Embed Size (px)
Citation preview

Wi
ID
a
ARRAA
KDEBCSDE
C
0d
Progress in Polymer Science 36 (2011) 1152– 1183
Contents lists available at ScienceDirect
Progress in Polymer Science
j ourna l ho me p ag e: www.elsev ier .com/ locate /ppolysc i
hen emulsification meets self-assembly: The role of emulsificationn directing block copolymer assembly
an Wyman, Gabriel Njikang, Guojun Liu ∗
epartment of Chemistry, Queen’s University, 90 Bader Lane, Kingston, Ontario, Canada K7L 3N6
r t i c l e i n f o
rticle history:eceived 31 January 2011eceived in revised form 26 April 2011ccepted 27 April 2011vailable online 26 May 2011
eywords:irected assemblymulsification
a b s t r a c t
Emulsification is used to generate spherical particles or droplets of immiscible liquids, whileblock copolymer self-assembly yields a wide variety of nanostructures. The combinationof these two methodologies can yield a variety of structures that would not be otherwiseobserved. The emulsification/solvent evaporation process provides a powerful means todirect block copolymer assembly. Various factors arising from the emulsification can directthe block copolymer assembly, such as confinement effects, interfacial tension, as well asother conditions. In this review, various emulsification techniques are discussed, such asoil-in-water emulsions, double emulsions, as well as the use of microfluidic devices. While
lock copolymersonfinement effectself-assemblyouble emulsions
emulsification-induced self-assembly may be used to control internal morphologies as wellas overall shapes of particles, it also lends a convenient method for controlling surfacestructures. Examples of exotic structures that may be obtained through the use of thesetechniques will be described. Also, ways in which morphologies may be controlled using
mulsification/solvent evaporation these methods will be discussed.© 2011 Elsevier Ltd. All rights reserved.
ontents
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1153
2. Block copolymer self-assembly in confined volumes . . . . . . . . . . . . . .2.1. Computer simulation results . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
2.2. Block copolymer particles derived from emulsion droplets
Abbreviations: AFM, atomic force microscopy; CTAB, cetyl trimethylammoniumDiameter/Periodicity); DN, decahydronaphthalene; FITC-Dextran, fluorescein ispoly(D,L-lactic acid); O/W, oil-in-water emulsion; PAA, poly(acrylic acid); PBacid); PCEA, poly-(2-cinnamoyloxyethyl acrylate); PCEMA, poly(2-cinnamoymethacrylate)-block-poly(glyceryl methacrylate); PEG, poly(ethylene glycol)poly(�-caprolactone); PFOB, perfluorooctyl bromide; PGMA, poly(glyceryl
block-poly(cinnamoyloxyethyl methacrylate)-block-poly(tert-butyl acrylate);
poly(isoprene)-block-poly(2-cinnamoyloxyethyl methacrylate); PI-b-PCEMA-b-poly(tert-butyl)acrylate; PI-b-PtBA, polyisoprene-block-poly(tert-butyl acrylate)oxide); PLGA, poly(lactide-co-glycolide); PLGA-b-PEO, poly(lactide-co-glycoliblock-poly(propylene oxide)-block-poly(ethylene oxide); PMMA, poly(methyl
polystyrene-block-polybutadiene; PSGMA, succinated poly(glyceryl methacrylateblock-poly(2-cinnamoylethyl methacrylate); PVA, poly(vinyl alcohol); r, moleculaRES, reticulo-endothelial system; SDS, sodium dodecyl sulfate; SEM, Scanning electransmission electron microscopy; THF, tet; W/O, water-in-oil emulsion; (W/O)/W∗ Corresponding author. Tel.: +1 613 533 6996; fax: +1 613 533 6669.
E-mail addresses: [email protected], [email protected] (G. Liu
079-6700/$ – see front matter © 2011 Elsevier Ltd. All rights reserved.oi:10.1016/j.progpolymsci.2011.04.005
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1155. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1155
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1156
bromide; DCM, dichloromethane; D/L0, confinement dimension (Particleothiocyanate-dextran; MPEG-b-PLA, methoxypolyethylene glycol-block-, poly(butadiene); PBA-b-PAA, poly(n-butyl acrylate)-block-poly(acrylicloxyethyl methacrylate); PCEMA-b-PGMA, poly(2-cinnamoyloxyethyl
; PEO, poly(ethylene oxide); PEO-b-PCL, poly(ethylene oxide)-block-methacrylate); PGMA-b-PCEMA-b-PtBA, poly(glyceryl methacrylate)-PI-b-PAA, polyisoprene-block-poly(tert-butyl acrylate); PI-b-PCEMA,
PtBA, polyisoprene-block-poly(cinnamoyloxyethyl methacrylate)-block-; PLA, poly(lactic acid); PLA-b-PEO, poly(lactic acid)-block-poly(ethylenede)-block-poly(ethylene oxide); Pluronic F108, poly(ethylene oxide)-methacrylate); PPO, poly(propylene oxide); PS, polystyrene; PS-b-PB,); PtBA, poly(tert-butyl acrylate); PtBA-b-PCEMA, poly(tert-butyl acrylate-r weight of a homopolymer with respect to that of its corresponding block;tron microscopy; STEM, Scanning transmission electron microscopy; TEM,, water-in-oil-in water emulsion; XPS, X-ray photoelectron spectroscopy.
).

I. Wyman et al. / Progress in Polymer Science 36 (2011) 1152– 1183 1153
2.3. Seeing morphologies of block copolymers confined within microspheres . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11572.4. Segregation behavior of block copolymer/homopolymer blends . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1158
2.4.1. Influence of copolymer/homopolymer blend composition. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11582.4.2. Influence of homopolymer molecular weight relative to that of the copolymer . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11612.4.3. Influence of D/L0 upon the morphologies of copolymer/homopolymer blends . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1162
3. Block copolymer vesicles and capsules prepared by emulsion techniques. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11633.1. Capsules from block copolymer assembly and emulsification . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11643.2. Controlled vesicle formation using microfluidic devices . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1165
4. Block copolymer self-assembly in 2D spaces . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11674.1. Assembly at the 2D interface . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11674.2. Influence of surfactant upon surface morphology . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1170
5. Emulsion as a tool to direct the formation of exotic architectures . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11725.1. Influence of interfacial tension on the formation of budding vesicles . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11725.2. Molecular containers and porous materials from block copolymer emulsion spheres . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1175
6. Perspectives and outlook . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11786.1. Microphase segregation within solid emulsion particles . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11786.2. Block copolymer vesicles through emulsification . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11796.3. Block copolymer assembly at 2D surfaces . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11796.4. Exotic and useful structures . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11796.5. Outlook . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1179Acknowledgements . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1179
. . . . . . . .
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .1. Introduction
Block copolymers consist of two or more chemically dis-tinct polymer blocks [1]. The simplest block copolymer isa diblock copolymer, AnBm, consisting of n consecutive Aunits and m consecutive B units. Scheme 1 shows the struc-tures of a diblock copolymer polyisoprene-block-poly(2-cinnamoyloxyethyl methacrylate) (PI-b-PCEMA) and atriblock copolymer poly(glyceryl methacrylate)-block-poly(2-cinnamoyloxyethyl methacrylate)-block-poly(tert-butyl acrylate) (PGMA-b-PCEMA-b-PtBA) [2,3].
In the absence of strong intermolecular interactions,
such as hydrogen bonding and electrostatic attraction,most polymers are incompatible above some criticalmolecular weights. In bulk or the solid state, thedifferent blocks of a block copolymer segregate orScheme 1. Block copolymers PI-b-PCEMA (top)
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1179
undergo self-assembly with the constituent blocks form-ing regularly-shaped and uniformly-sized domains thatare periodically spaced. For coil–coil diblock copolymersAnBm, the shape of the segregated domains of the minorityblock is governed by its volume fraction, �, and by blockincompatibility. Fig. 1 shows the equilibrium morpholo-gies documented for coil–coil diblock copolymers [4–6].At a volume fraction of ∼20%, the minority block forms abody-centered cubic spherical phase in the matrix of themajority block. It changes to hexagonally packed cylin-ders at a volume fraction of ∼30%. Alternating lamellaeare formed at approximately equal volume fractions for
the two blocks. At a volume fraction of ∼38%, the minor-ity block forms gyroid or perforated layers at moderateand high incompatibility, respectively. These interestingmorphological transitions have been established experi-and PGMA-b-PCEMA-b-PtBA (bottom).

1154 I. Wyman et al. / Progress in Polymer Science 36 (2011) 1152– 1183
F includea erved aR Chemic
mteottt
abvbluoaomsdsb
scbamsArthsfmmb[
nbpf
vcad
ig. 1. Bulk segregation patterns of diblock copolymers [4]. These phases
lternating lamellae. Progressing from left to right, these phases are obseprinted with permission from Reference [4]. Copyright 1995 American
entally [7–11] and can be accounted for by statisticalhermodynamic theories [12–20]. Furthermore, the small-st dimension of a segregated domain, e.g., the diameterf a cylinder, is proportional to the two-thirds power ofhe molar mass of the minority block, and can typically beuned from ∼5 to ∼50 nm by changing the molar mass ofhe block [1].
In analogy to their bulk behavior, diblock copolymerslso self-assemble in block-selective solvents, which solu-ilize one but not the other block, forming micelles witharious shapes [21]. If the soluble block is long, the insolu-le block aggregates to produce spherical micelles. As the
ength of the soluble block is decreased relative to the insol-ble block, cylindrical micelles or vesicles, and micelles ofther shapes can be formed, as first demonstrated by Zhangnd Eisenberg [21,22]. From a single diblock copolymer,ne can also effect morphological transitions of copoly-er micelles by preparing micelles in different selective
olvents. Normally, the transition from spherical to cylin-rical and vesicular micelles is accomplished by usingolvents that are increasingly poor for the core or insolublelock.
ABC triblock copolymers, AnBmCl, can also undergoelf-assembly in bulk or block-selective solvents. Triblockopolymers have many more block segregation patterns inulk than diblock copolymers, and some of the patternsre very intricate and visually striking [1]. The shapes oficelles formed by triblock copolymers in block-selective
olvents are also greatly diversified. Cylindrical micelles ofBC triblock copolymers alone have included variationsanging from straight cylinders to segmented cylinders,wisted cylinders, single helices, double helices, and tripleelices, etc. Fig. 2 shows a transmission electron micro-copic (TEM) image and a TEM tomography image of helicesormed from the self-assembly of an ABC triblock copoly-
er in a good solvent for A, a poor solvent for B, and aarginal solvent for C. Such a structure resembles the dou-
le helix structure seen in DNA, and is highly sophisticated23].
Due to the synthetic challenges involved, there haveot been many studies on the self-assembly of ABCD tetra-lock copolymers [24–28]. The number of self-assembledatterns in either bulk or selective solvents should increaseurther for tetra- and penta-block copolymers.
Block copolymer self-assembly is robust, and can yield
arious nanostructured materials for a wide range of appli-ations. A variety of these applications are highlighted inrecent review by Kim et al. [29]. The shape diversity ofiscrete nanoobjects produced in block-selective solvents,
(from left to right) spherical, cylindrical, gyroid, perforated lamellae, ands the block distributions of the copolymers are increasingly symmetric.al Society.
for example, should facilitate their applications in areassuch as nanofabrication [3,30–37], lithography [38–40],cell cultures [41], and drug delivery [42–44]. Despite therobustness of the self-assembly strategy, there are certainlimitations. For example, the cylindrical domains formedfrom the minority block of a diblock copolymer in bulkare packed with hexagonal ordering within grains of thesize of micrometers. From grain to grain the orientation ischanged. In the case of block copolymer self-assembly inblock-selective solvents, the smallest dimension, e.g., thecross-sectional diameter of a strand in the double helixshown in Fig. 2b, of a self-assembled structure is typicallybetween several and tens of nanometers. While possible[22,45,46], it is not straightforward to produce compositestructures, e.g., spheres with composite internal structures,from solution self-assembly of block copolymers.
A wide variety of block copolymer architectures can alsobe prepared through directed assembly [47]. In this review,directed assembly refers to block copolymer self-assemblyunder external constraints, control, or influence. These con-straints can be a specific set of restricting conditions, e.g.,confined volumes or 2D or 1D spaces, that are used for theself-assembly process to take place. External influence canbe exerted by using external fields [48,49], including elec-tric or magnetic fields. These influences can even includea solvent evaporation front [50], or a tailored substrate[51]. External control can also be exerted by adding foreignreagents into a system. For example, Pochan and Wooleyadded multiamines into their solvents to introduce inter-actions with the coronal poly(acrylic acid) chains of themicelles [52,53]. Using this strategy, they have been ableto produce interesting and exotic structures.
While a number of reviews have described varioustopics of emulsification [54–59], block copolymer self-assembly [1,6,21,60–65], and directed assembly [47,66],this review will focus on the intersection between thesetopics. More specifically, we will describe how emulsifica-tion can be used to direct block copolymer assembly, par-ticularly through the emulsification/evaporation approach.Emulsification provides a powerful platform from whichone can direct block copolymer assembly. This is especiallytrue if the organic phase is subsequently evaporated, thusforcing the block copolymer to collapse either within anemulsion droplet, or along its surface. Significant progresshas been made in recent years towards refining this tech-
nique. While the emulsion droplets serve as templates todirect the assembly, in some cases researchers have com-bined this with other external stimuli to achieve evenhigher degrees of control. Through these studies, a wide
I. Wyman et al. / Progress in Polymer Science 36 (2011) 1152– 1183 1155
of PBM
Fig. 2. TEM (left) and TEM tomography (right) images of double helicesGmbH & Co. KGaA. Reproduced with permission.variety of exciting, and potentially useful, block copolymerassemblies have been prepared. This review will attemptto highlight the developments that have been made in thisarea. In Section 2, we will discuss the creation of confinedvolumes for block copolymer assembly. Vesicle formationthrough block copolymer assembly along the interfaces ofemulsion droplets will be described in Section 3. Blockcopolymer assembly at the oil/water interface on spheri-cal 2D surfaces, and how this may provide surface controlwill be discussed in Section 4. The preparation of exoticand potentially useful structures through emulsificationwill be described in Section 5. We will summarize ourconclusions and present our perspectives on this topic inSection 6.
2. Block copolymer self-assembly in confinedvolumes
Block copolymer confinement can be achieved throughvarious routes and to varying degrees. The most attentionhas focussed on 1D block copolymer confinement in thinfilms, which has been highlighted in numerous reviews[67–69]. A recent review has also described progress intheir 2D confinement within cylinders [70]. Stewart-Sloanand Thomas recently reviewed experimental and theoreti-cal aspects of 1D, 2D, and 3D block copolymer confinement[71]. The 3D confinement of block copolymers has beenachieved using aerosol droplets, initially by Thomas et al.[72], and more recently by Zhang et al. [73,74]. 3D blockcopolymer confinement has also been accomplished using3D templates by Manners and coworkers [75,76], whileYabu and coworkers have confined block copolymers usinga novel solvent evaporation method [77–82]. The abovemethods have yielded diverse morphologies that oftencould not be achieved in bulk. Another route towards 3Dconfinement is through emulsification.
Emulsification is the process of breaking up a contin-uous organic (or aqueous) phase and dispersing, with the
aid of a surfactant, the resultant oil (or water) droplets inan aqueous (or oil) medium [83]. If the dispersed dropletscontain a block copolymer, solvent evaporation from thedroplets causes them to shrink, and causes an increaseA250-b-PCEMA160-b-PtBA160 [23]. Ref. [23]. Copyright Wiley-VCH Verlag
of copolymer concentration. This could eventually lead tosolidification of the copolymer and segregation of the dif-ferent blocks within the confined volumes.
If the diameter (D) of the confining sphere in which ablock copolymer resides is small (e.g., if D is comparableto, or smaller than, the periodicity (L0) of regular domainsin a block copolymer), the final block-segregated structureformed by the copolymer will differ from that found inbulk or the solid state, where D can be viewed as infinitelylarge in the case of a bulk solid. Consequently, the self-assembly of block copolymers in confined volumes can leadto novel and interesting structures. Even if D is consider-ably larger than L0, one can still obtain novel and complexblock copolymer structures by the combined use of emul-sification and block copolymer self-assembly [84].
2.1. Computer simulation results
Computer simulation methods have been used to studythe effect of spherical and cylindrical confinements onthe assembly of block copolymers. Pan and coworkersobserved concentric structures when they used a MonteCarlo simulation to study the self-assembly of symmet-ric diblock copolymers under spherical and cylindricalconfinement [85], with these morphologies arising whenboundary interactions were favored by one block morethan by the other. It was also predicted that different phasesegregated structures could be designed by simply adjust-ing the boundary shapes and boundary-block copolymerinteractions.
Fraaije and Sevink used a self-consistent-field modelto study the directed assembly of diblock copolymers innanodroplets [86]. By varying the copolymer block ratiosunder a fixed drop radius, a series of unique structureswere found. Microphase separation and morphologies ofasymmetric and symmetric diblock copolymers confinedinside nanospheres with various sizes and interfacial ener-gies have been studied by Feng et al. [87] using dissipative
particle dynamics. They noted that the morphologies ofthe copolymers within the nanospheres are strongly influ-enced by both the sizes and the surface properties of thenanospheres. Recent studies using annealing Monte Carlo
1156 I. Wyman et al. / Progress in Polymer Science 36 (2011) 1152– 1183
Fig. 3. Cross-sectional view of self-assembled morphologies predicted by Monte Carlo simulation for symmetrical diblock copolymers under sphericalconfinement at various D/L0 and ̨ values [88]. At ̨ = 1, the surface preference for the A block (shown in red) is the strongest, while at ̨ = 0, no preferentialinteraction between the surface and a particular block is observed. The structures below and above the dotted line correspond to ̨ values on the scalesa trate thet erential
l ith per
scscfisaacRtcbtficn
sbb(dsw
t the bottom and top of the diagram, respectively. This helps to demonshrough relatively small changes of the degree of confinement and prefegend, the reader is referred to the web version of the article.) Reprinted w
imulations on the self-assembly of symmetric diblockopolymers in spherical nanopores [88], and real-spaceelf-consistent field calculations on the self-assembly ofylinder-forming diblock copolymers under spherical con-nements [89] have also revealed a rich variety of noveltructures that are not possible in the bulk state. The resultslso show that self-assembly is largely governed by inter-ctions between the confinement surface and the polymerhains, and the dimension of the confinement space (Fig. 3).ecently Li et al. [90] used real-space self-consistent fieldheory calculations to model spherically confined blockopolymers with fixed degrees of confinement but varyinglock volume ratios and Flory-Huggins interaction parame-ers. Alternatively, they also kept the latter two parametersxed to model spherically confined cylinder-forming blockopolymers under various degrees of confinement, withovel structures being predicted in both scenarios [90].
Many of the above-mentioned simulation studies havehown that major parameters controlling the assembly oflock copolymers in confined geometries are whether onelock interacts preferentially with the boundary surface
˛) other another block, and the copolymer confinementimension (D/L0). This can be highlighted by Fig. 3, whichhows the diversity of structures predicted by Yu et al. [88]hen these variables are altered among a series of symmet-dramatic range of block copolymer morphologies that may be acquiredinteractions. (For interpretation of the references to color in this figuremission from Reference [88]. Copyright 2007 American Chemical Society.
ric diblock copolymers. In the bulk phase, the copolymerwould form a lamellar structure, while a vast array ofmorphologies may be obtained under the influences of con-finement as well as preferential surface interactions.
2.2. Block copolymer particles derived from emulsiondroplets
The use of the emulsification/solvent evaporation tech-nique to prepare copolymer microspheres began a fewdecades ago, with the use of poly(lactic acid) (PLA)-basedcopolymers to prepare biodegradable microcapsules forthe controlled release of drugs. Ogawa et al. [91] dis-solved leuprolide acetate in a mixture of water and gelatineto obtain the aqueous phase. The oil phase, which con-sisted of a solution of poly(lactic acid)-co-poly(glycolicacid) (PLGA) in dichloromethane (DCM), was slowly addedto the aqueous phase under vigorous stirring to generatewater-in-oil (W/O) emulsion droplets. This W/O emulsionwas subsequently poured into a stirred aqueous solutioncontaining poly(vinyl alcohol) as surfactant to produce
water-in-oil-in-water ((W/O)/W) emulsions. The DCM waslater evaporated from the emulsion, leading to collapseof the copolymer and capsule formation. A general sum-mary of this procedure is shown in Scheme 2. This double
I. Wyman et al. / Progress in Polymer Science 36 (2011) 1152– 1183 1157
mulsion, a water-in-oil-in-water emulsion, and subsequent formation of vesicles
Scheme 3. Preparation of block copolymer microspheres (copoly-mer = PtBA-b-PCEMA or PI-b-PtBA) by O/W emulsification (A → B) andevaporation of the organic phase (B → C). Following this, crosslinking wasperformed to permanently lock the structure. This could be accomplished
Scheme 2. Schematic diagram showing the preparation of a water-in-oil eafter evaporation of the organic solvent.
emulsion solvent evaporation technique, which was firstemployed by Vrancken et al. [92] to prepare homopoly-mer microspheres and later by Ouchi et al. [93] to preparePLA-based copolymer microcapsules, was primarily usedfor the preparation of biodegradable microcapsules andmicrospheres for drug encapsulation and release studies.No microphase separation within the microcapsules orinterfacially-driven self-assembly of the block copolymersat the 2D oil–water interface was ever reported.
2.3. Seeing morphologies of block copolymers confinedwithin microspheres
Liu and coworkers [94] were the first to report blocksegregation in copolymer microspheres generated fromemulsification. In their system they used two diblockcopolymers, including poly(tert-butyl acrylate)390-block-poly(2-cinnamoyloxyethyl methacrylate)420(PtBA390-b-PCEMA420) and poly(2-cinnamoyloxyethylmethacrylate)32-block-poly(glyceryl methacrylate)176(PCEMA32-b-PGMA176). The former copolymer was usedto form the microsphere core, while the latter was used asa surfactant to stabilize the emulsion-droplets, and later
the microspheres after oil-phase solvent evaporation. ThePCEMA-b-PGMA surfactant was dissolved into a minimalamount of methanol, and water was then added to forman aqueous phase (Scheme 3). PtBA390-b-PCEMA420 waseither by photolysis of the PCEMA block (PtBA-b-PCEMA) or exposing thePI block to S2Cl2 (PI-b-PtBA) (C → D). The PtBA domains could also be con-verted into PAA by hydrolysis (D → E) [94]. Adapted with permission fromReference [94]. Copyright 2001 American Chemical Society.

