Embed Size (px)
Citation preview

letter
nature genetics volume 20 september 1998 43
The paleontology of intergene retrotransposons of maize
Phillip SanMiguel1,2, Brandon S. Gaut3, Alexander Tikhonov1, Yuko Nakajima1 & Jeffrey L. Bennetzen1,2
1Department of Biological Sciences and 2Genetics Program, Purdue University, West Lafayette, Indiana 47907-1392, USA. 3Department of Ecology andEvolutionary Biology, University of California, Irvine, California 92697-2525, USA. Correspondence should be addressed to J.L.B.(e-mail: [email protected]).
Retrotransposons, transposable elements related to animalretroviruses, are found in all eukaryotes investigated and makeup the majority of many plant genomes1–5. Their ubiquity pointsto their importance, especially in their contribution to the large-scale structure of complex genomes. The nature and frequencyof retro-element appearance, activation and amplification arepoorly understood in all higher eukaryotes. Here we employ anovel approach to determine the insertion dates for 17 of 23retrotransposons found near the maize adh1 gene, and two oth-ers from unlinked sites in the maize genome, by comparison oflong terminal repeat (LTR) divergences with the sequence diver-gence between adh1 in maize and sorghum. All retrotransposonsexamined have inserted within the last six million years, most inthe last three million years. The structure of the adh1 regionappears to be standard relative to the other gene-containingregions of the maize genome, thus suggesting that retrotranspo-son insertions have increased the size of the maize genome fromapproximately 1200 Mb to 2400 Mb in the last three millionyears. Furthermore, the results indicate an increased mutationrate in retrotransposons compared with genes.
The 240-kb interval of the maize genome containing adh1 islargely composed of clusters of retrotransposons insertedbetween low copy-number loci5. Each retrotransposon can bethought of as a ‘stratum’ that originated at a later time than theDNA flanking it. It is possible to date these strata becausesequence divergence between the initially identical LTRs of aretrotransposon should be proportional to the time that haselapsed since its insertion. At least 23 members of 11 families ofretrotransposons are found in the 240-kb region containingmaize adh1, accounting for over 160 kb of DNA (Fig. 1).
The key to dating the insertion of these elements is their LTRs—characteristic features that flank the internal region of a retro-transposon. Every family of retrotransposons has different (non-cross-hybridizing) LTRs, and elements within a family can varyappreciably (0−50%) in their LTR sequences. Due to the natureof the transposition process, the two LTRs of a single retrotrans-poson are usually identical at the time of its insertion into thehost genome6. As time passes, nucleotide substitutions causesequence divergence between the two LTRs. If the substitutionrate is known, then the date of insertion can be estimated fromthe amount of divergence between the two LTRs.
We determined the sequence for both LTRs of 17 retrotrans-posons in the region flanking adh1. The substitutions discoveredby comparisons of the two LTRs with each of these retrotrans-posons and two other retrotransposons from elsewhere in themaize genome are listed (Table 1). The data indicate substantialvariation in the amount of sequence divergence between theLTRs of individual elements (Table 1). To account for the poten-tial mutation rate differences at various sites, including thosedeoxycytidines likely to be 5-methylated, the gamma-correctedKimura two parameter method (γ-K2P) of distance estimationwas used. These distance estimates are largely consistent with the
Fig. 1 Spatial arrangement of maize adh1-Fregion retrotransposons. Short lines, retrotrans-posons; black, internal domains; grey, LTRs; openarrows, position of retrotransposon insertion;long line, low-copy sequence into which theretrotransposons inserted; arrows, putative direc-tions of transcription for genes in this region.
