Embed Size (px)
Citation preview

1
COMBUSTION AND GASIFICATION RATES OF LIGNOCELLULOSIC CHARS
(submitted for publication in “Progress in Energy and Combustion Science”
Colomba Di Blasi
Dipartimento di Ingegneria Chimica, Università degli Studi di Napoli "Federico II"
Tel: 39-081-7682232; Fax: 39-081-2391800;e-mail:[email protected]

2
Abstract This review critically examines the state of the art of rate laws and kinetic constants for the gasification, with carbon dioxide and steam, and the combustion of chars produced from wood and biomass, including a brief outline about yields and composition of pyrolysis products. The analysis also gives space to the role played by various factors, such as heating rate, temperature and pressure of the pyrolysis stage, feedstock and content/composition of the inorganic matter, on char reactivity. Finally, directions for future research are suggested. Contents 1. Introduction 2. Char yields and reactivity
2.1 Products yields from wood and biomass pyrolysis 2.2 Composition of volatile products of wood pyrolysis 2.3 Char reactivity 2.4 Kinetic and mass-transfer control 2.5 Factors affecting char reactivity
3. Chemical kinetics of char combustion and gasification 3.1 Kinetic and structural effects during char conversion 3.2 Mechanisms and rate laws of char gasification 3.3 Mechanisms and rate laws of char combustion 3.4 Structural models of char conversion 3.5 Kinetics of wood and biomass combustion Conclusions
References 1. Introduction Biomass is a renewable fuel and the fourth largest following coal, oil and natural gas. Compared with these fossil fuels, it has the advantages of being neutral in regard to the emissions of the green-house gas carbon dioxide, as this participates in biomass growth through the photosynthesis reactions, and reducing pollutant species generation, given the low sulfur and nitrogen contents. Thermal and chemical processes of biomass conversion consist of direct combustion, to generate heat and electricity, pyrolysis and gasification, to produce mainly liquid and gaseous fuels to be processed afterwards. Pyrolysis also plays an important role as the first chemical step in gasification and combustion. Indeed, it occurs at lower temperatures and, in numerous practical applications, solid conversion can be seen as a two-stage process: pyrolysis (or devolatilization) and slow heterogeneous conversion of char. Pyrolysis conditions highly affect the yields of char and its reactivity in combustion and gasification. Hence, low amounts of chars, associated with high yields of gaseous and vapor-phase (tar) products, and high char reactivity are key variables for the capacity of gasifiers and combustors. The heterogeneous reaction rates of char conversion are useful for design and development purposes as, to provide detailed process simulation, kinetic mechanisms should be coupled with the description of heat, mass and momentum transfer. Therefore, the intrinsic rate, that is, the rate of the reaction step free from heat and mass transfer limitations should be investigated. A huge amount of literature has been produced on the reactivity and the heterogeneous kinetics of coal and coal char gasification and combustion (reviews on this subject include, among others, Refs. [1-4]). The number of studies on wood/biomass char reactivity is comparatively much smaller. Many analogies exist between the two classes of combustibles but lignocellulosic chars are far more reactive than coals. As an example, the rate of steam gasification of biomass is about 4-10 times greater than

3
that of lignite, as a consequence of peculiar chemico-physical properties [5]. The volatile content of lignocellulosic fuels (typically 80-90%) is at least twice that of coal. The hydrogen/carbon and oxygen/carbon molar ratios vary between 1.3-1.5 and 0.5-0.6, respectively (versus 0.8-0.9 and 0.1-0.3 for coals). Wood chars have porosities with values from 40 to 50% and pore sizes between 20 and 30µm, whereas coals have porosities ranging from 2 to 18% and pore size around 5Å. Furthermore, the ash content is very low and the pore structure is highly directional, typical of that of wood and its intra-fiber cavities [6]. The production and properties of lignocellulosic chars have been reviewed in the comprehensive analysis by Antal and Gronli [7] and specific information on the combustion behavior can be found in [8-10]. However, kinetic modeling of the gasification and combustion reactions has not been given significant consideration. In this review the structural and kinetic contributions in the models of char reactivity and the kinetic parameters currently available are examined and compared. An outline is also provided of the main factors affecting char yield and reactivity taking into account the possible interactions between heat/mass transfer and chemical reactions. 2. Char yields and reactivity In this section the influences of the thermal conditions on the pyrolysis products of lignocellulosic fuels are summarized and commented. The conditions are also discussed of combustion and gasification for the experiments motivated by kinetic analysis. Finally, a critical examination is provided about how char reactivity is affected by heating rate, temperature, pressure and residence time during pyrolysis and mineral matter content of the sample. 2.1 Product yields from wood and biomass pyrolysis Pyrolysis of lignocellulosic materials gives rise to a huge number of chemical compounds which, for engineering applications, are lumped into three groups: permanent gases, a pyrolytic liquid (bio-oil/tar) and char [11-13]. They result from both primary decomposition of the solid fuel and secondary reactions of volatile condensable organic products into low-molecular weight gases and char, as they are transported through the particle and the reaction environment. Extensive information is available on product yields and composition from wood (poplar, beech, oak, maple, etc.) for fast pyrolysis carried out through fluidized-bed reactors (see, for instance, [14-21]) or other devices [22] and single thick particles or packed beds (see, for instance, [23-30]). The yields of pyrolysis products have also been evaluated for several agricultural biomasses (see, for instance, [31-39]) but, in this case, a detailed chemical characterization is provided only for the gaseous products. The yields of the three classes of pyrolysis products depend mostly on the particle heating rate and temperature of both particle and reacting environment although other factors, such as amount and composition of ash catalytically active, may also play an important role [40-43]. Fast heating rates, temperatures below 750-800K and short vapor residence times (usually 0.5-2s) are applied to maximize the yields of primary tars [44] and thus bio-oil [22]. This process is commonly indicated as fast pyrolysis, but the definition of the thermal conditions is, at a certain extent, only qualitative because the actual heating rate and temperature experienced by particles during conversion cannot be easily evaluated. Indeed, apart from the influences of the particle properties (size, moisture content, initial density, etc.), the external heat transfer rate depends not only on the conversion unit but key variables are also the reactor design, the feeding modality and the operating conditions. Moreover particle conversion is a highly unsteady process, so that the thermal conditions inside and outside the particle vary continuously in time [13]. For the higher temperatures of gasification, secondary reactions [44,45] of primary tar vapors become active which cause cracking and polymerization. However, owing to physical or geometrical limitations of the reactor and the chemical limitations of the reactions involved

4
[46], tars are not completely converted and, to avoid fouling and corrosion problems during gas utilization, cleaning and conditioning are required. Conversion of relatively thin (below 6mm) particles in fluidized-bed reactors with short residence times of volatiles [14-20] can be considered to occur under conditions of fast heating whereas the experiments conducted with single thick particles (for instance, see [29] for 4cm thick cylinders) and packed-bed reactors (for instance, see [30] for a reactor diameter of 6cm) can be considered examples representative of slow heating rates. For comparison purposes, the yields of the three classes of pyrolysis products, as measured by these two groups of studies and expressed as percent of the initial dry wood mass, are reported in Figs. 1-3 as functions of the external (heating) temperature, Te, (solid and dashed lines are used to indicate the range of variation, for a given temperature, of the yields for fast and slow pyrolysis, respectively). In principle, differences can be due to reactor design (external heat transfer rates), operating conditions (volatile residence times, particle characteristics) and wood type. However, the first two factors are predominant, given that the differences between wood species belonging to the standard hardwood or softwood categories are relatively small [29]. In general, high lignin (softwoods) and/or extractive contents, which are associated with a high carbon content, result in higher yields of char. As the heating temperature increases, the char yields initially decrease, as a result of a competition between the primary reactions of char and volatile formation, with the latter becoming successively more favored [13]. Then, at high temperatures, they tend to become constant, because the variation in the actual degradation temperature of the solid (in relation to the external heating temperature) becomes small, due to the narrow range of temperatures characteristic of biomass pyrolysis, and heat transfer resistance through the packed bed or the particle. Moreover the temperature directly and indirectly, through variations in the release rate and residence time of volatile products, also influences the activity of secondary reactions (formation of secondary char, permanent gases and light organic compounds) [13]. The yields of liquid products present a maximum, caused by both primary volatile formation and secondary degradation of tar vapors enhanced by high temperatures. In accordance, the yield of gas continuously increases with temperature. Although the trends shown by the product yields on dependence of the heating temperature are the same, the reduction in the heating rate, from the set of fluidized-bed data to those of single thick particles and packed-bed reactors examined here, plays an important role from the quantitative point of view. It can be seen that the rates of increase (low temperature) of the liquid yields are much faster for fluidized-bed conversion, because the smaller particle sizes and the faster external heat transfer rates actually give rise to faster heating rates of the solid and higher temperatures for the primary decomposition reactions. In this way, the devolatilization reactions are successively more favored with respect to char formation. It can also be observed that, for high external heating temperatures, the rate of decrease in the yields of liquid products is again faster for the fluidized-bed data. This is because the distribution of volatile products is mainly dictated by extra-particle secondary reactions, which occur in the reacting environment at temperatures much higher than those of the vapor phase in the single-particle reactor or in the free-board region of the fixed-bed reactor. In this case, secondary reactions essentially take place only across the particle and the packed bed (for the first, structural failure, fissures and cracks highly reduce the intra-particle residence times of vapor-phase species). High temperatures favor the cracking of tar vapors, which result in a rapid decrease in the yields of liquid products and an increase in the gas yields. The comparison between the two sets of data also shows that, for temperatures below 750K when the activity of secondary reactions for gas formation is negligible, the yields of gas are small and roughly constant. In other words, the variations in the yields of liquid products are compensated by variations in the yields of char only.

5
From the quantitative point of view, results plotted in Figs. 1-3 show that, for fast pyrolysis, maximum liquid yields are attained for heating temperatures of about 750-800K and vary between 65-75%. For slow pyrolysis, maximum values are maintained over a wider temperature range (750-950K) and are lower (45-55%). The char yields are significantly affected by both heating rate and external temperature with ranges of values roughly between 28-8% (fast pyrolysis) and 40-20% (slow pyrolysis). As anticipated, char is a product of both primary and secondary reactions, although the quantitative understanding of the latter process is very limited [7,47,48]. Boroson and coworkers [49], in their experiments on homogeneous tar cracking, do not report significant yields of char and suggest that its formation may require high concentrations of tar vapors (re-polymerization of low-volatility intermediates) or participation of the condensed phase (liquid-phase reactions of tar). In another study [50], where the conversion of primary tars induced by contact with char is investigated, it is reported that heterogeneous conversion corresponds to about 14±7% of tar. Furthermore, small quantities of coke, generated from tar conversion, are deposited on the char bed, as can be understood by observing the reduction in the surface areas of chars responsible for the different extent of tar conversion. It is postulated that a tar fraction (about 35%) exists which is highly resistant to vapor-phase cracking for temperatures above 873K, contains oxygen and is probably more aromatic than the whole tar. Lignin is indicated as the major source of this tar fraction. However, it is well known that an important source for secondary char is also represented by vapor-phase reactions of carbohydrates, specifically levoglucosan [51-54]. High concentration (pressure) of tar vapors, prolonged vapor-phase residence time in the reaction zone and high temperature affect positively the formation of secondary char. Some information on this process can also be gained from the studies on the devolatilization behavior of bio-oil samples subjected to moderate heating (5K/min up to 600K) [55-57], where char yields are roughly 25-40% of the initial oil mass. Secondary char formation is directly linked with the pyrolytic lignin content of the oil but the cellulose oil (and thus carbohydrates) gives rise to yields of secondary char about twice higher than those obtained from the wood and biomass oils [58]. Product yields as functions of temperature show the same qualitative trends for wood and biomass, but quantitative differences are large. Biomass, especially agricultural residues, always give rise to larger yields of char (a comparison between wood and various agricultural biomasses is provided in Fig.4 for a packed bed exposed to several heating temperatures [35]), attributed to higher lignin (and carbon) contents and/or larger amounts of inorganics which favor charring reactions and displace towards lower temperatures the beginning of the decomposition process [31,34,35]. As a consequence of the higher char yields, liquid yields are lower whereas the variations in the gas yields are generally small. In general variations in the product yields are the result of differences in both chemical and physical properties, although the former seem to have a predominant role. On the other hand, the size and density of the particles (and that of the bed in the case of packed-bed reactors) are the main factors affecting the pyrolysis time [35]. 2.2 Composition of volatile products of wood pyrolysis Detailed chemical analyses are available of both gaseous and liquid products of wood pyrolysis. The pyrolysis gas consists mainly of CO2, CO, CH4 and lower amounts of H2 and C2 hydrocarbons [14-20,29,30]. Gas composition also remains qualitatively the same for biomass, although generally characterized by higher yields of CO2 [31,34,35]. The yields of CO2 and CO for slow and fast pyrolysis of wood are compared in Figs.5-6. At low temperatures, the evolution of CO and CO2 (and water vapor) is due mainly to the degradation of extractives and hemicellulose and the activity of the first path in cellulose degradation, leading to gas and char formation [11,12,59]. As temperature increases, the formation of tar vapors from cellulose becomes predominant, while lignin degradation also attains fast rates. This process is largely responsible for char formation and the evolution of CO2,

6
CO, CH4 and H2. From the quantitative point of view, the hardwoods (lower lignin contents) are characterized by CO2 yields higher than those of softwoods, whereas the yields of CO and CH4 are comparable. As already observed, for low temperatures, the heating rates of the solid exert a weak influence on the yields of the gaseous species. The chief differences between the two sets of data are seen at high temperatures and result from differences in the residence times of tar vapors which modify the activity of secondary reactions. The effects of secondary reactions of tar decomposition on the gas yields and composition have been investigated by several researchers (among others, [45,47-54,60-64]) and a review about the global kinetics of tar cracking is available [13]. It is observed that carbon dioxide is a product of both primary and secondary reactions (it can account up to 14% of tar converted [45,49]). Carbon monoxide and methane are produced in small quantities from primary reactions. A large part of their production, with acetylene, ethylene, ethane and hydrogen, arises from secondary decomposition. In particular, the yields of carbon monoxide and methane present an almost linear increase with temperature for values above 950K [45]. As carbon monoxide may account for about 50-70% of tar conversion (methane and ethylene account for 11 and 12%, respectively), it is suggested [49] that its concentration can be considered as an indicator for the extent of secondary reaction activity. However, Morf and coworkers [45] observe that the yield of hydrogen may be even better for such a scope given that it presents an exponential increase with temperature. High temperatures of the reaction environment also make possible a contribution from methanation and water gas shift reactions and interactions between the tar species and other reactants on the composition of the gas [45,64]. The yields of water are significantly affected by the particle heating rate, as shown in Fig.6, where results obtained for wood pyrolysis in fluidized-bed reactors [14-16,19] and a fixed-bed reactor [30] are reported. It can be seen that, in the former case, they are roughly comprised between 10-18% of the initial dry wood mass, whereas in the latter case they are between 17-25%. Water is also a product of the reactions of tar degradation and so its yield increases with temperature [20,65]. Hence, similar to the CO and H2 yields, it has also been indicated as a means to evaluate the extent of secondary reaction activity and the quality of the pyrolysis oil. A qualitative analysis of the composition of the wood pyrolysis liquids, obtained for different thermal conditions, is presented by Evans and Milne [66,67]. The nature of liquid products from pyrolysis and gasification depend not only from the biomass type, but mainly from the conversion technology, that is, the severity of the thermal treatment [45,66-68] and the presence of char and other catalysts [49,50,69-71]. Primary vapors (oxygenates) are associated with reaction temperatures below 673-773K, followed by hydrocarbons or secondary tars for temperatures up to 1123K and aromatic or tertiary tars above 1123-1273K. In this way, liquids produced from fast pyrolysis reactors and updraft gasifiers [30] usually present a chemical composition typical of primary products. On the other hand, secondary and tertiary compounds of tar are predominant in the high temperature fluidized-bed, entrained-bed and downdraft gasifers. Chemico-physical characterization of liquids generated from low-temperature processes (primary tars) has received significant attention and a review on these aspects has been recently produced [72], given their strategic importance as substitute fuel oil or for chemicals production [73]. From a chemical point of view, wood fast pyrolysis oils typically are a mixture of 30% water, 30% phenolics, 20% aldehydes and ketons, 15% alcohols and 10% miscellaneous compounds [73]. Studies on cellulose and other model compounds focused on product analysis report that levoglucosan, acetic acid, formic acid and hydroxyacetaldehyde are the chief products. Production of the latter is enhanced by the presence of alkali-metallic salts to the cellulose samples and occurs mainly at the expense of levoglucosan. Mild acid wash of cellulose or wood, by partial removal of the inorganic matter content, causes an increase the yields of levoglucosan. Pyrolysis products are qualitatively similar from both cellulose and

7
hemicellulose. The hydroxy-phenolics, guaiacols and syringols are derived from the lignin building blocks. Although the majority of studies on wood fast pyrolysis examines the chemical composition of the liquids only for optimal conditions (maximum liquid yield), some information is available [30] for the yields of about forty compounds on dependence of the heating temperature for slow pyrolysis of beech wood in a packed-bed reactor (yields already shown in Fig.3). The most abundant species, over a range of heating temperature of 600-900K, are acetic acid (4.8-5.5%), hydroxypropanone (1.1-1.6%), hydroxyacetaldehyde (0.7-2.3%), levoglucosan (0.2-0.8%), formic acid (0.4-0.6%), syringol (0.3-0.4%) and 2-furaldehyde (0.3-0.35%) (Fig.7). All the classes of compounds present an initial increase with temperature, reflecting the primary degradation of wood components, but secondary reactions in the vapor phase also take place, as testified by the maxima reached by furans, guaiacols and syringols. It is also evidenced [30] that faster heating rates enhance the formation of levoglucosan and hydroxyacetaldehyde with a diminution in the yields of carboxylic acids. Compared with low-temperature pyrolysis, a minor number of studies is focused on tars generated from high-temperature gasification of wood [44,45,66,67,74-77]. The transition from primary products to phenolic compounds and aromatic hydrocarbons, precursors of particulate matter formation, on dependence of the thermal severity is examined by Evans and Milne [44]. The authors suggest that "the decision to run a gasification system at high severity to crack tars should be balanced by a consideration of the tars that remain. The condensed aromatics in tertiary tars are harder to remove than the larger amount of primary or secondary tars produced under less severe gasification conditions". Although, as anticipated above, the yields of pyrolysis products have also been evaluated for several agricultural biomasses, in this case, information on the chemical composition of the pyrolysis liquids is not provided or limited only to the identification of the chief functional groups by means of Fourier transform infrared (FT-IR) techniques. The majority of these analyses is focused on one biomass only and no comparison is given with wood that can be considered as a sort of reference fuel. Furthermore, pyrolysis tests of the various feedstocks have been executed with different reactor configurations and experimental conditions. Hence, the consequent variations in the actual conversion conditions, which cannot be easily quantified, hinder the comparison between the samples examined. 2.3 Char reactivity Solid reactivity studies, useful for kinetic analysis, are mainly based on thermogravimetric measurements. Given the small sample amounts and the relatively slow heating rates, the weight loss versus temperature curves show several sequential zones, as in the example for waste wood exposed to nitrogen, air or carbon dioxide of Figs.9A-9B obtained with the data provided in [78]. The first zone of rapid (temperatures below 630K) weight loss (conversion up to 65%) is the pyrolysis (or devolatilization) stage, whose characteristics are affected by the presence of oxygen in the reaction environment. Char oxidation, adjoining solid pyrolysis, is completed at about 750K while char gasification with CO2 does not occur significantly for temperatures below 1000K. Rates of steam gasification are faster than those with CO2 (factors of about 2-5 for wood char [79,80]) and the gas has a higher calorific value. In any case, char gasification rates are always much slower than char oxidation rates which, in turn, are slower than solid devolatilization rates (for instance, see also [81]). Hence, combustion and especially gasification kinetics of wood and biomass is generally investigated with separate experiments for solid devolatilization and char conversion. Char conversion is more complicated than solid devolatilization as it is an heterogeneous process where the surface is the location of the chemical reactions [82]. Weight loss curves of lignocellulosic fuels in air, measured under dynamic conditions, are reported by several studies [83-98]. As anticipated in Figs. 9A-9B, two sequential zones appear corresponding to solid devolatilization and char oxidation. Also, from the qualitative point of view, the features of the