1158 I. Wyman et al. / Progress in Polymer Science 36 (2011) 1152– 1183
Fig. 4. TEM images of thin film cross-sections of PtBA390-b-PCEMA420 microspheres that were prepared using dichloromethane (a) or toluene (b) as theo ylindrical the imaA
dfseswaaaP
feteTcfabsdtd
ttasho
2c
imTmc
rganic phase. In image (a) the circles or ellipses (c) correspond to PtBA cight stripes (d) represent PtBA cylinders that are lying flat in the plane ofmerican Chemical Society.
issolved into DCM and mixed with the aqueous sur-actant solution. This mixture was stirred and sonicated,ubsequently yielding an O/W emulsion. Subsequentvaporation of the DCM by mild heating at 50 ◦C led to theolidification of PtBA390-b-PCEMA420. These solid spheresere then photo-crosslinked to lock in their structure,
nd subsequently centrifuged from the solution. TEMnalysis of thin sections of the microspheres revealed that
PCEMA matrix interwoven with hexagonally-packedtBA cylinders filled the cores of these spheres (Fig. 4a).
In bulk phase the PtBA domains of PtBA390-b-PCEMA420ormed cylindrical structures with a similar average diam-ter to those prepared by emulsification [94,95]. However,he degree of ordering among the structures confined inmulsion droplets differed from those prepared in bulk.he orientations of the PtBA cylinders varied inside theonfined spheres (∼2 �m in diameter), with the cylindersacing different directions in one region of the sphere thannother. Meanwhile, the analogous PtBA cylinders in theulk films were straighter than those prepared by emul-ification, and aligned in the same direction over longeristances [95]. The cylinders in the spheres bent to adapto the confined volume encountered within the emulsion-roplets.
The internal block segregation pattern changed whenhe emulsion droplets were prepared using DCM instead ofoluene [94]. Because of the higher boiling point of toluene,
higher temperature (90 ◦C) was used for the evaporationtage. Under this set of conditions, the resultant particlesad an interior consisting of onion-like alternating layersf PtBA and PCEMA (Fig. 4b).
.4. Segregation behavior of blockopolymer/homopolymer blends
If a block copolymer is mixed with one of its correspond-ng homopolymers, swelling of the existing morphologies
ay result, leading to a morphological transition [96,97].his can also occur within block copolymer/homopolymericrospheres prepared from the emulsification proto-
ol. Factors such as the weight (or volume) ratios
l domains that were cut perpendicularly to the axis of the cylinders. Thege [94]. Reprinted with permission from Reference [94]. Copyright 2001
between the amount of copolymer and homopolymer(or between the homopolymer and its correspondingcopolymer block) present within a blend, as well as therelative molecular weight ratios between the homopoly-mer and its corresponding copolymer block can affectthe morphology. Therefore, adjusting the compositionof copolymer/homopolymer blends within an emulsionsphere provides a means to direct the assembly in a highlycontrolled manner.
2.4.1. Influence of copolymer/homopolymer blendcomposition
One of the earliest groups to study the influence ofcopolymer/homopolymer blends on the internal mor-phologies of microspheres formed via emulsion dropletswas that of Liu and coworkers [98]. The morphologies ofthe PtBA domains within emulsion spheres could be alteredby adding PtBA homopolymer (hPtBA) to polyisoprene-block-poly(tert-butyl acrylate) (PI-b-PtBA). These emulsionspheres were prepared using an O/W emulsion very similarto that described earlier for the PtBA-b-PCEMA micro-spheres and shown in Scheme 3. In this case, the oilphase consisted of a PI980-b-PtBA200/hPtBA110 blend dis-solved in DCM. Meanwhile, the aqueous phase containedpolyisoprene-block-poly(acrylic acid) (PI-b-PAA), whichstabilized the oil droplets. Subsequent DCM evaporationcaused the block copolymer to collapse, thus yielding themicrospheres, which were collected and dried. The PIdomains of these spheres were then crosslinked with S2Cl2,to lock in their structure. The PtBA block could be convertedinto PAA by hydrolysis, yielding porous microspheres.
The internal morphologies changed with hPtBA addi-tion, which essentially increased the total PtBA volumefraction. Without hPtBA, the internal morphologies of thePI-b-PtBA microspheres consisted of mixtures of PtBAspheres and cylinders surrounded by PI (Fig. 5a). With smallamounts of hPtBA, the PtBA domains (with a volume frac-
tion of 39%) formed worm-like structures (Fig. 5b), whichLiu and coworkers attributed to a gyroid-like morphology[98]. Thus, the morphologies apparently paralleled bulkbehavior, [1,4,99–101], with decreasing curvature between
I. Wyman et al. / Progress in Polymer Science 36 (2011) 1152– 1183 1159
Fig. 5. TEM images of microsphere cross sections composed of PI-b-PtBA/hPtBA blends. The microspheres shown in (a) were prepared in the absence ofhPtBA and had an overall PtBA volume fraction of 25%. The microspheres shown in (b) were prepared from a blend of PI-b-PtBA and had an overall PtBA
volumewith S2C
volume content of 39%. The microspheres shown in (c) had an overall PtBAstained the PI domains. The PI blocks of these samples were crosslinked
John Wiley and Sons.
the two blocks as the block distribution became more bal-anced, or symmetrical. However, when more hPtBA waspresent, yielding an overall PtBA volume fraction of 54%,the PtBA domains had an ill-defined morphology (Fig. 5c).This did not correspond with the lamellar morphology thatwould be anticipated for a copolymer in bulk with a sym-metric block distribution. Considering that the D/L0 valuesamong these large microspheres were above 20, it is lesslikely that the assembly of these structures was directed byconfinement. The high weight fraction of the homopoly-mer (40% relative to the blend) in this latter sample mayhave played a role instead. Previous researchers observedmacrophase segregation among copolymer/homopolymerblends when large amounts of homopolymer were present[100].
Onion-like morphologies have been observed whencopolymer/homopolymer blends were confined withinemulsion droplets. Significant insight into these systemshas been provided by Okubo and coworkers [102,103].They [102] studied a blend consisting of the blockcopolymer polystyrene-block-poly(methyl methacrylate)(PS-b-PMMA) and its homopolymers, with combinationssuch as PS-b-PMMA/hPS, PS-b-PMMA/hPMMA, and PS-b-PMMA/hPS/hPMMA. In all cases the molecular weight of thehomopolymer was lower than its corresponding copoly-mer block, as previous researchers had shown that thehomopolymer should be no longer than its correspond-ing copolymer block to give a miscible blend [104,105].Typically, a solution of PS-b-PMMA along with homopoly-mer(s), were dissolved into toluene. This organic phase wasthen mixed with an aqueous phase containing SDS as a sur-factant, generating an O/W emulsion. The emulsion wasthen placed in an open vessel to evaporate the tolueneunder continuous stirring [102,103].
In their study, Okubo et al. [102] varied the weightratio between the copolymer and the homopolymer. Inthis comparison, they used a lamella-forming symmet-ric block copolymer. Without homopolymer, the resultantparticles had onion-like interior morphologies. When a
blend of the copolymer and homopolymer had a PS-b-PMMA/hPS weight ratio of 80/20, the particles obtainedhad various internal structures, including cylinder-like andbicontinuous gyroid morphologies (Fig. 6a). Meanwhile, ifcontent of 54%. These samples were stained with OsO4, which selectivelyl2 [98]. Reprinted with permission from Reference [98]. Copyright 2003
the ratio of hPS was increased further, to a weight ratioof 50/50, a “sea-island” interior structure was obtained,with the “sea” composed of PS and the “islands” consist-ing of PMMA domains (Fig. 6b). When the homopolymerwas changed to hPMMA (Fig. 6c and d), a similar generaltransition from lamellar to “sea island” structures occurred[102].
Okubo and coworkers [103] also quantified the relation-ships between the thickness of onion-like layers withinemulsion spheres and copolymer/homopolymer blendcomposition, and also between lamellar thickness andcopolymer molecular weight. Consistent with their ear-lier results [102], the layer thickness within these lamellarstructures increased as the volume fractions of PS-b-PMMAwere decreased with respect to its homopolymers, hPS andhPMMA (Fig. 7). In particular, they observed that if thehomopolymers had lower molecular weights than theircorresponding copolymer blocks, the layer thickness wasproportional to the –1/3 power of the volume fractionof the homopolymer within the blend [103]. This trendwas consistent with earlier predictions by Hashimoto etal. [100]. Within the copolymer, the volume ratios of thePS and PMMA blocks were equal, and also the blendswere prepared with equal amounts of each homopolymer.Therefore, changes arose from variation of the copoly-mer/homopolymer volume ratios, rather than changes ofrelative PS or PMMA content within the blend. Onion-like morphologies were maintained throughout this series,although the concentric layers became broken when thecopolymer volume was reduced to 0.1 [103]. In their ear-lier report, they attributed this to insufficient copolymer toform complete concentric layers at low copolymer volumefractions [102].
Changing the amount of homopolymer within a copoly-mer/homopolymer blend has a significant influence uponthe resultant assembly structure. If a given homopolymerhA is added to an AB diblock copolymer, and the blenddoes not undergo macrophase segregation, hA addition cancause the blend to behave analogously to an AB copolymer
with a larger proportion of the A block [106]. In effect, theadded homopolymer “mimics” its corresponding block, anddirects the blend’s morphological assembly. With only oneblock copolymer and a corresponding homopolymer (or
1160 I. Wyman et al. / Progress in Polymer Science 36 (2011) 1152– 1183
F w/w) PSP ticles w(
aogwc
FT0fP
ig. 6. TEM thin cross-sectional images of particles composed of 80/20 (MMA/hPMMA (c), and 50/50 (w/w) PS-b-PMMA/hPMMA (d). These par2005), with permission from Elsevier.
lternatively a homopolymer corresponding to each block),ne may potentially obtain a similar range of morpholo-
ies to that observed among a series of copolymers with aide range of block ratios, simply by adjusting the blendomposition. Considering the synthetic demands required
ig. 7. TEM images of thinly sliced cross-sections of onion-like particles obtained
he volume fractions among the hPS/PS-b-PMMA/hPMMA blends were: (a) 0/1.45/0.1/0.45. The samples were stained with RuO4 and the darker regions are
ractions of the homopolymers increased the layers became thicker, and eventuaS regions and the lighter regions are PMMA domains. Reprinted with permission
-b-PMMA/hPS (a), 50/50 (w/w) PS-b-PMMA/hPS (b), 80/20 (w/w) PS-b-ere stained with RuO4 vapor [102]. Reprinted from Ref. [102], Copyright
to generate such a wide library of block copolymers withvarious block ratios, copolymer/homopolymer blends pro-
vide an attractive alternative. Similarly, blends can providea facile means to direct the assembly of a block copolymerthrough emulsification.from O/W emulsion droplets containing hPS/PS-b-PMMA/hPMMA blends./0, (b) 0.1/0.8/0.1, (c) 0.2/0.6/0.2, (d) 0.3/0.4/0.3, (e) 0.4/0.2/0.4, and (f)PS domains, while the lighter regions are PMMA layers. As the volumelly broken layers were observed [103]. The darker regions correspond to
from Reference [103]. Copyright 2009 American Chemical Society.

