Embed Size (px)
Citation preview

Cellular Viability Imaging Using Dynamic Light
Scattering Optical Coherence Tomography
by
Julia Seungmi Lee
B.S. Mechanical Engineering and Industrial Engineering,
Korea University; Seoul, Korea, 2010
M.S. Mechanical Engineering, Korea University; Seoul, South Korea, 2012
M.S. Engineering, Brown University; Providence, RI, 2014
A dissertation submitted in partial fulfillment of the
requirements for the degree of Doctor of Philosophy
in the School of Engineering at Brown University
PROVIDENCE, RHODE ISLAND
May 2018

© Copyright 2018 by Julia Seungmi Lee

iii
This dissertation by Julia Seungmi Lee is accepted in its present form
by the School of Engineering as satisfying the
dissertation requirement for the degree of Doctor of Philosophy.
Date_____________
_________________________________________ Jonghwan Lee, Ph.D., Advisor
Recommended to the Graduate Council
Date_____________
Date_____________
_________________________________________ Diane Hoffman-Kim., Ph.D., Reader __________________________________________ Thomas R. Powers, Ph.D., Reader
Approved by the Graduate Council
Date_____________
___________________________________________ Andrew G. Campbell, Dean of the Graduate School

iv
Vitae
Julia Lee, after graduating from Daewon Foreign Language High School, attended Korea
University in Seoul, Korea, from which she graduated with a Bachelor of Science in
Mechanical Engineering and Industrial Engineering in 2010, then a Master of Science in
Mechanical Engineering in 2012. She completed her doctorate in engineering at Brown
University in 2018, collecting a Master of Science in Engineering in 2014 en route.

v
Acknowledgement
First and foremost, I would like to express my deep gratitude to my advisor, Jonghwan Lee,
for his guidance and support. Thanks to all the members in Lee lab, with special thanks to
Kyungsik Eom, Collin Polucha, and Madison Kuhn for help during the experiments; to our
collaborators in Morgan Lab, with special thanks to Blanche Ip, Ben Wilks, and Kali
Manning for providing samples; to Dr. Jefferey Morgan for guidance in delivering my
research to the audiences.
Deep gratitude and love to my friends and family.

vi
To my family and family-to-be

vii
Abstract of Cellular Viability Imaging Using Dynamic Light Scattering Optical
Coherence Tomography, by Julia Seungmi Lee, Ph.D., Brown University, May 2018.
Cellular viability represents whether a cell is performing normal functions, relating to
intracellular energy synthesis. Accurately quantifying the cellular viability would facilitate
novel studies on how pathological environments affect the functioning of cells in various
diseases. Nevertheless, technologies for monitoring the cellular viability in live tissue
models are currently lacking. This study aims at testing a recently developed technology,
which integrates dynamic light scattering and optical coherence tomography (called DLS-
OCT), to image the cellular viability with single-cell resolution. DLS analyzes fluctuations
in light scattered by particles to measure diffusion or flow of the particles, and OCT uses
coherence gating to collect light only scattered from a small volume for high-resolution
structural imaging. Integrating the two technologies, DLS-OCT constructs high-resolution
diffusion coefficient and flow velocity 3D maps. It is known that the motion of intracellular
organelles, often called intracellular motility, resembles a random walk in the confined
cytoplasm space, thus it can be quantified by the diffusion coefficient. Since the
intracellular motility is correlated with the cell’s metabolism level, the diffusion coefficient
map of DLS-OCT is expected to enable us to image the cellular viability. Here, the DLS-
OCT imaging of cellular viability was validated by characterizing responses of the
measured intracellular motility to the environmental conditions such as the temperature
and pH, in animal retinal explant samples. First, we characterized our new OCT system
and optimized scanning sequences and processing procedures for DLS-OCT data, to match
the dynamic range of our DLS-OCT measurement with the typical range of intracellular

viii
motility. Both numerical simulation and phantom experiments were performed for
optimization. Second, methods required for animal retinal explant experiments were
established, and DLS-OCT data from retinal tissue while manipulating the cellular viability
were acquired and analyzed, to test the technical hypothesis that DLS-OCT-measured
intracellular motility of neurons significantly diminishes when the cellular viability levels
are out of the physiological ranges. Similar operations were performed to tissue spheroids
with additional morphological measurements. As a result, we measured individual cells’
healthiness for tempered conditions, which will enlighten studying cells’ healthiness
during disease progress or therapeutic treatment in stroke, epilepsy, and Alzheimer’s
disease among others.

ix
Contents
1........................................................................................................................................... 1
INTRODUCTION .............................................................................................................. 1
1.1. Biological Background ......................................................................................... 2
Cellular Structures ........................................................................................ 2
Cellular Metabolism...................................................................................... 3
Intracellular Motility ..................................................................................... 6
Effects of Environmental Change and Chemical Treatment on Intracellular Motility ....................................................................................................................... 7
1.2. Technical Background.......................................................................................... 8
Previous Techniques to Measure Intracellular Motility................................ 8
Previous Techniques to Measure Intracellular Diffusion Coefficient ........ 10
1.3. Objective and strategy ........................................................................................ 13
Previous studies with OCT to measure dynamics....................................... 14
2......................................................................................................................................... 15
DYNAMIC LIGHT SCATTERING OPTICAL COHERENCE TOMOGRAPHY ........ 15
2.1. Introduction ........................................................................................................ 16
2.2. Theoretical background ...................................................................................... 17
Optical Coherence Tomography ................................................................. 17
Dynamic Light Scattering ........................................................................... 20
Dynamic Light Scattering Optical Coherence Tomography (DLS-OCT) .. 21
2.3. DLS-OCT simulation ......................................................................................... 24
Motivation for Simulation........................................................................... 24
Procedures of Simulation ............................................................................ 25
Simulation Results ...................................................................................... 25
Discussion on Numerical Simulation.......................................................... 28
2.4. Experimental setup ............................................................................................. 30
Spectral-Domain Optical Coherence Tomography (SD-OCT) System ...... 30
DLS-OCT .................................................................................................... 31
Simultaneous fluorescence imaging ........................................................... 32
Data Acquisition and Processing ................................................................ 33
2.5. Results and discussions ...................................................................................... 35

x
Characterization of the System ................................................................... 35
Phantom measurement of diffusion coefficient .......................................... 38
Simultaneous OCT and fluorescence imaging ............................................ 41
3......................................................................................................................................... 44
CELLULAR VIABILITY MEASUREMENT OF RETINAL NEURONAL CELLS.... 44
3.1. Introduction ........................................................................................................ 45
3.2. Experimental methods ........................................................................................ 46
Retinal dissection and handling .................................................................. 46
Image acquisition ........................................................................................ 48
Drug induce and temperature change ......................................................... 53
3.3. Results and discussions ...................................................................................... 54
Fluorescence imaging ................................................................................. 54
Diffusion coefficient ................................................................................... 55
Decorrelation............................................................................................... 68
3.4. Conclusions ........................................................................................................ 80
4......................................................................................................................................... 81
CELLULAR VIABILITY MEASUREMENT OF TISSUE SPHEROIDS...................... 81
4.1. Introduction ........................................................................................................ 82
Fibroblast .................................................................................................... 83
HEP G2 ....................................................................................................... 84
4.2. Experimental methods ........................................................................................ 84
Microscopic imaging .................................................................................. 84
Macroscopic imaging .................................................................................. 84
4.3. Results and discussions ...................................................................................... 93
Viability metric observations ...................................................................... 93
Morphological observations........................................................................ 99
4.4. Conclusions ...................................................................................................... 110
5....................................................................................................................................... 112
CONCLUSION ............................................................................................................... 112

xi
List of Tables
Table 1 Effect of environmental change and chemical treatment on the cell ......................7
Table 2 Previous methods to measure intracellular motion .................................................9
Table 3 Previous methods to measure diffusion coefficient ..............................................12

xii
List of Figures
Figure 1 Cellular structure. ..................................................................................................3
Figure 2 Schematics of interferometers. (a) Schematic of Michelson interferometer. (b)
Schematic of time-domain optical coherence tomography. ...............................................18
Figure 3 Conceptual illustration of the DLS-OCT theory. (a) Particles within the OCT
resolution volume can be categorized into three groups: static, flowing or diffusing, and
entering or exiting particles. For clarification, entering/exiting particles enter or exit out of
the voxel during a single measurement time step, resulting in stochastic fluctuations of the
OCT signal. (b) The general behavior of the field autocorrelation function in the complex
plane predicted by our model. MS and MF are approximately proportional to the fractions
of static and flowing/diffusing particles, respectively, weighted by their scattering cross-
sections [30]. ......................................................................................................................23
Figure 4 Process of numerical simulation for validating the DLS-OCT theory. True values
of the diffusion coefficient and velocity were determined by fitting the mean square
displacement (MSD) of the numerical position data. ........................................................25
Figure 5 Examples of the autocorrelation functions. Examples of the autocorrelation
functions from the numerical position data and the fitting results are plotted. Here, D = 0.3
μm2/s. .................................................................................................................................26
Figure 6 Results of numerical DLS-OCT measurements. Results of numerical DLS-OCT
measurements with the optimum measurement parameters (nt, τmax) and over the
appropriate ranges of the dynamics parameters (Nf and v). The performance was tested for
a total of 1,050 combinations (6 number densities, 5 diffusion coefficients, 5 velocities and

xiii
7 flow angles). ME= 1 - MS - MF. The points represent the mean and error from other
combinations, while fixing the parameter of investigation. ...............................................27
Figure 7 The error in measurements of D and MF·D. (a) D (b) MfD over the whole range
(c) Magnified view of the box in (b). .................................................................................28
Figure 8 Examples of the autocorrelation functions with conventional theory. Examples of
the autocorrelation functions of the numerical OCT data of the conventional theory. Here,
v = 1 mm/s, and vz = 0 mm/s. ............................................................................................29
Figure 9 Examples of the autocorrelation functions with our new theory. Examples of the
autocorrelation functions of the numerical OCT data follows our new theory than the
conventional one. Here, v = 0.1 mm/s, and vz = 0 mm/s. ..................................................30
Figure 10 Schematic of simultaneous imaging system. .....................................................33
Figure 11 Lateral resolution calculation using Rayleigh Criterion. (a) Maximum intensity
projection of the 3D volume (1024 pixels X 512 pixels X 512 pixels) image from SD-OCT
signal. (b) Magnified view of the area of interest marked in (a). (c) The intensity of the
white line in (b). .................................................................................................................37
Figure 12 Normalized intensity plot of USAF target. Normalized intensity plot of USAF
target per line width with Rayleigh criterion as a comparison (Line). ..............................38
Figure 13 Static sample measurement. Diffusion coefficient, velocity, and coefficient of
determination for the static sample measurement with the optimal fitting condition. .......39
Figure 14 Phantom sample measurements of diffusion coefficients. Microspheres with
diameter 0.05um, 0.07um, 0.1um and 0.15um were used. The circles with error bar indicate
the measurements of the sample, the line indicates the theoretical diffusion coefficient. .41

xiv
Figure 15 Simultaneous imaging of fluorescent microbeads. Acquired image of fluorescent
microbeads from OCT signal (a) and fluorescence imaging (b) using 40x lens. Red arrows
indicate the corresponding figures in both images. ...........................................................42
Figure 16 Images of 1951 USAF target. Images of 1951 USAF target from OCT signal (a)
and OCT camera (b) using 40x lens. White dotted circles indicate the corresponding area.
............................................................................................................................................43
Figure 17 Dark box setup for image acquisition. Retina imaging chamber is located below
SD-OCT for image acquisition. SD-OCT system and the retina chamber are inside a dark
box......................................................................................................................................49
Figure 18 Retina imaging chamber. (a) Assembled retina imaging chamber and filter holder
design in Solidworks. (b) 3D-printed product of the model attached to the breadboard. The
image shows the needles used for perfusion supply system. .............................................50
Figure 19 Schematic of the retina chamber. The perfusion medium bubbled with Oxygen
with 5% CO2 then travels through the heater to be supplied to the retina chamber. Waste
medium is sucked with vacuum on the other end of the chamber. ....................................51
Figure 20 Comparison of cell images in fluorescence imaging and SD-OCT imaging. ...55
Figure 21 Baseline measurement of diffusion coefficient. ................................................56
Figure 22 Diffusion coefficient of the retinal neural cells to different temperatures.. ......57
Figure 24 Diffusion coefficient of the retinal neural cells to exposure of 2.2M of ethanol in
the media. ...........................................................................................................................59
Figure 25 Diffusion coefficient of the retinal neural cells to exposure of hypotonic
condition.. ..........................................................................................................................61

xv
Figure 26 Diffusion coefficient of the retinal neural cells to exposure of hypertonic
condition. ...........................................................................................................................62
Figure 27 Diffusion coefficient of the retinal neural cells to exposure of pH 8. ...............64
Figure 28 Diffusion coefficient of the retinal neural cells to exposure of pH 3.5. ............65
Figure 29 Comparison of diffusion coefficient of the retinal neural cells to the pilocarpine.
............................................................................................................................................67
Figure 30 Intensity, decorrelation, and relative decorrelation maps. .................................69
Figure 31 Decorrelation response to different temperatures ..............................................71
Figure 32 Decorrelation response to ethanol .....................................................................73
Figure 33 Decorrelation response to osmolarity ................................................................75
Figure 34 Decorrelation response to pH ............................................................................77
Figure 35 Decorrelation response to pilocarpine ...............................................................79
Figure 36 Maximum intensity projections of tissue spheroid inside. ................................86
Figure 37 Maximum intensity projections of HEP G2 spheroids .....................................87
Figure 38 Binary image after thresholding ........................................................................88
Figure 39 Mask array .........................................................................................................89
Figure 40 Post -mask array ................................................................................................90
Figure 41 Maximum intensity projection of tissue array in final form..............................91
Figure 42 En face images of the differential array. ...........................................................92
Figure 43 Maximum intensity projection of tissue array where a spheroid is selected. ....92
Figure 44 Maximum intensity projection of individual spheroids ....................................94
Figure 45 Intensity, decorrelation, and relative decorrelation at 14 pixels (48.44 um) below
the tissue equator................................................................................................................95

xvi
Figure 46 Intensity, decorrelation, and relative decorrelation at the tissue equator. .........96
Figure 47 Maximum intensity projection of individual spheroids with different seeding
numbers in yz plane. ..........................................................................................................97
Figure 48 Maximum intensity projection of individual spheroids with different seeding
numbers in yz plane. ..........................................................................................................98
Figure 49 Maximum projections of intensity, decorrelation, and relative decorrelation at
the surface of the tissue and at 6 pixels (20.76 um) below the tissue surface. ..................99
Figure 50 Maximum intensity projection of individual spheroids with different seeding
numbers in xy and yz planes. ...........................................................................................100
Figure 51 Maximum intensity projection of individual spheroids with different DIV in xy
and xz planes. ...................................................................................................................101
Figure 52 Surface area to volume ratio with error bars ...................................................102
Figure 53 Height to width ratio (HWR) to seeding numbers.. .........................................103
Figure 54 Tissue volume plotted to seeding numbers.. ...................................................104
Figure 55 Cellular volume (volume of a cell) is plotted to seeding numbers. .................105
Figure 56 Maximum intensity projection of individual spheroids with different seeding
numbers in xy and yz planes. ...........................................................................................106
Figure 57 Surface area to volume ratio with error bars of samples to the seeding numbers.
..........................................................................................................................................107
Figure 58 Height to width ratio (HWR) to seeding numbers. ..........................................108
Figure 59 Tissue volume (mm3) is plotted to seeding numbers.. .....................................109
Figure 60 Cellular volume (mm3, volume of a cell) is plotted to seeding numbers. .......110

1
1.
INTRODUCTION

2
1.1. Biological Background
Cellular Structures
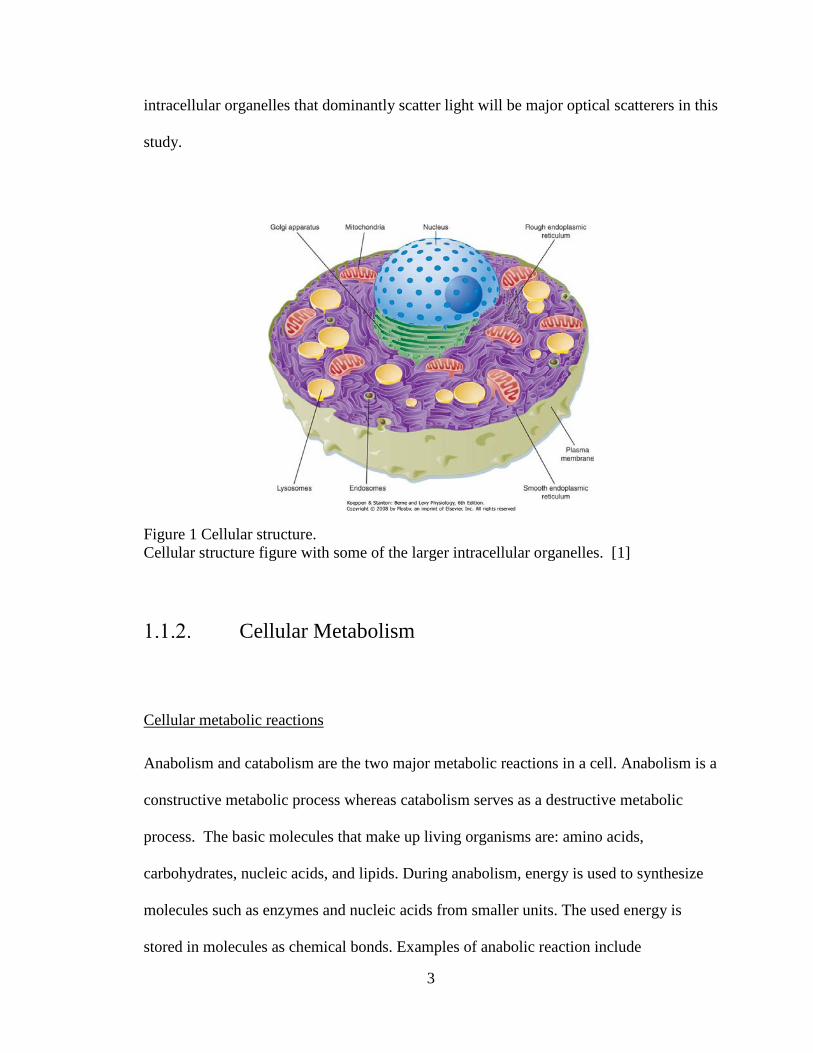
A cell is the smallest unit of life in structure and function. The basic structure of a cell
consists of a membrane layer which encloses the cytoplasm, wherein many intracellular
organelles are found. Intracellular organelles are subunits of the cell, the name of which
derives from its function to the cell that are analogous to the organs to the body.
Relatively large intracellular organelles found in a eukaryotic cell, the most commonly
found cell type in an animal body, include: Nucleus, mitochondria, vacuole, Golgi
apparatus, endoplasmic reticulum, flagellum, and chloroplast. The largest organelle is
nucleus, with the size of 10 μm approximately. Nucleus controls all activities of the cell
and maintains DNA, thus changes its size during the cell division. Following nucleus,
mitochondria, lysosome, and vacuoles are the next large organelles, exhibiting sizes of
~1μm. Mitochondria produces energy from glycolysis while vacuole helps maintain
homeostasis.
There are also numerous small organelles, each exhibiting important functions to
maintain a healthy cell. In the optical regime where we use the center wavelength of
1,300 nm, however, these small organelles will much less contribute to the ensemble
scattering of light, compared to the relatively larger organelles. According to Mie
scattering theory, large optical scattering occurs when the size of a scatterer is
comparable to the wavelength in the order of magnitude. Thus, the relatively large
3
intracellular organelles that dominantly scatter light will be major optical scatterers in this
study.
Figure 1 Cellular structure. Cellular structure figure with some of the larger intracellular organelles. [1]
Cellular Metabolism
Cellular metabolic reactions
Anabolism and catabolism are the two major metabolic reactions in a cell. Anabolism is a
constructive metabolic process whereas catabolism serves as a destructive metabolic
process. The basic molecules that make up living organisms are: amino acids,
carbohydrates, nucleic acids, and lipids. During anabolism, energy is used to synthesize
molecules such as enzymes and nucleic acids from smaller units. The used energy is
stored in molecules as chemical bonds. Examples of anabolic reaction include

4
photosynthesis during which carbon dioxide and water are synthesized into glucose and
oxygen. On the other hand, the cell breaks down complex molecules into simple
molecules and release the stored energy during catabolism. Glycolysis is an example of
catabolic reactions, where glucose is converted into pyruvate, while releasing adenosine
5'-triphosphate (ATP). The released energy and broken-down smaller molecules are used
during anabolic reactions, in turn, thus the two metabolic reactions balance each other.
Cellular respiration: glycolysis and oxidation
Cellular respiration is a set of catabolic reactions that takes place in a cell, to provide
ATP from biochemical energy and nutrients. There are two types of respiration: aerobic
respiration and anaerobic respiration. Aerobic respiration consumes oxygen to produce
energy whereas anaerobic respiration which does not require oxygen but produce less
energy compared to the aerobic respiration. Here, we focus on aerobic respiration
because anaerobic respiration occurs less in animals (muscle contraction from vigorous
exercise or in some microorganisms with no supply of oxygen). During aerobic
respiration, glucose and oxygen reacts to produce carbon dioxide, water, and energy as
ATP. Simplified reaction equation of cellular respiration is:
6O2 + C6H12O6 → 6CO2 + 6H2O + Energy ( 1 )
Glycolysis and Oxidation are the two main mechanisms which give rises to the
cellular respiration in an animal metabolism.

5
Glycolysis is an oxygen independent metabolic pathway comprising of series of
reactions to convert glucose (C6H12O6) into pyruvate (CH3COCOO−+ H+). Glycolysis
produces a small amount of ATP, as it is in the initial stage of the cellular metabolism.
C6H12O6 + 2 NAD+ + 2 ADP + 2 Pi
→ 2 Pyruvate + 2 NADH + 2 H+ + 2 ATP + 2 H2O ( 2 )
In an aerobic cell, oxidation, a process during which glucose is disposed using O2
is performed in mitochondria, where pyruvate is transported. The pyruvates are oxidized
to CO2 by O2, generating large amount of ATP from the conversion of glucose to CO2.
The complete aerobic respiration is:
C6H12O6 + 6O2 + 30 H+ + 30 ADP + 30 Pi → 6 CO2 + 36 H2O + 30 ATP ( 3 )
In some cases, no oxygen is used during the disposal of pyruvate, which is called
anaerobic respiration. Pyruvate is converted into lactic acid, producing only 10% of the
energy produced during aerobic oxidation.
Cellular viability
The above metabolic reactions are critical to normal functioning of the cell. Thus, the
cellular viability, which represents the degree of how the cell is healthy or viable, should
be closely related with the metabolic reactions occurring in it. This concept of cellular

6
viability is widely utilized in studies using cell assays on the effect of drug treatment as
well as cytotoxicity tests of chemicals on cells [2–4].
Intracellular Motility
The cellular metabolic reactions are also associated with mechanical motions occurring
inside the cell. Intracellular organelles are known to move in the confined intracellular
space. Such movements of intracellular organelles exhibit interesting characteristics.
1. Active movements consuming ATP: The movements are not passive Brownian
motions resulting from the collision with water molecules. The large
intracellular organelles are transported by active intracellular mechanical
processes involving microtubules and cytoskeletons [5,6]. The molecular origin
for the transition from the passive random to active intracellular motion is
closely related to the physiological state and condition of the cells [7].
2. Diffusion-like trajectories: Although the organelle movements result from the
active processes using ATP, their trajectories, interestingly, resemble random
walks probably because of the confined space of the cytoplasm and/or highly
complicated intracellular structure.
Based on these characteristics, it is possible to quantify the degree of the
intracellular organelle movement via the diffusion coefficient. Thus, we hypothesize that
the diffusion coefficient value can be a surrogate of the cellular viability because it would

7
become very low when the cell is dead. Although the large intracellular organelles can
exhibit free diffusion due to the collision with water molecules when the cell is dead,
such diffusion may be much smaller in the confined cytoplasm than in medium. This is
an open question and will be tested through the present study.
Effects of Environmental Change and Chemical
Treatment on Intracellular Motility
Studies on the effects of environmental changes and chemical treatment on intracellular
motility are tabulated below (Table 1) [3, 6–12] . The intracellular motility was
measured using various methods outlined in the next section. As for environmental
changes, the pH level, temperature, and osmolarity have been shown to lead a change in
the intracellular motility. Chemical treatment shown to result in a change in the
intracellular motility are generally those that either inhibit activity of the intracellular
components such as actin filaments and microtubules so that the structural change in
cytoplasm can occur. Structural change can give rise to movement of the organelles
formerly represented as bending motion, which can be detected as intracellular motility
change. Inhibiting glycolysis also influenced intracellular motility, supporting our
hypothesis that the observed intracellular motility can represent the metabolism-related
cellular viability.
Table 1 Effect of environmental change and chemical treatment on the cell
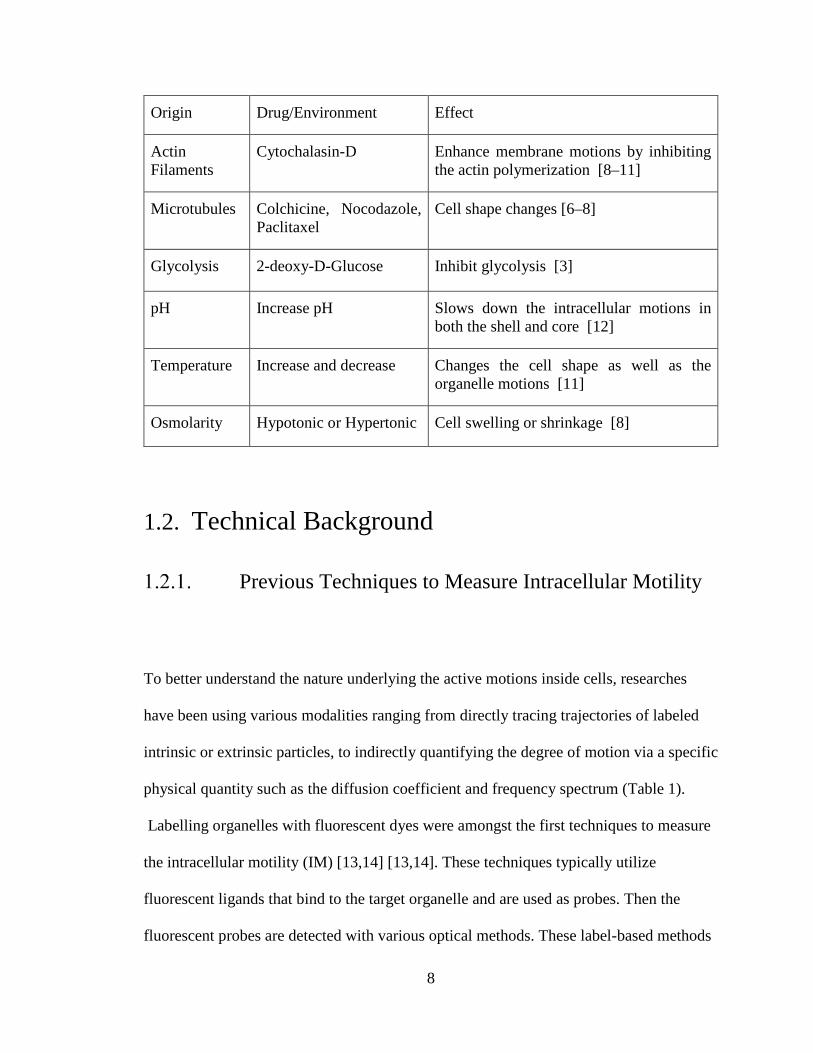
8
Origin Drug/Environment Effect
Actin Filaments
Cytochalasin-D Enhance membrane motions by inhibiting the actin polymerization [8–11]
Microtubules Colchicine, Nocodazole, Paclitaxel
Cell shape changes [6–8]
Glycolysis 2-deoxy-D-Glucose Inhibit glycolysis [3]
pH Increase pH Slows down the intracellular motions in both the shell and core [12]
Temperature Increase and decrease Changes the cell shape as well as the organelle motions [11]
Osmolarity Hypotonic or Hypertonic Cell swelling or shrinkage [8]
1.2. Technical Background
Previous Techniques to Measure Intracellular Motility
To better understand the nature underlying the active motions inside cells, researches
have been using various modalities ranging from directly tracing trajectories of labeled
intrinsic or extrinsic particles, to indirectly quantifying the degree of motion via a specific
physical quantity such as the diffusion coefficient and frequency spectrum (Table 1).
Labelling organelles with fluorescent dyes were amongst the first techniques to measure
the intracellular motility (IM) [13,14] [13,14]. These techniques typically utilize
fluorescent ligands that bind to the target organelle and are used as probes. Then the
fluorescent probes are detected with various optical methods. These label-based methods

