Embed Size (px)
Citation preview

Vitamin D receptor is essential for normalkeratinocyte stem cell functionLuisella Cianferotti, Megan Cox, Kristi Skorija, and Marie B. Demay*
Endocrine Unit, Massachusetts General Hospital and Harvard Medical School, Boston, MA 02114
Communicated by John T. Potts, Massachusetts General Hospital, Charlestown, MA, March 29, 2007 (received for review December 13, 2006)
The major physiological role of the vitamin D receptor (VDR) is themaintenance of mineral ion homeostasis. Mutation of the VDR, inhumans and mice, results in alopecia. Unlike the effects of the VDRon mineral ion homeostasis, the actions of the VDR that preventalopecia are ligand-independent. Although absence of the VDRdoes not prevent the development of a keratinocyte stem cell nichein the bulge region of the hair follicle, it results in an inability ofthese stem cells to regenerate the lower portion of the hair folliclein vivo and impairs keratinocyte stem cell colony formation in vitro.VDR ablation is associated with a gradual decrease in keratinocytestem cells, accompanied by an increase in sebaceous activity, aphenotype analogous to that seen with impaired canonical Wntsignaling. Transient gene expression assays demonstrate that thecooperative transcriptional effects of �-catenin and Lef1 are abol-ished in keratinocytes isolated from VDR-null mice, revealing a rolefor the unliganded VDR in canonical Wnt signaling. Thus, absenceof the VDR impairs canonical Wnt signaling in keratinocytes andleads to the development of alopecia due to a defect in keratino-cyte stem cells.
alopecia � knockout � Wnt signaling � Lef
A lopecia is a feature of hereditary vitamin D-resistant rickets(1), the molecular basis of which is a mutation of the vitamin
D receptor (VDR) (2). Alopecia is also seen in mice with targetedablation of the VDR (3–6); however, it is not observed in vitaminD-deficient humans or rodents, suggesting that the absence ofligand and the absence of the receptor have different effects on thecells responsible for the cyclic regeneration of the hair follicle (7).Hair follicle development proceeds normally in VDR-null mice(7–9). However, once the morphogenic period ends, during thesecond week of life, VDR-null mice cannot initiate hair cycles.Thus, when hair is lost, it does not regrow and alopecia ensues.
The hair follicle is an organ composed of epidermal keratinocytesand mesodermal dermal papilla cells (10). VDR expression in theepidermal keratinocyte compartment of the hair follicle, but not themesodermal dermal papilla, is required to prevent the developmentof alopecia (7, 11, 12). Primary neonatal keratinocytes from VDR-null mice have no detectable abnormality in proliferation oracquisition of markers of differentiation (8). Furthermore, neonatalkeratinocytes lacking the VDR are able to recapitulate hair folliclemorphogenesis when implanted into a nude mouse host, along withdermal papilla cells. However, neither the follicles reconstitutedusing keratinocytes lacking the VDR nor the native follicles of theVDR-null mice are able to undergo postmorphogenic hair cycling(7). Thus, the alopecia observed in VDR-null mice is secondary toan intrinsic keratinocyte defect that is first evident after the periodof hair follicle morphogenesis is complete in the second week ofpostnatal life. Consistent with the observation that alopecia is notobserved in states of vitamin D deficiency, keratinocyte-specificexpression of a mutant VDR, incapable of ligand binding orligand-dependent transactivation, maintains postnatal hair cyclingin the VDR-null mice (13).
Hair follicle morphogenesis and the regulation of postnatal haircycling are dependent on reciprocal interactions between thekeratinocyte and dermal papilla components of the hair follicle.Postnatally, the epidermal component of the hair follicle, below the
sebaceous gland and the bulge, undergoes cycles of growth, apo-ptosis, and quiescence (14). Keratinocyte stem cells in the bulge arethought to respond to signals from the mesodermal dermal papilla,resulting in the initiation of a new hair cycle (15). In addition toregenerating the lower portion of the hair follicle, these bulge stemcells also play a role in epidermal regeneration and are capable ofdifferentiating into sebocytes that make up the sebaceous glands(16–18).
One of the most extensively studied pathways in hair folliclebiology is the Wnt signaling pathway (19). Keratinocyte-specificdeletion of �-catenin results in impaired follicle development andlack of hair regrowth after hair follicle morphogenesis (20), whereasoverexpression of a constitutively active �-catenin results in de novohair follicle morphogenesis (21, 22). This same canonical Wntsignaling pathway has been shown to modulate nuclear receptor-dependent gene expression (23–26). Like mutations of the VDR,mutations of the nuclear corepressor Hairless (27) result in alopeciain humans (28) and mice (29). Although the mechanism by whichHairless mutations result in alopecia is still under active investiga-tion, it has been demonstrated that, in addition to interacting withthe VDR (30) and suppressing VDR-mediated transactivation inkeratinocytes (31), Hairless promotes canonical Wnt signaling (32).
Because the VDR is critical for the maintenance of cutaneoushomeostasis and because the effects of the unliganded VDR oncanonical Wnt signaling have not been extensively examined,studies were undertaken to address the hypothesis that the ligand-independent actions of the VDR intersect with this key signalingpathway to regulate postmorphogenic hair cycles.
