Embed Size (px)
Citation preview

Supported by educational grants from
Genentech, Inc., and Boehringer Ingelheim Pharmaceuticals, Inc.
A CME/CE Certified Supplement to
Jointly provided by
A Current Practice Snapshot of IPF Diagnosis and ManagementWhere We Are and Where We Need to Be
Faculty Jonathan H. Chung, MDAssociate Professor of RadiologyAssociate Section Chief Thoracic RadiologyUniversity of ChicagoChicago, Illinois
Gregory P. Cosgrove, MD, FCCPChief Medical OfficerPulmonary Fibrosis FoundationChicago, Illinois Associate Professor of Medicine National Jewish Health and University of Colorado, DenverDenver, Colorado
Kristin L. DeSimone, MDClinical Assistant ProfessorSidney Kimmel Medical College Thomas Jefferson UniversityPhiladelphia, Pennsylvania
IntroductionWhat Is IPF?Identifying and Diagnosing IPFMultidisciplinary Care for IPFPost-Test and Evaluation
Original Release Date: December 1, 2016
Expiration Date: December 31, 2017
Estimated Time to Complete Activity: 1.5 hours
©iStock.com
/Liderina/Brian McEntire

Table of ContentsIntroduction ......................................................................... 4What Is IPF? ........................................................................ 4Pathophysiology ................................................................................. 4Risk Factors for IPF ............................................................................ 5The Tragedy of Delayed Diagnosis .................................................... 5
Identifying and Diagnosing IPF ...................................... 6Diagnostic Approach .......................................................................... 6The Role of Imaging ........................................................................... 6Differential Diagnosis ......................................................................... 8The Role of Primary Care: Timely Referral ........................................ 9
A Current Practice Snapshot: Infographic ................10Multidisciplinary Care for IPF .......................................12Common Comorbidities ...................................................................12Prognostic Factors ...........................................................................13Nonpharmacologic Therapies .........................................................13Pharmacologic Treatment ................................................................14Communicating With the Patient .....................................................17
Summary .............................................................................17 Post-Test and Evaluation ...............................................19
This continuing education supplement was developed from interviews with the faculty. It is the final activity in the three-part curriculum, Improving the Diagnosis and Management of IPF Through Simulation and Peer Benchmarking. The supplement content brings together the key teaching points from the previous two online simulations, which may be found at: http://www.globalacademycme.com/supplements/clinical-decision-making- in-ipf-management.html.
The faculty acknowledge the editorial assistance of Global Academy for Medical Education, LLC, and Josh Kilbridge, medical writer, in the development of this supplement.
Neither the editors of CHEST® Physician nor the Editorial Advisory Board nor the reporting staff contributed to its content. The ideas and opinions expressed are those of the faculty and do not necessarily reflect the views of the supporters, Global Academy for Medical Education, Postgraduate Institute for Medicine, or the Publisher. Please refer to the official prescribing information for each product for discussion of approved indications, contraindications, and warnings.
Participants have an implied responsibility to use the newly acquired information to enhance patient outcomes and their own professional development. The information presented in this activity is not meant to serve as a guideline for patient management. Any procedures, medications, or other courses of diagnosis or treatment discussed or suggested in this activity should not be used by clinicians without evaluation of their patient’s conditions and possible contraindications and/or dangers in use, review of any applicable manufacturer’s product information, and comparison with recommendations of other authorities.
Copyright © 2016 by Global Academy for Medical Education, LLC, Frontline Medical Communications Inc., and its Licensors. All rights reserved. No part of this publication may be reproduced or transmitted in any form, by any means, without prior written permission of the Publisher. Global Academy for Medical Education, LLC, will not assume responsibility for damages, loss, or claims of any kind arising from or related to the information contained in this publication, including any claims related to the products, drugs, or services mentioned herein.
2 A Current Practice Snapshot of IPF Diagnosis and Management: Where We Are and Where We Need to Be
A Current Practice Snapshot of IPF Diagnosis and ManagementWhere We Are and Where We Need to Be
Faculty Jonathan H. Chung, MDAssociate Professor of RadiologyAssociate Section Chief Thoracic RadiologyUniversity of ChicagoChicago, Illinois
Gregory P. Cosgrove, MD, FCCPChief Medical OfficerPulmonary Fibrosis FoundationChicago, Illinois Associate Professor of Medicine National Jewish Health and University of Colorado, DenverDenver, Colorado
Kristin L. DeSimone, MDClinical Assistant ProfessorSidney Kimmel Medical CollegeThomas Jefferson UniversityPhiladelphia, Pennsylvania
A CME/CE Certified Supplement to

Original Release Date: December 1, 2016 Expiration Date: December 31, 2017 Estimated Time to Complete Activity: 1.5 hours
Method of ParticipationParticipants should read the activity information, review the activity in its entirety, and complete the online post-test and evaluation. Upon completing this activity as designed and achieving a passing score on the post-test, you will be directed to a Web page that will allow you to receive your certificate of credit via e-mail or you may print it out at that time.The online post-test and evaluation can be accessed at: http://tinyurl.com/IPFSup16Inquiries may be directed to Global Academy for Medical Education at [email protected] or (973) 290-8225.
Accreditation StatementsPhysicians: This activity has been planned and implemented in accordance with the accreditation requirements and policies of the Accreditation Council for Continuing Medical Education (ACCME) through the joint providership of Postgraduate Institute for Medicine and Global Academy for Medical Education. The Postgraduate Institute for Medicine is accredited by the ACCME to provide continuing medical education for physicians.Postgraduate Institute for Medicine designates this enduring material for a maximum of 1.5 AMA PRA Category 1 Credit(s)™. Physicians should claim only the credit commensurate with the extent of their participation in the activity.Nurses: Postgraduate Institute for Medicine is accredited as a provider of continuing nursing education by the American Nurses Credentialing Center’s Commission on Accreditation.This educational activity for 1.5 contact hours is provided by Postgraduate Institute for Medicine.
Target AudienceThis journal supplement is intended for pulmonologists, family practi-tioners, internists, nurse practitioners, physician assistants, and other clinicians who treat patients with idiopathic pulmonary fibrosis (IPF).
Educational NeedsThe identification and management of patients with IPF is replete with challenges. The symptoms of IPF are common to a variety of pulmonary conditions and easily missed by patients and clinicians. Suspicion of IPF often does not arise for at least several months after the onset of symptoms. Indeed, a key and common reason contributing to delayed diagnosis is the lack of expertise in IPF among community physicians. Primary care clinicians and pulmonary therapists may be unaware of the core signs, symptoms, and tests for IPF and its differential diagnosis. The early symptoms of IPF, dyspnea on exertion and dry cough, are often ignored by patients and primary care physicians and are attributed to aging or smoking. Because IPF most commonly occurs in men, older individuals (>50 years), and those with a history of smoking, clinicians may be focused on more common causes of pulmonary symptoms, especially in the community setting.
Studies of IPF also found that a multidisciplinary approach, including radiologists, pathologists, and pulmonologists, improved diagnostic agreement at both academic and community sites. A study from Europe found good accuracy for the diagnosis of IPF at expert centers (approaching 90%), but the level of agreement within the expert teams was only fair to moderate. Together, these findings suggest that a multi-disciplinary team is essential, even in tertiary care settings. Indeed, the American Thoracic Society (ATS) guidelines specifically recommend a
multidisciplinary approach to diagnosis and management. The availability of new therapies shown to slow disease progression
highlights the need for earlier diagnosis and intervention in IPF. It has been suggested that a “window of opportunity” may exist during which treatment can promote optimal outcomes. Furthermore, delays in diagnosis may limit treatment options, as well as increase costs, reduce patient quality of life, and impact survival.
Learning ObjectivesAt the conclusion of this program, participants should be better able to: • Utilize the signs, symptoms, and epidemiology of IPF to better
recognize patients in need of specialty care• Employ a multidisciplinary care team approach to IPF to enhance
patient outcomes • Provide timely referral of patients with IPF to tertiary care to
improve survival • Review clinical trial data supporting the efficacy and safety of
pirfenidone and nintedanib • Select appropriate pharmacologic therapy for patients with IPF • Communicate with patients with IPF and provide effective disease-
state education
Disclosure DeclarationsPostgraduate Institute for Medicine (PIM) requires instructors, planners, managers, and other individuals who are in a position to control the content of this activity to disclose any real or apparent conflict of interest (COI) they may have as related to the content of this activity. All identified COIs are thoroughly vetted and resolved according to PIM policy. PIM is committed to providing its learners with high-quality CME/CE activities and related materials that promote improvements or quality in healthcare and not a specific proprietary business interest of a commercial interest.
The faculty reported the following financial relationships or relation-ships to products or devices they or their spouse/life partner have with commercial interests related to the content of this CME/CE activity:
Gregory P. Cosgrove, MD, FCCP: Dr Cosgrove has been a consultant for Boehringer Ingelheim, Genentech, Global Blood Therapeutics, and Veracyte.
Jonathan H. Chung, MD: Dr Chung has no relevant financial relation-ships to disclose.
Kristin L. DeSimone, MD: Dr DeSimone has no relevant financial rela-tionships to disclose.
Global Academy for Medical Education Staff: Sylvia H. Reitman, MBA, DipEd; Mike LoPresti; Shirley V. Jones, MBA; Ron Schaumburg; and Josh Kilbridge hereby state that they or their spouse/life partner do not have any financial relationships or relationships to products or devices with any commercial interest related to the content of this activity of any amount during the past 12 months.
Postgraduate Institute for Medicine: The following planners and managers, Trace Hutchison, PharmD; Samantha Mattiucci, PharmD, CHCP; Judi Smelker-Mitchek, MBA, MSN, RN; and Jan Schultz, MSN, RN, CHCP, have nothing to disclose.
Off-Label/Investigational Use DisclosureThis educational activity may contain discussion of published and/or investigational uses of agents that are not indicated by the US Food and Drug Administration. The planners of this activity do not recommend the use of any agent outside of the labeled indications.
A Current Practice Snapshot of IPF Diagnosis and Management: Where We Are and Where We Need to Be • globalacademycme.com/pulmonology 3
This activity is not an official program of the American College of Chest Physicians (CHEST) and accordingly is not accredited by CHEST®.