8
devolatilization stage remain unchanged with respect to those already extensively discussed in previous reviews [13,59,99]. In quantitative terms, the presence of oxygen has been observed [86,90,91,95] to anticipate and to increase the devolatilization peaks attributable to the components of lignocellulosic fuels. A comparison between curves obtained in air and inert atmosphere shows that the hemicellulose shoulder and the cellulose peak occur at lower temperatures (about 15K and 25-35K lower, respectively). Moreover, the cellulose peaks are about 1.5 times higher. In the absence of oxygen, the peak rate is followed by a wide region of very low values, essentially as a result of lignin decomposition [59,99]. In the presence of oxygen, given the relatively high temperatures, it is plausible that the activity of the combustion reactions is already important even before the second peak in the weight loss rate is observed. Char combustion also takes place according to a multi-step process [100-107]. Weight loss starts at relatively low temperatures even in inert atmospheres with the formation of gas (mainly CO and CO2) and small amounts of aerosols [106] (char devolatilization). More specifically, low-temperature cellulosic chars [100] undergo devolatilization according to three sequential stages, corresponding to the pyrolysis of partially decomposed and intact glycosyl units and decomposition of paraffinic and carbonyl groups. Chars obtained at temperatures above 673K only show the last stage (for a comprehensive analysis of these aspects see Antal and Gronli [7]). Moreover, fast heating rates during pyrolysis and higher temperatures during conversion reduce the importance of devolatilization with respect to oxidation during char conversion [104]. Char devolatilization, though not accompanied by significant weight loss, produces a more stable and relatively ordered carbon structure [100,108]. Moreover, it may be an important route for the formation of aromatic compounds [109] for low temperature processes such as smoldering combustion and slow pyrolysis. Indeed, for cellulosic chars, aromatic compounds (benzene, toluene, naphthalene, anthracene) are detected between 673-873K, whereas above 873K methane, benzene, hydrogen and carbon monoxide also appear. A first stage of devolatilization is also observed [110,111] during the combustion of biomass chars for conditions simulating pulverized fuel-fired boilers (nominal particle size 75-106 µm and gas temperature around 1600K). It leads to the removal of amorphous material and the release of a large part of the H and O content of the char (formation of CO, CO2, H2O and minor amounts of hydrocarbons and other gases). Successively, combustion takes place together with transformations of the inorganic constituents, that is, vaporization (especially Na and K), surface migration and coalescence, and the incorporation of metals (particularly Ca) into silicate structures. Finally, towards the conclusion of the conversion process, inorganic constituents are transformed from amorphous phases to crystalline forms. On the whole, a decrease in the char reactivity during conversion is observed, as the more reactive carbon is preferentially depleted and the transformation of the inorganic constituents reduce their catalytic activity. Contrary to the case of biomass or char oxidation where thermogravimetric curves are measured under dynamic conditions, static (isothermal) data are mostly produced for the analysis of the gasification kinetics which essentially considers char samples. The experiments foresee a period of char pre-heating (in N2) up to high temperatures (1273K), a retention period (10 min) and cooling down (25K/min) to the desired temperature still in an inert environment (figures are those in [112]). Finally, the gasification tests are carried out under isothermal conditions at selected temperatures, to provide data for kinetic analysis. The char gasification rate depends on temperature and concentration of reactants and varies with the conversion degree (for instance, [112,113]). Furthermore, the presence of CO and H2 inhibits char gasification in atmospheres bearing CO2 [112,114-116,] and H2O [114,115,117,118], respectively. These effects may be quantitatively very important. For instance, the presence of 30% CO

9
or H2 in the gas reduces the char reactivity by a factor of about 10 or 15 for CO2 or H2O gasification with no CO or H2 content [116,119]. The overall kinetics of char conversion is measured via the reactivity [1,112,116,119,120], R,:
tX
XtM
MR
∂∂
−=
∂∂−=
111
where M is the mass of the organic portion of the sample, dM/dt is the conversion rate and X is the degree of conversion
∞−−
=MMMM
X0
0
(M0 and M∞ are the initial and final values of the sample mass). It is recognized that the heterogeneous rates of char conversion are determined by the total surface area, the number of active sites per unit surface area, and the local gaseous reactant concentration. Consequently reactivity depends on three chief characteristics of the sample: chemical structure, inorganic constituents and porosity. To understand the reactivity of solid/gas reactions, the detailed interactions between the two phases should be taken into consideration. For a given position of the surface within the char particle, the local heterogeneous reaction rate is determined by the local value of the gas concentration [1] and it is a measure of the intrinsic surface rate of the solid/gas reaction (the intrinsic rate is per unit internal surface area whereas the overall rate (eqn.1) is per unit particle (or sample) mass). For lignocellulosic fuels the reactivity generally increases with conversion, as shown in Fig.8 for biomass chars while undergoing combustion for the conditions typical of dynamic thermal analysis [121]. However, a maximum or a minimum may also appear in the reactivity versus conversion curves. 2.4 Kinetic and mass-transfer control When neglecting the devolatilization stage, the reactivity of a solid particle is governed [118] by: 1) film diffusion of oxidizing/gasifying agent, 2) diffusion through the ash layer and the particle, 3) adsorption onto the reaction surface, 4) chemical reaction, 5) desorption of product gas from the surface, 6) diffusion of product gas through the particle and the ash layer, 7) film diffusion back into the ambient gas. Apart from the surface chemical reaction (item 4), the remaining are mass-transport steps. Based on the Thiele modulus (ratio of the overall reaction rate to internal diffusion rate) and the effectiveness factor (ratio of the actual reaction rate to that which would occur if all the surface throughout the internal pores were exposed to the gaseous reactant at the same conditions as that existing at the external surface of the particle) [122], three main regimes are introduced in solid conversion [1,8,9,82,123-126]. For the regime I, the Thiele modulus should be small and the effectiveness factor should be ideally unity [3]. This regime is established for low temperatures and char particles so small that the diffusion rate is much faster than the chemical reaction rate (kinetic control). Conversion occurs throughout the particle with changes in density but with a constant size (conversion time and reaction rate independent of the particle size). Diffusion effects during char gasification may be important even for the conditions typical of thermogravimetric analysis [127-130]. An extensive analysis [129] about the effects of particle sizes (0.06-2.1mm olive stone particles), CO2 partial pressure (20-50kPa) and temperature (1073-1223K) shows that they cannot be neglected unless very small-sized particles (0.06mm) and moderate temperatures are considered (typically below 1023K [131], 1173K [132], 1223K [129]). Also, given that the role played by diffusion is a function of the conversion degree, it is suggested that,
(1)
(2)

10
before kinetic evaluation, an assessment of these effects should be made for the entire range of conversions and operating conditions. Once the particle size is increased, it is likely that the rate of gaseous species diffusion in the pores becomes comparable to the reaction rate, leading to a limited gas penetration into the char. When intra-particle mass transfer is the controlling process (regime II) there is only a limited penetration of the gaseous reagent into the particle, the Thiele modulus is much greater than unity and the effectiveness factor much less than unity. Conversion is confined to the exterior surface of the particle, the size diminishes with no apparent change in density. Consequently the reaction rate is proportional to the external surface of the particle and the conversion time to the particle diameter. For control by external mass transfer from the bulk gas to the external surface of the particle (regime III), where the reaction takes place, the char shrinks at constant density. The reaction rate is proportional to the external surface of the particle and a mass transfer coefficient. Heat transfer may also play an important role in char conversion, as the exothermicity and endothermicity of combustion and gasification, respectively, may enhance the temperature gradient between surface and core of the particle, especially for thick particles and severe heating [121,133]. Both mass and heat transfer effects are enhanced by high temperatures. In fact, given the exponential increase in the reaction rate, in the absence of an adequate increase in the supply of the gaseous reagent and a reduction (combustion) or an increase (gasification) in the rate of external heat supply, the differences in temperature and gas concentration between the bulk of the gas and the reaction sites also increase. The high exothermicity of char combustion also causes uncertainty in both particle temperature and active site concentration [1] and may give rise to reaction rates much faster than the typical response time of measuring devices and feedback control systems in kinetic analysis [121]. Also, the attempts made to reproduce the high external heating rates encountered in combustion at a laboratory scale [134] "would result in mass/heat transfer controlled conditions for the laboratory equipment of the kinetic measurements, and the results would reflect the hydrodynamics of that specific piece of equipment instead of the true combustion rate". The occurrence of homogeneous reactions in the gas phase surrounding the particle depends on temperature, pressure and size of the particle, thus further complicating the interpretation of the data. CO and CO2 are products of char oxidation but their yields vary with the operating conditions. In particular, CO formation is favored at high temperatures, whereas CO2 may be a product of both char or CO oxidation. The ratio CO/CO2 is generally correlated by an Arrhenius rate expression [1]. 2.4 Factors affecting char reactivity Char reactivity is affected by the morphological structure which, for a given fuel, is especially influenced by the release rate of volatiles, that is, the pyrolysis conditions, and the amount and composition of inorganic matter [135]. The effects of pyrolysis conditions on the properties and reactivity in air of lignocellulosic chars have been investigated in several studies in relation to the following parameters: heating rate, temperature, residence time and pressure. High heating rates during pyrolysis produce a more reactive char for both oxidation [134,136] and gasification [79,113,114,137-140]. From the quantitative point of view, variations in the heating rate during pyrolysis between 1 and 300K/s for a final temperature of 873K result in a time needed for complete oxidation of small-sized pine wood char particles decreased by about one order of magnitude [134]. As for gasification with steam or carbon dioxide, chars produced from birch wood at fast (free-fall reactor) and slow (thermobalance) heating rate present a variation in the reactivity by factors of about two-three [137]. Also, for char particles produced from 10mm thick beech wood, the times for

11
complete gasification (20% steam in N2 at 1200K) vary in a ratio of 1-2.6 for external heating rates between 2.6-900K/min during pyrolysis [79]. While, for slow heating rates, volatile pyrolysis products are released through the natural porosity and no major change takes place in the particle morphology [141], for fast heating rates the original cellular structure is lost [142] as a consequence of melting phenomena [138-140]. Fast volatile release produces substantial internal overpressure and coalescence of the smaller pores, leading to large internal cavities and a more open structure of both wood [136] and lignin [138]. Hence, for pyrolysis carried out at atmospheric pressure, chars produced at slow heating rates mainly consist of a micropore structure whereas those obtained with high heating rates mainly present macropores [136,138-140]. An analysis of the literature review on coal chars [79] leads to the conclusion that the surface area developed by mesopores and macropores is a better indicator for the reactive surface than the total surface area also including the contribution of micropores (below 2nm) which probably do not participate in the reaction. Therefore, the increase of the char reactivity with the heating rate during pyrolysis can be explained by the occurrence of gasification reactions mainly on the surface of large pores [138] which may also be associated with a higher total surface area [139-140] and/or a higher concentration of active sites [138]. It is, however, important to point out that the reported surface area values are affected by the measurement technique (N2, Ar or CO2 adsorption or Hg intrusion) [79]. The increase in the maximum rate of weight loss and volatile yield observed at high heating rates during pyrolysis also make shorter the residence time of tar vapors in the pores, thus reducing the activity of condensation reactions [142] and preventing char agglomeration and condensation of fragments on the char surface [138]. These fragments provide lesser active reaction site concentration than char formed from primary reactions of wood decomposition [113]. The reduced activity of condensation reactions during pyrolysis of wood blocks subjected to microwave heating [143], which results in the absence carbonaceous deposits over the microporous char structure, is also observed to produce chars with a higher specific surface area (factors of about 2.5 compared with conventional heating). Some considerations can also be made in relation to the physical structure of chars produced from primary and secondary reactions, taking as representative of the latter, those produced from bio-oil devolatilization [55-58]. In this case it can be observed that microporosity is completely absent and the solid surface of the char is compact, glossy and brittle with the presence of intact or burst bubbles. Moreover thermogravimetric measurements [56] show that the peak of the combustion reaction is reduced (with respect to a primary char produced under fast heating) up to factors of about 2.5 and retarded by about 30-60K. In terms of elemental composition, rapid heating during pyrolysis causes a weak diminution in the C content (to the advantage of H and O) of the char [7], but differences tend to disappear at high temperatures. The observation of the char microstructure confirms the presence of larger amounts of oxygen containing groups [142] and, in general, higher contents of hydrogen and oxygen [136] which are related to the availability of active sites and thus to enhanced reactivity [1]. In particular, char oxidation experiments [136] show that the CO/CO2 ratio is always lower for char produced at slow heating rates (the more ordered the char structure the lower the oxygen content of the char). A smaller amount of the surface is covered with stable carbon-oxygen C(O) complexes in favor of reactive complexes, so that a faster rate of CO2 formation is established (and a lower CO/CO2). Moreover, fast decomposition seems to produce defective carbon microcrystallites having a higher concentration of active reaction sites [113]. The heating rate established during pyrolysis is also a function of the particle size so that, for thick particles, the heating conditions are different on the surface and the more internal zones [114]. Hence, it is easily understandable that the reactivity decreases for char produced from thick [144] or densified [145] samples. Variations in the final pyrolysis temperature between 773-873K (values of interest in fast pyrolysis) or the retention time (20-100s) do not appear to play an

12
important role on the combustion reactivity of small sized char particles [134]. The negligible role played by this range of final temperatures is also found [104] for chars produced from thick particles of different wood species. Higher temperatures (above 1073K) cause a significant diminution in the char reactivity as reported for both combustion [136,146,147] and gasification [113]. Indeed, the specific surface area slightly increases with the devolatilization temperature up to values of 1078K and then decreases, owing to structural ordering and micropore coalescence, with thermal deactivation (or thermal annealing) of the char [136]. Similar to the influences of pyrolysis conditions on char reactivity, a high heating rate during char pre-heating (before attaining the gasification temperature) also increases the reactivity [148], but prolonged exposure of char to high temperature reduces the reactivity by thermal annealing [149]. The C content of char becomes successively higher as the pyrolysis temperature is increased. For instance, for pine wood and fast pyrolysis [19], it is comprised between about 78 and 89% as the heating temperature is varied from 722 to 835K. Hence, the increased structural ordering of the carbon matrix with temperature [109] is responsible for lowering the concentration of reaction sites [113]. Increasing the solid residence time provokes effects similar to temperature, again improving structural ordering in the resulting char [113,150]. Similar results are also reported for black liquor char [149] with a reduction in the porosity (and surface area) and a rearrangement of the char structure at a molecular level. Furthermore, in the presence of CO, it is suggested that deposition of solid carbon, as a consequence of the reaction 2CO�C+CO2, causes a decrease in the initial rate of gasification. Decreasing the (external) pressure during pyrolysis gives rise to higher reactivities of wood chars in carbon dioxide bearing atmospheres [139,140,148]. From the morphological side, larger cavities with thinner walls inside the char particles are formed, probably also owing to swelling and, at high heating rates, formation of clusters associated with melting and fusion [139,140]. The increase in the reactivity with the decrease in the external pressure during pyrolysis is attributed to a larger surface area and a reduced graphitization in the char structure [139,140] or a higher content of oxygen [148]. Furthermore, it should be noted that this result is consistent with the lower reactivity of char generated by secondary reactions of tar in the vapor phase, whose amount is highly reduced for pyrolysis carried at low pressures [113] and, in general, by short residence times of tar vapors inside the pyrolysing particle [13]. Char obtained by vacuum pyrolysis is also a more suitable feedstock for activation with steam compared to char produced at atmospheric conditions, again as a consequence of the limited presence of carbonaceous deposits on the surface and inside the char particles, resulting from the partial adsorption of hydrocarbons formed during the devolatilization stage [151]. Indigenous inorganic matter may act as catalyst and improve the efficiency of biomass gasification and combustion, although the catalytic activity is affected by metal dispersion (sintering), depending among others on the pyrolysis conditions [152] and the mode of their occurrence (as minerals (mainly in coals), salts or organically bound compounds (biomass)) [147]. Alkali metals, such as sodium and especially potassium, which is present in larger amounts, are known to be active catalysts for the oxidation reactions [147,153]. The oxides and salts of alkaline (Na, K) and alkaline earth (Ca, Mg) metals are also good catalysts for steam gasification of carbon [5]. A significant number of char samples of different origin show gasification rates in CO2 linearly dependent on the concentration of the dominant metals in ash (potassium and calcium) [154]. Alkali metals also catalyze the polymerization reactions of tar vapors, during pyrolysis, thus increasing the yields of chars which also become less porous and accessible to gaseous reagents [135]. A confirmation of the important role played by mineral matter on the reactivity of char is obtained by examining the effects of pre-treatments consisting of water or mild acid leaching, which cause partial demineralization (see, for instance, [42,155,156]). Chars produced from leached biomass present a higher surface area with respect to those generated from untreated samples [105]. However, oxidation

13
and gasification reactivities are significantly reduced [42,105,147,157] (for instance, factors of 30-40 are reported for straw char combustion [147]). In other words, water- or acid-soluble minerals have a higher influence on char reactivity than the surface area does. The ratio between carbon and the catalytically active substances is another important parameter for the char reactivity in relation to both gasification [157,158] and oxidation [121]. Indeed, the char reactivity increases with conversion, at low and intermediate levels, essentially as a consequence of the increase of the surface area during the course of gasification. At high conversion levels (beyond 70%) a very steep rise is generally observed, which cannot be plausibly explained by the development of the surface area, but may be attributed to an increased catalytic effect of the metallic constituents (mainly potassium and lower amounts of sodium) of ash, following the increase of their concentration during char conversion (see Fig.8). Inorganic matter may also cause an increase in the char reactivity only up to certain temperatures, followed by a diminution owing to modifications in the chemical occurrence of the catalysts and/or changes in the char structure [147,152,153,159]. For peat char [114], the steam gasification rate increases to a maximum before decreasing as a function of conversion, as a consequence of the progressive deactivation of active substances such as calcium. Deactivation can result from sintering and agglomeration, the formation of new mineral compounds or the occurrence of reactions between the mineral compounds and the gasifying agents. The changes in the oxidation rate versus temperature observed for chars from pectins/uronic acids are also attributed [160] to modification in the alkali metals. These bound in pectins initially as the salt of uronic acid group decompose (at 573-673K) to produce a metal complex in the char which apparently inhibits/shields the carbon char from oxidation. Temperatures above 873K are required to decompose the metal-char complex and to permit the oxidation of surrounding carbon. Although ash elements can exert a catalytic role on the reactivity of char, their presence can decrease the porosity to such an extent that the active surface area is also highly decreased [135]. For instance, in the case of bark char [107], the large presence of calcium and transparent solidified material over the porous surface and the reduced porosity, with respect to wood char, certainly contribute to diminish the extension of the active surface area and, through this, the apparent reactivity of the material. Indeed, it is easily understandable that the more internal structure becomes hardly accessible to the gaseous reagents for the heterogeneous reactions. Differences between the reactivity of chars obtained from different wood species are small [104,119]. In relation to this issue, De Groot and Shafizadeh [161] write that "the distinction between hardwoods and softwoods is apparently insignificant, apart from the catalytic effects of the ash content", again pointing out the importance of inorganic matter naturally present in lignocellulosic fuels. Probably this is also the main factor responsible for the large differences exhibited by biomass (agricultural residues) char with respect to wood char for both combustion [121] and gasification [112,120]. 3. Chemical kinetics of biomass/char combustion and gasification In this section, the structural and kinetic contributions in the mathematical description of char reactivity are evidenced. Then mechanisms, rate laws and structural models are presented including a survey of the kinetic parameters currently available for the processes of char gasification and biomass/char combustion. As the carbon-hydrogen reaction is too slow for most practical applications [1], only gasification with CO2 and H2O is examined. 3.1 Kinetic and structural effects during char conversion The reactivity R, that is, the rate per unit particle (or sample) mass, as defined by eqn.(1) is usually measured for the evaluation of the overall kinetics of char conversion. Models of char reactivity are

14
often indicated as structural or volumetric [162]. Structural-type models try to describe the internal solid matrix (grain models) or the internal pore structure (pore development models) and the modifications taking place during char conversion, so requiring significant effort for the independent determination of model parameters. In the volumetric-type models, changes in the porous structure during conversion are described by means of empirical correlations where porosity or pore properties do not appear explicitly (the conversion, X, is the sole variable introduced in almost all the models). In this review a distinction between the two types of models is not considered. Indeed, the reactivity, which is a function of temperature, concentration of gaseous reactants and degree of solid conversion, can always be expressed [120] by means of a chemical kinetic term, rc (T,Pi), accounting for temperature and reactant partial pressure effects, and a structural term, rs (X), implicitly or explicitly assumed to describe the effects of available internal surface (actual surface over initial surface, S(X)/S0), available active or reactive sites (Cf) and pore evolution (porosity, pore size and shape), and to be invariant over the temperature-pressure domain:
( ) ( )XrPTrR sic ×= ,
The assumption of invariant structural profile is not always valid as evidenced [120], for instance, when considering the effects exerted by temperature and partial pressures of gaseous species on the activation of catalytic sites. Therefore, a range of temperatures and partial pressures of gasifying agents should always be specified for the validity of this assumption. Only if the assumption of invariant structural profile is approximately valid, a reference profile (Rref) can be used to eliminate the structural effects from reactivity and to introduce a normalized reactivity (Rn) representative of intrinsic kinetics:
crefn rRRR ==
Given the dependence on conversion, a specific conversion value is selected for Rref, typically the initial 10 or 50% (the latter value has been selected by the most recent investigations [112,116,119,120], with R(X)=R50F(X), where F(50%)=1 and R50 is the representative reactivity) or a normalization of the structural term is made with respect to the maximum value [105]. As anticipated, the conversion of a char particle results from the interaction of several physical processes, among which the most important are mass transfer through the pores within the particle and mass transfer of the gaseous reagents to the exterior of the particle, and intrinsic kinetics of the reaction. In order to evaluate the intrinsic chemical kinetics, conversion should occur in the regime I and thus to separate the contributions of chemistry and transport phenomena, reaction conditions (low temperatures and high flow rates of the gaseous reactant), sample characteristics (low amount of mass, small particle size) and sample position in the reaction environment (for instance, crucible location of the thermogravimetric system [127,128]) should be adequately chosen. 3.2 Mechanisms and rate laws of char gasification The carbon dioxide gasification of char has been extensively studied and the following oxygen exchange mechanism is postulated [1,116]:
( )( ) 2
2
12
COCCOOC
COOCCOC
fk
kf
+→+
+→+
( ) fk CCOOC +→ 3
where k1, k2 and k3 are the usual Arrhenius rate constants, Cf represents an active carbon site and C(O) a carbon-oxygen complex. The presence of CO produces an inhibiting effect by lowering the steady-
(3)
(4)
(a1)
(a2)
(a3)

