) The xanthine skeleton of
caffeine is derived from purine nucleotides The initial step in caffeine biosynthesis is
the methylation of xanthosine by a SAM- dependent N-methyltransferase In addition
to experiments with radiolabeled precursors substrate specificities of native (Kato et
al 1999) and recombinant N-methyltransferases (Kato et al 2000 Ogawa et al
2001 Mizuno et al 2003a Mizuno et al 2003b Uefuji et al 2003) strongly
suggest that the major route to caffeine is a xanthosine rarr7-methylxanthosinrarr 7-
methylxanthine rarr theobromine rarr caffeine pathway (Figure 12) Although the
information has been obtained mainly from coffee (C arabica) and tea (Camellia
sinensis) the available evidence indicates that the pathway is essentially the same in
other purine alkaloid-forming plants such as mateacute (Ashihara 1993) and cacao
(Koyama et al 2003 Yoneyama et al 2006)
The first step in the caffeine biosynthetic pathway from xanthosine is
conversion of xanthosine to 7-methylxanthosine (Figure 12) This reaction is
catalyzed by the 7-methylxanthosine synthase (xanthosine 7 N-methyltransferase EC
211158) The genes encoding for 7-methylxanthosine synthase CmXRS1 (AB
034699) and CaXMT1 (AB 048793) were isolated from the C arabica (Mizuno et
al 2003a Uefuji et al 2003) The recombinant proteins obtained from these genes
exhibit 7-methylxanthosine synthase activity in vitro Xanthosine monophosphate
(XMP) was not converted to 7-methylxanthosine by the recombinant enzymes thus
inclusion of 7-methy-XMP was proposed (Schulthess et al 1996)
The second step of caffeine biosynthesis involves a nucleosidase which
catalyses the hydrolysis of 7-methylxanthosine Although N-methylnucleosidase (EC
32225) was partially purified from tea leaves (Negishi et al 1988) recent detailed
structural studies on coffee 7-methylxanthosine synthase suggested that the methyl
transfer and nucleoside cleavage may be coupled and catalyzed by a single enzyme
(McCarthy amp McCarthy 2007)
Chapter 1 Introduction and Review of Literature
9
The last two steps of the caffeine synthesis are also catalyzed by a SAM-
dependent N-methyltransferase(s) which is different from the N-methyltransferase
enzyme that catalyzes the first step in the pathway Native N-methyltransferase
activities have been detected in crude and partially purified extracts from tea and
coffee plants (Ashihara amp Suzuki 2004) and a highly purified preparation has been
obtained from young tea leaves (Kato et al 1999) The enzyme was assigned the
name caffeine synthase (EC 211160) that catalyses the last two steps of caffeine
biosynthesis viz the conversion of 7-methylxanthine to caffeine via theobromine
(Figure 12) The gene encoding caffeine synthase was first cloned from young tea
leaves (Kato et al 2000) Since then several genes encoding N-methyltransferases
with different substrate specificity have been reported
Activity of the recombinant theobromine synthase (CTS1 and CaMXMT EC
211159) is specific for the conversion of 7-methylxanthine to theobromine In
contrast the recombinant caffeine synthases (CCS1 and CaDMXMT1) can utilize
paraxanthine theobromine and 7-methylxanthine as shown with tea caffeine synthase
(TCS1) (Mohanpuria et al 2011) Although paraxanthine is the most active substrate
of this recombinant enzyme only limited amounts of paraxanthine are synthesized in
coffee cells hence in vivo caffeine synthase is principally involved in the conversion
of 7-methylxanthine to caffeine via theobromine
Radiolabelled tracer experiments with theobromine accumulating cacao and
Chinese tea (Camellia ptilophylla) showed a limited conversion of theobromine to
caffeine N-methyltransferase which catalyzed the conversion of 7-methylxanthine to
theobromine was detected in crude extracts from C ptilophylla but conversion of
theobromine to caffeine was not observed (Ashihara et al 1998) Genes homologous
to caffeine synthase have been isolated from several theobromine accumulating plants
including cacao (Yoneyama et al 2006) The recombinant enzymes derived from
these genes have 3 N-methyltransferase activity suggesting that these theobromine
accumulating plants contain specific theobromine synthase Caffeine does not occur
in C ptilophylla (Ashihara et al 1998) although it is present in leaves and fruits of
cacao along with theobromine (Koyama et al 2003 Zheng amp Ashihara 2004)
Chapter 1 Introduction and Review of Literature
10
Figure 12 Pathways for the biosynthesis of caffeine
The major pathway is indicated by solid lines (Mizuno et al 2003b Uefuji et al 2003) Alternative pathways in coffee and tea plants are indicated
by dashed lines(Kato et al 1996 Schulthess et al 1996) Enzymes (1) 7-methylxanthosine synthase (2) N-methylnucleosidase (3) Theobromine
synthase (4) Caffeine synthase
N
N
N
N7
H3C
3
1
O
O
Caffeine
N
NH
N
N7
3
O
O
Theobromine
N
N
N
N
H
H
7
O
O
N
N
N
N
H
H
O
O
N
N
N
N
H
H
O
O
Xanthosine
N
N
N
N
H
H
O
O
N
N
N
N
H
H
O
ON
N
N
N
H
H3C
71
O
O
Paraxanthine
CH3
CH3
CH3
CH3CH3
7-methylxanthine
Rib P
Rib
(1)
(4)
Rib
+
CH3
7-methyl
xanthosine
(2)
(3)
SAM SAH
SAM SAH SAM SAH
Xanthosine mono
phosphate
Rib P
7-methyl xanthosine
monophosphate
+
CH3 CH3
Inosine 5-mono
phosphate
Adenosine 5-mono
phosphate
SAM SAH
Guanosine
Guanosine 5-mono
phosphate
Chapter 1 Introduction and Review of Literature
11
181 Source of Xanthosine for caffeine biosynthesis
Xanthosine the initial substrate of purine alkaloid synthesis is supplied by at least four
different pathways de novo purine biosynthesis (de novo route) the degradation
pathways of adenine nucleotides (AMP route) and guanine nucleotides (GMP route)
and the S-adenosyl-L-methionine (SAM) cycle (SAM route) (Figure 13) Purine
nucleotides are synthesized by de novo and salvage pathways (Ashihara amp Crozier
1999 Stasolla et al 2003 Zrenner et al 2006) The de novo synthesis of non purine
precursors like CO2 10-formyltetrahydrofolate 5-phosphoribosyl-1-pyrophosphate and
the amino acids Glycine glutamine and aspartate results in the production of purine
nucleotides
Figure 13 Xanthosine biosynthesis Xanthosine for purine alkaloid biosynthesis is supplied by at
least four routes from adenosine released from the SAM cycle (SAM route) from IMP
originating from de novo purine synthesis (de novo route) from the cellular adenine
nucleotide pool (AMP route) and from the guanine nucleotide pool (GMP route)
Adapted from (Ashihara et al 2008)
The utilization of IMP for caffeine biosynthetic pathway which is formed by the
de novo purine biosynthetic pathway for caffeine biosynthetic pathways was
demonstrated in young tea leaves using 15
N-glycine and 14
C-labelled precursors and
inhibitors of de novo purine biosynthesis (Ito amp Ashihara 1999) Xanthosine is formed
by an IMP rarr XMP rarr xanthosine pathway IMP dehydrogenase (EC 111205) and 5prime-
nucleotidase (EC 3135) catalyze these reactions The inhibition of IMP dehydrogen-
ase by ribavirin inhibits both caffeine and gunanine nucleotide biosynthesis in tea and
coffee leaves (Keya et al 2003)
Chapter 1 Introduction and Review of Literature
12
The portion of xanthosine used for caffeine biosynthesis is derived from adenine
and guanine nucleotide pools which are produce de novo by salvage pathways there are
several potential pathways for xanthosine synthesis from AMP but the AMP rarr IMP
rarr XMPrarr xanthosine route is predominant All three enzymes involved in the
conversion AMP deaminase (EC 3546) IMP dehydrogenase and 5prime-nucleosidase
have been detected in tea leaves (Koshiishi et al 2001) Xanthosine for caffeine
biosynthesis can also be produced from guanine nucleotides by GMPrarrguanosine
rarrxanthosine pathway Guanosine deaminase (EC 35415) activity is found in cell free
extracts from young tea leaves (Negishi et al 1994) but GMP deaminase activity was
not detected in plants (Stasolla et al 2003 Zrenner et al 2006) The absence of GMP
deaminase activity suggests that a GMP rarr IMP rarr XMP rarr xanthosine route is not
functional in plants
The use of radiolabelled purine bases and nucleosides for the study of caffeine
biosynthetic pathway was facilitated by the fact that exogenous purine nucleotides are
rapidly hydrolyzed by the plant tissues forming purine nucleosides andor bases which
are then utilized in pathways which in most instances indirectly lead to xanthosine
Most of these purine compounds including adenine adenosine inosine hypoxanthine
and guanine are converted to their respective nucleotides by salvage enzymes
including adenine phosphoribosyl transferase (EC 2427) hypoxanthineguanine
phosphoribosyl transferase (EC 2428) adenosine kinase (EC 27120) and
inosineguanosine kinase (EC 2421) which usually have high activities in plant cells
The resultant nucleotides then enter the purine alkaloid biosynthetic pathway In the
case of guanosine it may be directly deaminated to xanthosine and utilized for caffeine
biosynthesis (Figure 13) (Ashihara et al 1997)
182 SAM cycle in plants
SAM is the methyl donor for the three methylation steps in the caffeine biosynthetic
pathway In this process SAM is converted to S-adenosyl-L-homocysteine (SAH)
which in turn is hydrolyzed to homocysteine and adenosine Homocysteine is recycled
via the SAM cycle to replenish SAM levels and adenosine is released from the cycle
Adenosine released from the cycle is converted to AMP directly andor indirectly via
adenine AMP is converted to xanthosine and used for caffeine biosynthesis Since
three moles of SAH are produced via the SAM cycle for each mole of caffeine that is
synthesized in theory this pathway has the capacity to be the sole source of both the
Chapter 1 Introduction and Review of Literature
13
purine skeleton and the methyl groups required for caffeine biosynthesis in young tea
leaves (Koshiishi et al 2001)
Methionine
S-Adenosyl-L-methionineHomocysteine
ATP
PPi+ Pi
Tetrahydrofolate
5-Methyl-
tetrahydrofolate
CH3+Adenosine
S-Adenosyl-L-
homocysteine
(xanthine
structure)(Methyl groups)
Caffeine
(1)
(2)(3)
(4)
Figure 14 The SAM cycle (activated methyl cycle) in plants (Ashihara amp Suzuki 2004)
Enzymes SAM synthetase SAM-dependent N-methyltransferases S-adenosyl-
homocysteine (SAH) hydrolase Methionine synthase Adenosine released from the cycle
is salvaged to adenine nucleotides and utilized both for purine structure of caffeine via
xanthosine and for re-synthesis of SAM via ATP
183 N-methyltransferase in caffeine biosynthesis Molecular cloning
The genes encoding for the enzymes involved in the caffeine biosynthesis were
successfully cloned from the coffee family (Ogawa et al 2001 Mizuno et al 2003a
Mizuno et al 2003b Uefuji et al 2003) The conserved amino acid regions of tea CS
(AB31280) the sequences of O-methyltransferases derived from plant origin and
Arabidopsis hypothetical proteins were made use of in designing primers for RT-PCR
and RACE technique Independent groups have isolated N-methyltransferase (NMT)
genes from coffee which have been classified based on the substrate specificity of the
recombinant proteins expressed in Ecoli (Table 15) PCR products thus obtained were
used as the probe to isolate full length cDNA from the library resulting in the
identification of all three N-methyltransferases involved in caffeine biosynthesis
(Figure 12) Several cDNA have been characterized from the coffee plants one for
xanthosine methyltransferase (XMTXRS) three for 7-methylxanthine methyltransferase
or theobromine synthase (MXMTCTS) and three for 37-dimethylxanthine
methyltransferase or caffeine synthase (DXMTCCS) (Table 15) (Kato amp Mizuno
2004)
A single gene encoding for xanthosine methyltransferase (XMTCmXRS1) was
identified which encoded for a polypeptide consisting of 372 amino acids with an
apparent molecular mass of 418kDa It is expressed almost uniformly in aerial tissues
Chapter 1 Introduction and Review of Literature
14
of C arabica including leaves floral buds and immature but not in mature beans
Three genes encoding 7-methylxanthine methyltransferase (theobromine synthase) have
been identified The number of amino acids in the putative polypeptides are 378 for
MXMT1 (427kDa) and 384 for MXMT2 and CTS2 (434kDa) They differ by insertion
or deletion of blocks of several residues in the C-terminal region Their catalytic
properties based on kinetic parameters such as Km values were different They are
expressed in young leaves floral buds and immature but not mature beans Three genes
were identified for 3 7-dimethylxanthine methyltransferase (caffeine synthase) DXMT
CCS1 and CtCS7 each encoding a 43 kDa polypeptide consisting of 384 amino acids
However their kinetic properties differ from each other such as DXMT and CCS1
showed Km values of 1200 and 157microM for theobromine respectively Expressions
profiles are also distinct DXMT being expressed exclusively in immature beans while
CCS1 expression is ubiquitous occurring in all tissues The presence of isoforms of
these enzymes with different properties suggests that caffeine is synthesized through
multiple pathways depending on availability and concentration of the substrate (Mizuno
et al 2001 Ogawa et al 2001 Mizuno et al 2003a Mizuno et al 2003b Uefuji et
al 2003) Genomic clones of theobromine synthase were obtained by PCR- RFLP
from C canephora (Satyanarayana et al 2005 Satyanarayana 2006) The genomic
clones are reported to have highly conserved exons and a variable third intron The
promoter for the theobromine synthase was cloned and characterized (Satyanarayana et
al 2005)
There is high degree of similarity in the coding region of coffee NMT genes
isolated so far though differences exist in the 3prime untranslated region (UTR) for these
genes The deduced amino acid sequence of these enzymes shows more than 85
homology between these genes Phylogenetic analysis indicates that they are more
closely related to C-methyltransferases including those for jasmonic acid salicylic acid
and benzoic acid than to other N-methyltransferases This suggests that coffee N-methyl
transferases belong to a distinct sub-group within plant methyltransferases Their
cellular localization determined by green fluorescent protein fusion method and β-
glucourodinase (GUS) showed that the all the three genes are localized in the cytoplasm
(Ogawa et al 2001 Vinod et al 2007) It was also shown that these were present
along with chlorogenic acid(Vinod et al 2007)
Chapter 1 Introduction and Review of Literature
15
Table 15 N-methyltransferases and their encoding genes involved in caffeine biosynthesis
Gene (Enzyme) Gene name Accession No Description Reference
Xanthosine methyltransferase
( 7-methylxanthosine synthase) (EC 211158)
CaXMT AB 048793 Immature fruit cDNA Screening (Uefuji et al 2003)
CmXRS1 AB 034699 Leaf cDNA library RACE (Mizuno et al 2003a)
CaMXMT1 AB 048794 Leaf cDNA library RACE (Ogawa et al 2001)
7-methylxanthine synthase transferase
(theobromine synthase) ( EC 211159)
CTS1 AB 034700 Immature fruit cDNA Screening (Uefuji et al 2003)
CaMXMT2 AB 984125 Leaf cDNA library Screening (Mizuno et al 2001)
CTS2 AB 054841 Leaf cDNA library Screening (Mizuno et al 2001)
37-methylxanthinemethyltransferase
(Caffeine synthase) (EC 211160)
CaDXMT1 AB 086414 Immature fruit cDNA Screening (Uefuji et al 2003)
CCS1 AB 0861414 Immature fruit cDNA Screening (Uefuji et al 2003)
CtCS7 AB 0864415 Immature fruit cDNA
NMT genomic clones
Theobromine synthase-1
CX10 Complete coding region PCR Satyanarayan 2006
CX8 Complete coding region PCR -do-
Theobromine synthase-1 PG-5 Promoter + coding region RAGE -do-
Theobromine synthase-1 PG-1 Promoter + coding region RAGE Satyanarayan 2006
PG-4 Promoter + coding region RAGE -do-
Theobromine synthase-1 NMT
(AY273814) Complete coding region PCR Kochko A et al 2010
Theobromine synthase-2 NMT
(AY362825) Complete coding region PCR Kochko A et al 2010
Chapter 1 Introduction and Review of Literature
16
19 Catabolism of caffeine
Caffeine is produced in young leaves and immature fruits and continues to accumulate
gradually during the maturation of these organs At the same time caffeine is slowly
degraded with the removal of the three methyl groups resulting in the formation of
xanthine Catabolism of caffeine was first reported in coffee leaves (Kalberer 1965)
Since then a number of tracer experiments using 14
C labelled purine alkaloids have
been reported (Suzuki amp Waller 1984 Ashihara et al 1996 Vitoria amp Mazzafera
1999 Mazzafera 2004) They demonstrated the major catabolic pathway is caffeine rarr
theophylline rarr 3-methylxanthine rarr xanthine Xanthine is further degraded by the
removal by the conventional purine catabolic pathway to CO2 and NH3 via uric acid
allatonin and allantoate (Figure 15) (Ashihara amp Crozier 1999 Stasolla et al 2003
Zrenner et al 2006) Caffeine catabolism usually begins with its conversion to
theophylline catalyzed by N7-demethylase The involvement of P450-dependent mono-
oxygenase activity for this reaction was suggested (Huber amp Baumann 1998
Mazzafera 2004) However the activity of this enzyme has not yet been determined in
cell free extracts [8-14
C] Theophylline degraded to CO2 at much faster rate than [8-14
C]
caffeine indicating that conversion of caffeine to theophylline is a major rate limiting
step of caffeine catabolism and a possible reason why caffeine accumulates in high
concentrations in tissues of C sinensis and C arabica It is widely believed that the
control of caffeine levels in plants is a function of the balance between rates of
synthesis and degradation and this balance seems to vary depending on the plant
species and the tissue developmental stage (Mazzafera 2004)
Chapter 1 Introduction and Review of Literature
17
Figure 15 Caffeine catabolic pathways Caffeine is mainly catabolised via theophylline and 3-
methylxanthine Xanthine is further degraded to CO2 and NH3 by the conventional
oxidative pathway Adapted from (Ashihara et al 2008)
Chapter 1 Introduction and Review of Literature
18
110 Genetic engineering of Coffee
Genetic transformation has potential applications in coffee agriculture for incorporating
genes which could provide desirable traits such as disease and insect resistance
drought and frost tolerance and herbicide resistance Transgenic technology can also be
used to increase nutritional value and improve cup quality produce varieties with
caffeine-free beans and for production of hybrid crops for molecular farming
Identification of target specific genes is one of the pre-requisites for developing
transgenic crops The availability of a large number of EST sequences in coffee and
initiation of coffee genome sequencing may speed up the gene discovery and accelerate
transgenic research efforts in coffee
1101 Insect Resistance
Production of insect resistant coffee plants is one of the major objectives of the
breeding programs The major pests attacking coffee include coffee berry borer (CBB
Hypothenemus hampei) white stem borer (WSB Xylotrechus quadripes) leaf miner
(Perileucoptera coffeela) and root nematodes (Meloidogyne spp and Pratylenchus
spp) The CBB is present in almost all the coffee growing countries and considered to
the most devastating pest in coffee To date there is no reported source of resistance to
CBB in the coffee gene pool Like CBB WSB is another serious pest in C arabica in
India and several other East Asian countries Both CBB and WSB belong to the order
Coleoptera For India controlling WSB is the biggest challenge and has therefore
become the highest research priority Robusta is generally resistant to WSB but the
interspecific Robusta Arabica hybrids are susceptible to WSB Although leaf miner is
not yet a serious pest in India and other East Asian countries it is an economically
important pest in East Africa and Brazil Effective chemical control of CBB and WSB
is difficult due to the nature of their life cycle inside the berry and stem respectively as
well as environmental concern regarding the use of pesticides Biological control
measures are adapted to combat these insect pests with varying degrees of success
Developing coffee plants resistant to these pests using genetic transformation
technology could be one of the alternative strategies to counter pest damage For insect
resistance several different classes of proteins from bacterial plant and animal sources
have been isolated and their insecticidal properties tested against many important pests
Amongst these proteins Bt toxins are most important and several transgenic crops
expressing Bt genes have been commercialized Coffee transgenic plants carrying a
Chapter 1 Introduction and Review of Literature
19
synthetic version of the cry1Ac gene have been produced (Leroy et al 2000 Mishra et
al 2002) Indeed this was the first report of an important agronomic trait being
introduced into a coffee plant The transgenic plants presented similar features in
growth and development compared to normal plants Transgenic plants highly resistant
to leaf miner under greenhouse conditions were tested under field conditions in French
Guyana for 4 years for pest resistance (Perthius et al 2005 Perthius et al 2006)
From 54 independent transformation events 70 of the events were resistant to leaf
miner Unfortunately the field trial was vandalized which led to the termination of the
experiment (Montagnon et al 2005)The effectiveness of Bt genes in controlling
coleopteran pests is well documented in corn and potato which indicates that Bt genes
might be effective against CBB The high toxicity of B thuringiensis sero var
israelensis against first in star larvae of CBB has been demonstrated (Mendez et al
2003) In another experiment an α-amylase inhibitor from Phaseolus vulgaris was
tested against CBB and found to have an inhibitory effect on its growth and
development (Grossi et al 2004)In addition to the CBB and WSB C arabica
varieties are also susceptible to endoparasitic root-knot nematode (Meloidogyne spp)
(Campos et al 1990 Mishra et al 2012)
So far 15 species have been reported to be parasites of coffee Controlling
nematodes is extremely difficult and currently seedling grafting with robusta rootstock
is followed as one of the control measure Sources of resistance specific to root-knot
nematodes have been identified in coffee trees (Bertrand et al 2001) and the Mex-1
gene conferring resistance to M exigua in C arabica is in the process of isolation (Noir
et al 2003)
1102 Tolerance to Abiotic Stress
In many coffee growing countries abiotic stress such as drought and frost are the major
climatic factors that limit coffee production Changes in climatic patterns due to global
climate change are considered increasingly important for coffee cultivation Drought
induces water stress in plants which affects vegetative growth and vigor and triggers
floral abnormalities and poor fruit set It also indirectly increases the incidence of pests
and diseases in the plants C arabica is generally more tolerant to water stress than C
canephora partly due to its extensive deep root system C racemosa is known to be a
good source of drought tolerance In India hybrids between C canephora and C
racemosa have been obtained and are currently under evaluation for drought tolerance
Chapter 1 Introduction and Review of Literature
20
In many coffee growing countries coffee is propagated in marginal areas where the
annual rainfall is below 1000thinspmm with prolonged dry spells of over 4-5 months In
those areas water shortage and unfavorable temperatures constitute major constraints
and the growth and productivity of C canephora are badly affected As with drought
periodic frost also affects coffee production in parts of Brazil The introduction of
drought and frost tolerant genes through genetic transformation would be of great
importance for alleviating these problems Research is now being carried out by several
groups to identify genes involved in biotic as well as abiotic stress The most promising
genetic engineering approach for drought tolerance includes the use of functional or
regulatory genes as well as the transfer of transcription factors In recent years plants
tolerant to high temperature and water stress have been the subject of intense research
(Chaves et al 2003 Chaves et al 2004 Coraggio et al 2004) For achieving drought
tolerance genes that have been targeted include those encoding enzymes involved in
detoxification or osmotic response metabolism enzymes active in signaling proteins
involved in the transport of metabolites and regulating the plant energy status (Chaves
et al 2003 Chaves et al 2004 Coraggio et al 2004) The dissection of molecular
mechanisms related to signal transduction and transcriptional regulation might help in
engineering drought tolerance in coffee
1103 Disease Resistance
Coffee leaf rust (CLR) caused by the fungus Hemileia vastatrix is the most important
disease in coffee with substantial loss to coffee production and productivity in all the
coffee growing countries In addition to leaf rust coffee berry disease (CBD) caused by
fungus Colletotrichum kahawae can be a devastating anthracnose leading to substantial
crop loss in Africa Several other fungal and bacterial diseases may also affect coffee
to a extent C arabica is more susceptible to many diseases than C canephora Though
most of the disease control measures rely upon chemical control they are more
expensive and labour intensive The long-term solution is the breeding of resistant
varieties which is the focus of many breeding programs However breeding for disease
resistant varieties is time consuming due to the perennial nature of coffee with its long
gestation period India has a long history of C arabica breeding especially for leaf rust
resistance and is the first country to demonstrate the existence of multiple races of leaf
rust Resistance to CLR is conditioned primarily by a number of major (SH) genes and
coffee genotypes are classified into different resistance groups based on their
Chapter 1 Introduction and Review of Literature
21
interaction with different rust races pathogen (van der Vossen 2001) Currently C
canephora provides the main source of resistance to pests and diseases including CLR
(H vastatrix) and CBD (C kahawae) and therefore used in breeding programs Other
diploid species like C liberica and C racemosa are being used as a source of resistance
to coffee leaf rust and coffee leaf miner respectively (Guerreiro et al 1999
Sreenivasan et al 1989) The development of coffee varieties resistant to major fungal
diseases such as CLR and CBD using transgenic technology will benefit the coffee
industry immensely During the last 15 years significant progress has been made in the
area of host-pathogen interactions (Staskawicz et al 1995 Feys et al 2000
Shivaprasad et al 2012) and many resistance genes involved in recognizing invading
pathogens have been identified and cloned (Takken et al 2000) A number of
signalling pathways induced because of pathogen infection have been dissected (Dong
et al 1998 Navarro et al 2006) Many antifungal compounds synthesized by plants to
combat fungal infection have been identified (Does et al 1998) Understanding the
specific induction of targeted pathways and identification of specific pathways
responsible for particular fungal resistance is important in order to employ this strategy
in transgenic technology The recent investigation of gene expression during coffee leaf
rust infection could provide an insight into the defence pathways operating in coffee
(Fernandez et al 2004 Nardi et al 2006) Efforts have been made to identify and
clone resistance genes from coffee for achieving durable resistance The genetic and
physical map of two resistance genes SH3gene conferring resistance to rust (Prakash et
al 2004) and the Ck1 gene conferring resistance to C kahawae CBD (Gichuru et al
2006) have been established These genes could be used for molecular marker assisted
breeding programs (Mishra et al 2012)
1104 Production of low-caffeine coffee
A widespread belief that the ingestion of caffeine can have adverse effects on
health resulted in the increased demand for decaffeinated coffee (Ashihara amp Crozier
2001) Several method were used to obtain decaffeinated plants by conventional
breeding techniques using low yielding varieties and mutants that accumulated
relatively low amounts of caffeine when compared to the commercially available ones
Low-caffeine and decaffeinated coffee represent around 10 of the coffee sales
around the world (Ribas et al 2006b) The industrial process for coffee decaffeination
can be expensive and affects the original flavour and aroma in coffee (David 2002)
Chapter 1 Introduction and Review of Literature
22
Transgenic coffee plants with suppressed caffeine synthesis using RNA interference
(RNAi) technology have been obtained (Ogita et al 2003 Ogita et al 2004) Specific
sequences in the 3prime untranslated region of the theobromine synthase gene (CaMXMT1)
were selected for construction of RNAi short and long fragments The caffeine and
theobromine content of the transgenic plants were reduced by ~ 70 when compared to
the untransformed plants In C canephora RNAi technology has also been employed
to silence the N-methyl transferase gene involved in caffeine biosynthesis (Vinod et al
2007) Promoter of an N-methyltransferase (NMT) gene involved in caffeine
biosynthesis was cloned (Satyanarayana et al 2005 Satyanarayana 2006) which will
be very useful for studying the regulation of caffeine biosynthesis
1105 Improvement in cup quality
Improvement in coffee cup quality requires elaborate knowledge of the chemical
constituents as well as the metabolic pathways involved in the elaboration of quality
The constituents of coffee beans include minerals proteins carbohydrates caffeine
chlorogenic acids (CGA) glycosides lipids and many volatile compounds that give
flavour to coffee by roasting Among these the role of three major constituents
sucrose CGA and trigonelline have been studied in coffee The sucrose content of
coffee bean is associated with coffee flavour the higher the sucrose content in green
beans the more intense will be the cup flavour (Clifford et al 1985 de Maria et al
1996) The sucrose content of C arabica (82-83) is higher than C canephora (33ndash
40) The sucrose amino acids ratio in green beans determines the profile of volatile
compounds Manipulating sucrose content in coffee bean is therefore important in
improving cup quality The sucrose synthase gene (CcSUS2) from C canephora has
been cloned and sequenced (Leroy et al 2005) This provides an opportunity to
manipulate the sucrose content in coffee Chlorogenic acids are products of
phenylpropanoid metabolism Chlorogenic acids are a group of hydroxycinnamoyl
quinic acids (HQA) formed by esterification between caffeic acids coumaric acids and
quinic acids (Clifford et al 1999) They are present in relatively large quantities in the
coffee bean and are the precursor of phenolic compounds in roasted beans C
canephora beans contain higher CGA (10) compared to C arabica beans (6-7)
CGA are known to have antioxidant properties as well as being associated with disease
resistance (Campa et al 2003) Genetic manipulations of genes involved in CGA
synthesis can serve either of these purpose by up- or down regulating the pathway
Chapter 1 Introduction and Review of Literature
23
Phenylalanine ammonia-lyase (PAL) catalyzes the first step of the phenylpropanoid
pathway leading to the synthesis of a wide range of chemical compounds including
flavonoids coumarins hydroxycinnamoyl esters and lignins (Hahlbrock amp Scheel
1989) Recently the full length cDNA and corresponding genomic sequences of PAL
from C canephora was isolated characterized and functionally validated (Mahesh et
al 2006 Parvatam et al 2006) This has opened up new possibilities for manipulating
the level of the PAL enzyme in coffee which in turn can be useful for improvement of
cup quality and manipulating antioxidant properties in coffee
1106 Fruit Ripening
Uniformity during fruit ripening is decisively related to cup quality in coffee and
consequently to the value of the product Fruits at the ideal ripening stage produce the
best organoleptic characteristics for coffee The presence of over ripened or green fruits
changes the acidity the bitterness and consequently the cup quality In order to
maximize uniform ripening of coffee fruits it is essential to control the action of genes
involved in the last step of maturation process Ethylene is known to trigger ripening
and increasing ethylene biosynthesis is associated with various stages of ripening
process (Protasio et al 2005) To control coffee fruit maturation two of the major
genes involved in ethylene biosynthesis namely ACC synthase and ACC oxidase
have been cloned (Protasio et al 2005 Neupane et al 1999) Introduction of the ACC
oxidase gene in antisense orientation have been achieved in both C arabica and C
canephora The effect of the transgene on ethylene production and fruit maturation has
yet to be reported The inhibition of genes downstream to the initial ethylene burst is
also an option to control coffee fruit maturation (Ribas et al 2006)
111 RNA interferenceRNAi
The regulation of gene expression at post transcriptional level has recently attracted
much attention because of the discovery of the phenomenon called RNA interference
(RNAi) RNAi based silencing techniques are more efficient than antisense mediated
gene silencing (Wesley et al 2001) The effect was first observed in plants wherein
silencing was observed when some copies of a transgene was in the forward orientation
with respect to the promoter and some in the reverse orientation This resulted in the
formation of double stranded RNA in the system The phenomenon in which
experimentally introduced double stranded RNA (dsRNA) leads to the loss of
expression of the corresponding cellular gene by sequence specific RNA degradation
Chapter 1 Introduction and Review of Literature
24
was termed as RNA interference or RNAi The protective effect of RNA silencing in
reducing virus infectivity supports the view that PTGS has evolved as a mechanism to
defend plants against virus infection and also to moderate the possible deleterious
genome-restructuring (insertional) activity of virus-like mobile genetic elements eg
retrotransposons A growing body of biochemical and genetic data has further
demonstrated that plants animals and yeasts share related mechanisms of specific
degradation of RNAs in which double-stranded forms of RNA are initiator molecules
(Brenstein et al 2001)
The key insight into the process of PGTS was provided by the experiments
conducted by Hamilton amp Baulcombe (1999) who identified the product of RNA
degradation as small RNA (siRNA) species of ~25nt of both sense and antisense
polarity SiRNA are formed and accumulate as double stranded RNA molecules of
defined chemical structures Both genetic and biochemical approaches were undertaken
to understand the basis of silencing Genetic screens were carried out in fungus
Neurospora carassa the alga Chalmydomonas reinhardtii and plant Arabidopsis
thaliana to search for detective mutants in PTGS
A combination of results obtained from several in vivo and in vitro experiments
have gelled into a two step mechanistic model for RNAiPTGS The first step referred
to as the RNAi initiating step involves binding of the RNA nucleases to a larges
dsRNA and its cleavage into discrete ~21-25nt RNA fragments This is ATP-
dependent reaction which involved RNase III - type endonucleases which make
staggered cuts in both strands of the dsRNA leaving 3prime overhang of 2 nucleotides with
5prime- phosphate 3prime-hydroxyl and no modification in the sugar phosphate backbone
(Hamilton amp Baulcombe 1999 Elbashir et al 2001) SiRNAs are the direct products
of dsRNA cleavage by the multi-domain RNase III enzyme DICER (Figure 16)
(Brenstein et al 2001 Karthikeyan et l 2013)
In the second step or commonly known as the effector step the double stranded
siRNAs produced in the first step bind to an RNAi specific protein complex to form a
RISC The complex undergoes activation in the presence of ATP so that the antisense
component of the unwound siRNA becomes exposed and allows the RISC to perform
the downstream RNAi reaction
Chapter 1 Introduction and Review of Literature
25
Several enzymes are involved in RNAi based on their role genes that encode for them
are classified into RNAi initiators RNAi effectors RNA dependent RNA polymerases
and silencing genes Two C elegans genes rdeI and rde4 (rde stands for lsquo RNAi
deficientrsquo) are believed to be involved in the initiation step of RNAi The C elegans
rde1 gene is a member of a large family of genes and is homologous to the Neurospora
qde2 (qde stands for ldquoquelling deficientrdquo) and Arabidopsis AGO1 (AGO stands for
agronaute AGO1 was previously identified to be involved in Arabidopsis
development) Although the functions of these genes are not clear a mammalian
member of RDE1 family has been identified as a translation initiation factor (Thakur
2005) Important genes for the effector step of PTGS in C elegans are rde2 and mut7
genes These genes were identified from heterozygous mutant worms that were unable
to transmit RNAi to their homozygous offsprings Worms with mutated rde2 or mut 7
genes show defective RNAi mut-7 gene encodes a protein with homology to the
nuclease domains of RNAase D and a protein implicated in Werner syndrome (a rapid
ageing disease in humans) (Grishok et al 2001 Kamath-Loeb et al 2012s)
Neurospora qde-1 Arabidopsis SDE-1SGS-2 and C elegans ego-1 appear to encode
RNA dependent RNA polymerase (RdRPs) It assumed that this is a proof that an RdRp
activity is required for RNAi Certainly the existence of an RdRp might explain the
remarkable efficiency of dsRNA induced silencing if it amplifies either the dsRNA
prior to cleavage or the siRNAs directly (Muers 2013) In C elegans ego-1 mutants
(ego stands for lsquoenhancer of glp-1) RNAi functions normally in somatic cells but is
defective in germline cells where ego-1 is primarily expressed In Arabidopsis SDE-
1SGS-2 mutants (SGS stands for suppressor of gene silencing) siRNA are produced
when dsRNA is introduced via an endogenously replicating RNA virus but not
introduced by a transgene It has been proposed that perhaps the viral RdRP is
substituting for the Arabidopsis enzyme in these mutants Random degradative PCR
model suggests that an RdRP uses the guide strand of an siRNA as a primer for the
target mRNA generating a dsRNA substrate for Dicer and thus more siRNAs
Several genes controlling RNA silencing in plants have been identified through
genetic screens of Arabidopsis mutants impaired in transgene induced RNA silencing
They encode a putative RNA-dependent RNA polymerase (SGS2SDE1) a coiled
protein (SGS3) a protein containing PAZ and Piwi domains (AGO1) and an RNA
helicase (SDE3) The putative SGS2SDE1 of Neurospora is related to QDE-1 and
Chapter 1 Introduction and Review of Literature
26
EGO-1 of C elegans whereas PAZPiwi protein AGO is related to QDE-2 of
Neurospora RDE-1of C elegans RNA helicase SDE3 is related to SMG-2 of C
elegans and Mut-6 of Chlamydomonas MUT-7 gene of C elegans encodes a protein
similar to Rnase D whereas the Drosophila DICER gene encode a protein similar to
RNase III An Arabidopsis ortholog of DICER gene has been identified called CAF
SIN1 SUS1(Thakur 2005 Poethig 2009)
Since the RNAi machinery is present constitutively within the eukaryotic cell it
is important to explore the metabolic advantages that are accorded by the RNAi related
proteins during the intrinsic normal growth of cells and development of organisms The
natural RNAi machinery not only keeps the mobile transposable elements from
disrupting the integrity of the genomes as suggested by the analysis of model organisms
like A thaliana C elegans D melanogaster (Tabara et al 1999 Wu-Scharf et al
2000 Aravin et al 2001 Hamilton et al 2002 Lisch 2009) but also participates in
development of organisms Genetic defects in Celegans RNAi genes ego1 and dicer
cause known specific developmental errors (Grishok et al 2000 Grishok et al 2001
Knight amp Bass 2001 Hall et al 2013) Similarly the Argonaute family of genes of A
thaliana (especially the ZWILLE proteins) is also responsible for plant architecture and
meristem development (Carmell et al 2002 Hamant and Pautot 2010) and the Dicer
homologue of A thaliana CAF1 is required for embryo development (Golden et al
2002 Nakano et al 2011) Thus genetic evidence illustrates the role of the RNAi
machinery as a controller of development related genes The mechanistic details of
these developmental processes are beginning to emerge
Chapter 1 Introduction and Review of Literature
27
Figure 16 Mechanism of gene silencing induced double stranded RNA The dicer enzyme to
produce siRNAs cleaves the RNA The siRNA generated bind to the nuclease complex
RISC The antisense component in the siRNA in the RISC guides the complex towards
the cognate mRNA resulting in endonucleolytic cleavage of the mRNA
Chapter 1 Introduction and Review of Literature
28
112 MicroRNA or MiRNA
In 1991 Ambros and co-workers first isolated a lin-4 mutant of Celegans which was
arrested at the first larval stage (Lee et al 1993) Later on the let-7 mutation was
isolated in the same system which was responsible for development through the fourth
larval stage Both lin-4 and let-7 encode short 22-nucleotide mature RNAs and were
called short temporal RNA because they control the temporal development program of
C elegans The mature lin-4 RNA defines (negatively regulates) the mRNA expression
of the lin-14 and lin-28 hetrochronic genes with the antisense mediated repression
mechanism of translation initiation and thus specifies the fate of cells during the first
three larval stages Recent studies have revealed that the short temporal RNAs are
actually members of a group of tiny RNAs (21to 28 nucleotides) called the micro-
RNAs (miRNA) (Bartel 2009) isolated members of which could easily run to a few
thousand which includes both those were identified computationally and by deep
sequencing Some of the components of the RNAi machinery have also been clearly
established as the effector proteins for the maturation of miRNAs
113 MicroRNAs in plants
1131 Discovery
MicroRNAs have been discovered by three basic approaches Direct cloning forward
genetic direct cloning and bioinformatic prediction followed by experimental
validation The most direct method of miRNA discovery is to isolate and clone small
RNAs from biological samples and several groups have used this approach to identify
plant miRNAs (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002 Xie et al 2003 Sunkar et al 2005 Williams et al 2013) The cloning methods
adapted were similar to those used to identify large numbers of animal miRNAs
(Lagos-Quintana et al 2001 Lau et al 2001 Aravin amp Tuschl 2005) which involve
isolating small RNAs followed by oligonucleotide adapter ligation reverse
transcription amplification and sequencing Some protocols incorporate methods to
enrich for Dicer cleavage products (ie molecules with 5prime-phosphates and 3prime-
hydroxyls) and to concatemerize the short cDNAs so that several may be identified in a
single sequencing read (Lau et al 2001 Llave et al 2002a Jagadeeswaran amp Sunkar
2013)The initial cloning experiments in Arabidopsis identified 19 miRNAs belonging
to 15 families (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002) although hundreds of other small RNAs such as siRNAs and RNA degradation
Chapter 1 Introduction and Review of Literature
29
fragments were also cloned through the direct cloning methods To select miRNAs
from the pool of small RNAs secondary structures of the RNA molecules were
examined in compliance with the acknowledged structural features of miRNAs
(Ambros et al 2003) The secondary structures were validated using several ad-hoc
software tools such as the Mfold or RNAfold programs (Reeder et al 2006) Finally
the existence of mature miRNAs was determined by RNA gel blot hybridization These
direct cloning methods were successful in isolating many miRNAs in Arabidopsis
(Reinhart et al 2002 Sunkar amp Zhu 2004) Rice (Sunkar et al 2005) Poplar (Lu et
al 2005) moss (Arazi et al 2005) and Tobacco (Billoud et al 2005)
Although miRNAs were first discovered through forward genetic screens in
worms (Lee et al 1993 Reinhart et al 2000)only few A thaliana miRNA gene
families have been discovered by this method (Chen 2008) in plants MiRNA
involvement in plant mutant phenotypes were not properly inferred till the cloning
experiments established that plant genomes contain numerous miRNAs (Park et al
2002 Reinhart et al 2002 Rhoades et al 2002) MiRNA loss-of-function allele has
been identified by forward genetic screens early extra petala1 is caused by a
transposon insertion ~160bp upstream of the predicted miR164c stem loop resulting in
flowers with extra petals (Baker et al 2005 Irish 2008) The fact that loss-of-function
miRNA mutants have been rarely discovered using forward genetics reflects small
target size for mutagenesis coupled with redundancy in nearly all evolutionarily
conserved plant miRNAs which are encoded by gene families (Table16) Family
members are likely to have overlapping functions buffering against loss at any single
miRNA locus Over expression screens can circumvent redundancy limitations At least
four plant miRNAs miR319 (also known as miR-JAW) miR172 (also known as EAT)
and miR166 were isolated in over expression screens for dominant mutants with
developmental abnormalities (Aukerman amp Sakai 2003 Palatnik et al 2003 Kim et
al 2005 Williams et al 2005b Schommer et al 2012) Mutations in miRNA target
sites which can prevent the entire family of miRNAs from repressing a target gene can
also circumvent redundant functions of miRNA family members
The dominant mutations in the HD-ZIP genes PHB PHV and REVOLUTA
(REV) in Arabidopsis and ROLLEDLEAF1 (RLD1) in Maize result in adaxialization of
leaves andor Vasculature (McConnell amp Barton 1998 McConnell et al 2001 Nelson
et al 2002 Zhong amp Ye 2004 Allen amp Millar 2012) and are all caused by mutations
Chapter 1 Introduction and Review of Literature
30
in miR166 complementary sites (Rhoades et al 2002 Emery et al 2003 Jones-
Rhoades amp Bartel 2004 Mallory et al 2004 Zhong amp Ye 2004)
In both plants and animals cloning was the initial means of large-scale
miRNA discovery (Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Reinhart et al 2002 Mendes et al 2012) However cloning is biased toward
RNAs that are expressed highly and broadly MiRNAs expressed at low levels or
only in specific cell types or in response to certain environmental stimuli were more
difficult to clone Sequence based biases in cloning procedures might also cause
certain miRNAs to be missed Because of these limitations bioinformatics approaches
to identify miRNAs have provided a useful complement to cloning
A straightforward use of bioinformatics has been carried out to find homologs
of known miRNAs both within the same genome and in the genomes of other species
(Pasquinelli et al 2000 Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Mendes et al 2009) A more difficult challenge is to identify miRNAs
unrelated to previously known miRNAs This was first accomplished for
vertebrate nematode and fly miRNAs using algorithms that search for conservation
of sequence and secondary structure (ie miRNA stem-loop precursors) between
species searching for patterns that are characteristic of miRNAs (Lai et al 2003 Lim
et al 2003 Lim et al 2005)
Most of the methods identified numerous miRNAs but are necessarily more
relaxed in terms of allowed structures Some approaches take advantage of the high
complementarity of plant miRNAs to target messages implementing the requirement
that the candidate has conserved complementarity to mRNAs (Jones-Rhoades amp Bartel
2004 Adai et al 2005) This additional filter has been useful for distinguishing
authentic plant miRNAs from false positives which later extended to mammalian
miRNA gene prediction (Xie et al 2005 Mendes et al 2012)
Another criteria used by most algorithms in the filters is evolutionary
conservation In plants mature miRNA sequences are conserved to a higher degree
than their precursor sequences For example Arabidopsis and rice diverged more than
130 million years ago (Friis et al 2004) but some miRNAs are highly conserved
in these plant species (Reinhart et al 2002) Furthermore various parameters for the
secondary structures such as free energy the number of paired residues within a
Chapter 1 Introduction and Review of Literature
31
miRNA or the number and size of bulges are also used to validate the predictions
(Jones-Rhoades amp Bartel 2004 Wang et al 2004 Adai et al 2005 Mendes et al
2012)
1132 Conserved microRNAs in Plants
Till date cloning genetics and bioinformatics screens have resulted in annotation of
approximately 184 potential Arabidopsis miRNAs (miRBase release 13) These
miRNAs seem to arise through gene duplication and subsequent diversification and
represent approximately 100 families (Maher et al 2006 Axtell 2013) Twenty one
families represented by 92 genes are clearly conserved in species beyond Arabidopsis
(Table 15) The presence of these miRNA families in other sequenced plant genomes
indicates that different plant species have an evolutionarily fluid set of miRNAs
(Willmann amp Poethig 2007) Recent studies on mosses one of the most ancient land
plants have shown that at least 14 miRNA families are conserved in eudicots and
mosses and therefore it appears that plant miRNAs share a common ancient origin
(Arazi et al 2005 Axtell amp Bartel 2005 Talmor-Neiman et al 2006 Zhang et al
2006 Axtell 2013) The number of members per family in a genome ranges from 1-32
with the exception of miR-430 which is represented by a cluster of ~80 loci in zebra
fish (Giraldez et al 2005)
In plants the number of members in each family correlates among examined
species certain families contain numerous members in all three species (eg miR156
miR166 miR169) whereas others consistently contain fewer genes (eg miR162
mir168 miR394) (Table 16) Although it is still unclear why certain miRNA are
encoded by more than one gene in plants (eg 12 genes encoding miR156) this
correlation might suggest functional significance of various miRNA family sizes Most
annotated plant miRNAs are conserved throughout flowering plants while a few
miRNAs are found only in a single sequenced genome and thus they could be
evolutionarily lsquoyoungrsquo miRNAs For example Arabidopsis has several miRNA
families that are not found in poplar or rice suggesting that miRNAs could be
generated continuously during evolution (Allen et al 2004a Rajagopalan et al
2006 Fahlgren et al 2007 Axtell 2012 de Alba et al 2013) Consistent with the
notion that non conserved miRNAs are of a more recent evolutionary origin these
miRNAs are mainly encoded by a single locus in the genome Within each family the
mature miRNA is always located in the same arm
Chapter 1 Introduction and Review of Literature
32
Table 16 MicroRNA families conserved in plants
miRNA
family
Arabidopsis Oryza Populus
miR156 12 12 11 miR159319 6 8 15 miR160 3 6 8 miR162 2 2 3 miR164 3 5 6 miR166 9 12 17 miR167 4 9 8 miR168 2 2 2 miR169 14 17 32 miR171 4 7 10 miR172 5 3 9 miR390 3 1 4 miR393 2 2 4 miR394 2 1 2 miR395 6 19 10 miR396 2 5 7 miR397 2 2 3 miR398 3 2 3 miR399 6 11 12 miR408 1 1 1 miR403 1 0 2 miR437 0 1 0 miR444 0 1 0 miR445 0 9 0 Total 92 127 169
All known miRNA families that are conserved between more than one plant species are listed
together with the number of genes identified in the sequenced genomes Rice miRNA families that have orthologs in maize but do not appear to have orthologs in the eudicots (Arabidopsis and Populus) are marked with an asterisk The following families contain miRNA genes annotated with
more than one number miR156 (miR156 and miR157) miR159319 (miR159 and miR319) miR166 (miR165 and miR166) miR171 (miR170 and miR171) and miR390 (miR390 and miR391)
of the stem loop (5prime- or 3prime) as it would be expected if the genes carry common ancestry
(Figure 17) The sequence of the mature miRNA and to some extent the segment on
the opposite arm of the hairpin to which it pairs is highly conserved between the
members of the same family (both within and between the species) while the sequence
secondary structure and length of the intervening ldquolooprdquo region can be highly divergent
within the same family
The patterns of paring and non pairing nucleotides are often conserved between
homologous miRNA stem loops from different species (Figure 17) The significance of
conserved mismatches is opined to guide DCL1 to cleave at the appropriate positions
along the stem loop
Chapter 1 Introduction and Review of Literature
33
Figure 17 Representative stem loop of miR164 Segments corresponding to the mature miRNAs
are shown in red (Adapted from Rajagopalan et al 2006)
Although many miRNAs are highly conserved in plants the degree of conservation in
most cases is quite low between plants and animals one exception is miR854 A recent
study has revealed that miR854 is conserved between Arabidopsis and animals and that
the Arabidopsis miR854 differs from the human miR854 only by a single nucleotide
(Arteaga-Vazquez et al 2006 Axtell 2012 de Alba et al 2013) The Arabidopsis
miR854 has multiple binding sites in the 3prime-untranslated region (UTR) of
OLIGOURIDYLATE-binding-PROTEIN mRNA 1bA (UBP1b) and represses the
translation of the UBP1b mRNA The animal miR854 is also complementary to a site in
the 3prime-UTR of the UBP1b homolog in animals However it is currently unclear how the
animal miR854 regulates its target mRNA Another distinction between plant and
animal miRNAs is the genomic organization of the miRNA genes While plant miRNA
genes are located predominantly in the intergenic regions animal miRNA genes tend to
localize in the intragenic regions both in the introns or exons of protein coding genes
Moreover miRNA genes are often clustered within a single precursor transcript
whereas individual plant miRNAs are derived from individual precursor RNAs (Bartel
Chapter 1 Introduction and Review of Literature
34
2004 Bonnet et al 2004 Kim 2005 Vaucheret et al 2006 Marco et al 2013)
1133 Non-conserved miRNA and challenges in annotation
Most miRNAs are conserved throughout flowering plants (Table 15) but many more
were found only in a single sequenced genome thus considered to be of a more recent
evolutionary origin The extended homology between few non conserved miRNAs
precursors and their target genes provides strong evidence that these relatively ldquoYoungrdquo
miRNAs arose from the duplication of the target gene segments (Allen et al 2004)
Several non-conserved miRNAs including miR161 miR163 miR173 miR447
miR475 and miR476 are known to direct cleavage of target transcripts (Allen et al
2004a Adai et al 2005 Sunkar et al 2005 de Alba et al 2013) It is difficult to
confidently predict targets for many because it is not possible to use complementary
site as a filter against false-positive target predictions It is also difficult to
confidently annotate non-conserved miRNAs as miRNA rather than siRNAs The
established minimal standard for miRNA annotation is a small RNA with detectable
expression and the potential to form a stem-loop when joined to flanking genomic
sequence (Kasschau et al 2003) In practice these requirements are too loose to
definitively categorize many small RNAs cloned from plants Many plant siRNAs are
detectable on blots (Vazquez et al 2004b) and hundreds of thousands of non-
miRNA genomic sequences can be predicted to fold into secondary structures that
resemble structures of plant miRNA precursors (Jones-Rhoades amp Bartel 2004)
Therefore without conservation of both sequence and secondary structure it is
difficult to be confident that a given cloned RNA originated from a stem-loop (ie is a
miRNA) rather than from a double-stranded RNA (ie is a siRNA) In fact many of
the thousands o f small RNAs cloned from Arabidopsis (Gustafson et al 2005) would
probably meet the literal requirements for annotation as miRNAs A few of these
sequences might be miRNAs but others that meet the literal criteria probably are not
so The challenges of annotating non conserved small RNAs were evidenced by
three related small silencing RNAs that were originally annotated as miRNAs
that turned out to be trans-acting siRNAs (tasiRNAs)(Kanno et al2013)
Although most miRNAs that are broadly conserved among flowering plants are
now being identified and experimentally validated (Table 16)(Jones-Rhoades amp
Bartel 2004) the challenges and ambiguities for classifying non-conserved miRNAs
prevent meaningful estimates of the total number of miRNA genes in Arabidopsis and
Chapter 1 Introduction and Review of Literature
35
other plant genomes It is possible that miRNAs might have escaped detection
because they are non-conserved and expressed in specific tissues or conditions and can
only be identified by using high- throughput sequencing
1134 MicroRNA biogenesis
11341 Transcription of microRNA precursor
Plant miRNAs are primarily found in the genomic regions that are not associated with
the protein coding genes (Reinhart et al 2002) and it appears that most if not all plant
miRNAs are produced from their own transcriptional units Plant miRNA genes are
occasionally clustered near each other in the genome suggesting transcription of
multiple miRNAs from a single primary transcript [eg the miR395 cluster (Grishok
et al 2001 Jones-Rhoades amp Bartel 2004b)] but this polycistronic arrangement of
miRNA genes appears far less frequently in plants than in animals (Bartel 2004)
Northern blot EST and mapping evidence indicate that plant miRNA primary
transcripts (also known as pri-miRNAs) as in animals are longer than needed to
encompass the miRNA stem-loops (Palatnik et al 2003 Jones-Rhoades amp Bartel
2004b) At least some of these pri-miRNA transcripts appear to be spliced
polyadenylated (Kurihara amp Watanabe 2004) and capped (Xie Z et al 2005) Two
rice miRNAs are contained within transcripts that contain exon junctions within the
presumptive stem-loop precursor implying that in these cases splicing is a prerequisite
for Dicer recognition (Sunkar et al 2005) Plant pri-miRNAs are over 1kb in length
and they are usually preceded by typical TATA box motifs They can also undergo
canonical splicing polyadenylation and capping All these observations indicates RNA
polymerase II is probably responsible for transcribing most plant miRNAs (Xie Z et
al 2005) as appears to be the case for many animal miRNAs Relatively little is
known about the regulation of miRNA transcription in plants but there is no reason
to suspect that this regulation would differ from that of protein-coding transcripts as
the bioinformatics analysis of the sequence upstream of the transcriptional start
sites of the miRNA genes showed the presence of putative binding motifs of
number of known transcription factors (Megraw et al 2006 Sethupathy 2013)
11342 MicroRNA processing and export
In plants maturation of miRNA is a step wise process A miRNA gene is transcribed to
pri-miRNA which is much longer than the pre-miRNA possessing the characteristic
stem-loop structure Like transcription of most protein-coding genes the miRNA
Chapter 1 Introduction and Review of Literature
36
transcription is processed by RNA polymerase II (Pol II) enzymes and the pri-
miRNAs are 5prime-capped and 3prime-polyadenylated (Lee et al 2004 Xie Z et al 2005)
Mature miRNAs are produced from pri-miRNAs through at least two sequential
processing steps by RNase III-type endonucleases In animals pri-miRNAs are first
processed at the bottom portion of the stem by Drosha a nuclear-localized RNase III
enzyme to liberate pre-miRNAs The pre-miRNAs are then exported to the cytoplasm
with the help of exportin-5The pre-miRNAs are further processed by Dicer another
RNaseIII enzyme to a duplex consisting of the miRNA and the antisense strands
(miRNA) Since plants do not have any Drosha homologs the two sequential
processing steps seem to be carried out by DCL1 a Dicer homolog in the nucleus
(Kurihara amp Watanabe 2004 de Alba et al 2013 Rogers amp Chen 2013)
The processing of pri-miRNAs to pre-miRNAs by DCL1 is assisted by two
other proteins HYPONASTY LEAVES1 (HYL1) and SERRATE (SE) HYL1 belongs to a
family of double-stranded RNA (dsRNA) binding protein and interacts with DCL1 in
vitro and in vivo (Lu amp Fedoroff 2000 Hiraguri et al 2005 de Alba et al 2013
Rogers amp Chen 2013) Loss-of-function mutations in the HYL1 gene result in
decreased accumulation of miRNAs and concomitant accumulation of pri-miRNAs
(Han et al 2004 Vazquez et al 2004a Kurihara et al 2006) SE a C2H2 zinc finger
protein interacts with DCL1 and HYL1 and plays a role in the processing of pri-
miRNAs to pre-miRNAs (Lobbes et al 2006 Yang L et al 2006) In addition the
nuclear cap-binding complex proteins CBP20 and CBP80 that are known as ABA
HYPER SENSITIVE 1(ABH1) have been shown to be required for proper miRNA
processing (Gregory et al 2008 Laubinger et al 2008) Recently it has been
demonstrated that DCL1 can release 21-nt RNAs from dsRNA as well as from
synthetic miR167 precursor RNAs in vitro However this cleavage is error prone and
inefficient in the absence of HYL1 and SE which accelerate the rate of DCL1-mediated
cleavage and promote accurate processing of miRNAs (Dong et al 2008 Rogers amp
Chen 2013)
11343 Methylation of the miRNAmiRNAduplex
After the miRNAmiRNAduplexes are released from the pre-miRNAs by the DCL1
activities the duplexes are methylated at the 2primeOH of the3prime-ends by HUA ENHANCER1
(HEN1) a small RNA methyltransferase (Yu et al 2005 Yang Z et al 2006 Rogers
amp Chen 2013 ) This step which is distinct from the RNase III-mediated processing
Chapter 1 Introduction and Review of Literature
37
step distinguishes the biogenesis of plant miRNAs from that of animal miRNAs
Mutations in the HEN1 gene were first isolated in a morphological screening for floral
development although the HEN1 mutants display pleiotropic phenotypes similar to the
developmental defects observed in the DCL1 mutants (Chen et al 2002 Park et al
2002 Rogers amp Chen 2013)
Figure 18 Biogenesis of microRNA
A Biogenesis of plant MicroRNA B Biogenesis of metazoananimal MicroRNA
The miRNA (red) is incorporated into RISC and the miRNA(blue) in degraded
This enzyme has two dsRNA-binding domains and nuclear localization signal It
is also conserved in fungi and is required for the processing of siRNAs as well as of
miRNAs (Park et al 2002 Boutet et al 2003 Xie et al 2003) Although the HEN1
protein is localized to the nucleus its methylation activity in the cytoplasm cannot be
ignored (Xie et al 2004 Rogers amp Chen 2013)
Chapter 1 Introduction and Review of Literature
38
The methylation of the miRNAmiRNA duplex is required to protect miRNAs
against the 3prime-end uridylation activity and subsequent degradation (Li et al 2005) In
the hen1 mutants accumulation of miRNAs is reduced to a lower level Also the
miRNAs appear to be heterogeneous in their sizes such that a ladder of bands rather
than a single species is observed for most miRNAs in the mutants (Li et al 2005)
MiRNA cloning and sequencing analyses revealed the presence of a number of U
residues at the 3prime-end of miRNAs in the hen1 mutants Moreover these nucleotides do
not correspond to the miRNAs in the pre-miRNAs indicating that these nucleotides are
added after the processing of the miRNAs from pre-miRNAs (Li et al 2005)
Uridylation of RNAs has also been proven to be correlated with RNA decay (Shen amp
Goodman 2004 Mullen amp Marzluff 2008) suggesting that uridylation attracts an
exonuclease which degrades miRNAs in plants
11344 MiRNA incorporation in to the RISC and its nuclear export
The methylated miRNAmiRNA duplexes undergo RISC assembly In this process
the miRNA of the duplexes is selectively incorporated into the RISC and the miRNA
appears to be removed and subsequently degraded (Hammond et al 2000 Hutvagner
amp Zamore 2002 Schwarz et al 2003 de Alba et al 2013 Rogers amp Chen 2013)
The ARGONAUTE 1(AGO1) proteins are core components of the RISC complex
They contain two structural domains the N-terminal PAZ domain that binds to the 3prime-
end of single-stranded RNAs and the C-terminal piwi domains that has a structure
similar to that of RNase H (Cerutti et al 2000 Parker et al 2004) Several AGO
proteins possess endonucleolytic activities within the PIWI domain and cleave target
mRNAs in the middle of the site complementary to miRNA (Liu et al 2004 Meister et
al 2004 Miyoshi et al 2005) Mutations in the AGO1 gene cause miRNA target
genes to be ectopically expressed and the levels of some miRNAs are reduced in the
mutants (Kidner amp Martienssen 2004 Vaucheret et al 2004 Kidner amp Martienssen
2005 Ronemus et al 2006) Moreover the phenotype of the AGO1 hypomorphic
alleles display defects in lateral organ polarity and leaf and flower morphology as
observed in the dcl1 mutants and null AGO1 alleles are embryonically lethal
supporting the close relationship between the AGO proteins and miRNA processing
(Kidner amp Martienssen 2004 Vaucheret et al 2004 de Alba et al 2013 Rogers amp
Chen 2013)
The production of miRNA miRNA duplexes occur in the nucleus while
Chapter 1 Introduction and Review of Literature
39
miRNA-RISC complexes are present in the cytoplasm where target mRNAs are
located This discordance of functional sites requires an export step during miRNA
biogenesis HASTY the Arabidopsis homolog of exportin-5 is thought to be involved
in miRNA nuclear export (Bollman et al 2003 Park et al 2005)The hasty mutant
exhibits pleiotropic defects in plant development and reduced accumulation of most
miRNAs as in the DCL1 or AGO1 mutants Howeverit is unclear whether the HASTY
proteins export the miRNA miRNA duplexes or the miRNA-RISC complexes in
plants
11345 Feedback regulation of miRNA biogenesis
MiRNA biogenesis is under feedback regulation such that the DCL1 and AGO1 genes
are themselves regulated by miRNAs DCL1 is itself a target of miR162 which leads to
the cleavage of the DCL1 mRNA (Xie et al 2003 de Alba et al 2013) Consistent
with this the levels of DCL1 mRNA are elevated in the mutants of miRNA processing
genes in which the miR162 abundance is reduced In addition there is a predicted
hairpin structure of miR838 in the 14th
intron of the DCL1 primary transcript
(Rajagopalan et al 2006) This prediction implies that DCL1-mediated processing
on this site can result in the cleavage of the DCL1 primary transcript into two truncated
fragments the N-terminal capped fragment and the C-terminal polyadenylated
fragment If this is the case it is possible that miRNA biogenesis competes with the
splicing event of the DCL1 pre-mRNA
The AGO1 is also regulated by miRNA There is a complementary site for
miR168 binding in the AGO1 mRNA and the transcript level of the AGO1 mRNA is
reduced through an mRNA cleavage mechanism (Vaucheret et al 2006 de Alba et al
2013 Rogers amp Chen 2013) indicating that the AGO1 mRNA levels are tightly
controlled by the levels of the AGO1 protein
1135 Mechanism of miRNA action
11351 MiRNA-directed mRNA cleavage
By forming the miRNA-RISC assembly miRNAs can direct the RISC to regulate gene
expression by two post transcriptional mechanisms mRNA cleavage or translational
repression (Bartel 2004 Jones-Rhoades et al 2006 Dalmay 2013) The degree
of miRNA-mRNA complementarity plays an important role for the
determination of the mechanism to be used A general rule is that while perfect or
Chapter 1 Introduction and Review of Literature
40
near-perfect complementarity induces mRNA cleavage central mismatches trigger
translational repression (Carrington amp Ambros 2003 Jones-Rhoades amp Bartel 2004)
Unlike most animal miRNAs plant miRNAs have near-perfect or perfect
complementarity to their targets and mRNA cleavage is assumed to be a predominant
mechanism by which miRNAs regulate gene expression in plants
In RNA cleavage mechanism miRNAs guide the AGO component of RISC to
cleave a single phosphodiester bond of the target mRNA within the miRNA-binding
site (Llave et al 2002b Tang et al 2003) The truncated fragments are then released
and degraded freeing the RISC to recognize and cleave another target mRNA This has
been validated by the detection of 3prime cleavage products that have 5prime ends that start at the
middle of the complementary regions The mRNA cleavage products are then further
degraded by other mechanisms The 3prime cleavage products of some miRNA targets are
degraded by the 5prime-3prime exonuclease XRN4 an Arabidopsis homolog of the major Yeast
mRNA degrading exonuclease Xrn1p It has been observed that the 3prime cleavage
products of some target mRNAs were accumulated in the xrn4 mutants (Souret et al
2004 Dalmay 2013) However the 3prime cleavage products of other miRNA targets do
not accumulate in the xrn4 mutants indicating that there are also XRN4 independent
mechanisms The 5prime cleavage products are typically untraceable by RNA gel blot
hybridization but can be detected by sensitive PCR based methods It has been found
that the 5prime cleavage products tend to obtain an oligo U tail at the 3prime ends This 3prime U tail
is correlated with shortening of the 5prime cleavage products from the 5prime end leading to the
conclusion that the 3prime uridylation causes 5 prime- 3 prime exonucleolytic decay of the 5prime cleavage
products (Shen amp Goodman 2004)
DCL1 HYL1 and SE interact with each other and are co-localized in the
nuclear bodies termed Dicing bodies or D-bodies in which some pri-miRNA shave
been found suggesting that D-bodies serve as a center for active miRNA processing
(Fang amp Spector 2007 Fujioka et al 2007 Song et al 2007) The miRNA
processing site appears to be distinct from the Cajal bodies that have previously been
found as a siRNA processing site (Pontes amp Pikaard 2008) The D-bodies include
SmD3 and SmB proteins that are found in the Cajal bodies However At collin an
additional marker for the Cajal bodies is not found in the D-bodies Moreover the D-
bodies are present in an At collin mutant lacking the Cajal bodies The pri-miRNAs of
miR171A and miR159A genes have been found to be co-localized with HYL1 and
Chapter 1 Introduction and Review of Literature
41
DCL1 in the D- bodies that are distinct from the Cajal bodies (Song et al 2007)
11352 MiRNA-directed translational repression
Translation repression is another mechanism exerted by plant miRNAs The regulatory
mechanism of miR172 which regulates flowering time and floral organ identity is
consistent with translation repression MiR172 over production does not affect the
transcript levels of APETALA2 (AP2) and AP2- like target mRNAs Instead the AP2
protein level is reduced in the miR172 over expressing mutant (Aukerman amp Sakai
2003 Chen 2004 Dalmay 2013) It was later shown that miR172 can also direct the
cleavage of the target mRNAs although the steady-state mRNA level is unchanged due
to feedback regulation (Schwab et al 2005 Jung et al 2007) It is clear that miR172
regulates its target genes primarily by translational repression rather than mRNA
cleavage Recently it has been reported that miR156157 and miR854 also regulate
their target genes through translational repression (Arteaga-Vazquez et al 2006
Gandikota et al 2007 Dalmay 2013) It was once thought that translational repression
is an exception to the rule However experimental evidence suggest a widespread
coexistence of translational repression and mRNA cleavage (Brodersen et al 2008)
1136 Regulatory roles of miRNAs in plant development
The miRNA target prediction has been a difficult task in order to obtain regulatory
targets without bringing in too many false predictions Plant growth and developmental
processes regulated by miRNAs are quite diverse and enormous Furthermore plant
miRNAs work cooperatively or antagonistically to establish a balanced regulation This
notion is well consistent with the findings that mutations in the regulators of miRNA
biogenesis transport and processing such as AGO1 DCL1 HYL1 HEN1 ABH1
and HASTY cause pleiotropic phenotypes (Mallory amp Vaucheret 2006 de Alba et al
2013) To date the roles of miRNAs have been mostly deduced from obtaining miRNA
over expressing transgenic plants or gain-of-function mutants in which miRNA-
resistant target genes are ectopically expressed
11361 Phase transition
Plants pass through a series of developmental phase transitions during their life span
beginning with seed germination continuing through vegetative phase change
reproductive phase change flowering initiation and finally seed production for the next
generation Because of their importance in understanding plant developmental
Chapter 1 Introduction and Review of Literature
42
processes genes regulating developmental transitions have been extensively studied
and underlying molecular mechanisms have been explored in various plant species
Majority of researches were mainly focused on vegetative phase change and
reproductive transition in Arabidopsis and Maize (Poethig 2003 Baurle amp Dean
2006) Particularly the mutations of the genes encoding various regulators of miRNA
biogenesis and transport such as HST SE and AGO exhibit altered juvenile to adult
vegetative phase change and vegetative to reproductive change (Hunter et al 2003
Peragine et al 2004 Vazquez et al 2004a Jones-Rhoades et al 2006 de Alba et al
2013)
Over-expression of the miR156a gene causes late flowering and delays
vegetative phase change (Wu amp Poethig 2006 Gandikota et al 2007 Wang et al
2008) It has been shown that miR156 negatively regulates a group of genes encoding
SQUAMOSA PROMOTER-BINDING PROTEIN-LIKE (Lewis et al 2007 de Alba et
al 2013 Rogers amp Chen 2013) transcription factors such as SPL3 SPL4 and SPL5
that promote flowering initiation In contrast transgenic plants over expressing
miR156-resistant SPL345 genes are early flowering Interestingly the endogenous
level of miR156 is temporally regulated In the early juvenile vegetative phase the
level of miR156 is very high but decreases rapidly before the onset of the adult juvenile
phase (Wu amp Poethig 2006)
The Arabidopsis early activation tagged (eat-D) mutant exhibits early flowering
with disrupted floral structure The mutant phenotypes are caused by over expression of
the miR172a-2gene (Aukerman amp Sakai 2003) MiR172 regulates a small group of
genes encoding APETALLA2 (AP2)-like transcription factors such as TARGET OF
EAT1 (TOE1) TOE2 SCHLAFMUTZE (SMZ) and SCHNARCHZAPFEN(SNZ)
TOE1 TOE2 SMZ and SNZ have been shown to participate in their productive phase
change by repressing a floral integrator FLOWERING LOCUS T (FT)(Jung et al
2007) Consistent with this toe1-2D 35STOE2 35SSMZ and 35SSNZ plants
exhibit late flowering (Jung et al 2007) MiR172 is also regulated temporally and
highly expressed in the adult vegetative phase both in Arabidopsis and maize (Chuck et
al 2007) Because the temporal expression patterns of miR156 and miR172 are
complementary to each other it has been suggested that the two miRNAs interact with
each other in regulating phase change (Chuck et al 2009 de Alba et al 2013) In
support of this view it has been shown that miR172 is down-regulated in the
Chapter 1 Introduction and Review of Literature
43
Corngrass1(Cg1) mutant in which miR156 is overproduced (Chuck et al 2007 de
Alba et al 2013) However there is no direct evidence for the direct linkage between
miR156 and miR172 It is also envisioned that miR156 and miR172 would not be
directly related For example miR156 regulates plastochron index that is the inverse of
leaf initiation rate and thus frequently used to analyze temporal patterns of plant
development In contrast miR172 does not affect plastochron (Wang et al 2008) The
down-regulation of miR172 in the maize miR156-over producing mutants would be
due to the prolonged juvenile vegetative phase in the mutants
Another miRNA that regulates flowering initiation is miR159 It regulates
MYB33 and MYB65 which are closely related to the barley GAMY B transcription
factor that responds to gibberellic acid (GA) The level of miR159 is elevated in GA-
treated seedlings but reduced in the GA biosynthetic ga1 mutant Transgenic plants
over producing miR159 exhibit delayed floral induction under short days (Achard et
al 2004) It is been suggested that MYB33 mediates GA signal to regulate LEAFY
(LFY) These observations indicate that miR159 plays an important role in the GA
signaling pathways that regulate proper floral induction under short days (Achard et al
2004)
11362 Floral development
Arabidopsis flowers consist of four distinct organs They arise in a characteristic
pattern within concentric rings called whorls from outside to inside four sepals four
petals six stamens and the central pistil consisting of two carpels Identities of the
floral organs are determined by three classes of regulatory genes classA classB and
class C genes (Krizek amp Fletcher 2005) The A and C class genes act antagonistically
to restrict their expression domains (Bowman et al 1991)
It is notable that miR172 also regulates floral architecture in addition to its role
in the timing of flowering initiation MiR172 inhibits the translation of the AP2 mRNA
(Chen 2004) Transgenic plants overproducing miR172 not only exhibit early
flowering but also show abnormal floral development which is quite similar to those
of the class A gene mutants such as the ap2 mutants (Aukerman amp Sakai 2003 de
Alba et al 2013) When the activity of the class A genes is inactivated that of the class
C genes such as AGAMOUS (AG) is elevated As a result the miR172- overproducing
transgenic plants exhibit no petals along with homeotic transformation of the sepals to
Chapter 1 Introduction and Review of Literature
44
the carpels (Aukerman amp Sakai 2003 Chen 2004)
Interestingly miR166 also regulates floral architecture The role of miR165166
in the regulation of meristem activity is closely linked to floral meristem formation and
organization (Zhao et al 2007 Miyashima et al 2013) Floral structure is severely
disrupted in the miR165166-overproducing mutants such as men1 and jba-1D in
which miR166 is overproduced (Williams et al 2005b Jung amp Park 2007) The men1
and jba-1D mutants have smaller gynoecium and reduced carpel number In an extreme
case the jba-1D homozygous plants lack visible carpels (Williams et al 2005b)
Similar phenotypes have been observed in the miR165 overproducers (Zhao et al
2007) It has been suggested that the phenotypes are possibly caused by altered AG
expression in the floral meristem (Jung amp Park 2007 Turchi et al 2013)
Other miRNAs also affect floral development Overproduction of miRNAs
involved in auxin signaling also affects floral architecture Transgenic plants
overproducing miR167 have defects in the formation of ovule and anther with reduced
fertility (Ru et al 2006) It has been shown that miR167 targets ARF6 and ARF8 that
play a role in gynoecium and stamen development and in regulating fertility (Ru et al
2006 Wu et al 2006) These observations suggest that auxin signals are also closely
related to floral organogenesis In addition transgenic plants over-producing miR164
and the cuc1cuc2 double mutants show fused sepals and reduced petals (Laufs et al
2004 Turchi et al 2013) suggesting that miR164 is also related to floral meristem
activity and boundary specification of the meristematic region
11363 Shoot apical meristem development
Shoot apical meristem comprises self-maintaining totipotent cells The shoot apical
meristem activity affects diverse aspects of plant developmental processes including
leaf development vascular development and lateral organ polarity (Bowman et al
2002) Because miR165166 plays a primary role in meristem formation
overproduction of miR165166 and multiple knockout mutants of its target genes
exhibit pleiotropic pheonotypes (McConnell et al 2001 Emery et al 2003 Kim et al
2005 Williams et al 2005b)
The targets of miR165166 include genes encoding the homeo domain-leucine
zipper (HD-ZIP) class III transcription factors such as PHABULOSA(PHB)
PHAVOLUTA (PHV) REVOLUTA(REV) ATHB15CORONA (CNA) and ATHB8
Chapter 1 Introduction and Review of Literature
45
PHB PHV and ATHB15 have overlapping functions in controlling meristem
development (Turchi et al 2013) Indeed the phb phv athb15 triple mutants have an
enlarged shoot apical meristem Consistent with this the miR166- overproducing men1
and jba-1D mutants exhibit enlarged shoot apical meristems (Kim et al 2005
Williams et al 2005b Turchi et al 2013) In the mutants the shoot apical meristems
are multiplied and large
The development of shoot apical meristems is also regulated by the WUSCHEL
(WUS)ndashCLAVATA (CLV) signaling pathway WUS positively regulates cell
proliferation In contrast CLV3 which is expressed in stem cells limits the WUS
expression and thus inhibits cell proliferation in the SAM (Sharma et al 2003)
Consistent with this notion WUS is expressed to a higher level in jba-1D
explaining the ectopic enlargement of the SAM in the mutant (Williams et al 2005)
While WUS is closely related to miR165166 in the shoot apical meristem
development the underlying molecular mechanisms are currently unclear It has been
observed that the miR166-over producing men1 plants have an arrested meristem
development (Jung amp Park 2007) In addition the heterozygous men1+ and the
homozygous men1 plants show different expression patterns of WUS and CLV3 It
appears that miR166 regulates WUS indirectly and influences the expressional balance
between WUS and CLV3 through a negative feedback loop (Jung amp Park 2007 de
Alba et al 2013)
The phv phb athb15 triple mutant has an enlarged shoot apical meristems the
rev phb phv mutant does not have functional shoot apical meristems (Prigge et al
2005) This distinction may be explained by the fact that while miR166 targets
primarily PHB PHV and ATHB15 miR165 targets all the five HD-ZIP III genes (Zhou
et al 2007) Consistent with this transgenic plants overproducing miR166 are distinct
from those overproducing miR165 The miR165-overproducing transgenic plants lack
functional shoot apical meristem and a pin-like structure is formed at the apical region
of the mutant These phenotypes are quite similar to rev phb phv triple mutant (Zhou et
al 2007 Liu et al 2013) The phenotypic distinction may because by the difference
of the cleavage efficiencies of the target mRNAs In accordance with this notion a few
35S miR166a transgenic lines exhibit no shoot apical meristems The men1
homozygous plants also show arrested meristem development (Jung amp Park 2007)
Chapter 1 Introduction and Review of Literature
46
MiR164 cleaves the CUC1 and CUC2 mRNAs as well as the NAC1 mRNA
CUC1 and CUC2 determines the organ boundary in the meristem Phenotypes of
transgenic plants that overproduce miR164 are similar to those of the cuc1cuc2 double
mutant that exhibits embryonic developmental defects such as cup-shaped cotyledons
and fused cotyledons (Laufs et al 2004) When a miR164-resistant CUC2 is
expressed the boundary domain is enlarged CUC also plays a role in embryonic shoot
meristem formation by regulating the SHOOT MERISTEMLESS gene It was therefore
concluded that miR164-mediated cleavage of CUC1 and CUC2 mRNAs determines the
sizes of the boundary domains in the shoot apical meristem and regulates separation of
the organs by restricting cell proliferation (Laufs et al 2004 de Alba et al 2013)
11364 Leaf development
The role of miR319 has been extensively studied in leaf development A dominant
jaw-D mutant overproducing miR319 shows uneven curved leaves with enlarged size
and five TCP genes which are closely related to the CINCINNATA (CIN) gene in
Antirrhinum majus are down regulated in the mutant suggesting that miR319 mediated
regulation of the TCP transcription factor genes are important for the final step of
leaf shaping (Palatnik et al 2003 Naz et al 2013) In addition to leaf shape and size
leaf polarity is also specified by miRNA MiR166-mediated cleavage of the HD-ZIPIII
mRNAs is a well to establish leaf polarity This discovery originates from the gain-of-
function mutants such as phb-1d and phv-1d wherein point mutations in the
complementary sites to miRNA helps the mRNA to evade or resist from miRNA-
mediated cleavage (McConnell et al 2001 Emery et al 2003 Naz et al 2013)
Plant leaves have a dorso-ventral polarity consisting of the adaxial (upper
surface) and abaxial (lower surface) sides The adaxial side is closer to the shoot
apical meristem Adaxial identity is specified by functionally redundant class III HD-
ZIP family genes such as PHB PHV and REV Ectopic expression of each genes
result in adaxialized leaves Dominant phb-1d and phv-1d mutants exhibit
transformation of the abaxial to adaxial fate and have rod-shaped leaves In contrast
miR165166-overproducing plants show pin-like cotyledons radicalized leaves and
abaxialized leaves (Chitwood et al 2007 Pattanayak et al 2013)
Leaf asymmetry is determined by miR166-mediated restriction of the
expression domains of the HD-ZIPIII genes The HD-ZIPIII genes are expressed in the
Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora
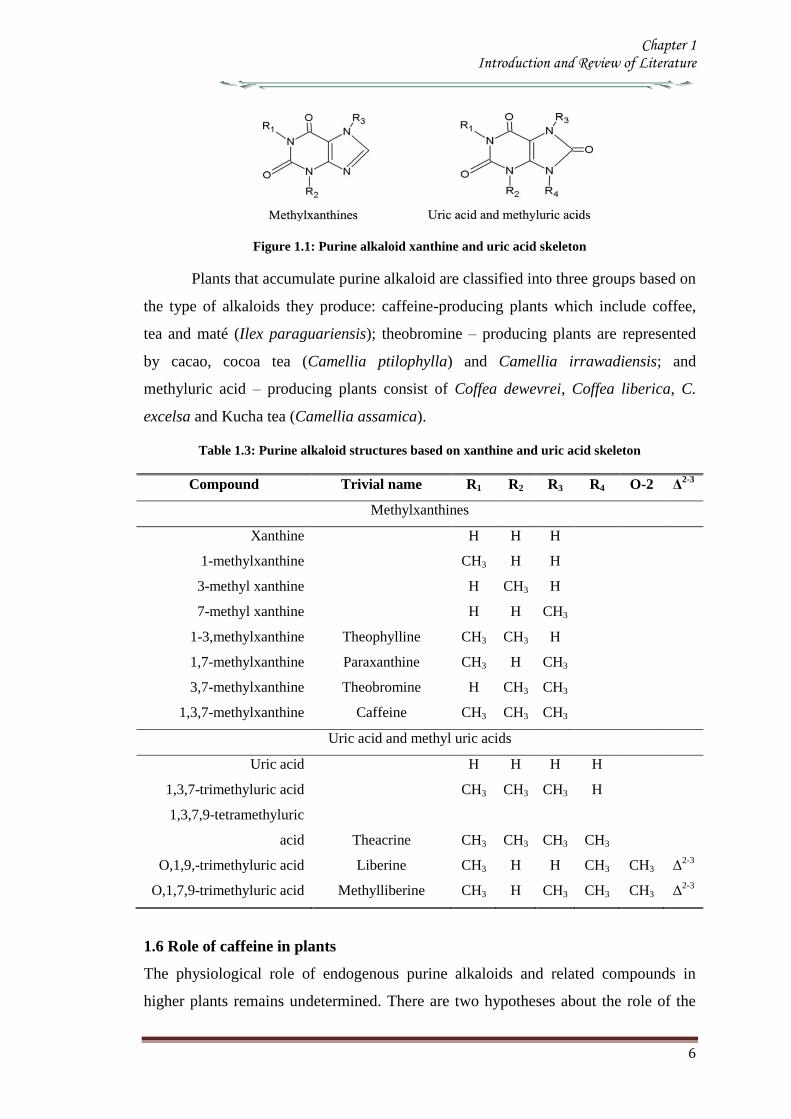
Chapter 1 Introduction and Review of Literature
6
Figure 11 Purine alkaloid xanthine and uric acid skeleton
Plants that accumulate purine alkaloid are classified into three groups based on
the type of alkaloids they produce caffeine-producing plants which include coffee
tea and mateacute (Ilex paraguariensis) theobromine ndash producing plants are represented
by cacao cocoa tea (Camellia ptilophylla) and Camellia irrawadiensis and
methyluric acid ndash producing plants consist of Coffea dewevrei Coffea liberica C
excelsa and Kucha tea (Camellia assamica)
Table 13 Purine alkaloid structures based on xanthine and uric acid skeleton
Compound Trivial name R1 R2 R3 R4 O-2 Δ2-3
Methylxanthines
Xanthine H H H
1-methylxanthine CH3 H H
3-methyl xanthine H CH3 H
7-methyl xanthine H H CH3
1-3methylxanthine Theophylline CH3 CH3 H
17-methylxanthine Paraxanthine CH3 H CH3
37-methylxanthine Theobromine H CH3 CH3
137-methylxanthine Caffeine CH3 CH3 CH3
Uric acid and methyl uric acids
Uric acid H H H H
137-trimethyluric acid CH3 CH3 CH3 H
1379-tetramethyluric
acid Theacrine CH3 CH3 CH3 CH3
O19-trimethyluric acid Liberine CH3 H H CH3 CH3 Δ2-3
O179-trimethyluric acid Methylliberine CH3 H CH3 CH3 CH3 Δ2-3
16 Role of caffeine in plants
The physiological role of endogenous purine alkaloids and related compounds in
higher plants remains undetermined There are two hypotheses about the role of the
Chapter 1 Introduction and Review of Literature
7
high concentrations of caffeine that accumulate in tea coffee and a few other plant
species The lsquoallelopathic theory or the auto toxic function theoryrsquo proposes that
caffeine in seed coats is released into the soil and inhibits the germination of seeds
around the parent plants (Anaya et al 2006)
The lsquochemical defense theoryrsquo proposes that caffeine in young leaves fruits
and flower buds acts to protect soft tissues from insect pests (Ashihara amp Crozier
2001 Ashihara et al 2008) It has been shown that spraying tomato leaves with a 1
solution of caffeine deters feeding by tobacco horn worms while treatment of
cabbage leaves and orchids with 001ndash01 solutions of caffeine acts as a neurotoxin
and kills or repels slugs and snails (Hollingsworth et al 2002) This work has now
been extended with convincing evidence for chemical defense theory has recently
been obtained with transgenic caffeine ndash producing tobacco plants (Uefuji et al 2005
Kim et al 2006)
17 Distribution of caffeine in plants
Caffeine has been found in 13 orders of the plant kingdom (Ashihara and Suzuki
2004 Anaya et al 2006) In some species the main purine alkaloid is theobromine or
methyl uric acids such as theacrine liberine and methylliberine rather than caffeine
(Ashihara amp Crozier 1999 Ashihara amp Crozier 2001)(Table 14)
Table 14 Distribution of purine alkaloids in plants
SNo Plant species Common
name Major alkaloid Plant parts
containing
alkaloids 1 Coffea arabica Arabica Caffeine Leaves Seeds
2 Coffea canephora Robusta Caffeine Leaves Seeds
3 Coffea liberica Caffeine Theacrine Liberine
Seeds Mature leaves
4 Coffea dewevrei Caffeine Theacrine Liberine
Seeds Mature leaves
5 Camellia sinensis Tea Caffeine Leaves
6 Camellia assamica Assam tea Caffeine Leaves
7 Camellia assamica var
kucha Kucha Theacrine Caffeine Leaves
8 Camellia irrawadensis Theobromine Leaves
9 Camellia ptilophylla Cocoa tea Theobromine Leaves
10 Theobroma cacao Cocoa Theobromine Seeds
11 Paullinia cupuna Guarana Caffeine Seeds
Chapter 1 Introduction and Review of Literature
8
12 Cola nitida Caffeine Seeds
13 Citrus sp Caffeine Pollen
14 Ilex paraguariensis Mate Caffeine Leaves
Adapted from (Ashihara amp Crozier 1999 Ashihara amp Crozier 2001)
18 Caffeine biosynthesis
The major biosynthetic pathway is a four step sequence consisting of three sequential
methylation and one nucleosidase reactions (Figure 12) The xanthine skeleton of
caffeine is derived from purine nucleotides The initial step in caffeine biosynthesis is
the methylation of xanthosine by a SAM- dependent N-methyltransferase In addition
to experiments with radiolabeled precursors substrate specificities of native (Kato et
al 1999) and recombinant N-methyltransferases (Kato et al 2000 Ogawa et al
2001 Mizuno et al 2003a Mizuno et al 2003b Uefuji et al 2003) strongly
suggest that the major route to caffeine is a xanthosine rarr7-methylxanthosinrarr 7-
methylxanthine rarr theobromine rarr caffeine pathway (Figure 12) Although the
information has been obtained mainly from coffee (C arabica) and tea (Camellia
sinensis) the available evidence indicates that the pathway is essentially the same in
other purine alkaloid-forming plants such as mateacute (Ashihara 1993) and cacao
(Koyama et al 2003 Yoneyama et al 2006)
The first step in the caffeine biosynthetic pathway from xanthosine is
conversion of xanthosine to 7-methylxanthosine (Figure 12) This reaction is
catalyzed by the 7-methylxanthosine synthase (xanthosine 7 N-methyltransferase EC
211158) The genes encoding for 7-methylxanthosine synthase CmXRS1 (AB
034699) and CaXMT1 (AB 048793) were isolated from the C arabica (Mizuno et
al 2003a Uefuji et al 2003) The recombinant proteins obtained from these genes
exhibit 7-methylxanthosine synthase activity in vitro Xanthosine monophosphate
(XMP) was not converted to 7-methylxanthosine by the recombinant enzymes thus
inclusion of 7-methy-XMP was proposed (Schulthess et al 1996)
The second step of caffeine biosynthesis involves a nucleosidase which
catalyses the hydrolysis of 7-methylxanthosine Although N-methylnucleosidase (EC
32225) was partially purified from tea leaves (Negishi et al 1988) recent detailed
structural studies on coffee 7-methylxanthosine synthase suggested that the methyl
transfer and nucleoside cleavage may be coupled and catalyzed by a single enzyme
(McCarthy amp McCarthy 2007)
Chapter 1 Introduction and Review of Literature
9
The last two steps of the caffeine synthesis are also catalyzed by a SAM-
dependent N-methyltransferase(s) which is different from the N-methyltransferase
enzyme that catalyzes the first step in the pathway Native N-methyltransferase
activities have been detected in crude and partially purified extracts from tea and
coffee plants (Ashihara amp Suzuki 2004) and a highly purified preparation has been
obtained from young tea leaves (Kato et al 1999) The enzyme was assigned the
name caffeine synthase (EC 211160) that catalyses the last two steps of caffeine
biosynthesis viz the conversion of 7-methylxanthine to caffeine via theobromine
(Figure 12) The gene encoding caffeine synthase was first cloned from young tea
leaves (Kato et al 2000) Since then several genes encoding N-methyltransferases
with different substrate specificity have been reported
Activity of the recombinant theobromine synthase (CTS1 and CaMXMT EC
211159) is specific for the conversion of 7-methylxanthine to theobromine In
contrast the recombinant caffeine synthases (CCS1 and CaDMXMT1) can utilize
paraxanthine theobromine and 7-methylxanthine as shown with tea caffeine synthase
(TCS1) (Mohanpuria et al 2011) Although paraxanthine is the most active substrate
of this recombinant enzyme only limited amounts of paraxanthine are synthesized in
coffee cells hence in vivo caffeine synthase is principally involved in the conversion
of 7-methylxanthine to caffeine via theobromine
Radiolabelled tracer experiments with theobromine accumulating cacao and
Chinese tea (Camellia ptilophylla) showed a limited conversion of theobromine to
caffeine N-methyltransferase which catalyzed the conversion of 7-methylxanthine to
theobromine was detected in crude extracts from C ptilophylla but conversion of
theobromine to caffeine was not observed (Ashihara et al 1998) Genes homologous
to caffeine synthase have been isolated from several theobromine accumulating plants
including cacao (Yoneyama et al 2006) The recombinant enzymes derived from
these genes have 3 N-methyltransferase activity suggesting that these theobromine
accumulating plants contain specific theobromine synthase Caffeine does not occur
in C ptilophylla (Ashihara et al 1998) although it is present in leaves and fruits of
cacao along with theobromine (Koyama et al 2003 Zheng amp Ashihara 2004)
Chapter 1 Introduction and Review of Literature
10
Figure 12 Pathways for the biosynthesis of caffeine
The major pathway is indicated by solid lines (Mizuno et al 2003b Uefuji et al 2003) Alternative pathways in coffee and tea plants are indicated
by dashed lines(Kato et al 1996 Schulthess et al 1996) Enzymes (1) 7-methylxanthosine synthase (2) N-methylnucleosidase (3) Theobromine
synthase (4) Caffeine synthase
N
N
N
N7
H3C
3
1
O
O
Caffeine
N
NH
N
N7
3
O
O
Theobromine
N
N
N
N
H
H
7
O
O
N
N
N
N
H
H
O
O
N
N
N
N
H
H
O
O
Xanthosine
N
N
N
N
H
H
O
O
N
N
N
N
H
H
O
ON
N
N
N
H
H3C
71
O
O
Paraxanthine
CH3
CH3
CH3
CH3CH3
7-methylxanthine
Rib P
Rib
(1)
(4)
Rib
+
CH3
7-methyl
xanthosine
(2)
(3)
SAM SAH
SAM SAH SAM SAH
Xanthosine mono
phosphate
Rib P
7-methyl xanthosine
monophosphate
+
CH3 CH3
Inosine 5-mono
phosphate
Adenosine 5-mono
phosphate
SAM SAH
Guanosine
Guanosine 5-mono
phosphate
Chapter 1 Introduction and Review of Literature
11
181 Source of Xanthosine for caffeine biosynthesis
Xanthosine the initial substrate of purine alkaloid synthesis is supplied by at least four
different pathways de novo purine biosynthesis (de novo route) the degradation
pathways of adenine nucleotides (AMP route) and guanine nucleotides (GMP route)
and the S-adenosyl-L-methionine (SAM) cycle (SAM route) (Figure 13) Purine
nucleotides are synthesized by de novo and salvage pathways (Ashihara amp Crozier
1999 Stasolla et al 2003 Zrenner et al 2006) The de novo synthesis of non purine
precursors like CO2 10-formyltetrahydrofolate 5-phosphoribosyl-1-pyrophosphate and
the amino acids Glycine glutamine and aspartate results in the production of purine
nucleotides
Figure 13 Xanthosine biosynthesis Xanthosine for purine alkaloid biosynthesis is supplied by at
least four routes from adenosine released from the SAM cycle (SAM route) from IMP
originating from de novo purine synthesis (de novo route) from the cellular adenine
nucleotide pool (AMP route) and from the guanine nucleotide pool (GMP route)
Adapted from (Ashihara et al 2008)
The utilization of IMP for caffeine biosynthetic pathway which is formed by the
de novo purine biosynthetic pathway for caffeine biosynthetic pathways was
demonstrated in young tea leaves using 15
N-glycine and 14
C-labelled precursors and
inhibitors of de novo purine biosynthesis (Ito amp Ashihara 1999) Xanthosine is formed
by an IMP rarr XMP rarr xanthosine pathway IMP dehydrogenase (EC 111205) and 5prime-
nucleotidase (EC 3135) catalyze these reactions The inhibition of IMP dehydrogen-
ase by ribavirin inhibits both caffeine and gunanine nucleotide biosynthesis in tea and
coffee leaves (Keya et al 2003)
Chapter 1 Introduction and Review of Literature
12
The portion of xanthosine used for caffeine biosynthesis is derived from adenine
and guanine nucleotide pools which are produce de novo by salvage pathways there are
several potential pathways for xanthosine synthesis from AMP but the AMP rarr IMP
rarr XMPrarr xanthosine route is predominant All three enzymes involved in the
conversion AMP deaminase (EC 3546) IMP dehydrogenase and 5prime-nucleosidase
have been detected in tea leaves (Koshiishi et al 2001) Xanthosine for caffeine
biosynthesis can also be produced from guanine nucleotides by GMPrarrguanosine
rarrxanthosine pathway Guanosine deaminase (EC 35415) activity is found in cell free
extracts from young tea leaves (Negishi et al 1994) but GMP deaminase activity was
not detected in plants (Stasolla et al 2003 Zrenner et al 2006) The absence of GMP
deaminase activity suggests that a GMP rarr IMP rarr XMP rarr xanthosine route is not
functional in plants
The use of radiolabelled purine bases and nucleosides for the study of caffeine
biosynthetic pathway was facilitated by the fact that exogenous purine nucleotides are
rapidly hydrolyzed by the plant tissues forming purine nucleosides andor bases which
are then utilized in pathways which in most instances indirectly lead to xanthosine
Most of these purine compounds including adenine adenosine inosine hypoxanthine
and guanine are converted to their respective nucleotides by salvage enzymes
including adenine phosphoribosyl transferase (EC 2427) hypoxanthineguanine
phosphoribosyl transferase (EC 2428) adenosine kinase (EC 27120) and
inosineguanosine kinase (EC 2421) which usually have high activities in plant cells
The resultant nucleotides then enter the purine alkaloid biosynthetic pathway In the
case of guanosine it may be directly deaminated to xanthosine and utilized for caffeine
biosynthesis (Figure 13) (Ashihara et al 1997)
182 SAM cycle in plants
SAM is the methyl donor for the three methylation steps in the caffeine biosynthetic
pathway In this process SAM is converted to S-adenosyl-L-homocysteine (SAH)
which in turn is hydrolyzed to homocysteine and adenosine Homocysteine is recycled
via the SAM cycle to replenish SAM levels and adenosine is released from the cycle
Adenosine released from the cycle is converted to AMP directly andor indirectly via
adenine AMP is converted to xanthosine and used for caffeine biosynthesis Since
three moles of SAH are produced via the SAM cycle for each mole of caffeine that is
synthesized in theory this pathway has the capacity to be the sole source of both the
Chapter 1 Introduction and Review of Literature
13
purine skeleton and the methyl groups required for caffeine biosynthesis in young tea
leaves (Koshiishi et al 2001)
Methionine
S-Adenosyl-L-methionineHomocysteine
ATP
PPi+ Pi
Tetrahydrofolate
5-Methyl-
tetrahydrofolate
CH3+Adenosine
S-Adenosyl-L-
homocysteine
(xanthine
structure)(Methyl groups)
Caffeine
(1)
(2)(3)
(4)
Figure 14 The SAM cycle (activated methyl cycle) in plants (Ashihara amp Suzuki 2004)
Enzymes SAM synthetase SAM-dependent N-methyltransferases S-adenosyl-
homocysteine (SAH) hydrolase Methionine synthase Adenosine released from the cycle
is salvaged to adenine nucleotides and utilized both for purine structure of caffeine via
xanthosine and for re-synthesis of SAM via ATP
183 N-methyltransferase in caffeine biosynthesis Molecular cloning
The genes encoding for the enzymes involved in the caffeine biosynthesis were
successfully cloned from the coffee family (Ogawa et al 2001 Mizuno et al 2003a
Mizuno et al 2003b Uefuji et al 2003) The conserved amino acid regions of tea CS
(AB31280) the sequences of O-methyltransferases derived from plant origin and
Arabidopsis hypothetical proteins were made use of in designing primers for RT-PCR
and RACE technique Independent groups have isolated N-methyltransferase (NMT)
genes from coffee which have been classified based on the substrate specificity of the
recombinant proteins expressed in Ecoli (Table 15) PCR products thus obtained were
used as the probe to isolate full length cDNA from the library resulting in the
identification of all three N-methyltransferases involved in caffeine biosynthesis
(Figure 12) Several cDNA have been characterized from the coffee plants one for
xanthosine methyltransferase (XMTXRS) three for 7-methylxanthine methyltransferase
or theobromine synthase (MXMTCTS) and three for 37-dimethylxanthine
methyltransferase or caffeine synthase (DXMTCCS) (Table 15) (Kato amp Mizuno
2004)
A single gene encoding for xanthosine methyltransferase (XMTCmXRS1) was
identified which encoded for a polypeptide consisting of 372 amino acids with an
apparent molecular mass of 418kDa It is expressed almost uniformly in aerial tissues
Chapter 1 Introduction and Review of Literature
14
of C arabica including leaves floral buds and immature but not in mature beans
Three genes encoding 7-methylxanthine methyltransferase (theobromine synthase) have
been identified The number of amino acids in the putative polypeptides are 378 for
MXMT1 (427kDa) and 384 for MXMT2 and CTS2 (434kDa) They differ by insertion
or deletion of blocks of several residues in the C-terminal region Their catalytic
properties based on kinetic parameters such as Km values were different They are
expressed in young leaves floral buds and immature but not mature beans Three genes
were identified for 3 7-dimethylxanthine methyltransferase (caffeine synthase) DXMT
CCS1 and CtCS7 each encoding a 43 kDa polypeptide consisting of 384 amino acids
However their kinetic properties differ from each other such as DXMT and CCS1
showed Km values of 1200 and 157microM for theobromine respectively Expressions
profiles are also distinct DXMT being expressed exclusively in immature beans while
CCS1 expression is ubiquitous occurring in all tissues The presence of isoforms of
these enzymes with different properties suggests that caffeine is synthesized through
multiple pathways depending on availability and concentration of the substrate (Mizuno
et al 2001 Ogawa et al 2001 Mizuno et al 2003a Mizuno et al 2003b Uefuji et
al 2003) Genomic clones of theobromine synthase were obtained by PCR- RFLP
from C canephora (Satyanarayana et al 2005 Satyanarayana 2006) The genomic
clones are reported to have highly conserved exons and a variable third intron The
promoter for the theobromine synthase was cloned and characterized (Satyanarayana et
al 2005)
There is high degree of similarity in the coding region of coffee NMT genes
isolated so far though differences exist in the 3prime untranslated region (UTR) for these
genes The deduced amino acid sequence of these enzymes shows more than 85
homology between these genes Phylogenetic analysis indicates that they are more
closely related to C-methyltransferases including those for jasmonic acid salicylic acid
and benzoic acid than to other N-methyltransferases This suggests that coffee N-methyl
transferases belong to a distinct sub-group within plant methyltransferases Their
cellular localization determined by green fluorescent protein fusion method and β-
glucourodinase (GUS) showed that the all the three genes are localized in the cytoplasm
(Ogawa et al 2001 Vinod et al 2007) It was also shown that these were present
along with chlorogenic acid(Vinod et al 2007)
Chapter 1 Introduction and Review of Literature
15
Table 15 N-methyltransferases and their encoding genes involved in caffeine biosynthesis
Gene (Enzyme) Gene name Accession No Description Reference
Xanthosine methyltransferase
( 7-methylxanthosine synthase) (EC 211158)
CaXMT AB 048793 Immature fruit cDNA Screening (Uefuji et al 2003)
CmXRS1 AB 034699 Leaf cDNA library RACE (Mizuno et al 2003a)
CaMXMT1 AB 048794 Leaf cDNA library RACE (Ogawa et al 2001)
7-methylxanthine synthase transferase
(theobromine synthase) ( EC 211159)
CTS1 AB 034700 Immature fruit cDNA Screening (Uefuji et al 2003)
CaMXMT2 AB 984125 Leaf cDNA library Screening (Mizuno et al 2001)
CTS2 AB 054841 Leaf cDNA library Screening (Mizuno et al 2001)
37-methylxanthinemethyltransferase
(Caffeine synthase) (EC 211160)
CaDXMT1 AB 086414 Immature fruit cDNA Screening (Uefuji et al 2003)
CCS1 AB 0861414 Immature fruit cDNA Screening (Uefuji et al 2003)
CtCS7 AB 0864415 Immature fruit cDNA
NMT genomic clones
Theobromine synthase-1
CX10 Complete coding region PCR Satyanarayan 2006
CX8 Complete coding region PCR -do-
Theobromine synthase-1 PG-5 Promoter + coding region RAGE -do-
Theobromine synthase-1 PG-1 Promoter + coding region RAGE Satyanarayan 2006
PG-4 Promoter + coding region RAGE -do-
Theobromine synthase-1 NMT
(AY273814) Complete coding region PCR Kochko A et al 2010
Theobromine synthase-2 NMT
(AY362825) Complete coding region PCR Kochko A et al 2010
Chapter 1 Introduction and Review of Literature
16
19 Catabolism of caffeine
Caffeine is produced in young leaves and immature fruits and continues to accumulate
gradually during the maturation of these organs At the same time caffeine is slowly
degraded with the removal of the three methyl groups resulting in the formation of
xanthine Catabolism of caffeine was first reported in coffee leaves (Kalberer 1965)
Since then a number of tracer experiments using 14
C labelled purine alkaloids have
been reported (Suzuki amp Waller 1984 Ashihara et al 1996 Vitoria amp Mazzafera
1999 Mazzafera 2004) They demonstrated the major catabolic pathway is caffeine rarr
theophylline rarr 3-methylxanthine rarr xanthine Xanthine is further degraded by the
removal by the conventional purine catabolic pathway to CO2 and NH3 via uric acid
allatonin and allantoate (Figure 15) (Ashihara amp Crozier 1999 Stasolla et al 2003
Zrenner et al 2006) Caffeine catabolism usually begins with its conversion to
theophylline catalyzed by N7-demethylase The involvement of P450-dependent mono-
oxygenase activity for this reaction was suggested (Huber amp Baumann 1998
Mazzafera 2004) However the activity of this enzyme has not yet been determined in
cell free extracts [8-14
C] Theophylline degraded to CO2 at much faster rate than [8-14
C]
caffeine indicating that conversion of caffeine to theophylline is a major rate limiting
step of caffeine catabolism and a possible reason why caffeine accumulates in high
concentrations in tissues of C sinensis and C arabica It is widely believed that the
control of caffeine levels in plants is a function of the balance between rates of
synthesis and degradation and this balance seems to vary depending on the plant
species and the tissue developmental stage (Mazzafera 2004)
Chapter 1 Introduction and Review of Literature
17
Figure 15 Caffeine catabolic pathways Caffeine is mainly catabolised via theophylline and 3-
methylxanthine Xanthine is further degraded to CO2 and NH3 by the conventional
oxidative pathway Adapted from (Ashihara et al 2008)
Chapter 1 Introduction and Review of Literature
18
110 Genetic engineering of Coffee
Genetic transformation has potential applications in coffee agriculture for incorporating
genes which could provide desirable traits such as disease and insect resistance
drought and frost tolerance and herbicide resistance Transgenic technology can also be
used to increase nutritional value and improve cup quality produce varieties with
caffeine-free beans and for production of hybrid crops for molecular farming
Identification of target specific genes is one of the pre-requisites for developing
transgenic crops The availability of a large number of EST sequences in coffee and
initiation of coffee genome sequencing may speed up the gene discovery and accelerate
transgenic research efforts in coffee
1101 Insect Resistance
Production of insect resistant coffee plants is one of the major objectives of the
breeding programs The major pests attacking coffee include coffee berry borer (CBB
Hypothenemus hampei) white stem borer (WSB Xylotrechus quadripes) leaf miner
(Perileucoptera coffeela) and root nematodes (Meloidogyne spp and Pratylenchus
spp) The CBB is present in almost all the coffee growing countries and considered to
the most devastating pest in coffee To date there is no reported source of resistance to
CBB in the coffee gene pool Like CBB WSB is another serious pest in C arabica in
India and several other East Asian countries Both CBB and WSB belong to the order
Coleoptera For India controlling WSB is the biggest challenge and has therefore
become the highest research priority Robusta is generally resistant to WSB but the
interspecific Robusta Arabica hybrids are susceptible to WSB Although leaf miner is
not yet a serious pest in India and other East Asian countries it is an economically
important pest in East Africa and Brazil Effective chemical control of CBB and WSB
is difficult due to the nature of their life cycle inside the berry and stem respectively as
well as environmental concern regarding the use of pesticides Biological control
measures are adapted to combat these insect pests with varying degrees of success
Developing coffee plants resistant to these pests using genetic transformation
technology could be one of the alternative strategies to counter pest damage For insect
resistance several different classes of proteins from bacterial plant and animal sources
have been isolated and their insecticidal properties tested against many important pests
Amongst these proteins Bt toxins are most important and several transgenic crops
expressing Bt genes have been commercialized Coffee transgenic plants carrying a
Chapter 1 Introduction and Review of Literature
19
synthetic version of the cry1Ac gene have been produced (Leroy et al 2000 Mishra et
al 2002) Indeed this was the first report of an important agronomic trait being
introduced into a coffee plant The transgenic plants presented similar features in
growth and development compared to normal plants Transgenic plants highly resistant
to leaf miner under greenhouse conditions were tested under field conditions in French
Guyana for 4 years for pest resistance (Perthius et al 2005 Perthius et al 2006)
From 54 independent transformation events 70 of the events were resistant to leaf
miner Unfortunately the field trial was vandalized which led to the termination of the
experiment (Montagnon et al 2005)The effectiveness of Bt genes in controlling
coleopteran pests is well documented in corn and potato which indicates that Bt genes
might be effective against CBB The high toxicity of B thuringiensis sero var
israelensis against first in star larvae of CBB has been demonstrated (Mendez et al
2003) In another experiment an α-amylase inhibitor from Phaseolus vulgaris was
tested against CBB and found to have an inhibitory effect on its growth and
development (Grossi et al 2004)In addition to the CBB and WSB C arabica
varieties are also susceptible to endoparasitic root-knot nematode (Meloidogyne spp)
(Campos et al 1990 Mishra et al 2012)
So far 15 species have been reported to be parasites of coffee Controlling
nematodes is extremely difficult and currently seedling grafting with robusta rootstock
is followed as one of the control measure Sources of resistance specific to root-knot
nematodes have been identified in coffee trees (Bertrand et al 2001) and the Mex-1
gene conferring resistance to M exigua in C arabica is in the process of isolation (Noir
et al 2003)
1102 Tolerance to Abiotic Stress
In many coffee growing countries abiotic stress such as drought and frost are the major
climatic factors that limit coffee production Changes in climatic patterns due to global
climate change are considered increasingly important for coffee cultivation Drought
induces water stress in plants which affects vegetative growth and vigor and triggers
floral abnormalities and poor fruit set It also indirectly increases the incidence of pests
and diseases in the plants C arabica is generally more tolerant to water stress than C
canephora partly due to its extensive deep root system C racemosa is known to be a
good source of drought tolerance In India hybrids between C canephora and C
racemosa have been obtained and are currently under evaluation for drought tolerance
Chapter 1 Introduction and Review of Literature
20
In many coffee growing countries coffee is propagated in marginal areas where the
annual rainfall is below 1000thinspmm with prolonged dry spells of over 4-5 months In
those areas water shortage and unfavorable temperatures constitute major constraints
and the growth and productivity of C canephora are badly affected As with drought
periodic frost also affects coffee production in parts of Brazil The introduction of
drought and frost tolerant genes through genetic transformation would be of great
importance for alleviating these problems Research is now being carried out by several
groups to identify genes involved in biotic as well as abiotic stress The most promising
genetic engineering approach for drought tolerance includes the use of functional or
regulatory genes as well as the transfer of transcription factors In recent years plants
tolerant to high temperature and water stress have been the subject of intense research
(Chaves et al 2003 Chaves et al 2004 Coraggio et al 2004) For achieving drought
tolerance genes that have been targeted include those encoding enzymes involved in
detoxification or osmotic response metabolism enzymes active in signaling proteins
involved in the transport of metabolites and regulating the plant energy status (Chaves
et al 2003 Chaves et al 2004 Coraggio et al 2004) The dissection of molecular
mechanisms related to signal transduction and transcriptional regulation might help in
engineering drought tolerance in coffee
1103 Disease Resistance
Coffee leaf rust (CLR) caused by the fungus Hemileia vastatrix is the most important
disease in coffee with substantial loss to coffee production and productivity in all the
coffee growing countries In addition to leaf rust coffee berry disease (CBD) caused by
fungus Colletotrichum kahawae can be a devastating anthracnose leading to substantial
crop loss in Africa Several other fungal and bacterial diseases may also affect coffee
to a extent C arabica is more susceptible to many diseases than C canephora Though
most of the disease control measures rely upon chemical control they are more
expensive and labour intensive The long-term solution is the breeding of resistant
varieties which is the focus of many breeding programs However breeding for disease
resistant varieties is time consuming due to the perennial nature of coffee with its long
gestation period India has a long history of C arabica breeding especially for leaf rust
resistance and is the first country to demonstrate the existence of multiple races of leaf
rust Resistance to CLR is conditioned primarily by a number of major (SH) genes and
coffee genotypes are classified into different resistance groups based on their
Chapter 1 Introduction and Review of Literature
21
interaction with different rust races pathogen (van der Vossen 2001) Currently C
canephora provides the main source of resistance to pests and diseases including CLR
(H vastatrix) and CBD (C kahawae) and therefore used in breeding programs Other
diploid species like C liberica and C racemosa are being used as a source of resistance
to coffee leaf rust and coffee leaf miner respectively (Guerreiro et al 1999
Sreenivasan et al 1989) The development of coffee varieties resistant to major fungal
diseases such as CLR and CBD using transgenic technology will benefit the coffee
industry immensely During the last 15 years significant progress has been made in the
area of host-pathogen interactions (Staskawicz et al 1995 Feys et al 2000
Shivaprasad et al 2012) and many resistance genes involved in recognizing invading
pathogens have been identified and cloned (Takken et al 2000) A number of
signalling pathways induced because of pathogen infection have been dissected (Dong
et al 1998 Navarro et al 2006) Many antifungal compounds synthesized by plants to
combat fungal infection have been identified (Does et al 1998) Understanding the
specific induction of targeted pathways and identification of specific pathways
responsible for particular fungal resistance is important in order to employ this strategy
in transgenic technology The recent investigation of gene expression during coffee leaf
rust infection could provide an insight into the defence pathways operating in coffee
(Fernandez et al 2004 Nardi et al 2006) Efforts have been made to identify and
clone resistance genes from coffee for achieving durable resistance The genetic and
physical map of two resistance genes SH3gene conferring resistance to rust (Prakash et
al 2004) and the Ck1 gene conferring resistance to C kahawae CBD (Gichuru et al
2006) have been established These genes could be used for molecular marker assisted
breeding programs (Mishra et al 2012)
1104 Production of low-caffeine coffee
A widespread belief that the ingestion of caffeine can have adverse effects on
health resulted in the increased demand for decaffeinated coffee (Ashihara amp Crozier
2001) Several method were used to obtain decaffeinated plants by conventional
breeding techniques using low yielding varieties and mutants that accumulated
relatively low amounts of caffeine when compared to the commercially available ones
Low-caffeine and decaffeinated coffee represent around 10 of the coffee sales
around the world (Ribas et al 2006b) The industrial process for coffee decaffeination
can be expensive and affects the original flavour and aroma in coffee (David 2002)
Chapter 1 Introduction and Review of Literature
22
Transgenic coffee plants with suppressed caffeine synthesis using RNA interference
(RNAi) technology have been obtained (Ogita et al 2003 Ogita et al 2004) Specific
sequences in the 3prime untranslated region of the theobromine synthase gene (CaMXMT1)
were selected for construction of RNAi short and long fragments The caffeine and
theobromine content of the transgenic plants were reduced by ~ 70 when compared to
the untransformed plants In C canephora RNAi technology has also been employed
to silence the N-methyl transferase gene involved in caffeine biosynthesis (Vinod et al
2007) Promoter of an N-methyltransferase (NMT) gene involved in caffeine
biosynthesis was cloned (Satyanarayana et al 2005 Satyanarayana 2006) which will
be very useful for studying the regulation of caffeine biosynthesis
1105 Improvement in cup quality
Improvement in coffee cup quality requires elaborate knowledge of the chemical
constituents as well as the metabolic pathways involved in the elaboration of quality
The constituents of coffee beans include minerals proteins carbohydrates caffeine
chlorogenic acids (CGA) glycosides lipids and many volatile compounds that give
flavour to coffee by roasting Among these the role of three major constituents
sucrose CGA and trigonelline have been studied in coffee The sucrose content of
coffee bean is associated with coffee flavour the higher the sucrose content in green
beans the more intense will be the cup flavour (Clifford et al 1985 de Maria et al
1996) The sucrose content of C arabica (82-83) is higher than C canephora (33ndash
40) The sucrose amino acids ratio in green beans determines the profile of volatile
compounds Manipulating sucrose content in coffee bean is therefore important in
improving cup quality The sucrose synthase gene (CcSUS2) from C canephora has
been cloned and sequenced (Leroy et al 2005) This provides an opportunity to
manipulate the sucrose content in coffee Chlorogenic acids are products of
phenylpropanoid metabolism Chlorogenic acids are a group of hydroxycinnamoyl
quinic acids (HQA) formed by esterification between caffeic acids coumaric acids and
quinic acids (Clifford et al 1999) They are present in relatively large quantities in the
coffee bean and are the precursor of phenolic compounds in roasted beans C
canephora beans contain higher CGA (10) compared to C arabica beans (6-7)
CGA are known to have antioxidant properties as well as being associated with disease
resistance (Campa et al 2003) Genetic manipulations of genes involved in CGA
synthesis can serve either of these purpose by up- or down regulating the pathway
Chapter 1 Introduction and Review of Literature
23
Phenylalanine ammonia-lyase (PAL) catalyzes the first step of the phenylpropanoid
pathway leading to the synthesis of a wide range of chemical compounds including
flavonoids coumarins hydroxycinnamoyl esters and lignins (Hahlbrock amp Scheel
1989) Recently the full length cDNA and corresponding genomic sequences of PAL
from C canephora was isolated characterized and functionally validated (Mahesh et
al 2006 Parvatam et al 2006) This has opened up new possibilities for manipulating
the level of the PAL enzyme in coffee which in turn can be useful for improvement of
cup quality and manipulating antioxidant properties in coffee
1106 Fruit Ripening
Uniformity during fruit ripening is decisively related to cup quality in coffee and
consequently to the value of the product Fruits at the ideal ripening stage produce the
best organoleptic characteristics for coffee The presence of over ripened or green fruits
changes the acidity the bitterness and consequently the cup quality In order to
maximize uniform ripening of coffee fruits it is essential to control the action of genes
involved in the last step of maturation process Ethylene is known to trigger ripening
and increasing ethylene biosynthesis is associated with various stages of ripening
process (Protasio et al 2005) To control coffee fruit maturation two of the major
genes involved in ethylene biosynthesis namely ACC synthase and ACC oxidase
have been cloned (Protasio et al 2005 Neupane et al 1999) Introduction of the ACC
oxidase gene in antisense orientation have been achieved in both C arabica and C
canephora The effect of the transgene on ethylene production and fruit maturation has
yet to be reported The inhibition of genes downstream to the initial ethylene burst is
also an option to control coffee fruit maturation (Ribas et al 2006)
111 RNA interferenceRNAi
The regulation of gene expression at post transcriptional level has recently attracted
much attention because of the discovery of the phenomenon called RNA interference
(RNAi) RNAi based silencing techniques are more efficient than antisense mediated
gene silencing (Wesley et al 2001) The effect was first observed in plants wherein
silencing was observed when some copies of a transgene was in the forward orientation
with respect to the promoter and some in the reverse orientation This resulted in the
formation of double stranded RNA in the system The phenomenon in which
experimentally introduced double stranded RNA (dsRNA) leads to the loss of
expression of the corresponding cellular gene by sequence specific RNA degradation
Chapter 1 Introduction and Review of Literature
24
was termed as RNA interference or RNAi The protective effect of RNA silencing in
reducing virus infectivity supports the view that PTGS has evolved as a mechanism to
defend plants against virus infection and also to moderate the possible deleterious
genome-restructuring (insertional) activity of virus-like mobile genetic elements eg
retrotransposons A growing body of biochemical and genetic data has further
demonstrated that plants animals and yeasts share related mechanisms of specific
degradation of RNAs in which double-stranded forms of RNA are initiator molecules
(Brenstein et al 2001)
The key insight into the process of PGTS was provided by the experiments
conducted by Hamilton amp Baulcombe (1999) who identified the product of RNA
degradation as small RNA (siRNA) species of ~25nt of both sense and antisense
polarity SiRNA are formed and accumulate as double stranded RNA molecules of
defined chemical structures Both genetic and biochemical approaches were undertaken
to understand the basis of silencing Genetic screens were carried out in fungus
Neurospora carassa the alga Chalmydomonas reinhardtii and plant Arabidopsis
thaliana to search for detective mutants in PTGS
A combination of results obtained from several in vivo and in vitro experiments
have gelled into a two step mechanistic model for RNAiPTGS The first step referred
to as the RNAi initiating step involves binding of the RNA nucleases to a larges
dsRNA and its cleavage into discrete ~21-25nt RNA fragments This is ATP-
dependent reaction which involved RNase III - type endonucleases which make
staggered cuts in both strands of the dsRNA leaving 3prime overhang of 2 nucleotides with
5prime- phosphate 3prime-hydroxyl and no modification in the sugar phosphate backbone
(Hamilton amp Baulcombe 1999 Elbashir et al 2001) SiRNAs are the direct products
of dsRNA cleavage by the multi-domain RNase III enzyme DICER (Figure 16)
(Brenstein et al 2001 Karthikeyan et l 2013)
In the second step or commonly known as the effector step the double stranded
siRNAs produced in the first step bind to an RNAi specific protein complex to form a
RISC The complex undergoes activation in the presence of ATP so that the antisense
component of the unwound siRNA becomes exposed and allows the RISC to perform
the downstream RNAi reaction
Chapter 1 Introduction and Review of Literature
25
Several enzymes are involved in RNAi based on their role genes that encode for them
are classified into RNAi initiators RNAi effectors RNA dependent RNA polymerases
and silencing genes Two C elegans genes rdeI and rde4 (rde stands for lsquo RNAi
deficientrsquo) are believed to be involved in the initiation step of RNAi The C elegans
rde1 gene is a member of a large family of genes and is homologous to the Neurospora
qde2 (qde stands for ldquoquelling deficientrdquo) and Arabidopsis AGO1 (AGO stands for
agronaute AGO1 was previously identified to be involved in Arabidopsis
development) Although the functions of these genes are not clear a mammalian
member of RDE1 family has been identified as a translation initiation factor (Thakur
2005) Important genes for the effector step of PTGS in C elegans are rde2 and mut7
genes These genes were identified from heterozygous mutant worms that were unable
to transmit RNAi to their homozygous offsprings Worms with mutated rde2 or mut 7
genes show defective RNAi mut-7 gene encodes a protein with homology to the
nuclease domains of RNAase D and a protein implicated in Werner syndrome (a rapid
ageing disease in humans) (Grishok et al 2001 Kamath-Loeb et al 2012s)
Neurospora qde-1 Arabidopsis SDE-1SGS-2 and C elegans ego-1 appear to encode
RNA dependent RNA polymerase (RdRPs) It assumed that this is a proof that an RdRp
activity is required for RNAi Certainly the existence of an RdRp might explain the
remarkable efficiency of dsRNA induced silencing if it amplifies either the dsRNA
prior to cleavage or the siRNAs directly (Muers 2013) In C elegans ego-1 mutants
(ego stands for lsquoenhancer of glp-1) RNAi functions normally in somatic cells but is
defective in germline cells where ego-1 is primarily expressed In Arabidopsis SDE-
1SGS-2 mutants (SGS stands for suppressor of gene silencing) siRNA are produced
when dsRNA is introduced via an endogenously replicating RNA virus but not
introduced by a transgene It has been proposed that perhaps the viral RdRP is
substituting for the Arabidopsis enzyme in these mutants Random degradative PCR
model suggests that an RdRP uses the guide strand of an siRNA as a primer for the
target mRNA generating a dsRNA substrate for Dicer and thus more siRNAs
Several genes controlling RNA silencing in plants have been identified through
genetic screens of Arabidopsis mutants impaired in transgene induced RNA silencing
They encode a putative RNA-dependent RNA polymerase (SGS2SDE1) a coiled
protein (SGS3) a protein containing PAZ and Piwi domains (AGO1) and an RNA
helicase (SDE3) The putative SGS2SDE1 of Neurospora is related to QDE-1 and
Chapter 1 Introduction and Review of Literature
26
EGO-1 of C elegans whereas PAZPiwi protein AGO is related to QDE-2 of
Neurospora RDE-1of C elegans RNA helicase SDE3 is related to SMG-2 of C
elegans and Mut-6 of Chlamydomonas MUT-7 gene of C elegans encodes a protein
similar to Rnase D whereas the Drosophila DICER gene encode a protein similar to
RNase III An Arabidopsis ortholog of DICER gene has been identified called CAF
SIN1 SUS1(Thakur 2005 Poethig 2009)
Since the RNAi machinery is present constitutively within the eukaryotic cell it
is important to explore the metabolic advantages that are accorded by the RNAi related
proteins during the intrinsic normal growth of cells and development of organisms The
natural RNAi machinery not only keeps the mobile transposable elements from
disrupting the integrity of the genomes as suggested by the analysis of model organisms
like A thaliana C elegans D melanogaster (Tabara et al 1999 Wu-Scharf et al
2000 Aravin et al 2001 Hamilton et al 2002 Lisch 2009) but also participates in
development of organisms Genetic defects in Celegans RNAi genes ego1 and dicer
cause known specific developmental errors (Grishok et al 2000 Grishok et al 2001
Knight amp Bass 2001 Hall et al 2013) Similarly the Argonaute family of genes of A
thaliana (especially the ZWILLE proteins) is also responsible for plant architecture and
meristem development (Carmell et al 2002 Hamant and Pautot 2010) and the Dicer
homologue of A thaliana CAF1 is required for embryo development (Golden et al
2002 Nakano et al 2011) Thus genetic evidence illustrates the role of the RNAi
machinery as a controller of development related genes The mechanistic details of
these developmental processes are beginning to emerge
Chapter 1 Introduction and Review of Literature
27
Figure 16 Mechanism of gene silencing induced double stranded RNA The dicer enzyme to
produce siRNAs cleaves the RNA The siRNA generated bind to the nuclease complex
RISC The antisense component in the siRNA in the RISC guides the complex towards
the cognate mRNA resulting in endonucleolytic cleavage of the mRNA
Chapter 1 Introduction and Review of Literature
28
112 MicroRNA or MiRNA
In 1991 Ambros and co-workers first isolated a lin-4 mutant of Celegans which was
arrested at the first larval stage (Lee et al 1993) Later on the let-7 mutation was
isolated in the same system which was responsible for development through the fourth
larval stage Both lin-4 and let-7 encode short 22-nucleotide mature RNAs and were
called short temporal RNA because they control the temporal development program of
C elegans The mature lin-4 RNA defines (negatively regulates) the mRNA expression
of the lin-14 and lin-28 hetrochronic genes with the antisense mediated repression
mechanism of translation initiation and thus specifies the fate of cells during the first
three larval stages Recent studies have revealed that the short temporal RNAs are
actually members of a group of tiny RNAs (21to 28 nucleotides) called the micro-
RNAs (miRNA) (Bartel 2009) isolated members of which could easily run to a few
thousand which includes both those were identified computationally and by deep
sequencing Some of the components of the RNAi machinery have also been clearly
established as the effector proteins for the maturation of miRNAs
113 MicroRNAs in plants
1131 Discovery
MicroRNAs have been discovered by three basic approaches Direct cloning forward
genetic direct cloning and bioinformatic prediction followed by experimental
validation The most direct method of miRNA discovery is to isolate and clone small
RNAs from biological samples and several groups have used this approach to identify
plant miRNAs (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002 Xie et al 2003 Sunkar et al 2005 Williams et al 2013) The cloning methods
adapted were similar to those used to identify large numbers of animal miRNAs
(Lagos-Quintana et al 2001 Lau et al 2001 Aravin amp Tuschl 2005) which involve
isolating small RNAs followed by oligonucleotide adapter ligation reverse
transcription amplification and sequencing Some protocols incorporate methods to
enrich for Dicer cleavage products (ie molecules with 5prime-phosphates and 3prime-
hydroxyls) and to concatemerize the short cDNAs so that several may be identified in a
single sequencing read (Lau et al 2001 Llave et al 2002a Jagadeeswaran amp Sunkar
2013)The initial cloning experiments in Arabidopsis identified 19 miRNAs belonging
to 15 families (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002) although hundreds of other small RNAs such as siRNAs and RNA degradation
Chapter 1 Introduction and Review of Literature
29
fragments were also cloned through the direct cloning methods To select miRNAs
from the pool of small RNAs secondary structures of the RNA molecules were
examined in compliance with the acknowledged structural features of miRNAs
(Ambros et al 2003) The secondary structures were validated using several ad-hoc
software tools such as the Mfold or RNAfold programs (Reeder et al 2006) Finally
the existence of mature miRNAs was determined by RNA gel blot hybridization These
direct cloning methods were successful in isolating many miRNAs in Arabidopsis
(Reinhart et al 2002 Sunkar amp Zhu 2004) Rice (Sunkar et al 2005) Poplar (Lu et
al 2005) moss (Arazi et al 2005) and Tobacco (Billoud et al 2005)
Although miRNAs were first discovered through forward genetic screens in
worms (Lee et al 1993 Reinhart et al 2000)only few A thaliana miRNA gene
families have been discovered by this method (Chen 2008) in plants MiRNA
involvement in plant mutant phenotypes were not properly inferred till the cloning
experiments established that plant genomes contain numerous miRNAs (Park et al
2002 Reinhart et al 2002 Rhoades et al 2002) MiRNA loss-of-function allele has
been identified by forward genetic screens early extra petala1 is caused by a
transposon insertion ~160bp upstream of the predicted miR164c stem loop resulting in
flowers with extra petals (Baker et al 2005 Irish 2008) The fact that loss-of-function
miRNA mutants have been rarely discovered using forward genetics reflects small
target size for mutagenesis coupled with redundancy in nearly all evolutionarily
conserved plant miRNAs which are encoded by gene families (Table16) Family
members are likely to have overlapping functions buffering against loss at any single
miRNA locus Over expression screens can circumvent redundancy limitations At least
four plant miRNAs miR319 (also known as miR-JAW) miR172 (also known as EAT)
and miR166 were isolated in over expression screens for dominant mutants with
developmental abnormalities (Aukerman amp Sakai 2003 Palatnik et al 2003 Kim et
al 2005 Williams et al 2005b Schommer et al 2012) Mutations in miRNA target
sites which can prevent the entire family of miRNAs from repressing a target gene can
also circumvent redundant functions of miRNA family members
The dominant mutations in the HD-ZIP genes PHB PHV and REVOLUTA
(REV) in Arabidopsis and ROLLEDLEAF1 (RLD1) in Maize result in adaxialization of
leaves andor Vasculature (McConnell amp Barton 1998 McConnell et al 2001 Nelson
et al 2002 Zhong amp Ye 2004 Allen amp Millar 2012) and are all caused by mutations
Chapter 1 Introduction and Review of Literature
30
in miR166 complementary sites (Rhoades et al 2002 Emery et al 2003 Jones-
Rhoades amp Bartel 2004 Mallory et al 2004 Zhong amp Ye 2004)
In both plants and animals cloning was the initial means of large-scale
miRNA discovery (Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Reinhart et al 2002 Mendes et al 2012) However cloning is biased toward
RNAs that are expressed highly and broadly MiRNAs expressed at low levels or
only in specific cell types or in response to certain environmental stimuli were more
difficult to clone Sequence based biases in cloning procedures might also cause
certain miRNAs to be missed Because of these limitations bioinformatics approaches
to identify miRNAs have provided a useful complement to cloning
A straightforward use of bioinformatics has been carried out to find homologs
of known miRNAs both within the same genome and in the genomes of other species
(Pasquinelli et al 2000 Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Mendes et al 2009) A more difficult challenge is to identify miRNAs
unrelated to previously known miRNAs This was first accomplished for
vertebrate nematode and fly miRNAs using algorithms that search for conservation
of sequence and secondary structure (ie miRNA stem-loop precursors) between
species searching for patterns that are characteristic of miRNAs (Lai et al 2003 Lim
et al 2003 Lim et al 2005)
Most of the methods identified numerous miRNAs but are necessarily more
relaxed in terms of allowed structures Some approaches take advantage of the high
complementarity of plant miRNAs to target messages implementing the requirement
that the candidate has conserved complementarity to mRNAs (Jones-Rhoades amp Bartel
2004 Adai et al 2005) This additional filter has been useful for distinguishing
authentic plant miRNAs from false positives which later extended to mammalian
miRNA gene prediction (Xie et al 2005 Mendes et al 2012)
Another criteria used by most algorithms in the filters is evolutionary
conservation In plants mature miRNA sequences are conserved to a higher degree
than their precursor sequences For example Arabidopsis and rice diverged more than
130 million years ago (Friis et al 2004) but some miRNAs are highly conserved
in these plant species (Reinhart et al 2002) Furthermore various parameters for the
secondary structures such as free energy the number of paired residues within a
Chapter 1 Introduction and Review of Literature
31
miRNA or the number and size of bulges are also used to validate the predictions
(Jones-Rhoades amp Bartel 2004 Wang et al 2004 Adai et al 2005 Mendes et al
2012)
1132 Conserved microRNAs in Plants
Till date cloning genetics and bioinformatics screens have resulted in annotation of
approximately 184 potential Arabidopsis miRNAs (miRBase release 13) These
miRNAs seem to arise through gene duplication and subsequent diversification and
represent approximately 100 families (Maher et al 2006 Axtell 2013) Twenty one
families represented by 92 genes are clearly conserved in species beyond Arabidopsis
(Table 15) The presence of these miRNA families in other sequenced plant genomes
indicates that different plant species have an evolutionarily fluid set of miRNAs
(Willmann amp Poethig 2007) Recent studies on mosses one of the most ancient land
plants have shown that at least 14 miRNA families are conserved in eudicots and
mosses and therefore it appears that plant miRNAs share a common ancient origin
(Arazi et al 2005 Axtell amp Bartel 2005 Talmor-Neiman et al 2006 Zhang et al
2006 Axtell 2013) The number of members per family in a genome ranges from 1-32
with the exception of miR-430 which is represented by a cluster of ~80 loci in zebra
fish (Giraldez et al 2005)
In plants the number of members in each family correlates among examined
species certain families contain numerous members in all three species (eg miR156
miR166 miR169) whereas others consistently contain fewer genes (eg miR162
mir168 miR394) (Table 16) Although it is still unclear why certain miRNA are
encoded by more than one gene in plants (eg 12 genes encoding miR156) this
correlation might suggest functional significance of various miRNA family sizes Most
annotated plant miRNAs are conserved throughout flowering plants while a few
miRNAs are found only in a single sequenced genome and thus they could be
evolutionarily lsquoyoungrsquo miRNAs For example Arabidopsis has several miRNA
families that are not found in poplar or rice suggesting that miRNAs could be
generated continuously during evolution (Allen et al 2004a Rajagopalan et al
2006 Fahlgren et al 2007 Axtell 2012 de Alba et al 2013) Consistent with the
notion that non conserved miRNAs are of a more recent evolutionary origin these
miRNAs are mainly encoded by a single locus in the genome Within each family the
mature miRNA is always located in the same arm
Chapter 1 Introduction and Review of Literature
32
Table 16 MicroRNA families conserved in plants
miRNA
family
Arabidopsis Oryza Populus
miR156 12 12 11 miR159319 6 8 15 miR160 3 6 8 miR162 2 2 3 miR164 3 5 6 miR166 9 12 17 miR167 4 9 8 miR168 2 2 2 miR169 14 17 32 miR171 4 7 10 miR172 5 3 9 miR390 3 1 4 miR393 2 2 4 miR394 2 1 2 miR395 6 19 10 miR396 2 5 7 miR397 2 2 3 miR398 3 2 3 miR399 6 11 12 miR408 1 1 1 miR403 1 0 2 miR437 0 1 0 miR444 0 1 0 miR445 0 9 0 Total 92 127 169
All known miRNA families that are conserved between more than one plant species are listed
together with the number of genes identified in the sequenced genomes Rice miRNA families that have orthologs in maize but do not appear to have orthologs in the eudicots (Arabidopsis and Populus) are marked with an asterisk The following families contain miRNA genes annotated with
more than one number miR156 (miR156 and miR157) miR159319 (miR159 and miR319) miR166 (miR165 and miR166) miR171 (miR170 and miR171) and miR390 (miR390 and miR391)
of the stem loop (5prime- or 3prime) as it would be expected if the genes carry common ancestry
(Figure 17) The sequence of the mature miRNA and to some extent the segment on
the opposite arm of the hairpin to which it pairs is highly conserved between the
members of the same family (both within and between the species) while the sequence
secondary structure and length of the intervening ldquolooprdquo region can be highly divergent
within the same family
The patterns of paring and non pairing nucleotides are often conserved between
homologous miRNA stem loops from different species (Figure 17) The significance of
conserved mismatches is opined to guide DCL1 to cleave at the appropriate positions
along the stem loop
Chapter 1 Introduction and Review of Literature
33
Figure 17 Representative stem loop of miR164 Segments corresponding to the mature miRNAs
are shown in red (Adapted from Rajagopalan et al 2006)
Although many miRNAs are highly conserved in plants the degree of conservation in
most cases is quite low between plants and animals one exception is miR854 A recent
study has revealed that miR854 is conserved between Arabidopsis and animals and that
the Arabidopsis miR854 differs from the human miR854 only by a single nucleotide
(Arteaga-Vazquez et al 2006 Axtell 2012 de Alba et al 2013) The Arabidopsis
miR854 has multiple binding sites in the 3prime-untranslated region (UTR) of
OLIGOURIDYLATE-binding-PROTEIN mRNA 1bA (UBP1b) and represses the
translation of the UBP1b mRNA The animal miR854 is also complementary to a site in
the 3prime-UTR of the UBP1b homolog in animals However it is currently unclear how the
animal miR854 regulates its target mRNA Another distinction between plant and
animal miRNAs is the genomic organization of the miRNA genes While plant miRNA
genes are located predominantly in the intergenic regions animal miRNA genes tend to
localize in the intragenic regions both in the introns or exons of protein coding genes
Moreover miRNA genes are often clustered within a single precursor transcript
whereas individual plant miRNAs are derived from individual precursor RNAs (Bartel
Chapter 1 Introduction and Review of Literature
34
2004 Bonnet et al 2004 Kim 2005 Vaucheret et al 2006 Marco et al 2013)
1133 Non-conserved miRNA and challenges in annotation
Most miRNAs are conserved throughout flowering plants (Table 15) but many more
were found only in a single sequenced genome thus considered to be of a more recent
evolutionary origin The extended homology between few non conserved miRNAs
precursors and their target genes provides strong evidence that these relatively ldquoYoungrdquo
miRNAs arose from the duplication of the target gene segments (Allen et al 2004)
Several non-conserved miRNAs including miR161 miR163 miR173 miR447
miR475 and miR476 are known to direct cleavage of target transcripts (Allen et al
2004a Adai et al 2005 Sunkar et al 2005 de Alba et al 2013) It is difficult to
confidently predict targets for many because it is not possible to use complementary
site as a filter against false-positive target predictions It is also difficult to
confidently annotate non-conserved miRNAs as miRNA rather than siRNAs The
established minimal standard for miRNA annotation is a small RNA with detectable
expression and the potential to form a stem-loop when joined to flanking genomic
sequence (Kasschau et al 2003) In practice these requirements are too loose to
definitively categorize many small RNAs cloned from plants Many plant siRNAs are
detectable on blots (Vazquez et al 2004b) and hundreds of thousands of non-
miRNA genomic sequences can be predicted to fold into secondary structures that
resemble structures of plant miRNA precursors (Jones-Rhoades amp Bartel 2004)
Therefore without conservation of both sequence and secondary structure it is
difficult to be confident that a given cloned RNA originated from a stem-loop (ie is a
miRNA) rather than from a double-stranded RNA (ie is a siRNA) In fact many of
the thousands o f small RNAs cloned from Arabidopsis (Gustafson et al 2005) would
probably meet the literal requirements for annotation as miRNAs A few of these
sequences might be miRNAs but others that meet the literal criteria probably are not
so The challenges of annotating non conserved small RNAs were evidenced by
three related small silencing RNAs that were originally annotated as miRNAs
that turned out to be trans-acting siRNAs (tasiRNAs)(Kanno et al2013)
Although most miRNAs that are broadly conserved among flowering plants are
now being identified and experimentally validated (Table 16)(Jones-Rhoades amp
Bartel 2004) the challenges and ambiguities for classifying non-conserved miRNAs
prevent meaningful estimates of the total number of miRNA genes in Arabidopsis and
Chapter 1 Introduction and Review of Literature
35
other plant genomes It is possible that miRNAs might have escaped detection
because they are non-conserved and expressed in specific tissues or conditions and can
only be identified by using high- throughput sequencing
1134 MicroRNA biogenesis
11341 Transcription of microRNA precursor
Plant miRNAs are primarily found in the genomic regions that are not associated with
the protein coding genes (Reinhart et al 2002) and it appears that most if not all plant
miRNAs are produced from their own transcriptional units Plant miRNA genes are
occasionally clustered near each other in the genome suggesting transcription of
multiple miRNAs from a single primary transcript [eg the miR395 cluster (Grishok
et al 2001 Jones-Rhoades amp Bartel 2004b)] but this polycistronic arrangement of
miRNA genes appears far less frequently in plants than in animals (Bartel 2004)
Northern blot EST and mapping evidence indicate that plant miRNA primary
transcripts (also known as pri-miRNAs) as in animals are longer than needed to
encompass the miRNA stem-loops (Palatnik et al 2003 Jones-Rhoades amp Bartel
2004b) At least some of these pri-miRNA transcripts appear to be spliced
polyadenylated (Kurihara amp Watanabe 2004) and capped (Xie Z et al 2005) Two
rice miRNAs are contained within transcripts that contain exon junctions within the
presumptive stem-loop precursor implying that in these cases splicing is a prerequisite
for Dicer recognition (Sunkar et al 2005) Plant pri-miRNAs are over 1kb in length
and they are usually preceded by typical TATA box motifs They can also undergo
canonical splicing polyadenylation and capping All these observations indicates RNA
polymerase II is probably responsible for transcribing most plant miRNAs (Xie Z et
al 2005) as appears to be the case for many animal miRNAs Relatively little is
known about the regulation of miRNA transcription in plants but there is no reason
to suspect that this regulation would differ from that of protein-coding transcripts as
the bioinformatics analysis of the sequence upstream of the transcriptional start
sites of the miRNA genes showed the presence of putative binding motifs of
number of known transcription factors (Megraw et al 2006 Sethupathy 2013)
11342 MicroRNA processing and export
In plants maturation of miRNA is a step wise process A miRNA gene is transcribed to
pri-miRNA which is much longer than the pre-miRNA possessing the characteristic
stem-loop structure Like transcription of most protein-coding genes the miRNA
Chapter 1 Introduction and Review of Literature
36
transcription is processed by RNA polymerase II (Pol II) enzymes and the pri-
miRNAs are 5prime-capped and 3prime-polyadenylated (Lee et al 2004 Xie Z et al 2005)
Mature miRNAs are produced from pri-miRNAs through at least two sequential
processing steps by RNase III-type endonucleases In animals pri-miRNAs are first
processed at the bottom portion of the stem by Drosha a nuclear-localized RNase III
enzyme to liberate pre-miRNAs The pre-miRNAs are then exported to the cytoplasm
with the help of exportin-5The pre-miRNAs are further processed by Dicer another
RNaseIII enzyme to a duplex consisting of the miRNA and the antisense strands
(miRNA) Since plants do not have any Drosha homologs the two sequential
processing steps seem to be carried out by DCL1 a Dicer homolog in the nucleus
(Kurihara amp Watanabe 2004 de Alba et al 2013 Rogers amp Chen 2013)
The processing of pri-miRNAs to pre-miRNAs by DCL1 is assisted by two
other proteins HYPONASTY LEAVES1 (HYL1) and SERRATE (SE) HYL1 belongs to a
family of double-stranded RNA (dsRNA) binding protein and interacts with DCL1 in
vitro and in vivo (Lu amp Fedoroff 2000 Hiraguri et al 2005 de Alba et al 2013
Rogers amp Chen 2013) Loss-of-function mutations in the HYL1 gene result in
decreased accumulation of miRNAs and concomitant accumulation of pri-miRNAs
(Han et al 2004 Vazquez et al 2004a Kurihara et al 2006) SE a C2H2 zinc finger
protein interacts with DCL1 and HYL1 and plays a role in the processing of pri-
miRNAs to pre-miRNAs (Lobbes et al 2006 Yang L et al 2006) In addition the
nuclear cap-binding complex proteins CBP20 and CBP80 that are known as ABA
HYPER SENSITIVE 1(ABH1) have been shown to be required for proper miRNA
processing (Gregory et al 2008 Laubinger et al 2008) Recently it has been
demonstrated that DCL1 can release 21-nt RNAs from dsRNA as well as from
synthetic miR167 precursor RNAs in vitro However this cleavage is error prone and
inefficient in the absence of HYL1 and SE which accelerate the rate of DCL1-mediated
cleavage and promote accurate processing of miRNAs (Dong et al 2008 Rogers amp
Chen 2013)
11343 Methylation of the miRNAmiRNAduplex
After the miRNAmiRNAduplexes are released from the pre-miRNAs by the DCL1
activities the duplexes are methylated at the 2primeOH of the3prime-ends by HUA ENHANCER1
(HEN1) a small RNA methyltransferase (Yu et al 2005 Yang Z et al 2006 Rogers
amp Chen 2013 ) This step which is distinct from the RNase III-mediated processing
Chapter 1 Introduction and Review of Literature
37
step distinguishes the biogenesis of plant miRNAs from that of animal miRNAs
Mutations in the HEN1 gene were first isolated in a morphological screening for floral
development although the HEN1 mutants display pleiotropic phenotypes similar to the
developmental defects observed in the DCL1 mutants (Chen et al 2002 Park et al
2002 Rogers amp Chen 2013)
Figure 18 Biogenesis of microRNA
A Biogenesis of plant MicroRNA B Biogenesis of metazoananimal MicroRNA
The miRNA (red) is incorporated into RISC and the miRNA(blue) in degraded
This enzyme has two dsRNA-binding domains and nuclear localization signal It
is also conserved in fungi and is required for the processing of siRNAs as well as of
miRNAs (Park et al 2002 Boutet et al 2003 Xie et al 2003) Although the HEN1
protein is localized to the nucleus its methylation activity in the cytoplasm cannot be
ignored (Xie et al 2004 Rogers amp Chen 2013)
Chapter 1 Introduction and Review of Literature
38
The methylation of the miRNAmiRNA duplex is required to protect miRNAs
against the 3prime-end uridylation activity and subsequent degradation (Li et al 2005) In
the hen1 mutants accumulation of miRNAs is reduced to a lower level Also the
miRNAs appear to be heterogeneous in their sizes such that a ladder of bands rather
than a single species is observed for most miRNAs in the mutants (Li et al 2005)
MiRNA cloning and sequencing analyses revealed the presence of a number of U
residues at the 3prime-end of miRNAs in the hen1 mutants Moreover these nucleotides do
not correspond to the miRNAs in the pre-miRNAs indicating that these nucleotides are
added after the processing of the miRNAs from pre-miRNAs (Li et al 2005)
Uridylation of RNAs has also been proven to be correlated with RNA decay (Shen amp
Goodman 2004 Mullen amp Marzluff 2008) suggesting that uridylation attracts an
exonuclease which degrades miRNAs in plants
11344 MiRNA incorporation in to the RISC and its nuclear export
The methylated miRNAmiRNA duplexes undergo RISC assembly In this process
the miRNA of the duplexes is selectively incorporated into the RISC and the miRNA
appears to be removed and subsequently degraded (Hammond et al 2000 Hutvagner
amp Zamore 2002 Schwarz et al 2003 de Alba et al 2013 Rogers amp Chen 2013)
The ARGONAUTE 1(AGO1) proteins are core components of the RISC complex
They contain two structural domains the N-terminal PAZ domain that binds to the 3prime-
end of single-stranded RNAs and the C-terminal piwi domains that has a structure
similar to that of RNase H (Cerutti et al 2000 Parker et al 2004) Several AGO
proteins possess endonucleolytic activities within the PIWI domain and cleave target
mRNAs in the middle of the site complementary to miRNA (Liu et al 2004 Meister et
al 2004 Miyoshi et al 2005) Mutations in the AGO1 gene cause miRNA target
genes to be ectopically expressed and the levels of some miRNAs are reduced in the
mutants (Kidner amp Martienssen 2004 Vaucheret et al 2004 Kidner amp Martienssen
2005 Ronemus et al 2006) Moreover the phenotype of the AGO1 hypomorphic
alleles display defects in lateral organ polarity and leaf and flower morphology as
observed in the dcl1 mutants and null AGO1 alleles are embryonically lethal
supporting the close relationship between the AGO proteins and miRNA processing
(Kidner amp Martienssen 2004 Vaucheret et al 2004 de Alba et al 2013 Rogers amp
Chen 2013)
The production of miRNA miRNA duplexes occur in the nucleus while
Chapter 1 Introduction and Review of Literature
39
miRNA-RISC complexes are present in the cytoplasm where target mRNAs are
located This discordance of functional sites requires an export step during miRNA
biogenesis HASTY the Arabidopsis homolog of exportin-5 is thought to be involved
in miRNA nuclear export (Bollman et al 2003 Park et al 2005)The hasty mutant
exhibits pleiotropic defects in plant development and reduced accumulation of most
miRNAs as in the DCL1 or AGO1 mutants Howeverit is unclear whether the HASTY
proteins export the miRNA miRNA duplexes or the miRNA-RISC complexes in
plants
11345 Feedback regulation of miRNA biogenesis
MiRNA biogenesis is under feedback regulation such that the DCL1 and AGO1 genes
are themselves regulated by miRNAs DCL1 is itself a target of miR162 which leads to
the cleavage of the DCL1 mRNA (Xie et al 2003 de Alba et al 2013) Consistent
with this the levels of DCL1 mRNA are elevated in the mutants of miRNA processing
genes in which the miR162 abundance is reduced In addition there is a predicted
hairpin structure of miR838 in the 14th
intron of the DCL1 primary transcript
(Rajagopalan et al 2006) This prediction implies that DCL1-mediated processing
on this site can result in the cleavage of the DCL1 primary transcript into two truncated
fragments the N-terminal capped fragment and the C-terminal polyadenylated
fragment If this is the case it is possible that miRNA biogenesis competes with the
splicing event of the DCL1 pre-mRNA
The AGO1 is also regulated by miRNA There is a complementary site for
miR168 binding in the AGO1 mRNA and the transcript level of the AGO1 mRNA is
reduced through an mRNA cleavage mechanism (Vaucheret et al 2006 de Alba et al
2013 Rogers amp Chen 2013) indicating that the AGO1 mRNA levels are tightly
controlled by the levels of the AGO1 protein
1135 Mechanism of miRNA action
11351 MiRNA-directed mRNA cleavage
By forming the miRNA-RISC assembly miRNAs can direct the RISC to regulate gene
expression by two post transcriptional mechanisms mRNA cleavage or translational
repression (Bartel 2004 Jones-Rhoades et al 2006 Dalmay 2013) The degree
of miRNA-mRNA complementarity plays an important role for the
determination of the mechanism to be used A general rule is that while perfect or
Chapter 1 Introduction and Review of Literature
40
near-perfect complementarity induces mRNA cleavage central mismatches trigger
translational repression (Carrington amp Ambros 2003 Jones-Rhoades amp Bartel 2004)
Unlike most animal miRNAs plant miRNAs have near-perfect or perfect
complementarity to their targets and mRNA cleavage is assumed to be a predominant
mechanism by which miRNAs regulate gene expression in plants
In RNA cleavage mechanism miRNAs guide the AGO component of RISC to
cleave a single phosphodiester bond of the target mRNA within the miRNA-binding
site (Llave et al 2002b Tang et al 2003) The truncated fragments are then released
and degraded freeing the RISC to recognize and cleave another target mRNA This has
been validated by the detection of 3prime cleavage products that have 5prime ends that start at the
middle of the complementary regions The mRNA cleavage products are then further
degraded by other mechanisms The 3prime cleavage products of some miRNA targets are
degraded by the 5prime-3prime exonuclease XRN4 an Arabidopsis homolog of the major Yeast
mRNA degrading exonuclease Xrn1p It has been observed that the 3prime cleavage
products of some target mRNAs were accumulated in the xrn4 mutants (Souret et al
2004 Dalmay 2013) However the 3prime cleavage products of other miRNA targets do
not accumulate in the xrn4 mutants indicating that there are also XRN4 independent
mechanisms The 5prime cleavage products are typically untraceable by RNA gel blot
hybridization but can be detected by sensitive PCR based methods It has been found
that the 5prime cleavage products tend to obtain an oligo U tail at the 3prime ends This 3prime U tail
is correlated with shortening of the 5prime cleavage products from the 5prime end leading to the
conclusion that the 3prime uridylation causes 5 prime- 3 prime exonucleolytic decay of the 5prime cleavage
products (Shen amp Goodman 2004)
DCL1 HYL1 and SE interact with each other and are co-localized in the
nuclear bodies termed Dicing bodies or D-bodies in which some pri-miRNA shave
been found suggesting that D-bodies serve as a center for active miRNA processing
(Fang amp Spector 2007 Fujioka et al 2007 Song et al 2007) The miRNA
processing site appears to be distinct from the Cajal bodies that have previously been
found as a siRNA processing site (Pontes amp Pikaard 2008) The D-bodies include
SmD3 and SmB proteins that are found in the Cajal bodies However At collin an
additional marker for the Cajal bodies is not found in the D-bodies Moreover the D-
bodies are present in an At collin mutant lacking the Cajal bodies The pri-miRNAs of
miR171A and miR159A genes have been found to be co-localized with HYL1 and
Chapter 1 Introduction and Review of Literature
41
DCL1 in the D- bodies that are distinct from the Cajal bodies (Song et al 2007)
11352 MiRNA-directed translational repression
Translation repression is another mechanism exerted by plant miRNAs The regulatory
mechanism of miR172 which regulates flowering time and floral organ identity is
consistent with translation repression MiR172 over production does not affect the
transcript levels of APETALA2 (AP2) and AP2- like target mRNAs Instead the AP2
protein level is reduced in the miR172 over expressing mutant (Aukerman amp Sakai
2003 Chen 2004 Dalmay 2013) It was later shown that miR172 can also direct the
cleavage of the target mRNAs although the steady-state mRNA level is unchanged due
to feedback regulation (Schwab et al 2005 Jung et al 2007) It is clear that miR172
regulates its target genes primarily by translational repression rather than mRNA
cleavage Recently it has been reported that miR156157 and miR854 also regulate
their target genes through translational repression (Arteaga-Vazquez et al 2006
Gandikota et al 2007 Dalmay 2013) It was once thought that translational repression
is an exception to the rule However experimental evidence suggest a widespread
coexistence of translational repression and mRNA cleavage (Brodersen et al 2008)
1136 Regulatory roles of miRNAs in plant development
The miRNA target prediction has been a difficult task in order to obtain regulatory
targets without bringing in too many false predictions Plant growth and developmental
processes regulated by miRNAs are quite diverse and enormous Furthermore plant
miRNAs work cooperatively or antagonistically to establish a balanced regulation This
notion is well consistent with the findings that mutations in the regulators of miRNA
biogenesis transport and processing such as AGO1 DCL1 HYL1 HEN1 ABH1
and HASTY cause pleiotropic phenotypes (Mallory amp Vaucheret 2006 de Alba et al
2013) To date the roles of miRNAs have been mostly deduced from obtaining miRNA
over expressing transgenic plants or gain-of-function mutants in which miRNA-
resistant target genes are ectopically expressed
11361 Phase transition
Plants pass through a series of developmental phase transitions during their life span
beginning with seed germination continuing through vegetative phase change
reproductive phase change flowering initiation and finally seed production for the next
generation Because of their importance in understanding plant developmental
Chapter 1 Introduction and Review of Literature
42
processes genes regulating developmental transitions have been extensively studied
and underlying molecular mechanisms have been explored in various plant species
Majority of researches were mainly focused on vegetative phase change and
reproductive transition in Arabidopsis and Maize (Poethig 2003 Baurle amp Dean
2006) Particularly the mutations of the genes encoding various regulators of miRNA
biogenesis and transport such as HST SE and AGO exhibit altered juvenile to adult
vegetative phase change and vegetative to reproductive change (Hunter et al 2003
Peragine et al 2004 Vazquez et al 2004a Jones-Rhoades et al 2006 de Alba et al
2013)
Over-expression of the miR156a gene causes late flowering and delays
vegetative phase change (Wu amp Poethig 2006 Gandikota et al 2007 Wang et al
2008) It has been shown that miR156 negatively regulates a group of genes encoding
SQUAMOSA PROMOTER-BINDING PROTEIN-LIKE (Lewis et al 2007 de Alba et
al 2013 Rogers amp Chen 2013) transcription factors such as SPL3 SPL4 and SPL5
that promote flowering initiation In contrast transgenic plants over expressing
miR156-resistant SPL345 genes are early flowering Interestingly the endogenous
level of miR156 is temporally regulated In the early juvenile vegetative phase the
level of miR156 is very high but decreases rapidly before the onset of the adult juvenile
phase (Wu amp Poethig 2006)
The Arabidopsis early activation tagged (eat-D) mutant exhibits early flowering
with disrupted floral structure The mutant phenotypes are caused by over expression of
the miR172a-2gene (Aukerman amp Sakai 2003) MiR172 regulates a small group of
genes encoding APETALLA2 (AP2)-like transcription factors such as TARGET OF
EAT1 (TOE1) TOE2 SCHLAFMUTZE (SMZ) and SCHNARCHZAPFEN(SNZ)
TOE1 TOE2 SMZ and SNZ have been shown to participate in their productive phase
change by repressing a floral integrator FLOWERING LOCUS T (FT)(Jung et al
2007) Consistent with this toe1-2D 35STOE2 35SSMZ and 35SSNZ plants
exhibit late flowering (Jung et al 2007) MiR172 is also regulated temporally and
highly expressed in the adult vegetative phase both in Arabidopsis and maize (Chuck et
al 2007) Because the temporal expression patterns of miR156 and miR172 are
complementary to each other it has been suggested that the two miRNAs interact with
each other in regulating phase change (Chuck et al 2009 de Alba et al 2013) In
support of this view it has been shown that miR172 is down-regulated in the
Chapter 1 Introduction and Review of Literature
43
Corngrass1(Cg1) mutant in which miR156 is overproduced (Chuck et al 2007 de
Alba et al 2013) However there is no direct evidence for the direct linkage between
miR156 and miR172 It is also envisioned that miR156 and miR172 would not be
directly related For example miR156 regulates plastochron index that is the inverse of
leaf initiation rate and thus frequently used to analyze temporal patterns of plant
development In contrast miR172 does not affect plastochron (Wang et al 2008) The
down-regulation of miR172 in the maize miR156-over producing mutants would be
due to the prolonged juvenile vegetative phase in the mutants
Another miRNA that regulates flowering initiation is miR159 It regulates
MYB33 and MYB65 which are closely related to the barley GAMY B transcription
factor that responds to gibberellic acid (GA) The level of miR159 is elevated in GA-
treated seedlings but reduced in the GA biosynthetic ga1 mutant Transgenic plants
over producing miR159 exhibit delayed floral induction under short days (Achard et
al 2004) It is been suggested that MYB33 mediates GA signal to regulate LEAFY
(LFY) These observations indicate that miR159 plays an important role in the GA
signaling pathways that regulate proper floral induction under short days (Achard et al
2004)
11362 Floral development
Arabidopsis flowers consist of four distinct organs They arise in a characteristic
pattern within concentric rings called whorls from outside to inside four sepals four
petals six stamens and the central pistil consisting of two carpels Identities of the
floral organs are determined by three classes of regulatory genes classA classB and
class C genes (Krizek amp Fletcher 2005) The A and C class genes act antagonistically
to restrict their expression domains (Bowman et al 1991)
It is notable that miR172 also regulates floral architecture in addition to its role
in the timing of flowering initiation MiR172 inhibits the translation of the AP2 mRNA
(Chen 2004) Transgenic plants overproducing miR172 not only exhibit early
flowering but also show abnormal floral development which is quite similar to those
of the class A gene mutants such as the ap2 mutants (Aukerman amp Sakai 2003 de
Alba et al 2013) When the activity of the class A genes is inactivated that of the class
C genes such as AGAMOUS (AG) is elevated As a result the miR172- overproducing
transgenic plants exhibit no petals along with homeotic transformation of the sepals to
Chapter 1 Introduction and Review of Literature
44
the carpels (Aukerman amp Sakai 2003 Chen 2004)
Interestingly miR166 also regulates floral architecture The role of miR165166
in the regulation of meristem activity is closely linked to floral meristem formation and
organization (Zhao et al 2007 Miyashima et al 2013) Floral structure is severely
disrupted in the miR165166-overproducing mutants such as men1 and jba-1D in
which miR166 is overproduced (Williams et al 2005b Jung amp Park 2007) The men1
and jba-1D mutants have smaller gynoecium and reduced carpel number In an extreme
case the jba-1D homozygous plants lack visible carpels (Williams et al 2005b)
Similar phenotypes have been observed in the miR165 overproducers (Zhao et al
2007) It has been suggested that the phenotypes are possibly caused by altered AG
expression in the floral meristem (Jung amp Park 2007 Turchi et al 2013)
Other miRNAs also affect floral development Overproduction of miRNAs
involved in auxin signaling also affects floral architecture Transgenic plants
overproducing miR167 have defects in the formation of ovule and anther with reduced
fertility (Ru et al 2006) It has been shown that miR167 targets ARF6 and ARF8 that
play a role in gynoecium and stamen development and in regulating fertility (Ru et al
2006 Wu et al 2006) These observations suggest that auxin signals are also closely
related to floral organogenesis In addition transgenic plants over-producing miR164
and the cuc1cuc2 double mutants show fused sepals and reduced petals (Laufs et al
2004 Turchi et al 2013) suggesting that miR164 is also related to floral meristem
activity and boundary specification of the meristematic region
11363 Shoot apical meristem development
Shoot apical meristem comprises self-maintaining totipotent cells The shoot apical
meristem activity affects diverse aspects of plant developmental processes including
leaf development vascular development and lateral organ polarity (Bowman et al
2002) Because miR165166 plays a primary role in meristem formation
overproduction of miR165166 and multiple knockout mutants of its target genes
exhibit pleiotropic pheonotypes (McConnell et al 2001 Emery et al 2003 Kim et al
2005 Williams et al 2005b)
The targets of miR165166 include genes encoding the homeo domain-leucine
zipper (HD-ZIP) class III transcription factors such as PHABULOSA(PHB)
PHAVOLUTA (PHV) REVOLUTA(REV) ATHB15CORONA (CNA) and ATHB8
Chapter 1 Introduction and Review of Literature
45
PHB PHV and ATHB15 have overlapping functions in controlling meristem
development (Turchi et al 2013) Indeed the phb phv athb15 triple mutants have an
enlarged shoot apical meristem Consistent with this the miR166- overproducing men1
and jba-1D mutants exhibit enlarged shoot apical meristems (Kim et al 2005
Williams et al 2005b Turchi et al 2013) In the mutants the shoot apical meristems
are multiplied and large
The development of shoot apical meristems is also regulated by the WUSCHEL
(WUS)ndashCLAVATA (CLV) signaling pathway WUS positively regulates cell
proliferation In contrast CLV3 which is expressed in stem cells limits the WUS
expression and thus inhibits cell proliferation in the SAM (Sharma et al 2003)
Consistent with this notion WUS is expressed to a higher level in jba-1D
explaining the ectopic enlargement of the SAM in the mutant (Williams et al 2005)
While WUS is closely related to miR165166 in the shoot apical meristem
development the underlying molecular mechanisms are currently unclear It has been
observed that the miR166-over producing men1 plants have an arrested meristem
development (Jung amp Park 2007) In addition the heterozygous men1+ and the
homozygous men1 plants show different expression patterns of WUS and CLV3 It
appears that miR166 regulates WUS indirectly and influences the expressional balance
between WUS and CLV3 through a negative feedback loop (Jung amp Park 2007 de
Alba et al 2013)
The phv phb athb15 triple mutant has an enlarged shoot apical meristems the
rev phb phv mutant does not have functional shoot apical meristems (Prigge et al
2005) This distinction may be explained by the fact that while miR166 targets
primarily PHB PHV and ATHB15 miR165 targets all the five HD-ZIP III genes (Zhou
et al 2007) Consistent with this transgenic plants overproducing miR166 are distinct
from those overproducing miR165 The miR165-overproducing transgenic plants lack
functional shoot apical meristem and a pin-like structure is formed at the apical region
of the mutant These phenotypes are quite similar to rev phb phv triple mutant (Zhou et
al 2007 Liu et al 2013) The phenotypic distinction may because by the difference
of the cleavage efficiencies of the target mRNAs In accordance with this notion a few
35S miR166a transgenic lines exhibit no shoot apical meristems The men1
homozygous plants also show arrested meristem development (Jung amp Park 2007)
Chapter 1 Introduction and Review of Literature
46
MiR164 cleaves the CUC1 and CUC2 mRNAs as well as the NAC1 mRNA
CUC1 and CUC2 determines the organ boundary in the meristem Phenotypes of
transgenic plants that overproduce miR164 are similar to those of the cuc1cuc2 double
mutant that exhibits embryonic developmental defects such as cup-shaped cotyledons
and fused cotyledons (Laufs et al 2004) When a miR164-resistant CUC2 is
expressed the boundary domain is enlarged CUC also plays a role in embryonic shoot
meristem formation by regulating the SHOOT MERISTEMLESS gene It was therefore
concluded that miR164-mediated cleavage of CUC1 and CUC2 mRNAs determines the
sizes of the boundary domains in the shoot apical meristem and regulates separation of
the organs by restricting cell proliferation (Laufs et al 2004 de Alba et al 2013)
11364 Leaf development
The role of miR319 has been extensively studied in leaf development A dominant
jaw-D mutant overproducing miR319 shows uneven curved leaves with enlarged size
and five TCP genes which are closely related to the CINCINNATA (CIN) gene in
Antirrhinum majus are down regulated in the mutant suggesting that miR319 mediated
regulation of the TCP transcription factor genes are important for the final step of
leaf shaping (Palatnik et al 2003 Naz et al 2013) In addition to leaf shape and size
leaf polarity is also specified by miRNA MiR166-mediated cleavage of the HD-ZIPIII
mRNAs is a well to establish leaf polarity This discovery originates from the gain-of-
function mutants such as phb-1d and phv-1d wherein point mutations in the
complementary sites to miRNA helps the mRNA to evade or resist from miRNA-
mediated cleavage (McConnell et al 2001 Emery et al 2003 Naz et al 2013)
Plant leaves have a dorso-ventral polarity consisting of the adaxial (upper
surface) and abaxial (lower surface) sides The adaxial side is closer to the shoot
apical meristem Adaxial identity is specified by functionally redundant class III HD-
ZIP family genes such as PHB PHV and REV Ectopic expression of each genes
result in adaxialized leaves Dominant phb-1d and phv-1d mutants exhibit
transformation of the abaxial to adaxial fate and have rod-shaped leaves In contrast
miR165166-overproducing plants show pin-like cotyledons radicalized leaves and
abaxialized leaves (Chitwood et al 2007 Pattanayak et al 2013)
Leaf asymmetry is determined by miR166-mediated restriction of the
expression domains of the HD-ZIPIII genes The HD-ZIPIII genes are expressed in the
Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
7
high concentrations of caffeine that accumulate in tea coffee and a few other plant
species The lsquoallelopathic theory or the auto toxic function theoryrsquo proposes that
caffeine in seed coats is released into the soil and inhibits the germination of seeds
around the parent plants (Anaya et al 2006)
The lsquochemical defense theoryrsquo proposes that caffeine in young leaves fruits
and flower buds acts to protect soft tissues from insect pests (Ashihara amp Crozier
2001 Ashihara et al 2008) It has been shown that spraying tomato leaves with a 1
solution of caffeine deters feeding by tobacco horn worms while treatment of
cabbage leaves and orchids with 001ndash01 solutions of caffeine acts as a neurotoxin
and kills or repels slugs and snails (Hollingsworth et al 2002) This work has now
been extended with convincing evidence for chemical defense theory has recently
been obtained with transgenic caffeine ndash producing tobacco plants (Uefuji et al 2005
Kim et al 2006)
17 Distribution of caffeine in plants
Caffeine has been found in 13 orders of the plant kingdom (Ashihara and Suzuki
2004 Anaya et al 2006) In some species the main purine alkaloid is theobromine or
methyl uric acids such as theacrine liberine and methylliberine rather than caffeine
(Ashihara amp Crozier 1999 Ashihara amp Crozier 2001)(Table 14)
Table 14 Distribution of purine alkaloids in plants
SNo Plant species Common
name Major alkaloid Plant parts
containing
alkaloids 1 Coffea arabica Arabica Caffeine Leaves Seeds
2 Coffea canephora Robusta Caffeine Leaves Seeds
3 Coffea liberica Caffeine Theacrine Liberine
Seeds Mature leaves
4 Coffea dewevrei Caffeine Theacrine Liberine
Seeds Mature leaves
5 Camellia sinensis Tea Caffeine Leaves
6 Camellia assamica Assam tea Caffeine Leaves
7 Camellia assamica var
kucha Kucha Theacrine Caffeine Leaves
8 Camellia irrawadensis Theobromine Leaves
9 Camellia ptilophylla Cocoa tea Theobromine Leaves
10 Theobroma cacao Cocoa Theobromine Seeds
11 Paullinia cupuna Guarana Caffeine Seeds
Chapter 1 Introduction and Review of Literature
8
12 Cola nitida Caffeine Seeds
13 Citrus sp Caffeine Pollen
14 Ilex paraguariensis Mate Caffeine Leaves
Adapted from (Ashihara amp Crozier 1999 Ashihara amp Crozier 2001)
18 Caffeine biosynthesis
The major biosynthetic pathway is a four step sequence consisting of three sequential
methylation and one nucleosidase reactions (Figure 12) The xanthine skeleton of
caffeine is derived from purine nucleotides The initial step in caffeine biosynthesis is
the methylation of xanthosine by a SAM- dependent N-methyltransferase In addition
to experiments with radiolabeled precursors substrate specificities of native (Kato et
al 1999) and recombinant N-methyltransferases (Kato et al 2000 Ogawa et al
2001 Mizuno et al 2003a Mizuno et al 2003b Uefuji et al 2003) strongly
suggest that the major route to caffeine is a xanthosine rarr7-methylxanthosinrarr 7-
methylxanthine rarr theobromine rarr caffeine pathway (Figure 12) Although the
information has been obtained mainly from coffee (C arabica) and tea (Camellia
sinensis) the available evidence indicates that the pathway is essentially the same in
other purine alkaloid-forming plants such as mateacute (Ashihara 1993) and cacao
(Koyama et al 2003 Yoneyama et al 2006)
The first step in the caffeine biosynthetic pathway from xanthosine is
conversion of xanthosine to 7-methylxanthosine (Figure 12) This reaction is
catalyzed by the 7-methylxanthosine synthase (xanthosine 7 N-methyltransferase EC
211158) The genes encoding for 7-methylxanthosine synthase CmXRS1 (AB
034699) and CaXMT1 (AB 048793) were isolated from the C arabica (Mizuno et
al 2003a Uefuji et al 2003) The recombinant proteins obtained from these genes
exhibit 7-methylxanthosine synthase activity in vitro Xanthosine monophosphate
(XMP) was not converted to 7-methylxanthosine by the recombinant enzymes thus
inclusion of 7-methy-XMP was proposed (Schulthess et al 1996)
The second step of caffeine biosynthesis involves a nucleosidase which
catalyses the hydrolysis of 7-methylxanthosine Although N-methylnucleosidase (EC
32225) was partially purified from tea leaves (Negishi et al 1988) recent detailed
structural studies on coffee 7-methylxanthosine synthase suggested that the methyl
transfer and nucleoside cleavage may be coupled and catalyzed by a single enzyme
(McCarthy amp McCarthy 2007)
Chapter 1 Introduction and Review of Literature
9
The last two steps of the caffeine synthesis are also catalyzed by a SAM-
dependent N-methyltransferase(s) which is different from the N-methyltransferase
enzyme that catalyzes the first step in the pathway Native N-methyltransferase
activities have been detected in crude and partially purified extracts from tea and
coffee plants (Ashihara amp Suzuki 2004) and a highly purified preparation has been
obtained from young tea leaves (Kato et al 1999) The enzyme was assigned the
name caffeine synthase (EC 211160) that catalyses the last two steps of caffeine
biosynthesis viz the conversion of 7-methylxanthine to caffeine via theobromine
(Figure 12) The gene encoding caffeine synthase was first cloned from young tea
leaves (Kato et al 2000) Since then several genes encoding N-methyltransferases
with different substrate specificity have been reported
Activity of the recombinant theobromine synthase (CTS1 and CaMXMT EC
211159) is specific for the conversion of 7-methylxanthine to theobromine In
contrast the recombinant caffeine synthases (CCS1 and CaDMXMT1) can utilize
paraxanthine theobromine and 7-methylxanthine as shown with tea caffeine synthase
(TCS1) (Mohanpuria et al 2011) Although paraxanthine is the most active substrate
of this recombinant enzyme only limited amounts of paraxanthine are synthesized in
coffee cells hence in vivo caffeine synthase is principally involved in the conversion
of 7-methylxanthine to caffeine via theobromine
Radiolabelled tracer experiments with theobromine accumulating cacao and
Chinese tea (Camellia ptilophylla) showed a limited conversion of theobromine to
caffeine N-methyltransferase which catalyzed the conversion of 7-methylxanthine to
theobromine was detected in crude extracts from C ptilophylla but conversion of
theobromine to caffeine was not observed (Ashihara et al 1998) Genes homologous
to caffeine synthase have been isolated from several theobromine accumulating plants
including cacao (Yoneyama et al 2006) The recombinant enzymes derived from
these genes have 3 N-methyltransferase activity suggesting that these theobromine
accumulating plants contain specific theobromine synthase Caffeine does not occur
in C ptilophylla (Ashihara et al 1998) although it is present in leaves and fruits of
cacao along with theobromine (Koyama et al 2003 Zheng amp Ashihara 2004)
Chapter 1 Introduction and Review of Literature
10
Figure 12 Pathways for the biosynthesis of caffeine
The major pathway is indicated by solid lines (Mizuno et al 2003b Uefuji et al 2003) Alternative pathways in coffee and tea plants are indicated
by dashed lines(Kato et al 1996 Schulthess et al 1996) Enzymes (1) 7-methylxanthosine synthase (2) N-methylnucleosidase (3) Theobromine
synthase (4) Caffeine synthase
N
N
N
N7
H3C
3
1
O
O
Caffeine
N
NH
N
N7
3
O
O
Theobromine
N
N
N
N
H
H
7
O
O
N
N
N
N
H
H
O
O
N
N
N
N
H
H
O
O
Xanthosine
N
N
N
N
H
H
O
O
N
N
N
N
H
H
O
ON
N
N
N
H
H3C
71
O
O
Paraxanthine
CH3
CH3
CH3
CH3CH3
7-methylxanthine
Rib P
Rib
(1)
(4)
Rib
+
CH3
7-methyl
xanthosine
(2)
(3)
SAM SAH
SAM SAH SAM SAH
Xanthosine mono
phosphate
Rib P
7-methyl xanthosine
monophosphate
+
CH3 CH3
Inosine 5-mono
phosphate
Adenosine 5-mono
phosphate
SAM SAH
Guanosine
Guanosine 5-mono
phosphate
Chapter 1 Introduction and Review of Literature
11
181 Source of Xanthosine for caffeine biosynthesis
Xanthosine the initial substrate of purine alkaloid synthesis is supplied by at least four
different pathways de novo purine biosynthesis (de novo route) the degradation
pathways of adenine nucleotides (AMP route) and guanine nucleotides (GMP route)
and the S-adenosyl-L-methionine (SAM) cycle (SAM route) (Figure 13) Purine
nucleotides are synthesized by de novo and salvage pathways (Ashihara amp Crozier
1999 Stasolla et al 2003 Zrenner et al 2006) The de novo synthesis of non purine
precursors like CO2 10-formyltetrahydrofolate 5-phosphoribosyl-1-pyrophosphate and
the amino acids Glycine glutamine and aspartate results in the production of purine
nucleotides
Figure 13 Xanthosine biosynthesis Xanthosine for purine alkaloid biosynthesis is supplied by at
least four routes from adenosine released from the SAM cycle (SAM route) from IMP
originating from de novo purine synthesis (de novo route) from the cellular adenine
nucleotide pool (AMP route) and from the guanine nucleotide pool (GMP route)
Adapted from (Ashihara et al 2008)
The utilization of IMP for caffeine biosynthetic pathway which is formed by the
de novo purine biosynthetic pathway for caffeine biosynthetic pathways was
demonstrated in young tea leaves using 15
N-glycine and 14
C-labelled precursors and
inhibitors of de novo purine biosynthesis (Ito amp Ashihara 1999) Xanthosine is formed
by an IMP rarr XMP rarr xanthosine pathway IMP dehydrogenase (EC 111205) and 5prime-
nucleotidase (EC 3135) catalyze these reactions The inhibition of IMP dehydrogen-
ase by ribavirin inhibits both caffeine and gunanine nucleotide biosynthesis in tea and
coffee leaves (Keya et al 2003)
Chapter 1 Introduction and Review of Literature
12
The portion of xanthosine used for caffeine biosynthesis is derived from adenine
and guanine nucleotide pools which are produce de novo by salvage pathways there are
several potential pathways for xanthosine synthesis from AMP but the AMP rarr IMP
rarr XMPrarr xanthosine route is predominant All three enzymes involved in the
conversion AMP deaminase (EC 3546) IMP dehydrogenase and 5prime-nucleosidase
have been detected in tea leaves (Koshiishi et al 2001) Xanthosine for caffeine
biosynthesis can also be produced from guanine nucleotides by GMPrarrguanosine
rarrxanthosine pathway Guanosine deaminase (EC 35415) activity is found in cell free
extracts from young tea leaves (Negishi et al 1994) but GMP deaminase activity was
not detected in plants (Stasolla et al 2003 Zrenner et al 2006) The absence of GMP
deaminase activity suggests that a GMP rarr IMP rarr XMP rarr xanthosine route is not
functional in plants
The use of radiolabelled purine bases and nucleosides for the study of caffeine
biosynthetic pathway was facilitated by the fact that exogenous purine nucleotides are
rapidly hydrolyzed by the plant tissues forming purine nucleosides andor bases which
are then utilized in pathways which in most instances indirectly lead to xanthosine
Most of these purine compounds including adenine adenosine inosine hypoxanthine
and guanine are converted to their respective nucleotides by salvage enzymes
including adenine phosphoribosyl transferase (EC 2427) hypoxanthineguanine
phosphoribosyl transferase (EC 2428) adenosine kinase (EC 27120) and
inosineguanosine kinase (EC 2421) which usually have high activities in plant cells
The resultant nucleotides then enter the purine alkaloid biosynthetic pathway In the
case of guanosine it may be directly deaminated to xanthosine and utilized for caffeine
biosynthesis (Figure 13) (Ashihara et al 1997)
182 SAM cycle in plants
SAM is the methyl donor for the three methylation steps in the caffeine biosynthetic
pathway In this process SAM is converted to S-adenosyl-L-homocysteine (SAH)
which in turn is hydrolyzed to homocysteine and adenosine Homocysteine is recycled
via the SAM cycle to replenish SAM levels and adenosine is released from the cycle
Adenosine released from the cycle is converted to AMP directly andor indirectly via
adenine AMP is converted to xanthosine and used for caffeine biosynthesis Since
three moles of SAH are produced via the SAM cycle for each mole of caffeine that is
synthesized in theory this pathway has the capacity to be the sole source of both the
Chapter 1 Introduction and Review of Literature
13
purine skeleton and the methyl groups required for caffeine biosynthesis in young tea
leaves (Koshiishi et al 2001)
Methionine
S-Adenosyl-L-methionineHomocysteine
ATP
PPi+ Pi
Tetrahydrofolate
5-Methyl-
tetrahydrofolate
CH3+Adenosine
S-Adenosyl-L-
homocysteine
(xanthine
structure)(Methyl groups)
Caffeine
(1)
(2)(3)
(4)
Figure 14 The SAM cycle (activated methyl cycle) in plants (Ashihara amp Suzuki 2004)
Enzymes SAM synthetase SAM-dependent N-methyltransferases S-adenosyl-
homocysteine (SAH) hydrolase Methionine synthase Adenosine released from the cycle
is salvaged to adenine nucleotides and utilized both for purine structure of caffeine via
xanthosine and for re-synthesis of SAM via ATP
183 N-methyltransferase in caffeine biosynthesis Molecular cloning
The genes encoding for the enzymes involved in the caffeine biosynthesis were
successfully cloned from the coffee family (Ogawa et al 2001 Mizuno et al 2003a
Mizuno et al 2003b Uefuji et al 2003) The conserved amino acid regions of tea CS
(AB31280) the sequences of O-methyltransferases derived from plant origin and
Arabidopsis hypothetical proteins were made use of in designing primers for RT-PCR
and RACE technique Independent groups have isolated N-methyltransferase (NMT)
genes from coffee which have been classified based on the substrate specificity of the
recombinant proteins expressed in Ecoli (Table 15) PCR products thus obtained were
used as the probe to isolate full length cDNA from the library resulting in the
identification of all three N-methyltransferases involved in caffeine biosynthesis
(Figure 12) Several cDNA have been characterized from the coffee plants one for
xanthosine methyltransferase (XMTXRS) three for 7-methylxanthine methyltransferase
or theobromine synthase (MXMTCTS) and three for 37-dimethylxanthine
methyltransferase or caffeine synthase (DXMTCCS) (Table 15) (Kato amp Mizuno
2004)
A single gene encoding for xanthosine methyltransferase (XMTCmXRS1) was
identified which encoded for a polypeptide consisting of 372 amino acids with an
apparent molecular mass of 418kDa It is expressed almost uniformly in aerial tissues
Chapter 1 Introduction and Review of Literature
14
of C arabica including leaves floral buds and immature but not in mature beans
Three genes encoding 7-methylxanthine methyltransferase (theobromine synthase) have
been identified The number of amino acids in the putative polypeptides are 378 for
MXMT1 (427kDa) and 384 for MXMT2 and CTS2 (434kDa) They differ by insertion
or deletion of blocks of several residues in the C-terminal region Their catalytic
properties based on kinetic parameters such as Km values were different They are
expressed in young leaves floral buds and immature but not mature beans Three genes
were identified for 3 7-dimethylxanthine methyltransferase (caffeine synthase) DXMT
CCS1 and CtCS7 each encoding a 43 kDa polypeptide consisting of 384 amino acids
However their kinetic properties differ from each other such as DXMT and CCS1
showed Km values of 1200 and 157microM for theobromine respectively Expressions
profiles are also distinct DXMT being expressed exclusively in immature beans while
CCS1 expression is ubiquitous occurring in all tissues The presence of isoforms of
these enzymes with different properties suggests that caffeine is synthesized through
multiple pathways depending on availability and concentration of the substrate (Mizuno
et al 2001 Ogawa et al 2001 Mizuno et al 2003a Mizuno et al 2003b Uefuji et
al 2003) Genomic clones of theobromine synthase were obtained by PCR- RFLP
from C canephora (Satyanarayana et al 2005 Satyanarayana 2006) The genomic
clones are reported to have highly conserved exons and a variable third intron The
promoter for the theobromine synthase was cloned and characterized (Satyanarayana et
al 2005)
There is high degree of similarity in the coding region of coffee NMT genes
isolated so far though differences exist in the 3prime untranslated region (UTR) for these
genes The deduced amino acid sequence of these enzymes shows more than 85
homology between these genes Phylogenetic analysis indicates that they are more
closely related to C-methyltransferases including those for jasmonic acid salicylic acid
and benzoic acid than to other N-methyltransferases This suggests that coffee N-methyl
transferases belong to a distinct sub-group within plant methyltransferases Their
cellular localization determined by green fluorescent protein fusion method and β-
glucourodinase (GUS) showed that the all the three genes are localized in the cytoplasm
(Ogawa et al 2001 Vinod et al 2007) It was also shown that these were present
along with chlorogenic acid(Vinod et al 2007)
Chapter 1 Introduction and Review of Literature
15
Table 15 N-methyltransferases and their encoding genes involved in caffeine biosynthesis
Gene (Enzyme) Gene name Accession No Description Reference
Xanthosine methyltransferase
( 7-methylxanthosine synthase) (EC 211158)
CaXMT AB 048793 Immature fruit cDNA Screening (Uefuji et al 2003)
CmXRS1 AB 034699 Leaf cDNA library RACE (Mizuno et al 2003a)
CaMXMT1 AB 048794 Leaf cDNA library RACE (Ogawa et al 2001)
7-methylxanthine synthase transferase
(theobromine synthase) ( EC 211159)
CTS1 AB 034700 Immature fruit cDNA Screening (Uefuji et al 2003)
CaMXMT2 AB 984125 Leaf cDNA library Screening (Mizuno et al 2001)
CTS2 AB 054841 Leaf cDNA library Screening (Mizuno et al 2001)
37-methylxanthinemethyltransferase
(Caffeine synthase) (EC 211160)
CaDXMT1 AB 086414 Immature fruit cDNA Screening (Uefuji et al 2003)
CCS1 AB 0861414 Immature fruit cDNA Screening (Uefuji et al 2003)
CtCS7 AB 0864415 Immature fruit cDNA
NMT genomic clones
Theobromine synthase-1
CX10 Complete coding region PCR Satyanarayan 2006
CX8 Complete coding region PCR -do-
Theobromine synthase-1 PG-5 Promoter + coding region RAGE -do-
Theobromine synthase-1 PG-1 Promoter + coding region RAGE Satyanarayan 2006
PG-4 Promoter + coding region RAGE -do-
Theobromine synthase-1 NMT
(AY273814) Complete coding region PCR Kochko A et al 2010
Theobromine synthase-2 NMT
(AY362825) Complete coding region PCR Kochko A et al 2010
Chapter 1 Introduction and Review of Literature
16
19 Catabolism of caffeine
Caffeine is produced in young leaves and immature fruits and continues to accumulate
gradually during the maturation of these organs At the same time caffeine is slowly
degraded with the removal of the three methyl groups resulting in the formation of
xanthine Catabolism of caffeine was first reported in coffee leaves (Kalberer 1965)
Since then a number of tracer experiments using 14
C labelled purine alkaloids have
been reported (Suzuki amp Waller 1984 Ashihara et al 1996 Vitoria amp Mazzafera
1999 Mazzafera 2004) They demonstrated the major catabolic pathway is caffeine rarr
theophylline rarr 3-methylxanthine rarr xanthine Xanthine is further degraded by the
removal by the conventional purine catabolic pathway to CO2 and NH3 via uric acid
allatonin and allantoate (Figure 15) (Ashihara amp Crozier 1999 Stasolla et al 2003
Zrenner et al 2006) Caffeine catabolism usually begins with its conversion to
theophylline catalyzed by N7-demethylase The involvement of P450-dependent mono-
oxygenase activity for this reaction was suggested (Huber amp Baumann 1998
Mazzafera 2004) However the activity of this enzyme has not yet been determined in
cell free extracts [8-14
C] Theophylline degraded to CO2 at much faster rate than [8-14
C]
caffeine indicating that conversion of caffeine to theophylline is a major rate limiting
step of caffeine catabolism and a possible reason why caffeine accumulates in high
concentrations in tissues of C sinensis and C arabica It is widely believed that the
control of caffeine levels in plants is a function of the balance between rates of
synthesis and degradation and this balance seems to vary depending on the plant
species and the tissue developmental stage (Mazzafera 2004)
Chapter 1 Introduction and Review of Literature
17
Figure 15 Caffeine catabolic pathways Caffeine is mainly catabolised via theophylline and 3-
methylxanthine Xanthine is further degraded to CO2 and NH3 by the conventional
oxidative pathway Adapted from (Ashihara et al 2008)
Chapter 1 Introduction and Review of Literature
18
110 Genetic engineering of Coffee
Genetic transformation has potential applications in coffee agriculture for incorporating
genes which could provide desirable traits such as disease and insect resistance
drought and frost tolerance and herbicide resistance Transgenic technology can also be
used to increase nutritional value and improve cup quality produce varieties with
caffeine-free beans and for production of hybrid crops for molecular farming
Identification of target specific genes is one of the pre-requisites for developing
transgenic crops The availability of a large number of EST sequences in coffee and
initiation of coffee genome sequencing may speed up the gene discovery and accelerate
transgenic research efforts in coffee
1101 Insect Resistance
Production of insect resistant coffee plants is one of the major objectives of the
breeding programs The major pests attacking coffee include coffee berry borer (CBB
Hypothenemus hampei) white stem borer (WSB Xylotrechus quadripes) leaf miner
(Perileucoptera coffeela) and root nematodes (Meloidogyne spp and Pratylenchus
spp) The CBB is present in almost all the coffee growing countries and considered to
the most devastating pest in coffee To date there is no reported source of resistance to
CBB in the coffee gene pool Like CBB WSB is another serious pest in C arabica in
India and several other East Asian countries Both CBB and WSB belong to the order
Coleoptera For India controlling WSB is the biggest challenge and has therefore
become the highest research priority Robusta is generally resistant to WSB but the
interspecific Robusta Arabica hybrids are susceptible to WSB Although leaf miner is
not yet a serious pest in India and other East Asian countries it is an economically
important pest in East Africa and Brazil Effective chemical control of CBB and WSB
is difficult due to the nature of their life cycle inside the berry and stem respectively as
well as environmental concern regarding the use of pesticides Biological control
measures are adapted to combat these insect pests with varying degrees of success
Developing coffee plants resistant to these pests using genetic transformation
technology could be one of the alternative strategies to counter pest damage For insect
resistance several different classes of proteins from bacterial plant and animal sources
have been isolated and their insecticidal properties tested against many important pests
Amongst these proteins Bt toxins are most important and several transgenic crops
expressing Bt genes have been commercialized Coffee transgenic plants carrying a
Chapter 1 Introduction and Review of Literature
19
synthetic version of the cry1Ac gene have been produced (Leroy et al 2000 Mishra et
al 2002) Indeed this was the first report of an important agronomic trait being
introduced into a coffee plant The transgenic plants presented similar features in
growth and development compared to normal plants Transgenic plants highly resistant
to leaf miner under greenhouse conditions were tested under field conditions in French
Guyana for 4 years for pest resistance (Perthius et al 2005 Perthius et al 2006)
From 54 independent transformation events 70 of the events were resistant to leaf
miner Unfortunately the field trial was vandalized which led to the termination of the
experiment (Montagnon et al 2005)The effectiveness of Bt genes in controlling
coleopteran pests is well documented in corn and potato which indicates that Bt genes
might be effective against CBB The high toxicity of B thuringiensis sero var
israelensis against first in star larvae of CBB has been demonstrated (Mendez et al
2003) In another experiment an α-amylase inhibitor from Phaseolus vulgaris was
tested against CBB and found to have an inhibitory effect on its growth and
development (Grossi et al 2004)In addition to the CBB and WSB C arabica
varieties are also susceptible to endoparasitic root-knot nematode (Meloidogyne spp)
(Campos et al 1990 Mishra et al 2012)
So far 15 species have been reported to be parasites of coffee Controlling
nematodes is extremely difficult and currently seedling grafting with robusta rootstock
is followed as one of the control measure Sources of resistance specific to root-knot
nematodes have been identified in coffee trees (Bertrand et al 2001) and the Mex-1
gene conferring resistance to M exigua in C arabica is in the process of isolation (Noir
et al 2003)
1102 Tolerance to Abiotic Stress
In many coffee growing countries abiotic stress such as drought and frost are the major
climatic factors that limit coffee production Changes in climatic patterns due to global
climate change are considered increasingly important for coffee cultivation Drought
induces water stress in plants which affects vegetative growth and vigor and triggers
floral abnormalities and poor fruit set It also indirectly increases the incidence of pests
and diseases in the plants C arabica is generally more tolerant to water stress than C
canephora partly due to its extensive deep root system C racemosa is known to be a
good source of drought tolerance In India hybrids between C canephora and C
racemosa have been obtained and are currently under evaluation for drought tolerance
Chapter 1 Introduction and Review of Literature
20
In many coffee growing countries coffee is propagated in marginal areas where the
annual rainfall is below 1000thinspmm with prolonged dry spells of over 4-5 months In
those areas water shortage and unfavorable temperatures constitute major constraints
and the growth and productivity of C canephora are badly affected As with drought
periodic frost also affects coffee production in parts of Brazil The introduction of
drought and frost tolerant genes through genetic transformation would be of great
importance for alleviating these problems Research is now being carried out by several
groups to identify genes involved in biotic as well as abiotic stress The most promising
genetic engineering approach for drought tolerance includes the use of functional or
regulatory genes as well as the transfer of transcription factors In recent years plants
tolerant to high temperature and water stress have been the subject of intense research
(Chaves et al 2003 Chaves et al 2004 Coraggio et al 2004) For achieving drought
tolerance genes that have been targeted include those encoding enzymes involved in
detoxification or osmotic response metabolism enzymes active in signaling proteins
involved in the transport of metabolites and regulating the plant energy status (Chaves
et al 2003 Chaves et al 2004 Coraggio et al 2004) The dissection of molecular
mechanisms related to signal transduction and transcriptional regulation might help in
engineering drought tolerance in coffee
1103 Disease Resistance
Coffee leaf rust (CLR) caused by the fungus Hemileia vastatrix is the most important
disease in coffee with substantial loss to coffee production and productivity in all the
coffee growing countries In addition to leaf rust coffee berry disease (CBD) caused by
fungus Colletotrichum kahawae can be a devastating anthracnose leading to substantial
crop loss in Africa Several other fungal and bacterial diseases may also affect coffee
to a extent C arabica is more susceptible to many diseases than C canephora Though
most of the disease control measures rely upon chemical control they are more
expensive and labour intensive The long-term solution is the breeding of resistant
varieties which is the focus of many breeding programs However breeding for disease
resistant varieties is time consuming due to the perennial nature of coffee with its long
gestation period India has a long history of C arabica breeding especially for leaf rust
resistance and is the first country to demonstrate the existence of multiple races of leaf
rust Resistance to CLR is conditioned primarily by a number of major (SH) genes and
coffee genotypes are classified into different resistance groups based on their
Chapter 1 Introduction and Review of Literature
21
interaction with different rust races pathogen (van der Vossen 2001) Currently C
canephora provides the main source of resistance to pests and diseases including CLR
(H vastatrix) and CBD (C kahawae) and therefore used in breeding programs Other
diploid species like C liberica and C racemosa are being used as a source of resistance
to coffee leaf rust and coffee leaf miner respectively (Guerreiro et al 1999
Sreenivasan et al 1989) The development of coffee varieties resistant to major fungal
diseases such as CLR and CBD using transgenic technology will benefit the coffee
industry immensely During the last 15 years significant progress has been made in the
area of host-pathogen interactions (Staskawicz et al 1995 Feys et al 2000
Shivaprasad et al 2012) and many resistance genes involved in recognizing invading
pathogens have been identified and cloned (Takken et al 2000) A number of
signalling pathways induced because of pathogen infection have been dissected (Dong
et al 1998 Navarro et al 2006) Many antifungal compounds synthesized by plants to
combat fungal infection have been identified (Does et al 1998) Understanding the
specific induction of targeted pathways and identification of specific pathways
responsible for particular fungal resistance is important in order to employ this strategy
in transgenic technology The recent investigation of gene expression during coffee leaf
rust infection could provide an insight into the defence pathways operating in coffee
(Fernandez et al 2004 Nardi et al 2006) Efforts have been made to identify and
clone resistance genes from coffee for achieving durable resistance The genetic and
physical map of two resistance genes SH3gene conferring resistance to rust (Prakash et
al 2004) and the Ck1 gene conferring resistance to C kahawae CBD (Gichuru et al
2006) have been established These genes could be used for molecular marker assisted
breeding programs (Mishra et al 2012)
1104 Production of low-caffeine coffee
A widespread belief that the ingestion of caffeine can have adverse effects on
health resulted in the increased demand for decaffeinated coffee (Ashihara amp Crozier
2001) Several method were used to obtain decaffeinated plants by conventional
breeding techniques using low yielding varieties and mutants that accumulated
relatively low amounts of caffeine when compared to the commercially available ones
Low-caffeine and decaffeinated coffee represent around 10 of the coffee sales
around the world (Ribas et al 2006b) The industrial process for coffee decaffeination
can be expensive and affects the original flavour and aroma in coffee (David 2002)
Chapter 1 Introduction and Review of Literature
22
Transgenic coffee plants with suppressed caffeine synthesis using RNA interference
(RNAi) technology have been obtained (Ogita et al 2003 Ogita et al 2004) Specific
sequences in the 3prime untranslated region of the theobromine synthase gene (CaMXMT1)
were selected for construction of RNAi short and long fragments The caffeine and
theobromine content of the transgenic plants were reduced by ~ 70 when compared to
the untransformed plants In C canephora RNAi technology has also been employed
to silence the N-methyl transferase gene involved in caffeine biosynthesis (Vinod et al
2007) Promoter of an N-methyltransferase (NMT) gene involved in caffeine
biosynthesis was cloned (Satyanarayana et al 2005 Satyanarayana 2006) which will
be very useful for studying the regulation of caffeine biosynthesis
1105 Improvement in cup quality
Improvement in coffee cup quality requires elaborate knowledge of the chemical
constituents as well as the metabolic pathways involved in the elaboration of quality
The constituents of coffee beans include minerals proteins carbohydrates caffeine
chlorogenic acids (CGA) glycosides lipids and many volatile compounds that give
flavour to coffee by roasting Among these the role of three major constituents
sucrose CGA and trigonelline have been studied in coffee The sucrose content of
coffee bean is associated with coffee flavour the higher the sucrose content in green
beans the more intense will be the cup flavour (Clifford et al 1985 de Maria et al
1996) The sucrose content of C arabica (82-83) is higher than C canephora (33ndash
40) The sucrose amino acids ratio in green beans determines the profile of volatile
compounds Manipulating sucrose content in coffee bean is therefore important in
improving cup quality The sucrose synthase gene (CcSUS2) from C canephora has
been cloned and sequenced (Leroy et al 2005) This provides an opportunity to
manipulate the sucrose content in coffee Chlorogenic acids are products of
phenylpropanoid metabolism Chlorogenic acids are a group of hydroxycinnamoyl
quinic acids (HQA) formed by esterification between caffeic acids coumaric acids and
quinic acids (Clifford et al 1999) They are present in relatively large quantities in the
coffee bean and are the precursor of phenolic compounds in roasted beans C
canephora beans contain higher CGA (10) compared to C arabica beans (6-7)
CGA are known to have antioxidant properties as well as being associated with disease
resistance (Campa et al 2003) Genetic manipulations of genes involved in CGA
synthesis can serve either of these purpose by up- or down regulating the pathway
Chapter 1 Introduction and Review of Literature
23
Phenylalanine ammonia-lyase (PAL) catalyzes the first step of the phenylpropanoid
pathway leading to the synthesis of a wide range of chemical compounds including
flavonoids coumarins hydroxycinnamoyl esters and lignins (Hahlbrock amp Scheel
1989) Recently the full length cDNA and corresponding genomic sequences of PAL
from C canephora was isolated characterized and functionally validated (Mahesh et
al 2006 Parvatam et al 2006) This has opened up new possibilities for manipulating
the level of the PAL enzyme in coffee which in turn can be useful for improvement of
cup quality and manipulating antioxidant properties in coffee
1106 Fruit Ripening
Uniformity during fruit ripening is decisively related to cup quality in coffee and
consequently to the value of the product Fruits at the ideal ripening stage produce the
best organoleptic characteristics for coffee The presence of over ripened or green fruits
changes the acidity the bitterness and consequently the cup quality In order to
maximize uniform ripening of coffee fruits it is essential to control the action of genes
involved in the last step of maturation process Ethylene is known to trigger ripening
and increasing ethylene biosynthesis is associated with various stages of ripening
process (Protasio et al 2005) To control coffee fruit maturation two of the major
genes involved in ethylene biosynthesis namely ACC synthase and ACC oxidase
have been cloned (Protasio et al 2005 Neupane et al 1999) Introduction of the ACC
oxidase gene in antisense orientation have been achieved in both C arabica and C
canephora The effect of the transgene on ethylene production and fruit maturation has
yet to be reported The inhibition of genes downstream to the initial ethylene burst is
also an option to control coffee fruit maturation (Ribas et al 2006)
111 RNA interferenceRNAi
The regulation of gene expression at post transcriptional level has recently attracted
much attention because of the discovery of the phenomenon called RNA interference
(RNAi) RNAi based silencing techniques are more efficient than antisense mediated
gene silencing (Wesley et al 2001) The effect was first observed in plants wherein
silencing was observed when some copies of a transgene was in the forward orientation
with respect to the promoter and some in the reverse orientation This resulted in the
formation of double stranded RNA in the system The phenomenon in which
experimentally introduced double stranded RNA (dsRNA) leads to the loss of
expression of the corresponding cellular gene by sequence specific RNA degradation
Chapter 1 Introduction and Review of Literature
24
was termed as RNA interference or RNAi The protective effect of RNA silencing in
reducing virus infectivity supports the view that PTGS has evolved as a mechanism to
defend plants against virus infection and also to moderate the possible deleterious
genome-restructuring (insertional) activity of virus-like mobile genetic elements eg
retrotransposons A growing body of biochemical and genetic data has further
demonstrated that plants animals and yeasts share related mechanisms of specific
degradation of RNAs in which double-stranded forms of RNA are initiator molecules
(Brenstein et al 2001)
The key insight into the process of PGTS was provided by the experiments
conducted by Hamilton amp Baulcombe (1999) who identified the product of RNA
degradation as small RNA (siRNA) species of ~25nt of both sense and antisense
polarity SiRNA are formed and accumulate as double stranded RNA molecules of
defined chemical structures Both genetic and biochemical approaches were undertaken
to understand the basis of silencing Genetic screens were carried out in fungus
Neurospora carassa the alga Chalmydomonas reinhardtii and plant Arabidopsis
thaliana to search for detective mutants in PTGS
A combination of results obtained from several in vivo and in vitro experiments
have gelled into a two step mechanistic model for RNAiPTGS The first step referred
to as the RNAi initiating step involves binding of the RNA nucleases to a larges
dsRNA and its cleavage into discrete ~21-25nt RNA fragments This is ATP-
dependent reaction which involved RNase III - type endonucleases which make
staggered cuts in both strands of the dsRNA leaving 3prime overhang of 2 nucleotides with
5prime- phosphate 3prime-hydroxyl and no modification in the sugar phosphate backbone
(Hamilton amp Baulcombe 1999 Elbashir et al 2001) SiRNAs are the direct products
of dsRNA cleavage by the multi-domain RNase III enzyme DICER (Figure 16)
(Brenstein et al 2001 Karthikeyan et l 2013)
In the second step or commonly known as the effector step the double stranded
siRNAs produced in the first step bind to an RNAi specific protein complex to form a
RISC The complex undergoes activation in the presence of ATP so that the antisense
component of the unwound siRNA becomes exposed and allows the RISC to perform
the downstream RNAi reaction
Chapter 1 Introduction and Review of Literature
25
Several enzymes are involved in RNAi based on their role genes that encode for them
are classified into RNAi initiators RNAi effectors RNA dependent RNA polymerases
and silencing genes Two C elegans genes rdeI and rde4 (rde stands for lsquo RNAi
deficientrsquo) are believed to be involved in the initiation step of RNAi The C elegans
rde1 gene is a member of a large family of genes and is homologous to the Neurospora
qde2 (qde stands for ldquoquelling deficientrdquo) and Arabidopsis AGO1 (AGO stands for
agronaute AGO1 was previously identified to be involved in Arabidopsis
development) Although the functions of these genes are not clear a mammalian
member of RDE1 family has been identified as a translation initiation factor (Thakur
2005) Important genes for the effector step of PTGS in C elegans are rde2 and mut7
genes These genes were identified from heterozygous mutant worms that were unable
to transmit RNAi to their homozygous offsprings Worms with mutated rde2 or mut 7
genes show defective RNAi mut-7 gene encodes a protein with homology to the
nuclease domains of RNAase D and a protein implicated in Werner syndrome (a rapid
ageing disease in humans) (Grishok et al 2001 Kamath-Loeb et al 2012s)
Neurospora qde-1 Arabidopsis SDE-1SGS-2 and C elegans ego-1 appear to encode
RNA dependent RNA polymerase (RdRPs) It assumed that this is a proof that an RdRp
activity is required for RNAi Certainly the existence of an RdRp might explain the
remarkable efficiency of dsRNA induced silencing if it amplifies either the dsRNA
prior to cleavage or the siRNAs directly (Muers 2013) In C elegans ego-1 mutants
(ego stands for lsquoenhancer of glp-1) RNAi functions normally in somatic cells but is
defective in germline cells where ego-1 is primarily expressed In Arabidopsis SDE-
1SGS-2 mutants (SGS stands for suppressor of gene silencing) siRNA are produced
when dsRNA is introduced via an endogenously replicating RNA virus but not
introduced by a transgene It has been proposed that perhaps the viral RdRP is
substituting for the Arabidopsis enzyme in these mutants Random degradative PCR
model suggests that an RdRP uses the guide strand of an siRNA as a primer for the
target mRNA generating a dsRNA substrate for Dicer and thus more siRNAs
Several genes controlling RNA silencing in plants have been identified through
genetic screens of Arabidopsis mutants impaired in transgene induced RNA silencing
They encode a putative RNA-dependent RNA polymerase (SGS2SDE1) a coiled
protein (SGS3) a protein containing PAZ and Piwi domains (AGO1) and an RNA
helicase (SDE3) The putative SGS2SDE1 of Neurospora is related to QDE-1 and
Chapter 1 Introduction and Review of Literature
26
EGO-1 of C elegans whereas PAZPiwi protein AGO is related to QDE-2 of
Neurospora RDE-1of C elegans RNA helicase SDE3 is related to SMG-2 of C
elegans and Mut-6 of Chlamydomonas MUT-7 gene of C elegans encodes a protein
similar to Rnase D whereas the Drosophila DICER gene encode a protein similar to
RNase III An Arabidopsis ortholog of DICER gene has been identified called CAF
SIN1 SUS1(Thakur 2005 Poethig 2009)
Since the RNAi machinery is present constitutively within the eukaryotic cell it
is important to explore the metabolic advantages that are accorded by the RNAi related
proteins during the intrinsic normal growth of cells and development of organisms The
natural RNAi machinery not only keeps the mobile transposable elements from
disrupting the integrity of the genomes as suggested by the analysis of model organisms
like A thaliana C elegans D melanogaster (Tabara et al 1999 Wu-Scharf et al
2000 Aravin et al 2001 Hamilton et al 2002 Lisch 2009) but also participates in
development of organisms Genetic defects in Celegans RNAi genes ego1 and dicer
cause known specific developmental errors (Grishok et al 2000 Grishok et al 2001
Knight amp Bass 2001 Hall et al 2013) Similarly the Argonaute family of genes of A
thaliana (especially the ZWILLE proteins) is also responsible for plant architecture and
meristem development (Carmell et al 2002 Hamant and Pautot 2010) and the Dicer
homologue of A thaliana CAF1 is required for embryo development (Golden et al
2002 Nakano et al 2011) Thus genetic evidence illustrates the role of the RNAi
machinery as a controller of development related genes The mechanistic details of
these developmental processes are beginning to emerge
Chapter 1 Introduction and Review of Literature
27
Figure 16 Mechanism of gene silencing induced double stranded RNA The dicer enzyme to
produce siRNAs cleaves the RNA The siRNA generated bind to the nuclease complex
RISC The antisense component in the siRNA in the RISC guides the complex towards
the cognate mRNA resulting in endonucleolytic cleavage of the mRNA
Chapter 1 Introduction and Review of Literature
28
112 MicroRNA or MiRNA
In 1991 Ambros and co-workers first isolated a lin-4 mutant of Celegans which was
arrested at the first larval stage (Lee et al 1993) Later on the let-7 mutation was
isolated in the same system which was responsible for development through the fourth
larval stage Both lin-4 and let-7 encode short 22-nucleotide mature RNAs and were
called short temporal RNA because they control the temporal development program of
C elegans The mature lin-4 RNA defines (negatively regulates) the mRNA expression
of the lin-14 and lin-28 hetrochronic genes with the antisense mediated repression
mechanism of translation initiation and thus specifies the fate of cells during the first
three larval stages Recent studies have revealed that the short temporal RNAs are
actually members of a group of tiny RNAs (21to 28 nucleotides) called the micro-
RNAs (miRNA) (Bartel 2009) isolated members of which could easily run to a few
thousand which includes both those were identified computationally and by deep
sequencing Some of the components of the RNAi machinery have also been clearly
established as the effector proteins for the maturation of miRNAs
113 MicroRNAs in plants
1131 Discovery
MicroRNAs have been discovered by three basic approaches Direct cloning forward
genetic direct cloning and bioinformatic prediction followed by experimental
validation The most direct method of miRNA discovery is to isolate and clone small
RNAs from biological samples and several groups have used this approach to identify
plant miRNAs (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002 Xie et al 2003 Sunkar et al 2005 Williams et al 2013) The cloning methods
adapted were similar to those used to identify large numbers of animal miRNAs
(Lagos-Quintana et al 2001 Lau et al 2001 Aravin amp Tuschl 2005) which involve
isolating small RNAs followed by oligonucleotide adapter ligation reverse
transcription amplification and sequencing Some protocols incorporate methods to
enrich for Dicer cleavage products (ie molecules with 5prime-phosphates and 3prime-
hydroxyls) and to concatemerize the short cDNAs so that several may be identified in a
single sequencing read (Lau et al 2001 Llave et al 2002a Jagadeeswaran amp Sunkar
2013)The initial cloning experiments in Arabidopsis identified 19 miRNAs belonging
to 15 families (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002) although hundreds of other small RNAs such as siRNAs and RNA degradation
Chapter 1 Introduction and Review of Literature
29
fragments were also cloned through the direct cloning methods To select miRNAs
from the pool of small RNAs secondary structures of the RNA molecules were
examined in compliance with the acknowledged structural features of miRNAs
(Ambros et al 2003) The secondary structures were validated using several ad-hoc
software tools such as the Mfold or RNAfold programs (Reeder et al 2006) Finally
the existence of mature miRNAs was determined by RNA gel blot hybridization These
direct cloning methods were successful in isolating many miRNAs in Arabidopsis
(Reinhart et al 2002 Sunkar amp Zhu 2004) Rice (Sunkar et al 2005) Poplar (Lu et
al 2005) moss (Arazi et al 2005) and Tobacco (Billoud et al 2005)
Although miRNAs were first discovered through forward genetic screens in
worms (Lee et al 1993 Reinhart et al 2000)only few A thaliana miRNA gene
families have been discovered by this method (Chen 2008) in plants MiRNA
involvement in plant mutant phenotypes were not properly inferred till the cloning
experiments established that plant genomes contain numerous miRNAs (Park et al
2002 Reinhart et al 2002 Rhoades et al 2002) MiRNA loss-of-function allele has
been identified by forward genetic screens early extra petala1 is caused by a
transposon insertion ~160bp upstream of the predicted miR164c stem loop resulting in
flowers with extra petals (Baker et al 2005 Irish 2008) The fact that loss-of-function
miRNA mutants have been rarely discovered using forward genetics reflects small
target size for mutagenesis coupled with redundancy in nearly all evolutionarily
conserved plant miRNAs which are encoded by gene families (Table16) Family
members are likely to have overlapping functions buffering against loss at any single
miRNA locus Over expression screens can circumvent redundancy limitations At least
four plant miRNAs miR319 (also known as miR-JAW) miR172 (also known as EAT)
and miR166 were isolated in over expression screens for dominant mutants with
developmental abnormalities (Aukerman amp Sakai 2003 Palatnik et al 2003 Kim et
al 2005 Williams et al 2005b Schommer et al 2012) Mutations in miRNA target
sites which can prevent the entire family of miRNAs from repressing a target gene can
also circumvent redundant functions of miRNA family members
The dominant mutations in the HD-ZIP genes PHB PHV and REVOLUTA
(REV) in Arabidopsis and ROLLEDLEAF1 (RLD1) in Maize result in adaxialization of
leaves andor Vasculature (McConnell amp Barton 1998 McConnell et al 2001 Nelson
et al 2002 Zhong amp Ye 2004 Allen amp Millar 2012) and are all caused by mutations
Chapter 1 Introduction and Review of Literature
30
in miR166 complementary sites (Rhoades et al 2002 Emery et al 2003 Jones-
Rhoades amp Bartel 2004 Mallory et al 2004 Zhong amp Ye 2004)
In both plants and animals cloning was the initial means of large-scale
miRNA discovery (Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Reinhart et al 2002 Mendes et al 2012) However cloning is biased toward
RNAs that are expressed highly and broadly MiRNAs expressed at low levels or
only in specific cell types or in response to certain environmental stimuli were more
difficult to clone Sequence based biases in cloning procedures might also cause
certain miRNAs to be missed Because of these limitations bioinformatics approaches
to identify miRNAs have provided a useful complement to cloning
A straightforward use of bioinformatics has been carried out to find homologs
of known miRNAs both within the same genome and in the genomes of other species
(Pasquinelli et al 2000 Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Mendes et al 2009) A more difficult challenge is to identify miRNAs
unrelated to previously known miRNAs This was first accomplished for
vertebrate nematode and fly miRNAs using algorithms that search for conservation
of sequence and secondary structure (ie miRNA stem-loop precursors) between
species searching for patterns that are characteristic of miRNAs (Lai et al 2003 Lim
et al 2003 Lim et al 2005)
Most of the methods identified numerous miRNAs but are necessarily more
relaxed in terms of allowed structures Some approaches take advantage of the high
complementarity of plant miRNAs to target messages implementing the requirement
that the candidate has conserved complementarity to mRNAs (Jones-Rhoades amp Bartel
2004 Adai et al 2005) This additional filter has been useful for distinguishing
authentic plant miRNAs from false positives which later extended to mammalian
miRNA gene prediction (Xie et al 2005 Mendes et al 2012)
Another criteria used by most algorithms in the filters is evolutionary
conservation In plants mature miRNA sequences are conserved to a higher degree
than their precursor sequences For example Arabidopsis and rice diverged more than
130 million years ago (Friis et al 2004) but some miRNAs are highly conserved
in these plant species (Reinhart et al 2002) Furthermore various parameters for the
secondary structures such as free energy the number of paired residues within a
Chapter 1 Introduction and Review of Literature
31
miRNA or the number and size of bulges are also used to validate the predictions
(Jones-Rhoades amp Bartel 2004 Wang et al 2004 Adai et al 2005 Mendes et al
2012)
1132 Conserved microRNAs in Plants
Till date cloning genetics and bioinformatics screens have resulted in annotation of
approximately 184 potential Arabidopsis miRNAs (miRBase release 13) These
miRNAs seem to arise through gene duplication and subsequent diversification and
represent approximately 100 families (Maher et al 2006 Axtell 2013) Twenty one
families represented by 92 genes are clearly conserved in species beyond Arabidopsis
(Table 15) The presence of these miRNA families in other sequenced plant genomes
indicates that different plant species have an evolutionarily fluid set of miRNAs
(Willmann amp Poethig 2007) Recent studies on mosses one of the most ancient land
plants have shown that at least 14 miRNA families are conserved in eudicots and
mosses and therefore it appears that plant miRNAs share a common ancient origin
(Arazi et al 2005 Axtell amp Bartel 2005 Talmor-Neiman et al 2006 Zhang et al
2006 Axtell 2013) The number of members per family in a genome ranges from 1-32
with the exception of miR-430 which is represented by a cluster of ~80 loci in zebra
fish (Giraldez et al 2005)
In plants the number of members in each family correlates among examined
species certain families contain numerous members in all three species (eg miR156
miR166 miR169) whereas others consistently contain fewer genes (eg miR162
mir168 miR394) (Table 16) Although it is still unclear why certain miRNA are
encoded by more than one gene in plants (eg 12 genes encoding miR156) this
correlation might suggest functional significance of various miRNA family sizes Most
annotated plant miRNAs are conserved throughout flowering plants while a few
miRNAs are found only in a single sequenced genome and thus they could be
evolutionarily lsquoyoungrsquo miRNAs For example Arabidopsis has several miRNA
families that are not found in poplar or rice suggesting that miRNAs could be
generated continuously during evolution (Allen et al 2004a Rajagopalan et al
2006 Fahlgren et al 2007 Axtell 2012 de Alba et al 2013) Consistent with the
notion that non conserved miRNAs are of a more recent evolutionary origin these
miRNAs are mainly encoded by a single locus in the genome Within each family the
mature miRNA is always located in the same arm
Chapter 1 Introduction and Review of Literature
32
Table 16 MicroRNA families conserved in plants
miRNA
family
Arabidopsis Oryza Populus
miR156 12 12 11 miR159319 6 8 15 miR160 3 6 8 miR162 2 2 3 miR164 3 5 6 miR166 9 12 17 miR167 4 9 8 miR168 2 2 2 miR169 14 17 32 miR171 4 7 10 miR172 5 3 9 miR390 3 1 4 miR393 2 2 4 miR394 2 1 2 miR395 6 19 10 miR396 2 5 7 miR397 2 2 3 miR398 3 2 3 miR399 6 11 12 miR408 1 1 1 miR403 1 0 2 miR437 0 1 0 miR444 0 1 0 miR445 0 9 0 Total 92 127 169
All known miRNA families that are conserved between more than one plant species are listed
together with the number of genes identified in the sequenced genomes Rice miRNA families that have orthologs in maize but do not appear to have orthologs in the eudicots (Arabidopsis and Populus) are marked with an asterisk The following families contain miRNA genes annotated with
more than one number miR156 (miR156 and miR157) miR159319 (miR159 and miR319) miR166 (miR165 and miR166) miR171 (miR170 and miR171) and miR390 (miR390 and miR391)
of the stem loop (5prime- or 3prime) as it would be expected if the genes carry common ancestry
(Figure 17) The sequence of the mature miRNA and to some extent the segment on
the opposite arm of the hairpin to which it pairs is highly conserved between the
members of the same family (both within and between the species) while the sequence
secondary structure and length of the intervening ldquolooprdquo region can be highly divergent
within the same family
The patterns of paring and non pairing nucleotides are often conserved between
homologous miRNA stem loops from different species (Figure 17) The significance of
conserved mismatches is opined to guide DCL1 to cleave at the appropriate positions
along the stem loop
Chapter 1 Introduction and Review of Literature
33
Figure 17 Representative stem loop of miR164 Segments corresponding to the mature miRNAs
are shown in red (Adapted from Rajagopalan et al 2006)
Although many miRNAs are highly conserved in plants the degree of conservation in
most cases is quite low between plants and animals one exception is miR854 A recent
study has revealed that miR854 is conserved between Arabidopsis and animals and that
the Arabidopsis miR854 differs from the human miR854 only by a single nucleotide
(Arteaga-Vazquez et al 2006 Axtell 2012 de Alba et al 2013) The Arabidopsis
miR854 has multiple binding sites in the 3prime-untranslated region (UTR) of
OLIGOURIDYLATE-binding-PROTEIN mRNA 1bA (UBP1b) and represses the
translation of the UBP1b mRNA The animal miR854 is also complementary to a site in
the 3prime-UTR of the UBP1b homolog in animals However it is currently unclear how the
animal miR854 regulates its target mRNA Another distinction between plant and
animal miRNAs is the genomic organization of the miRNA genes While plant miRNA
genes are located predominantly in the intergenic regions animal miRNA genes tend to
localize in the intragenic regions both in the introns or exons of protein coding genes
Moreover miRNA genes are often clustered within a single precursor transcript
whereas individual plant miRNAs are derived from individual precursor RNAs (Bartel
Chapter 1 Introduction and Review of Literature
34
2004 Bonnet et al 2004 Kim 2005 Vaucheret et al 2006 Marco et al 2013)
1133 Non-conserved miRNA and challenges in annotation
Most miRNAs are conserved throughout flowering plants (Table 15) but many more
were found only in a single sequenced genome thus considered to be of a more recent
evolutionary origin The extended homology between few non conserved miRNAs
precursors and their target genes provides strong evidence that these relatively ldquoYoungrdquo
miRNAs arose from the duplication of the target gene segments (Allen et al 2004)
Several non-conserved miRNAs including miR161 miR163 miR173 miR447
miR475 and miR476 are known to direct cleavage of target transcripts (Allen et al
2004a Adai et al 2005 Sunkar et al 2005 de Alba et al 2013) It is difficult to
confidently predict targets for many because it is not possible to use complementary
site as a filter against false-positive target predictions It is also difficult to
confidently annotate non-conserved miRNAs as miRNA rather than siRNAs The
established minimal standard for miRNA annotation is a small RNA with detectable
expression and the potential to form a stem-loop when joined to flanking genomic
sequence (Kasschau et al 2003) In practice these requirements are too loose to
definitively categorize many small RNAs cloned from plants Many plant siRNAs are
detectable on blots (Vazquez et al 2004b) and hundreds of thousands of non-
miRNA genomic sequences can be predicted to fold into secondary structures that
resemble structures of plant miRNA precursors (Jones-Rhoades amp Bartel 2004)
Therefore without conservation of both sequence and secondary structure it is
difficult to be confident that a given cloned RNA originated from a stem-loop (ie is a
miRNA) rather than from a double-stranded RNA (ie is a siRNA) In fact many of
the thousands o f small RNAs cloned from Arabidopsis (Gustafson et al 2005) would
probably meet the literal requirements for annotation as miRNAs A few of these
sequences might be miRNAs but others that meet the literal criteria probably are not
so The challenges of annotating non conserved small RNAs were evidenced by
three related small silencing RNAs that were originally annotated as miRNAs
that turned out to be trans-acting siRNAs (tasiRNAs)(Kanno et al2013)
Although most miRNAs that are broadly conserved among flowering plants are
now being identified and experimentally validated (Table 16)(Jones-Rhoades amp
Bartel 2004) the challenges and ambiguities for classifying non-conserved miRNAs
prevent meaningful estimates of the total number of miRNA genes in Arabidopsis and
Chapter 1 Introduction and Review of Literature
35
other plant genomes It is possible that miRNAs might have escaped detection
because they are non-conserved and expressed in specific tissues or conditions and can
only be identified by using high- throughput sequencing
1134 MicroRNA biogenesis
11341 Transcription of microRNA precursor
Plant miRNAs are primarily found in the genomic regions that are not associated with
the protein coding genes (Reinhart et al 2002) and it appears that most if not all plant
miRNAs are produced from their own transcriptional units Plant miRNA genes are
occasionally clustered near each other in the genome suggesting transcription of
multiple miRNAs from a single primary transcript [eg the miR395 cluster (Grishok
et al 2001 Jones-Rhoades amp Bartel 2004b)] but this polycistronic arrangement of
miRNA genes appears far less frequently in plants than in animals (Bartel 2004)
Northern blot EST and mapping evidence indicate that plant miRNA primary
transcripts (also known as pri-miRNAs) as in animals are longer than needed to
encompass the miRNA stem-loops (Palatnik et al 2003 Jones-Rhoades amp Bartel
2004b) At least some of these pri-miRNA transcripts appear to be spliced
polyadenylated (Kurihara amp Watanabe 2004) and capped (Xie Z et al 2005) Two
rice miRNAs are contained within transcripts that contain exon junctions within the
presumptive stem-loop precursor implying that in these cases splicing is a prerequisite
for Dicer recognition (Sunkar et al 2005) Plant pri-miRNAs are over 1kb in length
and they are usually preceded by typical TATA box motifs They can also undergo
canonical splicing polyadenylation and capping All these observations indicates RNA
polymerase II is probably responsible for transcribing most plant miRNAs (Xie Z et
al 2005) as appears to be the case for many animal miRNAs Relatively little is
known about the regulation of miRNA transcription in plants but there is no reason
to suspect that this regulation would differ from that of protein-coding transcripts as
the bioinformatics analysis of the sequence upstream of the transcriptional start
sites of the miRNA genes showed the presence of putative binding motifs of
number of known transcription factors (Megraw et al 2006 Sethupathy 2013)
11342 MicroRNA processing and export
In plants maturation of miRNA is a step wise process A miRNA gene is transcribed to
pri-miRNA which is much longer than the pre-miRNA possessing the characteristic
stem-loop structure Like transcription of most protein-coding genes the miRNA
Chapter 1 Introduction and Review of Literature
36
transcription is processed by RNA polymerase II (Pol II) enzymes and the pri-
miRNAs are 5prime-capped and 3prime-polyadenylated (Lee et al 2004 Xie Z et al 2005)
Mature miRNAs are produced from pri-miRNAs through at least two sequential
processing steps by RNase III-type endonucleases In animals pri-miRNAs are first
processed at the bottom portion of the stem by Drosha a nuclear-localized RNase III
enzyme to liberate pre-miRNAs The pre-miRNAs are then exported to the cytoplasm
with the help of exportin-5The pre-miRNAs are further processed by Dicer another
RNaseIII enzyme to a duplex consisting of the miRNA and the antisense strands
(miRNA) Since plants do not have any Drosha homologs the two sequential
processing steps seem to be carried out by DCL1 a Dicer homolog in the nucleus
(Kurihara amp Watanabe 2004 de Alba et al 2013 Rogers amp Chen 2013)
The processing of pri-miRNAs to pre-miRNAs by DCL1 is assisted by two
other proteins HYPONASTY LEAVES1 (HYL1) and SERRATE (SE) HYL1 belongs to a
family of double-stranded RNA (dsRNA) binding protein and interacts with DCL1 in
vitro and in vivo (Lu amp Fedoroff 2000 Hiraguri et al 2005 de Alba et al 2013
Rogers amp Chen 2013) Loss-of-function mutations in the HYL1 gene result in
decreased accumulation of miRNAs and concomitant accumulation of pri-miRNAs
(Han et al 2004 Vazquez et al 2004a Kurihara et al 2006) SE a C2H2 zinc finger
protein interacts with DCL1 and HYL1 and plays a role in the processing of pri-
miRNAs to pre-miRNAs (Lobbes et al 2006 Yang L et al 2006) In addition the
nuclear cap-binding complex proteins CBP20 and CBP80 that are known as ABA
HYPER SENSITIVE 1(ABH1) have been shown to be required for proper miRNA
processing (Gregory et al 2008 Laubinger et al 2008) Recently it has been
demonstrated that DCL1 can release 21-nt RNAs from dsRNA as well as from
synthetic miR167 precursor RNAs in vitro However this cleavage is error prone and
inefficient in the absence of HYL1 and SE which accelerate the rate of DCL1-mediated
cleavage and promote accurate processing of miRNAs (Dong et al 2008 Rogers amp
Chen 2013)
11343 Methylation of the miRNAmiRNAduplex
After the miRNAmiRNAduplexes are released from the pre-miRNAs by the DCL1
activities the duplexes are methylated at the 2primeOH of the3prime-ends by HUA ENHANCER1
(HEN1) a small RNA methyltransferase (Yu et al 2005 Yang Z et al 2006 Rogers
amp Chen 2013 ) This step which is distinct from the RNase III-mediated processing
Chapter 1 Introduction and Review of Literature
37
step distinguishes the biogenesis of plant miRNAs from that of animal miRNAs
Mutations in the HEN1 gene were first isolated in a morphological screening for floral
development although the HEN1 mutants display pleiotropic phenotypes similar to the
developmental defects observed in the DCL1 mutants (Chen et al 2002 Park et al
2002 Rogers amp Chen 2013)
Figure 18 Biogenesis of microRNA
A Biogenesis of plant MicroRNA B Biogenesis of metazoananimal MicroRNA
The miRNA (red) is incorporated into RISC and the miRNA(blue) in degraded
This enzyme has two dsRNA-binding domains and nuclear localization signal It
is also conserved in fungi and is required for the processing of siRNAs as well as of
miRNAs (Park et al 2002 Boutet et al 2003 Xie et al 2003) Although the HEN1
protein is localized to the nucleus its methylation activity in the cytoplasm cannot be
ignored (Xie et al 2004 Rogers amp Chen 2013)
Chapter 1 Introduction and Review of Literature
38
The methylation of the miRNAmiRNA duplex is required to protect miRNAs
against the 3prime-end uridylation activity and subsequent degradation (Li et al 2005) In
the hen1 mutants accumulation of miRNAs is reduced to a lower level Also the
miRNAs appear to be heterogeneous in their sizes such that a ladder of bands rather
than a single species is observed for most miRNAs in the mutants (Li et al 2005)
MiRNA cloning and sequencing analyses revealed the presence of a number of U
residues at the 3prime-end of miRNAs in the hen1 mutants Moreover these nucleotides do
not correspond to the miRNAs in the pre-miRNAs indicating that these nucleotides are
added after the processing of the miRNAs from pre-miRNAs (Li et al 2005)
Uridylation of RNAs has also been proven to be correlated with RNA decay (Shen amp
Goodman 2004 Mullen amp Marzluff 2008) suggesting that uridylation attracts an
exonuclease which degrades miRNAs in plants
11344 MiRNA incorporation in to the RISC and its nuclear export
The methylated miRNAmiRNA duplexes undergo RISC assembly In this process
the miRNA of the duplexes is selectively incorporated into the RISC and the miRNA
appears to be removed and subsequently degraded (Hammond et al 2000 Hutvagner
amp Zamore 2002 Schwarz et al 2003 de Alba et al 2013 Rogers amp Chen 2013)
The ARGONAUTE 1(AGO1) proteins are core components of the RISC complex
They contain two structural domains the N-terminal PAZ domain that binds to the 3prime-
end of single-stranded RNAs and the C-terminal piwi domains that has a structure
similar to that of RNase H (Cerutti et al 2000 Parker et al 2004) Several AGO
proteins possess endonucleolytic activities within the PIWI domain and cleave target
mRNAs in the middle of the site complementary to miRNA (Liu et al 2004 Meister et
al 2004 Miyoshi et al 2005) Mutations in the AGO1 gene cause miRNA target
genes to be ectopically expressed and the levels of some miRNAs are reduced in the
mutants (Kidner amp Martienssen 2004 Vaucheret et al 2004 Kidner amp Martienssen
2005 Ronemus et al 2006) Moreover the phenotype of the AGO1 hypomorphic
alleles display defects in lateral organ polarity and leaf and flower morphology as
observed in the dcl1 mutants and null AGO1 alleles are embryonically lethal
supporting the close relationship between the AGO proteins and miRNA processing
(Kidner amp Martienssen 2004 Vaucheret et al 2004 de Alba et al 2013 Rogers amp
Chen 2013)
The production of miRNA miRNA duplexes occur in the nucleus while
Chapter 1 Introduction and Review of Literature
39
miRNA-RISC complexes are present in the cytoplasm where target mRNAs are
located This discordance of functional sites requires an export step during miRNA
biogenesis HASTY the Arabidopsis homolog of exportin-5 is thought to be involved
in miRNA nuclear export (Bollman et al 2003 Park et al 2005)The hasty mutant
exhibits pleiotropic defects in plant development and reduced accumulation of most
miRNAs as in the DCL1 or AGO1 mutants Howeverit is unclear whether the HASTY
proteins export the miRNA miRNA duplexes or the miRNA-RISC complexes in
plants
11345 Feedback regulation of miRNA biogenesis
MiRNA biogenesis is under feedback regulation such that the DCL1 and AGO1 genes
are themselves regulated by miRNAs DCL1 is itself a target of miR162 which leads to
the cleavage of the DCL1 mRNA (Xie et al 2003 de Alba et al 2013) Consistent
with this the levels of DCL1 mRNA are elevated in the mutants of miRNA processing
genes in which the miR162 abundance is reduced In addition there is a predicted
hairpin structure of miR838 in the 14th
intron of the DCL1 primary transcript
(Rajagopalan et al 2006) This prediction implies that DCL1-mediated processing
on this site can result in the cleavage of the DCL1 primary transcript into two truncated
fragments the N-terminal capped fragment and the C-terminal polyadenylated
fragment If this is the case it is possible that miRNA biogenesis competes with the
splicing event of the DCL1 pre-mRNA
The AGO1 is also regulated by miRNA There is a complementary site for
miR168 binding in the AGO1 mRNA and the transcript level of the AGO1 mRNA is
reduced through an mRNA cleavage mechanism (Vaucheret et al 2006 de Alba et al
2013 Rogers amp Chen 2013) indicating that the AGO1 mRNA levels are tightly
controlled by the levels of the AGO1 protein
1135 Mechanism of miRNA action
11351 MiRNA-directed mRNA cleavage
By forming the miRNA-RISC assembly miRNAs can direct the RISC to regulate gene
expression by two post transcriptional mechanisms mRNA cleavage or translational
repression (Bartel 2004 Jones-Rhoades et al 2006 Dalmay 2013) The degree
of miRNA-mRNA complementarity plays an important role for the
determination of the mechanism to be used A general rule is that while perfect or
Chapter 1 Introduction and Review of Literature
40
near-perfect complementarity induces mRNA cleavage central mismatches trigger
translational repression (Carrington amp Ambros 2003 Jones-Rhoades amp Bartel 2004)
Unlike most animal miRNAs plant miRNAs have near-perfect or perfect
complementarity to their targets and mRNA cleavage is assumed to be a predominant
mechanism by which miRNAs regulate gene expression in plants
In RNA cleavage mechanism miRNAs guide the AGO component of RISC to
cleave a single phosphodiester bond of the target mRNA within the miRNA-binding
site (Llave et al 2002b Tang et al 2003) The truncated fragments are then released
and degraded freeing the RISC to recognize and cleave another target mRNA This has
been validated by the detection of 3prime cleavage products that have 5prime ends that start at the
middle of the complementary regions The mRNA cleavage products are then further
degraded by other mechanisms The 3prime cleavage products of some miRNA targets are
degraded by the 5prime-3prime exonuclease XRN4 an Arabidopsis homolog of the major Yeast
mRNA degrading exonuclease Xrn1p It has been observed that the 3prime cleavage
products of some target mRNAs were accumulated in the xrn4 mutants (Souret et al
2004 Dalmay 2013) However the 3prime cleavage products of other miRNA targets do
not accumulate in the xrn4 mutants indicating that there are also XRN4 independent
mechanisms The 5prime cleavage products are typically untraceable by RNA gel blot
hybridization but can be detected by sensitive PCR based methods It has been found
that the 5prime cleavage products tend to obtain an oligo U tail at the 3prime ends This 3prime U tail
is correlated with shortening of the 5prime cleavage products from the 5prime end leading to the
conclusion that the 3prime uridylation causes 5 prime- 3 prime exonucleolytic decay of the 5prime cleavage
products (Shen amp Goodman 2004)
DCL1 HYL1 and SE interact with each other and are co-localized in the
nuclear bodies termed Dicing bodies or D-bodies in which some pri-miRNA shave
been found suggesting that D-bodies serve as a center for active miRNA processing
(Fang amp Spector 2007 Fujioka et al 2007 Song et al 2007) The miRNA
processing site appears to be distinct from the Cajal bodies that have previously been
found as a siRNA processing site (Pontes amp Pikaard 2008) The D-bodies include
SmD3 and SmB proteins that are found in the Cajal bodies However At collin an
additional marker for the Cajal bodies is not found in the D-bodies Moreover the D-
bodies are present in an At collin mutant lacking the Cajal bodies The pri-miRNAs of
miR171A and miR159A genes have been found to be co-localized with HYL1 and
Chapter 1 Introduction and Review of Literature
41
DCL1 in the D- bodies that are distinct from the Cajal bodies (Song et al 2007)
11352 MiRNA-directed translational repression
Translation repression is another mechanism exerted by plant miRNAs The regulatory
mechanism of miR172 which regulates flowering time and floral organ identity is
consistent with translation repression MiR172 over production does not affect the
transcript levels of APETALA2 (AP2) and AP2- like target mRNAs Instead the AP2
protein level is reduced in the miR172 over expressing mutant (Aukerman amp Sakai
2003 Chen 2004 Dalmay 2013) It was later shown that miR172 can also direct the
cleavage of the target mRNAs although the steady-state mRNA level is unchanged due
to feedback regulation (Schwab et al 2005 Jung et al 2007) It is clear that miR172
regulates its target genes primarily by translational repression rather than mRNA
cleavage Recently it has been reported that miR156157 and miR854 also regulate
their target genes through translational repression (Arteaga-Vazquez et al 2006
Gandikota et al 2007 Dalmay 2013) It was once thought that translational repression
is an exception to the rule However experimental evidence suggest a widespread
coexistence of translational repression and mRNA cleavage (Brodersen et al 2008)
1136 Regulatory roles of miRNAs in plant development
The miRNA target prediction has been a difficult task in order to obtain regulatory
targets without bringing in too many false predictions Plant growth and developmental
processes regulated by miRNAs are quite diverse and enormous Furthermore plant
miRNAs work cooperatively or antagonistically to establish a balanced regulation This
notion is well consistent with the findings that mutations in the regulators of miRNA
biogenesis transport and processing such as AGO1 DCL1 HYL1 HEN1 ABH1
and HASTY cause pleiotropic phenotypes (Mallory amp Vaucheret 2006 de Alba et al
2013) To date the roles of miRNAs have been mostly deduced from obtaining miRNA
over expressing transgenic plants or gain-of-function mutants in which miRNA-
resistant target genes are ectopically expressed
11361 Phase transition
Plants pass through a series of developmental phase transitions during their life span
beginning with seed germination continuing through vegetative phase change
reproductive phase change flowering initiation and finally seed production for the next
generation Because of their importance in understanding plant developmental
Chapter 1 Introduction and Review of Literature
42
processes genes regulating developmental transitions have been extensively studied
and underlying molecular mechanisms have been explored in various plant species
Majority of researches were mainly focused on vegetative phase change and
reproductive transition in Arabidopsis and Maize (Poethig 2003 Baurle amp Dean
2006) Particularly the mutations of the genes encoding various regulators of miRNA
biogenesis and transport such as HST SE and AGO exhibit altered juvenile to adult
vegetative phase change and vegetative to reproductive change (Hunter et al 2003
Peragine et al 2004 Vazquez et al 2004a Jones-Rhoades et al 2006 de Alba et al
2013)
Over-expression of the miR156a gene causes late flowering and delays
vegetative phase change (Wu amp Poethig 2006 Gandikota et al 2007 Wang et al
2008) It has been shown that miR156 negatively regulates a group of genes encoding
SQUAMOSA PROMOTER-BINDING PROTEIN-LIKE (Lewis et al 2007 de Alba et
al 2013 Rogers amp Chen 2013) transcription factors such as SPL3 SPL4 and SPL5
that promote flowering initiation In contrast transgenic plants over expressing
miR156-resistant SPL345 genes are early flowering Interestingly the endogenous
level of miR156 is temporally regulated In the early juvenile vegetative phase the
level of miR156 is very high but decreases rapidly before the onset of the adult juvenile
phase (Wu amp Poethig 2006)
The Arabidopsis early activation tagged (eat-D) mutant exhibits early flowering
with disrupted floral structure The mutant phenotypes are caused by over expression of
the miR172a-2gene (Aukerman amp Sakai 2003) MiR172 regulates a small group of
genes encoding APETALLA2 (AP2)-like transcription factors such as TARGET OF
EAT1 (TOE1) TOE2 SCHLAFMUTZE (SMZ) and SCHNARCHZAPFEN(SNZ)
TOE1 TOE2 SMZ and SNZ have been shown to participate in their productive phase
change by repressing a floral integrator FLOWERING LOCUS T (FT)(Jung et al
2007) Consistent with this toe1-2D 35STOE2 35SSMZ and 35SSNZ plants
exhibit late flowering (Jung et al 2007) MiR172 is also regulated temporally and
highly expressed in the adult vegetative phase both in Arabidopsis and maize (Chuck et
al 2007) Because the temporal expression patterns of miR156 and miR172 are
complementary to each other it has been suggested that the two miRNAs interact with
each other in regulating phase change (Chuck et al 2009 de Alba et al 2013) In
support of this view it has been shown that miR172 is down-regulated in the
Chapter 1 Introduction and Review of Literature
43
Corngrass1(Cg1) mutant in which miR156 is overproduced (Chuck et al 2007 de
Alba et al 2013) However there is no direct evidence for the direct linkage between
miR156 and miR172 It is also envisioned that miR156 and miR172 would not be
directly related For example miR156 regulates plastochron index that is the inverse of
leaf initiation rate and thus frequently used to analyze temporal patterns of plant
development In contrast miR172 does not affect plastochron (Wang et al 2008) The
down-regulation of miR172 in the maize miR156-over producing mutants would be
due to the prolonged juvenile vegetative phase in the mutants
Another miRNA that regulates flowering initiation is miR159 It regulates
MYB33 and MYB65 which are closely related to the barley GAMY B transcription
factor that responds to gibberellic acid (GA) The level of miR159 is elevated in GA-
treated seedlings but reduced in the GA biosynthetic ga1 mutant Transgenic plants
over producing miR159 exhibit delayed floral induction under short days (Achard et
al 2004) It is been suggested that MYB33 mediates GA signal to regulate LEAFY
(LFY) These observations indicate that miR159 plays an important role in the GA
signaling pathways that regulate proper floral induction under short days (Achard et al
2004)
11362 Floral development
Arabidopsis flowers consist of four distinct organs They arise in a characteristic
pattern within concentric rings called whorls from outside to inside four sepals four
petals six stamens and the central pistil consisting of two carpels Identities of the
floral organs are determined by three classes of regulatory genes classA classB and
class C genes (Krizek amp Fletcher 2005) The A and C class genes act antagonistically
to restrict their expression domains (Bowman et al 1991)
It is notable that miR172 also regulates floral architecture in addition to its role
in the timing of flowering initiation MiR172 inhibits the translation of the AP2 mRNA
(Chen 2004) Transgenic plants overproducing miR172 not only exhibit early
flowering but also show abnormal floral development which is quite similar to those
of the class A gene mutants such as the ap2 mutants (Aukerman amp Sakai 2003 de
Alba et al 2013) When the activity of the class A genes is inactivated that of the class
C genes such as AGAMOUS (AG) is elevated As a result the miR172- overproducing
transgenic plants exhibit no petals along with homeotic transformation of the sepals to
Chapter 1 Introduction and Review of Literature
44
the carpels (Aukerman amp Sakai 2003 Chen 2004)
Interestingly miR166 also regulates floral architecture The role of miR165166
in the regulation of meristem activity is closely linked to floral meristem formation and
organization (Zhao et al 2007 Miyashima et al 2013) Floral structure is severely
disrupted in the miR165166-overproducing mutants such as men1 and jba-1D in
which miR166 is overproduced (Williams et al 2005b Jung amp Park 2007) The men1
and jba-1D mutants have smaller gynoecium and reduced carpel number In an extreme
case the jba-1D homozygous plants lack visible carpels (Williams et al 2005b)
Similar phenotypes have been observed in the miR165 overproducers (Zhao et al
2007) It has been suggested that the phenotypes are possibly caused by altered AG
expression in the floral meristem (Jung amp Park 2007 Turchi et al 2013)
Other miRNAs also affect floral development Overproduction of miRNAs
involved in auxin signaling also affects floral architecture Transgenic plants
overproducing miR167 have defects in the formation of ovule and anther with reduced
fertility (Ru et al 2006) It has been shown that miR167 targets ARF6 and ARF8 that
play a role in gynoecium and stamen development and in regulating fertility (Ru et al
2006 Wu et al 2006) These observations suggest that auxin signals are also closely
related to floral organogenesis In addition transgenic plants over-producing miR164
and the cuc1cuc2 double mutants show fused sepals and reduced petals (Laufs et al
2004 Turchi et al 2013) suggesting that miR164 is also related to floral meristem
activity and boundary specification of the meristematic region
11363 Shoot apical meristem development
Shoot apical meristem comprises self-maintaining totipotent cells The shoot apical
meristem activity affects diverse aspects of plant developmental processes including
leaf development vascular development and lateral organ polarity (Bowman et al
2002) Because miR165166 plays a primary role in meristem formation
overproduction of miR165166 and multiple knockout mutants of its target genes
exhibit pleiotropic pheonotypes (McConnell et al 2001 Emery et al 2003 Kim et al
2005 Williams et al 2005b)
The targets of miR165166 include genes encoding the homeo domain-leucine
zipper (HD-ZIP) class III transcription factors such as PHABULOSA(PHB)
PHAVOLUTA (PHV) REVOLUTA(REV) ATHB15CORONA (CNA) and ATHB8
Chapter 1 Introduction and Review of Literature
45
PHB PHV and ATHB15 have overlapping functions in controlling meristem
development (Turchi et al 2013) Indeed the phb phv athb15 triple mutants have an
enlarged shoot apical meristem Consistent with this the miR166- overproducing men1
