Embed Size (px)
Citation preview

MOLECULAR CARCINOGENESIS
Tolfenamic Acid Inhibits Neuroblastoma CellProliferation and Induces Apoptosis: A NovelTherapeutic Agent for Neuroblastoma
Don Eslin,1,2* Umesh T. Sankpal,1 Chris Lee,1 Robert M. Sutphin,1,2 Pius Maliakal,1,3 Erika Currier,4
Giselle Sholler,5 Moeez Khan,1 and Riyaz Basha1,3,6*1MD Anderson Cancer Center Orlando, Orlando, Florida2Arnold Palmer Hospital for Children, Orlando, Florida3Florida State University College of Medicine, Orlando, Florida4Vermont Cancer Center, University of Vermont College of Medicine, Burlington, Vermont5Department of Pediatric Oncology, Van Andel Research Institute, Grand Rapids, Michigan6University of Central Florida College of Medicine, Orlando, Florida
Current therapeutic options for recurrent neuroblastoma have poor outcomes that warrant the development ofnovel therapeutic strategies. Specificity protein (Sp) transcription factors regulate several genes involved in cell prolifer-
ation, survival, and angiogenesis. Sp1 regulates genes believed to be important determinants of the biological behav-ior of neuroblastoma. Tolfenamic acid (TA), a non-steroidal anti-inflammatory drug, is known to induce thedegradation of Sp proteins and may serve as a novel anti-cancer agent. The objective of this investigation was toexamine the anti-cancer activity of TA using established human neuroblastoma cell lines. We tested the anti-prolifer-
ative effect of TA using SH-SY5Y, CHLA90, LA1 55n, SHEP, Be2c, CMP 13Y, and SMS KCNR cell lines. Cells weretreated with TA (0/25/50/100 mM) and cell viability was measured at 24, 48, and 72 h post-treatment. Selectedneuroblastoma cell lines were treated with 50 mM TA for 24 and 48 h and tested for cell apoptosis using Annexin-V
staining. Caspase activity was measured with caspase 3/7 Glo kit. Cell lysates were prepared and the expression ofSp1, survivin, and c-PARP were evaluated through Western blot analysis. TA significantly inhibited the growth ofneuroblastoma cells in a dose/time-dependent manner and significantly decreased Sp1 and survivin expression. Apart
from cell cycle (G0/G1) arrest, TA caused significant increase in the apoptotic cell population, caspase 3/7 activity, andc-PARP expression. These results show that TA effectively inhibits neuroblastoma cell growth potentially through sup-pressing mitosis, Sp1, and survivin expression, and inducing apoptosis. These results show TA as a novel therapeutic
agent for neuroblastoma. � 2011 Wiley Periodicals, Inc.
Key words: pediatric tumors; NSAID; survivin; transcription factors; Sp1
INTRODUCTION
Neuroblastoma is an embryonal tumor of the pe-ripheral sympathetic nervous system found ininfants and children. It is the most common extra-cranial solid malignancy in childhood andaccounts for more than 15% of deaths due tochildhood cancers [1–4]. Neuroblastoma is a veryaggressive malignancy with approximately 70% ofhigh-risk patients presenting with distant metasta-sis at the time of diagnosis [5]. High-risk neuro-blastomas are rapidly progressive and relapse iscommon with currently no known curative thera-py [6]. The standard treatment options for this ma-lignancy include surgery, chemotherapy, radiationtherapy, bone marrow transplant, and biologictherapy. Neuroblastomas manifest extraordinaryclinical and biological heterogeneity and despiteintensive therapy, the mortality rate remains morethan 50% [5–7]. The side effects of standard thera-py can cause acute and long-term damage and tox-icity in various biological functions and can lead
to the development of secondary tumors [5].Therefore, there is an urgent need in identifyingnovel strategies in neuroblastoma that improveoutcome with less toxicity.Specificity protein (Sp) transcription factors play
a critical role in growth and metastasis of manycancers and there is strong evidence that Sp1 ex-pression is a negative prognostic factor for survival
Additional Supporting Information may be found in the onlineversion of this article.
Abbreviations: Sp, specificity protein; NSAIDs, non-steroidal anti-inflammatory drugs; TA, tolfenamic acid; IAP, inhibitor of apoptosisproteins; c-PARP, cleaved poly ADP-ribose polymerase; DMSO,dimethyl sulfoxide; PBS, phosphate-buffered saline; BCA, bicincho-ninic acid; PI, propidium iodide.
*Correspondence to: MD Anderson Cancer Center Orlando,Orlando, FL.
Received 21 August 2011; Revised 14 November 2011; Accept-ed 2 December 2011
DOI 10.1002/mc.21866
Published online in Wiley Online Library(wileyonlinelibrary.com).
� 2011 WILEY PERIODICALS, INC.

in some cancer patients [8–11]. Previous studiesfrom our group and others demonstrated the criti-cal role of Sp proteins on growth and angiogenesisof cancer cells while targeting the degradation ofthese proteins has shown potential for the treat-ment of certain adult cancers [8,9,12–14]. SinceSp1 mediates the expression of several genesthat appear to be important determinants of thebiological behavior of neuroblastoma [15–19],identifying small molecules that can induce thedegradation of Sp proteins will represent a novelmechanism-based approach for the treatment ofneuroblastoma. Small molecules such as non-ste-roidal anti-inflammatory drugs (NSAIDs) havebeen extensively studied for their anti-cancer activ-ity in several cancers [20–22]. The research fromour laboratory and others has discerned that tolfe-namic acid (TA), a NSAID is a novel anti-canceragent in adult malignancies such as pancreatic,lung, and ovarian cancer models [8,12,23]. Thesestudies showed that TA induces the degradation ofspecific Sp transcription factors, Sp1, Sp3, and Sp4[8]. These Sp proteins regulate the expression ofcritical genes associated with a number of cellularand molecular processes leading to the develop-ment of human cancers. The expression of Sp pro-teins, especially Sp1, is often associated withaggressive disease and poor prognosis in a numberof malignancies [9,24]. Sp proteins also mediatethe regulation of a number of genes that controlcell proliferation and survival [8]. A number ofstudies have revealed that Sp proteins regulate sev-eral important genes associated with neuroblasto-ma including MYCN and TrkA [15–19]. Currently,TA is used for treating migraine headaches and isknown to penetrate into the brain. This drug wasrecently tested in an animal model for the neuro-degenerative disorder, Alzheimer’s disease [25].This research group also confirmed the ability ofTA to penetrate into brain using computational, invitro and in vivo models [26].
