Embed Size (px)
Citation preview

APPLIED AND ENVIRONMENTAL MICROBIOLOGY, July 2007, p. 4286–4293 Vol. 73, No. 130099-2240/07/$08.00�0 doi:10.1128/AEM.00119-07Copyright © 2007, American Society for Microbiology. All Rights Reserved.
A Rapid and Efficient Method for Cloning Genes of Type IIRestriction-Modification Systems by Use of a Killer Plasmid�
Iwona Mruk and Tadeusz Kaczorowski*Department of Microbiology, University of Gdansk, Kladki 24, 80-822 Gdansk, Poland
Received 17 January 2007/Accepted 18 April 2007
We present a method for cloning restriction-modification (R-M) systems that is based on the use of a lethalplasmid (pKILLER). The plasmid carries a functional gene for a restriction endonuclease having the sameDNA specificity as the R-M system of interest. The first step is the standard preparation of a representative,plasmid-borne genomic library. Then this library is transformed with the killer plasmid. The only survivingbacteria are those which carry the gene specifying a protective DNA methyltransferase. Conceptually, this invivo selection approach resembles earlier methods in which a plasmid library was selected in vitro by digestionwith a suitable restriction endonuclease, but it is much more efficient than those methods. The new method wassuccessfully used to clone two R-M systems, BstZ1II from Bacillus stearothermophilus 14P and Csp231I fromCitrobacter sp. strain RFL231, both isospecific to the prototype HindIII R-M system.
In the biology of microorganisms, the restriction-modifica-tion (R-M) systems play the important role of “molecularwardens” by protecting bacteria against phage infections. Onthe other hand, the enzymatic constituents of these systemshave also made great contributions to the field of recombinantDNA technology and have served as attractive models for thestudy of DNA-protein interactions due to the exact mode oftarget recognition (33, 34, 37). However, practical applicationsare limited almost solely to the enzymes belonging to type IIR-M systems. Each of these systems is composed of two enti-ties that act independently: a restriction endonuclease (ENase)that recognizes and cleaves a specific short nucleotide se-quence (4 to 8 bp) in DNA and a methyltransferase (MTase)that modifies the same sequence in order to protect the hostgenomic DNA against the action of the cognate restrictionenzyme. As both genes coding for R-M systems are usuallylocalized in close proximity (55), cloning an MTase gene oftenresults in the cloning of a matching ENase gene.
Until now, more than 400 type II R-M systems have beencloned (38) by the use of either (i) the “Hungarian trick” (45),which selects clones carrying the gene coding for a specificDNA MTase that confers resistance to digestion by the cog-nate restriction enzyme; (ii) methods based on the detection ofSOS response induction in Escherichia coli (10, 32); or (iii) adirect search through genomic databases to identify open read-ing frames coding for conserved amino acid sequence motifscharacteristic of DNA MTases, followed by PCR-based clon-ing (27).
Our research is focused on the nature of the isospecificityphenomenon among type II R-M systems. We would like toknow (i) how similar the genes coding for isospecific enzymesare, (ii) whether is it possible to map their functional domains,(iii) whether the systems recognize a cognate sequence in the
same way, (iv) what their mode of action is, and (v) how thosesystems spread among bacteria.
For the model in our study, we decided to use a group ofsystems isospecific to HindIII, an R-M system from Haemo-philus influenzae Rd (30, 39). This group consists of over 30R-M systems isolated from different bacteria, all of them rec-ognizing the same specific palindromic sequence: 5�-AAGCTT-3� (38). To date, except for HindIII (28), only two systems,EcoVIII from Escherichia coli E1585-68 (15, 26) and LlaCIfrom Lactococcus lactis subsp. cremoris W15, have been clonedand sequenced (22).
The phenomenon of isospecificity among type II R-M sys-tems is interesting in many ways. First, it poses the intriguingquestion of how genes that code for functional homologsevolve in bacteria that are often phylogenetically remote. Inaddition, the structural analysis of these enzymes can help toidentify particular protein motifs responsible for catalysis andtarget recognition and, consequently, may lead to the designingof enzymes with novel/tailored specificities in the future. At themoment, the small number of cloned isospecific systems makessuch studies difficult. Therefore, we have developed a simplecloning method to speed up our research efforts. The methodis based on the use of the killer plasmid, which simplifies theselection of clones carrying the gene coding for a specific DNAMTase. In this paper, we report the successful application ofthis approach to the cloning of two new HindIII-isospecificR-M systems: BstZ1II of Bacillus stearothermophilus 14P andCsp231I of Citrobacter sp. strain RFL231.
MATERIALS AND METHODS
Bacterial strains and plasmids. The bacterial strains used in this study, Ba-cillus stearothermophilus 14P (BstZ1II R� M�, where R is endonuclease activityand M is DNA methyltransferase activity) and Citrobacter sp. strain RFL231(Csp231I R� M�), were kindly provided by E. Raleigh, New England Biolabs,and A. Janulaitis, MBI Fermentas, Lithuania, respectively. Batch cultures of B.stearothermophilus 14P were grown at 55°C. The strains of E. coli used wereDH5�, MM294 (40), and MMS1999 (MM294 attB::ecoVIIIRM bla), obtainedfrom M. Sektas, Department of Microbiology, University of Gdansk, Poland.Bacteria were cultivated in LB or LA medium (40) that was supplemented, whenrequired and as appropriate, with the following antibiotics: ampicillin (Ap), 100
* Corresponding author. Mailing address: Department of Microbi-ology, University of Gdansk, Kladki 24, 80-822 Gdansk, Poland.Phone: (48-58) 305-6242. Fax: (48-58) 320-2031. E-mail: [email protected].
� Published ahead of print on 27 April 2007.
4286
on January 8, 2019 by guesthttp://aem
.asm.org/
Dow
nloaded from

�g/ml; tetracycline (Tc), 15 �g/ml; and chloramphenicol (Cm), 30 �g/ml. Thefollowing plasmids were used in this work: pKRP10 as a source for the cat (Cmr)cassette (35), and pGEM3Zf(�), pSP72 (Promega), and pBR322 (6) as vectorsfor cloning experiments.
DNA isolation and manipulation. For molecular cloning, we used standardtechniques (40). Recombinant plasmids were transformed into an appropriate E.coli strain and verified by restriction analysis and automated DNA sequencing.Genomic DNA was isolated as described previously (40) or with the use of aGenomic Mini AX bacterial kit (A&A Biotechnology). Restriction ENases andDNA-modifying enzymes were purchased from New England Biolabs, Epicentre,or MBI Fermentas. Enzymatic reactions were carried out under conditionssuggested by the suppliers. PCRs were performed with DyNAzyme II DNApolymerase from Thermus brockianus (Finnzymes Inc.).
Genomic library construction. Ten micrograms of genomic DNA from Bacil-lus stearothermophilus 14P or Citrobacter sp. strain RFL231 was partially digestedwith EcoRI, cloned into the EcoRI site of pBR322, and introduced into E. coliMM294. The resultant transformants were selected for resistance by plating onLA medium with Ap and Tc.
