Embed Size (px)
Citation preview

September 21, 20122012; doi: 10.1101/cshperspect.a006320 originally published onlineCold Spring Harb Perspect Med
Frank M. LaFerla and Kim N. Green Animal Models of Alzheimer Disease
Subject Collection The Biology of Alzheimer Disease
Animal Models of Alzheimer DiseaseFrank M. LaFerla and Kim N. Green
Alzheimer Disease in 2020
Dennis J. SelkoeDavid M. Holtzman, Eckhard Mandelkow and
-Peptide Clearance in Alzheimer DiseaseβNeurovascular Dysfunction and Faulty Amyloid
ZlokovicAbhay P. Sagare, Robert D. Bell and Berislav V.
The Genetics of Alzheimer DiseaseRudolph E. Tanzi
-ProteinβTreatment Strategies Targeting Amyloid
PangalosDale Schenk, Guriqbal S. Basi and Menelas N.
Fluid Biomarkers in Alzheimer DiseaseKaj Blennow, Henrik Zetterberg and Anne M. Fagan
DiseaseLysosomal System in Alzheimer−Autophagic
Proteasome System and the−The Ubiquitin
Ralph NixonYasuo Ihara, Maho Morishima-Kawashima and
Epidemiology of Alzheimer DiseaseRichard Mayeux and Yaakov Stern
Network Dysfunction-Protein: Synaptic andβNeurotoxicity of Amyloid
Lennart Mucke and Dennis J. SelkoeNeurofibrillary DegenerationBiochemistry and Cell Biology of Tau Protein in
Eva-Maria Mandelkow and Eckhard Mandelkow-ProteinβProteolytic Degradation of Amyloid
Takaomi Saido and Malcolm A. Leissring Deposits in Alzheimer Disease-Protein and AmyloidβBiochemistry of Amyloid
Colin L. Masters and Dennis J. SelkoeBrain Imaging in Alzheimer Disease
al.Keith A. Johnson, Nick C. Fox, Reisa A. Sperling, et Disease
The Neuropsychological Profile of Alzheimer
SalmonSandra Weintraub, Alissa H. Wicklund and David P.
Pharmacologic Treatment for Alzheimer DiseaseSymptomatic and Nonamyloid/Tau Based
SchneiderPaul S. Aisen, Jeffrey Cummings and Lon S.
Normal Biology and Roles in Alzheimer DiseaseApolipoprotein E and Apolipoprotein E Receptors:
David M. Holtzman, Joachim Herz and Guojun Bu
http://perspectivesinmedicine.cshlp.org/cgi/collection/ For additional articles in this collection, see
Copyright © 2012 Cold Spring Harbor Laboratory Press; all rights reserved
Spring Harbor Laboratory Press at NEW YORK UNIVERSITY on October 15, 2014 - Published by Coldhttp://perspectivesinmedicine.cshlp.org/Downloaded from
Spring Harbor Laboratory Press at NEW YORK UNIVERSITY on October 15, 2014 - Published by Coldhttp://perspectivesinmedicine.cshlp.org/Downloaded from

Animal Models of Alzheimer Disease
Frank M. LaFerla and Kim N. Green
Institute for Memory Impairments and Neurological Disorders, Department of Neurobiology and Behavior,University of California, Irvine, Irvine, California 92697-4545
Correspondence: [email protected]
Significant insights into the function of genes associated with Alzheimer disease and relateddementias have occurred through studying genetically modified animals. Although none ofthe existing models fully reproduces the complete spectrum of this insidious human disease,critical aspects of Alzheimer pathology and disease processes can be experimentallyrecapitulated. Genetically modified animal models have helped advance our understandingof the underlying mechanisms of disease and have proven to be invaluable in the preclinicalevaluation of potential therapeutic interventions. Continuing refinement and evolution toyield the next generation of animal models will facilitate successes in producing greatertranslational concordance between preclinical studies and human clinical trials and even-tually lead to the introduction of novel therapies into clinical practice.
Alzheimer disease (AD), the most commoncause of dementia, accounts for approxi-
mately two-thirds of all dementia cases andafflicts more than 35 million individuals world-wide, including more than 5.4 million Ameri-cans. It is a relentlessly progressive disorderthat typically manifests initially by severe lossof memory, particularly of episodic memory.At present, the disorder is not curable, therebyincreasing the urgency of developing and char-acterizing relevant animal models to facili-tate translational research and preclinical drugdevelopment.
Research progress over the past two decades,including the elucidation of AD susceptibilityand causative genes as well as other proteinsinvolved in the pathogenic process, has pro-foundly facilitated the development of geneti-cally altered mouse models (see http://www.alzforum.org/res/com/tra for a listing of cur-
rently available models). Animal models haveplayed a major role in defining critical dis-ease-related mechanisms and have been at theforefront of evaluating novel therapeutic ap-proaches, with many treatments currently inclinical trial owing their origins to studies ini-tially performed in mice. Nevertheless, thereare significant translational issues that havebeen raised of late, as there has been somepotential discordance between preclinical drugstudies and human clinical trials.
ASPECTS OF HUMAN AD MODELEDIN TRANSGENIC MICE
The vast majority of AD cases are sporadic(sAD), and the causes underlying these casesremain unknown. Neuropathologically, AD ischaracterized by the accumulation of amyloid-b (Ab) plaques and neurofibrillary tangles, in
Editors: Dennis J. Selkoe, Eckhard Mandelkow, and David M. Holtzman
Additional Perspectives on The Biology of Alzheimer Disease available at www.perspectivesinmedicine.org
Copyright # 2012 Cold Spring Harbor Laboratory Press; all rights reserved; doi: 10.1101/cshperspect.a006320
Cite this article as Cold Spring Harb Perspect Med 2012;2:a006320
1
ww
w.p
ersp
ecti
vesi
nm
edic
ine.
org
Spring Harbor Laboratory Press at NEW YORK UNIVERSITY on October 15, 2014 - Published by Coldhttp://perspectivesinmedicine.cshlp.org/Downloaded from

addition to widespread synaptic loss, inflam-mation and oxidative damage, and neuronaldeath. Notably, the neuropathology and clinicalphenotype are generally indistinguishable in theearly-onset familial versus the sporadic form ofthe disease, with the biggest difference being theage of onset (Selkoe 2002). Because the etiologyof idiopathic AD is unknown, animal modelshave relied on the utilization of genetic muta-tions associated with familial AD (fAD), withthe rationale that the events downstream ofthe initial trigger are quite similar. These geneticmodels have still been invaluable in determin-ing the molecular mechanisms of disease pro-gression and for testing potential therapeutics.Although no single mouse model recapitulatesall of the aspects of the disease spectrum, eachmodel allows for in-depth analysis of one ortwo components of the disease, which is notreadily possible or ethical with human patientsor samples.
Transgenic mice overproducing mutantAPP develop pathology that is similar to thatfound in the human brain; importantly, Abaccumulation into extracellular plaques occursand is age-dependent—in other words, despiteconstant Ab production, plaques only occur inmid to late adulthood in the majority of theseanimals. Notably, plaque formation is accel-erated when the longer Ab42 is preferentiallycleaved from APP, as this peptide is more prone
to aggregation than Ab40 and leads to earlierand more severe cognitive decline (reviewed inFindeis 2007). The importance of Ab42 to dis-ease progression was highlighted by showingthat elevated levels of Ab40, the shorter, morecommon form of Ab, actually prevented theformation of Ab pathology in the widely usedTg2576 mouse model (McGowan et al. 2005).On the contrary, elevated levels of Ab42 mark-edly exacerbated pathology in the same mousemodel.
Ab plaques found in the brains of AD trans-genic mice are structurally similar to thosefound in the human brain; they initiate as dif-fuse plaques consisting mainly of Ab42, developa dense Ab42 core, and then incorporate Ab40,as well as numerous other non-Ab componentssuch as ubiquitin and a-synuclein (Yang et al.2000). As in the human brain, these plaquesstain positive with both thioflavin and Congored, and show similar fibrillar structures by mi-croscopy (Fig. 1).
