Embed Size (px)
Citation preview

Angiogenesis, Metastasis, and the Cellular Microenvironment
TGF-b1 Induces Endothelial Cell Apoptosis by Shifting VEGFActivation of p38MAPK from the Prosurvival p38b toProapoptotic p38a
Giovanni Ferrari1, Vitaly Terushkin2, Martin J. Wolff2, Xiaodong Zhang2, Cristina Valacca2, Paolo Poggio1,Giuseppe Pintucci2, and Paolo Mignatti2,3
AbstractTGF-b1 and VEGF, both angiogenesis inducers, have opposing effects on vascular endothelial cells. TGF-b1
induces apoptosis; VEGF induces survival. We have previously shown that TGF-b1 induces endothelial cellexpression of VEGF, which mediates TGF-b1 induction of apoptosis through activation of p38mitogen-activatedprotein kinase (MAPK). Because VEGF activates p38MAPK but protects the cells from apoptosis, this findingsuggested that TGF-b1 converts p38MAPK signaling from prosurvival to proapoptotic. Four isoforms of p38MAPK
—a, b, g , and d—have been identified. Therefore, we hypothesized that different p38MAPK isoforms controlendothelial cell apoptosis or survival, and that TGF-b1 directs VEGF activation of p38MAPK from a prosurvival to aproapoptotic isoform.Here, we report that cultured endothelial cells express p38a, b, and g . VEGF activates p38b,whereas TGF-b1 activates p38a. TGF-b1 treatment rapidly induces p38a activation and apoptosis. Subsequently,p38a activation is downregulated, p38b is activated, and the surviving cells become refractory to TGF-b1induction of apoptosis and proliferate. Gene silencing of p38a blocks TGF-b1 induction of apoptosis, whereasdownregulation of p38b or p38g expression results in massive apoptosis. Thus, in endothelial cells p38amediatesapoptotic signaling, whereas p38b and p38g transduce survival signaling. TGF-b1 activation of p38a is mediatedby VEGF, which in the absence of TGF-b1 activates p38b. Therefore, these results show that TGF-b1 inducesendothelial cell apoptosis by shifting VEGF signaling from the prosurvival p38b to the proapoptotic p38a. MolCancer Res; 10(5); 605–14. �2012 AACR.
IntroductionAngiogenesis, the formation of capillaries from preexisting
blood vessel, is mediated by a variety of cytokines and growthfactors with paracrine or autocrine modes of action. VEGFand TGF-b1, potent angiogenesis inducers, act on vascularendothelial cells through different mechanisms (1–3).VEGF (VEGF A), the prototype member of a family of 4
growth factors (VEGF A–D), upregulates endothelial cellproliferation and migration and protects endothelial cellsfrom apoptosis (1). VEGF exerts its activity through 2tyrosine kinase receptors, VEGFR-1 (flt-1) and VEGFR-2(flk-1). VEGFR-2 has been implicated in endothelial cellproliferation and survival, and VEGFR-1 in chemotaxis
and vascular permeability (1, 4). Protein kinase B (Akt) andmitogen-activated protein kinases (MAPK) are componentsof the signaling mechanism activated by VEGFR-2 (5).TGF-b1, the prototype member of a superfamily of 5
multifunctional growth factors, is a potent proliferationinhibitor for most cell types, and an important regulator oftissue morphogenesis (3, 6). TGF-b1 induces vessel for-mation in vitro and in vivo (7–11); however, it inhibitsendothelial cell proliferation andmigration (12) and down-regulates VEGFR-2 expression (13, 14). Notably, TGF-b1induces endothelial cell apoptosis (12, 15) by inhibitingexpression of the antiapoptotic protein Bcl-2 (16) andactivating p38 MAPK (p38MAPK; ref. 17).Thus, although both VEGF and TGF-b1 induce angio-
genesis, they have opposing effects on endothelial cells.Remarkably, TGF-b1 is a potent inducer of apoptosis,whereas VEGF protects the cells from apoptosis. It hastherefore been proposed that TGF-b1 induces angiogenesisin vivo through an indirect mechanism, by recruiting inflam-matory cells that in turn secrete VEGF and/or other angio-genesis inducers. However, TGF-b1 induces endothelial cellexpression of VEGF in vitro and in vivo (18–20) and,surprisingly, VEGF is required for induction of endothelialcell apoptosis through VEGFR-2 activation of p38MAPK
(19, 20).
Authors' Affiliations: 1Division of Cardiovascular Surgery, Department ofSurgery, University of Pennsylvania School of Medicine, Philadelphia,Pennsylvania; Departments of 2Cardiothoracic Surgery and 3Cell Biology,New York University School of Medicine, New York, New York
Note: Supplementary data for this article are available at Molecular CancerResearch Online (http://mcr.aacrjournals.org/).
Corresponding Author: Paolo Mignatti, New York University School ofMedicine, 550First Avenue,NBV16W15,NewYork,NY10016.Phone: 212-263-1478; Fax: 212-263-3161; E-mail: [email protected]
doi: 10.1158/1541-7786.MCR-11-0507
�2012 American Association for Cancer Research.
MolecularCancer
Research
www.aacrjournals.org 605
on July 12, 2019. © 2012 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst April 20, 2012; DOI: 10.1158/1541-7786.MCR-11-0507

p38MAPK, a class of MAPK activated by environmentalstresses, growth factors and cytokines, controls cell functionsincluding proliferation and apoptosis, differentiation, andsenescence (21, 22). Four isoforms of p38MAPK —a, b, g ,and d—have been identified in mammalian cells. Despitemore than 60% sequence homology and more than 90%identity within their kinase domains, the p38MAPK isoformsshow notable differences in tissue expression, upstreamactivators, and downstream effectors. p38a and p38b areubiquitous; p38g and p38d are tissue specific (22). Inaddition, the p38 isoforms have been described in differentcell compartments, in which they can have opposing effectson the same substrate (21). The specific function(s) of theindividual isoforms in physiology and pathology are largelyunknown. The genetic deficiency of p38a in mice results inembryonic lethality, with aberrant placental developmentand abnormal angiogenesis in the yolk sac and embryo (23).In contrast, disruption of the other isoforms generates noapparent phenotype (21). Numerous studies have implicat-ed p38MAPK in induction of endothelial cell apoptosis by avariety of agents (24–27). However, the p38 isoformsinvolved have not been characterized.Here, we report that in vascular endothelial cells, p38a
mediates proapoptotic signaling from inducers of apoptosissuch as TGF-b1, whereas p38b relays survival signaling fromprosurvival factors including VEGF, and that TGF-b1induces endothelial cell apoptosis by converting VEGFsignaling from the prosurvival p38b to the proapoptoticp38a.
Materials and MethodsMaterialsHuman purified or recombinant TGF-b1, recombinant
human VEGF, and antibodies to p38a, p38g , and p38dwere purchased from R&D Systems; antibodies to humancleaved caspase 3 and cleaved PARP from Cell SignalingTechnologies; mouse and rabbit nonimmune immunoglob-ulin G (IgG) from Sigma-Aldrich; and antibodies to p38,a-tubulin, and cyclin E from Santa Cruz Biotechnology,Inc.. Human recombinant fibroblast growth factor-2 (FGF-2) was purchased from Gibco BRL; antibody to active p38from Promega Corp.; monoclonal antibody to p38b fromZymed Laboratories; recombinant p38a from R&D Sys-tems; p38 b from Biovision; and p38g from Abnova.