Oligonucleotides and DNA sequencing. Oligonucleotide synthesis was per-formed by Proligo. The following primers were used for PCR (all listed 5� to 3�):for vectorette PCR, VecTOP (GATCAGGCTGGAGATGT AGCAGATTGAGATATTCGTTATAGTTTACCTATCCCGACCGAGCATG), VecBOT (CATGCTCGGTCGGGATAGGCACTGGTCTAGAGGGTTAGGTTCCTGCTACATCTCCAGCCTGATC), VecPRIM (AGGCACTGGTCTAGAGGGTTAGGTTC), and BST4 (CATTCTGTTAC CATAGCC); for inverse PCR, BST12(GAAATGTTTCAGCAGTAC), and BST13 (AAACTATGC TATATTTTATAC).
DNA sequence analysis was performed using the ABI Prism BigDye Termi-nator cycle sequencing ready reaction kit, version 2.0 (Applied Biosystems), asrecommended by the manufacturer. The DNA template amounts for the se-quencing reactions were 50 ng for the PCR fragment, 300 ng for plasmid DNA,and 3 �g in the case of the chromosomal DNA of Citrobacter sp. strain RFL231.The cycling temperature profile for the sequencing of the chromosomal DNAwas as follows: 5 min at 96°C; 90 cycles of 96°C for 1.5 min, 50°C for 1 min, 60°Cfor 3 min; and extension at 72°C for 5 min. Products were analyzed using the ABIPRISM 310 automated sequencer (Perkin-Elmer, Applied Biosystems).
Vectorette and inverse PCR. Two PCR techniques were performed in order toreconstruct the cloned 5� end of a 10-kb EcoRI DNA fragment from B. stearo-thermophilus 14P: vectorette PCR (1, 4) and inverse PCR (I-PCR) (18, 29). Thevectorette unit was produced by hybridizing 500 pmol VecTOP with 500 pmolVecBOT in 50 �l containing 33 mM Tris-acetate (pH 7.8), 66 mM potassiumacetate, 10 mM magnesium acetate, and 0.5 mM dithiothreitol; heating themixture to 80°C; and then gradually cooling it to room temperature.
Approximately 1 �g of purified genomic DNA from B. stearothermophilus 14Pwas digested with SspI or RsaI to produce blunt ends. Vectorette libraries wereconstructed by ligating the corresponding compatible vectorette units to theobtained DNA fragments. The ligation mixture (10 �l) contained 10 pmol of thevectorette units, the fragmented genomic DNA (1 �g), 33 mM Tris-acetate (pH7.8), 66 mM potassium acetate, 10 mM magnesium acetate, 0.5 mM dithiothre-itol, 1 mM ATP, and 12 U of T4 DNA ligase. After incubation at 20°C for 1.5 h,the reaction mixture was inactivated by heating (60°C, 10 min). Vectorettelibraries were amplified in 50-�l reaction mixtures containing 50 pmol of specificprimer (BST4), 50 pmol of vectorette primer (VecTOP), 10 mM Tris-HCl, (pH8.3), 1.5 mM MgCl2, 150 �M deoxynucleoside triphosphates, and 5 U ofDyNAzyme II DNA polymerase. The PCR was run at 96°C for 1 min, 56°C for 1min, and 72°C for 2 min for 30 cycles, followed by extension at 72°C for 10 min.
The I-PCR includes digestion of target genomic DNA, self-ligation of theDNA fragment, and amplification of the circularized fragment by PCR. Onemicrogram of genomic DNA from Bacillus stearothermophilus 14P was digestedwith HinfI enzyme and self-ligated as previously described. PCRs were carriedout using 50-�l volumes of the following ingredients: the total DNA from theprevious step, 50 pmol of primer BST12, 50 pmol of primer BST13, 10 mMTris-HCl, (pH 8.3), 1 mM magnesium chloride, 500 �M deoxynucleoside triphos-phates, and 5 U of DNA polymerase. The PCRs were run with the followingcycling conditions: 96°C for 1 min; 30 cycles of 96°C for 1 min, 56°C for 1 min,and 72°C for 3 min; and a final step at 72°C for 10 min. The PCR productsobtained as a result of the two methods were separated by 7% polyacrylamide gelelectrophoresis, eluted, and analyzed by DNA sequencing.
Expression of genes coding for R.BstZ1II and R.Csp231I in E. coli. To expressthe gene coding for BstZ1II ENase in E. coli cells, the EcoRI–BamHI fragment(4.3 kb, R� M�) was subcloned into pSP72. After digestion with EcoRI andEcoRV, the resultant plasmid, pSPBst4.3 (R� M�), was used for cloning a475-bp DNA fragment containing the 5� end of bstZ1IIR. This fragment was
amplified from B. stearothermophilus genomic DNA by PCR (primers 5�-CATTCTGTTACCATAGCC-3� and 5�-GAGAATTCCTGTATAAGTTG-3�; under-lining indicates EcoRI site) and then processed with EcoRI. The constructobtained, pBstZ1II (R� M�), was introduced into E. coli MM294. The cloningprocedure was successful only if the cells were carrying the plasmid bearing theM.EcoVIII gene (pEcoVIIIM1), which has the same specificity as M.BstZ1IIIM.Thus, MTase was preexpressed in competent cells as described for the BamHIand DdeI R-M system (7, 13).
For the expression of the gene coding for R.Csp231I, a 2.4-kb EcoRI-BglIIDNA fragment was cloned into pSP72 double digested with EcoRI and BglII togenerate pSPCsp3.1 (Csp231I R� M�). Then a 0.53-kb DNA fragment codingfor C.Csp231I as well as the N-terminal portion of the R.Csp231I was amplifiedby PCR from the genomic DNA of Citrobacter sp. strain RFL231 (primers5�-AGGAATTCTTAGCAAAAGTG-3� and 5�-ATGATCACTAAACCAACG-3�), cleaved with EcoRI, ligated into EcoRI-digested pSPCsp3.1, and introducedinto premethylated competent E. coli MM294(pEcoVIIIM1) cells. The sequenceof the newly generated plasmid (pCsp231I, R� M�) was confirmed.
To determine the activity of R.BstZ1II or R.Csp231I, E. coli strains containingthe different ENase gene-bearing plasmids were grown and lysed by sonication orby incubation with lysozyme (0.4 mg/ml). Restriction activity was assayed in a20-�l reaction mixture containing 0.5 �g of � DNA, 10 mM Tris-HCl (pH 7.9),50 mM NaCl, 10 mM MgCl2, 1 mM dithiothreitol, and a solution of cell extractcontaining the enzyme (15 min at 37°C for R.Csp231I or 15 min at 55°C forR.BstZ1II). The products of � DNA digestion were analyzed on 1% agarose gels.
