Embed Size (px)
Citation preview

50
CHAPTER - 3
STUDIES IN THE SYNTHESIS
OF
MONTELUKAST SODIUM

51
3.1 - INTRODUCTION
The newer generation of leukotriene antagonists, such as ICI 204,219
(or Accollate), the quinolones MK-571 and RG-12,525, ONO-1078
(prankulast) and SK&F 104,353 are more promising1 as enumerated in
introduction chapter-1.
Leukotrienes constitute a group of locally acting hormones produced
in living systems from arachidonic acid. Major leukotrienes are
Leukotriene B4 (abbreviated as LTB4), LTC4, LTD4, and LTE4.
Biosynthesis of these leukotrienes begins with the action of 5-
lipoxygenase on arachidonic acid to produce the epoxide known as LTA4.
This is converted by further enzymatic transfermations to other
leukotrienes.24
A number of compounds of Formula (I) in which A represents
optionally substituted heterocycle, and pharmaceutically acceptable salts
thereof, have been disclosed as leukotriene antagonists and inhibitors of
leukotriene biosynthesis.24
The sodium salt of Montelukast (25) is a leukotriene receptor
antagonist (LTD4). It is useful in treatment of asthma, inflammation,

52
allergies, angina, cerebral spasm, glomerular nephritis, hepatitis,
endotoxemia, uveitis and allograft rejection.26
Bhupathy et al reported compounds of Formula (I) in which A
represents optionally substituted quinoline; more specifically disclosed is
the compound in which A represents 7-chloro-2-quinolinyl. U.S. Patent
document no. 5,270,324 reported two compounds of Formula (I) in which
A represents 6-fluoro- or 6,7-difluoro-2-quinolinyl. In the European
patent publication EP 0604,114 A1 there is disclosed compounds in
which A is halo-substituted thieno[2,3-b]pyridine, particularly 2,3-
dichlorothieno[2,3-b]pyridin-5-yl.27
Table-3.1: PRODUCT PROFILE OF MONTELUKAST SODIUM 26
Generic Name Montelukast sodium
Brand Name Singulair
Active Ingredient Montelukast
Innovator Merck & Co Inc
Marketed by Merck & Co Inc
Chemical Name [R]-1-[[[1-[3-[2-(7-chloro-2-quinolinyl)
ethenyl]phenyl]-3-[2-(1-hydroxy-1-methyl ethyl)-
phenyl]propyl]thio]methyl]cyclopropane acetic acid,
monosodium salt
Chemical C35H35ClNNaO3S

53
Formula
Molecular Weight 608.18
Chemical
Structure
CAS Registry No 151767-02-1
Physical
description
Hygroscopic, optically active, white to off-white
powder.
Solubility Freely soluble in ethanol, MeOH, and water; and
practically insoluble in acetonitrile.
3.2 – LITERATURE REVIEW
Many synthetic processes for preparation of Montelukast and its salts
are reported in literature. Some of them are discussed here under.
Belley et al 24 reported certain substituted quinoline compounds,
including (25), methods for their preparation, and methods of
pharmaceutical formulations using these compounds. The process
disclosed in Belley et al included preparation of (26), its reaction with
(27) in presence of hydrazine, cesium carbonate in acetonitrile as solvent
to provide (28). The protected compound (28) was reacted with

54
pyridinium p-toluene sulfonate in presence of NaOH in MeOH+THF to
afford (29) followed by its conversion to (25) (Scheme-3.1).
Scheme-3.1:
Cyclopropyl intermediate (27), which is used in scheme-3.1, was
prepared as per scheme-3.2.24
Scheme-3.2:
Bhupathy et al 27 reported a process for the preparation of (25) and
certain process intermediates. The process involved generation of
dilithium dianion of (36) followed by condensation with mesylate of (35)
to afford (29), which was further converted to (25) via dicyclohexyl amine
salt (37) (Scheme-3.3).
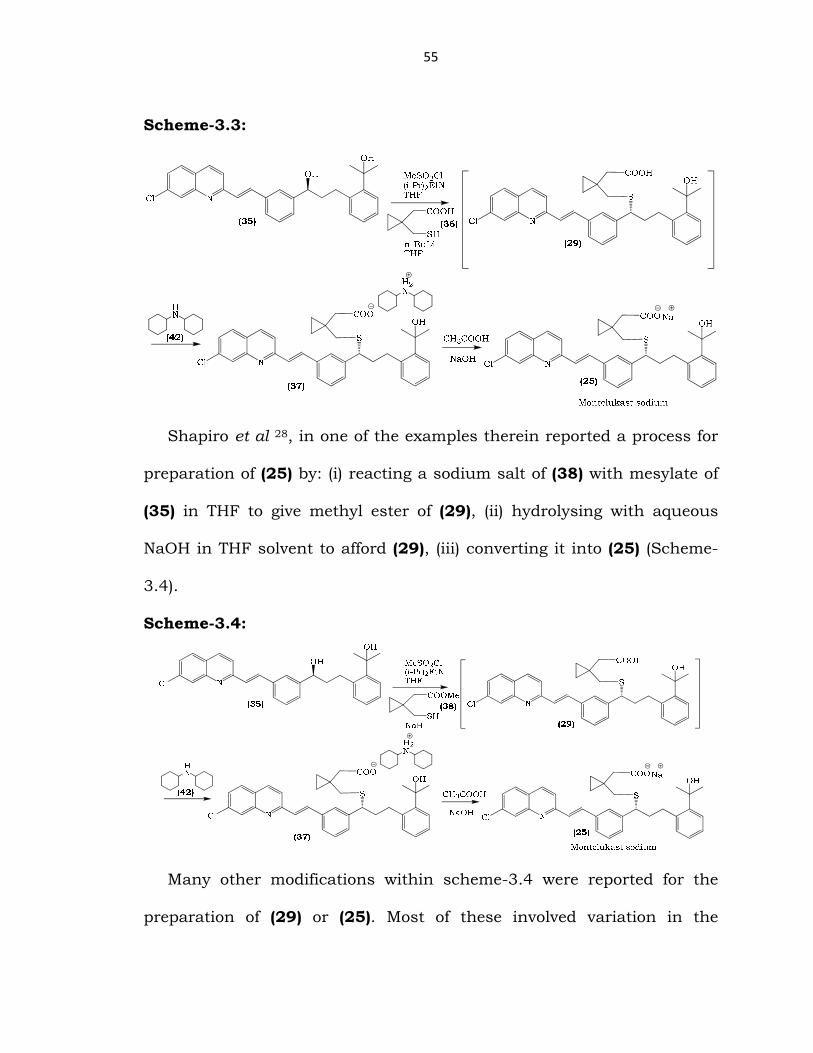
55
Scheme-3.3:
Shapiro et al 28, in one of the examples therein reported a process for
preparation of (25) by: (i) reacting a sodium salt of (38) with mesylate of
(35) in THF to give methyl ester of (29), (ii) hydrolysing with aqueous
NaOH in THF solvent to afford (29), (iii) converting it into (25) (Scheme-
3.4).
Scheme-3.4:
Many other modifications within scheme-3.4 were reported for the
preparation of (29) or (25). Most of these involved variation in the

56
functional group -COOH group present on cyclopropyl intermediate, such
as -COOCH3, amine salt of -COOH group instead of alkali metal salt etc.
Few other schemes involved other amine salts of (29) such as
adamentane salt etc.
Various other synthetic approaches were reported in literature.29-51
3.3 - PRESENT WORK
In view of the importance of (25) in drug therapy as enumerated above
and in chapter-1; and also in view of its market potential as evident from
its worldwide sales and worldwide consumption given in the abstract of
the thesis; though several synthetic processes for preparation of (25) are
reported in literature as discussed above, there is a continuing need for
new processes for the preparation of (29) and its salts more specifically
(25).
The objective of the present work is to study novel synthetic
approaches to provide cost effective, eco-friendly process for the
preparation of (25), which is well suited for commercial scale up.

57
3.4 – RESULTS AND DISCUSSION
In accordance with the above objective, two different synthetic
approaches for the preparation of (25) were developed.
3.4.1 - FIRST NOVEL SYNTHETIC APPROACH FOR THE
PREPARATION OF MONTELUKAST SODIUM OF THE PRESENT
WORK
The present work provides a first novel synthetic route for the
preparation of (29) and its salts, which was published as Reguri et al 52
and Chandra et al (ii) (Scheme-3.5).
Scheme-3.5:

58
In the process of developing the first novel synthetic approach, the
process was initially worked out by: (i) reducing (39) using (-)-DIP-Cl as a
chiral reducing reagent to provide corresponding hydroxyl ester, (ii)
mesylating the hydroxy ester with MsCl in presence of DIPEA, (iv)
reacting the resulting mesylate with disodium salt of (36) that is
prepared by treating it with sodium methoxide, to provide an ester, (v)
converting it into DCHA salt by treating with DCHA (42) in acetone to
provide dicyclohexyl amine salt of the ester, (vi) reacting the ester with
methyl magnesium chloride under Grignard’s reaction conditions until
the starting material is substantially consumed as observed by TLC to
provide the corresponding tertiary alcohol, which was expected to be
(25).
All the reactions were monitored by TLC and the final compound was
analyzed by High performance liquid chromatography (HPLC) using
chiral column to check the optical purity of the compound. It was
observed from the chromatograph that the retention time of the major
component was not matching with the retention of the (25) working
standard available at the time of development of the process. It rather
matched with the retention time of (65), the unwanted S-isomer of (25),
while the required isomer was R-isomer. When the reasons for this result
were investigated, the following facts were identified.

59
It was observed from many reported synthetic schemes discussed
before in this chapter that the reaction of S-isomer of mesylate with
mercaptan must happen in SN2 (substitution nucleophilic bi-molecular)
fashion, thereby it under goes inversion in configuration i.e., S- to R-
whereby the resulting compound after reaction would be R-isomer. But
in the instant case, though the starting hydroxy ester was an S-isomer,
the final compound was found to be having S-configuration as its
retention time matched with the retention time of unwanted isomer (65)
by chiral HPLC. This was further confirmed by its SR of –980 (c=1 in
chloroform). Whereas (25) has a SR of +1030 (c=1 in chloroform).
From the above discussion, it was evident that when S-isomer of
hydroxy ester (60) was used, the final product of the process was (65)
i.e., S-isomer and not the required R-isomer (25). The reason for this
could be due to anchiemeric affect of neighbouring carbomethoxy group
participation Hence, the synthetic scheme was modified to start from R-
isomer of hydroxy ester (40) to get required R-isomer (25).
Therefore, the process was developed by: (i) reducing (39) using (+)-
DIP-Cl as a chiral reducing reagent in DCM solvent followed by
quenching the reaction mixture with aqueous ammonia and isolating the
resulting precipitate, (ii) purifying it in a mixture of MeOH and water to
provide pure R-isomer (40), (iii) mesylating the resulting purified hydroxy

60
ester (40) with MsCl in presence of DIPEA in DCM solvent to provide the
mesylate (41), (iv) reacting the mesylate (41) with disodium salt of (36)
that is prepared by treating it with sodium methoxide, in a mixture of
DCM and DMF to provide an ester analog of (29), (v) converting it into
dicyclohexyl amine salt by treating with DCHA (42) in acetone solvent to
provide (43), (vi) reacting the free acid of (43) with methyl magnesium
chloride in THF under Grignard’s reaction conditions in toluene solvent
to get (29).
As is well known, the Grignard’s reaction of a carboxylic acid ester
involves a two step conversion. The first step of reaction involves
conversion of ester group into the corresponding methyl ketone with one
mole of Grignard’s reagent. The second step of reaction involves
conversion of the methyl ketone obtained in the first step into the
corresponding tertiary alcohol with another mole of Grignard’s reagent.
In the present case also there was conversion of an ester group into the
corresponding tertiary alcohol by Grignard’s reaction. However, it was
observed that the rate of first step of conversion of ester group into the
corresponding methyl ketone was faster whereas the rate of second step
of conversion of the methyl ketone into the corresponding tertiary alcohol
was slower when it was carried out within in the same reaction mixture
either by using excess moles of Grignard’s reagent from the beginning or
by using excess moles during the course of reaction in the conversion of

61
(43) to (29). During the reaction, three spots were observed in TLC
corresponding to ester, ketone and alcohol respectively. The reaction was
stopped after disappearance of ester by TLC and was quenched with aq.
acetic acid, layers were separated and the organic layer was washed aq.
sodium bicarbonate solution followed by washing with water. After
removing water azeotropically, the resulting solution containing ketone
and alcohol was reacted with Grignard’s reagent once again to consume
the ketone. Quenched with aq. acetic acid, layers were separated, organic
layer was washed with aq. sodium bicarbonate solution followed by
water. Solvent was removed by distillation under reduced pressure. The
resulting crude (29) had a purity of about 96% as measured by HPLC. It
was purified by repeated recrystallizations from toluene to get pure
crystalline (29). The purity of the crystalline (29) after 5th
recrystallization was about 98% by HPLC. It was observed that the purity
though increased after repeated recrystallizations, in order to achieve a
purity of about 99% or more, it was required to convert (29) into (44).
Therefore, the pure crystalline (29) was treated with TBA (45) in a
mixture of acetone and IPA; and its purification by repeated
recrystallization from a mixture of acetone and IPA gave a pure
crystalline (44). The said (44) was then suspended in DCM, treated with
aq. acetic acid, stirred to get a clear biphasic medium wherein all the
solids were dissolved. Organic layer was separated and washed with

62
water to get a solution of (29) in DCM, followed by treatment with
methanolic NaOH solution to get a solution of (25) in a mixture of DCM
and MeOH. The solvent was distilled completely under reduced pressure
and the resulting crude was dissolved in minimum quantity of toluene,
the solution was added to n-heptane and stirred to get a precipitate. The
solid precipitate was filtered, washed with n-heptane and dried under
reduced pressure to provide (25). The resulting (25) had a purity of
about 99% as measured by HPLC and an optical purity of about 99.8%
as measured by chiral HPLC method. It had a SR of +1030 (c=1 in
chloroform).
3.4.2 - SECOND NOVEL SYNTHETIC APPROACH FOR THE
PREPARATION OF MONTELUKAST SODIUM OF THE PRESENT
WORK
The present work further provides a second novel synthetic approach
for the preparation of (29) and its salts published as Sundaram et al 53
and Chandra et al. (iii)
In the second novel synthetic approach for the preparation of (25) of
the present work, (10) was one of the key starting materials from which
further process steps were evolved. Another key intermediate involved
was (50).
The process of the second novel synthetic approach for the
preparation of (25) involved: (i) mesylation of (35) using MsCl in presence

63
of DIPEA in acetonitrile solvent; (ii) condensation of the resulting
mesylate in a mixture acetonitrile and DMF with lithium salt of (46) that
was prepared by treating it with n-butyl lithium to provide (47); (iii)
hydrolysis of the resulting compound with aqueous NaOH to provide
(29); (iv) converting it into (44) by treating with TBA (45) in acetone and
purifying the isolated (29) by recrystallization from acetone; (v)
converting the purified (44) into (25) (Scheme-3.6).
Scheme-3.6:
When a mixture of (46) and (49) was used, the resulting product in
step-1 of scheme-3.6 would be a mixture of (47) and (50) (Scheme-3.7).

64
Scheme-3.7:
NCl
OH
OH
CN
SH
n-BuLiCH3CNDMF
NCl
S
OHCN
MsClDIPEADCM
(46)
(35)
(49)
NCl
OSO2Me
OH
(48)
NCl
OSO2Me
OH
(48)+ +
CONH2
SH
(47)(50)
+ NCl
S
OHCONH2
3.4.3 - STUDIES IN THE SYNTHESIS OF STARTING MATERIALS OF
THE FIRST AND SECOND NOVEL SYNTHETIC APPROACHES FOR
THE PREPARATION MONTELUKAST SODIUM OF THE PRESENT
WORK
(39) and (36) were the key starting materials in the process of first
novel synthetic approach of the present work; and (35) and {(46) or a
mixture of (46) and (49)} were the key starting materials in the process of
the second novel synthetic approach of the present work. The synthetic
processes for the preparation compounds (39), (36), (35), {(46) or a
mixture of (46) and (49)} were broadly selected based on the respective
synthetic processes disclosed in Belley et al.