15
state concentration of C(O) by the reaction a2. The gasification rate, in accordance with reactions a1-a3 and applying the steady-state assumption for the C(O) complex, can be expressed as:
( ) ( ) 23132
21
1 COCO
COc PkkPkk
Pkr
++=
where PCO2 and PCO are the partial pressure of CO2 and CO. When the CO concentrations are small and/or the inhibiting effect exerted by this species is not taken into account, a simple global model can be applied:
COCOC 22 →+ , ( ) nCOc PRTEAr 2exp−=
(A is the pre-exponential factor, E the activation energy, R the universal gas constant and n an empirical parameter). The steam gasification of char also takes place according to several steps as summarized in [1,119,163]. Gasification with steam is more complex than CO2 gasification because of the action of H2, CO2 and CO following the equilibrium of the water gas shift reaction CO+H2O=CO2+H2 (this reaction usually proceeds very slowly in the absence of catalysts or inorganic matter in biomass). The amount of CH4 formed at atmospheric pressure is small (usually neglected) and is thought to be due to a side reaction between carbon and hydrogen [164]. At high pressures, the amount of methane, formed as a consequence of the direct attack of steam on certain carbon atoms, is considerable and cannot be ignored. Steam gasification of carbon takes place according to two basic mechanisms indicated as the oxygen exchange mechanism and the hydrogen inhibition mechanism [119,163]. The steps to be considered are:
( )( ) OHCHOC
HOCOHC
fk
kf
22
2
21
2
+→+
+→+
( ) fk CCOOC +→ 3
( )24
2 HCHC kf →+
( ) 25
2 HCHC fk +→
( )HCHC kf →+ 6
221
( ) 27
21
HCHC fk +→
(k1-k7 are Arrhenius rate constants). The oxygen exchange mechanism consists of steps b1-b3. The hydrogen inhibition mechanism may consist either of steps b1, b3 and b4-b5 or steps b1, b3 and b6-b7. For the oxygen exchange mechanism, hydrogen inhibition is due to the equilibrium of the dissociation reactions b1-b2. For the first hydrogen inhibition mechanism, the formation of the C(H)2 complex is the reason of the inhibition. In the other case, a dissociative chemisorption of H2 on the active sites occurs and in this way the active sites become not accessible for the oxygen transfer with steam. In all cases, the surface rate equation is formally identical [119]:
( ) ( )2231
21
1 HOH
OHw pfPkk
Pkr
++=
(a5)
(a4,6)
(b1)
(b2)
(b3)
(b4)
(b5)
(b6)
(b7)
(7)

16
where PH2O and PH2 are the partial pressures of H2O and H2, respectively, and f(pH2) depends on the selected mechanism. The following expressions are obtained for the oxygen exchange
( ) 23
22 HH P
kk
pf =
the hydrogen inhibition by formation of the C(H)2 complex
( ) 25
42 HH P
kk
pf =
and the hydrogen inhibition by formation of C(H) complex
( ) 5.02
7
62 HH P
kk
pf =
Similar to CO2 gasification, the majority of kinetic analyses simply considers a global model for char gasification with steam:
22 HCOOHC +→+ , ( ) nOHc PRTEAr 2exp −=
The expressions of the char conversion rates with parameter values [112,113,115-120,129,132,133,146,148,157,158,161,162,165-169] are summarized in Tables 1-2 for CO2 and H2O gasification, respectively. The temperature and pressure applied during gasification, the origin and size of the particles and the pyrolysis conditions are also reported. To facilitate the comparison, a proper re-examination of the original expressions has been made to model the progress of the reaction in terms of dX/dt, to include a dependence on the partial pressure of the gasifying agent (instead of the molar concentration) and to express the structural contribution (in square brackets in Tables 1-2) by means of the conversion, X, so that the general expression takes the form:
( )XrKPdtdX
Si ×= , ( )RTEAK ii −= exp , OHCOi 22 ,=
Several authors [112,116,117,119,120,129,131,132,146,162,165] propose a rate of char conversion with a kinetic contribution and a structural term, but simple pure kinetic laws have also been used [113,115,118,133,157,158,161,166-169]. In some cases [113,115,117,118,133,157,158,166,167] the pre-exponential factor also incorporates the partial pressure effects (not explicitly described in the reactivity model), so that the kinetic parameters have a more limited range of validity. Tables 1-2 show that the experiments have been carried out at atmospheric pressure (apart from [148] for CO2 and [117] for H2O), within a range of partial pressures of the gasifying agent. The presence of CO [112,116,133] or H2 [117,119], to investigate the inhibition on the CO2 and H2O gasification, can be found only in a few experiments. The results obtained by Groeneveld and Van Swaaij [162], using a mixture of CO2 and H2O, include a power-law dependence of the kinetic rate on the sum of the CO2 and H2O partial pressure. The variability of the pre-exponential factor, over two orders of magnitude, probably takes into account the changes in the composition of the gaseous mixture. Experiments carried out to determine the gasification rates generally foresee the production of isothermal data, according to the procedure described above (except for the dynamic analysis by Cozzani [146]) with significantly different ranges of temperatures (comprised between 973-1280K for CO2 and 823-1273 for H2O). A large majority of the studies is focused on wood but some consideration is also given to agricultural biomasses such as coconut shells [133], rice husks [132], straw [120,167], olive stones [112,129], grapefruit peels [157], and refuse derived fuel (RDF) [146]. The char samples
(8)
(9)
(10)
(11)
(12)

17
analyzed are mainly generated under conditions of slow pyrolysis justified by the use of thick particles in gasifiers (between 6-200mm for fixed-bed reactors and 3-50mm for fluidized bed reactors [68]). The thermal conditions during pyrolysis are varied in [113,117] but their effects are modeled only in terms of kinetic parameters and not explicitly by the expression of the gasification rate. An attempt in this direction is the treatment proposed by McCarthy [170] where, apart from the usual Arrhenius dependence on temperature, the effective rate constant also considers a contribution of the form exp(-Ed/RTp), where Ed is the energy of thermal deactivation and Tp the temperature of char preparation. An examination of the results of the kinetic evaluations (Tables 1-2) shows that the exponent of the gaseous reactant pressure is around 0.4-0.6 and 0.4-1 for CO2 and H2O gasification, respectively. The corresponding activation energies vary between 88-250kJ/mol (with a large part of the values around 200-250kJ/mol) and 143-237 kJ/mol (with a large part of the values around 180-200kJ/mol). The lower E and the higher n values evaluated for H2O gasification are probably representative of the higher rates with respect to those of CO2 gasification [79,80]. Apart from a very few exceptions, it appears that high activation energies are typically computed for the process of char gasification. In fact, a value of 210kJ/mol is also proposed by an empirical correlation [6] for the CO2 gasification rate of thick (10-34mm) char particles (apart from the Arrhenius temperature law a power law dependence is also postulated on the CO2 concentration (exponent equal to 0.71) and the initial particle radius (exponent equal to -0.81)). In accordance with the analysis about the factors affecting the char reactivity, the kinetic parameters are found to depend on the pyrolysis conditions [113,117,148,157,158,166] and the ash content/composition of the samples [157]. Also, the estimated values of the kinetic parameters for the global one-step reaction appear to be altered by the composition of the gaseous mixture [117]. As expected, when a structural term is not explicitly included in the reactivity expression, a dependence on the conversion degree is also evidenced [157,158]. However, while all the investigations are concord on the qualitative effects exerted by various parameters on the measured char reactivity, univocal trends in the estimated values of the kinetic parameters (E,n) cannot be observed. Thus the diminution in the activation energy (from 218 to 138kJ/mol) as the heating rate is decreased or the temperature is increased during pyrolysis, reported by Kumar and Gupta [113] and attributed to an increased reactivity, does not find a confirmation in the evaluations of Ref. [158]. Similarly, the parameters for the CO2 gasification kinetics of wheat straw char are roughly the same as wood [120] although it is well known that herbaceous biomass presents high alkali metal contents which catalyze the gasification reactions. On the other hand, the much lower activation energies (133-143kJ/mol) of chars from olive residues are attributed [112,129] to the high potassium content of the ashes. Moreover, water extracted char, where the alkali metal content is presumably low, present activation energies lower than those of untreated char in [157]. The estimated values of the activation energy do not show a clear trend on dependence of conversion [157,158]. Finally, Barrio and coworkers [119] examine the influence of the reactivity definition on the computation of the kinetic parameters. It is found that the selection of a representative reactivity value does not affect the estimated activation energies and reaction orders. Given that the definitions of representative reactivity are related to a fixed degree of conversion, the differences appear as a multiplying factor incorporated into the pre-exponential factors. In this regard the accuracy of the calculations might also be affected. From the analysis of the data of Tables 1-2 it can be understood that, in addition to the differences in the experimental technique, operating conditions and sample characteristics, the method of mathematical analysis and heat/mass transfer intrusions (these especially related to the particle size/mass and the reaction temperature) may significantly affect the computed values of the model parameters. It is explicitly stated that very small particles (below 0.06mm) are used only in a few cases [116,119,120,129], while other studies are based on particle sizes of about 0.4-0.8mm or even up to

18
several milliliters [113,118]. Furthermore, as already noticed, gasification temperatures above 1173K [129,132] introduce non-negligible mass transfer limitations. To carry out a comparison in terms of actual values of the kinetic contribution in the reactivity, Figures 10-11 propose an Arrhenius plot for rc computed over the temperature range 800-1250K at atmospheric pressure of the gasifying agent (when all the model parameters are available). The results by Bhat and coworkers [132] are excluded because higher by several orders of magnitude in comparison with the other cases, whereas the lower and upper boundaries provided for the pre-exponential factor by Groenenveld and van Swaaij [162] are referred to CO2 and H2O gasification, respectively. Apart from the expected role played the activation energy values (for instance, see the much slower slope of the Arrhenius plot for the kinetics with low activation energies [112,129,148] compared with that of the group of high activation energies in Fig.10), it can be observed that the actual kinetic contributions are significantly different. For instance, given a temperature of 1000K, rc varies between about 3x10-5s-1 [162] to 1.3s-1[148] for CO2 gasification and between 5x10-6s-1 [169] to 2x10-2s-1 [131] for H2O gasification. The more complicated Langmuir-Hinshelwood kinetics is considered only in a few cases for both CO2 [82,112,116,171-173] and H2O [82,119,168] gasification. The agreement between predictions and measurements of the reactivity versus the partial pressure of the gasifying agent at the various temperatures is improved by these more complicated mechanisms [119]. Not all the studies provide an estimate of the kinetic parameters, but an examination of the activation energies for the reaction mechanism a1-a3 of CO2 gasification, as collected by Barrio and Hustad [116] and taking into account successive work [112], shows that E2 always assumes very low or negative values, a consequence of a kinetic constant for the reaction a2 practically independent of the temperature. As for the other two reactions, E1 is in the range 60-245kJ/mol and E3 in the range 120-421kJ/mol. The higher values of the latter support the speculation that the gasification of carbon is the limiting step in the mechanism a1-a3. Some evaluations of the Langmuir-Hinshelwood kinetics of char gasification with steam, based on the oxygen exchange mechanism b1-b3, have been compared [119]. In this case the activation energies are quite high for all the reactions of the mechanism b1-b3 (E1=149-214kJ/mol), E2=140-280kJ/mol, E3=225-273kJ/mol) but the third reaction (carbon gasification) presents slightly higher values again indicating that carbon gasification is the slowest step. 3.3 Mechanisms and rate laws of char combustion The reactions of carbon and oxygen have been extensively investigated for coal char ([1,4] and Refs.1-11 reported by Bews and coworkers [3]) but still present aspects that are poorly understood. However, it can be assumed that the mechanisms of char combustion summarized by Hurt and Calo [4] for coal chars are also, in principle, applicable for those originated from lignocellulosic fuels. The most widely used treatment is based on a simple global reaction
22 COCOOC →+ , ( ) nOc PRTEkr 20 exp−=
where PO2 is the partial pressure of O2. In reality, the carbon-oxygen reaction consists of a series of adsorption and desorption processes including the following [4,174]:
( )OCOC kf 22 1
2 →+
( ) COOC k→ 2
( ) fk CCOOC +→ 232
( ) ( )OCCOCOOOC k +→+ 24
2
( )OCCOCOC kf +→+ 5
2
(13)
(c1)
(c2)
(c3)
(c4)
(c5)

19
where k1-k5 are Arrhenius rate constants. The reaction c1 represents the chemisorption of oxygen on active sites and the reactions c2 the formation of CO through desorption (similar to the gasification mechanisms, Cf is the number of active or available sites and C(O) a carbon-oxygen complex which occupies the site). Formation of CO2 by surface reaction is also observed (reaction c3) or by interaction of the gas phase oxygen with surface complexes (reaction c4) which may or may not involve the generation of a new complex C(O) on the product side. The reaction rate of CO2 and carbon (reaction c5) is much slower than that of O2 and carbon. Therefore, reaction c5 is usually excluded as a significant step, when considering the carbon-oxygen reaction. Semi-global mechanisms have been modeled in terms of kinetic laws [4] of the Langmuir-Hinshelwood form. The simplest of these, taking into account reactions c1-c2, is:
221
221
kPkPkk
rO
Oc +
=
whose parameters, however, have not yet been evaluated for the chars of interest in this review. A one-step global reaction is used by several authors to describe the process up to complete conversion [86,104,107,121,122,134,147,175,176] but multi-step models have also been proposed. The additional steps concern the low-temperature devolatilization of char [104,107] or both devolatilization and combustion of char [105,107], but none of the reactions can be referred to the mechanism c1-c5 previously introduced. Parallel reactions are always proposed but a comparison with series reactions [177] shows that kinetic evaluation produces invariant activation energies and pre-exponential factors (small variations in the amounts of volatiles released for each step). The expressions for the rates of char conversion with the estimated kinetic parameters for the one-step models and the quantitatively most important combustion reaction of the multi-step models (the parameter νc represents the corresponding amount of volatiles generated in terms of solid mass fraction) are summarized in Table 3 (the structural contribution is again enclosed by square brackets). Information is also provided about the conditions of char production and combustion. Several authors [104,107,121,147,176] incorporate the dependence of the reactivity on the oxidant partial pressure into the pre-exponential factor, so that the results obtained are rigorously valid only for the gaseous mixture (air) used in the experiments. Feedstocks include wood, agricultural biomass, RDF and wood components (cellulose, lignin). Both moderate [86,104,107,121,122,147,175] and fast [104,134] heating rates have been applied during pyrolysis. Flash carbonization [178] is used for the samples studied in [105]. The char combustion experiments have been conducted either under static [134,175] or dynamic [86,104,105,107,121,122,147,176] conditions with maximum temperatures between 623-1273K (typically a value of 873K is used). Finally, the majority of the experiments examines air or oxygen concentrations below the air values. Significant variation is shown by kinetic parameters, with activation energies roughly comprised between 76-229kJ/mol for the chief combustion reaction. Quantitative differences are caused by the same factors as already pointed out for the gasification kinetics (biomass properties, operating conditions and devices of the experiments, pyrolysis conditions, amount and composition of ashes). However, it is known that, for dynamic thermogravimetric experiments, differential (versus integral) data should be used in kinetic evaluations because the details of the weight loss process are better shown [13]. Moreover, low values of the activation energies are generally produced when a single-step reaction is applied to fit a temperature dependence that arises from the occurrence of different reactions in different temperature intervals [105]. Another aspect to be taken into account is that, apart from the most recent studies [104,105,107], kinetic evaluations have been made by means of one experiments only so that it cannot be excluded that the results are affected by compensation effects.
(14)

20
Specific investigations on the variability of the kinetic parameters confirm that the use of integral measurements and the disregard of the devolatilization stage give rise to low values of the activation energy of the combustion reaction. More specifically, a n-order global reaction, which produces a poor description of the differential curves, requires relatively low activation energies (around 100kJ/mol) for both wood char [104] and oak bark char [107]. The combination of a global one step reaction of char combustion with a first-order reaction for the devolatilization stage results in accurate predictions of both integral and differential weight loss curves [104] and a higher activation energy of the combustion reaction. As shown by the example for pine wood char (produced by slow pyrolysis) in Fig.11, the devolatilization stage foresees the release of a relatively small amount of volatile matter (generally between 16-20%), but this model is accurate and presents an activation energy of the combustion reaction of 183 kJ/mol (versus 114.5 kJ/mol for the one-step global reaction) [104]. The devolatilization stage is well described by relatively low activation energies [104,105,107] and an increase in the number of reaction steps (from one to two) does not improve visibly the accuracy of the predictions [104]. Excluding the cases of low activation energies estimated by means of the one-step global models and the integral data [121,134,175,176], Table 3 shows activation energies of the combustion reaction comprised in the range 140-230kJ/mol, with the upper boundary comparable with the values reported for the low-temperature zone of coal char or graphite combustion [3]. The differences in the char reactivity deriving from the wood species can be taken into account essentially by the pre-exponential factor [104]. For bark char [107], the combustion reaction is associated with a lower activation energy (169 versus 182kJ/mol) which may be partly explained by the differences in the morphological structure of wood and bark chars. A clear trend in the activation energies, introduced by demineralization of the biomass samples, cannot be seen from the values reported for straw [147] or corncobs [105], although a diminution in the reactivity is evident. Moreover, the activation energy of both the one-step combustion reaction and that of the combustion stage in multi-step models become significantly higher for the chars generated from thin wood particles in fluidized-bed reactors (167 and 229kJ/mol, respectively) [104]. On the other hand, an increase [147] or a slight decrease [105] in the activation energy of the combustion step is reported as the pyrolysis temperature is increased (and the reactivity decreases). Hence, a clear trend in the estimated kinetic parameters is not shown as the char reactivity increases as a consequence of fast heating rates and/or low temperatures during pyrolysis. Diffusion effects and deviations of the actual conversion temperature with respect to the programmed (assigned) value may also play an important role in the estimated values of the activation energies. For conversion in the regime II and a Thiele modulus above 3 it is indicated [126] that the activation energy of the combustion reaction approaches half the intrinsic value. Moreover, for the regime III, the observed activation energy is small and the observed reaction order with respect to oxygen is unity. It is worth noting that evaluations of the Thiele modulus for pulverized bagasse char lead Luo and Stanmore [122] to conclude that the combustion reaction, for the conditions of their study, occurs in the regime II. However, this conclusion is not supported by the high activation energy estimated (180kJ/mol). The reaction mechanism c1-c5 can be used to justify the variation in the reaction order with respect to PO2 as the reaction conditions are varied [134]. The reaction order with respect to PO2 will be 1 when the reaction c1 is the rate-limiting step (temperatures above 973-1073K). When the reactions c2 and c3 are rate-controlling (temperatures below 573-673K), a zero-order dependence is found. A competition usually exists between c1 and c2-c3, so that the reaction order is something between 1 and 0, as confirmed by the values listed in Table 3 and roughly comprised between 0.5 and 1. To carry out a comparison in terms of the actual values of the kinetic contribution in the reactivity, Figure 13 proposes an Arrhenius plot for rc computed over the temperature range 650-950K at

21
atmospheric pressure for the analyses where all the model parameters are available. When various feedstock or pyrolysis conditions are used only a few are taken as representative of the results (i.e. pine wood char [121,176], slow pyrolysis [104]). Also, the results obtained [147] for the straw char generated at high pyrolysis temperatures are not included in the plot because they produce rc values lower about two orders of magnitude with respect to those obtained at low pyrolysis temperatures (and comparable with the pyrolysis temperatures of other studies). It can be seen that the actual rc values vary over a range of about two orders of magnitude (given a reaction temperature of 800K, they are comprised between about 0.03 and 1.3s-1). 3.4 Structural models of char conversion In the models of lignocellulosic char reactivity, a kinetic term can be straightly identified while the structural contribution takes different expressions. As it appears from Tables 1-3 (structural contribution in square brackets), which summarize the reactivity expressions proposed by several authors for lignocellulosic char conversion in the presence of CO2, H2O and O2, a structural term is absent in some cases [113,115,118,133,147,157,158,161,166-169]. The large majority of the structural models assumes a simple linear [86,117,122,131,132,146,162] or a power law [121,104,107,134,176] dependence on the char mass fraction (in the latter case exponents are in the range 0.4-2). A 5-6th order polynomial in the conversion is proposed in [112,116,119,120,129]. In this category can also be included the empirical functions with adjustable parameters proposed for CO2 gasification of poplar char [148] and combustion of corncob char [105]. Moreover, models originally formulated for coal have been used in [165] for CO2 gasification of beech wood char (this model is re-expressed in Table 1 in terms of conversion, X) and in [175] for the interpretation of the oxidation curves of hardwood and lignin char (in this case the Bhatia and Perlmutter model [179] is used, which refers to the ratio S(X)/S0, that is, the relative change in the available (reactive) surface area during reaction). An investigation about the performances of the most popular structural models of coal char conversion when applied to describe pine wood char oxidation [134] shows that none of these produces a better description of the measured conversion curves than the simple power-law model. A few attempts have also been made to modify structural models proposed for the heterogeneous reactions of coal to take into account the peculiarities of biomass/wood chars. In general, these models do not include any kinetic law but are somewhat empirical in that they use adjustable parameters to take into account the variations in the char reactivity with temperature and/or catalysts such as in [180,181]. In these studies the isothermal thermogravimetric data of CO2 gasification of fir wood char are used to modify the Bathia and Perlmutter model [179], describing the changes in the char reactivity exclusively in terms of structural variations. In particular, modifications have been introduced to predict the higher reactivity (with respect to the predictions of the random pore models) at high conversion (beyond 70%). The parameters of the empirical model vary with the reaction temperature and the pre-treatments of the sample. 3.5 Kinetics of wood/biomass combustion As already pointed out, as a consequence of the small sample mass and the mild thermal conditions applied to reduce the effects of heat and mass transfer intrusions and to control the reaction exothermicity, thermogravimetric curves of wood or biomass combustion [83-98,107] show two main reaction zones, corresponding to solid devolatilization and char oxidation, respectively. The mathematical treatments of the data always consider devolatilization models for the first stage, that is, only the rate of volatile release is described [13]. Moreover, a highly simplified approach is commonly used based on two global reactions for the first and second zone of the weight loss curves (a power-law dependence on the solid mass fraction takes usually into account structural effects)