Computational analysis of DNA and proteins. Nucleotide and protein se-quences were searched for in the GenBank database (http://www.ncbi.nlm.nih.gov) using the BLAST program (2). Protein sequences were aligned by using theCLUSTAL W program (47), accessible through the European BioinformaticsInstitute server (http://www.ebi.ac.uk). Genes coding for the BstZ1II andCsp231I R-M systems were analyzed with DNASIS software (Hitachi SoftwareEngineering).
Nucleotide sequence accession numbers. The nucleotide sequences of genescoding for the BstZ1II and Csp231I R-M systems have been deposited in theGenBank database under accession numbers AY789018 and AY787793, respec-tively.
RESULTS
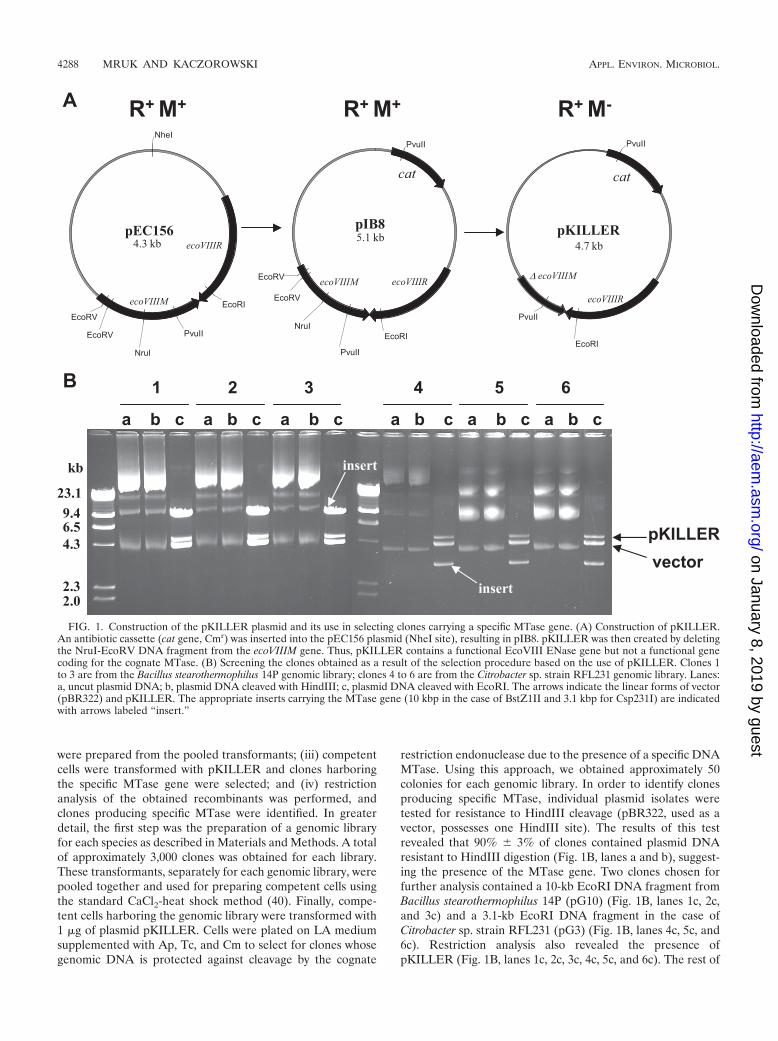
Construction of the lethal plasmid pKILLER. PlasmidpKILLER was constructed from a naturally occurring plasmid,pEC156, that carries the genes of the EcoVIII R-M system (25,26). In the first step of the plasmid construction, a 0.8-kb XbaIfragment (cat gene, Cmr) derived from pKRP10 (35) wascloned into the pEC156 plasmid linearized with NheI. Theobtained pEC156 derivative (pIB8, EcoVIII R� M�) (Fig.1A), after removal of the EcoRV-NruI DNA fragment (354bp) from the ecoVIIIM gene, was ligated, resulting in a finalconstruct. Thus, pKILLER (EcoVIII R� M�; 4.7 kb) containsa functional gene coding for EcoVIII ENase and an inactivegene coding for the cognate MTase (Fig. 1A). To demonstrateits efficiency in selecting clones carrying a gene for specificMTase, the pKILLER plasmid was introduced into E. colistrain MM294, used as a host in cloning experiments. As ex-pected, no transformants were obtained due to the lethal effectof the pKILLER plasmid which, inside the bacterial cell, func-tions as a Trojan horse. Therefore, to maintain and propagatepKILLER in bacteria, E. coli MMS1999, which contains achromosomal copy of the gene coding for EcoVIII MTase, wasused as a host.
Cloning strategy. The cloning procedure was carried outseparately for Bacillus stearothermophilus 14P and Citrobactersp. strain RFL231. In short, the steps involved in our methodwere as follows: (i) the genomic library was constructed bydigesting DNA with the suitable restriction enzyme, ligatingDNA fragments to the plasmid vector, and introducing therecombinant plasmids into E. coli cells; (ii) competent cells
VOL. 73, 2007 A METHOD FOR CLONING GENES OF TYPE II R-M SYSTEMS 4287
on January 8, 2019 by guesthttp://aem
.asm.org/
Dow
nloaded from
were prepared from the pooled transformants; (iii) competentcells were transformed with pKILLER and clones harboringthe specific MTase gene were selected; and (iv) restrictionanalysis of the obtained recombinants was performed, andclones producing specific MTase were identified. In greaterdetail, the first step was the preparation of a genomic libraryfor each species as described in Materials and Methods. A totalof approximately 3,000 clones was obtained for each library.These transformants, separately for each genomic library, werepooled together and used for preparing competent cells usingthe standard CaCl2-heat shock method (40). Finally, compe-tent cells harboring the genomic library were transformed with1 �g of plasmid pKILLER. Cells were plated on LA mediumsupplemented with Ap, Tc, and Cm to select for clones whosegenomic DNA is protected against cleavage by the cognate
restriction endonuclease due to the presence of a specific DNAMTase. Using this approach, we obtained approximately 50colonies for each genomic library. In order to identify clonesproducing specific MTase, individual plasmid isolates weretested for resistance to HindIII cleavage (pBR322, used as avector, possesses one HindIII site). The results of this testrevealed that 90% � 3% of clones contained plasmid DNAresistant to HindIII digestion (Fig. 1B, lanes a and b), suggest-ing the presence of the MTase gene. Two clones chosen forfurther analysis contained a 10-kb EcoRI DNA fragment fromBacillus stearothermophilus 14P (pG10) (Fig. 1B, lanes 1c, 2c,and 3c) and a 3.1-kb EcoRI DNA fragment in the case ofCitrobacter sp. strain RFL231 (pG3) (Fig. 1B, lanes 4c, 5c, and6c). Restriction analysis also revealed the presence ofpKILLER (Fig. 1B, lanes 1c, 2c, 3c, 4c, 5c, and 6c). The rest of
FIG. 1. Construction of the pKILLER plasmid and its use in selecting clones carrying a specific MTase gene. (A) Construction of pKILLER.An antibiotic cassette (cat gene, Cmr) was inserted into the pEC156 plasmid (NheI site), resulting in pIB8. pKILLER was then created by deletingthe NruI-EcoRV DNA fragment from the ecoVIIIM gene. Thus, pKILLER contains a functional EcoVIII ENase gene but not a functional genecoding for the cognate MTase. (B) Screening the clones obtained as a result of the selection procedure based on the use of pKILLER. Clones 1to 3 are from the Bacillus stearothermophilus 14P genomic library; clones 4 to 6 are from the Citrobacter sp. strain RFL231 genomic library. Lanes:a, uncut plasmid DNA; b, plasmid DNA cleaved with HindIII; c, plasmid DNA cleaved with EcoRI. The arrows indicate the linear forms of vector(pBR322) and pKILLER. The appropriate inserts carrying the MTase gene (10 kbp in the case of BstZ1II and 3.1 kbp for Csp231I) are indicatedwith arrows labeled “insert.”