ResultsMutations of the VDR result in alopecia in both humans and mice.In addition to a defect in postmorphogenic hair cycling, theVDR-null mice develop lipid-laden dermal cysts and an increase inoil-red-O-stained sebaceous glands with age (Fig. 1B). Neither ofthese cutaneous abnormalities is seen in the VDR-null mice withkeratinocyte-specific expression of a VDR transgene (Fig. 1C),demonstrating that this phenotype is a direct consequence ofimpaired VDR action in the keratinocyte. Because keratinocytestem cells (KSCs) in the bulge of the hair follicle are capable ofdifferentiating into sebocytes, epidermal keratinocytes, and folliclekeratinocytes (16–18), and because transcriptional profiling of thebulge stem cells demonstrates enhanced expression of the VDR (16,18), investigations were undertaken to determine whether thecutaneous consequences of VDR ablation were due to a defect inkeratinocyte stem cell self-renewal and/or lineage specification.When plated at low density onto irradiated feeder layers andcultured in media with numerous mineral and vitamin supplementsfor 2 weeks, �5% of epidermal keratinocytes form small colonies
Author contributions: L.C. and M.C. contributed equally to this work; M.B.D. designedresearch; L.C., M.C., K.S., and M.B.D. performed research; L.C., M.C., K.S., and M.B.D.analyzed data; and M.B.D. wrote the paper.
The authors declare no conflict of interest.
Abbreviations: AR, androgren receptor; KSC, keratinocyte stem cell; PPAR, peroxisomeproliferator-activated receptor; VDR, vitamin D receptor.
*To whom correspondence should be addressed. E-mail: [email protected].
© 2007 by The National Academy of Sciences of the USA
9428–9433 � PNAS � May 29, 2007 � vol. 104 � no. 22 www.pnas.org�cgi�doi�10.1073�pnas.0702884104
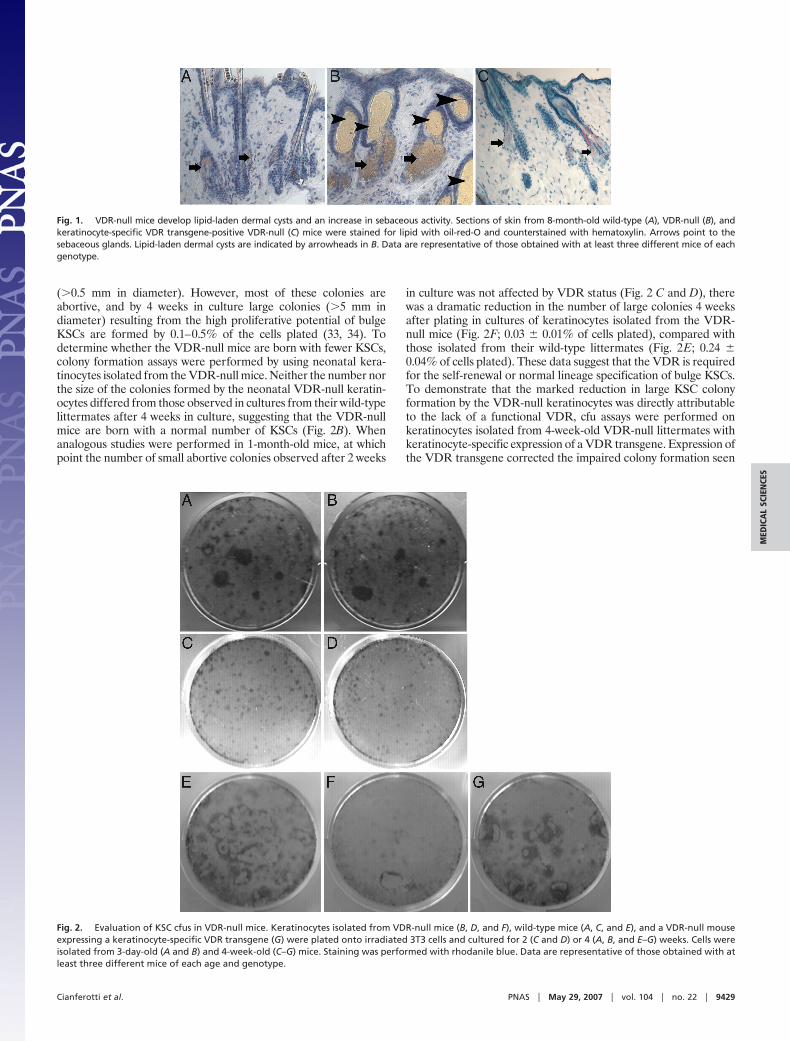
(�0.5 mm in diameter). However, most of these colonies areabortive, and by 4 weeks in culture large colonies (�5 mm indiameter) resulting from the high proliferative potential of bulgeKSCs are formed by 0.1–0.5% of the cells plated (33, 34). Todetermine whether the VDR-null mice are born with fewer KSCs,colony formation assays were performed by using neonatal kera-tinocytes isolated from the VDR-null mice. Neither the number northe size of the colonies formed by the neonatal VDR-null keratin-ocytes differed from those observed in cultures from their wild-typelittermates after 4 weeks in culture, suggesting that the VDR-nullmice are born with a normal number of KSCs (Fig. 2B). Whenanalogous studies were performed in 1-month-old mice, at whichpoint the number of small abortive colonies observed after 2 weeks
in culture was not affected by VDR status (Fig. 2 C and D), therewas a dramatic reduction in the number of large colonies 4 weeksafter plating in cultures of keratinocytes isolated from the VDR-null mice (Fig. 2F; 0.03 � 0.01% of cells plated), compared withthose isolated from their wild-type littermates (Fig. 2E; 0.24 �0.04% of cells plated). These data suggest that the VDR is requiredfor the self-renewal or normal lineage specification of bulge KSCs.To demonstrate that the marked reduction in large KSC colonyformation by the VDR-null keratinocytes was directly attributableto the lack of a functional VDR, cfu assays were performed onkeratinocytes isolated from 4-week-old VDR-null littermates withkeratinocyte-specific expression of a VDR transgene. Expression ofthe VDR transgene corrected the impaired colony formation seen
Fig. 1. VDR-null mice develop lipid-laden dermal cysts and an increase in sebaceous activity. Sections of skin from 8-month-old wild-type (A), VDR-null (B), andkeratinocyte-specific VDR transgene-positive VDR-null (C) mice were stained for lipid with oil-red-O and counterstained with hematoxylin. Arrows point to thesebaceous glands. Lipid-laden dermal cysts are indicated by arrowheads in B. Data are representative of those obtained with at least three different mice of eachgenotype.