and jba-1D mutants exhibit enlarged shoot apical meristems (Kim et al 2005
Williams et al 2005b Turchi et al 2013) In the mutants the shoot apical meristems
are multiplied and large
The development of shoot apical meristems is also regulated by the WUSCHEL
(WUS)ndashCLAVATA (CLV) signaling pathway WUS positively regulates cell
proliferation In contrast CLV3 which is expressed in stem cells limits the WUS
expression and thus inhibits cell proliferation in the SAM (Sharma et al 2003)
Consistent with this notion WUS is expressed to a higher level in jba-1D
explaining the ectopic enlargement of the SAM in the mutant (Williams et al 2005)
While WUS is closely related to miR165166 in the shoot apical meristem
development the underlying molecular mechanisms are currently unclear It has been
observed that the miR166-over producing men1 plants have an arrested meristem
development (Jung amp Park 2007) In addition the heterozygous men1+ and the
homozygous men1 plants show different expression patterns of WUS and CLV3 It
appears that miR166 regulates WUS indirectly and influences the expressional balance
between WUS and CLV3 through a negative feedback loop (Jung amp Park 2007 de
Alba et al 2013)
The phv phb athb15 triple mutant has an enlarged shoot apical meristems the
rev phb phv mutant does not have functional shoot apical meristems (Prigge et al
2005) This distinction may be explained by the fact that while miR166 targets
primarily PHB PHV and ATHB15 miR165 targets all the five HD-ZIP III genes (Zhou
et al 2007) Consistent with this transgenic plants overproducing miR166 are distinct
from those overproducing miR165 The miR165-overproducing transgenic plants lack
functional shoot apical meristem and a pin-like structure is formed at the apical region
of the mutant These phenotypes are quite similar to rev phb phv triple mutant (Zhou et
al 2007 Liu et al 2013) The phenotypic distinction may because by the difference
of the cleavage efficiencies of the target mRNAs In accordance with this notion a few
35S miR166a transgenic lines exhibit no shoot apical meristems The men1
homozygous plants also show arrested meristem development (Jung amp Park 2007)
Chapter 1 Introduction and Review of Literature
46
MiR164 cleaves the CUC1 and CUC2 mRNAs as well as the NAC1 mRNA
CUC1 and CUC2 determines the organ boundary in the meristem Phenotypes of
transgenic plants that overproduce miR164 are similar to those of the cuc1cuc2 double
mutant that exhibits embryonic developmental defects such as cup-shaped cotyledons
and fused cotyledons (Laufs et al 2004) When a miR164-resistant CUC2 is
expressed the boundary domain is enlarged CUC also plays a role in embryonic shoot
meristem formation by regulating the SHOOT MERISTEMLESS gene It was therefore
concluded that miR164-mediated cleavage of CUC1 and CUC2 mRNAs determines the
sizes of the boundary domains in the shoot apical meristem and regulates separation of
the organs by restricting cell proliferation (Laufs et al 2004 de Alba et al 2013)
11364 Leaf development
The role of miR319 has been extensively studied in leaf development A dominant
jaw-D mutant overproducing miR319 shows uneven curved leaves with enlarged size
and five TCP genes which are closely related to the CINCINNATA (CIN) gene in
Antirrhinum majus are down regulated in the mutant suggesting that miR319 mediated
regulation of the TCP transcription factor genes are important for the final step of
leaf shaping (Palatnik et al 2003 Naz et al 2013) In addition to leaf shape and size
leaf polarity is also specified by miRNA MiR166-mediated cleavage of the HD-ZIPIII
mRNAs is a well to establish leaf polarity This discovery originates from the gain-of-
function mutants such as phb-1d and phv-1d wherein point mutations in the
complementary sites to miRNA helps the mRNA to evade or resist from miRNA-
mediated cleavage (McConnell et al 2001 Emery et al 2003 Naz et al 2013)
Plant leaves have a dorso-ventral polarity consisting of the adaxial (upper
surface) and abaxial (lower surface) sides The adaxial side is closer to the shoot
apical meristem Adaxial identity is specified by functionally redundant class III HD-
ZIP family genes such as PHB PHV and REV Ectopic expression of each genes
result in adaxialized leaves Dominant phb-1d and phv-1d mutants exhibit
transformation of the abaxial to adaxial fate and have rod-shaped leaves In contrast
miR165166-overproducing plants show pin-like cotyledons radicalized leaves and
abaxialized leaves (Chitwood et al 2007 Pattanayak et al 2013)
Leaf asymmetry is determined by miR166-mediated restriction of the
expression domains of the HD-ZIPIII genes The HD-ZIPIII genes are expressed in the
Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
8
12 Cola nitida Caffeine Seeds
13 Citrus sp Caffeine Pollen
14 Ilex paraguariensis Mate Caffeine Leaves
Adapted from (Ashihara amp Crozier 1999 Ashihara amp Crozier 2001)
18 Caffeine biosynthesis
The major biosynthetic pathway is a four step sequence consisting of three sequential
methylation and one nucleosidase reactions (Figure 12) The xanthine skeleton of
caffeine is derived from purine nucleotides The initial step in caffeine biosynthesis is
the methylation of xanthosine by a SAM- dependent N-methyltransferase In addition
to experiments with radiolabeled precursors substrate specificities of native (Kato et
al 1999) and recombinant N-methyltransferases (Kato et al 2000 Ogawa et al
2001 Mizuno et al 2003a Mizuno et al 2003b Uefuji et al 2003) strongly
suggest that the major route to caffeine is a xanthosine rarr7-methylxanthosinrarr 7-
methylxanthine rarr theobromine rarr caffeine pathway (Figure 12) Although the
information has been obtained mainly from coffee (C arabica) and tea (Camellia
sinensis) the available evidence indicates that the pathway is essentially the same in
other purine alkaloid-forming plants such as mateacute (Ashihara 1993) and cacao
(Koyama et al 2003 Yoneyama et al 2006)
The first step in the caffeine biosynthetic pathway from xanthosine is
conversion of xanthosine to 7-methylxanthosine (Figure 12) This reaction is
catalyzed by the 7-methylxanthosine synthase (xanthosine 7 N-methyltransferase EC
211158) The genes encoding for 7-methylxanthosine synthase CmXRS1 (AB
034699) and CaXMT1 (AB 048793) were isolated from the C arabica (Mizuno et
al 2003a Uefuji et al 2003) The recombinant proteins obtained from these genes
exhibit 7-methylxanthosine synthase activity in vitro Xanthosine monophosphate
(XMP) was not converted to 7-methylxanthosine by the recombinant enzymes thus
inclusion of 7-methy-XMP was proposed (Schulthess et al 1996)
The second step of caffeine biosynthesis involves a nucleosidase which
catalyses the hydrolysis of 7-methylxanthosine Although N-methylnucleosidase (EC
32225) was partially purified from tea leaves (Negishi et al 1988) recent detailed
structural studies on coffee 7-methylxanthosine synthase suggested that the methyl
transfer and nucleoside cleavage may be coupled and catalyzed by a single enzyme
(McCarthy amp McCarthy 2007)
Chapter 1 Introduction and Review of Literature
9
The last two steps of the caffeine synthesis are also catalyzed by a SAM-
dependent N-methyltransferase(s) which is different from the N-methyltransferase
enzyme that catalyzes the first step in the pathway Native N-methyltransferase
activities have been detected in crude and partially purified extracts from tea and
coffee plants (Ashihara amp Suzuki 2004) and a highly purified preparation has been
obtained from young tea leaves (Kato et al 1999) The enzyme was assigned the
name caffeine synthase (EC 211160) that catalyses the last two steps of caffeine
biosynthesis viz the conversion of 7-methylxanthine to caffeine via theobromine
(Figure 12) The gene encoding caffeine synthase was first cloned from young tea
leaves (Kato et al 2000) Since then several genes encoding N-methyltransferases
with different substrate specificity have been reported
Activity of the recombinant theobromine synthase (CTS1 and CaMXMT EC
211159) is specific for the conversion of 7-methylxanthine to theobromine In
contrast the recombinant caffeine synthases (CCS1 and CaDMXMT1) can utilize
paraxanthine theobromine and 7-methylxanthine as shown with tea caffeine synthase
(TCS1) (Mohanpuria et al 2011) Although paraxanthine is the most active substrate
of this recombinant enzyme only limited amounts of paraxanthine are synthesized in
coffee cells hence in vivo caffeine synthase is principally involved in the conversion
of 7-methylxanthine to caffeine via theobromine
Radiolabelled tracer experiments with theobromine accumulating cacao and
Chinese tea (Camellia ptilophylla) showed a limited conversion of theobromine to
caffeine N-methyltransferase which catalyzed the conversion of 7-methylxanthine to
theobromine was detected in crude extracts from C ptilophylla but conversion of
theobromine to caffeine was not observed (Ashihara et al 1998) Genes homologous
to caffeine synthase have been isolated from several theobromine accumulating plants
including cacao (Yoneyama et al 2006) The recombinant enzymes derived from
these genes have 3 N-methyltransferase activity suggesting that these theobromine
accumulating plants contain specific theobromine synthase Caffeine does not occur
in C ptilophylla (Ashihara et al 1998) although it is present in leaves and fruits of
cacao along with theobromine (Koyama et al 2003 Zheng amp Ashihara 2004)
Chapter 1 Introduction and Review of Literature
10
Figure 12 Pathways for the biosynthesis of caffeine
The major pathway is indicated by solid lines (Mizuno et al 2003b Uefuji et al 2003) Alternative pathways in coffee and tea plants are indicated
by dashed lines(Kato et al 1996 Schulthess et al 1996) Enzymes (1) 7-methylxanthosine synthase (2) N-methylnucleosidase (3) Theobromine
synthase (4) Caffeine synthase
N
N
N
N7
H3C
3
1
O
O
Caffeine
N
NH
N
N7
3
O
O
Theobromine
N
N
N
N
H
H
7
O
O
N
N
N
N
H
H
O
O
N
N
N
N
H
H
O
O
Xanthosine
N
N
N
N
H
H
O
O
N
N
N
N
H
H
O
ON
N
N
N
H
H3C
71
O
O
Paraxanthine
CH3
CH3
CH3
CH3CH3
7-methylxanthine
Rib P
Rib
(1)
(4)
Rib
+
CH3
7-methyl
xanthosine
(2)
(3)
SAM SAH
SAM SAH SAM SAH
Xanthosine mono
phosphate
Rib P
7-methyl xanthosine
monophosphate
+
CH3 CH3
Inosine 5-mono
phosphate
Adenosine 5-mono
phosphate
SAM SAH
Guanosine
Guanosine 5-mono
phosphate
Chapter 1 Introduction and Review of Literature
11
181 Source of Xanthosine for caffeine biosynthesis
Xanthosine the initial substrate of purine alkaloid synthesis is supplied by at least four
different pathways de novo purine biosynthesis (de novo route) the degradation
pathways of adenine nucleotides (AMP route) and guanine nucleotides (GMP route)
and the S-adenosyl-L-methionine (SAM) cycle (SAM route) (Figure 13) Purine
nucleotides are synthesized by de novo and salvage pathways (Ashihara amp Crozier
1999 Stasolla et al 2003 Zrenner et al 2006) The de novo synthesis of non purine
precursors like CO2 10-formyltetrahydrofolate 5-phosphoribosyl-1-pyrophosphate and
the amino acids Glycine glutamine and aspartate results in the production of purine
nucleotides
Figure 13 Xanthosine biosynthesis Xanthosine for purine alkaloid biosynthesis is supplied by at
least four routes from adenosine released from the SAM cycle (SAM route) from IMP
originating from de novo purine synthesis (de novo route) from the cellular adenine
nucleotide pool (AMP route) and from the guanine nucleotide pool (GMP route)
Adapted from (Ashihara et al 2008)
The utilization of IMP for caffeine biosynthetic pathway which is formed by the
de novo purine biosynthetic pathway for caffeine biosynthetic pathways was
demonstrated in young tea leaves using 15
N-glycine and 14
C-labelled precursors and
inhibitors of de novo purine biosynthesis (Ito amp Ashihara 1999) Xanthosine is formed
by an IMP rarr XMP rarr xanthosine pathway IMP dehydrogenase (EC 111205) and 5prime-
nucleotidase (EC 3135) catalyze these reactions The inhibition of IMP dehydrogen-
ase by ribavirin inhibits both caffeine and gunanine nucleotide biosynthesis in tea and
coffee leaves (Keya et al 2003)
Chapter 1 Introduction and Review of Literature
12
The portion of xanthosine used for caffeine biosynthesis is derived from adenine
and guanine nucleotide pools which are produce de novo by salvage pathways there are
several potential pathways for xanthosine synthesis from AMP but the AMP rarr IMP
rarr XMPrarr xanthosine route is predominant All three enzymes involved in the
conversion AMP deaminase (EC 3546) IMP dehydrogenase and 5prime-nucleosidase
have been detected in tea leaves (Koshiishi et al 2001) Xanthosine for caffeine
biosynthesis can also be produced from guanine nucleotides by GMPrarrguanosine
rarrxanthosine pathway Guanosine deaminase (EC 35415) activity is found in cell free
extracts from young tea leaves (Negishi et al 1994) but GMP deaminase activity was
not detected in plants (Stasolla et al 2003 Zrenner et al 2006) The absence of GMP
deaminase activity suggests that a GMP rarr IMP rarr XMP rarr xanthosine route is not
functional in plants
The use of radiolabelled purine bases and nucleosides for the study of caffeine
biosynthetic pathway was facilitated by the fact that exogenous purine nucleotides are
rapidly hydrolyzed by the plant tissues forming purine nucleosides andor bases which
are then utilized in pathways which in most instances indirectly lead to xanthosine
Most of these purine compounds including adenine adenosine inosine hypoxanthine
and guanine are converted to their respective nucleotides by salvage enzymes
including adenine phosphoribosyl transferase (EC 2427) hypoxanthineguanine
phosphoribosyl transferase (EC 2428) adenosine kinase (EC 27120) and
inosineguanosine kinase (EC 2421) which usually have high activities in plant cells
The resultant nucleotides then enter the purine alkaloid biosynthetic pathway In the
case of guanosine it may be directly deaminated to xanthosine and utilized for caffeine
biosynthesis (Figure 13) (Ashihara et al 1997)
182 SAM cycle in plants
SAM is the methyl donor for the three methylation steps in the caffeine biosynthetic
pathway In this process SAM is converted to S-adenosyl-L-homocysteine (SAH)
which in turn is hydrolyzed to homocysteine and adenosine Homocysteine is recycled
via the SAM cycle to replenish SAM levels and adenosine is released from the cycle
Adenosine released from the cycle is converted to AMP directly andor indirectly via
adenine AMP is converted to xanthosine and used for caffeine biosynthesis Since
three moles of SAH are produced via the SAM cycle for each mole of caffeine that is
synthesized in theory this pathway has the capacity to be the sole source of both the
Chapter 1 Introduction and Review of Literature
13
purine skeleton and the methyl groups required for caffeine biosynthesis in young tea
leaves (Koshiishi et al 2001)
Methionine
S-Adenosyl-L-methionineHomocysteine
ATP
PPi+ Pi
Tetrahydrofolate
5-Methyl-
tetrahydrofolate
CH3+Adenosine
S-Adenosyl-L-
homocysteine
(xanthine
structure)(Methyl groups)
Caffeine
(1)
(2)(3)
(4)
Figure 14 The SAM cycle (activated methyl cycle) in plants (Ashihara amp Suzuki 2004)
Enzymes SAM synthetase SAM-dependent N-methyltransferases S-adenosyl-
homocysteine (SAH) hydrolase Methionine synthase Adenosine released from the cycle
is salvaged to adenine nucleotides and utilized both for purine structure of caffeine via
xanthosine and for re-synthesis of SAM via ATP
183 N-methyltransferase in caffeine biosynthesis Molecular cloning
The genes encoding for the enzymes involved in the caffeine biosynthesis were
successfully cloned from the coffee family (Ogawa et al 2001 Mizuno et al 2003a
Mizuno et al 2003b Uefuji et al 2003) The conserved amino acid regions of tea CS
(AB31280) the sequences of O-methyltransferases derived from plant origin and
Arabidopsis hypothetical proteins were made use of in designing primers for RT-PCR
and RACE technique Independent groups have isolated N-methyltransferase (NMT)
genes from coffee which have been classified based on the substrate specificity of the
recombinant proteins expressed in Ecoli (Table 15) PCR products thus obtained were
used as the probe to isolate full length cDNA from the library resulting in the
identification of all three N-methyltransferases involved in caffeine biosynthesis
(Figure 12) Several cDNA have been characterized from the coffee plants one for
xanthosine methyltransferase (XMTXRS) three for 7-methylxanthine methyltransferase
or theobromine synthase (MXMTCTS) and three for 37-dimethylxanthine
methyltransferase or caffeine synthase (DXMTCCS) (Table 15) (Kato amp Mizuno
2004)
A single gene encoding for xanthosine methyltransferase (XMTCmXRS1) was
identified which encoded for a polypeptide consisting of 372 amino acids with an
apparent molecular mass of 418kDa It is expressed almost uniformly in aerial tissues
Chapter 1 Introduction and Review of Literature
14
of C arabica including leaves floral buds and immature but not in mature beans
Three genes encoding 7-methylxanthine methyltransferase (theobromine synthase) have
been identified The number of amino acids in the putative polypeptides are 378 for
MXMT1 (427kDa) and 384 for MXMT2 and CTS2 (434kDa) They differ by insertion
or deletion of blocks of several residues in the C-terminal region Their catalytic
properties based on kinetic parameters such as Km values were different They are
expressed in young leaves floral buds and immature but not mature beans Three genes
were identified for 3 7-dimethylxanthine methyltransferase (caffeine synthase) DXMT
CCS1 and CtCS7 each encoding a 43 kDa polypeptide consisting of 384 amino acids
However their kinetic properties differ from each other such as DXMT and CCS1
showed Km values of 1200 and 157microM for theobromine respectively Expressions
profiles are also distinct DXMT being expressed exclusively in immature beans while
CCS1 expression is ubiquitous occurring in all tissues The presence of isoforms of
these enzymes with different properties suggests that caffeine is synthesized through
multiple pathways depending on availability and concentration of the substrate (Mizuno
et al 2001 Ogawa et al 2001 Mizuno et al 2003a Mizuno et al 2003b Uefuji et
al 2003) Genomic clones of theobromine synthase were obtained by PCR- RFLP
from C canephora (Satyanarayana et al 2005 Satyanarayana 2006) The genomic
clones are reported to have highly conserved exons and a variable third intron The
promoter for the theobromine synthase was cloned and characterized (Satyanarayana et
al 2005)
There is high degree of similarity in the coding region of coffee NMT genes
isolated so far though differences exist in the 3prime untranslated region (UTR) for these
genes The deduced amino acid sequence of these enzymes shows more than 85
homology between these genes Phylogenetic analysis indicates that they are more
closely related to C-methyltransferases including those for jasmonic acid salicylic acid
and benzoic acid than to other N-methyltransferases This suggests that coffee N-methyl
transferases belong to a distinct sub-group within plant methyltransferases Their
cellular localization determined by green fluorescent protein fusion method and β-
glucourodinase (GUS) showed that the all the three genes are localized in the cytoplasm
(Ogawa et al 2001 Vinod et al 2007) It was also shown that these were present
along with chlorogenic acid(Vinod et al 2007)
Chapter 1 Introduction and Review of Literature
15
Table 15 N-methyltransferases and their encoding genes involved in caffeine biosynthesis
Gene (Enzyme) Gene name Accession No Description Reference
Xanthosine methyltransferase
( 7-methylxanthosine synthase) (EC 211158)
CaXMT AB 048793 Immature fruit cDNA Screening (Uefuji et al 2003)
CmXRS1 AB 034699 Leaf cDNA library RACE (Mizuno et al 2003a)
CaMXMT1 AB 048794 Leaf cDNA library RACE (Ogawa et al 2001)
7-methylxanthine synthase transferase
(theobromine synthase) ( EC 211159)
CTS1 AB 034700 Immature fruit cDNA Screening (Uefuji et al 2003)
CaMXMT2 AB 984125 Leaf cDNA library Screening (Mizuno et al 2001)
CTS2 AB 054841 Leaf cDNA library Screening (Mizuno et al 2001)
37-methylxanthinemethyltransferase
(Caffeine synthase) (EC 211160)
CaDXMT1 AB 086414 Immature fruit cDNA Screening (Uefuji et al 2003)
CCS1 AB 0861414 Immature fruit cDNA Screening (Uefuji et al 2003)
CtCS7 AB 0864415 Immature fruit cDNA
NMT genomic clones
Theobromine synthase-1
CX10 Complete coding region PCR Satyanarayan 2006
CX8 Complete coding region PCR -do-
Theobromine synthase-1 PG-5 Promoter + coding region RAGE -do-
Theobromine synthase-1 PG-1 Promoter + coding region RAGE Satyanarayan 2006
PG-4 Promoter + coding region RAGE -do-
Theobromine synthase-1 NMT
(AY273814) Complete coding region PCR Kochko A et al 2010
Theobromine synthase-2 NMT
(AY362825) Complete coding region PCR Kochko A et al 2010
Chapter 1 Introduction and Review of Literature
16
19 Catabolism of caffeine
Caffeine is produced in young leaves and immature fruits and continues to accumulate
gradually during the maturation of these organs At the same time caffeine is slowly
degraded with the removal of the three methyl groups resulting in the formation of
xanthine Catabolism of caffeine was first reported in coffee leaves (Kalberer 1965)
Since then a number of tracer experiments using 14
C labelled purine alkaloids have
been reported (Suzuki amp Waller 1984 Ashihara et al 1996 Vitoria amp Mazzafera
1999 Mazzafera 2004) They demonstrated the major catabolic pathway is caffeine rarr
theophylline rarr 3-methylxanthine rarr xanthine Xanthine is further degraded by the
removal by the conventional purine catabolic pathway to CO2 and NH3 via uric acid
allatonin and allantoate (Figure 15) (Ashihara amp Crozier 1999 Stasolla et al 2003
Zrenner et al 2006) Caffeine catabolism usually begins with its conversion to
theophylline catalyzed by N7-demethylase The involvement of P450-dependent mono-
oxygenase activity for this reaction was suggested (Huber amp Baumann 1998
Mazzafera 2004) However the activity of this enzyme has not yet been determined in
cell free extracts [8-14
C] Theophylline degraded to CO2 at much faster rate than [8-14
C]
caffeine indicating that conversion of caffeine to theophylline is a major rate limiting
step of caffeine catabolism and a possible reason why caffeine accumulates in high
concentrations in tissues of C sinensis and C arabica It is widely believed that the
control of caffeine levels in plants is a function of the balance between rates of
synthesis and degradation and this balance seems to vary depending on the plant
species and the tissue developmental stage (Mazzafera 2004)
Chapter 1 Introduction and Review of Literature
17
Figure 15 Caffeine catabolic pathways Caffeine is mainly catabolised via theophylline and 3-
methylxanthine Xanthine is further degraded to CO2 and NH3 by the conventional
oxidative pathway Adapted from (Ashihara et al 2008)
Chapter 1 Introduction and Review of Literature
18
110 Genetic engineering of Coffee
Genetic transformation has potential applications in coffee agriculture for incorporating
genes which could provide desirable traits such as disease and insect resistance
drought and frost tolerance and herbicide resistance Transgenic technology can also be
used to increase nutritional value and improve cup quality produce varieties with
caffeine-free beans and for production of hybrid crops for molecular farming
Identification of target specific genes is one of the pre-requisites for developing
transgenic crops The availability of a large number of EST sequences in coffee and
initiation of coffee genome sequencing may speed up the gene discovery and accelerate
transgenic research efforts in coffee
1101 Insect Resistance
Production of insect resistant coffee plants is one of the major objectives of the
breeding programs The major pests attacking coffee include coffee berry borer (CBB
Hypothenemus hampei) white stem borer (WSB Xylotrechus quadripes) leaf miner
(Perileucoptera coffeela) and root nematodes (Meloidogyne spp and Pratylenchus
spp) The CBB is present in almost all the coffee growing countries and considered to
the most devastating pest in coffee To date there is no reported source of resistance to
CBB in the coffee gene pool Like CBB WSB is another serious pest in C arabica in
India and several other East Asian countries Both CBB and WSB belong to the order
Coleoptera For India controlling WSB is the biggest challenge and has therefore
become the highest research priority Robusta is generally resistant to WSB but the
interspecific Robusta Arabica hybrids are susceptible to WSB Although leaf miner is
not yet a serious pest in India and other East Asian countries it is an economically
important pest in East Africa and Brazil Effective chemical control of CBB and WSB
is difficult due to the nature of their life cycle inside the berry and stem respectively as
well as environmental concern regarding the use of pesticides Biological control
measures are adapted to combat these insect pests with varying degrees of success
Developing coffee plants resistant to these pests using genetic transformation
technology could be one of the alternative strategies to counter pest damage For insect
resistance several different classes of proteins from bacterial plant and animal sources
have been isolated and their insecticidal properties tested against many important pests
Amongst these proteins Bt toxins are most important and several transgenic crops
expressing Bt genes have been commercialized Coffee transgenic plants carrying a
Chapter 1 Introduction and Review of Literature
19
synthetic version of the cry1Ac gene have been produced (Leroy et al 2000 Mishra et
al 2002) Indeed this was the first report of an important agronomic trait being
introduced into a coffee plant The transgenic plants presented similar features in
growth and development compared to normal plants Transgenic plants highly resistant
to leaf miner under greenhouse conditions were tested under field conditions in French
Guyana for 4 years for pest resistance (Perthius et al 2005 Perthius et al 2006)
From 54 independent transformation events 70 of the events were resistant to leaf
miner Unfortunately the field trial was vandalized which led to the termination of the
experiment (Montagnon et al 2005)The effectiveness of Bt genes in controlling
coleopteran pests is well documented in corn and potato which indicates that Bt genes
might be effective against CBB The high toxicity of B thuringiensis sero var
israelensis against first in star larvae of CBB has been demonstrated (Mendez et al
2003) In another experiment an α-amylase inhibitor from Phaseolus vulgaris was
tested against CBB and found to have an inhibitory effect on its growth and
development (Grossi et al 2004)In addition to the CBB and WSB C arabica
varieties are also susceptible to endoparasitic root-knot nematode (Meloidogyne spp)
(Campos et al 1990 Mishra et al 2012)
So far 15 species have been reported to be parasites of coffee Controlling
nematodes is extremely difficult and currently seedling grafting with robusta rootstock
is followed as one of the control measure Sources of resistance specific to root-knot
nematodes have been identified in coffee trees (Bertrand et al 2001) and the Mex-1
gene conferring resistance to M exigua in C arabica is in the process of isolation (Noir
et al 2003)
1102 Tolerance to Abiotic Stress
In many coffee growing countries abiotic stress such as drought and frost are the major
climatic factors that limit coffee production Changes in climatic patterns due to global
climate change are considered increasingly important for coffee cultivation Drought
induces water stress in plants which affects vegetative growth and vigor and triggers
floral abnormalities and poor fruit set It also indirectly increases the incidence of pests
and diseases in the plants C arabica is generally more tolerant to water stress than C
canephora partly due to its extensive deep root system C racemosa is known to be a
good source of drought tolerance In India hybrids between C canephora and C
racemosa have been obtained and are currently under evaluation for drought tolerance
Chapter 1 Introduction and Review of Literature
20
In many coffee growing countries coffee is propagated in marginal areas where the
annual rainfall is below 1000thinspmm with prolonged dry spells of over 4-5 months In
those areas water shortage and unfavorable temperatures constitute major constraints
and the growth and productivity of C canephora are badly affected As with drought
periodic frost also affects coffee production in parts of Brazil The introduction of
drought and frost tolerant genes through genetic transformation would be of great
importance for alleviating these problems Research is now being carried out by several
groups to identify genes involved in biotic as well as abiotic stress The most promising
genetic engineering approach for drought tolerance includes the use of functional or
regulatory genes as well as the transfer of transcription factors In recent years plants
tolerant to high temperature and water stress have been the subject of intense research
(Chaves et al 2003 Chaves et al 2004 Coraggio et al 2004) For achieving drought
tolerance genes that have been targeted include those encoding enzymes involved in
detoxification or osmotic response metabolism enzymes active in signaling proteins
involved in the transport of metabolites and regulating the plant energy status (Chaves
et al 2003 Chaves et al 2004 Coraggio et al 2004) The dissection of molecular
mechanisms related to signal transduction and transcriptional regulation might help in
engineering drought tolerance in coffee
1103 Disease Resistance
Coffee leaf rust (CLR) caused by the fungus Hemileia vastatrix is the most important
disease in coffee with substantial loss to coffee production and productivity in all the
coffee growing countries In addition to leaf rust coffee berry disease (CBD) caused by
fungus Colletotrichum kahawae can be a devastating anthracnose leading to substantial
crop loss in Africa Several other fungal and bacterial diseases may also affect coffee
to a extent C arabica is more susceptible to many diseases than C canephora Though
most of the disease control measures rely upon chemical control they are more
expensive and labour intensive The long-term solution is the breeding of resistant
varieties which is the focus of many breeding programs However breeding for disease
resistant varieties is time consuming due to the perennial nature of coffee with its long
gestation period India has a long history of C arabica breeding especially for leaf rust
resistance and is the first country to demonstrate the existence of multiple races of leaf
rust Resistance to CLR is conditioned primarily by a number of major (SH) genes and
coffee genotypes are classified into different resistance groups based on their
Chapter 1 Introduction and Review of Literature
21
interaction with different rust races pathogen (van der Vossen 2001) Currently C
canephora provides the main source of resistance to pests and diseases including CLR
(H vastatrix) and CBD (C kahawae) and therefore used in breeding programs Other
diploid species like C liberica and C racemosa are being used as a source of resistance
to coffee leaf rust and coffee leaf miner respectively (Guerreiro et al 1999
Sreenivasan et al 1989) The development of coffee varieties resistant to major fungal
diseases such as CLR and CBD using transgenic technology will benefit the coffee
industry immensely During the last 15 years significant progress has been made in the
area of host-pathogen interactions (Staskawicz et al 1995 Feys et al 2000
Shivaprasad et al 2012) and many resistance genes involved in recognizing invading
pathogens have been identified and cloned (Takken et al 2000) A number of
signalling pathways induced because of pathogen infection have been dissected (Dong
et al 1998 Navarro et al 2006) Many antifungal compounds synthesized by plants to
combat fungal infection have been identified (Does et al 1998) Understanding the
specific induction of targeted pathways and identification of specific pathways
responsible for particular fungal resistance is important in order to employ this strategy
in transgenic technology The recent investigation of gene expression during coffee leaf
rust infection could provide an insight into the defence pathways operating in coffee
(Fernandez et al 2004 Nardi et al 2006) Efforts have been made to identify and
clone resistance genes from coffee for achieving durable resistance The genetic and
physical map of two resistance genes SH3gene conferring resistance to rust (Prakash et
al 2004) and the Ck1 gene conferring resistance to C kahawae CBD (Gichuru et al
2006) have been established These genes could be used for molecular marker assisted
breeding programs (Mishra et al 2012)
1104 Production of low-caffeine coffee
A widespread belief that the ingestion of caffeine can have adverse effects on
health resulted in the increased demand for decaffeinated coffee (Ashihara amp Crozier
2001) Several method were used to obtain decaffeinated plants by conventional
breeding techniques using low yielding varieties and mutants that accumulated
relatively low amounts of caffeine when compared to the commercially available ones
Low-caffeine and decaffeinated coffee represent around 10 of the coffee sales
around the world (Ribas et al 2006b) The industrial process for coffee decaffeination
can be expensive and affects the original flavour and aroma in coffee (David 2002)
Chapter 1 Introduction and Review of Literature
22
Transgenic coffee plants with suppressed caffeine synthesis using RNA interference
(RNAi) technology have been obtained (Ogita et al 2003 Ogita et al 2004) Specific
sequences in the 3prime untranslated region of the theobromine synthase gene (CaMXMT1)
were selected for construction of RNAi short and long fragments The caffeine and
theobromine content of the transgenic plants were reduced by ~ 70 when compared to
the untransformed plants In C canephora RNAi technology has also been employed
to silence the N-methyl transferase gene involved in caffeine biosynthesis (Vinod et al
2007) Promoter of an N-methyltransferase (NMT) gene involved in caffeine
biosynthesis was cloned (Satyanarayana et al 2005 Satyanarayana 2006) which will
be very useful for studying the regulation of caffeine biosynthesis
1105 Improvement in cup quality
Improvement in coffee cup quality requires elaborate knowledge of the chemical
constituents as well as the metabolic pathways involved in the elaboration of quality
The constituents of coffee beans include minerals proteins carbohydrates caffeine
chlorogenic acids (CGA) glycosides lipids and many volatile compounds that give
flavour to coffee by roasting Among these the role of three major constituents
sucrose CGA and trigonelline have been studied in coffee The sucrose content of
coffee bean is associated with coffee flavour the higher the sucrose content in green
beans the more intense will be the cup flavour (Clifford et al 1985 de Maria et al
1996) The sucrose content of C arabica (82-83) is higher than C canephora (33ndash
40) The sucrose amino acids ratio in green beans determines the profile of volatile
compounds Manipulating sucrose content in coffee bean is therefore important in
improving cup quality The sucrose synthase gene (CcSUS2) from C canephora has
been cloned and sequenced (Leroy et al 2005) This provides an opportunity to
manipulate the sucrose content in coffee Chlorogenic acids are products of
phenylpropanoid metabolism Chlorogenic acids are a group of hydroxycinnamoyl
quinic acids (HQA) formed by esterification between caffeic acids coumaric acids and
quinic acids (Clifford et al 1999) They are present in relatively large quantities in the
coffee bean and are the precursor of phenolic compounds in roasted beans C
canephora beans contain higher CGA (10) compared to C arabica beans (6-7)
CGA are known to have antioxidant properties as well as being associated with disease
resistance (Campa et al 2003) Genetic manipulations of genes involved in CGA
synthesis can serve either of these purpose by up- or down regulating the pathway
Chapter 1 Introduction and Review of Literature
23
Phenylalanine ammonia-lyase (PAL) catalyzes the first step of the phenylpropanoid
pathway leading to the synthesis of a wide range of chemical compounds including
flavonoids coumarins hydroxycinnamoyl esters and lignins (Hahlbrock amp Scheel
1989) Recently the full length cDNA and corresponding genomic sequences of PAL
from C canephora was isolated characterized and functionally validated (Mahesh et
al 2006 Parvatam et al 2006) This has opened up new possibilities for manipulating
the level of the PAL enzyme in coffee which in turn can be useful for improvement of
cup quality and manipulating antioxidant properties in coffee
1106 Fruit Ripening
Uniformity during fruit ripening is decisively related to cup quality in coffee and
consequently to the value of the product Fruits at the ideal ripening stage produce the
best organoleptic characteristics for coffee The presence of over ripened or green fruits
changes the acidity the bitterness and consequently the cup quality In order to
maximize uniform ripening of coffee fruits it is essential to control the action of genes
involved in the last step of maturation process Ethylene is known to trigger ripening
and increasing ethylene biosynthesis is associated with various stages of ripening
process (Protasio et al 2005) To control coffee fruit maturation two of the major
genes involved in ethylene biosynthesis namely ACC synthase and ACC oxidase
have been cloned (Protasio et al 2005 Neupane et al 1999) Introduction of the ACC
oxidase gene in antisense orientation have been achieved in both C arabica and C
canephora The effect of the transgene on ethylene production and fruit maturation has
yet to be reported The inhibition of genes downstream to the initial ethylene burst is
also an option to control coffee fruit maturation (Ribas et al 2006)
111 RNA interferenceRNAi
The regulation of gene expression at post transcriptional level has recently attracted
much attention because of the discovery of the phenomenon called RNA interference
(RNAi) RNAi based silencing techniques are more efficient than antisense mediated
gene silencing (Wesley et al 2001) The effect was first observed in plants wherein
silencing was observed when some copies of a transgene was in the forward orientation
with respect to the promoter and some in the reverse orientation This resulted in the
formation of double stranded RNA in the system The phenomenon in which
experimentally introduced double stranded RNA (dsRNA) leads to the loss of
expression of the corresponding cellular gene by sequence specific RNA degradation
Chapter 1 Introduction and Review of Literature
24
was termed as RNA interference or RNAi The protective effect of RNA silencing in
reducing virus infectivity supports the view that PTGS has evolved as a mechanism to
defend plants against virus infection and also to moderate the possible deleterious
genome-restructuring (insertional) activity of virus-like mobile genetic elements eg
retrotransposons A growing body of biochemical and genetic data has further
demonstrated that plants animals and yeasts share related mechanisms of specific
degradation of RNAs in which double-stranded forms of RNA are initiator molecules
(Brenstein et al 2001)
The key insight into the process of PGTS was provided by the experiments
conducted by Hamilton amp Baulcombe (1999) who identified the product of RNA
degradation as small RNA (siRNA) species of ~25nt of both sense and antisense
polarity SiRNA are formed and accumulate as double stranded RNA molecules of
defined chemical structures Both genetic and biochemical approaches were undertaken
to understand the basis of silencing Genetic screens were carried out in fungus
Neurospora carassa the alga Chalmydomonas reinhardtii and plant Arabidopsis
thaliana to search for detective mutants in PTGS
A combination of results obtained from several in vivo and in vitro experiments
have gelled into a two step mechanistic model for RNAiPTGS The first step referred
to as the RNAi initiating step involves binding of the RNA nucleases to a larges
dsRNA and its cleavage into discrete ~21-25nt RNA fragments This is ATP-
dependent reaction which involved RNase III - type endonucleases which make
staggered cuts in both strands of the dsRNA leaving 3prime overhang of 2 nucleotides with
5prime- phosphate 3prime-hydroxyl and no modification in the sugar phosphate backbone
(Hamilton amp Baulcombe 1999 Elbashir et al 2001) SiRNAs are the direct products
of dsRNA cleavage by the multi-domain RNase III enzyme DICER (Figure 16)
(Brenstein et al 2001 Karthikeyan et l 2013)
In the second step or commonly known as the effector step the double stranded
siRNAs produced in the first step bind to an RNAi specific protein complex to form a
RISC The complex undergoes activation in the presence of ATP so that the antisense
component of the unwound siRNA becomes exposed and allows the RISC to perform
the downstream RNAi reaction
Chapter 1 Introduction and Review of Literature
25
Several enzymes are involved in RNAi based on their role genes that encode for them
are classified into RNAi initiators RNAi effectors RNA dependent RNA polymerases
and silencing genes Two C elegans genes rdeI and rde4 (rde stands for lsquo RNAi
deficientrsquo) are believed to be involved in the initiation step of RNAi The C elegans
rde1 gene is a member of a large family of genes and is homologous to the Neurospora
qde2 (qde stands for ldquoquelling deficientrdquo) and Arabidopsis AGO1 (AGO stands for
agronaute AGO1 was previously identified to be involved in Arabidopsis
development) Although the functions of these genes are not clear a mammalian
member of RDE1 family has been identified as a translation initiation factor (Thakur
2005) Important genes for the effector step of PTGS in C elegans are rde2 and mut7
genes These genes were identified from heterozygous mutant worms that were unable
to transmit RNAi to their homozygous offsprings Worms with mutated rde2 or mut 7
genes show defective RNAi mut-7 gene encodes a protein with homology to the
nuclease domains of RNAase D and a protein implicated in Werner syndrome (a rapid
ageing disease in humans) (Grishok et al 2001 Kamath-Loeb et al 2012s)
Neurospora qde-1 Arabidopsis SDE-1SGS-2 and C elegans ego-1 appear to encode
RNA dependent RNA polymerase (RdRPs) It assumed that this is a proof that an RdRp
activity is required for RNAi Certainly the existence of an RdRp might explain the
remarkable efficiency of dsRNA induced silencing if it amplifies either the dsRNA
prior to cleavage or the siRNAs directly (Muers 2013) In C elegans ego-1 mutants
(ego stands for lsquoenhancer of glp-1) RNAi functions normally in somatic cells but is
defective in germline cells where ego-1 is primarily expressed In Arabidopsis SDE-
1SGS-2 mutants (SGS stands for suppressor of gene silencing) siRNA are produced
when dsRNA is introduced via an endogenously replicating RNA virus but not
introduced by a transgene It has been proposed that perhaps the viral RdRP is
substituting for the Arabidopsis enzyme in these mutants Random degradative PCR
model suggests that an RdRP uses the guide strand of an siRNA as a primer for the
target mRNA generating a dsRNA substrate for Dicer and thus more siRNAs
Several genes controlling RNA silencing in plants have been identified through
genetic screens of Arabidopsis mutants impaired in transgene induced RNA silencing
They encode a putative RNA-dependent RNA polymerase (SGS2SDE1) a coiled
protein (SGS3) a protein containing PAZ and Piwi domains (AGO1) and an RNA
helicase (SDE3) The putative SGS2SDE1 of Neurospora is related to QDE-1 and
Chapter 1 Introduction and Review of Literature
26
EGO-1 of C elegans whereas PAZPiwi protein AGO is related to QDE-2 of
Neurospora RDE-1of C elegans RNA helicase SDE3 is related to SMG-2 of C
elegans and Mut-6 of Chlamydomonas MUT-7 gene of C elegans encodes a protein
similar to Rnase D whereas the Drosophila DICER gene encode a protein similar to
RNase III An Arabidopsis ortholog of DICER gene has been identified called CAF
SIN1 SUS1(Thakur 2005 Poethig 2009)
Since the RNAi machinery is present constitutively within the eukaryotic cell it
is important to explore the metabolic advantages that are accorded by the RNAi related
proteins during the intrinsic normal growth of cells and development of organisms The
natural RNAi machinery not only keeps the mobile transposable elements from
disrupting the integrity of the genomes as suggested by the analysis of model organisms
like A thaliana C elegans D melanogaster (Tabara et al 1999 Wu-Scharf et al
2000 Aravin et al 2001 Hamilton et al 2002 Lisch 2009) but also participates in
development of organisms Genetic defects in Celegans RNAi genes ego1 and dicer
cause known specific developmental errors (Grishok et al 2000 Grishok et al 2001
Knight amp Bass 2001 Hall et al 2013) Similarly the Argonaute family of genes of A
thaliana (especially the ZWILLE proteins) is also responsible for plant architecture and
meristem development (Carmell et al 2002 Hamant and Pautot 2010) and the Dicer
homologue of A thaliana CAF1 is required for embryo development (Golden et al
2002 Nakano et al 2011) Thus genetic evidence illustrates the role of the RNAi
machinery as a controller of development related genes The mechanistic details of
these developmental processes are beginning to emerge
Chapter 1 Introduction and Review of Literature
27
Figure 16 Mechanism of gene silencing induced double stranded RNA The dicer enzyme to
produce siRNAs cleaves the RNA The siRNA generated bind to the nuclease complex
RISC The antisense component in the siRNA in the RISC guides the complex towards
the cognate mRNA resulting in endonucleolytic cleavage of the mRNA
Chapter 1 Introduction and Review of Literature
28
112 MicroRNA or MiRNA
In 1991 Ambros and co-workers first isolated a lin-4 mutant of Celegans which was
arrested at the first larval stage (Lee et al 1993) Later on the let-7 mutation was
isolated in the same system which was responsible for development through the fourth
larval stage Both lin-4 and let-7 encode short 22-nucleotide mature RNAs and were
called short temporal RNA because they control the temporal development program of
C elegans The mature lin-4 RNA defines (negatively regulates) the mRNA expression
of the lin-14 and lin-28 hetrochronic genes with the antisense mediated repression
mechanism of translation initiation and thus specifies the fate of cells during the first
three larval stages Recent studies have revealed that the short temporal RNAs are
actually members of a group of tiny RNAs (21to 28 nucleotides) called the micro-
RNAs (miRNA) (Bartel 2009) isolated members of which could easily run to a few
thousand which includes both those were identified computationally and by deep
sequencing Some of the components of the RNAi machinery have also been clearly
established as the effector proteins for the maturation of miRNAs
113 MicroRNAs in plants
1131 Discovery
MicroRNAs have been discovered by three basic approaches Direct cloning forward
genetic direct cloning and bioinformatic prediction followed by experimental
validation The most direct method of miRNA discovery is to isolate and clone small
RNAs from biological samples and several groups have used this approach to identify
plant miRNAs (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002 Xie et al 2003 Sunkar et al 2005 Williams et al 2013) The cloning methods
adapted were similar to those used to identify large numbers of animal miRNAs
(Lagos-Quintana et al 2001 Lau et al 2001 Aravin amp Tuschl 2005) which involve
isolating small RNAs followed by oligonucleotide adapter ligation reverse
transcription amplification and sequencing Some protocols incorporate methods to
enrich for Dicer cleavage products (ie molecules with 5prime-phosphates and 3prime-
hydroxyls) and to concatemerize the short cDNAs so that several may be identified in a
single sequencing read (Lau et al 2001 Llave et al 2002a Jagadeeswaran amp Sunkar
2013)The initial cloning experiments in Arabidopsis identified 19 miRNAs belonging
to 15 families (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002) although hundreds of other small RNAs such as siRNAs and RNA degradation
Chapter 1 Introduction and Review of Literature
29
fragments were also cloned through the direct cloning methods To select miRNAs
from the pool of small RNAs secondary structures of the RNA molecules were
examined in compliance with the acknowledged structural features of miRNAs
(Ambros et al 2003) The secondary structures were validated using several ad-hoc
software tools such as the Mfold or RNAfold programs (Reeder et al 2006) Finally
the existence of mature miRNAs was determined by RNA gel blot hybridization These
direct cloning methods were successful in isolating many miRNAs in Arabidopsis
(Reinhart et al 2002 Sunkar amp Zhu 2004) Rice (Sunkar et al 2005) Poplar (Lu et
al 2005) moss (Arazi et al 2005) and Tobacco (Billoud et al 2005)
Although miRNAs were first discovered through forward genetic screens in
worms (Lee et al 1993 Reinhart et al 2000)only few A thaliana miRNA gene
families have been discovered by this method (Chen 2008) in plants MiRNA
involvement in plant mutant phenotypes were not properly inferred till the cloning
experiments established that plant genomes contain numerous miRNAs (Park et al
2002 Reinhart et al 2002 Rhoades et al 2002) MiRNA loss-of-function allele has
been identified by forward genetic screens early extra petala1 is caused by a
transposon insertion ~160bp upstream of the predicted miR164c stem loop resulting in
flowers with extra petals (Baker et al 2005 Irish 2008) The fact that loss-of-function
miRNA mutants have been rarely discovered using forward genetics reflects small
target size for mutagenesis coupled with redundancy in nearly all evolutionarily
conserved plant miRNAs which are encoded by gene families (Table16) Family
members are likely to have overlapping functions buffering against loss at any single
miRNA locus Over expression screens can circumvent redundancy limitations At least
four plant miRNAs miR319 (also known as miR-JAW) miR172 (also known as EAT)
and miR166 were isolated in over expression screens for dominant mutants with
developmental abnormalities (Aukerman amp Sakai 2003 Palatnik et al 2003 Kim et
al 2005 Williams et al 2005b Schommer et al 2012) Mutations in miRNA target
sites which can prevent the entire family of miRNAs from repressing a target gene can
also circumvent redundant functions of miRNA family members
The dominant mutations in the HD-ZIP genes PHB PHV and REVOLUTA
(REV) in Arabidopsis and ROLLEDLEAF1 (RLD1) in Maize result in adaxialization of
leaves andor Vasculature (McConnell amp Barton 1998 McConnell et al 2001 Nelson
et al 2002 Zhong amp Ye 2004 Allen amp Millar 2012) and are all caused by mutations
Chapter 1 Introduction and Review of Literature
30
in miR166 complementary sites (Rhoades et al 2002 Emery et al 2003 Jones-
Rhoades amp Bartel 2004 Mallory et al 2004 Zhong amp Ye 2004)
In both plants and animals cloning was the initial means of large-scale
miRNA discovery (Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Reinhart et al 2002 Mendes et al 2012) However cloning is biased toward
RNAs that are expressed highly and broadly MiRNAs expressed at low levels or
only in specific cell types or in response to certain environmental stimuli were more
difficult to clone Sequence based biases in cloning procedures might also cause
certain miRNAs to be missed Because of these limitations bioinformatics approaches
to identify miRNAs have provided a useful complement to cloning
A straightforward use of bioinformatics has been carried out to find homologs
of known miRNAs both within the same genome and in the genomes of other species
(Pasquinelli et al 2000 Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Mendes et al 2009) A more difficult challenge is to identify miRNAs
unrelated to previously known miRNAs This was first accomplished for
vertebrate nematode and fly miRNAs using algorithms that search for conservation
of sequence and secondary structure (ie miRNA stem-loop precursors) between
species searching for patterns that are characteristic of miRNAs (Lai et al 2003 Lim
et al 2003 Lim et al 2005)
Most of the methods identified numerous miRNAs but are necessarily more
relaxed in terms of allowed structures Some approaches take advantage of the high
complementarity of plant miRNAs to target messages implementing the requirement
that the candidate has conserved complementarity to mRNAs (Jones-Rhoades amp Bartel
2004 Adai et al 2005) This additional filter has been useful for distinguishing
authentic plant miRNAs from false positives which later extended to mammalian
miRNA gene prediction (Xie et al 2005 Mendes et al 2012)
Another criteria used by most algorithms in the filters is evolutionary
conservation In plants mature miRNA sequences are conserved to a higher degree
than their precursor sequences For example Arabidopsis and rice diverged more than
130 million years ago (Friis et al 2004) but some miRNAs are highly conserved
in these plant species (Reinhart et al 2002) Furthermore various parameters for the
secondary structures such as free energy the number of paired residues within a
Chapter 1 Introduction and Review of Literature
31
miRNA or the number and size of bulges are also used to validate the predictions
(Jones-Rhoades amp Bartel 2004 Wang et al 2004 Adai et al 2005 Mendes et al
2012)
1132 Conserved microRNAs in Plants
Till date cloning genetics and bioinformatics screens have resulted in annotation of
approximately 184 potential Arabidopsis miRNAs (miRBase release 13) These
miRNAs seem to arise through gene duplication and subsequent diversification and
represent approximately 100 families (Maher et al 2006 Axtell 2013) Twenty one
families represented by 92 genes are clearly conserved in species beyond Arabidopsis
(Table 15) The presence of these miRNA families in other sequenced plant genomes
indicates that different plant species have an evolutionarily fluid set of miRNAs
(Willmann amp Poethig 2007) Recent studies on mosses one of the most ancient land
plants have shown that at least 14 miRNA families are conserved in eudicots and
mosses and therefore it appears that plant miRNAs share a common ancient origin
(Arazi et al 2005 Axtell amp Bartel 2005 Talmor-Neiman et al 2006 Zhang et al
2006 Axtell 2013) The number of members per family in a genome ranges from 1-32
with the exception of miR-430 which is represented by a cluster of ~80 loci in zebra
fish (Giraldez et al 2005)
In plants the number of members in each family correlates among examined
species certain families contain numerous members in all three species (eg miR156
miR166 miR169) whereas others consistently contain fewer genes (eg miR162
mir168 miR394) (Table 16) Although it is still unclear why certain miRNA are
encoded by more than one gene in plants (eg 12 genes encoding miR156) this
correlation might suggest functional significance of various miRNA family sizes Most
annotated plant miRNAs are conserved throughout flowering plants while a few
miRNAs are found only in a single sequenced genome and thus they could be
evolutionarily lsquoyoungrsquo miRNAs For example Arabidopsis has several miRNA
families that are not found in poplar or rice suggesting that miRNAs could be
generated continuously during evolution (Allen et al 2004a Rajagopalan et al
2006 Fahlgren et al 2007 Axtell 2012 de Alba et al 2013) Consistent with the
notion that non conserved miRNAs are of a more recent evolutionary origin these
miRNAs are mainly encoded by a single locus in the genome Within each family the
mature miRNA is always located in the same arm
Chapter 1 Introduction and Review of Literature
32
Table 16 MicroRNA families conserved in plants
miRNA
family
Arabidopsis Oryza Populus
miR156 12 12 11 miR159319 6 8 15 miR160 3 6 8 miR162 2 2 3 miR164 3 5 6 miR166 9 12 17 miR167 4 9 8 miR168 2 2 2 miR169 14 17 32 miR171 4 7 10 miR172 5 3 9 miR390 3 1 4 miR393 2 2 4 miR394 2 1 2 miR395 6 19 10 miR396 2 5 7 miR397 2 2 3 miR398 3 2 3 miR399 6 11 12 miR408 1 1 1 miR403 1 0 2 miR437 0 1 0 miR444 0 1 0 miR445 0 9 0 Total 92 127 169
All known miRNA families that are conserved between more than one plant species are listed
together with the number of genes identified in the sequenced genomes Rice miRNA families that have orthologs in maize but do not appear to have orthologs in the eudicots (Arabidopsis and Populus) are marked with an asterisk The following families contain miRNA genes annotated with
more than one number miR156 (miR156 and miR157) miR159319 (miR159 and miR319) miR166 (miR165 and miR166) miR171 (miR170 and miR171) and miR390 (miR390 and miR391)
of the stem loop (5prime- or 3prime) as it would be expected if the genes carry common ancestry
(Figure 17) The sequence of the mature miRNA and to some extent the segment on
the opposite arm of the hairpin to which it pairs is highly conserved between the
members of the same family (both within and between the species) while the sequence
secondary structure and length of the intervening ldquolooprdquo region can be highly divergent
within the same family
The patterns of paring and non pairing nucleotides are often conserved between
homologous miRNA stem loops from different species (Figure 17) The significance of
conserved mismatches is opined to guide DCL1 to cleave at the appropriate positions
along the stem loop
Chapter 1 Introduction and Review of Literature
33
Figure 17 Representative stem loop of miR164 Segments corresponding to the mature miRNAs
are shown in red (Adapted from Rajagopalan et al 2006)
Although many miRNAs are highly conserved in plants the degree of conservation in
most cases is quite low between plants and animals one exception is miR854 A recent
study has revealed that miR854 is conserved between Arabidopsis and animals and that
the Arabidopsis miR854 differs from the human miR854 only by a single nucleotide
(Arteaga-Vazquez et al 2006 Axtell 2012 de Alba et al 2013) The Arabidopsis
miR854 has multiple binding sites in the 3prime-untranslated region (UTR) of
OLIGOURIDYLATE-binding-PROTEIN mRNA 1bA (UBP1b) and represses the
translation of the UBP1b mRNA The animal miR854 is also complementary to a site in
the 3prime-UTR of the UBP1b homolog in animals However it is currently unclear how the
animal miR854 regulates its target mRNA Another distinction between plant and
animal miRNAs is the genomic organization of the miRNA genes While plant miRNA
genes are located predominantly in the intergenic regions animal miRNA genes tend to
localize in the intragenic regions both in the introns or exons of protein coding genes
Moreover miRNA genes are often clustered within a single precursor transcript
whereas individual plant miRNAs are derived from individual precursor RNAs (Bartel
Chapter 1 Introduction and Review of Literature
34
2004 Bonnet et al 2004 Kim 2005 Vaucheret et al 2006 Marco et al 2013)
1133 Non-conserved miRNA and challenges in annotation
Most miRNAs are conserved throughout flowering plants (Table 15) but many more
were found only in a single sequenced genome thus considered to be of a more recent
evolutionary origin The extended homology between few non conserved miRNAs
precursors and their target genes provides strong evidence that these relatively ldquoYoungrdquo
miRNAs arose from the duplication of the target gene segments (Allen et al 2004)
Several non-conserved miRNAs including miR161 miR163 miR173 miR447
miR475 and miR476 are known to direct cleavage of target transcripts (Allen et al
2004a Adai et al 2005 Sunkar et al 2005 de Alba et al 2013) It is difficult to
confidently predict targets for many because it is not possible to use complementary
site as a filter against false-positive target predictions It is also difficult to
confidently annotate non-conserved miRNAs as miRNA rather than siRNAs The
established minimal standard for miRNA annotation is a small RNA with detectable
expression and the potential to form a stem-loop when joined to flanking genomic
sequence (Kasschau et al 2003) In practice these requirements are too loose to
definitively categorize many small RNAs cloned from plants Many plant siRNAs are
detectable on blots (Vazquez et al 2004b) and hundreds of thousands of non-
miRNA genomic sequences can be predicted to fold into secondary structures that
resemble structures of plant miRNA precursors (Jones-Rhoades amp Bartel 2004)
Therefore without conservation of both sequence and secondary structure it is
difficult to be confident that a given cloned RNA originated from a stem-loop (ie is a
miRNA) rather than from a double-stranded RNA (ie is a siRNA) In fact many of
the thousands o f small RNAs cloned from Arabidopsis (Gustafson et al 2005) would
probably meet the literal requirements for annotation as miRNAs A few of these
sequences might be miRNAs but others that meet the literal criteria probably are not
so The challenges of annotating non conserved small RNAs were evidenced by
three related small silencing RNAs that were originally annotated as miRNAs
that turned out to be trans-acting siRNAs (tasiRNAs)(Kanno et al2013)
Although most miRNAs that are broadly conserved among flowering plants are
now being identified and experimentally validated (Table 16)(Jones-Rhoades amp
Bartel 2004) the challenges and ambiguities for classifying non-conserved miRNAs
prevent meaningful estimates of the total number of miRNA genes in Arabidopsis and
Chapter 1 Introduction and Review of Literature
35
other plant genomes It is possible that miRNAs might have escaped detection
because they are non-conserved and expressed in specific tissues or conditions and can
only be identified by using high- throughput sequencing
1134 MicroRNA biogenesis
11341 Transcription of microRNA precursor
Plant miRNAs are primarily found in the genomic regions that are not associated with
the protein coding genes (Reinhart et al 2002) and it appears that most if not all plant
miRNAs are produced from their own transcriptional units Plant miRNA genes are
occasionally clustered near each other in the genome suggesting transcription of
multiple miRNAs from a single primary transcript [eg the miR395 cluster (Grishok
et al 2001 Jones-Rhoades amp Bartel 2004b)] but this polycistronic arrangement of
miRNA genes appears far less frequently in plants than in animals (Bartel 2004)
Northern blot EST and mapping evidence indicate that plant miRNA primary
transcripts (also known as pri-miRNAs) as in animals are longer than needed to
encompass the miRNA stem-loops (Palatnik et al 2003 Jones-Rhoades amp Bartel
2004b) At least some of these pri-miRNA transcripts appear to be spliced
polyadenylated (Kurihara amp Watanabe 2004) and capped (Xie Z et al 2005) Two
rice miRNAs are contained within transcripts that contain exon junctions within the
presumptive stem-loop precursor implying that in these cases splicing is a prerequisite
for Dicer recognition (Sunkar et al 2005) Plant pri-miRNAs are over 1kb in length
and they are usually preceded by typical TATA box motifs They can also undergo
canonical splicing polyadenylation and capping All these observations indicates RNA
polymerase II is probably responsible for transcribing most plant miRNAs (Xie Z et
al 2005) as appears to be the case for many animal miRNAs Relatively little is
known about the regulation of miRNA transcription in plants but there is no reason
to suspect that this regulation would differ from that of protein-coding transcripts as
the bioinformatics analysis of the sequence upstream of the transcriptional start
sites of the miRNA genes showed the presence of putative binding motifs of
number of known transcription factors (Megraw et al 2006 Sethupathy 2013)
11342 MicroRNA processing and export
In plants maturation of miRNA is a step wise process A miRNA gene is transcribed to
pri-miRNA which is much longer than the pre-miRNA possessing the characteristic
stem-loop structure Like transcription of most protein-coding genes the miRNA
Chapter 1 Introduction and Review of Literature
36
transcription is processed by RNA polymerase II (Pol II) enzymes and the pri-
miRNAs are 5prime-capped and 3prime-polyadenylated (Lee et al 2004 Xie Z et al 2005)
Mature miRNAs are produced from pri-miRNAs through at least two sequential
processing steps by RNase III-type endonucleases In animals pri-miRNAs are first
processed at the bottom portion of the stem by Drosha a nuclear-localized RNase III
enzyme to liberate pre-miRNAs The pre-miRNAs are then exported to the cytoplasm
with the help of exportin-5The pre-miRNAs are further processed by Dicer another
RNaseIII enzyme to a duplex consisting of the miRNA and the antisense strands
(miRNA) Since plants do not have any Drosha homologs the two sequential
processing steps seem to be carried out by DCL1 a Dicer homolog in the nucleus
(Kurihara amp Watanabe 2004 de Alba et al 2013 Rogers amp Chen 2013)
The processing of pri-miRNAs to pre-miRNAs by DCL1 is assisted by two
other proteins HYPONASTY LEAVES1 (HYL1) and SERRATE (SE) HYL1 belongs to a
family of double-stranded RNA (dsRNA) binding protein and interacts with DCL1 in
vitro and in vivo (Lu amp Fedoroff 2000 Hiraguri et al 2005 de Alba et al 2013
Rogers amp Chen 2013) Loss-of-function mutations in the HYL1 gene result in
decreased accumulation of miRNAs and concomitant accumulation of pri-miRNAs
(Han et al 2004 Vazquez et al 2004a Kurihara et al 2006) SE a C2H2 zinc finger
protein interacts with DCL1 and HYL1 and plays a role in the processing of pri-
miRNAs to pre-miRNAs (Lobbes et al 2006 Yang L et al 2006) In addition the
nuclear cap-binding complex proteins CBP20 and CBP80 that are known as ABA
HYPER SENSITIVE 1(ABH1) have been shown to be required for proper miRNA
processing (Gregory et al 2008 Laubinger et al 2008) Recently it has been
demonstrated that DCL1 can release 21-nt RNAs from dsRNA as well as from
synthetic miR167 precursor RNAs in vitro However this cleavage is error prone and
inefficient in the absence of HYL1 and SE which accelerate the rate of DCL1-mediated
cleavage and promote accurate processing of miRNAs (Dong et al 2008 Rogers amp
Chen 2013)
11343 Methylation of the miRNAmiRNAduplex
After the miRNAmiRNAduplexes are released from the pre-miRNAs by the DCL1
activities the duplexes are methylated at the 2primeOH of the3prime-ends by HUA ENHANCER1
(HEN1) a small RNA methyltransferase (Yu et al 2005 Yang Z et al 2006 Rogers
amp Chen 2013 ) This step which is distinct from the RNase III-mediated processing
Chapter 1 Introduction and Review of Literature
37
step distinguishes the biogenesis of plant miRNAs from that of animal miRNAs
Mutations in the HEN1 gene were first isolated in a morphological screening for floral
development although the HEN1 mutants display pleiotropic phenotypes similar to the
developmental defects observed in the DCL1 mutants (Chen et al 2002 Park et al
2002 Rogers amp Chen 2013)
Figure 18 Biogenesis of microRNA
A Biogenesis of plant MicroRNA B Biogenesis of metazoananimal MicroRNA
The miRNA (red) is incorporated into RISC and the miRNA(blue) in degraded
This enzyme has two dsRNA-binding domains and nuclear localization signal It
is also conserved in fungi and is required for the processing of siRNAs as well as of
miRNAs (Park et al 2002 Boutet et al 2003 Xie et al 2003) Although the HEN1
protein is localized to the nucleus its methylation activity in the cytoplasm cannot be
ignored (Xie et al 2004 Rogers amp Chen 2013)
Chapter 1 Introduction and Review of Literature
38
The methylation of the miRNAmiRNA duplex is required to protect miRNAs
against the 3prime-end uridylation activity and subsequent degradation (Li et al 2005) In
the hen1 mutants accumulation of miRNAs is reduced to a lower level Also the
miRNAs appear to be heterogeneous in their sizes such that a ladder of bands rather
than a single species is observed for most miRNAs in the mutants (Li et al 2005)
MiRNA cloning and sequencing analyses revealed the presence of a number of U
residues at the 3prime-end of miRNAs in the hen1 mutants Moreover these nucleotides do
not correspond to the miRNAs in the pre-miRNAs indicating that these nucleotides are
added after the processing of the miRNAs from pre-miRNAs (Li et al 2005)
Uridylation of RNAs has also been proven to be correlated with RNA decay (Shen amp
Goodman 2004 Mullen amp Marzluff 2008) suggesting that uridylation attracts an
exonuclease which degrades miRNAs in plants
11344 MiRNA incorporation in to the RISC and its nuclear export
The methylated miRNAmiRNA duplexes undergo RISC assembly In this process
the miRNA of the duplexes is selectively incorporated into the RISC and the miRNA
appears to be removed and subsequently degraded (Hammond et al 2000 Hutvagner
amp Zamore 2002 Schwarz et al 2003 de Alba et al 2013 Rogers amp Chen 2013)
The ARGONAUTE 1(AGO1) proteins are core components of the RISC complex
They contain two structural domains the N-terminal PAZ domain that binds to the 3prime-
end of single-stranded RNAs and the C-terminal piwi domains that has a structure
similar to that of RNase H (Cerutti et al 2000 Parker et al 2004) Several AGO
proteins possess endonucleolytic activities within the PIWI domain and cleave target
mRNAs in the middle of the site complementary to miRNA (Liu et al 2004 Meister et
al 2004 Miyoshi et al 2005) Mutations in the AGO1 gene cause miRNA target
genes to be ectopically expressed and the levels of some miRNAs are reduced in the
mutants (Kidner amp Martienssen 2004 Vaucheret et al 2004 Kidner amp Martienssen
2005 Ronemus et al 2006) Moreover the phenotype of the AGO1 hypomorphic
alleles display defects in lateral organ polarity and leaf and flower morphology as
observed in the dcl1 mutants and null AGO1 alleles are embryonically lethal
supporting the close relationship between the AGO proteins and miRNA processing
(Kidner amp Martienssen 2004 Vaucheret et al 2004 de Alba et al 2013 Rogers amp
Chen 2013)
The production of miRNA miRNA duplexes occur in the nucleus while
Chapter 1 Introduction and Review of Literature
39
miRNA-RISC complexes are present in the cytoplasm where target mRNAs are
located This discordance of functional sites requires an export step during miRNA
biogenesis HASTY the Arabidopsis homolog of exportin-5 is thought to be involved
in miRNA nuclear export (Bollman et al 2003 Park et al 2005)The hasty mutant
exhibits pleiotropic defects in plant development and reduced accumulation of most
miRNAs as in the DCL1 or AGO1 mutants Howeverit is unclear whether the HASTY
proteins export the miRNA miRNA duplexes or the miRNA-RISC complexes in
plants
11345 Feedback regulation of miRNA biogenesis
MiRNA biogenesis is under feedback regulation such that the DCL1 and AGO1 genes
are themselves regulated by miRNAs DCL1 is itself a target of miR162 which leads to
the cleavage of the DCL1 mRNA (Xie et al 2003 de Alba et al 2013) Consistent
with this the levels of DCL1 mRNA are elevated in the mutants of miRNA processing
genes in which the miR162 abundance is reduced In addition there is a predicted
hairpin structure of miR838 in the 14th
intron of the DCL1 primary transcript
(Rajagopalan et al 2006) This prediction implies that DCL1-mediated processing
on this site can result in the cleavage of the DCL1 primary transcript into two truncated
fragments the N-terminal capped fragment and the C-terminal polyadenylated
fragment If this is the case it is possible that miRNA biogenesis competes with the
splicing event of the DCL1 pre-mRNA
The AGO1 is also regulated by miRNA There is a complementary site for
miR168 binding in the AGO1 mRNA and the transcript level of the AGO1 mRNA is
reduced through an mRNA cleavage mechanism (Vaucheret et al 2006 de Alba et al
2013 Rogers amp Chen 2013) indicating that the AGO1 mRNA levels are tightly
controlled by the levels of the AGO1 protein
1135 Mechanism of miRNA action
11351 MiRNA-directed mRNA cleavage
By forming the miRNA-RISC assembly miRNAs can direct the RISC to regulate gene
expression by two post transcriptional mechanisms mRNA cleavage or translational
repression (Bartel 2004 Jones-Rhoades et al 2006 Dalmay 2013) The degree
of miRNA-mRNA complementarity plays an important role for the
determination of the mechanism to be used A general rule is that while perfect or
Chapter 1 Introduction and Review of Literature
40
near-perfect complementarity induces mRNA cleavage central mismatches trigger
translational repression (Carrington amp Ambros 2003 Jones-Rhoades amp Bartel 2004)
Unlike most animal miRNAs plant miRNAs have near-perfect or perfect
complementarity to their targets and mRNA cleavage is assumed to be a predominant
mechanism by which miRNAs regulate gene expression in plants
In RNA cleavage mechanism miRNAs guide the AGO component of RISC to
cleave a single phosphodiester bond of the target mRNA within the miRNA-binding
site (Llave et al 2002b Tang et al 2003) The truncated fragments are then released
and degraded freeing the RISC to recognize and cleave another target mRNA This has
been validated by the detection of 3prime cleavage products that have 5prime ends that start at the
middle of the complementary regions The mRNA cleavage products are then further
degraded by other mechanisms The 3prime cleavage products of some miRNA targets are
degraded by the 5prime-3prime exonuclease XRN4 an Arabidopsis homolog of the major Yeast
mRNA degrading exonuclease Xrn1p It has been observed that the 3prime cleavage
products of some target mRNAs were accumulated in the xrn4 mutants (Souret et al
2004 Dalmay 2013) However the 3prime cleavage products of other miRNA targets do
not accumulate in the xrn4 mutants indicating that there are also XRN4 independent
mechanisms The 5prime cleavage products are typically untraceable by RNA gel blot
hybridization but can be detected by sensitive PCR based methods It has been found
that the 5prime cleavage products tend to obtain an oligo U tail at the 3prime ends This 3prime U tail
is correlated with shortening of the 5prime cleavage products from the 5prime end leading to the
conclusion that the 3prime uridylation causes 5 prime- 3 prime exonucleolytic decay of the 5prime cleavage
products (Shen amp Goodman 2004)
DCL1 HYL1 and SE interact with each other and are co-localized in the
nuclear bodies termed Dicing bodies or D-bodies in which some pri-miRNA shave
been found suggesting that D-bodies serve as a center for active miRNA processing
(Fang amp Spector 2007 Fujioka et al 2007 Song et al 2007) The miRNA
processing site appears to be distinct from the Cajal bodies that have previously been
found as a siRNA processing site (Pontes amp Pikaard 2008) The D-bodies include
SmD3 and SmB proteins that are found in the Cajal bodies However At collin an
additional marker for the Cajal bodies is not found in the D-bodies Moreover the D-
bodies are present in an At collin mutant lacking the Cajal bodies The pri-miRNAs of
miR171A and miR159A genes have been found to be co-localized with HYL1 and
Chapter 1 Introduction and Review of Literature
41
DCL1 in the D- bodies that are distinct from the Cajal bodies (Song et al 2007)
11352 MiRNA-directed translational repression
Translation repression is another mechanism exerted by plant miRNAs The regulatory
mechanism of miR172 which regulates flowering time and floral organ identity is
consistent with translation repression MiR172 over production does not affect the
transcript levels of APETALA2 (AP2) and AP2- like target mRNAs Instead the AP2
protein level is reduced in the miR172 over expressing mutant (Aukerman amp Sakai
2003 Chen 2004 Dalmay 2013) It was later shown that miR172 can also direct the
cleavage of the target mRNAs although the steady-state mRNA level is unchanged due
to feedback regulation (Schwab et al 2005 Jung et al 2007) It is clear that miR172
regulates its target genes primarily by translational repression rather than mRNA
cleavage Recently it has been reported that miR156157 and miR854 also regulate
their target genes through translational repression (Arteaga-Vazquez et al 2006
Gandikota et al 2007 Dalmay 2013) It was once thought that translational repression
is an exception to the rule However experimental evidence suggest a widespread
coexistence of translational repression and mRNA cleavage (Brodersen et al 2008)
1136 Regulatory roles of miRNAs in plant development
The miRNA target prediction has been a difficult task in order to obtain regulatory
targets without bringing in too many false predictions Plant growth and developmental
processes regulated by miRNAs are quite diverse and enormous Furthermore plant
miRNAs work cooperatively or antagonistically to establish a balanced regulation This
notion is well consistent with the findings that mutations in the regulators of miRNA
biogenesis transport and processing such as AGO1 DCL1 HYL1 HEN1 ABH1
and HASTY cause pleiotropic phenotypes (Mallory amp Vaucheret 2006 de Alba et al
2013) To date the roles of miRNAs have been mostly deduced from obtaining miRNA
over expressing transgenic plants or gain-of-function mutants in which miRNA-
resistant target genes are ectopically expressed
11361 Phase transition
Plants pass through a series of developmental phase transitions during their life span
beginning with seed germination continuing through vegetative phase change
reproductive phase change flowering initiation and finally seed production for the next
generation Because of their importance in understanding plant developmental
Chapter 1 Introduction and Review of Literature
42
processes genes regulating developmental transitions have been extensively studied
and underlying molecular mechanisms have been explored in various plant species
Majority of researches were mainly focused on vegetative phase change and
reproductive transition in Arabidopsis and Maize (Poethig 2003 Baurle amp Dean
2006) Particularly the mutations of the genes encoding various regulators of miRNA
biogenesis and transport such as HST SE and AGO exhibit altered juvenile to adult
vegetative phase change and vegetative to reproductive change (Hunter et al 2003
Peragine et al 2004 Vazquez et al 2004a Jones-Rhoades et al 2006 de Alba et al
2013)
Over-expression of the miR156a gene causes late flowering and delays
vegetative phase change (Wu amp Poethig 2006 Gandikota et al 2007 Wang et al
2008) It has been shown that miR156 negatively regulates a group of genes encoding
SQUAMOSA PROMOTER-BINDING PROTEIN-LIKE (Lewis et al 2007 de Alba et
al 2013 Rogers amp Chen 2013) transcription factors such as SPL3 SPL4 and SPL5
that promote flowering initiation In contrast transgenic plants over expressing
miR156-resistant SPL345 genes are early flowering Interestingly the endogenous
level of miR156 is temporally regulated In the early juvenile vegetative phase the
level of miR156 is very high but decreases rapidly before the onset of the adult juvenile
phase (Wu amp Poethig 2006)
The Arabidopsis early activation tagged (eat-D) mutant exhibits early flowering
with disrupted floral structure The mutant phenotypes are caused by over expression of
the miR172a-2gene (Aukerman amp Sakai 2003) MiR172 regulates a small group of
genes encoding APETALLA2 (AP2)-like transcription factors such as TARGET OF
EAT1 (TOE1) TOE2 SCHLAFMUTZE (SMZ) and SCHNARCHZAPFEN(SNZ)
TOE1 TOE2 SMZ and SNZ have been shown to participate in their productive phase
change by repressing a floral integrator FLOWERING LOCUS T (FT)(Jung et al
2007) Consistent with this toe1-2D 35STOE2 35SSMZ and 35SSNZ plants
exhibit late flowering (Jung et al 2007) MiR172 is also regulated temporally and
highly expressed in the adult vegetative phase both in Arabidopsis and maize (Chuck et
al 2007) Because the temporal expression patterns of miR156 and miR172 are
complementary to each other it has been suggested that the two miRNAs interact with
each other in regulating phase change (Chuck et al 2009 de Alba et al 2013) In
support of this view it has been shown that miR172 is down-regulated in the
Chapter 1 Introduction and Review of Literature
43
Corngrass1(Cg1) mutant in which miR156 is overproduced (Chuck et al 2007 de
Alba et al 2013) However there is no direct evidence for the direct linkage between
miR156 and miR172 It is also envisioned that miR156 and miR172 would not be
directly related For example miR156 regulates plastochron index that is the inverse of
leaf initiation rate and thus frequently used to analyze temporal patterns of plant
development In contrast miR172 does not affect plastochron (Wang et al 2008) The
down-regulation of miR172 in the maize miR156-over producing mutants would be
due to the prolonged juvenile vegetative phase in the mutants
Another miRNA that regulates flowering initiation is miR159 It regulates
MYB33 and MYB65 which are closely related to the barley GAMY B transcription
factor that responds to gibberellic acid (GA) The level of miR159 is elevated in GA-
treated seedlings but reduced in the GA biosynthetic ga1 mutant Transgenic plants
over producing miR159 exhibit delayed floral induction under short days (Achard et
al 2004) It is been suggested that MYB33 mediates GA signal to regulate LEAFY
(LFY) These observations indicate that miR159 plays an important role in the GA
signaling pathways that regulate proper floral induction under short days (Achard et al
2004)
11362 Floral development
Arabidopsis flowers consist of four distinct organs They arise in a characteristic
pattern within concentric rings called whorls from outside to inside four sepals four
petals six stamens and the central pistil consisting of two carpels Identities of the
floral organs are determined by three classes of regulatory genes classA classB and
class C genes (Krizek amp Fletcher 2005) The A and C class genes act antagonistically
to restrict their expression domains (Bowman et al 1991)
It is notable that miR172 also regulates floral architecture in addition to its role
in the timing of flowering initiation MiR172 inhibits the translation of the AP2 mRNA
(Chen 2004) Transgenic plants overproducing miR172 not only exhibit early
flowering but also show abnormal floral development which is quite similar to those
of the class A gene mutants such as the ap2 mutants (Aukerman amp Sakai 2003 de
Alba et al 2013) When the activity of the class A genes is inactivated that of the class
C genes such as AGAMOUS (AG) is elevated As a result the miR172- overproducing
transgenic plants exhibit no petals along with homeotic transformation of the sepals to
Chapter 1 Introduction and Review of Literature
44
the carpels (Aukerman amp Sakai 2003 Chen 2004)
Interestingly miR166 also regulates floral architecture The role of miR165166
in the regulation of meristem activity is closely linked to floral meristem formation and
organization (Zhao et al 2007 Miyashima et al 2013) Floral structure is severely
disrupted in the miR165166-overproducing mutants such as men1 and jba-1D in
which miR166 is overproduced (Williams et al 2005b Jung amp Park 2007) The men1
and jba-1D mutants have smaller gynoecium and reduced carpel number In an extreme
case the jba-1D homozygous plants lack visible carpels (Williams et al 2005b)
Similar phenotypes have been observed in the miR165 overproducers (Zhao et al
2007) It has been suggested that the phenotypes are possibly caused by altered AG
expression in the floral meristem (Jung amp Park 2007 Turchi et al 2013)
Other miRNAs also affect floral development Overproduction of miRNAs
involved in auxin signaling also affects floral architecture Transgenic plants
overproducing miR167 have defects in the formation of ovule and anther with reduced
fertility (Ru et al 2006) It has been shown that miR167 targets ARF6 and ARF8 that
play a role in gynoecium and stamen development and in regulating fertility (Ru et al
2006 Wu et al 2006) These observations suggest that auxin signals are also closely
related to floral organogenesis In addition transgenic plants over-producing miR164
and the cuc1cuc2 double mutants show fused sepals and reduced petals (Laufs et al
2004 Turchi et al 2013) suggesting that miR164 is also related to floral meristem
activity and boundary specification of the meristematic region
11363 Shoot apical meristem development
Shoot apical meristem comprises self-maintaining totipotent cells The shoot apical
meristem activity affects diverse aspects of plant developmental processes including
leaf development vascular development and lateral organ polarity (Bowman et al
2002) Because miR165166 plays a primary role in meristem formation
overproduction of miR165166 and multiple knockout mutants of its target genes
exhibit pleiotropic pheonotypes (McConnell et al 2001 Emery et al 2003 Kim et al
2005 Williams et al 2005b)
The targets of miR165166 include genes encoding the homeo domain-leucine
zipper (HD-ZIP) class III transcription factors such as PHABULOSA(PHB)
PHAVOLUTA (PHV) REVOLUTA(REV) ATHB15CORONA (CNA) and ATHB8
Chapter 1 Introduction and Review of Literature
45
PHB PHV and ATHB15 have overlapping functions in controlling meristem
development (Turchi et al 2013) Indeed the phb phv athb15 triple mutants have an
enlarged shoot apical meristem Consistent with this the miR166- overproducing men1
and jba-1D mutants exhibit enlarged shoot apical meristems (Kim et al 2005
Williams et al 2005b Turchi et al 2013) In the mutants the shoot apical meristems
are multiplied and large
The development of shoot apical meristems is also regulated by the WUSCHEL
(WUS)ndashCLAVATA (CLV) signaling pathway WUS positively regulates cell
proliferation In contrast CLV3 which is expressed in stem cells limits the WUS
expression and thus inhibits cell proliferation in the SAM (Sharma et al 2003)
Consistent with this notion WUS is expressed to a higher level in jba-1D
explaining the ectopic enlargement of the SAM in the mutant (Williams et al 2005)
While WUS is closely related to miR165166 in the shoot apical meristem
development the underlying molecular mechanisms are currently unclear It has been
observed that the miR166-over producing men1 plants have an arrested meristem
development (Jung amp Park 2007) In addition the heterozygous men1+ and the
homozygous men1 plants show different expression patterns of WUS and CLV3 It
appears that miR166 regulates WUS indirectly and influences the expressional balance
between WUS and CLV3 through a negative feedback loop (Jung amp Park 2007 de
Alba et al 2013)
The phv phb athb15 triple mutant has an enlarged shoot apical meristems the
rev phb phv mutant does not have functional shoot apical meristems (Prigge et al
2005) This distinction may be explained by the fact that while miR166 targets
primarily PHB PHV and ATHB15 miR165 targets all the five HD-ZIP III genes (Zhou
et al 2007) Consistent with this transgenic plants overproducing miR166 are distinct
from those overproducing miR165 The miR165-overproducing transgenic plants lack
functional shoot apical meristem and a pin-like structure is formed at the apical region
of the mutant These phenotypes are quite similar to rev phb phv triple mutant (Zhou et
al 2007 Liu et al 2013) The phenotypic distinction may because by the difference
of the cleavage efficiencies of the target mRNAs In accordance with this notion a few
35S miR166a transgenic lines exhibit no shoot apical meristems The men1
homozygous plants also show arrested meristem development (Jung amp Park 2007)
Chapter 1 Introduction and Review of Literature
46
MiR164 cleaves the CUC1 and CUC2 mRNAs as well as the NAC1 mRNA
CUC1 and CUC2 determines the organ boundary in the meristem Phenotypes of
transgenic plants that overproduce miR164 are similar to those of the cuc1cuc2 double
mutant that exhibits embryonic developmental defects such as cup-shaped cotyledons
and fused cotyledons (Laufs et al 2004) When a miR164-resistant CUC2 is
expressed the boundary domain is enlarged CUC also plays a role in embryonic shoot
meristem formation by regulating the SHOOT MERISTEMLESS gene It was therefore
concluded that miR164-mediated cleavage of CUC1 and CUC2 mRNAs determines the
sizes of the boundary domains in the shoot apical meristem and regulates separation of
the organs by restricting cell proliferation (Laufs et al 2004 de Alba et al 2013)
11364 Leaf development
The role of miR319 has been extensively studied in leaf development A dominant
jaw-D mutant overproducing miR319 shows uneven curved leaves with enlarged size
and five TCP genes which are closely related to the CINCINNATA (CIN) gene in
Antirrhinum majus are down regulated in the mutant suggesting that miR319 mediated
regulation of the TCP transcription factor genes are important for the final step of
leaf shaping (Palatnik et al 2003 Naz et al 2013) In addition to leaf shape and size
leaf polarity is also specified by miRNA MiR166-mediated cleavage of the HD-ZIPIII
mRNAs is a well to establish leaf polarity This discovery originates from the gain-of-
function mutants such as phb-1d and phv-1d wherein point mutations in the
complementary sites to miRNA helps the mRNA to evade or resist from miRNA-
mediated cleavage (McConnell et al 2001 Emery et al 2003 Naz et al 2013)
Plant leaves have a dorso-ventral polarity consisting of the adaxial (upper
surface) and abaxial (lower surface) sides The adaxial side is closer to the shoot
apical meristem Adaxial identity is specified by functionally redundant class III HD-
ZIP family genes such as PHB PHV and REV Ectopic expression of each genes
result in adaxialized leaves Dominant phb-1d and phv-1d mutants exhibit
transformation of the abaxial to adaxial fate and have rod-shaped leaves In contrast
miR165166-overproducing plants show pin-like cotyledons radicalized leaves and
abaxialized leaves (Chitwood et al 2007 Pattanayak et al 2013)
Leaf asymmetry is determined by miR166-mediated restriction of the
expression domains of the HD-ZIPIII genes The HD-ZIPIII genes are expressed in the
Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
9
The last two steps of the caffeine synthesis are also catalyzed by a SAM-
dependent N-methyltransferase(s) which is different from the N-methyltransferase
enzyme that catalyzes the first step in the pathway Native N-methyltransferase
activities have been detected in crude and partially purified extracts from tea and
coffee plants (Ashihara amp Suzuki 2004) and a highly purified preparation has been
obtained from young tea leaves (Kato et al 1999) The enzyme was assigned the
name caffeine synthase (EC 211160) that catalyses the last two steps of caffeine
biosynthesis viz the conversion of 7-methylxanthine to caffeine via theobromine
(Figure 12) The gene encoding caffeine synthase was first cloned from young tea
leaves (Kato et al 2000) Since then several genes encoding N-methyltransferases
with different substrate specificity have been reported
Activity of the recombinant theobromine synthase (CTS1 and CaMXMT EC
211159) is specific for the conversion of 7-methylxanthine to theobromine In
contrast the recombinant caffeine synthases (CCS1 and CaDMXMT1) can utilize
paraxanthine theobromine and 7-methylxanthine as shown with tea caffeine synthase
(TCS1) (Mohanpuria et al 2011) Although paraxanthine is the most active substrate
of this recombinant enzyme only limited amounts of paraxanthine are synthesized in
coffee cells hence in vivo caffeine synthase is principally involved in the conversion
of 7-methylxanthine to caffeine via theobromine
Radiolabelled tracer experiments with theobromine accumulating cacao and
Chinese tea (Camellia ptilophylla) showed a limited conversion of theobromine to
caffeine N-methyltransferase which catalyzed the conversion of 7-methylxanthine to
theobromine was detected in crude extracts from C ptilophylla but conversion of
theobromine to caffeine was not observed (Ashihara et al 1998) Genes homologous
to caffeine synthase have been isolated from several theobromine accumulating plants
including cacao (Yoneyama et al 2006) The recombinant enzymes derived from
these genes have 3 N-methyltransferase activity suggesting that these theobromine
accumulating plants contain specific theobromine synthase Caffeine does not occur
in C ptilophylla (Ashihara et al 1998) although it is present in leaves and fruits of
cacao along with theobromine (Koyama et al 2003 Zheng amp Ashihara 2004)
Chapter 1 Introduction and Review of Literature
10
Figure 12 Pathways for the biosynthesis of caffeine
The major pathway is indicated by solid lines (Mizuno et al 2003b Uefuji et al 2003) Alternative pathways in coffee and tea plants are indicated
by dashed lines(Kato et al 1996 Schulthess et al 1996) Enzymes (1) 7-methylxanthosine synthase (2) N-methylnucleosidase (3) Theobromine
synthase (4) Caffeine synthase
N
N
N
N7
H3C
3
1
O
O
Caffeine
N
NH
N
N7
3
O
O
Theobromine
N
N
N
N
H
H
7
O
O
N
N
N
N
H
H
O
O
N
N
N
N
H
H
O
O
Xanthosine
N
N
N
N
H
H
O
O
N
N
N
N
H
H
O
ON
N
N
N
H
H3C
71
O
O
Paraxanthine
CH3
CH3
CH3
CH3CH3
7-methylxanthine
Rib P
Rib
(1)
(4)
Rib
+
CH3
7-methyl
xanthosine
(2)
(3)
SAM SAH
SAM SAH SAM SAH
Xanthosine mono
phosphate
Rib P
7-methyl xanthosine
monophosphate
+
CH3 CH3
Inosine 5-mono
phosphate
Adenosine 5-mono
phosphate
SAM SAH
Guanosine
Guanosine 5-mono
phosphate
Chapter 1 Introduction and Review of Literature
11
181 Source of Xanthosine for caffeine biosynthesis
Xanthosine the initial substrate of purine alkaloid synthesis is supplied by at least four
different pathways de novo purine biosynthesis (de novo route) the degradation
pathways of adenine nucleotides (AMP route) and guanine nucleotides (GMP route)
and the S-adenosyl-L-methionine (SAM) cycle (SAM route) (Figure 13) Purine
nucleotides are synthesized by de novo and salvage pathways (Ashihara amp Crozier
1999 Stasolla et al 2003 Zrenner et al 2006) The de novo synthesis of non purine
precursors like CO2 10-formyltetrahydrofolate 5-phosphoribosyl-1-pyrophosphate and
the amino acids Glycine glutamine and aspartate results in the production of purine
nucleotides
Figure 13 Xanthosine biosynthesis Xanthosine for purine alkaloid biosynthesis is supplied by at
least four routes from adenosine released from the SAM cycle (SAM route) from IMP
originating from de novo purine synthesis (de novo route) from the cellular adenine
nucleotide pool (AMP route) and from the guanine nucleotide pool (GMP route)
Adapted from (Ashihara et al 2008)
The utilization of IMP for caffeine biosynthetic pathway which is formed by the
de novo purine biosynthetic pathway for caffeine biosynthetic pathways was
demonstrated in young tea leaves using 15
N-glycine and 14
C-labelled precursors and
inhibitors of de novo purine biosynthesis (Ito amp Ashihara 1999) Xanthosine is formed
by an IMP rarr XMP rarr xanthosine pathway IMP dehydrogenase (EC 111205) and 5prime-
nucleotidase (EC 3135) catalyze these reactions The inhibition of IMP dehydrogen-
ase by ribavirin inhibits both caffeine and gunanine nucleotide biosynthesis in tea and
coffee leaves (Keya et al 2003)
Chapter 1 Introduction and Review of Literature
12
The portion of xanthosine used for caffeine biosynthesis is derived from adenine
and guanine nucleotide pools which are produce de novo by salvage pathways there are
several potential pathways for xanthosine synthesis from AMP but the AMP rarr IMP
rarr XMPrarr xanthosine route is predominant All three enzymes involved in the
conversion AMP deaminase (EC 3546) IMP dehydrogenase and 5prime-nucleosidase
have been detected in tea leaves (Koshiishi et al 2001) Xanthosine for caffeine
biosynthesis can also be produced from guanine nucleotides by GMPrarrguanosine
rarrxanthosine pathway Guanosine deaminase (EC 35415) activity is found in cell free
extracts from young tea leaves (Negishi et al 1994) but GMP deaminase activity was
not detected in plants (Stasolla et al 2003 Zrenner et al 2006) The absence of GMP
deaminase activity suggests that a GMP rarr IMP rarr XMP rarr xanthosine route is not
functional in plants
The use of radiolabelled purine bases and nucleosides for the study of caffeine
biosynthetic pathway was facilitated by the fact that exogenous purine nucleotides are
rapidly hydrolyzed by the plant tissues forming purine nucleosides andor bases which
are then utilized in pathways which in most instances indirectly lead to xanthosine
Most of these purine compounds including adenine adenosine inosine hypoxanthine
and guanine are converted to their respective nucleotides by salvage enzymes
including adenine phosphoribosyl transferase (EC 2427) hypoxanthineguanine
phosphoribosyl transferase (EC 2428) adenosine kinase (EC 27120) and
inosineguanosine kinase (EC 2421) which usually have high activities in plant cells
The resultant nucleotides then enter the purine alkaloid biosynthetic pathway In the
case of guanosine it may be directly deaminated to xanthosine and utilized for caffeine
biosynthesis (Figure 13) (Ashihara et al 1997)
182 SAM cycle in plants
SAM is the methyl donor for the three methylation steps in the caffeine biosynthetic
pathway In this process SAM is converted to S-adenosyl-L-homocysteine (SAH)
which in turn is hydrolyzed to homocysteine and adenosine Homocysteine is recycled
via the SAM cycle to replenish SAM levels and adenosine is released from the cycle
Adenosine released from the cycle is converted to AMP directly andor indirectly via
adenine AMP is converted to xanthosine and used for caffeine biosynthesis Since
three moles of SAH are produced via the SAM cycle for each mole of caffeine that is
synthesized in theory this pathway has the capacity to be the sole source of both the
Chapter 1 Introduction and Review of Literature
13
purine skeleton and the methyl groups required for caffeine biosynthesis in young tea
leaves (Koshiishi et al 2001)
Methionine
S-Adenosyl-L-methionineHomocysteine
ATP
PPi+ Pi
Tetrahydrofolate
5-Methyl-
tetrahydrofolate
CH3+Adenosine
S-Adenosyl-L-
homocysteine
(xanthine
structure)(Methyl groups)
Caffeine
(1)
(2)(3)
(4)
Figure 14 The SAM cycle (activated methyl cycle) in plants (Ashihara amp Suzuki 2004)
Enzymes SAM synthetase SAM-dependent N-methyltransferases S-adenosyl-
homocysteine (SAH) hydrolase Methionine synthase Adenosine released from the cycle
is salvaged to adenine nucleotides and utilized both for purine structure of caffeine via
xanthosine and for re-synthesis of SAM via ATP
183 N-methyltransferase in caffeine biosynthesis Molecular cloning
The genes encoding for the enzymes involved in the caffeine biosynthesis were
successfully cloned from the coffee family (Ogawa et al 2001 Mizuno et al 2003a
Mizuno et al 2003b Uefuji et al 2003) The conserved amino acid regions of tea CS
(AB31280) the sequences of O-methyltransferases derived from plant origin and
Arabidopsis hypothetical proteins were made use of in designing primers for RT-PCR
and RACE technique Independent groups have isolated N-methyltransferase (NMT)
genes from coffee which have been classified based on the substrate specificity of the
recombinant proteins expressed in Ecoli (Table 15) PCR products thus obtained were
used as the probe to isolate full length cDNA from the library resulting in the
identification of all three N-methyltransferases involved in caffeine biosynthesis
(Figure 12) Several cDNA have been characterized from the coffee plants one for
xanthosine methyltransferase (XMTXRS) three for 7-methylxanthine methyltransferase
or theobromine synthase (MXMTCTS) and three for 37-dimethylxanthine
methyltransferase or caffeine synthase (DXMTCCS) (Table 15) (Kato amp Mizuno
2004)
A single gene encoding for xanthosine methyltransferase (XMTCmXRS1) was
identified which encoded for a polypeptide consisting of 372 amino acids with an
apparent molecular mass of 418kDa It is expressed almost uniformly in aerial tissues
Chapter 1 Introduction and Review of Literature
14
of C arabica including leaves floral buds and immature but not in mature beans
Three genes encoding 7-methylxanthine methyltransferase (theobromine synthase) have
been identified The number of amino acids in the putative polypeptides are 378 for
MXMT1 (427kDa) and 384 for MXMT2 and CTS2 (434kDa) They differ by insertion
or deletion of blocks of several residues in the C-terminal region Their catalytic
properties based on kinetic parameters such as Km values were different They are
expressed in young leaves floral buds and immature but not mature beans Three genes
were identified for 3 7-dimethylxanthine methyltransferase (caffeine synthase) DXMT
CCS1 and CtCS7 each encoding a 43 kDa polypeptide consisting of 384 amino acids
However their kinetic properties differ from each other such as DXMT and CCS1
showed Km values of 1200 and 157microM for theobromine respectively Expressions
profiles are also distinct DXMT being expressed exclusively in immature beans while
CCS1 expression is ubiquitous occurring in all tissues The presence of isoforms of
these enzymes with different properties suggests that caffeine is synthesized through
multiple pathways depending on availability and concentration of the substrate (Mizuno
et al 2001 Ogawa et al 2001 Mizuno et al 2003a Mizuno et al 2003b Uefuji et
al 2003) Genomic clones of theobromine synthase were obtained by PCR- RFLP
from C canephora (Satyanarayana et al 2005 Satyanarayana 2006) The genomic
clones are reported to have highly conserved exons and a variable third intron The
promoter for the theobromine synthase was cloned and characterized (Satyanarayana et
al 2005)
There is high degree of similarity in the coding region of coffee NMT genes
isolated so far though differences exist in the 3prime untranslated region (UTR) for these
genes The deduced amino acid sequence of these enzymes shows more than 85
homology between these genes Phylogenetic analysis indicates that they are more
closely related to C-methyltransferases including those for jasmonic acid salicylic acid
and benzoic acid than to other N-methyltransferases This suggests that coffee N-methyl
transferases belong to a distinct sub-group within plant methyltransferases Their
cellular localization determined by green fluorescent protein fusion method and β-
glucourodinase (GUS) showed that the all the three genes are localized in the cytoplasm
(Ogawa et al 2001 Vinod et al 2007) It was also shown that these were present
along with chlorogenic acid(Vinod et al 2007)
Chapter 1 Introduction and Review of Literature
15
Table 15 N-methyltransferases and their encoding genes involved in caffeine biosynthesis
Gene (Enzyme) Gene name Accession No Description Reference
Xanthosine methyltransferase
( 7-methylxanthosine synthase) (EC 211158)
CaXMT AB 048793 Immature fruit cDNA Screening (Uefuji et al 2003)
CmXRS1 AB 034699 Leaf cDNA library RACE (Mizuno et al 2003a)
CaMXMT1 AB 048794 Leaf cDNA library RACE (Ogawa et al 2001)
7-methylxanthine synthase transferase
(theobromine synthase) ( EC 211159)
CTS1 AB 034700 Immature fruit cDNA Screening (Uefuji et al 2003)
CaMXMT2 AB 984125 Leaf cDNA library Screening (Mizuno et al 2001)
CTS2 AB 054841 Leaf cDNA library Screening (Mizuno et al 2001)
37-methylxanthinemethyltransferase
(Caffeine synthase) (EC 211160)
CaDXMT1 AB 086414 Immature fruit cDNA Screening (Uefuji et al 2003)
CCS1 AB 0861414 Immature fruit cDNA Screening (Uefuji et al 2003)
CtCS7 AB 0864415 Immature fruit cDNA
NMT genomic clones
Theobromine synthase-1
CX10 Complete coding region PCR Satyanarayan 2006
CX8 Complete coding region PCR -do-
Theobromine synthase-1 PG-5 Promoter + coding region RAGE -do-
Theobromine synthase-1 PG-1 Promoter + coding region RAGE Satyanarayan 2006
PG-4 Promoter + coding region RAGE -do-
Theobromine synthase-1 NMT
(AY273814) Complete coding region PCR Kochko A et al 2010
Theobromine synthase-2 NMT
(AY362825) Complete coding region PCR Kochko A et al 2010
Chapter 1 Introduction and Review of Literature
16
19 Catabolism of caffeine
Caffeine is produced in young leaves and immature fruits and continues to accumulate
gradually during the maturation of these organs At the same time caffeine is slowly
degraded with the removal of the three methyl groups resulting in the formation of
xanthine Catabolism of caffeine was first reported in coffee leaves (Kalberer 1965)
Since then a number of tracer experiments using 14
C labelled purine alkaloids have
been reported (Suzuki amp Waller 1984 Ashihara et al 1996 Vitoria amp Mazzafera
1999 Mazzafera 2004) They demonstrated the major catabolic pathway is caffeine rarr
theophylline rarr 3-methylxanthine rarr xanthine Xanthine is further degraded by the
removal by the conventional purine catabolic pathway to CO2 and NH3 via uric acid
allatonin and allantoate (Figure 15) (Ashihara amp Crozier 1999 Stasolla et al 2003
Zrenner et al 2006) Caffeine catabolism usually begins with its conversion to
theophylline catalyzed by N7-demethylase The involvement of P450-dependent mono-
oxygenase activity for this reaction was suggested (Huber amp Baumann 1998
Mazzafera 2004) However the activity of this enzyme has not yet been determined in
cell free extracts [8-14
C] Theophylline degraded to CO2 at much faster rate than [8-14
C]
caffeine indicating that conversion of caffeine to theophylline is a major rate limiting
step of caffeine catabolism and a possible reason why caffeine accumulates in high
concentrations in tissues of C sinensis and C arabica It is widely believed that the
control of caffeine levels in plants is a function of the balance between rates of
synthesis and degradation and this balance seems to vary depending on the plant
species and the tissue developmental stage (Mazzafera 2004)
Chapter 1 Introduction and Review of Literature
17
Figure 15 Caffeine catabolic pathways Caffeine is mainly catabolised via theophylline and 3-
methylxanthine Xanthine is further degraded to CO2 and NH3 by the conventional
oxidative pathway Adapted from (Ashihara et al 2008)
Chapter 1 Introduction and Review of Literature
18
110 Genetic engineering of Coffee
Genetic transformation has potential applications in coffee agriculture for incorporating
genes which could provide desirable traits such as disease and insect resistance
drought and frost tolerance and herbicide resistance Transgenic technology can also be
used to increase nutritional value and improve cup quality produce varieties with
caffeine-free beans and for production of hybrid crops for molecular farming
Identification of target specific genes is one of the pre-requisites for developing
transgenic crops The availability of a large number of EST sequences in coffee and
initiation of coffee genome sequencing may speed up the gene discovery and accelerate
transgenic research efforts in coffee
1101 Insect Resistance
Production of insect resistant coffee plants is one of the major objectives of the
breeding programs The major pests attacking coffee include coffee berry borer (CBB
Hypothenemus hampei) white stem borer (WSB Xylotrechus quadripes) leaf miner
(Perileucoptera coffeela) and root nematodes (Meloidogyne spp and Pratylenchus
spp) The CBB is present in almost all the coffee growing countries and considered to
the most devastating pest in coffee To date there is no reported source of resistance to
CBB in the coffee gene pool Like CBB WSB is another serious pest in C arabica in
India and several other East Asian countries Both CBB and WSB belong to the order
Coleoptera For India controlling WSB is the biggest challenge and has therefore
become the highest research priority Robusta is generally resistant to WSB but the
interspecific Robusta Arabica hybrids are susceptible to WSB Although leaf miner is
not yet a serious pest in India and other East Asian countries it is an economically
important pest in East Africa and Brazil Effective chemical control of CBB and WSB
is difficult due to the nature of their life cycle inside the berry and stem respectively as
well as environmental concern regarding the use of pesticides Biological control
measures are adapted to combat these insect pests with varying degrees of success
Developing coffee plants resistant to these pests using genetic transformation
technology could be one of the alternative strategies to counter pest damage For insect
resistance several different classes of proteins from bacterial plant and animal sources
have been isolated and their insecticidal properties tested against many important pests
Amongst these proteins Bt toxins are most important and several transgenic crops
expressing Bt genes have been commercialized Coffee transgenic plants carrying a
Chapter 1 Introduction and Review of Literature
19
synthetic version of the cry1Ac gene have been produced (Leroy et al 2000 Mishra et
al 2002) Indeed this was the first report of an important agronomic trait being
introduced into a coffee plant The transgenic plants presented similar features in
growth and development compared to normal plants Transgenic plants highly resistant
to leaf miner under greenhouse conditions were tested under field conditions in French
Guyana for 4 years for pest resistance (Perthius et al 2005 Perthius et al 2006)
From 54 independent transformation events 70 of the events were resistant to leaf
miner Unfortunately the field trial was vandalized which led to the termination of the
experiment (Montagnon et al 2005)The effectiveness of Bt genes in controlling
coleopteran pests is well documented in corn and potato which indicates that Bt genes
might be effective against CBB The high toxicity of B thuringiensis sero var
israelensis against first in star larvae of CBB has been demonstrated (Mendez et al
2003) In another experiment an α-amylase inhibitor from Phaseolus vulgaris was
tested against CBB and found to have an inhibitory effect on its growth and
development (Grossi et al 2004)In addition to the CBB and WSB C arabica
varieties are also susceptible to endoparasitic root-knot nematode (Meloidogyne spp)
(Campos et al 1990 Mishra et al 2012)
So far 15 species have been reported to be parasites of coffee Controlling
nematodes is extremely difficult and currently seedling grafting with robusta rootstock
is followed as one of the control measure Sources of resistance specific to root-knot
nematodes have been identified in coffee trees (Bertrand et al 2001) and the Mex-1
gene conferring resistance to M exigua in C arabica is in the process of isolation (Noir
et al 2003)
1102 Tolerance to Abiotic Stress
In many coffee growing countries abiotic stress such as drought and frost are the major
climatic factors that limit coffee production Changes in climatic patterns due to global
climate change are considered increasingly important for coffee cultivation Drought
induces water stress in plants which affects vegetative growth and vigor and triggers
floral abnormalities and poor fruit set It also indirectly increases the incidence of pests
and diseases in the plants C arabica is generally more tolerant to water stress than C
canephora partly due to its extensive deep root system C racemosa is known to be a
good source of drought tolerance In India hybrids between C canephora and C
racemosa have been obtained and are currently under evaluation for drought tolerance
Chapter 1 Introduction and Review of Literature
20
In many coffee growing countries coffee is propagated in marginal areas where the
annual rainfall is below 1000thinspmm with prolonged dry spells of over 4-5 months In
those areas water shortage and unfavorable temperatures constitute major constraints
and the growth and productivity of C canephora are badly affected As with drought
periodic frost also affects coffee production in parts of Brazil The introduction of
drought and frost tolerant genes through genetic transformation would be of great
importance for alleviating these problems Research is now being carried out by several
groups to identify genes involved in biotic as well as abiotic stress The most promising
genetic engineering approach for drought tolerance includes the use of functional or
regulatory genes as well as the transfer of transcription factors In recent years plants
tolerant to high temperature and water stress have been the subject of intense research
(Chaves et al 2003 Chaves et al 2004 Coraggio et al 2004) For achieving drought
tolerance genes that have been targeted include those encoding enzymes involved in
detoxification or osmotic response metabolism enzymes active in signaling proteins
involved in the transport of metabolites and regulating the plant energy status (Chaves
et al 2003 Chaves et al 2004 Coraggio et al 2004) The dissection of molecular
mechanisms related to signal transduction and transcriptional regulation might help in
engineering drought tolerance in coffee
1103 Disease Resistance
Coffee leaf rust (CLR) caused by the fungus Hemileia vastatrix is the most important
disease in coffee with substantial loss to coffee production and productivity in all the
coffee growing countries In addition to leaf rust coffee berry disease (CBD) caused by
fungus Colletotrichum kahawae can be a devastating anthracnose leading to substantial
crop loss in Africa Several other fungal and bacterial diseases may also affect coffee
to a extent C arabica is more susceptible to many diseases than C canephora Though
most of the disease control measures rely upon chemical control they are more
expensive and labour intensive The long-term solution is the breeding of resistant
varieties which is the focus of many breeding programs However breeding for disease
resistant varieties is time consuming due to the perennial nature of coffee with its long
gestation period India has a long history of C arabica breeding especially for leaf rust
resistance and is the first country to demonstrate the existence of multiple races of leaf
rust Resistance to CLR is conditioned primarily by a number of major (SH) genes and
coffee genotypes are classified into different resistance groups based on their
Chapter 1 Introduction and Review of Literature
21
interaction with different rust races pathogen (van der Vossen 2001) Currently C
canephora provides the main source of resistance to pests and diseases including CLR
(H vastatrix) and CBD (C kahawae) and therefore used in breeding programs Other
diploid species like C liberica and C racemosa are being used as a source of resistance
to coffee leaf rust and coffee leaf miner respectively (Guerreiro et al 1999
Sreenivasan et al 1989) The development of coffee varieties resistant to major fungal
diseases such as CLR and CBD using transgenic technology will benefit the coffee
industry immensely During the last 15 years significant progress has been made in the
area of host-pathogen interactions (Staskawicz et al 1995 Feys et al 2000
Shivaprasad et al 2012) and many resistance genes involved in recognizing invading
pathogens have been identified and cloned (Takken et al 2000) A number of
signalling pathways induced because of pathogen infection have been dissected (Dong
et al 1998 Navarro et al 2006) Many antifungal compounds synthesized by plants to
combat fungal infection have been identified (Does et al 1998) Understanding the
specific induction of targeted pathways and identification of specific pathways
responsible for particular fungal resistance is important in order to employ this strategy
in transgenic technology The recent investigation of gene expression during coffee leaf
rust infection could provide an insight into the defence pathways operating in coffee
(Fernandez et al 2004 Nardi et al 2006) Efforts have been made to identify and
clone resistance genes from coffee for achieving durable resistance The genetic and
physical map of two resistance genes SH3gene conferring resistance to rust (Prakash et
al 2004) and the Ck1 gene conferring resistance to C kahawae CBD (Gichuru et al
2006) have been established These genes could be used for molecular marker assisted
breeding programs (Mishra et al 2012)
1104 Production of low-caffeine coffee
A widespread belief that the ingestion of caffeine can have adverse effects on
health resulted in the increased demand for decaffeinated coffee (Ashihara amp Crozier
2001) Several method were used to obtain decaffeinated plants by conventional
breeding techniques using low yielding varieties and mutants that accumulated
relatively low amounts of caffeine when compared to the commercially available ones
Low-caffeine and decaffeinated coffee represent around 10 of the coffee sales
around the world (Ribas et al 2006b) The industrial process for coffee decaffeination
can be expensive and affects the original flavour and aroma in coffee (David 2002)
Chapter 1 Introduction and Review of Literature
22
Transgenic coffee plants with suppressed caffeine synthesis using RNA interference
(RNAi) technology have been obtained (Ogita et al 2003 Ogita et al 2004) Specific
sequences in the 3prime untranslated region of the theobromine synthase gene (CaMXMT1)
were selected for construction of RNAi short and long fragments The caffeine and
theobromine content of the transgenic plants were reduced by ~ 70 when compared to
the untransformed plants In C canephora RNAi technology has also been employed
to silence the N-methyl transferase gene involved in caffeine biosynthesis (Vinod et al
2007) Promoter of an N-methyltransferase (NMT) gene involved in caffeine
biosynthesis was cloned (Satyanarayana et al 2005 Satyanarayana 2006) which will
be very useful for studying the regulation of caffeine biosynthesis
1105 Improvement in cup quality
Improvement in coffee cup quality requires elaborate knowledge of the chemical
constituents as well as the metabolic pathways involved in the elaboration of quality
The constituents of coffee beans include minerals proteins carbohydrates caffeine
chlorogenic acids (CGA) glycosides lipids and many volatile compounds that give
flavour to coffee by roasting Among these the role of three major constituents
sucrose CGA and trigonelline have been studied in coffee The sucrose content of
coffee bean is associated with coffee flavour the higher the sucrose content in green
beans the more intense will be the cup flavour (Clifford et al 1985 de Maria et al
1996) The sucrose content of C arabica (82-83) is higher than C canephora (33ndash
40) The sucrose amino acids ratio in green beans determines the profile of volatile
compounds Manipulating sucrose content in coffee bean is therefore important in
improving cup quality The sucrose synthase gene (CcSUS2) from C canephora has
been cloned and sequenced (Leroy et al 2005) This provides an opportunity to
manipulate the sucrose content in coffee Chlorogenic acids are products of
phenylpropanoid metabolism Chlorogenic acids are a group of hydroxycinnamoyl
quinic acids (HQA) formed by esterification between caffeic acids coumaric acids and
quinic acids (Clifford et al 1999) They are present in relatively large quantities in the
coffee bean and are the precursor of phenolic compounds in roasted beans C
canephora beans contain higher CGA (10) compared to C arabica beans (6-7)
CGA are known to have antioxidant properties as well as being associated with disease
resistance (Campa et al 2003) Genetic manipulations of genes involved in CGA
synthesis can serve either of these purpose by up- or down regulating the pathway
Chapter 1 Introduction and Review of Literature
23
Phenylalanine ammonia-lyase (PAL) catalyzes the first step of the phenylpropanoid
pathway leading to the synthesis of a wide range of chemical compounds including
flavonoids coumarins hydroxycinnamoyl esters and lignins (Hahlbrock amp Scheel
1989) Recently the full length cDNA and corresponding genomic sequences of PAL
from C canephora was isolated characterized and functionally validated (Mahesh et
al 2006 Parvatam et al 2006) This has opened up new possibilities for manipulating
the level of the PAL enzyme in coffee which in turn can be useful for improvement of
cup quality and manipulating antioxidant properties in coffee
1106 Fruit Ripening
Uniformity during fruit ripening is decisively related to cup quality in coffee and
consequently to the value of the product Fruits at the ideal ripening stage produce the
best organoleptic characteristics for coffee The presence of over ripened or green fruits
changes the acidity the bitterness and consequently the cup quality In order to
maximize uniform ripening of coffee fruits it is essential to control the action of genes
involved in the last step of maturation process Ethylene is known to trigger ripening
and increasing ethylene biosynthesis is associated with various stages of ripening
process (Protasio et al 2005) To control coffee fruit maturation two of the major
genes involved in ethylene biosynthesis namely ACC synthase and ACC oxidase
have been cloned (Protasio et al 2005 Neupane et al 1999) Introduction of the ACC
oxidase gene in antisense orientation have been achieved in both C arabica and C
canephora The effect of the transgene on ethylene production and fruit maturation has
yet to be reported The inhibition of genes downstream to the initial ethylene burst is
also an option to control coffee fruit maturation (Ribas et al 2006)
111 RNA interferenceRNAi
The regulation of gene expression at post transcriptional level has recently attracted
much attention because of the discovery of the phenomenon called RNA interference
(RNAi) RNAi based silencing techniques are more efficient than antisense mediated
gene silencing (Wesley et al 2001) The effect was first observed in plants wherein
silencing was observed when some copies of a transgene was in the forward orientation
with respect to the promoter and some in the reverse orientation This resulted in the
formation of double stranded RNA in the system The phenomenon in which
experimentally introduced double stranded RNA (dsRNA) leads to the loss of
expression of the corresponding cellular gene by sequence specific RNA degradation
Chapter 1 Introduction and Review of Literature
24
was termed as RNA interference or RNAi The protective effect of RNA silencing in
reducing virus infectivity supports the view that PTGS has evolved as a mechanism to
defend plants against virus infection and also to moderate the possible deleterious
genome-restructuring (insertional) activity of virus-like mobile genetic elements eg
retrotransposons A growing body of biochemical and genetic data has further
demonstrated that plants animals and yeasts share related mechanisms of specific
degradation of RNAs in which double-stranded forms of RNA are initiator molecules
(Brenstein et al 2001)
The key insight into the process of PGTS was provided by the experiments
conducted by Hamilton amp Baulcombe (1999) who identified the product of RNA
degradation as small RNA (siRNA) species of ~25nt of both sense and antisense
polarity SiRNA are formed and accumulate as double stranded RNA molecules of
defined chemical structures Both genetic and biochemical approaches were undertaken
to understand the basis of silencing Genetic screens were carried out in fungus
Neurospora carassa the alga Chalmydomonas reinhardtii and plant Arabidopsis
thaliana to search for detective mutants in PTGS
A combination of results obtained from several in vivo and in vitro experiments
have gelled into a two step mechanistic model for RNAiPTGS The first step referred
to as the RNAi initiating step involves binding of the RNA nucleases to a larges
dsRNA and its cleavage into discrete ~21-25nt RNA fragments This is ATP-
dependent reaction which involved RNase III - type endonucleases which make
staggered cuts in both strands of the dsRNA leaving 3prime overhang of 2 nucleotides with
5prime- phosphate 3prime-hydroxyl and no modification in the sugar phosphate backbone
(Hamilton amp Baulcombe 1999 Elbashir et al 2001) SiRNAs are the direct products
of dsRNA cleavage by the multi-domain RNase III enzyme DICER (Figure 16)
(Brenstein et al 2001 Karthikeyan et l 2013)
In the second step or commonly known as the effector step the double stranded
siRNAs produced in the first step bind to an RNAi specific protein complex to form a
RISC The complex undergoes activation in the presence of ATP so that the antisense
component of the unwound siRNA becomes exposed and allows the RISC to perform
the downstream RNAi reaction
Chapter 1 Introduction and Review of Literature
25
Several enzymes are involved in RNAi based on their role genes that encode for them
are classified into RNAi initiators RNAi effectors RNA dependent RNA polymerases
and silencing genes Two C elegans genes rdeI and rde4 (rde stands for lsquo RNAi
deficientrsquo) are believed to be involved in the initiation step of RNAi The C elegans
rde1 gene is a member of a large family of genes and is homologous to the Neurospora
qde2 (qde stands for ldquoquelling deficientrdquo) and Arabidopsis AGO1 (AGO stands for
agronaute AGO1 was previously identified to be involved in Arabidopsis
development) Although the functions of these genes are not clear a mammalian
member of RDE1 family has been identified as a translation initiation factor (Thakur
2005) Important genes for the effector step of PTGS in C elegans are rde2 and mut7
genes These genes were identified from heterozygous mutant worms that were unable
to transmit RNAi to their homozygous offsprings Worms with mutated rde2 or mut 7
genes show defective RNAi mut-7 gene encodes a protein with homology to the
nuclease domains of RNAase D and a protein implicated in Werner syndrome (a rapid
ageing disease in humans) (Grishok et al 2001 Kamath-Loeb et al 2012s)
Neurospora qde-1 Arabidopsis SDE-1SGS-2 and C elegans ego-1 appear to encode
RNA dependent RNA polymerase (RdRPs) It assumed that this is a proof that an RdRp
activity is required for RNAi Certainly the existence of an RdRp might explain the
remarkable efficiency of dsRNA induced silencing if it amplifies either the dsRNA
prior to cleavage or the siRNAs directly (Muers 2013) In C elegans ego-1 mutants
(ego stands for lsquoenhancer of glp-1) RNAi functions normally in somatic cells but is
defective in germline cells where ego-1 is primarily expressed In Arabidopsis SDE-
1SGS-2 mutants (SGS stands for suppressor of gene silencing) siRNA are produced
when dsRNA is introduced via an endogenously replicating RNA virus but not
introduced by a transgene It has been proposed that perhaps the viral RdRP is
substituting for the Arabidopsis enzyme in these mutants Random degradative PCR
model suggests that an RdRP uses the guide strand of an siRNA as a primer for the
target mRNA generating a dsRNA substrate for Dicer and thus more siRNAs
Several genes controlling RNA silencing in plants have been identified through
genetic screens of Arabidopsis mutants impaired in transgene induced RNA silencing
They encode a putative RNA-dependent RNA polymerase (SGS2SDE1) a coiled
protein (SGS3) a protein containing PAZ and Piwi domains (AGO1) and an RNA
helicase (SDE3) The putative SGS2SDE1 of Neurospora is related to QDE-1 and
Chapter 1 Introduction and Review of Literature
26
EGO-1 of C elegans whereas PAZPiwi protein AGO is related to QDE-2 of
Neurospora RDE-1of C elegans RNA helicase SDE3 is related to SMG-2 of C
elegans and Mut-6 of Chlamydomonas MUT-7 gene of C elegans encodes a protein
similar to Rnase D whereas the Drosophila DICER gene encode a protein similar to
RNase III An Arabidopsis ortholog of DICER gene has been identified called CAF
SIN1 SUS1(Thakur 2005 Poethig 2009)
Since the RNAi machinery is present constitutively within the eukaryotic cell it
is important to explore the metabolic advantages that are accorded by the RNAi related
proteins during the intrinsic normal growth of cells and development of organisms The
natural RNAi machinery not only keeps the mobile transposable elements from
disrupting the integrity of the genomes as suggested by the analysis of model organisms
like A thaliana C elegans D melanogaster (Tabara et al 1999 Wu-Scharf et al
2000 Aravin et al 2001 Hamilton et al 2002 Lisch 2009) but also participates in
development of organisms Genetic defects in Celegans RNAi genes ego1 and dicer
cause known specific developmental errors (Grishok et al 2000 Grishok et al 2001
Knight amp Bass 2001 Hall et al 2013) Similarly the Argonaute family of genes of A
thaliana (especially the ZWILLE proteins) is also responsible for plant architecture and
meristem development (Carmell et al 2002 Hamant and Pautot 2010) and the Dicer
homologue of A thaliana CAF1 is required for embryo development (Golden et al
2002 Nakano et al 2011) Thus genetic evidence illustrates the role of the RNAi
machinery as a controller of development related genes The mechanistic details of
these developmental processes are beginning to emerge
Chapter 1 Introduction and Review of Literature
27
Figure 16 Mechanism of gene silencing induced double stranded RNA The dicer enzyme to
produce siRNAs cleaves the RNA The siRNA generated bind to the nuclease complex
RISC The antisense component in the siRNA in the RISC guides the complex towards
the cognate mRNA resulting in endonucleolytic cleavage of the mRNA
Chapter 1 Introduction and Review of Literature
28
112 MicroRNA or MiRNA
In 1991 Ambros and co-workers first isolated a lin-4 mutant of Celegans which was
arrested at the first larval stage (Lee et al 1993) Later on the let-7 mutation was
isolated in the same system which was responsible for development through the fourth
larval stage Both lin-4 and let-7 encode short 22-nucleotide mature RNAs and were
called short temporal RNA because they control the temporal development program of
C elegans The mature lin-4 RNA defines (negatively regulates) the mRNA expression
of the lin-14 and lin-28 hetrochronic genes with the antisense mediated repression
mechanism of translation initiation and thus specifies the fate of cells during the first
three larval stages Recent studies have revealed that the short temporal RNAs are
actually members of a group of tiny RNAs (21to 28 nucleotides) called the micro-
RNAs (miRNA) (Bartel 2009) isolated members of which could easily run to a few
thousand which includes both those were identified computationally and by deep
sequencing Some of the components of the RNAi machinery have also been clearly
established as the effector proteins for the maturation of miRNAs
113 MicroRNAs in plants
1131 Discovery
MicroRNAs have been discovered by three basic approaches Direct cloning forward
genetic direct cloning and bioinformatic prediction followed by experimental
validation The most direct method of miRNA discovery is to isolate and clone small
RNAs from biological samples and several groups have used this approach to identify
plant miRNAs (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002 Xie et al 2003 Sunkar et al 2005 Williams et al 2013) The cloning methods
adapted were similar to those used to identify large numbers of animal miRNAs
(Lagos-Quintana et al 2001 Lau et al 2001 Aravin amp Tuschl 2005) which involve
isolating small RNAs followed by oligonucleotide adapter ligation reverse
transcription amplification and sequencing Some protocols incorporate methods to
enrich for Dicer cleavage products (ie molecules with 5prime-phosphates and 3prime-
hydroxyls) and to concatemerize the short cDNAs so that several may be identified in a
single sequencing read (Lau et al 2001 Llave et al 2002a Jagadeeswaran amp Sunkar
2013)The initial cloning experiments in Arabidopsis identified 19 miRNAs belonging
to 15 families (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002) although hundreds of other small RNAs such as siRNAs and RNA degradation
Chapter 1 Introduction and Review of Literature
29
fragments were also cloned through the direct cloning methods To select miRNAs
from the pool of small RNAs secondary structures of the RNA molecules were
examined in compliance with the acknowledged structural features of miRNAs
(Ambros et al 2003) The secondary structures were validated using several ad-hoc
software tools such as the Mfold or RNAfold programs (Reeder et al 2006) Finally
the existence of mature miRNAs was determined by RNA gel blot hybridization These
direct cloning methods were successful in isolating many miRNAs in Arabidopsis
(Reinhart et al 2002 Sunkar amp Zhu 2004) Rice (Sunkar et al 2005) Poplar (Lu et
al 2005) moss (Arazi et al 2005) and Tobacco (Billoud et al 2005)
Although miRNAs were first discovered through forward genetic screens in
worms (Lee et al 1993 Reinhart et al 2000)only few A thaliana miRNA gene
families have been discovered by this method (Chen 2008) in plants MiRNA
involvement in plant mutant phenotypes were not properly inferred till the cloning
experiments established that plant genomes contain numerous miRNAs (Park et al
2002 Reinhart et al 2002 Rhoades et al 2002) MiRNA loss-of-function allele has
been identified by forward genetic screens early extra petala1 is caused by a
transposon insertion ~160bp upstream of the predicted miR164c stem loop resulting in
flowers with extra petals (Baker et al 2005 Irish 2008) The fact that loss-of-function
miRNA mutants have been rarely discovered using forward genetics reflects small
target size for mutagenesis coupled with redundancy in nearly all evolutionarily
conserved plant miRNAs which are encoded by gene families (Table16) Family
members are likely to have overlapping functions buffering against loss at any single
miRNA locus Over expression screens can circumvent redundancy limitations At least
four plant miRNAs miR319 (also known as miR-JAW) miR172 (also known as EAT)
and miR166 were isolated in over expression screens for dominant mutants with
developmental abnormalities (Aukerman amp Sakai 2003 Palatnik et al 2003 Kim et
al 2005 Williams et al 2005b Schommer et al 2012) Mutations in miRNA target
sites which can prevent the entire family of miRNAs from repressing a target gene can
also circumvent redundant functions of miRNA family members
The dominant mutations in the HD-ZIP genes PHB PHV and REVOLUTA
(REV) in Arabidopsis and ROLLEDLEAF1 (RLD1) in Maize result in adaxialization of
leaves andor Vasculature (McConnell amp Barton 1998 McConnell et al 2001 Nelson
et al 2002 Zhong amp Ye 2004 Allen amp Millar 2012) and are all caused by mutations
Chapter 1 Introduction and Review of Literature
30
in miR166 complementary sites (Rhoades et al 2002 Emery et al 2003 Jones-
Rhoades amp Bartel 2004 Mallory et al 2004 Zhong amp Ye 2004)
In both plants and animals cloning was the initial means of large-scale
miRNA discovery (Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Reinhart et al 2002 Mendes et al 2012) However cloning is biased toward
RNAs that are expressed highly and broadly MiRNAs expressed at low levels or
only in specific cell types or in response to certain environmental stimuli were more
difficult to clone Sequence based biases in cloning procedures might also cause
certain miRNAs to be missed Because of these limitations bioinformatics approaches
to identify miRNAs have provided a useful complement to cloning
A straightforward use of bioinformatics has been carried out to find homologs
of known miRNAs both within the same genome and in the genomes of other species
(Pasquinelli et al 2000 Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Mendes et al 2009) A more difficult challenge is to identify miRNAs
unrelated to previously known miRNAs This was first accomplished for
vertebrate nematode and fly miRNAs using algorithms that search for conservation
of sequence and secondary structure (ie miRNA stem-loop precursors) between
species searching for patterns that are characteristic of miRNAs (Lai et al 2003 Lim
et al 2003 Lim et al 2005)
Most of the methods identified numerous miRNAs but are necessarily more
relaxed in terms of allowed structures Some approaches take advantage of the high
complementarity of plant miRNAs to target messages implementing the requirement
that the candidate has conserved complementarity to mRNAs (Jones-Rhoades amp Bartel
2004 Adai et al 2005) This additional filter has been useful for distinguishing
authentic plant miRNAs from false positives which later extended to mammalian
miRNA gene prediction (Xie et al 2005 Mendes et al 2012)
Another criteria used by most algorithms in the filters is evolutionary
conservation In plants mature miRNA sequences are conserved to a higher degree
than their precursor sequences For example Arabidopsis and rice diverged more than
130 million years ago (Friis et al 2004) but some miRNAs are highly conserved
in these plant species (Reinhart et al 2002) Furthermore various parameters for the
secondary structures such as free energy the number of paired residues within a
Chapter 1 Introduction and Review of Literature
31
miRNA or the number and size of bulges are also used to validate the predictions
(Jones-Rhoades amp Bartel 2004 Wang et al 2004 Adai et al 2005 Mendes et al
2012)
1132 Conserved microRNAs in Plants
Till date cloning genetics and bioinformatics screens have resulted in annotation of
approximately 184 potential Arabidopsis miRNAs (miRBase release 13) These
miRNAs seem to arise through gene duplication and subsequent diversification and
represent approximately 100 families (Maher et al 2006 Axtell 2013) Twenty one
families represented by 92 genes are clearly conserved in species beyond Arabidopsis
(Table 15) The presence of these miRNA families in other sequenced plant genomes
indicates that different plant species have an evolutionarily fluid set of miRNAs
(Willmann amp Poethig 2007) Recent studies on mosses one of the most ancient land
plants have shown that at least 14 miRNA families are conserved in eudicots and
mosses and therefore it appears that plant miRNAs share a common ancient origin
(Arazi et al 2005 Axtell amp Bartel 2005 Talmor-Neiman et al 2006 Zhang et al
2006 Axtell 2013) The number of members per family in a genome ranges from 1-32
with the exception of miR-430 which is represented by a cluster of ~80 loci in zebra
fish (Giraldez et al 2005)
In plants the number of members in each family correlates among examined
species certain families contain numerous members in all three species (eg miR156
miR166 miR169) whereas others consistently contain fewer genes (eg miR162
mir168 miR394) (Table 16) Although it is still unclear why certain miRNA are
encoded by more than one gene in plants (eg 12 genes encoding miR156) this
correlation might suggest functional significance of various miRNA family sizes Most
annotated plant miRNAs are conserved throughout flowering plants while a few
miRNAs are found only in a single sequenced genome and thus they could be
evolutionarily lsquoyoungrsquo miRNAs For example Arabidopsis has several miRNA
families that are not found in poplar or rice suggesting that miRNAs could be
generated continuously during evolution (Allen et al 2004a Rajagopalan et al
2006 Fahlgren et al 2007 Axtell 2012 de Alba et al 2013) Consistent with the
notion that non conserved miRNAs are of a more recent evolutionary origin these
miRNAs are mainly encoded by a single locus in the genome Within each family the
mature miRNA is always located in the same arm
Chapter 1 Introduction and Review of Literature
32
Table 16 MicroRNA families conserved in plants
miRNA
family
Arabidopsis Oryza Populus
miR156 12 12 11 miR159319 6 8 15 miR160 3 6 8 miR162 2 2 3 miR164 3 5 6 miR166 9 12 17 miR167 4 9 8 miR168 2 2 2 miR169 14 17 32 miR171 4 7 10 miR172 5 3 9 miR390 3 1 4 miR393 2 2 4 miR394 2 1 2 miR395 6 19 10 miR396 2 5 7 miR397 2 2 3 miR398 3 2 3 miR399 6 11 12 miR408 1 1 1 miR403 1 0 2 miR437 0 1 0 miR444 0 1 0 miR445 0 9 0 Total 92 127 169
All known miRNA families that are conserved between more than one plant species are listed
together with the number of genes identified in the sequenced genomes Rice miRNA families that have orthologs in maize but do not appear to have orthologs in the eudicots (Arabidopsis and Populus) are marked with an asterisk The following families contain miRNA genes annotated with
more than one number miR156 (miR156 and miR157) miR159319 (miR159 and miR319) miR166 (miR165 and miR166) miR171 (miR170 and miR171) and miR390 (miR390 and miR391)
of the stem loop (5prime- or 3prime) as it would be expected if the genes carry common ancestry
(Figure 17) The sequence of the mature miRNA and to some extent the segment on
the opposite arm of the hairpin to which it pairs is highly conserved between the
members of the same family (both within and between the species) while the sequence
secondary structure and length of the intervening ldquolooprdquo region can be highly divergent
within the same family
The patterns of paring and non pairing nucleotides are often conserved between
homologous miRNA stem loops from different species (Figure 17) The significance of
conserved mismatches is opined to guide DCL1 to cleave at the appropriate positions
along the stem loop
Chapter 1 Introduction and Review of Literature
33
Figure 17 Representative stem loop of miR164 Segments corresponding to the mature miRNAs
are shown in red (Adapted from Rajagopalan et al 2006)
Although many miRNAs are highly conserved in plants the degree of conservation in
most cases is quite low between plants and animals one exception is miR854 A recent
study has revealed that miR854 is conserved between Arabidopsis and animals and that
the Arabidopsis miR854 differs from the human miR854 only by a single nucleotide
(Arteaga-Vazquez et al 2006 Axtell 2012 de Alba et al 2013) The Arabidopsis
miR854 has multiple binding sites in the 3prime-untranslated region (UTR) of
OLIGOURIDYLATE-binding-PROTEIN mRNA 1bA (UBP1b) and represses the
translation of the UBP1b mRNA The animal miR854 is also complementary to a site in
the 3prime-UTR of the UBP1b homolog in animals However it is currently unclear how the
animal miR854 regulates its target mRNA Another distinction between plant and
animal miRNAs is the genomic organization of the miRNA genes While plant miRNA
genes are located predominantly in the intergenic regions animal miRNA genes tend to
localize in the intragenic regions both in the introns or exons of protein coding genes
Moreover miRNA genes are often clustered within a single precursor transcript
whereas individual plant miRNAs are derived from individual precursor RNAs (Bartel
Chapter 1 Introduction and Review of Literature
34
2004 Bonnet et al 2004 Kim 2005 Vaucheret et al 2006 Marco et al 2013)
1133 Non-conserved miRNA and challenges in annotation
Most miRNAs are conserved throughout flowering plants (Table 15) but many more
were found only in a single sequenced genome thus considered to be of a more recent
evolutionary origin The extended homology between few non conserved miRNAs
precursors and their target genes provides strong evidence that these relatively ldquoYoungrdquo
miRNAs arose from the duplication of the target gene segments (Allen et al 2004)
Several non-conserved miRNAs including miR161 miR163 miR173 miR447
miR475 and miR476 are known to direct cleavage of target transcripts (Allen et al
2004a Adai et al 2005 Sunkar et al 2005 de Alba et al 2013) It is difficult to
confidently predict targets for many because it is not possible to use complementary
site as a filter against false-positive target predictions It is also difficult to
confidently annotate non-conserved miRNAs as miRNA rather than siRNAs The
established minimal standard for miRNA annotation is a small RNA with detectable
expression and the potential to form a stem-loop when joined to flanking genomic
sequence (Kasschau et al 2003) In practice these requirements are too loose to
definitively categorize many small RNAs cloned from plants Many plant siRNAs are
detectable on blots (Vazquez et al 2004b) and hundreds of thousands of non-
miRNA genomic sequences can be predicted to fold into secondary structures that
resemble structures of plant miRNA precursors (Jones-Rhoades amp Bartel 2004)
Therefore without conservation of both sequence and secondary structure it is
difficult to be confident that a given cloned RNA originated from a stem-loop (ie is a
miRNA) rather than from a double-stranded RNA (ie is a siRNA) In fact many of
the thousands o f small RNAs cloned from Arabidopsis (Gustafson et al 2005) would
probably meet the literal requirements for annotation as miRNAs A few of these
sequences might be miRNAs but others that meet the literal criteria probably are not
so The challenges of annotating non conserved small RNAs were evidenced by
three related small silencing RNAs that were originally annotated as miRNAs
that turned out to be trans-acting siRNAs (tasiRNAs)(Kanno et al2013)
Although most miRNAs that are broadly conserved among flowering plants are
now being identified and experimentally validated (Table 16)(Jones-Rhoades amp
Bartel 2004) the challenges and ambiguities for classifying non-conserved miRNAs
prevent meaningful estimates of the total number of miRNA genes in Arabidopsis and
Chapter 1 Introduction and Review of Literature
35
other plant genomes It is possible that miRNAs might have escaped detection
because they are non-conserved and expressed in specific tissues or conditions and can
only be identified by using high- throughput sequencing
1134 MicroRNA biogenesis
11341 Transcription of microRNA precursor
Plant miRNAs are primarily found in the genomic regions that are not associated with
the protein coding genes (Reinhart et al 2002) and it appears that most if not all plant
miRNAs are produced from their own transcriptional units Plant miRNA genes are
occasionally clustered near each other in the genome suggesting transcription of
multiple miRNAs from a single primary transcript [eg the miR395 cluster (Grishok
et al 2001 Jones-Rhoades amp Bartel 2004b)] but this polycistronic arrangement of
miRNA genes appears far less frequently in plants than in animals (Bartel 2004)
Northern blot EST and mapping evidence indicate that plant miRNA primary
transcripts (also known as pri-miRNAs) as in animals are longer than needed to
encompass the miRNA stem-loops (Palatnik et al 2003 Jones-Rhoades amp Bartel
2004b) At least some of these pri-miRNA transcripts appear to be spliced
polyadenylated (Kurihara amp Watanabe 2004) and capped (Xie Z et al 2005) Two
rice miRNAs are contained within transcripts that contain exon junctions within the
presumptive stem-loop precursor implying that in these cases splicing is a prerequisite
for Dicer recognition (Sunkar et al 2005) Plant pri-miRNAs are over 1kb in length
and they are usually preceded by typical TATA box motifs They can also undergo
canonical splicing polyadenylation and capping All these observations indicates RNA
polymerase II is probably responsible for transcribing most plant miRNAs (Xie Z et
al 2005) as appears to be the case for many animal miRNAs Relatively little is
known about the regulation of miRNA transcription in plants but there is no reason
to suspect that this regulation would differ from that of protein-coding transcripts as
the bioinformatics analysis of the sequence upstream of the transcriptional start
sites of the miRNA genes showed the presence of putative binding motifs of
number of known transcription factors (Megraw et al 2006 Sethupathy 2013)
11342 MicroRNA processing and export
In plants maturation of miRNA is a step wise process A miRNA gene is transcribed to
pri-miRNA which is much longer than the pre-miRNA possessing the characteristic
stem-loop structure Like transcription of most protein-coding genes the miRNA
Chapter 1 Introduction and Review of Literature
36
transcription is processed by RNA polymerase II (Pol II) enzymes and the pri-
miRNAs are 5prime-capped and 3prime-polyadenylated (Lee et al 2004 Xie Z et al 2005)
Mature miRNAs are produced from pri-miRNAs through at least two sequential
processing steps by RNase III-type endonucleases In animals pri-miRNAs are first
processed at the bottom portion of the stem by Drosha a nuclear-localized RNase III
enzyme to liberate pre-miRNAs The pre-miRNAs are then exported to the cytoplasm
with the help of exportin-5The pre-miRNAs are further processed by Dicer another
RNaseIII enzyme to a duplex consisting of the miRNA and the antisense strands
(miRNA) Since plants do not have any Drosha homologs the two sequential
processing steps seem to be carried out by DCL1 a Dicer homolog in the nucleus
(Kurihara amp Watanabe 2004 de Alba et al 2013 Rogers amp Chen 2013)
The processing of pri-miRNAs to pre-miRNAs by DCL1 is assisted by two
other proteins HYPONASTY LEAVES1 (HYL1) and SERRATE (SE) HYL1 belongs to a
family of double-stranded RNA (dsRNA) binding protein and interacts with DCL1 in
vitro and in vivo (Lu amp Fedoroff 2000 Hiraguri et al 2005 de Alba et al 2013
Rogers amp Chen 2013) Loss-of-function mutations in the HYL1 gene result in
decreased accumulation of miRNAs and concomitant accumulation of pri-miRNAs
(Han et al 2004 Vazquez et al 2004a Kurihara et al 2006) SE a C2H2 zinc finger
protein interacts with DCL1 and HYL1 and plays a role in the processing of pri-
miRNAs to pre-miRNAs (Lobbes et al 2006 Yang L et al 2006) In addition the
nuclear cap-binding complex proteins CBP20 and CBP80 that are known as ABA
HYPER SENSITIVE 1(ABH1) have been shown to be required for proper miRNA
processing (Gregory et al 2008 Laubinger et al 2008) Recently it has been
demonstrated that DCL1 can release 21-nt RNAs from dsRNA as well as from
synthetic miR167 precursor RNAs in vitro However this cleavage is error prone and
inefficient in the absence of HYL1 and SE which accelerate the rate of DCL1-mediated
cleavage and promote accurate processing of miRNAs (Dong et al 2008 Rogers amp
Chen 2013)
11343 Methylation of the miRNAmiRNAduplex
After the miRNAmiRNAduplexes are released from the pre-miRNAs by the DCL1
activities the duplexes are methylated at the 2primeOH of the3prime-ends by HUA ENHANCER1
(HEN1) a small RNA methyltransferase (Yu et al 2005 Yang Z et al 2006 Rogers
amp Chen 2013 ) This step which is distinct from the RNase III-mediated processing
Chapter 1 Introduction and Review of Literature
37
step distinguishes the biogenesis of plant miRNAs from that of animal miRNAs
Mutations in the HEN1 gene were first isolated in a morphological screening for floral
development although the HEN1 mutants display pleiotropic phenotypes similar to the
developmental defects observed in the DCL1 mutants (Chen et al 2002 Park et al
2002 Rogers amp Chen 2013)
Figure 18 Biogenesis of microRNA
A Biogenesis of plant MicroRNA B Biogenesis of metazoananimal MicroRNA
The miRNA (red) is incorporated into RISC and the miRNA(blue) in degraded
This enzyme has two dsRNA-binding domains and nuclear localization signal It
is also conserved in fungi and is required for the processing of siRNAs as well as of
miRNAs (Park et al 2002 Boutet et al 2003 Xie et al 2003) Although the HEN1
protein is localized to the nucleus its methylation activity in the cytoplasm cannot be
ignored (Xie et al 2004 Rogers amp Chen 2013)
Chapter 1 Introduction and Review of Literature
38
The methylation of the miRNAmiRNA duplex is required to protect miRNAs
against the 3prime-end uridylation activity and subsequent degradation (Li et al 2005) In
the hen1 mutants accumulation of miRNAs is reduced to a lower level Also the
miRNAs appear to be heterogeneous in their sizes such that a ladder of bands rather
than a single species is observed for most miRNAs in the mutants (Li et al 2005)
MiRNA cloning and sequencing analyses revealed the presence of a number of U
residues at the 3prime-end of miRNAs in the hen1 mutants Moreover these nucleotides do
not correspond to the miRNAs in the pre-miRNAs indicating that these nucleotides are
added after the processing of the miRNAs from pre-miRNAs (Li et al 2005)
Uridylation of RNAs has also been proven to be correlated with RNA decay (Shen amp
Goodman 2004 Mullen amp Marzluff 2008) suggesting that uridylation attracts an
exonuclease which degrades miRNAs in plants
11344 MiRNA incorporation in to the RISC and its nuclear export
The methylated miRNAmiRNA duplexes undergo RISC assembly In this process
the miRNA of the duplexes is selectively incorporated into the RISC and the miRNA
appears to be removed and subsequently degraded (Hammond et al 2000 Hutvagner
amp Zamore 2002 Schwarz et al 2003 de Alba et al 2013 Rogers amp Chen 2013)
The ARGONAUTE 1(AGO1) proteins are core components of the RISC complex
They contain two structural domains the N-terminal PAZ domain that binds to the 3prime-
end of single-stranded RNAs and the C-terminal piwi domains that has a structure
similar to that of RNase H (Cerutti et al 2000 Parker et al 2004) Several AGO
proteins possess endonucleolytic activities within the PIWI domain and cleave target
mRNAs in the middle of the site complementary to miRNA (Liu et al 2004 Meister et
al 2004 Miyoshi et al 2005) Mutations in the AGO1 gene cause miRNA target
genes to be ectopically expressed and the levels of some miRNAs are reduced in the
mutants (Kidner amp Martienssen 2004 Vaucheret et al 2004 Kidner amp Martienssen
2005 Ronemus et al 2006) Moreover the phenotype of the AGO1 hypomorphic
alleles display defects in lateral organ polarity and leaf and flower morphology as
observed in the dcl1 mutants and null AGO1 alleles are embryonically lethal
supporting the close relationship between the AGO proteins and miRNA processing
(Kidner amp Martienssen 2004 Vaucheret et al 2004 de Alba et al 2013 Rogers amp
Chen 2013)
The production of miRNA miRNA duplexes occur in the nucleus while
Chapter 1 Introduction and Review of Literature
39
miRNA-RISC complexes are present in the cytoplasm where target mRNAs are
located This discordance of functional sites requires an export step during miRNA
biogenesis HASTY the Arabidopsis homolog of exportin-5 is thought to be involved
in miRNA nuclear export (Bollman et al 2003 Park et al 2005)The hasty mutant
exhibits pleiotropic defects in plant development and reduced accumulation of most
miRNAs as in the DCL1 or AGO1 mutants Howeverit is unclear whether the HASTY
proteins export the miRNA miRNA duplexes or the miRNA-RISC complexes in
plants
11345 Feedback regulation of miRNA biogenesis
MiRNA biogenesis is under feedback regulation such that the DCL1 and AGO1 genes
are themselves regulated by miRNAs DCL1 is itself a target of miR162 which leads to
the cleavage of the DCL1 mRNA (Xie et al 2003 de Alba et al 2013) Consistent
with this the levels of DCL1 mRNA are elevated in the mutants of miRNA processing
genes in which the miR162 abundance is reduced In addition there is a predicted
hairpin structure of miR838 in the 14th
intron of the DCL1 primary transcript
(Rajagopalan et al 2006) This prediction implies that DCL1-mediated processing
on this site can result in the cleavage of the DCL1 primary transcript into two truncated
fragments the N-terminal capped fragment and the C-terminal polyadenylated
fragment If this is the case it is possible that miRNA biogenesis competes with the
splicing event of the DCL1 pre-mRNA
The AGO1 is also regulated by miRNA There is a complementary site for
miR168 binding in the AGO1 mRNA and the transcript level of the AGO1 mRNA is
reduced through an mRNA cleavage mechanism (Vaucheret et al 2006 de Alba et al
2013 Rogers amp Chen 2013) indicating that the AGO1 mRNA levels are tightly
controlled by the levels of the AGO1 protein
1135 Mechanism of miRNA action
11351 MiRNA-directed mRNA cleavage
By forming the miRNA-RISC assembly miRNAs can direct the RISC to regulate gene
expression by two post transcriptional mechanisms mRNA cleavage or translational
repression (Bartel 2004 Jones-Rhoades et al 2006 Dalmay 2013) The degree
of miRNA-mRNA complementarity plays an important role for the
determination of the mechanism to be used A general rule is that while perfect or
Chapter 1 Introduction and Review of Literature
40
near-perfect complementarity induces mRNA cleavage central mismatches trigger
translational repression (Carrington amp Ambros 2003 Jones-Rhoades amp Bartel 2004)
Unlike most animal miRNAs plant miRNAs have near-perfect or perfect
complementarity to their targets and mRNA cleavage is assumed to be a predominant
mechanism by which miRNAs regulate gene expression in plants
In RNA cleavage mechanism miRNAs guide the AGO component of RISC to
cleave a single phosphodiester bond of the target mRNA within the miRNA-binding
site (Llave et al 2002b Tang et al 2003) The truncated fragments are then released
and degraded freeing the RISC to recognize and cleave another target mRNA This has
been validated by the detection of 3prime cleavage products that have 5prime ends that start at the
middle of the complementary regions The mRNA cleavage products are then further
degraded by other mechanisms The 3prime cleavage products of some miRNA targets are
degraded by the 5prime-3prime exonuclease XRN4 an Arabidopsis homolog of the major Yeast
mRNA degrading exonuclease Xrn1p It has been observed that the 3prime cleavage
products of some target mRNAs were accumulated in the xrn4 mutants (Souret et al
2004 Dalmay 2013) However the 3prime cleavage products of other miRNA targets do
not accumulate in the xrn4 mutants indicating that there are also XRN4 independent
mechanisms The 5prime cleavage products are typically untraceable by RNA gel blot
hybridization but can be detected by sensitive PCR based methods It has been found
that the 5prime cleavage products tend to obtain an oligo U tail at the 3prime ends This 3prime U tail
is correlated with shortening of the 5prime cleavage products from the 5prime end leading to the
conclusion that the 3prime uridylation causes 5 prime- 3 prime exonucleolytic decay of the 5prime cleavage
products (Shen amp Goodman 2004)
DCL1 HYL1 and SE interact with each other and are co-localized in the
nuclear bodies termed Dicing bodies or D-bodies in which some pri-miRNA shave
been found suggesting that D-bodies serve as a center for active miRNA processing
(Fang amp Spector 2007 Fujioka et al 2007 Song et al 2007) The miRNA
processing site appears to be distinct from the Cajal bodies that have previously been
found as a siRNA processing site (Pontes amp Pikaard 2008) The D-bodies include
SmD3 and SmB proteins that are found in the Cajal bodies However At collin an
additional marker for the Cajal bodies is not found in the D-bodies Moreover the D-
bodies are present in an At collin mutant lacking the Cajal bodies The pri-miRNAs of
miR171A and miR159A genes have been found to be co-localized with HYL1 and
Chapter 1 Introduction and Review of Literature
41
DCL1 in the D- bodies that are distinct from the Cajal bodies (Song et al 2007)
11352 MiRNA-directed translational repression
Translation repression is another mechanism exerted by plant miRNAs The regulatory
mechanism of miR172 which regulates flowering time and floral organ identity is
consistent with translation repression MiR172 over production does not affect the
transcript levels of APETALA2 (AP2) and AP2- like target mRNAs Instead the AP2
protein level is reduced in the miR172 over expressing mutant (Aukerman amp Sakai
2003 Chen 2004 Dalmay 2013) It was later shown that miR172 can also direct the
cleavage of the target mRNAs although the steady-state mRNA level is unchanged due
to feedback regulation (Schwab et al 2005 Jung et al 2007) It is clear that miR172
regulates its target genes primarily by translational repression rather than mRNA
cleavage Recently it has been reported that miR156157 and miR854 also regulate
their target genes through translational repression (Arteaga-Vazquez et al 2006
Gandikota et al 2007 Dalmay 2013) It was once thought that translational repression
is an exception to the rule However experimental evidence suggest a widespread
coexistence of translational repression and mRNA cleavage (Brodersen et al 2008)
1136 Regulatory roles of miRNAs in plant development
The miRNA target prediction has been a difficult task in order to obtain regulatory
targets without bringing in too many false predictions Plant growth and developmental
processes regulated by miRNAs are quite diverse and enormous Furthermore plant
miRNAs work cooperatively or antagonistically to establish a balanced regulation This
notion is well consistent with the findings that mutations in the regulators of miRNA
biogenesis transport and processing such as AGO1 DCL1 HYL1 HEN1 ABH1
and HASTY cause pleiotropic phenotypes (Mallory amp Vaucheret 2006 de Alba et al
2013) To date the roles of miRNAs have been mostly deduced from obtaining miRNA
over expressing transgenic plants or gain-of-function mutants in which miRNA-
resistant target genes are ectopically expressed
11361 Phase transition
Plants pass through a series of developmental phase transitions during their life span
beginning with seed germination continuing through vegetative phase change
reproductive phase change flowering initiation and finally seed production for the next
generation Because of their importance in understanding plant developmental
Chapter 1 Introduction and Review of Literature
42
processes genes regulating developmental transitions have been extensively studied
and underlying molecular mechanisms have been explored in various plant species
Majority of researches were mainly focused on vegetative phase change and
reproductive transition in Arabidopsis and Maize (Poethig 2003 Baurle amp Dean
2006) Particularly the mutations of the genes encoding various regulators of miRNA
biogenesis and transport such as HST SE and AGO exhibit altered juvenile to adult
vegetative phase change and vegetative to reproductive change (Hunter et al 2003
Peragine et al 2004 Vazquez et al 2004a Jones-Rhoades et al 2006 de Alba et al
2013)
Over-expression of the miR156a gene causes late flowering and delays
vegetative phase change (Wu amp Poethig 2006 Gandikota et al 2007 Wang et al
2008) It has been shown that miR156 negatively regulates a group of genes encoding
SQUAMOSA PROMOTER-BINDING PROTEIN-LIKE (Lewis et al 2007 de Alba et
al 2013 Rogers amp Chen 2013) transcription factors such as SPL3 SPL4 and SPL5
that promote flowering initiation In contrast transgenic plants over expressing
miR156-resistant SPL345 genes are early flowering Interestingly the endogenous
level of miR156 is temporally regulated In the early juvenile vegetative phase the
level of miR156 is very high but decreases rapidly before the onset of the adult juvenile
phase (Wu amp Poethig 2006)
The Arabidopsis early activation tagged (eat-D) mutant exhibits early flowering
with disrupted floral structure The mutant phenotypes are caused by over expression of
the miR172a-2gene (Aukerman amp Sakai 2003) MiR172 regulates a small group of
genes encoding APETALLA2 (AP2)-like transcription factors such as TARGET OF
EAT1 (TOE1) TOE2 SCHLAFMUTZE (SMZ) and SCHNARCHZAPFEN(SNZ)
TOE1 TOE2 SMZ and SNZ have been shown to participate in their productive phase
change by repressing a floral integrator FLOWERING LOCUS T (FT)(Jung et al
2007) Consistent with this toe1-2D 35STOE2 35SSMZ and 35SSNZ plants
exhibit late flowering (Jung et al 2007) MiR172 is also regulated temporally and
highly expressed in the adult vegetative phase both in Arabidopsis and maize (Chuck et
al 2007) Because the temporal expression patterns of miR156 and miR172 are
complementary to each other it has been suggested that the two miRNAs interact with
each other in regulating phase change (Chuck et al 2009 de Alba et al 2013) In
support of this view it has been shown that miR172 is down-regulated in the
Chapter 1 Introduction and Review of Literature
43
Corngrass1(Cg1) mutant in which miR156 is overproduced (Chuck et al 2007 de
Alba et al 2013) However there is no direct evidence for the direct linkage between
miR156 and miR172 It is also envisioned that miR156 and miR172 would not be
directly related For example miR156 regulates plastochron index that is the inverse of
leaf initiation rate and thus frequently used to analyze temporal patterns of plant
development In contrast miR172 does not affect plastochron (Wang et al 2008) The
down-regulation of miR172 in the maize miR156-over producing mutants would be
due to the prolonged juvenile vegetative phase in the mutants
Another miRNA that regulates flowering initiation is miR159 It regulates
MYB33 and MYB65 which are closely related to the barley GAMY B transcription
factor that responds to gibberellic acid (GA) The level of miR159 is elevated in GA-
treated seedlings but reduced in the GA biosynthetic ga1 mutant Transgenic plants
over producing miR159 exhibit delayed floral induction under short days (Achard et
al 2004) It is been suggested that MYB33 mediates GA signal to regulate LEAFY
(LFY) These observations indicate that miR159 plays an important role in the GA
signaling pathways that regulate proper floral induction under short days (Achard et al
2004)
11362 Floral development
Arabidopsis flowers consist of four distinct organs They arise in a characteristic
pattern within concentric rings called whorls from outside to inside four sepals four
petals six stamens and the central pistil consisting of two carpels Identities of the
floral organs are determined by three classes of regulatory genes classA classB and
class C genes (Krizek amp Fletcher 2005) The A and C class genes act antagonistically
to restrict their expression domains (Bowman et al 1991)
It is notable that miR172 also regulates floral architecture in addition to its role
in the timing of flowering initiation MiR172 inhibits the translation of the AP2 mRNA
(Chen 2004) Transgenic plants overproducing miR172 not only exhibit early
flowering but also show abnormal floral development which is quite similar to those
of the class A gene mutants such as the ap2 mutants (Aukerman amp Sakai 2003 de
Alba et al 2013) When the activity of the class A genes is inactivated that of the class
C genes such as AGAMOUS (AG) is elevated As a result the miR172- overproducing
transgenic plants exhibit no petals along with homeotic transformation of the sepals to
Chapter 1 Introduction and Review of Literature
44
the carpels (Aukerman amp Sakai 2003 Chen 2004)
Interestingly miR166 also regulates floral architecture The role of miR165166
in the regulation of meristem activity is closely linked to floral meristem formation and
organization (Zhao et al 2007 Miyashima et al 2013) Floral structure is severely
disrupted in the miR165166-overproducing mutants such as men1 and jba-1D in
which miR166 is overproduced (Williams et al 2005b Jung amp Park 2007) The men1
and jba-1D mutants have smaller gynoecium and reduced carpel number In an extreme
case the jba-1D homozygous plants lack visible carpels (Williams et al 2005b)
Similar phenotypes have been observed in the miR165 overproducers (Zhao et al
2007) It has been suggested that the phenotypes are possibly caused by altered AG
expression in the floral meristem (Jung amp Park 2007 Turchi et al 2013)
Other miRNAs also affect floral development Overproduction of miRNAs
involved in auxin signaling also affects floral architecture Transgenic plants
overproducing miR167 have defects in the formation of ovule and anther with reduced
fertility (Ru et al 2006) It has been shown that miR167 targets ARF6 and ARF8 that
play a role in gynoecium and stamen development and in regulating fertility (Ru et al
2006 Wu et al 2006) These observations suggest that auxin signals are also closely
related to floral organogenesis In addition transgenic plants over-producing miR164
and the cuc1cuc2 double mutants show fused sepals and reduced petals (Laufs et al
2004 Turchi et al 2013) suggesting that miR164 is also related to floral meristem
activity and boundary specification of the meristematic region
11363 Shoot apical meristem development
Shoot apical meristem comprises self-maintaining totipotent cells The shoot apical
meristem activity affects diverse aspects of plant developmental processes including
leaf development vascular development and lateral organ polarity (Bowman et al
2002) Because miR165166 plays a primary role in meristem formation
overproduction of miR165166 and multiple knockout mutants of its target genes
exhibit pleiotropic pheonotypes (McConnell et al 2001 Emery et al 2003 Kim et al
2005 Williams et al 2005b)
The targets of miR165166 include genes encoding the homeo domain-leucine
zipper (HD-ZIP) class III transcription factors such as PHABULOSA(PHB)
PHAVOLUTA (PHV) REVOLUTA(REV) ATHB15CORONA (CNA) and ATHB8
Chapter 1 Introduction and Review of Literature
45
PHB PHV and ATHB15 have overlapping functions in controlling meristem
development (Turchi et al 2013) Indeed the phb phv athb15 triple mutants have an
enlarged shoot apical meristem Consistent with this the miR166- overproducing men1
and jba-1D mutants exhibit enlarged shoot apical meristems (Kim et al 2005
Williams et al 2005b Turchi et al 2013) In the mutants the shoot apical meristems
are multiplied and large
The development of shoot apical meristems is also regulated by the WUSCHEL
(WUS)ndashCLAVATA (CLV) signaling pathway WUS positively regulates cell
proliferation In contrast CLV3 which is expressed in stem cells limits the WUS
expression and thus inhibits cell proliferation in the SAM (Sharma et al 2003)
Consistent with this notion WUS is expressed to a higher level in jba-1D
explaining the ectopic enlargement of the SAM in the mutant (Williams et al 2005)
While WUS is closely related to miR165166 in the shoot apical meristem
development the underlying molecular mechanisms are currently unclear It has been
observed that the miR166-over producing men1 plants have an arrested meristem
development (Jung amp Park 2007) In addition the heterozygous men1+ and the
homozygous men1 plants show different expression patterns of WUS and CLV3 It
appears that miR166 regulates WUS indirectly and influences the expressional balance
between WUS and CLV3 through a negative feedback loop (Jung amp Park 2007 de
Alba et al 2013)
The phv phb athb15 triple mutant has an enlarged shoot apical meristems the
rev phb phv mutant does not have functional shoot apical meristems (Prigge et al
2005) This distinction may be explained by the fact that while miR166 targets
primarily PHB PHV and ATHB15 miR165 targets all the five HD-ZIP III genes (Zhou
et al 2007) Consistent with this transgenic plants overproducing miR166 are distinct
from those overproducing miR165 The miR165-overproducing transgenic plants lack
functional shoot apical meristem and a pin-like structure is formed at the apical region
of the mutant These phenotypes are quite similar to rev phb phv triple mutant (Zhou et
al 2007 Liu et al 2013) The phenotypic distinction may because by the difference
of the cleavage efficiencies of the target mRNAs In accordance with this notion a few
35S miR166a transgenic lines exhibit no shoot apical meristems The men1
homozygous plants also show arrested meristem development (Jung amp Park 2007)
Chapter 1 Introduction and Review of Literature
46
MiR164 cleaves the CUC1 and CUC2 mRNAs as well as the NAC1 mRNA
CUC1 and CUC2 determines the organ boundary in the meristem Phenotypes of
transgenic plants that overproduce miR164 are similar to those of the cuc1cuc2 double
mutant that exhibits embryonic developmental defects such as cup-shaped cotyledons
and fused cotyledons (Laufs et al 2004) When a miR164-resistant CUC2 is
expressed the boundary domain is enlarged CUC also plays a role in embryonic shoot
meristem formation by regulating the SHOOT MERISTEMLESS gene It was therefore
concluded that miR164-mediated cleavage of CUC1 and CUC2 mRNAs determines the
sizes of the boundary domains in the shoot apical meristem and regulates separation of
the organs by restricting cell proliferation (Laufs et al 2004 de Alba et al 2013)
11364 Leaf development
The role of miR319 has been extensively studied in leaf development A dominant
jaw-D mutant overproducing miR319 shows uneven curved leaves with enlarged size
and five TCP genes which are closely related to the CINCINNATA (CIN) gene in
Antirrhinum majus are down regulated in the mutant suggesting that miR319 mediated
regulation of the TCP transcription factor genes are important for the final step of
leaf shaping (Palatnik et al 2003 Naz et al 2013) In addition to leaf shape and size
leaf polarity is also specified by miRNA MiR166-mediated cleavage of the HD-ZIPIII
mRNAs is a well to establish leaf polarity This discovery originates from the gain-of-
function mutants such as phb-1d and phv-1d wherein point mutations in the
complementary sites to miRNA helps the mRNA to evade or resist from miRNA-
mediated cleavage (McConnell et al 2001 Emery et al 2003 Naz et al 2013)
Plant leaves have a dorso-ventral polarity consisting of the adaxial (upper
surface) and abaxial (lower surface) sides The adaxial side is closer to the shoot
apical meristem Adaxial identity is specified by functionally redundant class III HD-
ZIP family genes such as PHB PHV and REV Ectopic expression of each genes
result in adaxialized leaves Dominant phb-1d and phv-1d mutants exhibit
transformation of the abaxial to adaxial fate and have rod-shaped leaves In contrast
miR165166-overproducing plants show pin-like cotyledons radicalized leaves and
abaxialized leaves (Chitwood et al 2007 Pattanayak et al 2013)
Leaf asymmetry is determined by miR166-mediated restriction of the
expression domains of the HD-ZIPIII genes The HD-ZIPIII genes are expressed in the
Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora
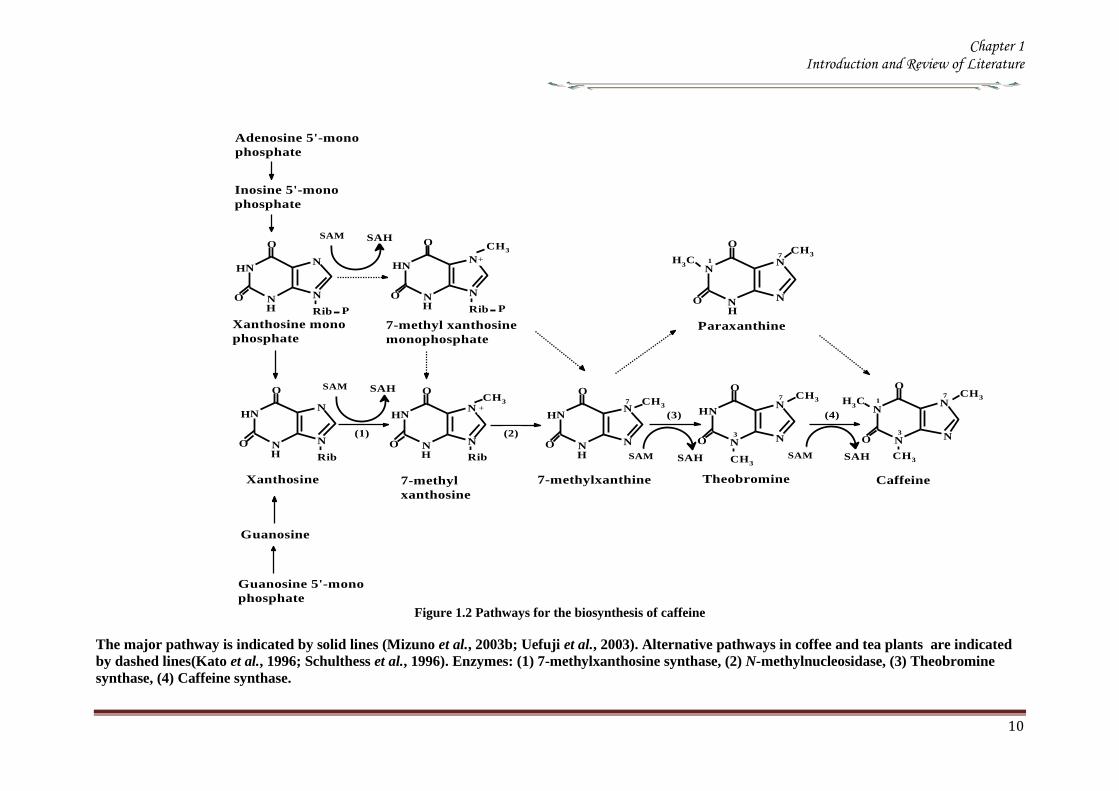
Chapter 1 Introduction and Review of Literature
10
Figure 12 Pathways for the biosynthesis of caffeine
The major pathway is indicated by solid lines (Mizuno et al 2003b Uefuji et al 2003) Alternative pathways in coffee and tea plants are indicated
by dashed lines(Kato et al 1996 Schulthess et al 1996) Enzymes (1) 7-methylxanthosine synthase (2) N-methylnucleosidase (3) Theobromine
synthase (4) Caffeine synthase
N
N
N
N7
H3C
3
1
O
O
Caffeine
N
NH
N
N7
3
O
O
Theobromine
N
N
N
N
H
H
7
O
O
N
N
N
N
H
H
O
O
N
N
N
N
H
H
O
O
Xanthosine
N
N
N
N
H
H
O
O
N
N
N
N
H
H
O
ON
N
N
N
H
H3C
71
O
O
Paraxanthine
CH3
CH3
CH3
CH3CH3
7-methylxanthine
Rib P
Rib
(1)
(4)
Rib
+
CH3
7-methyl
xanthosine
(2)
(3)
SAM SAH
SAM SAH SAM SAH
Xanthosine mono
phosphate
Rib P
7-methyl xanthosine
monophosphate
+
CH3 CH3
Inosine 5-mono
phosphate
Adenosine 5-mono
phosphate
SAM SAH
Guanosine
Guanosine 5-mono
phosphate
Chapter 1 Introduction and Review of Literature
11
181 Source of Xanthosine for caffeine biosynthesis
Xanthosine the initial substrate of purine alkaloid synthesis is supplied by at least four
different pathways de novo purine biosynthesis (de novo route) the degradation
pathways of adenine nucleotides (AMP route) and guanine nucleotides (GMP route)
and the S-adenosyl-L-methionine (SAM) cycle (SAM route) (Figure 13) Purine
nucleotides are synthesized by de novo and salvage pathways (Ashihara amp Crozier
1999 Stasolla et al 2003 Zrenner et al 2006) The de novo synthesis of non purine
precursors like CO2 10-formyltetrahydrofolate 5-phosphoribosyl-1-pyrophosphate and
the amino acids Glycine glutamine and aspartate results in the production of purine
nucleotides
Figure 13 Xanthosine biosynthesis Xanthosine for purine alkaloid biosynthesis is supplied by at
least four routes from adenosine released from the SAM cycle (SAM route) from IMP
originating from de novo purine synthesis (de novo route) from the cellular adenine
nucleotide pool (AMP route) and from the guanine nucleotide pool (GMP route)
Adapted from (Ashihara et al 2008)
The utilization of IMP for caffeine biosynthetic pathway which is formed by the
de novo purine biosynthetic pathway for caffeine biosynthetic pathways was
demonstrated in young tea leaves using 15
N-glycine and 14
C-labelled precursors and
inhibitors of de novo purine biosynthesis (Ito amp Ashihara 1999) Xanthosine is formed
by an IMP rarr XMP rarr xanthosine pathway IMP dehydrogenase (EC 111205) and 5prime-
nucleotidase (EC 3135) catalyze these reactions The inhibition of IMP dehydrogen-
ase by ribavirin inhibits both caffeine and gunanine nucleotide biosynthesis in tea and
coffee leaves (Keya et al 2003)
Chapter 1 Introduction and Review of Literature
12
The portion of xanthosine used for caffeine biosynthesis is derived from adenine
and guanine nucleotide pools which are produce de novo by salvage pathways there are
several potential pathways for xanthosine synthesis from AMP but the AMP rarr IMP
rarr XMPrarr xanthosine route is predominant All three enzymes involved in the
conversion AMP deaminase (EC 3546) IMP dehydrogenase and 5prime-nucleosidase
have been detected in tea leaves (Koshiishi et al 2001) Xanthosine for caffeine
biosynthesis can also be produced from guanine nucleotides by GMPrarrguanosine
rarrxanthosine pathway Guanosine deaminase (EC 35415) activity is found in cell free
extracts from young tea leaves (Negishi et al 1994) but GMP deaminase activity was
not detected in plants (Stasolla et al 2003 Zrenner et al 2006) The absence of GMP
deaminase activity suggests that a GMP rarr IMP rarr XMP rarr xanthosine route is not
functional in plants
The use of radiolabelled purine bases and nucleosides for the study of caffeine
biosynthetic pathway was facilitated by the fact that exogenous purine nucleotides are
rapidly hydrolyzed by the plant tissues forming purine nucleosides andor bases which
are then utilized in pathways which in most instances indirectly lead to xanthosine
Most of these purine compounds including adenine adenosine inosine hypoxanthine
and guanine are converted to their respective nucleotides by salvage enzymes
including adenine phosphoribosyl transferase (EC 2427) hypoxanthineguanine
phosphoribosyl transferase (EC 2428) adenosine kinase (EC 27120) and
inosineguanosine kinase (EC 2421) which usually have high activities in plant cells
The resultant nucleotides then enter the purine alkaloid biosynthetic pathway In the
case of guanosine it may be directly deaminated to xanthosine and utilized for caffeine
biosynthesis (Figure 13) (Ashihara et al 1997)
182 SAM cycle in plants
SAM is the methyl donor for the three methylation steps in the caffeine biosynthetic
pathway In this process SAM is converted to S-adenosyl-L-homocysteine (SAH)
which in turn is hydrolyzed to homocysteine and adenosine Homocysteine is recycled
via the SAM cycle to replenish SAM levels and adenosine is released from the cycle
Adenosine released from the cycle is converted to AMP directly andor indirectly via
adenine AMP is converted to xanthosine and used for caffeine biosynthesis Since
three moles of SAH are produced via the SAM cycle for each mole of caffeine that is
synthesized in theory this pathway has the capacity to be the sole source of both the
Chapter 1 Introduction and Review of Literature
13
purine skeleton and the methyl groups required for caffeine biosynthesis in young tea
leaves (Koshiishi et al 2001)
Methionine
S-Adenosyl-L-methionineHomocysteine
ATP
PPi+ Pi
Tetrahydrofolate
5-Methyl-
tetrahydrofolate
CH3+Adenosine
S-Adenosyl-L-
homocysteine
(xanthine
structure)(Methyl groups)
Caffeine
(1)
(2)(3)
(4)
Figure 14 The SAM cycle (activated methyl cycle) in plants (Ashihara amp Suzuki 2004)
Enzymes SAM synthetase SAM-dependent N-methyltransferases S-adenosyl-
homocysteine (SAH) hydrolase Methionine synthase Adenosine released from the cycle
is salvaged to adenine nucleotides and utilized both for purine structure of caffeine via
xanthosine and for re-synthesis of SAM via ATP
183 N-methyltransferase in caffeine biosynthesis Molecular cloning
The genes encoding for the enzymes involved in the caffeine biosynthesis were
successfully cloned from the coffee family (Ogawa et al 2001 Mizuno et al 2003a
Mizuno et al 2003b Uefuji et al 2003) The conserved amino acid regions of tea CS
(AB31280) the sequences of O-methyltransferases derived from plant origin and
Arabidopsis hypothetical proteins were made use of in designing primers for RT-PCR
and RACE technique Independent groups have isolated N-methyltransferase (NMT)
genes from coffee which have been classified based on the substrate specificity of the
recombinant proteins expressed in Ecoli (Table 15) PCR products thus obtained were
used as the probe to isolate full length cDNA from the library resulting in the
identification of all three N-methyltransferases involved in caffeine biosynthesis
(Figure 12) Several cDNA have been characterized from the coffee plants one for
xanthosine methyltransferase (XMTXRS) three for 7-methylxanthine methyltransferase
or theobromine synthase (MXMTCTS) and three for 37-dimethylxanthine
methyltransferase or caffeine synthase (DXMTCCS) (Table 15) (Kato amp Mizuno
2004)
A single gene encoding for xanthosine methyltransferase (XMTCmXRS1) was
identified which encoded for a polypeptide consisting of 372 amino acids with an
apparent molecular mass of 418kDa It is expressed almost uniformly in aerial tissues
Chapter 1 Introduction and Review of Literature
14
of C arabica including leaves floral buds and immature but not in mature beans
Three genes encoding 7-methylxanthine methyltransferase (theobromine synthase) have
been identified The number of amino acids in the putative polypeptides are 378 for
MXMT1 (427kDa) and 384 for MXMT2 and CTS2 (434kDa) They differ by insertion
or deletion of blocks of several residues in the C-terminal region Their catalytic
properties based on kinetic parameters such as Km values were different They are
expressed in young leaves floral buds and immature but not mature beans Three genes
were identified for 3 7-dimethylxanthine methyltransferase (caffeine synthase) DXMT
CCS1 and CtCS7 each encoding a 43 kDa polypeptide consisting of 384 amino acids
However their kinetic properties differ from each other such as DXMT and CCS1
showed Km values of 1200 and 157microM for theobromine respectively Expressions
profiles are also distinct DXMT being expressed exclusively in immature beans while
CCS1 expression is ubiquitous occurring in all tissues The presence of isoforms of
these enzymes with different properties suggests that caffeine is synthesized through
multiple pathways depending on availability and concentration of the substrate (Mizuno
et al 2001 Ogawa et al 2001 Mizuno et al 2003a Mizuno et al 2003b Uefuji et
al 2003) Genomic clones of theobromine synthase were obtained by PCR- RFLP
from C canephora (Satyanarayana et al 2005 Satyanarayana 2006) The genomic
clones are reported to have highly conserved exons and a variable third intron The
promoter for the theobromine synthase was cloned and characterized (Satyanarayana et
al 2005)
There is high degree of similarity in the coding region of coffee NMT genes
isolated so far though differences exist in the 3prime untranslated region (UTR) for these
genes The deduced amino acid sequence of these enzymes shows more than 85
homology between these genes Phylogenetic analysis indicates that they are more
closely related to C-methyltransferases including those for jasmonic acid salicylic acid
and benzoic acid than to other N-methyltransferases This suggests that coffee N-methyl
transferases belong to a distinct sub-group within plant methyltransferases Their
cellular localization determined by green fluorescent protein fusion method and β-
glucourodinase (GUS) showed that the all the three genes are localized in the cytoplasm
(Ogawa et al 2001 Vinod et al 2007) It was also shown that these were present
along with chlorogenic acid(Vinod et al 2007)
Chapter 1 Introduction and Review of Literature
15
Table 15 N-methyltransferases and their encoding genes involved in caffeine biosynthesis
Gene (Enzyme) Gene name Accession No Description Reference
Xanthosine methyltransferase
( 7-methylxanthosine synthase) (EC 211158)
CaXMT AB 048793 Immature fruit cDNA Screening (Uefuji et al 2003)
CmXRS1 AB 034699 Leaf cDNA library RACE (Mizuno et al 2003a)
CaMXMT1 AB 048794 Leaf cDNA library RACE (Ogawa et al 2001)
7-methylxanthine synthase transferase
(theobromine synthase) ( EC 211159)
CTS1 AB 034700 Immature fruit cDNA Screening (Uefuji et al 2003)
CaMXMT2 AB 984125 Leaf cDNA library Screening (Mizuno et al 2001)
CTS2 AB 054841 Leaf cDNA library Screening (Mizuno et al 2001)
37-methylxanthinemethyltransferase
(Caffeine synthase) (EC 211160)
CaDXMT1 AB 086414 Immature fruit cDNA Screening (Uefuji et al 2003)
CCS1 AB 0861414 Immature fruit cDNA Screening (Uefuji et al 2003)
CtCS7 AB 0864415 Immature fruit cDNA
NMT genomic clones
Theobromine synthase-1
CX10 Complete coding region PCR Satyanarayan 2006
CX8 Complete coding region PCR -do-
Theobromine synthase-1 PG-5 Promoter + coding region RAGE -do-
Theobromine synthase-1 PG-1 Promoter + coding region RAGE Satyanarayan 2006
PG-4 Promoter + coding region RAGE -do-
Theobromine synthase-1 NMT
(AY273814) Complete coding region PCR Kochko A et al 2010
Theobromine synthase-2 NMT
(AY362825) Complete coding region PCR Kochko A et al 2010
Chapter 1 Introduction and Review of Literature
16
19 Catabolism of caffeine
Caffeine is produced in young leaves and immature fruits and continues to accumulate
gradually during the maturation of these organs At the same time caffeine is slowly
degraded with the removal of the three methyl groups resulting in the formation of
xanthine Catabolism of caffeine was first reported in coffee leaves (Kalberer 1965)
Since then a number of tracer experiments using 14
C labelled purine alkaloids have
been reported (Suzuki amp Waller 1984 Ashihara et al 1996 Vitoria amp Mazzafera
1999 Mazzafera 2004) They demonstrated the major catabolic pathway is caffeine rarr
theophylline rarr 3-methylxanthine rarr xanthine Xanthine is further degraded by the
removal by the conventional purine catabolic pathway to CO2 and NH3 via uric acid
allatonin and allantoate (Figure 15) (Ashihara amp Crozier 1999 Stasolla et al 2003
Zrenner et al 2006) Caffeine catabolism usually begins with its conversion to
theophylline catalyzed by N7-demethylase The involvement of P450-dependent mono-
oxygenase activity for this reaction was suggested (Huber amp Baumann 1998
Mazzafera 2004) However the activity of this enzyme has not yet been determined in
cell free extracts [8-14
C] Theophylline degraded to CO2 at much faster rate than [8-14
C]
caffeine indicating that conversion of caffeine to theophylline is a major rate limiting
step of caffeine catabolism and a possible reason why caffeine accumulates in high
concentrations in tissues of C sinensis and C arabica It is widely believed that the
control of caffeine levels in plants is a function of the balance between rates of
synthesis and degradation and this balance seems to vary depending on the plant
species and the tissue developmental stage (Mazzafera 2004)
Chapter 1 Introduction and Review of Literature
17
Figure 15 Caffeine catabolic pathways Caffeine is mainly catabolised via theophylline and 3-
methylxanthine Xanthine is further degraded to CO2 and NH3 by the conventional
oxidative pathway Adapted from (Ashihara et al 2008)
Chapter 1 Introduction and Review of Literature
18
110 Genetic engineering of Coffee
Genetic transformation has potential applications in coffee agriculture for incorporating
genes which could provide desirable traits such as disease and insect resistance
drought and frost tolerance and herbicide resistance Transgenic technology can also be
used to increase nutritional value and improve cup quality produce varieties with
caffeine-free beans and for production of hybrid crops for molecular farming
Identification of target specific genes is one of the pre-requisites for developing
transgenic crops The availability of a large number of EST sequences in coffee and
initiation of coffee genome sequencing may speed up the gene discovery and accelerate
transgenic research efforts in coffee
1101 Insect Resistance
Production of insect resistant coffee plants is one of the major objectives of the
breeding programs The major pests attacking coffee include coffee berry borer (CBB
Hypothenemus hampei) white stem borer (WSB Xylotrechus quadripes) leaf miner
(Perileucoptera coffeela) and root nematodes (Meloidogyne spp and Pratylenchus
spp) The CBB is present in almost all the coffee growing countries and considered to
the most devastating pest in coffee To date there is no reported source of resistance to
CBB in the coffee gene pool Like CBB WSB is another serious pest in C arabica in
India and several other East Asian countries Both CBB and WSB belong to the order
Coleoptera For India controlling WSB is the biggest challenge and has therefore
become the highest research priority Robusta is generally resistant to WSB but the
interspecific Robusta Arabica hybrids are susceptible to WSB Although leaf miner is
not yet a serious pest in India and other East Asian countries it is an economically
important pest in East Africa and Brazil Effective chemical control of CBB and WSB
is difficult due to the nature of their life cycle inside the berry and stem respectively as
well as environmental concern regarding the use of pesticides Biological control
measures are adapted to combat these insect pests with varying degrees of success
Developing coffee plants resistant to these pests using genetic transformation
technology could be one of the alternative strategies to counter pest damage For insect
resistance several different classes of proteins from bacterial plant and animal sources
have been isolated and their insecticidal properties tested against many important pests
Amongst these proteins Bt toxins are most important and several transgenic crops
expressing Bt genes have been commercialized Coffee transgenic plants carrying a
Chapter 1 Introduction and Review of Literature
19
synthetic version of the cry1Ac gene have been produced (Leroy et al 2000 Mishra et
al 2002) Indeed this was the first report of an important agronomic trait being
introduced into a coffee plant The transgenic plants presented similar features in
growth and development compared to normal plants Transgenic plants highly resistant
to leaf miner under greenhouse conditions were tested under field conditions in French
Guyana for 4 years for pest resistance (Perthius et al 2005 Perthius et al 2006)
From 54 independent transformation events 70 of the events were resistant to leaf
miner Unfortunately the field trial was vandalized which led to the termination of the
experiment (Montagnon et al 2005)The effectiveness of Bt genes in controlling
coleopteran pests is well documented in corn and potato which indicates that Bt genes
might be effective against CBB The high toxicity of B thuringiensis sero var
israelensis against first in star larvae of CBB has been demonstrated (Mendez et al
2003) In another experiment an α-amylase inhibitor from Phaseolus vulgaris was
tested against CBB and found to have an inhibitory effect on its growth and
development (Grossi et al 2004)In addition to the CBB and WSB C arabica
varieties are also susceptible to endoparasitic root-knot nematode (Meloidogyne spp)
(Campos et al 1990 Mishra et al 2012)
So far 15 species have been reported to be parasites of coffee Controlling
nematodes is extremely difficult and currently seedling grafting with robusta rootstock
is followed as one of the control measure Sources of resistance specific to root-knot
nematodes have been identified in coffee trees (Bertrand et al 2001) and the Mex-1
gene conferring resistance to M exigua in C arabica is in the process of isolation (Noir
et al 2003)
1102 Tolerance to Abiotic Stress
In many coffee growing countries abiotic stress such as drought and frost are the major
climatic factors that limit coffee production Changes in climatic patterns due to global
climate change are considered increasingly important for coffee cultivation Drought
induces water stress in plants which affects vegetative growth and vigor and triggers
floral abnormalities and poor fruit set It also indirectly increases the incidence of pests
and diseases in the plants C arabica is generally more tolerant to water stress than C
canephora partly due to its extensive deep root system C racemosa is known to be a
good source of drought tolerance In India hybrids between C canephora and C
racemosa have been obtained and are currently under evaluation for drought tolerance
Chapter 1 Introduction and Review of Literature
20
In many coffee growing countries coffee is propagated in marginal areas where the
annual rainfall is below 1000thinspmm with prolonged dry spells of over 4-5 months In
those areas water shortage and unfavorable temperatures constitute major constraints
and the growth and productivity of C canephora are badly affected As with drought
periodic frost also affects coffee production in parts of Brazil The introduction of
drought and frost tolerant genes through genetic transformation would be of great
importance for alleviating these problems Research is now being carried out by several
groups to identify genes involved in biotic as well as abiotic stress The most promising
genetic engineering approach for drought tolerance includes the use of functional or
regulatory genes as well as the transfer of transcription factors In recent years plants
tolerant to high temperature and water stress have been the subject of intense research
(Chaves et al 2003 Chaves et al 2004 Coraggio et al 2004) For achieving drought
tolerance genes that have been targeted include those encoding enzymes involved in
detoxification or osmotic response metabolism enzymes active in signaling proteins
involved in the transport of metabolites and regulating the plant energy status (Chaves
et al 2003 Chaves et al 2004 Coraggio et al 2004) The dissection of molecular
mechanisms related to signal transduction and transcriptional regulation might help in
engineering drought tolerance in coffee
1103 Disease Resistance
Coffee leaf rust (CLR) caused by the fungus Hemileia vastatrix is the most important
disease in coffee with substantial loss to coffee production and productivity in all the
coffee growing countries In addition to leaf rust coffee berry disease (CBD) caused by
fungus Colletotrichum kahawae can be a devastating anthracnose leading to substantial
crop loss in Africa Several other fungal and bacterial diseases may also affect coffee
to a extent C arabica is more susceptible to many diseases than C canephora Though
most of the disease control measures rely upon chemical control they are more
expensive and labour intensive The long-term solution is the breeding of resistant
varieties which is the focus of many breeding programs However breeding for disease
resistant varieties is time consuming due to the perennial nature of coffee with its long
gestation period India has a long history of C arabica breeding especially for leaf rust
resistance and is the first country to demonstrate the existence of multiple races of leaf
rust Resistance to CLR is conditioned primarily by a number of major (SH) genes and
coffee genotypes are classified into different resistance groups based on their
Chapter 1 Introduction and Review of Literature
21
interaction with different rust races pathogen (van der Vossen 2001) Currently C
canephora provides the main source of resistance to pests and diseases including CLR
(H vastatrix) and CBD (C kahawae) and therefore used in breeding programs Other
diploid species like C liberica and C racemosa are being used as a source of resistance
to coffee leaf rust and coffee leaf miner respectively (Guerreiro et al 1999
Sreenivasan et al 1989) The development of coffee varieties resistant to major fungal
diseases such as CLR and CBD using transgenic technology will benefit the coffee
industry immensely During the last 15 years significant progress has been made in the
area of host-pathogen interactions (Staskawicz et al 1995 Feys et al 2000
Shivaprasad et al 2012) and many resistance genes involved in recognizing invading
pathogens have been identified and cloned (Takken et al 2000) A number of
signalling pathways induced because of pathogen infection have been dissected (Dong
et al 1998 Navarro et al 2006) Many antifungal compounds synthesized by plants to
combat fungal infection have been identified (Does et al 1998) Understanding the
specific induction of targeted pathways and identification of specific pathways
responsible for particular fungal resistance is important in order to employ this strategy
in transgenic technology The recent investigation of gene expression during coffee leaf
rust infection could provide an insight into the defence pathways operating in coffee
(Fernandez et al 2004 Nardi et al 2006) Efforts have been made to identify and
clone resistance genes from coffee for achieving durable resistance The genetic and
physical map of two resistance genes SH3gene conferring resistance to rust (Prakash et
al 2004) and the Ck1 gene conferring resistance to C kahawae CBD (Gichuru et al
2006) have been established These genes could be used for molecular marker assisted
breeding programs (Mishra et al 2012)
1104 Production of low-caffeine coffee
A widespread belief that the ingestion of caffeine can have adverse effects on
health resulted in the increased demand for decaffeinated coffee (Ashihara amp Crozier
2001) Several method were used to obtain decaffeinated plants by conventional
breeding techniques using low yielding varieties and mutants that accumulated
relatively low amounts of caffeine when compared to the commercially available ones
Low-caffeine and decaffeinated coffee represent around 10 of the coffee sales
around the world (Ribas et al 2006b) The industrial process for coffee decaffeination
can be expensive and affects the original flavour and aroma in coffee (David 2002)
Chapter 1 Introduction and Review of Literature
22
Transgenic coffee plants with suppressed caffeine synthesis using RNA interference
(RNAi) technology have been obtained (Ogita et al 2003 Ogita et al 2004) Specific
sequences in the 3prime untranslated region of the theobromine synthase gene (CaMXMT1)
were selected for construction of RNAi short and long fragments The caffeine and
theobromine content of the transgenic plants were reduced by ~ 70 when compared to
the untransformed plants In C canephora RNAi technology has also been employed
to silence the N-methyl transferase gene involved in caffeine biosynthesis (Vinod et al
2007) Promoter of an N-methyltransferase (NMT) gene involved in caffeine
biosynthesis was cloned (Satyanarayana et al 2005 Satyanarayana 2006) which will
be very useful for studying the regulation of caffeine biosynthesis
1105 Improvement in cup quality
Improvement in coffee cup quality requires elaborate knowledge of the chemical
constituents as well as the metabolic pathways involved in the elaboration of quality
The constituents of coffee beans include minerals proteins carbohydrates caffeine
chlorogenic acids (CGA) glycosides lipids and many volatile compounds that give
flavour to coffee by roasting Among these the role of three major constituents
sucrose CGA and trigonelline have been studied in coffee The sucrose content of
coffee bean is associated with coffee flavour the higher the sucrose content in green
beans the more intense will be the cup flavour (Clifford et al 1985 de Maria et al
1996) The sucrose content of C arabica (82-83) is higher than C canephora (33ndash
40) The sucrose amino acids ratio in green beans determines the profile of volatile
compounds Manipulating sucrose content in coffee bean is therefore important in
improving cup quality The sucrose synthase gene (CcSUS2) from C canephora has
been cloned and sequenced (Leroy et al 2005) This provides an opportunity to
manipulate the sucrose content in coffee Chlorogenic acids are products of
phenylpropanoid metabolism Chlorogenic acids are a group of hydroxycinnamoyl
quinic acids (HQA) formed by esterification between caffeic acids coumaric acids and
quinic acids (Clifford et al 1999) They are present in relatively large quantities in the
coffee bean and are the precursor of phenolic compounds in roasted beans C
canephora beans contain higher CGA (10) compared to C arabica beans (6-7)
CGA are known to have antioxidant properties as well as being associated with disease
resistance (Campa et al 2003) Genetic manipulations of genes involved in CGA
synthesis can serve either of these purpose by up- or down regulating the pathway
Chapter 1 Introduction and Review of Literature
23
Phenylalanine ammonia-lyase (PAL) catalyzes the first step of the phenylpropanoid
pathway leading to the synthesis of a wide range of chemical compounds including
flavonoids coumarins hydroxycinnamoyl esters and lignins (Hahlbrock amp Scheel
1989) Recently the full length cDNA and corresponding genomic sequences of PAL
from C canephora was isolated characterized and functionally validated (Mahesh et
al 2006 Parvatam et al 2006) This has opened up new possibilities for manipulating
the level of the PAL enzyme in coffee which in turn can be useful for improvement of
cup quality and manipulating antioxidant properties in coffee
1106 Fruit Ripening
Uniformity during fruit ripening is decisively related to cup quality in coffee and
consequently to the value of the product Fruits at the ideal ripening stage produce the
best organoleptic characteristics for coffee The presence of over ripened or green fruits
changes the acidity the bitterness and consequently the cup quality In order to
maximize uniform ripening of coffee fruits it is essential to control the action of genes
involved in the last step of maturation process Ethylene is known to trigger ripening
and increasing ethylene biosynthesis is associated with various stages of ripening
process (Protasio et al 2005) To control coffee fruit maturation two of the major
genes involved in ethylene biosynthesis namely ACC synthase and ACC oxidase
have been cloned (Protasio et al 2005 Neupane et al 1999) Introduction of the ACC
oxidase gene in antisense orientation have been achieved in both C arabica and C
canephora The effect of the transgene on ethylene production and fruit maturation has
yet to be reported The inhibition of genes downstream to the initial ethylene burst is
also an option to control coffee fruit maturation (Ribas et al 2006)
111 RNA interferenceRNAi
The regulation of gene expression at post transcriptional level has recently attracted
much attention because of the discovery of the phenomenon called RNA interference
(RNAi) RNAi based silencing techniques are more efficient than antisense mediated
gene silencing (Wesley et al 2001) The effect was first observed in plants wherein
silencing was observed when some copies of a transgene was in the forward orientation
with respect to the promoter and some in the reverse orientation This resulted in the
formation of double stranded RNA in the system The phenomenon in which
experimentally introduced double stranded RNA (dsRNA) leads to the loss of
expression of the corresponding cellular gene by sequence specific RNA degradation
Chapter 1 Introduction and Review of Literature
24
was termed as RNA interference or RNAi The protective effect of RNA silencing in
reducing virus infectivity supports the view that PTGS has evolved as a mechanism to
defend plants against virus infection and also to moderate the possible deleterious
genome-restructuring (insertional) activity of virus-like mobile genetic elements eg
retrotransposons A growing body of biochemical and genetic data has further
demonstrated that plants animals and yeasts share related mechanisms of specific
degradation of RNAs in which double-stranded forms of RNA are initiator molecules
(Brenstein et al 2001)
The key insight into the process of PGTS was provided by the experiments
conducted by Hamilton amp Baulcombe (1999) who identified the product of RNA
degradation as small RNA (siRNA) species of ~25nt of both sense and antisense
polarity SiRNA are formed and accumulate as double stranded RNA molecules of
defined chemical structures Both genetic and biochemical approaches were undertaken
to understand the basis of silencing Genetic screens were carried out in fungus
Neurospora carassa the alga Chalmydomonas reinhardtii and plant Arabidopsis
thaliana to search for detective mutants in PTGS
A combination of results obtained from several in vivo and in vitro experiments
have gelled into a two step mechanistic model for RNAiPTGS The first step referred
to as the RNAi initiating step involves binding of the RNA nucleases to a larges
dsRNA and its cleavage into discrete ~21-25nt RNA fragments This is ATP-
dependent reaction which involved RNase III - type endonucleases which make
staggered cuts in both strands of the dsRNA leaving 3prime overhang of 2 nucleotides with
5prime- phosphate 3prime-hydroxyl and no modification in the sugar phosphate backbone
(Hamilton amp Baulcombe 1999 Elbashir et al 2001) SiRNAs are the direct products
of dsRNA cleavage by the multi-domain RNase III enzyme DICER (Figure 16)
(Brenstein et al 2001 Karthikeyan et l 2013)
In the second step or commonly known as the effector step the double stranded
siRNAs produced in the first step bind to an RNAi specific protein complex to form a
RISC The complex undergoes activation in the presence of ATP so that the antisense
component of the unwound siRNA becomes exposed and allows the RISC to perform
the downstream RNAi reaction
Chapter 1 Introduction and Review of Literature
25
Several enzymes are involved in RNAi based on their role genes that encode for them
are classified into RNAi initiators RNAi effectors RNA dependent RNA polymerases
and silencing genes Two C elegans genes rdeI and rde4 (rde stands for lsquo RNAi
deficientrsquo) are believed to be involved in the initiation step of RNAi The C elegans
rde1 gene is a member of a large family of genes and is homologous to the Neurospora
qde2 (qde stands for ldquoquelling deficientrdquo) and Arabidopsis AGO1 (AGO stands for
agronaute AGO1 was previously identified to be involved in Arabidopsis
development) Although the functions of these genes are not clear a mammalian
member of RDE1 family has been identified as a translation initiation factor (Thakur
2005) Important genes for the effector step of PTGS in C elegans are rde2 and mut7
genes These genes were identified from heterozygous mutant worms that were unable
to transmit RNAi to their homozygous offsprings Worms with mutated rde2 or mut 7
genes show defective RNAi mut-7 gene encodes a protein with homology to the
nuclease domains of RNAase D and a protein implicated in Werner syndrome (a rapid
ageing disease in humans) (Grishok et al 2001 Kamath-Loeb et al 2012s)
Neurospora qde-1 Arabidopsis SDE-1SGS-2 and C elegans ego-1 appear to encode
RNA dependent RNA polymerase (RdRPs) It assumed that this is a proof that an RdRp
activity is required for RNAi Certainly the existence of an RdRp might explain the
remarkable efficiency of dsRNA induced silencing if it amplifies either the dsRNA
prior to cleavage or the siRNAs directly (Muers 2013) In C elegans ego-1 mutants
(ego stands for lsquoenhancer of glp-1) RNAi functions normally in somatic cells but is
defective in germline cells where ego-1 is primarily expressed In Arabidopsis SDE-
1SGS-2 mutants (SGS stands for suppressor of gene silencing) siRNA are produced
when dsRNA is introduced via an endogenously replicating RNA virus but not
introduced by a transgene It has been proposed that perhaps the viral RdRP is
substituting for the Arabidopsis enzyme in these mutants Random degradative PCR
model suggests that an RdRP uses the guide strand of an siRNA as a primer for the
target mRNA generating a dsRNA substrate for Dicer and thus more siRNAs
Several genes controlling RNA silencing in plants have been identified through
genetic screens of Arabidopsis mutants impaired in transgene induced RNA silencing
They encode a putative RNA-dependent RNA polymerase (SGS2SDE1) a coiled
protein (SGS3) a protein containing PAZ and Piwi domains (AGO1) and an RNA
helicase (SDE3) The putative SGS2SDE1 of Neurospora is related to QDE-1 and
Chapter 1 Introduction and Review of Literature
26
EGO-1 of C elegans whereas PAZPiwi protein AGO is related to QDE-2 of
Neurospora RDE-1of C elegans RNA helicase SDE3 is related to SMG-2 of C
elegans and Mut-6 of Chlamydomonas MUT-7 gene of C elegans encodes a protein
similar to Rnase D whereas the Drosophila DICER gene encode a protein similar to
RNase III An Arabidopsis ortholog of DICER gene has been identified called CAF
SIN1 SUS1(Thakur 2005 Poethig 2009)
Since the RNAi machinery is present constitutively within the eukaryotic cell it
is important to explore the metabolic advantages that are accorded by the RNAi related
proteins during the intrinsic normal growth of cells and development of organisms The
natural RNAi machinery not only keeps the mobile transposable elements from
disrupting the integrity of the genomes as suggested by the analysis of model organisms
like A thaliana C elegans D melanogaster (Tabara et al 1999 Wu-Scharf et al
2000 Aravin et al 2001 Hamilton et al 2002 Lisch 2009) but also participates in
development of organisms Genetic defects in Celegans RNAi genes ego1 and dicer
cause known specific developmental errors (Grishok et al 2000 Grishok et al 2001
Knight amp Bass 2001 Hall et al 2013) Similarly the Argonaute family of genes of A
thaliana (especially the ZWILLE proteins) is also responsible for plant architecture and
meristem development (Carmell et al 2002 Hamant and Pautot 2010) and the Dicer
homologue of A thaliana CAF1 is required for embryo development (Golden et al
2002 Nakano et al 2011) Thus genetic evidence illustrates the role of the RNAi
machinery as a controller of development related genes The mechanistic details of
these developmental processes are beginning to emerge
Chapter 1 Introduction and Review of Literature
27
Figure 16 Mechanism of gene silencing induced double stranded RNA The dicer enzyme to
produce siRNAs cleaves the RNA The siRNA generated bind to the nuclease complex
RISC The antisense component in the siRNA in the RISC guides the complex towards
the cognate mRNA resulting in endonucleolytic cleavage of the mRNA
Chapter 1 Introduction and Review of Literature
28
112 MicroRNA or MiRNA
In 1991 Ambros and co-workers first isolated a lin-4 mutant of Celegans which was
arrested at the first larval stage (Lee et al 1993) Later on the let-7 mutation was
isolated in the same system which was responsible for development through the fourth
larval stage Both lin-4 and let-7 encode short 22-nucleotide mature RNAs and were
called short temporal RNA because they control the temporal development program of
C elegans The mature lin-4 RNA defines (negatively regulates) the mRNA expression
of the lin-14 and lin-28 hetrochronic genes with the antisense mediated repression
mechanism of translation initiation and thus specifies the fate of cells during the first
three larval stages Recent studies have revealed that the short temporal RNAs are
actually members of a group of tiny RNAs (21to 28 nucleotides) called the micro-
RNAs (miRNA) (Bartel 2009) isolated members of which could easily run to a few
thousand which includes both those were identified computationally and by deep
sequencing Some of the components of the RNAi machinery have also been clearly
established as the effector proteins for the maturation of miRNAs
113 MicroRNAs in plants
1131 Discovery
MicroRNAs have been discovered by three basic approaches Direct cloning forward
genetic direct cloning and bioinformatic prediction followed by experimental
validation The most direct method of miRNA discovery is to isolate and clone small
RNAs from biological samples and several groups have used this approach to identify
plant miRNAs (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002 Xie et al 2003 Sunkar et al 2005 Williams et al 2013) The cloning methods
adapted were similar to those used to identify large numbers of animal miRNAs
(Lagos-Quintana et al 2001 Lau et al 2001 Aravin amp Tuschl 2005) which involve
isolating small RNAs followed by oligonucleotide adapter ligation reverse
transcription amplification and sequencing Some protocols incorporate methods to
enrich for Dicer cleavage products (ie molecules with 5prime-phosphates and 3prime-
hydroxyls) and to concatemerize the short cDNAs so that several may be identified in a
single sequencing read (Lau et al 2001 Llave et al 2002a Jagadeeswaran amp Sunkar
2013)The initial cloning experiments in Arabidopsis identified 19 miRNAs belonging
to 15 families (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002) although hundreds of other small RNAs such as siRNAs and RNA degradation
Chapter 1 Introduction and Review of Literature
29
fragments were also cloned through the direct cloning methods To select miRNAs
from the pool of small RNAs secondary structures of the RNA molecules were
examined in compliance with the acknowledged structural features of miRNAs
(Ambros et al 2003) The secondary structures were validated using several ad-hoc
software tools such as the Mfold or RNAfold programs (Reeder et al 2006) Finally
the existence of mature miRNAs was determined by RNA gel blot hybridization These
direct cloning methods were successful in isolating many miRNAs in Arabidopsis
(Reinhart et al 2002 Sunkar amp Zhu 2004) Rice (Sunkar et al 2005) Poplar (Lu et
al 2005) moss (Arazi et al 2005) and Tobacco (Billoud et al 2005)
Although miRNAs were first discovered through forward genetic screens in
worms (Lee et al 1993 Reinhart et al 2000)only few A thaliana miRNA gene
families have been discovered by this method (Chen 2008) in plants MiRNA
involvement in plant mutant phenotypes were not properly inferred till the cloning
experiments established that plant genomes contain numerous miRNAs (Park et al
2002 Reinhart et al 2002 Rhoades et al 2002) MiRNA loss-of-function allele has
been identified by forward genetic screens early extra petala1 is caused by a
transposon insertion ~160bp upstream of the predicted miR164c stem loop resulting in
flowers with extra petals (Baker et al 2005 Irish 2008) The fact that loss-of-function
miRNA mutants have been rarely discovered using forward genetics reflects small
target size for mutagenesis coupled with redundancy in nearly all evolutionarily
conserved plant miRNAs which are encoded by gene families (Table16) Family
members are likely to have overlapping functions buffering against loss at any single
miRNA locus Over expression screens can circumvent redundancy limitations At least
four plant miRNAs miR319 (also known as miR-JAW) miR172 (also known as EAT)
and miR166 were isolated in over expression screens for dominant mutants with
developmental abnormalities (Aukerman amp Sakai 2003 Palatnik et al 2003 Kim et
al 2005 Williams et al 2005b Schommer et al 2012) Mutations in miRNA target
sites which can prevent the entire family of miRNAs from repressing a target gene can
also circumvent redundant functions of miRNA family members
The dominant mutations in the HD-ZIP genes PHB PHV and REVOLUTA
(REV) in Arabidopsis and ROLLEDLEAF1 (RLD1) in Maize result in adaxialization of
leaves andor Vasculature (McConnell amp Barton 1998 McConnell et al 2001 Nelson
et al 2002 Zhong amp Ye 2004 Allen amp Millar 2012) and are all caused by mutations
Chapter 1 Introduction and Review of Literature
30
in miR166 complementary sites (Rhoades et al 2002 Emery et al 2003 Jones-
Rhoades amp Bartel 2004 Mallory et al 2004 Zhong amp Ye 2004)
In both plants and animals cloning was the initial means of large-scale
miRNA discovery (Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Reinhart et al 2002 Mendes et al 2012) However cloning is biased toward
RNAs that are expressed highly and broadly MiRNAs expressed at low levels or
only in specific cell types or in response to certain environmental stimuli were more
difficult to clone Sequence based biases in cloning procedures might also cause
certain miRNAs to be missed Because of these limitations bioinformatics approaches
to identify miRNAs have provided a useful complement to cloning
A straightforward use of bioinformatics has been carried out to find homologs
of known miRNAs both within the same genome and in the genomes of other species
(Pasquinelli et al 2000 Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Mendes et al 2009) A more difficult challenge is to identify miRNAs
unrelated to previously known miRNAs This was first accomplished for
vertebrate nematode and fly miRNAs using algorithms that search for conservation
of sequence and secondary structure (ie miRNA stem-loop precursors) between
species searching for patterns that are characteristic of miRNAs (Lai et al 2003 Lim
et al 2003 Lim et al 2005)
Most of the methods identified numerous miRNAs but are necessarily more
relaxed in terms of allowed structures Some approaches take advantage of the high
complementarity of plant miRNAs to target messages implementing the requirement
that the candidate has conserved complementarity to mRNAs (Jones-Rhoades amp Bartel
2004 Adai et al 2005) This additional filter has been useful for distinguishing
authentic plant miRNAs from false positives which later extended to mammalian
miRNA gene prediction (Xie et al 2005 Mendes et al 2012)
Another criteria used by most algorithms in the filters is evolutionary
conservation In plants mature miRNA sequences are conserved to a higher degree
than their precursor sequences For example Arabidopsis and rice diverged more than
130 million years ago (Friis et al 2004) but some miRNAs are highly conserved
in these plant species (Reinhart et al 2002) Furthermore various parameters for the
secondary structures such as free energy the number of paired residues within a
Chapter 1 Introduction and Review of Literature
31
miRNA or the number and size of bulges are also used to validate the predictions
(Jones-Rhoades amp Bartel 2004 Wang et al 2004 Adai et al 2005 Mendes et al
2012)
1132 Conserved microRNAs in Plants
Till date cloning genetics and bioinformatics screens have resulted in annotation of
approximately 184 potential Arabidopsis miRNAs (miRBase release 13) These
miRNAs seem to arise through gene duplication and subsequent diversification and
represent approximately 100 families (Maher et al 2006 Axtell 2013) Twenty one
families represented by 92 genes are clearly conserved in species beyond Arabidopsis
(Table 15) The presence of these miRNA families in other sequenced plant genomes
indicates that different plant species have an evolutionarily fluid set of miRNAs
(Willmann amp Poethig 2007) Recent studies on mosses one of the most ancient land
plants have shown that at least 14 miRNA families are conserved in eudicots and
mosses and therefore it appears that plant miRNAs share a common ancient origin
(Arazi et al 2005 Axtell amp Bartel 2005 Talmor-Neiman et al 2006 Zhang et al
2006 Axtell 2013) The number of members per family in a genome ranges from 1-32
with the exception of miR-430 which is represented by a cluster of ~80 loci in zebra
fish (Giraldez et al 2005)
In plants the number of members in each family correlates among examined
species certain families contain numerous members in all three species (eg miR156
miR166 miR169) whereas others consistently contain fewer genes (eg miR162
mir168 miR394) (Table 16) Although it is still unclear why certain miRNA are
encoded by more than one gene in plants (eg 12 genes encoding miR156) this
correlation might suggest functional significance of various miRNA family sizes Most
annotated plant miRNAs are conserved throughout flowering plants while a few
miRNAs are found only in a single sequenced genome and thus they could be
evolutionarily lsquoyoungrsquo miRNAs For example Arabidopsis has several miRNA
families that are not found in poplar or rice suggesting that miRNAs could be
generated continuously during evolution (Allen et al 2004a Rajagopalan et al
2006 Fahlgren et al 2007 Axtell 2012 de Alba et al 2013) Consistent with the
notion that non conserved miRNAs are of a more recent evolutionary origin these
miRNAs are mainly encoded by a single locus in the genome Within each family the
mature miRNA is always located in the same arm
Chapter 1 Introduction and Review of Literature
32
Table 16 MicroRNA families conserved in plants
miRNA
family
Arabidopsis Oryza Populus
miR156 12 12 11 miR159319 6 8 15 miR160 3 6 8 miR162 2 2 3 miR164 3 5 6 miR166 9 12 17 miR167 4 9 8 miR168 2 2 2 miR169 14 17 32 miR171 4 7 10 miR172 5 3 9 miR390 3 1 4 miR393 2 2 4 miR394 2 1 2 miR395 6 19 10 miR396 2 5 7 miR397 2 2 3 miR398 3 2 3 miR399 6 11 12 miR408 1 1 1 miR403 1 0 2 miR437 0 1 0 miR444 0 1 0 miR445 0 9 0 Total 92 127 169
All known miRNA families that are conserved between more than one plant species are listed
together with the number of genes identified in the sequenced genomes Rice miRNA families that have orthologs in maize but do not appear to have orthologs in the eudicots (Arabidopsis and Populus) are marked with an asterisk The following families contain miRNA genes annotated with
more than one number miR156 (miR156 and miR157) miR159319 (miR159 and miR319) miR166 (miR165 and miR166) miR171 (miR170 and miR171) and miR390 (miR390 and miR391)
of the stem loop (5prime- or 3prime) as it would be expected if the genes carry common ancestry
(Figure 17) The sequence of the mature miRNA and to some extent the segment on
the opposite arm of the hairpin to which it pairs is highly conserved between the
members of the same family (both within and between the species) while the sequence
secondary structure and length of the intervening ldquolooprdquo region can be highly divergent
within the same family
The patterns of paring and non pairing nucleotides are often conserved between
homologous miRNA stem loops from different species (Figure 17) The significance of
conserved mismatches is opined to guide DCL1 to cleave at the appropriate positions
along the stem loop
Chapter 1 Introduction and Review of Literature
33
Figure 17 Representative stem loop of miR164 Segments corresponding to the mature miRNAs
are shown in red (Adapted from Rajagopalan et al 2006)
Although many miRNAs are highly conserved in plants the degree of conservation in
most cases is quite low between plants and animals one exception is miR854 A recent
study has revealed that miR854 is conserved between Arabidopsis and animals and that
the Arabidopsis miR854 differs from the human miR854 only by a single nucleotide
(Arteaga-Vazquez et al 2006 Axtell 2012 de Alba et al 2013) The Arabidopsis
miR854 has multiple binding sites in the 3prime-untranslated region (UTR) of
OLIGOURIDYLATE-binding-PROTEIN mRNA 1bA (UBP1b) and represses the
translation of the UBP1b mRNA The animal miR854 is also complementary to a site in
the 3prime-UTR of the UBP1b homolog in animals However it is currently unclear how the
animal miR854 regulates its target mRNA Another distinction between plant and
animal miRNAs is the genomic organization of the miRNA genes While plant miRNA
genes are located predominantly in the intergenic regions animal miRNA genes tend to
localize in the intragenic regions both in the introns or exons of protein coding genes
Moreover miRNA genes are often clustered within a single precursor transcript
whereas individual plant miRNAs are derived from individual precursor RNAs (Bartel
Chapter 1 Introduction and Review of Literature
34
2004 Bonnet et al 2004 Kim 2005 Vaucheret et al 2006 Marco et al 2013)
1133 Non-conserved miRNA and challenges in annotation
Most miRNAs are conserved throughout flowering plants (Table 15) but many more
were found only in a single sequenced genome thus considered to be of a more recent
evolutionary origin The extended homology between few non conserved miRNAs
precursors and their target genes provides strong evidence that these relatively ldquoYoungrdquo
miRNAs arose from the duplication of the target gene segments (Allen et al 2004)
Several non-conserved miRNAs including miR161 miR163 miR173 miR447
miR475 and miR476 are known to direct cleavage of target transcripts (Allen et al
2004a Adai et al 2005 Sunkar et al 2005 de Alba et al 2013) It is difficult to
confidently predict targets for many because it is not possible to use complementary
site as a filter against false-positive target predictions It is also difficult to
confidently annotate non-conserved miRNAs as miRNA rather than siRNAs The
established minimal standard for miRNA annotation is a small RNA with detectable
expression and the potential to form a stem-loop when joined to flanking genomic
sequence (Kasschau et al 2003) In practice these requirements are too loose to
definitively categorize many small RNAs cloned from plants Many plant siRNAs are
detectable on blots (Vazquez et al 2004b) and hundreds of thousands of non-
miRNA genomic sequences can be predicted to fold into secondary structures that
resemble structures of plant miRNA precursors (Jones-Rhoades amp Bartel 2004)
Therefore without conservation of both sequence and secondary structure it is
difficult to be confident that a given cloned RNA originated from a stem-loop (ie is a
miRNA) rather than from a double-stranded RNA (ie is a siRNA) In fact many of
the thousands o f small RNAs cloned from Arabidopsis (Gustafson et al 2005) would
probably meet the literal requirements for annotation as miRNAs A few of these
sequences might be miRNAs but others that meet the literal criteria probably are not
so The challenges of annotating non conserved small RNAs were evidenced by
three related small silencing RNAs that were originally annotated as miRNAs
that turned out to be trans-acting siRNAs (tasiRNAs)(Kanno et al2013)
Although most miRNAs that are broadly conserved among flowering plants are
now being identified and experimentally validated (Table 16)(Jones-Rhoades amp
Bartel 2004) the challenges and ambiguities for classifying non-conserved miRNAs
prevent meaningful estimates of the total number of miRNA genes in Arabidopsis and
Chapter 1 Introduction and Review of Literature
35
other plant genomes It is possible that miRNAs might have escaped detection
because they are non-conserved and expressed in specific tissues or conditions and can
only be identified by using high- throughput sequencing
1134 MicroRNA biogenesis
11341 Transcription of microRNA precursor
Plant miRNAs are primarily found in the genomic regions that are not associated with
the protein coding genes (Reinhart et al 2002) and it appears that most if not all plant
miRNAs are produced from their own transcriptional units Plant miRNA genes are
occasionally clustered near each other in the genome suggesting transcription of
multiple miRNAs from a single primary transcript [eg the miR395 cluster (Grishok
et al 2001 Jones-Rhoades amp Bartel 2004b)] but this polycistronic arrangement of
miRNA genes appears far less frequently in plants than in animals (Bartel 2004)
Northern blot EST and mapping evidence indicate that plant miRNA primary
transcripts (also known as pri-miRNAs) as in animals are longer than needed to
encompass the miRNA stem-loops (Palatnik et al 2003 Jones-Rhoades amp Bartel
2004b) At least some of these pri-miRNA transcripts appear to be spliced
polyadenylated (Kurihara amp Watanabe 2004) and capped (Xie Z et al 2005) Two
rice miRNAs are contained within transcripts that contain exon junctions within the
presumptive stem-loop precursor implying that in these cases splicing is a prerequisite
for Dicer recognition (Sunkar et al 2005) Plant pri-miRNAs are over 1kb in length
and they are usually preceded by typical TATA box motifs They can also undergo
canonical splicing polyadenylation and capping All these observations indicates RNA
polymerase II is probably responsible for transcribing most plant miRNAs (Xie Z et
al 2005) as appears to be the case for many animal miRNAs Relatively little is
known about the regulation of miRNA transcription in plants but there is no reason
to suspect that this regulation would differ from that of protein-coding transcripts as
the bioinformatics analysis of the sequence upstream of the transcriptional start
sites of the miRNA genes showed the presence of putative binding motifs of
number of known transcription factors (Megraw et al 2006 Sethupathy 2013)
11342 MicroRNA processing and export
In plants maturation of miRNA is a step wise process A miRNA gene is transcribed to
pri-miRNA which is much longer than the pre-miRNA possessing the characteristic
stem-loop structure Like transcription of most protein-coding genes the miRNA
Chapter 1 Introduction and Review of Literature
36
transcription is processed by RNA polymerase II (Pol II) enzymes and the pri-
miRNAs are 5prime-capped and 3prime-polyadenylated (Lee et al 2004 Xie Z et al 2005)
Mature miRNAs are produced from pri-miRNAs through at least two sequential
processing steps by RNase III-type endonucleases In animals pri-miRNAs are first
processed at the bottom portion of the stem by Drosha a nuclear-localized RNase III
enzyme to liberate pre-miRNAs The pre-miRNAs are then exported to the cytoplasm
with the help of exportin-5The pre-miRNAs are further processed by Dicer another
RNaseIII enzyme to a duplex consisting of the miRNA and the antisense strands
(miRNA) Since plants do not have any Drosha homologs the two sequential
processing steps seem to be carried out by DCL1 a Dicer homolog in the nucleus
(Kurihara amp Watanabe 2004 de Alba et al 2013 Rogers amp Chen 2013)
The processing of pri-miRNAs to pre-miRNAs by DCL1 is assisted by two
other proteins HYPONASTY LEAVES1 (HYL1) and SERRATE (SE) HYL1 belongs to a
family of double-stranded RNA (dsRNA) binding protein and interacts with DCL1 in
vitro and in vivo (Lu amp Fedoroff 2000 Hiraguri et al 2005 de Alba et al 2013
Rogers amp Chen 2013) Loss-of-function mutations in the HYL1 gene result in
decreased accumulation of miRNAs and concomitant accumulation of pri-miRNAs
(Han et al 2004 Vazquez et al 2004a Kurihara et al 2006) SE a C2H2 zinc finger
protein interacts with DCL1 and HYL1 and plays a role in the processing of pri-
miRNAs to pre-miRNAs (Lobbes et al 2006 Yang L et al 2006) In addition the
nuclear cap-binding complex proteins CBP20 and CBP80 that are known as ABA
HYPER SENSITIVE 1(ABH1) have been shown to be required for proper miRNA
processing (Gregory et al 2008 Laubinger et al 2008) Recently it has been
demonstrated that DCL1 can release 21-nt RNAs from dsRNA as well as from
synthetic miR167 precursor RNAs in vitro However this cleavage is error prone and
inefficient in the absence of HYL1 and SE which accelerate the rate of DCL1-mediated
cleavage and promote accurate processing of miRNAs (Dong et al 2008 Rogers amp
Chen 2013)
11343 Methylation of the miRNAmiRNAduplex
After the miRNAmiRNAduplexes are released from the pre-miRNAs by the DCL1
activities the duplexes are methylated at the 2primeOH of the3prime-ends by HUA ENHANCER1
(HEN1) a small RNA methyltransferase (Yu et al 2005 Yang Z et al 2006 Rogers
amp Chen 2013 ) This step which is distinct from the RNase III-mediated processing
Chapter 1 Introduction and Review of Literature
37
step distinguishes the biogenesis of plant miRNAs from that of animal miRNAs
Mutations in the HEN1 gene were first isolated in a morphological screening for floral
development although the HEN1 mutants display pleiotropic phenotypes similar to the
developmental defects observed in the DCL1 mutants (Chen et al 2002 Park et al
2002 Rogers amp Chen 2013)
Figure 18 Biogenesis of microRNA
A Biogenesis of plant MicroRNA B Biogenesis of metazoananimal MicroRNA
The miRNA (red) is incorporated into RISC and the miRNA(blue) in degraded
This enzyme has two dsRNA-binding domains and nuclear localization signal It
is also conserved in fungi and is required for the processing of siRNAs as well as of
miRNAs (Park et al 2002 Boutet et al 2003 Xie et al 2003) Although the HEN1
protein is localized to the nucleus its methylation activity in the cytoplasm cannot be
ignored (Xie et al 2004 Rogers amp Chen 2013)
Chapter 1 Introduction and Review of Literature
38
The methylation of the miRNAmiRNA duplex is required to protect miRNAs
against the 3prime-end uridylation activity and subsequent degradation (Li et al 2005) In
the hen1 mutants accumulation of miRNAs is reduced to a lower level Also the
miRNAs appear to be heterogeneous in their sizes such that a ladder of bands rather
than a single species is observed for most miRNAs in the mutants (Li et al 2005)
MiRNA cloning and sequencing analyses revealed the presence of a number of U
residues at the 3prime-end of miRNAs in the hen1 mutants Moreover these nucleotides do
not correspond to the miRNAs in the pre-miRNAs indicating that these nucleotides are
added after the processing of the miRNAs from pre-miRNAs (Li et al 2005)
Uridylation of RNAs has also been proven to be correlated with RNA decay (Shen amp
Goodman 2004 Mullen amp Marzluff 2008) suggesting that uridylation attracts an
exonuclease which degrades miRNAs in plants
11344 MiRNA incorporation in to the RISC and its nuclear export
The methylated miRNAmiRNA duplexes undergo RISC assembly In this process
the miRNA of the duplexes is selectively incorporated into the RISC and the miRNA
appears to be removed and subsequently degraded (Hammond et al 2000 Hutvagner
amp Zamore 2002 Schwarz et al 2003 de Alba et al 2013 Rogers amp Chen 2013)
The ARGONAUTE 1(AGO1) proteins are core components of the RISC complex
They contain two structural domains the N-terminal PAZ domain that binds to the 3prime-
end of single-stranded RNAs and the C-terminal piwi domains that has a structure
similar to that of RNase H (Cerutti et al 2000 Parker et al 2004) Several AGO
proteins possess endonucleolytic activities within the PIWI domain and cleave target
mRNAs in the middle of the site complementary to miRNA (Liu et al 2004 Meister et
al 2004 Miyoshi et al 2005) Mutations in the AGO1 gene cause miRNA target
genes to be ectopically expressed and the levels of some miRNAs are reduced in the
mutants (Kidner amp Martienssen 2004 Vaucheret et al 2004 Kidner amp Martienssen
2005 Ronemus et al 2006) Moreover the phenotype of the AGO1 hypomorphic
alleles display defects in lateral organ polarity and leaf and flower morphology as
observed in the dcl1 mutants and null AGO1 alleles are embryonically lethal
supporting the close relationship between the AGO proteins and miRNA processing
(Kidner amp Martienssen 2004 Vaucheret et al 2004 de Alba et al 2013 Rogers amp
Chen 2013)
The production of miRNA miRNA duplexes occur in the nucleus while
Chapter 1 Introduction and Review of Literature
39
miRNA-RISC complexes are present in the cytoplasm where target mRNAs are
located This discordance of functional sites requires an export step during miRNA
biogenesis HASTY the Arabidopsis homolog of exportin-5 is thought to be involved
in miRNA nuclear export (Bollman et al 2003 Park et al 2005)The hasty mutant
exhibits pleiotropic defects in plant development and reduced accumulation of most
miRNAs as in the DCL1 or AGO1 mutants Howeverit is unclear whether the HASTY
proteins export the miRNA miRNA duplexes or the miRNA-RISC complexes in
plants
11345 Feedback regulation of miRNA biogenesis
MiRNA biogenesis is under feedback regulation such that the DCL1 and AGO1 genes
are themselves regulated by miRNAs DCL1 is itself a target of miR162 which leads to
the cleavage of the DCL1 mRNA (Xie et al 2003 de Alba et al 2013) Consistent
with this the levels of DCL1 mRNA are elevated in the mutants of miRNA processing
genes in which the miR162 abundance is reduced In addition there is a predicted
hairpin structure of miR838 in the 14th
intron of the DCL1 primary transcript
(Rajagopalan et al 2006) This prediction implies that DCL1-mediated processing
on this site can result in the cleavage of the DCL1 primary transcript into two truncated
fragments the N-terminal capped fragment and the C-terminal polyadenylated
fragment If this is the case it is possible that miRNA biogenesis competes with the
splicing event of the DCL1 pre-mRNA
The AGO1 is also regulated by miRNA There is a complementary site for
miR168 binding in the AGO1 mRNA and the transcript level of the AGO1 mRNA is
reduced through an mRNA cleavage mechanism (Vaucheret et al 2006 de Alba et al
2013 Rogers amp Chen 2013) indicating that the AGO1 mRNA levels are tightly
controlled by the levels of the AGO1 protein
1135 Mechanism of miRNA action
11351 MiRNA-directed mRNA cleavage
By forming the miRNA-RISC assembly miRNAs can direct the RISC to regulate gene
expression by two post transcriptional mechanisms mRNA cleavage or translational
repression (Bartel 2004 Jones-Rhoades et al 2006 Dalmay 2013) The degree
of miRNA-mRNA complementarity plays an important role for the
determination of the mechanism to be used A general rule is that while perfect or
Chapter 1 Introduction and Review of Literature
40
near-perfect complementarity induces mRNA cleavage central mismatches trigger
translational repression (Carrington amp Ambros 2003 Jones-Rhoades amp Bartel 2004)
Unlike most animal miRNAs plant miRNAs have near-perfect or perfect
complementarity to their targets and mRNA cleavage is assumed to be a predominant
mechanism by which miRNAs regulate gene expression in plants
In RNA cleavage mechanism miRNAs guide the AGO component of RISC to
cleave a single phosphodiester bond of the target mRNA within the miRNA-binding
site (Llave et al 2002b Tang et al 2003) The truncated fragments are then released
and degraded freeing the RISC to recognize and cleave another target mRNA This has
been validated by the detection of 3prime cleavage products that have 5prime ends that start at the
middle of the complementary regions The mRNA cleavage products are then further
degraded by other mechanisms The 3prime cleavage products of some miRNA targets are
degraded by the 5prime-3prime exonuclease XRN4 an Arabidopsis homolog of the major Yeast
mRNA degrading exonuclease Xrn1p It has been observed that the 3prime cleavage
products of some target mRNAs were accumulated in the xrn4 mutants (Souret et al
2004 Dalmay 2013) However the 3prime cleavage products of other miRNA targets do
not accumulate in the xrn4 mutants indicating that there are also XRN4 independent
mechanisms The 5prime cleavage products are typically untraceable by RNA gel blot
hybridization but can be detected by sensitive PCR based methods It has been found
that the 5prime cleavage products tend to obtain an oligo U tail at the 3prime ends This 3prime U tail
is correlated with shortening of the 5prime cleavage products from the 5prime end leading to the
conclusion that the 3prime uridylation causes 5 prime- 3 prime exonucleolytic decay of the 5prime cleavage
products (Shen amp Goodman 2004)
DCL1 HYL1 and SE interact with each other and are co-localized in the
nuclear bodies termed Dicing bodies or D-bodies in which some pri-miRNA shave
been found suggesting that D-bodies serve as a center for active miRNA processing
(Fang amp Spector 2007 Fujioka et al 2007 Song et al 2007) The miRNA
processing site appears to be distinct from the Cajal bodies that have previously been
found as a siRNA processing site (Pontes amp Pikaard 2008) The D-bodies include
SmD3 and SmB proteins that are found in the Cajal bodies However At collin an
additional marker for the Cajal bodies is not found in the D-bodies Moreover the D-
bodies are present in an At collin mutant lacking the Cajal bodies The pri-miRNAs of
miR171A and miR159A genes have been found to be co-localized with HYL1 and
Chapter 1 Introduction and Review of Literature
41
DCL1 in the D- bodies that are distinct from the Cajal bodies (Song et al 2007)
11352 MiRNA-directed translational repression
Translation repression is another mechanism exerted by plant miRNAs The regulatory
mechanism of miR172 which regulates flowering time and floral organ identity is
consistent with translation repression MiR172 over production does not affect the
transcript levels of APETALA2 (AP2) and AP2- like target mRNAs Instead the AP2
protein level is reduced in the miR172 over expressing mutant (Aukerman amp Sakai
2003 Chen 2004 Dalmay 2013) It was later shown that miR172 can also direct the
cleavage of the target mRNAs although the steady-state mRNA level is unchanged due
to feedback regulation (Schwab et al 2005 Jung et al 2007) It is clear that miR172
regulates its target genes primarily by translational repression rather than mRNA
cleavage Recently it has been reported that miR156157 and miR854 also regulate
their target genes through translational repression (Arteaga-Vazquez et al 2006
Gandikota et al 2007 Dalmay 2013) It was once thought that translational repression
is an exception to the rule However experimental evidence suggest a widespread
coexistence of translational repression and mRNA cleavage (Brodersen et al 2008)
1136 Regulatory roles of miRNAs in plant development
The miRNA target prediction has been a difficult task in order to obtain regulatory
targets without bringing in too many false predictions Plant growth and developmental
processes regulated by miRNAs are quite diverse and enormous Furthermore plant
miRNAs work cooperatively or antagonistically to establish a balanced regulation This
notion is well consistent with the findings that mutations in the regulators of miRNA
biogenesis transport and processing such as AGO1 DCL1 HYL1 HEN1 ABH1
and HASTY cause pleiotropic phenotypes (Mallory amp Vaucheret 2006 de Alba et al
2013) To date the roles of miRNAs have been mostly deduced from obtaining miRNA
over expressing transgenic plants or gain-of-function mutants in which miRNA-
resistant target genes are ectopically expressed
11361 Phase transition
Plants pass through a series of developmental phase transitions during their life span
beginning with seed germination continuing through vegetative phase change
reproductive phase change flowering initiation and finally seed production for the next
generation Because of their importance in understanding plant developmental
Chapter 1 Introduction and Review of Literature
42
processes genes regulating developmental transitions have been extensively studied
and underlying molecular mechanisms have been explored in various plant species
Majority of researches were mainly focused on vegetative phase change and
reproductive transition in Arabidopsis and Maize (Poethig 2003 Baurle amp Dean
2006) Particularly the mutations of the genes encoding various regulators of miRNA
biogenesis and transport such as HST SE and AGO exhibit altered juvenile to adult
vegetative phase change and vegetative to reproductive change (Hunter et al 2003
Peragine et al 2004 Vazquez et al 2004a Jones-Rhoades et al 2006 de Alba et al
2013)
Over-expression of the miR156a gene causes late flowering and delays
vegetative phase change (Wu amp Poethig 2006 Gandikota et al 2007 Wang et al
2008) It has been shown that miR156 negatively regulates a group of genes encoding
SQUAMOSA PROMOTER-BINDING PROTEIN-LIKE (Lewis et al 2007 de Alba et
al 2013 Rogers amp Chen 2013) transcription factors such as SPL3 SPL4 and SPL5
that promote flowering initiation In contrast transgenic plants over expressing
miR156-resistant SPL345 genes are early flowering Interestingly the endogenous
level of miR156 is temporally regulated In the early juvenile vegetative phase the
level of miR156 is very high but decreases rapidly before the onset of the adult juvenile
phase (Wu amp Poethig 2006)
The Arabidopsis early activation tagged (eat-D) mutant exhibits early flowering
with disrupted floral structure The mutant phenotypes are caused by over expression of
the miR172a-2gene (Aukerman amp Sakai 2003) MiR172 regulates a small group of
genes encoding APETALLA2 (AP2)-like transcription factors such as TARGET OF
EAT1 (TOE1) TOE2 SCHLAFMUTZE (SMZ) and SCHNARCHZAPFEN(SNZ)
TOE1 TOE2 SMZ and SNZ have been shown to participate in their productive phase
change by repressing a floral integrator FLOWERING LOCUS T (FT)(Jung et al
2007) Consistent with this toe1-2D 35STOE2 35SSMZ and 35SSNZ plants
exhibit late flowering (Jung et al 2007) MiR172 is also regulated temporally and
highly expressed in the adult vegetative phase both in Arabidopsis and maize (Chuck et
al 2007) Because the temporal expression patterns of miR156 and miR172 are
complementary to each other it has been suggested that the two miRNAs interact with
each other in regulating phase change (Chuck et al 2009 de Alba et al 2013) In
support of this view it has been shown that miR172 is down-regulated in the
Chapter 1 Introduction and Review of Literature
43
Corngrass1(Cg1) mutant in which miR156 is overproduced (Chuck et al 2007 de
Alba et al 2013) However there is no direct evidence for the direct linkage between
miR156 and miR172 It is also envisioned that miR156 and miR172 would not be
directly related For example miR156 regulates plastochron index that is the inverse of
leaf initiation rate and thus frequently used to analyze temporal patterns of plant
development In contrast miR172 does not affect plastochron (Wang et al 2008) The
down-regulation of miR172 in the maize miR156-over producing mutants would be
due to the prolonged juvenile vegetative phase in the mutants
Another miRNA that regulates flowering initiation is miR159 It regulates
MYB33 and MYB65 which are closely related to the barley GAMY B transcription
factor that responds to gibberellic acid (GA) The level of miR159 is elevated in GA-
treated seedlings but reduced in the GA biosynthetic ga1 mutant Transgenic plants
over producing miR159 exhibit delayed floral induction under short days (Achard et
al 2004) It is been suggested that MYB33 mediates GA signal to regulate LEAFY
(LFY) These observations indicate that miR159 plays an important role in the GA
signaling pathways that regulate proper floral induction under short days (Achard et al
2004)
11362 Floral development
Arabidopsis flowers consist of four distinct organs They arise in a characteristic
pattern within concentric rings called whorls from outside to inside four sepals four
petals six stamens and the central pistil consisting of two carpels Identities of the
floral organs are determined by three classes of regulatory genes classA classB and
class C genes (Krizek amp Fletcher 2005) The A and C class genes act antagonistically
to restrict their expression domains (Bowman et al 1991)
It is notable that miR172 also regulates floral architecture in addition to its role
in the timing of flowering initiation MiR172 inhibits the translation of the AP2 mRNA
(Chen 2004) Transgenic plants overproducing miR172 not only exhibit early
flowering but also show abnormal floral development which is quite similar to those
of the class A gene mutants such as the ap2 mutants (Aukerman amp Sakai 2003 de
Alba et al 2013) When the activity of the class A genes is inactivated that of the class
C genes such as AGAMOUS (AG) is elevated As a result the miR172- overproducing
transgenic plants exhibit no petals along with homeotic transformation of the sepals to
Chapter 1 Introduction and Review of Literature
44
the carpels (Aukerman amp Sakai 2003 Chen 2004)
Interestingly miR166 also regulates floral architecture The role of miR165166
in the regulation of meristem activity is closely linked to floral meristem formation and
organization (Zhao et al 2007 Miyashima et al 2013) Floral structure is severely
disrupted in the miR165166-overproducing mutants such as men1 and jba-1D in
which miR166 is overproduced (Williams et al 2005b Jung amp Park 2007) The men1
and jba-1D mutants have smaller gynoecium and reduced carpel number In an extreme
case the jba-1D homozygous plants lack visible carpels (Williams et al 2005b)
Similar phenotypes have been observed in the miR165 overproducers (Zhao et al
2007) It has been suggested that the phenotypes are possibly caused by altered AG
expression in the floral meristem (Jung amp Park 2007 Turchi et al 2013)
Other miRNAs also affect floral development Overproduction of miRNAs
involved in auxin signaling also affects floral architecture Transgenic plants
overproducing miR167 have defects in the formation of ovule and anther with reduced
fertility (Ru et al 2006) It has been shown that miR167 targets ARF6 and ARF8 that
play a role in gynoecium and stamen development and in regulating fertility (Ru et al
2006 Wu et al 2006) These observations suggest that auxin signals are also closely
related to floral organogenesis In addition transgenic plants over-producing miR164
and the cuc1cuc2 double mutants show fused sepals and reduced petals (Laufs et al
2004 Turchi et al 2013) suggesting that miR164 is also related to floral meristem
activity and boundary specification of the meristematic region
11363 Shoot apical meristem development
Shoot apical meristem comprises self-maintaining totipotent cells The shoot apical
meristem activity affects diverse aspects of plant developmental processes including
leaf development vascular development and lateral organ polarity (Bowman et al
2002) Because miR165166 plays a primary role in meristem formation
overproduction of miR165166 and multiple knockout mutants of its target genes
exhibit pleiotropic pheonotypes (McConnell et al 2001 Emery et al 2003 Kim et al
2005 Williams et al 2005b)
The targets of miR165166 include genes encoding the homeo domain-leucine
zipper (HD-ZIP) class III transcription factors such as PHABULOSA(PHB)
PHAVOLUTA (PHV) REVOLUTA(REV) ATHB15CORONA (CNA) and ATHB8
Chapter 1 Introduction and Review of Literature
45
PHB PHV and ATHB15 have overlapping functions in controlling meristem
development (Turchi et al 2013) Indeed the phb phv athb15 triple mutants have an
enlarged shoot apical meristem Consistent with this the miR166- overproducing men1
and jba-1D mutants exhibit enlarged shoot apical meristems (Kim et al 2005
Williams et al 2005b Turchi et al 2013) In the mutants the shoot apical meristems
are multiplied and large
The development of shoot apical meristems is also regulated by the WUSCHEL
(WUS)ndashCLAVATA (CLV) signaling pathway WUS positively regulates cell
proliferation In contrast CLV3 which is expressed in stem cells limits the WUS
expression and thus inhibits cell proliferation in the SAM (Sharma et al 2003)
Consistent with this notion WUS is expressed to a higher level in jba-1D
explaining the ectopic enlargement of the SAM in the mutant (Williams et al 2005)
While WUS is closely related to miR165166 in the shoot apical meristem
development the underlying molecular mechanisms are currently unclear It has been
observed that the miR166-over producing men1 plants have an arrested meristem
development (Jung amp Park 2007) In addition the heterozygous men1+ and the
homozygous men1 plants show different expression patterns of WUS and CLV3 It
appears that miR166 regulates WUS indirectly and influences the expressional balance
between WUS and CLV3 through a negative feedback loop (Jung amp Park 2007 de
Alba et al 2013)
The phv phb athb15 triple mutant has an enlarged shoot apical meristems the
rev phb phv mutant does not have functional shoot apical meristems (Prigge et al
2005) This distinction may be explained by the fact that while miR166 targets
primarily PHB PHV and ATHB15 miR165 targets all the five HD-ZIP III genes (Zhou
et al 2007) Consistent with this transgenic plants overproducing miR166 are distinct
from those overproducing miR165 The miR165-overproducing transgenic plants lack
functional shoot apical meristem and a pin-like structure is formed at the apical region
of the mutant These phenotypes are quite similar to rev phb phv triple mutant (Zhou et
al 2007 Liu et al 2013) The phenotypic distinction may because by the difference
of the cleavage efficiencies of the target mRNAs In accordance with this notion a few
35S miR166a transgenic lines exhibit no shoot apical meristems The men1
homozygous plants also show arrested meristem development (Jung amp Park 2007)
Chapter 1 Introduction and Review of Literature
46
MiR164 cleaves the CUC1 and CUC2 mRNAs as well as the NAC1 mRNA
CUC1 and CUC2 determines the organ boundary in the meristem Phenotypes of
transgenic plants that overproduce miR164 are similar to those of the cuc1cuc2 double
mutant that exhibits embryonic developmental defects such as cup-shaped cotyledons
and fused cotyledons (Laufs et al 2004) When a miR164-resistant CUC2 is
expressed the boundary domain is enlarged CUC also plays a role in embryonic shoot
meristem formation by regulating the SHOOT MERISTEMLESS gene It was therefore
concluded that miR164-mediated cleavage of CUC1 and CUC2 mRNAs determines the
sizes of the boundary domains in the shoot apical meristem and regulates separation of
the organs by restricting cell proliferation (Laufs et al 2004 de Alba et al 2013)
11364 Leaf development
The role of miR319 has been extensively studied in leaf development A dominant
jaw-D mutant overproducing miR319 shows uneven curved leaves with enlarged size
and five TCP genes which are closely related to the CINCINNATA (CIN) gene in
Antirrhinum majus are down regulated in the mutant suggesting that miR319 mediated
regulation of the TCP transcription factor genes are important for the final step of
leaf shaping (Palatnik et al 2003 Naz et al 2013) In addition to leaf shape and size
leaf polarity is also specified by miRNA MiR166-mediated cleavage of the HD-ZIPIII
mRNAs is a well to establish leaf polarity This discovery originates from the gain-of-
function mutants such as phb-1d and phv-1d wherein point mutations in the
complementary sites to miRNA helps the mRNA to evade or resist from miRNA-
mediated cleavage (McConnell et al 2001 Emery et al 2003 Naz et al 2013)
Plant leaves have a dorso-ventral polarity consisting of the adaxial (upper
surface) and abaxial (lower surface) sides The adaxial side is closer to the shoot
apical meristem Adaxial identity is specified by functionally redundant class III HD-
ZIP family genes such as PHB PHV and REV Ectopic expression of each genes
result in adaxialized leaves Dominant phb-1d and phv-1d mutants exhibit
transformation of the abaxial to adaxial fate and have rod-shaped leaves In contrast
miR165166-overproducing plants show pin-like cotyledons radicalized leaves and
abaxialized leaves (Chitwood et al 2007 Pattanayak et al 2013)
Leaf asymmetry is determined by miR166-mediated restriction of the
expression domains of the HD-ZIPIII genes The HD-ZIPIII genes are expressed in the
Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora
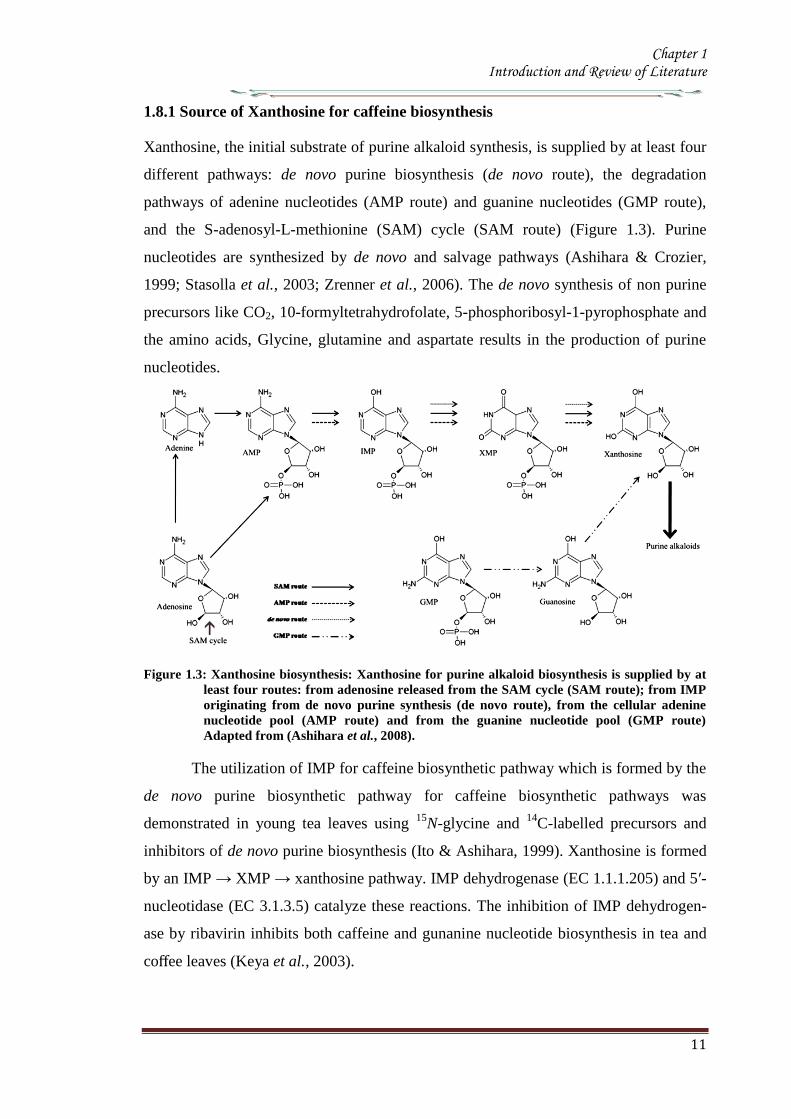
Chapter 1 Introduction and Review of Literature
11
181 Source of Xanthosine for caffeine biosynthesis
Xanthosine the initial substrate of purine alkaloid synthesis is supplied by at least four
different pathways de novo purine biosynthesis (de novo route) the degradation
pathways of adenine nucleotides (AMP route) and guanine nucleotides (GMP route)
and the S-adenosyl-L-methionine (SAM) cycle (SAM route) (Figure 13) Purine
nucleotides are synthesized by de novo and salvage pathways (Ashihara amp Crozier
1999 Stasolla et al 2003 Zrenner et al 2006) The de novo synthesis of non purine
precursors like CO2 10-formyltetrahydrofolate 5-phosphoribosyl-1-pyrophosphate and
the amino acids Glycine glutamine and aspartate results in the production of purine
nucleotides
Figure 13 Xanthosine biosynthesis Xanthosine for purine alkaloid biosynthesis is supplied by at
least four routes from adenosine released from the SAM cycle (SAM route) from IMP
originating from de novo purine synthesis (de novo route) from the cellular adenine
nucleotide pool (AMP route) and from the guanine nucleotide pool (GMP route)
Adapted from (Ashihara et al 2008)
The utilization of IMP for caffeine biosynthetic pathway which is formed by the
de novo purine biosynthetic pathway for caffeine biosynthetic pathways was
demonstrated in young tea leaves using 15
N-glycine and 14
C-labelled precursors and
inhibitors of de novo purine biosynthesis (Ito amp Ashihara 1999) Xanthosine is formed
by an IMP rarr XMP rarr xanthosine pathway IMP dehydrogenase (EC 111205) and 5prime-
nucleotidase (EC 3135) catalyze these reactions The inhibition of IMP dehydrogen-
ase by ribavirin inhibits both caffeine and gunanine nucleotide biosynthesis in tea and
coffee leaves (Keya et al 2003)
Chapter 1 Introduction and Review of Literature
12
The portion of xanthosine used for caffeine biosynthesis is derived from adenine
and guanine nucleotide pools which are produce de novo by salvage pathways there are
several potential pathways for xanthosine synthesis from AMP but the AMP rarr IMP
rarr XMPrarr xanthosine route is predominant All three enzymes involved in the
conversion AMP deaminase (EC 3546) IMP dehydrogenase and 5prime-nucleosidase
have been detected in tea leaves (Koshiishi et al 2001) Xanthosine for caffeine
biosynthesis can also be produced from guanine nucleotides by GMPrarrguanosine
rarrxanthosine pathway Guanosine deaminase (EC 35415) activity is found in cell free
extracts from young tea leaves (Negishi et al 1994) but GMP deaminase activity was
not detected in plants (Stasolla et al 2003 Zrenner et al 2006) The absence of GMP
deaminase activity suggests that a GMP rarr IMP rarr XMP rarr xanthosine route is not
functional in plants
The use of radiolabelled purine bases and nucleosides for the study of caffeine