4288 MRUK AND KACZOROWSKI APPL. ENVIRON. MICROBIOL.
on January 8, 2019 by guesthttp://aem
.asm.org/
Dow
nloaded from
the analyzed clones (�10%) have not possessed a gene forMTase or shown EcoVIII ENase activity (data not shown).This, however, contradicts our earlier experiments and might beexplained by mutations of unknown origin within the ecoVIIIRgene. Plasmid pKILLER was removed from selected clonesafter they were grown for a few generations without selectionfor resistance (Cm).
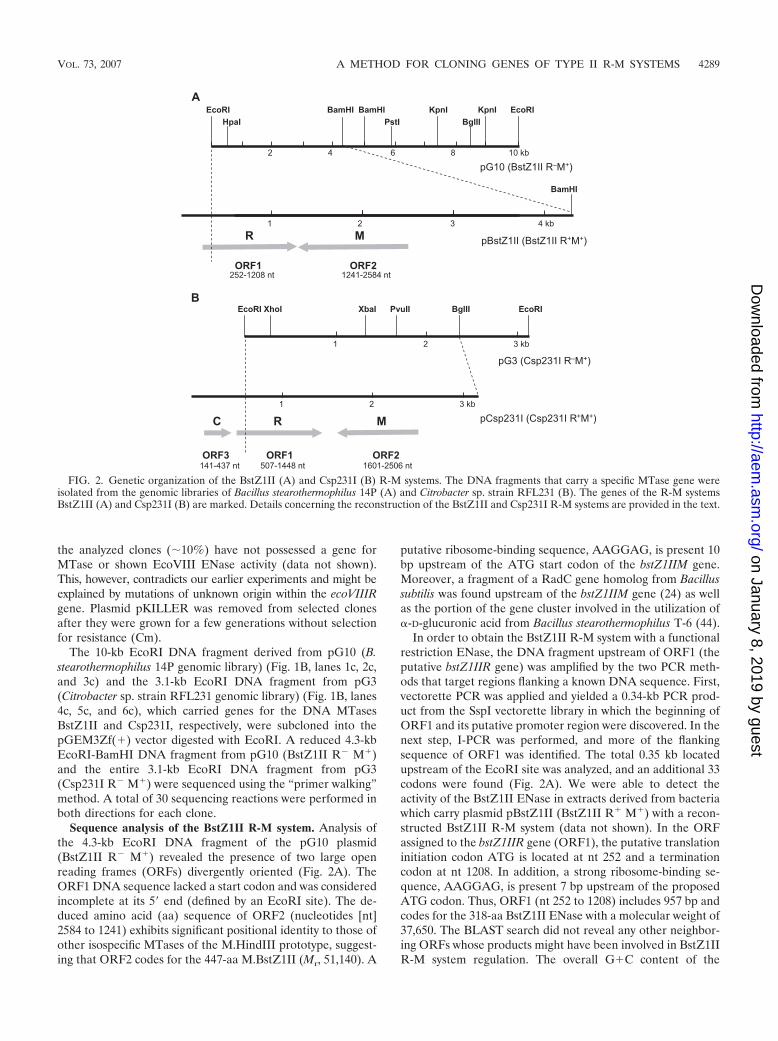
The 10-kb EcoRI DNA fragment derived from pG10 (B.stearothermophilus 14P genomic library) (Fig. 1B, lanes 1c, 2c,and 3c) and the 3.1-kb EcoRI DNA fragment from pG3(Citrobacter sp. strain RFL231 genomic library) (Fig. 1B, lanes4c, 5c, and 6c), which carried genes for the DNA MTasesBstZ1II and Csp231I, respectively, were subcloned into thepGEM3Zf(�) vector digested with EcoRI. A reduced 4.3-kbEcoRI-BamHI DNA fragment from pG10 (BstZ1II R� M�)and the entire 3.1-kb EcoRI DNA fragment from pG3(Csp231I R� M�) were sequenced using the “primer walking”method. A total of 30 sequencing reactions were performed inboth directions for each clone.
Sequence analysis of the BstZ1II R-M system. Analysis ofthe 4.3-kb EcoRI DNA fragment of the pG10 plasmid(BstZ1II R� M�) revealed the presence of two large openreading frames (ORFs) divergently oriented (Fig. 2A). TheORF1 DNA sequence lacked a start codon and was consideredincomplete at its 5� end (defined by an EcoRI site). The de-duced amino acid (aa) sequence of ORF2 (nucleotides [nt]2584 to 1241) exhibits significant positional identity to those ofother isospecific MTases of the M.HindIII prototype, suggest-ing that ORF2 codes for the 447-aa M.BstZ1II (Mr, 51,140). A
putative ribosome-binding sequence, AAGGAG, is present 10bp upstream of the ATG start codon of the bstZ1IIM gene.Moreover, a fragment of a RadC gene homolog from Bacillussubtilis was found upstream of the bstZ1IIM gene (24) as wellas the portion of the gene cluster involved in the utilization of�-D-glucuronic acid from Bacillus stearothermophilus T-6 (44).
