Embed Size (px)
Citation preview


II | Índice
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
I. HEMATOLOGÍA ..................................................................................................................... 1 A. CARACTERÍSTICAS GENERALES DE LA SANGRE ................................................................................... 2 B. PROPIEDADES FISICOQUÍMICAS .................................................................................................................... 3 C. COMPONENTES DE LA SANGRE ...................................................................................................................... 3
1. Plasma ............................................................................................................................................................. 3 2. Elementos formes ..................................................................................................................................... 4
D. FISIOLOGÍA GENERAL DE LA HEMOSTASIA ...........................................................................................26 1. Etapas de la hemostasia .......................................................................................................................29 2. Alteraciones de la coagulación .........................................................................................................33 3. Trombosis ...................................................................................................................................................34 4. Anticoagulantes ........................................................................................................................................35 5. Pruebas de coagulación ........................................................................................................................35
E. GRUPOS SANGUÍNEOS ........................................................................................................................................38 1. Antígenos y anticuerpos de la membrana del hematíe........................................................38 2. Herencia de los antígenos A y B .......................................................................................................39 3. Sistema RH ..................................................................................................................................................39 4. Transfusión sanguínea .........................................................................................................................41
II. PATOLOGÍA EN HEMATOLOGÍA .................................................................................... 52 A. TRASTORNOS HEMORRÁGICOS.....................................................................................................................52
1. Evaluación de la hemostasia y pruebas diagnósticas ...........................................................52 2. Alteraciones de las plaquetas: trombopenias y trombopatías ........................................54
B. POLICITEMIA VERA ..............................................................................................................................................69 1. Etiología........................................................................................................................................................69 2. Clínica ............................................................................................................................................................69 3. Diagnóstico .................................................................................................................................................69 4. Evolución .....................................................................................................................................................70 5. Pronóstico ...................................................................................................................................................70 6. Tratamiento y evolución natural .....................................................................................................70
C. ERITROCITOSIS O POLIGLOBULIA ...............................................................................................................71 1. Etiología........................................................................................................................................................71 2. Riesgos y consecuencias de la eritrocitosis: ..............................................................................71
D. ANEMIAS ....................................................................................................................................................................71 1. Anemia ferropriva o ferropénica ....................................................................................................75 2. Anemias megaloblásticas ....................................................................................................................75 3. Anemia de las enfermedades crónicas .........................................................................................77 4. Anemia aplástica o aplásica ...............................................................................................................78
E. LEUCEMIA AGUDA ................................................................................................................................................80 1. Clasificación................................................................................................................................................80 2. Frecuencia de presentación ...............................................................................................................80

III | Índice
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
3. Alteraciones genéticas ..........................................................................................................................81 4. Clínica ............................................................................................................................................................82 5. Diagnóstico .................................................................................................................................................82 6. Tratamiento................................................................................................................................................82 7. Factores de riesgo ...................................................................................................................................83 8. Pronóstico ...................................................................................................................................................84
F. LINFOMA NO HODGKIN .....................................................................................................................................84 1. Linfoma linfocítico pequeño ..............................................................................................................86 2. Linfoma MALT B de bajo grado ........................................................................................................86 3. Linfoma difuso de células B ...............................................................................................................86 4. Linfoma periférico de células T ........................................................................................................87 5. Linfoma/leucemia de células T del adulto .................................................................................87

HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
HEMATOLOGÍA
I. HEMATOLOGÍA La supervivencia y la función de cada célula del organismo están supeditadas a recibir en cada momento los nutrientes, hormonas y fermentos necesarios y poder eliminar sus desechos metabólicos.
La sangre el principal de los fluidos orgánicos, es el medio ideal para esta misión ya que tiene acceso a cada rincón de nuestra economía, interrelacionando los diversos tejidos y aparatos. Para ello necesita un cauce, los vasos sanguíneos, y un motor, el corazón.
La tarea del sistema cardiovascular-sanguíneo es completada y complementada por el sistema linfático, otro sistema vascular que utiliza como vehículo la linfa y que se relaciona funcionalmente con los ganglios linfáticos y los órganos linfoides. Entre éstos estudiaremos especialmente el timo y el bazo, órganos que participan, entre otras, en las tareas defensivas que nuestro cuerpo organiza contra la agresión. Todos sabemos el interés y la demostrada importancia que la inmunología ha cobrado en los últimos años para explicar muchas de las funciones defensivas del organismo y las enfermedades debidas a sus alteraciones; por ello, hemos querido incluir en un apartado específico algunas nociones sobre inmunidad que consideramos de gran interés.
La sangre puede considerarse como un tejido conectivo especializado, en el que la sustancia intercelular es un líquido y no existe componente fibroso.
En los mamíferos, la mayor parte de los elementos formes de la sangre no están representados por células «verdaderas» sino por elementos anucleados (glóbulos rojos, eritrocitos o hematíes) y fragmentos de citoplasma (plaquetas).
Las células verdaderas (leucocitos o glóbulos blancos) son sólo una pequeña parte de los elementos formes y su presencia es transitoria, usando la sangre como vehículo para su diseminación a otros órganos y tejidos, a los que migran para realizar sus funciones.
Únicamente los eritrocitos y las plaquetas llevan a cabo su misión en el interior del sistema vascular sanguíneo.
Ninguno de los elementos formes se duplica en el torrente sanguíneo. Cuando se destruyen, nuevos elementos son agregados desde los tejidos especiales «formadores de sangre» (hematopoyéticos), localizados fuera de la circulación.
La cantidad total de sangre representa una proporción bastante constante de la masa corporal total, siendo generalmente entre el 6 y el 8 % del peso total del cuerpo, es decir, un volumen aproximado de 5-6 I en el varón y 4,5-5,5 1 en la mujer.

2 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
Figura 1. Distribución normal de la sangre en el adulto.
A. CARACTERÍSTICAS GENERALES DE LA SANGRE La sangre, fluido opaco y turbio, presenta una coloración que oscila entre el escarlata brillante (cuando está oxigenada: sangre arterial) y el rojo oscuro o púrpura (cuando es pobre en oxígeno: sangre venosa).
Las funciones desempeñadas por este tejido revisten gran importancia para el mantenimiento de la homeostasia:
• Absorción en el intestino de los principios inmediatos que ingresan con la dieta. • Transporte de oxígeno, asimilado en los pulmones, que es llevado a cabo por los hematíes. • Transporte de sustancias de desecho que serán eliminadas por el pulmón (en forma de
CO2) y el riñón (en forma de urea y ácido úrico). • Transporte de hormonas. • Regulación de la temperatura corporal.

3 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
• Regulación del pH, función que, en parte, será llevada a cabo por las proteínas, debido a su carácter anfótero, y, en parte, por sustancias disueltas en la sangre y que actúan como tampón, entre las que cabe destacar los bicarbonatos y los fosfatos.
• Función defensiva, que será de dos tipos: Inmunidad humoral, sistema específico de defensa frente a agentes extraños
(antígenos) y llevado a cabo por los anticuerpos circulantes (gammaglobulinas plasmáticas).
Inmunidad celular, en que las células (linfocitos) son las encargadas de la defensa frente a infecciones o tejidos extraños (por ejemplo trasplantes).
B. PROPIEDADES FISICOQUÍMICAS Para que la sangre cumpla adecuadamente sus funciones deben mantenerse constantes sus características fisicoquímicas, entre las que caben destacar:
• Isotonía: corresponde a una solución salina al 0,9 %. • Isohidria: concentración constante de hidrogeniones (pH), que oscila entre 7,38 y 7,44. • Peso específico: depende del contenido en hematíes, pero oscila entre 1.050 y 1.064. • Viscosidad: depende también de la concentración de hematíes y proteínas plasmáticas.
La viscosidad relativa de la sangre total referida al agua (=1) es de 3,6-5,4; la del plasma, de 1,9-2,3, y la del suero de 1,7-2. Se debe aclarar que plasma y suero no son términos equivalentes, aunque ambos representen el componente líquido de la sangre: el plasma se obtiene de la sangre después de un tratamiento anticoagulante y contiene todos los componentes de la parte líquida, mientras que el suero se obtiene a partir de la sangre coagulada o desfibrinada, por lo que no contiene fibrinógeno, ni los factores de coagulación II, V y VIII. Esta característica resulta de gran importancia en la circulación sanguínea, pues un aumento en la viscosidad implica que el corazón tenga que aumentar su fuerza de contracción y, por otra parte, favorece la formación de trombos.
C. COMPONENTES DE LA SANGRE Si bien a primera vista la sangre puede parecer homogénea, después de dejarla en reposo o centrifugarla (previamente tratada con anticoagulante), es posible observar en realidad su carácter heterogéneo, pues aparecen: un sobrenadante, el líquido denominado plasma (aproximadamente el 55 %), y un sedimento, los elementos formes (células) (alrededor del 45 %).
1. Plasma Líquido claro ligeramente amarillento, compuesto en un 90 % por agua; el resto lo constituyen iones , los principales son Na+, Cl-, K+, Ca–, Mg-, y moléculas orgánicas, como glúcidos, lípidos y proteínas, entre las cuales cabe destacar:

4 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
Albúmina (pm 69.000) Es la más abundante de las proteínas plasmáticas, con una concentración de 3,5-5 g/100 ml. Sintetizada por el hígado, desempeña funciones de gran importancia:
• Transporte de aniones y cationes. • Regulación de la presión oncótica o presión osmótica ejercida por las soluciones
proteicas; si la concentración de albúmina disminuye, es más fácil la aparición de edemas. • Transporte de ácidos grasos, bilirrubina y fármacos poco solubles.
Globulinas (pm entre 150.000 y 900.000) Los valores normales en el plasma oscilan alrededor de 2,5-3,5 g/100 ml. La fracción globulínica se subdivide en numerosos componentes, entre los que cabe destacar: alfa-1, alfa-2, beta y gamma.
Estos componentes presentan la propiedad de migrar con diferente velocidad al ser sometidos a la acción de un campo eléctrico. Mediante la técnica de electroforesis es posible diferenciar y evaluar las diferentes fracciones.
Las globulinas alfa y beta se encargan del transporte de sustancias como hierro, cobre, triglicéridos, vitaminas y hormonas.
Entre las gammaglobulinas se encuentran los anticuerpos o inmunoglobulinas, que, como se ha mencionado con anterioridad, participan en el mecanismo de defensa del organismo. Esta fracción proteica es sintetizada por linfocitos de la médula ósea, el bazo y los ganglios linfáticos (linfocitos B), a diferencia de las restantes proteínas plasmáticas que son formadas en el hígado a razón de 15-20 g/día.
Fibrinógeno (pm 340.000) Proteína plasmática soluble, sintetizada por el hígado, que resulta esencial en el complejo mecanismo de la coagulación. Los valores normales en plasma son de 200-400 mg/100 ml.
2. Elementos formes Hematíes (eritrocitos o glóbulos rojos) En los mamíferos el glóbulo rojo normal es una célula anucleada con forma de disco bicóncavo de aproximadamente 7,5 µm de diámetro, 2 µm de espesor en la periferia y 1 µm en el centro y cuyo contenido en un 30-35 % se compone de hemoglobina (pigmento rojo de la sangre).

5 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
PORCENTAJE
Albúmina Globulina alfa-1 Globulina alfa-2 Globulina beta
Gammaglobulina
50 - 60 4,2 - 7,2 6,8 - 12 9,3 - 15 13 - 23
Tabla 1. Valores normales relativos a las diferentes proteínas plasmáticas respecto al total
Esta forma bicóncava y el elevado grado de elasticidad de la membrana, unidos a la existencia de un medio intracelular constante, permiten al glóbulo rojo atravesar, sin lesionarse, capilares de pequeño calibre. Toda modificación de la forma, aumento de la rigidez de la membrana o de la viscosidad de la hemoglobina contribuye a disminuir dicha plasticidad, lo cual conduce a un trastorno circulatorio y favorece la destrucción de los hematíes.
En el adulto, los hematíes son elaborados, al igual que el resto de las células sanguíneas, en la médula ósea y, después de pasar por diferentes fases de maduración (expulsan el núcleo y son incorporados al torrente circulatorio. En el feto también son órganos hematopoyéticos (productores de sangre) el saco vitelino, el hígado y el bazo.
Los valores normales en el hombre oscilan alrededor de 4,5-6 x 106 y en la mujer de 4-5,5 x 106 hematíes por cada mm3 de sangre, aunque a lo largo de la vida esta cifra varía, siendo mayor en el nacimiento (aproximadamente 6 x 106).
La función del glóbulo rojo es asegurar el transporte y mantenimiento en estado funcional de la hemoglobina, la cual, a su vez, es la encargada del transporte de oxígeno (98 % del oxígeno circulante) y de una parte del anhídrido carbónico.
Aunque el hematíe es una célula anucleada, tiene una actividad metabólica importante, ya que en su membrana se produce la glucólisis (anaerobia) que libera el ATP necesario para que la membrana pueda permanecer intacta ante soluciones hipotónicas o hipertónicas (funcionamiento correcto de la bomba de sodio).

6 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
Figura 2. Elementos figurados de la sangre.
Eritropoyesis Se entiende como tal al conjunto de procesos que llevan a la formación de eritrocitos o glóbulos rojos, se caracteriza por ser:
• Un fenómeno permanente: cada día 1/120 de los glóbulos rojos llegan al final de su vida y son destruidos. Como compensación la eritropoyesis ha de mantener el equilibrio poniendo en circulación (en el adulto) los glóbulos rojos contenidos en 25-50 ml de sangre, aproximadamente.

7 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
• Un fenómeno adaptativo: en caso de necesidad, la actividad eritropoyética puede multiplicarse por 7 u 8. Cada elemento celular puede, por división, originar dos elementos iguales a sí mismo o, por el contrario, madurar y diferenciarse en otros elementos más avanzados de la escala hematopoyética. Todas las células sanguíneas se originan del mesénquima no diferenciado: células madre o pluripotenciales, que pueden pertenecer a la estirpe mieloide (hematíes, polinucleares y monocitos) o a la linfoide (linfocitos). Las células primitivas de cada familia tienen características morfológicas similares y, en general, no se pueden diferenciar sólo por su aspecto.
Figura 3. Medidas del eritrocito.
A medida que las células primitivas cambian a formas celulares más maduras, se producen alteraciones en las características del núcleo y del citoplasma:
• Las células disminuyen de tamaño. • El volumen absoluto y relativo del núcleo se reduce (y en los eritrocitos desaparece). • Condensación de la cromatina nuclear. • En el citoplasma se va atenuando la intensidad de la tinción desde un azul intenso
(basofilía) en las células inmaduras hasta cierto color rojizo (acidófila) en las células maduras.
• Aparición de granulación específica (en determinadas líneas celulares).
Se denomina línea eritroblástica al conjunto de células que se diferencian hacia la síntesis de hemoglobina, finalizando en el glóbulo rojo o hematíe. En el hombre la línea eritroblástica está localizada en la médula ósea y representa el 10-30 % de las células medulares.
Por orden de maduración creciente se distinguen: proeritroblasto → eritroblasto basófilo → eritroblastopolicromatófilo → eritroblasto ortocromático → reticulocito → glóbulo rojo o hematíe.
En la sangre periférica de un individuo sano y normal se encuentran hematíes y unos pocos reticulocitos (alrededor del 0,5-2,5 % del recuento total de hematíes). El nombre de reticulocito proviene de la observación de las células al microscopio, pues se ve una estructura parecida a una red o retícula, que corresponde a restos de ARN. El número de reticulocitos en sangre periférica es un indicador del grado de actividad eritropoyética medular.

8 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
En condiciones normales los reticulocitos permanecen en la médula 2-3 días y terminan su maduración en aproximadamente 24 horas, ya en sangre periférica. Un aumento en el porcentaje de reticulocitos indica que la liberación de hematíes al torrente sanguíneo es más rápida de lo normal:
• Después de una pérdida aguda de sangre, el recuento de reticulocitos puede llegar al 10 %, ya que es una respuesta fisiológica a una mayor necesidad de hematíes.
• En una anemia ferropénica, al inicio del tratamiento con suplementos de hierro, el recuento de reticulocitos puede aumentar hasta el 30 %, lo cual es un signo alentador de que la médula ósea responde bien.
Por el contrario, una disminución de reticulocitos, especialmente después de una hemorragia, indica una respuesta anormal de la médula ósea. También en la anemia perniciosa (por déficit de vitamina B12) la producción ineficaz de hematíes determina un recuento bajo de reticulocitos.
1) Control de la eritropoyesis
La regulación de la eritropoyesis se realiza principalmente a través de un factor humoral específico (eritropoyetina), de factores humorales inespecíficos (tiroxina y andrógenos) y también del sistema nervioso central (SNC) particularmente del hipotálamo.
La síntesis de eritropoyetina, llevada a cabo sobre todo por el riñón, es regulada por la oxigenación hística; la hipoxia determina que la concentración de eritropoyetina se eleve a niveles que pueden detectarse en sangre, lo cual promueve un aumento en la producción de hematíes en la médula ósea. Por el contrario, la hiperoxigenación o un aumento en la masa globular circulante deprimen la síntesis de eritropoyetina (mecanismo de feedback o retroalimentación).
La función de esta hormona es promover la diferenciación de las células madre en proeritroblastos; también aumenta la velocidad de síntesis de la hemoglobina y acelera la salida de los reticulocitos.
2) Factores exógenos necesarios para la maduración del eritrocito
La eritropoyesis requiere simultáneamente la síntesis de ADN y de hemoglobina. Para sintetizar ADN, el organismo debe contener una cantidad suficiente de vitamina 13,2 y ácido fólico que ingresan con la dieta:
• Vitamina B2 (cianocobalamina): está presente en hígado, riñón, carnes y productos lácteos.
• Ácido fólico: se encuentra en legumbres verdes, cereales, hígado y en las carnes.
Para la síntesis de hemoglobina es necesario el hierro que proviene de la alimentación, principalmente de legumbres, espinacas, frutos secos, chocolate, etc. También se requiere, para la síntesis del grupo hem de la hemoglobina, la presencia de vitamina B6 (piridoxina), cuyas fuentes naturales son la yema de huevo, las carnes, los pescados, el hígado y la leche.
Las cantidades necesarias de estos elementos están aseguradas con una dieta equilibrada.

9 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
Figura 4. Etapas en la maduración del eritrocito.
Hemoglobina La hemoglobina es una proteína heterogénea que se origina durante el desarrollo del eritroblasto. Es el componente de los glóbulos rojos responsable del transporte de gases, principalmente oxígeno, razón por la que se la denomina pigmento respiratorio; contiene hierro y da el color rojo a la sangre.
Cada molécula de hemoglobina consta de cuatro cadenas proteínicas (dos llamadas alfa y betados), cada una de las cuales «envuelve» un grupo prostético (no proteínico) portador de

10 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
oxígeno. Dicho grupo prostético, denominado «hem», consta de un complejo de hierro ferroso (Fe–) unido a un anillo de porfirina. Cada átomo de hierro puede llevar una molécula de oxígeno.
Figura 5. Regulación hormonal de la eritropoyesis.
La composición y la secuencia de los aminoácidos en la cadena peptídica confiere a la molécula especificidad de especie y determina el tipo de hemoglobina; en el ser humano la hemoglobina presente en los glóbulos rojos fetales difiere de la de los adultos en la ordenación de los aminoáci-dos y en la composición de las cadenas beta.
La hemoglobina normal del adulto humano es conocida como hemoglobina A (Hb A: 97-99 %), pero también existen pequeñas cantidades de una hemoglobina menor denominada Hb A2 (1-3 %) e indicios de hemoglobina fetal (Hb F).
Íntimamente ligada a la Hb A, y de gran interés para el control clínico de la diabetes sacarina, está la fracción Hb A1, o Hbglucosilada.
El estudio de este parámetro constituye una gran ayuda para el conocimiento del estado glucémico habitual, con independencia de las fluctuaciones más o menos esporádicas de la glucosa.
1) Funciones de la hemoglobina
Como pigmento respiratorio de los glóbulos rojos, la hemoglobina asegura varias funciones, pero la principal es el transporte de oxígeno desde los pulmones hasta los tejidos. Cada molécula de hemoglobina fija cuatro moléculas de oxígeno sobre el hierro, formando oxihemoglobina.
Cuando la concentración o presión parcial de oxígeno (PO2) es elevada, como ocurre en los pulmones, todas las moléculas de hemoglobina se saturan de oxígeno, y cuando la PO2 disminuye, como sucede en los tejidos, la hemoglobina libera progresivamente su oxígeno de acuerdo con una cinética sigmoide característica que se denomina curva de disociación de la hemoglobina. También la hemoglobina transporta una pequeña parte de anhídrido carbónico (alrededor del 30

11 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
%) desde los tejidos a los pulmones, para ser eliminado. El resto es transportado por el plasma en forma de bicarbonato.
2) Hemoglobinopatías.
Son trastornos hereditarios que resultan de mutaciones en los genes de la globina que alteran la secuencia de aminoácidos (estructura de la proteína) o la velocidad de síntesis de una cadena de globina dada. Según este criterio se distinguen:
• Hemoglobinopatías estructurales: debidas a una estructura anormal de la globina (por ejemplo anemia drepanocítica).
• Talasemias: por una síntesis anormal de la globina que, según afecten una cadena u otra, se clasifican en alfatalasemias y betatalasemias.
La consecuencia de estas alteraciones es que los hematíes son más frágiles y se destruyen fácilmente (hemolisis).
Dado que la hemoglobina es un componente de los hematíes, cualquier enfermedad que produzca una disminución en el número de éstos, también ocasionará una disminución de la tasa de hemoglobina, encontrándose entre las causas más frecuentes: la pérdida de sangre, las anemias hemolíticas y cualquier hipofunción de la médula ósea.
3) Valores normales de hemoglobina.
Varían según la edad y el sexo; son más elevados al nacimiento y especialmente bajos en la mujer embarazada, teniendo normalmente el hombre un contenido hemoglobínico superior al de la mujer.
Metabolismo del hierro El hierro es un metal fundamental en el metabolismo celular, ya que forma parte de la estructura de gran número de proteínas, entre las que se encuentra la hemoglobina. En el organismo hay 4-5 g de hierro, presentes en dos formas:
1) Compuestos hemínicos (que contienen «hem»)
• Hemoglobina: normalmente es el mayor compartimiento de hierro del organismo, alrededor del 67 %.
• Mioglobina: una pequeña parte del hierro (aproximadamente el 3 %) está contenido en las células de la musculatura esquelética y cardíaca. Citocromos y enzimas: aproximadamente el 0,2 %.
2) Compuestos no hemínicos, dedicados al transporte y almacenamiento
• Transferrina o siderofilina: proteína mediante la cual el hierro es transportado por el plasma y distribuido por todo el organismo. Cuando llega a la médula ósea, penetra en los eritroblastos, donde es utilizado para la síntesis de hemoglobina. Si no es usado

12 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
inmediatamente, se deposita en forma de ferritina en el citoplasma del propio eritroblasto o de los macrófagos del sistema mononuclear fagocítico (SMF) especialmente de la médula ósea y del hígado, formando el hierro de reserva. La concentración de transferrina en el plasma suele valorarse mediante la medición de la capacidad total de fijación del hierro (CTFH).
• Ferritina: proteína hidrosoluble que almacena hierro, por lo que constituye una reserva de la que se puede disponer rápidamente.
• Hemosiderina: es un derivado insoluble en agua de la ferritina, lo que implica un intercambio de hierro más lento y del que no se puede disponer con tanta rapidez.
PÉRDIDA Y APORTE DE HIERRO Normalmente, las pérdidas diarias de hierro son escasas, del orden de 1 mg/día. Se producen a través de las células de descamación, faneras y heces y, en menor cantidad, por el sudor y la orina. Dichas pérdidas son ampliamente compensadas por el hierro ingerido en la alimentación diaria (10-25 mg), aunque de la cantidad ingerida sólo se absorbe el 10-20 %, eliminándose el resto por las heces.
Hay que señalar que estas necesidades de hierro se encuentran aumentadas de manera fisiológica durante el embarazo, en el lactante y en la adolescencia.
El contenido en hierro de los distintos alimentos es variable, siendo los más ricos el hígado, las ostras, las legumbres, el chocolate y los frutos secos; contienen menor cantidad la carne de ternera, el cordero, el cerdo, el pollo y los pescados; los cereales aportan poco hierro y las frutas carecen de él.
La absorción de hierro se realiza en todo el tracto intestinal, pero es más eficiente en el duodeno. Para ser absorbido tiene que encontrarse en forma de hierro ferroso (Fe-) y, además, ser liberado de las proteínas alimentarias, por lo cual una disminución en la secreción de pepsina en el estómago, conduce a una disminución de la absorción de hierro.
Una vez que el hierro es absorbido en el intestino, se fija sobre la transferrina, que lo transportará hasta el lugar de utilización (médula ósea) o será almacenado como ferritina.
Eritrocateresis El glóbulo rojo normal vive un promedio de 120 días y muere por envejecimiento; al ser una célula anucleada, no puede renovar su stock de enzimas y, como resultado, se produce una alteración de la membrana, siendo retenido por las células macrófagas que lo fagocitan y destruyen.
En condiciones normales dicha hemólisis se produce en los capilares del sistema reticuloendotelial (médula ósea, hígado y bazo), donde la hemoglobina es digerida:
• La globina es degradada en aminoácidos sin destino particular. • El grupo hem es transformado en una serie de pigmentos que son liberados al plasma en
forma de bilirrubina libre (no conjugada o indirecta), la cual, fijada a la albúmina, es transportada a las células hepáticas; una vez aquí, una enzima (glucuroniltransferasa)