9
have been advanced from directly tracing the particles to quantifying other useful
properties such as applied force. To directly measure the applied forces, endogenous
cytoskeletal microtubules are used as probes. Due to its local bending motion, the
microtubules’ amplitude is useful in determining the applied force.
However, these methods are invasive in nature, posing several problems such as
phototoxicity, photobleaching, eventually making a non-viable condition for the sample.
To overcome such limitation, methods which do not include dyes have been advanced to
enable label-free measurements of IM. By analyzing light scattered from cells, quantities
such as frequency fluctuations and mean square displacement have been suggested to
represent intracellular movements.
Table 2 Previous methods to measure intracellular motion
Quantified measurement Method
Label-based technique
Velocity Photon Correlation Spectroscopy of fluorescence-labeled organelles [14]
Velocity1 Time-lapse video image intensification microscopy [13]
Mean square displacement, Creep compliance
Fluorescence correlation spectroscopy of fluorescence-labeled injected extrinsic particles [15,16]
Diffusion coefficient fluorescence recovery after photo-bleaching of fluorescence-labeled injected extrinsic particles [17,18]
Diffusion coefficient Fluorescence-labeled injected extrinsic particles [19,20]
1 Direct tracing of particles for measurement

10
Diffusion coefficient Fluorescence correlation spectroscopy of fluorescence-labeled injected extrinsic particles [8]
Label-free technique
Mean square displacement Image Correlation spectroscopy [21]
Frequency fluctuations Holographic spectroscopy [12]
Force spectrum Optical Tweezer/FSM [22]
amplitude (motility) and time scale (autocorrelation decay time)
OCT [23,24]
Signal Contrast, Autocorrelation.
OCT [25]
Phase shifts (optical path length variations)
Phase contrast microscopy [26,27]
Mean square displacement Quantitative phase microscopy [28]
Intensity autocorrelation function
Photon Correlation Spectroscopy [29]
Previous Techniques to Measure Intracellular Diffusion
Coefficient
Table 3 lists the studies done in pursuit of measuring diffusion coefficients from the inside
of cells. The techniques can be categorized into three types; direct measurement,
measurements with transfect, and measurements with probe particles. Although
measurements with probe particles provide information of how diffusive the cytoplasm is,
and how it varies with drug treatment or environmental changes, they measure diffusion of

11
the probe particles, not the organelles. Compared to these extrinsic probe particle-based
methods, transfection-based methods attach fluorescent proteins to specific intracellular
organelles so that they measure the motion of the target organelles. Then, various optical
methods such as fluorescence correlation spectroscopy are used to measure the diffusion
coefficient of the target organelles. Then, various optical methods such as fluorescence
correlation spectroscopy are used to measure the diffusion coefficient of the target
organelles. Such method provides us with diffusion coefficient measurement, however, due
to the destructive nature from labelling, direct measurement techniques that uses neither
probe particles nor transfection is desired. Direct measurement techniques use neither
probe particles nor transfection. In this study, we will use our recently developed technique,
termed dynamic light scattering optical coherence tomography (DLS-OCT; see the next
chapter for details of this technique), to directly measure the diffusion coefficient of
intracellular organelles without the aid of fluorescence transfection or extrinsic probe
particles.
While many of the techniques listed in Table 2 are suitable for tracing relative
changes in the intracellular motility using arbitrary unit, the quantitative techniques listed
in Table 3 quantify intracellular motility to an absolute value of the diffusion coefficient.
This absolute measurement enables us to compare intracellular motility across different
specimen (not only tracing temporal relative changes over the same specimen), and
technically make the measurement free from a specific configuration of a measurement
system (wavelength, temporal resolution, etc).

12
Table 3 Previous methods to measure diffusion coefficient
Measured particles
Method Cell type Diffusion coefficient
[μm2/s]
Temperature [°C]
Organelles DLS-OCT [30,31]
living rodent cortex
0.1-10
Transfected organelles
k-space Image Correlation Spectroscopy [32]
MDCK GII cells cultured then, transfected with basolateral aquaporin-3 (AQP3-EGFP) and EGFP-AQP4 on collagen and E-cadherin:Fc surfaces : extracellular
0.014-0.044 37
Fluorescent correlation spectroscopy (FCS) [22]
Transfected HeLa cells with GFP (nano-sized proteins)
5-35 37
Extrinsic particles
Pulse-gradient spin–echo (s-PGSE) NMR technique [33]
Rat/Chicken erythrocytes Rat liver mitochondria In fluids (Not in a cell), intrinsic or extrinsic compounds used as probes
710-1810 25
Fluorescence recovery after photobleaching (FRAP)
Human neonatal foreskin diploid fibroblast cells grown in medium loaded with the indicated probe [34]
0.9-1 22

13
Swiss 3T3 cells cultured dextrans in cytoplasm as probes (with its size (2-40nm) being a factor for different diffusion coefficient) [17,18]
0.33~29 37
BG-9 human diploid fibroblast cells were cultured, and microinjected with probes [8]
25-47.2 37
Spot photobleaching
Fluorescence labeled DNA microinjected in HeLa cells cytoplasm [35]
3-50 23
Fluorescence Correlation Microscopy
Fluorescence labeled amino dextrans microinjected in cultured cells [36]
17-280 37
1.3. Objective and strategy
The objective of this study is to validate the DLS-OCT imaging of cellular viability by
characterizing responses of the measured intracellular motility to the environmental
conditions such as the temperature and pH, in animal retinal explant samples. As a
strategy, first step is to characterize our new OCT system and optimize scanning
sequences and processing procedures for DLS-OCT data, to match the dynamic range of

14
our DLS-OCT measurement with the typical range of intracellular motility. Both
numerical simulation and phantom experiments are performed for this optimization. Next
step is to establish methods required for animal retinal explant experiments, building
imaging chamber and the perfusion delivery and drainage system. Next, DLS-OCT data
from retinal tissue with different environmental conditions are performed and analyzed,
to test the technical hypothesis that DLS-OCT-measured intracellular motility of neurons
will significantly diminish. Similar approach is applied to tissue spheroids to test the
applicability of the method. Morphological observation is also performed with tissue
spheroid, to show the valuable potential of our system.
Previous studies with OCT to measure dynamics
Although OCT has been used in many studies especially for its ability to produce
tomographic information, measurement of dynamics of the sample has been limited.
Angiography and doppler has been widely used with OCT [37–42] to quantify blood flow
as flow index, morphological metric such as diameter and volume deformation [43] has
also been used as to show the dynamics. However, measurement of universally used
metrics such as diffusion coefficient or velocity have been lacking. Thus, providing
means to measure the universal quantity will aid in studies using OCT.

15
2.
DYNAMIC LIGHT SCATTERING
OPTICAL COHERENCE
TOMOGRAPHY

16
2.1. Introduction
Optical coherence tomography (OCT) is an emerging technology for label-free, three-
dimensional imaging of tissue structures with micrometer-scale resolution [44]. It has
been widely used for both clinical medicine including ophthalmology [45] and basic
research for neuroscience [46] and cancer biology [47]. Although its unparalleled
capability for structural imaging has been demonstrated to simultaneously achieve both
high spatial resolution and relatively large imaging depth, its capability for dynamic
imaging has been relatively less developed.
Dynamic light scattering (DLS) has been used for a long time to measure
diffusion or flow of particles. It analyzes fluctuations in light scattered by particles,
specifically its autocorrelation function. DLS has been mainly used for measurement of
the diffusion coefficient of gas molecules or micro-scale particles in a bulk sample, so it
has been challenging to apply DLS techniques for the measurement in biological
specimen which exhibit highly heterogeneous dynamics in the micrometer scale.
Since OCT uses coherence gating to collect light only scattered from a small volume for
the high-resolution structural imaging, integrating OCT with DLS could construct high-
resolution 3D maps of the diffusion coefficient and flow velocity, even in biological
specimen. The integration of two technologies has been demonstrated, termed DLS-OCT,
to provide us with a unique means to measure the diffusion coefficient and the flow
velocities at the same time, with the unprecedented resolution, and without the need of
extrinsic contrasts.

17
Previously, DLS-OCT imaging of the animal brain found high diffusion
coefficients in neurons; however, it is unclear whether the observed high diffusion
coefficients indeed represent cellular viability. Thus, there is a critical need to validate
the promising measurement capability in order to make the technology as a useful toolkit
and deploy it for a broad range of biomedical research.
2.2. Theoretical background
Optical Coherence Tomography
Michelson interferometer
The Michelson interferometer is one of the most commonly used interferometers for
scientific research, since it was first introduced in the late 19th century [48]. In a
Michelson interferometer, monochromatic light source travels through a beam splitter
and split into two arms. The split light beams travel to mirrors on each arm and reflect to
the beam splitter and then to a detector, where two light beams are interfered. Intensity of
the interference is measured with the detector z), and the interference signal is highly
sensitive to the relative positions of the mirrors so that the interferometer can detect a
slight movement of a mirror, generally down to 1/1,000 of the wavelength. The Laser
Interferometer Gravitational-Wave Observatory (LIGO) [49] is one of the latest
applications of this sensitive Michelson interferometer, which has successfully measured
a tiny signal of gravitational waves in 2016.

18
Figure 2 Schematics of interferometers. (a) Schematic of Michelson interferometer. (b) Schematic of time-domain optical coherence tomography.

19
Optical Coherence Tomography (OCT)
The principle of OCT is based on the Michelson interferometer. OCT analyzes the
interference between the light beam reflected or back-scattered from a sample and
another beam reflected from a mirror (called the reference mirror) as shown in Figure
2(b). When using a light source with low coherence (e.g., broadband light), the
interference only occurs when the photons scattered from the sample have the same
optical path length to those reflected from the reference mirror, and thus the detector can
measure the photons only scattered from the sample at a specific depth. By varying the
reference beam path length, OCT can explore different depths of the sample to obtain its
axial profile of reflectivity. Finally, by repeating this acquisition while scanning the probe
beam in the lateral directions, OCT can reconstruct a three-dimensional map of the
optical reflectivity of the sample. This is how the initial version of OCT works, called
time-domain OCT (TD-OCT). In summary, TD-OCT measures the axial reflectivity
profile, called A-scan profile, through scanning the optical path length of the sample
beam in time by moving the reference mirror.
In the last decade, OCT has been advanced to several different versions, including
spectral-domain OCT (SD-OCT), one of the most widely used versions. SD-OCT
replaces the detector with a spectrometer, to measure the interference pattern as a
function of the wavelength. Mathematically, the axial profile of the sample reflectivity
can be obtained using the inverse Fourier transform of this interference pattern. Thus,
SD-OCT does not need to move the reference mirror, dramatically increasing the imaging

20
speed and signal-to-noise ratio [50,51]. Furthermore, SD-OCT enables us to obtain the
optical reflectivity as the complex numbers (including the phase information of the
sample’s reflectivity), which was not possible with TD-OCT. In this thesis, we use a
latest high-speed SD-OCT system (Thorlabs Telesto III; 147,000 A-scan/s; 1,310-nm
center wavelength; 170-nm bandwidth; 3.5-um axial resolution).
Dynamic Light Scattering
Optical scattering theories
Depending on the size and shape of the scatterers, corresponding light scattering theories
are applied. For a spherical particle, Mie theory is applied. However, when the particle
size is much smaller than the wavelength of the incident beam, the Mie theory can be
approximated to Rayleigh scattering. In Rayleigh scattering, light scatters forward and
backwards in symmetry.
Photons are known to scatter most strongly by particles whose size is similar to
the light wavelength in the order of magnitude and whose refractive index mismatches
that of the surrounding medium. The refractive indices of common tissue components are
1.35-1.36 for extracellular fluid, 1.36-1.375 for cytoplasm, 1.38-1.41 for nuclei,
mitochondria and organelles. Generally, cell nuclei and mitochondria are strong scatterers
for visual and near-infrared light [52].
Fundamentals of dynamic light scattering analysis

21
When scatterers move, light scattered from them exhibits fluctuations. DLS is a set of
theories to describe how the time-varying light scattering results from dynamics of the
scatterers. In particular, it is well known that the autocorrelation function of the time-
varying electric field of scattered light shows an exponential decay when scatterers
exhibit Brownian motions, with the exponential decay constant being proportional to the
diffusion coefficient of the Brownian motion [53,54].
DLS has been used to measure the diffusion coefficient for various particles such
as proteins, polymers and nanoparticles [55], but mostly in bulky samples without high-
resolution 3D imaging capability. The previous methods were more like ensemble-
averaged point measurements.
Dynamic Light Scattering Optical Coherence Tomography
(DLS-OCT)
DLS model for the integration with OCT
With the assumption that static and moving particles are mixed in an OCT resolution
volume and that the moving particles can exhibit either translational or diffusive motion,
the 4D (space and time lag) field autocorrelation function of the 4D (space and time) SD-
OCT signal is given as [30]:
𝑔𝑔(𝒓𝒓, 𝜏𝜏) = ⟨𝑅𝑅∗(𝒓𝒓,𝑡𝑡)𝑅𝑅(𝒓𝒓,𝑡𝑡+𝜏𝜏)⟩𝑡𝑡⟨𝑅𝑅∗(𝒓𝒓,𝑡𝑡)𝑅𝑅(𝒓𝒓,𝑡𝑡)⟩𝑡𝑡
( 4 )

22
= 𝑀𝑀𝑆𝑆(𝒓𝒓) + 𝑀𝑀𝐹𝐹(𝒓𝒓)𝑒𝑒−ℎ𝑡𝑡2𝑣𝑣𝑡𝑡2(𝒓𝒓)𝜏𝜏2−ℎ2𝑣𝑣𝑧𝑧2(𝒓𝒓)𝜏𝜏2𝑒𝑒−𝑞𝑞2𝐷𝐷(𝒓𝒓)𝜏𝜏𝑒𝑒𝑖𝑖𝑞𝑞𝑣𝑣𝑧𝑧(𝒓𝒓)𝜏𝜏 + [1 −𝑀𝑀𝑆𝑆(𝒓𝒓)−𝑀𝑀𝐹𝐹(𝒓𝒓)]𝛿𝛿(𝜏𝜏)
Where R(r,t) is the reflectivity at given position r(x,y,z) and time t, and MS(r),
MF(r), vt(r), vz(r), and D(r) are the parameters of particle dynamics to be estimated for
each position. MS is the composition ratio of static particles, MF is that of
flowing/diffusing particles, vt is the transverse component of the flow velocity, vz is the
axial component, and D is the diffusion coefficient. The other parameters are defined in
the citation [30].
The general behavior of this autocorrelation function is illustrated in Figure 3(b).
For given voxel, the static term (MS) is the center of rotation, and MF is the initial
amplitude of rotation, which we assume do not vary during a short correlation time (in
the order of millisecond). The axial velocity-dependent phase term (𝑒𝑒𝑖𝑖𝑞𝑞𝑣𝑣𝑧𝑧(𝒓𝒓)𝜏𝜏) determines
the speed of rotation and the diffusion-oriented decay term (𝑒𝑒−𝑞𝑞2𝐷𝐷(𝒓𝒓)𝜏𝜏) determines decay
of the amplitude of rotation. However, the decay of the amplitude of rotation is not
simply an exponential decay in that it is also affected by the velocity-dependent term
(𝑒𝑒−ℎ𝑡𝑡2𝑣𝑣𝑡𝑡2(𝒓𝒓)𝜏𝜏2−ℎ2𝑣𝑣𝑧𝑧2(𝒓𝒓)𝜏𝜏2). By taking this term into account, we can estimate the transverse
component of the flow velocity as well as the axial component provided by the phase
term.

23
Figure 3 Conceptual illustration of the DLS-OCT theory. (a) Particles within the OCT resolution volume can be categorized into three groups: static, flowing or diffusing, and entering or exiting particles. For clarification, entering/exiting particles enter or exit out of the voxel during a single measurement time step, resulting in stochastic fluctuations of the OCT signal. (b) The general behavior of the field autocorrelation function in the complex plane predicted by our model. MS and MF are approximately proportional to the fractions of static and flowing/diffusing particles, respectively, weighted by their scattering cross-sections [30].
Previous results using DLS-OCT
Results from rodent brain imaging [31] showed that we can measure the cerebral blood
flow velocities. Also, in the data, it is found that neurons exhibit higher diffusion
coefficients than neighboring tissue. This data suggests the feasibility of label-free,
quantitative imaging of intracellular motility. Whether the relatively high diffusion
coefficients found from cells indeed represent cellular viability, however, needs
validation, which is one the major objectives of this thesis.

24
2.3. DLS-OCT simulation
Motivation for Simulation
We performed numerical simulations to meet two technical needs. First, we need to
optimize the scanning sequence parameters of the number of A-scans at each position (nt)
and the maximum time lag in the autocorrelation function (maxlag) toward accurate
measurement of the diffusion coefficients (D) over the range of intracellular motility
reported in the literature (Table 3). We chose five values of D in the range of 0.3-30
μm2/s, with even intervals in log scale.
Second, we need to determine the extent to which ranges of the particle dynamics-
related parameters our measurement of the diffusion coefficient will be robust, including
the number density of diffusing particles (Nf) and the velocity of flowing particles (v).
When particles exhibit a mixture of random walk and translational flow (e.g., flowing
Brownian particles), and when the velocity of the flow is too large compared to the
diffusive motion, the distinctive measurement of the diffusion coefficient against the
dominating flow velocity will not be sufficiently robust. Similarly, when the number
density of diffusing particles (Nf) in the imaging volume is too small (e.g., only 2
particles exhibit random walks while the other 98 particles do not move), the signal will
be very weak so that the measurement of the diffusion coefficient (of the small number of
the diffusing particles) will not be robust.

25
Procedures of Simulation
Process of numerical validation of our DLS-OCT measurement is summarized in Figure 4.
We generated two-dimensional (transverse and axial) position data of 100 particles for 100
time steps (∆t = 6.8 us) from the dynamic parameters. Position data were used to generate
SD-OCT signals, with which we obtained the first-order autocorrelation data. The
autocorrelation data generated for the combinations of parameters were fitted to determine
five independent coefficients (MS, MF, Vt, Vz and D), which minimizes the sum of squared
residuals (i.e. maximizing the coefficient of determination, R2). Finally, we compared
these estimated results with the true values calculated from the dynamic parameters,
position data and the SD-OCT signal.
Figure 4 Process of numerical simulation for validating the DLS-OCT theory. True values of the diffusion coefficient and velocity were determined by fitting the mean square displacement (MSD) of the numerical position data.
Simulation Results

26
Examples of the position data and corresponding OCT signals are displayed in Figure
5. They have a stochastic nature due to the random walk.
First, we confirmed that the behavior of g(τ) predicted by our theory (the Fig.
3(b)) is very close to that of the autocorrelation function of the numerically-generated
OCT signal, over a wide range of the dynamic parameters of NS, NF, NE, θ, v and D.
Several examples are shown in Figure 5.
Figure 5 Examples of the autocorrelation functions. Examples of the autocorrelation functions from the numerical position data and the fitting results are plotted. Here, D = 0.3 μm2/s.
The optimization was performed by iteratively selecting the set of measurement
parameters (nt , maxlag) toward minimization of error between the estimated and true
values of D, while exploring different ranges of the affecting dynamics parameters (Nf,

27
v). In detail, the criteria for the optimization was the root mean square (RMS) error of
MF⋅D, instead of D, because a small Mf can make the estimation result in an unreasonably
large D. We explored the values of nt = 100, 200 and 400 and maxlag = nt /2 and nt /4. As
a result, the measurement of MF⋅D was the most accurate with nt = 100 and maxlag =
nt/4, and it was robust over the ranges of Nf > 0.6 and v < 1 mm/s (Figure 6), leading to
the RMS error of 1.7%. This result implies that our analysis of the autocorrelation
function of the OCT signal, when acquired over nt= 100 and maxlag = nt/4, would
provide an accurate measurement of the diffusion coefficient (with less error than 2%)
over the range of 0.3-30 μm2/s, as long as more than 60% of the particles in the OCT
voxel exhibit a canonical random walk motions mixed with a smaller translational flow
than 1 mm/s.
Figure 6 Results of numerical DLS-OCT measurements. Results of numerical DLS-OCT measurements with the optimum measurement parameters (nt, τmax) and over the appropriate ranges of the dynamics parameters (Nf and v). The performance was tested for a total of 1,050 combinations (6 number densities, 5

28
diffusion coefficients, 5 velocities and 7 flow angles). ME= 1 - MS - MF. The points represent the mean and error from other combinations, while fixing the parameter of investigation.
Figure 7 The error in measurements of D and MF·D. (a) D (b) MfD over the whole range (c) Magnified view of the box in (b).
Discussion on Numerical Simulation
One of the major differences of our theory against previous ones is the g(τ) rotates in the
complex plane, not around the origin (which is assumed in most of previous theories), but
around a non-zero point (i.e., Ms in our theory). This was confirmed in the examples in
Figure 8, but we further investigated the decay of the magnitude of g. When the
magnitude of g(τ) relative to the origin point was plotted according to the previous
theories, the decay pattern was occasionally neither exponential (pure diffusion),
Gaussian (pure flow), nor a mixture of two (Figure 9). In contrast, when the magnitude is
plotted relative to the Ms point as predicted by our theory, the decay pattern was
relatively canonical (i.e., exhibited a mixture of exponential and Gaussian decays)
(Figure 9). This result supports that our novel approach enables accurate measurement of

29
the diffusion coefficient even when moving particles are mixed with static particles
within an OCT resolution volume. Other advantages include that we can estimate the
relative number density of moving particles (MF) and how the particle motions are close
to either canonical diffusive or translative motions (R2). When particles exhibit
oscillating motions for instance, it will result in a low R2 because our model did not
include such oscillating motions.
Figure 8 Examples of the autocorrelation functions with conventional theory. Examples of the autocorrelation functions of the numerical OCT data of the conventional theory. Here, v = 1 mm/s, and vz = 0 mm/s.

30
Figure 9 Examples of the autocorrelation functions with our new theory. Examples of the autocorrelation functions of the numerical OCT data follows our new theory than the conventional one. Here, v = 0.1 mm/s, and vz = 0 mm/s.
2.4. Experimental setup
Spectral-Domain Optical Coherence Tomography (SD-
OCT) System
A spectral-domain optical coherence tomography (SD-OCT) system (Thorlabs, Inc) is
used in this study. The SD-OCT system is optimized for DLS-OCT imaging. A large-
bandwidth near-infrared light source with center wavelength of 1310 nm, and wavelength
bandwidth of 170 nm is employed in the system for a large imaging depth and high
spatial resolution. The maximum imaging depth is 2.5 mm and the axial resolution is 3.46

31
um. The lateral resolution is determined by the objective lens in use. The 40x objective
lens (1-U2M587, LUMPLFLN 40XW, Olympus America, Inc) was used for cellular
imaging with its lateral resolution of 0.78 um. The scanning speed of the system is
147,000 A-scans/s.
DLS-OCT
Using the OCT system, we built LabVIEW software to control the system and acquire
data. Acquired data were post-processed using our lab software libraries in MATLAB.
The LabVIEW software includes a scanning sequence for DLS-OCT, and the software
libraries include a processing algorithm for DLS-OCT data. The published DLS-OCT
sequence and algorithm have been implemented with the previous OCT system with 47
kHz, where 100 A-scans were repeated, and the max time lag was 25 time points (~2 ms
range) [30]. In this study, we use the new OCT system with 147 kHz. Since the speed
(i.e., time lag sampling) and the number of A-scans (time lag range) affect the dynamic
range and accuracy of DLS measurement, we needed to optimize the scanning parameters
and processing algorithms for the new faster OCT system. For this reason, numerical
simulation was performed as in the previous section for the optimization and then
phantom measurements were conducted for calibration.

32
Simultaneous fluorescence imaging
To validate OCT imaging of individual cells (either by structural or dynamic contrast),
we built and integrated a component for simultaneous fluorescence imaging to the OCT
system. Figure 10 shows the schematic of the simultaneous imaging system. Blue light
with nominal wavelength of 490 nm from a mounted LED and a T-Cube LED Driver
(M490L4 and LEDD1B, Thorlabs) travels through the diffuser (ACL2520UDG6,
Aspheric Condenser Lens, Thorlabs) to the excitation filter (MF469-24, GFP Excitation
Filter, Thorlabs) and dichroic beamsplitter (69-899, Dichroic Longpass Filter, Edmund
Optics) where light direction is changed to the sample. The reflected light from the
sample travels through the beamsplitter and emission filter (FELH0500, Premium
Longpass Filter, Thorlabs) to the probe where the reflected lights are send to the
computer for real-time imaging and acquisition. The optics components were chosen so
that the OCT signal will neither crosstalk with the LED light nor be blocked.