I. Wyman et al. / Progress in Polymer Science 36 (2011) 1152– 1183 1161
Fig. 8. TEM images of blend particles of PS-b-PB and hPS, with a hPS weight fraction of 50%. The molecular weight of hPS is 9.6 × 104 g mol–1, whichis approximately double the molecular weight of the corresponding PS block (r ∼ 2). The particle shown in (a) has a diameter of 340 nm and consistsof one spherical lamella and one hemispherical lamella. The particle in image (b) has a diameter of 400 nm and consists of two spherical lamellae andone hemispherical lamella. The particle shown in image (c) has a diameter of 420 nm and consists of three spherical lamellae. The spherical lamellae in
ed with
these images are off-center and unevenly spaced. The samples were stainReference [107]. Copyright 2007 American Chemical Society.2.4.2. Influence of homopolymer molecular weightrelative to that of the copolymer
The molecular weight ratio between a homopolymerand its corresponding copolymer block can also influ-ence the morphology of the blend. An extensive studyexploring various factors influencing the morphologiesof emulsion particles composed of PS-b-PB (polystyrene-block-polybutadiene) and hPS was conducted by Jeon et al.[107]. The two copolymer blocks were approximately sym-metrical, with a PS block weight fraction of 55%. Theyvaried the degree of confinement, the weight fraction ofthe homopolymer, and the molecular weight of hPS rel-ative to that of the PS block. To prepare the emulsion,PS-b-PB and hPS were dissolved in toluene and mixed withan aqueous solution containing the triblock copolymerpoly(ethylene oxide)-block-poly(propylene oxide)-block-poly(ethylene oxide) (PEO-b-PPO-b-PEO, or Pluronic F108)as a stabilizer. Once the O/W emulsion was formed, thetoluene was evaporated.
While the molecular weight of the copolymer remainedconstant throughout the study, the molecular weight of hPSwas varied [107]. Three molecular weight regimes werestudied, including when the molecular weight of hPS wasless than (r < 1), similar to (r ∼ 1), or greater than (r > 1) thatof the corresponding copolymer’s PS block. Within eachregime, the effects of changes of the weight fraction of hPSand the degree of confinement were compared [107].
When the molecular weight of hPS was less than that ofthe PS block (r < 1), and the weight fraction of hPS relative tothe PS block was increased, Jeon et al. [107] observed thatthe internal morphologies changed gradually from concen-tric lamellae (onion-like layers), to perforated lamellae tocylinders, and eventually to spheres. This behavior paral-lels the general trend observed among emulsion spheres byOkubo et al. [102]. The spherically confined cylinders struc-
tures were generally distorted to yield internal structuressuch as circular helices or stacked hoops, to accommodatetheir confinement within the spheres. Blends of homopoly-mers and copolymers are generally miscible when theOsO4, which selectively stains PB [107]. Reprinted with permission from
homopolymer has a lower molecular weight than its corre-sponding copolymer block [99,108]. In this regime, addinghomopolymer can have a similar morphological effect asincreasing the volume ratio of the corresponding copoly-mer block.
Jeon et al. [107] also studied PS-b-PB/hPS blends wherethe molecular weight of hPS was approximately equalto that of the PS block (r ∼ 1). In these circumstances,the homopolymer should be soluble in the correspondingdomains, but unable to reach the interfaces of the domain.Consequently, hPS should occupy the central region ofthe PS domain where it is isolated from the interfaces[108]. In addition, the PS domain should swell upon theaddition of hPS [99]. A general trend among these parti-cles was that their internal morphologies changed fromonion-like lamellae to perforated lamellae, and cylindricalmorphologies with increasing homopolymer weight frac-tions. However, many of the structures had unexpectedmorphologies, and the behavior varied considerably withdiffering particle diameters. The morphological transitionsseemed to be delayed somewhat in the smaller and moreconfined spheres, apparently due to surface effects. Insome cases, combinations of onion-like lamellae and per-forated lamellae were observed in the same particle, withlamellae near the surface and perforated lamellae near thecore. The conversion from lamellar to perforated lamellarmorphologies apparently began near the cores of the par-ticles, which were more isolated from the surface. Variousexotic structures were obtained at higher homopolymerweight fractions, such as hoop-shaped, tetragon-shaped,figure-eight, and pretzel-shaped internal morphologies.These latter structures were apparently combinations ofhoops that became fused together. The wide range of mor-phologies was attributed to the combination of microphaseand macrophase segregations occurring under this regime,
which is further enhanced by the varying degrees of con-finement [107].PS-b-PB/hPS blends with hPS molecular weights exceed-ing that of the corresponding PS block (r > 1) were also

1 olymer S
ipoUbrotodpd5ogtMmeoa4wrtdtt[
ictcimlmibs
2c
tcbgcyiwwoltsWp(
162 I. Wyman et al. / Progress in P
nvestigated by Jeon et al. [107]. Jeon et al. used hPS sam-les with molecular weights approximately double (r ∼ 2)r quadruple (r ∼ 4) that of the corresponding PS block.nder these conditions, the hPS and its corresponding PSlock can readily phase separate from one another. When
∼ 2, lamellar morphologies were maintained, regardlessf the hPS weight fraction. However, as the weight frac-ion increased beyond 32%, random segregation of hPSccurred which caused these lamellar structures to becomeeformed. The rings of the onion-like structure wereushed off-center, so that the rings were no longer evenlyistributed. Also, when the weight fraction of hPS was0%, the outer layer was often extremely deformed, withne portion of the circular layer becoming flattened toive a hemispherical shape (Fig. 8). The PS layers becamehicker as the hPS weight fraction was increased as well.
eanwhile, when r ∼ 4, the lamellar structure was alsoaintained as the weight fraction of hPS was varied. How-
ver, the increasing thickness of the PS layer was notbserved once the homopolymer weight fraction reachednd exceeded 40%. When the weight fraction was less than0%, hPS segregated itself randomly. Meanwhile, once theeight fraction of hPS was above 40%, the hPS became seg-
egated in the outer regions of the blend particles ratherhan the interior. Unlike the case when r ∼ 2, if r ∼ 4 theistance between the layers did not change significantly ashe weight fraction of hPS was varied. Jeon et al. attributedhis behavior to the macrophase segregation of hPS107].
The molecular weight of a homopolymer significantlynfluences how its presence may direct the assembly of aopolymer/homopolymer blend. If the molecular weight ofhe homopolymer is well below that of the correspondingopolymer block, it can readily become incorporated intots matching block’s domains [106]. Meanwhile, homopoly-
ers with relatively high molecular weights are moreikely to undergo macrophase segregation. When the
olecular weights of the homopolymer and its match-ng copolymer block are comparable, a combination oflending and macrophase segregation can occur, which canometimes yield unexpected morphologies.
.4.3. Influence of D/L0 upon the morphologies ofopolymer/homopolymer blends
Jeon et al. [107] also examined confinement effects onhe morphologies of PS-b-PB/hPS blends. Under greateronfinement, the formation of a single internal morphologyecomes more favorable than a mixture of morpholo-ies [107]. They attributed this to the greater interfacialurvatures observed in more confined droplets, whichield stronger capillary forces. The interface had greaternfluence upon the arrangement of the interior domains
hen D/L0 is small. When the diameter of the particlesas 240 nm and the D/L0 value was approximately 4, the
nion-like morphology was formed with only the inneramella being perforated. This behavior was attributed tohe weaker interfacial forces observed deep inside the
phere than by the layers that are closer to the surface.hen the D/L0 value was increased to 5, the interior mor-hology consisted purely of concentric perforated lamellaeFig. 9).
cience 36 (2011) 1152– 1183
Besides the internal morphologies, the overall particleshape could also be altered. Two competing influences canaffect the shape of the particles prepared by emulsification[107]. Capillary forces act to compress the matter withinthe particle, and these forces are spherically symmetric.Meanwhile, the free energy that arises from the morpholo-gies of the interior domains can also affect the overallshape of the particle. This latter force can cause a parti-cle to become distorted from a spherical shape if it is notconsistent with the morphology of the interior domains.Depending on the shape of the internal morphology, spher-ical confinement may involve high entropic costs. Unlikeunder 1D or 2D confinement, there are no unconfined direc-tions available along which a spherically confined polymerchain may realign itself to relieve frustration [89]. However,Jeon et al. observed that spherically confined copolymersmay relieve this strain by distorting the overall shape ofthe confining particle [107]. This can occur when the inte-rior morphology consists of hexagonally packed cylindersor helices, as well as stacked discs. If spherical particlesare distorted into elliptical shapes, with the helices alignedwith the major axis of the ellipse, the shape of the nanopar-ticle can better match the entropic demands of the interiormorphologies. Such a shape distortion may occur providedthat it is not outweighed by the capillary forces, which favora spherical particle. At larger D/L0 values, spherical parti-cles were more common than distorted particles due to thedominance of capillary forces over the entropy needs of theentrapped polymer chains [107].
When sufficient hPS had been added so that the PS-b-PBcopolymer would normally form cylindrical structures inbulk, the range of morphologies varied significantly withD/L0 values. For example, when the hPS volume fractionwas 51% and the D/L0 values were increased, the inter-nal morphology generally changed from a single sphere(D/L0 = 2), to stacked discs and hoops (D/L0 ∼ 2.5–7.0), tohelices (D/L0 ∼ 7–15), and towards loosely coiled cylinders(D/L0 ∼ 17) (Fig. 10). In general, the number of inter-nal structures (such as hoops or discs) within a particleincreased with increasing D/L0. The general trend fromdiscs towards helices or stacked discs as the confinementof cylinder-forming block copolymers was reduced paral-lels behavior observed by Russell and coworkers amongcylindrically confined PS-b-PB copolymers [109]. Cautionmay be needed if one attempts to draw universal trends onthe morphological behaviors of spherically confined blockcopolymers at this point. The behaviors of spherically con-fined copolymers are generally more complex than thoseunder 1D or 2D confinement [89]. This is particularly trueamong asymmetric or non-lamellar block copolymers.
As described earlier, the effects of adjusting con-finement can generate a diverse array of copolymermorphologies [88]. The combined influence of altering con-finement and blend composition can extend this varietyeven further, while also providing another tool to directthe assembly of a copolymer/homopolymer blend. In addi-tion to confinement, contributing factors which direct the
assembly can include the weight fraction of the homopoly-mer within the blend, as well as the molecular weight ofthe homopolymer relative to the corresponding block ofthe copolymer. The behavior of a confined polymer can
I. Wyman et al. / Progress in Polymer Science 36 (2011) 1152– 1183 1163
s showiwas 34%
ella is
Refere
Fig. 9. TEM images of PS-b-PB/hPS blend particles (a and c) and diagramhPS in these blends was 1.0 × 104 g mol–1 and the weight fraction of hPS
blend particle with D/L0 ∼4. The outer lamella is solid while the inner lamas a TEM image (c) and drawing (d) [107]. Reprinted with permission from
vary depending if the confining surface is more compatible
with one copolymer block over another, or if the surface hasno preference [71]. This combination of factors can directthe assembly to generate complex structures that would bedifficult or impossible to obtain by other means.Fig. 10. TEM images of thin cross-sections of particles that were prepared by emmolecular weight of hPS was 104 g mol–1). Images (a) shows a particle with a D/L0
with a D/L0 value of 3.3 which contains three discs and two hoops, and (c) shows[107]. Reprinted with permission from Reference [107]. Copyright 2007 American
ng their corresponding morphologies (c and d). The molecular weight of. A TEM image (a) and schematic drawing (b) show the morphology of a
perforated. A blend particle with D/L0 ∼5 and perforated layers is shownnce [107]. Copyright 2007 American Chemical Society.
3. Block copolymer vesicles and capsules prepared
by emulsion techniquesThe preparation of capsules derived from block copoly-mers has attracted significant attention in recent years.
ulsion from a PS-b-PB/hPS blend with a hPS weight fraction of 51% (thevalue of 2.5 which consists of two discs and one hoop, (b) shows a particle
a particle with a D/L0 value of 7, which contains several discs and hoops Chemical Society.

1164 I. Wyman et al. / Progress in Polymer Science 36 (2011) 1152– 1183
Scheme 4. Schematic diagram of vesicles based on the triblock copolymer PGMA-b-PCEMA-b-PtBA [118]. The PGMA, PCEMA, and PtBA domains are shownin red, green, and black, respectively. The solutions are mixed to form an O/W emulsion (A → B). The copolymer aggregated at the oil/water interface, withthe PtBA block being directed towards the interior of the oil droplets and the hydrophilic PGMA block being directed outward towards the aqueous phase.E block,
b the refev pyright
Od[mcotapbsvomeca
3e
tpbstdSatioDPcc
vaporation of dichloromethane (B → C) led to the collapse of the PCEMAy photo-crosslinking the PCEMA domains (C → D). (For interpretation ofersion of the article.) Adapted with permission from Reference [118]. Co
ne factor driving this is their potential applications asrug delivery vehicles [110]. Numerous recent reviews111–117] highlight some of the progress that has been
ade involving polymer-based capsules. Interesting vesi-le systems have been prepared through the combined usef double emulsion techniques, triblock copolymers, andernary solvents [118]. Vesicles having an aqueous cavitynd whose walls are based on polymers are often calledolymersomes, and are analogous to liposomes, which areased on phospholipids. Normally, polymersomes are moretable than liposomes, due to the thicker membranes pro-ided by the polymer [119–121]. While polymersomes areften prepared using film rehydration [121] or electrofor-ation [119] techniques, they have also been prepared by
mulsification methods, such as double emulsions. In someases, the double emulsions used to prepare polymersomesre formed with the use of microfluidic devices [122–124].
.1. Capsules from block copolymer assembly andmulsification
Zheng and Liu have prepared vesicle-like capsules fromhe triblock copolymer poly(glyceryl methacrylate)-block-oly(2-cinnamoyloxyethyl methacrylate)-block-poly(tert-utyl acrylate) [118]. This copolymer was dissolved in amall amount of a methanol and dichloromethane mix-ure. It was then added into a stirred oil/water mixture ofecahydronaphthalene (DN), DCM, and water (Scheme 4).ince the PGMA block was water-soluble, and the PCEMAnd PtBA blocks were soluble in the organic phase, theriblock copolymer assembled quickly at the oil/waternterface to stabilize the oil droplets. The DCM was evap-rated by mild heating, yielding droplets containing only
N within their interiors. This led to the collapse of theCEMA block. The PCEMA block was subsequently photo-rosslinked to produce vesicles with DN trapped in theiravities. The central PCEMA blocks formed the walls ofand formation of the capsule walls. These walls were structurally lockedrences to color in this scheme caption, the reader is referred to the web
2007 American Chemical Society.
the vesicles, while the PtBA blocks were directed inwardstowards the organic phase, and the PGMA blocks wereprojected outward into the aqueous phase. Therefore,this assembly process was driven by the need for theamphiphilic copolymer to stabilize the emulsion-droplets,with the oil/water interface effectively acting as a tem-plate. Subsequent crosslinking of the PCEMA block servedto lock in these structures. While polymersomes entrapaqueous cores, nanocapsules may encapsulate hydropho-bic liquids [125]. The media occupying a polymersomecavity and surrounding a polymersome are generally sim-ilar, both being aqueous. Meanwhile, the liquid within ananocapsule’s core may differ from the continuous phase.A hydrophilic guest can be entrapped within the aqueouscore of a polymersome. It has also been demonstrated thatpolymersomes may simultaneously carry both hydropho-bic and hydrophilic guests, which are located within thepolymersome wall and aqueous core, respectively [126].Meanwhile, a nanocapsule may encapsulate a hydropho-bic guest within its oil-filled core. If a nanocapsule’s liquidcore differs from the surrounding media, a guest may bedriven to occupy the cavity, particularly if it is more sol-uble in the core-filling solvent than in the continuousphase [44,127]. In addition, while a hydrophobic guestmay be dissolved as a solution within the organic sol-vent of the vesicle cavity, in other cases the active guestmay occupy the cavity as a neat liquid. For example, poly-isobutylcyanoacrylate nanocapsules had been preparedwhich encapsulated lipidiol, a radiological tracer, as theentrapped oil [128]. More recently, lipidiol has also beenentrapped within PEO-b-PPO-b-PEO nanocapsules [129].The anticancer-drug paclitaxel could also be dissolved inthe entrapped lipidiol compartment in this latter example.
Block copolymer vesicles can also be prepared from(W/O)/W double emulsions. In a typical preparation, ablock copolymer is dissolved in a volatile organic phase,which is then mixed with water to yield a W/O emul-