Our aim was to exploit the strategy of targetingSp proteins using TA in the treatment of neuro-blastoma. In this study, we examined the anti-pro-liferative activity of TA using 7 neuroblastoma celllines and evaluated the ability of TA to induce apo-ptosis, and cell cycle arrest using selected cell lines.Apoptosis in mammalian cells primarily takesplace through extrinsic and intrinsic pathways.These two apoptotic pathways converge on thedownstream effector caspases 3 and 7 [27]. A num-ber of studies have suggested that survivin opposesboth intrinsic and extrinsic mediators of apoptosis[28,29]. Survivin is a member of the inhibitor ofapoptosis proteins (IAP) family that has functionalroles in both cell division and apoptosis control[27,30] and is often found to be up-regulated incancers including neuroblastoma [31–33]. Interest-ingly, Sp1 is a key transcription factor that
regulates survivin expression [34–36] and previousstudies have shown that Sp1 is the primary targetfor TA [12–14,37]. Hence, apart from a criticalmember of the Sp family (Sp1), we also studied theimpact of this drug on the expression of survivinand key markers of apoptosis such as the expres-sion of cleaved poly ADP-ribose polymerase(c-PARP) and the activation of caspase 3/7 to un-derstand the mechanism of the anti-cancer activityof TA in neuroblastoma.
MATERIALS AND METHODS
Cell Lines and Chemicals
CHLA-90, LA1-55n, SMS-KCNR, SH-SY5Y, SHEP,Be2c, CMP 13Y are neuroblastoma cell lines de-rived from aggressive childhood tumors. Thesecells were grown in RPMI 1640 media (ATCC) sup-plemented with fetal bovine serum and 1% antibi-otic (penstrep) and maintained at 378C with 5%CO2 as previously described [38].Antibodies for Sp1 and survivin were obtained
from Santa Cruz Biotechnology (Santa Cruz, CA),c-PARP from Cell Signaling Technology (Danvers,MA), and b-actin from Sigma Chemical Co. (St.Louis, MO). Annexin-V/7-AAD kit for apoptosiswas purchased from BD Biosciences (San Jose, CA).Tolfenamic acid, dimethyl sulfoxide (DMSO), andprotease inhibitor were purchased from Sigma.Phosphate-buffered saline (PBS) was purchasedfrom Mediatch (Manassas, VA), and cell lysis bufferwas obtained from Invitrogen (Carlsbad, CA).Bicinchoninic acid (BCA) protein assay kit andSupersignal West Dura were purchased from Pierce(Rockford, IL) and Caspase-Glo 3/7 CellTiter Glokits were obtained from Promega (Madison, WI).
Western Immunoblot
Protein extracts were prepared from control(DMSO) and TA-treated cells and the expression ofproteins of interest was determined through West-ern blot. SH-SY5Y and LA1 55n cells were culturedin 100-mm dish and treated with 50 mM tolfe-namic acid. Cells were collected by trypsinizationat 24 h or 48 h post-treatment, washed once withPBS and re-suspended in cell lysis buffer contain-ing protease inhibitor. The cells were incubated at48C for 30 min with mixing every 5 min followedby centrifugation at 12,000 rpm for 15 min at 48C.Protein content in the supernatant was estimatedusing BCA protein assay kit. Protein extracts (25–30 mg protein) were boiled with loading buffer andseparated using a 10 or 12% SDS–polyacrylamidegels and transferred to nitrocellulose membranes.The blots were blocked with blocking buffer [5%(w/v) non-fat dry milk in 10 mmol/L Tris (pH 7.5),10 mmol/L sodium chloride, and 0.1% Tween 20]for 1 h at room temperature. Blots were incubatedwith the primary antibody for 2 h at room
2 ESLIN ET AL.
Molecular Carcinogenesis
temperature or overnight at 48C and processedwith appropriate HRP-conjugated secondary anti-body. Blots were developed using Supersignal WestDura.
Cell Viability
Cell viability of neuroblastoma cells was assessedwith CellTiter Glo kit (Promega) according toinstructions provided by the supplier. Briefly, cells(4,000/well in 100 ml) were plated in 96-wellwhite walled clear bottom plates (Lonza, Basel,Switzerland) and incubated for 24 h at 378C in acell culture incubator. Cells were then treated withincreasing concentration of tolfenamic acid (25,50, and 100 mM) for 24, 48, and 72 h. At the endof the incubation period, 100 ml of assay reagentwas added, mixed and the plate was incubated inthe dark for 15 min. Luminescence values wereobtained from each well using FLUOstar Optima(BMG LABTECH) plate reader. All treatments wereperformed in triplicate and the data were normal-ized to control (DMSO-treated) cells and plotted aspercent viable cells versus drug concentration.