33
Figure 10 Schematic of simultaneous imaging system.
Data Acquisition and Processing
SD-OCT system is used to acquire the OCT signal of the sample. SD-OCT system
parameters were (nt and maxlag) optimized in agreement to the result of the DLS-OCT
simulation. Data acquisition was performed using a labview file “Main.vi” in OCT
computer. Focusing was done with using “2D” and “3D” function in the file. Using “2D”
function, real-time B-scan views were shown, with which focal depth was set. With “3D”

34
function, real-time tomography was shown. After confirming the view of interest, data
was acquired using “ACQUIRE” function. In the next three sections, set of data
parameters used for each purpose is described. In the result sections, data gathered in a
different manner from the standard procedure will be explained as needed.
Reconstruction of a tomography
Three -dimensional SD-OCT signal data sized 512 pixels(W) X 512 pixels(H) X 1024
pixels(D) with transverse resolution of 0.5 um and axial resolution of 3.46 um was taken
using the 40x lens. A-scan and B-scan were both 1 and 4 identical volumes were taken
for volume averaging. The signal data was then reconstructed using
“Main_Reconstruct.m”. Data specific identification information was changed in the code.
For larger volume, three -dimensional SD-OCT signal data sized 512 pixels(W) X
512 pixels(H) X 1024 pixels(D) with transverse resolution of 20 um and axial resolution
of 3.46 um was taken using the LSM03 lens (Scan Lens, Thorlabs) . A-scan and B-scan
were both 1 and 4 identical volumes were taken for volume averaging. The signal data
was then reconstructed using “Main_Reconstruct.m”. Data specific identification
information was changed in the code.
Reconstruction of a diffusion coefficient data
Three -dimensional SD-OCT signal data sized 64 pixels(W) X 256 pixels(H) X 1024
pixels(D) with transverse resolution of 0.5 um and axial resolution of 3.46 um was taken

35
using the 40x lens. A-scans were repeated at a fixed position for 128 times and was
moved to next scanning position to scan a 3D volume of the sample. B-scan was 1 and 2
identical volumes were taken for volume averaging. 4 mosaic volumes were taken in a
row using MX = 4, x center =0.048mm. The signal data was then reconstructed using
“Main_ReconstructDLS.m”. Data specific identification information was changed in the
code. The depth of the data to be reconstructed was shown in the code. After confirming
the depth of interest, the signal data is then reconstructed and fitted to determine the five
independent coefficients (MS, MF, Vt, Vz and D).
Reconstruction of a decorrelation data
Three -dimensional SD-OCT signal data sized 512 pixels(W) X 512 pixels(H) X 1024
pixels(D) with transverse resolution of 0.5 um and axial resolution of 3.46 um was taken
using the 40x lens. B-scan was repeated 2 times over time interval of 10 ms and 20
ms. and 2 identical volumes were taken for volume averaging. The signal data was then
reconstructed using “Main_ReconstructAngio.m”. Data specific identification
information was changed in the code.
2.5. Results and discussions
Characterization of the System
Our DLS-OCT measurement algorithm includes the parameter of h and ht as in Eq.4,
which are functions of the spatial resolution [30]. Thus, it is important to accurately

36
measure the actual resolution of our system prior to DLS-OCT imaging of biological
specimen.
A 40X objective lens (water-immersed; 1-U2M587, LUMPLFLN 40XW,
Olympus America, Inc) is used with our SD-OCT system for cellular resolution. The
manufacturer specifications for the numerical aperture (NA) is 0.8, and working distance
is 3.3 mm. When used with our light source (center wavelength = 1.3 μm), the theoretical
lateral resolution is:
𝑟𝑟 = 0.61 𝜆𝜆1.3𝑁𝑁𝑁𝑁
= 0.76 (µ𝑚𝑚) ( 5 )
To determine the actual resolution with our OCT system, we measured the resolution with
a resolution target (1951 USAF Hi-Resolution Target, Edmund Optics). Figure 11(a)
shows the maximum intensity projection of the 3D image data (512 pixels X 512 pixels X
1024 pixels, width, depth, and height respectively, with transverse resolution of 0.5 um and
axial resolution of 3.46 um). Here, we applied Rayleigh criterion [56] to calculate the
lateral resolution. To determine the lateral resolution from the recorded data, intensity plots
of the line pairs in the targets were investigated. Selected area in Figure 11(a) was the
narrowest line pairs that was resolved according to the Rayleigh criterion. The lines had
width of 0.87 μm, which closely follow the theoretical resolution than the theoretical lateral
resolution.

37
𝐼𝐼0 = 𝐼𝐼 8𝜋𝜋2
( 6 )
Figure 11 Lateral resolution calculation using Rayleigh Criterion. (a) Maximum intensity projection of the 3D volume (1024 pixels X 512 pixels X 512 pixels) image from SD-OCT signal. (b) Magnified view of the area of interest marked in (a). (c) The intensity of the white line in (b).
Figure 12 shows the Rayleigh criterion and I/I0 for different line pair width. Line with
0.87 um as its width is the narrowest line to be resolved according to the criteria as
discussed above. Line with width of 0.78 um does not satisfy the criteria.

38
Figure 12 Normalized intensity plot of USAF target. Normalized intensity plot of USAF target per line width with Rayleigh criterion as a comparison (Line).
Phantom measurement of diffusion coefficient
We performed phantom (standard sample) experiments to validate that DLS-OCT can
accurately measure the diffusion coefficient, using previously optimized parameters from
the simulation following the standard procedure described in section 2.4.4.2.
Diffusion coefficient measurement from static sample
Diffuse reflectance standard (WS-1, Ocean Optics) was used as a static sample. Given
that true values of the parameters should be MS =1, MF, Vt, Vz, D =0, the data collected
from this sample was used to determine the kernel size for ensemble averaging of
autocorrelation function and whether to compensate for global phase shift. Median filter

39
and gaussian filter sizes were also determined using the data. The performance was tested
for a total of 24 combinations (2 kernel sizes for ensemble averaging, 2 options for global
phase shift, 2 window sizes for median filter and 3 window sizes for gaussian filter). The
optimal combination was determined such that it results in the minimum D. We found
that D was minimum as 0.054± 0.024 um2/s (R2=0.97) when the kernel size for ensemble
average was a 3 by 3 by 3 array with uniform values that sum up to 1, global phase shift
was not compensated, median filter size of 3 and gaussian filter size of 5 for the static
sample. The resulting en face plots of diffusion coefficient, velocity, and coefficient of
determination for the static sample with optimal conditions are shown in Figure 13. In
consequence, our DLS-OCT technique has the baseline noise of 0.054 um2/s in the
diffusion coefficient measurement.
Figure 13 Static sample measurement. Diffusion coefficient, velocity, and coefficient of determination for the static sample measurement with the optimal fitting condition.

40
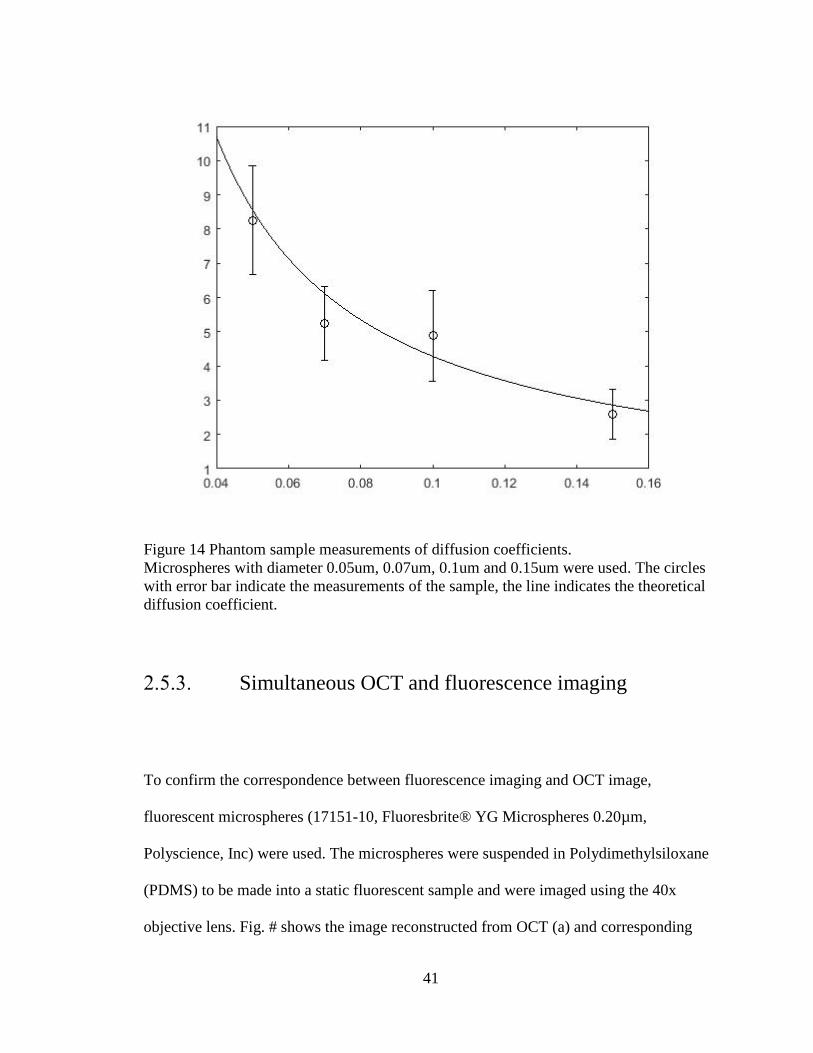
Microspheres as standard samples
Microspheres were used to measure the diffusion coefficient using our methods. The
diffusion coefficient and diameters of the particle have an inversely linear relationship as
given by Stokes-Einstein equation. Thus, we used microspheres with different sizes to
validate the diffusion coefficient measurement. Standard procedure described in section
2.4.4.2 was used to collect and process the data. Monodisperse polystyrene microspheres
in 2.5% solids (w/v) aqueous suspension (Polysciences, Inc.) with diameters of 0.05 μm,
0.07um, 0.1 μm and 0.15um were used. We compared these estimated results with the
theoretical values of diffusion coefficient as given by Stokes-Einstein equation. Our OCT
system with the fitting algorithm have measuring range of 2.8um3/s to 8.1 um3/s, which
includes the diffusion coefficients of previously measured biological tissues (Table 3). As
shown in Fig. #, DLS-OCT-measured diffusion coefficients followed the general trend in
the theory. Note that microspheres have packed structure, which limited light penetration.
This prevented measuring the diffusion coefficient beyond our lower bound.
41
Figure 14 Phantom sample measurements of diffusion coefficients. Microspheres with diameter 0.05um, 0.07um, 0.1um and 0.15um were used. The circles with error bar indicate the measurements of the sample, the line indicates the theoretical diffusion coefficient.
Simultaneous OCT and fluorescence imaging
To confirm the correspondence between fluorescence imaging and OCT image,
fluorescent microspheres (17151-10, Fluoresbrite® YG Microspheres 0.20µm,
Polyscience, Inc) were used. The microspheres were suspended in Polydimethylsiloxane
(PDMS) to be made into a static fluorescent sample and were imaged using the 40x
objective lens. Fig. # shows the image reconstructed from OCT (a) and corresponding

42
image with fluorescence imaging. Red arrows indicate the corresponding figures in each
image. Note that more fluorescent lump appears in the fluorescence imaging due to the
characteristic difference of the imaging methods. The fluorescence imaging captures all
the features in the focal depth whereas the OCT captures a three-dimensional data and
shows each slice. Here in Figure 15 (a), maximum intensities from 5 neighboring depth
layers in focal depth (corresponding to 17.3 um) were projected to make a maximum
intensity projection. It can be inferred that within those 5 layers, the five distinctive
lumps were present.
Figure 15 Simultaneous imaging of fluorescent microbeads. Acquired image of fluorescent microbeads from OCT signal (a) and fluorescence imaging (b) using 40x lens. Red arrows indicate the corresponding figures in both images.
USAF target was also used to further confirm the correspondence between the
images. Here, LED was not used. Figure 16 shows the two images taken from OCT and
the camera with 40x lens. From these images, we determined the mapping function

43
between the OCT and fluorescence microscope. When OCT imaged the area of 32.5 um X
80 um around the center with 65 pixel X 160 pixel, the microscope image (60 pixel X 120
pixel) could be best overlapped to the OCT MIP image when transformed by a mapping
function of x’ = 0.94x-0.59 and y’ = -0.41y+0.88.
Figure 16 Images of 1951 USAF target. Images of 1951 USAF target from OCT signal (a) and OCT camera (b) using 40x lens. White dotted circles indicate the corresponding area.

44
3.
CELLULAR VIABILITY
MEASUREMENT OF
RETINAL NEURONAL CELLS

45
3.1. Introduction
Mouse retina has served an important role for studies of genetics and investigating
diseases and treatments [57,58] owing to its structure being layers of different types of
neurons being apparent a homogeneous within the layer. We chose the mouse retina as a
biological sample on which we validate the cellular viability measurement, because of the
unique laminar cell-type distribution and because our lab routinely dissects the retina
from mice.
As shown in the Table 1, changing environmental condition and drug
delivery can affect the motions inside a cell. The perturbation can be such that the
cell cannot survive or decrease in viability, which will either slow down or speed up
the motions, reflected on our diffusion coefficient measurement as a result. With our
hypothesis that DLS-OCT-measured intracellular motility of neurons will
significantly diminish when the cellular viability levels are out of the physiological
ranges, condition change was induced. From previous studies [10,12,59,60],
commonly used environmental changes were chosen to be change factor for our
experiment, the changes include temperature, ethanol treatment, osmolarity change,
and pH change. Lowering the temperature slows down molecular mobility [61], with
our hypothesis, lowering the temperature will decrease the diffusion coefficient
measurement. Ethanol, which can have cytotoxic effect, can cause complex
inhibition towards multiple cellular functions. However, it can also facilitate the
malfunction of the cells effectively. Osmolarity change influences cell

46
viability [62,63], however intracellular motility response to the osmotic stress is little
known. During hypotonic stress, cell swelling occurs, giving the molecules in
cytoplasm freedom to move, whereas during hypertonic stress, cell desiccation
occurs. The diffusion coefficient response to each condition was studied. Lowering
pH brings down viability [64,65], sometimes killing cells. This will also affect the
diffusion coefficient. Pilocarpine delivery was also performed, as a drug that targets
on affecting the cytoskeleton reorganization [66]. These modifications in active
cytoplasmic motion will have a significant impact on the diffusion coefficient
variance.
3.2. Experimental methods
Retinal dissection and handling
Mice were placed in a closed anesthesia chamber, and anesthesia is induced using 3-5%
isoflurane with mixture of 100% oxygen in air. Immediately after the initiation of
anesthesia, pharmaceutical grade Dexamethasone (0.2 mg/kg, IP for) is administered to
decrease inflammation, and buprenorphine (0.05-0.1 mg/kg IP) is administered to ensure
pain relief. The animal is examined to ensure that the level of anesthesia is sufficient to
prevent hindpaw reflexes, whisking behavior and corneal reflexes. Living whole retinas
or retinal slices were harvested directly from the euthanized animal and maintained in
vitro so that we can study neurons electrophysiologically or by functional imaging. The
surgical sites were clipped to remove fur and the site was cleaned using betadine

47
alternated with alcohol and repeated three times in total. The detailed method was:
Puncture the eye. Cut out the cornea. cut around the eye at the limbus, very close to the
limbus (retina is attached to the eye cup into 2 places: at the limbus and the optic disk), to
release the retina. After the cut around the limbus, place a forcep in the optic disk to fix
the eyecup, now gently with the other forceps (preferably blunt tip), go around the edges
to free the retina from the eyecup. Now the retina is fixed only at the optic disk, with the
forceps used to fix the optic disk, pressurize the area so that the optic nerve is damaged
and frees the retina or go from beneath the retina and hold the optic nerve and try to pluck
the retina gently. During surgery, the animal was monitored for anesthetic depth and
well-being by monitoring the body temperature, the respiration (character, depth,
frequency) and the pedal reflex. All the procedures were performed under the approval by
IACUC (1510000167).
The dissection is done in a dark room to prevent light exposure to the neurons,
then the sample is put on a filter (Millicell Cell Culture Insert, EMD Millipore Corp.),
sucked from the other side to be fixed at the surface. Then the sample and the filter are
placed in an imaging chamber covered entirely, so that during the delivery to the imaging
dark box, light exposure is minimized.
Fluorescence imaging preparation
For simultaneous OCT and fluorescence imaging, genetically encoded calcium indicator
(GCaMP) transgenic mice were used for green fluorescent protein (GFP)
excitation [67]. Wild-type mice were also used by staining the neuronal cells with

48
Calcium indicator (Oregon Green, Thermo Fischer Scientific). To prepare a wild-type
mouse retina for fluorescence imaging, the mouse was kept in dark for adaptation for an
hour during which AMES medium was prepared and bobbled. After the adaptation, eyes
were enucleated from the mouse and cornea, vitreous, and lens were removed. The
eyecup was to be left with sclera then the eyecup. The eyecup was incubated in papain
(Worthington Biochemical Corporation) and Ames solution for 20 minutes for removal
of the inner limiting layer while retinal neuronal cells are facing up. After 20 minutes in
papain solution, the supernatant was removed, and the eyecup was incubated in
ovomucoid for another 10 minutes to inhibit the papain enzyme destroying the neuronal
cells. After treating in ovomucoid, remove the supernatant and wash the eyecup using
AMES medium. The eyecup now can either be treated in calcium indicator before
dissection or be dissected and treated in calcium indicator for 30 minutes. The retinal
tissue is ready after washing the calcium indicator. During the process, 95% O2 and 5%
CO2 gas was supplied to the incubator while maintaining the temperature at 36.5°C.
Image acquisition
Images acquisition system
For the image acquisition of the mouse retina, a dark box is prepared (Figure 17). From
the dissection to the imaging, the retina was not exposed to light, because light exposure
can stimulate neurons, resulting in functional change in the sample [68,69]. The box with
size of 15” X 15” X 20” was assembled with construction rails, then covered with black
poster board and blackout fabrics on an Aluminum breadboard. SD-OCT system is

49
located inside the box with the fibers and cables connected to the computer accessed
through a slit in the bottom of the dark box. The slit is then covered with the blackout
fabric to prevent lights from penetrating. The frontal area of the box is covered with
extra-long blackout fabric for flexibility of the access to the interior of the box. Tubings
for supplying and draining the perfusion system are also accessed in the same manner.
Figure 17 Dark box setup for image acquisition. Retina imaging chamber is located below SD-OCT for image acquisition. SD-OCT system and the retina chamber are inside a dark box.
Retina imaging chamber and the filter holder were designed with Solidworks
(Dassault systemes) (Figure 18(a)) and printed with a 3D printer (B9Creator v1.2,
B9Creations. LLC) (Figure 18 (b)). The retina imaging chamber has two open walls on
each side so that the perfusion can be supplied from one end of the chamber and the
waste can be drained on the other end of the chamber without disturbing the imaging area
by minimizing the perturbation of the fluid. On the bottom of the chamber at the center,

50
there are three legs where the filter holder is placed. The three legs prevent the filter
holder from floating inside the chamber. The filter holder holds a filter, on which
dissected retina is placed.
Figure 18 Retina imaging chamber. (a) Assembled retina imaging chamber and filter holder design in Solidworks. (b) 3D-printed product of the model attached to the breadboard. The image shows the needles used for perfusion supply system.
Detailed perfusion supply and drainage system is shown in Figure 19. Ames
medium [70] (Sigma-Aldrich Corp.) in a borosilicate glass aspirator (Kimble) is bobbled
by O2 (5% CO2). Ames medium is prepared by dissolving one bottle of AMES medium,
1.9 g of NaHCO3, and 1.8 g of glucose to 1 L of distilled water. The medium goes
through a 3-way tubing connected to the bottom outlet, one of which can be used for drug
release, and the other goes through a syringe pump (AL-1000, WPI Inc.) for precise
controlling. The controlled flow then goes through a heater (SH-27B, Warner
instrument), controlled by a heater controller (TC-324C, Warner instrument) with a
sensor probe inside the chamber to maintain a desired temperature. Except for the set of
experiment with the temperature variance, we choose the temperature to be maintained at

51
33℃ [71,72], for the retina tissue to be active and healthy, which we will call normal
condition group. The temperature measured at the sensor often lower than the real
temperature in the tissue, because of the distance between the tube heater and the sensor
probe. The Ames medium travels inside the chamber to the other side of the chamber
where fluid outlet is located. A suction needle connected to a tubing with vacuum is
placed at a fixed position, so that the needle sucks any fluid that overflows the set level,
thus the amount of fluid in the chamber is kept at constant.
Figure 19 Schematic of the retina chamber. The perfusion medium bubbled with Oxygen with 5% CO2 then travels through the heater to be supplied to the retina chamber. Waste medium is sucked with vacuum on the other end of the chamber.

52
SD-OCT
As discussed in the section 2.4.4.2, standard procedures were taken for diffusion
coefficient measurement. After the fitting, 12 cells were selected manually from the
diffusion coefficient map, then cellular regions were chosen with the same three-
dimensional size of 5 pixels X 25 pixels X 25 pixels (17.3 um X 12.5 um X 12.5 um).
The average D value of each region of interest (ROI; representing each cell) was
computed, leading to 12 mean diffusion coefficient values. Then the 12 means were
averaged again to represent the diffusion coefficient (with an error bar; standard
deviation) of the data set. Note that these D values can be lower than the exact values in
the cytoplasm because the rectangular 3D ROI can involve the nucleus and extracellular
space in addition to the cytoplasm.
For decorrelation measurement, standard procedures were taken as discussed in
section 2.4.4.3. Although the decorrelation only provides a qualitative means to estimate
the intracellular motility, its relative changes in response to environmental manipulations
may give us another insight. We acquired the decorrelation data from the same samples
and in the same condition whenever we took the DLS-OCT data. Intensity of the tissues
can vary greatly which can affect decorrelation. Generally, higher intensity induces
higher decorrelation. Thus, we invite the concept of relative decorrelation, to normalize
the variance among the measurements. Here, relative decorrelation of a voxel is
decorrelation value divided by intensity value of a voxel. In most cases, average of the
intensity, decorrelation, and the relative decorrelation of the area of focus was calculated
from MIP and plotted in the figure. The area of focus had a size of 256 pixels X 256

53
pixels (128um X 128 um), chosen to minimize the intensity variance within the area.
Note that for experiments where osmolarity was changed, 12 cells were selected
manually, then cellular voxel data was generated with the size of 5 pixels X 25 pixels X
25 pixels (17.3 um X 12.5 um X 12.5 um). The average values of each cell were
computed to generate 12 average intensity, decorrelation, and relative decorrelation
values. Then the 12 averages were averaged again to represent each metrics.
Drug induce and temperature change
Temperature, osmolarity changes can be done simply by changing the level of the
components in our perfusion system. The temperature was varied by changing the heater
settings. To establish the osmotic pressure between the cell and the surrounding fluid was
controlled by changing the concentration of Sodium hydroxide in the Ames medium. PH
change was induced by undersupplying 5% CO2 and 95% O2 gas or adding hydrogen
chloride. Ethanol treatment (2.2 M) was also induced for its cytotoxic effect. Pilocarpine
was used to induce change in cytoskeleton.

54
3.3. Results and discussions
Fluorescence imaging
Fluorescence imaging using GCaMP mice were performed to confirm the detection of the
cells in the metrics used. Figure 20(a-b) shows the fluorescence imaging result and the
decorrelation map to the corresponding area comparison. The red arrows in Figure 20(a)
have pattern that is also found in Figure 20(b). Part of the cells seen in the fluorescence
imaging is absent in the decorrelation map due to the difference of the imaging
techniques. Fluorescence imaging does not have the ability to penetrate the cells and
show the images as slices, thus the depth at which individual cells are present in the
image vary within the focal depth. On the other hand, with the tomographic map obtained
from SD-OCT shows the cells at certain depth. In addition, the use of 40x objective
causes confocal effect, which can distort the position of the cells. The diffusion
coefficient map was compared with the fluorescence imaging in the same manner (Figure
20 (c-d)). Fluorescence imaging identified the existence of cells within our field of view,
qualitatively in terms of sizes as well [73–77].

55
Figure 20 Comparison of cell images in fluorescence imaging and SD-OCT imaging. (a) Fluorescence imaging of the neurons. (b) Decorrelation map obtained using SD-OCT of the same areas as (a). (c) Fluorescence imaging of the neurons. (d) Diffusion coefficient map obtained using SD-OCT of the same areas as (c). The red arrows demonstrate the corresponding cell figures in both imaging methods.
Diffusion coefficient
Diffusion coefficient in cytoplasm was measured higher than that of the nucleus, due to the
higher movement in the cytoplasm whereas there is in the nucleus (Figure 21 (a)). Although
there are movements in the nucleus as well as between the nucleus and the cytoplasm such
as RNA transport [78], diffusion aroused from those movements were not captured
possibly due to its small size. The results in the normal-condition group varied across the
different samples from 0.2 um3/s to 30 um3/s. This large variance can arise from the
difference in the viability between the dissected samples [79,80]. During the dissection,
damages can be done physically to the neural cells and its functions. Therefore, rather than
comparing averages between the groups, we analyzed the relative changes within the same
sample, induced by environmental manipulation or chemical treatment, as detailed in the
previous section.

56
Maximum intensity projections in xy and yz planes Figure 21(b) and maximum
diffusion coefficient projections in xy and yz planes are shown in Figure 21(c), with the
window size of 17 pixels (8.5um) in x and y-axis and 7 pixels in z axis (24.22um).
Figure 21 Baseline measurement of diffusion coefficient. (a) Baseline measurement of diffusion coefficient in the normal-condition group. (b) Maximum intensity projections in xy, yz, and xz planes. (c) Maximum diffusion coefficient projections in xy, yz, and xz planes. Scale bar: 5um
Temperature
Diffusion coefficient change in reaction to the temperature change from viable body
temperature around 36.5°C to 13°C is shown in Figure 22. The diffusion coefficient was
measured after 30 minutes of exposure to the changed temperature. Viability of the cells
and tissues worsens as temperature deviates from body temperature. Diffusion coefficient

57
also decreased as temperature decreased. Diffusion coefficient is related to the
intracellular motility which originate from active movements in the cytoplasm. It is
reported that routine metabolic rate decreases as temperature changes [81]. Intracellular
motility retard from the temperature drop was observed as diffusion coefficient drop
resultantly.
Figure 22 Diffusion coefficient of the retinal neural cells to different temperatures. (a) Diffusion coefficient map of the retinal neural cells at normal condition. (b) Diffusion coefficient map of the retinal neural cells at 15°C. Scale bar: 10um (c) Red bar indicates the diffusion coefficient of the retinal neural cells at 31°C. Blue bar indicates the diffusion coefficient at 15°C.

58
Ethanol
Diffusion coefficient change in exposure of 2.2M ethanol in the media was measured
with respect to the time of exposure (Figure 22). Within 30 minutes of the exposure to the
ethanol, the diffusion coefficient decreased. However, after 60 minutes of exposure, the
diffusion coefficient increased. Ethanol was used for its cytotoxic effect. Ethanol can
induce cell death by mechanisms such as DNA synthesis inhibition or fragmentation, and
oxidative stress increase resulting in reduction of metabolic function. It is expected that
when used in high dose, ethanol can induce cell membrane rupture and necrosis [82] and
cell viability decreases after alcohol treatment [83]. From our result, active movements in
the cytoplasm decreased initially, then increased in time. The initial decrease in the active
movements can be due to the toxicity of the ethanol, while increase in movement with
prolonged exposure to the ethanol may have arose from necrosis-related movements.