I. Wyman et al. / Progress in Polymer S
Scheme 5. Formation of a polymersome from a double emulsion. Theblock copolymer is dissolved in the organic layer, and becomes localized atthe oil–water interfaces, with the hydrophobic block (blue) projected intothe oil phase and the hydrophilic block (red) projected towards the waterphases. The oil phase becomes thinner during evaporation, and eventuallythe hydrophobic copolymer block collapses, thus forming the polymer-some wall. The hydrophilic block can extend into the aqueous phases,which are both surrounding the polymersome and inside its cavity [124].
(For interpretation of the references to color in this scheme caption, thereader is referred to the web version of the article.) Reprinted with permis-sion from Reference [124]. Copyright 2006 American Chemical Society.sion. Following this, the W/O emulsion is then added toan aqueous phase, to yield a (W/O)/W emulsion uponmixing. The organic phase is then evaporated from the(W/O)/W emulsion. As this occurs, the copolymer becomesmore concentrated and aggregates along the oil–waterinterfaces, and eventually the hydrophobic block collapses(Scheme 5). This yields water-filled capsules whose wallsare composed of the copolymer. The hydrophilic block willnormally be projected into the aqueous phase both insideand outside the polymersome (thus acting as a corona),while the collapsed hydrophobic block forms the “core” ofthe polymersome wall.
Recently Shim et al. [130] prepared microcapsulesfrom blends of the copolymer poly(styrene)-block-poly(butadiene)-block-poly(styrene) (PS-b-PB-b-PS) andhPS using this general approach (Scheme 6). The microcap-sules could be separated to isolate those of a desired sizethrough selective sedimentation. Since both the PS and PBblocks were hydrophobic, they both collapsed and wereconfined within the capsule wall as the organic phase wasevaporated. Therefore the structures of the capsule wallsdiffered from that typically seen among polymersomes,as there were no hydrophilic blocks extending into theaqueous phase. Furthermore, microphase segregation ofthe PS and PB domains occurred within these thin walls.The internal morphology could be tuned by varying thehPS content within the PS-b-PB-b-PS/hPS blends. WithouthPS, the internal morphology of the polymersome wallsconsisted of PS cylinders surrounded by PB. IncreasinghPS content within the copolymer/homopolymer blendresulted in morphological changes from PS cylinders, tolamellae, to PB cylinders, and finally to PB spheres. Amongthese morphologies, the lamellae and the cylinders weremainly aligned parallel with the surfaces of the capsulewalls. Because the PB domains were more flexible thanthose of PS, addition of hPS also changed the mechanicalproperties of the polymersomes. The capsule walls were
more rigid if they were prepared from blends with higherhPS content. The (W/O)/W emulsion droplets served astemplates to direct the overall assembly of the capsules.Meanwhile, the blend composition played a key role incience 36 (2011) 1152– 1183 1165
determining the internal morphology of the capsule walls,as well as the flexibility of the capsules.
3.2. Controlled vesicle formation using microfluidicdevices
In recent years, the development of microfluidic deviceshas aided research in various areas of chemistry and biology[131]. Microfluidic devices have also been applied towardsemulsion chemistry [132], and this technology has beenapplied to preparing unique diblock copolymer assemblies[122–124,133]. Key advantages of microfluidic devices arethe monodispersity of the resultant particles, and the easeof adjusting the droplet diameters. Because the D/L0 valueshave a strong influence on the morphologies of the blockcopolymers, control of the droplet diameters can be usedto tune the morphology of the diblock copolymer. In con-trast to traditional double emulsions, which involve twomain steps, a microfluidic device can allow the prepara-tion of a double emulsion in one step [123]. In commonwith traditional double emulsions, the confinement of thecopolymer at the interfaces between the oil phase and thewater phase is used to direct the assembly of the poly-mersomes. Because the size distribution of the (W/O)/Wdroplets obtained using microfluidic devices are narrow,monodisperse polymersomes can be prepared using thesedouble emulsions as templates.
Similar to their traditional double emulsion counter-parts, double emulsions prepared by microfluidic devicescan also yield polymersomes upon organic phase evap-oration. Since the diameters of these droplets can bereadily controlled, the diameters of the resultant polymer-somes can be adjusted also. In 2005, Weitz and coworkers[122,123] used a microfluidic device to prepare a (W/O)/Wdouble emulsion, which yielded polymersomes composedof the diblock copolymer poly(n-butyl acrylate)-block-poly(acrylic acid) (PBA-b-PAA). This device consisted of twotubes with circular cross-sections that were inserted intoa glass tube with a square cross-section (Fig. 11a). Oneof these circular tubes served as an injection tube, whilethe other formed a collection tube. An inner aqueous fluidflowed through the injection tube, while an intermediateorganic solution flowed in the same direction in the regionsurrounding this tube. The copolymer was dissolved in thisintermediate organic phase. Meanwhile, an outer aqueoussolvent flowed through the square capillary surroundingthe collection tube from the opposite direction. Once thesefluids met, the outer fluid reversed its direction to flowalong with the other two fluids. These three solvent phaseswere focused through a narrow opening into the collectiontube, and formed droplets after passing through this con-stricted opening. The size of the droplets could be adjustedby changing the size of this opening.
Weitz and coworkers noted that the diblock copolymerplayed a critical role in stabilizing (W/O)/W emulsion-droplets generated by these devices. Without the copoly-mer, the interior droplet broke through the intermediate
organic phase and merged with the outer aqueous phase[122]. Therefore, the assembly of these polymersomes isdirected by demand for the amphiphilic copolymer toreduce the overall interfacial tension between the oil and
1166 I. Wyman et al. / Progress in Polymer Science 36 (2011) 1152– 1183
Scheme 6. Preparation of PS-b-PB-b-PS/hPS capsules from a (W/O)/W emulsification. This diagram highlights the solvent evaporation stage. PEO-b-PPO-b-PEO was used as a stabilizer. The PS-b-PB-b-PS/hPS blend became localized within the capsule wall, and the internal morphology was dependent uponthe hPS content in the blend [130]. Reprinted with permission from Reference [130]. Copyright 2010 American Chemical Society.
Fig. 11. A diagram of the microcapillary system used by Weitz and coworkers to prepare double emulsion droplets (a). The inner and outer fluids areaqueous phases, while the middle fluid is the organic phase [133]. A more recent device developed by Weitz and coworkers is shown in image (b), whichallows loading of two inner aqueous fluids forming separate droplets, and subsequently multicompartment polymersomes. The two inner aqueous fluidsmay contain different hydrophilic guests, which will be isolated from one another. Conceptual (c) and overlays (d and e) of optical and fluorescencemicroscope images of PEG-b-PLA polymersomes with aqueous compartments loaded with FITC-Dextran (green) and PEG (white in image c, grey in imagesd and e) [135]. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.) Image (a) reprintedfrom Reference [133]. Copyright 2008 American Chemical Society. Images (b–e) reprinted from: Ref. [135]. Copyright 2011 Wiley-VCH Verlag GmbH & Co.KGaA. Reproduced with permission.

olymer S
I. Wyman et al. / Progress in Pwater phases. In addition, the interior aqueous dropletserves as a template to determine the size of the polymer-some shell [123].
The number of vesicles formed within the droplets canvary, depending on whether the intermediate organic orthe inner aqueous fluid forms droplets first. If the inner-most aqueous phase forms droplets first, multiple waterdroplets will be suspended inside a larger oil droplet. Onthe other hand, if the interior and intermediate solutionsform droplets simultaneously, the resultant dropletswill contain one inner aqueous droplet surrounded byan organic middle phase and a (W/O)/W double emul-sion is thus formed. The organic phase is subsequentlyevaporated, and the organic layer becomes thinner, andeventually forms a membrane composed of the diblockcopolymer. Weitz and coworkers [122] noted that thethickness of the vesicle membrane varied depending onthe concentration of the diblock copolymer. Weitz andcoworkers [134] recently demonstrated that adjusting thesolvent content of the organic phase provides another levelof control. Their organic phase here was a solvent mixturewith a volatile good solvent (chloroform) and less volatilepoor solvent (hexane) for the PEG-b-PLA copolymer [134].The more volatile chloroform is preferentially lost to thecontinuous phase, leading to a dewetting process yieldingpolymersomes. At lower chloroform volume ratios, thetwo copolymer monolayers at the middle-outer and inner-middle interfaces were more likely to adhere together toyield polymersomes, expelling remaining organic solventmixture in the process. However, if the organic phase hadinsufficient chloroform, the copolymer would precipitatewithout yielding polymersomes. Therefore, by adjustingthe solvent ratios in the organic phase, they could there-fore control how readily a double emulsion would yieldpolymersomes, and also the strength of the polymersomewalls [134].
Recently, Weitz and coworkers [135] prepared non-spherical, multi-compartment polymersomes of PEG-b-PLA, which could encapsulate different hydrophilic guestswithin the different inner aqueous droplets. This wasaccomplished by modifying their microcapillary system sothat it incorporated two parallel injection tubes (Fig. 11b)delivering the inner aqueous phase [135,136], rather thanone such tube. This system allows the loading of dif-ferent hydrophilic guests into separate inner aqueousdroplets of the double emulsion and hence the resul-tant polymersomes, without cross-contamination, whichthey demonstrated using aqueous phases carrying PEG andFITC-Dextran (Fig. 11c–e). Weitz and coworkers [135] sug-gested that these polymersomes could be useful for carry-ing reagents that need to be isolated from one another untilan appointed time, when the reagents could be mixed bybreaking down the polymersomes. The inner droplets andthe surrounding diblock copolymer membranes were clus-tered together, giving these particles their non-sphericalshape. Various approaches to prepare non-spherical par-ticles through microfluidic devices have recently been
highlighted in a review by Weitz and coworkers [137].Polymersomes were also prepared by microfluidicdevices incorporating cross-junctions or T-junctions, asdemonstrated by Weitz and coworkers [138], and by
cience 36 (2011) 1152– 1183 1167
Colin and coworkers [139], respectively. The cross-junction device used by Weitz was composed ofpoly(dimethylsiloxane) (PDMS) which was coated withglass to improve its resistance to organic solvents, andcould also allow subsequent functionalization to controlthe hydrophobicity of the channels, and hence their wet-tibility. This could help to prevent fouling of the devices byblock copolymer deposition [138]. Meanwhile, the deviceused by Colin and coworkers was made up of fused sil-ica, which had good compatibility with organic solvents. Inboth systems, the inner aqueous phase was fed through themain channel or capillary, and the middle organic and outeraqueous phases were fed perpendicularly into the mainchannel to create the double emulsions. The device used byWeitz and coworkers [138] used two middle phase organiccross junctions, so that the organic solvent mixture could betuned. Meanwhile, Colin and coworkers showed that theycould obtain a high degree of control over their resultantpolymersomes by adjusting the relative flow rates of theinner, intermediate, and outer phases, which allowed tun-ing of the number of inner droplets, as well as the diametersof the inner droplets and the overall polymersomes [139].
Although the principles of emulsification prepared bymicrocapillary devices parallel those of traditional emul-sions, a key benefit of this technology is the level of controlprovided. These devices effectively behave as templates tofurther direct the assembly of the polymersomes. Micro-capillary systems can generate monodisperse droplets ofa particular diameter, thus yielding monodisperse poly-mersomes. In addition, they allow one to readily controlwhether the double emulsion droplets encapsulate a singlesmaller droplet, or multiple droplets. A current limitationof microfluidic devices is the quantity of material that canbe prepared. Weitz and coworkers have suggested thatthis hurdle may be overcome through the use of paralleldevices [137].
4. Block copolymer self-assembly in 2D spaces
While emulsification provides an effective method todirect block copolymer assembly inside particles, it canalso be used to direct the self-assembly of block copoly-mers on their surfaces. With this approach, one of theblocks of a given copolymer should be soluble in a sol-vent used for the emulsification, while the other block(s)should be insoluble in that solvent. As observed duringvesicle formation from double emulsions, the amphiphilicblock copolymer is driven to the interface between theoil and water phases. However, in these cases the assem-bly generally occurs along one interface instead of two, oron the surface of a structure. Confining self-assembly ontothe surfaces of emulsion-droplets can yield structures withinteresting surface morphologies, and allow control of thesurface composition. Sometimes the surfactant can deter-mine the surface composition, even if it is not incorporatedinto the final structure.
4.1. Assembly at the 2D interface
Liu and coworkers demonstrated that emulsificationcould be used to prepare spheres having segregateddomains on their surfaces [140]. They used a combination
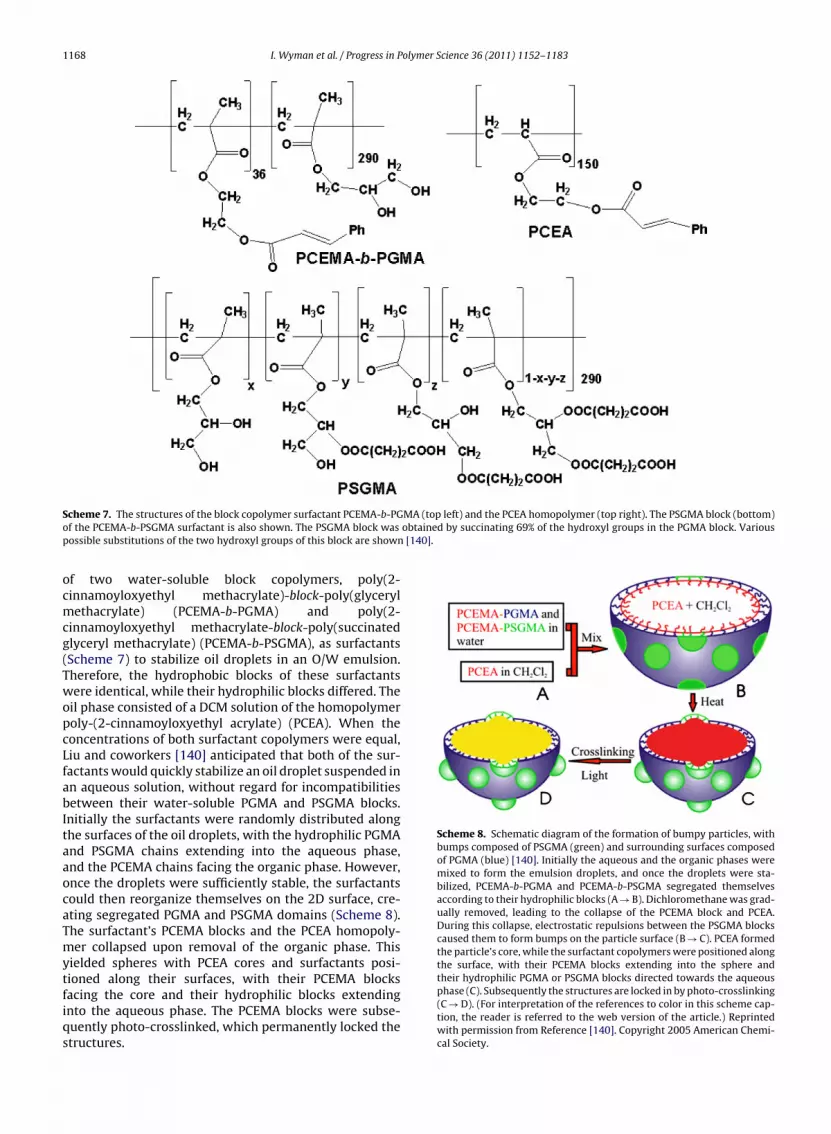
1168 I. Wyman et al. / Progress in Polymer Science 36 (2011) 1152– 1183
S MA (top left) and the PCEA homopolymer (top right). The PSGMA block (bottom)o obtained by succinating 69% of the hydroxyl groups in the PGMA block. Variousp [140].
ocmcg(TwopcLfabItaaocaTmytfiqs
Scheme 8. Schematic diagram of the formation of bumpy particles, withbumps composed of PSGMA (green) and surrounding surfaces composedof PGMA (blue) [140]. Initially the aqueous and the organic phases weremixed to form the emulsion droplets, and once the droplets were sta-bilized, PCEMA-b-PGMA and PCEMA-b-PSGMA segregated themselvesaccording to their hydrophilic blocks (A → B). Dichloromethane was grad-ually removed, leading to the collapse of the PCEMA block and PCEA.During this collapse, electrostatic repulsions between the PSGMA blockscaused them to form bumps on the particle surface (B → C). PCEA formedthe particle’s core, while the surfactant copolymers were positioned alongthe surface, with their PCEMA blocks extending into the sphere andtheir hydrophilic PGMA or PSGMA blocks directed towards the aqueous
cheme 7. The structures of the block copolymer surfactant PCEMA-b-PGf the PCEMA-b-PSGMA surfactant is also shown. The PSGMA block was
ossible substitutions of the two hydroxyl groups of this block are shown
f two water-soluble block copolymers, poly(2-innamoyloxyethyl methacrylate)-block-poly(glycerylethacrylate) (PCEMA-b-PGMA) and poly(2-
innamoyloxyethyl methacrylate-block-poly(succinatedlyceryl methacrylate) (PCEMA-b-PSGMA), as surfactantsScheme 7) to stabilize oil droplets in an O/W emulsion.herefore, the hydrophobic blocks of these surfactantsere identical, while their hydrophilic blocks differed. The
il phase consisted of a DCM solution of the homopolymeroly-(2-cinnamoyloxyethyl acrylate) (PCEA). When theoncentrations of both surfactant copolymers were equal,iu and coworkers [140] anticipated that both of the sur-actants would quickly stabilize an oil droplet suspended inn aqueous solution, without regard for incompatibilitiesetween their water-soluble PGMA and PSGMA blocks.
nitially the surfactants were randomly distributed alonghe surfaces of the oil droplets, with the hydrophilic PGMAnd PSGMA chains extending into the aqueous phase,nd the PCEMA chains facing the organic phase. However,nce the droplets were sufficiently stable, the surfactantsould then reorganize themselves on the 2D surface, cre-ting segregated PGMA and PSGMA domains (Scheme 8).he surfactant’s PCEMA blocks and the PCEA homopoly-er collapsed upon removal of the organic phase. This
ielded spheres with PCEA cores and surfactants posi-ioned along their surfaces, with their PCEMA blocks
acing the core and their hydrophilic blocks extendingnto the aqueous phase. The PCEMA blocks were subse-uently photo-crosslinked, which permanently locked thetructures.phase (C). Subsequently the structures are locked in by photo-crosslinking(C → D). (For interpretation of the references to color in this scheme cap-tion, the reader is referred to the web version of the article.) Reprintedwith permission from Reference [140]. Copyright 2005 American Chemi-cal Society.

