Embed Size (px)
Citation preview

Wound-induced calcium waves in alveolar type II cells
LEE E. HINMAN,1 GREG J. BEILMAN,2,3 KRISTINE E. GROEHLER,2,3 AND PAUL J. SAMMAK1
Departments of 1Pharmacology and 2Surgery, University of Minnesota,Minneapolis 55455; and 3North Trauma Institute, Robbinsdale, Minnesota 55422
Hinman, Lee E., Greg J. Beilman, Kristine E.Groehler, and Paul J. Sammak. Wound-induced calciumwaves in alveolar type II cells. Am. J. Physiol. 273 (Lung Cell.Mol. Physiol. 17): L1242–L1248, 1997.—Alveolar type IIepithelial (ATII) cells repopulate the alveolus after acute lunginjury. We hypothesized that injury would initiate signals innearby survivors. When rat ATII monolayers were wounded,elevations in intracellular free Ca21 concentration ([Ca21]i)began at the edge of the wound and propagated outward as awave for at least 300 µm. The [Ca21]i wave was due to bothinflux of extracellular Ca21 and release of intracellular Ca21
stores. Reducing Ca21 influx with brief treatments of ethyleneglycol-bis(b-aminoethyl ether)-N,N,N8,N8-tetraacetic acid orGd31 reduced both the amplitude and the apparent speed.Draining intracellular Ca21 stores by pretreatment withcyclopiazonic acid eliminated the [Ca21]i wave. Therefore, the[Ca21]i wave depended critically on intracellular Ca21 stores.[Ca21]i elevations propagated over a break in the monolayer,suggesting that extracellular pathways were involved. Fur-thermore, extracellular factors from injured cells elevated[Ca21]i in uninjured cultures. We conclude that woundingproduces a [Ca21]i wave in surviving cells and part of thisresponse is mediated by soluble factors released into theextracellular space during injury.
alveolar epithelial cells; acute lung injury; mechanical injury;intercellular signaling; intracellular calcium stores
SURVIVAL AFTER ACUTE lung injury is dependent on therestoration of the integrity of the alveolar-capillaryinterface. Preservation of lung function requires thereestablishment of an intact epithelial barrier (24).Cells that are primarily responsible for the reestablish-ment of this barrier are alveolar type II (ATII) cells.ATII cells repopulate the alveolus after lung injury,restoring barrier function, alveolar fluid absorption,and the synthesis of pulmonary surfactant and surfac-tant proteins (9, 18). The ability of the ATII cells torespond quickly to injury is critical to the prevention oflater development of fibrosis (26). For ATII cells toinitiate processes such as proliferation to restore theepithelial lining, they must first detect the onset ofalveolar injury. The mode of transmission of such asignal and the type of signal (positive or negative) iscurrently unknown. To determine which cellular signal-ing systems might underlie rapid detection of nearbyinjury, we have investigated some of the earliest signal-ing responses of ATII cells to nearby injury. These earlysignals may begin the cascade of events that regulatethe healing response after injury.
Mechanical injury produces immediate Ca21 signalsin several cell types. Enomoto et al. (8) and Sammak etal. (27) have found that cell rupture releases factorsthat can stimulate elevations in intracellular free Ca21
concentrations ([Ca21]i) in endothelial and epithelialcells. It has also been shown that gentle stimulation
with microneedles (30, 34) or shear stress (33) elevates[Ca21]i in single cells. Gentle mechanical stretching,without membrane rupture, leads to propagation of[Ca21]i waves in monolayers of endothelial cells as wellas tracheal, mammary, lens, and ATII epithelial cells(6, 8, 25, 27, 30, 38). Therefore, we hypothesize thatelevations in [Ca21]i might be involved in the ATIIresponse to injury.
If [Ca21]i were elevated during injury, it might regu-late cell behavior such as motility, proliferation, andalveolar fluid absorption and secretion. It is known thatelevations in [Ca21]i lead to secretion of pulmonarysurfactant and surfactant proteins (5). Although thesignals that cause ATII cell motility, proliferation, anddifferentiation into type I cells are still unknown, it hasbeen shown in other cell types that [Ca21]i influencesmotile speed (2, 11, 12, 19, 20; P. J. Sammak, P. O. T.Tran, L. E. Hinman, Q.-H. P. Tran, G. M. Unger, andR. L. Bellrichard, unpublished observations), prolifera-tion (1, 3, 19, 21, 22, 36, 38), and differentiation (13, 15).
Because we could not directly observe the alveolus insitu, we wounded primary cultures of rat ATII cells.Confluent monolayers of ATII cells were wounded bymaking a scratch in the monolayer with an 18-gaugeneedle held in a micromanipulator. This model systemallowed us to look for [Ca21]i signals that originatedduring and immediately after wounding and how thesesignals might be propagated throughout the alveolus.This model also allows us to look at signals that aregenerated after injury that results in immediate cellrupture.
The property of intercellular communication is funda-mental to any multicellular system, including the lung.Intercellular communication could be used to coordi-nate a response to many stimuli, including wounding.[Ca21]i-dependent intercellular communication has beenstudied in respiratory tract cilia where it coordinatesciliary beat frequency (17) and in endothelial cellswhere it stimulates movement of endothelial cell mono-layers during wound closure (Sammak et al., unpub-lished observations).
In the current study, we show that wounding ATIIcells produces an [Ca21]i wave that is initiated at thesite of injury and propagates throughout the monolayerfor several hundred micrometers. In addition, we findthat the signal for the [Ca21]i wave is carried, at least inpart, via diffusion through the extracellular media. Thesource of the Ca21 for the [Ca21]i wave was from bothintracellular and extracellular Ca21 pools; however, the[Ca21]i wave was totally dependent on the filling ofintracellular Ca21 stores. These findings suggest thatATII cells can detect wounding in nearby cells asmonitored by increases in [Ca21]i. It is plausible thatthese changes in [Ca21]i may have an important role in
1040-0605/97 $5.00 Copyright r 1997 the American Physiological SocietyL1242

the determination of the response of ATII cells afterwounding.
