Embed Size (px)
Citation preview

NEAFS CE-DNA Workshop (Butler and McCord) Sept 29-30, 2004
http://www.cstl.nist.gov/biotech/strbase/NISTpub.htm 1
Capillary Electrophoresis in DNA Analysis
NEAFS WorkshopMystic, CT
September 29-30, 2004Dr. John M. Butler
Dr. Bruce R. McCord
Data Interpretation
Outline for Workshop
• Introductions• STR Analysis• Introduction to CE and ABI 310• Data Interpretation• Additional Topics – Real-time PCR and miniSTRs• Higher Throughput Approaches• Troubleshooting the ABI 310 (Participant Roundtable)• Additional Topics – Y-STRs, validation, accuracy• Review and Test
Mixture of dye-labeled PCR products from
multiplex PCR reaction
Sample Separation
Sample DetectionCCD Panel (with virtual filters)
Argon ion LASER (488 nm)
ColorSeparationFluorescence
ABI Prism spectrograph
Capillary
Sample Injection
SizeSeparation
Processing with GeneScan/Genotyper software
Sample Interpretation
Butler, J.M. (2005) Forensic DNA Typing, 2nd Edition, Figure 13.8, © Elsevier Science/Academic Press
Steps in STR Typing
ABI 310
480 and 9600GeneAmp 9700
NIST Instruments Used for STR Typing
ABI 3100
FMBIO III Gel Imager System
PowerPlex 16 BIO
Penta E
D18S51
D21S11
TH01
D3S1358
Penta D
CSF1PO
D7S820
D13S317
D5S818
D16S539
TPOX
D8S1179
VWA
FGA
Amelogenin
Profiler Plus™
COfiler™
SGM Plus™
Green I
Profiler™
Blue
TH01
Amel D16S539D7S820
CSF1POTPOX
D3S1358
D16S539 D18S51D21S11
Amel
Amel
D3S1358
D3S1358
D18S51D21S11
D8S1179
D7S820
D13S317D5S818
D19S433 D2S1338
FGAvWA
vWA
FGA
TH01
D3S1358 vWA FGA
D7S820D5S818D13S317
TH01CSF1POTPOX
D8S1179
vWATH01 CSF1PO
TPOXAmel FGAD3S1358
Amel
PCR Product Size (bp)We work with all of the different commercial STR kits
We work with all of the different commercial STR kits

NEAFS CE-DNA Workshop (Butler and McCord) Sept 29-30, 2004
http://www.cstl.nist.gov/biotech/strbase/NISTpub.htm 2
Identifiler™ kit multiplex STR result
AMELD3
TH01
TPOX
D2
D19
FGAD21D18
CSFD16
D7
D13D5 VWA
D8
PowerPlex® 16 kit multiplex STR result
AMEL
D3 TH01TPOX
Penta D
Penta E
FGA
D21 D18 CSF
D16
D7D13
D5
VWA
D8
Same DNA Sample with Two Commercial 16plex STR Typing Kits
NIST SRM 2391b Component 1
U.S. Population Study with Identifiler STR Kit
Identifiler™
J. Forensic Sci. 48(4):908-911
0.00000
0.10000
0.20000
0.30000
0.40000
0.50000
5 6 7 8 9 9.3 10 11
Allele
Freq
uenc
y
302 Caucasians 258 African Americans 140 HispanicsTH01302 Caucasians 258 African Americans 140 Hispanics
5 0.00166 0.003886 0.23179 0.12403 0.214297 0.19040 0.42054 0.278578 0.08444 0.19380 0.096439 0.11424 0.15116 0.15000
9.3 0.36755 0.10465 0.2464310 0.00828 0.00194 0.0142911 0.00166
TH01 Allele Frequencies
5 µL PCR protocol on ABI 3100Reagents cost only $3.53 per sample
700 U.S. samples examined
http://www.cstl.nist.gov/biotech/strbase/NISTpop.htm
Sample Interpretation Overview
• Data collection on analytical instrument (e.g., ABI 310 or ABI 3100)
• Peak identification and sizing
• Correlation of peak sizes to STR allele repeat number through comparison with previously or concurrently run allelic ladder
• Review of “called” alleles with editing where needed according to laboratory-specific interpretation guidelines
• Transfer of final reviewed allele calls from STR profile to table of genotypes
Done completely or with aid of computer program(s)
Data Collection
Peak Identification
Data Review by Analyst/Examiner
Color Separation
Peak Sizing
Comparison to Allelic Ladder
Confirmation of Results by Second Analyst/Examiner
Genotype Assignment to Alleles
GeneScansoftware
Genotypersoftware
Internal sizing standard
(e.g., GS500-ROX)
Matrix file
Allelic ladder sample
Steps in STR Genotyping ProcessData Collection
software
Expert Systems under Development
(e.g., True Allele)
Adapted from Butler (2001) Forensic DNA Typing, Figure 13.1
D3S1358 16,17vWA 17,18Amel X,Y (male)D8S1179 12,14
COfiler STR data
GeneScan view
Genotyperview
Allele call (repeat number)determined by comparison of peak size (bp) to allelic ladder allele peak sizes run under the same electrophoretic conditions
Peak height in relative fluorescence units (RFUs)
Three Possible Outcomes
• Match – Peaks between the compared STR profiles have the same genotypes and no unexplainable differences exist between the samples. Statistical evaluation of the significanceof the match is usually reported with the match report.