Introduction Idiopathic pulmonary fibrosis (IPF) is a rare but lethal lung disease with a prognosis worse than many cancers. The disease is characterized by insidious onset and a progressive decline in lung function due to fibrosis of the lung parenchyma. Although rare enough to be considered an orphan disease, IPF still accounts for 15,000 to 40,000 deaths per year in the United States—and the incidence and mortality of IPF are increasing.1-4
The prognosis of IPF is poor, with median survival estimates of 3.8 years or less from diagnosis.4 There is currently no cure for IPF, but two new drugs were recently approved by the US Food and Drug Administration (FDA): nintedanib and pirfenidone. In clinical trials, these agents slowed the progression of disease in many patients, but they have not yet been shown to improve survival.5,6 The only intervention so far shown to extend survival in patients with IPF is lung transplantation.7 However, the availability of these new therapies highlights the need for earlier diagnosis and intervention in IPF. It has been proposed that a “window of opportunity” may exist during which treatment can promote optimal outcomes.8
Unfortunately, the gradual onset and nonspecific symptoms of IPF contribute to a high rate of missed or delayed diagnosis, leaving many patients untreated or inappropriately treated. Furthermore, delays in diagnosis may limit treatment options, increase costs, reduce patient quality of life, and decrease survival.9,10 In 2011 and 2015, updated guidelines for the management of IPF were published by an international coalition of societies, including the American Thoracic Society (ATS).7,11 These guidelines provide clinicians with evidence-based recommendations for the diagnosis and management of IPF. As valuable as the guidelines are, the limited evidence available means that IPF remains a clinical challenge for primary and specialty providers.
Persistent gaps in the care of patients with IPF include delayed or inaccurate diagnosis, delayed referral to specialty care, limited use of recommended multidisciplinary care, variable approaches to management, and poor patient-provider commu-nication. This monograph attempts to address these gaps by reviewing the latest research into IPF and the key features of the disease to facilitate recognition of IPF in primary care, timely referral to specialists, and optimal management to maximize quality and quantity of life. Highlighted throughout the monograph are the experiences of real patients with IPF and their caregivers, as elicited through personal interviews. These individuals report their stories of delayed diagnosis, frustrations with the healthcare system, and preferences and hopes for therapy. Finally, data are included from an online clinical simulation to illustrate the real-world decisions and performance of both primary care and specialty clinicians in the diagnosis and manage-ment of this challenging disease. This online simulation program includes images from diagnostic studies and audio and video commentary from leading experts in IPF.
IPF is the most common and lethal of the idiopathic interstitial pneumonias, a subgroup of the family of intersti-
tial lung diseases (ILDs) (Figure).12 ILDs involve inflammation of the interstitium, or the tissue and spaces around the bronchi/ bronchioles and alveoli of the lungs. IPF accounts for about 20% of all ILDs and is the most common and severe of the idio-pathic interstitial pneumonias.12
IPF is clinically characterized by nonspe-cific symptoms, such as dyspnea on exertion and dry cough, and requires specific inves-tigations (eg, high-resolution computed tomography [HRCT]) for diagnosis. The disease is associated with aging (median age of onset, 65-70 years) and is more common in
men and patients with a history of smoking.7 Features of IPF that cloud understanding of
the disease include its variable natural history, a high rate of complicating comorbidities, a paucity of accurate indicators of disease progression, and limited insight into its pathogenesis.13,14 Other factors contributing to suboptimal management include variation among providers in the application of guide-line recommendations and in the analysis of histologic and radiographic studies.14
Pathophysiology The pathophysiology of IPF is poorly under-stood but likely involves multiple pathways and complex interactions between genetic,
epigenetic, metabolic, and environmental factors.15 Indeed, the diversity of presen-tations, findings, and courses of disease in IPF suggests that fibrosis results from multiple pathogenic pathways, each of which may be influenced by endoge- nous and environmental factors.16 This multifactorial pathogenesis may explain the often poor results from clinical trials of agents that target individual mediators or pathways in IPF.15
The current model for the pathogen-esis of IPF involves abnormal wound healing in response to persistent or recur-rent injuries by environmental factors (eg , cigarette smoke, microaspirations,
4 globalacademycme.com/pulmonology • A Current Practice Snapshot of IPF Diagnosis and Management: Where We Are and Where We Need to Be
What Is IPF?

infection, dusts) against a background of genetic susceptibility.15,17 The result is an imbalance between pro-fibrotic and anti-fibrotic mediators, leading to excessive deposition of extracellular matrix compo-nents in the interstitium and alveolar spaces of the lungs. Progressive interstitial fibrosis and scarring destroy the lung microarchi-tecture and compromise lung function.15 As a result, gas exchange is impaired. Although oxygen tension may be normal or near normal in IPF in the initial stages, diffusing capacity of the lungs for carbon monoxide (DLCO) is often reduced.
Risk Factors for IPFAlthough IPF is an idiopathic disease whose causes remain unknown, several potential risk factors have been iden-tified, including cigarette smoking , environmental exposures, infection, gastro-esophageal reflux, and genetic factors.7 The association between smoking and IPF is strong, especially among patients with a smoking history of >20 pack-years. Increased risk for IPF has also been described in association with exposure to certain dusts (eg , metal, wood, and agricultural dusts) and microbial agents (eg, Epstein-Barr virus, cytomegalovirus, and human herpesvirus-7 and -8). Investigation into these associations continues, and the role of exposures to dusts or microbial agents in IPF remains putative.7
Gastroesophageal reflux is very common in patients with IPF and is often asymptom-atic. An intriguing mechanistic association
between microaspiration and IPF has been proposed, but no clear causative connection has yet been established.7 As noted, genetic factors likely play a role in susceptibility to IPF, and a small fraction of cases (<5%) involve familial forms of IPF (ie, affecting two or more family members).7 Several genetic factors have been investigated in sporadic IPF, but no genetic markers have yet been validated.7
The Tragedy of Delayed Diagnosis The identification of patients with IPF remains a major clinical challenge, especially in the primary care setting. The symptoms of IPF are common to a variety of condi-tions and are easily missed by patients and clinicians. Consideration for IPF often does not arise for at least several months after the onset of symptoms. Because IPF most commonly occurs in men, older indi-viduals (>50 years), and those with a history of smoking, clinicians may focus on more common causes of pulmonary symptoms in these patients, especially in the community setting. Indeed, the early symptoms of IPF, dyspnea on exertion and dry cough, are often ignored by patients and their physicians or attributed to aging or smoking history.18
A central diagnostic challenge in IPF is its differentiation from a wide range of conditions (see Figure), some of which may be nearly indistinguishable from IPF without a thorough history, examination, and appropriate imaging. The results of a poor understanding of the diagnosis
of IPF are evident in clinical studies.19 The importance of accurate diagnosis is highlighted by differences in treatment approach. For example, immunosup-pressive therapy may be appropriate for hypersensitivity pneumonitis, but has been associated with increased risk for death in patients with IPF.20
PATIENT EXPERIENCES Delayed Diagnosis An 84-year-old man:“I’d say five years ago, I noticed a little shortness of breath. I went to my family physician…. He said, ‘As you get older, your lungs get leathery, like a bellows on a firing forge….’ So I waited a couple of months, and I went to see a pulmonary physician. I told him I had been a smoker and quit 10-15 years prior. He had me walking around his office and said, ‘You probably have a little COPD [chronic obstructive pulmonary disease].’ I walked around the perimeter of his hallway until he was tired of taking a record of it! He said, ‘You quit smoking in time and it didn’t do any damage to you….’”
“[Then] I’m back in to see the pulmonary physician again, and he gave me a breathing test. At the end, he said, ‘You have enough lung capacity to last you until you are 120 years old….’ [Later] I was having trouble walking from the living room into the bathroom. I couldn’t even wash my face, I was out of breath so much. I went back in and…saw his PA. She didn’t take any x-rays, but she said, ‘[You've] got pneumonia.’ She gave me this stuff people breathe for COPD and an antibiotic….
“I went back to the pulmonary doctor. He put down his diagnosis: ‘Pulmonary nodules and COPD….’ I went back again in April and— here the words come on his diagnosis: ‘COPD, post-inflammatory fibrosis.’ Even on my second visit we had talked about fibrosis, and he said, ‘That’s just some kind of stuff in your lungs.’ I didn’t have any big concerns until I went back to him and he said IPF gave me my breathing problems. He still hadn’t given me any treatment.”
Figure. Classification of Interstitial Lung Diseases (ILDs)12
ILDs
Known etiology Granulomatous IIPs Miscellaneous
•LAM•Histiocytosis XIPF
•Sarcoidosis•Hypersensitivity pneumonitis
•Connective tissue disease•Drugs•Occupational exposures
Non-IPF IIP• Nonspecific interstitial pneumonia• Cryptogenic organization pneumonia• Respiratory bronchiolitis ILD• Desquamative interstitial pneumonia• Acute interstitial pneumonia• Lymphocytic interstitial pneumonia• Idiopathic pleuroparenchymal
fibroelastosis
A Current Practice Snapshot of IPF Diagnosis and Management: Where We Are and Where We Need to Be • globalacademycme.com/pulmonology 5
Histiocytosis X=pulmonary Langerhans cell histiocytosis; IIP=idiopathic interstitial pneumonia; IPF=idiopathic pulmonary fibrosis; LAM=lymphangioleiomyomatosis.

Diagnostic Approach As noted, the typical clinical presentation of IPF is nonspecific:
progressive dyspnea on exertion and dry cough. Features that may increase suspicion for IPF include male gender, age >50 years, history of smoking (current or former), history of gastroesophageal reflux disease (GERD), and family history of IPF (or pulmonary disease of unknown etiology).7 In studies, the age range of patients with IPF is 55 to 80 years, and older age (ie, >70 years) is the most powerful clinical predictor of IPF in a patient with idio-pathic ILD.17 Conversely, the probability of IPF in a patient younger than 50 years is much lower.17,21
In the absence of a family history of IPF it remains more likely to have a more common disorder, such as asthma, COPD, pneumonia, lung cancer, or heart disease than IPF. But recognizing the presentation and risk factors for IPF is the first step in correctly diagnosing IPF.
Physical Examination On physical examination, patients with IPF demonstrate fine bibasilar crackles on auscultation. These inspiratory crackles,
often called “Velcro® crackles” because of their characteristic sound, are nearly constant in IPF and develop early in the course of the disease.22 To identify bibasilar crackles, auscultation must be performed over all lung fields, especially the lower fields; a cursory examination may miss this critical feature of IPF. Crackles on auscul-tation—which should never be considered normal—may result from a variety of diseases, including pneumonia, heart failure, IPF, or other pulmonary disease. Although bibasilar crackles are not specific for IPF, their presence in an age-appropriate patient should prompt a thorough evalu-ation, including use of HRCT. In fact, bibasilar crackles in an older male patient likely signify IPF.23
Other elements of the physical examina-tion may also influence the likelihood of IPF. For example, finger clubbing is present in 25% to 50% of patients with IPF. Such findings as weight loss or muscle wasting are not common in IPF and may suggest other diseases.7 Examination of the joints and skin may identify features consistent with alternative diagnoses, such as connective tissue disease. A cardiac examination is appropriate to identify features consistent with heart failure or other conditions.
Investigations Studies that can help to rule out common causes of presenting symptoms and increase suspicion for IPF include pulmo-nary function tests (PFTs), oxygen saturation, and imaging. Findings on PFTs may suggest asthma or COPD but are not diagnostic for IPF. Typically, patients with IPF demonstrate a restrictive pattern on PFTs. However, COPD may be comorbid with IPF in some patients (such as those with a history of smoking ), and a PFT pattern that suggests COPD does not necessarily rule out IPF. When IPF is a possibility, DLCO should be included with the PFTs; DLCO is often reduced in patients with IPF due to impaired gas
exchange. Finally, some PFT findings, including DLCO, have prognostic utility following a diagnosis of IPF.7
Oxygen saturation is a standard test for patients who present with pulmonary or cardiac symptoms. However, because patients with IPF may have normal or near-normal oxygen saturation at rest, clinicians should consider a simple functional test of oxygen saturation. The 6-minute walk test (6MWT) is a validated test that can easily be performed in the primary care setting by having patients walk as far as they can within 6 minutes (or until the onset of symptoms). The 6MWT can be performed in a hallway that has been measured and marked at regular intervals to deter-mine the total distance walked within 6 minutes, or before the patient becomes symptomatic. A clinician walks with the patient to monitor oxygen saturation and symptoms. Metrics for the 6MWT include total distance walked, oxygen saturation on pulse oximetry, heart rate, and onset of symptoms such as dyspnea or chest pain. As noted by ATS guidelines, desaturation (ie, oxygen saturation <88%) on func-tion indicates the need for supplemental oxygen and may inform prognosis in IPF.7
The Role of Imaging The Right Test Can Be DiagnosticImaging is the most critical study in the diagnosis of IPF. Plain chest radiography is likely the first choice of many primary care clinicians and may identify infiltrates (suggesting pneumonia), cardiomegaly (suggesting heart failure), or nodules or masses (suggesting lung cancer). In more advanced cases of IPF, plain radiography may identify evidence of reticular changes. However, plain radiography is neither sensitive nor specific for the diagnosis of IPF, and it often identifies no abnormalities in early stages of the disease.
As a side note, plain radiography is also no longer recommended for routine lung cancer screening. Guidelines from multiple
Identifying and Diagnosing IPF
6 globalacademycme.com/pulmonology • A Current Practice Snapshot of IPF Diagnosis and Management: Where We Are and Where We Need to Be
CLINICAL PEARLS Recognizing the Key Clinical
Features of IPF• Symptoms: Dry cough and
dyspnea on exertion • Age: >50 years; median age,
65-70 years• Gender: Male predominance• Smoking history: Especially
≥20 pack-year history • Bibasilar crackles: Nearly
constant in IPF; sound like Velcro® being pulled apart
• Finger clubbing: Occurs in 25% to 50% of cases
IPF=idiopathic pulmonary fibrosis.