65
It is also the objective of the present work to provide an improved
process for the preparation of compounds (39), (36), (35), {(46) or a
mixture of (46) and (49)}.
3.4.3.1 - Improved process for the preparation of (39) and an
improved process for the preparation of (35) from (39):
Synthetic processes disclosed in Belley et al and few other prior art
references were taken as the basis for preparation of different
intermediates of (39) and incorporated various improvements such as
avoiding hazardous reagents, costly raw materials and solvents,
simplifying reaction and work up procedures, purifying the intermediates
thereby providing cost effective, eco-friendly and commercially viable
process for each of the intermediate.
The sequence of steps for the preparation of (39) (scheme-3.8) started
from reaction of (51) with (52) in presence of Ac2O in toluene solvent. In
this reaction the major by-product was a dimer (66), which was formed
due to reaction of the required product of the reaction (53) with one more
mole of (51). In Tung et al 32, the process for preparation of (53) involved
1.5 molar equivalents of (52) per mole of (51) in excess Ac2O in xylene
solvent. In an objective to minimize the quantity of Ac2O whose excess
usage was not preferable on a commercial scale, the process was
optimized with 1.2 molar equivalents of (52) and just 1.9 molar
equivalents of Ac2O per mole of (51) in toluene solvent at reflux

66
condition. Reaction was monitored by TLC (mobile phase: Ethyl acetate :
Hexanes – 1:4). After completion of reaction, the isolated wet compound
containing (53) and the dimer (66) was suspended in ethyl acetate and
heated to reflux. The mixture was filtered, filtrate containing pure (53)
was concentrated, cooled to room temperature and added hexanes to
precipitate out the compound completely. The resulting yield was
improved with good quality.
The next step was Grignard’s reaction of (53) with methyl magnesium
chloride in THF in toluene, precipitation of compound by adding aqueous
NH4Cl solution to provide the alcohol (54). The improvement in this step
was the replacement of expensive methyl magnesium bromide and
aqueous acetic acid reported in Balley et al for this reaction with
comparatively inexpensive methyl magnesium chloride and aqueous
NH4Cl respectively, which improved the yield to about 80% with good
quality and was void of extraction and void of column chromatography
for purification.
The next step was oxidation of the alcohol (54) to provide the
corresponding methyl ketone (55). The oxidizing agent used was
manganese (IV) dioxide. Initially, when the process reported in Belley et
al was reproduced exactly, the reaction did not go to completion as
monitored by TLC (mobile phase: Ethyl acetate : Hexanes – 1: 4). To
complete the reaction, the solvent was changed from ethyl acetate to

67
DCM but there was not much improvement. Then it was thought that the
activity of manganese (IV) dioxide must be playing a role in the reaction
and hence, tried with activated manganese (IV) dioxide. Though there
was slight improvement in the reactivity, still the problem was not
resolved completely as various commercial lots of manganese (IV) dioxide
lots had varying activity and therefore the activity of given lot to be used
for a given experiment had become highly unpredictable, which was not
a preferable situation to make the process commercially viable.
Therefore, it was thought to work on the temperature of the reaction by
the right solvent choice as the reaction medium. In the reported
processes, reaction was conducted at room temperature and the reaction
time used run through several hours such as 20 or more hours
depending on the activity of the manganese (IV) dioxide lot used. To
overcome this problem, the reaction was conducted by using manganese
(IV) dioxide of a randomly chosen commercial lot by not taking its activity
into consideration in toluene as solvent at reflux temperature. The
reaction completed within about 5 hours. The reaction mixture was
filtered hot to remove reduced manganese dioxide, the filtrate was
concentrated and the solid was isolated to provide the methyl ketone (55)
in quantitative yield with good quality.
The next step involved conversion of the methyl ketone (55) into the β-
keto ester (56) by condensation with dimethyl carbonate. Reported

68
procedure involved sodium hydride base and THF solvent. As both base
and solvent were not preferable on commercial scale, they were replaced
by sodium methoxide powder and 1,4-dioxan respectively, both of which
are safe to handle on large scale and inexpensive. After completion of the
reaction (monitored by TLC; Mobile phase: Ethyl acetate : Hexanes – 1:
4), the reaction mixture was cooled to room temperature and was
quenched with slow addition of water. The separated solid was filtered
and the wet solid was washed with MeOH with stirring to provide the β-
keto ester (56) with quantitative yield and good quality.
The next step was the condensation of the β-keto ester (56) with (57)
to get a diester (58). The reported procedure involves NaH base and THF
solvent. As both base and solvent are not preferable on commercial scale,
they were replaced by K2CO3 and DMF respectively, as both of which are
safe to handle on large scale and inexpensive. The reaction proceeded
very smoothly with good conversion rate. However, as observed by TLC
(mobile phase: Ethyl acetate : Hexanes – 1: 4), three product spots were
observed, which corresponded to the diester (58), the keto acid (59) and
the keto ester (39) respectively. The reaction mixture after substantial
disappearance of β-keto ester (56), was quenched with aqueous sodium
acetate and the resulting compound was filtered and washed with MeOH
with stirring to provide diester (58) with quantitative yield and good
quality.

69
In the next step, the diester (58) was hydrolyzed with a mixture of
acetic acid and aqueous HCl to provide the corresponding keto acid (59).
Reaction was monitored by TLC (mobile phase: Ethyl acetate : Hexanes –
1: 4) only for substantial disappearance of the diester (58). In this step
two product spots were observed corresponding to the keto acid (59) and
the keto ester (39) respectively, with the major spot being keto ester (39),
which infers that the major reaction in this step is decarboxylation of
carboxylic group attached to aliphatic chain alone with the retention of
the ester group attached to aromatic ring with a minor reaction of
decarboxylation and hydrolysis to lead to the keto acid (59) as the minor
product. The resulting isolated wet compound was stirred in MeOH to get
keto acid (59) with quantitative yield and good quality.
In the next step, the keto acid (59) containing the keto ester (39) as
the major component was converted to the keto ester (39) by
esterification. Reported procedure involved the said conversion with
methyl iodide in presence of potassium carbonate in acetone solvent. The
reaction mixture after completion of reaction (monitored by TLC; mobile
phase: Ethyl acetate : Hexanes – 1: 4) was filtered to remove potassium
carbonate. Acetone from the filtrate was distilled completely. The
resulting mass was dissolved in minimum quantity of chloroform and
keto ester (39) was isolated in quantitative yield and good quality. The

70
said keto ester (39) was used as the starting material in the first novel
synthetic approach of the present work.
Next step (scheme-3.9) was the chiral reduction of the keto ester (39)
with (-)-DIP-Cl to get the hydroxy ester (60), which was the S-isomer of
(40). Reported procedure involved THF as solvent and diethanolamine
was used for quenching the reaction mixture. The reaction temperature
in the reported procedure was -250C. The process was simplified by
replacing the THF solvent with DCM, using aqueous ammonia in place of
diethanolamine for quenching the reaction mixture and optimizing the
reaction temperature to -5 to 00C. Reaction was monitored by TLC
(mobile phase: Ethyl acetate : Hexanes – 1 : 4 and one drop of aq.
ammonia). The resulting hydroxy ester (60) carried α-pinene as the side
product of the reaction that was the starting material for the preparation
of (-)-DIP-Cl. Due to the presence of α-pinene along with the obtained
hydroxy ester (60), it was denatured with gumminess. To get a pure
hydroxy ester (60), it was purified by suspending the gummy solid in
MeOH whereby the gummy insoluble solid separated out, the mixture
was filtered, water was added to the clear filtrate to provide the pure
hydroxy ester (60) in quantitative yield and good quality.
Next step was the Grignard’s reaction of the hydroxy ester (60) to get
the diol (35). The reported procedure involved usage of methyl
magnesium bromide and a mixture of Toluene and THF as reaction

71
medium. The reaction conditions were simplified by replacing expensive
methyl magnesium bromide with inexpensive methyl magnesium
chloride and by using only toluene as the solvent without using
additional THF. That means THF associated with methyl magnesium
chloride was sufficient for the required polarity for the reaction. Reaction
was monitored by TLC (mobile phase: Ethyl acetate : Hexanes – 1 : 4 and
one drop of aq. ammonia). The resulting diol (35) was purified by
recrystallization from toluene.
Hydroxy ester (40) i.e., R-isomer was the required intermediate in the
first novel synthetic approach of the present work rather than hydroxy
ester (60) to get Montelukast having R-configuration, which was the
required isomer rather than S-isomer (65). Hydroxy ester (40) was
prepared by following substantially the similar procedure as that of
hydroxy ester (60) discussed above except that (+)-DIP-Cl was used
instead of (-)-DIP-Cl as the required isomer of hydroxy ester is the R-
isomer.
Scheme-3.8:

72
Scheme-3.9:
The process for preparation of hydroxy ester (40) from keto ester (39)
according to the present work is given in Scheme-3.10.
Scheme-3.10:
NCl
O COOCH3
(+)-DIP chlorideDCM
NCl
OH COOCH3
(39)
(40)
3.4.3.2 - Improved process for the preparation of (46) and a mixture
of (46) and (49):
(46) was prepared from (61) by hydrolysis with methanolic NaOH at a
temperature of -15 to -120C (Scheme-3.11).

73
A mixture of (46) and (49) was prepared by hydrolysis of (61) with
potassium hydroxide in a mixture of MeOH and water at a temperature
of 500C (Scheme-3.11).
Scheme-3.11:
3.4.4 - RESULTS AND DISCUSSION ON IMPURITIES OF
MONTELUKAST SODIUM OBTAINED THE PRESENT WORK AND ITS
INTERMEDIATES
The following impurities were identified in (25) and its intermediates
by LC-MS method or by spiking method by HPLC. The said impurities
were prepared by the procedures described here under in the
experimental section for the preparation of impurities of (25) and were
characterized.
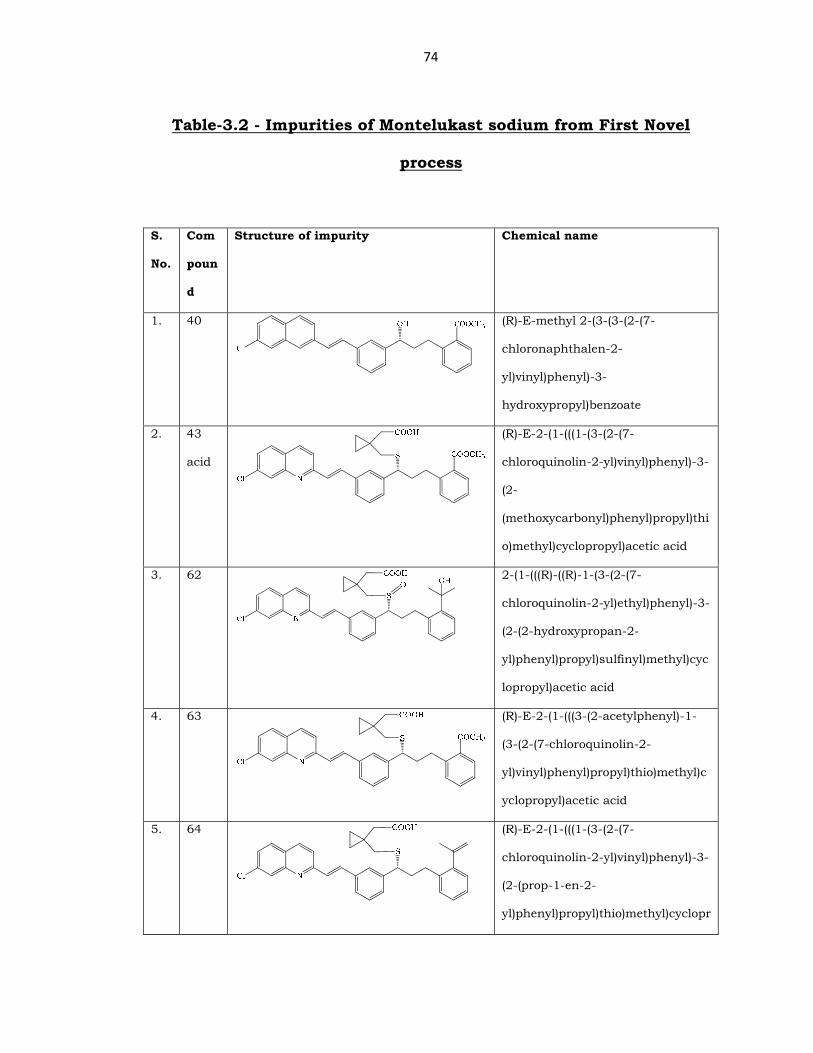
74
Table-3.2 - Impurities of Montelukast sodium from First Novel
process
S.
No.
Com
poun
d
Structure of impurity Chemical name
1. 40
(R)-E-methyl 2-(3-(3-(2-(7-
chloronaphthalen-2-
yl)vinyl)phenyl)-3-
hydroxypropyl)benzoate
2. 43
acid
(R)-E-2-(1-(((1-(3-(2-(7-
chloroquinolin-2-yl)vinyl)phenyl)-3-
(2-
(methoxycarbonyl)phenyl)propyl)thi
o)methyl)cyclopropyl)acetic acid
3. 62
2-(1-(((R)-((R)-1-(3-(2-(7-
chloroquinolin-2-yl)ethyl)phenyl)-3-
(2-(2-hydroxypropan-2-
yl)phenyl)propyl)sulfinyl)methyl)cyc
lopropyl)acetic acid
4. 63
(R)-E-2-(1-(((3-(2-acetylphenyl)-1-
(3-(2-(7-chloroquinolin-2-
yl)vinyl)phenyl)propyl)thio)methyl)c
yclopropyl)acetic acid
5. 64
(R)-E-2-(1-(((1-(3-(2-(7-
chloroquinolin-2-yl)vinyl)phenyl)-3-
(2-(prop-1-en-2-
yl)phenyl)propyl)thio)methyl)cyclopr
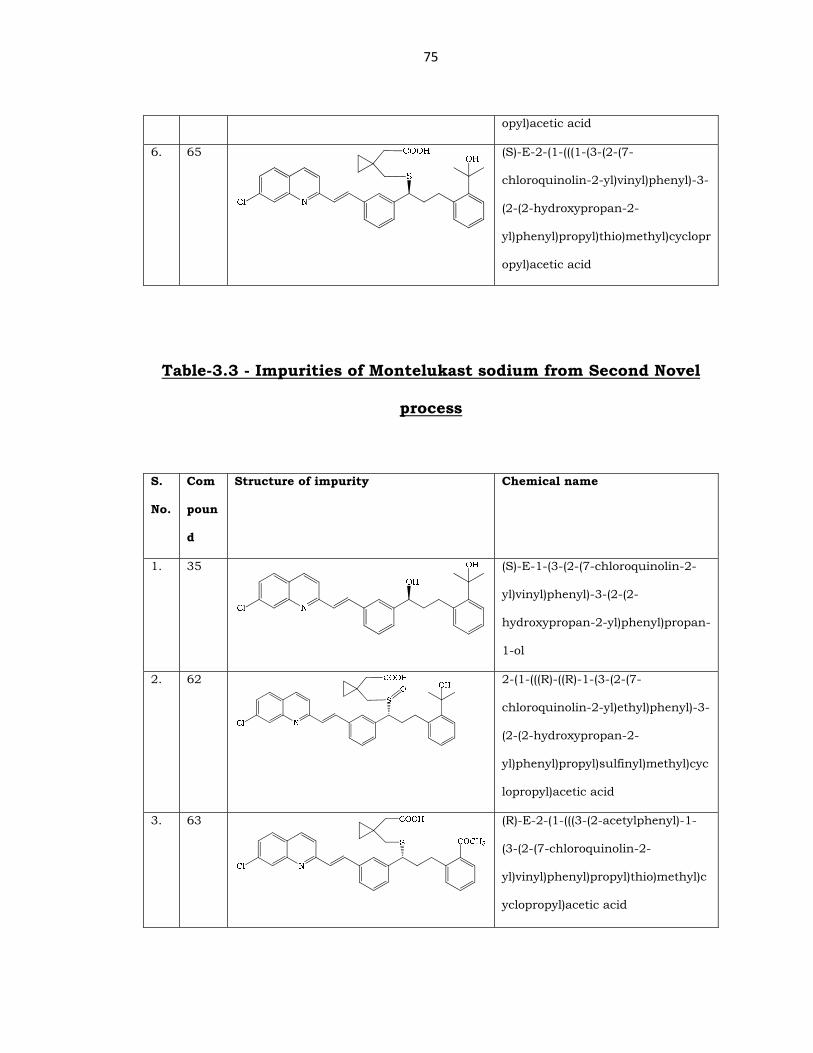
75
opyl)acetic acid
6. 65
(S)-E-2-(1-(((1-(3-(2-(7-
chloroquinolin-2-yl)vinyl)phenyl)-3-
(2-(2-hydroxypropan-2-
yl)phenyl)propyl)thio)methyl)cyclopr
opyl)acetic acid
Table-3.3 - Impurities of Montelukast sodium from Second Novel
process
S.
No.
Com
poun
d
Structure of impurity Chemical name
1. 35
(S)-E-1-(3-(2-(7-chloroquinolin-2-
yl)vinyl)phenyl)-3-(2-(2-
hydroxypropan-2-yl)phenyl)propan-
1-ol
2. 62
2-(1-(((R)-((R)-1-(3-(2-(7-
chloroquinolin-2-yl)ethyl)phenyl)-3-
(2-(2-hydroxypropan-2-
yl)phenyl)propyl)sulfinyl)methyl)cyc
lopropyl)acetic acid
3. 63
(R)-E-2-(1-(((3-(2-acetylphenyl)-1-
(3-(2-(7-chloroquinolin-2-
yl)vinyl)phenyl)propyl)thio)methyl)c
yclopropyl)acetic acid
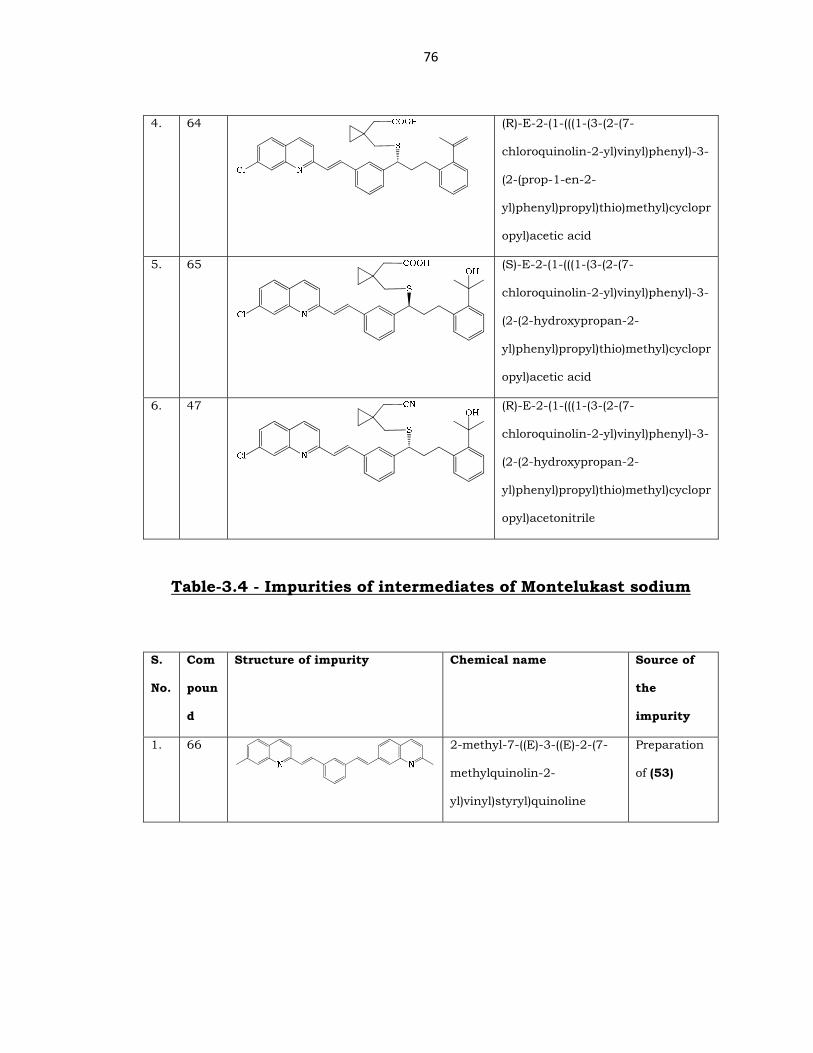
76
4. 64
(R)-E-2-(1-(((1-(3-(2-(7-
chloroquinolin-2-yl)vinyl)phenyl)-3-
(2-(prop-1-en-2-
yl)phenyl)propyl)thio)methyl)cyclopr
opyl)acetic acid
5. 65
(S)-E-2-(1-(((1-(3-(2-(7-
chloroquinolin-2-yl)vinyl)phenyl)-3-
(2-(2-hydroxypropan-2-
yl)phenyl)propyl)thio)methyl)cyclopr
opyl)acetic acid
6. 47
(R)-E-2-(1-(((1-(3-(2-(7-
chloroquinolin-2-yl)vinyl)phenyl)-3-
(2-(2-hydroxypropan-2-
yl)phenyl)propyl)thio)methyl)cyclopr
opyl)acetonitrile
Table-3.4 - Impurities of intermediates of Montelukast sodium
S.
No.
Com
poun
d
Structure of impurity Chemical name Source of
the
impurity
1. 66
2-methyl-7-((E)-3-((E)-2-(7-
methylquinolin-2-
yl)vinyl)styryl)quinoline
Preparation
of (53)