biosynthetic pathway was facilitated by the fact that exogenous purine nucleotides are
rapidly hydrolyzed by the plant tissues forming purine nucleosides andor bases which
are then utilized in pathways which in most instances indirectly lead to xanthosine
Most of these purine compounds including adenine adenosine inosine hypoxanthine
and guanine are converted to their respective nucleotides by salvage enzymes
including adenine phosphoribosyl transferase (EC 2427) hypoxanthineguanine
phosphoribosyl transferase (EC 2428) adenosine kinase (EC 27120) and
inosineguanosine kinase (EC 2421) which usually have high activities in plant cells
The resultant nucleotides then enter the purine alkaloid biosynthetic pathway In the
case of guanosine it may be directly deaminated to xanthosine and utilized for caffeine
biosynthesis (Figure 13) (Ashihara et al 1997)
182 SAM cycle in plants
SAM is the methyl donor for the three methylation steps in the caffeine biosynthetic
pathway In this process SAM is converted to S-adenosyl-L-homocysteine (SAH)
which in turn is hydrolyzed to homocysteine and adenosine Homocysteine is recycled
via the SAM cycle to replenish SAM levels and adenosine is released from the cycle
Adenosine released from the cycle is converted to AMP directly andor indirectly via
adenine AMP is converted to xanthosine and used for caffeine biosynthesis Since
three moles of SAH are produced via the SAM cycle for each mole of caffeine that is
synthesized in theory this pathway has the capacity to be the sole source of both the
Chapter 1 Introduction and Review of Literature
13
purine skeleton and the methyl groups required for caffeine biosynthesis in young tea
leaves (Koshiishi et al 2001)
Methionine
S-Adenosyl-L-methionineHomocysteine
ATP
PPi+ Pi
Tetrahydrofolate
5-Methyl-
tetrahydrofolate
CH3+Adenosine
S-Adenosyl-L-
homocysteine
(xanthine
structure)(Methyl groups)
Caffeine
(1)
(2)(3)
(4)
Figure 14 The SAM cycle (activated methyl cycle) in plants (Ashihara amp Suzuki 2004)
Enzymes SAM synthetase SAM-dependent N-methyltransferases S-adenosyl-
homocysteine (SAH) hydrolase Methionine synthase Adenosine released from the cycle
is salvaged to adenine nucleotides and utilized both for purine structure of caffeine via
xanthosine and for re-synthesis of SAM via ATP
183 N-methyltransferase in caffeine biosynthesis Molecular cloning
The genes encoding for the enzymes involved in the caffeine biosynthesis were
successfully cloned from the coffee family (Ogawa et al 2001 Mizuno et al 2003a
Mizuno et al 2003b Uefuji et al 2003) The conserved amino acid regions of tea CS
(AB31280) the sequences of O-methyltransferases derived from plant origin and
Arabidopsis hypothetical proteins were made use of in designing primers for RT-PCR
and RACE technique Independent groups have isolated N-methyltransferase (NMT)
genes from coffee which have been classified based on the substrate specificity of the
recombinant proteins expressed in Ecoli (Table 15) PCR products thus obtained were
used as the probe to isolate full length cDNA from the library resulting in the
identification of all three N-methyltransferases involved in caffeine biosynthesis
(Figure 12) Several cDNA have been characterized from the coffee plants one for
xanthosine methyltransferase (XMTXRS) three for 7-methylxanthine methyltransferase
or theobromine synthase (MXMTCTS) and three for 37-dimethylxanthine
methyltransferase or caffeine synthase (DXMTCCS) (Table 15) (Kato amp Mizuno
2004)
A single gene encoding for xanthosine methyltransferase (XMTCmXRS1) was
identified which encoded for a polypeptide consisting of 372 amino acids with an
apparent molecular mass of 418kDa It is expressed almost uniformly in aerial tissues
Chapter 1 Introduction and Review of Literature
14
of C arabica including leaves floral buds and immature but not in mature beans
Three genes encoding 7-methylxanthine methyltransferase (theobromine synthase) have
been identified The number of amino acids in the putative polypeptides are 378 for
MXMT1 (427kDa) and 384 for MXMT2 and CTS2 (434kDa) They differ by insertion
or deletion of blocks of several residues in the C-terminal region Their catalytic
properties based on kinetic parameters such as Km values were different They are
expressed in young leaves floral buds and immature but not mature beans Three genes
were identified for 3 7-dimethylxanthine methyltransferase (caffeine synthase) DXMT
CCS1 and CtCS7 each encoding a 43 kDa polypeptide consisting of 384 amino acids
However their kinetic properties differ from each other such as DXMT and CCS1
showed Km values of 1200 and 157microM for theobromine respectively Expressions
profiles are also distinct DXMT being expressed exclusively in immature beans while
CCS1 expression is ubiquitous occurring in all tissues The presence of isoforms of
these enzymes with different properties suggests that caffeine is synthesized through
multiple pathways depending on availability and concentration of the substrate (Mizuno
et al 2001 Ogawa et al 2001 Mizuno et al 2003a Mizuno et al 2003b Uefuji et
al 2003) Genomic clones of theobromine synthase were obtained by PCR- RFLP
from C canephora (Satyanarayana et al 2005 Satyanarayana 2006) The genomic
clones are reported to have highly conserved exons and a variable third intron The
promoter for the theobromine synthase was cloned and characterized (Satyanarayana et
al 2005)
There is high degree of similarity in the coding region of coffee NMT genes
isolated so far though differences exist in the 3prime untranslated region (UTR) for these
genes The deduced amino acid sequence of these enzymes shows more than 85
homology between these genes Phylogenetic analysis indicates that they are more
closely related to C-methyltransferases including those for jasmonic acid salicylic acid
and benzoic acid than to other N-methyltransferases This suggests that coffee N-methyl
transferases belong to a distinct sub-group within plant methyltransferases Their
cellular localization determined by green fluorescent protein fusion method and β-
glucourodinase (GUS) showed that the all the three genes are localized in the cytoplasm
(Ogawa et al 2001 Vinod et al 2007) It was also shown that these were present
along with chlorogenic acid(Vinod et al 2007)
Chapter 1 Introduction and Review of Literature
15
Table 15 N-methyltransferases and their encoding genes involved in caffeine biosynthesis
Gene (Enzyme) Gene name Accession No Description Reference
Xanthosine methyltransferase
( 7-methylxanthosine synthase) (EC 211158)
CaXMT AB 048793 Immature fruit cDNA Screening (Uefuji et al 2003)
CmXRS1 AB 034699 Leaf cDNA library RACE (Mizuno et al 2003a)
CaMXMT1 AB 048794 Leaf cDNA library RACE (Ogawa et al 2001)
7-methylxanthine synthase transferase
(theobromine synthase) ( EC 211159)
CTS1 AB 034700 Immature fruit cDNA Screening (Uefuji et al 2003)
CaMXMT2 AB 984125 Leaf cDNA library Screening (Mizuno et al 2001)
CTS2 AB 054841 Leaf cDNA library Screening (Mizuno et al 2001)
37-methylxanthinemethyltransferase
(Caffeine synthase) (EC 211160)
CaDXMT1 AB 086414 Immature fruit cDNA Screening (Uefuji et al 2003)
CCS1 AB 0861414 Immature fruit cDNA Screening (Uefuji et al 2003)
CtCS7 AB 0864415 Immature fruit cDNA
NMT genomic clones
Theobromine synthase-1
CX10 Complete coding region PCR Satyanarayan 2006
CX8 Complete coding region PCR -do-
Theobromine synthase-1 PG-5 Promoter + coding region RAGE -do-
Theobromine synthase-1 PG-1 Promoter + coding region RAGE Satyanarayan 2006
PG-4 Promoter + coding region RAGE -do-
Theobromine synthase-1 NMT
(AY273814) Complete coding region PCR Kochko A et al 2010
Theobromine synthase-2 NMT
(AY362825) Complete coding region PCR Kochko A et al 2010
Chapter 1 Introduction and Review of Literature
16
19 Catabolism of caffeine
Caffeine is produced in young leaves and immature fruits and continues to accumulate
gradually during the maturation of these organs At the same time caffeine is slowly
degraded with the removal of the three methyl groups resulting in the formation of
xanthine Catabolism of caffeine was first reported in coffee leaves (Kalberer 1965)
Since then a number of tracer experiments using 14
C labelled purine alkaloids have
been reported (Suzuki amp Waller 1984 Ashihara et al 1996 Vitoria amp Mazzafera
1999 Mazzafera 2004) They demonstrated the major catabolic pathway is caffeine rarr
theophylline rarr 3-methylxanthine rarr xanthine Xanthine is further degraded by the
removal by the conventional purine catabolic pathway to CO2 and NH3 via uric acid
allatonin and allantoate (Figure 15) (Ashihara amp Crozier 1999 Stasolla et al 2003
Zrenner et al 2006) Caffeine catabolism usually begins with its conversion to
theophylline catalyzed by N7-demethylase The involvement of P450-dependent mono-
oxygenase activity for this reaction was suggested (Huber amp Baumann 1998
Mazzafera 2004) However the activity of this enzyme has not yet been determined in
cell free extracts [8-14
C] Theophylline degraded to CO2 at much faster rate than [8-14
C]
caffeine indicating that conversion of caffeine to theophylline is a major rate limiting
step of caffeine catabolism and a possible reason why caffeine accumulates in high
concentrations in tissues of C sinensis and C arabica It is widely believed that the
control of caffeine levels in plants is a function of the balance between rates of
synthesis and degradation and this balance seems to vary depending on the plant
species and the tissue developmental stage (Mazzafera 2004)
Chapter 1 Introduction and Review of Literature
17
Figure 15 Caffeine catabolic pathways Caffeine is mainly catabolised via theophylline and 3-
methylxanthine Xanthine is further degraded to CO2 and NH3 by the conventional
oxidative pathway Adapted from (Ashihara et al 2008)
Chapter 1 Introduction and Review of Literature
18
110 Genetic engineering of Coffee
Genetic transformation has potential applications in coffee agriculture for incorporating
genes which could provide desirable traits such as disease and insect resistance
drought and frost tolerance and herbicide resistance Transgenic technology can also be
used to increase nutritional value and improve cup quality produce varieties with
caffeine-free beans and for production of hybrid crops for molecular farming
Identification of target specific genes is one of the pre-requisites for developing
transgenic crops The availability of a large number of EST sequences in coffee and
initiation of coffee genome sequencing may speed up the gene discovery and accelerate
transgenic research efforts in coffee
1101 Insect Resistance
Production of insect resistant coffee plants is one of the major objectives of the
breeding programs The major pests attacking coffee include coffee berry borer (CBB
Hypothenemus hampei) white stem borer (WSB Xylotrechus quadripes) leaf miner
(Perileucoptera coffeela) and root nematodes (Meloidogyne spp and Pratylenchus
spp) The CBB is present in almost all the coffee growing countries and considered to
the most devastating pest in coffee To date there is no reported source of resistance to
CBB in the coffee gene pool Like CBB WSB is another serious pest in C arabica in
India and several other East Asian countries Both CBB and WSB belong to the order
Coleoptera For India controlling WSB is the biggest challenge and has therefore
become the highest research priority Robusta is generally resistant to WSB but the
interspecific Robusta Arabica hybrids are susceptible to WSB Although leaf miner is
not yet a serious pest in India and other East Asian countries it is an economically
important pest in East Africa and Brazil Effective chemical control of CBB and WSB
is difficult due to the nature of their life cycle inside the berry and stem respectively as
well as environmental concern regarding the use of pesticides Biological control
measures are adapted to combat these insect pests with varying degrees of success
Developing coffee plants resistant to these pests using genetic transformation
technology could be one of the alternative strategies to counter pest damage For insect
resistance several different classes of proteins from bacterial plant and animal sources
have been isolated and their insecticidal properties tested against many important pests
Amongst these proteins Bt toxins are most important and several transgenic crops
expressing Bt genes have been commercialized Coffee transgenic plants carrying a
Chapter 1 Introduction and Review of Literature
19
synthetic version of the cry1Ac gene have been produced (Leroy et al 2000 Mishra et
al 2002) Indeed this was the first report of an important agronomic trait being
introduced into a coffee plant The transgenic plants presented similar features in
growth and development compared to normal plants Transgenic plants highly resistant
to leaf miner under greenhouse conditions were tested under field conditions in French
Guyana for 4 years for pest resistance (Perthius et al 2005 Perthius et al 2006)
From 54 independent transformation events 70 of the events were resistant to leaf
miner Unfortunately the field trial was vandalized which led to the termination of the
experiment (Montagnon et al 2005)The effectiveness of Bt genes in controlling
coleopteran pests is well documented in corn and potato which indicates that Bt genes
might be effective against CBB The high toxicity of B thuringiensis sero var
israelensis against first in star larvae of CBB has been demonstrated (Mendez et al
2003) In another experiment an α-amylase inhibitor from Phaseolus vulgaris was
tested against CBB and found to have an inhibitory effect on its growth and
development (Grossi et al 2004)In addition to the CBB and WSB C arabica
varieties are also susceptible to endoparasitic root-knot nematode (Meloidogyne spp)
(Campos et al 1990 Mishra et al 2012)
So far 15 species have been reported to be parasites of coffee Controlling
nematodes is extremely difficult and currently seedling grafting with robusta rootstock
is followed as one of the control measure Sources of resistance specific to root-knot
nematodes have been identified in coffee trees (Bertrand et al 2001) and the Mex-1
gene conferring resistance to M exigua in C arabica is in the process of isolation (Noir
et al 2003)
1102 Tolerance to Abiotic Stress
In many coffee growing countries abiotic stress such as drought and frost are the major
climatic factors that limit coffee production Changes in climatic patterns due to global
climate change are considered increasingly important for coffee cultivation Drought
induces water stress in plants which affects vegetative growth and vigor and triggers
floral abnormalities and poor fruit set It also indirectly increases the incidence of pests
and diseases in the plants C arabica is generally more tolerant to water stress than C
canephora partly due to its extensive deep root system C racemosa is known to be a
good source of drought tolerance In India hybrids between C canephora and C
racemosa have been obtained and are currently under evaluation for drought tolerance
Chapter 1 Introduction and Review of Literature
20
In many coffee growing countries coffee is propagated in marginal areas where the
annual rainfall is below 1000thinspmm with prolonged dry spells of over 4-5 months In
those areas water shortage and unfavorable temperatures constitute major constraints
and the growth and productivity of C canephora are badly affected As with drought
periodic frost also affects coffee production in parts of Brazil The introduction of
drought and frost tolerant genes through genetic transformation would be of great
importance for alleviating these problems Research is now being carried out by several
groups to identify genes involved in biotic as well as abiotic stress The most promising
genetic engineering approach for drought tolerance includes the use of functional or
regulatory genes as well as the transfer of transcription factors In recent years plants
tolerant to high temperature and water stress have been the subject of intense research
(Chaves et al 2003 Chaves et al 2004 Coraggio et al 2004) For achieving drought
tolerance genes that have been targeted include those encoding enzymes involved in
detoxification or osmotic response metabolism enzymes active in signaling proteins
involved in the transport of metabolites and regulating the plant energy status (Chaves
et al 2003 Chaves et al 2004 Coraggio et al 2004) The dissection of molecular
mechanisms related to signal transduction and transcriptional regulation might help in
engineering drought tolerance in coffee
1103 Disease Resistance
Coffee leaf rust (CLR) caused by the fungus Hemileia vastatrix is the most important
disease in coffee with substantial loss to coffee production and productivity in all the
coffee growing countries In addition to leaf rust coffee berry disease (CBD) caused by
fungus Colletotrichum kahawae can be a devastating anthracnose leading to substantial
crop loss in Africa Several other fungal and bacterial diseases may also affect coffee
to a extent C arabica is more susceptible to many diseases than C canephora Though
most of the disease control measures rely upon chemical control they are more
expensive and labour intensive The long-term solution is the breeding of resistant
varieties which is the focus of many breeding programs However breeding for disease
resistant varieties is time consuming due to the perennial nature of coffee with its long
gestation period India has a long history of C arabica breeding especially for leaf rust
resistance and is the first country to demonstrate the existence of multiple races of leaf
rust Resistance to CLR is conditioned primarily by a number of major (SH) genes and
coffee genotypes are classified into different resistance groups based on their
Chapter 1 Introduction and Review of Literature
21
interaction with different rust races pathogen (van der Vossen 2001) Currently C
canephora provides the main source of resistance to pests and diseases including CLR
(H vastatrix) and CBD (C kahawae) and therefore used in breeding programs Other
diploid species like C liberica and C racemosa are being used as a source of resistance
to coffee leaf rust and coffee leaf miner respectively (Guerreiro et al 1999
Sreenivasan et al 1989) The development of coffee varieties resistant to major fungal
diseases such as CLR and CBD using transgenic technology will benefit the coffee
industry immensely During the last 15 years significant progress has been made in the
area of host-pathogen interactions (Staskawicz et al 1995 Feys et al 2000
Shivaprasad et al 2012) and many resistance genes involved in recognizing invading
pathogens have been identified and cloned (Takken et al 2000) A number of
signalling pathways induced because of pathogen infection have been dissected (Dong
et al 1998 Navarro et al 2006) Many antifungal compounds synthesized by plants to
combat fungal infection have been identified (Does et al 1998) Understanding the
specific induction of targeted pathways and identification of specific pathways
responsible for particular fungal resistance is important in order to employ this strategy
in transgenic technology The recent investigation of gene expression during coffee leaf
rust infection could provide an insight into the defence pathways operating in coffee
(Fernandez et al 2004 Nardi et al 2006) Efforts have been made to identify and
clone resistance genes from coffee for achieving durable resistance The genetic and
physical map of two resistance genes SH3gene conferring resistance to rust (Prakash et
al 2004) and the Ck1 gene conferring resistance to C kahawae CBD (Gichuru et al
2006) have been established These genes could be used for molecular marker assisted
breeding programs (Mishra et al 2012)
1104 Production of low-caffeine coffee
A widespread belief that the ingestion of caffeine can have adverse effects on
health resulted in the increased demand for decaffeinated coffee (Ashihara amp Crozier
2001) Several method were used to obtain decaffeinated plants by conventional
breeding techniques using low yielding varieties and mutants that accumulated
relatively low amounts of caffeine when compared to the commercially available ones
Low-caffeine and decaffeinated coffee represent around 10 of the coffee sales
around the world (Ribas et al 2006b) The industrial process for coffee decaffeination
can be expensive and affects the original flavour and aroma in coffee (David 2002)
Chapter 1 Introduction and Review of Literature
22
Transgenic coffee plants with suppressed caffeine synthesis using RNA interference
(RNAi) technology have been obtained (Ogita et al 2003 Ogita et al 2004) Specific
sequences in the 3prime untranslated region of the theobromine synthase gene (CaMXMT1)
were selected for construction of RNAi short and long fragments The caffeine and
theobromine content of the transgenic plants were reduced by ~ 70 when compared to
the untransformed plants In C canephora RNAi technology has also been employed
to silence the N-methyl transferase gene involved in caffeine biosynthesis (Vinod et al
2007) Promoter of an N-methyltransferase (NMT) gene involved in caffeine
biosynthesis was cloned (Satyanarayana et al 2005 Satyanarayana 2006) which will
be very useful for studying the regulation of caffeine biosynthesis
1105 Improvement in cup quality
Improvement in coffee cup quality requires elaborate knowledge of the chemical
constituents as well as the metabolic pathways involved in the elaboration of quality
The constituents of coffee beans include minerals proteins carbohydrates caffeine
chlorogenic acids (CGA) glycosides lipids and many volatile compounds that give
flavour to coffee by roasting Among these the role of three major constituents
sucrose CGA and trigonelline have been studied in coffee The sucrose content of
coffee bean is associated with coffee flavour the higher the sucrose content in green
beans the more intense will be the cup flavour (Clifford et al 1985 de Maria et al
1996) The sucrose content of C arabica (82-83) is higher than C canephora (33ndash
40) The sucrose amino acids ratio in green beans determines the profile of volatile
compounds Manipulating sucrose content in coffee bean is therefore important in
improving cup quality The sucrose synthase gene (CcSUS2) from C canephora has
been cloned and sequenced (Leroy et al 2005) This provides an opportunity to
manipulate the sucrose content in coffee Chlorogenic acids are products of
phenylpropanoid metabolism Chlorogenic acids are a group of hydroxycinnamoyl
quinic acids (HQA) formed by esterification between caffeic acids coumaric acids and
quinic acids (Clifford et al 1999) They are present in relatively large quantities in the
coffee bean and are the precursor of phenolic compounds in roasted beans C
canephora beans contain higher CGA (10) compared to C arabica beans (6-7)
CGA are known to have antioxidant properties as well as being associated with disease
resistance (Campa et al 2003) Genetic manipulations of genes involved in CGA
synthesis can serve either of these purpose by up- or down regulating the pathway
Chapter 1 Introduction and Review of Literature
23
Phenylalanine ammonia-lyase (PAL) catalyzes the first step of the phenylpropanoid
pathway leading to the synthesis of a wide range of chemical compounds including
flavonoids coumarins hydroxycinnamoyl esters and lignins (Hahlbrock amp Scheel
1989) Recently the full length cDNA and corresponding genomic sequences of PAL
from C canephora was isolated characterized and functionally validated (Mahesh et
al 2006 Parvatam et al 2006) This has opened up new possibilities for manipulating
the level of the PAL enzyme in coffee which in turn can be useful for improvement of
cup quality and manipulating antioxidant properties in coffee
1106 Fruit Ripening
Uniformity during fruit ripening is decisively related to cup quality in coffee and
consequently to the value of the product Fruits at the ideal ripening stage produce the
best organoleptic characteristics for coffee The presence of over ripened or green fruits
changes the acidity the bitterness and consequently the cup quality In order to
maximize uniform ripening of coffee fruits it is essential to control the action of genes
involved in the last step of maturation process Ethylene is known to trigger ripening
and increasing ethylene biosynthesis is associated with various stages of ripening
process (Protasio et al 2005) To control coffee fruit maturation two of the major
genes involved in ethylene biosynthesis namely ACC synthase and ACC oxidase
have been cloned (Protasio et al 2005 Neupane et al 1999) Introduction of the ACC
oxidase gene in antisense orientation have been achieved in both C arabica and C
canephora The effect of the transgene on ethylene production and fruit maturation has
yet to be reported The inhibition of genes downstream to the initial ethylene burst is
also an option to control coffee fruit maturation (Ribas et al 2006)
111 RNA interferenceRNAi
The regulation of gene expression at post transcriptional level has recently attracted
much attention because of the discovery of the phenomenon called RNA interference
(RNAi) RNAi based silencing techniques are more efficient than antisense mediated
gene silencing (Wesley et al 2001) The effect was first observed in plants wherein
silencing was observed when some copies of a transgene was in the forward orientation
with respect to the promoter and some in the reverse orientation This resulted in the
formation of double stranded RNA in the system The phenomenon in which
experimentally introduced double stranded RNA (dsRNA) leads to the loss of
expression of the corresponding cellular gene by sequence specific RNA degradation
Chapter 1 Introduction and Review of Literature
24
was termed as RNA interference or RNAi The protective effect of RNA silencing in
reducing virus infectivity supports the view that PTGS has evolved as a mechanism to
defend plants against virus infection and also to moderate the possible deleterious
genome-restructuring (insertional) activity of virus-like mobile genetic elements eg
retrotransposons A growing body of biochemical and genetic data has further
demonstrated that plants animals and yeasts share related mechanisms of specific
degradation of RNAs in which double-stranded forms of RNA are initiator molecules
(Brenstein et al 2001)
The key insight into the process of PGTS was provided by the experiments
conducted by Hamilton amp Baulcombe (1999) who identified the product of RNA
degradation as small RNA (siRNA) species of ~25nt of both sense and antisense
polarity SiRNA are formed and accumulate as double stranded RNA molecules of
defined chemical structures Both genetic and biochemical approaches were undertaken
to understand the basis of silencing Genetic screens were carried out in fungus
Neurospora carassa the alga Chalmydomonas reinhardtii and plant Arabidopsis
thaliana to search for detective mutants in PTGS
A combination of results obtained from several in vivo and in vitro experiments
have gelled into a two step mechanistic model for RNAiPTGS The first step referred
to as the RNAi initiating step involves binding of the RNA nucleases to a larges
dsRNA and its cleavage into discrete ~21-25nt RNA fragments This is ATP-
dependent reaction which involved RNase III - type endonucleases which make
staggered cuts in both strands of the dsRNA leaving 3prime overhang of 2 nucleotides with
5prime- phosphate 3prime-hydroxyl and no modification in the sugar phosphate backbone
(Hamilton amp Baulcombe 1999 Elbashir et al 2001) SiRNAs are the direct products
of dsRNA cleavage by the multi-domain RNase III enzyme DICER (Figure 16)
(Brenstein et al 2001 Karthikeyan et l 2013)
In the second step or commonly known as the effector step the double stranded
siRNAs produced in the first step bind to an RNAi specific protein complex to form a
RISC The complex undergoes activation in the presence of ATP so that the antisense
component of the unwound siRNA becomes exposed and allows the RISC to perform
the downstream RNAi reaction
Chapter 1 Introduction and Review of Literature
25
Several enzymes are involved in RNAi based on their role genes that encode for them
are classified into RNAi initiators RNAi effectors RNA dependent RNA polymerases
and silencing genes Two C elegans genes rdeI and rde4 (rde stands for lsquo RNAi
deficientrsquo) are believed to be involved in the initiation step of RNAi The C elegans
rde1 gene is a member of a large family of genes and is homologous to the Neurospora
qde2 (qde stands for ldquoquelling deficientrdquo) and Arabidopsis AGO1 (AGO stands for
agronaute AGO1 was previously identified to be involved in Arabidopsis
development) Although the functions of these genes are not clear a mammalian
member of RDE1 family has been identified as a translation initiation factor (Thakur
2005) Important genes for the effector step of PTGS in C elegans are rde2 and mut7
genes These genes were identified from heterozygous mutant worms that were unable
to transmit RNAi to their homozygous offsprings Worms with mutated rde2 or mut 7
genes show defective RNAi mut-7 gene encodes a protein with homology to the
nuclease domains of RNAase D and a protein implicated in Werner syndrome (a rapid
ageing disease in humans) (Grishok et al 2001 Kamath-Loeb et al 2012s)
Neurospora qde-1 Arabidopsis SDE-1SGS-2 and C elegans ego-1 appear to encode
RNA dependent RNA polymerase (RdRPs) It assumed that this is a proof that an RdRp
activity is required for RNAi Certainly the existence of an RdRp might explain the
remarkable efficiency of dsRNA induced silencing if it amplifies either the dsRNA
prior to cleavage or the siRNAs directly (Muers 2013) In C elegans ego-1 mutants
(ego stands for lsquoenhancer of glp-1) RNAi functions normally in somatic cells but is
defective in germline cells where ego-1 is primarily expressed In Arabidopsis SDE-
1SGS-2 mutants (SGS stands for suppressor of gene silencing) siRNA are produced
when dsRNA is introduced via an endogenously replicating RNA virus but not
introduced by a transgene It has been proposed that perhaps the viral RdRP is
substituting for the Arabidopsis enzyme in these mutants Random degradative PCR
model suggests that an RdRP uses the guide strand of an siRNA as a primer for the
target mRNA generating a dsRNA substrate for Dicer and thus more siRNAs
Several genes controlling RNA silencing in plants have been identified through
genetic screens of Arabidopsis mutants impaired in transgene induced RNA silencing
They encode a putative RNA-dependent RNA polymerase (SGS2SDE1) a coiled
protein (SGS3) a protein containing PAZ and Piwi domains (AGO1) and an RNA
helicase (SDE3) The putative SGS2SDE1 of Neurospora is related to QDE-1 and
Chapter 1 Introduction and Review of Literature
26
EGO-1 of C elegans whereas PAZPiwi protein AGO is related to QDE-2 of
Neurospora RDE-1of C elegans RNA helicase SDE3 is related to SMG-2 of C
elegans and Mut-6 of Chlamydomonas MUT-7 gene of C elegans encodes a protein
similar to Rnase D whereas the Drosophila DICER gene encode a protein similar to
RNase III An Arabidopsis ortholog of DICER gene has been identified called CAF
SIN1 SUS1(Thakur 2005 Poethig 2009)
Since the RNAi machinery is present constitutively within the eukaryotic cell it
is important to explore the metabolic advantages that are accorded by the RNAi related
proteins during the intrinsic normal growth of cells and development of organisms The
natural RNAi machinery not only keeps the mobile transposable elements from
disrupting the integrity of the genomes as suggested by the analysis of model organisms
like A thaliana C elegans D melanogaster (Tabara et al 1999 Wu-Scharf et al
2000 Aravin et al 2001 Hamilton et al 2002 Lisch 2009) but also participates in
development of organisms Genetic defects in Celegans RNAi genes ego1 and dicer
cause known specific developmental errors (Grishok et al 2000 Grishok et al 2001
Knight amp Bass 2001 Hall et al 2013) Similarly the Argonaute family of genes of A
thaliana (especially the ZWILLE proteins) is also responsible for plant architecture and
meristem development (Carmell et al 2002 Hamant and Pautot 2010) and the Dicer
homologue of A thaliana CAF1 is required for embryo development (Golden et al
2002 Nakano et al 2011) Thus genetic evidence illustrates the role of the RNAi
machinery as a controller of development related genes The mechanistic details of
these developmental processes are beginning to emerge
Chapter 1 Introduction and Review of Literature
27
Figure 16 Mechanism of gene silencing induced double stranded RNA The dicer enzyme to
produce siRNAs cleaves the RNA The siRNA generated bind to the nuclease complex
RISC The antisense component in the siRNA in the RISC guides the complex towards
the cognate mRNA resulting in endonucleolytic cleavage of the mRNA
Chapter 1 Introduction and Review of Literature
28
112 MicroRNA or MiRNA
In 1991 Ambros and co-workers first isolated a lin-4 mutant of Celegans which was
arrested at the first larval stage (Lee et al 1993) Later on the let-7 mutation was
isolated in the same system which was responsible for development through the fourth
larval stage Both lin-4 and let-7 encode short 22-nucleotide mature RNAs and were
called short temporal RNA because they control the temporal development program of
C elegans The mature lin-4 RNA defines (negatively regulates) the mRNA expression
of the lin-14 and lin-28 hetrochronic genes with the antisense mediated repression
mechanism of translation initiation and thus specifies the fate of cells during the first
three larval stages Recent studies have revealed that the short temporal RNAs are
actually members of a group of tiny RNAs (21to 28 nucleotides) called the micro-
RNAs (miRNA) (Bartel 2009) isolated members of which could easily run to a few
thousand which includes both those were identified computationally and by deep
sequencing Some of the components of the RNAi machinery have also been clearly
established as the effector proteins for the maturation of miRNAs
113 MicroRNAs in plants
1131 Discovery
MicroRNAs have been discovered by three basic approaches Direct cloning forward
genetic direct cloning and bioinformatic prediction followed by experimental
validation The most direct method of miRNA discovery is to isolate and clone small
RNAs from biological samples and several groups have used this approach to identify
plant miRNAs (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002 Xie et al 2003 Sunkar et al 2005 Williams et al 2013) The cloning methods
adapted were similar to those used to identify large numbers of animal miRNAs
(Lagos-Quintana et al 2001 Lau et al 2001 Aravin amp Tuschl 2005) which involve
isolating small RNAs followed by oligonucleotide adapter ligation reverse
transcription amplification and sequencing Some protocols incorporate methods to
enrich for Dicer cleavage products (ie molecules with 5prime-phosphates and 3prime-
hydroxyls) and to concatemerize the short cDNAs so that several may be identified in a
single sequencing read (Lau et al 2001 Llave et al 2002a Jagadeeswaran amp Sunkar
2013)The initial cloning experiments in Arabidopsis identified 19 miRNAs belonging
to 15 families (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002) although hundreds of other small RNAs such as siRNAs and RNA degradation
Chapter 1 Introduction and Review of Literature
29
fragments were also cloned through the direct cloning methods To select miRNAs
from the pool of small RNAs secondary structures of the RNA molecules were
examined in compliance with the acknowledged structural features of miRNAs
(Ambros et al 2003) The secondary structures were validated using several ad-hoc
software tools such as the Mfold or RNAfold programs (Reeder et al 2006) Finally
the existence of mature miRNAs was determined by RNA gel blot hybridization These
direct cloning methods were successful in isolating many miRNAs in Arabidopsis
(Reinhart et al 2002 Sunkar amp Zhu 2004) Rice (Sunkar et al 2005) Poplar (Lu et
al 2005) moss (Arazi et al 2005) and Tobacco (Billoud et al 2005)
Although miRNAs were first discovered through forward genetic screens in
worms (Lee et al 1993 Reinhart et al 2000)only few A thaliana miRNA gene
families have been discovered by this method (Chen 2008) in plants MiRNA
involvement in plant mutant phenotypes were not properly inferred till the cloning
experiments established that plant genomes contain numerous miRNAs (Park et al
2002 Reinhart et al 2002 Rhoades et al 2002) MiRNA loss-of-function allele has
been identified by forward genetic screens early extra petala1 is caused by a
transposon insertion ~160bp upstream of the predicted miR164c stem loop resulting in
flowers with extra petals (Baker et al 2005 Irish 2008) The fact that loss-of-function
miRNA mutants have been rarely discovered using forward genetics reflects small
target size for mutagenesis coupled with redundancy in nearly all evolutionarily
conserved plant miRNAs which are encoded by gene families (Table16) Family
members are likely to have overlapping functions buffering against loss at any single
miRNA locus Over expression screens can circumvent redundancy limitations At least
four plant miRNAs miR319 (also known as miR-JAW) miR172 (also known as EAT)
and miR166 were isolated in over expression screens for dominant mutants with
developmental abnormalities (Aukerman amp Sakai 2003 Palatnik et al 2003 Kim et
al 2005 Williams et al 2005b Schommer et al 2012) Mutations in miRNA target
sites which can prevent the entire family of miRNAs from repressing a target gene can
also circumvent redundant functions of miRNA family members
The dominant mutations in the HD-ZIP genes PHB PHV and REVOLUTA
(REV) in Arabidopsis and ROLLEDLEAF1 (RLD1) in Maize result in adaxialization of
leaves andor Vasculature (McConnell amp Barton 1998 McConnell et al 2001 Nelson
et al 2002 Zhong amp Ye 2004 Allen amp Millar 2012) and are all caused by mutations
Chapter 1 Introduction and Review of Literature
30
in miR166 complementary sites (Rhoades et al 2002 Emery et al 2003 Jones-
Rhoades amp Bartel 2004 Mallory et al 2004 Zhong amp Ye 2004)
In both plants and animals cloning was the initial means of large-scale
miRNA discovery (Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Reinhart et al 2002 Mendes et al 2012) However cloning is biased toward
RNAs that are expressed highly and broadly MiRNAs expressed at low levels or
only in specific cell types or in response to certain environmental stimuli were more
difficult to clone Sequence based biases in cloning procedures might also cause
certain miRNAs to be missed Because of these limitations bioinformatics approaches
to identify miRNAs have provided a useful complement to cloning
A straightforward use of bioinformatics has been carried out to find homologs
of known miRNAs both within the same genome and in the genomes of other species
(Pasquinelli et al 2000 Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Mendes et al 2009) A more difficult challenge is to identify miRNAs
unrelated to previously known miRNAs This was first accomplished for
vertebrate nematode and fly miRNAs using algorithms that search for conservation
of sequence and secondary structure (ie miRNA stem-loop precursors) between
species searching for patterns that are characteristic of miRNAs (Lai et al 2003 Lim
et al 2003 Lim et al 2005)
Most of the methods identified numerous miRNAs but are necessarily more
relaxed in terms of allowed structures Some approaches take advantage of the high
complementarity of plant miRNAs to target messages implementing the requirement
that the candidate has conserved complementarity to mRNAs (Jones-Rhoades amp Bartel
2004 Adai et al 2005) This additional filter has been useful for distinguishing
authentic plant miRNAs from false positives which later extended to mammalian
miRNA gene prediction (Xie et al 2005 Mendes et al 2012)
Another criteria used by most algorithms in the filters is evolutionary
conservation In plants mature miRNA sequences are conserved to a higher degree
than their precursor sequences For example Arabidopsis and rice diverged more than
130 million years ago (Friis et al 2004) but some miRNAs are highly conserved
in these plant species (Reinhart et al 2002) Furthermore various parameters for the
secondary structures such as free energy the number of paired residues within a
Chapter 1 Introduction and Review of Literature
31
miRNA or the number and size of bulges are also used to validate the predictions
(Jones-Rhoades amp Bartel 2004 Wang et al 2004 Adai et al 2005 Mendes et al
2012)
1132 Conserved microRNAs in Plants
Till date cloning genetics and bioinformatics screens have resulted in annotation of
approximately 184 potential Arabidopsis miRNAs (miRBase release 13) These
miRNAs seem to arise through gene duplication and subsequent diversification and
represent approximately 100 families (Maher et al 2006 Axtell 2013) Twenty one
families represented by 92 genes are clearly conserved in species beyond Arabidopsis
(Table 15) The presence of these miRNA families in other sequenced plant genomes
indicates that different plant species have an evolutionarily fluid set of miRNAs
(Willmann amp Poethig 2007) Recent studies on mosses one of the most ancient land
plants have shown that at least 14 miRNA families are conserved in eudicots and
mosses and therefore it appears that plant miRNAs share a common ancient origin
(Arazi et al 2005 Axtell amp Bartel 2005 Talmor-Neiman et al 2006 Zhang et al
2006 Axtell 2013) The number of members per family in a genome ranges from 1-32
with the exception of miR-430 which is represented by a cluster of ~80 loci in zebra
fish (Giraldez et al 2005)
In plants the number of members in each family correlates among examined
species certain families contain numerous members in all three species (eg miR156
miR166 miR169) whereas others consistently contain fewer genes (eg miR162
mir168 miR394) (Table 16) Although it is still unclear why certain miRNA are
encoded by more than one gene in plants (eg 12 genes encoding miR156) this
correlation might suggest functional significance of various miRNA family sizes Most
annotated plant miRNAs are conserved throughout flowering plants while a few
miRNAs are found only in a single sequenced genome and thus they could be
evolutionarily lsquoyoungrsquo miRNAs For example Arabidopsis has several miRNA
families that are not found in poplar or rice suggesting that miRNAs could be
generated continuously during evolution (Allen et al 2004a Rajagopalan et al
2006 Fahlgren et al 2007 Axtell 2012 de Alba et al 2013) Consistent with the
notion that non conserved miRNAs are of a more recent evolutionary origin these
miRNAs are mainly encoded by a single locus in the genome Within each family the
mature miRNA is always located in the same arm
Chapter 1 Introduction and Review of Literature
32
Table 16 MicroRNA families conserved in plants
miRNA
family
Arabidopsis Oryza Populus
miR156 12 12 11 miR159319 6 8 15 miR160 3 6 8 miR162 2 2 3 miR164 3 5 6 miR166 9 12 17 miR167 4 9 8 miR168 2 2 2 miR169 14 17 32 miR171 4 7 10 miR172 5 3 9 miR390 3 1 4 miR393 2 2 4 miR394 2 1 2 miR395 6 19 10 miR396 2 5 7 miR397 2 2 3 miR398 3 2 3 miR399 6 11 12 miR408 1 1 1 miR403 1 0 2 miR437 0 1 0 miR444 0 1 0 miR445 0 9 0 Total 92 127 169
All known miRNA families that are conserved between more than one plant species are listed
together with the number of genes identified in the sequenced genomes Rice miRNA families that have orthologs in maize but do not appear to have orthologs in the eudicots (Arabidopsis and Populus) are marked with an asterisk The following families contain miRNA genes annotated with
more than one number miR156 (miR156 and miR157) miR159319 (miR159 and miR319) miR166 (miR165 and miR166) miR171 (miR170 and miR171) and miR390 (miR390 and miR391)
of the stem loop (5prime- or 3prime) as it would be expected if the genes carry common ancestry
(Figure 17) The sequence of the mature miRNA and to some extent the segment on
the opposite arm of the hairpin to which it pairs is highly conserved between the
members of the same family (both within and between the species) while the sequence
secondary structure and length of the intervening ldquolooprdquo region can be highly divergent
within the same family
The patterns of paring and non pairing nucleotides are often conserved between
homologous miRNA stem loops from different species (Figure 17) The significance of
conserved mismatches is opined to guide DCL1 to cleave at the appropriate positions
along the stem loop
Chapter 1 Introduction and Review of Literature
33
Figure 17 Representative stem loop of miR164 Segments corresponding to the mature miRNAs
are shown in red (Adapted from Rajagopalan et al 2006)
Although many miRNAs are highly conserved in plants the degree of conservation in
most cases is quite low between plants and animals one exception is miR854 A recent
study has revealed that miR854 is conserved between Arabidopsis and animals and that
the Arabidopsis miR854 differs from the human miR854 only by a single nucleotide
(Arteaga-Vazquez et al 2006 Axtell 2012 de Alba et al 2013) The Arabidopsis
miR854 has multiple binding sites in the 3prime-untranslated region (UTR) of
OLIGOURIDYLATE-binding-PROTEIN mRNA 1bA (UBP1b) and represses the
translation of the UBP1b mRNA The animal miR854 is also complementary to a site in
the 3prime-UTR of the UBP1b homolog in animals However it is currently unclear how the
animal miR854 regulates its target mRNA Another distinction between plant and
animal miRNAs is the genomic organization of the miRNA genes While plant miRNA
genes are located predominantly in the intergenic regions animal miRNA genes tend to
localize in the intragenic regions both in the introns or exons of protein coding genes
Moreover miRNA genes are often clustered within a single precursor transcript
whereas individual plant miRNAs are derived from individual precursor RNAs (Bartel
Chapter 1 Introduction and Review of Literature
34
2004 Bonnet et al 2004 Kim 2005 Vaucheret et al 2006 Marco et al 2013)
1133 Non-conserved miRNA and challenges in annotation
Most miRNAs are conserved throughout flowering plants (Table 15) but many more
were found only in a single sequenced genome thus considered to be of a more recent
evolutionary origin The extended homology between few non conserved miRNAs
precursors and their target genes provides strong evidence that these relatively ldquoYoungrdquo
miRNAs arose from the duplication of the target gene segments (Allen et al 2004)
Several non-conserved miRNAs including miR161 miR163 miR173 miR447
miR475 and miR476 are known to direct cleavage of target transcripts (Allen et al
2004a Adai et al 2005 Sunkar et al 2005 de Alba et al 2013) It is difficult to
confidently predict targets for many because it is not possible to use complementary
site as a filter against false-positive target predictions It is also difficult to
confidently annotate non-conserved miRNAs as miRNA rather than siRNAs The
established minimal standard for miRNA annotation is a small RNA with detectable
expression and the potential to form a stem-loop when joined to flanking genomic
sequence (Kasschau et al 2003) In practice these requirements are too loose to
definitively categorize many small RNAs cloned from plants Many plant siRNAs are
detectable on blots (Vazquez et al 2004b) and hundreds of thousands of non-
miRNA genomic sequences can be predicted to fold into secondary structures that
resemble structures of plant miRNA precursors (Jones-Rhoades amp Bartel 2004)
Therefore without conservation of both sequence and secondary structure it is
difficult to be confident that a given cloned RNA originated from a stem-loop (ie is a
miRNA) rather than from a double-stranded RNA (ie is a siRNA) In fact many of
the thousands o f small RNAs cloned from Arabidopsis (Gustafson et al 2005) would
probably meet the literal requirements for annotation as miRNAs A few of these
sequences might be miRNAs but others that meet the literal criteria probably are not
so The challenges of annotating non conserved small RNAs were evidenced by
three related small silencing RNAs that were originally annotated as miRNAs
that turned out to be trans-acting siRNAs (tasiRNAs)(Kanno et al2013)
Although most miRNAs that are broadly conserved among flowering plants are
now being identified and experimentally validated (Table 16)(Jones-Rhoades amp
Bartel 2004) the challenges and ambiguities for classifying non-conserved miRNAs
prevent meaningful estimates of the total number of miRNA genes in Arabidopsis and
Chapter 1 Introduction and Review of Literature
35
other plant genomes It is possible that miRNAs might have escaped detection
because they are non-conserved and expressed in specific tissues or conditions and can
only be identified by using high- throughput sequencing
1134 MicroRNA biogenesis
11341 Transcription of microRNA precursor
Plant miRNAs are primarily found in the genomic regions that are not associated with
the protein coding genes (Reinhart et al 2002) and it appears that most if not all plant
miRNAs are produced from their own transcriptional units Plant miRNA genes are
occasionally clustered near each other in the genome suggesting transcription of
multiple miRNAs from a single primary transcript [eg the miR395 cluster (Grishok
et al 2001 Jones-Rhoades amp Bartel 2004b)] but this polycistronic arrangement of
miRNA genes appears far less frequently in plants than in animals (Bartel 2004)
Northern blot EST and mapping evidence indicate that plant miRNA primary
transcripts (also known as pri-miRNAs) as in animals are longer than needed to
encompass the miRNA stem-loops (Palatnik et al 2003 Jones-Rhoades amp Bartel
2004b) At least some of these pri-miRNA transcripts appear to be spliced
polyadenylated (Kurihara amp Watanabe 2004) and capped (Xie Z et al 2005) Two
rice miRNAs are contained within transcripts that contain exon junctions within the
presumptive stem-loop precursor implying that in these cases splicing is a prerequisite
for Dicer recognition (Sunkar et al 2005) Plant pri-miRNAs are over 1kb in length
and they are usually preceded by typical TATA box motifs They can also undergo
canonical splicing polyadenylation and capping All these observations indicates RNA
polymerase II is probably responsible for transcribing most plant miRNAs (Xie Z et
al 2005) as appears to be the case for many animal miRNAs Relatively little is
known about the regulation of miRNA transcription in plants but there is no reason
to suspect that this regulation would differ from that of protein-coding transcripts as
the bioinformatics analysis of the sequence upstream of the transcriptional start
sites of the miRNA genes showed the presence of putative binding motifs of
number of known transcription factors (Megraw et al 2006 Sethupathy 2013)
11342 MicroRNA processing and export
In plants maturation of miRNA is a step wise process A miRNA gene is transcribed to
pri-miRNA which is much longer than the pre-miRNA possessing the characteristic
stem-loop structure Like transcription of most protein-coding genes the miRNA
Chapter 1 Introduction and Review of Literature
36
transcription is processed by RNA polymerase II (Pol II) enzymes and the pri-
miRNAs are 5prime-capped and 3prime-polyadenylated (Lee et al 2004 Xie Z et al 2005)
Mature miRNAs are produced from pri-miRNAs through at least two sequential
processing steps by RNase III-type endonucleases In animals pri-miRNAs are first
processed at the bottom portion of the stem by Drosha a nuclear-localized RNase III
enzyme to liberate pre-miRNAs The pre-miRNAs are then exported to the cytoplasm
with the help of exportin-5The pre-miRNAs are further processed by Dicer another
RNaseIII enzyme to a duplex consisting of the miRNA and the antisense strands
(miRNA) Since plants do not have any Drosha homologs the two sequential
processing steps seem to be carried out by DCL1 a Dicer homolog in the nucleus
(Kurihara amp Watanabe 2004 de Alba et al 2013 Rogers amp Chen 2013)
The processing of pri-miRNAs to pre-miRNAs by DCL1 is assisted by two
other proteins HYPONASTY LEAVES1 (HYL1) and SERRATE (SE) HYL1 belongs to a
family of double-stranded RNA (dsRNA) binding protein and interacts with DCL1 in
vitro and in vivo (Lu amp Fedoroff 2000 Hiraguri et al 2005 de Alba et al 2013
Rogers amp Chen 2013) Loss-of-function mutations in the HYL1 gene result in
decreased accumulation of miRNAs and concomitant accumulation of pri-miRNAs
(Han et al 2004 Vazquez et al 2004a Kurihara et al 2006) SE a C2H2 zinc finger
protein interacts with DCL1 and HYL1 and plays a role in the processing of pri-
miRNAs to pre-miRNAs (Lobbes et al 2006 Yang L et al 2006) In addition the
nuclear cap-binding complex proteins CBP20 and CBP80 that are known as ABA
HYPER SENSITIVE 1(ABH1) have been shown to be required for proper miRNA
processing (Gregory et al 2008 Laubinger et al 2008) Recently it has been
demonstrated that DCL1 can release 21-nt RNAs from dsRNA as well as from
synthetic miR167 precursor RNAs in vitro However this cleavage is error prone and
inefficient in the absence of HYL1 and SE which accelerate the rate of DCL1-mediated
cleavage and promote accurate processing of miRNAs (Dong et al 2008 Rogers amp
Chen 2013)
11343 Methylation of the miRNAmiRNAduplex
After the miRNAmiRNAduplexes are released from the pre-miRNAs by the DCL1
activities the duplexes are methylated at the 2primeOH of the3prime-ends by HUA ENHANCER1
(HEN1) a small RNA methyltransferase (Yu et al 2005 Yang Z et al 2006 Rogers
amp Chen 2013 ) This step which is distinct from the RNase III-mediated processing
Chapter 1 Introduction and Review of Literature
37
step distinguishes the biogenesis of plant miRNAs from that of animal miRNAs
Mutations in the HEN1 gene were first isolated in a morphological screening for floral
development although the HEN1 mutants display pleiotropic phenotypes similar to the
developmental defects observed in the DCL1 mutants (Chen et al 2002 Park et al
2002 Rogers amp Chen 2013)
Figure 18 Biogenesis of microRNA
A Biogenesis of plant MicroRNA B Biogenesis of metazoananimal MicroRNA
The miRNA (red) is incorporated into RISC and the miRNA(blue) in degraded
This enzyme has two dsRNA-binding domains and nuclear localization signal It
is also conserved in fungi and is required for the processing of siRNAs as well as of
miRNAs (Park et al 2002 Boutet et al 2003 Xie et al 2003) Although the HEN1
protein is localized to the nucleus its methylation activity in the cytoplasm cannot be
ignored (Xie et al 2004 Rogers amp Chen 2013)
Chapter 1 Introduction and Review of Literature
38
The methylation of the miRNAmiRNA duplex is required to protect miRNAs
against the 3prime-end uridylation activity and subsequent degradation (Li et al 2005) In
the hen1 mutants accumulation of miRNAs is reduced to a lower level Also the
miRNAs appear to be heterogeneous in their sizes such that a ladder of bands rather
than a single species is observed for most miRNAs in the mutants (Li et al 2005)
MiRNA cloning and sequencing analyses revealed the presence of a number of U
residues at the 3prime-end of miRNAs in the hen1 mutants Moreover these nucleotides do
not correspond to the miRNAs in the pre-miRNAs indicating that these nucleotides are
added after the processing of the miRNAs from pre-miRNAs (Li et al 2005)
Uridylation of RNAs has also been proven to be correlated with RNA decay (Shen amp
Goodman 2004 Mullen amp Marzluff 2008) suggesting that uridylation attracts an
exonuclease which degrades miRNAs in plants
11344 MiRNA incorporation in to the RISC and its nuclear export
The methylated miRNAmiRNA duplexes undergo RISC assembly In this process
the miRNA of the duplexes is selectively incorporated into the RISC and the miRNA
appears to be removed and subsequently degraded (Hammond et al 2000 Hutvagner
amp Zamore 2002 Schwarz et al 2003 de Alba et al 2013 Rogers amp Chen 2013)
The ARGONAUTE 1(AGO1) proteins are core components of the RISC complex
They contain two structural domains the N-terminal PAZ domain that binds to the 3prime-
end of single-stranded RNAs and the C-terminal piwi domains that has a structure
similar to that of RNase H (Cerutti et al 2000 Parker et al 2004) Several AGO
proteins possess endonucleolytic activities within the PIWI domain and cleave target
mRNAs in the middle of the site complementary to miRNA (Liu et al 2004 Meister et
al 2004 Miyoshi et al 2005) Mutations in the AGO1 gene cause miRNA target
genes to be ectopically expressed and the levels of some miRNAs are reduced in the
mutants (Kidner amp Martienssen 2004 Vaucheret et al 2004 Kidner amp Martienssen
2005 Ronemus et al 2006) Moreover the phenotype of the AGO1 hypomorphic
alleles display defects in lateral organ polarity and leaf and flower morphology as
observed in the dcl1 mutants and null AGO1 alleles are embryonically lethal
supporting the close relationship between the AGO proteins and miRNA processing
(Kidner amp Martienssen 2004 Vaucheret et al 2004 de Alba et al 2013 Rogers amp
Chen 2013)
The production of miRNA miRNA duplexes occur in the nucleus while
Chapter 1 Introduction and Review of Literature
39
miRNA-RISC complexes are present in the cytoplasm where target mRNAs are
located This discordance of functional sites requires an export step during miRNA
biogenesis HASTY the Arabidopsis homolog of exportin-5 is thought to be involved
in miRNA nuclear export (Bollman et al 2003 Park et al 2005)The hasty mutant
exhibits pleiotropic defects in plant development and reduced accumulation of most
miRNAs as in the DCL1 or AGO1 mutants Howeverit is unclear whether the HASTY
proteins export the miRNA miRNA duplexes or the miRNA-RISC complexes in
plants
11345 Feedback regulation of miRNA biogenesis
MiRNA biogenesis is under feedback regulation such that the DCL1 and AGO1 genes
are themselves regulated by miRNAs DCL1 is itself a target of miR162 which leads to
the cleavage of the DCL1 mRNA (Xie et al 2003 de Alba et al 2013) Consistent
with this the levels of DCL1 mRNA are elevated in the mutants of miRNA processing
genes in which the miR162 abundance is reduced In addition there is a predicted
hairpin structure of miR838 in the 14th
intron of the DCL1 primary transcript
(Rajagopalan et al 2006) This prediction implies that DCL1-mediated processing
on this site can result in the cleavage of the DCL1 primary transcript into two truncated
fragments the N-terminal capped fragment and the C-terminal polyadenylated
fragment If this is the case it is possible that miRNA biogenesis competes with the
splicing event of the DCL1 pre-mRNA
The AGO1 is also regulated by miRNA There is a complementary site for
miR168 binding in the AGO1 mRNA and the transcript level of the AGO1 mRNA is
reduced through an mRNA cleavage mechanism (Vaucheret et al 2006 de Alba et al
2013 Rogers amp Chen 2013) indicating that the AGO1 mRNA levels are tightly
controlled by the levels of the AGO1 protein
1135 Mechanism of miRNA action
11351 MiRNA-directed mRNA cleavage
By forming the miRNA-RISC assembly miRNAs can direct the RISC to regulate gene
expression by two post transcriptional mechanisms mRNA cleavage or translational
repression (Bartel 2004 Jones-Rhoades et al 2006 Dalmay 2013) The degree
of miRNA-mRNA complementarity plays an important role for the
determination of the mechanism to be used A general rule is that while perfect or
Chapter 1 Introduction and Review of Literature
40
near-perfect complementarity induces mRNA cleavage central mismatches trigger
translational repression (Carrington amp Ambros 2003 Jones-Rhoades amp Bartel 2004)
Unlike most animal miRNAs plant miRNAs have near-perfect or perfect
complementarity to their targets and mRNA cleavage is assumed to be a predominant
mechanism by which miRNAs regulate gene expression in plants
In RNA cleavage mechanism miRNAs guide the AGO component of RISC to
cleave a single phosphodiester bond of the target mRNA within the miRNA-binding
site (Llave et al 2002b Tang et al 2003) The truncated fragments are then released
and degraded freeing the RISC to recognize and cleave another target mRNA This has
been validated by the detection of 3prime cleavage products that have 5prime ends that start at the
middle of the complementary regions The mRNA cleavage products are then further
degraded by other mechanisms The 3prime cleavage products of some miRNA targets are
degraded by the 5prime-3prime exonuclease XRN4 an Arabidopsis homolog of the major Yeast
mRNA degrading exonuclease Xrn1p It has been observed that the 3prime cleavage
products of some target mRNAs were accumulated in the xrn4 mutants (Souret et al
2004 Dalmay 2013) However the 3prime cleavage products of other miRNA targets do
not accumulate in the xrn4 mutants indicating that there are also XRN4 independent
mechanisms The 5prime cleavage products are typically untraceable by RNA gel blot
hybridization but can be detected by sensitive PCR based methods It has been found
that the 5prime cleavage products tend to obtain an oligo U tail at the 3prime ends This 3prime U tail
is correlated with shortening of the 5prime cleavage products from the 5prime end leading to the
conclusion that the 3prime uridylation causes 5 prime- 3 prime exonucleolytic decay of the 5prime cleavage
products (Shen amp Goodman 2004)
DCL1 HYL1 and SE interact with each other and are co-localized in the
nuclear bodies termed Dicing bodies or D-bodies in which some pri-miRNA shave
been found suggesting that D-bodies serve as a center for active miRNA processing
(Fang amp Spector 2007 Fujioka et al 2007 Song et al 2007) The miRNA
processing site appears to be distinct from the Cajal bodies that have previously been
found as a siRNA processing site (Pontes amp Pikaard 2008) The D-bodies include
SmD3 and SmB proteins that are found in the Cajal bodies However At collin an
additional marker for the Cajal bodies is not found in the D-bodies Moreover the D-
bodies are present in an At collin mutant lacking the Cajal bodies The pri-miRNAs of
miR171A and miR159A genes have been found to be co-localized with HYL1 and
Chapter 1 Introduction and Review of Literature
41
DCL1 in the D- bodies that are distinct from the Cajal bodies (Song et al 2007)
11352 MiRNA-directed translational repression
Translation repression is another mechanism exerted by plant miRNAs The regulatory
mechanism of miR172 which regulates flowering time and floral organ identity is
consistent with translation repression MiR172 over production does not affect the
transcript levels of APETALA2 (AP2) and AP2- like target mRNAs Instead the AP2
protein level is reduced in the miR172 over expressing mutant (Aukerman amp Sakai
2003 Chen 2004 Dalmay 2013) It was later shown that miR172 can also direct the
cleavage of the target mRNAs although the steady-state mRNA level is unchanged due
to feedback regulation (Schwab et al 2005 Jung et al 2007) It is clear that miR172
regulates its target genes primarily by translational repression rather than mRNA
cleavage Recently it has been reported that miR156157 and miR854 also regulate
their target genes through translational repression (Arteaga-Vazquez et al 2006
Gandikota et al 2007 Dalmay 2013) It was once thought that translational repression
is an exception to the rule However experimental evidence suggest a widespread
coexistence of translational repression and mRNA cleavage (Brodersen et al 2008)
1136 Regulatory roles of miRNAs in plant development
The miRNA target prediction has been a difficult task in order to obtain regulatory
targets without bringing in too many false predictions Plant growth and developmental
processes regulated by miRNAs are quite diverse and enormous Furthermore plant
miRNAs work cooperatively or antagonistically to establish a balanced regulation This
notion is well consistent with the findings that mutations in the regulators of miRNA
biogenesis transport and processing such as AGO1 DCL1 HYL1 HEN1 ABH1
and HASTY cause pleiotropic phenotypes (Mallory amp Vaucheret 2006 de Alba et al
2013) To date the roles of miRNAs have been mostly deduced from obtaining miRNA
over expressing transgenic plants or gain-of-function mutants in which miRNA-
resistant target genes are ectopically expressed
11361 Phase transition
Plants pass through a series of developmental phase transitions during their life span
beginning with seed germination continuing through vegetative phase change
reproductive phase change flowering initiation and finally seed production for the next
generation Because of their importance in understanding plant developmental
Chapter 1 Introduction and Review of Literature
42
processes genes regulating developmental transitions have been extensively studied
and underlying molecular mechanisms have been explored in various plant species
Majority of researches were mainly focused on vegetative phase change and
reproductive transition in Arabidopsis and Maize (Poethig 2003 Baurle amp Dean
2006) Particularly the mutations of the genes encoding various regulators of miRNA
biogenesis and transport such as HST SE and AGO exhibit altered juvenile to adult
vegetative phase change and vegetative to reproductive change (Hunter et al 2003
Peragine et al 2004 Vazquez et al 2004a Jones-Rhoades et al 2006 de Alba et al
2013)
Over-expression of the miR156a gene causes late flowering and delays
vegetative phase change (Wu amp Poethig 2006 Gandikota et al 2007 Wang et al
2008) It has been shown that miR156 negatively regulates a group of genes encoding
SQUAMOSA PROMOTER-BINDING PROTEIN-LIKE (Lewis et al 2007 de Alba et
al 2013 Rogers amp Chen 2013) transcription factors such as SPL3 SPL4 and SPL5
that promote flowering initiation In contrast transgenic plants over expressing
miR156-resistant SPL345 genes are early flowering Interestingly the endogenous
level of miR156 is temporally regulated In the early juvenile vegetative phase the
level of miR156 is very high but decreases rapidly before the onset of the adult juvenile
phase (Wu amp Poethig 2006)
The Arabidopsis early activation tagged (eat-D) mutant exhibits early flowering
with disrupted floral structure The mutant phenotypes are caused by over expression of
the miR172a-2gene (Aukerman amp Sakai 2003) MiR172 regulates a small group of
genes encoding APETALLA2 (AP2)-like transcription factors such as TARGET OF
EAT1 (TOE1) TOE2 SCHLAFMUTZE (SMZ) and SCHNARCHZAPFEN(SNZ)
TOE1 TOE2 SMZ and SNZ have been shown to participate in their productive phase
change by repressing a floral integrator FLOWERING LOCUS T (FT)(Jung et al
2007) Consistent with this toe1-2D 35STOE2 35SSMZ and 35SSNZ plants
exhibit late flowering (Jung et al 2007) MiR172 is also regulated temporally and
highly expressed in the adult vegetative phase both in Arabidopsis and maize (Chuck et
al 2007) Because the temporal expression patterns of miR156 and miR172 are
complementary to each other it has been suggested that the two miRNAs interact with
each other in regulating phase change (Chuck et al 2009 de Alba et al 2013) In
support of this view it has been shown that miR172 is down-regulated in the
Chapter 1 Introduction and Review of Literature
43
Corngrass1(Cg1) mutant in which miR156 is overproduced (Chuck et al 2007 de
Alba et al 2013) However there is no direct evidence for the direct linkage between
miR156 and miR172 It is also envisioned that miR156 and miR172 would not be
directly related For example miR156 regulates plastochron index that is the inverse of
leaf initiation rate and thus frequently used to analyze temporal patterns of plant
development In contrast miR172 does not affect plastochron (Wang et al 2008) The
down-regulation of miR172 in the maize miR156-over producing mutants would be
due to the prolonged juvenile vegetative phase in the mutants
Another miRNA that regulates flowering initiation is miR159 It regulates
MYB33 and MYB65 which are closely related to the barley GAMY B transcription
factor that responds to gibberellic acid (GA) The level of miR159 is elevated in GA-
treated seedlings but reduced in the GA biosynthetic ga1 mutant Transgenic plants
over producing miR159 exhibit delayed floral induction under short days (Achard et
al 2004) It is been suggested that MYB33 mediates GA signal to regulate LEAFY
(LFY) These observations indicate that miR159 plays an important role in the GA
signaling pathways that regulate proper floral induction under short days (Achard et al
2004)
11362 Floral development
Arabidopsis flowers consist of four distinct organs They arise in a characteristic
pattern within concentric rings called whorls from outside to inside four sepals four
petals six stamens and the central pistil consisting of two carpels Identities of the
floral organs are determined by three classes of regulatory genes classA classB and
class C genes (Krizek amp Fletcher 2005) The A and C class genes act antagonistically
to restrict their expression domains (Bowman et al 1991)
It is notable that miR172 also regulates floral architecture in addition to its role
in the timing of flowering initiation MiR172 inhibits the translation of the AP2 mRNA
(Chen 2004) Transgenic plants overproducing miR172 not only exhibit early
flowering but also show abnormal floral development which is quite similar to those
of the class A gene mutants such as the ap2 mutants (Aukerman amp Sakai 2003 de
Alba et al 2013) When the activity of the class A genes is inactivated that of the class
C genes such as AGAMOUS (AG) is elevated As a result the miR172- overproducing
transgenic plants exhibit no petals along with homeotic transformation of the sepals to
Chapter 1 Introduction and Review of Literature
44
the carpels (Aukerman amp Sakai 2003 Chen 2004)
Interestingly miR166 also regulates floral architecture The role of miR165166
in the regulation of meristem activity is closely linked to floral meristem formation and
organization (Zhao et al 2007 Miyashima et al 2013) Floral structure is severely
disrupted in the miR165166-overproducing mutants such as men1 and jba-1D in
which miR166 is overproduced (Williams et al 2005b Jung amp Park 2007) The men1
and jba-1D mutants have smaller gynoecium and reduced carpel number In an extreme
case the jba-1D homozygous plants lack visible carpels (Williams et al 2005b)
Similar phenotypes have been observed in the miR165 overproducers (Zhao et al
2007) It has been suggested that the phenotypes are possibly caused by altered AG
expression in the floral meristem (Jung amp Park 2007 Turchi et al 2013)
Other miRNAs also affect floral development Overproduction of miRNAs
involved in auxin signaling also affects floral architecture Transgenic plants
overproducing miR167 have defects in the formation of ovule and anther with reduced
fertility (Ru et al 2006) It has been shown that miR167 targets ARF6 and ARF8 that
play a role in gynoecium and stamen development and in regulating fertility (Ru et al
2006 Wu et al 2006) These observations suggest that auxin signals are also closely
related to floral organogenesis In addition transgenic plants over-producing miR164
and the cuc1cuc2 double mutants show fused sepals and reduced petals (Laufs et al
2004 Turchi et al 2013) suggesting that miR164 is also related to floral meristem
activity and boundary specification of the meristematic region
11363 Shoot apical meristem development
Shoot apical meristem comprises self-maintaining totipotent cells The shoot apical
meristem activity affects diverse aspects of plant developmental processes including
leaf development vascular development and lateral organ polarity (Bowman et al
2002) Because miR165166 plays a primary role in meristem formation
overproduction of miR165166 and multiple knockout mutants of its target genes
exhibit pleiotropic pheonotypes (McConnell et al 2001 Emery et al 2003 Kim et al
2005 Williams et al 2005b)
The targets of miR165166 include genes encoding the homeo domain-leucine
zipper (HD-ZIP) class III transcription factors such as PHABULOSA(PHB)
PHAVOLUTA (PHV) REVOLUTA(REV) ATHB15CORONA (CNA) and ATHB8
Chapter 1 Introduction and Review of Literature
45
PHB PHV and ATHB15 have overlapping functions in controlling meristem
development (Turchi et al 2013) Indeed the phb phv athb15 triple mutants have an
enlarged shoot apical meristem Consistent with this the miR166- overproducing men1
and jba-1D mutants exhibit enlarged shoot apical meristems (Kim et al 2005
Williams et al 2005b Turchi et al 2013) In the mutants the shoot apical meristems
are multiplied and large
The development of shoot apical meristems is also regulated by the WUSCHEL
(WUS)ndashCLAVATA (CLV) signaling pathway WUS positively regulates cell
proliferation In contrast CLV3 which is expressed in stem cells limits the WUS
expression and thus inhibits cell proliferation in the SAM (Sharma et al 2003)
Consistent with this notion WUS is expressed to a higher level in jba-1D
explaining the ectopic enlargement of the SAM in the mutant (Williams et al 2005)
While WUS is closely related to miR165166 in the shoot apical meristem
development the underlying molecular mechanisms are currently unclear It has been
observed that the miR166-over producing men1 plants have an arrested meristem
development (Jung amp Park 2007) In addition the heterozygous men1+ and the
homozygous men1 plants show different expression patterns of WUS and CLV3 It
appears that miR166 regulates WUS indirectly and influences the expressional balance
between WUS and CLV3 through a negative feedback loop (Jung amp Park 2007 de
Alba et al 2013)
The phv phb athb15 triple mutant has an enlarged shoot apical meristems the
rev phb phv mutant does not have functional shoot apical meristems (Prigge et al
2005) This distinction may be explained by the fact that while miR166 targets
primarily PHB PHV and ATHB15 miR165 targets all the five HD-ZIP III genes (Zhou
et al 2007) Consistent with this transgenic plants overproducing miR166 are distinct
from those overproducing miR165 The miR165-overproducing transgenic plants lack
functional shoot apical meristem and a pin-like structure is formed at the apical region
of the mutant These phenotypes are quite similar to rev phb phv triple mutant (Zhou et
al 2007 Liu et al 2013) The phenotypic distinction may because by the difference
of the cleavage efficiencies of the target mRNAs In accordance with this notion a few
35S miR166a transgenic lines exhibit no shoot apical meristems The men1
homozygous plants also show arrested meristem development (Jung amp Park 2007)
Chapter 1 Introduction and Review of Literature
46
MiR164 cleaves the CUC1 and CUC2 mRNAs as well as the NAC1 mRNA
CUC1 and CUC2 determines the organ boundary in the meristem Phenotypes of
transgenic plants that overproduce miR164 are similar to those of the cuc1cuc2 double
mutant that exhibits embryonic developmental defects such as cup-shaped cotyledons
and fused cotyledons (Laufs et al 2004) When a miR164-resistant CUC2 is
expressed the boundary domain is enlarged CUC also plays a role in embryonic shoot
meristem formation by regulating the SHOOT MERISTEMLESS gene It was therefore
concluded that miR164-mediated cleavage of CUC1 and CUC2 mRNAs determines the
sizes of the boundary domains in the shoot apical meristem and regulates separation of
the organs by restricting cell proliferation (Laufs et al 2004 de Alba et al 2013)
11364 Leaf development
The role of miR319 has been extensively studied in leaf development A dominant
jaw-D mutant overproducing miR319 shows uneven curved leaves with enlarged size
and five TCP genes which are closely related to the CINCINNATA (CIN) gene in
Antirrhinum majus are down regulated in the mutant suggesting that miR319 mediated
regulation of the TCP transcription factor genes are important for the final step of
leaf shaping (Palatnik et al 2003 Naz et al 2013) In addition to leaf shape and size
leaf polarity is also specified by miRNA MiR166-mediated cleavage of the HD-ZIPIII
mRNAs is a well to establish leaf polarity This discovery originates from the gain-of-
function mutants such as phb-1d and phv-1d wherein point mutations in the
complementary sites to miRNA helps the mRNA to evade or resist from miRNA-
mediated cleavage (McConnell et al 2001 Emery et al 2003 Naz et al 2013)
Plant leaves have a dorso-ventral polarity consisting of the adaxial (upper
surface) and abaxial (lower surface) sides The adaxial side is closer to the shoot
apical meristem Adaxial identity is specified by functionally redundant class III HD-
ZIP family genes such as PHB PHV and REV Ectopic expression of each genes
result in adaxialized leaves Dominant phb-1d and phv-1d mutants exhibit
transformation of the abaxial to adaxial fate and have rod-shaped leaves In contrast
miR165166-overproducing plants show pin-like cotyledons radicalized leaves and
abaxialized leaves (Chitwood et al 2007 Pattanayak et al 2013)
Leaf asymmetry is determined by miR166-mediated restriction of the
expression domains of the HD-ZIPIII genes The HD-ZIPIII genes are expressed in the
Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
12
The portion of xanthosine used for caffeine biosynthesis is derived from adenine
and guanine nucleotide pools which are produce de novo by salvage pathways there are
several potential pathways for xanthosine synthesis from AMP but the AMP rarr IMP
rarr XMPrarr xanthosine route is predominant All three enzymes involved in the
conversion AMP deaminase (EC 3546) IMP dehydrogenase and 5prime-nucleosidase
have been detected in tea leaves (Koshiishi et al 2001) Xanthosine for caffeine
biosynthesis can also be produced from guanine nucleotides by GMPrarrguanosine
rarrxanthosine pathway Guanosine deaminase (EC 35415) activity is found in cell free
extracts from young tea leaves (Negishi et al 1994) but GMP deaminase activity was
not detected in plants (Stasolla et al 2003 Zrenner et al 2006) The absence of GMP
deaminase activity suggests that a GMP rarr IMP rarr XMP rarr xanthosine route is not
functional in plants
The use of radiolabelled purine bases and nucleosides for the study of caffeine
biosynthetic pathway was facilitated by the fact that exogenous purine nucleotides are
rapidly hydrolyzed by the plant tissues forming purine nucleosides andor bases which
are then utilized in pathways which in most instances indirectly lead to xanthosine
Most of these purine compounds including adenine adenosine inosine hypoxanthine
and guanine are converted to their respective nucleotides by salvage enzymes
including adenine phosphoribosyl transferase (EC 2427) hypoxanthineguanine
phosphoribosyl transferase (EC 2428) adenosine kinase (EC 27120) and
inosineguanosine kinase (EC 2421) which usually have high activities in plant cells
The resultant nucleotides then enter the purine alkaloid biosynthetic pathway In the
case of guanosine it may be directly deaminated to xanthosine and utilized for caffeine
biosynthesis (Figure 13) (Ashihara et al 1997)
182 SAM cycle in plants
SAM is the methyl donor for the three methylation steps in the caffeine biosynthetic
pathway In this process SAM is converted to S-adenosyl-L-homocysteine (SAH)
which in turn is hydrolyzed to homocysteine and adenosine Homocysteine is recycled
via the SAM cycle to replenish SAM levels and adenosine is released from the cycle
Adenosine released from the cycle is converted to AMP directly andor indirectly via
adenine AMP is converted to xanthosine and used for caffeine biosynthesis Since
three moles of SAH are produced via the SAM cycle for each mole of caffeine that is
synthesized in theory this pathway has the capacity to be the sole source of both the
Chapter 1 Introduction and Review of Literature
13
purine skeleton and the methyl groups required for caffeine biosynthesis in young tea
leaves (Koshiishi et al 2001)
Methionine
S-Adenosyl-L-methionineHomocysteine
ATP
PPi+ Pi
Tetrahydrofolate
5-Methyl-
tetrahydrofolate
CH3+Adenosine
S-Adenosyl-L-
homocysteine
(xanthine
structure)(Methyl groups)
Caffeine
(1)
(2)(3)
(4)
Figure 14 The SAM cycle (activated methyl cycle) in plants (Ashihara amp Suzuki 2004)
Enzymes SAM synthetase SAM-dependent N-methyltransferases S-adenosyl-
homocysteine (SAH) hydrolase Methionine synthase Adenosine released from the cycle
is salvaged to adenine nucleotides and utilized both for purine structure of caffeine via
xanthosine and for re-synthesis of SAM via ATP
183 N-methyltransferase in caffeine biosynthesis Molecular cloning
The genes encoding for the enzymes involved in the caffeine biosynthesis were
successfully cloned from the coffee family (Ogawa et al 2001 Mizuno et al 2003a
Mizuno et al 2003b Uefuji et al 2003) The conserved amino acid regions of tea CS
(AB31280) the sequences of O-methyltransferases derived from plant origin and
Arabidopsis hypothetical proteins were made use of in designing primers for RT-PCR
and RACE technique Independent groups have isolated N-methyltransferase (NMT)
genes from coffee which have been classified based on the substrate specificity of the
recombinant proteins expressed in Ecoli (Table 15) PCR products thus obtained were
used as the probe to isolate full length cDNA from the library resulting in the
identification of all three N-methyltransferases involved in caffeine biosynthesis
(Figure 12) Several cDNA have been characterized from the coffee plants one for
xanthosine methyltransferase (XMTXRS) three for 7-methylxanthine methyltransferase
or theobromine synthase (MXMTCTS) and three for 37-dimethylxanthine
methyltransferase or caffeine synthase (DXMTCCS) (Table 15) (Kato amp Mizuno
2004)
A single gene encoding for xanthosine methyltransferase (XMTCmXRS1) was
identified which encoded for a polypeptide consisting of 372 amino acids with an
apparent molecular mass of 418kDa It is expressed almost uniformly in aerial tissues
Chapter 1 Introduction and Review of Literature
14
of C arabica including leaves floral buds and immature but not in mature beans
Three genes encoding 7-methylxanthine methyltransferase (theobromine synthase) have
been identified The number of amino acids in the putative polypeptides are 378 for
MXMT1 (427kDa) and 384 for MXMT2 and CTS2 (434kDa) They differ by insertion
or deletion of blocks of several residues in the C-terminal region Their catalytic
properties based on kinetic parameters such as Km values were different They are
expressed in young leaves floral buds and immature but not mature beans Three genes
were identified for 3 7-dimethylxanthine methyltransferase (caffeine synthase) DXMT
CCS1 and CtCS7 each encoding a 43 kDa polypeptide consisting of 384 amino acids
However their kinetic properties differ from each other such as DXMT and CCS1
showed Km values of 1200 and 157microM for theobromine respectively Expressions
profiles are also distinct DXMT being expressed exclusively in immature beans while
CCS1 expression is ubiquitous occurring in all tissues The presence of isoforms of
these enzymes with different properties suggests that caffeine is synthesized through
multiple pathways depending on availability and concentration of the substrate (Mizuno
et al 2001 Ogawa et al 2001 Mizuno et al 2003a Mizuno et al 2003b Uefuji et
al 2003) Genomic clones of theobromine synthase were obtained by PCR- RFLP
from C canephora (Satyanarayana et al 2005 Satyanarayana 2006) The genomic
clones are reported to have highly conserved exons and a variable third intron The
promoter for the theobromine synthase was cloned and characterized (Satyanarayana et
al 2005)
There is high degree of similarity in the coding region of coffee NMT genes
isolated so far though differences exist in the 3prime untranslated region (UTR) for these
genes The deduced amino acid sequence of these enzymes shows more than 85
homology between these genes Phylogenetic analysis indicates that they are more
closely related to C-methyltransferases including those for jasmonic acid salicylic acid
and benzoic acid than to other N-methyltransferases This suggests that coffee N-methyl
transferases belong to a distinct sub-group within plant methyltransferases Their
cellular localization determined by green fluorescent protein fusion method and β-
glucourodinase (GUS) showed that the all the three genes are localized in the cytoplasm
(Ogawa et al 2001 Vinod et al 2007) It was also shown that these were present
along with chlorogenic acid(Vinod et al 2007)
Chapter 1 Introduction and Review of Literature
15
Table 15 N-methyltransferases and their encoding genes involved in caffeine biosynthesis
Gene (Enzyme) Gene name Accession No Description Reference
Xanthosine methyltransferase
( 7-methylxanthosine synthase) (EC 211158)
CaXMT AB 048793 Immature fruit cDNA Screening (Uefuji et al 2003)
CmXRS1 AB 034699 Leaf cDNA library RACE (Mizuno et al 2003a)
CaMXMT1 AB 048794 Leaf cDNA library RACE (Ogawa et al 2001)
7-methylxanthine synthase transferase
(theobromine synthase) ( EC 211159)
CTS1 AB 034700 Immature fruit cDNA Screening (Uefuji et al 2003)
CaMXMT2 AB 984125 Leaf cDNA library Screening (Mizuno et al 2001)
CTS2 AB 054841 Leaf cDNA library Screening (Mizuno et al 2001)
37-methylxanthinemethyltransferase
(Caffeine synthase) (EC 211160)
CaDXMT1 AB 086414 Immature fruit cDNA Screening (Uefuji et al 2003)
CCS1 AB 0861414 Immature fruit cDNA Screening (Uefuji et al 2003)
CtCS7 AB 0864415 Immature fruit cDNA
NMT genomic clones
Theobromine synthase-1
CX10 Complete coding region PCR Satyanarayan 2006
CX8 Complete coding region PCR -do-
Theobromine synthase-1 PG-5 Promoter + coding region RAGE -do-
Theobromine synthase-1 PG-1 Promoter + coding region RAGE Satyanarayan 2006
PG-4 Promoter + coding region RAGE -do-
Theobromine synthase-1 NMT
(AY273814) Complete coding region PCR Kochko A et al 2010
Theobromine synthase-2 NMT
(AY362825) Complete coding region PCR Kochko A et al 2010
Chapter 1 Introduction and Review of Literature
16
19 Catabolism of caffeine
Caffeine is produced in young leaves and immature fruits and continues to accumulate
gradually during the maturation of these organs At the same time caffeine is slowly
degraded with the removal of the three methyl groups resulting in the formation of
xanthine Catabolism of caffeine was first reported in coffee leaves (Kalberer 1965)
Since then a number of tracer experiments using 14
C labelled purine alkaloids have
been reported (Suzuki amp Waller 1984 Ashihara et al 1996 Vitoria amp Mazzafera
1999 Mazzafera 2004) They demonstrated the major catabolic pathway is caffeine rarr
theophylline rarr 3-methylxanthine rarr xanthine Xanthine is further degraded by the
removal by the conventional purine catabolic pathway to CO2 and NH3 via uric acid
allatonin and allantoate (Figure 15) (Ashihara amp Crozier 1999 Stasolla et al 2003
Zrenner et al 2006) Caffeine catabolism usually begins with its conversion to
theophylline catalyzed by N7-demethylase The involvement of P450-dependent mono-
oxygenase activity for this reaction was suggested (Huber amp Baumann 1998
Mazzafera 2004) However the activity of this enzyme has not yet been determined in
cell free extracts [8-14
C] Theophylline degraded to CO2 at much faster rate than [8-14
C]
caffeine indicating that conversion of caffeine to theophylline is a major rate limiting
step of caffeine catabolism and a possible reason why caffeine accumulates in high
concentrations in tissues of C sinensis and C arabica It is widely believed that the
control of caffeine levels in plants is a function of the balance between rates of
synthesis and degradation and this balance seems to vary depending on the plant
species and the tissue developmental stage (Mazzafera 2004)
Chapter 1 Introduction and Review of Literature
17
Figure 15 Caffeine catabolic pathways Caffeine is mainly catabolised via theophylline and 3-
methylxanthine Xanthine is further degraded to CO2 and NH3 by the conventional
oxidative pathway Adapted from (Ashihara et al 2008)
Chapter 1 Introduction and Review of Literature
18
110 Genetic engineering of Coffee
Genetic transformation has potential applications in coffee agriculture for incorporating
genes which could provide desirable traits such as disease and insect resistance
drought and frost tolerance and herbicide resistance Transgenic technology can also be
used to increase nutritional value and improve cup quality produce varieties with
caffeine-free beans and for production of hybrid crops for molecular farming
Identification of target specific genes is one of the pre-requisites for developing
transgenic crops The availability of a large number of EST sequences in coffee and
initiation of coffee genome sequencing may speed up the gene discovery and accelerate
transgenic research efforts in coffee
1101 Insect Resistance
Production of insect resistant coffee plants is one of the major objectives of the
breeding programs The major pests attacking coffee include coffee berry borer (CBB
Hypothenemus hampei) white stem borer (WSB Xylotrechus quadripes) leaf miner
(Perileucoptera coffeela) and root nematodes (Meloidogyne spp and Pratylenchus
spp) The CBB is present in almost all the coffee growing countries and considered to
the most devastating pest in coffee To date there is no reported source of resistance to
CBB in the coffee gene pool Like CBB WSB is another serious pest in C arabica in
India and several other East Asian countries Both CBB and WSB belong to the order
Coleoptera For India controlling WSB is the biggest challenge and has therefore
become the highest research priority Robusta is generally resistant to WSB but the
interspecific Robusta Arabica hybrids are susceptible to WSB Although leaf miner is
not yet a serious pest in India and other East Asian countries it is an economically
important pest in East Africa and Brazil Effective chemical control of CBB and WSB
is difficult due to the nature of their life cycle inside the berry and stem respectively as
well as environmental concern regarding the use of pesticides Biological control
measures are adapted to combat these insect pests with varying degrees of success
Developing coffee plants resistant to these pests using genetic transformation
technology could be one of the alternative strategies to counter pest damage For insect
resistance several different classes of proteins from bacterial plant and animal sources
have been isolated and their insecticidal properties tested against many important pests
Amongst these proteins Bt toxins are most important and several transgenic crops
expressing Bt genes have been commercialized Coffee transgenic plants carrying a
Chapter 1 Introduction and Review of Literature
19
synthetic version of the cry1Ac gene have been produced (Leroy et al 2000 Mishra et
al 2002) Indeed this was the first report of an important agronomic trait being
introduced into a coffee plant The transgenic plants presented similar features in
growth and development compared to normal plants Transgenic plants highly resistant
to leaf miner under greenhouse conditions were tested under field conditions in French
Guyana for 4 years for pest resistance (Perthius et al 2005 Perthius et al 2006)
From 54 independent transformation events 70 of the events were resistant to leaf
miner Unfortunately the field trial was vandalized which led to the termination of the
experiment (Montagnon et al 2005)The effectiveness of Bt genes in controlling
coleopteran pests is well documented in corn and potato which indicates that Bt genes
might be effective against CBB The high toxicity of B thuringiensis sero var
israelensis against first in star larvae of CBB has been demonstrated (Mendez et al
2003) In another experiment an α-amylase inhibitor from Phaseolus vulgaris was
tested against CBB and found to have an inhibitory effect on its growth and
development (Grossi et al 2004)In addition to the CBB and WSB C arabica
varieties are also susceptible to endoparasitic root-knot nematode (Meloidogyne spp)
(Campos et al 1990 Mishra et al 2012)
So far 15 species have been reported to be parasites of coffee Controlling
nematodes is extremely difficult and currently seedling grafting with robusta rootstock
is followed as one of the control measure Sources of resistance specific to root-knot
nematodes have been identified in coffee trees (Bertrand et al 2001) and the Mex-1
gene conferring resistance to M exigua in C arabica is in the process of isolation (Noir
et al 2003)
1102 Tolerance to Abiotic Stress
In many coffee growing countries abiotic stress such as drought and frost are the major
climatic factors that limit coffee production Changes in climatic patterns due to global
climate change are considered increasingly important for coffee cultivation Drought
induces water stress in plants which affects vegetative growth and vigor and triggers
floral abnormalities and poor fruit set It also indirectly increases the incidence of pests
and diseases in the plants C arabica is generally more tolerant to water stress than C
canephora partly due to its extensive deep root system C racemosa is known to be a
good source of drought tolerance In India hybrids between C canephora and C
racemosa have been obtained and are currently under evaluation for drought tolerance
Chapter 1 Introduction and Review of Literature
20
In many coffee growing countries coffee is propagated in marginal areas where the
annual rainfall is below 1000thinspmm with prolonged dry spells of over 4-5 months In
those areas water shortage and unfavorable temperatures constitute major constraints
and the growth and productivity of C canephora are badly affected As with drought
periodic frost also affects coffee production in parts of Brazil The introduction of
drought and frost tolerant genes through genetic transformation would be of great
importance for alleviating these problems Research is now being carried out by several
groups to identify genes involved in biotic as well as abiotic stress The most promising
genetic engineering approach for drought tolerance includes the use of functional or
regulatory genes as well as the transfer of transcription factors In recent years plants
tolerant to high temperature and water stress have been the subject of intense research
(Chaves et al 2003 Chaves et al 2004 Coraggio et al 2004) For achieving drought
tolerance genes that have been targeted include those encoding enzymes involved in
detoxification or osmotic response metabolism enzymes active in signaling proteins
involved in the transport of metabolites and regulating the plant energy status (Chaves
et al 2003 Chaves et al 2004 Coraggio et al 2004) The dissection of molecular
mechanisms related to signal transduction and transcriptional regulation might help in
engineering drought tolerance in coffee
1103 Disease Resistance
Coffee leaf rust (CLR) caused by the fungus Hemileia vastatrix is the most important
disease in coffee with substantial loss to coffee production and productivity in all the
coffee growing countries In addition to leaf rust coffee berry disease (CBD) caused by
fungus Colletotrichum kahawae can be a devastating anthracnose leading to substantial
crop loss in Africa Several other fungal and bacterial diseases may also affect coffee
to a extent C arabica is more susceptible to many diseases than C canephora Though
most of the disease control measures rely upon chemical control they are more
expensive and labour intensive The long-term solution is the breeding of resistant
varieties which is the focus of many breeding programs However breeding for disease
resistant varieties is time consuming due to the perennial nature of coffee with its long
gestation period India has a long history of C arabica breeding especially for leaf rust
resistance and is the first country to demonstrate the existence of multiple races of leaf
rust Resistance to CLR is conditioned primarily by a number of major (SH) genes and
coffee genotypes are classified into different resistance groups based on their
Chapter 1 Introduction and Review of Literature
21
interaction with different rust races pathogen (van der Vossen 2001) Currently C
canephora provides the main source of resistance to pests and diseases including CLR
(H vastatrix) and CBD (C kahawae) and therefore used in breeding programs Other
diploid species like C liberica and C racemosa are being used as a source of resistance
to coffee leaf rust and coffee leaf miner respectively (Guerreiro et al 1999
Sreenivasan et al 1989) The development of coffee varieties resistant to major fungal
diseases such as CLR and CBD using transgenic technology will benefit the coffee
industry immensely During the last 15 years significant progress has been made in the
area of host-pathogen interactions (Staskawicz et al 1995 Feys et al 2000
Shivaprasad et al 2012) and many resistance genes involved in recognizing invading
pathogens have been identified and cloned (Takken et al 2000) A number of
signalling pathways induced because of pathogen infection have been dissected (Dong
et al 1998 Navarro et al 2006) Many antifungal compounds synthesized by plants to
combat fungal infection have been identified (Does et al 1998) Understanding the
specific induction of targeted pathways and identification of specific pathways
responsible for particular fungal resistance is important in order to employ this strategy
in transgenic technology The recent investigation of gene expression during coffee leaf
rust infection could provide an insight into the defence pathways operating in coffee
(Fernandez et al 2004 Nardi et al 2006) Efforts have been made to identify and
clone resistance genes from coffee for achieving durable resistance The genetic and
physical map of two resistance genes SH3gene conferring resistance to rust (Prakash et
al 2004) and the Ck1 gene conferring resistance to C kahawae CBD (Gichuru et al
2006) have been established These genes could be used for molecular marker assisted
breeding programs (Mishra et al 2012)
1104 Production of low-caffeine coffee
A widespread belief that the ingestion of caffeine can have adverse effects on
health resulted in the increased demand for decaffeinated coffee (Ashihara amp Crozier
2001) Several method were used to obtain decaffeinated plants by conventional
breeding techniques using low yielding varieties and mutants that accumulated
relatively low amounts of caffeine when compared to the commercially available ones
Low-caffeine and decaffeinated coffee represent around 10 of the coffee sales
around the world (Ribas et al 2006b) The industrial process for coffee decaffeination
can be expensive and affects the original flavour and aroma in coffee (David 2002)
Chapter 1 Introduction and Review of Literature
22
Transgenic coffee plants with suppressed caffeine synthesis using RNA interference
(RNAi) technology have been obtained (Ogita et al 2003 Ogita et al 2004) Specific
sequences in the 3prime untranslated region of the theobromine synthase gene (CaMXMT1)
were selected for construction of RNAi short and long fragments The caffeine and
theobromine content of the transgenic plants were reduced by ~ 70 when compared to
the untransformed plants In C canephora RNAi technology has also been employed
to silence the N-methyl transferase gene involved in caffeine biosynthesis (Vinod et al
2007) Promoter of an N-methyltransferase (NMT) gene involved in caffeine
biosynthesis was cloned (Satyanarayana et al 2005 Satyanarayana 2006) which will
be very useful for studying the regulation of caffeine biosynthesis
1105 Improvement in cup quality
Improvement in coffee cup quality requires elaborate knowledge of the chemical
constituents as well as the metabolic pathways involved in the elaboration of quality
The constituents of coffee beans include minerals proteins carbohydrates caffeine
chlorogenic acids (CGA) glycosides lipids and many volatile compounds that give
flavour to coffee by roasting Among these the role of three major constituents
sucrose CGA and trigonelline have been studied in coffee The sucrose content of
coffee bean is associated with coffee flavour the higher the sucrose content in green
beans the more intense will be the cup flavour (Clifford et al 1985 de Maria et al
1996) The sucrose content of C arabica (82-83) is higher than C canephora (33ndash
40) The sucrose amino acids ratio in green beans determines the profile of volatile
compounds Manipulating sucrose content in coffee bean is therefore important in
improving cup quality The sucrose synthase gene (CcSUS2) from C canephora has
been cloned and sequenced (Leroy et al 2005) This provides an opportunity to
manipulate the sucrose content in coffee Chlorogenic acids are products of
phenylpropanoid metabolism Chlorogenic acids are a group of hydroxycinnamoyl
quinic acids (HQA) formed by esterification between caffeic acids coumaric acids and
quinic acids (Clifford et al 1999) They are present in relatively large quantities in the
coffee bean and are the precursor of phenolic compounds in roasted beans C
canephora beans contain higher CGA (10) compared to C arabica beans (6-7)
CGA are known to have antioxidant properties as well as being associated with disease
resistance (Campa et al 2003) Genetic manipulations of genes involved in CGA
synthesis can serve either of these purpose by up- or down regulating the pathway
Chapter 1 Introduction and Review of Literature
23
Phenylalanine ammonia-lyase (PAL) catalyzes the first step of the phenylpropanoid
pathway leading to the synthesis of a wide range of chemical compounds including
flavonoids coumarins hydroxycinnamoyl esters and lignins (Hahlbrock amp Scheel
1989) Recently the full length cDNA and corresponding genomic sequences of PAL
from C canephora was isolated characterized and functionally validated (Mahesh et
al 2006 Parvatam et al 2006) This has opened up new possibilities for manipulating
the level of the PAL enzyme in coffee which in turn can be useful for improvement of
cup quality and manipulating antioxidant properties in coffee
1106 Fruit Ripening
Uniformity during fruit ripening is decisively related to cup quality in coffee and
consequently to the value of the product Fruits at the ideal ripening stage produce the
best organoleptic characteristics for coffee The presence of over ripened or green fruits
changes the acidity the bitterness and consequently the cup quality In order to
maximize uniform ripening of coffee fruits it is essential to control the action of genes
involved in the last step of maturation process Ethylene is known to trigger ripening
and increasing ethylene biosynthesis is associated with various stages of ripening
process (Protasio et al 2005) To control coffee fruit maturation two of the major
genes involved in ethylene biosynthesis namely ACC synthase and ACC oxidase
have been cloned (Protasio et al 2005 Neupane et al 1999) Introduction of the ACC
oxidase gene in antisense orientation have been achieved in both C arabica and C
canephora The effect of the transgene on ethylene production and fruit maturation has
yet to be reported The inhibition of genes downstream to the initial ethylene burst is
also an option to control coffee fruit maturation (Ribas et al 2006)
111 RNA interferenceRNAi
The regulation of gene expression at post transcriptional level has recently attracted
much attention because of the discovery of the phenomenon called RNA interference
(RNAi) RNAi based silencing techniques are more efficient than antisense mediated
gene silencing (Wesley et al 2001) The effect was first observed in plants wherein
silencing was observed when some copies of a transgene was in the forward orientation
with respect to the promoter and some in the reverse orientation This resulted in the
formation of double stranded RNA in the system The phenomenon in which
experimentally introduced double stranded RNA (dsRNA) leads to the loss of
expression of the corresponding cellular gene by sequence specific RNA degradation
Chapter 1 Introduction and Review of Literature
24
was termed as RNA interference or RNAi The protective effect of RNA silencing in
reducing virus infectivity supports the view that PTGS has evolved as a mechanism to
defend plants against virus infection and also to moderate the possible deleterious
genome-restructuring (insertional) activity of virus-like mobile genetic elements eg
retrotransposons A growing body of biochemical and genetic data has further
demonstrated that plants animals and yeasts share related mechanisms of specific
degradation of RNAs in which double-stranded forms of RNA are initiator molecules
(Brenstein et al 2001)
The key insight into the process of PGTS was provided by the experiments
conducted by Hamilton amp Baulcombe (1999) who identified the product of RNA
degradation as small RNA (siRNA) species of ~25nt of both sense and antisense
polarity SiRNA are formed and accumulate as double stranded RNA molecules of
defined chemical structures Both genetic and biochemical approaches were undertaken
to understand the basis of silencing Genetic screens were carried out in fungus
Neurospora carassa the alga Chalmydomonas reinhardtii and plant Arabidopsis
thaliana to search for detective mutants in PTGS
A combination of results obtained from several in vivo and in vitro experiments
have gelled into a two step mechanistic model for RNAiPTGS The first step referred
to as the RNAi initiating step involves binding of the RNA nucleases to a larges
dsRNA and its cleavage into discrete ~21-25nt RNA fragments This is ATP-
dependent reaction which involved RNase III - type endonucleases which make
staggered cuts in both strands of the dsRNA leaving 3prime overhang of 2 nucleotides with
5prime- phosphate 3prime-hydroxyl and no modification in the sugar phosphate backbone
(Hamilton amp Baulcombe 1999 Elbashir et al 2001) SiRNAs are the direct products
of dsRNA cleavage by the multi-domain RNase III enzyme DICER (Figure 16)
(Brenstein et al 2001 Karthikeyan et l 2013)
In the second step or commonly known as the effector step the double stranded
siRNAs produced in the first step bind to an RNAi specific protein complex to form a
RISC The complex undergoes activation in the presence of ATP so that the antisense
component of the unwound siRNA becomes exposed and allows the RISC to perform
the downstream RNAi reaction
Chapter 1 Introduction and Review of Literature
25
Several enzymes are involved in RNAi based on their role genes that encode for them
are classified into RNAi initiators RNAi effectors RNA dependent RNA polymerases
and silencing genes Two C elegans genes rdeI and rde4 (rde stands for lsquo RNAi
deficientrsquo) are believed to be involved in the initiation step of RNAi The C elegans
rde1 gene is a member of a large family of genes and is homologous to the Neurospora
qde2 (qde stands for ldquoquelling deficientrdquo) and Arabidopsis AGO1 (AGO stands for
agronaute AGO1 was previously identified to be involved in Arabidopsis
development) Although the functions of these genes are not clear a mammalian
member of RDE1 family has been identified as a translation initiation factor (Thakur
2005) Important genes for the effector step of PTGS in C elegans are rde2 and mut7
genes These genes were identified from heterozygous mutant worms that were unable
to transmit RNAi to their homozygous offsprings Worms with mutated rde2 or mut 7
genes show defective RNAi mut-7 gene encodes a protein with homology to the
nuclease domains of RNAase D and a protein implicated in Werner syndrome (a rapid
ageing disease in humans) (Grishok et al 2001 Kamath-Loeb et al 2012s)
Neurospora qde-1 Arabidopsis SDE-1SGS-2 and C elegans ego-1 appear to encode
RNA dependent RNA polymerase (RdRPs) It assumed that this is a proof that an RdRp
activity is required for RNAi Certainly the existence of an RdRp might explain the
remarkable efficiency of dsRNA induced silencing if it amplifies either the dsRNA
prior to cleavage or the siRNAs directly (Muers 2013) In C elegans ego-1 mutants
(ego stands for lsquoenhancer of glp-1) RNAi functions normally in somatic cells but is
defective in germline cells where ego-1 is primarily expressed In Arabidopsis SDE-
1SGS-2 mutants (SGS stands for suppressor of gene silencing) siRNA are produced
when dsRNA is introduced via an endogenously replicating RNA virus but not
introduced by a transgene It has been proposed that perhaps the viral RdRP is
substituting for the Arabidopsis enzyme in these mutants Random degradative PCR
model suggests that an RdRP uses the guide strand of an siRNA as a primer for the
target mRNA generating a dsRNA substrate for Dicer and thus more siRNAs
Several genes controlling RNA silencing in plants have been identified through
genetic screens of Arabidopsis mutants impaired in transgene induced RNA silencing
They encode a putative RNA-dependent RNA polymerase (SGS2SDE1) a coiled
protein (SGS3) a protein containing PAZ and Piwi domains (AGO1) and an RNA
helicase (SDE3) The putative SGS2SDE1 of Neurospora is related to QDE-1 and
Chapter 1 Introduction and Review of Literature
26
EGO-1 of C elegans whereas PAZPiwi protein AGO is related to QDE-2 of
Neurospora RDE-1of C elegans RNA helicase SDE3 is related to SMG-2 of C
elegans and Mut-6 of Chlamydomonas MUT-7 gene of C elegans encodes a protein
similar to Rnase D whereas the Drosophila DICER gene encode a protein similar to
RNase III An Arabidopsis ortholog of DICER gene has been identified called CAF
SIN1 SUS1(Thakur 2005 Poethig 2009)
Since the RNAi machinery is present constitutively within the eukaryotic cell it
is important to explore the metabolic advantages that are accorded by the RNAi related
proteins during the intrinsic normal growth of cells and development of organisms The
natural RNAi machinery not only keeps the mobile transposable elements from
disrupting the integrity of the genomes as suggested by the analysis of model organisms
like A thaliana C elegans D melanogaster (Tabara et al 1999 Wu-Scharf et al
2000 Aravin et al 2001 Hamilton et al 2002 Lisch 2009) but also participates in
development of organisms Genetic defects in Celegans RNAi genes ego1 and dicer
cause known specific developmental errors (Grishok et al 2000 Grishok et al 2001
Knight amp Bass 2001 Hall et al 2013) Similarly the Argonaute family of genes of A
thaliana (especially the ZWILLE proteins) is also responsible for plant architecture and
meristem development (Carmell et al 2002 Hamant and Pautot 2010) and the Dicer
homologue of A thaliana CAF1 is required for embryo development (Golden et al
2002 Nakano et al 2011) Thus genetic evidence illustrates the role of the RNAi
machinery as a controller of development related genes The mechanistic details of
these developmental processes are beginning to emerge
Chapter 1 Introduction and Review of Literature
27
Figure 16 Mechanism of gene silencing induced double stranded RNA The dicer enzyme to
produce siRNAs cleaves the RNA The siRNA generated bind to the nuclease complex
RISC The antisense component in the siRNA in the RISC guides the complex towards
the cognate mRNA resulting in endonucleolytic cleavage of the mRNA
Chapter 1 Introduction and Review of Literature
28
112 MicroRNA or MiRNA
In 1991 Ambros and co-workers first isolated a lin-4 mutant of Celegans which was
arrested at the first larval stage (Lee et al 1993) Later on the let-7 mutation was
isolated in the same system which was responsible for development through the fourth
larval stage Both lin-4 and let-7 encode short 22-nucleotide mature RNAs and were
called short temporal RNA because they control the temporal development program of
C elegans The mature lin-4 RNA defines (negatively regulates) the mRNA expression
of the lin-14 and lin-28 hetrochronic genes with the antisense mediated repression
mechanism of translation initiation and thus specifies the fate of cells during the first
three larval stages Recent studies have revealed that the short temporal RNAs are
actually members of a group of tiny RNAs (21to 28 nucleotides) called the micro-
RNAs (miRNA) (Bartel 2009) isolated members of which could easily run to a few
thousand which includes both those were identified computationally and by deep
sequencing Some of the components of the RNAi machinery have also been clearly
established as the effector proteins for the maturation of miRNAs
113 MicroRNAs in plants
1131 Discovery
MicroRNAs have been discovered by three basic approaches Direct cloning forward
genetic direct cloning and bioinformatic prediction followed by experimental
validation The most direct method of miRNA discovery is to isolate and clone small
RNAs from biological samples and several groups have used this approach to identify
plant miRNAs (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002 Xie et al 2003 Sunkar et al 2005 Williams et al 2013) The cloning methods
adapted were similar to those used to identify large numbers of animal miRNAs
(Lagos-Quintana et al 2001 Lau et al 2001 Aravin amp Tuschl 2005) which involve
isolating small RNAs followed by oligonucleotide adapter ligation reverse
transcription amplification and sequencing Some protocols incorporate methods to
enrich for Dicer cleavage products (ie molecules with 5prime-phosphates and 3prime-
hydroxyls) and to concatemerize the short cDNAs so that several may be identified in a
single sequencing read (Lau et al 2001 Llave et al 2002a Jagadeeswaran amp Sunkar
2013)The initial cloning experiments in Arabidopsis identified 19 miRNAs belonging
to 15 families (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002) although hundreds of other small RNAs such as siRNAs and RNA degradation
Chapter 1 Introduction and Review of Literature
29
fragments were also cloned through the direct cloning methods To select miRNAs
from the pool of small RNAs secondary structures of the RNA molecules were
examined in compliance with the acknowledged structural features of miRNAs
(Ambros et al 2003) The secondary structures were validated using several ad-hoc
software tools such as the Mfold or RNAfold programs (Reeder et al 2006) Finally
the existence of mature miRNAs was determined by RNA gel blot hybridization These
direct cloning methods were successful in isolating many miRNAs in Arabidopsis
(Reinhart et al 2002 Sunkar amp Zhu 2004) Rice (Sunkar et al 2005) Poplar (Lu et
al 2005) moss (Arazi et al 2005) and Tobacco (Billoud et al 2005)
Although miRNAs were first discovered through forward genetic screens in
worms (Lee et al 1993 Reinhart et al 2000)only few A thaliana miRNA gene
families have been discovered by this method (Chen 2008) in plants MiRNA
involvement in plant mutant phenotypes were not properly inferred till the cloning
experiments established that plant genomes contain numerous miRNAs (Park et al
2002 Reinhart et al 2002 Rhoades et al 2002) MiRNA loss-of-function allele has
been identified by forward genetic screens early extra petala1 is caused by a
transposon insertion ~160bp upstream of the predicted miR164c stem loop resulting in
flowers with extra petals (Baker et al 2005 Irish 2008) The fact that loss-of-function
miRNA mutants have been rarely discovered using forward genetics reflects small
target size for mutagenesis coupled with redundancy in nearly all evolutionarily
conserved plant miRNAs which are encoded by gene families (Table16) Family
members are likely to have overlapping functions buffering against loss at any single
miRNA locus Over expression screens can circumvent redundancy limitations At least
four plant miRNAs miR319 (also known as miR-JAW) miR172 (also known as EAT)
and miR166 were isolated in over expression screens for dominant mutants with
developmental abnormalities (Aukerman amp Sakai 2003 Palatnik et al 2003 Kim et
al 2005 Williams et al 2005b Schommer et al 2012) Mutations in miRNA target
sites which can prevent the entire family of miRNAs from repressing a target gene can
also circumvent redundant functions of miRNA family members
The dominant mutations in the HD-ZIP genes PHB PHV and REVOLUTA
(REV) in Arabidopsis and ROLLEDLEAF1 (RLD1) in Maize result in adaxialization of
leaves andor Vasculature (McConnell amp Barton 1998 McConnell et al 2001 Nelson
et al 2002 Zhong amp Ye 2004 Allen amp Millar 2012) and are all caused by mutations
Chapter 1 Introduction and Review of Literature
30
in miR166 complementary sites (Rhoades et al 2002 Emery et al 2003 Jones-
Rhoades amp Bartel 2004 Mallory et al 2004 Zhong amp Ye 2004)
In both plants and animals cloning was the initial means of large-scale
miRNA discovery (Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Reinhart et al 2002 Mendes et al 2012) However cloning is biased toward
RNAs that are expressed highly and broadly MiRNAs expressed at low levels or
only in specific cell types or in response to certain environmental stimuli were more
difficult to clone Sequence based biases in cloning procedures might also cause
certain miRNAs to be missed Because of these limitations bioinformatics approaches
to identify miRNAs have provided a useful complement to cloning
A straightforward use of bioinformatics has been carried out to find homologs
of known miRNAs both within the same genome and in the genomes of other species
(Pasquinelli et al 2000 Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Mendes et al 2009) A more difficult challenge is to identify miRNAs
unrelated to previously known miRNAs This was first accomplished for
vertebrate nematode and fly miRNAs using algorithms that search for conservation
of sequence and secondary structure (ie miRNA stem-loop precursors) between
species searching for patterns that are characteristic of miRNAs (Lai et al 2003 Lim
et al 2003 Lim et al 2005)
Most of the methods identified numerous miRNAs but are necessarily more
relaxed in terms of allowed structures Some approaches take advantage of the high
complementarity of plant miRNAs to target messages implementing the requirement
that the candidate has conserved complementarity to mRNAs (Jones-Rhoades amp Bartel
2004 Adai et al 2005) This additional filter has been useful for distinguishing
authentic plant miRNAs from false positives which later extended to mammalian
miRNA gene prediction (Xie et al 2005 Mendes et al 2012)
Another criteria used by most algorithms in the filters is evolutionary
conservation In plants mature miRNA sequences are conserved to a higher degree
than their precursor sequences For example Arabidopsis and rice diverged more than
130 million years ago (Friis et al 2004) but some miRNAs are highly conserved
in these plant species (Reinhart et al 2002) Furthermore various parameters for the
secondary structures such as free energy the number of paired residues within a
Chapter 1 Introduction and Review of Literature
31
miRNA or the number and size of bulges are also used to validate the predictions
(Jones-Rhoades amp Bartel 2004 Wang et al 2004 Adai et al 2005 Mendes et al
2012)
1132 Conserved microRNAs in Plants
Till date cloning genetics and bioinformatics screens have resulted in annotation of
approximately 184 potential Arabidopsis miRNAs (miRBase release 13) These
miRNAs seem to arise through gene duplication and subsequent diversification and
represent approximately 100 families (Maher et al 2006 Axtell 2013) Twenty one
families represented by 92 genes are clearly conserved in species beyond Arabidopsis
(Table 15) The presence of these miRNA families in other sequenced plant genomes
indicates that different plant species have an evolutionarily fluid set of miRNAs
(Willmann amp Poethig 2007) Recent studies on mosses one of the most ancient land
plants have shown that at least 14 miRNA families are conserved in eudicots and
mosses and therefore it appears that plant miRNAs share a common ancient origin
(Arazi et al 2005 Axtell amp Bartel 2005 Talmor-Neiman et al 2006 Zhang et al
2006 Axtell 2013) The number of members per family in a genome ranges from 1-32
with the exception of miR-430 which is represented by a cluster of ~80 loci in zebra
fish (Giraldez et al 2005)
In plants the number of members in each family correlates among examined
species certain families contain numerous members in all three species (eg miR156
miR166 miR169) whereas others consistently contain fewer genes (eg miR162
mir168 miR394) (Table 16) Although it is still unclear why certain miRNA are
encoded by more than one gene in plants (eg 12 genes encoding miR156) this
correlation might suggest functional significance of various miRNA family sizes Most
annotated plant miRNAs are conserved throughout flowering plants while a few
miRNAs are found only in a single sequenced genome and thus they could be
evolutionarily lsquoyoungrsquo miRNAs For example Arabidopsis has several miRNA
families that are not found in poplar or rice suggesting that miRNAs could be
generated continuously during evolution (Allen et al 2004a Rajagopalan et al
2006 Fahlgren et al 2007 Axtell 2012 de Alba et al 2013) Consistent with the
notion that non conserved miRNAs are of a more recent evolutionary origin these
miRNAs are mainly encoded by a single locus in the genome Within each family the
mature miRNA is always located in the same arm
Chapter 1 Introduction and Review of Literature
32
Table 16 MicroRNA families conserved in plants
miRNA
family
Arabidopsis Oryza Populus
miR156 12 12 11 miR159319 6 8 15 miR160 3 6 8 miR162 2 2 3 miR164 3 5 6 miR166 9 12 17 miR167 4 9 8 miR168 2 2 2 miR169 14 17 32 miR171 4 7 10 miR172 5 3 9 miR390 3 1 4 miR393 2 2 4 miR394 2 1 2 miR395 6 19 10 miR396 2 5 7 miR397 2 2 3 miR398 3 2 3 miR399 6 11 12 miR408 1 1 1 miR403 1 0 2 miR437 0 1 0 miR444 0 1 0 miR445 0 9 0 Total 92 127 169
All known miRNA families that are conserved between more than one plant species are listed
together with the number of genes identified in the sequenced genomes Rice miRNA families that have orthologs in maize but do not appear to have orthologs in the eudicots (Arabidopsis and Populus) are marked with an asterisk The following families contain miRNA genes annotated with
more than one number miR156 (miR156 and miR157) miR159319 (miR159 and miR319) miR166 (miR165 and miR166) miR171 (miR170 and miR171) and miR390 (miR390 and miR391)
of the stem loop (5prime- or 3prime) as it would be expected if the genes carry common ancestry
(Figure 17) The sequence of the mature miRNA and to some extent the segment on
the opposite arm of the hairpin to which it pairs is highly conserved between the
members of the same family (both within and between the species) while the sequence
secondary structure and length of the intervening ldquolooprdquo region can be highly divergent
within the same family
The patterns of paring and non pairing nucleotides are often conserved between
homologous miRNA stem loops from different species (Figure 17) The significance of
conserved mismatches is opined to guide DCL1 to cleave at the appropriate positions
along the stem loop
Chapter 1 Introduction and Review of Literature
33
Figure 17 Representative stem loop of miR164 Segments corresponding to the mature miRNAs
are shown in red (Adapted from Rajagopalan et al 2006)
Although many miRNAs are highly conserved in plants the degree of conservation in
most cases is quite low between plants and animals one exception is miR854 A recent
study has revealed that miR854 is conserved between Arabidopsis and animals and that
the Arabidopsis miR854 differs from the human miR854 only by a single nucleotide
(Arteaga-Vazquez et al 2006 Axtell 2012 de Alba et al 2013) The Arabidopsis
miR854 has multiple binding sites in the 3prime-untranslated region (UTR) of
OLIGOURIDYLATE-binding-PROTEIN mRNA 1bA (UBP1b) and represses the
translation of the UBP1b mRNA The animal miR854 is also complementary to a site in
the 3prime-UTR of the UBP1b homolog in animals However it is currently unclear how the
animal miR854 regulates its target mRNA Another distinction between plant and
animal miRNAs is the genomic organization of the miRNA genes While plant miRNA
genes are located predominantly in the intergenic regions animal miRNA genes tend to
localize in the intragenic regions both in the introns or exons of protein coding genes
Moreover miRNA genes are often clustered within a single precursor transcript
whereas individual plant miRNAs are derived from individual precursor RNAs (Bartel
Chapter 1 Introduction and Review of Literature
34
2004 Bonnet et al 2004 Kim 2005 Vaucheret et al 2006 Marco et al 2013)
1133 Non-conserved miRNA and challenges in annotation
Most miRNAs are conserved throughout flowering plants (Table 15) but many more
were found only in a single sequenced genome thus considered to be of a more recent
evolutionary origin The extended homology between few non conserved miRNAs
precursors and their target genes provides strong evidence that these relatively ldquoYoungrdquo
miRNAs arose from the duplication of the target gene segments (Allen et al 2004)
Several non-conserved miRNAs including miR161 miR163 miR173 miR447
miR475 and miR476 are known to direct cleavage of target transcripts (Allen et al
2004a Adai et al 2005 Sunkar et al 2005 de Alba et al 2013) It is difficult to
confidently predict targets for many because it is not possible to use complementary
site as a filter against false-positive target predictions It is also difficult to
confidently annotate non-conserved miRNAs as miRNA rather than siRNAs The
established minimal standard for miRNA annotation is a small RNA with detectable
expression and the potential to form a stem-loop when joined to flanking genomic
sequence (Kasschau et al 2003) In practice these requirements are too loose to
definitively categorize many small RNAs cloned from plants Many plant siRNAs are
detectable on blots (Vazquez et al 2004b) and hundreds of thousands of non-
miRNA genomic sequences can be predicted to fold into secondary structures that
resemble structures of plant miRNA precursors (Jones-Rhoades amp Bartel 2004)
Therefore without conservation of both sequence and secondary structure it is
difficult to be confident that a given cloned RNA originated from a stem-loop (ie is a
miRNA) rather than from a double-stranded RNA (ie is a siRNA) In fact many of
the thousands o f small RNAs cloned from Arabidopsis (Gustafson et al 2005) would
probably meet the literal requirements for annotation as miRNAs A few of these
sequences might be miRNAs but others that meet the literal criteria probably are not
so The challenges of annotating non conserved small RNAs were evidenced by
three related small silencing RNAs that were originally annotated as miRNAs
that turned out to be trans-acting siRNAs (tasiRNAs)(Kanno et al2013)
Although most miRNAs that are broadly conserved among flowering plants are
now being identified and experimentally validated (Table 16)(Jones-Rhoades amp
Bartel 2004) the challenges and ambiguities for classifying non-conserved miRNAs
prevent meaningful estimates of the total number of miRNA genes in Arabidopsis and
Chapter 1 Introduction and Review of Literature
35
other plant genomes It is possible that miRNAs might have escaped detection
because they are non-conserved and expressed in specific tissues or conditions and can
only be identified by using high- throughput sequencing
1134 MicroRNA biogenesis
11341 Transcription of microRNA precursor
Plant miRNAs are primarily found in the genomic regions that are not associated with
the protein coding genes (Reinhart et al 2002) and it appears that most if not all plant
miRNAs are produced from their own transcriptional units Plant miRNA genes are
occasionally clustered near each other in the genome suggesting transcription of
multiple miRNAs from a single primary transcript [eg the miR395 cluster (Grishok
et al 2001 Jones-Rhoades amp Bartel 2004b)] but this polycistronic arrangement of
miRNA genes appears far less frequently in plants than in animals (Bartel 2004)
Northern blot EST and mapping evidence indicate that plant miRNA primary
transcripts (also known as pri-miRNAs) as in animals are longer than needed to
encompass the miRNA stem-loops (Palatnik et al 2003 Jones-Rhoades amp Bartel
2004b) At least some of these pri-miRNA transcripts appear to be spliced
polyadenylated (Kurihara amp Watanabe 2004) and capped (Xie Z et al 2005) Two
rice miRNAs are contained within transcripts that contain exon junctions within the
presumptive stem-loop precursor implying that in these cases splicing is a prerequisite
for Dicer recognition (Sunkar et al 2005) Plant pri-miRNAs are over 1kb in length
and they are usually preceded by typical TATA box motifs They can also undergo
canonical splicing polyadenylation and capping All these observations indicates RNA
polymerase II is probably responsible for transcribing most plant miRNAs (Xie Z et
al 2005) as appears to be the case for many animal miRNAs Relatively little is
known about the regulation of miRNA transcription in plants but there is no reason
to suspect that this regulation would differ from that of protein-coding transcripts as
the bioinformatics analysis of the sequence upstream of the transcriptional start
sites of the miRNA genes showed the presence of putative binding motifs of
number of known transcription factors (Megraw et al 2006 Sethupathy 2013)
11342 MicroRNA processing and export
In plants maturation of miRNA is a step wise process A miRNA gene is transcribed to
pri-miRNA which is much longer than the pre-miRNA possessing the characteristic
stem-loop structure Like transcription of most protein-coding genes the miRNA
Chapter 1 Introduction and Review of Literature
36
transcription is processed by RNA polymerase II (Pol II) enzymes and the pri-
miRNAs are 5prime-capped and 3prime-polyadenylated (Lee et al 2004 Xie Z et al 2005)
Mature miRNAs are produced from pri-miRNAs through at least two sequential
processing steps by RNase III-type endonucleases In animals pri-miRNAs are first
processed at the bottom portion of the stem by Drosha a nuclear-localized RNase III
enzyme to liberate pre-miRNAs The pre-miRNAs are then exported to the cytoplasm
with the help of exportin-5The pre-miRNAs are further processed by Dicer another
RNaseIII enzyme to a duplex consisting of the miRNA and the antisense strands
(miRNA) Since plants do not have any Drosha homologs the two sequential
processing steps seem to be carried out by DCL1 a Dicer homolog in the nucleus
(Kurihara amp Watanabe 2004 de Alba et al 2013 Rogers amp Chen 2013)
The processing of pri-miRNAs to pre-miRNAs by DCL1 is assisted by two
other proteins HYPONASTY LEAVES1 (HYL1) and SERRATE (SE) HYL1 belongs to a
family of double-stranded RNA (dsRNA) binding protein and interacts with DCL1 in
vitro and in vivo (Lu amp Fedoroff 2000 Hiraguri et al 2005 de Alba et al 2013
Rogers amp Chen 2013) Loss-of-function mutations in the HYL1 gene result in
decreased accumulation of miRNAs and concomitant accumulation of pri-miRNAs
(Han et al 2004 Vazquez et al 2004a Kurihara et al 2006) SE a C2H2 zinc finger
protein interacts with DCL1 and HYL1 and plays a role in the processing of pri-
miRNAs to pre-miRNAs (Lobbes et al 2006 Yang L et al 2006) In addition the
nuclear cap-binding complex proteins CBP20 and CBP80 that are known as ABA
HYPER SENSITIVE 1(ABH1) have been shown to be required for proper miRNA
processing (Gregory et al 2008 Laubinger et al 2008) Recently it has been
demonstrated that DCL1 can release 21-nt RNAs from dsRNA as well as from
synthetic miR167 precursor RNAs in vitro However this cleavage is error prone and
inefficient in the absence of HYL1 and SE which accelerate the rate of DCL1-mediated
cleavage and promote accurate processing of miRNAs (Dong et al 2008 Rogers amp
Chen 2013)
11343 Methylation of the miRNAmiRNAduplex
After the miRNAmiRNAduplexes are released from the pre-miRNAs by the DCL1
activities the duplexes are methylated at the 2primeOH of the3prime-ends by HUA ENHANCER1
(HEN1) a small RNA methyltransferase (Yu et al 2005 Yang Z et al 2006 Rogers
amp Chen 2013 ) This step which is distinct from the RNase III-mediated processing
Chapter 1 Introduction and Review of Literature
37
step distinguishes the biogenesis of plant miRNAs from that of animal miRNAs
Mutations in the HEN1 gene were first isolated in a morphological screening for floral
development although the HEN1 mutants display pleiotropic phenotypes similar to the
developmental defects observed in the DCL1 mutants (Chen et al 2002 Park et al
2002 Rogers amp Chen 2013)
Figure 18 Biogenesis of microRNA
A Biogenesis of plant MicroRNA B Biogenesis of metazoananimal MicroRNA
The miRNA (red) is incorporated into RISC and the miRNA(blue) in degraded
This enzyme has two dsRNA-binding domains and nuclear localization signal It
is also conserved in fungi and is required for the processing of siRNAs as well as of
miRNAs (Park et al 2002 Boutet et al 2003 Xie et al 2003) Although the HEN1
protein is localized to the nucleus its methylation activity in the cytoplasm cannot be
ignored (Xie et al 2004 Rogers amp Chen 2013)
Chapter 1 Introduction and Review of Literature
38
The methylation of the miRNAmiRNA duplex is required to protect miRNAs
against the 3prime-end uridylation activity and subsequent degradation (Li et al 2005) In
the hen1 mutants accumulation of miRNAs is reduced to a lower level Also the
miRNAs appear to be heterogeneous in their sizes such that a ladder of bands rather
than a single species is observed for most miRNAs in the mutants (Li et al 2005)
MiRNA cloning and sequencing analyses revealed the presence of a number of U
residues at the 3prime-end of miRNAs in the hen1 mutants Moreover these nucleotides do
not correspond to the miRNAs in the pre-miRNAs indicating that these nucleotides are
added after the processing of the miRNAs from pre-miRNAs (Li et al 2005)
Uridylation of RNAs has also been proven to be correlated with RNA decay (Shen amp
Goodman 2004 Mullen amp Marzluff 2008) suggesting that uridylation attracts an
exonuclease which degrades miRNAs in plants
11344 MiRNA incorporation in to the RISC and its nuclear export
The methylated miRNAmiRNA duplexes undergo RISC assembly In this process
the miRNA of the duplexes is selectively incorporated into the RISC and the miRNA
appears to be removed and subsequently degraded (Hammond et al 2000 Hutvagner
amp Zamore 2002 Schwarz et al 2003 de Alba et al 2013 Rogers amp Chen 2013)
The ARGONAUTE 1(AGO1) proteins are core components of the RISC complex
They contain two structural domains the N-terminal PAZ domain that binds to the 3prime-
end of single-stranded RNAs and the C-terminal piwi domains that has a structure
similar to that of RNase H (Cerutti et al 2000 Parker et al 2004) Several AGO
proteins possess endonucleolytic activities within the PIWI domain and cleave target
mRNAs in the middle of the site complementary to miRNA (Liu et al 2004 Meister et
al 2004 Miyoshi et al 2005) Mutations in the AGO1 gene cause miRNA target
genes to be ectopically expressed and the levels of some miRNAs are reduced in the
mutants (Kidner amp Martienssen 2004 Vaucheret et al 2004 Kidner amp Martienssen
2005 Ronemus et al 2006) Moreover the phenotype of the AGO1 hypomorphic
alleles display defects in lateral organ polarity and leaf and flower morphology as
observed in the dcl1 mutants and null AGO1 alleles are embryonically lethal
supporting the close relationship between the AGO proteins and miRNA processing
(Kidner amp Martienssen 2004 Vaucheret et al 2004 de Alba et al 2013 Rogers amp
Chen 2013)
The production of miRNA miRNA duplexes occur in the nucleus while
Chapter 1 Introduction and Review of Literature
39
miRNA-RISC complexes are present in the cytoplasm where target mRNAs are
located This discordance of functional sites requires an export step during miRNA
biogenesis HASTY the Arabidopsis homolog of exportin-5 is thought to be involved
in miRNA nuclear export (Bollman et al 2003 Park et al 2005)The hasty mutant
exhibits pleiotropic defects in plant development and reduced accumulation of most
miRNAs as in the DCL1 or AGO1 mutants Howeverit is unclear whether the HASTY
proteins export the miRNA miRNA duplexes or the miRNA-RISC complexes in
plants
11345 Feedback regulation of miRNA biogenesis
MiRNA biogenesis is under feedback regulation such that the DCL1 and AGO1 genes
are themselves regulated by miRNAs DCL1 is itself a target of miR162 which leads to
the cleavage of the DCL1 mRNA (Xie et al 2003 de Alba et al 2013) Consistent
with this the levels of DCL1 mRNA are elevated in the mutants of miRNA processing
genes in which the miR162 abundance is reduced In addition there is a predicted
hairpin structure of miR838 in the 14th
intron of the DCL1 primary transcript
(Rajagopalan et al 2006) This prediction implies that DCL1-mediated processing
on this site can result in the cleavage of the DCL1 primary transcript into two truncated
fragments the N-terminal capped fragment and the C-terminal polyadenylated
fragment If this is the case it is possible that miRNA biogenesis competes with the
splicing event of the DCL1 pre-mRNA
The AGO1 is also regulated by miRNA There is a complementary site for
miR168 binding in the AGO1 mRNA and the transcript level of the AGO1 mRNA is
reduced through an mRNA cleavage mechanism (Vaucheret et al 2006 de Alba et al
2013 Rogers amp Chen 2013) indicating that the AGO1 mRNA levels are tightly
controlled by the levels of the AGO1 protein
1135 Mechanism of miRNA action
11351 MiRNA-directed mRNA cleavage
By forming the miRNA-RISC assembly miRNAs can direct the RISC to regulate gene
expression by two post transcriptional mechanisms mRNA cleavage or translational
repression (Bartel 2004 Jones-Rhoades et al 2006 Dalmay 2013) The degree
of miRNA-mRNA complementarity plays an important role for the
determination of the mechanism to be used A general rule is that while perfect or
Chapter 1 Introduction and Review of Literature
40
near-perfect complementarity induces mRNA cleavage central mismatches trigger
translational repression (Carrington amp Ambros 2003 Jones-Rhoades amp Bartel 2004)
Unlike most animal miRNAs plant miRNAs have near-perfect or perfect
complementarity to their targets and mRNA cleavage is assumed to be a predominant
mechanism by which miRNAs regulate gene expression in plants
In RNA cleavage mechanism miRNAs guide the AGO component of RISC to
cleave a single phosphodiester bond of the target mRNA within the miRNA-binding
site (Llave et al 2002b Tang et al 2003) The truncated fragments are then released
and degraded freeing the RISC to recognize and cleave another target mRNA This has
been validated by the detection of 3prime cleavage products that have 5prime ends that start at the
middle of the complementary regions The mRNA cleavage products are then further
degraded by other mechanisms The 3prime cleavage products of some miRNA targets are
degraded by the 5prime-3prime exonuclease XRN4 an Arabidopsis homolog of the major Yeast
mRNA degrading exonuclease Xrn1p It has been observed that the 3prime cleavage
products of some target mRNAs were accumulated in the xrn4 mutants (Souret et al
2004 Dalmay 2013) However the 3prime cleavage products of other miRNA targets do
not accumulate in the xrn4 mutants indicating that there are also XRN4 independent
mechanisms The 5prime cleavage products are typically untraceable by RNA gel blot
hybridization but can be detected by sensitive PCR based methods It has been found
that the 5prime cleavage products tend to obtain an oligo U tail at the 3prime ends This 3prime U tail
is correlated with shortening of the 5prime cleavage products from the 5prime end leading to the
conclusion that the 3prime uridylation causes 5 prime- 3 prime exonucleolytic decay of the 5prime cleavage
products (Shen amp Goodman 2004)
DCL1 HYL1 and SE interact with each other and are co-localized in the
nuclear bodies termed Dicing bodies or D-bodies in which some pri-miRNA shave
been found suggesting that D-bodies serve as a center for active miRNA processing
(Fang amp Spector 2007 Fujioka et al 2007 Song et al 2007) The miRNA
processing site appears to be distinct from the Cajal bodies that have previously been
found as a siRNA processing site (Pontes amp Pikaard 2008) The D-bodies include
SmD3 and SmB proteins that are found in the Cajal bodies However At collin an
additional marker for the Cajal bodies is not found in the D-bodies Moreover the D-
bodies are present in an At collin mutant lacking the Cajal bodies The pri-miRNAs of
miR171A and miR159A genes have been found to be co-localized with HYL1 and
Chapter 1 Introduction and Review of Literature
41
DCL1 in the D- bodies that are distinct from the Cajal bodies (Song et al 2007)
11352 MiRNA-directed translational repression
Translation repression is another mechanism exerted by plant miRNAs The regulatory
mechanism of miR172 which regulates flowering time and floral organ identity is
consistent with translation repression MiR172 over production does not affect the
transcript levels of APETALA2 (AP2) and AP2- like target mRNAs Instead the AP2
protein level is reduced in the miR172 over expressing mutant (Aukerman amp Sakai
2003 Chen 2004 Dalmay 2013) It was later shown that miR172 can also direct the
cleavage of the target mRNAs although the steady-state mRNA level is unchanged due
to feedback regulation (Schwab et al 2005 Jung et al 2007) It is clear that miR172
regulates its target genes primarily by translational repression rather than mRNA
cleavage Recently it has been reported that miR156157 and miR854 also regulate
their target genes through translational repression (Arteaga-Vazquez et al 2006
Gandikota et al 2007 Dalmay 2013) It was once thought that translational repression
is an exception to the rule However experimental evidence suggest a widespread
coexistence of translational repression and mRNA cleavage (Brodersen et al 2008)
1136 Regulatory roles of miRNAs in plant development
The miRNA target prediction has been a difficult task in order to obtain regulatory
targets without bringing in too many false predictions Plant growth and developmental
processes regulated by miRNAs are quite diverse and enormous Furthermore plant
miRNAs work cooperatively or antagonistically to establish a balanced regulation This
notion is well consistent with the findings that mutations in the regulators of miRNA
biogenesis transport and processing such as AGO1 DCL1 HYL1 HEN1 ABH1
and HASTY cause pleiotropic phenotypes (Mallory amp Vaucheret 2006 de Alba et al
2013) To date the roles of miRNAs have been mostly deduced from obtaining miRNA
over expressing transgenic plants or gain-of-function mutants in which miRNA-
resistant target genes are ectopically expressed
11361 Phase transition
Plants pass through a series of developmental phase transitions during their life span
beginning with seed germination continuing through vegetative phase change
reproductive phase change flowering initiation and finally seed production for the next
generation Because of their importance in understanding plant developmental
Chapter 1 Introduction and Review of Literature
42
processes genes regulating developmental transitions have been extensively studied
and underlying molecular mechanisms have been explored in various plant species
Majority of researches were mainly focused on vegetative phase change and
reproductive transition in Arabidopsis and Maize (Poethig 2003 Baurle amp Dean
2006) Particularly the mutations of the genes encoding various regulators of miRNA
biogenesis and transport such as HST SE and AGO exhibit altered juvenile to adult
vegetative phase change and vegetative to reproductive change (Hunter et al 2003
Peragine et al 2004 Vazquez et al 2004a Jones-Rhoades et al 2006 de Alba et al
2013)
Over-expression of the miR156a gene causes late flowering and delays
vegetative phase change (Wu amp Poethig 2006 Gandikota et al 2007 Wang et al
2008) It has been shown that miR156 negatively regulates a group of genes encoding
SQUAMOSA PROMOTER-BINDING PROTEIN-LIKE (Lewis et al 2007 de Alba et
al 2013 Rogers amp Chen 2013) transcription factors such as SPL3 SPL4 and SPL5
that promote flowering initiation In contrast transgenic plants over expressing
miR156-resistant SPL345 genes are early flowering Interestingly the endogenous
level of miR156 is temporally regulated In the early juvenile vegetative phase the
level of miR156 is very high but decreases rapidly before the onset of the adult juvenile
phase (Wu amp Poethig 2006)
The Arabidopsis early activation tagged (eat-D) mutant exhibits early flowering
with disrupted floral structure The mutant phenotypes are caused by over expression of
the miR172a-2gene (Aukerman amp Sakai 2003) MiR172 regulates a small group of
genes encoding APETALLA2 (AP2)-like transcription factors such as TARGET OF
EAT1 (TOE1) TOE2 SCHLAFMUTZE (SMZ) and SCHNARCHZAPFEN(SNZ)
TOE1 TOE2 SMZ and SNZ have been shown to participate in their productive phase
change by repressing a floral integrator FLOWERING LOCUS T (FT)(Jung et al
2007) Consistent with this toe1-2D 35STOE2 35SSMZ and 35SSNZ plants
exhibit late flowering (Jung et al 2007) MiR172 is also regulated temporally and
highly expressed in the adult vegetative phase both in Arabidopsis and maize (Chuck et
al 2007) Because the temporal expression patterns of miR156 and miR172 are
complementary to each other it has been suggested that the two miRNAs interact with
each other in regulating phase change (Chuck et al 2009 de Alba et al 2013) In
support of this view it has been shown that miR172 is down-regulated in the
Chapter 1 Introduction and Review of Literature
43
Corngrass1(Cg1) mutant in which miR156 is overproduced (Chuck et al 2007 de
Alba et al 2013) However there is no direct evidence for the direct linkage between
miR156 and miR172 It is also envisioned that miR156 and miR172 would not be
directly related For example miR156 regulates plastochron index that is the inverse of
leaf initiation rate and thus frequently used to analyze temporal patterns of plant
development In contrast miR172 does not affect plastochron (Wang et al 2008) The
down-regulation of miR172 in the maize miR156-over producing mutants would be
due to the prolonged juvenile vegetative phase in the mutants
Another miRNA that regulates flowering initiation is miR159 It regulates
MYB33 and MYB65 which are closely related to the barley GAMY B transcription
factor that responds to gibberellic acid (GA) The level of miR159 is elevated in GA-
treated seedlings but reduced in the GA biosynthetic ga1 mutant Transgenic plants
over producing miR159 exhibit delayed floral induction under short days (Achard et
al 2004) It is been suggested that MYB33 mediates GA signal to regulate LEAFY
(LFY) These observations indicate that miR159 plays an important role in the GA
signaling pathways that regulate proper floral induction under short days (Achard et al
2004)
11362 Floral development
Arabidopsis flowers consist of four distinct organs They arise in a characteristic
pattern within concentric rings called whorls from outside to inside four sepals four
petals six stamens and the central pistil consisting of two carpels Identities of the
floral organs are determined by three classes of regulatory genes classA classB and
class C genes (Krizek amp Fletcher 2005) The A and C class genes act antagonistically
to restrict their expression domains (Bowman et al 1991)
It is notable that miR172 also regulates floral architecture in addition to its role
in the timing of flowering initiation MiR172 inhibits the translation of the AP2 mRNA
(Chen 2004) Transgenic plants overproducing miR172 not only exhibit early
flowering but also show abnormal floral development which is quite similar to those
of the class A gene mutants such as the ap2 mutants (Aukerman amp Sakai 2003 de
Alba et al 2013) When the activity of the class A genes is inactivated that of the class
C genes such as AGAMOUS (AG) is elevated As a result the miR172- overproducing
transgenic plants exhibit no petals along with homeotic transformation of the sepals to
Chapter 1 Introduction and Review of Literature
44
the carpels (Aukerman amp Sakai 2003 Chen 2004)
Interestingly miR166 also regulates floral architecture The role of miR165166
in the regulation of meristem activity is closely linked to floral meristem formation and
organization (Zhao et al 2007 Miyashima et al 2013) Floral structure is severely
disrupted in the miR165166-overproducing mutants such as men1 and jba-1D in
which miR166 is overproduced (Williams et al 2005b Jung amp Park 2007) The men1
and jba-1D mutants have smaller gynoecium and reduced carpel number In an extreme
case the jba-1D homozygous plants lack visible carpels (Williams et al 2005b)
Similar phenotypes have been observed in the miR165 overproducers (Zhao et al
2007) It has been suggested that the phenotypes are possibly caused by altered AG
expression in the floral meristem (Jung amp Park 2007 Turchi et al 2013)
Other miRNAs also affect floral development Overproduction of miRNAs
involved in auxin signaling also affects floral architecture Transgenic plants
overproducing miR167 have defects in the formation of ovule and anther with reduced
fertility (Ru et al 2006) It has been shown that miR167 targets ARF6 and ARF8 that
play a role in gynoecium and stamen development and in regulating fertility (Ru et al
2006 Wu et al 2006) These observations suggest that auxin signals are also closely
related to floral organogenesis In addition transgenic plants over-producing miR164
and the cuc1cuc2 double mutants show fused sepals and reduced petals (Laufs et al
2004 Turchi et al 2013) suggesting that miR164 is also related to floral meristem
activity and boundary specification of the meristematic region
11363 Shoot apical meristem development
Shoot apical meristem comprises self-maintaining totipotent cells The shoot apical
meristem activity affects diverse aspects of plant developmental processes including
leaf development vascular development and lateral organ polarity (Bowman et al
2002) Because miR165166 plays a primary role in meristem formation
overproduction of miR165166 and multiple knockout mutants of its target genes
exhibit pleiotropic pheonotypes (McConnell et al 2001 Emery et al 2003 Kim et al
2005 Williams et al 2005b)
The targets of miR165166 include genes encoding the homeo domain-leucine
zipper (HD-ZIP) class III transcription factors such as PHABULOSA(PHB)
PHAVOLUTA (PHV) REVOLUTA(REV) ATHB15CORONA (CNA) and ATHB8
Chapter 1 Introduction and Review of Literature
45
PHB PHV and ATHB15 have overlapping functions in controlling meristem
development (Turchi et al 2013) Indeed the phb phv athb15 triple mutants have an
enlarged shoot apical meristem Consistent with this the miR166- overproducing men1
and jba-1D mutants exhibit enlarged shoot apical meristems (Kim et al 2005
Williams et al 2005b Turchi et al 2013) In the mutants the shoot apical meristems
are multiplied and large
The development of shoot apical meristems is also regulated by the WUSCHEL
(WUS)ndashCLAVATA (CLV) signaling pathway WUS positively regulates cell
proliferation In contrast CLV3 which is expressed in stem cells limits the WUS
expression and thus inhibits cell proliferation in the SAM (Sharma et al 2003)
Consistent with this notion WUS is expressed to a higher level in jba-1D
explaining the ectopic enlargement of the SAM in the mutant (Williams et al 2005)
While WUS is closely related to miR165166 in the shoot apical meristem
development the underlying molecular mechanisms are currently unclear It has been
observed that the miR166-over producing men1 plants have an arrested meristem
development (Jung amp Park 2007) In addition the heterozygous men1+ and the
homozygous men1 plants show different expression patterns of WUS and CLV3 It
appears that miR166 regulates WUS indirectly and influences the expressional balance
between WUS and CLV3 through a negative feedback loop (Jung amp Park 2007 de
Alba et al 2013)
The phv phb athb15 triple mutant has an enlarged shoot apical meristems the
rev phb phv mutant does not have functional shoot apical meristems (Prigge et al
2005) This distinction may be explained by the fact that while miR166 targets
primarily PHB PHV and ATHB15 miR165 targets all the five HD-ZIP III genes (Zhou
et al 2007) Consistent with this transgenic plants overproducing miR166 are distinct
from those overproducing miR165 The miR165-overproducing transgenic plants lack
functional shoot apical meristem and a pin-like structure is formed at the apical region
of the mutant These phenotypes are quite similar to rev phb phv triple mutant (Zhou et
al 2007 Liu et al 2013) The phenotypic distinction may because by the difference
of the cleavage efficiencies of the target mRNAs In accordance with this notion a few
35S miR166a transgenic lines exhibit no shoot apical meristems The men1
homozygous plants also show arrested meristem development (Jung amp Park 2007)
Chapter 1 Introduction and Review of Literature
46
MiR164 cleaves the CUC1 and CUC2 mRNAs as well as the NAC1 mRNA
CUC1 and CUC2 determines the organ boundary in the meristem Phenotypes of
transgenic plants that overproduce miR164 are similar to those of the cuc1cuc2 double
mutant that exhibits embryonic developmental defects such as cup-shaped cotyledons
and fused cotyledons (Laufs et al 2004) When a miR164-resistant CUC2 is
expressed the boundary domain is enlarged CUC also plays a role in embryonic shoot
meristem formation by regulating the SHOOT MERISTEMLESS gene It was therefore
concluded that miR164-mediated cleavage of CUC1 and CUC2 mRNAs determines the
sizes of the boundary domains in the shoot apical meristem and regulates separation of
the organs by restricting cell proliferation (Laufs et al 2004 de Alba et al 2013)
11364 Leaf development
The role of miR319 has been extensively studied in leaf development A dominant
jaw-D mutant overproducing miR319 shows uneven curved leaves with enlarged size
and five TCP genes which are closely related to the CINCINNATA (CIN) gene in
Antirrhinum majus are down regulated in the mutant suggesting that miR319 mediated
regulation of the TCP transcription factor genes are important for the final step of
leaf shaping (Palatnik et al 2003 Naz et al 2013) In addition to leaf shape and size
leaf polarity is also specified by miRNA MiR166-mediated cleavage of the HD-ZIPIII
mRNAs is a well to establish leaf polarity This discovery originates from the gain-of-
function mutants such as phb-1d and phv-1d wherein point mutations in the
complementary sites to miRNA helps the mRNA to evade or resist from miRNA-
mediated cleavage (McConnell et al 2001 Emery et al 2003 Naz et al 2013)
Plant leaves have a dorso-ventral polarity consisting of the adaxial (upper
surface) and abaxial (lower surface) sides The adaxial side is closer to the shoot
apical meristem Adaxial identity is specified by functionally redundant class III HD-
ZIP family genes such as PHB PHV and REV Ectopic expression of each genes
result in adaxialized leaves Dominant phb-1d and phv-1d mutants exhibit
transformation of the abaxial to adaxial fate and have rod-shaped leaves In contrast
miR165166-overproducing plants show pin-like cotyledons radicalized leaves and
abaxialized leaves (Chitwood et al 2007 Pattanayak et al 2013)
Leaf asymmetry is determined by miR166-mediated restriction of the
expression domains of the HD-ZIPIII genes The HD-ZIPIII genes are expressed in the
Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
13
purine skeleton and the methyl groups required for caffeine biosynthesis in young tea
leaves (Koshiishi et al 2001)
Methionine
S-Adenosyl-L-methionineHomocysteine
ATP
PPi+ Pi
Tetrahydrofolate
5-Methyl-
tetrahydrofolate
CH3+Adenosine
S-Adenosyl-L-
homocysteine
(xanthine
structure)(Methyl groups)
Caffeine
(1)
(2)(3)
(4)
Figure 14 The SAM cycle (activated methyl cycle) in plants (Ashihara amp Suzuki 2004)
Enzymes SAM synthetase SAM-dependent N-methyltransferases S-adenosyl-
homocysteine (SAH) hydrolase Methionine synthase Adenosine released from the cycle
is salvaged to adenine nucleotides and utilized both for purine structure of caffeine via
xanthosine and for re-synthesis of SAM via ATP
183 N-methyltransferase in caffeine biosynthesis Molecular cloning
The genes encoding for the enzymes involved in the caffeine biosynthesis were
successfully cloned from the coffee family (Ogawa et al 2001 Mizuno et al 2003a
Mizuno et al 2003b Uefuji et al 2003) The conserved amino acid regions of tea CS
(AB31280) the sequences of O-methyltransferases derived from plant origin and
Arabidopsis hypothetical proteins were made use of in designing primers for RT-PCR
and RACE technique Independent groups have isolated N-methyltransferase (NMT)
genes from coffee which have been classified based on the substrate specificity of the
recombinant proteins expressed in Ecoli (Table 15) PCR products thus obtained were
used as the probe to isolate full length cDNA from the library resulting in the
identification of all three N-methyltransferases involved in caffeine biosynthesis
(Figure 12) Several cDNA have been characterized from the coffee plants one for
xanthosine methyltransferase (XMTXRS) three for 7-methylxanthine methyltransferase
or theobromine synthase (MXMTCTS) and three for 37-dimethylxanthine
methyltransferase or caffeine synthase (DXMTCCS) (Table 15) (Kato amp Mizuno
2004)
A single gene encoding for xanthosine methyltransferase (XMTCmXRS1) was
identified which encoded for a polypeptide consisting of 372 amino acids with an
apparent molecular mass of 418kDa It is expressed almost uniformly in aerial tissues
Chapter 1 Introduction and Review of Literature
14
of C arabica including leaves floral buds and immature but not in mature beans
Three genes encoding 7-methylxanthine methyltransferase (theobromine synthase) have
been identified The number of amino acids in the putative polypeptides are 378 for
MXMT1 (427kDa) and 384 for MXMT2 and CTS2 (434kDa) They differ by insertion
or deletion of blocks of several residues in the C-terminal region Their catalytic
properties based on kinetic parameters such as Km values were different They are
expressed in young leaves floral buds and immature but not mature beans Three genes
were identified for 3 7-dimethylxanthine methyltransferase (caffeine synthase) DXMT
CCS1 and CtCS7 each encoding a 43 kDa polypeptide consisting of 384 amino acids
However their kinetic properties differ from each other such as DXMT and CCS1
showed Km values of 1200 and 157microM for theobromine respectively Expressions
profiles are also distinct DXMT being expressed exclusively in immature beans while
CCS1 expression is ubiquitous occurring in all tissues The presence of isoforms of
these enzymes with different properties suggests that caffeine is synthesized through
multiple pathways depending on availability and concentration of the substrate (Mizuno
et al 2001 Ogawa et al 2001 Mizuno et al 2003a Mizuno et al 2003b Uefuji et
al 2003) Genomic clones of theobromine synthase were obtained by PCR- RFLP
from C canephora (Satyanarayana et al 2005 Satyanarayana 2006) The genomic
clones are reported to have highly conserved exons and a variable third intron The
promoter for the theobromine synthase was cloned and characterized (Satyanarayana et
al 2005)
There is high degree of similarity in the coding region of coffee NMT genes
isolated so far though differences exist in the 3prime untranslated region (UTR) for these
genes The deduced amino acid sequence of these enzymes shows more than 85
homology between these genes Phylogenetic analysis indicates that they are more
closely related to C-methyltransferases including those for jasmonic acid salicylic acid
and benzoic acid than to other N-methyltransferases This suggests that coffee N-methyl
transferases belong to a distinct sub-group within plant methyltransferases Their
cellular localization determined by green fluorescent protein fusion method and β-
glucourodinase (GUS) showed that the all the three genes are localized in the cytoplasm
(Ogawa et al 2001 Vinod et al 2007) It was also shown that these were present
along with chlorogenic acid(Vinod et al 2007)
Chapter 1 Introduction and Review of Literature
15
Table 15 N-methyltransferases and their encoding genes involved in caffeine biosynthesis
Gene (Enzyme) Gene name Accession No Description Reference
Xanthosine methyltransferase
( 7-methylxanthosine synthase) (EC 211158)
CaXMT AB 048793 Immature fruit cDNA Screening (Uefuji et al 2003)
CmXRS1 AB 034699 Leaf cDNA library RACE (Mizuno et al 2003a)
CaMXMT1 AB 048794 Leaf cDNA library RACE (Ogawa et al 2001)
7-methylxanthine synthase transferase
(theobromine synthase) ( EC 211159)
CTS1 AB 034700 Immature fruit cDNA Screening (Uefuji et al 2003)
CaMXMT2 AB 984125 Leaf cDNA library Screening (Mizuno et al 2001)
CTS2 AB 054841 Leaf cDNA library Screening (Mizuno et al 2001)
37-methylxanthinemethyltransferase
(Caffeine synthase) (EC 211160)
CaDXMT1 AB 086414 Immature fruit cDNA Screening (Uefuji et al 2003)
CCS1 AB 0861414 Immature fruit cDNA Screening (Uefuji et al 2003)
CtCS7 AB 0864415 Immature fruit cDNA
NMT genomic clones
Theobromine synthase-1
CX10 Complete coding region PCR Satyanarayan 2006
CX8 Complete coding region PCR -do-
Theobromine synthase-1 PG-5 Promoter + coding region RAGE -do-
Theobromine synthase-1 PG-1 Promoter + coding region RAGE Satyanarayan 2006
PG-4 Promoter + coding region RAGE -do-
Theobromine synthase-1 NMT
(AY273814) Complete coding region PCR Kochko A et al 2010
Theobromine synthase-2 NMT
(AY362825) Complete coding region PCR Kochko A et al 2010
Chapter 1 Introduction and Review of Literature
16
19 Catabolism of caffeine
Caffeine is produced in young leaves and immature fruits and continues to accumulate
gradually during the maturation of these organs At the same time caffeine is slowly
degraded with the removal of the three methyl groups resulting in the formation of
xanthine Catabolism of caffeine was first reported in coffee leaves (Kalberer 1965)
Since then a number of tracer experiments using 14
C labelled purine alkaloids have
been reported (Suzuki amp Waller 1984 Ashihara et al 1996 Vitoria amp Mazzafera
1999 Mazzafera 2004) They demonstrated the major catabolic pathway is caffeine rarr
theophylline rarr 3-methylxanthine rarr xanthine Xanthine is further degraded by the
removal by the conventional purine catabolic pathway to CO2 and NH3 via uric acid
allatonin and allantoate (Figure 15) (Ashihara amp Crozier 1999 Stasolla et al 2003
Zrenner et al 2006) Caffeine catabolism usually begins with its conversion to
theophylline catalyzed by N7-demethylase The involvement of P450-dependent mono-
oxygenase activity for this reaction was suggested (Huber amp Baumann 1998
Mazzafera 2004) However the activity of this enzyme has not yet been determined in
cell free extracts [8-14
C] Theophylline degraded to CO2 at much faster rate than [8-14
C]
caffeine indicating that conversion of caffeine to theophylline is a major rate limiting
step of caffeine catabolism and a possible reason why caffeine accumulates in high
concentrations in tissues of C sinensis and C arabica It is widely believed that the
control of caffeine levels in plants is a function of the balance between rates of
synthesis and degradation and this balance seems to vary depending on the plant
species and the tissue developmental stage (Mazzafera 2004)
Chapter 1 Introduction and Review of Literature
17
Figure 15 Caffeine catabolic pathways Caffeine is mainly catabolised via theophylline and 3-
methylxanthine Xanthine is further degraded to CO2 and NH3 by the conventional
oxidative pathway Adapted from (Ashihara et al 2008)
Chapter 1 Introduction and Review of Literature
18
110 Genetic engineering of Coffee
Genetic transformation has potential applications in coffee agriculture for incorporating
genes which could provide desirable traits such as disease and insect resistance
drought and frost tolerance and herbicide resistance Transgenic technology can also be
used to increase nutritional value and improve cup quality produce varieties with
caffeine-free beans and for production of hybrid crops for molecular farming
Identification of target specific genes is one of the pre-requisites for developing
transgenic crops The availability of a large number of EST sequences in coffee and
initiation of coffee genome sequencing may speed up the gene discovery and accelerate
transgenic research efforts in coffee
1101 Insect Resistance
Production of insect resistant coffee plants is one of the major objectives of the
breeding programs The major pests attacking coffee include coffee berry borer (CBB
Hypothenemus hampei) white stem borer (WSB Xylotrechus quadripes) leaf miner
(Perileucoptera coffeela) and root nematodes (Meloidogyne spp and Pratylenchus
spp) The CBB is present in almost all the coffee growing countries and considered to
the most devastating pest in coffee To date there is no reported source of resistance to
CBB in the coffee gene pool Like CBB WSB is another serious pest in C arabica in
India and several other East Asian countries Both CBB and WSB belong to the order
Coleoptera For India controlling WSB is the biggest challenge and has therefore
become the highest research priority Robusta is generally resistant to WSB but the
interspecific Robusta Arabica hybrids are susceptible to WSB Although leaf miner is
not yet a serious pest in India and other East Asian countries it is an economically
important pest in East Africa and Brazil Effective chemical control of CBB and WSB
is difficult due to the nature of their life cycle inside the berry and stem respectively as
well as environmental concern regarding the use of pesticides Biological control
measures are adapted to combat these insect pests with varying degrees of success
Developing coffee plants resistant to these pests using genetic transformation
technology could be one of the alternative strategies to counter pest damage For insect
resistance several different classes of proteins from bacterial plant and animal sources
have been isolated and their insecticidal properties tested against many important pests
Amongst these proteins Bt toxins are most important and several transgenic crops
expressing Bt genes have been commercialized Coffee transgenic plants carrying a
Chapter 1 Introduction and Review of Literature
19
synthetic version of the cry1Ac gene have been produced (Leroy et al 2000 Mishra et
al 2002) Indeed this was the first report of an important agronomic trait being
introduced into a coffee plant The transgenic plants presented similar features in
growth and development compared to normal plants Transgenic plants highly resistant
to leaf miner under greenhouse conditions were tested under field conditions in French
Guyana for 4 years for pest resistance (Perthius et al 2005 Perthius et al 2006)
From 54 independent transformation events 70 of the events were resistant to leaf
miner Unfortunately the field trial was vandalized which led to the termination of the
experiment (Montagnon et al 2005)The effectiveness of Bt genes in controlling
coleopteran pests is well documented in corn and potato which indicates that Bt genes
might be effective against CBB The high toxicity of B thuringiensis sero var
israelensis against first in star larvae of CBB has been demonstrated (Mendez et al
2003) In another experiment an α-amylase inhibitor from Phaseolus vulgaris was
tested against CBB and found to have an inhibitory effect on its growth and
development (Grossi et al 2004)In addition to the CBB and WSB C arabica
varieties are also susceptible to endoparasitic root-knot nematode (Meloidogyne spp)
(Campos et al 1990 Mishra et al 2012)
So far 15 species have been reported to be parasites of coffee Controlling
nematodes is extremely difficult and currently seedling grafting with robusta rootstock
is followed as one of the control measure Sources of resistance specific to root-knot
nematodes have been identified in coffee trees (Bertrand et al 2001) and the Mex-1
gene conferring resistance to M exigua in C arabica is in the process of isolation (Noir
et al 2003)
1102 Tolerance to Abiotic Stress
In many coffee growing countries abiotic stress such as drought and frost are the major
climatic factors that limit coffee production Changes in climatic patterns due to global
climate change are considered increasingly important for coffee cultivation Drought
induces water stress in plants which affects vegetative growth and vigor and triggers
floral abnormalities and poor fruit set It also indirectly increases the incidence of pests
and diseases in the plants C arabica is generally more tolerant to water stress than C
canephora partly due to its extensive deep root system C racemosa is known to be a
good source of drought tolerance In India hybrids between C canephora and C
racemosa have been obtained and are currently under evaluation for drought tolerance
Chapter 1 Introduction and Review of Literature
20
In many coffee growing countries coffee is propagated in marginal areas where the
annual rainfall is below 1000thinspmm with prolonged dry spells of over 4-5 months In
those areas water shortage and unfavorable temperatures constitute major constraints
and the growth and productivity of C canephora are badly affected As with drought
periodic frost also affects coffee production in parts of Brazil The introduction of
drought and frost tolerant genes through genetic transformation would be of great
importance for alleviating these problems Research is now being carried out by several
groups to identify genes involved in biotic as well as abiotic stress The most promising
genetic engineering approach for drought tolerance includes the use of functional or
regulatory genes as well as the transfer of transcription factors In recent years plants
tolerant to high temperature and water stress have been the subject of intense research
(Chaves et al 2003 Chaves et al 2004 Coraggio et al 2004) For achieving drought
tolerance genes that have been targeted include those encoding enzymes involved in
detoxification or osmotic response metabolism enzymes active in signaling proteins
involved in the transport of metabolites and regulating the plant energy status (Chaves
et al 2003 Chaves et al 2004 Coraggio et al 2004) The dissection of molecular
mechanisms related to signal transduction and transcriptional regulation might help in
engineering drought tolerance in coffee
1103 Disease Resistance
Coffee leaf rust (CLR) caused by the fungus Hemileia vastatrix is the most important
disease in coffee with substantial loss to coffee production and productivity in all the
coffee growing countries In addition to leaf rust coffee berry disease (CBD) caused by
fungus Colletotrichum kahawae can be a devastating anthracnose leading to substantial
crop loss in Africa Several other fungal and bacterial diseases may also affect coffee
to a extent C arabica is more susceptible to many diseases than C canephora Though
most of the disease control measures rely upon chemical control they are more
expensive and labour intensive The long-term solution is the breeding of resistant
varieties which is the focus of many breeding programs However breeding for disease
resistant varieties is time consuming due to the perennial nature of coffee with its long
gestation period India has a long history of C arabica breeding especially for leaf rust
resistance and is the first country to demonstrate the existence of multiple races of leaf
rust Resistance to CLR is conditioned primarily by a number of major (SH) genes and
coffee genotypes are classified into different resistance groups based on their
Chapter 1 Introduction and Review of Literature
21
interaction with different rust races pathogen (van der Vossen 2001) Currently C
canephora provides the main source of resistance to pests and diseases including CLR
(H vastatrix) and CBD (C kahawae) and therefore used in breeding programs Other
diploid species like C liberica and C racemosa are being used as a source of resistance
to coffee leaf rust and coffee leaf miner respectively (Guerreiro et al 1999
Sreenivasan et al 1989) The development of coffee varieties resistant to major fungal
diseases such as CLR and CBD using transgenic technology will benefit the coffee
industry immensely During the last 15 years significant progress has been made in the
area of host-pathogen interactions (Staskawicz et al 1995 Feys et al 2000
Shivaprasad et al 2012) and many resistance genes involved in recognizing invading
pathogens have been identified and cloned (Takken et al 2000) A number of
signalling pathways induced because of pathogen infection have been dissected (Dong
et al 1998 Navarro et al 2006) Many antifungal compounds synthesized by plants to
combat fungal infection have been identified (Does et al 1998) Understanding the
specific induction of targeted pathways and identification of specific pathways
responsible for particular fungal resistance is important in order to employ this strategy
in transgenic technology The recent investigation of gene expression during coffee leaf
rust infection could provide an insight into the defence pathways operating in coffee
(Fernandez et al 2004 Nardi et al 2006) Efforts have been made to identify and
clone resistance genes from coffee for achieving durable resistance The genetic and
physical map of two resistance genes SH3gene conferring resistance to rust (Prakash et
al 2004) and the Ck1 gene conferring resistance to C kahawae CBD (Gichuru et al
2006) have been established These genes could be used for molecular marker assisted
breeding programs (Mishra et al 2012)
1104 Production of low-caffeine coffee
A widespread belief that the ingestion of caffeine can have adverse effects on
health resulted in the increased demand for decaffeinated coffee (Ashihara amp Crozier
2001) Several method were used to obtain decaffeinated plants by conventional
breeding techniques using low yielding varieties and mutants that accumulated
relatively low amounts of caffeine when compared to the commercially available ones
Low-caffeine and decaffeinated coffee represent around 10 of the coffee sales
around the world (Ribas et al 2006b) The industrial process for coffee decaffeination
can be expensive and affects the original flavour and aroma in coffee (David 2002)
Chapter 1 Introduction and Review of Literature
22
Transgenic coffee plants with suppressed caffeine synthesis using RNA interference
(RNAi) technology have been obtained (Ogita et al 2003 Ogita et al 2004) Specific
sequences in the 3prime untranslated region of the theobromine synthase gene (CaMXMT1)
were selected for construction of RNAi short and long fragments The caffeine and
theobromine content of the transgenic plants were reduced by ~ 70 when compared to
the untransformed plants In C canephora RNAi technology has also been employed
to silence the N-methyl transferase gene involved in caffeine biosynthesis (Vinod et al
2007) Promoter of an N-methyltransferase (NMT) gene involved in caffeine
biosynthesis was cloned (Satyanarayana et al 2005 Satyanarayana 2006) which will
be very useful for studying the regulation of caffeine biosynthesis
1105 Improvement in cup quality
Improvement in coffee cup quality requires elaborate knowledge of the chemical
constituents as well as the metabolic pathways involved in the elaboration of quality
The constituents of coffee beans include minerals proteins carbohydrates caffeine
chlorogenic acids (CGA) glycosides lipids and many volatile compounds that give
flavour to coffee by roasting Among these the role of three major constituents
sucrose CGA and trigonelline have been studied in coffee The sucrose content of
coffee bean is associated with coffee flavour the higher the sucrose content in green
beans the more intense will be the cup flavour (Clifford et al 1985 de Maria et al
1996) The sucrose content of C arabica (82-83) is higher than C canephora (33ndash
40) The sucrose amino acids ratio in green beans determines the profile of volatile
compounds Manipulating sucrose content in coffee bean is therefore important in
improving cup quality The sucrose synthase gene (CcSUS2) from C canephora has
been cloned and sequenced (Leroy et al 2005) This provides an opportunity to
manipulate the sucrose content in coffee Chlorogenic acids are products of
phenylpropanoid metabolism Chlorogenic acids are a group of hydroxycinnamoyl
quinic acids (HQA) formed by esterification between caffeic acids coumaric acids and
quinic acids (Clifford et al 1999) They are present in relatively large quantities in the
coffee bean and are the precursor of phenolic compounds in roasted beans C
canephora beans contain higher CGA (10) compared to C arabica beans (6-7)
CGA are known to have antioxidant properties as well as being associated with disease
resistance (Campa et al 2003) Genetic manipulations of genes involved in CGA
synthesis can serve either of these purpose by up- or down regulating the pathway
Chapter 1 Introduction and Review of Literature
23
Phenylalanine ammonia-lyase (PAL) catalyzes the first step of the phenylpropanoid
pathway leading to the synthesis of a wide range of chemical compounds including
flavonoids coumarins hydroxycinnamoyl esters and lignins (Hahlbrock amp Scheel
1989) Recently the full length cDNA and corresponding genomic sequences of PAL
from C canephora was isolated characterized and functionally validated (Mahesh et
al 2006 Parvatam et al 2006) This has opened up new possibilities for manipulating
the level of the PAL enzyme in coffee which in turn can be useful for improvement of
cup quality and manipulating antioxidant properties in coffee
1106 Fruit Ripening
Uniformity during fruit ripening is decisively related to cup quality in coffee and
consequently to the value of the product Fruits at the ideal ripening stage produce the
best organoleptic characteristics for coffee The presence of over ripened or green fruits
changes the acidity the bitterness and consequently the cup quality In order to
maximize uniform ripening of coffee fruits it is essential to control the action of genes
involved in the last step of maturation process Ethylene is known to trigger ripening
and increasing ethylene biosynthesis is associated with various stages of ripening
process (Protasio et al 2005) To control coffee fruit maturation two of the major
genes involved in ethylene biosynthesis namely ACC synthase and ACC oxidase
have been cloned (Protasio et al 2005 Neupane et al 1999) Introduction of the ACC
oxidase gene in antisense orientation have been achieved in both C arabica and C
canephora The effect of the transgene on ethylene production and fruit maturation has
yet to be reported The inhibition of genes downstream to the initial ethylene burst is
also an option to control coffee fruit maturation (Ribas et al 2006)
111 RNA interferenceRNAi
The regulation of gene expression at post transcriptional level has recently attracted
much attention because of the discovery of the phenomenon called RNA interference
(RNAi) RNAi based silencing techniques are more efficient than antisense mediated
gene silencing (Wesley et al 2001) The effect was first observed in plants wherein
silencing was observed when some copies of a transgene was in the forward orientation
with respect to the promoter and some in the reverse orientation This resulted in the
formation of double stranded RNA in the system The phenomenon in which
experimentally introduced double stranded RNA (dsRNA) leads to the loss of
expression of the corresponding cellular gene by sequence specific RNA degradation
Chapter 1 Introduction and Review of Literature
24
was termed as RNA interference or RNAi The protective effect of RNA silencing in
reducing virus infectivity supports the view that PTGS has evolved as a mechanism to
defend plants against virus infection and also to moderate the possible deleterious
genome-restructuring (insertional) activity of virus-like mobile genetic elements eg
retrotransposons A growing body of biochemical and genetic data has further
demonstrated that plants animals and yeasts share related mechanisms of specific
degradation of RNAs in which double-stranded forms of RNA are initiator molecules
(Brenstein et al 2001)
The key insight into the process of PGTS was provided by the experiments
conducted by Hamilton amp Baulcombe (1999) who identified the product of RNA
degradation as small RNA (siRNA) species of ~25nt of both sense and antisense
polarity SiRNA are formed and accumulate as double stranded RNA molecules of
defined chemical structures Both genetic and biochemical approaches were undertaken
to understand the basis of silencing Genetic screens were carried out in fungus
Neurospora carassa the alga Chalmydomonas reinhardtii and plant Arabidopsis
thaliana to search for detective mutants in PTGS
A combination of results obtained from several in vivo and in vitro experiments
have gelled into a two step mechanistic model for RNAiPTGS The first step referred
to as the RNAi initiating step involves binding of the RNA nucleases to a larges
dsRNA and its cleavage into discrete ~21-25nt RNA fragments This is ATP-
dependent reaction which involved RNase III - type endonucleases which make
staggered cuts in both strands of the dsRNA leaving 3prime overhang of 2 nucleotides with
5prime- phosphate 3prime-hydroxyl and no modification in the sugar phosphate backbone
(Hamilton amp Baulcombe 1999 Elbashir et al 2001) SiRNAs are the direct products
of dsRNA cleavage by the multi-domain RNase III enzyme DICER (Figure 16)
(Brenstein et al 2001 Karthikeyan et l 2013)
In the second step or commonly known as the effector step the double stranded
siRNAs produced in the first step bind to an RNAi specific protein complex to form a
RISC The complex undergoes activation in the presence of ATP so that the antisense
component of the unwound siRNA becomes exposed and allows the RISC to perform
the downstream RNAi reaction
Chapter 1 Introduction and Review of Literature
25
Several enzymes are involved in RNAi based on their role genes that encode for them
are classified into RNAi initiators RNAi effectors RNA dependent RNA polymerases
and silencing genes Two C elegans genes rdeI and rde4 (rde stands for lsquo RNAi
deficientrsquo) are believed to be involved in the initiation step of RNAi The C elegans
rde1 gene is a member of a large family of genes and is homologous to the Neurospora
qde2 (qde stands for ldquoquelling deficientrdquo) and Arabidopsis AGO1 (AGO stands for
agronaute AGO1 was previously identified to be involved in Arabidopsis
development) Although the functions of these genes are not clear a mammalian
member of RDE1 family has been identified as a translation initiation factor (Thakur
2005) Important genes for the effector step of PTGS in C elegans are rde2 and mut7
genes These genes were identified from heterozygous mutant worms that were unable
to transmit RNAi to their homozygous offsprings Worms with mutated rde2 or mut 7
genes show defective RNAi mut-7 gene encodes a protein with homology to the
nuclease domains of RNAase D and a protein implicated in Werner syndrome (a rapid
ageing disease in humans) (Grishok et al 2001 Kamath-Loeb et al 2012s)
Neurospora qde-1 Arabidopsis SDE-1SGS-2 and C elegans ego-1 appear to encode
RNA dependent RNA polymerase (RdRPs) It assumed that this is a proof that an RdRp
activity is required for RNAi Certainly the existence of an RdRp might explain the
remarkable efficiency of dsRNA induced silencing if it amplifies either the dsRNA
prior to cleavage or the siRNAs directly (Muers 2013) In C elegans ego-1 mutants
(ego stands for lsquoenhancer of glp-1) RNAi functions normally in somatic cells but is
defective in germline cells where ego-1 is primarily expressed In Arabidopsis SDE-
1SGS-2 mutants (SGS stands for suppressor of gene silencing) siRNA are produced
when dsRNA is introduced via an endogenously replicating RNA virus but not
introduced by a transgene It has been proposed that perhaps the viral RdRP is
substituting for the Arabidopsis enzyme in these mutants Random degradative PCR
model suggests that an RdRP uses the guide strand of an siRNA as a primer for the
target mRNA generating a dsRNA substrate for Dicer and thus more siRNAs
Several genes controlling RNA silencing in plants have been identified through
genetic screens of Arabidopsis mutants impaired in transgene induced RNA silencing
They encode a putative RNA-dependent RNA polymerase (SGS2SDE1) a coiled
protein (SGS3) a protein containing PAZ and Piwi domains (AGO1) and an RNA
helicase (SDE3) The putative SGS2SDE1 of Neurospora is related to QDE-1 and
Chapter 1 Introduction and Review of Literature
26
EGO-1 of C elegans whereas PAZPiwi protein AGO is related to QDE-2 of
Neurospora RDE-1of C elegans RNA helicase SDE3 is related to SMG-2 of C
elegans and Mut-6 of Chlamydomonas MUT-7 gene of C elegans encodes a protein
similar to Rnase D whereas the Drosophila DICER gene encode a protein similar to
RNase III An Arabidopsis ortholog of DICER gene has been identified called CAF
SIN1 SUS1(Thakur 2005 Poethig 2009)
Since the RNAi machinery is present constitutively within the eukaryotic cell it
is important to explore the metabolic advantages that are accorded by the RNAi related
proteins during the intrinsic normal growth of cells and development of organisms The
natural RNAi machinery not only keeps the mobile transposable elements from
disrupting the integrity of the genomes as suggested by the analysis of model organisms
like A thaliana C elegans D melanogaster (Tabara et al 1999 Wu-Scharf et al
2000 Aravin et al 2001 Hamilton et al 2002 Lisch 2009) but also participates in
development of organisms Genetic defects in Celegans RNAi genes ego1 and dicer
cause known specific developmental errors (Grishok et al 2000 Grishok et al 2001
Knight amp Bass 2001 Hall et al 2013) Similarly the Argonaute family of genes of A
thaliana (especially the ZWILLE proteins) is also responsible for plant architecture and
meristem development (Carmell et al 2002 Hamant and Pautot 2010) and the Dicer
homologue of A thaliana CAF1 is required for embryo development (Golden et al
2002 Nakano et al 2011) Thus genetic evidence illustrates the role of the RNAi
machinery as a controller of development related genes The mechanistic details of
these developmental processes are beginning to emerge
Chapter 1 Introduction and Review of Literature
27
Figure 16 Mechanism of gene silencing induced double stranded RNA The dicer enzyme to
produce siRNAs cleaves the RNA The siRNA generated bind to the nuclease complex
RISC The antisense component in the siRNA in the RISC guides the complex towards
the cognate mRNA resulting in endonucleolytic cleavage of the mRNA
Chapter 1 Introduction and Review of Literature
28
112 MicroRNA or MiRNA
In 1991 Ambros and co-workers first isolated a lin-4 mutant of Celegans which was
arrested at the first larval stage (Lee et al 1993) Later on the let-7 mutation was
isolated in the same system which was responsible for development through the fourth
larval stage Both lin-4 and let-7 encode short 22-nucleotide mature RNAs and were
called short temporal RNA because they control the temporal development program of
C elegans The mature lin-4 RNA defines (negatively regulates) the mRNA expression
of the lin-14 and lin-28 hetrochronic genes with the antisense mediated repression
mechanism of translation initiation and thus specifies the fate of cells during the first
three larval stages Recent studies have revealed that the short temporal RNAs are
actually members of a group of tiny RNAs (21to 28 nucleotides) called the micro-
RNAs (miRNA) (Bartel 2009) isolated members of which could easily run to a few
thousand which includes both those were identified computationally and by deep
sequencing Some of the components of the RNAi machinery have also been clearly
established as the effector proteins for the maturation of miRNAs
113 MicroRNAs in plants
1131 Discovery
MicroRNAs have been discovered by three basic approaches Direct cloning forward
genetic direct cloning and bioinformatic prediction followed by experimental
validation The most direct method of miRNA discovery is to isolate and clone small
RNAs from biological samples and several groups have used this approach to identify
plant miRNAs (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002 Xie et al 2003 Sunkar et al 2005 Williams et al 2013) The cloning methods
adapted were similar to those used to identify large numbers of animal miRNAs
(Lagos-Quintana et al 2001 Lau et al 2001 Aravin amp Tuschl 2005) which involve
isolating small RNAs followed by oligonucleotide adapter ligation reverse
transcription amplification and sequencing Some protocols incorporate methods to
enrich for Dicer cleavage products (ie molecules with 5prime-phosphates and 3prime-
hydroxyls) and to concatemerize the short cDNAs so that several may be identified in a
single sequencing read (Lau et al 2001 Llave et al 2002a Jagadeeswaran amp Sunkar
2013)The initial cloning experiments in Arabidopsis identified 19 miRNAs belonging
to 15 families (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002) although hundreds of other small RNAs such as siRNAs and RNA degradation
Chapter 1 Introduction and Review of Literature
29
fragments were also cloned through the direct cloning methods To select miRNAs
from the pool of small RNAs secondary structures of the RNA molecules were
examined in compliance with the acknowledged structural features of miRNAs
(Ambros et al 2003) The secondary structures were validated using several ad-hoc
software tools such as the Mfold or RNAfold programs (Reeder et al 2006) Finally
the existence of mature miRNAs was determined by RNA gel blot hybridization These
direct cloning methods were successful in isolating many miRNAs in Arabidopsis
(Reinhart et al 2002 Sunkar amp Zhu 2004) Rice (Sunkar et al 2005) Poplar (Lu et
al 2005) moss (Arazi et al 2005) and Tobacco (Billoud et al 2005)
Although miRNAs were first discovered through forward genetic screens in
worms (Lee et al 1993 Reinhart et al 2000)only few A thaliana miRNA gene
families have been discovered by this method (Chen 2008) in plants MiRNA
involvement in plant mutant phenotypes were not properly inferred till the cloning
experiments established that plant genomes contain numerous miRNAs (Park et al
2002 Reinhart et al 2002 Rhoades et al 2002) MiRNA loss-of-function allele has
been identified by forward genetic screens early extra petala1 is caused by a
transposon insertion ~160bp upstream of the predicted miR164c stem loop resulting in
flowers with extra petals (Baker et al 2005 Irish 2008) The fact that loss-of-function
miRNA mutants have been rarely discovered using forward genetics reflects small
target size for mutagenesis coupled with redundancy in nearly all evolutionarily
conserved plant miRNAs which are encoded by gene families (Table16) Family
members are likely to have overlapping functions buffering against loss at any single
miRNA locus Over expression screens can circumvent redundancy limitations At least
four plant miRNAs miR319 (also known as miR-JAW) miR172 (also known as EAT)
and miR166 were isolated in over expression screens for dominant mutants with
developmental abnormalities (Aukerman amp Sakai 2003 Palatnik et al 2003 Kim et
al 2005 Williams et al 2005b Schommer et al 2012) Mutations in miRNA target
sites which can prevent the entire family of miRNAs from repressing a target gene can
also circumvent redundant functions of miRNA family members
The dominant mutations in the HD-ZIP genes PHB PHV and REVOLUTA
(REV) in Arabidopsis and ROLLEDLEAF1 (RLD1) in Maize result in adaxialization of
leaves andor Vasculature (McConnell amp Barton 1998 McConnell et al 2001 Nelson
et al 2002 Zhong amp Ye 2004 Allen amp Millar 2012) and are all caused by mutations
Chapter 1 Introduction and Review of Literature
30
in miR166 complementary sites (Rhoades et al 2002 Emery et al 2003 Jones-
Rhoades amp Bartel 2004 Mallory et al 2004 Zhong amp Ye 2004)
In both plants and animals cloning was the initial means of large-scale
miRNA discovery (Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Reinhart et al 2002 Mendes et al 2012) However cloning is biased toward
RNAs that are expressed highly and broadly MiRNAs expressed at low levels or
only in specific cell types or in response to certain environmental stimuli were more
difficult to clone Sequence based biases in cloning procedures might also cause
certain miRNAs to be missed Because of these limitations bioinformatics approaches
to identify miRNAs have provided a useful complement to cloning
A straightforward use of bioinformatics has been carried out to find homologs
of known miRNAs both within the same genome and in the genomes of other species
(Pasquinelli et al 2000 Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Mendes et al 2009) A more difficult challenge is to identify miRNAs
unrelated to previously known miRNAs This was first accomplished for
vertebrate nematode and fly miRNAs using algorithms that search for conservation
of sequence and secondary structure (ie miRNA stem-loop precursors) between
species searching for patterns that are characteristic of miRNAs (Lai et al 2003 Lim
et al 2003 Lim et al 2005)
Most of the methods identified numerous miRNAs but are necessarily more
relaxed in terms of allowed structures Some approaches take advantage of the high
complementarity of plant miRNAs to target messages implementing the requirement
that the candidate has conserved complementarity to mRNAs (Jones-Rhoades amp Bartel
2004 Adai et al 2005) This additional filter has been useful for distinguishing
authentic plant miRNAs from false positives which later extended to mammalian
miRNA gene prediction (Xie et al 2005 Mendes et al 2012)
Another criteria used by most algorithms in the filters is evolutionary
conservation In plants mature miRNA sequences are conserved to a higher degree
than their precursor sequences For example Arabidopsis and rice diverged more than
130 million years ago (Friis et al 2004) but some miRNAs are highly conserved
in these plant species (Reinhart et al 2002) Furthermore various parameters for the
secondary structures such as free energy the number of paired residues within a
Chapter 1 Introduction and Review of Literature
31
miRNA or the number and size of bulges are also used to validate the predictions
(Jones-Rhoades amp Bartel 2004 Wang et al 2004 Adai et al 2005 Mendes et al
2012)
1132 Conserved microRNAs in Plants
Till date cloning genetics and bioinformatics screens have resulted in annotation of
approximately 184 potential Arabidopsis miRNAs (miRBase release 13) These
miRNAs seem to arise through gene duplication and subsequent diversification and
represent approximately 100 families (Maher et al 2006 Axtell 2013) Twenty one
families represented by 92 genes are clearly conserved in species beyond Arabidopsis
(Table 15) The presence of these miRNA families in other sequenced plant genomes
indicates that different plant species have an evolutionarily fluid set of miRNAs
(Willmann amp Poethig 2007) Recent studies on mosses one of the most ancient land
plants have shown that at least 14 miRNA families are conserved in eudicots and
mosses and therefore it appears that plant miRNAs share a common ancient origin
(Arazi et al 2005 Axtell amp Bartel 2005 Talmor-Neiman et al 2006 Zhang et al
2006 Axtell 2013) The number of members per family in a genome ranges from 1-32
with the exception of miR-430 which is represented by a cluster of ~80 loci in zebra
fish (Giraldez et al 2005)
In plants the number of members in each family correlates among examined
species certain families contain numerous members in all three species (eg miR156
miR166 miR169) whereas others consistently contain fewer genes (eg miR162
mir168 miR394) (Table 16) Although it is still unclear why certain miRNA are
encoded by more than one gene in plants (eg 12 genes encoding miR156) this
correlation might suggest functional significance of various miRNA family sizes Most
annotated plant miRNAs are conserved throughout flowering plants while a few
miRNAs are found only in a single sequenced genome and thus they could be
evolutionarily lsquoyoungrsquo miRNAs For example Arabidopsis has several miRNA
families that are not found in poplar or rice suggesting that miRNAs could be
generated continuously during evolution (Allen et al 2004a Rajagopalan et al
2006 Fahlgren et al 2007 Axtell 2012 de Alba et al 2013) Consistent with the
notion that non conserved miRNAs are of a more recent evolutionary origin these
miRNAs are mainly encoded by a single locus in the genome Within each family the
mature miRNA is always located in the same arm
Chapter 1 Introduction and Review of Literature
32
Table 16 MicroRNA families conserved in plants
miRNA
family
Arabidopsis Oryza Populus
miR156 12 12 11 miR159319 6 8 15 miR160 3 6 8 miR162 2 2 3 miR164 3 5 6 miR166 9 12 17 miR167 4 9 8 miR168 2 2 2 miR169 14 17 32 miR171 4 7 10 miR172 5 3 9 miR390 3 1 4 miR393 2 2 4 miR394 2 1 2 miR395 6 19 10 miR396 2 5 7 miR397 2 2 3 miR398 3 2 3 miR399 6 11 12 miR408 1 1 1 miR403 1 0 2 miR437 0 1 0 miR444 0 1 0 miR445 0 9 0 Total 92 127 169
All known miRNA families that are conserved between more than one plant species are listed
together with the number of genes identified in the sequenced genomes Rice miRNA families that have orthologs in maize but do not appear to have orthologs in the eudicots (Arabidopsis and Populus) are marked with an asterisk The following families contain miRNA genes annotated with
more than one number miR156 (miR156 and miR157) miR159319 (miR159 and miR319) miR166 (miR165 and miR166) miR171 (miR170 and miR171) and miR390 (miR390 and miR391)
of the stem loop (5prime- or 3prime) as it would be expected if the genes carry common ancestry
(Figure 17) The sequence of the mature miRNA and to some extent the segment on
the opposite arm of the hairpin to which it pairs is highly conserved between the
members of the same family (both within and between the species) while the sequence
secondary structure and length of the intervening ldquolooprdquo region can be highly divergent
within the same family
The patterns of paring and non pairing nucleotides are often conserved between
homologous miRNA stem loops from different species (Figure 17) The significance of
conserved mismatches is opined to guide DCL1 to cleave at the appropriate positions
along the stem loop
Chapter 1 Introduction and Review of Literature
33
Figure 17 Representative stem loop of miR164 Segments corresponding to the mature miRNAs
are shown in red (Adapted from Rajagopalan et al 2006)
Although many miRNAs are highly conserved in plants the degree of conservation in
most cases is quite low between plants and animals one exception is miR854 A recent
study has revealed that miR854 is conserved between Arabidopsis and animals and that
the Arabidopsis miR854 differs from the human miR854 only by a single nucleotide
(Arteaga-Vazquez et al 2006 Axtell 2012 de Alba et al 2013) The Arabidopsis
miR854 has multiple binding sites in the 3prime-untranslated region (UTR) of
OLIGOURIDYLATE-binding-PROTEIN mRNA 1bA (UBP1b) and represses the
translation of the UBP1b mRNA The animal miR854 is also complementary to a site in
the 3prime-UTR of the UBP1b homolog in animals However it is currently unclear how the
animal miR854 regulates its target mRNA Another distinction between plant and
animal miRNAs is the genomic organization of the miRNA genes While plant miRNA
genes are located predominantly in the intergenic regions animal miRNA genes tend to
localize in the intragenic regions both in the introns or exons of protein coding genes
Moreover miRNA genes are often clustered within a single precursor transcript
whereas individual plant miRNAs are derived from individual precursor RNAs (Bartel
Chapter 1 Introduction and Review of Literature
34
2004 Bonnet et al 2004 Kim 2005 Vaucheret et al 2006 Marco et al 2013)
1133 Non-conserved miRNA and challenges in annotation
Most miRNAs are conserved throughout flowering plants (Table 15) but many more
were found only in a single sequenced genome thus considered to be of a more recent
evolutionary origin The extended homology between few non conserved miRNAs
precursors and their target genes provides strong evidence that these relatively ldquoYoungrdquo
miRNAs arose from the duplication of the target gene segments (Allen et al 2004)
Several non-conserved miRNAs including miR161 miR163 miR173 miR447
miR475 and miR476 are known to direct cleavage of target transcripts (Allen et al
2004a Adai et al 2005 Sunkar et al 2005 de Alba et al 2013) It is difficult to
confidently predict targets for many because it is not possible to use complementary
site as a filter against false-positive target predictions It is also difficult to
confidently annotate non-conserved miRNAs as miRNA rather than siRNAs The
established minimal standard for miRNA annotation is a small RNA with detectable
expression and the potential to form a stem-loop when joined to flanking genomic
sequence (Kasschau et al 2003) In practice these requirements are too loose to
definitively categorize many small RNAs cloned from plants Many plant siRNAs are
detectable on blots (Vazquez et al 2004b) and hundreds of thousands of non-
miRNA genomic sequences can be predicted to fold into secondary structures that
resemble structures of plant miRNA precursors (Jones-Rhoades amp Bartel 2004)
Therefore without conservation of both sequence and secondary structure it is
difficult to be confident that a given cloned RNA originated from a stem-loop (ie is a
miRNA) rather than from a double-stranded RNA (ie is a siRNA) In fact many of
the thousands o f small RNAs cloned from Arabidopsis (Gustafson et al 2005) would
probably meet the literal requirements for annotation as miRNAs A few of these
sequences might be miRNAs but others that meet the literal criteria probably are not
so The challenges of annotating non conserved small RNAs were evidenced by
three related small silencing RNAs that were originally annotated as miRNAs
that turned out to be trans-acting siRNAs (tasiRNAs)(Kanno et al2013)
Although most miRNAs that are broadly conserved among flowering plants are
now being identified and experimentally validated (Table 16)(Jones-Rhoades amp
Bartel 2004) the challenges and ambiguities for classifying non-conserved miRNAs
prevent meaningful estimates of the total number of miRNA genes in Arabidopsis and
Chapter 1 Introduction and Review of Literature
35
other plant genomes It is possible that miRNAs might have escaped detection
because they are non-conserved and expressed in specific tissues or conditions and can
only be identified by using high- throughput sequencing
1134 MicroRNA biogenesis
11341 Transcription of microRNA precursor
Plant miRNAs are primarily found in the genomic regions that are not associated with
the protein coding genes (Reinhart et al 2002) and it appears that most if not all plant
miRNAs are produced from their own transcriptional units Plant miRNA genes are
occasionally clustered near each other in the genome suggesting transcription of
multiple miRNAs from a single primary transcript [eg the miR395 cluster (Grishok
et al 2001 Jones-Rhoades amp Bartel 2004b)] but this polycistronic arrangement of
miRNA genes appears far less frequently in plants than in animals (Bartel 2004)
Northern blot EST and mapping evidence indicate that plant miRNA primary
transcripts (also known as pri-miRNAs) as in animals are longer than needed to
encompass the miRNA stem-loops (Palatnik et al 2003 Jones-Rhoades amp Bartel
2004b) At least some of these pri-miRNA transcripts appear to be spliced
polyadenylated (Kurihara amp Watanabe 2004) and capped (Xie Z et al 2005) Two
rice miRNAs are contained within transcripts that contain exon junctions within the
presumptive stem-loop precursor implying that in these cases splicing is a prerequisite
for Dicer recognition (Sunkar et al 2005) Plant pri-miRNAs are over 1kb in length
and they are usually preceded by typical TATA box motifs They can also undergo
canonical splicing polyadenylation and capping All these observations indicates RNA
polymerase II is probably responsible for transcribing most plant miRNAs (Xie Z et
al 2005) as appears to be the case for many animal miRNAs Relatively little is
known about the regulation of miRNA transcription in plants but there is no reason
to suspect that this regulation would differ from that of protein-coding transcripts as
the bioinformatics analysis of the sequence upstream of the transcriptional start
sites of the miRNA genes showed the presence of putative binding motifs of
number of known transcription factors (Megraw et al 2006 Sethupathy 2013)
11342 MicroRNA processing and export
In plants maturation of miRNA is a step wise process A miRNA gene is transcribed to
pri-miRNA which is much longer than the pre-miRNA possessing the characteristic
stem-loop structure Like transcription of most protein-coding genes the miRNA
Chapter 1 Introduction and Review of Literature
36
transcription is processed by RNA polymerase II (Pol II) enzymes and the pri-
miRNAs are 5prime-capped and 3prime-polyadenylated (Lee et al 2004 Xie Z et al 2005)
Mature miRNAs are produced from pri-miRNAs through at least two sequential
processing steps by RNase III-type endonucleases In animals pri-miRNAs are first
processed at the bottom portion of the stem by Drosha a nuclear-localized RNase III
enzyme to liberate pre-miRNAs The pre-miRNAs are then exported to the cytoplasm
with the help of exportin-5The pre-miRNAs are further processed by Dicer another
RNaseIII enzyme to a duplex consisting of the miRNA and the antisense strands
(miRNA) Since plants do not have any Drosha homologs the two sequential
processing steps seem to be carried out by DCL1 a Dicer homolog in the nucleus
(Kurihara amp Watanabe 2004 de Alba et al 2013 Rogers amp Chen 2013)
The processing of pri-miRNAs to pre-miRNAs by DCL1 is assisted by two
other proteins HYPONASTY LEAVES1 (HYL1) and SERRATE (SE) HYL1 belongs to a
family of double-stranded RNA (dsRNA) binding protein and interacts with DCL1 in
vitro and in vivo (Lu amp Fedoroff 2000 Hiraguri et al 2005 de Alba et al 2013
Rogers amp Chen 2013) Loss-of-function mutations in the HYL1 gene result in
decreased accumulation of miRNAs and concomitant accumulation of pri-miRNAs
(Han et al 2004 Vazquez et al 2004a Kurihara et al 2006) SE a C2H2 zinc finger
protein interacts with DCL1 and HYL1 and plays a role in the processing of pri-
miRNAs to pre-miRNAs (Lobbes et al 2006 Yang L et al 2006) In addition the
nuclear cap-binding complex proteins CBP20 and CBP80 that are known as ABA
HYPER SENSITIVE 1(ABH1) have been shown to be required for proper miRNA
processing (Gregory et al 2008 Laubinger et al 2008) Recently it has been
demonstrated that DCL1 can release 21-nt RNAs from dsRNA as well as from
synthetic miR167 precursor RNAs in vitro However this cleavage is error prone and
inefficient in the absence of HYL1 and SE which accelerate the rate of DCL1-mediated
cleavage and promote accurate processing of miRNAs (Dong et al 2008 Rogers amp
Chen 2013)
11343 Methylation of the miRNAmiRNAduplex
After the miRNAmiRNAduplexes are released from the pre-miRNAs by the DCL1
activities the duplexes are methylated at the 2primeOH of the3prime-ends by HUA ENHANCER1
(HEN1) a small RNA methyltransferase (Yu et al 2005 Yang Z et al 2006 Rogers
amp Chen 2013 ) This step which is distinct from the RNase III-mediated processing
Chapter 1 Introduction and Review of Literature
37
step distinguishes the biogenesis of plant miRNAs from that of animal miRNAs
Mutations in the HEN1 gene were first isolated in a morphological screening for floral
development although the HEN1 mutants display pleiotropic phenotypes similar to the
developmental defects observed in the DCL1 mutants (Chen et al 2002 Park et al
2002 Rogers amp Chen 2013)
Figure 18 Biogenesis of microRNA
A Biogenesis of plant MicroRNA B Biogenesis of metazoananimal MicroRNA
The miRNA (red) is incorporated into RISC and the miRNA(blue) in degraded
This enzyme has two dsRNA-binding domains and nuclear localization signal It
is also conserved in fungi and is required for the processing of siRNAs as well as of
miRNAs (Park et al 2002 Boutet et al 2003 Xie et al 2003) Although the HEN1
protein is localized to the nucleus its methylation activity in the cytoplasm cannot be
ignored (Xie et al 2004 Rogers amp Chen 2013)
Chapter 1 Introduction and Review of Literature
38
The methylation of the miRNAmiRNA duplex is required to protect miRNAs
against the 3prime-end uridylation activity and subsequent degradation (Li et al 2005) In
the hen1 mutants accumulation of miRNAs is reduced to a lower level Also the
miRNAs appear to be heterogeneous in their sizes such that a ladder of bands rather
than a single species is observed for most miRNAs in the mutants (Li et al 2005)
MiRNA cloning and sequencing analyses revealed the presence of a number of U
residues at the 3prime-end of miRNAs in the hen1 mutants Moreover these nucleotides do
not correspond to the miRNAs in the pre-miRNAs indicating that these nucleotides are
added after the processing of the miRNAs from pre-miRNAs (Li et al 2005)
Uridylation of RNAs has also been proven to be correlated with RNA decay (Shen amp
Goodman 2004 Mullen amp Marzluff 2008) suggesting that uridylation attracts an
exonuclease which degrades miRNAs in plants
11344 MiRNA incorporation in to the RISC and its nuclear export
The methylated miRNAmiRNA duplexes undergo RISC assembly In this process
the miRNA of the duplexes is selectively incorporated into the RISC and the miRNA
appears to be removed and subsequently degraded (Hammond et al 2000 Hutvagner
amp Zamore 2002 Schwarz et al 2003 de Alba et al 2013 Rogers amp Chen 2013)
The ARGONAUTE 1(AGO1) proteins are core components of the RISC complex
They contain two structural domains the N-terminal PAZ domain that binds to the 3prime-
end of single-stranded RNAs and the C-terminal piwi domains that has a structure
similar to that of RNase H (Cerutti et al 2000 Parker et al 2004) Several AGO
proteins possess endonucleolytic activities within the PIWI domain and cleave target
mRNAs in the middle of the site complementary to miRNA (Liu et al 2004 Meister et
al 2004 Miyoshi et al 2005) Mutations in the AGO1 gene cause miRNA target
genes to be ectopically expressed and the levels of some miRNAs are reduced in the
mutants (Kidner amp Martienssen 2004 Vaucheret et al 2004 Kidner amp Martienssen
2005 Ronemus et al 2006) Moreover the phenotype of the AGO1 hypomorphic
alleles display defects in lateral organ polarity and leaf and flower morphology as
observed in the dcl1 mutants and null AGO1 alleles are embryonically lethal
supporting the close relationship between the AGO proteins and miRNA processing
(Kidner amp Martienssen 2004 Vaucheret et al 2004 de Alba et al 2013 Rogers amp
Chen 2013)
The production of miRNA miRNA duplexes occur in the nucleus while
Chapter 1 Introduction and Review of Literature
39
miRNA-RISC complexes are present in the cytoplasm where target mRNAs are
located This discordance of functional sites requires an export step during miRNA
biogenesis HASTY the Arabidopsis homolog of exportin-5 is thought to be involved
in miRNA nuclear export (Bollman et al 2003 Park et al 2005)The hasty mutant
exhibits pleiotropic defects in plant development and reduced accumulation of most
miRNAs as in the DCL1 or AGO1 mutants Howeverit is unclear whether the HASTY
proteins export the miRNA miRNA duplexes or the miRNA-RISC complexes in
plants
11345 Feedback regulation of miRNA biogenesis
MiRNA biogenesis is under feedback regulation such that the DCL1 and AGO1 genes
are themselves regulated by miRNAs DCL1 is itself a target of miR162 which leads to
the cleavage of the DCL1 mRNA (Xie et al 2003 de Alba et al 2013) Consistent
with this the levels of DCL1 mRNA are elevated in the mutants of miRNA processing
genes in which the miR162 abundance is reduced In addition there is a predicted
hairpin structure of miR838 in the 14th
intron of the DCL1 primary transcript
(Rajagopalan et al 2006) This prediction implies that DCL1-mediated processing
on this site can result in the cleavage of the DCL1 primary transcript into two truncated
fragments the N-terminal capped fragment and the C-terminal polyadenylated
fragment If this is the case it is possible that miRNA biogenesis competes with the
splicing event of the DCL1 pre-mRNA
The AGO1 is also regulated by miRNA There is a complementary site for
miR168 binding in the AGO1 mRNA and the transcript level of the AGO1 mRNA is
reduced through an mRNA cleavage mechanism (Vaucheret et al 2006 de Alba et al
2013 Rogers amp Chen 2013) indicating that the AGO1 mRNA levels are tightly
controlled by the levels of the AGO1 protein
1135 Mechanism of miRNA action
11351 MiRNA-directed mRNA cleavage
By forming the miRNA-RISC assembly miRNAs can direct the RISC to regulate gene
expression by two post transcriptional mechanisms mRNA cleavage or translational
repression (Bartel 2004 Jones-Rhoades et al 2006 Dalmay 2013) The degree
of miRNA-mRNA complementarity plays an important role for the
determination of the mechanism to be used A general rule is that while perfect or
Chapter 1 Introduction and Review of Literature
40
near-perfect complementarity induces mRNA cleavage central mismatches trigger
translational repression (Carrington amp Ambros 2003 Jones-Rhoades amp Bartel 2004)
Unlike most animal miRNAs plant miRNAs have near-perfect or perfect
complementarity to their targets and mRNA cleavage is assumed to be a predominant
mechanism by which miRNAs regulate gene expression in plants
In RNA cleavage mechanism miRNAs guide the AGO component of RISC to
cleave a single phosphodiester bond of the target mRNA within the miRNA-binding
site (Llave et al 2002b Tang et al 2003) The truncated fragments are then released
and degraded freeing the RISC to recognize and cleave another target mRNA This has
been validated by the detection of 3prime cleavage products that have 5prime ends that start at the
middle of the complementary regions The mRNA cleavage products are then further
degraded by other mechanisms The 3prime cleavage products of some miRNA targets are
degraded by the 5prime-3prime exonuclease XRN4 an Arabidopsis homolog of the major Yeast
mRNA degrading exonuclease Xrn1p It has been observed that the 3prime cleavage
products of some target mRNAs were accumulated in the xrn4 mutants (Souret et al
2004 Dalmay 2013) However the 3prime cleavage products of other miRNA targets do
not accumulate in the xrn4 mutants indicating that there are also XRN4 independent
mechanisms The 5prime cleavage products are typically untraceable by RNA gel blot
hybridization but can be detected by sensitive PCR based methods It has been found
that the 5prime cleavage products tend to obtain an oligo U tail at the 3prime ends This 3prime U tail
is correlated with shortening of the 5prime cleavage products from the 5prime end leading to the
conclusion that the 3prime uridylation causes 5 prime- 3 prime exonucleolytic decay of the 5prime cleavage
products (Shen amp Goodman 2004)
DCL1 HYL1 and SE interact with each other and are co-localized in the
nuclear bodies termed Dicing bodies or D-bodies in which some pri-miRNA shave
been found suggesting that D-bodies serve as a center for active miRNA processing
(Fang amp Spector 2007 Fujioka et al 2007 Song et al 2007) The miRNA
processing site appears to be distinct from the Cajal bodies that have previously been
found as a siRNA processing site (Pontes amp Pikaard 2008) The D-bodies include
SmD3 and SmB proteins that are found in the Cajal bodies However At collin an
additional marker for the Cajal bodies is not found in the D-bodies Moreover the D-
bodies are present in an At collin mutant lacking the Cajal bodies The pri-miRNAs of
miR171A and miR159A genes have been found to be co-localized with HYL1 and
Chapter 1 Introduction and Review of Literature
41
DCL1 in the D- bodies that are distinct from the Cajal bodies (Song et al 2007)
11352 MiRNA-directed translational repression
Translation repression is another mechanism exerted by plant miRNAs The regulatory
mechanism of miR172 which regulates flowering time and floral organ identity is
consistent with translation repression MiR172 over production does not affect the
transcript levels of APETALA2 (AP2) and AP2- like target mRNAs Instead the AP2
protein level is reduced in the miR172 over expressing mutant (Aukerman amp Sakai
2003 Chen 2004 Dalmay 2013) It was later shown that miR172 can also direct the
cleavage of the target mRNAs although the steady-state mRNA level is unchanged due
to feedback regulation (Schwab et al 2005 Jung et al 2007) It is clear that miR172
regulates its target genes primarily by translational repression rather than mRNA
cleavage Recently it has been reported that miR156157 and miR854 also regulate
their target genes through translational repression (Arteaga-Vazquez et al 2006
Gandikota et al 2007 Dalmay 2013) It was once thought that translational repression
is an exception to the rule However experimental evidence suggest a widespread
coexistence of translational repression and mRNA cleavage (Brodersen et al 2008)
1136 Regulatory roles of miRNAs in plant development
The miRNA target prediction has been a difficult task in order to obtain regulatory
targets without bringing in too many false predictions Plant growth and developmental
processes regulated by miRNAs are quite diverse and enormous Furthermore plant
miRNAs work cooperatively or antagonistically to establish a balanced regulation This
notion is well consistent with the findings that mutations in the regulators of miRNA
biogenesis transport and processing such as AGO1 DCL1 HYL1 HEN1 ABH1
and HASTY cause pleiotropic phenotypes (Mallory amp Vaucheret 2006 de Alba et al
2013) To date the roles of miRNAs have been mostly deduced from obtaining miRNA
over expressing transgenic plants or gain-of-function mutants in which miRNA-
resistant target genes are ectopically expressed
11361 Phase transition
Plants pass through a series of developmental phase transitions during their life span
beginning with seed germination continuing through vegetative phase change
reproductive phase change flowering initiation and finally seed production for the next
generation Because of their importance in understanding plant developmental
Chapter 1 Introduction and Review of Literature
42
processes genes regulating developmental transitions have been extensively studied
and underlying molecular mechanisms have been explored in various plant species
Majority of researches were mainly focused on vegetative phase change and
reproductive transition in Arabidopsis and Maize (Poethig 2003 Baurle amp Dean
2006) Particularly the mutations of the genes encoding various regulators of miRNA
biogenesis and transport such as HST SE and AGO exhibit altered juvenile to adult
vegetative phase change and vegetative to reproductive change (Hunter et al 2003
Peragine et al 2004 Vazquez et al 2004a Jones-Rhoades et al 2006 de Alba et al
2013)
Over-expression of the miR156a gene causes late flowering and delays
vegetative phase change (Wu amp Poethig 2006 Gandikota et al 2007 Wang et al
2008) It has been shown that miR156 negatively regulates a group of genes encoding
SQUAMOSA PROMOTER-BINDING PROTEIN-LIKE (Lewis et al 2007 de Alba et
al 2013 Rogers amp Chen 2013) transcription factors such as SPL3 SPL4 and SPL5
that promote flowering initiation In contrast transgenic plants over expressing
miR156-resistant SPL345 genes are early flowering Interestingly the endogenous
level of miR156 is temporally regulated In the early juvenile vegetative phase the
level of miR156 is very high but decreases rapidly before the onset of the adult juvenile
phase (Wu amp Poethig 2006)
The Arabidopsis early activation tagged (eat-D) mutant exhibits early flowering
with disrupted floral structure The mutant phenotypes are caused by over expression of
the miR172a-2gene (Aukerman amp Sakai 2003) MiR172 regulates a small group of
genes encoding APETALLA2 (AP2)-like transcription factors such as TARGET OF
EAT1 (TOE1) TOE2 SCHLAFMUTZE (SMZ) and SCHNARCHZAPFEN(SNZ)
TOE1 TOE2 SMZ and SNZ have been shown to participate in their productive phase
change by repressing a floral integrator FLOWERING LOCUS T (FT)(Jung et al
2007) Consistent with this toe1-2D 35STOE2 35SSMZ and 35SSNZ plants
exhibit late flowering (Jung et al 2007) MiR172 is also regulated temporally and
highly expressed in the adult vegetative phase both in Arabidopsis and maize (Chuck et
al 2007) Because the temporal expression patterns of miR156 and miR172 are
complementary to each other it has been suggested that the two miRNAs interact with
each other in regulating phase change (Chuck et al 2009 de Alba et al 2013) In
support of this view it has been shown that miR172 is down-regulated in the
Chapter 1 Introduction and Review of Literature
43
Corngrass1(Cg1) mutant in which miR156 is overproduced (Chuck et al 2007 de
Alba et al 2013) However there is no direct evidence for the direct linkage between
miR156 and miR172 It is also envisioned that miR156 and miR172 would not be
directly related For example miR156 regulates plastochron index that is the inverse of
leaf initiation rate and thus frequently used to analyze temporal patterns of plant
development In contrast miR172 does not affect plastochron (Wang et al 2008) The
down-regulation of miR172 in the maize miR156-over producing mutants would be
due to the prolonged juvenile vegetative phase in the mutants
Another miRNA that regulates flowering initiation is miR159 It regulates
MYB33 and MYB65 which are closely related to the barley GAMY B transcription
factor that responds to gibberellic acid (GA) The level of miR159 is elevated in GA-
treated seedlings but reduced in the GA biosynthetic ga1 mutant Transgenic plants
over producing miR159 exhibit delayed floral induction under short days (Achard et
al 2004) It is been suggested that MYB33 mediates GA signal to regulate LEAFY
(LFY) These observations indicate that miR159 plays an important role in the GA
signaling pathways that regulate proper floral induction under short days (Achard et al
2004)
11362 Floral development
Arabidopsis flowers consist of four distinct organs They arise in a characteristic
pattern within concentric rings called whorls from outside to inside four sepals four
petals six stamens and the central pistil consisting of two carpels Identities of the
floral organs are determined by three classes of regulatory genes classA classB and
class C genes (Krizek amp Fletcher 2005) The A and C class genes act antagonistically
to restrict their expression domains (Bowman et al 1991)
It is notable that miR172 also regulates floral architecture in addition to its role
in the timing of flowering initiation MiR172 inhibits the translation of the AP2 mRNA
(Chen 2004) Transgenic plants overproducing miR172 not only exhibit early
flowering but also show abnormal floral development which is quite similar to those
of the class A gene mutants such as the ap2 mutants (Aukerman amp Sakai 2003 de
Alba et al 2013) When the activity of the class A genes is inactivated that of the class
C genes such as AGAMOUS (AG) is elevated As a result the miR172- overproducing
transgenic plants exhibit no petals along with homeotic transformation of the sepals to
Chapter 1 Introduction and Review of Literature
44
the carpels (Aukerman amp Sakai 2003 Chen 2004)
Interestingly miR166 also regulates floral architecture The role of miR165166
in the regulation of meristem activity is closely linked to floral meristem formation and
organization (Zhao et al 2007 Miyashima et al 2013) Floral structure is severely
disrupted in the miR165166-overproducing mutants such as men1 and jba-1D in
which miR166 is overproduced (Williams et al 2005b Jung amp Park 2007) The men1
and jba-1D mutants have smaller gynoecium and reduced carpel number In an extreme
case the jba-1D homozygous plants lack visible carpels (Williams et al 2005b)
Similar phenotypes have been observed in the miR165 overproducers (Zhao et al
2007) It has been suggested that the phenotypes are possibly caused by altered AG
expression in the floral meristem (Jung amp Park 2007 Turchi et al 2013)
Other miRNAs also affect floral development Overproduction of miRNAs
involved in auxin signaling also affects floral architecture Transgenic plants
overproducing miR167 have defects in the formation of ovule and anther with reduced
fertility (Ru et al 2006) It has been shown that miR167 targets ARF6 and ARF8 that
play a role in gynoecium and stamen development and in regulating fertility (Ru et al
2006 Wu et al 2006) These observations suggest that auxin signals are also closely
related to floral organogenesis In addition transgenic plants over-producing miR164
and the cuc1cuc2 double mutants show fused sepals and reduced petals (Laufs et al
2004 Turchi et al 2013) suggesting that miR164 is also related to floral meristem
activity and boundary specification of the meristematic region
11363 Shoot apical meristem development
Shoot apical meristem comprises self-maintaining totipotent cells The shoot apical
meristem activity affects diverse aspects of plant developmental processes including
leaf development vascular development and lateral organ polarity (Bowman et al
2002) Because miR165166 plays a primary role in meristem formation
overproduction of miR165166 and multiple knockout mutants of its target genes
exhibit pleiotropic pheonotypes (McConnell et al 2001 Emery et al 2003 Kim et al
2005 Williams et al 2005b)
The targets of miR165166 include genes encoding the homeo domain-leucine
zipper (HD-ZIP) class III transcription factors such as PHABULOSA(PHB)
PHAVOLUTA (PHV) REVOLUTA(REV) ATHB15CORONA (CNA) and ATHB8
Chapter 1 Introduction and Review of Literature
45
PHB PHV and ATHB15 have overlapping functions in controlling meristem
development (Turchi et al 2013) Indeed the phb phv athb15 triple mutants have an
enlarged shoot apical meristem Consistent with this the miR166- overproducing men1
and jba-1D mutants exhibit enlarged shoot apical meristems (Kim et al 2005
Williams et al 2005b Turchi et al 2013) In the mutants the shoot apical meristems
are multiplied and large
The development of shoot apical meristems is also regulated by the WUSCHEL
(WUS)ndashCLAVATA (CLV) signaling pathway WUS positively regulates cell
proliferation In contrast CLV3 which is expressed in stem cells limits the WUS
expression and thus inhibits cell proliferation in the SAM (Sharma et al 2003)
Consistent with this notion WUS is expressed to a higher level in jba-1D
explaining the ectopic enlargement of the SAM in the mutant (Williams et al 2005)
While WUS is closely related to miR165166 in the shoot apical meristem
development the underlying molecular mechanisms are currently unclear It has been
observed that the miR166-over producing men1 plants have an arrested meristem
development (Jung amp Park 2007) In addition the heterozygous men1+ and the
homozygous men1 plants show different expression patterns of WUS and CLV3 It
appears that miR166 regulates WUS indirectly and influences the expressional balance
between WUS and CLV3 through a negative feedback loop (Jung amp Park 2007 de
Alba et al 2013)
The phv phb athb15 triple mutant has an enlarged shoot apical meristems the
rev phb phv mutant does not have functional shoot apical meristems (Prigge et al
2005) This distinction may be explained by the fact that while miR166 targets
primarily PHB PHV and ATHB15 miR165 targets all the five HD-ZIP III genes (Zhou
et al 2007) Consistent with this transgenic plants overproducing miR166 are distinct
from those overproducing miR165 The miR165-overproducing transgenic plants lack
functional shoot apical meristem and a pin-like structure is formed at the apical region
of the mutant These phenotypes are quite similar to rev phb phv triple mutant (Zhou et
al 2007 Liu et al 2013) The phenotypic distinction may because by the difference
of the cleavage efficiencies of the target mRNAs In accordance with this notion a few
35S miR166a transgenic lines exhibit no shoot apical meristems The men1
homozygous plants also show arrested meristem development (Jung amp Park 2007)
Chapter 1 Introduction and Review of Literature
46
MiR164 cleaves the CUC1 and CUC2 mRNAs as well as the NAC1 mRNA
CUC1 and CUC2 determines the organ boundary in the meristem Phenotypes of
transgenic plants that overproduce miR164 are similar to those of the cuc1cuc2 double
mutant that exhibits embryonic developmental defects such as cup-shaped cotyledons
and fused cotyledons (Laufs et al 2004) When a miR164-resistant CUC2 is
expressed the boundary domain is enlarged CUC also plays a role in embryonic shoot
meristem formation by regulating the SHOOT MERISTEMLESS gene It was therefore
concluded that miR164-mediated cleavage of CUC1 and CUC2 mRNAs determines the
sizes of the boundary domains in the shoot apical meristem and regulates separation of
the organs by restricting cell proliferation (Laufs et al 2004 de Alba et al 2013)
11364 Leaf development
The role of miR319 has been extensively studied in leaf development A dominant
jaw-D mutant overproducing miR319 shows uneven curved leaves with enlarged size
and five TCP genes which are closely related to the CINCINNATA (CIN) gene in
Antirrhinum majus are down regulated in the mutant suggesting that miR319 mediated
regulation of the TCP transcription factor genes are important for the final step of
leaf shaping (Palatnik et al 2003 Naz et al 2013) In addition to leaf shape and size
leaf polarity is also specified by miRNA MiR166-mediated cleavage of the HD-ZIPIII
mRNAs is a well to establish leaf polarity This discovery originates from the gain-of-
function mutants such as phb-1d and phv-1d wherein point mutations in the
complementary sites to miRNA helps the mRNA to evade or resist from miRNA-
mediated cleavage (McConnell et al 2001 Emery et al 2003 Naz et al 2013)
Plant leaves have a dorso-ventral polarity consisting of the adaxial (upper
surface) and abaxial (lower surface) sides The adaxial side is closer to the shoot
apical meristem Adaxial identity is specified by functionally redundant class III HD-
ZIP family genes such as PHB PHV and REV Ectopic expression of each genes
result in adaxialized leaves Dominant phb-1d and phv-1d mutants exhibit
transformation of the abaxial to adaxial fate and have rod-shaped leaves In contrast
miR165166-overproducing plants show pin-like cotyledons radicalized leaves and
abaxialized leaves (Chitwood et al 2007 Pattanayak et al 2013)
Leaf asymmetry is determined by miR166-mediated restriction of the
expression domains of the HD-ZIPIII genes The HD-ZIPIII genes are expressed in the
Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
14
of C arabica including leaves floral buds and immature but not in mature beans
Three genes encoding 7-methylxanthine methyltransferase (theobromine synthase) have
been identified The number of amino acids in the putative polypeptides are 378 for
MXMT1 (427kDa) and 384 for MXMT2 and CTS2 (434kDa) They differ by insertion
or deletion of blocks of several residues in the C-terminal region Their catalytic
properties based on kinetic parameters such as Km values were different They are
expressed in young leaves floral buds and immature but not mature beans Three genes
were identified for 3 7-dimethylxanthine methyltransferase (caffeine synthase) DXMT
CCS1 and CtCS7 each encoding a 43 kDa polypeptide consisting of 384 amino acids
However their kinetic properties differ from each other such as DXMT and CCS1
showed Km values of 1200 and 157microM for theobromine respectively Expressions
profiles are also distinct DXMT being expressed exclusively in immature beans while
CCS1 expression is ubiquitous occurring in all tissues The presence of isoforms of
these enzymes with different properties suggests that caffeine is synthesized through
multiple pathways depending on availability and concentration of the substrate (Mizuno
et al 2001 Ogawa et al 2001 Mizuno et al 2003a Mizuno et al 2003b Uefuji et
al 2003) Genomic clones of theobromine synthase were obtained by PCR- RFLP
from C canephora (Satyanarayana et al 2005 Satyanarayana 2006) The genomic
clones are reported to have highly conserved exons and a variable third intron The
promoter for the theobromine synthase was cloned and characterized (Satyanarayana et
al 2005)
There is high degree of similarity in the coding region of coffee NMT genes
isolated so far though differences exist in the 3prime untranslated region (UTR) for these
genes The deduced amino acid sequence of these enzymes shows more than 85
homology between these genes Phylogenetic analysis indicates that they are more
closely related to C-methyltransferases including those for jasmonic acid salicylic acid
and benzoic acid than to other N-methyltransferases This suggests that coffee N-methyl
transferases belong to a distinct sub-group within plant methyltransferases Their
cellular localization determined by green fluorescent protein fusion method and β-
glucourodinase (GUS) showed that the all the three genes are localized in the cytoplasm
(Ogawa et al 2001 Vinod et al 2007) It was also shown that these were present
along with chlorogenic acid(Vinod et al 2007)
Chapter 1 Introduction and Review of Literature
15
Table 15 N-methyltransferases and their encoding genes involved in caffeine biosynthesis
Gene (Enzyme) Gene name Accession No Description Reference
Xanthosine methyltransferase
( 7-methylxanthosine synthase) (EC 211158)
CaXMT AB 048793 Immature fruit cDNA Screening (Uefuji et al 2003)
CmXRS1 AB 034699 Leaf cDNA library RACE (Mizuno et al 2003a)
CaMXMT1 AB 048794 Leaf cDNA library RACE (Ogawa et al 2001)
7-methylxanthine synthase transferase
(theobromine synthase) ( EC 211159)
CTS1 AB 034700 Immature fruit cDNA Screening (Uefuji et al 2003)
CaMXMT2 AB 984125 Leaf cDNA library Screening (Mizuno et al 2001)
CTS2 AB 054841 Leaf cDNA library Screening (Mizuno et al 2001)
37-methylxanthinemethyltransferase
(Caffeine synthase) (EC 211160)
CaDXMT1 AB 086414 Immature fruit cDNA Screening (Uefuji et al 2003)
CCS1 AB 0861414 Immature fruit cDNA Screening (Uefuji et al 2003)
CtCS7 AB 0864415 Immature fruit cDNA
NMT genomic clones
Theobromine synthase-1
CX10 Complete coding region PCR Satyanarayan 2006
CX8 Complete coding region PCR -do-
Theobromine synthase-1 PG-5 Promoter + coding region RAGE -do-
Theobromine synthase-1 PG-1 Promoter + coding region RAGE Satyanarayan 2006
PG-4 Promoter + coding region RAGE -do-
Theobromine synthase-1 NMT
(AY273814) Complete coding region PCR Kochko A et al 2010
Theobromine synthase-2 NMT
(AY362825) Complete coding region PCR Kochko A et al 2010
Chapter 1 Introduction and Review of Literature
16
19 Catabolism of caffeine
Caffeine is produced in young leaves and immature fruits and continues to accumulate
gradually during the maturation of these organs At the same time caffeine is slowly
degraded with the removal of the three methyl groups resulting in the formation of
xanthine Catabolism of caffeine was first reported in coffee leaves (Kalberer 1965)
Since then a number of tracer experiments using 14
C labelled purine alkaloids have
been reported (Suzuki amp Waller 1984 Ashihara et al 1996 Vitoria amp Mazzafera
1999 Mazzafera 2004) They demonstrated the major catabolic pathway is caffeine rarr
theophylline rarr 3-methylxanthine rarr xanthine Xanthine is further degraded by the
removal by the conventional purine catabolic pathway to CO2 and NH3 via uric acid
allatonin and allantoate (Figure 15) (Ashihara amp Crozier 1999 Stasolla et al 2003
Zrenner et al 2006) Caffeine catabolism usually begins with its conversion to
theophylline catalyzed by N7-demethylase The involvement of P450-dependent mono-
oxygenase activity for this reaction was suggested (Huber amp Baumann 1998
Mazzafera 2004) However the activity of this enzyme has not yet been determined in
cell free extracts [8-14
C] Theophylline degraded to CO2 at much faster rate than [8-14
C]
caffeine indicating that conversion of caffeine to theophylline is a major rate limiting
step of caffeine catabolism and a possible reason why caffeine accumulates in high
concentrations in tissues of C sinensis and C arabica It is widely believed that the
control of caffeine levels in plants is a function of the balance between rates of
synthesis and degradation and this balance seems to vary depending on the plant
species and the tissue developmental stage (Mazzafera 2004)
Chapter 1 Introduction and Review of Literature
17
Figure 15 Caffeine catabolic pathways Caffeine is mainly catabolised via theophylline and 3-
methylxanthine Xanthine is further degraded to CO2 and NH3 by the conventional
oxidative pathway Adapted from (Ashihara et al 2008)
Chapter 1 Introduction and Review of Literature
18
110 Genetic engineering of Coffee
Genetic transformation has potential applications in coffee agriculture for incorporating
genes which could provide desirable traits such as disease and insect resistance
drought and frost tolerance and herbicide resistance Transgenic technology can also be
used to increase nutritional value and improve cup quality produce varieties with
caffeine-free beans and for production of hybrid crops for molecular farming
Identification of target specific genes is one of the pre-requisites for developing
transgenic crops The availability of a large number of EST sequences in coffee and
initiation of coffee genome sequencing may speed up the gene discovery and accelerate
transgenic research efforts in coffee
1101 Insect Resistance
Production of insect resistant coffee plants is one of the major objectives of the
breeding programs The major pests attacking coffee include coffee berry borer (CBB
Hypothenemus hampei) white stem borer (WSB Xylotrechus quadripes) leaf miner
(Perileucoptera coffeela) and root nematodes (Meloidogyne spp and Pratylenchus
spp) The CBB is present in almost all the coffee growing countries and considered to
the most devastating pest in coffee To date there is no reported source of resistance to
CBB in the coffee gene pool Like CBB WSB is another serious pest in C arabica in
India and several other East Asian countries Both CBB and WSB belong to the order
Coleoptera For India controlling WSB is the biggest challenge and has therefore
become the highest research priority Robusta is generally resistant to WSB but the
interspecific Robusta Arabica hybrids are susceptible to WSB Although leaf miner is
not yet a serious pest in India and other East Asian countries it is an economically
important pest in East Africa and Brazil Effective chemical control of CBB and WSB
is difficult due to the nature of their life cycle inside the berry and stem respectively as
well as environmental concern regarding the use of pesticides Biological control
measures are adapted to combat these insect pests with varying degrees of success
Developing coffee plants resistant to these pests using genetic transformation
technology could be one of the alternative strategies to counter pest damage For insect
resistance several different classes of proteins from bacterial plant and animal sources
have been isolated and their insecticidal properties tested against many important pests
Amongst these proteins Bt toxins are most important and several transgenic crops
expressing Bt genes have been commercialized Coffee transgenic plants carrying a
Chapter 1 Introduction and Review of Literature
19
synthetic version of the cry1Ac gene have been produced (Leroy et al 2000 Mishra et
al 2002) Indeed this was the first report of an important agronomic trait being
introduced into a coffee plant The transgenic plants presented similar features in
growth and development compared to normal plants Transgenic plants highly resistant
to leaf miner under greenhouse conditions were tested under field conditions in French
Guyana for 4 years for pest resistance (Perthius et al 2005 Perthius et al 2006)
From 54 independent transformation events 70 of the events were resistant to leaf
miner Unfortunately the field trial was vandalized which led to the termination of the
experiment (Montagnon et al 2005)The effectiveness of Bt genes in controlling
coleopteran pests is well documented in corn and potato which indicates that Bt genes
might be effective against CBB The high toxicity of B thuringiensis sero var
israelensis against first in star larvae of CBB has been demonstrated (Mendez et al
2003) In another experiment an α-amylase inhibitor from Phaseolus vulgaris was
tested against CBB and found to have an inhibitory effect on its growth and
development (Grossi et al 2004)In addition to the CBB and WSB C arabica
varieties are also susceptible to endoparasitic root-knot nematode (Meloidogyne spp)
(Campos et al 1990 Mishra et al 2012)
So far 15 species have been reported to be parasites of coffee Controlling
nematodes is extremely difficult and currently seedling grafting with robusta rootstock
is followed as one of the control measure Sources of resistance specific to root-knot
nematodes have been identified in coffee trees (Bertrand et al 2001) and the Mex-1
gene conferring resistance to M exigua in C arabica is in the process of isolation (Noir
et al 2003)
1102 Tolerance to Abiotic Stress
In many coffee growing countries abiotic stress such as drought and frost are the major
climatic factors that limit coffee production Changes in climatic patterns due to global
climate change are considered increasingly important for coffee cultivation Drought
induces water stress in plants which affects vegetative growth and vigor and triggers
floral abnormalities and poor fruit set It also indirectly increases the incidence of pests
and diseases in the plants C arabica is generally more tolerant to water stress than C
canephora partly due to its extensive deep root system C racemosa is known to be a
good source of drought tolerance In India hybrids between C canephora and C
racemosa have been obtained and are currently under evaluation for drought tolerance
Chapter 1 Introduction and Review of Literature
20
In many coffee growing countries coffee is propagated in marginal areas where the
annual rainfall is below 1000thinspmm with prolonged dry spells of over 4-5 months In
those areas water shortage and unfavorable temperatures constitute major constraints
and the growth and productivity of C canephora are badly affected As with drought
periodic frost also affects coffee production in parts of Brazil The introduction of
drought and frost tolerant genes through genetic transformation would be of great
importance for alleviating these problems Research is now being carried out by several
groups to identify genes involved in biotic as well as abiotic stress The most promising
genetic engineering approach for drought tolerance includes the use of functional or
regulatory genes as well as the transfer of transcription factors In recent years plants
tolerant to high temperature and water stress have been the subject of intense research
(Chaves et al 2003 Chaves et al 2004 Coraggio et al 2004) For achieving drought
tolerance genes that have been targeted include those encoding enzymes involved in
detoxification or osmotic response metabolism enzymes active in signaling proteins
involved in the transport of metabolites and regulating the plant energy status (Chaves
et al 2003 Chaves et al 2004 Coraggio et al 2004) The dissection of molecular
mechanisms related to signal transduction and transcriptional regulation might help in
engineering drought tolerance in coffee
1103 Disease Resistance
Coffee leaf rust (CLR) caused by the fungus Hemileia vastatrix is the most important
disease in coffee with substantial loss to coffee production and productivity in all the
coffee growing countries In addition to leaf rust coffee berry disease (CBD) caused by
fungus Colletotrichum kahawae can be a devastating anthracnose leading to substantial
crop loss in Africa Several other fungal and bacterial diseases may also affect coffee
to a extent C arabica is more susceptible to many diseases than C canephora Though
most of the disease control measures rely upon chemical control they are more
expensive and labour intensive The long-term solution is the breeding of resistant
varieties which is the focus of many breeding programs However breeding for disease
resistant varieties is time consuming due to the perennial nature of coffee with its long
gestation period India has a long history of C arabica breeding especially for leaf rust
resistance and is the first country to demonstrate the existence of multiple races of leaf
rust Resistance to CLR is conditioned primarily by a number of major (SH) genes and
coffee genotypes are classified into different resistance groups based on their
Chapter 1 Introduction and Review of Literature
21
interaction with different rust races pathogen (van der Vossen 2001) Currently C
canephora provides the main source of resistance to pests and diseases including CLR
(H vastatrix) and CBD (C kahawae) and therefore used in breeding programs Other
diploid species like C liberica and C racemosa are being used as a source of resistance
to coffee leaf rust and coffee leaf miner respectively (Guerreiro et al 1999
Sreenivasan et al 1989) The development of coffee varieties resistant to major fungal
diseases such as CLR and CBD using transgenic technology will benefit the coffee
industry immensely During the last 15 years significant progress has been made in the
area of host-pathogen interactions (Staskawicz et al 1995 Feys et al 2000
Shivaprasad et al 2012) and many resistance genes involved in recognizing invading
pathogens have been identified and cloned (Takken et al 2000) A number of
signalling pathways induced because of pathogen infection have been dissected (Dong
et al 1998 Navarro et al 2006) Many antifungal compounds synthesized by plants to
combat fungal infection have been identified (Does et al 1998) Understanding the
specific induction of targeted pathways and identification of specific pathways
responsible for particular fungal resistance is important in order to employ this strategy
in transgenic technology The recent investigation of gene expression during coffee leaf
rust infection could provide an insight into the defence pathways operating in coffee
(Fernandez et al 2004 Nardi et al 2006) Efforts have been made to identify and
clone resistance genes from coffee for achieving durable resistance The genetic and
physical map of two resistance genes SH3gene conferring resistance to rust (Prakash et
al 2004) and the Ck1 gene conferring resistance to C kahawae CBD (Gichuru et al
2006) have been established These genes could be used for molecular marker assisted
breeding programs (Mishra et al 2012)
1104 Production of low-caffeine coffee
A widespread belief that the ingestion of caffeine can have adverse effects on
health resulted in the increased demand for decaffeinated coffee (Ashihara amp Crozier
2001) Several method were used to obtain decaffeinated plants by conventional
breeding techniques using low yielding varieties and mutants that accumulated
relatively low amounts of caffeine when compared to the commercially available ones
Low-caffeine and decaffeinated coffee represent around 10 of the coffee sales
around the world (Ribas et al 2006b) The industrial process for coffee decaffeination
can be expensive and affects the original flavour and aroma in coffee (David 2002)
Chapter 1 Introduction and Review of Literature
22
Transgenic coffee plants with suppressed caffeine synthesis using RNA interference
(RNAi) technology have been obtained (Ogita et al 2003 Ogita et al 2004) Specific
sequences in the 3prime untranslated region of the theobromine synthase gene (CaMXMT1)
were selected for construction of RNAi short and long fragments The caffeine and
theobromine content of the transgenic plants were reduced by ~ 70 when compared to
the untransformed plants In C canephora RNAi technology has also been employed
to silence the N-methyl transferase gene involved in caffeine biosynthesis (Vinod et al
2007) Promoter of an N-methyltransferase (NMT) gene involved in caffeine
biosynthesis was cloned (Satyanarayana et al 2005 Satyanarayana 2006) which will
be very useful for studying the regulation of caffeine biosynthesis
1105 Improvement in cup quality
Improvement in coffee cup quality requires elaborate knowledge of the chemical
constituents as well as the metabolic pathways involved in the elaboration of quality
The constituents of coffee beans include minerals proteins carbohydrates caffeine
chlorogenic acids (CGA) glycosides lipids and many volatile compounds that give
flavour to coffee by roasting Among these the role of three major constituents
sucrose CGA and trigonelline have been studied in coffee The sucrose content of
coffee bean is associated with coffee flavour the higher the sucrose content in green
beans the more intense will be the cup flavour (Clifford et al 1985 de Maria et al
1996) The sucrose content of C arabica (82-83) is higher than C canephora (33ndash
40) The sucrose amino acids ratio in green beans determines the profile of volatile
compounds Manipulating sucrose content in coffee bean is therefore important in
improving cup quality The sucrose synthase gene (CcSUS2) from C canephora has
been cloned and sequenced (Leroy et al 2005) This provides an opportunity to
manipulate the sucrose content in coffee Chlorogenic acids are products of
phenylpropanoid metabolism Chlorogenic acids are a group of hydroxycinnamoyl
quinic acids (HQA) formed by esterification between caffeic acids coumaric acids and
quinic acids (Clifford et al 1999) They are present in relatively large quantities in the
coffee bean and are the precursor of phenolic compounds in roasted beans C
canephora beans contain higher CGA (10) compared to C arabica beans (6-7)
CGA are known to have antioxidant properties as well as being associated with disease
resistance (Campa et al 2003) Genetic manipulations of genes involved in CGA
synthesis can serve either of these purpose by up- or down regulating the pathway
Chapter 1 Introduction and Review of Literature
23
Phenylalanine ammonia-lyase (PAL) catalyzes the first step of the phenylpropanoid
pathway leading to the synthesis of a wide range of chemical compounds including
flavonoids coumarins hydroxycinnamoyl esters and lignins (Hahlbrock amp Scheel
1989) Recently the full length cDNA and corresponding genomic sequences of PAL
from C canephora was isolated characterized and functionally validated (Mahesh et
al 2006 Parvatam et al 2006) This has opened up new possibilities for manipulating
the level of the PAL enzyme in coffee which in turn can be useful for improvement of
cup quality and manipulating antioxidant properties in coffee
1106 Fruit Ripening
Uniformity during fruit ripening is decisively related to cup quality in coffee and
consequently to the value of the product Fruits at the ideal ripening stage produce the
best organoleptic characteristics for coffee The presence of over ripened or green fruits
changes the acidity the bitterness and consequently the cup quality In order to
maximize uniform ripening of coffee fruits it is essential to control the action of genes
involved in the last step of maturation process Ethylene is known to trigger ripening
and increasing ethylene biosynthesis is associated with various stages of ripening
process (Protasio et al 2005) To control coffee fruit maturation two of the major
genes involved in ethylene biosynthesis namely ACC synthase and ACC oxidase
have been cloned (Protasio et al 2005 Neupane et al 1999) Introduction of the ACC
oxidase gene in antisense orientation have been achieved in both C arabica and C
canephora The effect of the transgene on ethylene production and fruit maturation has
yet to be reported The inhibition of genes downstream to the initial ethylene burst is
also an option to control coffee fruit maturation (Ribas et al 2006)
111 RNA interferenceRNAi
The regulation of gene expression at post transcriptional level has recently attracted
much attention because of the discovery of the phenomenon called RNA interference
(RNAi) RNAi based silencing techniques are more efficient than antisense mediated
gene silencing (Wesley et al 2001) The effect was first observed in plants wherein
silencing was observed when some copies of a transgene was in the forward orientation
with respect to the promoter and some in the reverse orientation This resulted in the
formation of double stranded RNA in the system The phenomenon in which
experimentally introduced double stranded RNA (dsRNA) leads to the loss of
expression of the corresponding cellular gene by sequence specific RNA degradation
Chapter 1 Introduction and Review of Literature
24
was termed as RNA interference or RNAi The protective effect of RNA silencing in
reducing virus infectivity supports the view that PTGS has evolved as a mechanism to
defend plants against virus infection and also to moderate the possible deleterious
genome-restructuring (insertional) activity of virus-like mobile genetic elements eg
retrotransposons A growing body of biochemical and genetic data has further
demonstrated that plants animals and yeasts share related mechanisms of specific
degradation of RNAs in which double-stranded forms of RNA are initiator molecules
(Brenstein et al 2001)
The key insight into the process of PGTS was provided by the experiments
conducted by Hamilton amp Baulcombe (1999) who identified the product of RNA
degradation as small RNA (siRNA) species of ~25nt of both sense and antisense
polarity SiRNA are formed and accumulate as double stranded RNA molecules of
defined chemical structures Both genetic and biochemical approaches were undertaken
to understand the basis of silencing Genetic screens were carried out in fungus
Neurospora carassa the alga Chalmydomonas reinhardtii and plant Arabidopsis
thaliana to search for detective mutants in PTGS
A combination of results obtained from several in vivo and in vitro experiments
have gelled into a two step mechanistic model for RNAiPTGS The first step referred
to as the RNAi initiating step involves binding of the RNA nucleases to a larges
dsRNA and its cleavage into discrete ~21-25nt RNA fragments This is ATP-
dependent reaction which involved RNase III - type endonucleases which make
staggered cuts in both strands of the dsRNA leaving 3prime overhang of 2 nucleotides with
5prime- phosphate 3prime-hydroxyl and no modification in the sugar phosphate backbone
(Hamilton amp Baulcombe 1999 Elbashir et al 2001) SiRNAs are the direct products
of dsRNA cleavage by the multi-domain RNase III enzyme DICER (Figure 16)
(Brenstein et al 2001 Karthikeyan et l 2013)
In the second step or commonly known as the effector step the double stranded
siRNAs produced in the first step bind to an RNAi specific protein complex to form a
RISC The complex undergoes activation in the presence of ATP so that the antisense
component of the unwound siRNA becomes exposed and allows the RISC to perform
the downstream RNAi reaction
Chapter 1 Introduction and Review of Literature
25
Several enzymes are involved in RNAi based on their role genes that encode for them
are classified into RNAi initiators RNAi effectors RNA dependent RNA polymerases
and silencing genes Two C elegans genes rdeI and rde4 (rde stands for lsquo RNAi
deficientrsquo) are believed to be involved in the initiation step of RNAi The C elegans
rde1 gene is a member of a large family of genes and is homologous to the Neurospora
qde2 (qde stands for ldquoquelling deficientrdquo) and Arabidopsis AGO1 (AGO stands for
agronaute AGO1 was previously identified to be involved in Arabidopsis
development) Although the functions of these genes are not clear a mammalian
member of RDE1 family has been identified as a translation initiation factor (Thakur
2005) Important genes for the effector step of PTGS in C elegans are rde2 and mut7
genes These genes were identified from heterozygous mutant worms that were unable
to transmit RNAi to their homozygous offsprings Worms with mutated rde2 or mut 7
genes show defective RNAi mut-7 gene encodes a protein with homology to the
nuclease domains of RNAase D and a protein implicated in Werner syndrome (a rapid
ageing disease in humans) (Grishok et al 2001 Kamath-Loeb et al 2012s)
Neurospora qde-1 Arabidopsis SDE-1SGS-2 and C elegans ego-1 appear to encode
RNA dependent RNA polymerase (RdRPs) It assumed that this is a proof that an RdRp
activity is required for RNAi Certainly the existence of an RdRp might explain the
remarkable efficiency of dsRNA induced silencing if it amplifies either the dsRNA
prior to cleavage or the siRNAs directly (Muers 2013) In C elegans ego-1 mutants
(ego stands for lsquoenhancer of glp-1) RNAi functions normally in somatic cells but is
defective in germline cells where ego-1 is primarily expressed In Arabidopsis SDE-
1SGS-2 mutants (SGS stands for suppressor of gene silencing) siRNA are produced
when dsRNA is introduced via an endogenously replicating RNA virus but not