13 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
transforma la bilirrubina libre en bilirrubina conjugada (directa), que es hidrosoluble y pasa a la bilis, eliminándose por la excreta como estercobilinógeno (da color a las heces), y en orina como urobilina. La tasa de bilirrubina libre o indirecta (en condiciones normales inferior a 11 mg/dl) es proporcional a la masa de hemoglobina liberada por la hemólisis; por lo tanto, un aumento de la bilirrubina libre puede estar ocasionado por un incremento en la hemólisis (enfermedades autoinmunes, intoxicaciones farmacológicas, incompatibilidad Rh o ABO en el recién nacido o por reacciones transfusionales). Una situación de hemólisis fisiológica ocurre con cierta frecuencia en el recién nacido, como una adaptación a su nuevo entorno. Consecuentemente aumenta la tasa de bilirrubina y se produce una ictericia fisiológica que remite a los pocos días.
• El hierro es rescatado y reciclado; el 80 % se incorpora de inmediato a la hemoglobina recién formada. Una parte del hierro restante es incorporado en compuestos de hierro de otros tejidos, pero la mayoría queda retenida en el sistema reticuloendotelial como ferritina.
1) Índices eritrocitarios primarios
RECUENTO DE HEMATÍES Es la determinación del número de hematíes por milímetro cúbico (mm3) de sangre.
• Hematocrito: determina el porcentaje de glóbulos rojos que hay en el plasma. El hematocrito es útil como medida de recuento de hematíes sólo en el caso de que la hidratación del individuo sea normal, ya que cualquier disminución del volumen plasmático produce un incremento en el valor hematocrito, aunque no esté aumentado el número de hematíes. Toda deshidratación grave (por ejemplo quemado) ocasiona un hematocrito anormalmente alto. Del mismo modo, un hematocrito disminuido puede deberse a una sobre hidratación del paciente, con un aumento del volumen plasmático, aunque, por lo general, la causa más frecuente de la disminución del valor hematocrito es una reducción real del número de hematíes. El hematocrito, sirve sobre todo para valorar la magnitud de la pérdida de sangre. Los valores de referencia se encuentran reflejados en la tabla 2.
• Hemoglobina: si cada hematíe contiene una cantidad normal de hemoglobina, el hematocrito será aproximadamente 3 veces el nivel de ésta. Por ejemplo, a un hematocrito del 45 % le corresponde una hemoglobina de 15 g/dl. Pero la hemoglobina no se puede calcular a partir del hematocrito cuando los hematíes poseen un tamaño o forma anormal ni cuando la hemoglobina se fabrique de forma anormal.
Varones 4,5 - 6 millones/mm3 Mujeres 4 - 5,5 millones/mm3 Embarazo Valores algo inferiores Recién nacidos 5,5 - 6 millones/mm3 (disminuye gradualmente) Niños 4,5 - 4,8 millones/mm3 (según la edad)
Tabla 2. Recuento normal de hematíes.

14 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
Cualquier enfermedad que disminuya el número de hematíes, reduce consecuentemente el nivel de hemoglobina. Entre las causas más frecuentes destacan hemorragias, anemias hemolíticas y cualquier depresión de la médula ósea. También en las hemoglobinopatías el valor de la hemoglobina está disminuido. El conocimiento de este parámetro es necesario para la valoración de varios tipos de anemia. Puede existir un nivel bajo de hemoglobina pero con un número de hematíes dentro de los valores normales , anemia por déficit de hierro o ferropénica, pero cada célula contiene menos hemoglobina de lo normal, denominándose por ello anemia hipocrómica (poco color); además, las células tienden a tener menor tamaño, por lo que la anemia también será microcítica.
VELOCIDAD DE SEDIMENTACIÓN GLOBULAR (VSG) Si se deja un tubo con sangre anticoagulada en reposo, después de cierto tiempo se observa que las células sedimentan formándose un «paquete» de hematíes en el fondo; a este proceso se lo denomina sedimentación globular, y el interés de su estudio radica en la velocidad a la que se produce, es decir, a la velocidad de sedimentación globular. Los resultados se expresan en milí-metros por hora y se estudia durante 2 horas.
Al ser la VSG una medida de agregabilidad de los hematíes, depende de una serie de factores, como: tamaño de los hematíes; diferencia de densidad entre hematíes y plasma; viscosidad plasmática, dependiente de la concentración de fibrinógeno y globulinas, y temperatura ambiente.
Un aumento de globulinas o de fibrinógeno en el plasma hará que las células se aglutinen y se depositen más rápidamente de lo normal. Lo mismo ocurre si las células tienen un tamaño menor de lo normal (microcitosis) o si hay pocas células (anemia). Por lo tanto, en ambos casos la VSG se encontrará elevada.
Por el contrario, si existe una macrocitosis o un aumento en el número de células (poliglobulia), éstas se depositan más lentamente.
Varones 45 - 52% Mujeres 37 - 48% Embarazo Disminuye sobre todo el tercer trimestre, al haber aumento en el volumen plasmático. Recién nacidos Hasta 45 - 60% Niños 36 - 44% en el primer año, luego varía con la edad
Tabla 3. Valores normales de hemoglobina.
Fisiológicamente, durante el embarazo, desde el 2°-3er mes y en el puerperio, la VSG está aumentada debido a la elevación que se produce de globulinas y fibrinógeno. También hay discretos aumentos no patológicos en el lactante de pocos meses y en la vejez.
El aumento patológico de la VSG (> 100) se debe en general a un proceso inflamatorio o a una lesión hística, siendo las causas más frecuentes las infecciones, las neoplasias y las enfermedades del tejido conectivo vascular. Es de gran utilidad en el control de la artritis reumatoide y de la enfermedad pélvica inflamatoria (EPI).

15 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
En general, una VSG disminuida no es clínicamente significativa ya que el límite inferior de normalidad es cero, pero la VSG está baja en los pacientes con policitemia vera-1 e hipoalbuminemia.
El aumento de la VSG se puede considerar en la segunda hora: ligero (hasta 30), moderado (30 a 50), intenso (50 a 100) o muy intenso (mayor de 100).
ÍNDICES ERITROCITARIOS SECUNDARIOS Son los que relacionan el hematocrito con el número de hematíes y la concentración de hemoglobina. Se los denomina secundarios por estar calculados a partir de los índices eritrocitarios primarios. Fundamentalmente son tres: volumen corpuscular medio (VCM), hemoglobina corpuscular media (HCM), y concentración de hemoglobina corpuscular media (CHCM).
Su determinación es de gran utilidad en la valoración y la orientación diagnósticas de los distintos tipos de anemias, dado que dichos parámetros indican si el tamaño de los hematíes y la concentración de hemoglobina son normales. Aunque actualmente la mayoría de los autoanaliza-dores utilizados en los laboratorios de hematología proporcionan estos índices de forma sistemática, no está de más indicar sus fórmulas para facilitar la explicación del significado de los resultados.
VOLUMEN CORPUSCULAR MEDIO (VCM) Describe el volumen medio del volumen de cada hematíe en µes cúbicos (pm’) o femtolitros (fl = 10-15 litros); las operaciones que hay que realizar para su obtención son las siguientes:
VCM = Hematocrito % x 10
Hematíes (millones/mm3)
Los valores normales se encuentran entre 80 y 96 fl. Al ser el VCM un indicador del tamaño del hematíe, un descenso por debajo de 80 fl indicará que los hematíes son más pequeños de lo normal: microcíticos (por ejemplo anemia ferropénica).
Síndrome mieloproliferativo en el que, sin que medie estímulo conocido alguno, se produce un aumento en la producción de elementos formes, en particular de la serie roja.
Por el contrario, si el VCM es superior a 96 fl, los hematíes son más grandes de lo normal: macrocíticos (por ejemplo anemias por déficit de ácido fólico, anemia perniciosa por déficit de vitamina B12).
Si el VCM está dentro de los límites normales, los hematíes son normocíticos; por ejemplo, en las anemias debidas a una pérdida de sangre o en las anemias hemolíticas se mantiene un VCM normal.

16 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
HEMOGLOBINA CORPUSCULAR MEDIA (HCM) Es la cantidad de hemoglobina presente en cada hematíe. El resultado se expresa en picogramos (pg = 10-12 gramos). El peso medio de la hemoglobina en la célula se obtiene dividiendo la hemoglobina entre el número de hematíes:
HCM = Hemoglobina (g/dl) x 10
Hematíes (millones/mm3)
Los valores de referencia se encuentran entre 27 y 32 pg.
CONCENTRACIÓN DE HEMOGLOBINA CORPUSCULAR MEDIA (CHCM) Expresa la proporción de cada hematíe que está ocupado por la hemoglobina. El resultado, por tanto, se expresa en porcentaje y el cálculo se realiza de la siguiente manera:
CHCM = Hemoglobina x 100
Hematocrito
Los valores de referencia están entre 32 y 36 %.
Con el estudio conjunto de estos dos últimos índices (HCM y CHCM) es posible determinar si los hematíes son hipocromos, hipercromos o normocromos (= color normal).
Una CHCM por debajo del 32 % (HCM < 27 pg) indica que los hematíes tienen una concentración baja de hemoglobina: hipocromía. A este tipo pertenecen la anemia ferropénica y ciertas anemias genéticas (talasemias).
En otros tipos de anemia, la concentración de hemoglobina es normal, en cuyo caso se llaman anemias normocrómicas.
Sólo algunas enfermedades genéticas raras cursan con hipercromía; CHCM > 36 %.
Anemia La medición de los glóbulos rojos se efectúa a través de tres valores del hemograma: número de hematíes, hematocrito y tasa de hemoglobina. Si estos tres valores se modificaran paralelamente, bastaría la medición de uno de ellos, pero en la práctica se observa que pueden evolucionar de forma disociada; así, por ejemplo, puede ocurrir una disminución del volumen de cada glóbulo rojo (VCM) sin que se modifique su número y, sin embargo, se encuentran disminuidos los valores de Hb y hematocrito.
Por el contrario, puede haber una disminución del número de glóbulos rojos, pero con un aumento en su volumen sin que disminuya la tasa de hemoglobina o el hematocrito (macrocitosis sin anemia).

17 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
Pero lo que en realidad tiene importancia para el organismo no es el número de glóbulos rojos, sino la cantidad de oxígeno que transportan y, por lo tanto, la tasa de hemoglobina; por ello, la anemia no se define como la disminución del número de glóbulos rojos, sino como la disminución de la tasa de hemoglobina por unidad de volumen de sangre por debajo de los valores fisiológicos.
Sin embargo, existen casos en los que la disminución de la tasa de hemoglobina puede ser consecuencia de una hemodilución, como causa de una hipervolemia plasmática, y no de una anemia verdadera. Dicha hemodilución puede producirse en distintas situaciones, como: trimestre del embarazo, hiperhidratación de cualquier origen (insuficiencia cardíaca congestiva, grandes esplenomegalias, hipertensión portal, etc.) entre otras.
Figura 6. Comparación de los datos obtenidos en una anemia microcítica y en una macrocitosis con un patrón de normalidad.
Si se conoce la proporción de hemoglobina contenida en los glóbulos rojos (según la HCM y la CHCM), es posible catalogar las anemias como hipercromas o hipocromas, y si se dispone del dato del carácter microcitario o macrocitario (según el VCM), se puede clasificar a las anemias en varios tipos, de los que haremos una somera clasificación:
1) Anemias hipocromas y microcíticas (VCM < 80 fl; HCM < 27 pg; CHCM < 32 %):
• Anemias ferropénicas (cursan con hiposideremia): Por pérdidas de sangre crónicas. Por deficiente aporte exógeno de hierro. Por malabsorción de hierro. Por aumento del consumo de hierro.
• Anemias sideroacrésticas (por déficit de utilización del hierro). Cursan con hipersideremia.

18 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
Por bloqueo de hierro en los tejidos. Por mala síntesis de hemoglobina (talasemias).
2) Anemias hipercromas y macrocíticas (VCM > 96 fl; HCM > 32 pg):
• Anemias carenciales de vitamina B12. • Anemias carenciales de ácido fólico.
3) Anemias normocíticas (VCM = 80-96 fl; HCM = 27-32 pg):
• Hemorragia aguda reciente. • Anemias hemolíticas. • Anemias aplásicas. • Anemias de las enfermedades crónicas (infecciosas, neoplásicas o sistémicas).
Poliglobulia Se define como el aumento de la masa globular total del organismo, con un aumento de los glóbulos rojos superior a 6 millones/mm3 o una tasa de hemoglobina superior a 18 g/dl en el varón (16 g/dl en la mujer) o un valor hematocrito superior al 52 % en el varón (48 % en la mujer).
Al igual que sucede con las falsas anemias (por hemodilución), puede haber seudopoliglobulias por hemoconcentración en cualquier deshidratación: hipovolemia por sudación profusa, diarreas, vómitos, shock en grandes quemados, etc.
Los desplazamientos a grandes alturas o el aumento del ejercicio físico producen un aumento fisiológico en el número de hematíes como respuesta a una mayor necesidad de oxígeno. Pero el número de hematíes puede aumentar por otras muchas razones patológicas, que son siempre debidas a una producción anómala por parte de la médula ósea y nunca a un aumento de la estancia de los hematíes en el torrente circulatorio. Podemos clasificar las poliglobulinas de la siguiente manera:
• Policitemia vera: enfermedad de etiología desconocida que cursa con un aumento verdadero de los hematíes.
• Poliglobulias secundarias: es decir, expresivas de una enfermedad de base, en las que el aumento de hematíes constituye un intento de compensar la hipoxia crónica provocada por la enfermedad (por ejemplo enfermedades pulmonares, niños con enfermedades cardíacas congénitas acompañadas de cianosis, etc.).
La consecuencia del aumento de la masa globular es la hiperviscosidad (por encima del 50-60 % de hematocrito), de lo que resulta:
• Un riesgo de trombosis considerable, por encima de 60 % de hematocrito, incrementado si además existe deshidratación.
• Alteración del transporte de oxígeno a los tejidos. • Existencia de vasodilatación periférica.

19 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
Leucocitos (glóbulos blancos) Los leucocitos son células sanguíneas incoloras (por ello también reciben el nombre de glóbulos blancos) dotadas de núcleo, lo que permite su fácil identificación morfológica respecto de los hematíes y las plaquetas. Son producidos por la médula ósea y los ganglios linfáticos (linfocitos) y, aunque circulan por la sangre, ejercen su función lejos de ella.
Sus funciones son diversas, pero siempre relacionadas con la defensa del organismo frente a diversas sustancias o agentes patógenos (fagocitosis e inmunidad).
Desde el punto de vista morfológico, los leucocitos pueden clasificarse en dos grupos:
1) Polinucleares
En realidad, se trata de un solo núcleo segmentado. Se clasifican en neutrófilos, eosinófilos o basófilos según la afinidad del citoplasma de estas células por los colorantes empleados para su tinción (eosina y azul de metileno).
2) Mononucleares
Poseen un único núcleo, de forma más o menos redondeada. Dentro de esta categoría se encuentran linfocitos y monocitos.
A partir de la observación al microscopio óptico, a los polinucleares también se los denominó granulocitos, dado que poseen granulaciones en su citoplasma, y a los mononucleares, por no poseerlas, agranulocitos, aunque posteriormente se pudo comprobar al microscopio electrónico que cualquier leucocito (incluidos linfocitos y monocitos) presenta algún tipo de granulación citoplasmática.
Generalmente se realizan dos determinaciones de leucocitos: por un lado, el recuento del número total en 1 mm de sangre y, por otro, el recuento diferencial, también denominada fórmula leucocitaria, consistente en determinar la proporción de cada uno de los cinco tipos de leucocitos (que están) presentes en una muestra de 100 leucocitos.
La normalidad en el recuento del número total de leucocitos se encuentra entre 4.000 y 11.000 leucocitos/mm3.
El recuento diferencial se da en porcentajes, lo que significa que el aumento en el porcentaje de un tipo de células implica una disminución en el porcentaje de otro tipo celular, aunque el número absoluto de las células de este segundo tipo no se haya alterado. Por ejemplo: un individuo posee un recuento total de 10.000 leucocitos/mm3, con un porcentaje de neutrófilos del 60 % (6.000 neutrófilos/mm) y de linfocitos del 30 % (3.000 linfocitos/mm3). Con una infección bacteriana, casi todo el aumento de leucocitos que experimente (supongamos que llega a 20.000 leucocitos/mm3) se efectuará a expensas de los neutrófilos, cuyo recuento diferencial será del 75 %, correspondiendo sólo el 15 % a los linfocitos. Sin embargo, esto no significa que haya un menor número de linfocitos, ya que su cifra absoluta (el 15 % de 20.000 es 3.000 leucocitos/mm3) es exactamente igual que antes de la infección. Anteriormente se ha mencionado que los valores normales de los leucocitos son de 4.000-11.000/mm3. No obstante, existen situaciones que, pese a ser fisiológicas, presentan un número más elevado de leucocitos, como las siguientes:

20 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
• Embarazo: leucocitosis de más de 16.000/mm3, debida sobre todo a un aumento de los neutrófilos, con un ligero aumento de los linfocitos.
• Infancia: en el recién nacido la fórmula sanguínea es similar a la del adulto, aunque el recuento total está por encima de los 18.000 leucocitos/mm3. Durante el primer mes se establece una fórmula con predominio linfocitario. El paso a la fórmula adulta se realiza entre los 4 y los 10 años.
FÓRMULA LEUCOCITARIA PORCENTAJE
Neutrófilos jóvenes (cayados) Neutrófilos maduros (segmentados)
Eosinófilos Basófilos Linfocitos Monocitos
3 -5 50 - 60
1 - 5 0 - 1
25 - 35 3 - 10
Tabla 4. Fórmula leucocitaria.
Polinucleares (granulocitos o segmentados) El proceso de maduración de los granulocitos, que dura unos 10 días, entraña cambios morfológicos genéricos similares a los ya mencionados respecto de la eritropoyesis: disminución del tamaño celular, condensación de la cromatina celular, etc.
Del mismo modo que los eritrocitos, los granulocitos proceden de la célula madre mieloide pluripotencial. La descripción del proceso madurativo del polinuclear neutrófilo se puede hacer extensiva al eosinófilo y al basófilo, por lo que se describe como ejemplo la maduración del neutrófilo.
De la célula madre deriva el mieloblasto, que es la célula más inmadura ya determinada para formar exclusivamente los tres tipos de granulocitos (neutrófilo, eosinófilo y basófilo). El mieloblasto puede dar origen a otros mieloblastos o bien madurar a promielocito, el cual a su vez, puede generar otros promielocitos o madurar a mielocito.
Hasta aquí (mieloblastos, promielocitos y mielocitos), al ser células capaces de dividirse, integran el denominado compartimiento medular de formación. El mielocito puede dividirse o bien madurar a metamielocito, el cual ya no es apto para la mitosis, sino que madura, obligatoriamente, a granulocito en banda o cayado, el cual, a su vez y como estadio final, llega a polinuclear maduro o segmentado.
Las células que no se dividen más (metamielocitos, cayados y segmentados) permanecen en la médula durante 4-5 días para acabar de madurar antes de pasar a la sangre, constituyendo el compartimiento de maduración y reserva. Ya en la sangre, los polinucleares neutrófilos (cayados y segmentados) no se encuentran todos ellos en circulación, sino que están divididos en dos sectores: el sector circulante y el sector marginal, formado por los polinucleares pegados a las paredes vasculares y que se reincorporan a la circulación según las necesidades.

21 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
Figura 7. Maduración de los leucocitos.
Tras una breve estancia en la sangre (aproximadamente 12 horas), el 50 % de los polinucleares neutrófilos de la sangre pasan a los tejidos, donde cumplen su misión.
1) Polinuclear neutrófilo
Su función esencial es la fagocitosis y destrucción de bacterias y cuerpos extraños. La fagocitosis es una función compleja que implica una serie de actividades sucesivas: la movilización hacia el agente que va a ser agredido, la fagocitosis propiamente dicha y, por último, la lisis.
El desplazamiento (diapédesis) se efectúa por movimientos ameboides que le permiten deformarse para poder pasar de los vasos a los tejidos. Esta movilización está dirigida por diversos factores quimiotácticos (especialmente los productos bacterianos) que atraen a los polinucleares neutrófilos y, una vez llegados a los tejidos, ingieren los cuerpos extraños a los que incluyen dentro de vacuolas intracitoplasmáticas. La fagocitosis es especialmente intensa para las partículas que están cubiertas por anticuerpos (opsonizadas).
Posteriormente, dentro de la vacuola de fagocitosis y gracias a la acción de diversas sustancias aún mal conocidas, entre ellas el peróxido de hidrógeno o agua oxigenada (1-1202), se produce la lisis de la membrana de la bacteria. Una vez muerta la bacteria, es atacada por las hidrolasas contenidas en los lisosomas de los neutrófilos, que se vierten en la vacuola de fagocitosis (desgranulación de los polinucleares neutrófilos), y es completamente destruida. Los polinucleares neutrófilos, vaciados de su granulación, mueren formando parte del pus.
Dado que cada granulocito sólo puede fagocitar un pequeño número de bacterias, también se lo ha denominado micrófago.