22
[84,85,88,93,94,97]. For both devolatilization and combustion, widely variable activation energies are reported although they are quite low (roughly 60-130kJ/mol for solid devolatilization and 30-150kJ/mol for char oxidation). As for the first step, this result stems primarily from the high simplification introduced by the one-step reaction model to describe a process which is complicated by the composite nature of the biomass fuels (the contribution of the main constituents in the thermogravimetric curves), the use of integral (versus differential) data and the possible presence of compensation effects, as extensively discussed for decomposition in inert atmospheres [13]. The larger variations between the kinetics for the combustion step can be probably due to the stronger role played by the nature of the samples especially by the content and composition of ashes which catalyze the reaction. Also, inaccuracies in the thermogravimetric measurements, owing to the very small mass of the sample after devolatilization, may be another cause for such discrepancies. Improvements are represented by models where the devolatilization stage is described by two [90,96], three [98] or four [107] reactions, similar to the kinetic models of biomass devolatilization in inert atmosphere describing the release of volatile matter from the main chemical pseudo-components [13]. In particular, results obtained for two wood species (beech and Douglas fir) [98] show, for the pseudo-components hemicellulose, cellulose and lignin, activation energies of 106, 226 and 114kJ/mol respectively, which compare well with the values already found in the absence of oxygen (the combustion step presents an activation energy of 183kJ/mol). Lower values of the activation energies of the four devolatilization reactions (70-100kJ/mol) have been estimated [107] for oak bark, most likely owing to a very peculiar chemical composition (the main components are extractives and lignin with small amounts of holocellulose) and the high ash content. 4. Conclusions The distribution among gases, organic condensable products, water and char from lignocellulosic fuel pyrolysis is highly affected by size and physico-chemical properties of the particles, external heating and reactor configuration. These can be summarized in terms of average heating rate/temperature of the solid and the reaction environment and residence time of the tar vapors in the hot reaction zone. The literature provides sufficient information for wood in relation to both yields and composition of products whereas considerable less work has been carried out for biomass and the results are essentially limited to product yields. As for char, in quantitative terms, given external heating temperatures of about 650-950K, pre-dried hardwoods and softwoods produce yields roughly comprised between 28-10% and 40-20%, depending on fast (fluidized bed reactors and particle sizes below 6mm) or slow (packed beds of small-sized particles or thick samples) pyrolysis. Biomass, in particular agricultural residues, produce higher char yields (up to factors of about two, on ash free basis, with respect to wood) originated from larger contents of lignin (and C) and alkali compounds (the latter catalyze charring reactions). Quantitative contributions of primary and secondary reactions on the amount of char formed are still unknown and further research work on these aspects is highly desirable. As a consequence of the much faster rates of solid pyrolysis compared with those of char conversion, char yield and reactivity are the main factors affecting the feed rate and size of combustors and gasifiers. On the other hand, the properties of the solid and the pyrolysis conditions not only affect the amount of char produced during pyrolysis but also its reactivity. The gasification and combustion reactivity of char decreases when temperature and retention time established during pyrolysis are augmented and/or the heating rate is diminished. Indeed, an increase in the structural ordering of the carbon matrix (thermal annealing) and a reduction in the oxygen containing groups is observed, in this way lowering the concentration of active sites. Moreover, under fast heating, particles undergo a

23
molten phase with the consequent creation of smoother surfaces and spherical cavities. The micro-pore network, a reproduction of the wood structure preserved by slow heating rates, is reduced owing to melting in favor of macro pores where heterogeneous reactions mainly occur. Conditions that favor the activity of intra-particle secondary reactions, such as high external pressures and slow heating rates (and rate of volatile matter release), cause the deposition of secondary char fragments on the primary char surfaces. Apart from a reduction in the specific surface area, it appears that secondary chars are less reactive than primary chars and, on global terms, the reactivity is reduced. However, the understanding of these features is essentially only qualitative and further research is needed to provide the basis for the formulation of reliable kinetic models. It is speculated that the nature of the lignocellulosic fuels does not affect significantly the char reactivity and the differences among various samples can be attributed essentially to the amount and composition of ashes, more specifically to the catalytic effects exerted on the heterogeneous reactions by some compounds (mainly alkaline and alkaline earth metals). Partial elimination of water- or acid-soluble minerals has been observed to reduce, at least by one order of magnitude, the oxidation reactivity of char despite an increase the specific surface area. In quantitative terms, the catalytic action varies during conversion owing to modifications in the ratio between carbon and the catalytically active substances, the chemical occurrence of the catalysts (for instance, deactivation resulting from sintering and agglomeration) and char structure. Three conversion regimes of char are usually considered, based on the Thiele modulus and the effectiveness factor. To evaluate the intrinsic kinetics of the heterogenous gasification and combustion reactions, experimental conditions should be established so as heat and mass transfer limitations are avoided (regime I). However, only a very few kinetic investigations give the due consideration to these aspects and, contrary to pyrolysis, researchers seem to be only partially aware of the possible flaws in the measurements and analysis of the data. A large part of the experiments, motivated by kinetic analysis, has been made using thermogravimetry under static (isothermal) conditions for gasification and dynamic (assigned heating rate) conditions for combustion. Measurements are generally expressed in terms of reactivity versus conversion and it is accepted that the mechanisms of char combustion and gasification (these taking into account the inhibiting effects exerted by CO (CO2 gasification) and H2 (steam gasification)) formulated by the extensive investigations about coal chars are also applicable for lignocellulosic fuels. However, the most widely used treatment is based on a simple global reaction where the reactivity is modeled by means of a chemical kinetic term, accounting for temperature and reactant partial pressure effects, and a structural contribution describing the effects of porosity evolution and available internal surface area or concentration of active sites. Structural models mainly assume a linear or a power-law dependence of the reactivity on conversion although the most recent studies on gasification propose 5-6 order polynomials. Specific structural models, originally developed for coal, have also been used for the interpretation of the conversion curves of lignocellulosic chars but, at least for combustion, they do not appear to produce better results than the simpler power-law model. The kinetic contribution is always modeled by the Arrhenius dependence on temperature and a power-law dependence on the partial pressure of the gaseous reagent. Moreover while the kinetic contribution is taken into account by all the reactivity models, in several cases the structural contribution is completely absent. Hence, the different formulation of the reactivity models is the first cause for the discrepancies among estimated values of the model parameters and numerical predictions. Variations can also be attributed to the differences introduced by the experimental techniques, the operating conditions, the sample characteristics (chemico-physical properties and pyrolysis conditions) and the method of kinetic analysis. A comparison of the predicted gasification reactivities, using those models which provide data for all the parameters, show values varying by about four orders of magnitude. There is quite good agreement

24
in relation to the high values of the activation energy with a large part comprised in the ranges 200-250kJ/mol and 180-200kJ/mol for CO2 and H2O gasification, respectively. The corresponding ranges of the exponent of the gaseous reactant pressure are about 0.4-0.6 and 0.4-1. However, despite the agreement among the various researches about the experimental trends shown by reactivity on various factors (pyrolysis conditions, content and composition of ashes, etc.), the estimated kinetic constants do not show an univocal trend on dependence of these same factors. Therefore, further research efforts are required for the formulation of global models of char gasification valid over wide ranges of experimental conditions which can reliably describe the influence of the key parameters on char reactivity. Also, the more complicated Langmuir-Hinshelwood kinetics is considered only in a few cases, often with the sole evaluation of the rate constants at a selected temperature. Early kinetic models of wood char combustion, using integral thermogravimetric dynamic data for parameter estimation, consist of a one-step global reaction characterized by relatively low values of the activation energy (below 100kJ/mol). These provide very poor predictions of differential thermogravimetric measurements and their parameters are not free from compensation effects. More recent investigations use multi-step models for the interpretation of differential thermogravimetric data, measured under different thermal conditions. In this way the so-called char devolatilization is taken into account (release of about 6-25% of volatile matter) followed by the actual combustion step (in a few cases this is described by two steps). This treatment produces accurate predictions and gives rise to much higher activation energies of the combustion reaction (140-230kJ/mol). However, similar to gasification, the trends of the estimated kinetic parameters on dependence of important factors affecting the char reactivity are not in agreement and differences in the predicted reactivities of at least two order of magnitude are calculated. These circumstances and the small number of studies currently available and focused only on a few specific feedstocks put into evidence the need of more comprehensive investigations also in this sector. Acknowledgments This review is part of the activities carried out in the framework of the European network ThermalNet (EIE-04-159), the partial support of which is gratefully acknowledged. References [1] Laurendeau, NM. Heterogeneous kinetics of coal char gasification and combustion. Progr Energy Combust Science 1978; 4: 221-270. [2] Smith IW. The combustion rates of coal chars: a review. In: Nineteenth symposium (international) on combustion. Pittsburgh, P: The Combustion Institute; 1982. p 1045-1065. [3] Bews IM, Hayhurst AN, Richardson SM, Taylor SG. The order, Arrhenius parameters, and mechanism of the reaction between gaseous oxygen and solid carbon. Combust Flame 2001; 124: 231-245. [4] Hurt RH, Calo JM. Semi-global intrinsic kinetics for char combustion modeling. Combust Flame 2001; 125: 1138-1149. [5] Encinar JM, Gonzalez JF, Rodriguez JJ, Ramiro MJ. Catalysed and uncatalysed steam gasification of Eucalyptus char: influence of variables and kinetic study. Fuel 2001; 80: 2025-2036. [6] Standish N, Tanjung AFA. Gasification of single wood charcoal particles in CO2. Fuel 1988; 67: 666-672. [7] Antal MJ, Gronli MG. The art, science and technology of charcoal production. Ind Eng Chem Res 2003; 42: 1619-1640. [8] Hurt RH. Structure, properties and reactivity of solid fuels. In: Twenty-seventh symposium (international) on combustion. Pittsburgh, P: The Combustion Institute; 1998. p. 2887-2904.

25
[9] Williams A, Pourkashanian M, Jones JM. Combustion of pulverized coal and biomass. Progr Energy Combust Science 27, 587-610, 2001. [10] Nussbaumer T. Combustion and co-combustion of biomass: fundamentals, technologies, and primary measures for emission reduction. Energy Fuels 2003; 17: 1510-1521. [11] Di Blasi C. Modeling and simulation of combustion processes of charring and non-charring solid fuels. Progr Energy Combust Science 1993; 19: 71-104. [12] Di Blasi C. The state of the art of transport models for charring solid degradation. Polym Int 2000; 49: 1133-1146. [13] Di Blasi C. Modeling chemical and physical processes of wood and biomass pyrolysis. Progr Energy Combust Science 2008; 34: 47-90. [14] Scott DS, Piskorz J. The flash pyrolysis of aspen-poplar wood. Can J Chem Eng 1982; 60: 666-674. [15] Scott DS, Piskorz J. The continuous flash pyrolysis of biomass. Can J Chem Eng 1984, 62: 404-412. [16] Scott, DS, Piskorz J, Bergougnou MA, Graham R, Overend RP. The role of temperature in the fast pyrolysis of cellulose and wood. Ind Eng Chem Res 1988; 27: 8-15. [17] Horne PA, Williams PT. Influence of temperature on the products from the flash pyrolysis of biomass. Fuel 1996; 75: 1051-1059. [18] Wehlte S, Meier D, Moltran J, Faix O. The impact of wood preservatives on the flash pyrolysis of biomass. In: Bridgwater AV, Boocock DGB, editors. Developments in Thermochemical Biomass Conversion. London: Blackie A&P; 1997. p. 206-29. [19] Peacocke CGV, Dick CM, Hague RA, Cooke LA, Bridgwater AV. Comparison of ablative and fluid bed fast pyrolysis: yields and analyses. In: Bridgwater AV, Boocock DGB, editors. Developments in Thermochemical Biomass Conversion. London: Blackie A&P; 1997. p. 191-205. [20] Gerdes C, Simon CM, Ollesch T, Meier D, Kaminsky W. Design, construction, and operation of a fast pyrolysis plant for biomass. Eng Life Sci 2 2002; 6:167-174. [21] Di Blasi C, Branca C. Temperatures of wood particles in a hot-sand bed fluidized by nitrogen. Energy Fuels 2003; 17: 247-254. [22] Bridgwater AV. Principles and practice of biomass fast pyrolysis processes for liquids. J Anal Appl Pyrol 1999; 51: 3-22. [23] Chan WR, Kelbon M, Krieger BB. Modeling and experimental verification of physical and chemical processes during pyrolysis of large biomass particle. Fuel 1985; 64: 1505-1513. [24] Figueiredo JL, Valenzuela C, Bernalte A, Encinar JM. Pyrolysis of holm-oak wood: influence of temperature and particle size. Fuel 1989; 68: 1012-1016. [25] Bilbao R, Millera A, Murillo M B. Temperature profiles and weight loss in the thermal decomposition of large spherical wood particles. Ind Eng Chem Res 1993; 32: 1811-1817. [26] Gronli MG. A theoretical and experimental study of the thermal degradation of biomass, PhD. Thesis, NTNU, Trondheim, Norway, 1996. [27] Di Blasi C, Gonzalez Hernandez E, Santoro A. Radiative pyrolysis of single moist wood particles. Ind Eng Chem Res 2000; 39: 873-882. [28] Di Blasi C, Branca C, Santoro A, Perez Bermudez RA. Weight loss dynamics of wood chips under fast radiative heating. J Anal Appl Pyrol 2001; 57: 77-90. [29] Di Blasi C, Branca C, Santoro A, Gonzalez Hernandez E. Pyrolytic behavior and products of some wood varieties. Combust Flame 2001; 124: 165-177. [30] Branca C, Giudicianni P, Di Blasi C. GC/MS characterization of liquids generated from low-temperature pyrolysis of wood. Ind Eng Chem Res 2003; 42: 3190-3202.

26
[31] Scott DS, Piskor J, Radlein D. Liquid products from the continuous flash pyrolysis of biomass. Ind Eng Chem Process Des Dev 1985; 24: 581-588. [32] Williams PT, Besler S. The pyrolysis of rice husks in a thermogravimetric analyzer and static batch reactor. Fuel 1993; 72: 151-159. [33] Encinar JM, Beltran FJ, Bernalte A, Ramiro A, Gonzalez JF. Pyrolysis of two agricultural residues: olive and grape bagasse. Influence of particle size and temperature. Biomass Bioenergy 1996; 11: 397-409. [34] Zanzi R, Sjostrom K, Bjornbom E. Rapid high-temperature pyrolysis of biomass in a free-fall reactor. Fuel 1996; 75: 545-550. [35] Di Blasi C, Signorelli G, Di Russo C, Rea G. Product distribution from pyrolysis of wood and agricultural residues. Ind Eng Chem Res 1999; 38: 2216-2224. [36] Gercel HF. Production and characterization of pyrolysis liquids from sunflower-pressed bagasse. Bioresource Technol 2002; 85: 113-117. [37] Blanco Lopez MC, Blanco CG, Martinez-Alonso A, Tascon JMD. Composition of gases released during olive stones pyrolysis. J Anal Appl Pyrol 2002; 65: 313-322. [38] Cao Q, Xie KC, Bao WR, Shen SG. Pyrolytic behavior of waste corn cob. Bioresource Technol 2004; 94: 83-89. [39] Gonzalez JF, Ramiro A, Gonzalez-Garcia CM, Ganan J, Encinar JM, Sabio E, Rubiales J. Pyrolysis of almond shells. Energy applications of fractions. Ind Eng Chem Res 2005; 44: 3003-3012. [40] Piskorz J, Radlein D, Scott DS, Czernik S. Liquid products from the fast pyrolysis of wood and cellulose. In: Bridgwater AV, Kuester JL, editors. Research in Thermochemical biomass conversion. London and New York: Elsevier Applied Science; 1988. p. 557-571. [41] Raveendran K, Ganesh A, Khilar K. Influence of mineral matter on biomass pyrolysis characteristics. Fuel 1995; 74: 1812-1822. [42] Di Blasi C, Branca C, D'Errico G. Degradation characteristics of straw and washed straw. Thermochim Acta 2000; 364: 133-142. [43] Piskorz J, Majerski P, Radlein D, Vladars-Usas A, Scott DS. Flash pyrolysis of cellulose for production of anhydro-oligomers. J Anal Appl Pyrol 2000; 56: 145-166. [44] Evans RJ, Milne TA. Chemistry of tar formation and maturation in the thermochemical conversion of biomass. In: Bridgwater AV, Boocock DGB, editors. Developments in thermochemical biomass conversion. London: Blackie Academic & Professional; 1997. p. 803-816. [45] Morf P, Hasler P, Nussbaumer T. Mechanisms and kinetics of homogeneous secondary reactions of tar from continuous pyrolysis of wood chips. Fuel 2002; 81: 843-853. [46] Bridgwater AV. The technical and economic feasibility of biomass gasification for power generation. Fuel 1995; 74: 631-653. [47] Antal MJ. Effects of reactor severity on the gas-phase pyrolysis of cellulose- and kraft lignin-derived volatile matter. Ind Eng Prod Res Dev 1983; 22: 366-375. [48] Antal MJ. A review of the vapor phase pyrolysis of biomass derived volatile matter. In: Overend RP, Milne TA, Mudge LK, editors. Fundamentals of biomass thermochemical conversion. London: Elsevier; 1985. p. 511-537. [49] Boroson ML, Howard JB, Longwell JP, Peters AW. Products yields and kinetics from the vapor phase cracking of wood pyrolysis tars. AIChE J 1989; 35: 120-128. [50] Boroson ML, Howard JB, Longwell JP, Peters WA. Heterogeneous cracking of wood pyrolysis tars over fresh wood char surfaces. Energy Fuels 1988; 3: 735-740. [51] Arseneau D. Competitive reactions in the thermal decomposition of cellulose. Can J Chem 1971; 49: 632-638.

27
[52] Mok WSL, Antal MJ. Effects of pressure on biomass pyrolysis. II. Heats of reactions of cellulose pyrolysis. Thermochim Acta 1983; 68: 165-186. [53] Milosavljevic I, Oja V, Suuberg EM. Thermal effects in cellulose pyrolysis: relationship to char formation. Ind Eng Chem Res 1996; 35: 653-662. [54] Shin E, Nimlos MR, Evan RJ. Kinetic analysis of the gas-phase pyrolysis of carbohydrates. Fuel 2001; 80: 1697-1709. [55] Branca C, Di Blasi C, Russo C. Devolatilization in the temperature range 300-600K of liquids derived from wood pyrolysis and gasification. Fuel 2005; 84: 37-45. [56] Branca C, Di Blasi C, Elefante R. Devolatilization and heterogeneous combustion of wood fast pyrolysis oils. Ind Eng Chem Res 2005; 44: 799-810. [57] Branca C, Di Blasi C. A multi-step mechanism for the devolatilization of biomass fast pyrolysis oils. Ind Eng Chem Res 2006; 45: 5891-5899, 2006. [58] Branca C, Di Blasi C, Elefante R. Devolatilization of conventional pyrolysis oils generated from biomass and cellulose. Energy Fuels 2006; 20: 2253-2261, 2006. [59] Antal MJ, Varhegyi G. Cellulose pyrolysis kinetics: the current state of knowledge. Ind Eng Chem Res 1995; 34: 703-717. [60] Diebold JP. The cracking kinetics of depolymerized biomass in a continuous tubular reactor. PhD Thesis T-3007 - Colorado School of Mines, Golden Co, 1985. [61] Liden AG, Berruti F, Scott DS. A kinetic model for the production of liquids from the flash pyrolysis of biomass. Chem Eng Comm 1988; 65: 207-221. [62] Graham RG, Bergougnou MA, Freel BA. The kinetics of vapor-phase cellulose fast pyrolysis reactions. Biomass Bioenergy 1994; 7: 33-47. [63] Rath J, Staudinger G. Cracking reactions of tar from pyrolysis of spruce wood. Fuel 2001; 80: 1379-1389. [64] Baumlin S, Broust F, Ferrer M, Meunier N, Marty E, Lede J. The continuous self stirred tank reactor: measurement of the cracking kinetics of biomass pyrolysis vapors. Chem Eng Sci 2005; 60: 41-55. [65] Kersten SRA, Wang X, Prins W, van Swaaij WPM. Biomass pyrolysis in a fluidized bed reactor. Part 1: Literature review and model simulations. Ind Eng Chem Res 2005; 44: 8773-8785. [66] Evans RJ, Milne TA. Molecular Characterization of the pyrolysis of biomass. 1. Fundamentals. Energy Fuels 1987; 1: 123-137. [67] Evans RJ, Milne TA. Molecular Characterization of the pyrolysis of biomass. 2. Applications. Energy Fuels 1987; 1: 311-319. [68] Buekens AG, Schoeters JG. Modelling of biomass gasification. In: Overend RP, Milne TA, Mudge LK, editors. Fundamentals of thermochemical biomass conversion. London: Elsevier; 1985. p. 619-689. [69] Sutton D, Kelleher B, Ross JHR. Review of literature on catalysts for biomass gasification. Fuel Process Technol 2001; 73: 155-173. [70] Devi L, Ptasinski KJ, Janssen FJJG. A review of the primary measures for tar elimination in biomass gasification processes. Biomass Bioenergy 2003; 24: 125-140. [71] Abu El-Rub Z, Bramer EA, Brem G. Review of catalysts for tar elimination in biomass gasification processes. Ind Eng Chem Res 2004; 43: 6911-6919. [72] Mohan D, Pittman CU, Steele P. Pyrolysis of wood/biomass for bio-oil: a critical review. Energy Fuels 20; 848-889, 2006. [73] Fernando S, Adhikari S, Chandrapal C, Murali N. Biorefineries: current status, challenges, and future direction. Energy Fuels 20; 1727-1737, 2006.