In order to obtain the BstZ1II R-M system with a functionalrestriction ENase, the DNA fragment upstream of ORF1 (theputative bstZ1IIR gene) was amplified by the two PCR meth-ods that target regions flanking a known DNA sequence. First,vectorette PCR was applied and yielded a 0.34-kb PCR prod-uct from the SspI vectorette library in which the beginning ofORF1 and its putative promoter region were discovered. In thenext step, I-PCR was performed, and more of the flankingsequence of ORF1 was identified. The total 0.35 kb locatedupstream of the EcoRI site was analyzed, and an additional 33codons were found (Fig. 2A). We were able to detect theactivity of the BstZ1II ENase in extracts derived from bacteriawhich carry plasmid pBstZ1II (BstZ1II R� M�) with a recon-structed BstZ1II R-M system (data not shown). In the ORFassigned to the bstZ1IIR gene (ORF1), the putative translationinitiation codon ATG is located at nt 252 and a terminationcodon at nt 1208. In addition, a strong ribosome-binding se-quence, AAGGAG, is present 7 bp upstream of the proposedATG codon. Thus, ORF1 (nt 252 to 1208) includes 957 bp andcodes for the 318-aa BstZ1II ENase with a molecular weight of37,650. The BLAST search did not reveal any other neighbor-ing ORFs whose products might have been involved in BstZ1IIR-M system regulation. The overall G�C content of the
FIG. 2. Genetic organization of the BstZ1II (A) and Csp231I (B) R-M systems. The DNA fragments that carry a specific MTase gene wereisolated from the genomic libraries of Bacillus stearothermophilus 14P (A) and Citrobacter sp. strain RFL231 (B). The genes of the R-M systemsBstZ1II (A) and Csp231I (B) are marked. Details concerning the reconstruction of the BstZ1II and Csp231I R-M systems are provided in the text.
VOL. 73, 2007 A METHOD FOR CLONING GENES OF TYPE II R-M SYSTEMS 4289
on January 8, 2019 by guesthttp://aem
.asm.org/
Dow
nloaded from

BstZ1II R-M system is 30.2% (29.9% for bstZ1IIR and 30.4%for bstZ1IIM), which is significantly lower than the averageG�C content of B. stearothermophilus genomic DNA (50.9%).
Sequence analysis of the Csp231I R-M system. Analysis ofthe 3.1-kb EcoRI DNA fragment from pG3 (Csp231I R� M�)revealed the presence of two large ORFs divergently oriented(Fig. 2B). However, ORF1 was interrupted at its 5� end (theEcoRI site), and this, as in the case of the BstZ1II R-M system,corresponds to the restriction ENase gene. ORF2 (nt 2506 to1601) codes for the 301-aa Csp231I MTase (Mr, 34,140). Theribosome-binding sequence, AGGA, is present 10 bp upstreamof the translational start codon. Upstream of the csp231IMgene, we have found an incomplete ORF consisting of 177 bpwith a predicted amino acid sequence that is 51% identical toOrfB, a RadC ortholog of Bacillus, neighboring the MTasegene of the BslI R-M system (14). The E. coli radC genespecifies a RecG-like DNA recombination/repair function as-sociated with the replication fork.
To obtain the complete nucleotide sequence of the csp231IRgene, the same PCR methods as those described for the re-construction of the BstZ1II R-M system were used. However,despite repeated efforts, these approaches failed, so we em-ployed DNA sequencing using genomic DNA from Citrobactersp. strain RFL231 as a template. In each of two attempts, areverse primer was designed for the clearest part of the se-quencing traces, and then sequencing of PCR products wasperformed. The total flanking sequence of ORF1 (0.6 kb) wasidentified (Fig. 2B). ORF1 (csp231IR) is 942 bp long (nt 507 to1448) and codes for a polypeptide of 313 aa (Mr, 36,680). Aputative ribosome-binding site was found upstream of thisgene (AAGGA, 6 bp upstream of the ATG codon). In addi-tion, R.Csp231I activity was detected in bacterial extracts ofcells harboring pCsp231I carrying the reconstructed Csp231IR-M system (data not shown).
Computational analysis of the DNA fragment carrying theCsp231I R-M system revealed a third ORF (nt 141 to 437)(Fig. 2B) located 510 bp upstream of csp231IR and coding fora protein of 98 aa (Mr, 11,360) that shows similarity to Cproteins that are involved in the regulation of some R-M sys-tems’ gene expression.
The overall G�C content of the genes coding for theCsp231I R-M system is 34.2% (31.3% for csp231IR, 36.3% forcsp231IM, and 36.7% for csp231IC), which is significantlylower than the average G�C content of Citrobacter genomicDNA (50.5%).
Analysis of the amino acid sequences. The presence anddistribution of nine conserved amino acid motifs and a targetrecognition domain in the enzyme structure suggest thatM.Csp231I and M.BstZ1II belong to m6N-adenine -class
MTases (Fig. 3A). These motifs can be grouped in three clus-ters which are responsible for three principal functions: (i)sequence-specific DNA recognition (target recognition do-main [TRD]), (ii) binding of methyl group donor S-adenosyl-methionine (motifs X, I, II, and III), and (iii) catalysis ofmethyl group transfer (motifs IV, V, VI, VII, and VIII) (9, 11,23, 54). The overall level of positional identity between ana-lyzed isospecific MTases is between 42 and 62%, with the leastsimilar pair being M.BstZ1I and M.LlaCI and the most similarpair being M.EcoVIII and M.LlaCI. It is striking thatM.BstZ1I contains an additional 68-aa fragment located be-tween motif VIII and TRD that is absent in other isospecificMTases (Fig. 3A). A BLASTP search revealed no homology ofthis 68-aa region with any protein in the GenBank database.We hypothesize that this 68-aa fragment of the M.BstZ1IImight function as an extended TRD that determines the addi-tional sequence-specific methylation as previously shown forother multispecific MTases isolated from Bacillus and itsphages (42, 48, 50, 53).
The alignment of the predicted amino acid sequence ofENases isospecific to HindIII (Fig. 3B) showed low positionalidentity, limited to the putative catalytic/Mg(II) binding se-quence motif (PD/EXnDXK) that is characteristic for restric-tion ENases and is usually located in the N-terminal portion ofeach enzyme (34). When the sequences were compared inpairs, the highest level of positional identity showed that en-zymes R.BstZ1II and R.LlaCI (with 41% identity) originatedfrom gram-positive bacteria. Surprisingly, the level of posi-tional identity for isospecific ENases from gram-negative bac-teria was much lower (e.g., R.EcoVIII identity to R.Csp231Iwas 10% and R.HindIII identity to Csp231I was 11%). Thedifferences in G�C content between the BstZ1II and Csp231IR-M systems and in the genomic DNA of their hosts suggestthat these genetic entities entered the genomes of B. stearo-thermophilus and Citrobacter sp. relatively recently, whichagrees with the notion that R-M systems behave like mobilegenetic elements (17).
In our previous report, we proposed a catalytic motif(D94X14DXK111) for R.HindIII based on some structural andfunctional constraints (25). Thus, the suggested putative cata-lytic motifs for R.BstZ1II and R.Csp231I are D100X12DXK115
and D149X12DXK164, respectively (Fig. 3B), and these motifsare located in a 20-aa region of substantial positional identitythat is present in all HindIII isoschizomers (Fig. 3B).