Work in transgenic mice has highlighted thedynamic nature of extracellular plaques and hasalso aided in the clarification of important ele-ments in both the brain environment and theAb peptide needed for aggregation of Ab intoplaques. Although formation of plaques in ADtransgenic mice is typically age-dependent (asis AD pathology in humans), plaque formationoccurs very quickly in the brains of older AD
A B C
D E
Figure 1. Visualization of amyloid plaques in 3xTg-AD mice with classical stains. 3xTg-AD mice develop diffuseand fibrillar plaques, as detected with antibody 6E10 (A and B), thioflavin-S (C), Congo red (D), and Gallyasstain (E).
F.M. LaFerla and K.N. Green
2 Cite this article as Cold Spring Harb Perspect Med 2012;2:a006320
ww
w.p
ersp
ecti
vesi
nm
edic
ine.
org
Spring Harbor Laboratory Press at NEW YORK UNIVERSITY on October 15, 2014 - Published by Coldhttp://perspectivesinmedicine.cshlp.org/Downloaded from

transgenic mice. This has been shown using awindow in the skull of APP transgenic mice(Meyer-Luehmann et al. 2008) and further sup-ported by data that plaque volume in aged ADtransgenic mice rapidly returns to high levelswithin 30 days following plaque removal byimmunotherapy (Oddo et al. 2004), in graftsof wild-type tissue into AD transgenic mousebrains (Meyer-Luehmann et al. 2003), and inthe brains of prepathologic AD transgenic micefollowing injection with brain extracts fromhuman AD brain or aged AD transgenic mouse(Meyer-Luehmann et al. 2006). These data indi-cate that the adult AD transgenic mouse brain isripe for the development of Ab pathology andthe latter study also suggests that the ability ofAb to act as a seed for aggregation is dependenton its source.
Most AD transgenic models exhibit mem-ory impairments, with the cognitive deficits ap-pearing to occur earlier than the appearance ofextracellular plaques. These observations ledto a search for earlier pathological species ofAb that could be mediating cognitive decline.Research shifted to identifying the precursorsto plaque formation and identifying how aggre-gation of Ab was crucial to its toxicity. This ledto the focus on soluble oligomeric Ab species—low-molecular-weight aggregates up to �150kDa consisting of two to 30 Ab peptides. As inAD transgenic mice, cognitive decline in hu-mans is not proportional to Ab plaque load(Terry et al. 1991), but does correlate with solu-ble Ab species (Wang et al. 1999). However, inhumans, unlike AD transgenic mice, cognitivedecline does not begin until there is a largequantity of Ab accumulation in the brain,including large amounts of amyloid plaquesand probably oligomers. The latest data nowindicate that soluble oligomeric species play acritical role in the pathogenicity of AD (forreviews, see Haass and Selkoe 2007; Walsh andSelkoe 2007). Evidence supporting involvementof soluble Ab oligomers in AD is present inhuman postmortem brain tissue (Naslund et al.2000; Kokubo et al. 2005); however, much ofthe evidence for the toxicity of oligomeric Aband its central part in AD has come directlyfrom the use of transgenic mouse models of AD.
Intraneuronal Ab has also gained experimentalsupport in recent years (LaFerla et al. 2007). Asin human AD and Down syndrome patients(who develop AD-like pathology by the fifthdecade), many APP AD transgenic mice exhibitintraneuronal amyloid accumulation. The ac-cumulation of intracellular Ab has been shownto precede extracellular deposition in both hu-man (Gyure et al. 2001; Mori et al. 2002) andsome mouse studies (Oddo et al. 2003b). Infact, it was found in transgenic mice that intra-neuronal Ab strongly correlates with initial def-icits on a hippocampal-based memory task(Billings et al. 2005). Data from transgenic ADmice also indicate that intraneuronal Ab ismore neurotoxic than extracellular Ab (Casaset al. 2004).
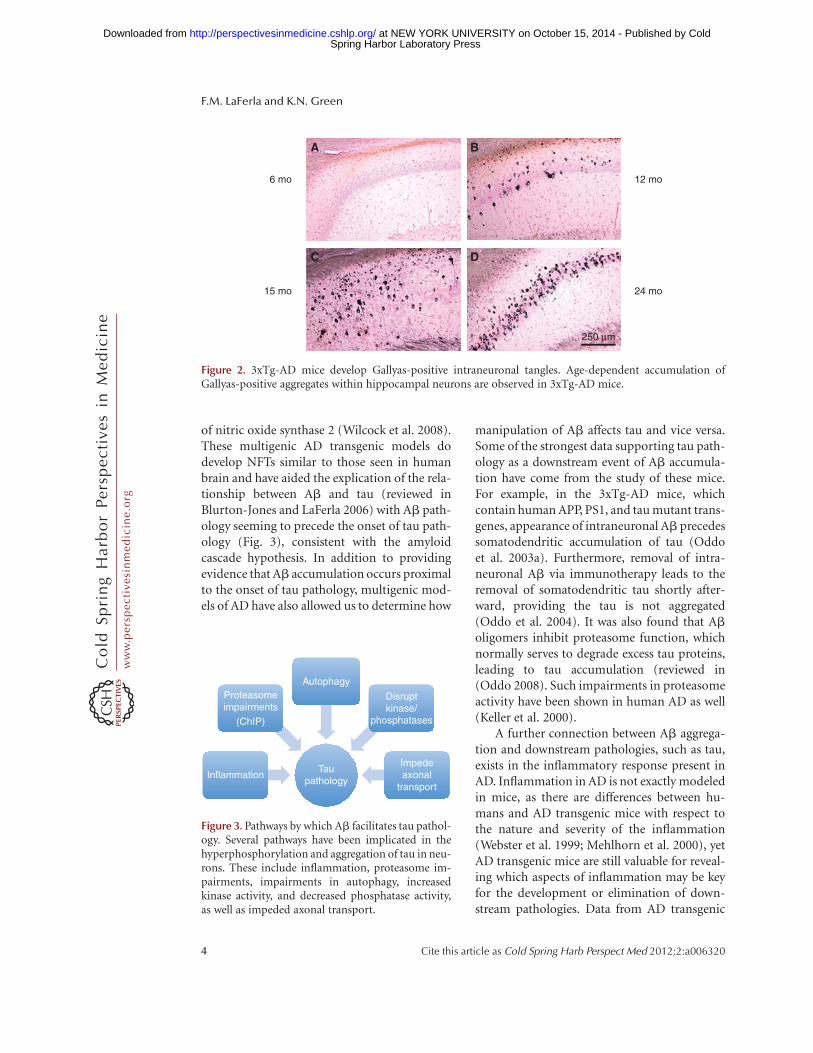
The other hallmark pathology of human ADare the intraneuronal aggregates of hyperphos-phorylated tau known as neurofibrillary tangles(NFTs) (Fig. 2). The amyloid cascade hypothe-sis predicts that tau hyperphosphorylation oc-curs as a downstream consequence of Ab accu-mulation. APP-overexpressing transgenic micehave provided evidence both for and againstthis. Unlike humans with AD, these mousemodels do not develop NFTs, yet many do showincreased tau hyperphosphorylation (reviewedin Gotz et al. 2007). This could be because (1)human Ab accumulation is not sufficient tocause NFT formation; (2) rodent tau has a dif-ferent structure and sequence that may not beprone to aggregate formation; (3) the life spanof mice is not prolonged enough to allow forenough hyperphosphorylation/aggregation asthese pathologies develop over decades in hu-mans; or (4) a combination of these. So whereasAb accumulation in APP overexpressing micedoes not lead to NFT formation, it should beremembered that these animals still developrobust cognitive decline and also undergo moresubtle alterations in tau that resemble the pre-cursors to NFTs in the human brain (most nota-bly hyperphosphorylated tau).
