Embed Size (px)
Citation preview

Case ScenariosCase ScenariosIEM IEM
Majid AlfadhelGeneticist and PediatricianGeneticist and Pediatrician
MD,MHSc,SSCMD,MHSc,SSC--PedPed, ABHS(CH), FCCMG, ABHS(CH), FCCMG
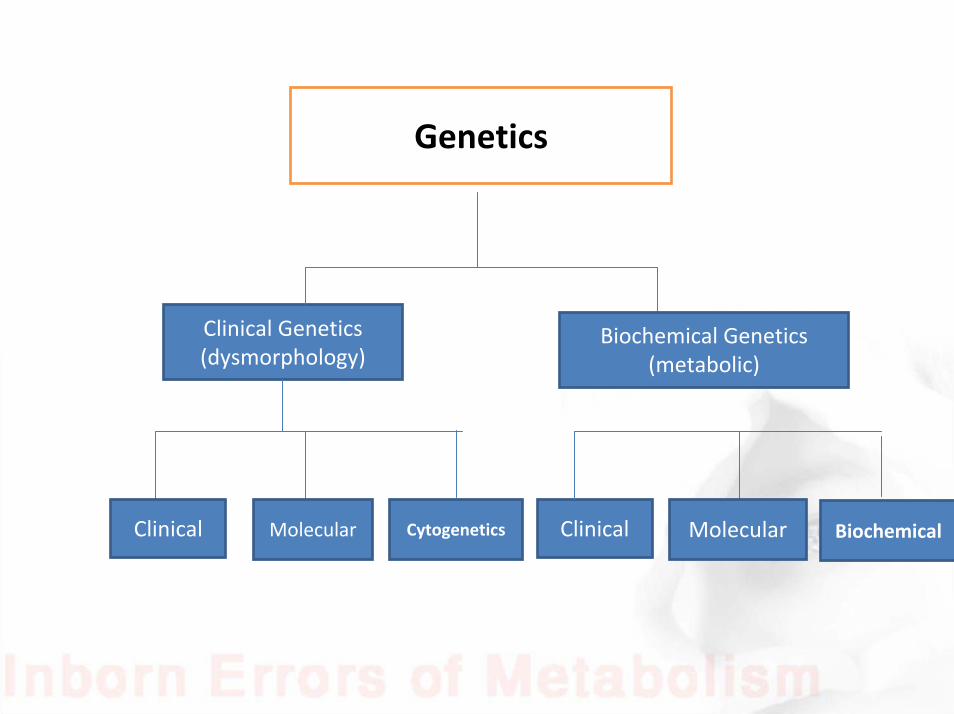
Genetics
Clinical Genetics (dysmorphology)
Biochemical Genetics (metabolic)
MolecularClinical Cytogenetics MolecularClinical Biochemical

How IEM happened
• Monogenic disease results from the absence or abnormality of an enzyme or its cofactor, leading to either accumulation or deficiency of a specific metabolite.
CofactorSubstrate Product
A outside A inside
A inside A outside

How IEM happened
• Monogenic disease result from the absence or abnormality of an enzyme or its cofactor, leading to either accumulation or deficiency of a specific metabolite.
CofactorSubstrate Product
A outside A inside
A outside A inside
Inborn Errors of Metabolism
A CB
D
BB BB
A1. deficiency
of product
accumulation of substrate
formation of unusual metabolites
(s)

Classification
• Acute vs chronic• Newborn, infancy, childhood, adult• Small molecules vs large molecules
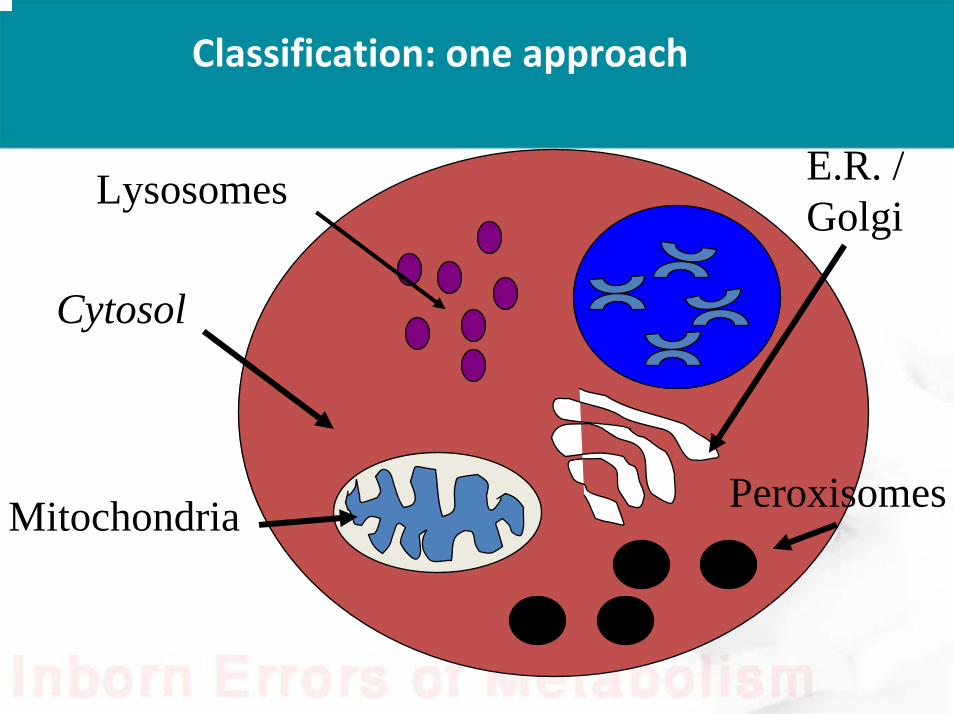
Classification: one approach
Lysosomes
Mitochondria Peroxisomes
E.R. / Golgi
Cytosol

Inborn Errors of Metabolism:a functional classification
I: Disorders of intermediary metabolism that give rise to “intoxication”(aka "small molecule" disorders)
II: Disorders of energy generation
III: Disorders of complex molecules
Reference: Inborn Metabolic Diseases, 3rd Ed. Fernandes...Saudubray