Moreover, a database search produced other putativeHindIII-isospecific ENases: BbrRORF307P from Bordetellabronchiseptica RB50 (Wellcome Trust Sanger Institute, UnitedKingdom; www.sanger.ac.uk) and BbrRORF307P isogensfrom Bordetella pertussis Tahoma I and Bordetella parapertussis
FIG. 3. Comparison of amino acid sequences of HindIII-isospecific R-M enzymes: (A) MTases, (B) ENases, and (C) putative regulatory Cproteins of Csp231I and EcoO109I R-M systems. The numbers on the right margin denote the amino acid positions relative to the N terminus.The conserved motifs of m6N-adenine MTases are boxed and denoted by Roman numerals. The position of the putative TRD is indicated. Theregion of pronounced similarity between all isospecific ENases is boxed. The amino acids of the putative catalytic/magnesium binding motifPD/EXnDXK and putative DNA binding motif RXXR are indicated. Sequences were aligned using the CLUSTAL W computer program.Asterisks indicate identical amino acids; colons and periods indicate very similar amino acids and somewhat similar amino acids, respectively;dashes indicate gaps in the aligned sequences. The accession numbers for the nucleotide sequences of the HindIII, EcoVIII, LlaCI, BstZ1II,Csp231, BbrRORF307, and EsaSS1092P R-M genes that have been deposited in the GenBank database are L15391, AF158026, AJ002064,AY789018, AY787793, BX640437, and AACY01401088, respectively.
VOL. 73, 2007 A METHOD FOR CLONING GENES OF TYPE II R-M SYSTEMS 4291
on January 8, 2019 by guesthttp://aem
.asm.org/
Dow
nloaded from

12822 (31). Further searching produced another R.HindIII-like ORF corresponding to the putative, although not com-plete, isospecific ENase EsaSS1092P derived from an unknownorganism isolated from the Sargasso Sea based on a whole-genome shotgun sequencing project (GenBank accession num-ber AACY01401088) (51). Amino acid sequences of theseenzymes show pronounced identity with respect to the 20-aaregion containing the catalytic/Mg(II) binding sequence motif(Fig. 3B).
The deduced amino acid sequence of Csp231I R-M ORF3(Fig. 2B) showed significant identity to the members of thehelix-turn-helix families of DNA-binding proteins. These alsoinclude C regulatory proteins of several R-M systems, such asPvuII (46), BamHI (8), HgiAI (19), BglII (3), BstLVI (50),Kpn2I (20), SmaI (12), BclI (41), and EcoRV (43). Therefore,ORF3 was designated csp231IC, which might produce a puta-tive control protein, C.Csp231I, for the Csp231I R-M genes.However, we were unable to find a conserved nucleotide se-quence that resembles the “C box” motif, which is character-istic of some C gene promoters in the upstream region (5, 36,52). The deduced amino acid sequence of the putative productof the csp231IC gene shows 57.1% (56/98 aa) identity and85.7% (84/98 aa) similarity to the C protein of the EcoO109IR-M system from E. coli H709c (Fig. 3C) (16). Furthermore,the specific binding sequence of C.EcoO109I [CTAAG(N)5CTTAG] has been identified 70 nt upstream of the initiationcodon, ATG, for csp231IC.
DISCUSSION
The method presented in this report comprises a new ap-proach for the cloning of genes constituting type II R-M sys-tems. Our approach, much like the “Hungarian trick” devel-oped in Pal Venetianer’s laboratory in Szeged, Hungary (45),is based on the assumption that DNA from clones carrying agene for a specific MTase is modified and becomes resistant todigestion by a cognate restriction ENase, which recognizes thesame specific sequence. The key difference between these twomethods is the procedure used for the selection of clones thatproduce the specific MTase. In the “Hungarian trick,” selec-tion involves in vitro digestion of the plasmid library containingcloned genomic DNA followed by its transformation into E.coli in order to recover the intact plasmids (45). If an excess ofENase was used, then the background of false-positive clones(with no MTase gene) should be minimal. In principal, how-ever, it is typical that several cycles of digestion and transfor-mation are needed to accomplish successful cloning. Morethan a hundred R-M systems have been cloned using thisapproach (21, 38, 55). In our procedure, to enhance the effi-ciency of the cloning of the R-M genes, the selection is madein vivo with the use of a lethal plasmid, pKILLER, whichcontains a functional ENase gene along with an inactive gene(deletion derivative) coding for the cognate MTase. The ap-plication of the pKILLER plasmid enables the efficient se-lection of clones carrying a gene for a specific MTase fromextensive genomic libraries with a low background of nonpro-ductive recombinants. However, the use of this method is lim-ited to cloning only isospecific R-M systems, not systems ofunknown specificity. A related, but more difficult, in vivo se-lection approach has been demonstrated in the cloning of the
ppu21IM gene (49). That method uses a temperature-sensitivemutant of the MTase gene in an otherwise intact R-M system.
We developed our approach in order to clone R-M systemsisospecific to HindIII. However, this idea could be also appliedto the construction of any killer plasmid with an appropriateENase gene. Another advantage of our method is that there isno need for the presence of a specific ENase site on theplasmid vector, as the chromosomal DNA is modified and thusbecomes resistant to the action of the ENase supplied by thekiller plasmid. In our opinion, the procedure is quite easy toperform, has a low background of nonproductive recombi-nants, and is less time-consuming than other cloning methods.
The effectiveness of this approach has been demonstrated bythe successful cloning of Csp231I and BstZ1II MTase genesfrom the genomic libraries of Citrobacter sp. strain RFL231and Bacillus stearothermophilus 14P, respectively. The detailedgenetic and biochemical analyses of both newly cloned R-Msystems are in progress.
ACKNOWLEDGMENTS
This paper is dedicated in memoriam to Anna J. Podhajska.We thank Elisabeth Raleigh and Richard J. Roberts (New England
Biolabs), Arvydas Janulaitis (Fermentas, Lithuania), and Slawek Sektas(Department of Microbiology, University of Gdansk, Poland) for bac-terial strains and plasmids and Robert Blumenthal and Robert Lintner(University of Toledo) for a critical reading of the manuscript.
This work was supported by grant 2P04B-013-30 from the Ministryof Science and Higher Education (Warsaw, Poland).
REFERENCES
1. Allen, M. J., A. Collick, and A. J. Jeffreys. 1994. Use of vectorette andsubvectorette PCR to isolate transgene flanking DNA. PCR Methods Appl.4:71–75.
2. Altschul, S. F., T. L. Madden, A. A. Schaffer, J. Zhang, Z. Zhang, W. Miller,and D. J. Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generationof protein database search programs. Nucleic Acids Res. 25:3389–3402.
3. Anton, B. P., D. F. Heiter, J. S. Benner, E. J. Hess, L. Greenough, L. S.Moran, B. E. Slatko, and J. E. Brooks. 1997. Cloning and characterization ofthe Bg/II restriction-modification system reveals a possible evolutionary foot-print. Gene 187:19–27.
4. Arnold, C., and I. J. Hodgson. 1991. Vectorette PCR: a novel approach togenomic walking. PCR Methods Appl. 1:39–42.
5. Bart, A., J. Dankert, and A. van der Ende. 1999. Operator sequences for theregulatory proteins of restriction modification systems. Mol. Microbiol. 31:1277–1278.