22 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
PARA SABER MÁS
Además de las infecciones bacterianas, la elevación del número de neutrófilos o neutrofilia puede deberse a varios procesos inflamatorios, estrés físico, necrosis hística, como el infarto agudo de miocardio, procesos neoplásicos o quemaduras graves. El parto y algunos fármacos y toxinas también pueden aumentar el número de neutrófilos. Asimismo, el estrés emocional puede producir neutrofilia, pero no tan elevado como el físico.
Aunque la mayoría de las infecciones bacterianas cursan con un aumento en el número de neutrófilos, algunas enfermedades, como la fiebre tifoidea o la brucelosis, provocan una disminución o neutropenia. También muchas enfermedades víricas cursan con neutropenia, como, por ejemplo, la hepatitis, el sarampión, la rubéola y la parotiditis.
La radioterapia y ciertos fármacos empleados sobre todo en el tratamiento del cáncer pueden provocar una depresión grave de la médula ósea.
También algunos antibióticos y psicofármacos pueden causar neutropenia.
2) Eosinófilos
Observado al microscopio óptico, el polinuclear eosinófilo es parecido al polinuclear neutrófilo, pero se diferencia porque poseen unas granulaciones más gruesas y de color anaranjado (tinción con eosina) y su núcleo generalmente es menos lobulado.
El polinuclear eosinófilo proviene de la médula ósea, donde existe una serie eosinófila análoga a la neutrófila, pero muy minoritaria. No se conoce con exactitud la duración de su vida en la sangre, pero probablemente es muy corta.
Al igual que el polinuclear neutrófilo, el eosinófilo está dotado de movilidad y capacidad de fagocitosis (los gránulos de su citoplasma son lisosomas). Sus funciones aún son mal conocidas, pero se sabe que fagocita especialmente los complejos antígeno-anticuerpo (Ag-Ac) y también los antígenos producidos en las reacciones alérgicas.
Entre las causas más frecuentes de eosinofilia (aumento de eosinófilos) se encuentran las reacciones alérgicas, como el asma, la fiebre del heno o la hipersensibilidad a los fármacos.
También algunas enfermedades cutáneas y neoplasias se asocian a un aumento de eosinófilos. Las infecciones parasitarias, como las helmintiasis (quistes hidatídicos, áscaris, triquinosis, etc.), son otras causas de eosinofilia.
Por otro lado, la disminución de eosinófilos, o eosinopenia, se produce ante niveles elevados de corticoides; por ejemplo, cabe esperar una disminución de eosinófilos en un paciente alérgico que inicia un tratamiento con corticoides.
3) Basófilos
También de origen medular, constituyen el tipo de leucocitos menos abundante en sangre periférica. Se caracterizan por poseer gruesas granulaciones basófilas (se tiñen con azul de

23 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
metileno) en el citoplasma e incluso cubriendo el núcleo, cuyo contenido principalmente es histamina y heparina. Se desconoce mucho sobre sus funciones, pero al parecer intervienen en las reacciones de hipersensibilidad retardada.
El mastocito o basófilo hístico es una célula algo diferente al basófilo sanguíneo, que se encuentra en los tejidos y desempeña un papel importante en las reacciones inflamatorias locales.
Aunque un aumento en el número de basófilos (basofilía) puede asociarse a ciertos estados de hipersensibilidad, lo más frecuente es su hallazgo en el curso de los síndromes mieloproliferativo crónicos (leucemia mieloide crónica y policitemia vera), en los que a veces se considera un signo de agudización de la enfermedad.
Mononucleares o agranulocitos
1) Monocitos
Célula de origen medular. Es la célula circulante de mayor tamaño y se caracteriza por tener un núcleo hendido pero no polilobulado. Se movilizan, junto con los neutrófilos como parte de la respuesta inflamatoria y constituyen una segunda línea de defensa contra las infecciones bacterianas. También desempeñan un papel importante en el procesamiento de los antígenos durante la respuesta inmunitaria.
Al contrario que los granulocitos, que son células terminales en el sentido de que no se dividen más, los monocitos son células intermedias destinadas a formar los macrófagos de los tejidos. El sistema de macrófagos hísticos se conoce como sistema reticuloendotelial.
Los monocitos atraviesan las paredes de las vénulas y de los capilares donde la circulación de la sangre es lenta y, al penetrar en los órganos, se transforman en macrófagos. Se produce un aumento de su capacidad fagocitaria y también del número de lisosomas, lo que tiene un gran significado funcional, pues el macrófago es mucho más activo en la fagocitosis que el monocito.
Los precursores monocíticos medulares junto con los monocitos y los macrófagos hísticos constituyen el denominado sistema mononuclear fagocítico (SMF), cuya función es la de eliminar, mediante fagocitosis, sustancias extrañas al organismo.
2) Linfocitos
Son células mononucleares de pequeño tamaño (6-8 µm de diámetro) que, al igual que los monocitos, carecen de granulación. Poseen un núcleo redondo que ocupa la mayor parte de la célula y un delgado halo de citoplasma, ligeramente basófilo, alrededor.
La linfopoyesis tiene una evolución y una regulación muy distintas a la línea mieloide (glóbulos rojos, polinucleares neutrófilos, basófilos y eosinófilos, monocitos y plaquetas). Existen también células madre linfoides, pero actualmente todavía son poco conocidas. Por orden de maduración creciente se distinguen: células madre linfoblastoprolinfocito linfocito maduro.
El tejido linfoide, además de estar presente en la médula, se halla en los ganglios linfáticos, el bazo, las placas de Peyer (intestino) y el timo.

24 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
Según las funciones que desempeñan se los clasifica en linfocitos B y linfocitos T.
Los linfocitos T se procesan y programan en el timo para tomar parte en los procesos de inmunidad celular, en los que la célula elabora agentes citolíticos no específicos. Intervienen en las reacciones alérgicas retardadas.
Los linfocitos B desempeñan un papel principal en los procesos de inmunidad humoral o inmediata, que es llevada a cabo por los anticuerpos.
Fisiológicamente, en el adulto los linfocitos son el segundo tipo de leucocitos más frecuente, después de los neutrófilos; en el niño de hasta 5-8 años el porcentaje de linfocitos es superior al de neutrófilos, pero hay situaciones patológicas en las que se produce un aumento (linfocitosis) o una disminución (linfopenia) en el número de linfocitos.
Se comprueba una linfocitosis en la mayoría de las enfermedades víricas como parotiditis o hepatitis infecciosa, en la mononucleosis infecciosa y a menudo también en la tuberculosis.
Las infecciones bacterianas crónicas también son causa de aumento del número de linfocitos, y el 90 % de todas las leucemias, tanto agudas como crónicas, son linfocíticas.
Por el contrario, cursan con linfopenia el síndrome de inmunodeficiencia adquirida (SIDA) y los tratamientos con fármacos inmunosupresores; incluso la desnutrición grave también puede reducir el número absoluto de linfocitos. Pero dado que el aumento de los neutrófilos ocurre por muchas razones, los recuentos bajos de linfocitos se explican a veces por los cambios producidos con los neutrófilos.
PARA SABER MÁS
Leucemia: Es la expresión en sangre periférica de una proliferación neoplásica de los leucocitos, ya sea de la serie mieloide (síndrome mieloproliferativo) o de la serie linfoide (síndrome linfoproliferativo). Según su evolución se las clasifica en leucemias agudas y crónicas, y atendiendo a la estirpe celular afectada se las divide en mieloides y linfoides (ambas pueden ser agudas o crónicas) o de células indiferenciadas (sólo agudas). Al igual que todas las neoplasias, las leucemias tienen una repercusión general en forma de astenia, anorexia y adelgazamiento. La infiltración leucémica de la médula ósea desplaza e inhibe a los tejidos eritropoyético, leucopoyético y trombopoyético normales, de lo cual puede derivar un síndrome anémico, una especial propensión a las infecciones y la diátesis hemorrágica. Además, la acumulación de células leucémicas en los órganos (hígado, bazo y ganglios linfáticos) los agranda y puede perturbar su función.
Plaquetas (trombocitos) Tienen su origen en la médula ósea. Como ya se indicó al inicio de este tema, no son «células verdaderas» sino que provienen de la fragmentación del citoplasma de los megacariocitos, que se forman por diferenciación de los megacarioblastos, los cuales derivan, a su vez, de una célula madre indiferenciada común a toda la serie mieloide.

25 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
Son las células sanguíneas de menor tamaño (2-4 de diámetro) y su vida media oscila alrededor de 4-8 días. Los valores normales son de 150.000-500.000 plaquetas/ mm3. Con valores inferiores a 150.000 plaquetas/mm3. Se habla de trombopenia, y por encima de 500.0000 plaquetas/ mm3, de trombocitosis.
3) Trombopenia
Cuando es de causa desconocida recibe el nombre de púrpura trombocitopénica idiopática (PTI). Otras veces la causa es más concreta: una infección vírica (es muy frecuente en el SIDA); los fármacos quimioterápicos y la radioterapia, que deprimen la función de la médula ósea, o situaciones en las que el bazo aumenta de tamaño (esple-nomegalia) o de función (hiperesplenismo).
Si una pérdida abundante de sangre es repuesta sólo con sangre conservada, al ser pobre en plaquetas, lleva rápidamente a una trombopenia intensa.
4) Trombocitosis.
Las trombocitosis permanentes se deben en general a una hiperproducción medular. En consecuencia, se ve considerablemente aumentado el riesgo de trombosis debido a la formación de agregados plaquetarios en la circulación, sobre todo cuando existen cifras superiores a 106 plaquetas/ mm3.
Las trombocitosis o trombocitemias pueden ser de origen desconocido (trombocitemia esencial), que es un síndrome mieloproliferativo con afección exclusiva de la línea plaquetaria.
También pueden producirse trombocitosis reactivas a diversas enfermedades infecciosas, síndromes inflamatorios y en casos avanzados de neoplasias. Otra causa de recuento elevado de plaquetas es la extirpación del bazo (esplenectomía).
FUNCIÓN PLAQUETARIA Entre las funciones de las plaquetas se incluyen:
• Prevenir la extravasación sanguínea en el vaso intacto. Aunque el mecanismo por el que ejercen esta función aún no es bien conocido, está demostrado que la falta de plaquetas tiene como consecuencia una tendencia a la hemorragia.
• Intervenir en la detención de la hemorragia al formar el trombo plaquetario. • Liberar, entre otras sustancias, el factor 3 plaquetario (F3P), fosfolípido que facilita el
encuentro y la posterior activación de los distintos factores plasmáticos de la coagulación. • Producir la retracción del coágulo de fibrina gracias a una proteína contráctil que existe
en la membrana plaquetaria.

26 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
D. FISIOLOGÍA GENERAL DE LA HEMOSTASIA La hemostasia es un proceso constituido por un conjunto de mecanismos biológicos relacionados entre sí, cuya misión consiste en prevenir la extravasación sanguínea espontánea, evitar un exceso de hemorragia en los vasos lesionados y mantener la fluidez de la sangre circulante.
El mantenimiento de una hemostasia normal depende del correcto funcionamiento de cuatro factores, de forma que la alteración de cualquiera de ellos es siempre causa de patología. Estos factores son:
• Reacción vascular. • Función plaquetaria. • Coagulación sanguínea. • Fibrinolisis.
Cuando se produce una lesión en un vaso, éste se contrae transitoriamente disminuyendo el paso de sangre a su través (reacción vascular). A continuación, las plaquetas circulantes se adhieren al colágeno del tejido subendotelial, que como consecuencia de la lesión se ha exteriorizado, y liberan ADP (adenosíndifosfato) facilitando la agregación y la formación de un trombo plaquetario (función plaquetaria). El trombo detiene momentáneamente la hemorragia mientras es reforzado primero y sustituido después por la formación de fibrina (coagulación sanguínea). Por último se inicia la cicatrización de la herida, y la fibrina va siendo digerida por un sistema enzimático, recuperando el vaso su estado originario (fibrinolisis).
La hemostasia deriva de la adecuada interacción de tres sistemas: la hemostasia primaria, hemostasia secundaria y sistema fibrinolítico.
• Hemostasia primaria: formación del tapón hemostático primario. Depende de la integridad vascular (endotelio y subendotelio) y funcionalidad plaquetaria (alteraciones cuantitativas o cualitativas). Cuando se produce una lesión en un vaso el primer mecanismo para detener la hemorragia es una vasoconstricción local refleja y a continuación la formación del tapón hemostático plaquetario. Adhesión plaquetaria: Las plaquetas se adhieren a las fibrillas de colágeno del subendotelio vascular mediante receptores de membrana: Gp Ia y Gp IIa (en endotelio) y GpIb/IX (en la membrana plaquetaria) formando un puente con el factor von Willebrand (vWF).

27 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
• Hemostasia secundaria: casi simultáneamente a la formación del tapón hemostático primario, se pone en marcha el proceso de coagulación dependiente de las proteína plasmáticas, y que consiste en la formación de fibrina soluble a partir de fibrinógeno plasmático. Clásicamente este conjunto de reacciones y activaciones de proteínas se ha interpretado como una cascada en donde se distinguían dos vías: en vía extrínseca e intrínseca. Actualmente se considera que ambas vías no son independientes en absoluto, ya que la vía extrínseca activa también al fX a través del fXI, considerándola como el inicio fisiológico de la coagulación. Sin embargo efectos didácticos y de pruebas diagnósticas, se sigue utilizando esta nomenclatura. Vía extrínseca o del Factor tisular:
Es una vía dependiente del Factor tisular (Tromboplastina) que forma un complejo con el Factor VII y el Calcio, convirtiendo al fVII en una proteasa activa que actúa sobre el factor X activándolo. Recientemente se ha visto la gran preponderancia de la vía del factor tisular en el mecanismo de la coagulación, surgiendo de este modo la llamada “hipótesis alterna o revisada del factor tisular”: el factor tisular es el mejor indicador de la puesta en marcha del proceso coagulativo, al formar un complejo con el FVII, activándolo (FVIIa). Al mismo tiempo el factor tisular hace de cofactor del FVIIa para que actúe sobre IX y X.
Vía intrínseca o sistema de contacto: El plasma contiene todos los elementos necesarios para la coagulación. En este caso la porción lipídica es el FP3. Los factores de contacto: fXII, Precalicreína, y cininógeno de alto peso molecular, se activan por el contacto con la piel, complejos Antígeno/anticuerpo, colágeno. El factor XIIa activa al XI y el XIa al IX, que forma complejo con el factor VIII, el FP3, y el Calcio (complejo protrombina) activando finalmente el factor X. Como ya se ha citado anteriormente, el factor XI también es activado por el factor VII (“hipótesis alterna del factor tisular”)
Vía común: El Factor Xa forma un complejo con el factor V y el Calcio que convierte la Protrombina en Trombina.
Fibrinogénesis: El papel fundamental de la Trombina es activar al factor XIII para actuar frente al Fibrinógeno convirtiéndolo en polímeros estables de Fibrina.
Fibrinolisis: La lisis del coágulo comienza inmediatamente después de la formación del coágulo. Sus activadores son tanto por parte de la vía extrínseca (factor tisular), como por la vía intrínseca, factor XII, así como otros exógenos: Urokinasa, tPA (activador tisular del plasminógeno). Los inhibidores del proceso de fibrinólisis ayudan a mantener el equilibrio hemostático y evitar los fenómenos trombóticos: Antitrombina, Proteína C, Proteína S.

28 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
FACTOR NOMBRE FORMA ACTIVA CARACTERISTICAS
I Fibrinógeno Fibrina Síntesis hepática. Sensible a la Trombina.
II Protrombina Trombina Síntesis hepática. Vitamino K dependiente.
III Tromboplastina (factor tisular) Cofactor
IV Calcio
V Proacelerina Cofactor Síntesis hepática. Sensible a la Trombina.
VII Proconvertina Serinproteasa Síntesis hepática. Vitamino K dependiente.
VIII/VIII:C Factor antihemofílico/Factor von Willebrand Cofactor Sensible a la Trombina.
IX Factor Christmas Serinproteasa Síntesis hepática. Vitamino K dependiente.
X Factor Stuart Serinproteasa Síntesis hepática. Vitamino K dependiente.
XI Serinproteasa Factor de contacto.
XII Factor Hageman Serinproteasa Factor de contacto.
XIII Estabilizador de la Fibrina Transglutaminasa Sensible a la Trombina.
Precalicreína Factor Fletcher Serinproteasa Factor de contacto.
Proteína C Antifibrinolítico Vitamino K dependiente.
Proteína S Cofactor de Prot C Antifibrinolítico Vitamino K dependiente.
Tabla 1. Factores de la coagulación

29 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
Figura 2. Esquema de la coagulación.
1. Etapas de la hemostasia Reacción vascular Cuando existe una lesión vascular, se produce una vasoconstricción local que ayuda a que la hemorragia disminuya. Esta vasoconstricción depende de ciertas reacciones hemodinámicas locales y de sustancias humorales liberadas por las plaquetas, por lo que la integridad estructural de los vasos es fundamental. Alteraciones de la estructura vascular, bien sean congénitas o adquiridas, hacen su manifestación con la aparición de petequias, equimosis y pequeñas hemorragias en las mucosas.
Tapón plaquetario Después de la rotura, ya sea espontánea o traumática, las plaquetas, al entrar en contacto con las fibras colágenas de las capas subendoteliales del vaso, se hinchan y se vuelven adherentes. Esta adhesión requiere la presencia de un componente de la coagulación denominado factor von Willebrand (FvW) que es una molécula del factor VIII, cuya ausencia da origen a la enfermedad de von Willebrand.
Las plaquetas liberan diversas sustancias (ADP, entre otras) que determinan la adhesión de las plaquetas entre sí, proceso que se designa agregación plaquetaria, y el grupo de plaquetas

30 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
constituye el tapón plaquetario primario, el cual, a no ser que se estabilice, se deshace espontáneamente.
Coagulación (propiamente dicha) La estabilización se lleva a cabo gracias a la activación secuencial de los factores de la coagulación que circulan en el plasma de forma inactivada.
La activación tiene como resultado la formación de una malla de fibrina alrededor del tapón plaquetario. Conviene señalar que la síntesis de dichos factores de coagulación, con excepción del factor VIII, se efectúa en el hígado, y que los factores II, VII, IX y X requieren la presencia de vitamina K para su elaboración (son vitamina K-dependientes).
FACTORES NOMBRE
I Fibrinógeno.
II Protrombina.
III Tromboplastina hística (factor hístico).
IV Calcio.
V Proacelerina.
VII Proconvertina.
VIII Factor antihemofílico A..
IX Factor Christmas. Factor antihemofílico B.
X Factor Stuart-Prower.
XI Antecedentes plasmático de la tromboplastina. Factor antihemofílico.
XII Factor Hageman.
XII Factor estabilizador de la fibrina.
Tabla 5. Factores de la coagulación.
El mecanismo de la coagulación encargado de la formación de la fibrina implica una «cascada» de reacciones complejas. El punto central de la fase de la coagulación es la conversión del fibrinógeno (proteína plasmática soluble) en fibrina insoluble. Fi-siológicamente este cambio sólo puede efectuarse gracias a la acción enzimática altamente específica de la trombina, que no circula en condiciones normales, sino que debe ser generada a partir de su preenzima la protrombina, proteína vitamina K-dependiente que se forma de manera continuada en el hígado.
La protrombina puede ser activada por cualquiera de los mecanismos, extrínseco o intrínseco, que se describen a continuación, pero ambos son necesarios para el funcionamiento normal de la hemostasia. Es probable que la vía intrínseca tenga mayor importancia, ya que los trastornos hemorrágicos debidos a deficiencia de factores extrínsecos de la coagulación son menos pronunciados que los debidos a factores intrínsecos, sobre todo los factores VIII y IX.

31 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
El mecanismo extrínseco se inicia con la rotura de los vasos sanguíneos y la exposición de la sangre a los tejidos desgarrados que rodean al vaso. De estos tejidos lesionados hay dos factores, el factor hístico (tromboplastina hística) y los fosfolípidos hísticos, que inician rápidamente una reacción en el plasma sanguíneo que hace que los factores V, VII y X reaccionen conjuntamente bajo la influencia catalizadora de los iones calcio (Ca+).
El mecanismo intrínseco se inicia con la alteración de la propia sangre al contacto con el colágeno y sigue con la serie de reacciones en cascada en la que intervienen los factores XII, XI, IX, VIII a la vez que los factores X y V. En la cascada, unos factores van activando a otros hasta llegar a formar el activador de protrombina, producto final común a las dos vías, a través del cual es posible la conversión de protrombina en trombina, que cataliza el paso de fibrinógeno a fibrina, cuyos filamentos encierran a los eritrocitos, los leucocitos, las plaquetas y el plasma para formar un coágulo definitivo.
Figura 8. Cascada de la coagulación sanguínea. F3P: factor 3 plaquetario.
Por último, el factor XIII estabiliza la fibrina, y la contracción provocada por la actomiosina (proteína contráctil) de las plaquetas adheridas a los filamentos de fibrina produce la retracción del coágulo.
Fibrinolisis Es el conjunto de procesos fisiológicos que tienen como finalidad la destrucción del trombo de fibrina, una vez que ha cumplido su función hemostática.
Consiste en transformar la fibrina insoluble en soluble mediante la acción de la plasmina. Ésta se forma a partir de un precursor plasmático inactivo, el plasminógeno, que es sintetizado por el

32 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
hígado. La activación del plasminógeno es llevada a cabo por activadores de distintos orígenes: hísticos, plasmáticos, enzimáticos (estreptocinasa, urocinasa).
El plasminógeno se incorpora en el coágulo de fibrina y se convierte en plasmina por acción de los activadores anteriormente nombrados; se produce así una fibrinolisis local, «cortándose» el coágulo insoluble en fragmentos (productos de degradación de la fibrina, PDF) capaces de inhibir la trombina.
Si la plasmina entra en la circulación, rápidamente es neutralizada por los inhibidores circulantes que en condiciones normales se encuentran en el plasma; por lo tanto, la actividad proteolítica está limitada a los lugares donde se encuentran los depósitos de fibrina.
Inhibidores fisiológicos de la coagulación Con el fin de que la coagulación se limite sólo a la zona vascular lesionada y no se extienda a todo el sistema circulatorio, existen inhibidores plasmáticos que neutralizan los factores de la coagulación activados.
La antitrombina III (AT-III), sintetizada por el hígado es el inhibidor fisiológico más importante de la coagulación y el cofactor fundamental de la heparina, sin el cual ésta no actúa. La AT-III no sólo inhibe a la trombina sino también a los factores IX, X, XI y XII activados.
PARA SABER MÁS
Los individuos que carecen de ella presentan especial predisposición a las trombosis venosas.

33 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
Figura 9. Etapas y cronología de la coagulación.
2. Alteraciones de la coagulación Hay que distinguir entre los síndromes hemorrágicos debidos a anomalías en la hemostasia primaria y las alteraciones de la coagulación propiamente dichas. Los primeros se caracterizan por trastornos hemorrágicos en piel y mucosas (petequias o equimosis según sea la extensión), mientras que en los trastornos de la coagulación las hemorragias son musculares y articulares.
Dentro de las anomalías en la hemostasia primaria se incluye:
• Trombopenias (ya analizadas al tratar sobre las plaquetas). • Enfermedad de von Willebrand.

34 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
• Trombopatías congénitas o adquiridas: anomalías en la función de las plaquetas.
Entre los síndromes hemorrágicos debidos a anomalías en la coagulación destacan:
Patología adquirida de la coagulación: • Déficit en la síntesis de factores de coagulación (lesiones hepáticas, avitaminosis K, etc.). • Fibrinólisis aguda. • Exceso de consumo, como ocurre en la coagulación intravascular diseminada (CID):
activación del sistema enzimático de la coagulación que lleva a la peligrosa formación de múltiples coágulos de fibrina. Esta activación puede producirse por diversas causas, como infecciones, desprendimiento prematuro de placenta, cirrosis, accidentes transfusionales, etc.
Déficit congénitos de factores de la coagulación. Dos de ellos son responsables de las hemofilias: déficit de factor VIII o hemofilia A (85 %) y déficit de factor IX o hemofilia B (15 %). La transmisión y los síntomas clínicos son idénticos en ambos casos, pero los exámenes biológicos y, sobre todo, el tratamiento son distintos.
La hemofilia se transmite por herencia recesiva ligada al sexo. Las mujeres transmiten el defecto, es decir, son portadoras, pero sólo los varones padecen la enfermedad. Las manifestaciones hemorrágicas pueden ser desde moderadas hasta graves dependiendo de la severidad del déficit.
No hay que olvidar los problemas sociales y psíquicos que se producen no sólo en el enfermo sino también en la familia, pues son importantes y requieren gran atención.
3. Trombosis Se puede definir como «una coagulación sanguínea en un momento y un lugar no deseados». La diferencia entre hemostasia y trombosis reside en que la primera es un proceso fisiológico (origina el coágulo hemostático), mientras que la segunda es patológica (formación del trombo).
Figura 10. Fibrinólisis. PDF: productos de degradación de la fibrina.
El trombo tiene gran similitud en su formación y en su estructura con el coágulo hemostático, pero presenta importantes diferencias: el coágulo es en gran medida extravascular, mientras que el trombo es intravascular; el coágulo hemostático se inicia siempre por lesión vascular, mientras

35 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
que hay otros estímulos para la formación del trombo, como factores que provocan adhesión y agregación de plaquetas, trombocitosis o factores que activan el proceso de la coagulación.
Las trombosis tienen consecuencias locales y a distancia:
• Consecuencias locales: Trombosis arteriales: al interrumpir el flujo sanguíneo provocan isquemia que
puede conducir a necrosis de los tejidos. Trombosis venosas: si la sangre no puede ser transportada por las venas
colaterales, provoca edemas por aumento de la presión hidrostática en los capilares.
• Consecuencias a distancia. Si se desprende un fragmento de trombo se constituye un émbolo, que se
trasladará con la corriente sanguínea hasta impactar en un vaso a distancia causando, al igual que los trombos, isquemia.
Los émbolos de origen venoso se detienen en las arterias pulmonares, produciendo embolias pulmonares, mientras que los de origen arterial se detienen en las arterias de la circulación sistémica.
4. Anticoagulantes Para evitar la coagulación de la sangre extraída por punción venosa, se pueden añadir varios anticoagulantes: citrato sódico, oxalato sódico y EDTA (ácido etilendiaminotetraacético).
Estas soluciones forman una unión compleja con el calcio que, como ya se ha señalado, es imprescindible para la coagulación.
Un inhibidor directo (de acción inmediata) es la heparina, sustancia propia del organismo, que se halla en las granulaciones basófilas. La heparina actúa activando y reforzando el efecto de la AT-III.
Como anticoagulantes indirectos existen los cumarínicos (la cumarina es una sustancia extraída de cierto tipo de trébol); de ellos el más usado es el Sintrom®, que actúa de forma indirecta sobre la coagulación por bloqueo de la vitamina K.
5. Pruebas de coagulación La exploración global de la coagulación sanguínea puede realizarse mediante varias pruebas que miden el tiempo que tarda en coagular el plasma sanguíneo. No sirven para el diagnóstico del déficit de un factor en particular, pero dan una idea general sobre el estado de la vía intrínseca y de la vía común. Estas pruebas son:
Tiempo de hemorragia (o de sangría) Es el tiempo que tarda en cohibirse una hemorragia provocada por una pequeña incisión en la piel (en general en el lóbulo de la oreja).