• Exclusion – The genotype comparison shows profile differences that can only be explained by the two samples originating from different sources.
• Inconclusive – The data does not support a conclusion as to whether the profiles match. This finding might be reported if two analysts remain in disagreement after review and discussion of the data and it is felt that insufficient information exists to support any conclusion.
Butler, J.M. (2001) Forensic DNA Typing, p. 202

NEAFS CE-DNA Workshop (Butler and McCord) Sept 29-30, 2004
http://www.cstl.nist.gov/biotech/strbase/NISTpub.htm 3
Suspect 1
Suspect 2
Evidence
Match (Inclusion)
Exclusion (No match)
Crime Scene STR Profile Compared to Two Suspects Comparison of Allelic Ladder to Samples to Convert Size into Allele Repeat Number
difference = - 0.02 bp
difference = + 0.05 bp
+/- 0.5 bp bin defined around each allele
Precision from Run-to-Run on ABI 310
Size deviation of 70 samples and two allelic ladders from one injection of allelic ladder on a single ABI PRISM 310 Genetic Analyzer run
From Identifiler User’s Manual
Data to be Reviewed from Electropherograms
Identifiler kit ladder and sample data
Final Answer DesiredWhat is the D8S1179 genotype?Black labels = allele call (repeat #)
Blue labels = peak size (bp)Red labels = peak heights (RFUs)
Bleedthrough of allele from another dye color
Identifiler sample data
Stutter product219/4315 = 0.051
(5.1% of allele)
Black labels = allele call (repeat #)Blue labels = peak size (bp)Red labels = peak heights (RFUs)
Heterozygote peak imbalance3031/4315 = 0.702 = 70.2%
Stutter product260/3031 = 0.086
(8.6% of allele)
“off-ladder” (OL) allele?Often due to noisy baseline or bleedthrough from another dye
color (can sometimes be eliminated by raising the peak threshold—
like to 100 RFU in this case)
“-A” peak due to incomplete adenylation
Usually “-A” peaks will remain fairly constant in amount across allele range for a locus while stutter products increase in amount with larger alleles (higher number of repeat units)
444/
4315
= 0
.103
298/
3031
= 0
.098
9.8%10.3%
Important information from this sampleD8S1179 genotype = 11,14
SWGDAM STR Interpretation Guidelineshttp://www.fbi.gov/hq/lab/fsc/backissu/july2000/strig.htm
The interpretation of results in casework is a matter of professional judgment and expertise. Not every situation can or should be covered by a preset rule. It is important that the laboratory develops and implements written guidelines for interpretation of analytical results. This document provides a framework for the laboratory to develop short tandem repeat (STR) interpretation guidelines. The laboratory's interpretation guidelines should be based upon validation studies, data from the literature, instrumentation used, and/or casework experience.

NEAFS CE-DNA Workshop (Butler and McCord) Sept 29-30, 2004
http://www.cstl.nist.gov/biotech/strbase/NISTpub.htm 4
1. Preliminary Evaluation of Data1.1. The laboratory should develop criteria to determine whether the results are of sufficient intensity/quality for interpretation purposes using methods appropriate for the detection platform. These criteria should be determined by evaluating data generated by the laboratory.
1.1.1. When quantitative results (e.g., peak amplitude) are used to evaluate STR profiles, the results should be examined to determine if they meet the laboratory's defined analytical and interpretational threshold(s).
1.1.1.1. The analytical threshold(s) is defined as the minimum and maximum intensity thresholds that are determined to assign alleles.
1.1.1.2. The interpretational threshold should be defined empirically.
1.1.2. When quantitative results are not used, the laboratory should establish criteria to interpret alleles based on visual inspection of gel images.