MATERIALS AND METHODS
Cell culture. ATII cells were harvested in a manner similarto that described by Dobbs et al. (7). Male Sprague-Dawleyrats weighing 250–275 g (Harlan Sprague Dawley, Indianapo-lis, IN) were used. Cells were plated on fibronectin-coatedglass coverslips at a concentration of 1 3 106 cells/coverslipand were grown for 2 days in 10% fetal calf serum (GIBCOBRL, Gaithersburg, MD) in 5% CO2 at 37°C. The viability ofcells by trypan blue exclusion was always .90%. Cells had.90% ATII morphology by light microscopy. Morphology wasconfirmed in representative samples by electron microscopy.
Ca21 imaging. Cells were loaded for 1 h at room tempera-ture with 2 µM acetoxymethyl ester of fura 2 (MolecularProbes, Eugene, OR) and 0.03% pluronic acid F-127 (Molecu-lar Probes). Both loading and imaging were done in Hanks’balanced salt solution (HBSS) with 20 mM N-2-hydroxyethyl-piperazine-N8-2-ethanesulfonic acid and without bicarbonateor phenol red. [Ca21]i measurements were made by fluores-cence ratio imaging of confluent monolayers at room tempera-ture in HBSS. The imaging system consisted of a Diaphot 300inverted microscope (Nikon, Melville, NY), a cooled charge-coupled device camera (PXL KAF 1400; Photometrics, Tuc-son, AZ), and a filter wheel (Mac 2000; Ludl Electronics,Hawthorne, NY), which were all controlled by IP Lab Spec-trum (Signal Analytics, Vienna, VA) on an Apple PowerMacintosh computer. Excitation light was provided by a150-W xenon arc lamp (Opti-Quip, Highland Mills, NY).Excitation light was selected with 340- and 385-nm band-pass filters (Chroma Technology, Battleboro, VT). Cells wereobserved with a Nikon CF fluor 320 objective with a numeri-cal aperture of 0.75.
Calibrations. Ratios of 340- to 385-nm intensity (R) werecalibrated to [Ca21]i by an in vitro approach similar toGrynkiewiez et al. (10). Briefly, 5 µM fura 2 in 10 mM Ca21
HBSS was imaged to record the ratio of fluorescence intensityat saturating Ca21 (Rmax). Five micromolar fura 2-free acid inCa21-free HBSS and 10 mM ethylene glycol-bis(b-aminoethylether)-N,N,N8,N8-tetraacetic acid (EGTA) were imaged torecord the ratio of fluorescence intensity at zero Ca21 (Rmin).The following formula was then used to generate a calibrationcurve
[Ca21]i 5 b ·Kd 1R 2 Rmin
Rmax 2 R2 (1)
where b is the ratio of 385-nm intensities at zero Ca21 overmaximal Ca21 and Kd is the dissociation constant at 224 nM(10).
Wave speeds were determined by measuring the distancein micrometers that the front edge of the wave had movedover three consecutive 5-s intervals. Distances were cali-brated with a microscope stage micrometer (Fisher, Pitts-burgh, PA)
Wounding. Cells were wounded with an 18-gauge needleheld in a micromanipulator (Narishige). The needle was heldat an ,45° angle to the coverslip. The needle was placed sothat it touched the coverslip but did not deflect the coverslip.Deflection was monitored by a loss of focus. The needle wasthen smoothly moved with the micromanipulator for a dis-tance of 2–3 mm at a rate of 2 mm/s, which removed a path ofcells that was 4–5 cells wide and 4–6 mm long.
Solutions. Ca21-EGTA solutions were prepared by addingEGTA (Sigma Chemical, St. Louis, MO) to Ca21-free HBSS(GIBCO) to a final concentration of 100 µM or 10 mM.
Cyclopiazonic acid (CPA; Calbiochem, La Jolla, CA) solutionswere prepared in HBSS to a final concentration of 10 µM.Gd31 solutions were freshly prepared by adding enoughgadolinium(III) chloride hexahydrate (Aldrich, Milwaukee,WI) to phosphate-free HBSS (chloride in place of phosphate toavoid precipitation with Gd31) to obtain a final concentrationof 100 µM.