I. Disorders of intermediary metabolism that give rise to “intoxication”
• Degradative pathways of amino acids
• Organic acidemias
• Urea cycle disorders
• Carbohydrate (“sugar”) intolerances
• Purine and pyrimidine disorders
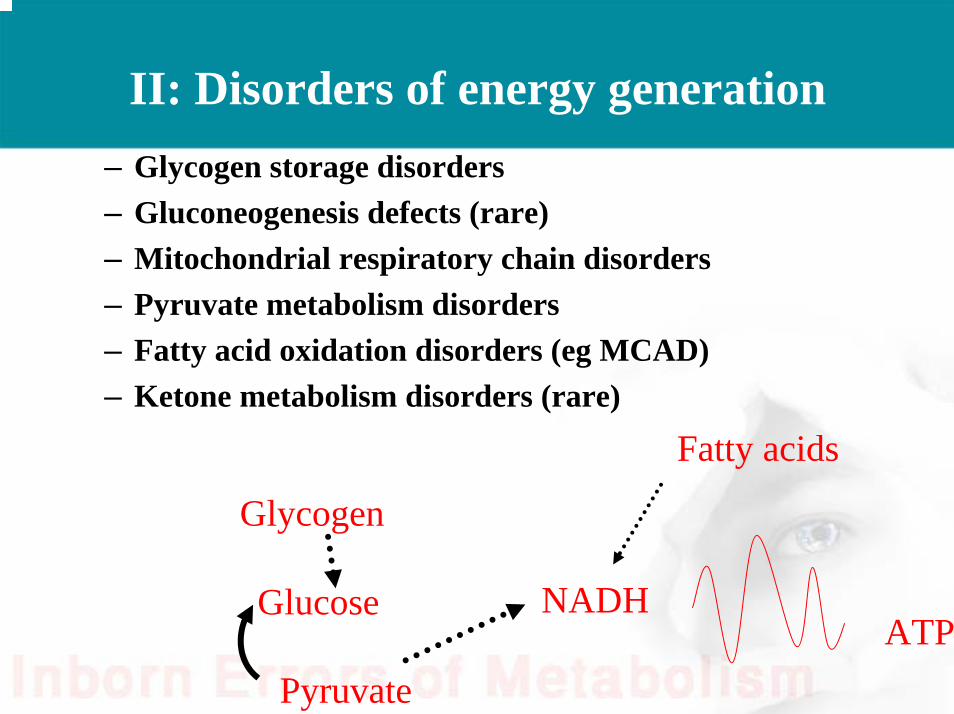
II: Disorders of energy generation– Glycogen storage disorders– Gluconeogenesis defects (rare)– Mitochondrial respiratory chain disorders– Pyruvate metabolism disorders – Fatty acid oxidation disorders (eg MCAD)– Ketone metabolism disorders (rare)
Glycogen
Glucose
Pyruvate
NADHATP
Fatty acids

III: Disorders of Complex Molecules
• Symptoms are permanent, progressive • Symptoms not related to food intake• Few abnormalities seen on "routine" chemistry
• Classification– Lysosomal storage disorders– Peroxisomal disorders– Congenital disorders of glycosylation (CDGS)– Inborn errors of cholesterol synthesis

Facts• IEM very heterogeneous groups of disorders
– Same disorder can present in neonate, infant, childhood and adult
– Same disorder has different outcome in 2 siblings– Poor correlation between clinical phenotype and genotype
• IEM not limited to pediatrics• IEM can be limited to a single organ and commonly
involves multiple organs• Many IEM disorders are treatable• Clinicians should have high index of suspicion to
recognize such disorders

Case ScenariosIEM

Case #1
• 3 days old infant presented to ER with poor feeding, vomiting, lethargy.

Case #1
• Product of FT NSVD.• Completely well until 3rd day of life.• Antenatal history: unremarkable.• Normal growth parameters.• History of fever but not documented.• Family history: unremarkable.• O/E : growth parameters: normal, CNS
:deep coma.

Case #1
• Investigations:– CBC: benign– Bun: <1, Creatinine:62, Na: 140 , K: 4 , Cl: 100,
co2: 18– Blood gas: PH: 7.48 (7.38-7.44)
Pco2: 20 (23-28 mmol/l)HCO3: 22 (21-28 mmol/l)
– Ammonia: 1000

Questions?
• What is ammonia?• How is it produced?• How to detox from the body ?• What are normal reference ranges?

Answers
• A compound of nitrogen and hydrogen with the formula NH3. It is a colourless gas with a characteristic pungent odor.
• Transamination of aminoacids and deamination of glutamate.
• Detoxified by:– Glutamine synthesis.– Urea Cycle.
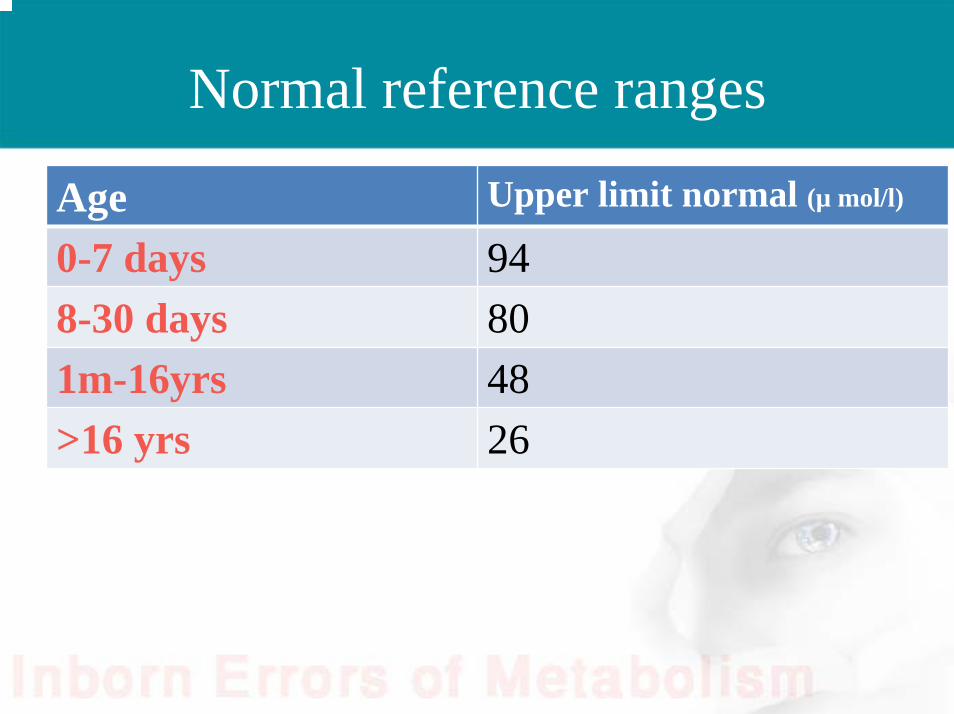
Normal reference ranges
Age Upper limit normal (µ mol/l)
0-7 days 948-30 days 801m-16yrs 48>16 yrs 26