introduced by a transgene It has been proposed that perhaps the viral RdRP is
substituting for the Arabidopsis enzyme in these mutants Random degradative PCR
model suggests that an RdRP uses the guide strand of an siRNA as a primer for the
target mRNA generating a dsRNA substrate for Dicer and thus more siRNAs
Several genes controlling RNA silencing in plants have been identified through
genetic screens of Arabidopsis mutants impaired in transgene induced RNA silencing
They encode a putative RNA-dependent RNA polymerase (SGS2SDE1) a coiled
protein (SGS3) a protein containing PAZ and Piwi domains (AGO1) and an RNA
helicase (SDE3) The putative SGS2SDE1 of Neurospora is related to QDE-1 and
Chapter 1 Introduction and Review of Literature
26
EGO-1 of C elegans whereas PAZPiwi protein AGO is related to QDE-2 of
Neurospora RDE-1of C elegans RNA helicase SDE3 is related to SMG-2 of C
elegans and Mut-6 of Chlamydomonas MUT-7 gene of C elegans encodes a protein
similar to Rnase D whereas the Drosophila DICER gene encode a protein similar to
RNase III An Arabidopsis ortholog of DICER gene has been identified called CAF
SIN1 SUS1(Thakur 2005 Poethig 2009)
Since the RNAi machinery is present constitutively within the eukaryotic cell it
is important to explore the metabolic advantages that are accorded by the RNAi related
proteins during the intrinsic normal growth of cells and development of organisms The
natural RNAi machinery not only keeps the mobile transposable elements from
disrupting the integrity of the genomes as suggested by the analysis of model organisms
like A thaliana C elegans D melanogaster (Tabara et al 1999 Wu-Scharf et al
2000 Aravin et al 2001 Hamilton et al 2002 Lisch 2009) but also participates in
development of organisms Genetic defects in Celegans RNAi genes ego1 and dicer
cause known specific developmental errors (Grishok et al 2000 Grishok et al 2001
Knight amp Bass 2001 Hall et al 2013) Similarly the Argonaute family of genes of A
thaliana (especially the ZWILLE proteins) is also responsible for plant architecture and
meristem development (Carmell et al 2002 Hamant and Pautot 2010) and the Dicer
homologue of A thaliana CAF1 is required for embryo development (Golden et al
2002 Nakano et al 2011) Thus genetic evidence illustrates the role of the RNAi
machinery as a controller of development related genes The mechanistic details of
these developmental processes are beginning to emerge
Chapter 1 Introduction and Review of Literature
27
Figure 16 Mechanism of gene silencing induced double stranded RNA The dicer enzyme to
produce siRNAs cleaves the RNA The siRNA generated bind to the nuclease complex
RISC The antisense component in the siRNA in the RISC guides the complex towards
the cognate mRNA resulting in endonucleolytic cleavage of the mRNA
Chapter 1 Introduction and Review of Literature
28
112 MicroRNA or MiRNA
In 1991 Ambros and co-workers first isolated a lin-4 mutant of Celegans which was
arrested at the first larval stage (Lee et al 1993) Later on the let-7 mutation was
isolated in the same system which was responsible for development through the fourth
larval stage Both lin-4 and let-7 encode short 22-nucleotide mature RNAs and were
called short temporal RNA because they control the temporal development program of
C elegans The mature lin-4 RNA defines (negatively regulates) the mRNA expression
of the lin-14 and lin-28 hetrochronic genes with the antisense mediated repression
mechanism of translation initiation and thus specifies the fate of cells during the first
three larval stages Recent studies have revealed that the short temporal RNAs are
actually members of a group of tiny RNAs (21to 28 nucleotides) called the micro-
RNAs (miRNA) (Bartel 2009) isolated members of which could easily run to a few
thousand which includes both those were identified computationally and by deep
sequencing Some of the components of the RNAi machinery have also been clearly
established as the effector proteins for the maturation of miRNAs
113 MicroRNAs in plants
1131 Discovery
MicroRNAs have been discovered by three basic approaches Direct cloning forward
genetic direct cloning and bioinformatic prediction followed by experimental
validation The most direct method of miRNA discovery is to isolate and clone small
RNAs from biological samples and several groups have used this approach to identify
plant miRNAs (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002 Xie et al 2003 Sunkar et al 2005 Williams et al 2013) The cloning methods
adapted were similar to those used to identify large numbers of animal miRNAs
(Lagos-Quintana et al 2001 Lau et al 2001 Aravin amp Tuschl 2005) which involve
isolating small RNAs followed by oligonucleotide adapter ligation reverse
transcription amplification and sequencing Some protocols incorporate methods to
enrich for Dicer cleavage products (ie molecules with 5prime-phosphates and 3prime-
hydroxyls) and to concatemerize the short cDNAs so that several may be identified in a
single sequencing read (Lau et al 2001 Llave et al 2002a Jagadeeswaran amp Sunkar
2013)The initial cloning experiments in Arabidopsis identified 19 miRNAs belonging
to 15 families (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002) although hundreds of other small RNAs such as siRNAs and RNA degradation
Chapter 1 Introduction and Review of Literature
29
fragments were also cloned through the direct cloning methods To select miRNAs
from the pool of small RNAs secondary structures of the RNA molecules were
examined in compliance with the acknowledged structural features of miRNAs
(Ambros et al 2003) The secondary structures were validated using several ad-hoc
software tools such as the Mfold or RNAfold programs (Reeder et al 2006) Finally
the existence of mature miRNAs was determined by RNA gel blot hybridization These
direct cloning methods were successful in isolating many miRNAs in Arabidopsis
(Reinhart et al 2002 Sunkar amp Zhu 2004) Rice (Sunkar et al 2005) Poplar (Lu et
al 2005) moss (Arazi et al 2005) and Tobacco (Billoud et al 2005)
Although miRNAs were first discovered through forward genetic screens in
worms (Lee et al 1993 Reinhart et al 2000)only few A thaliana miRNA gene
families have been discovered by this method (Chen 2008) in plants MiRNA
involvement in plant mutant phenotypes were not properly inferred till the cloning
experiments established that plant genomes contain numerous miRNAs (Park et al
2002 Reinhart et al 2002 Rhoades et al 2002) MiRNA loss-of-function allele has
been identified by forward genetic screens early extra petala1 is caused by a
transposon insertion ~160bp upstream of the predicted miR164c stem loop resulting in
flowers with extra petals (Baker et al 2005 Irish 2008) The fact that loss-of-function
miRNA mutants have been rarely discovered using forward genetics reflects small
target size for mutagenesis coupled with redundancy in nearly all evolutionarily
conserved plant miRNAs which are encoded by gene families (Table16) Family
members are likely to have overlapping functions buffering against loss at any single
miRNA locus Over expression screens can circumvent redundancy limitations At least
four plant miRNAs miR319 (also known as miR-JAW) miR172 (also known as EAT)
and miR166 were isolated in over expression screens for dominant mutants with
developmental abnormalities (Aukerman amp Sakai 2003 Palatnik et al 2003 Kim et
al 2005 Williams et al 2005b Schommer et al 2012) Mutations in miRNA target
sites which can prevent the entire family of miRNAs from repressing a target gene can
also circumvent redundant functions of miRNA family members
The dominant mutations in the HD-ZIP genes PHB PHV and REVOLUTA
(REV) in Arabidopsis and ROLLEDLEAF1 (RLD1) in Maize result in adaxialization of
leaves andor Vasculature (McConnell amp Barton 1998 McConnell et al 2001 Nelson
et al 2002 Zhong amp Ye 2004 Allen amp Millar 2012) and are all caused by mutations
Chapter 1 Introduction and Review of Literature
30
in miR166 complementary sites (Rhoades et al 2002 Emery et al 2003 Jones-
Rhoades amp Bartel 2004 Mallory et al 2004 Zhong amp Ye 2004)
In both plants and animals cloning was the initial means of large-scale
miRNA discovery (Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Reinhart et al 2002 Mendes et al 2012) However cloning is biased toward
RNAs that are expressed highly and broadly MiRNAs expressed at low levels or
only in specific cell types or in response to certain environmental stimuli were more
difficult to clone Sequence based biases in cloning procedures might also cause
certain miRNAs to be missed Because of these limitations bioinformatics approaches
to identify miRNAs have provided a useful complement to cloning
A straightforward use of bioinformatics has been carried out to find homologs
of known miRNAs both within the same genome and in the genomes of other species
(Pasquinelli et al 2000 Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Mendes et al 2009) A more difficult challenge is to identify miRNAs
unrelated to previously known miRNAs This was first accomplished for
vertebrate nematode and fly miRNAs using algorithms that search for conservation
of sequence and secondary structure (ie miRNA stem-loop precursors) between
species searching for patterns that are characteristic of miRNAs (Lai et al 2003 Lim
et al 2003 Lim et al 2005)
Most of the methods identified numerous miRNAs but are necessarily more
relaxed in terms of allowed structures Some approaches take advantage of the high
complementarity of plant miRNAs to target messages implementing the requirement
that the candidate has conserved complementarity to mRNAs (Jones-Rhoades amp Bartel
2004 Adai et al 2005) This additional filter has been useful for distinguishing
authentic plant miRNAs from false positives which later extended to mammalian
miRNA gene prediction (Xie et al 2005 Mendes et al 2012)
Another criteria used by most algorithms in the filters is evolutionary
conservation In plants mature miRNA sequences are conserved to a higher degree
than their precursor sequences For example Arabidopsis and rice diverged more than
130 million years ago (Friis et al 2004) but some miRNAs are highly conserved
in these plant species (Reinhart et al 2002) Furthermore various parameters for the
secondary structures such as free energy the number of paired residues within a
Chapter 1 Introduction and Review of Literature
31
miRNA or the number and size of bulges are also used to validate the predictions
(Jones-Rhoades amp Bartel 2004 Wang et al 2004 Adai et al 2005 Mendes et al
2012)
1132 Conserved microRNAs in Plants
Till date cloning genetics and bioinformatics screens have resulted in annotation of
approximately 184 potential Arabidopsis miRNAs (miRBase release 13) These
miRNAs seem to arise through gene duplication and subsequent diversification and
represent approximately 100 families (Maher et al 2006 Axtell 2013) Twenty one
families represented by 92 genes are clearly conserved in species beyond Arabidopsis
(Table 15) The presence of these miRNA families in other sequenced plant genomes
indicates that different plant species have an evolutionarily fluid set of miRNAs
(Willmann amp Poethig 2007) Recent studies on mosses one of the most ancient land
plants have shown that at least 14 miRNA families are conserved in eudicots and
mosses and therefore it appears that plant miRNAs share a common ancient origin
(Arazi et al 2005 Axtell amp Bartel 2005 Talmor-Neiman et al 2006 Zhang et al
2006 Axtell 2013) The number of members per family in a genome ranges from 1-32
with the exception of miR-430 which is represented by a cluster of ~80 loci in zebra
fish (Giraldez et al 2005)
In plants the number of members in each family correlates among examined
species certain families contain numerous members in all three species (eg miR156
miR166 miR169) whereas others consistently contain fewer genes (eg miR162
mir168 miR394) (Table 16) Although it is still unclear why certain miRNA are
encoded by more than one gene in plants (eg 12 genes encoding miR156) this
correlation might suggest functional significance of various miRNA family sizes Most
annotated plant miRNAs are conserved throughout flowering plants while a few
miRNAs are found only in a single sequenced genome and thus they could be
evolutionarily lsquoyoungrsquo miRNAs For example Arabidopsis has several miRNA
families that are not found in poplar or rice suggesting that miRNAs could be
generated continuously during evolution (Allen et al 2004a Rajagopalan et al
2006 Fahlgren et al 2007 Axtell 2012 de Alba et al 2013) Consistent with the
notion that non conserved miRNAs are of a more recent evolutionary origin these
miRNAs are mainly encoded by a single locus in the genome Within each family the
mature miRNA is always located in the same arm
Chapter 1 Introduction and Review of Literature
32
Table 16 MicroRNA families conserved in plants
miRNA
family
Arabidopsis Oryza Populus
miR156 12 12 11 miR159319 6 8 15 miR160 3 6 8 miR162 2 2 3 miR164 3 5 6 miR166 9 12 17 miR167 4 9 8 miR168 2 2 2 miR169 14 17 32 miR171 4 7 10 miR172 5 3 9 miR390 3 1 4 miR393 2 2 4 miR394 2 1 2 miR395 6 19 10 miR396 2 5 7 miR397 2 2 3 miR398 3 2 3 miR399 6 11 12 miR408 1 1 1 miR403 1 0 2 miR437 0 1 0 miR444 0 1 0 miR445 0 9 0 Total 92 127 169
All known miRNA families that are conserved between more than one plant species are listed
together with the number of genes identified in the sequenced genomes Rice miRNA families that have orthologs in maize but do not appear to have orthologs in the eudicots (Arabidopsis and Populus) are marked with an asterisk The following families contain miRNA genes annotated with
more than one number miR156 (miR156 and miR157) miR159319 (miR159 and miR319) miR166 (miR165 and miR166) miR171 (miR170 and miR171) and miR390 (miR390 and miR391)
of the stem loop (5prime- or 3prime) as it would be expected if the genes carry common ancestry
(Figure 17) The sequence of the mature miRNA and to some extent the segment on
the opposite arm of the hairpin to which it pairs is highly conserved between the
members of the same family (both within and between the species) while the sequence
secondary structure and length of the intervening ldquolooprdquo region can be highly divergent
within the same family
The patterns of paring and non pairing nucleotides are often conserved between
homologous miRNA stem loops from different species (Figure 17) The significance of
conserved mismatches is opined to guide DCL1 to cleave at the appropriate positions
along the stem loop
Chapter 1 Introduction and Review of Literature
33
Figure 17 Representative stem loop of miR164 Segments corresponding to the mature miRNAs
are shown in red (Adapted from Rajagopalan et al 2006)
Although many miRNAs are highly conserved in plants the degree of conservation in
most cases is quite low between plants and animals one exception is miR854 A recent
study has revealed that miR854 is conserved between Arabidopsis and animals and that
the Arabidopsis miR854 differs from the human miR854 only by a single nucleotide
(Arteaga-Vazquez et al 2006 Axtell 2012 de Alba et al 2013) The Arabidopsis
miR854 has multiple binding sites in the 3prime-untranslated region (UTR) of
OLIGOURIDYLATE-binding-PROTEIN mRNA 1bA (UBP1b) and represses the
translation of the UBP1b mRNA The animal miR854 is also complementary to a site in
the 3prime-UTR of the UBP1b homolog in animals However it is currently unclear how the
animal miR854 regulates its target mRNA Another distinction between plant and
animal miRNAs is the genomic organization of the miRNA genes While plant miRNA
genes are located predominantly in the intergenic regions animal miRNA genes tend to
localize in the intragenic regions both in the introns or exons of protein coding genes
Moreover miRNA genes are often clustered within a single precursor transcript
whereas individual plant miRNAs are derived from individual precursor RNAs (Bartel
Chapter 1 Introduction and Review of Literature
34
2004 Bonnet et al 2004 Kim 2005 Vaucheret et al 2006 Marco et al 2013)
1133 Non-conserved miRNA and challenges in annotation
Most miRNAs are conserved throughout flowering plants (Table 15) but many more
were found only in a single sequenced genome thus considered to be of a more recent
evolutionary origin The extended homology between few non conserved miRNAs
precursors and their target genes provides strong evidence that these relatively ldquoYoungrdquo
miRNAs arose from the duplication of the target gene segments (Allen et al 2004)
Several non-conserved miRNAs including miR161 miR163 miR173 miR447
miR475 and miR476 are known to direct cleavage of target transcripts (Allen et al
2004a Adai et al 2005 Sunkar et al 2005 de Alba et al 2013) It is difficult to
confidently predict targets for many because it is not possible to use complementary
site as a filter against false-positive target predictions It is also difficult to
confidently annotate non-conserved miRNAs as miRNA rather than siRNAs The
established minimal standard for miRNA annotation is a small RNA with detectable
expression and the potential to form a stem-loop when joined to flanking genomic
sequence (Kasschau et al 2003) In practice these requirements are too loose to
definitively categorize many small RNAs cloned from plants Many plant siRNAs are
detectable on blots (Vazquez et al 2004b) and hundreds of thousands of non-
miRNA genomic sequences can be predicted to fold into secondary structures that
resemble structures of plant miRNA precursors (Jones-Rhoades amp Bartel 2004)
Therefore without conservation of both sequence and secondary structure it is
difficult to be confident that a given cloned RNA originated from a stem-loop (ie is a
miRNA) rather than from a double-stranded RNA (ie is a siRNA) In fact many of
the thousands o f small RNAs cloned from Arabidopsis (Gustafson et al 2005) would
probably meet the literal requirements for annotation as miRNAs A few of these
sequences might be miRNAs but others that meet the literal criteria probably are not
so The challenges of annotating non conserved small RNAs were evidenced by
three related small silencing RNAs that were originally annotated as miRNAs
that turned out to be trans-acting siRNAs (tasiRNAs)(Kanno et al2013)
Although most miRNAs that are broadly conserved among flowering plants are
now being identified and experimentally validated (Table 16)(Jones-Rhoades amp
Bartel 2004) the challenges and ambiguities for classifying non-conserved miRNAs
prevent meaningful estimates of the total number of miRNA genes in Arabidopsis and
Chapter 1 Introduction and Review of Literature
35
other plant genomes It is possible that miRNAs might have escaped detection
because they are non-conserved and expressed in specific tissues or conditions and can
only be identified by using high- throughput sequencing
1134 MicroRNA biogenesis
11341 Transcription of microRNA precursor
Plant miRNAs are primarily found in the genomic regions that are not associated with
the protein coding genes (Reinhart et al 2002) and it appears that most if not all plant
miRNAs are produced from their own transcriptional units Plant miRNA genes are
occasionally clustered near each other in the genome suggesting transcription of
multiple miRNAs from a single primary transcript [eg the miR395 cluster (Grishok
et al 2001 Jones-Rhoades amp Bartel 2004b)] but this polycistronic arrangement of
miRNA genes appears far less frequently in plants than in animals (Bartel 2004)
Northern blot EST and mapping evidence indicate that plant miRNA primary
transcripts (also known as pri-miRNAs) as in animals are longer than needed to
encompass the miRNA stem-loops (Palatnik et al 2003 Jones-Rhoades amp Bartel
2004b) At least some of these pri-miRNA transcripts appear to be spliced
polyadenylated (Kurihara amp Watanabe 2004) and capped (Xie Z et al 2005) Two
rice miRNAs are contained within transcripts that contain exon junctions within the
presumptive stem-loop precursor implying that in these cases splicing is a prerequisite
for Dicer recognition (Sunkar et al 2005) Plant pri-miRNAs are over 1kb in length
and they are usually preceded by typical TATA box motifs They can also undergo
canonical splicing polyadenylation and capping All these observations indicates RNA
polymerase II is probably responsible for transcribing most plant miRNAs (Xie Z et
al 2005) as appears to be the case for many animal miRNAs Relatively little is
known about the regulation of miRNA transcription in plants but there is no reason
to suspect that this regulation would differ from that of protein-coding transcripts as
the bioinformatics analysis of the sequence upstream of the transcriptional start
sites of the miRNA genes showed the presence of putative binding motifs of
number of known transcription factors (Megraw et al 2006 Sethupathy 2013)
11342 MicroRNA processing and export
In plants maturation of miRNA is a step wise process A miRNA gene is transcribed to
pri-miRNA which is much longer than the pre-miRNA possessing the characteristic
stem-loop structure Like transcription of most protein-coding genes the miRNA
Chapter 1 Introduction and Review of Literature
36
transcription is processed by RNA polymerase II (Pol II) enzymes and the pri-
miRNAs are 5prime-capped and 3prime-polyadenylated (Lee et al 2004 Xie Z et al 2005)
Mature miRNAs are produced from pri-miRNAs through at least two sequential
processing steps by RNase III-type endonucleases In animals pri-miRNAs are first
processed at the bottom portion of the stem by Drosha a nuclear-localized RNase III
enzyme to liberate pre-miRNAs The pre-miRNAs are then exported to the cytoplasm
with the help of exportin-5The pre-miRNAs are further processed by Dicer another
RNaseIII enzyme to a duplex consisting of the miRNA and the antisense strands
(miRNA) Since plants do not have any Drosha homologs the two sequential
processing steps seem to be carried out by DCL1 a Dicer homolog in the nucleus
(Kurihara amp Watanabe 2004 de Alba et al 2013 Rogers amp Chen 2013)
The processing of pri-miRNAs to pre-miRNAs by DCL1 is assisted by two
other proteins HYPONASTY LEAVES1 (HYL1) and SERRATE (SE) HYL1 belongs to a
family of double-stranded RNA (dsRNA) binding protein and interacts with DCL1 in
vitro and in vivo (Lu amp Fedoroff 2000 Hiraguri et al 2005 de Alba et al 2013
Rogers amp Chen 2013) Loss-of-function mutations in the HYL1 gene result in
decreased accumulation of miRNAs and concomitant accumulation of pri-miRNAs
(Han et al 2004 Vazquez et al 2004a Kurihara et al 2006) SE a C2H2 zinc finger
protein interacts with DCL1 and HYL1 and plays a role in the processing of pri-
miRNAs to pre-miRNAs (Lobbes et al 2006 Yang L et al 2006) In addition the
nuclear cap-binding complex proteins CBP20 and CBP80 that are known as ABA
HYPER SENSITIVE 1(ABH1) have been shown to be required for proper miRNA
processing (Gregory et al 2008 Laubinger et al 2008) Recently it has been
demonstrated that DCL1 can release 21-nt RNAs from dsRNA as well as from
synthetic miR167 precursor RNAs in vitro However this cleavage is error prone and
inefficient in the absence of HYL1 and SE which accelerate the rate of DCL1-mediated
cleavage and promote accurate processing of miRNAs (Dong et al 2008 Rogers amp
Chen 2013)
11343 Methylation of the miRNAmiRNAduplex
After the miRNAmiRNAduplexes are released from the pre-miRNAs by the DCL1
activities the duplexes are methylated at the 2primeOH of the3prime-ends by HUA ENHANCER1
(HEN1) a small RNA methyltransferase (Yu et al 2005 Yang Z et al 2006 Rogers
amp Chen 2013 ) This step which is distinct from the RNase III-mediated processing
Chapter 1 Introduction and Review of Literature
37
step distinguishes the biogenesis of plant miRNAs from that of animal miRNAs
Mutations in the HEN1 gene were first isolated in a morphological screening for floral
development although the HEN1 mutants display pleiotropic phenotypes similar to the
developmental defects observed in the DCL1 mutants (Chen et al 2002 Park et al
2002 Rogers amp Chen 2013)
Figure 18 Biogenesis of microRNA
A Biogenesis of plant MicroRNA B Biogenesis of metazoananimal MicroRNA
The miRNA (red) is incorporated into RISC and the miRNA(blue) in degraded
This enzyme has two dsRNA-binding domains and nuclear localization signal It
is also conserved in fungi and is required for the processing of siRNAs as well as of
miRNAs (Park et al 2002 Boutet et al 2003 Xie et al 2003) Although the HEN1
protein is localized to the nucleus its methylation activity in the cytoplasm cannot be
ignored (Xie et al 2004 Rogers amp Chen 2013)
Chapter 1 Introduction and Review of Literature
38
The methylation of the miRNAmiRNA duplex is required to protect miRNAs
against the 3prime-end uridylation activity and subsequent degradation (Li et al 2005) In
the hen1 mutants accumulation of miRNAs is reduced to a lower level Also the
miRNAs appear to be heterogeneous in their sizes such that a ladder of bands rather
than a single species is observed for most miRNAs in the mutants (Li et al 2005)
MiRNA cloning and sequencing analyses revealed the presence of a number of U
residues at the 3prime-end of miRNAs in the hen1 mutants Moreover these nucleotides do
not correspond to the miRNAs in the pre-miRNAs indicating that these nucleotides are
added after the processing of the miRNAs from pre-miRNAs (Li et al 2005)
Uridylation of RNAs has also been proven to be correlated with RNA decay (Shen amp
Goodman 2004 Mullen amp Marzluff 2008) suggesting that uridylation attracts an
exonuclease which degrades miRNAs in plants
11344 MiRNA incorporation in to the RISC and its nuclear export
The methylated miRNAmiRNA duplexes undergo RISC assembly In this process
the miRNA of the duplexes is selectively incorporated into the RISC and the miRNA
appears to be removed and subsequently degraded (Hammond et al 2000 Hutvagner
amp Zamore 2002 Schwarz et al 2003 de Alba et al 2013 Rogers amp Chen 2013)
The ARGONAUTE 1(AGO1) proteins are core components of the RISC complex
They contain two structural domains the N-terminal PAZ domain that binds to the 3prime-
end of single-stranded RNAs and the C-terminal piwi domains that has a structure
similar to that of RNase H (Cerutti et al 2000 Parker et al 2004) Several AGO
proteins possess endonucleolytic activities within the PIWI domain and cleave target
mRNAs in the middle of the site complementary to miRNA (Liu et al 2004 Meister et
al 2004 Miyoshi et al 2005) Mutations in the AGO1 gene cause miRNA target
genes to be ectopically expressed and the levels of some miRNAs are reduced in the
mutants (Kidner amp Martienssen 2004 Vaucheret et al 2004 Kidner amp Martienssen
2005 Ronemus et al 2006) Moreover the phenotype of the AGO1 hypomorphic
alleles display defects in lateral organ polarity and leaf and flower morphology as
observed in the dcl1 mutants and null AGO1 alleles are embryonically lethal
supporting the close relationship between the AGO proteins and miRNA processing
(Kidner amp Martienssen 2004 Vaucheret et al 2004 de Alba et al 2013 Rogers amp
Chen 2013)
The production of miRNA miRNA duplexes occur in the nucleus while
Chapter 1 Introduction and Review of Literature
39
miRNA-RISC complexes are present in the cytoplasm where target mRNAs are
located This discordance of functional sites requires an export step during miRNA
biogenesis HASTY the Arabidopsis homolog of exportin-5 is thought to be involved
in miRNA nuclear export (Bollman et al 2003 Park et al 2005)The hasty mutant
exhibits pleiotropic defects in plant development and reduced accumulation of most
miRNAs as in the DCL1 or AGO1 mutants Howeverit is unclear whether the HASTY
proteins export the miRNA miRNA duplexes or the miRNA-RISC complexes in
plants
11345 Feedback regulation of miRNA biogenesis
MiRNA biogenesis is under feedback regulation such that the DCL1 and AGO1 genes
are themselves regulated by miRNAs DCL1 is itself a target of miR162 which leads to
the cleavage of the DCL1 mRNA (Xie et al 2003 de Alba et al 2013) Consistent
with this the levels of DCL1 mRNA are elevated in the mutants of miRNA processing
genes in which the miR162 abundance is reduced In addition there is a predicted
hairpin structure of miR838 in the 14th
intron of the DCL1 primary transcript
(Rajagopalan et al 2006) This prediction implies that DCL1-mediated processing
on this site can result in the cleavage of the DCL1 primary transcript into two truncated
fragments the N-terminal capped fragment and the C-terminal polyadenylated
fragment If this is the case it is possible that miRNA biogenesis competes with the
splicing event of the DCL1 pre-mRNA
The AGO1 is also regulated by miRNA There is a complementary site for
miR168 binding in the AGO1 mRNA and the transcript level of the AGO1 mRNA is
reduced through an mRNA cleavage mechanism (Vaucheret et al 2006 de Alba et al
2013 Rogers amp Chen 2013) indicating that the AGO1 mRNA levels are tightly
controlled by the levels of the AGO1 protein
1135 Mechanism of miRNA action
11351 MiRNA-directed mRNA cleavage
By forming the miRNA-RISC assembly miRNAs can direct the RISC to regulate gene
expression by two post transcriptional mechanisms mRNA cleavage or translational
repression (Bartel 2004 Jones-Rhoades et al 2006 Dalmay 2013) The degree
of miRNA-mRNA complementarity plays an important role for the
determination of the mechanism to be used A general rule is that while perfect or
Chapter 1 Introduction and Review of Literature
40
near-perfect complementarity induces mRNA cleavage central mismatches trigger
translational repression (Carrington amp Ambros 2003 Jones-Rhoades amp Bartel 2004)
Unlike most animal miRNAs plant miRNAs have near-perfect or perfect
complementarity to their targets and mRNA cleavage is assumed to be a predominant
mechanism by which miRNAs regulate gene expression in plants
In RNA cleavage mechanism miRNAs guide the AGO component of RISC to
cleave a single phosphodiester bond of the target mRNA within the miRNA-binding
site (Llave et al 2002b Tang et al 2003) The truncated fragments are then released
and degraded freeing the RISC to recognize and cleave another target mRNA This has
been validated by the detection of 3prime cleavage products that have 5prime ends that start at the
middle of the complementary regions The mRNA cleavage products are then further
degraded by other mechanisms The 3prime cleavage products of some miRNA targets are
degraded by the 5prime-3prime exonuclease XRN4 an Arabidopsis homolog of the major Yeast
mRNA degrading exonuclease Xrn1p It has been observed that the 3prime cleavage
products of some target mRNAs were accumulated in the xrn4 mutants (Souret et al
2004 Dalmay 2013) However the 3prime cleavage products of other miRNA targets do
not accumulate in the xrn4 mutants indicating that there are also XRN4 independent
mechanisms The 5prime cleavage products are typically untraceable by RNA gel blot
hybridization but can be detected by sensitive PCR based methods It has been found
that the 5prime cleavage products tend to obtain an oligo U tail at the 3prime ends This 3prime U tail
is correlated with shortening of the 5prime cleavage products from the 5prime end leading to the
conclusion that the 3prime uridylation causes 5 prime- 3 prime exonucleolytic decay of the 5prime cleavage
products (Shen amp Goodman 2004)
DCL1 HYL1 and SE interact with each other and are co-localized in the
nuclear bodies termed Dicing bodies or D-bodies in which some pri-miRNA shave
been found suggesting that D-bodies serve as a center for active miRNA processing
(Fang amp Spector 2007 Fujioka et al 2007 Song et al 2007) The miRNA
processing site appears to be distinct from the Cajal bodies that have previously been
found as a siRNA processing site (Pontes amp Pikaard 2008) The D-bodies include
SmD3 and SmB proteins that are found in the Cajal bodies However At collin an
additional marker for the Cajal bodies is not found in the D-bodies Moreover the D-
bodies are present in an At collin mutant lacking the Cajal bodies The pri-miRNAs of
miR171A and miR159A genes have been found to be co-localized with HYL1 and
Chapter 1 Introduction and Review of Literature
41
DCL1 in the D- bodies that are distinct from the Cajal bodies (Song et al 2007)
11352 MiRNA-directed translational repression
Translation repression is another mechanism exerted by plant miRNAs The regulatory
mechanism of miR172 which regulates flowering time and floral organ identity is
consistent with translation repression MiR172 over production does not affect the
transcript levels of APETALA2 (AP2) and AP2- like target mRNAs Instead the AP2
protein level is reduced in the miR172 over expressing mutant (Aukerman amp Sakai
2003 Chen 2004 Dalmay 2013) It was later shown that miR172 can also direct the
cleavage of the target mRNAs although the steady-state mRNA level is unchanged due
to feedback regulation (Schwab et al 2005 Jung et al 2007) It is clear that miR172
regulates its target genes primarily by translational repression rather than mRNA
cleavage Recently it has been reported that miR156157 and miR854 also regulate
their target genes through translational repression (Arteaga-Vazquez et al 2006
Gandikota et al 2007 Dalmay 2013) It was once thought that translational repression
is an exception to the rule However experimental evidence suggest a widespread
coexistence of translational repression and mRNA cleavage (Brodersen et al 2008)
1136 Regulatory roles of miRNAs in plant development
The miRNA target prediction has been a difficult task in order to obtain regulatory
targets without bringing in too many false predictions Plant growth and developmental
processes regulated by miRNAs are quite diverse and enormous Furthermore plant
miRNAs work cooperatively or antagonistically to establish a balanced regulation This
notion is well consistent with the findings that mutations in the regulators of miRNA
biogenesis transport and processing such as AGO1 DCL1 HYL1 HEN1 ABH1
and HASTY cause pleiotropic phenotypes (Mallory amp Vaucheret 2006 de Alba et al
2013) To date the roles of miRNAs have been mostly deduced from obtaining miRNA
over expressing transgenic plants or gain-of-function mutants in which miRNA-
resistant target genes are ectopically expressed
11361 Phase transition
Plants pass through a series of developmental phase transitions during their life span
beginning with seed germination continuing through vegetative phase change
reproductive phase change flowering initiation and finally seed production for the next
generation Because of their importance in understanding plant developmental
Chapter 1 Introduction and Review of Literature
42
processes genes regulating developmental transitions have been extensively studied
and underlying molecular mechanisms have been explored in various plant species
Majority of researches were mainly focused on vegetative phase change and
reproductive transition in Arabidopsis and Maize (Poethig 2003 Baurle amp Dean
2006) Particularly the mutations of the genes encoding various regulators of miRNA
biogenesis and transport such as HST SE and AGO exhibit altered juvenile to adult
vegetative phase change and vegetative to reproductive change (Hunter et al 2003
Peragine et al 2004 Vazquez et al 2004a Jones-Rhoades et al 2006 de Alba et al
2013)
Over-expression of the miR156a gene causes late flowering and delays
vegetative phase change (Wu amp Poethig 2006 Gandikota et al 2007 Wang et al
2008) It has been shown that miR156 negatively regulates a group of genes encoding
SQUAMOSA PROMOTER-BINDING PROTEIN-LIKE (Lewis et al 2007 de Alba et
al 2013 Rogers amp Chen 2013) transcription factors such as SPL3 SPL4 and SPL5
that promote flowering initiation In contrast transgenic plants over expressing
miR156-resistant SPL345 genes are early flowering Interestingly the endogenous
level of miR156 is temporally regulated In the early juvenile vegetative phase the
level of miR156 is very high but decreases rapidly before the onset of the adult juvenile
phase (Wu amp Poethig 2006)
The Arabidopsis early activation tagged (eat-D) mutant exhibits early flowering
with disrupted floral structure The mutant phenotypes are caused by over expression of
the miR172a-2gene (Aukerman amp Sakai 2003) MiR172 regulates a small group of
genes encoding APETALLA2 (AP2)-like transcription factors such as TARGET OF
EAT1 (TOE1) TOE2 SCHLAFMUTZE (SMZ) and SCHNARCHZAPFEN(SNZ)
TOE1 TOE2 SMZ and SNZ have been shown to participate in their productive phase
change by repressing a floral integrator FLOWERING LOCUS T (FT)(Jung et al
2007) Consistent with this toe1-2D 35STOE2 35SSMZ and 35SSNZ plants
exhibit late flowering (Jung et al 2007) MiR172 is also regulated temporally and
highly expressed in the adult vegetative phase both in Arabidopsis and maize (Chuck et
al 2007) Because the temporal expression patterns of miR156 and miR172 are
complementary to each other it has been suggested that the two miRNAs interact with
each other in regulating phase change (Chuck et al 2009 de Alba et al 2013) In
support of this view it has been shown that miR172 is down-regulated in the
Chapter 1 Introduction and Review of Literature
43
Corngrass1(Cg1) mutant in which miR156 is overproduced (Chuck et al 2007 de
Alba et al 2013) However there is no direct evidence for the direct linkage between
miR156 and miR172 It is also envisioned that miR156 and miR172 would not be
directly related For example miR156 regulates plastochron index that is the inverse of
leaf initiation rate and thus frequently used to analyze temporal patterns of plant
development In contrast miR172 does not affect plastochron (Wang et al 2008) The
down-regulation of miR172 in the maize miR156-over producing mutants would be
due to the prolonged juvenile vegetative phase in the mutants
Another miRNA that regulates flowering initiation is miR159 It regulates
MYB33 and MYB65 which are closely related to the barley GAMY B transcription
factor that responds to gibberellic acid (GA) The level of miR159 is elevated in GA-
treated seedlings but reduced in the GA biosynthetic ga1 mutant Transgenic plants
over producing miR159 exhibit delayed floral induction under short days (Achard et
al 2004) It is been suggested that MYB33 mediates GA signal to regulate LEAFY
(LFY) These observations indicate that miR159 plays an important role in the GA
signaling pathways that regulate proper floral induction under short days (Achard et al
2004)
11362 Floral development
Arabidopsis flowers consist of four distinct organs They arise in a characteristic
pattern within concentric rings called whorls from outside to inside four sepals four
petals six stamens and the central pistil consisting of two carpels Identities of the
floral organs are determined by three classes of regulatory genes classA classB and
class C genes (Krizek amp Fletcher 2005) The A and C class genes act antagonistically
to restrict their expression domains (Bowman et al 1991)
It is notable that miR172 also regulates floral architecture in addition to its role
in the timing of flowering initiation MiR172 inhibits the translation of the AP2 mRNA
(Chen 2004) Transgenic plants overproducing miR172 not only exhibit early
flowering but also show abnormal floral development which is quite similar to those
of the class A gene mutants such as the ap2 mutants (Aukerman amp Sakai 2003 de
Alba et al 2013) When the activity of the class A genes is inactivated that of the class
C genes such as AGAMOUS (AG) is elevated As a result the miR172- overproducing
transgenic plants exhibit no petals along with homeotic transformation of the sepals to
Chapter 1 Introduction and Review of Literature
44
the carpels (Aukerman amp Sakai 2003 Chen 2004)
Interestingly miR166 also regulates floral architecture The role of miR165166
in the regulation of meristem activity is closely linked to floral meristem formation and
organization (Zhao et al 2007 Miyashima et al 2013) Floral structure is severely
disrupted in the miR165166-overproducing mutants such as men1 and jba-1D in
which miR166 is overproduced (Williams et al 2005b Jung amp Park 2007) The men1
and jba-1D mutants have smaller gynoecium and reduced carpel number In an extreme
case the jba-1D homozygous plants lack visible carpels (Williams et al 2005b)
Similar phenotypes have been observed in the miR165 overproducers (Zhao et al
2007) It has been suggested that the phenotypes are possibly caused by altered AG
expression in the floral meristem (Jung amp Park 2007 Turchi et al 2013)
Other miRNAs also affect floral development Overproduction of miRNAs
involved in auxin signaling also affects floral architecture Transgenic plants
overproducing miR167 have defects in the formation of ovule and anther with reduced
fertility (Ru et al 2006) It has been shown that miR167 targets ARF6 and ARF8 that
play a role in gynoecium and stamen development and in regulating fertility (Ru et al
2006 Wu et al 2006) These observations suggest that auxin signals are also closely
related to floral organogenesis In addition transgenic plants over-producing miR164
and the cuc1cuc2 double mutants show fused sepals and reduced petals (Laufs et al
2004 Turchi et al 2013) suggesting that miR164 is also related to floral meristem
activity and boundary specification of the meristematic region
11363 Shoot apical meristem development
Shoot apical meristem comprises self-maintaining totipotent cells The shoot apical
meristem activity affects diverse aspects of plant developmental processes including
leaf development vascular development and lateral organ polarity (Bowman et al
2002) Because miR165166 plays a primary role in meristem formation
overproduction of miR165166 and multiple knockout mutants of its target genes
exhibit pleiotropic pheonotypes (McConnell et al 2001 Emery et al 2003 Kim et al
2005 Williams et al 2005b)
The targets of miR165166 include genes encoding the homeo domain-leucine
zipper (HD-ZIP) class III transcription factors such as PHABULOSA(PHB)
PHAVOLUTA (PHV) REVOLUTA(REV) ATHB15CORONA (CNA) and ATHB8
Chapter 1 Introduction and Review of Literature
45
PHB PHV and ATHB15 have overlapping functions in controlling meristem
development (Turchi et al 2013) Indeed the phb phv athb15 triple mutants have an
enlarged shoot apical meristem Consistent with this the miR166- overproducing men1
and jba-1D mutants exhibit enlarged shoot apical meristems (Kim et al 2005
Williams et al 2005b Turchi et al 2013) In the mutants the shoot apical meristems
are multiplied and large
The development of shoot apical meristems is also regulated by the WUSCHEL
(WUS)ndashCLAVATA (CLV) signaling pathway WUS positively regulates cell
proliferation In contrast CLV3 which is expressed in stem cells limits the WUS
expression and thus inhibits cell proliferation in the SAM (Sharma et al 2003)
Consistent with this notion WUS is expressed to a higher level in jba-1D
explaining the ectopic enlargement of the SAM in the mutant (Williams et al 2005)
While WUS is closely related to miR165166 in the shoot apical meristem
development the underlying molecular mechanisms are currently unclear It has been
observed that the miR166-over producing men1 plants have an arrested meristem
development (Jung amp Park 2007) In addition the heterozygous men1+ and the
homozygous men1 plants show different expression patterns of WUS and CLV3 It
appears that miR166 regulates WUS indirectly and influences the expressional balance
between WUS and CLV3 through a negative feedback loop (Jung amp Park 2007 de
Alba et al 2013)
The phv phb athb15 triple mutant has an enlarged shoot apical meristems the
rev phb phv mutant does not have functional shoot apical meristems (Prigge et al
2005) This distinction may be explained by the fact that while miR166 targets
primarily PHB PHV and ATHB15 miR165 targets all the five HD-ZIP III genes (Zhou
et al 2007) Consistent with this transgenic plants overproducing miR166 are distinct
from those overproducing miR165 The miR165-overproducing transgenic plants lack
functional shoot apical meristem and a pin-like structure is formed at the apical region
of the mutant These phenotypes are quite similar to rev phb phv triple mutant (Zhou et
al 2007 Liu et al 2013) The phenotypic distinction may because by the difference
of the cleavage efficiencies of the target mRNAs In accordance with this notion a few
35S miR166a transgenic lines exhibit no shoot apical meristems The men1
homozygous plants also show arrested meristem development (Jung amp Park 2007)
Chapter 1 Introduction and Review of Literature
46
MiR164 cleaves the CUC1 and CUC2 mRNAs as well as the NAC1 mRNA
CUC1 and CUC2 determines the organ boundary in the meristem Phenotypes of
transgenic plants that overproduce miR164 are similar to those of the cuc1cuc2 double
mutant that exhibits embryonic developmental defects such as cup-shaped cotyledons
and fused cotyledons (Laufs et al 2004) When a miR164-resistant CUC2 is
expressed the boundary domain is enlarged CUC also plays a role in embryonic shoot
meristem formation by regulating the SHOOT MERISTEMLESS gene It was therefore
concluded that miR164-mediated cleavage of CUC1 and CUC2 mRNAs determines the
sizes of the boundary domains in the shoot apical meristem and regulates separation of
the organs by restricting cell proliferation (Laufs et al 2004 de Alba et al 2013)
11364 Leaf development
The role of miR319 has been extensively studied in leaf development A dominant
jaw-D mutant overproducing miR319 shows uneven curved leaves with enlarged size
and five TCP genes which are closely related to the CINCINNATA (CIN) gene in
Antirrhinum majus are down regulated in the mutant suggesting that miR319 mediated
regulation of the TCP transcription factor genes are important for the final step of
leaf shaping (Palatnik et al 2003 Naz et al 2013) In addition to leaf shape and size
leaf polarity is also specified by miRNA MiR166-mediated cleavage of the HD-ZIPIII
mRNAs is a well to establish leaf polarity This discovery originates from the gain-of-
function mutants such as phb-1d and phv-1d wherein point mutations in the
complementary sites to miRNA helps the mRNA to evade or resist from miRNA-
mediated cleavage (McConnell et al 2001 Emery et al 2003 Naz et al 2013)
Plant leaves have a dorso-ventral polarity consisting of the adaxial (upper
surface) and abaxial (lower surface) sides The adaxial side is closer to the shoot
apical meristem Adaxial identity is specified by functionally redundant class III HD-
ZIP family genes such as PHB PHV and REV Ectopic expression of each genes
result in adaxialized leaves Dominant phb-1d and phv-1d mutants exhibit
transformation of the abaxial to adaxial fate and have rod-shaped leaves In contrast
miR165166-overproducing plants show pin-like cotyledons radicalized leaves and
abaxialized leaves (Chitwood et al 2007 Pattanayak et al 2013)
Leaf asymmetry is determined by miR166-mediated restriction of the
expression domains of the HD-ZIPIII genes The HD-ZIPIII genes are expressed in the
Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora
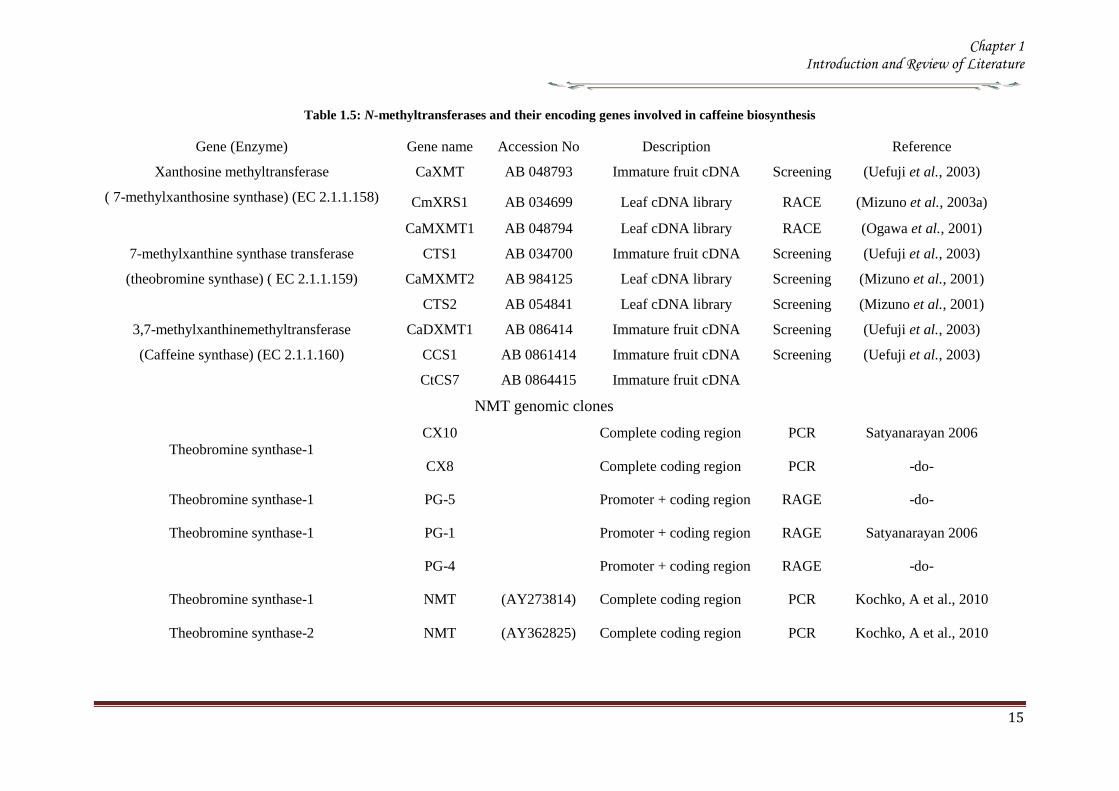
Chapter 1 Introduction and Review of Literature
15
Table 15 N-methyltransferases and their encoding genes involved in caffeine biosynthesis
Gene (Enzyme) Gene name Accession No Description Reference
Xanthosine methyltransferase
( 7-methylxanthosine synthase) (EC 211158)
CaXMT AB 048793 Immature fruit cDNA Screening (Uefuji et al 2003)
CmXRS1 AB 034699 Leaf cDNA library RACE (Mizuno et al 2003a)
CaMXMT1 AB 048794 Leaf cDNA library RACE (Ogawa et al 2001)
7-methylxanthine synthase transferase
(theobromine synthase) ( EC 211159)
CTS1 AB 034700 Immature fruit cDNA Screening (Uefuji et al 2003)
CaMXMT2 AB 984125 Leaf cDNA library Screening (Mizuno et al 2001)
CTS2 AB 054841 Leaf cDNA library Screening (Mizuno et al 2001)
37-methylxanthinemethyltransferase
(Caffeine synthase) (EC 211160)
CaDXMT1 AB 086414 Immature fruit cDNA Screening (Uefuji et al 2003)
CCS1 AB 0861414 Immature fruit cDNA Screening (Uefuji et al 2003)
CtCS7 AB 0864415 Immature fruit cDNA
NMT genomic clones
Theobromine synthase-1
CX10 Complete coding region PCR Satyanarayan 2006
CX8 Complete coding region PCR -do-
Theobromine synthase-1 PG-5 Promoter + coding region RAGE -do-
Theobromine synthase-1 PG-1 Promoter + coding region RAGE Satyanarayan 2006
PG-4 Promoter + coding region RAGE -do-
Theobromine synthase-1 NMT
(AY273814) Complete coding region PCR Kochko A et al 2010
Theobromine synthase-2 NMT
(AY362825) Complete coding region PCR Kochko A et al 2010
Chapter 1 Introduction and Review of Literature
16
19 Catabolism of caffeine
Caffeine is produced in young leaves and immature fruits and continues to accumulate
gradually during the maturation of these organs At the same time caffeine is slowly
degraded with the removal of the three methyl groups resulting in the formation of
xanthine Catabolism of caffeine was first reported in coffee leaves (Kalberer 1965)
Since then a number of tracer experiments using 14
C labelled purine alkaloids have
been reported (Suzuki amp Waller 1984 Ashihara et al 1996 Vitoria amp Mazzafera
1999 Mazzafera 2004) They demonstrated the major catabolic pathway is caffeine rarr
theophylline rarr 3-methylxanthine rarr xanthine Xanthine is further degraded by the
removal by the conventional purine catabolic pathway to CO2 and NH3 via uric acid
allatonin and allantoate (Figure 15) (Ashihara amp Crozier 1999 Stasolla et al 2003
Zrenner et al 2006) Caffeine catabolism usually begins with its conversion to
theophylline catalyzed by N7-demethylase The involvement of P450-dependent mono-
oxygenase activity for this reaction was suggested (Huber amp Baumann 1998
Mazzafera 2004) However the activity of this enzyme has not yet been determined in
cell free extracts [8-14
C] Theophylline degraded to CO2 at much faster rate than [8-14
C]
caffeine indicating that conversion of caffeine to theophylline is a major rate limiting
step of caffeine catabolism and a possible reason why caffeine accumulates in high
concentrations in tissues of C sinensis and C arabica It is widely believed that the
control of caffeine levels in plants is a function of the balance between rates of
synthesis and degradation and this balance seems to vary depending on the plant
species and the tissue developmental stage (Mazzafera 2004)
Chapter 1 Introduction and Review of Literature
17
Figure 15 Caffeine catabolic pathways Caffeine is mainly catabolised via theophylline and 3-
methylxanthine Xanthine is further degraded to CO2 and NH3 by the conventional
oxidative pathway Adapted from (Ashihara et al 2008)
Chapter 1 Introduction and Review of Literature
18
110 Genetic engineering of Coffee
Genetic transformation has potential applications in coffee agriculture for incorporating
genes which could provide desirable traits such as disease and insect resistance
drought and frost tolerance and herbicide resistance Transgenic technology can also be
used to increase nutritional value and improve cup quality produce varieties with
caffeine-free beans and for production of hybrid crops for molecular farming
Identification of target specific genes is one of the pre-requisites for developing
transgenic crops The availability of a large number of EST sequences in coffee and
initiation of coffee genome sequencing may speed up the gene discovery and accelerate
transgenic research efforts in coffee
1101 Insect Resistance
Production of insect resistant coffee plants is one of the major objectives of the
breeding programs The major pests attacking coffee include coffee berry borer (CBB
Hypothenemus hampei) white stem borer (WSB Xylotrechus quadripes) leaf miner
(Perileucoptera coffeela) and root nematodes (Meloidogyne spp and Pratylenchus
spp) The CBB is present in almost all the coffee growing countries and considered to
the most devastating pest in coffee To date there is no reported source of resistance to
CBB in the coffee gene pool Like CBB WSB is another serious pest in C arabica in
India and several other East Asian countries Both CBB and WSB belong to the order
Coleoptera For India controlling WSB is the biggest challenge and has therefore
become the highest research priority Robusta is generally resistant to WSB but the
interspecific Robusta Arabica hybrids are susceptible to WSB Although leaf miner is
not yet a serious pest in India and other East Asian countries it is an economically
important pest in East Africa and Brazil Effective chemical control of CBB and WSB
is difficult due to the nature of their life cycle inside the berry and stem respectively as
well as environmental concern regarding the use of pesticides Biological control
measures are adapted to combat these insect pests with varying degrees of success
Developing coffee plants resistant to these pests using genetic transformation
technology could be one of the alternative strategies to counter pest damage For insect
resistance several different classes of proteins from bacterial plant and animal sources
have been isolated and their insecticidal properties tested against many important pests
Amongst these proteins Bt toxins are most important and several transgenic crops
expressing Bt genes have been commercialized Coffee transgenic plants carrying a
Chapter 1 Introduction and Review of Literature
19
synthetic version of the cry1Ac gene have been produced (Leroy et al 2000 Mishra et
al 2002) Indeed this was the first report of an important agronomic trait being
introduced into a coffee plant The transgenic plants presented similar features in
growth and development compared to normal plants Transgenic plants highly resistant
to leaf miner under greenhouse conditions were tested under field conditions in French
Guyana for 4 years for pest resistance (Perthius et al 2005 Perthius et al 2006)
From 54 independent transformation events 70 of the events were resistant to leaf
miner Unfortunately the field trial was vandalized which led to the termination of the
experiment (Montagnon et al 2005)The effectiveness of Bt genes in controlling
coleopteran pests is well documented in corn and potato which indicates that Bt genes
might be effective against CBB The high toxicity of B thuringiensis sero var
israelensis against first in star larvae of CBB has been demonstrated (Mendez et al
2003) In another experiment an α-amylase inhibitor from Phaseolus vulgaris was
tested against CBB and found to have an inhibitory effect on its growth and
development (Grossi et al 2004)In addition to the CBB and WSB C arabica
varieties are also susceptible to endoparasitic root-knot nematode (Meloidogyne spp)
(Campos et al 1990 Mishra et al 2012)
So far 15 species have been reported to be parasites of coffee Controlling
nematodes is extremely difficult and currently seedling grafting with robusta rootstock
is followed as one of the control measure Sources of resistance specific to root-knot
nematodes have been identified in coffee trees (Bertrand et al 2001) and the Mex-1
gene conferring resistance to M exigua in C arabica is in the process of isolation (Noir
et al 2003)
1102 Tolerance to Abiotic Stress
In many coffee growing countries abiotic stress such as drought and frost are the major
climatic factors that limit coffee production Changes in climatic patterns due to global
climate change are considered increasingly important for coffee cultivation Drought
induces water stress in plants which affects vegetative growth and vigor and triggers
floral abnormalities and poor fruit set It also indirectly increases the incidence of pests
and diseases in the plants C arabica is generally more tolerant to water stress than C
canephora partly due to its extensive deep root system C racemosa is known to be a
good source of drought tolerance In India hybrids between C canephora and C
racemosa have been obtained and are currently under evaluation for drought tolerance
Chapter 1 Introduction and Review of Literature
20
In many coffee growing countries coffee is propagated in marginal areas where the
annual rainfall is below 1000thinspmm with prolonged dry spells of over 4-5 months In
those areas water shortage and unfavorable temperatures constitute major constraints
and the growth and productivity of C canephora are badly affected As with drought
periodic frost also affects coffee production in parts of Brazil The introduction of
drought and frost tolerant genes through genetic transformation would be of great
importance for alleviating these problems Research is now being carried out by several
groups to identify genes involved in biotic as well as abiotic stress The most promising
genetic engineering approach for drought tolerance includes the use of functional or
regulatory genes as well as the transfer of transcription factors In recent years plants
tolerant to high temperature and water stress have been the subject of intense research
(Chaves et al 2003 Chaves et al 2004 Coraggio et al 2004) For achieving drought
tolerance genes that have been targeted include those encoding enzymes involved in
detoxification or osmotic response metabolism enzymes active in signaling proteins
involved in the transport of metabolites and regulating the plant energy status (Chaves
et al 2003 Chaves et al 2004 Coraggio et al 2004) The dissection of molecular
mechanisms related to signal transduction and transcriptional regulation might help in
engineering drought tolerance in coffee
1103 Disease Resistance
Coffee leaf rust (CLR) caused by the fungus Hemileia vastatrix is the most important
disease in coffee with substantial loss to coffee production and productivity in all the
coffee growing countries In addition to leaf rust coffee berry disease (CBD) caused by
fungus Colletotrichum kahawae can be a devastating anthracnose leading to substantial
crop loss in Africa Several other fungal and bacterial diseases may also affect coffee
to a extent C arabica is more susceptible to many diseases than C canephora Though
most of the disease control measures rely upon chemical control they are more
expensive and labour intensive The long-term solution is the breeding of resistant
varieties which is the focus of many breeding programs However breeding for disease
resistant varieties is time consuming due to the perennial nature of coffee with its long
gestation period India has a long history of C arabica breeding especially for leaf rust
resistance and is the first country to demonstrate the existence of multiple races of leaf
rust Resistance to CLR is conditioned primarily by a number of major (SH) genes and
coffee genotypes are classified into different resistance groups based on their
Chapter 1 Introduction and Review of Literature
21
interaction with different rust races pathogen (van der Vossen 2001) Currently C
canephora provides the main source of resistance to pests and diseases including CLR
(H vastatrix) and CBD (C kahawae) and therefore used in breeding programs Other
diploid species like C liberica and C racemosa are being used as a source of resistance
to coffee leaf rust and coffee leaf miner respectively (Guerreiro et al 1999
Sreenivasan et al 1989) The development of coffee varieties resistant to major fungal
diseases such as CLR and CBD using transgenic technology will benefit the coffee
industry immensely During the last 15 years significant progress has been made in the
area of host-pathogen interactions (Staskawicz et al 1995 Feys et al 2000
Shivaprasad et al 2012) and many resistance genes involved in recognizing invading
pathogens have been identified and cloned (Takken et al 2000) A number of
signalling pathways induced because of pathogen infection have been dissected (Dong
et al 1998 Navarro et al 2006) Many antifungal compounds synthesized by plants to
combat fungal infection have been identified (Does et al 1998) Understanding the
specific induction of targeted pathways and identification of specific pathways
responsible for particular fungal resistance is important in order to employ this strategy
in transgenic technology The recent investigation of gene expression during coffee leaf
rust infection could provide an insight into the defence pathways operating in coffee
(Fernandez et al 2004 Nardi et al 2006) Efforts have been made to identify and
clone resistance genes from coffee for achieving durable resistance The genetic and
physical map of two resistance genes SH3gene conferring resistance to rust (Prakash et
al 2004) and the Ck1 gene conferring resistance to C kahawae CBD (Gichuru et al
2006) have been established These genes could be used for molecular marker assisted
breeding programs (Mishra et al 2012)
1104 Production of low-caffeine coffee
A widespread belief that the ingestion of caffeine can have adverse effects on
health resulted in the increased demand for decaffeinated coffee (Ashihara amp Crozier
2001) Several method were used to obtain decaffeinated plants by conventional
breeding techniques using low yielding varieties and mutants that accumulated
relatively low amounts of caffeine when compared to the commercially available ones
Low-caffeine and decaffeinated coffee represent around 10 of the coffee sales
around the world (Ribas et al 2006b) The industrial process for coffee decaffeination
can be expensive and affects the original flavour and aroma in coffee (David 2002)
Chapter 1 Introduction and Review of Literature
22
Transgenic coffee plants with suppressed caffeine synthesis using RNA interference
(RNAi) technology have been obtained (Ogita et al 2003 Ogita et al 2004) Specific
sequences in the 3prime untranslated region of the theobromine synthase gene (CaMXMT1)
were selected for construction of RNAi short and long fragments The caffeine and
theobromine content of the transgenic plants were reduced by ~ 70 when compared to
the untransformed plants In C canephora RNAi technology has also been employed
to silence the N-methyl transferase gene involved in caffeine biosynthesis (Vinod et al
2007) Promoter of an N-methyltransferase (NMT) gene involved in caffeine
biosynthesis was cloned (Satyanarayana et al 2005 Satyanarayana 2006) which will
be very useful for studying the regulation of caffeine biosynthesis
1105 Improvement in cup quality
Improvement in coffee cup quality requires elaborate knowledge of the chemical
constituents as well as the metabolic pathways involved in the elaboration of quality
The constituents of coffee beans include minerals proteins carbohydrates caffeine
chlorogenic acids (CGA) glycosides lipids and many volatile compounds that give
flavour to coffee by roasting Among these the role of three major constituents
sucrose CGA and trigonelline have been studied in coffee The sucrose content of
coffee bean is associated with coffee flavour the higher the sucrose content in green
beans the more intense will be the cup flavour (Clifford et al 1985 de Maria et al
1996) The sucrose content of C arabica (82-83) is higher than C canephora (33ndash
40) The sucrose amino acids ratio in green beans determines the profile of volatile
compounds Manipulating sucrose content in coffee bean is therefore important in
improving cup quality The sucrose synthase gene (CcSUS2) from C canephora has
been cloned and sequenced (Leroy et al 2005) This provides an opportunity to
manipulate the sucrose content in coffee Chlorogenic acids are products of
phenylpropanoid metabolism Chlorogenic acids are a group of hydroxycinnamoyl
quinic acids (HQA) formed by esterification between caffeic acids coumaric acids and
quinic acids (Clifford et al 1999) They are present in relatively large quantities in the
coffee bean and are the precursor of phenolic compounds in roasted beans C
canephora beans contain higher CGA (10) compared to C arabica beans (6-7)
CGA are known to have antioxidant properties as well as being associated with disease
resistance (Campa et al 2003) Genetic manipulations of genes involved in CGA
synthesis can serve either of these purpose by up- or down regulating the pathway
Chapter 1 Introduction and Review of Literature
23
Phenylalanine ammonia-lyase (PAL) catalyzes the first step of the phenylpropanoid
pathway leading to the synthesis of a wide range of chemical compounds including
flavonoids coumarins hydroxycinnamoyl esters and lignins (Hahlbrock amp Scheel
1989) Recently the full length cDNA and corresponding genomic sequences of PAL
from C canephora was isolated characterized and functionally validated (Mahesh et
al 2006 Parvatam et al 2006) This has opened up new possibilities for manipulating
the level of the PAL enzyme in coffee which in turn can be useful for improvement of
cup quality and manipulating antioxidant properties in coffee
1106 Fruit Ripening
Uniformity during fruit ripening is decisively related to cup quality in coffee and
consequently to the value of the product Fruits at the ideal ripening stage produce the
best organoleptic characteristics for coffee The presence of over ripened or green fruits
changes the acidity the bitterness and consequently the cup quality In order to
maximize uniform ripening of coffee fruits it is essential to control the action of genes
involved in the last step of maturation process Ethylene is known to trigger ripening
and increasing ethylene biosynthesis is associated with various stages of ripening
process (Protasio et al 2005) To control coffee fruit maturation two of the major
genes involved in ethylene biosynthesis namely ACC synthase and ACC oxidase
have been cloned (Protasio et al 2005 Neupane et al 1999) Introduction of the ACC
oxidase gene in antisense orientation have been achieved in both C arabica and C
canephora The effect of the transgene on ethylene production and fruit maturation has
yet to be reported The inhibition of genes downstream to the initial ethylene burst is
also an option to control coffee fruit maturation (Ribas et al 2006)
111 RNA interferenceRNAi
The regulation of gene expression at post transcriptional level has recently attracted
much attention because of the discovery of the phenomenon called RNA interference
(RNAi) RNAi based silencing techniques are more efficient than antisense mediated
gene silencing (Wesley et al 2001) The effect was first observed in plants wherein
silencing was observed when some copies of a transgene was in the forward orientation
with respect to the promoter and some in the reverse orientation This resulted in the
formation of double stranded RNA in the system The phenomenon in which
experimentally introduced double stranded RNA (dsRNA) leads to the loss of
expression of the corresponding cellular gene by sequence specific RNA degradation
Chapter 1 Introduction and Review of Literature
24
was termed as RNA interference or RNAi The protective effect of RNA silencing in
reducing virus infectivity supports the view that PTGS has evolved as a mechanism to
defend plants against virus infection and also to moderate the possible deleterious
genome-restructuring (insertional) activity of virus-like mobile genetic elements eg
retrotransposons A growing body of biochemical and genetic data has further
demonstrated that plants animals and yeasts share related mechanisms of specific
degradation of RNAs in which double-stranded forms of RNA are initiator molecules
(Brenstein et al 2001)
The key insight into the process of PGTS was provided by the experiments
conducted by Hamilton amp Baulcombe (1999) who identified the product of RNA
degradation as small RNA (siRNA) species of ~25nt of both sense and antisense
polarity SiRNA are formed and accumulate as double stranded RNA molecules of
defined chemical structures Both genetic and biochemical approaches were undertaken
to understand the basis of silencing Genetic screens were carried out in fungus
Neurospora carassa the alga Chalmydomonas reinhardtii and plant Arabidopsis
thaliana to search for detective mutants in PTGS
A combination of results obtained from several in vivo and in vitro experiments
have gelled into a two step mechanistic model for RNAiPTGS The first step referred
to as the RNAi initiating step involves binding of the RNA nucleases to a larges
dsRNA and its cleavage into discrete ~21-25nt RNA fragments This is ATP-
dependent reaction which involved RNase III - type endonucleases which make
staggered cuts in both strands of the dsRNA leaving 3prime overhang of 2 nucleotides with
5prime- phosphate 3prime-hydroxyl and no modification in the sugar phosphate backbone
(Hamilton amp Baulcombe 1999 Elbashir et al 2001) SiRNAs are the direct products
of dsRNA cleavage by the multi-domain RNase III enzyme DICER (Figure 16)
(Brenstein et al 2001 Karthikeyan et l 2013)
In the second step or commonly known as the effector step the double stranded
siRNAs produced in the first step bind to an RNAi specific protein complex to form a
RISC The complex undergoes activation in the presence of ATP so that the antisense
component of the unwound siRNA becomes exposed and allows the RISC to perform
the downstream RNAi reaction
Chapter 1 Introduction and Review of Literature
25
Several enzymes are involved in RNAi based on their role genes that encode for them
are classified into RNAi initiators RNAi effectors RNA dependent RNA polymerases
and silencing genes Two C elegans genes rdeI and rde4 (rde stands for lsquo RNAi
deficientrsquo) are believed to be involved in the initiation step of RNAi The C elegans
rde1 gene is a member of a large family of genes and is homologous to the Neurospora
qde2 (qde stands for ldquoquelling deficientrdquo) and Arabidopsis AGO1 (AGO stands for
agronaute AGO1 was previously identified to be involved in Arabidopsis
development) Although the functions of these genes are not clear a mammalian
member of RDE1 family has been identified as a translation initiation factor (Thakur
2005) Important genes for the effector step of PTGS in C elegans are rde2 and mut7
genes These genes were identified from heterozygous mutant worms that were unable
to transmit RNAi to their homozygous offsprings Worms with mutated rde2 or mut 7
genes show defective RNAi mut-7 gene encodes a protein with homology to the
nuclease domains of RNAase D and a protein implicated in Werner syndrome (a rapid
ageing disease in humans) (Grishok et al 2001 Kamath-Loeb et al 2012s)
Neurospora qde-1 Arabidopsis SDE-1SGS-2 and C elegans ego-1 appear to encode
RNA dependent RNA polymerase (RdRPs) It assumed that this is a proof that an RdRp
activity is required for RNAi Certainly the existence of an RdRp might explain the
remarkable efficiency of dsRNA induced silencing if it amplifies either the dsRNA
prior to cleavage or the siRNAs directly (Muers 2013) In C elegans ego-1 mutants
(ego stands for lsquoenhancer of glp-1) RNAi functions normally in somatic cells but is
defective in germline cells where ego-1 is primarily expressed In Arabidopsis SDE-
1SGS-2 mutants (SGS stands for suppressor of gene silencing) siRNA are produced
when dsRNA is introduced via an endogenously replicating RNA virus but not
introduced by a transgene It has been proposed that perhaps the viral RdRP is
substituting for the Arabidopsis enzyme in these mutants Random degradative PCR
model suggests that an RdRP uses the guide strand of an siRNA as a primer for the
target mRNA generating a dsRNA substrate for Dicer and thus more siRNAs
Several genes controlling RNA silencing in plants have been identified through
genetic screens of Arabidopsis mutants impaired in transgene induced RNA silencing
They encode a putative RNA-dependent RNA polymerase (SGS2SDE1) a coiled
protein (SGS3) a protein containing PAZ and Piwi domains (AGO1) and an RNA
helicase (SDE3) The putative SGS2SDE1 of Neurospora is related to QDE-1 and
Chapter 1 Introduction and Review of Literature
26
EGO-1 of C elegans whereas PAZPiwi protein AGO is related to QDE-2 of
Neurospora RDE-1of C elegans RNA helicase SDE3 is related to SMG-2 of C
elegans and Mut-6 of Chlamydomonas MUT-7 gene of C elegans encodes a protein
similar to Rnase D whereas the Drosophila DICER gene encode a protein similar to
RNase III An Arabidopsis ortholog of DICER gene has been identified called CAF
SIN1 SUS1(Thakur 2005 Poethig 2009)
Since the RNAi machinery is present constitutively within the eukaryotic cell it
is important to explore the metabolic advantages that are accorded by the RNAi related
proteins during the intrinsic normal growth of cells and development of organisms The
natural RNAi machinery not only keeps the mobile transposable elements from
disrupting the integrity of the genomes as suggested by the analysis of model organisms
like A thaliana C elegans D melanogaster (Tabara et al 1999 Wu-Scharf et al
2000 Aravin et al 2001 Hamilton et al 2002 Lisch 2009) but also participates in
development of organisms Genetic defects in Celegans RNAi genes ego1 and dicer
cause known specific developmental errors (Grishok et al 2000 Grishok et al 2001
Knight amp Bass 2001 Hall et al 2013) Similarly the Argonaute family of genes of A
thaliana (especially the ZWILLE proteins) is also responsible for plant architecture and
meristem development (Carmell et al 2002 Hamant and Pautot 2010) and the Dicer
homologue of A thaliana CAF1 is required for embryo development (Golden et al
2002 Nakano et al 2011) Thus genetic evidence illustrates the role of the RNAi
machinery as a controller of development related genes The mechanistic details of
these developmental processes are beginning to emerge
Chapter 1 Introduction and Review of Literature
27
Figure 16 Mechanism of gene silencing induced double stranded RNA The dicer enzyme to
produce siRNAs cleaves the RNA The siRNA generated bind to the nuclease complex
RISC The antisense component in the siRNA in the RISC guides the complex towards
the cognate mRNA resulting in endonucleolytic cleavage of the mRNA
Chapter 1 Introduction and Review of Literature
28
112 MicroRNA or MiRNA
In 1991 Ambros and co-workers first isolated a lin-4 mutant of Celegans which was
arrested at the first larval stage (Lee et al 1993) Later on the let-7 mutation was
isolated in the same system which was responsible for development through the fourth
larval stage Both lin-4 and let-7 encode short 22-nucleotide mature RNAs and were
called short temporal RNA because they control the temporal development program of
C elegans The mature lin-4 RNA defines (negatively regulates) the mRNA expression
of the lin-14 and lin-28 hetrochronic genes with the antisense mediated repression
mechanism of translation initiation and thus specifies the fate of cells during the first
three larval stages Recent studies have revealed that the short temporal RNAs are
actually members of a group of tiny RNAs (21to 28 nucleotides) called the micro-
RNAs (miRNA) (Bartel 2009) isolated members of which could easily run to a few
thousand which includes both those were identified computationally and by deep
sequencing Some of the components of the RNAi machinery have also been clearly
established as the effector proteins for the maturation of miRNAs
113 MicroRNAs in plants
1131 Discovery
MicroRNAs have been discovered by three basic approaches Direct cloning forward
genetic direct cloning and bioinformatic prediction followed by experimental
validation The most direct method of miRNA discovery is to isolate and clone small
RNAs from biological samples and several groups have used this approach to identify
plant miRNAs (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002 Xie et al 2003 Sunkar et al 2005 Williams et al 2013) The cloning methods
adapted were similar to those used to identify large numbers of animal miRNAs
(Lagos-Quintana et al 2001 Lau et al 2001 Aravin amp Tuschl 2005) which involve
isolating small RNAs followed by oligonucleotide adapter ligation reverse
transcription amplification and sequencing Some protocols incorporate methods to
enrich for Dicer cleavage products (ie molecules with 5prime-phosphates and 3prime-
hydroxyls) and to concatemerize the short cDNAs so that several may be identified in a
single sequencing read (Lau et al 2001 Llave et al 2002a Jagadeeswaran amp Sunkar
2013)The initial cloning experiments in Arabidopsis identified 19 miRNAs belonging
to 15 families (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002) although hundreds of other small RNAs such as siRNAs and RNA degradation
Chapter 1 Introduction and Review of Literature
29
fragments were also cloned through the direct cloning methods To select miRNAs
from the pool of small RNAs secondary structures of the RNA molecules were
examined in compliance with the acknowledged structural features of miRNAs
(Ambros et al 2003) The secondary structures were validated using several ad-hoc
software tools such as the Mfold or RNAfold programs (Reeder et al 2006) Finally
the existence of mature miRNAs was determined by RNA gel blot hybridization These
direct cloning methods were successful in isolating many miRNAs in Arabidopsis
(Reinhart et al 2002 Sunkar amp Zhu 2004) Rice (Sunkar et al 2005) Poplar (Lu et
al 2005) moss (Arazi et al 2005) and Tobacco (Billoud et al 2005)
Although miRNAs were first discovered through forward genetic screens in
worms (Lee et al 1993 Reinhart et al 2000)only few A thaliana miRNA gene
families have been discovered by this method (Chen 2008) in plants MiRNA
involvement in plant mutant phenotypes were not properly inferred till the cloning
experiments established that plant genomes contain numerous miRNAs (Park et al
2002 Reinhart et al 2002 Rhoades et al 2002) MiRNA loss-of-function allele has
been identified by forward genetic screens early extra petala1 is caused by a
transposon insertion ~160bp upstream of the predicted miR164c stem loop resulting in
flowers with extra petals (Baker et al 2005 Irish 2008) The fact that loss-of-function
miRNA mutants have been rarely discovered using forward genetics reflects small
target size for mutagenesis coupled with redundancy in nearly all evolutionarily
conserved plant miRNAs which are encoded by gene families (Table16) Family
members are likely to have overlapping functions buffering against loss at any single
miRNA locus Over expression screens can circumvent redundancy limitations At least
four plant miRNAs miR319 (also known as miR-JAW) miR172 (also known as EAT)
and miR166 were isolated in over expression screens for dominant mutants with
developmental abnormalities (Aukerman amp Sakai 2003 Palatnik et al 2003 Kim et
al 2005 Williams et al 2005b Schommer et al 2012) Mutations in miRNA target
sites which can prevent the entire family of miRNAs from repressing a target gene can
also circumvent redundant functions of miRNA family members
The dominant mutations in the HD-ZIP genes PHB PHV and REVOLUTA
(REV) in Arabidopsis and ROLLEDLEAF1 (RLD1) in Maize result in adaxialization of
leaves andor Vasculature (McConnell amp Barton 1998 McConnell et al 2001 Nelson
et al 2002 Zhong amp Ye 2004 Allen amp Millar 2012) and are all caused by mutations
Chapter 1 Introduction and Review of Literature
30
in miR166 complementary sites (Rhoades et al 2002 Emery et al 2003 Jones-
Rhoades amp Bartel 2004 Mallory et al 2004 Zhong amp Ye 2004)
In both plants and animals cloning was the initial means of large-scale
miRNA discovery (Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Reinhart et al 2002 Mendes et al 2012) However cloning is biased toward
RNAs that are expressed highly and broadly MiRNAs expressed at low levels or
only in specific cell types or in response to certain environmental stimuli were more
difficult to clone Sequence based biases in cloning procedures might also cause
certain miRNAs to be missed Because of these limitations bioinformatics approaches
to identify miRNAs have provided a useful complement to cloning
A straightforward use of bioinformatics has been carried out to find homologs
of known miRNAs both within the same genome and in the genomes of other species
(Pasquinelli et al 2000 Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Mendes et al 2009) A more difficult challenge is to identify miRNAs
unrelated to previously known miRNAs This was first accomplished for
vertebrate nematode and fly miRNAs using algorithms that search for conservation
of sequence and secondary structure (ie miRNA stem-loop precursors) between
species searching for patterns that are characteristic of miRNAs (Lai et al 2003 Lim
et al 2003 Lim et al 2005)
Most of the methods identified numerous miRNAs but are necessarily more
relaxed in terms of allowed structures Some approaches take advantage of the high
complementarity of plant miRNAs to target messages implementing the requirement
that the candidate has conserved complementarity to mRNAs (Jones-Rhoades amp Bartel
2004 Adai et al 2005) This additional filter has been useful for distinguishing
authentic plant miRNAs from false positives which later extended to mammalian
miRNA gene prediction (Xie et al 2005 Mendes et al 2012)
Another criteria used by most algorithms in the filters is evolutionary
conservation In plants mature miRNA sequences are conserved to a higher degree
than their precursor sequences For example Arabidopsis and rice diverged more than
130 million years ago (Friis et al 2004) but some miRNAs are highly conserved
in these plant species (Reinhart et al 2002) Furthermore various parameters for the
secondary structures such as free energy the number of paired residues within a
Chapter 1 Introduction and Review of Literature
31
miRNA or the number and size of bulges are also used to validate the predictions
(Jones-Rhoades amp Bartel 2004 Wang et al 2004 Adai et al 2005 Mendes et al
2012)
1132 Conserved microRNAs in Plants
Till date cloning genetics and bioinformatics screens have resulted in annotation of
approximately 184 potential Arabidopsis miRNAs (miRBase release 13) These
miRNAs seem to arise through gene duplication and subsequent diversification and
represent approximately 100 families (Maher et al 2006 Axtell 2013) Twenty one
families represented by 92 genes are clearly conserved in species beyond Arabidopsis
(Table 15) The presence of these miRNA families in other sequenced plant genomes
indicates that different plant species have an evolutionarily fluid set of miRNAs
(Willmann amp Poethig 2007) Recent studies on mosses one of the most ancient land
plants have shown that at least 14 miRNA families are conserved in eudicots and
mosses and therefore it appears that plant miRNAs share a common ancient origin
(Arazi et al 2005 Axtell amp Bartel 2005 Talmor-Neiman et al 2006 Zhang et al
2006 Axtell 2013) The number of members per family in a genome ranges from 1-32
with the exception of miR-430 which is represented by a cluster of ~80 loci in zebra
fish (Giraldez et al 2005)
In plants the number of members in each family correlates among examined
species certain families contain numerous members in all three species (eg miR156
miR166 miR169) whereas others consistently contain fewer genes (eg miR162
mir168 miR394) (Table 16) Although it is still unclear why certain miRNA are
encoded by more than one gene in plants (eg 12 genes encoding miR156) this
correlation might suggest functional significance of various miRNA family sizes Most
annotated plant miRNAs are conserved throughout flowering plants while a few
miRNAs are found only in a single sequenced genome and thus they could be
evolutionarily lsquoyoungrsquo miRNAs For example Arabidopsis has several miRNA
families that are not found in poplar or rice suggesting that miRNAs could be
generated continuously during evolution (Allen et al 2004a Rajagopalan et al
2006 Fahlgren et al 2007 Axtell 2012 de Alba et al 2013) Consistent with the
notion that non conserved miRNAs are of a more recent evolutionary origin these
miRNAs are mainly encoded by a single locus in the genome Within each family the
mature miRNA is always located in the same arm
Chapter 1 Introduction and Review of Literature
32
Table 16 MicroRNA families conserved in plants
miRNA
family
Arabidopsis Oryza Populus
miR156 12 12 11 miR159319 6 8 15 miR160 3 6 8 miR162 2 2 3 miR164 3 5 6 miR166 9 12 17 miR167 4 9 8 miR168 2 2 2 miR169 14 17 32 miR171 4 7 10 miR172 5 3 9 miR390 3 1 4 miR393 2 2 4 miR394 2 1 2 miR395 6 19 10 miR396 2 5 7 miR397 2 2 3 miR398 3 2 3 miR399 6 11 12 miR408 1 1 1 miR403 1 0 2 miR437 0 1 0 miR444 0 1 0 miR445 0 9 0 Total 92 127 169
All known miRNA families that are conserved between more than one plant species are listed
together with the number of genes identified in the sequenced genomes Rice miRNA families that have orthologs in maize but do not appear to have orthologs in the eudicots (Arabidopsis and Populus) are marked with an asterisk The following families contain miRNA genes annotated with
more than one number miR156 (miR156 and miR157) miR159319 (miR159 and miR319) miR166 (miR165 and miR166) miR171 (miR170 and miR171) and miR390 (miR390 and miR391)
of the stem loop (5prime- or 3prime) as it would be expected if the genes carry common ancestry
(Figure 17) The sequence of the mature miRNA and to some extent the segment on
the opposite arm of the hairpin to which it pairs is highly conserved between the
members of the same family (both within and between the species) while the sequence
secondary structure and length of the intervening ldquolooprdquo region can be highly divergent
within the same family
The patterns of paring and non pairing nucleotides are often conserved between
homologous miRNA stem loops from different species (Figure 17) The significance of
conserved mismatches is opined to guide DCL1 to cleave at the appropriate positions
along the stem loop
Chapter 1 Introduction and Review of Literature
33
Figure 17 Representative stem loop of miR164 Segments corresponding to the mature miRNAs
are shown in red (Adapted from Rajagopalan et al 2006)
Although many miRNAs are highly conserved in plants the degree of conservation in
most cases is quite low between plants and animals one exception is miR854 A recent
study has revealed that miR854 is conserved between Arabidopsis and animals and that
the Arabidopsis miR854 differs from the human miR854 only by a single nucleotide
(Arteaga-Vazquez et al 2006 Axtell 2012 de Alba et al 2013) The Arabidopsis
miR854 has multiple binding sites in the 3prime-untranslated region (UTR) of
OLIGOURIDYLATE-binding-PROTEIN mRNA 1bA (UBP1b) and represses the
translation of the UBP1b mRNA The animal miR854 is also complementary to a site in
the 3prime-UTR of the UBP1b homolog in animals However it is currently unclear how the
animal miR854 regulates its target mRNA Another distinction between plant and
animal miRNAs is the genomic organization of the miRNA genes While plant miRNA
genes are located predominantly in the intergenic regions animal miRNA genes tend to
localize in the intragenic regions both in the introns or exons of protein coding genes
Moreover miRNA genes are often clustered within a single precursor transcript
whereas individual plant miRNAs are derived from individual precursor RNAs (Bartel
Chapter 1 Introduction and Review of Literature
34
2004 Bonnet et al 2004 Kim 2005 Vaucheret et al 2006 Marco et al 2013)
1133 Non-conserved miRNA and challenges in annotation
Most miRNAs are conserved throughout flowering plants (Table 15) but many more
were found only in a single sequenced genome thus considered to be of a more recent
evolutionary origin The extended homology between few non conserved miRNAs
precursors and their target genes provides strong evidence that these relatively ldquoYoungrdquo
miRNAs arose from the duplication of the target gene segments (Allen et al 2004)
Several non-conserved miRNAs including miR161 miR163 miR173 miR447
miR475 and miR476 are known to direct cleavage of target transcripts (Allen et al
2004a Adai et al 2005 Sunkar et al 2005 de Alba et al 2013) It is difficult to
confidently predict targets for many because it is not possible to use complementary
site as a filter against false-positive target predictions It is also difficult to
confidently annotate non-conserved miRNAs as miRNA rather than siRNAs The
established minimal standard for miRNA annotation is a small RNA with detectable
expression and the potential to form a stem-loop when joined to flanking genomic
sequence (Kasschau et al 2003) In practice these requirements are too loose to
definitively categorize many small RNAs cloned from plants Many plant siRNAs are
detectable on blots (Vazquez et al 2004b) and hundreds of thousands of non-
miRNA genomic sequences can be predicted to fold into secondary structures that
resemble structures of plant miRNA precursors (Jones-Rhoades amp Bartel 2004)
Therefore without conservation of both sequence and secondary structure it is
difficult to be confident that a given cloned RNA originated from a stem-loop (ie is a
miRNA) rather than from a double-stranded RNA (ie is a siRNA) In fact many of
the thousands o f small RNAs cloned from Arabidopsis (Gustafson et al 2005) would
probably meet the literal requirements for annotation as miRNAs A few of these
sequences might be miRNAs but others that meet the literal criteria probably are not
so The challenges of annotating non conserved small RNAs were evidenced by
three related small silencing RNAs that were originally annotated as miRNAs
that turned out to be trans-acting siRNAs (tasiRNAs)(Kanno et al2013)
Although most miRNAs that are broadly conserved among flowering plants are
now being identified and experimentally validated (Table 16)(Jones-Rhoades amp
Bartel 2004) the challenges and ambiguities for classifying non-conserved miRNAs
prevent meaningful estimates of the total number of miRNA genes in Arabidopsis and
Chapter 1 Introduction and Review of Literature
35
other plant genomes It is possible that miRNAs might have escaped detection
because they are non-conserved and expressed in specific tissues or conditions and can
only be identified by using high- throughput sequencing
1134 MicroRNA biogenesis
11341 Transcription of microRNA precursor
Plant miRNAs are primarily found in the genomic regions that are not associated with
the protein coding genes (Reinhart et al 2002) and it appears that most if not all plant
miRNAs are produced from their own transcriptional units Plant miRNA genes are
occasionally clustered near each other in the genome suggesting transcription of
multiple miRNAs from a single primary transcript [eg the miR395 cluster (Grishok
et al 2001 Jones-Rhoades amp Bartel 2004b)] but this polycistronic arrangement of
miRNA genes appears far less frequently in plants than in animals (Bartel 2004)
Northern blot EST and mapping evidence indicate that plant miRNA primary
transcripts (also known as pri-miRNAs) as in animals are longer than needed to
encompass the miRNA stem-loops (Palatnik et al 2003 Jones-Rhoades amp Bartel
2004b) At least some of these pri-miRNA transcripts appear to be spliced
polyadenylated (Kurihara amp Watanabe 2004) and capped (Xie Z et al 2005) Two
rice miRNAs are contained within transcripts that contain exon junctions within the
presumptive stem-loop precursor implying that in these cases splicing is a prerequisite
for Dicer recognition (Sunkar et al 2005) Plant pri-miRNAs are over 1kb in length
and they are usually preceded by typical TATA box motifs They can also undergo
canonical splicing polyadenylation and capping All these observations indicates RNA
polymerase II is probably responsible for transcribing most plant miRNAs (Xie Z et
al 2005) as appears to be the case for many animal miRNAs Relatively little is
known about the regulation of miRNA transcription in plants but there is no reason
to suspect that this regulation would differ from that of protein-coding transcripts as
the bioinformatics analysis of the sequence upstream of the transcriptional start
sites of the miRNA genes showed the presence of putative binding motifs of
number of known transcription factors (Megraw et al 2006 Sethupathy 2013)
11342 MicroRNA processing and export
In plants maturation of miRNA is a step wise process A miRNA gene is transcribed to
pri-miRNA which is much longer than the pre-miRNA possessing the characteristic
stem-loop structure Like transcription of most protein-coding genes the miRNA
Chapter 1 Introduction and Review of Literature
36
transcription is processed by RNA polymerase II (Pol II) enzymes and the pri-
miRNAs are 5prime-capped and 3prime-polyadenylated (Lee et al 2004 Xie Z et al 2005)
Mature miRNAs are produced from pri-miRNAs through at least two sequential
processing steps by RNase III-type endonucleases In animals pri-miRNAs are first
processed at the bottom portion of the stem by Drosha a nuclear-localized RNase III
enzyme to liberate pre-miRNAs The pre-miRNAs are then exported to the cytoplasm
with the help of exportin-5The pre-miRNAs are further processed by Dicer another
RNaseIII enzyme to a duplex consisting of the miRNA and the antisense strands
(miRNA) Since plants do not have any Drosha homologs the two sequential
processing steps seem to be carried out by DCL1 a Dicer homolog in the nucleus
(Kurihara amp Watanabe 2004 de Alba et al 2013 Rogers amp Chen 2013)
The processing of pri-miRNAs to pre-miRNAs by DCL1 is assisted by two
other proteins HYPONASTY LEAVES1 (HYL1) and SERRATE (SE) HYL1 belongs to a
family of double-stranded RNA (dsRNA) binding protein and interacts with DCL1 in
vitro and in vivo (Lu amp Fedoroff 2000 Hiraguri et al 2005 de Alba et al 2013
Rogers amp Chen 2013) Loss-of-function mutations in the HYL1 gene result in
decreased accumulation of miRNAs and concomitant accumulation of pri-miRNAs
(Han et al 2004 Vazquez et al 2004a Kurihara et al 2006) SE a C2H2 zinc finger
protein interacts with DCL1 and HYL1 and plays a role in the processing of pri-
miRNAs to pre-miRNAs (Lobbes et al 2006 Yang L et al 2006) In addition the
nuclear cap-binding complex proteins CBP20 and CBP80 that are known as ABA
HYPER SENSITIVE 1(ABH1) have been shown to be required for proper miRNA
processing (Gregory et al 2008 Laubinger et al 2008) Recently it has been
demonstrated that DCL1 can release 21-nt RNAs from dsRNA as well as from
synthetic miR167 precursor RNAs in vitro However this cleavage is error prone and
inefficient in the absence of HYL1 and SE which accelerate the rate of DCL1-mediated
cleavage and promote accurate processing of miRNAs (Dong et al 2008 Rogers amp
Chen 2013)
11343 Methylation of the miRNAmiRNAduplex
After the miRNAmiRNAduplexes are released from the pre-miRNAs by the DCL1
activities the duplexes are methylated at the 2primeOH of the3prime-ends by HUA ENHANCER1
(HEN1) a small RNA methyltransferase (Yu et al 2005 Yang Z et al 2006 Rogers
amp Chen 2013 ) This step which is distinct from the RNase III-mediated processing
Chapter 1 Introduction and Review of Literature
37
step distinguishes the biogenesis of plant miRNAs from that of animal miRNAs
Mutations in the HEN1 gene were first isolated in a morphological screening for floral
development although the HEN1 mutants display pleiotropic phenotypes similar to the
developmental defects observed in the DCL1 mutants (Chen et al 2002 Park et al
2002 Rogers amp Chen 2013)
Figure 18 Biogenesis of microRNA
A Biogenesis of plant MicroRNA B Biogenesis of metazoananimal MicroRNA
The miRNA (red) is incorporated into RISC and the miRNA(blue) in degraded
This enzyme has two dsRNA-binding domains and nuclear localization signal It
is also conserved in fungi and is required for the processing of siRNAs as well as of
miRNAs (Park et al 2002 Boutet et al 2003 Xie et al 2003) Although the HEN1
protein is localized to the nucleus its methylation activity in the cytoplasm cannot be
ignored (Xie et al 2004 Rogers amp Chen 2013)
Chapter 1 Introduction and Review of Literature
38
The methylation of the miRNAmiRNA duplex is required to protect miRNAs
against the 3prime-end uridylation activity and subsequent degradation (Li et al 2005) In
the hen1 mutants accumulation of miRNAs is reduced to a lower level Also the
miRNAs appear to be heterogeneous in their sizes such that a ladder of bands rather
than a single species is observed for most miRNAs in the mutants (Li et al 2005)
MiRNA cloning and sequencing analyses revealed the presence of a number of U
residues at the 3prime-end of miRNAs in the hen1 mutants Moreover these nucleotides do
not correspond to the miRNAs in the pre-miRNAs indicating that these nucleotides are
added after the processing of the miRNAs from pre-miRNAs (Li et al 2005)
Uridylation of RNAs has also been proven to be correlated with RNA decay (Shen amp
Goodman 2004 Mullen amp Marzluff 2008) suggesting that uridylation attracts an
exonuclease which degrades miRNAs in plants
11344 MiRNA incorporation in to the RISC and its nuclear export
The methylated miRNAmiRNA duplexes undergo RISC assembly In this process
the miRNA of the duplexes is selectively incorporated into the RISC and the miRNA
appears to be removed and subsequently degraded (Hammond et al 2000 Hutvagner
amp Zamore 2002 Schwarz et al 2003 de Alba et al 2013 Rogers amp Chen 2013)
The ARGONAUTE 1(AGO1) proteins are core components of the RISC complex
They contain two structural domains the N-terminal PAZ domain that binds to the 3prime-
end of single-stranded RNAs and the C-terminal piwi domains that has a structure
similar to that of RNase H (Cerutti et al 2000 Parker et al 2004) Several AGO
proteins possess endonucleolytic activities within the PIWI domain and cleave target
mRNAs in the middle of the site complementary to miRNA (Liu et al 2004 Meister et
al 2004 Miyoshi et al 2005) Mutations in the AGO1 gene cause miRNA target
genes to be ectopically expressed and the levels of some miRNAs are reduced in the
mutants (Kidner amp Martienssen 2004 Vaucheret et al 2004 Kidner amp Martienssen
2005 Ronemus et al 2006) Moreover the phenotype of the AGO1 hypomorphic
alleles display defects in lateral organ polarity and leaf and flower morphology as
observed in the dcl1 mutants and null AGO1 alleles are embryonically lethal
supporting the close relationship between the AGO proteins and miRNA processing
(Kidner amp Martienssen 2004 Vaucheret et al 2004 de Alba et al 2013 Rogers amp
Chen 2013)
The production of miRNA miRNA duplexes occur in the nucleus while
Chapter 1 Introduction and Review of Literature
39
miRNA-RISC complexes are present in the cytoplasm where target mRNAs are
located This discordance of functional sites requires an export step during miRNA
biogenesis HASTY the Arabidopsis homolog of exportin-5 is thought to be involved
in miRNA nuclear export (Bollman et al 2003 Park et al 2005)The hasty mutant
exhibits pleiotropic defects in plant development and reduced accumulation of most
miRNAs as in the DCL1 or AGO1 mutants Howeverit is unclear whether the HASTY
proteins export the miRNA miRNA duplexes or the miRNA-RISC complexes in
plants
11345 Feedback regulation of miRNA biogenesis
MiRNA biogenesis is under feedback regulation such that the DCL1 and AGO1 genes
are themselves regulated by miRNAs DCL1 is itself a target of miR162 which leads to
the cleavage of the DCL1 mRNA (Xie et al 2003 de Alba et al 2013) Consistent
with this the levels of DCL1 mRNA are elevated in the mutants of miRNA processing
genes in which the miR162 abundance is reduced In addition there is a predicted
hairpin structure of miR838 in the 14th
intron of the DCL1 primary transcript
(Rajagopalan et al 2006) This prediction implies that DCL1-mediated processing
on this site can result in the cleavage of the DCL1 primary transcript into two truncated
fragments the N-terminal capped fragment and the C-terminal polyadenylated
fragment If this is the case it is possible that miRNA biogenesis competes with the
splicing event of the DCL1 pre-mRNA
The AGO1 is also regulated by miRNA There is a complementary site for
miR168 binding in the AGO1 mRNA and the transcript level of the AGO1 mRNA is
reduced through an mRNA cleavage mechanism (Vaucheret et al 2006 de Alba et al
2013 Rogers amp Chen 2013) indicating that the AGO1 mRNA levels are tightly
controlled by the levels of the AGO1 protein
1135 Mechanism of miRNA action
11351 MiRNA-directed mRNA cleavage
By forming the miRNA-RISC assembly miRNAs can direct the RISC to regulate gene
expression by two post transcriptional mechanisms mRNA cleavage or translational
repression (Bartel 2004 Jones-Rhoades et al 2006 Dalmay 2013) The degree
of miRNA-mRNA complementarity plays an important role for the
determination of the mechanism to be used A general rule is that while perfect or
Chapter 1 Introduction and Review of Literature
40
near-perfect complementarity induces mRNA cleavage central mismatches trigger
translational repression (Carrington amp Ambros 2003 Jones-Rhoades amp Bartel 2004)
Unlike most animal miRNAs plant miRNAs have near-perfect or perfect
complementarity to their targets and mRNA cleavage is assumed to be a predominant
mechanism by which miRNAs regulate gene expression in plants
In RNA cleavage mechanism miRNAs guide the AGO component of RISC to
cleave a single phosphodiester bond of the target mRNA within the miRNA-binding
site (Llave et al 2002b Tang et al 2003) The truncated fragments are then released
and degraded freeing the RISC to recognize and cleave another target mRNA This has
been validated by the detection of 3prime cleavage products that have 5prime ends that start at the
middle of the complementary regions The mRNA cleavage products are then further
degraded by other mechanisms The 3prime cleavage products of some miRNA targets are
degraded by the 5prime-3prime exonuclease XRN4 an Arabidopsis homolog of the major Yeast
mRNA degrading exonuclease Xrn1p It has been observed that the 3prime cleavage
products of some target mRNAs were accumulated in the xrn4 mutants (Souret et al
2004 Dalmay 2013) However the 3prime cleavage products of other miRNA targets do
not accumulate in the xrn4 mutants indicating that there are also XRN4 independent
mechanisms The 5prime cleavage products are typically untraceable by RNA gel blot
hybridization but can be detected by sensitive PCR based methods It has been found
that the 5prime cleavage products tend to obtain an oligo U tail at the 3prime ends This 3prime U tail
is correlated with shortening of the 5prime cleavage products from the 5prime end leading to the
conclusion that the 3prime uridylation causes 5 prime- 3 prime exonucleolytic decay of the 5prime cleavage
products (Shen amp Goodman 2004)
DCL1 HYL1 and SE interact with each other and are co-localized in the
nuclear bodies termed Dicing bodies or D-bodies in which some pri-miRNA shave
been found suggesting that D-bodies serve as a center for active miRNA processing
(Fang amp Spector 2007 Fujioka et al 2007 Song et al 2007) The miRNA
processing site appears to be distinct from the Cajal bodies that have previously been
found as a siRNA processing site (Pontes amp Pikaard 2008) The D-bodies include
SmD3 and SmB proteins that are found in the Cajal bodies However At collin an
additional marker for the Cajal bodies is not found in the D-bodies Moreover the D-
bodies are present in an At collin mutant lacking the Cajal bodies The pri-miRNAs of
miR171A and miR159A genes have been found to be co-localized with HYL1 and
Chapter 1 Introduction and Review of Literature
41
DCL1 in the D- bodies that are distinct from the Cajal bodies (Song et al 2007)
11352 MiRNA-directed translational repression
Translation repression is another mechanism exerted by plant miRNAs The regulatory
mechanism of miR172 which regulates flowering time and floral organ identity is
consistent with translation repression MiR172 over production does not affect the
transcript levels of APETALA2 (AP2) and AP2- like target mRNAs Instead the AP2
protein level is reduced in the miR172 over expressing mutant (Aukerman amp Sakai
2003 Chen 2004 Dalmay 2013) It was later shown that miR172 can also direct the
cleavage of the target mRNAs although the steady-state mRNA level is unchanged due
to feedback regulation (Schwab et al 2005 Jung et al 2007) It is clear that miR172
regulates its target genes primarily by translational repression rather than mRNA
cleavage Recently it has been reported that miR156157 and miR854 also regulate
their target genes through translational repression (Arteaga-Vazquez et al 2006
Gandikota et al 2007 Dalmay 2013) It was once thought that translational repression
is an exception to the rule However experimental evidence suggest a widespread
coexistence of translational repression and mRNA cleavage (Brodersen et al 2008)
1136 Regulatory roles of miRNAs in plant development
The miRNA target prediction has been a difficult task in order to obtain regulatory
targets without bringing in too many false predictions Plant growth and developmental
processes regulated by miRNAs are quite diverse and enormous Furthermore plant
miRNAs work cooperatively or antagonistically to establish a balanced regulation This
notion is well consistent with the findings that mutations in the regulators of miRNA
biogenesis transport and processing such as AGO1 DCL1 HYL1 HEN1 ABH1
and HASTY cause pleiotropic phenotypes (Mallory amp Vaucheret 2006 de Alba et al
2013) To date the roles of miRNAs have been mostly deduced from obtaining miRNA
over expressing transgenic plants or gain-of-function mutants in which miRNA-
resistant target genes are ectopically expressed
11361 Phase transition
Plants pass through a series of developmental phase transitions during their life span
beginning with seed germination continuing through vegetative phase change
reproductive phase change flowering initiation and finally seed production for the next
generation Because of their importance in understanding plant developmental
Chapter 1 Introduction and Review of Literature
42
processes genes regulating developmental transitions have been extensively studied
and underlying molecular mechanisms have been explored in various plant species
Majority of researches were mainly focused on vegetative phase change and
reproductive transition in Arabidopsis and Maize (Poethig 2003 Baurle amp Dean
2006) Particularly the mutations of the genes encoding various regulators of miRNA
biogenesis and transport such as HST SE and AGO exhibit altered juvenile to adult
vegetative phase change and vegetative to reproductive change (Hunter et al 2003
Peragine et al 2004 Vazquez et al 2004a Jones-Rhoades et al 2006 de Alba et al
2013)
Over-expression of the miR156a gene causes late flowering and delays
vegetative phase change (Wu amp Poethig 2006 Gandikota et al 2007 Wang et al
2008) It has been shown that miR156 negatively regulates a group of genes encoding
SQUAMOSA PROMOTER-BINDING PROTEIN-LIKE (Lewis et al 2007 de Alba et
al 2013 Rogers amp Chen 2013) transcription factors such as SPL3 SPL4 and SPL5
that promote flowering initiation In contrast transgenic plants over expressing
miR156-resistant SPL345 genes are early flowering Interestingly the endogenous
level of miR156 is temporally regulated In the early juvenile vegetative phase the
level of miR156 is very high but decreases rapidly before the onset of the adult juvenile
phase (Wu amp Poethig 2006)
The Arabidopsis early activation tagged (eat-D) mutant exhibits early flowering
with disrupted floral structure The mutant phenotypes are caused by over expression of
the miR172a-2gene (Aukerman amp Sakai 2003) MiR172 regulates a small group of
genes encoding APETALLA2 (AP2)-like transcription factors such as TARGET OF
EAT1 (TOE1) TOE2 SCHLAFMUTZE (SMZ) and SCHNARCHZAPFEN(SNZ)
TOE1 TOE2 SMZ and SNZ have been shown to participate in their productive phase
change by repressing a floral integrator FLOWERING LOCUS T (FT)(Jung et al
2007) Consistent with this toe1-2D 35STOE2 35SSMZ and 35SSNZ plants
exhibit late flowering (Jung et al 2007) MiR172 is also regulated temporally and
highly expressed in the adult vegetative phase both in Arabidopsis and maize (Chuck et
al 2007) Because the temporal expression patterns of miR156 and miR172 are
complementary to each other it has been suggested that the two miRNAs interact with
each other in regulating phase change (Chuck et al 2009 de Alba et al 2013) In
support of this view it has been shown that miR172 is down-regulated in the
Chapter 1 Introduction and Review of Literature
43
Corngrass1(Cg1) mutant in which miR156 is overproduced (Chuck et al 2007 de
Alba et al 2013) However there is no direct evidence for the direct linkage between
miR156 and miR172 It is also envisioned that miR156 and miR172 would not be
directly related For example miR156 regulates plastochron index that is the inverse of
leaf initiation rate and thus frequently used to analyze temporal patterns of plant
development In contrast miR172 does not affect plastochron (Wang et al 2008) The
down-regulation of miR172 in the maize miR156-over producing mutants would be
due to the prolonged juvenile vegetative phase in the mutants
Another miRNA that regulates flowering initiation is miR159 It regulates
MYB33 and MYB65 which are closely related to the barley GAMY B transcription
factor that responds to gibberellic acid (GA) The level of miR159 is elevated in GA-
treated seedlings but reduced in the GA biosynthetic ga1 mutant Transgenic plants
over producing miR159 exhibit delayed floral induction under short days (Achard et
al 2004) It is been suggested that MYB33 mediates GA signal to regulate LEAFY
(LFY) These observations indicate that miR159 plays an important role in the GA
signaling pathways that regulate proper floral induction under short days (Achard et al
2004)
11362 Floral development
Arabidopsis flowers consist of four distinct organs They arise in a characteristic
pattern within concentric rings called whorls from outside to inside four sepals four
petals six stamens and the central pistil consisting of two carpels Identities of the
floral organs are determined by three classes of regulatory genes classA classB and
class C genes (Krizek amp Fletcher 2005) The A and C class genes act antagonistically
to restrict their expression domains (Bowman et al 1991)
It is notable that miR172 also regulates floral architecture in addition to its role
in the timing of flowering initiation MiR172 inhibits the translation of the AP2 mRNA
(Chen 2004) Transgenic plants overproducing miR172 not only exhibit early
flowering but also show abnormal floral development which is quite similar to those
of the class A gene mutants such as the ap2 mutants (Aukerman amp Sakai 2003 de
Alba et al 2013) When the activity of the class A genes is inactivated that of the class
C genes such as AGAMOUS (AG) is elevated As a result the miR172- overproducing
transgenic plants exhibit no petals along with homeotic transformation of the sepals to
Chapter 1 Introduction and Review of Literature
44
the carpels (Aukerman amp Sakai 2003 Chen 2004)
Interestingly miR166 also regulates floral architecture The role of miR165166
in the regulation of meristem activity is closely linked to floral meristem formation and
organization (Zhao et al 2007 Miyashima et al 2013) Floral structure is severely
disrupted in the miR165166-overproducing mutants such as men1 and jba-1D in
which miR166 is overproduced (Williams et al 2005b Jung amp Park 2007) The men1
and jba-1D mutants have smaller gynoecium and reduced carpel number In an extreme
case the jba-1D homozygous plants lack visible carpels (Williams et al 2005b)
Similar phenotypes have been observed in the miR165 overproducers (Zhao et al
2007) It has been suggested that the phenotypes are possibly caused by altered AG
expression in the floral meristem (Jung amp Park 2007 Turchi et al 2013)
Other miRNAs also affect floral development Overproduction of miRNAs
involved in auxin signaling also affects floral architecture Transgenic plants
overproducing miR167 have defects in the formation of ovule and anther with reduced
fertility (Ru et al 2006) It has been shown that miR167 targets ARF6 and ARF8 that
play a role in gynoecium and stamen development and in regulating fertility (Ru et al
2006 Wu et al 2006) These observations suggest that auxin signals are also closely
related to floral organogenesis In addition transgenic plants over-producing miR164
and the cuc1cuc2 double mutants show fused sepals and reduced petals (Laufs et al
2004 Turchi et al 2013) suggesting that miR164 is also related to floral meristem
activity and boundary specification of the meristematic region
11363 Shoot apical meristem development
Shoot apical meristem comprises self-maintaining totipotent cells The shoot apical
meristem activity affects diverse aspects of plant developmental processes including
leaf development vascular development and lateral organ polarity (Bowman et al
2002) Because miR165166 plays a primary role in meristem formation
overproduction of miR165166 and multiple knockout mutants of its target genes
exhibit pleiotropic pheonotypes (McConnell et al 2001 Emery et al 2003 Kim et al
2005 Williams et al 2005b)
The targets of miR165166 include genes encoding the homeo domain-leucine
zipper (HD-ZIP) class III transcription factors such as PHABULOSA(PHB)
PHAVOLUTA (PHV) REVOLUTA(REV) ATHB15CORONA (CNA) and ATHB8
Chapter 1 Introduction and Review of Literature
45
PHB PHV and ATHB15 have overlapping functions in controlling meristem
development (Turchi et al 2013) Indeed the phb phv athb15 triple mutants have an
enlarged shoot apical meristem Consistent with this the miR166- overproducing men1
and jba-1D mutants exhibit enlarged shoot apical meristems (Kim et al 2005
Williams et al 2005b Turchi et al 2013) In the mutants the shoot apical meristems
are multiplied and large
The development of shoot apical meristems is also regulated by the WUSCHEL
(WUS)ndashCLAVATA (CLV) signaling pathway WUS positively regulates cell
proliferation In contrast CLV3 which is expressed in stem cells limits the WUS
expression and thus inhibits cell proliferation in the SAM (Sharma et al 2003)
Consistent with this notion WUS is expressed to a higher level in jba-1D
explaining the ectopic enlargement of the SAM in the mutant (Williams et al 2005)
While WUS is closely related to miR165166 in the shoot apical meristem
development the underlying molecular mechanisms are currently unclear It has been
observed that the miR166-over producing men1 plants have an arrested meristem
development (Jung amp Park 2007) In addition the heterozygous men1+ and the
homozygous men1 plants show different expression patterns of WUS and CLV3 It
appears that miR166 regulates WUS indirectly and influences the expressional balance
between WUS and CLV3 through a negative feedback loop (Jung amp Park 2007 de
Alba et al 2013)
The phv phb athb15 triple mutant has an enlarged shoot apical meristems the
rev phb phv mutant does not have functional shoot apical meristems (Prigge et al
2005) This distinction may be explained by the fact that while miR166 targets
primarily PHB PHV and ATHB15 miR165 targets all the five HD-ZIP III genes (Zhou
et al 2007) Consistent with this transgenic plants overproducing miR166 are distinct
from those overproducing miR165 The miR165-overproducing transgenic plants lack
functional shoot apical meristem and a pin-like structure is formed at the apical region
of the mutant These phenotypes are quite similar to rev phb phv triple mutant (Zhou et
al 2007 Liu et al 2013) The phenotypic distinction may because by the difference
of the cleavage efficiencies of the target mRNAs In accordance with this notion a few
35S miR166a transgenic lines exhibit no shoot apical meristems The men1
homozygous plants also show arrested meristem development (Jung amp Park 2007)
Chapter 1 Introduction and Review of Literature
46
MiR164 cleaves the CUC1 and CUC2 mRNAs as well as the NAC1 mRNA
CUC1 and CUC2 determines the organ boundary in the meristem Phenotypes of
transgenic plants that overproduce miR164 are similar to those of the cuc1cuc2 double
mutant that exhibits embryonic developmental defects such as cup-shaped cotyledons
and fused cotyledons (Laufs et al 2004) When a miR164-resistant CUC2 is
expressed the boundary domain is enlarged CUC also plays a role in embryonic shoot
meristem formation by regulating the SHOOT MERISTEMLESS gene It was therefore
concluded that miR164-mediated cleavage of CUC1 and CUC2 mRNAs determines the
sizes of the boundary domains in the shoot apical meristem and regulates separation of
the organs by restricting cell proliferation (Laufs et al 2004 de Alba et al 2013)
11364 Leaf development
The role of miR319 has been extensively studied in leaf development A dominant
jaw-D mutant overproducing miR319 shows uneven curved leaves with enlarged size
and five TCP genes which are closely related to the CINCINNATA (CIN) gene in
Antirrhinum majus are down regulated in the mutant suggesting that miR319 mediated
regulation of the TCP transcription factor genes are important for the final step of
leaf shaping (Palatnik et al 2003 Naz et al 2013) In addition to leaf shape and size
leaf polarity is also specified by miRNA MiR166-mediated cleavage of the HD-ZIPIII
mRNAs is a well to establish leaf polarity This discovery originates from the gain-of-
function mutants such as phb-1d and phv-1d wherein point mutations in the
complementary sites to miRNA helps the mRNA to evade or resist from miRNA-
mediated cleavage (McConnell et al 2001 Emery et al 2003 Naz et al 2013)
Plant leaves have a dorso-ventral polarity consisting of the adaxial (upper
surface) and abaxial (lower surface) sides The adaxial side is closer to the shoot
apical meristem Adaxial identity is specified by functionally redundant class III HD-
ZIP family genes such as PHB PHV and REV Ectopic expression of each genes
result in adaxialized leaves Dominant phb-1d and phv-1d mutants exhibit
transformation of the abaxial to adaxial fate and have rod-shaped leaves In contrast
miR165166-overproducing plants show pin-like cotyledons radicalized leaves and
abaxialized leaves (Chitwood et al 2007 Pattanayak et al 2013)
Leaf asymmetry is determined by miR166-mediated restriction of the
expression domains of the HD-ZIPIII genes The HD-ZIPIII genes are expressed in the
Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
16
19 Catabolism of caffeine
Caffeine is produced in young leaves and immature fruits and continues to accumulate
gradually during the maturation of these organs At the same time caffeine is slowly
degraded with the removal of the three methyl groups resulting in the formation of
xanthine Catabolism of caffeine was first reported in coffee leaves (Kalberer 1965)
Since then a number of tracer experiments using 14
C labelled purine alkaloids have
been reported (Suzuki amp Waller 1984 Ashihara et al 1996 Vitoria amp Mazzafera
1999 Mazzafera 2004) They demonstrated the major catabolic pathway is caffeine rarr
theophylline rarr 3-methylxanthine rarr xanthine Xanthine is further degraded by the
removal by the conventional purine catabolic pathway to CO2 and NH3 via uric acid
allatonin and allantoate (Figure 15) (Ashihara amp Crozier 1999 Stasolla et al 2003
Zrenner et al 2006) Caffeine catabolism usually begins with its conversion to
theophylline catalyzed by N7-demethylase The involvement of P450-dependent mono-
oxygenase activity for this reaction was suggested (Huber amp Baumann 1998
Mazzafera 2004) However the activity of this enzyme has not yet been determined in
cell free extracts [8-14
C] Theophylline degraded to CO2 at much faster rate than [8-14
C]
caffeine indicating that conversion of caffeine to theophylline is a major rate limiting
step of caffeine catabolism and a possible reason why caffeine accumulates in high
concentrations in tissues of C sinensis and C arabica It is widely believed that the
control of caffeine levels in plants is a function of the balance between rates of
synthesis and degradation and this balance seems to vary depending on the plant
species and the tissue developmental stage (Mazzafera 2004)
Chapter 1 Introduction and Review of Literature
17
Figure 15 Caffeine catabolic pathways Caffeine is mainly catabolised via theophylline and 3-
methylxanthine Xanthine is further degraded to CO2 and NH3 by the conventional
oxidative pathway Adapted from (Ashihara et al 2008)
Chapter 1 Introduction and Review of Literature
18
110 Genetic engineering of Coffee
Genetic transformation has potential applications in coffee agriculture for incorporating
genes which could provide desirable traits such as disease and insect resistance
drought and frost tolerance and herbicide resistance Transgenic technology can also be
used to increase nutritional value and improve cup quality produce varieties with
caffeine-free beans and for production of hybrid crops for molecular farming
Identification of target specific genes is one of the pre-requisites for developing
transgenic crops The availability of a large number of EST sequences in coffee and
initiation of coffee genome sequencing may speed up the gene discovery and accelerate
transgenic research efforts in coffee
1101 Insect Resistance
Production of insect resistant coffee plants is one of the major objectives of the
breeding programs The major pests attacking coffee include coffee berry borer (CBB
Hypothenemus hampei) white stem borer (WSB Xylotrechus quadripes) leaf miner
(Perileucoptera coffeela) and root nematodes (Meloidogyne spp and Pratylenchus
spp) The CBB is present in almost all the coffee growing countries and considered to
the most devastating pest in coffee To date there is no reported source of resistance to
CBB in the coffee gene pool Like CBB WSB is another serious pest in C arabica in
India and several other East Asian countries Both CBB and WSB belong to the order
Coleoptera For India controlling WSB is the biggest challenge and has therefore
become the highest research priority Robusta is generally resistant to WSB but the
interspecific Robusta Arabica hybrids are susceptible to WSB Although leaf miner is
not yet a serious pest in India and other East Asian countries it is an economically
important pest in East Africa and Brazil Effective chemical control of CBB and WSB
is difficult due to the nature of their life cycle inside the berry and stem respectively as
well as environmental concern regarding the use of pesticides Biological control
measures are adapted to combat these insect pests with varying degrees of success
Developing coffee plants resistant to these pests using genetic transformation
technology could be one of the alternative strategies to counter pest damage For insect
resistance several different classes of proteins from bacterial plant and animal sources
have been isolated and their insecticidal properties tested against many important pests
Amongst these proteins Bt toxins are most important and several transgenic crops
expressing Bt genes have been commercialized Coffee transgenic plants carrying a
Chapter 1 Introduction and Review of Literature
19
synthetic version of the cry1Ac gene have been produced (Leroy et al 2000 Mishra et
al 2002) Indeed this was the first report of an important agronomic trait being
introduced into a coffee plant The transgenic plants presented similar features in
growth and development compared to normal plants Transgenic plants highly resistant
to leaf miner under greenhouse conditions were tested under field conditions in French
Guyana for 4 years for pest resistance (Perthius et al 2005 Perthius et al 2006)
From 54 independent transformation events 70 of the events were resistant to leaf
miner Unfortunately the field trial was vandalized which led to the termination of the
experiment (Montagnon et al 2005)The effectiveness of Bt genes in controlling
coleopteran pests is well documented in corn and potato which indicates that Bt genes
might be effective against CBB The high toxicity of B thuringiensis sero var
israelensis against first in star larvae of CBB has been demonstrated (Mendez et al
2003) In another experiment an α-amylase inhibitor from Phaseolus vulgaris was
tested against CBB and found to have an inhibitory effect on its growth and
development (Grossi et al 2004)In addition to the CBB and WSB C arabica
varieties are also susceptible to endoparasitic root-knot nematode (Meloidogyne spp)
(Campos et al 1990 Mishra et al 2012)
So far 15 species have been reported to be parasites of coffee Controlling
nematodes is extremely difficult and currently seedling grafting with robusta rootstock
is followed as one of the control measure Sources of resistance specific to root-knot
nematodes have been identified in coffee trees (Bertrand et al 2001) and the Mex-1
gene conferring resistance to M exigua in C arabica is in the process of isolation (Noir
et al 2003)
1102 Tolerance to Abiotic Stress
In many coffee growing countries abiotic stress such as drought and frost are the major
climatic factors that limit coffee production Changes in climatic patterns due to global
climate change are considered increasingly important for coffee cultivation Drought
induces water stress in plants which affects vegetative growth and vigor and triggers
floral abnormalities and poor fruit set It also indirectly increases the incidence of pests
and diseases in the plants C arabica is generally more tolerant to water stress than C
canephora partly due to its extensive deep root system C racemosa is known to be a
good source of drought tolerance In India hybrids between C canephora and C
racemosa have been obtained and are currently under evaluation for drought tolerance
Chapter 1 Introduction and Review of Literature
20
In many coffee growing countries coffee is propagated in marginal areas where the
annual rainfall is below 1000thinspmm with prolonged dry spells of over 4-5 months In
those areas water shortage and unfavorable temperatures constitute major constraints
and the growth and productivity of C canephora are badly affected As with drought
periodic frost also affects coffee production in parts of Brazil The introduction of
drought and frost tolerant genes through genetic transformation would be of great
importance for alleviating these problems Research is now being carried out by several
groups to identify genes involved in biotic as well as abiotic stress The most promising
genetic engineering approach for drought tolerance includes the use of functional or
regulatory genes as well as the transfer of transcription factors In recent years plants
tolerant to high temperature and water stress have been the subject of intense research
(Chaves et al 2003 Chaves et al 2004 Coraggio et al 2004) For achieving drought
tolerance genes that have been targeted include those encoding enzymes involved in
detoxification or osmotic response metabolism enzymes active in signaling proteins
involved in the transport of metabolites and regulating the plant energy status (Chaves
et al 2003 Chaves et al 2004 Coraggio et al 2004) The dissection of molecular
mechanisms related to signal transduction and transcriptional regulation might help in
engineering drought tolerance in coffee
1103 Disease Resistance
Coffee leaf rust (CLR) caused by the fungus Hemileia vastatrix is the most important
disease in coffee with substantial loss to coffee production and productivity in all the
coffee growing countries In addition to leaf rust coffee berry disease (CBD) caused by
fungus Colletotrichum kahawae can be a devastating anthracnose leading to substantial
crop loss in Africa Several other fungal and bacterial diseases may also affect coffee
to a extent C arabica is more susceptible to many diseases than C canephora Though
most of the disease control measures rely upon chemical control they are more
expensive and labour intensive The long-term solution is the breeding of resistant
varieties which is the focus of many breeding programs However breeding for disease
resistant varieties is time consuming due to the perennial nature of coffee with its long
gestation period India has a long history of C arabica breeding especially for leaf rust
resistance and is the first country to demonstrate the existence of multiple races of leaf
rust Resistance to CLR is conditioned primarily by a number of major (SH) genes and
coffee genotypes are classified into different resistance groups based on their
Chapter 1 Introduction and Review of Literature
21
interaction with different rust races pathogen (van der Vossen 2001) Currently C
canephora provides the main source of resistance to pests and diseases including CLR
(H vastatrix) and CBD (C kahawae) and therefore used in breeding programs Other
diploid species like C liberica and C racemosa are being used as a source of resistance
to coffee leaf rust and coffee leaf miner respectively (Guerreiro et al 1999
Sreenivasan et al 1989) The development of coffee varieties resistant to major fungal
diseases such as CLR and CBD using transgenic technology will benefit the coffee
industry immensely During the last 15 years significant progress has been made in the
area of host-pathogen interactions (Staskawicz et al 1995 Feys et al 2000
Shivaprasad et al 2012) and many resistance genes involved in recognizing invading
pathogens have been identified and cloned (Takken et al 2000) A number of
signalling pathways induced because of pathogen infection have been dissected (Dong
et al 1998 Navarro et al 2006) Many antifungal compounds synthesized by plants to
combat fungal infection have been identified (Does et al 1998) Understanding the
specific induction of targeted pathways and identification of specific pathways
responsible for particular fungal resistance is important in order to employ this strategy
in transgenic technology The recent investigation of gene expression during coffee leaf
rust infection could provide an insight into the defence pathways operating in coffee
(Fernandez et al 2004 Nardi et al 2006) Efforts have been made to identify and
clone resistance genes from coffee for achieving durable resistance The genetic and
physical map of two resistance genes SH3gene conferring resistance to rust (Prakash et
al 2004) and the Ck1 gene conferring resistance to C kahawae CBD (Gichuru et al
2006) have been established These genes could be used for molecular marker assisted
breeding programs (Mishra et al 2012)
1104 Production of low-caffeine coffee
A widespread belief that the ingestion of caffeine can have adverse effects on
health resulted in the increased demand for decaffeinated coffee (Ashihara amp Crozier
2001) Several method were used to obtain decaffeinated plants by conventional
breeding techniques using low yielding varieties and mutants that accumulated
relatively low amounts of caffeine when compared to the commercially available ones
Low-caffeine and decaffeinated coffee represent around 10 of the coffee sales
around the world (Ribas et al 2006b) The industrial process for coffee decaffeination
can be expensive and affects the original flavour and aroma in coffee (David 2002)
Chapter 1 Introduction and Review of Literature
22
Transgenic coffee plants with suppressed caffeine synthesis using RNA interference
(RNAi) technology have been obtained (Ogita et al 2003 Ogita et al 2004) Specific
sequences in the 3prime untranslated region of the theobromine synthase gene (CaMXMT1)
were selected for construction of RNAi short and long fragments The caffeine and
theobromine content of the transgenic plants were reduced by ~ 70 when compared to
the untransformed plants In C canephora RNAi technology has also been employed
to silence the N-methyl transferase gene involved in caffeine biosynthesis (Vinod et al
2007) Promoter of an N-methyltransferase (NMT) gene involved in caffeine
biosynthesis was cloned (Satyanarayana et al 2005 Satyanarayana 2006) which will
be very useful for studying the regulation of caffeine biosynthesis
1105 Improvement in cup quality
Improvement in coffee cup quality requires elaborate knowledge of the chemical
constituents as well as the metabolic pathways involved in the elaboration of quality
The constituents of coffee beans include minerals proteins carbohydrates caffeine
chlorogenic acids (CGA) glycosides lipids and many volatile compounds that give
flavour to coffee by roasting Among these the role of three major constituents
sucrose CGA and trigonelline have been studied in coffee The sucrose content of
coffee bean is associated with coffee flavour the higher the sucrose content in green
beans the more intense will be the cup flavour (Clifford et al 1985 de Maria et al
1996) The sucrose content of C arabica (82-83) is higher than C canephora (33ndash
40) The sucrose amino acids ratio in green beans determines the profile of volatile
compounds Manipulating sucrose content in coffee bean is therefore important in
improving cup quality The sucrose synthase gene (CcSUS2) from C canephora has
been cloned and sequenced (Leroy et al 2005) This provides an opportunity to
manipulate the sucrose content in coffee Chlorogenic acids are products of
phenylpropanoid metabolism Chlorogenic acids are a group of hydroxycinnamoyl
quinic acids (HQA) formed by esterification between caffeic acids coumaric acids and
quinic acids (Clifford et al 1999) They are present in relatively large quantities in the
coffee bean and are the precursor of phenolic compounds in roasted beans C
canephora beans contain higher CGA (10) compared to C arabica beans (6-7)
CGA are known to have antioxidant properties as well as being associated with disease
resistance (Campa et al 2003) Genetic manipulations of genes involved in CGA
synthesis can serve either of these purpose by up- or down regulating the pathway
Chapter 1 Introduction and Review of Literature
23
Phenylalanine ammonia-lyase (PAL) catalyzes the first step of the phenylpropanoid
pathway leading to the synthesis of a wide range of chemical compounds including
flavonoids coumarins hydroxycinnamoyl esters and lignins (Hahlbrock amp Scheel
1989) Recently the full length cDNA and corresponding genomic sequences of PAL
from C canephora was isolated characterized and functionally validated (Mahesh et
al 2006 Parvatam et al 2006) This has opened up new possibilities for manipulating
the level of the PAL enzyme in coffee which in turn can be useful for improvement of
cup quality and manipulating antioxidant properties in coffee
1106 Fruit Ripening
Uniformity during fruit ripening is decisively related to cup quality in coffee and
consequently to the value of the product Fruits at the ideal ripening stage produce the
best organoleptic characteristics for coffee The presence of over ripened or green fruits
changes the acidity the bitterness and consequently the cup quality In order to
maximize uniform ripening of coffee fruits it is essential to control the action of genes
involved in the last step of maturation process Ethylene is known to trigger ripening
and increasing ethylene biosynthesis is associated with various stages of ripening
process (Protasio et al 2005) To control coffee fruit maturation two of the major
genes involved in ethylene biosynthesis namely ACC synthase and ACC oxidase
have been cloned (Protasio et al 2005 Neupane et al 1999) Introduction of the ACC
oxidase gene in antisense orientation have been achieved in both C arabica and C
canephora The effect of the transgene on ethylene production and fruit maturation has
yet to be reported The inhibition of genes downstream to the initial ethylene burst is
also an option to control coffee fruit maturation (Ribas et al 2006)
111 RNA interferenceRNAi
The regulation of gene expression at post transcriptional level has recently attracted
much attention because of the discovery of the phenomenon called RNA interference
(RNAi) RNAi based silencing techniques are more efficient than antisense mediated
gene silencing (Wesley et al 2001) The effect was first observed in plants wherein
silencing was observed when some copies of a transgene was in the forward orientation
with respect to the promoter and some in the reverse orientation This resulted in the
formation of double stranded RNA in the system The phenomenon in which
experimentally introduced double stranded RNA (dsRNA) leads to the loss of
expression of the corresponding cellular gene by sequence specific RNA degradation
Chapter 1 Introduction and Review of Literature
24
was termed as RNA interference or RNAi The protective effect of RNA silencing in
reducing virus infectivity supports the view that PTGS has evolved as a mechanism to
defend plants against virus infection and also to moderate the possible deleterious
genome-restructuring (insertional) activity of virus-like mobile genetic elements eg
retrotransposons A growing body of biochemical and genetic data has further
demonstrated that plants animals and yeasts share related mechanisms of specific
degradation of RNAs in which double-stranded forms of RNA are initiator molecules
(Brenstein et al 2001)
The key insight into the process of PGTS was provided by the experiments
conducted by Hamilton amp Baulcombe (1999) who identified the product of RNA
degradation as small RNA (siRNA) species of ~25nt of both sense and antisense
polarity SiRNA are formed and accumulate as double stranded RNA molecules of
defined chemical structures Both genetic and biochemical approaches were undertaken
to understand the basis of silencing Genetic screens were carried out in fungus
Neurospora carassa the alga Chalmydomonas reinhardtii and plant Arabidopsis
thaliana to search for detective mutants in PTGS
A combination of results obtained from several in vivo and in vitro experiments
have gelled into a two step mechanistic model for RNAiPTGS The first step referred
to as the RNAi initiating step involves binding of the RNA nucleases to a larges
dsRNA and its cleavage into discrete ~21-25nt RNA fragments This is ATP-
dependent reaction which involved RNase III - type endonucleases which make
staggered cuts in both strands of the dsRNA leaving 3prime overhang of 2 nucleotides with
5prime- phosphate 3prime-hydroxyl and no modification in the sugar phosphate backbone
(Hamilton amp Baulcombe 1999 Elbashir et al 2001) SiRNAs are the direct products
of dsRNA cleavage by the multi-domain RNase III enzyme DICER (Figure 16)
(Brenstein et al 2001 Karthikeyan et l 2013)
In the second step or commonly known as the effector step the double stranded
siRNAs produced in the first step bind to an RNAi specific protein complex to form a
RISC The complex undergoes activation in the presence of ATP so that the antisense
component of the unwound siRNA becomes exposed and allows the RISC to perform
the downstream RNAi reaction
Chapter 1 Introduction and Review of Literature
25
Several enzymes are involved in RNAi based on their role genes that encode for them
are classified into RNAi initiators RNAi effectors RNA dependent RNA polymerases
and silencing genes Two C elegans genes rdeI and rde4 (rde stands for lsquo RNAi
deficientrsquo) are believed to be involved in the initiation step of RNAi The C elegans
rde1 gene is a member of a large family of genes and is homologous to the Neurospora
qde2 (qde stands for ldquoquelling deficientrdquo) and Arabidopsis AGO1 (AGO stands for
agronaute AGO1 was previously identified to be involved in Arabidopsis
development) Although the functions of these genes are not clear a mammalian
member of RDE1 family has been identified as a translation initiation factor (Thakur
2005) Important genes for the effector step of PTGS in C elegans are rde2 and mut7
genes These genes were identified from heterozygous mutant worms that were unable
to transmit RNAi to their homozygous offsprings Worms with mutated rde2 or mut 7
genes show defective RNAi mut-7 gene encodes a protein with homology to the
nuclease domains of RNAase D and a protein implicated in Werner syndrome (a rapid
ageing disease in humans) (Grishok et al 2001 Kamath-Loeb et al 2012s)
Neurospora qde-1 Arabidopsis SDE-1SGS-2 and C elegans ego-1 appear to encode
RNA dependent RNA polymerase (RdRPs) It assumed that this is a proof that an RdRp
activity is required for RNAi Certainly the existence of an RdRp might explain the
remarkable efficiency of dsRNA induced silencing if it amplifies either the dsRNA
prior to cleavage or the siRNAs directly (Muers 2013) In C elegans ego-1 mutants
(ego stands for lsquoenhancer of glp-1) RNAi functions normally in somatic cells but is
defective in germline cells where ego-1 is primarily expressed In Arabidopsis SDE-
1SGS-2 mutants (SGS stands for suppressor of gene silencing) siRNA are produced
when dsRNA is introduced via an endogenously replicating RNA virus but not
introduced by a transgene It has been proposed that perhaps the viral RdRP is
substituting for the Arabidopsis enzyme in these mutants Random degradative PCR
model suggests that an RdRP uses the guide strand of an siRNA as a primer for the
target mRNA generating a dsRNA substrate for Dicer and thus more siRNAs
Several genes controlling RNA silencing in plants have been identified through
genetic screens of Arabidopsis mutants impaired in transgene induced RNA silencing
They encode a putative RNA-dependent RNA polymerase (SGS2SDE1) a coiled
protein (SGS3) a protein containing PAZ and Piwi domains (AGO1) and an RNA
helicase (SDE3) The putative SGS2SDE1 of Neurospora is related to QDE-1 and
Chapter 1 Introduction and Review of Literature
26
EGO-1 of C elegans whereas PAZPiwi protein AGO is related to QDE-2 of
Neurospora RDE-1of C elegans RNA helicase SDE3 is related to SMG-2 of C
elegans and Mut-6 of Chlamydomonas MUT-7 gene of C elegans encodes a protein
similar to Rnase D whereas the Drosophila DICER gene encode a protein similar to
RNase III An Arabidopsis ortholog of DICER gene has been identified called CAF
SIN1 SUS1(Thakur 2005 Poethig 2009)
Since the RNAi machinery is present constitutively within the eukaryotic cell it
is important to explore the metabolic advantages that are accorded by the RNAi related
proteins during the intrinsic normal growth of cells and development of organisms The
natural RNAi machinery not only keeps the mobile transposable elements from
disrupting the integrity of the genomes as suggested by the analysis of model organisms
like A thaliana C elegans D melanogaster (Tabara et al 1999 Wu-Scharf et al
2000 Aravin et al 2001 Hamilton et al 2002 Lisch 2009) but also participates in
development of organisms Genetic defects in Celegans RNAi genes ego1 and dicer
cause known specific developmental errors (Grishok et al 2000 Grishok et al 2001
Knight amp Bass 2001 Hall et al 2013) Similarly the Argonaute family of genes of A
thaliana (especially the ZWILLE proteins) is also responsible for plant architecture and
meristem development (Carmell et al 2002 Hamant and Pautot 2010) and the Dicer
homologue of A thaliana CAF1 is required for embryo development (Golden et al
2002 Nakano et al 2011) Thus genetic evidence illustrates the role of the RNAi
machinery as a controller of development related genes The mechanistic details of
these developmental processes are beginning to emerge
Chapter 1 Introduction and Review of Literature
27
Figure 16 Mechanism of gene silencing induced double stranded RNA The dicer enzyme to
produce siRNAs cleaves the RNA The siRNA generated bind to the nuclease complex
RISC The antisense component in the siRNA in the RISC guides the complex towards
the cognate mRNA resulting in endonucleolytic cleavage of the mRNA
Chapter 1 Introduction and Review of Literature
28
112 MicroRNA or MiRNA
In 1991 Ambros and co-workers first isolated a lin-4 mutant of Celegans which was
arrested at the first larval stage (Lee et al 1993) Later on the let-7 mutation was
isolated in the same system which was responsible for development through the fourth
larval stage Both lin-4 and let-7 encode short 22-nucleotide mature RNAs and were
called short temporal RNA because they control the temporal development program of
C elegans The mature lin-4 RNA defines (negatively regulates) the mRNA expression
of the lin-14 and lin-28 hetrochronic genes with the antisense mediated repression
mechanism of translation initiation and thus specifies the fate of cells during the first
three larval stages Recent studies have revealed that the short temporal RNAs are
actually members of a group of tiny RNAs (21to 28 nucleotides) called the micro-
RNAs (miRNA) (Bartel 2009) isolated members of which could easily run to a few
thousand which includes both those were identified computationally and by deep
sequencing Some of the components of the RNAi machinery have also been clearly
established as the effector proteins for the maturation of miRNAs
113 MicroRNAs in plants
1131 Discovery
MicroRNAs have been discovered by three basic approaches Direct cloning forward
genetic direct cloning and bioinformatic prediction followed by experimental
validation The most direct method of miRNA discovery is to isolate and clone small
RNAs from biological samples and several groups have used this approach to identify
plant miRNAs (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002 Xie et al 2003 Sunkar et al 2005 Williams et al 2013) The cloning methods
adapted were similar to those used to identify large numbers of animal miRNAs
(Lagos-Quintana et al 2001 Lau et al 2001 Aravin amp Tuschl 2005) which involve
isolating small RNAs followed by oligonucleotide adapter ligation reverse
transcription amplification and sequencing Some protocols incorporate methods to
enrich for Dicer cleavage products (ie molecules with 5prime-phosphates and 3prime-
hydroxyls) and to concatemerize the short cDNAs so that several may be identified in a
single sequencing read (Lau et al 2001 Llave et al 2002a Jagadeeswaran amp Sunkar
2013)The initial cloning experiments in Arabidopsis identified 19 miRNAs belonging
to 15 families (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002) although hundreds of other small RNAs such as siRNAs and RNA degradation
Chapter 1 Introduction and Review of Literature
29
fragments were also cloned through the direct cloning methods To select miRNAs
from the pool of small RNAs secondary structures of the RNA molecules were
examined in compliance with the acknowledged structural features of miRNAs
(Ambros et al 2003) The secondary structures were validated using several ad-hoc
software tools such as the Mfold or RNAfold programs (Reeder et al 2006) Finally
the existence of mature miRNAs was determined by RNA gel blot hybridization These
direct cloning methods were successful in isolating many miRNAs in Arabidopsis
(Reinhart et al 2002 Sunkar amp Zhu 2004) Rice (Sunkar et al 2005) Poplar (Lu et
al 2005) moss (Arazi et al 2005) and Tobacco (Billoud et al 2005)
Although miRNAs were first discovered through forward genetic screens in
worms (Lee et al 1993 Reinhart et al 2000)only few A thaliana miRNA gene
families have been discovered by this method (Chen 2008) in plants MiRNA
involvement in plant mutant phenotypes were not properly inferred till the cloning
experiments established that plant genomes contain numerous miRNAs (Park et al
2002 Reinhart et al 2002 Rhoades et al 2002) MiRNA loss-of-function allele has
been identified by forward genetic screens early extra petala1 is caused by a
transposon insertion ~160bp upstream of the predicted miR164c stem loop resulting in
flowers with extra petals (Baker et al 2005 Irish 2008) The fact that loss-of-function
miRNA mutants have been rarely discovered using forward genetics reflects small
target size for mutagenesis coupled with redundancy in nearly all evolutionarily
conserved plant miRNAs which are encoded by gene families (Table16) Family
members are likely to have overlapping functions buffering against loss at any single
miRNA locus Over expression screens can circumvent redundancy limitations At least
four plant miRNAs miR319 (also known as miR-JAW) miR172 (also known as EAT)
and miR166 were isolated in over expression screens for dominant mutants with
developmental abnormalities (Aukerman amp Sakai 2003 Palatnik et al 2003 Kim et
al 2005 Williams et al 2005b Schommer et al 2012) Mutations in miRNA target
sites which can prevent the entire family of miRNAs from repressing a target gene can
also circumvent redundant functions of miRNA family members
The dominant mutations in the HD-ZIP genes PHB PHV and REVOLUTA
(REV) in Arabidopsis and ROLLEDLEAF1 (RLD1) in Maize result in adaxialization of
leaves andor Vasculature (McConnell amp Barton 1998 McConnell et al 2001 Nelson
et al 2002 Zhong amp Ye 2004 Allen amp Millar 2012) and are all caused by mutations
Chapter 1 Introduction and Review of Literature
30
in miR166 complementary sites (Rhoades et al 2002 Emery et al 2003 Jones-
Rhoades amp Bartel 2004 Mallory et al 2004 Zhong amp Ye 2004)
In both plants and animals cloning was the initial means of large-scale
miRNA discovery (Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Reinhart et al 2002 Mendes et al 2012) However cloning is biased toward
RNAs that are expressed highly and broadly MiRNAs expressed at low levels or
only in specific cell types or in response to certain environmental stimuli were more
difficult to clone Sequence based biases in cloning procedures might also cause
certain miRNAs to be missed Because of these limitations bioinformatics approaches
to identify miRNAs have provided a useful complement to cloning
A straightforward use of bioinformatics has been carried out to find homologs
of known miRNAs both within the same genome and in the genomes of other species
(Pasquinelli et al 2000 Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Mendes et al 2009) A more difficult challenge is to identify miRNAs
unrelated to previously known miRNAs This was first accomplished for
vertebrate nematode and fly miRNAs using algorithms that search for conservation
of sequence and secondary structure (ie miRNA stem-loop precursors) between
species searching for patterns that are characteristic of miRNAs (Lai et al 2003 Lim
et al 2003 Lim et al 2005)
Most of the methods identified numerous miRNAs but are necessarily more
relaxed in terms of allowed structures Some approaches take advantage of the high
complementarity of plant miRNAs to target messages implementing the requirement
that the candidate has conserved complementarity to mRNAs (Jones-Rhoades amp Bartel
2004 Adai et al 2005) This additional filter has been useful for distinguishing
authentic plant miRNAs from false positives which later extended to mammalian
miRNA gene prediction (Xie et al 2005 Mendes et al 2012)
Another criteria used by most algorithms in the filters is evolutionary
conservation In plants mature miRNA sequences are conserved to a higher degree
than their precursor sequences For example Arabidopsis and rice diverged more than
130 million years ago (Friis et al 2004) but some miRNAs are highly conserved
in these plant species (Reinhart et al 2002) Furthermore various parameters for the
secondary structures such as free energy the number of paired residues within a
Chapter 1 Introduction and Review of Literature
31
miRNA or the number and size of bulges are also used to validate the predictions
(Jones-Rhoades amp Bartel 2004 Wang et al 2004 Adai et al 2005 Mendes et al
2012)
1132 Conserved microRNAs in Plants
Till date cloning genetics and bioinformatics screens have resulted in annotation of
approximately 184 potential Arabidopsis miRNAs (miRBase release 13) These
miRNAs seem to arise through gene duplication and subsequent diversification and
represent approximately 100 families (Maher et al 2006 Axtell 2013) Twenty one
families represented by 92 genes are clearly conserved in species beyond Arabidopsis
(Table 15) The presence of these miRNA families in other sequenced plant genomes
indicates that different plant species have an evolutionarily fluid set of miRNAs
(Willmann amp Poethig 2007) Recent studies on mosses one of the most ancient land
plants have shown that at least 14 miRNA families are conserved in eudicots and
mosses and therefore it appears that plant miRNAs share a common ancient origin
(Arazi et al 2005 Axtell amp Bartel 2005 Talmor-Neiman et al 2006 Zhang et al
2006 Axtell 2013) The number of members per family in a genome ranges from 1-32
with the exception of miR-430 which is represented by a cluster of ~80 loci in zebra
fish (Giraldez et al 2005)
In plants the number of members in each family correlates among examined
species certain families contain numerous members in all three species (eg miR156
miR166 miR169) whereas others consistently contain fewer genes (eg miR162
mir168 miR394) (Table 16) Although it is still unclear why certain miRNA are
encoded by more than one gene in plants (eg 12 genes encoding miR156) this
correlation might suggest functional significance of various miRNA family sizes Most
annotated plant miRNAs are conserved throughout flowering plants while a few
miRNAs are found only in a single sequenced genome and thus they could be
evolutionarily lsquoyoungrsquo miRNAs For example Arabidopsis has several miRNA
families that are not found in poplar or rice suggesting that miRNAs could be
generated continuously during evolution (Allen et al 2004a Rajagopalan et al
2006 Fahlgren et al 2007 Axtell 2012 de Alba et al 2013) Consistent with the
notion that non conserved miRNAs are of a more recent evolutionary origin these
miRNAs are mainly encoded by a single locus in the genome Within each family the
mature miRNA is always located in the same arm
Chapter 1 Introduction and Review of Literature
32
Table 16 MicroRNA families conserved in plants
miRNA
family
Arabidopsis Oryza Populus
miR156 12 12 11 miR159319 6 8 15 miR160 3 6 8 miR162 2 2 3 miR164 3 5 6 miR166 9 12 17 miR167 4 9 8 miR168 2 2 2 miR169 14 17 32 miR171 4 7 10 miR172 5 3 9 miR390 3 1 4 miR393 2 2 4 miR394 2 1 2 miR395 6 19 10 miR396 2 5 7 miR397 2 2 3 miR398 3 2 3 miR399 6 11 12 miR408 1 1 1 miR403 1 0 2 miR437 0 1 0 miR444 0 1 0 miR445 0 9 0 Total 92 127 169
All known miRNA families that are conserved between more than one plant species are listed
together with the number of genes identified in the sequenced genomes Rice miRNA families that have orthologs in maize but do not appear to have orthologs in the eudicots (Arabidopsis and Populus) are marked with an asterisk The following families contain miRNA genes annotated with
more than one number miR156 (miR156 and miR157) miR159319 (miR159 and miR319) miR166 (miR165 and miR166) miR171 (miR170 and miR171) and miR390 (miR390 and miR391)
of the stem loop (5prime- or 3prime) as it would be expected if the genes carry common ancestry
(Figure 17) The sequence of the mature miRNA and to some extent the segment on
the opposite arm of the hairpin to which it pairs is highly conserved between the
members of the same family (both within and between the species) while the sequence
secondary structure and length of the intervening ldquolooprdquo region can be highly divergent
within the same family
The patterns of paring and non pairing nucleotides are often conserved between
homologous miRNA stem loops from different species (Figure 17) The significance of
conserved mismatches is opined to guide DCL1 to cleave at the appropriate positions
along the stem loop
Chapter 1 Introduction and Review of Literature
33
Figure 17 Representative stem loop of miR164 Segments corresponding to the mature miRNAs
are shown in red (Adapted from Rajagopalan et al 2006)
Although many miRNAs are highly conserved in plants the degree of conservation in
most cases is quite low between plants and animals one exception is miR854 A recent
study has revealed that miR854 is conserved between Arabidopsis and animals and that
the Arabidopsis miR854 differs from the human miR854 only by a single nucleotide
(Arteaga-Vazquez et al 2006 Axtell 2012 de Alba et al 2013) The Arabidopsis
miR854 has multiple binding sites in the 3prime-untranslated region (UTR) of
OLIGOURIDYLATE-binding-PROTEIN mRNA 1bA (UBP1b) and represses the
translation of the UBP1b mRNA The animal miR854 is also complementary to a site in
the 3prime-UTR of the UBP1b homolog in animals However it is currently unclear how the
animal miR854 regulates its target mRNA Another distinction between plant and
animal miRNAs is the genomic organization of the miRNA genes While plant miRNA
genes are located predominantly in the intergenic regions animal miRNA genes tend to
localize in the intragenic regions both in the introns or exons of protein coding genes
Moreover miRNA genes are often clustered within a single precursor transcript
whereas individual plant miRNAs are derived from individual precursor RNAs (Bartel
Chapter 1 Introduction and Review of Literature
34
2004 Bonnet et al 2004 Kim 2005 Vaucheret et al 2006 Marco et al 2013)
1133 Non-conserved miRNA and challenges in annotation
Most miRNAs are conserved throughout flowering plants (Table 15) but many more
were found only in a single sequenced genome thus considered to be of a more recent
evolutionary origin The extended homology between few non conserved miRNAs
precursors and their target genes provides strong evidence that these relatively ldquoYoungrdquo
miRNAs arose from the duplication of the target gene segments (Allen et al 2004)
Several non-conserved miRNAs including miR161 miR163 miR173 miR447
miR475 and miR476 are known to direct cleavage of target transcripts (Allen et al
2004a Adai et al 2005 Sunkar et al 2005 de Alba et al 2013) It is difficult to
confidently predict targets for many because it is not possible to use complementary
site as a filter against false-positive target predictions It is also difficult to
confidently annotate non-conserved miRNAs as miRNA rather than siRNAs The
established minimal standard for miRNA annotation is a small RNA with detectable
expression and the potential to form a stem-loop when joined to flanking genomic
sequence (Kasschau et al 2003) In practice these requirements are too loose to
definitively categorize many small RNAs cloned from plants Many plant siRNAs are
detectable on blots (Vazquez et al 2004b) and hundreds of thousands of non-
miRNA genomic sequences can be predicted to fold into secondary structures that
resemble structures of plant miRNA precursors (Jones-Rhoades amp Bartel 2004)
Therefore without conservation of both sequence and secondary structure it is
difficult to be confident that a given cloned RNA originated from a stem-loop (ie is a
miRNA) rather than from a double-stranded RNA (ie is a siRNA) In fact many of
the thousands o f small RNAs cloned from Arabidopsis (Gustafson et al 2005) would
probably meet the literal requirements for annotation as miRNAs A few of these
sequences might be miRNAs but others that meet the literal criteria probably are not
so The challenges of annotating non conserved small RNAs were evidenced by
three related small silencing RNAs that were originally annotated as miRNAs
that turned out to be trans-acting siRNAs (tasiRNAs)(Kanno et al2013)
Although most miRNAs that are broadly conserved among flowering plants are
now being identified and experimentally validated (Table 16)(Jones-Rhoades amp
Bartel 2004) the challenges and ambiguities for classifying non-conserved miRNAs
prevent meaningful estimates of the total number of miRNA genes in Arabidopsis and
Chapter 1 Introduction and Review of Literature
35
other plant genomes It is possible that miRNAs might have escaped detection
because they are non-conserved and expressed in specific tissues or conditions and can
only be identified by using high- throughput sequencing
1134 MicroRNA biogenesis
11341 Transcription of microRNA precursor
Plant miRNAs are primarily found in the genomic regions that are not associated with
the protein coding genes (Reinhart et al 2002) and it appears that most if not all plant
miRNAs are produced from their own transcriptional units Plant miRNA genes are
occasionally clustered near each other in the genome suggesting transcription of
multiple miRNAs from a single primary transcript [eg the miR395 cluster (Grishok
et al 2001 Jones-Rhoades amp Bartel 2004b)] but this polycistronic arrangement of
miRNA genes appears far less frequently in plants than in animals (Bartel 2004)
Northern blot EST and mapping evidence indicate that plant miRNA primary
transcripts (also known as pri-miRNAs) as in animals are longer than needed to
encompass the miRNA stem-loops (Palatnik et al 2003 Jones-Rhoades amp Bartel
2004b) At least some of these pri-miRNA transcripts appear to be spliced
polyadenylated (Kurihara amp Watanabe 2004) and capped (Xie Z et al 2005) Two
rice miRNAs are contained within transcripts that contain exon junctions within the
presumptive stem-loop precursor implying that in these cases splicing is a prerequisite
for Dicer recognition (Sunkar et al 2005) Plant pri-miRNAs are over 1kb in length
and they are usually preceded by typical TATA box motifs They can also undergo
canonical splicing polyadenylation and capping All these observations indicates RNA
polymerase II is probably responsible for transcribing most plant miRNAs (Xie Z et
al 2005) as appears to be the case for many animal miRNAs Relatively little is
known about the regulation of miRNA transcription in plants but there is no reason
to suspect that this regulation would differ from that of protein-coding transcripts as
the bioinformatics analysis of the sequence upstream of the transcriptional start
sites of the miRNA genes showed the presence of putative binding motifs of
number of known transcription factors (Megraw et al 2006 Sethupathy 2013)
11342 MicroRNA processing and export
In plants maturation of miRNA is a step wise process A miRNA gene is transcribed to
pri-miRNA which is much longer than the pre-miRNA possessing the characteristic
stem-loop structure Like transcription of most protein-coding genes the miRNA
Chapter 1 Introduction and Review of Literature
36
transcription is processed by RNA polymerase II (Pol II) enzymes and the pri-
miRNAs are 5prime-capped and 3prime-polyadenylated (Lee et al 2004 Xie Z et al 2005)
Mature miRNAs are produced from pri-miRNAs through at least two sequential
processing steps by RNase III-type endonucleases In animals pri-miRNAs are first
processed at the bottom portion of the stem by Drosha a nuclear-localized RNase III
enzyme to liberate pre-miRNAs The pre-miRNAs are then exported to the cytoplasm
with the help of exportin-5The pre-miRNAs are further processed by Dicer another
RNaseIII enzyme to a duplex consisting of the miRNA and the antisense strands
(miRNA) Since plants do not have any Drosha homologs the two sequential
processing steps seem to be carried out by DCL1 a Dicer homolog in the nucleus
(Kurihara amp Watanabe 2004 de Alba et al 2013 Rogers amp Chen 2013)
The processing of pri-miRNAs to pre-miRNAs by DCL1 is assisted by two
other proteins HYPONASTY LEAVES1 (HYL1) and SERRATE (SE) HYL1 belongs to a
family of double-stranded RNA (dsRNA) binding protein and interacts with DCL1 in
vitro and in vivo (Lu amp Fedoroff 2000 Hiraguri et al 2005 de Alba et al 2013
Rogers amp Chen 2013) Loss-of-function mutations in the HYL1 gene result in
decreased accumulation of miRNAs and concomitant accumulation of pri-miRNAs
(Han et al 2004 Vazquez et al 2004a Kurihara et al 2006) SE a C2H2 zinc finger
protein interacts with DCL1 and HYL1 and plays a role in the processing of pri-
miRNAs to pre-miRNAs (Lobbes et al 2006 Yang L et al 2006) In addition the
nuclear cap-binding complex proteins CBP20 and CBP80 that are known as ABA
HYPER SENSITIVE 1(ABH1) have been shown to be required for proper miRNA
processing (Gregory et al 2008 Laubinger et al 2008) Recently it has been
demonstrated that DCL1 can release 21-nt RNAs from dsRNA as well as from
synthetic miR167 precursor RNAs in vitro However this cleavage is error prone and
inefficient in the absence of HYL1 and SE which accelerate the rate of DCL1-mediated
cleavage and promote accurate processing of miRNAs (Dong et al 2008 Rogers amp
Chen 2013)
11343 Methylation of the miRNAmiRNAduplex
After the miRNAmiRNAduplexes are released from the pre-miRNAs by the DCL1
activities the duplexes are methylated at the 2primeOH of the3prime-ends by HUA ENHANCER1
(HEN1) a small RNA methyltransferase (Yu et al 2005 Yang Z et al 2006 Rogers
amp Chen 2013 ) This step which is distinct from the RNase III-mediated processing
Chapter 1 Introduction and Review of Literature
37
step distinguishes the biogenesis of plant miRNAs from that of animal miRNAs
Mutations in the HEN1 gene were first isolated in a morphological screening for floral
development although the HEN1 mutants display pleiotropic phenotypes similar to the
developmental defects observed in the DCL1 mutants (Chen et al 2002 Park et al
2002 Rogers amp Chen 2013)
Figure 18 Biogenesis of microRNA
A Biogenesis of plant MicroRNA B Biogenesis of metazoananimal MicroRNA
The miRNA (red) is incorporated into RISC and the miRNA(blue) in degraded
This enzyme has two dsRNA-binding domains and nuclear localization signal It
is also conserved in fungi and is required for the processing of siRNAs as well as of
miRNAs (Park et al 2002 Boutet et al 2003 Xie et al 2003) Although the HEN1
protein is localized to the nucleus its methylation activity in the cytoplasm cannot be
ignored (Xie et al 2004 Rogers amp Chen 2013)
Chapter 1 Introduction and Review of Literature
38
The methylation of the miRNAmiRNA duplex is required to protect miRNAs
against the 3prime-end uridylation activity and subsequent degradation (Li et al 2005) In
the hen1 mutants accumulation of miRNAs is reduced to a lower level Also the
miRNAs appear to be heterogeneous in their sizes such that a ladder of bands rather
than a single species is observed for most miRNAs in the mutants (Li et al 2005)
MiRNA cloning and sequencing analyses revealed the presence of a number of U
residues at the 3prime-end of miRNAs in the hen1 mutants Moreover these nucleotides do
not correspond to the miRNAs in the pre-miRNAs indicating that these nucleotides are
added after the processing of the miRNAs from pre-miRNAs (Li et al 2005)
Uridylation of RNAs has also been proven to be correlated with RNA decay (Shen amp
Goodman 2004 Mullen amp Marzluff 2008) suggesting that uridylation attracts an
exonuclease which degrades miRNAs in plants
11344 MiRNA incorporation in to the RISC and its nuclear export
The methylated miRNAmiRNA duplexes undergo RISC assembly In this process
the miRNA of the duplexes is selectively incorporated into the RISC and the miRNA
appears to be removed and subsequently degraded (Hammond et al 2000 Hutvagner
amp Zamore 2002 Schwarz et al 2003 de Alba et al 2013 Rogers amp Chen 2013)
The ARGONAUTE 1(AGO1) proteins are core components of the RISC complex
They contain two structural domains the N-terminal PAZ domain that binds to the 3prime-
end of single-stranded RNAs and the C-terminal piwi domains that has a structure
similar to that of RNase H (Cerutti et al 2000 Parker et al 2004) Several AGO
proteins possess endonucleolytic activities within the PIWI domain and cleave target
mRNAs in the middle of the site complementary to miRNA (Liu et al 2004 Meister et
al 2004 Miyoshi et al 2005) Mutations in the AGO1 gene cause miRNA target
genes to be ectopically expressed and the levels of some miRNAs are reduced in the
mutants (Kidner amp Martienssen 2004 Vaucheret et al 2004 Kidner amp Martienssen
2005 Ronemus et al 2006) Moreover the phenotype of the AGO1 hypomorphic
alleles display defects in lateral organ polarity and leaf and flower morphology as
observed in the dcl1 mutants and null AGO1 alleles are embryonically lethal
supporting the close relationship between the AGO proteins and miRNA processing
(Kidner amp Martienssen 2004 Vaucheret et al 2004 de Alba et al 2013 Rogers amp
Chen 2013)
The production of miRNA miRNA duplexes occur in the nucleus while
Chapter 1 Introduction and Review of Literature
39
miRNA-RISC complexes are present in the cytoplasm where target mRNAs are
located This discordance of functional sites requires an export step during miRNA
biogenesis HASTY the Arabidopsis homolog of exportin-5 is thought to be involved
in miRNA nuclear export (Bollman et al 2003 Park et al 2005)The hasty mutant
exhibits pleiotropic defects in plant development and reduced accumulation of most
miRNAs as in the DCL1 or AGO1 mutants Howeverit is unclear whether the HASTY
proteins export the miRNA miRNA duplexes or the miRNA-RISC complexes in
plants
11345 Feedback regulation of miRNA biogenesis
MiRNA biogenesis is under feedback regulation such that the DCL1 and AGO1 genes
are themselves regulated by miRNAs DCL1 is itself a target of miR162 which leads to
the cleavage of the DCL1 mRNA (Xie et al 2003 de Alba et al 2013) Consistent
with this the levels of DCL1 mRNA are elevated in the mutants of miRNA processing
genes in which the miR162 abundance is reduced In addition there is a predicted
hairpin structure of miR838 in the 14th
intron of the DCL1 primary transcript
(Rajagopalan et al 2006) This prediction implies that DCL1-mediated processing
on this site can result in the cleavage of the DCL1 primary transcript into two truncated
fragments the N-terminal capped fragment and the C-terminal polyadenylated
fragment If this is the case it is possible that miRNA biogenesis competes with the
splicing event of the DCL1 pre-mRNA
The AGO1 is also regulated by miRNA There is a complementary site for
miR168 binding in the AGO1 mRNA and the transcript level of the AGO1 mRNA is
reduced through an mRNA cleavage mechanism (Vaucheret et al 2006 de Alba et al
2013 Rogers amp Chen 2013) indicating that the AGO1 mRNA levels are tightly
controlled by the levels of the AGO1 protein
1135 Mechanism of miRNA action
11351 MiRNA-directed mRNA cleavage
By forming the miRNA-RISC assembly miRNAs can direct the RISC to regulate gene
expression by two post transcriptional mechanisms mRNA cleavage or translational
repression (Bartel 2004 Jones-Rhoades et al 2006 Dalmay 2013) The degree
of miRNA-mRNA complementarity plays an important role for the
determination of the mechanism to be used A general rule is that while perfect or
Chapter 1 Introduction and Review of Literature
40
near-perfect complementarity induces mRNA cleavage central mismatches trigger
translational repression (Carrington amp Ambros 2003 Jones-Rhoades amp Bartel 2004)
Unlike most animal miRNAs plant miRNAs have near-perfect or perfect
complementarity to their targets and mRNA cleavage is assumed to be a predominant
mechanism by which miRNAs regulate gene expression in plants
In RNA cleavage mechanism miRNAs guide the AGO component of RISC to
cleave a single phosphodiester bond of the target mRNA within the miRNA-binding
site (Llave et al 2002b Tang et al 2003) The truncated fragments are then released
and degraded freeing the RISC to recognize and cleave another target mRNA This has
been validated by the detection of 3prime cleavage products that have 5prime ends that start at the
middle of the complementary regions The mRNA cleavage products are then further
degraded by other mechanisms The 3prime cleavage products of some miRNA targets are
degraded by the 5prime-3prime exonuclease XRN4 an Arabidopsis homolog of the major Yeast
mRNA degrading exonuclease Xrn1p It has been observed that the 3prime cleavage
products of some target mRNAs were accumulated in the xrn4 mutants (Souret et al
2004 Dalmay 2013) However the 3prime cleavage products of other miRNA targets do
not accumulate in the xrn4 mutants indicating that there are also XRN4 independent
mechanisms The 5prime cleavage products are typically untraceable by RNA gel blot
hybridization but can be detected by sensitive PCR based methods It has been found
that the 5prime cleavage products tend to obtain an oligo U tail at the 3prime ends This 3prime U tail
is correlated with shortening of the 5prime cleavage products from the 5prime end leading to the
conclusion that the 3prime uridylation causes 5 prime- 3 prime exonucleolytic decay of the 5prime cleavage
products (Shen amp Goodman 2004)
DCL1 HYL1 and SE interact with each other and are co-localized in the
nuclear bodies termed Dicing bodies or D-bodies in which some pri-miRNA shave
been found suggesting that D-bodies serve as a center for active miRNA processing
(Fang amp Spector 2007 Fujioka et al 2007 Song et al 2007) The miRNA
processing site appears to be distinct from the Cajal bodies that have previously been
found as a siRNA processing site (Pontes amp Pikaard 2008) The D-bodies include
SmD3 and SmB proteins that are found in the Cajal bodies However At collin an
additional marker for the Cajal bodies is not found in the D-bodies Moreover the D-
bodies are present in an At collin mutant lacking the Cajal bodies The pri-miRNAs of
miR171A and miR159A genes have been found to be co-localized with HYL1 and
Chapter 1 Introduction and Review of Literature
41
DCL1 in the D- bodies that are distinct from the Cajal bodies (Song et al 2007)
11352 MiRNA-directed translational repression
Translation repression is another mechanism exerted by plant miRNAs The regulatory
mechanism of miR172 which regulates flowering time and floral organ identity is
consistent with translation repression MiR172 over production does not affect the
transcript levels of APETALA2 (AP2) and AP2- like target mRNAs Instead the AP2
protein level is reduced in the miR172 over expressing mutant (Aukerman amp Sakai
2003 Chen 2004 Dalmay 2013) It was later shown that miR172 can also direct the
cleavage of the target mRNAs although the steady-state mRNA level is unchanged due
to feedback regulation (Schwab et al 2005 Jung et al 2007) It is clear that miR172
regulates its target genes primarily by translational repression rather than mRNA
cleavage Recently it has been reported that miR156157 and miR854 also regulate
their target genes through translational repression (Arteaga-Vazquez et al 2006
Gandikota et al 2007 Dalmay 2013) It was once thought that translational repression
is an exception to the rule However experimental evidence suggest a widespread
coexistence of translational repression and mRNA cleavage (Brodersen et al 2008)
1136 Regulatory roles of miRNAs in plant development
The miRNA target prediction has been a difficult task in order to obtain regulatory
targets without bringing in too many false predictions Plant growth and developmental
processes regulated by miRNAs are quite diverse and enormous Furthermore plant
miRNAs work cooperatively or antagonistically to establish a balanced regulation This
notion is well consistent with the findings that mutations in the regulators of miRNA
biogenesis transport and processing such as AGO1 DCL1 HYL1 HEN1 ABH1
and HASTY cause pleiotropic phenotypes (Mallory amp Vaucheret 2006 de Alba et al
2013) To date the roles of miRNAs have been mostly deduced from obtaining miRNA
over expressing transgenic plants or gain-of-function mutants in which miRNA-
resistant target genes are ectopically expressed
11361 Phase transition
Plants pass through a series of developmental phase transitions during their life span
beginning with seed germination continuing through vegetative phase change
reproductive phase change flowering initiation and finally seed production for the next
generation Because of their importance in understanding plant developmental
Chapter 1 Introduction and Review of Literature
42
processes genes regulating developmental transitions have been extensively studied
and underlying molecular mechanisms have been explored in various plant species
Majority of researches were mainly focused on vegetative phase change and
reproductive transition in Arabidopsis and Maize (Poethig 2003 Baurle amp Dean
2006) Particularly the mutations of the genes encoding various regulators of miRNA
biogenesis and transport such as HST SE and AGO exhibit altered juvenile to adult
vegetative phase change and vegetative to reproductive change (Hunter et al 2003
Peragine et al 2004 Vazquez et al 2004a Jones-Rhoades et al 2006 de Alba et al
2013)
Over-expression of the miR156a gene causes late flowering and delays
vegetative phase change (Wu amp Poethig 2006 Gandikota et al 2007 Wang et al
2008) It has been shown that miR156 negatively regulates a group of genes encoding
SQUAMOSA PROMOTER-BINDING PROTEIN-LIKE (Lewis et al 2007 de Alba et
al 2013 Rogers amp Chen 2013) transcription factors such as SPL3 SPL4 and SPL5
that promote flowering initiation In contrast transgenic plants over expressing
miR156-resistant SPL345 genes are early flowering Interestingly the endogenous
level of miR156 is temporally regulated In the early juvenile vegetative phase the
level of miR156 is very high but decreases rapidly before the onset of the adult juvenile
phase (Wu amp Poethig 2006)
The Arabidopsis early activation tagged (eat-D) mutant exhibits early flowering
with disrupted floral structure The mutant phenotypes are caused by over expression of
the miR172a-2gene (Aukerman amp Sakai 2003) MiR172 regulates a small group of
genes encoding APETALLA2 (AP2)-like transcription factors such as TARGET OF
EAT1 (TOE1) TOE2 SCHLAFMUTZE (SMZ) and SCHNARCHZAPFEN(SNZ)
TOE1 TOE2 SMZ and SNZ have been shown to participate in their productive phase
change by repressing a floral integrator FLOWERING LOCUS T (FT)(Jung et al
2007) Consistent with this toe1-2D 35STOE2 35SSMZ and 35SSNZ plants
exhibit late flowering (Jung et al 2007) MiR172 is also regulated temporally and
highly expressed in the adult vegetative phase both in Arabidopsis and maize (Chuck et
al 2007) Because the temporal expression patterns of miR156 and miR172 are
complementary to each other it has been suggested that the two miRNAs interact with
each other in regulating phase change (Chuck et al 2009 de Alba et al 2013) In
support of this view it has been shown that miR172 is down-regulated in the
Chapter 1 Introduction and Review of Literature
43
Corngrass1(Cg1) mutant in which miR156 is overproduced (Chuck et al 2007 de
Alba et al 2013) However there is no direct evidence for the direct linkage between
miR156 and miR172 It is also envisioned that miR156 and miR172 would not be
directly related For example miR156 regulates plastochron index that is the inverse of
leaf initiation rate and thus frequently used to analyze temporal patterns of plant
development In contrast miR172 does not affect plastochron (Wang et al 2008) The
down-regulation of miR172 in the maize miR156-over producing mutants would be
due to the prolonged juvenile vegetative phase in the mutants
Another miRNA that regulates flowering initiation is miR159 It regulates
MYB33 and MYB65 which are closely related to the barley GAMY B transcription
factor that responds to gibberellic acid (GA) The level of miR159 is elevated in GA-
treated seedlings but reduced in the GA biosynthetic ga1 mutant Transgenic plants
over producing miR159 exhibit delayed floral induction under short days (Achard et
al 2004) It is been suggested that MYB33 mediates GA signal to regulate LEAFY
(LFY) These observations indicate that miR159 plays an important role in the GA
signaling pathways that regulate proper floral induction under short days (Achard et al
2004)
11362 Floral development
Arabidopsis flowers consist of four distinct organs They arise in a characteristic
pattern within concentric rings called whorls from outside to inside four sepals four
petals six stamens and the central pistil consisting of two carpels Identities of the
floral organs are determined by three classes of regulatory genes classA classB and
class C genes (Krizek amp Fletcher 2005) The A and C class genes act antagonistically
to restrict their expression domains (Bowman et al 1991)
It is notable that miR172 also regulates floral architecture in addition to its role
in the timing of flowering initiation MiR172 inhibits the translation of the AP2 mRNA
(Chen 2004) Transgenic plants overproducing miR172 not only exhibit early
flowering but also show abnormal floral development which is quite similar to those
of the class A gene mutants such as the ap2 mutants (Aukerman amp Sakai 2003 de
Alba et al 2013) When the activity of the class A genes is inactivated that of the class
C genes such as AGAMOUS (AG) is elevated As a result the miR172- overproducing
transgenic plants exhibit no petals along with homeotic transformation of the sepals to
Chapter 1 Introduction and Review of Literature
44
the carpels (Aukerman amp Sakai 2003 Chen 2004)
Interestingly miR166 also regulates floral architecture The role of miR165166
in the regulation of meristem activity is closely linked to floral meristem formation and
organization (Zhao et al 2007 Miyashima et al 2013) Floral structure is severely
disrupted in the miR165166-overproducing mutants such as men1 and jba-1D in
which miR166 is overproduced (Williams et al 2005b Jung amp Park 2007) The men1
and jba-1D mutants have smaller gynoecium and reduced carpel number In an extreme
case the jba-1D homozygous plants lack visible carpels (Williams et al 2005b)
Similar phenotypes have been observed in the miR165 overproducers (Zhao et al
2007) It has been suggested that the phenotypes are possibly caused by altered AG
expression in the floral meristem (Jung amp Park 2007 Turchi et al 2013)
Other miRNAs also affect floral development Overproduction of miRNAs
involved in auxin signaling also affects floral architecture Transgenic plants
overproducing miR167 have defects in the formation of ovule and anther with reduced
fertility (Ru et al 2006) It has been shown that miR167 targets ARF6 and ARF8 that
play a role in gynoecium and stamen development and in regulating fertility (Ru et al
2006 Wu et al 2006) These observations suggest that auxin signals are also closely
related to floral organogenesis In addition transgenic plants over-producing miR164
and the cuc1cuc2 double mutants show fused sepals and reduced petals (Laufs et al
2004 Turchi et al 2013) suggesting that miR164 is also related to floral meristem
activity and boundary specification of the meristematic region
11363 Shoot apical meristem development
Shoot apical meristem comprises self-maintaining totipotent cells The shoot apical
meristem activity affects diverse aspects of plant developmental processes including
leaf development vascular development and lateral organ polarity (Bowman et al
2002) Because miR165166 plays a primary role in meristem formation
overproduction of miR165166 and multiple knockout mutants of its target genes
exhibit pleiotropic pheonotypes (McConnell et al 2001 Emery et al 2003 Kim et al
2005 Williams et al 2005b)
The targets of miR165166 include genes encoding the homeo domain-leucine
zipper (HD-ZIP) class III transcription factors such as PHABULOSA(PHB)
PHAVOLUTA (PHV) REVOLUTA(REV) ATHB15CORONA (CNA) and ATHB8
Chapter 1 Introduction and Review of Literature
45
PHB PHV and ATHB15 have overlapping functions in controlling meristem
development (Turchi et al 2013) Indeed the phb phv athb15 triple mutants have an
enlarged shoot apical meristem Consistent with this the miR166- overproducing men1
and jba-1D mutants exhibit enlarged shoot apical meristems (Kim et al 2005
Williams et al 2005b Turchi et al 2013) In the mutants the shoot apical meristems
are multiplied and large
The development of shoot apical meristems is also regulated by the WUSCHEL
(WUS)ndashCLAVATA (CLV) signaling pathway WUS positively regulates cell
proliferation In contrast CLV3 which is expressed in stem cells limits the WUS
expression and thus inhibits cell proliferation in the SAM (Sharma et al 2003)
Consistent with this notion WUS is expressed to a higher level in jba-1D
explaining the ectopic enlargement of the SAM in the mutant (Williams et al 2005)
While WUS is closely related to miR165166 in the shoot apical meristem
development the underlying molecular mechanisms are currently unclear It has been
observed that the miR166-over producing men1 plants have an arrested meristem
development (Jung amp Park 2007) In addition the heterozygous men1+ and the
homozygous men1 plants show different expression patterns of WUS and CLV3 It
appears that miR166 regulates WUS indirectly and influences the expressional balance
between WUS and CLV3 through a negative feedback loop (Jung amp Park 2007 de
Alba et al 2013)
The phv phb athb15 triple mutant has an enlarged shoot apical meristems the
rev phb phv mutant does not have functional shoot apical meristems (Prigge et al
2005) This distinction may be explained by the fact that while miR166 targets
primarily PHB PHV and ATHB15 miR165 targets all the five HD-ZIP III genes (Zhou
et al 2007) Consistent with this transgenic plants overproducing miR166 are distinct
from those overproducing miR165 The miR165-overproducing transgenic plants lack
functional shoot apical meristem and a pin-like structure is formed at the apical region
of the mutant These phenotypes are quite similar to rev phb phv triple mutant (Zhou et
al 2007 Liu et al 2013) The phenotypic distinction may because by the difference
of the cleavage efficiencies of the target mRNAs In accordance with this notion a few
35S miR166a transgenic lines exhibit no shoot apical meristems The men1
homozygous plants also show arrested meristem development (Jung amp Park 2007)
Chapter 1 Introduction and Review of Literature
46
MiR164 cleaves the CUC1 and CUC2 mRNAs as well as the NAC1 mRNA
CUC1 and CUC2 determines the organ boundary in the meristem Phenotypes of
transgenic plants that overproduce miR164 are similar to those of the cuc1cuc2 double
mutant that exhibits embryonic developmental defects such as cup-shaped cotyledons
and fused cotyledons (Laufs et al 2004) When a miR164-resistant CUC2 is
expressed the boundary domain is enlarged CUC also plays a role in embryonic shoot
meristem formation by regulating the SHOOT MERISTEMLESS gene It was therefore
concluded that miR164-mediated cleavage of CUC1 and CUC2 mRNAs determines the
sizes of the boundary domains in the shoot apical meristem and regulates separation of
the organs by restricting cell proliferation (Laufs et al 2004 de Alba et al 2013)
11364 Leaf development
The role of miR319 has been extensively studied in leaf development A dominant
jaw-D mutant overproducing miR319 shows uneven curved leaves with enlarged size
and five TCP genes which are closely related to the CINCINNATA (CIN) gene in
Antirrhinum majus are down regulated in the mutant suggesting that miR319 mediated
regulation of the TCP transcription factor genes are important for the final step of
leaf shaping (Palatnik et al 2003 Naz et al 2013) In addition to leaf shape and size
leaf polarity is also specified by miRNA MiR166-mediated cleavage of the HD-ZIPIII
mRNAs is a well to establish leaf polarity This discovery originates from the gain-of-
function mutants such as phb-1d and phv-1d wherein point mutations in the
complementary sites to miRNA helps the mRNA to evade or resist from miRNA-
mediated cleavage (McConnell et al 2001 Emery et al 2003 Naz et al 2013)
Plant leaves have a dorso-ventral polarity consisting of the adaxial (upper
surface) and abaxial (lower surface) sides The adaxial side is closer to the shoot
apical meristem Adaxial identity is specified by functionally redundant class III HD-
ZIP family genes such as PHB PHV and REV Ectopic expression of each genes
result in adaxialized leaves Dominant phb-1d and phv-1d mutants exhibit
transformation of the abaxial to adaxial fate and have rod-shaped leaves In contrast
miR165166-overproducing plants show pin-like cotyledons radicalized leaves and
abaxialized leaves (Chitwood et al 2007 Pattanayak et al 2013)
Leaf asymmetry is determined by miR166-mediated restriction of the
expression domains of the HD-ZIPIII genes The HD-ZIPIII genes are expressed in the
Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora
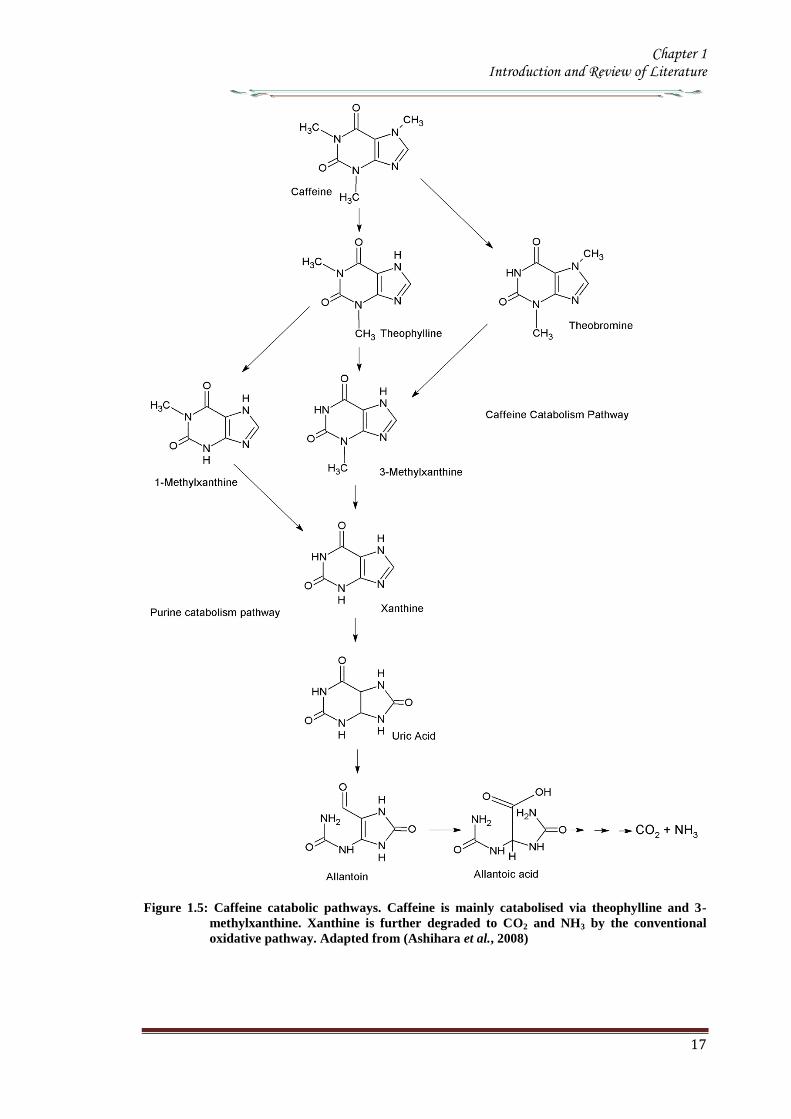
Chapter 1 Introduction and Review of Literature
17
Figure 15 Caffeine catabolic pathways Caffeine is mainly catabolised via theophylline and 3-
methylxanthine Xanthine is further degraded to CO2 and NH3 by the conventional
oxidative pathway Adapted from (Ashihara et al 2008)
Chapter 1 Introduction and Review of Literature
18
110 Genetic engineering of Coffee
Genetic transformation has potential applications in coffee agriculture for incorporating
genes which could provide desirable traits such as disease and insect resistance
drought and frost tolerance and herbicide resistance Transgenic technology can also be
used to increase nutritional value and improve cup quality produce varieties with
caffeine-free beans and for production of hybrid crops for molecular farming
Identification of target specific genes is one of the pre-requisites for developing
transgenic crops The availability of a large number of EST sequences in coffee and
initiation of coffee genome sequencing may speed up the gene discovery and accelerate
transgenic research efforts in coffee
1101 Insect Resistance
Production of insect resistant coffee plants is one of the major objectives of the
breeding programs The major pests attacking coffee include coffee berry borer (CBB
Hypothenemus hampei) white stem borer (WSB Xylotrechus quadripes) leaf miner
(Perileucoptera coffeela) and root nematodes (Meloidogyne spp and Pratylenchus
spp) The CBB is present in almost all the coffee growing countries and considered to
the most devastating pest in coffee To date there is no reported source of resistance to
CBB in the coffee gene pool Like CBB WSB is another serious pest in C arabica in
India and several other East Asian countries Both CBB and WSB belong to the order
Coleoptera For India controlling WSB is the biggest challenge and has therefore
become the highest research priority Robusta is generally resistant to WSB but the
interspecific Robusta Arabica hybrids are susceptible to WSB Although leaf miner is
not yet a serious pest in India and other East Asian countries it is an economically
important pest in East Africa and Brazil Effective chemical control of CBB and WSB
is difficult due to the nature of their life cycle inside the berry and stem respectively as
well as environmental concern regarding the use of pesticides Biological control
measures are adapted to combat these insect pests with varying degrees of success
Developing coffee plants resistant to these pests using genetic transformation
technology could be one of the alternative strategies to counter pest damage For insect
resistance several different classes of proteins from bacterial plant and animal sources
have been isolated and their insecticidal properties tested against many important pests
Amongst these proteins Bt toxins are most important and several transgenic crops
expressing Bt genes have been commercialized Coffee transgenic plants carrying a
Chapter 1 Introduction and Review of Literature
19
synthetic version of the cry1Ac gene have been produced (Leroy et al 2000 Mishra et
al 2002) Indeed this was the first report of an important agronomic trait being
introduced into a coffee plant The transgenic plants presented similar features in
growth and development compared to normal plants Transgenic plants highly resistant
to leaf miner under greenhouse conditions were tested under field conditions in French
Guyana for 4 years for pest resistance (Perthius et al 2005 Perthius et al 2006)
From 54 independent transformation events 70 of the events were resistant to leaf
miner Unfortunately the field trial was vandalized which led to the termination of the
experiment (Montagnon et al 2005)The effectiveness of Bt genes in controlling
coleopteran pests is well documented in corn and potato which indicates that Bt genes
might be effective against CBB The high toxicity of B thuringiensis sero var
israelensis against first in star larvae of CBB has been demonstrated (Mendez et al
2003) In another experiment an α-amylase inhibitor from Phaseolus vulgaris was
tested against CBB and found to have an inhibitory effect on its growth and
development (Grossi et al 2004)In addition to the CBB and WSB C arabica
varieties are also susceptible to endoparasitic root-knot nematode (Meloidogyne spp)
(Campos et al 1990 Mishra et al 2012)
So far 15 species have been reported to be parasites of coffee Controlling
nematodes is extremely difficult and currently seedling grafting with robusta rootstock
is followed as one of the control measure Sources of resistance specific to root-knot
nematodes have been identified in coffee trees (Bertrand et al 2001) and the Mex-1
gene conferring resistance to M exigua in C arabica is in the process of isolation (Noir
et al 2003)
1102 Tolerance to Abiotic Stress
In many coffee growing countries abiotic stress such as drought and frost are the major
climatic factors that limit coffee production Changes in climatic patterns due to global
climate change are considered increasingly important for coffee cultivation Drought
induces water stress in plants which affects vegetative growth and vigor and triggers
floral abnormalities and poor fruit set It also indirectly increases the incidence of pests
and diseases in the plants C arabica is generally more tolerant to water stress than C
canephora partly due to its extensive deep root system C racemosa is known to be a
good source of drought tolerance In India hybrids between C canephora and C
racemosa have been obtained and are currently under evaluation for drought tolerance
Chapter 1 Introduction and Review of Literature
20
In many coffee growing countries coffee is propagated in marginal areas where the
annual rainfall is below 1000thinspmm with prolonged dry spells of over 4-5 months In
those areas water shortage and unfavorable temperatures constitute major constraints
and the growth and productivity of C canephora are badly affected As with drought
periodic frost also affects coffee production in parts of Brazil The introduction of
drought and frost tolerant genes through genetic transformation would be of great
importance for alleviating these problems Research is now being carried out by several
groups to identify genes involved in biotic as well as abiotic stress The most promising
genetic engineering approach for drought tolerance includes the use of functional or
regulatory genes as well as the transfer of transcription factors In recent years plants
tolerant to high temperature and water stress have been the subject of intense research
(Chaves et al 2003 Chaves et al 2004 Coraggio et al 2004) For achieving drought
tolerance genes that have been targeted include those encoding enzymes involved in
detoxification or osmotic response metabolism enzymes active in signaling proteins
involved in the transport of metabolites and regulating the plant energy status (Chaves
et al 2003 Chaves et al 2004 Coraggio et al 2004) The dissection of molecular
mechanisms related to signal transduction and transcriptional regulation might help in
engineering drought tolerance in coffee
1103 Disease Resistance
Coffee leaf rust (CLR) caused by the fungus Hemileia vastatrix is the most important
disease in coffee with substantial loss to coffee production and productivity in all the
coffee growing countries In addition to leaf rust coffee berry disease (CBD) caused by
fungus Colletotrichum kahawae can be a devastating anthracnose leading to substantial
crop loss in Africa Several other fungal and bacterial diseases may also affect coffee
to a extent C arabica is more susceptible to many diseases than C canephora Though
most of the disease control measures rely upon chemical control they are more
expensive and labour intensive The long-term solution is the breeding of resistant
varieties which is the focus of many breeding programs However breeding for disease
resistant varieties is time consuming due to the perennial nature of coffee with its long
gestation period India has a long history of C arabica breeding especially for leaf rust
resistance and is the first country to demonstrate the existence of multiple races of leaf
rust Resistance to CLR is conditioned primarily by a number of major (SH) genes and
coffee genotypes are classified into different resistance groups based on their
Chapter 1 Introduction and Review of Literature
21
interaction with different rust races pathogen (van der Vossen 2001) Currently C
canephora provides the main source of resistance to pests and diseases including CLR
(H vastatrix) and CBD (C kahawae) and therefore used in breeding programs Other
diploid species like C liberica and C racemosa are being used as a source of resistance
to coffee leaf rust and coffee leaf miner respectively (Guerreiro et al 1999
Sreenivasan et al 1989) The development of coffee varieties resistant to major fungal
diseases such as CLR and CBD using transgenic technology will benefit the coffee
industry immensely During the last 15 years significant progress has been made in the
area of host-pathogen interactions (Staskawicz et al 1995 Feys et al 2000
Shivaprasad et al 2012) and many resistance genes involved in recognizing invading
pathogens have been identified and cloned (Takken et al 2000) A number of
signalling pathways induced because of pathogen infection have been dissected (Dong
et al 1998 Navarro et al 2006) Many antifungal compounds synthesized by plants to
combat fungal infection have been identified (Does et al 1998) Understanding the
specific induction of targeted pathways and identification of specific pathways
responsible for particular fungal resistance is important in order to employ this strategy
in transgenic technology The recent investigation of gene expression during coffee leaf
rust infection could provide an insight into the defence pathways operating in coffee
(Fernandez et al 2004 Nardi et al 2006) Efforts have been made to identify and
clone resistance genes from coffee for achieving durable resistance The genetic and
physical map of two resistance genes SH3gene conferring resistance to rust (Prakash et
al 2004) and the Ck1 gene conferring resistance to C kahawae CBD (Gichuru et al
2006) have been established These genes could be used for molecular marker assisted
breeding programs (Mishra et al 2012)
1104 Production of low-caffeine coffee
A widespread belief that the ingestion of caffeine can have adverse effects on
health resulted in the increased demand for decaffeinated coffee (Ashihara amp Crozier
2001) Several method were used to obtain decaffeinated plants by conventional
breeding techniques using low yielding varieties and mutants that accumulated
relatively low amounts of caffeine when compared to the commercially available ones
Low-caffeine and decaffeinated coffee represent around 10 of the coffee sales
around the world (Ribas et al 2006b) The industrial process for coffee decaffeination
can be expensive and affects the original flavour and aroma in coffee (David 2002)
Chapter 1 Introduction and Review of Literature
22
Transgenic coffee plants with suppressed caffeine synthesis using RNA interference
(RNAi) technology have been obtained (Ogita et al 2003 Ogita et al 2004) Specific
sequences in the 3prime untranslated region of the theobromine synthase gene (CaMXMT1)
were selected for construction of RNAi short and long fragments The caffeine and
theobromine content of the transgenic plants were reduced by ~ 70 when compared to
the untransformed plants In C canephora RNAi technology has also been employed
to silence the N-methyl transferase gene involved in caffeine biosynthesis (Vinod et al
2007) Promoter of an N-methyltransferase (NMT) gene involved in caffeine
biosynthesis was cloned (Satyanarayana et al 2005 Satyanarayana 2006) which will
be very useful for studying the regulation of caffeine biosynthesis
1105 Improvement in cup quality
Improvement in coffee cup quality requires elaborate knowledge of the chemical
constituents as well as the metabolic pathways involved in the elaboration of quality
The constituents of coffee beans include minerals proteins carbohydrates caffeine
chlorogenic acids (CGA) glycosides lipids and many volatile compounds that give
flavour to coffee by roasting Among these the role of three major constituents
sucrose CGA and trigonelline have been studied in coffee The sucrose content of
coffee bean is associated with coffee flavour the higher the sucrose content in green
beans the more intense will be the cup flavour (Clifford et al 1985 de Maria et al
1996) The sucrose content of C arabica (82-83) is higher than C canephora (33ndash
40) The sucrose amino acids ratio in green beans determines the profile of volatile
compounds Manipulating sucrose content in coffee bean is therefore important in
improving cup quality The sucrose synthase gene (CcSUS2) from C canephora has
been cloned and sequenced (Leroy et al 2005) This provides an opportunity to
manipulate the sucrose content in coffee Chlorogenic acids are products of
phenylpropanoid metabolism Chlorogenic acids are a group of hydroxycinnamoyl
quinic acids (HQA) formed by esterification between caffeic acids coumaric acids and
quinic acids (Clifford et al 1999) They are present in relatively large quantities in the
coffee bean and are the precursor of phenolic compounds in roasted beans C
canephora beans contain higher CGA (10) compared to C arabica beans (6-7)
CGA are known to have antioxidant properties as well as being associated with disease
resistance (Campa et al 2003) Genetic manipulations of genes involved in CGA
synthesis can serve either of these purpose by up- or down regulating the pathway
Chapter 1 Introduction and Review of Literature
23
Phenylalanine ammonia-lyase (PAL) catalyzes the first step of the phenylpropanoid
pathway leading to the synthesis of a wide range of chemical compounds including
flavonoids coumarins hydroxycinnamoyl esters and lignins (Hahlbrock amp Scheel
1989) Recently the full length cDNA and corresponding genomic sequences of PAL
from C canephora was isolated characterized and functionally validated (Mahesh et
al 2006 Parvatam et al 2006) This has opened up new possibilities for manipulating
the level of the PAL enzyme in coffee which in turn can be useful for improvement of
cup quality and manipulating antioxidant properties in coffee
1106 Fruit Ripening
Uniformity during fruit ripening is decisively related to cup quality in coffee and
consequently to the value of the product Fruits at the ideal ripening stage produce the
best organoleptic characteristics for coffee The presence of over ripened or green fruits
changes the acidity the bitterness and consequently the cup quality In order to
maximize uniform ripening of coffee fruits it is essential to control the action of genes
involved in the last step of maturation process Ethylene is known to trigger ripening
and increasing ethylene biosynthesis is associated with various stages of ripening
process (Protasio et al 2005) To control coffee fruit maturation two of the major
genes involved in ethylene biosynthesis namely ACC synthase and ACC oxidase
have been cloned (Protasio et al 2005 Neupane et al 1999) Introduction of the ACC
oxidase gene in antisense orientation have been achieved in both C arabica and C
canephora The effect of the transgene on ethylene production and fruit maturation has
yet to be reported The inhibition of genes downstream to the initial ethylene burst is
also an option to control coffee fruit maturation (Ribas et al 2006)
111 RNA interferenceRNAi
The regulation of gene expression at post transcriptional level has recently attracted
much attention because of the discovery of the phenomenon called RNA interference
(RNAi) RNAi based silencing techniques are more efficient than antisense mediated
gene silencing (Wesley et al 2001) The effect was first observed in plants wherein
silencing was observed when some copies of a transgene was in the forward orientation
with respect to the promoter and some in the reverse orientation This resulted in the
formation of double stranded RNA in the system The phenomenon in which
experimentally introduced double stranded RNA (dsRNA) leads to the loss of
expression of the corresponding cellular gene by sequence specific RNA degradation
Chapter 1 Introduction and Review of Literature
24
was termed as RNA interference or RNAi The protective effect of RNA silencing in
reducing virus infectivity supports the view that PTGS has evolved as a mechanism to
defend plants against virus infection and also to moderate the possible deleterious
genome-restructuring (insertional) activity of virus-like mobile genetic elements eg
retrotransposons A growing body of biochemical and genetic data has further
demonstrated that plants animals and yeasts share related mechanisms of specific
degradation of RNAs in which double-stranded forms of RNA are initiator molecules
(Brenstein et al 2001)
The key insight into the process of PGTS was provided by the experiments
conducted by Hamilton amp Baulcombe (1999) who identified the product of RNA
degradation as small RNA (siRNA) species of ~25nt of both sense and antisense
polarity SiRNA are formed and accumulate as double stranded RNA molecules of
defined chemical structures Both genetic and biochemical approaches were undertaken
to understand the basis of silencing Genetic screens were carried out in fungus
Neurospora carassa the alga Chalmydomonas reinhardtii and plant Arabidopsis
thaliana to search for detective mutants in PTGS
A combination of results obtained from several in vivo and in vitro experiments
have gelled into a two step mechanistic model for RNAiPTGS The first step referred
to as the RNAi initiating step involves binding of the RNA nucleases to a larges
dsRNA and its cleavage into discrete ~21-25nt RNA fragments This is ATP-
dependent reaction which involved RNase III - type endonucleases which make
staggered cuts in both strands of the dsRNA leaving 3prime overhang of 2 nucleotides with
5prime- phosphate 3prime-hydroxyl and no modification in the sugar phosphate backbone
(Hamilton amp Baulcombe 1999 Elbashir et al 2001) SiRNAs are the direct products
of dsRNA cleavage by the multi-domain RNase III enzyme DICER (Figure 16)
(Brenstein et al 2001 Karthikeyan et l 2013)
In the second step or commonly known as the effector step the double stranded
siRNAs produced in the first step bind to an RNAi specific protein complex to form a
RISC The complex undergoes activation in the presence of ATP so that the antisense
component of the unwound siRNA becomes exposed and allows the RISC to perform
the downstream RNAi reaction
Chapter 1 Introduction and Review of Literature
25
Several enzymes are involved in RNAi based on their role genes that encode for them
are classified into RNAi initiators RNAi effectors RNA dependent RNA polymerases
and silencing genes Two C elegans genes rdeI and rde4 (rde stands for lsquo RNAi
deficientrsquo) are believed to be involved in the initiation step of RNAi The C elegans
rde1 gene is a member of a large family of genes and is homologous to the Neurospora
qde2 (qde stands for ldquoquelling deficientrdquo) and Arabidopsis AGO1 (AGO stands for
agronaute AGO1 was previously identified to be involved in Arabidopsis
development) Although the functions of these genes are not clear a mammalian
member of RDE1 family has been identified as a translation initiation factor (Thakur
2005) Important genes for the effector step of PTGS in C elegans are rde2 and mut7
genes These genes were identified from heterozygous mutant worms that were unable
to transmit RNAi to their homozygous offsprings Worms with mutated rde2 or mut 7
genes show defective RNAi mut-7 gene encodes a protein with homology to the
nuclease domains of RNAase D and a protein implicated in Werner syndrome (a rapid
ageing disease in humans) (Grishok et al 2001 Kamath-Loeb et al 2012s)
Neurospora qde-1 Arabidopsis SDE-1SGS-2 and C elegans ego-1 appear to encode
RNA dependent RNA polymerase (RdRPs) It assumed that this is a proof that an RdRp
activity is required for RNAi Certainly the existence of an RdRp might explain the
remarkable efficiency of dsRNA induced silencing if it amplifies either the dsRNA
prior to cleavage or the siRNAs directly (Muers 2013) In C elegans ego-1 mutants
(ego stands for lsquoenhancer of glp-1) RNAi functions normally in somatic cells but is
defective in germline cells where ego-1 is primarily expressed In Arabidopsis SDE-
1SGS-2 mutants (SGS stands for suppressor of gene silencing) siRNA are produced
when dsRNA is introduced via an endogenously replicating RNA virus but not
introduced by a transgene It has been proposed that perhaps the viral RdRP is
substituting for the Arabidopsis enzyme in these mutants Random degradative PCR
model suggests that an RdRP uses the guide strand of an siRNA as a primer for the
target mRNA generating a dsRNA substrate for Dicer and thus more siRNAs
Several genes controlling RNA silencing in plants have been identified through
genetic screens of Arabidopsis mutants impaired in transgene induced RNA silencing
They encode a putative RNA-dependent RNA polymerase (SGS2SDE1) a coiled
protein (SGS3) a protein containing PAZ and Piwi domains (AGO1) and an RNA
helicase (SDE3) The putative SGS2SDE1 of Neurospora is related to QDE-1 and
Chapter 1 Introduction and Review of Literature
26
EGO-1 of C elegans whereas PAZPiwi protein AGO is related to QDE-2 of
Neurospora RDE-1of C elegans RNA helicase SDE3 is related to SMG-2 of C
elegans and Mut-6 of Chlamydomonas MUT-7 gene of C elegans encodes a protein
similar to Rnase D whereas the Drosophila DICER gene encode a protein similar to
RNase III An Arabidopsis ortholog of DICER gene has been identified called CAF
SIN1 SUS1(Thakur 2005 Poethig 2009)
Since the RNAi machinery is present constitutively within the eukaryotic cell it
is important to explore the metabolic advantages that are accorded by the RNAi related
proteins during the intrinsic normal growth of cells and development of organisms The
natural RNAi machinery not only keeps the mobile transposable elements from
disrupting the integrity of the genomes as suggested by the analysis of model organisms
like A thaliana C elegans D melanogaster (Tabara et al 1999 Wu-Scharf et al
2000 Aravin et al 2001 Hamilton et al 2002 Lisch 2009) but also participates in
development of organisms Genetic defects in Celegans RNAi genes ego1 and dicer
cause known specific developmental errors (Grishok et al 2000 Grishok et al 2001
Knight amp Bass 2001 Hall et al 2013) Similarly the Argonaute family of genes of A
thaliana (especially the ZWILLE proteins) is also responsible for plant architecture and
meristem development (Carmell et al 2002 Hamant and Pautot 2010) and the Dicer
homologue of A thaliana CAF1 is required for embryo development (Golden et al
2002 Nakano et al 2011) Thus genetic evidence illustrates the role of the RNAi
machinery as a controller of development related genes The mechanistic details of
these developmental processes are beginning to emerge
Chapter 1 Introduction and Review of Literature
27
Figure 16 Mechanism of gene silencing induced double stranded RNA The dicer enzyme to
produce siRNAs cleaves the RNA The siRNA generated bind to the nuclease complex
RISC The antisense component in the siRNA in the RISC guides the complex towards
the cognate mRNA resulting in endonucleolytic cleavage of the mRNA
Chapter 1 Introduction and Review of Literature
28
112 MicroRNA or MiRNA
In 1991 Ambros and co-workers first isolated a lin-4 mutant of Celegans which was
arrested at the first larval stage (Lee et al 1993) Later on the let-7 mutation was
isolated in the same system which was responsible for development through the fourth
larval stage Both lin-4 and let-7 encode short 22-nucleotide mature RNAs and were
called short temporal RNA because they control the temporal development program of
C elegans The mature lin-4 RNA defines (negatively regulates) the mRNA expression
of the lin-14 and lin-28 hetrochronic genes with the antisense mediated repression
mechanism of translation initiation and thus specifies the fate of cells during the first
three larval stages Recent studies have revealed that the short temporal RNAs are
actually members of a group of tiny RNAs (21to 28 nucleotides) called the micro-
RNAs (miRNA) (Bartel 2009) isolated members of which could easily run to a few
thousand which includes both those were identified computationally and by deep
sequencing Some of the components of the RNAi machinery have also been clearly
established as the effector proteins for the maturation of miRNAs
113 MicroRNAs in plants
1131 Discovery
MicroRNAs have been discovered by three basic approaches Direct cloning forward
genetic direct cloning and bioinformatic prediction followed by experimental
validation The most direct method of miRNA discovery is to isolate and clone small
RNAs from biological samples and several groups have used this approach to identify
plant miRNAs (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002 Xie et al 2003 Sunkar et al 2005 Williams et al 2013) The cloning methods
adapted were similar to those used to identify large numbers of animal miRNAs
(Lagos-Quintana et al 2001 Lau et al 2001 Aravin amp Tuschl 2005) which involve
isolating small RNAs followed by oligonucleotide adapter ligation reverse
transcription amplification and sequencing Some protocols incorporate methods to
enrich for Dicer cleavage products (ie molecules with 5prime-phosphates and 3prime-
hydroxyls) and to concatemerize the short cDNAs so that several may be identified in a
single sequencing read (Lau et al 2001 Llave et al 2002a Jagadeeswaran amp Sunkar
2013)The initial cloning experiments in Arabidopsis identified 19 miRNAs belonging
to 15 families (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002) although hundreds of other small RNAs such as siRNAs and RNA degradation
Chapter 1 Introduction and Review of Literature
29
fragments were also cloned through the direct cloning methods To select miRNAs
from the pool of small RNAs secondary structures of the RNA molecules were
examined in compliance with the acknowledged structural features of miRNAs
(Ambros et al 2003) The secondary structures were validated using several ad-hoc
software tools such as the Mfold or RNAfold programs (Reeder et al 2006) Finally
the existence of mature miRNAs was determined by RNA gel blot hybridization These
direct cloning methods were successful in isolating many miRNAs in Arabidopsis
(Reinhart et al 2002 Sunkar amp Zhu 2004) Rice (Sunkar et al 2005) Poplar (Lu et
al 2005) moss (Arazi et al 2005) and Tobacco (Billoud et al 2005)
Although miRNAs were first discovered through forward genetic screens in
worms (Lee et al 1993 Reinhart et al 2000)only few A thaliana miRNA gene
families have been discovered by this method (Chen 2008) in plants MiRNA
involvement in plant mutant phenotypes were not properly inferred till the cloning
experiments established that plant genomes contain numerous miRNAs (Park et al
2002 Reinhart et al 2002 Rhoades et al 2002) MiRNA loss-of-function allele has
been identified by forward genetic screens early extra petala1 is caused by a
transposon insertion ~160bp upstream of the predicted miR164c stem loop resulting in
flowers with extra petals (Baker et al 2005 Irish 2008) The fact that loss-of-function
miRNA mutants have been rarely discovered using forward genetics reflects small
target size for mutagenesis coupled with redundancy in nearly all evolutionarily
conserved plant miRNAs which are encoded by gene families (Table16) Family
members are likely to have overlapping functions buffering against loss at any single
miRNA locus Over expression screens can circumvent redundancy limitations At least
four plant miRNAs miR319 (also known as miR-JAW) miR172 (also known as EAT)
and miR166 were isolated in over expression screens for dominant mutants with
developmental abnormalities (Aukerman amp Sakai 2003 Palatnik et al 2003 Kim et
al 2005 Williams et al 2005b Schommer et al 2012) Mutations in miRNA target
sites which can prevent the entire family of miRNAs from repressing a target gene can
also circumvent redundant functions of miRNA family members
The dominant mutations in the HD-ZIP genes PHB PHV and REVOLUTA
(REV) in Arabidopsis and ROLLEDLEAF1 (RLD1) in Maize result in adaxialization of
leaves andor Vasculature (McConnell amp Barton 1998 McConnell et al 2001 Nelson
et al 2002 Zhong amp Ye 2004 Allen amp Millar 2012) and are all caused by mutations
Chapter 1 Introduction and Review of Literature
30
in miR166 complementary sites (Rhoades et al 2002 Emery et al 2003 Jones-
Rhoades amp Bartel 2004 Mallory et al 2004 Zhong amp Ye 2004)
In both plants and animals cloning was the initial means of large-scale
miRNA discovery (Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Reinhart et al 2002 Mendes et al 2012) However cloning is biased toward
RNAs that are expressed highly and broadly MiRNAs expressed at low levels or
only in specific cell types or in response to certain environmental stimuli were more
difficult to clone Sequence based biases in cloning procedures might also cause
certain miRNAs to be missed Because of these limitations bioinformatics approaches
to identify miRNAs have provided a useful complement to cloning
A straightforward use of bioinformatics has been carried out to find homologs
of known miRNAs both within the same genome and in the genomes of other species
(Pasquinelli et al 2000 Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Mendes et al 2009) A more difficult challenge is to identify miRNAs
unrelated to previously known miRNAs This was first accomplished for
vertebrate nematode and fly miRNAs using algorithms that search for conservation
of sequence and secondary structure (ie miRNA stem-loop precursors) between
species searching for patterns that are characteristic of miRNAs (Lai et al 2003 Lim
et al 2003 Lim et al 2005)
Most of the methods identified numerous miRNAs but are necessarily more
relaxed in terms of allowed structures Some approaches take advantage of the high
complementarity of plant miRNAs to target messages implementing the requirement
that the candidate has conserved complementarity to mRNAs (Jones-Rhoades amp Bartel
2004 Adai et al 2005) This additional filter has been useful for distinguishing
authentic plant miRNAs from false positives which later extended to mammalian
miRNA gene prediction (Xie et al 2005 Mendes et al 2012)
Another criteria used by most algorithms in the filters is evolutionary
conservation In plants mature miRNA sequences are conserved to a higher degree
than their precursor sequences For example Arabidopsis and rice diverged more than
130 million years ago (Friis et al 2004) but some miRNAs are highly conserved
in these plant species (Reinhart et al 2002) Furthermore various parameters for the
secondary structures such as free energy the number of paired residues within a
Chapter 1 Introduction and Review of Literature
31
miRNA or the number and size of bulges are also used to validate the predictions
(Jones-Rhoades amp Bartel 2004 Wang et al 2004 Adai et al 2005 Mendes et al
2012)
1132 Conserved microRNAs in Plants
Till date cloning genetics and bioinformatics screens have resulted in annotation of
approximately 184 potential Arabidopsis miRNAs (miRBase release 13) These
miRNAs seem to arise through gene duplication and subsequent diversification and
represent approximately 100 families (Maher et al 2006 Axtell 2013) Twenty one
families represented by 92 genes are clearly conserved in species beyond Arabidopsis
(Table 15) The presence of these miRNA families in other sequenced plant genomes
indicates that different plant species have an evolutionarily fluid set of miRNAs
(Willmann amp Poethig 2007) Recent studies on mosses one of the most ancient land
plants have shown that at least 14 miRNA families are conserved in eudicots and
mosses and therefore it appears that plant miRNAs share a common ancient origin
(Arazi et al 2005 Axtell amp Bartel 2005 Talmor-Neiman et al 2006 Zhang et al
2006 Axtell 2013) The number of members per family in a genome ranges from 1-32
with the exception of miR-430 which is represented by a cluster of ~80 loci in zebra
fish (Giraldez et al 2005)
In plants the number of members in each family correlates among examined
species certain families contain numerous members in all three species (eg miR156
miR166 miR169) whereas others consistently contain fewer genes (eg miR162
mir168 miR394) (Table 16) Although it is still unclear why certain miRNA are
encoded by more than one gene in plants (eg 12 genes encoding miR156) this
correlation might suggest functional significance of various miRNA family sizes Most
annotated plant miRNAs are conserved throughout flowering plants while a few
miRNAs are found only in a single sequenced genome and thus they could be
evolutionarily lsquoyoungrsquo miRNAs For example Arabidopsis has several miRNA
families that are not found in poplar or rice suggesting that miRNAs could be
generated continuously during evolution (Allen et al 2004a Rajagopalan et al
2006 Fahlgren et al 2007 Axtell 2012 de Alba et al 2013) Consistent with the
notion that non conserved miRNAs are of a more recent evolutionary origin these
miRNAs are mainly encoded by a single locus in the genome Within each family the
mature miRNA is always located in the same arm
Chapter 1 Introduction and Review of Literature
32
Table 16 MicroRNA families conserved in plants
miRNA
family
Arabidopsis Oryza Populus
miR156 12 12 11 miR159319 6 8 15 miR160 3 6 8 miR162 2 2 3 miR164 3 5 6 miR166 9 12 17 miR167 4 9 8 miR168 2 2 2 miR169 14 17 32 miR171 4 7 10 miR172 5 3 9 miR390 3 1 4 miR393 2 2 4 miR394 2 1 2 miR395 6 19 10 miR396 2 5 7 miR397 2 2 3 miR398 3 2 3 miR399 6 11 12 miR408 1 1 1 miR403 1 0 2 miR437 0 1 0 miR444 0 1 0 miR445 0 9 0 Total 92 127 169
All known miRNA families that are conserved between more than one plant species are listed
together with the number of genes identified in the sequenced genomes Rice miRNA families that have orthologs in maize but do not appear to have orthologs in the eudicots (Arabidopsis and Populus) are marked with an asterisk The following families contain miRNA genes annotated with
more than one number miR156 (miR156 and miR157) miR159319 (miR159 and miR319) miR166 (miR165 and miR166) miR171 (miR170 and miR171) and miR390 (miR390 and miR391)
of the stem loop (5prime- or 3prime) as it would be expected if the genes carry common ancestry
(Figure 17) The sequence of the mature miRNA and to some extent the segment on
the opposite arm of the hairpin to which it pairs is highly conserved between the
members of the same family (both within and between the species) while the sequence
secondary structure and length of the intervening ldquolooprdquo region can be highly divergent
within the same family
The patterns of paring and non pairing nucleotides are often conserved between
homologous miRNA stem loops from different species (Figure 17) The significance of
conserved mismatches is opined to guide DCL1 to cleave at the appropriate positions
along the stem loop
Chapter 1 Introduction and Review of Literature
33
Figure 17 Representative stem loop of miR164 Segments corresponding to the mature miRNAs
are shown in red (Adapted from Rajagopalan et al 2006)
Although many miRNAs are highly conserved in plants the degree of conservation in
most cases is quite low between plants and animals one exception is miR854 A recent
study has revealed that miR854 is conserved between Arabidopsis and animals and that
the Arabidopsis miR854 differs from the human miR854 only by a single nucleotide
(Arteaga-Vazquez et al 2006 Axtell 2012 de Alba et al 2013) The Arabidopsis
miR854 has multiple binding sites in the 3prime-untranslated region (UTR) of
OLIGOURIDYLATE-binding-PROTEIN mRNA 1bA (UBP1b) and represses the
translation of the UBP1b mRNA The animal miR854 is also complementary to a site in
the 3prime-UTR of the UBP1b homolog in animals However it is currently unclear how the
animal miR854 regulates its target mRNA Another distinction between plant and
animal miRNAs is the genomic organization of the miRNA genes While plant miRNA
genes are located predominantly in the intergenic regions animal miRNA genes tend to
localize in the intragenic regions both in the introns or exons of protein coding genes
Moreover miRNA genes are often clustered within a single precursor transcript
whereas individual plant miRNAs are derived from individual precursor RNAs (Bartel
Chapter 1 Introduction and Review of Literature
34
2004 Bonnet et al 2004 Kim 2005 Vaucheret et al 2006 Marco et al 2013)
1133 Non-conserved miRNA and challenges in annotation
Most miRNAs are conserved throughout flowering plants (Table 15) but many more
were found only in a single sequenced genome thus considered to be of a more recent
evolutionary origin The extended homology between few non conserved miRNAs
precursors and their target genes provides strong evidence that these relatively ldquoYoungrdquo
miRNAs arose from the duplication of the target gene segments (Allen et al 2004)
Several non-conserved miRNAs including miR161 miR163 miR173 miR447
miR475 and miR476 are known to direct cleavage of target transcripts (Allen et al
2004a Adai et al 2005 Sunkar et al 2005 de Alba et al 2013) It is difficult to
confidently predict targets for many because it is not possible to use complementary
site as a filter against false-positive target predictions It is also difficult to
confidently annotate non-conserved miRNAs as miRNA rather than siRNAs The
established minimal standard for miRNA annotation is a small RNA with detectable
expression and the potential to form a stem-loop when joined to flanking genomic
sequence (Kasschau et al 2003) In practice these requirements are too loose to
definitively categorize many small RNAs cloned from plants Many plant siRNAs are
detectable on blots (Vazquez et al 2004b) and hundreds of thousands of non-
miRNA genomic sequences can be predicted to fold into secondary structures that
resemble structures of plant miRNA precursors (Jones-Rhoades amp Bartel 2004)
Therefore without conservation of both sequence and secondary structure it is
difficult to be confident that a given cloned RNA originated from a stem-loop (ie is a
miRNA) rather than from a double-stranded RNA (ie is a siRNA) In fact many of
the thousands o f small RNAs cloned from Arabidopsis (Gustafson et al 2005) would
probably meet the literal requirements for annotation as miRNAs A few of these
sequences might be miRNAs but others that meet the literal criteria probably are not
so The challenges of annotating non conserved small RNAs were evidenced by
three related small silencing RNAs that were originally annotated as miRNAs
that turned out to be trans-acting siRNAs (tasiRNAs)(Kanno et al2013)
Although most miRNAs that are broadly conserved among flowering plants are
now being identified and experimentally validated (Table 16)(Jones-Rhoades amp
Bartel 2004) the challenges and ambiguities for classifying non-conserved miRNAs
prevent meaningful estimates of the total number of miRNA genes in Arabidopsis and
Chapter 1 Introduction and Review of Literature
35
other plant genomes It is possible that miRNAs might have escaped detection
because they are non-conserved and expressed in specific tissues or conditions and can
only be identified by using high- throughput sequencing
1134 MicroRNA biogenesis
11341 Transcription of microRNA precursor
Plant miRNAs are primarily found in the genomic regions that are not associated with
the protein coding genes (Reinhart et al 2002) and it appears that most if not all plant
miRNAs are produced from their own transcriptional units Plant miRNA genes are
occasionally clustered near each other in the genome suggesting transcription of
multiple miRNAs from a single primary transcript [eg the miR395 cluster (Grishok
et al 2001 Jones-Rhoades amp Bartel 2004b)] but this polycistronic arrangement of
miRNA genes appears far less frequently in plants than in animals (Bartel 2004)
Northern blot EST and mapping evidence indicate that plant miRNA primary
transcripts (also known as pri-miRNAs) as in animals are longer than needed to
encompass the miRNA stem-loops (Palatnik et al 2003 Jones-Rhoades amp Bartel
2004b) At least some of these pri-miRNA transcripts appear to be spliced
polyadenylated (Kurihara amp Watanabe 2004) and capped (Xie Z et al 2005) Two
rice miRNAs are contained within transcripts that contain exon junctions within the
presumptive stem-loop precursor implying that in these cases splicing is a prerequisite
for Dicer recognition (Sunkar et al 2005) Plant pri-miRNAs are over 1kb in length
and they are usually preceded by typical TATA box motifs They can also undergo
canonical splicing polyadenylation and capping All these observations indicates RNA
polymerase II is probably responsible for transcribing most plant miRNAs (Xie Z et
al 2005) as appears to be the case for many animal miRNAs Relatively little is
known about the regulation of miRNA transcription in plants but there is no reason
to suspect that this regulation would differ from that of protein-coding transcripts as
the bioinformatics analysis of the sequence upstream of the transcriptional start
sites of the miRNA genes showed the presence of putative binding motifs of
number of known transcription factors (Megraw et al 2006 Sethupathy 2013)
11342 MicroRNA processing and export
In plants maturation of miRNA is a step wise process A miRNA gene is transcribed to
pri-miRNA which is much longer than the pre-miRNA possessing the characteristic
stem-loop structure Like transcription of most protein-coding genes the miRNA
Chapter 1 Introduction and Review of Literature
36
transcription is processed by RNA polymerase II (Pol II) enzymes and the pri-
miRNAs are 5prime-capped and 3prime-polyadenylated (Lee et al 2004 Xie Z et al 2005)
Mature miRNAs are produced from pri-miRNAs through at least two sequential
processing steps by RNase III-type endonucleases In animals pri-miRNAs are first
processed at the bottom portion of the stem by Drosha a nuclear-localized RNase III
enzyme to liberate pre-miRNAs The pre-miRNAs are then exported to the cytoplasm
with the help of exportin-5The pre-miRNAs are further processed by Dicer another
RNaseIII enzyme to a duplex consisting of the miRNA and the antisense strands
(miRNA) Since plants do not have any Drosha homologs the two sequential
processing steps seem to be carried out by DCL1 a Dicer homolog in the nucleus
(Kurihara amp Watanabe 2004 de Alba et al 2013 Rogers amp Chen 2013)
The processing of pri-miRNAs to pre-miRNAs by DCL1 is assisted by two
other proteins HYPONASTY LEAVES1 (HYL1) and SERRATE (SE) HYL1 belongs to a
family of double-stranded RNA (dsRNA) binding protein and interacts with DCL1 in
vitro and in vivo (Lu amp Fedoroff 2000 Hiraguri et al 2005 de Alba et al 2013
Rogers amp Chen 2013) Loss-of-function mutations in the HYL1 gene result in
decreased accumulation of miRNAs and concomitant accumulation of pri-miRNAs
(Han et al 2004 Vazquez et al 2004a Kurihara et al 2006) SE a C2H2 zinc finger
protein interacts with DCL1 and HYL1 and plays a role in the processing of pri-
miRNAs to pre-miRNAs (Lobbes et al 2006 Yang L et al 2006) In addition the
nuclear cap-binding complex proteins CBP20 and CBP80 that are known as ABA
HYPER SENSITIVE 1(ABH1) have been shown to be required for proper miRNA
processing (Gregory et al 2008 Laubinger et al 2008) Recently it has been
demonstrated that DCL1 can release 21-nt RNAs from dsRNA as well as from
synthetic miR167 precursor RNAs in vitro However this cleavage is error prone and
inefficient in the absence of HYL1 and SE which accelerate the rate of DCL1-mediated
cleavage and promote accurate processing of miRNAs (Dong et al 2008 Rogers amp
Chen 2013)
11343 Methylation of the miRNAmiRNAduplex
After the miRNAmiRNAduplexes are released from the pre-miRNAs by the DCL1
activities the duplexes are methylated at the 2primeOH of the3prime-ends by HUA ENHANCER1
(HEN1) a small RNA methyltransferase (Yu et al 2005 Yang Z et al 2006 Rogers
amp Chen 2013 ) This step which is distinct from the RNase III-mediated processing
Chapter 1 Introduction and Review of Literature
37
step distinguishes the biogenesis of plant miRNAs from that of animal miRNAs
Mutations in the HEN1 gene were first isolated in a morphological screening for floral
development although the HEN1 mutants display pleiotropic phenotypes similar to the
developmental defects observed in the DCL1 mutants (Chen et al 2002 Park et al
2002 Rogers amp Chen 2013)
Figure 18 Biogenesis of microRNA
A Biogenesis of plant MicroRNA B Biogenesis of metazoananimal MicroRNA
The miRNA (red) is incorporated into RISC and the miRNA(blue) in degraded
This enzyme has two dsRNA-binding domains and nuclear localization signal It
is also conserved in fungi and is required for the processing of siRNAs as well as of
miRNAs (Park et al 2002 Boutet et al 2003 Xie et al 2003) Although the HEN1
protein is localized to the nucleus its methylation activity in the cytoplasm cannot be
ignored (Xie et al 2004 Rogers amp Chen 2013)
Chapter 1 Introduction and Review of Literature
38
The methylation of the miRNAmiRNA duplex is required to protect miRNAs
against the 3prime-end uridylation activity and subsequent degradation (Li et al 2005) In
the hen1 mutants accumulation of miRNAs is reduced to a lower level Also the
miRNAs appear to be heterogeneous in their sizes such that a ladder of bands rather
than a single species is observed for most miRNAs in the mutants (Li et al 2005)
MiRNA cloning and sequencing analyses revealed the presence of a number of U
residues at the 3prime-end of miRNAs in the hen1 mutants Moreover these nucleotides do
not correspond to the miRNAs in the pre-miRNAs indicating that these nucleotides are
added after the processing of the miRNAs from pre-miRNAs (Li et al 2005)
Uridylation of RNAs has also been proven to be correlated with RNA decay (Shen amp
Goodman 2004 Mullen amp Marzluff 2008) suggesting that uridylation attracts an
exonuclease which degrades miRNAs in plants
11344 MiRNA incorporation in to the RISC and its nuclear export
The methylated miRNAmiRNA duplexes undergo RISC assembly In this process
the miRNA of the duplexes is selectively incorporated into the RISC and the miRNA
appears to be removed and subsequently degraded (Hammond et al 2000 Hutvagner
amp Zamore 2002 Schwarz et al 2003 de Alba et al 2013 Rogers amp Chen 2013)
The ARGONAUTE 1(AGO1) proteins are core components of the RISC complex
They contain two structural domains the N-terminal PAZ domain that binds to the 3prime-
end of single-stranded RNAs and the C-terminal piwi domains that has a structure
similar to that of RNase H (Cerutti et al 2000 Parker et al 2004) Several AGO
proteins possess endonucleolytic activities within the PIWI domain and cleave target
mRNAs in the middle of the site complementary to miRNA (Liu et al 2004 Meister et
al 2004 Miyoshi et al 2005) Mutations in the AGO1 gene cause miRNA target
genes to be ectopically expressed and the levels of some miRNAs are reduced in the
mutants (Kidner amp Martienssen 2004 Vaucheret et al 2004 Kidner amp Martienssen
2005 Ronemus et al 2006) Moreover the phenotype of the AGO1 hypomorphic
alleles display defects in lateral organ polarity and leaf and flower morphology as
observed in the dcl1 mutants and null AGO1 alleles are embryonically lethal
supporting the close relationship between the AGO proteins and miRNA processing
(Kidner amp Martienssen 2004 Vaucheret et al 2004 de Alba et al 2013 Rogers amp
Chen 2013)
The production of miRNA miRNA duplexes occur in the nucleus while
Chapter 1 Introduction and Review of Literature
39
miRNA-RISC complexes are present in the cytoplasm where target mRNAs are
located This discordance of functional sites requires an export step during miRNA
biogenesis HASTY the Arabidopsis homolog of exportin-5 is thought to be involved
in miRNA nuclear export (Bollman et al 2003 Park et al 2005)The hasty mutant
exhibits pleiotropic defects in plant development and reduced accumulation of most
miRNAs as in the DCL1 or AGO1 mutants Howeverit is unclear whether the HASTY
proteins export the miRNA miRNA duplexes or the miRNA-RISC complexes in
plants
11345 Feedback regulation of miRNA biogenesis
MiRNA biogenesis is under feedback regulation such that the DCL1 and AGO1 genes
are themselves regulated by miRNAs DCL1 is itself a target of miR162 which leads to
the cleavage of the DCL1 mRNA (Xie et al 2003 de Alba et al 2013) Consistent
with this the levels of DCL1 mRNA are elevated in the mutants of miRNA processing
genes in which the miR162 abundance is reduced In addition there is a predicted
hairpin structure of miR838 in the 14th
intron of the DCL1 primary transcript
(Rajagopalan et al 2006) This prediction implies that DCL1-mediated processing
on this site can result in the cleavage of the DCL1 primary transcript into two truncated
fragments the N-terminal capped fragment and the C-terminal polyadenylated
fragment If this is the case it is possible that miRNA biogenesis competes with the
splicing event of the DCL1 pre-mRNA
The AGO1 is also regulated by miRNA There is a complementary site for
miR168 binding in the AGO1 mRNA and the transcript level of the AGO1 mRNA is
reduced through an mRNA cleavage mechanism (Vaucheret et al 2006 de Alba et al
2013 Rogers amp Chen 2013) indicating that the AGO1 mRNA levels are tightly
controlled by the levels of the AGO1 protein
1135 Mechanism of miRNA action
11351 MiRNA-directed mRNA cleavage
By forming the miRNA-RISC assembly miRNAs can direct the RISC to regulate gene
expression by two post transcriptional mechanisms mRNA cleavage or translational
repression (Bartel 2004 Jones-Rhoades et al 2006 Dalmay 2013) The degree
of miRNA-mRNA complementarity plays an important role for the
determination of the mechanism to be used A general rule is that while perfect or
Chapter 1 Introduction and Review of Literature
40
near-perfect complementarity induces mRNA cleavage central mismatches trigger
translational repression (Carrington amp Ambros 2003 Jones-Rhoades amp Bartel 2004)
Unlike most animal miRNAs plant miRNAs have near-perfect or perfect
complementarity to their targets and mRNA cleavage is assumed to be a predominant
mechanism by which miRNAs regulate gene expression in plants
In RNA cleavage mechanism miRNAs guide the AGO component of RISC to
cleave a single phosphodiester bond of the target mRNA within the miRNA-binding
site (Llave et al 2002b Tang et al 2003) The truncated fragments are then released
and degraded freeing the RISC to recognize and cleave another target mRNA This has
been validated by the detection of 3prime cleavage products that have 5prime ends that start at the
middle of the complementary regions The mRNA cleavage products are then further
degraded by other mechanisms The 3prime cleavage products of some miRNA targets are
degraded by the 5prime-3prime exonuclease XRN4 an Arabidopsis homolog of the major Yeast
mRNA degrading exonuclease Xrn1p It has been observed that the 3prime cleavage
products of some target mRNAs were accumulated in the xrn4 mutants (Souret et al
2004 Dalmay 2013) However the 3prime cleavage products of other miRNA targets do
not accumulate in the xrn4 mutants indicating that there are also XRN4 independent
mechanisms The 5prime cleavage products are typically untraceable by RNA gel blot
hybridization but can be detected by sensitive PCR based methods It has been found
that the 5prime cleavage products tend to obtain an oligo U tail at the 3prime ends This 3prime U tail
is correlated with shortening of the 5prime cleavage products from the 5prime end leading to the
conclusion that the 3prime uridylation causes 5 prime- 3 prime exonucleolytic decay of the 5prime cleavage
products (Shen amp Goodman 2004)
DCL1 HYL1 and SE interact with each other and are co-localized in the
nuclear bodies termed Dicing bodies or D-bodies in which some pri-miRNA shave
been found suggesting that D-bodies serve as a center for active miRNA processing
(Fang amp Spector 2007 Fujioka et al 2007 Song et al 2007) The miRNA
processing site appears to be distinct from the Cajal bodies that have previously been
found as a siRNA processing site (Pontes amp Pikaard 2008) The D-bodies include
SmD3 and SmB proteins that are found in the Cajal bodies However At collin an
additional marker for the Cajal bodies is not found in the D-bodies Moreover the D-
bodies are present in an At collin mutant lacking the Cajal bodies The pri-miRNAs of
miR171A and miR159A genes have been found to be co-localized with HYL1 and
Chapter 1 Introduction and Review of Literature
41
DCL1 in the D- bodies that are distinct from the Cajal bodies (Song et al 2007)
11352 MiRNA-directed translational repression
Translation repression is another mechanism exerted by plant miRNAs The regulatory
mechanism of miR172 which regulates flowering time and floral organ identity is
consistent with translation repression MiR172 over production does not affect the
transcript levels of APETALA2 (AP2) and AP2- like target mRNAs Instead the AP2
protein level is reduced in the miR172 over expressing mutant (Aukerman amp Sakai
2003 Chen 2004 Dalmay 2013) It was later shown that miR172 can also direct the
cleavage of the target mRNAs although the steady-state mRNA level is unchanged due
to feedback regulation (Schwab et al 2005 Jung et al 2007) It is clear that miR172
regulates its target genes primarily by translational repression rather than mRNA
cleavage Recently it has been reported that miR156157 and miR854 also regulate
their target genes through translational repression (Arteaga-Vazquez et al 2006
Gandikota et al 2007 Dalmay 2013) It was once thought that translational repression
is an exception to the rule However experimental evidence suggest a widespread
coexistence of translational repression and mRNA cleavage (Brodersen et al 2008)
1136 Regulatory roles of miRNAs in plant development
The miRNA target prediction has been a difficult task in order to obtain regulatory
targets without bringing in too many false predictions Plant growth and developmental
processes regulated by miRNAs are quite diverse and enormous Furthermore plant
miRNAs work cooperatively or antagonistically to establish a balanced regulation This
notion is well consistent with the findings that mutations in the regulators of miRNA
biogenesis transport and processing such as AGO1 DCL1 HYL1 HEN1 ABH1
and HASTY cause pleiotropic phenotypes (Mallory amp Vaucheret 2006 de Alba et al
2013) To date the roles of miRNAs have been mostly deduced from obtaining miRNA
over expressing transgenic plants or gain-of-function mutants in which miRNA-
resistant target genes are ectopically expressed
11361 Phase transition
Plants pass through a series of developmental phase transitions during their life span
beginning with seed germination continuing through vegetative phase change
reproductive phase change flowering initiation and finally seed production for the next
generation Because of their importance in understanding plant developmental
Chapter 1 Introduction and Review of Literature
42
processes genes regulating developmental transitions have been extensively studied
and underlying molecular mechanisms have been explored in various plant species
Majority of researches were mainly focused on vegetative phase change and
reproductive transition in Arabidopsis and Maize (Poethig 2003 Baurle amp Dean
2006) Particularly the mutations of the genes encoding various regulators of miRNA
biogenesis and transport such as HST SE and AGO exhibit altered juvenile to adult
vegetative phase change and vegetative to reproductive change (Hunter et al 2003
Peragine et al 2004 Vazquez et al 2004a Jones-Rhoades et al 2006 de Alba et al
2013)
Over-expression of the miR156a gene causes late flowering and delays
vegetative phase change (Wu amp Poethig 2006 Gandikota et al 2007 Wang et al
2008) It has been shown that miR156 negatively regulates a group of genes encoding
SQUAMOSA PROMOTER-BINDING PROTEIN-LIKE (Lewis et al 2007 de Alba et
al 2013 Rogers amp Chen 2013) transcription factors such as SPL3 SPL4 and SPL5
that promote flowering initiation In contrast transgenic plants over expressing
miR156-resistant SPL345 genes are early flowering Interestingly the endogenous
level of miR156 is temporally regulated In the early juvenile vegetative phase the
level of miR156 is very high but decreases rapidly before the onset of the adult juvenile
phase (Wu amp Poethig 2006)
The Arabidopsis early activation tagged (eat-D) mutant exhibits early flowering
with disrupted floral structure The mutant phenotypes are caused by over expression of
the miR172a-2gene (Aukerman amp Sakai 2003) MiR172 regulates a small group of
genes encoding APETALLA2 (AP2)-like transcription factors such as TARGET OF
EAT1 (TOE1) TOE2 SCHLAFMUTZE (SMZ) and SCHNARCHZAPFEN(SNZ)
TOE1 TOE2 SMZ and SNZ have been shown to participate in their productive phase
change by repressing a floral integrator FLOWERING LOCUS T (FT)(Jung et al
2007) Consistent with this toe1-2D 35STOE2 35SSMZ and 35SSNZ plants
exhibit late flowering (Jung et al 2007) MiR172 is also regulated temporally and
highly expressed in the adult vegetative phase both in Arabidopsis and maize (Chuck et
al 2007) Because the temporal expression patterns of miR156 and miR172 are
complementary to each other it has been suggested that the two miRNAs interact with
each other in regulating phase change (Chuck et al 2009 de Alba et al 2013) In
support of this view it has been shown that miR172 is down-regulated in the
Chapter 1 Introduction and Review of Literature
43
Corngrass1(Cg1) mutant in which miR156 is overproduced (Chuck et al 2007 de
Alba et al 2013) However there is no direct evidence for the direct linkage between
miR156 and miR172 It is also envisioned that miR156 and miR172 would not be
directly related For example miR156 regulates plastochron index that is the inverse of
leaf initiation rate and thus frequently used to analyze temporal patterns of plant
development In contrast miR172 does not affect plastochron (Wang et al 2008) The
down-regulation of miR172 in the maize miR156-over producing mutants would be
due to the prolonged juvenile vegetative phase in the mutants
Another miRNA that regulates flowering initiation is miR159 It regulates
MYB33 and MYB65 which are closely related to the barley GAMY B transcription
factor that responds to gibberellic acid (GA) The level of miR159 is elevated in GA-
treated seedlings but reduced in the GA biosynthetic ga1 mutant Transgenic plants
over producing miR159 exhibit delayed floral induction under short days (Achard et
al 2004) It is been suggested that MYB33 mediates GA signal to regulate LEAFY
(LFY) These observations indicate that miR159 plays an important role in the GA
signaling pathways that regulate proper floral induction under short days (Achard et al
2004)
11362 Floral development
Arabidopsis flowers consist of four distinct organs They arise in a characteristic
pattern within concentric rings called whorls from outside to inside four sepals four
petals six stamens and the central pistil consisting of two carpels Identities of the
floral organs are determined by three classes of regulatory genes classA classB and
class C genes (Krizek amp Fletcher 2005) The A and C class genes act antagonistically
to restrict their expression domains (Bowman et al 1991)
It is notable that miR172 also regulates floral architecture in addition to its role
in the timing of flowering initiation MiR172 inhibits the translation of the AP2 mRNA
(Chen 2004) Transgenic plants overproducing miR172 not only exhibit early
flowering but also show abnormal floral development which is quite similar to those
of the class A gene mutants such as the ap2 mutants (Aukerman amp Sakai 2003 de
Alba et al 2013) When the activity of the class A genes is inactivated that of the class
C genes such as AGAMOUS (AG) is elevated As a result the miR172- overproducing
transgenic plants exhibit no petals along with homeotic transformation of the sepals to
Chapter 1 Introduction and Review of Literature
44
the carpels (Aukerman amp Sakai 2003 Chen 2004)
Interestingly miR166 also regulates floral architecture The role of miR165166
in the regulation of meristem activity is closely linked to floral meristem formation and
organization (Zhao et al 2007 Miyashima et al 2013) Floral structure is severely
disrupted in the miR165166-overproducing mutants such as men1 and jba-1D in
which miR166 is overproduced (Williams et al 2005b Jung amp Park 2007) The men1
and jba-1D mutants have smaller gynoecium and reduced carpel number In an extreme
case the jba-1D homozygous plants lack visible carpels (Williams et al 2005b)
Similar phenotypes have been observed in the miR165 overproducers (Zhao et al
2007) It has been suggested that the phenotypes are possibly caused by altered AG
expression in the floral meristem (Jung amp Park 2007 Turchi et al 2013)
Other miRNAs also affect floral development Overproduction of miRNAs
involved in auxin signaling also affects floral architecture Transgenic plants
overproducing miR167 have defects in the formation of ovule and anther with reduced
fertility (Ru et al 2006) It has been shown that miR167 targets ARF6 and ARF8 that
play a role in gynoecium and stamen development and in regulating fertility (Ru et al
2006 Wu et al 2006) These observations suggest that auxin signals are also closely
related to floral organogenesis In addition transgenic plants over-producing miR164
and the cuc1cuc2 double mutants show fused sepals and reduced petals (Laufs et al
2004 Turchi et al 2013) suggesting that miR164 is also related to floral meristem
activity and boundary specification of the meristematic region
11363 Shoot apical meristem development
Shoot apical meristem comprises self-maintaining totipotent cells The shoot apical
meristem activity affects diverse aspects of plant developmental processes including
leaf development vascular development and lateral organ polarity (Bowman et al
2002) Because miR165166 plays a primary role in meristem formation
overproduction of miR165166 and multiple knockout mutants of its target genes
exhibit pleiotropic pheonotypes (McConnell et al 2001 Emery et al 2003 Kim et al
2005 Williams et al 2005b)
The targets of miR165166 include genes encoding the homeo domain-leucine
zipper (HD-ZIP) class III transcription factors such as PHABULOSA(PHB)
PHAVOLUTA (PHV) REVOLUTA(REV) ATHB15CORONA (CNA) and ATHB8
Chapter 1 Introduction and Review of Literature
45
PHB PHV and ATHB15 have overlapping functions in controlling meristem
development (Turchi et al 2013) Indeed the phb phv athb15 triple mutants have an
enlarged shoot apical meristem Consistent with this the miR166- overproducing men1
and jba-1D mutants exhibit enlarged shoot apical meristems (Kim et al 2005
Williams et al 2005b Turchi et al 2013) In the mutants the shoot apical meristems
are multiplied and large
The development of shoot apical meristems is also regulated by the WUSCHEL
(WUS)ndashCLAVATA (CLV) signaling pathway WUS positively regulates cell
proliferation In contrast CLV3 which is expressed in stem cells limits the WUS
expression and thus inhibits cell proliferation in the SAM (Sharma et al 2003)
Consistent with this notion WUS is expressed to a higher level in jba-1D
explaining the ectopic enlargement of the SAM in the mutant (Williams et al 2005)
While WUS is closely related to miR165166 in the shoot apical meristem
development the underlying molecular mechanisms are currently unclear It has been
observed that the miR166-over producing men1 plants have an arrested meristem
development (Jung amp Park 2007) In addition the heterozygous men1+ and the
homozygous men1 plants show different expression patterns of WUS and CLV3 It
appears that miR166 regulates WUS indirectly and influences the expressional balance
between WUS and CLV3 through a negative feedback loop (Jung amp Park 2007 de
Alba et al 2013)
The phv phb athb15 triple mutant has an enlarged shoot apical meristems the
rev phb phv mutant does not have functional shoot apical meristems (Prigge et al
2005) This distinction may be explained by the fact that while miR166 targets
primarily PHB PHV and ATHB15 miR165 targets all the five HD-ZIP III genes (Zhou
et al 2007) Consistent with this transgenic plants overproducing miR166 are distinct
from those overproducing miR165 The miR165-overproducing transgenic plants lack
functional shoot apical meristem and a pin-like structure is formed at the apical region
of the mutant These phenotypes are quite similar to rev phb phv triple mutant (Zhou et
al 2007 Liu et al 2013) The phenotypic distinction may because by the difference
of the cleavage efficiencies of the target mRNAs In accordance with this notion a few
35S miR166a transgenic lines exhibit no shoot apical meristems The men1
homozygous plants also show arrested meristem development (Jung amp Park 2007)
Chapter 1 Introduction and Review of Literature
46
MiR164 cleaves the CUC1 and CUC2 mRNAs as well as the NAC1 mRNA
CUC1 and CUC2 determines the organ boundary in the meristem Phenotypes of
transgenic plants that overproduce miR164 are similar to those of the cuc1cuc2 double
mutant that exhibits embryonic developmental defects such as cup-shaped cotyledons
and fused cotyledons (Laufs et al 2004) When a miR164-resistant CUC2 is
expressed the boundary domain is enlarged CUC also plays a role in embryonic shoot
meristem formation by regulating the SHOOT MERISTEMLESS gene It was therefore
concluded that miR164-mediated cleavage of CUC1 and CUC2 mRNAs determines the
sizes of the boundary domains in the shoot apical meristem and regulates separation of
the organs by restricting cell proliferation (Laufs et al 2004 de Alba et al 2013)
11364 Leaf development
The role of miR319 has been extensively studied in leaf development A dominant
jaw-D mutant overproducing miR319 shows uneven curved leaves with enlarged size
and five TCP genes which are closely related to the CINCINNATA (CIN) gene in
Antirrhinum majus are down regulated in the mutant suggesting that miR319 mediated
regulation of the TCP transcription factor genes are important for the final step of
leaf shaping (Palatnik et al 2003 Naz et al 2013) In addition to leaf shape and size
leaf polarity is also specified by miRNA MiR166-mediated cleavage of the HD-ZIPIII
mRNAs is a well to establish leaf polarity This discovery originates from the gain-of-
function mutants such as phb-1d and phv-1d wherein point mutations in the
complementary sites to miRNA helps the mRNA to evade or resist from miRNA-
mediated cleavage (McConnell et al 2001 Emery et al 2003 Naz et al 2013)
Plant leaves have a dorso-ventral polarity consisting of the adaxial (upper
surface) and abaxial (lower surface) sides The adaxial side is closer to the shoot
apical meristem Adaxial identity is specified by functionally redundant class III HD-
ZIP family genes such as PHB PHV and REV Ectopic expression of each genes
result in adaxialized leaves Dominant phb-1d and phv-1d mutants exhibit
transformation of the abaxial to adaxial fate and have rod-shaped leaves In contrast
miR165166-overproducing plants show pin-like cotyledons radicalized leaves and
abaxialized leaves (Chitwood et al 2007 Pattanayak et al 2013)
Leaf asymmetry is determined by miR166-mediated restriction of the
expression domains of the HD-ZIPIII genes The HD-ZIPIII genes are expressed in the
Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
18
110 Genetic engineering of Coffee
Genetic transformation has potential applications in coffee agriculture for incorporating
genes which could provide desirable traits such as disease and insect resistance
drought and frost tolerance and herbicide resistance Transgenic technology can also be
used to increase nutritional value and improve cup quality produce varieties with
caffeine-free beans and for production of hybrid crops for molecular farming
Identification of target specific genes is one of the pre-requisites for developing
transgenic crops The availability of a large number of EST sequences in coffee and
initiation of coffee genome sequencing may speed up the gene discovery and accelerate
transgenic research efforts in coffee
1101 Insect Resistance
Production of insect resistant coffee plants is one of the major objectives of the
breeding programs The major pests attacking coffee include coffee berry borer (CBB
Hypothenemus hampei) white stem borer (WSB Xylotrechus quadripes) leaf miner
(Perileucoptera coffeela) and root nematodes (Meloidogyne spp and Pratylenchus
spp) The CBB is present in almost all the coffee growing countries and considered to
the most devastating pest in coffee To date there is no reported source of resistance to
CBB in the coffee gene pool Like CBB WSB is another serious pest in C arabica in
India and several other East Asian countries Both CBB and WSB belong to the order
Coleoptera For India controlling WSB is the biggest challenge and has therefore
become the highest research priority Robusta is generally resistant to WSB but the
interspecific Robusta Arabica hybrids are susceptible to WSB Although leaf miner is
not yet a serious pest in India and other East Asian countries it is an economically
important pest in East Africa and Brazil Effective chemical control of CBB and WSB
is difficult due to the nature of their life cycle inside the berry and stem respectively as
well as environmental concern regarding the use of pesticides Biological control
measures are adapted to combat these insect pests with varying degrees of success