59
Figure 23 Diffusion coefficient of the retinal neural cells to exposure of 2.2M of ethanol in the media. (a) Diffusion coefficient map of the retinal neural cells at normal condition. (b) Diffusion coefficient map of the retinal neural cells 30 minutes after alcohol treatment. Scale bar: 10um (c) Red bar indicates the diffusion coefficient of the retinal neural cells at 31°C. Blue bar indicates the diffusion coefficient when the tissue was exposed 30 minutes and 60 minutes, respectively.
Osmolarity
AMES media used for in this study has isotonic osmolarity of 329 mOsm designed to
provide a physiological osmolarity. The change in osmolarity can induce either cell
swelling under hypotonic condition or cell shrinkage under hypertonic condition, both of
which can give rise to the change in diffusion coefficient.

60
The temperature was maintained at normal condition at 0 minute. The temperature
and CO2 level were maintained all throughout the experiment, except when pH change
was induced. Ames media with higher or lower osmolarity was supplied to reach either
hypertonic or hypotonic condition.
Exposure to the hypotonic stress (200 Osm) lead to increase in diffusion
coefficient in time (Figure 24(c)). The diffusion coefficient increase in hypotonic
condition may have relation to the active movements of the organelles because the
reconstructing of the membranes to prevent the rupture of the membranes draws extra
membranes and intracellular reserves to the surface [84,85].
Decrease in diffusion coefficient was observed in exposure to the hypertonic
stress (500 Osm) in time (Figure 25(c)). Viability decline due to osmotic stress has been
studied [86,87], which may be source of the diffusion coefficient decrease seen here.

61
Figure 24 Diffusion coefficient of the retinal neural cells to exposure of hypotonic condition. (a) Diffusion coefficient map of the retinal neural cells before exposure to the hypotonic media. (b) Diffusion coefficient map of the retinal neural cells 65 minutes after exposure to the hypotonic media. Scale bar: 10um (c) Diffusion coefficient of the retinal neural cells with errorbar to the duration of exposure of hypotonic condition of 200 Osm at 31°C. The retinal tissue was exposed to the hypotonic condition at 0 minutes and the diffusion coefficient was measured at 15 minutes, and 65 minutes, respectively.
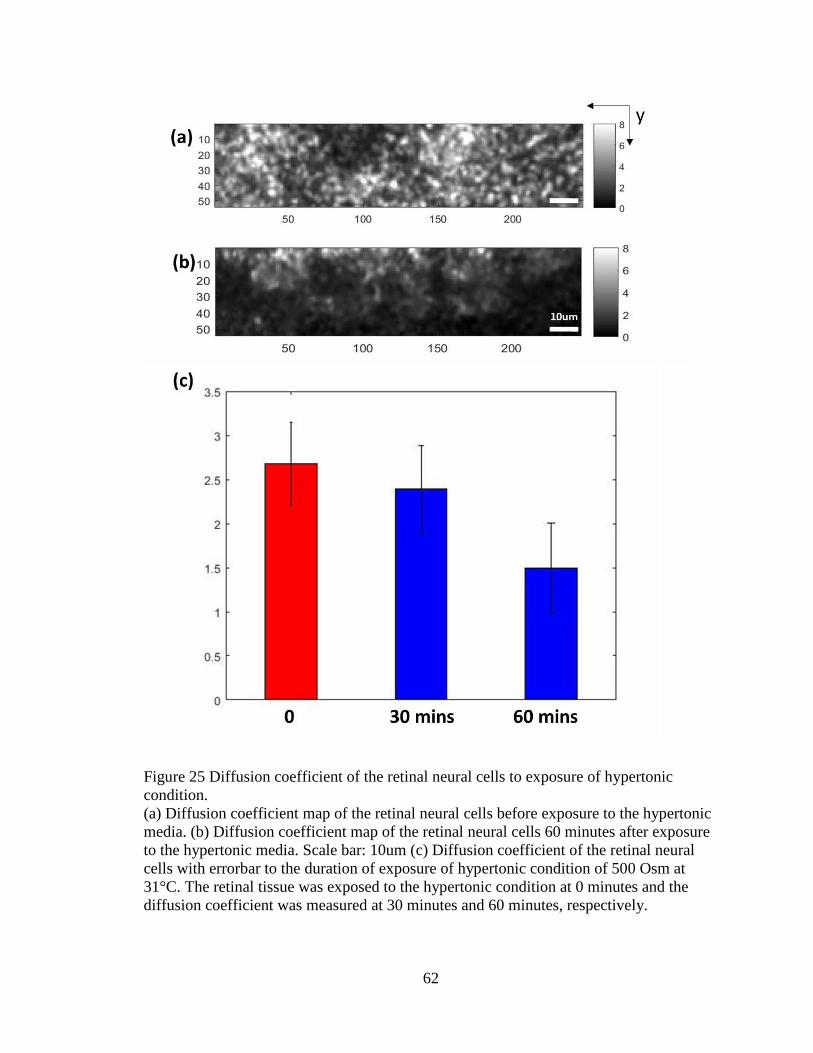
62
Figure 25 Diffusion coefficient of the retinal neural cells to exposure of hypertonic condition. (a) Diffusion coefficient map of the retinal neural cells before exposure to the hypertonic media. (b) Diffusion coefficient map of the retinal neural cells 60 minutes after exposure to the hypertonic media. Scale bar: 10um (c) Diffusion coefficient of the retinal neural cells with errorbar to the duration of exposure of hypertonic condition of 500 Osm at 31°C. The retinal tissue was exposed to the hypertonic condition at 0 minutes and the diffusion coefficient was measured at 30 minutes and 60 minutes, respectively.

63
pH change
pH at controlled condition is maintained at pH 7.5. CO2 level in the media was changed
to increase the pH to 8. Tracking the diffusion coefficient over time showed decrease in
metric. The decreased amount was within the range of error, the decrease is feasible in
that when tissues are exposed to higher pH, oxygen consumption rate decreases thus
slowing the oxygen transport activity [88].
Tracking the diffusion coefficient over time showed decrease in metric when pH
decrease was induced after 10 minutes and remained at the decreased value. Diffusion
coefficient decrease may come from low rate of oxygen consumption in the cells. Also,
highly acidic condition may have destructed the cells, dissolving the organelles. After 1
hour, calcium indicator in the GCaMP mouse was deactivated, also suggesting that cell
functions have been inhibited [89].

64
Figure 26 Diffusion coefficient of the retinal neural cells to exposure of pH 8. (a) Diffusion coefficient map of the retinal neural cells before exposure to the higher pH. (b) Diffusion coefficient map of the retinal neural cells 60 minutes after exposure to the higher pH. Scale bar: 10um (c) Diffusion coefficient of the retinal neural cells with errorbar to the duration of exposure of increased pH of 8 at 31°C. The retinal tissue was exposed to the higher pH at 0 minutes and the diffusion coefficient was measured at 30 minutes and 60 minutes, respectively.

65
Figure 27 Diffusion coefficient of the retinal neural cells to exposure of pH 3.5. (a) Diffusion coefficient map of the retinal neural cells before exposure to the lower pH. (b) Diffusion coefficient map of the retinal neural cells 60 minutes after exposure to the lower pH. (c) Diffusion coefficient map of the retinal neural cells 60 minutes after exposure to the lower pH with different color limits to reveal the cell shapes. Scale bar: 10um. (d)Diffusion coefficient of the retinal neural cells with errorbar to the duration of exposure of decreased pH of 3.5 at 31°C. The retinal tissue was exposed to the lower pH at 0 minutes and the diffusion coefficient was measured at 30 minutes and 60 minutes, respectively.

66
Pilocarpine
Pilocarpine is known to induced epilepsy, and its association with actin cytoskeleton
reorganization has been reported [66]. The actin cytoskeleton reorganization can give rise
to a change in the intracellular motility. Diffusion coefficient change after 30 minutes of
exposure to the 10% pilocarpine in the media was measured and compared to the viable
condition (Figure 28). The diffusion coefficient increased in response to the pilocarpine
exposure, which can arise from the known active movements from actin cytoskeleton.

67
Figure 28 Comparison of diffusion coefficient of the retinal neural cells to the pilocarpine. (a) Diffusion coefficient map of the retinal neural cells at normal condition. (b) Diffusion coefficient map of the retinal neural cells 30 minutes after exposure to the pilocarpine. (c) Red bar indicates the diffusion coefficient of the retinal neural cells at 31°C. Blue bar indicates the diffusion coefficient when tissue is treated with pilocarpine for 30 minutes.

68
Decorrelation
This section presents the result from the decorrelation imaging. Figure 29 shows the
intensity, decorrelation, and relative decorrelation maps of 5 neighboring axial layers are
shown by averaging the layers, and cellular mappings of each metrics.

69
Figure 29 Intensity, decorrelation, and relative decorrelation maps. Intensity (a), decorrelation (b), and relative decorrelation (c) maps of 5 neighboring axial layers are shown by averaging the layers. Scale bar: 10 um. Cellular maps of Intensity (c), decorrelation (d), and relative decorrelation (e) are shown in xy, yz, and xz planes. Scale bar: 5 um.

70
Temperature
Figure 30 shows the intensity, decorrelation, and relative decorrelation response to the
temperature change. The relative decorrelations of the lower temperature were lower in
all cases and time steps. It can be inferred that there were more signal change and/or
movements in the tissue at viable condition of 33°C. More decorrelation is gained over
longer b-scan rate as shown in the figure. However, between 17 ms and 37 ms, there was
little change, thus time steps of 10 ms and 20 ms will be used.
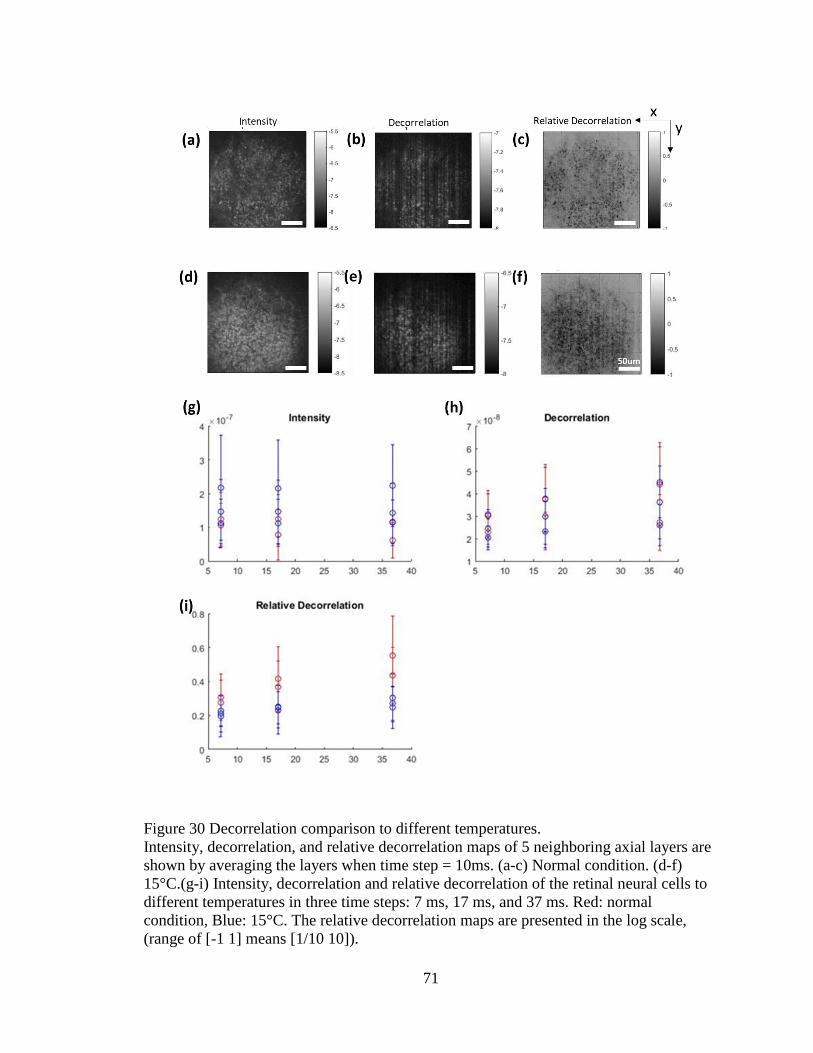
71
Figure 30 Decorrelation comparison to different temperatures. Intensity, decorrelation, and relative decorrelation maps of 5 neighboring axial layers are shown by averaging the layers when time step = 10ms. (a-c) Normal condition. (d-f) 15°C.(g-i) Intensity, decorrelation and relative decorrelation of the retinal neural cells to different temperatures in three time steps: 7 ms, 17 ms, and 37 ms. Red: normal condition, Blue: 15°C. The relative decorrelation maps are presented in the log scale, (range of [-1 1] means [1/10 10]).

72
Ethanol
Figure 31 shows the intensity, decorrelation, and relative decorrelation response to
exposure to the 2.2M ethanol for 40 minutes. The relative decorrelations after exposure to
the ethanol was lower than the viable condition, which agrees with the diffusion
coefficient measurement. However, to investigate if the movement indeed increases in
one hour of exposure, measurements of each metric over a longer duration of time at
frequent interval should be followed.

73
Figure 31 Intensity, decorrelation, and relative decorrelation maps of 5 neighboring axial layers are shown by averaging the layers when time step = 10ms. (a-c) Normal condition. (d-f) after treating with 2.2M alcohol for 30 minutes. (g-i) Intensity, decorrelation and relative decorrelation of the retinal neural cells to different temperatures in two time steps: 10 ms and 20 ms. Red: normal condition, Orange: after treatment. The relative decorrelation maps are presented in the log scale, (range of [-1 1] means [1/10 10]).

74
Osmolarity
Figure 32 shows average of 12 selected cells from relative decorrelation data of each
data set. Relative decorrelation of the tissues’ response to the hypotonic stress over time
is plotted in Figure 32 Relative decorrelation increased over the duration of the hypotonic
condition, which agrees with the diffusion coefficient measurement.

75
Figure 32 Intensity, decorrelation, and relative decorrelation maps of 5 neighboring axial layers are shown by averaging the layers when time step = 10ms. (a-c) Normal condition. (d-f) after being in hypotonic condition for 65 minutes. (g-i) Intensity, decorrelation and relative decorrelation of the retinal neural cells to different temperatures in two time steps: 10 ms and 20 ms over exposure time. Red: normal condition, blue: hypotonic. Here, x-axis represents the exposure time to the hypotonic condition. The relative decorrelation maps are presented in the log scale, (range of [-1 1] means [1/10 10]).

76
pH
Figure 33 shows relative decorrelation data of each data set. Relative decorrelation of the
tissues’ response to the lower pH over time is plotted in Figure 33. Relative decorrelation
decreased after 10 minutes from the exposure and stayed in the lower decorrelation
throughout one hour. This result agrees with the diffusion coefficient measurement as a
response to the extreme acidic condition.

77
Figure 33 Intensity, decorrelation, and relative decorrelation maps of 5 neighboring axial layers are shown by averaging the layers when time step = 10ms. (a-c) Normal condition. (d-f) after exposure to low pH (pH 3.5) condition at 10 minutes. (g-i) Intensity, decorrelation and relative decorrelation of the retinal neural cells to different temperatures in two time steps: 10 ms and 20 ms. Red: normal condition, blue: after exposure to low pH condition at 0, 10, 20, 30, 40, 50, and 60 minutes, respectively. Here, x-axis represents the exposure time to the hypotonic condition. The relative decorrelation maps are presented in the log scale, (range of [-1 1] means [1/10 10]).

78
Pilocarpine
Treatment of 10 % Pilocarpine in the media brought increase in the relative decorrelation.
Under the same treatment, increase in diffusion coefficient was also observed. It can be
inferred that the actin cytoskeleton reorganization brings the signal change, which can
arise from more movement or optical property change in the cells.
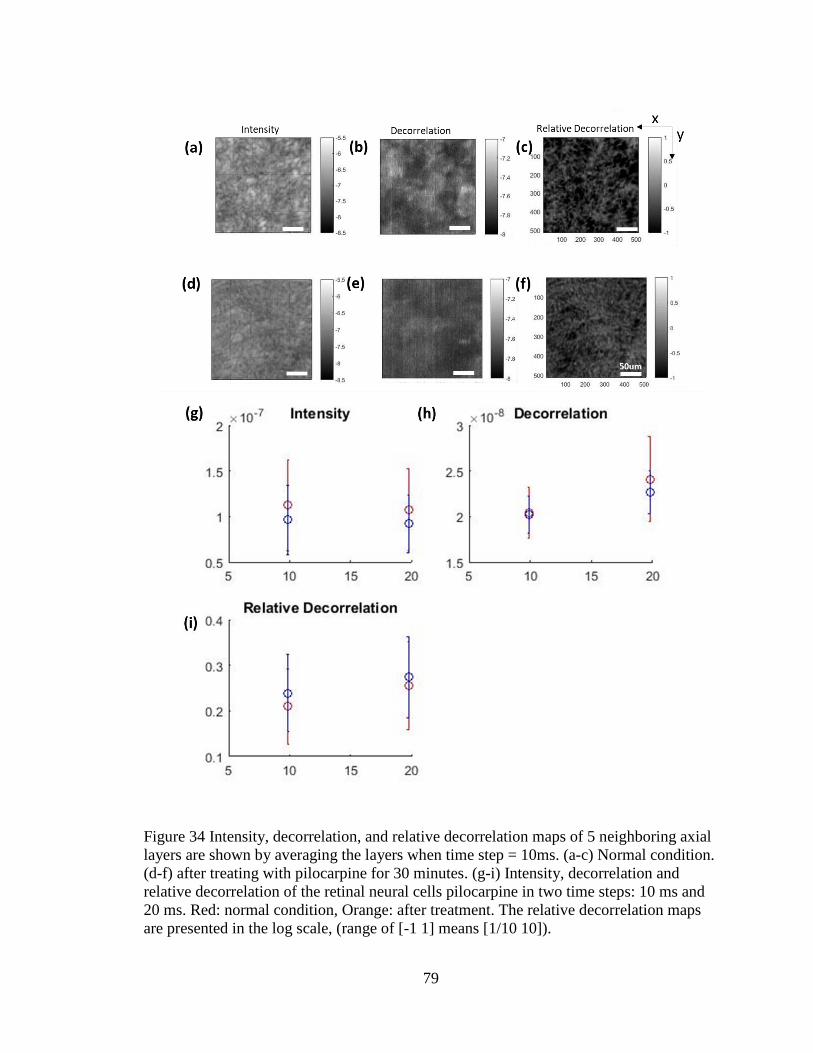
79
Figure 34 Intensity, decorrelation, and relative decorrelation maps of 5 neighboring axial layers are shown by averaging the layers when time step = 10ms. (a-c) Normal condition. (d-f) after treating with pilocarpine for 30 minutes. (g-i) Intensity, decorrelation and relative decorrelation of the retinal neural cells pilocarpine in two time steps: 10 ms and 20 ms. Red: normal condition, Orange: after treatment. The relative decorrelation maps are presented in the log scale, (range of [-1 1] means [1/10 10]).

80
3.4. Conclusions
Diffusion coefficient measurement and mapping was performed using ex vivo retina tissue.
Patterns observed in the D map were consistent with what we expect from the cellular
structure. And simultaneous imaging confirmed that the patterns truly represent
intracellular motility. We were also able to image intracellular motility even through
scattering tissues. When viability is manipulated using the conditions with well-known
effects, IM exhibited consistent changes with the known effect, except the long-term effect
of ethanol. However, the diffusion coefficient measurement at viable condition as a
baseline varied across the tissues, which may have arisen from difference in initial cellular
viability of the tissues. Studying the variance of the viability between samples will
strengthen the effectiveness of the diffusion coefficient measurement. The additional
metric relative decorrelation measurements also changed with changed conditions of the
tissue. Further studying into the matter will aid in understanding the change in cytoplasm
as signal intensity change or active movements inside.

81
4.
CELLULAR VIABILITY
MEASUREMENT OF TISSUE
SPHEROIDS

82
4.1. Introduction
Three-dimensional tissue spheroids are widely used to investigate the living organisms
owing to its benefit of simplifying the system. Measuring the viability of these tissue
spheroids has been in center of attention for many researches [90–93]. From chemical
assays to histology, many techniques have been applied. However, status of art still
requires destructive process to the tissues. Here we test the versatility of DLS-OCT by
investigating its capability in tissue spheroid viability imaging. In addition to the
diffusion coefficient, we test other metrics such as decorrelation and surface area to
volume ratio to gain knowledge of the relationships between viability and spheroid
structure.
Three -dimensional tissue spheroids are extremely useful tool to understand
behavior of organs or tumoroids, thus have been used in numerous studies in cancer
research, drug discovery. Tissue spheroids provide an environment well controlled for
research, compared to the in vivo samples in a host environment. It is also beneficial in
that the testing subject have flexibility in terms of testing procedures or availability
compared to living subjects. Especially when it comes to understanding tumoroids, the
similarity of the three-dimensional tissue spheroids to the tumor cells in vivo, such as the
presence of stroma and extracellular matrix (ECM) lead to the popularity of the tissue
spheroids.

83
Different cell types have different characteristics when they are seeded to form a
spheroid [94–96]. Unlike monolayer tissues which have non-distinctive forms, the shapes
of three-dimensional spheroid can range from round, mass, grape-like to stellate structure
groups [95]. While the shape of microwells in which the cells are seeded can affect the
structure of the tissue the seeded cells form, the seeding number also can affect the formed
shape. Final shape can also change as the day in vitro (DIV) increases, due to gravity,
necrotic core, surface tension, cytoskeleton movement, and debris because of the dead cells.
Cell shape difference as well as its growth and the physiological change including ECM
can result in optical property variance [97]. To study on how different cell types and shapes
are observed under our imaging technique and further image the viability difference, we
use two different types of cells.
Fibroblast
Fibroblast is known to take part in wound healing mechanism and it is one of stromal
cells which synthesizes the ECM and collagen. Being a key component in tumor
development with its relation to the tumor cell metastasis [98–101], proliferation [102]
and maintaining its shape, fibroblast has been a popular subject of study. In this study,
Fibroblast spheroids with seeding number of 1,000 cells/spheroid, 4,000 cells/spheroid,
and, 6,000 cells/spheroid were used (Morgan Lab). DIV 1 and DIV 14 samples were
studied. Fibroblast spheroid samples forms a mass-shaped spheroid with the current
seeding density.

84
HEP G2
Hep G2 is a cell line derived from a human liver carcinoma, which is known to exhibit
various morphological and functional in vitro, thus used for metabolic studies such as
lipoprotein mechanism, cholesterol synthesis and transport, and bile acids
synthesis [103]. In this study, Hep G2 cell spheroids with seeding number of 2,000
cells/spheroid, 4,000 cells/spheroid, and 8,000 cells/spheroid were used (Morgan Lab).
All the samples were DIV 1. Hep G2 spheroid samples forms a mass-shaped spheroid
with the current seeding density, however the shape at DIV 1 diverges from shape of
fibroblast which also forms a mass in that HEP G2 spheroids develop craters in the
center. The crater size differs across different seeding density.
4.2. Experimental methods
Microscopic imaging
Diffusion coefficient and decorrelation measurement methods are consistent with sections
3.3.2 and 3.3.3.
Macroscopic imaging

85
For macroscopic imaging, three -dimensional SD-OCT signal data sized 512 pixels(W) X
512 pixels(H) X 1024 pixels(D) with transverse resolution of 20 um and axial resolution
of 3.46 um were taken using LSM03 lens (Thorlabs). The signal data is then
reconstructed to generate macroscopic three-dimensional data that captures all the wells
in a 5 X 7 microwell (Mircotissues) each sized 800 um in diameter and in height which
holds a volume of 75 ul.
Surface area, volume, and dimensions measurement
A graphical user interface is written in matlab to easily gather the measurements. Once
raw data is reconstructed to obtain three-dimensional intensity data (see section 2.4.4.1),
the intensity array is loaded and shown as maximum intensity plots in all directions in the
Figure 35.

86
Figure 35 Maximum intensity projections in yz (a), xz (b), and x- and y axis (c) of a microwell with tissue spheroid inside. The tissue spheroids shown are HEP G2 spheroids with seeding number of 8,000 at DIV 1. Scale bar : 500um
The data is then cut in axial direction to obtain the three-dimensional array with
tissues inside (Figure 36(a)). Next, the data is cut in transverse direction to disregard any
unnecessary voxels (Figure 36 (b)‚ the maximum intensity plots of the resulting array are

87
shown in the Figure 36 (c-e) to ensure the user that the tissues are not cut in any
direction.
Figure 36 Maximum intensity projections in yz (a) and xy planes (b). The vertical and horizontal line in shows the user where they have chosen to cut eh arrays. Maximum intensity projections in yz (c), xz (d), and xy planes (e) of cropped array. The tissue spheroids shown are HEP G2 spheroids with seeding number of 8,000 at DIV 1. Scale bar: 500 um
With the updated intensity array, a threshold value is selected to make a binary
array of the tissues. Figure 37 (a-b) show the maximum intensity plot of the binary array
when higher threshold is applied. Although unnecessary voxels presenting defects or
debris are disregarded, tissues are also not fully included. When lower threshold is
applied (Figure 37 (c-d), the tissues are enclosed, but many voxels of the agarose

88
microwell structure are included as well. To address this issue, masking technique is
employed.
Figure 37 Maximum intensity projections in xy plane (a,c) and xz plane (b,d) of the binary array obtained from applying a threshold intensity value to select the voxels where tissues are present. High threshold neglects the area where tissue is present (a,b), while low threshold performs poorly in distinguishing the tissue and the microwell. The tissue spheroids shown are HEP G2 spheroids with seeding number of 8,000 at DIV 1. Scale bar: 500 um
From the maximum intensity plot, voxels above the tissues are present are chosen
(Figure 38(a)) to represent the agarose microwell structure (Figure 38 (b-c)) which will
be masked out from the tissue binary array. Thresholds for the intensity are carefully
chosen to obtain the mask while not including the area where tissues are present. The
mask shown in here (Figure 38 (d)) is subtracted from a same sized array of ones, then
multiplied by the tissue array to get the masked-out result.