Table 1 • Composition of LTR and intron divergent sites
Sitesa Tb Vc ∆d TCG e T/V (T-TCG)/VLTRsKake-1 173 0 0 0 0 — —Grande-Zm1 623 0 1 0 0 0.0 0.0Opie-2 1272 1 2 1 1 0.5 0.0PREM-2f 1307 3 1 0 3 3.0 0.0Cinful-1 586 1 1 0 0 1.0 1.0Opie-3 1260 7 1 2 6 7.0 1.0Huck-2 1588 16 3 6 11 5.3 1.7Fourf 1142 19 1 3 10 19.0 9.0Milt 719 12 2 5 7 6.0 2.5Ji-1 1255 22 3 9 16 7.3 2.0Ji-4 1274 19 7 2 15 2.7 0.6Opie-1 974 16 4 4 10 4.0 1.5Ji-3 1159 18 9 4 15 2.0 0.3Reina 312 7 1 2 4 7.0 3.0Huck-1 1400 32 7 3 27 4.6 0.7Victim 101 2 1 1 2 2.0 0.0Zeon-1f 650 20 2 2 9 10.0 5.5Ji-6 1175 56 14 6 32 4.0 1.7Tekay 2742 110 57 21 60 1.9 0.9total 19704 361 117 63 228 3.1 1.1
IntronsS/F allelesg 1636 17 14 9 5 1.2 0.9sorghum/maizeh 1510 111 72 44 45 1.5 0.9total 3146 128 86 53 50 1.5 0.9aEach insertion/deletion (indel) was counted as a single site. bTransitions.cTransversions. dIndels, including retrotransposon insertions. eTransitions thatoccurred at CG or CNG sites. fElements not from the adh1 region. gMaize adh1-F allele introns compared with adh1-S allele introns. hComparison of sorghumand maize adh1 introns.
© 1998 Nature America Inc. • http://genetics.nature.com©
199
8 N
atu
re A
mer
ica
Inc.
• h
ttp
://g
enet
ics.
nat
ure
.co
m
letter
44 nature genetics volume 20 september 1998
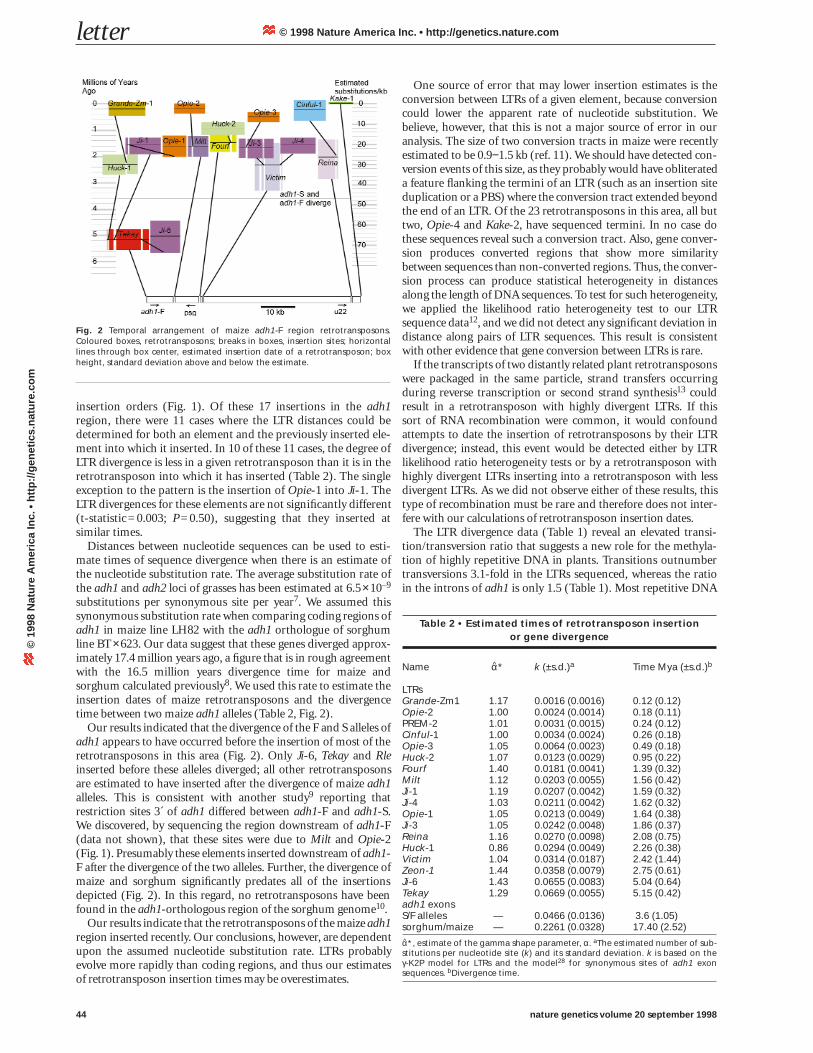
insertion orders (Fig. 1). Of these 17 insertions in the adh1region, there were 11 cases where the LTR distances could bedetermined for both an element and the previously inserted ele-ment into which it inserted. In 10 of these 11 cases, the degree ofLTR divergence is less in a given retrotransposon than it is in theretrotransposon into which it has inserted (Table 2). The singleexception to the pattern is the insertion of Opie-1 into Ji-1. TheLTR divergences for these elements are not significantly different(t-statistic=0.003; P=0.50), suggesting that they inserted atsimilar times.