Cells and mediaBovine capillary endothelial cells (BCE) were as described
(28) and used at passages 6 to 15. Human umbilical veinendothelial (HUVE; Clonetics) cells were grown in medium(EBM2; Clonetics) containing 2% fetal calf serum (FCS)and the growth supplements provided by the company, andwere used at passages 3 to 5. BCE or HUVE cells werestarved overnight in their medium supplemented with 0.5%donor calf serum (DCS) or FCS, respectively, after whicheither TGF-b1 (1 ng/mL) or VEGF (30 ng/mL) or FGF-2(10 ng/mL) was added and incubation was continued for theindicated time. The time of addition of TGF-b1 was
considered as time 0. Western blotting was carried out asdescribed (19). Bromodeoxyuridine (BrdU) uptake wascarried out as described (19).Immunoprecipitation. Cells were lysed in 10 mmol/L
Tris-HCl pH 7.4 containing 150mmol/LNaCl, 1%TritonX-100, 1 mmol/L Pefabloc (Roche), 1 mmol/L leupeptin, 1mmol/L Na3VO4, and 2 mmol/L CaCl2. (plus 100 mmol/Lperoxyvanadate for phosphorylation studies). One hundredmicrograms of cell extract protein was precleared at 4�C for30 minutes with 0.5 mg of nonimmune IgG coupled to 10mL of protein AþG agarose beads (Santa Cruz Biotechnol-ogy). Precleared extracts were centrifuged for 60 seconds at300 � g, and the supernatant was immunoprecipitatedovernight at 4�C with 10 mL of protein AþG agarose beadsand 0.75mg of antibody per 100mg of protein. After washing3 times with lysis buffer, the beads were boiled in reducingLaemmli buffer for 5 minutes and loaded onto SDS/PAGEgels.siRNA transfection. Subconfluent HUVE cells were
transiently transfected as described (19) and used for theexperiments 48 hours after transfection.Reverse transcription PCR. The following primers
were synthesized by IDT DNA technologies based on thepublished sequences: glyceraldehyde-3-phosphate dehydro-genase: (forward 50-CCC ACT CTT CCA CCT TCG-30;reverse 5-TCC TTG GAG GCC ATG TAG GCC AT-30);p38a: (forward 50-GCA GGG ACC TTC TCA TAG AT-30; reverse 50-GAG GGA TAG CCT CAG ACC-30); p38b:(forward 50-CTG CAA GGA AAG GCC CTC-30; reverse50-CAG GCA ATG CCT CAC TGC-30); p38g : (forward50-GAT TAC TGGGAAGAT CCT G-30; reverse 50-CGTCAC AGA GCC GTC TCC-30); p38d: (forward: 50-GACACT CTT CAA GGG CAA G-30; reverse 50-GCC ATCAAT CAC TGC AGC-30). cDNA was synthesized from 1mg of total RNA with SuperScript II RT (Invitrogen) andoligo-dT 30 primer. cDNA (2 mL) was amplified by PCR asdescribed.Proximity ligation assay. HUVE cells grown on gela-
tin-coated glass coverslips were treated with TGF-b1 (1 ng/mL) or control medium for 6 or 72 hours and fixed. The cellswere then incubated with isoform-specific antibodies top38a or p38b together with antibody to phospho-p38overnight at 4�C with gentle agitation. PLUS and MINUSsecondary Proximity ligation assay (PLA; ref. 29) probesagainst rabbit and mouse IgG (Olink Bioscience) wereadded, and the cells were incubated at 37�C for 1 hourwith gentle agitation, followed by incubation with ligationmix for 30 minutes at 37�C. Amplification mix was thenapplied for 100 minutes at 37�C. The coverslips weremounted on microscope slides with Doulink MountingMedium with DAPI, and the cells photographed under afluorescence microscope.In vitro angiogenesis assay in 3D collagen gel. Colla-
gen type I (BectonDickinson) was placed into 24-well tissueculture plates (40 mL per well) and allowed to gel for 30minutes at 37�C.HUVE cells (2.5� 104 cells per well) wereseeded into the wells and incubated at 37�C for 1 hour ingrowth medium. Subsequently, the medium was removed
Ferrari et al.
Mol Cancer Res; 10(5) May 2012 Molecular Cancer Research606
on July 12, 2019. © 2012 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst April 20, 2012; DOI: 10.1158/1541-7786.MCR-11-0507

and 40mL per well of collagenwas added. After incubation at37�C for 30minutes, 100mLofmedium supplementedwith0.5% FCS and 1 ng/mL of TGF-b1 was added, and theplates were incubated at 37�C (30). TGF-b1 (1 ng/mL) wasadded every other day. After 5 to 7 days, the culture mediumwas removed; the cultures were washed with PBS, stainedwith toluidine blue, and photographed under an invertedmicroscope. The number of tube-like structures forminganastomoses was counted as described (20).
Statistical analysisThe Student t tests on the equality of means were carried
out using Stata 8.
ResultsWe have previously shown that TGF-b1 induces endo-
thelial cell apoptosis in vitro and in vivo through VEGF/VEGFR-2 activation of p38MAPK (19, 20). In the absence ofTGF-b1, VEGF activates p38MAPK but protects endothelialcells from apoptosis (19). Therefore, our finding raised thequestion: how does TGF-b1 convert VEGF signaling fromanti- to proapoptotic? Because p38MAPK exists in 4 isoformswith different biological functions, we hypothesized thatTGF-b1 shifts VEGF activation of p38MAPK from 1 isoformto another.To investigate this hypothesis, we characterized endothe-