I. Wyman et al. / Progress in Polymer Science 36 (2011) 1152– 1183 1169
Fig. 12. TEM (a) and AFM phase-contrast (b) images of PCEA microspheres with surfaces consisting of PGMA and PSGMA domains. The bumpy microspheresshown in the TEM image were prepared in the presence of CuCl2 and stained with uranyl acetate. The flat regions of the surface correspond to the PGMAdomains, while the bumps on the surface are occupied by the PSGMA domains (a). The microspheres shown in image (b) were prepared in the presence of
ce of Naission fr
formed a role in directing the assembly of these hierarchicalstructures.
NaCl instead of CuCl2. Although the bumpiness was reduced in the presenthe PGMA and PSGMA domains had occurred [140]. Reprinted with perm
TEM and atomic force microscopy (AFM) images(Fig. 12) indicated that the PGMA and PSGMA domainshad segregated themselves along the 2D surfaces of thedroplets, thus forming bumps on the surfaces of thesespheres. The bumps consisted of PSGMA while the flat sur-face regions were composed of PGMA. These bumps arosefrom electrostatic repulsions between the carboxyl groupsof the PSGMA chains as they became more crowded in theabsence of DCM [140]. The bumps could be made taller orshorter by adding either CuCl2 or NaCl, respectively, intothe aqueous phase during the emulsion preparation. Theseadditives helped tune the assembly by bridging the PSGMAchains (CuCl2) or by screening the electrostatic repulsionsbetween those chains (NaCl).
ABC triblock copolymers can also form hierarchi-cal assemblies on the surfaces of emulsion droplets.Liu and coworkers [141] prepared cylindrical andspherical micelle-like aggregates of the triblock copoly-mer polyisoprene110-block-poly(2-cinnamoyloxyethylmethacrylate)150-block-poly(tert-butyl)acrylate320 (PI110-b-PCEMA150-b-PtBA320, Scheme 9). From these micelle-likeaggregates, they then prepared hierarchical superaggre-gates of these structures by allowing them to congregateon the emulsion droplet surfaces.
The preparations of the spherical and the cylindricalmicelle-like aggregates differed somewhat (Scheme 10)[141]. To prepare cylindrical micelles, the copolymer washeated in the block-selective solvent decahydronaptha-lene, which is selective for the PtBA and PI blocks. Sphericalmicelles were prepared by initially dissolving the copoly-mer in a good solvent such as DCM, and subsequentlyadding DN to this solution. After this, the DCM solventwas removed, causing the PCEMA block to collapse. These
cylindrical or spherical micelle-like aggregates essentiallyserved as building blocks for the hierarchical structuressubsequently prepared on the surfaces of the emulsiondroplets.Cl, the contrast visible in the lighter regions suggests that segregation ofom Reference [140]. Copyright 2005 American Chemical Society.
Once either the cylindrical or spherical micelles wereformed, a second block-selective solvent (methanol) wasadded to the micellar solutions that were dispersed intoDN, and the mixture was stirred [141]. Methanol is not mis-cible with DN, and it is selective for only the PtBA block.Therefore, the micelles aggregated on the surfaces of themethanol droplets, with their PtBA blocks stretched intothe methanol droplets, and the PI blocks projected out intothe surrounding DN solution. While many emulsions utilizean organic and aqueous phase, in this example both of theimmiscible phases were organic solvents. The micelle-likeaggregates formed flower-like superaggregates and rib-bon cage-shaped structures at 22 and 52 ◦C, respectively(Fig. 13). These structures formed at their correspondingtemperatures regardless of whether they had spherical orcylindrical micelles as precursors, suggesting that thesewere equilibrium structures. Therefore, temperature per-
Scheme 9. Structure of polyisoprene110-block-poly(2-cinnamoyl-oxyethyl methacrylate)150-block-poly(tert-butyl)acrylate320 (PI110-b-PCEMA150-b-PtBA320).

1170 I. Wyman et al. / Progress in Polymer Science 36 (2011) 1152– 1183
Scheme 10. Schematic diagram summarizing the preparation of spherical and cylindrical micelle-like aggregates from PI-b-PCEMA-b-PtBA, followed byemulsification between two poorly miscible solvents (DN and methanol) to yield flower-like superaggregates or ribbon cage structures by assembly at22 or 52 ◦C, respectively. Flower-like superaggregates or ribbon cage structures were obtained through assembly at their corresponding temperatures,regardless of whether spherical or cylindrical micelle-like aggregates were used [141]. The inset image shows cylindrical micelle-like aggregates before( faces of
t A domat
4
rptbdrcfia
ceesptsnssoatft
A) and after (B) assembly in methanol. The micelles aggregate at the surhe PI (blue) and PtBA (light blue) chains extend outward from the PCEMhe reader is referred to the web version of the article.)
.2. Influence of surfactant upon surface morphology
Surfactants play a critical role in emulsification, as theyeduce the interfacial tension between the oil and waterhases. This in turn helps the emulsion system overcomehe penalty arising from the increased overall surface areaetween the two phases as the diameters of emulsion-roplets are reduced. In addition to this more traditionalole, surfactants can also direct the assembly of blockopolymer emulsions, particularly by determining the sur-ace composition of these structures. This can occur evenf the surfactant itself is not incorporated into the finalssembly structure.
As suggested above, the nature of the surfactantan determine which copolymer block will form thexposed surface of an emulsion particle. Okubo and work-rs [142], observed this when they varied the aqueousurfactants stabilizing oil droplets of polystyrene-block-oly(methyl methacrylate) (PS-b-PMMA) dissolved inoluene. The surfactants included either sodium dodecylulfate (SDS), poly(vinyl alcohol) (PVA), poly(oxyethyleneonyl phenyl ether) (Emulgen 911), or poly(oxyethyleneorbitan monooleate) (Tween 80). Regardless of whichurfactant was used, spherical particles composed ofnion-like alternating PS and PMMA layers were obtainedfter the toluene was evaporated. However, the domain
hat formed the surface layer differed according to the sur-actant, with the block that was more compatible withhe chosen surfactant forming the surface layer. Whenthe DN droplets. The central cylinder is composed of PCEMA (red) whilein. (For interpretation of the references to color in this scheme caption,
either SDS or PVA was used, PMMA formed the surfacelayers of the resultant particles. Meanwhile, if either Emul-gen 911 or Tween 80 was used, the resultant particlesurfaces were composed of PS. Analysis of interfacial ten-sion indicated that the surface layers were determined bywhichever block provided the least surface tension withthe surfactant-bearing aqueous phase. These experimentalobservations that the block most compatible with the sur-rounding formed the particle surface were consistent withthe Monte Carlo simulations by Yu et al. [88].
Jeon et al. [143] observed similar behavior amongemulsion particles composed of polystyrene-block-poly(butadiene) (PS-b-PB) and the polystyrenehomopolymer (hPS). These particles were also preparedfrom O/W emulsions, with the oil droplets contain-ing the copolymer/homopolymer blend dissolved intotoluene, which was subsequently evaporated to yieldthe particles. The oil droplets in the O/W were stabi-lized by the amphiphilic block copolymer surfactantspolystyrene-block-poly(ethylene oxide) (PS-b-PEO) andpolybutadiene-block-poly(ethylene oxide) (PB-b-PEO). Inagreement with the results of Okubo and workers [142],the block that was most compatible with the prevailingsurfactant formed the surface layer. Therefore, whenPS-b-PEO was used as the surfactant, the surfaces of theresultant particles were composed of PS (Fig. 14a–c).
Meanwhile, if PB-b-PEO was used to stabilize the oildroplets, the outer layer of the particles was composed ofPB (Fig. 14i–k). Because the PS-b-PB copolymer was sym-
I. Wyman et al. / Progress in Polymer Science 36 (2011) 1152– 1183 1171
superagg ribboon from
Fig. 13. TEM images of spherical micelle-like aggregates (a) and floweryAlso shown are cylindrical micelle-like aggregates (c) and correspondinspecimens were stained with RuO4 for 2 h [141]. Reprinted with permissi
metric, onion-like lamellar structures (Fig. 14a and i) wereobtained in the absence of hPS. The internal morphologieschanged to cylindrical PB helices (Fig. 14b and j) and PBspheres (Fig. 14c and k) as the volume fraction of hPS wasincreased.
The morphologies became more complex when mix-tures of the PS-b-PEO and PB-b-PEO surfactants were used[143]. The surface morphologies, and even the overall par-ticle shapes, varied significantly in this case. In the absenceof hPS, shapes of the particles changed from PB-coveredspheres (Fig. 14i), to tulip-bulb shaped structures (Fig. 14f),to striped elliptical particles (Fig. 14g), to reversed tulip-bulb shaped particles (Fig. 14d), and finally to PS-coveredspheres (Fig. 14a) as the surfactant mixture was changedfrom solely PB-b-PEO, to mixtures of the two surfactantswith increasing PS-b-PEO content, until the surfactant mix-ture was composed solely of PS-b-PEO. The surfactant
composition directed the surface morphology and shapesof the droplets, with a mixture of PS and PB domains beingexposed when a mixture of surfactants was present. Thetulip-bulb shape formed because the symmetric PS-b-PBgregates (b) formed at 22 ◦C two days after the initial addition of MeOH.n cages formed at 52 ◦C three days after the addition of MeOH (d). All
Reference [141]. Copyright 2008 American Chemical Society.
copolymer could not maintain alternating PS and PB lay-ers over a spherical particle due to the lack of either PS-(Fig. 14f) or PB-bearing surfactant (Fig. 14d). When the sur-factant mixture was composed of an approximately equalmixture of PS-b-PEO and PB-b-PEO, the particles acquiredan elliptical shape (Fig. 14g). This shape was attributedto anisotropic interfacial energy [144], as well as entropy[145]. The surfaces that were covered by alternating PS andPB domains had lower curvatures to avoid loss of entropydue to bending of the polymer chains. At the ends of theellipse, the curvature was higher, and only one polymerdomain occupied these regions.
The combined effect of simultaneously using mixedsurfactants and varying the composition of the PS-b-PB/hPS blend was also studied. In these cases oblatediscs which were axially penetrated by PB cylinders(Fig. 14e) were observed when both the surfactant and
copolymer/homopolymer blend consisted of intermediatemixtures. As hPS content was increased further, the minorPB domain changed from cylinders to spheres, yieldingspherical PB domains surrounded by a PS matrix (Fig. 14h).
1172 I. Wyman et al. / Progress in Polymer Science 36 (2011) 1152– 1183
Fig. 14. Graphical summary of the combined role of surfactant composition and homopolymer volume fraction on directing the assembly of PS-b-PB/hPSb he blens locks. TP aA. Rep
Ta
ccboudotcpbdca
5e
lesiptwstgw
lends. The horizontal axis represents the hPS volume fraction within turfactants PS-b-PEO and PB-b-PEO out of the total volume of PS and PB bB domains [143]. Ref. [143]. Copyright Wiley-VCH Verlag GmbH & Co. KG
hese PB spheres were distributed throughout the interior,nd also exposed to the surfaces of the particles.
These reports helped demonstrate how emulsion parti-le surfaces could be modified by changing the surfactantomposition. The copolymer block that is most compati-le with the prevailing surfactant will generally form theuter layer [142,143]. When a mixture of surfactants wassed, with each surfactant being more compatible with aifferent copolymer block, a diverse array of structures wasbtained. Both of the copolymer blocks became exposed tohe surface, and the overall shapes of the resultant parti-les varied also. Careful choice of the surfactant can thusrovide a potent means to direct block copolymer assem-ly. As demonstrated by Jeon et al. [143], this structuraliversity could be extended further by also varying theopolymer/homopolymer blend composition, providing andditional dimension of control over the assembly.
. Emulsion as a tool to direct the formation ofxotic architectures
Emulsification can yield a variety of complex micelle-ike block copolymer aggregates, as well as particles withxciting applications. Some examples of these complextructures have been described earlier in this review,ncluding the flower-like and ribbon cage structures pre-ared by Hu et al. [141], as well as the non-sphericalulip-bulb shaped particles observed by Jeon et al. [143]hen they used mixed surfactants to stabilize their emul-
ions. This section will highlight approaches that have ledo the preparation of complex block copolymer aggre-ates. Some of the structures described in this sectionere obtained by combining emulsification techniques
d, while the vertical axis represents the PS volume fraction among thehe lighter domains represent PS domains, while the darker domains areroduced with permission.
with the use of copolymer/homopolymer blends, micro-capillary systems, or also by obtaining kinetically trappedvesicles from the breakdown of larger emulsion dropletsinto smaller emulsion droplets. These approaches mayoffer high degrees of control, so that one can readily tunethe morphologies of their target structures.
5.1. Influence of interfacial tension on the formation ofbudding vesicles
Emulsion droplets can become unstable and break downinto smaller droplets if the interfacial tension between theemulsion droplet and the continuous phase is reduced.This situation has been utilized to prepare block copoly-mer assemblies [146,147], and the process is similar tothat of a spontaneous emulsion [148,149]. As the solventis gradually removed from an emulsion droplet, the blockcopolymer’s concentration increases inside the droplet.This causes the amphiphilic block copolymer to aggregateat the oil/water interface, thus behaving as a surfactant,and causing the interfacial tension between the droplet andcontinuous phase to decrease [150]. As a result, the dropletsmay eventually break down to form smaller droplets(which have a larger total surface area per given volume),and sometimes release the copolymer into the solutionas micelle-like aggregates. Geng and Discher [151,152]used this approach to prepare worm-shaped micelles frompoly(ethylene oxide)-block-poly(�-caprolactone) (PEO-b-PCL). Hayward and coworkers [146,147,153] have also
utilized this method to prepare micelles from PS-b-PEO/hPS blends.Zhu and Hayward prepared emulsions by initiallydissolving PS-b-PEO into chloroform [146]. In some

olymer S
I. Wyman et al. / Progress in Pexperiments they dissolved the copolymer by itself, whilein other cases the copolymer was mixed with hPS as ablend. An O/W emulsion was prepared by mixing thisoil phase by hand with an aqueous solution containingPVA surfactant. The droplet diameters in this hand-shakenemulsion were large, ranging between 5 and 100 �m. As theorganic phase was gradually evaporated and the polymerconcentration increased, the droplet surfaces roughened,and subsequently the droplets broke down into smallerdroplets (with diameters below 1 �m) and threads. Amongthe PS-b-PEO systems, as the chloroform was removed,the equilibrium apparently favored formation of PS-b-PEOaggregates that were dispersed into water [146]. The PSblock formed the core while the water-soluble PEO blockformed the corona.
When Zhu and Hayward varied the weight fractionof the PEO block among their diblock copolymers, themorphologies of the micelles ranged from predominantlyspherical micelles when the PEO weight fraction was65%, to predominantly worm-like micelles when the PEOweight fraction was 34%. This trend agrees with obser-vations by Geng and Discher [152], who noted that withincreasing hydrophilic PEO volume fractions among theirPEO-b-PCL copolymers, spherical micelles became favoredover worm-like micelles. This behavior also paralleledthat reported Jain and Bates [154], when they variedthe weight fraction of PEO among a series of PB-b-PEOcopolymers. Meanwhile, the diameters and morphologiesof the worm-like micelles could be controlled by adjust-ing the homopolymer content within the PS-b-PEO/hPSblends [146]. When the weight percentage of hPS was 70%,pearl necklace structures were observed, which apparentlyformed intermediate structures between cylinders andspheres. Once the weight percentage of hPS was increasedfurther to 80%, spherical micelles could be seen along withthe pearl necklace structures (Fig. 15).
In a related study, Zhu and Hayward [147] used amicrofluidic device (Fig. 16) to prepare O/W emulsiondroplets containing PS-b-PEO. As the chloroform solventwas subsequently evaporated from the copolymer-bearingoil droplets, “budding vesicles” formed on the dropletsurfaces (Fig. 17a). Zhu and Hayward suggested that theformation of these budding vesicles parallels the bud-ding mechanisms of lipid and block copolymer vesicles[147,155]. These results were compared to those in a PS-b-PEO/hPS blend. When hPS was present at a 50% weightfraction, dendritic particles formed, which had narrowcylindrical arms reaching out from their surfaces (Fig. 17b).These arms apparently formed during the initial stagesof interfacial instability, and become trapped due to thepresence of the homopolymer. As proposed by Zhu andHayward, the hPS effectively behaved as a selective solventfor the PS block, and thus affected its interfacial interactionsand curvature. The homopolymer also slowed the kineticsof the system by replacing solvent molecules with polymerchains, thus slowing the breakdown of the original dropletinto smaller droplets [146].
The weight fraction of the PEO block among PEO-b-PS copolymers also influenced the breakdown of theemulsion droplets into smaller droplets [147]. While bud-ding vesicles formed among emulsion droplets containing
cience 36 (2011) 1152– 1183 1173
copolymers with larger PEO weight fractions (such as 35%),they were not seen when the copolymer had lower PEOweight fractions (15%) when spherical particles with roughsurfaces formed instead. These rough surfaces providedincreased surface areas, indicating that the interfacial ten-sion had decreased, but not to the point where buddingvesicles were generated. Budding vesicle formation wastherefore is inhibited at lower PEO weight fractions. Thereduced hydrophilic PEO weight fraction may have coun-teracted any decreases of interfacial tension that wouldhave been observed otherwise. The influence of a blockcopolymer’s composition upon the interfacial tension ofthe droplets and the subsequent influence on the resultantmorphologies was recently described as part of a review byHayward and Pochan [150].
Hayward and coworkers [153] observed that varyingthe surfactant concentration also affected the interfacialtension of droplets containing PS-b-PEO in a similar man-ner as changing the weight fraction of the PEO block(Fig. 18). With increasing SDS concentration, the resul-tant morphologies of PS-b-PEO (with molecular weights of3.7 × 104 and 6.5 × 103 g mol–1 for the PS and PEO blocks,respectively) particles yielded either spherical particleswith rough surfaces (0.1 mg/mL SDS), budding vesicles(0.2–0.7 mg/mL SDS), and eventually released the copoly-mer as worm-like vesicles (1 mg/mL SDS). At higherSDS concentrations (∼5 mg/mL), spherical micelles werefavored over worm-like micelles [153]. A similar trendwas observed when PVA was used as the surfactant, butmuch higher concentrations were required to obtain a sim-ilar effect as produced by SDS. As mentioned earlier, thegroups of Okubo and workers [142] and Jeon et al. [143]have demonstrated that surfactant composition can directthe morphology of emulsion droplets. It is apparent fromHayward and coworkers’ [153] results that surfactant con-centration may direct this assembly also. Therefore, notonly choice of surfactant, but also its quantity, may directemulsion particle assembly.
Surface protrusions were also observed by Tsapis andcoworkers [156], among O/W emulsions incorporatingblends of poly(lactide-co-glycolide)-block-poly(ethyleneglycol) (PLGA-b-PEG) and poly(lactide-co-glycolide)(hPLGA). The copolymer/homopolymer blends were dis-solved in the oil phase, which was a mixture of DCM andperfluorooctyl bromide (PFOB), and the oil phase wassubsequently evaporated. During the evaporation, DCMwas removed, while the less volatile PFOB remained andphase segregated from the rest of the oil phase as a PFOBdroplet. Unlike DCM, the PFOB was a poor solvent forthe copolymer, and hence the copolymer blend becameconcentrated in the remaining DCM surrounding the PFOBdroplet, thus forming the capsule wall. Therefore, the PFOBdroplets eventually formed the capsule cores. Despite itssolubility in water, it was noted that PEG actually favorsDCM [157,158]. However, at a critical point during theevaporation, insufficient DCM remained to solubilize thePEG chains, which then extended into the aqueous phase.
The interfacial area increases with increasing PLGA-b-PEGconcentration to provide greater contact between theaqueous phase and the PEG blocks. Thus, the need toincrease the surface area of the droplets in order to expose
1174 I. Wyman et al. / Progress in Polymer Science 36 (2011) 1152– 1183
Fig. 15. Bright field TEM images of PS-b-PEO/hPS blends with hPS weight fractions of 60% (a), 70% (b), and 80% (c). At a hPS weight fraction of 60%, a mixtureof cylinders and pearl necklace structures are present. The pearl necklace structures apparently correspond to an intermediate morphology betweencylindrical and spherical structures. The pearl necklace structures became dominant at a weight fraction of 70% hPS. When the weight fraction of hPS wasi e presenA
ttmPhcwmsh
FTW
ncreased further to 80%, both pearl necklace and spherical structures wermerican Chemical Society.
he PEG domains to the aqueous phase apparently drovehe assembly of these protruding structures. The surface
orphologies of the capsules varied depending upon theLGA-b-PEG/hPLGA blend content, with more extensiveair-like protrusions growing from capsules with higheropolymer content (Fig. 19). This behavior was consistent
ith observations by Gref et al. [159], who reported thaticroparticles composed of hPLA and PLA-b-PEG hadmooth and rough surfaces when they were made up ofPLA and PLA-b-PEG, respectively.
ig. 16. Microfluidic device used by Zhu and Hayward to prepare uniform chlorofhe aqueous phase was a 1/1 (v/v) mixture of water and glycerol, and PVA wailey-VCH Verlag GmbH & Co. KGaA. Reproduced with permission.
t [146]. Reprinted with permission from Reference [146]. Copyright 2008
Understanding the transitions that occur with changinginterfacial tension at the surfaces of emulsion droplets canprovide a helpful tool for particle design, and may allowone to tune the surface areas and morphologies of the par-ticles generated. As described above, one can kineticallytrap the breakdown of emulsion droplets at various stages.
This can be accomplished by controlling factors such as theblock distribution within an amphiphilic copolymer, theconcentration and choice of surfactant, the nature of thecopolymer/homopolymer blend, or the length of the evap-orm droplets containing PS-b-PEO suspended in the aqueous phase [147].s dissolved in this phase to stabilize the droplets. Ref. [147]. Copyright