1.2. The laboratory should develop criteria to evaluate internal lane size standards and/or allelic ladders.
1.3. Controls are required to assess analytical procedures.
1.3.1. The laboratory should establish criteria for evaluation of the following controls, including but not limited to: reagent blank, amplification blank, and positive control.
1.3.2. The laboratory should develop criteria for the interpretation and documentation of results in the event that the controls do not perform as expected.
1.4. A laboratory using STR multiplexes that contain redundant loci should establish criteria regarding the concordance of such data.
http://www.fbi.gov/hq/lab/fsc/backissu/july2000/strig.htm
SWGDAM STR Interpretation Guidelines
2. Designation2.1. The laboratory should establish criteria to assign allele designations to appropriate peaks or bands.
2.1.1. Locus Designation: The laboratory should establish criteria to address locus assignment for alleles.
2.1.2. Allele Designation: The laboratory should designate alleles in accordance with Combined DNA Index System (CODIS) recommendations.
2.1.2.1. Whenever possible, allele designation should be based operationally on the number of repeat sequences contained within the allele and by comparison to an allelic ladder.
2.1.2.2. The designation of alleles containing an incomplete repeat motif (i.e., an off-ladder allele falling within the range spanned by the ladder alleles) should include the number of complete repeats and, separated by a decimal point, the number of base pairs in the incomplete repeat (e.g., FGA 18.2 allele).
2.1.2.3. If an allele falls above the largest or below the smallest allele of the allelic ladder, the allele should be designated as either greater than (>) or less than (<) the respective ladder allele, or when appropriate interpolation can be used.
2.2. Artifacts can occur and should be noted. These may include, but are not limited to, the following: pull-up, stutter, and nontemplate nucleotide addition. The laboratory should establish guidelines based on empirical data (obtained internally or externally) to address the interpretation of these and other artifacts.
http://www.fbi.gov/hq/lab/fsc/backissu/july2000/strig.htm
SWGDAM STR Interpretation Guidelines
3. Interpretation of Results3.1. The laboratory should define conditions in which the data would lead to the conclusion that the source of the DNA is either from a single person or more than one person. This may be accomplished by an examination of the number of alleles at each locus, peak height ratios, and/or band intensities.
3.1.1. Single Contributor: A sample may be considered to be from a single contributor when the observed number of alleles at each locus and the signal intensity ratios of alleles at a locus are consistent with a profile from a single contributor. All loci should be evaluated in making this determination.
3.1.2. Mixtures With Major/Minor Contributors: A sample may be considered to consist of a mixture of major and minor contributors if there is a distinct contrast in signal intensities among the alleles. The difference is evaluated on a case-by-case context. All loci should be evaluated in making this determination.
3.1.3. Mixtures With a Known Contributor(s): In some cases, when one of the contributors (e.g., the victim) is known, the genetic profile of the unknown contributor may be inferred. Depending on the profiles in the specific instance, this can be accomplished by subtracting the contribution of the known donor from the mixed profile.
3.1.4. Mixtures With Indistinguishable Contributors: When major or minor contributors cannot be distinguished because of similarity in signal intensities or the presence of shared or masked alleles, individuals may still be included or excluded as possible contributors.
3.2. The laboratory should have guidelines for interpretation of partial profiles (i.e., profiles with fewer loci than tested) that may arise from degraded or limited quantity DNA or from the presence of polymerase chain reaction (PCR) inhibitors.
3.3. The laboratory should establish guidelines to interpret profiles that exhibit potential stochastic effects (e.g., allele dropout and/or substantial imbalance of alleles).
http://www.fbi.gov/hq/lab/fsc/backissu/july2000/strig.htm
SWGDAM STR Interpretation Guidelines
4. Conclusions
4.1. The laboratory should prepare guidelines for formulating conclusions resulting from comparisons of single source samples and mixtures with known reference samples.
4.1.1. General categories of conclusions include, but are not limited to: inclusion or match, exclusion or nonmatch, inconclusive or uninterpretable, and no results.
http://www.fbi.gov/hq/lab/fsc/backissu/july2000/strig.htm
SWGDAM STR Interpretation Guidelines
5. Statistical Interpretation
5.1. The source of the population database(s) used should be documented. Relevant population(s) for which the frequency will be calculated should be identified.
5.2. The formulas used in calculating the frequency of a DNA profile should be defined for the following:
5.2.1. Heterozygote profiles
5.2.2. Homozygote profiles
5.2.3. Composite profiles (i.e., multiple locus profiles)
5.2.4. Minimum allele frequencies
5.2.5. Mixture calculations
5.2.6. Biological relationships, where appropriate
5.3. When used, criteria for the declaration of source attribution should be documented.
http://www.fbi.gov/hq/lab/fsc/backissu/july2000/strig.htm
SWGDAM STR Interpretation Guidelines
6. References/Suggested Readings
Committee on DNA Forensic Science, National Research Council. An Update: The Evaluation of Forensic DNA Evidence. National Academy Press, Washington, DC, 1996.