89
Figure 38 (a) Maximum intensity projections in yz plane where users select the arrays to use as a mask. Maximum intensity projections in xy and yz planes (b,c) of the selected mask array. Binary two-dimensional array obtained from applying a threshold intensity value to select the voxels where microwell is present. The tissue spheroids shown are HEP G2 spheroids with seeding number of 8,000 at DIV 1. Scale bar: 500 um
The resulting array of the masking is shown as maximum intensity plot in Figure
39(a-b). There are still defects that are not masked out with the process. The user can
remove the unwanted defects manually by selecting rectangular area from the plot
(Figure 39 (c)), which is also achievable with y- and z- plot. Note that two tissues in the
far-right column are also manually masked. Due to confocal effect, tissues in the edge of
the field of view are sometimes not captured with the comparably high intensity as the

90
other tissues are. These tissues are removed from the array to get an accurate measure of
the data.
Figure 39 Maximum intensity projection of tissue array in xy plane (a) and xz plane (b) after masking is applied. Manual selecting of the masking shown in maximum intensity projection of tissue array in xy plane (c). The tissue spheroids shown are HEP G2 spheroids with seeding number of 8,000 at DIV 1. Scale bar: 500 um
Resulting array is shown in Figure 40 (a-b). From this binary array, number of
ones are counted and multiplied by the voxel size (20 um X 20 um X 3.46 um) to

91
calculate the volume of all tissues. The tissue counts are also recorded to obtain average
volume of the individual cell.
Figure 40 Maximum intensity projection of tissue array in xy plane (a) and xz plane (b) in final form. The tissue spheroids shown are HEP G2 spheroids with seeding number of 8,000 at DIV 1. Scale bar : 500um
Surface area is calculated by counting the differential sign change of the binary
array. Differential array of the current binary array is obtained. Differential will occur
when there is change in value, which can be interpreted as either tissue to ambient fluid
or ambient fluid to tissue, thus the surface area of the tissue. These changes are summed
and multiplied by the diagonal length of a pixel in x- and z- direction. Figure 41 shows en
face images of the differential array at 138 um from the of the top of the array, in the
center, and 138 um from the of the bottom of the array, respectively.

92
Figure 41 En face images of the differential array at 138 um from the of the top of the array, in the center, and 138 um from the of the bottom of the array, respectively (a-c).
To investigate if the tissues form a spherical shape, height to width ratio (HWR)
of the individual tissues were also measured. Users select center of a tissue spheroid, then
two maximum intensity plots in en face and b-scan appear where two ends of width
(Figure 42(b)) and height ((Figure 42 (c)) are chosen. Eight tissue spheroids are chosen
per each set.
Figure 42 Maximum intensity projection of tissue array in xy plane (a) where a spheroid is selected. Selected tissue is shown in (b-c) as maximum intensity projections of tissue

93
array in xy and yz planes. The two windows collect the height and width data from manual selection of the user. The tissue spheroids shown are HEP G2 spheroids with seeding number of 8,000 at DIV 1. Scale bar: 500um
4.3. Results and discussions
Viability metric observations
Fibroblast
Diffusion coefficient and decorrelation measurement were performed with tissue
spheroids. Figure 43 shows intensity, decorrelation, and relative decorrelation of the
fibroblast tissue at the equator of three different seeding numbers. In all cases, individual
cells are not seen when the focal depth is in the equator. Cell shapes are seen in the rim of
the spheroids, where the depth of tissue for light penetration is the smallest.

94
Figure 43 Maximum intensity projection of individual spheroids with different seeding numbers in yz plane. Maximum projections of 5 neighboring layers (17.3 um) at tissue equator of intensity, decorrelation, and relative decorrelation are shown in the column below each seeding numbers. The tissue spheroids shown are fibroblast spheroids with seeding numbers of 1,000 (a-d), 4,000 (e-h), and 16,000 (i-l) at DIV 1. Scale bar: 100 um
Figure 44 shows the intensity, decorrelation, and relative decorrelation of the
same data, but at different axial position (14 pixels (47.6 um) from Figure 44(i-l)). Due to
the confocal effect, the focus is in the rim of the spheroid (Figure 44(a)). Cell shape in the
rim where the intensity is the highest appears in all metrics. Mapping of the three metrics
in retinal tissue layers (Chapter.3) demonstrated the cell existence when the penetration
depth was up to 200 um. The largest spheroid has diameter around 400 um, thus at the
equator, penetration depth is 200 um, comparable to the retinal tissue layer penetration
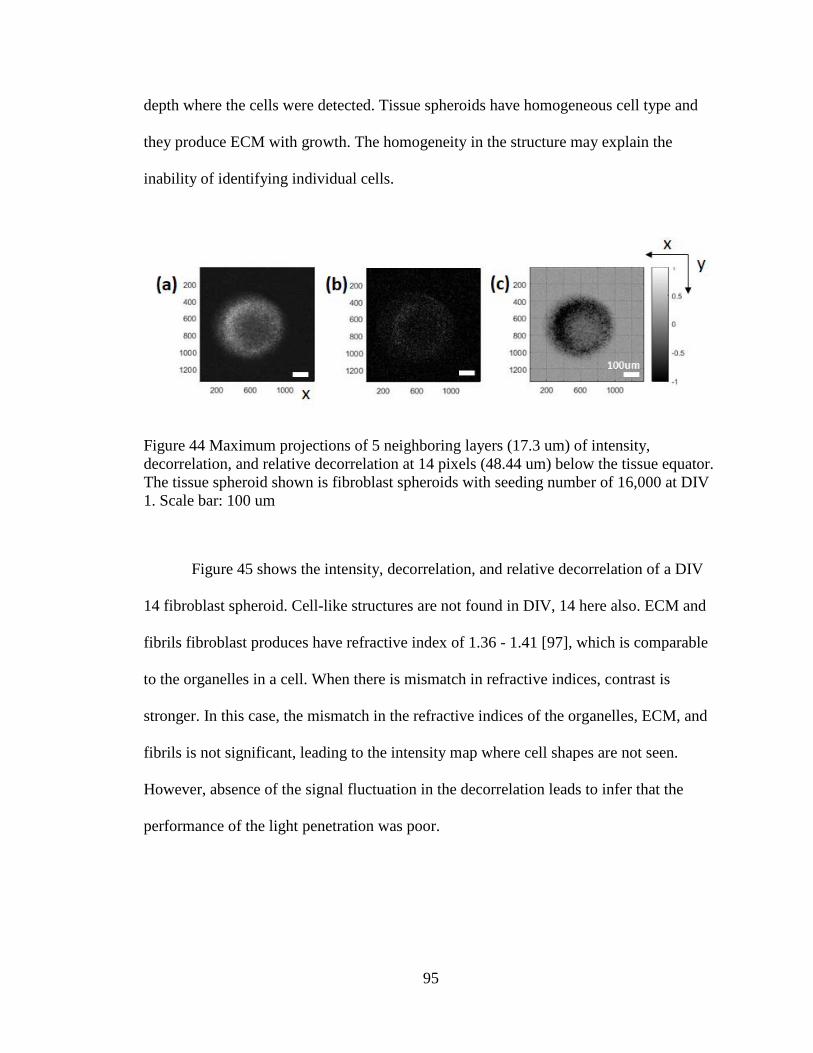
95
depth where the cells were detected. Tissue spheroids have homogeneous cell type and
they produce ECM with growth. The homogeneity in the structure may explain the
inability of identifying individual cells.
Figure 44 Maximum projections of 5 neighboring layers (17.3 um) of intensity, decorrelation, and relative decorrelation at 14 pixels (48.44 um) below the tissue equator. The tissue spheroid shown is fibroblast spheroids with seeding number of 16,000 at DIV 1. Scale bar: 100 um
Figure 45 shows the intensity, decorrelation, and relative decorrelation of a DIV
14 fibroblast spheroid. Cell-like structures are not found in DIV, 14 here also. ECM and
fibrils fibroblast produces have refractive index of 1.36 - 1.41 [97], which is comparable
to the organelles in a cell. When there is mismatch in refractive indices, contrast is
stronger. In this case, the mismatch in the refractive indices of the organelles, ECM, and
fibrils is not significant, leading to the intensity map where cell shapes are not seen.
However, absence of the signal fluctuation in the decorrelation leads to infer that the
performance of the light penetration was poor.

96
Figure 45 Maximum projections of 5 neighboring layers (17.3 um) of intensity, decorrelation, and relative decorrelation at the tissue equator. The tissue spheroid shown is fibroblast spheroids with seeding number of 8,000 at DIV 14. Scale bar: 100 um
HEP G2
Figure 46 shows intensity, decorrelation, and relative decorrelation of the HEP G2
tissue at the equator of three different seeding numbers. Individual cells are seen when
the focal depth is in the equator. Similar to the fibroblast tissues, cell shapes are seen
better in the rim of the spheroids, where the depth of tissue for light penetration is the
smallest.

97
Figure 46 Maximum intensity projection of individual spheroids with different seeding numbers in yz plane. Maximum projections of 5 neighboring layers (17.3 um) at tissue equator of intensity, decorrelation, and relative decorrelation are shown in the column below each seeding numbers. The tissue spheroids shown are fibroblast spheroids with seeding numbers of 1,000 (a-d), 4,000 (e-h), and 16,000 (i-l) at DIV 1. Scale bar: 100 um
HEP G2 tissues do not form a sphere, rather it forms a shape with comparatively
flat top. Figure 47 shows the intensity, decorrelation, and relative decorrelation of a DIV
1 HEP G2 spheroid. Cell shapes are detected in all three metrics at the surface.
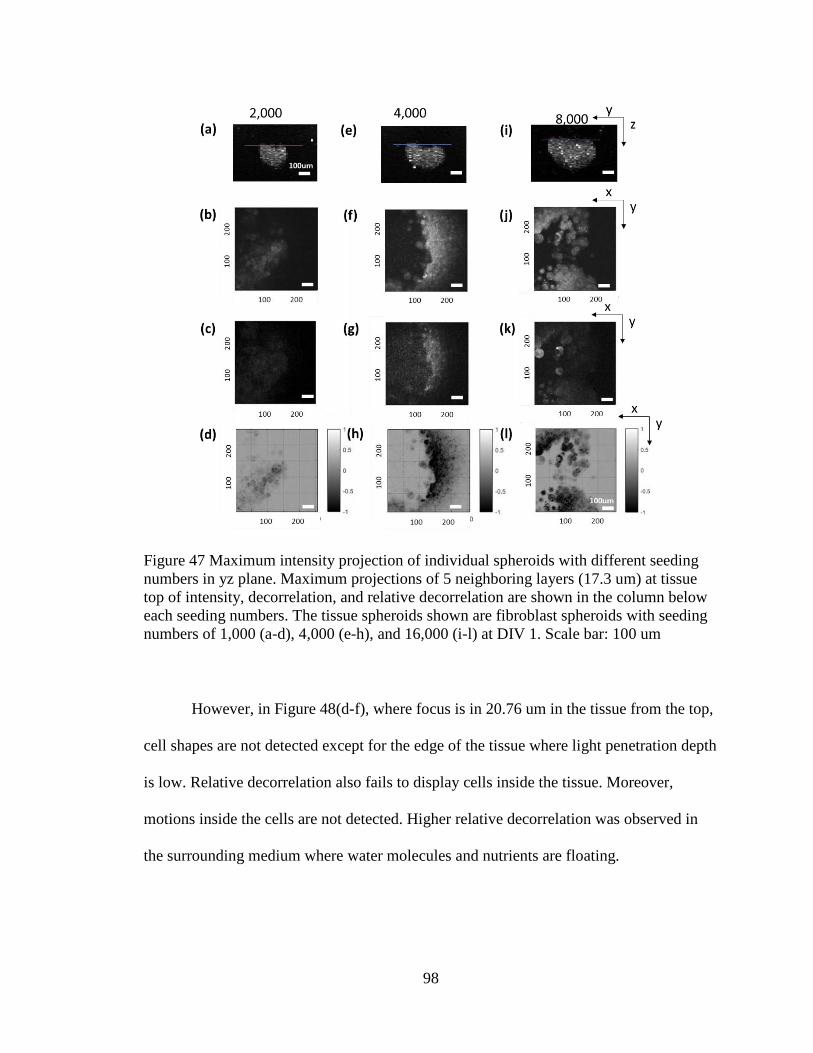
98
Figure 47 Maximum intensity projection of individual spheroids with different seeding numbers in yz plane. Maximum projections of 5 neighboring layers (17.3 um) at tissue top of intensity, decorrelation, and relative decorrelation are shown in the column below each seeding numbers. The tissue spheroids shown are fibroblast spheroids with seeding numbers of 1,000 (a-d), 4,000 (e-h), and 16,000 (i-l) at DIV 1. Scale bar: 100 um
However, in Figure 48(d-f), where focus is in 20.76 um in the tissue from the top,
cell shapes are not detected except for the edge of the tissue where light penetration depth
is low. Relative decorrelation also fails to display cells inside the tissue. Moreover,
motions inside the cells are not detected. Higher relative decorrelation was observed in
the surrounding medium where water molecules and nutrients are floating.

99
Figure 48 Maximum projections of 5 neighboring layers (17.3 um) of intensity, decorrelation, and relative decorrelation at the surface of the tissue and at 6 pixels (20.76 um) below the tissue surface. The tissue spheroid shown is HEP G2 spheroids with seeding number of 8,000 at DIV 1. Scale bar : 100 um
Morphological observations
Fibroblast Spheroids
Three different seeding numbers yielded difference in sizing, moreover, DIV 24 samples
showed difference in the shapes due to the debris around the spheroid. Figure 49 (a-b, c-
d, e-f) shows maximum intensity projections in xy and yz planes of a fibroblast spheroid
with seeding number of 1,000, 4,000, and 16,000 at DIV 1.

100
Figure 49 Maximum intensity projection of individual spheroids with different seeding numbers in xy and yz planes. The tissue spheroids shown are fibroblast spheroids with seeding numbers of 1,000 (a-b), 4,000 (c-d), and 16,000 (e-f) at DIV 1. Scale bar: 100 um
Figure 50(a-b) shows maximum intensity projections in xy and yz plane of a fibroblast
spheroid with seeding number of 16,000 and DIV 1. Figure 50 (c-d) shows maximum
intensity projections in xy and yz plane of a fibroblast spheroid with seeding number of
16,000 and DIV 14. DIV 1 spheroid is a prolate ellipsoid without any debris around the
spheroid whereas DIV 14 spheroid has a main body that is spherical and has a band around
the bottom of the spheroid that is debris from the main body.

101
Figure 50 Maximum intensity projection of individual spheroids with different DIV in xy and xz planes. The tissue spheroids shown are fibroblast spheroids with seeding numbers of 16,000 at DIV 1 (a-b) and DIV 14 (c-d). Scale bar: 100um
Surface area to volume ratio
Surface area to volume ratio (SVR) in 1/mm^3 of the fibroblast tissue spheroids of
different seeding numbers per individual tissue spheroids are shown in Fig. #. Two
separate samples of DIV 1 do behave similarly. However, there deviation is large. SVR
does not decrease linearly, due to its proportionality to the inverse of diameter of a
spheroid. To better understand the morphology of the tissue, Height to width ratio
(HWR) is measured.

102
Figure 51 Surface area to volume ratio with error bars of DIV 1 and DIV 14 samples to the seeding numbers (1,000, 4,000 and 16,000). Red and blue: DIV 1 of separate samples. Yellow: DIV 14 samples
HWR of the spheroids are plotted to investigate the shape of the spheroid. A
Perfect spheroid have HWR of 1. The HWR plotted shows that the tissues are prolate
ellipsoids except for the 2,000 spheroids with DIV 14 which may be an oblate ellipsoid.
In all seeding numbers, HWRs of DIV 14 were lower than that of DIV 1. This can be due
to the shape change (Figure 52) from having debris around the main body which became
spherical. The shapes of the tissue spheroids are determined by many factors such as
surface tension from interaction of the neighboring cells [104], the effect from
cytoskeleton [105], as well as the capability of the penetration of the nutrients. Fibroblast

103
cell type formed prolate ellipsoid in most cases to optimize the mechanical as well as
biological conditions [106].
Figure 52 Height to width ratio (HWR) to seeding numbers. The average HWR is fitted across the seeding numbers within the set of samples.
Individual tissue volume
Individual tissue volumes per each seeding numbers are plotted and fitted using least
square fit (Figure 53). Single spheroid volumes were higher in DIV 1 in larger seeding
numbers (4,000 and 16,000). Single volume for seeding number of 2,000 was higher in
older sample (DIV 14). General trend in all the set of samples showed a linear
relationship to the seeding numbers. However, the slope of the fitted lines for DIV 24
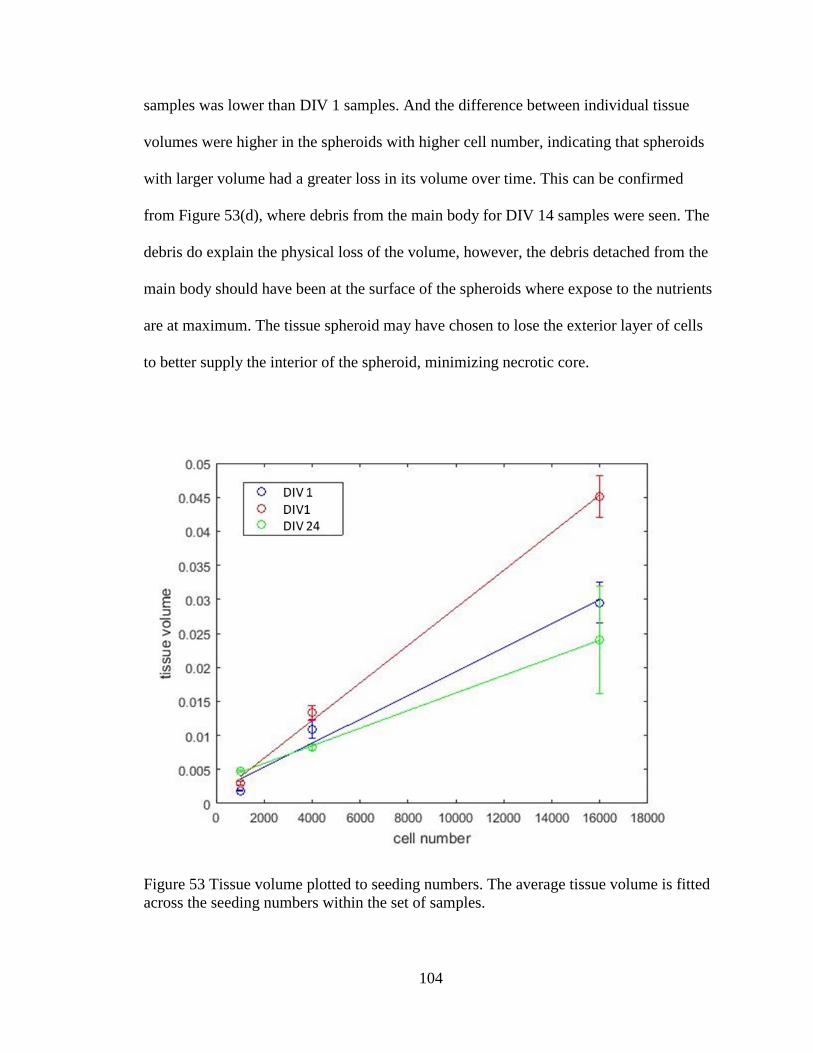
104
samples was lower than DIV 1 samples. And the difference between individual tissue
volumes were higher in the spheroids with higher cell number, indicating that spheroids
with larger volume had a greater loss in its volume over time. This can be confirmed
from Figure 53(d), where debris from the main body for DIV 14 samples were seen. The
debris do explain the physical loss of the volume, however, the debris detached from the
main body should have been at the surface of the spheroids where expose to the nutrients
are at maximum. The tissue spheroid may have chosen to lose the exterior layer of cells
to better supply the interior of the spheroid, minimizing necrotic core.
Figure 53 Tissue volume plotted to seeding numbers. The average tissue volume is fitted across the seeding numbers within the set of samples.
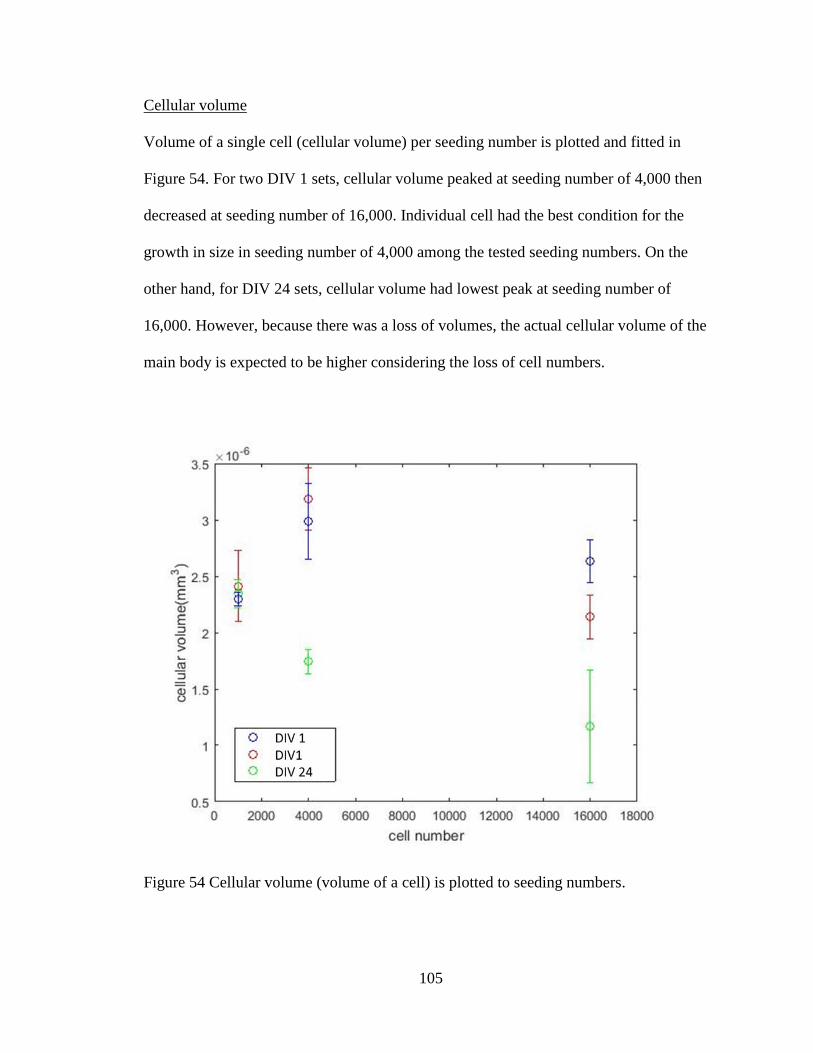
105
Cellular volume
Volume of a single cell (cellular volume) per seeding number is plotted and fitted in
Figure 54. For two DIV 1 sets, cellular volume peaked at seeding number of 4,000 then
decreased at seeding number of 16,000. Individual cell had the best condition for the
growth in size in seeding number of 4,000 among the tested seeding numbers. On the
other hand, for DIV 24 sets, cellular volume had lowest peak at seeding number of
16,000. However, because there was a loss of volumes, the actual cellular volume of the
main body is expected to be higher considering the loss of cell numbers.
Figure 54 Cellular volume (volume of a cell) is plotted to seeding numbers.

106
HEP G2 spheroids
Three different seeding numbers yielded difference in sizing. Figure 55(a-b, c-d, e-f)
shows maximum intensity projections in xy and yz planes of a fibroblast spheroid with
seeding number of 2,000, 4,000, and 8,000 at DIV 1. Hep G2 spheroids are oblate across
all the seeding numbers with a crater in the center shaping itself into a bowl-like
structure. The crater was largest in the spheroid with seeding number of 8,000.
Figure 55 Maximum intensity projection of individual spheroids with different seeding numbers in xy and yz planes. The tissue spheroids shown are HEP G2 spheroids with seeding numbers of 2,000 (a-b), 4,000 (c-d), and 8,000 (e-f) at DIV 1. Scale bar: 100um
Surface area to volume ratio
Surface area to volume ratio (SVR) in 1/mm3 of the HEP G2 tissue spheroids of different
seeding numbers per individual tissue spheroids are shown in Figure 56. SVR does not
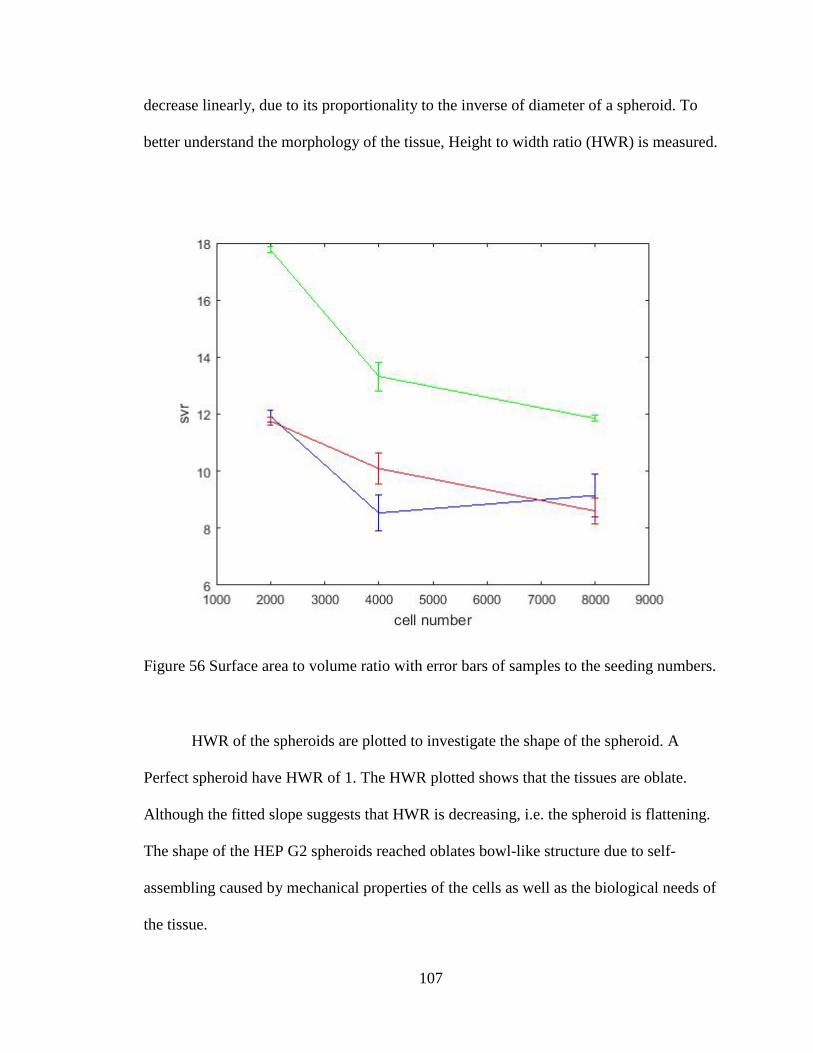
107
decrease linearly, due to its proportionality to the inverse of diameter of a spheroid. To
better understand the morphology of the tissue, Height to width ratio (HWR) is measured.
Figure 56 Surface area to volume ratio with error bars of samples to the seeding numbers.
HWR of the spheroids are plotted to investigate the shape of the spheroid. A
Perfect spheroid have HWR of 1. The HWR plotted shows that the tissues are oblate.
Although the fitted slope suggests that HWR is decreasing, i.e. the spheroid is flattening.
The shape of the HEP G2 spheroids reached oblates bowl-like structure due to self-
assembling caused by mechanical properties of the cells as well as the biological needs of
the tissue.