Fig. 2. Evaluation of KSC cfus in VDR-null mice. Keratinocytes isolated from VDR-null mice (B, D, and F), wild-type mice (A, C, and E), and a VDR-null mouseexpressing a keratinocyte-specific VDR transgene (G) were plated onto irradiated 3T3 cells and cultured for 2 (C and D) or 4 (A, B, and E–G) weeks. Cells wereisolated from 3-day-old (A and B) and 4-week-old (C–G) mice. Staining was performed with rhodanile blue. Data are representative of those obtained with atleast three different mice of each age and genotype.
Cianferotti et al. PNAS � May 29, 2007 � vol. 104 � no. 22 � 9429
MED
ICA
LSC
IEN
CES

in the transgene-negative VDR-null mice (Fig. 2G). Thus, colonyformation by KSCs isolated from neonatal VDR-null mice isindistinguishable from that of their wild-type littermates; however,by 4 weeks of age, in the absence of the VDR, markedly impairedKSC colony formation is observed.
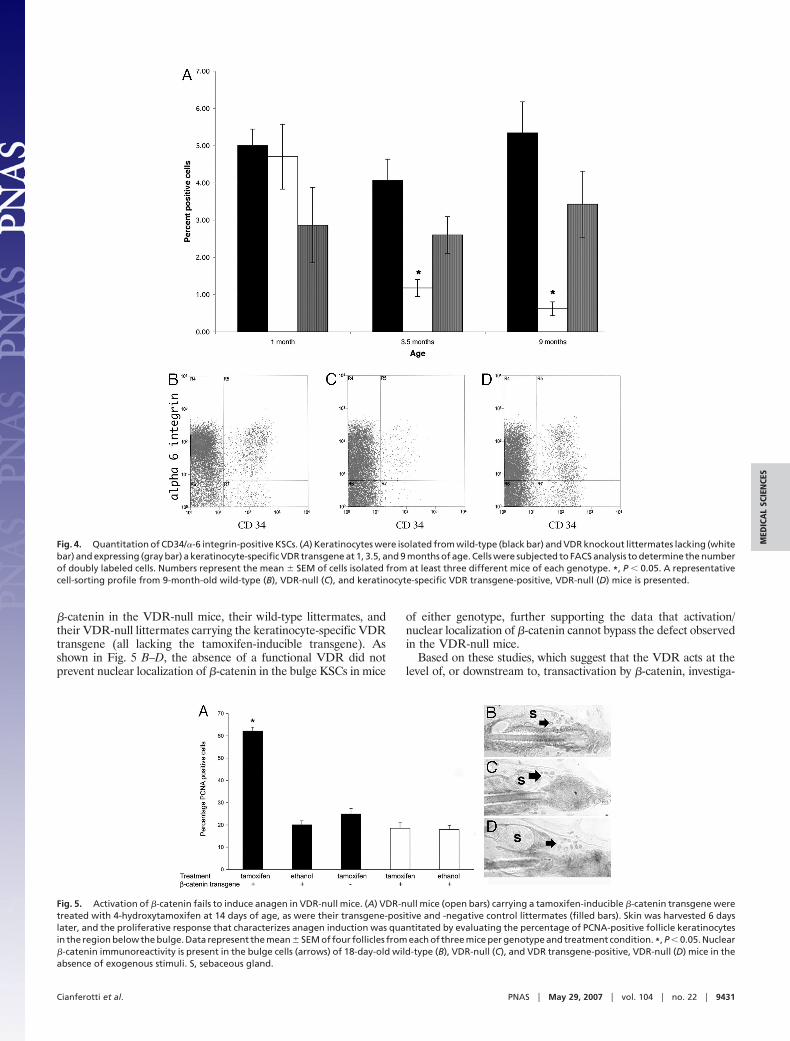
Like other stem cells, the KSCs reside in a special niche knownas the bulge region of the hair follicle. This bulge is not presentduring hair follicle morphogenesis, but is observed at the time ofinitiation of the first postnatal hair cycle at �3 weeks of age (35).Analogous to other stem cells, including those of the hematopoeitic,myogenic, and endothelial lineages, murine bulge stem cells expressthe cell surface marker CD34 (36). Therefore immunohistochem-istry was performed to address whether absence of the VDRinterfered with the formation of this stem cell niche in the hairfollicle. These studies demonstrate that neither the histologicalappearance nor the CD34 immunoreactivity of the budge is alteredin 4-week-old VDR-null mice (Fig. 3B), compared with that of theirwild-type littermates (Fig. 3A) or the VDR-null mice expressing akeratinocyte-specific VDR transgene (Fig. 3C). However, by 9months of age, while CD34 immunoreactivity was preserved in thebulge region of the hair follicles of the wild-type mice (Fig. 3D), itwas not present in VDR-null mice (Fig. 3E). Correlating with thepreservation of postmorphogenic hair cycles, the knockout miceexpressing the keratinocyte-specific VDR transgene retained CD34immunoreactivity in the follicle bulge (Fig. 3F).