Cell Cycle Analysis
SH-SY5Y and LA1 55n were plated in 100 mmdish and treated with 50 mM tolfenamic acid. Cellswere harvested at 24 h or 48 h post-treatment andprocessed for cell cycle analysis. Cells were washedonce with PBS and fixed in cold 70% ethanol for24 h at 48C. Fixed cells were then centrifuged andthe cell pellets were washed with PBS. Then thecells were resuspended in propidium iodide (PI)buffer (0.20 mg/ml PI, 20 mg/ml RNAse A in PBS)and incubated for 15 min at room temperature,in the dark. Cell cycle distribution was evaluatedusing BD FACSCalibur flow cytometer. Data wereanalyzed using FlowJo software (Tree Star, Inc.,Ashland, OR) and represented as percent cell inG0/G1, S, and G2/M [12].
Cell Apoptosis Using Flow Cytometry
Apoptotic cells were measured using Annexin V-PE/7-AAD Apoptosis detection kit (BD Biosciences).Briefly, cells were harvested after treatment withtolfenamic acid and incubated with annexin V-PEantibody and 7-AAD for 15 min in 1� bindingbuffer. SH-SY5Y and LA1 55n cells were analyzedusing BD FACSCalibur flow cytometer and datawere analyzed using FlowJo software (Tree Star,Inc., Ashland, OR).
Caspase Activation Assay
The effect of TA on activated caspases was evalu-ated using caspase-Glo 3/7 kit (Promega) accordingto the instructions provided by the supplier.Briefly, LA1 55n and SH-SY5Y cells (4,000/well in100 ml) were plated in a 96-well white walled clearbottom plate (Lonza). Twenty-four hours after
seeding, cells were treated with DMSO (control) or35 or 50 mM TA. Following 24 and 48 h post-treat-ment, the assay reagent (100 ml/well) was added,mixed and the plates were incubated in the darkfor 30–60 min. After the incubation luminescencewas measured in a Optima plate reader. All thetreatments were performed in triplicate and thedata were presented as mean � SD.
RESULTS
Tolfenamic Acid Inhibits Neuroblastoma Cell GrowthIn Vitro
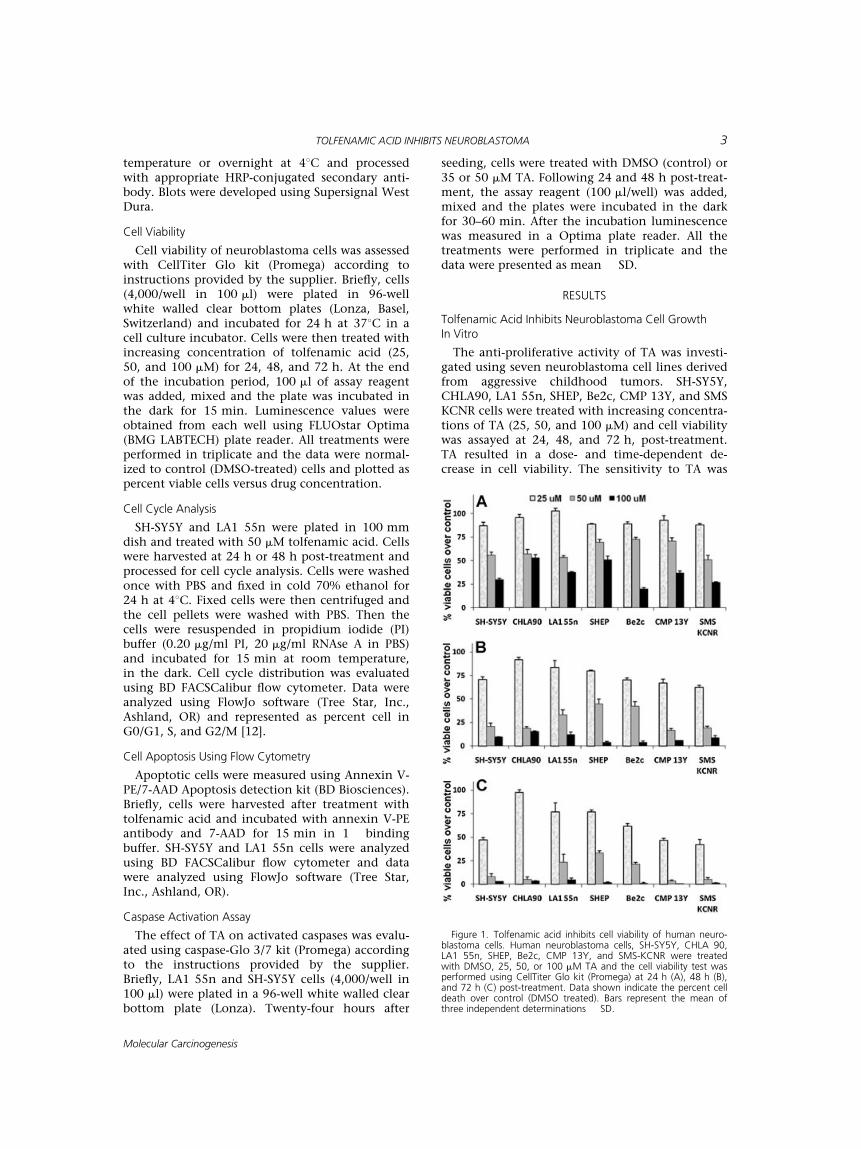
The anti-proliferative activity of TA was investi-gated using seven neuroblastoma cell lines derivedfrom aggressive childhood tumors. SH-SY5Y,CHLA90, LA1 55n, SHEP, Be2c, CMP 13Y, and SMSKCNR cells were treated with increasing concentra-tions of TA (25, 50, and 100 mM) and cell viabilitywas assayed at 24, 48, and 72 h, post-treatment.TA resulted in a dose- and time-dependent de-crease in cell viability. The sensitivity to TA was
Figure 1. Tolfenamic acid inhibits cell viability of human neuro-blastoma cells. Human neuroblastoma cells, SH-SY5Y, CHLA 90,LA1 55n, SHEP, Be2c, CMP 13Y, and SMS-KCNR were treatedwith DMSO, 25, 50, or 100 mM TA and the cell viability test wasperformed using CellTiter Glo kit (Promega) at 24 h (A), 48 h (B),and 72 h (C) post-treatment. Data shown indicate the percent celldeath over control (DMSO treated). Bars represent the mean ofthree independent determinations � SD.