108
Figure 57 Height to width ratio (HWR) to seeding numbers. The average HWR is fitted across the seeding numbers within the set of samples.
Individual tissue volume
Individual tissue volumes per each seeding numbers are plotted and fitted using least
square fit (Figure 58). General trend in all the set of samples showed a linear relationship
to the seeding numbers. Slope of the fitting across different set of samples also matched.
One sample with seeding number of 4,000 behaves disparately with a very high standard
deviation.

109
Figure 58 Tissue volume (mm3) is plotted to seeding numbers. The average tissue volume is fitted across the seeding numbers within the set of samples.
Cellular volume
Volume of a single cell (cellular volume) per seeding number is plotted and fitted in
Figure 59. Cellular volume change in HEP G2 cells did not have a significant trend
within the sets of samples we measured.

110
Figure 59 Cellular volume (mm3, volume of a cell) is plotted to seeding numbers. The average cellular volume is fitted across the seeding numbers within the set of samples.
4.4. Conclusions
Two types of homogeneous tissue spheroids were studied to understand the applicability
of the SD-OCT. Our metric of interest diffusion coefficient and decorrelation was used to
quantify the viability of the tissue spheroid. Although penetrating depth of the
homogeneous tissues are comparatively low compared to the retinal tissue, the metrics
gave insights to the viability of the cells at the surface of the tissue. Further investigation
using our technique may reveal adaptable applicability. Combining this result with

111
histology and fluorescence microscopy will further guide us to the linkage of our metrics
to the conventional methods.
Morphological observation using SD-OCT revealed the adaptability for
measuring volumetric and surficial measures of the tissues. Our method of measurement
dismisses biased measurement done by directly measuring. On top of gathering the
volumetric information at once, it will further improve the accuracy of the measurements
by automating most of analysis.
Overall, measuring volumetric and surficial metric was enabled using our label-
free and noninvasive technique. Viability imaging was not as successful as ex vivo tissue,
nevertheless, it guided us in understanding the surficial cellular viability.

112
5.
CONCLUSION

113
In this study, our newly developed label-free, noninvasive technique using SD-OCT, dynamic light
scattering optical coherence tomography (DLS-OCT) was optimized and validated for accurate 3D
mapping of diffusion coefficient without any labelling. We demonstrated cellular viability imaging
of retinal neural cells and tissue spheroids, as well as volumetric and surficial measurement on
geography of tissue spheroids.
To validate that our diffusion coefficient measurement of intracellular motility
indeed represents cellular viability, we triggered the change of the health in the sample by
changing the environmental conditions and releasing certain chemicals known to affect the
organelles and the structure of the cytoplasm. We saw change when such change was
induced, when the condition change to the viability was mild, the diffusion coefficient
change was also subtle. When change to the viability was harsh, the diffusion coefficient
change was also large. Further study in linking the cytoplasmic change known with the
diffusion coefficient will guide us in understanding the motional change each cytoplasmic
change induces. Decorrelation was also studied using various conditions induced.
Diffusion coefficient and decorrelation change (relative decorrelation, specifically) have
similarity although the absolute comparison was not available. Here, intracellular motility
(IM) was quantitatively imaged with cellular resolution through scattering tissue and
validated to represent cellular viability.
Measurement of geographical and volumetric metric of tissues is also promising in
that it is done without destructing the tissues. Using our technique, longitudinal study of
specific tissue spheroids will be possible. This will fortify the observations we saw

114
statistically. Similar to biological tissue, when viability was manipulated, IM exhibited a
decrease as expected from the known effect. In addition, morphological measurement using
SD-OCT revealed the adaptability for measuring volumetric and surficial measures of the
tissues. Therefore, it can be concluded that the viability of the engineered tissue can also
be assessed through IM as well as morphological measurement.
To Conclude, this research has addressed the current limitations in the existing methods
for tissue viability assessment. The technique developed through and findings obtained
from this research are expected to address the need to nondestructively measure the
viability of biological and engineered tissue. Such nondestructive viability assessment
will have a range of applications, from disease studies to drug discovery and tissue
production.

115
REFERENCES
1. R. M. Berne, B. M. Koeppen, and B. A. Stanton, Berne & Levy Physiology
(Mosby/Elsevier, 2010).
2. O. Kepp, L. Galluzzi, M. Lipinski, J. Yuan, and G. Kroemer, "Cell death assays for
drug discovery," Nat. Rev. Drug Discov. 10, 221–237 (2011).
3. D. T. Vistica, P. Skehan, D. Scudiero, A. Monks, A. Pittman, and M. R. Boyd,
"Tetrazolium-based Assays for Cellular Viability: A Critical Examination of
Selected Parameters Affecting Formazan Production," CANCER Res. 51, 2515–
2520 (1991).
4. S. R. Khetani and S. N. Bhatia, "Microscale culture of human liver cells for drug
development," Nat. Biotechnol. 26, 120–126 (2008).
5. A. Ridley and R. Heald, "Cell structure and dynamics," Curr. Opin. Cell Biol. 23,
1–3 (2011).
6. C. M. Waterman-Storer and E. D. Salmon, "Actomyosin-based Retrograde Flow of
Microtubules in the Lamella of Migrating Epithelial Cells Influences Microtubule
Dynamic Instability and Turnover and Is Associated with Microtubule Breakage and
Treadmilling," J. Cell Biol. 139, 417–434 (1997).
7. C. Joo, C. L. Evans, T. Stepinac, T. Hasan, and J. F. de Boer, "Diffusive and
directional intracellular dynamics measured by field-based dynamic light
scattering," Opt. Express 18, 2858 (2010).

116
8. K. Jacobson and J. Wojcieszyn, "The translational mobility of substances within the
cytoplasmic matrix.," Proc. Natl. Acad. Sci. U. S. A. 81, 6747–6751 (1984).
9. P. A. Valberg and H. A. Feldman, "Magnetic particle motions within living cells.
Measurement of cytoplasmic viscosity and motile activity.," Biophys. J. 52, 551–61
(1987).
10. D. Merrill, R. An, J. Turek, and D. D. Nolte, "Digital holography of intracellular
dynamics to probe tissue physiology.," Appl. Opt. 54, A89-97 (2015).
11. P. A. Valberg and D. F. Albertini, "Cytoplasmic motions, rheology, and structure
probed by a novel magnetic particle method.," J. Cell Biol. 101, 130–40 (1985).
12. D. D. Nolte, R. An, J. Turek, and K. Jeong, "Holographic tissue dynamics
spectroscopy.," J. Biomed. Opt. 16, 87004 (2011).
13. B. Herman, "A time-lapse video image intensification analysis of cytoplasmic
organelle movements during endosome translocation," J. Cell Biol. 98, 565–576
(1984).
14. C. P. Brangwynne, F. C. MacKintosh, and D. A. Weitz, "Force fluctuations and
polymerization dynamics of intracellular microtubules.," Proc. Natl. Acad. Sci. U.
S. A. 104, 16128–33 (2007).
15. Y. Tseng, T. P. Kole, and D. Wirtz, "Micromechanical mapping of live cells by
multiple-particle-tracking microrheology," Biophys. J. 83, 3162–3176 (2002).
16. P. Panorchan, J. S. H. Lee, T. P. Kole, Y. Tseng, and D. Wirtz, "Microrheology and
ROCK signaling of human endothelial cells embedded in a 3D matrix.," Biophys. J.
91, 3499–507 (2006).

117
17. K. Luby-phelps, P. E. Castles, D. L. Taylort, and F. Lannit, "Hindered diffusion of
inert tracer particles in the cytoplasm of mouse 3T3 cells," 84, 4910–4913 (1987).
18. K. Luby-Phelps, D. L. Taylor, and F. Lanni, "Probing the structure of cytoplasm," J.
Cell Biol. 102, 2015–2022 (1986).
19. E. L. Elson and D. Magde, "Fluorescence correlation spectroscopy. I. Conceptual
basis and theory," Biopolymers 13, 1–27 (1974).
20. H. Chen, E. R. Farkas, and W. W. Webb, "Chapter 1 In Vivo Applications of
Fluorescence Correlation Spectroscopy," Methods Cell Biol. 89, 3–35 (2008).
21. M. Prummer, D. Kling, V. Trefzer, T. Enderle, S. Zoffmann, and M. Prunotto, "A
random motility assay based on image correlation spectroscopy.," Biophys. J. 104,
2362–72 (2013).
22. M. Guo, A. J. Ehrlicher, M. H. Jensen, M. Renz, J. R. Moore, R. D. Goldman, J.
Lippincott-Schwartz, F. C. Mackintosh, and D. A. Weitz, "Probing the stochastic,
motor-driven properties of the cytoplasm using force spectrum microscopy," Cell
158, 822–832 (2014).
23. A. L. Oldenburg, R. K. Chhetri, J. M. Cooper, W.-C. Wu, M. A. Troester, and J. B.
Tracy, "Motility-, autocorrelation-, and polarization-sensitive optical coherence
tomography discriminates cells and gold nanorods within 3D tissue cultures," Opt.
Lett. 38, 2923 (2013).
24. A. L. Oldenburg, X. Yu, T. Gilliss, O. Alabi, R. M. Taylor, and M. A. Troester,
"Inverse-power-law behavior of cellular motility reveals stromal–epithelial cell
interactions in 3D co-culture by OCT fluctuation spectroscopy," Optica 2, 877

118
(2015).
25. C. Apelian, F. Harms, O. Thouvenin, and A. C. Boccara, "Dynamic full field optical
coherence tomography: subcellular metabolic contrast revealed in tissues by
interferometric signals temporal analysis," Biomed. Opt. Express 7, 1511 (2016).
26. N. T. Shaked and A. Wax, "Quantitative Phase Microscopy of Live Biological Cell
Dynamics," in Advanced Phase Measurement Methods in Optics and Imaging
(Springer Berlin Heidelberg, 2010), pp. 283–288.
27. G. Popescu, Y. Park, W. Choi, R. R. Dasari, M. S. Feld, and K. Badizadegan,
"Imaging red blood cell dynamics by quantitative phase microscopy," Blood Cells,
Mol. Dis. 41, 10–16 (2008).
28. T. Yamauchi, H. Iwai, and Y. Yamashita, "Label-free imaging of intracellular
motility by low-coherent quantitative phase microscopy," Opt. Express 19, 5536
(2011).
29. D. B. Sattelle, D. J. Green, and K. H. Langley, "Subcellular Motions in Nitella
flexilis studied by Photon Correlation Spectroscopy," Phys. Scr. 19, 471–475 (1979).
30. J. Lee, W. Wu, J. Y. Jiang, B. Zhu, and D. A. Boas, "Dynamic light scattering optical
coherence tomography," Opt. Express 20, 22262–22277 (2012).
31. J. Lee, H. Radhakrishnan, W. Wu, A. Daneshmand, M. Climov, C. Ayata, and D. A.
Boas, "Quantitative imaging of cerebral blood flow velocity and intracellular
motility using dynamic light scattering-optical coherence tomography," J. Cereb.
Blood Flow Metab. 33, 819–825 (2013).
32. S. Marlar, E. C. Arnspang, G. A. Pedersen, J. S. Koffman, and L. N. Nejsum,

119
"Measuring localization and diffusion coefficients of basolateral proteins in lateral
versus basal membranes using functionalized substrates and kICS analysis,"
Biochim. Biophys. Acta - Biomembr. 1838, 2404–2411 (2014).
33. a I. García-Pérez, E. A. López-Beltrán, P. Klüner, J. Luque, P. Ballesteros, and S.
Cerdán, "Molecular crowding and viscosity as determinants of translational
diffusion of metabolites in subcellular organelles.," Arch. Biochem. Biophys. 362,
329–38 (1999).
34. J. W. Wojcieszyn, R. A. Schlegelt, E.-S. Wut, and K. A. Jacobson, "Diffusion of
injected macromolecules within the cytoplasm of living cells (erythrocyte-mediated
microinjection/photobleaching/human fibroblast/cytoplasmic
viscosity/cytoskeleton)," Cell Biol. 78, 4407–4410 (1981).
35. G. L. Lukacs, P. Haggie, O. Seksek, D. Lechardeur, N. Freedman, and A. S.
Verkman, "Size-dependent DNA mobility in cytoplasm and nucleus," J. Biol. Chem.
275, 1625–1629 (2000).
36. R. Brock, M. A. Hink, and T. M. Jovin, "Fluorescence correlation microscopy of
cells in the presence of autofluorescence.," Biophys. J. 75, 2547–57 (1998).
37. S. Yazdanfar, A. M. Rollins, and J. A. Izatt, "In Vivo Imaging of Human Retinal
Flow Dynamics by Color Doppler Optical Coherence Tomography," Arch.
Ophthalmol. 121, 235 (2003).
38. J. A. Izatt, M. D. Kulkarni, S. Yazdanfar, J. K. Barton, and A. J. Welch, "In vivo
bidirectional color Doppler flow imaging of picoliter blood volumes using optical
coherence tomography," Opt. Lett. 22, 1439 (1997).

120
39. Y. Chen, A. D. Aguirre, L. Ruvinskaya, A. Devor, D. A. Boas, and J. G. Fujimoto,
"Optical coherence tomography (OCT) reveals depth-resolved dynamics during
functional brain activation," J. Neurosci. Methods 178, 162–173 (2009).
40. S. B. Ploner, E. M. Moult, W. Choi, N. K. Waheed, B. Lee, E. A. Novais, E. D. Cole,
B. Potsaid, L. Husvogt, J. Schottenhamml, A. Maier, P. J. Rosenfeld, J. S. Duker, J.
Hornegger, and J. G. Fujimoto, "TOWARD QUANTITATIVE OPTICAL
COHERENCE TOMOGRAPHY ANGIOGRAPHY," Retina 36, S118–S126 (2016).
41. D. Jaque, L. Martínez Maestro, B. del Rosal, P. Haro-Gonzalez, A. Benayas, J. L.
Plaza, E. Martín Rodríguez, J. García Solé, B. Hildebrandt, P. Wust, O. Ahlers, A.
Dieing, G. Sreenivasa, T. Kerner, R. Felix, H. Riess, J. L. R. Roti, A. Chicheł, J.
Skowronek, M. Kubaszewska, M. Kanikowski, P. Wust, B. Hildebrandt, G.
Sreenivasa, B. Rau, J. Gellermann, H. Riess, R. Felix, P. M. Schlag, A. Burlaka, S.
Lukin, S. Prylutska, O. Remeniak, Y. Prylutskyy, M. Shuba, S. Maksimenko, U.
Ritter, P. Scharff, N. P. O’Grady, P. S. Barie, J. G. Bartlett, T. Bleck, G. Garvey, J.
Jacobi, P. Linden, D. G. Maki, M. Nam, W. Pasculle, J. Van der Zee, R. W. Habash,
R. Bansal, D. Krewski, H. T. Alhafid, R. W. Habash, R. Bansal, D. Krewski, H. T.
Alhafid, M. Johannsen, U. Gneveckow, L. Eckelt, A. Feussner, N. Waldofner, R.
Scholz, S. Deger, P. Wust, S. A. Loening, A. Jordan, J. Mendecki, E. Friedenthal,
C. Botstein, R. Paglione, F. Sterzer, G. Hegyi, G. P. Szigeti, A. Szasz, F. Zhou, D.
Xing, Z. Ou, B. Wu, D. E. Resasco, W. R. Chen, J. W. Fisher, S. Sarkar, C. F.
Buchanan, C. S. Szot, J. Whitney, H. C. Hatcher, S. V. Torti, C. G. Rylander, M. N.
Rylander, T. Hironobu, N. Takuro, N. Ayuko, N. Yasuro, Y. Sunao, C. W. Song, S.
S. Chen, J. Humphrey, J. R. Oleson, T. V. Samulski, K. A. Leopold, S. T. Clegg, M.

121
W. Dewhirst, R. K. Dodge, S. L. George, R. W. Habash, R. Bansal, D. Krewski, H.
T. Alhafid, P. Chakravarty, R. Marches, N. S. Zimmerman, A. D. Swafford, P. Bajaj,
I. H. Musselman, P. Pantano, R. K. Draper, E. S. Vitetta, R. W. Habash, R. Bansal,
D. Krewski, H. T. Alhafid, S. V. Torti, F. Byrne, O. Whelan, N. Levi, B. Ucer, M.
Schmid, F. M. Torti, S. Akman, J. Liu, P. M. Ajayan, O. Nalamasu, D. L. Carroll,
S. N. Goldberg, M. C. Stein, G. S. Gazelle, R. G. Sheiman, J. B. Kruskal, M. E.
Clouse, C. J. Diederich, T. Corwin, G. Lindberg, O. Traxer, M. T. Gettman, T. G.
Smith, M. S. Pearle, J. A. Cadeddu, F. Litvack, W. S. Grundfest, T. Papaioannou, F.
W. Mohr, A. T. Jakubowski, J. S. Forrester, S. Rastegar, M. Motamedi, A. J. Welch,
L. J. Hayes, A. J. Welch, M. Motamedi, S. Rastegar, G. L. Lecarpentier, D. Jansen,
S. A. Sapareto, W. C. Dewey, A. Busetti, M. Soncin, E. Reddi, M. A. Rodgers, M.
E. Kenney, G. Jori, J. R. Lepock, K.-H. Cheng, H. Al-qysi, I. Sim, C. J. Koch, J.
Kruuv, J. R. Lepock, H. E. Frey, A. M. Rodahl, J. Kruuv, A. E. Caccamo, S.
Desenzani, L. Belloni, A. F. Borghetti, S. Bettuzzi, K. J. Henle, L. A. Dethlefsen, B.
W. Henderson, V. H. Fingar, M. Ahmed, L. E. Gerweck, V. K. Pustovalov, L. G.
Astafyeva, W. Fritzsche, M. P. V. Shekhar, C. L. Garrett, D. O. Draper, K. L. Knight,
N. A. Armar, G. C. L. Lachelin, U. G. Longo, M. Ronga, N. Maffulli, H. I. Bicher,
F. W. Hetzel, T. S. Sandhu, S. Frinak, P. Vaupel, M. O’Hara, T. O’Brien, J. G. Short,
P. F. Turner, A. I. Minchinton, I. F. Tannock, S. H. van Rijt, P. J. Sadler, Y. Lee, S.
L. Auh, Y. Wang, B. Burnette, Y. Wang, Y. Meng, M. Beckett, R. Sharma, R. Chin,
T. Tu, A. A. Lugade, J. P. Moran, S. A. Gerber, R. C. Rose, J. G. Frelinger, E. M.
Lord, T. J. Dougherty, C. J. Gomer, B. W. Henderson, G. Jori, D. Kessel, M.
Korbelik, J. Moan, Q. Peng, M. P. Carey, T. G. Burish, J. E. Johnson, L. M. Nail, D.

122
Lauver, K. B. King, H. Keys, M. Clarke, R. Collins, S. Darby, C. Davies, P.
Elphinstone, E. Evans, J. Godwin, R. Gray, C. Hicks, S. James, I. Takahashi, Y. Emi,
S. Hasuda, Y. Kakeji, Y. Maehara, K. Sugimachi, E. L. Jones, J. R. Oleson, L. R.
Prosnitz, T. V. Samulski, Z. Vujaskovic, D. Yu, L. L. Sanders, M. W. Dewhirst, J.
B. Marmor, D. Pounds, T. B. Postic, G. M. Hahn, L. O. Svaasand, T. Boerslid, M.
Oeveraasen, A. Y. Cheung, A. Neyzari, W. Hurter, F. Reinbold, W. Lorenz, J.
Delannoy, D. LeBihan, D. Hoult, R. Levin, B. S. Trembly, B. E. Lyons, R. H. Britt,
J. W. Strohbehn, M. Johannsen, B. Thiesen, P. Wust, A. Jordan, M. Salloum, R. Ma,
D. Weeks, L. Zhu, G. Kong, R. D. Braun, M. W. Dewhirst, S. Mornet, S. Vasseur,
F. Grasset, E. Duguet, Q. A. Pankhurst, J. Connolly, S. Jones, J. Dobson, R. Hergt,
S. Dutz, R. Müller, M. Zeisberger, B. Thiesen, A. Jordan, A. Ito, M. Shinkai, H.
Honda, T. Kobayashi, R. K. Gilchrist, R. Medal, W. D. Shorey, R. C. Hanselman, J.
C. Parrott, C. B. Taylor, M. Johannsen, U. Gneveckow, K. Taymoorian, B. Thiesen,
N. Waldofner, R. Scholz, K. Jung, A. Jordan, P. Wust, S. A. Loening, B. A. Moffat,
G. R. Reddy, P. McConville, D. E. Hall, T. L. Chenevert, R. R. Kopelman, M.
Philbert, R. Weissleder, A. Rehemtulla, B. D. Ross, D. Pissuwan, S. M. Valenzuela,
M. B. Cortie, X. H. Huang, P. K. Jain, I. H. El-Sayed, M. A. El-Sayed, X. Huang, I.
H. El-Sayed, W. Qian, M. A. El-Sayed, P. K. Jain, X. Huang, I. H. El-Sayed, M. A.
El-Sayed, A. M. Smith, M. C. Mancini, S. Nie, R. Weissleder, K. König, F.
Helmchen, W. Denk, M. P. Melancon, M. Zhou, C. Li, G. S. Terentyuk, G. N.
Maslyakova, L. V. Suleymanova, N. G. Khlebtsov, B. N. Khlebtsov, M. Olivo, C.
Y. Fu, V. Raghavan, W. K. Lau, M. A. Albota, C. Xu, W. W. Webb, J. G.
Teeguarden, P. M. Hinderliter, G. Orr, B. D. Thrall, J. G. Pounds, M. S. Nikolic, M.

123
Krack, V. Aleksandrovic, A. Kornowski, S. Förster, H. Weller, X. Shi, S. Wang, H.
Sun, J. R. Baker, W. W. Yu, E. Chang, R. Drezek, V. L. Colvin, N. R. Jana, C.
Earhart, J. Y. Ying, J. -s. Choi, Y. -w. Jun, S.-I. Yeon, H. C. Kim, J.-S. Shin, J.
Cheon, X. Hong, Z. Wang, W. Cai, F. Lu, J. Zhang, Y. Yang, N. Ma, Y. Liu, J. R.
McCarthy, E. Korngold, R. Weissleder, F. A. Jaffer, A. Barhoumi, R. Huschka, R.
Bardhan, M. W. Knight, N. J. Halas, U. Rocha, K. U. Kumar, C. Jacinto, I. Villa, F.
Sanz-Rodríguez, M. del C. I. de la Cruz, A. Juarranz, E. Carrasco, F. C. J. M. van
Veggel, E. Bovero, J. G. Solé, D. Jaque, H. Huang, S. Delikanli, H. Zeng, D. M.
Ferkey, A. Pralle, L. Cheng, K. Yang, Q. Chen, Z. Liu, L. M. Maestro, P. Haro-
Gonzalez, B. del Rosal, J. Ramiro, A. J. Caamano, E. Carrasco, A. Juarranz, F. Sanz-
Rodriguez, J. G. Sole, D. Jaque, S. Link, M. A. El-Sayed, S. Link, M. A. El-Sayed,
G. Mie, J. F. Lovell, C. S. Jin, E. Huynh, H. Jin, C. Kim, S. K. Ghosh, T. Pal, P. K.
Jain, I. H. El-Sayed, M. A. El-Sayed, S. E. Skrabalak, J. Chen, L. Au, X. Lu, X. Li,
Y. Xia, E. Carbo-Argibay, B. Rodriguez-Gonzalez, J. Pacifico, I. Pastoriza-Santos,
J. Perez-Juste, L. M. Liz-Marzan, J. Pérez-Juste, I. Pastoriza-Santos, L. M. Liz-
Marzán, P. Mulvaney, P. K. Jain, K. S. Lee, I. H. El-Sayed, M. A. El-Sayed, L.
Maestro, P. Haro-González, J. Coello, D. Jaque, E. Prodan, P. Nordlander, N. J.
Halas, S. J. Oldenburg, J. B. Jackson, S. L. Westcott, N. J. Halas, A. M. Fales, H.
Yuan, T. Vo-Dinh, R. Rodríguez-Oliveros, J. A. Sánchez-Gil, H. Yuan, C. G.
Khoury, C. M. Wilson, G. A. Grant, A. J. Bennett, T. Vo-Dinh, S. Trigari, A. Rindi,
G. Margheri, S. Sottini, G. Dellepiane, E. Giorgetti, J. Chen, B. Wiley, Z. Y. Li, D.
Campbell, F. Saeki, H. Cang, L. Au, J. Lee, X. Li, Y. Xia, M. F. Tsai, S. H. Chang,
F. Y. Cheng, V. Shanmugam, Y. S. Cheng, C. H. Su, C. S. Yeh, Y. Jin, J. Wang, H.

124
Ke, S. Wang, Z. Dai, H. Ke, J. Wang, Z. Dai, Y. Jin, E. Qu, Z. Xing, C. Guo, X.
Yue, J. Liu, H. Ke, J. Wang, S. Tong, Y. Jin, S. Wang, E. Qu, G. Bao, Z. Dai, H. Ke,
X. Yue, J. Wang, S. Xing, Q. Zhang, Z. Dai, J. Tian, S. Wang, Y. Jin, S. C. Boca,
M. Potara, A. M. Gabudean, A. Juhem, P. L. Baldeck, S. Astilean, G. Fu, W. Liu, S.
Feng, X. Yue, M. Shokouhimehr, E. S. Soehnlen, A. Khitrin, S. Basu, S. P. D. Huang,
M. Shokouhimehr, E. S. Soehnlen, J. H. Hao, M. Griswold, C. Flask, X. D. Fan, J.
P. Basilion, S. Basu, S. P. D. Huang, N. H. Levi-Polyachenko, E. J. Merkel, B. T.
Jones, D. L. Carroll, J. H. Stewart, S. J. Yu, M. W. Kang, H. C. Chang, K. M. Chen,
Y. C. Yu, G. Kucsko, P. C. Maurer, N. Y. Yao, M. Kubo, H. J. Noh, S. Iijima, J. T.
Robinson, S. M. Tabakman, Y. Y. Liang, H. L. Wang, H. S. Casalongue, D. Vinh,
H. J. Dai, X. Sun, Z. Liu, K. Welsher, J. Robinson, A. Goodwin, S. Zaric, H. Dai, F.
Wang, G. Dukovic, L. E. Brus, T. F. Heinz, M. S. Strano, S. K. Doorn, E. H. Haroz,
C. Kittrell, R. H. Hauge, R. E. Smalley, L. Van Hove, T. Stöckli, Z. L. Wang, J. M.
Bonard, P. Sadelmann, A. Chatelain, A. V. Naumov, S. Ghosh, D. A. Tsyboulski, S.
M. Bachilo, R. B. Weisman, M. J. O’Connell, S. M. Bachilo, C. B. Huffman, V. C.
Moore, M. S. Strano, E. H. Haroz, K. L. Rialon, P. J. Boul, W. H. Noon, C. Kittrell,
J. Ma, R. H. Hauge, R. B. Weisman, R. E. Smalley, F. Wang, G. Dukovic, L. E.
Brus, T. F. Heinz, J. Crochet, M. Clemens, T. Hertel, S.-Y. Ju, W. P. Kopcha, F.
Papadimitrakopoulos, M. Lin, K. W. K. Shung, B. Zhao, M. E. Itkis, S. Niyogi, H.
Hu, J. Zhang, R. C. Haddon, F. Schöppler, C. Mann, T. C. Hain, F. M. Neubauer, G.
Privitera, F. Bonaccorso, D. Chu, A. C. Ferrari, T. Hertel, Q. Cheng, S. Debnath, E.
Gregan, H. J. Byrne, A. Burke, X. Ding, R. Singh, R. A. Kraft, N. Levi-Polyachenko,
M. N. Rylander, C. Szot, C. Buchanan, J. Whitney, J. Fisher, H. C. Hatcher, J. R.