Because these bulge cells make up a small percentage of thekeratinocyte population in the skin of mice and are characterizedby the expression of both CD34 and �-6 integrin, cell sorting was
performed to address whether the marked impairment of colonyformation in the keratinocytes of the 28-day-old VDR-null micewas due to a decrease in the number of KSCs residing in the bulgeor a functional abnormality of these cells. Consistent with thenormal CD34 immunoreactivity (Fig. 3) of the bulge area in theVDR-null mice at 1 month of age, the number of doubly labeledcells detected by FACS analysis at this age was not significantlyaltered (Fig. 4A). These data strongly suggest that the KSCs in theVDR-null mice have an altered lineage progression or an alteredability to proliferate (or self-renew or survive) because they areunable to give rise to large stem cell colonies in vitro when placedin culture and are unable to generate functional hair follicles in vivoat a point in time when their numbers are apparently unaffected. Itis notable that, with aging, there is a progressive decline in thenumber of doubly labeled cells in the VDR-null mice due to amarked decrease in CD34-positive cells (Fig. 4 A and C), confirm-ing the lack of CD34 immunoreactivity seen at 9 months of age inthe skin of the VDR-null mice (Fig. 3E). These data suggest thatKSC self-renewal is impaired by the lack of a functional VDR. Todetermine whether the lack of VDR expression specifically in thekeratinocyte component of the hair follicle is responsible for thisreduction in CD34/�-6 integrin-positive cells with age, the KSCnumber was evaluated in VDR-null mice expressing the K-14 VDRtransgene. As indicated in Fig. 4 A and D, the number of doublylabeled KSCs in VDR-null mice expressing the K-14-VDR trans-gene is not significantly different from that of their wild-typelittermates at 1, 3.5, or 9 months of age.
These studies demonstrate that the absence of the VDR leads toa functional abnormality in KSCs, evidenced by impaired colonyformation in vitro and an inability to regenerate the lower portionof the hair follicle in vivo by 1 month of age, a time when the numberof CD34/�-6 integrin-positive stem cells is normal. However, as themice age, the KSC number decreases, and the mice developlipid-laden dermal cysts as well as an increase in sebaceous glands,a phenotype reminiscent of that observed with impaired Wntsignaling. The canonical Wnt signaling pathway has been shown toplay a critical role in the lineage determination of KSCs. Attenu-ation of Wnt signaling and overexpression of a dominant-negativeLef1 impair follicle morphogenesis and result in enhanced sebocytedifferentiation (37, 38). In contrast, high levels of Wnt signaling dueto constitutive overexpression of �-catenin promote hair follicleformation (20–22). Similarly, induction of a constitutively active�-catenin transgene induces anagen in postmorphogenic hair fol-licles. To evaluate whether induction of �-catenin could bypass theabnormality in the VDR-null keratinocytes, mice expressing akeratinocyte-specific, tamoxifen-inducible, constitutively active�-catenin transgene (39) were mated into the VDR-null back-ground. VDR-null and control mice carrying this transgene weretreated with tamoxifen, to induce transgene expression, at 14 daysof age. Studies were specifically performed at this age because it isbefore the onset of the first postmorphogenic anagen and beforethe observed decrease in CD34/�-6 integrin-positive KSCs in theVDR-null mice. Induction of the transgene with 4-hydroxytamox-ifen failed to induced a proliferative response characteristic of earlyanagen in the hair follicle keratinocytes of the VDR-null micecarrying the transgene and in control littermates lacking thetransgene. A marked increase in proliferating hair follicle keratin-ocytes, below the bulge region, was observed in the �-catenintransgene-positive control mice in response to tamoxifen treatment(Fig. 5). Treatment with vehicle (ethanol) failed to induce aproliferative response. Analogous studies performed at 14 and 18days demonstrated that induction of the transgene did not lead tohair regrowth in either shaved or waxed VDR-null mice. These datademonstrate that activation of �-catenin cannot bypass the haircycling defect in the VDR-null mice at this age, suggesting thatnuclear �-catenin is appropriately present in the bulge KSCs in theabsence of induction of this transgene. Therefore, immunohisto-chemistry was performed to evaluate the cellular distribution of
Fig. 3. Absence of the VDR does not prevent formation of the hair follicle stemcell niche. Immunohistochemistry with an anti-CD34 antibody was performed toevaluate formation of the KSC niche in the bulge region of the hair follicle.Sections were obtained from wild-type mice (A and D), VDR-null mice (B and E),and VDR-null mice expressing a keratinocyte-specific VDR transgene (C and F) at4 weeks (A–C) and 9 months (D–F) of age. (D–F) Images are overexposed to permitevaluation of the section from the 9-month-old VDR-null mouse, which fails toreveal membrane-associated fluorescence. Data are representative of those ob-tained with at least three different mice of each age and genotype.
9430 � www.pnas.org�cgi�doi�10.1073�pnas.0702884104 Cianferotti et al.