I. Wyman et al. / Progress in Polymer Science 36 (2011) 1152– 1183 1175
Fig. 17. SEM images of (a) PS-b-PEO budding vesicles which formed during chloroform evaporation from emulsion droplets and (b) dendritic particles of aform fr
yright W
blend of PS-b-PEO and hPS which formed during the evaporation of chloroobtained after the samples were stained with RuO4 [147]. Ref. [147]. Coporation time. Careful control of these conditions may yieldparticles with a desired surface area or surface roughnessthrough directed assembly.
5.2. Molecular containers and porous materials fromblock copolymer emulsion spheres
The preparation of supramolecular hosts and porousmaterials is of great interest, particularly if they can be usedas hosts for smaller molecules within the void spaces ontheir surfaces or in their interiors [160,161]. In this way,these materials have potential applications for targetedremoval of toxins or unwanted compounds, or also for drugdelivery systems [125]. They could also be potentially usedas catalytic materials. In some cases, the functional groupsof the polymer lining the void spaces may also serve tofacilitate binding between the host material and the targetmolecule.
Liu and coworkers demonstrated that emulsificationcould be used to generate porous microspheres basedon block copolymers such as PtBA-b-PCEMA or PI-b-PtBA(which were described earlier in Sections 2.3 and 2.4.1,respectively) after the subsequent hydrolysis of the PtBAblock to yield PAA [94,98,162]. The interiors of these micro-spheres contained void channels which were lined withPAA domains that could bind to cations such as Cu2+, Fe3+,Mn2+, and Pd2+ (Fig. 20). The void spaces among the PAA-b-PCEMA microspheres arose from the loss of the tert-butylgroup from the PtBA domains during the hydrolysis [94].
When PCEMA-b-PAA microspheres were stirred with anaqueous Pd(NO3)2 solution, Liu and coworkers [162] deter-mined that as much as 151 mg of Pd2+ could be incorpo-rated into the PAA-lined channels per gram of microsphere.Furthermore, when they reduced the Pd2+ occupying themicrospheres, the spheres became catalytically active, and
could catalyze the hydrogenation of methyl methacrylate(MMA) to methyl 2-methylpropionate. The catalytic per-formance of the microspheres was compared with that ofPd black, and the conversion of MMA was more efficientom the emulsion droplets. The inset images of (a) and (b) are TEM imagesiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission.
when the Pd-bearing spheres were used as catalysts thanwhen Pd black was used.
Porous PI-b-PAA microspheres were prepared in a sim-ilar manner [98]. In this case, the microspheres wereprepared using a PI-b-PtBA/hPtBA blend. Consequently,hPAA could be removed after the hydrolysis step to yieldlarger void spaces within the spheres. In addition, thenature and size of the void regions and PAA domains couldbe tuned by adjusting the amount of hPtBA present duringthe emulsification (Fig. 21). Transition metals such as Fe3+
and Cu2+ could bind to the PAA-lined channels through thePAA carboxyl groups. Therefore, in addition to providingvoid spaces, these microspheres also directed metal load-ing through attractive interactions with the PAA domains.
As mentioned earlier, the preparation of block copoly-mer microspheres through emulsification began with thework of Ogawa, for the purpose of preparing drug deliverysystems [91]. The development of block copolymer-baseddrug delivery vehicles and biomedical devices has gen-erated significant attention over the years, and has beendescribed in numerous reviews [42,115,125,163–170].While a variety of blocks are used, many of these systemsutilize blocks such as PEG and PLA (or their deriva-tives) as hydrophilic and hydrophobic blocks, respectively[159]. Particles coated with PEG often have longer cir-culation times in living systems [110,125,171], due tothe inhibited uptake of PEG-bearing nanoparticles by thereticulo-endothelial system (RES) [125]. This feature canallow PEG-coated particles to circulate long enough todeliver drugs to well-hidden targets, such as hard to reachtumors.
As suggested above, a block-copolymer based deliveryvehicle may enhance a drug’s effectiveness. For exam-ple, Onishi and coworkers [172] loaded the anti-tumordrug camptothecin into micelles of methoxypolyethyleneglycol-block-poly(D,L-lactic acid) (MPEG-b-PLA) following
an O/W emulsion. Both the copolymer and the drug weredissolved into the oil phase, which was composed ofDCM. The PLA block collapsed as the DCM was subse-quently evaporated, to form the resultant particle’s core,
1176 I. Wyman et al. / Progress in Polymer Science 36 (2011) 1152– 1183
Fig. 18. Various images showing the influence of SDS concentration upon the morphologies of droplets containing PS-b-PEO. SEM images are shown inparts (a–c) and bright field TEM images are shown in images (d–f). When the SDS concentration was 0.1 mg/mL, spherical particles with rough surfaceswere observed (a). Meanwhile, when the surfactant was increased to 0.5 (b) and 0.7 mg/mL (c) budding vesicles formed. The copolymer escaped from thedroplets to form worm-like micelles of the copolymer when the SDS concentration was 1.0 mg/mL (d). A combination of worm-like and spherical micelleswere observed when the concentration of SDS was 2.4 mg/mL (e). Predominantly spherical micelles formed when the concentration of SDS was increasedto 5.0 mg/mL (f) [153]. Ref. [153]––Reproduced by permission of the Royal Society of Chemistry.

I. Wyman et al. / Progress in Polymer Science 36 (2011) 1152– 1183 1177
Fig. 19. Bright field images of emulsion-droplets containing PLGA-b-PEO/hPLGA blends during the evaporation of dichloromethane. After approximately1 h, PFOB begins to phase segregate from the rest of the oil phase, and becomes visible (second image from the left). The volume of the PFOB dropletincreases during the evaporation process. Approximately 2.5 h after evaporation began, the surface of the droplet begins to roughen. This effect is enhancedwith increasing weight fractions of the copolymer. The weight fractions of PLGA-b-PEO within the blend shown in the three images at the top right andbottom right are 5% and 100%, respectively [156]. Ref. [156]––Reproduced by permission of the Royal Society of Chemistry.
Fig. 20. TEM images of PCEMA-b-PtBA microspheres prior to Pd loading (a). Microspheres that have been loaded with 27% (b) and 63% (c) weight percentagesof Pd are also shown [162]. Reprinted with permission from Reference [162]. Copyright 2001 American Chemical Society.

1178 I. Wyman et al. / Progress in Polymer Science 36 (2011) 1152– 1183
Fig. 21. TEM images of spheres prepared from PI-b-PtBA/hPtBA blends after the hydrolysis of PtBA to PAA. The spheres on the left initially had a 32% volumefraction of PtBA (including both hPtBA and the PtBA block), while the spheres on the left initially had a 54% volume fraction of PtBA the blend. The samplesw g after ht le the w[ n Wiley
wtdoTctotdcdmi
tpclvibeutbdppheboaiWiue
ere not stained, and the lighter regions represent void spaces remainino the PI domains, the light grey regions correspond to PAA domains, whi98]. Reprinted with permission from Reference [98]. Copyright 2003 Joh
hile the MPEG blocks extended into the aqueous solu-ion to form the corona. The hydrophobic effect helpedirect the host–guest encapsulation, with camptothecinccupying the hydrophobic PLA domains of the particles.he activity of these particles against sarcoma 180 tumorells was studied by injecting tumor-bearing mice withhe camptothecin-loaded MPEG-b-PLA nanoparticles, andther mice with free solutions of the drug. The camp-othecin was retained in the blood plasma for a longeruration among the mice injected with the nanoparti-les than among those injected with free solutions of therug. In addition, the volumes of the tumors in theseice decreased more dramatically compared with those
njected with the free drug [172].The confinement-induced self-assembly that is attained
hrough emulsification provides a powerful means to pre-are a variety of exotic structures. Choosing a block thatan be readily modified so that it may consequently col-apse or occupy a smaller volume, can yield particles withoid spaces [94,162]. Larger void spaces may be obtainedf a homopolymer is incorporated with the copolymer as alend, especially if the homopolymer can be subsequentlyxtracted from the particles [98]. In this scenario, the vol-me of the void spaces may be controlled by adjustinghe homopolymer content. While the preparation of thelock copolymer particles themselves are directed by con-itions such as confinement, interfacial tension, or otherhenomena, the incorporation of guest species into a hostarticle can also arise through directed assembly. Thisost–guest assembly may be driven by the hydrophobicffect, or other supramolecular activity such as hydrogenonding, coordination bonding, electrostatic interactions,r �–� interactions. The block copolymer particle can acts a template to induce guest incorporation either withints interior or onto its surfaces through directed assembly.
hile these structures are often complex and fascinatingn their own right, they also show very strong promise andtility for applications, such as for catalysts and drug deliv-ry systems.
ydrolysis of PtBA and extraction of PtBA. The darker regions correspondhite regions likely correspond to void spaces left after extraction of hPAA
and Sons.
6. Perspectives and outlook
The combination of block copolymer self-assembly andemulsification can provide a rich variety of morphologiesand exciting applications. Complex architectures have beenobtained both within the interiors of emulsion droplets,and also on droplet surfaces. A common paradigm followedfor many preparations of block copolymer assembliesthrough emulsification is to dissolve the copolymer in avolatile organic solvent and mix this oil phase with an aque-ous phase, which normally contains a surfactant to stabilizethe emulsion droplets. These immiscible phases are thenmixed together, normally by stirring or sonication, to forman emulsion. Organic solvent is then removed from theemulsion droplets via evaporation to induce collapse of atleast one of the copolymer’s blocks, leading to assemblyof the copolymer. Amphiphilic block copolymers are nat-urally well-adapted towards emulsification. In fact, theircombination of hydrophilic and hydrophobic blocks allowthem to behave as surfactants, thus sometimes allowingemulsion stabilization without requiring use of additionalsurfactant [119]. In addition, their amphiphilic nature canyield a variety of exotic morphologies through microphasesegregation, depending on the prevailing conditions.
6.1. Microphase segregation within solid emulsionparticles
If both blocks collapse during solvent evaporation froman emulsion droplet, they may form the core of the resul-tant emulsion sphere and undergo microphase segregation.As seen in Section 2, there are a number of ways in whichtheir morphology may be controlled. Since the blocks areconfined within the emulsion sphere, D/L0 can play animportant role on influencing the architecture [88,107]. As
is the case in bulk, the volume fractions of the copolymerblocks is a major factor in determining the morphology ofthe copolymer assembly. A popular way to tune the mor-phology is through the use of copolymer/homopolymer
olymer S
I. Wyman et al. / Progress in Pblends. Adjusting the homopolymer content may have asimilar effect as changing the block ratio of the copoly-mer, and may thus induce a morphological transition.As is the case in bulk, incremental changes of copoly-mer/homopolymer blend composition can allow one tofine tune the morphologies produced through emulsifica-tion [102]. Macrophase segregation may also occur in thepresence of a homopolymer, particularly as its molecularweight becomes comparable with its corresponding blocksor if large amounts of homopolymer are present. Withinemulsion spheres, a combination of these factors may influ-ence the final morphology so that it will be unique fromthose observed either in bulk, or under a single influence.
6.2. Block copolymer vesicles through emulsification
If the block copolymer assembles at the interface sur-rounding an emulsion droplet, and at least one of theblocks collapses upon solvent evaporation, the copolymercan form a vesicle. Water-filled polymersomes may be pre-pared if the copolymer is initially dissolved in the oil phaseof a (W/O)/W double emulsion [123]. The inner aqueousdroplet effectively serves as a template, with the copoly-mer behaving as a surfactant and assembling along thedroplet surface, as well as at the interface between the oilphase and the surrounding aqueous solution as the organicsolvent is removed. The use of microfluidic devices allowsproducion of monodisperse particles with easily controlleddiameters. Alternatively, nanocapsules with oil-filled coresmay be prepared by O/W emulsion [118]. The oil phase willoften be a mixture of two organic solvents, one of whichis volatile, and the other non-volatile. At least one of theblocks should be insoluble in the non-volatile solvent. Uponevaporation of the volatile solvent, the non-volatile solventremains to form the oil-filled core of the capsule. The oil-filled core has a different composition than the surroundingsolution. This feature differs from that of polymersomes,where both the vesicle core and the surrounding solutionare aqueous media. In both cases, the inner droplet acts asa template for copolymer assembly.
6.3. Block copolymer assembly at 2D surfaces
Block copolymer assembly may also occur on the sur-face of emulsion spheres with solid cores. The directionof these assemblies are quite similar to those involved informing vesicles, with the block-copolymer assembly tak-ing place at the interface between the surface of a dropletor a surface and the surrounding media. In contrast todouble emulsions, this assembly normally occurs at oneinterface rather than two. However, hierarchical assemblymay also be observed, such as the flower-like superag-gregates and ribbon-cage structures reported by Liu andcoworkers [141]. One may prepare particles with patchy orbumpy surfaces if two copolymers with similar hydropho-bic blocks and different hydrophilic blocks assemble on anO/W droplet surface, and subsequently segregate them-
selves according to their hydrophilic block [140].The surfactant may direct the assembly, even if it is notincorporated into the final assembly structure. If a surfac-tant is more compatible with one copolymer block than
cience 36 (2011) 1152– 1183 1179
another, emulsion spheres with uniform surfaces result,with the choice of surfactant dictating the surface compo-sition [142]. Alternatively, a mixture of surfactants, whereeach surfactant is more compatible with a different copoly-mer block, can yield particles with more than one of theblocks exposed to the surface, and may even produce non-spherical particles [143].
6.4. Exotic and useful structures
A diverse variety of block copolymer architectures areavailable through emulsification, some of which would bedifficult or impossible to obtain through other approaches.The combination of directing influences that one maymanipulate in block copolymer emulsification providesvirtually endless possibilities. In addition to fascinatingmorphologies, emulsification can also yield block copoly-mer assemblies with various applications. In fact, emulsionparticles derived from copolymers were first prepared byOgawa et al. [91], for the purpose of developing drug deliv-ery vehicles. Since that time, this has been a major area ofresearch involving these particles. In addition to particleswith solid cores, vesicles and polymersomes have attractedgreat interest as well [173]. Catalytic materials may be pre-pared through this route also, such as porous emulsionspheres [162]. Control of surface roughness via assembly ofblock copolymer surfactants [140], or by interfacial insta-bility [146], provide promising routes towards developingcatalytic surfaces.
6.5. Outlook
Given the developments made in recent years, thepotential for many further discoveries and applicationsto be developed in the future. Recent systematic stud-ies [103,107,123,142,143,146] summarized in this reviewhave furthered our understanding of block copolymerassembly occurring during the emulsification/solventevaporation process, and how the assembly can be con-trolled. With this knowledge in hand, emulsification canprovide a powerful method to tailor block copolymer archi-tectures according to desired structures or applications.Many of the assembly systems described have been basedon diblock copolymers, although triblock copolymers haveused as well [118,130,141]. Given the greater morpho-logical diversity of ABC triblock copolymers, their furtheruse (as well as tetrablock and pentablock copolymers)in emulsification may generate highly complex assemblystructures.
Acknowledgements
NSERC of Canada is gratefully acknowledged for fundingthis work. Guojun Liu wishes to thank the Canada ResearchChair program for a senior Canada Research Chair positionin Materials Science.
References
[1] Bates FS, Fredrickson GH. Block copolymers–designer soft materi-als. Phys Today 1999;52:32–8.