125
D’Agostino, N. D. Kock, P. M. Ajayan, D. L. Carroll, S. Akman, F. M. Torti, S. V.
Torti, S. H. Jeong, K. K. Kim, S. J. Jeong, K. H. An, S. H. Lee, Y. H. Lee, S. H.
Kim, G. W. Mulholland, M. R. Zachariah, L. M. Maestro, E. M. Rodriguez, F.
Vetrone, R. Naccache, H. L. Ramirez, D. Jaque, J. A. Capobianco, J. G. Solé, I. L.
Medintz, H. T. Uyeda, E. R. Goldman, H. Mattoussi, N. Lewinski, V. Colvin, R.
Drezek, K. T. Yong, W. C. Law, R. Hu, L. Ye, L. Liu, M. T. Swihart, P. N. Prasad,
L. M. Maestro, P. Haro-González, M. C. I. la Cruz, F. SanzRodríguez, Á. Juarranz,
J. G. Solé, D. Jaque, S. B. Lakshmanan, X. Zou, M. Hossu, L. Ma, C. Yang, W.
Chen, Q. Tian, M. Tang, Y. Sun, R. Zou, Z. Chen, M. Zhu, S. Yang, J. Wang, J.
Wang, J. Hu, A. M. Derfus, W. C. W. Chan, S. N. Bhatia, Y. Li, W. Lu, Q. Huang,
C. Li, W. Chen, V. M. García, P. K. Nair, M. T. S. Nair, C. M. Hessel, V. P. Pattani,
M. Rasch, M. G. Panthani, B. Koo, J. W. Tunnell, B. A. Korgel, Y. Zhao, H. Pan,
Y. Lou, X. Qiu, J. Zhu, C. Burda, J. M. Luther, P. K. Jain, T. Ewers, A. P. Alivisatos,
T. N. Lambert, N. L. Andrews, H. Gerung, T. J. Boyle, J. M. Oliver, B. S. Wilson,
S. M. Han, G. Chen, T. Y. Ohulchanskyy, S. Liu, W.-C. Law, F. Wu, M. T. Swihart,
H. Ågren, P. N. Prasad, G. Chen, J. Shen, T. Y. Ohulchanskyy, N. J. Patel, A.
Kutikov, Z. Li, J. Song, R. K. Pandey, H. Ågren, P. N. Prasad, G. Han, F. Vetrone,
R. Naccache, A. J. de la Fuente, F. Sanz-Rodriguez, A. Blazquez-Castro, E. M.
Rodriguez, D. Jaque, J. G. Sole, J. A. Capobianco, D. Jaque, F. Vetrone, A. Gnach,
A. Bednarkiewicz, A. A. Ansari, S. P. Singh, N. Singh, B. D. Malhotra, D.
Wawrzynczyk, A. Bednarkiewicz, M. Nyk, W. Strek, M. Samoc, A. Bednarkiewicz,
D. Wawrzynczyk, M. Nyk, W. Strek, V. K. Tikhomirov, K. Driesen, V. D.
Rodriguez, P. Gredin, M. Mortier, U. Rocha, C. J. da Silva, W. F. Silva, I. Guedes,

126
A. Benayas, L. M. Maestro, M. A. Elias, E. Bovero, F. C. J. M. van Veggel, J. A. G.
Solé, D. Jaque, M. Ge, J. Rong, X. Fang, A. Zhang, Y. Lu, C. Zhou, A. G. Cullis, L.
T. Canham, P. D. J. Calcott, S. P. Low, N. H. Voelcker, L. T. Canham, K. A.
Williams, L. Xiao, L. Gu, S. B. Howell, M. J. Sailor, E. J. Anglin, L. Cheng, W. R.
Freeman, M. J. Sailor, E. Secret, K. Smith, V. Dubljevic, E. Moore, P. Macardle, B.
Delalat, M. L. Rogers, T. G. Johns, J. O. Durand, F. Cunin, N. H. Voelcker, J.-H.
Park, L. Gu, G. von Maltzahn, E. Ruoslahti, S. N. Bhatia, M. J. Sailor, D. Gallach,
G. R. Sánchez, A. M. Noval, M. M. Silván, G. Ceccone, R. J. M. Palma, V. T. Costa,
J. M. M. Duart, C. Lee, H. Kim, C. Hong, M. Kim, S. S. Hong, D. H. Lee, W. I. Lee,
C. Lee, C. Hong, J. Lee, M. Son, S.-S. Hong, I. Sagnes, A. Halimaoui, G. Vincent,
P. A. Badoz, J. Yu, D. Javier, M. A. Yaseen, N. Nitin, R. Richards-Kortum, J. Yang,
J. Choi, D. Bang, E. Kim, E. K. Lim, H. Park, J. S. Suh, K. Lee, K. H. Yoo, E. K.
Kim, Y. M. Huh, S. Haam, S. Sharifi, S. Behzadi, S. Laurent, M. L. Forrest, P.
Stroeve, H. Shinohara, A. Tanaka, T. Kitai, N. Yanabu, T. Inomoto, M. Zheng, C.
Yue, Y. Ma, P. Gong, P. Zhao, C. Zheng, Z. Sheng, P. Zhang, Z. Wang, L. Cai, B.
Jung, A. L. N. Rao, B. Anvari, S. Gupta, M. R. Chatni, A. L. N. Rao, V. I. Vullev,
L. V. Wang, B. Anvari, B. Bahmani, D. Bacon, B. Anvari, Y. Ma, S. Tong, G. Bao,
C. Gao, Z. Dai, C.-Y. Li, W.-Y. Chiu, C.-F. Lee, O. P. Dimitriev, L. Cheng, W. He,
H. Gong, C. Wang, Q. Chen, Z. Cheng, Z. Liu, M. Soncin, A. Busetti, F. Fusi, G.
Jori, M. A. J. Rodgers, C. S. Jin, J. F. Lovell, J. Chen, G. Zheng, L. M. Maestro, P.
Haro-González, A. Sánchez-Iglesias, L. M. Liz-Marzán, J. A. G. Sole, D. Jaque, Z.
Zha, X. Yue, Q. Ren, Z. Dai, Z. Zha, J. Wang, S. Zhang, S. Wang, E. Qu, Y. Zhang,
Z. Dai, Y. Ma, X. Liang, S. Tong, G. Bao, Q. Ren, Z. Dai, J. Turkevich, P. C.

127
Stevenson, J. Hillier, G. Frens, W. S. Sutherland, J. D. Winefordner, K. C. Grabar,
R. G. Freeman, M. B. Hommer, M. J. Natan, Z. Liang, J. Zhang, L. Wang, S. Song,
C. Fan, M. Brust, M. Walker, D. Bethell, D. J. Schiffrin, R. Whyman, J. L. Plaza, V.
Carcelen, X. Wang, J. Zhuang, Q. Peng, Y. D. Li, C. J. Murphy, N. R. Jana, B.
Nikoobakht, M. A. El-Sayed, E. B. Dickerson, E. C. Dreaden, X. H. Huang, I. H.
El-Sayed, H. H. Chu, S. Pushpanketh, J. F. McDonald, M. A. El-Sayed, F. M.
Veronese, G. Pasut, J. R. Cole, N. A. Mirin, M. W. Knight, G. P. Goodrich, N. J.
Halas, G. P. Goodrich, L. Bao, K. Gill-Sharp, K. L. Sang, J. Wang, Q. Zhang, W.
Li, L. P. Wen, J. Chen, Y. Xia, S. E. Skrabalak, J. Chen, Y. Sun, X. Lu, L. Au, J.
Chen, M. Yang, Q. Zhang, E. C. Cho, C. M. Cobley, C. Kim, C. Glaus, L. V. Wang,
M. J. Welch, Y. Xia, M. Hu, J. Chen, Z.-Y. Li, L. Au, G. V. Hartland, X. Li, M.
Marquez, Y. Xia, C. Journet, W. K. Maser, P. Bernier, A. Loiseau, M. L.
delaChapelle, M. C. Altay, S. Eroglu, T. Guo, P. Nikolaev, A. Thess, D. T. Colbert,
R. E. Smalley, M. Paradise, T. Goswami, H. Dai, Y. Uemura, K. Baba, H. Ohe, Y.
Ohzuno, Y. Hatate, A.-C. Dupuis, S. McCaldin, M. Bououdina, D. M. Grant, G. S.
Walker, A. Thess, R. Lee, P. Nikolaev, H. J. Dai, P. Petit, A. Valizadeh, H. Mikaeili,
M. Samiei, S. M. Farkhani, N. Zarghami, M. Kouhi, A. Akbarzadeh, S. Davaran, D.
Bera, L. Qian, T.-K. Tseng, P. H. Holloway, L. Y. Zhang, L. Wen, J. J. Zhang, L. L.
Hu, N. Bogdan, E. M. Rodriguez, F. Sanz-Rodriguez, M. C. I. de la Cruz, A.
Juarranz, D. Jaque, J. G. Sole, J. A. Capobianco, V. Mahalingam, F. Vetrone, R.
Naccache, A. Speghini, J. A. Capobianco, W. Zhang, Z. Guo, D. Huang, Z. Liu, X.
Guo, H. Zhong, X. Teng, Y. Zhu, W. Wei, S. Wang, J. Huang, R. Naccache, W. Hu,
A. I. Y. Tok, Y. Han, Q. Zhang, Q. Fan, W. Huang, J. A. Capobianco, L. Huang, F.

128
Vetrone, R. Naccache, V. Mahalingam, C. G. Morgan, J. A. Capobianco, F. Wang,
D. K. Chatterjee, Z. Q. Li, Y. Zhang, X. P. Fan, M. Q. Wang, H. X. Mai, Y. W.
Zhang, R. Si, Z. G. Yan, L. D. Sun, L. P. You, C. H. Yan, Y. Su, L. Li, G. Li, J.
Zhuang, L. Liang, H. H. Y. Sung, X. Yang, M. Wu, I. D. Williams, S. Feng, Q. Su,
F. Wang, D. Banerjee, Y. Liu, X. Chen, X. Liu, W. Zhou, X. Liu, J. Ji, M.
Aeschbacher, C. A. Reinhardt, G. Zbinden, E. C. Cho, L. Au, Q. Zhang, Y. Xia, L.
Tong, Y. Zhao, T. B. Huff, M. N. Hansen, A. Wei, J. X. Cheng, T. B. Huff, L. Tong,
Y. Zhao, M. N. Hansen, J. X. Cheng, A. Wei, M. J. Waring, C. H. Chou, C. D. Chen,
C. R. C. Wang, P. Prentice, A. Cuschierp, K. Dholakia, M. Prausnitz, P. Campbell,
X. Huang, P. K. Jain, I. H. El-Sayed, M. A. El-Sayed, M. Longmire, P. L. Choyke,
H. Kobayashi, A. K. Gupta, R. R. Naregalkar, V. D. Vaidya, M. Gupta, X. H. Gao,
Y. Y. Cui, R. M. Levenson, L. W. K. Chung, S. M. Nie, I. H. El-Sayed, X. Huang,
M. A. El-Sayed, M. S. Yavuz, Y. Cheng, J. Chen, C. M. Cobley, Q. Zhang, M.
Rycenga, J. Xie, C. Kim, K. H. Song, A. G. Schwartz, L. V. Wang, Y. Xia, A. S.
Hoffman, B. D. Chithrani, A. A. Ghazani, W. C. W. Chan, B. D. Chithrani, W. C.
W. Chan, F. Osaki, T. Kanamori, S. Sando, T. Sera, Y. Aoyama, J. D. Trono, K.
Mizuno, N. Yusa, T. Matsukawa, K. Yokoyama, M. Uesaka, J. Davda, V.
Labhasetwar, X. Xing, X. He, J. Peng, K. Wang, W. Tan, S. J. H. Soenen, E. Illyes,
D. Vercauteren, K. Braeckmans, Z. Majer, H. Y. Nam, S. M. Kwon, H. Chung, S.-
Y. Lee, S.-H. Kwon, H. Jeon, Y. Kim, J. H. Park, J. Kim, S. Her, Y.-K. Oh, I. C.
Kwon, K. Kim, S. Y. Jeong, J. L. Goldstein, R. G. W. Anderson, M. S. Brown, L.
Tong, Y. Lu, R. J. Lee, J.-X. Cheng, N. Tran, T. J. Webster, Y. Zhang, W. Chen, J.
Zhang, J. Liu, G. Chen, C. Pope, N. Khlebtsov, L. Dykman, N. Zeng, A. B. Murphy,

129
L. Tong, Q. Wei, A. Wei, J. X. Cheng, G. T. Charras, M. Coughlin, T. J. Mitchison,
L. Mahadevan, J. Hagmann, M. M. Burger, D. Dagan, S. Link, C. Burda, M. B.
Mohamed, B. Nikoobakht, M. A. El-Sayed, V. Kotaidis, A. Plech, S. Link, Z. L.
Wang, M. A. El-Sayed, S. Link, C. Burda, B. Nikoobakht, M. A. El-Sayed, H.
Petrova, J. P. Juste, I. Pastoriza-Santos, G. V. Hartland, L. M. Liz-Marzan, M. B.
Mohamed, K. Z. Ismail, S. Link, M. A. El-Sayed, H. Kang, B. Jia, J. Li, D. Morrish,
M. Gu, E. Y. Hleb, J. H. Hafner, J. N. Myers, E. Y. Hanna, B. C. Rostro, S. A.
Zhdanok, D. O. Lapotko, E. Y. Lukianova-Hleb, D. O. Lapotko, E. Y. Lukianova-
Hleb, X. Y. Ren, J. A. Zasadzinski, X. W. Wu, D. O. Lapotko, Z. Li, P. Huang, X.
Zhang, J. Lin, S. Yang, B. Liu, F. Gao, P. Xi, Q. Ren, D. Cui, D. O. Lapotko, E. Y.
Lukianova-Hleb, A. A. Oraevsky, B. P. Danysh, P. E. Constantinou, E. Y.
Lukianova-Hleb, D. O. Lapotko, D. D. Carson, E. Y. Lukianova-Hleb, A. Belyanin,
S. Kashinath, X. W. Wu, D. O. Lapotko, E. Lukianova-Hleb, Y. Hu, L. Latterini, L.
Tarpani, S. Lee, R. A. Drezek, J. H. Hafner, D. O. Lapotko, H. Maeda, A. Iyer, G.
Khaled, J. Fang, H. Maeda, H. Wang, W. Su, S. Wang, X. Wang, Z. Liao, H. K.
Moon, S. H. Lee, H. C. Choi, R. R. Anderson, J. A. Parrish, N. N. Huang, H. Q.
Wang, J. H. Zhao, H. Lui, M. Korbelik, H. S. Zeng, L. C. Kennedy, L. R. Bickford,
N. A. Lewinski, A. J. Coughlin, Y. Hu, E. S. Day, J. L. West, R. A. Drezek, F. Alexis,
E. Pridgen, L. K. Molnar, O. C. Farokhzad, L. Balogh, S. S. Nigavekar, B. M. Nair,
W. Lesniak, C. Zhang, L. Y. Sung, M. S. Kariapper, A. El-Jawahri, M. Llanes, B.
Bolton, F. Mamou, W. Tan, A. Hutson, L. Minc, M. K. Khan, Z. Liu, C. Davis, W.
B. Cai, L. He, X. Y. Chen, H. J. Dai, F. Sousa, S. Mandal, C. Garrovo, A. Astolfo,
A. Bonifacio, D. Latawiec, R. H. Menk, F. Arfelli, S. Huewel, G. Legname, H. J.

130
Galla, S. Krol, G. Zhang, Z. Yang, W. Lu, R. Zhang, Q. Huang, M. Tian, L. Li, D.
Liang, C. Li, S. D. Perrault, C. Walkey, T. Jennings, H. C. Fischer, W. C. Chan, B.
Kim, G. Han, B. J. Toley, C. K. Kim, V. M. Rotello, N. S. Forbes, K. Welsher, S. P.
Sherlock, H. Dai, J. T. Robinson, G. Hong, Y. Liang, B. Zhang, O. K. Yaghi, H. Dai,
B. J. Darien, P. A. Sims, K. T. Kruseelliott, T. S. Homan, R. J. Cashwell, A. J.
Cooley, R. M. Albrecht, G. Hong, J. T. Robinson, Y. Zhang, S. Diao, A. L. Antaris,
Q. Wang, H. Dai, K. Yang, S. Zhang, G. Zhang, X. Sun, S. T. Lee, Z. Liu, Y. Zhang,
G. Hong, Y. Zhang, G. Chen, F. Li, H. Dai, Q. Wang, J. Chen, C. Glaus, R. Laforest,
Q. Zhang, M. Yang, E. Day, P. A. Thompson, L. Zhang, N. A. Lewinski, N. Ahmed,
R. Atkinson, M. Zhang, P. Diagaradjane, S. Peddibhotla, and A. Contreras,
"Nanoparticles for photothermal therapies," Nanoscale 6, 9494 (2014).
42. A. D. Pechauer, Y. Jia, L. Liu, S. S. Gao, C. Jiang, and D. Huang, "Optical
Coherence Tomography Angiography of Peripapillary Retinal Blood Flow
Response to Hyperoxia," Investig. Opthalmology Vis. Sci. 56, 3287 (2015).
43. C. Dorronsoro, D. Pascual, P. Pérez-Merino, S. Kling, and S. Marcos, "Dynamic
OCT measurement of corneal deformation by an air puff in normal and cross-linked
corneas," Biomed. Opt. Express 3, 473 (2012).
44. D. Huang, E. A. Swanson, C. P. Lin, J. S. Schuman, W. G. Stinson, W. Chang, M.
R. Hee, T. Flotte, K. Gregory, C. A. Puliafito, and J. G. Fujimoto, "Optical
coherence tomography.," Science 254, 1178–81 (1991).
45. W. Drexler, U. Morgner, R. Ghanta, and F. Kärtner, "Ultrahigh-resolution
ophthalmic optical coherence tomography," Nat. Med. (2001).
46. S. A. Boppart, B. E. Bouma, M. E. Brezinski, G. J. Tearney, and J. G. Fujimoto,

131
"Imaging developing neural morphology using optical coherence tomography," J.
Neurosci. Methods 70, 65–72 (1996).
47. G. J. Tearney, M. E. Brezinski, B. E. Bouma, S. A. Boppart, C. Pitris, J. F. Southern,
and J. G. Fujimoto, "In vivo endoscopic optical biopsy with optical coherence
tomography.," Science 276, 2037–2039 (1997).
48. A. A. Michelson and E. W. Morley, "On the Relative Motion of the Earth and the
Luminiferous Ether," Am. J. Sci. 34, 333–345 (1881).
49. B. P. Abbott, R. Abbott, T. D. Abbott, M. R. Abernathy, F. Acernese, K. Ackley, C.
Adams, T. Adams, P. Addesso, R. X. Adhikari, V. B. Adya, C. Affeldt, M. Agathos,
K. Agatsuma, N. Aggarwal, O. D. Aguiar, L. Aiello, A. Ain, P. Ajith, B. Allen, A.
Allocca, P. A. Altin, S. B. Anderson, W. G. Anderson, K. Arai, M. A. Arain, M. C.
Araya, C. C. Arceneaux, J. S. Areeda, N. Arnaud, K. G. Arun, S. Ascenzi, G. Ashton,
M. Ast, S. M. Aston, P. Astone, P. Aufmuth, C. Aulbert, S. Babak, P. Bacon, M. K.
M. Bader, P. T. Baker, F. Baldaccini, G. Ballardin, S. W. Ballmer, J. C. Barayoga,
S. E. Barclay, B. C. Barish, D. Barker, F. Barone, B. Barr, L. Barsotti, M. Barsuglia,
D. Barta, J. Bartlett, M. A. Barton, I. Bartos, R. Bassiri, A. Basti, J. C. Batch, C.
Baune, V. Bavigadda, M. Bazzan, B. Behnke, M. Bejger, C. Belczynski, A. S. Bell,
C. J. Bell, B. K. Berger, J. Bergman, G. Bergmann, C. P. L. Berry, D. Bersanetti, A.
Bertolini, J. Betzwieser, S. Bhagwat, R. Bhandare, I. A. Bilenko, G. Billingsley, J.
Birch, R. Birney, O. Birnholtz, S. Biscans, A. Bisht, M. Bitossi, C. Biwer, M. A.
Bizouard, J. K. Blackburn, C. D. Blair, D. G. Blair, R. M. Blair, S. Bloemen, O.
Bock, T. P. Bodiya, M. Boer, G. Bogaert, C. Bogan, A. Bohe, P. Bojtos, C. Bond,
F. Bondu, R. Bonnand, B. A. Boom, R. Bork, V. Boschi, S. Bose, Y. Bouffanais, A.

132
Bozzi, C. Bradaschia, P. R. Brady, V. B. Braginsky, M. Branchesi, J. E. Brau, T.
Briant, A. Brillet, M. Brinkmann, V. Brisson, P. Brockill, A. F. Brooks, D. A. Brown,
D. D. Brown, N. M. Brown, C. C. Buchanan, A. Buikema, T. Bulik, H. J. Bulten, A.
Buonanno, D. Buskulic, C. Buy, R. L. Byer, M. Cabero, L. Cadonati, G. Cagnoli, C.
Cahillane, J. C. Bustillo, T. Callister, E. Calloni, J. B. Camp, K. C. Cannon, J. Cao,
C. D. Capano, E. Capocasa, F. Carbognani, S. Caride, J. C. Diaz, C. Casentini, S.
Caudill, M. Cavaglià, F. Cavalier, R. Cavalieri, G. Cella, C. B. Cepeda, L. C. Baiardi,
G. Cerretani, E. Cesarini, R. Chakraborty, T. Chalermsongsak, S. J. Chamberlin, M.
Chan, S. Chao, P. Charlton, E. Chassande-Mottin, H. Y. Chen, Y. Chen, C. Cheng,
A. Chincarini, A. Chiummo, H. S. Cho, M. Cho, J. H. Chow, N. Christensen, Q.
Chu, S. Chua, S. Chung, G. Ciani, F. Clara, J. A. Clark, F. Cleva, E. Coccia, P.-F.
Cohadon, A. Colla, C. G. Collette, L. Cominsky, M. Constancio, A. Conte, L. Conti,
D. Cook, T. R. Corbitt, N. Cornish, A. Corsi, S. Cortese, C. A. Costa, M. W.
Coughlin, S. B. Coughlin, J.-P. Coulon, S. T. Countryman, P. Couvares, E. E.
Cowan, D. M. Coward, M. J. Cowart, D. C. Coyne, R. Coyne, K. Craig, J. D. E.
Creighton, T. D. Creighton, J. Cripe, S. G. Crowder, A. M. Cruise, A. Cumming, L.
Cunningham, E. Cuoco, T. D. Canton, S. L. Danilishin, S. D’Antonio, K. Danzmann,
N. S. Darman, C. F. Da Silva Costa, V. Dattilo, I. Dave, H. P. Daveloza, M. Davier,
G. S. Davies, E. J. Daw, R. Day, S. De, D. DeBra, G. Debreczeni, J. Degallaix, M.
De Laurentis, S. Deléglise, W. Del Pozzo, T. Denker, T. Dent, H. Dereli, V.
Dergachev, R. T. DeRosa, R. De Rosa, R. DeSalvo, S. Dhurandhar, M. C. Díaz, L.
Di Fiore, M. Di Giovanni, A. Di Lieto, S. Di Pace, I. Di Palma, A. Di Virgilio, G.
Dojcinoski, V. Dolique, F. Donovan, K. L. Dooley, S. Doravari, R. Douglas, T. P.