Distances between nucleotide sequences can be used to esti-mate times of sequence divergence when there is an estimate ofthe nucleotide substitution rate. The average substitution rate ofthe adh1 and adh2 loci of grasses has been estimated at 6.5×10–9
substitutions per synonymous site per year7. We assumed thissynonymous substitution rate when comparing coding regions ofadh1 in maize line LH82 with the adh1 orthologue of sorghumline BT×623. Our data suggest that these genes diverged approx-imately 17.4 million years ago, a figure that is in rough agreementwith the 16.5 million years divergence time for maize andsorghum calculated previously8. We used this rate to estimate theinsertion dates of maize retrotransposons and the divergencetime between two maize adh1 alleles (Table 2, Fig. 2).
Our results indicated that the divergence of the F and S alleles ofadh1 appears to have occurred before the insertion of most of theretrotransposons in this area (Fig. 2). Only Ji-6, Tekay and Rleinserted before these alleles diverged; all other retrotransposonsare estimated to have inserted after the divergence of maize adh1alleles. This is consistent with another study9 reporting thatrestriction sites 3´ of adh1 differed between adh1-F and adh1-S.We discovered, by sequencing the region downstream of adh1-F(data not shown), that these sites were due to Milt and Opie-2(Fig. 1). Presumably these elements inserted downstream of adh1-F after the divergence of the two alleles. Further, the divergence ofmaize and sorghum significantly predates all of the insertionsdepicted (Fig. 2). In this regard, no retrotransposons have beenfound in the adh1-orthologous region of the sorghum genome10.
Our results indicate that the retrotransposons of the maize adh1region inserted recently. Our conclusions, however, are dependentupon the assumed nucleotide substitution rate. LTRs probablyevolve more rapidly than coding regions, and thus our estimatesof retrotransposon insertion times may be overestimates.
One source of error that may lower insertion estimates is theconversion between LTRs of a given element, because conversioncould lower the apparent rate of nucleotide substitution. Webelieve, however, that this is not a major source of error in ouranalysis. The size of two conversion tracts in maize were recentlyestimated to be 0.9−1.5 kb (ref. 11). We should have detected con-version events of this size, as they probably would have obliterateda feature flanking the termini of an LTR (such as an insertion siteduplication or a PBS) where the conversion tract extended beyondthe end of an LTR. Of the 23 retrotransposons in this area, all buttwo, Opie-4 and Kake-2, have sequenced termini. In no case dothese sequences reveal such a conversion tract. Also, gene conver-sion produces converted regions that show more similaritybetween sequences than non-converted regions. Thus, the conver-sion process can produce statistical heterogeneity in distancesalong the length of DNA sequences. To test for such heterogeneity,we applied the likelihood ratio heterogeneity test to our LTRsequence data12, and we did not detect any significant deviation indistance along pairs of LTR sequences. This result is consistentwith other evidence that gene conversion between LTRs is rare.
If the transcripts of two distantly related plant retrotransposonswere packaged in the same particle, strand transfers occurringduring reverse transcription or second strand synthesis13 couldresult in a retrotransposon with highly divergent LTRs. If thissort of RNA recombination were common, it would confoundattempts to date the insertion of retrotransposons by their LTRdivergence; instead, this event would be detected either by LTRlikelihood ratio heterogeneity tests or by a retrotransposon withhighly divergent LTRs inserting into a retrotransposon with lessdivergent LTRs. As we did not observe either of these results, thistype of recombination must be rare and therefore does not inter-fere with our calculations of retrotransposon insertion dates.