DNA Advisory Board. Quality assurance standards for convicted offender DNA databasinglaboratories (approved April 1999), Forensic Science Communications (July 2000) 2. Available at www.fbi.gov/programs/lab/fsc/backissu/july2000/codispre.htm
DNA Advisory Board. Quality assurance standards for forensic DNA testing laboratories (approved October 1998), Forensic Science Communications (July 2000) 2. Available at www.fbi.gov/programs/lab/fsc/backissu/july2000/codispre.htm
DNA Commission, ISFH. DNA recommendations: 1994 report concerning further recommendations regarding PCR-based polymorphisms in STR (short tandem repeat) systems, Forensic Science International (1994) 69:103–104.
Federal Bureau of Investigation. National DNA Index System (NDIS) Procedures Manual.U.S. Department of Justice, Washington, DC, February 1999 (revised).
http://www.fbi.gov/hq/lab/fsc/backissu/july2000/strig.htm
SWGDAM STR Interpretation Guidelines

NEAFS CE-DNA Workshop (Butler and McCord) Sept 29-30, 2004
http://www.cstl.nist.gov/biotech/strbase/NISTpub.htm 5
GeneScan and Genotyper Review of STR Data
• GeneScan– Apply matrix, size standard, and analysis parameters– Remove the peak designation from the “250 bp” peak in the
GS500 ROX/LIZ internal size standard– Confirm that all alleles in the allelic ladder have been
designated as peaks
• Genotyper– Scan sizes of all peaks in the internal size standard to
confirm that they are correct (especially the 340 bp peak)– Run genotyping macro (Kazaam or Power)– Review electropherograms and edit data primarily through
removing labels on peaks determined not to be alleles– Create a table of final allele calls for export
GeneScan® Software
• Calls peaks (based on threshold values)• Separates colors with matrix file• Sizes peaks with internal size standard
Macintosh
2.1
3.1
3.1.2 (5-dye)
Windows NT
3.7 (5-dye)
3.7.1 (5-dye)
ABI manual is P/N 4303189
GeneScan Software Screen Views
Raw Data
Sample Information
Size Curve
Instrument Performance Indicators (Current, etc.)
Analyzed Data
From Applied Biosystems User Bulletin, ABI Prism GeneScan Analysis Software for the Windows NT Operating System
Flowchart of Steps Used by GeneScan Software to
Produce Analyzed STR Data
Baseline Subtraction
Peaks are filtered to produce normalized data
Analysis Parameters
NEAFS CE-DNA Workshop (Butler and McCord) Sept 29-30, 2004
http://www.cstl.nist.gov/biotech/strbase/NISTpub.htm 6
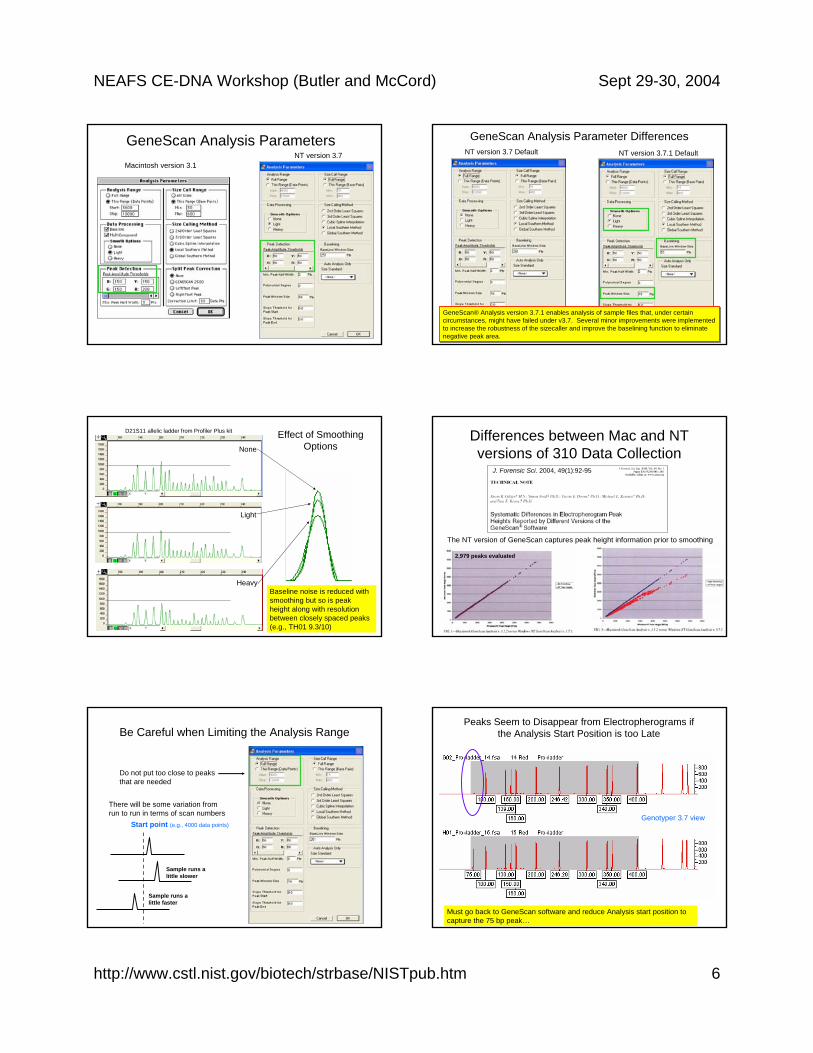
GeneScan Analysis ParametersMacintosh version 3.1
NT version 3.7 NT version 3.7.1 DefaultNT version 3.7 Default
GeneScan Analysis Parameter Differences
GeneScan® Analysis version 3.7.1 enables analysis of sample files that, under certain circumstances, might have failed under v3.7. Several minor improvements were implemented to increase the robustness of the sizecaller and improve the baselining function to eliminate negative peak area.
GeneScan® Analysis version 3.7.1 enables analysis of sample files that, under certain circumstances, might have failed under v3.7. Several minor improvements were implemented to increase the robustness of the sizecaller and improve the baselining function to eliminate negative peak area.