TOLFENAMIC ACID INHIBITS NEUROBLASTOMA 3
Molecular Carcinogenesis
more prominent at later time points and higherdosage (Figure 1; Supplementary data Table 1).TA-induced cell growth inhibition was 20–40%within 48 h with 25 mM, 60–80% with 50 mM, and85–95% with 100 mM. At 72 h post-treatment, thecell death was 40–60% with 25 mM, 70–90% with50 mM, and >95% with 100 mM. The IC50 valuesof these cell lines ranged between 40 and 50 mM(SH-SY5Y: 43.9 mM; CHLA90: 50.7 mM; LA1 55n:51.5 mM; SHEP: 50.9 mM; Be2c: 40.9 mM; CMP13Y: 47.9 mM; SMS KCNR: 41.1 mM) at 48 h post-treatment. These results show that neuroblastomacells are sensitive to even low/therapeutic concen-trations of TA and has anti-proliferative activity onall tested neuroblastoma cell lines. The plasmaconcentration of TA in human studies has beenmeasured at 40 mM following therapeutic doses ofthe drug. Since the effect of TA is very low at25 mM we have conducted dose and time responsestudies using 35 and 50 mM of TA.
Tolfenamic Acid Inhibits the Expression of Sp1and Survivin
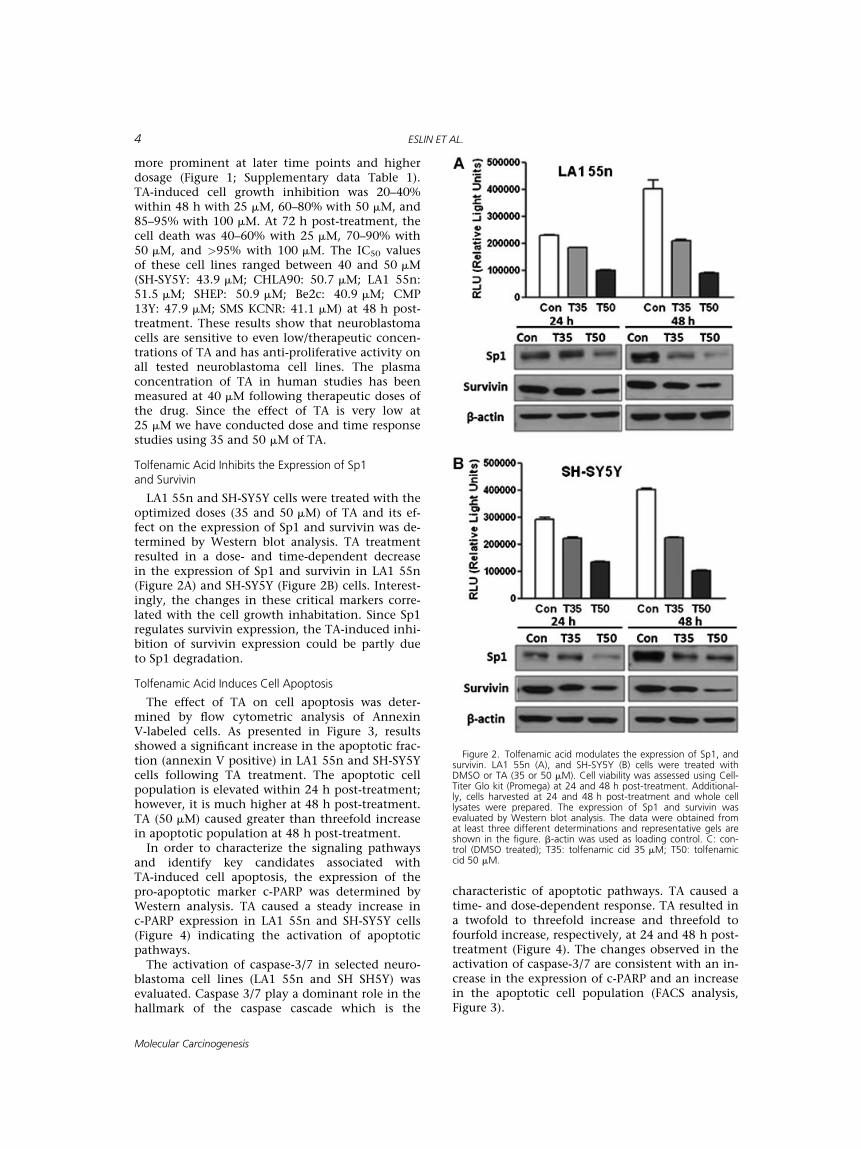
LA1 55n and SH-SY5Y cells were treated with theoptimized doses (35 and 50 mM) of TA and its ef-fect on the expression of Sp1 and survivin was de-termined by Western blot analysis. TA treatmentresulted in a dose- and time-dependent decreasein the expression of Sp1 and survivin in LA1 55n(Figure 2A) and SH-SY5Y (Figure 2B) cells. Interest-ingly, the changes in these critical markers corre-lated with the cell growth inhabitation. Since Sp1regulates survivin expression, the TA-induced inhi-bition of survivin expression could be partly dueto Sp1 degradation.