77
3.5 – EXPERIMENTAL SECTION
3.5.1 - Experimental Section For The First Novel Process For The
Preparation Of Montelukast Sodium (25):
Preparation of (40) {R (+)-hydroxy ester}:
100 grams (0.219 mol) of (39) was dissolved in 500 ml of DCM. 180
ml of (+)-DIP-Cl (0.346 mol, 1.6 equivalents) was taken separately in 500
ml of DCM and cooled to -50C with stirring. Above (39) solution in DCM
was added to (+)-DIP-Cl in DCM at -5 to 00C slowly drop wise. After
addition, the reaction mixture was aged at -5 to 00C for 10 hours.
Reaction was monitored by TLC (mobile phase: Ethyl acetate : Hexanes –
1:4 and one drop of aq. ammonia). After the completion of reaction, the
reaction mixture was quenched with aqueous ammonia and stirred 60
minutes. Aqueous sodium chloride solution was added and stirred for 30
minutes. Layers were separated and the organic layer was washed with
aqueous sodium chloride solution. DCM was distilled from the organic
layer.
Purification of (40):
The resulting crude (40) obtained above was dissolved in 1200 ml of
MeOH and filtered the insoluble solids. 50 ml water was added slowly
drop wise to the filtrate and continued stirring for 2 hours. The separated
solid was filtered, washed with a mixture of MeOH and water and dried
78
at 500C to yield 80 grams (yield: 80%) of (40). (Purity by HPLC: 98%). SR:
of +340 (c=1 in chloroform).
Characterization of (40):
Other than SR, the following characterization data confirmed the
structure of (40).
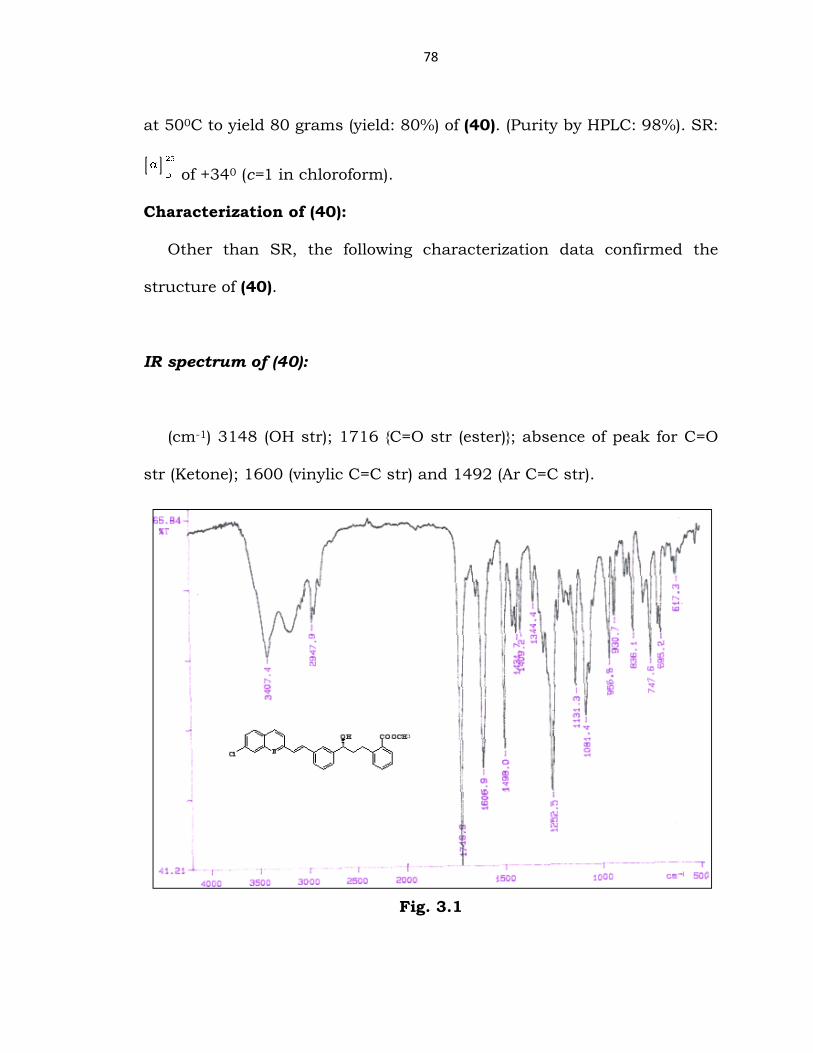
IR spectrum of (40):
(cm-1) 3148 (OH str); 1716 {C=O str (ester)}; absence of peak for C=O
str (Ketone); 1600 (vinylic C=C str) and 1492 (Ar C=C str).
Fig. 3.1
79
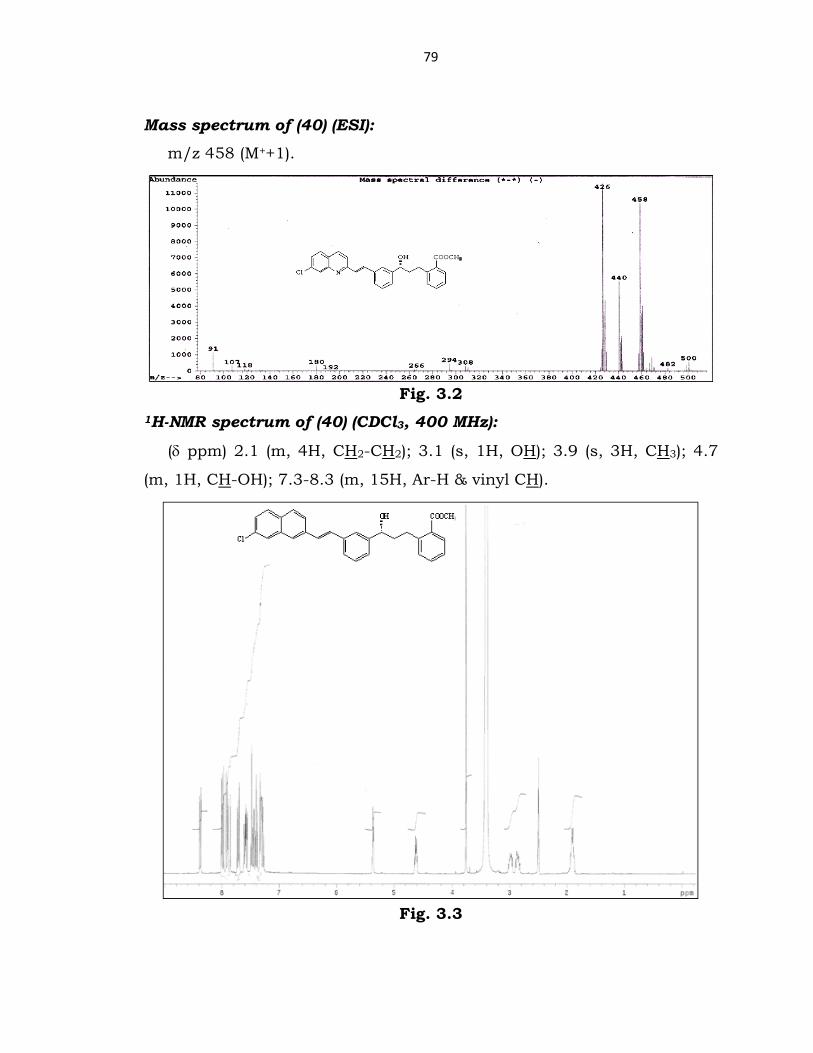
Mass spectrum of (40) (ESI):
m/z 458 (M++1).
Fig. 3.2
1H-NMR spectrum of (40) (CDCl3, 400 MHz):
(δ ppm) 2.1 (m, 4H, CH2-CH2); 3.1 (s, 1H, OH); 3.9 (s, 3H, CH3); 4.7
(m, 1H, CH-OH); 7.3-8.3 (m, 15H, Ar-H & vinyl CH).
Fig. 3.3
80
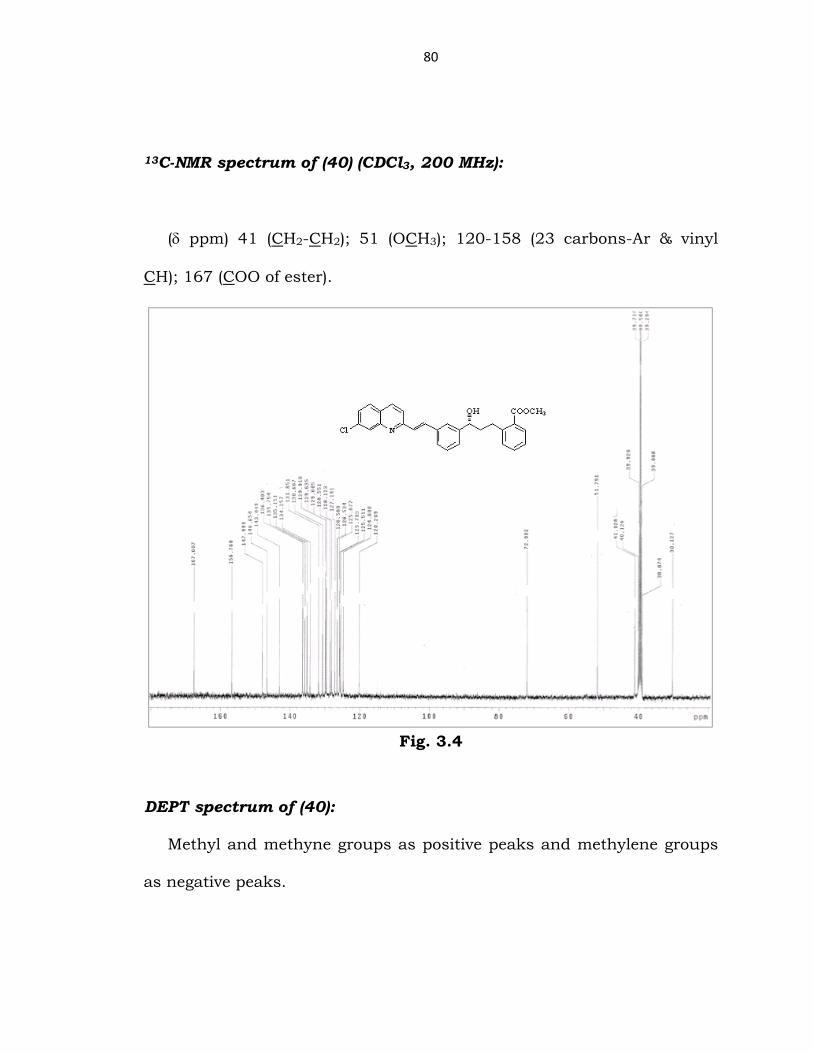
13C-NMR spectrum of (40) (CDCl3, 200 MHz):
(δ ppm) 41 (CH2-CH2); 51 (OCH3); 120-158 (23 carbons-Ar & vinyl
CH); 167 (COO of ester).
Fig. 3.4
DEPT spectrum of (40):
Methyl and methyne groups as positive peaks and methylene groups
as negative peaks.

81
Fig. 3.5
Preparation of (43):
A stirred mixture of 100 grams (0.219 mol) of (40) and 500 ml of
toluene was heated to reflux and water was removed by azeotropic
distillation using Dean-Stark apparatus. The mixture was cooled to 500C
and the remaining solvent was distilled under reduced pressure. The
residue was re-dissolved in 200 ml of DCM at ambient temperature and
the solvent was distilled again under reduced pressure. The residue was
re-dissolved in 1000 ml of DCM and the mixture was cooled to 0-50C.
57.5 ml (0.328 mol) of DIPEA were added at once to the stirred
mixture; and the reaction mass was stirred at 0-50C for 15-30 minutes.
22 ml (0.284 mol) of MsCl were added dropwise at 0-50C with stirring.
After the addition was completed, the cooling was discontinued, and the
reaction mass was maintained at 25-350C until reaction completion. 600
ml of water was added and the mass was stirred for another 30 minutes.