lial cell expression of the p38 isoforms, and the effect ofTGF-b1 and VEGF on their activation. By reverse tran-scriptase PCR (RT-PCR) endothelial cells expressed p38a,b, and g mRNAs, whereas p38d mRNA was barely detect-able (Fig. 1A). Western blotting with p38MAPK isoform-specific antibodies showed expression of the correspondingproteins (Fig. 1B and C).Neither VEGF nor TGF-b1 treatment of endothelial cells
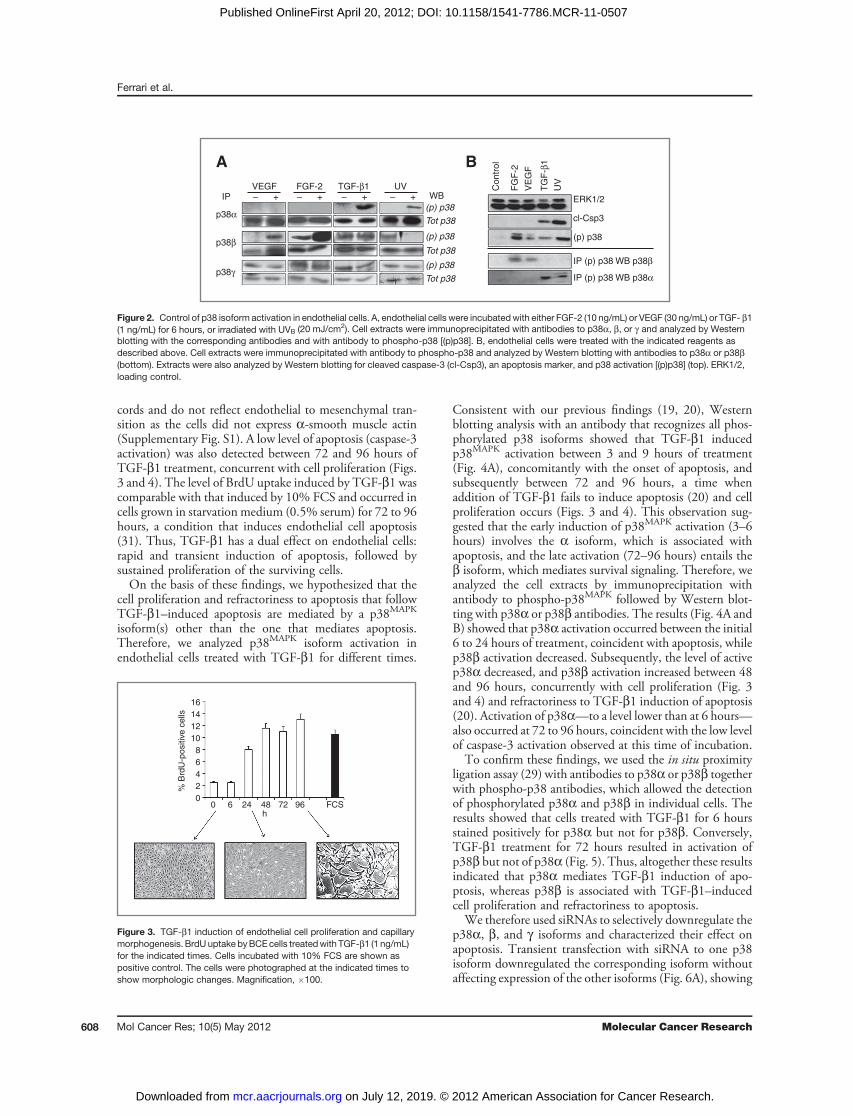
significantly altered p38a, b, or g expression (Fig. 1D),suggesting that the activity of these kinases is regulated atposttranscriptional level. Isoform-specific antibodies tophosphorylated p38 do not exist because the isoforms'phosphorylation sequences are identical. Therefore, weimmunoprecipitated extracts of TGF-b1- or VEGF-treatedcells with antibodies to the individual p38 isoforms andanalyzed the immunoprecipitates by Western blotting withan antibody that recognizes all phosphorylated isoforms.The results (Fig. 2A) showed that untreated endothelial cellshad no active p38a but showed comparable levels of activep38b and p38g . Treatment with TGF-b1 selectively acti-vated p38a. Conversely, VEGF selectively activated p38b.FGF-2, which protects endothelial cells from apoptosis,strongly upregulated p38b activation; in contrast, UVBirradiation, which induces apoptosis, activated p38a.Although the anti-p38a and p38b antibodies did not
show cross-reactivity by Western blotting (Fig. 1C), theymight cross-react by immunoprecipitation. Therefore, wecarried out reverse experiments in which cell extracts wereimmunoprecipitated with antibody to phospho-p38 and theimmunoprecipitates analyzed by Western blotting withantibodies to p38a or p38b. The results (Fig. 2B) showed
that treatment with FGF-2 or VEGF selectively activatedp38b, whereas TGF-b1 or UVB irradiation activated p38a.Western blotting analysis of caspase 3 activation, a marker ofapoptosis, showed that TGF-b1 and UVB induced celldeath, whereas FGF-2 and VEGF had no such effect (Fig.2B). Therefore, these findings suggested that in endothelialcells p38amediates apoptotic signaling, whereas p38b relaysprosurvival signaling. Our previous studies (19, 20) haveshown that in endothelial cells TGF-b1 activation ofp38MAPK is abolished by downregulation of VEGFR-2,showing that endothelial cell VEGF mediates p38MAPK
activation byTGF-b1. In addition,TGF-b1 does not induceapoptosis in endothelial cells that do not express VEGF inresponse toTGF-b1 (19). Because in the absence of TGF-b1VEGF activates p38b, our results suggested that TGF-b1induces endothelial cell apoptosis by shifting VEGF activa-tion of p38MAPK from the b to the a isoform.The apoptotic effect of TGF-b1 is rapid (3–12 hours) and
followed by refractoriness of the surviving cells to TGF-b1induction of apoptosis up to 96 hours (20). We found thatTGF-b1 treatment of endothelial cells for 72 to 96 hoursresulted in increased BrdU uptake and cyclin E expressionbetween 24 and 96 hours (Figs. 3 and 4), coincident with theformation of capillary-like structures (Fig. 3; refs. 20, 7).These structures are likely representative of precapillary
p38α
p38β
p38γ
r p3
8α
r p3
8β
r p3
8γ
p38δ
p38α
p38β
p38γ
p38δ
p38α
p38β
p38γ
p38α
p38β
p38γ δ-Tubulin
Coomassie
HUVEC
A
C
B
D 15 min 6 h
HU
VE
C
CO
NT
RO
L
FG
F-2
VE
GF
TG
F-β
1
TG
F-β
1
VE
GF
BC
E
MC
F-7
Figure 1. Endothelial cell expression of p38 isoforms. A, RT-PCR analysisof HUVE cell expression of p38MAPK isoforms. GAPDH is shown as aloading control in the bottom. B, Western blotting analysis of HUVE andBCE cell extracts expression of p38MAPK isoforms. Human MCF-7mammary carcinoma cells are shown as a control for p38d. The blot wasprobed sequentially with the different antibodies. C, recombinant (r)purified p38a, p38b, and p38g analyzed by Western blotting withantibodies to the 3 isoforms. Coomassie blue staining of the SDS/PAGEgel is shown as a loading control. D, Western blot analysis of p38 isoformexpression inHUVEcells incubatedwith either FGF-2 (10 ng/mL) or VEGF(30 ng/mL) or TGF-b1 (1 ng/mL) for the indicated times. Cells receiving noaddition (CONTROL) were used as control. The blot was probedsequentially with antibodies to p38a, b, and g and to a-tubulin as aloading control. These experiments were repeated twice withcomparable results.
Control of Endothelial Cell Apoptosis by p38MAPK Isoforms
www.aacrjournals.org Mol Cancer Res; 10(5) May 2012 607
on July 12, 2019. © 2012 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst April 20, 2012; DOI: 10.1158/1541-7786.MCR-11-0507
cords and do not reflect endothelial to mesenchymal tran-sition as the cells did not express a-smooth muscle actin(Supplementary Fig. S1). A low level of apoptosis (caspase-3activation) was also detected between 72 and 96 hours ofTGF-b1 treatment, concurrent with cell proliferation (Figs.3 and 4). The level of BrdU uptake induced by TGF-b1 wascomparable with that induced by 10% FCS and occurred incells grown in starvation medium (0.5% serum) for 72 to 96hours, a condition that induces endothelial cell apoptosis(31). Thus, TGF-b1 has a dual effect on endothelial cells:rapid and transient induction of apoptosis, followed bysustained proliferation of the surviving cells.On the basis of these findings, we hypothesized that the