To model the NFTs seen in human AD it hasbeen necessary to develop transgenic mice thatexpress further gene alterations in addition tomutated APP such as mutated human tau(Lewis et al. 2001; Oddo et al. 2003b) or removal
Animal Models of Alzheimer Disease
Cite this article as Cold Spring Harb Perspect Med 2012;2:a006320 3
ww
w.p
ersp
ecti
vesi
nm
edic
ine.
org
Spring Harbor Laboratory Press at NEW YORK UNIVERSITY on October 15, 2014 - Published by Coldhttp://perspectivesinmedicine.cshlp.org/Downloaded from
of nitric oxide synthase 2 (Wilcock et al. 2008).These multigenic AD transgenic models dodevelop NFTs similar to those seen in humanbrain and have aided the explication of the rela-tionship between Ab and tau (reviewed inBlurton-Jones and LaFerla 2006) with Ab path-ology seeming to precede the onset of tau path-ology (Fig. 3), consistent with the amyloidcascade hypothesis. In addition to providingevidence that Ab accumulation occurs proximalto the onset of tau pathology, multigenic mod-els of AD have also allowed us to determine how
manipulation of Ab affects tau and vice versa.Some of the strongest data supporting tau path-ology as a downstream event of Ab accumula-tion have come from the study of these mice.For example, in the 3xTg-AD mice, whichcontain human APP, PS1, and tau mutant trans-genes, appearance of intraneuronal Ab precedessomatodendritic accumulation of tau (Oddoet al. 2003a). Furthermore, removal of intra-neuronal Ab via immunotherapy leads to theremoval of somatodendritic tau shortly after-ward, providing the tau is not aggregated(Oddo et al. 2004). It was also found that Aboligomers inhibit proteasome function, whichnormally serves to degrade excess tau proteins,leading to tau accumulation (reviewed in(Oddo 2008). Such impairments in proteasomeactivity have been shown in human AD as well(Keller et al. 2000).
A further connection between Ab aggrega-tion and downstream pathologies, such as tau,exists in the inflammatory response present inAD. Inflammation in AD is not exactly modeledin mice, as there are differences between hu-mans and AD transgenic mice with respect tothe nature and severity of the inflammation(Webster et al. 1999; Mehlhorn et al. 2000), yetAD transgenic mice are still valuable for reveal-ing which aspects of inflammation may be keyfor the development or elimination of down-stream pathologies. Data from AD transgenic
6 mo
A B
C D
12 mo
15 mo 24 mo
250 μm
Figure 2. 3xTg-AD mice develop Gallyas-positive intraneuronal tangles. Age-dependent accumulation ofGallyas-positive aggregates within hippocampal neurons are observed in 3xTg-AD mice.
Proteasomeimpairments
(ChIP)
Autophagy
Disruptkinase/
phosphatases
Taupathology
InflammationImpedeaxonal
transport
Figure 3. Pathways by which Ab facilitates tau pathol-ogy. Several pathways have been implicated in thehyperphosphorylation and aggregation of tau in neu-rons. These include inflammation, proteasome im-pairments, impairments in autophagy, increasedkinase activity, and decreased phosphatase activity,as well as impeded axonal transport.
F.M. LaFerla and K.N. Green
4 Cite this article as Cold Spring Harb Perspect Med 2012;2:a006320
ww
w.p
ersp
ecti
vesi
nm
edic
ine.
org
Spring Harbor Laboratory Press at NEW YORK UNIVERSITY on October 15, 2014 - Published by Coldhttp://perspectivesinmedicine.cshlp.org/Downloaded from

mice indicate that inflammation, including ac-tivation of complement and various cytokines,occurs downstream from the aggregation of Ab(reviewed in Akiyama et al. 2000), and morespecifically, in association with fibrillar Ab(Kitazawa et al. 2005). Many of these inflamma-tory mediators that are up-regulated by Ab canserve to increase tau pathology (reviewed inBlurton-Jones and LaFerla 2006). For example,activation of Cdk5 following an inflammatoryresponse leads to tau hyperphosphorylation(Kitazawa et al. 2005). There is also produc-tion of reactive oxygen species as a result ofthis inflammatory response (Steele et al. 2007),which is damaging to cell membranes and mayfurther exacerbate the inflammatory response.In both humans and AD transgenic mice, Abplaques are surrounded by activated microgliaand astrocytes; thus even as the activation ofthe inflammatory response in AD can lead tothe detrimental effects discussed above, acti-vated microglia act in a beneficial manner byattempting to phagocytose Ab plaques (Wyss-Coray and Mucke 2002). In support of thehypothesis that inflammation may have favor-able effects in AD, acute inflammation, asbrought about by treatment with lipopolysac-charide (LPS), has been shown to clear Abplaques (DiCarlo et al. 2001) in AD transgenicmice, whereas more chronic LPS treatment po-tentiates tau pathology (Kitazawa et al. 2005).Active and passive immunotherapy strategiesusing Ab or antibodies against Ab, respectively,have also proven useful in reducing plaque and,subsequently, tangle pathology as well as cog-nitive deficits in AD transgenic mice. The stim-ulation of microglia is one mechanism thatappears to be involved in the reduction ofplaque burden (reviewed in Morgan 2006)and further supports the idea that an inflamma-tory response in the AD brain has some positiveeffects.
MODELS BASED ON GENE ABLATION
In addition to transgenic mouse models, whichoverproduce and recapitulate the Ab and taupathologies that are associated with AD, nu-merous genetically modified mice have been
produced that lack genes associated with thisdisorder. Although these mice do not recapitu-late the human pathological phenotype per se,they have proven useful in elucidating the mo-lecular mechanisms underlying the pathology,as well as identifying some of the other pathwaysthat are the targets of crucial drugs. Specifically,knockout mice have been made of the APP secre-tases (BACE, presenilin [1 and 2], and ADAM 10and 17; Shen et al. 1997; Herreman et al. 1999;Luo et al. 2001; Hartmann et al. 2002; Lee et al.2003). In addition, both APP and tau knockoutmice have proven invaluable in understandingdisease progression, as well as in identifyingphysiological roles for the APP protein (Zhenget al. 1996; Takei et al. 2000).
The presenilins were identified as being acrucial component of g-secretase in 1995, andtheir presence necessary for the production ofAb. As such, the presenilins and the g-secretasecomplex quickly became the primary smallmolecular drug target for AD. Presenilin 1knockout mice were produced in 1997, and itwas shown that homozygous knockout of PS1was lethal, with developmental defects in boththe central nervous system (CNS) and skeletalsystems (Shen et al. 1997). Hence, the produc-tion of these mice were the first indicationsthat presenilin, and the g-secretase complex,had vital roles outside of the production ofAb, and that inhibiting it may lead to unde-sirable off-target effects. Despite these earlyindications, and a wealth of subsequent publi-cations showing numerous substrates for theg-secretase complex (Beel and Sanders 2008),regulating many signaling pathways—includingsuppression of skin cancer (Zhang et al. 2007),as well as roles in calcium dyshomeostasis(Green and LaFerla 2008) and autophagy (Leeet al. 2010; Neely et al. 2011)—efforts pro-gressed toward developing g-secretase inhibi-tors and a great number of highly specificcompounds were identified. These inhibitorshave recently been tested in phase III clinical tri-als, and consistent with the wealth of dataobtained from presenilin knockout mice, oneof these was found to cause increased cognitivedecline, and increased the incidence of skincancer (Schor 2011). As such these inhibitor
Animal Models of Alzheimer Disease
Cite this article as Cold Spring Harb Perspect Med 2012;2:a006320 5
ww
w.p
ersp
ecti
vesi
nm
edic
ine.
org
Spring Harbor Laboratory Press at NEW YORK UNIVERSITY on October 15, 2014 - Published by Coldhttp://perspectivesinmedicine.cshlp.org/Downloaded from

programs have largely been abandoned in pref-erence for more APP selective g-secretase inhib-itors, or g-secretase modulators, which do notinhibit the g-secretase complex, but alter whereit cleaves APP to generate less aggregate-pronespecies of Ab.