RESULTS
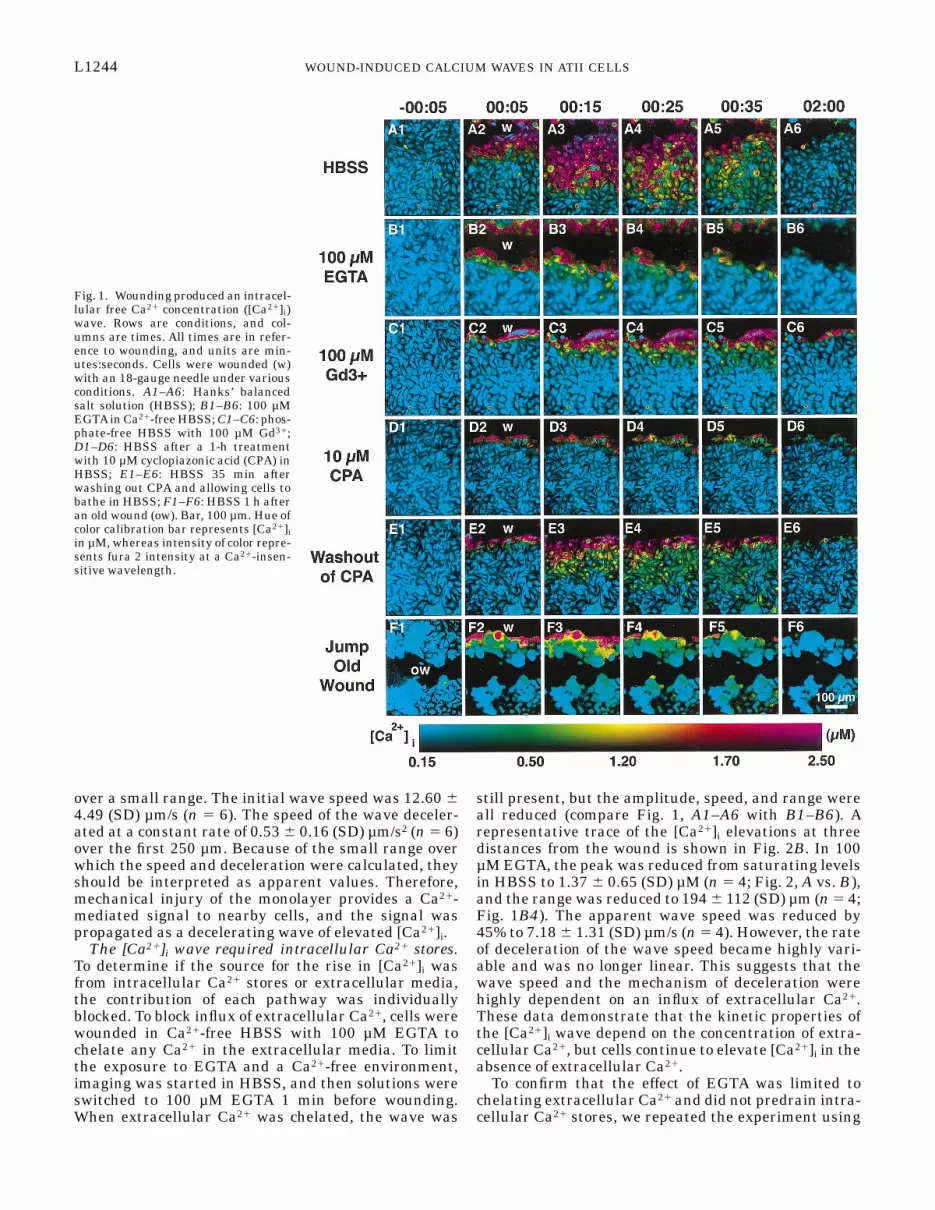
Wounding caused an elevation in [Ca21]i. To assess ifwounding elicited a [Ca21]i response, confluent monolay-ers of ATII cells were loaded with fura 2 and werewounded with an 18-gauge needle. The [Ca21]i was thenmeasured by fluorescence ratio imaging at 5-s inter-vals. Before wounding, the monolayer had an averagebasal [Ca21]i of 0.23 6 0.05 (SD) µM (n 5 31 monolay-ers). Immediately after the cells were wounded, [Ca21]iwas elevated in the cells at the wound edge (Fig. 1A2).After 15 s, the cells farthest from the wound edge alsohad elevated [Ca21]i (Fig. 1A3). Twenty-five secondspostwounding, cells up to 250 µm from the wound hadelevated [Ca21]i (Fig. 1A4). Thirty-five seconds post-wounding, cells had begun to return to baseline (Fig.1A5). After 2 min, nearly all cells in the field of viewhad returned to baseline. The magnitude of the eleva-tion was greatest at the wounded edge (.2.5 µM; Fig.1A3) and dropped off with increasing distance from thewound edge.
A few cells at the edge of the wound remainedelevated (Fig. 1A6) after cells away from the edge hadreturned to baseline. Persistent elevation of [Ca21]i ischaracteristic of cell injury (34). We have shown inother cell types that cells injured in this way usually die1 h after injury (27). Because we were interested in howmechanical injury was communicated to the survivingcells, we chose to exclude the injured cells from ourmeasurements. Therefore, subsequent analysis of[Ca21]i dynamics was performed with data from cells inthe second row and farther from the wound edge.
The elevation traveled such that cells that wereequidistant from the wound began elevating at similartimes. The wave was observed to travel out of the fieldof view (,300 µm) in every experiment. The amplitudeof the wave decreased with increasing distance fromthe wound until elevation in [Ca21]i could no longer beobserved. A representative trace of the [Ca21]i eleva-tions at three distances from the wound is shown in Fig.2A. The peak [Ca21]i in the second row of cells wasmeasured at 6.5 µM in this experiment. This value is.25 times the Kd of fura 2, and, as such, it should beconsidered to be an [Ca21]i of greater than or equivalentto 2.5 µM, which was a practical upper limit for fura 2in our experiments. After traveling 100 µm, the peakelevation was reduced to 2.5 µM (Fig. 2A), and after 250µm, it was reduced to 1 µM (Fig. 2A). On average, thepeak elevation in [Ca21]i in the second row of cells was3.57 6 1.71 (SD) µM (n 5 12); again, this should be readthat fura 2 was consistently saturated and at or aboveits reliable range.
Because image pairs were only collected at 5-s inter-vals, measurements on the speed of the wave were done
L1243WOUND-INDUCED CALCIUM WAVES IN ATII CELLS
over a small range. The initial wave speed was 12.60 64.49 (SD) µm/s (n 5 6). The speed of the wave deceler-ated at a constant rate of 0.53 6 0.16 (SD) µm/s2 (n 5 6)over the first 250 µm. Because of the small range overwhich the speed and deceleration were calculated, theyshould be interpreted as apparent values. Therefore,mechanical injury of the monolayer provides a Ca21-mediated signal to nearby cells, and the signal waspropagated as a decelerating wave of elevated [Ca21]i.