cell proliferation and refractoriness to apoptosis that followTGF-b1–induced apoptosis are mediated by a p38MAPK
isoform(s) other than the one that mediates apoptosis.Therefore, we analyzed p38MAPK isoform activation inendothelial cells treated with TGF-b1 for different times.
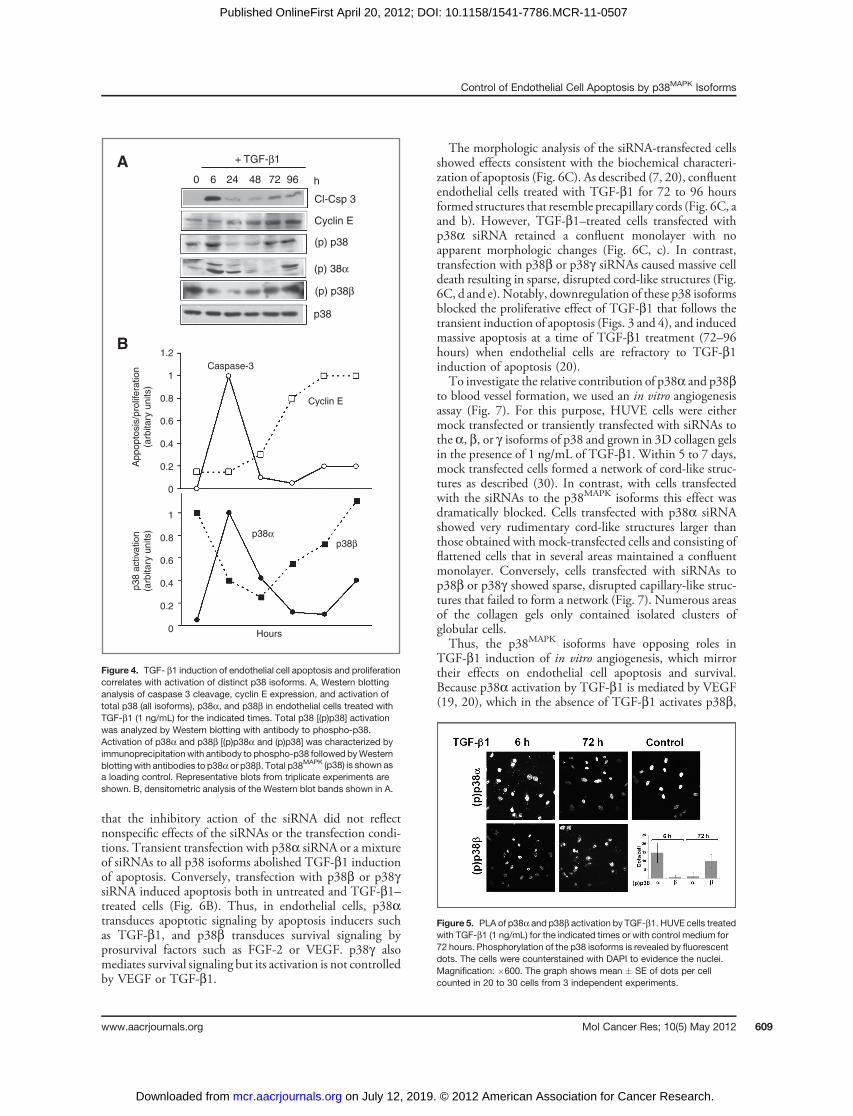
Consistent with our previous findings (19, 20), Westernblotting analysis with an antibody that recognizes all phos-phorylated p38 isoforms showed that TGF-b1 inducedp38MAPK activation between 3 and 9 hours of treatment(Fig. 4A), concomitantly with the onset of apoptosis, andsubsequently between 72 and 96 hours, a time whenaddition of TGF-b1 fails to induce apoptosis (20) and cellproliferation occurs (Figs. 3 and 4). This observation sug-gested that the early induction of p38MAPK activation (3–6hours) involves the a isoform, which is associated withapoptosis, and the late activation (72–96 hours) entails theb isoform, which mediates survival signaling. Therefore, weanalyzed the cell extracts by immunoprecipitation withantibody to phospho-p38MAPK followed by Western blot-ting with p38a or p38b antibodies. The results (Fig. 4A andB) showed that p38a activation occurred between the initial6 to 24 hours of treatment, coincident with apoptosis, whilep38b activation decreased. Subsequently, the level of activep38a decreased, and p38b activation increased between 48and 96 hours, concurrently with cell proliferation (Fig. 3and 4) and refractoriness to TGF-b1 induction of apoptosis(20). Activation of p38a—to a level lower than at 6 hours—also occurred at 72 to 96 hours, coincident with the low levelof caspase-3 activation observed at this time of incubation.To confirm these findings, we used the in situ proximity
ligation assay (29) with antibodies to p38a or p38b togetherwith phospho-p38 antibodies, which allowed the detectionof phosphorylated p38a and p38b in individual cells. Theresults showed that cells treated with TGF-b1 for 6 hoursstained positively for p38a but not for p38b. Conversely,TGF-b1 treatment for 72 hours resulted in activation ofp38b but not of p38a (Fig. 5). Thus, altogether these resultsindicated that p38a mediates TGF-b1 induction of apo-ptosis, whereas p38b is associated with TGF-b1–inducedcell proliferation and refractoriness to apoptosis.We therefore used siRNAs to selectively downregulate the
p38a, b, and g isoforms and characterized their effect onapoptosis. Transient transfection with siRNA to one p38isoform downregulated the corresponding isoform withoutaffecting expression of the other isoforms (Fig. 6A), showing
p38α
TGF-β1 UVWB ERK1/2
cl-Csp3(p) p38
(p) p38
IP (p) p38 WB p38β
IP (p) p38 WB p38α
(p) p38
(p) p38
Tot p38
Tot p38
Tot p38
VEGF FGF-2 TG
F-β
1
UV
Con
trol
VE
GF
FG
F-2
– + – + – + – +IP
A B
p38β
p38γ
Figure 2. Control of p38 isoform activation in endothelial cells. A, endothelial cells were incubatedwith either FGF-2 (10 ng/mL) or VEGF (30 ng/mL) or TGF- b1(1 ng/mL) for 6 hours, or irradiated with UVB (20 mJ/cm2). Cell extracts were immunoprecipitated with antibodies to p38a, b, or g and analyzed by Westernblotting with the corresponding antibodies and with antibody to phospho-p38 [(p)p38]. B, endothelial cells were treated with the indicated reagents asdescribed above. Cell extracts were immunoprecipitated with antibody to phospho-p38 and analyzed by Western blotting with antibodies to p38a or p38b(bottom). Extracts were also analyzed by Western blotting for cleaved caspase-3 (cl-Csp3), an apoptosis marker, and p38 activation [(p)p38] (top). ERK1/2,loading control.
1614121086420
0 6 24 48 72 96 FCSh
% B
rdU
-pos
itive
cel
ls
Figure 3. TGF-b1 induction of endothelial cell proliferation and capillarymorphogenesis. BrdUuptake byBCEcells treatedwith TGF-b1 (1 ng/mL)for the indicated times. Cells incubated with 10% FCS are shown aspositive control. The cells were photographed at the indicated times toshow morphologic changes. Magnification, �100.
Ferrari et al.
Mol Cancer Res; 10(5) May 2012 Molecular Cancer Research608
on July 12, 2019. © 2012 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst April 20, 2012; DOI: 10.1158/1541-7786.MCR-11-0507
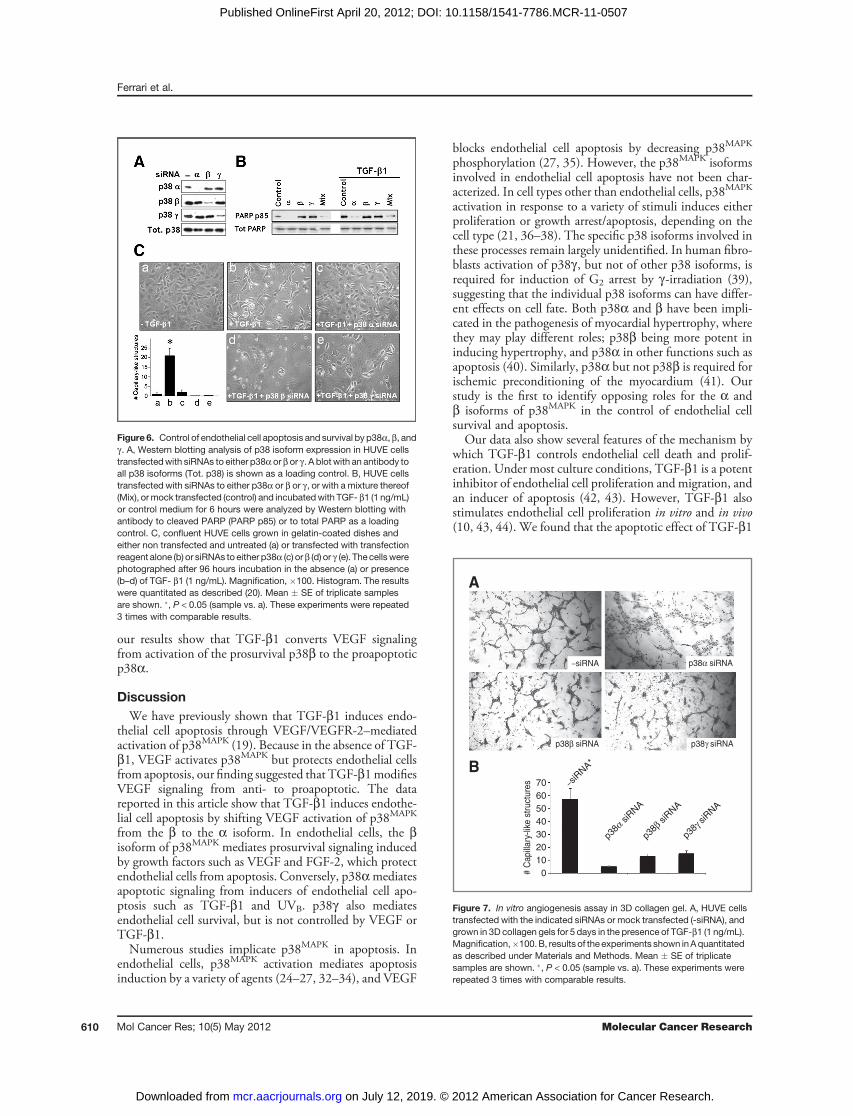
that the inhibitory action of the siRNA did not reflectnonspecific effects of the siRNAs or the transfection condi-tions. Transient transfection with p38a siRNA or a mixtureof siRNAs to all p38 isoforms abolished TGF-b1 inductionof apoptosis. Conversely, transfection with p38b or p38gsiRNA induced apoptosis both in untreated and TGF-b1–treated cells (Fig. 6B). Thus, in endothelial cells, p38atransduces apoptotic signaling by apoptosis inducers suchas TGF-b1, and p38b transduces survival signaling byprosurvival factors such as FGF-2 or VEGF. p38g alsomediates survival signaling but its activation is not controlledby VEGF or TGF-b1.