�-catenin in the VDR-null mice, their wild-type littermates, andtheir VDR-null littermates carrying the keratinocyte-specific VDRtransgene (all lacking the tamoxifen-inducible transgene). Asshown in Fig. 5 B–D, the absence of a functional VDR did notprevent nuclear localization of �-catenin in the bulge KSCs in mice
of either genotype, further supporting the data that activation/nuclear localization of �-catenin cannot bypass the defect observedin the VDR-null mice.
Based on these studies, which suggest that the VDR acts at thelevel of, or downstream to, transactivation by �-catenin, investiga-
Fig. 4. Quantitation of CD34/�-6 integrin-positive KSCs. (A) Keratinocytes were isolated from wild-type (black bar) and VDR knockout littermates lacking (whitebar) and expressing (gray bar) a keratinocyte-specific VDR transgene at 1, 3.5, and 9 months of age. Cells were subjected to FACS analysis to determine the numberof doubly labeled cells. Numbers represent the mean � SEM of cells isolated from at least three different mice of each genotype. *, P � 0.05. A representativecell-sorting profile from 9-month-old wild-type (B), VDR-null (C), and keratinocyte-specific VDR transgene-positive, VDR-null (D) mice is presented.
Fig. 5. Activation of �-catenin fails to induce anagen in VDR-null mice. (A) VDR-null mice (open bars) carrying a tamoxifen-inducible �-catenin transgene weretreated with 4-hydroxytamoxifen at 14 days of age, as were their transgene-positive and -negative control littermates (filled bars). Skin was harvested 6 dayslater, and the proliferative response that characterizes anagen induction was quantitated by evaluating the percentage of PCNA-positive follicle keratinocytesin the region below the bulge. Data represent the mean � SEM of four follicles from each of three mice per genotype and treatment condition. *, P � 0.05. Nuclear�-catenin immunoreactivity is present in the bulge cells (arrows) of 18-day-old wild-type (B), VDR-null (C), and VDR transgene-positive, VDR-null (D) mice in theabsence of exogenous stimuli. S, sebaceous gland.
Cianferotti et al. PNAS � May 29, 2007 � vol. 104 � no. 22 � 9431
MED
ICA
LSC
IEN
CES
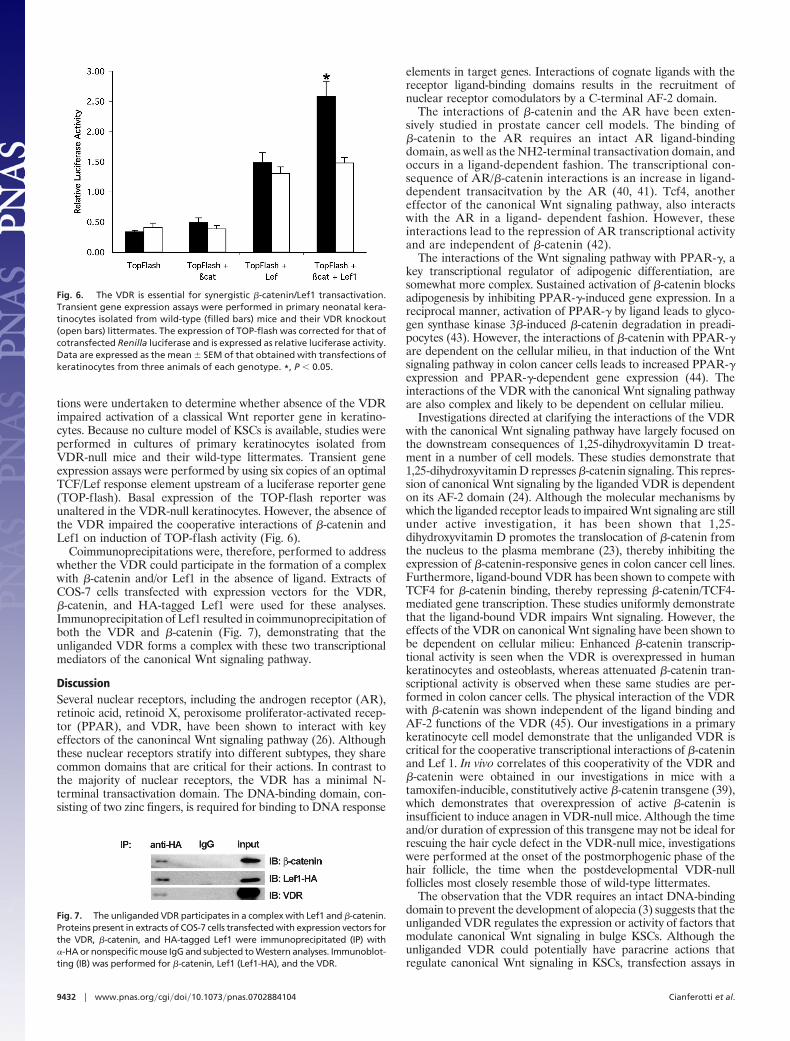
tions were undertaken to determine whether absence of the VDRimpaired activation of a classical Wnt reporter gene in keratino-cytes. Because no culture model of KSCs is available, studies wereperformed in cultures of primary keratinocytes isolated fromVDR-null mice and their wild-type littermates. Transient geneexpression assays were performed by using six copies of an optimalTCF/Lef response element upstream of a luciferase reporter gene(TOP-flash). Basal expression of the TOP-flash reporter wasunaltered in the VDR-null keratinocytes. However, the absence ofthe VDR impaired the cooperative interactions of �-catenin andLef1 on induction of TOP-flash activity (Fig. 6).