The [Ca21]i wave required intracellular Ca21 stores.To determine if the source for the rise in [Ca21]i wasfrom intracellular Ca21 stores or extracellular media,the contribution of each pathway was individuallyblocked. To block influx of extracellular Ca21, cells werewounded in Ca21-free HBSS with 100 µM EGTA tochelate any Ca21 in the extracellular media. To limitthe exposure to EGTA and a Ca21-free environment,imaging was started in HBSS, and then solutions wereswitched to 100 µM EGTA 1 min before wounding.When extracellular Ca21 was chelated, the wave was
still present, but the amplitude, speed, and range wereall reduced (compare Fig. 1, A1–A6 with B1–B6). Arepresentative trace of the [Ca21]i elevations at threedistances from the wound is shown in Fig. 2B. In 100µM EGTA, the peak was reduced from saturating levelsin HBSS to 1.37 6 0.65 (SD) µM (n 5 4; Fig. 2, A vs. B),and the range was reduced to 194 6 112 (SD) µm (n 5 4;Fig. 1B4). The apparent wave speed was reduced by45% to 7.18 6 1.31 (SD) µm/s (n 5 4). However, the rateof deceleration of the wave speed became highly vari-able and was no longer linear. This suggests that thewave speed and the mechanism of deceleration werehighly dependent on an influx of extracellular Ca21.These data demonstrate that the kinetic properties ofthe [Ca21]i wave depend on the concentration of extra-cellular Ca21, but cells continue to elevate [Ca21]i in theabsence of extracellular Ca21.
To confirm that the effect of EGTA was limited tochelating extracellular Ca21 and did not predrain intra-cellular Ca21 stores, we repeated the experiment using
Fig. 1. Wounding produced an intracel-lular free Ca21 concentration ([Ca21]i)wave. Rows are conditions, and col-umns are times. All times are in refer-ence to wounding, and units are min-utes:seconds. Cells were wounded (w)with an 18-gauge needle under variousconditions. A1–A6: Hanks’ balancedsalt solution (HBSS); B1–B6: 100 µMEGTAin Ca21-free HBSS; C1–C6: phos-phate-free HBSS with 100 µM Gd31;D1–D6: HBSS after a 1-h treatmentwith 10 µM cyclopiazonic acid (CPA) inHBSS; E1–E6: HBSS 35 min afterwashing out CPA and allowing cells tobathe in HBSS; F1–F6: HBSS 1 h afteran old wound (ow). Bar, 100 µm. Hue ofcolor calibration bar represents [Ca21]iin µM, whereas intensity of color repre-sents fura 2 intensity at a Ca21-insen-sitive wavelength.
L1244 WOUND-INDUCED CALCIUM WAVES IN ATII CELLS

Gd31 instead of EGTA. Gd31 is an effective and reliableCa21 channel blocker that has been used with successin other cell lines (Ref. 23 and Sammak et al., unpub-lished observations). When cells were wounded inphosphate-free HBSS with 1.3 mM Ca21 and 100 µMGd31, the wave was still present, but the peak eleva-tion, speed, and range were all reduced (compare Fig. 1,A1–A6 with C1–C6). A representative trace of the[Ca21]i elevations at three distances from the wound isshown in Fig. 2C. In 100 µM Gd31, the peak elevationwas reduced from saturating levels in HBSS to 1.42 60.69 (SD) µM (n 5 6; Fig. 2, A vs. C). The range wasreduced to 74.6 6 22.3 (SD) µm (n 5 3; Fig. 1, A5 vs.C5), and the apparent wave speed was reduced by 75%to 3.38 6 0.39 (SD) µm/s (n 5 3). The rate of decelera-tion in wave speed could not be measured because oflimits in the rate that data could be acquired. Inagreement with the EGTAexperiments, the Gd31 experi-ments suggest that, whereas extracellular Ca21 is asignificant source of Ca21 for the [Ca21]i wave, it is notrequired for the phenomenon.
To test if intracellular Ca21 stores were required forthe [Ca21]i wave, we emptied intracellular Ca21 stores
by pretreatment with CPA, an endoplasmic reticulumCa21-adenosinetriphosphatase (SERCA) inhibitor. Byinhibiting the SERCA pump, intracellular stores can nolonger be filled and the Ca21 leaks out into the cyto-plasm, eventually emptying the stores. After a 1-htreatment with CPA, [Ca21]i had returned to levels seenin untreated cells. Treated cultures were then wounded,and [Ca21]i was recorded (Fig. 1, D1–D6). A representa-tive trace of the [Ca21]i elevations at three distancesfrom the wound is shown in Fig. 2D. After CPA treat-ment, the peak elevation in the second row of cells wasreduced from saturating levels in HBSS to 0.35 6 0.06(SD) µM (n 5 3; Fig. 2, A vs. D). Even more dramaticwas the abolishment of wave propagation beyond thesecond row of cells (Fig. 1, A2–A6 vs. Fig. 2, D2–D6). Toensure that CPA did not permanently damage the cells,CPA was subsequently washed out, and the cells wereallowed to refill intracellular Ca21 stores in HBSS for35 min. Cultures were wounded again, and [Ca21]ielevations were observed that were qualitatively simi-lar to untreated cultures (compare Fig. 1, A1–A6 withD1–D6, and compare Fig. 2, A with E). The reversibil-
Fig. 2. Representative traces of the [Ca21]iresponse of cells vs. time. Solid line, [Ca21]iresponse of second row of cells; dashedline, [Ca21]i response in cells that were 100µm from wound edge; stippled line, [Ca21]iresponse of cells that were 250 µm fromwound edge. A: cells were wounded inHBSS, and [Ca21]i in second row of cellsquickly rose to .2.5 µM and came back tobaseline within 2 min. Response propa-gated through monolayer as a wave as canbe seen by delayed peak response at 100and 250 µm. B: cells wounded in Ca21-freeHBSS with 100 µM EGTA had a [Ca21]iresponse that was reduced but was stillpresent. Range of wave was reduced as canbe seen by the lack of elevation at 250 µm.C: cells that were wounded in 100 µM Gd31
had their [Ca21]i response reduced. Rangeof wave was reduced as can be seen by thelack of elevation at 200 µm. D: cells treatedwith 10 µM CPA for 1 h before woundingexhibited almost no elevation in [Ca21]i,and there was no propagation of the smallelevation. E: 35 min after CPA was washedout and cells were placed in HBSS at roomtemperature, [Ca21]i elevations could againbe seen that were qualitatively similar tountreated cultures. F: an old wound wasmade 1 h before second wound. Only tracesof the second row of cells and cells 250 µmaway from the wound are shown becausethe cells at 100 µm were removed by theold wound.