The LTR divergence data (Table 1) reveal an elevated transi-tion/transversion ratio that suggests a new role for the methyla-tion of highly repetitive DNA in plants. Transitions outnumbertransversions 3.1-fold in the LTRs sequenced, whereas the ratioin the introns of adh1 is only 1.5 (Table 1). Most repetitive DNA
Table 2 • Estimated times of retrotransposon insertionor gene divergence
Name α̂* k (±s.d.)a Time Mya (±s.d.)b
LTRsGrande-Zm1 1.17 0.0016 (0.0016) 0.12 (0.12)Opie-2 1.00 0.0024 (0.0014) 0.18 (0.11)PREM-2 1.01 0.0031 (0.0015) 0.24 (0.12)Cinful-1 1.00 0.0034 (0.0024) 0.26 (0.18)Opie-3 1.05 0.0064 (0.0023) 0.49 (0.18)Huck-2 1.07 0.0123 (0.0029) 0.95 (0.22)Fourf 1.40 0.0181 (0.0041) 1.39 (0.32)Milt 1.12 0.0203 (0.0055) 1.56 (0.42)Ji-1 1.19 0.0207 (0.0042) 1.59 (0.32)Ji-4 1.03 0.0211 (0.0042) 1.62 (0.32)Opie-1 1.05 0.0213 (0.0049) 1.64 (0.38)Ji-3 1.05 0.0242 (0.0048) 1.86 (0.37)Reina 1.16 0.0270 (0.0098) 2.08 (0.75)Huck-1 0.86 0.0294 (0.0049) 2.26 (0.38)Victim 1.04 0.0314 (0.0187) 2.42 (1.44)Zeon-1 1.44 0.0358 (0.0079) 2.75 (0.61)Ji-6 1.43 0.0655 (0.0083) 5.04 (0.64)Tekay 1.29 0.0669 (0.0055) 5.15 (0.42)adh1 exonsS/F alleles — 0.0466 (0.0136) 3.6 (1.05)sorghum/maize — 0.2261 (0.0328) 17.40 (2.52)
α̂*, estimate of the gamma shape parameter, α. aThe estimated number of sub-stitutions per nucleotide site (k) and its standard deviation. k is based on theγ-K2P model for LTRs and the model28 for synonymous sites of adh1 exonsequences. bDivergence time.
Fig. 2 Temporal arrangement of maize adh1-F region retrotransposons.Coloured boxes, retrotransposons; breaks in boxes, insertion sites; horizontallines through box center, estimated insertion date of a retrotransposon; boxheight, standard deviation above and below the estimate.
© 1998 Nature America Inc. • http://genetics.nature.com©
199
8 N
atu
re A
mer
ica
Inc.
• h
ttp
://g
enet
ics.
nat
ure
.co
m

letter
nature genetics volume 20 september 1998 45
in maize, including retrotransposons, are extensively methylatedat the 5 position in deoxycytidine in most or all tissues14. Oneexpected consequence of heavy deoxycytidine-methylation is anelevated transition rate15. In plants, most deoxycytidine methyla-tion occurs at CG or CNG sites16. If transitions that occurred atthese sites are omitted, then the transition-to-transversion ratiobecomes 1.1 for LTRs and 0.9 for introns (Table 1). Thus, deami-nation of 5-methyl deoxycytidine to thymidine in retrotrans-posons may have resulted in a mutation rate almost 1.5 times thatof other non-coding DNA (such as introns).
Although it occurs at a much lower rate, the apparentincreased genetic instability of methylated repetitive DNA inmaize is reminiscent of RIPing in Neurospora crassa17. This ele-vated mutation rate, whether the result of chemical deaminationor an active/enzymatic process, could help to control these ele-ments by interrupting the reading frames that produce geneproducts necessary for their transposition. Hence, deoxycytidinemethylation in plants may serve double duty in the inactivationof potential DNA parasites18 by contributing to transcriptionalinactivation and heterochromaticity of the sequences and byenhancing the rate of inactivating mutations.
Our data suggest that an explosive increase in the size of theadh1 region occurred in the last three million years. Of the 23identified retrotransposons identified, composing over 160 kb ofthe 240-kb adh1-F region, all arrived since sorghum and maizediverged less than 20 million years ago. Further, with the possibleexception of Rle, all probably inserted during the last six millionyears. Synonymous sites probably have more constraints on per-missible mutations than would methylated, presumably hete-rochromatic, neutral sites of intergene retrotransposons. Hence,we feel that a synonymous site mutational clock may be runningmore slowly than a clock for these retrotransposons and that,therefore, six million years is an upper estimate.