organizations, including the American Cancer Society and the US Preventive Services Task Force, currently recom-mend low-dose CT for annual screening of asymptomatic patients at high risk for lung cancer, defined as patients aged 55 to 80 years with a ≥30 pack-year smoking history who currently smoke or who quit within the last 15 years.24,25 The guideline authors note low-dose CT offers greater sensitivity compared to plain radiography and reduced exposure to ionizing radiation compared to conventional CT.
However, neither low-dose nor conven-tional CT provides sufficient detail to definitely identify the hallmark radio-graphic feature of IPF: a pattern of findings called a usual interstitial pneumonia (UIP) pattern. In cases with clinical suspi-cion for IPF, the gold-standard imaging study is HRCT.7 HRCT has 90% to 100% positive-predictive value for a pathologic UIP pattern (ie, a pattern confirmed by biopsy with histolog y). According
to current guidelines, this UIP pattern is a required criterion for a radiographic diagnosis of IPF. Features on HRCT that are consistent or inconsistent with a UIP pattern on HRCT are illustrated in the Table.7 In about two-thirds of cases, a diag-nosis of IPF can be made based on clinical history and a finding of UIP on HRCT.15
Although HRCT is considered the gold-standard test and is recommended by guidelines, the diagnostic features on HRCT are found in only about half of all patients with IPF, further increasing the difficulty of diagnosis.26 In fact, the typical honeycomb features of IPF on HRCT are a relatively late manifestation of the disease.20 In a patient with bibasilar crackles or other potential signs of IPF who does not have clear honeycombing on HRCT, a lung biopsy may be considered to support an earlier diagnosis.23 When lung biopsies are not possible, either due to the patient’s condition or resistance to an inva-sive test, patients are left with a diagnosis of
suspected IPF, potentially preventing the initiation of disease-appropriate therapy.10
Further complicating diagnosis, a UIP pattern may also be found in other disor-ders.15 Even in specialty centers, a diagnosis of unclassifiable ILD is made in about 10% of patients who present with progressive pulmonary fibrosis.27 For these reasons, evaluation of suspected IPF by a multidisci-plinary team is recommended by guidelines.
Table. HRCT Criteria for Usual Interstitial Pneumonia (UIP) Pattern7
UIP Pattern (all 4) Possible UIP (all 3) Inconsistent With UIP (any)
• Subpleural, basal predominance
• Reticular abnormality
• Honeycombing with or without traction bronchiectasis
• Absence of features inconsistent with UIP pattern (third column)
• Subpleural, basal predominance
• Reticular abnormality
• Absence of features inconsistent with UIP pattern (third column)
• Upper or mid-lung predominance
• Peribronchovascular predominance
• Extensive ground-glass abnormalities
• Profuse micronodules
• Discrete cysts
• Diffuse mosaic attenuation/air-trapping
• Consolidation in bronchopulmonary segment(s)/lobe(s)
HRCT=high-resolution computed tomography.
PATIENT EXPERIENCES Doing the Right TestsA 67-year-old man:“We circled everything for about two years. I saw a cardiologist, an ENT, an allergist—I was diagnosed with everything from sinus problems to allergies. I had multiple sessions of bronchitis and pneumonia. I went through two hernia operations because the coughing was so violent. The GP was trying to figure it out, but I was getting more and more sick.
“Then I saw an infectious disease doctor. He would ask questions, sit and think, scribble, ask more questions, circling around. After 45 minutes, he dragged me by the hand up a flight of stairs with an oximeter on my finger and nailed it. He said, ‘I think you have pulmonary fibrosis.’ He called my insurance company to get me a high-res CT scan, and got me an appointment with a pulmonologist the next day. The pulmonologist wanted a lung biopsy. Within the next week and a half, a lung biopsy confirmed IPF.”
A Current Practice Snapshot of IPF Diagnosis and Management: Where We Are and Where We Need to Be • globalacademycme.com/pulmonology 7
CLINICAL PEARLS ATS-Required Diagnostic
Criteria for IPF7
• Exclusion of other known causes of ILD (eg, environmental exposures, connective tissue disease, drug toxicity)
• Presence of a UIP pattern on HRCT
• Combinations of HRCT and surgical lung biopsy pattern
ATS=American Thoracic Society; HRCT=high-resolution computed tomography; ILD=interstitial lung disease; IPF=idiopathic pulmonary fibrosis; UIP=usual interstitial pneumonia.
CLINICAL PEARLS Use of Surgical Lung Biopsy
in ILD• Surgical lung biopsy may
contribute to diagnosis in patients with uncertain HRCT findings
• Most patients with a possible UIP pattern on HRCT have IPF
• The technique is associated with risk for mortality
• Patient health status and preferences must be considered
HRCT=high-resolution computed tomography; ILD=interstitial lung disease; IPF=idiopathic pulmonary fibrosis; UIP=usual interstitial pneumonia.

When Imaging May Not Be Enough: The Roles of Biopsy and Bronchial Alveolar LavageWhen a diagnosis of a UIP pattern is made with high confidence based on HRCT, a lung biopsy is not required, and the diag-nosis of IPF is made based on clinical and radiographic features. In some situations, such as when HRCT findings are incon-clusive but suggest UIP, further testing may be required. This testing may include bronchoscopy, surgical lung biopsy, and/or bronchial alveolar lavage. In these cases, the care team, including the pulmon-ologist, radiologist, and/or pathologist, should review and discuss the necessary tests and findings.
Surgical lung biopsy is an invasive procedure with attributable morbidity and mortality; depending on the study, mortality rates of 1% to 6% (or even higher in nonelective cases) have been reported.28-30 The risks and benefits of pursuing a surgical treatment must be weighed for each patient. Histology can be used to confirm a diagnosis of IPF or differ-entiate uncertain diagnoses. However, as noted, HRCT findings have 90% to 100% positive-predictive value for a pathologic UIP pattern, and surgical lung biopsy is not essential to confirm a UIP pattern identi-fied on HRCT.
In fact, the appropriate role of surgical lung biopsy in the diagnosis of IPF remains debatable. ATS guidelines suggest a role for surgical lung biopsy when HRCT findings for a UIP pattern are uncertain. However, small studies suggest that a possible UIP on HRCT may still have high specificity for IPF. For example, a retrospective study found that patient age >65 years and an HRCT finding of reticular abnormalities in the absence of honeycombing (the hall-mark radiologic feature of IPF) had >95% likelihood of discriminating IPF from other interstitial pneumonias.21 In other words, older age and HRCT findings of possible UIP predicted a diagnosis of IPF in >95% of cases in this study, suggesting that surgical lung biopsy would be unnec-essary in the vast majority of cases in which HRCT findings were neither inconsistent nor definitive for UIP.
A more recent study goes even further. In this retrospective study of 241 individuals enrolled in an IPF treatment trial, investiga-tors found that 94% of patients with possible UIP on HRCT had histologic evidence of UIP on biopsy—but 95% of patients with HRCT findings inconsistent with UIP still had histologic evidence of UIP on biopsy.31
Because patients in this study were already diagnosed with IPF and enrolled in an IPF clinical trial, the findings cannot be general-ized to the broader population of patients with IPF or ILD. Nevertheless, the findings do suggest that, in patients with clinical features consistent with IPF (ie, male, history of smoking, older age) and lacking alternative etiologies, IPF remains a likely diagnosis, regardless of HRCT findings.32
Bronchial alveolar lavage (BAL) also can contribute to the diagnosis of ILD when radiographic findings are not definitive. According to ATS guidelines for IPF, it is unclear whether BAL contributes signifi-cantly to the specificity of diagnosis.7 The ATS has also published guidelines for the use of BAL in the diagnosis of ILD.33 These guidelines note that BAL may be useful in the evaluation of patients with possible ILD when confidence in a UIP pattern on HRCT is lacking. Findings that may aid in the differential diagnosis include a predominantly inflammatory pattern, such as increased lymphocytes, eosinophils, or neutrophils, on BAL. For example, promi-nent lymphocytosis (>40%) may suggest hypersensitivity pneumonitis. However, a normal cell profile on BAL does not exclude microscopic abnormalities, and BAL alone is insufficient to diagnose most forms of ILD.7,33
Differential DiagnosisAs outlined by ATS guidelines, the diag-nosis of IPF requires the exclusion of other forms of ILD (see Figures 1 and 2).7 This differential diagnosis relies on serologic blood tests, a detailed patient medical history, and identification of any relevant environmental exposures.
Serologic blood tests are performed to identify or rule out connective tissue disease. A typical serology panel for a patient with suspected ILD includes antinuclear anti-bodies (ANAs), rheumatoid factor (RF), and anti–cyclic citrullinated peptide (anti-CCP) antibodies.7 Some patients with IPF may have mild elevations in ANAs and/or
Figure 1. Tests and Procedures That May Increase Suspicion for IPF
6-minute walk test: Simple functional testof oxygen saturation and symptoms
Spirometry/pulmonary function tests: Restrictive pattern, reduced diffusing capacity of the lungs for carbon monoxide
Pulse oximetry (with functional test): Desaturation on activity
Plain radiography: Rule out commonconditions (eg, pneumonia, heart failure)
HRCT: Gold-standard imaging test for IPF
8 globalacademycme.com/pulmonology • A Current Practice Snapshot of IPF Diagnosis and Management: Where We Are and Where We Need to Be
CLINICAL PEARLS Features That Suggest
a Need for Referral• Unexplained dry cough and
dyspnea on exertion in a clinically appropriate patient (eg, older man, history of smoking)
• Bibasilar crackles on lung auscultation
• Significant desaturation on oximetry (eg, <90%)
• Atypical findings on non-HRCT imaging (eg, bibasilar reticular abnormalities)
• HRCT findings suggesting UIP
• Lack of response to empiric therapies (eg, bronchodilators, antibiotics, inhaled corticosteroids)
HRCT=high-resolution computed tomography; UIP=usual interstitial pneumonia.
HRCT=high-resolution computed tomography; IPF=idiopathic pulmonary fibrosis.