28
[74] Kinoshita CM, Wang Y, Zhou J. Tar formation under different biomass gasification conditions. J Anal Appl Pyrol 1994; 29: 169-181. [75] Brage C, Yu Q, Sjostrom K. Characteristics of evolution of tar from wood pyrolysis in a fixed bed reactor. Fuel 1996; 75: 213-219. [76] Brage C, Yu Q, Chen G, Sjostrom K. Tar evolution profiles obtained from gasification of biomass and coal. Biomass Bioenergy 2000; 18:87-91. [77] Padban N, Wang W, Ye Z, Bjerle I, Odenbrand I. Tar formation in pressurized fluidized bed air gasification of woody biomass. Energy Fuels 2000; 14: 603-611. [78] Reina J, Velo E, Manya J, Puigjaner L. Thermogravimetric studies of waste wood in nitrogen, air and carbon dioxide atmospheres. In: Bridgwater AV, Boocock DGB, editors. Science in thermal and chemical biomass conversion. Newbury Berks (UK): CPL Press; 2006. pp. 1228-1238. [79] Mermoud F, Salvador S, Van de Steene L, Golfier F. Influence of the pyrolysis heating rate on the steam gasification rate of large wood char particles. Fuel 2006; 85: 1473-1482. [80] Mermoud F, Golfier F, Salvador S, Van de Steene L, Dirion JL. Experimental and numerical study of steam gasification of a single charcoal particle. Combust Flame 2006; 145: 59-79. [81] Henrich E, Burkle S, Meza-Renken ZI, Rumple S. Combustion and gasification kinetics of pyrolysis chars from waste and biomass. J Anal Appl Pyrol 1999; 49: 221-241. [82] Klose W, Wolki M. On the intrinsic reaction rate of biomass char gasification with carbon dioxide and steam. Fuel 2005; 84: 885-892. [83] Vovelle C, Mellottee H, Delbourgo R. Kinetics of the thermal degradation of cellulose and wood in inert and oxidative atmospheres. In: Nineteenth symposium (international) on combustion. Pittsburgh, P: The Combustion Institute; 1982. p. 797-805. [84] Aho M, Houtari J. The effects of atmosphere on pyrolysis of solid fuels produced in Finland. In: Overend RP, Milne TA, Mudge LK, editors. Fundamentals of thermochemical biomass conversion. London: Elsevier; 1985. p. 429-435. [85] Cordero T, Rodriguez-Maroto JM, Garcia F, Rodriguez JJ. Thermal decomposition of wood in oxidizing atmosphere. A kinetic study from non-isothermal TGA experiments. Thermochim Acta 1991; 191: 161-178. [86] Kashiwagi T, Nambu H. Global kinetic constants for thermal oxidative degradation of a cellulosic sample. Combust Flame 1992; 88: 345-368. [87] Ghaly AE, Ergudenler A. Reaction kinetics of rye straw for thermochemical conversion. Can J Chem Eng 1994; 72: 651-656. [88] Momoh M, Eboatu AN, Kolawole EG, Horrocks AR. Thermogravimetric studies of the pyrolytic behaviour in air of selected tropical timbers. Fire Mater 1996; 20: 173-181. [89] Ghetti P, Ricca L, Angelini L. Thermal analysis of biomass and corresponding pyrolysis products. Fuel 1996; 75: 565-573. [90] Bilbao R, Mastral JF, Aldea ME, Ceamanos J. The influence of the percentage of oxygen in the atmosphere on the thermal decomposition of lignocellulosic materials. J Anal Appl Pyrol 1997; 42: 189-202. [91] Simkovic I, Csomorova K. Flame-retarding properties of ion-exchanging groups introduced into beech sawdust. Fire Mater 1998; 22: 149-154. [92] Di Blasi C, Branca C. Global degradation kinetics of wood and agricultural residues in air. Can J Chem Eng 1999; 77: 555-561. [93] Mansaray KG, Ghaly AE. Determination of kinetic parameters of rice husks in oxygen using thermogravimetric analysis. Biomass Bioenergy 1999; 17: 19-31. [94] Liu NA, Fan W, Dobashi R, Huang L. Kinetic modeling of thermal decomposition of natural cellulosic materials in air atmosphere. J Anal Appl Pyrol 2002; 63: 303-325.

29
[95] Meszaros E, Jakab E, Varhegyi G, Marosvolgyi B. Thermal behavior of biomass plant materials. Chemia I Inzynieria Ekologiczna 2002; 9: 33-41. [96] Juahiainen J, Conesa JA, Font R, Martin-Gullon I. Kinetics of the pyrolysis and combustion of olive oil solid waste. J Anal Appl Pyrol 2004; 72: 9-15. [97] Calvo LF, Otero M, Jenkins BM, Moran A, Garcia AI. Heating process characteristics and kinetics of rice straw in different atmospheres. Fuel Process Technol 2004; 85: 279-291. [98] Branca C, Di Blasi C. Global intrinsic kinetics of wood oxidation. Fuel 2004; 83: 81-87. [99] Varhegyi G, Antal MJ, Jakab E, Szabo P. Kinetic modeling of biomass pyrolysis. J Anal Appl Pyrol 1996; 42: 73-87. [100] Shafizadeh F, Sekiguchi Y. Oxidation of chars during smoldering combustion of cellulosic materials. Combust Flame 1984; 55: 171-179. [101] Marcilla A, Conesa JA, Asensia M, Garcia-Garcia SM. Thermal treatment and foaming of chars obtained from almond shells: kinetic study. Fuel 2000; 79: 829-836. [102] Varhegyi G, Szabo P, Antal MJ, Dai X. Kinetic modeling of the gasification of biomass charcoals. In: Kyritsis S, Beenackers A A C M, Helm P, Grassi A, Chiaramonti D, editors. Proc.of the 1st World Conference on Biomass for Energy and Industry. London: James & James (Science Publisher) Ltd; 2001. p 1783-1785. [103] Varhegyi G, Szabo P, Antal MJ. Kinetics of charcoal devolatilization. Energy Fuels 2002; 16: 724-731. [104] Branca C, Di Blasi C. Devolatilization and combustion kinetics of wood chars. Energy Fuels 2003; 17: 1609-1615. [105] Varhegyi G, Meszaros E, Antal MJ, Bourke J, Jakab E. Combustion kinetics of corncob charcoal and partially demineralized corncob charcoal in the kinetic regime. Ind Eng Chem Res 2006; 45: 4962-4970. [106] Boateng AA. Characterization and thermal conversion of charcoal derived from fluidized-bed fast pyrolysis oil production of switchgrass. Ind Eng Chem Res 46: 8857-8862, 2007. [107] Branca C, Iannace A, Di Blasi C. Devolatilization and combustion kinetics of Quercus Cerris bark. Energy Fuels 2007; 21: 1078-1084. [108] Sharma RK, Wooten JB, Baliga VL, Hajaligol MR. Characterization of chars from biomass derived materials: pectin chars. Fuel 2001; 80: 1825-1836. [109] Hajaligol M, Waymack B, Kellogg D. Low temperature formation of aromatic hydrocarbon from pyrolysis of cellulosic materials. Fuel 2001; 80: 1799-1807. [110] Wornat MJ, Hurt RH, Yang NYC, Headley TJ. Structural and compositional transformations of biomass chars during combustion. Combust Flame 1995; 100: 131-143. [111] Wornat MJ, Hurt RH, Davis KA, Yang NYC. Single-particle combustion of two biomass chars. In: Twenty-sixth symposium (international) on combustion. Pittsburgh, P: The Combustion Institute; 1996. p. 3075-3083. [112] Ollero P, Serrera A, Arjona R, Alcantarilla S. The CO2 gasification kinetics of olive residue. Biomass Bioenergy 2003; 24: 151-161. [113] Kumar M, Gupta RC. Influence of carbonization conditions on the gasification of acacia and eucalyptus wood chars by carbon dioxide. Fuel 1994; 73: 1922-1925. [114] Moilanen A, Muhlen HJ. Characterization of gasification reactivity of peat char in pressurized conditions. Effect of product gas inhibition and inorganic material. Fuel 1996; 75: 1279-1285. [115] Moilanen A, Saviharju K. Gasification reactivities of biomass fuels in pressurized conditions and product gas mixtures. In: Bridgwater AV, Boocock DGB, editors. Developments in thermochemical biomass conversion. London: Blackie A. & P.; 1997. p. 828-837.

30
[116] Barrio M, Hustad JE. CO2 gasification of birch char and the effect of CO inhibition on the calculation of chemical kinetics. In: Bridgwater AV, editor. Progress in thermochemical biomass conversion. Oxford: Blackwell Science Ltd.; 2001. p. 47-60. [117] Nandi SP, Onischak M. Gasification of chars obtained from maple and jack pine woods, In: Overend RP, Milne TA, Mudge LK, editors. Fundamentals of biomass thermochemical conversion. London: Elsevier; 1985. p. 567-587. [118] Moilanen A, Saviharju K, Harju T. Steam gasification reactivities of various fuel chars. In: Bridgwater AV, editor. Advances in thermochemical biomass conversion. Glasgow: Blackie A & P Professional; 1994. p. 131-141. [119] Barrio M, Gobel B, Risnes H, Henriksen U, Hustad JE, Sorensen LH. Steam gasification of wood char and the effect of hydrogen inhibition on the chemical kinetics. In: Bridgwater AV, editor. Progress in themochemical biomass conversion. Oxford: Blackwell Science Ltd.; 2001. p. 32-46. [120] Risnes H, Sorensen LH, Hustad JE. CO2 reactivity of chars from wheat, spruce and coal. In: : Bridgwater AV, editor. Progress in themochemical biomass conversion. Oxford: Blackwell Science Ltd.; 2001. p. 61-72. [121] Di Blasi C, Buonanno F, Branca C. Reactivities of some biomass chars in air. Carbon 1999; 37: 1227-1238. [122] Luo M, Stanmore B. The combustion characteristics of char from pulverized bagasse. Fuel 1992; 71: 1074-1076. [123] Walker PL. Char properties and gasification. In: Bridgwater AV, Kuester JL, editors. Research in thermochemical biomass conversion. London and New York: Elsevier Applied Science; 1988. p. 485-509. [124] Basu P, Fraser SA. Circulating fluidized bed boilers, Butterworth-Heinemann, Stoneham, MA, p. 95, 1991. [125] Winter F, Prah ME, Hofbauer H. Temperatures in a fuel particle burning in a fluidized bed: the effect of drying, devolatilization and char combustion. Combust Flame 1997; 108: 302-314. [126] Dennis JS, Lambert RJ, Milne AJ, Scott SA, Hayhurst AN. The kinetics of combustion of chars derived from sewage sludge. Fuel 2005; 84: 117-126. [127] Ollero P, Serrera A, Arjona R, Alcantarilla S. Diffusional effects in TGA gasification experiments for kinetic determination. Fuel 2002; 81: 1989-2000. [128] Gomez-Barea A, Ollero P, Arjona R. Reaction-diffusion model of TGA gasification experiments for estimating diffusional effects. Fuel 2005; 84: 1695-1704. [129] Gomez-Barea A, Ollero P, Fernandez-Baco C. Diffusional effects in CO2 gasification experiments with single biomass char particles. 1. Experimental investigation. Energy Fuels 2006; 20: 2202-2210. [130] Gomez-Barea A, Ollero P, Villanueva A. Diffusional effects in CO2 gasification experiments with single biomass char particles. 1. Theoretical predictions. Energy Fuels 2006; 20: 2211-2222. [131] Hemati M, Laguerie C. Determination of the kinetics of the wood sawdust steam gasification of charcoal in a thermobalance. Entropie 1988; 142: 29-40. [132] Bhat A, Bheemaresetti JVM, Rao TR. Kinetics of rice husk char gasification. Energy Convers Manage 2001; 42: 2061-2069. [133] Bandyopadhyay D, Chakraborti N, Ghosh A. Heat and mass transfer limitations in gasification of carbon by carbon dioxide. Steel Research 1991; 62: 143-151. [134] Janse AMC, de Jonge HG, Prins W, van Swaaij WPM. The combustion kinetics of char obtained by flash pyrolysis of pine wood. Ind Eng Chem Res 1998; 37: 3909-3918. [135] Ravendraan K, Ganesh A. Adsorption characteristics and pore-development of biomass-pyrolysis char. Fuel 1998; 77: 769-781.

31
[136] Guerrero M, Ruiz MP, Alzueta MU, Bilbao R, Millera A. Pyrolysis of eucalyptus at different heating rates: studies of char characterization on oxidative reactivity. J Anal Appl Pyrol 2005; 74: 307-314. [137] Chen G, Yu Q, Sjostrom K. Reactivity of char from pyrolysis of birch wood. J Anal Appl Pyrol 1997; 40-41: 491-499. [138] Fushimi C, Araki K, Yamaguchi Y, Tsutsumi A. Effect of heating rate on steam gasification of biomass. 1. Reactivity of char. Ind Eng Chem Res 2003; 42: 3922-3928. [139] Cetin E, Moghtaderi B, Gupta R, Wall TF. Influence of pyrolysis conditions on the structure and gasification reactivity of biomass chars. Fuel 2004; 83: 2139-2150. [140] Cetin E, Gupta R, Moghtaderi B. Effect of pyrolysis pressure and heating rate on radiate pine char structure and apparent reactivity. Fuel 2005; 84: 1328-1334. [141] Della Rocca PA, Cerrella EG, Bonelli PR, Cuckierman AL. Pyrolysis of hardwoods residues: on kinetics and char characterization. Biomass Bioenergy 1999; 16: 79-88. [142] Kurosaki F, Ishimaru K, Hata T, Bronsveld P, Kobayashi E, Imamura Y. Microstructure of wood charcoal prepared by flash heating. Carbon 2003; 41: 3057-3062. [143] Miura M, Kaga H, Sakurai A, Kakuchi T, Takahashi K. Rapid pyrolysis of wood block by microwave heating. J Anal Appl Pyrol 2004; 71: 187-199. [144] Moilanen A, Vepsalainen J, Kurkela E, Konttinen J. Gasification reactivity of large biomass pieces. In: Bridgwater AV and Boocock DGB, editors. Science in thermal and chemical biomass conversion. Newbury Berks (UK): CPL Press; 2006. p. 509-517. [145] Erlich C, Bjornbon E, Bolado D, Giner M, Fransson TH. Pyrolysis and gasification of pellets from sugar cane bagasse and wood. Fuel 2006; 85: 1535-1540. [146] Cozzani V. Reactivity in oxygen and carbon dioxide of char formed in the pyrolysis of refuse-derived fuel. Ind Eng Chem Res 2000; 39: 864-872. [147] Zolin A, Jensen A, Jensen PA, Frandsen F, Dam-Johansen K. The influence of inorganic materials on the thermal deactivation of fuel chars. Energy Fuels 2001; 15: 1110-1122. [148] Plante P, Roy C, Chornet E. CO2 gasification of wood charcoals derived from vacuum and atmospheric pyrolysis. Can J Chem Eng 1988; 66: 307-312. [149] Whitty K, Backman R, Hupa M. Influence of char formation conditions on pressurized black liquor gasification rates. Carbon 1998; 36: 1683-1692. [150] Pindoria RV, Megaritis A, Messenbock RC, Dugwell DR, Kandiyoti R. Comparison of the pyrolysis and gasification of biomass: effect of reacting gas atmosphere and pressure on Eucalyptus wood. Fuel 1998; 77: 1247-1251. [151] Cao N, Darmstadt H, Soutric F, Roy C. Theremogravimetric study on the steam activation of charcoals obtained by vacuum and atmospheric pyrolysis of softwood bark residues. Carbon 2002; 40: 471-479. [152] Cazorla-Amoros D, Linares-Solano A, Salinas-Martinez de Lecea C, Yamashita H, Kyotani T, Tomita A, Nomura M. XAFS and thermogravimetric study of the sintering of calcium supported on carbon. Energy Fuels 1993; 7: 139-145. [153] Kannan MP, Richards GN. Potassium catalysis in air gasification of cellulosic chars. Fuel 1990; 69: 999-1006. [154] Kannan MP, Richards GN. Gasification of biomass chars in carbon dioxide: dependence of gasification rate on the indigenous metal content. Fuel 1990; 69: 747-753. [155] Jenkins BM, Bakker RR, Wei JB, On the properties of washed straw. Biomass Bioenergy 1996; 10:177-200. [156] Davidsson KO, Korsgren JG, Pettersson JBC, Jaglid U. The effects of fuel washing techniques on alkali release from biomass. Fuel 81: 137-142, 2002.

32
[157] Marquez-Montesinos F, Cordero T, Rodriguez-Mirasol J, Rodriguez JJ. CO2 and steam gasification of a grapefruit skin char. Fuel 2002; 81: 423-429. [158] Tancredi N, Cordero T, Rodriguez-Mirasol J, Rodriguez JJ. CO2 gasification of eucalyptus wood chars. Fuel 1996; 75: 1505-1508. [159] Ganga Devi T, Kannan MP. Calcium catalysis in air gasification of cellulosic chars. Fuel 1998; 77: 1825-1830. [160] Waymack BE, Belote J, Baliga VL, Hajaligol MR. Effects of metal salts on char oxidation in pectins/uronic acids and other acid derivative carbohydrates. Fuel 2004; 83: 1505-1518. [161] De Groot WF, Shafizadeh F. Kinetics of Douglas Fir and Cottonwood chars by carbon dioxide. Fuel 1984; 63: 210-216. [162] Groeneveld MJ, van Swaaij WPM. Gasification of char particles with CO2 and H2O. Chem Eng Sci 1980; 35: 307-313. [163] Huttinger KJ, Merdes WF. The carbon-steam reaction at elevated pressure: formations of product gases and hydrogen inhibitions. Carbon 1992; 30: 883-894. [164] Blackwood JD, McGrory F. The carbon-steam reaction at high pressure, Aust J Chem 1958; 11: 16-23. [165] Van Den Aarsen FG, Beenackers AACM, Van Swaaij WPM. Wood pyrolysis and carbon dioxide char gasification kinetics in a fluidized bed. In: Overend RP, Milne TA, Mudge LK, editors. Fundamentals of biomass thermochemical conversion. London: Elsevier; 1985. p. 691-715. [166] Rodriguez-Mirasol J, Cordero T, Rodriguez JJ. CO2-reactivity of eucalyptus kraft lignin chars. Carbon 1993; 31: 53-61. [167] Rensfeldt E, Blomkuist G, Ekstrom S, Espenas BG, Liinanki L. Basic gasification studies for development of biomass medium-btu gasification processes. In: Energy from biomass and wastes Chicago: IGT, 1978; Paper No. 27, p. 466-494. [168] Hawley MC, Boyd M, Anderson C, DeVera A. Gasification of wood char and effects of intraparticle transport. Fuel 1983; 62: 213-216. [169] Kojima T, Assavadakorn P, Furusawa T. Measurement and evaluation of gasification knetics of sawdust char with steam in an experimental fluidized bed. Fuel Process Technol 1993; 36: 201-207. [170] McCarthy DJ. Changes in oxyreactivity of carbons due to heat treatment and prehydrogeneation. Carbon 1981; 19: 297-301. [171] Gadsby J, Long FJ, Sleightholm P, Sykes KW. The mechanism of the carbon dioxide-carbon reaction. Proc. Of the Royal Society of London. Series A. Mathematical and Physical Sciences 193:357-376, 1948. [172] Edrich R, Bradley T, Graboski MS. The gasification of ponderosa pine charcoal. In: Overend RP, Milne TA, Mudge LK , editors. Fundamentals of thermochemical biomass conversion. London: Elsevier; 1985. p. 557-599. [173] Bandyopadhyay D, Gosh A. Validity of rate equation based on Langmuir-Hinshelwood mechanism for gasification of carbon - a reappraisal. Steel Research 1996; 67: 79-86. [174] Tseng HP, Edgar TF. Combustion behaviour of bituminous and anthracite coal char between 425 and 900C. Fuel 1985; 64: 373-379. [175] Magnaterra M, Fusco JR, Ochoa J, Cuckierman AL. Kinetic study of the reaction of different hardwood sawdust chars with oxygen, chemical and structural characterization of the samples. In: Bridgwater AV, editor. Advances in thermochemical biomass conversion. Glasgow: Blackie A. & P.; 1994. p 116-130. [176] Adanez J, de Diego LF, Garcia-Labiano F, Abad A, Abadanes JC. Determination of biomass char combustion reactivities for FBC applications by a combined method. Ind Eng Chem Res 2001; 40: 4317-4323.