Heavy
None
Light
Effect of Smoothing Options
Baseline noise is reduced with smoothing but so is peak height along with resolution between closely spaced peaks (e.g., TH01 9.3/10)
D21S11 allelic ladder from Profiler Plus kit Differences between Mac and NT versions of 310 Data Collection
J. Forensic Sci. 2004, 49(1):92-95
The NT version of GeneScan captures peak height information prior to smoothing
2,979 peaks evaluated
Be Careful when Limiting the Analysis Range
Do not put too close to peaks that are needed
Start point (e.g., 4000 data points)
There will be some variation from run to run in terms of scan numbers
Sample runs a little slower
Sample runs a little faster
Peaks Seem to Disappear from Electropherograms if the Analysis Start Position is too Late
Genotyper 3.7 view
Must go back to GeneScan software and reduce Analysis start position to capture the 75 bp peak…

NEAFS CE-DNA Workshop (Butler and McCord) Sept 29-30, 2004
http://www.cstl.nist.gov/biotech/strbase/NISTpub.htm 7
Genotyper 3.7 view
Peak appears out of +/-0.5 bp sizing window due to loss of 75 bp peak from the GS500 ROX sizing standard
Appearance of “poor precision” may be due to failure to include a peak in the internal size standard for that sample
Be careful about the stop point for Be careful about the stop point for data collection and analysisdata collection and analysis
Approx. 1 minute difference in these runsApprox. 1 minute difference in these runs
If a stop point of 25 minutes was used, the 400 bp peak would not be collected or analyzed…
There will be some variation from run to run in terms of scan numbers
Data from Debbie Hobson (2001) FBI Laboratory
Internal Size Standards and Sizing Algorithms
DNA fragment peaks in sample
DNA Size
Data Point
147.32 bp147.32 bp
165.05 bp165.05 bp
100
139150
160
200
250
DNA fragment peaks are sized based on the sizing curve produced from the points on the internal size standard
Process of Sizing DNA Fragments Using an Internal Standard
Butler, J.M. (2001) Forensic DNA Typing, Figure 13.2(b), ©Academic Press
Internal Sizing StandardsGS500 ROX (Applied Biosystems)
LTI 50-500 ROX (Life Technologies-no longer available)
ILS600 CXR (Promega)
GeneFlo 625 ROX (CHIMERx-no longer available?)
“250 peak” not included in calculations

NEAFS CE-DNA Workshop (Butler and McCord) Sept 29-30, 2004
http://www.cstl.nist.gov/biotech/strbase/NISTpub.htm 8
Local Southern Sizing Algorithm• Local Southern is commonly used but may not be the best in all
situations• Local Southern involves using 2 peak above and 2 peaks below
an unknown peak from the internal size standard to make a calculated DNA size
Size of STR Allele
X1 X2 X1 + X2
2=
Internal size standard
STR Allele
Elder, J. K. and Southern, E. M. (1983) Measurement of DNA length by gel electrophoresis II: Comparison of methods for relating mobility to fragment length, Analytical Biochemistry 128:227-231.