36 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
Dado que es el coágulo provisional de plaquetas el que ocluye los pequeños vasos sangrantes, el tiempo de hemorragia sirve para la exploración tanto del número de plaquetas como de su eficiencia y también de la contractilidad capilar.
Los valores normales, con la técnica habitual (Duke) se encuentran entre 2-5 min.
Tiempo de coagulación Es el tiempo que tarda en coagular la sangre in vitro. Se consideran valores normales tiempos de 5-10 min.
Indica el estado de los factores plasmáticos que intervienen en el mecanismo de la coagulación (protrombina, fibrinógeno, etc.) o que la dificultan (antitrombina, etc.). Hay que señalar, sin embargo, que es una prueba tosca, poco sensible y de escaso valor, pues pueden existir valores normales en muchos casos en los que existe alteración de los factores de coagulación anteriormente mencionados.
Tiempo de protrombina (TP) Además de medir la protrombina (factor II), mide otros factores elaborados en el hígado que requieren, en su mayoría, vitamina K para su fabricación: factores V, VII y X. Por lo tanto, el TP permite medir la vía extrínseca y la vía común.
El TP se puede usar para detectar la falta de factores de coagulación a causa de una disfunción hepática o por ausencia de vitamina K.
Es la prueba única y específica empleada para medir la eficacia de los fármacos anticoagulantes cumarínicos (Sintrom®), aunque el empleo de heparina a grandes dosis también puede alterar el TP.
El TP suele expresarse en porcentaje del contenido normal de protrombina que corresponde al tiempo normal, considerándose normales cifras del 85-110 % de actividad. Cifras inferiores al 30 % provocan síntomas clínicos.
Varias alteraciones patológicas causan un alargamiento en el TP, entre las que se encuentran la cirrosis hepática y la CID.
También hay sustancias que producen un alargamiento del TP, como alcohol, salicilatos, antibióticos, etc.
Una disminución o acortamiento del TP puede asociarse a fármacos como anticonceptivos orales, digital, vitamina K. También puede estar acortado en algún tipo de neoplasia, pero no reviste significado clínico como factor diagnóstico.
Tiempo de tromboplastina parcial (TTP) También se utiliza una variante, que es el tiempo de tromboplastina parcial activado (TTPA), cuyos valores normales son de 25-38 seg.

37 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
Resulta de gran utilidad para detectar la presencia de trastornos hemorrágicos provocados por defecto o deficiencia de factores circulantes que componen el sistema intrínseco.
Otro objetivo del TTP es el control de pacientes en tratamiento con heparina, ya que ésta produce un alargamiento en el TTP. Sin embargo, una prolongación del TTP en un paciente que no está en tratamiento con heparina puede significar un trastorno hemorrágico (para saber qué factor es el deficitario se requiere efectuar otras pruebas más complejas).
No hay que olvidar que medicamentos como los antiagregantes plaquetarios (salicilatos, dipiridamol, etc.) producen un alargamiento del TTP.
También en la CID se prolonga el TTP.
Un TTP disminuido no reviste un significado diagnóstico, pero puede ser un índice de hipercoagulabilidad.
Tiempo de trombina (TT) Mide el tiempo que tarda la sangre en coagular cuando se le añade trombina a la muestra. Por lo tanto, explora el paso de fibrinógeno a fibrina. Si un coágulo no se forma inmediatamente, existe un déficit de fibrinógeno. También el tratamiento con heparina impide la coagulación de la sangre.
Este parámetro también se utiliza para el control de pacientes que reciben terapia fibrinolítica (estreptocinasa, urocinasa).
Los valores normales están en 20 ± 2 seg.
Fibrinógeno Proteína plasmática elaborada por el hígado, que no requiere la presencia de vitamina K para su formación. Su única función es, al parecer, la formación de fibrina como producto final de la coagulación.
Niveles bajos de fibrinógeno pueden ser consecuencia de trastornos genéticos o de una enfermedad hepática grave, pero en general la disminución de fibrinógeno se debe a una CID, también conocida como coagulopatía de consumo, pues es un sobreestímulo patológico del proceso de coagulación. Como ya se señaló al tratar la coagulación, la CID no es un trastorno primario, sino secundario a otras enfermedades graves: lesiones hísticas extensas sirven de estímulo para el mecanismo de la coagulación, originando la formación patológica de pequeños trombos en la microcirculación; paradójicamente, el paciente empieza a sangrar porque los factores de coagulación están agotados.
Además de encontrarse el fibrinógeno disminuido y el TP y el TTP alargados, el recuento plaquetario está descendido.

38 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
E. GRUPOS SANGUÍNEOS
1. Antígenos y anticuerpos de la membrana del hematíe Se pueden establecer diferentes grupos sanguíneos en función de los factores antigénicos que aparecen en la membrana del eritrocito. Existen, por tanto, numerosos sistemas de grupos sanguíneos, de los cuales los más importantes son:
• El sistema ABO, que permite diferenciar cuatro grupos sanguíneos: A, B, AB y O. • El sistema Rhesus, conocido como Rh.
En ambos casos se trata de antígenos fijados por la herencia (siguiendo las leyes de Mendel), que se encuentran de forma especial en la membrana de los eritrocitos pero que también se hallan en membranas celulares de otros tejidos: glándulas salivales, hígado, páncreas, semen, líquido amniótico.
Los antígenos eritrocitarios se denominan aglutinógenos, ya que con los anticuerpos correspondientes, las aglutininas, dan origen a la agrupación de los glóbulos rojos: aglutinación. Las aglutininas (anticuerpos) están en el plasma y pueden existir de forma natural, es decir, ser heredadas, sin que se haya tenido nunca contacto con otros grupos de sangre.
Cada persona nace con un tipo de aglutinógeno A, B, AB o O, y tiene, de forma natural, los anticuerpos frente al resto de los antígenos que no posee; por ejemplo, individuos que tengan sangre del grupo A (aglutinógeno A en los eritrocitos), siempre tienen un título de anticuerpos contra el aglutinógeno B: aglutinina anti-B. Por tanto, si su plasma se mezcla con células del grupo B (por ejemplo en una transfusión), las aglutininas antiB (del receptor) y los aglutinógenos B (del donante) reaccionan haciendo que se produzca una aglutinación y posteriormente una destrucción de los hematíes (hemólisis), lo que provoca una liberación de hemoglobina al torrente sanguíneo, que puede causar lesiones graves de los túbulos renales, insuficiencia renal e, incluso, la muerte.
Del mismo modo, los individuos del grupo B tienen aglutininas antiA; los sujetos del grupo O tienen aglutininas anti-A y anti-B, y los sujetos del grupo AB no tienen aglutininas circulantes, por lo que se los denomina receptores universales: pueden recibir sangre de todos los demás grupos ya que no poseen anticuerpos contra ninguno de ellos. Igualmente, a los individuos del grupo O se los denomina donantes universales, porque no poseen antígenos A ni antígenos B en sus hematíes y al confrontar, por ejemplo, a un receptor del grupo A (que posee aglutininas anti-B) con sangre de un donante del grupo O (que no tiene antígeno B) no se produce aglutinación .

39 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
Figura 10. Aglutinación de los hematíes en caso de incompatibilidad de grupos sanguí-neos.
En el sistema ABO, las aglutininas, además de poseerse de forma natural, también pueden ser formadas por exposición a los glóbulos rojos de otro individuo, ya sea a través de una transfusión o durante el parto, cuando algunos eritrocitos fetales cruzan la placenta y entran en la circulación materna. En cambio, en el sistema Rhesus o sistema Rh y otros sistemas (Kell, Lewis, Duffy, etc.), las aglutininas son producidas siempre por exposición a eritrocitos extraños.
2. Herencia de los antígenos A y B Los antígenos A y B son transmitidos como alelomorfos mendelianos y son dominantes, es decir, un individuo con sangre del grupo A, por ejemplo, puede haber heredado el antígeno A de cada progenitor o bien un antígeno A de uno de ellos y un antígeno O del otro progenitor. En otras palabras, un individuo cuyo fenotipo es A (recordemos que el fenotipo es la expresión de los caracteres que se manifiestan) puede tener dos genotipos (conjunto de todos los genes de dicho individuo): genotipo AA (homocigótico) o genotipo AO (heterocigótico). Por ello, cuando se conocen los grupos sanguíneos de los padres, pueden formularse los genotipos posibles de los hijos.
3. Sistema RH En realidad es un sistema compuesto por muchos antígenos (C, D, E), entre los cuales el antígeno D es con diferencia el más antigénico, y el término Rh-positivo (Rh+), como generalmente se usa, significa que el individuo posee aglutinógeno D.
El factor Rh debe su nombre a la investigación original de este factor en un mono, el macacus Rhesus. Se inyectaban eritrocitos de monos Rhesus a cobayas, y éstos reaccionaban formando anticuerpos, a los que se denominó anticuerpos Rhesus; con éstos se pueden aglutinar los glóbulos rojos de aproximadamente el 85 % de nuestra población, es decir, que el 85 % es Rh+, tiene antígeno Rh o antígeno D en sus hematíes y, por lo tanto, carece de anticuerpos (aglutininas anti-D) frente al factor Rh. El 15 % restante no posee antígeno Rh en sus hematíes, por lo que sólo desarrollarán anticuerpos frente al mismo si se produce una sensibilización por transfusión con sangre que contenga factor Rh.

40 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
POBLACIÓN DE EUROPA CENT. (%) GRUPO SANGUÍNEO AGLUTINÓGENO
(AG) AGLUTININAS (AC)
46 O Ninguno Anti-A y Anti-B
41 A A Anti-B
9 B B Anti-A
4 AB AB Ninguno
Tabla 6. Frecuencia en la distribución de grupos sanguíneos. Antígenos y anticuerpos presentes en cada uno de ellos.
Figura 11. Herencia de los grupos sanguíneos.
Cuando se inyectan hematíes que contienen factor Rh (Rh+) a una persona que no posee dicho factor en sus hematíes (Rh-), ésta irá desarrollando lentamente aglutininas anti-Rh, cuya máxima concentración se alcanza al cabo de 2-4 meses, con lo cual se dice que la persona Rh está sensibilizada al factor Rh; un posterior contacto con este antígeno (por ejemplo otra transfusión) provocará una reacción hemolítica porque los anticuerpos formados anteriormente atacarán a los hematíes Rh+ que se están administrando en ese momento. La gravedad de esta reacción hemolítica suele ser menor que la producida en la incompatibilidad ABO, pero es de gran importancia evitarla.
El factor Rh también ha de tenerse en cuenta en las mujeres embarazadas, ya que si la madre es Rh- y el padre Rh+, hay muchas posibilidades de que el niño sea Rh+ (85 %), lo cual puede plantear problemas. En el primer embarazo no ocurre nada; el feto no se afecta porque la madre no ha tenido tiempo de fabricar los anticuerpos anti-Rh (anti-D), pero durante el nacimiento o en un aborto, a través de la herida que se produce por el desprendimiento placentario, pasan hematíes fetales a la circulación materna y desencadenan la producción general de anticuerpos antiRh (anti-D). En posteriores embarazos de fetos Rh+, dichos anticuerpos pasan a la circulación fetal a través de la placenta y producen la aglutinación y hemólisis de los glóbulos rojos Rh+ del feto, con lo que éste sufre una falta de oxígeno o bien nace con una grave ictericia hemolítica o muere antes en el claustro materno. En el primer caso el niño nace extremadamente anémico a causa de la destrucción de los hematíes, además de con una intensa ictericia debida a la conversión de la hemoglobina liberada de los eritrocitos destruidos, en bilirrubina (que da el color amarillo a la piel).

41 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
Probablemente es necesaria una exaguinotransfusión, es decir, cambiar toda la sangre del niño por sangre Rh- que no contenga aglutininas anti-Rh.
Para evitar esta complicación, inmediatamente después del nacimiento, se administra a la madre inmunoglobulinas anti-Rh (es decir, anti cuerpos anti-D), cuya actuación se basa en el principio de la inmunidad pasiva. La inyección de inmunoglobulinas proporciona el incremento temporal de anticuerpos necesario para bloquear al antígeno proveniente del feto, de modo que la madre no es estimulada para iniciar la producción de sus propios anticuerpos.
4. Transfusión sanguínea Los modernos métodos de recogida de sangre, conservación, procesado y almacenamiento han conducido a una amplia disponibilidad de sangre y de sus componentes para la transfusión.
Donación de sangre En nuestro país las donaciones de sangre dependen exclusivamente de los donantes voluntarios. Éstos han de estar sanos, por lo que siempre hay que efectuar antes de la donación una breve exploración física: toma de tensión arterial y pulso, control de los niveles de hemoglobina, etc., y una historia de los antecedentes médicos ocurridos desde la última donación: si ha sufrido alguna enfermedad, intervención quirúrgica, si toma medicamentos, etc.
El donante pasa luego a la sala de extracción, donde se prepara asépticamente la zona de la venotomía (en general en la flexura del antebrazo, venas mediana cefálica o mediana basílica). La sangre es recogida en sistemas de plástico, que tienen adosadas otra serie de bolsas colaterales que permiten la posterior separación de los componentes sanguíneos, siempre mediante un sistema cerrado que evita las posibles contaminaciones bacterianas.
Se extraen aproximadamente 450 ml; cuando termina la donación, el donante debe permanecer en reposo durante un corto periodo (5-10 min) antes de incorporarse.
La mayoría de los individuos toleran bien la donación, pero algunas personas pueden presentar una reacción vasovagal con mareo, hipotensión, sudación, etc.
Existe un tipo de donación denominada donación autóloga, consistente en la donación de sangre de un paciente para su propio uso en una posterior intervención (no antes de 72 horas), lo cual ha llegado a ser una cuestión de interés a causa de la actual preocupación existente debido a las posibles enfermedades adquiridas a través de las transfusiones.
Otro tipo de donación es la hemaféresis, que consiste en una recogida selectiva de cualquiera de los componentes de la sangre mediante separadores celulares. Se usa para recoger leucocitos, plasma o plaquetas que son separados mediante centrifugación, recogiéndose la fracción deseada y devolviéndose el resto al donante. Este donante habrá de cumplir los requisitos expuestos anteriormente y, además, se le efectuará previamente un estudio completo de enfermedades transmisibles (hepatitis, SIDA, sífilis, etc.).
Se utilizan las venas antecubitales de ambos brazos; por una de ellas fluye la sangre, que será procesada en una unidad de separación celular, donde se recoge la fracción deseada, y el resto es reinfundido al donante a través de la otra vena antecubital.

42 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
Este proceso dura unas 2 horas o más hasta que se obtiene el volumen necesario del componente deseado.
Anticoagulantes y conservantes Desde el momento de la donación, dentro de las bolsas de plástico de recogida de sangre hay soluciones para evitar que se produzca la coagulación y también para aportar nutrientes a las células durante todo el almacenamiento.
Los anticoagulantes que contienen citrato inhiben los pasos dependientes del ión calcio en la vía de la coagulación; el almacenamiento a baja temperatura (1-5 °C) enlentece la glucólisis, mientras que la presencia de dextrosa permite la generación de ATP.
El uso de citrato-fosfato-dextrosa (CPD) como anticoagulante preserva los hematíes durante 21 días a una temperatura entre 1 y 5 °C. Si a este anticoagulante además se le añade adenina (CPD-A), se proporciona un sustrato para que los hematíes puedan sintetizar ATP, lo que permite un almacenamiento algo más prolongado.
Transfusión
1) Pruebas de compatibilidad (pruebas cruzadas)
El análisis que se realiza antes de cualquier transfusión incluye varios pasos para asegurar que los hematíes del donante son compatibles con el receptor. El primero y uno de los más importantes es la correcta identificación del paciente y de la muestra de su sangre; por ello, no se debe recoger petición alguna de transfusión que no haya sido correctamente rellenada, ni muestra de sangre alguna que no esté debidamente identificada con nombre, dos apellidos y habitación del paciente.
Cuando la muestra llega al laboratorio se debe verificar si existen registros antiguos del paciente que recojan el grupo sanguíneo y Rh y la historia transfusional (si ha presentado alguna vez reacciones transfusionales, etc.).
La muestra es analizada en cuanto al grupo ABO y Rh; se valora si existen en el suero del paciente anticuerpos irregulares y autoanticuerpos; también se efectúa un enfrentamiento de los glóbulos rojos del donante con el suero del receptor (prueba cruzada propiamente dicha). Con todo ello se detectará la mayor parte de los anticuerpos que pudieran existir en el suero del receptor dirigidos contra los glóbulos rojos del donante.
En la gran mayoría de casos el examen de anticuerpos será negativo y las pruebas cruzadas compatibles, pero si el examen de anticuerpos resulta positivo o las pruebas cruzadas incompatibles, hay que proceder a la identificación de las causas.
2) Sangre no contrastada
En el caso de una urgencia extremadamente grave, puede ser necesario transfundir sangre antes de haber terminado de realizar las pruebas de compatibilidad; en este caso, el médico se verá obligado a contrapesar la necesidad urgente de la transfusión con el riesgo de transfundir sangre incompatible.

43 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
Si se ha tenido tiempo de realizar el tipado ABO y Rh, el paciente debe recibir la sangre del tipo específico. Si no se ha realizado análisis, el paciente recibirá hematíes del grupo O y Rh (negativo) hasta que se estudie el grupo exacto; una vez determinado éste, se usará la sangre específica para continuar las transfusiones posteriores.
3) Administración de las transfusiones
• Identificación del donante y del receptor: reviste gran importancia para una transfusión segura identificar correctamente al receptor y la unidad de sangre del donante que se le va a administrar. Aunque la última comprobación se efectúa al lado de la cama del paciente, previamente se habrá desarrollado una serie de etapas desde el momento inicial de tipado de la sangre y las pruebas cruzadas de compatibilidad, en las que es necesaria una estricta observancia de la identidad tanto del paciente como del donante. La mayoría de las reacciones hemolíticas se deben a errores burocráticos responsables de que el paciente reciba una unidad de sangre incorrecta.
• Grosor de la aguja de perfusión: una aguja de calibre grueso (18 gauges) proporciona una velocidad de flujo adecuada para la sangre total o para sus componentes. Se pueden usar agujas de menor calibre, pero la transfusión será más lenta. Las agujas más finas (23 G) pueden producir hemólisis de los glóbulos rojos si la perfusión se realiza bajo presión (manguito de presión para acelerar la velocidad de goteo).
• Líquidos compatibles: el único líquido que se puede administrar con la sangre es suero fisiológico. Si fuera necesaria la dilución de un concentrado de hematíes (pues éste puede llegar a ser viscoso y, por consiguiente, enlentecer la velocidad de perfusión), se utilizará suero fisiológico para disminuir la viscosidad. Las soluciones que contienen calcio (por ejemplo Ringer lactato) no deben añadirse a ningún producto sanguíneo, ya que pueden producir coagulación. Las soluciones que contengan glucosa (o dextrosa) deben evitarse ya que forman grumos de hematíes. En general no deben mezclarse con la sangre otras soluciones intravenosas ni medicamentos.
• Velocidad de infusión: dependerá de la volemia del paciente y del estado del corazón. Si se requiere administrar rápidamente una transfusión, una aguja de grueso calibre y un aparato externo de presión pueden aumentar la velocidad, pero el tiempo de transfusión rutinario de una unidad es de aproximadamente 2-3 horas; de cualquier modo, nunca debe durar más de 4 horas debido a la posibilidad de crecimiento bacteriano cuando la unidad permanece mucho tiempo a temperatura ambiente. Si el paciente no puede tolerar bien el aporte de un volumen extra, la transfusión debe ser lenta, pudiéndose dispensar la unidad de sangre en varias fracciones
• Cuidados al paciente: antes de llevar a cabo la transfusión se deben tomar las constantes basales al paciente. La enfermera que la administre debe permanecer con el paciente durante el período inicial para asegurarse de que no se producen reacciones adversas. Durante la transfusión y una vez concluida se debe preguntar al paciente acerca de posibles síntomas de reacción transfusional: dificultad respiratoria, náuseas, dolor abdominal, etc.

44 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
4) Calentamiento de la sangre
La transfusión rápida de grandes volúmenes de sangre fría (conservada en refrigerador a una temperatura de 1-5 “C) provoca un descenso de la temperatura central del paciente y puede ocasionar arritmias cardíacas. Por ello, se pueden evitar problemas si la sangre se calienta previamente a 37 “C mediante calentadores mecánicos.
Es importante controlar que la temperatura no exceda de 37°C pues por encima de esta temperatura la sangre puede hemolizarse.
5) Filtros
La sangre y sus componentes (plasma y plaquetas) siempre han de administrarse mediante un equipo de perfusión dotado de un filtro de malla que sea capaz de atrapar los coágulos que hayan podido formarse durante el almacenamiento de la unidad.
También existen otros filtros (denominados de microagregados) que retienen los pequeños residuos, células degeneradas y la fibrina. Suele usarse cuando se transfunden grandes volúmenes de sangre o bien si el paciente ha presentado con anterioridad algún tipo de problema transfusional, aunque sea mínimo: escalofríos, tiritona, etc.
Productos sanguíneos a transfundir
1) Sangre total
Se denomina como tal al producto de una sangría de 450 ml a la que se le han añadido 63 ml de conservante. Se almacena en nevera controlada a 1-5 °C y tiene hematocrito medio del 40 %.
Los factores de coagulación lábiles (factores V y VIII) y las plaquetas disminuyen durante el almacenamiento y los niveles de los factores estables (factores II, VII, IX, X y XI) se mantienen.
La sangre total se puede emplear siempre que el paciente sangre activamente, como sucede en las hemorragias masivas agudas (traumatismos por accidentes o hemorragias gastrointestinales). Pero hay que tener en cuenta que las plaquetas y los niveles de los factores de coagulación disminuyen durante el almacenamiento, por lo que un paciente que haya sido transfundido masivamente (transfusión masiva = transfusión de un volumen igual a la volemia del paciente en un tiempo de 24 horas), si recibe sangre almacenada de más de 5 días puede desarrollar una trombocitopenia o un déficit de factor VIII. También hay que vigilar la disminución de calcio ionizado, pues pueden llegar a producirse alteraciones electrocardiográficas y, si es necesario, hay que administrar sales de calcio (gluconato cálcico).
2) Concentrado de hematíes
Son preparados a partir de sangre total, mediante centrifugación; la cantidad resultante es de aproximadamente 250-300 ml con un hematocrito del 70-80 %.
Los glóbulos rojos de los concentrados de hematíes restauran la capacidad de transporte de oxígeno en los pacientes con anemia debida a pérdida de sangre de forma aguda o crónica, por

45 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
hemólisis, por insuficiencia de la médula ósea o por eritropoyesis ineficaz, y también son el producto de elección en los pacientes que, precisando hematíes, están normovolémicos.
El propósito de la transfusión es mejorar el estado del paciente, al aliviar los síntomas provocados por la anemia (debilidad, sensación de falta de aire al respirar, etc.) y restaurar la capacidad de transporte de oxígeno de los glóbulos rojos.
Como orientación cabe decir que por cada unidad transfundida se debe esperar un aumento del 3 % en el hematocrito y de 1 g/dl en la hemoglobina.
3) Plaquetas
Los concentrados de plaquetas consisten en plasma rico en plaquetas, separado de la sangre dentro de las 6 horas siguientes a su extracción. Se obtiene mediante centrifugación de la sangre total y el tiempo de conservación oscila alrededor de 3-5 días.
Las plaquetas se usan en el tratamiento de las hemorragias que cursan con trombopenia, en pacientes con insuficiencia de la médula ósea o en los que han sufrido una transfusión masiva para contrarrestar la trombopeniadilucional que se produce.
SANGRE TOTAL
CONCENTRADO DE HEMATÍES
Volumen total 500 ml 300 ml
Volumen de hematíes 200 ml 200 ml
Volumen de plasma 300 ml 100 ml
Hematócrito 40% 70%
Tabla 7. Sangre total y concentrado de hematíes.
El propósito de este tipo de transfusión es la prevención o la detención de una hemorragia, pero evitando la sensibilización. Dado que los antígenos ABO se expresan sobre las plaquetas, habrá que procurar siempre transfundir plaquetas compatibles en cuanto a dicho sistema.
4) Plasma fresco congelado
Se obtiene mediante separación del plasma de la sangre total (un volumen aproximado de 200-250 ml) y congelación antes de transcurridas 6 horas de su recogida.
Contiene niveles óptimos de factores de la coagulación, tanto de los lábiles (factores V y VIII) como de los estables (factores II, VII, IX y X).
Las condiciones de almacenamiento son a 18 °C o incluso menos y tienen una caducidad de 12 meses. Para poder utilizarlo debe descongelarse a una temperatura de 30-37 °C y transfundirse antes de transcurridas 12 horas desde su descongelación.
Siempre que sea posible, los pacientes deben recibir plasma compatible en cuanto al grupo sanguíneo.