Question
• List 4 metabolic genetics causes of hyperammonemia?
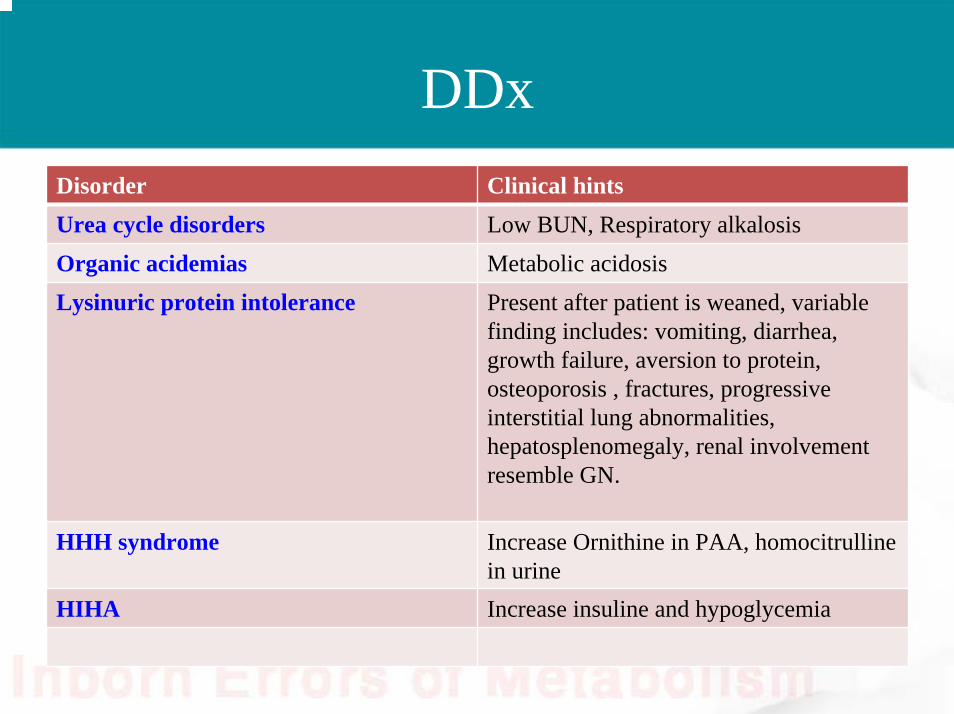
DDxDisorder Clinical hintsUrea cycle disorders Low BUN, Respiratory alkalosisOrganic acidemias Metabolic acidosisLysinuric protein intolerance Present after patient is weaned, variable
finding includes: vomiting, diarrhea, growth failure, aversion to protein, osteoporosis , fractures, progressive interstitial lung abnormalities, hepatosplenomegaly, renal involvement resemble GN.
HHH syndrome Increase Ornithine in PAA, homocitrullinein urine
HIHA Increase insuline and hypoglycemia

DDxDisorder Clinical hintsFAOD 4months-4yrs, acute hypoglycemic
encephalopathy and liver dysfunction precipitated by prolong fasting or URTI
Pyruvate dehydrogenase deficiency Lactic acidosis, psycomotor retardation, hyppotonia, epilepsy
Pyruvate carboxylase deficiency Lactic acidosis, psycomotor retardation, hyppotonia, epilepsy
Transient hyperammonemia of newborn
Preterm, RDS

Clinical differences THAN vs UCD
Clinical finding Clinical finding THANTHAN UCDUCD
HistoryHistory Sick immediately after Sick immediately after birthbirth
Sick after the first daySick after the first day
CXRCXR AbnormalAbnormal NormalNormal
Gestational ageGestational age Preterm Preterm Term Term
Birth weightBirth weight < 2500< 2500 >2500>2500
ComaComa < 48 h< 48 h > 48 h> 48 h
NH3NH3 > 1500> 1500 <1500<1500
Blood gasBlood gas AcidosisAcidosis AlkalosisAlkalosis

• PAA profile :showed decrease citrulline and arginine while glutamine is increased.
• UOA :showed increase orotic acid excretion.

Question?
• What is the diagnosis of the previous case?

Diagnosis
Ornithinetranscarbamylasedeficiency


Urea cycle disorders
• Characterized by the triad of hyperammonemia, encephalopathy and respiratory alkalosis

Question?
• List 4 enzyme deficiencies causing urea cycle disorders?

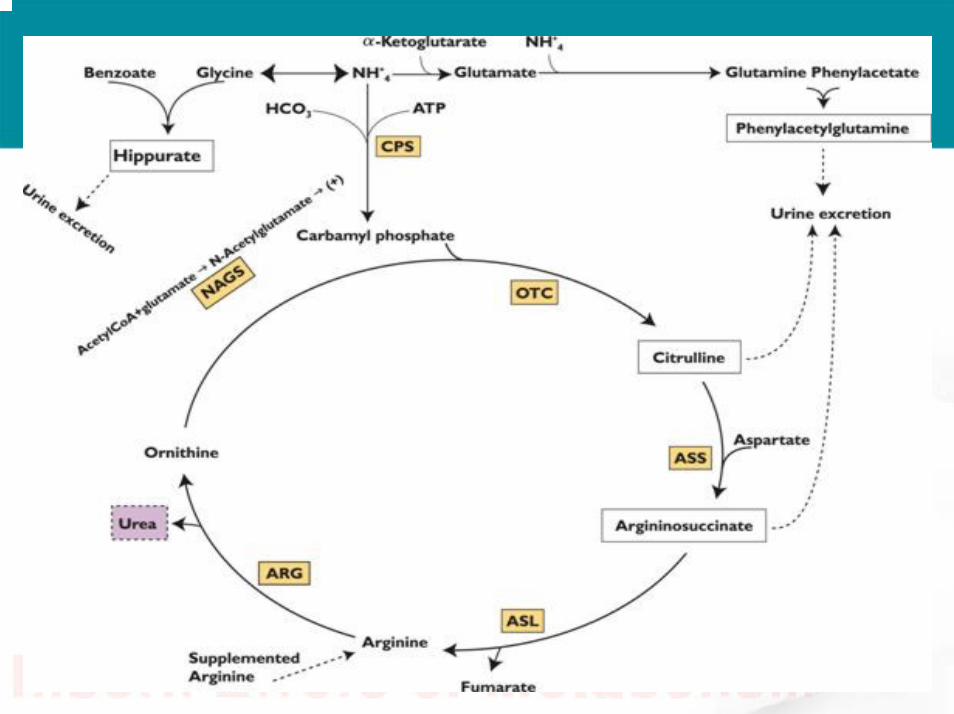
•• Enzyme deficiency include :Enzyme deficiency include :– N- acetylglutamate synthase (NAGS)– Carbamyl phosphate synthase(CPS)– Ornithine transcarbamylase (OTC)– Argininosuccinic acid synthatase(ASS)– Argininosuccinic acid lyase(ASL)– Arginase.

Genetics
• all are autosomal recessive except OTC deficiency X-linked dominant

Classical neonatal form
• Most babies of normal birth weight and are initially healthy but after short intervals , they become unwell, common early symptoms are poor feeding ,vomiting , lethargy ,irritability that progress to seizure ,deep coma hyperventilation, respiratory alkalosis and even death if not treated .