Normally the 250 bp peak is not included because it does not always migrate uniformly compared to the other peaks in the size standard
Global Southern vs Local Southern MethodsGlobal Southern Sizing
There is value in Global Southern Sizing for STRs when temperature variations within a run exist:– Hartzell, B., Graham, K., McCord,B. (2003) Forensic Sci. Int.
133:228-234– Klein et al. (2003) Forensic Sci. Comm. 5(1)
• Global Southern Method: Generates best-fit curve from all matched fragments in the size standard
• Local Southern Method: Generates best-fit curve from only nearby internal lane standard data points (usually two peaks on either side of the unknown)
Abstract
Early studies have established the Local Southern algorithm as a precise tool for sizing DNA fragments. As a result, the Local Southern algorithm of the PE Applied Biosystems' software, GeneScan® Analysis (PE Applied Biosystems, Foster City, California), is the manufacturer's recommended method for sizing short tandem repeats (STRs). However, this recommendation is made with the warning that size estimates may be imprecise if any of the standard fragments run anomalously. Specifically, the GeneScan®-500 (GS-500) internal standard fragments of 250 and 340 bases in length run anomalously under non-optimal conditions on the ABI Prism® 310 Genetic Analyzer (PE Applied Biosystems, Foster City, California).
The California Department of Justice DNA Laboratory currently uses the GS-500-size standard without the 250-base standard assigned and the Local Southern method to size AmpFlSTR® Profiler Plus™ alleles. However, even with the manufacturer's recommended instrument running conditions, studies in this laboratory demonstrate that ambient temperature variation over the course of a 310 run can result in anomalous migration of GS-500 standard fragments. When ambient temperature varies, a simple analysis method change can improve precision.
This study suggests that the Global Southern method may provide improved precision over the Local Southern method when using the GS-500 internal standard with the ABI Prism® 310 Genetic Analyzer. In addition, this study shows that precision for fragments greater than 300 bases is further improved by excluding the 340-base GS-500 fragment in conjunction with using the Global Southern method. When ambient temperature shifts occur, this sizing method change should reduce the number of sample reruns necessary.
Klein et al. (2003) Forensic Sci. Comm. 5(1) Addressing Ambient Temperature Variation Effects on Sizing Precision of AmpFlSTR® Profiler Plus™ Alleles Detected on the ABI Prism® 310 Genetic Analyzer
http://www.fbi.gov/hq/lab/fsc/backissu/jan2003/klein.htm
Thoughts on Size Standards• Be consistent in use if you want to be able to compare
data over time
• All size standards I have tested work
• Allele sizes are different with different internal sizing standards
• GS500 has a large “hole” in its sizing ability when using the local Southern algorithm for medium-sized STR alleles because of the 250 bp peak that cannot be used; also must be run out to 450 bp to be able to type large FGA alleles with ABI kits

NEAFS CE-DNA Workshop (Butler and McCord) Sept 29-30, 2004
http://www.cstl.nist.gov/biotech/strbase/NISTpub.htm 9
Create New Project (Load sample files)Establish Matrix Define Size Standard PeaksSet Analysis Parameters
Minimum peak height (e.g., 50 RFU)Scan range evaluated (best to avoid primer region)Sizing algorithm options (Local Southern is default)
Data Review WindowsAnalysis Control WindowResults Control Window
Steps for Processing Samples:Install matrix onto each (highlighted) sampleSelect appropriate size standard (or create a new one)Check analysis parameter values (or select pre-defined analysis parameter file)Click “Analyze” button
Examine data in Analysis Control by double clicking on sample file name (this allows you to pull up files in all colors and select or de-select color of interest)
Summary of Steps in GeneScan Data Analysis
Confirm that all allele peaks are called in the allelic ladder
Failure to have these alleles designated will result in Genotyper macro failure and inability to type samples
Peak threshold too high so that some alleles are not designated as peaks by the software
Effect of Peak Detection Threshold Genotyper Software
• Converts GeneScan sized peaks into genotype calls using macros
• Genotyping performed by comparison of allele sizes in allelic ladder to sample alleles
Macintosh
2.0
2.5
2.5.2 (5-dye)
Windows NT
3.7 (5-dye)
ABI manual is P/N 904648
Summary of Genotyper Data Analysis
Open template file containing typing macro specific for STR kit used
Import PROCESSED/REVIEWED GeneScan files