Tolfenamic Acid Induces Cell Apoptosis
The effect of TA on cell apoptosis was deter-mined by flow cytometric analysis of AnnexinV-labeled cells. As presented in Figure 3, resultsshowed a significant increase in the apoptotic frac-tion (annexin V positive) in LA1 55n and SH-SY5Ycells following TA treatment. The apoptotic cellpopulation is elevated within 24 h post-treatment;however, it is much higher at 48 h post-treatment.TA (50 mM) caused greater than threefold increasein apoptotic population at 48 h post-treatment.
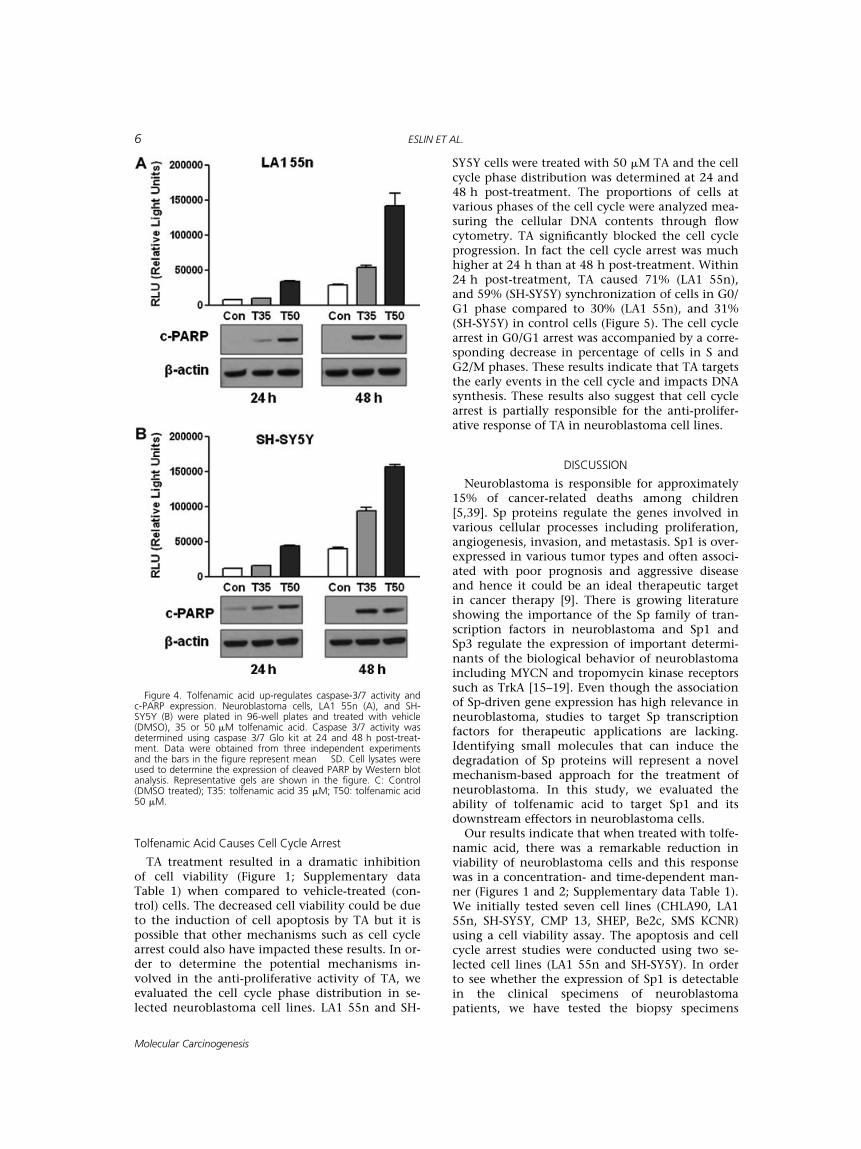
In order to characterize the signaling pathwaysand identify key candidates associated withTA-induced cell apoptosis, the expression of thepro-apoptotic marker c-PARP was determined byWestern analysis. TA caused a steady increase inc-PARP expression in LA1 55n and SH-SY5Y cells(Figure 4) indicating the activation of apoptoticpathways.
The activation of caspase-3/7 in selected neuro-blastoma cell lines (LA1 55n and SH SH5Y) wasevaluated. Caspase 3/7 play a dominant role in thehallmark of the caspase cascade which is the
characteristic of apoptotic pathways. TA caused atime- and dose-dependent response. TA resulted ina twofold to threefold increase and threefold tofourfold increase, respectively, at 24 and 48 h post-treatment (Figure 4). The changes observed in theactivation of caspase-3/7 are consistent with an in-crease in the expression of c-PARP and an increasein the apoptotic cell population (FACS analysis,Figure 3).
Figure 2. Tolfenamic acid modulates the expression of Sp1, andsurvivin. LA1 55n (A), and SH-SY5Y (B) cells were treated withDMSO or TA (35 or 50 mM). Cell viability was assessed using Cell-Titer Glo kit (Promega) at 24 and 48 h post-treatment. Additional-ly, cells harvested at 24 and 48 h post-treatment and whole celllysates were prepared. The expression of Sp1 and survivin wasevaluated by Western blot analysis. The data were obtained fromat least three different determinations and representative gels areshown in the figure. b-actin was used as loading control. C: con-trol (DMSO treated); T35: tolfenamic cid 35 mM; T50: tolfenamiccid 50 mM.