Coimmunoprecipitations were, therefore, performed to addresswhether the VDR could participate in the formation of a complexwith �-catenin and/or Lef1 in the absence of ligand. Extracts ofCOS-7 cells transfected with expression vectors for the VDR,�-catenin, and HA-tagged Lef1 were used for these analyses.Immunoprecipitation of Lef1 resulted in coimmunoprecipitation ofboth the VDR and �-catenin (Fig. 7), demonstrating that theunliganded VDR forms a complex with these two transcriptionalmediators of the canonical Wnt signaling pathway.
DiscussionSeveral nuclear receptors, including the androgen receptor (AR),retinoic acid, retinoid X, peroxisome proliferator-activated recep-tor (PPAR), and VDR, have been shown to interact with keyeffectors of the canonincal Wnt signaling pathway (26). Althoughthese nuclear receptors stratify into different subtypes, they sharecommon domains that are critical for their actions. In contrast tothe majority of nuclear receptors, the VDR has a minimal N-terminal transactivation domain. The DNA-binding domain, con-sisting of two zinc fingers, is required for binding to DNA response
elements in target genes. Interactions of cognate ligands with thereceptor ligand-binding domains results in the recruitment ofnuclear receptor comodulators by a C-terminal AF-2 domain.
The interactions of �-catenin and the AR have been exten-sively studied in prostate cancer cell models. The binding of�-catenin to the AR requires an intact AR ligand-bindingdomain, as well as the NH2-terminal transactivation domain, andoccurs in a ligand-dependent fashion. The transcriptional con-sequence of AR/�-catenin interactions is an increase in ligand-dependent transacitvation by the AR (40, 41). Tcf4, anothereffector of the canonical Wnt signaling pathway, also interactswith the AR in a ligand- dependent fashion. However, theseinteractions lead to the repression of AR transcriptional activityand are independent of �-catenin (42).
The interactions of the Wnt signaling pathway with PPAR-�, akey transcriptional regulator of adipogenic differentiation, aresomewhat more complex. Sustained activation of �-catenin blocksadipogenesis by inhibiting PPAR-�-induced gene expression. In areciprocal manner, activation of PPAR-� by ligand leads to glyco-gen synthase kinase 3�-induced �-catenin degradation in preadi-pocytes (43). However, the interactions of �-catenin with PPAR-�are dependent on the cellular milieu, in that induction of the Wntsignaling pathway in colon cancer cells leads to increased PPAR-�expression and PPAR-�-dependent gene expression (44). Theinteractions of the VDR with the canonical Wnt signaling pathwayare also complex and likely to be dependent on cellular milieu.
Investigations directed at clarifying the interactions of the VDRwith the canonical Wnt signaling pathway have largely focused onthe downstream consequences of 1,25-dihydroxyvitamin D treat-ment in a number of cell models. These studies demonstrate that1,25-dihydroxyvitamin D represses �-catenin signaling. This repres-sion of canonical Wnt signaling by the liganded VDR is dependenton its AF-2 domain (24). Although the molecular mechanisms bywhich the liganded receptor leads to impaired Wnt signaling are stillunder active investigation, it has been shown that 1,25-dihydroxyvitamin D promotes the translocation of �-catenin fromthe nucleus to the plasma membrane (23), thereby inhibiting theexpression of �-catenin-responsive genes in colon cancer cell lines.Furthermore, ligand-bound VDR has been shown to compete withTCF4 for �-catenin binding, thereby repressing �-catenin/TCF4-mediated gene transcription. These studies uniformly demonstratethat the ligand-bound VDR impairs Wnt signaling. However, theeffects of the VDR on canonical Wnt signaling have been shown tobe dependent on cellular milieu: Enhanced �-catenin transcrip-tional activity is seen when the VDR is overexpressed in humankeratinocytes and osteoblasts, whereas attenuated �-catenin tran-scriptional activity is observed when these same studies are per-formed in colon cancer cells. The physical interaction of the VDRwith �-catenin was shown independent of the ligand binding andAF-2 functions of the VDR (45). Our investigations in a primarykeratinocyte cell model demonstrate that the unliganded VDR iscritical for the cooperative transcriptional interactions of �-cateninand Lef 1. In vivo correlates of this cooperativity of the VDR and�-catenin were obtained in our investigations in mice with atamoxifen-inducible, constitutively active �-catenin transgene (39),which demonstrates that overexpression of active �-catenin isinsufficient to induce anagen in VDR-null mice. Although the timeand/or duration of expression of this transgene may not be ideal forrescuing the hair cycle defect in the VDR-null mice, investigationswere performed at the onset of the postmorphogenic phase of thehair follicle, the time when the postdevelopmental VDR-nullfollicles most closely resemble those of wild-type littermates.
The observation that the VDR requires an intact DNA-bindingdomain to prevent the development of alopecia (3) suggests that theunliganded VDR regulates the expression or activity of factors thatmodulate canonical Wnt signaling in bulge KSCs. Although theunliganded VDR could potentially have paracrine actions thatregulate canonical Wnt signaling in KSCs, transfection assays in
Fig. 6. The VDR is essential for synergistic �-catenin/Lef1 transactivation.Transient gene expression assays were performed in primary neonatal kera-tinocytes isolated from wild-type (filled bars) mice and their VDR knockout(open bars) littermates. The expression of TOP-flash was corrected for that ofcotransfected Renilla luciferase and is expressed as relative luciferase activity.Data are expressed as the mean � SEM of that obtained with transfections ofkeratinocytes from three animals of each genotype. *, P � 0.05.