Developing coffee plants resistant to these pests using genetic transformation
technology could be one of the alternative strategies to counter pest damage For insect
resistance several different classes of proteins from bacterial plant and animal sources
have been isolated and their insecticidal properties tested against many important pests
Amongst these proteins Bt toxins are most important and several transgenic crops
expressing Bt genes have been commercialized Coffee transgenic plants carrying a
Chapter 1 Introduction and Review of Literature
19
synthetic version of the cry1Ac gene have been produced (Leroy et al 2000 Mishra et
al 2002) Indeed this was the first report of an important agronomic trait being
introduced into a coffee plant The transgenic plants presented similar features in
growth and development compared to normal plants Transgenic plants highly resistant
to leaf miner under greenhouse conditions were tested under field conditions in French
Guyana for 4 years for pest resistance (Perthius et al 2005 Perthius et al 2006)
From 54 independent transformation events 70 of the events were resistant to leaf
miner Unfortunately the field trial was vandalized which led to the termination of the
experiment (Montagnon et al 2005)The effectiveness of Bt genes in controlling
coleopteran pests is well documented in corn and potato which indicates that Bt genes
might be effective against CBB The high toxicity of B thuringiensis sero var
israelensis against first in star larvae of CBB has been demonstrated (Mendez et al
2003) In another experiment an α-amylase inhibitor from Phaseolus vulgaris was
tested against CBB and found to have an inhibitory effect on its growth and
development (Grossi et al 2004)In addition to the CBB and WSB C arabica
varieties are also susceptible to endoparasitic root-knot nematode (Meloidogyne spp)
(Campos et al 1990 Mishra et al 2012)
So far 15 species have been reported to be parasites of coffee Controlling
nematodes is extremely difficult and currently seedling grafting with robusta rootstock
is followed as one of the control measure Sources of resistance specific to root-knot
nematodes have been identified in coffee trees (Bertrand et al 2001) and the Mex-1
gene conferring resistance to M exigua in C arabica is in the process of isolation (Noir
et al 2003)
1102 Tolerance to Abiotic Stress
In many coffee growing countries abiotic stress such as drought and frost are the major
climatic factors that limit coffee production Changes in climatic patterns due to global
climate change are considered increasingly important for coffee cultivation Drought
induces water stress in plants which affects vegetative growth and vigor and triggers
floral abnormalities and poor fruit set It also indirectly increases the incidence of pests
and diseases in the plants C arabica is generally more tolerant to water stress than C
canephora partly due to its extensive deep root system C racemosa is known to be a
good source of drought tolerance In India hybrids between C canephora and C
racemosa have been obtained and are currently under evaluation for drought tolerance
Chapter 1 Introduction and Review of Literature
20
In many coffee growing countries coffee is propagated in marginal areas where the
annual rainfall is below 1000thinspmm with prolonged dry spells of over 4-5 months In
those areas water shortage and unfavorable temperatures constitute major constraints
and the growth and productivity of C canephora are badly affected As with drought
periodic frost also affects coffee production in parts of Brazil The introduction of
drought and frost tolerant genes through genetic transformation would be of great
importance for alleviating these problems Research is now being carried out by several
groups to identify genes involved in biotic as well as abiotic stress The most promising
genetic engineering approach for drought tolerance includes the use of functional or
regulatory genes as well as the transfer of transcription factors In recent years plants
tolerant to high temperature and water stress have been the subject of intense research
(Chaves et al 2003 Chaves et al 2004 Coraggio et al 2004) For achieving drought
tolerance genes that have been targeted include those encoding enzymes involved in
detoxification or osmotic response metabolism enzymes active in signaling proteins
involved in the transport of metabolites and regulating the plant energy status (Chaves
et al 2003 Chaves et al 2004 Coraggio et al 2004) The dissection of molecular
mechanisms related to signal transduction and transcriptional regulation might help in
engineering drought tolerance in coffee
1103 Disease Resistance
Coffee leaf rust (CLR) caused by the fungus Hemileia vastatrix is the most important
disease in coffee with substantial loss to coffee production and productivity in all the
coffee growing countries In addition to leaf rust coffee berry disease (CBD) caused by
fungus Colletotrichum kahawae can be a devastating anthracnose leading to substantial
crop loss in Africa Several other fungal and bacterial diseases may also affect coffee
to a extent C arabica is more susceptible to many diseases than C canephora Though
most of the disease control measures rely upon chemical control they are more
expensive and labour intensive The long-term solution is the breeding of resistant
varieties which is the focus of many breeding programs However breeding for disease
resistant varieties is time consuming due to the perennial nature of coffee with its long
gestation period India has a long history of C arabica breeding especially for leaf rust
resistance and is the first country to demonstrate the existence of multiple races of leaf
rust Resistance to CLR is conditioned primarily by a number of major (SH) genes and
coffee genotypes are classified into different resistance groups based on their
Chapter 1 Introduction and Review of Literature
21
interaction with different rust races pathogen (van der Vossen 2001) Currently C
canephora provides the main source of resistance to pests and diseases including CLR
(H vastatrix) and CBD (C kahawae) and therefore used in breeding programs Other
diploid species like C liberica and C racemosa are being used as a source of resistance
to coffee leaf rust and coffee leaf miner respectively (Guerreiro et al 1999
Sreenivasan et al 1989) The development of coffee varieties resistant to major fungal
diseases such as CLR and CBD using transgenic technology will benefit the coffee
industry immensely During the last 15 years significant progress has been made in the
area of host-pathogen interactions (Staskawicz et al 1995 Feys et al 2000
Shivaprasad et al 2012) and many resistance genes involved in recognizing invading
pathogens have been identified and cloned (Takken et al 2000) A number of
signalling pathways induced because of pathogen infection have been dissected (Dong
et al 1998 Navarro et al 2006) Many antifungal compounds synthesized by plants to
combat fungal infection have been identified (Does et al 1998) Understanding the
specific induction of targeted pathways and identification of specific pathways
responsible for particular fungal resistance is important in order to employ this strategy
in transgenic technology The recent investigation of gene expression during coffee leaf
rust infection could provide an insight into the defence pathways operating in coffee
(Fernandez et al 2004 Nardi et al 2006) Efforts have been made to identify and
clone resistance genes from coffee for achieving durable resistance The genetic and
physical map of two resistance genes SH3gene conferring resistance to rust (Prakash et
al 2004) and the Ck1 gene conferring resistance to C kahawae CBD (Gichuru et al
2006) have been established These genes could be used for molecular marker assisted
breeding programs (Mishra et al 2012)
1104 Production of low-caffeine coffee
A widespread belief that the ingestion of caffeine can have adverse effects on
health resulted in the increased demand for decaffeinated coffee (Ashihara amp Crozier
2001) Several method were used to obtain decaffeinated plants by conventional
breeding techniques using low yielding varieties and mutants that accumulated
relatively low amounts of caffeine when compared to the commercially available ones
Low-caffeine and decaffeinated coffee represent around 10 of the coffee sales
around the world (Ribas et al 2006b) The industrial process for coffee decaffeination
can be expensive and affects the original flavour and aroma in coffee (David 2002)
Chapter 1 Introduction and Review of Literature
22
Transgenic coffee plants with suppressed caffeine synthesis using RNA interference
(RNAi) technology have been obtained (Ogita et al 2003 Ogita et al 2004) Specific
sequences in the 3prime untranslated region of the theobromine synthase gene (CaMXMT1)
were selected for construction of RNAi short and long fragments The caffeine and
theobromine content of the transgenic plants were reduced by ~ 70 when compared to
the untransformed plants In C canephora RNAi technology has also been employed
to silence the N-methyl transferase gene involved in caffeine biosynthesis (Vinod et al
2007) Promoter of an N-methyltransferase (NMT) gene involved in caffeine
biosynthesis was cloned (Satyanarayana et al 2005 Satyanarayana 2006) which will
be very useful for studying the regulation of caffeine biosynthesis
1105 Improvement in cup quality
Improvement in coffee cup quality requires elaborate knowledge of the chemical
constituents as well as the metabolic pathways involved in the elaboration of quality
The constituents of coffee beans include minerals proteins carbohydrates caffeine
chlorogenic acids (CGA) glycosides lipids and many volatile compounds that give
flavour to coffee by roasting Among these the role of three major constituents
sucrose CGA and trigonelline have been studied in coffee The sucrose content of
coffee bean is associated with coffee flavour the higher the sucrose content in green
beans the more intense will be the cup flavour (Clifford et al 1985 de Maria et al
1996) The sucrose content of C arabica (82-83) is higher than C canephora (33ndash
40) The sucrose amino acids ratio in green beans determines the profile of volatile
compounds Manipulating sucrose content in coffee bean is therefore important in
improving cup quality The sucrose synthase gene (CcSUS2) from C canephora has
been cloned and sequenced (Leroy et al 2005) This provides an opportunity to
manipulate the sucrose content in coffee Chlorogenic acids are products of
phenylpropanoid metabolism Chlorogenic acids are a group of hydroxycinnamoyl
quinic acids (HQA) formed by esterification between caffeic acids coumaric acids and
quinic acids (Clifford et al 1999) They are present in relatively large quantities in the
coffee bean and are the precursor of phenolic compounds in roasted beans C
canephora beans contain higher CGA (10) compared to C arabica beans (6-7)
CGA are known to have antioxidant properties as well as being associated with disease
resistance (Campa et al 2003) Genetic manipulations of genes involved in CGA
synthesis can serve either of these purpose by up- or down regulating the pathway
Chapter 1 Introduction and Review of Literature
23
Phenylalanine ammonia-lyase (PAL) catalyzes the first step of the phenylpropanoid
pathway leading to the synthesis of a wide range of chemical compounds including
flavonoids coumarins hydroxycinnamoyl esters and lignins (Hahlbrock amp Scheel
1989) Recently the full length cDNA and corresponding genomic sequences of PAL
from C canephora was isolated characterized and functionally validated (Mahesh et
al 2006 Parvatam et al 2006) This has opened up new possibilities for manipulating
the level of the PAL enzyme in coffee which in turn can be useful for improvement of
cup quality and manipulating antioxidant properties in coffee
1106 Fruit Ripening
Uniformity during fruit ripening is decisively related to cup quality in coffee and
consequently to the value of the product Fruits at the ideal ripening stage produce the
best organoleptic characteristics for coffee The presence of over ripened or green fruits
changes the acidity the bitterness and consequently the cup quality In order to
maximize uniform ripening of coffee fruits it is essential to control the action of genes
involved in the last step of maturation process Ethylene is known to trigger ripening
and increasing ethylene biosynthesis is associated with various stages of ripening
process (Protasio et al 2005) To control coffee fruit maturation two of the major
genes involved in ethylene biosynthesis namely ACC synthase and ACC oxidase
have been cloned (Protasio et al 2005 Neupane et al 1999) Introduction of the ACC
oxidase gene in antisense orientation have been achieved in both C arabica and C
canephora The effect of the transgene on ethylene production and fruit maturation has
yet to be reported The inhibition of genes downstream to the initial ethylene burst is
also an option to control coffee fruit maturation (Ribas et al 2006)
111 RNA interferenceRNAi
The regulation of gene expression at post transcriptional level has recently attracted
much attention because of the discovery of the phenomenon called RNA interference
(RNAi) RNAi based silencing techniques are more efficient than antisense mediated
gene silencing (Wesley et al 2001) The effect was first observed in plants wherein
silencing was observed when some copies of a transgene was in the forward orientation
with respect to the promoter and some in the reverse orientation This resulted in the
formation of double stranded RNA in the system The phenomenon in which
experimentally introduced double stranded RNA (dsRNA) leads to the loss of
expression of the corresponding cellular gene by sequence specific RNA degradation
Chapter 1 Introduction and Review of Literature
24
was termed as RNA interference or RNAi The protective effect of RNA silencing in
reducing virus infectivity supports the view that PTGS has evolved as a mechanism to
defend plants against virus infection and also to moderate the possible deleterious
genome-restructuring (insertional) activity of virus-like mobile genetic elements eg
retrotransposons A growing body of biochemical and genetic data has further
demonstrated that plants animals and yeasts share related mechanisms of specific
degradation of RNAs in which double-stranded forms of RNA are initiator molecules
(Brenstein et al 2001)
The key insight into the process of PGTS was provided by the experiments
conducted by Hamilton amp Baulcombe (1999) who identified the product of RNA
degradation as small RNA (siRNA) species of ~25nt of both sense and antisense
polarity SiRNA are formed and accumulate as double stranded RNA molecules of
defined chemical structures Both genetic and biochemical approaches were undertaken
to understand the basis of silencing Genetic screens were carried out in fungus
Neurospora carassa the alga Chalmydomonas reinhardtii and plant Arabidopsis
thaliana to search for detective mutants in PTGS
A combination of results obtained from several in vivo and in vitro experiments
have gelled into a two step mechanistic model for RNAiPTGS The first step referred
to as the RNAi initiating step involves binding of the RNA nucleases to a larges
dsRNA and its cleavage into discrete ~21-25nt RNA fragments This is ATP-
dependent reaction which involved RNase III - type endonucleases which make
staggered cuts in both strands of the dsRNA leaving 3prime overhang of 2 nucleotides with
5prime- phosphate 3prime-hydroxyl and no modification in the sugar phosphate backbone
(Hamilton amp Baulcombe 1999 Elbashir et al 2001) SiRNAs are the direct products
of dsRNA cleavage by the multi-domain RNase III enzyme DICER (Figure 16)
(Brenstein et al 2001 Karthikeyan et l 2013)
In the second step or commonly known as the effector step the double stranded
siRNAs produced in the first step bind to an RNAi specific protein complex to form a
RISC The complex undergoes activation in the presence of ATP so that the antisense
component of the unwound siRNA becomes exposed and allows the RISC to perform
the downstream RNAi reaction
Chapter 1 Introduction and Review of Literature
25
Several enzymes are involved in RNAi based on their role genes that encode for them
are classified into RNAi initiators RNAi effectors RNA dependent RNA polymerases
and silencing genes Two C elegans genes rdeI and rde4 (rde stands for lsquo RNAi
deficientrsquo) are believed to be involved in the initiation step of RNAi The C elegans
rde1 gene is a member of a large family of genes and is homologous to the Neurospora
qde2 (qde stands for ldquoquelling deficientrdquo) and Arabidopsis AGO1 (AGO stands for
agronaute AGO1 was previously identified to be involved in Arabidopsis
development) Although the functions of these genes are not clear a mammalian
member of RDE1 family has been identified as a translation initiation factor (Thakur
2005) Important genes for the effector step of PTGS in C elegans are rde2 and mut7
genes These genes were identified from heterozygous mutant worms that were unable
to transmit RNAi to their homozygous offsprings Worms with mutated rde2 or mut 7
genes show defective RNAi mut-7 gene encodes a protein with homology to the
nuclease domains of RNAase D and a protein implicated in Werner syndrome (a rapid
ageing disease in humans) (Grishok et al 2001 Kamath-Loeb et al 2012s)
Neurospora qde-1 Arabidopsis SDE-1SGS-2 and C elegans ego-1 appear to encode
RNA dependent RNA polymerase (RdRPs) It assumed that this is a proof that an RdRp
activity is required for RNAi Certainly the existence of an RdRp might explain the
remarkable efficiency of dsRNA induced silencing if it amplifies either the dsRNA
prior to cleavage or the siRNAs directly (Muers 2013) In C elegans ego-1 mutants
(ego stands for lsquoenhancer of glp-1) RNAi functions normally in somatic cells but is
defective in germline cells where ego-1 is primarily expressed In Arabidopsis SDE-
1SGS-2 mutants (SGS stands for suppressor of gene silencing) siRNA are produced
when dsRNA is introduced via an endogenously replicating RNA virus but not
introduced by a transgene It has been proposed that perhaps the viral RdRP is
substituting for the Arabidopsis enzyme in these mutants Random degradative PCR
model suggests that an RdRP uses the guide strand of an siRNA as a primer for the
target mRNA generating a dsRNA substrate for Dicer and thus more siRNAs
Several genes controlling RNA silencing in plants have been identified through
genetic screens of Arabidopsis mutants impaired in transgene induced RNA silencing
They encode a putative RNA-dependent RNA polymerase (SGS2SDE1) a coiled
protein (SGS3) a protein containing PAZ and Piwi domains (AGO1) and an RNA
helicase (SDE3) The putative SGS2SDE1 of Neurospora is related to QDE-1 and
Chapter 1 Introduction and Review of Literature
26
EGO-1 of C elegans whereas PAZPiwi protein AGO is related to QDE-2 of
Neurospora RDE-1of C elegans RNA helicase SDE3 is related to SMG-2 of C
elegans and Mut-6 of Chlamydomonas MUT-7 gene of C elegans encodes a protein
similar to Rnase D whereas the Drosophila DICER gene encode a protein similar to
RNase III An Arabidopsis ortholog of DICER gene has been identified called CAF
SIN1 SUS1(Thakur 2005 Poethig 2009)
Since the RNAi machinery is present constitutively within the eukaryotic cell it
is important to explore the metabolic advantages that are accorded by the RNAi related
proteins during the intrinsic normal growth of cells and development of organisms The
natural RNAi machinery not only keeps the mobile transposable elements from
disrupting the integrity of the genomes as suggested by the analysis of model organisms
like A thaliana C elegans D melanogaster (Tabara et al 1999 Wu-Scharf et al
2000 Aravin et al 2001 Hamilton et al 2002 Lisch 2009) but also participates in
development of organisms Genetic defects in Celegans RNAi genes ego1 and dicer
cause known specific developmental errors (Grishok et al 2000 Grishok et al 2001
Knight amp Bass 2001 Hall et al 2013) Similarly the Argonaute family of genes of A
thaliana (especially the ZWILLE proteins) is also responsible for plant architecture and
meristem development (Carmell et al 2002 Hamant and Pautot 2010) and the Dicer
homologue of A thaliana CAF1 is required for embryo development (Golden et al
2002 Nakano et al 2011) Thus genetic evidence illustrates the role of the RNAi
machinery as a controller of development related genes The mechanistic details of
these developmental processes are beginning to emerge
Chapter 1 Introduction and Review of Literature
27
Figure 16 Mechanism of gene silencing induced double stranded RNA The dicer enzyme to
produce siRNAs cleaves the RNA The siRNA generated bind to the nuclease complex
RISC The antisense component in the siRNA in the RISC guides the complex towards
the cognate mRNA resulting in endonucleolytic cleavage of the mRNA
Chapter 1 Introduction and Review of Literature
28
112 MicroRNA or MiRNA
In 1991 Ambros and co-workers first isolated a lin-4 mutant of Celegans which was
arrested at the first larval stage (Lee et al 1993) Later on the let-7 mutation was
isolated in the same system which was responsible for development through the fourth
larval stage Both lin-4 and let-7 encode short 22-nucleotide mature RNAs and were
called short temporal RNA because they control the temporal development program of
C elegans The mature lin-4 RNA defines (negatively regulates) the mRNA expression
of the lin-14 and lin-28 hetrochronic genes with the antisense mediated repression
mechanism of translation initiation and thus specifies the fate of cells during the first
three larval stages Recent studies have revealed that the short temporal RNAs are
actually members of a group of tiny RNAs (21to 28 nucleotides) called the micro-
RNAs (miRNA) (Bartel 2009) isolated members of which could easily run to a few
thousand which includes both those were identified computationally and by deep
sequencing Some of the components of the RNAi machinery have also been clearly
established as the effector proteins for the maturation of miRNAs
113 MicroRNAs in plants
1131 Discovery
MicroRNAs have been discovered by three basic approaches Direct cloning forward
genetic direct cloning and bioinformatic prediction followed by experimental
validation The most direct method of miRNA discovery is to isolate and clone small
RNAs from biological samples and several groups have used this approach to identify
plant miRNAs (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002 Xie et al 2003 Sunkar et al 2005 Williams et al 2013) The cloning methods
adapted were similar to those used to identify large numbers of animal miRNAs
(Lagos-Quintana et al 2001 Lau et al 2001 Aravin amp Tuschl 2005) which involve
isolating small RNAs followed by oligonucleotide adapter ligation reverse
transcription amplification and sequencing Some protocols incorporate methods to
enrich for Dicer cleavage products (ie molecules with 5prime-phosphates and 3prime-
hydroxyls) and to concatemerize the short cDNAs so that several may be identified in a
single sequencing read (Lau et al 2001 Llave et al 2002a Jagadeeswaran amp Sunkar
2013)The initial cloning experiments in Arabidopsis identified 19 miRNAs belonging
to 15 families (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002) although hundreds of other small RNAs such as siRNAs and RNA degradation
Chapter 1 Introduction and Review of Literature
29
fragments were also cloned through the direct cloning methods To select miRNAs
from the pool of small RNAs secondary structures of the RNA molecules were
examined in compliance with the acknowledged structural features of miRNAs
(Ambros et al 2003) The secondary structures were validated using several ad-hoc
software tools such as the Mfold or RNAfold programs (Reeder et al 2006) Finally
the existence of mature miRNAs was determined by RNA gel blot hybridization These
direct cloning methods were successful in isolating many miRNAs in Arabidopsis
(Reinhart et al 2002 Sunkar amp Zhu 2004) Rice (Sunkar et al 2005) Poplar (Lu et
al 2005) moss (Arazi et al 2005) and Tobacco (Billoud et al 2005)
Although miRNAs were first discovered through forward genetic screens in
worms (Lee et al 1993 Reinhart et al 2000)only few A thaliana miRNA gene
families have been discovered by this method (Chen 2008) in plants MiRNA
involvement in plant mutant phenotypes were not properly inferred till the cloning
experiments established that plant genomes contain numerous miRNAs (Park et al
2002 Reinhart et al 2002 Rhoades et al 2002) MiRNA loss-of-function allele has
been identified by forward genetic screens early extra petala1 is caused by a
transposon insertion ~160bp upstream of the predicted miR164c stem loop resulting in
flowers with extra petals (Baker et al 2005 Irish 2008) The fact that loss-of-function
miRNA mutants have been rarely discovered using forward genetics reflects small
target size for mutagenesis coupled with redundancy in nearly all evolutionarily
conserved plant miRNAs which are encoded by gene families (Table16) Family
members are likely to have overlapping functions buffering against loss at any single
miRNA locus Over expression screens can circumvent redundancy limitations At least
four plant miRNAs miR319 (also known as miR-JAW) miR172 (also known as EAT)
and miR166 were isolated in over expression screens for dominant mutants with
developmental abnormalities (Aukerman amp Sakai 2003 Palatnik et al 2003 Kim et
al 2005 Williams et al 2005b Schommer et al 2012) Mutations in miRNA target
sites which can prevent the entire family of miRNAs from repressing a target gene can
also circumvent redundant functions of miRNA family members
The dominant mutations in the HD-ZIP genes PHB PHV and REVOLUTA
(REV) in Arabidopsis and ROLLEDLEAF1 (RLD1) in Maize result in adaxialization of
leaves andor Vasculature (McConnell amp Barton 1998 McConnell et al 2001 Nelson
et al 2002 Zhong amp Ye 2004 Allen amp Millar 2012) and are all caused by mutations
Chapter 1 Introduction and Review of Literature
30
in miR166 complementary sites (Rhoades et al 2002 Emery et al 2003 Jones-
Rhoades amp Bartel 2004 Mallory et al 2004 Zhong amp Ye 2004)
In both plants and animals cloning was the initial means of large-scale
miRNA discovery (Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Reinhart et al 2002 Mendes et al 2012) However cloning is biased toward
RNAs that are expressed highly and broadly MiRNAs expressed at low levels or
only in specific cell types or in response to certain environmental stimuli were more
difficult to clone Sequence based biases in cloning procedures might also cause
certain miRNAs to be missed Because of these limitations bioinformatics approaches
to identify miRNAs have provided a useful complement to cloning
A straightforward use of bioinformatics has been carried out to find homologs
of known miRNAs both within the same genome and in the genomes of other species
(Pasquinelli et al 2000 Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Mendes et al 2009) A more difficult challenge is to identify miRNAs
unrelated to previously known miRNAs This was first accomplished for
vertebrate nematode and fly miRNAs using algorithms that search for conservation
of sequence and secondary structure (ie miRNA stem-loop precursors) between
species searching for patterns that are characteristic of miRNAs (Lai et al 2003 Lim
et al 2003 Lim et al 2005)
Most of the methods identified numerous miRNAs but are necessarily more
relaxed in terms of allowed structures Some approaches take advantage of the high
complementarity of plant miRNAs to target messages implementing the requirement
that the candidate has conserved complementarity to mRNAs (Jones-Rhoades amp Bartel
2004 Adai et al 2005) This additional filter has been useful for distinguishing
authentic plant miRNAs from false positives which later extended to mammalian
miRNA gene prediction (Xie et al 2005 Mendes et al 2012)
Another criteria used by most algorithms in the filters is evolutionary
conservation In plants mature miRNA sequences are conserved to a higher degree
than their precursor sequences For example Arabidopsis and rice diverged more than
130 million years ago (Friis et al 2004) but some miRNAs are highly conserved
in these plant species (Reinhart et al 2002) Furthermore various parameters for the
secondary structures such as free energy the number of paired residues within a
Chapter 1 Introduction and Review of Literature
31
miRNA or the number and size of bulges are also used to validate the predictions
(Jones-Rhoades amp Bartel 2004 Wang et al 2004 Adai et al 2005 Mendes et al
2012)
1132 Conserved microRNAs in Plants
Till date cloning genetics and bioinformatics screens have resulted in annotation of
approximately 184 potential Arabidopsis miRNAs (miRBase release 13) These
miRNAs seem to arise through gene duplication and subsequent diversification and
represent approximately 100 families (Maher et al 2006 Axtell 2013) Twenty one
families represented by 92 genes are clearly conserved in species beyond Arabidopsis
(Table 15) The presence of these miRNA families in other sequenced plant genomes
indicates that different plant species have an evolutionarily fluid set of miRNAs
(Willmann amp Poethig 2007) Recent studies on mosses one of the most ancient land
plants have shown that at least 14 miRNA families are conserved in eudicots and
mosses and therefore it appears that plant miRNAs share a common ancient origin
(Arazi et al 2005 Axtell amp Bartel 2005 Talmor-Neiman et al 2006 Zhang et al
2006 Axtell 2013) The number of members per family in a genome ranges from 1-32
with the exception of miR-430 which is represented by a cluster of ~80 loci in zebra
fish (Giraldez et al 2005)
In plants the number of members in each family correlates among examined
species certain families contain numerous members in all three species (eg miR156
miR166 miR169) whereas others consistently contain fewer genes (eg miR162
mir168 miR394) (Table 16) Although it is still unclear why certain miRNA are
encoded by more than one gene in plants (eg 12 genes encoding miR156) this
correlation might suggest functional significance of various miRNA family sizes Most
annotated plant miRNAs are conserved throughout flowering plants while a few
miRNAs are found only in a single sequenced genome and thus they could be
evolutionarily lsquoyoungrsquo miRNAs For example Arabidopsis has several miRNA
families that are not found in poplar or rice suggesting that miRNAs could be
generated continuously during evolution (Allen et al 2004a Rajagopalan et al
2006 Fahlgren et al 2007 Axtell 2012 de Alba et al 2013) Consistent with the
notion that non conserved miRNAs are of a more recent evolutionary origin these
miRNAs are mainly encoded by a single locus in the genome Within each family the
mature miRNA is always located in the same arm
Chapter 1 Introduction and Review of Literature
32
Table 16 MicroRNA families conserved in plants
miRNA
family
Arabidopsis Oryza Populus
miR156 12 12 11 miR159319 6 8 15 miR160 3 6 8 miR162 2 2 3 miR164 3 5 6 miR166 9 12 17 miR167 4 9 8 miR168 2 2 2 miR169 14 17 32 miR171 4 7 10 miR172 5 3 9 miR390 3 1 4 miR393 2 2 4 miR394 2 1 2 miR395 6 19 10 miR396 2 5 7 miR397 2 2 3 miR398 3 2 3 miR399 6 11 12 miR408 1 1 1 miR403 1 0 2 miR437 0 1 0 miR444 0 1 0 miR445 0 9 0 Total 92 127 169
All known miRNA families that are conserved between more than one plant species are listed
together with the number of genes identified in the sequenced genomes Rice miRNA families that have orthologs in maize but do not appear to have orthologs in the eudicots (Arabidopsis and Populus) are marked with an asterisk The following families contain miRNA genes annotated with
more than one number miR156 (miR156 and miR157) miR159319 (miR159 and miR319) miR166 (miR165 and miR166) miR171 (miR170 and miR171) and miR390 (miR390 and miR391)
of the stem loop (5prime- or 3prime) as it would be expected if the genes carry common ancestry
(Figure 17) The sequence of the mature miRNA and to some extent the segment on
the opposite arm of the hairpin to which it pairs is highly conserved between the
members of the same family (both within and between the species) while the sequence
secondary structure and length of the intervening ldquolooprdquo region can be highly divergent
within the same family
The patterns of paring and non pairing nucleotides are often conserved between
homologous miRNA stem loops from different species (Figure 17) The significance of
conserved mismatches is opined to guide DCL1 to cleave at the appropriate positions
along the stem loop
Chapter 1 Introduction and Review of Literature
33
Figure 17 Representative stem loop of miR164 Segments corresponding to the mature miRNAs
are shown in red (Adapted from Rajagopalan et al 2006)
Although many miRNAs are highly conserved in plants the degree of conservation in
most cases is quite low between plants and animals one exception is miR854 A recent
study has revealed that miR854 is conserved between Arabidopsis and animals and that
the Arabidopsis miR854 differs from the human miR854 only by a single nucleotide
(Arteaga-Vazquez et al 2006 Axtell 2012 de Alba et al 2013) The Arabidopsis
miR854 has multiple binding sites in the 3prime-untranslated region (UTR) of
OLIGOURIDYLATE-binding-PROTEIN mRNA 1bA (UBP1b) and represses the
translation of the UBP1b mRNA The animal miR854 is also complementary to a site in
the 3prime-UTR of the UBP1b homolog in animals However it is currently unclear how the
animal miR854 regulates its target mRNA Another distinction between plant and
animal miRNAs is the genomic organization of the miRNA genes While plant miRNA
genes are located predominantly in the intergenic regions animal miRNA genes tend to
localize in the intragenic regions both in the introns or exons of protein coding genes
Moreover miRNA genes are often clustered within a single precursor transcript
whereas individual plant miRNAs are derived from individual precursor RNAs (Bartel
Chapter 1 Introduction and Review of Literature
34
2004 Bonnet et al 2004 Kim 2005 Vaucheret et al 2006 Marco et al 2013)
1133 Non-conserved miRNA and challenges in annotation
Most miRNAs are conserved throughout flowering plants (Table 15) but many more
were found only in a single sequenced genome thus considered to be of a more recent
evolutionary origin The extended homology between few non conserved miRNAs
precursors and their target genes provides strong evidence that these relatively ldquoYoungrdquo
miRNAs arose from the duplication of the target gene segments (Allen et al 2004)
Several non-conserved miRNAs including miR161 miR163 miR173 miR447
miR475 and miR476 are known to direct cleavage of target transcripts (Allen et al
2004a Adai et al 2005 Sunkar et al 2005 de Alba et al 2013) It is difficult to
confidently predict targets for many because it is not possible to use complementary
site as a filter against false-positive target predictions It is also difficult to
confidently annotate non-conserved miRNAs as miRNA rather than siRNAs The
established minimal standard for miRNA annotation is a small RNA with detectable
expression and the potential to form a stem-loop when joined to flanking genomic
sequence (Kasschau et al 2003) In practice these requirements are too loose to
definitively categorize many small RNAs cloned from plants Many plant siRNAs are
detectable on blots (Vazquez et al 2004b) and hundreds of thousands of non-
miRNA genomic sequences can be predicted to fold into secondary structures that
resemble structures of plant miRNA precursors (Jones-Rhoades amp Bartel 2004)
Therefore without conservation of both sequence and secondary structure it is
difficult to be confident that a given cloned RNA originated from a stem-loop (ie is a
miRNA) rather than from a double-stranded RNA (ie is a siRNA) In fact many of
the thousands o f small RNAs cloned from Arabidopsis (Gustafson et al 2005) would
probably meet the literal requirements for annotation as miRNAs A few of these
sequences might be miRNAs but others that meet the literal criteria probably are not
so The challenges of annotating non conserved small RNAs were evidenced by
three related small silencing RNAs that were originally annotated as miRNAs
that turned out to be trans-acting siRNAs (tasiRNAs)(Kanno et al2013)
Although most miRNAs that are broadly conserved among flowering plants are
now being identified and experimentally validated (Table 16)(Jones-Rhoades amp
Bartel 2004) the challenges and ambiguities for classifying non-conserved miRNAs
prevent meaningful estimates of the total number of miRNA genes in Arabidopsis and
Chapter 1 Introduction and Review of Literature
35
other plant genomes It is possible that miRNAs might have escaped detection
because they are non-conserved and expressed in specific tissues or conditions and can
only be identified by using high- throughput sequencing
1134 MicroRNA biogenesis
11341 Transcription of microRNA precursor
Plant miRNAs are primarily found in the genomic regions that are not associated with
the protein coding genes (Reinhart et al 2002) and it appears that most if not all plant
miRNAs are produced from their own transcriptional units Plant miRNA genes are
occasionally clustered near each other in the genome suggesting transcription of
multiple miRNAs from a single primary transcript [eg the miR395 cluster (Grishok
et al 2001 Jones-Rhoades amp Bartel 2004b)] but this polycistronic arrangement of
miRNA genes appears far less frequently in plants than in animals (Bartel 2004)
Northern blot EST and mapping evidence indicate that plant miRNA primary
transcripts (also known as pri-miRNAs) as in animals are longer than needed to
encompass the miRNA stem-loops (Palatnik et al 2003 Jones-Rhoades amp Bartel
2004b) At least some of these pri-miRNA transcripts appear to be spliced
polyadenylated (Kurihara amp Watanabe 2004) and capped (Xie Z et al 2005) Two
rice miRNAs are contained within transcripts that contain exon junctions within the
presumptive stem-loop precursor implying that in these cases splicing is a prerequisite
for Dicer recognition (Sunkar et al 2005) Plant pri-miRNAs are over 1kb in length
and they are usually preceded by typical TATA box motifs They can also undergo
canonical splicing polyadenylation and capping All these observations indicates RNA
polymerase II is probably responsible for transcribing most plant miRNAs (Xie Z et
al 2005) as appears to be the case for many animal miRNAs Relatively little is
known about the regulation of miRNA transcription in plants but there is no reason
to suspect that this regulation would differ from that of protein-coding transcripts as
the bioinformatics analysis of the sequence upstream of the transcriptional start
sites of the miRNA genes showed the presence of putative binding motifs of
number of known transcription factors (Megraw et al 2006 Sethupathy 2013)
11342 MicroRNA processing and export
In plants maturation of miRNA is a step wise process A miRNA gene is transcribed to
pri-miRNA which is much longer than the pre-miRNA possessing the characteristic
stem-loop structure Like transcription of most protein-coding genes the miRNA
Chapter 1 Introduction and Review of Literature
36
transcription is processed by RNA polymerase II (Pol II) enzymes and the pri-
miRNAs are 5prime-capped and 3prime-polyadenylated (Lee et al 2004 Xie Z et al 2005)
Mature miRNAs are produced from pri-miRNAs through at least two sequential
processing steps by RNase III-type endonucleases In animals pri-miRNAs are first
processed at the bottom portion of the stem by Drosha a nuclear-localized RNase III
enzyme to liberate pre-miRNAs The pre-miRNAs are then exported to the cytoplasm
with the help of exportin-5The pre-miRNAs are further processed by Dicer another
RNaseIII enzyme to a duplex consisting of the miRNA and the antisense strands
(miRNA) Since plants do not have any Drosha homologs the two sequential
processing steps seem to be carried out by DCL1 a Dicer homolog in the nucleus
(Kurihara amp Watanabe 2004 de Alba et al 2013 Rogers amp Chen 2013)
The processing of pri-miRNAs to pre-miRNAs by DCL1 is assisted by two
other proteins HYPONASTY LEAVES1 (HYL1) and SERRATE (SE) HYL1 belongs to a
family of double-stranded RNA (dsRNA) binding protein and interacts with DCL1 in
vitro and in vivo (Lu amp Fedoroff 2000 Hiraguri et al 2005 de Alba et al 2013
Rogers amp Chen 2013) Loss-of-function mutations in the HYL1 gene result in
decreased accumulation of miRNAs and concomitant accumulation of pri-miRNAs
(Han et al 2004 Vazquez et al 2004a Kurihara et al 2006) SE a C2H2 zinc finger
protein interacts with DCL1 and HYL1 and plays a role in the processing of pri-
miRNAs to pre-miRNAs (Lobbes et al 2006 Yang L et al 2006) In addition the
nuclear cap-binding complex proteins CBP20 and CBP80 that are known as ABA
HYPER SENSITIVE 1(ABH1) have been shown to be required for proper miRNA
processing (Gregory et al 2008 Laubinger et al 2008) Recently it has been
demonstrated that DCL1 can release 21-nt RNAs from dsRNA as well as from
synthetic miR167 precursor RNAs in vitro However this cleavage is error prone and
inefficient in the absence of HYL1 and SE which accelerate the rate of DCL1-mediated
cleavage and promote accurate processing of miRNAs (Dong et al 2008 Rogers amp
Chen 2013)
11343 Methylation of the miRNAmiRNAduplex
After the miRNAmiRNAduplexes are released from the pre-miRNAs by the DCL1
activities the duplexes are methylated at the 2primeOH of the3prime-ends by HUA ENHANCER1
(HEN1) a small RNA methyltransferase (Yu et al 2005 Yang Z et al 2006 Rogers
amp Chen 2013 ) This step which is distinct from the RNase III-mediated processing
Chapter 1 Introduction and Review of Literature
37
step distinguishes the biogenesis of plant miRNAs from that of animal miRNAs
Mutations in the HEN1 gene were first isolated in a morphological screening for floral
development although the HEN1 mutants display pleiotropic phenotypes similar to the
developmental defects observed in the DCL1 mutants (Chen et al 2002 Park et al
2002 Rogers amp Chen 2013)
Figure 18 Biogenesis of microRNA
A Biogenesis of plant MicroRNA B Biogenesis of metazoananimal MicroRNA
The miRNA (red) is incorporated into RISC and the miRNA(blue) in degraded
This enzyme has two dsRNA-binding domains and nuclear localization signal It
is also conserved in fungi and is required for the processing of siRNAs as well as of
miRNAs (Park et al 2002 Boutet et al 2003 Xie et al 2003) Although the HEN1
protein is localized to the nucleus its methylation activity in the cytoplasm cannot be
ignored (Xie et al 2004 Rogers amp Chen 2013)
Chapter 1 Introduction and Review of Literature
38
The methylation of the miRNAmiRNA duplex is required to protect miRNAs
against the 3prime-end uridylation activity and subsequent degradation (Li et al 2005) In
the hen1 mutants accumulation of miRNAs is reduced to a lower level Also the
miRNAs appear to be heterogeneous in their sizes such that a ladder of bands rather
than a single species is observed for most miRNAs in the mutants (Li et al 2005)
MiRNA cloning and sequencing analyses revealed the presence of a number of U
residues at the 3prime-end of miRNAs in the hen1 mutants Moreover these nucleotides do
not correspond to the miRNAs in the pre-miRNAs indicating that these nucleotides are
added after the processing of the miRNAs from pre-miRNAs (Li et al 2005)
Uridylation of RNAs has also been proven to be correlated with RNA decay (Shen amp
Goodman 2004 Mullen amp Marzluff 2008) suggesting that uridylation attracts an
exonuclease which degrades miRNAs in plants
11344 MiRNA incorporation in to the RISC and its nuclear export
The methylated miRNAmiRNA duplexes undergo RISC assembly In this process
the miRNA of the duplexes is selectively incorporated into the RISC and the miRNA
appears to be removed and subsequently degraded (Hammond et al 2000 Hutvagner
amp Zamore 2002 Schwarz et al 2003 de Alba et al 2013 Rogers amp Chen 2013)
The ARGONAUTE 1(AGO1) proteins are core components of the RISC complex
They contain two structural domains the N-terminal PAZ domain that binds to the 3prime-
end of single-stranded RNAs and the C-terminal piwi domains that has a structure
similar to that of RNase H (Cerutti et al 2000 Parker et al 2004) Several AGO
proteins possess endonucleolytic activities within the PIWI domain and cleave target
mRNAs in the middle of the site complementary to miRNA (Liu et al 2004 Meister et
al 2004 Miyoshi et al 2005) Mutations in the AGO1 gene cause miRNA target
genes to be ectopically expressed and the levels of some miRNAs are reduced in the
mutants (Kidner amp Martienssen 2004 Vaucheret et al 2004 Kidner amp Martienssen
2005 Ronemus et al 2006) Moreover the phenotype of the AGO1 hypomorphic
alleles display defects in lateral organ polarity and leaf and flower morphology as
observed in the dcl1 mutants and null AGO1 alleles are embryonically lethal
supporting the close relationship between the AGO proteins and miRNA processing
(Kidner amp Martienssen 2004 Vaucheret et al 2004 de Alba et al 2013 Rogers amp
Chen 2013)
The production of miRNA miRNA duplexes occur in the nucleus while
Chapter 1 Introduction and Review of Literature
39
miRNA-RISC complexes are present in the cytoplasm where target mRNAs are
located This discordance of functional sites requires an export step during miRNA
biogenesis HASTY the Arabidopsis homolog of exportin-5 is thought to be involved
in miRNA nuclear export (Bollman et al 2003 Park et al 2005)The hasty mutant
exhibits pleiotropic defects in plant development and reduced accumulation of most
miRNAs as in the DCL1 or AGO1 mutants Howeverit is unclear whether the HASTY
proteins export the miRNA miRNA duplexes or the miRNA-RISC complexes in
plants
11345 Feedback regulation of miRNA biogenesis
MiRNA biogenesis is under feedback regulation such that the DCL1 and AGO1 genes
are themselves regulated by miRNAs DCL1 is itself a target of miR162 which leads to
the cleavage of the DCL1 mRNA (Xie et al 2003 de Alba et al 2013) Consistent
with this the levels of DCL1 mRNA are elevated in the mutants of miRNA processing
genes in which the miR162 abundance is reduced In addition there is a predicted
hairpin structure of miR838 in the 14th
intron of the DCL1 primary transcript
(Rajagopalan et al 2006) This prediction implies that DCL1-mediated processing
on this site can result in the cleavage of the DCL1 primary transcript into two truncated
fragments the N-terminal capped fragment and the C-terminal polyadenylated
fragment If this is the case it is possible that miRNA biogenesis competes with the
splicing event of the DCL1 pre-mRNA
The AGO1 is also regulated by miRNA There is a complementary site for
miR168 binding in the AGO1 mRNA and the transcript level of the AGO1 mRNA is
reduced through an mRNA cleavage mechanism (Vaucheret et al 2006 de Alba et al
2013 Rogers amp Chen 2013) indicating that the AGO1 mRNA levels are tightly
controlled by the levels of the AGO1 protein
1135 Mechanism of miRNA action
11351 MiRNA-directed mRNA cleavage
By forming the miRNA-RISC assembly miRNAs can direct the RISC to regulate gene
expression by two post transcriptional mechanisms mRNA cleavage or translational
repression (Bartel 2004 Jones-Rhoades et al 2006 Dalmay 2013) The degree
of miRNA-mRNA complementarity plays an important role for the
determination of the mechanism to be used A general rule is that while perfect or
Chapter 1 Introduction and Review of Literature
40
near-perfect complementarity induces mRNA cleavage central mismatches trigger
translational repression (Carrington amp Ambros 2003 Jones-Rhoades amp Bartel 2004)
Unlike most animal miRNAs plant miRNAs have near-perfect or perfect
complementarity to their targets and mRNA cleavage is assumed to be a predominant
mechanism by which miRNAs regulate gene expression in plants
In RNA cleavage mechanism miRNAs guide the AGO component of RISC to
cleave a single phosphodiester bond of the target mRNA within the miRNA-binding
site (Llave et al 2002b Tang et al 2003) The truncated fragments are then released
and degraded freeing the RISC to recognize and cleave another target mRNA This has
been validated by the detection of 3prime cleavage products that have 5prime ends that start at the
middle of the complementary regions The mRNA cleavage products are then further
degraded by other mechanisms The 3prime cleavage products of some miRNA targets are
degraded by the 5prime-3prime exonuclease XRN4 an Arabidopsis homolog of the major Yeast
mRNA degrading exonuclease Xrn1p It has been observed that the 3prime cleavage
products of some target mRNAs were accumulated in the xrn4 mutants (Souret et al
2004 Dalmay 2013) However the 3prime cleavage products of other miRNA targets do
not accumulate in the xrn4 mutants indicating that there are also XRN4 independent
mechanisms The 5prime cleavage products are typically untraceable by RNA gel blot
hybridization but can be detected by sensitive PCR based methods It has been found
that the 5prime cleavage products tend to obtain an oligo U tail at the 3prime ends This 3prime U tail
is correlated with shortening of the 5prime cleavage products from the 5prime end leading to the
conclusion that the 3prime uridylation causes 5 prime- 3 prime exonucleolytic decay of the 5prime cleavage
products (Shen amp Goodman 2004)
DCL1 HYL1 and SE interact with each other and are co-localized in the
nuclear bodies termed Dicing bodies or D-bodies in which some pri-miRNA shave
been found suggesting that D-bodies serve as a center for active miRNA processing
(Fang amp Spector 2007 Fujioka et al 2007 Song et al 2007) The miRNA
processing site appears to be distinct from the Cajal bodies that have previously been
found as a siRNA processing site (Pontes amp Pikaard 2008) The D-bodies include
SmD3 and SmB proteins that are found in the Cajal bodies However At collin an
additional marker for the Cajal bodies is not found in the D-bodies Moreover the D-
bodies are present in an At collin mutant lacking the Cajal bodies The pri-miRNAs of
miR171A and miR159A genes have been found to be co-localized with HYL1 and
Chapter 1 Introduction and Review of Literature
41
DCL1 in the D- bodies that are distinct from the Cajal bodies (Song et al 2007)
11352 MiRNA-directed translational repression
Translation repression is another mechanism exerted by plant miRNAs The regulatory
mechanism of miR172 which regulates flowering time and floral organ identity is
consistent with translation repression MiR172 over production does not affect the
transcript levels of APETALA2 (AP2) and AP2- like target mRNAs Instead the AP2
protein level is reduced in the miR172 over expressing mutant (Aukerman amp Sakai
2003 Chen 2004 Dalmay 2013) It was later shown that miR172 can also direct the
cleavage of the target mRNAs although the steady-state mRNA level is unchanged due
to feedback regulation (Schwab et al 2005 Jung et al 2007) It is clear that miR172
regulates its target genes primarily by translational repression rather than mRNA
cleavage Recently it has been reported that miR156157 and miR854 also regulate
their target genes through translational repression (Arteaga-Vazquez et al 2006
Gandikota et al 2007 Dalmay 2013) It was once thought that translational repression
is an exception to the rule However experimental evidence suggest a widespread
coexistence of translational repression and mRNA cleavage (Brodersen et al 2008)
1136 Regulatory roles of miRNAs in plant development
The miRNA target prediction has been a difficult task in order to obtain regulatory
targets without bringing in too many false predictions Plant growth and developmental
processes regulated by miRNAs are quite diverse and enormous Furthermore plant
miRNAs work cooperatively or antagonistically to establish a balanced regulation This
notion is well consistent with the findings that mutations in the regulators of miRNA
biogenesis transport and processing such as AGO1 DCL1 HYL1 HEN1 ABH1
and HASTY cause pleiotropic phenotypes (Mallory amp Vaucheret 2006 de Alba et al
2013) To date the roles of miRNAs have been mostly deduced from obtaining miRNA
over expressing transgenic plants or gain-of-function mutants in which miRNA-
resistant target genes are ectopically expressed
11361 Phase transition
Plants pass through a series of developmental phase transitions during their life span
beginning with seed germination continuing through vegetative phase change
reproductive phase change flowering initiation and finally seed production for the next
generation Because of their importance in understanding plant developmental
Chapter 1 Introduction and Review of Literature
42
processes genes regulating developmental transitions have been extensively studied
and underlying molecular mechanisms have been explored in various plant species
Majority of researches were mainly focused on vegetative phase change and
reproductive transition in Arabidopsis and Maize (Poethig 2003 Baurle amp Dean
2006) Particularly the mutations of the genes encoding various regulators of miRNA
biogenesis and transport such as HST SE and AGO exhibit altered juvenile to adult
vegetative phase change and vegetative to reproductive change (Hunter et al 2003
Peragine et al 2004 Vazquez et al 2004a Jones-Rhoades et al 2006 de Alba et al
2013)
Over-expression of the miR156a gene causes late flowering and delays
vegetative phase change (Wu amp Poethig 2006 Gandikota et al 2007 Wang et al
2008) It has been shown that miR156 negatively regulates a group of genes encoding
SQUAMOSA PROMOTER-BINDING PROTEIN-LIKE (Lewis et al 2007 de Alba et
al 2013 Rogers amp Chen 2013) transcription factors such as SPL3 SPL4 and SPL5
that promote flowering initiation In contrast transgenic plants over expressing
miR156-resistant SPL345 genes are early flowering Interestingly the endogenous
level of miR156 is temporally regulated In the early juvenile vegetative phase the
level of miR156 is very high but decreases rapidly before the onset of the adult juvenile
phase (Wu amp Poethig 2006)
The Arabidopsis early activation tagged (eat-D) mutant exhibits early flowering
with disrupted floral structure The mutant phenotypes are caused by over expression of
the miR172a-2gene (Aukerman amp Sakai 2003) MiR172 regulates a small group of
genes encoding APETALLA2 (AP2)-like transcription factors such as TARGET OF
EAT1 (TOE1) TOE2 SCHLAFMUTZE (SMZ) and SCHNARCHZAPFEN(SNZ)
TOE1 TOE2 SMZ and SNZ have been shown to participate in their productive phase
change by repressing a floral integrator FLOWERING LOCUS T (FT)(Jung et al
2007) Consistent with this toe1-2D 35STOE2 35SSMZ and 35SSNZ plants
exhibit late flowering (Jung et al 2007) MiR172 is also regulated temporally and
highly expressed in the adult vegetative phase both in Arabidopsis and maize (Chuck et
al 2007) Because the temporal expression patterns of miR156 and miR172 are
complementary to each other it has been suggested that the two miRNAs interact with
each other in regulating phase change (Chuck et al 2009 de Alba et al 2013) In
support of this view it has been shown that miR172 is down-regulated in the
Chapter 1 Introduction and Review of Literature
43
Corngrass1(Cg1) mutant in which miR156 is overproduced (Chuck et al 2007 de
Alba et al 2013) However there is no direct evidence for the direct linkage between
miR156 and miR172 It is also envisioned that miR156 and miR172 would not be
directly related For example miR156 regulates plastochron index that is the inverse of
leaf initiation rate and thus frequently used to analyze temporal patterns of plant
development In contrast miR172 does not affect plastochron (Wang et al 2008) The
down-regulation of miR172 in the maize miR156-over producing mutants would be
due to the prolonged juvenile vegetative phase in the mutants
Another miRNA that regulates flowering initiation is miR159 It regulates
MYB33 and MYB65 which are closely related to the barley GAMY B transcription
factor that responds to gibberellic acid (GA) The level of miR159 is elevated in GA-
treated seedlings but reduced in the GA biosynthetic ga1 mutant Transgenic plants
over producing miR159 exhibit delayed floral induction under short days (Achard et
al 2004) It is been suggested that MYB33 mediates GA signal to regulate LEAFY
(LFY) These observations indicate that miR159 plays an important role in the GA
signaling pathways that regulate proper floral induction under short days (Achard et al
2004)
11362 Floral development
Arabidopsis flowers consist of four distinct organs They arise in a characteristic
pattern within concentric rings called whorls from outside to inside four sepals four
petals six stamens and the central pistil consisting of two carpels Identities of the
floral organs are determined by three classes of regulatory genes classA classB and
class C genes (Krizek amp Fletcher 2005) The A and C class genes act antagonistically
to restrict their expression domains (Bowman et al 1991)
It is notable that miR172 also regulates floral architecture in addition to its role
in the timing of flowering initiation MiR172 inhibits the translation of the AP2 mRNA
(Chen 2004) Transgenic plants overproducing miR172 not only exhibit early
flowering but also show abnormal floral development which is quite similar to those
of the class A gene mutants such as the ap2 mutants (Aukerman amp Sakai 2003 de
Alba et al 2013) When the activity of the class A genes is inactivated that of the class
C genes such as AGAMOUS (AG) is elevated As a result the miR172- overproducing
transgenic plants exhibit no petals along with homeotic transformation of the sepals to
Chapter 1 Introduction and Review of Literature
44
the carpels (Aukerman amp Sakai 2003 Chen 2004)
Interestingly miR166 also regulates floral architecture The role of miR165166
in the regulation of meristem activity is closely linked to floral meristem formation and
organization (Zhao et al 2007 Miyashima et al 2013) Floral structure is severely
disrupted in the miR165166-overproducing mutants such as men1 and jba-1D in
which miR166 is overproduced (Williams et al 2005b Jung amp Park 2007) The men1
and jba-1D mutants have smaller gynoecium and reduced carpel number In an extreme
case the jba-1D homozygous plants lack visible carpels (Williams et al 2005b)
Similar phenotypes have been observed in the miR165 overproducers (Zhao et al
2007) It has been suggested that the phenotypes are possibly caused by altered AG
expression in the floral meristem (Jung amp Park 2007 Turchi et al 2013)
Other miRNAs also affect floral development Overproduction of miRNAs
involved in auxin signaling also affects floral architecture Transgenic plants
overproducing miR167 have defects in the formation of ovule and anther with reduced
fertility (Ru et al 2006) It has been shown that miR167 targets ARF6 and ARF8 that
play a role in gynoecium and stamen development and in regulating fertility (Ru et al
2006 Wu et al 2006) These observations suggest that auxin signals are also closely
related to floral organogenesis In addition transgenic plants over-producing miR164
and the cuc1cuc2 double mutants show fused sepals and reduced petals (Laufs et al
2004 Turchi et al 2013) suggesting that miR164 is also related to floral meristem
activity and boundary specification of the meristematic region
11363 Shoot apical meristem development
Shoot apical meristem comprises self-maintaining totipotent cells The shoot apical
meristem activity affects diverse aspects of plant developmental processes including
leaf development vascular development and lateral organ polarity (Bowman et al
2002) Because miR165166 plays a primary role in meristem formation
overproduction of miR165166 and multiple knockout mutants of its target genes
exhibit pleiotropic pheonotypes (McConnell et al 2001 Emery et al 2003 Kim et al
2005 Williams et al 2005b)
The targets of miR165166 include genes encoding the homeo domain-leucine
zipper (HD-ZIP) class III transcription factors such as PHABULOSA(PHB)
PHAVOLUTA (PHV) REVOLUTA(REV) ATHB15CORONA (CNA) and ATHB8
Chapter 1 Introduction and Review of Literature
45
PHB PHV and ATHB15 have overlapping functions in controlling meristem
development (Turchi et al 2013) Indeed the phb phv athb15 triple mutants have an
enlarged shoot apical meristem Consistent with this the miR166- overproducing men1
and jba-1D mutants exhibit enlarged shoot apical meristems (Kim et al 2005
Williams et al 2005b Turchi et al 2013) In the mutants the shoot apical meristems
are multiplied and large
The development of shoot apical meristems is also regulated by the WUSCHEL
(WUS)ndashCLAVATA (CLV) signaling pathway WUS positively regulates cell
proliferation In contrast CLV3 which is expressed in stem cells limits the WUS
expression and thus inhibits cell proliferation in the SAM (Sharma et al 2003)
Consistent with this notion WUS is expressed to a higher level in jba-1D
explaining the ectopic enlargement of the SAM in the mutant (Williams et al 2005)
While WUS is closely related to miR165166 in the shoot apical meristem
development the underlying molecular mechanisms are currently unclear It has been
observed that the miR166-over producing men1 plants have an arrested meristem
development (Jung amp Park 2007) In addition the heterozygous men1+ and the
homozygous men1 plants show different expression patterns of WUS and CLV3 It
appears that miR166 regulates WUS indirectly and influences the expressional balance
between WUS and CLV3 through a negative feedback loop (Jung amp Park 2007 de
Alba et al 2013)
The phv phb athb15 triple mutant has an enlarged shoot apical meristems the
rev phb phv mutant does not have functional shoot apical meristems (Prigge et al
2005) This distinction may be explained by the fact that while miR166 targets
primarily PHB PHV and ATHB15 miR165 targets all the five HD-ZIP III genes (Zhou
et al 2007) Consistent with this transgenic plants overproducing miR166 are distinct
from those overproducing miR165 The miR165-overproducing transgenic plants lack
functional shoot apical meristem and a pin-like structure is formed at the apical region
of the mutant These phenotypes are quite similar to rev phb phv triple mutant (Zhou et
al 2007 Liu et al 2013) The phenotypic distinction may because by the difference
of the cleavage efficiencies of the target mRNAs In accordance with this notion a few
35S miR166a transgenic lines exhibit no shoot apical meristems The men1
homozygous plants also show arrested meristem development (Jung amp Park 2007)
Chapter 1 Introduction and Review of Literature
46
MiR164 cleaves the CUC1 and CUC2 mRNAs as well as the NAC1 mRNA
CUC1 and CUC2 determines the organ boundary in the meristem Phenotypes of
transgenic plants that overproduce miR164 are similar to those of the cuc1cuc2 double
mutant that exhibits embryonic developmental defects such as cup-shaped cotyledons
and fused cotyledons (Laufs et al 2004) When a miR164-resistant CUC2 is
expressed the boundary domain is enlarged CUC also plays a role in embryonic shoot
meristem formation by regulating the SHOOT MERISTEMLESS gene It was therefore
concluded that miR164-mediated cleavage of CUC1 and CUC2 mRNAs determines the
sizes of the boundary domains in the shoot apical meristem and regulates separation of
the organs by restricting cell proliferation (Laufs et al 2004 de Alba et al 2013)
11364 Leaf development
The role of miR319 has been extensively studied in leaf development A dominant
jaw-D mutant overproducing miR319 shows uneven curved leaves with enlarged size
and five TCP genes which are closely related to the CINCINNATA (CIN) gene in
Antirrhinum majus are down regulated in the mutant suggesting that miR319 mediated
regulation of the TCP transcription factor genes are important for the final step of
leaf shaping (Palatnik et al 2003 Naz et al 2013) In addition to leaf shape and size
leaf polarity is also specified by miRNA MiR166-mediated cleavage of the HD-ZIPIII
mRNAs is a well to establish leaf polarity This discovery originates from the gain-of-
function mutants such as phb-1d and phv-1d wherein point mutations in the
complementary sites to miRNA helps the mRNA to evade or resist from miRNA-
mediated cleavage (McConnell et al 2001 Emery et al 2003 Naz et al 2013)
Plant leaves have a dorso-ventral polarity consisting of the adaxial (upper
surface) and abaxial (lower surface) sides The adaxial side is closer to the shoot
apical meristem Adaxial identity is specified by functionally redundant class III HD-
ZIP family genes such as PHB PHV and REV Ectopic expression of each genes
result in adaxialized leaves Dominant phb-1d and phv-1d mutants exhibit
transformation of the abaxial to adaxial fate and have rod-shaped leaves In contrast
miR165166-overproducing plants show pin-like cotyledons radicalized leaves and
abaxialized leaves (Chitwood et al 2007 Pattanayak et al 2013)
Leaf asymmetry is determined by miR166-mediated restriction of the
expression domains of the HD-ZIPIII genes The HD-ZIPIII genes are expressed in the
Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
19
synthetic version of the cry1Ac gene have been produced (Leroy et al 2000 Mishra et
al 2002) Indeed this was the first report of an important agronomic trait being
introduced into a coffee plant The transgenic plants presented similar features in
growth and development compared to normal plants Transgenic plants highly resistant
to leaf miner under greenhouse conditions were tested under field conditions in French
Guyana for 4 years for pest resistance (Perthius et al 2005 Perthius et al 2006)
From 54 independent transformation events 70 of the events were resistant to leaf
miner Unfortunately the field trial was vandalized which led to the termination of the
experiment (Montagnon et al 2005)The effectiveness of Bt genes in controlling
coleopteran pests is well documented in corn and potato which indicates that Bt genes
might be effective against CBB The high toxicity of B thuringiensis sero var
israelensis against first in star larvae of CBB has been demonstrated (Mendez et al
2003) In another experiment an α-amylase inhibitor from Phaseolus vulgaris was
tested against CBB and found to have an inhibitory effect on its growth and
development (Grossi et al 2004)In addition to the CBB and WSB C arabica
varieties are also susceptible to endoparasitic root-knot nematode (Meloidogyne spp)
(Campos et al 1990 Mishra et al 2012)
So far 15 species have been reported to be parasites of coffee Controlling
nematodes is extremely difficult and currently seedling grafting with robusta rootstock
is followed as one of the control measure Sources of resistance specific to root-knot
nematodes have been identified in coffee trees (Bertrand et al 2001) and the Mex-1
gene conferring resistance to M exigua in C arabica is in the process of isolation (Noir
et al 2003)
1102 Tolerance to Abiotic Stress
In many coffee growing countries abiotic stress such as drought and frost are the major
climatic factors that limit coffee production Changes in climatic patterns due to global
climate change are considered increasingly important for coffee cultivation Drought
induces water stress in plants which affects vegetative growth and vigor and triggers
floral abnormalities and poor fruit set It also indirectly increases the incidence of pests
and diseases in the plants C arabica is generally more tolerant to water stress than C
canephora partly due to its extensive deep root system C racemosa is known to be a
good source of drought tolerance In India hybrids between C canephora and C
racemosa have been obtained and are currently under evaluation for drought tolerance
Chapter 1 Introduction and Review of Literature
20
In many coffee growing countries coffee is propagated in marginal areas where the
annual rainfall is below 1000thinspmm with prolonged dry spells of over 4-5 months In
those areas water shortage and unfavorable temperatures constitute major constraints
and the growth and productivity of C canephora are badly affected As with drought
periodic frost also affects coffee production in parts of Brazil The introduction of
drought and frost tolerant genes through genetic transformation would be of great
importance for alleviating these problems Research is now being carried out by several
groups to identify genes involved in biotic as well as abiotic stress The most promising
genetic engineering approach for drought tolerance includes the use of functional or
regulatory genes as well as the transfer of transcription factors In recent years plants
tolerant to high temperature and water stress have been the subject of intense research
(Chaves et al 2003 Chaves et al 2004 Coraggio et al 2004) For achieving drought
tolerance genes that have been targeted include those encoding enzymes involved in
detoxification or osmotic response metabolism enzymes active in signaling proteins
involved in the transport of metabolites and regulating the plant energy status (Chaves
et al 2003 Chaves et al 2004 Coraggio et al 2004) The dissection of molecular
mechanisms related to signal transduction and transcriptional regulation might help in
engineering drought tolerance in coffee
1103 Disease Resistance
Coffee leaf rust (CLR) caused by the fungus Hemileia vastatrix is the most important
disease in coffee with substantial loss to coffee production and productivity in all the
coffee growing countries In addition to leaf rust coffee berry disease (CBD) caused by
fungus Colletotrichum kahawae can be a devastating anthracnose leading to substantial
crop loss in Africa Several other fungal and bacterial diseases may also affect coffee
to a extent C arabica is more susceptible to many diseases than C canephora Though
most of the disease control measures rely upon chemical control they are more
expensive and labour intensive The long-term solution is the breeding of resistant
varieties which is the focus of many breeding programs However breeding for disease
resistant varieties is time consuming due to the perennial nature of coffee with its long
gestation period India has a long history of C arabica breeding especially for leaf rust
resistance and is the first country to demonstrate the existence of multiple races of leaf
rust Resistance to CLR is conditioned primarily by a number of major (SH) genes and
coffee genotypes are classified into different resistance groups based on their
Chapter 1 Introduction and Review of Literature
21
interaction with different rust races pathogen (van der Vossen 2001) Currently C
canephora provides the main source of resistance to pests and diseases including CLR
(H vastatrix) and CBD (C kahawae) and therefore used in breeding programs Other
diploid species like C liberica and C racemosa are being used as a source of resistance
to coffee leaf rust and coffee leaf miner respectively (Guerreiro et al 1999
Sreenivasan et al 1989) The development of coffee varieties resistant to major fungal
diseases such as CLR and CBD using transgenic technology will benefit the coffee
industry immensely During the last 15 years significant progress has been made in the
area of host-pathogen interactions (Staskawicz et al 1995 Feys et al 2000
Shivaprasad et al 2012) and many resistance genes involved in recognizing invading
pathogens have been identified and cloned (Takken et al 2000) A number of
signalling pathways induced because of pathogen infection have been dissected (Dong
et al 1998 Navarro et al 2006) Many antifungal compounds synthesized by plants to
combat fungal infection have been identified (Does et al 1998) Understanding the
specific induction of targeted pathways and identification of specific pathways
responsible for particular fungal resistance is important in order to employ this strategy
in transgenic technology The recent investigation of gene expression during coffee leaf
rust infection could provide an insight into the defence pathways operating in coffee
(Fernandez et al 2004 Nardi et al 2006) Efforts have been made to identify and
clone resistance genes from coffee for achieving durable resistance The genetic and
physical map of two resistance genes SH3gene conferring resistance to rust (Prakash et
al 2004) and the Ck1 gene conferring resistance to C kahawae CBD (Gichuru et al
2006) have been established These genes could be used for molecular marker assisted
breeding programs (Mishra et al 2012)
1104 Production of low-caffeine coffee
A widespread belief that the ingestion of caffeine can have adverse effects on
health resulted in the increased demand for decaffeinated coffee (Ashihara amp Crozier
2001) Several method were used to obtain decaffeinated plants by conventional
breeding techniques using low yielding varieties and mutants that accumulated
relatively low amounts of caffeine when compared to the commercially available ones
Low-caffeine and decaffeinated coffee represent around 10 of the coffee sales
around the world (Ribas et al 2006b) The industrial process for coffee decaffeination
can be expensive and affects the original flavour and aroma in coffee (David 2002)
Chapter 1 Introduction and Review of Literature
22
Transgenic coffee plants with suppressed caffeine synthesis using RNA interference
(RNAi) technology have been obtained (Ogita et al 2003 Ogita et al 2004) Specific
sequences in the 3prime untranslated region of the theobromine synthase gene (CaMXMT1)
were selected for construction of RNAi short and long fragments The caffeine and
theobromine content of the transgenic plants were reduced by ~ 70 when compared to
the untransformed plants In C canephora RNAi technology has also been employed
to silence the N-methyl transferase gene involved in caffeine biosynthesis (Vinod et al
2007) Promoter of an N-methyltransferase (NMT) gene involved in caffeine
biosynthesis was cloned (Satyanarayana et al 2005 Satyanarayana 2006) which will
be very useful for studying the regulation of caffeine biosynthesis
1105 Improvement in cup quality
Improvement in coffee cup quality requires elaborate knowledge of the chemical
constituents as well as the metabolic pathways involved in the elaboration of quality
The constituents of coffee beans include minerals proteins carbohydrates caffeine
chlorogenic acids (CGA) glycosides lipids and many volatile compounds that give
flavour to coffee by roasting Among these the role of three major constituents
sucrose CGA and trigonelline have been studied in coffee The sucrose content of
coffee bean is associated with coffee flavour the higher the sucrose content in green
beans the more intense will be the cup flavour (Clifford et al 1985 de Maria et al
1996) The sucrose content of C arabica (82-83) is higher than C canephora (33ndash
40) The sucrose amino acids ratio in green beans determines the profile of volatile
compounds Manipulating sucrose content in coffee bean is therefore important in
improving cup quality The sucrose synthase gene (CcSUS2) from C canephora has
been cloned and sequenced (Leroy et al 2005) This provides an opportunity to
manipulate the sucrose content in coffee Chlorogenic acids are products of
phenylpropanoid metabolism Chlorogenic acids are a group of hydroxycinnamoyl
quinic acids (HQA) formed by esterification between caffeic acids coumaric acids and
quinic acids (Clifford et al 1999) They are present in relatively large quantities in the
coffee bean and are the precursor of phenolic compounds in roasted beans C
canephora beans contain higher CGA (10) compared to C arabica beans (6-7)
CGA are known to have antioxidant properties as well as being associated with disease
resistance (Campa et al 2003) Genetic manipulations of genes involved in CGA
synthesis can serve either of these purpose by up- or down regulating the pathway
Chapter 1 Introduction and Review of Literature
23
Phenylalanine ammonia-lyase (PAL) catalyzes the first step of the phenylpropanoid
pathway leading to the synthesis of a wide range of chemical compounds including
flavonoids coumarins hydroxycinnamoyl esters and lignins (Hahlbrock amp Scheel
1989) Recently the full length cDNA and corresponding genomic sequences of PAL
from C canephora was isolated characterized and functionally validated (Mahesh et
al 2006 Parvatam et al 2006) This has opened up new possibilities for manipulating
the level of the PAL enzyme in coffee which in turn can be useful for improvement of
cup quality and manipulating antioxidant properties in coffee
1106 Fruit Ripening
Uniformity during fruit ripening is decisively related to cup quality in coffee and
consequently to the value of the product Fruits at the ideal ripening stage produce the
best organoleptic characteristics for coffee The presence of over ripened or green fruits
changes the acidity the bitterness and consequently the cup quality In order to
maximize uniform ripening of coffee fruits it is essential to control the action of genes
involved in the last step of maturation process Ethylene is known to trigger ripening
and increasing ethylene biosynthesis is associated with various stages of ripening
process (Protasio et al 2005) To control coffee fruit maturation two of the major
genes involved in ethylene biosynthesis namely ACC synthase and ACC oxidase
have been cloned (Protasio et al 2005 Neupane et al 1999) Introduction of the ACC
oxidase gene in antisense orientation have been achieved in both C arabica and C
canephora The effect of the transgene on ethylene production and fruit maturation has
yet to be reported The inhibition of genes downstream to the initial ethylene burst is
also an option to control coffee fruit maturation (Ribas et al 2006)
111 RNA interferenceRNAi
The regulation of gene expression at post transcriptional level has recently attracted
much attention because of the discovery of the phenomenon called RNA interference
(RNAi) RNAi based silencing techniques are more efficient than antisense mediated
gene silencing (Wesley et al 2001) The effect was first observed in plants wherein
silencing was observed when some copies of a transgene was in the forward orientation
with respect to the promoter and some in the reverse orientation This resulted in the
formation of double stranded RNA in the system The phenomenon in which
experimentally introduced double stranded RNA (dsRNA) leads to the loss of
expression of the corresponding cellular gene by sequence specific RNA degradation
Chapter 1 Introduction and Review of Literature
24
was termed as RNA interference or RNAi The protective effect of RNA silencing in
reducing virus infectivity supports the view that PTGS has evolved as a mechanism to
defend plants against virus infection and also to moderate the possible deleterious
genome-restructuring (insertional) activity of virus-like mobile genetic elements eg
retrotransposons A growing body of biochemical and genetic data has further
demonstrated that plants animals and yeasts share related mechanisms of specific
degradation of RNAs in which double-stranded forms of RNA are initiator molecules
(Brenstein et al 2001)
The key insight into the process of PGTS was provided by the experiments
conducted by Hamilton amp Baulcombe (1999) who identified the product of RNA
degradation as small RNA (siRNA) species of ~25nt of both sense and antisense
polarity SiRNA are formed and accumulate as double stranded RNA molecules of
defined chemical structures Both genetic and biochemical approaches were undertaken
to understand the basis of silencing Genetic screens were carried out in fungus
Neurospora carassa the alga Chalmydomonas reinhardtii and plant Arabidopsis
thaliana to search for detective mutants in PTGS
A combination of results obtained from several in vivo and in vitro experiments
have gelled into a two step mechanistic model for RNAiPTGS The first step referred
to as the RNAi initiating step involves binding of the RNA nucleases to a larges
dsRNA and its cleavage into discrete ~21-25nt RNA fragments This is ATP-
dependent reaction which involved RNase III - type endonucleases which make
staggered cuts in both strands of the dsRNA leaving 3prime overhang of 2 nucleotides with
5prime- phosphate 3prime-hydroxyl and no modification in the sugar phosphate backbone
(Hamilton amp Baulcombe 1999 Elbashir et al 2001) SiRNAs are the direct products
of dsRNA cleavage by the multi-domain RNase III enzyme DICER (Figure 16)
(Brenstein et al 2001 Karthikeyan et l 2013)
In the second step or commonly known as the effector step the double stranded
siRNAs produced in the first step bind to an RNAi specific protein complex to form a
RISC The complex undergoes activation in the presence of ATP so that the antisense
component of the unwound siRNA becomes exposed and allows the RISC to perform
the downstream RNAi reaction
Chapter 1 Introduction and Review of Literature
25
Several enzymes are involved in RNAi based on their role genes that encode for them
are classified into RNAi initiators RNAi effectors RNA dependent RNA polymerases
and silencing genes Two C elegans genes rdeI and rde4 (rde stands for lsquo RNAi
deficientrsquo) are believed to be involved in the initiation step of RNAi The C elegans
rde1 gene is a member of a large family of genes and is homologous to the Neurospora
qde2 (qde stands for ldquoquelling deficientrdquo) and Arabidopsis AGO1 (AGO stands for
agronaute AGO1 was previously identified to be involved in Arabidopsis
development) Although the functions of these genes are not clear a mammalian
member of RDE1 family has been identified as a translation initiation factor (Thakur
2005) Important genes for the effector step of PTGS in C elegans are rde2 and mut7
genes These genes were identified from heterozygous mutant worms that were unable
to transmit RNAi to their homozygous offsprings Worms with mutated rde2 or mut 7
genes show defective RNAi mut-7 gene encodes a protein with homology to the
nuclease domains of RNAase D and a protein implicated in Werner syndrome (a rapid
ageing disease in humans) (Grishok et al 2001 Kamath-Loeb et al 2012s)
Neurospora qde-1 Arabidopsis SDE-1SGS-2 and C elegans ego-1 appear to encode
RNA dependent RNA polymerase (RdRPs) It assumed that this is a proof that an RdRp
activity is required for RNAi Certainly the existence of an RdRp might explain the
remarkable efficiency of dsRNA induced silencing if it amplifies either the dsRNA
prior to cleavage or the siRNAs directly (Muers 2013) In C elegans ego-1 mutants
(ego stands for lsquoenhancer of glp-1) RNAi functions normally in somatic cells but is
defective in germline cells where ego-1 is primarily expressed In Arabidopsis SDE-
1SGS-2 mutants (SGS stands for suppressor of gene silencing) siRNA are produced
when dsRNA is introduced via an endogenously replicating RNA virus but not
introduced by a transgene It has been proposed that perhaps the viral RdRP is
substituting for the Arabidopsis enzyme in these mutants Random degradative PCR
model suggests that an RdRP uses the guide strand of an siRNA as a primer for the
target mRNA generating a dsRNA substrate for Dicer and thus more siRNAs
Several genes controlling RNA silencing in plants have been identified through
genetic screens of Arabidopsis mutants impaired in transgene induced RNA silencing
They encode a putative RNA-dependent RNA polymerase (SGS2SDE1) a coiled
protein (SGS3) a protein containing PAZ and Piwi domains (AGO1) and an RNA
helicase (SDE3) The putative SGS2SDE1 of Neurospora is related to QDE-1 and
Chapter 1 Introduction and Review of Literature
26
EGO-1 of C elegans whereas PAZPiwi protein AGO is related to QDE-2 of
Neurospora RDE-1of C elegans RNA helicase SDE3 is related to SMG-2 of C
elegans and Mut-6 of Chlamydomonas MUT-7 gene of C elegans encodes a protein
similar to Rnase D whereas the Drosophila DICER gene encode a protein similar to
RNase III An Arabidopsis ortholog of DICER gene has been identified called CAF
SIN1 SUS1(Thakur 2005 Poethig 2009)
Since the RNAi machinery is present constitutively within the eukaryotic cell it
is important to explore the metabolic advantages that are accorded by the RNAi related
proteins during the intrinsic normal growth of cells and development of organisms The
natural RNAi machinery not only keeps the mobile transposable elements from
disrupting the integrity of the genomes as suggested by the analysis of model organisms
like A thaliana C elegans D melanogaster (Tabara et al 1999 Wu-Scharf et al
2000 Aravin et al 2001 Hamilton et al 2002 Lisch 2009) but also participates in
development of organisms Genetic defects in Celegans RNAi genes ego1 and dicer
cause known specific developmental errors (Grishok et al 2000 Grishok et al 2001
Knight amp Bass 2001 Hall et al 2013) Similarly the Argonaute family of genes of A
thaliana (especially the ZWILLE proteins) is also responsible for plant architecture and
meristem development (Carmell et al 2002 Hamant and Pautot 2010) and the Dicer
homologue of A thaliana CAF1 is required for embryo development (Golden et al
2002 Nakano et al 2011) Thus genetic evidence illustrates the role of the RNAi
machinery as a controller of development related genes The mechanistic details of
these developmental processes are beginning to emerge
Chapter 1 Introduction and Review of Literature
27
Figure 16 Mechanism of gene silencing induced double stranded RNA The dicer enzyme to
produce siRNAs cleaves the RNA The siRNA generated bind to the nuclease complex
RISC The antisense component in the siRNA in the RISC guides the complex towards
the cognate mRNA resulting in endonucleolytic cleavage of the mRNA
Chapter 1 Introduction and Review of Literature
28
112 MicroRNA or MiRNA
In 1991 Ambros and co-workers first isolated a lin-4 mutant of Celegans which was
arrested at the first larval stage (Lee et al 1993) Later on the let-7 mutation was
isolated in the same system which was responsible for development through the fourth
larval stage Both lin-4 and let-7 encode short 22-nucleotide mature RNAs and were
called short temporal RNA because they control the temporal development program of
C elegans The mature lin-4 RNA defines (negatively regulates) the mRNA expression
of the lin-14 and lin-28 hetrochronic genes with the antisense mediated repression
mechanism of translation initiation and thus specifies the fate of cells during the first
three larval stages Recent studies have revealed that the short temporal RNAs are
actually members of a group of tiny RNAs (21to 28 nucleotides) called the micro-
RNAs (miRNA) (Bartel 2009) isolated members of which could easily run to a few
thousand which includes both those were identified computationally and by deep
sequencing Some of the components of the RNAi machinery have also been clearly
established as the effector proteins for the maturation of miRNAs
113 MicroRNAs in plants
1131 Discovery
MicroRNAs have been discovered by three basic approaches Direct cloning forward
genetic direct cloning and bioinformatic prediction followed by experimental
validation The most direct method of miRNA discovery is to isolate and clone small
RNAs from biological samples and several groups have used this approach to identify
plant miRNAs (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002 Xie et al 2003 Sunkar et al 2005 Williams et al 2013) The cloning methods
adapted were similar to those used to identify large numbers of animal miRNAs
(Lagos-Quintana et al 2001 Lau et al 2001 Aravin amp Tuschl 2005) which involve
isolating small RNAs followed by oligonucleotide adapter ligation reverse
transcription amplification and sequencing Some protocols incorporate methods to
enrich for Dicer cleavage products (ie molecules with 5prime-phosphates and 3prime-
hydroxyls) and to concatemerize the short cDNAs so that several may be identified in a
single sequencing read (Lau et al 2001 Llave et al 2002a Jagadeeswaran amp Sunkar
2013)The initial cloning experiments in Arabidopsis identified 19 miRNAs belonging
to 15 families (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002) although hundreds of other small RNAs such as siRNAs and RNA degradation
Chapter 1 Introduction and Review of Literature
29
fragments were also cloned through the direct cloning methods To select miRNAs
from the pool of small RNAs secondary structures of the RNA molecules were
examined in compliance with the acknowledged structural features of miRNAs
(Ambros et al 2003) The secondary structures were validated using several ad-hoc
software tools such as the Mfold or RNAfold programs (Reeder et al 2006) Finally
the existence of mature miRNAs was determined by RNA gel blot hybridization These
direct cloning methods were successful in isolating many miRNAs in Arabidopsis
(Reinhart et al 2002 Sunkar amp Zhu 2004) Rice (Sunkar et al 2005) Poplar (Lu et
al 2005) moss (Arazi et al 2005) and Tobacco (Billoud et al 2005)
Although miRNAs were first discovered through forward genetic screens in
worms (Lee et al 1993 Reinhart et al 2000)only few A thaliana miRNA gene
families have been discovered by this method (Chen 2008) in plants MiRNA
involvement in plant mutant phenotypes were not properly inferred till the cloning
experiments established that plant genomes contain numerous miRNAs (Park et al
2002 Reinhart et al 2002 Rhoades et al 2002) MiRNA loss-of-function allele has
been identified by forward genetic screens early extra petala1 is caused by a
transposon insertion ~160bp upstream of the predicted miR164c stem loop resulting in
flowers with extra petals (Baker et al 2005 Irish 2008) The fact that loss-of-function
miRNA mutants have been rarely discovered using forward genetics reflects small
target size for mutagenesis coupled with redundancy in nearly all evolutionarily
conserved plant miRNAs which are encoded by gene families (Table16) Family
members are likely to have overlapping functions buffering against loss at any single
miRNA locus Over expression screens can circumvent redundancy limitations At least
four plant miRNAs miR319 (also known as miR-JAW) miR172 (also known as EAT)
and miR166 were isolated in over expression screens for dominant mutants with
developmental abnormalities (Aukerman amp Sakai 2003 Palatnik et al 2003 Kim et
al 2005 Williams et al 2005b Schommer et al 2012) Mutations in miRNA target
sites which can prevent the entire family of miRNAs from repressing a target gene can
also circumvent redundant functions of miRNA family members
The dominant mutations in the HD-ZIP genes PHB PHV and REVOLUTA
(REV) in Arabidopsis and ROLLEDLEAF1 (RLD1) in Maize result in adaxialization of
leaves andor Vasculature (McConnell amp Barton 1998 McConnell et al 2001 Nelson
et al 2002 Zhong amp Ye 2004 Allen amp Millar 2012) and are all caused by mutations
Chapter 1 Introduction and Review of Literature
30
in miR166 complementary sites (Rhoades et al 2002 Emery et al 2003 Jones-
Rhoades amp Bartel 2004 Mallory et al 2004 Zhong amp Ye 2004)
In both plants and animals cloning was the initial means of large-scale
miRNA discovery (Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Reinhart et al 2002 Mendes et al 2012) However cloning is biased toward
RNAs that are expressed highly and broadly MiRNAs expressed at low levels or
only in specific cell types or in response to certain environmental stimuli were more
difficult to clone Sequence based biases in cloning procedures might also cause
certain miRNAs to be missed Because of these limitations bioinformatics approaches
to identify miRNAs have provided a useful complement to cloning
A straightforward use of bioinformatics has been carried out to find homologs
of known miRNAs both within the same genome and in the genomes of other species
(Pasquinelli et al 2000 Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Mendes et al 2009) A more difficult challenge is to identify miRNAs
unrelated to previously known miRNAs This was first accomplished for
vertebrate nematode and fly miRNAs using algorithms that search for conservation
of sequence and secondary structure (ie miRNA stem-loop precursors) between
species searching for patterns that are characteristic of miRNAs (Lai et al 2003 Lim
et al 2003 Lim et al 2005)
Most of the methods identified numerous miRNAs but are necessarily more
relaxed in terms of allowed structures Some approaches take advantage of the high
complementarity of plant miRNAs to target messages implementing the requirement
that the candidate has conserved complementarity to mRNAs (Jones-Rhoades amp Bartel
2004 Adai et al 2005) This additional filter has been useful for distinguishing
authentic plant miRNAs from false positives which later extended to mammalian
miRNA gene prediction (Xie et al 2005 Mendes et al 2012)
Another criteria used by most algorithms in the filters is evolutionary
conservation In plants mature miRNA sequences are conserved to a higher degree
than their precursor sequences For example Arabidopsis and rice diverged more than
130 million years ago (Friis et al 2004) but some miRNAs are highly conserved
in these plant species (Reinhart et al 2002) Furthermore various parameters for the
secondary structures such as free energy the number of paired residues within a
Chapter 1 Introduction and Review of Literature
31
miRNA or the number and size of bulges are also used to validate the predictions
(Jones-Rhoades amp Bartel 2004 Wang et al 2004 Adai et al 2005 Mendes et al
2012)
1132 Conserved microRNAs in Plants
Till date cloning genetics and bioinformatics screens have resulted in annotation of
approximately 184 potential Arabidopsis miRNAs (miRBase release 13) These
miRNAs seem to arise through gene duplication and subsequent diversification and
represent approximately 100 families (Maher et al 2006 Axtell 2013) Twenty one
families represented by 92 genes are clearly conserved in species beyond Arabidopsis
(Table 15) The presence of these miRNA families in other sequenced plant genomes
indicates that different plant species have an evolutionarily fluid set of miRNAs
(Willmann amp Poethig 2007) Recent studies on mosses one of the most ancient land
plants have shown that at least 14 miRNA families are conserved in eudicots and
mosses and therefore it appears that plant miRNAs share a common ancient origin
(Arazi et al 2005 Axtell amp Bartel 2005 Talmor-Neiman et al 2006 Zhang et al
2006 Axtell 2013) The number of members per family in a genome ranges from 1-32
with the exception of miR-430 which is represented by a cluster of ~80 loci in zebra
fish (Giraldez et al 2005)
In plants the number of members in each family correlates among examined
species certain families contain numerous members in all three species (eg miR156
miR166 miR169) whereas others consistently contain fewer genes (eg miR162
mir168 miR394) (Table 16) Although it is still unclear why certain miRNA are
encoded by more than one gene in plants (eg 12 genes encoding miR156) this
correlation might suggest functional significance of various miRNA family sizes Most
annotated plant miRNAs are conserved throughout flowering plants while a few
miRNAs are found only in a single sequenced genome and thus they could be
evolutionarily lsquoyoungrsquo miRNAs For example Arabidopsis has several miRNA
families that are not found in poplar or rice suggesting that miRNAs could be
generated continuously during evolution (Allen et al 2004a Rajagopalan et al
2006 Fahlgren et al 2007 Axtell 2012 de Alba et al 2013) Consistent with the
notion that non conserved miRNAs are of a more recent evolutionary origin these
miRNAs are mainly encoded by a single locus in the genome Within each family the
mature miRNA is always located in the same arm
Chapter 1 Introduction and Review of Literature
32
Table 16 MicroRNA families conserved in plants
miRNA
family
Arabidopsis Oryza Populus
miR156 12 12 11 miR159319 6 8 15 miR160 3 6 8 miR162 2 2 3 miR164 3 5 6 miR166 9 12 17 miR167 4 9 8 miR168 2 2 2 miR169 14 17 32 miR171 4 7 10 miR172 5 3 9 miR390 3 1 4 miR393 2 2 4 miR394 2 1 2 miR395 6 19 10 miR396 2 5 7 miR397 2 2 3 miR398 3 2 3 miR399 6 11 12 miR408 1 1 1 miR403 1 0 2 miR437 0 1 0 miR444 0 1 0 miR445 0 9 0 Total 92 127 169
All known miRNA families that are conserved between more than one plant species are listed
together with the number of genes identified in the sequenced genomes Rice miRNA families that have orthologs in maize but do not appear to have orthologs in the eudicots (Arabidopsis and Populus) are marked with an asterisk The following families contain miRNA genes annotated with
more than one number miR156 (miR156 and miR157) miR159319 (miR159 and miR319) miR166 (miR165 and miR166) miR171 (miR170 and miR171) and miR390 (miR390 and miR391)
of the stem loop (5prime- or 3prime) as it would be expected if the genes carry common ancestry
(Figure 17) The sequence of the mature miRNA and to some extent the segment on
the opposite arm of the hairpin to which it pairs is highly conserved between the
members of the same family (both within and between the species) while the sequence
secondary structure and length of the intervening ldquolooprdquo region can be highly divergent
within the same family
The patterns of paring and non pairing nucleotides are often conserved between
homologous miRNA stem loops from different species (Figure 17) The significance of
conserved mismatches is opined to guide DCL1 to cleave at the appropriate positions
along the stem loop
Chapter 1 Introduction and Review of Literature
33
Figure 17 Representative stem loop of miR164 Segments corresponding to the mature miRNAs
are shown in red (Adapted from Rajagopalan et al 2006)
Although many miRNAs are highly conserved in plants the degree of conservation in
most cases is quite low between plants and animals one exception is miR854 A recent
study has revealed that miR854 is conserved between Arabidopsis and animals and that
the Arabidopsis miR854 differs from the human miR854 only by a single nucleotide
(Arteaga-Vazquez et al 2006 Axtell 2012 de Alba et al 2013) The Arabidopsis
miR854 has multiple binding sites in the 3prime-untranslated region (UTR) of
OLIGOURIDYLATE-binding-PROTEIN mRNA 1bA (UBP1b) and represses the
translation of the UBP1b mRNA The animal miR854 is also complementary to a site in
the 3prime-UTR of the UBP1b homolog in animals However it is currently unclear how the
animal miR854 regulates its target mRNA Another distinction between plant and
animal miRNAs is the genomic organization of the miRNA genes While plant miRNA
genes are located predominantly in the intergenic regions animal miRNA genes tend to
localize in the intragenic regions both in the introns or exons of protein coding genes
Moreover miRNA genes are often clustered within a single precursor transcript
whereas individual plant miRNAs are derived from individual precursor RNAs (Bartel
Chapter 1 Introduction and Review of Literature
34
2004 Bonnet et al 2004 Kim 2005 Vaucheret et al 2006 Marco et al 2013)
1133 Non-conserved miRNA and challenges in annotation
Most miRNAs are conserved throughout flowering plants (Table 15) but many more
were found only in a single sequenced genome thus considered to be of a more recent
evolutionary origin The extended homology between few non conserved miRNAs
precursors and their target genes provides strong evidence that these relatively ldquoYoungrdquo
miRNAs arose from the duplication of the target gene segments (Allen et al 2004)
Several non-conserved miRNAs including miR161 miR163 miR173 miR447
miR475 and miR476 are known to direct cleavage of target transcripts (Allen et al
2004a Adai et al 2005 Sunkar et al 2005 de Alba et al 2013) It is difficult to
confidently predict targets for many because it is not possible to use complementary
site as a filter against false-positive target predictions It is also difficult to
confidently annotate non-conserved miRNAs as miRNA rather than siRNAs The
established minimal standard for miRNA annotation is a small RNA with detectable
expression and the potential to form a stem-loop when joined to flanking genomic
sequence (Kasschau et al 2003) In practice these requirements are too loose to
definitively categorize many small RNAs cloned from plants Many plant siRNAs are
detectable on blots (Vazquez et al 2004b) and hundreds of thousands of non-
miRNA genomic sequences can be predicted to fold into secondary structures that
resemble structures of plant miRNA precursors (Jones-Rhoades amp Bartel 2004)
Therefore without conservation of both sequence and secondary structure it is
difficult to be confident that a given cloned RNA originated from a stem-loop (ie is a
miRNA) rather than from a double-stranded RNA (ie is a siRNA) In fact many of
the thousands o f small RNAs cloned from Arabidopsis (Gustafson et al 2005) would
probably meet the literal requirements for annotation as miRNAs A few of these
sequences might be miRNAs but others that meet the literal criteria probably are not
so The challenges of annotating non conserved small RNAs were evidenced by
three related small silencing RNAs that were originally annotated as miRNAs
that turned out to be trans-acting siRNAs (tasiRNAs)(Kanno et al2013)
Although most miRNAs that are broadly conserved among flowering plants are
now being identified and experimentally validated (Table 16)(Jones-Rhoades amp
Bartel 2004) the challenges and ambiguities for classifying non-conserved miRNAs
prevent meaningful estimates of the total number of miRNA genes in Arabidopsis and
Chapter 1 Introduction and Review of Literature
35
other plant genomes It is possible that miRNAs might have escaped detection
because they are non-conserved and expressed in specific tissues or conditions and can
only be identified by using high- throughput sequencing
1134 MicroRNA biogenesis
11341 Transcription of microRNA precursor
Plant miRNAs are primarily found in the genomic regions that are not associated with
the protein coding genes (Reinhart et al 2002) and it appears that most if not all plant
miRNAs are produced from their own transcriptional units Plant miRNA genes are
occasionally clustered near each other in the genome suggesting transcription of
multiple miRNAs from a single primary transcript [eg the miR395 cluster (Grishok
et al 2001 Jones-Rhoades amp Bartel 2004b)] but this polycistronic arrangement of
miRNA genes appears far less frequently in plants than in animals (Bartel 2004)
Northern blot EST and mapping evidence indicate that plant miRNA primary
transcripts (also known as pri-miRNAs) as in animals are longer than needed to
encompass the miRNA stem-loops (Palatnik et al 2003 Jones-Rhoades amp Bartel
2004b) At least some of these pri-miRNA transcripts appear to be spliced
polyadenylated (Kurihara amp Watanabe 2004) and capped (Xie Z et al 2005) Two
rice miRNAs are contained within transcripts that contain exon junctions within the
presumptive stem-loop precursor implying that in these cases splicing is a prerequisite
for Dicer recognition (Sunkar et al 2005) Plant pri-miRNAs are over 1kb in length
and they are usually preceded by typical TATA box motifs They can also undergo
canonical splicing polyadenylation and capping All these observations indicates RNA
polymerase II is probably responsible for transcribing most plant miRNAs (Xie Z et
al 2005) as appears to be the case for many animal miRNAs Relatively little is
known about the regulation of miRNA transcription in plants but there is no reason
to suspect that this regulation would differ from that of protein-coding transcripts as
the bioinformatics analysis of the sequence upstream of the transcriptional start
sites of the miRNA genes showed the presence of putative binding motifs of
number of known transcription factors (Megraw et al 2006 Sethupathy 2013)
11342 MicroRNA processing and export
In plants maturation of miRNA is a step wise process A miRNA gene is transcribed to
pri-miRNA which is much longer than the pre-miRNA possessing the characteristic
stem-loop structure Like transcription of most protein-coding genes the miRNA
Chapter 1 Introduction and Review of Literature
36
transcription is processed by RNA polymerase II (Pol II) enzymes and the pri-
miRNAs are 5prime-capped and 3prime-polyadenylated (Lee et al 2004 Xie Z et al 2005)
Mature miRNAs are produced from pri-miRNAs through at least two sequential
processing steps by RNase III-type endonucleases In animals pri-miRNAs are first
processed at the bottom portion of the stem by Drosha a nuclear-localized RNase III
enzyme to liberate pre-miRNAs The pre-miRNAs are then exported to the cytoplasm
with the help of exportin-5The pre-miRNAs are further processed by Dicer another
RNaseIII enzyme to a duplex consisting of the miRNA and the antisense strands
(miRNA) Since plants do not have any Drosha homologs the two sequential
processing steps seem to be carried out by DCL1 a Dicer homolog in the nucleus
(Kurihara amp Watanabe 2004 de Alba et al 2013 Rogers amp Chen 2013)
The processing of pri-miRNAs to pre-miRNAs by DCL1 is assisted by two
other proteins HYPONASTY LEAVES1 (HYL1) and SERRATE (SE) HYL1 belongs to a
family of double-stranded RNA (dsRNA) binding protein and interacts with DCL1 in
vitro and in vivo (Lu amp Fedoroff 2000 Hiraguri et al 2005 de Alba et al 2013
Rogers amp Chen 2013) Loss-of-function mutations in the HYL1 gene result in
decreased accumulation of miRNAs and concomitant accumulation of pri-miRNAs
(Han et al 2004 Vazquez et al 2004a Kurihara et al 2006) SE a C2H2 zinc finger
protein interacts with DCL1 and HYL1 and plays a role in the processing of pri-
miRNAs to pre-miRNAs (Lobbes et al 2006 Yang L et al 2006) In addition the
nuclear cap-binding complex proteins CBP20 and CBP80 that are known as ABA
HYPER SENSITIVE 1(ABH1) have been shown to be required for proper miRNA
processing (Gregory et al 2008 Laubinger et al 2008) Recently it has been
demonstrated that DCL1 can release 21-nt RNAs from dsRNA as well as from
synthetic miR167 precursor RNAs in vitro However this cleavage is error prone and
inefficient in the absence of HYL1 and SE which accelerate the rate of DCL1-mediated
cleavage and promote accurate processing of miRNAs (Dong et al 2008 Rogers amp
Chen 2013)
11343 Methylation of the miRNAmiRNAduplex
After the miRNAmiRNAduplexes are released from the pre-miRNAs by the DCL1
activities the duplexes are methylated at the 2primeOH of the3prime-ends by HUA ENHANCER1
(HEN1) a small RNA methyltransferase (Yu et al 2005 Yang Z et al 2006 Rogers
amp Chen 2013 ) This step which is distinct from the RNase III-mediated processing
Chapter 1 Introduction and Review of Literature
37
step distinguishes the biogenesis of plant miRNAs from that of animal miRNAs
Mutations in the HEN1 gene were first isolated in a morphological screening for floral
development although the HEN1 mutants display pleiotropic phenotypes similar to the
developmental defects observed in the DCL1 mutants (Chen et al 2002 Park et al
2002 Rogers amp Chen 2013)
Figure 18 Biogenesis of microRNA
A Biogenesis of plant MicroRNA B Biogenesis of metazoananimal MicroRNA
The miRNA (red) is incorporated into RISC and the miRNA(blue) in degraded
This enzyme has two dsRNA-binding domains and nuclear localization signal It
is also conserved in fungi and is required for the processing of siRNAs as well as of
miRNAs (Park et al 2002 Boutet et al 2003 Xie et al 2003) Although the HEN1
protein is localized to the nucleus its methylation activity in the cytoplasm cannot be
ignored (Xie et al 2004 Rogers amp Chen 2013)
Chapter 1 Introduction and Review of Literature
38
The methylation of the miRNAmiRNA duplex is required to protect miRNAs
against the 3prime-end uridylation activity and subsequent degradation (Li et al 2005) In
the hen1 mutants accumulation of miRNAs is reduced to a lower level Also the
miRNAs appear to be heterogeneous in their sizes such that a ladder of bands rather
than a single species is observed for most miRNAs in the mutants (Li et al 2005)
MiRNA cloning and sequencing analyses revealed the presence of a number of U
residues at the 3prime-end of miRNAs in the hen1 mutants Moreover these nucleotides do
not correspond to the miRNAs in the pre-miRNAs indicating that these nucleotides are
added after the processing of the miRNAs from pre-miRNAs (Li et al 2005)
Uridylation of RNAs has also been proven to be correlated with RNA decay (Shen amp
Goodman 2004 Mullen amp Marzluff 2008) suggesting that uridylation attracts an
exonuclease which degrades miRNAs in plants
11344 MiRNA incorporation in to the RISC and its nuclear export
The methylated miRNAmiRNA duplexes undergo RISC assembly In this process
the miRNA of the duplexes is selectively incorporated into the RISC and the miRNA
appears to be removed and subsequently degraded (Hammond et al 2000 Hutvagner
amp Zamore 2002 Schwarz et al 2003 de Alba et al 2013 Rogers amp Chen 2013)
The ARGONAUTE 1(AGO1) proteins are core components of the RISC complex
They contain two structural domains the N-terminal PAZ domain that binds to the 3prime-
end of single-stranded RNAs and the C-terminal piwi domains that has a structure
similar to that of RNase H (Cerutti et al 2000 Parker et al 2004) Several AGO
proteins possess endonucleolytic activities within the PIWI domain and cleave target
mRNAs in the middle of the site complementary to miRNA (Liu et al 2004 Meister et
al 2004 Miyoshi et al 2005) Mutations in the AGO1 gene cause miRNA target
genes to be ectopically expressed and the levels of some miRNAs are reduced in the
mutants (Kidner amp Martienssen 2004 Vaucheret et al 2004 Kidner amp Martienssen
2005 Ronemus et al 2006) Moreover the phenotype of the AGO1 hypomorphic
alleles display defects in lateral organ polarity and leaf and flower morphology as
observed in the dcl1 mutants and null AGO1 alleles are embryonically lethal
supporting the close relationship between the AGO proteins and miRNA processing
(Kidner amp Martienssen 2004 Vaucheret et al 2004 de Alba et al 2013 Rogers amp
Chen 2013)
The production of miRNA miRNA duplexes occur in the nucleus while
Chapter 1 Introduction and Review of Literature
39
miRNA-RISC complexes are present in the cytoplasm where target mRNAs are
located This discordance of functional sites requires an export step during miRNA
biogenesis HASTY the Arabidopsis homolog of exportin-5 is thought to be involved
in miRNA nuclear export (Bollman et al 2003 Park et al 2005)The hasty mutant
exhibits pleiotropic defects in plant development and reduced accumulation of most
miRNAs as in the DCL1 or AGO1 mutants Howeverit is unclear whether the HASTY
proteins export the miRNA miRNA duplexes or the miRNA-RISC complexes in
plants
11345 Feedback regulation of miRNA biogenesis
MiRNA biogenesis is under feedback regulation such that the DCL1 and AGO1 genes
are themselves regulated by miRNAs DCL1 is itself a target of miR162 which leads to
the cleavage of the DCL1 mRNA (Xie et al 2003 de Alba et al 2013) Consistent
with this the levels of DCL1 mRNA are elevated in the mutants of miRNA processing
genes in which the miR162 abundance is reduced In addition there is a predicted
hairpin structure of miR838 in the 14th
intron of the DCL1 primary transcript
(Rajagopalan et al 2006) This prediction implies that DCL1-mediated processing
on this site can result in the cleavage of the DCL1 primary transcript into two truncated
fragments the N-terminal capped fragment and the C-terminal polyadenylated
fragment If this is the case it is possible that miRNA biogenesis competes with the
splicing event of the DCL1 pre-mRNA
The AGO1 is also regulated by miRNA There is a complementary site for
miR168 binding in the AGO1 mRNA and the transcript level of the AGO1 mRNA is
reduced through an mRNA cleavage mechanism (Vaucheret et al 2006 de Alba et al
2013 Rogers amp Chen 2013) indicating that the AGO1 mRNA levels are tightly
controlled by the levels of the AGO1 protein
1135 Mechanism of miRNA action
11351 MiRNA-directed mRNA cleavage
By forming the miRNA-RISC assembly miRNAs can direct the RISC to regulate gene
expression by two post transcriptional mechanisms mRNA cleavage or translational
repression (Bartel 2004 Jones-Rhoades et al 2006 Dalmay 2013) The degree
of miRNA-mRNA complementarity plays an important role for the
determination of the mechanism to be used A general rule is that while perfect or
Chapter 1 Introduction and Review of Literature
40
near-perfect complementarity induces mRNA cleavage central mismatches trigger
translational repression (Carrington amp Ambros 2003 Jones-Rhoades amp Bartel 2004)
Unlike most animal miRNAs plant miRNAs have near-perfect or perfect
complementarity to their targets and mRNA cleavage is assumed to be a predominant
mechanism by which miRNAs regulate gene expression in plants
In RNA cleavage mechanism miRNAs guide the AGO component of RISC to
cleave a single phosphodiester bond of the target mRNA within the miRNA-binding
site (Llave et al 2002b Tang et al 2003) The truncated fragments are then released
and degraded freeing the RISC to recognize and cleave another target mRNA This has
been validated by the detection of 3prime cleavage products that have 5prime ends that start at the
middle of the complementary regions The mRNA cleavage products are then further
degraded by other mechanisms The 3prime cleavage products of some miRNA targets are
degraded by the 5prime-3prime exonuclease XRN4 an Arabidopsis homolog of the major Yeast
mRNA degrading exonuclease Xrn1p It has been observed that the 3prime cleavage
products of some target mRNAs were accumulated in the xrn4 mutants (Souret et al
2004 Dalmay 2013) However the 3prime cleavage products of other miRNA targets do
not accumulate in the xrn4 mutants indicating that there are also XRN4 independent
mechanisms The 5prime cleavage products are typically untraceable by RNA gel blot
hybridization but can be detected by sensitive PCR based methods It has been found
that the 5prime cleavage products tend to obtain an oligo U tail at the 3prime ends This 3prime U tail
is correlated with shortening of the 5prime cleavage products from the 5prime end leading to the
conclusion that the 3prime uridylation causes 5 prime- 3 prime exonucleolytic decay of the 5prime cleavage
products (Shen amp Goodman 2004)
DCL1 HYL1 and SE interact with each other and are co-localized in the
nuclear bodies termed Dicing bodies or D-bodies in which some pri-miRNA shave
been found suggesting that D-bodies serve as a center for active miRNA processing
(Fang amp Spector 2007 Fujioka et al 2007 Song et al 2007) The miRNA
processing site appears to be distinct from the Cajal bodies that have previously been
found as a siRNA processing site (Pontes amp Pikaard 2008) The D-bodies include
SmD3 and SmB proteins that are found in the Cajal bodies However At collin an
additional marker for the Cajal bodies is not found in the D-bodies Moreover the D-
bodies are present in an At collin mutant lacking the Cajal bodies The pri-miRNAs of
miR171A and miR159A genes have been found to be co-localized with HYL1 and
Chapter 1 Introduction and Review of Literature
41
DCL1 in the D- bodies that are distinct from the Cajal bodies (Song et al 2007)
11352 MiRNA-directed translational repression
Translation repression is another mechanism exerted by plant miRNAs The regulatory
mechanism of miR172 which regulates flowering time and floral organ identity is
consistent with translation repression MiR172 over production does not affect the
transcript levels of APETALA2 (AP2) and AP2- like target mRNAs Instead the AP2
protein level is reduced in the miR172 over expressing mutant (Aukerman amp Sakai
2003 Chen 2004 Dalmay 2013) It was later shown that miR172 can also direct the
cleavage of the target mRNAs although the steady-state mRNA level is unchanged due
to feedback regulation (Schwab et al 2005 Jung et al 2007) It is clear that miR172
regulates its target genes primarily by translational repression rather than mRNA
cleavage Recently it has been reported that miR156157 and miR854 also regulate
their target genes through translational repression (Arteaga-Vazquez et al 2006
Gandikota et al 2007 Dalmay 2013) It was once thought that translational repression
is an exception to the rule However experimental evidence suggest a widespread
coexistence of translational repression and mRNA cleavage (Brodersen et al 2008)
1136 Regulatory roles of miRNAs in plant development
The miRNA target prediction has been a difficult task in order to obtain regulatory
targets without bringing in too many false predictions Plant growth and developmental
processes regulated by miRNAs are quite diverse and enormous Furthermore plant
miRNAs work cooperatively or antagonistically to establish a balanced regulation This
notion is well consistent with the findings that mutations in the regulators of miRNA
biogenesis transport and processing such as AGO1 DCL1 HYL1 HEN1 ABH1
and HASTY cause pleiotropic phenotypes (Mallory amp Vaucheret 2006 de Alba et al
2013) To date the roles of miRNAs have been mostly deduced from obtaining miRNA
over expressing transgenic plants or gain-of-function mutants in which miRNA-
resistant target genes are ectopically expressed
11361 Phase transition
Plants pass through a series of developmental phase transitions during their life span
beginning with seed germination continuing through vegetative phase change
reproductive phase change flowering initiation and finally seed production for the next
generation Because of their importance in understanding plant developmental
Chapter 1 Introduction and Review of Literature
42
processes genes regulating developmental transitions have been extensively studied
and underlying molecular mechanisms have been explored in various plant species
Majority of researches were mainly focused on vegetative phase change and
reproductive transition in Arabidopsis and Maize (Poethig 2003 Baurle amp Dean
2006) Particularly the mutations of the genes encoding various regulators of miRNA
biogenesis and transport such as HST SE and AGO exhibit altered juvenile to adult
vegetative phase change and vegetative to reproductive change (Hunter et al 2003
Peragine et al 2004 Vazquez et al 2004a Jones-Rhoades et al 2006 de Alba et al
2013)
Over-expression of the miR156a gene causes late flowering and delays
vegetative phase change (Wu amp Poethig 2006 Gandikota et al 2007 Wang et al
2008) It has been shown that miR156 negatively regulates a group of genes encoding
SQUAMOSA PROMOTER-BINDING PROTEIN-LIKE (Lewis et al 2007 de Alba et
al 2013 Rogers amp Chen 2013) transcription factors such as SPL3 SPL4 and SPL5
that promote flowering initiation In contrast transgenic plants over expressing
miR156-resistant SPL345 genes are early flowering Interestingly the endogenous
level of miR156 is temporally regulated In the early juvenile vegetative phase the
level of miR156 is very high but decreases rapidly before the onset of the adult juvenile
phase (Wu amp Poethig 2006)
The Arabidopsis early activation tagged (eat-D) mutant exhibits early flowering
with disrupted floral structure The mutant phenotypes are caused by over expression of
the miR172a-2gene (Aukerman amp Sakai 2003) MiR172 regulates a small group of
genes encoding APETALLA2 (AP2)-like transcription factors such as TARGET OF
EAT1 (TOE1) TOE2 SCHLAFMUTZE (SMZ) and SCHNARCHZAPFEN(SNZ)
TOE1 TOE2 SMZ and SNZ have been shown to participate in their productive phase
change by repressing a floral integrator FLOWERING LOCUS T (FT)(Jung et al
2007) Consistent with this toe1-2D 35STOE2 35SSMZ and 35SSNZ plants
exhibit late flowering (Jung et al 2007) MiR172 is also regulated temporally and
highly expressed in the adult vegetative phase both in Arabidopsis and maize (Chuck et
al 2007) Because the temporal expression patterns of miR156 and miR172 are
complementary to each other it has been suggested that the two miRNAs interact with
each other in regulating phase change (Chuck et al 2009 de Alba et al 2013) In
support of this view it has been shown that miR172 is down-regulated in the
Chapter 1 Introduction and Review of Literature
43
Corngrass1(Cg1) mutant in which miR156 is overproduced (Chuck et al 2007 de
Alba et al 2013) However there is no direct evidence for the direct linkage between
miR156 and miR172 It is also envisioned that miR156 and miR172 would not be
directly related For example miR156 regulates plastochron index that is the inverse of
leaf initiation rate and thus frequently used to analyze temporal patterns of plant
development In contrast miR172 does not affect plastochron (Wang et al 2008) The
down-regulation of miR172 in the maize miR156-over producing mutants would be
due to the prolonged juvenile vegetative phase in the mutants
Another miRNA that regulates flowering initiation is miR159 It regulates
MYB33 and MYB65 which are closely related to the barley GAMY B transcription
factor that responds to gibberellic acid (GA) The level of miR159 is elevated in GA-
treated seedlings but reduced in the GA biosynthetic ga1 mutant Transgenic plants
over producing miR159 exhibit delayed floral induction under short days (Achard et
al 2004) It is been suggested that MYB33 mediates GA signal to regulate LEAFY
(LFY) These observations indicate that miR159 plays an important role in the GA
signaling pathways that regulate proper floral induction under short days (Achard et al
2004)
11362 Floral development
Arabidopsis flowers consist of four distinct organs They arise in a characteristic
pattern within concentric rings called whorls from outside to inside four sepals four
petals six stamens and the central pistil consisting of two carpels Identities of the
floral organs are determined by three classes of regulatory genes classA classB and
class C genes (Krizek amp Fletcher 2005) The A and C class genes act antagonistically
to restrict their expression domains (Bowman et al 1991)
It is notable that miR172 also regulates floral architecture in addition to its role
in the timing of flowering initiation MiR172 inhibits the translation of the AP2 mRNA
(Chen 2004) Transgenic plants overproducing miR172 not only exhibit early
flowering but also show abnormal floral development which is quite similar to those
of the class A gene mutants such as the ap2 mutants (Aukerman amp Sakai 2003 de
Alba et al 2013) When the activity of the class A genes is inactivated that of the class
C genes such as AGAMOUS (AG) is elevated As a result the miR172- overproducing
transgenic plants exhibit no petals along with homeotic transformation of the sepals to
Chapter 1 Introduction and Review of Literature
44
the carpels (Aukerman amp Sakai 2003 Chen 2004)
Interestingly miR166 also regulates floral architecture The role of miR165166
in the regulation of meristem activity is closely linked to floral meristem formation and
organization (Zhao et al 2007 Miyashima et al 2013) Floral structure is severely
disrupted in the miR165166-overproducing mutants such as men1 and jba-1D in
which miR166 is overproduced (Williams et al 2005b Jung amp Park 2007) The men1
and jba-1D mutants have smaller gynoecium and reduced carpel number In an extreme
case the jba-1D homozygous plants lack visible carpels (Williams et al 2005b)
Similar phenotypes have been observed in the miR165 overproducers (Zhao et al
2007) It has been suggested that the phenotypes are possibly caused by altered AG
expression in the floral meristem (Jung amp Park 2007 Turchi et al 2013)
Other miRNAs also affect floral development Overproduction of miRNAs
involved in auxin signaling also affects floral architecture Transgenic plants
overproducing miR167 have defects in the formation of ovule and anther with reduced
fertility (Ru et al 2006) It has been shown that miR167 targets ARF6 and ARF8 that
play a role in gynoecium and stamen development and in regulating fertility (Ru et al
2006 Wu et al 2006) These observations suggest that auxin signals are also closely
related to floral organogenesis In addition transgenic plants over-producing miR164
and the cuc1cuc2 double mutants show fused sepals and reduced petals (Laufs et al
2004 Turchi et al 2013) suggesting that miR164 is also related to floral meristem
activity and boundary specification of the meristematic region
11363 Shoot apical meristem development
Shoot apical meristem comprises self-maintaining totipotent cells The shoot apical
meristem activity affects diverse aspects of plant developmental processes including
leaf development vascular development and lateral organ polarity (Bowman et al
2002) Because miR165166 plays a primary role in meristem formation
overproduction of miR165166 and multiple knockout mutants of its target genes
exhibit pleiotropic pheonotypes (McConnell et al 2001 Emery et al 2003 Kim et al
2005 Williams et al 2005b)
The targets of miR165166 include genes encoding the homeo domain-leucine
zipper (HD-ZIP) class III transcription factors such as PHABULOSA(PHB)
PHAVOLUTA (PHV) REVOLUTA(REV) ATHB15CORONA (CNA) and ATHB8
Chapter 1 Introduction and Review of Literature
45
PHB PHV and ATHB15 have overlapping functions in controlling meristem
development (Turchi et al 2013) Indeed the phb phv athb15 triple mutants have an
enlarged shoot apical meristem Consistent with this the miR166- overproducing men1
and jba-1D mutants exhibit enlarged shoot apical meristems (Kim et al 2005
Williams et al 2005b Turchi et al 2013) In the mutants the shoot apical meristems
are multiplied and large
The development of shoot apical meristems is also regulated by the WUSCHEL
(WUS)ndashCLAVATA (CLV) signaling pathway WUS positively regulates cell
proliferation In contrast CLV3 which is expressed in stem cells limits the WUS
expression and thus inhibits cell proliferation in the SAM (Sharma et al 2003)
Consistent with this notion WUS is expressed to a higher level in jba-1D
explaining the ectopic enlargement of the SAM in the mutant (Williams et al 2005)
While WUS is closely related to miR165166 in the shoot apical meristem
development the underlying molecular mechanisms are currently unclear It has been
observed that the miR166-over producing men1 plants have an arrested meristem
development (Jung amp Park 2007) In addition the heterozygous men1+ and the
homozygous men1 plants show different expression patterns of WUS and CLV3 It
appears that miR166 regulates WUS indirectly and influences the expressional balance
between WUS and CLV3 through a negative feedback loop (Jung amp Park 2007 de
Alba et al 2013)
The phv phb athb15 triple mutant has an enlarged shoot apical meristems the
rev phb phv mutant does not have functional shoot apical meristems (Prigge et al
2005) This distinction may be explained by the fact that while miR166 targets
primarily PHB PHV and ATHB15 miR165 targets all the five HD-ZIP III genes (Zhou
et al 2007) Consistent with this transgenic plants overproducing miR166 are distinct
from those overproducing miR165 The miR165-overproducing transgenic plants lack
functional shoot apical meristem and a pin-like structure is formed at the apical region
of the mutant These phenotypes are quite similar to rev phb phv triple mutant (Zhou et
al 2007 Liu et al 2013) The phenotypic distinction may because by the difference
of the cleavage efficiencies of the target mRNAs In accordance with this notion a few
35S miR166a transgenic lines exhibit no shoot apical meristems The men1
homozygous plants also show arrested meristem development (Jung amp Park 2007)
Chapter 1 Introduction and Review of Literature
46
MiR164 cleaves the CUC1 and CUC2 mRNAs as well as the NAC1 mRNA
CUC1 and CUC2 determines the organ boundary in the meristem Phenotypes of
transgenic plants that overproduce miR164 are similar to those of the cuc1cuc2 double
mutant that exhibits embryonic developmental defects such as cup-shaped cotyledons
and fused cotyledons (Laufs et al 2004) When a miR164-resistant CUC2 is
expressed the boundary domain is enlarged CUC also plays a role in embryonic shoot
meristem formation by regulating the SHOOT MERISTEMLESS gene It was therefore
concluded that miR164-mediated cleavage of CUC1 and CUC2 mRNAs determines the
sizes of the boundary domains in the shoot apical meristem and regulates separation of
the organs by restricting cell proliferation (Laufs et al 2004 de Alba et al 2013)
11364 Leaf development
The role of miR319 has been extensively studied in leaf development A dominant
jaw-D mutant overproducing miR319 shows uneven curved leaves with enlarged size
and five TCP genes which are closely related to the CINCINNATA (CIN) gene in
Antirrhinum majus are down regulated in the mutant suggesting that miR319 mediated
regulation of the TCP transcription factor genes are important for the final step of
leaf shaping (Palatnik et al 2003 Naz et al 2013) In addition to leaf shape and size
leaf polarity is also specified by miRNA MiR166-mediated cleavage of the HD-ZIPIII
mRNAs is a well to establish leaf polarity This discovery originates from the gain-of-
function mutants such as phb-1d and phv-1d wherein point mutations in the
complementary sites to miRNA helps the mRNA to evade or resist from miRNA-
mediated cleavage (McConnell et al 2001 Emery et al 2003 Naz et al 2013)
Plant leaves have a dorso-ventral polarity consisting of the adaxial (upper
surface) and abaxial (lower surface) sides The adaxial side is closer to the shoot
apical meristem Adaxial identity is specified by functionally redundant class III HD-
ZIP family genes such as PHB PHV and REV Ectopic expression of each genes
result in adaxialized leaves Dominant phb-1d and phv-1d mutants exhibit
transformation of the abaxial to adaxial fate and have rod-shaped leaves In contrast
miR165166-overproducing plants show pin-like cotyledons radicalized leaves and
abaxialized leaves (Chitwood et al 2007 Pattanayak et al 2013)
Leaf asymmetry is determined by miR166-mediated restriction of the
expression domains of the HD-ZIPIII genes The HD-ZIPIII genes are expressed in the
Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
20
In many coffee growing countries coffee is propagated in marginal areas where the
annual rainfall is below 1000thinspmm with prolonged dry spells of over 4-5 months In
those areas water shortage and unfavorable temperatures constitute major constraints
and the growth and productivity of C canephora are badly affected As with drought
periodic frost also affects coffee production in parts of Brazil The introduction of
drought and frost tolerant genes through genetic transformation would be of great
importance for alleviating these problems Research is now being carried out by several
groups to identify genes involved in biotic as well as abiotic stress The most promising
genetic engineering approach for drought tolerance includes the use of functional or
regulatory genes as well as the transfer of transcription factors In recent years plants
tolerant to high temperature and water stress have been the subject of intense research
(Chaves et al 2003 Chaves et al 2004 Coraggio et al 2004) For achieving drought
tolerance genes that have been targeted include those encoding enzymes involved in
detoxification or osmotic response metabolism enzymes active in signaling proteins
involved in the transport of metabolites and regulating the plant energy status (Chaves
et al 2003 Chaves et al 2004 Coraggio et al 2004) The dissection of molecular
mechanisms related to signal transduction and transcriptional regulation might help in
engineering drought tolerance in coffee
1103 Disease Resistance
Coffee leaf rust (CLR) caused by the fungus Hemileia vastatrix is the most important
disease in coffee with substantial loss to coffee production and productivity in all the
coffee growing countries In addition to leaf rust coffee berry disease (CBD) caused by
fungus Colletotrichum kahawae can be a devastating anthracnose leading to substantial
crop loss in Africa Several other fungal and bacterial diseases may also affect coffee
to a extent C arabica is more susceptible to many diseases than C canephora Though
most of the disease control measures rely upon chemical control they are more
expensive and labour intensive The long-term solution is the breeding of resistant
varieties which is the focus of many breeding programs However breeding for disease
resistant varieties is time consuming due to the perennial nature of coffee with its long
gestation period India has a long history of C arabica breeding especially for leaf rust
resistance and is the first country to demonstrate the existence of multiple races of leaf
rust Resistance to CLR is conditioned primarily by a number of major (SH) genes and
coffee genotypes are classified into different resistance groups based on their
Chapter 1 Introduction and Review of Literature
21
interaction with different rust races pathogen (van der Vossen 2001) Currently C
canephora provides the main source of resistance to pests and diseases including CLR
(H vastatrix) and CBD (C kahawae) and therefore used in breeding programs Other
diploid species like C liberica and C racemosa are being used as a source of resistance
to coffee leaf rust and coffee leaf miner respectively (Guerreiro et al 1999
Sreenivasan et al 1989) The development of coffee varieties resistant to major fungal
diseases such as CLR and CBD using transgenic technology will benefit the coffee
industry immensely During the last 15 years significant progress has been made in the
area of host-pathogen interactions (Staskawicz et al 1995 Feys et al 2000
Shivaprasad et al 2012) and many resistance genes involved in recognizing invading
pathogens have been identified and cloned (Takken et al 2000) A number of
signalling pathways induced because of pathogen infection have been dissected (Dong
et al 1998 Navarro et al 2006) Many antifungal compounds synthesized by plants to
combat fungal infection have been identified (Does et al 1998) Understanding the
specific induction of targeted pathways and identification of specific pathways
responsible for particular fungal resistance is important in order to employ this strategy
in transgenic technology The recent investigation of gene expression during coffee leaf
rust infection could provide an insight into the defence pathways operating in coffee
(Fernandez et al 2004 Nardi et al 2006) Efforts have been made to identify and
clone resistance genes from coffee for achieving durable resistance The genetic and
physical map of two resistance genes SH3gene conferring resistance to rust (Prakash et
al 2004) and the Ck1 gene conferring resistance to C kahawae CBD (Gichuru et al
2006) have been established These genes could be used for molecular marker assisted
breeding programs (Mishra et al 2012)
1104 Production of low-caffeine coffee
A widespread belief that the ingestion of caffeine can have adverse effects on
health resulted in the increased demand for decaffeinated coffee (Ashihara amp Crozier
2001) Several method were used to obtain decaffeinated plants by conventional
breeding techniques using low yielding varieties and mutants that accumulated
relatively low amounts of caffeine when compared to the commercially available ones
Low-caffeine and decaffeinated coffee represent around 10 of the coffee sales
around the world (Ribas et al 2006b) The industrial process for coffee decaffeination
can be expensive and affects the original flavour and aroma in coffee (David 2002)
Chapter 1 Introduction and Review of Literature
22
Transgenic coffee plants with suppressed caffeine synthesis using RNA interference
(RNAi) technology have been obtained (Ogita et al 2003 Ogita et al 2004) Specific
sequences in the 3prime untranslated region of the theobromine synthase gene (CaMXMT1)
were selected for construction of RNAi short and long fragments The caffeine and
theobromine content of the transgenic plants were reduced by ~ 70 when compared to
the untransformed plants In C canephora RNAi technology has also been employed
to silence the N-methyl transferase gene involved in caffeine biosynthesis (Vinod et al
2007) Promoter of an N-methyltransferase (NMT) gene involved in caffeine
biosynthesis was cloned (Satyanarayana et al 2005 Satyanarayana 2006) which will
be very useful for studying the regulation of caffeine biosynthesis
1105 Improvement in cup quality
Improvement in coffee cup quality requires elaborate knowledge of the chemical
constituents as well as the metabolic pathways involved in the elaboration of quality
The constituents of coffee beans include minerals proteins carbohydrates caffeine
chlorogenic acids (CGA) glycosides lipids and many volatile compounds that give
flavour to coffee by roasting Among these the role of three major constituents
sucrose CGA and trigonelline have been studied in coffee The sucrose content of
coffee bean is associated with coffee flavour the higher the sucrose content in green
beans the more intense will be the cup flavour (Clifford et al 1985 de Maria et al
1996) The sucrose content of C arabica (82-83) is higher than C canephora (33ndash
40) The sucrose amino acids ratio in green beans determines the profile of volatile
compounds Manipulating sucrose content in coffee bean is therefore important in
improving cup quality The sucrose synthase gene (CcSUS2) from C canephora has
been cloned and sequenced (Leroy et al 2005) This provides an opportunity to
manipulate the sucrose content in coffee Chlorogenic acids are products of
phenylpropanoid metabolism Chlorogenic acids are a group of hydroxycinnamoyl
quinic acids (HQA) formed by esterification between caffeic acids coumaric acids and
quinic acids (Clifford et al 1999) They are present in relatively large quantities in the
coffee bean and are the precursor of phenolic compounds in roasted beans C
canephora beans contain higher CGA (10) compared to C arabica beans (6-7)
CGA are known to have antioxidant properties as well as being associated with disease
resistance (Campa et al 2003) Genetic manipulations of genes involved in CGA
synthesis can serve either of these purpose by up- or down regulating the pathway
Chapter 1 Introduction and Review of Literature
23
Phenylalanine ammonia-lyase (PAL) catalyzes the first step of the phenylpropanoid
pathway leading to the synthesis of a wide range of chemical compounds including
flavonoids coumarins hydroxycinnamoyl esters and lignins (Hahlbrock amp Scheel
1989) Recently the full length cDNA and corresponding genomic sequences of PAL
from C canephora was isolated characterized and functionally validated (Mahesh et
al 2006 Parvatam et al 2006) This has opened up new possibilities for manipulating
the level of the PAL enzyme in coffee which in turn can be useful for improvement of
cup quality and manipulating antioxidant properties in coffee
1106 Fruit Ripening
Uniformity during fruit ripening is decisively related to cup quality in coffee and
consequently to the value of the product Fruits at the ideal ripening stage produce the
best organoleptic characteristics for coffee The presence of over ripened or green fruits
changes the acidity the bitterness and consequently the cup quality In order to
maximize uniform ripening of coffee fruits it is essential to control the action of genes
involved in the last step of maturation process Ethylene is known to trigger ripening
and increasing ethylene biosynthesis is associated with various stages of ripening
process (Protasio et al 2005) To control coffee fruit maturation two of the major
genes involved in ethylene biosynthesis namely ACC synthase and ACC oxidase
have been cloned (Protasio et al 2005 Neupane et al 1999) Introduction of the ACC
oxidase gene in antisense orientation have been achieved in both C arabica and C
canephora The effect of the transgene on ethylene production and fruit maturation has
yet to be reported The inhibition of genes downstream to the initial ethylene burst is
also an option to control coffee fruit maturation (Ribas et al 2006)
111 RNA interferenceRNAi
The regulation of gene expression at post transcriptional level has recently attracted
much attention because of the discovery of the phenomenon called RNA interference
(RNAi) RNAi based silencing techniques are more efficient than antisense mediated
gene silencing (Wesley et al 2001) The effect was first observed in plants wherein
silencing was observed when some copies of a transgene was in the forward orientation
with respect to the promoter and some in the reverse orientation This resulted in the
formation of double stranded RNA in the system The phenomenon in which
experimentally introduced double stranded RNA (dsRNA) leads to the loss of
expression of the corresponding cellular gene by sequence specific RNA degradation
Chapter 1 Introduction and Review of Literature
24
was termed as RNA interference or RNAi The protective effect of RNA silencing in
reducing virus infectivity supports the view that PTGS has evolved as a mechanism to
defend plants against virus infection and also to moderate the possible deleterious
genome-restructuring (insertional) activity of virus-like mobile genetic elements eg
retrotransposons A growing body of biochemical and genetic data has further
demonstrated that plants animals and yeasts share related mechanisms of specific
degradation of RNAs in which double-stranded forms of RNA are initiator molecules
(Brenstein et al 2001)
The key insight into the process of PGTS was provided by the experiments
conducted by Hamilton amp Baulcombe (1999) who identified the product of RNA
degradation as small RNA (siRNA) species of ~25nt of both sense and antisense
polarity SiRNA are formed and accumulate as double stranded RNA molecules of
defined chemical structures Both genetic and biochemical approaches were undertaken
to understand the basis of silencing Genetic screens were carried out in fungus
Neurospora carassa the alga Chalmydomonas reinhardtii and plant Arabidopsis
thaliana to search for detective mutants in PTGS
A combination of results obtained from several in vivo and in vitro experiments
have gelled into a two step mechanistic model for RNAiPTGS The first step referred
to as the RNAi initiating step involves binding of the RNA nucleases to a larges
dsRNA and its cleavage into discrete ~21-25nt RNA fragments This is ATP-
dependent reaction which involved RNase III - type endonucleases which make
staggered cuts in both strands of the dsRNA leaving 3prime overhang of 2 nucleotides with
5prime- phosphate 3prime-hydroxyl and no modification in the sugar phosphate backbone
(Hamilton amp Baulcombe 1999 Elbashir et al 2001) SiRNAs are the direct products
of dsRNA cleavage by the multi-domain RNase III enzyme DICER (Figure 16)
(Brenstein et al 2001 Karthikeyan et l 2013)
In the second step or commonly known as the effector step the double stranded
siRNAs produced in the first step bind to an RNAi specific protein complex to form a
RISC The complex undergoes activation in the presence of ATP so that the antisense
component of the unwound siRNA becomes exposed and allows the RISC to perform
the downstream RNAi reaction
Chapter 1 Introduction and Review of Literature
25
Several enzymes are involved in RNAi based on their role genes that encode for them
are classified into RNAi initiators RNAi effectors RNA dependent RNA polymerases
and silencing genes Two C elegans genes rdeI and rde4 (rde stands for lsquo RNAi
deficientrsquo) are believed to be involved in the initiation step of RNAi The C elegans
rde1 gene is a member of a large family of genes and is homologous to the Neurospora
qde2 (qde stands for ldquoquelling deficientrdquo) and Arabidopsis AGO1 (AGO stands for
agronaute AGO1 was previously identified to be involved in Arabidopsis
development) Although the functions of these genes are not clear a mammalian
member of RDE1 family has been identified as a translation initiation factor (Thakur
2005) Important genes for the effector step of PTGS in C elegans are rde2 and mut7
genes These genes were identified from heterozygous mutant worms that were unable
to transmit RNAi to their homozygous offsprings Worms with mutated rde2 or mut 7
genes show defective RNAi mut-7 gene encodes a protein with homology to the
nuclease domains of RNAase D and a protein implicated in Werner syndrome (a rapid
ageing disease in humans) (Grishok et al 2001 Kamath-Loeb et al 2012s)
Neurospora qde-1 Arabidopsis SDE-1SGS-2 and C elegans ego-1 appear to encode
RNA dependent RNA polymerase (RdRPs) It assumed that this is a proof that an RdRp
activity is required for RNAi Certainly the existence of an RdRp might explain the
remarkable efficiency of dsRNA induced silencing if it amplifies either the dsRNA
prior to cleavage or the siRNAs directly (Muers 2013) In C elegans ego-1 mutants
(ego stands for lsquoenhancer of glp-1) RNAi functions normally in somatic cells but is
defective in germline cells where ego-1 is primarily expressed In Arabidopsis SDE-
1SGS-2 mutants (SGS stands for suppressor of gene silencing) siRNA are produced
when dsRNA is introduced via an endogenously replicating RNA virus but not
introduced by a transgene It has been proposed that perhaps the viral RdRP is
substituting for the Arabidopsis enzyme in these mutants Random degradative PCR
model suggests that an RdRP uses the guide strand of an siRNA as a primer for the
target mRNA generating a dsRNA substrate for Dicer and thus more siRNAs
Several genes controlling RNA silencing in plants have been identified through
genetic screens of Arabidopsis mutants impaired in transgene induced RNA silencing
They encode a putative RNA-dependent RNA polymerase (SGS2SDE1) a coiled
protein (SGS3) a protein containing PAZ and Piwi domains (AGO1) and an RNA
helicase (SDE3) The putative SGS2SDE1 of Neurospora is related to QDE-1 and
Chapter 1 Introduction and Review of Literature
26
EGO-1 of C elegans whereas PAZPiwi protein AGO is related to QDE-2 of
Neurospora RDE-1of C elegans RNA helicase SDE3 is related to SMG-2 of C
elegans and Mut-6 of Chlamydomonas MUT-7 gene of C elegans encodes a protein
similar to Rnase D whereas the Drosophila DICER gene encode a protein similar to
RNase III An Arabidopsis ortholog of DICER gene has been identified called CAF
SIN1 SUS1(Thakur 2005 Poethig 2009)
Since the RNAi machinery is present constitutively within the eukaryotic cell it
is important to explore the metabolic advantages that are accorded by the RNAi related
proteins during the intrinsic normal growth of cells and development of organisms The
natural RNAi machinery not only keeps the mobile transposable elements from
disrupting the integrity of the genomes as suggested by the analysis of model organisms
like A thaliana C elegans D melanogaster (Tabara et al 1999 Wu-Scharf et al
2000 Aravin et al 2001 Hamilton et al 2002 Lisch 2009) but also participates in
development of organisms Genetic defects in Celegans RNAi genes ego1 and dicer
cause known specific developmental errors (Grishok et al 2000 Grishok et al 2001
Knight amp Bass 2001 Hall et al 2013) Similarly the Argonaute family of genes of A
thaliana (especially the ZWILLE proteins) is also responsible for plant architecture and
meristem development (Carmell et al 2002 Hamant and Pautot 2010) and the Dicer
homologue of A thaliana CAF1 is required for embryo development (Golden et al
2002 Nakano et al 2011) Thus genetic evidence illustrates the role of the RNAi
machinery as a controller of development related genes The mechanistic details of
these developmental processes are beginning to emerge
Chapter 1 Introduction and Review of Literature
27
Figure 16 Mechanism of gene silencing induced double stranded RNA The dicer enzyme to
produce siRNAs cleaves the RNA The siRNA generated bind to the nuclease complex
RISC The antisense component in the siRNA in the RISC guides the complex towards
the cognate mRNA resulting in endonucleolytic cleavage of the mRNA
Chapter 1 Introduction and Review of Literature
28
112 MicroRNA or MiRNA
In 1991 Ambros and co-workers first isolated a lin-4 mutant of Celegans which was
arrested at the first larval stage (Lee et al 1993) Later on the let-7 mutation was
isolated in the same system which was responsible for development through the fourth
larval stage Both lin-4 and let-7 encode short 22-nucleotide mature RNAs and were
called short temporal RNA because they control the temporal development program of
C elegans The mature lin-4 RNA defines (negatively regulates) the mRNA expression
of the lin-14 and lin-28 hetrochronic genes with the antisense mediated repression
mechanism of translation initiation and thus specifies the fate of cells during the first
three larval stages Recent studies have revealed that the short temporal RNAs are
actually members of a group of tiny RNAs (21to 28 nucleotides) called the micro-
RNAs (miRNA) (Bartel 2009) isolated members of which could easily run to a few
thousand which includes both those were identified computationally and by deep
sequencing Some of the components of the RNAi machinery have also been clearly
established as the effector proteins for the maturation of miRNAs
113 MicroRNAs in plants
1131 Discovery
MicroRNAs have been discovered by three basic approaches Direct cloning forward
genetic direct cloning and bioinformatic prediction followed by experimental
validation The most direct method of miRNA discovery is to isolate and clone small
RNAs from biological samples and several groups have used this approach to identify
plant miRNAs (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002 Xie et al 2003 Sunkar et al 2005 Williams et al 2013) The cloning methods
adapted were similar to those used to identify large numbers of animal miRNAs
(Lagos-Quintana et al 2001 Lau et al 2001 Aravin amp Tuschl 2005) which involve
isolating small RNAs followed by oligonucleotide adapter ligation reverse
transcription amplification and sequencing Some protocols incorporate methods to
enrich for Dicer cleavage products (ie molecules with 5prime-phosphates and 3prime-
hydroxyls) and to concatemerize the short cDNAs so that several may be identified in a
single sequencing read (Lau et al 2001 Llave et al 2002a Jagadeeswaran amp Sunkar
2013)The initial cloning experiments in Arabidopsis identified 19 miRNAs belonging
to 15 families (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002) although hundreds of other small RNAs such as siRNAs and RNA degradation
Chapter 1 Introduction and Review of Literature
29
fragments were also cloned through the direct cloning methods To select miRNAs
from the pool of small RNAs secondary structures of the RNA molecules were
examined in compliance with the acknowledged structural features of miRNAs
(Ambros et al 2003) The secondary structures were validated using several ad-hoc
software tools such as the Mfold or RNAfold programs (Reeder et al 2006) Finally
the existence of mature miRNAs was determined by RNA gel blot hybridization These
direct cloning methods were successful in isolating many miRNAs in Arabidopsis
(Reinhart et al 2002 Sunkar amp Zhu 2004) Rice (Sunkar et al 2005) Poplar (Lu et
al 2005) moss (Arazi et al 2005) and Tobacco (Billoud et al 2005)
Although miRNAs were first discovered through forward genetic screens in
worms (Lee et al 1993 Reinhart et al 2000)only few A thaliana miRNA gene
families have been discovered by this method (Chen 2008) in plants MiRNA
involvement in plant mutant phenotypes were not properly inferred till the cloning
experiments established that plant genomes contain numerous miRNAs (Park et al
2002 Reinhart et al 2002 Rhoades et al 2002) MiRNA loss-of-function allele has
been identified by forward genetic screens early extra petala1 is caused by a
transposon insertion ~160bp upstream of the predicted miR164c stem loop resulting in
flowers with extra petals (Baker et al 2005 Irish 2008) The fact that loss-of-function
miRNA mutants have been rarely discovered using forward genetics reflects small
target size for mutagenesis coupled with redundancy in nearly all evolutionarily
conserved plant miRNAs which are encoded by gene families (Table16) Family
members are likely to have overlapping functions buffering against loss at any single
miRNA locus Over expression screens can circumvent redundancy limitations At least
four plant miRNAs miR319 (also known as miR-JAW) miR172 (also known as EAT)
and miR166 were isolated in over expression screens for dominant mutants with
developmental abnormalities (Aukerman amp Sakai 2003 Palatnik et al 2003 Kim et
al 2005 Williams et al 2005b Schommer et al 2012) Mutations in miRNA target
sites which can prevent the entire family of miRNAs from repressing a target gene can
also circumvent redundant functions of miRNA family members
The dominant mutations in the HD-ZIP genes PHB PHV and REVOLUTA
(REV) in Arabidopsis and ROLLEDLEAF1 (RLD1) in Maize result in adaxialization of
leaves andor Vasculature (McConnell amp Barton 1998 McConnell et al 2001 Nelson
et al 2002 Zhong amp Ye 2004 Allen amp Millar 2012) and are all caused by mutations
Chapter 1 Introduction and Review of Literature
30
in miR166 complementary sites (Rhoades et al 2002 Emery et al 2003 Jones-
Rhoades amp Bartel 2004 Mallory et al 2004 Zhong amp Ye 2004)
In both plants and animals cloning was the initial means of large-scale
miRNA discovery (Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Reinhart et al 2002 Mendes et al 2012) However cloning is biased toward
RNAs that are expressed highly and broadly MiRNAs expressed at low levels or
only in specific cell types or in response to certain environmental stimuli were more
difficult to clone Sequence based biases in cloning procedures might also cause
certain miRNAs to be missed Because of these limitations bioinformatics approaches
to identify miRNAs have provided a useful complement to cloning
A straightforward use of bioinformatics has been carried out to find homologs
of known miRNAs both within the same genome and in the genomes of other species
(Pasquinelli et al 2000 Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Mendes et al 2009) A more difficult challenge is to identify miRNAs
unrelated to previously known miRNAs This was first accomplished for
vertebrate nematode and fly miRNAs using algorithms that search for conservation
of sequence and secondary structure (ie miRNA stem-loop precursors) between
species searching for patterns that are characteristic of miRNAs (Lai et al 2003 Lim
et al 2003 Lim et al 2005)
Most of the methods identified numerous miRNAs but are necessarily more
relaxed in terms of allowed structures Some approaches take advantage of the high
complementarity of plant miRNAs to target messages implementing the requirement
that the candidate has conserved complementarity to mRNAs (Jones-Rhoades amp Bartel
2004 Adai et al 2005) This additional filter has been useful for distinguishing
authentic plant miRNAs from false positives which later extended to mammalian
miRNA gene prediction (Xie et al 2005 Mendes et al 2012)
Another criteria used by most algorithms in the filters is evolutionary
conservation In plants mature miRNA sequences are conserved to a higher degree
than their precursor sequences For example Arabidopsis and rice diverged more than
130 million years ago (Friis et al 2004) but some miRNAs are highly conserved
in these plant species (Reinhart et al 2002) Furthermore various parameters for the
secondary structures such as free energy the number of paired residues within a
Chapter 1 Introduction and Review of Literature
31
miRNA or the number and size of bulges are also used to validate the predictions
(Jones-Rhoades amp Bartel 2004 Wang et al 2004 Adai et al 2005 Mendes et al
2012)
1132 Conserved microRNAs in Plants
Till date cloning genetics and bioinformatics screens have resulted in annotation of
approximately 184 potential Arabidopsis miRNAs (miRBase release 13) These
miRNAs seem to arise through gene duplication and subsequent diversification and
represent approximately 100 families (Maher et al 2006 Axtell 2013) Twenty one
families represented by 92 genes are clearly conserved in species beyond Arabidopsis
(Table 15) The presence of these miRNA families in other sequenced plant genomes
indicates that different plant species have an evolutionarily fluid set of miRNAs
(Willmann amp Poethig 2007) Recent studies on mosses one of the most ancient land
plants have shown that at least 14 miRNA families are conserved in eudicots and
mosses and therefore it appears that plant miRNAs share a common ancient origin
(Arazi et al 2005 Axtell amp Bartel 2005 Talmor-Neiman et al 2006 Zhang et al
2006 Axtell 2013) The number of members per family in a genome ranges from 1-32
with the exception of miR-430 which is represented by a cluster of ~80 loci in zebra
fish (Giraldez et al 2005)
In plants the number of members in each family correlates among examined
species certain families contain numerous members in all three species (eg miR156
miR166 miR169) whereas others consistently contain fewer genes (eg miR162
mir168 miR394) (Table 16) Although it is still unclear why certain miRNA are
encoded by more than one gene in plants (eg 12 genes encoding miR156) this
correlation might suggest functional significance of various miRNA family sizes Most
annotated plant miRNAs are conserved throughout flowering plants while a few
miRNAs are found only in a single sequenced genome and thus they could be
evolutionarily lsquoyoungrsquo miRNAs For example Arabidopsis has several miRNA
families that are not found in poplar or rice suggesting that miRNAs could be
generated continuously during evolution (Allen et al 2004a Rajagopalan et al
2006 Fahlgren et al 2007 Axtell 2012 de Alba et al 2013) Consistent with the
notion that non conserved miRNAs are of a more recent evolutionary origin these
miRNAs are mainly encoded by a single locus in the genome Within each family the
mature miRNA is always located in the same arm
Chapter 1 Introduction and Review of Literature
32
Table 16 MicroRNA families conserved in plants
miRNA
family
Arabidopsis Oryza Populus
miR156 12 12 11 miR159319 6 8 15 miR160 3 6 8 miR162 2 2 3 miR164 3 5 6 miR166 9 12 17 miR167 4 9 8 miR168 2 2 2 miR169 14 17 32 miR171 4 7 10 miR172 5 3 9 miR390 3 1 4 miR393 2 2 4 miR394 2 1 2 miR395 6 19 10 miR396 2 5 7 miR397 2 2 3 miR398 3 2 3 miR399 6 11 12 miR408 1 1 1 miR403 1 0 2 miR437 0 1 0 miR444 0 1 0 miR445 0 9 0 Total 92 127 169
All known miRNA families that are conserved between more than one plant species are listed
together with the number of genes identified in the sequenced genomes Rice miRNA families that have orthologs in maize but do not appear to have orthologs in the eudicots (Arabidopsis and Populus) are marked with an asterisk The following families contain miRNA genes annotated with
more than one number miR156 (miR156 and miR157) miR159319 (miR159 and miR319) miR166 (miR165 and miR166) miR171 (miR170 and miR171) and miR390 (miR390 and miR391)
of the stem loop (5prime- or 3prime) as it would be expected if the genes carry common ancestry
(Figure 17) The sequence of the mature miRNA and to some extent the segment on
the opposite arm of the hairpin to which it pairs is highly conserved between the
members of the same family (both within and between the species) while the sequence
secondary structure and length of the intervening ldquolooprdquo region can be highly divergent
within the same family
The patterns of paring and non pairing nucleotides are often conserved between
homologous miRNA stem loops from different species (Figure 17) The significance of
conserved mismatches is opined to guide DCL1 to cleave at the appropriate positions
along the stem loop
Chapter 1 Introduction and Review of Literature
33
Figure 17 Representative stem loop of miR164 Segments corresponding to the mature miRNAs
are shown in red (Adapted from Rajagopalan et al 2006)
Although many miRNAs are highly conserved in plants the degree of conservation in
most cases is quite low between plants and animals one exception is miR854 A recent
study has revealed that miR854 is conserved between Arabidopsis and animals and that
the Arabidopsis miR854 differs from the human miR854 only by a single nucleotide
(Arteaga-Vazquez et al 2006 Axtell 2012 de Alba et al 2013) The Arabidopsis
miR854 has multiple binding sites in the 3prime-untranslated region (UTR) of
OLIGOURIDYLATE-binding-PROTEIN mRNA 1bA (UBP1b) and represses the
translation of the UBP1b mRNA The animal miR854 is also complementary to a site in
the 3prime-UTR of the UBP1b homolog in animals However it is currently unclear how the
animal miR854 regulates its target mRNA Another distinction between plant and
animal miRNAs is the genomic organization of the miRNA genes While plant miRNA
genes are located predominantly in the intergenic regions animal miRNA genes tend to
localize in the intragenic regions both in the introns or exons of protein coding genes
Moreover miRNA genes are often clustered within a single precursor transcript
whereas individual plant miRNAs are derived from individual precursor RNAs (Bartel
Chapter 1 Introduction and Review of Literature
34
2004 Bonnet et al 2004 Kim 2005 Vaucheret et al 2006 Marco et al 2013)
1133 Non-conserved miRNA and challenges in annotation
Most miRNAs are conserved throughout flowering plants (Table 15) but many more
were found only in a single sequenced genome thus considered to be of a more recent
evolutionary origin The extended homology between few non conserved miRNAs
precursors and their target genes provides strong evidence that these relatively ldquoYoungrdquo
miRNAs arose from the duplication of the target gene segments (Allen et al 2004)
Several non-conserved miRNAs including miR161 miR163 miR173 miR447
miR475 and miR476 are known to direct cleavage of target transcripts (Allen et al
2004a Adai et al 2005 Sunkar et al 2005 de Alba et al 2013) It is difficult to
confidently predict targets for many because it is not possible to use complementary
site as a filter against false-positive target predictions It is also difficult to
confidently annotate non-conserved miRNAs as miRNA rather than siRNAs The
established minimal standard for miRNA annotation is a small RNA with detectable
expression and the potential to form a stem-loop when joined to flanking genomic
sequence (Kasschau et al 2003) In practice these requirements are too loose to
definitively categorize many small RNAs cloned from plants Many plant siRNAs are
detectable on blots (Vazquez et al 2004b) and hundreds of thousands of non-
miRNA genomic sequences can be predicted to fold into secondary structures that
resemble structures of plant miRNA precursors (Jones-Rhoades amp Bartel 2004)
Therefore without conservation of both sequence and secondary structure it is
difficult to be confident that a given cloned RNA originated from a stem-loop (ie is a
miRNA) rather than from a double-stranded RNA (ie is a siRNA) In fact many of
the thousands o f small RNAs cloned from Arabidopsis (Gustafson et al 2005) would
probably meet the literal requirements for annotation as miRNAs A few of these
sequences might be miRNAs but others that meet the literal criteria probably are not
so The challenges of annotating non conserved small RNAs were evidenced by
three related small silencing RNAs that were originally annotated as miRNAs
that turned out to be trans-acting siRNAs (tasiRNAs)(Kanno et al2013)
Although most miRNAs that are broadly conserved among flowering plants are
now being identified and experimentally validated (Table 16)(Jones-Rhoades amp
Bartel 2004) the challenges and ambiguities for classifying non-conserved miRNAs
prevent meaningful estimates of the total number of miRNA genes in Arabidopsis and
Chapter 1 Introduction and Review of Literature
35
other plant genomes It is possible that miRNAs might have escaped detection
because they are non-conserved and expressed in specific tissues or conditions and can
only be identified by using high- throughput sequencing
1134 MicroRNA biogenesis
11341 Transcription of microRNA precursor
Plant miRNAs are primarily found in the genomic regions that are not associated with
the protein coding genes (Reinhart et al 2002) and it appears that most if not all plant
miRNAs are produced from their own transcriptional units Plant miRNA genes are
occasionally clustered near each other in the genome suggesting transcription of
multiple miRNAs from a single primary transcript [eg the miR395 cluster (Grishok
et al 2001 Jones-Rhoades amp Bartel 2004b)] but this polycistronic arrangement of
miRNA genes appears far less frequently in plants than in animals (Bartel 2004)
Northern blot EST and mapping evidence indicate that plant miRNA primary
transcripts (also known as pri-miRNAs) as in animals are longer than needed to
encompass the miRNA stem-loops (Palatnik et al 2003 Jones-Rhoades amp Bartel
2004b) At least some of these pri-miRNA transcripts appear to be spliced
polyadenylated (Kurihara amp Watanabe 2004) and capped (Xie Z et al 2005) Two
rice miRNAs are contained within transcripts that contain exon junctions within the
presumptive stem-loop precursor implying that in these cases splicing is a prerequisite
for Dicer recognition (Sunkar et al 2005) Plant pri-miRNAs are over 1kb in length
and they are usually preceded by typical TATA box motifs They can also undergo
canonical splicing polyadenylation and capping All these observations indicates RNA
polymerase II is probably responsible for transcribing most plant miRNAs (Xie Z et
al 2005) as appears to be the case for many animal miRNAs Relatively little is
known about the regulation of miRNA transcription in plants but there is no reason
to suspect that this regulation would differ from that of protein-coding transcripts as
the bioinformatics analysis of the sequence upstream of the transcriptional start
sites of the miRNA genes showed the presence of putative binding motifs of
number of known transcription factors (Megraw et al 2006 Sethupathy 2013)
11342 MicroRNA processing and export
In plants maturation of miRNA is a step wise process A miRNA gene is transcribed to
pri-miRNA which is much longer than the pre-miRNA possessing the characteristic
stem-loop structure Like transcription of most protein-coding genes the miRNA
Chapter 1 Introduction and Review of Literature
36
transcription is processed by RNA polymerase II (Pol II) enzymes and the pri-
miRNAs are 5prime-capped and 3prime-polyadenylated (Lee et al 2004 Xie Z et al 2005)
Mature miRNAs are produced from pri-miRNAs through at least two sequential
processing steps by RNase III-type endonucleases In animals pri-miRNAs are first
processed at the bottom portion of the stem by Drosha a nuclear-localized RNase III
enzyme to liberate pre-miRNAs The pre-miRNAs are then exported to the cytoplasm
with the help of exportin-5The pre-miRNAs are further processed by Dicer another
RNaseIII enzyme to a duplex consisting of the miRNA and the antisense strands
(miRNA) Since plants do not have any Drosha homologs the two sequential
processing steps seem to be carried out by DCL1 a Dicer homolog in the nucleus
(Kurihara amp Watanabe 2004 de Alba et al 2013 Rogers amp Chen 2013)
The processing of pri-miRNAs to pre-miRNAs by DCL1 is assisted by two
other proteins HYPONASTY LEAVES1 (HYL1) and SERRATE (SE) HYL1 belongs to a
family of double-stranded RNA (dsRNA) binding protein and interacts with DCL1 in
vitro and in vivo (Lu amp Fedoroff 2000 Hiraguri et al 2005 de Alba et al 2013
Rogers amp Chen 2013) Loss-of-function mutations in the HYL1 gene result in
decreased accumulation of miRNAs and concomitant accumulation of pri-miRNAs
(Han et al 2004 Vazquez et al 2004a Kurihara et al 2006) SE a C2H2 zinc finger
protein interacts with DCL1 and HYL1 and plays a role in the processing of pri-
miRNAs to pre-miRNAs (Lobbes et al 2006 Yang L et al 2006) In addition the
nuclear cap-binding complex proteins CBP20 and CBP80 that are known as ABA
HYPER SENSITIVE 1(ABH1) have been shown to be required for proper miRNA
processing (Gregory et al 2008 Laubinger et al 2008) Recently it has been
demonstrated that DCL1 can release 21-nt RNAs from dsRNA as well as from
synthetic miR167 precursor RNAs in vitro However this cleavage is error prone and
inefficient in the absence of HYL1 and SE which accelerate the rate of DCL1-mediated
cleavage and promote accurate processing of miRNAs (Dong et al 2008 Rogers amp
Chen 2013)
11343 Methylation of the miRNAmiRNAduplex
After the miRNAmiRNAduplexes are released from the pre-miRNAs by the DCL1
activities the duplexes are methylated at the 2primeOH of the3prime-ends by HUA ENHANCER1
(HEN1) a small RNA methyltransferase (Yu et al 2005 Yang Z et al 2006 Rogers
amp Chen 2013 ) This step which is distinct from the RNase III-mediated processing
Chapter 1 Introduction and Review of Literature
37
step distinguishes the biogenesis of plant miRNAs from that of animal miRNAs
Mutations in the HEN1 gene were first isolated in a morphological screening for floral
development although the HEN1 mutants display pleiotropic phenotypes similar to the
developmental defects observed in the DCL1 mutants (Chen et al 2002 Park et al
2002 Rogers amp Chen 2013)
Figure 18 Biogenesis of microRNA
A Biogenesis of plant MicroRNA B Biogenesis of metazoananimal MicroRNA
The miRNA (red) is incorporated into RISC and the miRNA(blue) in degraded
This enzyme has two dsRNA-binding domains and nuclear localization signal It
is also conserved in fungi and is required for the processing of siRNAs as well as of
miRNAs (Park et al 2002 Boutet et al 2003 Xie et al 2003) Although the HEN1
protein is localized to the nucleus its methylation activity in the cytoplasm cannot be
ignored (Xie et al 2004 Rogers amp Chen 2013)
Chapter 1 Introduction and Review of Literature
38
The methylation of the miRNAmiRNA duplex is required to protect miRNAs
against the 3prime-end uridylation activity and subsequent degradation (Li et al 2005) In
the hen1 mutants accumulation of miRNAs is reduced to a lower level Also the
miRNAs appear to be heterogeneous in their sizes such that a ladder of bands rather
than a single species is observed for most miRNAs in the mutants (Li et al 2005)
MiRNA cloning and sequencing analyses revealed the presence of a number of U
residues at the 3prime-end of miRNAs in the hen1 mutants Moreover these nucleotides do
not correspond to the miRNAs in the pre-miRNAs indicating that these nucleotides are
added after the processing of the miRNAs from pre-miRNAs (Li et al 2005)
Uridylation of RNAs has also been proven to be correlated with RNA decay (Shen amp
Goodman 2004 Mullen amp Marzluff 2008) suggesting that uridylation attracts an
exonuclease which degrades miRNAs in plants
11344 MiRNA incorporation in to the RISC and its nuclear export
The methylated miRNAmiRNA duplexes undergo RISC assembly In this process
the miRNA of the duplexes is selectively incorporated into the RISC and the miRNA
appears to be removed and subsequently degraded (Hammond et al 2000 Hutvagner
amp Zamore 2002 Schwarz et al 2003 de Alba et al 2013 Rogers amp Chen 2013)
The ARGONAUTE 1(AGO1) proteins are core components of the RISC complex
They contain two structural domains the N-terminal PAZ domain that binds to the 3prime-
end of single-stranded RNAs and the C-terminal piwi domains that has a structure
similar to that of RNase H (Cerutti et al 2000 Parker et al 2004) Several AGO
proteins possess endonucleolytic activities within the PIWI domain and cleave target
mRNAs in the middle of the site complementary to miRNA (Liu et al 2004 Meister et
al 2004 Miyoshi et al 2005) Mutations in the AGO1 gene cause miRNA target
genes to be ectopically expressed and the levels of some miRNAs are reduced in the
mutants (Kidner amp Martienssen 2004 Vaucheret et al 2004 Kidner amp Martienssen
2005 Ronemus et al 2006) Moreover the phenotype of the AGO1 hypomorphic
alleles display defects in lateral organ polarity and leaf and flower morphology as
observed in the dcl1 mutants and null AGO1 alleles are embryonically lethal
supporting the close relationship between the AGO proteins and miRNA processing
(Kidner amp Martienssen 2004 Vaucheret et al 2004 de Alba et al 2013 Rogers amp
Chen 2013)
The production of miRNA miRNA duplexes occur in the nucleus while
Chapter 1 Introduction and Review of Literature
39
miRNA-RISC complexes are present in the cytoplasm where target mRNAs are
located This discordance of functional sites requires an export step during miRNA
biogenesis HASTY the Arabidopsis homolog of exportin-5 is thought to be involved
in miRNA nuclear export (Bollman et al 2003 Park et al 2005)The hasty mutant
exhibits pleiotropic defects in plant development and reduced accumulation of most
miRNAs as in the DCL1 or AGO1 mutants Howeverit is unclear whether the HASTY
proteins export the miRNA miRNA duplexes or the miRNA-RISC complexes in
plants
11345 Feedback regulation of miRNA biogenesis
MiRNA biogenesis is under feedback regulation such that the DCL1 and AGO1 genes
are themselves regulated by miRNAs DCL1 is itself a target of miR162 which leads to
the cleavage of the DCL1 mRNA (Xie et al 2003 de Alba et al 2013) Consistent
with this the levels of DCL1 mRNA are elevated in the mutants of miRNA processing
genes in which the miR162 abundance is reduced In addition there is a predicted
hairpin structure of miR838 in the 14th
intron of the DCL1 primary transcript
(Rajagopalan et al 2006) This prediction implies that DCL1-mediated processing
on this site can result in the cleavage of the DCL1 primary transcript into two truncated
fragments the N-terminal capped fragment and the C-terminal polyadenylated
fragment If this is the case it is possible that miRNA biogenesis competes with the
splicing event of the DCL1 pre-mRNA
The AGO1 is also regulated by miRNA There is a complementary site for
miR168 binding in the AGO1 mRNA and the transcript level of the AGO1 mRNA is
reduced through an mRNA cleavage mechanism (Vaucheret et al 2006 de Alba et al
2013 Rogers amp Chen 2013) indicating that the AGO1 mRNA levels are tightly
controlled by the levels of the AGO1 protein
1135 Mechanism of miRNA action
11351 MiRNA-directed mRNA cleavage
By forming the miRNA-RISC assembly miRNAs can direct the RISC to regulate gene
expression by two post transcriptional mechanisms mRNA cleavage or translational
repression (Bartel 2004 Jones-Rhoades et al 2006 Dalmay 2013) The degree
of miRNA-mRNA complementarity plays an important role for the
determination of the mechanism to be used A general rule is that while perfect or
Chapter 1 Introduction and Review of Literature
40
near-perfect complementarity induces mRNA cleavage central mismatches trigger
translational repression (Carrington amp Ambros 2003 Jones-Rhoades amp Bartel 2004)
Unlike most animal miRNAs plant miRNAs have near-perfect or perfect
complementarity to their targets and mRNA cleavage is assumed to be a predominant
mechanism by which miRNAs regulate gene expression in plants
In RNA cleavage mechanism miRNAs guide the AGO component of RISC to
cleave a single phosphodiester bond of the target mRNA within the miRNA-binding
site (Llave et al 2002b Tang et al 2003) The truncated fragments are then released
and degraded freeing the RISC to recognize and cleave another target mRNA This has
been validated by the detection of 3prime cleavage products that have 5prime ends that start at the
middle of the complementary regions The mRNA cleavage products are then further
degraded by other mechanisms The 3prime cleavage products of some miRNA targets are
degraded by the 5prime-3prime exonuclease XRN4 an Arabidopsis homolog of the major Yeast
mRNA degrading exonuclease Xrn1p It has been observed that the 3prime cleavage
products of some target mRNAs were accumulated in the xrn4 mutants (Souret et al
2004 Dalmay 2013) However the 3prime cleavage products of other miRNA targets do
not accumulate in the xrn4 mutants indicating that there are also XRN4 independent
mechanisms The 5prime cleavage products are typically untraceable by RNA gel blot
hybridization but can be detected by sensitive PCR based methods It has been found
that the 5prime cleavage products tend to obtain an oligo U tail at the 3prime ends This 3prime U tail
is correlated with shortening of the 5prime cleavage products from the 5prime end leading to the
conclusion that the 3prime uridylation causes 5 prime- 3 prime exonucleolytic decay of the 5prime cleavage
products (Shen amp Goodman 2004)
DCL1 HYL1 and SE interact with each other and are co-localized in the
nuclear bodies termed Dicing bodies or D-bodies in which some pri-miRNA shave
been found suggesting that D-bodies serve as a center for active miRNA processing
(Fang amp Spector 2007 Fujioka et al 2007 Song et al 2007) The miRNA
processing site appears to be distinct from the Cajal bodies that have previously been
found as a siRNA processing site (Pontes amp Pikaard 2008) The D-bodies include
SmD3 and SmB proteins that are found in the Cajal bodies However At collin an
additional marker for the Cajal bodies is not found in the D-bodies Moreover the D-
bodies are present in an At collin mutant lacking the Cajal bodies The pri-miRNAs of
miR171A and miR159A genes have been found to be co-localized with HYL1 and
Chapter 1 Introduction and Review of Literature
41
DCL1 in the D- bodies that are distinct from the Cajal bodies (Song et al 2007)
11352 MiRNA-directed translational repression
Translation repression is another mechanism exerted by plant miRNAs The regulatory
mechanism of miR172 which regulates flowering time and floral organ identity is
consistent with translation repression MiR172 over production does not affect the
transcript levels of APETALA2 (AP2) and AP2- like target mRNAs Instead the AP2
protein level is reduced in the miR172 over expressing mutant (Aukerman amp Sakai
2003 Chen 2004 Dalmay 2013) It was later shown that miR172 can also direct the
cleavage of the target mRNAs although the steady-state mRNA level is unchanged due
to feedback regulation (Schwab et al 2005 Jung et al 2007) It is clear that miR172
regulates its target genes primarily by translational repression rather than mRNA
cleavage Recently it has been reported that miR156157 and miR854 also regulate
their target genes through translational repression (Arteaga-Vazquez et al 2006
Gandikota et al 2007 Dalmay 2013) It was once thought that translational repression
is an exception to the rule However experimental evidence suggest a widespread
coexistence of translational repression and mRNA cleavage (Brodersen et al 2008)
1136 Regulatory roles of miRNAs in plant development
The miRNA target prediction has been a difficult task in order to obtain regulatory
targets without bringing in too many false predictions Plant growth and developmental
processes regulated by miRNAs are quite diverse and enormous Furthermore plant
miRNAs work cooperatively or antagonistically to establish a balanced regulation This
notion is well consistent with the findings that mutations in the regulators of miRNA
biogenesis transport and processing such as AGO1 DCL1 HYL1 HEN1 ABH1
and HASTY cause pleiotropic phenotypes (Mallory amp Vaucheret 2006 de Alba et al
2013) To date the roles of miRNAs have been mostly deduced from obtaining miRNA
over expressing transgenic plants or gain-of-function mutants in which miRNA-
resistant target genes are ectopically expressed
11361 Phase transition
Plants pass through a series of developmental phase transitions during their life span
beginning with seed germination continuing through vegetative phase change
reproductive phase change flowering initiation and finally seed production for the next
generation Because of their importance in understanding plant developmental
Chapter 1 Introduction and Review of Literature
42
processes genes regulating developmental transitions have been extensively studied
and underlying molecular mechanisms have been explored in various plant species
Majority of researches were mainly focused on vegetative phase change and
reproductive transition in Arabidopsis and Maize (Poethig 2003 Baurle amp Dean
2006) Particularly the mutations of the genes encoding various regulators of miRNA
biogenesis and transport such as HST SE and AGO exhibit altered juvenile to adult
vegetative phase change and vegetative to reproductive change (Hunter et al 2003
Peragine et al 2004 Vazquez et al 2004a Jones-Rhoades et al 2006 de Alba et al
2013)
Over-expression of the miR156a gene causes late flowering and delays
vegetative phase change (Wu amp Poethig 2006 Gandikota et al 2007 Wang et al
2008) It has been shown that miR156 negatively regulates a group of genes encoding
SQUAMOSA PROMOTER-BINDING PROTEIN-LIKE (Lewis et al 2007 de Alba et
al 2013 Rogers amp Chen 2013) transcription factors such as SPL3 SPL4 and SPL5
that promote flowering initiation In contrast transgenic plants over expressing
miR156-resistant SPL345 genes are early flowering Interestingly the endogenous
level of miR156 is temporally regulated In the early juvenile vegetative phase the
level of miR156 is very high but decreases rapidly before the onset of the adult juvenile
phase (Wu amp Poethig 2006)
The Arabidopsis early activation tagged (eat-D) mutant exhibits early flowering
with disrupted floral structure The mutant phenotypes are caused by over expression of
the miR172a-2gene (Aukerman amp Sakai 2003) MiR172 regulates a small group of
genes encoding APETALLA2 (AP2)-like transcription factors such as TARGET OF
EAT1 (TOE1) TOE2 SCHLAFMUTZE (SMZ) and SCHNARCHZAPFEN(SNZ)
TOE1 TOE2 SMZ and SNZ have been shown to participate in their productive phase
change by repressing a floral integrator FLOWERING LOCUS T (FT)(Jung et al
2007) Consistent with this toe1-2D 35STOE2 35SSMZ and 35SSNZ plants
exhibit late flowering (Jung et al 2007) MiR172 is also regulated temporally and
highly expressed in the adult vegetative phase both in Arabidopsis and maize (Chuck et
al 2007) Because the temporal expression patterns of miR156 and miR172 are
complementary to each other it has been suggested that the two miRNAs interact with
each other in regulating phase change (Chuck et al 2009 de Alba et al 2013) In
support of this view it has been shown that miR172 is down-regulated in the
Chapter 1 Introduction and Review of Literature
43
Corngrass1(Cg1) mutant in which miR156 is overproduced (Chuck et al 2007 de
Alba et al 2013) However there is no direct evidence for the direct linkage between
miR156 and miR172 It is also envisioned that miR156 and miR172 would not be
directly related For example miR156 regulates plastochron index that is the inverse of
leaf initiation rate and thus frequently used to analyze temporal patterns of plant
development In contrast miR172 does not affect plastochron (Wang et al 2008) The
down-regulation of miR172 in the maize miR156-over producing mutants would be
due to the prolonged juvenile vegetative phase in the mutants
Another miRNA that regulates flowering initiation is miR159 It regulates
MYB33 and MYB65 which are closely related to the barley GAMY B transcription
factor that responds to gibberellic acid (GA) The level of miR159 is elevated in GA-
treated seedlings but reduced in the GA biosynthetic ga1 mutant Transgenic plants
over producing miR159 exhibit delayed floral induction under short days (Achard et
al 2004) It is been suggested that MYB33 mediates GA signal to regulate LEAFY
(LFY) These observations indicate that miR159 plays an important role in the GA
signaling pathways that regulate proper floral induction under short days (Achard et al
2004)
11362 Floral development
Arabidopsis flowers consist of four distinct organs They arise in a characteristic
pattern within concentric rings called whorls from outside to inside four sepals four
petals six stamens and the central pistil consisting of two carpels Identities of the
floral organs are determined by three classes of regulatory genes classA classB and
class C genes (Krizek amp Fletcher 2005) The A and C class genes act antagonistically
to restrict their expression domains (Bowman et al 1991)
It is notable that miR172 also regulates floral architecture in addition to its role
in the timing of flowering initiation MiR172 inhibits the translation of the AP2 mRNA
(Chen 2004) Transgenic plants overproducing miR172 not only exhibit early
flowering but also show abnormal floral development which is quite similar to those
of the class A gene mutants such as the ap2 mutants (Aukerman amp Sakai 2003 de
Alba et al 2013) When the activity of the class A genes is inactivated that of the class
C genes such as AGAMOUS (AG) is elevated As a result the miR172- overproducing
transgenic plants exhibit no petals along with homeotic transformation of the sepals to
Chapter 1 Introduction and Review of Literature
44
the carpels (Aukerman amp Sakai 2003 Chen 2004)
Interestingly miR166 also regulates floral architecture The role of miR165166
in the regulation of meristem activity is closely linked to floral meristem formation and
organization (Zhao et al 2007 Miyashima et al 2013) Floral structure is severely
disrupted in the miR165166-overproducing mutants such as men1 and jba-1D in
which miR166 is overproduced (Williams et al 2005b Jung amp Park 2007) The men1
and jba-1D mutants have smaller gynoecium and reduced carpel number In an extreme
case the jba-1D homozygous plants lack visible carpels (Williams et al 2005b)
Similar phenotypes have been observed in the miR165 overproducers (Zhao et al
2007) It has been suggested that the phenotypes are possibly caused by altered AG
expression in the floral meristem (Jung amp Park 2007 Turchi et al 2013)
Other miRNAs also affect floral development Overproduction of miRNAs
involved in auxin signaling also affects floral architecture Transgenic plants
overproducing miR167 have defects in the formation of ovule and anther with reduced
fertility (Ru et al 2006) It has been shown that miR167 targets ARF6 and ARF8 that
play a role in gynoecium and stamen development and in regulating fertility (Ru et al
2006 Wu et al 2006) These observations suggest that auxin signals are also closely
related to floral organogenesis In addition transgenic plants over-producing miR164
and the cuc1cuc2 double mutants show fused sepals and reduced petals (Laufs et al
2004 Turchi et al 2013) suggesting that miR164 is also related to floral meristem
activity and boundary specification of the meristematic region
11363 Shoot apical meristem development
Shoot apical meristem comprises self-maintaining totipotent cells The shoot apical
meristem activity affects diverse aspects of plant developmental processes including
leaf development vascular development and lateral organ polarity (Bowman et al
2002) Because miR165166 plays a primary role in meristem formation
overproduction of miR165166 and multiple knockout mutants of its target genes
exhibit pleiotropic pheonotypes (McConnell et al 2001 Emery et al 2003 Kim et al
2005 Williams et al 2005b)
The targets of miR165166 include genes encoding the homeo domain-leucine
zipper (HD-ZIP) class III transcription factors such as PHABULOSA(PHB)
PHAVOLUTA (PHV) REVOLUTA(REV) ATHB15CORONA (CNA) and ATHB8
Chapter 1 Introduction and Review of Literature
45
PHB PHV and ATHB15 have overlapping functions in controlling meristem
development (Turchi et al 2013) Indeed the phb phv athb15 triple mutants have an
enlarged shoot apical meristem Consistent with this the miR166- overproducing men1
and jba-1D mutants exhibit enlarged shoot apical meristems (Kim et al 2005
Williams et al 2005b Turchi et al 2013) In the mutants the shoot apical meristems
are multiplied and large
The development of shoot apical meristems is also regulated by the WUSCHEL
(WUS)ndashCLAVATA (CLV) signaling pathway WUS positively regulates cell
proliferation In contrast CLV3 which is expressed in stem cells limits the WUS
expression and thus inhibits cell proliferation in the SAM (Sharma et al 2003)
Consistent with this notion WUS is expressed to a higher level in jba-1D
explaining the ectopic enlargement of the SAM in the mutant (Williams et al 2005)
While WUS is closely related to miR165166 in the shoot apical meristem
development the underlying molecular mechanisms are currently unclear It has been
observed that the miR166-over producing men1 plants have an arrested meristem
development (Jung amp Park 2007) In addition the heterozygous men1+ and the
homozygous men1 plants show different expression patterns of WUS and CLV3 It
appears that miR166 regulates WUS indirectly and influences the expressional balance
between WUS and CLV3 through a negative feedback loop (Jung amp Park 2007 de
Alba et al 2013)
The phv phb athb15 triple mutant has an enlarged shoot apical meristems the
rev phb phv mutant does not have functional shoot apical meristems (Prigge et al
2005) This distinction may be explained by the fact that while miR166 targets
primarily PHB PHV and ATHB15 miR165 targets all the five HD-ZIP III genes (Zhou
et al 2007) Consistent with this transgenic plants overproducing miR166 are distinct
from those overproducing miR165 The miR165-overproducing transgenic plants lack
functional shoot apical meristem and a pin-like structure is formed at the apical region
of the mutant These phenotypes are quite similar to rev phb phv triple mutant (Zhou et
al 2007 Liu et al 2013) The phenotypic distinction may because by the difference
of the cleavage efficiencies of the target mRNAs In accordance with this notion a few
35S miR166a transgenic lines exhibit no shoot apical meristems The men1
homozygous plants also show arrested meristem development (Jung amp Park 2007)
Chapter 1 Introduction and Review of Literature
46
MiR164 cleaves the CUC1 and CUC2 mRNAs as well as the NAC1 mRNA
CUC1 and CUC2 determines the organ boundary in the meristem Phenotypes of
transgenic plants that overproduce miR164 are similar to those of the cuc1cuc2 double
mutant that exhibits embryonic developmental defects such as cup-shaped cotyledons
and fused cotyledons (Laufs et al 2004) When a miR164-resistant CUC2 is
expressed the boundary domain is enlarged CUC also plays a role in embryonic shoot
meristem formation by regulating the SHOOT MERISTEMLESS gene It was therefore
concluded that miR164-mediated cleavage of CUC1 and CUC2 mRNAs determines the
sizes of the boundary domains in the shoot apical meristem and regulates separation of
the organs by restricting cell proliferation (Laufs et al 2004 de Alba et al 2013)
11364 Leaf development
The role of miR319 has been extensively studied in leaf development A dominant
jaw-D mutant overproducing miR319 shows uneven curved leaves with enlarged size
and five TCP genes which are closely related to the CINCINNATA (CIN) gene in
Antirrhinum majus are down regulated in the mutant suggesting that miR319 mediated
regulation of the TCP transcription factor genes are important for the final step of
leaf shaping (Palatnik et al 2003 Naz et al 2013) In addition to leaf shape and size
leaf polarity is also specified by miRNA MiR166-mediated cleavage of the HD-ZIPIII
mRNAs is a well to establish leaf polarity This discovery originates from the gain-of-
function mutants such as phb-1d and phv-1d wherein point mutations in the
complementary sites to miRNA helps the mRNA to evade or resist from miRNA-
mediated cleavage (McConnell et al 2001 Emery et al 2003 Naz et al 2013)
Plant leaves have a dorso-ventral polarity consisting of the adaxial (upper
surface) and abaxial (lower surface) sides The adaxial side is closer to the shoot
apical meristem Adaxial identity is specified by functionally redundant class III HD-
ZIP family genes such as PHB PHV and REV Ectopic expression of each genes
result in adaxialized leaves Dominant phb-1d and phv-1d mutants exhibit
transformation of the abaxial to adaxial fate and have rod-shaped leaves In contrast
miR165166-overproducing plants show pin-like cotyledons radicalized leaves and
abaxialized leaves (Chitwood et al 2007 Pattanayak et al 2013)
Leaf asymmetry is determined by miR166-mediated restriction of the
expression domains of the HD-ZIPIII genes The HD-ZIPIII genes are expressed in the
Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
21
interaction with different rust races pathogen (van der Vossen 2001) Currently C
canephora provides the main source of resistance to pests and diseases including CLR
(H vastatrix) and CBD (C kahawae) and therefore used in breeding programs Other
diploid species like C liberica and C racemosa are being used as a source of resistance
to coffee leaf rust and coffee leaf miner respectively (Guerreiro et al 1999
Sreenivasan et al 1989) The development of coffee varieties resistant to major fungal
diseases such as CLR and CBD using transgenic technology will benefit the coffee
industry immensely During the last 15 years significant progress has been made in the
area of host-pathogen interactions (Staskawicz et al 1995 Feys et al 2000
Shivaprasad et al 2012) and many resistance genes involved in recognizing invading
pathogens have been identified and cloned (Takken et al 2000) A number of
signalling pathways induced because of pathogen infection have been dissected (Dong
et al 1998 Navarro et al 2006) Many antifungal compounds synthesized by plants to
combat fungal infection have been identified (Does et al 1998) Understanding the
specific induction of targeted pathways and identification of specific pathways
responsible for particular fungal resistance is important in order to employ this strategy
in transgenic technology The recent investigation of gene expression during coffee leaf
rust infection could provide an insight into the defence pathways operating in coffee
(Fernandez et al 2004 Nardi et al 2006) Efforts have been made to identify and
clone resistance genes from coffee for achieving durable resistance The genetic and
physical map of two resistance genes SH3gene conferring resistance to rust (Prakash et
al 2004) and the Ck1 gene conferring resistance to C kahawae CBD (Gichuru et al
2006) have been established These genes could be used for molecular marker assisted
breeding programs (Mishra et al 2012)
1104 Production of low-caffeine coffee
A widespread belief that the ingestion of caffeine can have adverse effects on
health resulted in the increased demand for decaffeinated coffee (Ashihara amp Crozier
2001) Several method were used to obtain decaffeinated plants by conventional
breeding techniques using low yielding varieties and mutants that accumulated
relatively low amounts of caffeine when compared to the commercially available ones
Low-caffeine and decaffeinated coffee represent around 10 of the coffee sales
around the world (Ribas et al 2006b) The industrial process for coffee decaffeination
can be expensive and affects the original flavour and aroma in coffee (David 2002)
Chapter 1 Introduction and Review of Literature
22
Transgenic coffee plants with suppressed caffeine synthesis using RNA interference
(RNAi) technology have been obtained (Ogita et al 2003 Ogita et al 2004) Specific
sequences in the 3prime untranslated region of the theobromine synthase gene (CaMXMT1)
were selected for construction of RNAi short and long fragments The caffeine and
theobromine content of the transgenic plants were reduced by ~ 70 when compared to
the untransformed plants In C canephora RNAi technology has also been employed
to silence the N-methyl transferase gene involved in caffeine biosynthesis (Vinod et al
2007) Promoter of an N-methyltransferase (NMT) gene involved in caffeine
biosynthesis was cloned (Satyanarayana et al 2005 Satyanarayana 2006) which will
be very useful for studying the regulation of caffeine biosynthesis
1105 Improvement in cup quality
Improvement in coffee cup quality requires elaborate knowledge of the chemical
constituents as well as the metabolic pathways involved in the elaboration of quality
The constituents of coffee beans include minerals proteins carbohydrates caffeine
chlorogenic acids (CGA) glycosides lipids and many volatile compounds that give
flavour to coffee by roasting Among these the role of three major constituents
sucrose CGA and trigonelline have been studied in coffee The sucrose content of
coffee bean is associated with coffee flavour the higher the sucrose content in green
beans the more intense will be the cup flavour (Clifford et al 1985 de Maria et al
1996) The sucrose content of C arabica (82-83) is higher than C canephora (33ndash
40) The sucrose amino acids ratio in green beans determines the profile of volatile
compounds Manipulating sucrose content in coffee bean is therefore important in
improving cup quality The sucrose synthase gene (CcSUS2) from C canephora has
been cloned and sequenced (Leroy et al 2005) This provides an opportunity to
manipulate the sucrose content in coffee Chlorogenic acids are products of
phenylpropanoid metabolism Chlorogenic acids are a group of hydroxycinnamoyl
quinic acids (HQA) formed by esterification between caffeic acids coumaric acids and
quinic acids (Clifford et al 1999) They are present in relatively large quantities in the
coffee bean and are the precursor of phenolic compounds in roasted beans C
canephora beans contain higher CGA (10) compared to C arabica beans (6-7)
CGA are known to have antioxidant properties as well as being associated with disease
resistance (Campa et al 2003) Genetic manipulations of genes involved in CGA
synthesis can serve either of these purpose by up- or down regulating the pathway
Chapter 1 Introduction and Review of Literature
23
Phenylalanine ammonia-lyase (PAL) catalyzes the first step of the phenylpropanoid
pathway leading to the synthesis of a wide range of chemical compounds including
flavonoids coumarins hydroxycinnamoyl esters and lignins (Hahlbrock amp Scheel
1989) Recently the full length cDNA and corresponding genomic sequences of PAL
from C canephora was isolated characterized and functionally validated (Mahesh et
al 2006 Parvatam et al 2006) This has opened up new possibilities for manipulating
the level of the PAL enzyme in coffee which in turn can be useful for improvement of
cup quality and manipulating antioxidant properties in coffee
1106 Fruit Ripening
Uniformity during fruit ripening is decisively related to cup quality in coffee and
consequently to the value of the product Fruits at the ideal ripening stage produce the
best organoleptic characteristics for coffee The presence of over ripened or green fruits
changes the acidity the bitterness and consequently the cup quality In order to
maximize uniform ripening of coffee fruits it is essential to control the action of genes
involved in the last step of maturation process Ethylene is known to trigger ripening
and increasing ethylene biosynthesis is associated with various stages of ripening
process (Protasio et al 2005) To control coffee fruit maturation two of the major
genes involved in ethylene biosynthesis namely ACC synthase and ACC oxidase
have been cloned (Protasio et al 2005 Neupane et al 1999) Introduction of the ACC
oxidase gene in antisense orientation have been achieved in both C arabica and C
canephora The effect of the transgene on ethylene production and fruit maturation has
yet to be reported The inhibition of genes downstream to the initial ethylene burst is
also an option to control coffee fruit maturation (Ribas et al 2006)
111 RNA interferenceRNAi
The regulation of gene expression at post transcriptional level has recently attracted
much attention because of the discovery of the phenomenon called RNA interference
(RNAi) RNAi based silencing techniques are more efficient than antisense mediated
gene silencing (Wesley et al 2001) The effect was first observed in plants wherein
silencing was observed when some copies of a transgene was in the forward orientation
with respect to the promoter and some in the reverse orientation This resulted in the
formation of double stranded RNA in the system The phenomenon in which
experimentally introduced double stranded RNA (dsRNA) leads to the loss of
expression of the corresponding cellular gene by sequence specific RNA degradation
Chapter 1 Introduction and Review of Literature
24
was termed as RNA interference or RNAi The protective effect of RNA silencing in
reducing virus infectivity supports the view that PTGS has evolved as a mechanism to
defend plants against virus infection and also to moderate the possible deleterious
genome-restructuring (insertional) activity of virus-like mobile genetic elements eg
retrotransposons A growing body of biochemical and genetic data has further
demonstrated that plants animals and yeasts share related mechanisms of specific
degradation of RNAs in which double-stranded forms of RNA are initiator molecules
(Brenstein et al 2001)
The key insight into the process of PGTS was provided by the experiments
conducted by Hamilton amp Baulcombe (1999) who identified the product of RNA
degradation as small RNA (siRNA) species of ~25nt of both sense and antisense
polarity SiRNA are formed and accumulate as double stranded RNA molecules of
defined chemical structures Both genetic and biochemical approaches were undertaken
to understand the basis of silencing Genetic screens were carried out in fungus
Neurospora carassa the alga Chalmydomonas reinhardtii and plant Arabidopsis
thaliana to search for detective mutants in PTGS
A combination of results obtained from several in vivo and in vitro experiments
have gelled into a two step mechanistic model for RNAiPTGS The first step referred
to as the RNAi initiating step involves binding of the RNA nucleases to a larges
dsRNA and its cleavage into discrete ~21-25nt RNA fragments This is ATP-
dependent reaction which involved RNase III - type endonucleases which make
staggered cuts in both strands of the dsRNA leaving 3prime overhang of 2 nucleotides with
5prime- phosphate 3prime-hydroxyl and no modification in the sugar phosphate backbone
(Hamilton amp Baulcombe 1999 Elbashir et al 2001) SiRNAs are the direct products
of dsRNA cleavage by the multi-domain RNase III enzyme DICER (Figure 16)
(Brenstein et al 2001 Karthikeyan et l 2013)
In the second step or commonly known as the effector step the double stranded
siRNAs produced in the first step bind to an RNAi specific protein complex to form a
RISC The complex undergoes activation in the presence of ATP so that the antisense
component of the unwound siRNA becomes exposed and allows the RISC to perform
the downstream RNAi reaction
Chapter 1 Introduction and Review of Literature
25
Several enzymes are involved in RNAi based on their role genes that encode for them
are classified into RNAi initiators RNAi effectors RNA dependent RNA polymerases
and silencing genes Two C elegans genes rdeI and rde4 (rde stands for lsquo RNAi
deficientrsquo) are believed to be involved in the initiation step of RNAi The C elegans
rde1 gene is a member of a large family of genes and is homologous to the Neurospora
qde2 (qde stands for ldquoquelling deficientrdquo) and Arabidopsis AGO1 (AGO stands for
agronaute AGO1 was previously identified to be involved in Arabidopsis
development) Although the functions of these genes are not clear a mammalian
member of RDE1 family has been identified as a translation initiation factor (Thakur
2005) Important genes for the effector step of PTGS in C elegans are rde2 and mut7
genes These genes were identified from heterozygous mutant worms that were unable
to transmit RNAi to their homozygous offsprings Worms with mutated rde2 or mut 7
genes show defective RNAi mut-7 gene encodes a protein with homology to the
nuclease domains of RNAase D and a protein implicated in Werner syndrome (a rapid
ageing disease in humans) (Grishok et al 2001 Kamath-Loeb et al 2012s)
Neurospora qde-1 Arabidopsis SDE-1SGS-2 and C elegans ego-1 appear to encode
RNA dependent RNA polymerase (RdRPs) It assumed that this is a proof that an RdRp
activity is required for RNAi Certainly the existence of an RdRp might explain the
remarkable efficiency of dsRNA induced silencing if it amplifies either the dsRNA
prior to cleavage or the siRNAs directly (Muers 2013) In C elegans ego-1 mutants
(ego stands for lsquoenhancer of glp-1) RNAi functions normally in somatic cells but is
defective in germline cells where ego-1 is primarily expressed In Arabidopsis SDE-
1SGS-2 mutants (SGS stands for suppressor of gene silencing) siRNA are produced
when dsRNA is introduced via an endogenously replicating RNA virus but not
introduced by a transgene It has been proposed that perhaps the viral RdRP is
substituting for the Arabidopsis enzyme in these mutants Random degradative PCR
model suggests that an RdRP uses the guide strand of an siRNA as a primer for the
target mRNA generating a dsRNA substrate for Dicer and thus more siRNAs
Several genes controlling RNA silencing in plants have been identified through
genetic screens of Arabidopsis mutants impaired in transgene induced RNA silencing
They encode a putative RNA-dependent RNA polymerase (SGS2SDE1) a coiled
protein (SGS3) a protein containing PAZ and Piwi domains (AGO1) and an RNA
helicase (SDE3) The putative SGS2SDE1 of Neurospora is related to QDE-1 and
Chapter 1 Introduction and Review of Literature
26
EGO-1 of C elegans whereas PAZPiwi protein AGO is related to QDE-2 of
Neurospora RDE-1of C elegans RNA helicase SDE3 is related to SMG-2 of C
elegans and Mut-6 of Chlamydomonas MUT-7 gene of C elegans encodes a protein
similar to Rnase D whereas the Drosophila DICER gene encode a protein similar to
RNase III An Arabidopsis ortholog of DICER gene has been identified called CAF
SIN1 SUS1(Thakur 2005 Poethig 2009)
Since the RNAi machinery is present constitutively within the eukaryotic cell it
is important to explore the metabolic advantages that are accorded by the RNAi related
proteins during the intrinsic normal growth of cells and development of organisms The
natural RNAi machinery not only keeps the mobile transposable elements from
disrupting the integrity of the genomes as suggested by the analysis of model organisms
like A thaliana C elegans D melanogaster (Tabara et al 1999 Wu-Scharf et al
2000 Aravin et al 2001 Hamilton et al 2002 Lisch 2009) but also participates in
development of organisms Genetic defects in Celegans RNAi genes ego1 and dicer
cause known specific developmental errors (Grishok et al 2000 Grishok et al 2001
Knight amp Bass 2001 Hall et al 2013) Similarly the Argonaute family of genes of A
thaliana (especially the ZWILLE proteins) is also responsible for plant architecture and
meristem development (Carmell et al 2002 Hamant and Pautot 2010) and the Dicer
homologue of A thaliana CAF1 is required for embryo development (Golden et al
2002 Nakano et al 2011) Thus genetic evidence illustrates the role of the RNAi
machinery as a controller of development related genes The mechanistic details of
these developmental processes are beginning to emerge
Chapter 1 Introduction and Review of Literature
27
Figure 16 Mechanism of gene silencing induced double stranded RNA The dicer enzyme to
produce siRNAs cleaves the RNA The siRNA generated bind to the nuclease complex
RISC The antisense component in the siRNA in the RISC guides the complex towards
the cognate mRNA resulting in endonucleolytic cleavage of the mRNA
Chapter 1 Introduction and Review of Literature
28
112 MicroRNA or MiRNA
In 1991 Ambros and co-workers first isolated a lin-4 mutant of Celegans which was
arrested at the first larval stage (Lee et al 1993) Later on the let-7 mutation was
isolated in the same system which was responsible for development through the fourth
larval stage Both lin-4 and let-7 encode short 22-nucleotide mature RNAs and were
called short temporal RNA because they control the temporal development program of
C elegans The mature lin-4 RNA defines (negatively regulates) the mRNA expression
of the lin-14 and lin-28 hetrochronic genes with the antisense mediated repression
mechanism of translation initiation and thus specifies the fate of cells during the first
three larval stages Recent studies have revealed that the short temporal RNAs are
actually members of a group of tiny RNAs (21to 28 nucleotides) called the micro-
RNAs (miRNA) (Bartel 2009) isolated members of which could easily run to a few
thousand which includes both those were identified computationally and by deep
sequencing Some of the components of the RNAi machinery have also been clearly
established as the effector proteins for the maturation of miRNAs
113 MicroRNAs in plants
1131 Discovery
MicroRNAs have been discovered by three basic approaches Direct cloning forward
genetic direct cloning and bioinformatic prediction followed by experimental
validation The most direct method of miRNA discovery is to isolate and clone small
RNAs from biological samples and several groups have used this approach to identify
plant miRNAs (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002 Xie et al 2003 Sunkar et al 2005 Williams et al 2013) The cloning methods
adapted were similar to those used to identify large numbers of animal miRNAs
(Lagos-Quintana et al 2001 Lau et al 2001 Aravin amp Tuschl 2005) which involve
isolating small RNAs followed by oligonucleotide adapter ligation reverse
transcription amplification and sequencing Some protocols incorporate methods to
enrich for Dicer cleavage products (ie molecules with 5prime-phosphates and 3prime-
hydroxyls) and to concatemerize the short cDNAs so that several may be identified in a
single sequencing read (Lau et al 2001 Llave et al 2002a Jagadeeswaran amp Sunkar
2013)The initial cloning experiments in Arabidopsis identified 19 miRNAs belonging
to 15 families (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002) although hundreds of other small RNAs such as siRNAs and RNA degradation
Chapter 1 Introduction and Review of Literature
29
fragments were also cloned through the direct cloning methods To select miRNAs
from the pool of small RNAs secondary structures of the RNA molecules were
examined in compliance with the acknowledged structural features of miRNAs
(Ambros et al 2003) The secondary structures were validated using several ad-hoc
software tools such as the Mfold or RNAfold programs (Reeder et al 2006) Finally
the existence of mature miRNAs was determined by RNA gel blot hybridization These
direct cloning methods were successful in isolating many miRNAs in Arabidopsis
(Reinhart et al 2002 Sunkar amp Zhu 2004) Rice (Sunkar et al 2005) Poplar (Lu et
al 2005) moss (Arazi et al 2005) and Tobacco (Billoud et al 2005)
Although miRNAs were first discovered through forward genetic screens in
worms (Lee et al 1993 Reinhart et al 2000)only few A thaliana miRNA gene
families have been discovered by this method (Chen 2008) in plants MiRNA
involvement in plant mutant phenotypes were not properly inferred till the cloning
experiments established that plant genomes contain numerous miRNAs (Park et al
2002 Reinhart et al 2002 Rhoades et al 2002) MiRNA loss-of-function allele has
been identified by forward genetic screens early extra petala1 is caused by a
transposon insertion ~160bp upstream of the predicted miR164c stem loop resulting in
flowers with extra petals (Baker et al 2005 Irish 2008) The fact that loss-of-function
miRNA mutants have been rarely discovered using forward genetics reflects small
target size for mutagenesis coupled with redundancy in nearly all evolutionarily
conserved plant miRNAs which are encoded by gene families (Table16) Family
members are likely to have overlapping functions buffering against loss at any single
miRNA locus Over expression screens can circumvent redundancy limitations At least
four plant miRNAs miR319 (also known as miR-JAW) miR172 (also known as EAT)
and miR166 were isolated in over expression screens for dominant mutants with
developmental abnormalities (Aukerman amp Sakai 2003 Palatnik et al 2003 Kim et
al 2005 Williams et al 2005b Schommer et al 2012) Mutations in miRNA target
sites which can prevent the entire family of miRNAs from repressing a target gene can
also circumvent redundant functions of miRNA family members
The dominant mutations in the HD-ZIP genes PHB PHV and REVOLUTA
(REV) in Arabidopsis and ROLLEDLEAF1 (RLD1) in Maize result in adaxialization of
leaves andor Vasculature (McConnell amp Barton 1998 McConnell et al 2001 Nelson
et al 2002 Zhong amp Ye 2004 Allen amp Millar 2012) and are all caused by mutations
Chapter 1 Introduction and Review of Literature
30
in miR166 complementary sites (Rhoades et al 2002 Emery et al 2003 Jones-
Rhoades amp Bartel 2004 Mallory et al 2004 Zhong amp Ye 2004)
In both plants and animals cloning was the initial means of large-scale
miRNA discovery (Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Reinhart et al 2002 Mendes et al 2012) However cloning is biased toward
RNAs that are expressed highly and broadly MiRNAs expressed at low levels or
only in specific cell types or in response to certain environmental stimuli were more
difficult to clone Sequence based biases in cloning procedures might also cause
certain miRNAs to be missed Because of these limitations bioinformatics approaches
to identify miRNAs have provided a useful complement to cloning
A straightforward use of bioinformatics has been carried out to find homologs
of known miRNAs both within the same genome and in the genomes of other species
(Pasquinelli et al 2000 Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Mendes et al 2009) A more difficult challenge is to identify miRNAs
unrelated to previously known miRNAs This was first accomplished for
vertebrate nematode and fly miRNAs using algorithms that search for conservation
of sequence and secondary structure (ie miRNA stem-loop precursors) between
species searching for patterns that are characteristic of miRNAs (Lai et al 2003 Lim
et al 2003 Lim et al 2005)
Most of the methods identified numerous miRNAs but are necessarily more
relaxed in terms of allowed structures Some approaches take advantage of the high
complementarity of plant miRNAs to target messages implementing the requirement
that the candidate has conserved complementarity to mRNAs (Jones-Rhoades amp Bartel
2004 Adai et al 2005) This additional filter has been useful for distinguishing
authentic plant miRNAs from false positives which later extended to mammalian
miRNA gene prediction (Xie et al 2005 Mendes et al 2012)
Another criteria used by most algorithms in the filters is evolutionary
conservation In plants mature miRNA sequences are conserved to a higher degree
than their precursor sequences For example Arabidopsis and rice diverged more than
130 million years ago (Friis et al 2004) but some miRNAs are highly conserved
in these plant species (Reinhart et al 2002) Furthermore various parameters for the
secondary structures such as free energy the number of paired residues within a
Chapter 1 Introduction and Review of Literature
31
miRNA or the number and size of bulges are also used to validate the predictions
(Jones-Rhoades amp Bartel 2004 Wang et al 2004 Adai et al 2005 Mendes et al
2012)
1132 Conserved microRNAs in Plants
Till date cloning genetics and bioinformatics screens have resulted in annotation of
approximately 184 potential Arabidopsis miRNAs (miRBase release 13) These
miRNAs seem to arise through gene duplication and subsequent diversification and
represent approximately 100 families (Maher et al 2006 Axtell 2013) Twenty one
families represented by 92 genes are clearly conserved in species beyond Arabidopsis
(Table 15) The presence of these miRNA families in other sequenced plant genomes
indicates that different plant species have an evolutionarily fluid set of miRNAs
(Willmann amp Poethig 2007) Recent studies on mosses one of the most ancient land
plants have shown that at least 14 miRNA families are conserved in eudicots and
mosses and therefore it appears that plant miRNAs share a common ancient origin
(Arazi et al 2005 Axtell amp Bartel 2005 Talmor-Neiman et al 2006 Zhang et al
2006 Axtell 2013) The number of members per family in a genome ranges from 1-32
with the exception of miR-430 which is represented by a cluster of ~80 loci in zebra
fish (Giraldez et al 2005)
In plants the number of members in each family correlates among examined
species certain families contain numerous members in all three species (eg miR156
miR166 miR169) whereas others consistently contain fewer genes (eg miR162
mir168 miR394) (Table 16) Although it is still unclear why certain miRNA are
encoded by more than one gene in plants (eg 12 genes encoding miR156) this
correlation might suggest functional significance of various miRNA family sizes Most
annotated plant miRNAs are conserved throughout flowering plants while a few
miRNAs are found only in a single sequenced genome and thus they could be
evolutionarily lsquoyoungrsquo miRNAs For example Arabidopsis has several miRNA
families that are not found in poplar or rice suggesting that miRNAs could be
generated continuously during evolution (Allen et al 2004a Rajagopalan et al
2006 Fahlgren et al 2007 Axtell 2012 de Alba et al 2013) Consistent with the
notion that non conserved miRNAs are of a more recent evolutionary origin these
miRNAs are mainly encoded by a single locus in the genome Within each family the
mature miRNA is always located in the same arm
Chapter 1 Introduction and Review of Literature
32
Table 16 MicroRNA families conserved in plants
miRNA
family
Arabidopsis Oryza Populus
miR156 12 12 11 miR159319 6 8 15 miR160 3 6 8 miR162 2 2 3 miR164 3 5 6 miR166 9 12 17 miR167 4 9 8 miR168 2 2 2 miR169 14 17 32 miR171 4 7 10 miR172 5 3 9 miR390 3 1 4 miR393 2 2 4 miR394 2 1 2 miR395 6 19 10 miR396 2 5 7 miR397 2 2 3 miR398 3 2 3 miR399 6 11 12 miR408 1 1 1 miR403 1 0 2 miR437 0 1 0 miR444 0 1 0 miR445 0 9 0 Total 92 127 169
All known miRNA families that are conserved between more than one plant species are listed
together with the number of genes identified in the sequenced genomes Rice miRNA families that have orthologs in maize but do not appear to have orthologs in the eudicots (Arabidopsis and Populus) are marked with an asterisk The following families contain miRNA genes annotated with
more than one number miR156 (miR156 and miR157) miR159319 (miR159 and miR319) miR166 (miR165 and miR166) miR171 (miR170 and miR171) and miR390 (miR390 and miR391)
of the stem loop (5prime- or 3prime) as it would be expected if the genes carry common ancestry
(Figure 17) The sequence of the mature miRNA and to some extent the segment on
the opposite arm of the hairpin to which it pairs is highly conserved between the
members of the same family (both within and between the species) while the sequence
secondary structure and length of the intervening ldquolooprdquo region can be highly divergent
within the same family
The patterns of paring and non pairing nucleotides are often conserved between
homologous miRNA stem loops from different species (Figure 17) The significance of
conserved mismatches is opined to guide DCL1 to cleave at the appropriate positions
along the stem loop
Chapter 1 Introduction and Review of Literature
33
Figure 17 Representative stem loop of miR164 Segments corresponding to the mature miRNAs
are shown in red (Adapted from Rajagopalan et al 2006)
Although many miRNAs are highly conserved in plants the degree of conservation in
most cases is quite low between plants and animals one exception is miR854 A recent
study has revealed that miR854 is conserved between Arabidopsis and animals and that
the Arabidopsis miR854 differs from the human miR854 only by a single nucleotide
(Arteaga-Vazquez et al 2006 Axtell 2012 de Alba et al 2013) The Arabidopsis
miR854 has multiple binding sites in the 3prime-untranslated region (UTR) of
OLIGOURIDYLATE-binding-PROTEIN mRNA 1bA (UBP1b) and represses the
translation of the UBP1b mRNA The animal miR854 is also complementary to a site in
the 3prime-UTR of the UBP1b homolog in animals However it is currently unclear how the
animal miR854 regulates its target mRNA Another distinction between plant and
animal miRNAs is the genomic organization of the miRNA genes While plant miRNA
genes are located predominantly in the intergenic regions animal miRNA genes tend to
localize in the intragenic regions both in the introns or exons of protein coding genes
Moreover miRNA genes are often clustered within a single precursor transcript
whereas individual plant miRNAs are derived from individual precursor RNAs (Bartel
Chapter 1 Introduction and Review of Literature
34
2004 Bonnet et al 2004 Kim 2005 Vaucheret et al 2006 Marco et al 2013)
1133 Non-conserved miRNA and challenges in annotation
Most miRNAs are conserved throughout flowering plants (Table 15) but many more
were found only in a single sequenced genome thus considered to be of a more recent
evolutionary origin The extended homology between few non conserved miRNAs
precursors and their target genes provides strong evidence that these relatively ldquoYoungrdquo
miRNAs arose from the duplication of the target gene segments (Allen et al 2004)
Several non-conserved miRNAs including miR161 miR163 miR173 miR447
miR475 and miR476 are known to direct cleavage of target transcripts (Allen et al
2004a Adai et al 2005 Sunkar et al 2005 de Alba et al 2013) It is difficult to
confidently predict targets for many because it is not possible to use complementary
site as a filter against false-positive target predictions It is also difficult to
confidently annotate non-conserved miRNAs as miRNA rather than siRNAs The
established minimal standard for miRNA annotation is a small RNA with detectable
expression and the potential to form a stem-loop when joined to flanking genomic
sequence (Kasschau et al 2003) In practice these requirements are too loose to
definitively categorize many small RNAs cloned from plants Many plant siRNAs are
detectable on blots (Vazquez et al 2004b) and hundreds of thousands of non-
miRNA genomic sequences can be predicted to fold into secondary structures that
resemble structures of plant miRNA precursors (Jones-Rhoades amp Bartel 2004)
Therefore without conservation of both sequence and secondary structure it is
difficult to be confident that a given cloned RNA originated from a stem-loop (ie is a
miRNA) rather than from a double-stranded RNA (ie is a siRNA) In fact many of
the thousands o f small RNAs cloned from Arabidopsis (Gustafson et al 2005) would
probably meet the literal requirements for annotation as miRNAs A few of these
sequences might be miRNAs but others that meet the literal criteria probably are not
so The challenges of annotating non conserved small RNAs were evidenced by
three related small silencing RNAs that were originally annotated as miRNAs
that turned out to be trans-acting siRNAs (tasiRNAs)(Kanno et al2013)
Although most miRNAs that are broadly conserved among flowering plants are
now being identified and experimentally validated (Table 16)(Jones-Rhoades amp
Bartel 2004) the challenges and ambiguities for classifying non-conserved miRNAs
prevent meaningful estimates of the total number of miRNA genes in Arabidopsis and
Chapter 1 Introduction and Review of Literature
35
other plant genomes It is possible that miRNAs might have escaped detection
because they are non-conserved and expressed in specific tissues or conditions and can
only be identified by using high- throughput sequencing
1134 MicroRNA biogenesis
11341 Transcription of microRNA precursor
Plant miRNAs are primarily found in the genomic regions that are not associated with
the protein coding genes (Reinhart et al 2002) and it appears that most if not all plant
miRNAs are produced from their own transcriptional units Plant miRNA genes are
occasionally clustered near each other in the genome suggesting transcription of
multiple miRNAs from a single primary transcript [eg the miR395 cluster (Grishok
et al 2001 Jones-Rhoades amp Bartel 2004b)] but this polycistronic arrangement of
miRNA genes appears far less frequently in plants than in animals (Bartel 2004)
Northern blot EST and mapping evidence indicate that plant miRNA primary
transcripts (also known as pri-miRNAs) as in animals are longer than needed to
encompass the miRNA stem-loops (Palatnik et al 2003 Jones-Rhoades amp Bartel
2004b) At least some of these pri-miRNA transcripts appear to be spliced
polyadenylated (Kurihara amp Watanabe 2004) and capped (Xie Z et al 2005) Two
rice miRNAs are contained within transcripts that contain exon junctions within the
presumptive stem-loop precursor implying that in these cases splicing is a prerequisite
for Dicer recognition (Sunkar et al 2005) Plant pri-miRNAs are over 1kb in length
and they are usually preceded by typical TATA box motifs They can also undergo
canonical splicing polyadenylation and capping All these observations indicates RNA
polymerase II is probably responsible for transcribing most plant miRNAs (Xie Z et
al 2005) as appears to be the case for many animal miRNAs Relatively little is
known about the regulation of miRNA transcription in plants but there is no reason
to suspect that this regulation would differ from that of protein-coding transcripts as
the bioinformatics analysis of the sequence upstream of the transcriptional start
sites of the miRNA genes showed the presence of putative binding motifs of
number of known transcription factors (Megraw et al 2006 Sethupathy 2013)
11342 MicroRNA processing and export
In plants maturation of miRNA is a step wise process A miRNA gene is transcribed to
pri-miRNA which is much longer than the pre-miRNA possessing the characteristic
stem-loop structure Like transcription of most protein-coding genes the miRNA
Chapter 1 Introduction and Review of Literature
36
transcription is processed by RNA polymerase II (Pol II) enzymes and the pri-
miRNAs are 5prime-capped and 3prime-polyadenylated (Lee et al 2004 Xie Z et al 2005)
Mature miRNAs are produced from pri-miRNAs through at least two sequential
processing steps by RNase III-type endonucleases In animals pri-miRNAs are first
processed at the bottom portion of the stem by Drosha a nuclear-localized RNase III
enzyme to liberate pre-miRNAs The pre-miRNAs are then exported to the cytoplasm
with the help of exportin-5The pre-miRNAs are further processed by Dicer another
RNaseIII enzyme to a duplex consisting of the miRNA and the antisense strands
(miRNA) Since plants do not have any Drosha homologs the two sequential
processing steps seem to be carried out by DCL1 a Dicer homolog in the nucleus
(Kurihara amp Watanabe 2004 de Alba et al 2013 Rogers amp Chen 2013)
The processing of pri-miRNAs to pre-miRNAs by DCL1 is assisted by two
other proteins HYPONASTY LEAVES1 (HYL1) and SERRATE (SE) HYL1 belongs to a
family of double-stranded RNA (dsRNA) binding protein and interacts with DCL1 in
vitro and in vivo (Lu amp Fedoroff 2000 Hiraguri et al 2005 de Alba et al 2013
Rogers amp Chen 2013) Loss-of-function mutations in the HYL1 gene result in
decreased accumulation of miRNAs and concomitant accumulation of pri-miRNAs
(Han et al 2004 Vazquez et al 2004a Kurihara et al 2006) SE a C2H2 zinc finger
protein interacts with DCL1 and HYL1 and plays a role in the processing of pri-
miRNAs to pre-miRNAs (Lobbes et al 2006 Yang L et al 2006) In addition the
nuclear cap-binding complex proteins CBP20 and CBP80 that are known as ABA
HYPER SENSITIVE 1(ABH1) have been shown to be required for proper miRNA
processing (Gregory et al 2008 Laubinger et al 2008) Recently it has been
demonstrated that DCL1 can release 21-nt RNAs from dsRNA as well as from
synthetic miR167 precursor RNAs in vitro However this cleavage is error prone and
inefficient in the absence of HYL1 and SE which accelerate the rate of DCL1-mediated
cleavage and promote accurate processing of miRNAs (Dong et al 2008 Rogers amp
Chen 2013)
11343 Methylation of the miRNAmiRNAduplex
After the miRNAmiRNAduplexes are released from the pre-miRNAs by the DCL1
activities the duplexes are methylated at the 2primeOH of the3prime-ends by HUA ENHANCER1
(HEN1) a small RNA methyltransferase (Yu et al 2005 Yang Z et al 2006 Rogers
amp Chen 2013 ) This step which is distinct from the RNase III-mediated processing
Chapter 1 Introduction and Review of Literature
37
step distinguishes the biogenesis of plant miRNAs from that of animal miRNAs
Mutations in the HEN1 gene were first isolated in a morphological screening for floral
development although the HEN1 mutants display pleiotropic phenotypes similar to the
developmental defects observed in the DCL1 mutants (Chen et al 2002 Park et al
2002 Rogers amp Chen 2013)
Figure 18 Biogenesis of microRNA
A Biogenesis of plant MicroRNA B Biogenesis of metazoananimal MicroRNA
The miRNA (red) is incorporated into RISC and the miRNA(blue) in degraded
This enzyme has two dsRNA-binding domains and nuclear localization signal It
is also conserved in fungi and is required for the processing of siRNAs as well as of
miRNAs (Park et al 2002 Boutet et al 2003 Xie et al 2003) Although the HEN1
protein is localized to the nucleus its methylation activity in the cytoplasm cannot be
ignored (Xie et al 2004 Rogers amp Chen 2013)
Chapter 1 Introduction and Review of Literature
38
The methylation of the miRNAmiRNA duplex is required to protect miRNAs
against the 3prime-end uridylation activity and subsequent degradation (Li et al 2005) In
the hen1 mutants accumulation of miRNAs is reduced to a lower level Also the
miRNAs appear to be heterogeneous in their sizes such that a ladder of bands rather
than a single species is observed for most miRNAs in the mutants (Li et al 2005)
MiRNA cloning and sequencing analyses revealed the presence of a number of U
residues at the 3prime-end of miRNAs in the hen1 mutants Moreover these nucleotides do
not correspond to the miRNAs in the pre-miRNAs indicating that these nucleotides are
added after the processing of the miRNAs from pre-miRNAs (Li et al 2005)
Uridylation of RNAs has also been proven to be correlated with RNA decay (Shen amp
Goodman 2004 Mullen amp Marzluff 2008) suggesting that uridylation attracts an
exonuclease which degrades miRNAs in plants
11344 MiRNA incorporation in to the RISC and its nuclear export
The methylated miRNAmiRNA duplexes undergo RISC assembly In this process
the miRNA of the duplexes is selectively incorporated into the RISC and the miRNA
appears to be removed and subsequently degraded (Hammond et al 2000 Hutvagner
amp Zamore 2002 Schwarz et al 2003 de Alba et al 2013 Rogers amp Chen 2013)
The ARGONAUTE 1(AGO1) proteins are core components of the RISC complex
They contain two structural domains the N-terminal PAZ domain that binds to the 3prime-
end of single-stranded RNAs and the C-terminal piwi domains that has a structure
similar to that of RNase H (Cerutti et al 2000 Parker et al 2004) Several AGO
proteins possess endonucleolytic activities within the PIWI domain and cleave target
mRNAs in the middle of the site complementary to miRNA (Liu et al 2004 Meister et
al 2004 Miyoshi et al 2005) Mutations in the AGO1 gene cause miRNA target
genes to be ectopically expressed and the levels of some miRNAs are reduced in the
mutants (Kidner amp Martienssen 2004 Vaucheret et al 2004 Kidner amp Martienssen
2005 Ronemus et al 2006) Moreover the phenotype of the AGO1 hypomorphic
alleles display defects in lateral organ polarity and leaf and flower morphology as
observed in the dcl1 mutants and null AGO1 alleles are embryonically lethal
supporting the close relationship between the AGO proteins and miRNA processing
(Kidner amp Martienssen 2004 Vaucheret et al 2004 de Alba et al 2013 Rogers amp
Chen 2013)
The production of miRNA miRNA duplexes occur in the nucleus while
Chapter 1 Introduction and Review of Literature
39
miRNA-RISC complexes are present in the cytoplasm where target mRNAs are
located This discordance of functional sites requires an export step during miRNA
biogenesis HASTY the Arabidopsis homolog of exportin-5 is thought to be involved
in miRNA nuclear export (Bollman et al 2003 Park et al 2005)The hasty mutant
exhibits pleiotropic defects in plant development and reduced accumulation of most
miRNAs as in the DCL1 or AGO1 mutants Howeverit is unclear whether the HASTY
proteins export the miRNA miRNA duplexes or the miRNA-RISC complexes in
plants
11345 Feedback regulation of miRNA biogenesis
MiRNA biogenesis is under feedback regulation such that the DCL1 and AGO1 genes
are themselves regulated by miRNAs DCL1 is itself a target of miR162 which leads to
the cleavage of the DCL1 mRNA (Xie et al 2003 de Alba et al 2013) Consistent
with this the levels of DCL1 mRNA are elevated in the mutants of miRNA processing
genes in which the miR162 abundance is reduced In addition there is a predicted
hairpin structure of miR838 in the 14th
intron of the DCL1 primary transcript
(Rajagopalan et al 2006) This prediction implies that DCL1-mediated processing
on this site can result in the cleavage of the DCL1 primary transcript into two truncated
fragments the N-terminal capped fragment and the C-terminal polyadenylated
fragment If this is the case it is possible that miRNA biogenesis competes with the
splicing event of the DCL1 pre-mRNA
The AGO1 is also regulated by miRNA There is a complementary site for
miR168 binding in the AGO1 mRNA and the transcript level of the AGO1 mRNA is
reduced through an mRNA cleavage mechanism (Vaucheret et al 2006 de Alba et al
2013 Rogers amp Chen 2013) indicating that the AGO1 mRNA levels are tightly
controlled by the levels of the AGO1 protein
1135 Mechanism of miRNA action
11351 MiRNA-directed mRNA cleavage
By forming the miRNA-RISC assembly miRNAs can direct the RISC to regulate gene
expression by two post transcriptional mechanisms mRNA cleavage or translational
repression (Bartel 2004 Jones-Rhoades et al 2006 Dalmay 2013) The degree
of miRNA-mRNA complementarity plays an important role for the
determination of the mechanism to be used A general rule is that while perfect or
Chapter 1 Introduction and Review of Literature
40
near-perfect complementarity induces mRNA cleavage central mismatches trigger
translational repression (Carrington amp Ambros 2003 Jones-Rhoades amp Bartel 2004)
Unlike most animal miRNAs plant miRNAs have near-perfect or perfect
complementarity to their targets and mRNA cleavage is assumed to be a predominant
mechanism by which miRNAs regulate gene expression in plants
In RNA cleavage mechanism miRNAs guide the AGO component of RISC to
cleave a single phosphodiester bond of the target mRNA within the miRNA-binding
site (Llave et al 2002b Tang et al 2003) The truncated fragments are then released
and degraded freeing the RISC to recognize and cleave another target mRNA This has
been validated by the detection of 3prime cleavage products that have 5prime ends that start at the
middle of the complementary regions The mRNA cleavage products are then further
degraded by other mechanisms The 3prime cleavage products of some miRNA targets are
degraded by the 5prime-3prime exonuclease XRN4 an Arabidopsis homolog of the major Yeast
mRNA degrading exonuclease Xrn1p It has been observed that the 3prime cleavage
products of some target mRNAs were accumulated in the xrn4 mutants (Souret et al
2004 Dalmay 2013) However the 3prime cleavage products of other miRNA targets do
not accumulate in the xrn4 mutants indicating that there are also XRN4 independent
mechanisms The 5prime cleavage products are typically untraceable by RNA gel blot
hybridization but can be detected by sensitive PCR based methods It has been found
that the 5prime cleavage products tend to obtain an oligo U tail at the 3prime ends This 3prime U tail
is correlated with shortening of the 5prime cleavage products from the 5prime end leading to the
conclusion that the 3prime uridylation causes 5 prime- 3 prime exonucleolytic decay of the 5prime cleavage
products (Shen amp Goodman 2004)
DCL1 HYL1 and SE interact with each other and are co-localized in the
nuclear bodies termed Dicing bodies or D-bodies in which some pri-miRNA shave
been found suggesting that D-bodies serve as a center for active miRNA processing
(Fang amp Spector 2007 Fujioka et al 2007 Song et al 2007) The miRNA
processing site appears to be distinct from the Cajal bodies that have previously been
found as a siRNA processing site (Pontes amp Pikaard 2008) The D-bodies include
SmD3 and SmB proteins that are found in the Cajal bodies However At collin an
additional marker for the Cajal bodies is not found in the D-bodies Moreover the D-
bodies are present in an At collin mutant lacking the Cajal bodies The pri-miRNAs of
miR171A and miR159A genes have been found to be co-localized with HYL1 and
Chapter 1 Introduction and Review of Literature
41
DCL1 in the D- bodies that are distinct from the Cajal bodies (Song et al 2007)
11352 MiRNA-directed translational repression
Translation repression is another mechanism exerted by plant miRNAs The regulatory
mechanism of miR172 which regulates flowering time and floral organ identity is
consistent with translation repression MiR172 over production does not affect the
transcript levels of APETALA2 (AP2) and AP2- like target mRNAs Instead the AP2
protein level is reduced in the miR172 over expressing mutant (Aukerman amp Sakai
2003 Chen 2004 Dalmay 2013) It was later shown that miR172 can also direct the
cleavage of the target mRNAs although the steady-state mRNA level is unchanged due
to feedback regulation (Schwab et al 2005 Jung et al 2007) It is clear that miR172
regulates its target genes primarily by translational repression rather than mRNA
cleavage Recently it has been reported that miR156157 and miR854 also regulate
their target genes through translational repression (Arteaga-Vazquez et al 2006
Gandikota et al 2007 Dalmay 2013) It was once thought that translational repression
is an exception to the rule However experimental evidence suggest a widespread
coexistence of translational repression and mRNA cleavage (Brodersen et al 2008)
1136 Regulatory roles of miRNAs in plant development
The miRNA target prediction has been a difficult task in order to obtain regulatory
targets without bringing in too many false predictions Plant growth and developmental
processes regulated by miRNAs are quite diverse and enormous Furthermore plant
miRNAs work cooperatively or antagonistically to establish a balanced regulation This
notion is well consistent with the findings that mutations in the regulators of miRNA
biogenesis transport and processing such as AGO1 DCL1 HYL1 HEN1 ABH1
and HASTY cause pleiotropic phenotypes (Mallory amp Vaucheret 2006 de Alba et al
2013) To date the roles of miRNAs have been mostly deduced from obtaining miRNA
over expressing transgenic plants or gain-of-function mutants in which miRNA-
resistant target genes are ectopically expressed
11361 Phase transition
Plants pass through a series of developmental phase transitions during their life span
beginning with seed germination continuing through vegetative phase change
reproductive phase change flowering initiation and finally seed production for the next
generation Because of their importance in understanding plant developmental
Chapter 1 Introduction and Review of Literature
42
processes genes regulating developmental transitions have been extensively studied
and underlying molecular mechanisms have been explored in various plant species
Majority of researches were mainly focused on vegetative phase change and
reproductive transition in Arabidopsis and Maize (Poethig 2003 Baurle amp Dean
2006) Particularly the mutations of the genes encoding various regulators of miRNA
biogenesis and transport such as HST SE and AGO exhibit altered juvenile to adult
vegetative phase change and vegetative to reproductive change (Hunter et al 2003
Peragine et al 2004 Vazquez et al 2004a Jones-Rhoades et al 2006 de Alba et al
2013)
Over-expression of the miR156a gene causes late flowering and delays
vegetative phase change (Wu amp Poethig 2006 Gandikota et al 2007 Wang et al
2008) It has been shown that miR156 negatively regulates a group of genes encoding
SQUAMOSA PROMOTER-BINDING PROTEIN-LIKE (Lewis et al 2007 de Alba et
al 2013 Rogers amp Chen 2013) transcription factors such as SPL3 SPL4 and SPL5
that promote flowering initiation In contrast transgenic plants over expressing
miR156-resistant SPL345 genes are early flowering Interestingly the endogenous
level of miR156 is temporally regulated In the early juvenile vegetative phase the
level of miR156 is very high but decreases rapidly before the onset of the adult juvenile
phase (Wu amp Poethig 2006)
The Arabidopsis early activation tagged (eat-D) mutant exhibits early flowering
with disrupted floral structure The mutant phenotypes are caused by over expression of
the miR172a-2gene (Aukerman amp Sakai 2003) MiR172 regulates a small group of
genes encoding APETALLA2 (AP2)-like transcription factors such as TARGET OF
EAT1 (TOE1) TOE2 SCHLAFMUTZE (SMZ) and SCHNARCHZAPFEN(SNZ)
TOE1 TOE2 SMZ and SNZ have been shown to participate in their productive phase
change by repressing a floral integrator FLOWERING LOCUS T (FT)(Jung et al
2007) Consistent with this toe1-2D 35STOE2 35SSMZ and 35SSNZ plants
exhibit late flowering (Jung et al 2007) MiR172 is also regulated temporally and
highly expressed in the adult vegetative phase both in Arabidopsis and maize (Chuck et
al 2007) Because the temporal expression patterns of miR156 and miR172 are
complementary to each other it has been suggested that the two miRNAs interact with
each other in regulating phase change (Chuck et al 2009 de Alba et al 2013) In
support of this view it has been shown that miR172 is down-regulated in the
Chapter 1 Introduction and Review of Literature
43
Corngrass1(Cg1) mutant in which miR156 is overproduced (Chuck et al 2007 de
Alba et al 2013) However there is no direct evidence for the direct linkage between
miR156 and miR172 It is also envisioned that miR156 and miR172 would not be
directly related For example miR156 regulates plastochron index that is the inverse of
leaf initiation rate and thus frequently used to analyze temporal patterns of plant
development In contrast miR172 does not affect plastochron (Wang et al 2008) The
down-regulation of miR172 in the maize miR156-over producing mutants would be
due to the prolonged juvenile vegetative phase in the mutants
Another miRNA that regulates flowering initiation is miR159 It regulates
MYB33 and MYB65 which are closely related to the barley GAMY B transcription
factor that responds to gibberellic acid (GA) The level of miR159 is elevated in GA-
treated seedlings but reduced in the GA biosynthetic ga1 mutant Transgenic plants
over producing miR159 exhibit delayed floral induction under short days (Achard et
al 2004) It is been suggested that MYB33 mediates GA signal to regulate LEAFY
(LFY) These observations indicate that miR159 plays an important role in the GA
signaling pathways that regulate proper floral induction under short days (Achard et al
2004)
11362 Floral development
Arabidopsis flowers consist of four distinct organs They arise in a characteristic
pattern within concentric rings called whorls from outside to inside four sepals four
petals six stamens and the central pistil consisting of two carpels Identities of the
floral organs are determined by three classes of regulatory genes classA classB and
class C genes (Krizek amp Fletcher 2005) The A and C class genes act antagonistically
to restrict their expression domains (Bowman et al 1991)
It is notable that miR172 also regulates floral architecture in addition to its role
in the timing of flowering initiation MiR172 inhibits the translation of the AP2 mRNA
(Chen 2004) Transgenic plants overproducing miR172 not only exhibit early
flowering but also show abnormal floral development which is quite similar to those
of the class A gene mutants such as the ap2 mutants (Aukerman amp Sakai 2003 de
Alba et al 2013) When the activity of the class A genes is inactivated that of the class
C genes such as AGAMOUS (AG) is elevated As a result the miR172- overproducing
transgenic plants exhibit no petals along with homeotic transformation of the sepals to
Chapter 1 Introduction and Review of Literature
44
the carpels (Aukerman amp Sakai 2003 Chen 2004)
Interestingly miR166 also regulates floral architecture The role of miR165166
in the regulation of meristem activity is closely linked to floral meristem formation and
organization (Zhao et al 2007 Miyashima et al 2013) Floral structure is severely
disrupted in the miR165166-overproducing mutants such as men1 and jba-1D in
which miR166 is overproduced (Williams et al 2005b Jung amp Park 2007) The men1
and jba-1D mutants have smaller gynoecium and reduced carpel number In an extreme
case the jba-1D homozygous plants lack visible carpels (Williams et al 2005b)
Similar phenotypes have been observed in the miR165 overproducers (Zhao et al
2007) It has been suggested that the phenotypes are possibly caused by altered AG
expression in the floral meristem (Jung amp Park 2007 Turchi et al 2013)
Other miRNAs also affect floral development Overproduction of miRNAs
involved in auxin signaling also affects floral architecture Transgenic plants
overproducing miR167 have defects in the formation of ovule and anther with reduced
fertility (Ru et al 2006) It has been shown that miR167 targets ARF6 and ARF8 that
play a role in gynoecium and stamen development and in regulating fertility (Ru et al
2006 Wu et al 2006) These observations suggest that auxin signals are also closely
related to floral organogenesis In addition transgenic plants over-producing miR164
and the cuc1cuc2 double mutants show fused sepals and reduced petals (Laufs et al
2004 Turchi et al 2013) suggesting that miR164 is also related to floral meristem
activity and boundary specification of the meristematic region
11363 Shoot apical meristem development
Shoot apical meristem comprises self-maintaining totipotent cells The shoot apical
meristem activity affects diverse aspects of plant developmental processes including
leaf development vascular development and lateral organ polarity (Bowman et al
2002) Because miR165166 plays a primary role in meristem formation
overproduction of miR165166 and multiple knockout mutants of its target genes
exhibit pleiotropic pheonotypes (McConnell et al 2001 Emery et al 2003 Kim et al
2005 Williams et al 2005b)
The targets of miR165166 include genes encoding the homeo domain-leucine
zipper (HD-ZIP) class III transcription factors such as PHABULOSA(PHB)
PHAVOLUTA (PHV) REVOLUTA(REV) ATHB15CORONA (CNA) and ATHB8
Chapter 1 Introduction and Review of Literature
45
PHB PHV and ATHB15 have overlapping functions in controlling meristem
development (Turchi et al 2013) Indeed the phb phv athb15 triple mutants have an
enlarged shoot apical meristem Consistent with this the miR166- overproducing men1
and jba-1D mutants exhibit enlarged shoot apical meristems (Kim et al 2005
Williams et al 2005b Turchi et al 2013) In the mutants the shoot apical meristems
are multiplied and large
The development of shoot apical meristems is also regulated by the WUSCHEL
(WUS)ndashCLAVATA (CLV) signaling pathway WUS positively regulates cell
proliferation In contrast CLV3 which is expressed in stem cells limits the WUS
expression and thus inhibits cell proliferation in the SAM (Sharma et al 2003)
Consistent with this notion WUS is expressed to a higher level in jba-1D
explaining the ectopic enlargement of the SAM in the mutant (Williams et al 2005)
While WUS is closely related to miR165166 in the shoot apical meristem
development the underlying molecular mechanisms are currently unclear It has been
observed that the miR166-over producing men1 plants have an arrested meristem
development (Jung amp Park 2007) In addition the heterozygous men1+ and the
homozygous men1 plants show different expression patterns of WUS and CLV3 It
appears that miR166 regulates WUS indirectly and influences the expressional balance
between WUS and CLV3 through a negative feedback loop (Jung amp Park 2007 de
Alba et al 2013)
The phv phb athb15 triple mutant has an enlarged shoot apical meristems the
rev phb phv mutant does not have functional shoot apical meristems (Prigge et al
2005) This distinction may be explained by the fact that while miR166 targets
primarily PHB PHV and ATHB15 miR165 targets all the five HD-ZIP III genes (Zhou
et al 2007) Consistent with this transgenic plants overproducing miR166 are distinct
from those overproducing miR165 The miR165-overproducing transgenic plants lack
functional shoot apical meristem and a pin-like structure is formed at the apical region
of the mutant These phenotypes are quite similar to rev phb phv triple mutant (Zhou et
al 2007 Liu et al 2013) The phenotypic distinction may because by the difference
of the cleavage efficiencies of the target mRNAs In accordance with this notion a few
35S miR166a transgenic lines exhibit no shoot apical meristems The men1
homozygous plants also show arrested meristem development (Jung amp Park 2007)
Chapter 1 Introduction and Review of Literature
46
MiR164 cleaves the CUC1 and CUC2 mRNAs as well as the NAC1 mRNA
CUC1 and CUC2 determines the organ boundary in the meristem Phenotypes of
transgenic plants that overproduce miR164 are similar to those of the cuc1cuc2 double
mutant that exhibits embryonic developmental defects such as cup-shaped cotyledons
and fused cotyledons (Laufs et al 2004) When a miR164-resistant CUC2 is
expressed the boundary domain is enlarged CUC also plays a role in embryonic shoot
meristem formation by regulating the SHOOT MERISTEMLESS gene It was therefore
concluded that miR164-mediated cleavage of CUC1 and CUC2 mRNAs determines the
sizes of the boundary domains in the shoot apical meristem and regulates separation of
the organs by restricting cell proliferation (Laufs et al 2004 de Alba et al 2013)
11364 Leaf development
The role of miR319 has been extensively studied in leaf development A dominant
jaw-D mutant overproducing miR319 shows uneven curved leaves with enlarged size
and five TCP genes which are closely related to the CINCINNATA (CIN) gene in
Antirrhinum majus are down regulated in the mutant suggesting that miR319 mediated
regulation of the TCP transcription factor genes are important for the final step of
leaf shaping (Palatnik et al 2003 Naz et al 2013) In addition to leaf shape and size
leaf polarity is also specified by miRNA MiR166-mediated cleavage of the HD-ZIPIII
mRNAs is a well to establish leaf polarity This discovery originates from the gain-of-
function mutants such as phb-1d and phv-1d wherein point mutations in the
complementary sites to miRNA helps the mRNA to evade or resist from miRNA-
mediated cleavage (McConnell et al 2001 Emery et al 2003 Naz et al 2013)
Plant leaves have a dorso-ventral polarity consisting of the adaxial (upper
surface) and abaxial (lower surface) sides The adaxial side is closer to the shoot
apical meristem Adaxial identity is specified by functionally redundant class III HD-
ZIP family genes such as PHB PHV and REV Ectopic expression of each genes
result in adaxialized leaves Dominant phb-1d and phv-1d mutants exhibit
transformation of the abaxial to adaxial fate and have rod-shaped leaves In contrast
miR165166-overproducing plants show pin-like cotyledons radicalized leaves and
abaxialized leaves (Chitwood et al 2007 Pattanayak et al 2013)
Leaf asymmetry is determined by miR166-mediated restriction of the
expression domains of the HD-ZIPIII genes The HD-ZIPIII genes are expressed in the
Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
22
Transgenic coffee plants with suppressed caffeine synthesis using RNA interference
(RNAi) technology have been obtained (Ogita et al 2003 Ogita et al 2004) Specific
sequences in the 3prime untranslated region of the theobromine synthase gene (CaMXMT1)
were selected for construction of RNAi short and long fragments The caffeine and
theobromine content of the transgenic plants were reduced by ~ 70 when compared to
the untransformed plants In C canephora RNAi technology has also been employed
to silence the N-methyl transferase gene involved in caffeine biosynthesis (Vinod et al
2007) Promoter of an N-methyltransferase (NMT) gene involved in caffeine
biosynthesis was cloned (Satyanarayana et al 2005 Satyanarayana 2006) which will
be very useful for studying the regulation of caffeine biosynthesis
1105 Improvement in cup quality
Improvement in coffee cup quality requires elaborate knowledge of the chemical
constituents as well as the metabolic pathways involved in the elaboration of quality
The constituents of coffee beans include minerals proteins carbohydrates caffeine
chlorogenic acids (CGA) glycosides lipids and many volatile compounds that give
flavour to coffee by roasting Among these the role of three major constituents
sucrose CGA and trigonelline have been studied in coffee The sucrose content of
coffee bean is associated with coffee flavour the higher the sucrose content in green
beans the more intense will be the cup flavour (Clifford et al 1985 de Maria et al
1996) The sucrose content of C arabica (82-83) is higher than C canephora (33ndash
40) The sucrose amino acids ratio in green beans determines the profile of volatile
compounds Manipulating sucrose content in coffee bean is therefore important in
improving cup quality The sucrose synthase gene (CcSUS2) from C canephora has
been cloned and sequenced (Leroy et al 2005) This provides an opportunity to
manipulate the sucrose content in coffee Chlorogenic acids are products of
phenylpropanoid metabolism Chlorogenic acids are a group of hydroxycinnamoyl
quinic acids (HQA) formed by esterification between caffeic acids coumaric acids and
quinic acids (Clifford et al 1999) They are present in relatively large quantities in the
coffee bean and are the precursor of phenolic compounds in roasted beans C
canephora beans contain higher CGA (10) compared to C arabica beans (6-7)
CGA are known to have antioxidant properties as well as being associated with disease
resistance (Campa et al 2003) Genetic manipulations of genes involved in CGA
synthesis can serve either of these purpose by up- or down regulating the pathway
Chapter 1 Introduction and Review of Literature
23
Phenylalanine ammonia-lyase (PAL) catalyzes the first step of the phenylpropanoid
pathway leading to the synthesis of a wide range of chemical compounds including
flavonoids coumarins hydroxycinnamoyl esters and lignins (Hahlbrock amp Scheel
1989) Recently the full length cDNA and corresponding genomic sequences of PAL
from C canephora was isolated characterized and functionally validated (Mahesh et
al 2006 Parvatam et al 2006) This has opened up new possibilities for manipulating
the level of the PAL enzyme in coffee which in turn can be useful for improvement of
cup quality and manipulating antioxidant properties in coffee
1106 Fruit Ripening
Uniformity during fruit ripening is decisively related to cup quality in coffee and
consequently to the value of the product Fruits at the ideal ripening stage produce the
best organoleptic characteristics for coffee The presence of over ripened or green fruits
changes the acidity the bitterness and consequently the cup quality In order to
maximize uniform ripening of coffee fruits it is essential to control the action of genes
involved in the last step of maturation process Ethylene is known to trigger ripening
and increasing ethylene biosynthesis is associated with various stages of ripening
process (Protasio et al 2005) To control coffee fruit maturation two of the major
genes involved in ethylene biosynthesis namely ACC synthase and ACC oxidase
have been cloned (Protasio et al 2005 Neupane et al 1999) Introduction of the ACC
oxidase gene in antisense orientation have been achieved in both C arabica and C
canephora The effect of the transgene on ethylene production and fruit maturation has
yet to be reported The inhibition of genes downstream to the initial ethylene burst is
also an option to control coffee fruit maturation (Ribas et al 2006)
111 RNA interferenceRNAi
The regulation of gene expression at post transcriptional level has recently attracted
much attention because of the discovery of the phenomenon called RNA interference
(RNAi) RNAi based silencing techniques are more efficient than antisense mediated
gene silencing (Wesley et al 2001) The effect was first observed in plants wherein
silencing was observed when some copies of a transgene was in the forward orientation
with respect to the promoter and some in the reverse orientation This resulted in the
formation of double stranded RNA in the system The phenomenon in which
experimentally introduced double stranded RNA (dsRNA) leads to the loss of
expression of the corresponding cellular gene by sequence specific RNA degradation
Chapter 1 Introduction and Review of Literature
24
was termed as RNA interference or RNAi The protective effect of RNA silencing in
reducing virus infectivity supports the view that PTGS has evolved as a mechanism to
defend plants against virus infection and also to moderate the possible deleterious
genome-restructuring (insertional) activity of virus-like mobile genetic elements eg
retrotransposons A growing body of biochemical and genetic data has further
demonstrated that plants animals and yeasts share related mechanisms of specific
degradation of RNAs in which double-stranded forms of RNA are initiator molecules