The morphologic analysis of the siRNA-transfected cellsshowed effects consistent with the biochemical characteri-zation of apoptosis (Fig. 6C). As described (7, 20), confluentendothelial cells treated with TGF-b1 for 72 to 96 hoursformed structures that resemble precapillary cords (Fig. 6C, aand b). However, TGF-b1–treated cells transfected withp38a siRNA retained a confluent monolayer with noapparent morphologic changes (Fig. 6C, c). In contrast,transfection with p38b or p38g siRNAs caused massive celldeath resulting in sparse, disrupted cord-like structures (Fig.6C, d and e).Notably, downregulation of these p38 isoformsblocked the proliferative effect of TGF-b1 that follows thetransient induction of apoptosis (Figs. 3 and 4), and inducedmassive apoptosis at a time of TGF-b1 treatment (72–96hours) when endothelial cells are refractory to TGF-b1induction of apoptosis (20).To investigate the relative contribution of p38a and p38b
to blood vessel formation, we used an in vitro angiogenesisassay (Fig. 7). For this purpose, HUVE cells were eithermock transfected or transiently transfected with siRNAs tothe a, b, or g isoforms of p38 and grown in 3D collagen gelsin the presence of 1 ng/mL of TGF-b1. Within 5 to 7 days,mock transfected cells formed a network of cord-like struc-tures as described (30). In contrast, with cells transfectedwith the siRNAs to the p38MAPK isoforms this effect wasdramatically blocked. Cells transfected with p38a siRNAshowed very rudimentary cord-like structures larger thanthose obtained with mock-transfected cells and consisting offlattened cells that in several areas maintained a confluentmonolayer. Conversely, cells transfected with siRNAs top38b or p38g showed sparse, disrupted capillary-like struc-tures that failed to form a network (Fig. 7). Numerous areasof the collagen gels only contained isolated clusters ofglobular cells.Thus, the p38MAPK isoforms have opposing roles in
TGF-b1 induction of in vitro angiogenesis, which mirrortheir effects on endothelial cell apoptosis and survival.Because p38a activation by TGF-b1 is mediated by VEGF(19, 20), which in the absence of TGF-b1 activates p38b,
A
B
p38α
Caspase-3
Cyclin E
p38β
1.2
1
0.8
0.6
0.4
0.2
0
App
opto
sis/
prol
ifera
tion
(arb
itary
uni
ts)
p38
activ
atio
n(a
rbita
ry u
nits
)
1
0.8
0.6
0.4
0.2
0 Hours
(p) 38α
(p) p38β
(p) p38
Cyclin E
Cl-Csp 3
h9672482460
+ TGF-β1
p38
Figure 4. TGF- b1 induction of endothelial cell apoptosis and proliferationcorrelates with activation of distinct p38 isoforms. A, Western blottinganalysis of caspase 3 cleavage, cyclin E expression, and activation oftotal p38 (all isoforms), p38a, and p38b in endothelial cells treated withTGF-b1 (1 ng/mL) for the indicated times. Total p38 [(p)p38] activationwas analyzed by Western blotting with antibody to phospho-p38.Activation of p38a and p38b [(p)p38a and (p)p38] was characterized byimmunoprecipitation with antibody to phospho-p38 followed byWesternblottingwith antibodies to p38a or p38b. Total p38MAPK (p38) is shown asa loading control. Representative blots from triplicate experiments areshown. B, densitometric analysis of the Western blot bands shown in A.
Figure 5. PLA of p38a and p38b activation by TGF-b1. HUVE cells treatedwith TGF-b1 (1 ng/mL) for the indicated times or with control medium for72 hours. Phosphorylation of the p38 isoforms is revealed by fluorescentdots. The cells were counterstained with DAPI to evidence the nuclei.Magnification: �600. The graph shows mean � SE of dots per cellcounted in 20 to 30 cells from 3 independent experiments.
Control of Endothelial Cell Apoptosis by p38MAPK Isoforms
www.aacrjournals.org Mol Cancer Res; 10(5) May 2012 609
on July 12, 2019. © 2012 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst April 20, 2012; DOI: 10.1158/1541-7786.MCR-11-0507
our results show that TGF-b1 converts VEGF signalingfrom activation of the prosurvival p38b to the proapoptoticp38a.
DiscussionWe have previously shown that TGF-b1 induces endo-
thelial cell apoptosis through VEGF/VEGFR-2–mediatedactivation of p38MAPK (19). Because in the absence of TGF-b1, VEGF activates p38MAPK but protects endothelial cellsfrom apoptosis, our finding suggested that TGF-b1modifiesVEGF signaling from anti- to proapoptotic. The datareported in this article show that TGF-b1 induces endothe-lial cell apoptosis by shifting VEGF activation of p38MAPK
from the b to the a isoform. In endothelial cells, the bisoform of p38MAPK mediates prosurvival signaling inducedby growth factors such as VEGF and FGF-2, which protectendothelial cells from apoptosis. Conversely, p38amediatesapoptotic signaling from inducers of endothelial cell apo-ptosis such as TGF-b1 and UVB. p38g also mediatesendothelial cell survival, but is not controlled by VEGF orTGF-b1.Numerous studies implicate p38MAPK in apoptosis. In
endothelial cells, p38MAPK activation mediates apoptosisinduction by a variety of agents (24–27, 32–34), and VEGF
blocks endothelial cell apoptosis by decreasing p38MAPK
phosphorylation (27, 35). However, the p38MAPK isoformsinvolved in endothelial cell apoptosis have not been char-acterized. In cell types other than endothelial cells, p38MAPK
activation in response to a variety of stimuli induces eitherproliferation or growth arrest/apoptosis, depending on thecell type (21, 36–38). The specific p38 isoforms involved inthese processes remain largely unidentified. In human fibro-blasts activation of p38g , but not of other p38 isoforms, isrequired for induction of G2 arrest by g-irradiation (39),suggesting that the individual p38 isoforms can have differ-ent effects on cell fate. Both p38a and b have been impli-cated in the pathogenesis of myocardial hypertrophy, wherethey may play different roles; p38b being more potent ininducing hypertrophy, and p38a in other functions such asapoptosis (40). Similarly, p38a but not p38b is required forischemic preconditioning of the myocardium (41). Ourstudy is the first to identify opposing roles for the a andb isoforms of p38MAPK in the control of endothelial cellsurvival and apoptosis.Our data also show several features of the mechanism by
which TGF-b1 controls endothelial cell death and prolif-eration. Under most culture conditions, TGF-b1 is a potentinhibitor of endothelial cell proliferation and migration, andan inducer of apoptosis (42, 43). However, TGF-b1 alsostimulates endothelial cell proliferation in vitro and in vivo(10, 43, 44). We found that the apoptotic effect of TGF-b1
706050403020100
p38α si
RNA
–siR
NA*
p38β si
RNA
p38γ s
iRNA
# C
apill
ary-
like
stru
ctur
es
p38β siRNA
–siRNA
A
B
p38γ siRNA
p38α siRNA
Figure 7. In vitro angiogenesis assay in 3D collagen gel. A, HUVE cellstransfected with the indicated siRNAs or mock transfected (-siRNA), andgrown in 3D collagen gels for 5 days in the presence of TGF-b1 (1 ng/mL).Magnification,�100.B, results of the experiments shown inAquantitatedas described under Materials and Methods. Mean � SE of triplicatesamples are shown. �, P < 0.05 (sample vs. a). These experiments wererepeated 3 times with comparable results.
Figure 6. Control of endothelial cell apoptosis and survival by p38a, b, andg . A, Western blotting analysis of p38 isoform expression in HUVE cellstransfectedwith siRNAs to either p38aor bor g . A blotwith an antibody toall p38 isoforms (Tot. p38) is shown as a loading control. B, HUVE cellstransfected with siRNAs to either p38a or b or g , or with a mixture thereof(Mix), ormock transfected (control) and incubatedwith TGF- b1 (1 ng/mL)or control medium for 6 hours were analyzed by Western blotting withantibody to cleaved PARP (PARP p85) or to total PARP as a loadingcontrol. C, confluent HUVE cells grown in gelatin-coated dishes andeither non transfected and untreated (a) or transfected with transfectionreagent alone (b) or siRNAs to either p38a (c) orb (d) or g (e). The cellswerephotographed after 96 hours incubation in the absence (a) or presence(b–d) of TGF- b1 (1 ng/mL). Magnification, �100. Histogram. The resultswere quantitated as described (20). Mean � SE of triplicate samplesare shown. �, P < 0.05 (sample vs. a). These experiments were repeated3 times with comparable results.