BACE1 is the sole b-secretase enzyme, andits activity is also crucial for the production ofAb. Several groups first identified it in 1999(Hussain et al. 1999; Sinha et al. 1999; Vassaret al. 1999), but its physiological functionswere unclear. As with presenilin and the g-sec-retase complex, its cloning and identificationmade the creation of BACE inhibitor programsa primary target for the treatment of AD.Contrary to presenilin-deficient mice, BACE1knockout mice were found to be healthy andviable with no obvious defects (Luo et al.2001; Roberds et al. 2001) and, importantly,no longer produced any Ab. Thus, BACE1appears a much more attractive drug targetthat the g-secretase complex owing to a lackof obvious deficits when its activity is ablated.As proof of concept that targeting of BACE1for the treatment of AD would be effective,BACE1 knockout mice were crossed with theTg2576 mice. The absence of BACE1 preventedcognitive decline in these animals and markedlyreduced Ab levels (Ohno et al. 2004). TheseBACE1 knockout mice have also been used toidentify substrates other than APP and havehighlighted b subunits of voltage-gated sodiumchannels (Dominguez et al. 2005; Wong et al.2005; Kim et al. 2011), klotho (Bloch et al.2009), and neuregulin-1 (Hu et al. 2006; Willemet al. 2006), which is essential for myelination ofboth peripheral and central neurons. Hence,BACE1 appears to have a role in the develop-mental myelination of the nervous system,with BACE1 knockout mice showing signifi-cantly reduced levels of myelination and myelinthickness (Hu et al. 2006; Willem et al. 2006).Further analyses revealed that BACE1 knockoutmice had increased pain sensitivity and reducedgrip strength (Hu et al. 2006). However, thesedeficits may be due to developmental issuesthat do not arise when BACE1 is ablated orinhibited in the developed/aged organism, andthus far, no conditional BACE1 knockout mice
exist to test this issue. Peripheral remyelinationhas been investigated in mature BACE1 knock-out mice and has been shown to be impaired(Hu et al. 2008), yet axonal regeneration is en-hanced following axotomy (Farah et al. 2011),suggesting a possible unwanted side effect ofsystemically administered BACE1 inhibitors.
Related to these deficits in myelination inBACE1 knockout a mouse, careful reanalysishas revealed subtle deficits in prepulse inhibi-tion, hypersensitivity to a glutamatergic psycho-stimulant, cognitive impairments, and reduceddendritic spine densities (Laird et al. 2005; Savo-nenko et al. 2008). Collectively, BACE1 knockoutmice have established BACE1 as a primary targetfor the treatment of AD with minimal off-targeteffects, but have highlighted its role in both mye-lination and regulation of the levels of voltage-gated sodium channels, suggesting that therecould be some deleterious side effects from inhib-iting BACE1 in the mature body/CNS. Regard-less, inhibiting BACE1 in the adult brain appearsto be far less problematic than inhibiting the pre-senilins and theg-secretase complex, and remainsa far safer target.
Given the information gleaned from BACE1knockout mice, why is it that a g-secretase in-hibitor has been through a phase III clinicaltrial, whereas no BACE1 inhibitor results havecome to light? This appears to be due to limita-tions of the compounds discovered thus far.BACE1 has a large active site, and it has beenchallenging to find compounds that cross theblood–brain barrier and are large enough toinhibit the active site without being brokendown by endopeptidases (Huang et al. 2009).However, some promising compounds havebeen reported to lower Ab levels in transgenicmice (Chang et al. 2004; Hussain et al. 2007;Fukumoto et al. 2010; Lerchner et al. 2010)and primates (Sankaranarayanan et al. 2009),and human clinical trials are ensuing.
Tau knockout mice have shed light on themechanism by which Ab induces cognitivedeficits in APP transgenic models. Curiously,plaque load does not correlate well with cogni-tive decline in AD patients (Nagy et al. 1995),whereas cognitive deficits are detected in mostAPP transgenic mice prior to plaque deposition
F.M. LaFerla and K.N. Green
6 Cite this article as Cold Spring Harb Perspect Med 2012;2:a006320
ww
w.p
ersp
ecti
vesi
nm
edic
ine.
org
Spring Harbor Laboratory Press at NEW YORK UNIVERSITY on October 15, 2014 - Published by Coldhttp://perspectivesinmedicine.cshlp.org/Downloaded from

(Billings et al. 2005). Furthermore, the involve-ment of tau in the pathogenesis of AD has neverbeen fully understood—mutations in APP orpresenilin lead to AD, but mutations in taulead to their own neurodegenerative diseases.Tau becomes hyperphosphorylated during theAD process, and aggregates into NFTs. Manylines of evidence indicate that the presence ofAb pathologies can activate kinases, down-regulate phosphatases, and impair degradationof tau, leading to tau pathology (Blurton-Jonesand LaFerla 2006). However, what is the conse-quence of these tau pathologies on cognitionand neuronal/synaptic loss, compared withthe consequences of Ab pathology alone? Nota-bly, it has been shown that crossing APP trans-genic mice onto a tau knockout backgroundprevents all cognitive deficits associated withthe presence of APP and Ab (Roberson et al.2007), including reduced spontaneous seizures(Palop et al. 2007) and long-term potentiation(LTP; Shipton et al. 2011). The absence of tauhas no effect on the development of Ab pathol-ogies, including plaque load. Hence, endoge-nous murine tau is necessary for APP and Abto mediate its effects on cognition, LTP andhyperexcitibility of neurons. These exciting dis-coveries have shed light on the crucial role thattau may play in directly mediating the effectsof Ab on cognition and other adverse effects.Notably, APP transgenic mice do not developextensive tau pathologies on their own—somehyperphosphorylation is seen, but the tauremains soluble and does not resemble theNTFs found in the human disease. Hence, sol-uble tau may be more important to the diseaseprocess than tau that has aggregated intoNFTs. During the disease process, the axonalprotein tau becomes mislocalized to the somaand dendrites. Accumulation of tau within thedendritic spines impairs synaptic function(Hoover et al. 2010), but only when the tau isphosphorylated. It has been shown that taupresent in the dendrites can target the proteinkinase Fyn to the postsynaptic membranes (Itt-ner et al. 2010). Once there, it phosphorylatesthe NR2b subunit of the NMDA (N-methyl-D-aspartate) receptor, causing it to stabilize withPSD-95. This stabilization leads to a greater
influx of calcium through the NMDA receptorand can be synaptotoxic. Notably, Ab oligomerscan interact with NMDA receptors and so it maybe the combined effects of both Ab and tau thatdrive the effects at the synapse and lead toimpaired cognition, LTP, hyperexcitibility, andchanges in dendritic spine morphology anddensity. Hence, the use of tau knockout mice,if the effects seen are relevant to human AD,has radically changed our understanding of therole of tau in disease progression, and the natureof the relationship between Ab and tau andtheir effects on cognition. Importantly, thesestudies and mice have highlighted an importantrole for tau outside of AD, but more generally inexcitotoxicity.
TRANSLATIONAL ISSUES
More than 15 years have passed since the firsttransgenic models were derived that recapitu-lated aspects of AD pathology. Somewhat sur-prisingly, since that time, no new therapies forAD have been approved and introduced intothe clinic, and the two currently approved drugclasses (acetylcholinesterase inhibitors and mem-antine) were not tested in these transgenicmice prior to the clinic. This stark reality begsthe question of how useful have these animalmodels actually been for the field? Have theybeen a distraction to the real issues facing anAD patient, or will their continued use bearfruits over the coming years? Why have somany therapies and interventions been success-ful in these models, but have universally failedwhen evaluated in clinic trials? Although anin-depth discussion of these questions is be-yond the scope of this review, we will touch onsome of the translational issues (Box 1).
The most widely used animal models inthe field are in fact models based on the geneticsof familial AD. Less than 1% of AD cases are dueto autosomal-dominant AD, rather than spo-radic AD, so the obvious initial question iswhether fAD and sAD are the same pheno-typical disease or are there subtle differencesbetween the pathologies that would allow atreatment to work in one but not the other.By all pathological counts, they are essentially
Animal Models of Alzheimer Disease
Cite this article as Cold Spring Harb Perspect Med 2012;2:a006320 7
ww
w.p
ersp
ecti
vesi
nm
edic
ine.
org
Spring Harbor Laboratory Press at NEW YORK UNIVERSITY on October 15, 2014 - Published by Coldhttp://perspectivesinmedicine.cshlp.org/Downloaded from

the same disease, with abundant plaque andtangle accumulations in the same brain areasas well as high levels of synaptic and neuronalloss. The differences appear to be what causesthe buildup of the pathologies in the first place.In fAD, a mutation in APP or PS1/2 causes theaccumulation of Ab, whereas the causes ofAb accumulation in sAD are unclear, but likelyto be a combination of genetic and environ-mental factors. Both are highly influenced byaging, with fAD manifesting at younger ages,and being more aggressive in its progression.A priori, one could postulate that fAD mightbe harder to treat than sAD, because of theaggressiveness of the pathology. Hence, trans-genic mice that model fAD, and do so in avery short time frame (1–2 years) should bethe hardest to treat, and should therefore trans-late to sAD very effectively. Obviously, theplethora of numerous failed clinical trials indi-cates that this is not the case, and so why do somany treatments show success in these aggres-sive mouse models of fAD and then fail inpatients with sAD?