(Brenstein et al 2001)
The key insight into the process of PGTS was provided by the experiments
conducted by Hamilton amp Baulcombe (1999) who identified the product of RNA
degradation as small RNA (siRNA) species of ~25nt of both sense and antisense
polarity SiRNA are formed and accumulate as double stranded RNA molecules of
defined chemical structures Both genetic and biochemical approaches were undertaken
to understand the basis of silencing Genetic screens were carried out in fungus
Neurospora carassa the alga Chalmydomonas reinhardtii and plant Arabidopsis
thaliana to search for detective mutants in PTGS
A combination of results obtained from several in vivo and in vitro experiments
have gelled into a two step mechanistic model for RNAiPTGS The first step referred
to as the RNAi initiating step involves binding of the RNA nucleases to a larges
dsRNA and its cleavage into discrete ~21-25nt RNA fragments This is ATP-
dependent reaction which involved RNase III - type endonucleases which make
staggered cuts in both strands of the dsRNA leaving 3prime overhang of 2 nucleotides with
5prime- phosphate 3prime-hydroxyl and no modification in the sugar phosphate backbone
(Hamilton amp Baulcombe 1999 Elbashir et al 2001) SiRNAs are the direct products
of dsRNA cleavage by the multi-domain RNase III enzyme DICER (Figure 16)
(Brenstein et al 2001 Karthikeyan et l 2013)
In the second step or commonly known as the effector step the double stranded
siRNAs produced in the first step bind to an RNAi specific protein complex to form a
RISC The complex undergoes activation in the presence of ATP so that the antisense
component of the unwound siRNA becomes exposed and allows the RISC to perform
the downstream RNAi reaction
Chapter 1 Introduction and Review of Literature
25
Several enzymes are involved in RNAi based on their role genes that encode for them
are classified into RNAi initiators RNAi effectors RNA dependent RNA polymerases
and silencing genes Two C elegans genes rdeI and rde4 (rde stands for lsquo RNAi
deficientrsquo) are believed to be involved in the initiation step of RNAi The C elegans
rde1 gene is a member of a large family of genes and is homologous to the Neurospora
qde2 (qde stands for ldquoquelling deficientrdquo) and Arabidopsis AGO1 (AGO stands for
agronaute AGO1 was previously identified to be involved in Arabidopsis
development) Although the functions of these genes are not clear a mammalian
member of RDE1 family has been identified as a translation initiation factor (Thakur
2005) Important genes for the effector step of PTGS in C elegans are rde2 and mut7
genes These genes were identified from heterozygous mutant worms that were unable
to transmit RNAi to their homozygous offsprings Worms with mutated rde2 or mut 7
genes show defective RNAi mut-7 gene encodes a protein with homology to the
nuclease domains of RNAase D and a protein implicated in Werner syndrome (a rapid
ageing disease in humans) (Grishok et al 2001 Kamath-Loeb et al 2012s)
Neurospora qde-1 Arabidopsis SDE-1SGS-2 and C elegans ego-1 appear to encode
RNA dependent RNA polymerase (RdRPs) It assumed that this is a proof that an RdRp
activity is required for RNAi Certainly the existence of an RdRp might explain the
remarkable efficiency of dsRNA induced silencing if it amplifies either the dsRNA
prior to cleavage or the siRNAs directly (Muers 2013) In C elegans ego-1 mutants
(ego stands for lsquoenhancer of glp-1) RNAi functions normally in somatic cells but is
defective in germline cells where ego-1 is primarily expressed In Arabidopsis SDE-
1SGS-2 mutants (SGS stands for suppressor of gene silencing) siRNA are produced
when dsRNA is introduced via an endogenously replicating RNA virus but not
introduced by a transgene It has been proposed that perhaps the viral RdRP is
substituting for the Arabidopsis enzyme in these mutants Random degradative PCR
model suggests that an RdRP uses the guide strand of an siRNA as a primer for the
target mRNA generating a dsRNA substrate for Dicer and thus more siRNAs
Several genes controlling RNA silencing in plants have been identified through
genetic screens of Arabidopsis mutants impaired in transgene induced RNA silencing
They encode a putative RNA-dependent RNA polymerase (SGS2SDE1) a coiled
protein (SGS3) a protein containing PAZ and Piwi domains (AGO1) and an RNA
helicase (SDE3) The putative SGS2SDE1 of Neurospora is related to QDE-1 and
Chapter 1 Introduction and Review of Literature
26
EGO-1 of C elegans whereas PAZPiwi protein AGO is related to QDE-2 of
Neurospora RDE-1of C elegans RNA helicase SDE3 is related to SMG-2 of C
elegans and Mut-6 of Chlamydomonas MUT-7 gene of C elegans encodes a protein
similar to Rnase D whereas the Drosophila DICER gene encode a protein similar to
RNase III An Arabidopsis ortholog of DICER gene has been identified called CAF
SIN1 SUS1(Thakur 2005 Poethig 2009)
Since the RNAi machinery is present constitutively within the eukaryotic cell it
is important to explore the metabolic advantages that are accorded by the RNAi related
proteins during the intrinsic normal growth of cells and development of organisms The
natural RNAi machinery not only keeps the mobile transposable elements from
disrupting the integrity of the genomes as suggested by the analysis of model organisms
like A thaliana C elegans D melanogaster (Tabara et al 1999 Wu-Scharf et al
2000 Aravin et al 2001 Hamilton et al 2002 Lisch 2009) but also participates in
development of organisms Genetic defects in Celegans RNAi genes ego1 and dicer
cause known specific developmental errors (Grishok et al 2000 Grishok et al 2001
Knight amp Bass 2001 Hall et al 2013) Similarly the Argonaute family of genes of A
thaliana (especially the ZWILLE proteins) is also responsible for plant architecture and
meristem development (Carmell et al 2002 Hamant and Pautot 2010) and the Dicer
homologue of A thaliana CAF1 is required for embryo development (Golden et al
2002 Nakano et al 2011) Thus genetic evidence illustrates the role of the RNAi
machinery as a controller of development related genes The mechanistic details of
these developmental processes are beginning to emerge
Chapter 1 Introduction and Review of Literature
27
Figure 16 Mechanism of gene silencing induced double stranded RNA The dicer enzyme to
produce siRNAs cleaves the RNA The siRNA generated bind to the nuclease complex
RISC The antisense component in the siRNA in the RISC guides the complex towards
the cognate mRNA resulting in endonucleolytic cleavage of the mRNA
Chapter 1 Introduction and Review of Literature
28
112 MicroRNA or MiRNA
In 1991 Ambros and co-workers first isolated a lin-4 mutant of Celegans which was
arrested at the first larval stage (Lee et al 1993) Later on the let-7 mutation was
isolated in the same system which was responsible for development through the fourth
larval stage Both lin-4 and let-7 encode short 22-nucleotide mature RNAs and were
called short temporal RNA because they control the temporal development program of
C elegans The mature lin-4 RNA defines (negatively regulates) the mRNA expression
of the lin-14 and lin-28 hetrochronic genes with the antisense mediated repression
mechanism of translation initiation and thus specifies the fate of cells during the first
three larval stages Recent studies have revealed that the short temporal RNAs are
actually members of a group of tiny RNAs (21to 28 nucleotides) called the micro-
RNAs (miRNA) (Bartel 2009) isolated members of which could easily run to a few
thousand which includes both those were identified computationally and by deep
sequencing Some of the components of the RNAi machinery have also been clearly
established as the effector proteins for the maturation of miRNAs
113 MicroRNAs in plants
1131 Discovery
MicroRNAs have been discovered by three basic approaches Direct cloning forward
genetic direct cloning and bioinformatic prediction followed by experimental
validation The most direct method of miRNA discovery is to isolate and clone small
RNAs from biological samples and several groups have used this approach to identify
plant miRNAs (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002 Xie et al 2003 Sunkar et al 2005 Williams et al 2013) The cloning methods
adapted were similar to those used to identify large numbers of animal miRNAs
(Lagos-Quintana et al 2001 Lau et al 2001 Aravin amp Tuschl 2005) which involve
isolating small RNAs followed by oligonucleotide adapter ligation reverse
transcription amplification and sequencing Some protocols incorporate methods to
enrich for Dicer cleavage products (ie molecules with 5prime-phosphates and 3prime-
hydroxyls) and to concatemerize the short cDNAs so that several may be identified in a
single sequencing read (Lau et al 2001 Llave et al 2002a Jagadeeswaran amp Sunkar
2013)The initial cloning experiments in Arabidopsis identified 19 miRNAs belonging
to 15 families (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002) although hundreds of other small RNAs such as siRNAs and RNA degradation
Chapter 1 Introduction and Review of Literature
29
fragments were also cloned through the direct cloning methods To select miRNAs
from the pool of small RNAs secondary structures of the RNA molecules were
examined in compliance with the acknowledged structural features of miRNAs
(Ambros et al 2003) The secondary structures were validated using several ad-hoc
software tools such as the Mfold or RNAfold programs (Reeder et al 2006) Finally
the existence of mature miRNAs was determined by RNA gel blot hybridization These
direct cloning methods were successful in isolating many miRNAs in Arabidopsis
(Reinhart et al 2002 Sunkar amp Zhu 2004) Rice (Sunkar et al 2005) Poplar (Lu et
al 2005) moss (Arazi et al 2005) and Tobacco (Billoud et al 2005)
Although miRNAs were first discovered through forward genetic screens in
worms (Lee et al 1993 Reinhart et al 2000)only few A thaliana miRNA gene
families have been discovered by this method (Chen 2008) in plants MiRNA
involvement in plant mutant phenotypes were not properly inferred till the cloning
experiments established that plant genomes contain numerous miRNAs (Park et al
2002 Reinhart et al 2002 Rhoades et al 2002) MiRNA loss-of-function allele has
been identified by forward genetic screens early extra petala1 is caused by a
transposon insertion ~160bp upstream of the predicted miR164c stem loop resulting in
flowers with extra petals (Baker et al 2005 Irish 2008) The fact that loss-of-function
miRNA mutants have been rarely discovered using forward genetics reflects small
target size for mutagenesis coupled with redundancy in nearly all evolutionarily
conserved plant miRNAs which are encoded by gene families (Table16) Family
members are likely to have overlapping functions buffering against loss at any single
miRNA locus Over expression screens can circumvent redundancy limitations At least
four plant miRNAs miR319 (also known as miR-JAW) miR172 (also known as EAT)
and miR166 were isolated in over expression screens for dominant mutants with
developmental abnormalities (Aukerman amp Sakai 2003 Palatnik et al 2003 Kim et
al 2005 Williams et al 2005b Schommer et al 2012) Mutations in miRNA target
sites which can prevent the entire family of miRNAs from repressing a target gene can
also circumvent redundant functions of miRNA family members
The dominant mutations in the HD-ZIP genes PHB PHV and REVOLUTA
(REV) in Arabidopsis and ROLLEDLEAF1 (RLD1) in Maize result in adaxialization of
leaves andor Vasculature (McConnell amp Barton 1998 McConnell et al 2001 Nelson
et al 2002 Zhong amp Ye 2004 Allen amp Millar 2012) and are all caused by mutations
Chapter 1 Introduction and Review of Literature
30
in miR166 complementary sites (Rhoades et al 2002 Emery et al 2003 Jones-
Rhoades amp Bartel 2004 Mallory et al 2004 Zhong amp Ye 2004)
In both plants and animals cloning was the initial means of large-scale
miRNA discovery (Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Reinhart et al 2002 Mendes et al 2012) However cloning is biased toward
RNAs that are expressed highly and broadly MiRNAs expressed at low levels or
only in specific cell types or in response to certain environmental stimuli were more
difficult to clone Sequence based biases in cloning procedures might also cause
certain miRNAs to be missed Because of these limitations bioinformatics approaches
to identify miRNAs have provided a useful complement to cloning
A straightforward use of bioinformatics has been carried out to find homologs
of known miRNAs both within the same genome and in the genomes of other species
(Pasquinelli et al 2000 Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Mendes et al 2009) A more difficult challenge is to identify miRNAs
unrelated to previously known miRNAs This was first accomplished for
vertebrate nematode and fly miRNAs using algorithms that search for conservation
of sequence and secondary structure (ie miRNA stem-loop precursors) between
species searching for patterns that are characteristic of miRNAs (Lai et al 2003 Lim
et al 2003 Lim et al 2005)
Most of the methods identified numerous miRNAs but are necessarily more
relaxed in terms of allowed structures Some approaches take advantage of the high
complementarity of plant miRNAs to target messages implementing the requirement
that the candidate has conserved complementarity to mRNAs (Jones-Rhoades amp Bartel
2004 Adai et al 2005) This additional filter has been useful for distinguishing
authentic plant miRNAs from false positives which later extended to mammalian
miRNA gene prediction (Xie et al 2005 Mendes et al 2012)
Another criteria used by most algorithms in the filters is evolutionary
conservation In plants mature miRNA sequences are conserved to a higher degree
than their precursor sequences For example Arabidopsis and rice diverged more than
130 million years ago (Friis et al 2004) but some miRNAs are highly conserved
in these plant species (Reinhart et al 2002) Furthermore various parameters for the
secondary structures such as free energy the number of paired residues within a
Chapter 1 Introduction and Review of Literature
31
miRNA or the number and size of bulges are also used to validate the predictions
(Jones-Rhoades amp Bartel 2004 Wang et al 2004 Adai et al 2005 Mendes et al
2012)
1132 Conserved microRNAs in Plants
Till date cloning genetics and bioinformatics screens have resulted in annotation of
approximately 184 potential Arabidopsis miRNAs (miRBase release 13) These
miRNAs seem to arise through gene duplication and subsequent diversification and
represent approximately 100 families (Maher et al 2006 Axtell 2013) Twenty one
families represented by 92 genes are clearly conserved in species beyond Arabidopsis
(Table 15) The presence of these miRNA families in other sequenced plant genomes
indicates that different plant species have an evolutionarily fluid set of miRNAs
(Willmann amp Poethig 2007) Recent studies on mosses one of the most ancient land
plants have shown that at least 14 miRNA families are conserved in eudicots and
mosses and therefore it appears that plant miRNAs share a common ancient origin
(Arazi et al 2005 Axtell amp Bartel 2005 Talmor-Neiman et al 2006 Zhang et al
2006 Axtell 2013) The number of members per family in a genome ranges from 1-32
with the exception of miR-430 which is represented by a cluster of ~80 loci in zebra
fish (Giraldez et al 2005)
In plants the number of members in each family correlates among examined
species certain families contain numerous members in all three species (eg miR156
miR166 miR169) whereas others consistently contain fewer genes (eg miR162
mir168 miR394) (Table 16) Although it is still unclear why certain miRNA are
encoded by more than one gene in plants (eg 12 genes encoding miR156) this
correlation might suggest functional significance of various miRNA family sizes Most
annotated plant miRNAs are conserved throughout flowering plants while a few
miRNAs are found only in a single sequenced genome and thus they could be
evolutionarily lsquoyoungrsquo miRNAs For example Arabidopsis has several miRNA
families that are not found in poplar or rice suggesting that miRNAs could be
generated continuously during evolution (Allen et al 2004a Rajagopalan et al
2006 Fahlgren et al 2007 Axtell 2012 de Alba et al 2013) Consistent with the
notion that non conserved miRNAs are of a more recent evolutionary origin these
miRNAs are mainly encoded by a single locus in the genome Within each family the
mature miRNA is always located in the same arm
Chapter 1 Introduction and Review of Literature
32
Table 16 MicroRNA families conserved in plants
miRNA
family
Arabidopsis Oryza Populus
miR156 12 12 11 miR159319 6 8 15 miR160 3 6 8 miR162 2 2 3 miR164 3 5 6 miR166 9 12 17 miR167 4 9 8 miR168 2 2 2 miR169 14 17 32 miR171 4 7 10 miR172 5 3 9 miR390 3 1 4 miR393 2 2 4 miR394 2 1 2 miR395 6 19 10 miR396 2 5 7 miR397 2 2 3 miR398 3 2 3 miR399 6 11 12 miR408 1 1 1 miR403 1 0 2 miR437 0 1 0 miR444 0 1 0 miR445 0 9 0 Total 92 127 169
All known miRNA families that are conserved between more than one plant species are listed
together with the number of genes identified in the sequenced genomes Rice miRNA families that have orthologs in maize but do not appear to have orthologs in the eudicots (Arabidopsis and Populus) are marked with an asterisk The following families contain miRNA genes annotated with
more than one number miR156 (miR156 and miR157) miR159319 (miR159 and miR319) miR166 (miR165 and miR166) miR171 (miR170 and miR171) and miR390 (miR390 and miR391)
of the stem loop (5prime- or 3prime) as it would be expected if the genes carry common ancestry
(Figure 17) The sequence of the mature miRNA and to some extent the segment on
the opposite arm of the hairpin to which it pairs is highly conserved between the
members of the same family (both within and between the species) while the sequence
secondary structure and length of the intervening ldquolooprdquo region can be highly divergent
within the same family
The patterns of paring and non pairing nucleotides are often conserved between
homologous miRNA stem loops from different species (Figure 17) The significance of
conserved mismatches is opined to guide DCL1 to cleave at the appropriate positions
along the stem loop
Chapter 1 Introduction and Review of Literature
33
Figure 17 Representative stem loop of miR164 Segments corresponding to the mature miRNAs
are shown in red (Adapted from Rajagopalan et al 2006)
Although many miRNAs are highly conserved in plants the degree of conservation in
most cases is quite low between plants and animals one exception is miR854 A recent
study has revealed that miR854 is conserved between Arabidopsis and animals and that
the Arabidopsis miR854 differs from the human miR854 only by a single nucleotide
(Arteaga-Vazquez et al 2006 Axtell 2012 de Alba et al 2013) The Arabidopsis
miR854 has multiple binding sites in the 3prime-untranslated region (UTR) of
OLIGOURIDYLATE-binding-PROTEIN mRNA 1bA (UBP1b) and represses the
translation of the UBP1b mRNA The animal miR854 is also complementary to a site in
the 3prime-UTR of the UBP1b homolog in animals However it is currently unclear how the
animal miR854 regulates its target mRNA Another distinction between plant and
animal miRNAs is the genomic organization of the miRNA genes While plant miRNA
genes are located predominantly in the intergenic regions animal miRNA genes tend to
localize in the intragenic regions both in the introns or exons of protein coding genes
Moreover miRNA genes are often clustered within a single precursor transcript
whereas individual plant miRNAs are derived from individual precursor RNAs (Bartel
Chapter 1 Introduction and Review of Literature
34
2004 Bonnet et al 2004 Kim 2005 Vaucheret et al 2006 Marco et al 2013)
1133 Non-conserved miRNA and challenges in annotation
Most miRNAs are conserved throughout flowering plants (Table 15) but many more
were found only in a single sequenced genome thus considered to be of a more recent
evolutionary origin The extended homology between few non conserved miRNAs
precursors and their target genes provides strong evidence that these relatively ldquoYoungrdquo
miRNAs arose from the duplication of the target gene segments (Allen et al 2004)
Several non-conserved miRNAs including miR161 miR163 miR173 miR447
miR475 and miR476 are known to direct cleavage of target transcripts (Allen et al
2004a Adai et al 2005 Sunkar et al 2005 de Alba et al 2013) It is difficult to
confidently predict targets for many because it is not possible to use complementary
site as a filter against false-positive target predictions It is also difficult to
confidently annotate non-conserved miRNAs as miRNA rather than siRNAs The
established minimal standard for miRNA annotation is a small RNA with detectable
expression and the potential to form a stem-loop when joined to flanking genomic
sequence (Kasschau et al 2003) In practice these requirements are too loose to
definitively categorize many small RNAs cloned from plants Many plant siRNAs are
detectable on blots (Vazquez et al 2004b) and hundreds of thousands of non-
miRNA genomic sequences can be predicted to fold into secondary structures that
resemble structures of plant miRNA precursors (Jones-Rhoades amp Bartel 2004)
Therefore without conservation of both sequence and secondary structure it is
difficult to be confident that a given cloned RNA originated from a stem-loop (ie is a
miRNA) rather than from a double-stranded RNA (ie is a siRNA) In fact many of
the thousands o f small RNAs cloned from Arabidopsis (Gustafson et al 2005) would
probably meet the literal requirements for annotation as miRNAs A few of these
sequences might be miRNAs but others that meet the literal criteria probably are not
so The challenges of annotating non conserved small RNAs were evidenced by
three related small silencing RNAs that were originally annotated as miRNAs
that turned out to be trans-acting siRNAs (tasiRNAs)(Kanno et al2013)
Although most miRNAs that are broadly conserved among flowering plants are
now being identified and experimentally validated (Table 16)(Jones-Rhoades amp
Bartel 2004) the challenges and ambiguities for classifying non-conserved miRNAs
prevent meaningful estimates of the total number of miRNA genes in Arabidopsis and
Chapter 1 Introduction and Review of Literature
35
other plant genomes It is possible that miRNAs might have escaped detection
because they are non-conserved and expressed in specific tissues or conditions and can
only be identified by using high- throughput sequencing
1134 MicroRNA biogenesis
11341 Transcription of microRNA precursor
Plant miRNAs are primarily found in the genomic regions that are not associated with
the protein coding genes (Reinhart et al 2002) and it appears that most if not all plant
miRNAs are produced from their own transcriptional units Plant miRNA genes are
occasionally clustered near each other in the genome suggesting transcription of
multiple miRNAs from a single primary transcript [eg the miR395 cluster (Grishok
et al 2001 Jones-Rhoades amp Bartel 2004b)] but this polycistronic arrangement of
miRNA genes appears far less frequently in plants than in animals (Bartel 2004)
Northern blot EST and mapping evidence indicate that plant miRNA primary
transcripts (also known as pri-miRNAs) as in animals are longer than needed to
encompass the miRNA stem-loops (Palatnik et al 2003 Jones-Rhoades amp Bartel
2004b) At least some of these pri-miRNA transcripts appear to be spliced
polyadenylated (Kurihara amp Watanabe 2004) and capped (Xie Z et al 2005) Two
rice miRNAs are contained within transcripts that contain exon junctions within the
presumptive stem-loop precursor implying that in these cases splicing is a prerequisite
for Dicer recognition (Sunkar et al 2005) Plant pri-miRNAs are over 1kb in length
and they are usually preceded by typical TATA box motifs They can also undergo
canonical splicing polyadenylation and capping All these observations indicates RNA
polymerase II is probably responsible for transcribing most plant miRNAs (Xie Z et
al 2005) as appears to be the case for many animal miRNAs Relatively little is
known about the regulation of miRNA transcription in plants but there is no reason
to suspect that this regulation would differ from that of protein-coding transcripts as
the bioinformatics analysis of the sequence upstream of the transcriptional start
sites of the miRNA genes showed the presence of putative binding motifs of
number of known transcription factors (Megraw et al 2006 Sethupathy 2013)
11342 MicroRNA processing and export
In plants maturation of miRNA is a step wise process A miRNA gene is transcribed to
pri-miRNA which is much longer than the pre-miRNA possessing the characteristic
stem-loop structure Like transcription of most protein-coding genes the miRNA
Chapter 1 Introduction and Review of Literature
36
transcription is processed by RNA polymerase II (Pol II) enzymes and the pri-
miRNAs are 5prime-capped and 3prime-polyadenylated (Lee et al 2004 Xie Z et al 2005)
Mature miRNAs are produced from pri-miRNAs through at least two sequential
processing steps by RNase III-type endonucleases In animals pri-miRNAs are first
processed at the bottom portion of the stem by Drosha a nuclear-localized RNase III
enzyme to liberate pre-miRNAs The pre-miRNAs are then exported to the cytoplasm
with the help of exportin-5The pre-miRNAs are further processed by Dicer another
RNaseIII enzyme to a duplex consisting of the miRNA and the antisense strands
(miRNA) Since plants do not have any Drosha homologs the two sequential
processing steps seem to be carried out by DCL1 a Dicer homolog in the nucleus
(Kurihara amp Watanabe 2004 de Alba et al 2013 Rogers amp Chen 2013)
The processing of pri-miRNAs to pre-miRNAs by DCL1 is assisted by two
other proteins HYPONASTY LEAVES1 (HYL1) and SERRATE (SE) HYL1 belongs to a
family of double-stranded RNA (dsRNA) binding protein and interacts with DCL1 in
vitro and in vivo (Lu amp Fedoroff 2000 Hiraguri et al 2005 de Alba et al 2013
Rogers amp Chen 2013) Loss-of-function mutations in the HYL1 gene result in
decreased accumulation of miRNAs and concomitant accumulation of pri-miRNAs
(Han et al 2004 Vazquez et al 2004a Kurihara et al 2006) SE a C2H2 zinc finger
protein interacts with DCL1 and HYL1 and plays a role in the processing of pri-
miRNAs to pre-miRNAs (Lobbes et al 2006 Yang L et al 2006) In addition the
nuclear cap-binding complex proteins CBP20 and CBP80 that are known as ABA
HYPER SENSITIVE 1(ABH1) have been shown to be required for proper miRNA
processing (Gregory et al 2008 Laubinger et al 2008) Recently it has been
demonstrated that DCL1 can release 21-nt RNAs from dsRNA as well as from
synthetic miR167 precursor RNAs in vitro However this cleavage is error prone and
inefficient in the absence of HYL1 and SE which accelerate the rate of DCL1-mediated
cleavage and promote accurate processing of miRNAs (Dong et al 2008 Rogers amp
Chen 2013)
11343 Methylation of the miRNAmiRNAduplex
After the miRNAmiRNAduplexes are released from the pre-miRNAs by the DCL1
activities the duplexes are methylated at the 2primeOH of the3prime-ends by HUA ENHANCER1
(HEN1) a small RNA methyltransferase (Yu et al 2005 Yang Z et al 2006 Rogers
amp Chen 2013 ) This step which is distinct from the RNase III-mediated processing
Chapter 1 Introduction and Review of Literature
37
step distinguishes the biogenesis of plant miRNAs from that of animal miRNAs
Mutations in the HEN1 gene were first isolated in a morphological screening for floral
development although the HEN1 mutants display pleiotropic phenotypes similar to the
developmental defects observed in the DCL1 mutants (Chen et al 2002 Park et al
2002 Rogers amp Chen 2013)
Figure 18 Biogenesis of microRNA
A Biogenesis of plant MicroRNA B Biogenesis of metazoananimal MicroRNA
The miRNA (red) is incorporated into RISC and the miRNA(blue) in degraded
This enzyme has two dsRNA-binding domains and nuclear localization signal It
is also conserved in fungi and is required for the processing of siRNAs as well as of
miRNAs (Park et al 2002 Boutet et al 2003 Xie et al 2003) Although the HEN1
protein is localized to the nucleus its methylation activity in the cytoplasm cannot be
ignored (Xie et al 2004 Rogers amp Chen 2013)
Chapter 1 Introduction and Review of Literature
38
The methylation of the miRNAmiRNA duplex is required to protect miRNAs
against the 3prime-end uridylation activity and subsequent degradation (Li et al 2005) In
the hen1 mutants accumulation of miRNAs is reduced to a lower level Also the
miRNAs appear to be heterogeneous in their sizes such that a ladder of bands rather
than a single species is observed for most miRNAs in the mutants (Li et al 2005)
MiRNA cloning and sequencing analyses revealed the presence of a number of U
residues at the 3prime-end of miRNAs in the hen1 mutants Moreover these nucleotides do
not correspond to the miRNAs in the pre-miRNAs indicating that these nucleotides are
added after the processing of the miRNAs from pre-miRNAs (Li et al 2005)
Uridylation of RNAs has also been proven to be correlated with RNA decay (Shen amp
Goodman 2004 Mullen amp Marzluff 2008) suggesting that uridylation attracts an
exonuclease which degrades miRNAs in plants
11344 MiRNA incorporation in to the RISC and its nuclear export
The methylated miRNAmiRNA duplexes undergo RISC assembly In this process
the miRNA of the duplexes is selectively incorporated into the RISC and the miRNA
appears to be removed and subsequently degraded (Hammond et al 2000 Hutvagner
amp Zamore 2002 Schwarz et al 2003 de Alba et al 2013 Rogers amp Chen 2013)
The ARGONAUTE 1(AGO1) proteins are core components of the RISC complex
They contain two structural domains the N-terminal PAZ domain that binds to the 3prime-
end of single-stranded RNAs and the C-terminal piwi domains that has a structure
similar to that of RNase H (Cerutti et al 2000 Parker et al 2004) Several AGO
proteins possess endonucleolytic activities within the PIWI domain and cleave target
mRNAs in the middle of the site complementary to miRNA (Liu et al 2004 Meister et
al 2004 Miyoshi et al 2005) Mutations in the AGO1 gene cause miRNA target
genes to be ectopically expressed and the levels of some miRNAs are reduced in the
mutants (Kidner amp Martienssen 2004 Vaucheret et al 2004 Kidner amp Martienssen
2005 Ronemus et al 2006) Moreover the phenotype of the AGO1 hypomorphic
alleles display defects in lateral organ polarity and leaf and flower morphology as
observed in the dcl1 mutants and null AGO1 alleles are embryonically lethal
supporting the close relationship between the AGO proteins and miRNA processing
(Kidner amp Martienssen 2004 Vaucheret et al 2004 de Alba et al 2013 Rogers amp
Chen 2013)
The production of miRNA miRNA duplexes occur in the nucleus while
Chapter 1 Introduction and Review of Literature
39
miRNA-RISC complexes are present in the cytoplasm where target mRNAs are
located This discordance of functional sites requires an export step during miRNA
biogenesis HASTY the Arabidopsis homolog of exportin-5 is thought to be involved
in miRNA nuclear export (Bollman et al 2003 Park et al 2005)The hasty mutant
exhibits pleiotropic defects in plant development and reduced accumulation of most
miRNAs as in the DCL1 or AGO1 mutants Howeverit is unclear whether the HASTY
proteins export the miRNA miRNA duplexes or the miRNA-RISC complexes in
plants
11345 Feedback regulation of miRNA biogenesis
MiRNA biogenesis is under feedback regulation such that the DCL1 and AGO1 genes
are themselves regulated by miRNAs DCL1 is itself a target of miR162 which leads to
the cleavage of the DCL1 mRNA (Xie et al 2003 de Alba et al 2013) Consistent
with this the levels of DCL1 mRNA are elevated in the mutants of miRNA processing
genes in which the miR162 abundance is reduced In addition there is a predicted
hairpin structure of miR838 in the 14th
intron of the DCL1 primary transcript
(Rajagopalan et al 2006) This prediction implies that DCL1-mediated processing
on this site can result in the cleavage of the DCL1 primary transcript into two truncated
fragments the N-terminal capped fragment and the C-terminal polyadenylated
fragment If this is the case it is possible that miRNA biogenesis competes with the
splicing event of the DCL1 pre-mRNA
The AGO1 is also regulated by miRNA There is a complementary site for
miR168 binding in the AGO1 mRNA and the transcript level of the AGO1 mRNA is
reduced through an mRNA cleavage mechanism (Vaucheret et al 2006 de Alba et al
2013 Rogers amp Chen 2013) indicating that the AGO1 mRNA levels are tightly
controlled by the levels of the AGO1 protein
1135 Mechanism of miRNA action
11351 MiRNA-directed mRNA cleavage
By forming the miRNA-RISC assembly miRNAs can direct the RISC to regulate gene
expression by two post transcriptional mechanisms mRNA cleavage or translational
repression (Bartel 2004 Jones-Rhoades et al 2006 Dalmay 2013) The degree
of miRNA-mRNA complementarity plays an important role for the
determination of the mechanism to be used A general rule is that while perfect or
Chapter 1 Introduction and Review of Literature
40
near-perfect complementarity induces mRNA cleavage central mismatches trigger
translational repression (Carrington amp Ambros 2003 Jones-Rhoades amp Bartel 2004)
Unlike most animal miRNAs plant miRNAs have near-perfect or perfect
complementarity to their targets and mRNA cleavage is assumed to be a predominant
mechanism by which miRNAs regulate gene expression in plants
In RNA cleavage mechanism miRNAs guide the AGO component of RISC to
cleave a single phosphodiester bond of the target mRNA within the miRNA-binding
site (Llave et al 2002b Tang et al 2003) The truncated fragments are then released
and degraded freeing the RISC to recognize and cleave another target mRNA This has
been validated by the detection of 3prime cleavage products that have 5prime ends that start at the
middle of the complementary regions The mRNA cleavage products are then further
degraded by other mechanisms The 3prime cleavage products of some miRNA targets are
degraded by the 5prime-3prime exonuclease XRN4 an Arabidopsis homolog of the major Yeast
mRNA degrading exonuclease Xrn1p It has been observed that the 3prime cleavage
products of some target mRNAs were accumulated in the xrn4 mutants (Souret et al
2004 Dalmay 2013) However the 3prime cleavage products of other miRNA targets do
not accumulate in the xrn4 mutants indicating that there are also XRN4 independent
mechanisms The 5prime cleavage products are typically untraceable by RNA gel blot
hybridization but can be detected by sensitive PCR based methods It has been found
that the 5prime cleavage products tend to obtain an oligo U tail at the 3prime ends This 3prime U tail
is correlated with shortening of the 5prime cleavage products from the 5prime end leading to the
conclusion that the 3prime uridylation causes 5 prime- 3 prime exonucleolytic decay of the 5prime cleavage
products (Shen amp Goodman 2004)
DCL1 HYL1 and SE interact with each other and are co-localized in the
nuclear bodies termed Dicing bodies or D-bodies in which some pri-miRNA shave
been found suggesting that D-bodies serve as a center for active miRNA processing
(Fang amp Spector 2007 Fujioka et al 2007 Song et al 2007) The miRNA
processing site appears to be distinct from the Cajal bodies that have previously been
found as a siRNA processing site (Pontes amp Pikaard 2008) The D-bodies include
SmD3 and SmB proteins that are found in the Cajal bodies However At collin an
additional marker for the Cajal bodies is not found in the D-bodies Moreover the D-
bodies are present in an At collin mutant lacking the Cajal bodies The pri-miRNAs of
miR171A and miR159A genes have been found to be co-localized with HYL1 and
Chapter 1 Introduction and Review of Literature
41
DCL1 in the D- bodies that are distinct from the Cajal bodies (Song et al 2007)
11352 MiRNA-directed translational repression
Translation repression is another mechanism exerted by plant miRNAs The regulatory
mechanism of miR172 which regulates flowering time and floral organ identity is
consistent with translation repression MiR172 over production does not affect the
transcript levels of APETALA2 (AP2) and AP2- like target mRNAs Instead the AP2
protein level is reduced in the miR172 over expressing mutant (Aukerman amp Sakai
2003 Chen 2004 Dalmay 2013) It was later shown that miR172 can also direct the
cleavage of the target mRNAs although the steady-state mRNA level is unchanged due
to feedback regulation (Schwab et al 2005 Jung et al 2007) It is clear that miR172
regulates its target genes primarily by translational repression rather than mRNA
cleavage Recently it has been reported that miR156157 and miR854 also regulate
their target genes through translational repression (Arteaga-Vazquez et al 2006
Gandikota et al 2007 Dalmay 2013) It was once thought that translational repression
is an exception to the rule However experimental evidence suggest a widespread
coexistence of translational repression and mRNA cleavage (Brodersen et al 2008)
1136 Regulatory roles of miRNAs in plant development
The miRNA target prediction has been a difficult task in order to obtain regulatory
targets without bringing in too many false predictions Plant growth and developmental
processes regulated by miRNAs are quite diverse and enormous Furthermore plant
miRNAs work cooperatively or antagonistically to establish a balanced regulation This
notion is well consistent with the findings that mutations in the regulators of miRNA
biogenesis transport and processing such as AGO1 DCL1 HYL1 HEN1 ABH1
and HASTY cause pleiotropic phenotypes (Mallory amp Vaucheret 2006 de Alba et al
2013) To date the roles of miRNAs have been mostly deduced from obtaining miRNA
over expressing transgenic plants or gain-of-function mutants in which miRNA-
resistant target genes are ectopically expressed
11361 Phase transition
Plants pass through a series of developmental phase transitions during their life span
beginning with seed germination continuing through vegetative phase change
reproductive phase change flowering initiation and finally seed production for the next
generation Because of their importance in understanding plant developmental
Chapter 1 Introduction and Review of Literature
42
processes genes regulating developmental transitions have been extensively studied
and underlying molecular mechanisms have been explored in various plant species
Majority of researches were mainly focused on vegetative phase change and
reproductive transition in Arabidopsis and Maize (Poethig 2003 Baurle amp Dean
2006) Particularly the mutations of the genes encoding various regulators of miRNA
biogenesis and transport such as HST SE and AGO exhibit altered juvenile to adult
vegetative phase change and vegetative to reproductive change (Hunter et al 2003
Peragine et al 2004 Vazquez et al 2004a Jones-Rhoades et al 2006 de Alba et al
2013)
Over-expression of the miR156a gene causes late flowering and delays
vegetative phase change (Wu amp Poethig 2006 Gandikota et al 2007 Wang et al
2008) It has been shown that miR156 negatively regulates a group of genes encoding
SQUAMOSA PROMOTER-BINDING PROTEIN-LIKE (Lewis et al 2007 de Alba et
al 2013 Rogers amp Chen 2013) transcription factors such as SPL3 SPL4 and SPL5
that promote flowering initiation In contrast transgenic plants over expressing
miR156-resistant SPL345 genes are early flowering Interestingly the endogenous
level of miR156 is temporally regulated In the early juvenile vegetative phase the
level of miR156 is very high but decreases rapidly before the onset of the adult juvenile
phase (Wu amp Poethig 2006)
The Arabidopsis early activation tagged (eat-D) mutant exhibits early flowering
with disrupted floral structure The mutant phenotypes are caused by over expression of
the miR172a-2gene (Aukerman amp Sakai 2003) MiR172 regulates a small group of
genes encoding APETALLA2 (AP2)-like transcription factors such as TARGET OF
EAT1 (TOE1) TOE2 SCHLAFMUTZE (SMZ) and SCHNARCHZAPFEN(SNZ)
TOE1 TOE2 SMZ and SNZ have been shown to participate in their productive phase
change by repressing a floral integrator FLOWERING LOCUS T (FT)(Jung et al
2007) Consistent with this toe1-2D 35STOE2 35SSMZ and 35SSNZ plants
exhibit late flowering (Jung et al 2007) MiR172 is also regulated temporally and
highly expressed in the adult vegetative phase both in Arabidopsis and maize (Chuck et
al 2007) Because the temporal expression patterns of miR156 and miR172 are
complementary to each other it has been suggested that the two miRNAs interact with
each other in regulating phase change (Chuck et al 2009 de Alba et al 2013) In
support of this view it has been shown that miR172 is down-regulated in the
Chapter 1 Introduction and Review of Literature
43
Corngrass1(Cg1) mutant in which miR156 is overproduced (Chuck et al 2007 de
Alba et al 2013) However there is no direct evidence for the direct linkage between
miR156 and miR172 It is also envisioned that miR156 and miR172 would not be
directly related For example miR156 regulates plastochron index that is the inverse of
leaf initiation rate and thus frequently used to analyze temporal patterns of plant
development In contrast miR172 does not affect plastochron (Wang et al 2008) The
down-regulation of miR172 in the maize miR156-over producing mutants would be
due to the prolonged juvenile vegetative phase in the mutants
Another miRNA that regulates flowering initiation is miR159 It regulates
MYB33 and MYB65 which are closely related to the barley GAMY B transcription
factor that responds to gibberellic acid (GA) The level of miR159 is elevated in GA-
treated seedlings but reduced in the GA biosynthetic ga1 mutant Transgenic plants
over producing miR159 exhibit delayed floral induction under short days (Achard et
al 2004) It is been suggested that MYB33 mediates GA signal to regulate LEAFY
(LFY) These observations indicate that miR159 plays an important role in the GA
signaling pathways that regulate proper floral induction under short days (Achard et al
2004)
11362 Floral development
Arabidopsis flowers consist of four distinct organs They arise in a characteristic
pattern within concentric rings called whorls from outside to inside four sepals four
petals six stamens and the central pistil consisting of two carpels Identities of the
floral organs are determined by three classes of regulatory genes classA classB and
class C genes (Krizek amp Fletcher 2005) The A and C class genes act antagonistically
to restrict their expression domains (Bowman et al 1991)
It is notable that miR172 also regulates floral architecture in addition to its role
in the timing of flowering initiation MiR172 inhibits the translation of the AP2 mRNA
(Chen 2004) Transgenic plants overproducing miR172 not only exhibit early
flowering but also show abnormal floral development which is quite similar to those
of the class A gene mutants such as the ap2 mutants (Aukerman amp Sakai 2003 de
Alba et al 2013) When the activity of the class A genes is inactivated that of the class
C genes such as AGAMOUS (AG) is elevated As a result the miR172- overproducing
transgenic plants exhibit no petals along with homeotic transformation of the sepals to
Chapter 1 Introduction and Review of Literature
44
the carpels (Aukerman amp Sakai 2003 Chen 2004)
Interestingly miR166 also regulates floral architecture The role of miR165166
in the regulation of meristem activity is closely linked to floral meristem formation and
organization (Zhao et al 2007 Miyashima et al 2013) Floral structure is severely
disrupted in the miR165166-overproducing mutants such as men1 and jba-1D in
which miR166 is overproduced (Williams et al 2005b Jung amp Park 2007) The men1
and jba-1D mutants have smaller gynoecium and reduced carpel number In an extreme
case the jba-1D homozygous plants lack visible carpels (Williams et al 2005b)
Similar phenotypes have been observed in the miR165 overproducers (Zhao et al
2007) It has been suggested that the phenotypes are possibly caused by altered AG
expression in the floral meristem (Jung amp Park 2007 Turchi et al 2013)
Other miRNAs also affect floral development Overproduction of miRNAs
involved in auxin signaling also affects floral architecture Transgenic plants
overproducing miR167 have defects in the formation of ovule and anther with reduced
fertility (Ru et al 2006) It has been shown that miR167 targets ARF6 and ARF8 that
play a role in gynoecium and stamen development and in regulating fertility (Ru et al
2006 Wu et al 2006) These observations suggest that auxin signals are also closely
related to floral organogenesis In addition transgenic plants over-producing miR164
and the cuc1cuc2 double mutants show fused sepals and reduced petals (Laufs et al
2004 Turchi et al 2013) suggesting that miR164 is also related to floral meristem
activity and boundary specification of the meristematic region
11363 Shoot apical meristem development
Shoot apical meristem comprises self-maintaining totipotent cells The shoot apical
meristem activity affects diverse aspects of plant developmental processes including
leaf development vascular development and lateral organ polarity (Bowman et al
2002) Because miR165166 plays a primary role in meristem formation
overproduction of miR165166 and multiple knockout mutants of its target genes
exhibit pleiotropic pheonotypes (McConnell et al 2001 Emery et al 2003 Kim et al
2005 Williams et al 2005b)
The targets of miR165166 include genes encoding the homeo domain-leucine
zipper (HD-ZIP) class III transcription factors such as PHABULOSA(PHB)
PHAVOLUTA (PHV) REVOLUTA(REV) ATHB15CORONA (CNA) and ATHB8
Chapter 1 Introduction and Review of Literature
45
PHB PHV and ATHB15 have overlapping functions in controlling meristem
development (Turchi et al 2013) Indeed the phb phv athb15 triple mutants have an
enlarged shoot apical meristem Consistent with this the miR166- overproducing men1
and jba-1D mutants exhibit enlarged shoot apical meristems (Kim et al 2005
Williams et al 2005b Turchi et al 2013) In the mutants the shoot apical meristems
are multiplied and large
The development of shoot apical meristems is also regulated by the WUSCHEL
(WUS)ndashCLAVATA (CLV) signaling pathway WUS positively regulates cell
proliferation In contrast CLV3 which is expressed in stem cells limits the WUS
expression and thus inhibits cell proliferation in the SAM (Sharma et al 2003)
Consistent with this notion WUS is expressed to a higher level in jba-1D
explaining the ectopic enlargement of the SAM in the mutant (Williams et al 2005)
While WUS is closely related to miR165166 in the shoot apical meristem
development the underlying molecular mechanisms are currently unclear It has been
observed that the miR166-over producing men1 plants have an arrested meristem
development (Jung amp Park 2007) In addition the heterozygous men1+ and the
homozygous men1 plants show different expression patterns of WUS and CLV3 It
appears that miR166 regulates WUS indirectly and influences the expressional balance
between WUS and CLV3 through a negative feedback loop (Jung amp Park 2007 de
Alba et al 2013)
The phv phb athb15 triple mutant has an enlarged shoot apical meristems the
rev phb phv mutant does not have functional shoot apical meristems (Prigge et al
2005) This distinction may be explained by the fact that while miR166 targets
primarily PHB PHV and ATHB15 miR165 targets all the five HD-ZIP III genes (Zhou
et al 2007) Consistent with this transgenic plants overproducing miR166 are distinct
from those overproducing miR165 The miR165-overproducing transgenic plants lack
functional shoot apical meristem and a pin-like structure is formed at the apical region
of the mutant These phenotypes are quite similar to rev phb phv triple mutant (Zhou et
al 2007 Liu et al 2013) The phenotypic distinction may because by the difference
of the cleavage efficiencies of the target mRNAs In accordance with this notion a few
35S miR166a transgenic lines exhibit no shoot apical meristems The men1
homozygous plants also show arrested meristem development (Jung amp Park 2007)
Chapter 1 Introduction and Review of Literature
46
MiR164 cleaves the CUC1 and CUC2 mRNAs as well as the NAC1 mRNA
CUC1 and CUC2 determines the organ boundary in the meristem Phenotypes of
transgenic plants that overproduce miR164 are similar to those of the cuc1cuc2 double
mutant that exhibits embryonic developmental defects such as cup-shaped cotyledons
and fused cotyledons (Laufs et al 2004) When a miR164-resistant CUC2 is
expressed the boundary domain is enlarged CUC also plays a role in embryonic shoot
meristem formation by regulating the SHOOT MERISTEMLESS gene It was therefore
concluded that miR164-mediated cleavage of CUC1 and CUC2 mRNAs determines the
sizes of the boundary domains in the shoot apical meristem and regulates separation of
the organs by restricting cell proliferation (Laufs et al 2004 de Alba et al 2013)
11364 Leaf development
The role of miR319 has been extensively studied in leaf development A dominant
jaw-D mutant overproducing miR319 shows uneven curved leaves with enlarged size
and five TCP genes which are closely related to the CINCINNATA (CIN) gene in
Antirrhinum majus are down regulated in the mutant suggesting that miR319 mediated
regulation of the TCP transcription factor genes are important for the final step of
leaf shaping (Palatnik et al 2003 Naz et al 2013) In addition to leaf shape and size
leaf polarity is also specified by miRNA MiR166-mediated cleavage of the HD-ZIPIII
mRNAs is a well to establish leaf polarity This discovery originates from the gain-of-
function mutants such as phb-1d and phv-1d wherein point mutations in the
complementary sites to miRNA helps the mRNA to evade or resist from miRNA-
mediated cleavage (McConnell et al 2001 Emery et al 2003 Naz et al 2013)
Plant leaves have a dorso-ventral polarity consisting of the adaxial (upper
surface) and abaxial (lower surface) sides The adaxial side is closer to the shoot
apical meristem Adaxial identity is specified by functionally redundant class III HD-
ZIP family genes such as PHB PHV and REV Ectopic expression of each genes
result in adaxialized leaves Dominant phb-1d and phv-1d mutants exhibit
transformation of the abaxial to adaxial fate and have rod-shaped leaves In contrast
miR165166-overproducing plants show pin-like cotyledons radicalized leaves and
abaxialized leaves (Chitwood et al 2007 Pattanayak et al 2013)
Leaf asymmetry is determined by miR166-mediated restriction of the
expression domains of the HD-ZIPIII genes The HD-ZIPIII genes are expressed in the
Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
23
Phenylalanine ammonia-lyase (PAL) catalyzes the first step of the phenylpropanoid
pathway leading to the synthesis of a wide range of chemical compounds including
flavonoids coumarins hydroxycinnamoyl esters and lignins (Hahlbrock amp Scheel
1989) Recently the full length cDNA and corresponding genomic sequences of PAL
from C canephora was isolated characterized and functionally validated (Mahesh et
al 2006 Parvatam et al 2006) This has opened up new possibilities for manipulating
the level of the PAL enzyme in coffee which in turn can be useful for improvement of
cup quality and manipulating antioxidant properties in coffee
1106 Fruit Ripening
Uniformity during fruit ripening is decisively related to cup quality in coffee and
consequently to the value of the product Fruits at the ideal ripening stage produce the
best organoleptic characteristics for coffee The presence of over ripened or green fruits
changes the acidity the bitterness and consequently the cup quality In order to
maximize uniform ripening of coffee fruits it is essential to control the action of genes
involved in the last step of maturation process Ethylene is known to trigger ripening
and increasing ethylene biosynthesis is associated with various stages of ripening
process (Protasio et al 2005) To control coffee fruit maturation two of the major
genes involved in ethylene biosynthesis namely ACC synthase and ACC oxidase
have been cloned (Protasio et al 2005 Neupane et al 1999) Introduction of the ACC
oxidase gene in antisense orientation have been achieved in both C arabica and C
canephora The effect of the transgene on ethylene production and fruit maturation has
yet to be reported The inhibition of genes downstream to the initial ethylene burst is
also an option to control coffee fruit maturation (Ribas et al 2006)
111 RNA interferenceRNAi
The regulation of gene expression at post transcriptional level has recently attracted
much attention because of the discovery of the phenomenon called RNA interference
(RNAi) RNAi based silencing techniques are more efficient than antisense mediated
gene silencing (Wesley et al 2001) The effect was first observed in plants wherein
silencing was observed when some copies of a transgene was in the forward orientation
with respect to the promoter and some in the reverse orientation This resulted in the
formation of double stranded RNA in the system The phenomenon in which
experimentally introduced double stranded RNA (dsRNA) leads to the loss of
expression of the corresponding cellular gene by sequence specific RNA degradation
Chapter 1 Introduction and Review of Literature
24
was termed as RNA interference or RNAi The protective effect of RNA silencing in
reducing virus infectivity supports the view that PTGS has evolved as a mechanism to
defend plants against virus infection and also to moderate the possible deleterious
genome-restructuring (insertional) activity of virus-like mobile genetic elements eg
retrotransposons A growing body of biochemical and genetic data has further
demonstrated that plants animals and yeasts share related mechanisms of specific
degradation of RNAs in which double-stranded forms of RNA are initiator molecules
(Brenstein et al 2001)
The key insight into the process of PGTS was provided by the experiments
conducted by Hamilton amp Baulcombe (1999) who identified the product of RNA
degradation as small RNA (siRNA) species of ~25nt of both sense and antisense
polarity SiRNA are formed and accumulate as double stranded RNA molecules of
defined chemical structures Both genetic and biochemical approaches were undertaken
to understand the basis of silencing Genetic screens were carried out in fungus
Neurospora carassa the alga Chalmydomonas reinhardtii and plant Arabidopsis
thaliana to search for detective mutants in PTGS
A combination of results obtained from several in vivo and in vitro experiments
have gelled into a two step mechanistic model for RNAiPTGS The first step referred
to as the RNAi initiating step involves binding of the RNA nucleases to a larges
dsRNA and its cleavage into discrete ~21-25nt RNA fragments This is ATP-
dependent reaction which involved RNase III - type endonucleases which make
staggered cuts in both strands of the dsRNA leaving 3prime overhang of 2 nucleotides with
5prime- phosphate 3prime-hydroxyl and no modification in the sugar phosphate backbone
(Hamilton amp Baulcombe 1999 Elbashir et al 2001) SiRNAs are the direct products
of dsRNA cleavage by the multi-domain RNase III enzyme DICER (Figure 16)
(Brenstein et al 2001 Karthikeyan et l 2013)
In the second step or commonly known as the effector step the double stranded
siRNAs produced in the first step bind to an RNAi specific protein complex to form a
RISC The complex undergoes activation in the presence of ATP so that the antisense
component of the unwound siRNA becomes exposed and allows the RISC to perform
the downstream RNAi reaction
Chapter 1 Introduction and Review of Literature
25
Several enzymes are involved in RNAi based on their role genes that encode for them
are classified into RNAi initiators RNAi effectors RNA dependent RNA polymerases
and silencing genes Two C elegans genes rdeI and rde4 (rde stands for lsquo RNAi
deficientrsquo) are believed to be involved in the initiation step of RNAi The C elegans
rde1 gene is a member of a large family of genes and is homologous to the Neurospora
qde2 (qde stands for ldquoquelling deficientrdquo) and Arabidopsis AGO1 (AGO stands for
agronaute AGO1 was previously identified to be involved in Arabidopsis
development) Although the functions of these genes are not clear a mammalian
member of RDE1 family has been identified as a translation initiation factor (Thakur
2005) Important genes for the effector step of PTGS in C elegans are rde2 and mut7
genes These genes were identified from heterozygous mutant worms that were unable
to transmit RNAi to their homozygous offsprings Worms with mutated rde2 or mut 7
genes show defective RNAi mut-7 gene encodes a protein with homology to the
nuclease domains of RNAase D and a protein implicated in Werner syndrome (a rapid
ageing disease in humans) (Grishok et al 2001 Kamath-Loeb et al 2012s)
Neurospora qde-1 Arabidopsis SDE-1SGS-2 and C elegans ego-1 appear to encode
RNA dependent RNA polymerase (RdRPs) It assumed that this is a proof that an RdRp
activity is required for RNAi Certainly the existence of an RdRp might explain the
remarkable efficiency of dsRNA induced silencing if it amplifies either the dsRNA
prior to cleavage or the siRNAs directly (Muers 2013) In C elegans ego-1 mutants
(ego stands for lsquoenhancer of glp-1) RNAi functions normally in somatic cells but is
defective in germline cells where ego-1 is primarily expressed In Arabidopsis SDE-
1SGS-2 mutants (SGS stands for suppressor of gene silencing) siRNA are produced
when dsRNA is introduced via an endogenously replicating RNA virus but not
introduced by a transgene It has been proposed that perhaps the viral RdRP is
substituting for the Arabidopsis enzyme in these mutants Random degradative PCR
model suggests that an RdRP uses the guide strand of an siRNA as a primer for the
target mRNA generating a dsRNA substrate for Dicer and thus more siRNAs
Several genes controlling RNA silencing in plants have been identified through
genetic screens of Arabidopsis mutants impaired in transgene induced RNA silencing
They encode a putative RNA-dependent RNA polymerase (SGS2SDE1) a coiled
protein (SGS3) a protein containing PAZ and Piwi domains (AGO1) and an RNA
helicase (SDE3) The putative SGS2SDE1 of Neurospora is related to QDE-1 and
Chapter 1 Introduction and Review of Literature
26
EGO-1 of C elegans whereas PAZPiwi protein AGO is related to QDE-2 of
Neurospora RDE-1of C elegans RNA helicase SDE3 is related to SMG-2 of C
elegans and Mut-6 of Chlamydomonas MUT-7 gene of C elegans encodes a protein
similar to Rnase D whereas the Drosophila DICER gene encode a protein similar to
RNase III An Arabidopsis ortholog of DICER gene has been identified called CAF
SIN1 SUS1(Thakur 2005 Poethig 2009)
Since the RNAi machinery is present constitutively within the eukaryotic cell it
is important to explore the metabolic advantages that are accorded by the RNAi related
proteins during the intrinsic normal growth of cells and development of organisms The
natural RNAi machinery not only keeps the mobile transposable elements from
disrupting the integrity of the genomes as suggested by the analysis of model organisms
like A thaliana C elegans D melanogaster (Tabara et al 1999 Wu-Scharf et al
2000 Aravin et al 2001 Hamilton et al 2002 Lisch 2009) but also participates in
development of organisms Genetic defects in Celegans RNAi genes ego1 and dicer
cause known specific developmental errors (Grishok et al 2000 Grishok et al 2001
Knight amp Bass 2001 Hall et al 2013) Similarly the Argonaute family of genes of A
thaliana (especially the ZWILLE proteins) is also responsible for plant architecture and
meristem development (Carmell et al 2002 Hamant and Pautot 2010) and the Dicer
homologue of A thaliana CAF1 is required for embryo development (Golden et al
2002 Nakano et al 2011) Thus genetic evidence illustrates the role of the RNAi
machinery as a controller of development related genes The mechanistic details of
these developmental processes are beginning to emerge
Chapter 1 Introduction and Review of Literature
27
Figure 16 Mechanism of gene silencing induced double stranded RNA The dicer enzyme to
produce siRNAs cleaves the RNA The siRNA generated bind to the nuclease complex
RISC The antisense component in the siRNA in the RISC guides the complex towards
the cognate mRNA resulting in endonucleolytic cleavage of the mRNA
Chapter 1 Introduction and Review of Literature
28
112 MicroRNA or MiRNA
In 1991 Ambros and co-workers first isolated a lin-4 mutant of Celegans which was
arrested at the first larval stage (Lee et al 1993) Later on the let-7 mutation was
isolated in the same system which was responsible for development through the fourth
larval stage Both lin-4 and let-7 encode short 22-nucleotide mature RNAs and were
called short temporal RNA because they control the temporal development program of
C elegans The mature lin-4 RNA defines (negatively regulates) the mRNA expression
of the lin-14 and lin-28 hetrochronic genes with the antisense mediated repression
mechanism of translation initiation and thus specifies the fate of cells during the first
three larval stages Recent studies have revealed that the short temporal RNAs are
actually members of a group of tiny RNAs (21to 28 nucleotides) called the micro-
RNAs (miRNA) (Bartel 2009) isolated members of which could easily run to a few
thousand which includes both those were identified computationally and by deep
sequencing Some of the components of the RNAi machinery have also been clearly
established as the effector proteins for the maturation of miRNAs
113 MicroRNAs in plants
1131 Discovery
MicroRNAs have been discovered by three basic approaches Direct cloning forward
genetic direct cloning and bioinformatic prediction followed by experimental
validation The most direct method of miRNA discovery is to isolate and clone small
RNAs from biological samples and several groups have used this approach to identify
plant miRNAs (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002 Xie et al 2003 Sunkar et al 2005 Williams et al 2013) The cloning methods
adapted were similar to those used to identify large numbers of animal miRNAs
(Lagos-Quintana et al 2001 Lau et al 2001 Aravin amp Tuschl 2005) which involve
isolating small RNAs followed by oligonucleotide adapter ligation reverse
transcription amplification and sequencing Some protocols incorporate methods to
enrich for Dicer cleavage products (ie molecules with 5prime-phosphates and 3prime-
hydroxyls) and to concatemerize the short cDNAs so that several may be identified in a
single sequencing read (Lau et al 2001 Llave et al 2002a Jagadeeswaran amp Sunkar
2013)The initial cloning experiments in Arabidopsis identified 19 miRNAs belonging
to 15 families (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002) although hundreds of other small RNAs such as siRNAs and RNA degradation
Chapter 1 Introduction and Review of Literature
29
fragments were also cloned through the direct cloning methods To select miRNAs
from the pool of small RNAs secondary structures of the RNA molecules were
examined in compliance with the acknowledged structural features of miRNAs
(Ambros et al 2003) The secondary structures were validated using several ad-hoc
software tools such as the Mfold or RNAfold programs (Reeder et al 2006) Finally
the existence of mature miRNAs was determined by RNA gel blot hybridization These
direct cloning methods were successful in isolating many miRNAs in Arabidopsis
(Reinhart et al 2002 Sunkar amp Zhu 2004) Rice (Sunkar et al 2005) Poplar (Lu et
al 2005) moss (Arazi et al 2005) and Tobacco (Billoud et al 2005)
Although miRNAs were first discovered through forward genetic screens in
worms (Lee et al 1993 Reinhart et al 2000)only few A thaliana miRNA gene
families have been discovered by this method (Chen 2008) in plants MiRNA
involvement in plant mutant phenotypes were not properly inferred till the cloning
experiments established that plant genomes contain numerous miRNAs (Park et al
2002 Reinhart et al 2002 Rhoades et al 2002) MiRNA loss-of-function allele has
been identified by forward genetic screens early extra petala1 is caused by a
transposon insertion ~160bp upstream of the predicted miR164c stem loop resulting in
flowers with extra petals (Baker et al 2005 Irish 2008) The fact that loss-of-function
miRNA mutants have been rarely discovered using forward genetics reflects small
target size for mutagenesis coupled with redundancy in nearly all evolutionarily
conserved plant miRNAs which are encoded by gene families (Table16) Family
members are likely to have overlapping functions buffering against loss at any single
miRNA locus Over expression screens can circumvent redundancy limitations At least
four plant miRNAs miR319 (also known as miR-JAW) miR172 (also known as EAT)
and miR166 were isolated in over expression screens for dominant mutants with
developmental abnormalities (Aukerman amp Sakai 2003 Palatnik et al 2003 Kim et
al 2005 Williams et al 2005b Schommer et al 2012) Mutations in miRNA target
sites which can prevent the entire family of miRNAs from repressing a target gene can
also circumvent redundant functions of miRNA family members
The dominant mutations in the HD-ZIP genes PHB PHV and REVOLUTA
(REV) in Arabidopsis and ROLLEDLEAF1 (RLD1) in Maize result in adaxialization of
leaves andor Vasculature (McConnell amp Barton 1998 McConnell et al 2001 Nelson
et al 2002 Zhong amp Ye 2004 Allen amp Millar 2012) and are all caused by mutations
Chapter 1 Introduction and Review of Literature
30
in miR166 complementary sites (Rhoades et al 2002 Emery et al 2003 Jones-
Rhoades amp Bartel 2004 Mallory et al 2004 Zhong amp Ye 2004)
In both plants and animals cloning was the initial means of large-scale
miRNA discovery (Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Reinhart et al 2002 Mendes et al 2012) However cloning is biased toward
RNAs that are expressed highly and broadly MiRNAs expressed at low levels or
only in specific cell types or in response to certain environmental stimuli were more
difficult to clone Sequence based biases in cloning procedures might also cause
certain miRNAs to be missed Because of these limitations bioinformatics approaches
to identify miRNAs have provided a useful complement to cloning
A straightforward use of bioinformatics has been carried out to find homologs
of known miRNAs both within the same genome and in the genomes of other species
(Pasquinelli et al 2000 Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Mendes et al 2009) A more difficult challenge is to identify miRNAs
unrelated to previously known miRNAs This was first accomplished for
vertebrate nematode and fly miRNAs using algorithms that search for conservation
of sequence and secondary structure (ie miRNA stem-loop precursors) between
species searching for patterns that are characteristic of miRNAs (Lai et al 2003 Lim
et al 2003 Lim et al 2005)
Most of the methods identified numerous miRNAs but are necessarily more
relaxed in terms of allowed structures Some approaches take advantage of the high
complementarity of plant miRNAs to target messages implementing the requirement
that the candidate has conserved complementarity to mRNAs (Jones-Rhoades amp Bartel
2004 Adai et al 2005) This additional filter has been useful for distinguishing
authentic plant miRNAs from false positives which later extended to mammalian
miRNA gene prediction (Xie et al 2005 Mendes et al 2012)
Another criteria used by most algorithms in the filters is evolutionary
conservation In plants mature miRNA sequences are conserved to a higher degree
than their precursor sequences For example Arabidopsis and rice diverged more than
130 million years ago (Friis et al 2004) but some miRNAs are highly conserved
in these plant species (Reinhart et al 2002) Furthermore various parameters for the
secondary structures such as free energy the number of paired residues within a
Chapter 1 Introduction and Review of Literature
31
miRNA or the number and size of bulges are also used to validate the predictions
(Jones-Rhoades amp Bartel 2004 Wang et al 2004 Adai et al 2005 Mendes et al
2012)
1132 Conserved microRNAs in Plants
Till date cloning genetics and bioinformatics screens have resulted in annotation of
approximately 184 potential Arabidopsis miRNAs (miRBase release 13) These
miRNAs seem to arise through gene duplication and subsequent diversification and
represent approximately 100 families (Maher et al 2006 Axtell 2013) Twenty one
families represented by 92 genes are clearly conserved in species beyond Arabidopsis
(Table 15) The presence of these miRNA families in other sequenced plant genomes
indicates that different plant species have an evolutionarily fluid set of miRNAs
(Willmann amp Poethig 2007) Recent studies on mosses one of the most ancient land
plants have shown that at least 14 miRNA families are conserved in eudicots and
mosses and therefore it appears that plant miRNAs share a common ancient origin
(Arazi et al 2005 Axtell amp Bartel 2005 Talmor-Neiman et al 2006 Zhang et al
2006 Axtell 2013) The number of members per family in a genome ranges from 1-32
with the exception of miR-430 which is represented by a cluster of ~80 loci in zebra
fish (Giraldez et al 2005)
In plants the number of members in each family correlates among examined
species certain families contain numerous members in all three species (eg miR156
miR166 miR169) whereas others consistently contain fewer genes (eg miR162
mir168 miR394) (Table 16) Although it is still unclear why certain miRNA are
encoded by more than one gene in plants (eg 12 genes encoding miR156) this
correlation might suggest functional significance of various miRNA family sizes Most
annotated plant miRNAs are conserved throughout flowering plants while a few
miRNAs are found only in a single sequenced genome and thus they could be
evolutionarily lsquoyoungrsquo miRNAs For example Arabidopsis has several miRNA
families that are not found in poplar or rice suggesting that miRNAs could be
generated continuously during evolution (Allen et al 2004a Rajagopalan et al
2006 Fahlgren et al 2007 Axtell 2012 de Alba et al 2013) Consistent with the
notion that non conserved miRNAs are of a more recent evolutionary origin these
miRNAs are mainly encoded by a single locus in the genome Within each family the
mature miRNA is always located in the same arm
Chapter 1 Introduction and Review of Literature
32
Table 16 MicroRNA families conserved in plants
miRNA
family
Arabidopsis Oryza Populus
miR156 12 12 11 miR159319 6 8 15 miR160 3 6 8 miR162 2 2 3 miR164 3 5 6 miR166 9 12 17 miR167 4 9 8 miR168 2 2 2 miR169 14 17 32 miR171 4 7 10 miR172 5 3 9 miR390 3 1 4 miR393 2 2 4 miR394 2 1 2 miR395 6 19 10 miR396 2 5 7 miR397 2 2 3 miR398 3 2 3 miR399 6 11 12 miR408 1 1 1 miR403 1 0 2 miR437 0 1 0 miR444 0 1 0 miR445 0 9 0 Total 92 127 169
All known miRNA families that are conserved between more than one plant species are listed
together with the number of genes identified in the sequenced genomes Rice miRNA families that have orthologs in maize but do not appear to have orthologs in the eudicots (Arabidopsis and Populus) are marked with an asterisk The following families contain miRNA genes annotated with
more than one number miR156 (miR156 and miR157) miR159319 (miR159 and miR319) miR166 (miR165 and miR166) miR171 (miR170 and miR171) and miR390 (miR390 and miR391)
of the stem loop (5prime- or 3prime) as it would be expected if the genes carry common ancestry
(Figure 17) The sequence of the mature miRNA and to some extent the segment on
the opposite arm of the hairpin to which it pairs is highly conserved between the
members of the same family (both within and between the species) while the sequence
secondary structure and length of the intervening ldquolooprdquo region can be highly divergent
within the same family
The patterns of paring and non pairing nucleotides are often conserved between
homologous miRNA stem loops from different species (Figure 17) The significance of
conserved mismatches is opined to guide DCL1 to cleave at the appropriate positions
along the stem loop
Chapter 1 Introduction and Review of Literature
33
Figure 17 Representative stem loop of miR164 Segments corresponding to the mature miRNAs
are shown in red (Adapted from Rajagopalan et al 2006)
Although many miRNAs are highly conserved in plants the degree of conservation in
most cases is quite low between plants and animals one exception is miR854 A recent
study has revealed that miR854 is conserved between Arabidopsis and animals and that
the Arabidopsis miR854 differs from the human miR854 only by a single nucleotide
(Arteaga-Vazquez et al 2006 Axtell 2012 de Alba et al 2013) The Arabidopsis
miR854 has multiple binding sites in the 3prime-untranslated region (UTR) of
OLIGOURIDYLATE-binding-PROTEIN mRNA 1bA (UBP1b) and represses the
translation of the UBP1b mRNA The animal miR854 is also complementary to a site in
the 3prime-UTR of the UBP1b homolog in animals However it is currently unclear how the
animal miR854 regulates its target mRNA Another distinction between plant and
animal miRNAs is the genomic organization of the miRNA genes While plant miRNA
genes are located predominantly in the intergenic regions animal miRNA genes tend to
localize in the intragenic regions both in the introns or exons of protein coding genes
Moreover miRNA genes are often clustered within a single precursor transcript
whereas individual plant miRNAs are derived from individual precursor RNAs (Bartel
Chapter 1 Introduction and Review of Literature
34
2004 Bonnet et al 2004 Kim 2005 Vaucheret et al 2006 Marco et al 2013)
1133 Non-conserved miRNA and challenges in annotation
Most miRNAs are conserved throughout flowering plants (Table 15) but many more
were found only in a single sequenced genome thus considered to be of a more recent
evolutionary origin The extended homology between few non conserved miRNAs
precursors and their target genes provides strong evidence that these relatively ldquoYoungrdquo
miRNAs arose from the duplication of the target gene segments (Allen et al 2004)
Several non-conserved miRNAs including miR161 miR163 miR173 miR447
miR475 and miR476 are known to direct cleavage of target transcripts (Allen et al
2004a Adai et al 2005 Sunkar et al 2005 de Alba et al 2013) It is difficult to
confidently predict targets for many because it is not possible to use complementary
site as a filter against false-positive target predictions It is also difficult to
confidently annotate non-conserved miRNAs as miRNA rather than siRNAs The
established minimal standard for miRNA annotation is a small RNA with detectable
expression and the potential to form a stem-loop when joined to flanking genomic
sequence (Kasschau et al 2003) In practice these requirements are too loose to
definitively categorize many small RNAs cloned from plants Many plant siRNAs are
detectable on blots (Vazquez et al 2004b) and hundreds of thousands of non-
miRNA genomic sequences can be predicted to fold into secondary structures that
resemble structures of plant miRNA precursors (Jones-Rhoades amp Bartel 2004)
Therefore without conservation of both sequence and secondary structure it is
difficult to be confident that a given cloned RNA originated from a stem-loop (ie is a
miRNA) rather than from a double-stranded RNA (ie is a siRNA) In fact many of
the thousands o f small RNAs cloned from Arabidopsis (Gustafson et al 2005) would
probably meet the literal requirements for annotation as miRNAs A few of these
sequences might be miRNAs but others that meet the literal criteria probably are not
so The challenges of annotating non conserved small RNAs were evidenced by
three related small silencing RNAs that were originally annotated as miRNAs
that turned out to be trans-acting siRNAs (tasiRNAs)(Kanno et al2013)
Although most miRNAs that are broadly conserved among flowering plants are
now being identified and experimentally validated (Table 16)(Jones-Rhoades amp
Bartel 2004) the challenges and ambiguities for classifying non-conserved miRNAs
prevent meaningful estimates of the total number of miRNA genes in Arabidopsis and
Chapter 1 Introduction and Review of Literature
35
other plant genomes It is possible that miRNAs might have escaped detection
because they are non-conserved and expressed in specific tissues or conditions and can
only be identified by using high- throughput sequencing
1134 MicroRNA biogenesis
11341 Transcription of microRNA precursor
Plant miRNAs are primarily found in the genomic regions that are not associated with
the protein coding genes (Reinhart et al 2002) and it appears that most if not all plant
miRNAs are produced from their own transcriptional units Plant miRNA genes are
occasionally clustered near each other in the genome suggesting transcription of
multiple miRNAs from a single primary transcript [eg the miR395 cluster (Grishok
et al 2001 Jones-Rhoades amp Bartel 2004b)] but this polycistronic arrangement of
miRNA genes appears far less frequently in plants than in animals (Bartel 2004)
Northern blot EST and mapping evidence indicate that plant miRNA primary
transcripts (also known as pri-miRNAs) as in animals are longer than needed to
encompass the miRNA stem-loops (Palatnik et al 2003 Jones-Rhoades amp Bartel
2004b) At least some of these pri-miRNA transcripts appear to be spliced
polyadenylated (Kurihara amp Watanabe 2004) and capped (Xie Z et al 2005) Two
rice miRNAs are contained within transcripts that contain exon junctions within the
presumptive stem-loop precursor implying that in these cases splicing is a prerequisite
for Dicer recognition (Sunkar et al 2005) Plant pri-miRNAs are over 1kb in length
and they are usually preceded by typical TATA box motifs They can also undergo
canonical splicing polyadenylation and capping All these observations indicates RNA
polymerase II is probably responsible for transcribing most plant miRNAs (Xie Z et
al 2005) as appears to be the case for many animal miRNAs Relatively little is
known about the regulation of miRNA transcription in plants but there is no reason
to suspect that this regulation would differ from that of protein-coding transcripts as
the bioinformatics analysis of the sequence upstream of the transcriptional start
sites of the miRNA genes showed the presence of putative binding motifs of
number of known transcription factors (Megraw et al 2006 Sethupathy 2013)
11342 MicroRNA processing and export
In plants maturation of miRNA is a step wise process A miRNA gene is transcribed to
pri-miRNA which is much longer than the pre-miRNA possessing the characteristic
stem-loop structure Like transcription of most protein-coding genes the miRNA
Chapter 1 Introduction and Review of Literature
36
transcription is processed by RNA polymerase II (Pol II) enzymes and the pri-
miRNAs are 5prime-capped and 3prime-polyadenylated (Lee et al 2004 Xie Z et al 2005)
Mature miRNAs are produced from pri-miRNAs through at least two sequential
processing steps by RNase III-type endonucleases In animals pri-miRNAs are first
processed at the bottom portion of the stem by Drosha a nuclear-localized RNase III
enzyme to liberate pre-miRNAs The pre-miRNAs are then exported to the cytoplasm
with the help of exportin-5The pre-miRNAs are further processed by Dicer another
RNaseIII enzyme to a duplex consisting of the miRNA and the antisense strands
(miRNA) Since plants do not have any Drosha homologs the two sequential
processing steps seem to be carried out by DCL1 a Dicer homolog in the nucleus
(Kurihara amp Watanabe 2004 de Alba et al 2013 Rogers amp Chen 2013)
The processing of pri-miRNAs to pre-miRNAs by DCL1 is assisted by two
other proteins HYPONASTY LEAVES1 (HYL1) and SERRATE (SE) HYL1 belongs to a
family of double-stranded RNA (dsRNA) binding protein and interacts with DCL1 in
vitro and in vivo (Lu amp Fedoroff 2000 Hiraguri et al 2005 de Alba et al 2013
Rogers amp Chen 2013) Loss-of-function mutations in the HYL1 gene result in
decreased accumulation of miRNAs and concomitant accumulation of pri-miRNAs
(Han et al 2004 Vazquez et al 2004a Kurihara et al 2006) SE a C2H2 zinc finger
protein interacts with DCL1 and HYL1 and plays a role in the processing of pri-
miRNAs to pre-miRNAs (Lobbes et al 2006 Yang L et al 2006) In addition the
nuclear cap-binding complex proteins CBP20 and CBP80 that are known as ABA
HYPER SENSITIVE 1(ABH1) have been shown to be required for proper miRNA
processing (Gregory et al 2008 Laubinger et al 2008) Recently it has been
demonstrated that DCL1 can release 21-nt RNAs from dsRNA as well as from
synthetic miR167 precursor RNAs in vitro However this cleavage is error prone and
inefficient in the absence of HYL1 and SE which accelerate the rate of DCL1-mediated
cleavage and promote accurate processing of miRNAs (Dong et al 2008 Rogers amp
Chen 2013)
11343 Methylation of the miRNAmiRNAduplex
After the miRNAmiRNAduplexes are released from the pre-miRNAs by the DCL1
activities the duplexes are methylated at the 2primeOH of the3prime-ends by HUA ENHANCER1
(HEN1) a small RNA methyltransferase (Yu et al 2005 Yang Z et al 2006 Rogers
amp Chen 2013 ) This step which is distinct from the RNase III-mediated processing
Chapter 1 Introduction and Review of Literature
37
step distinguishes the biogenesis of plant miRNAs from that of animal miRNAs
Mutations in the HEN1 gene were first isolated in a morphological screening for floral
development although the HEN1 mutants display pleiotropic phenotypes similar to the
developmental defects observed in the DCL1 mutants (Chen et al 2002 Park et al
2002 Rogers amp Chen 2013)
Figure 18 Biogenesis of microRNA
A Biogenesis of plant MicroRNA B Biogenesis of metazoananimal MicroRNA
The miRNA (red) is incorporated into RISC and the miRNA(blue) in degraded
This enzyme has two dsRNA-binding domains and nuclear localization signal It
is also conserved in fungi and is required for the processing of siRNAs as well as of
miRNAs (Park et al 2002 Boutet et al 2003 Xie et al 2003) Although the HEN1
protein is localized to the nucleus its methylation activity in the cytoplasm cannot be
ignored (Xie et al 2004 Rogers amp Chen 2013)
Chapter 1 Introduction and Review of Literature
38
The methylation of the miRNAmiRNA duplex is required to protect miRNAs
against the 3prime-end uridylation activity and subsequent degradation (Li et al 2005) In
the hen1 mutants accumulation of miRNAs is reduced to a lower level Also the
miRNAs appear to be heterogeneous in their sizes such that a ladder of bands rather
than a single species is observed for most miRNAs in the mutants (Li et al 2005)
MiRNA cloning and sequencing analyses revealed the presence of a number of U
residues at the 3prime-end of miRNAs in the hen1 mutants Moreover these nucleotides do
not correspond to the miRNAs in the pre-miRNAs indicating that these nucleotides are
added after the processing of the miRNAs from pre-miRNAs (Li et al 2005)
Uridylation of RNAs has also been proven to be correlated with RNA decay (Shen amp
Goodman 2004 Mullen amp Marzluff 2008) suggesting that uridylation attracts an
exonuclease which degrades miRNAs in plants
11344 MiRNA incorporation in to the RISC and its nuclear export
The methylated miRNAmiRNA duplexes undergo RISC assembly In this process
the miRNA of the duplexes is selectively incorporated into the RISC and the miRNA
appears to be removed and subsequently degraded (Hammond et al 2000 Hutvagner
amp Zamore 2002 Schwarz et al 2003 de Alba et al 2013 Rogers amp Chen 2013)
The ARGONAUTE 1(AGO1) proteins are core components of the RISC complex
They contain two structural domains the N-terminal PAZ domain that binds to the 3prime-
end of single-stranded RNAs and the C-terminal piwi domains that has a structure
similar to that of RNase H (Cerutti et al 2000 Parker et al 2004) Several AGO
proteins possess endonucleolytic activities within the PIWI domain and cleave target
mRNAs in the middle of the site complementary to miRNA (Liu et al 2004 Meister et
al 2004 Miyoshi et al 2005) Mutations in the AGO1 gene cause miRNA target
genes to be ectopically expressed and the levels of some miRNAs are reduced in the
mutants (Kidner amp Martienssen 2004 Vaucheret et al 2004 Kidner amp Martienssen
2005 Ronemus et al 2006) Moreover the phenotype of the AGO1 hypomorphic
alleles display defects in lateral organ polarity and leaf and flower morphology as
observed in the dcl1 mutants and null AGO1 alleles are embryonically lethal
supporting the close relationship between the AGO proteins and miRNA processing
(Kidner amp Martienssen 2004 Vaucheret et al 2004 de Alba et al 2013 Rogers amp
Chen 2013)
The production of miRNA miRNA duplexes occur in the nucleus while
Chapter 1 Introduction and Review of Literature
39
miRNA-RISC complexes are present in the cytoplasm where target mRNAs are
located This discordance of functional sites requires an export step during miRNA
biogenesis HASTY the Arabidopsis homolog of exportin-5 is thought to be involved
in miRNA nuclear export (Bollman et al 2003 Park et al 2005)The hasty mutant
exhibits pleiotropic defects in plant development and reduced accumulation of most
miRNAs as in the DCL1 or AGO1 mutants Howeverit is unclear whether the HASTY
proteins export the miRNA miRNA duplexes or the miRNA-RISC complexes in
plants
11345 Feedback regulation of miRNA biogenesis
MiRNA biogenesis is under feedback regulation such that the DCL1 and AGO1 genes
are themselves regulated by miRNAs DCL1 is itself a target of miR162 which leads to
the cleavage of the DCL1 mRNA (Xie et al 2003 de Alba et al 2013) Consistent
with this the levels of DCL1 mRNA are elevated in the mutants of miRNA processing
genes in which the miR162 abundance is reduced In addition there is a predicted
hairpin structure of miR838 in the 14th
intron of the DCL1 primary transcript
(Rajagopalan et al 2006) This prediction implies that DCL1-mediated processing
on this site can result in the cleavage of the DCL1 primary transcript into two truncated
fragments the N-terminal capped fragment and the C-terminal polyadenylated
fragment If this is the case it is possible that miRNA biogenesis competes with the
splicing event of the DCL1 pre-mRNA
The AGO1 is also regulated by miRNA There is a complementary site for
miR168 binding in the AGO1 mRNA and the transcript level of the AGO1 mRNA is
reduced through an mRNA cleavage mechanism (Vaucheret et al 2006 de Alba et al
2013 Rogers amp Chen 2013) indicating that the AGO1 mRNA levels are tightly
controlled by the levels of the AGO1 protein
1135 Mechanism of miRNA action
11351 MiRNA-directed mRNA cleavage
By forming the miRNA-RISC assembly miRNAs can direct the RISC to regulate gene
expression by two post transcriptional mechanisms mRNA cleavage or translational
repression (Bartel 2004 Jones-Rhoades et al 2006 Dalmay 2013) The degree
of miRNA-mRNA complementarity plays an important role for the
determination of the mechanism to be used A general rule is that while perfect or
Chapter 1 Introduction and Review of Literature
40
near-perfect complementarity induces mRNA cleavage central mismatches trigger
translational repression (Carrington amp Ambros 2003 Jones-Rhoades amp Bartel 2004)
Unlike most animal miRNAs plant miRNAs have near-perfect or perfect
complementarity to their targets and mRNA cleavage is assumed to be a predominant
mechanism by which miRNAs regulate gene expression in plants
In RNA cleavage mechanism miRNAs guide the AGO component of RISC to
cleave a single phosphodiester bond of the target mRNA within the miRNA-binding
site (Llave et al 2002b Tang et al 2003) The truncated fragments are then released
and degraded freeing the RISC to recognize and cleave another target mRNA This has
been validated by the detection of 3prime cleavage products that have 5prime ends that start at the
middle of the complementary regions The mRNA cleavage products are then further
degraded by other mechanisms The 3prime cleavage products of some miRNA targets are
degraded by the 5prime-3prime exonuclease XRN4 an Arabidopsis homolog of the major Yeast
mRNA degrading exonuclease Xrn1p It has been observed that the 3prime cleavage
products of some target mRNAs were accumulated in the xrn4 mutants (Souret et al
2004 Dalmay 2013) However the 3prime cleavage products of other miRNA targets do
not accumulate in the xrn4 mutants indicating that there are also XRN4 independent
mechanisms The 5prime cleavage products are typically untraceable by RNA gel blot
hybridization but can be detected by sensitive PCR based methods It has been found
that the 5prime cleavage products tend to obtain an oligo U tail at the 3prime ends This 3prime U tail
is correlated with shortening of the 5prime cleavage products from the 5prime end leading to the
conclusion that the 3prime uridylation causes 5 prime- 3 prime exonucleolytic decay of the 5prime cleavage
products (Shen amp Goodman 2004)
DCL1 HYL1 and SE interact with each other and are co-localized in the
nuclear bodies termed Dicing bodies or D-bodies in which some pri-miRNA shave
been found suggesting that D-bodies serve as a center for active miRNA processing
(Fang amp Spector 2007 Fujioka et al 2007 Song et al 2007) The miRNA
processing site appears to be distinct from the Cajal bodies that have previously been
found as a siRNA processing site (Pontes amp Pikaard 2008) The D-bodies include
SmD3 and SmB proteins that are found in the Cajal bodies However At collin an
additional marker for the Cajal bodies is not found in the D-bodies Moreover the D-
bodies are present in an At collin mutant lacking the Cajal bodies The pri-miRNAs of
miR171A and miR159A genes have been found to be co-localized with HYL1 and
Chapter 1 Introduction and Review of Literature
41
DCL1 in the D- bodies that are distinct from the Cajal bodies (Song et al 2007)
11352 MiRNA-directed translational repression
Translation repression is another mechanism exerted by plant miRNAs The regulatory
mechanism of miR172 which regulates flowering time and floral organ identity is
consistent with translation repression MiR172 over production does not affect the
transcript levels of APETALA2 (AP2) and AP2- like target mRNAs Instead the AP2
protein level is reduced in the miR172 over expressing mutant (Aukerman amp Sakai
2003 Chen 2004 Dalmay 2013) It was later shown that miR172 can also direct the
cleavage of the target mRNAs although the steady-state mRNA level is unchanged due
to feedback regulation (Schwab et al 2005 Jung et al 2007) It is clear that miR172
regulates its target genes primarily by translational repression rather than mRNA
cleavage Recently it has been reported that miR156157 and miR854 also regulate
their target genes through translational repression (Arteaga-Vazquez et al 2006
Gandikota et al 2007 Dalmay 2013) It was once thought that translational repression
is an exception to the rule However experimental evidence suggest a widespread
coexistence of translational repression and mRNA cleavage (Brodersen et al 2008)
1136 Regulatory roles of miRNAs in plant development
The miRNA target prediction has been a difficult task in order to obtain regulatory
targets without bringing in too many false predictions Plant growth and developmental
processes regulated by miRNAs are quite diverse and enormous Furthermore plant
miRNAs work cooperatively or antagonistically to establish a balanced regulation This
notion is well consistent with the findings that mutations in the regulators of miRNA
biogenesis transport and processing such as AGO1 DCL1 HYL1 HEN1 ABH1
and HASTY cause pleiotropic phenotypes (Mallory amp Vaucheret 2006 de Alba et al
2013) To date the roles of miRNAs have been mostly deduced from obtaining miRNA
over expressing transgenic plants or gain-of-function mutants in which miRNA-
resistant target genes are ectopically expressed
11361 Phase transition
Plants pass through a series of developmental phase transitions during their life span
beginning with seed germination continuing through vegetative phase change
reproductive phase change flowering initiation and finally seed production for the next
generation Because of their importance in understanding plant developmental
Chapter 1 Introduction and Review of Literature
42
processes genes regulating developmental transitions have been extensively studied
and underlying molecular mechanisms have been explored in various plant species
Majority of researches were mainly focused on vegetative phase change and
reproductive transition in Arabidopsis and Maize (Poethig 2003 Baurle amp Dean
2006) Particularly the mutations of the genes encoding various regulators of miRNA
biogenesis and transport such as HST SE and AGO exhibit altered juvenile to adult
vegetative phase change and vegetative to reproductive change (Hunter et al 2003
Peragine et al 2004 Vazquez et al 2004a Jones-Rhoades et al 2006 de Alba et al
2013)
Over-expression of the miR156a gene causes late flowering and delays
vegetative phase change (Wu amp Poethig 2006 Gandikota et al 2007 Wang et al
2008) It has been shown that miR156 negatively regulates a group of genes encoding
SQUAMOSA PROMOTER-BINDING PROTEIN-LIKE (Lewis et al 2007 de Alba et
al 2013 Rogers amp Chen 2013) transcription factors such as SPL3 SPL4 and SPL5
that promote flowering initiation In contrast transgenic plants over expressing
miR156-resistant SPL345 genes are early flowering Interestingly the endogenous
level of miR156 is temporally regulated In the early juvenile vegetative phase the
level of miR156 is very high but decreases rapidly before the onset of the adult juvenile
phase (Wu amp Poethig 2006)
The Arabidopsis early activation tagged (eat-D) mutant exhibits early flowering
with disrupted floral structure The mutant phenotypes are caused by over expression of
the miR172a-2gene (Aukerman amp Sakai 2003) MiR172 regulates a small group of
genes encoding APETALLA2 (AP2)-like transcription factors such as TARGET OF
EAT1 (TOE1) TOE2 SCHLAFMUTZE (SMZ) and SCHNARCHZAPFEN(SNZ)
TOE1 TOE2 SMZ and SNZ have been shown to participate in their productive phase
change by repressing a floral integrator FLOWERING LOCUS T (FT)(Jung et al
2007) Consistent with this toe1-2D 35STOE2 35SSMZ and 35SSNZ plants
exhibit late flowering (Jung et al 2007) MiR172 is also regulated temporally and
highly expressed in the adult vegetative phase both in Arabidopsis and maize (Chuck et
al 2007) Because the temporal expression patterns of miR156 and miR172 are
complementary to each other it has been suggested that the two miRNAs interact with
each other in regulating phase change (Chuck et al 2009 de Alba et al 2013) In
support of this view it has been shown that miR172 is down-regulated in the
Chapter 1 Introduction and Review of Literature
43
Corngrass1(Cg1) mutant in which miR156 is overproduced (Chuck et al 2007 de
Alba et al 2013) However there is no direct evidence for the direct linkage between
miR156 and miR172 It is also envisioned that miR156 and miR172 would not be
directly related For example miR156 regulates plastochron index that is the inverse of
leaf initiation rate and thus frequently used to analyze temporal patterns of plant
development In contrast miR172 does not affect plastochron (Wang et al 2008) The
down-regulation of miR172 in the maize miR156-over producing mutants would be
due to the prolonged juvenile vegetative phase in the mutants
Another miRNA that regulates flowering initiation is miR159 It regulates
MYB33 and MYB65 which are closely related to the barley GAMY B transcription
factor that responds to gibberellic acid (GA) The level of miR159 is elevated in GA-
treated seedlings but reduced in the GA biosynthetic ga1 mutant Transgenic plants
over producing miR159 exhibit delayed floral induction under short days (Achard et
al 2004) It is been suggested that MYB33 mediates GA signal to regulate LEAFY
(LFY) These observations indicate that miR159 plays an important role in the GA
signaling pathways that regulate proper floral induction under short days (Achard et al
2004)
11362 Floral development
Arabidopsis flowers consist of four distinct organs They arise in a characteristic
pattern within concentric rings called whorls from outside to inside four sepals four
petals six stamens and the central pistil consisting of two carpels Identities of the
floral organs are determined by three classes of regulatory genes classA classB and
class C genes (Krizek amp Fletcher 2005) The A and C class genes act antagonistically
to restrict their expression domains (Bowman et al 1991)
It is notable that miR172 also regulates floral architecture in addition to its role
in the timing of flowering initiation MiR172 inhibits the translation of the AP2 mRNA
(Chen 2004) Transgenic plants overproducing miR172 not only exhibit early
flowering but also show abnormal floral development which is quite similar to those
of the class A gene mutants such as the ap2 mutants (Aukerman amp Sakai 2003 de
Alba et al 2013) When the activity of the class A genes is inactivated that of the class
C genes such as AGAMOUS (AG) is elevated As a result the miR172- overproducing
transgenic plants exhibit no petals along with homeotic transformation of the sepals to
Chapter 1 Introduction and Review of Literature
44
the carpels (Aukerman amp Sakai 2003 Chen 2004)
Interestingly miR166 also regulates floral architecture The role of miR165166
in the regulation of meristem activity is closely linked to floral meristem formation and
organization (Zhao et al 2007 Miyashima et al 2013) Floral structure is severely
disrupted in the miR165166-overproducing mutants such as men1 and jba-1D in
which miR166 is overproduced (Williams et al 2005b Jung amp Park 2007) The men1
and jba-1D mutants have smaller gynoecium and reduced carpel number In an extreme
case the jba-1D homozygous plants lack visible carpels (Williams et al 2005b)
Similar phenotypes have been observed in the miR165 overproducers (Zhao et al
2007) It has been suggested that the phenotypes are possibly caused by altered AG
expression in the floral meristem (Jung amp Park 2007 Turchi et al 2013)
Other miRNAs also affect floral development Overproduction of miRNAs
involved in auxin signaling also affects floral architecture Transgenic plants
overproducing miR167 have defects in the formation of ovule and anther with reduced
fertility (Ru et al 2006) It has been shown that miR167 targets ARF6 and ARF8 that
play a role in gynoecium and stamen development and in regulating fertility (Ru et al
2006 Wu et al 2006) These observations suggest that auxin signals are also closely
related to floral organogenesis In addition transgenic plants over-producing miR164
and the cuc1cuc2 double mutants show fused sepals and reduced petals (Laufs et al
2004 Turchi et al 2013) suggesting that miR164 is also related to floral meristem
activity and boundary specification of the meristematic region
11363 Shoot apical meristem development
Shoot apical meristem comprises self-maintaining totipotent cells The shoot apical
meristem activity affects diverse aspects of plant developmental processes including
leaf development vascular development and lateral organ polarity (Bowman et al
2002) Because miR165166 plays a primary role in meristem formation
overproduction of miR165166 and multiple knockout mutants of its target genes
exhibit pleiotropic pheonotypes (McConnell et al 2001 Emery et al 2003 Kim et al
2005 Williams et al 2005b)
The targets of miR165166 include genes encoding the homeo domain-leucine
zipper (HD-ZIP) class III transcription factors such as PHABULOSA(PHB)
PHAVOLUTA (PHV) REVOLUTA(REV) ATHB15CORONA (CNA) and ATHB8
Chapter 1 Introduction and Review of Literature
45
PHB PHV and ATHB15 have overlapping functions in controlling meristem
development (Turchi et al 2013) Indeed the phb phv athb15 triple mutants have an
enlarged shoot apical meristem Consistent with this the miR166- overproducing men1
and jba-1D mutants exhibit enlarged shoot apical meristems (Kim et al 2005
Williams et al 2005b Turchi et al 2013) In the mutants the shoot apical meristems
are multiplied and large
The development of shoot apical meristems is also regulated by the WUSCHEL
(WUS)ndashCLAVATA (CLV) signaling pathway WUS positively regulates cell
proliferation In contrast CLV3 which is expressed in stem cells limits the WUS
expression and thus inhibits cell proliferation in the SAM (Sharma et al 2003)
Consistent with this notion WUS is expressed to a higher level in jba-1D
explaining the ectopic enlargement of the SAM in the mutant (Williams et al 2005)
While WUS is closely related to miR165166 in the shoot apical meristem
development the underlying molecular mechanisms are currently unclear It has been
observed that the miR166-over producing men1 plants have an arrested meristem
development (Jung amp Park 2007) In addition the heterozygous men1+ and the
homozygous men1 plants show different expression patterns of WUS and CLV3 It
appears that miR166 regulates WUS indirectly and influences the expressional balance
between WUS and CLV3 through a negative feedback loop (Jung amp Park 2007 de
Alba et al 2013)
The phv phb athb15 triple mutant has an enlarged shoot apical meristems the
rev phb phv mutant does not have functional shoot apical meristems (Prigge et al
2005) This distinction may be explained by the fact that while miR166 targets
primarily PHB PHV and ATHB15 miR165 targets all the five HD-ZIP III genes (Zhou
et al 2007) Consistent with this transgenic plants overproducing miR166 are distinct
from those overproducing miR165 The miR165-overproducing transgenic plants lack
functional shoot apical meristem and a pin-like structure is formed at the apical region
of the mutant These phenotypes are quite similar to rev phb phv triple mutant (Zhou et
al 2007 Liu et al 2013) The phenotypic distinction may because by the difference
of the cleavage efficiencies of the target mRNAs In accordance with this notion a few
35S miR166a transgenic lines exhibit no shoot apical meristems The men1
homozygous plants also show arrested meristem development (Jung amp Park 2007)
Chapter 1 Introduction and Review of Literature
46
MiR164 cleaves the CUC1 and CUC2 mRNAs as well as the NAC1 mRNA
CUC1 and CUC2 determines the organ boundary in the meristem Phenotypes of
transgenic plants that overproduce miR164 are similar to those of the cuc1cuc2 double
mutant that exhibits embryonic developmental defects such as cup-shaped cotyledons
and fused cotyledons (Laufs et al 2004) When a miR164-resistant CUC2 is
expressed the boundary domain is enlarged CUC also plays a role in embryonic shoot
meristem formation by regulating the SHOOT MERISTEMLESS gene It was therefore
concluded that miR164-mediated cleavage of CUC1 and CUC2 mRNAs determines the
sizes of the boundary domains in the shoot apical meristem and regulates separation of
the organs by restricting cell proliferation (Laufs et al 2004 de Alba et al 2013)
11364 Leaf development
The role of miR319 has been extensively studied in leaf development A dominant
jaw-D mutant overproducing miR319 shows uneven curved leaves with enlarged size
and five TCP genes which are closely related to the CINCINNATA (CIN) gene in
Antirrhinum majus are down regulated in the mutant suggesting that miR319 mediated
regulation of the TCP transcription factor genes are important for the final step of
leaf shaping (Palatnik et al 2003 Naz et al 2013) In addition to leaf shape and size
leaf polarity is also specified by miRNA MiR166-mediated cleavage of the HD-ZIPIII
mRNAs is a well to establish leaf polarity This discovery originates from the gain-of-
function mutants such as phb-1d and phv-1d wherein point mutations in the
complementary sites to miRNA helps the mRNA to evade or resist from miRNA-
mediated cleavage (McConnell et al 2001 Emery et al 2003 Naz et al 2013)
Plant leaves have a dorso-ventral polarity consisting of the adaxial (upper
surface) and abaxial (lower surface) sides The adaxial side is closer to the shoot
apical meristem Adaxial identity is specified by functionally redundant class III HD-
ZIP family genes such as PHB PHV and REV Ectopic expression of each genes
result in adaxialized leaves Dominant phb-1d and phv-1d mutants exhibit
transformation of the abaxial to adaxial fate and have rod-shaped leaves In contrast
miR165166-overproducing plants show pin-like cotyledons radicalized leaves and
abaxialized leaves (Chitwood et al 2007 Pattanayak et al 2013)
Leaf asymmetry is determined by miR166-mediated restriction of the
expression domains of the HD-ZIPIII genes The HD-ZIPIII genes are expressed in the
Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
24
was termed as RNA interference or RNAi The protective effect of RNA silencing in
reducing virus infectivity supports the view that PTGS has evolved as a mechanism to
defend plants against virus infection and also to moderate the possible deleterious
genome-restructuring (insertional) activity of virus-like mobile genetic elements eg
retrotransposons A growing body of biochemical and genetic data has further
demonstrated that plants animals and yeasts share related mechanisms of specific
degradation of RNAs in which double-stranded forms of RNA are initiator molecules
(Brenstein et al 2001)
The key insight into the process of PGTS was provided by the experiments
conducted by Hamilton amp Baulcombe (1999) who identified the product of RNA
degradation as small RNA (siRNA) species of ~25nt of both sense and antisense
polarity SiRNA are formed and accumulate as double stranded RNA molecules of
defined chemical structures Both genetic and biochemical approaches were undertaken
to understand the basis of silencing Genetic screens were carried out in fungus
Neurospora carassa the alga Chalmydomonas reinhardtii and plant Arabidopsis
thaliana to search for detective mutants in PTGS
A combination of results obtained from several in vivo and in vitro experiments
have gelled into a two step mechanistic model for RNAiPTGS The first step referred
to as the RNAi initiating step involves binding of the RNA nucleases to a larges
dsRNA and its cleavage into discrete ~21-25nt RNA fragments This is ATP-
dependent reaction which involved RNase III - type endonucleases which make
staggered cuts in both strands of the dsRNA leaving 3prime overhang of 2 nucleotides with
5prime- phosphate 3prime-hydroxyl and no modification in the sugar phosphate backbone
(Hamilton amp Baulcombe 1999 Elbashir et al 2001) SiRNAs are the direct products
of dsRNA cleavage by the multi-domain RNase III enzyme DICER (Figure 16)
(Brenstein et al 2001 Karthikeyan et l 2013)
In the second step or commonly known as the effector step the double stranded
siRNAs produced in the first step bind to an RNAi specific protein complex to form a
RISC The complex undergoes activation in the presence of ATP so that the antisense
component of the unwound siRNA becomes exposed and allows the RISC to perform
the downstream RNAi reaction
Chapter 1 Introduction and Review of Literature
25
Several enzymes are involved in RNAi based on their role genes that encode for them
are classified into RNAi initiators RNAi effectors RNA dependent RNA polymerases
and silencing genes Two C elegans genes rdeI and rde4 (rde stands for lsquo RNAi
deficientrsquo) are believed to be involved in the initiation step of RNAi The C elegans
rde1 gene is a member of a large family of genes and is homologous to the Neurospora
qde2 (qde stands for ldquoquelling deficientrdquo) and Arabidopsis AGO1 (AGO stands for
agronaute AGO1 was previously identified to be involved in Arabidopsis
development) Although the functions of these genes are not clear a mammalian
member of RDE1 family has been identified as a translation initiation factor (Thakur
2005) Important genes for the effector step of PTGS in C elegans are rde2 and mut7
genes These genes were identified from heterozygous mutant worms that were unable
to transmit RNAi to their homozygous offsprings Worms with mutated rde2 or mut 7
genes show defective RNAi mut-7 gene encodes a protein with homology to the
nuclease domains of RNAase D and a protein implicated in Werner syndrome (a rapid
ageing disease in humans) (Grishok et al 2001 Kamath-Loeb et al 2012s)
Neurospora qde-1 Arabidopsis SDE-1SGS-2 and C elegans ego-1 appear to encode
RNA dependent RNA polymerase (RdRPs) It assumed that this is a proof that an RdRp
activity is required for RNAi Certainly the existence of an RdRp might explain the
remarkable efficiency of dsRNA induced silencing if it amplifies either the dsRNA
prior to cleavage or the siRNAs directly (Muers 2013) In C elegans ego-1 mutants
(ego stands for lsquoenhancer of glp-1) RNAi functions normally in somatic cells but is
defective in germline cells where ego-1 is primarily expressed In Arabidopsis SDE-
1SGS-2 mutants (SGS stands for suppressor of gene silencing) siRNA are produced
when dsRNA is introduced via an endogenously replicating RNA virus but not
introduced by a transgene It has been proposed that perhaps the viral RdRP is
substituting for the Arabidopsis enzyme in these mutants Random degradative PCR
model suggests that an RdRP uses the guide strand of an siRNA as a primer for the
target mRNA generating a dsRNA substrate for Dicer and thus more siRNAs
Several genes controlling RNA silencing in plants have been identified through
genetic screens of Arabidopsis mutants impaired in transgene induced RNA silencing
They encode a putative RNA-dependent RNA polymerase (SGS2SDE1) a coiled
protein (SGS3) a protein containing PAZ and Piwi domains (AGO1) and an RNA
helicase (SDE3) The putative SGS2SDE1 of Neurospora is related to QDE-1 and
Chapter 1 Introduction and Review of Literature
26
EGO-1 of C elegans whereas PAZPiwi protein AGO is related to QDE-2 of
Neurospora RDE-1of C elegans RNA helicase SDE3 is related to SMG-2 of C
elegans and Mut-6 of Chlamydomonas MUT-7 gene of C elegans encodes a protein
similar to Rnase D whereas the Drosophila DICER gene encode a protein similar to
RNase III An Arabidopsis ortholog of DICER gene has been identified called CAF
SIN1 SUS1(Thakur 2005 Poethig 2009)
Since the RNAi machinery is present constitutively within the eukaryotic cell it
is important to explore the metabolic advantages that are accorded by the RNAi related
proteins during the intrinsic normal growth of cells and development of organisms The
natural RNAi machinery not only keeps the mobile transposable elements from
disrupting the integrity of the genomes as suggested by the analysis of model organisms
like A thaliana C elegans D melanogaster (Tabara et al 1999 Wu-Scharf et al
2000 Aravin et al 2001 Hamilton et al 2002 Lisch 2009) but also participates in
development of organisms Genetic defects in Celegans RNAi genes ego1 and dicer
cause known specific developmental errors (Grishok et al 2000 Grishok et al 2001
Knight amp Bass 2001 Hall et al 2013) Similarly the Argonaute family of genes of A
thaliana (especially the ZWILLE proteins) is also responsible for plant architecture and
meristem development (Carmell et al 2002 Hamant and Pautot 2010) and the Dicer
homologue of A thaliana CAF1 is required for embryo development (Golden et al
2002 Nakano et al 2011) Thus genetic evidence illustrates the role of the RNAi
machinery as a controller of development related genes The mechanistic details of
these developmental processes are beginning to emerge
Chapter 1 Introduction and Review of Literature
27
Figure 16 Mechanism of gene silencing induced double stranded RNA The dicer enzyme to
produce siRNAs cleaves the RNA The siRNA generated bind to the nuclease complex
RISC The antisense component in the siRNA in the RISC guides the complex towards
the cognate mRNA resulting in endonucleolytic cleavage of the mRNA
Chapter 1 Introduction and Review of Literature
28
112 MicroRNA or MiRNA
In 1991 Ambros and co-workers first isolated a lin-4 mutant of Celegans which was
arrested at the first larval stage (Lee et al 1993) Later on the let-7 mutation was
isolated in the same system which was responsible for development through the fourth
larval stage Both lin-4 and let-7 encode short 22-nucleotide mature RNAs and were
called short temporal RNA because they control the temporal development program of
C elegans The mature lin-4 RNA defines (negatively regulates) the mRNA expression
of the lin-14 and lin-28 hetrochronic genes with the antisense mediated repression
mechanism of translation initiation and thus specifies the fate of cells during the first
three larval stages Recent studies have revealed that the short temporal RNAs are
actually members of a group of tiny RNAs (21to 28 nucleotides) called the micro-
RNAs (miRNA) (Bartel 2009) isolated members of which could easily run to a few
thousand which includes both those were identified computationally and by deep
sequencing Some of the components of the RNAi machinery have also been clearly
established as the effector proteins for the maturation of miRNAs
113 MicroRNAs in plants
1131 Discovery
MicroRNAs have been discovered by three basic approaches Direct cloning forward
genetic direct cloning and bioinformatic prediction followed by experimental
validation The most direct method of miRNA discovery is to isolate and clone small
RNAs from biological samples and several groups have used this approach to identify
plant miRNAs (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002 Xie et al 2003 Sunkar et al 2005 Williams et al 2013) The cloning methods
adapted were similar to those used to identify large numbers of animal miRNAs
(Lagos-Quintana et al 2001 Lau et al 2001 Aravin amp Tuschl 2005) which involve
isolating small RNAs followed by oligonucleotide adapter ligation reverse
transcription amplification and sequencing Some protocols incorporate methods to
enrich for Dicer cleavage products (ie molecules with 5prime-phosphates and 3prime-
hydroxyls) and to concatemerize the short cDNAs so that several may be identified in a
single sequencing read (Lau et al 2001 Llave et al 2002a Jagadeeswaran amp Sunkar
2013)The initial cloning experiments in Arabidopsis identified 19 miRNAs belonging
to 15 families (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002) although hundreds of other small RNAs such as siRNAs and RNA degradation
Chapter 1 Introduction and Review of Literature
29
fragments were also cloned through the direct cloning methods To select miRNAs
from the pool of small RNAs secondary structures of the RNA molecules were
examined in compliance with the acknowledged structural features of miRNAs
(Ambros et al 2003) The secondary structures were validated using several ad-hoc
software tools such as the Mfold or RNAfold programs (Reeder et al 2006) Finally
the existence of mature miRNAs was determined by RNA gel blot hybridization These
direct cloning methods were successful in isolating many miRNAs in Arabidopsis
(Reinhart et al 2002 Sunkar amp Zhu 2004) Rice (Sunkar et al 2005) Poplar (Lu et
al 2005) moss (Arazi et al 2005) and Tobacco (Billoud et al 2005)
Although miRNAs were first discovered through forward genetic screens in
worms (Lee et al 1993 Reinhart et al 2000)only few A thaliana miRNA gene
families have been discovered by this method (Chen 2008) in plants MiRNA
involvement in plant mutant phenotypes were not properly inferred till the cloning
experiments established that plant genomes contain numerous miRNAs (Park et al
2002 Reinhart et al 2002 Rhoades et al 2002) MiRNA loss-of-function allele has
been identified by forward genetic screens early extra petala1 is caused by a
transposon insertion ~160bp upstream of the predicted miR164c stem loop resulting in
flowers with extra petals (Baker et al 2005 Irish 2008) The fact that loss-of-function
miRNA mutants have been rarely discovered using forward genetics reflects small
target size for mutagenesis coupled with redundancy in nearly all evolutionarily
conserved plant miRNAs which are encoded by gene families (Table16) Family
members are likely to have overlapping functions buffering against loss at any single
miRNA locus Over expression screens can circumvent redundancy limitations At least
four plant miRNAs miR319 (also known as miR-JAW) miR172 (also known as EAT)
and miR166 were isolated in over expression screens for dominant mutants with
developmental abnormalities (Aukerman amp Sakai 2003 Palatnik et al 2003 Kim et
al 2005 Williams et al 2005b Schommer et al 2012) Mutations in miRNA target
sites which can prevent the entire family of miRNAs from repressing a target gene can
also circumvent redundant functions of miRNA family members
The dominant mutations in the HD-ZIP genes PHB PHV and REVOLUTA
(REV) in Arabidopsis and ROLLEDLEAF1 (RLD1) in Maize result in adaxialization of
leaves andor Vasculature (McConnell amp Barton 1998 McConnell et al 2001 Nelson
et al 2002 Zhong amp Ye 2004 Allen amp Millar 2012) and are all caused by mutations
Chapter 1 Introduction and Review of Literature
30
in miR166 complementary sites (Rhoades et al 2002 Emery et al 2003 Jones-
Rhoades amp Bartel 2004 Mallory et al 2004 Zhong amp Ye 2004)
In both plants and animals cloning was the initial means of large-scale
miRNA discovery (Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Reinhart et al 2002 Mendes et al 2012) However cloning is biased toward
RNAs that are expressed highly and broadly MiRNAs expressed at low levels or
only in specific cell types or in response to certain environmental stimuli were more
difficult to clone Sequence based biases in cloning procedures might also cause
certain miRNAs to be missed Because of these limitations bioinformatics approaches
to identify miRNAs have provided a useful complement to cloning
A straightforward use of bioinformatics has been carried out to find homologs
of known miRNAs both within the same genome and in the genomes of other species
(Pasquinelli et al 2000 Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Mendes et al 2009) A more difficult challenge is to identify miRNAs
unrelated to previously known miRNAs This was first accomplished for
vertebrate nematode and fly miRNAs using algorithms that search for conservation
of sequence and secondary structure (ie miRNA stem-loop precursors) between
species searching for patterns that are characteristic of miRNAs (Lai et al 2003 Lim
et al 2003 Lim et al 2005)
Most of the methods identified numerous miRNAs but are necessarily more
relaxed in terms of allowed structures Some approaches take advantage of the high
complementarity of plant miRNAs to target messages implementing the requirement
that the candidate has conserved complementarity to mRNAs (Jones-Rhoades amp Bartel
2004 Adai et al 2005) This additional filter has been useful for distinguishing
authentic plant miRNAs from false positives which later extended to mammalian
miRNA gene prediction (Xie et al 2005 Mendes et al 2012)
Another criteria used by most algorithms in the filters is evolutionary
conservation In plants mature miRNA sequences are conserved to a higher degree
than their precursor sequences For example Arabidopsis and rice diverged more than
130 million years ago (Friis et al 2004) but some miRNAs are highly conserved
in these plant species (Reinhart et al 2002) Furthermore various parameters for the
secondary structures such as free energy the number of paired residues within a
Chapter 1 Introduction and Review of Literature
31
miRNA or the number and size of bulges are also used to validate the predictions
(Jones-Rhoades amp Bartel 2004 Wang et al 2004 Adai et al 2005 Mendes et al
2012)
1132 Conserved microRNAs in Plants
Till date cloning genetics and bioinformatics screens have resulted in annotation of
approximately 184 potential Arabidopsis miRNAs (miRBase release 13) These
miRNAs seem to arise through gene duplication and subsequent diversification and
represent approximately 100 families (Maher et al 2006 Axtell 2013) Twenty one
families represented by 92 genes are clearly conserved in species beyond Arabidopsis
(Table 15) The presence of these miRNA families in other sequenced plant genomes
indicates that different plant species have an evolutionarily fluid set of miRNAs
(Willmann amp Poethig 2007) Recent studies on mosses one of the most ancient land
plants have shown that at least 14 miRNA families are conserved in eudicots and
mosses and therefore it appears that plant miRNAs share a common ancient origin
(Arazi et al 2005 Axtell amp Bartel 2005 Talmor-Neiman et al 2006 Zhang et al
2006 Axtell 2013) The number of members per family in a genome ranges from 1-32
with the exception of miR-430 which is represented by a cluster of ~80 loci in zebra
fish (Giraldez et al 2005)
In plants the number of members in each family correlates among examined
species certain families contain numerous members in all three species (eg miR156
miR166 miR169) whereas others consistently contain fewer genes (eg miR162
mir168 miR394) (Table 16) Although it is still unclear why certain miRNA are
encoded by more than one gene in plants (eg 12 genes encoding miR156) this
correlation might suggest functional significance of various miRNA family sizes Most
annotated plant miRNAs are conserved throughout flowering plants while a few
miRNAs are found only in a single sequenced genome and thus they could be
evolutionarily lsquoyoungrsquo miRNAs For example Arabidopsis has several miRNA
families that are not found in poplar or rice suggesting that miRNAs could be
generated continuously during evolution (Allen et al 2004a Rajagopalan et al
2006 Fahlgren et al 2007 Axtell 2012 de Alba et al 2013) Consistent with the
notion that non conserved miRNAs are of a more recent evolutionary origin these
miRNAs are mainly encoded by a single locus in the genome Within each family the
mature miRNA is always located in the same arm
Chapter 1 Introduction and Review of Literature
32
Table 16 MicroRNA families conserved in plants
miRNA
family
Arabidopsis Oryza Populus
miR156 12 12 11 miR159319 6 8 15 miR160 3 6 8 miR162 2 2 3 miR164 3 5 6 miR166 9 12 17 miR167 4 9 8 miR168 2 2 2 miR169 14 17 32 miR171 4 7 10 miR172 5 3 9 miR390 3 1 4 miR393 2 2 4 miR394 2 1 2 miR395 6 19 10 miR396 2 5 7 miR397 2 2 3 miR398 3 2 3 miR399 6 11 12 miR408 1 1 1 miR403 1 0 2 miR437 0 1 0 miR444 0 1 0 miR445 0 9 0 Total 92 127 169
All known miRNA families that are conserved between more than one plant species are listed
together with the number of genes identified in the sequenced genomes Rice miRNA families that have orthologs in maize but do not appear to have orthologs in the eudicots (Arabidopsis and Populus) are marked with an asterisk The following families contain miRNA genes annotated with
more than one number miR156 (miR156 and miR157) miR159319 (miR159 and miR319) miR166 (miR165 and miR166) miR171 (miR170 and miR171) and miR390 (miR390 and miR391)
of the stem loop (5prime- or 3prime) as it would be expected if the genes carry common ancestry
(Figure 17) The sequence of the mature miRNA and to some extent the segment on
the opposite arm of the hairpin to which it pairs is highly conserved between the
members of the same family (both within and between the species) while the sequence
secondary structure and length of the intervening ldquolooprdquo region can be highly divergent
within the same family
The patterns of paring and non pairing nucleotides are often conserved between
homologous miRNA stem loops from different species (Figure 17) The significance of
conserved mismatches is opined to guide DCL1 to cleave at the appropriate positions
along the stem loop
Chapter 1 Introduction and Review of Literature
33
Figure 17 Representative stem loop of miR164 Segments corresponding to the mature miRNAs
are shown in red (Adapted from Rajagopalan et al 2006)
Although many miRNAs are highly conserved in plants the degree of conservation in
most cases is quite low between plants and animals one exception is miR854 A recent
study has revealed that miR854 is conserved between Arabidopsis and animals and that
the Arabidopsis miR854 differs from the human miR854 only by a single nucleotide
(Arteaga-Vazquez et al 2006 Axtell 2012 de Alba et al 2013) The Arabidopsis
miR854 has multiple binding sites in the 3prime-untranslated region (UTR) of
OLIGOURIDYLATE-binding-PROTEIN mRNA 1bA (UBP1b) and represses the
translation of the UBP1b mRNA The animal miR854 is also complementary to a site in
the 3prime-UTR of the UBP1b homolog in animals However it is currently unclear how the
animal miR854 regulates its target mRNA Another distinction between plant and
animal miRNAs is the genomic organization of the miRNA genes While plant miRNA
genes are located predominantly in the intergenic regions animal miRNA genes tend to
localize in the intragenic regions both in the introns or exons of protein coding genes
Moreover miRNA genes are often clustered within a single precursor transcript
whereas individual plant miRNAs are derived from individual precursor RNAs (Bartel
Chapter 1 Introduction and Review of Literature
34
2004 Bonnet et al 2004 Kim 2005 Vaucheret et al 2006 Marco et al 2013)
1133 Non-conserved miRNA and challenges in annotation
Most miRNAs are conserved throughout flowering plants (Table 15) but many more
were found only in a single sequenced genome thus considered to be of a more recent
evolutionary origin The extended homology between few non conserved miRNAs
precursors and their target genes provides strong evidence that these relatively ldquoYoungrdquo
miRNAs arose from the duplication of the target gene segments (Allen et al 2004)
Several non-conserved miRNAs including miR161 miR163 miR173 miR447
miR475 and miR476 are known to direct cleavage of target transcripts (Allen et al
2004a Adai et al 2005 Sunkar et al 2005 de Alba et al 2013) It is difficult to
confidently predict targets for many because it is not possible to use complementary
site as a filter against false-positive target predictions It is also difficult to
confidently annotate non-conserved miRNAs as miRNA rather than siRNAs The
established minimal standard for miRNA annotation is a small RNA with detectable
expression and the potential to form a stem-loop when joined to flanking genomic
sequence (Kasschau et al 2003) In practice these requirements are too loose to
definitively categorize many small RNAs cloned from plants Many plant siRNAs are
detectable on blots (Vazquez et al 2004b) and hundreds of thousands of non-
miRNA genomic sequences can be predicted to fold into secondary structures that
resemble structures of plant miRNA precursors (Jones-Rhoades amp Bartel 2004)
Therefore without conservation of both sequence and secondary structure it is
difficult to be confident that a given cloned RNA originated from a stem-loop (ie is a
miRNA) rather than from a double-stranded RNA (ie is a siRNA) In fact many of
the thousands o f small RNAs cloned from Arabidopsis (Gustafson et al 2005) would
probably meet the literal requirements for annotation as miRNAs A few of these
sequences might be miRNAs but others that meet the literal criteria probably are not
so The challenges of annotating non conserved small RNAs were evidenced by
three related small silencing RNAs that were originally annotated as miRNAs
that turned out to be trans-acting siRNAs (tasiRNAs)(Kanno et al2013)
Although most miRNAs that are broadly conserved among flowering plants are
now being identified and experimentally validated (Table 16)(Jones-Rhoades amp
Bartel 2004) the challenges and ambiguities for classifying non-conserved miRNAs
prevent meaningful estimates of the total number of miRNA genes in Arabidopsis and
Chapter 1 Introduction and Review of Literature
35
other plant genomes It is possible that miRNAs might have escaped detection
because they are non-conserved and expressed in specific tissues or conditions and can
only be identified by using high- throughput sequencing
1134 MicroRNA biogenesis
11341 Transcription of microRNA precursor
Plant miRNAs are primarily found in the genomic regions that are not associated with
the protein coding genes (Reinhart et al 2002) and it appears that most if not all plant
miRNAs are produced from their own transcriptional units Plant miRNA genes are
occasionally clustered near each other in the genome suggesting transcription of
multiple miRNAs from a single primary transcript [eg the miR395 cluster (Grishok
et al 2001 Jones-Rhoades amp Bartel 2004b)] but this polycistronic arrangement of
miRNA genes appears far less frequently in plants than in animals (Bartel 2004)
Northern blot EST and mapping evidence indicate that plant miRNA primary
transcripts (also known as pri-miRNAs) as in animals are longer than needed to
encompass the miRNA stem-loops (Palatnik et al 2003 Jones-Rhoades amp Bartel
2004b) At least some of these pri-miRNA transcripts appear to be spliced
polyadenylated (Kurihara amp Watanabe 2004) and capped (Xie Z et al 2005) Two
rice miRNAs are contained within transcripts that contain exon junctions within the
presumptive stem-loop precursor implying that in these cases splicing is a prerequisite
for Dicer recognition (Sunkar et al 2005) Plant pri-miRNAs are over 1kb in length
and they are usually preceded by typical TATA box motifs They can also undergo
canonical splicing polyadenylation and capping All these observations indicates RNA
polymerase II is probably responsible for transcribing most plant miRNAs (Xie Z et
al 2005) as appears to be the case for many animal miRNAs Relatively little is
known about the regulation of miRNA transcription in plants but there is no reason
to suspect that this regulation would differ from that of protein-coding transcripts as
the bioinformatics analysis of the sequence upstream of the transcriptional start
sites of the miRNA genes showed the presence of putative binding motifs of
number of known transcription factors (Megraw et al 2006 Sethupathy 2013)
11342 MicroRNA processing and export
In plants maturation of miRNA is a step wise process A miRNA gene is transcribed to
pri-miRNA which is much longer than the pre-miRNA possessing the characteristic
stem-loop structure Like transcription of most protein-coding genes the miRNA
Chapter 1 Introduction and Review of Literature
36
transcription is processed by RNA polymerase II (Pol II) enzymes and the pri-
miRNAs are 5prime-capped and 3prime-polyadenylated (Lee et al 2004 Xie Z et al 2005)
Mature miRNAs are produced from pri-miRNAs through at least two sequential
processing steps by RNase III-type endonucleases In animals pri-miRNAs are first
processed at the bottom portion of the stem by Drosha a nuclear-localized RNase III
enzyme to liberate pre-miRNAs The pre-miRNAs are then exported to the cytoplasm
with the help of exportin-5The pre-miRNAs are further processed by Dicer another
RNaseIII enzyme to a duplex consisting of the miRNA and the antisense strands
(miRNA) Since plants do not have any Drosha homologs the two sequential
processing steps seem to be carried out by DCL1 a Dicer homolog in the nucleus
(Kurihara amp Watanabe 2004 de Alba et al 2013 Rogers amp Chen 2013)
The processing of pri-miRNAs to pre-miRNAs by DCL1 is assisted by two
other proteins HYPONASTY LEAVES1 (HYL1) and SERRATE (SE) HYL1 belongs to a
family of double-stranded RNA (dsRNA) binding protein and interacts with DCL1 in
vitro and in vivo (Lu amp Fedoroff 2000 Hiraguri et al 2005 de Alba et al 2013
Rogers amp Chen 2013) Loss-of-function mutations in the HYL1 gene result in
decreased accumulation of miRNAs and concomitant accumulation of pri-miRNAs
(Han et al 2004 Vazquez et al 2004a Kurihara et al 2006) SE a C2H2 zinc finger
protein interacts with DCL1 and HYL1 and plays a role in the processing of pri-
miRNAs to pre-miRNAs (Lobbes et al 2006 Yang L et al 2006) In addition the
nuclear cap-binding complex proteins CBP20 and CBP80 that are known as ABA
HYPER SENSITIVE 1(ABH1) have been shown to be required for proper miRNA
processing (Gregory et al 2008 Laubinger et al 2008) Recently it has been
demonstrated that DCL1 can release 21-nt RNAs from dsRNA as well as from
synthetic miR167 precursor RNAs in vitro However this cleavage is error prone and
inefficient in the absence of HYL1 and SE which accelerate the rate of DCL1-mediated
cleavage and promote accurate processing of miRNAs (Dong et al 2008 Rogers amp
Chen 2013)
11343 Methylation of the miRNAmiRNAduplex
After the miRNAmiRNAduplexes are released from the pre-miRNAs by the DCL1
activities the duplexes are methylated at the 2primeOH of the3prime-ends by HUA ENHANCER1
(HEN1) a small RNA methyltransferase (Yu et al 2005 Yang Z et al 2006 Rogers
amp Chen 2013 ) This step which is distinct from the RNase III-mediated processing
Chapter 1 Introduction and Review of Literature
37
step distinguishes the biogenesis of plant miRNAs from that of animal miRNAs
Mutations in the HEN1 gene were first isolated in a morphological screening for floral
development although the HEN1 mutants display pleiotropic phenotypes similar to the
developmental defects observed in the DCL1 mutants (Chen et al 2002 Park et al
2002 Rogers amp Chen 2013)
Figure 18 Biogenesis of microRNA
A Biogenesis of plant MicroRNA B Biogenesis of metazoananimal MicroRNA
The miRNA (red) is incorporated into RISC and the miRNA(blue) in degraded
This enzyme has two dsRNA-binding domains and nuclear localization signal It
is also conserved in fungi and is required for the processing of siRNAs as well as of
miRNAs (Park et al 2002 Boutet et al 2003 Xie et al 2003) Although the HEN1
protein is localized to the nucleus its methylation activity in the cytoplasm cannot be
ignored (Xie et al 2004 Rogers amp Chen 2013)
Chapter 1 Introduction and Review of Literature
38
The methylation of the miRNAmiRNA duplex is required to protect miRNAs
against the 3prime-end uridylation activity and subsequent degradation (Li et al 2005) In
the hen1 mutants accumulation of miRNAs is reduced to a lower level Also the
miRNAs appear to be heterogeneous in their sizes such that a ladder of bands rather
than a single species is observed for most miRNAs in the mutants (Li et al 2005)
MiRNA cloning and sequencing analyses revealed the presence of a number of U
residues at the 3prime-end of miRNAs in the hen1 mutants Moreover these nucleotides do
not correspond to the miRNAs in the pre-miRNAs indicating that these nucleotides are
added after the processing of the miRNAs from pre-miRNAs (Li et al 2005)
Uridylation of RNAs has also been proven to be correlated with RNA decay (Shen amp
Goodman 2004 Mullen amp Marzluff 2008) suggesting that uridylation attracts an
exonuclease which degrades miRNAs in plants
11344 MiRNA incorporation in to the RISC and its nuclear export
The methylated miRNAmiRNA duplexes undergo RISC assembly In this process
the miRNA of the duplexes is selectively incorporated into the RISC and the miRNA
appears to be removed and subsequently degraded (Hammond et al 2000 Hutvagner
amp Zamore 2002 Schwarz et al 2003 de Alba et al 2013 Rogers amp Chen 2013)
The ARGONAUTE 1(AGO1) proteins are core components of the RISC complex
They contain two structural domains the N-terminal PAZ domain that binds to the 3prime-
end of single-stranded RNAs and the C-terminal piwi domains that has a structure
similar to that of RNase H (Cerutti et al 2000 Parker et al 2004) Several AGO
proteins possess endonucleolytic activities within the PIWI domain and cleave target
mRNAs in the middle of the site complementary to miRNA (Liu et al 2004 Meister et
al 2004 Miyoshi et al 2005) Mutations in the AGO1 gene cause miRNA target
genes to be ectopically expressed and the levels of some miRNAs are reduced in the
mutants (Kidner amp Martienssen 2004 Vaucheret et al 2004 Kidner amp Martienssen
2005 Ronemus et al 2006) Moreover the phenotype of the AGO1 hypomorphic
alleles display defects in lateral organ polarity and leaf and flower morphology as
observed in the dcl1 mutants and null AGO1 alleles are embryonically lethal
supporting the close relationship between the AGO proteins and miRNA processing
(Kidner amp Martienssen 2004 Vaucheret et al 2004 de Alba et al 2013 Rogers amp
Chen 2013)
The production of miRNA miRNA duplexes occur in the nucleus while
Chapter 1 Introduction and Review of Literature
39
miRNA-RISC complexes are present in the cytoplasm where target mRNAs are
located This discordance of functional sites requires an export step during miRNA
biogenesis HASTY the Arabidopsis homolog of exportin-5 is thought to be involved
in miRNA nuclear export (Bollman et al 2003 Park et al 2005)The hasty mutant
exhibits pleiotropic defects in plant development and reduced accumulation of most
miRNAs as in the DCL1 or AGO1 mutants Howeverit is unclear whether the HASTY
proteins export the miRNA miRNA duplexes or the miRNA-RISC complexes in
plants
11345 Feedback regulation of miRNA biogenesis
MiRNA biogenesis is under feedback regulation such that the DCL1 and AGO1 genes
are themselves regulated by miRNAs DCL1 is itself a target of miR162 which leads to
the cleavage of the DCL1 mRNA (Xie et al 2003 de Alba et al 2013) Consistent
with this the levels of DCL1 mRNA are elevated in the mutants of miRNA processing
genes in which the miR162 abundance is reduced In addition there is a predicted
hairpin structure of miR838 in the 14th
intron of the DCL1 primary transcript
(Rajagopalan et al 2006) This prediction implies that DCL1-mediated processing
on this site can result in the cleavage of the DCL1 primary transcript into two truncated
fragments the N-terminal capped fragment and the C-terminal polyadenylated
fragment If this is the case it is possible that miRNA biogenesis competes with the
splicing event of the DCL1 pre-mRNA
The AGO1 is also regulated by miRNA There is a complementary site for
miR168 binding in the AGO1 mRNA and the transcript level of the AGO1 mRNA is
reduced through an mRNA cleavage mechanism (Vaucheret et al 2006 de Alba et al
2013 Rogers amp Chen 2013) indicating that the AGO1 mRNA levels are tightly
controlled by the levels of the AGO1 protein
1135 Mechanism of miRNA action
11351 MiRNA-directed mRNA cleavage
By forming the miRNA-RISC assembly miRNAs can direct the RISC to regulate gene
expression by two post transcriptional mechanisms mRNA cleavage or translational
repression (Bartel 2004 Jones-Rhoades et al 2006 Dalmay 2013) The degree
of miRNA-mRNA complementarity plays an important role for the
determination of the mechanism to be used A general rule is that while perfect or
Chapter 1 Introduction and Review of Literature
40
near-perfect complementarity induces mRNA cleavage central mismatches trigger
translational repression (Carrington amp Ambros 2003 Jones-Rhoades amp Bartel 2004)
Unlike most animal miRNAs plant miRNAs have near-perfect or perfect
complementarity to their targets and mRNA cleavage is assumed to be a predominant
mechanism by which miRNAs regulate gene expression in plants
In RNA cleavage mechanism miRNAs guide the AGO component of RISC to
cleave a single phosphodiester bond of the target mRNA within the miRNA-binding
site (Llave et al 2002b Tang et al 2003) The truncated fragments are then released
and degraded freeing the RISC to recognize and cleave another target mRNA This has
been validated by the detection of 3prime cleavage products that have 5prime ends that start at the
middle of the complementary regions The mRNA cleavage products are then further
degraded by other mechanisms The 3prime cleavage products of some miRNA targets are
degraded by the 5prime-3prime exonuclease XRN4 an Arabidopsis homolog of the major Yeast
mRNA degrading exonuclease Xrn1p It has been observed that the 3prime cleavage
products of some target mRNAs were accumulated in the xrn4 mutants (Souret et al
2004 Dalmay 2013) However the 3prime cleavage products of other miRNA targets do
not accumulate in the xrn4 mutants indicating that there are also XRN4 independent
mechanisms The 5prime cleavage products are typically untraceable by RNA gel blot
hybridization but can be detected by sensitive PCR based methods It has been found
that the 5prime cleavage products tend to obtain an oligo U tail at the 3prime ends This 3prime U tail
is correlated with shortening of the 5prime cleavage products from the 5prime end leading to the
conclusion that the 3prime uridylation causes 5 prime- 3 prime exonucleolytic decay of the 5prime cleavage
products (Shen amp Goodman 2004)
DCL1 HYL1 and SE interact with each other and are co-localized in the
nuclear bodies termed Dicing bodies or D-bodies in which some pri-miRNA shave
been found suggesting that D-bodies serve as a center for active miRNA processing
(Fang amp Spector 2007 Fujioka et al 2007 Song et al 2007) The miRNA
processing site appears to be distinct from the Cajal bodies that have previously been
found as a siRNA processing site (Pontes amp Pikaard 2008) The D-bodies include
SmD3 and SmB proteins that are found in the Cajal bodies However At collin an
additional marker for the Cajal bodies is not found in the D-bodies Moreover the D-
bodies are present in an At collin mutant lacking the Cajal bodies The pri-miRNAs of
miR171A and miR159A genes have been found to be co-localized with HYL1 and
Chapter 1 Introduction and Review of Literature
41
DCL1 in the D- bodies that are distinct from the Cajal bodies (Song et al 2007)
11352 MiRNA-directed translational repression
Translation repression is another mechanism exerted by plant miRNAs The regulatory
mechanism of miR172 which regulates flowering time and floral organ identity is
consistent with translation repression MiR172 over production does not affect the
transcript levels of APETALA2 (AP2) and AP2- like target mRNAs Instead the AP2
protein level is reduced in the miR172 over expressing mutant (Aukerman amp Sakai
2003 Chen 2004 Dalmay 2013) It was later shown that miR172 can also direct the
cleavage of the target mRNAs although the steady-state mRNA level is unchanged due
to feedback regulation (Schwab et al 2005 Jung et al 2007) It is clear that miR172
regulates its target genes primarily by translational repression rather than mRNA
cleavage Recently it has been reported that miR156157 and miR854 also regulate
their target genes through translational repression (Arteaga-Vazquez et al 2006
Gandikota et al 2007 Dalmay 2013) It was once thought that translational repression
is an exception to the rule However experimental evidence suggest a widespread
coexistence of translational repression and mRNA cleavage (Brodersen et al 2008)
1136 Regulatory roles of miRNAs in plant development
The miRNA target prediction has been a difficult task in order to obtain regulatory
targets without bringing in too many false predictions Plant growth and developmental
processes regulated by miRNAs are quite diverse and enormous Furthermore plant
miRNAs work cooperatively or antagonistically to establish a balanced regulation This
notion is well consistent with the findings that mutations in the regulators of miRNA
biogenesis transport and processing such as AGO1 DCL1 HYL1 HEN1 ABH1
and HASTY cause pleiotropic phenotypes (Mallory amp Vaucheret 2006 de Alba et al
2013) To date the roles of miRNAs have been mostly deduced from obtaining miRNA
over expressing transgenic plants or gain-of-function mutants in which miRNA-
resistant target genes are ectopically expressed
11361 Phase transition
Plants pass through a series of developmental phase transitions during their life span
beginning with seed germination continuing through vegetative phase change
reproductive phase change flowering initiation and finally seed production for the next
generation Because of their importance in understanding plant developmental
Chapter 1 Introduction and Review of Literature
42
processes genes regulating developmental transitions have been extensively studied
and underlying molecular mechanisms have been explored in various plant species
Majority of researches were mainly focused on vegetative phase change and
reproductive transition in Arabidopsis and Maize (Poethig 2003 Baurle amp Dean
2006) Particularly the mutations of the genes encoding various regulators of miRNA
biogenesis and transport such as HST SE and AGO exhibit altered juvenile to adult
vegetative phase change and vegetative to reproductive change (Hunter et al 2003
Peragine et al 2004 Vazquez et al 2004a Jones-Rhoades et al 2006 de Alba et al
2013)
Over-expression of the miR156a gene causes late flowering and delays
vegetative phase change (Wu amp Poethig 2006 Gandikota et al 2007 Wang et al
2008) It has been shown that miR156 negatively regulates a group of genes encoding
SQUAMOSA PROMOTER-BINDING PROTEIN-LIKE (Lewis et al 2007 de Alba et
al 2013 Rogers amp Chen 2013) transcription factors such as SPL3 SPL4 and SPL5
that promote flowering initiation In contrast transgenic plants over expressing
miR156-resistant SPL345 genes are early flowering Interestingly the endogenous
level of miR156 is temporally regulated In the early juvenile vegetative phase the
level of miR156 is very high but decreases rapidly before the onset of the adult juvenile
phase (Wu amp Poethig 2006)
The Arabidopsis early activation tagged (eat-D) mutant exhibits early flowering
with disrupted floral structure The mutant phenotypes are caused by over expression of
the miR172a-2gene (Aukerman amp Sakai 2003) MiR172 regulates a small group of
genes encoding APETALLA2 (AP2)-like transcription factors such as TARGET OF
EAT1 (TOE1) TOE2 SCHLAFMUTZE (SMZ) and SCHNARCHZAPFEN(SNZ)
TOE1 TOE2 SMZ and SNZ have been shown to participate in their productive phase
change by repressing a floral integrator FLOWERING LOCUS T (FT)(Jung et al
2007) Consistent with this toe1-2D 35STOE2 35SSMZ and 35SSNZ plants
exhibit late flowering (Jung et al 2007) MiR172 is also regulated temporally and
highly expressed in the adult vegetative phase both in Arabidopsis and maize (Chuck et
al 2007) Because the temporal expression patterns of miR156 and miR172 are
complementary to each other it has been suggested that the two miRNAs interact with
each other in regulating phase change (Chuck et al 2009 de Alba et al 2013) In
support of this view it has been shown that miR172 is down-regulated in the
Chapter 1 Introduction and Review of Literature
43
Corngrass1(Cg1) mutant in which miR156 is overproduced (Chuck et al 2007 de
Alba et al 2013) However there is no direct evidence for the direct linkage between
miR156 and miR172 It is also envisioned that miR156 and miR172 would not be
directly related For example miR156 regulates plastochron index that is the inverse of
leaf initiation rate and thus frequently used to analyze temporal patterns of plant
development In contrast miR172 does not affect plastochron (Wang et al 2008) The
down-regulation of miR172 in the maize miR156-over producing mutants would be
due to the prolonged juvenile vegetative phase in the mutants
Another miRNA that regulates flowering initiation is miR159 It regulates
MYB33 and MYB65 which are closely related to the barley GAMY B transcription
factor that responds to gibberellic acid (GA) The level of miR159 is elevated in GA-
treated seedlings but reduced in the GA biosynthetic ga1 mutant Transgenic plants
over producing miR159 exhibit delayed floral induction under short days (Achard et
al 2004) It is been suggested that MYB33 mediates GA signal to regulate LEAFY
(LFY) These observations indicate that miR159 plays an important role in the GA
signaling pathways that regulate proper floral induction under short days (Achard et al
2004)
11362 Floral development
Arabidopsis flowers consist of four distinct organs They arise in a characteristic
pattern within concentric rings called whorls from outside to inside four sepals four
petals six stamens and the central pistil consisting of two carpels Identities of the
floral organs are determined by three classes of regulatory genes classA classB and
class C genes (Krizek amp Fletcher 2005) The A and C class genes act antagonistically
to restrict their expression domains (Bowman et al 1991)
It is notable that miR172 also regulates floral architecture in addition to its role
in the timing of flowering initiation MiR172 inhibits the translation of the AP2 mRNA
(Chen 2004) Transgenic plants overproducing miR172 not only exhibit early
flowering but also show abnormal floral development which is quite similar to those
of the class A gene mutants such as the ap2 mutants (Aukerman amp Sakai 2003 de
Alba et al 2013) When the activity of the class A genes is inactivated that of the class
C genes such as AGAMOUS (AG) is elevated As a result the miR172- overproducing
transgenic plants exhibit no petals along with homeotic transformation of the sepals to
Chapter 1 Introduction and Review of Literature
44
the carpels (Aukerman amp Sakai 2003 Chen 2004)
Interestingly miR166 also regulates floral architecture The role of miR165166
in the regulation of meristem activity is closely linked to floral meristem formation and
organization (Zhao et al 2007 Miyashima et al 2013) Floral structure is severely
disrupted in the miR165166-overproducing mutants such as men1 and jba-1D in
which miR166 is overproduced (Williams et al 2005b Jung amp Park 2007) The men1
and jba-1D mutants have smaller gynoecium and reduced carpel number In an extreme
case the jba-1D homozygous plants lack visible carpels (Williams et al 2005b)
Similar phenotypes have been observed in the miR165 overproducers (Zhao et al
2007) It has been suggested that the phenotypes are possibly caused by altered AG
expression in the floral meristem (Jung amp Park 2007 Turchi et al 2013)
Other miRNAs also affect floral development Overproduction of miRNAs
involved in auxin signaling also affects floral architecture Transgenic plants
overproducing miR167 have defects in the formation of ovule and anther with reduced
fertility (Ru et al 2006) It has been shown that miR167 targets ARF6 and ARF8 that
play a role in gynoecium and stamen development and in regulating fertility (Ru et al
2006 Wu et al 2006) These observations suggest that auxin signals are also closely
related to floral organogenesis In addition transgenic plants over-producing miR164
and the cuc1cuc2 double mutants show fused sepals and reduced petals (Laufs et al
2004 Turchi et al 2013) suggesting that miR164 is also related to floral meristem
activity and boundary specification of the meristematic region
11363 Shoot apical meristem development
Shoot apical meristem comprises self-maintaining totipotent cells The shoot apical
meristem activity affects diverse aspects of plant developmental processes including
leaf development vascular development and lateral organ polarity (Bowman et al
2002) Because miR165166 plays a primary role in meristem formation
overproduction of miR165166 and multiple knockout mutants of its target genes
exhibit pleiotropic pheonotypes (McConnell et al 2001 Emery et al 2003 Kim et al
2005 Williams et al 2005b)
The targets of miR165166 include genes encoding the homeo domain-leucine
zipper (HD-ZIP) class III transcription factors such as PHABULOSA(PHB)
PHAVOLUTA (PHV) REVOLUTA(REV) ATHB15CORONA (CNA) and ATHB8
Chapter 1 Introduction and Review of Literature
45
PHB PHV and ATHB15 have overlapping functions in controlling meristem
development (Turchi et al 2013) Indeed the phb phv athb15 triple mutants have an
enlarged shoot apical meristem Consistent with this the miR166- overproducing men1
and jba-1D mutants exhibit enlarged shoot apical meristems (Kim et al 2005
Williams et al 2005b Turchi et al 2013) In the mutants the shoot apical meristems
are multiplied and large
The development of shoot apical meristems is also regulated by the WUSCHEL
(WUS)ndashCLAVATA (CLV) signaling pathway WUS positively regulates cell
proliferation In contrast CLV3 which is expressed in stem cells limits the WUS
expression and thus inhibits cell proliferation in the SAM (Sharma et al 2003)
Consistent with this notion WUS is expressed to a higher level in jba-1D
explaining the ectopic enlargement of the SAM in the mutant (Williams et al 2005)
While WUS is closely related to miR165166 in the shoot apical meristem
development the underlying molecular mechanisms are currently unclear It has been
observed that the miR166-over producing men1 plants have an arrested meristem
development (Jung amp Park 2007) In addition the heterozygous men1+ and the
homozygous men1 plants show different expression patterns of WUS and CLV3 It
appears that miR166 regulates WUS indirectly and influences the expressional balance
between WUS and CLV3 through a negative feedback loop (Jung amp Park 2007 de
Alba et al 2013)
The phv phb athb15 triple mutant has an enlarged shoot apical meristems the
rev phb phv mutant does not have functional shoot apical meristems (Prigge et al
2005) This distinction may be explained by the fact that while miR166 targets
primarily PHB PHV and ATHB15 miR165 targets all the five HD-ZIP III genes (Zhou
et al 2007) Consistent with this transgenic plants overproducing miR166 are distinct
from those overproducing miR165 The miR165-overproducing transgenic plants lack
functional shoot apical meristem and a pin-like structure is formed at the apical region
of the mutant These phenotypes are quite similar to rev phb phv triple mutant (Zhou et
al 2007 Liu et al 2013) The phenotypic distinction may because by the difference
of the cleavage efficiencies of the target mRNAs In accordance with this notion a few
35S miR166a transgenic lines exhibit no shoot apical meristems The men1
homozygous plants also show arrested meristem development (Jung amp Park 2007)
Chapter 1 Introduction and Review of Literature
46
MiR164 cleaves the CUC1 and CUC2 mRNAs as well as the NAC1 mRNA
CUC1 and CUC2 determines the organ boundary in the meristem Phenotypes of
transgenic plants that overproduce miR164 are similar to those of the cuc1cuc2 double
mutant that exhibits embryonic developmental defects such as cup-shaped cotyledons
and fused cotyledons (Laufs et al 2004) When a miR164-resistant CUC2 is
expressed the boundary domain is enlarged CUC also plays a role in embryonic shoot
meristem formation by regulating the SHOOT MERISTEMLESS gene It was therefore
concluded that miR164-mediated cleavage of CUC1 and CUC2 mRNAs determines the
sizes of the boundary domains in the shoot apical meristem and regulates separation of
the organs by restricting cell proliferation (Laufs et al 2004 de Alba et al 2013)
11364 Leaf development
The role of miR319 has been extensively studied in leaf development A dominant
jaw-D mutant overproducing miR319 shows uneven curved leaves with enlarged size
and five TCP genes which are closely related to the CINCINNATA (CIN) gene in
Antirrhinum majus are down regulated in the mutant suggesting that miR319 mediated
regulation of the TCP transcription factor genes are important for the final step of
leaf shaping (Palatnik et al 2003 Naz et al 2013) In addition to leaf shape and size
leaf polarity is also specified by miRNA MiR166-mediated cleavage of the HD-ZIPIII
mRNAs is a well to establish leaf polarity This discovery originates from the gain-of-
function mutants such as phb-1d and phv-1d wherein point mutations in the
complementary sites to miRNA helps the mRNA to evade or resist from miRNA-
mediated cleavage (McConnell et al 2001 Emery et al 2003 Naz et al 2013)
Plant leaves have a dorso-ventral polarity consisting of the adaxial (upper
surface) and abaxial (lower surface) sides The adaxial side is closer to the shoot
apical meristem Adaxial identity is specified by functionally redundant class III HD-
ZIP family genes such as PHB PHV and REV Ectopic expression of each genes
result in adaxialized leaves Dominant phb-1d and phv-1d mutants exhibit
transformation of the abaxial to adaxial fate and have rod-shaped leaves In contrast
miR165166-overproducing plants show pin-like cotyledons radicalized leaves and
abaxialized leaves (Chitwood et al 2007 Pattanayak et al 2013)
Leaf asymmetry is determined by miR166-mediated restriction of the
expression domains of the HD-ZIPIII genes The HD-ZIPIII genes are expressed in the
Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
25
Several enzymes are involved in RNAi based on their role genes that encode for them
are classified into RNAi initiators RNAi effectors RNA dependent RNA polymerases
and silencing genes Two C elegans genes rdeI and rde4 (rde stands for lsquo RNAi
deficientrsquo) are believed to be involved in the initiation step of RNAi The C elegans
rde1 gene is a member of a large family of genes and is homologous to the Neurospora
qde2 (qde stands for ldquoquelling deficientrdquo) and Arabidopsis AGO1 (AGO stands for
agronaute AGO1 was previously identified to be involved in Arabidopsis
development) Although the functions of these genes are not clear a mammalian
member of RDE1 family has been identified as a translation initiation factor (Thakur
2005) Important genes for the effector step of PTGS in C elegans are rde2 and mut7
genes These genes were identified from heterozygous mutant worms that were unable
to transmit RNAi to their homozygous offsprings Worms with mutated rde2 or mut 7
genes show defective RNAi mut-7 gene encodes a protein with homology to the
nuclease domains of RNAase D and a protein implicated in Werner syndrome (a rapid
ageing disease in humans) (Grishok et al 2001 Kamath-Loeb et al 2012s)
Neurospora qde-1 Arabidopsis SDE-1SGS-2 and C elegans ego-1 appear to encode
RNA dependent RNA polymerase (RdRPs) It assumed that this is a proof that an RdRp
activity is required for RNAi Certainly the existence of an RdRp might explain the
remarkable efficiency of dsRNA induced silencing if it amplifies either the dsRNA
prior to cleavage or the siRNAs directly (Muers 2013) In C elegans ego-1 mutants
(ego stands for lsquoenhancer of glp-1) RNAi functions normally in somatic cells but is
defective in germline cells where ego-1 is primarily expressed In Arabidopsis SDE-
1SGS-2 mutants (SGS stands for suppressor of gene silencing) siRNA are produced
when dsRNA is introduced via an endogenously replicating RNA virus but not
introduced by a transgene It has been proposed that perhaps the viral RdRP is
substituting for the Arabidopsis enzyme in these mutants Random degradative PCR
model suggests that an RdRP uses the guide strand of an siRNA as a primer for the
target mRNA generating a dsRNA substrate for Dicer and thus more siRNAs
Several genes controlling RNA silencing in plants have been identified through
genetic screens of Arabidopsis mutants impaired in transgene induced RNA silencing
They encode a putative RNA-dependent RNA polymerase (SGS2SDE1) a coiled
protein (SGS3) a protein containing PAZ and Piwi domains (AGO1) and an RNA
helicase (SDE3) The putative SGS2SDE1 of Neurospora is related to QDE-1 and
Chapter 1 Introduction and Review of Literature
26
EGO-1 of C elegans whereas PAZPiwi protein AGO is related to QDE-2 of
Neurospora RDE-1of C elegans RNA helicase SDE3 is related to SMG-2 of C
elegans and Mut-6 of Chlamydomonas MUT-7 gene of C elegans encodes a protein
similar to Rnase D whereas the Drosophila DICER gene encode a protein similar to
RNase III An Arabidopsis ortholog of DICER gene has been identified called CAF
SIN1 SUS1(Thakur 2005 Poethig 2009)
Since the RNAi machinery is present constitutively within the eukaryotic cell it
is important to explore the metabolic advantages that are accorded by the RNAi related
proteins during the intrinsic normal growth of cells and development of organisms The
natural RNAi machinery not only keeps the mobile transposable elements from
disrupting the integrity of the genomes as suggested by the analysis of model organisms
like A thaliana C elegans D melanogaster (Tabara et al 1999 Wu-Scharf et al
2000 Aravin et al 2001 Hamilton et al 2002 Lisch 2009) but also participates in
development of organisms Genetic defects in Celegans RNAi genes ego1 and dicer
cause known specific developmental errors (Grishok et al 2000 Grishok et al 2001
Knight amp Bass 2001 Hall et al 2013) Similarly the Argonaute family of genes of A
thaliana (especially the ZWILLE proteins) is also responsible for plant architecture and
meristem development (Carmell et al 2002 Hamant and Pautot 2010) and the Dicer
homologue of A thaliana CAF1 is required for embryo development (Golden et al
2002 Nakano et al 2011) Thus genetic evidence illustrates the role of the RNAi
machinery as a controller of development related genes The mechanistic details of
these developmental processes are beginning to emerge
Chapter 1 Introduction and Review of Literature
27
Figure 16 Mechanism of gene silencing induced double stranded RNA The dicer enzyme to
produce siRNAs cleaves the RNA The siRNA generated bind to the nuclease complex
RISC The antisense component in the siRNA in the RISC guides the complex towards
the cognate mRNA resulting in endonucleolytic cleavage of the mRNA
Chapter 1 Introduction and Review of Literature
28
112 MicroRNA or MiRNA
In 1991 Ambros and co-workers first isolated a lin-4 mutant of Celegans which was
arrested at the first larval stage (Lee et al 1993) Later on the let-7 mutation was
isolated in the same system which was responsible for development through the fourth
larval stage Both lin-4 and let-7 encode short 22-nucleotide mature RNAs and were
called short temporal RNA because they control the temporal development program of
C elegans The mature lin-4 RNA defines (negatively regulates) the mRNA expression
of the lin-14 and lin-28 hetrochronic genes with the antisense mediated repression
mechanism of translation initiation and thus specifies the fate of cells during the first
three larval stages Recent studies have revealed that the short temporal RNAs are
actually members of a group of tiny RNAs (21to 28 nucleotides) called the micro-
RNAs (miRNA) (Bartel 2009) isolated members of which could easily run to a few
thousand which includes both those were identified computationally and by deep
sequencing Some of the components of the RNAi machinery have also been clearly
established as the effector proteins for the maturation of miRNAs
113 MicroRNAs in plants
1131 Discovery
MicroRNAs have been discovered by three basic approaches Direct cloning forward
genetic direct cloning and bioinformatic prediction followed by experimental
validation The most direct method of miRNA discovery is to isolate and clone small
RNAs from biological samples and several groups have used this approach to identify
plant miRNAs (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002 Xie et al 2003 Sunkar et al 2005 Williams et al 2013) The cloning methods
adapted were similar to those used to identify large numbers of animal miRNAs
(Lagos-Quintana et al 2001 Lau et al 2001 Aravin amp Tuschl 2005) which involve
isolating small RNAs followed by oligonucleotide adapter ligation reverse
transcription amplification and sequencing Some protocols incorporate methods to
enrich for Dicer cleavage products (ie molecules with 5prime-phosphates and 3prime-
hydroxyls) and to concatemerize the short cDNAs so that several may be identified in a
single sequencing read (Lau et al 2001 Llave et al 2002a Jagadeeswaran amp Sunkar
2013)The initial cloning experiments in Arabidopsis identified 19 miRNAs belonging
to 15 families (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002) although hundreds of other small RNAs such as siRNAs and RNA degradation
Chapter 1 Introduction and Review of Literature
29
fragments were also cloned through the direct cloning methods To select miRNAs
from the pool of small RNAs secondary structures of the RNA molecules were
examined in compliance with the acknowledged structural features of miRNAs
(Ambros et al 2003) The secondary structures were validated using several ad-hoc
software tools such as the Mfold or RNAfold programs (Reeder et al 2006) Finally
the existence of mature miRNAs was determined by RNA gel blot hybridization These
direct cloning methods were successful in isolating many miRNAs in Arabidopsis
(Reinhart et al 2002 Sunkar amp Zhu 2004) Rice (Sunkar et al 2005) Poplar (Lu et
al 2005) moss (Arazi et al 2005) and Tobacco (Billoud et al 2005)
Although miRNAs were first discovered through forward genetic screens in
worms (Lee et al 1993 Reinhart et al 2000)only few A thaliana miRNA gene
families have been discovered by this method (Chen 2008) in plants MiRNA
involvement in plant mutant phenotypes were not properly inferred till the cloning
experiments established that plant genomes contain numerous miRNAs (Park et al
2002 Reinhart et al 2002 Rhoades et al 2002) MiRNA loss-of-function allele has
been identified by forward genetic screens early extra petala1 is caused by a
transposon insertion ~160bp upstream of the predicted miR164c stem loop resulting in
flowers with extra petals (Baker et al 2005 Irish 2008) The fact that loss-of-function
miRNA mutants have been rarely discovered using forward genetics reflects small
target size for mutagenesis coupled with redundancy in nearly all evolutionarily
conserved plant miRNAs which are encoded by gene families (Table16) Family
members are likely to have overlapping functions buffering against loss at any single
miRNA locus Over expression screens can circumvent redundancy limitations At least
four plant miRNAs miR319 (also known as miR-JAW) miR172 (also known as EAT)
and miR166 were isolated in over expression screens for dominant mutants with
developmental abnormalities (Aukerman amp Sakai 2003 Palatnik et al 2003 Kim et
al 2005 Williams et al 2005b Schommer et al 2012) Mutations in miRNA target
sites which can prevent the entire family of miRNAs from repressing a target gene can
also circumvent redundant functions of miRNA family members
The dominant mutations in the HD-ZIP genes PHB PHV and REVOLUTA
(REV) in Arabidopsis and ROLLEDLEAF1 (RLD1) in Maize result in adaxialization of
leaves andor Vasculature (McConnell amp Barton 1998 McConnell et al 2001 Nelson
et al 2002 Zhong amp Ye 2004 Allen amp Millar 2012) and are all caused by mutations
Chapter 1 Introduction and Review of Literature
30
in miR166 complementary sites (Rhoades et al 2002 Emery et al 2003 Jones-
Rhoades amp Bartel 2004 Mallory et al 2004 Zhong amp Ye 2004)
In both plants and animals cloning was the initial means of large-scale
miRNA discovery (Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Reinhart et al 2002 Mendes et al 2012) However cloning is biased toward
RNAs that are expressed highly and broadly MiRNAs expressed at low levels or
only in specific cell types or in response to certain environmental stimuli were more
difficult to clone Sequence based biases in cloning procedures might also cause
certain miRNAs to be missed Because of these limitations bioinformatics approaches
to identify miRNAs have provided a useful complement to cloning
A straightforward use of bioinformatics has been carried out to find homologs
of known miRNAs both within the same genome and in the genomes of other species
(Pasquinelli et al 2000 Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Mendes et al 2009) A more difficult challenge is to identify miRNAs
unrelated to previously known miRNAs This was first accomplished for
vertebrate nematode and fly miRNAs using algorithms that search for conservation
of sequence and secondary structure (ie miRNA stem-loop precursors) between
species searching for patterns that are characteristic of miRNAs (Lai et al 2003 Lim
et al 2003 Lim et al 2005)
Most of the methods identified numerous miRNAs but are necessarily more
relaxed in terms of allowed structures Some approaches take advantage of the high
complementarity of plant miRNAs to target messages implementing the requirement
that the candidate has conserved complementarity to mRNAs (Jones-Rhoades amp Bartel
2004 Adai et al 2005) This additional filter has been useful for distinguishing
authentic plant miRNAs from false positives which later extended to mammalian
miRNA gene prediction (Xie et al 2005 Mendes et al 2012)
Another criteria used by most algorithms in the filters is evolutionary
conservation In plants mature miRNA sequences are conserved to a higher degree
than their precursor sequences For example Arabidopsis and rice diverged more than
130 million years ago (Friis et al 2004) but some miRNAs are highly conserved
in these plant species (Reinhart et al 2002) Furthermore various parameters for the
secondary structures such as free energy the number of paired residues within a
Chapter 1 Introduction and Review of Literature
31
miRNA or the number and size of bulges are also used to validate the predictions
(Jones-Rhoades amp Bartel 2004 Wang et al 2004 Adai et al 2005 Mendes et al
2012)
1132 Conserved microRNAs in Plants
Till date cloning genetics and bioinformatics screens have resulted in annotation of
approximately 184 potential Arabidopsis miRNAs (miRBase release 13) These
miRNAs seem to arise through gene duplication and subsequent diversification and
represent approximately 100 families (Maher et al 2006 Axtell 2013) Twenty one
families represented by 92 genes are clearly conserved in species beyond Arabidopsis
(Table 15) The presence of these miRNA families in other sequenced plant genomes
indicates that different plant species have an evolutionarily fluid set of miRNAs
(Willmann amp Poethig 2007) Recent studies on mosses one of the most ancient land
plants have shown that at least 14 miRNA families are conserved in eudicots and
mosses and therefore it appears that plant miRNAs share a common ancient origin
(Arazi et al 2005 Axtell amp Bartel 2005 Talmor-Neiman et al 2006 Zhang et al
2006 Axtell 2013) The number of members per family in a genome ranges from 1-32
with the exception of miR-430 which is represented by a cluster of ~80 loci in zebra
fish (Giraldez et al 2005)
In plants the number of members in each family correlates among examined
species certain families contain numerous members in all three species (eg miR156
miR166 miR169) whereas others consistently contain fewer genes (eg miR162
mir168 miR394) (Table 16) Although it is still unclear why certain miRNA are
encoded by more than one gene in plants (eg 12 genes encoding miR156) this
correlation might suggest functional significance of various miRNA family sizes Most
annotated plant miRNAs are conserved throughout flowering plants while a few
miRNAs are found only in a single sequenced genome and thus they could be
evolutionarily lsquoyoungrsquo miRNAs For example Arabidopsis has several miRNA
families that are not found in poplar or rice suggesting that miRNAs could be
generated continuously during evolution (Allen et al 2004a Rajagopalan et al
2006 Fahlgren et al 2007 Axtell 2012 de Alba et al 2013) Consistent with the
notion that non conserved miRNAs are of a more recent evolutionary origin these
miRNAs are mainly encoded by a single locus in the genome Within each family the
mature miRNA is always located in the same arm
Chapter 1 Introduction and Review of Literature
32
Table 16 MicroRNA families conserved in plants
miRNA
family
Arabidopsis Oryza Populus
miR156 12 12 11 miR159319 6 8 15 miR160 3 6 8 miR162 2 2 3 miR164 3 5 6 miR166 9 12 17 miR167 4 9 8 miR168 2 2 2 miR169 14 17 32 miR171 4 7 10 miR172 5 3 9 miR390 3 1 4 miR393 2 2 4 miR394 2 1 2 miR395 6 19 10 miR396 2 5 7 miR397 2 2 3 miR398 3 2 3 miR399 6 11 12 miR408 1 1 1 miR403 1 0 2 miR437 0 1 0 miR444 0 1 0 miR445 0 9 0 Total 92 127 169
All known miRNA families that are conserved between more than one plant species are listed
together with the number of genes identified in the sequenced genomes Rice miRNA families that have orthologs in maize but do not appear to have orthologs in the eudicots (Arabidopsis and Populus) are marked with an asterisk The following families contain miRNA genes annotated with
more than one number miR156 (miR156 and miR157) miR159319 (miR159 and miR319) miR166 (miR165 and miR166) miR171 (miR170 and miR171) and miR390 (miR390 and miR391)
of the stem loop (5prime- or 3prime) as it would be expected if the genes carry common ancestry
(Figure 17) The sequence of the mature miRNA and to some extent the segment on
the opposite arm of the hairpin to which it pairs is highly conserved between the
members of the same family (both within and between the species) while the sequence
secondary structure and length of the intervening ldquolooprdquo region can be highly divergent
within the same family
The patterns of paring and non pairing nucleotides are often conserved between
homologous miRNA stem loops from different species (Figure 17) The significance of
conserved mismatches is opined to guide DCL1 to cleave at the appropriate positions
along the stem loop
Chapter 1 Introduction and Review of Literature
33
Figure 17 Representative stem loop of miR164 Segments corresponding to the mature miRNAs
are shown in red (Adapted from Rajagopalan et al 2006)
Although many miRNAs are highly conserved in plants the degree of conservation in
most cases is quite low between plants and animals one exception is miR854 A recent
study has revealed that miR854 is conserved between Arabidopsis and animals and that
the Arabidopsis miR854 differs from the human miR854 only by a single nucleotide
(Arteaga-Vazquez et al 2006 Axtell 2012 de Alba et al 2013) The Arabidopsis
miR854 has multiple binding sites in the 3prime-untranslated region (UTR) of
OLIGOURIDYLATE-binding-PROTEIN mRNA 1bA (UBP1b) and represses the
translation of the UBP1b mRNA The animal miR854 is also complementary to a site in
the 3prime-UTR of the UBP1b homolog in animals However it is currently unclear how the
animal miR854 regulates its target mRNA Another distinction between plant and
animal miRNAs is the genomic organization of the miRNA genes While plant miRNA
genes are located predominantly in the intergenic regions animal miRNA genes tend to
localize in the intragenic regions both in the introns or exons of protein coding genes
Moreover miRNA genes are often clustered within a single precursor transcript
whereas individual plant miRNAs are derived from individual precursor RNAs (Bartel
Chapter 1 Introduction and Review of Literature
34
2004 Bonnet et al 2004 Kim 2005 Vaucheret et al 2006 Marco et al 2013)
1133 Non-conserved miRNA and challenges in annotation
Most miRNAs are conserved throughout flowering plants (Table 15) but many more
were found only in a single sequenced genome thus considered to be of a more recent
evolutionary origin The extended homology between few non conserved miRNAs
precursors and their target genes provides strong evidence that these relatively ldquoYoungrdquo
miRNAs arose from the duplication of the target gene segments (Allen et al 2004)
Several non-conserved miRNAs including miR161 miR163 miR173 miR447
miR475 and miR476 are known to direct cleavage of target transcripts (Allen et al
2004a Adai et al 2005 Sunkar et al 2005 de Alba et al 2013) It is difficult to
confidently predict targets for many because it is not possible to use complementary
site as a filter against false-positive target predictions It is also difficult to
confidently annotate non-conserved miRNAs as miRNA rather than siRNAs The
established minimal standard for miRNA annotation is a small RNA with detectable
expression and the potential to form a stem-loop when joined to flanking genomic
sequence (Kasschau et al 2003) In practice these requirements are too loose to
definitively categorize many small RNAs cloned from plants Many plant siRNAs are
detectable on blots (Vazquez et al 2004b) and hundreds of thousands of non-
miRNA genomic sequences can be predicted to fold into secondary structures that
resemble structures of plant miRNA precursors (Jones-Rhoades amp Bartel 2004)
Therefore without conservation of both sequence and secondary structure it is
difficult to be confident that a given cloned RNA originated from a stem-loop (ie is a
miRNA) rather than from a double-stranded RNA (ie is a siRNA) In fact many of
the thousands o f small RNAs cloned from Arabidopsis (Gustafson et al 2005) would
probably meet the literal requirements for annotation as miRNAs A few of these
sequences might be miRNAs but others that meet the literal criteria probably are not
so The challenges of annotating non conserved small RNAs were evidenced by
three related small silencing RNAs that were originally annotated as miRNAs
that turned out to be trans-acting siRNAs (tasiRNAs)(Kanno et al2013)
Although most miRNAs that are broadly conserved among flowering plants are
now being identified and experimentally validated (Table 16)(Jones-Rhoades amp
Bartel 2004) the challenges and ambiguities for classifying non-conserved miRNAs
prevent meaningful estimates of the total number of miRNA genes in Arabidopsis and
Chapter 1 Introduction and Review of Literature
35
other plant genomes It is possible that miRNAs might have escaped detection
because they are non-conserved and expressed in specific tissues or conditions and can
only be identified by using high- throughput sequencing
1134 MicroRNA biogenesis
11341 Transcription of microRNA precursor
Plant miRNAs are primarily found in the genomic regions that are not associated with
the protein coding genes (Reinhart et al 2002) and it appears that most if not all plant
miRNAs are produced from their own transcriptional units Plant miRNA genes are
occasionally clustered near each other in the genome suggesting transcription of
multiple miRNAs from a single primary transcript [eg the miR395 cluster (Grishok
et al 2001 Jones-Rhoades amp Bartel 2004b)] but this polycistronic arrangement of
miRNA genes appears far less frequently in plants than in animals (Bartel 2004)
Northern blot EST and mapping evidence indicate that plant miRNA primary
transcripts (also known as pri-miRNAs) as in animals are longer than needed to
encompass the miRNA stem-loops (Palatnik et al 2003 Jones-Rhoades amp Bartel
2004b) At least some of these pri-miRNA transcripts appear to be spliced
polyadenylated (Kurihara amp Watanabe 2004) and capped (Xie Z et al 2005) Two
rice miRNAs are contained within transcripts that contain exon junctions within the
presumptive stem-loop precursor implying that in these cases splicing is a prerequisite
for Dicer recognition (Sunkar et al 2005) Plant pri-miRNAs are over 1kb in length
and they are usually preceded by typical TATA box motifs They can also undergo
canonical splicing polyadenylation and capping All these observations indicates RNA
polymerase II is probably responsible for transcribing most plant miRNAs (Xie Z et
al 2005) as appears to be the case for many animal miRNAs Relatively little is
known about the regulation of miRNA transcription in plants but there is no reason
to suspect that this regulation would differ from that of protein-coding transcripts as
the bioinformatics analysis of the sequence upstream of the transcriptional start
sites of the miRNA genes showed the presence of putative binding motifs of
number of known transcription factors (Megraw et al 2006 Sethupathy 2013)
11342 MicroRNA processing and export
In plants maturation of miRNA is a step wise process A miRNA gene is transcribed to
pri-miRNA which is much longer than the pre-miRNA possessing the characteristic
stem-loop structure Like transcription of most protein-coding genes the miRNA
Chapter 1 Introduction and Review of Literature
36
transcription is processed by RNA polymerase II (Pol II) enzymes and the pri-
miRNAs are 5prime-capped and 3prime-polyadenylated (Lee et al 2004 Xie Z et al 2005)
Mature miRNAs are produced from pri-miRNAs through at least two sequential
processing steps by RNase III-type endonucleases In animals pri-miRNAs are first
processed at the bottom portion of the stem by Drosha a nuclear-localized RNase III
enzyme to liberate pre-miRNAs The pre-miRNAs are then exported to the cytoplasm
with the help of exportin-5The pre-miRNAs are further processed by Dicer another
RNaseIII enzyme to a duplex consisting of the miRNA and the antisense strands
(miRNA) Since plants do not have any Drosha homologs the two sequential
processing steps seem to be carried out by DCL1 a Dicer homolog in the nucleus
(Kurihara amp Watanabe 2004 de Alba et al 2013 Rogers amp Chen 2013)
The processing of pri-miRNAs to pre-miRNAs by DCL1 is assisted by two
other proteins HYPONASTY LEAVES1 (HYL1) and SERRATE (SE) HYL1 belongs to a
family of double-stranded RNA (dsRNA) binding protein and interacts with DCL1 in
vitro and in vivo (Lu amp Fedoroff 2000 Hiraguri et al 2005 de Alba et al 2013
Rogers amp Chen 2013) Loss-of-function mutations in the HYL1 gene result in
decreased accumulation of miRNAs and concomitant accumulation of pri-miRNAs
(Han et al 2004 Vazquez et al 2004a Kurihara et al 2006) SE a C2H2 zinc finger
protein interacts with DCL1 and HYL1 and plays a role in the processing of pri-
miRNAs to pre-miRNAs (Lobbes et al 2006 Yang L et al 2006) In addition the
nuclear cap-binding complex proteins CBP20 and CBP80 that are known as ABA
HYPER SENSITIVE 1(ABH1) have been shown to be required for proper miRNA
processing (Gregory et al 2008 Laubinger et al 2008) Recently it has been
demonstrated that DCL1 can release 21-nt RNAs from dsRNA as well as from
synthetic miR167 precursor RNAs in vitro However this cleavage is error prone and
inefficient in the absence of HYL1 and SE which accelerate the rate of DCL1-mediated
cleavage and promote accurate processing of miRNAs (Dong et al 2008 Rogers amp
Chen 2013)
11343 Methylation of the miRNAmiRNAduplex
After the miRNAmiRNAduplexes are released from the pre-miRNAs by the DCL1
activities the duplexes are methylated at the 2primeOH of the3prime-ends by HUA ENHANCER1
(HEN1) a small RNA methyltransferase (Yu et al 2005 Yang Z et al 2006 Rogers
amp Chen 2013 ) This step which is distinct from the RNase III-mediated processing
Chapter 1 Introduction and Review of Literature
37
step distinguishes the biogenesis of plant miRNAs from that of animal miRNAs
Mutations in the HEN1 gene were first isolated in a morphological screening for floral
development although the HEN1 mutants display pleiotropic phenotypes similar to the
developmental defects observed in the DCL1 mutants (Chen et al 2002 Park et al
2002 Rogers amp Chen 2013)
Figure 18 Biogenesis of microRNA
A Biogenesis of plant MicroRNA B Biogenesis of metazoananimal MicroRNA
The miRNA (red) is incorporated into RISC and the miRNA(blue) in degraded
This enzyme has two dsRNA-binding domains and nuclear localization signal It
is also conserved in fungi and is required for the processing of siRNAs as well as of
miRNAs (Park et al 2002 Boutet et al 2003 Xie et al 2003) Although the HEN1
protein is localized to the nucleus its methylation activity in the cytoplasm cannot be
ignored (Xie et al 2004 Rogers amp Chen 2013)
Chapter 1 Introduction and Review of Literature
38
The methylation of the miRNAmiRNA duplex is required to protect miRNAs
against the 3prime-end uridylation activity and subsequent degradation (Li et al 2005) In
the hen1 mutants accumulation of miRNAs is reduced to a lower level Also the
miRNAs appear to be heterogeneous in their sizes such that a ladder of bands rather
than a single species is observed for most miRNAs in the mutants (Li et al 2005)
MiRNA cloning and sequencing analyses revealed the presence of a number of U
residues at the 3prime-end of miRNAs in the hen1 mutants Moreover these nucleotides do
not correspond to the miRNAs in the pre-miRNAs indicating that these nucleotides are
added after the processing of the miRNAs from pre-miRNAs (Li et al 2005)
Uridylation of RNAs has also been proven to be correlated with RNA decay (Shen amp
Goodman 2004 Mullen amp Marzluff 2008) suggesting that uridylation attracts an
exonuclease which degrades miRNAs in plants
11344 MiRNA incorporation in to the RISC and its nuclear export
The methylated miRNAmiRNA duplexes undergo RISC assembly In this process
the miRNA of the duplexes is selectively incorporated into the RISC and the miRNA
appears to be removed and subsequently degraded (Hammond et al 2000 Hutvagner
amp Zamore 2002 Schwarz et al 2003 de Alba et al 2013 Rogers amp Chen 2013)
The ARGONAUTE 1(AGO1) proteins are core components of the RISC complex
They contain two structural domains the N-terminal PAZ domain that binds to the 3prime-
end of single-stranded RNAs and the C-terminal piwi domains that has a structure
similar to that of RNase H (Cerutti et al 2000 Parker et al 2004) Several AGO
proteins possess endonucleolytic activities within the PIWI domain and cleave target
mRNAs in the middle of the site complementary to miRNA (Liu et al 2004 Meister et
al 2004 Miyoshi et al 2005) Mutations in the AGO1 gene cause miRNA target
genes to be ectopically expressed and the levels of some miRNAs are reduced in the
mutants (Kidner amp Martienssen 2004 Vaucheret et al 2004 Kidner amp Martienssen
2005 Ronemus et al 2006) Moreover the phenotype of the AGO1 hypomorphic
alleles display defects in lateral organ polarity and leaf and flower morphology as
observed in the dcl1 mutants and null AGO1 alleles are embryonically lethal
supporting the close relationship between the AGO proteins and miRNA processing
(Kidner amp Martienssen 2004 Vaucheret et al 2004 de Alba et al 2013 Rogers amp
Chen 2013)
The production of miRNA miRNA duplexes occur in the nucleus while
Chapter 1 Introduction and Review of Literature
39
miRNA-RISC complexes are present in the cytoplasm where target mRNAs are
located This discordance of functional sites requires an export step during miRNA
biogenesis HASTY the Arabidopsis homolog of exportin-5 is thought to be involved
in miRNA nuclear export (Bollman et al 2003 Park et al 2005)The hasty mutant
exhibits pleiotropic defects in plant development and reduced accumulation of most
miRNAs as in the DCL1 or AGO1 mutants Howeverit is unclear whether the HASTY
proteins export the miRNA miRNA duplexes or the miRNA-RISC complexes in
plants
11345 Feedback regulation of miRNA biogenesis
MiRNA biogenesis is under feedback regulation such that the DCL1 and AGO1 genes
are themselves regulated by miRNAs DCL1 is itself a target of miR162 which leads to
the cleavage of the DCL1 mRNA (Xie et al 2003 de Alba et al 2013) Consistent
with this the levels of DCL1 mRNA are elevated in the mutants of miRNA processing
genes in which the miR162 abundance is reduced In addition there is a predicted
hairpin structure of miR838 in the 14th
intron of the DCL1 primary transcript
(Rajagopalan et al 2006) This prediction implies that DCL1-mediated processing
on this site can result in the cleavage of the DCL1 primary transcript into two truncated
fragments the N-terminal capped fragment and the C-terminal polyadenylated
fragment If this is the case it is possible that miRNA biogenesis competes with the
splicing event of the DCL1 pre-mRNA
The AGO1 is also regulated by miRNA There is a complementary site for
miR168 binding in the AGO1 mRNA and the transcript level of the AGO1 mRNA is
reduced through an mRNA cleavage mechanism (Vaucheret et al 2006 de Alba et al
2013 Rogers amp Chen 2013) indicating that the AGO1 mRNA levels are tightly
controlled by the levels of the AGO1 protein
1135 Mechanism of miRNA action
11351 MiRNA-directed mRNA cleavage
By forming the miRNA-RISC assembly miRNAs can direct the RISC to regulate gene
expression by two post transcriptional mechanisms mRNA cleavage or translational
repression (Bartel 2004 Jones-Rhoades et al 2006 Dalmay 2013) The degree
of miRNA-mRNA complementarity plays an important role for the
determination of the mechanism to be used A general rule is that while perfect or
Chapter 1 Introduction and Review of Literature
40
near-perfect complementarity induces mRNA cleavage central mismatches trigger
translational repression (Carrington amp Ambros 2003 Jones-Rhoades amp Bartel 2004)
Unlike most animal miRNAs plant miRNAs have near-perfect or perfect
complementarity to their targets and mRNA cleavage is assumed to be a predominant
mechanism by which miRNAs regulate gene expression in plants
In RNA cleavage mechanism miRNAs guide the AGO component of RISC to
cleave a single phosphodiester bond of the target mRNA within the miRNA-binding
site (Llave et al 2002b Tang et al 2003) The truncated fragments are then released
and degraded freeing the RISC to recognize and cleave another target mRNA This has
been validated by the detection of 3prime cleavage products that have 5prime ends that start at the
middle of the complementary regions The mRNA cleavage products are then further
degraded by other mechanisms The 3prime cleavage products of some miRNA targets are
degraded by the 5prime-3prime exonuclease XRN4 an Arabidopsis homolog of the major Yeast
mRNA degrading exonuclease Xrn1p It has been observed that the 3prime cleavage
products of some target mRNAs were accumulated in the xrn4 mutants (Souret et al
2004 Dalmay 2013) However the 3prime cleavage products of other miRNA targets do
not accumulate in the xrn4 mutants indicating that there are also XRN4 independent
mechanisms The 5prime cleavage products are typically untraceable by RNA gel blot
hybridization but can be detected by sensitive PCR based methods It has been found
that the 5prime cleavage products tend to obtain an oligo U tail at the 3prime ends This 3prime U tail
is correlated with shortening of the 5prime cleavage products from the 5prime end leading to the
conclusion that the 3prime uridylation causes 5 prime- 3 prime exonucleolytic decay of the 5prime cleavage
products (Shen amp Goodman 2004)
DCL1 HYL1 and SE interact with each other and are co-localized in the
nuclear bodies termed Dicing bodies or D-bodies in which some pri-miRNA shave
been found suggesting that D-bodies serve as a center for active miRNA processing
(Fang amp Spector 2007 Fujioka et al 2007 Song et al 2007) The miRNA
processing site appears to be distinct from the Cajal bodies that have previously been
found as a siRNA processing site (Pontes amp Pikaard 2008) The D-bodies include
SmD3 and SmB proteins that are found in the Cajal bodies However At collin an
additional marker for the Cajal bodies is not found in the D-bodies Moreover the D-
bodies are present in an At collin mutant lacking the Cajal bodies The pri-miRNAs of
miR171A and miR159A genes have been found to be co-localized with HYL1 and
Chapter 1 Introduction and Review of Literature
41
DCL1 in the D- bodies that are distinct from the Cajal bodies (Song et al 2007)
11352 MiRNA-directed translational repression
Translation repression is another mechanism exerted by plant miRNAs The regulatory
mechanism of miR172 which regulates flowering time and floral organ identity is
consistent with translation repression MiR172 over production does not affect the
transcript levels of APETALA2 (AP2) and AP2- like target mRNAs Instead the AP2
protein level is reduced in the miR172 over expressing mutant (Aukerman amp Sakai
2003 Chen 2004 Dalmay 2013) It was later shown that miR172 can also direct the
cleavage of the target mRNAs although the steady-state mRNA level is unchanged due
to feedback regulation (Schwab et al 2005 Jung et al 2007) It is clear that miR172
regulates its target genes primarily by translational repression rather than mRNA
cleavage Recently it has been reported that miR156157 and miR854 also regulate
their target genes through translational repression (Arteaga-Vazquez et al 2006
Gandikota et al 2007 Dalmay 2013) It was once thought that translational repression
is an exception to the rule However experimental evidence suggest a widespread
coexistence of translational repression and mRNA cleavage (Brodersen et al 2008)
1136 Regulatory roles of miRNAs in plant development
The miRNA target prediction has been a difficult task in order to obtain regulatory
targets without bringing in too many false predictions Plant growth and developmental
processes regulated by miRNAs are quite diverse and enormous Furthermore plant
miRNAs work cooperatively or antagonistically to establish a balanced regulation This
notion is well consistent with the findings that mutations in the regulators of miRNA
biogenesis transport and processing such as AGO1 DCL1 HYL1 HEN1 ABH1
and HASTY cause pleiotropic phenotypes (Mallory amp Vaucheret 2006 de Alba et al
2013) To date the roles of miRNAs have been mostly deduced from obtaining miRNA
over expressing transgenic plants or gain-of-function mutants in which miRNA-
resistant target genes are ectopically expressed
11361 Phase transition
Plants pass through a series of developmental phase transitions during their life span
beginning with seed germination continuing through vegetative phase change
reproductive phase change flowering initiation and finally seed production for the next
generation Because of their importance in understanding plant developmental
Chapter 1 Introduction and Review of Literature
42
processes genes regulating developmental transitions have been extensively studied
and underlying molecular mechanisms have been explored in various plant species
Majority of researches were mainly focused on vegetative phase change and
reproductive transition in Arabidopsis and Maize (Poethig 2003 Baurle amp Dean
2006) Particularly the mutations of the genes encoding various regulators of miRNA
biogenesis and transport such as HST SE and AGO exhibit altered juvenile to adult
vegetative phase change and vegetative to reproductive change (Hunter et al 2003
Peragine et al 2004 Vazquez et al 2004a Jones-Rhoades et al 2006 de Alba et al
2013)
Over-expression of the miR156a gene causes late flowering and delays
vegetative phase change (Wu amp Poethig 2006 Gandikota et al 2007 Wang et al
2008) It has been shown that miR156 negatively regulates a group of genes encoding
SQUAMOSA PROMOTER-BINDING PROTEIN-LIKE (Lewis et al 2007 de Alba et
al 2013 Rogers amp Chen 2013) transcription factors such as SPL3 SPL4 and SPL5
that promote flowering initiation In contrast transgenic plants over expressing
miR156-resistant SPL345 genes are early flowering Interestingly the endogenous
level of miR156 is temporally regulated In the early juvenile vegetative phase the
level of miR156 is very high but decreases rapidly before the onset of the adult juvenile
phase (Wu amp Poethig 2006)
The Arabidopsis early activation tagged (eat-D) mutant exhibits early flowering
with disrupted floral structure The mutant phenotypes are caused by over expression of
the miR172a-2gene (Aukerman amp Sakai 2003) MiR172 regulates a small group of
genes encoding APETALLA2 (AP2)-like transcription factors such as TARGET OF
EAT1 (TOE1) TOE2 SCHLAFMUTZE (SMZ) and SCHNARCHZAPFEN(SNZ)
TOE1 TOE2 SMZ and SNZ have been shown to participate in their productive phase
change by repressing a floral integrator FLOWERING LOCUS T (FT)(Jung et al
2007) Consistent with this toe1-2D 35STOE2 35SSMZ and 35SSNZ plants
exhibit late flowering (Jung et al 2007) MiR172 is also regulated temporally and
highly expressed in the adult vegetative phase both in Arabidopsis and maize (Chuck et
al 2007) Because the temporal expression patterns of miR156 and miR172 are
complementary to each other it has been suggested that the two miRNAs interact with
each other in regulating phase change (Chuck et al 2009 de Alba et al 2013) In
support of this view it has been shown that miR172 is down-regulated in the
Chapter 1 Introduction and Review of Literature
43
Corngrass1(Cg1) mutant in which miR156 is overproduced (Chuck et al 2007 de
Alba et al 2013) However there is no direct evidence for the direct linkage between
miR156 and miR172 It is also envisioned that miR156 and miR172 would not be
directly related For example miR156 regulates plastochron index that is the inverse of
leaf initiation rate and thus frequently used to analyze temporal patterns of plant
development In contrast miR172 does not affect plastochron (Wang et al 2008) The
down-regulation of miR172 in the maize miR156-over producing mutants would be
due to the prolonged juvenile vegetative phase in the mutants
Another miRNA that regulates flowering initiation is miR159 It regulates
MYB33 and MYB65 which are closely related to the barley GAMY B transcription
factor that responds to gibberellic acid (GA) The level of miR159 is elevated in GA-
treated seedlings but reduced in the GA biosynthetic ga1 mutant Transgenic plants
over producing miR159 exhibit delayed floral induction under short days (Achard et
al 2004) It is been suggested that MYB33 mediates GA signal to regulate LEAFY
(LFY) These observations indicate that miR159 plays an important role in the GA
signaling pathways that regulate proper floral induction under short days (Achard et al
2004)
11362 Floral development
Arabidopsis flowers consist of four distinct organs They arise in a characteristic
pattern within concentric rings called whorls from outside to inside four sepals four
petals six stamens and the central pistil consisting of two carpels Identities of the
floral organs are determined by three classes of regulatory genes classA classB and
class C genes (Krizek amp Fletcher 2005) The A and C class genes act antagonistically
to restrict their expression domains (Bowman et al 1991)
It is notable that miR172 also regulates floral architecture in addition to its role
in the timing of flowering initiation MiR172 inhibits the translation of the AP2 mRNA
(Chen 2004) Transgenic plants overproducing miR172 not only exhibit early
flowering but also show abnormal floral development which is quite similar to those
of the class A gene mutants such as the ap2 mutants (Aukerman amp Sakai 2003 de
Alba et al 2013) When the activity of the class A genes is inactivated that of the class
C genes such as AGAMOUS (AG) is elevated As a result the miR172- overproducing
transgenic plants exhibit no petals along with homeotic transformation of the sepals to
Chapter 1 Introduction and Review of Literature
44
the carpels (Aukerman amp Sakai 2003 Chen 2004)
Interestingly miR166 also regulates floral architecture The role of miR165166
in the regulation of meristem activity is closely linked to floral meristem formation and
organization (Zhao et al 2007 Miyashima et al 2013) Floral structure is severely
disrupted in the miR165166-overproducing mutants such as men1 and jba-1D in
which miR166 is overproduced (Williams et al 2005b Jung amp Park 2007) The men1
and jba-1D mutants have smaller gynoecium and reduced carpel number In an extreme
case the jba-1D homozygous plants lack visible carpels (Williams et al 2005b)
Similar phenotypes have been observed in the miR165 overproducers (Zhao et al
2007) It has been suggested that the phenotypes are possibly caused by altered AG
expression in the floral meristem (Jung amp Park 2007 Turchi et al 2013)
Other miRNAs also affect floral development Overproduction of miRNAs
involved in auxin signaling also affects floral architecture Transgenic plants
overproducing miR167 have defects in the formation of ovule and anther with reduced
fertility (Ru et al 2006) It has been shown that miR167 targets ARF6 and ARF8 that
play a role in gynoecium and stamen development and in regulating fertility (Ru et al
2006 Wu et al 2006) These observations suggest that auxin signals are also closely
related to floral organogenesis In addition transgenic plants over-producing miR164
and the cuc1cuc2 double mutants show fused sepals and reduced petals (Laufs et al
2004 Turchi et al 2013) suggesting that miR164 is also related to floral meristem
activity and boundary specification of the meristematic region
11363 Shoot apical meristem development
Shoot apical meristem comprises self-maintaining totipotent cells The shoot apical
meristem activity affects diverse aspects of plant developmental processes including
leaf development vascular development and lateral organ polarity (Bowman et al
2002) Because miR165166 plays a primary role in meristem formation
overproduction of miR165166 and multiple knockout mutants of its target genes
exhibit pleiotropic pheonotypes (McConnell et al 2001 Emery et al 2003 Kim et al
2005 Williams et al 2005b)
The targets of miR165166 include genes encoding the homeo domain-leucine
zipper (HD-ZIP) class III transcription factors such as PHABULOSA(PHB)
PHAVOLUTA (PHV) REVOLUTA(REV) ATHB15CORONA (CNA) and ATHB8
Chapter 1 Introduction and Review of Literature
45
PHB PHV and ATHB15 have overlapping functions in controlling meristem
development (Turchi et al 2013) Indeed the phb phv athb15 triple mutants have an
enlarged shoot apical meristem Consistent with this the miR166- overproducing men1
and jba-1D mutants exhibit enlarged shoot apical meristems (Kim et al 2005
Williams et al 2005b Turchi et al 2013) In the mutants the shoot apical meristems
are multiplied and large
The development of shoot apical meristems is also regulated by the WUSCHEL
(WUS)ndashCLAVATA (CLV) signaling pathway WUS positively regulates cell
proliferation In contrast CLV3 which is expressed in stem cells limits the WUS
expression and thus inhibits cell proliferation in the SAM (Sharma et al 2003)
Consistent with this notion WUS is expressed to a higher level in jba-1D
explaining the ectopic enlargement of the SAM in the mutant (Williams et al 2005)
While WUS is closely related to miR165166 in the shoot apical meristem
development the underlying molecular mechanisms are currently unclear It has been
observed that the miR166-over producing men1 plants have an arrested meristem
development (Jung amp Park 2007) In addition the heterozygous men1+ and the
homozygous men1 plants show different expression patterns of WUS and CLV3 It
appears that miR166 regulates WUS indirectly and influences the expressional balance
between WUS and CLV3 through a negative feedback loop (Jung amp Park 2007 de
Alba et al 2013)
The phv phb athb15 triple mutant has an enlarged shoot apical meristems the
rev phb phv mutant does not have functional shoot apical meristems (Prigge et al
2005) This distinction may be explained by the fact that while miR166 targets
primarily PHB PHV and ATHB15 miR165 targets all the five HD-ZIP III genes (Zhou
et al 2007) Consistent with this transgenic plants overproducing miR166 are distinct
from those overproducing miR165 The miR165-overproducing transgenic plants lack
functional shoot apical meristem and a pin-like structure is formed at the apical region
of the mutant These phenotypes are quite similar to rev phb phv triple mutant (Zhou et
al 2007 Liu et al 2013) The phenotypic distinction may because by the difference
of the cleavage efficiencies of the target mRNAs In accordance with this notion a few
35S miR166a transgenic lines exhibit no shoot apical meristems The men1
homozygous plants also show arrested meristem development (Jung amp Park 2007)
Chapter 1 Introduction and Review of Literature
46
MiR164 cleaves the CUC1 and CUC2 mRNAs as well as the NAC1 mRNA
CUC1 and CUC2 determines the organ boundary in the meristem Phenotypes of
transgenic plants that overproduce miR164 are similar to those of the cuc1cuc2 double
mutant that exhibits embryonic developmental defects such as cup-shaped cotyledons
and fused cotyledons (Laufs et al 2004) When a miR164-resistant CUC2 is
expressed the boundary domain is enlarged CUC also plays a role in embryonic shoot
meristem formation by regulating the SHOOT MERISTEMLESS gene It was therefore
concluded that miR164-mediated cleavage of CUC1 and CUC2 mRNAs determines the
sizes of the boundary domains in the shoot apical meristem and regulates separation of
the organs by restricting cell proliferation (Laufs et al 2004 de Alba et al 2013)
11364 Leaf development
The role of miR319 has been extensively studied in leaf development A dominant
jaw-D mutant overproducing miR319 shows uneven curved leaves with enlarged size
and five TCP genes which are closely related to the CINCINNATA (CIN) gene in
Antirrhinum majus are down regulated in the mutant suggesting that miR319 mediated
regulation of the TCP transcription factor genes are important for the final step of
leaf shaping (Palatnik et al 2003 Naz et al 2013) In addition to leaf shape and size
leaf polarity is also specified by miRNA MiR166-mediated cleavage of the HD-ZIPIII
mRNAs is a well to establish leaf polarity This discovery originates from the gain-of-
function mutants such as phb-1d and phv-1d wherein point mutations in the
complementary sites to miRNA helps the mRNA to evade or resist from miRNA-
mediated cleavage (McConnell et al 2001 Emery et al 2003 Naz et al 2013)
Plant leaves have a dorso-ventral polarity consisting of the adaxial (upper
surface) and abaxial (lower surface) sides The adaxial side is closer to the shoot
apical meristem Adaxial identity is specified by functionally redundant class III HD-
ZIP family genes such as PHB PHV and REV Ectopic expression of each genes
result in adaxialized leaves Dominant phb-1d and phv-1d mutants exhibit
transformation of the abaxial to adaxial fate and have rod-shaped leaves In contrast
miR165166-overproducing plants show pin-like cotyledons radicalized leaves and
abaxialized leaves (Chitwood et al 2007 Pattanayak et al 2013)
Leaf asymmetry is determined by miR166-mediated restriction of the
expression domains of the HD-ZIPIII genes The HD-ZIPIII genes are expressed in the
Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
26
EGO-1 of C elegans whereas PAZPiwi protein AGO is related to QDE-2 of
Neurospora RDE-1of C elegans RNA helicase SDE3 is related to SMG-2 of C
elegans and Mut-6 of Chlamydomonas MUT-7 gene of C elegans encodes a protein
similar to Rnase D whereas the Drosophila DICER gene encode a protein similar to
RNase III An Arabidopsis ortholog of DICER gene has been identified called CAF
SIN1 SUS1(Thakur 2005 Poethig 2009)
Since the RNAi machinery is present constitutively within the eukaryotic cell it
is important to explore the metabolic advantages that are accorded by the RNAi related
proteins during the intrinsic normal growth of cells and development of organisms The
natural RNAi machinery not only keeps the mobile transposable elements from
disrupting the integrity of the genomes as suggested by the analysis of model organisms
like A thaliana C elegans D melanogaster (Tabara et al 1999 Wu-Scharf et al
2000 Aravin et al 2001 Hamilton et al 2002 Lisch 2009) but also participates in
development of organisms Genetic defects in Celegans RNAi genes ego1 and dicer
cause known specific developmental errors (Grishok et al 2000 Grishok et al 2001
Knight amp Bass 2001 Hall et al 2013) Similarly the Argonaute family of genes of A
thaliana (especially the ZWILLE proteins) is also responsible for plant architecture and
meristem development (Carmell et al 2002 Hamant and Pautot 2010) and the Dicer
homologue of A thaliana CAF1 is required for embryo development (Golden et al
2002 Nakano et al 2011) Thus genetic evidence illustrates the role of the RNAi
machinery as a controller of development related genes The mechanistic details of
these developmental processes are beginning to emerge
Chapter 1 Introduction and Review of Literature
27
Figure 16 Mechanism of gene silencing induced double stranded RNA The dicer enzyme to
produce siRNAs cleaves the RNA The siRNA generated bind to the nuclease complex
RISC The antisense component in the siRNA in the RISC guides the complex towards
the cognate mRNA resulting in endonucleolytic cleavage of the mRNA
Chapter 1 Introduction and Review of Literature
28
112 MicroRNA or MiRNA
In 1991 Ambros and co-workers first isolated a lin-4 mutant of Celegans which was
arrested at the first larval stage (Lee et al 1993) Later on the let-7 mutation was
isolated in the same system which was responsible for development through the fourth
larval stage Both lin-4 and let-7 encode short 22-nucleotide mature RNAs and were
called short temporal RNA because they control the temporal development program of
C elegans The mature lin-4 RNA defines (negatively regulates) the mRNA expression
of the lin-14 and lin-28 hetrochronic genes with the antisense mediated repression
mechanism of translation initiation and thus specifies the fate of cells during the first
three larval stages Recent studies have revealed that the short temporal RNAs are
actually members of a group of tiny RNAs (21to 28 nucleotides) called the micro-
RNAs (miRNA) (Bartel 2009) isolated members of which could easily run to a few
thousand which includes both those were identified computationally and by deep
sequencing Some of the components of the RNAi machinery have also been clearly
established as the effector proteins for the maturation of miRNAs
113 MicroRNAs in plants
1131 Discovery
MicroRNAs have been discovered by three basic approaches Direct cloning forward
genetic direct cloning and bioinformatic prediction followed by experimental
validation The most direct method of miRNA discovery is to isolate and clone small
RNAs from biological samples and several groups have used this approach to identify
plant miRNAs (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002 Xie et al 2003 Sunkar et al 2005 Williams et al 2013) The cloning methods
adapted were similar to those used to identify large numbers of animal miRNAs
(Lagos-Quintana et al 2001 Lau et al 2001 Aravin amp Tuschl 2005) which involve
isolating small RNAs followed by oligonucleotide adapter ligation reverse
transcription amplification and sequencing Some protocols incorporate methods to
enrich for Dicer cleavage products (ie molecules with 5prime-phosphates and 3prime-
hydroxyls) and to concatemerize the short cDNAs so that several may be identified in a
single sequencing read (Lau et al 2001 Llave et al 2002a Jagadeeswaran amp Sunkar
2013)The initial cloning experiments in Arabidopsis identified 19 miRNAs belonging
to 15 families (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002) although hundreds of other small RNAs such as siRNAs and RNA degradation
Chapter 1 Introduction and Review of Literature
29
fragments were also cloned through the direct cloning methods To select miRNAs
from the pool of small RNAs secondary structures of the RNA molecules were
examined in compliance with the acknowledged structural features of miRNAs
(Ambros et al 2003) The secondary structures were validated using several ad-hoc
software tools such as the Mfold or RNAfold programs (Reeder et al 2006) Finally
the existence of mature miRNAs was determined by RNA gel blot hybridization These
direct cloning methods were successful in isolating many miRNAs in Arabidopsis
(Reinhart et al 2002 Sunkar amp Zhu 2004) Rice (Sunkar et al 2005) Poplar (Lu et
al 2005) moss (Arazi et al 2005) and Tobacco (Billoud et al 2005)
Although miRNAs were first discovered through forward genetic screens in
worms (Lee et al 1993 Reinhart et al 2000)only few A thaliana miRNA gene
families have been discovered by this method (Chen 2008) in plants MiRNA
involvement in plant mutant phenotypes were not properly inferred till the cloning
experiments established that plant genomes contain numerous miRNAs (Park et al
2002 Reinhart et al 2002 Rhoades et al 2002) MiRNA loss-of-function allele has
been identified by forward genetic screens early extra petala1 is caused by a
transposon insertion ~160bp upstream of the predicted miR164c stem loop resulting in
flowers with extra petals (Baker et al 2005 Irish 2008) The fact that loss-of-function
miRNA mutants have been rarely discovered using forward genetics reflects small
target size for mutagenesis coupled with redundancy in nearly all evolutionarily
conserved plant miRNAs which are encoded by gene families (Table16) Family
members are likely to have overlapping functions buffering against loss at any single
miRNA locus Over expression screens can circumvent redundancy limitations At least
four plant miRNAs miR319 (also known as miR-JAW) miR172 (also known as EAT)
and miR166 were isolated in over expression screens for dominant mutants with
developmental abnormalities (Aukerman amp Sakai 2003 Palatnik et al 2003 Kim et
al 2005 Williams et al 2005b Schommer et al 2012) Mutations in miRNA target
sites which can prevent the entire family of miRNAs from repressing a target gene can
also circumvent redundant functions of miRNA family members
The dominant mutations in the HD-ZIP genes PHB PHV and REVOLUTA
(REV) in Arabidopsis and ROLLEDLEAF1 (RLD1) in Maize result in adaxialization of
leaves andor Vasculature (McConnell amp Barton 1998 McConnell et al 2001 Nelson
et al 2002 Zhong amp Ye 2004 Allen amp Millar 2012) and are all caused by mutations
Chapter 1 Introduction and Review of Literature
30
in miR166 complementary sites (Rhoades et al 2002 Emery et al 2003 Jones-
Rhoades amp Bartel 2004 Mallory et al 2004 Zhong amp Ye 2004)
In both plants and animals cloning was the initial means of large-scale
miRNA discovery (Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Reinhart et al 2002 Mendes et al 2012) However cloning is biased toward
RNAs that are expressed highly and broadly MiRNAs expressed at low levels or
only in specific cell types or in response to certain environmental stimuli were more
difficult to clone Sequence based biases in cloning procedures might also cause
certain miRNAs to be missed Because of these limitations bioinformatics approaches
to identify miRNAs have provided a useful complement to cloning
A straightforward use of bioinformatics has been carried out to find homologs
of known miRNAs both within the same genome and in the genomes of other species
(Pasquinelli et al 2000 Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Mendes et al 2009) A more difficult challenge is to identify miRNAs
unrelated to previously known miRNAs This was first accomplished for
vertebrate nematode and fly miRNAs using algorithms that search for conservation
of sequence and secondary structure (ie miRNA stem-loop precursors) between
species searching for patterns that are characteristic of miRNAs (Lai et al 2003 Lim
et al 2003 Lim et al 2005)
Most of the methods identified numerous miRNAs but are necessarily more
relaxed in terms of allowed structures Some approaches take advantage of the high
complementarity of plant miRNAs to target messages implementing the requirement
that the candidate has conserved complementarity to mRNAs (Jones-Rhoades amp Bartel
2004 Adai et al 2005) This additional filter has been useful for distinguishing
authentic plant miRNAs from false positives which later extended to mammalian
miRNA gene prediction (Xie et al 2005 Mendes et al 2012)
Another criteria used by most algorithms in the filters is evolutionary
conservation In plants mature miRNA sequences are conserved to a higher degree
than their precursor sequences For example Arabidopsis and rice diverged more than
130 million years ago (Friis et al 2004) but some miRNAs are highly conserved
in these plant species (Reinhart et al 2002) Furthermore various parameters for the
secondary structures such as free energy the number of paired residues within a
Chapter 1 Introduction and Review of Literature
31
miRNA or the number and size of bulges are also used to validate the predictions
(Jones-Rhoades amp Bartel 2004 Wang et al 2004 Adai et al 2005 Mendes et al
2012)
1132 Conserved microRNAs in Plants
Till date cloning genetics and bioinformatics screens have resulted in annotation of
approximately 184 potential Arabidopsis miRNAs (miRBase release 13) These
miRNAs seem to arise through gene duplication and subsequent diversification and
represent approximately 100 families (Maher et al 2006 Axtell 2013) Twenty one
families represented by 92 genes are clearly conserved in species beyond Arabidopsis
(Table 15) The presence of these miRNA families in other sequenced plant genomes
indicates that different plant species have an evolutionarily fluid set of miRNAs
(Willmann amp Poethig 2007) Recent studies on mosses one of the most ancient land
plants have shown that at least 14 miRNA families are conserved in eudicots and
mosses and therefore it appears that plant miRNAs share a common ancient origin
(Arazi et al 2005 Axtell amp Bartel 2005 Talmor-Neiman et al 2006 Zhang et al
2006 Axtell 2013) The number of members per family in a genome ranges from 1-32
with the exception of miR-430 which is represented by a cluster of ~80 loci in zebra
fish (Giraldez et al 2005)
In plants the number of members in each family correlates among examined
species certain families contain numerous members in all three species (eg miR156
miR166 miR169) whereas others consistently contain fewer genes (eg miR162
mir168 miR394) (Table 16) Although it is still unclear why certain miRNA are
encoded by more than one gene in plants (eg 12 genes encoding miR156) this
correlation might suggest functional significance of various miRNA family sizes Most
annotated plant miRNAs are conserved throughout flowering plants while a few
miRNAs are found only in a single sequenced genome and thus they could be
evolutionarily lsquoyoungrsquo miRNAs For example Arabidopsis has several miRNA
families that are not found in poplar or rice suggesting that miRNAs could be
generated continuously during evolution (Allen et al 2004a Rajagopalan et al
2006 Fahlgren et al 2007 Axtell 2012 de Alba et al 2013) Consistent with the
notion that non conserved miRNAs are of a more recent evolutionary origin these
miRNAs are mainly encoded by a single locus in the genome Within each family the
mature miRNA is always located in the same arm
Chapter 1 Introduction and Review of Literature
32
Table 16 MicroRNA families conserved in plants
miRNA
family
Arabidopsis Oryza Populus
miR156 12 12 11 miR159319 6 8 15 miR160 3 6 8 miR162 2 2 3 miR164 3 5 6 miR166 9 12 17 miR167 4 9 8 miR168 2 2 2 miR169 14 17 32 miR171 4 7 10 miR172 5 3 9 miR390 3 1 4 miR393 2 2 4 miR394 2 1 2 miR395 6 19 10 miR396 2 5 7 miR397 2 2 3 miR398 3 2 3 miR399 6 11 12 miR408 1 1 1 miR403 1 0 2 miR437 0 1 0 miR444 0 1 0 miR445 0 9 0 Total 92 127 169
All known miRNA families that are conserved between more than one plant species are listed
together with the number of genes identified in the sequenced genomes Rice miRNA families that have orthologs in maize but do not appear to have orthologs in the eudicots (Arabidopsis and Populus) are marked with an asterisk The following families contain miRNA genes annotated with
more than one number miR156 (miR156 and miR157) miR159319 (miR159 and miR319) miR166 (miR165 and miR166) miR171 (miR170 and miR171) and miR390 (miR390 and miR391)
of the stem loop (5prime- or 3prime) as it would be expected if the genes carry common ancestry
(Figure 17) The sequence of the mature miRNA and to some extent the segment on
the opposite arm of the hairpin to which it pairs is highly conserved between the
members of the same family (both within and between the species) while the sequence
secondary structure and length of the intervening ldquolooprdquo region can be highly divergent
within the same family
The patterns of paring and non pairing nucleotides are often conserved between
homologous miRNA stem loops from different species (Figure 17) The significance of
conserved mismatches is opined to guide DCL1 to cleave at the appropriate positions
along the stem loop
Chapter 1 Introduction and Review of Literature
33
Figure 17 Representative stem loop of miR164 Segments corresponding to the mature miRNAs
are shown in red (Adapted from Rajagopalan et al 2006)
Although many miRNAs are highly conserved in plants the degree of conservation in
most cases is quite low between plants and animals one exception is miR854 A recent
study has revealed that miR854 is conserved between Arabidopsis and animals and that
the Arabidopsis miR854 differs from the human miR854 only by a single nucleotide
(Arteaga-Vazquez et al 2006 Axtell 2012 de Alba et al 2013) The Arabidopsis
miR854 has multiple binding sites in the 3prime-untranslated region (UTR) of
OLIGOURIDYLATE-binding-PROTEIN mRNA 1bA (UBP1b) and represses the
translation of the UBP1b mRNA The animal miR854 is also complementary to a site in
the 3prime-UTR of the UBP1b homolog in animals However it is currently unclear how the
animal miR854 regulates its target mRNA Another distinction between plant and
animal miRNAs is the genomic organization of the miRNA genes While plant miRNA
genes are located predominantly in the intergenic regions animal miRNA genes tend to
localize in the intragenic regions both in the introns or exons of protein coding genes
Moreover miRNA genes are often clustered within a single precursor transcript
whereas individual plant miRNAs are derived from individual precursor RNAs (Bartel
Chapter 1 Introduction and Review of Literature
34
2004 Bonnet et al 2004 Kim 2005 Vaucheret et al 2006 Marco et al 2013)
1133 Non-conserved miRNA and challenges in annotation
Most miRNAs are conserved throughout flowering plants (Table 15) but many more
were found only in a single sequenced genome thus considered to be of a more recent
evolutionary origin The extended homology between few non conserved miRNAs
precursors and their target genes provides strong evidence that these relatively ldquoYoungrdquo
miRNAs arose from the duplication of the target gene segments (Allen et al 2004)
Several non-conserved miRNAs including miR161 miR163 miR173 miR447
miR475 and miR476 are known to direct cleavage of target transcripts (Allen et al
2004a Adai et al 2005 Sunkar et al 2005 de Alba et al 2013) It is difficult to
confidently predict targets for many because it is not possible to use complementary
site as a filter against false-positive target predictions It is also difficult to
confidently annotate non-conserved miRNAs as miRNA rather than siRNAs The
established minimal standard for miRNA annotation is a small RNA with detectable
expression and the potential to form a stem-loop when joined to flanking genomic
sequence (Kasschau et al 2003) In practice these requirements are too loose to
definitively categorize many small RNAs cloned from plants Many plant siRNAs are
detectable on blots (Vazquez et al 2004b) and hundreds of thousands of non-
miRNA genomic sequences can be predicted to fold into secondary structures that
resemble structures of plant miRNA precursors (Jones-Rhoades amp Bartel 2004)
Therefore without conservation of both sequence and secondary structure it is
difficult to be confident that a given cloned RNA originated from a stem-loop (ie is a
miRNA) rather than from a double-stranded RNA (ie is a siRNA) In fact many of
the thousands o f small RNAs cloned from Arabidopsis (Gustafson et al 2005) would
probably meet the literal requirements for annotation as miRNAs A few of these
sequences might be miRNAs but others that meet the literal criteria probably are not
so The challenges of annotating non conserved small RNAs were evidenced by
three related small silencing RNAs that were originally annotated as miRNAs
that turned out to be trans-acting siRNAs (tasiRNAs)(Kanno et al2013)
Although most miRNAs that are broadly conserved among flowering plants are
now being identified and experimentally validated (Table 16)(Jones-Rhoades amp
Bartel 2004) the challenges and ambiguities for classifying non-conserved miRNAs
prevent meaningful estimates of the total number of miRNA genes in Arabidopsis and
Chapter 1 Introduction and Review of Literature
35
other plant genomes It is possible that miRNAs might have escaped detection
because they are non-conserved and expressed in specific tissues or conditions and can
only be identified by using high- throughput sequencing
1134 MicroRNA biogenesis
11341 Transcription of microRNA precursor
Plant miRNAs are primarily found in the genomic regions that are not associated with
the protein coding genes (Reinhart et al 2002) and it appears that most if not all plant
miRNAs are produced from their own transcriptional units Plant miRNA genes are
occasionally clustered near each other in the genome suggesting transcription of
multiple miRNAs from a single primary transcript [eg the miR395 cluster (Grishok
et al 2001 Jones-Rhoades amp Bartel 2004b)] but this polycistronic arrangement of
miRNA genes appears far less frequently in plants than in animals (Bartel 2004)
Northern blot EST and mapping evidence indicate that plant miRNA primary
transcripts (also known as pri-miRNAs) as in animals are longer than needed to
encompass the miRNA stem-loops (Palatnik et al 2003 Jones-Rhoades amp Bartel
2004b) At least some of these pri-miRNA transcripts appear to be spliced
polyadenylated (Kurihara amp Watanabe 2004) and capped (Xie Z et al 2005) Two
rice miRNAs are contained within transcripts that contain exon junctions within the
presumptive stem-loop precursor implying that in these cases splicing is a prerequisite
for Dicer recognition (Sunkar et al 2005) Plant pri-miRNAs are over 1kb in length
and they are usually preceded by typical TATA box motifs They can also undergo
canonical splicing polyadenylation and capping All these observations indicates RNA
polymerase II is probably responsible for transcribing most plant miRNAs (Xie Z et
al 2005) as appears to be the case for many animal miRNAs Relatively little is
known about the regulation of miRNA transcription in plants but there is no reason
to suspect that this regulation would differ from that of protein-coding transcripts as
the bioinformatics analysis of the sequence upstream of the transcriptional start
sites of the miRNA genes showed the presence of putative binding motifs of
number of known transcription factors (Megraw et al 2006 Sethupathy 2013)
11342 MicroRNA processing and export
In plants maturation of miRNA is a step wise process A miRNA gene is transcribed to
pri-miRNA which is much longer than the pre-miRNA possessing the characteristic
stem-loop structure Like transcription of most protein-coding genes the miRNA
Chapter 1 Introduction and Review of Literature
36
transcription is processed by RNA polymerase II (Pol II) enzymes and the pri-
miRNAs are 5prime-capped and 3prime-polyadenylated (Lee et al 2004 Xie Z et al 2005)
Mature miRNAs are produced from pri-miRNAs through at least two sequential
processing steps by RNase III-type endonucleases In animals pri-miRNAs are first
processed at the bottom portion of the stem by Drosha a nuclear-localized RNase III
enzyme to liberate pre-miRNAs The pre-miRNAs are then exported to the cytoplasm
with the help of exportin-5The pre-miRNAs are further processed by Dicer another
RNaseIII enzyme to a duplex consisting of the miRNA and the antisense strands
(miRNA) Since plants do not have any Drosha homologs the two sequential
processing steps seem to be carried out by DCL1 a Dicer homolog in the nucleus
(Kurihara amp Watanabe 2004 de Alba et al 2013 Rogers amp Chen 2013)
The processing of pri-miRNAs to pre-miRNAs by DCL1 is assisted by two
other proteins HYPONASTY LEAVES1 (HYL1) and SERRATE (SE) HYL1 belongs to a
family of double-stranded RNA (dsRNA) binding protein and interacts with DCL1 in
vitro and in vivo (Lu amp Fedoroff 2000 Hiraguri et al 2005 de Alba et al 2013
Rogers amp Chen 2013) Loss-of-function mutations in the HYL1 gene result in
decreased accumulation of miRNAs and concomitant accumulation of pri-miRNAs
(Han et al 2004 Vazquez et al 2004a Kurihara et al 2006) SE a C2H2 zinc finger
protein interacts with DCL1 and HYL1 and plays a role in the processing of pri-
miRNAs to pre-miRNAs (Lobbes et al 2006 Yang L et al 2006) In addition the
nuclear cap-binding complex proteins CBP20 and CBP80 that are known as ABA
HYPER SENSITIVE 1(ABH1) have been shown to be required for proper miRNA
processing (Gregory et al 2008 Laubinger et al 2008) Recently it has been
demonstrated that DCL1 can release 21-nt RNAs from dsRNA as well as from
synthetic miR167 precursor RNAs in vitro However this cleavage is error prone and
inefficient in the absence of HYL1 and SE which accelerate the rate of DCL1-mediated
cleavage and promote accurate processing of miRNAs (Dong et al 2008 Rogers amp
Chen 2013)
11343 Methylation of the miRNAmiRNAduplex
After the miRNAmiRNAduplexes are released from the pre-miRNAs by the DCL1
activities the duplexes are methylated at the 2primeOH of the3prime-ends by HUA ENHANCER1
(HEN1) a small RNA methyltransferase (Yu et al 2005 Yang Z et al 2006 Rogers
amp Chen 2013 ) This step which is distinct from the RNase III-mediated processing
Chapter 1 Introduction and Review of Literature
37
step distinguishes the biogenesis of plant miRNAs from that of animal miRNAs
Mutations in the HEN1 gene were first isolated in a morphological screening for floral
development although the HEN1 mutants display pleiotropic phenotypes similar to the
developmental defects observed in the DCL1 mutants (Chen et al 2002 Park et al
2002 Rogers amp Chen 2013)
Figure 18 Biogenesis of microRNA
A Biogenesis of plant MicroRNA B Biogenesis of metazoananimal MicroRNA
The miRNA (red) is incorporated into RISC and the miRNA(blue) in degraded
This enzyme has two dsRNA-binding domains and nuclear localization signal It
is also conserved in fungi and is required for the processing of siRNAs as well as of
miRNAs (Park et al 2002 Boutet et al 2003 Xie et al 2003) Although the HEN1
protein is localized to the nucleus its methylation activity in the cytoplasm cannot be
ignored (Xie et al 2004 Rogers amp Chen 2013)
Chapter 1 Introduction and Review of Literature
38
The methylation of the miRNAmiRNA duplex is required to protect miRNAs
against the 3prime-end uridylation activity and subsequent degradation (Li et al 2005) In
the hen1 mutants accumulation of miRNAs is reduced to a lower level Also the
miRNAs appear to be heterogeneous in their sizes such that a ladder of bands rather
than a single species is observed for most miRNAs in the mutants (Li et al 2005)
MiRNA cloning and sequencing analyses revealed the presence of a number of U
residues at the 3prime-end of miRNAs in the hen1 mutants Moreover these nucleotides do
not correspond to the miRNAs in the pre-miRNAs indicating that these nucleotides are
added after the processing of the miRNAs from pre-miRNAs (Li et al 2005)
Uridylation of RNAs has also been proven to be correlated with RNA decay (Shen amp
Goodman 2004 Mullen amp Marzluff 2008) suggesting that uridylation attracts an
exonuclease which degrades miRNAs in plants
11344 MiRNA incorporation in to the RISC and its nuclear export
The methylated miRNAmiRNA duplexes undergo RISC assembly In this process
the miRNA of the duplexes is selectively incorporated into the RISC and the miRNA
appears to be removed and subsequently degraded (Hammond et al 2000 Hutvagner
amp Zamore 2002 Schwarz et al 2003 de Alba et al 2013 Rogers amp Chen 2013)
The ARGONAUTE 1(AGO1) proteins are core components of the RISC complex
They contain two structural domains the N-terminal PAZ domain that binds to the 3prime-
end of single-stranded RNAs and the C-terminal piwi domains that has a structure
similar to that of RNase H (Cerutti et al 2000 Parker et al 2004) Several AGO
proteins possess endonucleolytic activities within the PIWI domain and cleave target
mRNAs in the middle of the site complementary to miRNA (Liu et al 2004 Meister et
al 2004 Miyoshi et al 2005) Mutations in the AGO1 gene cause miRNA target
genes to be ectopically expressed and the levels of some miRNAs are reduced in the
mutants (Kidner amp Martienssen 2004 Vaucheret et al 2004 Kidner amp Martienssen
2005 Ronemus et al 2006) Moreover the phenotype of the AGO1 hypomorphic
alleles display defects in lateral organ polarity and leaf and flower morphology as
observed in the dcl1 mutants and null AGO1 alleles are embryonically lethal
supporting the close relationship between the AGO proteins and miRNA processing
(Kidner amp Martienssen 2004 Vaucheret et al 2004 de Alba et al 2013 Rogers amp
Chen 2013)
The production of miRNA miRNA duplexes occur in the nucleus while
Chapter 1 Introduction and Review of Literature
39
miRNA-RISC complexes are present in the cytoplasm where target mRNAs are
located This discordance of functional sites requires an export step during miRNA
biogenesis HASTY the Arabidopsis homolog of exportin-5 is thought to be involved
in miRNA nuclear export (Bollman et al 2003 Park et al 2005)The hasty mutant
exhibits pleiotropic defects in plant development and reduced accumulation of most
miRNAs as in the DCL1 or AGO1 mutants Howeverit is unclear whether the HASTY
proteins export the miRNA miRNA duplexes or the miRNA-RISC complexes in
plants
11345 Feedback regulation of miRNA biogenesis
MiRNA biogenesis is under feedback regulation such that the DCL1 and AGO1 genes
are themselves regulated by miRNAs DCL1 is itself a target of miR162 which leads to
the cleavage of the DCL1 mRNA (Xie et al 2003 de Alba et al 2013) Consistent
with this the levels of DCL1 mRNA are elevated in the mutants of miRNA processing
genes in which the miR162 abundance is reduced In addition there is a predicted
hairpin structure of miR838 in the 14th
intron of the DCL1 primary transcript
(Rajagopalan et al 2006) This prediction implies that DCL1-mediated processing
on this site can result in the cleavage of the DCL1 primary transcript into two truncated
fragments the N-terminal capped fragment and the C-terminal polyadenylated
fragment If this is the case it is possible that miRNA biogenesis competes with the
splicing event of the DCL1 pre-mRNA
The AGO1 is also regulated by miRNA There is a complementary site for
miR168 binding in the AGO1 mRNA and the transcript level of the AGO1 mRNA is
reduced through an mRNA cleavage mechanism (Vaucheret et al 2006 de Alba et al
2013 Rogers amp Chen 2013) indicating that the AGO1 mRNA levels are tightly
controlled by the levels of the AGO1 protein
1135 Mechanism of miRNA action
11351 MiRNA-directed mRNA cleavage
By forming the miRNA-RISC assembly miRNAs can direct the RISC to regulate gene
expression by two post transcriptional mechanisms mRNA cleavage or translational
repression (Bartel 2004 Jones-Rhoades et al 2006 Dalmay 2013) The degree
of miRNA-mRNA complementarity plays an important role for the
determination of the mechanism to be used A general rule is that while perfect or
Chapter 1 Introduction and Review of Literature
40
near-perfect complementarity induces mRNA cleavage central mismatches trigger
translational repression (Carrington amp Ambros 2003 Jones-Rhoades amp Bartel 2004)
Unlike most animal miRNAs plant miRNAs have near-perfect or perfect
complementarity to their targets and mRNA cleavage is assumed to be a predominant
mechanism by which miRNAs regulate gene expression in plants
In RNA cleavage mechanism miRNAs guide the AGO component of RISC to
cleave a single phosphodiester bond of the target mRNA within the miRNA-binding
site (Llave et al 2002b Tang et al 2003) The truncated fragments are then released
and degraded freeing the RISC to recognize and cleave another target mRNA This has
been validated by the detection of 3prime cleavage products that have 5prime ends that start at the
middle of the complementary regions The mRNA cleavage products are then further
degraded by other mechanisms The 3prime cleavage products of some miRNA targets are
degraded by the 5prime-3prime exonuclease XRN4 an Arabidopsis homolog of the major Yeast
mRNA degrading exonuclease Xrn1p It has been observed that the 3prime cleavage
products of some target mRNAs were accumulated in the xrn4 mutants (Souret et al
2004 Dalmay 2013) However the 3prime cleavage products of other miRNA targets do
not accumulate in the xrn4 mutants indicating that there are also XRN4 independent
mechanisms The 5prime cleavage products are typically untraceable by RNA gel blot
hybridization but can be detected by sensitive PCR based methods It has been found
that the 5prime cleavage products tend to obtain an oligo U tail at the 3prime ends This 3prime U tail
is correlated with shortening of the 5prime cleavage products from the 5prime end leading to the
conclusion that the 3prime uridylation causes 5 prime- 3 prime exonucleolytic decay of the 5prime cleavage
products (Shen amp Goodman 2004)
DCL1 HYL1 and SE interact with each other and are co-localized in the
nuclear bodies termed Dicing bodies or D-bodies in which some pri-miRNA shave
been found suggesting that D-bodies serve as a center for active miRNA processing
(Fang amp Spector 2007 Fujioka et al 2007 Song et al 2007) The miRNA
processing site appears to be distinct from the Cajal bodies that have previously been
found as a siRNA processing site (Pontes amp Pikaard 2008) The D-bodies include
SmD3 and SmB proteins that are found in the Cajal bodies However At collin an
additional marker for the Cajal bodies is not found in the D-bodies Moreover the D-
bodies are present in an At collin mutant lacking the Cajal bodies The pri-miRNAs of
miR171A and miR159A genes have been found to be co-localized with HYL1 and
Chapter 1 Introduction and Review of Literature
41
DCL1 in the D- bodies that are distinct from the Cajal bodies (Song et al 2007)
11352 MiRNA-directed translational repression
Translation repression is another mechanism exerted by plant miRNAs The regulatory
mechanism of miR172 which regulates flowering time and floral organ identity is
consistent with translation repression MiR172 over production does not affect the
transcript levels of APETALA2 (AP2) and AP2- like target mRNAs Instead the AP2
protein level is reduced in the miR172 over expressing mutant (Aukerman amp Sakai
2003 Chen 2004 Dalmay 2013) It was later shown that miR172 can also direct the
cleavage of the target mRNAs although the steady-state mRNA level is unchanged due
to feedback regulation (Schwab et al 2005 Jung et al 2007) It is clear that miR172
regulates its target genes primarily by translational repression rather than mRNA
cleavage Recently it has been reported that miR156157 and miR854 also regulate
their target genes through translational repression (Arteaga-Vazquez et al 2006
Gandikota et al 2007 Dalmay 2013) It was once thought that translational repression
is an exception to the rule However experimental evidence suggest a widespread
coexistence of translational repression and mRNA cleavage (Brodersen et al 2008)
1136 Regulatory roles of miRNAs in plant development
The miRNA target prediction has been a difficult task in order to obtain regulatory
targets without bringing in too many false predictions Plant growth and developmental
processes regulated by miRNAs are quite diverse and enormous Furthermore plant
miRNAs work cooperatively or antagonistically to establish a balanced regulation This
notion is well consistent with the findings that mutations in the regulators of miRNA
biogenesis transport and processing such as AGO1 DCL1 HYL1 HEN1 ABH1
and HASTY cause pleiotropic phenotypes (Mallory amp Vaucheret 2006 de Alba et al
2013) To date the roles of miRNAs have been mostly deduced from obtaining miRNA
over expressing transgenic plants or gain-of-function mutants in which miRNA-
resistant target genes are ectopically expressed
11361 Phase transition
Plants pass through a series of developmental phase transitions during their life span
beginning with seed germination continuing through vegetative phase change
reproductive phase change flowering initiation and finally seed production for the next
generation Because of their importance in understanding plant developmental
Chapter 1 Introduction and Review of Literature
42
processes genes regulating developmental transitions have been extensively studied
and underlying molecular mechanisms have been explored in various plant species
Majority of researches were mainly focused on vegetative phase change and
reproductive transition in Arabidopsis and Maize (Poethig 2003 Baurle amp Dean
2006) Particularly the mutations of the genes encoding various regulators of miRNA
biogenesis and transport such as HST SE and AGO exhibit altered juvenile to adult
vegetative phase change and vegetative to reproductive change (Hunter et al 2003
Peragine et al 2004 Vazquez et al 2004a Jones-Rhoades et al 2006 de Alba et al
2013)
Over-expression of the miR156a gene causes late flowering and delays
vegetative phase change (Wu amp Poethig 2006 Gandikota et al 2007 Wang et al
2008) It has been shown that miR156 negatively regulates a group of genes encoding
SQUAMOSA PROMOTER-BINDING PROTEIN-LIKE (Lewis et al 2007 de Alba et
al 2013 Rogers amp Chen 2013) transcription factors such as SPL3 SPL4 and SPL5
that promote flowering initiation In contrast transgenic plants over expressing
miR156-resistant SPL345 genes are early flowering Interestingly the endogenous
level of miR156 is temporally regulated In the early juvenile vegetative phase the
level of miR156 is very high but decreases rapidly before the onset of the adult juvenile
phase (Wu amp Poethig 2006)
The Arabidopsis early activation tagged (eat-D) mutant exhibits early flowering
with disrupted floral structure The mutant phenotypes are caused by over expression of
the miR172a-2gene (Aukerman amp Sakai 2003) MiR172 regulates a small group of
genes encoding APETALLA2 (AP2)-like transcription factors such as TARGET OF
EAT1 (TOE1) TOE2 SCHLAFMUTZE (SMZ) and SCHNARCHZAPFEN(SNZ)
TOE1 TOE2 SMZ and SNZ have been shown to participate in their productive phase
change by repressing a floral integrator FLOWERING LOCUS T (FT)(Jung et al
2007) Consistent with this toe1-2D 35STOE2 35SSMZ and 35SSNZ plants
exhibit late flowering (Jung et al 2007) MiR172 is also regulated temporally and
highly expressed in the adult vegetative phase both in Arabidopsis and maize (Chuck et
al 2007) Because the temporal expression patterns of miR156 and miR172 are
complementary to each other it has been suggested that the two miRNAs interact with
each other in regulating phase change (Chuck et al 2009 de Alba et al 2013) In
support of this view it has been shown that miR172 is down-regulated in the
Chapter 1 Introduction and Review of Literature
43
Corngrass1(Cg1) mutant in which miR156 is overproduced (Chuck et al 2007 de
Alba et al 2013) However there is no direct evidence for the direct linkage between
miR156 and miR172 It is also envisioned that miR156 and miR172 would not be
directly related For example miR156 regulates plastochron index that is the inverse of
leaf initiation rate and thus frequently used to analyze temporal patterns of plant
development In contrast miR172 does not affect plastochron (Wang et al 2008) The
down-regulation of miR172 in the maize miR156-over producing mutants would be
due to the prolonged juvenile vegetative phase in the mutants
Another miRNA that regulates flowering initiation is miR159 It regulates
MYB33 and MYB65 which are closely related to the barley GAMY B transcription
factor that responds to gibberellic acid (GA) The level of miR159 is elevated in GA-
treated seedlings but reduced in the GA biosynthetic ga1 mutant Transgenic plants
over producing miR159 exhibit delayed floral induction under short days (Achard et
al 2004) It is been suggested that MYB33 mediates GA signal to regulate LEAFY
(LFY) These observations indicate that miR159 plays an important role in the GA
signaling pathways that regulate proper floral induction under short days (Achard et al
2004)
11362 Floral development
Arabidopsis flowers consist of four distinct organs They arise in a characteristic
pattern within concentric rings called whorls from outside to inside four sepals four
petals six stamens and the central pistil consisting of two carpels Identities of the
floral organs are determined by three classes of regulatory genes classA classB and
class C genes (Krizek amp Fletcher 2005) The A and C class genes act antagonistically
to restrict their expression domains (Bowman et al 1991)
It is notable that miR172 also regulates floral architecture in addition to its role
in the timing of flowering initiation MiR172 inhibits the translation of the AP2 mRNA
(Chen 2004) Transgenic plants overproducing miR172 not only exhibit early
flowering but also show abnormal floral development which is quite similar to those
of the class A gene mutants such as the ap2 mutants (Aukerman amp Sakai 2003 de
Alba et al 2013) When the activity of the class A genes is inactivated that of the class
C genes such as AGAMOUS (AG) is elevated As a result the miR172- overproducing
transgenic plants exhibit no petals along with homeotic transformation of the sepals to
Chapter 1 Introduction and Review of Literature
44
the carpels (Aukerman amp Sakai 2003 Chen 2004)
Interestingly miR166 also regulates floral architecture The role of miR165166
in the regulation of meristem activity is closely linked to floral meristem formation and
organization (Zhao et al 2007 Miyashima et al 2013) Floral structure is severely
disrupted in the miR165166-overproducing mutants such as men1 and jba-1D in
which miR166 is overproduced (Williams et al 2005b Jung amp Park 2007) The men1
and jba-1D mutants have smaller gynoecium and reduced carpel number In an extreme
case the jba-1D homozygous plants lack visible carpels (Williams et al 2005b)
Similar phenotypes have been observed in the miR165 overproducers (Zhao et al
2007) It has been suggested that the phenotypes are possibly caused by altered AG
expression in the floral meristem (Jung amp Park 2007 Turchi et al 2013)
Other miRNAs also affect floral development Overproduction of miRNAs
involved in auxin signaling also affects floral architecture Transgenic plants
overproducing miR167 have defects in the formation of ovule and anther with reduced
fertility (Ru et al 2006) It has been shown that miR167 targets ARF6 and ARF8 that
play a role in gynoecium and stamen development and in regulating fertility (Ru et al
2006 Wu et al 2006) These observations suggest that auxin signals are also closely
related to floral organogenesis In addition transgenic plants over-producing miR164
and the cuc1cuc2 double mutants show fused sepals and reduced petals (Laufs et al
2004 Turchi et al 2013) suggesting that miR164 is also related to floral meristem
activity and boundary specification of the meristematic region
11363 Shoot apical meristem development
Shoot apical meristem comprises self-maintaining totipotent cells The shoot apical
meristem activity affects diverse aspects of plant developmental processes including
leaf development vascular development and lateral organ polarity (Bowman et al
2002) Because miR165166 plays a primary role in meristem formation
overproduction of miR165166 and multiple knockout mutants of its target genes
exhibit pleiotropic pheonotypes (McConnell et al 2001 Emery et al 2003 Kim et al
2005 Williams et al 2005b)
The targets of miR165166 include genes encoding the homeo domain-leucine
zipper (HD-ZIP) class III transcription factors such as PHABULOSA(PHB)
PHAVOLUTA (PHV) REVOLUTA(REV) ATHB15CORONA (CNA) and ATHB8
Chapter 1 Introduction and Review of Literature
45
PHB PHV and ATHB15 have overlapping functions in controlling meristem
development (Turchi et al 2013) Indeed the phb phv athb15 triple mutants have an
enlarged shoot apical meristem Consistent with this the miR166- overproducing men1
and jba-1D mutants exhibit enlarged shoot apical meristems (Kim et al 2005
Williams et al 2005b Turchi et al 2013) In the mutants the shoot apical meristems
are multiplied and large
The development of shoot apical meristems is also regulated by the WUSCHEL
(WUS)ndashCLAVATA (CLV) signaling pathway WUS positively regulates cell
proliferation In contrast CLV3 which is expressed in stem cells limits the WUS
expression and thus inhibits cell proliferation in the SAM (Sharma et al 2003)
Consistent with this notion WUS is expressed to a higher level in jba-1D
explaining the ectopic enlargement of the SAM in the mutant (Williams et al 2005)
While WUS is closely related to miR165166 in the shoot apical meristem
development the underlying molecular mechanisms are currently unclear It has been
observed that the miR166-over producing men1 plants have an arrested meristem
development (Jung amp Park 2007) In addition the heterozygous men1+ and the
homozygous men1 plants show different expression patterns of WUS and CLV3 It
appears that miR166 regulates WUS indirectly and influences the expressional balance
between WUS and CLV3 through a negative feedback loop (Jung amp Park 2007 de
Alba et al 2013)
The phv phb athb15 triple mutant has an enlarged shoot apical meristems the
rev phb phv mutant does not have functional shoot apical meristems (Prigge et al
2005) This distinction may be explained by the fact that while miR166 targets
primarily PHB PHV and ATHB15 miR165 targets all the five HD-ZIP III genes (Zhou
et al 2007) Consistent with this transgenic plants overproducing miR166 are distinct
from those overproducing miR165 The miR165-overproducing transgenic plants lack
functional shoot apical meristem and a pin-like structure is formed at the apical region
of the mutant These phenotypes are quite similar to rev phb phv triple mutant (Zhou et
al 2007 Liu et al 2013) The phenotypic distinction may because by the difference
of the cleavage efficiencies of the target mRNAs In accordance with this notion a few
35S miR166a transgenic lines exhibit no shoot apical meristems The men1
homozygous plants also show arrested meristem development (Jung amp Park 2007)
Chapter 1 Introduction and Review of Literature
46
MiR164 cleaves the CUC1 and CUC2 mRNAs as well as the NAC1 mRNA
CUC1 and CUC2 determines the organ boundary in the meristem Phenotypes of
transgenic plants that overproduce miR164 are similar to those of the cuc1cuc2 double
mutant that exhibits embryonic developmental defects such as cup-shaped cotyledons
and fused cotyledons (Laufs et al 2004) When a miR164-resistant CUC2 is
expressed the boundary domain is enlarged CUC also plays a role in embryonic shoot
meristem formation by regulating the SHOOT MERISTEMLESS gene It was therefore
concluded that miR164-mediated cleavage of CUC1 and CUC2 mRNAs determines the
sizes of the boundary domains in the shoot apical meristem and regulates separation of
the organs by restricting cell proliferation (Laufs et al 2004 de Alba et al 2013)
11364 Leaf development
The role of miR319 has been extensively studied in leaf development A dominant
jaw-D mutant overproducing miR319 shows uneven curved leaves with enlarged size
and five TCP genes which are closely related to the CINCINNATA (CIN) gene in
Antirrhinum majus are down regulated in the mutant suggesting that miR319 mediated
regulation of the TCP transcription factor genes are important for the final step of
leaf shaping (Palatnik et al 2003 Naz et al 2013) In addition to leaf shape and size
leaf polarity is also specified by miRNA MiR166-mediated cleavage of the HD-ZIPIII
mRNAs is a well to establish leaf polarity This discovery originates from the gain-of-
function mutants such as phb-1d and phv-1d wherein point mutations in the
complementary sites to miRNA helps the mRNA to evade or resist from miRNA-
mediated cleavage (McConnell et al 2001 Emery et al 2003 Naz et al 2013)
Plant leaves have a dorso-ventral polarity consisting of the adaxial (upper
surface) and abaxial (lower surface) sides The adaxial side is closer to the shoot
apical meristem Adaxial identity is specified by functionally redundant class III HD-
ZIP family genes such as PHB PHV and REV Ectopic expression of each genes
result in adaxialized leaves Dominant phb-1d and phv-1d mutants exhibit
transformation of the abaxial to adaxial fate and have rod-shaped leaves In contrast
miR165166-overproducing plants show pin-like cotyledons radicalized leaves and
abaxialized leaves (Chitwood et al 2007 Pattanayak et al 2013)
Leaf asymmetry is determined by miR166-mediated restriction of the
expression domains of the HD-ZIPIII genes The HD-ZIPIII genes are expressed in the
Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
27
Figure 16 Mechanism of gene silencing induced double stranded RNA The dicer enzyme to
produce siRNAs cleaves the RNA The siRNA generated bind to the nuclease complex
RISC The antisense component in the siRNA in the RISC guides the complex towards
the cognate mRNA resulting in endonucleolytic cleavage of the mRNA
Chapter 1 Introduction and Review of Literature
28
112 MicroRNA or MiRNA
In 1991 Ambros and co-workers first isolated a lin-4 mutant of Celegans which was
arrested at the first larval stage (Lee et al 1993) Later on the let-7 mutation was
isolated in the same system which was responsible for development through the fourth
larval stage Both lin-4 and let-7 encode short 22-nucleotide mature RNAs and were
called short temporal RNA because they control the temporal development program of
C elegans The mature lin-4 RNA defines (negatively regulates) the mRNA expression
of the lin-14 and lin-28 hetrochronic genes with the antisense mediated repression
mechanism of translation initiation and thus specifies the fate of cells during the first
three larval stages Recent studies have revealed that the short temporal RNAs are
actually members of a group of tiny RNAs (21to 28 nucleotides) called the micro-
RNAs (miRNA) (Bartel 2009) isolated members of which could easily run to a few
thousand which includes both those were identified computationally and by deep
sequencing Some of the components of the RNAi machinery have also been clearly
established as the effector proteins for the maturation of miRNAs
113 MicroRNAs in plants
1131 Discovery
MicroRNAs have been discovered by three basic approaches Direct cloning forward
genetic direct cloning and bioinformatic prediction followed by experimental
validation The most direct method of miRNA discovery is to isolate and clone small
RNAs from biological samples and several groups have used this approach to identify
plant miRNAs (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002 Xie et al 2003 Sunkar et al 2005 Williams et al 2013) The cloning methods
adapted were similar to those used to identify large numbers of animal miRNAs
(Lagos-Quintana et al 2001 Lau et al 2001 Aravin amp Tuschl 2005) which involve
isolating small RNAs followed by oligonucleotide adapter ligation reverse
transcription amplification and sequencing Some protocols incorporate methods to
enrich for Dicer cleavage products (ie molecules with 5prime-phosphates and 3prime-
hydroxyls) and to concatemerize the short cDNAs so that several may be identified in a
single sequencing read (Lau et al 2001 Llave et al 2002a Jagadeeswaran amp Sunkar
2013)The initial cloning experiments in Arabidopsis identified 19 miRNAs belonging
to 15 families (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002) although hundreds of other small RNAs such as siRNAs and RNA degradation
Chapter 1 Introduction and Review of Literature
29
fragments were also cloned through the direct cloning methods To select miRNAs
from the pool of small RNAs secondary structures of the RNA molecules were
examined in compliance with the acknowledged structural features of miRNAs
(Ambros et al 2003) The secondary structures were validated using several ad-hoc
software tools such as the Mfold or RNAfold programs (Reeder et al 2006) Finally
the existence of mature miRNAs was determined by RNA gel blot hybridization These
direct cloning methods were successful in isolating many miRNAs in Arabidopsis
(Reinhart et al 2002 Sunkar amp Zhu 2004) Rice (Sunkar et al 2005) Poplar (Lu et
al 2005) moss (Arazi et al 2005) and Tobacco (Billoud et al 2005)
Although miRNAs were first discovered through forward genetic screens in
worms (Lee et al 1993 Reinhart et al 2000)only few A thaliana miRNA gene
families have been discovered by this method (Chen 2008) in plants MiRNA
involvement in plant mutant phenotypes were not properly inferred till the cloning
experiments established that plant genomes contain numerous miRNAs (Park et al
2002 Reinhart et al 2002 Rhoades et al 2002) MiRNA loss-of-function allele has
been identified by forward genetic screens early extra petala1 is caused by a
transposon insertion ~160bp upstream of the predicted miR164c stem loop resulting in
flowers with extra petals (Baker et al 2005 Irish 2008) The fact that loss-of-function
miRNA mutants have been rarely discovered using forward genetics reflects small
target size for mutagenesis coupled with redundancy in nearly all evolutionarily
conserved plant miRNAs which are encoded by gene families (Table16) Family
members are likely to have overlapping functions buffering against loss at any single
miRNA locus Over expression screens can circumvent redundancy limitations At least
four plant miRNAs miR319 (also known as miR-JAW) miR172 (also known as EAT)
and miR166 were isolated in over expression screens for dominant mutants with
developmental abnormalities (Aukerman amp Sakai 2003 Palatnik et al 2003 Kim et
al 2005 Williams et al 2005b Schommer et al 2012) Mutations in miRNA target
sites which can prevent the entire family of miRNAs from repressing a target gene can
also circumvent redundant functions of miRNA family members
The dominant mutations in the HD-ZIP genes PHB PHV and REVOLUTA
(REV) in Arabidopsis and ROLLEDLEAF1 (RLD1) in Maize result in adaxialization of
leaves andor Vasculature (McConnell amp Barton 1998 McConnell et al 2001 Nelson
et al 2002 Zhong amp Ye 2004 Allen amp Millar 2012) and are all caused by mutations
Chapter 1 Introduction and Review of Literature
30
in miR166 complementary sites (Rhoades et al 2002 Emery et al 2003 Jones-
Rhoades amp Bartel 2004 Mallory et al 2004 Zhong amp Ye 2004)