RF without any clinical features of connec-tive tissue disease. Positive findings on these studies should trigger further investigation for features of connective tissue disease (eg, arthritis, Raynaud’s phenomenon, skin changes, abnormal esophageal motility).7
A thorough medical history may identify exposure to medications associated with ILD. Examples include certain chemo-therapeutic agents, antiarrhythmic drugs (eg , amiodarone), antimicrobial agents (eg, nitrofurantoin), and immunosuppres-sive agents.7 Exposure to environmental toxins should also be identified. Examples of domestic and occupational exposures associated with ILD include prolonged exposure to organic proteins (eg, mold, bird dander) as well as asbestos and dust from coal, silica, cotton, and hard metals.7
An unmet need in the diagnosis and management of IPF is the development and validation of diagnostic, prognostic, and predictive biomarkers specific to IPF. Validated biomarkers could aid in the identification of patients with IPF and, potentially, risk stratification and the selec-tion of appropriate therapies.15
The Role of Primary Care: Timely ReferralEven for specialists, IPF can be challenging to diagnose and manage. The role of the primary care clinician is not necessarily to confirm a diagnosis of IPF but rather to recognize features that suggest an atypical or serious pulmonary disease and then
refer the patient to a specialist, preferably one with experience in ILD.
Too often, the challenges of recognizing the features of IPF in primary care result in delayed referral and diagnosis. In one cohort study, the median delay between onset of dyspnea and initial evaluation in tertiary care was 2.2 years—an especially long time, considering that the median survival of patients with IPF is 3.8 years.9 A longer referral delay in this study was associated with increased risk of death, independent of disease severity.9 Findings such as these highlight the need for timely recognition of features that suggest an ILD (or IPF specifically) and immediate referral to a specialist.
The need for specialty care in IPF is highlighted by studies that demonstrate diagnostic inconsistencies among physicians. For example, a retrospective study found a lack of agreement between community and academic physicians in the diagnosis of paren-chymal lung diseases, including IPF.34 In this study, the likelihood of reaching a diagnosis of IPF differed between community and academic settings. The study also found that a multidisciplinary approach, including radi-ologists, pathologists, and pulmonologists, improved diagnostic agreement at both academic and community sites.34 These data highlight the benefits of referral and multi-disciplinary collaboration (Figure 3).
A Current Practice Snapshot of IPF Diagnosis and Management: Where We Are and Where We Need to Be • globalacademycme.com/pulmonology 9
PATIENT EXPERIENCES The Importance of Specialty CareA 67-year-old man:“If I had to give advice to someone having symptoms that might be IPF, I’d tell them to get to a pulmonolo-gist. And if you’re not convinced of the answer, see a second doctor. I think waiting can let you be robbed of some of your life. I don’t have time to ignore that flower; I don’t have time to waste. Stop and pull the precious parts of life out— a kid, a cat, a really good book—savor it. That’s what they need a correct diagnosis for.”
Figure 2. Diagnostic Algorithm for IPF, as Described by ATS Guidelines7
IPF
Suspected IPF
Identifiable causes for ILD?
HRCT
Surgical lung biopsy
IPF/Not IPF Not IPF
UIP
No
Yes
Not UIP
Possible UIP; inconsistent with UIP
MDD
Figure 3. The Multidisciplinary Team
Pulmonologist PathologistPatient With IPF
RadiologistPCP
The challenges of diagnosis and early referral indicate the need for the evaluation of possible IPF by an experienced multidisciplinary team.
ATS=American Thoracic Society; HRCT=high-resolution computed tomography; ILD=interstitial lung disease; IPF=idiopathic pulmonary fibrosis; MDD=multidisciplinary discussion; UIP=usual interstitial pneumonia.
IPF=idiopathic pulmonary fibrosis; PCP=primary care physician.

10 globalacademycme.com/pulmonology • A Current Practice Snapshot of IPF Diagnosis and Management: Where We Are and Where We Need to Be
A Current Practice Snapshot of Idiopathic Pulmonary Fibrosis Diagnosis and Management: Where We Are and Where We Need to BeThis infographic was developed using a mix of data from scientific sources and two currently available CME/CE simulation activities designed to educate clinicians and also identify trends in idiopathic pulmonary fibrosis (IPF) diagnosis and management. A “Current Practice Snapshot” was created from the simulation data to enable a comparison of current practice against the IPF evidence base and IPF guidelines (both presented in this supplement). Primary care physicians and pulmonologists participated in each simulation, providing an additional layer of data for analysis.
Onset ofsymptoms
Initial evaluationin tertiary care
Death?
Year 1 Year 2 Year 3 Year 4 Year 5
1 Delayed Diagnosis Is the Tragedyof IPF
IPF is the most common and severe of the idiopathic interstitial pneumo-nias. Identification of patients with IPF remains a major clinical challenge, especially in the primary care setting. The symptoms of IPF are common to a variety of conditions and are easily missed by patients and clinicians, and suspicion for IPF does not arise for several months.
6-minute walk test: Simple functional testof oxygen saturation and symptoms
Spirometry/pulmonary function tests: Restrictive pattern, reduced diffusing capacity of the lungs for carbon monoxide
Pulse oximetry (with functional test): Desaturation on activity
Plain radiography: Rule out commonconditions (eg, pneumonia, heart failure)
High-resolution computed tomography: Gold-standard imaging test for IPF
3 Specific Investigations Are Importantto Increase Suspicion for IPF
Age: >50 years;median age, 65-70 years
Bibasilar crackles(sound like Velcro®
being pulled apart)
Finger clubbing:Occurs in 25%-50%
Dry cough
Dyspneaon exertion
Environmentalexposures
Infection
Gastro-esophageal
reflux disease(GERD)
Malepredominance
Geneticfactors
Smoking history,especially ≥20
pack-years
2 Recognizing a Progressive ClinicalPattern Is the First Step in CorrectlyDiagnosing IPF
Causes of IPF remain unknown, but several potential risk factors have been identified. Typical clinical presentation is nonspecific: progressive dyspnea on exertion and dry cough.
Risk Factors & Symptoms
4 High-Resolution Computed Tomography (HRCT) Is Underutilized in Diagnosing IPFIn cases with clinical suspicion for IPF, the gold-standard imaging study is HRCT.
Clinicians are much more likely to order HRCT later in the diagnostic process– a reasonable and appropriate choice, but one that lengthens the time to confirmed diagnosis and delays the start of treatment.
Diagnostic algorithm for IPF, as described by the American Thoracic Society guidelines.1ILD=interstitial lung disease; MDD=multidisciplinary discussion; PCP=primary care physician; UIP=usual interstitial pneumonia.
94% of Pulmonologistsn=53
86% of PCPsn=29
Where we are:In the simulation case, HRCT was chosen at diagnosis by:
In a simulation case, HRCT was chosen at management by:
13% of PCPsn=227
53% of Pulmonologistsn=40
Where we need to be:
Suspected IPF
Yes
No
UIPNot UIP
Not IPFIPF/Not IPF
MDD
Surgical lung biopsy
IPF
Possible UIP; inconsistent with UIP
HRCT
Identifiable causes for ILD?
94% of Pulmonologistsn=53
86% of PCPsn=29
Where we are:In the simulation case, HRCT was chosen at diagnosis by:
In a simulation case, HRCT was chosen at management by:
13% of PCPsn=227
53% of Pulmonologistsn=40
Where we need to be:
Suspected IPF
Yes
No
UIPNot UIP
Not IPFIPF/Not IPF
MDD
Surgical lung biopsy
IPF
Possible UIP; inconsistent with UIP
HRCT
Identifiable causes for ILD?
A Current Practice Snapshot of IPF Diagnosis and Management: Where We Are and Where We Need to Be • globalacademycme.com/pulmonology 11
Please scan the QR code on the left or go to: http://bit.ly/IPF_info for an in-depth, interactive version of this infographic.Reference: 1. Travis WD, Costabel U, Hansell DM, et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188:733-748.
Where we are: Where we need to be:
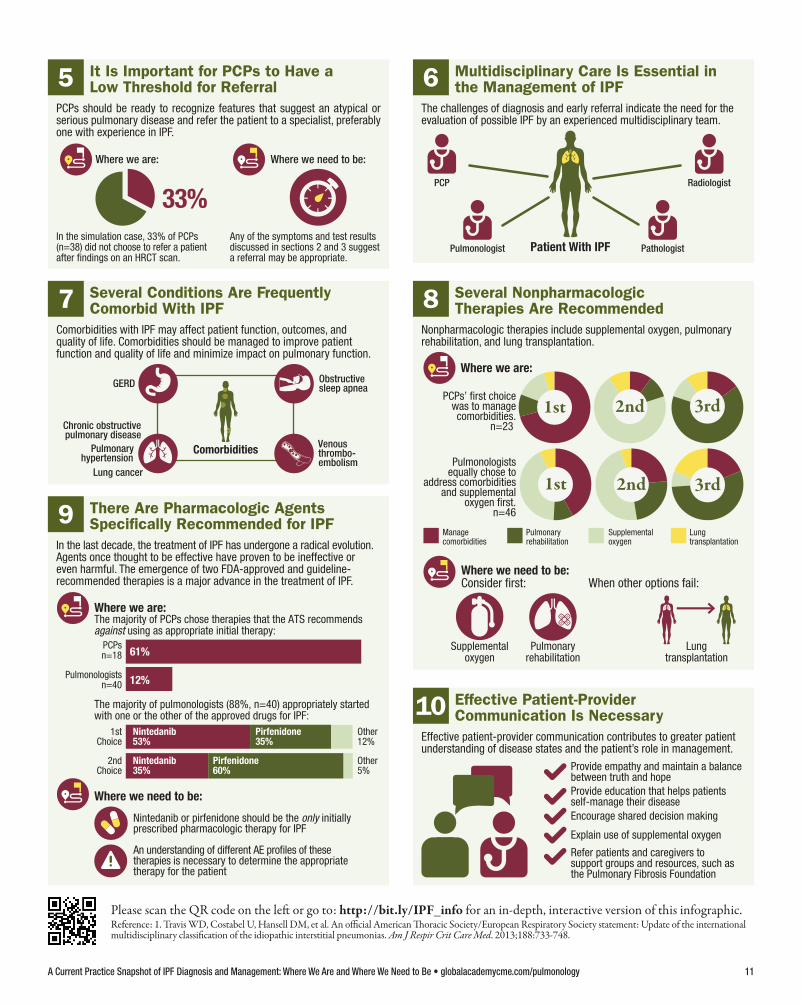
33%
5 It Is Important for PCPs to Have aLow Threshold for Referral
PCPs should be ready to recognize features that suggest an atypical or serious pulmonary disease and refer the patient to a specialist, preferably one with experience in IPF.
PCP Radiologist
PathologistPatient With IPFPulmonologist
6 Multidisciplinary Care Is Essential inthe Management of IPF
The challenges of diagnosis and early referral indicate the need for theevaluation of possible IPF by an experienced multidisciplinary team.
Comorbidities
GERD
Chronic obstructivepulmonary disease
Obstructive sleep apnea
Lung cancer
Venous thrombo-embolism
Pulmonaryhypertension
7 Several Conditions Are FrequentlyComorbid With IPF
Comorbidities with IPF may affect patient function, outcomes, andquality of life. Comorbidities should be managed to improve patientfunction and quality of life and minimize impact on pulmonary function.
Where we are:
Where we need to be:
Lung transplantation
Managecomorbidities
Pulmonaryrehabilitation
Supplementaloxygen
When other options fail:
Pulmonaryrehabilitation
Lungtransplantation
Supplementaloxygen
PCPs’ first choicewas to managecomorbidities.
n=23
Pulmonologistsequally chose to
address comorbiditiesand supplemental
oxygen first.n=46
1st
1st 2nd
2nd 3rd
3rd
Consider first:
8 Several NonpharmacologicTherapies Are Recommended
Nonpharmacologic therapies include supplemental oxygen, pulmonaryrehabilitation, and lung transplantation.
Where we are:
PCPsn=18
Pulmonologistsn=40
The majority of PCPs chose therapies that the ATS recommendsagainst using as appropriate initial therapy:
Where we need to be:
61%
12%
The majority of pulmonologists (88%, n=40) appropriately startedwith one or the other of the approved drugs for IPF:
1stChoice
2ndChoice
An understanding of different AE profiles of these therapies is necessary to determine the appropriate therapy for the patient
Nintedanib or pirfenidone should be the only initiallyprescribed pharmacologic therapy for IPF
Nintedanib 53%
Other5%
Other12%
Nintedanib35%
Pirfenidone35%
Pirfenidone60%
9 There Are Pharmacologic AgentsSpecifically Recommended for IPF
In the last decade, the treatment of IPF has undergone a radical evolution. Agents once thought to be effective have proven to be ineffective or even harmful. The emergence of two FDA-approved and guideline- recommended therapies is a major advance in the treatment of IPF.
Provide empathy and maintain a balancebetween truth and hope Provide education that helps patientsself-manage their disease
Refer patients and caregivers to support groups and resources, such as the Pulmonary Fibrosis Foundation
Explain use of supplemental oxygen
Encourage shared decision making
10 Effective Patient-ProviderCommunication Is Necessary
Effective patient-provider communication contributes to greater patientunderstanding of disease states and the patient’s role in management.
In the simulation case, 33% of PCPs (n=38) did not choose to refer a patient after findings on an HRCT scan.
Any of the symptoms and test resultsdiscussed in sections 2 and 3 suggest a referral may be appropriate.