82
The organic and aqueous layers were separated, and the aqueous
layer was extracted with 200 ml of DCM. The combined organic layers
were washed with water (3 X 600 ml). DCM was distilled off
atmospherically, followed by distillation under reduced pressure at a
temperature of below 500C. The resulting residue was re-dissolved in
toluene (200 ml), which was again distilled off under reduced pressure at
45-500C to obtain a residue of the mesylate (41).
38.3 grams (0.262 mol) of (36) and 450 ml of MeOH were stirred until
clear dissolution at 25-350C for 60 minutes. Added 29.34 grams (0.524
mol) of NaOMe powder slowly. A mixture of the crude mesylate (41)
obtained above, DCM and DMF (450 ml) were added to this disodium salt
of (36), and the resulting reaction mass was stirred for clear dissolution
at 25-350C. The reaction mass was heated and maintained at reflux
temperature for 2-3 hours. 450 ml of water was charged to the reaction
mixture and continued stirring for 15 minutes. The organic and aqueous
layers were separated; the aqueous layer extracted with 200 ml of DCM.
The combined organic layer was washed with a mixture of NaCl (37.5
grams) and water (400 ml) solution, then washed with a solution of acetic
acid (45 ml) in water (400 ml), followed by a water wash (4 X 400 ml).
The solvents were distilled off under atmospherically from the organic
layer; followed by distillation under reduced pressure at 45-500C. The
obtained residue was dissolved in 200 ml acetone; and acetone was

83
distilled off under reduced pressure at 45-500C. Thus obtained residual
crude product was re-dissolved in 500 ml acetone at 25-350C. 52 ml
(0.262 mol) of DCHA (42) were added to the solution of the crude residue
at 25-350C; and the mass was stirred at 25-350C until a solid separated.
Solid was filtered, the wet compound was taken into 400 ml of acetone,
and heated to reflux. The mass was maintained at reflux for 1-2 hours
and then cooled to 25-350C; stirring continued for 4-5 hours. The
resulting solid was filtered and washed with 50 ml of acetone. The solid
was dried in an oven at 45-500C to afford the 49.7 grams of (43).
Purification of (43):
49 grams (0.0639 mol) of (43) and 490 ml of acetone were charged
into a round bottomed flask, and the mixture was heated to reflux. The
mass was maintained at reflux for 1-2 hours, cooled to 25-350C slowly
under stirring, and maintained at 25-350C for another 4-5 hours. The
separated solid was filtered; washed with acetone (49 ml) and dried at
50-550C to afford 44.7 grams of purified (43).
Characterization of (43):
IR spectrum of (43):
(cm-1) 3431 (N-H str); 2668, 2530, 2380 (+N-H str); 3056 (Ar C-H str);
2926 & 2854 (aliphatic C-H str); 1721 (C=O str); 1605 (-COO- Asymm.
Str); 1533 (C=C str); 1496 (Ar C=C str); 697 (C-S str).

84
NCl
COOCH3
COO
S
H2N
Fig. 3.6
Mass spectrum of (43) (ES-MS):
m/z 767.8 (DCHA salt).
Fig. 3.7
85
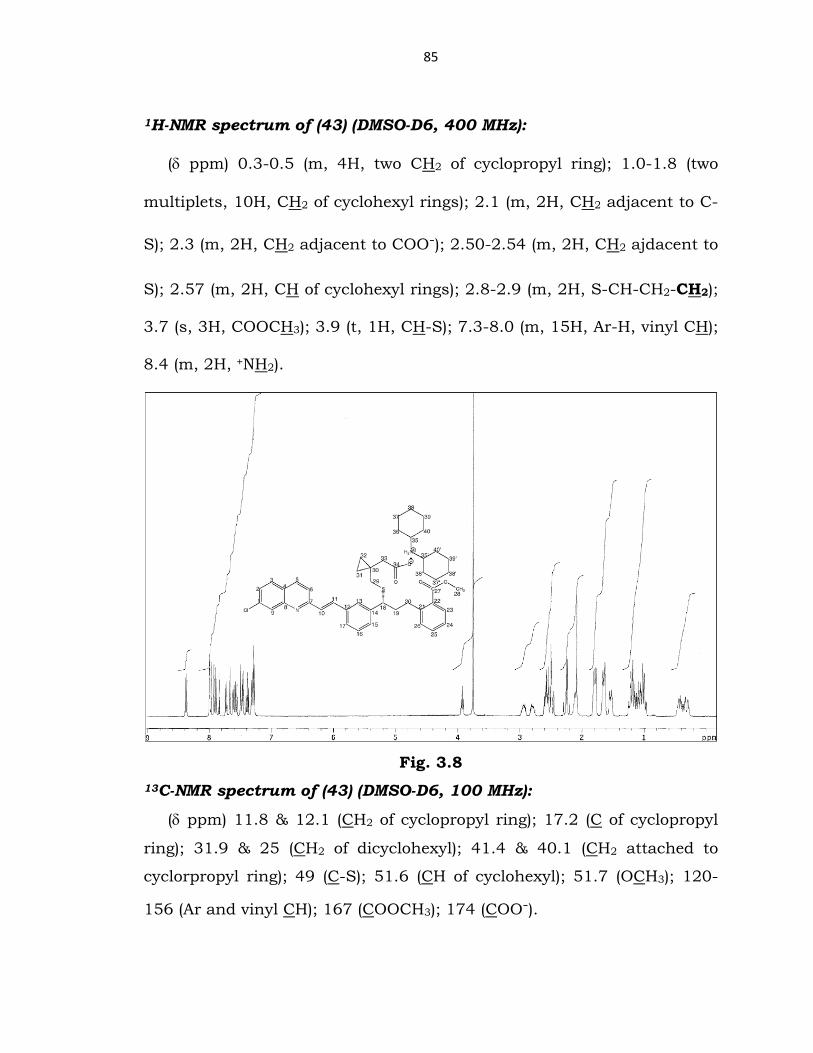
1H-NMR spectrum of (43) (DMSO-D6, 400 MHz):
(δ ppm) 0.3-0.5 (m, 4H, two CH2 of cyclopropyl ring); 1.0-1.8 (two
multiplets, 10H, CH2 of cyclohexyl rings); 2.1 (m, 2H, CH2 adjacent to C-
S); 2.3 (m, 2H, CH2 adjacent to COO-); 2.50-2.54 (m, 2H, CH2 ajdacent to
S); 2.57 (m, 2H, CH of cyclohexyl rings); 2.8-2.9 (m, 2H, S-CH-CH2-CH2);
3.7 (s, 3H, COOCH3); 3.9 (t, 1H, CH-S); 7.3-8.0 (m, 15H, Ar-H, vinyl CH);
8.4 (m, 2H, +NH2).
Fig. 3.8
13C-NMR spectrum of (43) (DMSO-D6, 100 MHz):
(δ ppm) 11.8 & 12.1 (CH2 of cyclopropyl ring); 17.2 (C of cyclopropyl
ring); 31.9 & 25 (CH2 of dicyclohexyl); 41.4 & 40.1 (CH2 attached to
cyclorpropyl ring); 49 (C-S); 51.6 (CH of cyclohexyl); 51.7 (OCH3); 120-
156 (Ar and vinyl CH); 167 (COOCH3); 174 (COO-).
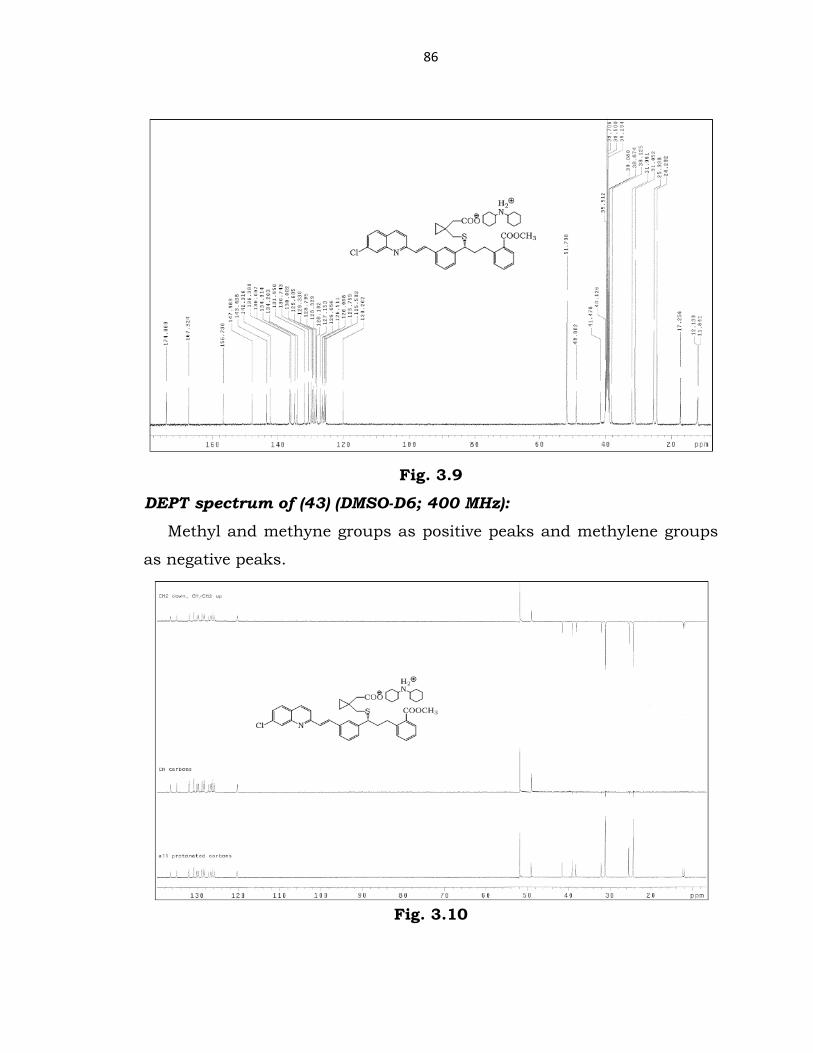
86
Fig. 3.9
DEPT spectrum of (43) (DMSO-D6; 400 MHz):
Methyl and methyne groups as positive peaks and methylene groups
as negative peaks.
Fig. 3.10

87
Preparation of (29):
100 grams (0.13 mol) of (43) and 1000 ml of toluene were charged to
a round bottomed flask, and stirred for about 5 minutes. A mixture of
acetic acid (15 ml) and water (500 ml) was added, and the mass was
stirred for another 30 minutes. The organic and aqueous layers were
separated. Organic layer was dried over anhydrous Na2SO4 after washing
with water (3 X 500 ml). The solvent was removed under reduced
pressure at a temperature below 500C. The resulting crude residue was
dissolved in a mixture of toluene (760 ml) and THF (760 ml); the solution
was transferred into a round bottomed flask and cooled to 00C under
nitrogen atmosphere. 261 ml of 3 M solution of methyl magnesium
chloride in THF were added dropwise during 2-3 hours at 0-50C. The
reaction mass was maintained at 0-50C for 6-7 hours, and cooled to 00C.
A mixture of acetic acid (90 ml) and water (750 ml) was slowly added at
below 150C for about one hour. The reaction mass was stirred at 25-350C
for another one hour until clear dissolution. The organic and aqueous
layers were separated. Organic layer was washed with 5% sodium
bicarbonate solution (2 X 750 ml), followed by a water wash (2 X 750 ml)
and dried over anhydrous Na2SO4. The solvent from the organic layer
was removed under reduced pressure. The resulting residue was treated
with additional amount of methyl magnesium chloride (50 ml) followed
by work-up in the same procedure.

88
The crude product was dissolved in toluene (100 ml) and stirred at
25-350C to separate a solid. The separated solid was filtered and washed
with toluene (30 ml). The wet solid and toluene (90 ml) were charged into
a round-bottomed flask, heated to 900C, and stirred for 30 minutes until
complete dissolution, cooled to 25-350C, and maintained for 6-10 hours.
The solid was filtered and washed with toluene (22 ml). The re-
precipitation process was repeated four to five times. The solid was dried
to afford about 17.4 grams of the purified (29).
Preparation of (44):
8.6 grams (0.0147 mol) of (29), 155 ml of acetone and 17 ml of IPA
were charged into a round bottomed flask and stirred at 25-350C until
clear dissolution. 2.3 ml (0.022 mol) of TBA (45) was added and the mass
was stirred at 25-350C. The separated solid was filtered, washed with
acetone (20 ml) and dried at 40-500C. The dried residue was re-
precipitated from a mixture of acetone (225 ml) and IPA (25 ml), affording
6 grams of (44). Characterization data of (44) is substantially in
accordance with the data discussed for (44) under experimental section
for second novel process for preparation of Montelukast sodium (25).
Preparation of (25):
(44) obtained above and 50 ml of DCM were mixed at 25-350C. A
mixture of 0.5 ml of acetic acid and 25 ml of water was added to the
mass, and stirred at 25-350C for 15 minutes. The organic and aqueous

89
layers were separated; the organic layer was washed with water (4 X 25
ml) and dried over Na2SO4. The solvent was removed under reduced
pressure at a temperature below 450C. 10 ml of MeOH were added to the
residue. The solvent was removed again under reduced pressure at a
temperature of below 450C. A mixture of 0.307 grams of freshly prepared
sodium pellets and 50 ml of MeOH was added to the residue at 25-350C.
0.5 grams of carbon were added and the mass was stirred for about 30
minutes at 25-350C. The carbon was filtered and washed with MeOH.
The filtrates were combined and the solvent was removed under reduced
pressure at a temperature below 450C. The residue was re-dissolved in
toluene (25 ml) and the solvent was removed again under reduced
pressure at a temperature below 450C. The residue was re-dissolved in
toluene (5 ml) and added to a pre-filtered n-heptane under nitrogen
atmosphere at 25-350C. The mixture was stirred at 25-350C for about 1
hour to form a precipitate, which was filtered and washed with n-heptane
(25 ml) under nitrogen atmosphere. The resulting solid was dried at 800C
to afford 3.2 grams of (25).
Characterization of (25):
Characterization data given here under for (25) obtained in the first
novel process is in agreement with the data discussed for (25) under
experimental section for second novel process for preparation of (25).

90
IR spectrum of (25):
(cm-1) 3350 (OH str); 1629 (C=O str); 2624 & 2536 (+N-H str); 1612
(C=C str); 1496 (aromatic C=C str); 697 (C-S str).
Mass spectrum of (25) (ES-MS):
m/z 586 corresponding to (29).
1H-NMR spectrum of (25):
(δ ppm) 0.4 (m, 4H, two CH2 groups of cyclopropyl ring); 1.3 (s, 6H,
CH3); 2.0-3.2 (m, 8H, all CH2 groups excluding CH2 groups of cyclopropyl
ring); 3.9 (t, 1H, CH-S); 5.2 (s, 1H, OH); 7.0-8.3 (m, 15H, Ar-H & vinyl
CH).
13C-NMR spectrum of (25):
(δ ppm) 12.2 (CH2 of cyclopropyl ring); 17.8 (C of cyclopropyl ring); 31
(CH3); 43 & 39 (CH2 attached to cyclorpropyl ring); 49 (C-S); 72 (t-C of t-
alcohol); 122-157 (Ar and vinyl CH); 174 (COO).
DEPT spectrum of (25):
Methyl and methyne groups as positive peaks and methylene groups
as negative peaks.