In both plants and animals cloning was the initial means of large-scale
miRNA discovery (Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Reinhart et al 2002 Mendes et al 2012) However cloning is biased toward
RNAs that are expressed highly and broadly MiRNAs expressed at low levels or
only in specific cell types or in response to certain environmental stimuli were more
difficult to clone Sequence based biases in cloning procedures might also cause
certain miRNAs to be missed Because of these limitations bioinformatics approaches
to identify miRNAs have provided a useful complement to cloning
A straightforward use of bioinformatics has been carried out to find homologs
of known miRNAs both within the same genome and in the genomes of other species
(Pasquinelli et al 2000 Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Mendes et al 2009) A more difficult challenge is to identify miRNAs
unrelated to previously known miRNAs This was first accomplished for
vertebrate nematode and fly miRNAs using algorithms that search for conservation
of sequence and secondary structure (ie miRNA stem-loop precursors) between
species searching for patterns that are characteristic of miRNAs (Lai et al 2003 Lim
et al 2003 Lim et al 2005)
Most of the methods identified numerous miRNAs but are necessarily more
relaxed in terms of allowed structures Some approaches take advantage of the high
complementarity of plant miRNAs to target messages implementing the requirement
that the candidate has conserved complementarity to mRNAs (Jones-Rhoades amp Bartel
2004 Adai et al 2005) This additional filter has been useful for distinguishing
authentic plant miRNAs from false positives which later extended to mammalian
miRNA gene prediction (Xie et al 2005 Mendes et al 2012)
Another criteria used by most algorithms in the filters is evolutionary
conservation In plants mature miRNA sequences are conserved to a higher degree
than their precursor sequences For example Arabidopsis and rice diverged more than
130 million years ago (Friis et al 2004) but some miRNAs are highly conserved
in these plant species (Reinhart et al 2002) Furthermore various parameters for the
secondary structures such as free energy the number of paired residues within a
Chapter 1 Introduction and Review of Literature
31
miRNA or the number and size of bulges are also used to validate the predictions
(Jones-Rhoades amp Bartel 2004 Wang et al 2004 Adai et al 2005 Mendes et al
2012)
1132 Conserved microRNAs in Plants
Till date cloning genetics and bioinformatics screens have resulted in annotation of
approximately 184 potential Arabidopsis miRNAs (miRBase release 13) These
miRNAs seem to arise through gene duplication and subsequent diversification and
represent approximately 100 families (Maher et al 2006 Axtell 2013) Twenty one
families represented by 92 genes are clearly conserved in species beyond Arabidopsis
(Table 15) The presence of these miRNA families in other sequenced plant genomes
indicates that different plant species have an evolutionarily fluid set of miRNAs
(Willmann amp Poethig 2007) Recent studies on mosses one of the most ancient land
plants have shown that at least 14 miRNA families are conserved in eudicots and
mosses and therefore it appears that plant miRNAs share a common ancient origin
(Arazi et al 2005 Axtell amp Bartel 2005 Talmor-Neiman et al 2006 Zhang et al
2006 Axtell 2013) The number of members per family in a genome ranges from 1-32
with the exception of miR-430 which is represented by a cluster of ~80 loci in zebra
fish (Giraldez et al 2005)
In plants the number of members in each family correlates among examined
species certain families contain numerous members in all three species (eg miR156
miR166 miR169) whereas others consistently contain fewer genes (eg miR162
mir168 miR394) (Table 16) Although it is still unclear why certain miRNA are
encoded by more than one gene in plants (eg 12 genes encoding miR156) this
correlation might suggest functional significance of various miRNA family sizes Most
annotated plant miRNAs are conserved throughout flowering plants while a few
miRNAs are found only in a single sequenced genome and thus they could be
evolutionarily lsquoyoungrsquo miRNAs For example Arabidopsis has several miRNA
families that are not found in poplar or rice suggesting that miRNAs could be
generated continuously during evolution (Allen et al 2004a Rajagopalan et al
2006 Fahlgren et al 2007 Axtell 2012 de Alba et al 2013) Consistent with the
notion that non conserved miRNAs are of a more recent evolutionary origin these
miRNAs are mainly encoded by a single locus in the genome Within each family the
mature miRNA is always located in the same arm
Chapter 1 Introduction and Review of Literature
32
Table 16 MicroRNA families conserved in plants
miRNA
family
Arabidopsis Oryza Populus
miR156 12 12 11 miR159319 6 8 15 miR160 3 6 8 miR162 2 2 3 miR164 3 5 6 miR166 9 12 17 miR167 4 9 8 miR168 2 2 2 miR169 14 17 32 miR171 4 7 10 miR172 5 3 9 miR390 3 1 4 miR393 2 2 4 miR394 2 1 2 miR395 6 19 10 miR396 2 5 7 miR397 2 2 3 miR398 3 2 3 miR399 6 11 12 miR408 1 1 1 miR403 1 0 2 miR437 0 1 0 miR444 0 1 0 miR445 0 9 0 Total 92 127 169
All known miRNA families that are conserved between more than one plant species are listed
together with the number of genes identified in the sequenced genomes Rice miRNA families that have orthologs in maize but do not appear to have orthologs in the eudicots (Arabidopsis and Populus) are marked with an asterisk The following families contain miRNA genes annotated with
more than one number miR156 (miR156 and miR157) miR159319 (miR159 and miR319) miR166 (miR165 and miR166) miR171 (miR170 and miR171) and miR390 (miR390 and miR391)
of the stem loop (5prime- or 3prime) as it would be expected if the genes carry common ancestry
(Figure 17) The sequence of the mature miRNA and to some extent the segment on
the opposite arm of the hairpin to which it pairs is highly conserved between the
members of the same family (both within and between the species) while the sequence
secondary structure and length of the intervening ldquolooprdquo region can be highly divergent
within the same family
The patterns of paring and non pairing nucleotides are often conserved between
homologous miRNA stem loops from different species (Figure 17) The significance of
conserved mismatches is opined to guide DCL1 to cleave at the appropriate positions
along the stem loop
Chapter 1 Introduction and Review of Literature
33
Figure 17 Representative stem loop of miR164 Segments corresponding to the mature miRNAs
are shown in red (Adapted from Rajagopalan et al 2006)
Although many miRNAs are highly conserved in plants the degree of conservation in
most cases is quite low between plants and animals one exception is miR854 A recent
study has revealed that miR854 is conserved between Arabidopsis and animals and that
the Arabidopsis miR854 differs from the human miR854 only by a single nucleotide
(Arteaga-Vazquez et al 2006 Axtell 2012 de Alba et al 2013) The Arabidopsis
miR854 has multiple binding sites in the 3prime-untranslated region (UTR) of
OLIGOURIDYLATE-binding-PROTEIN mRNA 1bA (UBP1b) and represses the
translation of the UBP1b mRNA The animal miR854 is also complementary to a site in
the 3prime-UTR of the UBP1b homolog in animals However it is currently unclear how the
animal miR854 regulates its target mRNA Another distinction between plant and
animal miRNAs is the genomic organization of the miRNA genes While plant miRNA
genes are located predominantly in the intergenic regions animal miRNA genes tend to
localize in the intragenic regions both in the introns or exons of protein coding genes
Moreover miRNA genes are often clustered within a single precursor transcript
whereas individual plant miRNAs are derived from individual precursor RNAs (Bartel
Chapter 1 Introduction and Review of Literature
34
2004 Bonnet et al 2004 Kim 2005 Vaucheret et al 2006 Marco et al 2013)
1133 Non-conserved miRNA and challenges in annotation
Most miRNAs are conserved throughout flowering plants (Table 15) but many more
were found only in a single sequenced genome thus considered to be of a more recent
evolutionary origin The extended homology between few non conserved miRNAs
precursors and their target genes provides strong evidence that these relatively ldquoYoungrdquo
miRNAs arose from the duplication of the target gene segments (Allen et al 2004)
Several non-conserved miRNAs including miR161 miR163 miR173 miR447
miR475 and miR476 are known to direct cleavage of target transcripts (Allen et al
2004a Adai et al 2005 Sunkar et al 2005 de Alba et al 2013) It is difficult to
confidently predict targets for many because it is not possible to use complementary
site as a filter against false-positive target predictions It is also difficult to
confidently annotate non-conserved miRNAs as miRNA rather than siRNAs The
established minimal standard for miRNA annotation is a small RNA with detectable
expression and the potential to form a stem-loop when joined to flanking genomic
sequence (Kasschau et al 2003) In practice these requirements are too loose to
definitively categorize many small RNAs cloned from plants Many plant siRNAs are
detectable on blots (Vazquez et al 2004b) and hundreds of thousands of non-
miRNA genomic sequences can be predicted to fold into secondary structures that
resemble structures of plant miRNA precursors (Jones-Rhoades amp Bartel 2004)
Therefore without conservation of both sequence and secondary structure it is
difficult to be confident that a given cloned RNA originated from a stem-loop (ie is a
miRNA) rather than from a double-stranded RNA (ie is a siRNA) In fact many of
the thousands o f small RNAs cloned from Arabidopsis (Gustafson et al 2005) would
probably meet the literal requirements for annotation as miRNAs A few of these
sequences might be miRNAs but others that meet the literal criteria probably are not
so The challenges of annotating non conserved small RNAs were evidenced by
three related small silencing RNAs that were originally annotated as miRNAs
that turned out to be trans-acting siRNAs (tasiRNAs)(Kanno et al2013)
Although most miRNAs that are broadly conserved among flowering plants are
now being identified and experimentally validated (Table 16)(Jones-Rhoades amp
Bartel 2004) the challenges and ambiguities for classifying non-conserved miRNAs
prevent meaningful estimates of the total number of miRNA genes in Arabidopsis and
Chapter 1 Introduction and Review of Literature
35
other plant genomes It is possible that miRNAs might have escaped detection
because they are non-conserved and expressed in specific tissues or conditions and can
only be identified by using high- throughput sequencing
1134 MicroRNA biogenesis
11341 Transcription of microRNA precursor
Plant miRNAs are primarily found in the genomic regions that are not associated with
the protein coding genes (Reinhart et al 2002) and it appears that most if not all plant
miRNAs are produced from their own transcriptional units Plant miRNA genes are
occasionally clustered near each other in the genome suggesting transcription of
multiple miRNAs from a single primary transcript [eg the miR395 cluster (Grishok
et al 2001 Jones-Rhoades amp Bartel 2004b)] but this polycistronic arrangement of
miRNA genes appears far less frequently in plants than in animals (Bartel 2004)
Northern blot EST and mapping evidence indicate that plant miRNA primary
transcripts (also known as pri-miRNAs) as in animals are longer than needed to
encompass the miRNA stem-loops (Palatnik et al 2003 Jones-Rhoades amp Bartel
2004b) At least some of these pri-miRNA transcripts appear to be spliced
polyadenylated (Kurihara amp Watanabe 2004) and capped (Xie Z et al 2005) Two
rice miRNAs are contained within transcripts that contain exon junctions within the
presumptive stem-loop precursor implying that in these cases splicing is a prerequisite
for Dicer recognition (Sunkar et al 2005) Plant pri-miRNAs are over 1kb in length
and they are usually preceded by typical TATA box motifs They can also undergo
canonical splicing polyadenylation and capping All these observations indicates RNA
polymerase II is probably responsible for transcribing most plant miRNAs (Xie Z et
al 2005) as appears to be the case for many animal miRNAs Relatively little is
known about the regulation of miRNA transcription in plants but there is no reason
to suspect that this regulation would differ from that of protein-coding transcripts as
the bioinformatics analysis of the sequence upstream of the transcriptional start
sites of the miRNA genes showed the presence of putative binding motifs of
number of known transcription factors (Megraw et al 2006 Sethupathy 2013)
11342 MicroRNA processing and export
In plants maturation of miRNA is a step wise process A miRNA gene is transcribed to
pri-miRNA which is much longer than the pre-miRNA possessing the characteristic
stem-loop structure Like transcription of most protein-coding genes the miRNA
Chapter 1 Introduction and Review of Literature
36
transcription is processed by RNA polymerase II (Pol II) enzymes and the pri-
miRNAs are 5prime-capped and 3prime-polyadenylated (Lee et al 2004 Xie Z et al 2005)
Mature miRNAs are produced from pri-miRNAs through at least two sequential
processing steps by RNase III-type endonucleases In animals pri-miRNAs are first
processed at the bottom portion of the stem by Drosha a nuclear-localized RNase III
enzyme to liberate pre-miRNAs The pre-miRNAs are then exported to the cytoplasm
with the help of exportin-5The pre-miRNAs are further processed by Dicer another
RNaseIII enzyme to a duplex consisting of the miRNA and the antisense strands
(miRNA) Since plants do not have any Drosha homologs the two sequential
processing steps seem to be carried out by DCL1 a Dicer homolog in the nucleus
(Kurihara amp Watanabe 2004 de Alba et al 2013 Rogers amp Chen 2013)
The processing of pri-miRNAs to pre-miRNAs by DCL1 is assisted by two
other proteins HYPONASTY LEAVES1 (HYL1) and SERRATE (SE) HYL1 belongs to a
family of double-stranded RNA (dsRNA) binding protein and interacts with DCL1 in
vitro and in vivo (Lu amp Fedoroff 2000 Hiraguri et al 2005 de Alba et al 2013
Rogers amp Chen 2013) Loss-of-function mutations in the HYL1 gene result in
decreased accumulation of miRNAs and concomitant accumulation of pri-miRNAs
(Han et al 2004 Vazquez et al 2004a Kurihara et al 2006) SE a C2H2 zinc finger
protein interacts with DCL1 and HYL1 and plays a role in the processing of pri-
miRNAs to pre-miRNAs (Lobbes et al 2006 Yang L et al 2006) In addition the
nuclear cap-binding complex proteins CBP20 and CBP80 that are known as ABA
HYPER SENSITIVE 1(ABH1) have been shown to be required for proper miRNA
processing (Gregory et al 2008 Laubinger et al 2008) Recently it has been
demonstrated that DCL1 can release 21-nt RNAs from dsRNA as well as from
synthetic miR167 precursor RNAs in vitro However this cleavage is error prone and
inefficient in the absence of HYL1 and SE which accelerate the rate of DCL1-mediated
cleavage and promote accurate processing of miRNAs (Dong et al 2008 Rogers amp
Chen 2013)
11343 Methylation of the miRNAmiRNAduplex
After the miRNAmiRNAduplexes are released from the pre-miRNAs by the DCL1
activities the duplexes are methylated at the 2primeOH of the3prime-ends by HUA ENHANCER1
(HEN1) a small RNA methyltransferase (Yu et al 2005 Yang Z et al 2006 Rogers
amp Chen 2013 ) This step which is distinct from the RNase III-mediated processing
Chapter 1 Introduction and Review of Literature
37
step distinguishes the biogenesis of plant miRNAs from that of animal miRNAs
Mutations in the HEN1 gene were first isolated in a morphological screening for floral
development although the HEN1 mutants display pleiotropic phenotypes similar to the
developmental defects observed in the DCL1 mutants (Chen et al 2002 Park et al
2002 Rogers amp Chen 2013)
Figure 18 Biogenesis of microRNA
A Biogenesis of plant MicroRNA B Biogenesis of metazoananimal MicroRNA
The miRNA (red) is incorporated into RISC and the miRNA(blue) in degraded
This enzyme has two dsRNA-binding domains and nuclear localization signal It
is also conserved in fungi and is required for the processing of siRNAs as well as of
miRNAs (Park et al 2002 Boutet et al 2003 Xie et al 2003) Although the HEN1
protein is localized to the nucleus its methylation activity in the cytoplasm cannot be
ignored (Xie et al 2004 Rogers amp Chen 2013)
Chapter 1 Introduction and Review of Literature
38
The methylation of the miRNAmiRNA duplex is required to protect miRNAs
against the 3prime-end uridylation activity and subsequent degradation (Li et al 2005) In
the hen1 mutants accumulation of miRNAs is reduced to a lower level Also the
miRNAs appear to be heterogeneous in their sizes such that a ladder of bands rather
than a single species is observed for most miRNAs in the mutants (Li et al 2005)
MiRNA cloning and sequencing analyses revealed the presence of a number of U
residues at the 3prime-end of miRNAs in the hen1 mutants Moreover these nucleotides do
not correspond to the miRNAs in the pre-miRNAs indicating that these nucleotides are
added after the processing of the miRNAs from pre-miRNAs (Li et al 2005)
Uridylation of RNAs has also been proven to be correlated with RNA decay (Shen amp
Goodman 2004 Mullen amp Marzluff 2008) suggesting that uridylation attracts an
exonuclease which degrades miRNAs in plants
11344 MiRNA incorporation in to the RISC and its nuclear export
The methylated miRNAmiRNA duplexes undergo RISC assembly In this process
the miRNA of the duplexes is selectively incorporated into the RISC and the miRNA
appears to be removed and subsequently degraded (Hammond et al 2000 Hutvagner
amp Zamore 2002 Schwarz et al 2003 de Alba et al 2013 Rogers amp Chen 2013)
The ARGONAUTE 1(AGO1) proteins are core components of the RISC complex
They contain two structural domains the N-terminal PAZ domain that binds to the 3prime-
end of single-stranded RNAs and the C-terminal piwi domains that has a structure
similar to that of RNase H (Cerutti et al 2000 Parker et al 2004) Several AGO
proteins possess endonucleolytic activities within the PIWI domain and cleave target
mRNAs in the middle of the site complementary to miRNA (Liu et al 2004 Meister et
al 2004 Miyoshi et al 2005) Mutations in the AGO1 gene cause miRNA target
genes to be ectopically expressed and the levels of some miRNAs are reduced in the
mutants (Kidner amp Martienssen 2004 Vaucheret et al 2004 Kidner amp Martienssen
2005 Ronemus et al 2006) Moreover the phenotype of the AGO1 hypomorphic
alleles display defects in lateral organ polarity and leaf and flower morphology as
observed in the dcl1 mutants and null AGO1 alleles are embryonically lethal
supporting the close relationship between the AGO proteins and miRNA processing
(Kidner amp Martienssen 2004 Vaucheret et al 2004 de Alba et al 2013 Rogers amp
Chen 2013)
The production of miRNA miRNA duplexes occur in the nucleus while
Chapter 1 Introduction and Review of Literature
39
miRNA-RISC complexes are present in the cytoplasm where target mRNAs are
located This discordance of functional sites requires an export step during miRNA
biogenesis HASTY the Arabidopsis homolog of exportin-5 is thought to be involved
in miRNA nuclear export (Bollman et al 2003 Park et al 2005)The hasty mutant
exhibits pleiotropic defects in plant development and reduced accumulation of most
miRNAs as in the DCL1 or AGO1 mutants Howeverit is unclear whether the HASTY
proteins export the miRNA miRNA duplexes or the miRNA-RISC complexes in
plants
11345 Feedback regulation of miRNA biogenesis
MiRNA biogenesis is under feedback regulation such that the DCL1 and AGO1 genes
are themselves regulated by miRNAs DCL1 is itself a target of miR162 which leads to
the cleavage of the DCL1 mRNA (Xie et al 2003 de Alba et al 2013) Consistent
with this the levels of DCL1 mRNA are elevated in the mutants of miRNA processing
genes in which the miR162 abundance is reduced In addition there is a predicted
hairpin structure of miR838 in the 14th
intron of the DCL1 primary transcript
(Rajagopalan et al 2006) This prediction implies that DCL1-mediated processing
on this site can result in the cleavage of the DCL1 primary transcript into two truncated
fragments the N-terminal capped fragment and the C-terminal polyadenylated
fragment If this is the case it is possible that miRNA biogenesis competes with the
splicing event of the DCL1 pre-mRNA
The AGO1 is also regulated by miRNA There is a complementary site for
miR168 binding in the AGO1 mRNA and the transcript level of the AGO1 mRNA is
reduced through an mRNA cleavage mechanism (Vaucheret et al 2006 de Alba et al
2013 Rogers amp Chen 2013) indicating that the AGO1 mRNA levels are tightly
controlled by the levels of the AGO1 protein
1135 Mechanism of miRNA action
11351 MiRNA-directed mRNA cleavage
By forming the miRNA-RISC assembly miRNAs can direct the RISC to regulate gene
expression by two post transcriptional mechanisms mRNA cleavage or translational
repression (Bartel 2004 Jones-Rhoades et al 2006 Dalmay 2013) The degree
of miRNA-mRNA complementarity plays an important role for the
determination of the mechanism to be used A general rule is that while perfect or
Chapter 1 Introduction and Review of Literature
40
near-perfect complementarity induces mRNA cleavage central mismatches trigger
translational repression (Carrington amp Ambros 2003 Jones-Rhoades amp Bartel 2004)
Unlike most animal miRNAs plant miRNAs have near-perfect or perfect
complementarity to their targets and mRNA cleavage is assumed to be a predominant
mechanism by which miRNAs regulate gene expression in plants
In RNA cleavage mechanism miRNAs guide the AGO component of RISC to
cleave a single phosphodiester bond of the target mRNA within the miRNA-binding
site (Llave et al 2002b Tang et al 2003) The truncated fragments are then released
and degraded freeing the RISC to recognize and cleave another target mRNA This has
been validated by the detection of 3prime cleavage products that have 5prime ends that start at the
middle of the complementary regions The mRNA cleavage products are then further
degraded by other mechanisms The 3prime cleavage products of some miRNA targets are
degraded by the 5prime-3prime exonuclease XRN4 an Arabidopsis homolog of the major Yeast
mRNA degrading exonuclease Xrn1p It has been observed that the 3prime cleavage
products of some target mRNAs were accumulated in the xrn4 mutants (Souret et al
2004 Dalmay 2013) However the 3prime cleavage products of other miRNA targets do
not accumulate in the xrn4 mutants indicating that there are also XRN4 independent
mechanisms The 5prime cleavage products are typically untraceable by RNA gel blot
hybridization but can be detected by sensitive PCR based methods It has been found
that the 5prime cleavage products tend to obtain an oligo U tail at the 3prime ends This 3prime U tail
is correlated with shortening of the 5prime cleavage products from the 5prime end leading to the
conclusion that the 3prime uridylation causes 5 prime- 3 prime exonucleolytic decay of the 5prime cleavage
products (Shen amp Goodman 2004)
DCL1 HYL1 and SE interact with each other and are co-localized in the
nuclear bodies termed Dicing bodies or D-bodies in which some pri-miRNA shave
been found suggesting that D-bodies serve as a center for active miRNA processing
(Fang amp Spector 2007 Fujioka et al 2007 Song et al 2007) The miRNA
processing site appears to be distinct from the Cajal bodies that have previously been
found as a siRNA processing site (Pontes amp Pikaard 2008) The D-bodies include
SmD3 and SmB proteins that are found in the Cajal bodies However At collin an
additional marker for the Cajal bodies is not found in the D-bodies Moreover the D-
bodies are present in an At collin mutant lacking the Cajal bodies The pri-miRNAs of
miR171A and miR159A genes have been found to be co-localized with HYL1 and
Chapter 1 Introduction and Review of Literature
41
DCL1 in the D- bodies that are distinct from the Cajal bodies (Song et al 2007)
11352 MiRNA-directed translational repression
Translation repression is another mechanism exerted by plant miRNAs The regulatory
mechanism of miR172 which regulates flowering time and floral organ identity is
consistent with translation repression MiR172 over production does not affect the
transcript levels of APETALA2 (AP2) and AP2- like target mRNAs Instead the AP2
protein level is reduced in the miR172 over expressing mutant (Aukerman amp Sakai
2003 Chen 2004 Dalmay 2013) It was later shown that miR172 can also direct the
cleavage of the target mRNAs although the steady-state mRNA level is unchanged due
to feedback regulation (Schwab et al 2005 Jung et al 2007) It is clear that miR172
regulates its target genes primarily by translational repression rather than mRNA
cleavage Recently it has been reported that miR156157 and miR854 also regulate
their target genes through translational repression (Arteaga-Vazquez et al 2006
Gandikota et al 2007 Dalmay 2013) It was once thought that translational repression
is an exception to the rule However experimental evidence suggest a widespread
coexistence of translational repression and mRNA cleavage (Brodersen et al 2008)
1136 Regulatory roles of miRNAs in plant development
The miRNA target prediction has been a difficult task in order to obtain regulatory
targets without bringing in too many false predictions Plant growth and developmental
processes regulated by miRNAs are quite diverse and enormous Furthermore plant
miRNAs work cooperatively or antagonistically to establish a balanced regulation This
notion is well consistent with the findings that mutations in the regulators of miRNA
biogenesis transport and processing such as AGO1 DCL1 HYL1 HEN1 ABH1
and HASTY cause pleiotropic phenotypes (Mallory amp Vaucheret 2006 de Alba et al
2013) To date the roles of miRNAs have been mostly deduced from obtaining miRNA
over expressing transgenic plants or gain-of-function mutants in which miRNA-
resistant target genes are ectopically expressed
11361 Phase transition
Plants pass through a series of developmental phase transitions during their life span
beginning with seed germination continuing through vegetative phase change
reproductive phase change flowering initiation and finally seed production for the next
generation Because of their importance in understanding plant developmental
Chapter 1 Introduction and Review of Literature
42
processes genes regulating developmental transitions have been extensively studied
and underlying molecular mechanisms have been explored in various plant species
Majority of researches were mainly focused on vegetative phase change and
reproductive transition in Arabidopsis and Maize (Poethig 2003 Baurle amp Dean
2006) Particularly the mutations of the genes encoding various regulators of miRNA
biogenesis and transport such as HST SE and AGO exhibit altered juvenile to adult
vegetative phase change and vegetative to reproductive change (Hunter et al 2003
Peragine et al 2004 Vazquez et al 2004a Jones-Rhoades et al 2006 de Alba et al
2013)
Over-expression of the miR156a gene causes late flowering and delays
vegetative phase change (Wu amp Poethig 2006 Gandikota et al 2007 Wang et al
2008) It has been shown that miR156 negatively regulates a group of genes encoding
SQUAMOSA PROMOTER-BINDING PROTEIN-LIKE (Lewis et al 2007 de Alba et
al 2013 Rogers amp Chen 2013) transcription factors such as SPL3 SPL4 and SPL5
that promote flowering initiation In contrast transgenic plants over expressing
miR156-resistant SPL345 genes are early flowering Interestingly the endogenous
level of miR156 is temporally regulated In the early juvenile vegetative phase the
level of miR156 is very high but decreases rapidly before the onset of the adult juvenile
phase (Wu amp Poethig 2006)
The Arabidopsis early activation tagged (eat-D) mutant exhibits early flowering
with disrupted floral structure The mutant phenotypes are caused by over expression of
the miR172a-2gene (Aukerman amp Sakai 2003) MiR172 regulates a small group of
genes encoding APETALLA2 (AP2)-like transcription factors such as TARGET OF
EAT1 (TOE1) TOE2 SCHLAFMUTZE (SMZ) and SCHNARCHZAPFEN(SNZ)
TOE1 TOE2 SMZ and SNZ have been shown to participate in their productive phase
change by repressing a floral integrator FLOWERING LOCUS T (FT)(Jung et al
2007) Consistent with this toe1-2D 35STOE2 35SSMZ and 35SSNZ plants
exhibit late flowering (Jung et al 2007) MiR172 is also regulated temporally and
highly expressed in the adult vegetative phase both in Arabidopsis and maize (Chuck et
al 2007) Because the temporal expression patterns of miR156 and miR172 are
complementary to each other it has been suggested that the two miRNAs interact with
each other in regulating phase change (Chuck et al 2009 de Alba et al 2013) In
support of this view it has been shown that miR172 is down-regulated in the
Chapter 1 Introduction and Review of Literature
43
Corngrass1(Cg1) mutant in which miR156 is overproduced (Chuck et al 2007 de
Alba et al 2013) However there is no direct evidence for the direct linkage between
miR156 and miR172 It is also envisioned that miR156 and miR172 would not be
directly related For example miR156 regulates plastochron index that is the inverse of
leaf initiation rate and thus frequently used to analyze temporal patterns of plant
development In contrast miR172 does not affect plastochron (Wang et al 2008) The
down-regulation of miR172 in the maize miR156-over producing mutants would be
due to the prolonged juvenile vegetative phase in the mutants
Another miRNA that regulates flowering initiation is miR159 It regulates
MYB33 and MYB65 which are closely related to the barley GAMY B transcription
factor that responds to gibberellic acid (GA) The level of miR159 is elevated in GA-
treated seedlings but reduced in the GA biosynthetic ga1 mutant Transgenic plants
over producing miR159 exhibit delayed floral induction under short days (Achard et
al 2004) It is been suggested that MYB33 mediates GA signal to regulate LEAFY
(LFY) These observations indicate that miR159 plays an important role in the GA
signaling pathways that regulate proper floral induction under short days (Achard et al
2004)
11362 Floral development
Arabidopsis flowers consist of four distinct organs They arise in a characteristic
pattern within concentric rings called whorls from outside to inside four sepals four
petals six stamens and the central pistil consisting of two carpels Identities of the
floral organs are determined by three classes of regulatory genes classA classB and
class C genes (Krizek amp Fletcher 2005) The A and C class genes act antagonistically
to restrict their expression domains (Bowman et al 1991)
It is notable that miR172 also regulates floral architecture in addition to its role
in the timing of flowering initiation MiR172 inhibits the translation of the AP2 mRNA
(Chen 2004) Transgenic plants overproducing miR172 not only exhibit early
flowering but also show abnormal floral development which is quite similar to those
of the class A gene mutants such as the ap2 mutants (Aukerman amp Sakai 2003 de
Alba et al 2013) When the activity of the class A genes is inactivated that of the class
C genes such as AGAMOUS (AG) is elevated As a result the miR172- overproducing
transgenic plants exhibit no petals along with homeotic transformation of the sepals to
Chapter 1 Introduction and Review of Literature
44
the carpels (Aukerman amp Sakai 2003 Chen 2004)
Interestingly miR166 also regulates floral architecture The role of miR165166
in the regulation of meristem activity is closely linked to floral meristem formation and
organization (Zhao et al 2007 Miyashima et al 2013) Floral structure is severely
disrupted in the miR165166-overproducing mutants such as men1 and jba-1D in
which miR166 is overproduced (Williams et al 2005b Jung amp Park 2007) The men1
and jba-1D mutants have smaller gynoecium and reduced carpel number In an extreme
case the jba-1D homozygous plants lack visible carpels (Williams et al 2005b)
Similar phenotypes have been observed in the miR165 overproducers (Zhao et al
2007) It has been suggested that the phenotypes are possibly caused by altered AG
expression in the floral meristem (Jung amp Park 2007 Turchi et al 2013)
Other miRNAs also affect floral development Overproduction of miRNAs
involved in auxin signaling also affects floral architecture Transgenic plants
overproducing miR167 have defects in the formation of ovule and anther with reduced
fertility (Ru et al 2006) It has been shown that miR167 targets ARF6 and ARF8 that
play a role in gynoecium and stamen development and in regulating fertility (Ru et al
2006 Wu et al 2006) These observations suggest that auxin signals are also closely
related to floral organogenesis In addition transgenic plants over-producing miR164
and the cuc1cuc2 double mutants show fused sepals and reduced petals (Laufs et al
2004 Turchi et al 2013) suggesting that miR164 is also related to floral meristem
activity and boundary specification of the meristematic region
11363 Shoot apical meristem development
Shoot apical meristem comprises self-maintaining totipotent cells The shoot apical
meristem activity affects diverse aspects of plant developmental processes including
leaf development vascular development and lateral organ polarity (Bowman et al
2002) Because miR165166 plays a primary role in meristem formation
overproduction of miR165166 and multiple knockout mutants of its target genes
exhibit pleiotropic pheonotypes (McConnell et al 2001 Emery et al 2003 Kim et al
2005 Williams et al 2005b)
The targets of miR165166 include genes encoding the homeo domain-leucine
zipper (HD-ZIP) class III transcription factors such as PHABULOSA(PHB)
PHAVOLUTA (PHV) REVOLUTA(REV) ATHB15CORONA (CNA) and ATHB8
Chapter 1 Introduction and Review of Literature
45
PHB PHV and ATHB15 have overlapping functions in controlling meristem
development (Turchi et al 2013) Indeed the phb phv athb15 triple mutants have an
enlarged shoot apical meristem Consistent with this the miR166- overproducing men1
and jba-1D mutants exhibit enlarged shoot apical meristems (Kim et al 2005
Williams et al 2005b Turchi et al 2013) In the mutants the shoot apical meristems
are multiplied and large
The development of shoot apical meristems is also regulated by the WUSCHEL
(WUS)ndashCLAVATA (CLV) signaling pathway WUS positively regulates cell
proliferation In contrast CLV3 which is expressed in stem cells limits the WUS
expression and thus inhibits cell proliferation in the SAM (Sharma et al 2003)
Consistent with this notion WUS is expressed to a higher level in jba-1D
explaining the ectopic enlargement of the SAM in the mutant (Williams et al 2005)
While WUS is closely related to miR165166 in the shoot apical meristem
development the underlying molecular mechanisms are currently unclear It has been
observed that the miR166-over producing men1 plants have an arrested meristem
development (Jung amp Park 2007) In addition the heterozygous men1+ and the
homozygous men1 plants show different expression patterns of WUS and CLV3 It
appears that miR166 regulates WUS indirectly and influences the expressional balance
between WUS and CLV3 through a negative feedback loop (Jung amp Park 2007 de
Alba et al 2013)
The phv phb athb15 triple mutant has an enlarged shoot apical meristems the
rev phb phv mutant does not have functional shoot apical meristems (Prigge et al
2005) This distinction may be explained by the fact that while miR166 targets
primarily PHB PHV and ATHB15 miR165 targets all the five HD-ZIP III genes (Zhou
et al 2007) Consistent with this transgenic plants overproducing miR166 are distinct
from those overproducing miR165 The miR165-overproducing transgenic plants lack
functional shoot apical meristem and a pin-like structure is formed at the apical region
of the mutant These phenotypes are quite similar to rev phb phv triple mutant (Zhou et
al 2007 Liu et al 2013) The phenotypic distinction may because by the difference
of the cleavage efficiencies of the target mRNAs In accordance with this notion a few
35S miR166a transgenic lines exhibit no shoot apical meristems The men1
homozygous plants also show arrested meristem development (Jung amp Park 2007)
Chapter 1 Introduction and Review of Literature
46
MiR164 cleaves the CUC1 and CUC2 mRNAs as well as the NAC1 mRNA
CUC1 and CUC2 determines the organ boundary in the meristem Phenotypes of
transgenic plants that overproduce miR164 are similar to those of the cuc1cuc2 double
mutant that exhibits embryonic developmental defects such as cup-shaped cotyledons
and fused cotyledons (Laufs et al 2004) When a miR164-resistant CUC2 is
expressed the boundary domain is enlarged CUC also plays a role in embryonic shoot
meristem formation by regulating the SHOOT MERISTEMLESS gene It was therefore
concluded that miR164-mediated cleavage of CUC1 and CUC2 mRNAs determines the
sizes of the boundary domains in the shoot apical meristem and regulates separation of
the organs by restricting cell proliferation (Laufs et al 2004 de Alba et al 2013)
11364 Leaf development
The role of miR319 has been extensively studied in leaf development A dominant
jaw-D mutant overproducing miR319 shows uneven curved leaves with enlarged size
and five TCP genes which are closely related to the CINCINNATA (CIN) gene in
Antirrhinum majus are down regulated in the mutant suggesting that miR319 mediated
regulation of the TCP transcription factor genes are important for the final step of
leaf shaping (Palatnik et al 2003 Naz et al 2013) In addition to leaf shape and size
leaf polarity is also specified by miRNA MiR166-mediated cleavage of the HD-ZIPIII
mRNAs is a well to establish leaf polarity This discovery originates from the gain-of-
function mutants such as phb-1d and phv-1d wherein point mutations in the
complementary sites to miRNA helps the mRNA to evade or resist from miRNA-
mediated cleavage (McConnell et al 2001 Emery et al 2003 Naz et al 2013)
Plant leaves have a dorso-ventral polarity consisting of the adaxial (upper
surface) and abaxial (lower surface) sides The adaxial side is closer to the shoot
apical meristem Adaxial identity is specified by functionally redundant class III HD-
ZIP family genes such as PHB PHV and REV Ectopic expression of each genes
result in adaxialized leaves Dominant phb-1d and phv-1d mutants exhibit
transformation of the abaxial to adaxial fate and have rod-shaped leaves In contrast
miR165166-overproducing plants show pin-like cotyledons radicalized leaves and
abaxialized leaves (Chitwood et al 2007 Pattanayak et al 2013)
Leaf asymmetry is determined by miR166-mediated restriction of the
expression domains of the HD-ZIPIII genes The HD-ZIPIII genes are expressed in the
Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
28
112 MicroRNA or MiRNA
In 1991 Ambros and co-workers first isolated a lin-4 mutant of Celegans which was
arrested at the first larval stage (Lee et al 1993) Later on the let-7 mutation was
isolated in the same system which was responsible for development through the fourth
larval stage Both lin-4 and let-7 encode short 22-nucleotide mature RNAs and were
called short temporal RNA because they control the temporal development program of
C elegans The mature lin-4 RNA defines (negatively regulates) the mRNA expression
of the lin-14 and lin-28 hetrochronic genes with the antisense mediated repression
mechanism of translation initiation and thus specifies the fate of cells during the first
three larval stages Recent studies have revealed that the short temporal RNAs are
actually members of a group of tiny RNAs (21to 28 nucleotides) called the micro-
RNAs (miRNA) (Bartel 2009) isolated members of which could easily run to a few
thousand which includes both those were identified computationally and by deep
sequencing Some of the components of the RNAi machinery have also been clearly
established as the effector proteins for the maturation of miRNAs
113 MicroRNAs in plants
1131 Discovery
MicroRNAs have been discovered by three basic approaches Direct cloning forward
genetic direct cloning and bioinformatic prediction followed by experimental
validation The most direct method of miRNA discovery is to isolate and clone small
RNAs from biological samples and several groups have used this approach to identify
plant miRNAs (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002 Xie et al 2003 Sunkar et al 2005 Williams et al 2013) The cloning methods
adapted were similar to those used to identify large numbers of animal miRNAs
(Lagos-Quintana et al 2001 Lau et al 2001 Aravin amp Tuschl 2005) which involve
isolating small RNAs followed by oligonucleotide adapter ligation reverse
transcription amplification and sequencing Some protocols incorporate methods to
enrich for Dicer cleavage products (ie molecules with 5prime-phosphates and 3prime-
hydroxyls) and to concatemerize the short cDNAs so that several may be identified in a
single sequencing read (Lau et al 2001 Llave et al 2002a Jagadeeswaran amp Sunkar
2013)The initial cloning experiments in Arabidopsis identified 19 miRNAs belonging
to 15 families (Llave et al 2002a Mette et al 2002 Park et al 2002 Reinhart et al
2002) although hundreds of other small RNAs such as siRNAs and RNA degradation
Chapter 1 Introduction and Review of Literature
29
fragments were also cloned through the direct cloning methods To select miRNAs
from the pool of small RNAs secondary structures of the RNA molecules were
examined in compliance with the acknowledged structural features of miRNAs
(Ambros et al 2003) The secondary structures were validated using several ad-hoc
software tools such as the Mfold or RNAfold programs (Reeder et al 2006) Finally
the existence of mature miRNAs was determined by RNA gel blot hybridization These
direct cloning methods were successful in isolating many miRNAs in Arabidopsis
(Reinhart et al 2002 Sunkar amp Zhu 2004) Rice (Sunkar et al 2005) Poplar (Lu et
al 2005) moss (Arazi et al 2005) and Tobacco (Billoud et al 2005)
Although miRNAs were first discovered through forward genetic screens in
worms (Lee et al 1993 Reinhart et al 2000)only few A thaliana miRNA gene
families have been discovered by this method (Chen 2008) in plants MiRNA
involvement in plant mutant phenotypes were not properly inferred till the cloning
experiments established that plant genomes contain numerous miRNAs (Park et al
2002 Reinhart et al 2002 Rhoades et al 2002) MiRNA loss-of-function allele has
been identified by forward genetic screens early extra petala1 is caused by a
transposon insertion ~160bp upstream of the predicted miR164c stem loop resulting in
flowers with extra petals (Baker et al 2005 Irish 2008) The fact that loss-of-function
miRNA mutants have been rarely discovered using forward genetics reflects small
target size for mutagenesis coupled with redundancy in nearly all evolutionarily
conserved plant miRNAs which are encoded by gene families (Table16) Family
members are likely to have overlapping functions buffering against loss at any single
miRNA locus Over expression screens can circumvent redundancy limitations At least
four plant miRNAs miR319 (also known as miR-JAW) miR172 (also known as EAT)
and miR166 were isolated in over expression screens for dominant mutants with
developmental abnormalities (Aukerman amp Sakai 2003 Palatnik et al 2003 Kim et
al 2005 Williams et al 2005b Schommer et al 2012) Mutations in miRNA target
sites which can prevent the entire family of miRNAs from repressing a target gene can
also circumvent redundant functions of miRNA family members
The dominant mutations in the HD-ZIP genes PHB PHV and REVOLUTA
(REV) in Arabidopsis and ROLLEDLEAF1 (RLD1) in Maize result in adaxialization of
leaves andor Vasculature (McConnell amp Barton 1998 McConnell et al 2001 Nelson
et al 2002 Zhong amp Ye 2004 Allen amp Millar 2012) and are all caused by mutations
Chapter 1 Introduction and Review of Literature
30
in miR166 complementary sites (Rhoades et al 2002 Emery et al 2003 Jones-
Rhoades amp Bartel 2004 Mallory et al 2004 Zhong amp Ye 2004)
In both plants and animals cloning was the initial means of large-scale
miRNA discovery (Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Reinhart et al 2002 Mendes et al 2012) However cloning is biased toward
RNAs that are expressed highly and broadly MiRNAs expressed at low levels or
only in specific cell types or in response to certain environmental stimuli were more
difficult to clone Sequence based biases in cloning procedures might also cause
certain miRNAs to be missed Because of these limitations bioinformatics approaches
to identify miRNAs have provided a useful complement to cloning
A straightforward use of bioinformatics has been carried out to find homologs
of known miRNAs both within the same genome and in the genomes of other species
(Pasquinelli et al 2000 Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Mendes et al 2009) A more difficult challenge is to identify miRNAs
unrelated to previously known miRNAs This was first accomplished for
vertebrate nematode and fly miRNAs using algorithms that search for conservation
of sequence and secondary structure (ie miRNA stem-loop precursors) between
species searching for patterns that are characteristic of miRNAs (Lai et al 2003 Lim
et al 2003 Lim et al 2005)
Most of the methods identified numerous miRNAs but are necessarily more
relaxed in terms of allowed structures Some approaches take advantage of the high
complementarity of plant miRNAs to target messages implementing the requirement
that the candidate has conserved complementarity to mRNAs (Jones-Rhoades amp Bartel
2004 Adai et al 2005) This additional filter has been useful for distinguishing
authentic plant miRNAs from false positives which later extended to mammalian
miRNA gene prediction (Xie et al 2005 Mendes et al 2012)
Another criteria used by most algorithms in the filters is evolutionary
conservation In plants mature miRNA sequences are conserved to a higher degree
than their precursor sequences For example Arabidopsis and rice diverged more than
130 million years ago (Friis et al 2004) but some miRNAs are highly conserved
in these plant species (Reinhart et al 2002) Furthermore various parameters for the
secondary structures such as free energy the number of paired residues within a
Chapter 1 Introduction and Review of Literature
31
miRNA or the number and size of bulges are also used to validate the predictions
(Jones-Rhoades amp Bartel 2004 Wang et al 2004 Adai et al 2005 Mendes et al
2012)
1132 Conserved microRNAs in Plants
Till date cloning genetics and bioinformatics screens have resulted in annotation of
approximately 184 potential Arabidopsis miRNAs (miRBase release 13) These
miRNAs seem to arise through gene duplication and subsequent diversification and
represent approximately 100 families (Maher et al 2006 Axtell 2013) Twenty one
families represented by 92 genes are clearly conserved in species beyond Arabidopsis
(Table 15) The presence of these miRNA families in other sequenced plant genomes
indicates that different plant species have an evolutionarily fluid set of miRNAs
(Willmann amp Poethig 2007) Recent studies on mosses one of the most ancient land
plants have shown that at least 14 miRNA families are conserved in eudicots and
mosses and therefore it appears that plant miRNAs share a common ancient origin
(Arazi et al 2005 Axtell amp Bartel 2005 Talmor-Neiman et al 2006 Zhang et al
2006 Axtell 2013) The number of members per family in a genome ranges from 1-32
with the exception of miR-430 which is represented by a cluster of ~80 loci in zebra
fish (Giraldez et al 2005)
In plants the number of members in each family correlates among examined
species certain families contain numerous members in all three species (eg miR156
miR166 miR169) whereas others consistently contain fewer genes (eg miR162
mir168 miR394) (Table 16) Although it is still unclear why certain miRNA are
encoded by more than one gene in plants (eg 12 genes encoding miR156) this
correlation might suggest functional significance of various miRNA family sizes Most
annotated plant miRNAs are conserved throughout flowering plants while a few
miRNAs are found only in a single sequenced genome and thus they could be
evolutionarily lsquoyoungrsquo miRNAs For example Arabidopsis has several miRNA
families that are not found in poplar or rice suggesting that miRNAs could be
generated continuously during evolution (Allen et al 2004a Rajagopalan et al
2006 Fahlgren et al 2007 Axtell 2012 de Alba et al 2013) Consistent with the
notion that non conserved miRNAs are of a more recent evolutionary origin these
miRNAs are mainly encoded by a single locus in the genome Within each family the
mature miRNA is always located in the same arm
Chapter 1 Introduction and Review of Literature
32
Table 16 MicroRNA families conserved in plants
miRNA
family
Arabidopsis Oryza Populus
miR156 12 12 11 miR159319 6 8 15 miR160 3 6 8 miR162 2 2 3 miR164 3 5 6 miR166 9 12 17 miR167 4 9 8 miR168 2 2 2 miR169 14 17 32 miR171 4 7 10 miR172 5 3 9 miR390 3 1 4 miR393 2 2 4 miR394 2 1 2 miR395 6 19 10 miR396 2 5 7 miR397 2 2 3 miR398 3 2 3 miR399 6 11 12 miR408 1 1 1 miR403 1 0 2 miR437 0 1 0 miR444 0 1 0 miR445 0 9 0 Total 92 127 169
All known miRNA families that are conserved between more than one plant species are listed
together with the number of genes identified in the sequenced genomes Rice miRNA families that have orthologs in maize but do not appear to have orthologs in the eudicots (Arabidopsis and Populus) are marked with an asterisk The following families contain miRNA genes annotated with
more than one number miR156 (miR156 and miR157) miR159319 (miR159 and miR319) miR166 (miR165 and miR166) miR171 (miR170 and miR171) and miR390 (miR390 and miR391)
of the stem loop (5prime- or 3prime) as it would be expected if the genes carry common ancestry
(Figure 17) The sequence of the mature miRNA and to some extent the segment on
the opposite arm of the hairpin to which it pairs is highly conserved between the
members of the same family (both within and between the species) while the sequence
secondary structure and length of the intervening ldquolooprdquo region can be highly divergent
within the same family
The patterns of paring and non pairing nucleotides are often conserved between
homologous miRNA stem loops from different species (Figure 17) The significance of
conserved mismatches is opined to guide DCL1 to cleave at the appropriate positions
along the stem loop
Chapter 1 Introduction and Review of Literature
33
Figure 17 Representative stem loop of miR164 Segments corresponding to the mature miRNAs
are shown in red (Adapted from Rajagopalan et al 2006)
Although many miRNAs are highly conserved in plants the degree of conservation in
most cases is quite low between plants and animals one exception is miR854 A recent
study has revealed that miR854 is conserved between Arabidopsis and animals and that
the Arabidopsis miR854 differs from the human miR854 only by a single nucleotide
(Arteaga-Vazquez et al 2006 Axtell 2012 de Alba et al 2013) The Arabidopsis
miR854 has multiple binding sites in the 3prime-untranslated region (UTR) of
OLIGOURIDYLATE-binding-PROTEIN mRNA 1bA (UBP1b) and represses the
translation of the UBP1b mRNA The animal miR854 is also complementary to a site in
the 3prime-UTR of the UBP1b homolog in animals However it is currently unclear how the
animal miR854 regulates its target mRNA Another distinction between plant and
animal miRNAs is the genomic organization of the miRNA genes While plant miRNA
genes are located predominantly in the intergenic regions animal miRNA genes tend to
localize in the intragenic regions both in the introns or exons of protein coding genes
Moreover miRNA genes are often clustered within a single precursor transcript
whereas individual plant miRNAs are derived from individual precursor RNAs (Bartel
Chapter 1 Introduction and Review of Literature
34
2004 Bonnet et al 2004 Kim 2005 Vaucheret et al 2006 Marco et al 2013)
1133 Non-conserved miRNA and challenges in annotation
Most miRNAs are conserved throughout flowering plants (Table 15) but many more
were found only in a single sequenced genome thus considered to be of a more recent
evolutionary origin The extended homology between few non conserved miRNAs
precursors and their target genes provides strong evidence that these relatively ldquoYoungrdquo
miRNAs arose from the duplication of the target gene segments (Allen et al 2004)
Several non-conserved miRNAs including miR161 miR163 miR173 miR447
miR475 and miR476 are known to direct cleavage of target transcripts (Allen et al
2004a Adai et al 2005 Sunkar et al 2005 de Alba et al 2013) It is difficult to
confidently predict targets for many because it is not possible to use complementary
site as a filter against false-positive target predictions It is also difficult to
confidently annotate non-conserved miRNAs as miRNA rather than siRNAs The
established minimal standard for miRNA annotation is a small RNA with detectable
expression and the potential to form a stem-loop when joined to flanking genomic
sequence (Kasschau et al 2003) In practice these requirements are too loose to
definitively categorize many small RNAs cloned from plants Many plant siRNAs are
detectable on blots (Vazquez et al 2004b) and hundreds of thousands of non-
miRNA genomic sequences can be predicted to fold into secondary structures that
resemble structures of plant miRNA precursors (Jones-Rhoades amp Bartel 2004)
Therefore without conservation of both sequence and secondary structure it is
difficult to be confident that a given cloned RNA originated from a stem-loop (ie is a
miRNA) rather than from a double-stranded RNA (ie is a siRNA) In fact many of
the thousands o f small RNAs cloned from Arabidopsis (Gustafson et al 2005) would
probably meet the literal requirements for annotation as miRNAs A few of these
sequences might be miRNAs but others that meet the literal criteria probably are not
so The challenges of annotating non conserved small RNAs were evidenced by
three related small silencing RNAs that were originally annotated as miRNAs
that turned out to be trans-acting siRNAs (tasiRNAs)(Kanno et al2013)
Although most miRNAs that are broadly conserved among flowering plants are
now being identified and experimentally validated (Table 16)(Jones-Rhoades amp
Bartel 2004) the challenges and ambiguities for classifying non-conserved miRNAs
prevent meaningful estimates of the total number of miRNA genes in Arabidopsis and
Chapter 1 Introduction and Review of Literature
35
other plant genomes It is possible that miRNAs might have escaped detection
because they are non-conserved and expressed in specific tissues or conditions and can
only be identified by using high- throughput sequencing
1134 MicroRNA biogenesis
11341 Transcription of microRNA precursor
Plant miRNAs are primarily found in the genomic regions that are not associated with
the protein coding genes (Reinhart et al 2002) and it appears that most if not all plant
miRNAs are produced from their own transcriptional units Plant miRNA genes are
occasionally clustered near each other in the genome suggesting transcription of
multiple miRNAs from a single primary transcript [eg the miR395 cluster (Grishok
et al 2001 Jones-Rhoades amp Bartel 2004b)] but this polycistronic arrangement of
miRNA genes appears far less frequently in plants than in animals (Bartel 2004)
Northern blot EST and mapping evidence indicate that plant miRNA primary
transcripts (also known as pri-miRNAs) as in animals are longer than needed to
encompass the miRNA stem-loops (Palatnik et al 2003 Jones-Rhoades amp Bartel
2004b) At least some of these pri-miRNA transcripts appear to be spliced
polyadenylated (Kurihara amp Watanabe 2004) and capped (Xie Z et al 2005) Two
rice miRNAs are contained within transcripts that contain exon junctions within the
presumptive stem-loop precursor implying that in these cases splicing is a prerequisite
for Dicer recognition (Sunkar et al 2005) Plant pri-miRNAs are over 1kb in length
and they are usually preceded by typical TATA box motifs They can also undergo
canonical splicing polyadenylation and capping All these observations indicates RNA
polymerase II is probably responsible for transcribing most plant miRNAs (Xie Z et
al 2005) as appears to be the case for many animal miRNAs Relatively little is
known about the regulation of miRNA transcription in plants but there is no reason
to suspect that this regulation would differ from that of protein-coding transcripts as
the bioinformatics analysis of the sequence upstream of the transcriptional start
sites of the miRNA genes showed the presence of putative binding motifs of
number of known transcription factors (Megraw et al 2006 Sethupathy 2013)
11342 MicroRNA processing and export
In plants maturation of miRNA is a step wise process A miRNA gene is transcribed to
pri-miRNA which is much longer than the pre-miRNA possessing the characteristic
stem-loop structure Like transcription of most protein-coding genes the miRNA
Chapter 1 Introduction and Review of Literature
36
transcription is processed by RNA polymerase II (Pol II) enzymes and the pri-
miRNAs are 5prime-capped and 3prime-polyadenylated (Lee et al 2004 Xie Z et al 2005)
Mature miRNAs are produced from pri-miRNAs through at least two sequential
processing steps by RNase III-type endonucleases In animals pri-miRNAs are first
processed at the bottom portion of the stem by Drosha a nuclear-localized RNase III
enzyme to liberate pre-miRNAs The pre-miRNAs are then exported to the cytoplasm
with the help of exportin-5The pre-miRNAs are further processed by Dicer another
RNaseIII enzyme to a duplex consisting of the miRNA and the antisense strands
(miRNA) Since plants do not have any Drosha homologs the two sequential
processing steps seem to be carried out by DCL1 a Dicer homolog in the nucleus
(Kurihara amp Watanabe 2004 de Alba et al 2013 Rogers amp Chen 2013)
The processing of pri-miRNAs to pre-miRNAs by DCL1 is assisted by two
other proteins HYPONASTY LEAVES1 (HYL1) and SERRATE (SE) HYL1 belongs to a
family of double-stranded RNA (dsRNA) binding protein and interacts with DCL1 in
vitro and in vivo (Lu amp Fedoroff 2000 Hiraguri et al 2005 de Alba et al 2013
Rogers amp Chen 2013) Loss-of-function mutations in the HYL1 gene result in
decreased accumulation of miRNAs and concomitant accumulation of pri-miRNAs
(Han et al 2004 Vazquez et al 2004a Kurihara et al 2006) SE a C2H2 zinc finger
protein interacts with DCL1 and HYL1 and plays a role in the processing of pri-
miRNAs to pre-miRNAs (Lobbes et al 2006 Yang L et al 2006) In addition the
nuclear cap-binding complex proteins CBP20 and CBP80 that are known as ABA
HYPER SENSITIVE 1(ABH1) have been shown to be required for proper miRNA
processing (Gregory et al 2008 Laubinger et al 2008) Recently it has been
demonstrated that DCL1 can release 21-nt RNAs from dsRNA as well as from
synthetic miR167 precursor RNAs in vitro However this cleavage is error prone and
inefficient in the absence of HYL1 and SE which accelerate the rate of DCL1-mediated
cleavage and promote accurate processing of miRNAs (Dong et al 2008 Rogers amp
Chen 2013)
11343 Methylation of the miRNAmiRNAduplex
After the miRNAmiRNAduplexes are released from the pre-miRNAs by the DCL1
activities the duplexes are methylated at the 2primeOH of the3prime-ends by HUA ENHANCER1
(HEN1) a small RNA methyltransferase (Yu et al 2005 Yang Z et al 2006 Rogers
amp Chen 2013 ) This step which is distinct from the RNase III-mediated processing
Chapter 1 Introduction and Review of Literature
37
step distinguishes the biogenesis of plant miRNAs from that of animal miRNAs
Mutations in the HEN1 gene were first isolated in a morphological screening for floral
development although the HEN1 mutants display pleiotropic phenotypes similar to the
developmental defects observed in the DCL1 mutants (Chen et al 2002 Park et al
2002 Rogers amp Chen 2013)
Figure 18 Biogenesis of microRNA
A Biogenesis of plant MicroRNA B Biogenesis of metazoananimal MicroRNA
The miRNA (red) is incorporated into RISC and the miRNA(blue) in degraded
This enzyme has two dsRNA-binding domains and nuclear localization signal It
is also conserved in fungi and is required for the processing of siRNAs as well as of
miRNAs (Park et al 2002 Boutet et al 2003 Xie et al 2003) Although the HEN1
protein is localized to the nucleus its methylation activity in the cytoplasm cannot be
ignored (Xie et al 2004 Rogers amp Chen 2013)
Chapter 1 Introduction and Review of Literature
38
The methylation of the miRNAmiRNA duplex is required to protect miRNAs
against the 3prime-end uridylation activity and subsequent degradation (Li et al 2005) In
the hen1 mutants accumulation of miRNAs is reduced to a lower level Also the
miRNAs appear to be heterogeneous in their sizes such that a ladder of bands rather
than a single species is observed for most miRNAs in the mutants (Li et al 2005)
MiRNA cloning and sequencing analyses revealed the presence of a number of U
residues at the 3prime-end of miRNAs in the hen1 mutants Moreover these nucleotides do
not correspond to the miRNAs in the pre-miRNAs indicating that these nucleotides are
added after the processing of the miRNAs from pre-miRNAs (Li et al 2005)
Uridylation of RNAs has also been proven to be correlated with RNA decay (Shen amp
Goodman 2004 Mullen amp Marzluff 2008) suggesting that uridylation attracts an
exonuclease which degrades miRNAs in plants
11344 MiRNA incorporation in to the RISC and its nuclear export
The methylated miRNAmiRNA duplexes undergo RISC assembly In this process
the miRNA of the duplexes is selectively incorporated into the RISC and the miRNA
appears to be removed and subsequently degraded (Hammond et al 2000 Hutvagner
amp Zamore 2002 Schwarz et al 2003 de Alba et al 2013 Rogers amp Chen 2013)
The ARGONAUTE 1(AGO1) proteins are core components of the RISC complex
They contain two structural domains the N-terminal PAZ domain that binds to the 3prime-
end of single-stranded RNAs and the C-terminal piwi domains that has a structure
similar to that of RNase H (Cerutti et al 2000 Parker et al 2004) Several AGO
proteins possess endonucleolytic activities within the PIWI domain and cleave target
mRNAs in the middle of the site complementary to miRNA (Liu et al 2004 Meister et
al 2004 Miyoshi et al 2005) Mutations in the AGO1 gene cause miRNA target
genes to be ectopically expressed and the levels of some miRNAs are reduced in the
mutants (Kidner amp Martienssen 2004 Vaucheret et al 2004 Kidner amp Martienssen
2005 Ronemus et al 2006) Moreover the phenotype of the AGO1 hypomorphic
alleles display defects in lateral organ polarity and leaf and flower morphology as
observed in the dcl1 mutants and null AGO1 alleles are embryonically lethal
supporting the close relationship between the AGO proteins and miRNA processing
(Kidner amp Martienssen 2004 Vaucheret et al 2004 de Alba et al 2013 Rogers amp
Chen 2013)
The production of miRNA miRNA duplexes occur in the nucleus while
Chapter 1 Introduction and Review of Literature
39
miRNA-RISC complexes are present in the cytoplasm where target mRNAs are
located This discordance of functional sites requires an export step during miRNA
biogenesis HASTY the Arabidopsis homolog of exportin-5 is thought to be involved
in miRNA nuclear export (Bollman et al 2003 Park et al 2005)The hasty mutant
exhibits pleiotropic defects in plant development and reduced accumulation of most
miRNAs as in the DCL1 or AGO1 mutants Howeverit is unclear whether the HASTY
proteins export the miRNA miRNA duplexes or the miRNA-RISC complexes in
plants
11345 Feedback regulation of miRNA biogenesis
MiRNA biogenesis is under feedback regulation such that the DCL1 and AGO1 genes
are themselves regulated by miRNAs DCL1 is itself a target of miR162 which leads to
the cleavage of the DCL1 mRNA (Xie et al 2003 de Alba et al 2013) Consistent
with this the levels of DCL1 mRNA are elevated in the mutants of miRNA processing
genes in which the miR162 abundance is reduced In addition there is a predicted
hairpin structure of miR838 in the 14th
intron of the DCL1 primary transcript
(Rajagopalan et al 2006) This prediction implies that DCL1-mediated processing
on this site can result in the cleavage of the DCL1 primary transcript into two truncated
fragments the N-terminal capped fragment and the C-terminal polyadenylated
fragment If this is the case it is possible that miRNA biogenesis competes with the
splicing event of the DCL1 pre-mRNA
The AGO1 is also regulated by miRNA There is a complementary site for
miR168 binding in the AGO1 mRNA and the transcript level of the AGO1 mRNA is
reduced through an mRNA cleavage mechanism (Vaucheret et al 2006 de Alba et al
2013 Rogers amp Chen 2013) indicating that the AGO1 mRNA levels are tightly
controlled by the levels of the AGO1 protein
1135 Mechanism of miRNA action
11351 MiRNA-directed mRNA cleavage
By forming the miRNA-RISC assembly miRNAs can direct the RISC to regulate gene
expression by two post transcriptional mechanisms mRNA cleavage or translational
repression (Bartel 2004 Jones-Rhoades et al 2006 Dalmay 2013) The degree
of miRNA-mRNA complementarity plays an important role for the
determination of the mechanism to be used A general rule is that while perfect or
Chapter 1 Introduction and Review of Literature
40
near-perfect complementarity induces mRNA cleavage central mismatches trigger
translational repression (Carrington amp Ambros 2003 Jones-Rhoades amp Bartel 2004)
Unlike most animal miRNAs plant miRNAs have near-perfect or perfect
complementarity to their targets and mRNA cleavage is assumed to be a predominant
mechanism by which miRNAs regulate gene expression in plants
In RNA cleavage mechanism miRNAs guide the AGO component of RISC to
cleave a single phosphodiester bond of the target mRNA within the miRNA-binding
site (Llave et al 2002b Tang et al 2003) The truncated fragments are then released
and degraded freeing the RISC to recognize and cleave another target mRNA This has
been validated by the detection of 3prime cleavage products that have 5prime ends that start at the
middle of the complementary regions The mRNA cleavage products are then further
degraded by other mechanisms The 3prime cleavage products of some miRNA targets are
degraded by the 5prime-3prime exonuclease XRN4 an Arabidopsis homolog of the major Yeast
mRNA degrading exonuclease Xrn1p It has been observed that the 3prime cleavage
products of some target mRNAs were accumulated in the xrn4 mutants (Souret et al
2004 Dalmay 2013) However the 3prime cleavage products of other miRNA targets do
not accumulate in the xrn4 mutants indicating that there are also XRN4 independent
mechanisms The 5prime cleavage products are typically untraceable by RNA gel blot
hybridization but can be detected by sensitive PCR based methods It has been found
that the 5prime cleavage products tend to obtain an oligo U tail at the 3prime ends This 3prime U tail
is correlated with shortening of the 5prime cleavage products from the 5prime end leading to the
conclusion that the 3prime uridylation causes 5 prime- 3 prime exonucleolytic decay of the 5prime cleavage
products (Shen amp Goodman 2004)
DCL1 HYL1 and SE interact with each other and are co-localized in the
nuclear bodies termed Dicing bodies or D-bodies in which some pri-miRNA shave
been found suggesting that D-bodies serve as a center for active miRNA processing
(Fang amp Spector 2007 Fujioka et al 2007 Song et al 2007) The miRNA
processing site appears to be distinct from the Cajal bodies that have previously been
found as a siRNA processing site (Pontes amp Pikaard 2008) The D-bodies include
SmD3 and SmB proteins that are found in the Cajal bodies However At collin an
additional marker for the Cajal bodies is not found in the D-bodies Moreover the D-
bodies are present in an At collin mutant lacking the Cajal bodies The pri-miRNAs of
miR171A and miR159A genes have been found to be co-localized with HYL1 and
Chapter 1 Introduction and Review of Literature
41
DCL1 in the D- bodies that are distinct from the Cajal bodies (Song et al 2007)
11352 MiRNA-directed translational repression
Translation repression is another mechanism exerted by plant miRNAs The regulatory
mechanism of miR172 which regulates flowering time and floral organ identity is
consistent with translation repression MiR172 over production does not affect the
transcript levels of APETALA2 (AP2) and AP2- like target mRNAs Instead the AP2
protein level is reduced in the miR172 over expressing mutant (Aukerman amp Sakai
2003 Chen 2004 Dalmay 2013) It was later shown that miR172 can also direct the
cleavage of the target mRNAs although the steady-state mRNA level is unchanged due
to feedback regulation (Schwab et al 2005 Jung et al 2007) It is clear that miR172
regulates its target genes primarily by translational repression rather than mRNA
cleavage Recently it has been reported that miR156157 and miR854 also regulate
their target genes through translational repression (Arteaga-Vazquez et al 2006
Gandikota et al 2007 Dalmay 2013) It was once thought that translational repression
is an exception to the rule However experimental evidence suggest a widespread
coexistence of translational repression and mRNA cleavage (Brodersen et al 2008)
1136 Regulatory roles of miRNAs in plant development
The miRNA target prediction has been a difficult task in order to obtain regulatory
targets without bringing in too many false predictions Plant growth and developmental
processes regulated by miRNAs are quite diverse and enormous Furthermore plant
miRNAs work cooperatively or antagonistically to establish a balanced regulation This
notion is well consistent with the findings that mutations in the regulators of miRNA
biogenesis transport and processing such as AGO1 DCL1 HYL1 HEN1 ABH1
and HASTY cause pleiotropic phenotypes (Mallory amp Vaucheret 2006 de Alba et al
2013) To date the roles of miRNAs have been mostly deduced from obtaining miRNA
over expressing transgenic plants or gain-of-function mutants in which miRNA-
resistant target genes are ectopically expressed
11361 Phase transition
Plants pass through a series of developmental phase transitions during their life span
beginning with seed germination continuing through vegetative phase change
reproductive phase change flowering initiation and finally seed production for the next
generation Because of their importance in understanding plant developmental
Chapter 1 Introduction and Review of Literature
42
processes genes regulating developmental transitions have been extensively studied
and underlying molecular mechanisms have been explored in various plant species
Majority of researches were mainly focused on vegetative phase change and
reproductive transition in Arabidopsis and Maize (Poethig 2003 Baurle amp Dean
2006) Particularly the mutations of the genes encoding various regulators of miRNA
biogenesis and transport such as HST SE and AGO exhibit altered juvenile to adult
vegetative phase change and vegetative to reproductive change (Hunter et al 2003
Peragine et al 2004 Vazquez et al 2004a Jones-Rhoades et al 2006 de Alba et al
2013)
Over-expression of the miR156a gene causes late flowering and delays
vegetative phase change (Wu amp Poethig 2006 Gandikota et al 2007 Wang et al
2008) It has been shown that miR156 negatively regulates a group of genes encoding
SQUAMOSA PROMOTER-BINDING PROTEIN-LIKE (Lewis et al 2007 de Alba et
al 2013 Rogers amp Chen 2013) transcription factors such as SPL3 SPL4 and SPL5
that promote flowering initiation In contrast transgenic plants over expressing
miR156-resistant SPL345 genes are early flowering Interestingly the endogenous
level of miR156 is temporally regulated In the early juvenile vegetative phase the
level of miR156 is very high but decreases rapidly before the onset of the adult juvenile
phase (Wu amp Poethig 2006)
The Arabidopsis early activation tagged (eat-D) mutant exhibits early flowering
with disrupted floral structure The mutant phenotypes are caused by over expression of
the miR172a-2gene (Aukerman amp Sakai 2003) MiR172 regulates a small group of
genes encoding APETALLA2 (AP2)-like transcription factors such as TARGET OF
EAT1 (TOE1) TOE2 SCHLAFMUTZE (SMZ) and SCHNARCHZAPFEN(SNZ)
TOE1 TOE2 SMZ and SNZ have been shown to participate in their productive phase
change by repressing a floral integrator FLOWERING LOCUS T (FT)(Jung et al
2007) Consistent with this toe1-2D 35STOE2 35SSMZ and 35SSNZ plants
exhibit late flowering (Jung et al 2007) MiR172 is also regulated temporally and
highly expressed in the adult vegetative phase both in Arabidopsis and maize (Chuck et
al 2007) Because the temporal expression patterns of miR156 and miR172 are
complementary to each other it has been suggested that the two miRNAs interact with
each other in regulating phase change (Chuck et al 2009 de Alba et al 2013) In
support of this view it has been shown that miR172 is down-regulated in the
Chapter 1 Introduction and Review of Literature
43
Corngrass1(Cg1) mutant in which miR156 is overproduced (Chuck et al 2007 de
Alba et al 2013) However there is no direct evidence for the direct linkage between
miR156 and miR172 It is also envisioned that miR156 and miR172 would not be
directly related For example miR156 regulates plastochron index that is the inverse of
leaf initiation rate and thus frequently used to analyze temporal patterns of plant
development In contrast miR172 does not affect plastochron (Wang et al 2008) The
down-regulation of miR172 in the maize miR156-over producing mutants would be
due to the prolonged juvenile vegetative phase in the mutants
Another miRNA that regulates flowering initiation is miR159 It regulates
MYB33 and MYB65 which are closely related to the barley GAMY B transcription
factor that responds to gibberellic acid (GA) The level of miR159 is elevated in GA-
treated seedlings but reduced in the GA biosynthetic ga1 mutant Transgenic plants
over producing miR159 exhibit delayed floral induction under short days (Achard et
al 2004) It is been suggested that MYB33 mediates GA signal to regulate LEAFY
(LFY) These observations indicate that miR159 plays an important role in the GA
signaling pathways that regulate proper floral induction under short days (Achard et al
2004)
11362 Floral development
Arabidopsis flowers consist of four distinct organs They arise in a characteristic
pattern within concentric rings called whorls from outside to inside four sepals four
petals six stamens and the central pistil consisting of two carpels Identities of the
floral organs are determined by three classes of regulatory genes classA classB and
class C genes (Krizek amp Fletcher 2005) The A and C class genes act antagonistically
to restrict their expression domains (Bowman et al 1991)
It is notable that miR172 also regulates floral architecture in addition to its role
in the timing of flowering initiation MiR172 inhibits the translation of the AP2 mRNA
(Chen 2004) Transgenic plants overproducing miR172 not only exhibit early
flowering but also show abnormal floral development which is quite similar to those
of the class A gene mutants such as the ap2 mutants (Aukerman amp Sakai 2003 de
Alba et al 2013) When the activity of the class A genes is inactivated that of the class
C genes such as AGAMOUS (AG) is elevated As a result the miR172- overproducing
transgenic plants exhibit no petals along with homeotic transformation of the sepals to
Chapter 1 Introduction and Review of Literature
44
the carpels (Aukerman amp Sakai 2003 Chen 2004)
Interestingly miR166 also regulates floral architecture The role of miR165166
in the regulation of meristem activity is closely linked to floral meristem formation and
organization (Zhao et al 2007 Miyashima et al 2013) Floral structure is severely
disrupted in the miR165166-overproducing mutants such as men1 and jba-1D in
which miR166 is overproduced (Williams et al 2005b Jung amp Park 2007) The men1
and jba-1D mutants have smaller gynoecium and reduced carpel number In an extreme
case the jba-1D homozygous plants lack visible carpels (Williams et al 2005b)
Similar phenotypes have been observed in the miR165 overproducers (Zhao et al
2007) It has been suggested that the phenotypes are possibly caused by altered AG
expression in the floral meristem (Jung amp Park 2007 Turchi et al 2013)
Other miRNAs also affect floral development Overproduction of miRNAs
involved in auxin signaling also affects floral architecture Transgenic plants
overproducing miR167 have defects in the formation of ovule and anther with reduced
fertility (Ru et al 2006) It has been shown that miR167 targets ARF6 and ARF8 that
play a role in gynoecium and stamen development and in regulating fertility (Ru et al
2006 Wu et al 2006) These observations suggest that auxin signals are also closely
related to floral organogenesis In addition transgenic plants over-producing miR164
and the cuc1cuc2 double mutants show fused sepals and reduced petals (Laufs et al
2004 Turchi et al 2013) suggesting that miR164 is also related to floral meristem
activity and boundary specification of the meristematic region
11363 Shoot apical meristem development
Shoot apical meristem comprises self-maintaining totipotent cells The shoot apical
meristem activity affects diverse aspects of plant developmental processes including
leaf development vascular development and lateral organ polarity (Bowman et al
2002) Because miR165166 plays a primary role in meristem formation
overproduction of miR165166 and multiple knockout mutants of its target genes
exhibit pleiotropic pheonotypes (McConnell et al 2001 Emery et al 2003 Kim et al
2005 Williams et al 2005b)
The targets of miR165166 include genes encoding the homeo domain-leucine
zipper (HD-ZIP) class III transcription factors such as PHABULOSA(PHB)
PHAVOLUTA (PHV) REVOLUTA(REV) ATHB15CORONA (CNA) and ATHB8
Chapter 1 Introduction and Review of Literature
45
PHB PHV and ATHB15 have overlapping functions in controlling meristem
development (Turchi et al 2013) Indeed the phb phv athb15 triple mutants have an
enlarged shoot apical meristem Consistent with this the miR166- overproducing men1
and jba-1D mutants exhibit enlarged shoot apical meristems (Kim et al 2005
Williams et al 2005b Turchi et al 2013) In the mutants the shoot apical meristems
are multiplied and large
The development of shoot apical meristems is also regulated by the WUSCHEL
(WUS)ndashCLAVATA (CLV) signaling pathway WUS positively regulates cell
proliferation In contrast CLV3 which is expressed in stem cells limits the WUS
expression and thus inhibits cell proliferation in the SAM (Sharma et al 2003)
Consistent with this notion WUS is expressed to a higher level in jba-1D
explaining the ectopic enlargement of the SAM in the mutant (Williams et al 2005)
While WUS is closely related to miR165166 in the shoot apical meristem
development the underlying molecular mechanisms are currently unclear It has been
observed that the miR166-over producing men1 plants have an arrested meristem
development (Jung amp Park 2007) In addition the heterozygous men1+ and the
homozygous men1 plants show different expression patterns of WUS and CLV3 It
appears that miR166 regulates WUS indirectly and influences the expressional balance
between WUS and CLV3 through a negative feedback loop (Jung amp Park 2007 de
Alba et al 2013)
The phv phb athb15 triple mutant has an enlarged shoot apical meristems the
rev phb phv mutant does not have functional shoot apical meristems (Prigge et al
2005) This distinction may be explained by the fact that while miR166 targets
primarily PHB PHV and ATHB15 miR165 targets all the five HD-ZIP III genes (Zhou
et al 2007) Consistent with this transgenic plants overproducing miR166 are distinct
from those overproducing miR165 The miR165-overproducing transgenic plants lack
functional shoot apical meristem and a pin-like structure is formed at the apical region
of the mutant These phenotypes are quite similar to rev phb phv triple mutant (Zhou et
al 2007 Liu et al 2013) The phenotypic distinction may because by the difference
of the cleavage efficiencies of the target mRNAs In accordance with this notion a few
35S miR166a transgenic lines exhibit no shoot apical meristems The men1
homozygous plants also show arrested meristem development (Jung amp Park 2007)
Chapter 1 Introduction and Review of Literature
46
MiR164 cleaves the CUC1 and CUC2 mRNAs as well as the NAC1 mRNA
CUC1 and CUC2 determines the organ boundary in the meristem Phenotypes of
transgenic plants that overproduce miR164 are similar to those of the cuc1cuc2 double
mutant that exhibits embryonic developmental defects such as cup-shaped cotyledons
and fused cotyledons (Laufs et al 2004) When a miR164-resistant CUC2 is
expressed the boundary domain is enlarged CUC also plays a role in embryonic shoot
meristem formation by regulating the SHOOT MERISTEMLESS gene It was therefore
concluded that miR164-mediated cleavage of CUC1 and CUC2 mRNAs determines the
sizes of the boundary domains in the shoot apical meristem and regulates separation of
the organs by restricting cell proliferation (Laufs et al 2004 de Alba et al 2013)
11364 Leaf development
The role of miR319 has been extensively studied in leaf development A dominant
jaw-D mutant overproducing miR319 shows uneven curved leaves with enlarged size
and five TCP genes which are closely related to the CINCINNATA (CIN) gene in
Antirrhinum majus are down regulated in the mutant suggesting that miR319 mediated
regulation of the TCP transcription factor genes are important for the final step of
leaf shaping (Palatnik et al 2003 Naz et al 2013) In addition to leaf shape and size
leaf polarity is also specified by miRNA MiR166-mediated cleavage of the HD-ZIPIII
mRNAs is a well to establish leaf polarity This discovery originates from the gain-of-
function mutants such as phb-1d and phv-1d wherein point mutations in the
complementary sites to miRNA helps the mRNA to evade or resist from miRNA-
mediated cleavage (McConnell et al 2001 Emery et al 2003 Naz et al 2013)
Plant leaves have a dorso-ventral polarity consisting of the adaxial (upper
surface) and abaxial (lower surface) sides The adaxial side is closer to the shoot
apical meristem Adaxial identity is specified by functionally redundant class III HD-
ZIP family genes such as PHB PHV and REV Ectopic expression of each genes
result in adaxialized leaves Dominant phb-1d and phv-1d mutants exhibit
transformation of the abaxial to adaxial fate and have rod-shaped leaves In contrast
miR165166-overproducing plants show pin-like cotyledons radicalized leaves and
abaxialized leaves (Chitwood et al 2007 Pattanayak et al 2013)
Leaf asymmetry is determined by miR166-mediated restriction of the
expression domains of the HD-ZIPIII genes The HD-ZIPIII genes are expressed in the
Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
29
fragments were also cloned through the direct cloning methods To select miRNAs
from the pool of small RNAs secondary structures of the RNA molecules were
examined in compliance with the acknowledged structural features of miRNAs
(Ambros et al 2003) The secondary structures were validated using several ad-hoc
software tools such as the Mfold or RNAfold programs (Reeder et al 2006) Finally
the existence of mature miRNAs was determined by RNA gel blot hybridization These
direct cloning methods were successful in isolating many miRNAs in Arabidopsis
(Reinhart et al 2002 Sunkar amp Zhu 2004) Rice (Sunkar et al 2005) Poplar (Lu et
al 2005) moss (Arazi et al 2005) and Tobacco (Billoud et al 2005)
Although miRNAs were first discovered through forward genetic screens in
worms (Lee et al 1993 Reinhart et al 2000)only few A thaliana miRNA gene
families have been discovered by this method (Chen 2008) in plants MiRNA
involvement in plant mutant phenotypes were not properly inferred till the cloning
experiments established that plant genomes contain numerous miRNAs (Park et al
2002 Reinhart et al 2002 Rhoades et al 2002) MiRNA loss-of-function allele has
been identified by forward genetic screens early extra petala1 is caused by a
transposon insertion ~160bp upstream of the predicted miR164c stem loop resulting in
flowers with extra petals (Baker et al 2005 Irish 2008) The fact that loss-of-function
miRNA mutants have been rarely discovered using forward genetics reflects small
target size for mutagenesis coupled with redundancy in nearly all evolutionarily
conserved plant miRNAs which are encoded by gene families (Table16) Family
members are likely to have overlapping functions buffering against loss at any single
miRNA locus Over expression screens can circumvent redundancy limitations At least
four plant miRNAs miR319 (also known as miR-JAW) miR172 (also known as EAT)
and miR166 were isolated in over expression screens for dominant mutants with
developmental abnormalities (Aukerman amp Sakai 2003 Palatnik et al 2003 Kim et
al 2005 Williams et al 2005b Schommer et al 2012) Mutations in miRNA target
sites which can prevent the entire family of miRNAs from repressing a target gene can
also circumvent redundant functions of miRNA family members
The dominant mutations in the HD-ZIP genes PHB PHV and REVOLUTA
(REV) in Arabidopsis and ROLLEDLEAF1 (RLD1) in Maize result in adaxialization of
leaves andor Vasculature (McConnell amp Barton 1998 McConnell et al 2001 Nelson
et al 2002 Zhong amp Ye 2004 Allen amp Millar 2012) and are all caused by mutations
Chapter 1 Introduction and Review of Literature
30
in miR166 complementary sites (Rhoades et al 2002 Emery et al 2003 Jones-
Rhoades amp Bartel 2004 Mallory et al 2004 Zhong amp Ye 2004)
In both plants and animals cloning was the initial means of large-scale
miRNA discovery (Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Reinhart et al 2002 Mendes et al 2012) However cloning is biased toward
RNAs that are expressed highly and broadly MiRNAs expressed at low levels or
only in specific cell types or in response to certain environmental stimuli were more
difficult to clone Sequence based biases in cloning procedures might also cause
certain miRNAs to be missed Because of these limitations bioinformatics approaches
to identify miRNAs have provided a useful complement to cloning
A straightforward use of bioinformatics has been carried out to find homologs
of known miRNAs both within the same genome and in the genomes of other species
(Pasquinelli et al 2000 Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Mendes et al 2009) A more difficult challenge is to identify miRNAs
unrelated to previously known miRNAs This was first accomplished for
vertebrate nematode and fly miRNAs using algorithms that search for conservation
of sequence and secondary structure (ie miRNA stem-loop precursors) between
species searching for patterns that are characteristic of miRNAs (Lai et al 2003 Lim
et al 2003 Lim et al 2005)
Most of the methods identified numerous miRNAs but are necessarily more
relaxed in terms of allowed structures Some approaches take advantage of the high
complementarity of plant miRNAs to target messages implementing the requirement
that the candidate has conserved complementarity to mRNAs (Jones-Rhoades amp Bartel
2004 Adai et al 2005) This additional filter has been useful for distinguishing
authentic plant miRNAs from false positives which later extended to mammalian
miRNA gene prediction (Xie et al 2005 Mendes et al 2012)
Another criteria used by most algorithms in the filters is evolutionary
conservation In plants mature miRNA sequences are conserved to a higher degree
than their precursor sequences For example Arabidopsis and rice diverged more than
130 million years ago (Friis et al 2004) but some miRNAs are highly conserved
in these plant species (Reinhart et al 2002) Furthermore various parameters for the
secondary structures such as free energy the number of paired residues within a
Chapter 1 Introduction and Review of Literature
31
miRNA or the number and size of bulges are also used to validate the predictions
(Jones-Rhoades amp Bartel 2004 Wang et al 2004 Adai et al 2005 Mendes et al
2012)
1132 Conserved microRNAs in Plants
Till date cloning genetics and bioinformatics screens have resulted in annotation of
approximately 184 potential Arabidopsis miRNAs (miRBase release 13) These
miRNAs seem to arise through gene duplication and subsequent diversification and
represent approximately 100 families (Maher et al 2006 Axtell 2013) Twenty one
families represented by 92 genes are clearly conserved in species beyond Arabidopsis
(Table 15) The presence of these miRNA families in other sequenced plant genomes
indicates that different plant species have an evolutionarily fluid set of miRNAs
(Willmann amp Poethig 2007) Recent studies on mosses one of the most ancient land
plants have shown that at least 14 miRNA families are conserved in eudicots and
mosses and therefore it appears that plant miRNAs share a common ancient origin
(Arazi et al 2005 Axtell amp Bartel 2005 Talmor-Neiman et al 2006 Zhang et al
2006 Axtell 2013) The number of members per family in a genome ranges from 1-32
with the exception of miR-430 which is represented by a cluster of ~80 loci in zebra
fish (Giraldez et al 2005)
In plants the number of members in each family correlates among examined
species certain families contain numerous members in all three species (eg miR156
miR166 miR169) whereas others consistently contain fewer genes (eg miR162
mir168 miR394) (Table 16) Although it is still unclear why certain miRNA are
encoded by more than one gene in plants (eg 12 genes encoding miR156) this
correlation might suggest functional significance of various miRNA family sizes Most
annotated plant miRNAs are conserved throughout flowering plants while a few
miRNAs are found only in a single sequenced genome and thus they could be
evolutionarily lsquoyoungrsquo miRNAs For example Arabidopsis has several miRNA
families that are not found in poplar or rice suggesting that miRNAs could be
generated continuously during evolution (Allen et al 2004a Rajagopalan et al
2006 Fahlgren et al 2007 Axtell 2012 de Alba et al 2013) Consistent with the
notion that non conserved miRNAs are of a more recent evolutionary origin these
miRNAs are mainly encoded by a single locus in the genome Within each family the
mature miRNA is always located in the same arm
Chapter 1 Introduction and Review of Literature
32
Table 16 MicroRNA families conserved in plants
miRNA
family
Arabidopsis Oryza Populus
miR156 12 12 11 miR159319 6 8 15 miR160 3 6 8 miR162 2 2 3 miR164 3 5 6 miR166 9 12 17 miR167 4 9 8 miR168 2 2 2 miR169 14 17 32 miR171 4 7 10 miR172 5 3 9 miR390 3 1 4 miR393 2 2 4 miR394 2 1 2 miR395 6 19 10 miR396 2 5 7 miR397 2 2 3 miR398 3 2 3 miR399 6 11 12 miR408 1 1 1 miR403 1 0 2 miR437 0 1 0 miR444 0 1 0 miR445 0 9 0 Total 92 127 169
All known miRNA families that are conserved between more than one plant species are listed
together with the number of genes identified in the sequenced genomes Rice miRNA families that have orthologs in maize but do not appear to have orthologs in the eudicots (Arabidopsis and Populus) are marked with an asterisk The following families contain miRNA genes annotated with
more than one number miR156 (miR156 and miR157) miR159319 (miR159 and miR319) miR166 (miR165 and miR166) miR171 (miR170 and miR171) and miR390 (miR390 and miR391)
of the stem loop (5prime- or 3prime) as it would be expected if the genes carry common ancestry
(Figure 17) The sequence of the mature miRNA and to some extent the segment on
the opposite arm of the hairpin to which it pairs is highly conserved between the
members of the same family (both within and between the species) while the sequence
secondary structure and length of the intervening ldquolooprdquo region can be highly divergent
within the same family
The patterns of paring and non pairing nucleotides are often conserved between
homologous miRNA stem loops from different species (Figure 17) The significance of
conserved mismatches is opined to guide DCL1 to cleave at the appropriate positions
along the stem loop
Chapter 1 Introduction and Review of Literature
33
Figure 17 Representative stem loop of miR164 Segments corresponding to the mature miRNAs
are shown in red (Adapted from Rajagopalan et al 2006)
Although many miRNAs are highly conserved in plants the degree of conservation in
most cases is quite low between plants and animals one exception is miR854 A recent
study has revealed that miR854 is conserved between Arabidopsis and animals and that
the Arabidopsis miR854 differs from the human miR854 only by a single nucleotide
(Arteaga-Vazquez et al 2006 Axtell 2012 de Alba et al 2013) The Arabidopsis
miR854 has multiple binding sites in the 3prime-untranslated region (UTR) of
OLIGOURIDYLATE-binding-PROTEIN mRNA 1bA (UBP1b) and represses the
translation of the UBP1b mRNA The animal miR854 is also complementary to a site in
the 3prime-UTR of the UBP1b homolog in animals However it is currently unclear how the
animal miR854 regulates its target mRNA Another distinction between plant and
animal miRNAs is the genomic organization of the miRNA genes While plant miRNA
genes are located predominantly in the intergenic regions animal miRNA genes tend to
localize in the intragenic regions both in the introns or exons of protein coding genes
Moreover miRNA genes are often clustered within a single precursor transcript
whereas individual plant miRNAs are derived from individual precursor RNAs (Bartel
Chapter 1 Introduction and Review of Literature
34
2004 Bonnet et al 2004 Kim 2005 Vaucheret et al 2006 Marco et al 2013)
1133 Non-conserved miRNA and challenges in annotation
Most miRNAs are conserved throughout flowering plants (Table 15) but many more
were found only in a single sequenced genome thus considered to be of a more recent
evolutionary origin The extended homology between few non conserved miRNAs
precursors and their target genes provides strong evidence that these relatively ldquoYoungrdquo
miRNAs arose from the duplication of the target gene segments (Allen et al 2004)
Several non-conserved miRNAs including miR161 miR163 miR173 miR447
miR475 and miR476 are known to direct cleavage of target transcripts (Allen et al
2004a Adai et al 2005 Sunkar et al 2005 de Alba et al 2013) It is difficult to
confidently predict targets for many because it is not possible to use complementary
site as a filter against false-positive target predictions It is also difficult to
confidently annotate non-conserved miRNAs as miRNA rather than siRNAs The
established minimal standard for miRNA annotation is a small RNA with detectable
expression and the potential to form a stem-loop when joined to flanking genomic
sequence (Kasschau et al 2003) In practice these requirements are too loose to
definitively categorize many small RNAs cloned from plants Many plant siRNAs are
detectable on blots (Vazquez et al 2004b) and hundreds of thousands of non-
miRNA genomic sequences can be predicted to fold into secondary structures that
resemble structures of plant miRNA precursors (Jones-Rhoades amp Bartel 2004)
Therefore without conservation of both sequence and secondary structure it is
difficult to be confident that a given cloned RNA originated from a stem-loop (ie is a
miRNA) rather than from a double-stranded RNA (ie is a siRNA) In fact many of
the thousands o f small RNAs cloned from Arabidopsis (Gustafson et al 2005) would
probably meet the literal requirements for annotation as miRNAs A few of these
sequences might be miRNAs but others that meet the literal criteria probably are not
so The challenges of annotating non conserved small RNAs were evidenced by
three related small silencing RNAs that were originally annotated as miRNAs
that turned out to be trans-acting siRNAs (tasiRNAs)(Kanno et al2013)
Although most miRNAs that are broadly conserved among flowering plants are
now being identified and experimentally validated (Table 16)(Jones-Rhoades amp
Bartel 2004) the challenges and ambiguities for classifying non-conserved miRNAs
prevent meaningful estimates of the total number of miRNA genes in Arabidopsis and
Chapter 1 Introduction and Review of Literature
35
other plant genomes It is possible that miRNAs might have escaped detection
because they are non-conserved and expressed in specific tissues or conditions and can
only be identified by using high- throughput sequencing
1134 MicroRNA biogenesis
11341 Transcription of microRNA precursor
Plant miRNAs are primarily found in the genomic regions that are not associated with
the protein coding genes (Reinhart et al 2002) and it appears that most if not all plant
miRNAs are produced from their own transcriptional units Plant miRNA genes are
occasionally clustered near each other in the genome suggesting transcription of
multiple miRNAs from a single primary transcript [eg the miR395 cluster (Grishok
et al 2001 Jones-Rhoades amp Bartel 2004b)] but this polycistronic arrangement of
miRNA genes appears far less frequently in plants than in animals (Bartel 2004)
Northern blot EST and mapping evidence indicate that plant miRNA primary
transcripts (also known as pri-miRNAs) as in animals are longer than needed to
encompass the miRNA stem-loops (Palatnik et al 2003 Jones-Rhoades amp Bartel
2004b) At least some of these pri-miRNA transcripts appear to be spliced
polyadenylated (Kurihara amp Watanabe 2004) and capped (Xie Z et al 2005) Two
rice miRNAs are contained within transcripts that contain exon junctions within the
presumptive stem-loop precursor implying that in these cases splicing is a prerequisite
for Dicer recognition (Sunkar et al 2005) Plant pri-miRNAs are over 1kb in length
and they are usually preceded by typical TATA box motifs They can also undergo
canonical splicing polyadenylation and capping All these observations indicates RNA
polymerase II is probably responsible for transcribing most plant miRNAs (Xie Z et
al 2005) as appears to be the case for many animal miRNAs Relatively little is
known about the regulation of miRNA transcription in plants but there is no reason
to suspect that this regulation would differ from that of protein-coding transcripts as
the bioinformatics analysis of the sequence upstream of the transcriptional start
sites of the miRNA genes showed the presence of putative binding motifs of
number of known transcription factors (Megraw et al 2006 Sethupathy 2013)
11342 MicroRNA processing and export
In plants maturation of miRNA is a step wise process A miRNA gene is transcribed to
pri-miRNA which is much longer than the pre-miRNA possessing the characteristic
stem-loop structure Like transcription of most protein-coding genes the miRNA
Chapter 1 Introduction and Review of Literature
36
transcription is processed by RNA polymerase II (Pol II) enzymes and the pri-
miRNAs are 5prime-capped and 3prime-polyadenylated (Lee et al 2004 Xie Z et al 2005)
Mature miRNAs are produced from pri-miRNAs through at least two sequential
processing steps by RNase III-type endonucleases In animals pri-miRNAs are first
processed at the bottom portion of the stem by Drosha a nuclear-localized RNase III
enzyme to liberate pre-miRNAs The pre-miRNAs are then exported to the cytoplasm
with the help of exportin-5The pre-miRNAs are further processed by Dicer another
RNaseIII enzyme to a duplex consisting of the miRNA and the antisense strands
(miRNA) Since plants do not have any Drosha homologs the two sequential
processing steps seem to be carried out by DCL1 a Dicer homolog in the nucleus
(Kurihara amp Watanabe 2004 de Alba et al 2013 Rogers amp Chen 2013)
The processing of pri-miRNAs to pre-miRNAs by DCL1 is assisted by two
other proteins HYPONASTY LEAVES1 (HYL1) and SERRATE (SE) HYL1 belongs to a
family of double-stranded RNA (dsRNA) binding protein and interacts with DCL1 in
vitro and in vivo (Lu amp Fedoroff 2000 Hiraguri et al 2005 de Alba et al 2013
Rogers amp Chen 2013) Loss-of-function mutations in the HYL1 gene result in
decreased accumulation of miRNAs and concomitant accumulation of pri-miRNAs
(Han et al 2004 Vazquez et al 2004a Kurihara et al 2006) SE a C2H2 zinc finger
protein interacts with DCL1 and HYL1 and plays a role in the processing of pri-
miRNAs to pre-miRNAs (Lobbes et al 2006 Yang L et al 2006) In addition the
nuclear cap-binding complex proteins CBP20 and CBP80 that are known as ABA
HYPER SENSITIVE 1(ABH1) have been shown to be required for proper miRNA
processing (Gregory et al 2008 Laubinger et al 2008) Recently it has been
demonstrated that DCL1 can release 21-nt RNAs from dsRNA as well as from
synthetic miR167 precursor RNAs in vitro However this cleavage is error prone and
inefficient in the absence of HYL1 and SE which accelerate the rate of DCL1-mediated
cleavage and promote accurate processing of miRNAs (Dong et al 2008 Rogers amp
Chen 2013)
11343 Methylation of the miRNAmiRNAduplex
After the miRNAmiRNAduplexes are released from the pre-miRNAs by the DCL1
activities the duplexes are methylated at the 2primeOH of the3prime-ends by HUA ENHANCER1
(HEN1) a small RNA methyltransferase (Yu et al 2005 Yang Z et al 2006 Rogers
amp Chen 2013 ) This step which is distinct from the RNase III-mediated processing
Chapter 1 Introduction and Review of Literature
37
step distinguishes the biogenesis of plant miRNAs from that of animal miRNAs
Mutations in the HEN1 gene were first isolated in a morphological screening for floral
development although the HEN1 mutants display pleiotropic phenotypes similar to the
developmental defects observed in the DCL1 mutants (Chen et al 2002 Park et al
2002 Rogers amp Chen 2013)
Figure 18 Biogenesis of microRNA
A Biogenesis of plant MicroRNA B Biogenesis of metazoananimal MicroRNA
The miRNA (red) is incorporated into RISC and the miRNA(blue) in degraded
This enzyme has two dsRNA-binding domains and nuclear localization signal It
is also conserved in fungi and is required for the processing of siRNAs as well as of
miRNAs (Park et al 2002 Boutet et al 2003 Xie et al 2003) Although the HEN1
protein is localized to the nucleus its methylation activity in the cytoplasm cannot be
ignored (Xie et al 2004 Rogers amp Chen 2013)
Chapter 1 Introduction and Review of Literature
38
The methylation of the miRNAmiRNA duplex is required to protect miRNAs
against the 3prime-end uridylation activity and subsequent degradation (Li et al 2005) In
the hen1 mutants accumulation of miRNAs is reduced to a lower level Also the
miRNAs appear to be heterogeneous in their sizes such that a ladder of bands rather
than a single species is observed for most miRNAs in the mutants (Li et al 2005)
MiRNA cloning and sequencing analyses revealed the presence of a number of U
residues at the 3prime-end of miRNAs in the hen1 mutants Moreover these nucleotides do
not correspond to the miRNAs in the pre-miRNAs indicating that these nucleotides are
added after the processing of the miRNAs from pre-miRNAs (Li et al 2005)
Uridylation of RNAs has also been proven to be correlated with RNA decay (Shen amp
Goodman 2004 Mullen amp Marzluff 2008) suggesting that uridylation attracts an
exonuclease which degrades miRNAs in plants
11344 MiRNA incorporation in to the RISC and its nuclear export
The methylated miRNAmiRNA duplexes undergo RISC assembly In this process
the miRNA of the duplexes is selectively incorporated into the RISC and the miRNA
appears to be removed and subsequently degraded (Hammond et al 2000 Hutvagner
amp Zamore 2002 Schwarz et al 2003 de Alba et al 2013 Rogers amp Chen 2013)
The ARGONAUTE 1(AGO1) proteins are core components of the RISC complex
They contain two structural domains the N-terminal PAZ domain that binds to the 3prime-
end of single-stranded RNAs and the C-terminal piwi domains that has a structure
similar to that of RNase H (Cerutti et al 2000 Parker et al 2004) Several AGO
proteins possess endonucleolytic activities within the PIWI domain and cleave target
mRNAs in the middle of the site complementary to miRNA (Liu et al 2004 Meister et
al 2004 Miyoshi et al 2005) Mutations in the AGO1 gene cause miRNA target
genes to be ectopically expressed and the levels of some miRNAs are reduced in the
mutants (Kidner amp Martienssen 2004 Vaucheret et al 2004 Kidner amp Martienssen
2005 Ronemus et al 2006) Moreover the phenotype of the AGO1 hypomorphic
alleles display defects in lateral organ polarity and leaf and flower morphology as
observed in the dcl1 mutants and null AGO1 alleles are embryonically lethal
supporting the close relationship between the AGO proteins and miRNA processing
(Kidner amp Martienssen 2004 Vaucheret et al 2004 de Alba et al 2013 Rogers amp
Chen 2013)
The production of miRNA miRNA duplexes occur in the nucleus while
Chapter 1 Introduction and Review of Literature
39
miRNA-RISC complexes are present in the cytoplasm where target mRNAs are
located This discordance of functional sites requires an export step during miRNA
biogenesis HASTY the Arabidopsis homolog of exportin-5 is thought to be involved
in miRNA nuclear export (Bollman et al 2003 Park et al 2005)The hasty mutant
exhibits pleiotropic defects in plant development and reduced accumulation of most
miRNAs as in the DCL1 or AGO1 mutants Howeverit is unclear whether the HASTY
proteins export the miRNA miRNA duplexes or the miRNA-RISC complexes in
plants
11345 Feedback regulation of miRNA biogenesis
MiRNA biogenesis is under feedback regulation such that the DCL1 and AGO1 genes
are themselves regulated by miRNAs DCL1 is itself a target of miR162 which leads to
the cleavage of the DCL1 mRNA (Xie et al 2003 de Alba et al 2013) Consistent
with this the levels of DCL1 mRNA are elevated in the mutants of miRNA processing
genes in which the miR162 abundance is reduced In addition there is a predicted
hairpin structure of miR838 in the 14th
intron of the DCL1 primary transcript
(Rajagopalan et al 2006) This prediction implies that DCL1-mediated processing
on this site can result in the cleavage of the DCL1 primary transcript into two truncated
fragments the N-terminal capped fragment and the C-terminal polyadenylated
fragment If this is the case it is possible that miRNA biogenesis competes with the
splicing event of the DCL1 pre-mRNA
The AGO1 is also regulated by miRNA There is a complementary site for
miR168 binding in the AGO1 mRNA and the transcript level of the AGO1 mRNA is
reduced through an mRNA cleavage mechanism (Vaucheret et al 2006 de Alba et al
2013 Rogers amp Chen 2013) indicating that the AGO1 mRNA levels are tightly
controlled by the levels of the AGO1 protein
1135 Mechanism of miRNA action
11351 MiRNA-directed mRNA cleavage
By forming the miRNA-RISC assembly miRNAs can direct the RISC to regulate gene
expression by two post transcriptional mechanisms mRNA cleavage or translational
repression (Bartel 2004 Jones-Rhoades et al 2006 Dalmay 2013) The degree
of miRNA-mRNA complementarity plays an important role for the
determination of the mechanism to be used A general rule is that while perfect or
Chapter 1 Introduction and Review of Literature
40
near-perfect complementarity induces mRNA cleavage central mismatches trigger
translational repression (Carrington amp Ambros 2003 Jones-Rhoades amp Bartel 2004)
Unlike most animal miRNAs plant miRNAs have near-perfect or perfect
complementarity to their targets and mRNA cleavage is assumed to be a predominant
mechanism by which miRNAs regulate gene expression in plants
In RNA cleavage mechanism miRNAs guide the AGO component of RISC to
cleave a single phosphodiester bond of the target mRNA within the miRNA-binding
site (Llave et al 2002b Tang et al 2003) The truncated fragments are then released
and degraded freeing the RISC to recognize and cleave another target mRNA This has
been validated by the detection of 3prime cleavage products that have 5prime ends that start at the
middle of the complementary regions The mRNA cleavage products are then further
degraded by other mechanisms The 3prime cleavage products of some miRNA targets are
degraded by the 5prime-3prime exonuclease XRN4 an Arabidopsis homolog of the major Yeast
mRNA degrading exonuclease Xrn1p It has been observed that the 3prime cleavage
products of some target mRNAs were accumulated in the xrn4 mutants (Souret et al
2004 Dalmay 2013) However the 3prime cleavage products of other miRNA targets do
not accumulate in the xrn4 mutants indicating that there are also XRN4 independent
mechanisms The 5prime cleavage products are typically untraceable by RNA gel blot
hybridization but can be detected by sensitive PCR based methods It has been found
that the 5prime cleavage products tend to obtain an oligo U tail at the 3prime ends This 3prime U tail
is correlated with shortening of the 5prime cleavage products from the 5prime end leading to the
conclusion that the 3prime uridylation causes 5 prime- 3 prime exonucleolytic decay of the 5prime cleavage
products (Shen amp Goodman 2004)
DCL1 HYL1 and SE interact with each other and are co-localized in the
nuclear bodies termed Dicing bodies or D-bodies in which some pri-miRNA shave
been found suggesting that D-bodies serve as a center for active miRNA processing
(Fang amp Spector 2007 Fujioka et al 2007 Song et al 2007) The miRNA
processing site appears to be distinct from the Cajal bodies that have previously been
found as a siRNA processing site (Pontes amp Pikaard 2008) The D-bodies include
SmD3 and SmB proteins that are found in the Cajal bodies However At collin an
additional marker for the Cajal bodies is not found in the D-bodies Moreover the D-
bodies are present in an At collin mutant lacking the Cajal bodies The pri-miRNAs of
miR171A and miR159A genes have been found to be co-localized with HYL1 and
Chapter 1 Introduction and Review of Literature
41
DCL1 in the D- bodies that are distinct from the Cajal bodies (Song et al 2007)
11352 MiRNA-directed translational repression
Translation repression is another mechanism exerted by plant miRNAs The regulatory
mechanism of miR172 which regulates flowering time and floral organ identity is
consistent with translation repression MiR172 over production does not affect the
transcript levels of APETALA2 (AP2) and AP2- like target mRNAs Instead the AP2
protein level is reduced in the miR172 over expressing mutant (Aukerman amp Sakai
2003 Chen 2004 Dalmay 2013) It was later shown that miR172 can also direct the
cleavage of the target mRNAs although the steady-state mRNA level is unchanged due
to feedback regulation (Schwab et al 2005 Jung et al 2007) It is clear that miR172
regulates its target genes primarily by translational repression rather than mRNA
cleavage Recently it has been reported that miR156157 and miR854 also regulate
their target genes through translational repression (Arteaga-Vazquez et al 2006
Gandikota et al 2007 Dalmay 2013) It was once thought that translational repression
is an exception to the rule However experimental evidence suggest a widespread
coexistence of translational repression and mRNA cleavage (Brodersen et al 2008)
1136 Regulatory roles of miRNAs in plant development
The miRNA target prediction has been a difficult task in order to obtain regulatory
targets without bringing in too many false predictions Plant growth and developmental
processes regulated by miRNAs are quite diverse and enormous Furthermore plant
miRNAs work cooperatively or antagonistically to establish a balanced regulation This
notion is well consistent with the findings that mutations in the regulators of miRNA
biogenesis transport and processing such as AGO1 DCL1 HYL1 HEN1 ABH1
and HASTY cause pleiotropic phenotypes (Mallory amp Vaucheret 2006 de Alba et al
2013) To date the roles of miRNAs have been mostly deduced from obtaining miRNA
over expressing transgenic plants or gain-of-function mutants in which miRNA-
resistant target genes are ectopically expressed
11361 Phase transition
Plants pass through a series of developmental phase transitions during their life span
beginning with seed germination continuing through vegetative phase change
reproductive phase change flowering initiation and finally seed production for the next
generation Because of their importance in understanding plant developmental
Chapter 1 Introduction and Review of Literature
42
processes genes regulating developmental transitions have been extensively studied
and underlying molecular mechanisms have been explored in various plant species
Majority of researches were mainly focused on vegetative phase change and
reproductive transition in Arabidopsis and Maize (Poethig 2003 Baurle amp Dean
2006) Particularly the mutations of the genes encoding various regulators of miRNA
biogenesis and transport such as HST SE and AGO exhibit altered juvenile to adult
vegetative phase change and vegetative to reproductive change (Hunter et al 2003
Peragine et al 2004 Vazquez et al 2004a Jones-Rhoades et al 2006 de Alba et al
2013)
Over-expression of the miR156a gene causes late flowering and delays
vegetative phase change (Wu amp Poethig 2006 Gandikota et al 2007 Wang et al
2008) It has been shown that miR156 negatively regulates a group of genes encoding
SQUAMOSA PROMOTER-BINDING PROTEIN-LIKE (Lewis et al 2007 de Alba et
al 2013 Rogers amp Chen 2013) transcription factors such as SPL3 SPL4 and SPL5
that promote flowering initiation In contrast transgenic plants over expressing
miR156-resistant SPL345 genes are early flowering Interestingly the endogenous
level of miR156 is temporally regulated In the early juvenile vegetative phase the
level of miR156 is very high but decreases rapidly before the onset of the adult juvenile
phase (Wu amp Poethig 2006)
The Arabidopsis early activation tagged (eat-D) mutant exhibits early flowering
with disrupted floral structure The mutant phenotypes are caused by over expression of
the miR172a-2gene (Aukerman amp Sakai 2003) MiR172 regulates a small group of
genes encoding APETALLA2 (AP2)-like transcription factors such as TARGET OF
EAT1 (TOE1) TOE2 SCHLAFMUTZE (SMZ) and SCHNARCHZAPFEN(SNZ)
TOE1 TOE2 SMZ and SNZ have been shown to participate in their productive phase
change by repressing a floral integrator FLOWERING LOCUS T (FT)(Jung et al
2007) Consistent with this toe1-2D 35STOE2 35SSMZ and 35SSNZ plants
exhibit late flowering (Jung et al 2007) MiR172 is also regulated temporally and
highly expressed in the adult vegetative phase both in Arabidopsis and maize (Chuck et
al 2007) Because the temporal expression patterns of miR156 and miR172 are
complementary to each other it has been suggested that the two miRNAs interact with
each other in regulating phase change (Chuck et al 2009 de Alba et al 2013) In
support of this view it has been shown that miR172 is down-regulated in the
Chapter 1 Introduction and Review of Literature
43
Corngrass1(Cg1) mutant in which miR156 is overproduced (Chuck et al 2007 de
Alba et al 2013) However there is no direct evidence for the direct linkage between
miR156 and miR172 It is also envisioned that miR156 and miR172 would not be
directly related For example miR156 regulates plastochron index that is the inverse of
leaf initiation rate and thus frequently used to analyze temporal patterns of plant
development In contrast miR172 does not affect plastochron (Wang et al 2008) The
down-regulation of miR172 in the maize miR156-over producing mutants would be
due to the prolonged juvenile vegetative phase in the mutants
Another miRNA that regulates flowering initiation is miR159 It regulates
MYB33 and MYB65 which are closely related to the barley GAMY B transcription
factor that responds to gibberellic acid (GA) The level of miR159 is elevated in GA-
treated seedlings but reduced in the GA biosynthetic ga1 mutant Transgenic plants
over producing miR159 exhibit delayed floral induction under short days (Achard et
al 2004) It is been suggested that MYB33 mediates GA signal to regulate LEAFY
(LFY) These observations indicate that miR159 plays an important role in the GA
signaling pathways that regulate proper floral induction under short days (Achard et al
2004)
11362 Floral development
Arabidopsis flowers consist of four distinct organs They arise in a characteristic
pattern within concentric rings called whorls from outside to inside four sepals four
petals six stamens and the central pistil consisting of two carpels Identities of the
floral organs are determined by three classes of regulatory genes classA classB and
class C genes (Krizek amp Fletcher 2005) The A and C class genes act antagonistically
to restrict their expression domains (Bowman et al 1991)
It is notable that miR172 also regulates floral architecture in addition to its role
in the timing of flowering initiation MiR172 inhibits the translation of the AP2 mRNA
(Chen 2004) Transgenic plants overproducing miR172 not only exhibit early
flowering but also show abnormal floral development which is quite similar to those
of the class A gene mutants such as the ap2 mutants (Aukerman amp Sakai 2003 de
Alba et al 2013) When the activity of the class A genes is inactivated that of the class
C genes such as AGAMOUS (AG) is elevated As a result the miR172- overproducing
transgenic plants exhibit no petals along with homeotic transformation of the sepals to
Chapter 1 Introduction and Review of Literature
44
the carpels (Aukerman amp Sakai 2003 Chen 2004)
Interestingly miR166 also regulates floral architecture The role of miR165166
in the regulation of meristem activity is closely linked to floral meristem formation and
organization (Zhao et al 2007 Miyashima et al 2013) Floral structure is severely
disrupted in the miR165166-overproducing mutants such as men1 and jba-1D in
which miR166 is overproduced (Williams et al 2005b Jung amp Park 2007) The men1
and jba-1D mutants have smaller gynoecium and reduced carpel number In an extreme
case the jba-1D homozygous plants lack visible carpels (Williams et al 2005b)
Similar phenotypes have been observed in the miR165 overproducers (Zhao et al
2007) It has been suggested that the phenotypes are possibly caused by altered AG
expression in the floral meristem (Jung amp Park 2007 Turchi et al 2013)
Other miRNAs also affect floral development Overproduction of miRNAs
involved in auxin signaling also affects floral architecture Transgenic plants
overproducing miR167 have defects in the formation of ovule and anther with reduced
fertility (Ru et al 2006) It has been shown that miR167 targets ARF6 and ARF8 that
play a role in gynoecium and stamen development and in regulating fertility (Ru et al
2006 Wu et al 2006) These observations suggest that auxin signals are also closely
related to floral organogenesis In addition transgenic plants over-producing miR164
and the cuc1cuc2 double mutants show fused sepals and reduced petals (Laufs et al
2004 Turchi et al 2013) suggesting that miR164 is also related to floral meristem
activity and boundary specification of the meristematic region
11363 Shoot apical meristem development
Shoot apical meristem comprises self-maintaining totipotent cells The shoot apical
meristem activity affects diverse aspects of plant developmental processes including
leaf development vascular development and lateral organ polarity (Bowman et al
2002) Because miR165166 plays a primary role in meristem formation
overproduction of miR165166 and multiple knockout mutants of its target genes
exhibit pleiotropic pheonotypes (McConnell et al 2001 Emery et al 2003 Kim et al
2005 Williams et al 2005b)
The targets of miR165166 include genes encoding the homeo domain-leucine
zipper (HD-ZIP) class III transcription factors such as PHABULOSA(PHB)
PHAVOLUTA (PHV) REVOLUTA(REV) ATHB15CORONA (CNA) and ATHB8
Chapter 1 Introduction and Review of Literature
45
PHB PHV and ATHB15 have overlapping functions in controlling meristem
development (Turchi et al 2013) Indeed the phb phv athb15 triple mutants have an
enlarged shoot apical meristem Consistent with this the miR166- overproducing men1
and jba-1D mutants exhibit enlarged shoot apical meristems (Kim et al 2005
Williams et al 2005b Turchi et al 2013) In the mutants the shoot apical meristems
are multiplied and large
The development of shoot apical meristems is also regulated by the WUSCHEL
(WUS)ndashCLAVATA (CLV) signaling pathway WUS positively regulates cell
proliferation In contrast CLV3 which is expressed in stem cells limits the WUS
expression and thus inhibits cell proliferation in the SAM (Sharma et al 2003)
Consistent with this notion WUS is expressed to a higher level in jba-1D
explaining the ectopic enlargement of the SAM in the mutant (Williams et al 2005)
While WUS is closely related to miR165166 in the shoot apical meristem
development the underlying molecular mechanisms are currently unclear It has been
observed that the miR166-over producing men1 plants have an arrested meristem
development (Jung amp Park 2007) In addition the heterozygous men1+ and the
homozygous men1 plants show different expression patterns of WUS and CLV3 It
appears that miR166 regulates WUS indirectly and influences the expressional balance
between WUS and CLV3 through a negative feedback loop (Jung amp Park 2007 de
Alba et al 2013)
The phv phb athb15 triple mutant has an enlarged shoot apical meristems the
rev phb phv mutant does not have functional shoot apical meristems (Prigge et al
2005) This distinction may be explained by the fact that while miR166 targets
primarily PHB PHV and ATHB15 miR165 targets all the five HD-ZIP III genes (Zhou
et al 2007) Consistent with this transgenic plants overproducing miR166 are distinct
from those overproducing miR165 The miR165-overproducing transgenic plants lack
functional shoot apical meristem and a pin-like structure is formed at the apical region
of the mutant These phenotypes are quite similar to rev phb phv triple mutant (Zhou et
al 2007 Liu et al 2013) The phenotypic distinction may because by the difference
of the cleavage efficiencies of the target mRNAs In accordance with this notion a few
35S miR166a transgenic lines exhibit no shoot apical meristems The men1
homozygous plants also show arrested meristem development (Jung amp Park 2007)
Chapter 1 Introduction and Review of Literature
46
MiR164 cleaves the CUC1 and CUC2 mRNAs as well as the NAC1 mRNA
CUC1 and CUC2 determines the organ boundary in the meristem Phenotypes of
transgenic plants that overproduce miR164 are similar to those of the cuc1cuc2 double
mutant that exhibits embryonic developmental defects such as cup-shaped cotyledons
and fused cotyledons (Laufs et al 2004) When a miR164-resistant CUC2 is
expressed the boundary domain is enlarged CUC also plays a role in embryonic shoot
meristem formation by regulating the SHOOT MERISTEMLESS gene It was therefore
concluded that miR164-mediated cleavage of CUC1 and CUC2 mRNAs determines the
sizes of the boundary domains in the shoot apical meristem and regulates separation of
the organs by restricting cell proliferation (Laufs et al 2004 de Alba et al 2013)
11364 Leaf development
The role of miR319 has been extensively studied in leaf development A dominant
jaw-D mutant overproducing miR319 shows uneven curved leaves with enlarged size
and five TCP genes which are closely related to the CINCINNATA (CIN) gene in
Antirrhinum majus are down regulated in the mutant suggesting that miR319 mediated
regulation of the TCP transcription factor genes are important for the final step of
leaf shaping (Palatnik et al 2003 Naz et al 2013) In addition to leaf shape and size
leaf polarity is also specified by miRNA MiR166-mediated cleavage of the HD-ZIPIII
mRNAs is a well to establish leaf polarity This discovery originates from the gain-of-
function mutants such as phb-1d and phv-1d wherein point mutations in the
complementary sites to miRNA helps the mRNA to evade or resist from miRNA-
mediated cleavage (McConnell et al 2001 Emery et al 2003 Naz et al 2013)
Plant leaves have a dorso-ventral polarity consisting of the adaxial (upper
surface) and abaxial (lower surface) sides The adaxial side is closer to the shoot
apical meristem Adaxial identity is specified by functionally redundant class III HD-
ZIP family genes such as PHB PHV and REV Ectopic expression of each genes
result in adaxialized leaves Dominant phb-1d and phv-1d mutants exhibit
transformation of the abaxial to adaxial fate and have rod-shaped leaves In contrast
miR165166-overproducing plants show pin-like cotyledons radicalized leaves and
abaxialized leaves (Chitwood et al 2007 Pattanayak et al 2013)
Leaf asymmetry is determined by miR166-mediated restriction of the
expression domains of the HD-ZIPIII genes The HD-ZIPIII genes are expressed in the
Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
30
in miR166 complementary sites (Rhoades et al 2002 Emery et al 2003 Jones-
Rhoades amp Bartel 2004 Mallory et al 2004 Zhong amp Ye 2004)
In both plants and animals cloning was the initial means of large-scale
miRNA discovery (Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Reinhart et al 2002 Mendes et al 2012) However cloning is biased toward
RNAs that are expressed highly and broadly MiRNAs expressed at low levels or
only in specific cell types or in response to certain environmental stimuli were more
difficult to clone Sequence based biases in cloning procedures might also cause
certain miRNAs to be missed Because of these limitations bioinformatics approaches
to identify miRNAs have provided a useful complement to cloning
A straightforward use of bioinformatics has been carried out to find homologs
of known miRNAs both within the same genome and in the genomes of other species
(Pasquinelli et al 2000 Lagos-Quintana et al 2001 Lau et al 2001 Lee amp Ambros
2001 Mendes et al 2009) A more difficult challenge is to identify miRNAs
unrelated to previously known miRNAs This was first accomplished for
vertebrate nematode and fly miRNAs using algorithms that search for conservation
of sequence and secondary structure (ie miRNA stem-loop precursors) between
species searching for patterns that are characteristic of miRNAs (Lai et al 2003 Lim
et al 2003 Lim et al 2005)
Most of the methods identified numerous miRNAs but are necessarily more
relaxed in terms of allowed structures Some approaches take advantage of the high
complementarity of plant miRNAs to target messages implementing the requirement
that the candidate has conserved complementarity to mRNAs (Jones-Rhoades amp Bartel
2004 Adai et al 2005) This additional filter has been useful for distinguishing
authentic plant miRNAs from false positives which later extended to mammalian
miRNA gene prediction (Xie et al 2005 Mendes et al 2012)
Another criteria used by most algorithms in the filters is evolutionary
conservation In plants mature miRNA sequences are conserved to a higher degree
than their precursor sequences For example Arabidopsis and rice diverged more than
130 million years ago (Friis et al 2004) but some miRNAs are highly conserved
in these plant species (Reinhart et al 2002) Furthermore various parameters for the
secondary structures such as free energy the number of paired residues within a
Chapter 1 Introduction and Review of Literature
31
miRNA or the number and size of bulges are also used to validate the predictions
(Jones-Rhoades amp Bartel 2004 Wang et al 2004 Adai et al 2005 Mendes et al
2012)
1132 Conserved microRNAs in Plants
Till date cloning genetics and bioinformatics screens have resulted in annotation of
approximately 184 potential Arabidopsis miRNAs (miRBase release 13) These
miRNAs seem to arise through gene duplication and subsequent diversification and
represent approximately 100 families (Maher et al 2006 Axtell 2013) Twenty one
families represented by 92 genes are clearly conserved in species beyond Arabidopsis
(Table 15) The presence of these miRNA families in other sequenced plant genomes
indicates that different plant species have an evolutionarily fluid set of miRNAs
(Willmann amp Poethig 2007) Recent studies on mosses one of the most ancient land
plants have shown that at least 14 miRNA families are conserved in eudicots and
mosses and therefore it appears that plant miRNAs share a common ancient origin
(Arazi et al 2005 Axtell amp Bartel 2005 Talmor-Neiman et al 2006 Zhang et al
2006 Axtell 2013) The number of members per family in a genome ranges from 1-32
with the exception of miR-430 which is represented by a cluster of ~80 loci in zebra
fish (Giraldez et al 2005)
In plants the number of members in each family correlates among examined
species certain families contain numerous members in all three species (eg miR156
miR166 miR169) whereas others consistently contain fewer genes (eg miR162
mir168 miR394) (Table 16) Although it is still unclear why certain miRNA are
encoded by more than one gene in plants (eg 12 genes encoding miR156) this
correlation might suggest functional significance of various miRNA family sizes Most
annotated plant miRNAs are conserved throughout flowering plants while a few
miRNAs are found only in a single sequenced genome and thus they could be
evolutionarily lsquoyoungrsquo miRNAs For example Arabidopsis has several miRNA
families that are not found in poplar or rice suggesting that miRNAs could be
generated continuously during evolution (Allen et al 2004a Rajagopalan et al
2006 Fahlgren et al 2007 Axtell 2012 de Alba et al 2013) Consistent with the
notion that non conserved miRNAs are of a more recent evolutionary origin these
miRNAs are mainly encoded by a single locus in the genome Within each family the
mature miRNA is always located in the same arm
Chapter 1 Introduction and Review of Literature
32
Table 16 MicroRNA families conserved in plants
miRNA
family
Arabidopsis Oryza Populus
miR156 12 12 11 miR159319 6 8 15 miR160 3 6 8 miR162 2 2 3 miR164 3 5 6 miR166 9 12 17 miR167 4 9 8 miR168 2 2 2 miR169 14 17 32 miR171 4 7 10 miR172 5 3 9 miR390 3 1 4 miR393 2 2 4 miR394 2 1 2 miR395 6 19 10 miR396 2 5 7 miR397 2 2 3 miR398 3 2 3 miR399 6 11 12 miR408 1 1 1 miR403 1 0 2 miR437 0 1 0 miR444 0 1 0 miR445 0 9 0 Total 92 127 169
All known miRNA families that are conserved between more than one plant species are listed
together with the number of genes identified in the sequenced genomes Rice miRNA families that have orthologs in maize but do not appear to have orthologs in the eudicots (Arabidopsis and Populus) are marked with an asterisk The following families contain miRNA genes annotated with
more than one number miR156 (miR156 and miR157) miR159319 (miR159 and miR319) miR166 (miR165 and miR166) miR171 (miR170 and miR171) and miR390 (miR390 and miR391)
of the stem loop (5prime- or 3prime) as it would be expected if the genes carry common ancestry
(Figure 17) The sequence of the mature miRNA and to some extent the segment on
the opposite arm of the hairpin to which it pairs is highly conserved between the
members of the same family (both within and between the species) while the sequence
secondary structure and length of the intervening ldquolooprdquo region can be highly divergent
within the same family
The patterns of paring and non pairing nucleotides are often conserved between
homologous miRNA stem loops from different species (Figure 17) The significance of
conserved mismatches is opined to guide DCL1 to cleave at the appropriate positions
along the stem loop
Chapter 1 Introduction and Review of Literature
33
Figure 17 Representative stem loop of miR164 Segments corresponding to the mature miRNAs
are shown in red (Adapted from Rajagopalan et al 2006)
Although many miRNAs are highly conserved in plants the degree of conservation in
most cases is quite low between plants and animals one exception is miR854 A recent
study has revealed that miR854 is conserved between Arabidopsis and animals and that
the Arabidopsis miR854 differs from the human miR854 only by a single nucleotide
(Arteaga-Vazquez et al 2006 Axtell 2012 de Alba et al 2013) The Arabidopsis
miR854 has multiple binding sites in the 3prime-untranslated region (UTR) of
OLIGOURIDYLATE-binding-PROTEIN mRNA 1bA (UBP1b) and represses the
translation of the UBP1b mRNA The animal miR854 is also complementary to a site in
the 3prime-UTR of the UBP1b homolog in animals However it is currently unclear how the
animal miR854 regulates its target mRNA Another distinction between plant and
animal miRNAs is the genomic organization of the miRNA genes While plant miRNA
genes are located predominantly in the intergenic regions animal miRNA genes tend to
localize in the intragenic regions both in the introns or exons of protein coding genes
Moreover miRNA genes are often clustered within a single precursor transcript
whereas individual plant miRNAs are derived from individual precursor RNAs (Bartel
Chapter 1 Introduction and Review of Literature
34
2004 Bonnet et al 2004 Kim 2005 Vaucheret et al 2006 Marco et al 2013)
1133 Non-conserved miRNA and challenges in annotation
Most miRNAs are conserved throughout flowering plants (Table 15) but many more
were found only in a single sequenced genome thus considered to be of a more recent
evolutionary origin The extended homology between few non conserved miRNAs
precursors and their target genes provides strong evidence that these relatively ldquoYoungrdquo
miRNAs arose from the duplication of the target gene segments (Allen et al 2004)
Several non-conserved miRNAs including miR161 miR163 miR173 miR447
miR475 and miR476 are known to direct cleavage of target transcripts (Allen et al
2004a Adai et al 2005 Sunkar et al 2005 de Alba et al 2013) It is difficult to
confidently predict targets for many because it is not possible to use complementary
site as a filter against false-positive target predictions It is also difficult to
confidently annotate non-conserved miRNAs as miRNA rather than siRNAs The
established minimal standard for miRNA annotation is a small RNA with detectable
expression and the potential to form a stem-loop when joined to flanking genomic
sequence (Kasschau et al 2003) In practice these requirements are too loose to
definitively categorize many small RNAs cloned from plants Many plant siRNAs are
detectable on blots (Vazquez et al 2004b) and hundreds of thousands of non-
miRNA genomic sequences can be predicted to fold into secondary structures that
resemble structures of plant miRNA precursors (Jones-Rhoades amp Bartel 2004)
Therefore without conservation of both sequence and secondary structure it is
difficult to be confident that a given cloned RNA originated from a stem-loop (ie is a
miRNA) rather than from a double-stranded RNA (ie is a siRNA) In fact many of
the thousands o f small RNAs cloned from Arabidopsis (Gustafson et al 2005) would
probably meet the literal requirements for annotation as miRNAs A few of these
sequences might be miRNAs but others that meet the literal criteria probably are not
so The challenges of annotating non conserved small RNAs were evidenced by
three related small silencing RNAs that were originally annotated as miRNAs
that turned out to be trans-acting siRNAs (tasiRNAs)(Kanno et al2013)
Although most miRNAs that are broadly conserved among flowering plants are
now being identified and experimentally validated (Table 16)(Jones-Rhoades amp
Bartel 2004) the challenges and ambiguities for classifying non-conserved miRNAs
prevent meaningful estimates of the total number of miRNA genes in Arabidopsis and
Chapter 1 Introduction and Review of Literature
35
other plant genomes It is possible that miRNAs might have escaped detection
because they are non-conserved and expressed in specific tissues or conditions and can
only be identified by using high- throughput sequencing
1134 MicroRNA biogenesis
11341 Transcription of microRNA precursor
Plant miRNAs are primarily found in the genomic regions that are not associated with
the protein coding genes (Reinhart et al 2002) and it appears that most if not all plant
miRNAs are produced from their own transcriptional units Plant miRNA genes are
occasionally clustered near each other in the genome suggesting transcription of
multiple miRNAs from a single primary transcript [eg the miR395 cluster (Grishok
et al 2001 Jones-Rhoades amp Bartel 2004b)] but this polycistronic arrangement of
miRNA genes appears far less frequently in plants than in animals (Bartel 2004)
Northern blot EST and mapping evidence indicate that plant miRNA primary
transcripts (also known as pri-miRNAs) as in animals are longer than needed to
encompass the miRNA stem-loops (Palatnik et al 2003 Jones-Rhoades amp Bartel
2004b) At least some of these pri-miRNA transcripts appear to be spliced
polyadenylated (Kurihara amp Watanabe 2004) and capped (Xie Z et al 2005) Two
rice miRNAs are contained within transcripts that contain exon junctions within the
presumptive stem-loop precursor implying that in these cases splicing is a prerequisite
for Dicer recognition (Sunkar et al 2005) Plant pri-miRNAs are over 1kb in length
and they are usually preceded by typical TATA box motifs They can also undergo
canonical splicing polyadenylation and capping All these observations indicates RNA
polymerase II is probably responsible for transcribing most plant miRNAs (Xie Z et
al 2005) as appears to be the case for many animal miRNAs Relatively little is
known about the regulation of miRNA transcription in plants but there is no reason
to suspect that this regulation would differ from that of protein-coding transcripts as
the bioinformatics analysis of the sequence upstream of the transcriptional start
sites of the miRNA genes showed the presence of putative binding motifs of
number of known transcription factors (Megraw et al 2006 Sethupathy 2013)
11342 MicroRNA processing and export
In plants maturation of miRNA is a step wise process A miRNA gene is transcribed to
pri-miRNA which is much longer than the pre-miRNA possessing the characteristic
stem-loop structure Like transcription of most protein-coding genes the miRNA
Chapter 1 Introduction and Review of Literature
36
transcription is processed by RNA polymerase II (Pol II) enzymes and the pri-
miRNAs are 5prime-capped and 3prime-polyadenylated (Lee et al 2004 Xie Z et al 2005)
Mature miRNAs are produced from pri-miRNAs through at least two sequential
processing steps by RNase III-type endonucleases In animals pri-miRNAs are first
processed at the bottom portion of the stem by Drosha a nuclear-localized RNase III
enzyme to liberate pre-miRNAs The pre-miRNAs are then exported to the cytoplasm
with the help of exportin-5The pre-miRNAs are further processed by Dicer another
RNaseIII enzyme to a duplex consisting of the miRNA and the antisense strands
(miRNA) Since plants do not have any Drosha homologs the two sequential
processing steps seem to be carried out by DCL1 a Dicer homolog in the nucleus
(Kurihara amp Watanabe 2004 de Alba et al 2013 Rogers amp Chen 2013)
The processing of pri-miRNAs to pre-miRNAs by DCL1 is assisted by two
other proteins HYPONASTY LEAVES1 (HYL1) and SERRATE (SE) HYL1 belongs to a
family of double-stranded RNA (dsRNA) binding protein and interacts with DCL1 in
vitro and in vivo (Lu amp Fedoroff 2000 Hiraguri et al 2005 de Alba et al 2013
Rogers amp Chen 2013) Loss-of-function mutations in the HYL1 gene result in
decreased accumulation of miRNAs and concomitant accumulation of pri-miRNAs
(Han et al 2004 Vazquez et al 2004a Kurihara et al 2006) SE a C2H2 zinc finger
protein interacts with DCL1 and HYL1 and plays a role in the processing of pri-
miRNAs to pre-miRNAs (Lobbes et al 2006 Yang L et al 2006) In addition the
nuclear cap-binding complex proteins CBP20 and CBP80 that are known as ABA
HYPER SENSITIVE 1(ABH1) have been shown to be required for proper miRNA
processing (Gregory et al 2008 Laubinger et al 2008) Recently it has been
demonstrated that DCL1 can release 21-nt RNAs from dsRNA as well as from
synthetic miR167 precursor RNAs in vitro However this cleavage is error prone and
inefficient in the absence of HYL1 and SE which accelerate the rate of DCL1-mediated
cleavage and promote accurate processing of miRNAs (Dong et al 2008 Rogers amp
Chen 2013)
11343 Methylation of the miRNAmiRNAduplex
After the miRNAmiRNAduplexes are released from the pre-miRNAs by the DCL1
activities the duplexes are methylated at the 2primeOH of the3prime-ends by HUA ENHANCER1
(HEN1) a small RNA methyltransferase (Yu et al 2005 Yang Z et al 2006 Rogers
amp Chen 2013 ) This step which is distinct from the RNase III-mediated processing
Chapter 1 Introduction and Review of Literature
37
step distinguishes the biogenesis of plant miRNAs from that of animal miRNAs
Mutations in the HEN1 gene were first isolated in a morphological screening for floral
development although the HEN1 mutants display pleiotropic phenotypes similar to the
developmental defects observed in the DCL1 mutants (Chen et al 2002 Park et al
2002 Rogers amp Chen 2013)
Figure 18 Biogenesis of microRNA
A Biogenesis of plant MicroRNA B Biogenesis of metazoananimal MicroRNA
The miRNA (red) is incorporated into RISC and the miRNA(blue) in degraded
This enzyme has two dsRNA-binding domains and nuclear localization signal It
is also conserved in fungi and is required for the processing of siRNAs as well as of
miRNAs (Park et al 2002 Boutet et al 2003 Xie et al 2003) Although the HEN1
protein is localized to the nucleus its methylation activity in the cytoplasm cannot be
ignored (Xie et al 2004 Rogers amp Chen 2013)
Chapter 1 Introduction and Review of Literature
38
The methylation of the miRNAmiRNA duplex is required to protect miRNAs
against the 3prime-end uridylation activity and subsequent degradation (Li et al 2005) In
the hen1 mutants accumulation of miRNAs is reduced to a lower level Also the
miRNAs appear to be heterogeneous in their sizes such that a ladder of bands rather
than a single species is observed for most miRNAs in the mutants (Li et al 2005)
MiRNA cloning and sequencing analyses revealed the presence of a number of U
residues at the 3prime-end of miRNAs in the hen1 mutants Moreover these nucleotides do
not correspond to the miRNAs in the pre-miRNAs indicating that these nucleotides are
added after the processing of the miRNAs from pre-miRNAs (Li et al 2005)
Uridylation of RNAs has also been proven to be correlated with RNA decay (Shen amp
Goodman 2004 Mullen amp Marzluff 2008) suggesting that uridylation attracts an
exonuclease which degrades miRNAs in plants
11344 MiRNA incorporation in to the RISC and its nuclear export
The methylated miRNAmiRNA duplexes undergo RISC assembly In this process
the miRNA of the duplexes is selectively incorporated into the RISC and the miRNA
appears to be removed and subsequently degraded (Hammond et al 2000 Hutvagner
amp Zamore 2002 Schwarz et al 2003 de Alba et al 2013 Rogers amp Chen 2013)
The ARGONAUTE 1(AGO1) proteins are core components of the RISC complex
They contain two structural domains the N-terminal PAZ domain that binds to the 3prime-
end of single-stranded RNAs and the C-terminal piwi domains that has a structure
similar to that of RNase H (Cerutti et al 2000 Parker et al 2004) Several AGO
proteins possess endonucleolytic activities within the PIWI domain and cleave target
mRNAs in the middle of the site complementary to miRNA (Liu et al 2004 Meister et
al 2004 Miyoshi et al 2005) Mutations in the AGO1 gene cause miRNA target
genes to be ectopically expressed and the levels of some miRNAs are reduced in the
mutants (Kidner amp Martienssen 2004 Vaucheret et al 2004 Kidner amp Martienssen
2005 Ronemus et al 2006) Moreover the phenotype of the AGO1 hypomorphic
alleles display defects in lateral organ polarity and leaf and flower morphology as
observed in the dcl1 mutants and null AGO1 alleles are embryonically lethal
supporting the close relationship between the AGO proteins and miRNA processing
(Kidner amp Martienssen 2004 Vaucheret et al 2004 de Alba et al 2013 Rogers amp
Chen 2013)
The production of miRNA miRNA duplexes occur in the nucleus while
Chapter 1 Introduction and Review of Literature
39
miRNA-RISC complexes are present in the cytoplasm where target mRNAs are
located This discordance of functional sites requires an export step during miRNA
biogenesis HASTY the Arabidopsis homolog of exportin-5 is thought to be involved
in miRNA nuclear export (Bollman et al 2003 Park et al 2005)The hasty mutant
exhibits pleiotropic defects in plant development and reduced accumulation of most
miRNAs as in the DCL1 or AGO1 mutants Howeverit is unclear whether the HASTY
proteins export the miRNA miRNA duplexes or the miRNA-RISC complexes in
plants
11345 Feedback regulation of miRNA biogenesis
MiRNA biogenesis is under feedback regulation such that the DCL1 and AGO1 genes
are themselves regulated by miRNAs DCL1 is itself a target of miR162 which leads to
the cleavage of the DCL1 mRNA (Xie et al 2003 de Alba et al 2013) Consistent
with this the levels of DCL1 mRNA are elevated in the mutants of miRNA processing
genes in which the miR162 abundance is reduced In addition there is a predicted
hairpin structure of miR838 in the 14th
intron of the DCL1 primary transcript
(Rajagopalan et al 2006) This prediction implies that DCL1-mediated processing
on this site can result in the cleavage of the DCL1 primary transcript into two truncated
fragments the N-terminal capped fragment and the C-terminal polyadenylated
fragment If this is the case it is possible that miRNA biogenesis competes with the
splicing event of the DCL1 pre-mRNA
The AGO1 is also regulated by miRNA There is a complementary site for
miR168 binding in the AGO1 mRNA and the transcript level of the AGO1 mRNA is
reduced through an mRNA cleavage mechanism (Vaucheret et al 2006 de Alba et al
2013 Rogers amp Chen 2013) indicating that the AGO1 mRNA levels are tightly
controlled by the levels of the AGO1 protein
1135 Mechanism of miRNA action
11351 MiRNA-directed mRNA cleavage
By forming the miRNA-RISC assembly miRNAs can direct the RISC to regulate gene
expression by two post transcriptional mechanisms mRNA cleavage or translational
repression (Bartel 2004 Jones-Rhoades et al 2006 Dalmay 2013) The degree
of miRNA-mRNA complementarity plays an important role for the
determination of the mechanism to be used A general rule is that while perfect or
Chapter 1 Introduction and Review of Literature
40
near-perfect complementarity induces mRNA cleavage central mismatches trigger
translational repression (Carrington amp Ambros 2003 Jones-Rhoades amp Bartel 2004)
Unlike most animal miRNAs plant miRNAs have near-perfect or perfect
complementarity to their targets and mRNA cleavage is assumed to be a predominant
mechanism by which miRNAs regulate gene expression in plants
In RNA cleavage mechanism miRNAs guide the AGO component of RISC to
cleave a single phosphodiester bond of the target mRNA within the miRNA-binding
site (Llave et al 2002b Tang et al 2003) The truncated fragments are then released
and degraded freeing the RISC to recognize and cleave another target mRNA This has
been validated by the detection of 3prime cleavage products that have 5prime ends that start at the
middle of the complementary regions The mRNA cleavage products are then further
degraded by other mechanisms The 3prime cleavage products of some miRNA targets are
degraded by the 5prime-3prime exonuclease XRN4 an Arabidopsis homolog of the major Yeast
mRNA degrading exonuclease Xrn1p It has been observed that the 3prime cleavage
products of some target mRNAs were accumulated in the xrn4 mutants (Souret et al
2004 Dalmay 2013) However the 3prime cleavage products of other miRNA targets do
not accumulate in the xrn4 mutants indicating that there are also XRN4 independent
mechanisms The 5prime cleavage products are typically untraceable by RNA gel blot
hybridization but can be detected by sensitive PCR based methods It has been found
that the 5prime cleavage products tend to obtain an oligo U tail at the 3prime ends This 3prime U tail
is correlated with shortening of the 5prime cleavage products from the 5prime end leading to the
conclusion that the 3prime uridylation causes 5 prime- 3 prime exonucleolytic decay of the 5prime cleavage
products (Shen amp Goodman 2004)
DCL1 HYL1 and SE interact with each other and are co-localized in the
nuclear bodies termed Dicing bodies or D-bodies in which some pri-miRNA shave
been found suggesting that D-bodies serve as a center for active miRNA processing
(Fang amp Spector 2007 Fujioka et al 2007 Song et al 2007) The miRNA
processing site appears to be distinct from the Cajal bodies that have previously been
found as a siRNA processing site (Pontes amp Pikaard 2008) The D-bodies include
SmD3 and SmB proteins that are found in the Cajal bodies However At collin an
additional marker for the Cajal bodies is not found in the D-bodies Moreover the D-
bodies are present in an At collin mutant lacking the Cajal bodies The pri-miRNAs of
miR171A and miR159A genes have been found to be co-localized with HYL1 and
Chapter 1 Introduction and Review of Literature
41
DCL1 in the D- bodies that are distinct from the Cajal bodies (Song et al 2007)
11352 MiRNA-directed translational repression
Translation repression is another mechanism exerted by plant miRNAs The regulatory
mechanism of miR172 which regulates flowering time and floral organ identity is
consistent with translation repression MiR172 over production does not affect the
transcript levels of APETALA2 (AP2) and AP2- like target mRNAs Instead the AP2
protein level is reduced in the miR172 over expressing mutant (Aukerman amp Sakai
2003 Chen 2004 Dalmay 2013) It was later shown that miR172 can also direct the
cleavage of the target mRNAs although the steady-state mRNA level is unchanged due
to feedback regulation (Schwab et al 2005 Jung et al 2007) It is clear that miR172
regulates its target genes primarily by translational repression rather than mRNA
cleavage Recently it has been reported that miR156157 and miR854 also regulate
their target genes through translational repression (Arteaga-Vazquez et al 2006
Gandikota et al 2007 Dalmay 2013) It was once thought that translational repression
is an exception to the rule However experimental evidence suggest a widespread
coexistence of translational repression and mRNA cleavage (Brodersen et al 2008)
1136 Regulatory roles of miRNAs in plant development
The miRNA target prediction has been a difficult task in order to obtain regulatory
targets without bringing in too many false predictions Plant growth and developmental
processes regulated by miRNAs are quite diverse and enormous Furthermore plant
miRNAs work cooperatively or antagonistically to establish a balanced regulation This
notion is well consistent with the findings that mutations in the regulators of miRNA
biogenesis transport and processing such as AGO1 DCL1 HYL1 HEN1 ABH1
and HASTY cause pleiotropic phenotypes (Mallory amp Vaucheret 2006 de Alba et al
2013) To date the roles of miRNAs have been mostly deduced from obtaining miRNA
over expressing transgenic plants or gain-of-function mutants in which miRNA-
resistant target genes are ectopically expressed
11361 Phase transition
Plants pass through a series of developmental phase transitions during their life span
beginning with seed germination continuing through vegetative phase change
reproductive phase change flowering initiation and finally seed production for the next
generation Because of their importance in understanding plant developmental
Chapter 1 Introduction and Review of Literature
42
processes genes regulating developmental transitions have been extensively studied
and underlying molecular mechanisms have been explored in various plant species
Majority of researches were mainly focused on vegetative phase change and
reproductive transition in Arabidopsis and Maize (Poethig 2003 Baurle amp Dean
2006) Particularly the mutations of the genes encoding various regulators of miRNA
biogenesis and transport such as HST SE and AGO exhibit altered juvenile to adult
vegetative phase change and vegetative to reproductive change (Hunter et al 2003
Peragine et al 2004 Vazquez et al 2004a Jones-Rhoades et al 2006 de Alba et al
2013)
Over-expression of the miR156a gene causes late flowering and delays
vegetative phase change (Wu amp Poethig 2006 Gandikota et al 2007 Wang et al
2008) It has been shown that miR156 negatively regulates a group of genes encoding
SQUAMOSA PROMOTER-BINDING PROTEIN-LIKE (Lewis et al 2007 de Alba et
al 2013 Rogers amp Chen 2013) transcription factors such as SPL3 SPL4 and SPL5
that promote flowering initiation In contrast transgenic plants over expressing
miR156-resistant SPL345 genes are early flowering Interestingly the endogenous
level of miR156 is temporally regulated In the early juvenile vegetative phase the
level of miR156 is very high but decreases rapidly before the onset of the adult juvenile
phase (Wu amp Poethig 2006)
The Arabidopsis early activation tagged (eat-D) mutant exhibits early flowering
with disrupted floral structure The mutant phenotypes are caused by over expression of
the miR172a-2gene (Aukerman amp Sakai 2003) MiR172 regulates a small group of
genes encoding APETALLA2 (AP2)-like transcription factors such as TARGET OF
EAT1 (TOE1) TOE2 SCHLAFMUTZE (SMZ) and SCHNARCHZAPFEN(SNZ)
TOE1 TOE2 SMZ and SNZ have been shown to participate in their productive phase
change by repressing a floral integrator FLOWERING LOCUS T (FT)(Jung et al
2007) Consistent with this toe1-2D 35STOE2 35SSMZ and 35SSNZ plants
exhibit late flowering (Jung et al 2007) MiR172 is also regulated temporally and
highly expressed in the adult vegetative phase both in Arabidopsis and maize (Chuck et
al 2007) Because the temporal expression patterns of miR156 and miR172 are
complementary to each other it has been suggested that the two miRNAs interact with
each other in regulating phase change (Chuck et al 2009 de Alba et al 2013) In
support of this view it has been shown that miR172 is down-regulated in the
Chapter 1 Introduction and Review of Literature
43
Corngrass1(Cg1) mutant in which miR156 is overproduced (Chuck et al 2007 de
Alba et al 2013) However there is no direct evidence for the direct linkage between
miR156 and miR172 It is also envisioned that miR156 and miR172 would not be
directly related For example miR156 regulates plastochron index that is the inverse of
leaf initiation rate and thus frequently used to analyze temporal patterns of plant
development In contrast miR172 does not affect plastochron (Wang et al 2008) The
down-regulation of miR172 in the maize miR156-over producing mutants would be
due to the prolonged juvenile vegetative phase in the mutants
Another miRNA that regulates flowering initiation is miR159 It regulates
MYB33 and MYB65 which are closely related to the barley GAMY B transcription
factor that responds to gibberellic acid (GA) The level of miR159 is elevated in GA-
treated seedlings but reduced in the GA biosynthetic ga1 mutant Transgenic plants
over producing miR159 exhibit delayed floral induction under short days (Achard et
al 2004) It is been suggested that MYB33 mediates GA signal to regulate LEAFY
(LFY) These observations indicate that miR159 plays an important role in the GA
signaling pathways that regulate proper floral induction under short days (Achard et al
2004)
11362 Floral development
Arabidopsis flowers consist of four distinct organs They arise in a characteristic
pattern within concentric rings called whorls from outside to inside four sepals four
petals six stamens and the central pistil consisting of two carpels Identities of the
floral organs are determined by three classes of regulatory genes classA classB and
class C genes (Krizek amp Fletcher 2005) The A and C class genes act antagonistically
to restrict their expression domains (Bowman et al 1991)
It is notable that miR172 also regulates floral architecture in addition to its role
in the timing of flowering initiation MiR172 inhibits the translation of the AP2 mRNA
(Chen 2004) Transgenic plants overproducing miR172 not only exhibit early
flowering but also show abnormal floral development which is quite similar to those
of the class A gene mutants such as the ap2 mutants (Aukerman amp Sakai 2003 de
Alba et al 2013) When the activity of the class A genes is inactivated that of the class
C genes such as AGAMOUS (AG) is elevated As a result the miR172- overproducing
transgenic plants exhibit no petals along with homeotic transformation of the sepals to
Chapter 1 Introduction and Review of Literature
44
the carpels (Aukerman amp Sakai 2003 Chen 2004)
Interestingly miR166 also regulates floral architecture The role of miR165166
in the regulation of meristem activity is closely linked to floral meristem formation and
organization (Zhao et al 2007 Miyashima et al 2013) Floral structure is severely
disrupted in the miR165166-overproducing mutants such as men1 and jba-1D in
which miR166 is overproduced (Williams et al 2005b Jung amp Park 2007) The men1
and jba-1D mutants have smaller gynoecium and reduced carpel number In an extreme
case the jba-1D homozygous plants lack visible carpels (Williams et al 2005b)
Similar phenotypes have been observed in the miR165 overproducers (Zhao et al
2007) It has been suggested that the phenotypes are possibly caused by altered AG
expression in the floral meristem (Jung amp Park 2007 Turchi et al 2013)
Other miRNAs also affect floral development Overproduction of miRNAs
involved in auxin signaling also affects floral architecture Transgenic plants
overproducing miR167 have defects in the formation of ovule and anther with reduced
fertility (Ru et al 2006) It has been shown that miR167 targets ARF6 and ARF8 that
play a role in gynoecium and stamen development and in regulating fertility (Ru et al
2006 Wu et al 2006) These observations suggest that auxin signals are also closely
related to floral organogenesis In addition transgenic plants over-producing miR164
and the cuc1cuc2 double mutants show fused sepals and reduced petals (Laufs et al
2004 Turchi et al 2013) suggesting that miR164 is also related to floral meristem
activity and boundary specification of the meristematic region
11363 Shoot apical meristem development
Shoot apical meristem comprises self-maintaining totipotent cells The shoot apical
meristem activity affects diverse aspects of plant developmental processes including
leaf development vascular development and lateral organ polarity (Bowman et al
2002) Because miR165166 plays a primary role in meristem formation
overproduction of miR165166 and multiple knockout mutants of its target genes
exhibit pleiotropic pheonotypes (McConnell et al 2001 Emery et al 2003 Kim et al
2005 Williams et al 2005b)
The targets of miR165166 include genes encoding the homeo domain-leucine
zipper (HD-ZIP) class III transcription factors such as PHABULOSA(PHB)
PHAVOLUTA (PHV) REVOLUTA(REV) ATHB15CORONA (CNA) and ATHB8
Chapter 1 Introduction and Review of Literature
45
PHB PHV and ATHB15 have overlapping functions in controlling meristem
development (Turchi et al 2013) Indeed the phb phv athb15 triple mutants have an
enlarged shoot apical meristem Consistent with this the miR166- overproducing men1
and jba-1D mutants exhibit enlarged shoot apical meristems (Kim et al 2005
Williams et al 2005b Turchi et al 2013) In the mutants the shoot apical meristems
are multiplied and large
The development of shoot apical meristems is also regulated by the WUSCHEL
(WUS)ndashCLAVATA (CLV) signaling pathway WUS positively regulates cell
proliferation In contrast CLV3 which is expressed in stem cells limits the WUS
expression and thus inhibits cell proliferation in the SAM (Sharma et al 2003)
Consistent with this notion WUS is expressed to a higher level in jba-1D
explaining the ectopic enlargement of the SAM in the mutant (Williams et al 2005)
While WUS is closely related to miR165166 in the shoot apical meristem
development the underlying molecular mechanisms are currently unclear It has been
observed that the miR166-over producing men1 plants have an arrested meristem
development (Jung amp Park 2007) In addition the heterozygous men1+ and the
homozygous men1 plants show different expression patterns of WUS and CLV3 It
appears that miR166 regulates WUS indirectly and influences the expressional balance
between WUS and CLV3 through a negative feedback loop (Jung amp Park 2007 de
Alba et al 2013)
The phv phb athb15 triple mutant has an enlarged shoot apical meristems the
rev phb phv mutant does not have functional shoot apical meristems (Prigge et al
2005) This distinction may be explained by the fact that while miR166 targets
primarily PHB PHV and ATHB15 miR165 targets all the five HD-ZIP III genes (Zhou
et al 2007) Consistent with this transgenic plants overproducing miR166 are distinct
from those overproducing miR165 The miR165-overproducing transgenic plants lack
functional shoot apical meristem and a pin-like structure is formed at the apical region
of the mutant These phenotypes are quite similar to rev phb phv triple mutant (Zhou et
al 2007 Liu et al 2013) The phenotypic distinction may because by the difference
of the cleavage efficiencies of the target mRNAs In accordance with this notion a few
35S miR166a transgenic lines exhibit no shoot apical meristems The men1
homozygous plants also show arrested meristem development (Jung amp Park 2007)
Chapter 1 Introduction and Review of Literature
46
MiR164 cleaves the CUC1 and CUC2 mRNAs as well as the NAC1 mRNA
CUC1 and CUC2 determines the organ boundary in the meristem Phenotypes of
transgenic plants that overproduce miR164 are similar to those of the cuc1cuc2 double
mutant that exhibits embryonic developmental defects such as cup-shaped cotyledons
and fused cotyledons (Laufs et al 2004) When a miR164-resistant CUC2 is
expressed the boundary domain is enlarged CUC also plays a role in embryonic shoot
meristem formation by regulating the SHOOT MERISTEMLESS gene It was therefore
concluded that miR164-mediated cleavage of CUC1 and CUC2 mRNAs determines the
sizes of the boundary domains in the shoot apical meristem and regulates separation of
the organs by restricting cell proliferation (Laufs et al 2004 de Alba et al 2013)
11364 Leaf development
The role of miR319 has been extensively studied in leaf development A dominant
jaw-D mutant overproducing miR319 shows uneven curved leaves with enlarged size
and five TCP genes which are closely related to the CINCINNATA (CIN) gene in
Antirrhinum majus are down regulated in the mutant suggesting that miR319 mediated
regulation of the TCP transcription factor genes are important for the final step of
leaf shaping (Palatnik et al 2003 Naz et al 2013) In addition to leaf shape and size
leaf polarity is also specified by miRNA MiR166-mediated cleavage of the HD-ZIPIII
mRNAs is a well to establish leaf polarity This discovery originates from the gain-of-
function mutants such as phb-1d and phv-1d wherein point mutations in the
complementary sites to miRNA helps the mRNA to evade or resist from miRNA-
mediated cleavage (McConnell et al 2001 Emery et al 2003 Naz et al 2013)
Plant leaves have a dorso-ventral polarity consisting of the adaxial (upper
surface) and abaxial (lower surface) sides The adaxial side is closer to the shoot
apical meristem Adaxial identity is specified by functionally redundant class III HD-
ZIP family genes such as PHB PHV and REV Ectopic expression of each genes
result in adaxialized leaves Dominant phb-1d and phv-1d mutants exhibit
transformation of the abaxial to adaxial fate and have rod-shaped leaves In contrast
miR165166-overproducing plants show pin-like cotyledons radicalized leaves and
abaxialized leaves (Chitwood et al 2007 Pattanayak et al 2013)
Leaf asymmetry is determined by miR166-mediated restriction of the
expression domains of the HD-ZIPIII genes The HD-ZIPIII genes are expressed in the
Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
31
miRNA or the number and size of bulges are also used to validate the predictions
(Jones-Rhoades amp Bartel 2004 Wang et al 2004 Adai et al 2005 Mendes et al
2012)
1132 Conserved microRNAs in Plants
Till date cloning genetics and bioinformatics screens have resulted in annotation of
approximately 184 potential Arabidopsis miRNAs (miRBase release 13) These
miRNAs seem to arise through gene duplication and subsequent diversification and
represent approximately 100 families (Maher et al 2006 Axtell 2013) Twenty one
families represented by 92 genes are clearly conserved in species beyond Arabidopsis
(Table 15) The presence of these miRNA families in other sequenced plant genomes
indicates that different plant species have an evolutionarily fluid set of miRNAs
(Willmann amp Poethig 2007) Recent studies on mosses one of the most ancient land
plants have shown that at least 14 miRNA families are conserved in eudicots and
mosses and therefore it appears that plant miRNAs share a common ancient origin
(Arazi et al 2005 Axtell amp Bartel 2005 Talmor-Neiman et al 2006 Zhang et al
2006 Axtell 2013) The number of members per family in a genome ranges from 1-32
with the exception of miR-430 which is represented by a cluster of ~80 loci in zebra
fish (Giraldez et al 2005)
In plants the number of members in each family correlates among examined
species certain families contain numerous members in all three species (eg miR156
miR166 miR169) whereas others consistently contain fewer genes (eg miR162
mir168 miR394) (Table 16) Although it is still unclear why certain miRNA are
encoded by more than one gene in plants (eg 12 genes encoding miR156) this
correlation might suggest functional significance of various miRNA family sizes Most
annotated plant miRNAs are conserved throughout flowering plants while a few
miRNAs are found only in a single sequenced genome and thus they could be
evolutionarily lsquoyoungrsquo miRNAs For example Arabidopsis has several miRNA
families that are not found in poplar or rice suggesting that miRNAs could be
generated continuously during evolution (Allen et al 2004a Rajagopalan et al
2006 Fahlgren et al 2007 Axtell 2012 de Alba et al 2013) Consistent with the
notion that non conserved miRNAs are of a more recent evolutionary origin these
miRNAs are mainly encoded by a single locus in the genome Within each family the
mature miRNA is always located in the same arm
Chapter 1 Introduction and Review of Literature
32
Table 16 MicroRNA families conserved in plants
miRNA
family
Arabidopsis Oryza Populus
miR156 12 12 11 miR159319 6 8 15 miR160 3 6 8 miR162 2 2 3 miR164 3 5 6 miR166 9 12 17 miR167 4 9 8 miR168 2 2 2 miR169 14 17 32 miR171 4 7 10 miR172 5 3 9 miR390 3 1 4 miR393 2 2 4 miR394 2 1 2 miR395 6 19 10 miR396 2 5 7 miR397 2 2 3 miR398 3 2 3 miR399 6 11 12 miR408 1 1 1 miR403 1 0 2 miR437 0 1 0 miR444 0 1 0 miR445 0 9 0 Total 92 127 169
All known miRNA families that are conserved between more than one plant species are listed
together with the number of genes identified in the sequenced genomes Rice miRNA families that have orthologs in maize but do not appear to have orthologs in the eudicots (Arabidopsis and Populus) are marked with an asterisk The following families contain miRNA genes annotated with
more than one number miR156 (miR156 and miR157) miR159319 (miR159 and miR319) miR166 (miR165 and miR166) miR171 (miR170 and miR171) and miR390 (miR390 and miR391)
of the stem loop (5prime- or 3prime) as it would be expected if the genes carry common ancestry
(Figure 17) The sequence of the mature miRNA and to some extent the segment on
the opposite arm of the hairpin to which it pairs is highly conserved between the
members of the same family (both within and between the species) while the sequence
secondary structure and length of the intervening ldquolooprdquo region can be highly divergent
within the same family
The patterns of paring and non pairing nucleotides are often conserved between
homologous miRNA stem loops from different species (Figure 17) The significance of
conserved mismatches is opined to guide DCL1 to cleave at the appropriate positions
along the stem loop
Chapter 1 Introduction and Review of Literature
33
Figure 17 Representative stem loop of miR164 Segments corresponding to the mature miRNAs
are shown in red (Adapted from Rajagopalan et al 2006)
Although many miRNAs are highly conserved in plants the degree of conservation in
most cases is quite low between plants and animals one exception is miR854 A recent
study has revealed that miR854 is conserved between Arabidopsis and animals and that
the Arabidopsis miR854 differs from the human miR854 only by a single nucleotide
(Arteaga-Vazquez et al 2006 Axtell 2012 de Alba et al 2013) The Arabidopsis
miR854 has multiple binding sites in the 3prime-untranslated region (UTR) of
OLIGOURIDYLATE-binding-PROTEIN mRNA 1bA (UBP1b) and represses the
translation of the UBP1b mRNA The animal miR854 is also complementary to a site in
the 3prime-UTR of the UBP1b homolog in animals However it is currently unclear how the
animal miR854 regulates its target mRNA Another distinction between plant and
animal miRNAs is the genomic organization of the miRNA genes While plant miRNA
genes are located predominantly in the intergenic regions animal miRNA genes tend to
localize in the intragenic regions both in the introns or exons of protein coding genes
Moreover miRNA genes are often clustered within a single precursor transcript
whereas individual plant miRNAs are derived from individual precursor RNAs (Bartel
Chapter 1 Introduction and Review of Literature
34
2004 Bonnet et al 2004 Kim 2005 Vaucheret et al 2006 Marco et al 2013)
1133 Non-conserved miRNA and challenges in annotation
Most miRNAs are conserved throughout flowering plants (Table 15) but many more
were found only in a single sequenced genome thus considered to be of a more recent
evolutionary origin The extended homology between few non conserved miRNAs
precursors and their target genes provides strong evidence that these relatively ldquoYoungrdquo
miRNAs arose from the duplication of the target gene segments (Allen et al 2004)
Several non-conserved miRNAs including miR161 miR163 miR173 miR447
miR475 and miR476 are known to direct cleavage of target transcripts (Allen et al
2004a Adai et al 2005 Sunkar et al 2005 de Alba et al 2013) It is difficult to
confidently predict targets for many because it is not possible to use complementary
site as a filter against false-positive target predictions It is also difficult to
confidently annotate non-conserved miRNAs as miRNA rather than siRNAs The
established minimal standard for miRNA annotation is a small RNA with detectable
expression and the potential to form a stem-loop when joined to flanking genomic
sequence (Kasschau et al 2003) In practice these requirements are too loose to
definitively categorize many small RNAs cloned from plants Many plant siRNAs are
detectable on blots (Vazquez et al 2004b) and hundreds of thousands of non-
miRNA genomic sequences can be predicted to fold into secondary structures that
resemble structures of plant miRNA precursors (Jones-Rhoades amp Bartel 2004)
Therefore without conservation of both sequence and secondary structure it is
difficult to be confident that a given cloned RNA originated from a stem-loop (ie is a
miRNA) rather than from a double-stranded RNA (ie is a siRNA) In fact many of
the thousands o f small RNAs cloned from Arabidopsis (Gustafson et al 2005) would
probably meet the literal requirements for annotation as miRNAs A few of these
sequences might be miRNAs but others that meet the literal criteria probably are not
so The challenges of annotating non conserved small RNAs were evidenced by
three related small silencing RNAs that were originally annotated as miRNAs
that turned out to be trans-acting siRNAs (tasiRNAs)(Kanno et al2013)
Although most miRNAs that are broadly conserved among flowering plants are
now being identified and experimentally validated (Table 16)(Jones-Rhoades amp
Bartel 2004) the challenges and ambiguities for classifying non-conserved miRNAs
prevent meaningful estimates of the total number of miRNA genes in Arabidopsis and
Chapter 1 Introduction and Review of Literature
35
other plant genomes It is possible that miRNAs might have escaped detection
because they are non-conserved and expressed in specific tissues or conditions and can
only be identified by using high- throughput sequencing
1134 MicroRNA biogenesis
11341 Transcription of microRNA precursor
Plant miRNAs are primarily found in the genomic regions that are not associated with
the protein coding genes (Reinhart et al 2002) and it appears that most if not all plant
miRNAs are produced from their own transcriptional units Plant miRNA genes are
occasionally clustered near each other in the genome suggesting transcription of
multiple miRNAs from a single primary transcript [eg the miR395 cluster (Grishok
et al 2001 Jones-Rhoades amp Bartel 2004b)] but this polycistronic arrangement of
miRNA genes appears far less frequently in plants than in animals (Bartel 2004)
Northern blot EST and mapping evidence indicate that plant miRNA primary
transcripts (also known as pri-miRNAs) as in animals are longer than needed to
encompass the miRNA stem-loops (Palatnik et al 2003 Jones-Rhoades amp Bartel
2004b) At least some of these pri-miRNA transcripts appear to be spliced
polyadenylated (Kurihara amp Watanabe 2004) and capped (Xie Z et al 2005) Two
rice miRNAs are contained within transcripts that contain exon junctions within the
presumptive stem-loop precursor implying that in these cases splicing is a prerequisite
for Dicer recognition (Sunkar et al 2005) Plant pri-miRNAs are over 1kb in length
and they are usually preceded by typical TATA box motifs They can also undergo
canonical splicing polyadenylation and capping All these observations indicates RNA
polymerase II is probably responsible for transcribing most plant miRNAs (Xie Z et
al 2005) as appears to be the case for many animal miRNAs Relatively little is
known about the regulation of miRNA transcription in plants but there is no reason
to suspect that this regulation would differ from that of protein-coding transcripts as
the bioinformatics analysis of the sequence upstream of the transcriptional start
sites of the miRNA genes showed the presence of putative binding motifs of
number of known transcription factors (Megraw et al 2006 Sethupathy 2013)
11342 MicroRNA processing and export
In plants maturation of miRNA is a step wise process A miRNA gene is transcribed to
pri-miRNA which is much longer than the pre-miRNA possessing the characteristic
stem-loop structure Like transcription of most protein-coding genes the miRNA
Chapter 1 Introduction and Review of Literature
36
transcription is processed by RNA polymerase II (Pol II) enzymes and the pri-
miRNAs are 5prime-capped and 3prime-polyadenylated (Lee et al 2004 Xie Z et al 2005)
Mature miRNAs are produced from pri-miRNAs through at least two sequential
processing steps by RNase III-type endonucleases In animals pri-miRNAs are first
processed at the bottom portion of the stem by Drosha a nuclear-localized RNase III
enzyme to liberate pre-miRNAs The pre-miRNAs are then exported to the cytoplasm
with the help of exportin-5The pre-miRNAs are further processed by Dicer another
RNaseIII enzyme to a duplex consisting of the miRNA and the antisense strands
(miRNA) Since plants do not have any Drosha homologs the two sequential
processing steps seem to be carried out by DCL1 a Dicer homolog in the nucleus
(Kurihara amp Watanabe 2004 de Alba et al 2013 Rogers amp Chen 2013)
The processing of pri-miRNAs to pre-miRNAs by DCL1 is assisted by two
other proteins HYPONASTY LEAVES1 (HYL1) and SERRATE (SE) HYL1 belongs to a
family of double-stranded RNA (dsRNA) binding protein and interacts with DCL1 in
vitro and in vivo (Lu amp Fedoroff 2000 Hiraguri et al 2005 de Alba et al 2013
Rogers amp Chen 2013) Loss-of-function mutations in the HYL1 gene result in
decreased accumulation of miRNAs and concomitant accumulation of pri-miRNAs
(Han et al 2004 Vazquez et al 2004a Kurihara et al 2006) SE a C2H2 zinc finger
protein interacts with DCL1 and HYL1 and plays a role in the processing of pri-
miRNAs to pre-miRNAs (Lobbes et al 2006 Yang L et al 2006) In addition the
nuclear cap-binding complex proteins CBP20 and CBP80 that are known as ABA
HYPER SENSITIVE 1(ABH1) have been shown to be required for proper miRNA
processing (Gregory et al 2008 Laubinger et al 2008) Recently it has been
demonstrated that DCL1 can release 21-nt RNAs from dsRNA as well as from
synthetic miR167 precursor RNAs in vitro However this cleavage is error prone and
inefficient in the absence of HYL1 and SE which accelerate the rate of DCL1-mediated
cleavage and promote accurate processing of miRNAs (Dong et al 2008 Rogers amp
Chen 2013)
11343 Methylation of the miRNAmiRNAduplex
After the miRNAmiRNAduplexes are released from the pre-miRNAs by the DCL1
activities the duplexes are methylated at the 2primeOH of the3prime-ends by HUA ENHANCER1
(HEN1) a small RNA methyltransferase (Yu et al 2005 Yang Z et al 2006 Rogers
amp Chen 2013 ) This step which is distinct from the RNase III-mediated processing
Chapter 1 Introduction and Review of Literature
37
step distinguishes the biogenesis of plant miRNAs from that of animal miRNAs
Mutations in the HEN1 gene were first isolated in a morphological screening for floral
development although the HEN1 mutants display pleiotropic phenotypes similar to the
developmental defects observed in the DCL1 mutants (Chen et al 2002 Park et al
2002 Rogers amp Chen 2013)
Figure 18 Biogenesis of microRNA
A Biogenesis of plant MicroRNA B Biogenesis of metazoananimal MicroRNA
The miRNA (red) is incorporated into RISC and the miRNA(blue) in degraded
This enzyme has two dsRNA-binding domains and nuclear localization signal It
is also conserved in fungi and is required for the processing of siRNAs as well as of
miRNAs (Park et al 2002 Boutet et al 2003 Xie et al 2003) Although the HEN1
protein is localized to the nucleus its methylation activity in the cytoplasm cannot be
ignored (Xie et al 2004 Rogers amp Chen 2013)
Chapter 1 Introduction and Review of Literature
38
The methylation of the miRNAmiRNA duplex is required to protect miRNAs
against the 3prime-end uridylation activity and subsequent degradation (Li et al 2005) In
the hen1 mutants accumulation of miRNAs is reduced to a lower level Also the
miRNAs appear to be heterogeneous in their sizes such that a ladder of bands rather
than a single species is observed for most miRNAs in the mutants (Li et al 2005)
MiRNA cloning and sequencing analyses revealed the presence of a number of U
residues at the 3prime-end of miRNAs in the hen1 mutants Moreover these nucleotides do
not correspond to the miRNAs in the pre-miRNAs indicating that these nucleotides are
added after the processing of the miRNAs from pre-miRNAs (Li et al 2005)
Uridylation of RNAs has also been proven to be correlated with RNA decay (Shen amp
Goodman 2004 Mullen amp Marzluff 2008) suggesting that uridylation attracts an
exonuclease which degrades miRNAs in plants
11344 MiRNA incorporation in to the RISC and its nuclear export
The methylated miRNAmiRNA duplexes undergo RISC assembly In this process
the miRNA of the duplexes is selectively incorporated into the RISC and the miRNA
appears to be removed and subsequently degraded (Hammond et al 2000 Hutvagner
amp Zamore 2002 Schwarz et al 2003 de Alba et al 2013 Rogers amp Chen 2013)
The ARGONAUTE 1(AGO1) proteins are core components of the RISC complex
They contain two structural domains the N-terminal PAZ domain that binds to the 3prime-
end of single-stranded RNAs and the C-terminal piwi domains that has a structure
similar to that of RNase H (Cerutti et al 2000 Parker et al 2004) Several AGO
proteins possess endonucleolytic activities within the PIWI domain and cleave target
mRNAs in the middle of the site complementary to miRNA (Liu et al 2004 Meister et
al 2004 Miyoshi et al 2005) Mutations in the AGO1 gene cause miRNA target
genes to be ectopically expressed and the levels of some miRNAs are reduced in the
mutants (Kidner amp Martienssen 2004 Vaucheret et al 2004 Kidner amp Martienssen
2005 Ronemus et al 2006) Moreover the phenotype of the AGO1 hypomorphic
alleles display defects in lateral organ polarity and leaf and flower morphology as
observed in the dcl1 mutants and null AGO1 alleles are embryonically lethal
supporting the close relationship between the AGO proteins and miRNA processing
(Kidner amp Martienssen 2004 Vaucheret et al 2004 de Alba et al 2013 Rogers amp
Chen 2013)
The production of miRNA miRNA duplexes occur in the nucleus while
Chapter 1 Introduction and Review of Literature
39
miRNA-RISC complexes are present in the cytoplasm where target mRNAs are
located This discordance of functional sites requires an export step during miRNA
biogenesis HASTY the Arabidopsis homolog of exportin-5 is thought to be involved
in miRNA nuclear export (Bollman et al 2003 Park et al 2005)The hasty mutant
exhibits pleiotropic defects in plant development and reduced accumulation of most
miRNAs as in the DCL1 or AGO1 mutants Howeverit is unclear whether the HASTY
proteins export the miRNA miRNA duplexes or the miRNA-RISC complexes in
plants
11345 Feedback regulation of miRNA biogenesis
MiRNA biogenesis is under feedback regulation such that the DCL1 and AGO1 genes
are themselves regulated by miRNAs DCL1 is itself a target of miR162 which leads to
the cleavage of the DCL1 mRNA (Xie et al 2003 de Alba et al 2013) Consistent
with this the levels of DCL1 mRNA are elevated in the mutants of miRNA processing
genes in which the miR162 abundance is reduced In addition there is a predicted
hairpin structure of miR838 in the 14th
intron of the DCL1 primary transcript
(Rajagopalan et al 2006) This prediction implies that DCL1-mediated processing
on this site can result in the cleavage of the DCL1 primary transcript into two truncated
fragments the N-terminal capped fragment and the C-terminal polyadenylated
fragment If this is the case it is possible that miRNA biogenesis competes with the
splicing event of the DCL1 pre-mRNA
The AGO1 is also regulated by miRNA There is a complementary site for
miR168 binding in the AGO1 mRNA and the transcript level of the AGO1 mRNA is
reduced through an mRNA cleavage mechanism (Vaucheret et al 2006 de Alba et al
2013 Rogers amp Chen 2013) indicating that the AGO1 mRNA levels are tightly
controlled by the levels of the AGO1 protein
1135 Mechanism of miRNA action
11351 MiRNA-directed mRNA cleavage
By forming the miRNA-RISC assembly miRNAs can direct the RISC to regulate gene
expression by two post transcriptional mechanisms mRNA cleavage or translational
repression (Bartel 2004 Jones-Rhoades et al 2006 Dalmay 2013) The degree
of miRNA-mRNA complementarity plays an important role for the
determination of the mechanism to be used A general rule is that while perfect or
Chapter 1 Introduction and Review of Literature
40
near-perfect complementarity induces mRNA cleavage central mismatches trigger
translational repression (Carrington amp Ambros 2003 Jones-Rhoades amp Bartel 2004)
Unlike most animal miRNAs plant miRNAs have near-perfect or perfect
complementarity to their targets and mRNA cleavage is assumed to be a predominant
mechanism by which miRNAs regulate gene expression in plants
In RNA cleavage mechanism miRNAs guide the AGO component of RISC to
cleave a single phosphodiester bond of the target mRNA within the miRNA-binding
site (Llave et al 2002b Tang et al 2003) The truncated fragments are then released
and degraded freeing the RISC to recognize and cleave another target mRNA This has
been validated by the detection of 3prime cleavage products that have 5prime ends that start at the
middle of the complementary regions The mRNA cleavage products are then further
degraded by other mechanisms The 3prime cleavage products of some miRNA targets are
degraded by the 5prime-3prime exonuclease XRN4 an Arabidopsis homolog of the major Yeast
mRNA degrading exonuclease Xrn1p It has been observed that the 3prime cleavage
products of some target mRNAs were accumulated in the xrn4 mutants (Souret et al
2004 Dalmay 2013) However the 3prime cleavage products of other miRNA targets do
not accumulate in the xrn4 mutants indicating that there are also XRN4 independent
mechanisms The 5prime cleavage products are typically untraceable by RNA gel blot
hybridization but can be detected by sensitive PCR based methods It has been found
that the 5prime cleavage products tend to obtain an oligo U tail at the 3prime ends This 3prime U tail
is correlated with shortening of the 5prime cleavage products from the 5prime end leading to the
conclusion that the 3prime uridylation causes 5 prime- 3 prime exonucleolytic decay of the 5prime cleavage
products (Shen amp Goodman 2004)
DCL1 HYL1 and SE interact with each other and are co-localized in the
nuclear bodies termed Dicing bodies or D-bodies in which some pri-miRNA shave
been found suggesting that D-bodies serve as a center for active miRNA processing
(Fang amp Spector 2007 Fujioka et al 2007 Song et al 2007) The miRNA
processing site appears to be distinct from the Cajal bodies that have previously been
found as a siRNA processing site (Pontes amp Pikaard 2008) The D-bodies include
SmD3 and SmB proteins that are found in the Cajal bodies However At collin an
additional marker for the Cajal bodies is not found in the D-bodies Moreover the D-
bodies are present in an At collin mutant lacking the Cajal bodies The pri-miRNAs of
miR171A and miR159A genes have been found to be co-localized with HYL1 and
Chapter 1 Introduction and Review of Literature
41
DCL1 in the D- bodies that are distinct from the Cajal bodies (Song et al 2007)
11352 MiRNA-directed translational repression
Translation repression is another mechanism exerted by plant miRNAs The regulatory
mechanism of miR172 which regulates flowering time and floral organ identity is
consistent with translation repression MiR172 over production does not affect the
transcript levels of APETALA2 (AP2) and AP2- like target mRNAs Instead the AP2
protein level is reduced in the miR172 over expressing mutant (Aukerman amp Sakai
2003 Chen 2004 Dalmay 2013) It was later shown that miR172 can also direct the
cleavage of the target mRNAs although the steady-state mRNA level is unchanged due
to feedback regulation (Schwab et al 2005 Jung et al 2007) It is clear that miR172
regulates its target genes primarily by translational repression rather than mRNA
cleavage Recently it has been reported that miR156157 and miR854 also regulate
their target genes through translational repression (Arteaga-Vazquez et al 2006
Gandikota et al 2007 Dalmay 2013) It was once thought that translational repression
is an exception to the rule However experimental evidence suggest a widespread
coexistence of translational repression and mRNA cleavage (Brodersen et al 2008)
1136 Regulatory roles of miRNAs in plant development
The miRNA target prediction has been a difficult task in order to obtain regulatory
targets without bringing in too many false predictions Plant growth and developmental
processes regulated by miRNAs are quite diverse and enormous Furthermore plant
miRNAs work cooperatively or antagonistically to establish a balanced regulation This
notion is well consistent with the findings that mutations in the regulators of miRNA
biogenesis transport and processing such as AGO1 DCL1 HYL1 HEN1 ABH1
and HASTY cause pleiotropic phenotypes (Mallory amp Vaucheret 2006 de Alba et al
2013) To date the roles of miRNAs have been mostly deduced from obtaining miRNA
over expressing transgenic plants or gain-of-function mutants in which miRNA-
resistant target genes are ectopically expressed
11361 Phase transition
Plants pass through a series of developmental phase transitions during their life span
beginning with seed germination continuing through vegetative phase change
reproductive phase change flowering initiation and finally seed production for the next
generation Because of their importance in understanding plant developmental
Chapter 1 Introduction and Review of Literature
42
processes genes regulating developmental transitions have been extensively studied
and underlying molecular mechanisms have been explored in various plant species
Majority of researches were mainly focused on vegetative phase change and
reproductive transition in Arabidopsis and Maize (Poethig 2003 Baurle amp Dean
2006) Particularly the mutations of the genes encoding various regulators of miRNA
biogenesis and transport such as HST SE and AGO exhibit altered juvenile to adult
vegetative phase change and vegetative to reproductive change (Hunter et al 2003
Peragine et al 2004 Vazquez et al 2004a Jones-Rhoades et al 2006 de Alba et al
2013)
Over-expression of the miR156a gene causes late flowering and delays
vegetative phase change (Wu amp Poethig 2006 Gandikota et al 2007 Wang et al
2008) It has been shown that miR156 negatively regulates a group of genes encoding
SQUAMOSA PROMOTER-BINDING PROTEIN-LIKE (Lewis et al 2007 de Alba et
al 2013 Rogers amp Chen 2013) transcription factors such as SPL3 SPL4 and SPL5
that promote flowering initiation In contrast transgenic plants over expressing
miR156-resistant SPL345 genes are early flowering Interestingly the endogenous
level of miR156 is temporally regulated In the early juvenile vegetative phase the
level of miR156 is very high but decreases rapidly before the onset of the adult juvenile
phase (Wu amp Poethig 2006)
The Arabidopsis early activation tagged (eat-D) mutant exhibits early flowering
with disrupted floral structure The mutant phenotypes are caused by over expression of
the miR172a-2gene (Aukerman amp Sakai 2003) MiR172 regulates a small group of
genes encoding APETALLA2 (AP2)-like transcription factors such as TARGET OF
EAT1 (TOE1) TOE2 SCHLAFMUTZE (SMZ) and SCHNARCHZAPFEN(SNZ)
TOE1 TOE2 SMZ and SNZ have been shown to participate in their productive phase
change by repressing a floral integrator FLOWERING LOCUS T (FT)(Jung et al
2007) Consistent with this toe1-2D 35STOE2 35SSMZ and 35SSNZ plants
exhibit late flowering (Jung et al 2007) MiR172 is also regulated temporally and
highly expressed in the adult vegetative phase both in Arabidopsis and maize (Chuck et
al 2007) Because the temporal expression patterns of miR156 and miR172 are
complementary to each other it has been suggested that the two miRNAs interact with
each other in regulating phase change (Chuck et al 2009 de Alba et al 2013) In
support of this view it has been shown that miR172 is down-regulated in the
Chapter 1 Introduction and Review of Literature
43
Corngrass1(Cg1) mutant in which miR156 is overproduced (Chuck et al 2007 de
Alba et al 2013) However there is no direct evidence for the direct linkage between
miR156 and miR172 It is also envisioned that miR156 and miR172 would not be
directly related For example miR156 regulates plastochron index that is the inverse of
leaf initiation rate and thus frequently used to analyze temporal patterns of plant
development In contrast miR172 does not affect plastochron (Wang et al 2008) The
down-regulation of miR172 in the maize miR156-over producing mutants would be
due to the prolonged juvenile vegetative phase in the mutants
Another miRNA that regulates flowering initiation is miR159 It regulates
MYB33 and MYB65 which are closely related to the barley GAMY B transcription
factor that responds to gibberellic acid (GA) The level of miR159 is elevated in GA-
treated seedlings but reduced in the GA biosynthetic ga1 mutant Transgenic plants
over producing miR159 exhibit delayed floral induction under short days (Achard et
al 2004) It is been suggested that MYB33 mediates GA signal to regulate LEAFY
(LFY) These observations indicate that miR159 plays an important role in the GA
signaling pathways that regulate proper floral induction under short days (Achard et al
2004)
11362 Floral development
Arabidopsis flowers consist of four distinct organs They arise in a characteristic
pattern within concentric rings called whorls from outside to inside four sepals four
petals six stamens and the central pistil consisting of two carpels Identities of the
floral organs are determined by three classes of regulatory genes classA classB and
class C genes (Krizek amp Fletcher 2005) The A and C class genes act antagonistically
to restrict their expression domains (Bowman et al 1991)
It is notable that miR172 also regulates floral architecture in addition to its role
in the timing of flowering initiation MiR172 inhibits the translation of the AP2 mRNA
(Chen 2004) Transgenic plants overproducing miR172 not only exhibit early
flowering but also show abnormal floral development which is quite similar to those
of the class A gene mutants such as the ap2 mutants (Aukerman amp Sakai 2003 de
Alba et al 2013) When the activity of the class A genes is inactivated that of the class
C genes such as AGAMOUS (AG) is elevated As a result the miR172- overproducing
transgenic plants exhibit no petals along with homeotic transformation of the sepals to
Chapter 1 Introduction and Review of Literature
44
the carpels (Aukerman amp Sakai 2003 Chen 2004)
Interestingly miR166 also regulates floral architecture The role of miR165166
in the regulation of meristem activity is closely linked to floral meristem formation and
organization (Zhao et al 2007 Miyashima et al 2013) Floral structure is severely
disrupted in the miR165166-overproducing mutants such as men1 and jba-1D in
which miR166 is overproduced (Williams et al 2005b Jung amp Park 2007) The men1
and jba-1D mutants have smaller gynoecium and reduced carpel number In an extreme
case the jba-1D homozygous plants lack visible carpels (Williams et al 2005b)
Similar phenotypes have been observed in the miR165 overproducers (Zhao et al
2007) It has been suggested that the phenotypes are possibly caused by altered AG
expression in the floral meristem (Jung amp Park 2007 Turchi et al 2013)
Other miRNAs also affect floral development Overproduction of miRNAs
involved in auxin signaling also affects floral architecture Transgenic plants
overproducing miR167 have defects in the formation of ovule and anther with reduced
fertility (Ru et al 2006) It has been shown that miR167 targets ARF6 and ARF8 that
play a role in gynoecium and stamen development and in regulating fertility (Ru et al
2006 Wu et al 2006) These observations suggest that auxin signals are also closely
related to floral organogenesis In addition transgenic plants over-producing miR164
and the cuc1cuc2 double mutants show fused sepals and reduced petals (Laufs et al
2004 Turchi et al 2013) suggesting that miR164 is also related to floral meristem
activity and boundary specification of the meristematic region
11363 Shoot apical meristem development
Shoot apical meristem comprises self-maintaining totipotent cells The shoot apical
meristem activity affects diverse aspects of plant developmental processes including
leaf development vascular development and lateral organ polarity (Bowman et al
2002) Because miR165166 plays a primary role in meristem formation
overproduction of miR165166 and multiple knockout mutants of its target genes
exhibit pleiotropic pheonotypes (McConnell et al 2001 Emery et al 2003 Kim et al
2005 Williams et al 2005b)
The targets of miR165166 include genes encoding the homeo domain-leucine
zipper (HD-ZIP) class III transcription factors such as PHABULOSA(PHB)
PHAVOLUTA (PHV) REVOLUTA(REV) ATHB15CORONA (CNA) and ATHB8
Chapter 1 Introduction and Review of Literature
45
PHB PHV and ATHB15 have overlapping functions in controlling meristem
development (Turchi et al 2013) Indeed the phb phv athb15 triple mutants have an
enlarged shoot apical meristem Consistent with this the miR166- overproducing men1
and jba-1D mutants exhibit enlarged shoot apical meristems (Kim et al 2005
Williams et al 2005b Turchi et al 2013) In the mutants the shoot apical meristems
are multiplied and large
The development of shoot apical meristems is also regulated by the WUSCHEL
(WUS)ndashCLAVATA (CLV) signaling pathway WUS positively regulates cell
proliferation In contrast CLV3 which is expressed in stem cells limits the WUS
expression and thus inhibits cell proliferation in the SAM (Sharma et al 2003)
Consistent with this notion WUS is expressed to a higher level in jba-1D
explaining the ectopic enlargement of the SAM in the mutant (Williams et al 2005)
While WUS is closely related to miR165166 in the shoot apical meristem
development the underlying molecular mechanisms are currently unclear It has been
observed that the miR166-over producing men1 plants have an arrested meristem
development (Jung amp Park 2007) In addition the heterozygous men1+ and the
homozygous men1 plants show different expression patterns of WUS and CLV3 It
appears that miR166 regulates WUS indirectly and influences the expressional balance
between WUS and CLV3 through a negative feedback loop (Jung amp Park 2007 de
Alba et al 2013)
The phv phb athb15 triple mutant has an enlarged shoot apical meristems the
rev phb phv mutant does not have functional shoot apical meristems (Prigge et al
2005) This distinction may be explained by the fact that while miR166 targets
primarily PHB PHV and ATHB15 miR165 targets all the five HD-ZIP III genes (Zhou
et al 2007) Consistent with this transgenic plants overproducing miR166 are distinct
from those overproducing miR165 The miR165-overproducing transgenic plants lack
functional shoot apical meristem and a pin-like structure is formed at the apical region
of the mutant These phenotypes are quite similar to rev phb phv triple mutant (Zhou et
al 2007 Liu et al 2013) The phenotypic distinction may because by the difference
of the cleavage efficiencies of the target mRNAs In accordance with this notion a few
35S miR166a transgenic lines exhibit no shoot apical meristems The men1
homozygous plants also show arrested meristem development (Jung amp Park 2007)
Chapter 1 Introduction and Review of Literature
46
MiR164 cleaves the CUC1 and CUC2 mRNAs as well as the NAC1 mRNA
CUC1 and CUC2 determines the organ boundary in the meristem Phenotypes of
transgenic plants that overproduce miR164 are similar to those of the cuc1cuc2 double
mutant that exhibits embryonic developmental defects such as cup-shaped cotyledons
and fused cotyledons (Laufs et al 2004) When a miR164-resistant CUC2 is
expressed the boundary domain is enlarged CUC also plays a role in embryonic shoot
meristem formation by regulating the SHOOT MERISTEMLESS gene It was therefore
concluded that miR164-mediated cleavage of CUC1 and CUC2 mRNAs determines the
sizes of the boundary domains in the shoot apical meristem and regulates separation of
the organs by restricting cell proliferation (Laufs et al 2004 de Alba et al 2013)
11364 Leaf development
The role of miR319 has been extensively studied in leaf development A dominant
jaw-D mutant overproducing miR319 shows uneven curved leaves with enlarged size
and five TCP genes which are closely related to the CINCINNATA (CIN) gene in
Antirrhinum majus are down regulated in the mutant suggesting that miR319 mediated
regulation of the TCP transcription factor genes are important for the final step of
leaf shaping (Palatnik et al 2003 Naz et al 2013) In addition to leaf shape and size
leaf polarity is also specified by miRNA MiR166-mediated cleavage of the HD-ZIPIII
mRNAs is a well to establish leaf polarity This discovery originates from the gain-of-
function mutants such as phb-1d and phv-1d wherein point mutations in the
complementary sites to miRNA helps the mRNA to evade or resist from miRNA-
mediated cleavage (McConnell et al 2001 Emery et al 2003 Naz et al 2013)
Plant leaves have a dorso-ventral polarity consisting of the adaxial (upper
surface) and abaxial (lower surface) sides The adaxial side is closer to the shoot
apical meristem Adaxial identity is specified by functionally redundant class III HD-
ZIP family genes such as PHB PHV and REV Ectopic expression of each genes
result in adaxialized leaves Dominant phb-1d and phv-1d mutants exhibit
transformation of the abaxial to adaxial fate and have rod-shaped leaves In contrast
miR165166-overproducing plants show pin-like cotyledons radicalized leaves and
abaxialized leaves (Chitwood et al 2007 Pattanayak et al 2013)
Leaf asymmetry is determined by miR166-mediated restriction of the
expression domains of the HD-ZIPIII genes The HD-ZIPIII genes are expressed in the
Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora
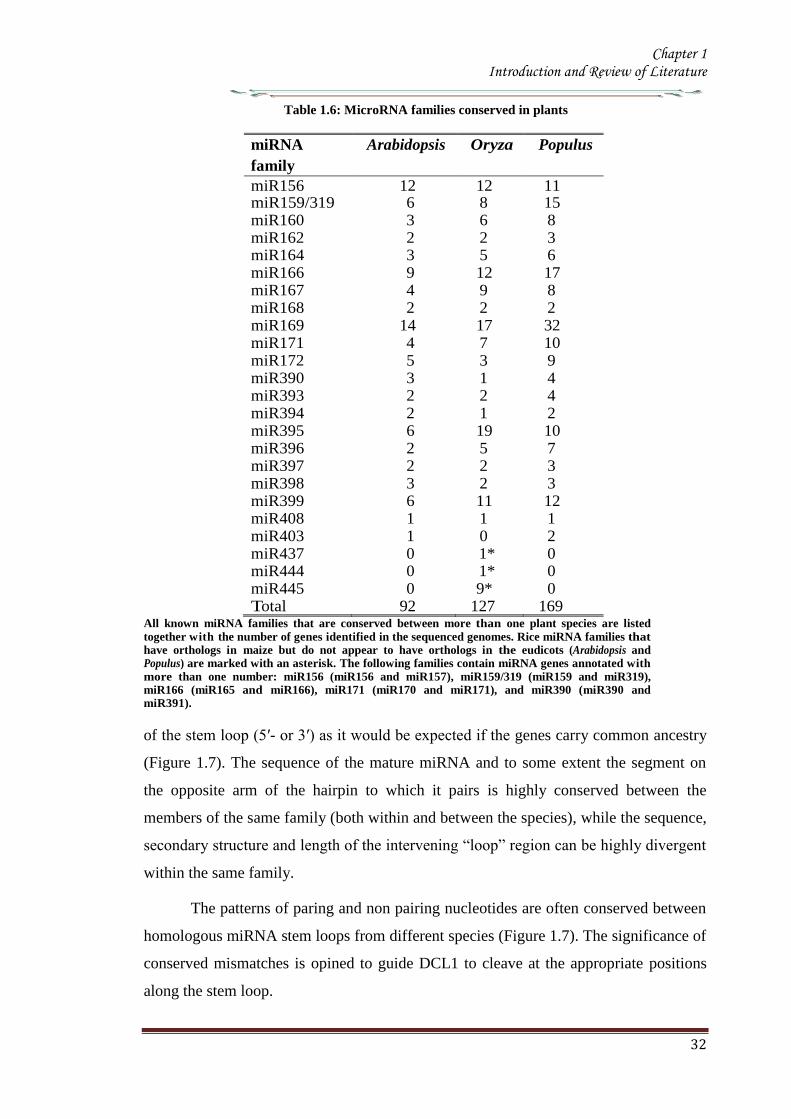
Chapter 1 Introduction and Review of Literature
32
Table 16 MicroRNA families conserved in plants
miRNA
family
Arabidopsis Oryza Populus
miR156 12 12 11 miR159319 6 8 15 miR160 3 6 8 miR162 2 2 3 miR164 3 5 6 miR166 9 12 17 miR167 4 9 8 miR168 2 2 2 miR169 14 17 32 miR171 4 7 10 miR172 5 3 9 miR390 3 1 4 miR393 2 2 4 miR394 2 1 2 miR395 6 19 10 miR396 2 5 7 miR397 2 2 3 miR398 3 2 3 miR399 6 11 12 miR408 1 1 1 miR403 1 0 2 miR437 0 1 0 miR444 0 1 0 miR445 0 9 0 Total 92 127 169
All known miRNA families that are conserved between more than one plant species are listed
together with the number of genes identified in the sequenced genomes Rice miRNA families that have orthologs in maize but do not appear to have orthologs in the eudicots (Arabidopsis and Populus) are marked with an asterisk The following families contain miRNA genes annotated with
more than one number miR156 (miR156 and miR157) miR159319 (miR159 and miR319) miR166 (miR165 and miR166) miR171 (miR170 and miR171) and miR390 (miR390 and miR391)
of the stem loop (5prime- or 3prime) as it would be expected if the genes carry common ancestry
(Figure 17) The sequence of the mature miRNA and to some extent the segment on
the opposite arm of the hairpin to which it pairs is highly conserved between the
members of the same family (both within and between the species) while the sequence
secondary structure and length of the intervening ldquolooprdquo region can be highly divergent
within the same family
The patterns of paring and non pairing nucleotides are often conserved between
homologous miRNA stem loops from different species (Figure 17) The significance of
conserved mismatches is opined to guide DCL1 to cleave at the appropriate positions
along the stem loop
Chapter 1 Introduction and Review of Literature
33
Figure 17 Representative stem loop of miR164 Segments corresponding to the mature miRNAs
are shown in red (Adapted from Rajagopalan et al 2006)
Although many miRNAs are highly conserved in plants the degree of conservation in
most cases is quite low between plants and animals one exception is miR854 A recent
study has revealed that miR854 is conserved between Arabidopsis and animals and that
the Arabidopsis miR854 differs from the human miR854 only by a single nucleotide
(Arteaga-Vazquez et al 2006 Axtell 2012 de Alba et al 2013) The Arabidopsis
miR854 has multiple binding sites in the 3prime-untranslated region (UTR) of
OLIGOURIDYLATE-binding-PROTEIN mRNA 1bA (UBP1b) and represses the
translation of the UBP1b mRNA The animal miR854 is also complementary to a site in
the 3prime-UTR of the UBP1b homolog in animals However it is currently unclear how the
animal miR854 regulates its target mRNA Another distinction between plant and
animal miRNAs is the genomic organization of the miRNA genes While plant miRNA
genes are located predominantly in the intergenic regions animal miRNA genes tend to
localize in the intragenic regions both in the introns or exons of protein coding genes
Moreover miRNA genes are often clustered within a single precursor transcript
whereas individual plant miRNAs are derived from individual precursor RNAs (Bartel
Chapter 1 Introduction and Review of Literature
34
2004 Bonnet et al 2004 Kim 2005 Vaucheret et al 2006 Marco et al 2013)
1133 Non-conserved miRNA and challenges in annotation
Most miRNAs are conserved throughout flowering plants (Table 15) but many more
were found only in a single sequenced genome thus considered to be of a more recent
evolutionary origin The extended homology between few non conserved miRNAs
precursors and their target genes provides strong evidence that these relatively ldquoYoungrdquo
miRNAs arose from the duplication of the target gene segments (Allen et al 2004)
Several non-conserved miRNAs including miR161 miR163 miR173 miR447
miR475 and miR476 are known to direct cleavage of target transcripts (Allen et al
2004a Adai et al 2005 Sunkar et al 2005 de Alba et al 2013) It is difficult to
confidently predict targets for many because it is not possible to use complementary
site as a filter against false-positive target predictions It is also difficult to
confidently annotate non-conserved miRNAs as miRNA rather than siRNAs The
established minimal standard for miRNA annotation is a small RNA with detectable
expression and the potential to form a stem-loop when joined to flanking genomic
sequence (Kasschau et al 2003) In practice these requirements are too loose to
definitively categorize many small RNAs cloned from plants Many plant siRNAs are
detectable on blots (Vazquez et al 2004b) and hundreds of thousands of non-
miRNA genomic sequences can be predicted to fold into secondary structures that
resemble structures of plant miRNA precursors (Jones-Rhoades amp Bartel 2004)
Therefore without conservation of both sequence and secondary structure it is
difficult to be confident that a given cloned RNA originated from a stem-loop (ie is a
miRNA) rather than from a double-stranded RNA (ie is a siRNA) In fact many of
the thousands o f small RNAs cloned from Arabidopsis (Gustafson et al 2005) would
probably meet the literal requirements for annotation as miRNAs A few of these
sequences might be miRNAs but others that meet the literal criteria probably are not
so The challenges of annotating non conserved small RNAs were evidenced by
three related small silencing RNAs that were originally annotated as miRNAs
that turned out to be trans-acting siRNAs (tasiRNAs)(Kanno et al2013)
Although most miRNAs that are broadly conserved among flowering plants are
now being identified and experimentally validated (Table 16)(Jones-Rhoades amp
Bartel 2004) the challenges and ambiguities for classifying non-conserved miRNAs
prevent meaningful estimates of the total number of miRNA genes in Arabidopsis and
Chapter 1 Introduction and Review of Literature
35
other plant genomes It is possible that miRNAs might have escaped detection
because they are non-conserved and expressed in specific tissues or conditions and can
only be identified by using high- throughput sequencing
1134 MicroRNA biogenesis
11341 Transcription of microRNA precursor
Plant miRNAs are primarily found in the genomic regions that are not associated with
the protein coding genes (Reinhart et al 2002) and it appears that most if not all plant
miRNAs are produced from their own transcriptional units Plant miRNA genes are
occasionally clustered near each other in the genome suggesting transcription of
multiple miRNAs from a single primary transcript [eg the miR395 cluster (Grishok
et al 2001 Jones-Rhoades amp Bartel 2004b)] but this polycistronic arrangement of
miRNA genes appears far less frequently in plants than in animals (Bartel 2004)
Northern blot EST and mapping evidence indicate that plant miRNA primary
transcripts (also known as pri-miRNAs) as in animals are longer than needed to
encompass the miRNA stem-loops (Palatnik et al 2003 Jones-Rhoades amp Bartel
2004b) At least some of these pri-miRNA transcripts appear to be spliced
polyadenylated (Kurihara amp Watanabe 2004) and capped (Xie Z et al 2005) Two
rice miRNAs are contained within transcripts that contain exon junctions within the
presumptive stem-loop precursor implying that in these cases splicing is a prerequisite
for Dicer recognition (Sunkar et al 2005) Plant pri-miRNAs are over 1kb in length
and they are usually preceded by typical TATA box motifs They can also undergo
canonical splicing polyadenylation and capping All these observations indicates RNA
polymerase II is probably responsible for transcribing most plant miRNAs (Xie Z et
al 2005) as appears to be the case for many animal miRNAs Relatively little is
known about the regulation of miRNA transcription in plants but there is no reason
to suspect that this regulation would differ from that of protein-coding transcripts as
the bioinformatics analysis of the sequence upstream of the transcriptional start
sites of the miRNA genes showed the presence of putative binding motifs of
number of known transcription factors (Megraw et al 2006 Sethupathy 2013)
11342 MicroRNA processing and export
In plants maturation of miRNA is a step wise process A miRNA gene is transcribed to
pri-miRNA which is much longer than the pre-miRNA possessing the characteristic
stem-loop structure Like transcription of most protein-coding genes the miRNA
Chapter 1 Introduction and Review of Literature
36
transcription is processed by RNA polymerase II (Pol II) enzymes and the pri-
miRNAs are 5prime-capped and 3prime-polyadenylated (Lee et al 2004 Xie Z et al 2005)
Mature miRNAs are produced from pri-miRNAs through at least two sequential
processing steps by RNase III-type endonucleases In animals pri-miRNAs are first
processed at the bottom portion of the stem by Drosha a nuclear-localized RNase III
enzyme to liberate pre-miRNAs The pre-miRNAs are then exported to the cytoplasm
with the help of exportin-5The pre-miRNAs are further processed by Dicer another
RNaseIII enzyme to a duplex consisting of the miRNA and the antisense strands
(miRNA) Since plants do not have any Drosha homologs the two sequential
processing steps seem to be carried out by DCL1 a Dicer homolog in the nucleus
(Kurihara amp Watanabe 2004 de Alba et al 2013 Rogers amp Chen 2013)
The processing of pri-miRNAs to pre-miRNAs by DCL1 is assisted by two
other proteins HYPONASTY LEAVES1 (HYL1) and SERRATE (SE) HYL1 belongs to a
family of double-stranded RNA (dsRNA) binding protein and interacts with DCL1 in
vitro and in vivo (Lu amp Fedoroff 2000 Hiraguri et al 2005 de Alba et al 2013
Rogers amp Chen 2013) Loss-of-function mutations in the HYL1 gene result in
decreased accumulation of miRNAs and concomitant accumulation of pri-miRNAs
(Han et al 2004 Vazquez et al 2004a Kurihara et al 2006) SE a C2H2 zinc finger
protein interacts with DCL1 and HYL1 and plays a role in the processing of pri-
miRNAs to pre-miRNAs (Lobbes et al 2006 Yang L et al 2006) In addition the
nuclear cap-binding complex proteins CBP20 and CBP80 that are known as ABA
HYPER SENSITIVE 1(ABH1) have been shown to be required for proper miRNA
processing (Gregory et al 2008 Laubinger et al 2008) Recently it has been
demonstrated that DCL1 can release 21-nt RNAs from dsRNA as well as from
synthetic miR167 precursor RNAs in vitro However this cleavage is error prone and
inefficient in the absence of HYL1 and SE which accelerate the rate of DCL1-mediated
cleavage and promote accurate processing of miRNAs (Dong et al 2008 Rogers amp
Chen 2013)
11343 Methylation of the miRNAmiRNAduplex
After the miRNAmiRNAduplexes are released from the pre-miRNAs by the DCL1
activities the duplexes are methylated at the 2primeOH of the3prime-ends by HUA ENHANCER1
(HEN1) a small RNA methyltransferase (Yu et al 2005 Yang Z et al 2006 Rogers
amp Chen 2013 ) This step which is distinct from the RNase III-mediated processing
Chapter 1 Introduction and Review of Literature
37
step distinguishes the biogenesis of plant miRNAs from that of animal miRNAs
Mutations in the HEN1 gene were first isolated in a morphological screening for floral
development although the HEN1 mutants display pleiotropic phenotypes similar to the
developmental defects observed in the DCL1 mutants (Chen et al 2002 Park et al
2002 Rogers amp Chen 2013)
Figure 18 Biogenesis of microRNA
A Biogenesis of plant MicroRNA B Biogenesis of metazoananimal MicroRNA
The miRNA (red) is incorporated into RISC and the miRNA(blue) in degraded
This enzyme has two dsRNA-binding domains and nuclear localization signal It
is also conserved in fungi and is required for the processing of siRNAs as well as of
miRNAs (Park et al 2002 Boutet et al 2003 Xie et al 2003) Although the HEN1
protein is localized to the nucleus its methylation activity in the cytoplasm cannot be
ignored (Xie et al 2004 Rogers amp Chen 2013)
Chapter 1 Introduction and Review of Literature
38
The methylation of the miRNAmiRNA duplex is required to protect miRNAs
against the 3prime-end uridylation activity and subsequent degradation (Li et al 2005) In
the hen1 mutants accumulation of miRNAs is reduced to a lower level Also the
miRNAs appear to be heterogeneous in their sizes such that a ladder of bands rather
than a single species is observed for most miRNAs in the mutants (Li et al 2005)
MiRNA cloning and sequencing analyses revealed the presence of a number of U
residues at the 3prime-end of miRNAs in the hen1 mutants Moreover these nucleotides do
not correspond to the miRNAs in the pre-miRNAs indicating that these nucleotides are
added after the processing of the miRNAs from pre-miRNAs (Li et al 2005)
Uridylation of RNAs has also been proven to be correlated with RNA decay (Shen amp
Goodman 2004 Mullen amp Marzluff 2008) suggesting that uridylation attracts an
exonuclease which degrades miRNAs in plants
11344 MiRNA incorporation in to the RISC and its nuclear export
The methylated miRNAmiRNA duplexes undergo RISC assembly In this process
the miRNA of the duplexes is selectively incorporated into the RISC and the miRNA
appears to be removed and subsequently degraded (Hammond et al 2000 Hutvagner
amp Zamore 2002 Schwarz et al 2003 de Alba et al 2013 Rogers amp Chen 2013)
The ARGONAUTE 1(AGO1) proteins are core components of the RISC complex
They contain two structural domains the N-terminal PAZ domain that binds to the 3prime-
end of single-stranded RNAs and the C-terminal piwi domains that has a structure
similar to that of RNase H (Cerutti et al 2000 Parker et al 2004) Several AGO
proteins possess endonucleolytic activities within the PIWI domain and cleave target
mRNAs in the middle of the site complementary to miRNA (Liu et al 2004 Meister et
al 2004 Miyoshi et al 2005) Mutations in the AGO1 gene cause miRNA target
genes to be ectopically expressed and the levels of some miRNAs are reduced in the
mutants (Kidner amp Martienssen 2004 Vaucheret et al 2004 Kidner amp Martienssen
2005 Ronemus et al 2006) Moreover the phenotype of the AGO1 hypomorphic
alleles display defects in lateral organ polarity and leaf and flower morphology as
observed in the dcl1 mutants and null AGO1 alleles are embryonically lethal
supporting the close relationship between the AGO proteins and miRNA processing
(Kidner amp Martienssen 2004 Vaucheret et al 2004 de Alba et al 2013 Rogers amp
Chen 2013)
The production of miRNA miRNA duplexes occur in the nucleus while
Chapter 1 Introduction and Review of Literature
39
miRNA-RISC complexes are present in the cytoplasm where target mRNAs are
located This discordance of functional sites requires an export step during miRNA
biogenesis HASTY the Arabidopsis homolog of exportin-5 is thought to be involved
in miRNA nuclear export (Bollman et al 2003 Park et al 2005)The hasty mutant
exhibits pleiotropic defects in plant development and reduced accumulation of most
miRNAs as in the DCL1 or AGO1 mutants Howeverit is unclear whether the HASTY
proteins export the miRNA miRNA duplexes or the miRNA-RISC complexes in
plants
11345 Feedback regulation of miRNA biogenesis
MiRNA biogenesis is under feedback regulation such that the DCL1 and AGO1 genes
are themselves regulated by miRNAs DCL1 is itself a target of miR162 which leads to
the cleavage of the DCL1 mRNA (Xie et al 2003 de Alba et al 2013) Consistent
with this the levels of DCL1 mRNA are elevated in the mutants of miRNA processing
genes in which the miR162 abundance is reduced In addition there is a predicted
hairpin structure of miR838 in the 14th
intron of the DCL1 primary transcript
(Rajagopalan et al 2006) This prediction implies that DCL1-mediated processing
on this site can result in the cleavage of the DCL1 primary transcript into two truncated
fragments the N-terminal capped fragment and the C-terminal polyadenylated
fragment If this is the case it is possible that miRNA biogenesis competes with the
splicing event of the DCL1 pre-mRNA
The AGO1 is also regulated by miRNA There is a complementary site for
miR168 binding in the AGO1 mRNA and the transcript level of the AGO1 mRNA is
reduced through an mRNA cleavage mechanism (Vaucheret et al 2006 de Alba et al
2013 Rogers amp Chen 2013) indicating that the AGO1 mRNA levels are tightly
controlled by the levels of the AGO1 protein
1135 Mechanism of miRNA action
11351 MiRNA-directed mRNA cleavage
By forming the miRNA-RISC assembly miRNAs can direct the RISC to regulate gene
expression by two post transcriptional mechanisms mRNA cleavage or translational
repression (Bartel 2004 Jones-Rhoades et al 2006 Dalmay 2013) The degree
of miRNA-mRNA complementarity plays an important role for the
determination of the mechanism to be used A general rule is that while perfect or
Chapter 1 Introduction and Review of Literature
40
near-perfect complementarity induces mRNA cleavage central mismatches trigger
translational repression (Carrington amp Ambros 2003 Jones-Rhoades amp Bartel 2004)
Unlike most animal miRNAs plant miRNAs have near-perfect or perfect
complementarity to their targets and mRNA cleavage is assumed to be a predominant
mechanism by which miRNAs regulate gene expression in plants
In RNA cleavage mechanism miRNAs guide the AGO component of RISC to
cleave a single phosphodiester bond of the target mRNA within the miRNA-binding
site (Llave et al 2002b Tang et al 2003) The truncated fragments are then released
and degraded freeing the RISC to recognize and cleave another target mRNA This has
been validated by the detection of 3prime cleavage products that have 5prime ends that start at the
middle of the complementary regions The mRNA cleavage products are then further
degraded by other mechanisms The 3prime cleavage products of some miRNA targets are
degraded by the 5prime-3prime exonuclease XRN4 an Arabidopsis homolog of the major Yeast
mRNA degrading exonuclease Xrn1p It has been observed that the 3prime cleavage
products of some target mRNAs were accumulated in the xrn4 mutants (Souret et al
2004 Dalmay 2013) However the 3prime cleavage products of other miRNA targets do
not accumulate in the xrn4 mutants indicating that there are also XRN4 independent
mechanisms The 5prime cleavage products are typically untraceable by RNA gel blot
hybridization but can be detected by sensitive PCR based methods It has been found
that the 5prime cleavage products tend to obtain an oligo U tail at the 3prime ends This 3prime U tail
is correlated with shortening of the 5prime cleavage products from the 5prime end leading to the
conclusion that the 3prime uridylation causes 5 prime- 3 prime exonucleolytic decay of the 5prime cleavage
products (Shen amp Goodman 2004)
DCL1 HYL1 and SE interact with each other and are co-localized in the
nuclear bodies termed Dicing bodies or D-bodies in which some pri-miRNA shave
been found suggesting that D-bodies serve as a center for active miRNA processing
(Fang amp Spector 2007 Fujioka et al 2007 Song et al 2007) The miRNA
processing site appears to be distinct from the Cajal bodies that have previously been
found as a siRNA processing site (Pontes amp Pikaard 2008) The D-bodies include
SmD3 and SmB proteins that are found in the Cajal bodies However At collin an
additional marker for the Cajal bodies is not found in the D-bodies Moreover the D-
bodies are present in an At collin mutant lacking the Cajal bodies The pri-miRNAs of
miR171A and miR159A genes have been found to be co-localized with HYL1 and
Chapter 1 Introduction and Review of Literature
41
DCL1 in the D- bodies that are distinct from the Cajal bodies (Song et al 2007)
11352 MiRNA-directed translational repression
Translation repression is another mechanism exerted by plant miRNAs The regulatory
mechanism of miR172 which regulates flowering time and floral organ identity is
consistent with translation repression MiR172 over production does not affect the
transcript levels of APETALA2 (AP2) and AP2- like target mRNAs Instead the AP2
protein level is reduced in the miR172 over expressing mutant (Aukerman amp Sakai
2003 Chen 2004 Dalmay 2013) It was later shown that miR172 can also direct the
cleavage of the target mRNAs although the steady-state mRNA level is unchanged due
to feedback regulation (Schwab et al 2005 Jung et al 2007) It is clear that miR172
regulates its target genes primarily by translational repression rather than mRNA
cleavage Recently it has been reported that miR156157 and miR854 also regulate
their target genes through translational repression (Arteaga-Vazquez et al 2006
Gandikota et al 2007 Dalmay 2013) It was once thought that translational repression
is an exception to the rule However experimental evidence suggest a widespread
coexistence of translational repression and mRNA cleavage (Brodersen et al 2008)
1136 Regulatory roles of miRNAs in plant development
The miRNA target prediction has been a difficult task in order to obtain regulatory
targets without bringing in too many false predictions Plant growth and developmental
processes regulated by miRNAs are quite diverse and enormous Furthermore plant
miRNAs work cooperatively or antagonistically to establish a balanced regulation This
notion is well consistent with the findings that mutations in the regulators of miRNA
biogenesis transport and processing such as AGO1 DCL1 HYL1 HEN1 ABH1
and HASTY cause pleiotropic phenotypes (Mallory amp Vaucheret 2006 de Alba et al
2013) To date the roles of miRNAs have been mostly deduced from obtaining miRNA
over expressing transgenic plants or gain-of-function mutants in which miRNA-
resistant target genes are ectopically expressed
11361 Phase transition
Plants pass through a series of developmental phase transitions during their life span
beginning with seed germination continuing through vegetative phase change
reproductive phase change flowering initiation and finally seed production for the next
generation Because of their importance in understanding plant developmental
Chapter 1 Introduction and Review of Literature
42
processes genes regulating developmental transitions have been extensively studied
and underlying molecular mechanisms have been explored in various plant species
Majority of researches were mainly focused on vegetative phase change and
reproductive transition in Arabidopsis and Maize (Poethig 2003 Baurle amp Dean
2006) Particularly the mutations of the genes encoding various regulators of miRNA
biogenesis and transport such as HST SE and AGO exhibit altered juvenile to adult
vegetative phase change and vegetative to reproductive change (Hunter et al 2003
Peragine et al 2004 Vazquez et al 2004a Jones-Rhoades et al 2006 de Alba et al
2013)
Over-expression of the miR156a gene causes late flowering and delays
vegetative phase change (Wu amp Poethig 2006 Gandikota et al 2007 Wang et al
2008) It has been shown that miR156 negatively regulates a group of genes encoding
SQUAMOSA PROMOTER-BINDING PROTEIN-LIKE (Lewis et al 2007 de Alba et
al 2013 Rogers amp Chen 2013) transcription factors such as SPL3 SPL4 and SPL5
that promote flowering initiation In contrast transgenic plants over expressing
miR156-resistant SPL345 genes are early flowering Interestingly the endogenous
level of miR156 is temporally regulated In the early juvenile vegetative phase the
level of miR156 is very high but decreases rapidly before the onset of the adult juvenile
phase (Wu amp Poethig 2006)
The Arabidopsis early activation tagged (eat-D) mutant exhibits early flowering
with disrupted floral structure The mutant phenotypes are caused by over expression of
the miR172a-2gene (Aukerman amp Sakai 2003) MiR172 regulates a small group of
genes encoding APETALLA2 (AP2)-like transcription factors such as TARGET OF
EAT1 (TOE1) TOE2 SCHLAFMUTZE (SMZ) and SCHNARCHZAPFEN(SNZ)
TOE1 TOE2 SMZ and SNZ have been shown to participate in their productive phase
change by repressing a floral integrator FLOWERING LOCUS T (FT)(Jung et al
2007) Consistent with this toe1-2D 35STOE2 35SSMZ and 35SSNZ plants
exhibit late flowering (Jung et al 2007) MiR172 is also regulated temporally and
highly expressed in the adult vegetative phase both in Arabidopsis and maize (Chuck et
al 2007) Because the temporal expression patterns of miR156 and miR172 are
complementary to each other it has been suggested that the two miRNAs interact with
each other in regulating phase change (Chuck et al 2009 de Alba et al 2013) In
support of this view it has been shown that miR172 is down-regulated in the
Chapter 1 Introduction and Review of Literature
43
Corngrass1(Cg1) mutant in which miR156 is overproduced (Chuck et al 2007 de
Alba et al 2013) However there is no direct evidence for the direct linkage between
miR156 and miR172 It is also envisioned that miR156 and miR172 would not be
directly related For example miR156 regulates plastochron index that is the inverse of
leaf initiation rate and thus frequently used to analyze temporal patterns of plant
development In contrast miR172 does not affect plastochron (Wang et al 2008) The
down-regulation of miR172 in the maize miR156-over producing mutants would be
due to the prolonged juvenile vegetative phase in the mutants
Another miRNA that regulates flowering initiation is miR159 It regulates
MYB33 and MYB65 which are closely related to the barley GAMY B transcription
factor that responds to gibberellic acid (GA) The level of miR159 is elevated in GA-
treated seedlings but reduced in the GA biosynthetic ga1 mutant Transgenic plants
over producing miR159 exhibit delayed floral induction under short days (Achard et
al 2004) It is been suggested that MYB33 mediates GA signal to regulate LEAFY
(LFY) These observations indicate that miR159 plays an important role in the GA
signaling pathways that regulate proper floral induction under short days (Achard et al
2004)
11362 Floral development
Arabidopsis flowers consist of four distinct organs They arise in a characteristic
pattern within concentric rings called whorls from outside to inside four sepals four
petals six stamens and the central pistil consisting of two carpels Identities of the
floral organs are determined by three classes of regulatory genes classA classB and
class C genes (Krizek amp Fletcher 2005) The A and C class genes act antagonistically
to restrict their expression domains (Bowman et al 1991)
It is notable that miR172 also regulates floral architecture in addition to its role
in the timing of flowering initiation MiR172 inhibits the translation of the AP2 mRNA
(Chen 2004) Transgenic plants overproducing miR172 not only exhibit early
flowering but also show abnormal floral development which is quite similar to those
of the class A gene mutants such as the ap2 mutants (Aukerman amp Sakai 2003 de
Alba et al 2013) When the activity of the class A genes is inactivated that of the class
C genes such as AGAMOUS (AG) is elevated As a result the miR172- overproducing
transgenic plants exhibit no petals along with homeotic transformation of the sepals to
Chapter 1 Introduction and Review of Literature
44
the carpels (Aukerman amp Sakai 2003 Chen 2004)
Interestingly miR166 also regulates floral architecture The role of miR165166
in the regulation of meristem activity is closely linked to floral meristem formation and
organization (Zhao et al 2007 Miyashima et al 2013) Floral structure is severely
disrupted in the miR165166-overproducing mutants such as men1 and jba-1D in
which miR166 is overproduced (Williams et al 2005b Jung amp Park 2007) The men1
and jba-1D mutants have smaller gynoecium and reduced carpel number In an extreme
case the jba-1D homozygous plants lack visible carpels (Williams et al 2005b)
Similar phenotypes have been observed in the miR165 overproducers (Zhao et al
2007) It has been suggested that the phenotypes are possibly caused by altered AG
expression in the floral meristem (Jung amp Park 2007 Turchi et al 2013)
Other miRNAs also affect floral development Overproduction of miRNAs
involved in auxin signaling also affects floral architecture Transgenic plants
overproducing miR167 have defects in the formation of ovule and anther with reduced
fertility (Ru et al 2006) It has been shown that miR167 targets ARF6 and ARF8 that
play a role in gynoecium and stamen development and in regulating fertility (Ru et al
2006 Wu et al 2006) These observations suggest that auxin signals are also closely
related to floral organogenesis In addition transgenic plants over-producing miR164
and the cuc1cuc2 double mutants show fused sepals and reduced petals (Laufs et al
2004 Turchi et al 2013) suggesting that miR164 is also related to floral meristem
activity and boundary specification of the meristematic region
11363 Shoot apical meristem development
Shoot apical meristem comprises self-maintaining totipotent cells The shoot apical
meristem activity affects diverse aspects of plant developmental processes including
leaf development vascular development and lateral organ polarity (Bowman et al
2002) Because miR165166 plays a primary role in meristem formation
overproduction of miR165166 and multiple knockout mutants of its target genes
exhibit pleiotropic pheonotypes (McConnell et al 2001 Emery et al 2003 Kim et al
2005 Williams et al 2005b)
The targets of miR165166 include genes encoding the homeo domain-leucine
zipper (HD-ZIP) class III transcription factors such as PHABULOSA(PHB)
PHAVOLUTA (PHV) REVOLUTA(REV) ATHB15CORONA (CNA) and ATHB8
Chapter 1 Introduction and Review of Literature
45
PHB PHV and ATHB15 have overlapping functions in controlling meristem
development (Turchi et al 2013) Indeed the phb phv athb15 triple mutants have an
enlarged shoot apical meristem Consistent with this the miR166- overproducing men1
and jba-1D mutants exhibit enlarged shoot apical meristems (Kim et al 2005
Williams et al 2005b Turchi et al 2013) In the mutants the shoot apical meristems
are multiplied and large
The development of shoot apical meristems is also regulated by the WUSCHEL
(WUS)ndashCLAVATA (CLV) signaling pathway WUS positively regulates cell
proliferation In contrast CLV3 which is expressed in stem cells limits the WUS
expression and thus inhibits cell proliferation in the SAM (Sharma et al 2003)
Consistent with this notion WUS is expressed to a higher level in jba-1D
explaining the ectopic enlargement of the SAM in the mutant (Williams et al 2005)
While WUS is closely related to miR165166 in the shoot apical meristem
development the underlying molecular mechanisms are currently unclear It has been
observed that the miR166-over producing men1 plants have an arrested meristem
development (Jung amp Park 2007) In addition the heterozygous men1+ and the
homozygous men1 plants show different expression patterns of WUS and CLV3 It
appears that miR166 regulates WUS indirectly and influences the expressional balance
between WUS and CLV3 through a negative feedback loop (Jung amp Park 2007 de
Alba et al 2013)
The phv phb athb15 triple mutant has an enlarged shoot apical meristems the
rev phb phv mutant does not have functional shoot apical meristems (Prigge et al
2005) This distinction may be explained by the fact that while miR166 targets
primarily PHB PHV and ATHB15 miR165 targets all the five HD-ZIP III genes (Zhou
et al 2007) Consistent with this transgenic plants overproducing miR166 are distinct
from those overproducing miR165 The miR165-overproducing transgenic plants lack
functional shoot apical meristem and a pin-like structure is formed at the apical region
of the mutant These phenotypes are quite similar to rev phb phv triple mutant (Zhou et
al 2007 Liu et al 2013) The phenotypic distinction may because by the difference
of the cleavage efficiencies of the target mRNAs In accordance with this notion a few
35S miR166a transgenic lines exhibit no shoot apical meristems The men1
homozygous plants also show arrested meristem development (Jung amp Park 2007)
Chapter 1 Introduction and Review of Literature
46
MiR164 cleaves the CUC1 and CUC2 mRNAs as well as the NAC1 mRNA
CUC1 and CUC2 determines the organ boundary in the meristem Phenotypes of
transgenic plants that overproduce miR164 are similar to those of the cuc1cuc2 double
mutant that exhibits embryonic developmental defects such as cup-shaped cotyledons
and fused cotyledons (Laufs et al 2004) When a miR164-resistant CUC2 is
expressed the boundary domain is enlarged CUC also plays a role in embryonic shoot
meristem formation by regulating the SHOOT MERISTEMLESS gene It was therefore
concluded that miR164-mediated cleavage of CUC1 and CUC2 mRNAs determines the
sizes of the boundary domains in the shoot apical meristem and regulates separation of
the organs by restricting cell proliferation (Laufs et al 2004 de Alba et al 2013)
11364 Leaf development
The role of miR319 has been extensively studied in leaf development A dominant
jaw-D mutant overproducing miR319 shows uneven curved leaves with enlarged size
and five TCP genes which are closely related to the CINCINNATA (CIN) gene in
Antirrhinum majus are down regulated in the mutant suggesting that miR319 mediated
regulation of the TCP transcription factor genes are important for the final step of
leaf shaping (Palatnik et al 2003 Naz et al 2013) In addition to leaf shape and size
leaf polarity is also specified by miRNA MiR166-mediated cleavage of the HD-ZIPIII
mRNAs is a well to establish leaf polarity This discovery originates from the gain-of-
function mutants such as phb-1d and phv-1d wherein point mutations in the
complementary sites to miRNA helps the mRNA to evade or resist from miRNA-
mediated cleavage (McConnell et al 2001 Emery et al 2003 Naz et al 2013)
Plant leaves have a dorso-ventral polarity consisting of the adaxial (upper
surface) and abaxial (lower surface) sides The adaxial side is closer to the shoot
apical meristem Adaxial identity is specified by functionally redundant class III HD-
ZIP family genes such as PHB PHV and REV Ectopic expression of each genes
result in adaxialized leaves Dominant phb-1d and phv-1d mutants exhibit
transformation of the abaxial to adaxial fate and have rod-shaped leaves In contrast
miR165166-overproducing plants show pin-like cotyledons radicalized leaves and
abaxialized leaves (Chitwood et al 2007 Pattanayak et al 2013)
Leaf asymmetry is determined by miR166-mediated restriction of the
expression domains of the HD-ZIPIII genes The HD-ZIPIII genes are expressed in the
Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora
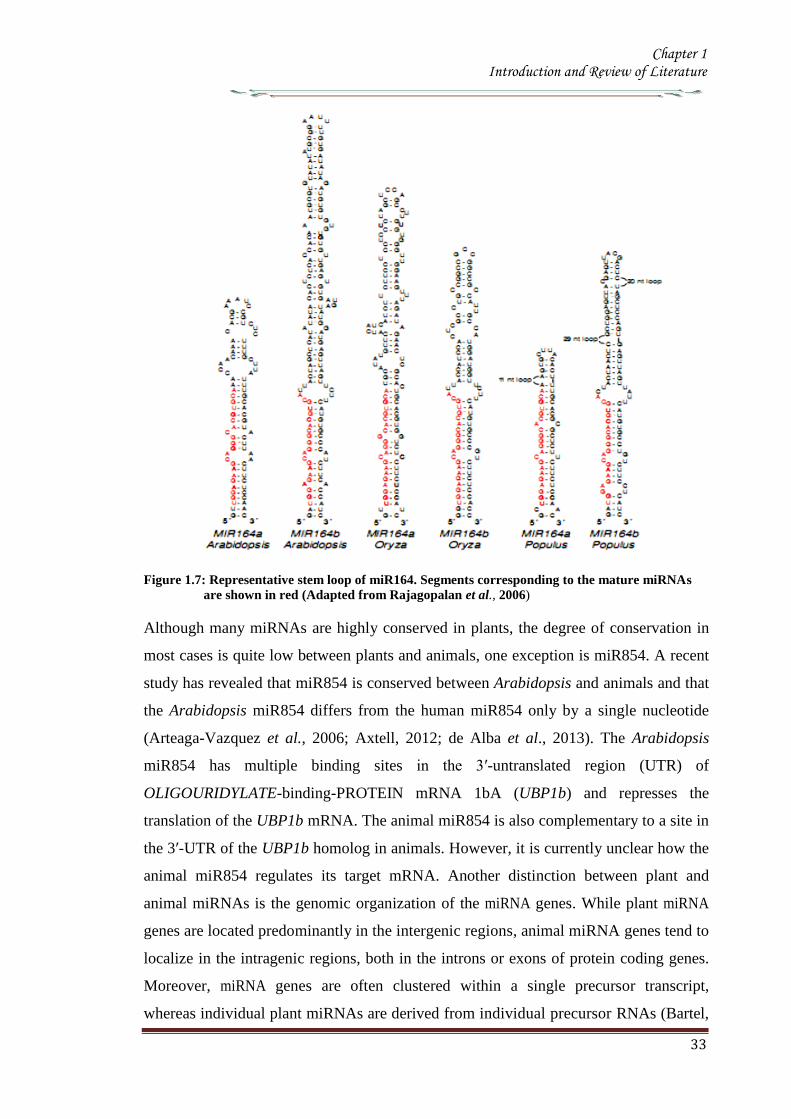
Chapter 1 Introduction and Review of Literature
33
Figure 17 Representative stem loop of miR164 Segments corresponding to the mature miRNAs
are shown in red (Adapted from Rajagopalan et al 2006)
Although many miRNAs are highly conserved in plants the degree of conservation in
most cases is quite low between plants and animals one exception is miR854 A recent
study has revealed that miR854 is conserved between Arabidopsis and animals and that
the Arabidopsis miR854 differs from the human miR854 only by a single nucleotide
(Arteaga-Vazquez et al 2006 Axtell 2012 de Alba et al 2013) The Arabidopsis
miR854 has multiple binding sites in the 3prime-untranslated region (UTR) of
OLIGOURIDYLATE-binding-PROTEIN mRNA 1bA (UBP1b) and represses the
translation of the UBP1b mRNA The animal miR854 is also complementary to a site in
the 3prime-UTR of the UBP1b homolog in animals However it is currently unclear how the
animal miR854 regulates its target mRNA Another distinction between plant and
animal miRNAs is the genomic organization of the miRNA genes While plant miRNA
genes are located predominantly in the intergenic regions animal miRNA genes tend to
localize in the intragenic regions both in the introns or exons of protein coding genes
Moreover miRNA genes are often clustered within a single precursor transcript
whereas individual plant miRNAs are derived from individual precursor RNAs (Bartel
Chapter 1 Introduction and Review of Literature
34
2004 Bonnet et al 2004 Kim 2005 Vaucheret et al 2006 Marco et al 2013)
1133 Non-conserved miRNA and challenges in annotation
Most miRNAs are conserved throughout flowering plants (Table 15) but many more
were found only in a single sequenced genome thus considered to be of a more recent
evolutionary origin The extended homology between few non conserved miRNAs
precursors and their target genes provides strong evidence that these relatively ldquoYoungrdquo
miRNAs arose from the duplication of the target gene segments (Allen et al 2004)
Several non-conserved miRNAs including miR161 miR163 miR173 miR447
miR475 and miR476 are known to direct cleavage of target transcripts (Allen et al
2004a Adai et al 2005 Sunkar et al 2005 de Alba et al 2013) It is difficult to
confidently predict targets for many because it is not possible to use complementary
site as a filter against false-positive target predictions It is also difficult to
confidently annotate non-conserved miRNAs as miRNA rather than siRNAs The
established minimal standard for miRNA annotation is a small RNA with detectable
expression and the potential to form a stem-loop when joined to flanking genomic
sequence (Kasschau et al 2003) In practice these requirements are too loose to
definitively categorize many small RNAs cloned from plants Many plant siRNAs are
detectable on blots (Vazquez et al 2004b) and hundreds of thousands of non-
miRNA genomic sequences can be predicted to fold into secondary structures that
resemble structures of plant miRNA precursors (Jones-Rhoades amp Bartel 2004)
Therefore without conservation of both sequence and secondary structure it is
difficult to be confident that a given cloned RNA originated from a stem-loop (ie is a
miRNA) rather than from a double-stranded RNA (ie is a siRNA) In fact many of
the thousands o f small RNAs cloned from Arabidopsis (Gustafson et al 2005) would
probably meet the literal requirements for annotation as miRNAs A few of these
sequences might be miRNAs but others that meet the literal criteria probably are not
so The challenges of annotating non conserved small RNAs were evidenced by
three related small silencing RNAs that were originally annotated as miRNAs
that turned out to be trans-acting siRNAs (tasiRNAs)(Kanno et al2013)
Although most miRNAs that are broadly conserved among flowering plants are
now being identified and experimentally validated (Table 16)(Jones-Rhoades amp
Bartel 2004) the challenges and ambiguities for classifying non-conserved miRNAs
prevent meaningful estimates of the total number of miRNA genes in Arabidopsis and
Chapter 1 Introduction and Review of Literature
35
other plant genomes It is possible that miRNAs might have escaped detection
because they are non-conserved and expressed in specific tissues or conditions and can
only be identified by using high- throughput sequencing
1134 MicroRNA biogenesis
11341 Transcription of microRNA precursor
Plant miRNAs are primarily found in the genomic regions that are not associated with
the protein coding genes (Reinhart et al 2002) and it appears that most if not all plant
miRNAs are produced from their own transcriptional units Plant miRNA genes are
occasionally clustered near each other in the genome suggesting transcription of
multiple miRNAs from a single primary transcript [eg the miR395 cluster (Grishok
et al 2001 Jones-Rhoades amp Bartel 2004b)] but this polycistronic arrangement of
miRNA genes appears far less frequently in plants than in animals (Bartel 2004)
Northern blot EST and mapping evidence indicate that plant miRNA primary
transcripts (also known as pri-miRNAs) as in animals are longer than needed to
encompass the miRNA stem-loops (Palatnik et al 2003 Jones-Rhoades amp Bartel
2004b) At least some of these pri-miRNA transcripts appear to be spliced
polyadenylated (Kurihara amp Watanabe 2004) and capped (Xie Z et al 2005) Two
rice miRNAs are contained within transcripts that contain exon junctions within the
presumptive stem-loop precursor implying that in these cases splicing is a prerequisite
for Dicer recognition (Sunkar et al 2005) Plant pri-miRNAs are over 1kb in length
and they are usually preceded by typical TATA box motifs They can also undergo
canonical splicing polyadenylation and capping All these observations indicates RNA
polymerase II is probably responsible for transcribing most plant miRNAs (Xie Z et
al 2005) as appears to be the case for many animal miRNAs Relatively little is
known about the regulation of miRNA transcription in plants but there is no reason
to suspect that this regulation would differ from that of protein-coding transcripts as
the bioinformatics analysis of the sequence upstream of the transcriptional start
sites of the miRNA genes showed the presence of putative binding motifs of
number of known transcription factors (Megraw et al 2006 Sethupathy 2013)
11342 MicroRNA processing and export
In plants maturation of miRNA is a step wise process A miRNA gene is transcribed to
pri-miRNA which is much longer than the pre-miRNA possessing the characteristic
stem-loop structure Like transcription of most protein-coding genes the miRNA
Chapter 1 Introduction and Review of Literature
36
transcription is processed by RNA polymerase II (Pol II) enzymes and the pri-
miRNAs are 5prime-capped and 3prime-polyadenylated (Lee et al 2004 Xie Z et al 2005)
Mature miRNAs are produced from pri-miRNAs through at least two sequential
processing steps by RNase III-type endonucleases In animals pri-miRNAs are first
processed at the bottom portion of the stem by Drosha a nuclear-localized RNase III
enzyme to liberate pre-miRNAs The pre-miRNAs are then exported to the cytoplasm
with the help of exportin-5The pre-miRNAs are further processed by Dicer another
RNaseIII enzyme to a duplex consisting of the miRNA and the antisense strands
(miRNA) Since plants do not have any Drosha homologs the two sequential
processing steps seem to be carried out by DCL1 a Dicer homolog in the nucleus
(Kurihara amp Watanabe 2004 de Alba et al 2013 Rogers amp Chen 2013)
The processing of pri-miRNAs to pre-miRNAs by DCL1 is assisted by two
other proteins HYPONASTY LEAVES1 (HYL1) and SERRATE (SE) HYL1 belongs to a
family of double-stranded RNA (dsRNA) binding protein and interacts with DCL1 in
vitro and in vivo (Lu amp Fedoroff 2000 Hiraguri et al 2005 de Alba et al 2013
Rogers amp Chen 2013) Loss-of-function mutations in the HYL1 gene result in
decreased accumulation of miRNAs and concomitant accumulation of pri-miRNAs
(Han et al 2004 Vazquez et al 2004a Kurihara et al 2006) SE a C2H2 zinc finger
protein interacts with DCL1 and HYL1 and plays a role in the processing of pri-
miRNAs to pre-miRNAs (Lobbes et al 2006 Yang L et al 2006) In addition the
nuclear cap-binding complex proteins CBP20 and CBP80 that are known as ABA
HYPER SENSITIVE 1(ABH1) have been shown to be required for proper miRNA
processing (Gregory et al 2008 Laubinger et al 2008) Recently it has been
demonstrated that DCL1 can release 21-nt RNAs from dsRNA as well as from
synthetic miR167 precursor RNAs in vitro However this cleavage is error prone and
inefficient in the absence of HYL1 and SE which accelerate the rate of DCL1-mediated
cleavage and promote accurate processing of miRNAs (Dong et al 2008 Rogers amp
Chen 2013)
11343 Methylation of the miRNAmiRNAduplex
After the miRNAmiRNAduplexes are released from the pre-miRNAs by the DCL1
activities the duplexes are methylated at the 2primeOH of the3prime-ends by HUA ENHANCER1
(HEN1) a small RNA methyltransferase (Yu et al 2005 Yang Z et al 2006 Rogers
amp Chen 2013 ) This step which is distinct from the RNase III-mediated processing
Chapter 1 Introduction and Review of Literature
37
step distinguishes the biogenesis of plant miRNAs from that of animal miRNAs
Mutations in the HEN1 gene were first isolated in a morphological screening for floral
development although the HEN1 mutants display pleiotropic phenotypes similar to the
developmental defects observed in the DCL1 mutants (Chen et al 2002 Park et al
2002 Rogers amp Chen 2013)
Figure 18 Biogenesis of microRNA
A Biogenesis of plant MicroRNA B Biogenesis of metazoananimal MicroRNA
The miRNA (red) is incorporated into RISC and the miRNA(blue) in degraded
This enzyme has two dsRNA-binding domains and nuclear localization signal It
is also conserved in fungi and is required for the processing of siRNAs as well as of
miRNAs (Park et al 2002 Boutet et al 2003 Xie et al 2003) Although the HEN1
protein is localized to the nucleus its methylation activity in the cytoplasm cannot be
ignored (Xie et al 2004 Rogers amp Chen 2013)
Chapter 1 Introduction and Review of Literature
38
The methylation of the miRNAmiRNA duplex is required to protect miRNAs
against the 3prime-end uridylation activity and subsequent degradation (Li et al 2005) In
the hen1 mutants accumulation of miRNAs is reduced to a lower level Also the
miRNAs appear to be heterogeneous in their sizes such that a ladder of bands rather
than a single species is observed for most miRNAs in the mutants (Li et al 2005)
MiRNA cloning and sequencing analyses revealed the presence of a number of U
residues at the 3prime-end of miRNAs in the hen1 mutants Moreover these nucleotides do
not correspond to the miRNAs in the pre-miRNAs indicating that these nucleotides are
added after the processing of the miRNAs from pre-miRNAs (Li et al 2005)
Uridylation of RNAs has also been proven to be correlated with RNA decay (Shen amp
Goodman 2004 Mullen amp Marzluff 2008) suggesting that uridylation attracts an
exonuclease which degrades miRNAs in plants
11344 MiRNA incorporation in to the RISC and its nuclear export
The methylated miRNAmiRNA duplexes undergo RISC assembly In this process
the miRNA of the duplexes is selectively incorporated into the RISC and the miRNA
appears to be removed and subsequently degraded (Hammond et al 2000 Hutvagner
amp Zamore 2002 Schwarz et al 2003 de Alba et al 2013 Rogers amp Chen 2013)
The ARGONAUTE 1(AGO1) proteins are core components of the RISC complex
They contain two structural domains the N-terminal PAZ domain that binds to the 3prime-
end of single-stranded RNAs and the C-terminal piwi domains that has a structure
similar to that of RNase H (Cerutti et al 2000 Parker et al 2004) Several AGO
proteins possess endonucleolytic activities within the PIWI domain and cleave target
mRNAs in the middle of the site complementary to miRNA (Liu et al 2004 Meister et
al 2004 Miyoshi et al 2005) Mutations in the AGO1 gene cause miRNA target
genes to be ectopically expressed and the levels of some miRNAs are reduced in the
mutants (Kidner amp Martienssen 2004 Vaucheret et al 2004 Kidner amp Martienssen
2005 Ronemus et al 2006) Moreover the phenotype of the AGO1 hypomorphic
alleles display defects in lateral organ polarity and leaf and flower morphology as
observed in the dcl1 mutants and null AGO1 alleles are embryonically lethal
supporting the close relationship between the AGO proteins and miRNA processing
(Kidner amp Martienssen 2004 Vaucheret et al 2004 de Alba et al 2013 Rogers amp
Chen 2013)
The production of miRNA miRNA duplexes occur in the nucleus while
Chapter 1 Introduction and Review of Literature
39
miRNA-RISC complexes are present in the cytoplasm where target mRNAs are
located This discordance of functional sites requires an export step during miRNA
biogenesis HASTY the Arabidopsis homolog of exportin-5 is thought to be involved
in miRNA nuclear export (Bollman et al 2003 Park et al 2005)The hasty mutant
exhibits pleiotropic defects in plant development and reduced accumulation of most
miRNAs as in the DCL1 or AGO1 mutants Howeverit is unclear whether the HASTY
proteins export the miRNA miRNA duplexes or the miRNA-RISC complexes in
plants
11345 Feedback regulation of miRNA biogenesis
MiRNA biogenesis is under feedback regulation such that the DCL1 and AGO1 genes
are themselves regulated by miRNAs DCL1 is itself a target of miR162 which leads to
the cleavage of the DCL1 mRNA (Xie et al 2003 de Alba et al 2013) Consistent
with this the levels of DCL1 mRNA are elevated in the mutants of miRNA processing
genes in which the miR162 abundance is reduced In addition there is a predicted
hairpin structure of miR838 in the 14th
intron of the DCL1 primary transcript
(Rajagopalan et al 2006) This prediction implies that DCL1-mediated processing
on this site can result in the cleavage of the DCL1 primary transcript into two truncated
fragments the N-terminal capped fragment and the C-terminal polyadenylated
fragment If this is the case it is possible that miRNA biogenesis competes with the
splicing event of the DCL1 pre-mRNA
The AGO1 is also regulated by miRNA There is a complementary site for
miR168 binding in the AGO1 mRNA and the transcript level of the AGO1 mRNA is
reduced through an mRNA cleavage mechanism (Vaucheret et al 2006 de Alba et al
2013 Rogers amp Chen 2013) indicating that the AGO1 mRNA levels are tightly
controlled by the levels of the AGO1 protein
1135 Mechanism of miRNA action
11351 MiRNA-directed mRNA cleavage
By forming the miRNA-RISC assembly miRNAs can direct the RISC to regulate gene
expression by two post transcriptional mechanisms mRNA cleavage or translational
repression (Bartel 2004 Jones-Rhoades et al 2006 Dalmay 2013) The degree
of miRNA-mRNA complementarity plays an important role for the
determination of the mechanism to be used A general rule is that while perfect or
Chapter 1 Introduction and Review of Literature
40
near-perfect complementarity induces mRNA cleavage central mismatches trigger
translational repression (Carrington amp Ambros 2003 Jones-Rhoades amp Bartel 2004)
Unlike most animal miRNAs plant miRNAs have near-perfect or perfect
complementarity to their targets and mRNA cleavage is assumed to be a predominant
mechanism by which miRNAs regulate gene expression in plants
In RNA cleavage mechanism miRNAs guide the AGO component of RISC to
cleave a single phosphodiester bond of the target mRNA within the miRNA-binding
site (Llave et al 2002b Tang et al 2003) The truncated fragments are then released
and degraded freeing the RISC to recognize and cleave another target mRNA This has
been validated by the detection of 3prime cleavage products that have 5prime ends that start at the
middle of the complementary regions The mRNA cleavage products are then further
degraded by other mechanisms The 3prime cleavage products of some miRNA targets are
degraded by the 5prime-3prime exonuclease XRN4 an Arabidopsis homolog of the major Yeast
mRNA degrading exonuclease Xrn1p It has been observed that the 3prime cleavage
products of some target mRNAs were accumulated in the xrn4 mutants (Souret et al
2004 Dalmay 2013) However the 3prime cleavage products of other miRNA targets do
not accumulate in the xrn4 mutants indicating that there are also XRN4 independent
mechanisms The 5prime cleavage products are typically untraceable by RNA gel blot
hybridization but can be detected by sensitive PCR based methods It has been found
that the 5prime cleavage products tend to obtain an oligo U tail at the 3prime ends This 3prime U tail
is correlated with shortening of the 5prime cleavage products from the 5prime end leading to the
conclusion that the 3prime uridylation causes 5 prime- 3 prime exonucleolytic decay of the 5prime cleavage
products (Shen amp Goodman 2004)
DCL1 HYL1 and SE interact with each other and are co-localized in the
nuclear bodies termed Dicing bodies or D-bodies in which some pri-miRNA shave
been found suggesting that D-bodies serve as a center for active miRNA processing
(Fang amp Spector 2007 Fujioka et al 2007 Song et al 2007) The miRNA
processing site appears to be distinct from the Cajal bodies that have previously been
found as a siRNA processing site (Pontes amp Pikaard 2008) The D-bodies include
SmD3 and SmB proteins that are found in the Cajal bodies However At collin an
additional marker for the Cajal bodies is not found in the D-bodies Moreover the D-
bodies are present in an At collin mutant lacking the Cajal bodies The pri-miRNAs of
miR171A and miR159A genes have been found to be co-localized with HYL1 and
Chapter 1 Introduction and Review of Literature
41
DCL1 in the D- bodies that are distinct from the Cajal bodies (Song et al 2007)
11352 MiRNA-directed translational repression
Translation repression is another mechanism exerted by plant miRNAs The regulatory
mechanism of miR172 which regulates flowering time and floral organ identity is
consistent with translation repression MiR172 over production does not affect the
transcript levels of APETALA2 (AP2) and AP2- like target mRNAs Instead the AP2
protein level is reduced in the miR172 over expressing mutant (Aukerman amp Sakai
2003 Chen 2004 Dalmay 2013) It was later shown that miR172 can also direct the
cleavage of the target mRNAs although the steady-state mRNA level is unchanged due
to feedback regulation (Schwab et al 2005 Jung et al 2007) It is clear that miR172
regulates its target genes primarily by translational repression rather than mRNA
cleavage Recently it has been reported that miR156157 and miR854 also regulate
their target genes through translational repression (Arteaga-Vazquez et al 2006
Gandikota et al 2007 Dalmay 2013) It was once thought that translational repression
is an exception to the rule However experimental evidence suggest a widespread
coexistence of translational repression and mRNA cleavage (Brodersen et al 2008)
1136 Regulatory roles of miRNAs in plant development
The miRNA target prediction has been a difficult task in order to obtain regulatory
targets without bringing in too many false predictions Plant growth and developmental
processes regulated by miRNAs are quite diverse and enormous Furthermore plant
miRNAs work cooperatively or antagonistically to establish a balanced regulation This
notion is well consistent with the findings that mutations in the regulators of miRNA
biogenesis transport and processing such as AGO1 DCL1 HYL1 HEN1 ABH1
and HASTY cause pleiotropic phenotypes (Mallory amp Vaucheret 2006 de Alba et al
2013) To date the roles of miRNAs have been mostly deduced from obtaining miRNA
over expressing transgenic plants or gain-of-function mutants in which miRNA-
resistant target genes are ectopically expressed
11361 Phase transition
Plants pass through a series of developmental phase transitions during their life span
beginning with seed germination continuing through vegetative phase change
reproductive phase change flowering initiation and finally seed production for the next
generation Because of their importance in understanding plant developmental
Chapter 1 Introduction and Review of Literature
42
processes genes regulating developmental transitions have been extensively studied
and underlying molecular mechanisms have been explored in various plant species
Majority of researches were mainly focused on vegetative phase change and
reproductive transition in Arabidopsis and Maize (Poethig 2003 Baurle amp Dean
2006) Particularly the mutations of the genes encoding various regulators of miRNA
biogenesis and transport such as HST SE and AGO exhibit altered juvenile to adult
vegetative phase change and vegetative to reproductive change (Hunter et al 2003
Peragine et al 2004 Vazquez et al 2004a Jones-Rhoades et al 2006 de Alba et al
2013)
Over-expression of the miR156a gene causes late flowering and delays
vegetative phase change (Wu amp Poethig 2006 Gandikota et al 2007 Wang et al
2008) It has been shown that miR156 negatively regulates a group of genes encoding
SQUAMOSA PROMOTER-BINDING PROTEIN-LIKE (Lewis et al 2007 de Alba et
al 2013 Rogers amp Chen 2013) transcription factors such as SPL3 SPL4 and SPL5
that promote flowering initiation In contrast transgenic plants over expressing
miR156-resistant SPL345 genes are early flowering Interestingly the endogenous
level of miR156 is temporally regulated In the early juvenile vegetative phase the
level of miR156 is very high but decreases rapidly before the onset of the adult juvenile
phase (Wu amp Poethig 2006)
The Arabidopsis early activation tagged (eat-D) mutant exhibits early flowering
with disrupted floral structure The mutant phenotypes are caused by over expression of
the miR172a-2gene (Aukerman amp Sakai 2003) MiR172 regulates a small group of
genes encoding APETALLA2 (AP2)-like transcription factors such as TARGET OF
EAT1 (TOE1) TOE2 SCHLAFMUTZE (SMZ) and SCHNARCHZAPFEN(SNZ)
TOE1 TOE2 SMZ and SNZ have been shown to participate in their productive phase
change by repressing a floral integrator FLOWERING LOCUS T (FT)(Jung et al
2007) Consistent with this toe1-2D 35STOE2 35SSMZ and 35SSNZ plants
exhibit late flowering (Jung et al 2007) MiR172 is also regulated temporally and