Other than the mutational Bs1 (ref. 19) and Hopscotch (ref. 20)elements that have identical LTRs, two additional maize retrotrans-posons have been completely sequenced: PREM2 (ref. 21) andZeon-1 (ref. 22). Using our LTR divergence analysis technique, wecalculated their insertion dates to have been approximately 0.2 and2.7 million years ago, respectively. As these dates are within the lastthree million years, similar to all but three or four of the 23 retro-transposon insertions in the adh1 region, the frequent insertions
near the adh1 gene may be a reasonable description for processesthat have occurred throughout the maize genome. Approximately120 kb of the 240-kb adh1 contig is composed of retrotransposonsthat inserted in the last three million years, suggesting that maizeDNA content increased from approximately 1200 Mb to its current2400 Mb in the last three million years due to retrotransposonarrival and/or amplification. Further studies are needed to deter-mine whether this explosive increase in retro-element content is aconsistent factor in the variations noted in plant genome size23.
MethodsEstimation of distance between LTRs. LTRs or introns were aligned usingGCG v. 8 (ref. 24) program GAP, with gap=1, len=0.2 and endweight para-meters set. (For a copy of our gapped sequences contact J.L.B.) The γ-K2P(ref. 25) model was used in the following manner. First, the shape para-meter α was estimated by maximum likelihood, using the program PAMLv. 1.2 (ref. 26). This estimate of α was used to estimate k and Var(k), thenumber of substitutions per site and its variance, under the γ-K2P model asimplemented in MEGA v. 1.0 (ref. 27).
Divergence estimation of adh1 genes and alleles. Divergence estimatesbetween adh1 genes were based on exon sequences. The number of basesubstitutions per synonymous site (ks) and its variance were estimatedusing described methods28.
Statistical test for conversion. The likelihood ratio heterogeneity test12,29 wasapplied to all pairs of LTR sequences, using partition lengths of 75−100 bp.A Bonferroni correction was used for an overall significance level of 0.05.
Pairwise comparison of estimated distances. Differences in divergencetimes were tested by pairwise comparison of estimated distances using at-test with infinite degrees of freedom. Significance values were adjustedfor an overall significance level of 0.05, using the Dunn-Sidak correction30.
GenBank accession numbers. LTRs, AF050436−AF050454; sorghum adh1coding sequence, AF050456; maize line LH82 adh1 coding sequence,AF050457.
AcknowledgementsWe thank S. Frank and S. Subramanian for technical assistance andZ. Avramova for useful discussions. This work was supported by grants fromthe USDA to J.L.B. and B.S.G.
Received 3 June; accepted 13 July, 1998.
1. Flavell, A.J. et al. Ty1-copia group retrotransposons are ubiquitous andheterogeneous in higher plants. Nucleic Acids Res. 20, 3639–3644 (1992).
2. Grandbastien, M.A. Retroelements in higher plants. Trends Genet. 8, 103–108(1992).
3. Voytas, D.F. et al. copia-like retrotransposons are ubiquitous among plants. Proc.Natl Acad. Sci. USA 89, 7124–7128 (1992).
4. Bennetzen, J.L. The contributions of retroelements to plant genomeorganization, function and evolution. Trends Microbiol. 4, 347–353 (1996).
5. SanMiguel, P. et al. Nested retrotransposons in the intergenic regions of themaize genome. Science 274, 765–768 (1996).
6. Lewin B. Genes VI. 603−604 (Oxford University Press, New York, 1997).7. Gaut, B.S., Morton, B.R., McCaig, B.C. & Clegg, M.T. Substitution rate comparisons
between grasses and palms: synonymous rate differences at the nuclear geneAdh parallel rate differences at the plastid gene rbcL. Proc. Natl Acad. Sci. USA 93,10274–10279 (1996).
8. Gaut, B.S. & Doebley, J.F. DNA sequence evidence for the segmental allotetraploidorigin of maize. Proc. Natl Acad. Sci. USA 94, 6809–6814 (1997).
9. Johns, M.A., Strommer, J.N. & Freeling, M. Exceptionally high levels of restrictionsite polymorphism in DNA near the maize Adh1 gene. Genetics 105, 733–743(1983).