Classical neonatal form
• the liver may be enlarged and serum level of transaminase are often elevated.
• In OTC deficiency the classical form are more common in male however small number of female may have also similar presentation

Late onset form• Less frequent.• Heterogenous presentation.• Recurrent or cyclic vomiting• Episodic Hyperammonemia• Develop. Delay ,MR,Psychatric sign.• Hepatomegally and elevated transaminase.• Asymptomatic

Additional clinical abnormalities
DiseaseDisease C/FC/F
ASS def.ASS def. Dysmorphic(MicrocephalyDysmorphic(Microcephaly,,brittle hairbrittle hair
ASL def.ASL def. Hepatomegaly,brittleHepatomegaly,brittle hairhair
Heterozygous OTCHeterozygous OTC Post partum Post partum hyperammonemiahyperammonemia
ArginseArginse def.def. Spastic Spastic diplegiadiplegiaAmmonia normal or mild Ammonia normal or mild
elevatedelevatedPMR,SeizurePMR,Seizure

• How to differentiate between different types of urea cycle disorders?
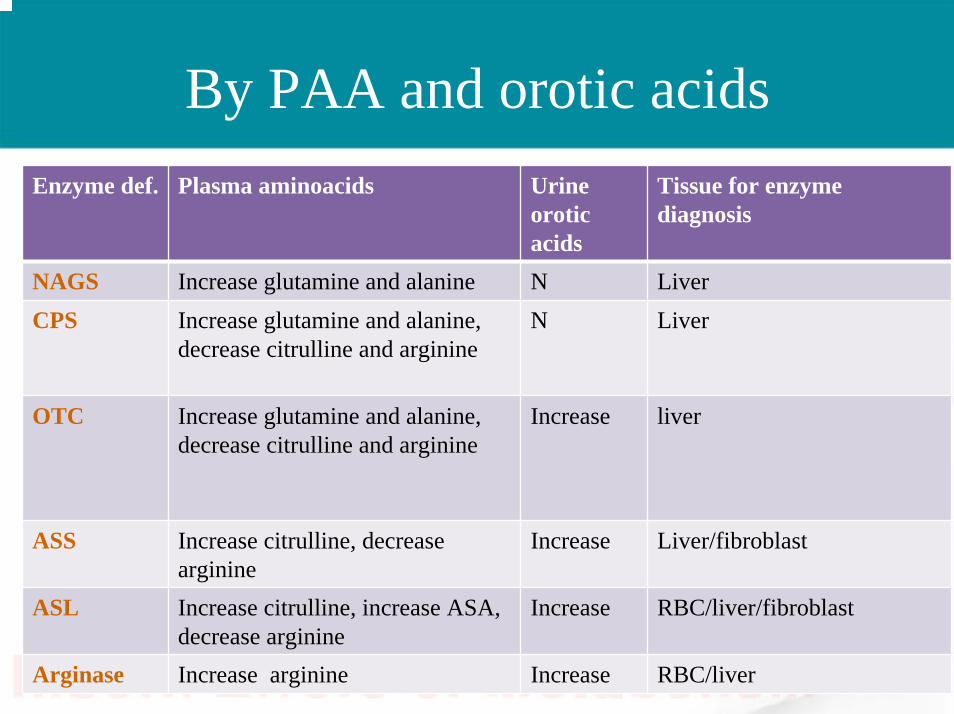
By PAA and orotic acidsEnzyme def. Plasma aminoacids Urine
oroticacids
Tissue for enzyme diagnosis
NAGS Increase glutamine and alanine N LiverCPS Increase glutamine and alanine,
decrease citrulline and arginineN Liver
OTC Increase glutamine and alanine, decrease citrulline and arginine
Increase liver
ASS Increase citrulline, decrease arginine
Increase Liver/fibroblast
ASL Increase citrulline, increase ASA, decrease arginine
Increase RBC/liver/fibroblast
Arginase Increase arginine Increase RBC/liver

Management
• A,B,C: The patient with urea cycle should have multilumen central line placed or umbilical catheter.
• Hydration • High caloric intake• Hyperammonemia scavengers• Dialysis

• It is crucial to take blood for plasma aminoacids and urine for orotic acids before start treatment

Question?
• What do we mean by high caloric intake?

Answer
• Covering 110% of the recommended daily allowance (RDA) in order to shut down endogenous protein breakage ( RDA: for newborn and infant: 110-120 kcal/kg/day, for 1-3 yrs:100, for 4-6yrs: 90, for 7-10yrs: 70, for 11-14 yrs: 50-55).

Questions
• How did you achieve high caloric intake?• Calculate the required fluids and lipids for
an infant if his weight: 10 kg?

Answers
• Dextrose + intralipids 20%• 60 X 0.34X 24/10= 50 kcal• 20gram of lipid= 200Kcal/10= 20 Kcal• Total= 60 Kcal• Is this enough?

• What are the indications for dialysis ?

Dialysis
Services Services Once you suspect to start Dialysis in next few hour please Once you suspect to start Dialysis in next few hour please consult ICU and Nephrology team immediatelyconsult ICU and Nephrology team immediately
IndicationIndication If Ammonia doesnIf Ammonia doesn’’t decrease tot decrease to< 200 < 200 umol/lumol/l within 4 hour from start therapywithin 4 hour from start therapy
HypeammonemicHypeammonemic comacomaAmmonia >400 initiallyAmmonia >400 initially
DiscontinueDiscontinue oo Ammonia < 200 Ammonia < 200 mmol/lmmol/loo Risk of rebound overcome by use of Risk of rebound overcome by use of
hemofiltrationhemofiltration..
MethodMethod Depends on the available expertise and equipment.Depends on the available expertise and equipment.
Best recommended Best recommended methodmethod
PumpPump--driven dialysis technique : continuous driven dialysis technique : continuous hemofiltrationhemofiltration with the same pump system, with the same pump system, egeg. CVVH. CVVH
Removal of Removal of cathetercatheter
After the plasma ammonia level has been stable for at After the plasma ammonia level has been stable for at least a day. least a day.
However, this should be balanced by the risk of However, this should be balanced by the risk of maintaining the patient in an maintaining the patient in an anticoagulatedanticoagulated statestate

Questions
• What are hyperammonemia scavengers?• Dosages?• Side effects?

ArginineArginine hydrochloride (200hydrochloride (200--600mg/kg) diluted in 600mg/kg) diluted in D10w given as bolus over 90 minute followed by D10w given as bolus over 90 minute followed by continuous IV infusion as maintenance.continuous IV infusion as maintenance.
ArginineArginine hydrochloride vial: 30 ml.hydrochloride vial: 30 ml.AmmonulAmmonul ((Sodium benzoate and Sodium Sodium benzoate and Sodium phenylacetatephenylacetate)) (250mg/kg or 5.5mg/m(250mg/kg or 5.5mg/m22 followed by followed by 250250--500mg/kg as continuous IV infusion as 500mg/kg as continuous IV infusion as maintenance.maintenance.Potassium can be given in the same line of Potassium can be given in the same line of AmmonulAmmonulinfusion*.infusion*.Reloading only in neonates with severe disorders or Reloading only in neonates with severe disorders or those who are undergoing dialysis, and should be those who are undergoing dialysis, and should be spaced at least 6 hours.spaced at least 6 hours.