Ferrari et al.
Mol Cancer Res; 10(5) May 2012 Molecular Cancer Research610
on July 12, 2019. © 2012 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst April 20, 2012; DOI: 10.1158/1541-7786.MCR-11-0507

on endothelial cells is rapid and transient, and is followed bya long period during which the surviving cells are refractoryto apoptosis induction by TGF-b1 (20) and proliferate.These opposing effects of TGF-b1 are mediated by theselective activation of 2 different p38 isoforms. The rapidinduction of apoptosis results from downregulation of p38band upregulation of p38a activation. Subsequently, down-regulation of p38a and parallel, sustained increase in p38bactivation provide survival signaling. On the basis of ourdata, we cannot conclude whether TGF-b1 induction ofendothelial cell proliferation is also mediated by p38bbecause siRNA-mediated downregulation of p38 b expres-sion results in rapid and massive apoptosis. Our previousfindings that TGF-b1 induces prolonged upregulation ofendothelial cell VEGF and FGF-2 expression and activatesextracellular signal-regulated kinase (ERK)1/2 by a VEGF/VEGFR-2-dependent mechanism (19) suggest that theproliferative effect of TGF-b1 could be mediated byERK1/2, while the sustained activation of p38b providesendothelial cells with survival signaling. Thus, TGF-b1controls both endothelial cell apoptosis and survival throughthe selective activation of the a and b isoforms of p38MAPK,respectively.The effect of TGF-b1 on endothelial cells is mediated by 2
type-I receptors (TGF-bRI): activin receptor–like kinase(ALK)-5, the predominant type I receptor that mediatesTGF-b1 responses, and ALK-1, whose expression is restrict-ed to endothelial cells. ALK-5 and ALK-1 mediate TGF-bsignaling through distinct Smad proteins: Smad2/Smad3and Smad1/Smad5, respectively (11). Although TGF-b1primarily activates Smad2 and Smad3, in primary endothe-lial cells it also activates Smad1 and Smad5 through aheterodimeric receptor complex consisting of ALK5 andALK1 (45). The ALK5 and ALK1 pathways mediate oppos-ing effects; ALK5 inhibits, whereas ALK1 stimulates cellmigration and proliferation (46). In addition, ALK5, but notALK1, mediates TGF-b1 induction of endothelial cellapoptosis (47). However, ALK-1/Smad1 signaling mediatesTGF-b1 induction of VEGF expression in endothelial cells(18), which is required for TGF-b1 induction of apoptosis(19). Based on our data, we cannot conclude whether ALK1and ALK5 activate different p38 isoforms. It is temptingto speculate that signaling by both receptors is requiredfor TGF-b1 induction of apoptosis, and that downregula-tion of ALK5 is responsible for the ensuing cell proliferation(Fig. 4; ref. 20). The relative contribution of TGFbRsto activation of the different isoforms of p38MAPK andcontrol of endothelial cell apoptosis warrants furtherinvestigation.Our conclusions are partly based on results of siRNA-
mediated gene silencing experiments. It has been reportedthat siRNAs can inhibit angiogenesis by a nonspecific effectmediated by toll-like receptor 3 (TLR3), a double-strandedRNA receptor that generates apoptotic signaling in endo-thelial cells (48, 49). This effect requires siRNAs more than21 nucleotides in length, and has been obtained withnonchemically modified molecules. We used 19-bp siRNAswith dinucleotide 30 DNA overhangs, a chemical modifica-
tion to prevent nonspecific effects. Indeed, in our studies, wedid not observe nonspecific apoptotic effects or p38MAPK
activation induced by our siRNAs. Our VEGFR-1 andVEGFR-2 siRNAs did not induce apoptosis (19, 20), nordid our siRNAs to p38a or a mixture of siRNAs to p38a, b,and g (Fig. 6B). Similarly, our siRNAs to VEGFR-1 andVEGFR-1, or control siRNA to lamin A did not inducep38MAPK activation (19). Therefore, our siRNAs did nothave nonspecific effects on p38MAPK activation or endothe-lial cell apoptosis mediated by TLR3.Our results are in contrast with previous data showing that
pharmacologic inhibition of p38MAPK blocks endothelial cellapoptosis (19, 27, 35). Because p38b and p38g mediatesurvival signaling, their inhibition should result in apoptosis.This discrepancy is explained by the fact that all currentlyavailable inhibitors of p38MAPK inhibit both p38a andp38b, and therefore abolish both pro- and antiapoptoticsignaling. In addition, the apoptotic effect of p38a activa-tion seems to be prevalent. Indeed, siRNA-mediated down-regulation of all the p38 isoforms expressed by endothelialcells blocks TGF-b1 induction of apoptosis as effectively asdownregulation of p38a alone. In contrast, selective inhi-bition of p38b or p38g expression results in apoptosis (Fig.6B and C).On the basis of our data, we cannot conclude whether the
proliferative effect of TGF-b1 on endothelial cells is medi-ated by VEGF or other endothelial cell-derived growthfactors. TGF-b1-induced upregulation of VEGF lasts atleast 24 h (19), a timing coincident with the onset of cellproliferation. Although TGF-b1 downregulates endothelialcell expression of VEGFR-2, this effect does not exceed 40–50% (13, 14). In addition, TGF-b1 upregulates endothelialcell expression of FGF-2 (19), which can also provide amitogenic stimulus. Therefore, both VEGF and FGF-2could mediate TGF-b1 induction of endothelial cell prolif-eration through sustained activation of the ERK1/2 or othersignaling pathways.The coordinated mechanism of control of endothelial cell
apoptosis, survival, and proliferation by TGF-b1—VEGFactivation of p38a and b has profound effects on in vitroangiogenesis. Downregulation of p38a-mediated apoptosisblocks the formation of capillary-like structures in 3Dcollagen gel. Similarly, induction of excess apoptosis bydownregulation of p38b—mediated survival signalingblocks endothelial cell sprouting and organization into acapillary-like network. These findings are consistent withour previous observation that inhibition of endothelial cellapoptosis blocks angiogenesis in vitro and in vivo (20).We have previously shown that VEGF mediates TGF-b1
induction of endothelial cell apoptosis through activation ofp38MAPK (19). This conclusion was based on our findingsthat inhibition of VEGF or downregulation of VEGFR-2blocks TGF-b1 activation of p38MAPK, and inhibition ofp38MAPK expression abrogates TGF-b1 induction of apo-ptosis (19). Our data show that VEGF activates the pro-survival p38b, whereas TGF-b1 activates the proapoptoticp38a. Because TGF-b1 activates p38a through VEGF,which in the absence of TGF-b1 activates p38b, our findings
Control of Endothelial Cell Apoptosis by p38MAPK Isoforms
www.aacrjournals.org Mol Cancer Res; 10(5) May 2012 611
on July 12, 2019. © 2012 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst April 20, 2012; DOI: 10.1158/1541-7786.MCR-11-0507

show that TGF-b1 shifts VEGF signaling from p38b top38a.Our results therefore generate the following model for the
control of endothelial cell apoptosis and survival by TGF-b1and VEGF (Fig. 8). In the absence of TGF-b1, VEGFbinding to VEGFR-2 induces endothelial cell activationof p38b and cell survival (Fig. 8, left). TGF-b1 inducesendothelial cell expression of VEGF and switches VEGF/VEGFR-2–activated signaling to the proapoptotic p38a.This effect involves crosstalk between TGF-b1 and VEGFsignaling pathways that can occur atmultiple levels upstreamof p38MAPK. VEGF/VEGFR-2–mediated activation ofp38a is necessary for TGF-b1 induction of endothelial cellapoptosis; however, parallel TGF-b1–initiated signalingacting in concert with p38a may also be required. Subse-quently (Fig. 8, right), the cells become refractory toTGF-b1, p38a activation is downregulated and p38b acti-vation upregulated. This effect results in abrogation ofapoptosis and increased prosurvival signaling.The signaling mechanism(s) that control the selective
activation of the a or b isoform of p38 are largelyunknown, as is the crosstalk between TGF-b1 and VEGFsignaling that mediates the shift of VEGF activation ofp38 from the b to the a isoform. Understanding thesemechanisms can shed light on the complex control ofvascular endothelial cell survival and death during angio-genesis. Importantly, our findings provide indications forthe development of novel pharmacologic approaches toinhibit tumor angiogenesis. We found that, whereas the
effect of TGF-b1 on apoptosis is transient (Fig. 4) andfollowed by refractoriness of the cells to TGF-b1 induc-tion of apoptosis (20), inhibition of p38b and/or p38gsignaling results in sustained induction of apoptosis byTGF-b1 (Fig. 6). Presently, all synthetic inhibitors of p38block both the a and the b isoforms. Novel inhibitors thatselectively inhibit p38b activity (or activation) could blockthe prosurvival effect of TGF-b1 (and VEGF) on endo-thelial cells which follow apoptosis induction. Theseinhibitors could thus transform the transient apoptoticeffect of TGF-b1 into a sustained, massive effect. Micegenetically deficient in p38b show no major phenotype ifunchallenged (50), indicating that selective inhibition ofp38b would represent a feasible and safe approach toantitumor angiogenesis therapy.