33
[177] Branca C, Di Blasi C. Parallel- and series-reaction mechanisms of wood and char combustion. Thermal Science 2004; 8: 51-63. [178] Nunoura T, Wade SR, Bourke JP, Antal MJ. Studies of the flash carbonization process. 1. Propagation of the flaming pyrolysis reaction and performance of a catalytic afterburner. Ind Eng Chem Res 2006, 45: 585-599. [179] Bhatia SK, Perlmutter DD. A random pore model for fluid-solid reactions: I. Isothermal kinetic control. AIChE J 1980; 26: 379-385. [180] Struis RPWJ, von Scala C, Stucki S, Prins R. Gasification reactivity of charcoal with CO2. Part I: conversion and structural phenomena. Chem Eng Sci 2002; 57: 3581-3592. [181] Struis RPWJ, von Scala C, Stucki S, Prins R. Gasification reactivity of charcoal with CO2. Part II: metal catalysis as a function of conversion. Chem Eng Sci 2002; 57: 3593-3602.

34
FIGURE CAPTIONS Fig.1 - Yields of char (% dry solid basis), on dependence of the heating temperature, Te, from fluidized-bed pyrolysis of hardwood (� [14-16]; ▽ [18]; △ [20]) and softwood (◯ [17], ◇ [19]; □ [18])
species, fixed-bed pyrolysis of beech wood (∗∗∗∗ [30]) and pyrolysis of thick (40mm diameter) cylinders [29] of hardwoods (■ beech; ▼ chestnut) and softwoods (● pine; ◆ Douglas fir; ▲ redwood) species. Fig.2 - Yields of liquids (% dry solid basis) on dependence of the heating temperature, Te, (conditions and References as in Fig.1). Fig.3 - Yields of gas (% dry solid basis) on dependence of the heating temperature, Te, (conditions and References as in Fig.1). Fig. 4 - Yields of char (% dry and ash-free solid basis), for a packed-bed of biomass or wood particles [35], on dependence of the heating temperature, Te. Fig. 5 - Yields of CO2 (% dry solid basis) on dependence of the heating temperature, Te, (conditions and References as in Fig.1). Fig. 6 - Yields of CO and H2O (% dry solid basis) on dependence of the heating temperature, Te, (conditions and References as in Fig.1). Fig. 7 - Yields of some organic compounds (% dry solid basis) from beech wood pyrolysis in a packed bed [30] on dependence of the heating temperature, Te. Fig. 8 - Reactivities of biomass and wood chars in air for a heating rate of 10K/min and a final temperature of 873K [121]. Fig. 9 - Conversion (A) and time derivative of conversion (B) as functions of temperature for waste wood heated in different atmospheres at 2K/min [78]. Fig. 10 - Arrhenius plot of the kinetic contribution, rc, of the reactivity for lignocellulosic char gasification with CO2 : a) cotton wood, b) Douglas fir, c) straw, d) spruce. Fig. 11 - Arrhenius plot of the kinetic contribution, rc, of the reactivity for lignocellulosic char gasification with H2O: a) straw, b) poplar, c) bark, d) beech, e) birch, f) maple, g) pine. Fig. 12 - Mass fraction and time derivative of the mass fraction versus time for pine wood char in air with a heating rate of 5K/min up to 873K as measured (symbols) and simulated (lines) (dashed lines refer to components) [104] (reaction steps 1 and 2 refer to char devolatilization and combustion, respectively). Fig. 13 - Arrhenius plot of the kinetic contribution, rc, of the reactivity for lignocellulosic char oxidation. Table 1 - Char gasification rates in CO2 bearing atmospheres with parameter values (the structural contribution is delimited by square brackets), temperature (Tg) and pressure (PCO2) during gasification, pyrolysis conditions and/or sample properties. Table 2 - Char gasification rates in H2O bearing atmospheres with parameter values (the structural contribution is delimited by square brackets), temperature (Tg) and pressure (PH2O) during gasifcation, pyrolysis conditions and/or sample properties. Table 3 - Char oxidation rates with parameter values (the structural contribution is delimited by square brackets) with heating rate, final temperature and volumetric fraction of O2 during oxidation, pyrolysis conditions and/or sample properties.
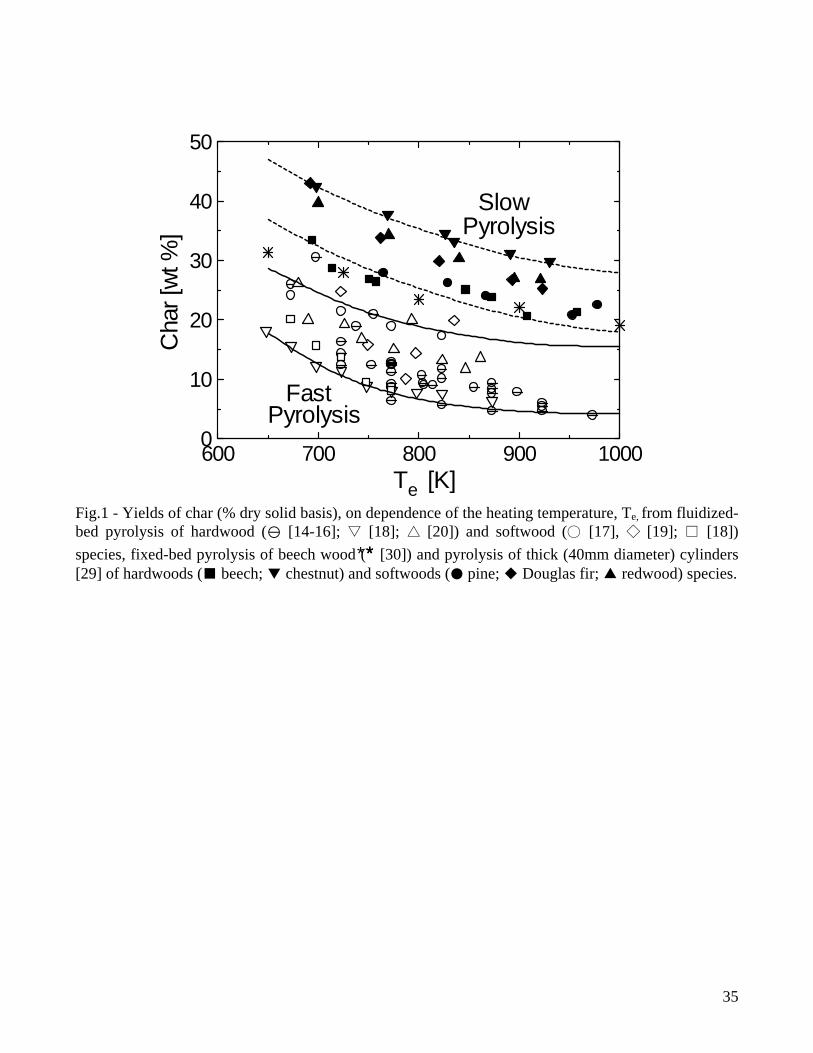
35
Fig.1 - Yields of char (% dry solid basis), on dependence of the heating temperature, Te, from fluidized-bed pyrolysis of hardwood (� [14-16]; ▽ [18]; △ [20]) and softwood (◯ [17], ◇ [19]; □ [18])
species, fixed-bed pyrolysis of beech wood (∗∗∗∗ [30]) and pyrolysis of thick (40mm diameter) cylinders [29] of hardwoods (■ beech; ▼ chestnut) and softwoods (● pine; ◆ Douglas fir; ▲ redwood) species.
600 700 800 900 10000
10
20
30
40
50
Pyrolysis
PyrolysisFast
SlowC
har [
wt %
]
Te [K]
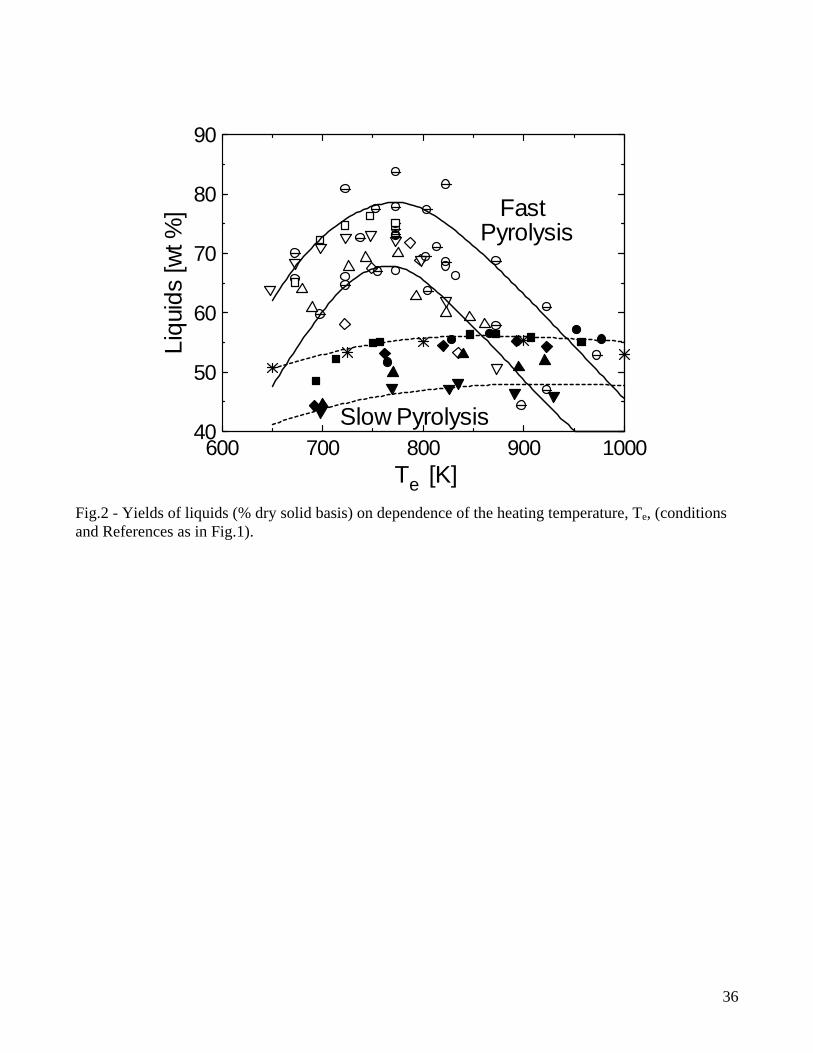
36
Fig.2 - Yields of liquids (% dry solid basis) on dependence of the heating temperature, Te, (conditions and References as in Fig.1).
600 700 800 900 100040
50
60
70
80
90
Pyrolysis
Slow Pyrolysis
FastLi
quid
s [w
t %]
Te [K]
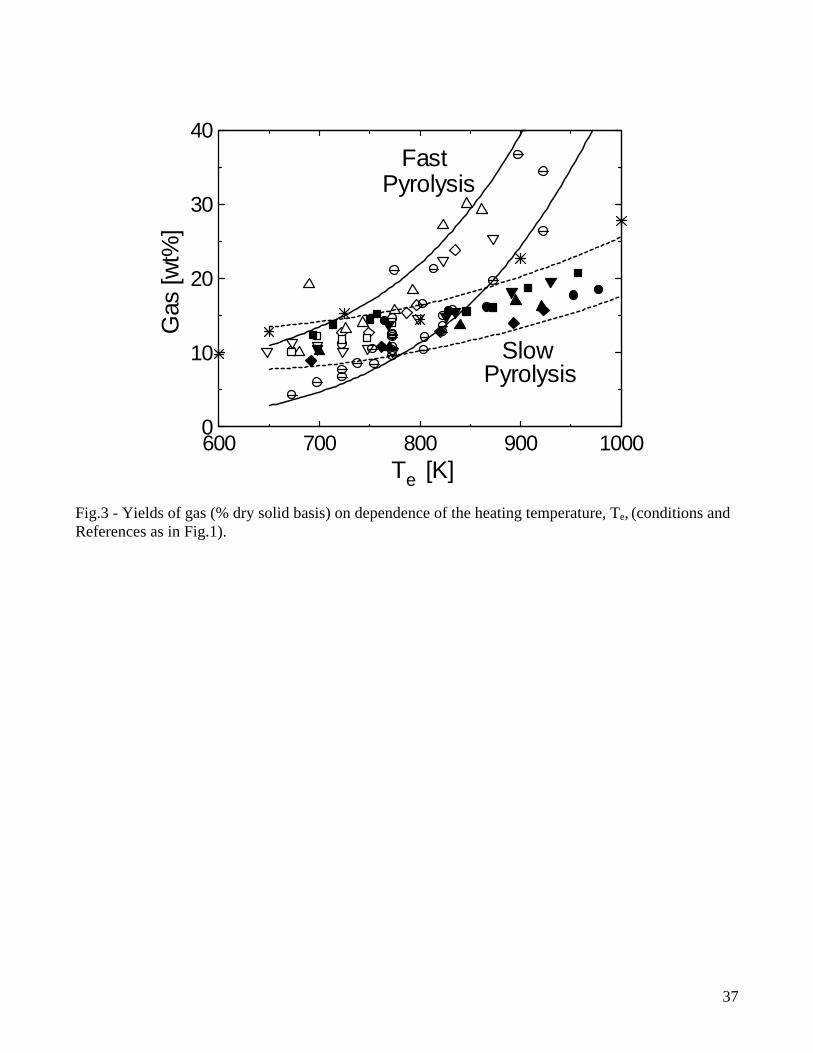
37
Fig.3 - Yields of gas (% dry solid basis) on dependence of the heating temperature, Te, (conditions and References as in Fig.1).
600 700 800 900 10000
10
20
30
40
Gas
[wt%
]
Te [K]
Pyrolysis
Pyrolysis
Fast
Slow
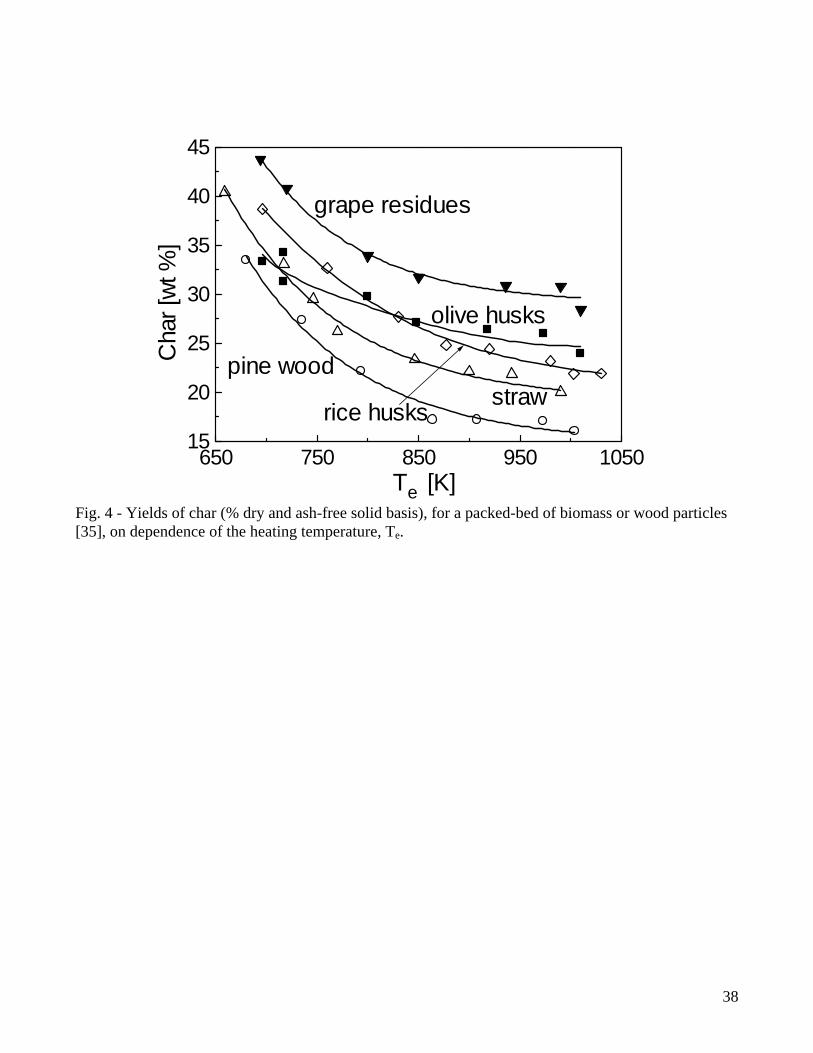
38
Fig. 4 - Yields of char (% dry and ash-free solid basis), for a packed-bed of biomass or wood particles [35], on dependence of the heating temperature, Te.
650 750 850 950 105015
20
25
30
35
40
45
strawpine wood
rice husks
olive husks
grape residues
Cha
r [w
t %]
Te [K]
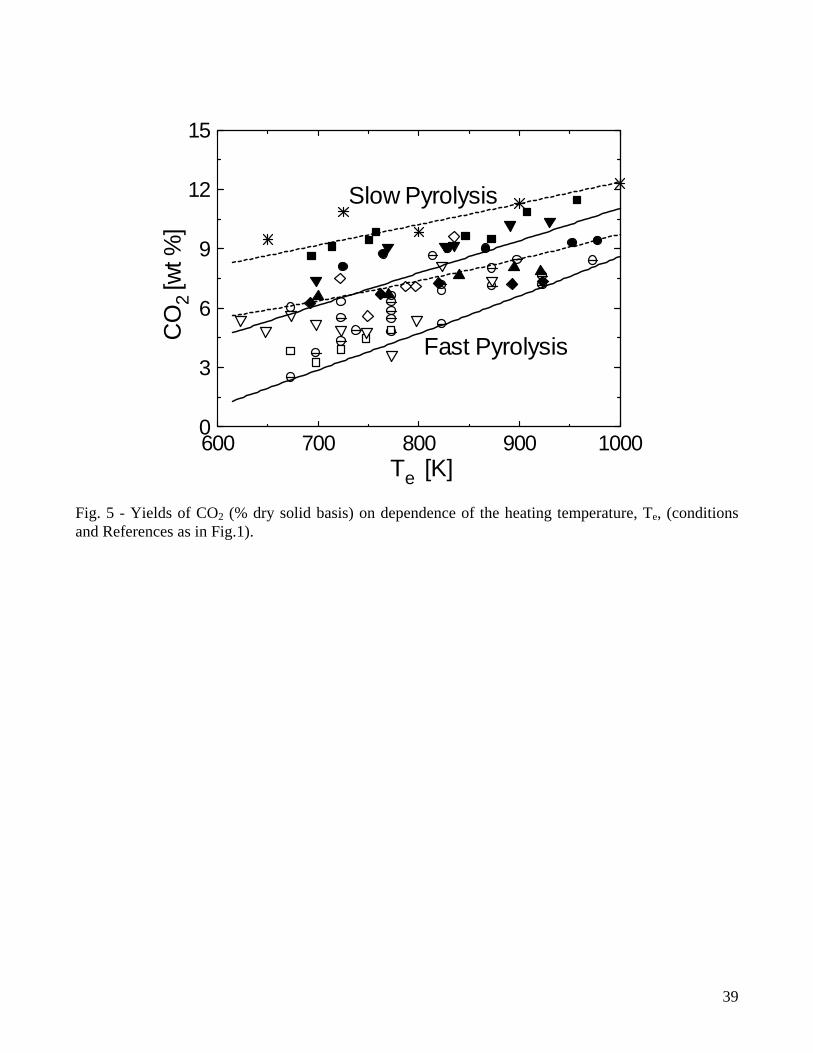
39
Fig. 5 - Yields of CO2 (% dry solid basis) on dependence of the heating temperature, Te, (conditions and References as in Fig.1).
600 700 800 900 10000
3
6
9
12
15
CO
2 [w
t %]
Te [K]
Slow Pyrolysis
Fast Pyrolysis
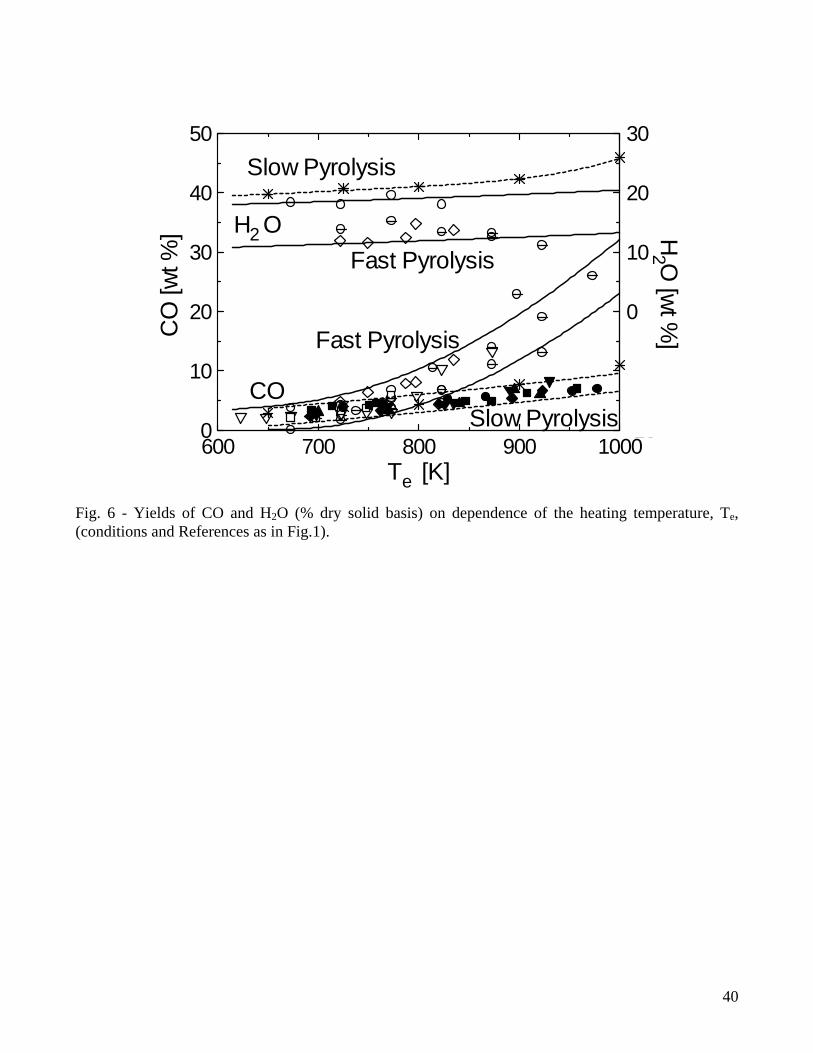
40
Fig. 6 - Yields of CO and H2O (% dry solid basis) on dependence of the heating temperature, Te, (conditions and References as in Fig.1).
600 700 800 900 10000
10
20
30
40
50
CO
[wt %
]
Te [K]
-20
-10
0
10
20
30
Fast Pyrolysis
Fast Pyrolysis
H2 O
CO
H2 O
[wt %
]Slow Pyrolysis
Slow Pyrolysis
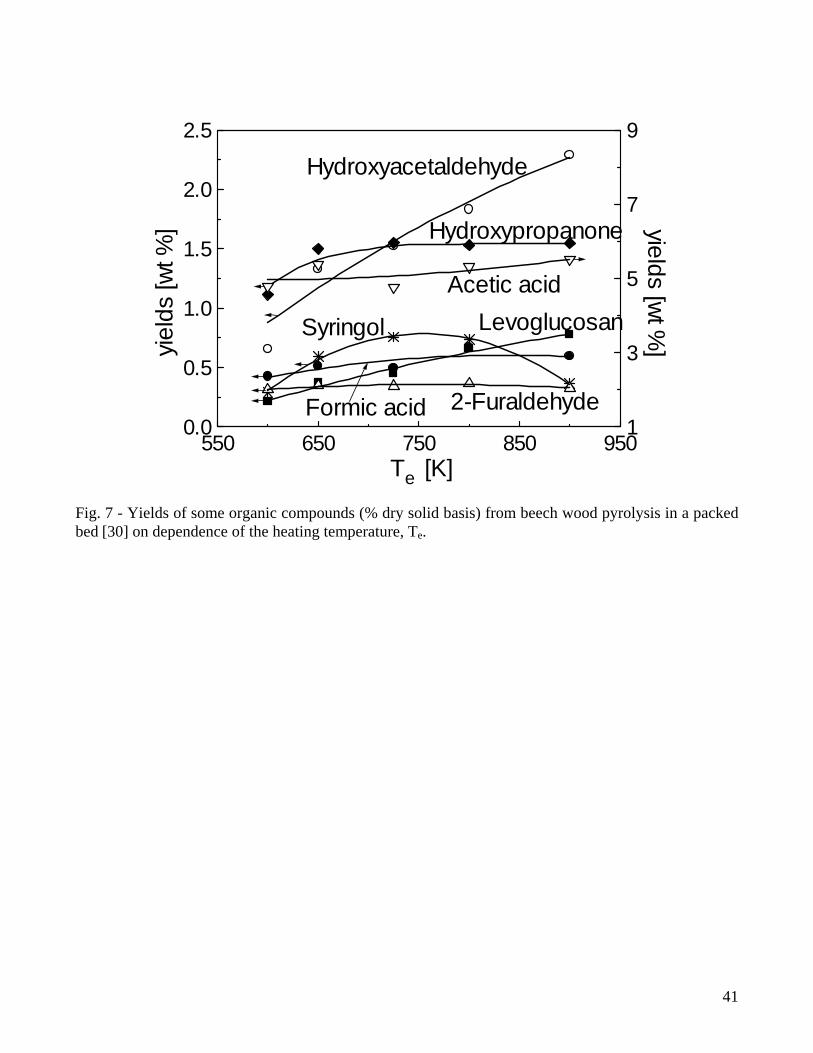
41
Fig. 7 - Yields of some organic compounds (% dry solid basis) from beech wood pyrolysis in a packed bed [30] on dependence of the heating temperature, Te.
550 650 750 850 9500.0
0.5
1.0
1.5
2.0
2.5
Syringol
2-FuraldehydeFormic acid
Acetic acid
Hydroxypropanone
Hydroxyacetaldehyde
Levoglucosan
yiel
ds [w
t %]
Te [K]
1
3
5
7
9yields [w
t %]
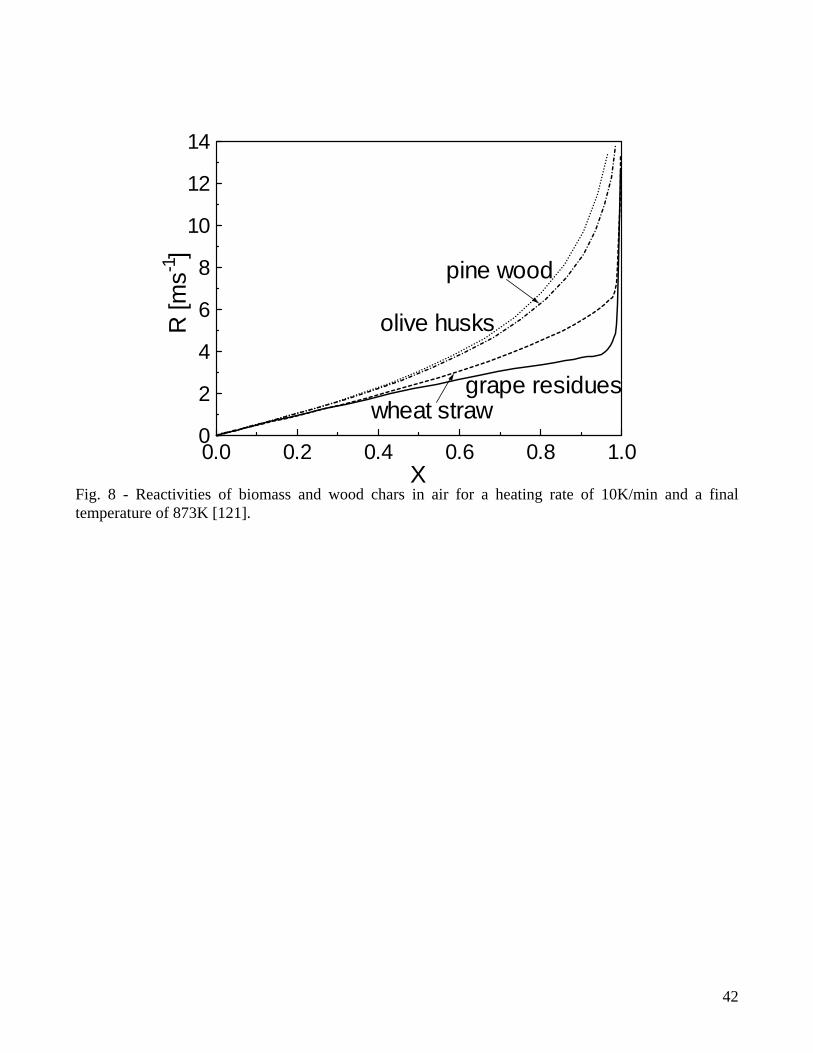
42
Fig. 8 - Reactivities of biomass and wood chars in air for a heating rate of 10K/min and a final temperature of 873K [121].
0.0 0.2 0.4 0.6 0.8 1.00
2
4
6
8
10
12
14
pine wood
olive husks
grape residueswheat straw
R [m
s-1]
X