Although there are numerous hypothesesthat may account for the discordance in resultsbetween preclinical animal models and humanclinical trials, no doubt one of the most signifi-cant may be that many AD models do not reca-pitulate the extensive neuronal loss observed inthe human condition. Human imaging studiesand clinical–pathological studies show thatpatients with mild–moderate AD already havenot only brain atrophy but also extensive neuro-nal loss in several brain regions. The therapies
that are being pioneered in mouse models areprimarily targeting the pathologies modeledand not dealing with the issue of extensive neu-ronal loss. Hence, many of these therapies maybe effective at preventing or clearing the pathol-ogy, and hence the disease, but are ineffectivein people in which the pathology has alreadydestroyed a huge proportion of the neuronsthat they need for memories and cognition.The acetylcholinesterase inhibitors were devel-oped following studies identifying cholinergicloss as being a highly important factor in ADcognitive decline.
This raises two pertinent questions: First,why do mice not recapitulate the neuronal lossseen in the disease, and, second, can we developmodels that do develop similar loss in whichtherapeutics can be evaluated? We believe thatthe fundamental reason why transgenic micedo not develop extensive neuronal loss, like hu-man AD patients, is the amount of time needed.In human AD, disease progresses over decades.During this time synaptic disturbances, suchas those measured in transgenic mice, couldeventually lead to neuronal death. The two yearsduring which we keep most transgenic APPmice may not be long enough for this to occur.Other issues also clearly play a role, such asbackground strains, such as the widely usedC57/Bl6, which may be more resistant to exci-totoxicity and heterogeneity of humans versusinbred mouse strains, as well as fundamentaldifferences between mice and humans. For ex-ample, it is well established that APP transgenicmice have cognitive decline prior to plaques,
BOX 1. TRANSLATIONAL CONCERNS WITH ANIMAL MODELS
† There is lack of concordance between preclinical models and human clinical trials.– Potential reasons: wrong targets, incomplete models, lack of variability among individuals in
the models, patients enrolled too late, comorbidities
† Humans enrolled in clinical trials are heterogenous, whereas most models utilize in-bred stains onmice.– Potential solution: evaluating novel treatments in multiple lines may help address this point
† There is lack of substantial cell and synaptic loss in the majority of rodent models, suggesting themodels better represent the prodromal phase of the disease.
† Models are of familial AD, and most people have sporadic AD.
F.M. LaFerla and K.N. Green
8 Cite this article as Cold Spring Harb Perspect Med 2012;2:a006320
ww
w.p
ersp
ecti
vesi
nm
edic
ine.
org
Spring Harbor Laboratory Press at NEW YORK UNIVERSITY on October 15, 2014 - Published by Coldhttp://perspectivesinmedicine.cshlp.org/Downloaded from

and prior to any measurable neuronal loss. Fur-thermore, Ab oligomers have been universallyshown to impair LTP in countless studies, andit is easy to then connect this to the cognitiveimpairments seen in these mice. Yet there isno evidence that Ab causes cognitive declinein humans prior to plaques (and neuronalloss). If Ab impairs LTP in the human brain inthe same, robust, fashion that it does in therodent brain then presumably humans wouldexperience cognitive decline in the absence ofboth plaques and neuronal loss whenever Aboligomers could first be detected (Kuo et al.1996; Tomic et al. 2009; Woltjer et al. 2009),and this cannot be explained by cognitivereserve. This has not yet been shown to occur,suggesting that it is possible that Ab may havedifferent modes of action in the rodent braincompared with that of a human.
So how do we develop therapies that targetthe neuronal loss and how do we then test andvalidate them in vivo, prior to the clinic? Clini-cal trials for AD are expensive—upward of $10–20 million for a well-powered phase II/III trial.Hence, only the most promising compoundscan be brought into the clinic, and every failureis costly and discouraging to alternative futuretrials. It should be noted that prevention trialshave not yet been feasible, in part owing to alack of biomarkers and the extreme expenseassociated with the numbers of people neededand the amount of time for which they wouldhave to be evaluated, and therefore trials haveonly used cognitive outcome measures in usu-ally mild–moderate AD patients—which meansthey already have extensive plaque and tangleloads, and that these have already caused exten-sive neuronal damage and loss, which in turncauses dementia. We have developed a novelapproach to this problem by using an induc-ible transgenic mouse model of neuronal loss(Yamasaki et al. 2007). We use the tetracycline-off system to drive expression of diptheria toxinA chains in neurons under control of the Calm-odulin Kinase II promoter. By withdrawingdoxycycline from the diet, we can specificallyablate neurons in regions of the brain that areimpacted in AD, and we can titrate that loss tolevels seen in the AD brain. Mice show cognitive
impairments, as expected, and can then be usedto identify treatments that can improve cogni-tion in the presence of extensive neuronal loss,such as that seen in AD patients. We are takingthe approach of combining therapy testing inthis model alongside a traditional APP/tautransgenic mouse, such as the 3xTg-AD, toidentify treatments that can improve cognitionin the presence of Ab and tau pathologies, butalso neuronal loss. This ensures that only thebest therapies will be selected and proposed asclinical candidates.
If 15 years of using transgenic mouse mod-els of AD have yielded no positive clinicalresults, then have these mice been a failure oreven a distraction from the real problems withAD? The answer is unequivocally no—manynovel approaches to reducing AD pathologyhave been discovered and developed in thesetransgenic mice, and will probably progressinto successful clinical trials when we findways to target prodromal stages of the diseasethrough biomarkers, or attempt prevention. Itis through these approaches that we will oneday be able to prevent the occurrence of the dis-ease as we age. For example, immunotherapywas developed in APP transgenic mice andcould not have been proven to clear pathologywithout them. Immunotherapy has progressedinto numerous clinical trials and has beenshown to reduce both Ab and tau levels inpatients (Boche et al. 2010), as shown in mousemodels (Schenk et al. 1999; Oddo et al. 2004).The effects on cognition have been mixed—benefits have been seen in patients without theapo14 allele, but not in those with apo14—which accounts for �60% of patients. As target-ing the plaques does nothing to address theextensive neuronal loss that has occurred inthese patients, hints of effects on cognition areextremely promising. We would predict, fromthis, that immunization as an AD preventa-tive may be effective. Furthermore, encephalitiscaused by immunotherapy in a small cohort ofpatients may not occur before abundant Abdeposits are found throughout the brain.
Other potential therapeutics developed inAD transgenic mice may yet show clinical suc-cess, either as preventatives or as treatments.
Animal Models of Alzheimer Disease
Cite this article as Cold Spring Harb Perspect Med 2012;2:a006320 9
ww
w.p
ersp
ecti
vesi
nm
edic
ine.
org
Spring Harbor Laboratory Press at NEW YORK UNIVERSITY on October 15, 2014 - Published by Coldhttp://perspectivesinmedicine.cshlp.org/Downloaded from

Some promising approaches include the cop-per/zinc chelator PBT2 (Adlard et al. 2008),which has shown efficacy in phase II clinicaltrials (Faux et al. 2010), and scyllo-inositol(McLaurin et al. 2006), which breaks up Aboligomers. Many companies are now explor-ing potential cognitive enhancers in AD trans-genics, such as a7 agonists (Marighetto et al.2008), phosphodiesterase inhibitors (Puzzoet al. 2009; Verhoest et al. 2009), H3 antagonists(Medhurst et al. 2007), and other approaches.Perhaps targeting cognitive decline in the pres-ence of pathology will be more successful thantargeting the pathology alone, which has beenthe trend of the past decade, or using a combi-nation approach.
CONCLUSIONS
What will the next generation of transgenicmodels of AD bring, and how can they be devel-oped to help develop therapeutics and preven-tatives for sporadic AD? A new era will beushered in as other types of animal models areproduced. For example, there may be advan-tages to moving away from mice and rats andgenetically modifying other smaller animal spe-cies, particularly those in which the endogenousAb sequence is identical to humans and those inwhich processing of tau is more closely alignedto humans. In addition, we need to find waysto model sporadic AD, rather than familialAD. This means that the animals will need todevelop pathology because they age, ratherthan because their genes program them to doso. Once such an animal is produced we willbe able to study which aspects of the aging proc-ess drive the pathology in the first place, andthen target them for prevention. We havealready proposed using alternative models ofneuronal loss to supplement AD pathologymodels, and think that this is also a goodapproach. Making such models will be chal-lenging and will require a great deal of invest-ment, both time and financial, and not allapproaches will work. However, we need toimprove on the current batch of AD transgenicsand look to future so that we will be able to treatand prevent this insidious disease.