6. Bolivar, F., R. L. Rodriguez, P. J. Greene, M. C. Betlach, H. L. Heyneker,and H. W. Boyer. 1977. Construction and characterization of new cloningvehicles. II. A multipurpose cloning system. Gene 2:95–113.
7. Brooks, J. E., J. S. Benner, D. F. Heiter, K. R. Silber, L. A. Sznyter, T.Jager-Quinton, L. S. Moran, B. E. Slatko, G. G. Wilson, and D. O. Nwankwo.1989. Cloning the BamHI restriction modification system. Nucleic AcidsRes. 17:979–997.
8. Brooks, J. E., P. D. Nathan, D. Landry, L. A. Sznyter, P. Waite-Rees, C. L.Ives, L. S. Moran, B. E. Slatko, and J. S. Benner. 1991. Characterization ofthe cloned BamHI restriction modification system: its nucleotide sequence,properties of the methylase, and expression in heterologous hosts. NucleicAcids Res. 19:841–850.
9. Bujnicki, J. M. 2001. Understanding the evolution of restriction-modifica-tion systems: clues from sequence and structure comparisons. Acta Biochim.Pol. 48:935–967.
10. Fomenkov, A., J. P. Xiao, D. Dila, E. Raleigh, and S. Y. Xu. 1994. The‘endo-blue method’ for direct cloning of restriction endonuclease genes in E.coli. Nucleic Acids Res. 22:2399–2403.
11. Gong, W., M. O’Gara, R. M. Blumenthal, and X. Cheng. 1997. Structure ofpvu II DNA-(cytosine N4) methyltransferase, an example of domain permu-tation and protein fold assignment. Nucleic Acids Res. 25:2702–2715.
12. Heidmann, S., W. Seifert, C. Kessler, and H. Domdey. 1989. Cloning, char-acterization and heterologous expression of the SmaI restriction-modifica-tion system. Nucleic Acids Res. 17:9783–9796.
13. Howard, K. A., C. Card, J. S. Benner, H. L. Callahan, R. Maunus, K. Silber,G. Wilson, and J. E. Brooks. 1986. Cloning the DdeI restriction-modificationsystem using a two-step method. Nucleic Acids Res. 14:7939–7951.
14. Hsieh, P.-C., J.-P. Xiao, D. O’Loane, and S.-Y. Xu. 2000. Cloning, expression,
4292 MRUK AND KACZOROWSKI APPL. ENVIRON. MICROBIOL.
on January 8, 2019 by guesthttp://aem
.asm.org/
Dow
nloaded from

and purification of a thermostable nonhomodimeric restriction enzyme, BslI.J. Bacteriol. 182:949–955.
15. Kaczorowski, T., and W. Szybalski. 1998. Genomic DNA sequencing bySPEL-6 primer walking using hexamer ligation. Gene 223:83–91.
16. Kita, K., J. Tsuda, and S. Y. Nakai. 2002. C.EcoO109I, a regulatory proteinfor production of EcoO109I restriction endonuclease, specifically binds toand bends DNA upstream of its translational start site. Nucleic Acids Res.30:3558–3565.
17. Kobayashi, I. 2001. Behavior of restriction-modification systems as selfishmobile elements and their impact on genome evolution. Nucleic Acids Res.29:3742–3756.
18. Kohda, T., and K. Taira. 2000. A simple and efficient method to determinethe terminal sequences of restriction fragments containing known sequences.DNA Res. 7:151–155.
19. Kroger, M., E. Blum, E. Deppe, A. Dusterhoft, D. Erdmann, S. Kilz, S.Meyer-Rogge, and D. Mostl. 1995. Organization and gene expression withinrestriction-modification systems of Herpetosiphon giganteus. Gene 157:43–47.
20. Lubys, A., S. Jurenaite, and A. Janulaitis. 1999. Structural organization andregulation of the plasmid-borne type II restriction-modification systemKpn2I from Klebsiella pneumoniae RFL2. Nucleic Acids Res. 27:4228–4234.
21. Lunnen, K. D., J. M. Barsomian, R. R. Camp, C. O. Card, S. Z. Chen, R.Croft, M. C. Looney, M. M. Meda, L. S. Moran, D. O. Nwankwo, et al. 1988.Cloning type-II restriction and modification genes. Gene 74:25–32.
22. Madsen, A., and J. Josephsen. 1998. Characterization of LlaCI, a newrestriction-modification system from Lactococcus lactis subsp. cremoris W15.Biol. Chem. 379:443–449.
23. Malone, T., R. M. Blumenthal, and X. Cheng. 1995. Structure-guided anal-ysis reveals nine sequence motifs conserved among DNA amino-methyl-transferases, and suggests a catalytic mechanism for these enzymes. J. Mol.Biol. 253:618–632.
24. Margolis, P. S., A. Driks, and R. Losick. 1993. Sporulation gene spoIIB fromBacillus subtilis. J. Bacteriol. 175:528–540.
25. Mruk, I., and T. Kaczorowski. 2003. Genetic organization and molecularanalysis of the EcoVIII restriction-modification system of Escherichia coliE1585-68 and its comparison with isospecific homologs. Appl. Environ. Mi-crobiol. 69:2638–2650.
26. Mruk, I., M. Sektas, and T. Kaczorowski. 2001. Characterization of pEC156,a ColE1-type plasmid from Escherichia coli E1585-68 that carries genes ofthe EcoVIII restriction-modification system. Plasmid 46:128–139.
27. Noren, C. J., R. J. Roberts, J. Patti, D. R. Byrd, and R. D. Morgan. March1999. Method for screening restriction endonucleases. U.S. patentWO9911821.
28. Nwankwo, D. O., L. S. Moran, B. E. Slatko, P. A. Waite-Rees, L. F. Dorner,J. S. Benner, and G. G. Wilson. 1994. Cloning, analysis and expression of theHindIII R-M-encoding genes. Gene 150:75–80.
29. Ochman, H., A. S. Gerber, and D. L. Hartl. 1988. Genetic applications of aninverse polymerase chain reaction. Genetics 120:621–623.
30. Old, R., K. Murray, and G. Boizes. 1975. Recognition sequence of restrictionendonuclease III from Hemophilus influenzae. J. Mol. Biol. 92:331–339.
31. Parkhill, J., M. Sebaihia, A. Preston, L. D. Murphy, N. Thomson, D. E.Harris, M. T. Holden, C. M. Churcher, S. D. Bentley, K. L. Mungall, A. M.Cerdeno-Tarraga, L. Temple, K. James, B. Harris, M. A. Quail, M. Acht-man, R. Atkin, S. Baker, D. Basham, N. Bason, I. Cherevach, T. Chilling-worth, M. Collins, A. Cronin, P. Davis, J. Doggett, T. Feltwell, A. Goble,N. Hamlin, H. Hauser, S. Holroyd, K. Jagels, S. Leather, S. Moule, H.Norberczak, S. O’Neil, D. Ormond, C. Price, E. Rabbinowitsch, S. Rutter, M.Sanders, D. Saunders, K. Seeger, S. Sharp, M. Simmonds, J. Skelton, R.Squares, S. Squares, K. Stevens, L. Unwin, S. Whitehead, B. G. Barrell, andD. J. Maskell. 2003. Comparative analysis of the genome sequences ofBordetella pertussis, Bordetella parapertussis and Bordetella bronchiseptica.Nat. Genet. 35:32–40.