46 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
5) Crioprecipitados
Se preparan a partir del plasma congelado dentro de las 6 horas siguientes a la extracción, congelándolo a 70 °C y dejándolo descongelar a 4 °C. El precipitado que se forma a modo de copos blancos es rico en factor VIII y fibrinógeno; puede conservarse a ,30 °C durante 12 meses.
6) Concentrado de leucocitos
Hay que señalar que se trata de una técnica sustitutiva poco corriente que requiere instalaciones muy especiales y que sólo tiene indicaciones limitadas a ciertos síndromes hematológicos graves.
Los leucocitos se recogen por aféresis y, como suele haber contaminación con glóbulos rojos, el producto ha de ser compatible con el receptor en cuanto a los sistemas ABO y Rh.
Dado que los granulocitos poseen una vida corta, las células deben ser transfundidas inmediatamente después de su recogida.
Pueden aplicarse a pacientes neutropénicos con infecciones que no mejoran con antibióticos, pero siempre debe sopesarse el beneficio de la transfusión de leucocitos con la posibilidad de reacciones pulmonares, infecciones por citomegalovirus y sensibilizaciones.
7) Concentrados de factores de coagulación
Se presentan como factores liofilizados y están indicados en pacientes con deficiencias congénitas de factores de la coagulación.
El concentrado de factor VIII es un preparado liofilizado procedente del plasma fresco congelado; contiene gran cantidad de factor VIII, junto con pequeñas cantidades de fibrinógeno y otras proteínas. Se utiliza en los pacientes con hemofilia A.
Los concentrados de factor IX se usan especialmente para el tratamiento de la hemofilia B. También es un preparado liofilizado que, además, contiene factores II, VII y X en forma concentrada.
8) Expansores del volumen
Los sustitutos del plasma preparados a partir de masas comunes de plasma humano son una fuente de volumen y de coloides. Entre ellos cabe citar:
• Albúmina, extraída de un pool de plasma y usada en el tratamiento de shock, quemaduras, cirugía, etc.
• Factor proteico del plasma (FPP), que contiene proteínas plasmáticas, albúmina y globulinas a y P. Se emplea en el tratamiento del shock y como fuente de proteínas por su efecto osmótico.
Las soluciones coloidales de productos sintéticos macromoleculares (no contienen plasma) que se utilizan como expansores del volumen sanguíneo no son tan efectivas como la albúmina; entre ellas se encuentran el dextrano 70 (Macrodex®) y el dextrano 40 (Rheomacrodex).

47 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
9) Inmunoglobulinas (Ig)
Son soluciones estériles de gammaglobulina específicas para ciertos anticuerpos, que son preparadas a partir de plasma. Se administran a través de inyecciones intramusculares. Algunas Ig utilizadas con frecuencia son:
• Ig anti-Rh. Se obtiene a partir de individuos Rh- que han producido anticuerpos anti-Rh (anti-D) por inmunización. Se utiliza para prevenir la sensibilización frente al antígeno Rh (antígeno D) en las mujeres Rh- que han tenido un aborto o un niño Rh+.
• Igantihepatitis B. Es un preparado de Ig sérica obtenida por fraccionamiento de plasma de donantes que posean un título elevado de anticuerpos contra el antígeno de superficie de la hepatitis B (AgHBs). La administración de esta Igantihepatitis B está indicada después de la exposición del sujeto a material infectado por el AgHBs. Para que resulte efectiva ha de ser administrada dentro de las 48 horas siguientes a la exposición. También está indicada en los recién nacidos cuyas madres son portadoras del AgHBs, pues se ha demostrado que esta medida reduce la incidencia de hepatitis neonatal.
• Ig antitetánica. Es la fracción de Ig sérica obtenida de individuos que han sido específicamente inmunizados con toxoide tetánico. Se aplica en la profilaxis de sujetos que presentan un riesgo elevado de contraer la enfermedad después de haber sufrido una herida potencialmente tetanígena y que no se hallan correctamente vacunados frente al tétanos.
• Igantivaricela zoster. Contiene anticuerpos contra la varicela zoster obtenidos del plasma de individuos recientemente afectados de herpes zoster. La administración de dichos anticuerpos específicos dentro de las 72 horas siguientes a la exposición, antes de que aparezcan los signos de la enfermedad, reduce la morbimortalidad de la varicela en los niños inmunodeficientes.
Reacciones adversas transfusionales Aunque la mayoría de las transfusiones son beneficiosas y seguras, hay pacientes que pueden experimentar algún tipo de reacción adversa frente a la sangre o a sus derivados. Estos efectos pueden llegar a ser graves y requieren una identificación inmediata y un tratamiento rápido.
1) Reacciones adversas inmediatas
REACCIÓN HEMOLÍTICA INMUNOMEDIADA La reacción hemolítica debida a una reacción inmunológica se inicia con reacciones antígeno-anticuerpo (Ag-Ac). En general se produce después de la administración de sangre ABO incompatible. La causa de este accidente con frecuencia es la incorrecta identificación del paciente o de su muestra de sangre.
El paciente suele quejarse de fiebre, disnea, dolor en el lugar de la perfusión, dolor lumbar. Entre los signos físicos se pueden observar hipotensión y hemoglobinuria, que en ocasiones evolucionan hacia una insuficiencia renal e incluso hacia la muerte. Si el paciente se encuentra anestesiado, el único signo que se observa es la hipotensión.

48 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
Ante cualquiera de estos síntomas se debe interrumpir la transfusión, pero manteniendo la vía con una perfusión de suero fisiológico, y avisar rápidamente al hematólogo. Hay que verificar al lado de la cama la etiqueta de la unidad de sangre y la identificación del paciente para comprobar si se ha cometido un error burocrático.
Se debe enviar al laboratorio una muestra de sangre del paciente junto con la unidad de sangre que se le estaba administrando, para realizar las pruebas pertinentes.
El tratamiento de las reacciones hemolíticas se basa en cuidados de mantenimiento hasta que se produzca la recuperación de las secuelas. Se deben mantener la presión sanguínea y la diuresis.
Otras causas no inmunes de hemólisis son los traumatismos mecánicos de los glóbulos rojos, bien por exceso de calor, perfusión con agujas de pequeño calibre o a presión, etc.
También se puede producir una lisis osmótica por contacto de los hematíes con soluciones hipotónicas.
REACCIÓN NO HEMOLÍTICA FEBRIL Cuando la temperatura del paciente aumenta en 1°C o más en asociación con la transfusión. También puede experimentar escalofríos intensos. Suele ocurrir sobre todo en pacientes politransfundidos o en mujeres que han tenido varios embarazos.
Al parecer, el mecanismo de producción de la reacción febril reside en que los anticuerpos del receptor reaccionan con los leucocitos o con antígenos plaquetarios del donante. Por ello, en transfusiones posteriores se debe utilizar sangre desleucotizada (con pocos leucocitos), lo cual se consigue por medio de la perfusión a través de filtros especiales que los retienen.
REACCIÓN ANAFILÁCTICA La característica notable de esta reacción es su inicio súbito tras la administración de unos cuantos mililitros de la transfusión.
Puede presentarse dificultad respiratoria, hipotensión, náuseas, dolor abdominal, vómitos, shock o pérdida de conciencia. Este tipo de reacción requiere siempre una actuación terapéutica rápida y eficiente.
Como en toda reacción anafiláctica, para corregir la hipotensión se administrarán líquidos intravenosos; también puede ser necesaria la administración de adrenalina (subcutánea o intravenosa) y corticoesteroides (intravenosos), que ayudan a mejorar el estado clínico.
Estas reacciones suelen producirse en pacientes con deficiencia de un tipo de inmunoglobulina (IgA) y cuyo suero contiene anticuerpos específicos anti-IgA, los cuales producen la reacción al entrar en contacto con la sangre transfundida que contiene cantidades normales de IgA.
REACCIONES ALÉRGICAS Asociadas a las transfusiones pueden producirse reacciones cutáneas limitadas, caracterizadas por prurito y exantema. Si éstos son controlados y no hay otros síntomas, se puede continuar con la transfusión.

49 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
Se cree que la reacción se debe a la presencia de alérgenos en el plasma donante que interaccionan con anticuerpos del receptor.
CONTAMINACIÓN BACTERIANA Una pequeña proporción de unidades de sangre o de componentes sanguíneos (3 %o) están contaminados con una pequeña cantidad de bacterias, lo cual no suele constituir problemas ya que la conservación de la mayor parte de los productos sanguíneos se realiza a temperaturas que inhiben el crecimiento de los microorganismos; no obstante, hay algunas especies de bacterias (ciertas cepas de Pseudomonas) que crecen a bajas temperaturas y pueden hallarse en grandes cantidades en el momento de la transfusión de la unidad de sangre o del derivado, causando una grave reacción en el receptor.
Los concentrados de plaquetas corren mayor riesgo de contaminación ya que deben mantenerse a 22 °C, lo que favorece el crecimiento de muchas especies bacterianas.
Las reacciones que provocan los productos infectados son fiebre, escalofríos, hipotensión, shock e incluso la muerte, lo cual puede llevar a confusión con un shock séptico. También pueden aparecer dolores musculares, calambres, vómitos, etc.
La contaminación es a menudo grave y requiere un tratamiento inmediato con antibióticos y corticoesteroides.
Para su prevención se recomienda utilizar una técnica aséptica para iniciar la transfusión; colgar la unidad de sangre en los primeros 30 min después de salir del banco de sangre y comprobar que se transfunde toda la unidad en un período no superior a 4 horas.
EMBOLIA GASEOSA Con el empleo de bolsas de plástico en la preparación y administración de sangre y derivados se ha eliminado la incidencia de las embolias gaseosas, pero con el objeto de evitar este tipo de accidentes nunca debe introducirse aire dentro de las bolsas ni del sistema de filtro.
2) Enfermedades infecciosas
Algunos individuos sanos poseen agentes infecciosos en su circulación, y la transfusión de su sangre puede dar como resultado la transmisión de la infección al receptor. Entre las enfermedades que se pueden transmitir destacan: hepatitis, SIDA, mononucleosis infecciosa, sífilis, infecciones bacterianas e infecciones por citomegalovirus.
HEPATITIS Es una complicación grave. Aunque la sangre del donante se analiza en cuanto al antígeno de superficie de la hepatitis B (AgHBs), persisten algunos casos asociados a la transfusión, pero el responsable de casi todos los casos de hepatitis no es el virus B sino el virus no A-no B (hepatitis C).
El cuadro clínico es variable; algunos pacientes presentan, semanas o meses después de la transfusión, náuseas, debilidad, dolores articulares y malestar, que revierten en un par de semanas; un pequeño porcentaje de enfermos, sin embargo, termina con una hepatopatía crónica.

50 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
SIDA El agente causal es un virus (retrovirus) que afecta el sistema inmunitario, incapacitando al huésped para la respuesta inmunológica normal. Se incluyen análisis para la detección de los anticuerpos frente al HIV (virus de la inmunodeficiencia humana), lo que ha proporcionado un incremento en la seguridad del aporte de la sangre y sus derivados.
3) Otras complicaciones
HIPERVOLEMIA Cuando un paciente recibe grandes cantidades de sangre y otros líquidos, puede producirse una sobrecarga circulatoria, que suele manifestarse con tos no productiva, taquicardia, disnea, cianosis y edema pulmonar. Si esto ocurriera, el tratamiento urgente consiste en interrumpir la transfusión y administrar oxígeno y, en ocasiones, incluso puede llegar a ser necesaria la práctica de una sangría terapéutica.
Para prevenir esta complicación, a los pacientes con insuficiencia cardíaca congestiva, edemas o trastornos de la función renal, si es necesario transfundirles y no son capaces de tolerar el volumen añadido, se utilizarán siempre concentrados de hematíes. Además, la transfusión debe efectuarse lentamente, con lo cual puede ser necesario dispensarla en fracciones, para así evitar una posible contaminación bacteriana.
SOBRECARGA DE HIERRO Los pacientes que reciben transfusiones de forma crónica (betatalasemias, anemias aplásicas) corren el riesgo de presentar una sobrecarga de hierro.
El hierro excedente se acumula en hígado, corazón y glándulas endocrinas (páncreas), produciéndose una disminución de su función: cirrosis hepática, insuficiencia cardíaca, diabetes, etc.
Los signos asociados a la transfusión pueden ser arritmias cardíacas o insuficiencia cardíaca; disfunción hepática y cirrosis, anomalías en la prueba tolerancia a la glucosa y diabetes.
TOXICIDAD POR CITRATO E HIPOCALCEMIA Es una complicación que puede producirse en casos de transfusión masiva o en recién nacidos sometidos a una exanguinotransfusión. El citrato presente como anticoagulante en la transfusión es metabolizado normalmente por el hígado, pero si la función hepática se encuentra alterada o sobrecargada, el citrato liga el calcio ionizado, produciéndose una disminución en sus niveles plasmáticos, que puede causar temblores musculares involuntarios o disfunción cardíaca.
La toxicidad del citrato se contrarresta con la administración intravenosa de gluconato cálcico.
HEMORRAGIA DEBIDA A LA DILUCIÓN DE LOS FACTORES DE COAGULACIÓN Los pacientes que reciben grandes volúmenes de sangre pueden presentar trastornos hemorrágicos debidos a la dilución de los factores de coagulación y las plaquetas, pues la sangre conservada contiene plaquetas que ya no son funcionales y niveles bajos de factores V y VIII.

51 | Hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
Esta complicación puede prevenirse administrando al paciente que recibe transfusiones masivas, concentrados de plaquetas, crioprecipitados y plasma fresco congelado.
HIPOTERMIA Como ya se ha señalado, las unidades de sangre son conservadas a 1-5 °C, por lo que la inyección rápida de grandes volúmenes de sangre fría pueden producir hipotermia, lo cual hace que aumente la afinidad de la hemoglobina por el oxígeno, con la consiguiente disminución de la liberación de dicho oxígeno en los tejidos. También pueden producirse arritmias cardíacas.
Por ello, si la sangre ha de ser transfundida con rapidez, debe conectarse a un calentador eléctrico que acondicione la temperatura de la sangre entre 30-37 °C antes de penetrar en el individuo.
HIPERPOTASEMIA El potasio sale de los hematíes durante el período de almacenamiento, pero en general esto no constituye un problema, a menos que el paciente presente hiperpotasemia antes de la transfusión.
MICROAGREGADOS También durante el período de almacenamiento los leucocitos y las plaquetas forman agregados microscópicos o microagregados, que son capaces de atravesar los filtros estándar utilizados en la perfusión sanguínea y causar reacciones adversas.
Se ha demostrado que las reacciones transfusionales relacionadas con los microagregados son variadas y de gran amplitud; sus efectos sobre el paciente dependerán de la cantidad de la infusión de microagregados y del estado del paciente.
Se han sugerido varios métodos para evitar los problemas relacionados con la formación de microagregados en sangre almacenada. Entre ellos se encuentra el microfiltrado, técnica no invasiva y de fácil uso que elimina eficazmente los coágulos.

52 | Patología en hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
II. PATOLOGÍA EN HEMATOLOGÍA
A. TRASTORNOS HEMORRÁGICOS
1. Evaluación de la hemostasia y pruebas diagnósticas Para el correcto enfoque diagnóstico de los trastornos de la coagulación es fundamental la realización de una buena anamnesis y exploración física minuciosa. Los datos clínicos, signos y síntomas, antecedentes personales hemorrágicos, o historia familiar de coagulopatías pueden resultar de gran ayuda para un primer enfoque diagnóstico.
Además también existe una completa batería de analíticas y pruebas complementarias, que permiten conocer el alcance y la gravedad de la enfermedad.
Anamnesis y exploración física La existencia de antecedentes familiares de enfermedades hemorrágicas (Hemofilia, Enfermedad de von Willebrand), puede orientar hacia trastornos hereditarios.
Se debe indagar acerca de los antecedentes personales de otros fenómenos hemorrágicos relacionados con intervenciones quirúrgicas, extracciones dentarias, partos, hemorragias mucosas espontáneas, existencia de hematomas o equimosis frecuentes…etc.
La edad de presentación del trastorno y su relación con otras enfermedades (infecciones, colagenosis, enfermedades hematológicas), toma de fármacos (aspirina, dicumarínicos), también puede ayudarnos en la búsqueda de la etiología.
Los trastornos de la hemostasia primaria: vasos y plaquetas, se manifiestan con clínica hemorrágica diferente de las coagulopatías por defectos de proteínas plasmáticas (hemostasia secundaria).
Así, en los trastornos plaquetarios la hemorragia suele ser inmediata (en los primeros minutos) y su localización más frecuente es en piel y mucosas: púrpuras, equimosis, epistaxis, gingivorragias (tras extracciones dentarias), hematuria.
Cuando se trata de coagulopatías por déficit de factores de la coagulación, la hemorragia puede tardar horas o incluso días en aparecer. La hemorragia suele afectar a articulaciones, músculos, órganos internos, y son de mayor cuantía generalmente.
La exploración física detallada es también muy importante, dependiendo del lugar y forma de sangrado podemos encontrar:
• Púrpura: Se trata de una hemorragia cutánea que se denominan petequias si son puntiformes y de pocos milímetros de diámetro, y equimosis si son de mayor tamaño (cardenales). Los hematomas son colecciones cutáneas palpables que afectan al tejido celular subcutáneo. Éstas no desaparecen con la vitropresión.

53 | Patología en hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
• Telangiectasias: dilataciones vasculares cutáneas en forma de pequeñas arañas con vitropresión positiva.
• Hemartrosis: hemorragia articular. Siempre tiene carácter patológico, e indica gravedad. -Mucosas: gingivorragias (encías), epistaxis (nariz), menorragia (uterina), hematuria (orina), rectorragias (sangre roja en las heces), hematemesis (vomitar sangre), hemoptisis (esputo con sangre). Estas hemorragias pueden obedecer a trastornos locales: cálculos, infecciones, úlceras pépticas, tumores… con lo que siempre hay que descartar estos diagnósticos en primer lugar.
Exploraciones complementarias
1) Pruebas básicas que pueden realizarse en urgencias
• Hemograma y recuento plaquetario: el número normal de plaquetas oscila entre 150-400 x 109/l. Recuentos mayores de 50 x 109 /l no suelen plantear problemas hemorrágicos.
• Morfología de plaquetas: para ello se solicita un frotis de sangre periférica. Sirve para descartar microagregados en las pseudotrombopenias, volumen aumentado en Bernard-Soulier, o síndromes mielodisplásicos.
• TTPA: Tiempo de tromboplastina parcial activado o de cefalina. Entre 25-35 segundos. Evalúa la integridad de la vía intrínseca y vía común (XII, XI, IX, VIII, X, V, II, I). Se altera por la acción de la heparina.
• TP (tiempo de protrombina) o Índice de Quick: Normal entre 10-15 seg. Valora la integridad de la vía extrínseca y común: VII, X, V, II, I. Aumenta por la acción de los anticoagulantes orales. Se ha establecido un parámetro normalizado para el control del tratamiento anticoagulante con dicumarínicos: INR, que permite comparar resultados de los diferentes laboratorios con reactivos distintos.
• TT (Tiempo de trombina). Permite explorar la cantidad y calidad de la fibrina y fibrinógeno. Oscila entre 20-30 seg.
• Determinación del Fibrinógeno (Método de Clauss): valora el Fibrinógeno funcional. Valores normales entre 2-4 g/l.
• Determinación del Dímero-D: Son productos de degradación del Fibrinógeno. Aumentan en estados de hiperactivación de la coagulación como CID, Tromboembolismos, hiperfibrinolisis.
2) Pruebas específicas programadas
• Trombopatías: Tiempo de hemorragia de “Ivy”: mide el tiempo en minutos (normal menor de 9
min.) que tarda en coagular una pequeña incisión con una microlanceta en el antebrazo al que se le aplica un manguito de presión. Valora la hemostasia primaria (trombopatías, Enfermedad de von Willebrand)
Estudio de agregación plaquetaria: con diferente agentes agregantes: ristocetina, ADP, colágeno. Se altera en ingesta de AAS, enfermedad de von Willebrand, Síndrome de Bernard-Soulier, trombastenia de Glanzmann.

54 | Patología en hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
Valoración del antígeno FvW: Mediante técnicas inmunológicas como ELISA, o RIA. • Coagulopatías:
Dosificación de la actividad funcional de los factores de la vía extrínseca (II, V, VII, X) e intrínseca (VIII, IX, XI, XII).
Valoración cualitativa del Factor XIII. Anticoagulantes circulantes: anticoagulante lúpico, inhibidores específicos del
factor VIII.
3) Otras pruebas
Métodos de Biología Molecular para detección de mutaciones en coagulopatías hereditarias: Mujeres portadoras, identificación de familiares asintomáticos.
TP TTPA TT DIAGNOSTICO
Normal Normal Normal Coagulación conservada. Sin síntomas hemorrágicos: Cuantificar Factor XIII, Factor von Willebrand, Pruebas de función plaquetaria.
Aumentado Normal Normal
Tratamiento con anticoagulantes orales. Déficit de factor VII. Déficit moderado de factores de la vía extrínseca: II, V, VII, X.
Normal Aumentado Normal
Muestra con Heparina/Tratamiento con Heparina. Anticoagulante lúpico. Alteración vía intrínseca: VIII, IX, XI, XII, precalicreína, cininógeno. Enf. Von Willebrand. Inhibidor específico.
Aumentado Aumentado Normal
Déficit aislado de II, V o X (vía común) o inhibidor específico. Déficit de vitamina K, Hepatópatas, Anticoagulantes orales. Síndrome hemorrágico del Recién Nacido.
Aumentado Aumentado Aumentado Hepatopatía severa, CID, Fibrinolisis sistemática, hipo o disfibrinogenemia.
Tabla 2. Resultados de las pruebas básicas de la coagulación y orientación diagnóstica.
2. Alteraciones de las plaquetas: trombopenias y trombopatías Trombopenias La cifra normal de plaquetas en un individuo sano oscila entre 150-400 x 109/l. Se define como trombopenia cifras inferiores a 150 x 109/l.
Los pacientes con recuentos mayores de 100 x 109/l plaquetas son asintomáticos y no poseen alteración del Tiempo de hemorragia. Entre 50-100 x 109/l, existe una pequeña alteración en el tiempo de hemorragia, sin embargo permanecen asintomáticos.