L1245WOUND-INDUCED CALCIUM WAVES IN ATII CELLS
ity of the effects of CPA suggested that CPA was specificto the SERCA and that it was nontoxic. These datasuggested that the release of Ca21 from intracellularstores was necessary for the [Ca21]i wave to occur.Therefore, wound-induced [Ca21]i signaling dependscritically on intracellular stores regardless of the extra-cellular Ca21 concentration.
An extracellular signal was sufficient to cause an[Ca21]i wave. Previously, we have found [Ca21]i wavesin endothelial cells that are totally dependent on extra-cellular communication (27). We examined whetherthis [Ca21]i wave had the same dependencies. To testthis, a confluent monolayer of cells was wounded toprovide a break in the monolayer that was devoid ofcells (Fig. 1F1). One hour after wounding, a secondwound parallel to the old wound was made. We foundthat the new wound produced elevations in [Ca21]i (Fig.1F2). A representative trace of the [Ca21]i elevations attwo distances from the wound is shown in Fig. 2F; the100-µm distance was omitted due to the fact that it wasin the middle of the old wound. Interestingly, cells onthe far side of the old wound also elevated [Ca21]i (Fig.1, F3–F5). The elevation on the far side of the oldwound was similar in amplitude and timing to thatobserved for a confluent monolayer at the same dis-tance from the wound (Fig. 1, A3–A5 vs. F3–F5, andFig. 2, A vs. F), although not all cells responded.Because the [Ca21]i wave was able to jump over a breakin the monolayer, we conclude that some signal for the[Ca21]i can be transmitted without direct cell-cell con-tact.
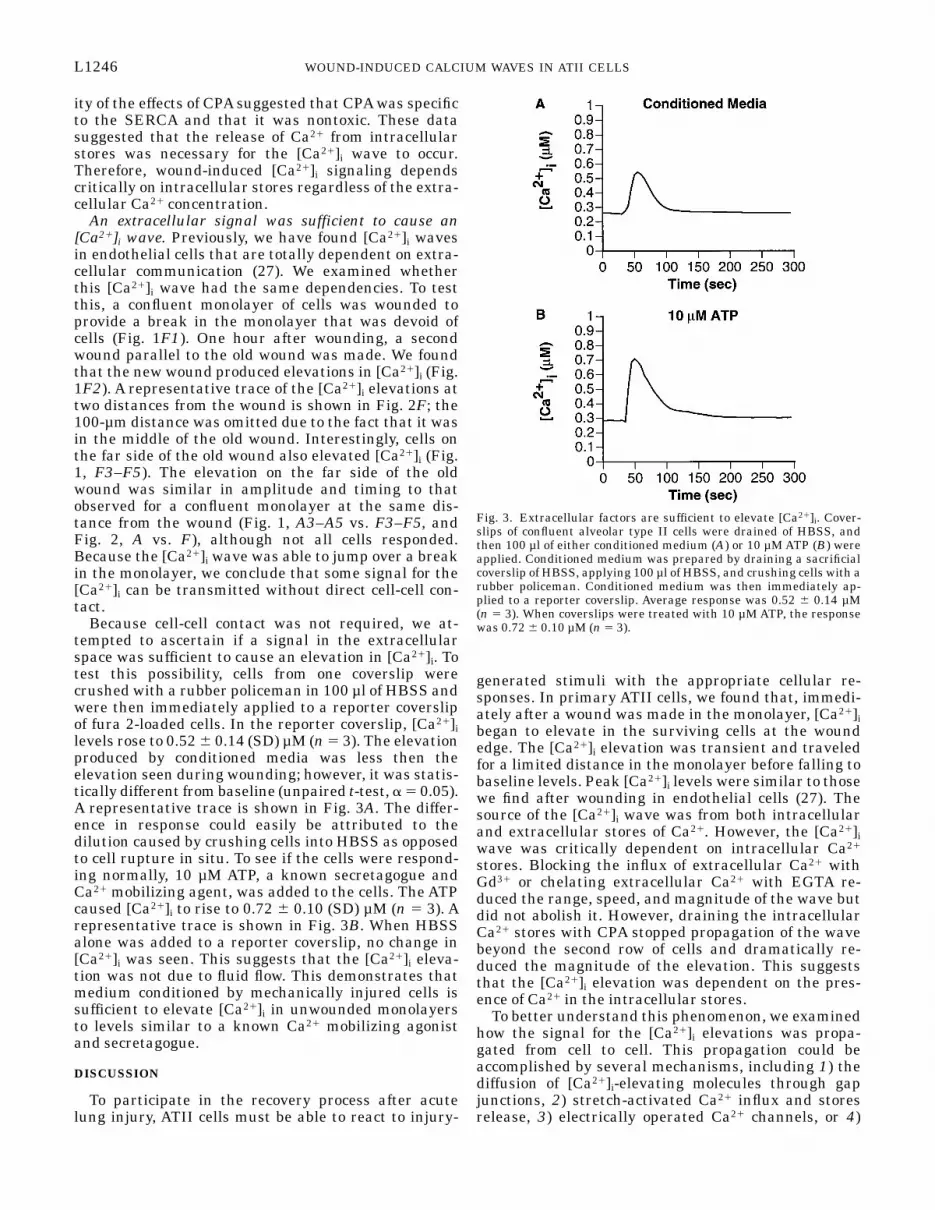
Because cell-cell contact was not required, we at-tempted to ascertain if a signal in the extracellularspace was sufficient to cause an elevation in [Ca21]i. Totest this possibility, cells from one coverslip werecrushed with a rubber policeman in 100 µl of HBSS andwere then immediately applied to a reporter coverslipof fura 2-loaded cells. In the reporter coverslip, [Ca21]ilevels rose to 0.52 6 0.14 (SD) µM (n 5 3). The elevationproduced by conditioned media was less then theelevation seen during wounding; however, it was statis-tically different from baseline (unpaired t-test, a 5 0.05).A representative trace is shown in Fig. 3A. The differ-ence in response could easily be attributed to thedilution caused by crushing cells into HBSS as opposedto cell rupture in situ. To see if the cells were respond-ing normally, 10 µM ATP, a known secretagogue andCa21 mobilizing agent, was added to the cells. The ATPcaused [Ca21]i to rise to 0.72 6 0.10 (SD) µM (n 5 3). Arepresentative trace is shown in Fig. 3B. When HBSSalone was added to a reporter coverslip, no change in[Ca21]i was seen. This suggests that the [Ca21]i eleva-tion was not due to fluid flow. This demonstrates thatmedium conditioned by mechanically injured cells issufficient to elevate [Ca21]i in unwounded monolayersto levels similar to a known Ca21 mobilizing agonistand secretagogue.