43
Fig. 9 - Conversion (A) and time derivative of conversion (B) as functions of temperature for waste wood heated in different atmospheres at 2K/min [78].
400 600 800 1000 12000
2
4
6
8
10B
Gas
ifica
tion
Com
bust
ion
N2
CO2
Air
dX/d
t x 1
0 3 [s
-1]
T [K]
0.0
0.2
0.4
0.6
0.8
1.0X
A
Gas
ifica
tion
Com
bust
ion
N2
CO2Air
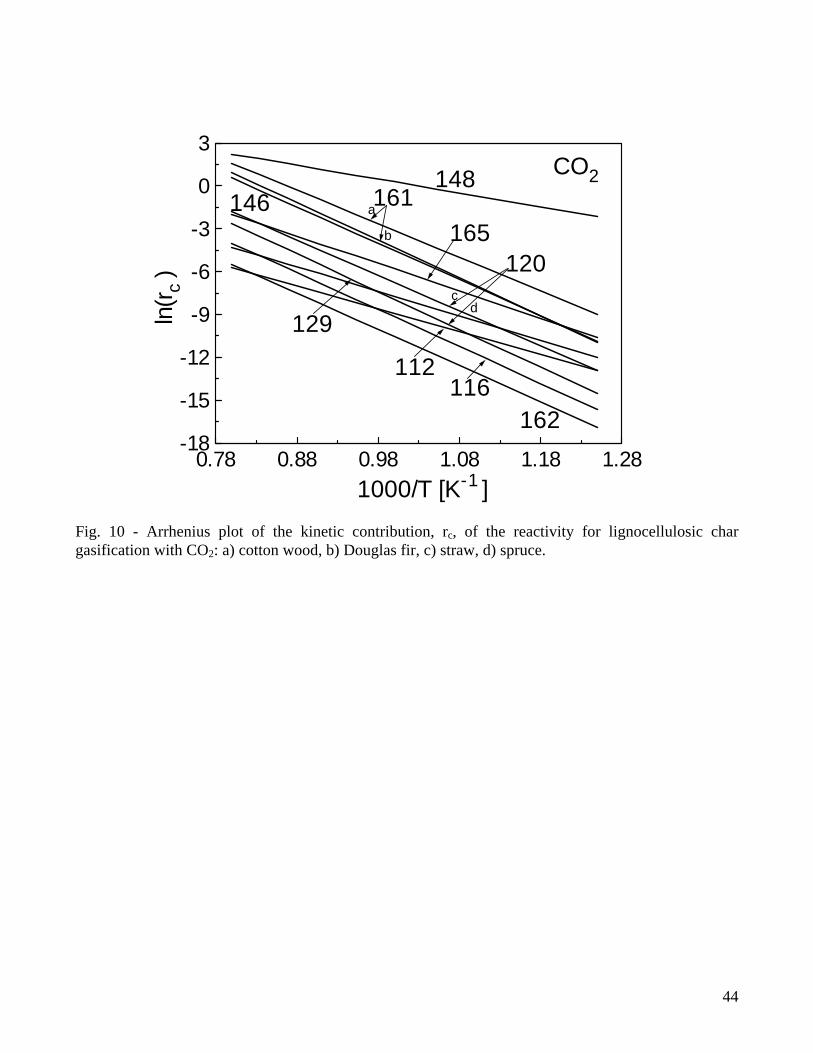
44
Fig. 10 - Arrhenius plot of the kinetic contribution, rc, of the reactivity for lignocellulosic char gasification with CO2: a) cotton wood, b) Douglas fir, c) straw, d) spruce.
0.78 0.88 0.98 1.08 1.18 1.28-18
-15
-12
-9
-6
-3
0
3
dc
b
a161
148 CO2
129
116112
120165
162
146
ln(r
c )
1000/T [K-1 ]