55 | Patología en hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
Según su etiología las trombopenias se clasifican en:
• Defectos en la producción: Debidas a fallo medular primario: aplasia medular, púrpura amegacariocítica
primaria, anemia de Fanconi, iatrogenia por quimioterapia o radioterapia. Infiltración de la médula ósea: Metástasis tumores sólidos, Leucemias, linfomas,
Fibrosis. • Aumento de la destrucción:
Destrucción no inmune: – Secuestro esplénico: Hipertensión portal, hipertrofia esplénica. – Prótesis valvulares cardiacas. – Vasculitis – Destrucción microangiopática: CID , Síndrome hemolítico urémico (SHU),
Púrpura trombótica trombocitopénica (PTT) – Hemangiomas gigantes: síndrome de Kasabach-Merrit
Destrucción inmune: TROMBOPENIAS INMUNES – Por autoanticuerpos frente a antígenos plaquetarios: Púrpura
Trombocitopénica Inmune (PTI), Púrpura transfusional, Isoinmunización neonatal.
– Anticuerpos frente a fármacos: Heparina, penicilinas, quinina, digoxina, valproato, interferon.
Enfermedades linfoproliferativas: Leucemia linfática crónica, Linfomas. Colagenosis: Lupus eritematosos sistémico, síndrome de Evans. Infecciones: virus, bacterias, sepsis.
SUPRESIÓN DE LA PRODUCCIÓN DESTRUCCIÓN INMUNE
Citarabina, Daunorrubicina, Ciclofosfamida, Busulfán, Metotrexate, Triacidas, Etanol, Estrógenos.
Heparina, Quinina, Quinidina, Sales de Oro, Indometacina, Sulfamidas, Rifampicina, Penicilina, AAS, Valproato, Carbamacepina, Carbamacepina, Arsénico, Cimetidina, Ranitidina, Furosemida, Tiacidas, Alfametildopa, Intraferon, Digoxina.
Tabla 3. Fármacos que pueden producir tombopenia por distintos mecanismos.
Trombopenias hereditarias Ante pacientes con trombopenias catalogadas de PTI que no responden al tratamiento esteroideo o esplenectomía, y con inicio en épocas tempranas, debe sospecharse la existencia de una trombopatía hereditaria.
Normalmente cursan con trombopenia leves o moderadas, raramente inferiores a 30 x 109/l, pero presentan historias de sangrado poco concordantes con las cifras de plaquetas.

56 | Patología en hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
Síndrome de Wiskott-Aldrich: Herencia recesiva ligada al cromosoma X. Cursa con deficiencia inmune en la maduración de los linfocitos y disminución de la vida media plaquetaria y agregación anormal. Asocia eczema e infecciones de repetición.
Síndrome de Bernard-Soulier: Herencia autosómica recesiva (AR). (Se estudiará en el apartado de trombopatías hereditarias)
Anomalía de May-Hegglin: Herencia autosómica dominante (AD). Asocia presencia de plaquetas gigantes con inclusiones azules (cuerpos de Döhle). No suelen necesitar tratamiento ya que raramente son sintomáticos.
Síndrome de Chediak-Higashi: Herencia AR. Trombopenia moderada con alteración de la agregación. Asocia albinismo, infecciones recurrentes, defecto en el almacenamiento de gránulos plaquetarios y grandes inclusiones leucocitarias
Púrpura trombopénica inmune (PTI) La PTI o Enfermedad de Werlhof es una trombopenia inmune idiopática producida por la adhesión de autoanticuerpos a la membrana de la plaqueta.
En su etiopatogenia intervienen la producción de autoanticuerpos que recubren las plaquetas, las cuales son captadas por el sistema mononuclear fagocítico y destruidas en su mayor parte por el bazo. Como respuesta compensatoria en la médula de estos pacientes se observa una hiperplasia de los megacariocitos.
1) Clínica y exploración Física
Es una enfermedad más frecuente en la infancia, apareciendo en muchas ocasiones tras una infección viral. En los niños acontece como un cuadro agudo con cifras muy baja de
plaquetas, y una recuperación en más de la mitad de los casos en cuatro a seis semanas, y más del 90% en los siguientes tres o seis meses. Menos del 10% de los casos aparece en pacientes adultos, mujeres en proporción 3:1, en estos casos suele cursar de forma crónica con recuentos variables.
Clínicamente puede cursar con sangrados cutáneo mucosos espontáneos como epistaxis, hematomas, gingivorragias (púrpura húmeda), pero también pueden ser asintomáticas (púrpura seca).
2) Datos de laboratorio
• Descenso moderado a grave del recuento plaquetario, con frecuencia menores de 50.000. • Tiempo de hemorragia alargado • TT, TP, Índice de Quick, TTPA se mantienen normales. • La batería analítica ha de ser amplia incluyendo: Anticuerpos antiplaquetarios, screening
de colagenosis, serología hepatitis y VIH… • Completar el estudio con pruebas de imagen: Radiografía de tórax, ecografía abdominal.

57 | Patología en hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
• Pruebas en Urgencias: hemograma y fórmula, frotis sanguíneo, pruebas básicas de coagulación
3) Diagnóstico diferencial
Se realiza tras descartar otras causas de trombopenia conocidas: colagenosis, infecciones, Hepatopatías, Sida, para diferenciarlo de otras hemopatías malignas en ocasiones se hace necesario practicar un aspirado de médula ósea.
4) Actuación en urgencias
Se realizarán las pruebas de laboratorio anteriormente citadas. Siempre hay que descartar la pseudotrombopenia por agregados mediante la realización de un frotis y observación al microscopio.
Si el paciente presenta trombopenia moderadas y no hay sangrado activo, puede enviarse al especialista para el estudio ambulatorio con carácter preferente.
Es conveniente recomendar la no realización de deportes o actividades violentas que puedan entrañar riesgos traumáticos e informar siempre de la contraindicación de la toma de aspirinas y AINES.
5) Tratamiento
El tratamiento de elección son los corticoides: prednisona a dosis de 1mg/Kg de peso y día, pero sólo cuando son sintomáticas o con cifras de plaquetas muy bajas (<50 x 109/l). Si no responden a corticoterapia como tratamiento de segunda línea se plantea la esplenectomía, siendo ésta eficaz en aproximadamente 60% de los casos. En los casos reiteradamente refractarios se utilizan inmunosupresores: ciclofosfamida, azatioprina, o inmunoglobulinas intravenosas. Hay que evitar la transfusión de plaquetas.
Trombopenias no inmunes microangiopáticas: PTT y SHU A/ La PTT y el SHU son dos síndromes que se consideran manifestaciones distintas de una misma entidad etiopatogénica: Trombopatía microangiopática.
En su patogenia se implica el daño endotelial de microarteriolas con formación de microtrombos de plaquetas ocasionando alteraciones funcionales en distintos órganos. B/ CLÍNICA Y
1) Diagnóstico
La pentada clínica característica consiste en: trombopenia, anemia hemolítica microangiopática, fiebre, manifestaciones neurológicas y fallo renal.
La anemia hemolítica microangiopática se caracteriza por la presencia de esquistocitos (fragmentos de hematíes) en el frotis de sangre periférica.

58 | Patología en hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
2) Tratamiento
La PTT es una enfermedad grave y a menudo fulminante, se asocia frecuentemente a déficit adquirido o congénito de proteasas encargadas de degradar los multímeros FvW de gran tamaño. Los síntomas neurológicos son muy llamativos. El tratamiento de elección consiste en la plasmaféresis y en la administración de plasma fresco congelado. Las transfusiones de plaquetas están contraindicadas en la PTT.
El SHU es una enfermedad propia de la infancia, con predominio del fallo renal e hipertensión, y escasos síntomas neurológicos. Típicamente puede ir precedida de un síndrome febril vírico o cuadro diarreico (E. Coli, Shigella). El tratamiento va dirigido a mantener la función renal precisando hemodiálisis hasta 75% de los casos, control de la hipertensión, transfusión de concentrados de hematíes y de plaquetas.
3) Actuación en urgencias
Ambas patologías requieren ingreso hospitalario y consulta al especialista: hematólogo, neurólogo o nefrólogo. Hay que empezar lo antes posible el tratamiento con hemofiltración si hay fallo renal y plasmaféresis diaria.
Trombopatías hereditarias
1) Clasificación etiopatogénica
Las trombopatías son defectos en la función plaquetaria, dependiendo del nivel en que se encuentre el defecto distinguimos:
• Defectos de adhesión: síndrome de Bernard-Soulier: herencia AR. Cursa con trombopenia moderada y plaquetas gigantes. Presentan asociada una alteración de la agregación a ristocetina secundaria a un defecto del complejo GPIb/IX del receptor superficial para el FvW. Suele manifestarse con hemorragias cutáneo-mucosas graves, requiriendo en ocasiones numerosas transfusiones.
• Defectos de agregación: trombastenia de Glanzmann: herencia AR. Presentan una alteración funcional a nivel del complejo GPIIb/IIIa. La agregación con ristocetina es normal, pero existe un defecto en la agregación con otros agonistas convencionales (fibrinógeno). Clínicamente se manifiestan hemorragias cutáneo mucosas de diferente intensidad según sean tipo I (ausencia de receptores) o tipo II.
• Defectos de secreción: Síndrome de la plaqueta gris: herencia AR. Presencia de gránulos á vacíos.
Se manifiesta por aumento del tiempo de sangría, hemorragias mucosas, trombopenia con plaquetas grandes, respuesta al tratamiento con Desmopresina. En casos graves transfusión de plaquetas.
Déficit de gránulos densos: Esta deficiencia se ha visto en asociación con enfermedades hereditarias distintas como el Síndrome de Hermansky-Pudlak y Síndrome de Chediak-Higashi, asociados a albinismo oculocutáneo. S de Wiskott-Aldrich.

59 | Patología en hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
– Síndrome de Ehler-Danlos. – Síndrome TAR (Trombopenia/agenesia de radio).
Generalmente estos pacientes presentan tendencias hemorrágicas moderadas con recuentos plaquetarios y tiempos de hemorragia normales. Se diagnostican por déficit de agregación con agregantes débiles, con respuesta normal a agregantes potentes.
Purpuras angiopáticas o vasculares Las púrpuras vasculares cursan generalmente con hemorragias leves cutáneas, y en ellas las pruebas básicas de coagulación y recuento plaquetario suelen ser normales.
CONGÉNITAS ADQUIRIDAS
Malformaciones vasculares: • Enfermedad de Rendu-Osler (Telangiectasia
hemorrágica hereditaria). • Enfermedad de Fabry (Angio queratoma corporal
difuso). • Ataxia-Telangiectasis. • Hemangioma cavernoso (S. de Kassabach-Merrit).
Alteración del tejido conjuntivo: • S. de Ehler-Danlos. • S. de Marfán. • Seudoxantoma elástico. • Osteogénesis imperfecta.
Púrpuras vasculares inmunes: • Enf. de Schönlein-Henoch. • Microangipotías trombóticas: PIT y SHU. • Fármacos: penicilina, sulfamidas, quinina,
tetraciclinas, AAS, Atropina. Alteración del tejido de soporte: • Escorbuto. • Púrpura caquéctica. • Púrpura senil. • Por corticoides. • Ammiloidosis.
Tabla. Clasificación de púrpuras vasculares.
De entre ellas, se exponen por su frecuencia e importancia clínica la PTT, SHU (ya explicadas) y la Púrpura de Schonlein-Henoch.
1) Púrpura anafilactoide de Schönlein-Henoch
PATOGENIA Y CLÍNICA Es un tipo de vasculitis que afecta a capilares de etiología alérgica desencadenado por procesos distintos: infecciones, fármacos, alimentos, toxinas endógenas…
De naturaleza benigna, afecta con mayor frecuencia a niños y jóvenes, y cura espontáneamente. Se manifiesta clínicamente por la aparición brusca de una púrpura palpable y pruriginosa de localización predominante en miembros inferiores y nalgas.
En ocasiones puede afectar a capilares de la mucosa digestiva, apareciendo sangrado intestinal y dolores abdominales. En 40% puede aparecer afectación renal, que raramente deviene en nefropatía crónica.

60 | Patología en hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
DIAGNÓSTICO El diagnóstico se basa en la clínica, característicamente no hay trombopenia ni alteración de la hemostasia. La biopsia cutánea muestra depósitos de IgA y complemento.
El diagnóstico diferencial se realiza púrpuras trombopénicas, y otros tipos de vasculitis más graves, así como colagenosis.
Es una enfermedad benigna que puede enviarse a estudio por especialista por vía ambulatoria, y que rara vez requiere ingreso hospitalario por sí misma.
TRATAMIENTO Sintomático: prurito, dolor abdominal; y de la causa desencadenante si se conoce.
Coagulopatías congénitas Las coagulopatias congénitas pueden deberse a déficit de la síntesis de los factores formadores de fibrina o a un incremento de la fibrinólisis:
DEFECTO DE FACTORES HIPERRFIBRINOLISIS
• Hemofilia A (VIII) • Hemofilia B (IX) • Enfermedad de Von Willebrand (WvW)
• Déficir de α2-antiplasmina. • Exceso de activador tisular del
plasminógeno.
Otros déficits menos frecuentes: • Fibrinófeno. • Protrombina. • Factor V, VII, X, XI, XII, Fletcher, XIII.
Tabla 5. Coagulopatías congénitas.

61 | Patología en hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
1) Hemofilia
ETIOPATOGENIA La hemofilia es una enfermedad hereditaria ligada al sexo. Se clasifica en dos tipos diferentes:
Hemofilia A ó clásica: deficiencia en la actividad del factor VIII (F VIII). Mucho más frecuente.
Hemofilia B o Enfermedad de Christmas: deficiencia en la actividad del factor IX.
HERENCIA Se transmite ligada al Cromosoma X, con lo que las mujeres son portadoras de la enfermedad sin padecerla (sólo en el caso excepcional de ser homocigotas), mientras que si lo hereda el hombre será un individuo enfermo.
Cuando la mujer es portadora y el hombre sano la descendencia masculina tiene un 50% de probabilidad de estar enfermo, y cada hija tiene un 50% de probabilidad de ser portadora. Cuando el hombre es el individuo enfermo y la mujer sana todos los hijos varones estarán sanos y todas las hijas mujeres serán portadoras.
Figura 5. Herencia ligada al sexo de la hemofilia. (X*: cromosoma afecto).
CLÍNICA La intensidad y frecuencia de los fenómenos hemorrágicos varía mucho de unos pacientes a otros dependiendo del déficit que posean.
Las manifestaciones hemorrágicas aparecen ante mínimas agresiones, suelen afectar a articulaciones, músculos, órganos internos, y sistema nervioso que son las de mayor gravedad. Las hemorragias cutáneo-mucosas son menos frecuentes en estos pacientes.
Las hemorragias más frecuentes son los hemartros (75%) en grandes articulaciones de los miembros: rodillas, codos, tobillos, hombros, muñecas. Dentro de las musculares cabe destacar el hematoma del psoas, que puede simular una apendicitis o hemartros de cadera. Las hemorragias articulares hay que tratarlas urgentemente para evitar lesiones crónicas degenerativas posteriores.
El porcentaje de déficit de factor permite una clasificación clínica:
• Grave:<1% de factor.

62 | Patología en hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
• Moderada: 1-5% de factor. • Leve: 5-40% de factor
DIAGNÓSTICO La clínica y la historia hemorrágica personal y familiar oriente hacia el diagnóstico inicial.
En las pruebas de coagulación existe un alargamiento del TTPA que se corrige añadiendo plasma normal. Es muy importante la cuantificación del déficit para calcular el tratamiento sustitutivo adecuado en caso de traumatismo o intervención quirúrgica.
En la actualidad las técnicas de Biología molecular son esenciales para el diagnóstico del paciente y la detección de familiares enfermos o portadoras, así como para detección prenatal de la enfermedad, mediante biopsia de vellosidad coriónica y amniocentesis.
TRATAMIENTO El tratamiento de la hemofilia se basa fundamentalmente en la prevención del sangrado y en la detención precoz de las hemorragias.
Tratamiento sustitutivo:
Se realiza mediante la administración de concentrados de factores VIII y IX. La dosis dependerá de la gravedad del déficit presentado. Un individuo normal posee una actividad del factor del 100%. Para que la hemostasia sea eficaz es necesario al menos unos niveles del 25-35% en Hemofilia A, y 20-25% en Hemofilia B.
En la Hemofilia A se emplean actualmente concentrados de F VIII de alta pureza, y F VIII recombinante que permite evitar el contagio de enfermedades víricas (VIH; hepatitis), que en décadas anteriores fueron desgraciadamente tan frecuentes.
Para la Hemofilia B se emplea F IX altamente purificado o concentrado plasmático de complejo protombínico (CCPT) inactivados víricamente.
Para el cálculo de la dosis hay que tener en cuenta el déficit, el peso del paciente y el nivel óptimo de factor deseado:
UI de F VIII = (Peso en Kg) x (% FVIII deseado) 2 UI de F IX= (Peso en Kg) x (% F IX deseado)

63 | Patología en hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
TIPO DE COMPLICACIÓN HEMORRÁGICA NIVEL DE F VIII DESEADO EN %
Hemorragia del sistema nervioso central 80 - 100
Cirugía mayor 50 - 100
Hemorragia gastrointestinal 50 - 100
Hematoma de psoas ilíaco 50 - 100
Hematuria 35 - 75
Hemartrosis y hematoma muscular 35 - 50
Tabla 6. Niveles de factor VIII deseados.
Dentro del tratamiento sustitutivo hay una modalidad para las hemofilias graves: la profilaxis primaria: Se trata de mantener niveles superiores al 1% de forma continuada mediante infusiones de factor 2-3 veces a la semana. Sin embargo no ha dejado de suscitar también controversias por el elevado coste, aparición de inhibidores más precoz, acceso venoso frecuente…
Tratamiento farmacológico:
En la hemofilia leve pueden emplearse los antifibrinolíticos sintéticos: Acido Tranexámico, Acido ε- amino caproico (EACA).
También se emplea la Desmopresina (DDAVP) vía intravenosa que aumenta los niveles de F VIII en 2 o 3 veces, no siendo eficaz en Hemofilias graves.
Como analgésico y antiinflamatorios puede utilizarse el Paracetamol, Ibuprofeno o los corticosteroides. La Aspirina está contraindicada.
Tratamiento de la hemofilia a con inhibidores
El 20-30 % de los pacientes desarrollan a lo largo de su vida anticuerpos frente al estímulo antigénico que supone la infusión de F VIII exógeno. En estos pacientes se han utilizado:
• FVIII a altas dosis. • FVIII porcino con éxito en pacientes que no presentan reacción cruzada con inhibidores a
FVIII porcino. • Concentrados de complejos protrombínicos (PCC, APCC). • FVII recombinante: última novedad en el tratamiento y se ha usado con éxito, ya que
se”bypasea” la vía intrínseca a través de la vía del factor tisular.
2) Enfermedad de Von Willebrand (EvW)
ETIOPATOGENIA Se trata de la coagulopatía congénita más frecuente. Afecta al 1% de la población. Dependiendo del tipo, se transmite con herencia autosómica dominante o recesiva.

64 | Patología en hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
Está causada por alteraciones genéticas: mutaciones, delecciones… en el gen del FvW (von Willebrand, brazo corto del cromosoma 12).
El FvW se sintetiza en el endotelio (de donde se libera al plasma) y en los megacariocitos (se almacena en los gránulos densos de las plaquetas).
El FvW circula por el plasma unido al FVIII para ralentizar su aclaramiento. A nivel plaquetario actúa facilitando la agregación y adhesión al interaccionar con la GPIIb/IIIa y GPIa.
CLASIFICACIÓN • Tipo 1: trastorno cualitativo. (Herencia AD) • Tipo 2: trastorno cualitativo:
2A: defectos del FvW con disminución de la función plaquetaria. 2B: incremento de afinidad del FvW a la GPIb. 2M: disminución de función plaquetaria sin afectar a los multímeros de alto peso
molecular. 2N (Normandía): defectos de la unión del FvW con el FVIII (herencia AD).
• Tipo 3: déficit absoluto de FvW (Herencia AR).
CLÍNICA Y DIAGNÓSTICO Las hemorragias son principalmente cutáneo-mucosas. En ocasiones puede haber sangrados gastrointestinales y, más raramente, hemartrosis o hematomas musculares.
En las exploraciones complementarias se descubre un alargamiento del Tiempo de hemorragia, sin embargo el TTPA se alargará sólo en los casos en que también esté disminuido el FVIII.
Existen otras pruebas específicas para la EvW como son: Análisis de multímeros de FvW, Actividad del Fvw cofactor Ristocetina, cuantificación de la actividad coagulante del FVIII, agregación plaquetaria.
TRATAMIENTO El tratamiento de la EvW se basa fundamentalmente en:
• Antifibrinolíticos: en casos de hemorragias leves. • Desmopresina (DDAVP) intravenosa. • Tratamiento sustitutivo:
Concentrados de FVIII ricos en FvW e inactivados víricamente. Concentrados de plaquetas (en tipo 3) FVIIr en situaciones graves (refuerza la vía del FT)
PARA SABER MÁS
http://www1.wfh.org/publication/files/pdf-1205.pdf

65 | Patología en hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
Coagulopatias adquiridas
1) Déficit de Factores dependientes de vitamina K
La vitamina K interviene en el proceso de metabolización hepática de ácido glutámico, cuando hay un defecto de la vitamina K, aunque existe síntesis de factores estos son inactivos. El déficit de vitamina K puede deberse a:
• Cúmarínicos (anticoagulantes orales): impiden la utilización de la vitamina K. • Antibióticos que destruyen la flora bacteriana que sintetiza la vitamina K: beta-lactámicos,
sulfamidas, amplio espectro. • Hepatopatías. • Falta de aporte alimentario (muy rara). • Enfermedad hemorrágica del recién nacido. • Falta de absorción: ictericia obstructiva, fístulas biliares.
Clínicamente cursa con hematomas cutáneos y hemorragias mucosas. Analíticamente hay alargamiento del TP y descenso del Índice de Quick, en casos graves también alargamiento del TTPA.
El tratamiento consiste en administración de vitamina K.
2) Enfermedad hepatocelular
Hay una disminución de los factores sintetizados por el hepatocito (excepto el VIII). Clínicamente es menos florida y se detecta en las analíticas de coagulación en donde hay alargamiento de TP con TTPA normal, y aumento de los PDF y Dímero-D.
El tratamiento se realiza con vitamina K o Plasma fresco congelado. En situaciones de gravedad.
3) Inhibidores adquiridos
En pacientes tratados con heparina, aparece anticoagulante antitrombínico circulante en plasma, también puede aparecer de forma excepcional antitrombina endógena en pacientes con mastocitosis generalizada.
Los inhibidores frente a todos los factores de la coagulación aparece en paciente politrasfundidos, con déficit congénito de factores (Hemofilia A y B).
La EvW adquirida se debe a la presencia de inhibidores frente al FvW en pacientes con enfermedades hematológicas como leucemia linfática crónica, otras neoplasias, lupus eritematoso sistémico.
El tratamiento sustitutivo es ineficaz, y se basa más bien en el control de la hemorragia, y erradicación del inhibidor con inmunotolorancia, gammaglobulina, plasmaféresis.

66 | Patología en hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
Coagulación intravascular diseminada (CID)
1) Etiopatogenia
Este síndrome se caracteriza por una activación generalizada de la coagulación a nivel de los pequeños vasos, debido a la masiva producción de trombina, produciéndose un consumo de factores y de plaquetas y una activación secundaria de la fibrinólisis.
La CID puede estar desencadenada por una serie de procesos muy heterogéneos, entre los que con más frecuencia pueden producirla se encuentran: Sepsis (meningococo, estafilococo), complicaciones obstétricas (desprendimiento de placenta, placenta previa), enfermedades neoplásicas, leucemias, inmunocomplejos circulantes.
2) Clínica y diagnóstico
Clínicamente se manifiesta con sangrados cutáneos y mucosos, o por heridas quirúrgicas, fiebre, hipoxia, coma, fallo renal, y un alargamiento de todos los tiempos de la coagulación, trombopenia e hipofibrinogenemia. Existe un aumento importante de los PDF y dímeros D.
3) Tratamiento
El tratamiento se basa en el etiológico principalmente y sustitutivo con transfusión de Plasma fresco congelado concentrados de plaquetas, fibrinógeno. El tratamiento con heparina, si bien se ha demostrado útil en las CID crónicas, sigue suponiendo gran controversia en la CID
La CID es un trastorno de la coagulación muy grave, donde puede ocurrir un fracaso multiórgánico y muerte si no se inicia tratamiento adecuado a la mayor brevedad. Son pacientes que no sólo hay que ingresar sino que tienen criterios de hacerlo en la unidad de cuidados intensivos, ya que requieren minuciosos y frecuentes controles.

67 | Patología en hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
Figura 7. Manejo terapéutico de trombopenia en urgencias.