• These medication should be given through central line, can be given through peripheral line on limited bases.
• Don’t exceed 500 mg/kg/dy of sodium benzoate and phenylacetate.

•• Note:Note:– Ammonul contain 30.5 mg/ml of sodium =1.3meq/ml– Concentration: Na benzoate 100 mg/ml ,phenylacetate 100
mg/ml.– Ammonul vial: 50 ml.

PharmacPharmacokineticsokinetics
NH3 NH3 Removal Removal
MOAMOAusesuses
1.5 hour1.5 hourMetabolism : Metabolism : liverliver
1 mole of 1 mole of sodium sodium benzoate benzoate remove 1mole remove 1mole of ammonia as of ammonia as glycineglycine
conjugate conjugate with glycine with glycine to form to form hippurichippuric acidacid
Acute ,some Acute ,some use it in use it in chronic if PB chronic if PB failfail
Sodium Sodium benzoatebenzoate
2 hour 2 hour Metabolism : via Metabolism : via acetylationacetylation in in liver and kidneyliver and kidney
1 mole of 1 mole of phenylaceatephenylaceateremove 2 mole of remove 2 mole of ammonia as ammonia as glutamineglutamine
conjugate with conjugate with glutamine to glutamine to form form phenylacetylglutaphenylacetylglutaminemine
AcuteAcuteSodium Sodium phenylacetatephenylacetate
1 mole of 1 mole of phenylaceatephenylaceateremove 2 mole of remove 2 mole of ammonia as ammonia as glutamineglutamine
ProdrugProdrug rapidly rapidly convert to PAGconvert to PAGchronicchronicSodium phenyl Sodium phenyl
buteratebuterate

Use in caution with
Congestive heart failureRenal or hepatic dysfunctionHyperbilirubenemiaSodium retention associated with edema

Adverse reaction
Sodium benzoate and sodium phenylacetate :• Cardiovascular: Hypotension (4%)• Dermatologic: Injection site reaction (3%)• Endocrine metabolic: Hyperglycemia (7%),
hypokalemia (7%)• Gastrointestinal: Vomiting (9%) , diarrhoea (3%)• CNS: Altered mental status (6%) , seizure (6%)
,cerebral oedema (5%) • Other: Fever (5%

Case#2
• Mohammed, 7 months old boy with Argininosuccinate Lyase deficiency admitted to PICU while you are luckily on-call that night. He is improving with hyperammonemia scavengers and high caloric intake. You are monitoring electrolytes, ammonia and blood gas q4 hourly. A nurse called you with glucose result of 20 mmol/l. What you will do?

Case#2–– Start Insulin infusion IV :Start Insulin infusion IV :
50 unit diluted in 500 ml NS to be run @ 1/4 -1/2 weight of the patient (0.025-0.05 unit /kg/hour). Usual dilution 0.1 unit/mlOnce infusion started measure glucocheck Q1 hourlyTitrate the insulin infusion rate according to the glucocheck result.The lowest rate through peripheral line is 2 ml/hour and central line 4 ml/hour to keep vein open.

Case#3
• Nojoud, 1 yr old with propionic acidemia. Admitted over the night with acute metabolic crises. Currently, on IVF D10% at 1and1/2 maintenance+ intralipid 20% (2 gram/kg). You are seeing her in the morning. Her electrolytes, ammonia, blood gas within normal. What will you do?

Case#3
• Call metabolic dietitian to start the propiomexformula if not started.
• Titrate IVF and D/C lipids

Case#4
• Saif, 4 years old with argininosuccinatelyase deficiency admitted to the general word with acute metabolic crises 3 days ago. You are preparing him for discharge, what are the parameters you are looking for before discharge?

Case#4
– Normal clinical status as home before crises.• Ammonia level normal.• Check the dosages of medications .

Case#5
• You are doing your daily morning round and one of your patients has urea cycle disorders. One of the junior residents ask you about the role of carnitine in such disorders?

Case#5
– Not believed to be beneficial

Case#6
– Lyan, 4 years old with argininosuccinate lyasedeficiency. Admitted overnight with metabolic crises, she is currently looking well but a nurse called you from general word with blood gas result of • PH: 7.46 (7.38-7.44) • Pco2: 25 (23-28 mmol/l) • HCO3: 15 (21-28 mmol/l). What you will do?

Case#6
• Check the dose of arginine hydrochloride. It could be side effect of medication
• Check hydration status and type of IVF.• Check electrolytes for hyperchloremia and
hypokalemia.• Repeat blood gas could be lab error.

Case#7
• You are seeing in the clinic a 1 year old boy with developmental delay and seizures. Opon examination: Microcephalic and his complexion is lighter than his brothers. His mother said that his urine smell musty. What is the most likely diagnosis?

Case#7
• Phenylketonuria (PKU)

Question?
• What lab investigations will you order?• Why does his urine smell musty?

Answers
• Plasma aminoacids• Because of increase concentration of
phenyllactic acid.

Abnormal odour

Case#8
• 3 days old newborn product of FT, NSVD, BW: 3Kg and AS: 9, 9, found to have poor feeding, lethargy started today.
• Ammonia: 250 (normal<72), blood gas: PH: 7.2
• PaCO2 : 22, HCO3 : 8. What is the most likely diagnosis

Case#8
• Most likely organic acid disorders:– Propionic acidemia– Methylmalonic acidemia

Question?
• What lab investigations will you order?

Answer
• Acylcarnitine profile (TMS)• Plasma aminoacids.• Urine for organic acids.

Organic acidemiasPoor feeding, vomiting, lethargy
metabolic acidosis , hypoglycemia
Ketosis No ketosis
No skin Skin involvement 1. HMG CO A lyase
deficiency2. FAOD3. GA II4. HMG CO A synthetase
def.
Multiple carboxylasedef.
No odour odour
MMAPPA
MSUDIVA

Case#9
• You are on-call and a Newborn Screening Lab called for positive newborn screening for Maple Syrup Urine Disease. What will you do?

Case#9
• Ask the lab to fax you the result.• Ask about the date of birth.• Call the family and take detailed history.• Call Biochemical Geneticist on-call.• Accordingly, you will decide to bring the baby
to ER or bring him tomorrow to repeat the test.

Question
• Which disorders are currently included in the National NBS program in Saudi Arabia?