Fig. 7. The unliganded VDR participates in a complex with Lef1 and �-catenin.Proteins present in extracts of COS-7 cells transfected with expression vectors forthe VDR, �-catenin, and HA-tagged Lef1 were immunoprecipitated (IP) with�-HA or nonspecific mouse IgG and subjected to Western analyses. Immunoblot-ting (IB) was performed for �-catenin, Lef1 (Lef1-HA), and the VDR.
9432 � www.pnas.org�cgi�doi�10.1073�pnas.0702884104 Cianferotti et al.

cultured keratinocytes suggest that its actions are, at least in part,autocrine. We were unable to detect the VDR in a complex with�-catenin and/or Lef1 on the TOP response element or on aclassical VDR on gel shift analyses. These data suggest that anadditional factor that is not ubiquitously expressed interacts with�-catenin /Lef1 and the VDR to promote differentiation of theKSCs into hair follicle keratinocytes, rather than sebocytes orepidermal keratinocytes. Restricted temporospatial expression ofthis presumptive factor would be consistent with the data in TOPgalmice demonstrating that activation of the canonical Wnt signalingpathway in the bulge KSCs is restricted to the period of anageninitiation (46). Thus, we propose that the unliganded VDR mod-ulates the activity of key effectors of the canonical Wnt signalingpathway in hair follicle KSCs to maintain keratinocyte stem cellself-renewal and to promote differentiation of these cells along thehair follicle keratinocyte lineage.
MethodsAnimal Maintenance. All studies were approved by the institutionalanimal care committee. Mice were exposed to a 12-h light/12-h darkcycle. The K5/S33Y �-catenin-estrogen receptor transgenic mice(39) were obtained from E. Fearon (University of Michigan, AnnArbor, MI). Treatment with tamoxifen or ethanol was performedas previously described (39).
Histology and Immunohistochemistry. Skin obtained from the dorsalregion was used for all investigations. Oil-red-O (3% in isopropa-nol) staining was performed at 37°C for 15 min. Anti-CD34antibodies were obtained from BD BioSciences (San Jose, CA).Proliferating cell nuclear antigen staining was performed by using
a Zymed (South San Francisco, CA) kit according to the manu-facturer’s instructions.
Cell Culture. Isolation and transfection of primary neonatal kera-tinocytes were performed with Lipofectamine (Invitrogen, Carls-bad CA) as previously described (13). Luciferase activity wasevaluated by using Stop and Glo (Promega, Madison, WI). Fireflyluciferase activity was normalized for a cotransfected Renilla lucif-erase control. Culture conditions for the evaluation of KSC cfushave been previously described (16, 34). Cells were sorted withFITC-labeled anti-CD34 and phycoerythrin-Cy5 conjugated anti-�-6 integrin antibodies from BD Biosciences (36).
Immunoprecipitation Analyses. COS-7 cells were transfected withexpression vectors for the VDR, �-catenin, and HA-tagged Lef1.Cell lysates were immunoprecipitated with a mouse a-HA antibody(BD Biosciences) and subjected to Western analyses. The sameantibody was used to detect immunoprecipitated Lef1. A mouseanti-�-catenin antibody (BD Biosciences) and 9A7 (AbCam) wereused to detect �-catenin and the VDR, respectively. Secondary goatanti-mouse and goat anti-rat antibodies were obtained from SantaCruz Technology (Santa Cruz, CA). Detection was performed byusing a chemiluminescence kit (Amersham Biosciences, Piscat-away, NJ).
The authors thank Dr. Eric Fearon for providing the K5/S33Y-�-catenin-ER transgenic mice and Dr. Hans Clevers (Netherlands Institutefor Developmental Biology, Utrecht, The Netherlands) for providing theHA-tagged Lef1 expression vector. This work was supported by NationalInstitutes of Health Grant DK 46974 (to M.B.D.).
1. Malloy PJ, Pike JW, Feldman D (1999) Endocr Rev 20:156–188.2. Hughes MR, Malloy PJ, Kieback DG, Kesterson RA, Pike JW, Feldman D,
O’Malley BW (1988) Science 242:1702–1705.3. Erben RG, Soegiarto DW, Weber K, Zeitz U, Lieberherr M, Gniadecki R,
Moller G, Adamski J, Balling R (2002) Mol Endocrinol 16:1524–1537.4. Li YC, Pirro AE, Amling M, Delling G, Baron R, Bronson R, Demay MB
(1997) Proc Natl Acad Sci USA 94:9831–9835.5. Yoshizawa T, Handa Y, Uematsu Y, Takeda S, Sekine K, Yoshihara Y,
Kawakami T, Alioka K, Sato H, Uchiyama Y, et al. (1997) Nat Genet16:391–396.
6. Van Cromphaut SJ, Dewerchin M, Hoenderop JG, Stockmans I, Van Herck E,Kato S, Bindels RJ, Collen D, Carmeliet P, Bouillon R, Carmeliet G (2001)Proc Natl Acad Sci USA 98:13324–13329.