REFERENCES
Adlard PA, Cherny RA, Finkelstein DI, Gautier E, Robb E,Cortes M, Volitakis I, Liu X, Smith JP, Perez K, et al.2008. Rapid restoration of cognition in Alzheimer’stransgenic mice with 8-hydroxy quinoline analogs isassociated with decreased interstitial Ab. Neuron 59:43–55.
Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM,Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL,et al. 2000. Inflammation and Alzheimer’s disease. Neu-robiol Aging 21: 383–421.
Beel AJ, Sanders CR. 2008. Substrate specificity of g-secre-tase and other intramembrane proteases. Cell Mol LifeSci 65: 1311–1334.
Billings LM, Oddo S, Green KN, McGaugh JL, LaFerla FM.2005. Intraneuronal Ab causes the onset of early Alz-heimer’s disease-related cognitive deficits in transgenicmice. Neuron 45: 675–688.
Bloch L, Sineshchekova O, Reichenbach D, Reiss K, Saftig P,Kuro-o M, Kaether C. 2009. Klotho is a substrate for a-,b- and g-secretase. FEBS Lett 583: 3221–3224.
Blurton-Jones M, LaFerla FM. 2006. Pathways by which Abfacilitates tau pathology. Curr Alzheimer Res 3: 437–448.
Boche D, Denham N, Holmes C, Nicoll JA. 2010. Neuropa-thology after active Ab42 immunotherapy: Implicationsfor Alzheimer’s disease pathogenesis. Acta Neuropathol120: 369–384.
Casas C, Sergeant N, Itier JM, Blanchard V, Wirths O, vander Kolk N, Vingtdeux V, van de Steeg E, Ret G, CantonT, et al. 2004. Massive CA1/2 neuronal loss with intra-neuronal and N-terminal truncated Ab42 accumulationin a novel Alzheimer transgenic model. Am J Pathol 165:1289–1300.
Chang WP, Koelsch G, Wong S, Downs D, Da H, WeerasenaV, Gordon B, Devasamudram T, Bilcer G, Ghosh AK, et al.2004. In vivo inhibition of Ab production by memapsin 2(b-secretase) inhibitors. J Neurochem 89: 1409–1416.
DiCarlo G, Wilcock D, Henderson D, Gordon M, MorganD. 2001. Intrahippocampal LPS injections reduce Abload in APP þ PS1 transgenic mice. Neurobiol Aging22: 1007–1012.
Dominguez D, Tournoy J, Hartmann D, Huth T, Cryns K,Deforce S, Serneels L, Camacho IE, Marjaux E, Craes-saerts K, et al. 2005. Phenotypic and biochemical analysesof BACE1- and BACE2-deficient mice. J Biol Chem 280:30797–30806.
Farah MH, Pan BH, Hoffman PN, Ferraris D, Tsukamoto T,Nguyen T, Wong PC, Price DL, Slusher BS, Griffin JW.2011. Reduced BACE1 activity enhances clearance ofmyelin debris and regeneration of axons in the injuredperipheral nervous system. J Neurosci 31: 5744–5754.
Faux NG, Ritchie CW, Gunn A, Rembach A, Tsatsanis A,Bedo J, Harrison J, Lannfelt L, Blennow K, ZetterbergH, et al. 2010. PBT2 rapidly improves cognition inAlzheimer’s disease: Additional phase II analyses. J Alz-heimers Dis 20: 509–516.
Findeis MA. 2007. The role of amyloid b peptide 42 in Alz-heimer’s disease. Pharmacol Ther 116: 266–286.
Fukumoto H, Takahashi H, Tarui N, Matsui J, Tomita T, Hir-ode M, Sagayama M, Maeda R, Kawamoto M, Hirai K,
F.M. LaFerla and K.N. Green
10 Cite this article as Cold Spring Harb Perspect Med 2012;2:a006320
ww
w.p
ersp
ecti
vesi
nm
edic
ine.
org
Spring Harbor Laboratory Press at NEW YORK UNIVERSITY on October 15, 2014 - Published by Coldhttp://perspectivesinmedicine.cshlp.org/Downloaded from

et al. 2010. A noncompetitive BACE1 inhibitor TAK-070ameliorates Ab pathology and behavioral deficits in amouse model of Alzheimer’s disease. J Neurosci 30:11157–11166.
Gotz J, Deters N, Doldissen A, Bokhari L, Ke Y, Wiesner A,Schonrock N, Ittner LM. 2007. A decade of tau transgenicanimal models and beyond. Brain Pathol 17: 91–103.
Green KN, LaFerla FM. 2008. Linking calcium to Ab andAlzheimer’s disease. Neuron 59: 190–194.
Gyure KA, Durham R, Stewart WF, Smialek JE, Troncoso JC.2001. Intraneuronal ab-amyloid precedes developmentof amyloid plaques in Down syndrome. Arch Pathol LabMed 125: 489–492.
Haass C, Selkoe DJ. 2007. Soluble protein oligomers in neu-rodegeneration: Lessons from the Alzheimer’s amyloidb-peptide. Nat Rev Mol Cell Biol 8: 101–112.
Hartmann D, de Strooper B, Serneels L, Craessaerts K, Her-reman A, Annaert W, Umans L, Lubke T, Lena Illert A,von Figura K, et al. 2002. The disintegrin/metallopro-tease ADAM 10 is essential for Notch signalling but notfor a-secretase activity in fibroblasts. Human Mol Genet11: 2615–2624.
Herreman A, Hartmann D, Annaert W, Saftig P, CraessaertsK, Serneels L, Umans L, Schrijvers V, Checler F, Vandersti-chele H, et al. 1999. Presenilin 2 deficiency causes a mildpulmonary phenotype and no changes in amyloid pre-cursor protein processing but enhances the embryoniclethal phenotype of presenilin 1 deficiency. Proc NatlAcad Sci 96: 11872–11877.
Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, GrantMK, Pitstick R, Carlson GA, Lanier LM, Yuan LL, et al.2010. Tau mislocalization to dendritic spines mediatessynaptic dysfunction independently of neurodegenera-tion. Neuron 68: 1067–1081.
Hu X, Hicks CW, He W, Wong P, Macklin WB, Trapp BD, YanR. 2006. Bace1 modulates myelination in the central andperipheral nervous system. Nat Neurosci 9: 1520–1525.
Hu X, He W, Diaconu C, Tang X, Kidd GJ, Macklin WB,Trapp BD, Yan R. 2008. Genetic deletion of BACE1 inmice affects remyelination of sciatic nerves. FASEB J 22:2970–2980.
Huang WH, Sheng R, Hu YZ. 2009. Progress in the develop-ment of nonpeptidomimetic BACE 1 inhibitors for Alz-heimer’s disease. Curr Med Chem 16: 1806–1820.
Hussain I, Powell D, Howlett DR, Tew DG, Meek TD, Chap-man C, Gloger IS, Murphy KE, Southan CD, Ryan DM,et al. 1999. Identification of a novel aspartic protease(Asp 2) as b-secretase. Mol Cell Neurosci 14: 419–427.
Hussain I, Hawkins J, Harrison D, Hille C, Wayne G, CutlerL, Buck T, Walter D, Demont E, Howes C, et al. 2007. Oraladministration of a potent and selective non-peptidicBACE-1 inhibitor decreases b-cleavage of amyloid pre-cursor protein and amyloid-b production in vivo. J Neu-rochem 100: 802–809.
Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J,Wolfing H, Chieng BC, Christie MJ, Napier IA, et al.2010. Dendritic function of tau mediates amyloid-b tox-icity in Alzheimer’s disease mouse models. Cell 142:387–397.
Keller JN, Hanni KB, Markesbery WR. 2000. Impaired pro-teasome function in Alzheimer’s disease. J Neurochem 75:436–439.