Check internal size standard peaks
Run Kazaam (or Power) macro
Review allelic ladder to confirm that all alleles are called correctly
Review sample data by dye color
Remove calls to stutter peaks or instrument artifacts
Create allele table
Export allele table
Rapid Genotyper Data Review
An overlay of electropherograms permits a rapid assessment of sizing precision and the number of alleles seen in a particular sample set…
PowerPlex 16 data viewed in Genotyper 3.7 software
TH01 alleles

NEAFS CE-DNA Workshop (Butler and McCord) Sept 29-30, 2004
http://www.cstl.nist.gov/biotech/strbase/NISTpub.htm 10
Dye blob
STR alleles
stutter
Pull-up (bleed-through)
spike
Blue channel
Green channel
Yellow channel
Red channel
Butler, J.M. (2005) Forensic DNA Typing, 2nd Edition, Figure 15.4, © Elsevier Science/Academic Press
Deciphering Artifacts from the True Alleles
D3S1358
Stutter products
6.0% 7.8%
Incomplete adenylation
D8S1179
-A
+A
-A
+A
Biological (PCR) artifacts
Dye Blobs (“Artifacts”)
DYS437HEX dye blob
Poor primer purity
• Free dye (not coupled to primer) can be injected into the CE capillary and interfere with detection of true STR alleles
• Dye blobs are wider and usually of less intensitythan true STR alleles (amount depends on the purity of the primers used)
• Dye blobs usually appear at an apparent size that is unique for each dye (e.g., FAM ~120 bp, PET ~100 bp)
Butler, J.M., Shen, Y., McCord, B.R. (2003) The development of reduced size STR amplicons as tools for analysis of degraded DNA. J. Forensic Sci 48(5) 1054-1064.
Filtered with Edge columns
Filtered with Edge columns
No Filtering (Straight from PCR)TH01
TPOXCSF1PO
D21S11
D7S820
FGA
TH01
TPOXCSF1PO
D21S11
D7S820
FGA
EDGE GEL FILTRATION CARTRIDGES
Removal of Dye Artifacts Following PCR Amplification
439 389II438
437391 389I
426YCAII
a/b390 385 a/b
393
392H4460
19388
448447
6FAM(blue)
VIC(green)
NED(Yellow)
PET(Red)
LIZ(Orange)
439 389II438437
391 389I
426390
385 a/b
393
392H4460 19
388
448447
100 bp139
200 250* 300150160
340350
YCAII a/b
Residual dye artifacts
100 bp139
200 250* 300150160
340350
Butler, J.M. (2005) Constructing STR multiplex assays. Methods in Molecular Biology: Forensic DNA Typing Protocols(Carracedo, A., ed.), Humana Press: Totowa, New Jersey, in press.
NIST Y-STR 20plex assay
Dye blob removal with Edge columns
Common Errors in Genotyping
• Genotyper table does not import the third allele in a tri-allelic pattern
• Bleedthrough (pull-up) between dye colors results in a peak that falls into a possible allele bin in an adjacent color
• Clicking off a peak label for a true allele by accident and failing to restore the label
• Accidentally clicking on a peak and inserting a label near the beginning of a locus size range that is not a true allele, whichcauses the second true allele to not show up in the final table of results since only two alleles are imported for each locus
GeneMapperIDv 3.1 Software
Since June 2004, Applied Biosystems no longer sells GeneScan and Genotyper software (they will support these programs until June 2009)…ABI is encouraging the adoption of GeneMapperID which replaces the functions of GeneScan and Genotyper.
Released in Nov 2003

NEAFS CE-DNA Workshop (Butler and McCord) Sept 29-30, 2004
http://www.cstl.nist.gov/biotech/strbase/NISTpub.htm 11
310 Data Collection Software and GeneMapperID
• Controls 310 run conditions• Translates light on CCD camera into
electropherogram (raw data)• Sample sheets and injection lists are created
Macintosh
1.0.2
1.2.2
2.1 (5-dye)
Windows NT
1.0 (5-dye)
2.0 (5-dye)
ABI manual is P/N 904958B
Only compatible with GeneMapper software (not back
compatible with GeneScan/Genotyper)
Use of GeneMapperID v3.1
• Projects• Kit, panel, marker, bin• Provides a flexible format and different
methods for viewing data• Can analyze results from multiple STR kits
within the same GeneMapper project file• Not designed to work with pentanucleotide
repeat loci (version 3.2 will be able)
The Project Window Analysis Parameters
Window
Algorithms:
Basic is the default for many applications
Advanced is similar to the Windows NT version of GeneScan
Classic is similar to Macintosh version of GeneScan
Sample Plot Window
GeneScan-like features
Genotyper-like features

NEAFS CE-DNA Workshop (Butler and McCord) Sept 29-30, 2004
http://www.cstl.nist.gov/biotech/strbase/NISTpub.htm 12
Review Multiple Loci from a Single Sample Review Multiple Samples at a Single Locus
Panel Manager Window
Replaces the need to create Genotyper macros
A kit is a collection of panels.A panel is a collection of markers.Bin sets are collections of the expected allele locations for markers contained with a kit.