10. Avramova, Z. et al. Gene identification in a complex chromosomal continuum bylocal genomic cross-referencing. Plant J. 10, 1163–1168 (1996).
11. Dooner, H.K. & Martinezferez, I.M. Recombination occurs uniformly within thebronze gene, a meiotic recombination hotspot in the maize genome. Plant Cell 9,1633–1646 (1997).
12. Gaut, B.S. & Clegg, M.T. Molecular evolution of the Adh1 locus in the genus Zea.Proc. Natl Acad. Sci. USA. 90, 5095–5099 (1993).
13. Hu, W.S. & Temin, H.M. Genetic consequences of packaging two RNA genomes inone retroviral particle: pseudodiploidy and high rate of genetic recombination.Proc. Natl Acad. Sci. USA 87, 1556–1560 (1990).
14. Bennetzen, J.L. et al. Active maize genes are unmodified and flanked by diverseclasses of modified, highly repetitive DNA. Genome 37, 565–576 (1994).
15. Coulondre, C., Miller, J.H., Farabaugh, P.J. & Gilbert, W. Molecular basis of base
substitution hotspots in Escherichia coli. Nature 274, 775–780 (1978).16. Gruenbaum, Y., Navey-Many, T., Cedar, H. & Razin, A. Sequence specificity of
methylation in higher plant DNA. Nature 292, 860–862 (1981).17. Selker, E.U. & Stevens, J.N. DNA methylation at asymmetric sites is associated with
numerous transition mutations. Proc. Natl Acad. Sci. USA 82, 8114–8118 (1985).18. Yoder, J.A. & Bestor, T.H. Genetic analysis of genomic methylation patterns in
plants and mammals. Biol. Chem. 377, 605–610 (1996).19. Jin, Y.K. & Bennetzen, J.L. Structure and coding properties of Bs1, a maize
retrovirus-like transposon. Proc. Natl Acad. Sci. USA 86, 6235–6239 (1989).20. White, S.E., Habera, L.F. & Wessler, S.R. Retrotransposons in the flanking regions
of normal plant genes: a role for copia-like elements in the evolution of genestructure and expression. Proc. Natl Acad. Sci. USA 91, 11792–11796 (1994).
21. Turcich, M.P. et al. PREM-2, a copia-type retroelement in maize is expressedpreferentially in early microspores. Sex. Plant Reprod. 9, 65–74 (1996).
22. Hu, W., Das, O.P. & Messing, J. Zeon-1, a member of a new maize retrotransposonfamily. Mol. Gen. Genet. 248, 471–480 (1995).
23. Flavell, R.B., Bennett, M.D., Smith, J.B. & Smith, D.B. Genome size and theproportion of repeated nucleotide sequence DNA in plants. Biochemical Genetics12, 257–269 (1974).
24. Devereux, J., Haeberli, P. & Smithies, O. A comprehensive set of sequence analysisprograms for the VAX. Nucleic Acids Res. 12, 387–395 (1984).
25. Kimura, M. A simple method for estimating evolutionary rates of basesubstitutions through comparative studies of nucleotide sequences. J. Mol. Evol.16, 111–120 (1980).
26. Yang, Z. PAML: a program package for phylogenetic analysis by maximumlikelihood. Comput. Appl. Biosci. 13, 555–556 (1997).
27. Kumar, S., Tamura, K. & Nei, M. MEGA: Molecular Evolutionary Genetics Analysissoftware for microcomputers. Comput. Appl. Biosci. 10, 189–191 (1994).
28. Nei, M. & Gojobori, T. Simple methods for estimating the numbers of synonymousand nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 3, 418–426 (1986).
29. Gaut, B.S. & Weir, B.S. Detecting substitution-rate heterogeneity among regionsof a nucleotide sequence. Mol. Biol. Evol. 11, 620–629 (1994).
30. Sokal, R.R. & Rohlf, F.J. Biometry. (Freeman, New York, 1995).
© 1998 Nature America Inc. • http://genetics.nature.com©
199
8 N
atu
re A
mer
ica
Inc.
• h
ttp
://g
enet
ics.
nat
ure
.co
m





