KAMC started NBS at 31 May 201116 disorders:
– Methylmalonic acidemia (MMA)
– Propionic acidemia (PA)
– Maple syrup Urine disease ( MSUD)
– Arginosuccinate lyase Deficiency (ASL)
– Citrullinemia (Cit)
– HMG‐CoA lyase def.
– Isovaleric acidemia (IVA)
– Glutaric acidemia (GA‐1)
– Betaketothiolase def. (BKT)
‐Medium Chain acyl coAdehydrogenaqse def. (MCAD)
– Phenylketonuria (PKU)– 3‐methylcrotonylcoa
carboxylase def. (3‐MCC)– Galactosemia– Biotinidase def.– Congenital adrenal
hyperplasia– Hypothyroidism

Case#9Case#9
• Baby is 8 days old and has poor feeding and crying all the time since today.
• You received the result from the lab, what amino acids you are looking for in this report?

Answer
• Leucine, Isoleucine and Valine

Case#9Case#9
• Leucine: 3400µmol/l (61-183)• Isoleucine: 900 µmol/l (27-80)• Valine: 780 (78-264)What you will do?

Answer
• Call the family to bring the baby to ER ASAP.• Call biochemical Genetics specialist.• Prepare IV team for peripheral and central IV
line insertion.• Call the pharmacy to prepare intralipid20%• Call the PICU team and Nephrology team for
dialysis.

Case#10Case#10
• A 9 months old child presented with coarse facial features and developmental delay. Eye examination showed corneal clouding, you ordered skeletal survey which showed:
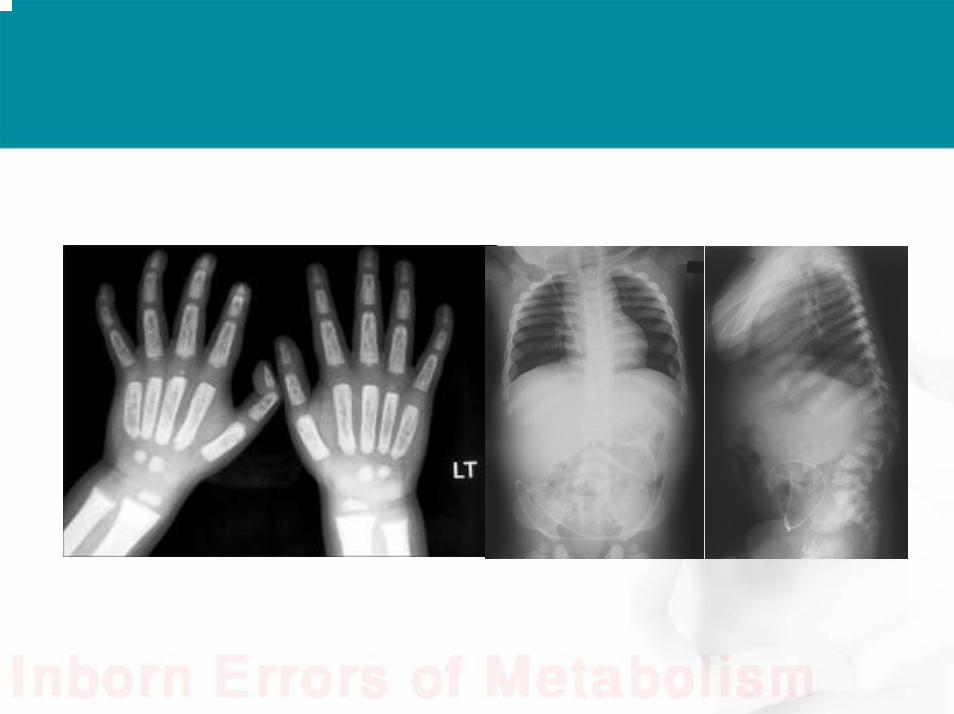

Answer• Dysostosis Multiplex: :Dysostosis: abnormal development
of the bone , Multiplex: multiple .• Spatulated ribs, Shallow acetabulum, vertebral bodies develop
peak-like projection on the lower anterior margin while upper part remains hypoplastic, tappering of terminal phalnges and widening at distal end and tapering at proximal ends of metacarpals
• What is DDx of dysostosis multiplex

DDx• Galactosialidosis• GM1 ganglisidosis• Mucopolysaccharidosis• Mucolipidosis I (Neuroaminidase Deficiency)• Mucolipidosis II ( I cell disease)• Mucolipidosis III• Multiple Sulfatase deficiency• Fucosidosis• Mannosidosis• Infantile sialic acid storage disorder• Geleophysic Dysplasia

Case#10Case#10
• You ordered urine for mucopolysaccharides as part of work-up for this patient and the result came back positive with high dermatan and heparan sulfatase, what is the most likely diagnosis?

• Hurler syndrome or MPSIH


Case#11Case#11• Lyan, an 18 months old girl with
severe developmental delay and coarse facial features as in the picture noted since birth.
• skeletal survey showed periostealreactions in the long bone. Lab investigations showed excessive excretion of oligosaccharides in the urine. What is the most likely diagnosis?

Case#11Case#11
• Mucolipidosis II or I cell disease.