Disclosure of Potential Conflicts of InterestNo potential conflicts of interest were disclosed.
Grant SupportThis work was supported by NIH grants R01 HL070203, R01 HL070203-03S1,
5R01CA136715, R21 RAG033735A; and funds fromDepartment of CardiothoracicSurgery, NYU School of Medicine, and Dr. Leo A. Shifrin and Roslyn Myers BreastCancer Discovery Fund (P. Mignatti), R21 AG028785 (G. Pintucci), RC1 HL10035(G. Ferrari).
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be herebymarked advertisement in accordance with18 U.S.C. Section 1734 solely to indicate this fact.
Received October 19, 2011; revised February 29, 2012; accepted March 14, 2012;published OnlineFirst April 20, 2012.
References1. Ferrara N. Vascular endothelial growth factor. Arterioscler Thromb
Vasc Biol 2009;29:789–91.2. Tian M, Neil JR, SchiemannWP. Transforming growth factor-beta and
the hallmarks of cancer. Cell Signal 2011;23:951–62.
3. Bierie B, Moses HL. Transforming growth factor beta (TGF-beta) andinflammation in cancer. Cytokine Growth Factor Rev 2010;21:49–59.
4. Ferrara N. Vascular endothelial growth factor: basic science andclinical progress. Endocr Rev 2004;25:581–611.
VEGF
Survival SurvivalApoptosis
flk-1
P
PPp38MAPK
p38MAPK p38MAPK
P P
P
P P P
VEGF VEGFTGF-β1 TGF-β1VEGF VEGF VEGF
β α βα
β α
flt-1flk-1
flt-1flk-1
flt-1
Figure 8. Control of endothelial cell apoptosis and survival by TGF- b1–VEGF interaction. In the absence of TGF- b1, VEGF induces activation of p38 b throughVEGFR-2 andpromotes endothelial cell survival (left). TGF- b1 induces VEGFexpression. Crosstalk between VEGF- and TGF-b1–specific signaling pathwaysconverts VEGF/VEGFR-2 activation of p38b into activation of the proapoptotic p38a. Activation of p38a is required for TGF- b1 induction of apoptosis;however, parallel TGF- b1signalingmayalsobenecessary. After the initial induction of apoptosis, endothelial cells become refractory to the apoptotic effect ofTGF- b1, and VEGF/VEGFR-2 signaling reverts to activation of the prosurvival p38 b (right).
Ferrari et al.
Mol Cancer Res; 10(5) May 2012 Molecular Cancer Research612
on July 12, 2019. © 2012 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst April 20, 2012; DOI: 10.1158/1541-7786.MCR-11-0507

5. Koch S, Tugues S, Li X, Gualandi L, Claesson-Welsh L. Signal trans-duction by vascular endothelial growth factor receptors. Biochem J2011;437:169–83.
6. Santibanez JF, Quintanilla M, BernabeuC. TGF-beta/TGF-beta recep-tor system and its role in physiological and pathological conditions.Clin Sci 2011;121:233–51.
7. Choi ME, Ballermann BJ. Inhibition of capillary morphogenesis andassociated apoptosis by dominant negative mutant transforminggrowth factor-beta receptors. J Biol Chem 1995;270:21144–50.
8. Madri JA, Pratt BM, Tucker AM. Phenotypic modulation of endothelialcells by transforming growth factor-beta depends upon the compo-sition and organization of the extracellular matrix. J Cell Biol1988;106:1375–84.
9. Roberts AB, Sporn MB, Assoian RK, Smith JM, Roche NS, WakefieldLM, et al. Transforming growth factor type beta: rapid induction offibrosis and angiogenesis in vivo and stimulation of collagen formationin vitro. Proc Natl Acad Sci U S A 1986;83:4167–71.
10. Yang EY, Moses HL. Transforming growth factor beta 1-inducedchanges in cell migration, proliferation, and angiogenesis in the chick-en chorioallantoic membrane. J Cell Biol 1990;111:731–41.
11. Orlova VV, Liu Z, Goumans MJ, Ten Dijke P. Controlling angiogenesisby two unique TGF-beta type I receptor signaling pathways. HistolHistopathol 2011;26:1219–30.
12. Pollman MJ, Naumovski L, Gibbons GH. Vascular cell apoptosis: celltype-specific modulation by transforming growth factor-beta1 inendothelial cells versus smooth muscle cells. Circulation 1999;99:2019–26.
13. Minami T, Rosenberg RD, Aird WC. Transforming growth factor-beta1-mediated inhibition of the flk-1/KDR gene is mediated by a 50-untranslated region palindromic GATA site. J Biol Chem 2001;276:5395–402.
14. Mandriota SJ, Menoud PA, Pepper MS. Transforming growth factorbeta 1 down-regulates vascular endothelial growth factor receptor 2/flk-1 expression in vascular endothelial cells. J Biol Chem 1996;271:11500–5.
15. Pollman MJ, Naumovski L, Gibbons GH. Endothelial cell apoptosis incapillary network remodeling. J Cell Physiol 1999;178:359–70.
16. Tsukada T, Eguchi K, Migita K, Kawabe Y, Kawakami A, Matsuoka N,et al. Transforming growth factor beta 1 induces apoptotic cell death incultured human umbilical vein endothelial cells with down-regulatedexpression of bcl-2. Biochem Biophys Res Commun 1995;210:1076–82.
17. HymanKM, Seghezzi G, Pintucci G, Stellari G, Kim JH,Grossi EA, et al.Transforming growth factor-beta1 induces apoptosis in vascularendothelial cells by activation of mitogen-activated protein kinase.Surgery 2002;132:173–79.
18. Bostrom K, Zebboudj AF, Yao Y, Lin TS, Torres A. Matrix GLA proteinstimulates VEGF expression through increased transforming growthfactor-beta1 activity in endothelial cells. J Biol Chem 2004;279:52904–13.
19. Ferrari G, Pintucci G, Seghezzi G, Hyman K, Galloway AC, Mignatti P.VEGF, a prosurvival factor, acts in concert with TGF-beta1 to induceendothelial cell apoptosis. Proc Natl Acad Sci U S A 2006;103:17260–5.
20. Ferrari G, Cook BD, Terushkin V, Pintucci G, Mignatti P. Transforminggrowth factor-beta 1 (TGF-beta1) induces angiogenesis through vas-cular endothelial growth factor (VEGF)-mediated apoptosis. J CellPhysiol 2009;219:449–58.
21. Cuenda A, Rousseau S. p38 MAP-kinases pathway regulation, func-tion and role in human diseases. Biochim Biophys Acta 2007;1773:1358–75.
22. Cuadrado A, Nebreda AR. Mechanisms and functions of p38 MAPKsignalling. Biochem J 2010;429:403–17.
23. Adams RH, Porras A, Alonso G, Jones M, Vintersten K, Panelli S, et al.Essential role of p38alpha MAP kinase in placental but not embryoniccardiovascular development. Mol Cell 2000;6:109–16.
24. Ohta T, Eguchi R, Suzuki A, Miyakaze S, Ayuzawa R, Kaji K. Hypoxia-induced apoptosis and tube breakdown are regulated by p38 MAPKbut not by caspase cascade in an in vitro capillary model composed ofhuman endothelial cells. J Cell Physiol 2007;211:673–81.