4 ESLIN ET AL.
Molecular Carcinogenesis
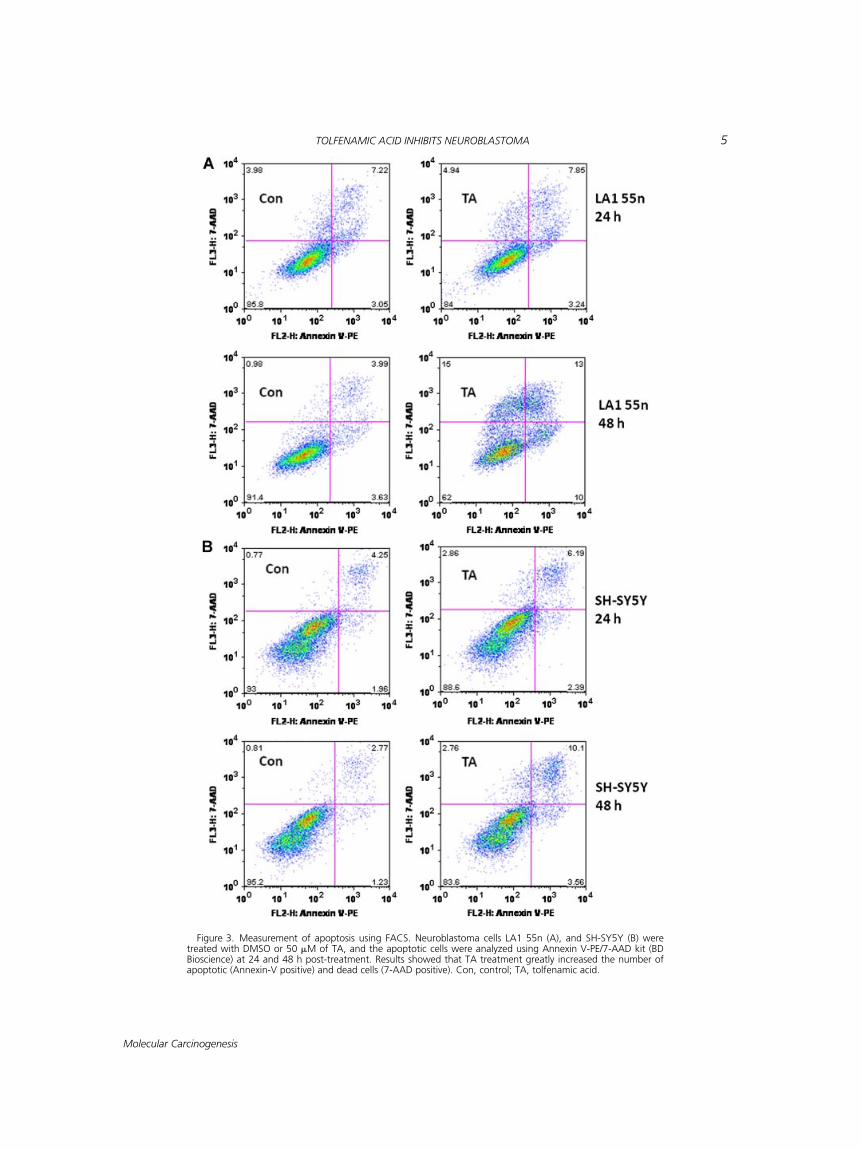
Figure 3. Measurement of apoptosis using FACS. Neuroblastoma cells LA1 55n (A), and SH-SY5Y (B) weretreated with DMSO or 50 mM of TA, and the apoptotic cells were analyzed using Annexin V-PE/7-AAD kit (BDBioscience) at 24 and 48 h post-treatment. Results showed that TA treatment greatly increased the number ofapoptotic (Annexin-V positive) and dead cells (7-AAD positive). Con, control; TA, tolfenamic acid.
TOLFENAMIC ACID INHIBITS NEUROBLASTOMA 5
Molecular Carcinogenesis
Tolfenamic Acid Causes Cell Cycle Arrest
TA treatment resulted in a dramatic inhibitionof cell viability (Figure 1; Supplementary dataTable 1) when compared to vehicle-treated (con-trol) cells. The decreased cell viability could be dueto the induction of cell apoptosis by TA but it ispossible that other mechanisms such as cell cyclearrest could also have impacted these results. In or-der to determine the potential mechanisms in-volved in the anti-proliferative activity of TA, weevaluated the cell cycle phase distribution in se-lected neuroblastoma cell lines. LA1 55n and SH-
SY5Y cells were treated with 50 mM TA and the cellcycle phase distribution was determined at 24 and48 h post-treatment. The proportions of cells atvarious phases of the cell cycle were analyzed mea-suring the cellular DNA contents through flowcytometry. TA significantly blocked the cell cycleprogression. In fact the cell cycle arrest was muchhigher at 24 h than at 48 h post-treatment. Within24 h post-treatment, TA caused 71% (LA1 55n),and 59% (SH-SY5Y) synchronization of cells in G0/G1 phase compared to 30% (LA1 55n), and 31%(SH-SY5Y) in control cells (Figure 5). The cell cyclearrest in G0/G1 arrest was accompanied by a corre-sponding decrease in percentage of cells in S andG2/M phases. These results indicate that TA targetsthe early events in the cell cycle and impacts DNAsynthesis. These results also suggest that cell cyclearrest is partially responsible for the anti-prolifer-ative response of TA in neuroblastoma cell lines.