DISCUSSION
To participate in the recovery process after acutelung injury, ATII cells must be able to react to injury-
generated stimuli with the appropriate cellular re-sponses. In primary ATII cells, we found that, immedi-ately after a wound was made in the monolayer, [Ca21]ibegan to elevate in the surviving cells at the woundedge. The [Ca21]i elevation was transient and traveledfor a limited distance in the monolayer before falling tobaseline levels. Peak [Ca21]i levels were similar to thosewe find after wounding in endothelial cells (27). Thesource of the [Ca21]i wave was from both intracellularand extracellular stores of Ca21. However, the [Ca21]iwave was critically dependent on intracellular Ca21
stores. Blocking the influx of extracellular Ca21 withGd31 or chelating extracellular Ca21 with EGTA re-duced the range, speed, and magnitude of the wave butdid not abolish it. However, draining the intracellularCa21 stores with CPA stopped propagation of the wavebeyond the second row of cells and dramatically re-duced the magnitude of the elevation. This suggeststhat the [Ca21]i elevation was dependent on the pres-ence of Ca21 in the intracellular stores.
To better understand this phenomenon, we examinedhow the signal for the [Ca21]i elevations was propa-gated from cell to cell. This propagation could beaccomplished by several mechanisms, including 1) thediffusion of [Ca21]i-elevating molecules through gapjunctions, 2) stretch-activated Ca21 influx and storesrelease, 3) electrically operated Ca21 channels, or 4)
Fig. 3. Extracellular factors are sufficient to elevate [Ca21]i. Cover-slips of confluent alveolar type II cells were drained of HBSS, andthen 100 µl of either conditioned medium (A) or 10 µM ATP (B) wereapplied. Conditioned medium was prepared by draining a sacrificialcoverslip of HBSS, applying 100 µl of HBSS, and crushing cells with arubber policeman. Conditioned medium was then immediately ap-plied to a reporter coverslip. Average response was 0.52 6 0.14 µM(n 5 3). When coverslips were treated with 10 µM ATP, the responsewas 0.72 6 0.10 µM (n 5 3).
L1246 WOUND-INDUCED CALCIUM WAVES IN ATII CELLS

diffusion of [Ca21]i-elevating molecules through theextracellular space.
In airway epithelial cells, it has been shown that[Ca21]i waves generated by mechanical stimulation arepropagated by diffusion of D-myo-inositol 1,4,5-trisphos-phate through gap junctions (29). If diffusion of anysignals through gap junctions was responsible for the[Ca21]i elevation, we would have expected the [Ca21]iwave to stop at a break in the monolayer. However, inour experiments the [Ca21]i elevation did cross thebreak in the monolayer as can be seen in Fig. 1, F1–F6.This experiment did not test for the presence of gap-junctional contributions, but it did demonstrate thatthey were not necessary for the [Ca21]i wave. Further-more, the wave had a similar speed and amplitude aftercrossing the barrier as when it traveled through aconfluent monolayer (compare Fig. 1, A5 with F5). Thisexperiment suggested that if gap junction transmissionwas present, then it was not critical to the timing andamplitude of the [Ca21]i wave.
The second possibility for propagation of the [Ca21]iwave was through stretch or electrical coupling. Thismodel has been observed in both airway epithelial cells(16) and endothelial cells (23). We performed our experi-ments in a manner that would minimize the contribu-tions of these pathways. During our experiments, weprevented flexing of the substrate by controlling theamount of pressure that was applied to the coverslip.We also tested to see if movement of the needle throughan area devoid of cells right next to reporter cells couldcause enough fluid flow to elevate [Ca21]i, which it didnot. It was still possible that the passage of the needlethrough the monolayer directly stretched the cells inthe plane of the monolayer during wounding. However,if this lateral stretch were the cause of the [Ca21]i wave,we would have expected the wave to stop at a break inthe monolayer because there would be no mechanismfor transfer of the stretch stimulus across the break.Therefore, stretch-activated channels were insufficientto account for ability of the [Ca21]i wave to jump overbreaks in the monolayer. A similar argument can bemade for the third possibility, electrically coupled prop-agation. Electrical propagation would also have re-quired direct cell-cell contacts for propagation. Theseexperiments did not directly test for the presence ofstretch or electrical coupling. It is quite likely that oneor both of these mechanisms was present, but neithermechanism alone could explain the jumping of thewave over the old wound.