25. Chai W, Liu Z. p38 mitogen-activated protein kinase mediates palmi-tate-induced apoptosis but not inhibitor of nuclear factor-kappaBdegradation in human coronary artery endothelial cells. Endocrinology2007;148:1622–8.
26. Kumar P, Miller AI, Polverini PJ. p38 MAPK mediates gamma-irradi-ation-induced endothelial cell apoptosis, and vascular endothelialgrowth factor protects endothelial cells through the phosphoinositide3-kinase-Akt-Bcl-2 pathway. J Biol Chem 2004;279:43352–60.
27. Gratton JP,Morales-Ruiz M, Kureishi Y, Fulton D,Walsh K, SessaWC.Akt down-regulation of p38 signaling provides a novel mechanism ofvascular endothelial growth factor-mediated cytoprotection in endo-thelial cells. J Biol Chem 2001;276:30359–65.
28. Seghezzi G, Patel S, Ren CJ, Gualandris A, Pintucci G, Robbins ES,et al. Fibroblast growth factor-2 (FGF-2) induces vascular endothelialgrowth factor (VEGF) expression in the endothelial cells of formingcapillaries: an autocrine mechanism contributing to angiogenesis. JCell Biol 1998;141:1659–73.
29. Soderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius KJ,Jarvius J, et al. Direct observation of individual endogenous proteincomplexes in situ by proximity ligation. Nat Methods 2006;3:995–1000.
30. Koh W, Stratman AN, Sacharidou A, Davis GE. In vitro three dimen-sional collagen matrix models of endothelial lumen formation duringvasculogenesis and angiogenesis. Methods Enzymol 2008;443:83–101.
31. Gerber H-P, Dixit V, Ferrara N. Vascular endothelial growth factorinduces expression of the antiapoptotic proteins Bcl-2 and A1 invascular endothelial cells. J Biol Chem 1998;273:13313–16.
32. Yue TL, Ni J, Romanic AM, Gu JL, Keller P, Wang C, et al. TL1, a noveltumor necrosis factor-like cytokine, induces apoptosis in endothelialcells. Involvement of activation of stress protein kinases (stress-acti-vated protein kinase and p38 mitogen-activated protein kinase) andcaspase-3-like protease. J Biol Chem 1999;274:1479–86.
33. Xu ZR, Hu L, Cheng LF, Qian Y, Yang YM. Dihydrotestosteroneprotects human vascular endothelial cells from H(2)O(2)-induced apo-ptosis through inhibition of caspase-3, caspase-9 and p38MAPK. EurJ Pharmacol 2010;643:254–9.
34. Jiang H, Liang C, Liu X, Jiang Q, He Z, Wu J, et al. Palmitic acidpromotes endothelial progenitor cells apoptosis via p38 and JNKmitogen-activated protein kinase pathways. Atherosclerosis 2010;210:71–7.
35. Yilmaz A, Kliche S, Mayr-Beyrle U, Fellbrich G, Waltenberger J. p38MAPK inhibition is critically involved in VEGFR-2-mediated endothelialcell survival. Biochem Biophys Res Commun 2003;306:730–6.
36. Ambrosino C, Nebreda AR. Cell cycle regulation by p38 MAP kinases.Biol Cell 2001;93:47–51.
37. Delston RB, Matatall KA, Sun Y, Onken MD, Harbour JW. p38 phos-phorylates Rb on Ser567 by a novel, cell cycle-independent mecha-nism that triggers Rb-Hdm2 interaction and apoptosis. Oncogene2011;30:588–99.
38. Smeeton J, Zhang X, Bulus N, Mernaugh G, Lange A, Karner CM, et al.Integrin-linked kinase regulates p38MAPK-dependent cell cycle arrestin ureteric bud development. Development 2010;137:3233–43.
39. Wang X, Mcgowan CH, Zhao M, He L, Downey JS, Fearns C, et al.Involvement of the MKK6-p38gamma cascade in gamma-radiation-induced cell cycle arrest. Mol Cell Biol 2000;20:4543–52.
40. NewL,Han J. The p38MAPkinase pathway and its biological function.Trends Cardiovasc Med 1998;8:220–8.
41. Sicard P, Clark JE, Jacquet S, Mohammadi S, Arthur JS, O'keefe SJ,et al. The activation of p38 alpha, and not p38 beta, mitogen-activatedprotein kinase is required for ischemic preconditioning. J Mol CellCardiol 2010;48:1324–8.
42. Sawdey M, Podor TJ, Loskutoff DJ. Regulation of type 1 plasminogenactivator inhibitor gene expression in culturedbovine aortic endothelialcells. Induction by transforming growth factor-beta, lipopolysaccha-ride, and tumor necrosis factor-alpha. J Biol Chem 1989;264:10396–401.
43. Madri JA, Bell L, Merwin JR. Modulation of vascular cell behaviorby transforming growth factors beta. Mol Reprod Dev 1992;32:121–6.
Control of Endothelial Cell Apoptosis by p38MAPK Isoforms
www.aacrjournals.org Mol Cancer Res; 10(5) May 2012 613
on July 12, 2019. © 2012 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst April 20, 2012; DOI: 10.1158/1541-7786.MCR-11-0507

44. Iruela-ArispeML, Sage EH. Endothelial cells exhibiting angiogenesis invitro proliferate in response to TGF-beta 1. J Cell Biochem 1993;52:414–30.
45. Goumans MJ, Valdimarsdottir G, Itoh S, Rosendahl A, Sideras P,Ten Dijke P. Balancing the activation state of the endotheliumvia two distinct TGF-beta type I receptors. Embo J 2002;21:1743–53.
46. GoumansMJ, Valdimarsdottir G, ItohS, Lebrin F, Larsson J,MummeryC, et al. Activin receptor-like kinase (ALK)1 is an antagonistic mediatorof lateral TGFbeta/ALK5 signaling. Mol Cell 2003;12:817–28.
47. Ota T, Fujii M, Sugizaki T, Ishii M, Miyazawa K, Aburatani H, et al.Targets of transcriptional regulation by two distinct type I receptors for
transforming growth factor-beta in human umbilical vein endothelialcells. J Cell Physiol 2002;193:299–318.
48. BergeM,Bonnin P, Sulpice E, Vilar J, AllanicD, Silvestre JS, et al. Smallinterfering RNAs induce target-independent inhibition of tumor growthand vasculature remodeling in a mouse model of hepatocellularcarcinoma. Am J Pathol 2010;177:3192–201.
49. Kleinman ME, Yamada K, Takeda A, Chandrasekaran V, Nozaki M,Baffi JZ, et al. Sequence- and target-independent angiogenesis sup-pression by siRNA via TLR3. Nature 2008;452:591–7.
50. BeardmoreVA,HintonHJ, Eftychi C, ApostolakiM, ArmakaM,DarraghJ, et al. Generation and characterization of p38beta (MAPK11) gene-targeted mice. Mol Cell Biol 2005;25:10454–64.
Ferrari et al.
Mol Cancer Res; 10(5) May 2012 Molecular Cancer Research614
on July 12, 2019. © 2012 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst April 20, 2012; DOI: 10.1158/1541-7786.MCR-11-0507

2012;10:605-614. Published OnlineFirst April 20, 2012.Mol Cancer Res Giovanni Ferrari, Vitaly Terushkin, Martin J. Wolff, et al.
αp38 to Proapoptoticβ from the Prosurvival p38MAPKActivation of p38
1 Induces Endothelial Cell Apoptosis by Shifting VEGFβTGF-
Updated version
10.1158/1541-7786.MCR-11-0507doi:
Access the most recent version of this article at:
Material
Supplementary
http://mcr.aacrjournals.org/content/suppl/2012/04/20/1541-7786.MCR-11-0507.DC1
Access the most recent supplemental material at:
Cited articles
http://mcr.aacrjournals.org/content/10/5/605.full#ref-list-1
This article cites 50 articles, 22 of which you can access for free at:
Citing articles
http://mcr.aacrjournals.org/content/10/5/605.full#related-urls
This article has been cited by 2 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://mcr.aacrjournals.org/content/10/5/605To request permission to re-use all or part of this article, use this link
on July 12, 2019. © 2012 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst April 20, 2012; DOI: 10.1158/1541-7786.MCR-11-0507