1 olymer S
180 I. Wyman et al. / Progress in P[2] Ding J, Liu GJ. Polyisoprene-block-poly(2-cinnamoylethylmethacrylate) vesicles and their aggregates. Macromolecules1997;30:655–7.
[3] Yan X, Liu G, Li Z. Preparation and phase segregation ofblock copolymer nanotube multiblocks. J Am Chem Soc2004;126:10059–66.
[4] Khandpur AK, Forster S, Bates FS, Hamley IW, Ryan AJ, Bras W, Alm-dal K, Mortensen K. Polyisoprene-polystyrene diblock copolymerphase diagram near the order-disorder transition. Macromolecules1995;28:8796–806.
[5] Hamley IW. Block copolymers. Oxford: Oxford University Press;1999.
[6] Hasegawa H, Hashimoto T. Self-assembly and morphology of blockcopolymer systems. In: Allen G, Aggarwal SL, Russo S, editors.Comprehensive polymer science. Second supplement. London:Pergamon Press; 1996. p. 497–539.
[7] Breiner U, Krappe U, Thomas EL, Stadler R. Structural characteriza-tion of the “knitting pattern” in polystyrene-block-poly(ethylene-co-butylene)-block-poly(methyl methacrylate) triblock copoly-mers. Macromolecules 1998;31:135–41.
[8] Hajduk DA, Harper PE, Grunner SM, Honeker CC, Kim G,Thomas EL, Fetters LJ. The gyroid: a new equilibrium morphol-ogy in weakly segregated diblock copolymers. Macromolecules1994;27:4063–75.
[9] Hashimoto T, Kawamura T, Harada M, Tanaka H. Small-anglescattering from hexagonally packed cylindrical particles withparacrystalline distortion. Macromolecules 1994;27:3063–72.
[10] Shefelbine TA, Vigild ME, Matsen MW, Hajduk DA, HillourerMA, Cussler EL, Bates FS. Core-shell gyroid morphology ina poly(isoprene-block-styrene-block-dimethylsiloxane) triblockcopolymer. J Am Chem Soc 1999;121:8457–65.
[11] Breiner U, Krappe U, Abetz V, Stadler R. Cylindrical morpholo-gies in asymmetric ABC triblock copolymers. Macromol Chem Phys1997;198:1051–83.
[12] Matsen MW, Schick M. Stable and unstable phases of a diblockcopolymer melt. Phys Rev Lett 1994;72:2660–3.
[13] Matsen MW, Schick M. Microphase separation in starblock copoly-mer melts. Macromolecules 1994;27:6761–7.
[14] Matsen MW, Schick M. Stable and unstable phases of a linear multi-block copolymer melt. Macromolecules 1994;27:7157–63.
[15] Leibler L. Theory of microphase separation in block copolymers.Macromolecules 1980;13:1602–17.
[16] Helfand E, Wasserman ZR. Block copolymer theory. 4. Narrow inter-phase approximation. Macromolecules 1976;9:879–88.
[17] Whitmore MD, Noolandi J. Self-consistent theory of block copoly-mer blends: neutral solvent. J Chem Phys 1990;93:2946–55.
[18] Semenov AN. Contribution to the theory of microphase layering inblock-copolymer melts. Sov Phys JETP 1985;61:733–42.
[19] Qi S, Wang ZG. Kinetics of phase transitions in weakly segregatedblock copolymers: pseudostable and transient states. Phys Rev E1997;55:1682–97.
[20] Matsen MW, Bates FS. Unifying weak- and strong-segregation blockcopolymer theories. Macromolecules 1996;29:1091–8.
[21] Cameron NS, Corbierre MK, Eisenberg A, Steacie EWR. Award Lec-ture: asymmetric amphiphilic block copolymers in solution: amorphological wonderland. Can J Chem 1999 1998;77:1311–26.
[22] Zhang L, Eisenberg A. Multiple morphologies of “crew-cut” aggre-gates of polystyrene-b-poly(acrylic acid) block copolymers. Science1995;268:1728–31.
[23] Dupont J, Liu GJ, Niihara KI, Kimoto R, Jinnai H. Self-assembled ABCtriblock copolymer double and triple helices. Angew Chem Int Ed2009;48:6144–7.
[24] Li Z, Liu GJ. Water-dispersible tetrablock copolymer synthesis,aggregation, nanotube preparation, and impregnation. Langmuir2003;19:10480–6.
[25] Gohy JF, Ott C, Hoeppener S, Schubert US. Multicompartmentmicelles from a metallo-supramolecular tetrablock quatercopoly-mer. Chem Commun 2009:6038–40.
[26] Hoogenboom R, Wiesbrock F, Leenen MAM, Thijs HML, Huang H,Fustin CA, Guillet P, Gohy JF, Schubert US. Synthesis and aqueousmicellization of amphiphilic tetrablock ter- and quarterpoly(2-oxazoline)s. Macromolecules 2007;40:2837–43.
[27] Fustin CA, Thijs-Lambermont HML, Hoeppener S, Hoogenboom R,Schubert US, Gohy JF. Multiple micellar morphologies from tri- and
tetrablock copoly(2-oxazoline)s in binary water-ethanol mixtures.J Polym Sci Part A Polym Chem 2010;48:3095–102.[28] Takahashi K, Hasegawa H, Hashimoto T, Bellas V, Iatrou H, Had-jichristidis N. Four-phase triple coaxial cylindrical microdomainmorphology in a linear tetrablock quaterpolymer of styrene, iso-
cience 36 (2011) 1152– 1183
prene, dimethylsiloxane, and 2-vinylpyridine. Macromolecules2002;35:4859–61.
[29] Kim HC, Park SM, Hinsberg W. Block copolymer based nanostruc-tures: materials, processes, and applications to electronics. ChemRev 2010;110:146–77.
[30] Thurn-Albrecht T, Schotter J, Kastle GA, Emley N, Shibauchi T,Krusin-Elbaum L, Guarini K, Black CT, Tuominen MT, RussellTP. Ultrahigh-density nanowire arrays grown in self-assembleddiblock copolymer templates. Science 2000;290:2126–9.
[31] Massey JA, Winnik MA, Manners I, Chan VZH, OstermannJM, Enchelmaier R, Spatz JP, Moller M. Fabrication of ori-ented nanoscopic ceramic lines from cylindrical micelles of anorganometallic polyferrocene block copolymer. J Am Chem Soc2001;123:3147–8.
[32] Liu GJ, Yan X, Li Z, Zhou J, Duncan S. End coupling of block copolymernanotubes to nanospheres. J Am Chem Soc 2003;125:14039–45.
[33] Huang W, Kim JB, Baker GL, Bruening ML. Preparation ofamphiphilic triblock copolymer brushes for surface patterning.Nanotechnology 2003;14:1075–80.
[34] Peng J, Xuan Y, Wang H, Yang Y, Li B, Han Y. Solvent-induced microphase separation in diblock copolymer thinfilms with reversibly switchable morphology. J Chem Phys2004;120:11163–70.
[35] Lazzari M, Scalarone D, Hoppe CE, Vazquez-Vazquez C, Lopez-Quintela MA. Tunable polyacrylonitrile-based micellar aggregatesas a potential tool for the fabrication of carbon nanofibers. ChemMater 2007;19:5818–20.
[36] Pietsch T, Gindy N, Mahltig B, Fahmi A. Fabrication of functionalnano-objects via self-assembly of nanostructured hybrid materials.J Polym Sci Part B Polym Phys 2010;48:1642–50.
[37] Quemener D, Bonniol G, Phan TNT, Gigmes D, Bertin D, Deratani A.Free-standing nanomaterials from block copolymer self-assembly.Macromolecules 2010;43:5060–5.
[38] Li Z, Zhao W, Liu Y, Rafailovich MH, Sokolov J, Khougaz K, EisenbergA, Lennox RB, Krausch G. Self-ordering of diblock copolymers fromsolution. J Am Chem Soc 1996;118:10892–3.
[39] Park M, Harrison C, Chaikin PM, Register RA, Adamson DH. Blockcopolymer lithography: periodic arrays of ∼1011 holes in 1 squarecentimeter. Science 1997;276:1401–4.
[40] Xu J, Park S, Wang S, Russell TP, Ocko BM, Checco A. Directedself-assembly of block copolymers on two-dimensional chemi-cal patterns fabricated by electro-oxidation nanolithography. AdvMater 2010;22:2268–72.
[41] Silva GA, Czeisler C, Niece KL, Beniash E, Harrington DA, KesslerJA, Stupp SI. Selective differentiation of neural progenitor cells byhigh-epitope density nanofibers. Science 2004;303:1352–5.
[42] Kataoka K, Harada A, Nagasaki Y. Block copolymer micelles for drugdelivery: design, characterization and biological significance. AdvDrug Delivery Rev 2001;47:113–31.
[43] Kabanov AV, Batrakova EA, Alakhov VY. Pluronic® block copoly-mers as novel therapeutics for drug and gene delivery. J ControlledRelease 2002;82:189–212.
[44] Vriezema DM, Aragones MC, Elemans JAAW, Cornelissen JJLM,Rowan AE, Nolte RJM. Self-assembled nanoreactors. Chem Rev2005;105:1445–89.
[45] Zhang L, Bartels C, Yu Y, Shen H, Eisenberg A. Mesosized crystal-like structure of hexagonally packed hollow hoops by solution self-assembly of diblock copolymers. Phys Rev Lett 1997;79:5034–7.
[46] Ding JF, Liu GJ. Growth and morphology change of polystyrene-block-poly(2-cinnamoylethyl methacrylate) particles insolvent-nonsolvent mixtures before precipitation. Macro-molecules 1999;32:8413–20.
[47] Darling SB. Directing the self-assembly of block copolymers. ProgPolym Sci 2007;32:1152–204.
[48] Elhadj S, Woody JW, Niu VS, Saraf RF. Orientation of self-assembled block copolymer cylinders perpendicular to electricfield in mesoscale film. Appl Phys Lett 2003;82:871–3.
[49] Olszowka V, Hund M, Kuntermann V, Scherdel S, Tsarkova L, BokerA, Krausch G. Large scale alignment of a lamellar block copolymerthin film via electric fields: a time resolved SFM study. Soft Matter2006;2:1089–94.
[50] Park S, Kim B, Xu J, Hofmann T, Ocko BM, Russell TP. Lateral orderingof cylindrical microdomains under solvent vapor. Macromolecules2009;42:1278–84.
[51] Cheng JY, Ross CA, Smith HI, Thomas EL. Templated self-assemblyof block copolymers: top-down helps bottom-up. Adv Mater2006;18:2505–21.
[52] Li Z, Chen Z, Cui H, Hales K, Qi K, Wooley KL, Pochan DJ.Disk morphology and disk-to-cylinder tunability of poly(acrylic

olymer S
I. Wyman et al. / Progress in Pacid)-b-poly(methyl acrylate)-b-polystyrene triblock copolymersolution-state assemblies. Langmuir 2005;21:7533–9.
[53] Hales K, Chen Z, Wooley KL, Pochan DJ. Nanoparticles with tunableinternal structure from triblock copolymers of PAA-b-PMA-b-PS.Nano Lett 2008;7:2023–6.
[54] Bibette J, Calderon FL, Poulin P. Emulsions: basic principles. RepProg Phys 1999;62:969–1033.
[55] Friberg SE. Some emulsion features. J Dispersion Sci Technol2007;28:1299–308.
[56] Leal-Calderon F, Thivilliers F, Schmitt V. Structured emulsions. CurrOpin Colloid Interface Sci 2007;12:206–12.
[57] Salager JL, Forgiarini A, Marquez L, Pena A, Pizzino A, RodriguezMP, Rondon-Gonzalez M. Using emulsion inversion in industrialprocesses. Adv Colloid Interface Sci 2004;108–109:259–72.
[58] Schwuger MJ, Stickdorn K, Schomacker R. Microemulsions in tech-nical processes. Chem Rev 1995;95:849–64.
[59] Solans C, Izquierdo P, Nolla J, Azemar N, Garcia-Celma MJ. Nano-emulsions. Curr Opin Colloid Interface Sci 2005;10:102–10.
[60] Matsen MW, Schick M. Self-assembly of block copolymers. CurrOpin Colloid Interface Sci 1996;1:329–36.
[61] Noolandi J, Shi AC. Self-assembly of block copolymers. Curr OpinColloid Interface Sci 1998;3:436–9.
[62] Riess G. Micellization of block copolymers. Prog Polym Sci2003;28:1107–70.
[63] Park C, Yoon J, Thomas EL. Enabling nanotechnology with selfassembled block copolymer patterns. Polymer 2003;44:6725–60.
[64] Smart T, Lomas H, Massignani M, Flores-Merino MV, PerezLR, Battaglia G. Block copolymer nanostructures. Nano Today2008;3:38–46.
[65] Kim JK, Yang SY, Lee Y, Kim Y. Functional nanomaterials based onblock copolymer self-assembly. Prog Polym Sci 2010;35:1325–49.
[66] Grzelczak M, Vermant J, Furst EM, Liz-Marzan LM. Directing theself-assembly of nanoparticles. ACS Nano 2010;4:3591–605.
[67] Fasolka MJ, Mayes AM. Block copolymer thin films: physics andapplications. Annu Rev Mater Res 2001;31:323–55.
[68] Hamley IW. Ordering in thin films of block copolymers: fundamen-tals to potential applications. Prog Polym Sci 2009;34:1161–210.
[69] Tsarkova L, Sevink GJA, Krausch G. Nanopattern evolution in blockcopolymer films: experiment, simulations and challenges. AdvPolym Sci 2010;227:33–73.
[70] Yu G, Cho W, Shin K. Polymers in nanotubes. In: Dunne LJ, Manos G,editors. Adsorption and phase behaviour in nanochannels and nan-otubes. Dordrecht: Springer Science + Business Media B.V.; 2010.p. 101–19.
[71] Stewart-Sloan CR, Thomas EL. Interplay of symmetries ofblock copolymers and confining geometries. Eur Polym J2011;47:630–46.
[72] Thomas EL, Reffner JR, Bellare J. A menagerie of interface structuresin copolymer systems. J Phys Colloq 1990;51:c7363–74.
[73] Zhang K, Yu X, Gao L, Chen Y, Yang Z. Mesostructured spheresof organic/inorganic hybrid from gelable block copolymers andarched nano-objects thereof. Langmuir 2008;24:6542–8.
[74] Zhang K, Gao L, Chen Y, Yang Z. Onionlike spherical polymercomposites with controlled dispersion of gold nanoclusters. ChemMater 2008;20:23–5.
[75] Arsenault AC, Rider DA, Tetreault N, Chen JIL, Coombs N, OzinGA, Manners I. Block copolymers under periodic, strong three-dimensional confinement. J Am Chem Soc 2005;127:9954–5.
[76] Rider DA, Chen JIL, Eloi JC, Arsenault AC, Russell TP, Ozin GA,Manners I. Controlling the morphologies of organometallic blockcopolymers in the 3-dimensional spatial confinement of col-loidal and inverse colloidal systems. Macromolecules 2008;41:2250–9.
[77] Yabu H, Higuchi T, Shimomura M. Unique phase-separationstructures of block copolymer nanoparticles. Adv Mater2005;17:2062–5.
[78] Yabu H, Higuchi T, Ijiro K, Shimomura M. Spontaneous formation ofpolymer nanoparticles by good-solvent evaporation as a nonequi-librium process. Chaos 2005;15, 047505/1-7.
[79] Higuchi T, Tajima A, Motoyoshi K, Yabu H, Shimomura M. Frustratedphases of block copolymers in nanoparticles. Angew Chem Int Ed2008;47:8044–6.
[80] Li L, Matsunaga K, Zhu J, Higuchi T, Yabu H, Shimomura M, Jin-nai H, Hayward RC, Russell TP. Solvent-driven evolution of block
copolymer morphology under 3D confinement. Macromolecules2010;43:7807–12.[81] Higuchi T, Tajima A, Yabu H, Shimomura M. Spontaneous forma-tion of polymer nanoparticles with inner micro-phase separationstructures. Soft Matter 2008;4:1302–5.
cience 36 (2011) 1152– 1183 1181
[82] Yabu H, Motoyoshi K, Higuchi T, Shimomura M. Hierarchical struc-tures in AB/BC type diblock-copolymer blend particles. Phys ChemChem Phys 2010;12:11944–7.
[83] Laidler KL, Meiser JH. Physical chemistry. 2nd Ed. Boston: HoughtonMifflin Company; 1995. pp. 862–863.
[84] Xiang H, Shin K, Kim T, Moon S, McCarthy TJ, Russell TP. The influ-ence of confinement and curvature on the morphology of blockcopolymers. J Polym Sci Part B Polym Phys 2005;43:3377–83.
[85] He X, Song M, Liang H, Pan C. Self-assembly of the symmetricdiblock copolymer in a confined state: Monte Carlo simulation. JChem Phys 2001;114:10510–3.
[86] Fraaije JGEM, Sevink GJA. Model for pattern formation in polymersurfactant nanodroplets. Macromolecules 2003;36:7891–3.
[87] Feng J, Liu H, Hu Y. Microphase separation of diblock copolymer ina nanosphere: dissipative particle dynamics approach. Fluid PhaseEquilib 2007;261:50–7.
[88] Yu B, Li B, Jin Q, Ding D, Shi AC. Self-assembly of symmetric diblockcopolymers confined in spherical nanopores. Macromolecules2007;40:9133–42.
[89] Chen P, Liang H, Shi AC. Microstructures of a cylinder-formingdiblock copolymer under spherical confinement. Macromolecules2008;41:8938–43.
[90] Li S, Chen P, Zhang L, Liang H. Geometric frustration phases ofdiblock copolymers in nanoparticles. Langmuir 2011;27:5081–9.
[91] Ogawa Y, Yamamoto M, Okada H, Yashiki T, Shimamoto T. A newtechnique to efficiently entrap leuprolide acetate into microcap-sules of polylactic acid or copoly(lactic/glycolic acid). Chem PharmBull 1988;36:1095–103.
[92] Vrancken MN. Process for encapsulating water and compounds inaqueous phase by evaporation. U.S. Patent 3,523,906; 1970.
[93] Ouchi T, Hamada A, Ohya Y. Biodegradable microspheres havingreactive groups prepared from L-lactic acid-depsipeptide copoly-mers. Macromol Chem Phys 1999;200:436–41.
[94] Lu Z, Liu GJ, Liu F. Block copolymer microspheres containingintricate nanometer-sized segregation patterns. Macromolecules2001;34:8814–7.
[95] Liu GJ, Ding J, Hashimoto T, Kimishima K, Winnik FM, Nigam S. Thinfilms with densely, regularly packed nanochannels: preparation,characterization, and applications. Chem Mater 1999;11:2230–40.
[96] Leibler L, Orland H, Wheeler JC. Theory of critical micelleconcentration for solutions of block copolymers. J Chem Phys1983;79:3550–7.
[97] Ruzette AV, Leibler L. Block copolymers in tomorrow’s plastics. NatMater 2005;4:19–31.
[98] Lu Z, Liu GJ, Liu F. Water-dispersible porous polyisoprene-block-poly(acrylic acid) microspheres. J Appl Polym Sci 2003;90:2785–93.
[99] Hashimoto T, Tanaka H, Hasegawa H. Ordered structure in mixturesof a block copolymer and homopolymers. 2. Effects of molecularweights of homopolymers. Macromolecules 1990;23:4378–86.
[100] Tanaka H, Hasegawa H, Hashimoto T. Ordered structure inmixtures of a block copolymer and homopolymers. 1. Solubiliza-tion of low molecular weight homopolymers. Macromolecules1991;24:240–51.
[101] Winey KI, Thomas EL, Fetters LJ. Ordered morphologies in binaryblends of diblock copolymer and homopolymer and character-ization of their intermaterial dividing surfaces. J Chem Phys1991;95:9367–75.
[102] Okubo M, Saito N, Takekoh R, Kobayashi H. Morphologyof polystyrene/polystyrene-block-poly(methyl methacry-late)/poly(methyl methacrylate) composite particles. Polymer2005;46:1151–6.
[103] Tanaka T, Saito N, Okubo M. Control of layer thickness of onionlikemultilayered composite polymer particles prepared by the solventevaporation method. Macromolecules 2009;42:7423–9.
[104] Hong KM, Noolandi J. Theory of phase equilibriums in systems con-taining block copolymers. Macromolecules 1983;16:1083–93.
[105] Roe RJ, Zin WC. Phase equilibria and transition in mixtures of ahomopolymer and a block copolymer. 2. Phase diagram. Macro-molecules 1984;17:189–94.
[106] Hashimoto H, Fujimora M, Hashimoto T, Kawai H. Domain-boundary structure of styrene-isoprene block copolymer filmscast from solutions. 7. Quantitative studies of solubilization ofhomopolymers in spherical domain systems. Macromolecules1981;14:844–51.
[107] Jeon SJ, Yi GR, Koo CM, Yang SM. Nanostructures insidecolloidal particles of block copolymer/homopolymer blends.Macromolecules 2007;40:8430–9.
[108] Matsen MW. Phase behavior of block copolymer/homopolymerblends. Macromolecules 1995;23:5765–73.