133
Downes, M. Drago, R. W. P. Drever, J. C. Driggers, Z. Du, M. Ducrot, S. E. Dwyer,
T. B. Edo, M. C. Edwards, A. Effler, H.-B. Eggenstein, P. Ehrens, J. Eichholz, S. S.
Eikenberry, W. Engels, R. C. Essick, T. Etzel, M. Evans, T. M. Evans, R. Everett,
M. Factourovich, V. Fafone, H. Fair, S. Fairhurst, X. Fan, Q. Fang, S. Farinon, B.
Farr, W. M. Farr, M. Favata, M. Fays, H. Fehrmann, M. M. Fejer, D. Feldbaum, I.
Ferrante, E. C. Ferreira, F. Ferrini, F. Fidecaro, L. S. Finn, I. Fiori, D. Fiorucci, R.
P. Fisher, R. Flaminio, M. Fletcher, H. Fong, J.-D. Fournier, S. Franco, S. Frasca, F.
Frasconi, M. Frede, Z. Frei, A. Freise, R. Frey, V. Frey, T. T. Fricke, P. Fritschel,
V. V. Frolov, P. Fulda, M. Fyffe, H. A. G. Gabbard, J. R. Gair, L. Gammaitoni, S.
G. Gaonkar, F. Garufi, A. Gatto, G. Gaur, N. Gehrels, G. Gemme, B. Gendre, E.
Genin, A. Gennai, J. George, L. Gergely, V. Germain, A. Ghosh, A. Ghosh, S.
Ghosh, J. A. Giaime, K. D. Giardina, A. Giazotto, K. Gill, A. Glaefke, J. R. Gleason,
E. Goetz, R. Goetz, L. Gondan, G. González, J. M. G. Castro, A. Gopakumar, N. A.
Gordon, M. L. Gorodetsky, S. E. Gossan, M. Gosselin, R. Gouaty, C. Graef, P. B.
Graff, M. Granata, A. Grant, S. Gras, C. Gray, G. Greco, A. C. Green, R. J. S.
Greenhalgh, P. Groot, H. Grote, S. Grunewald, G. M. Guidi, X. Guo, A. Gupta, M.
K. Gupta, K. E. Gushwa, E. K. Gustafson, R. Gustafson, J. J. Hacker, B. R. Hall, E.
D. Hall, G. Hammond, M. Haney, M. M. Hanke, J. Hanks, C. Hanna, M. D. Hannam,
J. Hanson, T. Hardwick, J. Harms, G. M. Harry, I. W. Harry, M. J. Hart, M. T.
Hartman, C.-J. Haster, K. Haughian, J. Healy, J. Heefner, A. Heidmann, M. C.
Heintze, G. Heinzel, H. Heitmann, P. Hello, G. Hemming, M. Hendry, I. S. Heng, J.
Hennig, A. W. Heptonstall, M. Heurs, S. Hild, D. Hoak, K. A. Hodge, D. Hofman,
S. E. Hollitt, K. Holt, D. E. Holz, P. Hopkins, D. J. Hosken, J. Hough, E. A. Houston,

134
E. J. Howell, Y. M. Hu, S. Huang, E. A. Huerta, D. Huet, B. Hughey, S. Husa, S. H.
Huttner, T. Huynh-Dinh, A. Idrisy, N. Indik, D. R. Ingram, R. Inta, H. N. Isa, J.-M.
Isac, M. Isi, G. Islas, T. Isogai, B. R. Iyer, K. Izumi, M. B. Jacobson, T. Jacqmin, H.
Jang, K. Jani, P. Jaranowski, S. Jawahar, F. Jiménez-Forteza, W. W. Johnson, N. K.
Johnson-McDaniel, D. I. Jones, R. Jones, R. J. G. Jonker, L. Ju, K. Haris, C. V.
Kalaghatgi, V. Kalogera, S. Kandhasamy, G. Kang, J. B. Kanner, S. Karki, M.
Kasprzack, E. Katsavounidis, W. Katzman, S. Kaufer, T. Kaur, K. Kawabe, F.
Kawazoe, F. Kéfélian, M. S. Kehl, D. Keitel, D. B. Kelley, W. Kells, R. Kennedy,
D. G. Keppel, J. S. Key, A. Khalaidovski, F. Y. Khalili, I. Khan, S. Khan, Z. Khan,
E. A. Khazanov, N. Kijbunchoo, C. Kim, J. Kim, K. Kim, N.-G. Kim, N. Kim, Y.-
M. Kim, E. J. King, P. J. King, D. L. Kinzel, J. S. Kissel, L. Kleybolte, S. Klimenko,
S. M. Koehlenbeck, K. Kokeyama, S. Koley, V. Kondrashov, A. Kontos, S. Koranda,
M. Korobko, W. Z. Korth, I. Kowalska, D. B. Kozak, V. Kringel, B. Krishnan, A.
Królak, C. Krueger, G. Kuehn, P. Kumar, R. Kumar, L. Kuo, A. Kutynia, P. Kwee,
B. D. Lackey, M. Landry, J. Lange, B. Lantz, P. D. Lasky, A. Lazzarini, C. Lazzaro,
P. Leaci, S. Leavey, E. O. Lebigot, C. H. Lee, H. K. Lee, H. M. Lee, K. Lee, A.
Lenon, M. Leonardi, J. R. Leong, N. Leroy, N. Letendre, Y. Levin, B. M. Levine,
T. G. F. Li, A. Libson, T. B. Littenberg, N. A. Lockerbie, J. Logue, A. L. Lombardi,
L. T. London, J. E. Lord, M. Lorenzini, V. Loriette, M. Lormand, G. Losurdo, J. D.
Lough, C. O. Lousto, G. Lovelace, H. Lück, A. P. Lundgren, J. Luo, R. Lynch, Y.
Ma, T. MacDonald, B. Machenschalk, M. MacInnis, D. M. Macleod, F. Magaña-
Sandoval, R. M. Magee, M. Mageswaran, E. Majorana, I. Maksimovic, V. Malvezzi,
N. Man, I. Mandel, V. Mandic, V. Mangano, G. L. Mansell, M. Manske, M.

135
Mantovani, F. Marchesoni, F. Marion, S. Márka, Z. Márka, A. S. Markosyan, E.
Maros, F. Martelli, L. Martellini, I. W. Martin, R. M. Martin, D. V. Martynov, J. N.
Marx, K. Mason, A. Masserot, T. J. Massinger, M. Masso-Reid, F. Matichard, L.
Matone, N. Mavalvala, N. Mazumder, G. Mazzolo, R. McCarthy, D. E. McClelland,
S. McCormick, S. C. McGuire, G. McIntyre, J. McIver, D. J. McManus, S. T.
McWilliams, D. Meacher, G. D. Meadors, J. Meidam, A. Melatos, G. Mendell, D.
Mendoza-Gandara, R. A. Mercer, E. Merilh, M. Merzougui, S. Meshkov, C.
Messenger, C. Messick, P. M. Meyers, F. Mezzani, H. Miao, C. Michel, H.
Middleton, E. E. Mikhailov, L. Milano, J. Miller, M. Millhouse, Y. Minenkov, J.
Ming, S. Mirshekari, C. Mishra, S. Mitra, V. P. Mitrofanov, G. Mitselmakher, R.
Mittleman, A. Moggi, M. Mohan, S. R. P. Mohapatra, M. Montani, B. C. Moore, C.
J. Moore, D. Moraru, G. Moreno, S. R. Morriss, K. Mossavi, B. Mours, C. M. Mow-
Lowry, C. L. Mueller, G. Mueller, A. W. Muir, A. Mukherjee, D. Mukherjee, S.
Mukherjee, N. Mukund, A. Mullavey, J. Munch, D. J. Murphy, P. G. Murray, A.
Mytidis, I. Nardecchia, L. Naticchioni, R. K. Nayak, V. Necula, K. Nedkova, G.
Nelemans, M. Neri, A. Neunzert, G. Newton, T. T. Nguyen, A. B. Nielsen, S.
Nissanke, A. Nitz, F. Nocera, D. Nolting, M. E. N. Normandin, L. K. Nuttall, J.
Oberling, E. Ochsner, J. O’Dell, E. Oelker, G. H. Ogin, J. J. Oh, S. H. Oh, F. Ohme,
M. Oliver, P. Oppermann, R. J. Oram, B. O’Reilly, R. O’Shaughnessy, C. D. Ott, D.
J. Ottaway, R. S. Ottens, H. Overmier, B. J. Owen, A. Pai, S. A. Pai, J. R. Palamos,
O. Palashov, C. Palomba, A. Pal-Singh, H. Pan, Y. Pan, C. Pankow, F. Pannarale,
B. C. Pant, F. Paoletti, A. Paoli, M. A. Papa, H. R. Paris, W. Parker, D. Pascucci, A.
Pasqualetti, R. Passaquieti, D. Passuello, B. Patricelli, Z. Patrick, B. L. Pearlstone,

136
M. Pedraza, R. Pedurand, L. Pekowsky, A. Pele, S. Penn, A. Perreca, H. P. Pfeiffer,
M. Phelps, O. Piccinni, M. Pichot, M. Pickenpack, F. Piergiovanni, V. Pierro, G.
Pillant, L. Pinard, I. M. Pinto, M. Pitkin, J. H. Poeld, R. Poggiani, P. Popolizio, A.
Post, J. Powell, J. Prasad, V. Predoi, S. S. Premachandra, T. Prestegard, L. R. Price,
M. Prijatelj, M. Principe, S. Privitera, R. Prix, G. A. Prodi, L. Prokhorov, O.
Puncken, M. Punturo, P. Puppo, M. Pürrer, H. Qi, J. Qin, V. Quetschke, E. A.
Quintero, R. Quitzow-James, F. J. Raab, D. S. Rabeling, H. Radkins, P. Raffai, S.
Raja, M. Rakhmanov, C. R. Ramet, P. Rapagnani, V. Raymond, M. Razzano, V. Re,
J. Read, C. M. Reed, T. Regimbau, L. Rei, S. Reid, D. H. Reitze, H. Rew, S. D.
Reyes, F. Ricci, K. Riles, N. A. Robertson, R. Robie, F. Robinet, A. Rocchi, L.
Rolland, J. G. Rollins, V. J. Roma, J. D. Romano, R. Romano, G. Romanov, J. H.
Romie, D. Rosińska, S. Rowan, A. Rüdiger, P. Ruggi, K. Ryan, S. Sachdev, T.
Sadecki, L. Sadeghian, L. Salconi, M. Saleem, F. Salemi, A. Samajdar, L. Sammut,
L. M. Sampson, E. J. Sanchez, V. Sandberg, B. Sandeen, G. H. Sanders, J. R.
Sanders, B. Sassolas, B. S. Sathyaprakash, P. R. Saulson, O. Sauter, R. L. Savage,
A. Sawadsky, P. Schale, R. Schilling, J. Schmidt, P. Schmidt, R. Schnabel, R. M. S.
Schofield, A. Schönbeck, E. Schreiber, D. Schuette, B. F. Schutz, J. Scott, S. M.
Scott, D. Sellers, A. S. Sengupta, D. Sentenac, V. Sequino, A. Sergeev, G. Serna, Y.
Setyawati, A. Sevigny, D. A. Shaddock, T. Shaffer, S. Shah, M. S. Shahriar, M.
Shaltev, Z. Shao, B. Shapiro, P. Shawhan, A. Sheperd, D. H. Shoemaker, D. M.
Shoemaker, K. Siellez, X. Siemens, D. Sigg, A. D. Silva, D. Simakov, A. Singer, L.
P. Singer, A. Singh, R. Singh, A. Singhal, A. M. Sintes, B. J. J. Slagmolen, J. R.
Smith, M. R. Smith, N. D. Smith, R. J. E. Smith, E. J. Son, B. Sorazu, F. Sorrentino,

137
T. Souradeep, A. K. Srivastava, A. Staley, M. Steinke, J. Steinlechner, S.
Steinlechner, D. Steinmeyer, B. C. Stephens, S. P. Stevenson, R. Stone, K. A. Strain,
N. Straniero, G. Stratta, N. A. Strauss, S. Strigin, R. Sturani, A. L. Stuver, T. Z.
Summerscales, L. Sun, P. J. Sutton, B. L. Swinkels, M. J. Szczepańczyk, M. Tacca,
D. Talukder, D. B. Tanner, M. Tápai, S. P. Tarabrin, A. Taracchini, R. Taylor, T.
Theeg, M. P. Thirugnanasambandam, E. G. Thomas, M. Thomas, P. Thomas, K. A.
Thorne, K. S. Thorne, E. Thrane, S. Tiwari, V. Tiwari, K. V. Tokmakov, C.
Tomlinson, M. Tonelli, C. V. Torres, C. I. Torrie, D. Töyrä, F. Travasso, G. Traylor,
D. Trifirò, M. C. Tringali, L. Trozzo, M. Tse, M. Turconi, D. Tuyenbayev, D.
Ugolini, C. S. Unnikrishnan, A. L. Urban, S. A. Usman, H. Vahlbruch, G. Vajente,
G. Valdes, M. Vallisneri, N. van Bakel, M. van Beuzekom, J. F. J. van den Brand,
C. Van Den Broeck, D. C. Vander-Hyde, L. van der Schaaf, J. V. van Heijningen,
A. A. van Veggel, M. Vardaro, S. Vass, M. Vasúth, R. Vaulin, A. Vecchio, G.
Vedovato, J. Veitch, P. J. Veitch, K. Venkateswara, D. Verkindt, F. Vetrano, A.
Viceré, S. Vinciguerra, D. J. Vine, J.-Y. Vinet, S. Vitale, T. Vo, H. Vocca, C.
Vorvick, D. Voss, W. D. Vousden, S. P. Vyatchanin, A. R. Wade, L. E. Wade, M.
Wade, S. J. Waldman, M. Walker, L. Wallace, S. Walsh, G. Wang, H. Wang, M.
Wang, X. Wang, Y. Wang, H. Ward, R. L. Ward, J. Warner, M. Was, B. Weaver,
L.-W. Wei, M. Weinert, A. J. Weinstein, R. Weiss, T. Welborn, L. Wen, P. Weßels,
T. Westphal, K. Wette, J. T. Whelan, S. E. Whitcomb, D. J. White, B. F. Whiting,
K. Wiesner, C. Wilkinson, P. A. Willems, L. Williams, R. D. Williams, A. R.
Williamson, J. L. Willis, B. Willke, M. H. Wimmer, L. Winkelmann, W. Winkler,
C. C. Wipf, A. G. Wiseman, H. Wittel, G. Woan, J. Worden, J. L. Wright, G. Wu,

138
J. Yablon, I. Yakushin, W. Yam, H. Yamamoto, C. C. Yancey, M. J. Yap, H. Yu,
M. Yvert, A. Zadrożny, L. Zangrando, M. Zanolin, J.-P. Zendri, M. Zevin, F. Zhang,
L. Zhang, M. Zhang, Y. Zhang, C. Zhao, M. Zhou, Z. Zhou, X. J. Zhu, M. E. Zucker,
S. E. Zuraw, and J. Zweizig, "Observation of Gravitational Waves from a Binary
Black Hole Merger," Phys. Rev. Lett. 116, 61102 (2016).
50. M. Choma, M. Sarunic, C. Yang, and J. Izatt, "Sensitivity advantage of swept source
and Fourier domain optical coherence tomography," Opt. Express 11, 2183 (2003).
51. J. F. De Boer, B. Cense, B. H. Park, M. C. Pierce, G. J. Tearney, and B. E. Bouma,
"Improved signal-to-noise ratio in spectral-domain compared with time-domain
optical coherence tomography," Opt. Lett. 28, 2067–2069 (2003).
52. J. R. Mourant, M. Canpolat, C. Brocker, O. Esponda-Ramos, T. M. Johnson, A.
Matanock, K. Stetter, and J. P. Freyer, "Light scattering from cells: the contribution
of the nucleus and the effects of proliferative status," J. Biomed. Opt. 5, 131 (2000).
53. R. Brown, "A brief account of microscopical observations made in the months of
June, July and August 1827, on the particles contained in the pollen of plants; and
on the general existence of active molecules in organic and inorganic bodies," Philos.
Mag. Ser. 2 4, 161–173 (1828).
54. A. Einstein, Investigations on the Theory of the Brownian Movement (Dover
Publications, 1956).
55. R. Dzakpasu and D. Axelrod, "Dynamic light scattering microscopy. A novel optical
technique to image submicroscopic motions. I: theory.," Biophys. J. 87, 1279–87
(2004).

139
56. L. R. Rayleigh, "Investigations in optics, with special reference to the spectroscope,"
Philos. Mag. Ser. 5 8, 261–274 (1879).
57. A. Ning, J. Cui, E. To, K. H. Ashe, and J. Matsubara, "Amyloid-beta deposits lead
to retinal degeneration in a mouse model of Alzheimer disease.," Invest.
Ophthalmol. Vis. Sci. 49, 5136–5143 (2008).
58. B. Liu, S. Rasool, Z. Yang, C. G. Glabe, S. S. Schreiber, J. Ge, and Z. Tan,
"Amyloid-peptide vaccinations reduce {beta}-amyloid plaques but exacerbate
vascular deposition and inflammation in the retina of Alzheimer’s transgenic mice.,"
Am. J. Pathol. 175, 2099–2110 (2009).
59. G. Lamouche, B. F. Kennedy, K. M. Kennedy, C.-E. Bisaillon, A. Curatolo, G.
Campbell, V. Pazos, and D. D. Sampson, "Review of tissue simulating phantoms
with controllable optical, mechanical and structural properties for use in optical
coherence tomography," Biomed. Opt. Express 3, 1381 (2012).
60. K. Jeong, J. J. Turek, and D. D. Nolte, "Speckle fluctuation spectroscopy of
intracellular motion in living tissue using coherence-domain digital holography.," J.
Biomed. Opt. 15, 30514 (2014).
61. J. Buitink, O. Leprince, M. A. Hemminga, and F. A. Hoekstra, "Molecular mobility
in the cytoplasm: an approach to describe and predict lifespan of dry germplasm.,"
Proc. Natl. Acad. Sci. U. S. A. 97, 2385–90 (2000).
62. Bishop, I. Villanueva, S. Gladem, and Bryant, "Medium Osmolarity Influences
Chondrocyte Survival During Photoencapsulation in Poly(ethylene glycol)
Hydrogels 1," J. Cell. Physiol. 11, 2385–2385 (2007).

140
63. E. H. Luh, S. R. Shackford, M. A. Shatos, and J. A. Pietropaoli, "The Effects of
Hyperosmolarity on the Viability and Function of Endothelial Cells 1," J. Surg. Res.
60, 122–128 (1996).
64. J. Overgaard, "Influence of extracellular ph on the viability and morphology of
tumor cells exposed to hyperthermia," J. Natl. Cancer Inst. 56, 1243–1250 (1976).
65. A. Jagielska, K. D. Wilhite, and K. J. Van Vliet, "Extracellular Acidic pH Inhibits
Oligodendrocyte Precursor Viability, Migration, and Differentiation," PLoS One 8,
e76048 (2013).
66. Y.-F. Zhang, T.-Q. Xiong, B.-H. Tan, Y. Song, S.-L. Li, L.-B. Yang, and Y.-C. Li,
"Pilocarpine-induced epilepsy is associated with actin cytoskeleton reorganization
in the mossy fiber-CA3 synapses," Epilepsy Res. 108, 379–389 (2014).
67. L. Tian, S. A. Hires, T. Mao, D. Huber, M. E. Chiappe, S. H. Chalasani, L. Petreanu,
J. Akerboom, S. A. McKinney, E. R. Schreiter, C. I. Bargmann, V. Jayaraman, K.
Svoboda, and L. L. Looger, "Imaging neural activity in worms, flies and mice with
improved GCaMP calcium indicators," Nat. Methods 6, 875–881 (2009).
68. M. A. Contín, M. M. Benedetto, M. L. Quinteros-Quintana, and M. E. Guido, "Light
pollution: the possible consequences of excessive illumination on retina," Eye 30,
255–263 (2016).
69. T.-Y. Yu, M. L. Acosta, S. Ready, Y.-L. Cheong, and M. Kalloniatis, "Light
exposure causes functional changes in the retina: increased photoreceptor cation
channel permeability, photoreceptor apoptosis, and altered retinal metabolic
function," J. Neurochem. 103, 714–724 (2007).

141
70. A. Ames and F. B. Nesbett, "In Vitro Retina as an Experimental Model of the Central
Nervous System," J. Neurochem. 37, 867–877 (1981).
71. K. H. Kim, M. Puoris’haag, G. N. Maguluri, Y. Umino, K. Cusato, R. B. Barlow,
and J. F. de Boer, "Monitoring mouse retinal degeneration with high-resolution
spectral-domain optical coherence tomography," J Vis 8, 17 1-11 (2008).
72. S.-K. Song, S.-W. Sun, W.-K. Ju, S.-J. Lin, A. H. Cross, and A. H. Neufeld,
"Diffusion tensor imaging detects and differentiates axon and myelin degeneration
in mouse optic nerve after retinal ischemia," Neuroimage 20, 1714–1722 (2003).
73. E. R. Büchi, "Cell death in the rat retina after a pressure-induced ischaemia-
reperfusion insult: An electron microscopic study. I. Ganglion cell layer and inner
nuclear layer," Exp. Eye Res. 55, 605–613 (1992).
74. L. Gan, M. Xiangts, L. Zhout, D. S. Wagner, W. H. Klein, J. Nathansst, and J. W.
Littlefield, "POU domain factor Brn-3b is required for the development of a large
set of retinal ganglion cells," Dev. Biol. 93, 3920–3925 (1996).
75. Y. Jian, R. J. Zawadzki, and M. V. Sarunic, "Adaptive optics optical coherence
tomography for in vivo mouse retinal imaging," J. Biomed. Opt. 18, 56007 (2013).
76. Y. Geng, A. Dubra, L. Yin, W. H. Merigan, R. Sharma, R. T. Libby, and D. R.
Williams, "Adaptive optics retinal imaging in the living mouse eye," Biomed. Opt.
Express 3, 715 (2012).
77. B. Lin, S. W. Wang, and R. H. Masland, "Retinal Ganglion Cell Type, Size, and
Spacing Can Be Specified Independent of Homotypic Dendritic Contacts," Neuron
43, 475–485 (2004).

142
78. T. Pederson, "Movement and localization of RNA in the cell nucleus.," FASEB J.
13 Suppl 2, S238-42 (1999).
79. L. J. Misantone and M. GERSHENBAUMand MARION, "Viability of retinal
ganglion cells after optic nerve crush in adult rats," J. Neurocytol. 13, 449–465
(1984).
80. Y. Liu, X. Xu, R. Tang, G. Chen, X. Lei, L. Gao, W. Li, and Y. Chen, "Viability of
primary cultured retinal neurons in a hyperglycemic condition.," Neural Regen. Res.
8, 410–9 (2013).
81. P. M. Schulte, "The effects of temperature on aerobic metabolism: towards a
mechanistic understanding of the responses of ectotherms to a changing
environment," J. Exp. Biol. 218, 1856–1866 (2015).
82. R. C. Baker and R. E. Kramer, "CYTOTOXICITY OF SHORT-CHAIN
ALCOHOLS," Annu. Rev. Pharmacol. Toxicol. 39, 127–150 (1999).
83. X. Cheng, J. Sherman, W. Murphy, E. Ratovitski, J. Canady, and M. Keidar, "The
Effect of Tuning Cold Plasma Composition on Glioblastoma Cell Viability," PLoS
One 9, e98652 (2014).
84. N. Groulx, F. Boudreault, S. N. Orlov, and R. Grygorczyk, "Membrane Reserves
and Hypotonic Cell Swelling," J. Membr. Biol. 214, 43–56 (2006).
85. P. L. Pratt, J. H. Bryce, and G. G. Stewart, "The Effects of Osmotic Pressure and
Ethanol on Yeast Viability and Morphology," J. Inst. Brew. 109, 218–228 (2003).
86. M. J. McCarthy, J. Baumber, P. H. Kass, and S. A. Meyers, "Osmotic stress induces
oxidative cell damage to rhesus macaque spermatozoa.," Biol. Reprod. 82, 644–51

143
(2010).
87. P. O’Herron, P. Y. Chhatbar, M. Levy, Z. Shen, A. E. Schramm, Z. Lu, and P. Kara,
"Neural correlates of single-vessel haemodynamic responses in vivo," Nature 534,
378–382 (2016).
88. Alfred E. Koehler and Raymond J. Reitzel, Rockefeller Institute for Medical
Research., and American Society for Biochemistry and Molecular Biology., "THE
EFFECT OF pH ON THE OXYGEN CONSUMPTION OF TISSUES," J. Biol.
Chem. 64, 739–751 (1925).
89. J. L. Perry, N. K. Ramachandran, B. Utama, and J. M. Hyser, "Use of genetically-
encoded calcium indicators for live cell calcium imaging and localization in virus-
infected cells.," Methods 90, 28–38 (2015).
90. M. Zanoni, F. Piccinini, C. Arienti, A. Zamagni, S. Santi, R. Polico, A. Bevilacqua,
and A. Tesei, "3D tumor spheroid models for in vitro therapeutic screening: a
systematic approach to enhance the biological relevance of data obtained," Sci. Rep.
6, 19103 (2016).
91. N. T. Elliott and F. Yuan, "A Review of Three-Dimensional In Vitro Tissue Models
for Drug Discovery and Transport Studies," J. Pharm. Sci. 100, 59–74 (2011).
92. C. M. West, "Size-dependent resistance of human tumour spheroids to
photodynamic treatment.," Br. J. Cancer 59, 510–4 (1989).
93. J. Bin Kim, R. Stein, and M. J. O’Hare, "Three-dimensional in vitro tissue culture
models of breast cancer — a review," Breast Cancer Res. Treat. 85, 281–291 (2004).
94. T.-M. Achilli, J. Meyer, and J. R. Morgan, "Advances in the formation, use and

144
understanding of multi-cellular spheroids.," Expert Opin. Biol. Ther. 12, 1347–60
(2012).
95. R. Edmondson, J. J. Broglie, A. F. Adcock, and L. Yang, "Three-Dimensional Cell
Culture Systems and Their Applications in Drug Discovery and Cell-Based
Biosensors," Assay Drug Dev. Technol. 12, 207–218 (2014).
96. P. A. Kenny, G. Y. Lee, C. A. Myers, R. M. Neve, J. R. Semeiks, P. T. Spellman,
K. Lorenz, E. H. Lee, M. H. Barcellos-Hoff, O. W. Petersen, J. W. Gray, and M. J.
Bissell, "The morphologies of breast cancer cell lines in three-dimensional assays
correlate with their profiles of gene expression.," Mol. Oncol. 1, 84–96 (2007).
97. D. W. Leonard and K. M. Meek, "Refractive Indices of the Collagen Fibrils and
Extrafibrillar Material of the Corneal Stroma," Biophys. J. 72, 1382–1387 (1997).
98. S. H. Kim, H. Y. Lee, S. P. Jung, S. Kim, J. E. Lee, S. J. Nam, and J. W. Bae, "Role
of secreted type I collagen derived from stromal cells in two breast cancer cell
lines.," Oncol. Lett. 8, 507–512 (2014).
99. F. Gattazzo, A. Urciuolo, and P. Bonaldo, "Extracellular matrix: a dynamic
microenvironment for stem cell niche.," Biochim. Biophys. Acta 1840, 2506–19
(2014).
100. M. A. Carlson and M. T. Longaker, "The fibroblast-populated collagen matrix as a
model of wound healing: a review of the evidence," Wound Repair Regen. 12, 134–
147 (2004).
101. T. R. Cox and J. T. Erler, "Remodeling and homeostasis of the extracellular matrix:
implications for fibrotic diseases and cancer.," Dis. Model. Mech. 4, 165–78 (2011).

145
102. C. Österholm, N. Lu, Å. Lidén, T. V. Karlsen, D. Gullberg, R. K. Reed, and M.
Kusche-Gullberg, "Fibroblast EXT1-Levels Influence Tumor Cell Proliferation and
Migration in Composite Spheroids," PLoS One 7, e41334 (2012).
103. N. B. Javitt, "Hep G2 cells as a resource for metabolic studies: lipoprotein,
cholesterol, and bile acids.," FASEB J. 4, 161–8 (1990).
104. A. Blumlein, N. Williams, and J. J. McManus, "The mechanical properties of
individual cell spheroids.," Sci. Rep. 7, 7346 (2017).
105. C. A. Czajka, A. N. Mehesz, T. C. Trusk, M. J. Yost, and C. J. Drake, "Scaffold-
free tissue engineering: organization of the tissue cytoskeleton and its effects on
tissue shape.," Ann. Biomed. Eng. 42, 1049–61 (2014).
106. M. Ingram, G. B. Techy, R. Saroufeem, O. Yazan, K. S. Narayan, T. J. Goodwin,
and G. F. Spaulding, "Three-dimensional growth patterns of various human tumor
cell lines in simulated microgravity of a NASA bioreactor," Vitr. Cell. Dev. Biol. -
Anim. 33, 459–466 (1997).


![Scattering rules in soliton cellular automata associated ...eprints.uthm.edu.my/9425/1/J4158_b1b2e8cecf823cd597df86bf69d… · Subsequently, in [2] new soliton cellular automata were](https://img.dokumen.tips/doc/110x75/5f06971e7e708231d418bf0b/scattering-rules-in-soliton-cellular-automata-associated-subsequently-in-2.jpg)


