91
3.5.2 - Experimental Section For The Second Novel Process For The
Preparation Of Montelukast Sodium (25):
Preparation of (47):
10 grams (0.0218 mol) of (35) was added in 50 ml of toluene, and the
mixture was heated to reflux. Reaction mixture was concentrated by
simultaneous azeotropic removal of water. 90 ml of acetonitrile was
added after room temperature was attained by the resulting mass and
was stirred at 50 600C for 30-45 minutes. Resulting mass was further
cooled to -10 to -150C. and 5.33 ml of DIPEA was added and was stirred
for about 30 minutes. 9.3 ml of MsCl was added and seeded with
mesylate of (35) and reaction mass was aged at -10 to -150C. for about 8
9 hours. The reaction mass was filtered and washed with acetonitrile
followed by hexanes to provide 10.0 g of mesylate of (35).
3.23 g (0.0254 mol) of (46) was dissolved in 40 ml of DMF and the
mixture was cooled to -10 to -150C. 31.75 ml of 3.4 M n-Butyl lithium
was added drop wise in reaction mass. 8 g of above obtained mesylate of
(35) was added to the reaction mass at -10 to -150C and the reaction
mass was aged at -10 to -150C. for about 6 8 hours. 50 ml of 15%
sodium chloride solution was added followed by 80 ml of toluene and the
reaction mass was stirred for about 30 minutes. Organic layer and
aqueous layer were separated. Aqueoues layer was extracted with
toluene. Water was added to combined organic layers and the pH was

92
adjusted to 5.0 using 5 ml of acetic acid and the reaction mass was
stirred at 25 350C for 30-40 minutes. Organic layer was washed with 64
ml of 5% NaHCO3 solution followed by water. 1 gm of carbon and Na2SO4
were added to the organic layer and stirred for 30 minutes. It was filtered
and washed with toluene followed by removal of solvent under reduced
pressure below 500C to afford 8 g of (47).
Characterization of (47):
IR spectrum of (47):
(cm-1) 3418 (O-H str); 2247 (C=N str); 3062 (Ar C-H str); 2957
(aliphatic C-H); 1635 (C=C str).
Fig. 3.11
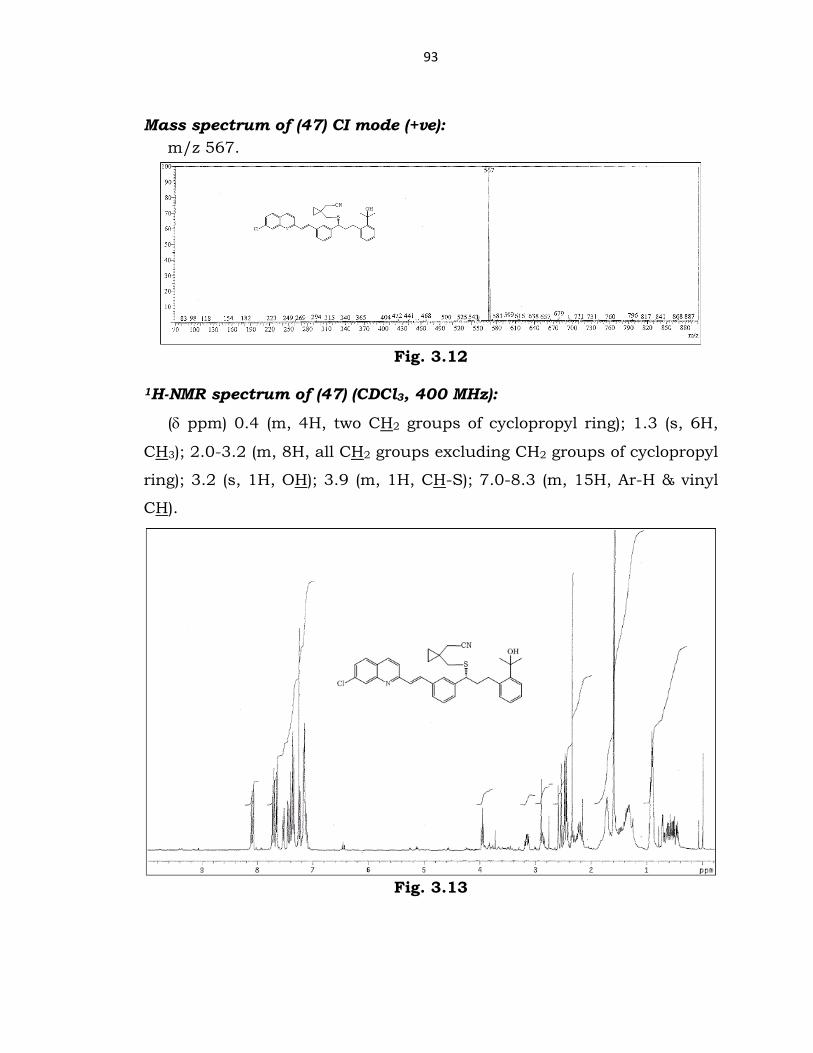
93
Mass spectrum of (47) CI mode (+ve): m/z 567.
Fig. 3.12
1H-NMR spectrum of (47) (CDCl3, 400 MHz):
(δ ppm) 0.4 (m, 4H, two CH2 groups of cyclopropyl ring); 1.3 (s, 6H,
CH3); 2.0-3.2 (m, 8H, all CH2 groups excluding CH2 groups of cyclopropyl
ring); 3.2 (s, 1H, OH); 3.9 (m, 1H, CH-S); 7.0-8.3 (m, 15H, Ar-H & vinyl
CH).
Fig. 3.13

94
Preparation of (44):
Method A:
65 g (0.114 mol) of (47) and 325 ml of caustic lye was added into
round bottom flask and further stirred and heated to reflux at 118-1220C
for 6 to 8 hours. 130 ml of water and 650 ml of toluene were added to the
reaction mass below 900C and stirred for 30 minutes. Separated the
layers and the aqueous layer was extracted with 325 ml of toluene at 60
- 700C. Combined organic layers were distilled under reduced pressure
below 500C and washed with 720 ml of n-heptane at 25-350C. 300 ml of
water and 200 ml of DCM were added to the reaction mass the pH was
adjusted to 5 with acetic acid. Layers were separated and the aqueous
layer was extracted with 200 ml of DCM. Combined organic layer was
washed with 1300 ml of water and distilled off solvent from organic layer
at atmospheric pressure followed by distillation under reduced pressure
below 500C to afford (29). 500 ml of acetone was added to the above
obtained crude and distilled of acetone under reduced pressure below
500C to remove the traces of DCM. 21 gm (0.287 mol) of TBA (45) was
added to the above reaction mass slowly at 25 - 300C and seeded. The
reaction mass was stirred till thick solid separation at 25-350C for 8 - 10
hours. The separated solid was filtered and washed with acetone. It was
then dried at 50 - 550C to afford 40 gm of (44).

95
Purification of (44):
30 gm of (44) was dissolved in 360 ml of acetone and heated to reflux
for 1 to 2 hours. Cooled to 250C and the reaction mixture was
maintained at the same temmperature for 10 hours. Solid was filtered,
washed with acetone and dried at 600C to afford 23.8 gm of purified (44).
Method B:
13.5 g (0.0238 mol) of (47), 94.5 ml of diethyleneglycol and a solution
of 10.7 g (0.19 mol) of KOH in 40 ml of water were added and refluxed for
24 hours. The reaction mass was cooled to room temperature and
washed with 325 ml of toluene. After addition of water (54 ml), the
product was extracted into ethyl acetate (472.5 ml). Organic layer was
washed with aqueous acetic acid and then with 50 ml of 5% of aqueous
NaHCO3 solution. The solvent was evaporated from the organic layer to
afford 7.5 g of (29). The obtained (29) was added into 45 ml of acetone. 2
ml of TBA (45) was added to reaction mass and stirred for about 10
hours. The separated solid was filtered and washed with acetone followed
by hexanes to get 4.3 g of (44).
(44) could be purified by recrystallization from solvents like ethyl
acetate, a mixture of IPA and acetonitrile or a mixture of MeOH and
acetonitrile as described above.

96
Method C:
The mixture of (47) and (50) was hydrolyzed by following the
procedure described above to afford (44).
Characterization of (44):
IR spectrum of (44):
(cm-1) 3360 (OH str); 1631 (C=O str); 2635 & 2554 (+N-H str); 1608
(C=C str); 1498 (Ar C=C str); 697 (C-S str).
Fig. 3.14
Mass spectrum of (44) (ESI):
m/z 586 correponding to (29).
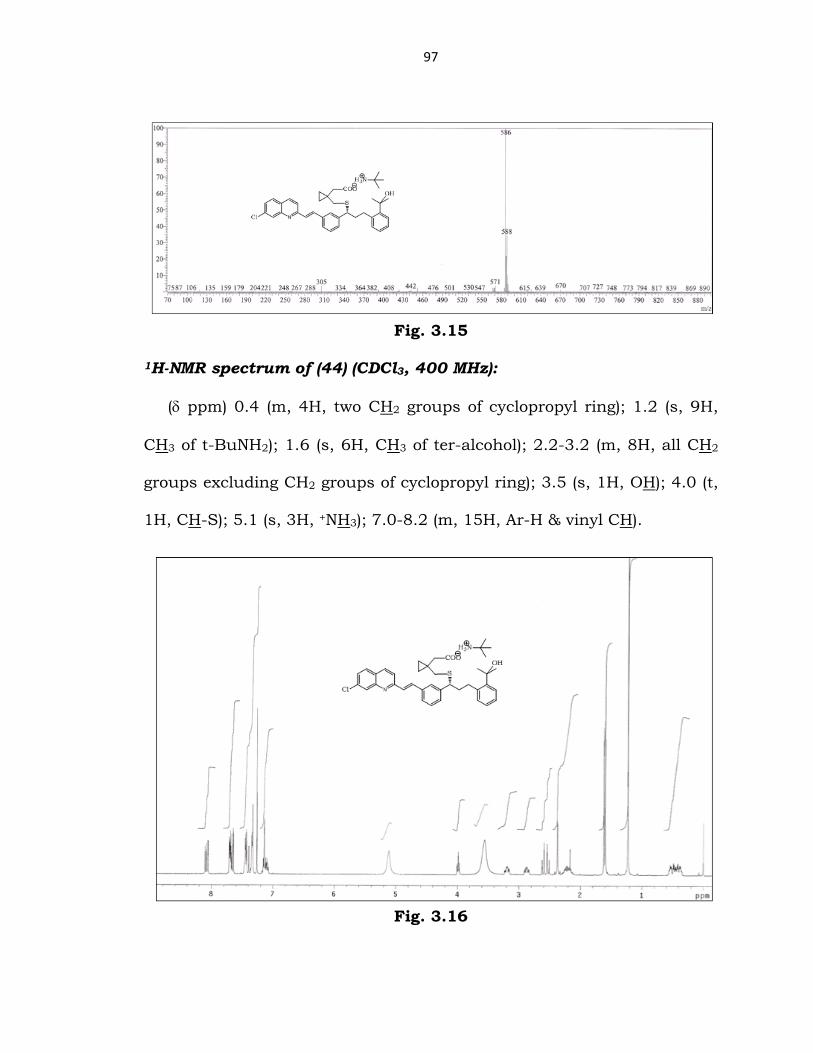
97
Fig. 3.15
1H-NMR spectrum of (44) (CDCl3, 400 MHz):
(δ ppm) 0.4 (m, 4H, two CH2 groups of cyclopropyl ring); 1.2 (s, 9H,
CH3 of t-BuNH2); 1.6 (s, 6H, CH3 of ter-alcohol); 2.2-3.2 (m, 8H, all CH2
groups excluding CH2 groups of cyclopropyl ring); 3.5 (s, 1H, OH); 4.0 (t,
1H, CH-S); 5.1 (s, 3H, +NH3); 7.0-8.2 (m, 15H, Ar-H & vinyl CH).
Fig. 3.16

98
Preparation of (25):
20.0 g (0.0303 mol) of (44) and DCM (50 ml) were added into round
bottom flask at 25 - 350C. Acetic acid (2.62 ml) and water (100 ml) were
added to the reaction mass and was stirred at 25 - 350C for 60 minutes.
Layers were separated and the aqueous layer was extracted with DCM
(40 ml). Organic layer was washed with water (4 X 25 ml); dried over
anhydrous sodium sulphate. Distilled off solvent completely from organic
layer under reduced pressure below 500C. Residual mass was dissolved
in MeOH (200 ml) and distilled off solvent completely under reduced
pressure below 500C. Residual mass was dissolved in 100 ml of MeOH.
Freshly prepared solution of NaOH (1.21 grams, 0.0303) pellets in MeOH
(100 ml) was added to the residual mass at 25 - 350C under nitrogen
blanketting and stirred for 30 minutes at 25-350C. Carbon (0.5 grams)
was added to reaction mass and stirred for 30 minutes at 25 - 350C.
Carbon was filtered and cake was washed with 25 ml of MeOH. Solvent
was distilled off completely under reduced pressure below 500C; the
obtained crude was dissolved in toluene (40 ml) and distilled off solvent
completely under reduced pressure below 500C. Finally crude was
dissolved in toluene (30 ml) and added to 200 ml of n-heptane under
nitrogen atmosphere at 25 - 350C. The reaction mass was maintained at
25 - 350C for 1 to 2 hours. The compound was filtered and washed with

99
n-heptane (40 ml) under nitrogen atmosphere and dried at 70 - 750C for
5 hours to afford 16.8 grams of (25) in amorphous form.
Characterization of (25):
IR spectrum of (25):
(cm-1) 3360 (OH str); 1631 (C=O str); 1608 (C=C str); 1498 (Ar C=C
str); 697 (C-S str).
Fig. 3.17
Mass spectrum of (25) (ESI):
m/z 608 (25) and m/z 586 (29).
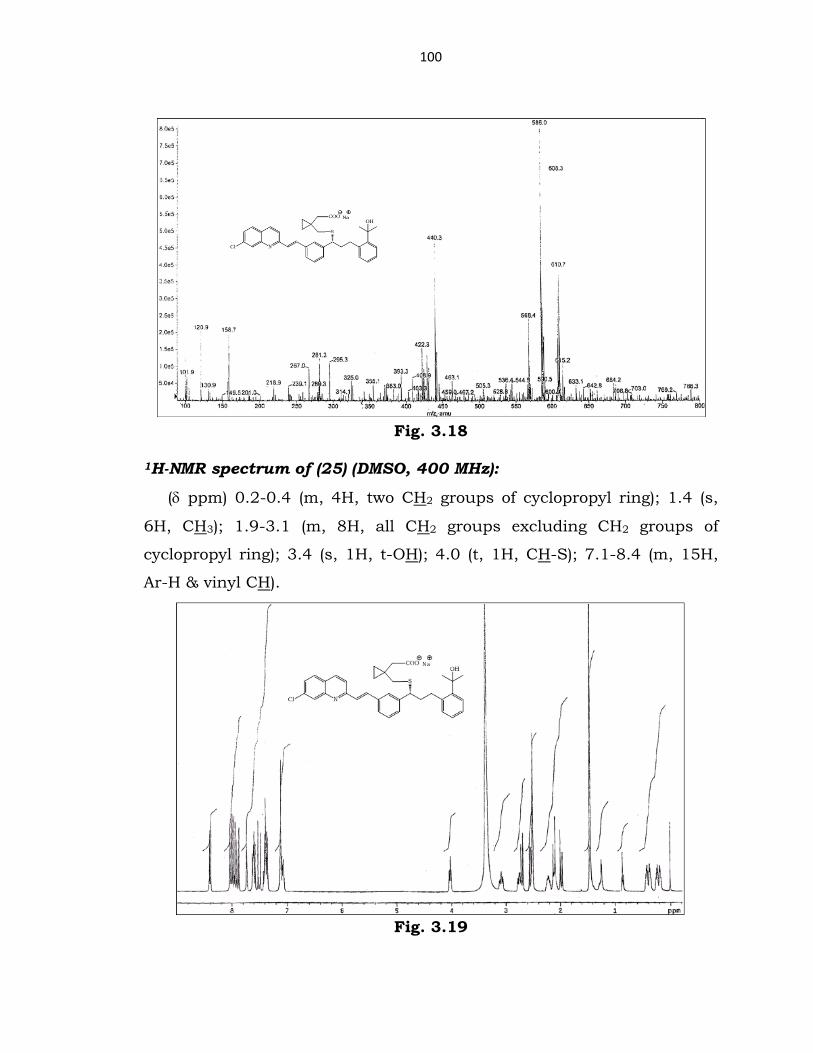
100
Fig. 3.18
1H-NMR spectrum of (25) (DMSO, 400 MHz):
(δ ppm) 0.2-0.4 (m, 4H, two CH2 groups of cyclopropyl ring); 1.4 (s,
6H, CH3); 1.9-3.1 (m, 8H, all CH2 groups excluding CH2 groups of
cyclopropyl ring); 3.4 (s, 1H, t-OH); 4.0 (t, 1H, CH-S); 7.1-8.4 (m, 15H,
Ar-H & vinyl CH).
Fig. 3.19

101
13C-NMR spectrum of (25) (DMSO-D6, 50 MHz):
(δ ppm) 175.9 (COO); 120-156 (Ar-C & vinyl CH); 71.6 (to t-C of t-
alcohol); 49.5 (C-S); 44 & 39.5 (CH2 attached to cyclorpropyl ring); 31.7
(CH3); 18.1 (C of cyclopropyl ring); 12.4 & 12.1 (CH2 of cyclopropyl ring).
Fig. 3.20
DEPT spectrum of (25) (DMSO-D6):
Methyl and methyne groups as positive peaks and methylene groups
as negative peaks.
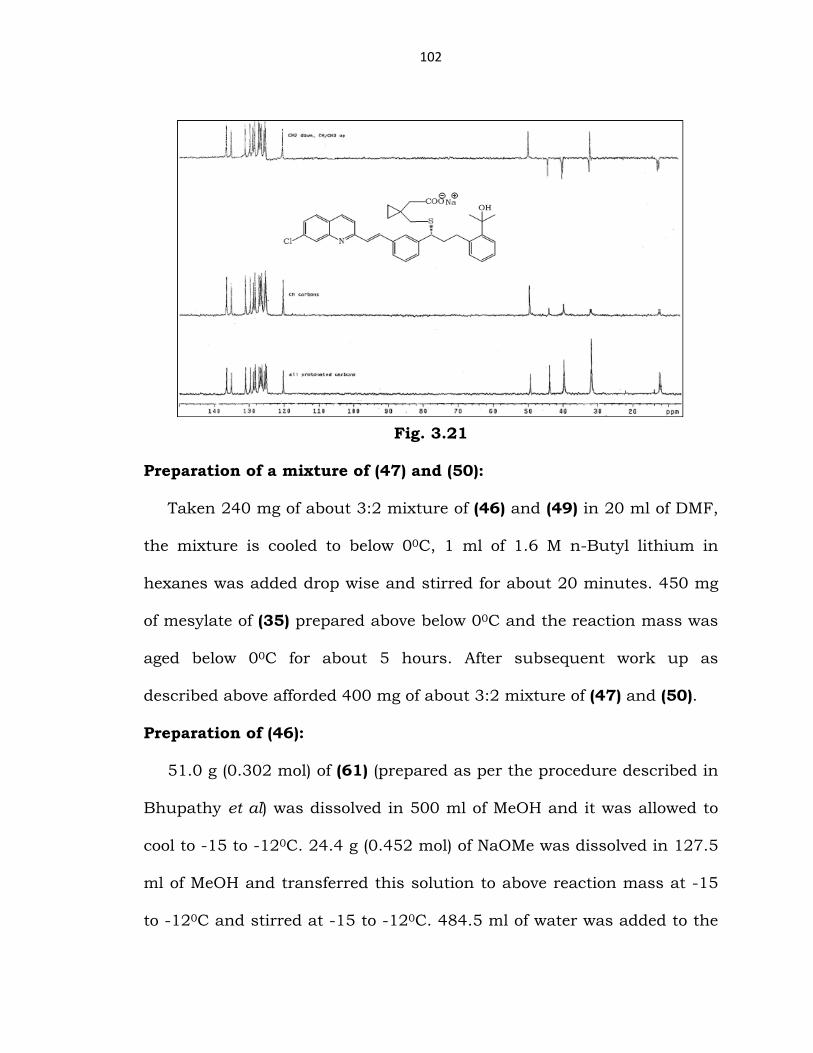
102
Fig. 3.21
Preparation of a mixture of (47) and (50):
Taken 240 mg of about 3:2 mixture of (46) and (49) in 20 ml of DMF,
the mixture is cooled to below 00C, 1 ml of 1.6 M n-Butyl lithium in
hexanes was added drop wise and stirred for about 20 minutes. 450 mg
of mesylate of (35) prepared above below 00C and the reaction mass was
aged below 00C for about 5 hours. After subsequent work up as
described above afforded 400 mg of about 3:2 mixture of (47) and (50).
Preparation of (46):
51.0 g (0.302 mol) of (61) (prepared as per the procedure described in
Bhupathy et al) was dissolved in 500 ml of MeOH and it was allowed to
cool to -15 to -120C. 24.4 g (0.452 mol) of NaOMe was dissolved in 127.5
ml of MeOH and transferred this solution to above reaction mass at -15
to -120C and stirred at -15 to -120C. 484.5 ml of water was added to the