Process Component Based Quality Values (PQVs)
Green squares = passYellow triangles = checkRed octagons = low quality data
ADO = Allele Display OverflowOS = OffscaleAE = Allele EditLPH = Low Peak HeightSPU = Spectral Pull-UpAN = Allele NumberBD = Broad PeakOVL = OverlapGQ = Genotype Quality
Expert Systems Under Development
• Difference between an “expert system” and a “quality assurance” tool
Examples• TrueAllele (Cybergenetics, Pittsburg, PA)
– Kadash, K. et al. (2004) J. Forensic Sci. 49(4):660-667• STRess2 (Forensic Science Service, UK)• OSIRIS (NCBI/NIJ, Steve Sherry’s team)
• CompareCalls (Myriad Genetics, Salt Lake City, UT)– Ryan, J.H. et al. (2004) J. Forensic Sci. 49(3):492-499
Model for Future Use of Expert Systems
NDIS Board Appendix B
Review of STR profile data

NEAFS CE-DNA Workshop (Butler and McCord) Sept 29-30, 2004
http://www.cstl.nist.gov/biotech/strbase/NISTpub.htm 13
TrueAllele
• TrueAllele® is designed to operate independently of other allele calling systems• A single reviewer with TrueAllele may be used in place of the current two-person
review process• Examined a dataset of 2,048 convicted offender STR profiles and found only
four samples with differences between TrueAllele and Genotyper at a single locus each (spikes, calling alleles outside of ladder range, and Genotyper missing the third allele in a tri-allelic pattern)
Journal of Forensic Sciences (July 2004), volume 49(2), pp. 660-667 Presenting Information Collected on the ABI 310
Steps in Creating PowerPoint Files from GeneScan and Genotyper Data
Steps for Putting STR Data into PowerPoint Presentations
• Open GeneScan or Genotyper file and put data into desired display format– It is better to keep image a medium size that can be
stretched within PowerPoint to make the lines thicker• Take a picture of the desired image on screen
– Windows: press “print screen”, paste image into Paint program, then crop portion of image desired
– Mac: shift-Apple-4 keys, draw box around desired image, open picture image under Macintosh hard drive
• Open PowerPoint and paste copied image from Paint• Label image within PowerPoint• Print as 2-per-page handout to have a nice size
annotated figure
Example with Profiler Plus Allelic Ladder Label Loci…
D3S1358 VWA FGA
Amelogenin
D8S1179 D21S11 D18S51
D5S818 D13S317 D7S820
GS500 ROX internal size standard
Image is too narrow for a nice balanced plot
Stretch the imageResults in thicker lines and more easily read numbers

NEAFS CE-DNA Workshop (Butler and McCord) Sept 29-30, 2004
http://www.cstl.nist.gov/biotech/strbase/NISTpub.htm 14
The same thing is done in Genotyper—display image in software, print screen, crop within Paint and paste into PowerPoint, stretch to fill slide, add labels as needed…
Example Identifiler Data Databank vs Casework Data Challenges
• Databank (single source samples)– Too much DNA may be added to the PCR reaction resulting
in pull-up between dye colors– Lots of data to review – often produced by contractors
• Casework (mixtures or low level samples)– Often limited DNA material to work with– Low copy number samples can result in allele dropout– Can produce complicated STR profiles to interpret
Common Casework Challenges
D3S1358 TH01 D13S317 D16S539 D2S1338
MIXTURES
DEGRADED DNA
D5S818D13S317
D7S820
D16S539 CSF1PO Penta D
From Butler, J.M. (2004) Short tandem repeat analysis for human identity testing. Current Protocols in Human Genetics, John Wiley & Sons, Hoboken, NJ, Unit 14.8, (Supplement 41), pp. 14.8.1-14.8.22
Loss of signal at larger size loci
More than two alleles at multiple loci
suspect
Crime scene evidence(MIXTURE of victim and perpetrator)
All of the suspect’s alleles are included in the evidentiary DNA profile
Forensic Casework DNA Profiles
Profiler Plus STR kit results





