12 globalacademycme.com/pulmonology • A Current Practice Snapshot of IPF Diagnosis and Management: Where We Are and Where We Need to Be
T he challenges described above indi-cate the need for the evaluation of possible ILD by an experienced
multidisciplinary team.35 The importance of multidisciplinary care is supported by guidelines and illustrated by studies of diag-nostic accuracy. For example, a study from Europe found high accuracy for the diag-nosis of IPF at expert centers (approaching 90%), but the level of agreement within the expert teams was only fair to moderate.36 ATS guidelines specifically recommend collaborative evaluation and management by a multidisciplinary team, including a pulmonologist, a radiologist, and, possibly, a pathologist.7
Ideally, the care team should have expe-rience with diagnosing and managing ILD. For example, radiologists without experience with ILD may not recognize the features of a UIP pattern on HRCT. Indeed, the distinctions between honey-combing (a hallmark feature of the UIP pattern) and traction bronchiectasis or concurrent emphysema and fibrosis can be challenging , even for experienced observers.37 When primary care clinicians recognize features that suggest a possible ILD, they should consider referring the patient to an ILD center, even before an HRCT is performed.
Common ComorbiditiesSeveral conditions are frequently comorbid with IPF and may affect patient function, outcomes, and quality of life. A recent retro-spective study of 272 patients with IPF found that 12% had no comorbidities, 58% had one to three comorbidities, and 30% had four to seven comorbidities.38 The most common comorbidities were cardiovascular, pulmo-nary, and oncologic. Predictably, median survival differed significantly by the number of comorbidities, ranging from 66 months among patients with no comorbidities to 48 months among those with one to three comorbidities and 35 months among those with four to seven comorbidities (P=0.004).
Common comorbidities include GERD, COPD, obstructive sleep apnea, and pulmo-nary hypertension (Figure 1). GERD is highly prevalent in IPF, occurring in as many as 90% of patients, and may be associated
with worsening or exacerbation of IPF.39 In more than 50% of patients with IPF, GERD may be clinically silent.39 Obstructive sleep apnea is similarly prevalent, occurring in up to 88% of cases.40 Pulmonary hyperten-sion is also common, with estimates ranging from one-third to more than three-quarters of patients with IPF, depending on the study and setting.41
All comorbidities should be managed to improve patient function and quality of life and minimize impact on pulmo-nary function. For example, some studies suggest that the treatment of GERD (even in asymptomatic patients) may improve outcomes in some patients with IPF.42 However, other studies have not supported this association.38 Current ATS guidelines recommend to consider treating GERD in most patients, even when reflux is asymp-tomatic.7,11 Some common comorbidities, such as COPD, may complicate the diag-nosis and treatment of IPF; these patients should be managed by a specialist with experience in IPF.
Because IPF is a progressive and fatal disease, some clinicians and patients may choose to ignore comorbid condi-tions. This choice may reflect a sense of fatalism about IPF or a well-meaning urge to minimize medical interventions. But such therapeutic nihilism sends the wrong signal to patients, suggesting that there is nothing medicine can offer them. It also ignores the potential to improve patient function and quality of life and could contribute to worsening of pulmonary function and progression of disease.
PATIENT EXPERIENCES Diagnostic Delays and the Need for a Multidisciplinary Team A 78-year-old man:“I had a CT scan [in] 2011, but I felt I had the beginnings of IPF 4 or 5 years prior when I started running and was suddenly wheezing. My primary care doctor of 30 years told me I had bronchitis/athletic asthma, and so I started taking inhalers. I went through a series of misdiagnoses for five years. I was a competitive swimmer doing 10Ks and martial arts, and I finally went in to a pulmonologist because my times for swimming were dropping. I had shortness of breath and weight loss—I dropped 50 pounds the year preceding my diagnosis—but the shortness of breath only bothered me because of my swimming times decreasing. I saw four different pulmonologists in four years trying to confirm my IPF diagnosis.”
Table. Features Associated With Increased Risk of Mortality in IPF7
Baseline Factors Longitudinal Factors
• Level of dyspnea
• DLCO <40% predicted
• Desaturation ≤88% during 6MWT
• Extent of honeycombing on HRCT
• Pulmonary hypertension
• Increase in level of dyspnea
• Decrease in FVC by ≥10% over time
• Decrease in DLCO ≥15% absolute value
• Worsening of fibrosis on HRCT
6MWT=6-minute walk test; DLCO=diffusing capacity for carbon monoxide; FVC=forced vital capacity; HRCT=high-resolution computed tomography; IPF=idiopathic pulmonary fibrosis.
Multidisciplinary Care for IPF

Prognostic Factors Although IPF is known to be a fatal disease, determining the prognosis for an indi-vidual patient remains challenging. Several possible natural histories of IPF have been described, ranging from relatively stable disease to rapid progression and death. Most patients experience a gradual wors-ening of lung function over the course of years. Some patients remain stable for long periods of time, while others experience a rapid progression of disease. A minority of patients (~5% to 10%) experience an unpre-dictable decline with increased shortness of breath and hypoxia with new infiltrates and opacities on imaging occurring over a period of 2 to 3 weeks, termed acute exacerbations, with significant morbidity and mortality.7
Validated biomarkers have not been established, and current prognostic models do not reliably predict disease course. Factors associated with increased risk of mortality in clinical studies are listed in the Table.7 An interesting recent study from Japan found that history of smoking had a paradoxical influence on survival.43 In this retrospective study, 32 patients with IPF who had never smoked had significantly shorter median survival compared to 66 patients with IPF who had smoked (18.5 vs 26.3 months; P<0.0001). Never-smokers
also had significantly more acute exacer-bations (50% vs 18.2%; P<0.0001). These findings suggest that different pathogenetic mechanisms may play a role in patients with and without a history of smoking, and that IPF in never-smokers may be a more aggres-sive form of disease. However, prospective studies are required to confirm these preliminary findings.
Telling Patients They Have IPFOnce a diagnosis of IPF is confirmed, it is time for the conversation that many clini-cians dread: telling the patient that he or she has a progressive and fatal disease. This discussion must be conducted with empathy and should maintain a delicate balance between truth and hope. Current evidence suggests that people diagnosed with IPF will live 2 to 5 years from the time of diagnosis.7 Some of the variation in this range may relate to the fact that a delayed diagnosis is common in IPF; as noted earlier, the average time from onset of symptoms to referral for specialty care is about 2 years.9
It is important to bear in mind that each patient’s experience of IPF may differ from that of others. For example, mortality statistics are based on broad populations of patients, which may combine older and younger patients, or patients who are untreated and those who have been treated aggressively. It is worth the effort to explain this to patients at the level of their understanding.
Furthermore, new therapies continue to be developed that can affect the course of the disease. The first therapies indicated for use in IPF were approved by the FDA as recently as 2014.
Nonpharmacologic Therapies Several nonpharmacologic therapies are recommended for many patients with IPF. These modalities include supplemental oxygen, pulmonary rehabilitation, and lung transplantation (see Figure 2).
Oxygen Therapy Oxygen therapy can improve the ability of patients with IPF to function. It is not clear whether supplemental oxygen affects survival in IPF, but it can improve patient function, sense of well-being, and quality of life. The ATS guidelines recommend long-term oxygen therapy for patients with IPF who demonstrate clinically significant hypoxemia, which they define as an oxygen saturation <88%. Supplemental oxygen may also help patients who have hypox-emia with activity to become more active. This is an important consideration, because patients with IPF are likely to decondition as their disease progresses.
A Current Practice Snapshot of IPF Diagnosis and Management: Where We Are and Where We Need to Be • globalacademycme.com/pulmonology 13
PATIENT EXPERIENCES Oxygen Therapy A 71-year-old woman:“The big thing is oxygen. That has become the biggest factor in having this illness. I’m 24/7 on oxygen, probably within six months of it having been prescribed.... I got a small carry-around at first, but [the supplier’s] idea of portable is not at all what I needed portable to be. Thank God I had some money so I could purchase things on my own, so I could have some sort of life. That has been trial and error and a frustration, because [my doctor] knows about using oxygen but she doesn’t understand the day to day and how it changes one’s life.”
PATIENT EXPERIENCES The Importance of FitnessA 63-year-old woman:“I would advise others to stay as healthy as possible. Get into a regimen of working out with oxygen, supervised if you can, to try to ward off advancement. It’s so individualized, you never know— it may never progress, or it may progress fast. Stay positive. A cure is coming. Stay healthy enough so that, when a cure comes, you can get it.”
Figure 1. Comorbidities Frequently Associated With IPF
Lung cancerComorbidities
GERD
COPD
Obstructive sleep apnea
Venous thromboembolismPulmonary hypertension
CLINICAL PEARLS Nonpharmacologic
Interventions for IPF7
• Oxygen therapy: Improves function; use especially when O2Sat is <88%
• Pulmonary rehabilitation: Improves patient function and quality of life
• Lung transplantation: Can improve survival in appropriate patients
IPF=idiopathic pulmonary fibrosis.
COPD=chronic obstructive pulmonary disease; GERD=gastroesophageal reflux disease; IPF=idiopathic pulmonary fibrosis.

14 globalacademycme.com/pulmonology • A Current Practice Snapshot of IPF Diagnosis and Management: Where We Are and Where We Need to Be
Despite the significant benefits of oxygen, patients may resist its use. Patients may cite the inconvenience of the required devices and the stigma of using oxygen in public. Cost may also be an issue for some patients. Finally, newly diagnosed patients may benefit from guidance on how to work with a supplier to obtain devices appro-priate for their lifestyle and clinical needs. When prescribing oxygen therapy, clini-cians should explain to patients what it is, why it will help them, and how to select the right oxygen system. By addressing issues such as convenience, stigma, and cost, clini-cians can help patients overcome barriers to the appropriate use of oxygen.
Pulmonary Rehabilitation Pulmonary rehabilitation programs for IPF may include aerobic conditioning, strength and flexibility training, nutritional guidance, education, and psychosocial support. In controlled trials, pulmonary
rehabilitation improved exercise capacity, lung function, symptoms, and quality of life in patients with IPF. These improvements may be greatest in patients with the lowest levels of baseline function. Accordingly, the 2011 ATS guidelines recommend pulmo-nary rehabilitation for most patients with IPF. These programs may last several weeks or a few months and can be repeated peri-odically—for example, yearly—to help patients maintain function.
Lung Transplantation Many patients with IPF will not be candi-dates for lung transplantation. However, appropriate patients may have reduced risk for mortality following transplantation. In clinical studies, lung transplantation is asso-ciated with 5-year survival rates of about 50%.44 Outcomes such as these suggest a tradeoff between risk for death due to IPF and the multiple risks of lung transplanta-tion, which include acute rejection, infection,
and cancer, among others. Nevertheless, for some patients with IPF, lung transplantation may increase both the quantity and quality of life.
The ATS guidelines encourage clini-cians to discuss lung transplantation soon after the diagnosis of IPF is confirmed. In general, younger, healthier patients may be considered for the procedure. Patients found to be potential candidates should be referred as soon as possible for evaluation.
Pharmacologic TreatmentIn the last decade, the treatment of IPF has undergone a radical evolution. Agents once thought to be effective in IPF have proven to be ineffective or even harmful. Conversely, the first two agents definitively shown to slow the progression of IPF were recently approved by the FDA.
Several agents once thought to be effec-tive should not be used in IPF. For example, the use of triple therapy—a combina-tion of prednisone, azathioprine, and N-acetylcysteine—was based on the hypoth-esis that IPF was primarily an inflammatory disease. In recent years, however, this triple therapy was proved to be not just ineffec-tive but potentially harmful.44 The use of N-acetylcysteine alone, another widely used therapeutic approach in IPF, was evaluated in an extension of the key clinical trial. This extension study also called into question the utility of N-acetylcysteine monotherapy for IPF.45 Neither of these approaches is currently recommended.7,11
Similarly, studies in animal models suggested potential efficacy for antico-agulants in IPF. However, a randomized, placebo-controlled trial in patients with IPF was terminated early due to a low probability of benefit and an increase in mortality rate in patients treated with warfarin.46 ATS guidelines currently recom-mend against the use of warfarin in patients with IPF.7,11
Finally, ambrisentan is a selective endo-thelin receptor antagonist that was evaluated for use in IPF. However, the major random-ized trial comparing this agent to placebo was terminated early due to lack of effi-cacy. In fact, some findings suggested that ambrisentan may have increased risk for disease progression and respiratory hospi-talizations.47 ATS guidelines currently recommend against the use of ambrisentan in IPF.7,11
Other agents specifically not recom- mended for the treatment of IPF by current ATS guidelines are imatinib (selective tyro-sine kinase inhibitor against platelet-derived
Figure 3. Medical Therapy for IPF
An understanding of the different adverse event profiles of these therapies is necessary to
determine the appropriate therapy for the patient
Nintedanib or pirfenidone should be the only initially prescribed pharmacologic therapy for IPF
Figure 2. Nonpharmacologic Therapies for IPF
Consider first: When other options fail:
Pulmonaryrehabilitation
Supplementaloxygen
Lungtransplantation
Nonpharmacologic therapies include supplemental oxygen, pulmonary rehabilitation, and lung transplantation.
In the last decade, the treatment of IPF has undergone a radical evolution. Agents once thought to be effective have proven to be ineffective or even harmful. The emergence of two FDA-approved and guideline-recommended therapies is a major advance in the treatment of IPF.
IPF=idiopathic pulmonary fibrosis.
FDA=US Food and Drug Administration; IPF=idiopathic pulmonary fibrosis.