1 olymer S
182 I. Wyman et al. / Progress in P[109] Dobriyal P, Xiang H, Kazuyuki M, Chen JT, Jinnai H, RussellTP. Cylindrical confined diblock copolymers. Macromolecules2009;42:9082–8.
[110] Gref R, Minamitake Y, Peracchia MT, Trubetskoy V, Torchilin V,Langer R. Biodegradable long-circulating polymeric nanospheres.Science 1994;263:1600–3.
[111] Blanazs A, Armes SP, Ryan AJ. Self-assembled block copolymeraggregates: from micelles to vesicles and their biological applica-tions. Macromol Rapid Commun 2009;30:267–77.
[112] Kita-Tokarczyk K, Grumelard J, Haefele T, Meier W. Block copoly-mer vesicles––using concepts from polymer chemistry to mimicbiomembranes. Polymer 2005;46:3540–63.
[113] Yow HN, Routh AF. Formation of liquid core-polymer shell micro-capsules. Soft Matter 2006;2:940–9.
[114] Discher DE, Ahmed F. Polymersomes. Annu Rev Biomed Eng2006;8:323–41.
[115] Mora-Huertas CE, Fessi H, Elaissari A. Polymer-based nanocapsulesfor drug delivery. Int J Pharm 2010;385:113–42.
[116] Discher DE, Ortiz V, Srinivas G, Klein ML, Kim Y, Christian D, CaiS, Photos P, Ahmed F. Emerging applications of polymersomes indelivery: from molecular dynamics to shrinkage of tumors. ProgPolym Sci 2007;32:838–57.
[117] Massignani M, Lomas H, Battaglia G. Polymersomes: a syntheticbiological approach to encapsulation and delivery. Adv Polym Sci2010;229:115–54.
[118] Zheng R, Liu GJ. Water-dispersible oil-filled ABC triblock copolymervesicles and nanochannels. Macromolecules 2007;40:5116–21.
[119] Discher BM, Won YY, Ege DS, Lee JCM, Bates FS, Discher DE, HammerDA. Polymersomes: tough vesicles made from diblock copolymers.Science 1999;284:1143–6.
[120] Cho HK, Cheong IW, Lee JM, Kim JH. Polymeric nanoparticles,micelles and polymersomes from amphiphilic block copolymer.Korean J Chem Eng 2010;27:731–40.
[121] Discher DE, Eisenberg A. Polymer vesicles. Science2002;297:967–73.
[122] Lorenceau E, Utada AS, Link DR, Cristobal G, Joanicot M, Weitz DA.Generation of polymersomes from double-emulsions. Langmuir2005;21:9183–6.
[123] Utada AS, Lorenceau E, Link DR, Kaplan PD, Stone HA, Weitz DA.Monodisperse double emulsions generated from a microcapillarydevice. Science 2005;308:537–41.
[124] Hayward RC, Utada AS, Dan N, Weitz DA. Dewetting instabilityduring the formation of polymersomes from block-copolymer-stabilized double emulsions. Langmuir 2006;22:4457–61.
[125] Letchford K, Burt H. A review of the formation of amphiphilic blockcopolymer nanoparticulate structures: micelles, nanospheres,nanocapsules and polymersomes. Eur J Pharm Biopharm2007;65:259–69.
[126] Jain JP, Kumar N. Self assembly of amphiphilic (PEG)3-PLA copoly-mer as polymersomes: preparation, characterization, and theirevaluation as drug carrier. Biomacromolecules 2010;11:1027–35.
[127] Ding J, Liu GJ. Water-soluble hollow nanospheres as potential drugcarriers. J Phys Chem B 1998;102:6107–13.
[128] Fallouh NAK, Roblot-Treupel L, Fessi H, Devissaguet JP, Puisieux F.Development of a new process for the manufacture of polyisobutyl-cyanoacrylate nanocapsules. Int J Pharm 1986;28:125–32.
[129] Bae KH, Lee Y, Park TG. Oil-encapsulating PEO-PPO-PEO/PEG shellcross-linked nanocapsules for target-specific delivery of paclitaxel.Biomacromolecules 2007;8:650–6.
[130] Shim JW, Kim SH, Jeon SJ, Yang SM, Yi GR. Microcapsules withtailored nanostructures by microphase separation of block copoly-mers. Chem Mater 2010;22:5593–600.
[131] Huebner A, Sharma S, Srisa-Art M, Hollfelder F, Edel JB, deMello AJ.Microdroplets: a sea of applications? Lab Chip 2008;8:1244–54.
[132] Seo M, Nie Z, Xu S, Mok M, Lewis PC, Graham R, Kumacheva E.Continuous microfluidic reactors for polymer particles. Langmuir2005;21:11614–22.
[133] Shum HC, Kim JW, Weitz DA. Microfluidic fabrication of monodis-perse biocompatible and biodegradable polymersomes withcontrolled permeability. J Am Chem Soc 2008;130:9543–9.
[134] Shum HC, Santanach-Carreras E, Kim JW, Ehrlicher A, Bibette J,Weitz DA. Dewetting-induced membrane formation by adhesionof amphiphile-laden devices. J Am Chem Soc 2011;133:4420–6.
[135] Shum HC, Zhao YJ, Kim SH, Weitz DA. Multicompartment
polymersomes from double emulsions. Angew Chem Int Ed2011;50:1648–51.[136] Sun BJ, Shum HC, Holtze C, Weitz DA. Microfluidic melt emulsi-fication for encapsulation and release of actives. ACS Appl MaterInterfaces 2010;2:3411–6.
cience 36 (2011) 1152– 1183
[137] Shum HC, Abate AR, Lee D, Studart AR, Wang W, Chen CH,Thiele J, Shah RK, Krummel A, Weitz DA. Droplet microfluidics forfabrication of non-spherical particles. Macromol Rapid Commun2010;31:108–18.
[138] Thiele J, Abate AR, Shum HC, Bachtler S, Forster S, Weitz DA.Fabrication of polymersomes using double emulsion templates inglass-coated stamped microfluidic devices. Small 2010;6:1723–7.
[139] Perro A, Nicolet C, Angly J, Lecommandoux S, Le Meins JF, ColinA. Mastering a double emulsion in a simple co-flow microfluidicto generate complex polymersomes. Langmuir 2011, online ASAParticle, doi:10.1021/la1037102.
[140] Zheng R, Liu GJ, Yan X. Polymer nano- and microsphereswith bumpy and chain-segregated surfaces. J Am Chem Soc2005;127:15358–9.
[141] Hu J, Liu GJ, Nijkang G. Hierarchical interfacial assembly of ABCtriblock copolymer. J Am Chem Soc 2008;130:3236–7.
[142] Saito N, Takekoh R, Nakatsuru R, Okubo M. Effect of stabilizer on for-mation of “onionlike” multilayered polystyrene-block-poly(methylmethacrylate) particles. Langmuir 2007;23:5978–83.
[143] Jeon SJ, Yi GR, Yang SM. Cooperative assembly of block copolymerswith deformable interfaces: toward nanostructured particles. AdvMater 2008;20:4103–8.
[144] Fredrickson GH, Binder K. Kinetics of metastable states in blockcopolymer melts. J Chem Phys 1989;91:7265–75.
[145] Sevink GJA, Zvelindovsky AV. Self-assembly of complex vesicles.Macromolecules 2005;38:7502–13.
[146] Zhu J, Hayward RC. Spontaneous generation of amphiphilic blockcopolymer micelles with multiple morphologies through interfa-cial instabilities. J Am Chem Soc 2008;130:7496–502.
[147] Zhu J, Hayward RC. Hierarchically structured microparticlesformed by interfacial instabilities of emulsion droplets con-taining amphiphilic block copolymers. Angew Chem Int Ed2008;47:2113–6.
[148] Lopez-Montilla JC, Herrera-Morales PE, Pandey S, Shah DO.Spontaneuous emulsification: mechanisms, physicochemicalaspects, modeling, and applications. J Dispersion Sci Technol2002;23:219–68.
[149] Lopez-Montilla JC, Herrera-Morales PE, Shah DO. New method toquantitatively determine the spontaneity of the emulsification pro-cess. Langmuir 2002;18:4258–62.
[150] Hayward RC, Pochan DJ. Tailored assemblies of block copoly-mers in solution: it is all about the process. Macromolecules2010;43:3577–84.
[151] Geng Y, Discher DE. Hydrolytic degradation of poly(ethyleneoxide)-block-polycaprolactone worm micelles. J Am Chem Soc2005;127:12780–1.
[152] Geng Y, Discher DE. Visualization of degradable worm micellebreakdown in relation to drug release. Polymer 2006;47:2519–25.
[153] Zhu J, Ferrer N, Hayward RC. Tuning the assembly of amphiphilicblock copolymers through instabilities of solvent/water interfacesin the presence of aqueous surfactants. Soft Matter 2009;5:2471–8.
[154] Jain S, Bates FS. On the origins of morphological complexity in blockcopolymer surfactants. Science 2003;300:460–4.
[155] Lee JCM, Bermudez H, Discher BM, Sheehan MA, Won YY, BatesFS, Discher DE. Preparation, stability, and in vitro performanceof vesicles made with diblock copolymers. Biotechnol Bioeng2001;73:135–45.
[156] Pisani E, Ringard C, Nicolas V, Raphael E, Rosilio V, Moine L, Fat-tal E, Tsapis N. Tuning microcapsules surface morphology usingblends of homo- and copolymers of PLGA and PLGA-PEG. Soft Mat-ter 2009;5:3054–60.
[157] Tomaszewski K, Szymanski A, Lukaszewski Z. Separation ofpoly(ethylene glycols) into fractions by sequential liquid-liquidextraction. Talanta 1999;50:299–306.
[158] Malzert-Freon A, Benoit JP, Boury F. Interactions betweenpoly(ethylene glycol) and protein in dichloromethane/water emul-sions: a study of interfacial properties. Eur J Pharm Biopharm2008;69:835–43.
[159] Gref R, Quellec P, Sanchez A, Calvo P, Dellacherie E, Alonso MJ.Development and characterization of CyA-loaded poly(lactic acid)-poly(ethylene glycol)PEG micro- and nanoparticles. Comparisonwith conventional PLA particulate carriers. Eur J Pharm Biopharm2001;51:111–8.
[160] Sellergren B, Allender CJ. Molecularly imprinted polymers: a
bridge to advanced drug delivery. Adv Drug Delivery Rev2005;57:1733–41.[161] Liu GJ, Zhou J. Diblock copolymer nanospheres with porouscores. 2. Porogen release and reuptake kinetics. Macromolecules2002;35:8167–72.

olymer S
I. Wyman et al. / Progress in P[162] Lu Z, Liu GJ, Phillips H, Hill JM, Chang J, Kydd RA. Palladiumnanoparticle catalyst prepared in poly(acrylic acid)-lined channelsof diblock copolymer nanospheres. Nano Lett 2001;1:683–7.
[163] Rosler A, Vandermeulen GWM, Klok HA. Advanced drug deliverydevices via self-assembly of amphiphilic block copolymers. AdvDrug Delivery Rev 2001;53:95–108.
[164] Lavasanifar A, Samuel J, Kwon GS. Poly(ethylene oxide)-block-poly(L-amino acid) micelles for drug delivery. Adv Drug DeliveryRev 2002;54:169–90.
[165] Harada A, Kataoka K. Supramolecular assemblies of block copoly-mers in aqueous media as nanocontainers relevant to biologicalapplications. Prog Polym Sci 2006;31:949–82.
[166] Sutton D, Nasongkla N, Blanco E, Gao J. Functionalized micel-
lar systems for cancer targeted drug delivery. Pharm Res2007;24:1029–46.[167] Batrakova EV, Kabanov AV. Pluronic block copolymers: evolutionof drug delivery concept from inert nanocarriers to biologicalresponse modifers. J Controlled Release 2008;130:98–106.
cience 36 (2011) 1152– 1183 1183
[168] Osada K, Christie RJ, Kataoka K. Polymeric micelles frompoly(ethylene glycol)-poly(amino acid) block copolymer for drugand gene delivery. J R Soc Interface 2009;6:S325–39.
[169] Tyrrell ZL, Shen Y, Radosz M. Fabrication of micellar nanoparticlesfor drug delivery through the self-assembly of block copolymers.Prog Polym Sci 2010;35:1128–43.
[170] Oh JK. Polylactide (PLA)-based amphiphilic block copolymers: syn-thesis, self-assembly, and biomedical applications. Soft Matter2011;7:5096–108.
[171] Vonarbourg A, Passirani C, Saulnier P, Benoit JP. Parametersinfluencing the stealthiness of colloidal drug delivery systems. Bio-materials 2006;27:4356–73.
[172] Miura H, Onishi H, Sasatsu M, Machida Y. Antitumor characteristics
of methoxypolyethylene glycol-poly(DL-lactic acid) nanoparticlescontaining camptothecin. J Controlled Release 2004;97:101–3.[173] Meng F, Zhong Z, Feijen J. Stimuli-responsive polymersomesfor programmed drug delivery. Biomacromolecules 2009;10:197–209.





