highly expressed in the adult vegetative phase both in Arabidopsis and maize (Chuck et
al 2007) Because the temporal expression patterns of miR156 and miR172 are
complementary to each other it has been suggested that the two miRNAs interact with
each other in regulating phase change (Chuck et al 2009 de Alba et al 2013) In
support of this view it has been shown that miR172 is down-regulated in the
Chapter 1 Introduction and Review of Literature
43
Corngrass1(Cg1) mutant in which miR156 is overproduced (Chuck et al 2007 de
Alba et al 2013) However there is no direct evidence for the direct linkage between
miR156 and miR172 It is also envisioned that miR156 and miR172 would not be
directly related For example miR156 regulates plastochron index that is the inverse of
leaf initiation rate and thus frequently used to analyze temporal patterns of plant
development In contrast miR172 does not affect plastochron (Wang et al 2008) The
down-regulation of miR172 in the maize miR156-over producing mutants would be
due to the prolonged juvenile vegetative phase in the mutants
Another miRNA that regulates flowering initiation is miR159 It regulates
MYB33 and MYB65 which are closely related to the barley GAMY B transcription
factor that responds to gibberellic acid (GA) The level of miR159 is elevated in GA-
treated seedlings but reduced in the GA biosynthetic ga1 mutant Transgenic plants
over producing miR159 exhibit delayed floral induction under short days (Achard et
al 2004) It is been suggested that MYB33 mediates GA signal to regulate LEAFY
(LFY) These observations indicate that miR159 plays an important role in the GA
signaling pathways that regulate proper floral induction under short days (Achard et al
2004)
11362 Floral development
Arabidopsis flowers consist of four distinct organs They arise in a characteristic
pattern within concentric rings called whorls from outside to inside four sepals four
petals six stamens and the central pistil consisting of two carpels Identities of the
floral organs are determined by three classes of regulatory genes classA classB and
class C genes (Krizek amp Fletcher 2005) The A and C class genes act antagonistically
to restrict their expression domains (Bowman et al 1991)
It is notable that miR172 also regulates floral architecture in addition to its role
in the timing of flowering initiation MiR172 inhibits the translation of the AP2 mRNA
(Chen 2004) Transgenic plants overproducing miR172 not only exhibit early
flowering but also show abnormal floral development which is quite similar to those
of the class A gene mutants such as the ap2 mutants (Aukerman amp Sakai 2003 de
Alba et al 2013) When the activity of the class A genes is inactivated that of the class
C genes such as AGAMOUS (AG) is elevated As a result the miR172- overproducing
transgenic plants exhibit no petals along with homeotic transformation of the sepals to
Chapter 1 Introduction and Review of Literature
44
the carpels (Aukerman amp Sakai 2003 Chen 2004)
Interestingly miR166 also regulates floral architecture The role of miR165166
in the regulation of meristem activity is closely linked to floral meristem formation and
organization (Zhao et al 2007 Miyashima et al 2013) Floral structure is severely
disrupted in the miR165166-overproducing mutants such as men1 and jba-1D in
which miR166 is overproduced (Williams et al 2005b Jung amp Park 2007) The men1
and jba-1D mutants have smaller gynoecium and reduced carpel number In an extreme
case the jba-1D homozygous plants lack visible carpels (Williams et al 2005b)
Similar phenotypes have been observed in the miR165 overproducers (Zhao et al
2007) It has been suggested that the phenotypes are possibly caused by altered AG
expression in the floral meristem (Jung amp Park 2007 Turchi et al 2013)
Other miRNAs also affect floral development Overproduction of miRNAs
involved in auxin signaling also affects floral architecture Transgenic plants
overproducing miR167 have defects in the formation of ovule and anther with reduced
fertility (Ru et al 2006) It has been shown that miR167 targets ARF6 and ARF8 that
play a role in gynoecium and stamen development and in regulating fertility (Ru et al
2006 Wu et al 2006) These observations suggest that auxin signals are also closely
related to floral organogenesis In addition transgenic plants over-producing miR164
and the cuc1cuc2 double mutants show fused sepals and reduced petals (Laufs et al
2004 Turchi et al 2013) suggesting that miR164 is also related to floral meristem
activity and boundary specification of the meristematic region
11363 Shoot apical meristem development
Shoot apical meristem comprises self-maintaining totipotent cells The shoot apical
meristem activity affects diverse aspects of plant developmental processes including
leaf development vascular development and lateral organ polarity (Bowman et al
2002) Because miR165166 plays a primary role in meristem formation
overproduction of miR165166 and multiple knockout mutants of its target genes
exhibit pleiotropic pheonotypes (McConnell et al 2001 Emery et al 2003 Kim et al
2005 Williams et al 2005b)
The targets of miR165166 include genes encoding the homeo domain-leucine
zipper (HD-ZIP) class III transcription factors such as PHABULOSA(PHB)
PHAVOLUTA (PHV) REVOLUTA(REV) ATHB15CORONA (CNA) and ATHB8
Chapter 1 Introduction and Review of Literature
45
PHB PHV and ATHB15 have overlapping functions in controlling meristem
development (Turchi et al 2013) Indeed the phb phv athb15 triple mutants have an
enlarged shoot apical meristem Consistent with this the miR166- overproducing men1
and jba-1D mutants exhibit enlarged shoot apical meristems (Kim et al 2005
Williams et al 2005b Turchi et al 2013) In the mutants the shoot apical meristems
are multiplied and large
The development of shoot apical meristems is also regulated by the WUSCHEL
(WUS)ndashCLAVATA (CLV) signaling pathway WUS positively regulates cell
proliferation In contrast CLV3 which is expressed in stem cells limits the WUS
expression and thus inhibits cell proliferation in the SAM (Sharma et al 2003)
Consistent with this notion WUS is expressed to a higher level in jba-1D
explaining the ectopic enlargement of the SAM in the mutant (Williams et al 2005)
While WUS is closely related to miR165166 in the shoot apical meristem
development the underlying molecular mechanisms are currently unclear It has been
observed that the miR166-over producing men1 plants have an arrested meristem
development (Jung amp Park 2007) In addition the heterozygous men1+ and the
homozygous men1 plants show different expression patterns of WUS and CLV3 It
appears that miR166 regulates WUS indirectly and influences the expressional balance
between WUS and CLV3 through a negative feedback loop (Jung amp Park 2007 de
Alba et al 2013)
The phv phb athb15 triple mutant has an enlarged shoot apical meristems the
rev phb phv mutant does not have functional shoot apical meristems (Prigge et al
2005) This distinction may be explained by the fact that while miR166 targets
primarily PHB PHV and ATHB15 miR165 targets all the five HD-ZIP III genes (Zhou
et al 2007) Consistent with this transgenic plants overproducing miR166 are distinct
from those overproducing miR165 The miR165-overproducing transgenic plants lack
functional shoot apical meristem and a pin-like structure is formed at the apical region
of the mutant These phenotypes are quite similar to rev phb phv triple mutant (Zhou et
al 2007 Liu et al 2013) The phenotypic distinction may because by the difference
of the cleavage efficiencies of the target mRNAs In accordance with this notion a few
35S miR166a transgenic lines exhibit no shoot apical meristems The men1
homozygous plants also show arrested meristem development (Jung amp Park 2007)
Chapter 1 Introduction and Review of Literature
46
MiR164 cleaves the CUC1 and CUC2 mRNAs as well as the NAC1 mRNA
CUC1 and CUC2 determines the organ boundary in the meristem Phenotypes of
transgenic plants that overproduce miR164 are similar to those of the cuc1cuc2 double
mutant that exhibits embryonic developmental defects such as cup-shaped cotyledons
and fused cotyledons (Laufs et al 2004) When a miR164-resistant CUC2 is
expressed the boundary domain is enlarged CUC also plays a role in embryonic shoot
meristem formation by regulating the SHOOT MERISTEMLESS gene It was therefore
concluded that miR164-mediated cleavage of CUC1 and CUC2 mRNAs determines the
sizes of the boundary domains in the shoot apical meristem and regulates separation of
the organs by restricting cell proliferation (Laufs et al 2004 de Alba et al 2013)
11364 Leaf development
The role of miR319 has been extensively studied in leaf development A dominant
jaw-D mutant overproducing miR319 shows uneven curved leaves with enlarged size
and five TCP genes which are closely related to the CINCINNATA (CIN) gene in
Antirrhinum majus are down regulated in the mutant suggesting that miR319 mediated
regulation of the TCP transcription factor genes are important for the final step of
leaf shaping (Palatnik et al 2003 Naz et al 2013) In addition to leaf shape and size
leaf polarity is also specified by miRNA MiR166-mediated cleavage of the HD-ZIPIII
mRNAs is a well to establish leaf polarity This discovery originates from the gain-of-
function mutants such as phb-1d and phv-1d wherein point mutations in the
complementary sites to miRNA helps the mRNA to evade or resist from miRNA-
mediated cleavage (McConnell et al 2001 Emery et al 2003 Naz et al 2013)
Plant leaves have a dorso-ventral polarity consisting of the adaxial (upper
surface) and abaxial (lower surface) sides The adaxial side is closer to the shoot
apical meristem Adaxial identity is specified by functionally redundant class III HD-
ZIP family genes such as PHB PHV and REV Ectopic expression of each genes
result in adaxialized leaves Dominant phb-1d and phv-1d mutants exhibit
transformation of the abaxial to adaxial fate and have rod-shaped leaves In contrast
miR165166-overproducing plants show pin-like cotyledons radicalized leaves and
abaxialized leaves (Chitwood et al 2007 Pattanayak et al 2013)
Leaf asymmetry is determined by miR166-mediated restriction of the
expression domains of the HD-ZIPIII genes The HD-ZIPIII genes are expressed in the
Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
34
2004 Bonnet et al 2004 Kim 2005 Vaucheret et al 2006 Marco et al 2013)
1133 Non-conserved miRNA and challenges in annotation
Most miRNAs are conserved throughout flowering plants (Table 15) but many more
were found only in a single sequenced genome thus considered to be of a more recent
evolutionary origin The extended homology between few non conserved miRNAs
precursors and their target genes provides strong evidence that these relatively ldquoYoungrdquo
miRNAs arose from the duplication of the target gene segments (Allen et al 2004)
Several non-conserved miRNAs including miR161 miR163 miR173 miR447
miR475 and miR476 are known to direct cleavage of target transcripts (Allen et al
2004a Adai et al 2005 Sunkar et al 2005 de Alba et al 2013) It is difficult to
confidently predict targets for many because it is not possible to use complementary
site as a filter against false-positive target predictions It is also difficult to
confidently annotate non-conserved miRNAs as miRNA rather than siRNAs The
established minimal standard for miRNA annotation is a small RNA with detectable
expression and the potential to form a stem-loop when joined to flanking genomic
sequence (Kasschau et al 2003) In practice these requirements are too loose to
definitively categorize many small RNAs cloned from plants Many plant siRNAs are
detectable on blots (Vazquez et al 2004b) and hundreds of thousands of non-
miRNA genomic sequences can be predicted to fold into secondary structures that
resemble structures of plant miRNA precursors (Jones-Rhoades amp Bartel 2004)
Therefore without conservation of both sequence and secondary structure it is
difficult to be confident that a given cloned RNA originated from a stem-loop (ie is a
miRNA) rather than from a double-stranded RNA (ie is a siRNA) In fact many of
the thousands o f small RNAs cloned from Arabidopsis (Gustafson et al 2005) would
probably meet the literal requirements for annotation as miRNAs A few of these
sequences might be miRNAs but others that meet the literal criteria probably are not
so The challenges of annotating non conserved small RNAs were evidenced by
three related small silencing RNAs that were originally annotated as miRNAs
that turned out to be trans-acting siRNAs (tasiRNAs)(Kanno et al2013)
Although most miRNAs that are broadly conserved among flowering plants are
now being identified and experimentally validated (Table 16)(Jones-Rhoades amp
Bartel 2004) the challenges and ambiguities for classifying non-conserved miRNAs
prevent meaningful estimates of the total number of miRNA genes in Arabidopsis and
Chapter 1 Introduction and Review of Literature
35
other plant genomes It is possible that miRNAs might have escaped detection
because they are non-conserved and expressed in specific tissues or conditions and can
only be identified by using high- throughput sequencing
1134 MicroRNA biogenesis
11341 Transcription of microRNA precursor
Plant miRNAs are primarily found in the genomic regions that are not associated with
the protein coding genes (Reinhart et al 2002) and it appears that most if not all plant
miRNAs are produced from their own transcriptional units Plant miRNA genes are
occasionally clustered near each other in the genome suggesting transcription of
multiple miRNAs from a single primary transcript [eg the miR395 cluster (Grishok
et al 2001 Jones-Rhoades amp Bartel 2004b)] but this polycistronic arrangement of
miRNA genes appears far less frequently in plants than in animals (Bartel 2004)
Northern blot EST and mapping evidence indicate that plant miRNA primary
transcripts (also known as pri-miRNAs) as in animals are longer than needed to
encompass the miRNA stem-loops (Palatnik et al 2003 Jones-Rhoades amp Bartel
2004b) At least some of these pri-miRNA transcripts appear to be spliced
polyadenylated (Kurihara amp Watanabe 2004) and capped (Xie Z et al 2005) Two
rice miRNAs are contained within transcripts that contain exon junctions within the
presumptive stem-loop precursor implying that in these cases splicing is a prerequisite
for Dicer recognition (Sunkar et al 2005) Plant pri-miRNAs are over 1kb in length
and they are usually preceded by typical TATA box motifs They can also undergo
canonical splicing polyadenylation and capping All these observations indicates RNA
polymerase II is probably responsible for transcribing most plant miRNAs (Xie Z et
al 2005) as appears to be the case for many animal miRNAs Relatively little is
known about the regulation of miRNA transcription in plants but there is no reason
to suspect that this regulation would differ from that of protein-coding transcripts as
the bioinformatics analysis of the sequence upstream of the transcriptional start
sites of the miRNA genes showed the presence of putative binding motifs of
number of known transcription factors (Megraw et al 2006 Sethupathy 2013)
11342 MicroRNA processing and export
In plants maturation of miRNA is a step wise process A miRNA gene is transcribed to
pri-miRNA which is much longer than the pre-miRNA possessing the characteristic
stem-loop structure Like transcription of most protein-coding genes the miRNA
Chapter 1 Introduction and Review of Literature
36
transcription is processed by RNA polymerase II (Pol II) enzymes and the pri-
miRNAs are 5prime-capped and 3prime-polyadenylated (Lee et al 2004 Xie Z et al 2005)
Mature miRNAs are produced from pri-miRNAs through at least two sequential
processing steps by RNase III-type endonucleases In animals pri-miRNAs are first
processed at the bottom portion of the stem by Drosha a nuclear-localized RNase III
enzyme to liberate pre-miRNAs The pre-miRNAs are then exported to the cytoplasm
with the help of exportin-5The pre-miRNAs are further processed by Dicer another
RNaseIII enzyme to a duplex consisting of the miRNA and the antisense strands
(miRNA) Since plants do not have any Drosha homologs the two sequential
processing steps seem to be carried out by DCL1 a Dicer homolog in the nucleus
(Kurihara amp Watanabe 2004 de Alba et al 2013 Rogers amp Chen 2013)
The processing of pri-miRNAs to pre-miRNAs by DCL1 is assisted by two
other proteins HYPONASTY LEAVES1 (HYL1) and SERRATE (SE) HYL1 belongs to a
family of double-stranded RNA (dsRNA) binding protein and interacts with DCL1 in
vitro and in vivo (Lu amp Fedoroff 2000 Hiraguri et al 2005 de Alba et al 2013
Rogers amp Chen 2013) Loss-of-function mutations in the HYL1 gene result in
decreased accumulation of miRNAs and concomitant accumulation of pri-miRNAs
(Han et al 2004 Vazquez et al 2004a Kurihara et al 2006) SE a C2H2 zinc finger
protein interacts with DCL1 and HYL1 and plays a role in the processing of pri-
miRNAs to pre-miRNAs (Lobbes et al 2006 Yang L et al 2006) In addition the
nuclear cap-binding complex proteins CBP20 and CBP80 that are known as ABA
HYPER SENSITIVE 1(ABH1) have been shown to be required for proper miRNA
processing (Gregory et al 2008 Laubinger et al 2008) Recently it has been
demonstrated that DCL1 can release 21-nt RNAs from dsRNA as well as from
synthetic miR167 precursor RNAs in vitro However this cleavage is error prone and
inefficient in the absence of HYL1 and SE which accelerate the rate of DCL1-mediated
cleavage and promote accurate processing of miRNAs (Dong et al 2008 Rogers amp
Chen 2013)
11343 Methylation of the miRNAmiRNAduplex
After the miRNAmiRNAduplexes are released from the pre-miRNAs by the DCL1
activities the duplexes are methylated at the 2primeOH of the3prime-ends by HUA ENHANCER1
(HEN1) a small RNA methyltransferase (Yu et al 2005 Yang Z et al 2006 Rogers
amp Chen 2013 ) This step which is distinct from the RNase III-mediated processing
Chapter 1 Introduction and Review of Literature
37
step distinguishes the biogenesis of plant miRNAs from that of animal miRNAs
Mutations in the HEN1 gene were first isolated in a morphological screening for floral
development although the HEN1 mutants display pleiotropic phenotypes similar to the
developmental defects observed in the DCL1 mutants (Chen et al 2002 Park et al
2002 Rogers amp Chen 2013)
Figure 18 Biogenesis of microRNA
A Biogenesis of plant MicroRNA B Biogenesis of metazoananimal MicroRNA
The miRNA (red) is incorporated into RISC and the miRNA(blue) in degraded
This enzyme has two dsRNA-binding domains and nuclear localization signal It
is also conserved in fungi and is required for the processing of siRNAs as well as of
miRNAs (Park et al 2002 Boutet et al 2003 Xie et al 2003) Although the HEN1
protein is localized to the nucleus its methylation activity in the cytoplasm cannot be
ignored (Xie et al 2004 Rogers amp Chen 2013)
Chapter 1 Introduction and Review of Literature
38
The methylation of the miRNAmiRNA duplex is required to protect miRNAs
against the 3prime-end uridylation activity and subsequent degradation (Li et al 2005) In
the hen1 mutants accumulation of miRNAs is reduced to a lower level Also the
miRNAs appear to be heterogeneous in their sizes such that a ladder of bands rather
than a single species is observed for most miRNAs in the mutants (Li et al 2005)
MiRNA cloning and sequencing analyses revealed the presence of a number of U
residues at the 3prime-end of miRNAs in the hen1 mutants Moreover these nucleotides do
not correspond to the miRNAs in the pre-miRNAs indicating that these nucleotides are
added after the processing of the miRNAs from pre-miRNAs (Li et al 2005)
Uridylation of RNAs has also been proven to be correlated with RNA decay (Shen amp
Goodman 2004 Mullen amp Marzluff 2008) suggesting that uridylation attracts an
exonuclease which degrades miRNAs in plants
11344 MiRNA incorporation in to the RISC and its nuclear export
The methylated miRNAmiRNA duplexes undergo RISC assembly In this process
the miRNA of the duplexes is selectively incorporated into the RISC and the miRNA
appears to be removed and subsequently degraded (Hammond et al 2000 Hutvagner
amp Zamore 2002 Schwarz et al 2003 de Alba et al 2013 Rogers amp Chen 2013)
The ARGONAUTE 1(AGO1) proteins are core components of the RISC complex
They contain two structural domains the N-terminal PAZ domain that binds to the 3prime-
end of single-stranded RNAs and the C-terminal piwi domains that has a structure
similar to that of RNase H (Cerutti et al 2000 Parker et al 2004) Several AGO
proteins possess endonucleolytic activities within the PIWI domain and cleave target
mRNAs in the middle of the site complementary to miRNA (Liu et al 2004 Meister et
al 2004 Miyoshi et al 2005) Mutations in the AGO1 gene cause miRNA target
genes to be ectopically expressed and the levels of some miRNAs are reduced in the
mutants (Kidner amp Martienssen 2004 Vaucheret et al 2004 Kidner amp Martienssen
2005 Ronemus et al 2006) Moreover the phenotype of the AGO1 hypomorphic
alleles display defects in lateral organ polarity and leaf and flower morphology as
observed in the dcl1 mutants and null AGO1 alleles are embryonically lethal
supporting the close relationship between the AGO proteins and miRNA processing
(Kidner amp Martienssen 2004 Vaucheret et al 2004 de Alba et al 2013 Rogers amp
Chen 2013)
The production of miRNA miRNA duplexes occur in the nucleus while
Chapter 1 Introduction and Review of Literature
39
miRNA-RISC complexes are present in the cytoplasm where target mRNAs are
located This discordance of functional sites requires an export step during miRNA
biogenesis HASTY the Arabidopsis homolog of exportin-5 is thought to be involved
in miRNA nuclear export (Bollman et al 2003 Park et al 2005)The hasty mutant
exhibits pleiotropic defects in plant development and reduced accumulation of most
miRNAs as in the DCL1 or AGO1 mutants Howeverit is unclear whether the HASTY
proteins export the miRNA miRNA duplexes or the miRNA-RISC complexes in
plants
11345 Feedback regulation of miRNA biogenesis
MiRNA biogenesis is under feedback regulation such that the DCL1 and AGO1 genes
are themselves regulated by miRNAs DCL1 is itself a target of miR162 which leads to
the cleavage of the DCL1 mRNA (Xie et al 2003 de Alba et al 2013) Consistent
with this the levels of DCL1 mRNA are elevated in the mutants of miRNA processing
genes in which the miR162 abundance is reduced In addition there is a predicted
hairpin structure of miR838 in the 14th
intron of the DCL1 primary transcript
(Rajagopalan et al 2006) This prediction implies that DCL1-mediated processing
on this site can result in the cleavage of the DCL1 primary transcript into two truncated
fragments the N-terminal capped fragment and the C-terminal polyadenylated
fragment If this is the case it is possible that miRNA biogenesis competes with the
splicing event of the DCL1 pre-mRNA
The AGO1 is also regulated by miRNA There is a complementary site for
miR168 binding in the AGO1 mRNA and the transcript level of the AGO1 mRNA is
reduced through an mRNA cleavage mechanism (Vaucheret et al 2006 de Alba et al
2013 Rogers amp Chen 2013) indicating that the AGO1 mRNA levels are tightly
controlled by the levels of the AGO1 protein
1135 Mechanism of miRNA action
11351 MiRNA-directed mRNA cleavage
By forming the miRNA-RISC assembly miRNAs can direct the RISC to regulate gene
expression by two post transcriptional mechanisms mRNA cleavage or translational
repression (Bartel 2004 Jones-Rhoades et al 2006 Dalmay 2013) The degree
of miRNA-mRNA complementarity plays an important role for the
determination of the mechanism to be used A general rule is that while perfect or
Chapter 1 Introduction and Review of Literature
40
near-perfect complementarity induces mRNA cleavage central mismatches trigger
translational repression (Carrington amp Ambros 2003 Jones-Rhoades amp Bartel 2004)
Unlike most animal miRNAs plant miRNAs have near-perfect or perfect
complementarity to their targets and mRNA cleavage is assumed to be a predominant
mechanism by which miRNAs regulate gene expression in plants
In RNA cleavage mechanism miRNAs guide the AGO component of RISC to
cleave a single phosphodiester bond of the target mRNA within the miRNA-binding
site (Llave et al 2002b Tang et al 2003) The truncated fragments are then released
and degraded freeing the RISC to recognize and cleave another target mRNA This has
been validated by the detection of 3prime cleavage products that have 5prime ends that start at the
middle of the complementary regions The mRNA cleavage products are then further
degraded by other mechanisms The 3prime cleavage products of some miRNA targets are
degraded by the 5prime-3prime exonuclease XRN4 an Arabidopsis homolog of the major Yeast
mRNA degrading exonuclease Xrn1p It has been observed that the 3prime cleavage
products of some target mRNAs were accumulated in the xrn4 mutants (Souret et al
2004 Dalmay 2013) However the 3prime cleavage products of other miRNA targets do
not accumulate in the xrn4 mutants indicating that there are also XRN4 independent
mechanisms The 5prime cleavage products are typically untraceable by RNA gel blot
hybridization but can be detected by sensitive PCR based methods It has been found
that the 5prime cleavage products tend to obtain an oligo U tail at the 3prime ends This 3prime U tail
is correlated with shortening of the 5prime cleavage products from the 5prime end leading to the
conclusion that the 3prime uridylation causes 5 prime- 3 prime exonucleolytic decay of the 5prime cleavage
products (Shen amp Goodman 2004)
DCL1 HYL1 and SE interact with each other and are co-localized in the
nuclear bodies termed Dicing bodies or D-bodies in which some pri-miRNA shave
been found suggesting that D-bodies serve as a center for active miRNA processing
(Fang amp Spector 2007 Fujioka et al 2007 Song et al 2007) The miRNA
processing site appears to be distinct from the Cajal bodies that have previously been
found as a siRNA processing site (Pontes amp Pikaard 2008) The D-bodies include
SmD3 and SmB proteins that are found in the Cajal bodies However At collin an
additional marker for the Cajal bodies is not found in the D-bodies Moreover the D-
bodies are present in an At collin mutant lacking the Cajal bodies The pri-miRNAs of
miR171A and miR159A genes have been found to be co-localized with HYL1 and
Chapter 1 Introduction and Review of Literature
41
DCL1 in the D- bodies that are distinct from the Cajal bodies (Song et al 2007)
11352 MiRNA-directed translational repression
Translation repression is another mechanism exerted by plant miRNAs The regulatory
mechanism of miR172 which regulates flowering time and floral organ identity is
consistent with translation repression MiR172 over production does not affect the
transcript levels of APETALA2 (AP2) and AP2- like target mRNAs Instead the AP2
protein level is reduced in the miR172 over expressing mutant (Aukerman amp Sakai
2003 Chen 2004 Dalmay 2013) It was later shown that miR172 can also direct the
cleavage of the target mRNAs although the steady-state mRNA level is unchanged due
to feedback regulation (Schwab et al 2005 Jung et al 2007) It is clear that miR172
regulates its target genes primarily by translational repression rather than mRNA
cleavage Recently it has been reported that miR156157 and miR854 also regulate
their target genes through translational repression (Arteaga-Vazquez et al 2006
Gandikota et al 2007 Dalmay 2013) It was once thought that translational repression
is an exception to the rule However experimental evidence suggest a widespread
coexistence of translational repression and mRNA cleavage (Brodersen et al 2008)
1136 Regulatory roles of miRNAs in plant development
The miRNA target prediction has been a difficult task in order to obtain regulatory
targets without bringing in too many false predictions Plant growth and developmental
processes regulated by miRNAs are quite diverse and enormous Furthermore plant
miRNAs work cooperatively or antagonistically to establish a balanced regulation This
notion is well consistent with the findings that mutations in the regulators of miRNA
biogenesis transport and processing such as AGO1 DCL1 HYL1 HEN1 ABH1
and HASTY cause pleiotropic phenotypes (Mallory amp Vaucheret 2006 de Alba et al
2013) To date the roles of miRNAs have been mostly deduced from obtaining miRNA
over expressing transgenic plants or gain-of-function mutants in which miRNA-
resistant target genes are ectopically expressed
11361 Phase transition
Plants pass through a series of developmental phase transitions during their life span
beginning with seed germination continuing through vegetative phase change
reproductive phase change flowering initiation and finally seed production for the next
generation Because of their importance in understanding plant developmental
Chapter 1 Introduction and Review of Literature
42
processes genes regulating developmental transitions have been extensively studied
and underlying molecular mechanisms have been explored in various plant species
Majority of researches were mainly focused on vegetative phase change and
reproductive transition in Arabidopsis and Maize (Poethig 2003 Baurle amp Dean
2006) Particularly the mutations of the genes encoding various regulators of miRNA
biogenesis and transport such as HST SE and AGO exhibit altered juvenile to adult
vegetative phase change and vegetative to reproductive change (Hunter et al 2003
Peragine et al 2004 Vazquez et al 2004a Jones-Rhoades et al 2006 de Alba et al
2013)
Over-expression of the miR156a gene causes late flowering and delays
vegetative phase change (Wu amp Poethig 2006 Gandikota et al 2007 Wang et al
2008) It has been shown that miR156 negatively regulates a group of genes encoding
SQUAMOSA PROMOTER-BINDING PROTEIN-LIKE (Lewis et al 2007 de Alba et
al 2013 Rogers amp Chen 2013) transcription factors such as SPL3 SPL4 and SPL5
that promote flowering initiation In contrast transgenic plants over expressing
miR156-resistant SPL345 genes are early flowering Interestingly the endogenous
level of miR156 is temporally regulated In the early juvenile vegetative phase the
level of miR156 is very high but decreases rapidly before the onset of the adult juvenile
phase (Wu amp Poethig 2006)
The Arabidopsis early activation tagged (eat-D) mutant exhibits early flowering
with disrupted floral structure The mutant phenotypes are caused by over expression of
the miR172a-2gene (Aukerman amp Sakai 2003) MiR172 regulates a small group of
genes encoding APETALLA2 (AP2)-like transcription factors such as TARGET OF
EAT1 (TOE1) TOE2 SCHLAFMUTZE (SMZ) and SCHNARCHZAPFEN(SNZ)
TOE1 TOE2 SMZ and SNZ have been shown to participate in their productive phase
change by repressing a floral integrator FLOWERING LOCUS T (FT)(Jung et al
2007) Consistent with this toe1-2D 35STOE2 35SSMZ and 35SSNZ plants
exhibit late flowering (Jung et al 2007) MiR172 is also regulated temporally and
highly expressed in the adult vegetative phase both in Arabidopsis and maize (Chuck et
al 2007) Because the temporal expression patterns of miR156 and miR172 are
complementary to each other it has been suggested that the two miRNAs interact with
each other in regulating phase change (Chuck et al 2009 de Alba et al 2013) In
support of this view it has been shown that miR172 is down-regulated in the
Chapter 1 Introduction and Review of Literature
43
Corngrass1(Cg1) mutant in which miR156 is overproduced (Chuck et al 2007 de
Alba et al 2013) However there is no direct evidence for the direct linkage between
miR156 and miR172 It is also envisioned that miR156 and miR172 would not be
directly related For example miR156 regulates plastochron index that is the inverse of
leaf initiation rate and thus frequently used to analyze temporal patterns of plant
development In contrast miR172 does not affect plastochron (Wang et al 2008) The
down-regulation of miR172 in the maize miR156-over producing mutants would be
due to the prolonged juvenile vegetative phase in the mutants
Another miRNA that regulates flowering initiation is miR159 It regulates
MYB33 and MYB65 which are closely related to the barley GAMY B transcription
factor that responds to gibberellic acid (GA) The level of miR159 is elevated in GA-
treated seedlings but reduced in the GA biosynthetic ga1 mutant Transgenic plants
over producing miR159 exhibit delayed floral induction under short days (Achard et
al 2004) It is been suggested that MYB33 mediates GA signal to regulate LEAFY
(LFY) These observations indicate that miR159 plays an important role in the GA
signaling pathways that regulate proper floral induction under short days (Achard et al
2004)
11362 Floral development
Arabidopsis flowers consist of four distinct organs They arise in a characteristic
pattern within concentric rings called whorls from outside to inside four sepals four
petals six stamens and the central pistil consisting of two carpels Identities of the
floral organs are determined by three classes of regulatory genes classA classB and
class C genes (Krizek amp Fletcher 2005) The A and C class genes act antagonistically
to restrict their expression domains (Bowman et al 1991)
It is notable that miR172 also regulates floral architecture in addition to its role
in the timing of flowering initiation MiR172 inhibits the translation of the AP2 mRNA
(Chen 2004) Transgenic plants overproducing miR172 not only exhibit early
flowering but also show abnormal floral development which is quite similar to those
of the class A gene mutants such as the ap2 mutants (Aukerman amp Sakai 2003 de
Alba et al 2013) When the activity of the class A genes is inactivated that of the class
C genes such as AGAMOUS (AG) is elevated As a result the miR172- overproducing
transgenic plants exhibit no petals along with homeotic transformation of the sepals to
Chapter 1 Introduction and Review of Literature
44
the carpels (Aukerman amp Sakai 2003 Chen 2004)
Interestingly miR166 also regulates floral architecture The role of miR165166
in the regulation of meristem activity is closely linked to floral meristem formation and
organization (Zhao et al 2007 Miyashima et al 2013) Floral structure is severely
disrupted in the miR165166-overproducing mutants such as men1 and jba-1D in
which miR166 is overproduced (Williams et al 2005b Jung amp Park 2007) The men1
and jba-1D mutants have smaller gynoecium and reduced carpel number In an extreme
case the jba-1D homozygous plants lack visible carpels (Williams et al 2005b)
Similar phenotypes have been observed in the miR165 overproducers (Zhao et al
2007) It has been suggested that the phenotypes are possibly caused by altered AG
expression in the floral meristem (Jung amp Park 2007 Turchi et al 2013)
Other miRNAs also affect floral development Overproduction of miRNAs
involved in auxin signaling also affects floral architecture Transgenic plants
overproducing miR167 have defects in the formation of ovule and anther with reduced
fertility (Ru et al 2006) It has been shown that miR167 targets ARF6 and ARF8 that
play a role in gynoecium and stamen development and in regulating fertility (Ru et al
2006 Wu et al 2006) These observations suggest that auxin signals are also closely
related to floral organogenesis In addition transgenic plants over-producing miR164
and the cuc1cuc2 double mutants show fused sepals and reduced petals (Laufs et al
2004 Turchi et al 2013) suggesting that miR164 is also related to floral meristem
activity and boundary specification of the meristematic region
11363 Shoot apical meristem development
Shoot apical meristem comprises self-maintaining totipotent cells The shoot apical
meristem activity affects diverse aspects of plant developmental processes including
leaf development vascular development and lateral organ polarity (Bowman et al
2002) Because miR165166 plays a primary role in meristem formation
overproduction of miR165166 and multiple knockout mutants of its target genes
exhibit pleiotropic pheonotypes (McConnell et al 2001 Emery et al 2003 Kim et al
2005 Williams et al 2005b)
The targets of miR165166 include genes encoding the homeo domain-leucine
zipper (HD-ZIP) class III transcription factors such as PHABULOSA(PHB)
PHAVOLUTA (PHV) REVOLUTA(REV) ATHB15CORONA (CNA) and ATHB8
Chapter 1 Introduction and Review of Literature
45
PHB PHV and ATHB15 have overlapping functions in controlling meristem
development (Turchi et al 2013) Indeed the phb phv athb15 triple mutants have an
enlarged shoot apical meristem Consistent with this the miR166- overproducing men1
and jba-1D mutants exhibit enlarged shoot apical meristems (Kim et al 2005
Williams et al 2005b Turchi et al 2013) In the mutants the shoot apical meristems
are multiplied and large
The development of shoot apical meristems is also regulated by the WUSCHEL
(WUS)ndashCLAVATA (CLV) signaling pathway WUS positively regulates cell
proliferation In contrast CLV3 which is expressed in stem cells limits the WUS
expression and thus inhibits cell proliferation in the SAM (Sharma et al 2003)
Consistent with this notion WUS is expressed to a higher level in jba-1D
explaining the ectopic enlargement of the SAM in the mutant (Williams et al 2005)
While WUS is closely related to miR165166 in the shoot apical meristem
development the underlying molecular mechanisms are currently unclear It has been
observed that the miR166-over producing men1 plants have an arrested meristem
development (Jung amp Park 2007) In addition the heterozygous men1+ and the
homozygous men1 plants show different expression patterns of WUS and CLV3 It
appears that miR166 regulates WUS indirectly and influences the expressional balance
between WUS and CLV3 through a negative feedback loop (Jung amp Park 2007 de
Alba et al 2013)
The phv phb athb15 triple mutant has an enlarged shoot apical meristems the
rev phb phv mutant does not have functional shoot apical meristems (Prigge et al
2005) This distinction may be explained by the fact that while miR166 targets
primarily PHB PHV and ATHB15 miR165 targets all the five HD-ZIP III genes (Zhou
et al 2007) Consistent with this transgenic plants overproducing miR166 are distinct
from those overproducing miR165 The miR165-overproducing transgenic plants lack
functional shoot apical meristem and a pin-like structure is formed at the apical region
of the mutant These phenotypes are quite similar to rev phb phv triple mutant (Zhou et
al 2007 Liu et al 2013) The phenotypic distinction may because by the difference
of the cleavage efficiencies of the target mRNAs In accordance with this notion a few
35S miR166a transgenic lines exhibit no shoot apical meristems The men1
homozygous plants also show arrested meristem development (Jung amp Park 2007)
Chapter 1 Introduction and Review of Literature
46
MiR164 cleaves the CUC1 and CUC2 mRNAs as well as the NAC1 mRNA
CUC1 and CUC2 determines the organ boundary in the meristem Phenotypes of
transgenic plants that overproduce miR164 are similar to those of the cuc1cuc2 double
mutant that exhibits embryonic developmental defects such as cup-shaped cotyledons
and fused cotyledons (Laufs et al 2004) When a miR164-resistant CUC2 is
expressed the boundary domain is enlarged CUC also plays a role in embryonic shoot
meristem formation by regulating the SHOOT MERISTEMLESS gene It was therefore
concluded that miR164-mediated cleavage of CUC1 and CUC2 mRNAs determines the
sizes of the boundary domains in the shoot apical meristem and regulates separation of
the organs by restricting cell proliferation (Laufs et al 2004 de Alba et al 2013)
11364 Leaf development
The role of miR319 has been extensively studied in leaf development A dominant
jaw-D mutant overproducing miR319 shows uneven curved leaves with enlarged size
and five TCP genes which are closely related to the CINCINNATA (CIN) gene in
Antirrhinum majus are down regulated in the mutant suggesting that miR319 mediated
regulation of the TCP transcription factor genes are important for the final step of
leaf shaping (Palatnik et al 2003 Naz et al 2013) In addition to leaf shape and size
leaf polarity is also specified by miRNA MiR166-mediated cleavage of the HD-ZIPIII
mRNAs is a well to establish leaf polarity This discovery originates from the gain-of-
function mutants such as phb-1d and phv-1d wherein point mutations in the
complementary sites to miRNA helps the mRNA to evade or resist from miRNA-
mediated cleavage (McConnell et al 2001 Emery et al 2003 Naz et al 2013)
Plant leaves have a dorso-ventral polarity consisting of the adaxial (upper
surface) and abaxial (lower surface) sides The adaxial side is closer to the shoot
apical meristem Adaxial identity is specified by functionally redundant class III HD-
ZIP family genes such as PHB PHV and REV Ectopic expression of each genes
result in adaxialized leaves Dominant phb-1d and phv-1d mutants exhibit
transformation of the abaxial to adaxial fate and have rod-shaped leaves In contrast
miR165166-overproducing plants show pin-like cotyledons radicalized leaves and
abaxialized leaves (Chitwood et al 2007 Pattanayak et al 2013)
Leaf asymmetry is determined by miR166-mediated restriction of the
expression domains of the HD-ZIPIII genes The HD-ZIPIII genes are expressed in the
Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
35
other plant genomes It is possible that miRNAs might have escaped detection
because they are non-conserved and expressed in specific tissues or conditions and can
only be identified by using high- throughput sequencing
1134 MicroRNA biogenesis
11341 Transcription of microRNA precursor
Plant miRNAs are primarily found in the genomic regions that are not associated with
the protein coding genes (Reinhart et al 2002) and it appears that most if not all plant
miRNAs are produced from their own transcriptional units Plant miRNA genes are
occasionally clustered near each other in the genome suggesting transcription of
multiple miRNAs from a single primary transcript [eg the miR395 cluster (Grishok
et al 2001 Jones-Rhoades amp Bartel 2004b)] but this polycistronic arrangement of
miRNA genes appears far less frequently in plants than in animals (Bartel 2004)
Northern blot EST and mapping evidence indicate that plant miRNA primary
transcripts (also known as pri-miRNAs) as in animals are longer than needed to
encompass the miRNA stem-loops (Palatnik et al 2003 Jones-Rhoades amp Bartel
2004b) At least some of these pri-miRNA transcripts appear to be spliced
polyadenylated (Kurihara amp Watanabe 2004) and capped (Xie Z et al 2005) Two
rice miRNAs are contained within transcripts that contain exon junctions within the
presumptive stem-loop precursor implying that in these cases splicing is a prerequisite
for Dicer recognition (Sunkar et al 2005) Plant pri-miRNAs are over 1kb in length
and they are usually preceded by typical TATA box motifs They can also undergo
canonical splicing polyadenylation and capping All these observations indicates RNA
polymerase II is probably responsible for transcribing most plant miRNAs (Xie Z et
al 2005) as appears to be the case for many animal miRNAs Relatively little is
known about the regulation of miRNA transcription in plants but there is no reason
to suspect that this regulation would differ from that of protein-coding transcripts as
the bioinformatics analysis of the sequence upstream of the transcriptional start
sites of the miRNA genes showed the presence of putative binding motifs of
number of known transcription factors (Megraw et al 2006 Sethupathy 2013)
11342 MicroRNA processing and export
In plants maturation of miRNA is a step wise process A miRNA gene is transcribed to
pri-miRNA which is much longer than the pre-miRNA possessing the characteristic
stem-loop structure Like transcription of most protein-coding genes the miRNA
Chapter 1 Introduction and Review of Literature
36
transcription is processed by RNA polymerase II (Pol II) enzymes and the pri-
miRNAs are 5prime-capped and 3prime-polyadenylated (Lee et al 2004 Xie Z et al 2005)
Mature miRNAs are produced from pri-miRNAs through at least two sequential
processing steps by RNase III-type endonucleases In animals pri-miRNAs are first
processed at the bottom portion of the stem by Drosha a nuclear-localized RNase III
enzyme to liberate pre-miRNAs The pre-miRNAs are then exported to the cytoplasm
with the help of exportin-5The pre-miRNAs are further processed by Dicer another
RNaseIII enzyme to a duplex consisting of the miRNA and the antisense strands
(miRNA) Since plants do not have any Drosha homologs the two sequential
processing steps seem to be carried out by DCL1 a Dicer homolog in the nucleus
(Kurihara amp Watanabe 2004 de Alba et al 2013 Rogers amp Chen 2013)
The processing of pri-miRNAs to pre-miRNAs by DCL1 is assisted by two
other proteins HYPONASTY LEAVES1 (HYL1) and SERRATE (SE) HYL1 belongs to a
family of double-stranded RNA (dsRNA) binding protein and interacts with DCL1 in
vitro and in vivo (Lu amp Fedoroff 2000 Hiraguri et al 2005 de Alba et al 2013
Rogers amp Chen 2013) Loss-of-function mutations in the HYL1 gene result in
decreased accumulation of miRNAs and concomitant accumulation of pri-miRNAs
(Han et al 2004 Vazquez et al 2004a Kurihara et al 2006) SE a C2H2 zinc finger
protein interacts with DCL1 and HYL1 and plays a role in the processing of pri-
miRNAs to pre-miRNAs (Lobbes et al 2006 Yang L et al 2006) In addition the
nuclear cap-binding complex proteins CBP20 and CBP80 that are known as ABA
HYPER SENSITIVE 1(ABH1) have been shown to be required for proper miRNA
processing (Gregory et al 2008 Laubinger et al 2008) Recently it has been
demonstrated that DCL1 can release 21-nt RNAs from dsRNA as well as from
synthetic miR167 precursor RNAs in vitro However this cleavage is error prone and
inefficient in the absence of HYL1 and SE which accelerate the rate of DCL1-mediated
cleavage and promote accurate processing of miRNAs (Dong et al 2008 Rogers amp
Chen 2013)
11343 Methylation of the miRNAmiRNAduplex
After the miRNAmiRNAduplexes are released from the pre-miRNAs by the DCL1
activities the duplexes are methylated at the 2primeOH of the3prime-ends by HUA ENHANCER1
(HEN1) a small RNA methyltransferase (Yu et al 2005 Yang Z et al 2006 Rogers
amp Chen 2013 ) This step which is distinct from the RNase III-mediated processing
Chapter 1 Introduction and Review of Literature
37
step distinguishes the biogenesis of plant miRNAs from that of animal miRNAs
Mutations in the HEN1 gene were first isolated in a morphological screening for floral
development although the HEN1 mutants display pleiotropic phenotypes similar to the
developmental defects observed in the DCL1 mutants (Chen et al 2002 Park et al
2002 Rogers amp Chen 2013)
Figure 18 Biogenesis of microRNA
A Biogenesis of plant MicroRNA B Biogenesis of metazoananimal MicroRNA
The miRNA (red) is incorporated into RISC and the miRNA(blue) in degraded
This enzyme has two dsRNA-binding domains and nuclear localization signal It
is also conserved in fungi and is required for the processing of siRNAs as well as of
miRNAs (Park et al 2002 Boutet et al 2003 Xie et al 2003) Although the HEN1
protein is localized to the nucleus its methylation activity in the cytoplasm cannot be
ignored (Xie et al 2004 Rogers amp Chen 2013)
Chapter 1 Introduction and Review of Literature
38
The methylation of the miRNAmiRNA duplex is required to protect miRNAs
against the 3prime-end uridylation activity and subsequent degradation (Li et al 2005) In
the hen1 mutants accumulation of miRNAs is reduced to a lower level Also the
miRNAs appear to be heterogeneous in their sizes such that a ladder of bands rather
than a single species is observed for most miRNAs in the mutants (Li et al 2005)
MiRNA cloning and sequencing analyses revealed the presence of a number of U
residues at the 3prime-end of miRNAs in the hen1 mutants Moreover these nucleotides do
not correspond to the miRNAs in the pre-miRNAs indicating that these nucleotides are
added after the processing of the miRNAs from pre-miRNAs (Li et al 2005)
Uridylation of RNAs has also been proven to be correlated with RNA decay (Shen amp
Goodman 2004 Mullen amp Marzluff 2008) suggesting that uridylation attracts an
exonuclease which degrades miRNAs in plants
11344 MiRNA incorporation in to the RISC and its nuclear export
The methylated miRNAmiRNA duplexes undergo RISC assembly In this process
the miRNA of the duplexes is selectively incorporated into the RISC and the miRNA
appears to be removed and subsequently degraded (Hammond et al 2000 Hutvagner
amp Zamore 2002 Schwarz et al 2003 de Alba et al 2013 Rogers amp Chen 2013)
The ARGONAUTE 1(AGO1) proteins are core components of the RISC complex
They contain two structural domains the N-terminal PAZ domain that binds to the 3prime-
end of single-stranded RNAs and the C-terminal piwi domains that has a structure
similar to that of RNase H (Cerutti et al 2000 Parker et al 2004) Several AGO
proteins possess endonucleolytic activities within the PIWI domain and cleave target
mRNAs in the middle of the site complementary to miRNA (Liu et al 2004 Meister et
al 2004 Miyoshi et al 2005) Mutations in the AGO1 gene cause miRNA target
genes to be ectopically expressed and the levels of some miRNAs are reduced in the
mutants (Kidner amp Martienssen 2004 Vaucheret et al 2004 Kidner amp Martienssen
2005 Ronemus et al 2006) Moreover the phenotype of the AGO1 hypomorphic
alleles display defects in lateral organ polarity and leaf and flower morphology as
observed in the dcl1 mutants and null AGO1 alleles are embryonically lethal
supporting the close relationship between the AGO proteins and miRNA processing
(Kidner amp Martienssen 2004 Vaucheret et al 2004 de Alba et al 2013 Rogers amp
Chen 2013)
The production of miRNA miRNA duplexes occur in the nucleus while
Chapter 1 Introduction and Review of Literature
39
miRNA-RISC complexes are present in the cytoplasm where target mRNAs are
located This discordance of functional sites requires an export step during miRNA
biogenesis HASTY the Arabidopsis homolog of exportin-5 is thought to be involved
in miRNA nuclear export (Bollman et al 2003 Park et al 2005)The hasty mutant
exhibits pleiotropic defects in plant development and reduced accumulation of most
miRNAs as in the DCL1 or AGO1 mutants Howeverit is unclear whether the HASTY
proteins export the miRNA miRNA duplexes or the miRNA-RISC complexes in
plants
11345 Feedback regulation of miRNA biogenesis
MiRNA biogenesis is under feedback regulation such that the DCL1 and AGO1 genes
are themselves regulated by miRNAs DCL1 is itself a target of miR162 which leads to
the cleavage of the DCL1 mRNA (Xie et al 2003 de Alba et al 2013) Consistent
with this the levels of DCL1 mRNA are elevated in the mutants of miRNA processing
genes in which the miR162 abundance is reduced In addition there is a predicted
hairpin structure of miR838 in the 14th
intron of the DCL1 primary transcript
(Rajagopalan et al 2006) This prediction implies that DCL1-mediated processing
on this site can result in the cleavage of the DCL1 primary transcript into two truncated
fragments the N-terminal capped fragment and the C-terminal polyadenylated
fragment If this is the case it is possible that miRNA biogenesis competes with the
splicing event of the DCL1 pre-mRNA
The AGO1 is also regulated by miRNA There is a complementary site for
miR168 binding in the AGO1 mRNA and the transcript level of the AGO1 mRNA is
reduced through an mRNA cleavage mechanism (Vaucheret et al 2006 de Alba et al
2013 Rogers amp Chen 2013) indicating that the AGO1 mRNA levels are tightly
controlled by the levels of the AGO1 protein
1135 Mechanism of miRNA action
11351 MiRNA-directed mRNA cleavage
By forming the miRNA-RISC assembly miRNAs can direct the RISC to regulate gene
expression by two post transcriptional mechanisms mRNA cleavage or translational
repression (Bartel 2004 Jones-Rhoades et al 2006 Dalmay 2013) The degree
of miRNA-mRNA complementarity plays an important role for the
determination of the mechanism to be used A general rule is that while perfect or
Chapter 1 Introduction and Review of Literature
40
near-perfect complementarity induces mRNA cleavage central mismatches trigger
translational repression (Carrington amp Ambros 2003 Jones-Rhoades amp Bartel 2004)
Unlike most animal miRNAs plant miRNAs have near-perfect or perfect
complementarity to their targets and mRNA cleavage is assumed to be a predominant
mechanism by which miRNAs regulate gene expression in plants
In RNA cleavage mechanism miRNAs guide the AGO component of RISC to
cleave a single phosphodiester bond of the target mRNA within the miRNA-binding
site (Llave et al 2002b Tang et al 2003) The truncated fragments are then released
and degraded freeing the RISC to recognize and cleave another target mRNA This has
been validated by the detection of 3prime cleavage products that have 5prime ends that start at the
middle of the complementary regions The mRNA cleavage products are then further
degraded by other mechanisms The 3prime cleavage products of some miRNA targets are
degraded by the 5prime-3prime exonuclease XRN4 an Arabidopsis homolog of the major Yeast
mRNA degrading exonuclease Xrn1p It has been observed that the 3prime cleavage
products of some target mRNAs were accumulated in the xrn4 mutants (Souret et al
2004 Dalmay 2013) However the 3prime cleavage products of other miRNA targets do
not accumulate in the xrn4 mutants indicating that there are also XRN4 independent
mechanisms The 5prime cleavage products are typically untraceable by RNA gel blot
hybridization but can be detected by sensitive PCR based methods It has been found
that the 5prime cleavage products tend to obtain an oligo U tail at the 3prime ends This 3prime U tail
is correlated with shortening of the 5prime cleavage products from the 5prime end leading to the
conclusion that the 3prime uridylation causes 5 prime- 3 prime exonucleolytic decay of the 5prime cleavage
products (Shen amp Goodman 2004)
DCL1 HYL1 and SE interact with each other and are co-localized in the
nuclear bodies termed Dicing bodies or D-bodies in which some pri-miRNA shave
been found suggesting that D-bodies serve as a center for active miRNA processing
(Fang amp Spector 2007 Fujioka et al 2007 Song et al 2007) The miRNA
processing site appears to be distinct from the Cajal bodies that have previously been
found as a siRNA processing site (Pontes amp Pikaard 2008) The D-bodies include
SmD3 and SmB proteins that are found in the Cajal bodies However At collin an
additional marker for the Cajal bodies is not found in the D-bodies Moreover the D-
bodies are present in an At collin mutant lacking the Cajal bodies The pri-miRNAs of
miR171A and miR159A genes have been found to be co-localized with HYL1 and
Chapter 1 Introduction and Review of Literature
41
DCL1 in the D- bodies that are distinct from the Cajal bodies (Song et al 2007)
11352 MiRNA-directed translational repression
Translation repression is another mechanism exerted by plant miRNAs The regulatory
mechanism of miR172 which regulates flowering time and floral organ identity is
consistent with translation repression MiR172 over production does not affect the
transcript levels of APETALA2 (AP2) and AP2- like target mRNAs Instead the AP2
protein level is reduced in the miR172 over expressing mutant (Aukerman amp Sakai
2003 Chen 2004 Dalmay 2013) It was later shown that miR172 can also direct the
cleavage of the target mRNAs although the steady-state mRNA level is unchanged due
to feedback regulation (Schwab et al 2005 Jung et al 2007) It is clear that miR172
regulates its target genes primarily by translational repression rather than mRNA
cleavage Recently it has been reported that miR156157 and miR854 also regulate
their target genes through translational repression (Arteaga-Vazquez et al 2006
Gandikota et al 2007 Dalmay 2013) It was once thought that translational repression
is an exception to the rule However experimental evidence suggest a widespread
coexistence of translational repression and mRNA cleavage (Brodersen et al 2008)
1136 Regulatory roles of miRNAs in plant development
The miRNA target prediction has been a difficult task in order to obtain regulatory
targets without bringing in too many false predictions Plant growth and developmental
processes regulated by miRNAs are quite diverse and enormous Furthermore plant
miRNAs work cooperatively or antagonistically to establish a balanced regulation This
notion is well consistent with the findings that mutations in the regulators of miRNA
biogenesis transport and processing such as AGO1 DCL1 HYL1 HEN1 ABH1
and HASTY cause pleiotropic phenotypes (Mallory amp Vaucheret 2006 de Alba et al
2013) To date the roles of miRNAs have been mostly deduced from obtaining miRNA
over expressing transgenic plants or gain-of-function mutants in which miRNA-
resistant target genes are ectopically expressed
11361 Phase transition
Plants pass through a series of developmental phase transitions during their life span
beginning with seed germination continuing through vegetative phase change
reproductive phase change flowering initiation and finally seed production for the next
generation Because of their importance in understanding plant developmental
Chapter 1 Introduction and Review of Literature
42
processes genes regulating developmental transitions have been extensively studied
and underlying molecular mechanisms have been explored in various plant species
Majority of researches were mainly focused on vegetative phase change and
reproductive transition in Arabidopsis and Maize (Poethig 2003 Baurle amp Dean
2006) Particularly the mutations of the genes encoding various regulators of miRNA
biogenesis and transport such as HST SE and AGO exhibit altered juvenile to adult
vegetative phase change and vegetative to reproductive change (Hunter et al 2003
Peragine et al 2004 Vazquez et al 2004a Jones-Rhoades et al 2006 de Alba et al
2013)
Over-expression of the miR156a gene causes late flowering and delays
vegetative phase change (Wu amp Poethig 2006 Gandikota et al 2007 Wang et al
2008) It has been shown that miR156 negatively regulates a group of genes encoding
SQUAMOSA PROMOTER-BINDING PROTEIN-LIKE (Lewis et al 2007 de Alba et
al 2013 Rogers amp Chen 2013) transcription factors such as SPL3 SPL4 and SPL5
that promote flowering initiation In contrast transgenic plants over expressing
miR156-resistant SPL345 genes are early flowering Interestingly the endogenous
level of miR156 is temporally regulated In the early juvenile vegetative phase the
level of miR156 is very high but decreases rapidly before the onset of the adult juvenile
phase (Wu amp Poethig 2006)
The Arabidopsis early activation tagged (eat-D) mutant exhibits early flowering
with disrupted floral structure The mutant phenotypes are caused by over expression of
the miR172a-2gene (Aukerman amp Sakai 2003) MiR172 regulates a small group of
genes encoding APETALLA2 (AP2)-like transcription factors such as TARGET OF
EAT1 (TOE1) TOE2 SCHLAFMUTZE (SMZ) and SCHNARCHZAPFEN(SNZ)
TOE1 TOE2 SMZ and SNZ have been shown to participate in their productive phase
change by repressing a floral integrator FLOWERING LOCUS T (FT)(Jung et al
2007) Consistent with this toe1-2D 35STOE2 35SSMZ and 35SSNZ plants
exhibit late flowering (Jung et al 2007) MiR172 is also regulated temporally and
highly expressed in the adult vegetative phase both in Arabidopsis and maize (Chuck et
al 2007) Because the temporal expression patterns of miR156 and miR172 are
complementary to each other it has been suggested that the two miRNAs interact with
each other in regulating phase change (Chuck et al 2009 de Alba et al 2013) In
support of this view it has been shown that miR172 is down-regulated in the
Chapter 1 Introduction and Review of Literature
43
Corngrass1(Cg1) mutant in which miR156 is overproduced (Chuck et al 2007 de
Alba et al 2013) However there is no direct evidence for the direct linkage between
miR156 and miR172 It is also envisioned that miR156 and miR172 would not be
directly related For example miR156 regulates plastochron index that is the inverse of
leaf initiation rate and thus frequently used to analyze temporal patterns of plant
development In contrast miR172 does not affect plastochron (Wang et al 2008) The
down-regulation of miR172 in the maize miR156-over producing mutants would be
due to the prolonged juvenile vegetative phase in the mutants
Another miRNA that regulates flowering initiation is miR159 It regulates
MYB33 and MYB65 which are closely related to the barley GAMY B transcription
factor that responds to gibberellic acid (GA) The level of miR159 is elevated in GA-
treated seedlings but reduced in the GA biosynthetic ga1 mutant Transgenic plants
over producing miR159 exhibit delayed floral induction under short days (Achard et
al 2004) It is been suggested that MYB33 mediates GA signal to regulate LEAFY
(LFY) These observations indicate that miR159 plays an important role in the GA
signaling pathways that regulate proper floral induction under short days (Achard et al
2004)
11362 Floral development
Arabidopsis flowers consist of four distinct organs They arise in a characteristic
pattern within concentric rings called whorls from outside to inside four sepals four
petals six stamens and the central pistil consisting of two carpels Identities of the
floral organs are determined by three classes of regulatory genes classA classB and
class C genes (Krizek amp Fletcher 2005) The A and C class genes act antagonistically
to restrict their expression domains (Bowman et al 1991)
It is notable that miR172 also regulates floral architecture in addition to its role
in the timing of flowering initiation MiR172 inhibits the translation of the AP2 mRNA
(Chen 2004) Transgenic plants overproducing miR172 not only exhibit early
flowering but also show abnormal floral development which is quite similar to those
of the class A gene mutants such as the ap2 mutants (Aukerman amp Sakai 2003 de
Alba et al 2013) When the activity of the class A genes is inactivated that of the class
C genes such as AGAMOUS (AG) is elevated As a result the miR172- overproducing
transgenic plants exhibit no petals along with homeotic transformation of the sepals to
Chapter 1 Introduction and Review of Literature
44
the carpels (Aukerman amp Sakai 2003 Chen 2004)
Interestingly miR166 also regulates floral architecture The role of miR165166
in the regulation of meristem activity is closely linked to floral meristem formation and
organization (Zhao et al 2007 Miyashima et al 2013) Floral structure is severely
disrupted in the miR165166-overproducing mutants such as men1 and jba-1D in
which miR166 is overproduced (Williams et al 2005b Jung amp Park 2007) The men1
and jba-1D mutants have smaller gynoecium and reduced carpel number In an extreme
case the jba-1D homozygous plants lack visible carpels (Williams et al 2005b)
Similar phenotypes have been observed in the miR165 overproducers (Zhao et al
2007) It has been suggested that the phenotypes are possibly caused by altered AG
expression in the floral meristem (Jung amp Park 2007 Turchi et al 2013)
Other miRNAs also affect floral development Overproduction of miRNAs
involved in auxin signaling also affects floral architecture Transgenic plants
overproducing miR167 have defects in the formation of ovule and anther with reduced
fertility (Ru et al 2006) It has been shown that miR167 targets ARF6 and ARF8 that
play a role in gynoecium and stamen development and in regulating fertility (Ru et al
2006 Wu et al 2006) These observations suggest that auxin signals are also closely
related to floral organogenesis In addition transgenic plants over-producing miR164
and the cuc1cuc2 double mutants show fused sepals and reduced petals (Laufs et al
2004 Turchi et al 2013) suggesting that miR164 is also related to floral meristem
activity and boundary specification of the meristematic region
11363 Shoot apical meristem development
Shoot apical meristem comprises self-maintaining totipotent cells The shoot apical
meristem activity affects diverse aspects of plant developmental processes including
leaf development vascular development and lateral organ polarity (Bowman et al
2002) Because miR165166 plays a primary role in meristem formation
overproduction of miR165166 and multiple knockout mutants of its target genes
exhibit pleiotropic pheonotypes (McConnell et al 2001 Emery et al 2003 Kim et al
2005 Williams et al 2005b)
The targets of miR165166 include genes encoding the homeo domain-leucine
zipper (HD-ZIP) class III transcription factors such as PHABULOSA(PHB)
PHAVOLUTA (PHV) REVOLUTA(REV) ATHB15CORONA (CNA) and ATHB8
Chapter 1 Introduction and Review of Literature
45
PHB PHV and ATHB15 have overlapping functions in controlling meristem
development (Turchi et al 2013) Indeed the phb phv athb15 triple mutants have an
enlarged shoot apical meristem Consistent with this the miR166- overproducing men1
and jba-1D mutants exhibit enlarged shoot apical meristems (Kim et al 2005
Williams et al 2005b Turchi et al 2013) In the mutants the shoot apical meristems
are multiplied and large
The development of shoot apical meristems is also regulated by the WUSCHEL
(WUS)ndashCLAVATA (CLV) signaling pathway WUS positively regulates cell
proliferation In contrast CLV3 which is expressed in stem cells limits the WUS
expression and thus inhibits cell proliferation in the SAM (Sharma et al 2003)
Consistent with this notion WUS is expressed to a higher level in jba-1D
explaining the ectopic enlargement of the SAM in the mutant (Williams et al 2005)
While WUS is closely related to miR165166 in the shoot apical meristem
development the underlying molecular mechanisms are currently unclear It has been
observed that the miR166-over producing men1 plants have an arrested meristem
development (Jung amp Park 2007) In addition the heterozygous men1+ and the
homozygous men1 plants show different expression patterns of WUS and CLV3 It
appears that miR166 regulates WUS indirectly and influences the expressional balance
between WUS and CLV3 through a negative feedback loop (Jung amp Park 2007 de
Alba et al 2013)
The phv phb athb15 triple mutant has an enlarged shoot apical meristems the
rev phb phv mutant does not have functional shoot apical meristems (Prigge et al
2005) This distinction may be explained by the fact that while miR166 targets
primarily PHB PHV and ATHB15 miR165 targets all the five HD-ZIP III genes (Zhou
et al 2007) Consistent with this transgenic plants overproducing miR166 are distinct
from those overproducing miR165 The miR165-overproducing transgenic plants lack
functional shoot apical meristem and a pin-like structure is formed at the apical region
of the mutant These phenotypes are quite similar to rev phb phv triple mutant (Zhou et
al 2007 Liu et al 2013) The phenotypic distinction may because by the difference
of the cleavage efficiencies of the target mRNAs In accordance with this notion a few
35S miR166a transgenic lines exhibit no shoot apical meristems The men1
homozygous plants also show arrested meristem development (Jung amp Park 2007)
Chapter 1 Introduction and Review of Literature
46
MiR164 cleaves the CUC1 and CUC2 mRNAs as well as the NAC1 mRNA
CUC1 and CUC2 determines the organ boundary in the meristem Phenotypes of
transgenic plants that overproduce miR164 are similar to those of the cuc1cuc2 double
mutant that exhibits embryonic developmental defects such as cup-shaped cotyledons
and fused cotyledons (Laufs et al 2004) When a miR164-resistant CUC2 is
expressed the boundary domain is enlarged CUC also plays a role in embryonic shoot
meristem formation by regulating the SHOOT MERISTEMLESS gene It was therefore
concluded that miR164-mediated cleavage of CUC1 and CUC2 mRNAs determines the
sizes of the boundary domains in the shoot apical meristem and regulates separation of
the organs by restricting cell proliferation (Laufs et al 2004 de Alba et al 2013)
11364 Leaf development
The role of miR319 has been extensively studied in leaf development A dominant
jaw-D mutant overproducing miR319 shows uneven curved leaves with enlarged size
and five TCP genes which are closely related to the CINCINNATA (CIN) gene in
Antirrhinum majus are down regulated in the mutant suggesting that miR319 mediated
regulation of the TCP transcription factor genes are important for the final step of
leaf shaping (Palatnik et al 2003 Naz et al 2013) In addition to leaf shape and size
leaf polarity is also specified by miRNA MiR166-mediated cleavage of the HD-ZIPIII
mRNAs is a well to establish leaf polarity This discovery originates from the gain-of-
function mutants such as phb-1d and phv-1d wherein point mutations in the
complementary sites to miRNA helps the mRNA to evade or resist from miRNA-
mediated cleavage (McConnell et al 2001 Emery et al 2003 Naz et al 2013)
Plant leaves have a dorso-ventral polarity consisting of the adaxial (upper
surface) and abaxial (lower surface) sides The adaxial side is closer to the shoot
apical meristem Adaxial identity is specified by functionally redundant class III HD-
ZIP family genes such as PHB PHV and REV Ectopic expression of each genes
result in adaxialized leaves Dominant phb-1d and phv-1d mutants exhibit
transformation of the abaxial to adaxial fate and have rod-shaped leaves In contrast
miR165166-overproducing plants show pin-like cotyledons radicalized leaves and
abaxialized leaves (Chitwood et al 2007 Pattanayak et al 2013)
Leaf asymmetry is determined by miR166-mediated restriction of the
expression domains of the HD-ZIPIII genes The HD-ZIPIII genes are expressed in the
Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
36
transcription is processed by RNA polymerase II (Pol II) enzymes and the pri-
miRNAs are 5prime-capped and 3prime-polyadenylated (Lee et al 2004 Xie Z et al 2005)
Mature miRNAs are produced from pri-miRNAs through at least two sequential
processing steps by RNase III-type endonucleases In animals pri-miRNAs are first
processed at the bottom portion of the stem by Drosha a nuclear-localized RNase III
enzyme to liberate pre-miRNAs The pre-miRNAs are then exported to the cytoplasm
with the help of exportin-5The pre-miRNAs are further processed by Dicer another
RNaseIII enzyme to a duplex consisting of the miRNA and the antisense strands
(miRNA) Since plants do not have any Drosha homologs the two sequential
processing steps seem to be carried out by DCL1 a Dicer homolog in the nucleus
(Kurihara amp Watanabe 2004 de Alba et al 2013 Rogers amp Chen 2013)
The processing of pri-miRNAs to pre-miRNAs by DCL1 is assisted by two
other proteins HYPONASTY LEAVES1 (HYL1) and SERRATE (SE) HYL1 belongs to a
family of double-stranded RNA (dsRNA) binding protein and interacts with DCL1 in
vitro and in vivo (Lu amp Fedoroff 2000 Hiraguri et al 2005 de Alba et al 2013
Rogers amp Chen 2013) Loss-of-function mutations in the HYL1 gene result in
decreased accumulation of miRNAs and concomitant accumulation of pri-miRNAs
(Han et al 2004 Vazquez et al 2004a Kurihara et al 2006) SE a C2H2 zinc finger
protein interacts with DCL1 and HYL1 and plays a role in the processing of pri-
miRNAs to pre-miRNAs (Lobbes et al 2006 Yang L et al 2006) In addition the
nuclear cap-binding complex proteins CBP20 and CBP80 that are known as ABA
HYPER SENSITIVE 1(ABH1) have been shown to be required for proper miRNA
processing (Gregory et al 2008 Laubinger et al 2008) Recently it has been
demonstrated that DCL1 can release 21-nt RNAs from dsRNA as well as from
synthetic miR167 precursor RNAs in vitro However this cleavage is error prone and
inefficient in the absence of HYL1 and SE which accelerate the rate of DCL1-mediated
cleavage and promote accurate processing of miRNAs (Dong et al 2008 Rogers amp
Chen 2013)
11343 Methylation of the miRNAmiRNAduplex
After the miRNAmiRNAduplexes are released from the pre-miRNAs by the DCL1
activities the duplexes are methylated at the 2primeOH of the3prime-ends by HUA ENHANCER1
(HEN1) a small RNA methyltransferase (Yu et al 2005 Yang Z et al 2006 Rogers
amp Chen 2013 ) This step which is distinct from the RNase III-mediated processing
Chapter 1 Introduction and Review of Literature
37
step distinguishes the biogenesis of plant miRNAs from that of animal miRNAs
Mutations in the HEN1 gene were first isolated in a morphological screening for floral
development although the HEN1 mutants display pleiotropic phenotypes similar to the
developmental defects observed in the DCL1 mutants (Chen et al 2002 Park et al
2002 Rogers amp Chen 2013)
Figure 18 Biogenesis of microRNA
A Biogenesis of plant MicroRNA B Biogenesis of metazoananimal MicroRNA
The miRNA (red) is incorporated into RISC and the miRNA(blue) in degraded
This enzyme has two dsRNA-binding domains and nuclear localization signal It
is also conserved in fungi and is required for the processing of siRNAs as well as of
miRNAs (Park et al 2002 Boutet et al 2003 Xie et al 2003) Although the HEN1
protein is localized to the nucleus its methylation activity in the cytoplasm cannot be
ignored (Xie et al 2004 Rogers amp Chen 2013)
Chapter 1 Introduction and Review of Literature
38
The methylation of the miRNAmiRNA duplex is required to protect miRNAs
against the 3prime-end uridylation activity and subsequent degradation (Li et al 2005) In
the hen1 mutants accumulation of miRNAs is reduced to a lower level Also the
miRNAs appear to be heterogeneous in their sizes such that a ladder of bands rather
than a single species is observed for most miRNAs in the mutants (Li et al 2005)
MiRNA cloning and sequencing analyses revealed the presence of a number of U
residues at the 3prime-end of miRNAs in the hen1 mutants Moreover these nucleotides do
not correspond to the miRNAs in the pre-miRNAs indicating that these nucleotides are
added after the processing of the miRNAs from pre-miRNAs (Li et al 2005)
Uridylation of RNAs has also been proven to be correlated with RNA decay (Shen amp
Goodman 2004 Mullen amp Marzluff 2008) suggesting that uridylation attracts an
exonuclease which degrades miRNAs in plants
11344 MiRNA incorporation in to the RISC and its nuclear export
The methylated miRNAmiRNA duplexes undergo RISC assembly In this process
the miRNA of the duplexes is selectively incorporated into the RISC and the miRNA
appears to be removed and subsequently degraded (Hammond et al 2000 Hutvagner
amp Zamore 2002 Schwarz et al 2003 de Alba et al 2013 Rogers amp Chen 2013)
The ARGONAUTE 1(AGO1) proteins are core components of the RISC complex
They contain two structural domains the N-terminal PAZ domain that binds to the 3prime-
end of single-stranded RNAs and the C-terminal piwi domains that has a structure
similar to that of RNase H (Cerutti et al 2000 Parker et al 2004) Several AGO
proteins possess endonucleolytic activities within the PIWI domain and cleave target
mRNAs in the middle of the site complementary to miRNA (Liu et al 2004 Meister et
al 2004 Miyoshi et al 2005) Mutations in the AGO1 gene cause miRNA target
genes to be ectopically expressed and the levels of some miRNAs are reduced in the
mutants (Kidner amp Martienssen 2004 Vaucheret et al 2004 Kidner amp Martienssen
2005 Ronemus et al 2006) Moreover the phenotype of the AGO1 hypomorphic
alleles display defects in lateral organ polarity and leaf and flower morphology as
observed in the dcl1 mutants and null AGO1 alleles are embryonically lethal
supporting the close relationship between the AGO proteins and miRNA processing
(Kidner amp Martienssen 2004 Vaucheret et al 2004 de Alba et al 2013 Rogers amp
Chen 2013)
The production of miRNA miRNA duplexes occur in the nucleus while
Chapter 1 Introduction and Review of Literature
39
miRNA-RISC complexes are present in the cytoplasm where target mRNAs are
located This discordance of functional sites requires an export step during miRNA
biogenesis HASTY the Arabidopsis homolog of exportin-5 is thought to be involved
in miRNA nuclear export (Bollman et al 2003 Park et al 2005)The hasty mutant
exhibits pleiotropic defects in plant development and reduced accumulation of most
miRNAs as in the DCL1 or AGO1 mutants Howeverit is unclear whether the HASTY
proteins export the miRNA miRNA duplexes or the miRNA-RISC complexes in
plants
11345 Feedback regulation of miRNA biogenesis
MiRNA biogenesis is under feedback regulation such that the DCL1 and AGO1 genes
are themselves regulated by miRNAs DCL1 is itself a target of miR162 which leads to
the cleavage of the DCL1 mRNA (Xie et al 2003 de Alba et al 2013) Consistent
with this the levels of DCL1 mRNA are elevated in the mutants of miRNA processing
genes in which the miR162 abundance is reduced In addition there is a predicted
hairpin structure of miR838 in the 14th
intron of the DCL1 primary transcript
(Rajagopalan et al 2006) This prediction implies that DCL1-mediated processing
on this site can result in the cleavage of the DCL1 primary transcript into two truncated
fragments the N-terminal capped fragment and the C-terminal polyadenylated
fragment If this is the case it is possible that miRNA biogenesis competes with the
splicing event of the DCL1 pre-mRNA
The AGO1 is also regulated by miRNA There is a complementary site for
miR168 binding in the AGO1 mRNA and the transcript level of the AGO1 mRNA is
reduced through an mRNA cleavage mechanism (Vaucheret et al 2006 de Alba et al
2013 Rogers amp Chen 2013) indicating that the AGO1 mRNA levels are tightly
controlled by the levels of the AGO1 protein
1135 Mechanism of miRNA action
11351 MiRNA-directed mRNA cleavage
By forming the miRNA-RISC assembly miRNAs can direct the RISC to regulate gene
expression by two post transcriptional mechanisms mRNA cleavage or translational
repression (Bartel 2004 Jones-Rhoades et al 2006 Dalmay 2013) The degree
of miRNA-mRNA complementarity plays an important role for the
determination of the mechanism to be used A general rule is that while perfect or
Chapter 1 Introduction and Review of Literature
40
near-perfect complementarity induces mRNA cleavage central mismatches trigger
translational repression (Carrington amp Ambros 2003 Jones-Rhoades amp Bartel 2004)
Unlike most animal miRNAs plant miRNAs have near-perfect or perfect
complementarity to their targets and mRNA cleavage is assumed to be a predominant
mechanism by which miRNAs regulate gene expression in plants
In RNA cleavage mechanism miRNAs guide the AGO component of RISC to
cleave a single phosphodiester bond of the target mRNA within the miRNA-binding
site (Llave et al 2002b Tang et al 2003) The truncated fragments are then released
and degraded freeing the RISC to recognize and cleave another target mRNA This has
been validated by the detection of 3prime cleavage products that have 5prime ends that start at the
middle of the complementary regions The mRNA cleavage products are then further
degraded by other mechanisms The 3prime cleavage products of some miRNA targets are
degraded by the 5prime-3prime exonuclease XRN4 an Arabidopsis homolog of the major Yeast
mRNA degrading exonuclease Xrn1p It has been observed that the 3prime cleavage
products of some target mRNAs were accumulated in the xrn4 mutants (Souret et al
2004 Dalmay 2013) However the 3prime cleavage products of other miRNA targets do
not accumulate in the xrn4 mutants indicating that there are also XRN4 independent
mechanisms The 5prime cleavage products are typically untraceable by RNA gel blot
hybridization but can be detected by sensitive PCR based methods It has been found
that the 5prime cleavage products tend to obtain an oligo U tail at the 3prime ends This 3prime U tail
is correlated with shortening of the 5prime cleavage products from the 5prime end leading to the
conclusion that the 3prime uridylation causes 5 prime- 3 prime exonucleolytic decay of the 5prime cleavage
products (Shen amp Goodman 2004)
DCL1 HYL1 and SE interact with each other and are co-localized in the
nuclear bodies termed Dicing bodies or D-bodies in which some pri-miRNA shave
been found suggesting that D-bodies serve as a center for active miRNA processing
(Fang amp Spector 2007 Fujioka et al 2007 Song et al 2007) The miRNA
processing site appears to be distinct from the Cajal bodies that have previously been
found as a siRNA processing site (Pontes amp Pikaard 2008) The D-bodies include
SmD3 and SmB proteins that are found in the Cajal bodies However At collin an
additional marker for the Cajal bodies is not found in the D-bodies Moreover the D-
bodies are present in an At collin mutant lacking the Cajal bodies The pri-miRNAs of
miR171A and miR159A genes have been found to be co-localized with HYL1 and
Chapter 1 Introduction and Review of Literature
41
DCL1 in the D- bodies that are distinct from the Cajal bodies (Song et al 2007)
11352 MiRNA-directed translational repression
Translation repression is another mechanism exerted by plant miRNAs The regulatory
mechanism of miR172 which regulates flowering time and floral organ identity is
consistent with translation repression MiR172 over production does not affect the
transcript levels of APETALA2 (AP2) and AP2- like target mRNAs Instead the AP2
protein level is reduced in the miR172 over expressing mutant (Aukerman amp Sakai
2003 Chen 2004 Dalmay 2013) It was later shown that miR172 can also direct the
cleavage of the target mRNAs although the steady-state mRNA level is unchanged due
to feedback regulation (Schwab et al 2005 Jung et al 2007) It is clear that miR172
regulates its target genes primarily by translational repression rather than mRNA
cleavage Recently it has been reported that miR156157 and miR854 also regulate
their target genes through translational repression (Arteaga-Vazquez et al 2006
Gandikota et al 2007 Dalmay 2013) It was once thought that translational repression
is an exception to the rule However experimental evidence suggest a widespread
coexistence of translational repression and mRNA cleavage (Brodersen et al 2008)
1136 Regulatory roles of miRNAs in plant development
The miRNA target prediction has been a difficult task in order to obtain regulatory
targets without bringing in too many false predictions Plant growth and developmental
processes regulated by miRNAs are quite diverse and enormous Furthermore plant
miRNAs work cooperatively or antagonistically to establish a balanced regulation This
notion is well consistent with the findings that mutations in the regulators of miRNA
biogenesis transport and processing such as AGO1 DCL1 HYL1 HEN1 ABH1
and HASTY cause pleiotropic phenotypes (Mallory amp Vaucheret 2006 de Alba et al
2013) To date the roles of miRNAs have been mostly deduced from obtaining miRNA
over expressing transgenic plants or gain-of-function mutants in which miRNA-
resistant target genes are ectopically expressed
11361 Phase transition
Plants pass through a series of developmental phase transitions during their life span
beginning with seed germination continuing through vegetative phase change
reproductive phase change flowering initiation and finally seed production for the next
generation Because of their importance in understanding plant developmental
Chapter 1 Introduction and Review of Literature
42
processes genes regulating developmental transitions have been extensively studied
and underlying molecular mechanisms have been explored in various plant species
Majority of researches were mainly focused on vegetative phase change and
reproductive transition in Arabidopsis and Maize (Poethig 2003 Baurle amp Dean
2006) Particularly the mutations of the genes encoding various regulators of miRNA
biogenesis and transport such as HST SE and AGO exhibit altered juvenile to adult
vegetative phase change and vegetative to reproductive change (Hunter et al 2003
Peragine et al 2004 Vazquez et al 2004a Jones-Rhoades et al 2006 de Alba et al
2013)
Over-expression of the miR156a gene causes late flowering and delays
vegetative phase change (Wu amp Poethig 2006 Gandikota et al 2007 Wang et al
2008) It has been shown that miR156 negatively regulates a group of genes encoding
SQUAMOSA PROMOTER-BINDING PROTEIN-LIKE (Lewis et al 2007 de Alba et
al 2013 Rogers amp Chen 2013) transcription factors such as SPL3 SPL4 and SPL5
that promote flowering initiation In contrast transgenic plants over expressing
miR156-resistant SPL345 genes are early flowering Interestingly the endogenous
level of miR156 is temporally regulated In the early juvenile vegetative phase the
level of miR156 is very high but decreases rapidly before the onset of the adult juvenile
phase (Wu amp Poethig 2006)
The Arabidopsis early activation tagged (eat-D) mutant exhibits early flowering
with disrupted floral structure The mutant phenotypes are caused by over expression of
the miR172a-2gene (Aukerman amp Sakai 2003) MiR172 regulates a small group of
genes encoding APETALLA2 (AP2)-like transcription factors such as TARGET OF
EAT1 (TOE1) TOE2 SCHLAFMUTZE (SMZ) and SCHNARCHZAPFEN(SNZ)
TOE1 TOE2 SMZ and SNZ have been shown to participate in their productive phase
change by repressing a floral integrator FLOWERING LOCUS T (FT)(Jung et al
2007) Consistent with this toe1-2D 35STOE2 35SSMZ and 35SSNZ plants
exhibit late flowering (Jung et al 2007) MiR172 is also regulated temporally and
highly expressed in the adult vegetative phase both in Arabidopsis and maize (Chuck et
al 2007) Because the temporal expression patterns of miR156 and miR172 are
complementary to each other it has been suggested that the two miRNAs interact with
each other in regulating phase change (Chuck et al 2009 de Alba et al 2013) In
support of this view it has been shown that miR172 is down-regulated in the
Chapter 1 Introduction and Review of Literature
43
Corngrass1(Cg1) mutant in which miR156 is overproduced (Chuck et al 2007 de
Alba et al 2013) However there is no direct evidence for the direct linkage between
miR156 and miR172 It is also envisioned that miR156 and miR172 would not be
directly related For example miR156 regulates plastochron index that is the inverse of
leaf initiation rate and thus frequently used to analyze temporal patterns of plant
development In contrast miR172 does not affect plastochron (Wang et al 2008) The
down-regulation of miR172 in the maize miR156-over producing mutants would be
due to the prolonged juvenile vegetative phase in the mutants
Another miRNA that regulates flowering initiation is miR159 It regulates
MYB33 and MYB65 which are closely related to the barley GAMY B transcription
factor that responds to gibberellic acid (GA) The level of miR159 is elevated in GA-
treated seedlings but reduced in the GA biosynthetic ga1 mutant Transgenic plants
over producing miR159 exhibit delayed floral induction under short days (Achard et
al 2004) It is been suggested that MYB33 mediates GA signal to regulate LEAFY
(LFY) These observations indicate that miR159 plays an important role in the GA
signaling pathways that regulate proper floral induction under short days (Achard et al
2004)
11362 Floral development
Arabidopsis flowers consist of four distinct organs They arise in a characteristic
pattern within concentric rings called whorls from outside to inside four sepals four
petals six stamens and the central pistil consisting of two carpels Identities of the
floral organs are determined by three classes of regulatory genes classA classB and
class C genes (Krizek amp Fletcher 2005) The A and C class genes act antagonistically
to restrict their expression domains (Bowman et al 1991)
It is notable that miR172 also regulates floral architecture in addition to its role
in the timing of flowering initiation MiR172 inhibits the translation of the AP2 mRNA
(Chen 2004) Transgenic plants overproducing miR172 not only exhibit early
flowering but also show abnormal floral development which is quite similar to those
of the class A gene mutants such as the ap2 mutants (Aukerman amp Sakai 2003 de
Alba et al 2013) When the activity of the class A genes is inactivated that of the class
C genes such as AGAMOUS (AG) is elevated As a result the miR172- overproducing
transgenic plants exhibit no petals along with homeotic transformation of the sepals to
Chapter 1 Introduction and Review of Literature
44
the carpels (Aukerman amp Sakai 2003 Chen 2004)
Interestingly miR166 also regulates floral architecture The role of miR165166
in the regulation of meristem activity is closely linked to floral meristem formation and
organization (Zhao et al 2007 Miyashima et al 2013) Floral structure is severely
disrupted in the miR165166-overproducing mutants such as men1 and jba-1D in
which miR166 is overproduced (Williams et al 2005b Jung amp Park 2007) The men1
and jba-1D mutants have smaller gynoecium and reduced carpel number In an extreme
case the jba-1D homozygous plants lack visible carpels (Williams et al 2005b)
Similar phenotypes have been observed in the miR165 overproducers (Zhao et al
2007) It has been suggested that the phenotypes are possibly caused by altered AG
expression in the floral meristem (Jung amp Park 2007 Turchi et al 2013)
Other miRNAs also affect floral development Overproduction of miRNAs
involved in auxin signaling also affects floral architecture Transgenic plants
overproducing miR167 have defects in the formation of ovule and anther with reduced
fertility (Ru et al 2006) It has been shown that miR167 targets ARF6 and ARF8 that
play a role in gynoecium and stamen development and in regulating fertility (Ru et al
2006 Wu et al 2006) These observations suggest that auxin signals are also closely
related to floral organogenesis In addition transgenic plants over-producing miR164
and the cuc1cuc2 double mutants show fused sepals and reduced petals (Laufs et al
2004 Turchi et al 2013) suggesting that miR164 is also related to floral meristem
activity and boundary specification of the meristematic region
11363 Shoot apical meristem development
Shoot apical meristem comprises self-maintaining totipotent cells The shoot apical
meristem activity affects diverse aspects of plant developmental processes including
leaf development vascular development and lateral organ polarity (Bowman et al
2002) Because miR165166 plays a primary role in meristem formation
overproduction of miR165166 and multiple knockout mutants of its target genes
exhibit pleiotropic pheonotypes (McConnell et al 2001 Emery et al 2003 Kim et al
2005 Williams et al 2005b)
The targets of miR165166 include genes encoding the homeo domain-leucine
zipper (HD-ZIP) class III transcription factors such as PHABULOSA(PHB)
PHAVOLUTA (PHV) REVOLUTA(REV) ATHB15CORONA (CNA) and ATHB8
Chapter 1 Introduction and Review of Literature
45
PHB PHV and ATHB15 have overlapping functions in controlling meristem
development (Turchi et al 2013) Indeed the phb phv athb15 triple mutants have an
enlarged shoot apical meristem Consistent with this the miR166- overproducing men1
and jba-1D mutants exhibit enlarged shoot apical meristems (Kim et al 2005
Williams et al 2005b Turchi et al 2013) In the mutants the shoot apical meristems
are multiplied and large
The development of shoot apical meristems is also regulated by the WUSCHEL
(WUS)ndashCLAVATA (CLV) signaling pathway WUS positively regulates cell
proliferation In contrast CLV3 which is expressed in stem cells limits the WUS
expression and thus inhibits cell proliferation in the SAM (Sharma et al 2003)
Consistent with this notion WUS is expressed to a higher level in jba-1D
explaining the ectopic enlargement of the SAM in the mutant (Williams et al 2005)
While WUS is closely related to miR165166 in the shoot apical meristem
development the underlying molecular mechanisms are currently unclear It has been
observed that the miR166-over producing men1 plants have an arrested meristem
development (Jung amp Park 2007) In addition the heterozygous men1+ and the
homozygous men1 plants show different expression patterns of WUS and CLV3 It
appears that miR166 regulates WUS indirectly and influences the expressional balance
between WUS and CLV3 through a negative feedback loop (Jung amp Park 2007 de
Alba et al 2013)
The phv phb athb15 triple mutant has an enlarged shoot apical meristems the
rev phb phv mutant does not have functional shoot apical meristems (Prigge et al
2005) This distinction may be explained by the fact that while miR166 targets
primarily PHB PHV and ATHB15 miR165 targets all the five HD-ZIP III genes (Zhou
et al 2007) Consistent with this transgenic plants overproducing miR166 are distinct
from those overproducing miR165 The miR165-overproducing transgenic plants lack
functional shoot apical meristem and a pin-like structure is formed at the apical region
of the mutant These phenotypes are quite similar to rev phb phv triple mutant (Zhou et
al 2007 Liu et al 2013) The phenotypic distinction may because by the difference
of the cleavage efficiencies of the target mRNAs In accordance with this notion a few
35S miR166a transgenic lines exhibit no shoot apical meristems The men1
homozygous plants also show arrested meristem development (Jung amp Park 2007)
Chapter 1 Introduction and Review of Literature
46
MiR164 cleaves the CUC1 and CUC2 mRNAs as well as the NAC1 mRNA
CUC1 and CUC2 determines the organ boundary in the meristem Phenotypes of
transgenic plants that overproduce miR164 are similar to those of the cuc1cuc2 double
mutant that exhibits embryonic developmental defects such as cup-shaped cotyledons
and fused cotyledons (Laufs et al 2004) When a miR164-resistant CUC2 is
expressed the boundary domain is enlarged CUC also plays a role in embryonic shoot
meristem formation by regulating the SHOOT MERISTEMLESS gene It was therefore
concluded that miR164-mediated cleavage of CUC1 and CUC2 mRNAs determines the
sizes of the boundary domains in the shoot apical meristem and regulates separation of
the organs by restricting cell proliferation (Laufs et al 2004 de Alba et al 2013)
11364 Leaf development
The role of miR319 has been extensively studied in leaf development A dominant
jaw-D mutant overproducing miR319 shows uneven curved leaves with enlarged size
and five TCP genes which are closely related to the CINCINNATA (CIN) gene in
Antirrhinum majus are down regulated in the mutant suggesting that miR319 mediated
regulation of the TCP transcription factor genes are important for the final step of
leaf shaping (Palatnik et al 2003 Naz et al 2013) In addition to leaf shape and size
leaf polarity is also specified by miRNA MiR166-mediated cleavage of the HD-ZIPIII
mRNAs is a well to establish leaf polarity This discovery originates from the gain-of-
function mutants such as phb-1d and phv-1d wherein point mutations in the
complementary sites to miRNA helps the mRNA to evade or resist from miRNA-
mediated cleavage (McConnell et al 2001 Emery et al 2003 Naz et al 2013)
Plant leaves have a dorso-ventral polarity consisting of the adaxial (upper
surface) and abaxial (lower surface) sides The adaxial side is closer to the shoot
apical meristem Adaxial identity is specified by functionally redundant class III HD-
ZIP family genes such as PHB PHV and REV Ectopic expression of each genes
result in adaxialized leaves Dominant phb-1d and phv-1d mutants exhibit
transformation of the abaxial to adaxial fate and have rod-shaped leaves In contrast
miR165166-overproducing plants show pin-like cotyledons radicalized leaves and
abaxialized leaves (Chitwood et al 2007 Pattanayak et al 2013)
Leaf asymmetry is determined by miR166-mediated restriction of the
expression domains of the HD-ZIPIII genes The HD-ZIPIII genes are expressed in the
Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora
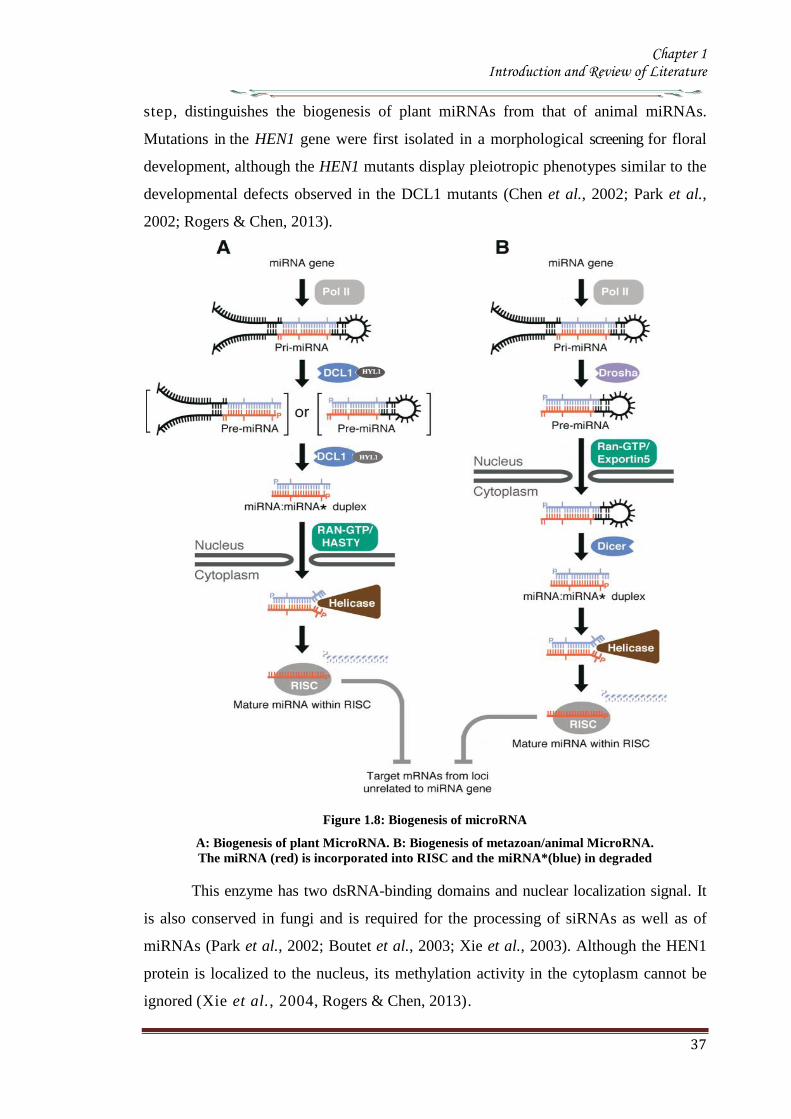
Chapter 1 Introduction and Review of Literature
37
step distinguishes the biogenesis of plant miRNAs from that of animal miRNAs
Mutations in the HEN1 gene were first isolated in a morphological screening for floral
development although the HEN1 mutants display pleiotropic phenotypes similar to the
developmental defects observed in the DCL1 mutants (Chen et al 2002 Park et al
2002 Rogers amp Chen 2013)
Figure 18 Biogenesis of microRNA
A Biogenesis of plant MicroRNA B Biogenesis of metazoananimal MicroRNA
The miRNA (red) is incorporated into RISC and the miRNA(blue) in degraded
This enzyme has two dsRNA-binding domains and nuclear localization signal It
is also conserved in fungi and is required for the processing of siRNAs as well as of
miRNAs (Park et al 2002 Boutet et al 2003 Xie et al 2003) Although the HEN1
protein is localized to the nucleus its methylation activity in the cytoplasm cannot be
ignored (Xie et al 2004 Rogers amp Chen 2013)
Chapter 1 Introduction and Review of Literature
38
The methylation of the miRNAmiRNA duplex is required to protect miRNAs
against the 3prime-end uridylation activity and subsequent degradation (Li et al 2005) In
the hen1 mutants accumulation of miRNAs is reduced to a lower level Also the
miRNAs appear to be heterogeneous in their sizes such that a ladder of bands rather
than a single species is observed for most miRNAs in the mutants (Li et al 2005)
MiRNA cloning and sequencing analyses revealed the presence of a number of U
residues at the 3prime-end of miRNAs in the hen1 mutants Moreover these nucleotides do
not correspond to the miRNAs in the pre-miRNAs indicating that these nucleotides are
added after the processing of the miRNAs from pre-miRNAs (Li et al 2005)
Uridylation of RNAs has also been proven to be correlated with RNA decay (Shen amp
Goodman 2004 Mullen amp Marzluff 2008) suggesting that uridylation attracts an
exonuclease which degrades miRNAs in plants
11344 MiRNA incorporation in to the RISC and its nuclear export
The methylated miRNAmiRNA duplexes undergo RISC assembly In this process
the miRNA of the duplexes is selectively incorporated into the RISC and the miRNA
appears to be removed and subsequently degraded (Hammond et al 2000 Hutvagner
amp Zamore 2002 Schwarz et al 2003 de Alba et al 2013 Rogers amp Chen 2013)
The ARGONAUTE 1(AGO1) proteins are core components of the RISC complex
They contain two structural domains the N-terminal PAZ domain that binds to the 3prime-
end of single-stranded RNAs and the C-terminal piwi domains that has a structure
similar to that of RNase H (Cerutti et al 2000 Parker et al 2004) Several AGO
proteins possess endonucleolytic activities within the PIWI domain and cleave target
mRNAs in the middle of the site complementary to miRNA (Liu et al 2004 Meister et
al 2004 Miyoshi et al 2005) Mutations in the AGO1 gene cause miRNA target
genes to be ectopically expressed and the levels of some miRNAs are reduced in the
mutants (Kidner amp Martienssen 2004 Vaucheret et al 2004 Kidner amp Martienssen
2005 Ronemus et al 2006) Moreover the phenotype of the AGO1 hypomorphic
alleles display defects in lateral organ polarity and leaf and flower morphology as
observed in the dcl1 mutants and null AGO1 alleles are embryonically lethal
supporting the close relationship between the AGO proteins and miRNA processing
(Kidner amp Martienssen 2004 Vaucheret et al 2004 de Alba et al 2013 Rogers amp
Chen 2013)
The production of miRNA miRNA duplexes occur in the nucleus while
Chapter 1 Introduction and Review of Literature
39
miRNA-RISC complexes are present in the cytoplasm where target mRNAs are
located This discordance of functional sites requires an export step during miRNA
biogenesis HASTY the Arabidopsis homolog of exportin-5 is thought to be involved
in miRNA nuclear export (Bollman et al 2003 Park et al 2005)The hasty mutant
exhibits pleiotropic defects in plant development and reduced accumulation of most
miRNAs as in the DCL1 or AGO1 mutants Howeverit is unclear whether the HASTY
proteins export the miRNA miRNA duplexes or the miRNA-RISC complexes in
plants
11345 Feedback regulation of miRNA biogenesis
MiRNA biogenesis is under feedback regulation such that the DCL1 and AGO1 genes
are themselves regulated by miRNAs DCL1 is itself a target of miR162 which leads to
the cleavage of the DCL1 mRNA (Xie et al 2003 de Alba et al 2013) Consistent
with this the levels of DCL1 mRNA are elevated in the mutants of miRNA processing
genes in which the miR162 abundance is reduced In addition there is a predicted
hairpin structure of miR838 in the 14th
intron of the DCL1 primary transcript
(Rajagopalan et al 2006) This prediction implies that DCL1-mediated processing
on this site can result in the cleavage of the DCL1 primary transcript into two truncated
fragments the N-terminal capped fragment and the C-terminal polyadenylated
fragment If this is the case it is possible that miRNA biogenesis competes with the
splicing event of the DCL1 pre-mRNA
The AGO1 is also regulated by miRNA There is a complementary site for
miR168 binding in the AGO1 mRNA and the transcript level of the AGO1 mRNA is
reduced through an mRNA cleavage mechanism (Vaucheret et al 2006 de Alba et al
2013 Rogers amp Chen 2013) indicating that the AGO1 mRNA levels are tightly
controlled by the levels of the AGO1 protein
1135 Mechanism of miRNA action
11351 MiRNA-directed mRNA cleavage
By forming the miRNA-RISC assembly miRNAs can direct the RISC to regulate gene
expression by two post transcriptional mechanisms mRNA cleavage or translational
repression (Bartel 2004 Jones-Rhoades et al 2006 Dalmay 2013) The degree
of miRNA-mRNA complementarity plays an important role for the
determination of the mechanism to be used A general rule is that while perfect or
Chapter 1 Introduction and Review of Literature
40
near-perfect complementarity induces mRNA cleavage central mismatches trigger
translational repression (Carrington amp Ambros 2003 Jones-Rhoades amp Bartel 2004)
Unlike most animal miRNAs plant miRNAs have near-perfect or perfect
complementarity to their targets and mRNA cleavage is assumed to be a predominant
mechanism by which miRNAs regulate gene expression in plants
In RNA cleavage mechanism miRNAs guide the AGO component of RISC to
cleave a single phosphodiester bond of the target mRNA within the miRNA-binding
site (Llave et al 2002b Tang et al 2003) The truncated fragments are then released
and degraded freeing the RISC to recognize and cleave another target mRNA This has
been validated by the detection of 3prime cleavage products that have 5prime ends that start at the
middle of the complementary regions The mRNA cleavage products are then further
degraded by other mechanisms The 3prime cleavage products of some miRNA targets are
degraded by the 5prime-3prime exonuclease XRN4 an Arabidopsis homolog of the major Yeast
mRNA degrading exonuclease Xrn1p It has been observed that the 3prime cleavage
products of some target mRNAs were accumulated in the xrn4 mutants (Souret et al
2004 Dalmay 2013) However the 3prime cleavage products of other miRNA targets do
not accumulate in the xrn4 mutants indicating that there are also XRN4 independent
mechanisms The 5prime cleavage products are typically untraceable by RNA gel blot
hybridization but can be detected by sensitive PCR based methods It has been found
that the 5prime cleavage products tend to obtain an oligo U tail at the 3prime ends This 3prime U tail
is correlated with shortening of the 5prime cleavage products from the 5prime end leading to the
conclusion that the 3prime uridylation causes 5 prime- 3 prime exonucleolytic decay of the 5prime cleavage
products (Shen amp Goodman 2004)
DCL1 HYL1 and SE interact with each other and are co-localized in the
nuclear bodies termed Dicing bodies or D-bodies in which some pri-miRNA shave
been found suggesting that D-bodies serve as a center for active miRNA processing
(Fang amp Spector 2007 Fujioka et al 2007 Song et al 2007) The miRNA
processing site appears to be distinct from the Cajal bodies that have previously been
found as a siRNA processing site (Pontes amp Pikaard 2008) The D-bodies include
SmD3 and SmB proteins that are found in the Cajal bodies However At collin an
additional marker for the Cajal bodies is not found in the D-bodies Moreover the D-
bodies are present in an At collin mutant lacking the Cajal bodies The pri-miRNAs of
miR171A and miR159A genes have been found to be co-localized with HYL1 and
Chapter 1 Introduction and Review of Literature
41
DCL1 in the D- bodies that are distinct from the Cajal bodies (Song et al 2007)
11352 MiRNA-directed translational repression
Translation repression is another mechanism exerted by plant miRNAs The regulatory
mechanism of miR172 which regulates flowering time and floral organ identity is
consistent with translation repression MiR172 over production does not affect the
transcript levels of APETALA2 (AP2) and AP2- like target mRNAs Instead the AP2
protein level is reduced in the miR172 over expressing mutant (Aukerman amp Sakai
2003 Chen 2004 Dalmay 2013) It was later shown that miR172 can also direct the
cleavage of the target mRNAs although the steady-state mRNA level is unchanged due
to feedback regulation (Schwab et al 2005 Jung et al 2007) It is clear that miR172
regulates its target genes primarily by translational repression rather than mRNA
cleavage Recently it has been reported that miR156157 and miR854 also regulate
their target genes through translational repression (Arteaga-Vazquez et al 2006
Gandikota et al 2007 Dalmay 2013) It was once thought that translational repression
is an exception to the rule However experimental evidence suggest a widespread
coexistence of translational repression and mRNA cleavage (Brodersen et al 2008)
1136 Regulatory roles of miRNAs in plant development
The miRNA target prediction has been a difficult task in order to obtain regulatory
targets without bringing in too many false predictions Plant growth and developmental
processes regulated by miRNAs are quite diverse and enormous Furthermore plant
miRNAs work cooperatively or antagonistically to establish a balanced regulation This
notion is well consistent with the findings that mutations in the regulators of miRNA
biogenesis transport and processing such as AGO1 DCL1 HYL1 HEN1 ABH1
and HASTY cause pleiotropic phenotypes (Mallory amp Vaucheret 2006 de Alba et al
2013) To date the roles of miRNAs have been mostly deduced from obtaining miRNA
over expressing transgenic plants or gain-of-function mutants in which miRNA-
resistant target genes are ectopically expressed
11361 Phase transition
Plants pass through a series of developmental phase transitions during their life span
beginning with seed germination continuing through vegetative phase change
reproductive phase change flowering initiation and finally seed production for the next
generation Because of their importance in understanding plant developmental
Chapter 1 Introduction and Review of Literature
42
processes genes regulating developmental transitions have been extensively studied
and underlying molecular mechanisms have been explored in various plant species
Majority of researches were mainly focused on vegetative phase change and
reproductive transition in Arabidopsis and Maize (Poethig 2003 Baurle amp Dean
2006) Particularly the mutations of the genes encoding various regulators of miRNA
biogenesis and transport such as HST SE and AGO exhibit altered juvenile to adult
vegetative phase change and vegetative to reproductive change (Hunter et al 2003
Peragine et al 2004 Vazquez et al 2004a Jones-Rhoades et al 2006 de Alba et al
2013)
Over-expression of the miR156a gene causes late flowering and delays
vegetative phase change (Wu amp Poethig 2006 Gandikota et al 2007 Wang et al
2008) It has been shown that miR156 negatively regulates a group of genes encoding
SQUAMOSA PROMOTER-BINDING PROTEIN-LIKE (Lewis et al 2007 de Alba et
al 2013 Rogers amp Chen 2013) transcription factors such as SPL3 SPL4 and SPL5
that promote flowering initiation In contrast transgenic plants over expressing
miR156-resistant SPL345 genes are early flowering Interestingly the endogenous
level of miR156 is temporally regulated In the early juvenile vegetative phase the
level of miR156 is very high but decreases rapidly before the onset of the adult juvenile
phase (Wu amp Poethig 2006)
The Arabidopsis early activation tagged (eat-D) mutant exhibits early flowering
with disrupted floral structure The mutant phenotypes are caused by over expression of
the miR172a-2gene (Aukerman amp Sakai 2003) MiR172 regulates a small group of
genes encoding APETALLA2 (AP2)-like transcription factors such as TARGET OF
EAT1 (TOE1) TOE2 SCHLAFMUTZE (SMZ) and SCHNARCHZAPFEN(SNZ)
TOE1 TOE2 SMZ and SNZ have been shown to participate in their productive phase
change by repressing a floral integrator FLOWERING LOCUS T (FT)(Jung et al
2007) Consistent with this toe1-2D 35STOE2 35SSMZ and 35SSNZ plants
exhibit late flowering (Jung et al 2007) MiR172 is also regulated temporally and
highly expressed in the adult vegetative phase both in Arabidopsis and maize (Chuck et
al 2007) Because the temporal expression patterns of miR156 and miR172 are
complementary to each other it has been suggested that the two miRNAs interact with
each other in regulating phase change (Chuck et al 2009 de Alba et al 2013) In
support of this view it has been shown that miR172 is down-regulated in the
Chapter 1 Introduction and Review of Literature
43
Corngrass1(Cg1) mutant in which miR156 is overproduced (Chuck et al 2007 de
Alba et al 2013) However there is no direct evidence for the direct linkage between
miR156 and miR172 It is also envisioned that miR156 and miR172 would not be
directly related For example miR156 regulates plastochron index that is the inverse of
leaf initiation rate and thus frequently used to analyze temporal patterns of plant
development In contrast miR172 does not affect plastochron (Wang et al 2008) The
down-regulation of miR172 in the maize miR156-over producing mutants would be
due to the prolonged juvenile vegetative phase in the mutants
Another miRNA that regulates flowering initiation is miR159 It regulates
MYB33 and MYB65 which are closely related to the barley GAMY B transcription
factor that responds to gibberellic acid (GA) The level of miR159 is elevated in GA-
treated seedlings but reduced in the GA biosynthetic ga1 mutant Transgenic plants
over producing miR159 exhibit delayed floral induction under short days (Achard et
al 2004) It is been suggested that MYB33 mediates GA signal to regulate LEAFY
(LFY) These observations indicate that miR159 plays an important role in the GA
signaling pathways that regulate proper floral induction under short days (Achard et al
2004)
11362 Floral development
Arabidopsis flowers consist of four distinct organs They arise in a characteristic
pattern within concentric rings called whorls from outside to inside four sepals four
petals six stamens and the central pistil consisting of two carpels Identities of the
floral organs are determined by three classes of regulatory genes classA classB and
class C genes (Krizek amp Fletcher 2005) The A and C class genes act antagonistically
to restrict their expression domains (Bowman et al 1991)
It is notable that miR172 also regulates floral architecture in addition to its role
in the timing of flowering initiation MiR172 inhibits the translation of the AP2 mRNA
(Chen 2004) Transgenic plants overproducing miR172 not only exhibit early
flowering but also show abnormal floral development which is quite similar to those
of the class A gene mutants such as the ap2 mutants (Aukerman amp Sakai 2003 de
Alba et al 2013) When the activity of the class A genes is inactivated that of the class
C genes such as AGAMOUS (AG) is elevated As a result the miR172- overproducing
transgenic plants exhibit no petals along with homeotic transformation of the sepals to
Chapter 1 Introduction and Review of Literature
44
the carpels (Aukerman amp Sakai 2003 Chen 2004)
Interestingly miR166 also regulates floral architecture The role of miR165166
in the regulation of meristem activity is closely linked to floral meristem formation and
organization (Zhao et al 2007 Miyashima et al 2013) Floral structure is severely
disrupted in the miR165166-overproducing mutants such as men1 and jba-1D in
which miR166 is overproduced (Williams et al 2005b Jung amp Park 2007) The men1
and jba-1D mutants have smaller gynoecium and reduced carpel number In an extreme
case the jba-1D homozygous plants lack visible carpels (Williams et al 2005b)
Similar phenotypes have been observed in the miR165 overproducers (Zhao et al
2007) It has been suggested that the phenotypes are possibly caused by altered AG
expression in the floral meristem (Jung amp Park 2007 Turchi et al 2013)
Other miRNAs also affect floral development Overproduction of miRNAs
involved in auxin signaling also affects floral architecture Transgenic plants
overproducing miR167 have defects in the formation of ovule and anther with reduced
fertility (Ru et al 2006) It has been shown that miR167 targets ARF6 and ARF8 that
play a role in gynoecium and stamen development and in regulating fertility (Ru et al
2006 Wu et al 2006) These observations suggest that auxin signals are also closely
related to floral organogenesis In addition transgenic plants over-producing miR164
and the cuc1cuc2 double mutants show fused sepals and reduced petals (Laufs et al
2004 Turchi et al 2013) suggesting that miR164 is also related to floral meristem
activity and boundary specification of the meristematic region
11363 Shoot apical meristem development
Shoot apical meristem comprises self-maintaining totipotent cells The shoot apical
meristem activity affects diverse aspects of plant developmental processes including
leaf development vascular development and lateral organ polarity (Bowman et al
2002) Because miR165166 plays a primary role in meristem formation
overproduction of miR165166 and multiple knockout mutants of its target genes
exhibit pleiotropic pheonotypes (McConnell et al 2001 Emery et al 2003 Kim et al
2005 Williams et al 2005b)
The targets of miR165166 include genes encoding the homeo domain-leucine
zipper (HD-ZIP) class III transcription factors such as PHABULOSA(PHB)
PHAVOLUTA (PHV) REVOLUTA(REV) ATHB15CORONA (CNA) and ATHB8
Chapter 1 Introduction and Review of Literature
45
PHB PHV and ATHB15 have overlapping functions in controlling meristem
development (Turchi et al 2013) Indeed the phb phv athb15 triple mutants have an
enlarged shoot apical meristem Consistent with this the miR166- overproducing men1
and jba-1D mutants exhibit enlarged shoot apical meristems (Kim et al 2005
Williams et al 2005b Turchi et al 2013) In the mutants the shoot apical meristems
are multiplied and large
The development of shoot apical meristems is also regulated by the WUSCHEL
(WUS)ndashCLAVATA (CLV) signaling pathway WUS positively regulates cell
proliferation In contrast CLV3 which is expressed in stem cells limits the WUS
expression and thus inhibits cell proliferation in the SAM (Sharma et al 2003)
Consistent with this notion WUS is expressed to a higher level in jba-1D
explaining the ectopic enlargement of the SAM in the mutant (Williams et al 2005)
While WUS is closely related to miR165166 in the shoot apical meristem
development the underlying molecular mechanisms are currently unclear It has been
observed that the miR166-over producing men1 plants have an arrested meristem
development (Jung amp Park 2007) In addition the heterozygous men1+ and the
homozygous men1 plants show different expression patterns of WUS and CLV3 It
appears that miR166 regulates WUS indirectly and influences the expressional balance
between WUS and CLV3 through a negative feedback loop (Jung amp Park 2007 de
Alba et al 2013)
The phv phb athb15 triple mutant has an enlarged shoot apical meristems the
rev phb phv mutant does not have functional shoot apical meristems (Prigge et al
2005) This distinction may be explained by the fact that while miR166 targets
primarily PHB PHV and ATHB15 miR165 targets all the five HD-ZIP III genes (Zhou
et al 2007) Consistent with this transgenic plants overproducing miR166 are distinct
from those overproducing miR165 The miR165-overproducing transgenic plants lack
functional shoot apical meristem and a pin-like structure is formed at the apical region
of the mutant These phenotypes are quite similar to rev phb phv triple mutant (Zhou et
al 2007 Liu et al 2013) The phenotypic distinction may because by the difference
of the cleavage efficiencies of the target mRNAs In accordance with this notion a few
35S miR166a transgenic lines exhibit no shoot apical meristems The men1
homozygous plants also show arrested meristem development (Jung amp Park 2007)
Chapter 1 Introduction and Review of Literature
46
MiR164 cleaves the CUC1 and CUC2 mRNAs as well as the NAC1 mRNA
CUC1 and CUC2 determines the organ boundary in the meristem Phenotypes of
transgenic plants that overproduce miR164 are similar to those of the cuc1cuc2 double
mutant that exhibits embryonic developmental defects such as cup-shaped cotyledons
and fused cotyledons (Laufs et al 2004) When a miR164-resistant CUC2 is
expressed the boundary domain is enlarged CUC also plays a role in embryonic shoot
meristem formation by regulating the SHOOT MERISTEMLESS gene It was therefore
concluded that miR164-mediated cleavage of CUC1 and CUC2 mRNAs determines the
sizes of the boundary domains in the shoot apical meristem and regulates separation of
the organs by restricting cell proliferation (Laufs et al 2004 de Alba et al 2013)
11364 Leaf development
The role of miR319 has been extensively studied in leaf development A dominant
jaw-D mutant overproducing miR319 shows uneven curved leaves with enlarged size
and five TCP genes which are closely related to the CINCINNATA (CIN) gene in
Antirrhinum majus are down regulated in the mutant suggesting that miR319 mediated
regulation of the TCP transcription factor genes are important for the final step of
leaf shaping (Palatnik et al 2003 Naz et al 2013) In addition to leaf shape and size
leaf polarity is also specified by miRNA MiR166-mediated cleavage of the HD-ZIPIII
mRNAs is a well to establish leaf polarity This discovery originates from the gain-of-
function mutants such as phb-1d and phv-1d wherein point mutations in the
complementary sites to miRNA helps the mRNA to evade or resist from miRNA-
mediated cleavage (McConnell et al 2001 Emery et al 2003 Naz et al 2013)
Plant leaves have a dorso-ventral polarity consisting of the adaxial (upper
surface) and abaxial (lower surface) sides The adaxial side is closer to the shoot
apical meristem Adaxial identity is specified by functionally redundant class III HD-
ZIP family genes such as PHB PHV and REV Ectopic expression of each genes
result in adaxialized leaves Dominant phb-1d and phv-1d mutants exhibit
transformation of the abaxial to adaxial fate and have rod-shaped leaves In contrast
miR165166-overproducing plants show pin-like cotyledons radicalized leaves and
abaxialized leaves (Chitwood et al 2007 Pattanayak et al 2013)
Leaf asymmetry is determined by miR166-mediated restriction of the
expression domains of the HD-ZIPIII genes The HD-ZIPIII genes are expressed in the
Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
38
The methylation of the miRNAmiRNA duplex is required to protect miRNAs
against the 3prime-end uridylation activity and subsequent degradation (Li et al 2005) In
the hen1 mutants accumulation of miRNAs is reduced to a lower level Also the
miRNAs appear to be heterogeneous in their sizes such that a ladder of bands rather
than a single species is observed for most miRNAs in the mutants (Li et al 2005)
MiRNA cloning and sequencing analyses revealed the presence of a number of U
residues at the 3prime-end of miRNAs in the hen1 mutants Moreover these nucleotides do
not correspond to the miRNAs in the pre-miRNAs indicating that these nucleotides are
added after the processing of the miRNAs from pre-miRNAs (Li et al 2005)
Uridylation of RNAs has also been proven to be correlated with RNA decay (Shen amp
Goodman 2004 Mullen amp Marzluff 2008) suggesting that uridylation attracts an
exonuclease which degrades miRNAs in plants
11344 MiRNA incorporation in to the RISC and its nuclear export
The methylated miRNAmiRNA duplexes undergo RISC assembly In this process
the miRNA of the duplexes is selectively incorporated into the RISC and the miRNA
appears to be removed and subsequently degraded (Hammond et al 2000 Hutvagner
amp Zamore 2002 Schwarz et al 2003 de Alba et al 2013 Rogers amp Chen 2013)
The ARGONAUTE 1(AGO1) proteins are core components of the RISC complex
They contain two structural domains the N-terminal PAZ domain that binds to the 3prime-
end of single-stranded RNAs and the C-terminal piwi domains that has a structure
similar to that of RNase H (Cerutti et al 2000 Parker et al 2004) Several AGO
proteins possess endonucleolytic activities within the PIWI domain and cleave target
mRNAs in the middle of the site complementary to miRNA (Liu et al 2004 Meister et
al 2004 Miyoshi et al 2005) Mutations in the AGO1 gene cause miRNA target
genes to be ectopically expressed and the levels of some miRNAs are reduced in the
mutants (Kidner amp Martienssen 2004 Vaucheret et al 2004 Kidner amp Martienssen
2005 Ronemus et al 2006) Moreover the phenotype of the AGO1 hypomorphic
alleles display defects in lateral organ polarity and leaf and flower morphology as
observed in the dcl1 mutants and null AGO1 alleles are embryonically lethal
supporting the close relationship between the AGO proteins and miRNA processing
(Kidner amp Martienssen 2004 Vaucheret et al 2004 de Alba et al 2013 Rogers amp
Chen 2013)
The production of miRNA miRNA duplexes occur in the nucleus while
Chapter 1 Introduction and Review of Literature
39
miRNA-RISC complexes are present in the cytoplasm where target mRNAs are
located This discordance of functional sites requires an export step during miRNA
biogenesis HASTY the Arabidopsis homolog of exportin-5 is thought to be involved
in miRNA nuclear export (Bollman et al 2003 Park et al 2005)The hasty mutant
exhibits pleiotropic defects in plant development and reduced accumulation of most
miRNAs as in the DCL1 or AGO1 mutants Howeverit is unclear whether the HASTY
proteins export the miRNA miRNA duplexes or the miRNA-RISC complexes in
plants
11345 Feedback regulation of miRNA biogenesis
MiRNA biogenesis is under feedback regulation such that the DCL1 and AGO1 genes
are themselves regulated by miRNAs DCL1 is itself a target of miR162 which leads to
the cleavage of the DCL1 mRNA (Xie et al 2003 de Alba et al 2013) Consistent
with this the levels of DCL1 mRNA are elevated in the mutants of miRNA processing
genes in which the miR162 abundance is reduced In addition there is a predicted
hairpin structure of miR838 in the 14th
intron of the DCL1 primary transcript
(Rajagopalan et al 2006) This prediction implies that DCL1-mediated processing
on this site can result in the cleavage of the DCL1 primary transcript into two truncated
fragments the N-terminal capped fragment and the C-terminal polyadenylated
fragment If this is the case it is possible that miRNA biogenesis competes with the
splicing event of the DCL1 pre-mRNA
The AGO1 is also regulated by miRNA There is a complementary site for
miR168 binding in the AGO1 mRNA and the transcript level of the AGO1 mRNA is
reduced through an mRNA cleavage mechanism (Vaucheret et al 2006 de Alba et al
2013 Rogers amp Chen 2013) indicating that the AGO1 mRNA levels are tightly
controlled by the levels of the AGO1 protein
1135 Mechanism of miRNA action
11351 MiRNA-directed mRNA cleavage
By forming the miRNA-RISC assembly miRNAs can direct the RISC to regulate gene
expression by two post transcriptional mechanisms mRNA cleavage or translational
repression (Bartel 2004 Jones-Rhoades et al 2006 Dalmay 2013) The degree
of miRNA-mRNA complementarity plays an important role for the
determination of the mechanism to be used A general rule is that while perfect or
Chapter 1 Introduction and Review of Literature
40
near-perfect complementarity induces mRNA cleavage central mismatches trigger
translational repression (Carrington amp Ambros 2003 Jones-Rhoades amp Bartel 2004)
Unlike most animal miRNAs plant miRNAs have near-perfect or perfect
complementarity to their targets and mRNA cleavage is assumed to be a predominant
mechanism by which miRNAs regulate gene expression in plants
In RNA cleavage mechanism miRNAs guide the AGO component of RISC to
cleave a single phosphodiester bond of the target mRNA within the miRNA-binding
site (Llave et al 2002b Tang et al 2003) The truncated fragments are then released
and degraded freeing the RISC to recognize and cleave another target mRNA This has
been validated by the detection of 3prime cleavage products that have 5prime ends that start at the
middle of the complementary regions The mRNA cleavage products are then further
degraded by other mechanisms The 3prime cleavage products of some miRNA targets are
degraded by the 5prime-3prime exonuclease XRN4 an Arabidopsis homolog of the major Yeast
mRNA degrading exonuclease Xrn1p It has been observed that the 3prime cleavage
products of some target mRNAs were accumulated in the xrn4 mutants (Souret et al
2004 Dalmay 2013) However the 3prime cleavage products of other miRNA targets do
not accumulate in the xrn4 mutants indicating that there are also XRN4 independent
mechanisms The 5prime cleavage products are typically untraceable by RNA gel blot
hybridization but can be detected by sensitive PCR based methods It has been found
that the 5prime cleavage products tend to obtain an oligo U tail at the 3prime ends This 3prime U tail
is correlated with shortening of the 5prime cleavage products from the 5prime end leading to the
conclusion that the 3prime uridylation causes 5 prime- 3 prime exonucleolytic decay of the 5prime cleavage
products (Shen amp Goodman 2004)
DCL1 HYL1 and SE interact with each other and are co-localized in the
nuclear bodies termed Dicing bodies or D-bodies in which some pri-miRNA shave
been found suggesting that D-bodies serve as a center for active miRNA processing
(Fang amp Spector 2007 Fujioka et al 2007 Song et al 2007) The miRNA
processing site appears to be distinct from the Cajal bodies that have previously been
found as a siRNA processing site (Pontes amp Pikaard 2008) The D-bodies include
SmD3 and SmB proteins that are found in the Cajal bodies However At collin an
additional marker for the Cajal bodies is not found in the D-bodies Moreover the D-
bodies are present in an At collin mutant lacking the Cajal bodies The pri-miRNAs of
miR171A and miR159A genes have been found to be co-localized with HYL1 and
Chapter 1 Introduction and Review of Literature
41
DCL1 in the D- bodies that are distinct from the Cajal bodies (Song et al 2007)
11352 MiRNA-directed translational repression
Translation repression is another mechanism exerted by plant miRNAs The regulatory
mechanism of miR172 which regulates flowering time and floral organ identity is
consistent with translation repression MiR172 over production does not affect the
transcript levels of APETALA2 (AP2) and AP2- like target mRNAs Instead the AP2
protein level is reduced in the miR172 over expressing mutant (Aukerman amp Sakai
2003 Chen 2004 Dalmay 2013) It was later shown that miR172 can also direct the
cleavage of the target mRNAs although the steady-state mRNA level is unchanged due
to feedback regulation (Schwab et al 2005 Jung et al 2007) It is clear that miR172
regulates its target genes primarily by translational repression rather than mRNA
cleavage Recently it has been reported that miR156157 and miR854 also regulate
their target genes through translational repression (Arteaga-Vazquez et al 2006
Gandikota et al 2007 Dalmay 2013) It was once thought that translational repression
is an exception to the rule However experimental evidence suggest a widespread
coexistence of translational repression and mRNA cleavage (Brodersen et al 2008)
1136 Regulatory roles of miRNAs in plant development
The miRNA target prediction has been a difficult task in order to obtain regulatory
targets without bringing in too many false predictions Plant growth and developmental
processes regulated by miRNAs are quite diverse and enormous Furthermore plant
miRNAs work cooperatively or antagonistically to establish a balanced regulation This
notion is well consistent with the findings that mutations in the regulators of miRNA
biogenesis transport and processing such as AGO1 DCL1 HYL1 HEN1 ABH1
and HASTY cause pleiotropic phenotypes (Mallory amp Vaucheret 2006 de Alba et al
2013) To date the roles of miRNAs have been mostly deduced from obtaining miRNA
over expressing transgenic plants or gain-of-function mutants in which miRNA-
resistant target genes are ectopically expressed
11361 Phase transition
Plants pass through a series of developmental phase transitions during their life span
beginning with seed germination continuing through vegetative phase change
reproductive phase change flowering initiation and finally seed production for the next
generation Because of their importance in understanding plant developmental
Chapter 1 Introduction and Review of Literature
42
processes genes regulating developmental transitions have been extensively studied
and underlying molecular mechanisms have been explored in various plant species
Majority of researches were mainly focused on vegetative phase change and
reproductive transition in Arabidopsis and Maize (Poethig 2003 Baurle amp Dean
2006) Particularly the mutations of the genes encoding various regulators of miRNA
biogenesis and transport such as HST SE and AGO exhibit altered juvenile to adult
vegetative phase change and vegetative to reproductive change (Hunter et al 2003
Peragine et al 2004 Vazquez et al 2004a Jones-Rhoades et al 2006 de Alba et al
2013)
Over-expression of the miR156a gene causes late flowering and delays
vegetative phase change (Wu amp Poethig 2006 Gandikota et al 2007 Wang et al
2008) It has been shown that miR156 negatively regulates a group of genes encoding
SQUAMOSA PROMOTER-BINDING PROTEIN-LIKE (Lewis et al 2007 de Alba et
al 2013 Rogers amp Chen 2013) transcription factors such as SPL3 SPL4 and SPL5
that promote flowering initiation In contrast transgenic plants over expressing
miR156-resistant SPL345 genes are early flowering Interestingly the endogenous
level of miR156 is temporally regulated In the early juvenile vegetative phase the
level of miR156 is very high but decreases rapidly before the onset of the adult juvenile
phase (Wu amp Poethig 2006)
The Arabidopsis early activation tagged (eat-D) mutant exhibits early flowering
with disrupted floral structure The mutant phenotypes are caused by over expression of
the miR172a-2gene (Aukerman amp Sakai 2003) MiR172 regulates a small group of
genes encoding APETALLA2 (AP2)-like transcription factors such as TARGET OF
EAT1 (TOE1) TOE2 SCHLAFMUTZE (SMZ) and SCHNARCHZAPFEN(SNZ)
TOE1 TOE2 SMZ and SNZ have been shown to participate in their productive phase
change by repressing a floral integrator FLOWERING LOCUS T (FT)(Jung et al
2007) Consistent with this toe1-2D 35STOE2 35SSMZ and 35SSNZ plants
exhibit late flowering (Jung et al 2007) MiR172 is also regulated temporally and
highly expressed in the adult vegetative phase both in Arabidopsis and maize (Chuck et
al 2007) Because the temporal expression patterns of miR156 and miR172 are
complementary to each other it has been suggested that the two miRNAs interact with
each other in regulating phase change (Chuck et al 2009 de Alba et al 2013) In
support of this view it has been shown that miR172 is down-regulated in the
Chapter 1 Introduction and Review of Literature
43
Corngrass1(Cg1) mutant in which miR156 is overproduced (Chuck et al 2007 de
Alba et al 2013) However there is no direct evidence for the direct linkage between
miR156 and miR172 It is also envisioned that miR156 and miR172 would not be
directly related For example miR156 regulates plastochron index that is the inverse of
leaf initiation rate and thus frequently used to analyze temporal patterns of plant
development In contrast miR172 does not affect plastochron (Wang et al 2008) The
down-regulation of miR172 in the maize miR156-over producing mutants would be
due to the prolonged juvenile vegetative phase in the mutants
Another miRNA that regulates flowering initiation is miR159 It regulates
MYB33 and MYB65 which are closely related to the barley GAMY B transcription
factor that responds to gibberellic acid (GA) The level of miR159 is elevated in GA-
treated seedlings but reduced in the GA biosynthetic ga1 mutant Transgenic plants
over producing miR159 exhibit delayed floral induction under short days (Achard et
al 2004) It is been suggested that MYB33 mediates GA signal to regulate LEAFY
(LFY) These observations indicate that miR159 plays an important role in the GA
signaling pathways that regulate proper floral induction under short days (Achard et al
2004)
11362 Floral development
Arabidopsis flowers consist of four distinct organs They arise in a characteristic
pattern within concentric rings called whorls from outside to inside four sepals four
petals six stamens and the central pistil consisting of two carpels Identities of the
floral organs are determined by three classes of regulatory genes classA classB and
class C genes (Krizek amp Fletcher 2005) The A and C class genes act antagonistically
to restrict their expression domains (Bowman et al 1991)
It is notable that miR172 also regulates floral architecture in addition to its role
in the timing of flowering initiation MiR172 inhibits the translation of the AP2 mRNA
(Chen 2004) Transgenic plants overproducing miR172 not only exhibit early
flowering but also show abnormal floral development which is quite similar to those
of the class A gene mutants such as the ap2 mutants (Aukerman amp Sakai 2003 de
Alba et al 2013) When the activity of the class A genes is inactivated that of the class
C genes such as AGAMOUS (AG) is elevated As a result the miR172- overproducing
transgenic plants exhibit no petals along with homeotic transformation of the sepals to
Chapter 1 Introduction and Review of Literature
44
the carpels (Aukerman amp Sakai 2003 Chen 2004)
Interestingly miR166 also regulates floral architecture The role of miR165166
in the regulation of meristem activity is closely linked to floral meristem formation and
organization (Zhao et al 2007 Miyashima et al 2013) Floral structure is severely
disrupted in the miR165166-overproducing mutants such as men1 and jba-1D in
which miR166 is overproduced (Williams et al 2005b Jung amp Park 2007) The men1
and jba-1D mutants have smaller gynoecium and reduced carpel number In an extreme
case the jba-1D homozygous plants lack visible carpels (Williams et al 2005b)
Similar phenotypes have been observed in the miR165 overproducers (Zhao et al
2007) It has been suggested that the phenotypes are possibly caused by altered AG
expression in the floral meristem (Jung amp Park 2007 Turchi et al 2013)
Other miRNAs also affect floral development Overproduction of miRNAs
involved in auxin signaling also affects floral architecture Transgenic plants
overproducing miR167 have defects in the formation of ovule and anther with reduced
fertility (Ru et al 2006) It has been shown that miR167 targets ARF6 and ARF8 that
play a role in gynoecium and stamen development and in regulating fertility (Ru et al
2006 Wu et al 2006) These observations suggest that auxin signals are also closely
related to floral organogenesis In addition transgenic plants over-producing miR164
and the cuc1cuc2 double mutants show fused sepals and reduced petals (Laufs et al
2004 Turchi et al 2013) suggesting that miR164 is also related to floral meristem
activity and boundary specification of the meristematic region
11363 Shoot apical meristem development
Shoot apical meristem comprises self-maintaining totipotent cells The shoot apical
meristem activity affects diverse aspects of plant developmental processes including
leaf development vascular development and lateral organ polarity (Bowman et al
2002) Because miR165166 plays a primary role in meristem formation
overproduction of miR165166 and multiple knockout mutants of its target genes
exhibit pleiotropic pheonotypes (McConnell et al 2001 Emery et al 2003 Kim et al
2005 Williams et al 2005b)
The targets of miR165166 include genes encoding the homeo domain-leucine
zipper (HD-ZIP) class III transcription factors such as PHABULOSA(PHB)
PHAVOLUTA (PHV) REVOLUTA(REV) ATHB15CORONA (CNA) and ATHB8
Chapter 1 Introduction and Review of Literature
45
PHB PHV and ATHB15 have overlapping functions in controlling meristem
development (Turchi et al 2013) Indeed the phb phv athb15 triple mutants have an
enlarged shoot apical meristem Consistent with this the miR166- overproducing men1
and jba-1D mutants exhibit enlarged shoot apical meristems (Kim et al 2005
Williams et al 2005b Turchi et al 2013) In the mutants the shoot apical meristems
are multiplied and large
The development of shoot apical meristems is also regulated by the WUSCHEL
(WUS)ndashCLAVATA (CLV) signaling pathway WUS positively regulates cell
proliferation In contrast CLV3 which is expressed in stem cells limits the WUS
expression and thus inhibits cell proliferation in the SAM (Sharma et al 2003)
Consistent with this notion WUS is expressed to a higher level in jba-1D
explaining the ectopic enlargement of the SAM in the mutant (Williams et al 2005)
While WUS is closely related to miR165166 in the shoot apical meristem
development the underlying molecular mechanisms are currently unclear It has been
observed that the miR166-over producing men1 plants have an arrested meristem
development (Jung amp Park 2007) In addition the heterozygous men1+ and the
homozygous men1 plants show different expression patterns of WUS and CLV3 It
appears that miR166 regulates WUS indirectly and influences the expressional balance
between WUS and CLV3 through a negative feedback loop (Jung amp Park 2007 de
Alba et al 2013)
The phv phb athb15 triple mutant has an enlarged shoot apical meristems the
rev phb phv mutant does not have functional shoot apical meristems (Prigge et al
2005) This distinction may be explained by the fact that while miR166 targets
primarily PHB PHV and ATHB15 miR165 targets all the five HD-ZIP III genes (Zhou
et al 2007) Consistent with this transgenic plants overproducing miR166 are distinct
from those overproducing miR165 The miR165-overproducing transgenic plants lack
functional shoot apical meristem and a pin-like structure is formed at the apical region
of the mutant These phenotypes are quite similar to rev phb phv triple mutant (Zhou et
al 2007 Liu et al 2013) The phenotypic distinction may because by the difference
of the cleavage efficiencies of the target mRNAs In accordance with this notion a few
35S miR166a transgenic lines exhibit no shoot apical meristems The men1
homozygous plants also show arrested meristem development (Jung amp Park 2007)
Chapter 1 Introduction and Review of Literature
46
MiR164 cleaves the CUC1 and CUC2 mRNAs as well as the NAC1 mRNA
CUC1 and CUC2 determines the organ boundary in the meristem Phenotypes of
transgenic plants that overproduce miR164 are similar to those of the cuc1cuc2 double
mutant that exhibits embryonic developmental defects such as cup-shaped cotyledons
and fused cotyledons (Laufs et al 2004) When a miR164-resistant CUC2 is
expressed the boundary domain is enlarged CUC also plays a role in embryonic shoot
meristem formation by regulating the SHOOT MERISTEMLESS gene It was therefore
concluded that miR164-mediated cleavage of CUC1 and CUC2 mRNAs determines the
sizes of the boundary domains in the shoot apical meristem and regulates separation of
the organs by restricting cell proliferation (Laufs et al 2004 de Alba et al 2013)
11364 Leaf development
The role of miR319 has been extensively studied in leaf development A dominant
jaw-D mutant overproducing miR319 shows uneven curved leaves with enlarged size
and five TCP genes which are closely related to the CINCINNATA (CIN) gene in
Antirrhinum majus are down regulated in the mutant suggesting that miR319 mediated
regulation of the TCP transcription factor genes are important for the final step of
leaf shaping (Palatnik et al 2003 Naz et al 2013) In addition to leaf shape and size
leaf polarity is also specified by miRNA MiR166-mediated cleavage of the HD-ZIPIII
mRNAs is a well to establish leaf polarity This discovery originates from the gain-of-
function mutants such as phb-1d and phv-1d wherein point mutations in the
complementary sites to miRNA helps the mRNA to evade or resist from miRNA-
mediated cleavage (McConnell et al 2001 Emery et al 2003 Naz et al 2013)
Plant leaves have a dorso-ventral polarity consisting of the adaxial (upper
surface) and abaxial (lower surface) sides The adaxial side is closer to the shoot
apical meristem Adaxial identity is specified by functionally redundant class III HD-
ZIP family genes such as PHB PHV and REV Ectopic expression of each genes
result in adaxialized leaves Dominant phb-1d and phv-1d mutants exhibit
transformation of the abaxial to adaxial fate and have rod-shaped leaves In contrast
miR165166-overproducing plants show pin-like cotyledons radicalized leaves and
abaxialized leaves (Chitwood et al 2007 Pattanayak et al 2013)
Leaf asymmetry is determined by miR166-mediated restriction of the
expression domains of the HD-ZIPIII genes The HD-ZIPIII genes are expressed in the
Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
39
miRNA-RISC complexes are present in the cytoplasm where target mRNAs are
located This discordance of functional sites requires an export step during miRNA
biogenesis HASTY the Arabidopsis homolog of exportin-5 is thought to be involved
in miRNA nuclear export (Bollman et al 2003 Park et al 2005)The hasty mutant
exhibits pleiotropic defects in plant development and reduced accumulation of most
miRNAs as in the DCL1 or AGO1 mutants Howeverit is unclear whether the HASTY
proteins export the miRNA miRNA duplexes or the miRNA-RISC complexes in
plants
11345 Feedback regulation of miRNA biogenesis
MiRNA biogenesis is under feedback regulation such that the DCL1 and AGO1 genes
are themselves regulated by miRNAs DCL1 is itself a target of miR162 which leads to
the cleavage of the DCL1 mRNA (Xie et al 2003 de Alba et al 2013) Consistent
with this the levels of DCL1 mRNA are elevated in the mutants of miRNA processing
genes in which the miR162 abundance is reduced In addition there is a predicted
hairpin structure of miR838 in the 14th
intron of the DCL1 primary transcript
(Rajagopalan et al 2006) This prediction implies that DCL1-mediated processing
on this site can result in the cleavage of the DCL1 primary transcript into two truncated
fragments the N-terminal capped fragment and the C-terminal polyadenylated
fragment If this is the case it is possible that miRNA biogenesis competes with the
splicing event of the DCL1 pre-mRNA
The AGO1 is also regulated by miRNA There is a complementary site for
miR168 binding in the AGO1 mRNA and the transcript level of the AGO1 mRNA is
reduced through an mRNA cleavage mechanism (Vaucheret et al 2006 de Alba et al
2013 Rogers amp Chen 2013) indicating that the AGO1 mRNA levels are tightly
controlled by the levels of the AGO1 protein
1135 Mechanism of miRNA action
11351 MiRNA-directed mRNA cleavage
By forming the miRNA-RISC assembly miRNAs can direct the RISC to regulate gene
expression by two post transcriptional mechanisms mRNA cleavage or translational
repression (Bartel 2004 Jones-Rhoades et al 2006 Dalmay 2013) The degree
of miRNA-mRNA complementarity plays an important role for the
determination of the mechanism to be used A general rule is that while perfect or
Chapter 1 Introduction and Review of Literature
40
near-perfect complementarity induces mRNA cleavage central mismatches trigger
translational repression (Carrington amp Ambros 2003 Jones-Rhoades amp Bartel 2004)
Unlike most animal miRNAs plant miRNAs have near-perfect or perfect
complementarity to their targets and mRNA cleavage is assumed to be a predominant
mechanism by which miRNAs regulate gene expression in plants
In RNA cleavage mechanism miRNAs guide the AGO component of RISC to
cleave a single phosphodiester bond of the target mRNA within the miRNA-binding
site (Llave et al 2002b Tang et al 2003) The truncated fragments are then released
and degraded freeing the RISC to recognize and cleave another target mRNA This has
been validated by the detection of 3prime cleavage products that have 5prime ends that start at the
middle of the complementary regions The mRNA cleavage products are then further
degraded by other mechanisms The 3prime cleavage products of some miRNA targets are
degraded by the 5prime-3prime exonuclease XRN4 an Arabidopsis homolog of the major Yeast
mRNA degrading exonuclease Xrn1p It has been observed that the 3prime cleavage
products of some target mRNAs were accumulated in the xrn4 mutants (Souret et al
2004 Dalmay 2013) However the 3prime cleavage products of other miRNA targets do
not accumulate in the xrn4 mutants indicating that there are also XRN4 independent
mechanisms The 5prime cleavage products are typically untraceable by RNA gel blot
hybridization but can be detected by sensitive PCR based methods It has been found
that the 5prime cleavage products tend to obtain an oligo U tail at the 3prime ends This 3prime U tail
is correlated with shortening of the 5prime cleavage products from the 5prime end leading to the
conclusion that the 3prime uridylation causes 5 prime- 3 prime exonucleolytic decay of the 5prime cleavage
products (Shen amp Goodman 2004)
DCL1 HYL1 and SE interact with each other and are co-localized in the
nuclear bodies termed Dicing bodies or D-bodies in which some pri-miRNA shave
been found suggesting that D-bodies serve as a center for active miRNA processing
(Fang amp Spector 2007 Fujioka et al 2007 Song et al 2007) The miRNA
processing site appears to be distinct from the Cajal bodies that have previously been
found as a siRNA processing site (Pontes amp Pikaard 2008) The D-bodies include
SmD3 and SmB proteins that are found in the Cajal bodies However At collin an
additional marker for the Cajal bodies is not found in the D-bodies Moreover the D-
bodies are present in an At collin mutant lacking the Cajal bodies The pri-miRNAs of
miR171A and miR159A genes have been found to be co-localized with HYL1 and
Chapter 1 Introduction and Review of Literature
41
DCL1 in the D- bodies that are distinct from the Cajal bodies (Song et al 2007)
11352 MiRNA-directed translational repression
Translation repression is another mechanism exerted by plant miRNAs The regulatory
mechanism of miR172 which regulates flowering time and floral organ identity is
consistent with translation repression MiR172 over production does not affect the
transcript levels of APETALA2 (AP2) and AP2- like target mRNAs Instead the AP2
protein level is reduced in the miR172 over expressing mutant (Aukerman amp Sakai
2003 Chen 2004 Dalmay 2013) It was later shown that miR172 can also direct the
cleavage of the target mRNAs although the steady-state mRNA level is unchanged due
to feedback regulation (Schwab et al 2005 Jung et al 2007) It is clear that miR172
regulates its target genes primarily by translational repression rather than mRNA
cleavage Recently it has been reported that miR156157 and miR854 also regulate
their target genes through translational repression (Arteaga-Vazquez et al 2006
Gandikota et al 2007 Dalmay 2013) It was once thought that translational repression
is an exception to the rule However experimental evidence suggest a widespread
coexistence of translational repression and mRNA cleavage (Brodersen et al 2008)
1136 Regulatory roles of miRNAs in plant development
The miRNA target prediction has been a difficult task in order to obtain regulatory
targets without bringing in too many false predictions Plant growth and developmental
processes regulated by miRNAs are quite diverse and enormous Furthermore plant
miRNAs work cooperatively or antagonistically to establish a balanced regulation This
notion is well consistent with the findings that mutations in the regulators of miRNA
biogenesis transport and processing such as AGO1 DCL1 HYL1 HEN1 ABH1
and HASTY cause pleiotropic phenotypes (Mallory amp Vaucheret 2006 de Alba et al
2013) To date the roles of miRNAs have been mostly deduced from obtaining miRNA
over expressing transgenic plants or gain-of-function mutants in which miRNA-
resistant target genes are ectopically expressed
11361 Phase transition
Plants pass through a series of developmental phase transitions during their life span
beginning with seed germination continuing through vegetative phase change
reproductive phase change flowering initiation and finally seed production for the next
generation Because of their importance in understanding plant developmental
Chapter 1 Introduction and Review of Literature
42
processes genes regulating developmental transitions have been extensively studied
and underlying molecular mechanisms have been explored in various plant species
Majority of researches were mainly focused on vegetative phase change and
reproductive transition in Arabidopsis and Maize (Poethig 2003 Baurle amp Dean
2006) Particularly the mutations of the genes encoding various regulators of miRNA
biogenesis and transport such as HST SE and AGO exhibit altered juvenile to adult
vegetative phase change and vegetative to reproductive change (Hunter et al 2003
Peragine et al 2004 Vazquez et al 2004a Jones-Rhoades et al 2006 de Alba et al
2013)
Over-expression of the miR156a gene causes late flowering and delays
vegetative phase change (Wu amp Poethig 2006 Gandikota et al 2007 Wang et al
2008) It has been shown that miR156 negatively regulates a group of genes encoding
SQUAMOSA PROMOTER-BINDING PROTEIN-LIKE (Lewis et al 2007 de Alba et
al 2013 Rogers amp Chen 2013) transcription factors such as SPL3 SPL4 and SPL5
that promote flowering initiation In contrast transgenic plants over expressing
miR156-resistant SPL345 genes are early flowering Interestingly the endogenous
level of miR156 is temporally regulated In the early juvenile vegetative phase the
level of miR156 is very high but decreases rapidly before the onset of the adult juvenile
phase (Wu amp Poethig 2006)
The Arabidopsis early activation tagged (eat-D) mutant exhibits early flowering
with disrupted floral structure The mutant phenotypes are caused by over expression of
the miR172a-2gene (Aukerman amp Sakai 2003) MiR172 regulates a small group of
genes encoding APETALLA2 (AP2)-like transcription factors such as TARGET OF
EAT1 (TOE1) TOE2 SCHLAFMUTZE (SMZ) and SCHNARCHZAPFEN(SNZ)
TOE1 TOE2 SMZ and SNZ have been shown to participate in their productive phase
change by repressing a floral integrator FLOWERING LOCUS T (FT)(Jung et al
2007) Consistent with this toe1-2D 35STOE2 35SSMZ and 35SSNZ plants
exhibit late flowering (Jung et al 2007) MiR172 is also regulated temporally and
highly expressed in the adult vegetative phase both in Arabidopsis and maize (Chuck et
al 2007) Because the temporal expression patterns of miR156 and miR172 are
complementary to each other it has been suggested that the two miRNAs interact with
each other in regulating phase change (Chuck et al 2009 de Alba et al 2013) In
support of this view it has been shown that miR172 is down-regulated in the
Chapter 1 Introduction and Review of Literature
43
Corngrass1(Cg1) mutant in which miR156 is overproduced (Chuck et al 2007 de
Alba et al 2013) However there is no direct evidence for the direct linkage between
miR156 and miR172 It is also envisioned that miR156 and miR172 would not be
directly related For example miR156 regulates plastochron index that is the inverse of
leaf initiation rate and thus frequently used to analyze temporal patterns of plant
development In contrast miR172 does not affect plastochron (Wang et al 2008) The
down-regulation of miR172 in the maize miR156-over producing mutants would be
due to the prolonged juvenile vegetative phase in the mutants
Another miRNA that regulates flowering initiation is miR159 It regulates
MYB33 and MYB65 which are closely related to the barley GAMY B transcription
factor that responds to gibberellic acid (GA) The level of miR159 is elevated in GA-
treated seedlings but reduced in the GA biosynthetic ga1 mutant Transgenic plants
over producing miR159 exhibit delayed floral induction under short days (Achard et
al 2004) It is been suggested that MYB33 mediates GA signal to regulate LEAFY
(LFY) These observations indicate that miR159 plays an important role in the GA
signaling pathways that regulate proper floral induction under short days (Achard et al
2004)
11362 Floral development
Arabidopsis flowers consist of four distinct organs They arise in a characteristic
pattern within concentric rings called whorls from outside to inside four sepals four
petals six stamens and the central pistil consisting of two carpels Identities of the
floral organs are determined by three classes of regulatory genes classA classB and
class C genes (Krizek amp Fletcher 2005) The A and C class genes act antagonistically
to restrict their expression domains (Bowman et al 1991)
It is notable that miR172 also regulates floral architecture in addition to its role
in the timing of flowering initiation MiR172 inhibits the translation of the AP2 mRNA
(Chen 2004) Transgenic plants overproducing miR172 not only exhibit early
flowering but also show abnormal floral development which is quite similar to those
of the class A gene mutants such as the ap2 mutants (Aukerman amp Sakai 2003 de
Alba et al 2013) When the activity of the class A genes is inactivated that of the class
C genes such as AGAMOUS (AG) is elevated As a result the miR172- overproducing
transgenic plants exhibit no petals along with homeotic transformation of the sepals to
Chapter 1 Introduction and Review of Literature
44
the carpels (Aukerman amp Sakai 2003 Chen 2004)
Interestingly miR166 also regulates floral architecture The role of miR165166
in the regulation of meristem activity is closely linked to floral meristem formation and
organization (Zhao et al 2007 Miyashima et al 2013) Floral structure is severely
disrupted in the miR165166-overproducing mutants such as men1 and jba-1D in
which miR166 is overproduced (Williams et al 2005b Jung amp Park 2007) The men1
and jba-1D mutants have smaller gynoecium and reduced carpel number In an extreme
case the jba-1D homozygous plants lack visible carpels (Williams et al 2005b)
Similar phenotypes have been observed in the miR165 overproducers (Zhao et al
2007) It has been suggested that the phenotypes are possibly caused by altered AG
expression in the floral meristem (Jung amp Park 2007 Turchi et al 2013)
Other miRNAs also affect floral development Overproduction of miRNAs
involved in auxin signaling also affects floral architecture Transgenic plants
overproducing miR167 have defects in the formation of ovule and anther with reduced
fertility (Ru et al 2006) It has been shown that miR167 targets ARF6 and ARF8 that
play a role in gynoecium and stamen development and in regulating fertility (Ru et al
2006 Wu et al 2006) These observations suggest that auxin signals are also closely
related to floral organogenesis In addition transgenic plants over-producing miR164
and the cuc1cuc2 double mutants show fused sepals and reduced petals (Laufs et al
2004 Turchi et al 2013) suggesting that miR164 is also related to floral meristem
activity and boundary specification of the meristematic region
11363 Shoot apical meristem development
Shoot apical meristem comprises self-maintaining totipotent cells The shoot apical
meristem activity affects diverse aspects of plant developmental processes including
leaf development vascular development and lateral organ polarity (Bowman et al
2002) Because miR165166 plays a primary role in meristem formation
overproduction of miR165166 and multiple knockout mutants of its target genes
exhibit pleiotropic pheonotypes (McConnell et al 2001 Emery et al 2003 Kim et al
2005 Williams et al 2005b)
The targets of miR165166 include genes encoding the homeo domain-leucine
zipper (HD-ZIP) class III transcription factors such as PHABULOSA(PHB)
PHAVOLUTA (PHV) REVOLUTA(REV) ATHB15CORONA (CNA) and ATHB8
Chapter 1 Introduction and Review of Literature
45
PHB PHV and ATHB15 have overlapping functions in controlling meristem
development (Turchi et al 2013) Indeed the phb phv athb15 triple mutants have an
enlarged shoot apical meristem Consistent with this the miR166- overproducing men1
and jba-1D mutants exhibit enlarged shoot apical meristems (Kim et al 2005
Williams et al 2005b Turchi et al 2013) In the mutants the shoot apical meristems
are multiplied and large
The development of shoot apical meristems is also regulated by the WUSCHEL
(WUS)ndashCLAVATA (CLV) signaling pathway WUS positively regulates cell
proliferation In contrast CLV3 which is expressed in stem cells limits the WUS
expression and thus inhibits cell proliferation in the SAM (Sharma et al 2003)
Consistent with this notion WUS is expressed to a higher level in jba-1D
explaining the ectopic enlargement of the SAM in the mutant (Williams et al 2005)
While WUS is closely related to miR165166 in the shoot apical meristem
development the underlying molecular mechanisms are currently unclear It has been
observed that the miR166-over producing men1 plants have an arrested meristem
development (Jung amp Park 2007) In addition the heterozygous men1+ and the
homozygous men1 plants show different expression patterns of WUS and CLV3 It
appears that miR166 regulates WUS indirectly and influences the expressional balance
between WUS and CLV3 through a negative feedback loop (Jung amp Park 2007 de
Alba et al 2013)
The phv phb athb15 triple mutant has an enlarged shoot apical meristems the
rev phb phv mutant does not have functional shoot apical meristems (Prigge et al
2005) This distinction may be explained by the fact that while miR166 targets
primarily PHB PHV and ATHB15 miR165 targets all the five HD-ZIP III genes (Zhou
et al 2007) Consistent with this transgenic plants overproducing miR166 are distinct
from those overproducing miR165 The miR165-overproducing transgenic plants lack
functional shoot apical meristem and a pin-like structure is formed at the apical region
of the mutant These phenotypes are quite similar to rev phb phv triple mutant (Zhou et
al 2007 Liu et al 2013) The phenotypic distinction may because by the difference
of the cleavage efficiencies of the target mRNAs In accordance with this notion a few
35S miR166a transgenic lines exhibit no shoot apical meristems The men1
homozygous plants also show arrested meristem development (Jung amp Park 2007)
Chapter 1 Introduction and Review of Literature
46
MiR164 cleaves the CUC1 and CUC2 mRNAs as well as the NAC1 mRNA
CUC1 and CUC2 determines the organ boundary in the meristem Phenotypes of
transgenic plants that overproduce miR164 are similar to those of the cuc1cuc2 double
mutant that exhibits embryonic developmental defects such as cup-shaped cotyledons
and fused cotyledons (Laufs et al 2004) When a miR164-resistant CUC2 is
expressed the boundary domain is enlarged CUC also plays a role in embryonic shoot
meristem formation by regulating the SHOOT MERISTEMLESS gene It was therefore
concluded that miR164-mediated cleavage of CUC1 and CUC2 mRNAs determines the
sizes of the boundary domains in the shoot apical meristem and regulates separation of
the organs by restricting cell proliferation (Laufs et al 2004 de Alba et al 2013)
11364 Leaf development
The role of miR319 has been extensively studied in leaf development A dominant
jaw-D mutant overproducing miR319 shows uneven curved leaves with enlarged size
and five TCP genes which are closely related to the CINCINNATA (CIN) gene in
Antirrhinum majus are down regulated in the mutant suggesting that miR319 mediated
regulation of the TCP transcription factor genes are important for the final step of
leaf shaping (Palatnik et al 2003 Naz et al 2013) In addition to leaf shape and size
leaf polarity is also specified by miRNA MiR166-mediated cleavage of the HD-ZIPIII
mRNAs is a well to establish leaf polarity This discovery originates from the gain-of-
function mutants such as phb-1d and phv-1d wherein point mutations in the
complementary sites to miRNA helps the mRNA to evade or resist from miRNA-
mediated cleavage (McConnell et al 2001 Emery et al 2003 Naz et al 2013)
Plant leaves have a dorso-ventral polarity consisting of the adaxial (upper
surface) and abaxial (lower surface) sides The adaxial side is closer to the shoot
apical meristem Adaxial identity is specified by functionally redundant class III HD-
ZIP family genes such as PHB PHV and REV Ectopic expression of each genes
result in adaxialized leaves Dominant phb-1d and phv-1d mutants exhibit
transformation of the abaxial to adaxial fate and have rod-shaped leaves In contrast
miR165166-overproducing plants show pin-like cotyledons radicalized leaves and
abaxialized leaves (Chitwood et al 2007 Pattanayak et al 2013)
Leaf asymmetry is determined by miR166-mediated restriction of the
expression domains of the HD-ZIPIII genes The HD-ZIPIII genes are expressed in the
Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
40
near-perfect complementarity induces mRNA cleavage central mismatches trigger
translational repression (Carrington amp Ambros 2003 Jones-Rhoades amp Bartel 2004)
Unlike most animal miRNAs plant miRNAs have near-perfect or perfect
complementarity to their targets and mRNA cleavage is assumed to be a predominant
mechanism by which miRNAs regulate gene expression in plants
In RNA cleavage mechanism miRNAs guide the AGO component of RISC to
cleave a single phosphodiester bond of the target mRNA within the miRNA-binding
site (Llave et al 2002b Tang et al 2003) The truncated fragments are then released
and degraded freeing the RISC to recognize and cleave another target mRNA This has
been validated by the detection of 3prime cleavage products that have 5prime ends that start at the
middle of the complementary regions The mRNA cleavage products are then further
degraded by other mechanisms The 3prime cleavage products of some miRNA targets are
degraded by the 5prime-3prime exonuclease XRN4 an Arabidopsis homolog of the major Yeast
mRNA degrading exonuclease Xrn1p It has been observed that the 3prime cleavage
products of some target mRNAs were accumulated in the xrn4 mutants (Souret et al
2004 Dalmay 2013) However the 3prime cleavage products of other miRNA targets do
not accumulate in the xrn4 mutants indicating that there are also XRN4 independent
mechanisms The 5prime cleavage products are typically untraceable by RNA gel blot
hybridization but can be detected by sensitive PCR based methods It has been found
that the 5prime cleavage products tend to obtain an oligo U tail at the 3prime ends This 3prime U tail
is correlated with shortening of the 5prime cleavage products from the 5prime end leading to the
conclusion that the 3prime uridylation causes 5 prime- 3 prime exonucleolytic decay of the 5prime cleavage
products (Shen amp Goodman 2004)
DCL1 HYL1 and SE interact with each other and are co-localized in the
nuclear bodies termed Dicing bodies or D-bodies in which some pri-miRNA shave
been found suggesting that D-bodies serve as a center for active miRNA processing
(Fang amp Spector 2007 Fujioka et al 2007 Song et al 2007) The miRNA
processing site appears to be distinct from the Cajal bodies that have previously been
found as a siRNA processing site (Pontes amp Pikaard 2008) The D-bodies include
SmD3 and SmB proteins that are found in the Cajal bodies However At collin an
additional marker for the Cajal bodies is not found in the D-bodies Moreover the D-
bodies are present in an At collin mutant lacking the Cajal bodies The pri-miRNAs of
miR171A and miR159A genes have been found to be co-localized with HYL1 and
Chapter 1 Introduction and Review of Literature
41
DCL1 in the D- bodies that are distinct from the Cajal bodies (Song et al 2007)
11352 MiRNA-directed translational repression
Translation repression is another mechanism exerted by plant miRNAs The regulatory
mechanism of miR172 which regulates flowering time and floral organ identity is
consistent with translation repression MiR172 over production does not affect the
transcript levels of APETALA2 (AP2) and AP2- like target mRNAs Instead the AP2
protein level is reduced in the miR172 over expressing mutant (Aukerman amp Sakai
2003 Chen 2004 Dalmay 2013) It was later shown that miR172 can also direct the
cleavage of the target mRNAs although the steady-state mRNA level is unchanged due
to feedback regulation (Schwab et al 2005 Jung et al 2007) It is clear that miR172
regulates its target genes primarily by translational repression rather than mRNA
cleavage Recently it has been reported that miR156157 and miR854 also regulate
their target genes through translational repression (Arteaga-Vazquez et al 2006
Gandikota et al 2007 Dalmay 2013) It was once thought that translational repression
is an exception to the rule However experimental evidence suggest a widespread
coexistence of translational repression and mRNA cleavage (Brodersen et al 2008)
1136 Regulatory roles of miRNAs in plant development
The miRNA target prediction has been a difficult task in order to obtain regulatory
targets without bringing in too many false predictions Plant growth and developmental
processes regulated by miRNAs are quite diverse and enormous Furthermore plant
miRNAs work cooperatively or antagonistically to establish a balanced regulation This
notion is well consistent with the findings that mutations in the regulators of miRNA
biogenesis transport and processing such as AGO1 DCL1 HYL1 HEN1 ABH1
and HASTY cause pleiotropic phenotypes (Mallory amp Vaucheret 2006 de Alba et al
2013) To date the roles of miRNAs have been mostly deduced from obtaining miRNA
over expressing transgenic plants or gain-of-function mutants in which miRNA-
resistant target genes are ectopically expressed
11361 Phase transition
Plants pass through a series of developmental phase transitions during their life span
beginning with seed germination continuing through vegetative phase change
reproductive phase change flowering initiation and finally seed production for the next
generation Because of their importance in understanding plant developmental
Chapter 1 Introduction and Review of Literature
42
processes genes regulating developmental transitions have been extensively studied
and underlying molecular mechanisms have been explored in various plant species
Majority of researches were mainly focused on vegetative phase change and
reproductive transition in Arabidopsis and Maize (Poethig 2003 Baurle amp Dean
2006) Particularly the mutations of the genes encoding various regulators of miRNA
biogenesis and transport such as HST SE and AGO exhibit altered juvenile to adult
vegetative phase change and vegetative to reproductive change (Hunter et al 2003
Peragine et al 2004 Vazquez et al 2004a Jones-Rhoades et al 2006 de Alba et al
2013)
Over-expression of the miR156a gene causes late flowering and delays
vegetative phase change (Wu amp Poethig 2006 Gandikota et al 2007 Wang et al
2008) It has been shown that miR156 negatively regulates a group of genes encoding
SQUAMOSA PROMOTER-BINDING PROTEIN-LIKE (Lewis et al 2007 de Alba et
al 2013 Rogers amp Chen 2013) transcription factors such as SPL3 SPL4 and SPL5
that promote flowering initiation In contrast transgenic plants over expressing
miR156-resistant SPL345 genes are early flowering Interestingly the endogenous
level of miR156 is temporally regulated In the early juvenile vegetative phase the
level of miR156 is very high but decreases rapidly before the onset of the adult juvenile
phase (Wu amp Poethig 2006)
The Arabidopsis early activation tagged (eat-D) mutant exhibits early flowering
with disrupted floral structure The mutant phenotypes are caused by over expression of
the miR172a-2gene (Aukerman amp Sakai 2003) MiR172 regulates a small group of
genes encoding APETALLA2 (AP2)-like transcription factors such as TARGET OF
EAT1 (TOE1) TOE2 SCHLAFMUTZE (SMZ) and SCHNARCHZAPFEN(SNZ)
TOE1 TOE2 SMZ and SNZ have been shown to participate in their productive phase
change by repressing a floral integrator FLOWERING LOCUS T (FT)(Jung et al
2007) Consistent with this toe1-2D 35STOE2 35SSMZ and 35SSNZ plants
exhibit late flowering (Jung et al 2007) MiR172 is also regulated temporally and
highly expressed in the adult vegetative phase both in Arabidopsis and maize (Chuck et
al 2007) Because the temporal expression patterns of miR156 and miR172 are
complementary to each other it has been suggested that the two miRNAs interact with
each other in regulating phase change (Chuck et al 2009 de Alba et al 2013) In
support of this view it has been shown that miR172 is down-regulated in the
Chapter 1 Introduction and Review of Literature
43
Corngrass1(Cg1) mutant in which miR156 is overproduced (Chuck et al 2007 de
Alba et al 2013) However there is no direct evidence for the direct linkage between
miR156 and miR172 It is also envisioned that miR156 and miR172 would not be
directly related For example miR156 regulates plastochron index that is the inverse of
leaf initiation rate and thus frequently used to analyze temporal patterns of plant
development In contrast miR172 does not affect plastochron (Wang et al 2008) The
down-regulation of miR172 in the maize miR156-over producing mutants would be
due to the prolonged juvenile vegetative phase in the mutants
Another miRNA that regulates flowering initiation is miR159 It regulates
MYB33 and MYB65 which are closely related to the barley GAMY B transcription
factor that responds to gibberellic acid (GA) The level of miR159 is elevated in GA-
treated seedlings but reduced in the GA biosynthetic ga1 mutant Transgenic plants
over producing miR159 exhibit delayed floral induction under short days (Achard et
al 2004) It is been suggested that MYB33 mediates GA signal to regulate LEAFY
(LFY) These observations indicate that miR159 plays an important role in the GA
signaling pathways that regulate proper floral induction under short days (Achard et al
2004)
11362 Floral development
Arabidopsis flowers consist of four distinct organs They arise in a characteristic
pattern within concentric rings called whorls from outside to inside four sepals four
petals six stamens and the central pistil consisting of two carpels Identities of the
floral organs are determined by three classes of regulatory genes classA classB and
class C genes (Krizek amp Fletcher 2005) The A and C class genes act antagonistically
to restrict their expression domains (Bowman et al 1991)
It is notable that miR172 also regulates floral architecture in addition to its role
in the timing of flowering initiation MiR172 inhibits the translation of the AP2 mRNA
(Chen 2004) Transgenic plants overproducing miR172 not only exhibit early
flowering but also show abnormal floral development which is quite similar to those
of the class A gene mutants such as the ap2 mutants (Aukerman amp Sakai 2003 de
Alba et al 2013) When the activity of the class A genes is inactivated that of the class
C genes such as AGAMOUS (AG) is elevated As a result the miR172- overproducing
transgenic plants exhibit no petals along with homeotic transformation of the sepals to
Chapter 1 Introduction and Review of Literature
44
the carpels (Aukerman amp Sakai 2003 Chen 2004)
Interestingly miR166 also regulates floral architecture The role of miR165166
in the regulation of meristem activity is closely linked to floral meristem formation and
organization (Zhao et al 2007 Miyashima et al 2013) Floral structure is severely
disrupted in the miR165166-overproducing mutants such as men1 and jba-1D in
which miR166 is overproduced (Williams et al 2005b Jung amp Park 2007) The men1
and jba-1D mutants have smaller gynoecium and reduced carpel number In an extreme
case the jba-1D homozygous plants lack visible carpels (Williams et al 2005b)
Similar phenotypes have been observed in the miR165 overproducers (Zhao et al
2007) It has been suggested that the phenotypes are possibly caused by altered AG
expression in the floral meristem (Jung amp Park 2007 Turchi et al 2013)
Other miRNAs also affect floral development Overproduction of miRNAs
involved in auxin signaling also affects floral architecture Transgenic plants
overproducing miR167 have defects in the formation of ovule and anther with reduced
fertility (Ru et al 2006) It has been shown that miR167 targets ARF6 and ARF8 that
play a role in gynoecium and stamen development and in regulating fertility (Ru et al
2006 Wu et al 2006) These observations suggest that auxin signals are also closely
related to floral organogenesis In addition transgenic plants over-producing miR164
and the cuc1cuc2 double mutants show fused sepals and reduced petals (Laufs et al
2004 Turchi et al 2013) suggesting that miR164 is also related to floral meristem
activity and boundary specification of the meristematic region
11363 Shoot apical meristem development
Shoot apical meristem comprises self-maintaining totipotent cells The shoot apical
meristem activity affects diverse aspects of plant developmental processes including
leaf development vascular development and lateral organ polarity (Bowman et al
2002) Because miR165166 plays a primary role in meristem formation
overproduction of miR165166 and multiple knockout mutants of its target genes
exhibit pleiotropic pheonotypes (McConnell et al 2001 Emery et al 2003 Kim et al
2005 Williams et al 2005b)
The targets of miR165166 include genes encoding the homeo domain-leucine
zipper (HD-ZIP) class III transcription factors such as PHABULOSA(PHB)
PHAVOLUTA (PHV) REVOLUTA(REV) ATHB15CORONA (CNA) and ATHB8
Chapter 1 Introduction and Review of Literature
45
PHB PHV and ATHB15 have overlapping functions in controlling meristem
development (Turchi et al 2013) Indeed the phb phv athb15 triple mutants have an
enlarged shoot apical meristem Consistent with this the miR166- overproducing men1
and jba-1D mutants exhibit enlarged shoot apical meristems (Kim et al 2005
Williams et al 2005b Turchi et al 2013) In the mutants the shoot apical meristems
are multiplied and large
The development of shoot apical meristems is also regulated by the WUSCHEL
(WUS)ndashCLAVATA (CLV) signaling pathway WUS positively regulates cell
proliferation In contrast CLV3 which is expressed in stem cells limits the WUS
expression and thus inhibits cell proliferation in the SAM (Sharma et al 2003)
Consistent with this notion WUS is expressed to a higher level in jba-1D
explaining the ectopic enlargement of the SAM in the mutant (Williams et al 2005)
While WUS is closely related to miR165166 in the shoot apical meristem
development the underlying molecular mechanisms are currently unclear It has been
observed that the miR166-over producing men1 plants have an arrested meristem
development (Jung amp Park 2007) In addition the heterozygous men1+ and the
homozygous men1 plants show different expression patterns of WUS and CLV3 It
appears that miR166 regulates WUS indirectly and influences the expressional balance
between WUS and CLV3 through a negative feedback loop (Jung amp Park 2007 de
Alba et al 2013)
The phv phb athb15 triple mutant has an enlarged shoot apical meristems the
rev phb phv mutant does not have functional shoot apical meristems (Prigge et al
2005) This distinction may be explained by the fact that while miR166 targets
primarily PHB PHV and ATHB15 miR165 targets all the five HD-ZIP III genes (Zhou
et al 2007) Consistent with this transgenic plants overproducing miR166 are distinct
from those overproducing miR165 The miR165-overproducing transgenic plants lack
functional shoot apical meristem and a pin-like structure is formed at the apical region
of the mutant These phenotypes are quite similar to rev phb phv triple mutant (Zhou et
al 2007 Liu et al 2013) The phenotypic distinction may because by the difference
of the cleavage efficiencies of the target mRNAs In accordance with this notion a few
35S miR166a transgenic lines exhibit no shoot apical meristems The men1
homozygous plants also show arrested meristem development (Jung amp Park 2007)
Chapter 1 Introduction and Review of Literature
46
MiR164 cleaves the CUC1 and CUC2 mRNAs as well as the NAC1 mRNA
CUC1 and CUC2 determines the organ boundary in the meristem Phenotypes of
transgenic plants that overproduce miR164 are similar to those of the cuc1cuc2 double
mutant that exhibits embryonic developmental defects such as cup-shaped cotyledons
and fused cotyledons (Laufs et al 2004) When a miR164-resistant CUC2 is
expressed the boundary domain is enlarged CUC also plays a role in embryonic shoot
meristem formation by regulating the SHOOT MERISTEMLESS gene It was therefore
concluded that miR164-mediated cleavage of CUC1 and CUC2 mRNAs determines the
sizes of the boundary domains in the shoot apical meristem and regulates separation of
the organs by restricting cell proliferation (Laufs et al 2004 de Alba et al 2013)
11364 Leaf development
The role of miR319 has been extensively studied in leaf development A dominant
jaw-D mutant overproducing miR319 shows uneven curved leaves with enlarged size
and five TCP genes which are closely related to the CINCINNATA (CIN) gene in
Antirrhinum majus are down regulated in the mutant suggesting that miR319 mediated
regulation of the TCP transcription factor genes are important for the final step of
leaf shaping (Palatnik et al 2003 Naz et al 2013) In addition to leaf shape and size
leaf polarity is also specified by miRNA MiR166-mediated cleavage of the HD-ZIPIII
mRNAs is a well to establish leaf polarity This discovery originates from the gain-of-
function mutants such as phb-1d and phv-1d wherein point mutations in the
complementary sites to miRNA helps the mRNA to evade or resist from miRNA-
mediated cleavage (McConnell et al 2001 Emery et al 2003 Naz et al 2013)
Plant leaves have a dorso-ventral polarity consisting of the adaxial (upper
surface) and abaxial (lower surface) sides The adaxial side is closer to the shoot
apical meristem Adaxial identity is specified by functionally redundant class III HD-
ZIP family genes such as PHB PHV and REV Ectopic expression of each genes
result in adaxialized leaves Dominant phb-1d and phv-1d mutants exhibit
transformation of the abaxial to adaxial fate and have rod-shaped leaves In contrast
miR165166-overproducing plants show pin-like cotyledons radicalized leaves and
abaxialized leaves (Chitwood et al 2007 Pattanayak et al 2013)
Leaf asymmetry is determined by miR166-mediated restriction of the
expression domains of the HD-ZIPIII genes The HD-ZIPIII genes are expressed in the
Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
41
DCL1 in the D- bodies that are distinct from the Cajal bodies (Song et al 2007)
11352 MiRNA-directed translational repression
Translation repression is another mechanism exerted by plant miRNAs The regulatory
mechanism of miR172 which regulates flowering time and floral organ identity is
consistent with translation repression MiR172 over production does not affect the
transcript levels of APETALA2 (AP2) and AP2- like target mRNAs Instead the AP2
protein level is reduced in the miR172 over expressing mutant (Aukerman amp Sakai
2003 Chen 2004 Dalmay 2013) It was later shown that miR172 can also direct the
cleavage of the target mRNAs although the steady-state mRNA level is unchanged due
to feedback regulation (Schwab et al 2005 Jung et al 2007) It is clear that miR172
regulates its target genes primarily by translational repression rather than mRNA
cleavage Recently it has been reported that miR156157 and miR854 also regulate
their target genes through translational repression (Arteaga-Vazquez et al 2006
Gandikota et al 2007 Dalmay 2013) It was once thought that translational repression
is an exception to the rule However experimental evidence suggest a widespread
coexistence of translational repression and mRNA cleavage (Brodersen et al 2008)
1136 Regulatory roles of miRNAs in plant development
The miRNA target prediction has been a difficult task in order to obtain regulatory
targets without bringing in too many false predictions Plant growth and developmental
processes regulated by miRNAs are quite diverse and enormous Furthermore plant
miRNAs work cooperatively or antagonistically to establish a balanced regulation This
notion is well consistent with the findings that mutations in the regulators of miRNA
biogenesis transport and processing such as AGO1 DCL1 HYL1 HEN1 ABH1
and HASTY cause pleiotropic phenotypes (Mallory amp Vaucheret 2006 de Alba et al
2013) To date the roles of miRNAs have been mostly deduced from obtaining miRNA
over expressing transgenic plants or gain-of-function mutants in which miRNA-
resistant target genes are ectopically expressed
11361 Phase transition
Plants pass through a series of developmental phase transitions during their life span
beginning with seed germination continuing through vegetative phase change
reproductive phase change flowering initiation and finally seed production for the next
generation Because of their importance in understanding plant developmental
Chapter 1 Introduction and Review of Literature
42
processes genes regulating developmental transitions have been extensively studied
and underlying molecular mechanisms have been explored in various plant species
Majority of researches were mainly focused on vegetative phase change and
reproductive transition in Arabidopsis and Maize (Poethig 2003 Baurle amp Dean
2006) Particularly the mutations of the genes encoding various regulators of miRNA
biogenesis and transport such as HST SE and AGO exhibit altered juvenile to adult
vegetative phase change and vegetative to reproductive change (Hunter et al 2003
Peragine et al 2004 Vazquez et al 2004a Jones-Rhoades et al 2006 de Alba et al
2013)
Over-expression of the miR156a gene causes late flowering and delays
vegetative phase change (Wu amp Poethig 2006 Gandikota et al 2007 Wang et al
2008) It has been shown that miR156 negatively regulates a group of genes encoding
SQUAMOSA PROMOTER-BINDING PROTEIN-LIKE (Lewis et al 2007 de Alba et
al 2013 Rogers amp Chen 2013) transcription factors such as SPL3 SPL4 and SPL5
that promote flowering initiation In contrast transgenic plants over expressing
miR156-resistant SPL345 genes are early flowering Interestingly the endogenous
level of miR156 is temporally regulated In the early juvenile vegetative phase the
level of miR156 is very high but decreases rapidly before the onset of the adult juvenile
phase (Wu amp Poethig 2006)
The Arabidopsis early activation tagged (eat-D) mutant exhibits early flowering
with disrupted floral structure The mutant phenotypes are caused by over expression of
the miR172a-2gene (Aukerman amp Sakai 2003) MiR172 regulates a small group of
genes encoding APETALLA2 (AP2)-like transcription factors such as TARGET OF
EAT1 (TOE1) TOE2 SCHLAFMUTZE (SMZ) and SCHNARCHZAPFEN(SNZ)
TOE1 TOE2 SMZ and SNZ have been shown to participate in their productive phase
change by repressing a floral integrator FLOWERING LOCUS T (FT)(Jung et al
2007) Consistent with this toe1-2D 35STOE2 35SSMZ and 35SSNZ plants
exhibit late flowering (Jung et al 2007) MiR172 is also regulated temporally and
highly expressed in the adult vegetative phase both in Arabidopsis and maize (Chuck et
al 2007) Because the temporal expression patterns of miR156 and miR172 are
complementary to each other it has been suggested that the two miRNAs interact with
each other in regulating phase change (Chuck et al 2009 de Alba et al 2013) In
support of this view it has been shown that miR172 is down-regulated in the
Chapter 1 Introduction and Review of Literature
43
Corngrass1(Cg1) mutant in which miR156 is overproduced (Chuck et al 2007 de
Alba et al 2013) However there is no direct evidence for the direct linkage between
miR156 and miR172 It is also envisioned that miR156 and miR172 would not be
directly related For example miR156 regulates plastochron index that is the inverse of
leaf initiation rate and thus frequently used to analyze temporal patterns of plant
development In contrast miR172 does not affect plastochron (Wang et al 2008) The
down-regulation of miR172 in the maize miR156-over producing mutants would be
due to the prolonged juvenile vegetative phase in the mutants
Another miRNA that regulates flowering initiation is miR159 It regulates
MYB33 and MYB65 which are closely related to the barley GAMY B transcription
factor that responds to gibberellic acid (GA) The level of miR159 is elevated in GA-
treated seedlings but reduced in the GA biosynthetic ga1 mutant Transgenic plants
over producing miR159 exhibit delayed floral induction under short days (Achard et
al 2004) It is been suggested that MYB33 mediates GA signal to regulate LEAFY
(LFY) These observations indicate that miR159 plays an important role in the GA
signaling pathways that regulate proper floral induction under short days (Achard et al
2004)
11362 Floral development
Arabidopsis flowers consist of four distinct organs They arise in a characteristic
pattern within concentric rings called whorls from outside to inside four sepals four
petals six stamens and the central pistil consisting of two carpels Identities of the
floral organs are determined by three classes of regulatory genes classA classB and
class C genes (Krizek amp Fletcher 2005) The A and C class genes act antagonistically
to restrict their expression domains (Bowman et al 1991)
It is notable that miR172 also regulates floral architecture in addition to its role
in the timing of flowering initiation MiR172 inhibits the translation of the AP2 mRNA
(Chen 2004) Transgenic plants overproducing miR172 not only exhibit early
flowering but also show abnormal floral development which is quite similar to those
of the class A gene mutants such as the ap2 mutants (Aukerman amp Sakai 2003 de
Alba et al 2013) When the activity of the class A genes is inactivated that of the class
C genes such as AGAMOUS (AG) is elevated As a result the miR172- overproducing
transgenic plants exhibit no petals along with homeotic transformation of the sepals to
Chapter 1 Introduction and Review of Literature
44
the carpels (Aukerman amp Sakai 2003 Chen 2004)
Interestingly miR166 also regulates floral architecture The role of miR165166
in the regulation of meristem activity is closely linked to floral meristem formation and
organization (Zhao et al 2007 Miyashima et al 2013) Floral structure is severely
disrupted in the miR165166-overproducing mutants such as men1 and jba-1D in
which miR166 is overproduced (Williams et al 2005b Jung amp Park 2007) The men1
and jba-1D mutants have smaller gynoecium and reduced carpel number In an extreme
case the jba-1D homozygous plants lack visible carpels (Williams et al 2005b)
Similar phenotypes have been observed in the miR165 overproducers (Zhao et al
2007) It has been suggested that the phenotypes are possibly caused by altered AG
expression in the floral meristem (Jung amp Park 2007 Turchi et al 2013)
Other miRNAs also affect floral development Overproduction of miRNAs
involved in auxin signaling also affects floral architecture Transgenic plants
overproducing miR167 have defects in the formation of ovule and anther with reduced
fertility (Ru et al 2006) It has been shown that miR167 targets ARF6 and ARF8 that
play a role in gynoecium and stamen development and in regulating fertility (Ru et al
2006 Wu et al 2006) These observations suggest that auxin signals are also closely
related to floral organogenesis In addition transgenic plants over-producing miR164
and the cuc1cuc2 double mutants show fused sepals and reduced petals (Laufs et al
2004 Turchi et al 2013) suggesting that miR164 is also related to floral meristem
activity and boundary specification of the meristematic region
11363 Shoot apical meristem development
Shoot apical meristem comprises self-maintaining totipotent cells The shoot apical
meristem activity affects diverse aspects of plant developmental processes including
leaf development vascular development and lateral organ polarity (Bowman et al
2002) Because miR165166 plays a primary role in meristem formation
overproduction of miR165166 and multiple knockout mutants of its target genes
exhibit pleiotropic pheonotypes (McConnell et al 2001 Emery et al 2003 Kim et al
2005 Williams et al 2005b)
The targets of miR165166 include genes encoding the homeo domain-leucine
zipper (HD-ZIP) class III transcription factors such as PHABULOSA(PHB)
PHAVOLUTA (PHV) REVOLUTA(REV) ATHB15CORONA (CNA) and ATHB8
Chapter 1 Introduction and Review of Literature
45
PHB PHV and ATHB15 have overlapping functions in controlling meristem
development (Turchi et al 2013) Indeed the phb phv athb15 triple mutants have an
enlarged shoot apical meristem Consistent with this the miR166- overproducing men1
and jba-1D mutants exhibit enlarged shoot apical meristems (Kim et al 2005
Williams et al 2005b Turchi et al 2013) In the mutants the shoot apical meristems
are multiplied and large
The development of shoot apical meristems is also regulated by the WUSCHEL
(WUS)ndashCLAVATA (CLV) signaling pathway WUS positively regulates cell
proliferation In contrast CLV3 which is expressed in stem cells limits the WUS
expression and thus inhibits cell proliferation in the SAM (Sharma et al 2003)
Consistent with this notion WUS is expressed to a higher level in jba-1D
explaining the ectopic enlargement of the SAM in the mutant (Williams et al 2005)
While WUS is closely related to miR165166 in the shoot apical meristem
development the underlying molecular mechanisms are currently unclear It has been
observed that the miR166-over producing men1 plants have an arrested meristem
development (Jung amp Park 2007) In addition the heterozygous men1+ and the
homozygous men1 plants show different expression patterns of WUS and CLV3 It
appears that miR166 regulates WUS indirectly and influences the expressional balance
between WUS and CLV3 through a negative feedback loop (Jung amp Park 2007 de
Alba et al 2013)
The phv phb athb15 triple mutant has an enlarged shoot apical meristems the
rev phb phv mutant does not have functional shoot apical meristems (Prigge et al
2005) This distinction may be explained by the fact that while miR166 targets
primarily PHB PHV and ATHB15 miR165 targets all the five HD-ZIP III genes (Zhou
et al 2007) Consistent with this transgenic plants overproducing miR166 are distinct
from those overproducing miR165 The miR165-overproducing transgenic plants lack
functional shoot apical meristem and a pin-like structure is formed at the apical region
of the mutant These phenotypes are quite similar to rev phb phv triple mutant (Zhou et
al 2007 Liu et al 2013) The phenotypic distinction may because by the difference
of the cleavage efficiencies of the target mRNAs In accordance with this notion a few
35S miR166a transgenic lines exhibit no shoot apical meristems The men1
homozygous plants also show arrested meristem development (Jung amp Park 2007)
Chapter 1 Introduction and Review of Literature
46
MiR164 cleaves the CUC1 and CUC2 mRNAs as well as the NAC1 mRNA
CUC1 and CUC2 determines the organ boundary in the meristem Phenotypes of
transgenic plants that overproduce miR164 are similar to those of the cuc1cuc2 double
mutant that exhibits embryonic developmental defects such as cup-shaped cotyledons
and fused cotyledons (Laufs et al 2004) When a miR164-resistant CUC2 is
expressed the boundary domain is enlarged CUC also plays a role in embryonic shoot
meristem formation by regulating the SHOOT MERISTEMLESS gene It was therefore
concluded that miR164-mediated cleavage of CUC1 and CUC2 mRNAs determines the
sizes of the boundary domains in the shoot apical meristem and regulates separation of
the organs by restricting cell proliferation (Laufs et al 2004 de Alba et al 2013)
11364 Leaf development
The role of miR319 has been extensively studied in leaf development A dominant
jaw-D mutant overproducing miR319 shows uneven curved leaves with enlarged size
and five TCP genes which are closely related to the CINCINNATA (CIN) gene in
Antirrhinum majus are down regulated in the mutant suggesting that miR319 mediated
regulation of the TCP transcription factor genes are important for the final step of
leaf shaping (Palatnik et al 2003 Naz et al 2013) In addition to leaf shape and size
leaf polarity is also specified by miRNA MiR166-mediated cleavage of the HD-ZIPIII
mRNAs is a well to establish leaf polarity This discovery originates from the gain-of-
function mutants such as phb-1d and phv-1d wherein point mutations in the
complementary sites to miRNA helps the mRNA to evade or resist from miRNA-
mediated cleavage (McConnell et al 2001 Emery et al 2003 Naz et al 2013)
Plant leaves have a dorso-ventral polarity consisting of the adaxial (upper
surface) and abaxial (lower surface) sides The adaxial side is closer to the shoot
apical meristem Adaxial identity is specified by functionally redundant class III HD-
ZIP family genes such as PHB PHV and REV Ectopic expression of each genes
result in adaxialized leaves Dominant phb-1d and phv-1d mutants exhibit
transformation of the abaxial to adaxial fate and have rod-shaped leaves In contrast
miR165166-overproducing plants show pin-like cotyledons radicalized leaves and
abaxialized leaves (Chitwood et al 2007 Pattanayak et al 2013)
Leaf asymmetry is determined by miR166-mediated restriction of the
expression domains of the HD-ZIPIII genes The HD-ZIPIII genes are expressed in the
Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
42
processes genes regulating developmental transitions have been extensively studied
and underlying molecular mechanisms have been explored in various plant species
Majority of researches were mainly focused on vegetative phase change and
reproductive transition in Arabidopsis and Maize (Poethig 2003 Baurle amp Dean
2006) Particularly the mutations of the genes encoding various regulators of miRNA
biogenesis and transport such as HST SE and AGO exhibit altered juvenile to adult
vegetative phase change and vegetative to reproductive change (Hunter et al 2003
Peragine et al 2004 Vazquez et al 2004a Jones-Rhoades et al 2006 de Alba et al
2013)
Over-expression of the miR156a gene causes late flowering and delays
vegetative phase change (Wu amp Poethig 2006 Gandikota et al 2007 Wang et al
2008) It has been shown that miR156 negatively regulates a group of genes encoding
SQUAMOSA PROMOTER-BINDING PROTEIN-LIKE (Lewis et al 2007 de Alba et
al 2013 Rogers amp Chen 2013) transcription factors such as SPL3 SPL4 and SPL5
that promote flowering initiation In contrast transgenic plants over expressing
miR156-resistant SPL345 genes are early flowering Interestingly the endogenous
level of miR156 is temporally regulated In the early juvenile vegetative phase the
level of miR156 is very high but decreases rapidly before the onset of the adult juvenile
phase (Wu amp Poethig 2006)
The Arabidopsis early activation tagged (eat-D) mutant exhibits early flowering
with disrupted floral structure The mutant phenotypes are caused by over expression of
the miR172a-2gene (Aukerman amp Sakai 2003) MiR172 regulates a small group of
genes encoding APETALLA2 (AP2)-like transcription factors such as TARGET OF
EAT1 (TOE1) TOE2 SCHLAFMUTZE (SMZ) and SCHNARCHZAPFEN(SNZ)
TOE1 TOE2 SMZ and SNZ have been shown to participate in their productive phase
change by repressing a floral integrator FLOWERING LOCUS T (FT)(Jung et al
2007) Consistent with this toe1-2D 35STOE2 35SSMZ and 35SSNZ plants
exhibit late flowering (Jung et al 2007) MiR172 is also regulated temporally and
highly expressed in the adult vegetative phase both in Arabidopsis and maize (Chuck et
al 2007) Because the temporal expression patterns of miR156 and miR172 are
complementary to each other it has been suggested that the two miRNAs interact with
each other in regulating phase change (Chuck et al 2009 de Alba et al 2013) In
support of this view it has been shown that miR172 is down-regulated in the
Chapter 1 Introduction and Review of Literature
43
Corngrass1(Cg1) mutant in which miR156 is overproduced (Chuck et al 2007 de
Alba et al 2013) However there is no direct evidence for the direct linkage between
miR156 and miR172 It is also envisioned that miR156 and miR172 would not be
directly related For example miR156 regulates plastochron index that is the inverse of
leaf initiation rate and thus frequently used to analyze temporal patterns of plant
development In contrast miR172 does not affect plastochron (Wang et al 2008) The
down-regulation of miR172 in the maize miR156-over producing mutants would be
due to the prolonged juvenile vegetative phase in the mutants
Another miRNA that regulates flowering initiation is miR159 It regulates
MYB33 and MYB65 which are closely related to the barley GAMY B transcription
factor that responds to gibberellic acid (GA) The level of miR159 is elevated in GA-
treated seedlings but reduced in the GA biosynthetic ga1 mutant Transgenic plants
over producing miR159 exhibit delayed floral induction under short days (Achard et
al 2004) It is been suggested that MYB33 mediates GA signal to regulate LEAFY
(LFY) These observations indicate that miR159 plays an important role in the GA
signaling pathways that regulate proper floral induction under short days (Achard et al
2004)
11362 Floral development
Arabidopsis flowers consist of four distinct organs They arise in a characteristic
pattern within concentric rings called whorls from outside to inside four sepals four
petals six stamens and the central pistil consisting of two carpels Identities of the
floral organs are determined by three classes of regulatory genes classA classB and
class C genes (Krizek amp Fletcher 2005) The A and C class genes act antagonistically
to restrict their expression domains (Bowman et al 1991)
It is notable that miR172 also regulates floral architecture in addition to its role
in the timing of flowering initiation MiR172 inhibits the translation of the AP2 mRNA
(Chen 2004) Transgenic plants overproducing miR172 not only exhibit early
flowering but also show abnormal floral development which is quite similar to those
of the class A gene mutants such as the ap2 mutants (Aukerman amp Sakai 2003 de
Alba et al 2013) When the activity of the class A genes is inactivated that of the class
C genes such as AGAMOUS (AG) is elevated As a result the miR172- overproducing
transgenic plants exhibit no petals along with homeotic transformation of the sepals to
Chapter 1 Introduction and Review of Literature
44
the carpels (Aukerman amp Sakai 2003 Chen 2004)
Interestingly miR166 also regulates floral architecture The role of miR165166
in the regulation of meristem activity is closely linked to floral meristem formation and
organization (Zhao et al 2007 Miyashima et al 2013) Floral structure is severely
disrupted in the miR165166-overproducing mutants such as men1 and jba-1D in
which miR166 is overproduced (Williams et al 2005b Jung amp Park 2007) The men1
and jba-1D mutants have smaller gynoecium and reduced carpel number In an extreme
case the jba-1D homozygous plants lack visible carpels (Williams et al 2005b)
Similar phenotypes have been observed in the miR165 overproducers (Zhao et al
2007) It has been suggested that the phenotypes are possibly caused by altered AG
expression in the floral meristem (Jung amp Park 2007 Turchi et al 2013)
Other miRNAs also affect floral development Overproduction of miRNAs
involved in auxin signaling also affects floral architecture Transgenic plants
overproducing miR167 have defects in the formation of ovule and anther with reduced
fertility (Ru et al 2006) It has been shown that miR167 targets ARF6 and ARF8 that
play a role in gynoecium and stamen development and in regulating fertility (Ru et al
2006 Wu et al 2006) These observations suggest that auxin signals are also closely
related to floral organogenesis In addition transgenic plants over-producing miR164
and the cuc1cuc2 double mutants show fused sepals and reduced petals (Laufs et al
2004 Turchi et al 2013) suggesting that miR164 is also related to floral meristem
activity and boundary specification of the meristematic region
11363 Shoot apical meristem development
Shoot apical meristem comprises self-maintaining totipotent cells The shoot apical
meristem activity affects diverse aspects of plant developmental processes including
leaf development vascular development and lateral organ polarity (Bowman et al
2002) Because miR165166 plays a primary role in meristem formation
overproduction of miR165166 and multiple knockout mutants of its target genes
exhibit pleiotropic pheonotypes (McConnell et al 2001 Emery et al 2003 Kim et al
2005 Williams et al 2005b)
The targets of miR165166 include genes encoding the homeo domain-leucine
zipper (HD-ZIP) class III transcription factors such as PHABULOSA(PHB)
PHAVOLUTA (PHV) REVOLUTA(REV) ATHB15CORONA (CNA) and ATHB8
Chapter 1 Introduction and Review of Literature
45
PHB PHV and ATHB15 have overlapping functions in controlling meristem
development (Turchi et al 2013) Indeed the phb phv athb15 triple mutants have an
enlarged shoot apical meristem Consistent with this the miR166- overproducing men1
and jba-1D mutants exhibit enlarged shoot apical meristems (Kim et al 2005
Williams et al 2005b Turchi et al 2013) In the mutants the shoot apical meristems
are multiplied and large
The development of shoot apical meristems is also regulated by the WUSCHEL
(WUS)ndashCLAVATA (CLV) signaling pathway WUS positively regulates cell
proliferation In contrast CLV3 which is expressed in stem cells limits the WUS
expression and thus inhibits cell proliferation in the SAM (Sharma et al 2003)
Consistent with this notion WUS is expressed to a higher level in jba-1D
explaining the ectopic enlargement of the SAM in the mutant (Williams et al 2005)
While WUS is closely related to miR165166 in the shoot apical meristem
development the underlying molecular mechanisms are currently unclear It has been
observed that the miR166-over producing men1 plants have an arrested meristem
development (Jung amp Park 2007) In addition the heterozygous men1+ and the
homozygous men1 plants show different expression patterns of WUS and CLV3 It
appears that miR166 regulates WUS indirectly and influences the expressional balance
between WUS and CLV3 through a negative feedback loop (Jung amp Park 2007 de
Alba et al 2013)
The phv phb athb15 triple mutant has an enlarged shoot apical meristems the
rev phb phv mutant does not have functional shoot apical meristems (Prigge et al
2005) This distinction may be explained by the fact that while miR166 targets
primarily PHB PHV and ATHB15 miR165 targets all the five HD-ZIP III genes (Zhou
et al 2007) Consistent with this transgenic plants overproducing miR166 are distinct
from those overproducing miR165 The miR165-overproducing transgenic plants lack
functional shoot apical meristem and a pin-like structure is formed at the apical region
of the mutant These phenotypes are quite similar to rev phb phv triple mutant (Zhou et
al 2007 Liu et al 2013) The phenotypic distinction may because by the difference
of the cleavage efficiencies of the target mRNAs In accordance with this notion a few
35S miR166a transgenic lines exhibit no shoot apical meristems The men1
homozygous plants also show arrested meristem development (Jung amp Park 2007)
Chapter 1 Introduction and Review of Literature
46
MiR164 cleaves the CUC1 and CUC2 mRNAs as well as the NAC1 mRNA
CUC1 and CUC2 determines the organ boundary in the meristem Phenotypes of
transgenic plants that overproduce miR164 are similar to those of the cuc1cuc2 double
mutant that exhibits embryonic developmental defects such as cup-shaped cotyledons
and fused cotyledons (Laufs et al 2004) When a miR164-resistant CUC2 is
expressed the boundary domain is enlarged CUC also plays a role in embryonic shoot
meristem formation by regulating the SHOOT MERISTEMLESS gene It was therefore
concluded that miR164-mediated cleavage of CUC1 and CUC2 mRNAs determines the
sizes of the boundary domains in the shoot apical meristem and regulates separation of
the organs by restricting cell proliferation (Laufs et al 2004 de Alba et al 2013)
11364 Leaf development
The role of miR319 has been extensively studied in leaf development A dominant
jaw-D mutant overproducing miR319 shows uneven curved leaves with enlarged size
and five TCP genes which are closely related to the CINCINNATA (CIN) gene in
Antirrhinum majus are down regulated in the mutant suggesting that miR319 mediated
regulation of the TCP transcription factor genes are important for the final step of
leaf shaping (Palatnik et al 2003 Naz et al 2013) In addition to leaf shape and size
leaf polarity is also specified by miRNA MiR166-mediated cleavage of the HD-ZIPIII
mRNAs is a well to establish leaf polarity This discovery originates from the gain-of-
function mutants such as phb-1d and phv-1d wherein point mutations in the
complementary sites to miRNA helps the mRNA to evade or resist from miRNA-
mediated cleavage (McConnell et al 2001 Emery et al 2003 Naz et al 2013)
Plant leaves have a dorso-ventral polarity consisting of the adaxial (upper
surface) and abaxial (lower surface) sides The adaxial side is closer to the shoot
apical meristem Adaxial identity is specified by functionally redundant class III HD-
ZIP family genes such as PHB PHV and REV Ectopic expression of each genes
result in adaxialized leaves Dominant phb-1d and phv-1d mutants exhibit
transformation of the abaxial to adaxial fate and have rod-shaped leaves In contrast
miR165166-overproducing plants show pin-like cotyledons radicalized leaves and
abaxialized leaves (Chitwood et al 2007 Pattanayak et al 2013)
Leaf asymmetry is determined by miR166-mediated restriction of the
expression domains of the HD-ZIPIII genes The HD-ZIPIII genes are expressed in the
Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
43
Corngrass1(Cg1) mutant in which miR156 is overproduced (Chuck et al 2007 de
Alba et al 2013) However there is no direct evidence for the direct linkage between
miR156 and miR172 It is also envisioned that miR156 and miR172 would not be
directly related For example miR156 regulates plastochron index that is the inverse of
leaf initiation rate and thus frequently used to analyze temporal patterns of plant
development In contrast miR172 does not affect plastochron (Wang et al 2008) The
down-regulation of miR172 in the maize miR156-over producing mutants would be
due to the prolonged juvenile vegetative phase in the mutants
Another miRNA that regulates flowering initiation is miR159 It regulates
MYB33 and MYB65 which are closely related to the barley GAMY B transcription
factor that responds to gibberellic acid (GA) The level of miR159 is elevated in GA-
treated seedlings but reduced in the GA biosynthetic ga1 mutant Transgenic plants
over producing miR159 exhibit delayed floral induction under short days (Achard et
al 2004) It is been suggested that MYB33 mediates GA signal to regulate LEAFY
(LFY) These observations indicate that miR159 plays an important role in the GA
signaling pathways that regulate proper floral induction under short days (Achard et al
2004)
11362 Floral development
Arabidopsis flowers consist of four distinct organs They arise in a characteristic
pattern within concentric rings called whorls from outside to inside four sepals four
petals six stamens and the central pistil consisting of two carpels Identities of the
floral organs are determined by three classes of regulatory genes classA classB and
class C genes (Krizek amp Fletcher 2005) The A and C class genes act antagonistically
to restrict their expression domains (Bowman et al 1991)
It is notable that miR172 also regulates floral architecture in addition to its role
in the timing of flowering initiation MiR172 inhibits the translation of the AP2 mRNA
(Chen 2004) Transgenic plants overproducing miR172 not only exhibit early
flowering but also show abnormal floral development which is quite similar to those
of the class A gene mutants such as the ap2 mutants (Aukerman amp Sakai 2003 de
Alba et al 2013) When the activity of the class A genes is inactivated that of the class
C genes such as AGAMOUS (AG) is elevated As a result the miR172- overproducing
transgenic plants exhibit no petals along with homeotic transformation of the sepals to
Chapter 1 Introduction and Review of Literature
44
the carpels (Aukerman amp Sakai 2003 Chen 2004)
Interestingly miR166 also regulates floral architecture The role of miR165166
in the regulation of meristem activity is closely linked to floral meristem formation and
organization (Zhao et al 2007 Miyashima et al 2013) Floral structure is severely
disrupted in the miR165166-overproducing mutants such as men1 and jba-1D in
which miR166 is overproduced (Williams et al 2005b Jung amp Park 2007) The men1
and jba-1D mutants have smaller gynoecium and reduced carpel number In an extreme
case the jba-1D homozygous plants lack visible carpels (Williams et al 2005b)
Similar phenotypes have been observed in the miR165 overproducers (Zhao et al
2007) It has been suggested that the phenotypes are possibly caused by altered AG
expression in the floral meristem (Jung amp Park 2007 Turchi et al 2013)
Other miRNAs also affect floral development Overproduction of miRNAs
involved in auxin signaling also affects floral architecture Transgenic plants
overproducing miR167 have defects in the formation of ovule and anther with reduced
fertility (Ru et al 2006) It has been shown that miR167 targets ARF6 and ARF8 that
play a role in gynoecium and stamen development and in regulating fertility (Ru et al
2006 Wu et al 2006) These observations suggest that auxin signals are also closely
related to floral organogenesis In addition transgenic plants over-producing miR164
and the cuc1cuc2 double mutants show fused sepals and reduced petals (Laufs et al
2004 Turchi et al 2013) suggesting that miR164 is also related to floral meristem
activity and boundary specification of the meristematic region
11363 Shoot apical meristem development
Shoot apical meristem comprises self-maintaining totipotent cells The shoot apical
meristem activity affects diverse aspects of plant developmental processes including
leaf development vascular development and lateral organ polarity (Bowman et al
2002) Because miR165166 plays a primary role in meristem formation
overproduction of miR165166 and multiple knockout mutants of its target genes
exhibit pleiotropic pheonotypes (McConnell et al 2001 Emery et al 2003 Kim et al
2005 Williams et al 2005b)
The targets of miR165166 include genes encoding the homeo domain-leucine
zipper (HD-ZIP) class III transcription factors such as PHABULOSA(PHB)
PHAVOLUTA (PHV) REVOLUTA(REV) ATHB15CORONA (CNA) and ATHB8
Chapter 1 Introduction and Review of Literature
45
PHB PHV and ATHB15 have overlapping functions in controlling meristem
development (Turchi et al 2013) Indeed the phb phv athb15 triple mutants have an
enlarged shoot apical meristem Consistent with this the miR166- overproducing men1
and jba-1D mutants exhibit enlarged shoot apical meristems (Kim et al 2005
Williams et al 2005b Turchi et al 2013) In the mutants the shoot apical meristems
are multiplied and large
The development of shoot apical meristems is also regulated by the WUSCHEL
(WUS)ndashCLAVATA (CLV) signaling pathway WUS positively regulates cell
proliferation In contrast CLV3 which is expressed in stem cells limits the WUS
expression and thus inhibits cell proliferation in the SAM (Sharma et al 2003)
Consistent with this notion WUS is expressed to a higher level in jba-1D
explaining the ectopic enlargement of the SAM in the mutant (Williams et al 2005)
While WUS is closely related to miR165166 in the shoot apical meristem
development the underlying molecular mechanisms are currently unclear It has been
observed that the miR166-over producing men1 plants have an arrested meristem
development (Jung amp Park 2007) In addition the heterozygous men1+ and the
homozygous men1 plants show different expression patterns of WUS and CLV3 It
appears that miR166 regulates WUS indirectly and influences the expressional balance
between WUS and CLV3 through a negative feedback loop (Jung amp Park 2007 de
Alba et al 2013)
The phv phb athb15 triple mutant has an enlarged shoot apical meristems the
rev phb phv mutant does not have functional shoot apical meristems (Prigge et al
2005) This distinction may be explained by the fact that while miR166 targets
primarily PHB PHV and ATHB15 miR165 targets all the five HD-ZIP III genes (Zhou
et al 2007) Consistent with this transgenic plants overproducing miR166 are distinct
from those overproducing miR165 The miR165-overproducing transgenic plants lack
functional shoot apical meristem and a pin-like structure is formed at the apical region
of the mutant These phenotypes are quite similar to rev phb phv triple mutant (Zhou et
al 2007 Liu et al 2013) The phenotypic distinction may because by the difference
of the cleavage efficiencies of the target mRNAs In accordance with this notion a few
35S miR166a transgenic lines exhibit no shoot apical meristems The men1
homozygous plants also show arrested meristem development (Jung amp Park 2007)
Chapter 1 Introduction and Review of Literature
46
MiR164 cleaves the CUC1 and CUC2 mRNAs as well as the NAC1 mRNA
CUC1 and CUC2 determines the organ boundary in the meristem Phenotypes of
transgenic plants that overproduce miR164 are similar to those of the cuc1cuc2 double
mutant that exhibits embryonic developmental defects such as cup-shaped cotyledons
and fused cotyledons (Laufs et al 2004) When a miR164-resistant CUC2 is
expressed the boundary domain is enlarged CUC also plays a role in embryonic shoot
meristem formation by regulating the SHOOT MERISTEMLESS gene It was therefore
concluded that miR164-mediated cleavage of CUC1 and CUC2 mRNAs determines the
sizes of the boundary domains in the shoot apical meristem and regulates separation of
the organs by restricting cell proliferation (Laufs et al 2004 de Alba et al 2013)
11364 Leaf development
The role of miR319 has been extensively studied in leaf development A dominant
jaw-D mutant overproducing miR319 shows uneven curved leaves with enlarged size
and five TCP genes which are closely related to the CINCINNATA (CIN) gene in
Antirrhinum majus are down regulated in the mutant suggesting that miR319 mediated
regulation of the TCP transcription factor genes are important for the final step of
leaf shaping (Palatnik et al 2003 Naz et al 2013) In addition to leaf shape and size
leaf polarity is also specified by miRNA MiR166-mediated cleavage of the HD-ZIPIII
mRNAs is a well to establish leaf polarity This discovery originates from the gain-of-
function mutants such as phb-1d and phv-1d wherein point mutations in the
complementary sites to miRNA helps the mRNA to evade or resist from miRNA-
mediated cleavage (McConnell et al 2001 Emery et al 2003 Naz et al 2013)
Plant leaves have a dorso-ventral polarity consisting of the adaxial (upper
surface) and abaxial (lower surface) sides The adaxial side is closer to the shoot
apical meristem Adaxial identity is specified by functionally redundant class III HD-
ZIP family genes such as PHB PHV and REV Ectopic expression of each genes
result in adaxialized leaves Dominant phb-1d and phv-1d mutants exhibit
transformation of the abaxial to adaxial fate and have rod-shaped leaves In contrast
miR165166-overproducing plants show pin-like cotyledons radicalized leaves and
abaxialized leaves (Chitwood et al 2007 Pattanayak et al 2013)
Leaf asymmetry is determined by miR166-mediated restriction of the
expression domains of the HD-ZIPIII genes The HD-ZIPIII genes are expressed in the
Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
44
the carpels (Aukerman amp Sakai 2003 Chen 2004)
Interestingly miR166 also regulates floral architecture The role of miR165166
in the regulation of meristem activity is closely linked to floral meristem formation and
organization (Zhao et al 2007 Miyashima et al 2013) Floral structure is severely
disrupted in the miR165166-overproducing mutants such as men1 and jba-1D in
which miR166 is overproduced (Williams et al 2005b Jung amp Park 2007) The men1
and jba-1D mutants have smaller gynoecium and reduced carpel number In an extreme
case the jba-1D homozygous plants lack visible carpels (Williams et al 2005b)
Similar phenotypes have been observed in the miR165 overproducers (Zhao et al
2007) It has been suggested that the phenotypes are possibly caused by altered AG
expression in the floral meristem (Jung amp Park 2007 Turchi et al 2013)
Other miRNAs also affect floral development Overproduction of miRNAs
involved in auxin signaling also affects floral architecture Transgenic plants
overproducing miR167 have defects in the formation of ovule and anther with reduced
fertility (Ru et al 2006) It has been shown that miR167 targets ARF6 and ARF8 that
play a role in gynoecium and stamen development and in regulating fertility (Ru et al
2006 Wu et al 2006) These observations suggest that auxin signals are also closely
related to floral organogenesis In addition transgenic plants over-producing miR164
and the cuc1cuc2 double mutants show fused sepals and reduced petals (Laufs et al
2004 Turchi et al 2013) suggesting that miR164 is also related to floral meristem
activity and boundary specification of the meristematic region
11363 Shoot apical meristem development
Shoot apical meristem comprises self-maintaining totipotent cells The shoot apical
meristem activity affects diverse aspects of plant developmental processes including
leaf development vascular development and lateral organ polarity (Bowman et al
2002) Because miR165166 plays a primary role in meristem formation
overproduction of miR165166 and multiple knockout mutants of its target genes
exhibit pleiotropic pheonotypes (McConnell et al 2001 Emery et al 2003 Kim et al
2005 Williams et al 2005b)
The targets of miR165166 include genes encoding the homeo domain-leucine
zipper (HD-ZIP) class III transcription factors such as PHABULOSA(PHB)
PHAVOLUTA (PHV) REVOLUTA(REV) ATHB15CORONA (CNA) and ATHB8
Chapter 1 Introduction and Review of Literature
45
PHB PHV and ATHB15 have overlapping functions in controlling meristem
development (Turchi et al 2013) Indeed the phb phv athb15 triple mutants have an
enlarged shoot apical meristem Consistent with this the miR166- overproducing men1
and jba-1D mutants exhibit enlarged shoot apical meristems (Kim et al 2005
Williams et al 2005b Turchi et al 2013) In the mutants the shoot apical meristems
are multiplied and large
The development of shoot apical meristems is also regulated by the WUSCHEL
(WUS)ndashCLAVATA (CLV) signaling pathway WUS positively regulates cell
proliferation In contrast CLV3 which is expressed in stem cells limits the WUS
expression and thus inhibits cell proliferation in the SAM (Sharma et al 2003)
Consistent with this notion WUS is expressed to a higher level in jba-1D
explaining the ectopic enlargement of the SAM in the mutant (Williams et al 2005)
While WUS is closely related to miR165166 in the shoot apical meristem
development the underlying molecular mechanisms are currently unclear It has been
observed that the miR166-over producing men1 plants have an arrested meristem
development (Jung amp Park 2007) In addition the heterozygous men1+ and the
homozygous men1 plants show different expression patterns of WUS and CLV3 It
appears that miR166 regulates WUS indirectly and influences the expressional balance
between WUS and CLV3 through a negative feedback loop (Jung amp Park 2007 de
Alba et al 2013)
The phv phb athb15 triple mutant has an enlarged shoot apical meristems the
rev phb phv mutant does not have functional shoot apical meristems (Prigge et al
2005) This distinction may be explained by the fact that while miR166 targets
primarily PHB PHV and ATHB15 miR165 targets all the five HD-ZIP III genes (Zhou
et al 2007) Consistent with this transgenic plants overproducing miR166 are distinct
from those overproducing miR165 The miR165-overproducing transgenic plants lack
functional shoot apical meristem and a pin-like structure is formed at the apical region
of the mutant These phenotypes are quite similar to rev phb phv triple mutant (Zhou et
al 2007 Liu et al 2013) The phenotypic distinction may because by the difference
of the cleavage efficiencies of the target mRNAs In accordance with this notion a few
35S miR166a transgenic lines exhibit no shoot apical meristems The men1
homozygous plants also show arrested meristem development (Jung amp Park 2007)
Chapter 1 Introduction and Review of Literature
46
MiR164 cleaves the CUC1 and CUC2 mRNAs as well as the NAC1 mRNA
CUC1 and CUC2 determines the organ boundary in the meristem Phenotypes of
transgenic plants that overproduce miR164 are similar to those of the cuc1cuc2 double
mutant that exhibits embryonic developmental defects such as cup-shaped cotyledons
and fused cotyledons (Laufs et al 2004) When a miR164-resistant CUC2 is
expressed the boundary domain is enlarged CUC also plays a role in embryonic shoot
meristem formation by regulating the SHOOT MERISTEMLESS gene It was therefore
concluded that miR164-mediated cleavage of CUC1 and CUC2 mRNAs determines the
sizes of the boundary domains in the shoot apical meristem and regulates separation of
the organs by restricting cell proliferation (Laufs et al 2004 de Alba et al 2013)
11364 Leaf development
The role of miR319 has been extensively studied in leaf development A dominant
jaw-D mutant overproducing miR319 shows uneven curved leaves with enlarged size
and five TCP genes which are closely related to the CINCINNATA (CIN) gene in
Antirrhinum majus are down regulated in the mutant suggesting that miR319 mediated
regulation of the TCP transcription factor genes are important for the final step of
leaf shaping (Palatnik et al 2003 Naz et al 2013) In addition to leaf shape and size
leaf polarity is also specified by miRNA MiR166-mediated cleavage of the HD-ZIPIII
mRNAs is a well to establish leaf polarity This discovery originates from the gain-of-
function mutants such as phb-1d and phv-1d wherein point mutations in the
complementary sites to miRNA helps the mRNA to evade or resist from miRNA-
mediated cleavage (McConnell et al 2001 Emery et al 2003 Naz et al 2013)
Plant leaves have a dorso-ventral polarity consisting of the adaxial (upper
surface) and abaxial (lower surface) sides The adaxial side is closer to the shoot
apical meristem Adaxial identity is specified by functionally redundant class III HD-
ZIP family genes such as PHB PHV and REV Ectopic expression of each genes
result in adaxialized leaves Dominant phb-1d and phv-1d mutants exhibit
transformation of the abaxial to adaxial fate and have rod-shaped leaves In contrast
miR165166-overproducing plants show pin-like cotyledons radicalized leaves and
abaxialized leaves (Chitwood et al 2007 Pattanayak et al 2013)
Leaf asymmetry is determined by miR166-mediated restriction of the
expression domains of the HD-ZIPIII genes The HD-ZIPIII genes are expressed in the
Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
45
PHB PHV and ATHB15 have overlapping functions in controlling meristem
development (Turchi et al 2013) Indeed the phb phv athb15 triple mutants have an
enlarged shoot apical meristem Consistent with this the miR166- overproducing men1
and jba-1D mutants exhibit enlarged shoot apical meristems (Kim et al 2005
Williams et al 2005b Turchi et al 2013) In the mutants the shoot apical meristems
are multiplied and large
The development of shoot apical meristems is also regulated by the WUSCHEL
(WUS)ndashCLAVATA (CLV) signaling pathway WUS positively regulates cell
proliferation In contrast CLV3 which is expressed in stem cells limits the WUS
expression and thus inhibits cell proliferation in the SAM (Sharma et al 2003)
Consistent with this notion WUS is expressed to a higher level in jba-1D
explaining the ectopic enlargement of the SAM in the mutant (Williams et al 2005)
While WUS is closely related to miR165166 in the shoot apical meristem
development the underlying molecular mechanisms are currently unclear It has been
observed that the miR166-over producing men1 plants have an arrested meristem
development (Jung amp Park 2007) In addition the heterozygous men1+ and the
homozygous men1 plants show different expression patterns of WUS and CLV3 It
appears that miR166 regulates WUS indirectly and influences the expressional balance
between WUS and CLV3 through a negative feedback loop (Jung amp Park 2007 de
Alba et al 2013)
The phv phb athb15 triple mutant has an enlarged shoot apical meristems the
rev phb phv mutant does not have functional shoot apical meristems (Prigge et al
2005) This distinction may be explained by the fact that while miR166 targets
primarily PHB PHV and ATHB15 miR165 targets all the five HD-ZIP III genes (Zhou
et al 2007) Consistent with this transgenic plants overproducing miR166 are distinct
from those overproducing miR165 The miR165-overproducing transgenic plants lack
functional shoot apical meristem and a pin-like structure is formed at the apical region
of the mutant These phenotypes are quite similar to rev phb phv triple mutant (Zhou et
al 2007 Liu et al 2013) The phenotypic distinction may because by the difference
of the cleavage efficiencies of the target mRNAs In accordance with this notion a few
35S miR166a transgenic lines exhibit no shoot apical meristems The men1
homozygous plants also show arrested meristem development (Jung amp Park 2007)
Chapter 1 Introduction and Review of Literature
46
MiR164 cleaves the CUC1 and CUC2 mRNAs as well as the NAC1 mRNA
CUC1 and CUC2 determines the organ boundary in the meristem Phenotypes of
transgenic plants that overproduce miR164 are similar to those of the cuc1cuc2 double
mutant that exhibits embryonic developmental defects such as cup-shaped cotyledons
and fused cotyledons (Laufs et al 2004) When a miR164-resistant CUC2 is
expressed the boundary domain is enlarged CUC also plays a role in embryonic shoot
meristem formation by regulating the SHOOT MERISTEMLESS gene It was therefore
concluded that miR164-mediated cleavage of CUC1 and CUC2 mRNAs determines the
sizes of the boundary domains in the shoot apical meristem and regulates separation of
the organs by restricting cell proliferation (Laufs et al 2004 de Alba et al 2013)
11364 Leaf development
The role of miR319 has been extensively studied in leaf development A dominant
jaw-D mutant overproducing miR319 shows uneven curved leaves with enlarged size
and five TCP genes which are closely related to the CINCINNATA (CIN) gene in
Antirrhinum majus are down regulated in the mutant suggesting that miR319 mediated
regulation of the TCP transcription factor genes are important for the final step of
leaf shaping (Palatnik et al 2003 Naz et al 2013) In addition to leaf shape and size
leaf polarity is also specified by miRNA MiR166-mediated cleavage of the HD-ZIPIII
mRNAs is a well to establish leaf polarity This discovery originates from the gain-of-
function mutants such as phb-1d and phv-1d wherein point mutations in the
complementary sites to miRNA helps the mRNA to evade or resist from miRNA-
mediated cleavage (McConnell et al 2001 Emery et al 2003 Naz et al 2013)
Plant leaves have a dorso-ventral polarity consisting of the adaxial (upper
surface) and abaxial (lower surface) sides The adaxial side is closer to the shoot
apical meristem Adaxial identity is specified by functionally redundant class III HD-
ZIP family genes such as PHB PHV and REV Ectopic expression of each genes
result in adaxialized leaves Dominant phb-1d and phv-1d mutants exhibit
transformation of the abaxial to adaxial fate and have rod-shaped leaves In contrast
miR165166-overproducing plants show pin-like cotyledons radicalized leaves and
abaxialized leaves (Chitwood et al 2007 Pattanayak et al 2013)
Leaf asymmetry is determined by miR166-mediated restriction of the
expression domains of the HD-ZIPIII genes The HD-ZIPIII genes are expressed in the
Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
46
MiR164 cleaves the CUC1 and CUC2 mRNAs as well as the NAC1 mRNA
CUC1 and CUC2 determines the organ boundary in the meristem Phenotypes of
transgenic plants that overproduce miR164 are similar to those of the cuc1cuc2 double
mutant that exhibits embryonic developmental defects such as cup-shaped cotyledons
and fused cotyledons (Laufs et al 2004) When a miR164-resistant CUC2 is
expressed the boundary domain is enlarged CUC also plays a role in embryonic shoot
meristem formation by regulating the SHOOT MERISTEMLESS gene It was therefore
concluded that miR164-mediated cleavage of CUC1 and CUC2 mRNAs determines the
sizes of the boundary domains in the shoot apical meristem and regulates separation of
the organs by restricting cell proliferation (Laufs et al 2004 de Alba et al 2013)
11364 Leaf development
The role of miR319 has been extensively studied in leaf development A dominant
jaw-D mutant overproducing miR319 shows uneven curved leaves with enlarged size
and five TCP genes which are closely related to the CINCINNATA (CIN) gene in
Antirrhinum majus are down regulated in the mutant suggesting that miR319 mediated
regulation of the TCP transcription factor genes are important for the final step of
leaf shaping (Palatnik et al 2003 Naz et al 2013) In addition to leaf shape and size
leaf polarity is also specified by miRNA MiR166-mediated cleavage of the HD-ZIPIII
mRNAs is a well to establish leaf polarity This discovery originates from the gain-of-
function mutants such as phb-1d and phv-1d wherein point mutations in the
complementary sites to miRNA helps the mRNA to evade or resist from miRNA-
mediated cleavage (McConnell et al 2001 Emery et al 2003 Naz et al 2013)
Plant leaves have a dorso-ventral polarity consisting of the adaxial (upper
surface) and abaxial (lower surface) sides The adaxial side is closer to the shoot
apical meristem Adaxial identity is specified by functionally redundant class III HD-
ZIP family genes such as PHB PHV and REV Ectopic expression of each genes
result in adaxialized leaves Dominant phb-1d and phv-1d mutants exhibit
transformation of the abaxial to adaxial fate and have rod-shaped leaves In contrast
miR165166-overproducing plants show pin-like cotyledons radicalized leaves and
abaxialized leaves (Chitwood et al 2007 Pattanayak et al 2013)
Leaf asymmetry is determined by miR166-mediated restriction of the
expression domains of the HD-ZIPIII genes The HD-ZIPIII genes are expressed in the
Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
47
meristem and on the adaxial side of leaves In contrast miR166 is expressed on the
abaxial side of leaves (Nogueira et al 2006) It is notable that miRNA not only
regulates mRNA abundance but also restricts the expression domains of the target
genes Spatial regulation of the HD-ZIPIII genes is defined by the gradient expression
of miR166165 (Kidner amp Timmermans 2007) Consistent with this several genes
that are involved in miRNA-mediated regulation show similar phenotypes with the
gain-of-function mutants of the miRNA targets In the ago1 mutant PHB expression
is expanded and leaves are adaxialized (Kidner amp Martienssen 2005) In addition a
mutation in the SERRATE (SE) gene which is involved in miRNA processing exhibits
increased PHB expression and adaxialized leaves (Byrne 2006 Pattanayak et al 2013)
Interestingly cell differentiation in the leave is also regulated by miRNAs
MiR824 that cleaves the AGL16 mRNA regulates stomatal patterning in Arabidopsis
Ectopic expression of a miRNA-resistant AGL16 exhibits increased stomatal
complexes as a result of earlier establishment of meristemoid It is now evident that
various miRNAs mediate diverse aspects of leaf development (Kutter et al 2007)
11365 Vascular development
The vascular system plays essential roles in transportation of water nutrient organic
materials and signalling molecules as well as serves to architectural maintenance
(Jung et al 2008) The xylem and phloem tissues are differentiated from the
procambium While xylem is localized on the adaxial side closer to the center phloem
is developed on the abaxial side closer to the peripheral zone The procambium is
localized between xylem and phloem (Jung et al 2008 Turchi et al 2013)
Patterning of the vascular bundles is closely linked to the organization of the abaxialndash
adaxial polarity
MiR165166 and its targets play an important role in vascular development
ATHB8 and ATHB15 expressed in the vascular tissue play a major role in the vascular
development The rev10d mutant shows an amphivasal bundle in which phloem is
surrounded by xylem The gain-of-function mutants of PHB and PHV also show
amphivasal bundles in the leaves (Baucher et al 2007) In contrast the phb phv rev
triple mutants exhibit a unique amphicribal bundle in which xylem is surrounded by
phloem (Prigge et al 2005 Turchi et al 2013) The miR166-overproducing men1
mutant is characterized by having fasciated stems due to enlarged meristem
Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
48
development and disrupted vascular patterning Although miR166 modulates all the
five HD-ZIPIII genes miR166 regulation of ATHB15 is critical for the vascular
development (Kim et al 2005) The number of vascular bundles is increased in the
men1 mutant and the ATHB15-antisense transgenic lines Lignified tissue is
significantly enlarged in the men1 vascular system The radial patterning of vascular
bundles and vascular polarity are remains unchanged in the mutant (Kim et al
2005) In addition only a few lines of the men1+ heterologous plants have
amphivasal bundles These abnormal bundles are also observed in the jba-1D mutant
(Williams et al 2005 de Alba et al 2013) It is therefore likely that ATHB15 plays a
role distinct from those of other HD-ZIPIII genes
In addition to the role in vascular polarity miR165166 also regulates xylem
differentiation (Kim et al 2005 Zhou et al 2007) While phloem formation is
marginally affected xylems are poorly developed in the transgenic plants over
expressing a miRNA-resistant ATHB15 On the other hand miR165 overproducers
exhibit inhibition of xylem differentiation (Zhou et al 2007) The phb phv athb15
triple mutant which has amphivasal bundles and the rev phb phv triple mutant which
has amphicribal bundles show opposed vascular phenotypes as observed in the shoot
apical meristem development of the mutants (Prigge et al 2005) These observations
may reflect the regulatory complexity of the HD-ZIPIII genes It has been suggested
that miRNA165166 modulates vascular development as well as vascular cell
differentiation by co-ordinately regulating the HD- ZIPIII activities (Kim et al 2005
Turchi et al 2013)
11366 Root development
Lateral root formation is an important agronomical trait that helps uptake of
nutrients and water from the soil MiRNA regulation of root development largely
depends on auxin signalling Auxin is critical for lateral root development and
adventitious root formation (Sorin et al 2005 De Smet et al 2006 Lavenus et al
2013) Several miRNAs participate in the modulation of auxin action in regulating
lateral root organization For example miR160 regulates Auxin Response Factor 17
(ARF17) through mRNA cleavage ARF17 regulates a subset of GH3 genes
encoding auxin-conjugating enzymes (Mallory et al 2005) It has been shown that the
reduced lateral root formation in the miR160-overproducing transgenic plants is
mediated by the ARF17-GH3 pathway (Mallory et al 2005) The ARF17-GH3
Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
49
pathway regulates both lateral and adventitious root formation suggesting that this
signalling module is important for auxin-mediated regulation of root development
The role of AGO1 in adventitious root formation is also consistent with the role
of miR160 in regulating lateral and adventitious root formation via auxin signaling The
ago1 mutant has defects in adventitious root formation (Sorin et al 2005) ARF17 is
highly expressed and several GH3 genes are down-regulated in the mutant As a result
both the levels of free and conjugated IAA levels are decreased and adventitious roots
are reduced in the mutant (Sorin et al 2005 de Alba et al 2013 Lavenus et al 2013)
Lateral root formation is elevated in the transgenic plants over expressing a
miR164-resistant NAC1 gene In contrast miR164 overproduction negatively regulates
NAC1 and thus lateral root formation NAC1 is mainly expressed in the root
particularly in the pericycle cells Therefore it may play a role in lateral root initiation
via auxin signalling pathway which involves AXR1 AXR2 and TIR1 (Guo et al
2005) While miRNAs are related with auxin signalling during lateral root
development it is also modulated by various environmental stress conditions (Deak amp
Malamy 2005) When plants are exposed to drought stress lateral root development
is severely suppressed (Deak amp Malamy 2005 de Alba et al 2013) ABA
treatments also show similar effects (Malamy 2005) It is well known that auxin and
ABA participate antagonistically in lateral root development despite a point of time for
interaction being unclear Consistent with this miR160 and miR164 might be mutually
linked directly or indirectly to ABA signalling
11367 Disease resistance
Plants are equipped to detect conserved molecular features of microbes termed as
Pathogen associated molecular patterns (PAMPs) PAMPS triggered immunity plays an
important role in the resistance of plants to numerous pathogens (Li et al 2010)
Studies have shown that flg22 induces the accumulation of miRNA 393 which
contributes to bacterial resistance by negatively regulating the mRNA levels of F-box
auxin receptors TIR1 AFB2 and AFB3 (Navarro et al 2006 Balmer amp Mauch-Mani
2013) Shivaprasad et al (2012) demonstrated that the miR4822118 family of
miRNAs target numerous NBS-LRR mRNAs encoding disease resistance proteins in
tomato
Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
50
11368 Stress responses
Accumulating evidence supports the role of miRNAs in plant response to abiotic stress
conditions Many miRNAs respond to nutrient deficiency While most miRNAs
regulate transcription factor genes miRNAs functioning in response to nutrient
deficiency mainly regulate genes encoding enzymes and transporters involved in plant
metabolism (Chiou 2007 Amaral et al 2013)
MiR398 regulates two genes encoding copper superoxide dismutases CSD1
and CSD2 Superoxide dismutases are involved in reactive oxygen species (ROS)
scavenging by converting O2oline t o H 2 O 2 (Yamasaki et al 2007) These enzymes
contain various metal cofactors which are used as a basis for their classification The
CSD enzymes contain copper as a cofactor CSD1 and CSD2 are found in the
cytoplasm and chloroplast stroma respectively MiR398 is induced by copper
deficiency and maintains the copper homeostasis by regulating CSD1 and CSD2
through mRNA cleavage (Yamasaki et al 2009) In addition miR398 also play
diverse roles in oxidative stress responses ROS is generated by various abiotic and
biotic responses and photosynthesis (Jagadeeswaran et al 2009) MiR398 is down-
regulated by Pseudomonas syringae infection ozone and salt stress which is a typical
response to oxidative stress and is thus influenced by ROS production Another role of
miR398 in ROS scavenging is sugar response Sugars induce miR398 independent of
copper (Dugas amp Bartel 2008) Sugars also inhibit photosynthesis which in turn
decreases ROS production Therefore lowered photosynthetic activity decreases the
requirement for the CSD activity (Dugas amp Bartel 2008) In contrast stress conditions
induce ROS production which up regulates the CSD activity to inactivate ROS Under
this condition miR398 is down regulated (Sunkar et al 2007 Amaral et al 2013)
MiR395 regulates sulphate assimilation and distribution by negatively
regulating genes encoding ATP sulphurylase (APS) and sulphate transporter Most
sulphate can be reduced and assimilated into amino acid cysteine by the APS activity
Sulphate is also converted into 5prime-adenylyl sulphate which is used to synthesize
sulphate esters by the cytosolic APS activity (Kawashima et al 2009)
In Arabidopsis two high-affinity sulphate transporters SULTR11 and
SULTR12 uptake sulphate from the soil to the root Two low affinity sulphate
transporters SULTR21 and SULTR22 modulate allocation of sulphate within a plant
Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
51
MiR395 targets the SULTR21 mRNA and regulates distribution of sulphate When the
availability of sulphate is increased miR395 levels are down-regulated Under this
circumstance where sulphate assimilation and allocation is required APS and
sulphate transporter genes are up regulated (Chiou 2007 Sunkar et al 2007)
In most cases a miRNA targets the members of the same gene family One
notable feature of miRNA targets is that a specific miRNA does not always target
genes belong to the same gene family eg miR395 The targets of miR395 include
members of the APS family (APS1 and APS4) and those of the SULTR family
(SULTR21) (Sunkar et al 2007) It is envisioned that miRNA regulation is not only
focused on the structural similarity of the target genes but also related to biological
function of the target genes Low phosphate induces miR399 which in turn down-
regulates a putative ubiquitin conjugating E2 enzyme gene UBC24 (Fujii et al 2005
Sunkar et al 2007) Unlike miR395 that regulates sulphate assimilation and allocation
miR399-mediated cleavage of UBC24 mRNA does not provide any clues as to the
cellular or molecular events occurring in response to low phosphate (Aung et al 2006
Chiou et al 2006) When miR399 is over-produced pyrophosphate transporter genes
such as PHT21 PHT32 and PHT33 exhibit altered expression patterns This implies
that the miR399-mediated pathway also regulates phosphate distribution within a plant
and maintains phosphate homeostasis (Lu amp Huang 2008 Rojas-Triana et al
2013)
MiR393 negatively regulates several F-box protein genes such as TIR1 and
AFBs to acquire resistance to pathogen infection (Navarro et al 2006) Auxin-
mediated growth suppression is an important mechanism to regulate plant adaptation to
environmental stress Growth suppression directs reallocation of metabolic resources
to resistance responses and provides a role for auxin in the fitness cost of induced
resistance in plants (Park et al 2007) MiR393 may play a role in growth suppression
and root architecture by cleaving the mRNA of F-box auxin receptor genes including
TIR1 AFB1 AFB2 and AFB3 (Vidal et al 2010) In addition miR393 is up
regulated by diverse abiotic stress conditions suggesting that antibacterial resistance
and stress resistance are modulated through the miR393 mediated auxin signalling
(Navarro et al 2006 Balmer amp Mauch-Mani 2013 Rojas-Triana et al 2013)
Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
52
11369 Growth hormone signalling
A few miRNAs have been confirmed to play a role in growth hormonal signalling
MiR160 and miR167 regulate the ARF genes in auxin signalling (Mallory et al 2005
Yang et al 2006) MiR393 regulates genes encoding F-box proteins such as TIR1 and
AFBs through mRNA cleavage (Navarro et al 2006) In addition miR164 targets the
NAC1 gene functioning in lateral root growth via auxin signalling (Guo et al 2005)
Another example is miR159 that mediates GA and ABA signalling by targeting a group
of MYB genes (Achard et al 2004 Reyes amp Chua 2007 Lu amp Huang 2008 Ding et
al 2013) Transgenic plants over expressing a miR160 resistant ARF17 which
contains a mutation in the miR160 complementary site exhibit altered phenotypes in
the root as well as in the shoot development (Mallory et al 2005) The phenotypic
alterations include leaf serration upward curling of leaves dwarfism and altered floral
structure and lateral organs These observations reflect the diverse roles of auxin in a
variety of plant growth and developmental processes Although expression of a few
GH3 genes is altered in the transgenic plants it is unclear whether the GH3 genes are
directly regulated by the miR160-ARF17 signals or influenced by altered auxin
signalling Although the role of miR167 in auxin signalling during floral development
is still elusive in Arabidopsis it has been shown that miR167 cleaves ARF6 and ARF8
mRNAs and regulates GH3 expression in rice culture cells (Yang et al 2006)
suggesting that miR167 signals would also regulate auxin homeostasis These
observations suggest that the miR167-ARF101617 signals and the miR160-ARF68
signals may share the same targets a group of the GH3 genes It is also envisioned that
auxin homeostasis is regulated co-ordinately through convergence of the signals from
separate miRNAs
ABA regulates seed germination lateral root development and stomatal aperture
in response to drought MiR159 is accumulated in plants treated with ABA or those
grown under drought (Lu amp Huang 2008) This miRNA regulates different target
genes in different developmental processes (Lu amp Huang 2008) MYB33 and MYB101
mRNAs are cleaved by miR159 and they regulate seed germination MiR159
overproducer is hyposensitive to ABA during seed germination which is also observed
in the myb33 and myb101 mutants (Reyes amp Chua 2007) In contrast transgenic plants
over expressing a miR159 resistant MYB33 and MYB101 show a hypersensitive
response to ABA However the miR159 mediated signals are independent of GA It
Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
53
has been suggested that GA-mediated miR159 signals control floral development and
flowering initiation (Achard et al 2004) which is functionally separated from the
ABA-mediated miR159 pathway (Reyes amp Chua 2007) Interestingly expression of
MYB65 another miR159 target is not detected during seed germination It may reflect
the functional distinction between ABA and GA responses MYB33 and MYB101
mediate ABA signalling whereas MYB33 and MYB65 mediate GA signalling (Reyes amp
Chua 2007)
114 ProteinndashmiRNA interactions
Most researches are focused primarily on miRNA biogenesis and miRNA regulation of
target genes However recent studies have shown that various transcriptional regulators
affect miRNA gene transcription In addition several proteins are known to modulate
miRNA stability and processing although the underlying molecular and biochemical
mechanisms are still largely unknown A large portion of plant miRNAs is transported
into the cytoplasm with the aid of HASTY (Bollman et al 2003 Park et al 2005)
The nucleo-cytoplasmic transport of miR172 is not influenced by the hasty mutation
(Park et al 2005) suggesting that specific protein-miRNA interactions would be
important for the combinational signalling
There are some other examples of transcriptional regulation of the miRNA
genes In response to the copper deficiency several miRNAs like miR397 miR398 and
miR408 show an increased level of transcription While transcription of the miRNA
genesis induced by copper deficiency the inductive effects disappear in the spl7
mutant Indeed the SPL7 transcription factor binds to the GTAC sequence within the
promoter of the miR398 gene (Yamasaki et al 2009) The miR399 transcription
is also regulated by the PHR1 transcription factor PHR1 acts upstream of miR399 by
directly binding to the GNATATNC sequence in the miR399 gene promoter (Bari et
al 2006)
Although several proteins have been shown to regulate miRNA processing the
overall regulatory scheme is still elusive In addition to the general regulators of
miRNA biogenesis and processing other RNA-binding proteins and regulatory proteins
that are involved in RNA metabolism would also regulate miRNA processing in plants
as observed in animal systems (Michlewski et al 2008 Rybak et al 2008)
Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
54
115 Kinship of RNAi and microRNA
Since miRNAs are derived from their double stranded precursors and are similar
in size to siRNAs the biogenesis of siRNAs and miRNAs is similar (Figure 19) Both
siRNAs and miRNAs are processed by Dicer activities in animals as well as in plants
(Hammond et al 2000 Grishok et al 2001 Bartel 2004) Human recombinant Dicer
is known to process pre-let7 RNA to mature let7 efficiently in vitro (Provost et al
2002) Bartelrsquos group has also shown that caf1 (dicer homologue) mutants of A
thaliana fail to process miRNAs (Reinhart et al 2002 Pluskota et al 2011) The
genetic and biochemical data point towards a strong interaction between Dicer and the
Argonaute group of proteins in C elegans and D melanogaster for processing the
miRNAs (Hammond et al 2001 Grad et al 2003) The similar interaction is also
present in plants between Dicer on one hand and PNH (zwillepinhead) on the other to
generate plant miRNAs (Golden et al 2002 Chen 2005 Jones-Rhoades et al 2006)
Additionally both forms of small RNA miRNAs and siRNAs were found to be
integrally associated with riboprotein complexes containing a member of the
PIWIPAZ domain family siRNAs in the RISC and miRNAs in the ribonucleoprotein
complexes (Hammond et al 2000) Evidences suggest that miRNA
microribonucleoprotein and the RISC complex are the same entity (Llave et al 2002b
Zhang et al 2007) The same or similar dicer and subsequent ribonucleoprotein
complexes are required to process mature forms of the miRNAs However in some
cases such as C elegans lin4 and let7 the 22-nucleotide form is processed from the 5prime
part of the stem and in other cases such as miR1 and miR58 maturation results from
the 3prime part of the precursors Thus there is a gene specificity of miRNA processing
andor stabilization (Lee amp Ambros 2001 Carthew amp Sontheimer 2009) Since the
biosynthetic pathways of miRNAs and siRNAs are similar the viral suppressors that
inhibit siRNA formation are also expected to interfere in the biogenesis of miRNAs A
detailed understanding of this suppression process may unravel the hitherto unknown
molecular basis of virus-induced development-related diseases in eukaryotes especially
in plants
Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
55
Figure 19 Kinship of miRNA and SiRNA biogenesis
An RNA-silencing suppressor PIHC-PRO of turnip mosaic virus induces a
number of developmental defects in the vegetative and reproductive organs of A
thaliana Many of these defects are reminiscent of observed defects in Dicer-like
mutants of A thaliana The PIHC-PRO suppressor interferes with the formation of
miR171 and as a result the downstream target mRNAs accumulate instead of being
cleaved causing developmental errors (Kasschau et al 2003) Thus it is interesting
that the counter defensive strategy of the viruses has evolved not only to protect the
viral RNA genome from the host degradative machinery but also to subvert the cellular
Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
56
development program in favour of the virus A viral suppressor of RNA silencing the
HC-PRO protein of potato virus Y has been found to differentially regulate the
accumulation of siRNAs and miRNAs in tobacco (Mallory et al 2002) The HC-PRO
protein prevents accumulation of siRNAs of the silenced genes and thus releases
silencing in a universal manner but the same protein helps accumulation of all
miRNAs tested namely miR167 miR164 and miR156 of tobacco in vivo This result
indicates that the dicing complexes for siRNA and miRNA may not be exactly similar
in biochemical features and as a result the biochemical functions of the complexes are
different in response to this particular HC-PRO protein However it is important to
mark the distinctions among the pathways leading to the formation as well as the
activities of siRNAs and miRNAs Although hundreds of miRNAs from various
organisms have been identified only about 3 of them are fully complementary to the
target mRNA sequences All known miRNAs are derived in vivo from double stranded
RNA precursors which are imperfectly annealed Since the biosynthesis and activities
of the miRNAs do not require perfect complementarity non canonical pathways of
RNAi are involved in the formation of miRNAs because usually RNAi requires
extensive complementarity of the dsRNA It is only because of this characteristic
mismatch between the sequences of miRNA and cognate mRNA that the in silico
identification of the target mRNA is so difficult (Rhoades et al 2002) The imperfect
nature of annealing between the two partners is viewed as the prime cause for
translational repression of the target mRNA (Pasquinelli 2002)
The mature miRNAs are always found in the single-stranded conformation in
nature for some unknown reason whereas siRNAs are double-stranded when detected
Third unlike siRNAs miRNAs enter riboprotein complexes with differing PPD
proteins (PAZ and Piwi domains) depending on the specificity of the miRNA or its
precursor with the cognate PPD proteins (Grad et al 2003) The sequence or structure
of miRNA or its precursor might ensure that it functions as a translational repressor and
not as a trigger of RNAi It is widely speculated that the siRNAs and miRNAs are
distinguished following their biosynthesis and these two are then allowed to form
related but distinct ribonucleoprotein complexes that target downstream substrates for
degradation or translation repression respectively This hypothesis is based on the
observation that siRNAs or exogenously supplied hairpin RNAs containing even a
Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
57
single mismatch with their substrate fail to repress the target mRNAs and do not simply
shift their regulatory mode to translation inhibition (Hannon 2002)
Not all RNAi pathway mutants are developmentally aberrant whereas micro-
RNA pathway mutants are expected to be defective in organism architecture and
development For example an RNA-dependent RNA polymerase-defective mutant of
A thaliana the sgs2sde1 mutant is unable to allow co-suppression of genes but is
perfectly normal in phenotypic development (Dalmay et al 2000)
116 Scope of the present investigation
The literature survey shows that coffee plants with minimal levels of caffeine have been
obtained by using specific region of the theobromine synthase gene in invert repeat
constructs The use of a region conserved for the NMT genes involved in caffeine
biosynthesis was reported earlier However the use of specific 3prime UTR region of the
theobromine synthase resulted in the spreading of silencing effect to non-specific genes
The use of conserved region which resulted in the reduction of caffeine theoretically
indicated silencing of all the NMT genes
The transformed somatic embryo needs to be regenerated and molecular and
biochemical analysis of individual plants is needed especially with regard to site of
integration and metabolic profiling of metabolites involved in caffeine biosynthesis
Metabolic profiling will enable an understanding of the specificity of silencing whether
any other metabolites are affected Knowledge of the site of integration will be
important in knowing whether integration of the transgene engenders a mutation in an
essential gene
The recent developments in the studies of gene regulation by small RNAs
especially those mediated by miRNA revealed that RNAi and the miRNA biogenesis
pathways shares a common pathway This lead to a hypothesis that RNAi production in
plants may affect the miRNA biosynthesis and this in turn may affect the gene
regulation indirectly It appeared that miRNA profiling in plants with normal and
transgenic plants would reveal something about the competition between the syntheses
of the two forms of RNA if such exists
Profiling of miRNAs can also give an insight into the regulatory mechanisms
present in coffee and their possible effect on caffeine biosynthesis They play an
important role in the development of plant and this evoked the significance of
Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora

Chapter 1 Introduction and Review of Literature
58
characterization of miRNAs in coffee plants in order to understand the various aspects
of gene regulation in caffeine metabolism
With these background knowledge the objectives of the present investigation
was laid as follows
1 Transformation of C canephora to produce minimal level caffeine lines
2 Characterization of transgenic plants for the transgene expression and
metabolic profiling of metabolites involved in caffeine biosynthesis
3 Studies on microRNA in C arabica and C canephora










![By: Leslie Harper. Classification [1] Kingdom: Plantae Subkingdom: Tracheobionta Superdivision: Spermatophyta Division: Magnoliophyta Class: Magnoliopsida](https://img.dokumen.tips/doc/110x75/56649ce15503460f949aca27/by-leslie-harper-classification-1-kingdom-plantae-subkingdom-tracheobionta.jpg)


