growth factor [PDGF] receptors), silde-nafil (phosphodiesterase type 5 [PDE-5] inhibitor), and macitentan and bosentan (endothelin receptor antagonists).11
Conversely, two new agents—pirfenidone and nintedanib—received FDA approval for the treatment of IPF in late 2014 (Figure 3). In separate phase 3 trials, both agents reduced the decline in lung func-tion that signals progression of disease.5,6 Clinicians who manage the care of patients with IPF need to be familiar with the use of these agents, including their side effect profiles and expectations regarding the benefits of treatment.
NintedanibNintedanib is an intracellular inhibitor of multiple tyrosine kinases. After a phase 2 trial suggested efficacy in IPF, a pair of randomized, placebo-controlled phase 3 trials were conducted.6 These studies (INPULSIS-1 and INPULSIS-2) were identical, 52-week studies that enrolled a total of 1,066 subjects with a diagnosis of IPF. To be eligible, subjects were required to have FVC ≥50% predicted, DLCO 30% to 79% predicted, and chest HRCT performed within the last 12 months. Subjects were randomized to 150 mg nin- tedanib twice daily or placebo. After 52 weeks, the primary endpoint (annual rate of decline in FVC) in INPULSIS-1 was –114.7 mL with nintedanib vs –239.9 mL with placebo (95% CI, 77.7 to 172.8; P<0.001); and in INPULSIS-2, –113.6 mL vs –207.3 mL (95% CI, 44.8 to 142.7; P<0.001). In INPULSIS-1, there was no significant difference between groups in the
time to first acute exacerbation (P=0.67), but in INPULSIS-2, there was a signifi-cant benefit with nintedanib compared to placebo (hazard ratio, 0.38; 95% CI, 0.19 to 0.77; P=0.005).
On the strength of these clinical trials, nintedanib was approved by the FDA for the treatment of IPF and is conditionally recommended by the updated 2015 ATS treatment guidelines.11
Pirfenidone Pirfenidone is an oral anti-fibrotic agent that has demonstrated efficacy for IPF in clinical trials. In an international, 52-week clinical trial (ASCEND), 555 subjects with IPF were randomized to pirfeni-done (2,403 mg per day) or placebo.5 Enrolled subjects had FVC 50% to 90% predicted, DLCO 30% to 90%, forced expiratory volume in the first second/FVC ≥0.80, and HRCT- and/or biopsy-confirmed diagnosis of IPF. At study end, the pirfenidone group had a relative reduc-tion of 47.9% in the proportion of subjects who had an absolute decline in FVC of 10% or more (or who died), compared to the placebo group. There was also a 132.5% relative increase in the propor-tion of patients with no decline in FVC (P<0.001) with pirfenidone vs placebo. Subjects in the pirfenidone group also had reduced decline in 6MWT distance (P=0.04), suggesting greater preservation of functional capacity. Finally, pirfeni-done improved progression-free survival compared to placebo (P<0.001), based on a prespecified analysis of pooled data from the ASCEND and CAPACITY trials.
On the strength of these clinical trials, pirfenidone was approved by the FDA for the treatment of IPF and is conditionally recommended by the updated 2015 ATS treatment guidelines.11
Selecting and Managing Pharmacologic TherapyThe emergence of two FDA-approved and guideline-recommended therapies is a major advance in the treatment of IPF. These agents provide options for patients today and hope for future advances in therapy. Ongoing studies may determine if these medications can also improve survival in this lethal disease. But patients must be treated if they are to receive these benefits.
Unfortunately, evidence suggests that a substantial proportion of patients with IPF receive no pharmacologic therapy at all.48,49
A Current Practice Snapshot of IPF Diagnosis and Management: Where We Are and Where We Need to Be • globalacademycme.com/pulmonology 15
PATIENT EXPERIENCES The Need for Patient-Level EducationA 74-year-old man:“When they told me I had IPF, it went 30,000 feet over my head. I didn’t have a clue what they were talking about. I saw three physicians that did not really explain what this was about. Or, they did say the usual: ‘Scarring of the lung, only remedy is lung transplant.’ So what? I was in LaLa land! They really didn’t explain in a way that connected with the patient. I found out from the Internet that it is a terminal disease. Instead of showing me pictures of lung terminology—tell me, what does it mean?
“I expected, I guess, my pulmo-nologist to sit me down and explain things....
“What I would expect to hear, with 20/20 hindsight: ‘You have IPF. It’s a scarring of the tissue, it cannot be corrected, it deteriorates, it’s terminal. This is what we can do: we can help you with this and that.’ So, educate me.”
PATIENT EXPERIENCES Communication Gaps A 71-year-old woman:“I could not walk very far and had shortness of breath. Shortly after, I was diagnosed with pneumonia. The doctor had me do a couple of x-rays.... All they said to me was, ‘It isn’t a tumor, but your lungs show signs of cracked glass.’ End of explanation.
“Based on what I know now, there were a number of tests they should’ve given me.... I didn’t have a pulmonary lung function test. No discussion of any treatment at all. No medication. No diagnosis. Nothing. No one thought it was important, so I didn’t really think about it. I went back to my doctor, and she didn’t say anything about what I should do.”
CLINICAL PEARLS Pharmacologic Interventions
for IPF7
FDA-approved and recommended: • Nintedanib• Pirfenidone NOT effective and NOT recommended: • Ambrisentan, macitentan, bosentan • Imatinib • N-acetylcysteine monotherapy • Prednisone monotherapy • Triple therapy (prednisone,
azathioprine, N-acetylcysteine)• Warfarin
FDA=US Food and Drug Administration; IPF=idiopathic pulmonary fibrosis.

For example, studies have found that many patients with IPF are managed with cortico-steroids, despite a strong recommendation against corticosteroid monotherapy in the ATS guidelines; in one study, for example, 49% of patients were treated with oral corticosteroids.49 A prospective study from Germany found that one-third of subjects with IPF were treated with pirfenidone, reflecting its recent approval in Europe prior to the study.48 However, none of the subjects were treated with nintedanib (not approved in Europe at the time), nearly a quarter received corticosteroids, one-third received acetylcysteine, and 17.9% received no treat-ment at all.48 Other surveys have reported similar wide variations in the treatment of patients with IPF, including inconsistent use of pirfenidone as monotherapy and in combination with other agents.49
The variation in treatment patterns described by these studies is disheartening. The data reveal frequent use of inappro-priate therapies and uncertainties regarding the use of approved therapies. An impor-tant response to these data is to ensure that appropriate therapies are discussed with the appropriate patients. Current data indi-cate that many patients in clinical trials experienced reduced progression of disease when treated with one of the approved therapies, and this hope should be offered to patients in clinical practice.
At this time, we have little evidence to guide selection between the two approved therapies for individual patients. No head-to-head studies have been performed comparing these two agents, and current evidence does not suggest significant differ-ences between the agents with regard to efficacy.50,51 Therefore, the key issues to consider when selecting between these agents include side effect profiles, risk for drug-drug interactions, and frequency of administration.
Side Effects and Interactions Both drugs were fairly well tolerated in clinical trials. However, some patients may experience side effects that they cannot tolerate. For example, pirfenidone was associated with risk for skin rash and gastro-intestinal events, such as nausea and reflux. Therefore, the agent should be taken with food, and patients should be counseled to avoid exposure to sunlight and sunlamps.52
In clinical studies, dose adjustments were also shown to improve treatment continua-tion in patients experiencing adverse events.52 For patients with GERD, pirfenidone may increase risk for reflux, and the presence of symptomatic GERD might be a reason to consider the alternative agent.
Pirfenidone is metabolized primarily by the CYP1A2 enzyme, and administra-tion of CYP1A2 inhibitors or inducers may affect pirfenidone serum drug levels. Strong inhibitors of CYP1A2, such as fluvoxamine, should be discontinued before initiating pirfenidone. Dose adjust-ments may be required when pirfenidone is coadministered with less-potent inhibi-tors of this enzyme.53
The most common side effects of nintedanib in trials were gastrointestinal, particularly diarrhea, which was reported by 62% of patients in studies.6,54 In most cases, the diarrhea was mild and did not require dose adjustment or discontinuation. However, nintedanib might not be the best option for patients with diarrhea at base-line. Because nintedanib is predominantly metabolized by the P-gp enzyme, coadmin-istration of P-gp inhibitors or inducers may alter serum levels of the drug.55
Finally, the agents differ with regard to administration. Pirfenidone is administered as three capsules three times per day. Nintedanib is administered as one capsule every 12 hours. Both agents should be taken with food.
The selection of specific therapy for IPF should be made using a shared decision making (SDM) model. In SDM, clinicians
16 globalacademycme.com/pulmonology • A Current Practice Snapshot of IPF Diagnosis and Management: Where We Are and Where We Need to Be
PATIENT EXPERIENCES Seeking Information and UnderstandingA 70-year-old man:“First thing we did, both of us, was get on the computer and read every-thing we could find about IPF. It’s not the best thing to do, to be honest— not without fully understanding the disease and treatments. If I focus on the right things, I’m fine. I made a mistake on focusing on the negative too soon before I really understood.”
A 67-year-old woman:“I read everything there was to read about IPF the minute I found out what I had. You couldn’t get me off the computer. But you find out that a lot of that stuff is not real.”
A 67-year-old man:“To someone newly diagnosed with IPF, I’d tell ‘em, Google it, but for God’s sake don’t get scared to death from the statistics. Those are old and not up to date. Unless you have an ‘expire by’ stamped on you some-where, grab today and don’t worry about tomorrow.”
PATIENT EXPERIENCES Support GroupsA 67-year-old man:“If [doctors are] handing out a diagnosis of a terminal disease, they should have a list of buddies: Here’s Joe Blow who has IPF, too; he said it’s okay to call. We can do things for each other that docs can’t do, and support each other, day and night, instead of just once a month.”
A 74-year-old man:“You have to realize—talking to a doctor, it’s not easy. You have to make appointments to see him, etc. So probably the smartest thing I did was to join a support group, because that’s where I learned much more than I learned from the physicians. I found out that there was no treatment except for two medications, but the best they can do is slow down the rate of damage.”
A 67-year-old woman:“The diagnosis was very traumatic. I did a lot of crying. I joined [a support group] and that’s helped a whole lot. I met some people locally, and we talk on the phone. I think it helps when you share.”
For further information on how to start a new PF support group or to get involved with a support group near you, contact the Pulmonary Fibrosis Foundation’s Patient Communication Center at 844.TalkPFF (844.825.5733) or [email protected]. Additional information is available at pulmonaryfibrosis.org.