103
reaction mass under stirring below 00C and the resulting aqueous mass
was washed with 1020 ml of heptane. Aqueous layer was acidified with
65.3 ml of acetic acid and stirred the reaction mixture below 00C for 30
minutes. Layers were separated and the aqueous layer was extracted
with 204 ml of DCM. Organic layer was washed with 102 ml of 5%
sodium bicarbonate followed by 459 ml of water. 5.1 gm of carbon and
sodium sulphate were added to combine organic layer and stirred for 30
minutes. Reaction mass was filtered over hyflow bed and washed with 51
ml of DCM followed by removal of solvent completely from organic layer
under reduced pressure below 500C afforded 27.0 g of (46).
Characterization of (46)
IR spectrum of (46):
(cm-1) 2923 (aliphatic C-H str); 2566 (S-H str) and 2248 (C N str).
Fig. 3.22

104
Mass spectrum of (46):
m/z 127 by GC-MS.
1H-NMR spectrum of (46) (CDCl3, 200 MHz):
(δ ppm) 0.6-0.7 (m, 4H, two CH2 groups of cyclopropyl ring); 1.4 (t,
1H, SH); 2.6-2.9 (m, 4H, CH2).
Fig. 3.23
Preparation of a mixture of (46) and (49):
2.5 g (0.0147 mol) of (61) (prepared as per the procedure described in
Bhupathy et al) was dissolved in 25 ml of MeOH and stirred at ambient
temperature. 2.5 g of KOH was dissolved in 10.0 ml of water and
transferred this solution to above reaction mass. The reaction mass was
then aged below 500C temperature until reaction was substantially

105
complete. Then 40 ml of water was added to reaction mass and washed
with 120 ml of hexanes. Aqueous phase was extracted with 160 ml of
ethyl acetate. Organic layer was then washed with aqueous acetic acid
followed by 5% sodium bicarbonate solution and then with water.
Evaporated the solvent from the organic layer to afford 600 mg of about
3:2 mixture of (46) and (49).
3.5.3 - Experimental Section For The Starting material (10):
Preparation of (60):
100 grams (0.219 mol) of (39) was dissolved in 500 ml of DCM. 180
ml of (-)-DIP-Cl (0.346 mol, 1.6 equivalents) was taken separately in 500
ml of DCM and cooled to -50C with stirring. Above (39) solution in DCM
was added to (-)-DIP-Cl in DCM at -5 to 00C slowly drop wise. After
addition, the reaction mixture was aged at -5 to 00C for 10 hours.
Reaction was monitored by TLC (mobile phase: Ethyl acetate : Hexanes –
1:4 and one drop of aq. ammonia). After the completion of reaction, the
reaction mixture was quenched with aqueous ammonia and stirred 60
minutes. Aqueous sodium chloride solution was added and stirred for 30
minutes. Layers were separated and the organic layer was washed with
aqueous sodium chloride solution. DCM was distilled from the organic
layer. The resulting crude was dissolved in 1200 ml of MeOH and filtered
to remove insoluble solids. To the filtrate 50 ml water was added slowly
drop wise and stirring was continued for 2 hours. The separated solid
106
was filtered, washed with a mixture of MeOH and water and dried at
500C to yield 80 grams (yield: 80%) of (60). (Purity by HPLC: 97%). SR:
of –32.90 (c=1 in chloroform).
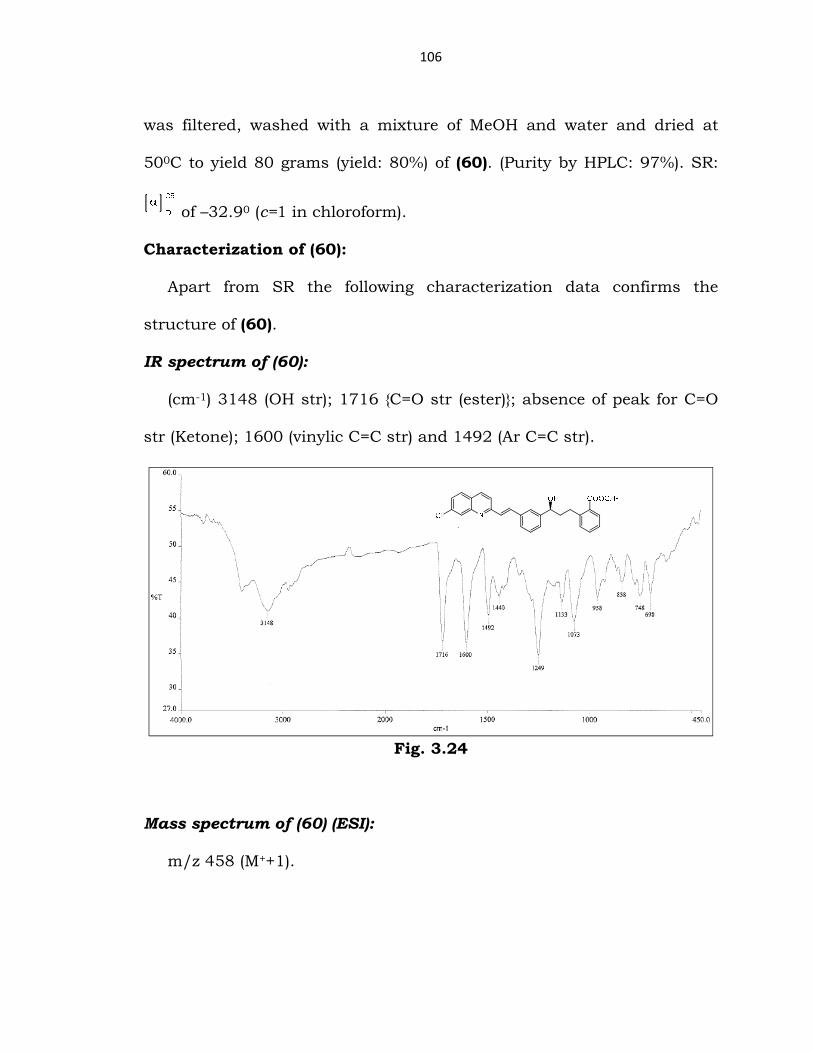
Characterization of (60):
Apart from SR the following characterization data confirms the
structure of (60).
IR spectrum of (60):
(cm-1) 3148 (OH str); 1716 {C=O str (ester)}; absence of peak for C=O
str (Ketone); 1600 (vinylic C=C str) and 1492 (Ar C=C str).
Fig. 3.24
Mass spectrum of (60) (ESI):
m/z 458 (M++1).
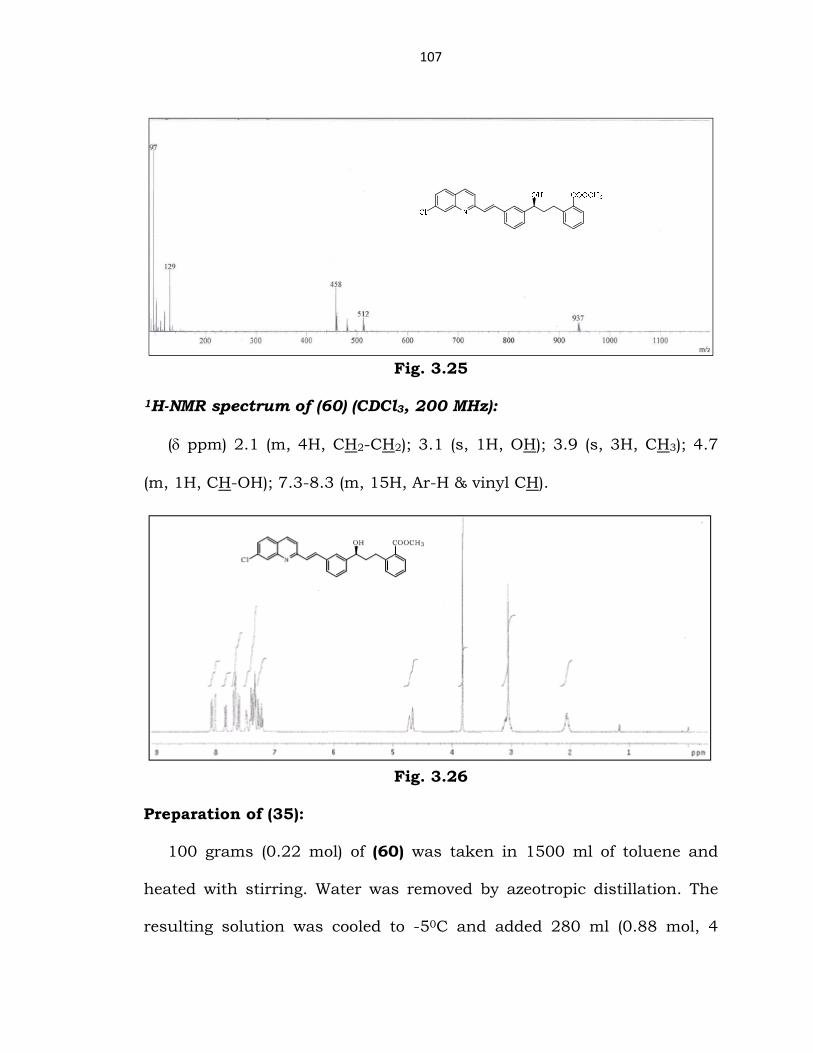
107
Fig. 3.25
1H-NMR spectrum of (60) (CDCl3, 200 MHz):
(δ ppm) 2.1 (m, 4H, CH2-CH2); 3.1 (s, 1H, OH); 3.9 (s, 3H, CH3); 4.7
(m, 1H, CH-OH); 7.3-8.3 (m, 15H, Ar-H & vinyl CH).
Fig. 3.26
Preparation of (35):
100 grams (0.22 mol) of (60) was taken in 1500 ml of toluene and
heated with stirring. Water was removed by azeotropic distillation. The
resulting solution was cooled to -50C and added 280 ml (0.88 mol, 4

108
equivalents) of methyl magnesium chloride in THF slowly at a
temperature of -5 to 00C. After addition was complete, the reaction
mixture was aged at -5 to 00C for 4 hours. Reaction was monitored by
TLC (mobile phase: Ethyl acetate : Hexanes – 1:4 and one drop of aq.
ammonia). After the completion of reaction, the reaction mixture was
quenched with aq. acetic acid and stirred one hour. Layers were
separated and the organic layer was washed with aq. sodium bicarbonate
solution followed by water. Water was removed from the organic phase by
azeotropic distillation. Concentrated the organic layer and cooled the
resulting solution to room temperature and stirred for 4 hours.
Separated solid was filtered and recrystallized from toluene to yield 70
grams (yield: 80%) of (35). (Purity by HPLC: 98%). SR: of –22.60 (c=1
in chloroform).
Characterization of (35):
IR spectrum of (35):
(cm-1) 3307 (OH str); absence of peak for C=O str (ester); 1607 (vinylic
C=C str); 1595 (OH bend); and 1492 (Ar C=C str).

109
Fig. 3.27
Mass spectrum of (35) (ESI):
m/z 458.
Fig. 3.28

110
1H-NMR spectrum of (35) (DMSO-D6, 200 MHz):
(δ ppm) 1.4 (s, 6H, CH3 of t-alcohol); 2.0 (m, 2H, CH2-CH2); 3.0 (m,
2H, CH2-CH2); 4.7 (m, 1H, CH-OH); 4.9 (s, 1H, OH of t-OH); 5.3 (s, 1H,
OH of CH-OH); 7.1-8.4 (m, 15H, Ar-H & vinyl CH).
Fig. 3.29
3.5.4 - EXPERIMENTAL SECTION FOR THE PREPARATION OF
IMPURITIES:
The impurities of (25) and its intermediates as given above in Tables-
3.3, 3.4 and 3.5 were prepared as described here under.
Preparation of impurities according to Table-3.3:
(40) & (43) are intermediates in the first novel process for the
preparation of (25). Their characterization data is in agreement with the
data for the same discussed herein above.
111
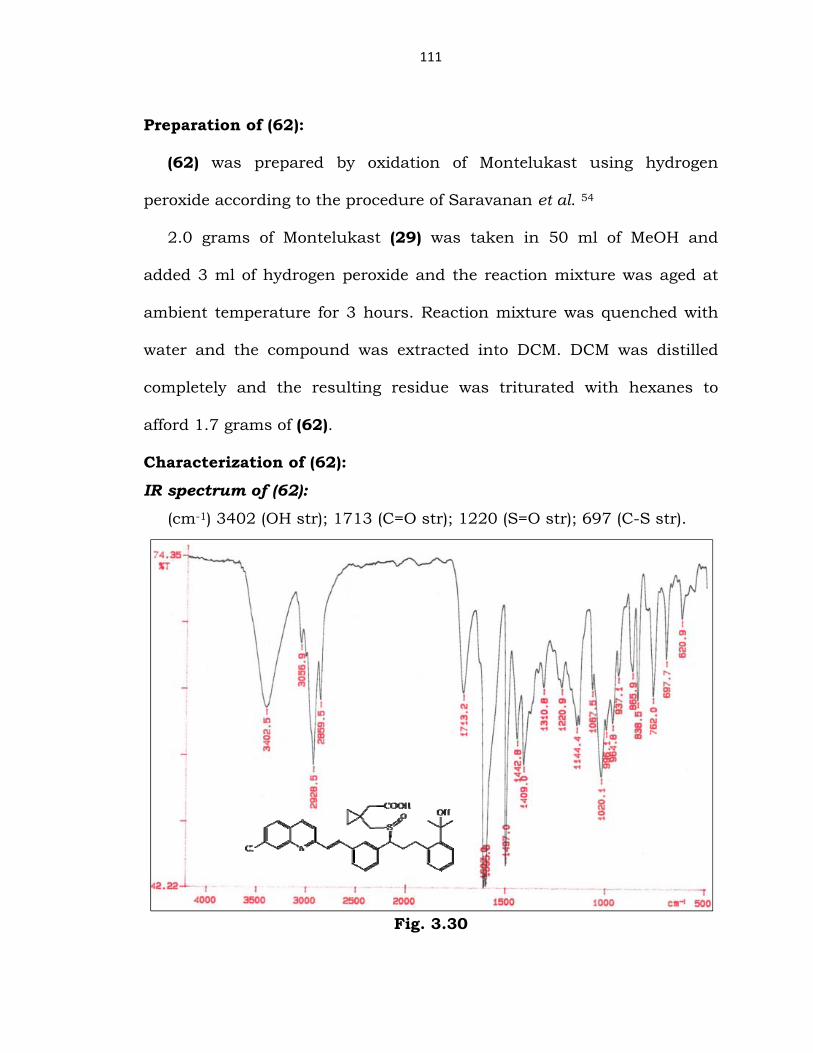
Preparation of (62):
(62) was prepared by oxidation of Montelukast using hydrogen
peroxide according to the procedure of Saravanan et al. 54
2.0 grams of Montelukast (29) was taken in 50 ml of MeOH and
added 3 ml of hydrogen peroxide and the reaction mixture was aged at
ambient temperature for 3 hours. Reaction mixture was quenched with
water and the compound was extracted into DCM. DCM was distilled
completely and the resulting residue was triturated with hexanes to
afford 1.7 grams of (62).
Characterization of (62):
IR spectrum of (62):
(cm-1) 3402 (OH str); 1713 (C=O str); 1220 (S=O str); 697 (C-S str).
Fig. 3.30