DISCUSSION
Neuroblastoma is responsible for approximately15% of cancer-related deaths among children[5,39]. Sp proteins regulate the genes involved invarious cellular processes including proliferation,angiogenesis, invasion, and metastasis. Sp1 is over-expressed in various tumor types and often associ-ated with poor prognosis and aggressive diseaseand hence it could be an ideal therapeutic targetin cancer therapy [9]. There is growing literatureshowing the importance of the Sp family of tran-scription factors in neuroblastoma and Sp1 andSp3 regulate the expression of important determi-nants of the biological behavior of neuroblastomaincluding MYCN and tropomycin kinase receptorssuch as TrkA [15–19]. Even though the associationof Sp-driven gene expression has high relevance inneuroblastoma, studies to target Sp transcriptionfactors for therapeutic applications are lacking.Identifying small molecules that can induce thedegradation of Sp proteins will represent a novelmechanism-based approach for the treatment ofneuroblastoma. In this study, we evaluated theability of tolfenamic acid to target Sp1 and itsdownstream effectors in neuroblastoma cells.Our results indicate that when treated with tolfe-
namic acid, there was a remarkable reduction inviability of neuroblastoma cells and this responsewas in a concentration- and time-dependent man-ner (Figures 1 and 2; Supplementary data Table 1).We initially tested seven cell lines (CHLA90, LA155n, SH-SY5Y, CMP 13, SHEP, Be2c, SMS KCNR)using a cell viability assay. The apoptosis and cellcycle arrest studies were conducted using two se-lected cell lines (LA1 55n and SH-SY5Y). In orderto see whether the expression of Sp1 is detectablein the clinical specimens of neuroblastomapatients, we have tested the biopsy specimens
Figure 4. Tolfenamic acid up-regulates caspase-3/7 activity andc-PARP expression. Neuroblastoma cells, LA1 55n (A), and SH-SY5Y (B) were plated in 96-well plates and treated with vehicle(DMSO), 35 or 50 mM tolfenamic acid. Caspase 3/7 activity wasdetermined using caspase 3/7 Glo kit at 24 and 48 h post-treat-ment. Data were obtained from three independent experimentsand the bars in the figure represent mean � SD. Cell lysates wereused to determine the expression of cleaved PARP by Western blotanalysis. Representative gels are shown in the figure. C: Control(DMSO treated); T35: tolfenamic acid 35 mM; T50: tolfenamic acid50 mM.
6 ESLIN ET AL.
Molecular Carcinogenesis
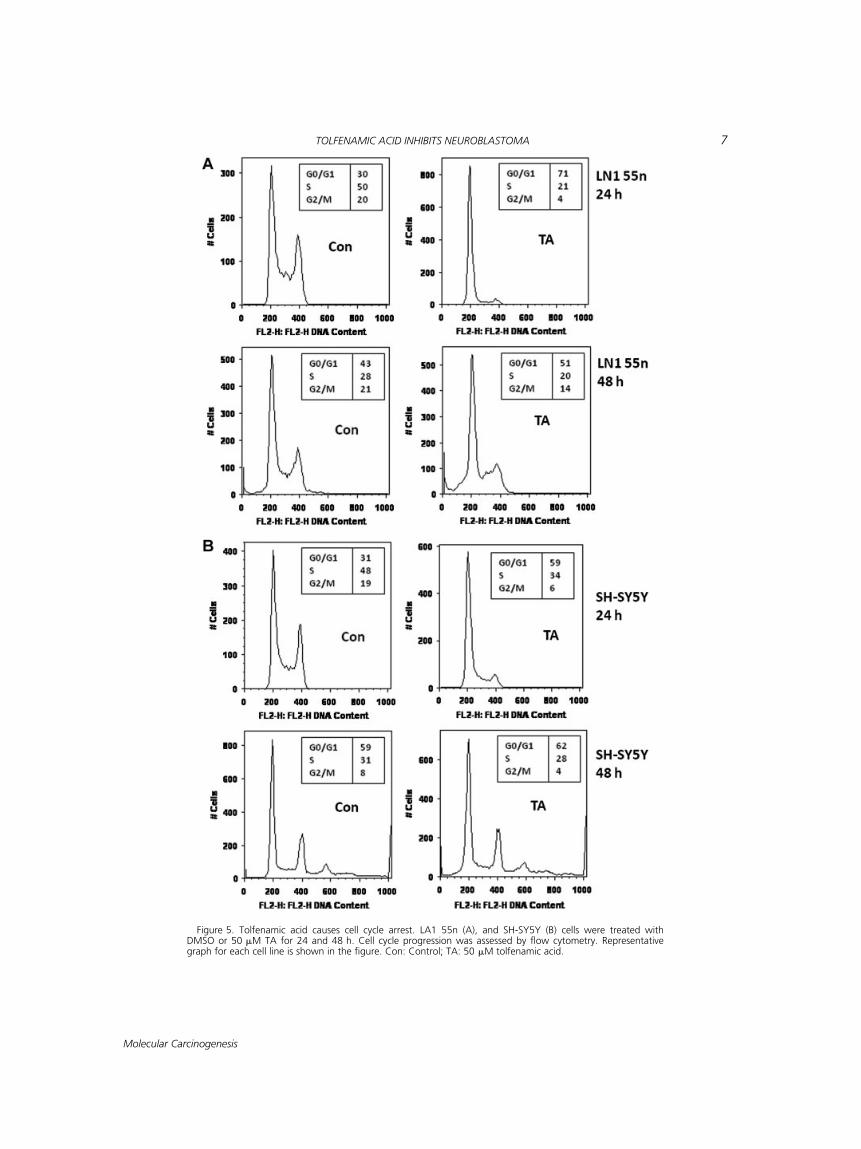
Figure 5. Tolfenamic acid causes cell cycle arrest. LA1 55n (A), and SH-SY5Y (B) cells were treated withDMSO or 50 mM TA for 24 and 48 h. Cell cycle progression was assessed by flow cytometry. Representativegraph for each cell line is shown in the figure. Con: Control; TA: 50 mM tolfenamic acid.
TOLFENAMIC ACID INHIBITS NEUROBLASTOMA 7
Molecular Carcinogenesis

obtained from a few neuroblastoma patients. Theimmunohistochemical analysis of these clinicalspecimens showed a distinct staining for Sp1 (Sup-plementary data Figure 1) further supporting theimportance of Sp1 in this malignancy.