A Current Practice Snapshot of IPF Diagnosis and Management: Where We Are and Where We Need to Be • globalacademycme.com/pulmonology 17
I n summary, IPF is a rare but fatal disease. Challenges in managing IPF include making a timely diagnosis, early referral to specialty care, discussing the disease with patients, and choosing appropriate treatment. The key take-home lessons for primary care prac-
titioners are to recognize features of the disease, perform a thorough evaluation (including PFTs, oximetry, and imaging), and refer all patients with features that suggest an atypical lung disease to a specialist, preferably one with experience in ILD.
In the specialty setting, the crucial test is HRCT; a typical UIP pattern on HRCT has 90% to 100% positive-predictive value for a UIP pattern and obviates the need for biopsy in most cases. Following the identification of a UIP pattern, a diagnosis of IPF must be confirmed by differentiating from other ILDs, as the treatment of IPF differs substantially from the management of other ILDs. Both nonpharmacologic (oxygen, rehabilitation, lung transplantation) and approved pharmacologic therapies (nintedanib, pirfenidone) should be considered and tailored to individual patients’ needs.
PATIENT EXPERIENCES Hearing the DiagnosisA 70-year-old man:“I went to my own doctor who did a chest x-ray and saw some funky stuff, so I went back to the pulmonologist, who suggested a CT scan and recommended that I see a different pulmonologist...who specializes in lung disease. I did a breathing test with the new pulmonologist. After that, he said, ‘You have idiopathic pulmonary fibrosis,’ and went on to describe it. All I really heard was, ‘blah blah blah, three years or less, blah blah blah.’”
Wife of patient with IPF:“When we went in for the results of the CT scan, we didn’t really know how serious of a nature we might be there to find out. A resident told us, ‘You are here today because you may have IPF.’ I grabbed my phone and said, ‘Can you spell that for me?’ and I was Googling it while he was saying it. The doctor walked in with a booklet, handed it to [my husband], and told him he had IPF. Everything he said after that was ‘Waa waaa waaaa waaaa…three years.’”
PATIENT EXPERIENCES Being Honest With the PatientA 70-year-old man:“Ten days [after a biopsy], I had the results: IPF. [The pulmonologist] didn’t try to sugarcoat it. He was not overly optimistic, but he was never negative, either—just realistic. His approach was, ‘This is it, ok? This is how we’re going to deal with it.’ He also said, ‘I don’t give timelines with life expectancy, especially where you’re at.’ He said, ‘A lot of those stats are based on old data. No one really knows how long you’re going to live, and I think when someone tells you how long, they are doing you a disservice.’”
Summary
share information with patients and then find agreement on goals and treatment approach. Patients’ preferences and beliefs regarding the disease and its treatment should be elicited using open-ended ques-tions. The final choice of treatment should be left to the well-informed patient.
Communicating With the PatientEffective patient-provider communication contributes to greater patient under-standing of disease states and the patient’s role in management. However, surveys suggest that patients would prefer improved communication with their clinicians.56
Patients with IPF have reported a lack of support as they attempt to understand the diagnosis and disease process.57 Patients have also cited a lack of information and educational resources regarding IPF and its management at the time of diagnosis.58,59
Effective education of patients diagnosed with IPF is essential to promote their ability to engage in self-management. But often patients are left with insufficient informa-tion, and many turn to the Internet for more. As clinicians know, this can be prob-lematic for everyone involved.
The impact of this lack of communication may be exacerbated by the fact that most patients with IPF see multiple clinicians before being diagnosed; 40% of patients
have consulted three or more physicians, and may have received conflicting informa-tion along the way.56
Methods for the improvement of patient-provider communication have been proposed and explored in the litera-ture.56 Basic principles include fostering mutual respect, harmonizing clinical goals, providing a supportive environment, and providing the right information with trans-parency and full disclosure. Additional approaches include patient-centered tech-niques such as maintaining eye contact, avoiding interruptions, encouraging patient participation, and soliciting the patient’s beliefs, values, and preferences. Providing information at a level the patient can under-stand is critical. Avoiding jargon can help patients better understand the medical aspects of the disease. Many patients may seek information from other sources, espe-cially the Internet. Information-seeking should not be discouraged, but many sources may leave patients with more ques-tions than answers.
Finally, clinicians should counsel all their patients with IPF about the valuable support groups that are available. These organizations, such as the Pulmonary Fibrosis Foundation, provide an under-standing community in which patients and clinicians can share their stories and learn about others’ experiences.

1. Nalysnyk L, Cid-Ruzafa J, Rotella P, Esser D. Incidence and prevalence of idiopathic pulmo-nary fibrosis: Review of the literature. Eur Respir Rev. 2012;21:355-361.
2. Hutchinson JP, McKeever TM, Fogarty AW, Navaratnam V, Hubbard RB. Increasing global mortality from idiopathic pulmonary fibrosis in the twenty-first century. Ann Am Thorac Soc. 2014;11:1176-1185.
3. Navaratnam V, Fogarty AW, Glendening R, McKeever T, Hubbard RB. The increasing secondary care burden of idiopathic pulmonary fibrosis: Hospital admission trends in England from 1998 to 2010. Chest. 2013;143:1078-1084.
4. Raghu G, Chen SY, Yeh WS, et al. Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: Incidence, prevalence, and survival, 2001-11. Lancet Respir Med. 2014;2:566-572.
5. King TE Jr, Bradford WZ, Castro-Bernardini S, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2083-2092.
6. Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmo-nary fibrosis. N Engl J Med. 2014;370:2071-2082.
7. Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183:788-824.
8. Ley B, Collard HR, King TE Jr. Clinical course and prediction of survival in idiopathic pulmo-nary fibrosis. Am J Respir Crit Care Med. 2011;183:431-440.
9. Lamas DJ, Kawut SM, Bagiella E, Philip N, Arcasoy SM, Lederer DJ. Delayed access and survival in idiopathic pulmonary fibrosis: A cohort study. Am J Respir Crit Care Med. 2011;184:842-847.
10. Cottin V, Richeldi L. Neglected evidence in idiopathic pulmonary fibrosis and the importance of early diagnosis and treatment. Eur Respir Rev. 2014;23:106-110.
11. Raghu G, Rochwerg B, Zhang Y, et al. An official ATS/ERS/JRS/ALAT clinical practice guideline: Treatment of idiopathic pulmonary fibrosis. An update of the 2011 clinical prac-tice guideline. Am J Respir Crit Care Med. 2015;192:e3-e19.
12. Travis WD, Costabel U, Hansell DM, et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188:733-748.
13. Loomis-King H, Flaherty KR, Moore BB. Pathogenesis, current treatments and future direc-tions for idiopathic pulmonary fibrosis. Curr Opin Pharmacol. 2013;13:377-385.
14. Richeldi L. Idiopathic pulmonary fibrosis: Current challenges and future perspectives. Eur Respir Rev. 2013;22:103-105.
15. Daccord C, Maher TM. Recent advances in understanding idiopathic pulmonary fibrosis. F1000Res. 2016;5:1046.
16. Wuyts WA, Antoniou KM, Borensztajn K, et al. Combination therapy: The future of manage-ment for idiopathic pulmonary fibrosis? Lancet Respir Med. 2014;2:933-942.
17. King TE Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet. 2011;378:1949-1961.
18. Spagnolo P, du Bois RM, Cottin V. Rare lung disease and orphan drug development. Lancet Respir Med. 2013;1:479-487.
19. Morell F, Villar A, Montero MÁ, et al. Chronic hypersensitivity pneumonitis in patients diag-nosed with idiopathic pulmonary fibrosis: A prospective case-cohort study. Lancet Respir Med. 2013;1:685-694.
20. Idiopathic Pulmonary Fibrosis Clinical Research Network, Raghu G, Anstrom KJ, King TE Jr, Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med. 2012;366:1968-1977.
21. Fell CD, Martinez FJ, Liu LX, et al. Clinical predictors of a diagnosis of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2010;181:832-837.
22. Cottin V, Cordier JF. Velcro crackles: The key for early diagnosis of idiopathic pulmonary fibrosis? Eur Respir J. 2012;40:519-521.
23. Cordier JF, Cottin V. Neglected evidence in idiopathic pulmonary fibrosis: From history to earlier diagnosis. Eur Respir J. 2013;42:916-923.
24. Moyer VA; US Preventive Services Task Force. Screening for lung cancer: US Preventive Services Task Force recommendation statement. Ann Intern Med. 2014;160:330-338.
25. Wender R, Fontham ET, Barrera E Jr, et al. American Cancer Society lung cancer screening guidelines. CA Cancer J Clin. 2013;63:107-117.
26. Hunninghake GW, Zimmerman MB, Schwartz DA, et al. Utility of a lung biopsy for the diagnosis of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2001;164:193-196.
27. Ryerson CJ, Urbania TH, Richeldi L, et al. Prevalence and prognosis of unclassifiable intersti-tial lung disease. Eur Respir J. 2013;42:750-757.
28. Hutchinson JP, Fogarty AW, McKeever TM, Hubbard RB. In-hospital mortality after surgical lung biopsy for interstitial lung disease in the United States. 2000 to 2011. Am J Respir Crit Care Med. 2016;193:1161-1167.
29. Rotolo N, Imperatori A, Dominioni L, et al. Efficacy and safety of surgical lung biopsy for interstitial disease. Experience of 161 consecutive patients from a single institution in Italy. Sarcoidosis Vasc Diffuse Lung Dis. 2015;32:251-258.
30. Han Q, Luo Q, Xie JX, et al. Diagnostic yield and postoperative mortality associated with surgical lung biopsy for evaluation of interstitial lung diseases: A systematic review and meta-analysis. J Thorac Cardiovasc Surg. 2015;149:1394-1401, e1.
31. Yagihashi K, Huckleberry J, Colby TV, et al. Radiologic-pathologic discordance in biopsy-proven usual interstitial pneumonia. Eur Respir J. 2016;47:1189-1197.
32. Spagnolo P, Sverzellati N, Rossi G. Is the imaging inconsistent with usual interstitial pneu-monia? Think about idiopathic pulmonary fibrosis then! Respirology. 2016;21:782-784.
33. Meyer KC, Raghu G, Baughman RP, et al. An official American Thoracic Society clinical practice guideline: The clinical utility of bronchoalveolar lavage cellular analysis in interstitial lung disease. Am J Respir Crit Care Med. 2012;185:1004-1014.
34. Flaherty KR, Andrei AC, King TE Jr, et al. Idiopathic interstitial pneumonia: Do community and academic physicians agree on diagnosis? Am J Respir Crit Care Med. 2007;175:1054-1060.
35. Chung JH, Lynch DA. The value of a multidisciplinary approach to the diagnosis of usual interstitial pneumonitis and idiopathic pulmonary fibrosis: Radiology, pathology, and clinical correlation. AJR Am J Roentgenol. 2016;206:463-471.
36. Thomeer M, Demedts M, Behr J, et al. Multidisciplinary interobserver agreement in the diagnosis of idiopathic pulmonary fibrosis. Eur Respir J. 2008;31:585-591.
37. Wells AU. The revised ATS/ERS/JRS/ALAT diagnostic criteria for idiopathic pulmo-nary fibrosis (IPF)—Practical implications. Respir Res. 2013;14(suppl 1):S2.
38. Kreuter M, Ehlers-Tenenbaum S, Palmowski K, et al. Impact of comorbidities on mortality in patients with idiopathic pulmonary fibrosis. PLoS One. 2016;11:e0151425.
39. Raghu G, Freudenberger TD, Yang S, et al. High prevalence of abnormal acid gastro-esophageal reflux in idiopathic pulmonary fibrosis. Eur Respir J. 2006;27:136-142.
40. Lancaster LH, Mason WR, Parnell JA, et al. Obstructive sleep apnea is common in idio-pathic pulmonary fibrosis. Chest. 2009;136:772-778.
41. Nathan SD, Shlobin OA, Ahmad S, et al. Serial development of pulmonary hypertension in patients with idiopathic pulmonary fibrosis. Respiration. 2008;76:288-294.
42. Lee CM, Lee DH, Ahn BK, et al. Protective effect of proton pump inhibitor for survival in patients with gastroesophageal reflux disease and idiopathic pulmonary fibrosis. J Neurogastroenterol Motil. 2016;22:444-451.
43. Kishaba T, Nagano H, Nei Y, Yamashiro S. Clinical characteristics of idiopathic pulmo-nary fibrosis patients according to their smoking status. J Thorac Dis. 2016;8:1112-1120.
44. Kistler KD, Nalysnyk L, Rotella P, Esser D. Lung transplantation in idiopathic pulmo-nary fibrosis: A systematic review of the literature. BMC Pulm Med. 2014;14:139.
45. Idiopathic Pulmonary Fibrosis Clinical Research Network, Martinez FJ, de Andrade JA, Anstrom KJ, King TE Jr, Raghu G. Randomized trial of acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2093-2101.
46. Noth I, Anstrom KJ, Calvert SB, et al. A placebo-controlled randomized trial of warfarin in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2012;186:88-95.
47. Raghu G, Behr J, Brown KK, et al. Treatment of idiopathic pulmonary fibrosis with ambrisentan: A parallel, randomized trial. Ann Intern Med. 2013;158:641-649.
48. Behr J, Kreuter M, Hoeper MM, et al. Management of patients with idiopathic pulmonary fibrosis in clinical practice: The INSIGHTS-IPF registry. Eur Respir J. 2015;46:186-196.
49. Cottin V, Cadranel J, Crestani B, et al. Management of idiopathic pulmonary fibrosis in France: A survey of 1244 pulmonologists. Respir Med. 2014;108:195-202.
50. Rogliani P, Calzetta L, Cavalli F, Matera MG, Cazzola M. Pirfenidone, nintedanib and N-acetylcysteine for the treatment of idiopathic pulmonary fibrosis: A systematic review and meta-analysis. Pulm Pharmacol Ther. 2016;40:95-103.
51. Canestaro WJ, Forrester SH, Raghu G, Ho L, Devine BE. Drug treatment of idio-pathic pulmonary fibrosis: Systematic review and network meta-analysis. Chest. 2016; 149:756-766.
52. Cottin V, Maher T. Long-term clinical and real-world experience with pirfenidone in the treatment of idiopathic pulmonary fibrosis. Eur Respir Rev. 2015;24:58-64.
53. Esbriet [prescribing information]. South San Francisco, CA: Genentech, Inc.; 2016.
54. Richeldi L, Cottin V, du Bois RM, et al. Nintedanib in patients with idiopathic pulmo-nary fibrosis: Combined evidence from the TOMORROW and INPULSIS(®) trials. Respir Med. 2016;113:74-79.
55. Ofev [prescribing information]. Ridgefield, CT: Boehringer Ingelheim Pharmaceuticals, Inc.; 2016.
56. Wuyts WA, Peccatori FA, Russell AM. Patient-centred management in idiopathic pulmonary fibrosis: Similar themes in three communication models. Eur Respir Rev. 2014;23:231-238.
57. Russell AM, Vancheri C, Maronati M, Giot C. A qualitative European survey of patients' perceptions of current management of idiopathic pulmonary fibrosis. Thorax. 2013;68(suppl 3):A166-A167.
58. Collard HR, Tino G, Noble PW, et al. Patient experiences with pulmonary fibrosis. Respir Med. 2007;101:1350-1354.
59. Schoenheit G, Becattelli I, Cohen AH. Living with idiopathic pulmonary fibrosis: An in-depth qualitative survey of European patients. Chron Respir Dis. 2011;8:225-231.
References
18 globalacademycme.com/pulmonology • A Current Practice Snapshot of IPF Diagnosis and Management: Where We Are and Where We Need to Be