32. Piekarowicz, A., R. Yuan, and D. C. Stein. 1991. A new method for the rapididentification of genes encoding restriction and modification enzymes. Nu-cleic Acids Res. 19:1831–1835.
33. Pingoud, A., and A. Jeltsch. 1997. Recognition and cleavage of DNA bytype-II restriction endonucleases. Eur. J. Biochem. 246:1–22.
34. Pingoud, A., and A. Jeltsch. 2001. Structure and function of type II restric-tion endonucleases. Nucleic Acids Res. 29:3705–3727.
35. Reece, K. S., and G. J. Phillips. 1995. New plasmids carrying antibiotic-resistance cassettes. Gene 165:141–142.
36. Rimseliene, R., R. Vaisvila, and A. Janulaitis. 1995. The eco72IC genespecifies a trans-acting factor which influences expression of both DNAmethyltransferase and endonuclease from the Eco72I restriction-modifica-tion system. Gene 157:217–219.
37. Roberts, R. J. 2005. How restriction enzymes became the workhorses ofmolecular biology. Proc. Natl. Acad. Sci. USA 102:5905–5908.
38. Roberts, R. J., T. Vincze, J. Posfai, and D. Macelis. 2007. REBASE—enzymes and genes for DNA restriction and modification. Nucleic Acids Res.35:D269–D270.
39. Roy, P. H., and H. O. Smith. 1973. DNA methylases of Hemophilus influen-zae Rd. I. Purification and properties. J. Mol. Biol. 81:427–444.
40. Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: alaboratory manual, 2nd ed. Cold Spring Harbor Laboratory, Cold SpringHarbor, NY.
41. Sawaya, M. R., Z. Zhu, F. Mersha, S. H. Chan, R. Dabur, S. Y. Xu, and G. K.Balendiran. 2005. Crystal structure of the restriction-modification systemcontrol element C.Bcll and mapping of its binding site. Structure 13:1837–1847.
42. Schumann, J., J. Walter, J. Willert, C. Wild, D. Koch, and T. A. Trautner.1996. M.BssHII, a multispecific cytosine-C5-DNA-methyltransferase withunusual target recognizing properties. J. Mol. Biol. 257:949–959.
43. Semenova, E., L. Minakhin, E. Bogdanova, M. Nagornykh, A. Vasilov, T.Heyduk, A. Solonin, M. Zakharova, and K. Severinov. 2005. Transcriptionregulation of the EcoRV restriction-modification system. Nucleic Acids Res.33:6942–6951.
44. Shulami, S., O. Gat, A. L. Sonenshein, and Y. Shoham. 1999. The glucuronicacid utilization gene cluster from Bacillus stearothermophilus T-6. J. Bacte-riol. 181:3695–3704.
45. Szomolanyi, E., A. Kiss, and P. Venetianer. 1980. Cloning the modificationmethylase gene of Bacillus sphaericus R in Escherichia coli. Gene 10:219–225.
46. Tao, T., J. C. Bourne, and R. M. Blumenthal. 1991. A family of regulatorygenes associated with type II restriction-modification systems. J. Bacteriol.173:1367–1375.
47. Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W:improving the sensitivity of progressive multiple sequence alignment throughsequence weighting, position-specific gap penalties and weight matrix choice.Nucleic Acids Res. 22:4673–4680.
48. Trautner, T. A., T. Balganesh, K. Wilke, M. Noyer-Weidner, E. Rauhut, R.Lauster, B. Behrens, and B. Pawlek. 1988. Organization of target-recogniz-ing domains in the multispecific DNA (cytosine-5)methyltransferases ofBacillus subtilis phages SPR and phi 3T. Gene 74:267.
49. Vaisvila, R., Z. Sliesaraviciute, S. Kulakauskas, and A. Janulaitis. 1995.Cloning of the ppu21IM gene using a in vivo selection method. Gene 157:55–57.
50. Vasquez, C. C., C. P. Saavedra, and S. E. Pichuantes. 2000. Nucleotidesequence of the gene encoding the BstLVI DNA methyltransferase: com-parison with other amino-DNA methyltransferases. Curr. Microbiol. 40:114–118.
51. Venter, J. C., K. Remington, J. F. Heidelberg, A. L. Halpern, D. Rusch, J. A.Eisen, D. Wu, I. Paulsen, K. E. Nelson, W. Nelson, D. E. Fouts, S. Levy, A. H.Knap, M. W. Lomas, K. Nealson, O. White, J. Peterson, J. Hoffman, R.Parsons, H. Baden-Tillson, C. Pfannkoch, Y. H. Rogers, and H. O. Smith.2004. Environmental genome shotgun sequencing of the Sargasso Sea. Sci-ence 304:66–74.
52. Vijesurier, R. M., L. Carlock, R. M. Blumenthal, and J. C. Dunbar. 2000.Role and mechanism of action of C � PvuII, a regulatory protein conservedamong restriction-modification systems. J. Bacteriol. 182:477–487.
53. Wilke, K., E. Rauhut, M. Noyer-Weidner, R. Lauster, B. Pawlek, B. Behrens,and T. A. Trautner. 1988. Sequential order of target-recognizing domains inmultispecific DNA-methyltransferases. EMBO J. 7:2601–2609.
54. Wilson, G. G. 1992. Amino acid sequence arrangements of DNA-methyl-transferases. Methods Enzymol. 216:259–279.
55. Wilson, G. G. 1991. Organization of restriction-modification systems. Nu-cleic Acids Res. 19:2539–2566.
VOL. 73, 2007 A METHOD FOR CLONING GENES OF TYPE II R-M SYSTEMS 4293
on January 8, 2019 by guesthttp://aem
.asm.org/
Dow
nloaded from



![Cold shock induction of recombinant Arctic environmental genes · Sub-cloning of genes into the pCold-II-based vectors was done by RF-cloning [22, 23], as described below. Several](https://img.dokumen.tips/doc/110x75/5f0c76ec7e708231d43588d8/cold-shock-induction-of-recombinant-arctic-environmental-genes-sub-cloning-of-genes.jpg)
![Enhanced biosurfactant production through cloning of … · Enhanced biosurfactant production through cloning of three genes and role of esterase ... agents [1,2,4,5]. ... New Delhi,](https://img.dokumen.tips/doc/110x75/5b5ad5b27f8b9a905c8cc115/enhanced-biosurfactant-production-through-cloning-of-enhanced-biosurfactant.jpg)

