112
Mass spectrum of (62) (ES-MS):
m/z 602.
Fig. 3.31
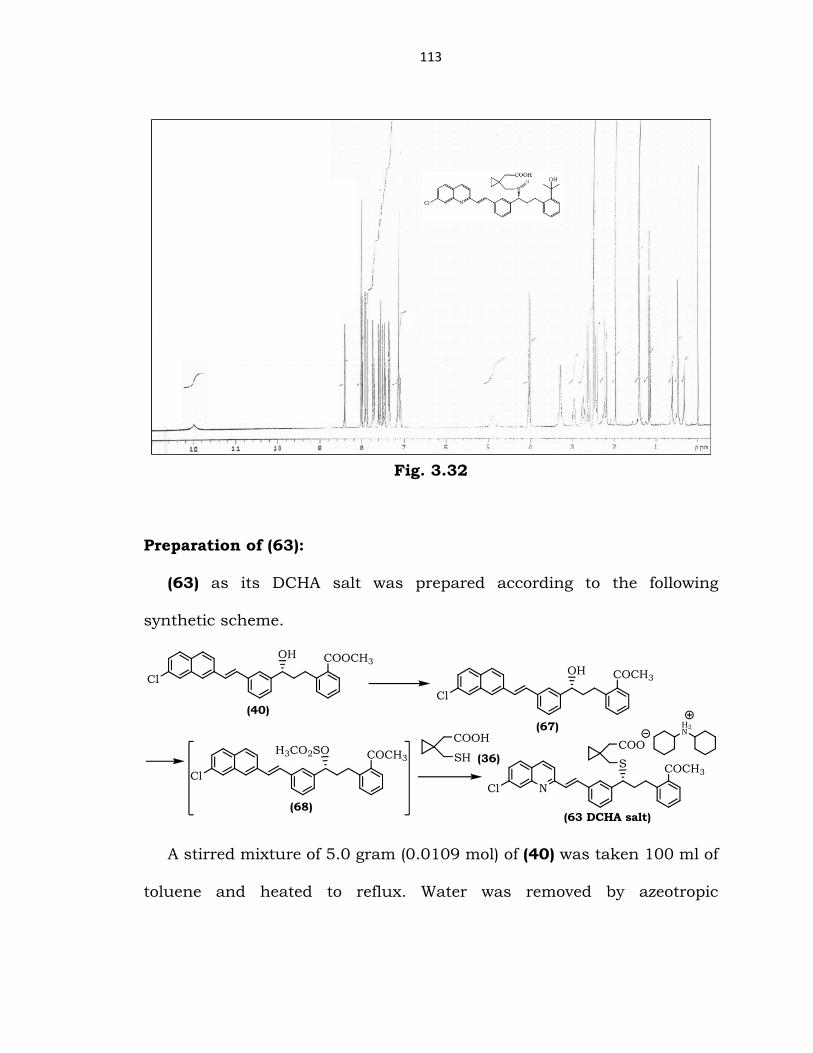
1H-NMR spectrum of (62) (DMSO-D6, 200 MHz):
(δ ppm) 0.3-0.6 (m, 4H, two CH2 groups of cyclopropyl ring); 12
(COOH); 1.4 (s, 6H, CH3); 2.0-2.9 (m, 8H, all CH2 groups excluding CH2
groups of cyclopropyl ring); 4.0 (t, 1H, CH-S); 4.9 (s, 1H, OH of t-alcohol);
7.1-8.4 (m, 15H, Ar-H & vinyl CH).
113
Fig. 3.32
Preparation of (63):
(63) as its DCHA salt was prepared according to the following
synthetic scheme.
Cl
OH COOCH3
(40)Cl
OH COCH3
(67)
Cl
H3CO2SO COCH3
(68)
COOH
SH (36)
NCl
COCH3
COOS
(63 DCHA salt)
H2N
A stirred mixture of 5.0 gram (0.0109 mol) of (40) was taken 100 ml of
toluene and heated to reflux. Water was removed by azeotropic

114
distillation and resulting mixture was cooled to -50C. 3.2 gram (0.0329, 3
molar equivalents) was added and stirred for 30 minutes. 3.8 ml of 1 M
ethyl magnesium bromide in THF (0.0285 mol, 2.6 molar equivalents)
was added drop wise. The reaction mixture was aged at -50C for 3 hours.
Reaction mixture was quenched with saturated aqueous ammonium
chloride solution and stirred for 30 minutes. Layers were separated.
Organic layer was washed with water and dried over anhyd.Na2SO4.
Toluene was distilled off completely and the resulting residue was
dissolved in 50 ml of THF and cooled to -50C. Added 36 ml of 1.5 M
methyl magnesium bromide in THF drop wise. Temperature was raised to
250C and was aged at that temperature for 3 hours. Reaction mixture
was quenched with aqueous NH4Cl solution and stirred at ambient
temperature for 30 minutes. Layers were separated and the aqueous
layer was extracted with toluene and the combined organic phases was
washed with water and dried over anhydrous Na2SO4. Solvents were
distilled completely to afford 4.5 grams of (67).
Above obtained (67) was mesylated using the procedure of (41) to
afford the corresponding mesylated (68), which was then converted to
(63) DCHA salt using the procedure of (43) given above in the
experimental section 3.5.1 except for using sodium methoxide powder
(1.2 molar equivalents to 41) in place of n-butyl lithium. The resulting

115
(63) in its free acid form was converted to DCHA salt by treating with
DCHA (42) in acetone to afford 1 gram of DCHA salt of (63).
Characterization of (63):
IR spectrum of (63):
(cm-1) 3421 (N-H str); 3057 (Ar C-H str); 2924 (aliphatic C-H str); 1706
(C=O str); 1608 (-COO- asymm. Str); 1560 (C=C str); 1498 (Ar C=C str);
697 (C-S str).
Fig. 3.33
Mass spectrum of (63) (ES-MS):
m/z 570 (63 acid).

116
Fig. 3.34
1H-NMR spectrum of (63) (CDCl3, 400 MHz):
(δ ppm) 0.3 (m, 4H, two CH2 groups of cyclopropyl ring); 1.3-3.0 (m,
33H, all CH2 groups excluding CH2 groups of cyclopropyl ring & CH
groups of dicyclohexyl and CH3); 3.9 (t, 1H, CH-S); 7.2-8.1 (m, 15H, Ar-H
& vinyl CH); 8.2 (m, 2H, +NH2).
Fig. 3.35

117
Preparation of (64):
5.0 grams of Montelukast acid was dissolved in 200 ml of chloroform
and added 0.8 ml of conc.H2SO4. Reaction mixture was aged at 500C for
6 hours. Reaction mixture was cooled to room temperature and
quenched with ice cooled water. Layers were separated and the organic
layer was washed with water followed by aqueous sodium bi-carbonate
solution. Chloroform was distilled completely to afford 4.6 grams of (64).
Characterization of (64):
IR spectrum of (64):
(cm-1) 3429 (OH str); 1713 (C=O str); 1608 (C=C str); 1499 (Ar C=C
str); 697 (C-S str).
Fig. 3.36
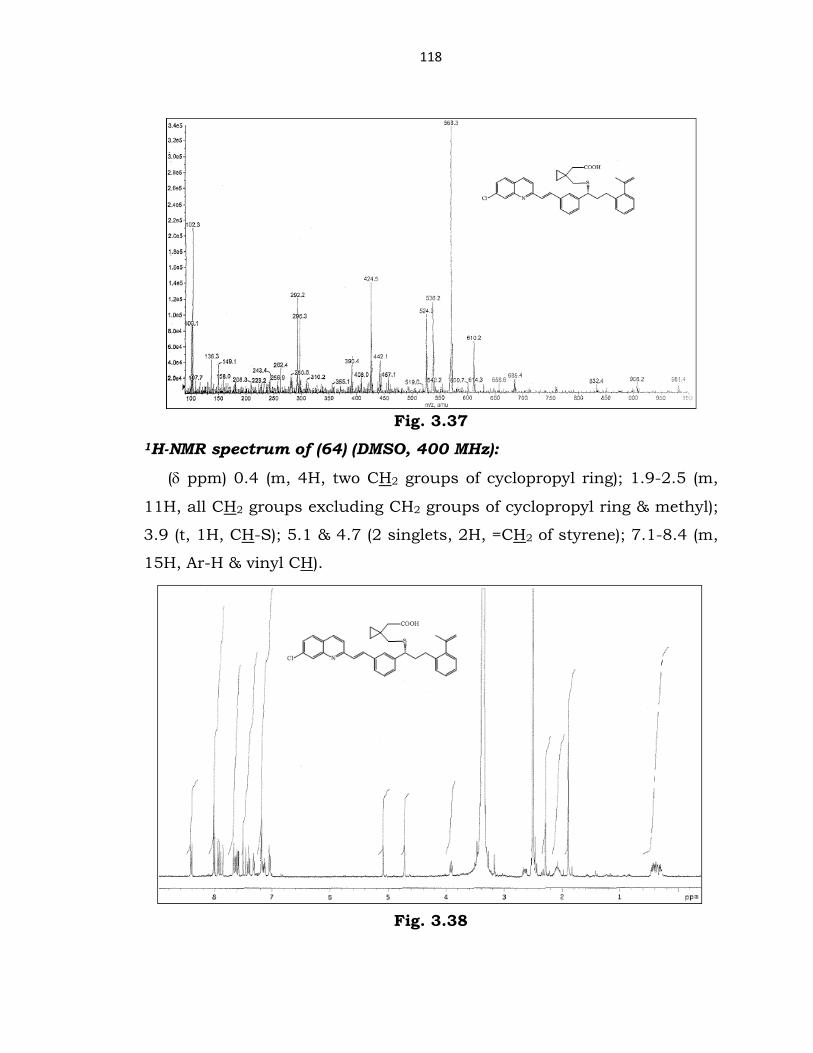
Mass spectrum of (64) (ESI):
m/z 568.
118
Fig. 3.37
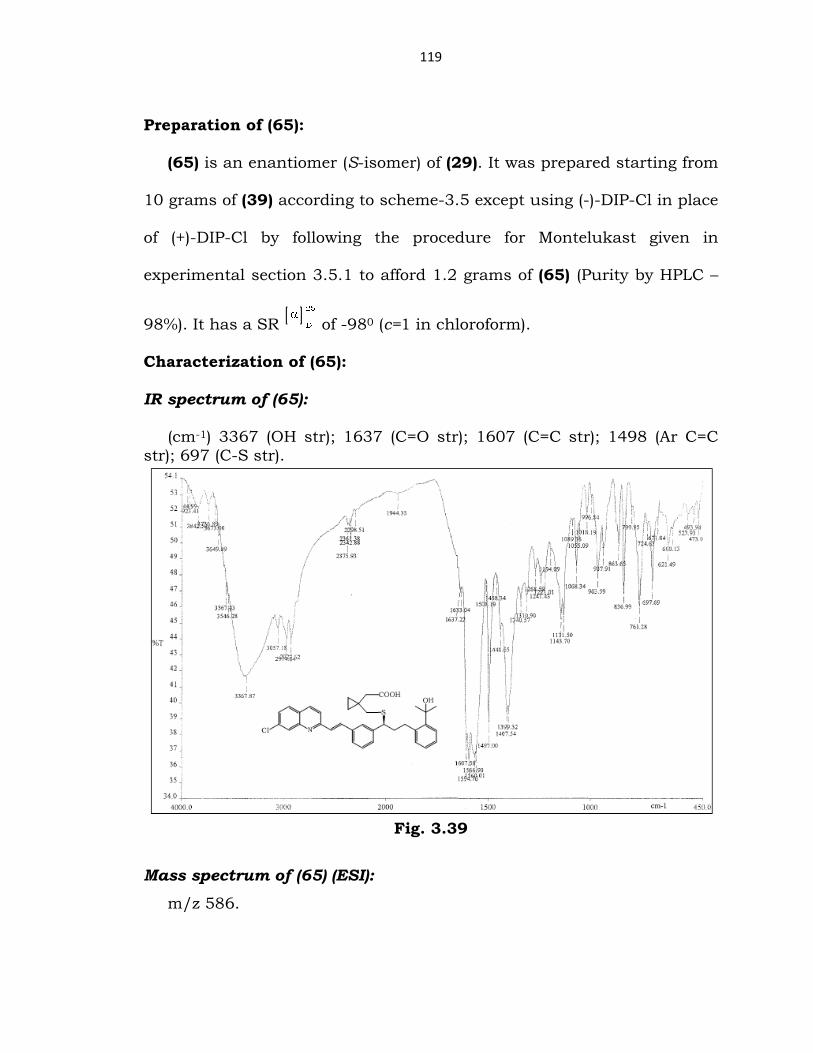
1H-NMR spectrum of (64) (DMSO, 400 MHz):
(δ ppm) 0.4 (m, 4H, two CH2 groups of cyclopropyl ring); 1.9-2.5 (m,
11H, all CH2 groups excluding CH2 groups of cyclopropyl ring & methyl);
3.9 (t, 1H, CH-S); 5.1 & 4.7 (2 singlets, 2H, =CH2 of styrene); 7.1-8.4 (m,
15H, Ar-H & vinyl CH).
Fig. 3.38
119
Preparation of (65):
(65) is an enantiomer (S-isomer) of (29). It was prepared starting from
10 grams of (39) according to scheme-3.5 except using (-)-DIP-Cl in place
of (+)-DIP-Cl by following the procedure for Montelukast given in
experimental section 3.5.1 to afford 1.2 grams of (65) (Purity by HPLC –
98%). It has a SR of -980 (c=1 in chloroform).
Characterization of (65):
IR spectrum of (65):
(cm-1) 3367 (OH str); 1637 (C=O str); 1607 (C=C str); 1498 (Ar C=C str); 697 (C-S str).
Fig. 3.39
Mass spectrum of (65) (ESI): m/z 586.
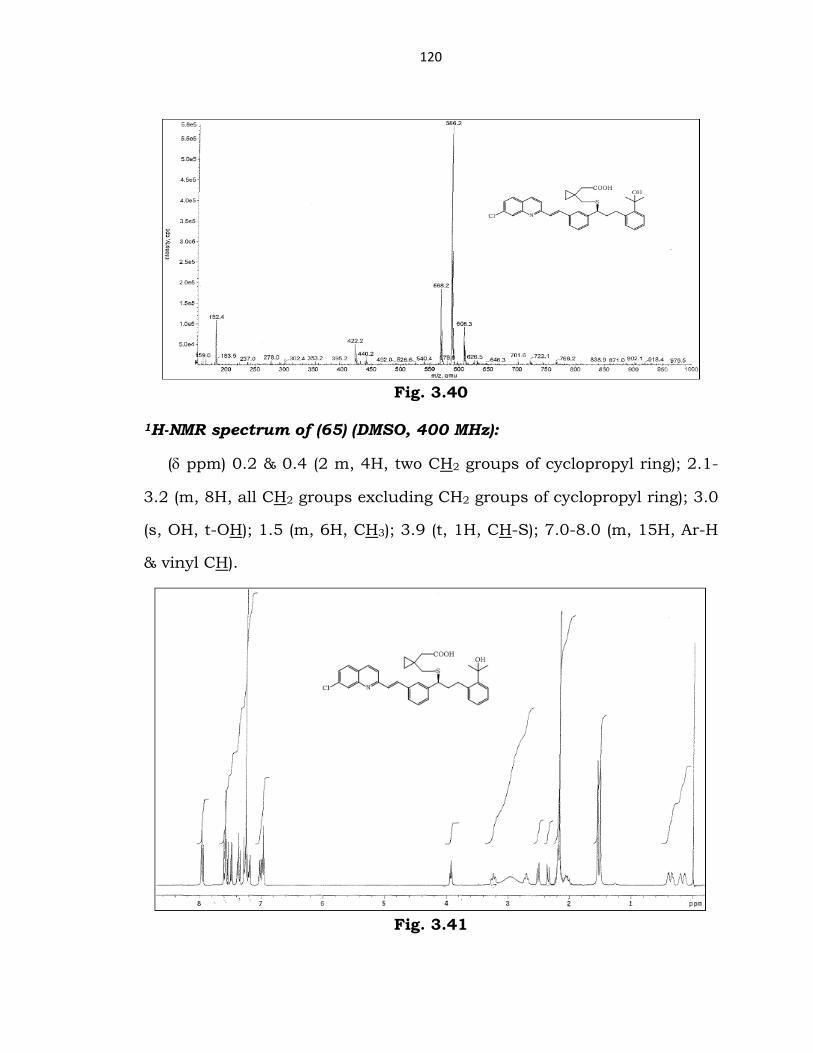
120
Fig. 3.40
1H-NMR spectrum of (65) (DMSO, 400 MHz):
(δ ppm) 0.2 & 0.4 (2 m, 4H, two CH2 groups of cyclopropyl ring); 2.1-
3.2 (m, 8H, all CH2 groups excluding CH2 groups of cyclopropyl ring); 3.0
(s, OH, t-OH); 1.5 (m, 6H, CH3); 3.9 (t, 1H, CH-S); 7.0-8.0 (m, 15H, Ar-H
& vinyl CH).
Fig. 3.41