Kim DY, Gersbacher MT, Inquimbert P, Kovacs DM. 2011.Reduced sodium channel Nav1.1 levels in BACE1-nullmice. J Biol Chem 286: 8106–8116.
Kitazawa M, Oddo S, Yamasaki TR, Green KN, LaFerla FM.2005. Lipopolysaccharide-induced inflammation exacer-bates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer’sdisease. J Neurosci 25: 8843–8853.
Kokubo H, Kayed R, Glabe CG, Yamaguchi H. 2005. SolubleAb oligomers ultrastructurally localize to cell processesand might be related to synaptic dysfunction in Alz-heimer’s disease brain. Brain Res 1031: 222–228.
Kuo YM, Emmerling MR, Vigo-Pelfrey C, Kasunic TC, Kirk-patrick JB, Murdoch GH, Ball MJ, Roher AE. 1996.Water-soluble Ab (N-40, N-42) oligomers in normal andAlzheimer disease brains. J Biol Chem 271: 4077–4081.
LaFerla FM, Green KN, Oddo S. 2007. Intracellularamyloid-b in Alzheimer’s disease. Nat Rev Neurosci 8:499–509.
Laird FM, Cai H, Savonenko AV, Farah MH, He K, Melni-kova T, Wen H, Chiang HC, Xu G, Koliatsos VE, et al.2005. BACE1, a major determinant of selective vulner-ability of the brain to amyloid-b amyloidogenesis, isessential for cognitive, emotional, and synaptic func-tions. J Neurosci 25: 11693–11709.
Lee DC, Sunnarborg SW, Hinkle CL, Myers TJ, StevensonMY, Russell WE, Castner BJ, Gerhart MJ, Paxton RJ, BlackRA, et al. 2003. TACE/ADAM17 processing of EGFRligands indicates a role as a physiological convertase.Ann NY Acad Sci 995: 22–38.
Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM,Wolfe DM, Martinez-Vicente M, Massey AC, Sovak G,et al. 2010. Lysosomal proteolysis and autophagy requirepresenilin 1 and are disrupted by Alzheimer-related PS1mutations. Cell 141: 1146–1158.
Lerchner A, Machauer R, Betschart C, Veenstra S, RueegerH, McCarthy C, Tintelnot-Blomley M, Jaton AL, RabeS, Desrayaud S, et al. 2010. Macrocyclic BACE-1 inhibi-tors acutely reduce Ab in brain after po application. Bio-org Med Chem Lett 20: 603–607.
Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, JonesG, Yen SH, Sahara N, Skipper L, Yager D, et al. 2001.Enhanced neurofibrillary degeneration in transgenicmice expressing mutant tau and APP. Science 293:1487–1491.
Luo Y, Bolon B, Kahn S, Bennett BD, Babu-Khan S, Denis P,Fan W, Kha H, Zhang J, Gong Y, et al. 2001. Mice deficientin BACE1, the Alzheimer’s b-secretase, have normal phe-notype and abolished b-amyloid generation. NatureNeurosci 4: 231–232.
Marighetto A, Valerio S, Desmedt A, Philippin JN, Trocme-Thibierge C, Morain P. 2008. Comparative effects of thea7 nicotinic partial agonist, S 24795, and the cholinester-ase inhibitor, donepezil, against aging-related deficits indeclarative and working memory in mice. Psychophar-macol (Berl) 197: 499–508.
McGowan E, Pickford F, Kim J, Onstead L, Eriksen J, Yu C,Skipper L, Murphy MP, Beard J, Das P, et al. 2005. Ab42 isessential for parenchymal and vascular amyloid deposi-tion in mice. Neuron 47: 191–199.
McLaurin J, Kierstead ME, Brown ME, Hawkes CA, Lamber-mon MH, Phinney AL, Darabie AA, Cousins JE, French
Animal Models of Alzheimer Disease
Cite this article as Cold Spring Harb Perspect Med 2012;2:a006320 11
ww
w.p
ersp
ecti
vesi
nm
edic
ine.
org
Spring Harbor Laboratory Press at NEW YORK UNIVERSITY on October 15, 2014 - Published by Coldhttp://perspectivesinmedicine.cshlp.org/Downloaded from

JE, Lan MF, et al. 2006. Cyclohexanehexol inhibitors ofAb aggregation prevent and reverse Alzheimer pheno-type in a mouse model. Nature Med 12: 801–808.
Medhurst AD, Atkins AR, Beresford IJ, Brackenborough K,Briggs MA, Calver AR, Cilia J, Cluderay JE, Crook B,Davis JB, et al. 2007. GSK189254, a novel H3 receptorantagonist that binds to histamine H3 receptors inAlzheimer’s disease brain and improves cognitive per-formance in preclinical models. J Pharmacol Exp Ther321: 1032–1045.
Mehlhorn G, Hollborn M, Schliebs R. 2000. Induction ofcytokines in glial cells surrounding cortical b-amyloidplaques in transgenic Tg2576 mice with Alzheimer path-ology. Int J Dev Neurosci 18: 423–431.
Meyer-Luehmann M, Stalder M, Herzig MC, Kaeser SA,Kohler E, Pfeifer M, Boncristiano S, Mathews PM,Mercken M, Abramowski D, et al. 2003. Extracellularamyloid formation and associated pathology in neuralgrafts. Nat Neurosci 6: 370–377.
Meyer-Luehmann M, Coomaraswamy J, Bolmont T, KaeserS, Schaefer C, Kilger E, Neuenschwander A, AbramowskiD, Frey P, Jaton AL, et al. 2006. Exogenous induction ofcerebral b-amyloidogenesis is governed by agent andhost. Science 313: 1781–1784.
Meyer-Luehmann M, Spires-Jones TL, Prada C, Garcia-Alloza M, de Calignon A, Rozkalne A, Koenigsknecht-Talboo J, Holtzman DM, Bacskai BJ, Hyman BT. 2008.Rapid appearance and local toxicity of amyloid-bplaques in a mouse model of Alzheimer’s disease. Nature451: 720–724.
Morgan D. 2006. Immunotherapy for Alzheimer’s disease.J Alzheimers Dis 9: 425–432.
Mori C, Spooner ET, Wisniewsk KE, Wisniewski TM, Yama-guch H, Saido TC, Tolan DR, Selkoe DJ, Lemere CA.2002. Intraneuronal Ab42 accumulation in Down syn-drome brain. Amyloid 9: 88–102.
Nagy Z, Esiri MM, Jobst KA, Morris JH, King EM, McDo-nald B, Litchfield S, Smith A, Barnetson L, Smith AD.1995. Relative roles of plaques and tangles in the demen-tia of Alzheimer’s disease: Correlations using three sets ofneuropathological criteria. Dementia 6: 21–31.
Naslund J, Haroutunian V, Mohs R, Davis KL, Davies P,Greengard P, Buxbaum JD. 2000. Correlation betweenelevated levels of amyloidb-peptide in the brain and cog-nitive decline. JAMA 283: 1571–1577.
Neely KM, Green KN, LaFerla FM. 2011. Presenilin is neces-sary for efficient proteolysis through the autophagy-lysosome system in a g-secretase-independent manner.J Neurosci 31: 2781–2791.
Oddo S. 2008. The ubiquitin–proteasome system in Alz-heimer’s disease. J Cell Mol Med 12: 363–373.
Oddo S, Caccamo A, Kitazawa M, Tseng BP, LaFerla FM.2003a. Amyloid deposition precedes tangle formationin a triple transgenic model of Alzheimer’s disease. Neu-robiol Aging 24: 1063–1070.
Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE,Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM.2003b. Triple-transgenic model of Alzheimer’s diseasewith plaques and tangles: Intracellular Ab and synapticdysfunction. Neuron 39: 409–421.
Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM. 2004.Ab immunotherapy leads to clearance of early, but not
late, hyperphosphorylated tau aggregates via the protea-some. Neuron 43: 321–332.
Ohno M, Sametsky EA, Younkin LH, Oakley H, YounkinSG, Citron M, Vassar R, Disterhoft JF. 2004. BACE1 defi-ciency rescues memory deficits and cholinergic dysfunc-tion in a mouse model of Alzheimer’s disease. Neuron 41:27–33.
Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-LyN, Yoo J, Ho KO, Yu GQ, Kreitzer A, et al. 2007.Aberrant excitatory neuronal activity and compensatoryremodeling of inhibitory hippocampal circuits in mousemodels of Alzheimer’s disease. Neuron 55: 697–711.
Puzzo D, Staniszewski A, Deng SX, Privitera L, Leznik E, LiuS, Zhang H, Feng Y, Palmeri A, Landry DW, et al. 2009.Phosphodiesterase 5 inhibition improves synaptic func-tion, memory, and amyloid-b load in an Alzheimer’s dis-ease mouse model. J Neurosci 29: 8075–8086.
Roberds SL, Anderson J, Basi G, Bienkowski MJ, BranstetterDG, Chen KS, Freedman SB, Frigon NL, Games D, Hu K,et al. 2001. BACE knockout mice are healthy despite lack-ing the primary b-secretase activity in brain: Implica-tions for Alzheimer’s disease therapeutics. Human MolGenet 10: 1317–1324.
Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH,Wu T, Gerstein H, Yu GQ, Mucke L. 2007. Reducingendogenous tau ameliorates amyloid b-induced deficitsin an Alzheimer’s disease mouse model. Science 316:750–754.
Sankaranarayanan S, Holahan MA, Colussi D, CrouthamelMC, Devanarayan V, Ellis J, Espeseth A, Gates AT, GrahamSL, Gregro AR, et al. 2009. First demonstration of cere-brospinal fluid and plasma A b lowering with oraladministration of a b-site amyloid precursor protein-cleaving enzyme 1 inhibitor in nonhuman primates.J Pharmacol Exp Ther 328: 131–140.
Savonenko AV, Melnikova T, Laird FM, Stewart KA, PriceDL, Wong PC. 2008. Alteration of BACE1-dependentNRG1/ErbB4 signaling and schizophrenia-like pheno-types in BACE1-null mice. Proc Natl Acad Sci 105:5585–5590.
Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H,Guido T, Hu K, Huang J, Johnson-Wood K, Khan K,et al. 1999. Immunization with amyloid-b attenuatesAlzheimer-disease-like pathology in the PDAPP mouse.Nature 400: 173–177.
Schor NF. 2011. What the halted phase III g-secretase inhib-itor trial may (or may not) be telling us. Ann Neurol 69:237–239.
Shen J, Bronson RT, Chen DF, Xia W, Selkoe DJ, Tonegawa S.1997. Skeletal and CNS defects in Presenilin-1-deficientmice. Cell 89: 629–639.
Shipton OA, Leitz JR, Dworzak J, Acton CE, Tunbridge EM,Denk F, Dawson HN, Vitek MP, Wade-Martins R, PaulsenO, et al. 2011. Tau protein is required for amyloidb-induced impairment of hippocampal long-termpotentiation. J Neurosci 31: 1688–1692.
Sinha S, Anderson JP, Barbour R, Basi GS, Caccavello R,Davis D, Doan M, Dovey HF, Frigon N, Hong J, et al.1999. Purification and cloning of amyloid precursorprotein b-secretase from human brain. Nature 402:537–540.
F.M. LaFerla and K.N. Green
12 Cite this article as Cold Spring Harb Perspect Med 2012;2:a006320
ww
w.p
ersp
ecti
vesi
nm
edic
ine.
org
Spring Harbor Laboratory Press at NEW YORK UNIVERSITY on October 15, 2014 - Published by Coldhttp://perspectivesinmedicine.cshlp.org/Downloaded from

Steele M, Stuchbury G, Munch G. 2007. The molecular basisof the prevention of Alzheimer’s disease through healthynutrition. Exp Gerontol 42: 28–36.
Takei Y, Teng J, Harada A, Hirokawa N. 2000. Defects in axo-nal elongation and neuronal migration in mice with dis-rupted tau and map1b genes. J Cell Biol 150: 989–1000.
Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, HillR, Hansen LA, Katzman R. 1991. Physical basis of cogni-tive alterations in Alzheimer’s disease: Synapse loss is themajor correlate of cognitive impairment. Ann Neurol 30:572–580.
Tomic JL, Pensalfini A, Head E, Glabe CG. 2009. Solublefibrillar oligomer levels are elevated in Alzheimer’sdisease brain and correlate with cognitive dysfunction.Neurobiol Dis 35: 352–358.
Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA,Denis P, Teplow DB, Ross S, Amarante P, Loeloff R,et al. 1999. b-Secretase cleavage of Alzheimer’s amyloidprecursor protein by the transmembrane aspartic pro-tease BACE. Science 286: 735–741.
Verhoest PR, Proulx-Lafrance C, Corman M, Chenard L,Helal CJ, Hou X, Kleiman R, Liu S, Marr E, MennitiFS, et al. 2009. Identification of a brain penetrantPDE9A inhibitor utilizing prospective design and chem-ical enablement as a rapid lead optimization strategy.J Med Chem 52: 7946–7949.
Walsh DM, Selkoe DJ. 2007. A b oligomers—A decade ofdiscovery. J Neurochem 101: 1172–1184.
Wang J, Dickson DW, Trojanowski JQ, Lee VM. 1999. Thelevels of soluble versus insoluble brain Ab distinguishAlzheimer’s disease from normal and pathologic aging.Exp Neurol 158: 328–337.
Webster SD, Tenner AJ, Poulos TL, Cribbs DH. 1999. Themouse C1q A-chain sequence alters b-amyloid-inducedcomplement activation. Neurobiol Aging 20: 297–304.
Wilcock DM, Lewis MR, Van Nostrand WE, Davis J, PrevitiML, Gharkholonarehe N, Vitek MP, Colton CA. 2008.Progression of amyloid pathology to Alzheimer’s disease
pathology in an amyloid precursor protein transgenicmouse model by removal of nitric oxide synthase 2.J Neurosci 28: 1537–1545.
Willem M, Garratt AN, Novak B, Citron M, Kaufmann S,Rittger A, DeStrooper B, Saftig P, Birchmeier C, HaassC. 2006. Control of peripheral nerve myelination by theb-secretase BACE1. Science 314: 664–666.
Woltjer RL, Sonnen JA, Sokal I, Rung LG, Yang W, KjerulfJD, Klingert D, Johnson C, Rhew I, Tsuang D, et al.2009. Quantitation and mapping of cerebral detergent-insoluble proteins in the elderly. Brain Pathol 19: 365–374.
Wong HK, Sakurai T, Oyama F, Kaneko K, Wada K, MiyazakiH, Kurosawa M, De Strooper B, Saftig P, Nukina N. 2005.b Subunits of voltage-gated sodium channels are novelsubstrates of b-site amyloid precursor protein-cleavingenzyme (BACE1) and g-secretase. J Biol Chem 280:23009–23017.
Wyss-Coray T, Mucke L. 2002. Inflammation in neurodege-nerative disease—A double-edged sword. Neuron 35:419–432.
Yamasaki TR, Blurton-Jones M, Morrissette DA, KitazawaM, Oddo S, LaFerla FM. 2007. Neural stem cells improvememory in an inducible mouse model of neuronal loss.J Neurosci 27: 11925–11933.
Yang F, Ueda K, Chen P, Ashe KH, Cole GM. 2000.Plaque-associated a-synuclein (NACP) pathology inaged transgenic mice expressing amyloid precursor pro-tein. Brain Res 853: 381–383.
Zhang YW, Wang R, Liu Q, Zhang H, Liao FF, Xu H. 2007.Presenilin/g-secretase-dependent processing of b-amy-loid precursor protein regulates EGF receptor expression.Proc Natl Acad Sci 104: 10613–10618.
Zheng H, Jiang M, Trumbauer ME, Hopkins R, Sirinath-singhji DJ, Stevens KA, Conner MW, Slunt HH, SisodiaSS, Chen HY, et al. 1996. Mice deficient for the amyloidprecursor protein gene. Ann NY Acad Sci 777: 421–426.
Animal Models of Alzheimer Disease
Cite this article as Cold Spring Harb Perspect Med 2012;2:a006320 13
ww
w.p
ersp
ecti
vesi
nm
edic
ine.
org
Spring Harbor Laboratory Press at NEW YORK UNIVERSITY on October 15, 2014 - Published by Coldhttp://perspectivesinmedicine.cshlp.org/Downloaded from





