7. Sakai Y, Kishimoto J, Demay M (2001) J Clin Invest 107:961–966.8. Sakai Y, Demay MB (2000) Endocrinology 141:2043–2049.9. Xie Z, Komuves L, Yu QC, Elalieh H, Ng DC, Leary C, Chang S, Crumrine
D, Yoshizawa T, Kato S, Bikle DD (2002) J Invest Dermatol 118:11–16.10. Paus R, Cotsarelis G (1999) N Engl J Med 341:491–497.11. Chen C, Sakai Y, Demay M (2001) Endocrinol 142:5386–5389.12. Kong J, Li XJ, Gavin D, Jiang Y, Li YC (2002) J Invest Dermatol 118:631–638.13. Skorija K, Cox M, Sisk JM, Dowd DR, MacDonald PN, Thompson CC, Demay
MB (2005) Mol Endocrinol 19:855–862.14. Dlugosz A (1999) J Clin Invest 104:851–853.15. Lavker RM, Sun TT, Oshima H, Barrandon Y, Akiyama M, Ferraris C,
Chevalier G, Favier B, Jahoda CA, Dhouailly D, et al. (2003) J Investig DermatolSymp Proc 8:28–38.
16. Morris RJ, Liu Y, Marles L, Yang Z, Trempus C, Li S, Lin JS, Sawicki JA,Cotsarelis G (2004) Nat Biotechnol 22:411–417.
17. Taylor G, Lehrer MS, Jensen PJ, Sun TT, Lavker RM (2000) Cell 102:451–461.18. Tumbar T, Guasch G, Greco V, Blanpain C, Lowry WE, Rendl M, Fuchs E
(2004) Science 303:359–363.19. Alonso L, Fuchs E (2003) Genes Dev 17:1189–1200.20. Huelsken J, Vogel R, Erdmann B, Cotsarelis G, Birchmeier W (2001) Cell
105:533–545.21. Gat U, DasGupta R, Degenstein L, Fuchs E (1998) Cell 95:605–614.22. Lo Celso C, Prowse DM, Watt FM (2004) Development (Cambridge, UK)
131:1787–1799.23. Palmer HG, Gonzalez-Sancho JM, Espada J, Berciano MT, Puig I, Baulida J,
Quintanilla M, Cano A, de Herreros AG, Lafarga M, Munoz A (2001) J CellBiol 154:369–387.
24. Shah S, Hecht A, Pestell R, Byers SW (2003) J Biol Chem 278:48137–48145.25. Shah S, Islam MN, Dakshanamurthy S, Rizvi I, Rao M, Herrell R, Zinser G,
Valrance M, Aranda A, Moras D, et al. (2006) Mol Cell 21:799–809.26. Mulholland DJ, Dedhar S, Coetzee GA, Nelson CC (2005) Endocr Rev
26:898–915.27. Thompson CC (1996) J Neurosci 16:7832–7840.28. Ahmad W, Faiyaz ul Haque M, Brancolini V, Tsou HC, ul Haque S, Lam H,
Aita VM, Owen J, deBlaquiere M, et al. (1998) Science 279:720–724.29. Sundberg JP, King LJ (1996) J Invest Dermatol 106:368–376.30. Hsieh JC, Sisk JM, Jurutka PW, Haussler CA, Slater SA, Haussler MR,
Thompson CC (2003) J Biol Chem 278:38665–38674.31. Xie Z, Chang S, Oda Y, Bikle DD (2006) Endocrinology 147:314–323.32. Beaudoin GM, III, Sisk JM, Coulombe PA, Thompson CC (2005) Proc Natl
Acad Sci USA 102:14653–14658.33. Morris RJ, Potten CS (1994) Cell Prolif 27:279–289.34. Wu WY, Morris RJ (2005) Methods Mol Biol 289:79–86.35. Blanpain C, Lowry WE, Geoghegan A, Polak L, Fuchs E (2004) Cell 118:635–
648.36. Trempus CS, Morris RJ, Bortner CD, Cotsarelis G, Faircloth RS, Reece JM,
Tennant RW (2003) J Invest Dermatol 120:501–511.37. Merrill BJ, Gat U, DasGupta R, Fuchs E (2001) Genes Dev 15:1688–1705.38. Niemann C, Unden AB, Lyle S, Zouboulis Ch C, Toftgard R, Watt FM (2003)
Proc Natl Acad Sci USA 100(Suppl 1):11873–11880.39. Van Mater D, Kolligs FT, Dlugosz AA, Fearon ER (2003) Genes Dev
17:1219–1224.40. Song LN, Herrell R, Byers S, Shah S, Wilson EM, Gelmann EP (2003) Mol Cell
Biol 23:1674–1687.41. Yang F, Li X, Sharma M, Sasaki CY, Longo DL, Lim B, Sun Z (2002) J Biol
Chem 277:11336–11344.42. Amir AL, Barua M, McKnight NC, Cheng S, Yuan X, Balk SP (2003) J Biol
Chem 278:30828–30834.43. Liu J, Farmer SR (2004) J Biol Chem 279:45020–45027.44. Jansson EA, Are A, Greicius G, Kuo IC, Kelly D, Arulampalam V, Pettersson
S (2005) Proc Natl Acad Sci USA 102:1460–1465.45. Jurutka PW, Hall N, Whitfield GK, Gurevich M, Barthel TK, Hsieh JC,
Haussler CA, Haussler MR (2004) J Bone Miner Res 19(S1):S543.46. DasGupta R, Fuchs E (1999) Development (Cambridge, UK) 126:4557–
4568.
Cianferotti et al. PNAS � May 29, 2007 � vol. 104 � no. 22 � 9433
MED
ICA
LSC
IEN
CES





