121
Preparation of impurities according to Table-3.4:
(35) was an intermediate of the second novel process for the
preparation of Montelukast sodium its characterization data was in
agreement with the data given for this compound herein above.
Process for preparation of (63), (64), (66) and (67) are described
above under the heading preparation of impurities according to Table-
3.3.
Preparation of impurities according to Table-3.4:
(66) was the dimer impurity that was formed during preparation of
(53) when a mole of 7-chloro-2-methyl quinoline reacts with (53). It was
isolated by filtering the hot suspension obtained during purification of
(53) from ethyl acetate as described in the experimental section 3.5.3 for
the preparation of intermediates of (25). The filtered solid is (66).
Characterization of (66):
IR spectrum of (66):
(cm-1) 2253 (C=N str); 1590 (vinylic C=C str); 1492 (Ar C=C str).
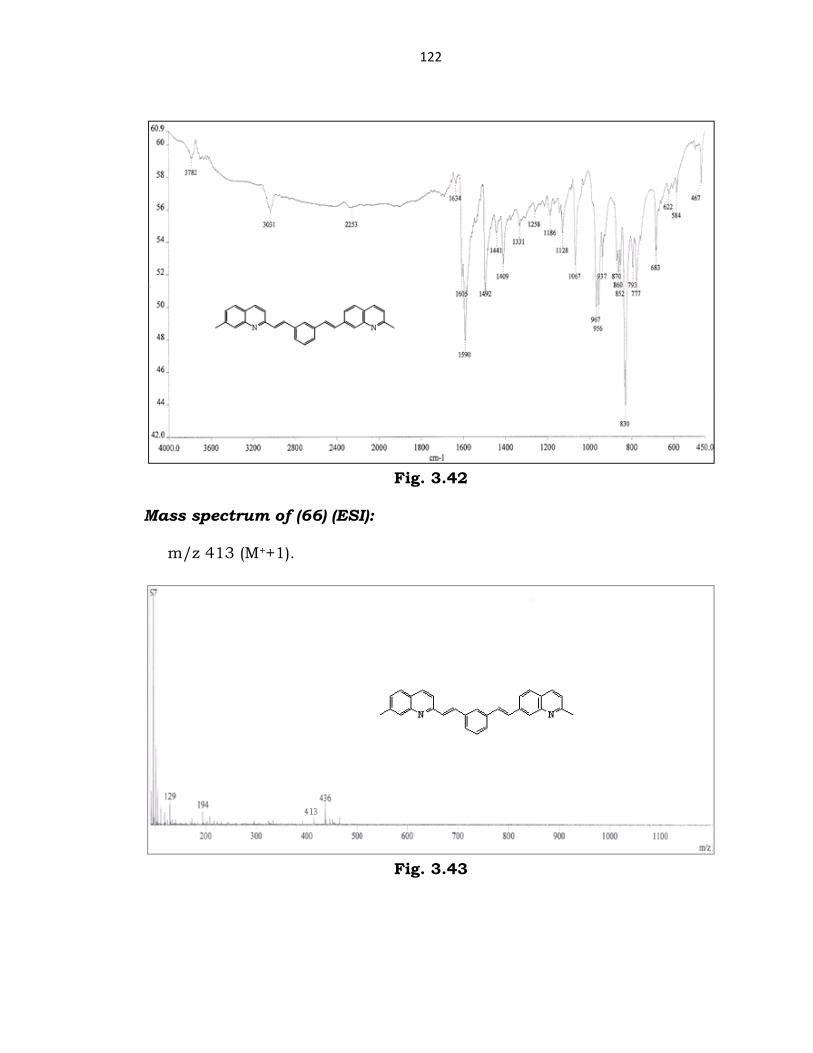
122
Fig. 3.42
Mass spectrum of (66) (ESI):
m/z 413 (M++1).
Fig. 3.43

123
3.6 - STUDY ON POLYMORPHISM OF MONTELUKAST AND ITS
SALTS
X-ray diffractometry is the most widely used analytical tool for the
determination of polymorphic forms of solids. Therefore, as part of the
present work, the polymorphic forms were determined using powder X-
ray diffraction pattern.
Definition of amorphous solid
Amorphous solids are the solids wherein the molecules are arranged
randomly in a three dimensional space. It is a non crystalline solid with
no well defined ordered structure. That means it lacks long range order
as a crystalline solid.
Hence, there would not be seen any sharp well defined peaks in a
typical X-ray diffraction pattern of an amorphous solid.
Definition of crystalline solid
Crystalline solids are the solids wherein the molecules are arranged in
a regular fashion in a three dimensional space. It will have long range
order. It will have well defined ordered three dimensional structures.
There would be seen sharp well defined peaks in a typical X-ray
diffraction pattern of a crystalline solid.

124
Study on polymorphic nature of Montelukast sodium (25) of the
present work:
(25) obtained in the process of the present work was confirmed to be
amorphous in nature as observed by its powder X-ray diffraction pattern
(Fig. 3.44), which did not have any well defined peaks. It was observed
as a hollow pattern without any sharp peaks.
Fig. 3.44
Study on polymorphic form of (44) of the present work:
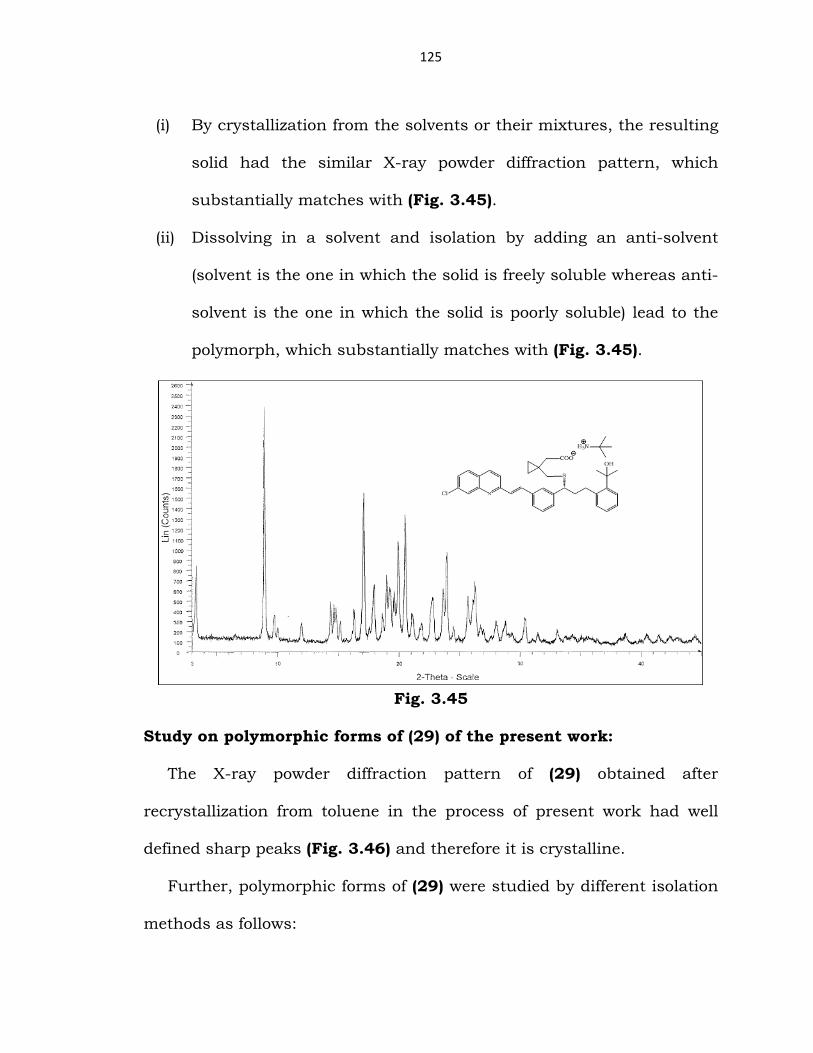
(44) obtained in the process of the present work was confirmed to be
crystalline in nature as observed by its powder X-ray diffraction pattern
(Fig. 3.45).
Further, polymorphic form of (44) was studied by different isolation
methods as follows:
125
(i) By crystallization from the solvents or their mixtures, the resulting
solid had the similar X-ray powder diffraction pattern, which
substantially matches with (Fig. 3.45).
(ii) Dissolving in a solvent and isolation by adding an anti-solvent
(solvent is the one in which the solid is freely soluble whereas anti-
solvent is the one in which the solid is poorly soluble) lead to the
polymorph, which substantially matches with (Fig. 3.45).
Fig. 3.45
Study on polymorphic forms of (29) of the present work:
The X-ray powder diffraction pattern of (29) obtained after
recrystallization from toluene in the process of present work had well
defined sharp peaks (Fig. 3.46) and therefore it is crystalline.
Further, polymorphic forms of (29) were studied by different isolation
methods as follows:

126
(i) After crystallization from most of the solvents or their mixtures, the
resulting solid had the similar X-ray powder diffraction pattern in
all cases, which substantially matched with (Fig. 3.46). Following
procedure was followed.
Taken 2 grams of (29) in 50 ml of MeOH and heated to dissolve.
After complete dissolution the solution was filtered at hot condition
and the filtrate was cooled to ambient temperature and aged for 10
hours. Separated solid was filtered and dried at 700C to afford (29)
whose X-ray powder diffraction pattern substantially matched with
(Fig. 3.46). Similarly, crystallization from the following solvents
was carried out and the X-ray powder diffraction pattern of the
resulting (29) substantially matched with (Fig. 3.46).
Table-3.5
Run Solvent XRPD
1. 2-Propanol (50 ml) Fig. 3.46
2. 1-Propanol (50 ml) Fig. 3.46
3. 1-Butanol (50 ml) Fig. 3.46
4. Ethyl acetate (50 ml) Fig. 3.46
5. Acetone (30 ml) Fig. 3.46
6. Acetonitrile (30 ml) Fig. 3.46
7. THF (20 ml) Fig. 3.46
127
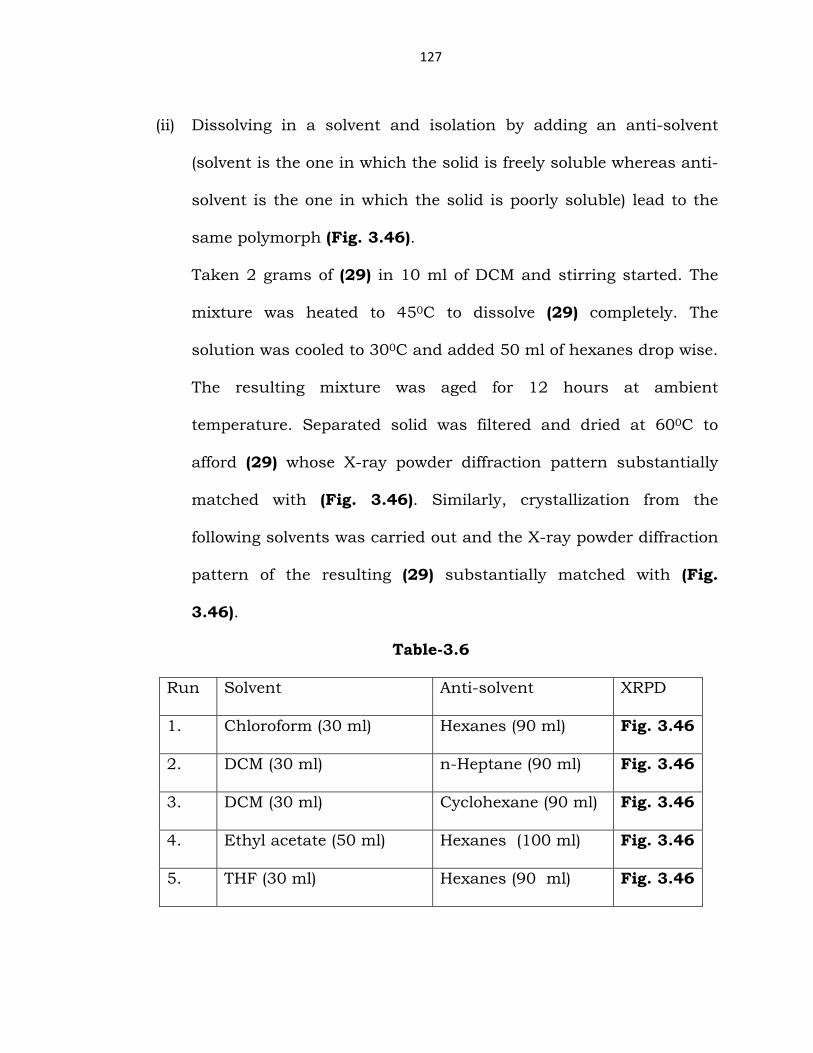
(ii) Dissolving in a solvent and isolation by adding an anti-solvent
(solvent is the one in which the solid is freely soluble whereas anti-
solvent is the one in which the solid is poorly soluble) lead to the
same polymorph (Fig. 3.46).
Taken 2 grams of (29) in 10 ml of DCM and stirring started. The
mixture was heated to 450C to dissolve (29) completely. The
solution was cooled to 300C and added 50 ml of hexanes drop wise.
The resulting mixture was aged for 12 hours at ambient
temperature. Separated solid was filtered and dried at 600C to
afford (29) whose X-ray powder diffraction pattern substantially
matched with (Fig. 3.46). Similarly, crystallization from the
following solvents was carried out and the X-ray powder diffraction
pattern of the resulting (29) substantially matched with (Fig.
3.46).
Table-3.6
Run Solvent Anti-solvent XRPD
1. Chloroform (30 ml) Hexanes (90 ml) Fig. 3.46
2. DCM (30 ml) n-Heptane (90 ml) Fig. 3.46
3. DCM (30 ml) Cyclohexane (90 ml) Fig. 3.46
4. Ethyl acetate (50 ml) Hexanes (100 ml) Fig. 3.46
5. THF (30 ml) Hexanes (90 ml) Fig. 3.46

128
(iii) Distilling off solvent completely under reduced pressure from a
solution of (29) in water miscible solvents such as alcohols,
acetonitrile and acetone lead the same polymorph (Fig. 3.46).
Taken 2 grams of (29) in 100 ml of MeOH and heated to reflux.
After complete dissolution, the solution was filtered at hot
condition and solvent was distilled off completely from the filtrate
under reduced pressure. The resulting solid was dried at 700C to
afford (29) whose X-ray powder diffraction pattern substantially
matched with (Fig. 3.46). Similar procedure was repeated with the
following solvents and the X-ray powder diffraction pattern of the
resulting (29) substantially matched with (Fig. 3.46).
Table-3.7
Run Solvent XRPD
1. 2-Propanol (100 ml) Fig. 3.46
2. 1-Propanol (100 ml) Fig. 3.46
3. Toluene (100 ml) Fig. 3.46
4. DCM (100 ml) Fig. 3.46
5. Chloroform (100 ml) Fig. 3.46
6. Ethyl acetate (100 ml) Fig. 3.46
7. THF (100 ml) Fig. 3.46

129
Fig. 3.46
3.7 – CONCLUSION
The processes described herein for the preparation of Montelukast
sodium (25) towards the objective of the present work avoided
disadvantages of the reported processes viz. series of protection-de-
protection steps resulting in a lengthy synthetic scheme rendering the
process expensive and hence leading to a commercially unviable process.
Reported processes involved undesirable hazardous and costly raw
materials such as ter-butyl dimethyl silane, dihydropyran, hydrazine,
pyridinium p-toluenesulfonate, cesium carbonate; and undesirable

130
reaction conditions such as low temperatures of -250C. The reported
processes involved tedious workups resulting in longer time cycle
rendering the process expensive and less eco friendly. Thus the
processes were not amenable for commercial scale up.
The objective of the present work was achieved by providing cost
effective, eco-friendly process, which was well suited for commercial scale
up. Montelukast sodium obtained in the present novel process had
>99.0% enantiomeric excess purity as determined by chiral HPLC and
resulted in amorphous form as characterized by X-ray powder diffraction.
Montelukast sodium obtained in the present process is free flowing and
non-solvated solid; and therefore it is well suited for pharmaceutical
applications. The process of the present work is cost effective, eco-
friendly and amenable for scale up.





