The fourth possibility for propagation of the wavewas the release of [Ca21 ]i-elevating molecules from thewound into the extracellular fluid. This mechanism hasbeen shown to be responsible for [Ca21]i waves inendothelial (27) and some epithelial (8) cells. If [Ca21]i-elevating molecules were released into the extracellu-lar fluid, we would expect the [Ca21]i wave to jump overa break in the monolayer. Not only did the wave jumpover the break in the monolayer, but it maintained asimilar magnitude and speed compared with an intactmonolayer. This is in agreement with the release of[Ca21]i-elevating molecules from the wound into the
extracellular fluid. Therefore, the release of [Ca21]i-elevating molecules from the wound into the extracellu-lar fluid is most consistent with our data. It is possiblethat the other signaling mechanisms have a role inmobilizing Ca21, but under our conditions they areinsufficient to explain our results. If release of [Ca21]i-elevating molecules from the wound into the extracellu-lar fluid were responsible for the wave, we would expectthe wave to exhibit kinetics consistent with diffusion.For simple one-dimensional diffusion, the distance thatthe wave front travels should be proportional to thesquare root of time (4), resulting in a time-dependentdecrease in velocity. When performed in the presence ofextracellular Ca21, our experimental data fit this gen-eral model. In the absence of extracellular Ca21, thewave speed became highly variable. One possible inter-pretation is that wave speed is dependent on the influxof Ca21 as well as on signaling molecule concentrationsuch that without an influx of Ca21 the extracellularsignaling molecules cannot elevate [Ca21]i in a reproduc-ible way (27).
The release of [Ca21]i-elevating molecules from thewound into the extracellular fluid would also explainwhy crushing cells and adding them back to an un-wounded reporter monolayer elevated [Ca21]i. Whencrushed cells were added back to reporter monolayers,the [Ca21]i elevation peaked at 0.52 6 0.14 µM. Thiselevation is modest compared with the elevation duringwounding. However, this value should not be comparedwith wounding because the concentration of [Ca21]i-elevating molecules that was applied was indetermin-able. Last, it should be noted that the crushed cellswere capable of producing an elevation in [Ca21]i thatwas comparable to 10 µM ATP, which is known to causesecretion of surfactant at these concentrations (5).
There are a number of functional implications for therole of Ca21 in signaling in the alveolus. The range ofpropagation of the Ca21 wave is comparable to alveolarsize (diameter of ,100 µm) and thus is appropriate forintercellular communication. Ca21 elevations have beendemonstrated in ATII cells in a number of modelsystems of surfactant production with secretagogues(14, 31, 32, 37). It is plausible that elevation of Ca21
through receptor-mediated (35) or other mechanisms isone of the first intracellular steps to increased surfac-tant secretion. Although the role of [Ca21]i in ATII cellproliferation and differentiation is currently unknown,changes in intracellular Ca21 are associated with in-creased proliferation in other cell types (1, 3, 19, 21, 36,38) and differentiation in other cell types (13, 15). Thisplaces the [Ca21]i wave at the right place and possiblythe right time to regulate both surfactant secretion andproliferation.
In summary, the present study demonstrates thatwounding confluent monolayers of ATII cells producedan [Ca21]i wave. This [Ca21]i wave involved the influx ofCa21 from the extracellular medium but was criticallydependent on the release of Ca21 from internal stores.The signal for the wave was partly carried in theextracellular medium, but other pathways might beinvolved in the [Ca21]i response. The role for the [Ca21]i
L1247WOUND-INDUCED CALCIUM WAVES IN ATII CELLS

wave has yet to be determined, but it is poised at aposition both temporally and spatially to coordinate thehealing response to acute lung injury.
This work as supported by National Science Foundation GrantMCB-9596838 and American Lung Association Grant RG-175-N (toP. J. Sammak) and The North Trauma Institute and NationalInstitutes of Health Grant BRSGS07RR06007–01 (to G. J. Beilman).
Address for reprint requests: P. J. Sammak, 3–249 Millard Hall,435 Delaware St. SE, Dept. of Pharmacology, Univ. of Minnesota,Minneapolis, MN 55455.
Received 27 May 1997; accepted in final form 5 September 1997.
REFERENCES
1. Berridge, M. J. Calcium signaling and cell proliferation. Bioes-says 17: 491–500, 1995.
2. Brundage, R. A., K. E. Fogarty, R. A. Tuft, and F. S. Fay.Chemotaxis of newt eosinophils: calcium regulation of chemotac-tic response. Am. J. Physiol. 265 (Cell Physiol. 34): C1527–C1543, 1993.
3. Byron, K. L., and M. L. Villereal. Mitogen-induced [Ca21]ichanges in individual human fibroblasts. Image analysis revealsasynchronous responses which are characteristic for differentmitogens. J. Biol. Chem. 264: 18234–18239, 1989.
4. Carrier, G. F., and C. E. Pearson. Partial Differential Equa-tions: Theory and Technique. New York: Academic, 1976.
5. Chander, A., N. Sen, A.-M. Wu, and A. R. Spitzer. Proteinkinase C in ATP regulation of lung surfactant secretion in type IIcells. Am. J. Physiol. 268 (Lung Cell. Mol. Physiol. 12): L108–L116, 1995.
6. Churchill, G. C., M. M. Atkinson, and C. F. Louis. Mechanicalstimulation initiates cell-to-cell calcium signaling in ovine lensepithelial cells. J. Cell Sci. 109: 355–365, 1996.
7. Dobbs, L. G., R. Gonzalez, and M. C. Williams. An improvedmethod for isolating type II cells in high yield and purity. Am.Rev. Respir. Dis. 134: 141–145, 1986.
8. Enomoto, K., K. Furuya, S. Yamagishi, T. Oka, and T.Maeno. The increase in the intracellular Ca21 concentrationinduced by mechanical stimulation is propagated via release ofpyrophosphorylated nucleotides in mammary epithelial cells.Pflugers Arch. 427: 533–542, 1994.
9. Finkelstein, J. N. Physiologic and toxicologic responses ofalveolar type II cells. Toxicology 60: 41–52, 1990.
10. Grynkiewicz, G., M. Poenie, and R. Y. Tsien. A new genera-tion of Ca21 indicators with greatly improved fluorescence prop-erties. J. Biol. Chem. 260: 3440–3450, 1985.
11. Hahn, K., R. DeBiasio, and D. L. Taylor. Patterns of elevatedfree calcium and calmodulin activation in living cells. Nature359: 736–738, 1992.