A Current Practice Snapshot of IPF Diagnosis and Management: Where We Are and Where We Need to Be • globalacademycme.com/pulmonology 19
1. Idiopathic pulmonary fibrosis (IPF) is associated with all of the following features EXCEPT:A. Older ageB. Female genderC. History of smokingD. Progressive dyspnea on exertion
2. All of the following findings on physical examination are consistent with idiopathic pulmonary fibrosis EXCEPT:A. Muscle wastingB. Finger clubbingC. Bibasilar crackles D. Normal joint examination
3. All of the following investigations should be considered for a patient presenting with features consistent with idiopathic pulmonary fibrosis EXCEPT:A. Low-dose computed tomography (CT)B. Cardiac workupC. 6-minute walk testD. Pulmonary function tests
4. In a patient with suspected idiopathic pulmonary fibrosis based on history, presentation, and clinical findings, which of the following tests would be the appropriate first step? A. Conventional CT B. High-resolution CTC. Surgical lung biopsyD. Plain chest radiography
5. All of the following conditions are commonly comorbid in patients with idiopathic pulmonary fibrosis EXCEPT:A. Asthma B. Obstructive sleep apnea C. Gastroesophageal reflux disease D. Chronic obstructive pulmonary disease
6. In the INPULSIS-1 and INPULSIS-2 studies, nintedanib was associated with which of the following outcomes compared to placebo?A. Significantly longer survival B. No significant differences in outcomes C. Significantly reduced rate of decline in forced vital
capacity (FVC)D. Significantly longer time to acute exacerbation
7. In a large-scale trial in patients with idiopathic pulmonary fibrosis, pirfenidone was associated with all of the following outcomes compared to placebo EXCEPT:A. Increased proportion of subjects with no decline in FVCB. Reduced proportion of subjects with a decline in FVC
of ≥10%C. Significantly reduced decline in 6-minute walk test
distance D. No significant differences between groups in
progression-free survival
8. In clinical trials, the common side effects of nintedanib included which of the following? A. Skin rashB. DiarrheaC. ConstipationD. Photosensitivity
9. When helping a patient with idiopathic pulmonary fibrosis select between the approved pharmacologic therapies, all of the following should be considered EXCEPT:A. Frequency of administrationB. Relative efficacy of each agentC. Potential drug-drug interactionsD. Side effect profiles of each agent
10. When counseling a patient with idiopathic pulmonary fibrosis, all of the following should be emphasized EXCEPT:A. Appropriate use of oxygen therapy
B. Evaluation for lung transplantation
C. Benefits of pulmonary rehabilitation
D. Estimates of life expectancy from clinical studies
A Current Practice Snapshot of IPF Diagnosis and Management: Where We Are and Where We Need to Be Post-TestOriginal Release Date: December 1, 2016 • Expiration Date: December 31, 2017 • Estimated Time to Complete Activity: 1.5 hours
To get instant CME/CE credits online, go to http://tinyurl.com/IPFSup16. Upon successful completion of the online test and evaluation form, you will be directed to a Web page that will allow you to receive your certificate of credit via e-mail.
If you have any questions or difficulties, please contact: Global Academy for Medical Education at [email protected] or (973) 290-8225.
FOR REVIEW PURPOSES ONLY. MUST BE COMPLETED ONLINE.
POST-TEST CME/CE QUESTIONS

Please indicate your profession/background:
MD/DO MSN/BSN/RN PA APN/NP PharmD/RPh Resident/Fellow Researcher Administrator StudentOther; specify ____________________________________________________
A Current Practice Snapshot of IPF Diagnosis and Management: Where We Are and Where We Need to Be Evaluation FormOriginal Release Date: December 1, 2016 • Expiration Date: December 31, 2017 • Estimated Time to Complete Activity: 1.5 hoursTo assist us in evaluating the effectiveness of this activity and to make recommendations for future educational offerings, please take a few moments to complete this evaluation form. Your response will help ensure that future programs are informative and meet the educational needs of all participants. CME/CE credit letters and long-term credit retention information will only be issued upon completion of the post-test and evaluation online at: http://tinyurl.com/IPFSup16.
If you do not feel confident that you can achieve the above objectives to some extent, please describe why not.______________________________________________________________
Based on the content of this activity, what will you do differently in the care of your patients/regarding your professional responsibilities? (check one)
Implement a change in my practice/workplace.
Seek additional information on this topic.
Do nothing differently. Current practice/job responsibilities reflect activity recommendations.
Do nothing differently as the content was not convincing.
Do nothing differently. System barriers prevent me from changing my practice/workplace.
If you anticipate changing one or more aspects of your practice/ professional responsibilities as a result of your participation in this activity, please briefly describe how you plan to do so.
______________________________________________________________
If you plan to change your practice/workplace, may we contact you in 2 months to see how you are progressing?
Yes. E-mail address: _____________________________________________No. I don’t plan to make a change.
If you are not able to effectively implement what you learned in this activity, please tell us what the system barriers are (eg, institutional systems, lack of resources, etc).
______________________________________________________________
LEARNING OBJECTIVES: Having completed this activity, you are better able to: Strongly Agree Agree Somewhat Agree Disagree Strongly Disagree
Utilize the signs, symptoms, and epidemiology of idiopathic pulmonary fibrosis (IPF) to better recognize patients in need of specialty care
5 4 3 2 1
Employ a multidisciplinary care team approach to IPF to enhance patient outcomes 5 4 3 2 1
Provide timely referral of patients with IPF to tertiary care to improve survival 5 4 3 2 1
Review clinical trial data supporting the efficacy and safety of pirfenidone and nintedanib 5 4 3 2 1
Select appropriate pharmacologic therapy for patients with IPF 5 4 3 2 1
Communicate with patients with IPF and provide effective disease-state education 5 4 3 2 1
What topics do you want to hear more about, and what issue(s) regarding your practice/professional responsibilities will they address?_____________________________________________________________
_____________________________________________________________
Please provide additional comments pertaining to this activity and any suggestions for improvement.__________________________________________________________________
__________________________________________________________________
OVERALL EVALUATION Strongly Agree Agree Somewhat Agree Disagree Strongly Disagree
The information presented increased my awareness/understanding of the subject. 5 4 3 2 1
The information presented will influence how I practice/do my job. 5 4 3 2 1
The information presented will help me improve patient care/my job performance. 5 4 3 2 1
The program was educationally sound and scientifically balanced. 5 4 3 2 1
Overall, the program met my expectations. 5 4 3 2 1
I would recommend this program to my colleagues. 5 4 3 2 1
Jonathan H. Chung, MDAuthor demonstrated current knowledge of the topic. 5 4 3 2 1
Author was organized in the written materials. 5 4 3 2 1
Gregory P. Cosgrove, MD, FCCPAuthor demonstrated current knowledge of the topic. 5 4 3 2 1
Author was organized in the written materials. 5 4 3 2 1
Kristin L. DeSimone, MDAuthor demonstrated current knowledge of the topic. 5 4 3 2 1
Author was organized in the written materials. 5 4 3 2 1
© 2016 Global Academy for Medical Education, LLC. All Rights Reserved.
FOR NOTES PURPOSES ONLY. MUST BE COMPLETED ONLINE.
The Postgraduate Institute for Medicine thanks you for your participation in this CME/CE activity. All information provided improves the scope and purpose of our programs and your patient care.





