Mucolipidosis
• 4 types• All are autosomal recessive disease
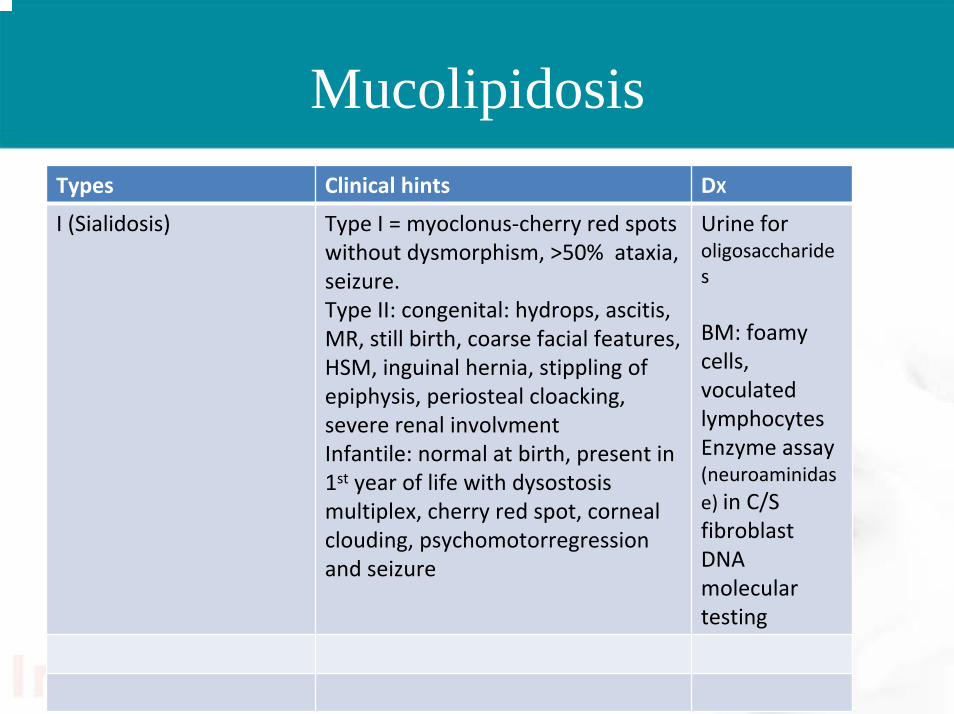
MucolipidosisTypes Clinical hints DX
I (Sialidosis) Type I = myoclonus‐cherry red spots without dysmorphism, >50% ataxia, seizure.Type II: congenital: hydrops, ascitis, MR, still birth, coarse facial features, HSM, inguinal hernia, stippling of epiphysis, periosteal cloacking, severe renal involvmentInfantile: normal at birth, present in 1st year of life with dysostosismultiplex, cherry red spot, corneal clouding, psychomotorregressionand seizure
Urine for oligosaccharides
BM: foamy cells, voculatedlymphocytesEnzyme assay (neuroaminidase) in C/S fibroblastDNA molecular testing
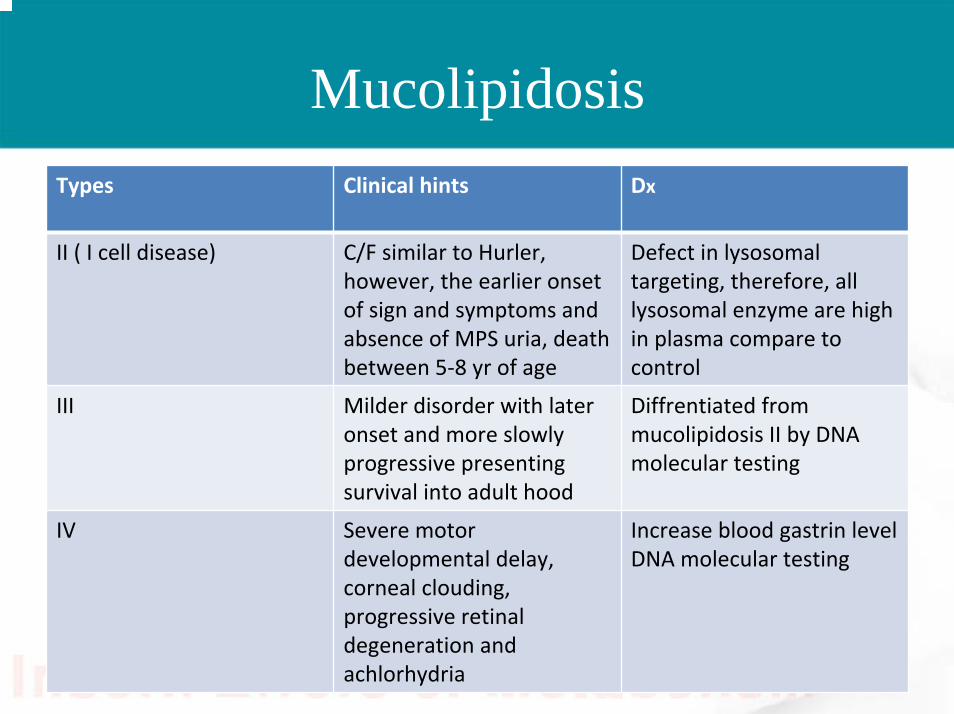
MucolipidosisTypes Clinical hints Dx
II ( I cell disease) C/F similar to Hurler, however, the earlier onset of sign and symptoms and absence of MPS uria, death between 5‐8 yr of age
Defect in lysosomaltargeting, therefore, all lysosomal enzyme are high in plasma compare to control
III Milder disorder with later onset and more slowly progressive presenting survival into adult hood
Diffrentiated from mucolipidosis II by DNA molecular testing
IV Severe motor developmental delay, corneal clouding, progressive retinal degeneration and achlorhydria
Increase blood gastrin levelDNA molecular testing

Case#12Case#12
• A 1 year old child presented to the ER with acute hepatic crises which is precipitated by URTI. He is febrile, irritable, has vomiting and hematemesis, his liver is enlarged and has jaundice, his blood glucose level is low and you started him on IVF D10%. He has high liver enzymes and coagulation abnormalities and smells like boiled cabbage.

Questions?
• What is the diagnosis?• What is the treatment of this condition?

Answers
• Tyrosinemia type I• Diet restricted in phenylalanine and tyrosine• NTBC (2-(nitro-4-trifluoromethylbenzoyl)-
1,3-cyclohexanedione).
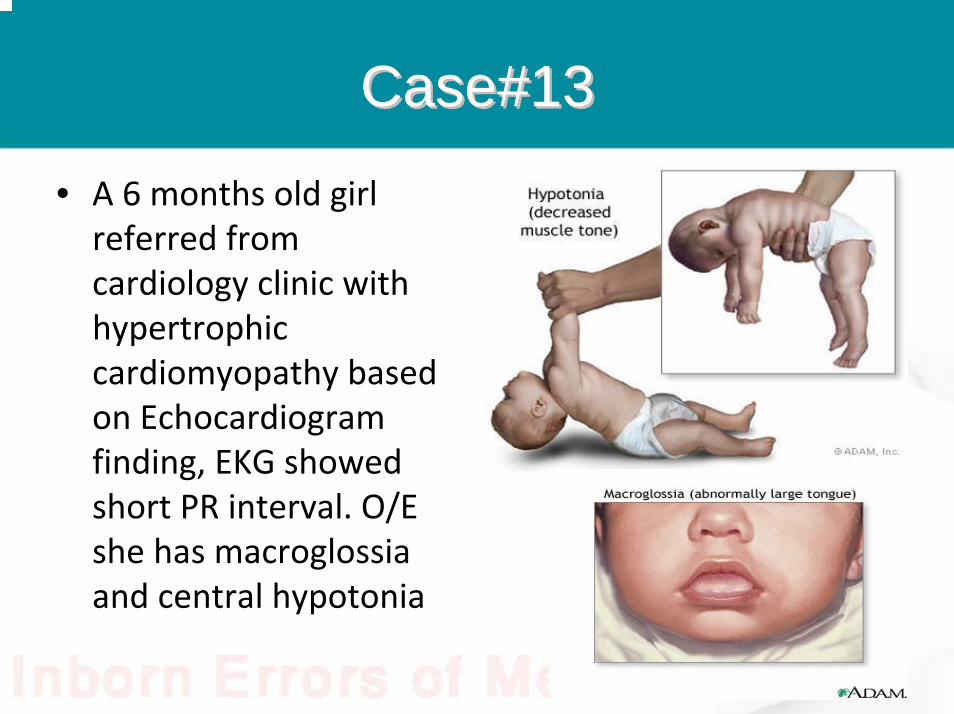
Case#13Case#13
• A 6 months old girl referred from cardiology clinic with hypertrophic cardiomyopathy based on Echocardiogram finding, EKG showed short PR interval. O/E she has macroglossiaand central hypotonia

Questions?Questions?
• What is the most likely diagnosis?• What is the enzyme deficient in this disorder?