45
Fig. 11 - Arrhenius plot of the kinetic contribution, rc, of the reactivity for lignocellulosic char gasification with H2O: a) straw, b) poplar, c) bark, d) beech, e) birch, f) maple, g) pine.
0.78 0.88 0.98 1.08 1.18 1.28-18
-15
-12
-9
-6
-3
0
3
gf
d
cb
e
a
169
167
162
Steam119
117
131
168
ln(r
c )
1000/T [K-1 ]
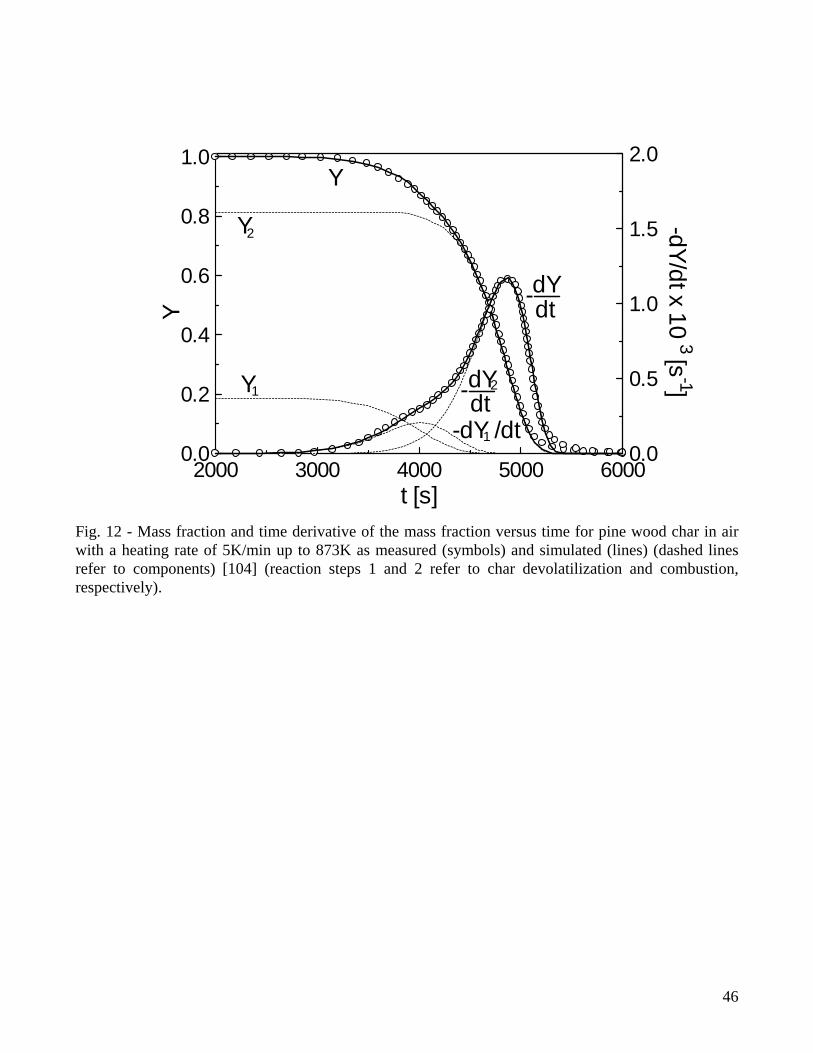
46
Fig. 12 - Mass fraction and time derivative of the mass fraction versus time for pine wood char in air with a heating rate of 5K/min up to 873K as measured (symbols) and simulated (lines) (dashed lines refer to components) [104] (reaction steps 1 and 2 refer to char devolatilization and combustion, respectively).
2000 3000 4000 5000 60000.0
0.2
0.4
0.6
0.8
1.0
1
2
Y
Y
1-dY /dt
2-dtdY
-dtdY
Y
-dY/dt x 10
3 [s-1]
Y
t [s]
0.0
0.5
1.0
1.5
2.0
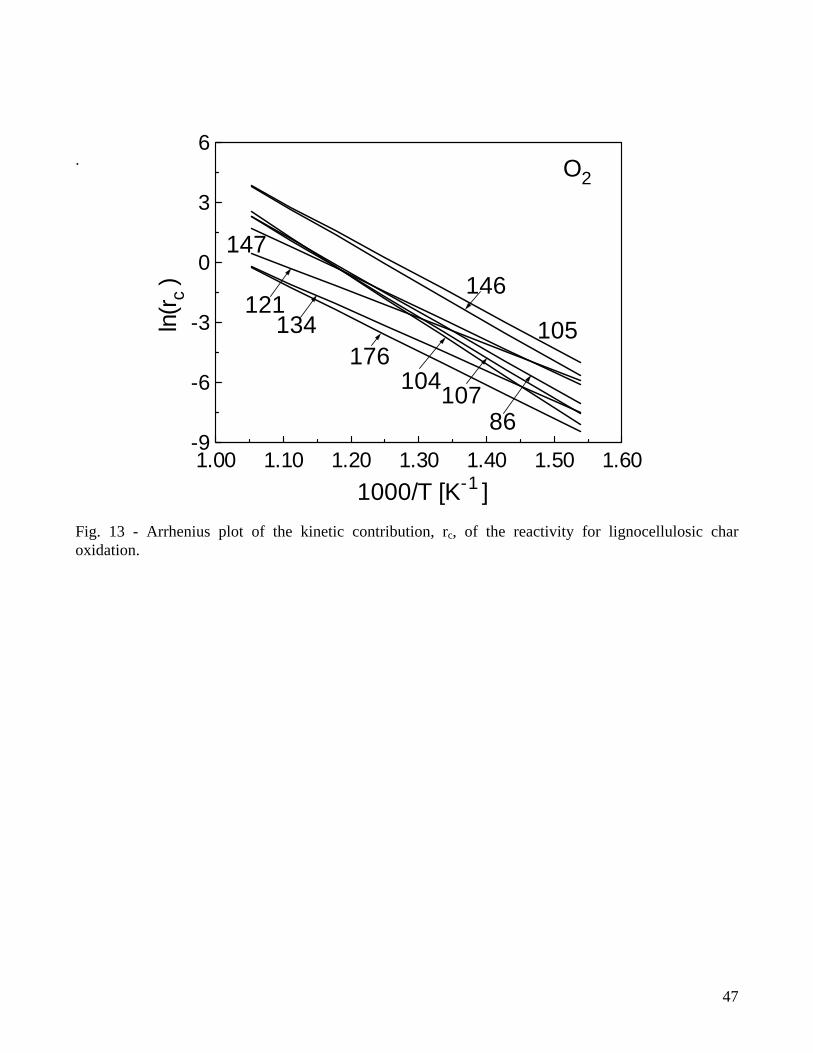
47
. Fig. 13 - Arrhenius plot of the kinetic contribution, rc, of the reactivity for lignocellulosic char oxidation.
1.00 1.10 1.20 1.30 1.40 1.50 1.60-9
-6
-3
0
3
6
104176
107
O2
121
147
105
86
134
146
ln(r
c )
1000/T [K-1 ]
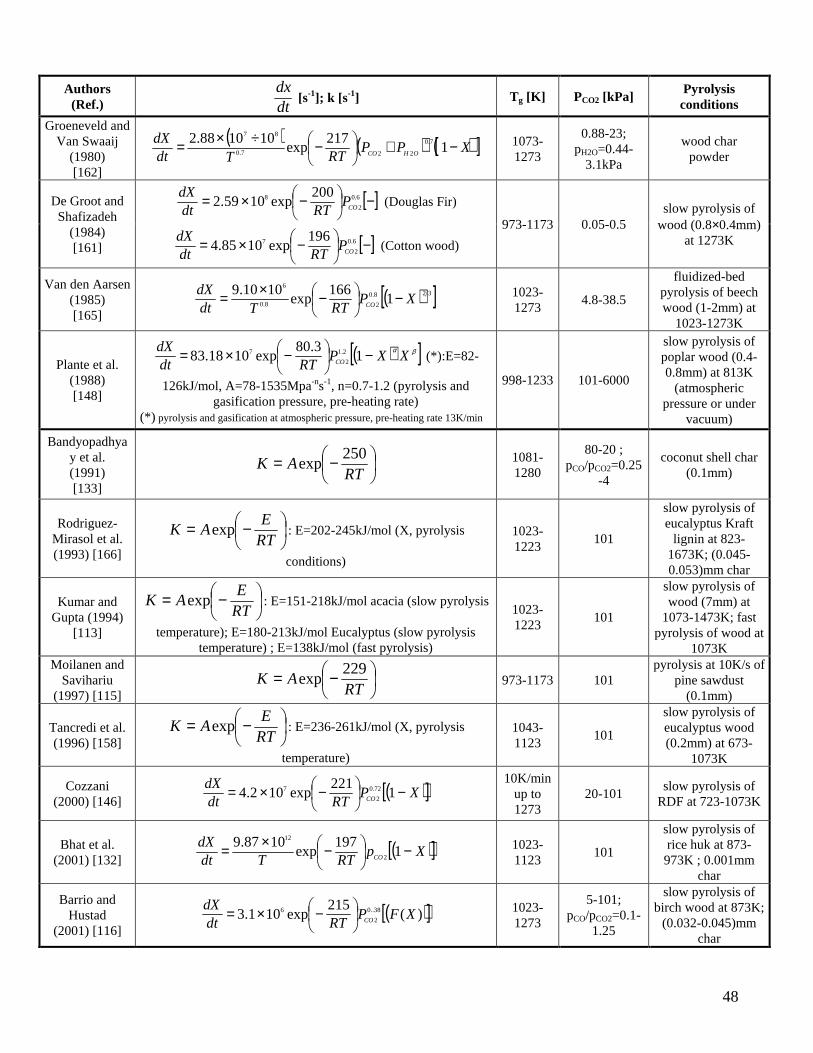
48
Authors (Ref.) dt
dx [s-1]; k [s-1] Tg [K] PCO2 [kPa]
Pyrolysis conditions
Groeneveld and Van Swaaij
(1980) [162]
( ) ( ) ( )[ ]XPPRTTdt
dXOHCO
−+
−÷×= 1217
exp101088.2 7.0
227.0
87
1073-1273
0.88-23; pH2O=0.44-
3.1kPa
wood char powder
De Groot and Shafizadeh
(1984) [161]
[ ]−
−×= 6.0
2
8 200exp1059.2
COP
RTdtdX
(Douglas Fir)
973-1173 0.05-0.5 slow pyrolysis of
wood (0.8×0.4mm) at 1273K [ ]−
−×= 6.0
2
7 196exp1085.4 COP
RTdtdX
(Cotton wood)
Van den Aarsen (1985) [165]
( )[ ]328.0
28.0
6
1166
exp1010.9
XPRTTdt
dXCO
−
−×= 1023-1273
4.8-38.5
fluidized-bed pyrolysis of beech wood (1-2mm) at
1023-1273K
Plante et al. (1988) [148]
( )[ ]βαXXP
RTdtdX
CO−
−×= 13.80
exp1018.83 2.1
2
7 (*):E=82-
126kJ/mol, A=78-1535Mpa-ns-1, n=0.7-1.2 (pyrolysis and gasification pressure, pre-heating rate)
(*) pyrolysis and gasification at atmospheric pressure, pre-heating rate 13K/min
998-1233 101-6000
slow pyrolysis of poplar wood (0.4-0.8mm) at 813K
(atmospheric pressure or under
vacuum)
Bandyopadhyay et al. (1991) [133]
−=RT
AK250
exp 1081-1280
80-20 ; pCO/pCO2=0.25
-4
coconut shell char (0.1mm)
Rodriguez-Mirasol et al. (1993) [166]
−=RTE
AK exp : E=202-245kJ/mol (X, pyrolysis
conditions)
1023-1223
101
slow pyrolysis of eucalyptus Kraft
lignin at 823-1673K; (0.045-0.053)mm char
Kumar and Gupta (1994)
[113]
−=RTE
AK exp : E=151-218kJ/mol acacia (slow pyrolysis
temperature); E=180-213kJ/mol Eucalyptus (slow pyrolysis temperature) ; E=138kJ/mol (fast pyrolysis)
1023-1223
101
slow pyrolysis of wood (7mm) at
1073-1473K; fast pyrolysis of wood at
1073K Moilanen and
Savihariu (1997) [115]
−=RT
AK229
exp 973-1173 101 pyrolysis at 10K/s of
pine sawdust (0.1mm)
Tancredi et al. (1996) [158]
−=RTE
AK exp : E=236-261kJ/mol (X, pyrolysis
temperature)
1043-1123
101
slow pyrolysis of eucalyptus wood (0.2mm) at 673-
1073K
Cozzani (2000) [146]
( )[ ]XPRTdt
dXCO
−
−×= 1221
exp102.4 72.0
2
7 10K/min
up to 1273
20-101 slow pyrolysis of
RDF at 723-1073K
Bhat et al. (2001) [132]
( )[ ]XpRTTdt
dXCO −
−×= 1197
exp1087.9
2
12
1023-1123
101
slow pyrolysis of rice huk at 873-973K ; 0.001mm
char
Barrio and Hustad
(2001) [116] ( )[ ])(
215exp101.3 38..0
2
6 XFPRTdt
dXCO
−×= 1023-1273
5-101; pCO/pCO2=0.1-
1.25
slow pyrolysis of birch wood at 873K;
(0.032-0.045)mm char
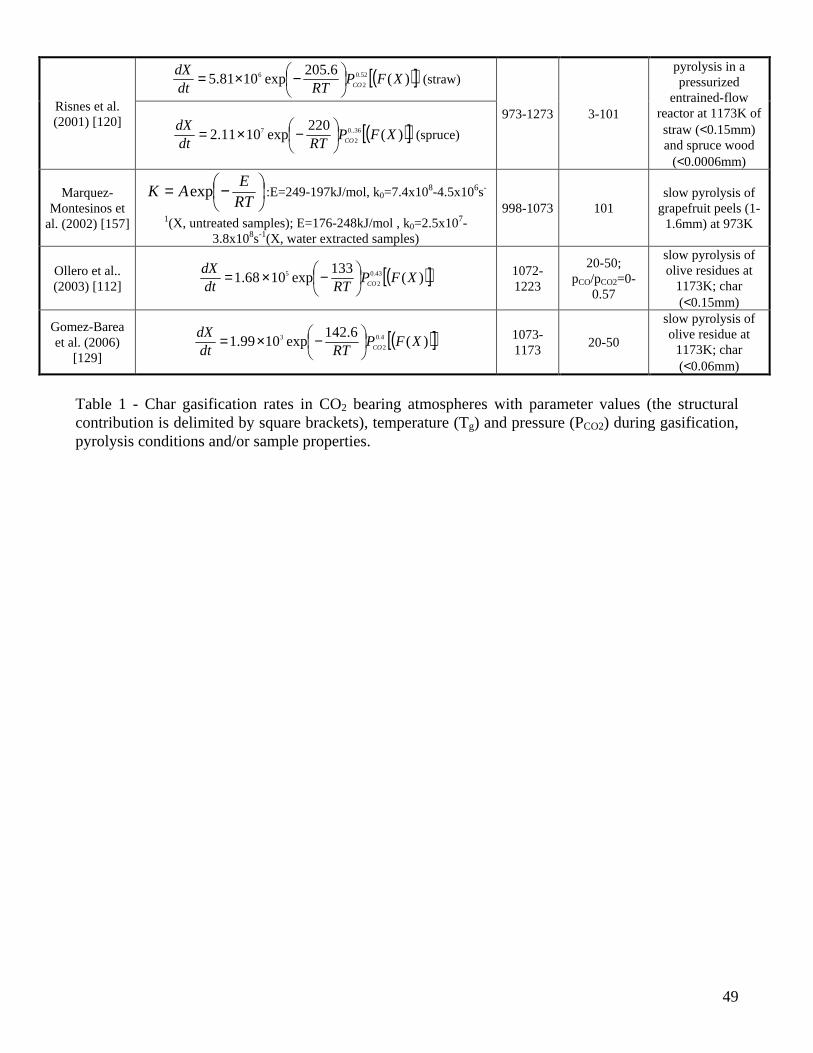
49
Risnes et al. (2001) [120]
( )[ ])(6.205
exp1081.5 52.0
2
6 XFPRTdt
dXCO
−×= (straw)
973-1273 3-101
pyrolysis in a pressurized
entrained-flow reactor at 1173K of straw (<0.15mm) and spruce wood
(<0.0006mm)
( )[ ])(220
exp1011.2 36..0
2
7 XFPRTdt
dXCO
−×= (spruce)
Marquez-Montesinos et
al. (2002) [157]
−=RTE
AK exp :E=249-197kJ/mol, k0=7.4x108-4.5x106s-
1(X, untreated samples); E=176-248kJ/mol , k0=2.5x107-3.8x108s-1(X, water extracted samples)
998-1073 101 slow pyrolysis of
grapefruit peels (1-1.6mm) at 973K
Ollero et al.. (2003) [112]
( )[ ])(133
exp1068.1 43.0
2
5 XFPRTdt
dXCO
−×= 1072-1223
20-50; pCO/pCO2=0-
0.57
slow pyrolysis of olive residues at
1173K; char (<0.15mm)
Gomez-Barea et al. (2006)
[129] ( )[ ])(
6.142exp1099.1 4.0
2
3 XFPRTdt
dXCO
−×= 1073-1173
20-50
slow pyrolysis of olive residue at
1173K; char (<0.06mm)
Table 1 - Char gasification rates in CO2 bearing atmospheres with parameter values (the structural contribution is delimited by square brackets), temperature (Tg) and pressure (PCO2) during gasification, pyrolysis conditions and/or sample properties.
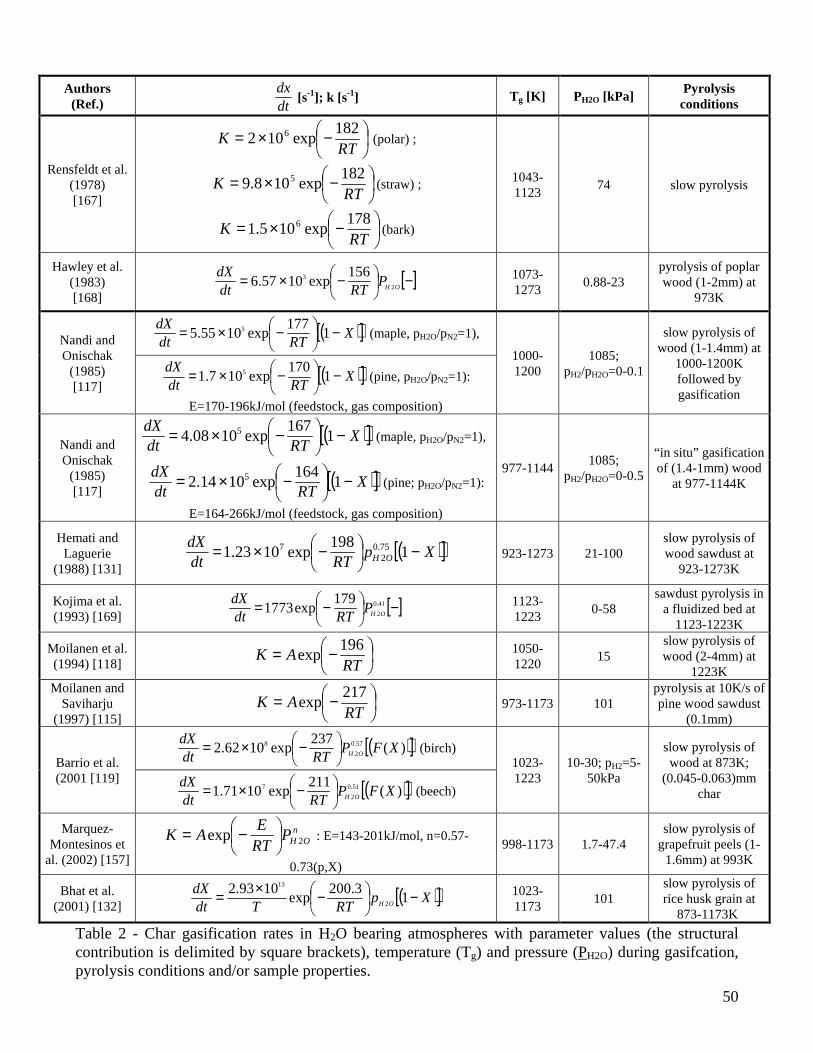
50
Authors (Ref.) dt
dx [s-1]; k [s-1] Tg [K] PH2O [kPa]
Pyrolysis conditions
Rensfeldt et al. (1978) [167]
−×=RT
K182
exp102 6 (polar) ;
−×=RT
K182
exp108.9 5 (straw) ;
−×=RT
K178
exp105.1 6 (bark)
1043-1123
74 slow pyrolysis
Hawley et al. (1983) [168]
[ ]−
−×= OHPRTdt
dX2
3 156exp1057.6 1073-
1273 0.88-23
pyrolysis of poplar wood (1-2mm) at
973K
Nandi and Onischak (1985) [117]
( )[ ]XRTdt
dX −
−×= 1177
exp1055.5 5 (maple, pH2O/pN2=1),
1000-1200
1085; pH2/pH2O=0-0.1
slow pyrolysis of wood (1-1.4mm) at
1000-1200K followed by gasification
( )[ ]XRTdt
dX −
−×= 1170
exp107.1 5 (pine, pH2O/pN2=1):
E=170-196kJ/mol (feedstock, gas composition)
Nandi and Onischak (1985) [117]
( )[ ]XRTdt
dX −
−×= 1167
exp1008.4 5 (maple, pH2O/pN2=1),
977-1144 1085;
pH2/pH2O=0-0.5
“in situ” gasification of (1.4-1mm) wood
at 977-1144K ( )[ ]XRTdt
dX −
−×= 1164
exp1014.2 5 (pine; pH2O/pN2=1):
E=164-266kJ/mol (feedstock, gas composition)
Hemati and Laguerie
(1988) [131] ( )[ ]Xp
RTdtdX
OH −
−×= 1198
exp1023.1 75.02
7 923-1273 21-100 slow pyrolysis of wood sawdust at
923-1273K
Kojima et al. (1993) [169]
[ ]−
−= 41.0
2
179exp1773 OHP
RTdtdX
1123-1223
0-58 sawdust pyrolysis in
a fluidized bed at 1123-1223K
Moilanen et al. (1994) [118]
−=RT
AK196
exp 1050-1220
15 slow pyrolysis of wood (2-4mm) at
1223K Moilanen and
Saviharju (1997) [115]
−=RT
AK217
exp 973-1173 101 pyrolysis at 10K/s of pine wood sawdust
(0.1mm)
Barrio et al. (2001 [119]
( )[ ])(237
exp1062.2 57.0
2
8 XFPRTdt
dXOH
−×= (birch) 1023-1223
10-30; pH2=5-50kPa
slow pyrolysis of wood at 873K;
(0.045-0.063)mm char ( )[ ])(
211exp1071.1 51.0
2
7 XFPRTdt
dXOH
−×= (beech)
Marquez-Montesinos et
al. (2002) [157]
nOHP
RTE
AK 2exp
−= : E=143-201kJ/mol, n=0.57-
0.73(p,X)
998-1173 1.7-47.4 slow pyrolysis of
grapefruit peels (1-1.6mm) at 993K
Bhat et al. (2001) [132]
( )[ ]XpRTTdt
dXOH
−
−×= 13.200
exp1093.2
2
13
1023-1173
101 slow pyrolysis of rice husk grain at
873-1173K
Table 2 - Char gasification rates in H2O bearing atmospheres with parameter values (the structural contribution is delimited by square brackets), temperature (Tg) and pressure (PH2O) during gasifcation, pyrolysis conditions and/or sample properties.
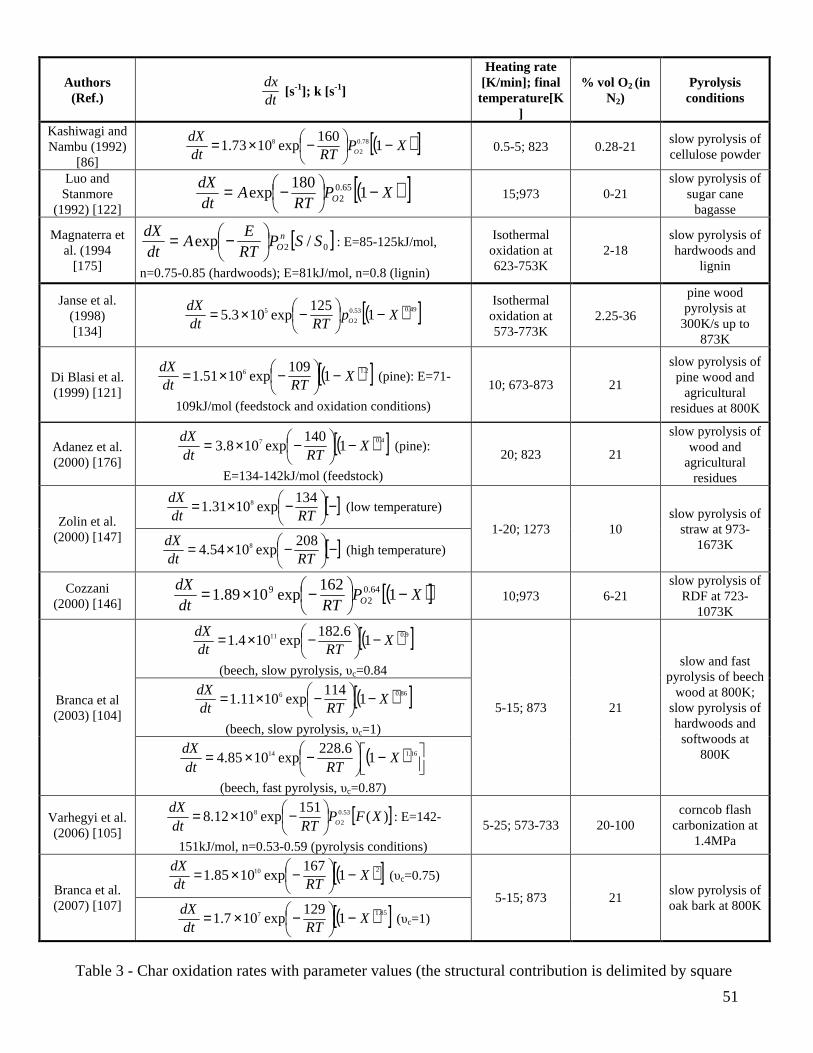
51
Authors (Ref.) dt
dx [s-1]; k [s-1]
Heating rate [K/min]; final temperature[K
]
% vol O2 (in N2)
Pyrolysis conditions
Kashiwagi and Nambu (1992)
[86] ( )[ ]XP
RTdtdX
O−
−×= 1160
exp1073.1 78.0
2
8 0.5-5; 823 0.28-21 slow pyrolysis of cellulose powder
Luo and Stanmore
(1992) [122] ( )[ ]XP
RTA
dtdX
O −
−= 1180
exp 65.02 15;973 0-21
slow pyrolysis of sugar cane
bagasse
Magnaterra et al. (1994
[175]
[ ]02 /exp SSPRTE
AdtdX n
O
−= : E=85-125kJ/mol,
n=0.75-0.85 (hardwoods); E=81kJ/mol, n=0.8 (lignin)
Isothermal oxidation at 623-753K
2-18 slow pyrolysis of hardwoods and
lignin
Janse et al. (1998) [134]
( )[ ]49.053.0
2
5 1125
exp103.5 XpRTdt
dXO −
−×= Isothermal oxidation at 573-773K
2.25-36
pine wood pyrolysis at 300K/s up to
873K
Di Blasi et al. (1999) [121]
( )[ ]2.16 1109
exp1051.1 XRTdt
dX −
−×= (pine): E=71-
109kJ/mol (feedstock and oxidation conditions)
10; 673-873 21
slow pyrolysis of pine wood and
agricultural residues at 800K
Adanez et al. (2000) [176]
( )[ ]4.07 1140
exp108.3 XRTdt
dX −
−×= (pine):
E=134-142kJ/mol (feedstock)
20; 823 21
slow pyrolysis of wood and
agricultural residues
Zolin et al. (2000) [147]
[ ]−
−×=RTdt
dX 134exp1031.1 8 (low temperature)
1-20; 1273 10 slow pyrolysis of
straw at 973-1673K [ ]−
−×=RTdt
dX 208exp1054.4 8 (high temperature)
Cozzani (2000) [146]
( )[ ]XPRTdt
dXO −
−×= 1162
exp1089.1 64.02
9 10;973 6-21 slow pyrolysis of
RDF at 723-1073K
Branca et al (2003) [104]
( )[ ]9.011 16.182
exp104.1 XRTdt
dX −
−×=
(beech, slow pyrolysis, υc=0.84
5-15; 873 21
slow and fast pyrolysis of beech
wood at 800K; slow pyrolysis of hardwoods and softwoods at
800K
( )[ ]86.06 1114
exp1011.1 XRTdt
dX −
−×=
(beech, slow pyrolysis, υc=1)
( )
−
−×= 16.114 16.228
exp1085.4 XRTdt
dX
(beech, fast pyrolysis, υc=0.87)
Varhegyi et al. (2006) [105]
[ ])(151
exp1012.8 53.0
2
8 XFPRTdt
dXO
−×= : E=142-
151kJ/mol, n=0.53-0.59 (pyrolysis conditions)
5-25; 573-733 20-100 corncob flash
carbonization at 1.4MPa
Branca et al. (2007) [107]
( )[ ]210 1167
exp1085.1 XRTdt
dX −
−×= (υc=0.75)
5-15; 873 21 slow pyrolysis of oak bark at 800K
( )[ ]85.17 1129
exp107.1 XRTdt
dX −
−×= (υc=1)
Table 3 - Char oxidation rates with parameter values (the structural contribution is delimited by square

52
brackets) with heating rate, final temperature and volumetric fraction of O2 during oxidation, pyrolysis conditions and/or sample properties.

53





