12. Herman, I. M. Molecular mechanisms regulating the vascularendothelial cell motile response to injury. J. Cardiovasc. Pharma-col. 22: S25–S36, 1993.
13. Jetten, A. M. Multistep process of squamous differentiation intracheobronchial epithelial cells in vitro: analogy with epidermaldifferentiation. Environ. Health Perspect. 80: 149–160, 1989.
14. Joyce, B. M., J. B. Rubins, M. P. Panchenko, J. Bernardo,M. P. Steele, L. Kolm, E. R. Simons, and B. F. Dickey.Mechanisms of mastoparan-stimulated surfactant secretion fromisolated pulmonary alveolar type 2 cells. J. Biol. Chem. 266:6859–6865, 1991.
15. Kasturi, L., N. Sizemore, R. L. Eckert, K. Martin, and E. A.Rorke. Calcium modulates cornified envelope formation, involu-crin content, and transglutaminase activity in cultured humanectocervical epithelial cells. Exp. Cell Res. 205: 84–90, 1993.
16. Kim, Y.-K., E. R. Dirksen, and M. J. Sanderson. Stretch-activated channels in airway epithelial cells. Am. J. Physiol. 265(Cell Physiol. 34): C1306–C1318, 1993.
17. Lansley, A. B., M. J. Sanderson, and E. R. Dirksen. Controlof the beat cycle of respiratory tract cilia by Ca21 and cAMP.Am. J. Physiol. 263 (Lung Cell. Mol. Physiol. 7): L232–L242,1992.
18. Marinelli, W. A., G. J. Walker Smith, and D. H. Ingbar.Inflammation and repair of the lung. In: Pulmonary and CriticalCare Medicine, edited by I. Bone. New York: Mosby Yearbook, 1996.
19. Marks, P. W., and F. R. Maxfield. Local and global changes incytosolic free calcium in neutrophils during chemotaxis andphagocytosis. Cell Calcium 11: 181–90, 1990.
20. Mascardo, R. N., and G. Eilon. The cysteine protease inhibitor,E-64, stimulates the polarization and locomotor responses ofendothelial cells to wounding. J. Pharmacol. Exp. Ther. 244:361–367, 1988.
21. Means, A. R. Calcium, calmodulin and cell cycle regulation.FEBS Lett. 347: 1–4, 1994.
22. Michikawa, T., A. Miyawaki, T. Furuichi, and K. Miko-shiba. Inositol 1,4,5-trisphosphate receptors and calcium signal-ing. Crit. Rev. Neurobiol. 10: 39–55, 1996.
23. Naruse, K., and M. Sokabe. Involvement of stretch-activatedion channels in Ca21 mobilization to mechanical stretch inendothelial cells. Am. J. Physiol. 264 (Cell Physiol. 33): C1037–C1044, 1993.
24. Novick, R. J., K. E. Gehman, I. S. Ali, and J. Lee. Lungpreservation: the importance of endothelial and alveolar type IIcell integrity. Ann. Thorac. Surg. 62: 302–314, 1996.
25. Palmer, R. K., D. I. Yule, D. S. Shewach, J. A. Willimas, andS. K. Fisher. Paracrine mediation of calcium signaling in humanSK-N-MCIXC neuroepithelialioma cells. Am. J. Physiol. 271(Cell Physiol. 40): C43–C53, 1996.
26. Perkett, E. A. Role of growth factors in lung repair and diseases.Curr. Opin. Pediatr. 7: 242–249, 1995.
27. Sammak, P. J., L. E. Hinman, P. O. T. Tran, M. D. Sjaastad,and T. E. Machen. How do injured cells communicate with thesurviving monolayer? J. Cell Sci. 116: 465–475, 1997.
29. Sanderson, M. J. Intercellular calcium waves mediated byinositol trisphosphate. Ciba Found. Symp. 188: 175–194, 1995.
30. Sanderson, M. J., A. C. Charles, S. Boitano, and E. R.Dirksen. Mechanisms and function of intercellular calciumsignaling. Mol. Cell. Endocrinol. 98: 173–187, 1994.
31. Sano, K., D. R. Voelker, and R. J. Mason. Effect of secreta-gogues on cytoplasmic free calcium in alveolar type II epithelialcells. Am. J. Physiol. 253 (Cell Physiol. 22): C679–C686, 1987.
32. Sen, N., M. M. Grunstein, and A. Chander. Stimulation oflung surfactant secretion by endothelin-1 from rat alveolar typeII cells. Am. J. Physiol. 266 (Lung Cell. Mol. Physiol. 10):L255–L262, 1994.
33. Shen, J., F. W. Luscinskas, A. Connolly, C. F. Dewey, Jr., andM. A. Gimbrone, Jr. Fluid shear stress modulates cytosolic freecalcium in vascular endothelial cells. Am. J. Physiol. 262 (CellPhysiol. 31): C384–C390, 1992.
34. Steinhardt, R. A., G. Bi, and J. M. Alderton. Cell membraneresealing by a vesicular mechanism similar to neurotransmitterrelease. Science 263: 390–393, 1994.
35. Strayer, D. S., R. Pinder, and A. Chander. Receptor-mediatedregulation of pulmonary surfactant secretion. Exp. Cell Res. 226:90–97, 1996.
36. Takuwa, N., W. Zhou, and Y. Takuwa. Calcium, calmodulinand cell cycle progression. Cell. Signal. 7: 93–104, 1995.
37. Voyno-Yasentskaya, T. A., L. G. Dobbs, and M. C. Williams.Regulation of ATP-dependent surfactant secretion and activationof second-messenger systems in alveolar type II cells. Am. J.Physiol. Suppl. 261: 105–109, 1991.
38. Wirtz, H. R., and L. G. Dobbs. Calcium mobilization andexocytosis after one mechanical stretch of lung epithelial cells.Science 250: 1266–1269, 1990.
L1248 WOUND-INDUCED CALCIUM WAVES IN ATII CELLS





