Answers
• GSD type II• Alpha glucosidase

GSDs
• 14 types• Liver types: I, III, IV, VI, IX, 0, Fanconi-
Bickel syndrome.• Muscle types: V, VII, phosphoglycerate kinase
def., X, XI, XII, XIII, XIV.• Cardiac involvement: II, IIb, III, Muscle
glycogen depletion syndromes.• Neurodegeneration: Lafora disease, adult
polyglucason disease.

GSDs
• Type IX classifications:– X- linked liver phosphorylase Kinase deficiency – Autosomal combined liver and muscle PKD– Autosomal liver PKD– X-linked muscle PKD– Autosomal muscle PKD– Heart limited PKD

Case#14Case#14
• You are on‐call at NICU and you received a baby who was just delivered with profound hypotonia and poor feeding and dysmorphic as in the picture (high forehead, large AF, hypoplastic supraorbitalridge, epicanthal fold, midface hypoplasia), skeletal survey: epiphysealstippling and renal U/S : renal cyst

Questions?• What is the most likely diagnosis?• Where is the defect located?

Answers
• Zellweger spectrum disorders• Peroxisomal biogenesis

Peroxisomal disorders
• What is peroxisomes?• What are their functions?• What is the classification of peroxisomal
disorders?

Answers• Peroxisomes organelles play a major role in the
oxidation of fatty acids for energy conversation.• Has multiple functions:
– Plasmalogen biosynthesis– FA beta-oxidation– Peroxisomal alpha oxidation– Glyoxylate detoxification– Hydrogen peroxide metabolism– L-pipecolic acid oxidation– Metabolism of bile acids– Cholesterol biosynthesis

Classification– Disorders of peroxisome import:
• Zellweger syndrome.• Neonatal ALD• Infantile Refsum disease• RCDP type I
– Disorders of single peroxisomal enzyme:• X-linked ALD• Bifunctional enzyme deficiency.• Acyl-Co A oxidase deficiency.• 2-methylacyl Co-A racemase deficiency.• Classic Refsum disease• RCDPII• RCDPIII• Acatalasemia• Peroxisomal thiolase deficiency• Glutaric aciduria type III

Questions
• What are the investigations you ordered for diagnosing a patient with suspected peroxisomal disorders?

Answer
• Very long chain fatty acids• Phytanic acid• Pristanic acid• Plasmalogen analysis in RBC• Dihydroxycholestanoic acid and trihydroxy
cholestanoic acid in plasma• Plasma and urine pipecolic acid
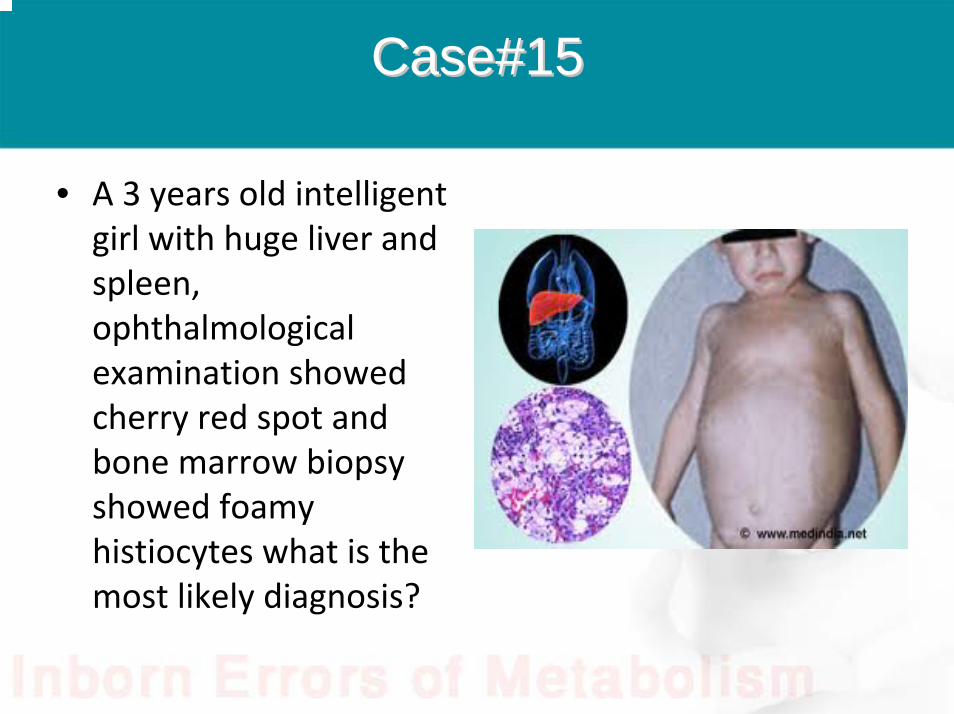
Case#15Case#15
• A 3 years old intelligent girl with huge liver and spleen, ophthalmological examination showed cherry red spot and bone marrow biopsy showed foamy histiocytes what is the most likely diagnosis?

Answer
• Niemann-pick disease type B

Niemann-pick vs GaucherNiemann-Pick Gaucher
Types A, B, C I,II,III, Perinatal lethal and cardiac
Enzyme deficient
Acid sphingomyelinase, C due to defect in lipid trafecking
Acid b-glucocerebrosidase
Cherry red spot
Present in A and B Absent
Treatment ERT for C ERT but not effective in neuronopathic form
Dx Enzyme assay in WBC and C/S fibroblast confirmed by DNA molecular testingFor type C: fillipin staining in C/S fibroblast
Enzyme assay in WBC and C/S fibroblast confirmed by DNA molecular testing

Niemann-Pick Gaucher
Clinical Type A: earlier onset, rapidly progressive, involvement of CNSType B: non-neronopathic, later onset, slowly progressiveBoth: HSM, diffuse lung infiltrate, recurrent chest infections, arthrogenic lipid profile and xanthomas.Type C: hydrops, neonatal ascitis, cholestatic jaundice, HSM, at childhood: ataxia, supranuclear vertical gasepalsy
TypeI: at any age, no CNS involvement, bone disease, splenomegaly > hepatomegaly, hematological manifistations and diffuse lung infiltrate.Type II: neronopathic form rapidly progress death by 2 years (supranuclearhorizontal gase palsy).Type III: subacuteneuronopathic form



![Open Access Genetic Control of -Cell Mass Homeostasis€¦ · Monogenic [MODY6] [146] PDX1 Transcription factor Insulin transcription [147], pancreas development [148] Monogenic [MODY4]](https://img.dokumen.tips/doc/110x75/6110925ed4eda8578404ac9a/open-access-genetic-control-of-cell-mass-homeostasis-monogenic-mody6-146-pdx1.jpg)

