68 | Patología en hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
Figura 6. Algoritmo diagnóstico de las diátesis hemorrágicas.

69 | Patología en hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
Figura 8. Manejo en urgencias del paciente con coagulopatía hemorragia.
B. POLICITEMIA VERA Es un desorden clonal, mieloproliferativo crónico (del tipo de la mielofibrosis idiopática, trombocitosis esencial o leucemia mieloide crónica), progresivo, caracterizado por aumento de la masa eritrocítica y usualmente leucocitosis, trombocitosis y esplenomegalia.
La incidencia de la Policitemia Vera es de 30 casos por cada 100.000 habitantes.
1. Etiología No se conocen las causas de este cuadro. En cuanto a la fisiopatología, los progenitores eritroides crecen sin eritropoyetina (EPO) con sobreproducción y son más resistentes a la apoptosis. Los progenitores con dominancia clonar inhiben a las células normales. Todo sugiere que el defecto está a nivel de la célula progenitora hematopoyética.
2. Clínica Se caracteriza por la siguiente sintomatología: esplenomegalia, plétora, hipertensión arterial, rubicundez, prurito, cefalea, debilidad, mareos, sudoración, alteraciones visuales, dolor epigástrico, pérdida de peso, gota, vértigos y acufenos.
3. Diagnóstico Deben aparecer 4 o más para llegar al diagnóstico de Policitemia Vera:
• Masa eritrocítica superior a 36 ml/kg en hombres y por encima de 32 ml/kg en mujeres (se mide con hematíes marcados Cr51).

70 | Patología en hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
• Saturación arteria de oxigeno superior a 92%. • Esplenomegalia. • Trombocitosis por encima de 400.000 y leucocitosis superior a 12.000. • Hipercelularidad en biopsia de médula ósea (BMO) con hiperplasia megacariocítica y
descenso en los depósitos de hierro. • Niveles séricos de eritropoyetina (EPO) bajos. • Proliferación anormal de colonias eritroides en cultivo en ausencia de eritropoyetina
(EPO) (eritrocitosis microcítica: Policitemia, Rasgo B talasémico, eritrocitosis hipóxica).
4. Evolución • Fase asintomática: esplenomegalia aislada, eritrocitosis, trombocitosis. • Fase eritrocítica: esplenomegalia, eritrocitosis, trombocitosis, leucocitosis, prurito,
trombosis, hemorragia. • Fase inactiva: No requiere flebotomías, ni quimioterapia, deficiencia en hierro. • Metaplasia mieloide post-policitémica: anemia, síntomas sistémicos (sudoración, fiebre,
disminución de peso), trombocitosis, trombopenia, esplenomegalia progresiva dura. • Leucemia aguda mieloide: anemia, infección, hemorragia.
5. Pronóstico La supervivencia es prolongada en la mayoría de los pacientes. La supervivencia promedio es de 10 años. Para su manejo, se requiere controlar la producción excesiva de eritrocitos y plaquetas. Puede evolucionar hacia mielofibrosis y/o leucemia aguda.
6. Tratamiento y evolución natural El objetivo del tratamiento es conseguir unos niveles de hemoglobina por debajo de 140g/L en hombres y de 120g/L en mujeres.
La técnica empleada: sangrías (flebotomías) hasta mantener hematocrito inferior al 45%, en casos de déficit de hierro, se puede realizar una sangría cada 3 semanas.
Se ha empleado fósforo radioactivo y Clorambucil, si bien conllevan alto riesgo de desarrollar cáncer.
La Hidroxiurea y el interferon alfa mantienen los niveles aceptables de hemoglobina y hematocrito y disminuyen la frecuencia necesaria de sangrías.
En casos refractarios a otras medidas terapéuticas: esplenectomía.

71 | Patología en hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
C. ERITROCITOSIS O POLIGLOBULIA
1. Etiología • Eritrocitosis relativa o espuria: hemoconcentración secundaria a deshidratación. • Eritrocitosis absoluta:
Hipoxia: intoxicación por monóxido de carbono (CO), altura (mal de alturas), enfermedad pulmonar crónica, hipoventilación mantenida, apnea del sueño, shunts cardiacos derecha-izquierda, defectos neurológicos del centro respiratorio.
Enfermedades renales: quistes, hidronefrosis, estenosis de la arteria renal, glomerulonefritis focal, trasplante renal.
Tumores: hipernefroma, hepatoma, hemangioblastoma cerebral, fibromioma uterino, tumores adrenales, meningioma, feocromocitoma.
Terapia androgénica. Síndrome de Barter. Policitemia familiar y congénita. Policitemia de Chuvash.
2. Riesgos y consecuencias de la eritrocitosis: La trombosis y la hemorragia (AINES), son causa de muerte en el 30% de los pacientes (síndrome de Budd-Chiari). Otras manifestaciones son de la eritrocitosis son:
• Anormalidades neurológicas vasculares, • Enfermedad vascular periférica, • Eritromelalgia (dolor quemante en las extremidades, dedos), • Mielofibrosis y metaplasia mieloide, • Ulcus gastroduodenal.
PARA SABER MÁS
http://www.touchoncology.com/articles/diagnosis-and-treatment-erythrocytosis http://www.haematologica.org/content/93/7/963.long
D. ANEMIAS Es la disminución de la cantidad de glóbulos rojos, del hematocrito o de la hemoglobina en sangre, con la consecuente disminución del transporte de oxígeno de la sangre.

72 | Patología en hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
Transporte de oxígeno a los tejidos
La extracción de oxígeno es de un 25% en condiciones normales, disminuye la presión parcial de oxígeno (PO2) de 100 a 40 mmHg, manteniendo la presión de difusión en el capilar para proveer suficiente oxígeno a un segmento de tejido con forma de cono truncado. La extracción mayor del 25% genera una hipoxia celular destructiva.
El cálculo aproximado sería: 1,34ml/gr de hemoglobinax 15gr de hemoglobina = 20ml de oxígeno/dl de sangre. Con débito cardíaco de 5.000 ml/min., el transporte de oxígeno al tejido es de 1.000 ml/min. Los requerimientos son sólo de 250ml.
Formación de glóbulos rojos
Se realiza en la médula ósea, donde existen 2 compartimentos:
• En el primero se encuentran las stem cell, se diferencian con la Eritropoyetina (determinada por la tensión de oxígeno en la sangre).
• En el segundo están los precursores eritroides, que maduran en 5 días.
Dentro de estos precursores se encuentran:
• Pronormoblasto. • Normoblasto basófilo. • Normoblasto policromático. • Normoblasto ortocromático. • Reticulocito. • Eritrocito maduro.
Se requiere: vitamina B12, hierro (Fe) y folato. En condiciones normales existen reservas para un mes de folato, para 2-3 años de vitamina B12 y respecto al hierro, hay reservas para sintetizar una pérdida de 250-300 gramos de hemoglobina. Cuando faltan algunos de los componentes se producen formas anormales de los glóbulos rojos:
• Déficit de vitamina B12y folato® Macrocitosis. • Déficit de hierro ®Microcitosis.
Ajustes compensatorios
• Menor afinidad de la hemoglobina por el oxígeno por un aumento del 2,3 difosfoglicerol (2,3 DPG).
• Apertura de capilares no usados y redistribución de flujo (desde piel, riñón y lechos mesentérico e iliaco a miocardio, cerebro y diafragma).
• Aumento del gasto cardiaco si la hemoglobina descienda por debajo de 7gr/dl; aumento de la función pulmonar.

73 | Patología en hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
• Aumento de la producción de glóbulos rojos por aumento de la eritropoyetina (EPO: aumenta la eritropoyesis, con aumento de reticulocitos).
• Aumento de la extracción de oxígeno en un 10-15%. • La hipoxia tisular no corregida: contribuye a la respuesta cardiovascular y eritropoyética.
Manifestaciones clínicas
Originadas por procesos de compensación:
• Palidez: por redistribución de flujo desde la piel. • Taquicardia, pulsatilidad aumentada, soplos funcionales, tinnitus: por hiperactividad
cardiaca. • Disnea de esfuerzo, ortopnea ocasional por aumento de la función pulmonar. • Sensibilidad o dolor en huesos hematopoyéticos por eritropoyesis compensadora. • Relacionados con la hipoxia tisular. • Musculares: angina, claudicación intermitente, calambres nocturnos, fatigabilidad. • Cerebrales: cefalea, falta de concentración, somnolencia.
Examen Físico: palidez de piel y mucosas, taquicardia, pulso amplio, soplo sistólico de eyección, discreto edema periférico.
Clasificación Morfológica
• Anemias microcíticas: deficiencia de hemoglobina: déficit de hierro, anemia de enfermedades crónicas, deficiencia de síntesis de globinas (Talasemias) o en síntesis del grupo Heme (sideroblástica).
• Anemias macrocíticas: anemias megaloblásticas, enfermedades hepáticas, hipotiroidismo, algunas anemias refractarias y mielodisplasias.
• Anemias Normocíticas: anemias aplásicas, anemias hemolíticas, etapas tempranas de otras anemias.
Clasificación funcional
• Hipoproliferativas: Índice reticulocitario bajo (inferior a 2). Daño de la médula ósea: aplasia (hereditaria, adquirida), fibrosis, infiltración. Disminución del estímulo: inflamación, insuficiencia renal, enfermedades
endocrinas (hipopituitarismo, hipotiroidismo, insuficiencia suprarrenal). Deficiencia de hierro: etapa inicial.
• Defectos de maduración: Índice reticulocitario bajo más alteraciones morfológicas. Defecto Citoplasmático (microcítica): deficiencia de hierro, rasgo talasémico,
anemia sideroblástica, anemias de enfermedades crónicas (ocasionalmente)

74 | Patología en hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
Defecto Nuclear (macrocítica): anemias megaloblásticas >115fl (déficit de vitaminas B12 y B9, mielodisplasia, anemias inducida por drogas), no megaloblásticas (hepatopatías, hipotiroidismo).
• Aumento de la destrucción o pérdida de glóbulos rojos. Índice reticulocitario alto (superior a 3). Hemolisis: inmune, mecánica, hereditaria, defectos de membrana adquiridos,
secundaria a infecciones. Hemorragia Aguda. Secuestro esplénico
Constantes hematológicas
• Volumen Corpuscular Medio (VCM): 80-100fl. • Hemoglobina Corpuscular Media (HCM): 27-34pg. • Concentración Media de Hemoglobina Corpuscular (HCMC): 31-36 gr/dl. • Índice reticulocitario=10 x (Hematocrito observado/Hematocrito real)/índice de
maduración.
Valores normales:
• Ferremia (sideremia): 50-150 ug/dl. • Ferritina: 10-250 ng/dl. • Transferrina: 250-450 ug/dl. • Porcentaje de saturación: 20-50% /TIBC: 300-370ug/dl. • Porcentaje de sideroblastos: 20-50%. • Depósitos de hierro: 4 gramos: hemoglobina 2,5 gramos, ferritina y hemosiderina
alcanzan 1 gramo y la mioglobina, citocromos, enzimas y transferrina sérica suponen aproximadamente 0,5 gramos.
• Requerimientos de hierro (Fe mg/día): hombre adulto: 1mg; mujer fértil: 1-2 mg. • Absorción de hierro: fundamentalmente duodenal, aumenta por el pH ácido, la vitamina
C, el tránsito intestinal lento y se absorbe mejor el hierro en estado Hem. Solo se absorbe el 10% del hierro de la dieta, pudiendo aumentar hasta 20% cuando existe déficit de ingesta o pérdidas, lo que equivale a 4 mg. Provocan menor absorción: ingesta de té, café, fibra, gastrectomía o SMA.
• Circulación del hierro: el hierro circula unido a transferrina; un tercio de la transferrina va ligada a hierro y en los dos tercios restantes se encuentra insaturada. La ferritina nos orienta sobre el depósito de hierro. En la médula ósea los eritroblastos tienen receptores para transferrina, una vez que el hierro pasa al citoplasma, se une a la protoporfirina para formar el grupo Hem. La protoporfirina libre eritrocitaria también se puede medir, porque cuando no hay hierro aumentan los niveles de protoporfirina.

75 | Patología en hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
1. Anemia ferropriva o ferropénica Es el tipo de anemia más frecuente.
Etiología • Disminución del aporte:
Dieta pobre en hierro: lactantes, desnutrición. Absorción deficiente: gastrectomía o SMA.
• Aumento de los requerimientos: Pérdidas sanguíneas: hemorragias ginecológicas, hemorragias digestivas (hernia
de hiato), hemorragias del aparato urinario o pulmonares, donantes de sangre, Policitemia vera, transfusiones, diátesis hemorrágica, hemosidenurias crónicas (hemólisis intravascular crónica, hemoglobinuria paroxística nocturna).
Crecimiento/Mujer fértil/Embarazo/Lactancia.
Clínica Síndrome anémico y Pica (tendencia a comer hielo, tierra, pasta de zapato), glositis, coiloniquia, queilitis angular, uñas quebradizas, atrofia papilar de la lengua, síndrome de Plummer Vinson: disfagia, glositis, membrana esofágica.
Laboratorio Se trata de una anemia microcítica hipocrómica, con frotis característico, plaquetas levemente elevadas.
Ferritina sérica baja, TIBC alta, porcentaje de saturación bajo, reticulocitos bajos.
El mielograma (diferencia sideroblastosis y talasemia) y la biopsia de médula son raramente necesarios. Otros: Ferremia (sideremia), Hemosiderina medular, Ferritina (es proteína de fase aguda).
Tratamiento • Identificar la causa. • Sulfato ferroso: 3 veces al día, antes de comer (100g de hierro elemental cada día). Control
en 10 días con en casos de insuficiencia renal. Después de normalizarse, 2-3 meses con suplementos de hierro. Los efectos secundarios son: diarreas, estreñimiento (constipación), nauseas, cólicos.
• Preparados de hierro de liberación prolongada: hierro parenteral.
2. Anemias megaloblásticas Se trata del defecto de síntesis del ADN. Relación ARN/ADN aumentada. La división celular retrasada acumula células en fase S del ciclo. 40-50% de células en fase S (normal: 20-25%). Eritropoyesis inefectiva: aumenta la muerte celular en médula ósea.

76 | Patología en hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
Defecto de ADN en deficiencias de b12 y folato
1) Patogenia
Deficiencia de cobalamina y folatos producen una reducción neta de 5,10, MTHF, lo que interrumpe la conversión de dUMP a dTMP mediado por timidilato sintetasa. El aumento de dUMP se traduce en aumento de dUTP (y consecuente reducción de dTTP).
ADN polimerasa no distingue entre ambos e incorpora erróneamente dUTP en el ADN. La ADN uracil-glicosilasa conoce el defecto y elimina dUTP del ADN; sin embargo, la carencia de dTTP impide su reparación. Los quiebres repetidos fragmentan el ADN, parte del cual escapa de la célula.
Además, se requiere factor intrínseco para la absorción de vitamina B12 (cianocobalamina).
2) Etiología
Dieta inadecuada, malabsorción (anemia perniciosa), gastrectomía, resecciones intestinales, enfermedad de Crohn.
3) Clínica
Síndrome anémico de inicio gradual, glositis, leve ictericia por eritropoyesis inefectiva, síntomas neurológicos (sensación vibratoria disminuida, postural, ataxia, parestesia, confusión y demencia), hiperhomocisteinemia.
4) Laboratorio
Anemia macrocítica, hipersegmentación de neutrófilos, pancitopenia, niveles bajos de B12 o B9, LDH elevada por la hemólisis.
5) Tratamiento
B12 intramuscular, control con endoscopia digestiva alta en pacientes con anemia perniciosa por asociación con Ca gástrico.
Deficiencia de ácido fólico El ácido fólico está en los vegetales. Los requerimientos diarios son de 100 microgramos, la dieta normal tiene 200-250 microgramos.
1) Etiología
Dieta deficiente, drogas (alcoholismo, antifolatos), malabsorción, necesidades aumentadas (embarazo, hemólisis).
Causas de macrocitosis: alcoholismo, hepatopatías, mixedema, drogas citotóxicas, reticulocitosis, mielodisplasia, embarazo, mieloma (por Roule).

77 | Patología en hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
2) Clínica
Indistinguible de la deficiencia de B12
3) Tratamiento
Suplemento con ácido fólico, si se sospecha déficit de B12, empezar con esta.
3. Anemia de las enfermedades crónicas Es la anemia más frecuente en los hospitalizados. Normocítica normocrómica o levemente hipocroma.
Patogenia Acortamiento de 20 a 30% de la vida globular: explicada por hiperactividad del sistema fagocítico mononuclear; y no por efecto intrínseco en los glóbulos rojos o hematíes.
Defecto en la reutilización del hierro: su entrega a transferrina (Tf) desde ferritina o hemosiderina de macrófago está disminuida, cayendo la concentración de hierro plasmático.
La apoferritina es la proteína de fase aguda, inmovilizando hierro en la célula. La lactoferrina (Lf) secretada por granulocitos activados, liga hierro con mayor afinidad que la transferrina y no hay receptores eritroblásticos que lo capten, volviendo a ser macrófagos. La transferrina es proteína de fase aguda negativa: cae su concentración por menor producción hepática y mayor secuestro por macrófagos. La ferritina también es proteína de fase aguda.
Existe una respuesta eritropoyética compensadora disminuida: interleuquina 1 (IL-1) y TNFa interfieren en producción de eritropoyetina (EPO). También hay resistencia eritroblástica a la acción de la eritropoyetina o EPO. La interleuquina 1 (IL-1) y TNFa suprimen formación de colonias BFU-E y CFU-E.
Etiología • Enfermedades Infecciosas crónicas: Tuberculosis, Endocarditis Infecciosa,
Osteomielitis, Infecciones Urinarias, Abscesos pulmonares, Infecciones micóticas, SIDA. • Enfermedades inflamatorias crónicas: Artritis Reumatoide, Lupus eritematoso
sistémico (LES), Colitis Ulcerosa, Infarto Agudo al miocardio, Sarcoidosis, Traumatismos extensos, Quemaduras extensas.
• Enfermedades Neoplásicas: Hematológicas (Leucemias, Linfomas, Mieloma), No hematológicas (Carcinomas metastásicos, Hipernefroma).
Clínica Está comandada por el cuadro de base, con anemia leve a moderada (hemoglobina entre 7 y 11gr, hematocrito entre 30 y 37%). Característicamente, se describe como normocítica y normocrómica, puede en 20 a 30% de los casos ser microcítica e hipocroma.

78 | Patología en hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
Exámenes de laboratorio El mielograma no es indispensable, pero apoya el diagnóstico: valorar hierro medular y más importante aún, descartar otra patología. Proteína C Reactiva elevada, velocidad de sedimentación elevada, haptoglobina elevada.
Diagnóstico diferencial Puede coexistir con otro tipo de anemia y ser enmascarada por ella. En la IR hay disminución de los niveles de eritropoyetina (EPO). El diagnóstico adecuado es fundamental por sus implicancias terapéuticas.
Tratamiento No hay tratamiento específico. Tratar la enfermedad de base. No está indicada la administración de hierro. Los pacientes con insuficiencia renal se benefician de la eritropoyetina (EPO).
4. Anemia aplástica o aplásica Es rara en Europa Occidental y USA (3 a 6 casos por millón por año). En China, Latinoamérica, México y Brasil es 4 veces más frecuente. Lo más probable es que no sea de origen genético sino que refleje exposición a tóxicos y/o virus.
La pancitopenia progresiva conlleva una mortalidad superior al 80% por año. Con trasplante de médula ósea e inmunosupresión mejora el pronóstico. Se observa en pacientes de cualquier edad, sobre todo entre 15-30 años y en los mayores de 60 años.
Etiología Defecto intrínseco o mecanismo inmunosupresor, lo que produce disminución de la las células CD34 (+) y CFU para proliferar y diferenciarse en la serie eritroide, granulocitos, megacariocitos en la médula ósea.
Diagnóstico y clínica Se caracteriza por pancitopenia e hipocelularidad de la médula ósea. No se encuentran células inmaduras en el frotis sanguíneo. Los síntomas son secundarios a la pancitopenia: palidez, tendencia a hemorragias mucocutáneas, infecciones (tardías). El cuadro clínico y evolución depende de la severidad de las citopenias.
• Anemia mielodisplásica: trastorno adquirido de las células hematopoyéticas pluripotenciales. Con el tiempo pueden desarrollar leucemia aguda, síndromes mielodisplásicos, hemoglobinuria paroxística nocturna (HPN)
• Anemia Aplástica o Aplásica Severa: granulocitos por debajo de 500 x ml, Plaquetas por debajo de 20.000 x ml, Reticulocitos corregidos bajo 1% (por debajo de 20.000ul). Biopsia de médula ósea marcadamente hipocelular (< 25%). Biopsia de médula ósea moderadamente hipocelular (25-30%).

79 | Patología en hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
• Anemia Aplástica o Aplásica Muy Severa: granulocitos por debajo de 200 e infección presente.
En el examen físico no se encuentra ni esplenomegalia ni hepatomegalia.
El diagnóstico se hace mediante biopsia de médula ósea (el mielograma no es concluyente).
Clasificación • Idiopática. • Hereditaria: Anemia de Fanconi, Disqueratosis Congénita, Anemia Aplástica Familiar,
Preleucemias, Trombocitopenia amegacariocítica, Blackfan-Diamond. • Adquiridas: Radiación, químicos, citotóxicos, benceno, solventes, insecticidas • Idiosincrásicas: cloranfenicol, sales de oro, antiinflamatorios no esteroideos (AINES),
anticonvulsivantes, antimetabolitos, antifúngicos, sulfamida, antitiroideo, antineoplásicas. • Virales: virus de Ebstein Barr, Hepatitis G, A, B, C, Parvovirus, virus de la
inmunodeficiencia humana (VIH), citomegalovirus (CMV). • Inmunológicas: Hipogamaglobulinemia, Timoma, Hemoglobinuria Paroxística Nocturna,
Embarazo, lupus eritematoso sistémico (LES) • Anemia mieloptísica: infiltración tumoral (1º o metástasis), granuloma, mielofibrosis,
enfermedades por depósitos, osteopetrosis.
Tratamiento Terapia de soporte: Transfusiones (plaquetas por debajo de 10.000 y hemoglobina por debajo de 7 + quelante de hierro), Antibióticos, Antivirales. Terapia definitiva: trasplante de médula ósea alogénico, inmunosupresión.
El pronóstico es de una supervivencia del 30% al año; si es leve, 80% al año.
Tratamiento en Anemia Aplástica Severa Terapias iniciales recomendadas:
• Trasplante de médula ósea alogénico: hermano compatible, donante relacionado no idéntico (5/6 locus de HLA). Indicación menores de 20 años y granulocitopenia con cifras de granulocitos inferiores a 200.
• Inmunosupresión: indicación: mayores de 20 años y granulocitopenia con cifras de granulocitos por superiores a 200. ATG (Timoglobulina), ALG (Linfoglobulina) y metilprednisolona, ciclosporina, terapia combinada para suprimir a los linfocitos que producen los anticuerpos autoinmune.
• Terapias alternativas o adyuvantes: factores de crecimiento hematopoyéticos: G-CSF, GM-CSF, EPO, TPO, IL-3, SCGF. Andrógenos. Trasplante de médula ósea con donante no relacionado.

80 | Patología en hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
E. LEUCEMIA AGUDA Se trata de una proliferación anormal de blastos (células inmaduras). Se reconocen bastantes alteraciones genéticas clonales que se repiten en todas las células anormales, sin comprometer a las no leucémicas.
La Leucemia Linfoide Aguda (LLA) es más típica en niños y la Leucemia Mieloide Aguda (LMA) es más frecuente en adultos (6-7ª década de la vida). Tiene concordancia entre gemelos y es más frecuente en desordenes congénitos (síndrome de Down, síndrome de Klinefelter). La incidencia en general es de 13 casos por cada 100.000 habitantes, con una proporción levemente superior de hombres frente al sexo femenino.
1. Clasificación Leucemias linfáticas agudas (lla)
• Según el tipo de precursores: Leucemia Linfática de precursores B: más común niños y curable. Leucemia Linfática de precursores T: CD10 negativo en el 20% de los casos.
• Clasificación morfológica: Leucemia Linfática tipo L1: células pequeñas de núcleo regular. Leucemia Linfática tipo L2: células grandes con pleomorfismo nuclear y nucleolo
prominente. Leucemia Linfática tipo L3: células con núcleo vesicular y citoplasma vacuolado.
Leucemias no linfáticas: Todos los casos expresan CD33 o bien expresan además CD13, 14 o 15.
• M0: Indiferenciada: 3%. • M1: Mieloblástica sin gránulos (10%). • M2: Mieloblástica gránulos (25%). • M3: Mieloblástica promielocítica (10%). • M4: Mielomonocítica (20%). • M5: Monocítica (20%). • M6: Eritroleucemia (5%). • M7: Megacariocítica (5%).
Existen otras leucemias, más raras, que no tienen marcadores (son células indiferenciadas).
2. Frecuencia de presentación La Leucemia Linfática B (LLAB), la Leucemia Linfática T (LLAT) y la Leucemia no Linfática aguda (LNLA) aumentan con la edad. Durante la primera y segunda décadas de la vida, la más frecuente

81 | Patología en hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
es la Leucemia Linfática B (LLAB). La Leucemia Linfática B (LLAB) pudiera tener relación con infección viral común en niños.
3. Alteraciones genéticas Hay alteraciones genéticas en el número de cromosomas en el 80% de Leucemias Linfáticas agudas y el 70% de las Leucemias no Linfáticas las presentan.
Alteraciones cromosómicas clonales • Hiperdiploide (más de 46 cromosomas):
Leucemia Linfática aguda: 25% (mejor pronóstico). Leucemia no linfática: 10%.
• Hipodiploide (menos de 46 cromosomas): Leucemia Linfática aguda: 5% (peor pronóstico). Leucemia no linfática: 20%.
• Pseudodiploide: Leucemia Linfática aguda: 45%. Leucemia no linfática: 40%.
• Translocaciones: Leucemia Linfática aguda: 40%. Leucemia no linfática: 35%.
• Otras (deleciones): Leucemia Linfática aguda: 10%. Leucemia no linfática: 3%.
Translocaciones cromosómicas • Leucemia linfoide aguda B:
T(12; 21) 20% - Genes involucrados: Tel-aml1. T (9; 22) 5% - Genes involucrados: Ph1-abl. T (4; 11) 3% - Genes involucrados: Af4-mll.
• Leucemia linfoide aguda T: T (1; 14) 25%. • Leucemia no linfoide aguda:
M2: T (8; 21) 25%. M3: T (15; 17) 100% - Genes involucrados: Pml-Rar. M4: T (16; 16) 25%. M5: T (4; 11) 30%.
Este listado de translocaciones cromosómicas orienta en el pronóstico y tratamiento. Leucemia aguda y core binding factor: proteína dimérica AML1-CBF b tiene que ver con la proliferación hematopoyética y patogénesis de la leucemia aguda.

82 | Patología en hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
4. Clínica • Síntomas derivados de la insuficiencia medular:
Anemia normocítica normocrómica: palidez, cansancio. Leucopenia con alteraciones morfológicas: infecciones con gérmenes habituales
pero con diseminación, candidiasis, úlceras orales, fiebre con o sin infección. Trombopenia: púrpura, hemorragia. En la leucemia no linfoide aguda tipo M3, por
ruptura de células y liberación de gránulos, se produce coagulación intravascular diseminada (CID), que origina hemorragia.
• Síntomas derivados de la proliferación celular en la leucemia linfoide aguda: hígado, bazo, riñones, adenopatías, infiltración testicular.
• Síndrome mielodisplásico (en leucemia mieloide aguda o LMA) con anemia refractaria. • Daño renal: infiltración tumoral, daño tubular por lizoenzimas celulares (hiponatremia,
hipopotasemia). • LDH y ácido úrico aumentados. • Historia corta de 3-4 meses, con tos, dolores óseos pero sin lesiones líticas, cefalea,
sudoración, masas localizadas en tejidos blandos, órganos y hueso - cloromas de leucemia no linfoide aguda M2 T (8; 21).
5. Diagnóstico Consiste en demostrar células leucémicas en médula ósea, sangre, infiltración extramedular.
• Hemograma: pancitopenia y blastos (se debe tener en cuenta que la anemia aplástica no tiene blastos).
• Mielograma: infiltración de médula ósea con un porcentaje de blastos superior al 25%. En la mononucleosis por virus de Ebstein-Barr (EBV) puede haberlos, pero en menos de un 25%.
• Inmunofenotipo: con citometría de flujo se puede confirmar el diagnóstico y asignar la estirpe celular (valor pronóstico).
• Citogenética: detección de alteraciones clonales diagnósticas y pronósticas. • Estudio de líquido cefalorraquídeo (LCR): la leucemia linfoide aguda (LLA) tiende a
invadir las meninges asintomáticamente y con ello hay peligro de afección de estructuras nerviosas; se debe hacer profilaxis para que no llegue al sistema nervioso central.
• Estudio de función hepática, renal, respiratoria y cardiovascular.
6. Tratamiento Objetivo: remisión de la enfermedad (se consigue el 85% de remisión completa en niños, pero en adultos el éxito es menor). Son sensibles a quimioterapia de combinación.
El tratamiento posterior está determinado por el tipo de leucemia, estado y edad del paciente y la respuesta precoz al tratamiento. El factor pronóstico más importante es el tratamiento. El éxito de remisión después de recaída es de 50-70%. Profilaxis para Pneumocistis carinii.

83 | Patología en hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
• Remisión: mielograma con menos de 5% de células blásticas en médula ósea y más de 1.500 neutrófilos y más de 100.000 plaquetas, sin blastos en sangre.
• Curación: remisión durante más de 5 años. • Recaída: vuelve la enfermedad después de haber terminado el tratamiento, por lo que se
necesita un tratamiento quimioterápico de mantenimiento a dosis bajas.
Tratamiento de la leucemia linfoide aguda (lla) • Inducción de la remisión: con Vincristina, Prednisona y Daunorrubicina o L-asparaginasa.
Se consigue un 90% de remisión completa. • Profilaxis de afección del sistema nervioso central: puede ser junto con la inducción. Se
usa Metotrexato intratecal. • Consolidación: con Citarabina, metotrexato, ciclofosfamida o doxorrubicina. • Mantenimiento: con Metotrexato o 6-mercaptopurina. Se puede suspender tras 3 años en
remisión completa. • Trasplante: Es el mejor tratamiento disponible, siendo mejor el heterotrasplante
esperando que se produzca el fenómeno de injerto vs huésped lo que eliminaría las células leucémicas residuales. En niños con leucemia linfoide aguda se utiliza el trasplante en la segunda remisión ya que en la primera se encuentran muy deteriorados.
• Trasplante alogénico: Pacientes de alto riesgo en primera remisión. Mala respuesta al tratamiento (no remite en el primer mes). Grupos citogenéticos de alto riesgo: T (9; 22), T (4; 11). Adultos con leucemia linfoide aguda nula y/o 100.000 blastos al diagnóstico.
Tratamiento de la leucemia mieloide aguda (lma) Consiste en inducir inmunosupresión con citarabina y daunorrubicina con lo que se logra remisión en un 80% de los casos. El tratamiento se repite hasta alcanzar remisión completa. La consolidación se hace de manera similar a la inducción repitiendo 3-4 veces. No se ha demostrado que sea beneficioso hacer profilaxis del sistema nervioso central.
Leucemia mieloide aguda (LMA) de riesgo citogenético favorable (M2, M3, M4): quimioterapia de inducción y quimioterapia de consolidación.
7. Factores de riesgo Constituyen parámetros para modular la intensidad del tratamiento e intensificar el mismo, así como el cuidado de apoyo en pacientes con pronóstico desfavorable.
Leucemia linfática aguda • Clínicos:
Edad: los menores de 1 año tienen alto riesgo si T (4-11).

84 | Patología en hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
Sexo: varones tienen peor pronóstico. Masa tumoral: recuento de blastos/mm3 al diagnóstico, a mayor recuento, peor
pronóstico. La respuesta precoz a la quimioterapia conlleva mejor pronóstico. • Biológicos:
Inmunofenotipo: antígeno común (mejor pronóstico). La leucemia linfática tipo L3: peor pronóstico.
Cariotipo: si hay más de 52 cromosomas, mejor pronóstico. Menor de 52 cromosomas: peor pronóstico.
La presencia de cromosoma filadelfia se asocia a peor pronóstico.
Leucemia no linfoblástica aguda: • Clínicos:
Edad: en general mejor en niños, pero muy dependiente del tratamiento. Masa tumoral: riesgo progresivo a mayor masa tumoral. Síndrome mielodisplásico previo: peor pronóstico.
• Biológicos: Subtipos de Leucemia no Linfoblástica Aguda: mejor pronóstico los subtipos M2,
M3 y M4. Citogenética: favorable las T (8; 21) (60% curables con quimioterapia), T (15; 17),
inv (16). Los casos con lesiones cromosómicas se asocian a peor pronóstico.
8. Pronóstico Leucemia Linfática Aguda: hay curación en un 25% de los adultos y en un 50-60% de los niños.
La Leucemia Mieloide Aguda: se consigue curación en menos de un tercio de los casos.
F. LINFOMA NO HODGKIN Corresponde a una entidad clínica cuyo desarrollo recién comienza en la actualidad. Sistemas de clasificación que hace 10 años agrupaban a todas las entidades clínicas han ido desapareciendo con el avance de los sistemas de diagnostico en particular la anatomopatologia y el uso de la citometría de flujo para determinar las características celulares.
Existen más de 20 subtipos de tumores que son incluidos en las clasificaciones actuales, cuyo diagnostico y tratamientos son independientes para cada uno de ellos.
La incidencia de estos tumores ha aumentado en los últimos 15 años a cifras cercanas al 50%, parcialmente en relación a pacientes jóvenes con SIDA, en quienes estos tumores tienen alta incidencia. Sin embargo existe un grupo de pacientes mayores de 50 años en quienes también se observa aumento de incidencia, sin que exista una causa clara.

85 | Patología en hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
Aunque los Linfoma no Hodgkin (LNH) se observan en todo el mundo, ciertos subtipos son más frecuentes en localización geográficas especificas – Linfoma de Burkitt África Tropical – Linfoma de Células T del Adulto Japón y el Caribe.
Etiología
Es desconocida, sin embargo ciertos estados de inmunodeficiencia adquiridos o congénitos, agentes infecciosos, agentes químicos y físicos, se han asociado a una mayor incidencia de estos tumores.
• Inmunodeficiencia: la permanente desregulación inmune, asociado a la exposición crónica de antígenos, aumenta el riesgo de desarrollar LNH secundario. Enfermedades Congénitas como: síndrome atáxico-telangectasia, síndrome Wiscott-Aldridge, síndrome linfoproliferativo asociado a cromosoma X, tienen una alta incidencia de LNH B de alto grado. De la misma forma, inmunodeficiencia asociado a drogas o infecciones también presentan mayor incidencia. Por ejemplo en pacientes post-transplante presentan un riesgo de entre 25-50% mayor de desarrollar LNH en comparación a población general. Caso similar se observa en pacientes VIH cuyo riesgo aumenta en casi 90-100 veces en comparación a la población general.
• Enfermedades autoinmunes: enfermedades tales como el síndrome de Sjögren, la tiroiditis de Hashimoto o la enfermedad celíaca, promueven el desarrollo de tejido linfoide asociado a mucosas (MALT) y aumentan el riesgo de LNH tipo B. No ha sido posible demostrar riesgo similar en pacientes con AR o LES.
• Agentes infecciosos (además de VIH) Helicobacter Pylori: la infección por este germen a nivel del estomago conlleva con
el desarrollo de gastritis crónica y MALT con alta asociación a Linfoma Gástrico. Virus Epstein-Barr: infecta e inmortaliza a los Linfocitos B, su infección tiene alta
asociación con el Linfoma de Burkitt y tumores de células B en pacientes inmunocomprometidos.
HTLV: retrovirus tipo C originalmente aislado en pacientes con Linfoma de Células T de adulto en USA y Japón. La infección por este virus endémico en ciertas regiones del Caribe y Japón, puede ir de síntomas gripales hasta el desarrollo de Linfomas y Leucemias en adultos.
• Agentes químicos y físicos: alta incidencia de Linfomas en pacientes con exposición a radiaciones intensas (bomba atómica o explosión de reactores), así mismo en pacientes sometidos a quimioterapia, especialmente con agentes alquilantes, el desarrollo de estos tumores es más frecuente.
Clasificación
La clasificación de este grupo de enfermedades ha sido en los últimos años uno de los puntos más conflictivos. La aparición de la citometría de flujo como método de diagnostico y tipificación de los tumores, permitió el descubrimiento de subtipos cuyo comportamiento era radicalmente diferente a lo que se estipulaba en clasificaciones anteriores.

86 | Patología en hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
Por este motivo, la WHO decidió crear una nueva clasificación que incluyera a los nuevos subtipos tumorales, publicada en el año 1997.
Neoplasias de Cels. B. Linfoma Linfoblástico precursor de Células B. Linfoma Linfocitico Pequeño Leucemia Prolinfocitica Linfoma Linfoplasmatico Linfoma Esplénico de zona marginal Leucemia de Células Velludas Linfoma Extranodal de la zona marginal Linfoma de las Células del Manto Linfoma Folicular Linfoma Nodal de la zona marginal Linfoma Difuso de Células Grandes Linfoma de Burkitt Plasmocitoma Mieloma Múltiple
Neoplasias de Cels. T. Leucemia Promielocitica Leucemia Linfocitica de Células Grandes Leucemia de Células NK Linfoma extranodal nasal Mucosis Fungoide Linfoma Primario Cutáneo Anaplastico de Células Grandes Linfoma Subcutáneo tipo Paniculitis Linfoma Intestinal Linfoma Hepato Esplénico Linfoma AngioInmunoblastico Linfoma Periférico Linfoma Anaplastico de Células Grandes , tipo sistémico Linfoma de Células T del Adulto.
1. Linfoma linfocítico pequeño En EEUU y Europa, sobre el 90% de estos tumores se manifiestan como leucemia crónica y solo un 5% solo desarrollan linfoma sin componentes leucémicos. Típicamente pacientes adulto mayor, con compromiso de medula ósea y periférico al momento del diagnóstico; se pueden observar además hepatoesplenomegalia y linfoadenopatias.
Aunque la diseminación de la enfermedad es el mejor predictor de sobrevida, las anomalías cromosómicas y de inmunofenotipo también influyen en la sobrevida final. Se observan frecuentemente en estos pacientes fenómenos asociados como anemia hemolítica o trombocitopenia. Cerca del 5% de estos pacientes tendrán transformación a linfomas agresivos el cual es fatal a los 3 años en el 95% de los pacientes. Sobrevida general es de aprox. 10 años.
2. Linfoma MALT B de bajo grado La mayoría de los linfomas gástricos de bajo grado y cerca del 50% de todas las neoplasias linfoides gástricas son del tipo MALT. Cerca del 40% de los linfomas orbitarios, tiroideos, pulmonares y de glándulas salivales corresponden a este tipo tumoral.
El sustrato para el desarrollo de este tipo de tumores corresponde a la inflamación crónica iniciada por una enfermedad auto inmune o infección (Síndrome de Sjögren, H. pylori). Por lo general, son tumores de buen pronóstico, con supervivencia cercana al 80% a los cinco años.
3. Linfoma difuso de células B Sobre un 30% de los LNH en adultos corresponden a este tipo histológico. Su edad de presentación media es de 60 años.
Estos pacientes presentan únicas o múltiples masas de crecimiento rápido es sitios nodales o extranodale (más frecuente el gástrico, aunque también se observa a nivel del SNC, hueso, riñón y testículo). Son tumores agresivos pero potencialmente curables con terapia agresiva.

87 | Patología en hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
4. Linfoma periférico de células T Corresponden aprox. al 10% de los LNH. Su forma de presentación corresponde inicialmente a una enfermedad diseminada, con eosinofilia, prurito, adenopatías, con compromiso hepático y esplénico.
El curso de la enfermedad por lo general es agresivo, es potencialmente curable con QT agresiva, pero tiende a la recaída con mayor frecuencia que el Linfoma Difuso de Cels. B.
5. Linfoma/leucemia de células T del adulto Pacientes con esta enfermedad en un 90% de los casos presentan títulos detectables de anticuerpos contra HTLV-1. Se presenta frecuentemente en Japón, aunque también se observan focos endémicos en el Caribe y EUA.
Su presentación más común corresponde a una enfermedad agresiva con leucocitosis, hepatoesplenomegalia, hipercalcemia y lesiones líticas a nivel óseo. Sobrevida por lo general es de pocos meses.
Menos frecuente se observa una forma crónica de esta enfermedad, sin leucocitosis ni hipercalcemia. Su sobrevida es algo mayor.
Etapificación En la actualidad no existe un sistema de etapificación propio para los LNH, de esta forma los pacientes son ubicados en diferentes estadios según la clasificación de Ann Arbor, utilizada originalmente para pacientes con Linfoma de Hodgkin:
• Etapa I: compromiso de una región linfática única (I) o un órgano extralinfatico único (IE).
• Etapa II: compromiso de dos o más regiones linfáticas en el mismo lado del diafragma (II) o compromiso localizado de un órgano extralinfatico (IIE).
• Etapa III: compromiso de regiones linfáticas en ambos lados del diafragma (III), compromiso localizado de órgano extralinfatico (IIIE), compromiso esplénico (IIIS) o ambos (IIIES).
• Etapa IV: compromiso difuso o diseminado de dos o más órganos extralinfaticos con o sin compromiso ganglionar.
La presencia de síntomas agregados se debe incluir como A, sin síntomas o B, con fiebre, sudoración, bajada de peso de del 10% de peso corporal.
Pronóstico Debido a que los patrones de diseminación de la Enfermedad de Hodgkin son distintos a los del LNH, la clasificación de Ann Arbor antes mencionada es poco específica para identificar a los subgrupos de pacientes con LNH.
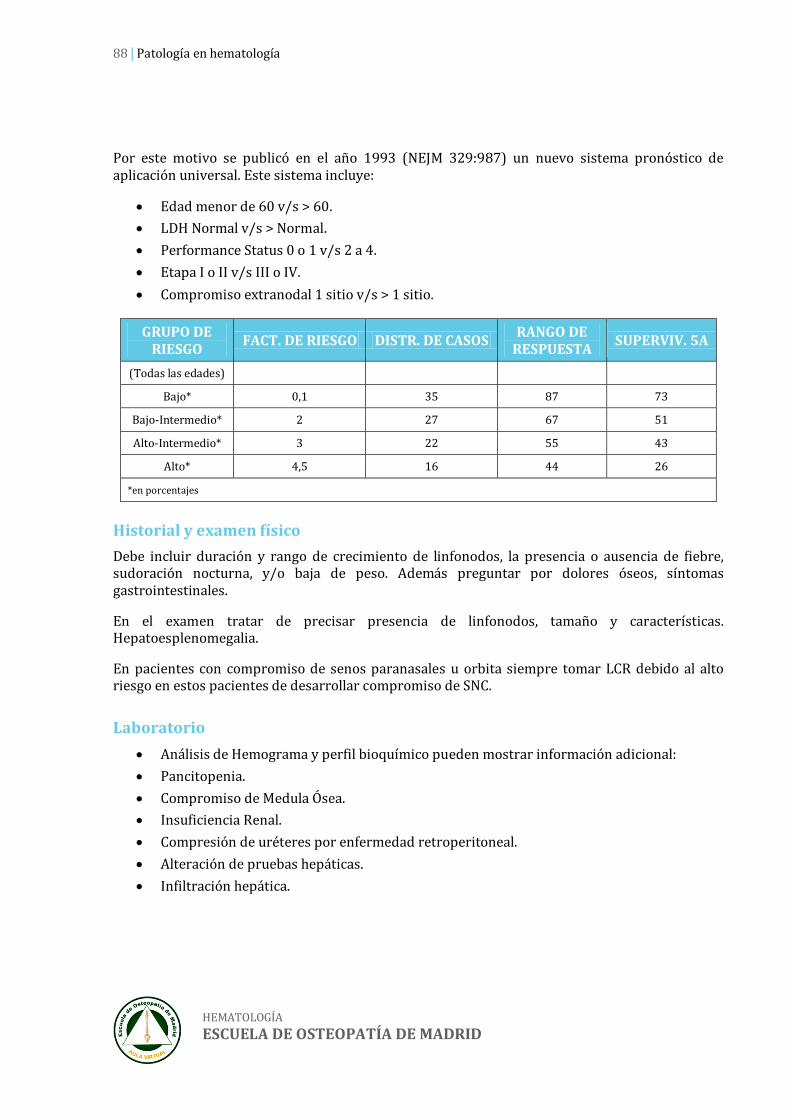
88 | Patología en hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
Por este motivo se publicó en el año 1993 (NEJM 329:987) un nuevo sistema pronóstico de aplicación universal. Este sistema incluye:
• Edad menor de 60 v/s > 60. • LDH Normal v/s > Normal. • Performance Status 0 o 1 v/s 2 a 4. • Etapa I o II v/s III o IV. • Compromiso extranodal 1 sitio v/s > 1 sitio.
GRUPO DE RIESGO FACT. DE RIESGO DISTR. DE CASOS RANGO DE
RESPUESTA SUPERVIV. 5A
(Todas las edades)
Bajo* 0,1 35 87 73
Bajo-Intermedio* 2 27 67 51
Alto-Intermedio* 3 22 55 43
Alto* 4,5 16 44 26
*en porcentajes
Historial y examen físico Debe incluir duración y rango de crecimiento de linfonodos, la presencia o ausencia de fiebre, sudoración nocturna, y/o baja de peso. Además preguntar por dolores óseos, síntomas gastrointestinales.
En el examen tratar de precisar presencia de linfonodos, tamaño y características. Hepatoesplenomegalia.
En pacientes con compromiso de senos paranasales u orbita siempre tomar LCR debido al alto riesgo en estos pacientes de desarrollar compromiso de SNC.
Laboratorio • Análisis de Hemograma y perfil bioquímico pueden mostrar información adicional: • Pancitopenia. • Compromiso de Medula Ósea. • Insuficiencia Renal. • Compresión de uréteres por enfermedad retroperitoneal. • Alteración de pruebas hepáticas. • Infiltración hepática.

89 | Patología en hematología
HEMATOLOGÍA ESCUELA DE OSTEOPATÍA DE MADRID
Imágenes En la actualidad la etapificación de este tipo de tumores se asocia directamente a lo observado en las imágenes, específicamente TAC, debido a la alta resolución y su capacidad de mostrar sitios comúnmente difíciles de observar en otro tipo de exámenes (mediastino, retroperitoneo).
• RNM: reservada para confirmar compromiso de Cerebro o Medula Espinal. • Cintigrama: utilizados principalmente en pacientes con alta sospecha en quienes no se
observa tumor evidente. El Cintigrama con Galio 67 es positivo en el 95% de los linfomas de alto grado y en cerca del 50% de los linfomas indolentes o de bajo grado.
• Biopsia de Medula Ósea: su utilidad actual está en la etapificación de este tipo de tumores, debido a que el compromiso de medula ósea es de peor pronóstico.
• Estudio Molecular: la citometría de flujo han permitido no solo confirmar o diagnosticar subtipos de linfomas gracias a la presencia de anticuerpos específicos a antígenos celulares, sino que permiten el seguimiento y la detección de enfermedad residual en pacientes en tratamiento o seguimiento. En la actualidad, esta técnica es parte del diagnostico en cualquier paciente con sospecha de LNH.
Tratamiento El acercamiento terapéutico a este tipo de Linfomas dependiera del tipo específico de tumor, el estadio de la enfermedad, el pronóstico y status del paciente.
El tratamiento para linfomas indolentes como el Linfoma Folicular, incluyen no dar terapia con seguimiento cercano, Radioterapia únicamente, Quimioterapia con monodrogas. Pacientes con Linfomas agresivos tales como el Linfoma Difuso de células B requieren de Quimioterapias con drogas múltiples asociado a Radioterapia. Agregándose en algunos casos la profilaxis a SNC.
El tipo de esquema específico y drogas utilizadas corresponde a enfrentamiento del especialista.
PARA SABER MÁS
Linfoma no Hodgkin: • Conceptos generales: http://www.medigraphic.com/pdfs/residente/rr-2013/rr131d.pdf • Guía de práctica clínica: http://www.guiasalud.es/GPC/GPC_528_Tratamiento_LH.pdf Linfoma de Hodgkin: http://www.seom.org/es/component/content/category/121?layout=blog