Research from our laboratory and others hasdemonstrated that tolfenamic acid inhibits cellproliferation and tumor growth in pancreatic[8,40], esophageal [41], lung [23], and ovarian can-cer [12] models. Consistent with these findings,Sp1 is significantly decreased following 24 and48 h post-treatment (Figure 2) suggesting thatthe effect of tolfenamic acid was at least in partmediated through the degradation of Sp proteinsultimately modulating the expression of Sp-dependent pro-apoptotic and/or survival genes inneuroblastoma. Survivin is involved in regulatingapoptosis and cell division and its over-expressedin several cancers is associated with poor prognosisprimarily through its ability to inhibit apoptosisand induce mitogenesis [42]. Expression of survi-vin is regulated by Sp1 and Sp3 transcriptionfactors [36,43]. TA caused a significant decrease inthe expression of survivin (Figure 2) potentiallythrough the degradation of Sp1.
This study has shown that the anti-proliferativeactivity of TA is partially due to its induction ofapoptosis. In order to test the effect of TA on cellapoptosis, we used flow cytometry and measuredthe apoptotic cell population. Flow cytometricanalysis using Annexin V showed a significant in-crease in Annexin V-stained cells following TA-treatment. Annexin-V staining revealed a threefoldto fivefold increase in the percentage of apoptoticcells when treated with TA (Figure 3). We furtheranalyzed the changes in the expressions ofcandidate proteins associated with cell apoptosisthrough Western blot analysis. Consistent withthe flow cytometry data, we observed that TA in-creased the expression of the pro-apoptotic markerc-PARP (Figure 4). To further confirm the apoptoticprocesses involved in TA-induced apoptosis in neu-roblastoma cells, we have evaluated the alterationsin the activity of caspase 3/7. TA resulted in three-fold to fourfold increase in caspase 3/7 activity(Figure 4) that is consistent with a decrease in cellviability (Figure 1; Supplementary data Table 1),and also the inhibition of survivin (Figure 2).
Unrestrained growth and deregulated cell apo-ptosis are among the primary reasons for theaggressive nature of neuroblastoma [33,44]. Apo-ptosis is primarily mediated via membrane deathreceptors and/or through activation/inactivationof pro- and anti-apoptotic proteins. These process-es direct the release of cytochrome c and form theapoptosome complex and ultimately result in cas-pase 9 cleavage and activation of effector caspases.Interestingly, the activities of effector caspases arecountered by the members of IAP family. A key
member of IAP family, survivin is often found up-regulated in a number of human cancers includingneuroblastoma [31–33]. Survivin is known to havefunctional roles in both cell division and apoptosiscontrol [27,30]. Since it is evident that survivininhibits effector caspases [33,45–47], TA-induceddown-regulation of survivin could be responsiblefor such a dramatic increase in effector caspases(3/7) and enhancing cell apoptosis in TA-treatedcell lines (Figure 4).TA treatment resulted in a dramatic inhibition
of cell viability of neuroblastoma cells (Figure 1;Supplmentary data Table 1). It is possible that mul-tiple mechanisms are involved in such a strong in-hibition and may be partially linked to cell cyclearrest. To investigate whether TA impacted the cellcycle, we have performed the cell cycle analysisusing flow cytometry and the results revealed thatTA is causing cell cycle arrest. Since the cell cyclearrest could be an early event in cell growth inhibi-tion, we have evaluated the cell cycle arrest at 24 hpost-treatment. As presented in Figure 5, a majori-ty of cells were arrested in G0/G1 phase with 24 htreatment with TA (50 mM). In eukaryotic cells, co-ordinated actions of various cellular proteins regu-late growth and drive the cell cycle into variousphases, finally, leading to cell division. From theseresults, it is clear that TA is acting at the earlyphases of the cell cycle keeping an increased num-ber in G0/G1 phase and decreasing the progressioninto further phases of the cell cycle (S and G2/M).In the mammalian tumorigenic process, G1 phaseis very important and impacts cell proliferation.Typically, the cells that enter into ‘‘S’’ phase auto-matically progress towards further stages of the cellcycle, and hence, the majority of anti-canceragents that affect the cell cycle target at ‘‘G1’’phase. The effect of TA on causing ‘‘G0/G1’’ cellcycle arrest could be attributed to its effects oncyclins, cyclin-dependent kinases (e.g., CDK2,CDK4), and CDK-inhibitors (p21/p27) which arecritical components of the cell cycle machinery[48–52]; however, the specific underlying mecha-nism of TA-induced cell cycle arrest in neuroblas-toma cells is as yet unknown but currently underinvestigation in our laboratory.As per the declaration from the suppliers and
other studies [53–55], similar to many NSAIDs,tolfenamic acid may cause gastrointestinal distur-bances including, nausea, vomiting, diarrhea,constipation, dyspepsia, and abdominal pain; how-ever, gastrointestinal bleeding/ulceration occursonly with very high oral doses. Other rarelyreported side effects of TA include headache, trem-or, fatigue, vertigo, euphoria, thrombocytopenia,anemia, and leucopenia. In animal studies, tolfe-namic acid showed the lowest gastro-ulcero-genicity among 11 tested NSAIDS includingketoprofen, naproxen and indomethacin [56]. The
8 ESLIN ET AL.
Molecular Carcinogenesis

pharmacokinetics of tolfenamic acid after intrave-nous as well as oral administration has been stud-ied earlier and is described by a two-compartmentmodel. The peak plasma concentrations areachieved in 1–2 h after oral administration. Theplasma concentration of TA was 40 mM in men fol-lowing the oral administration of the therapeuticdose (800 mg) [57]. Due to variations in humanstudies and laboratory experiments, the optimizedconcentration was 50 mM for in vitro studies[8,12,23]. With limited side effects and a therapeu-tic response at 35–50 mM concentration, which iswithin the known obtainable plasma concentra-tions (40 mM), research on this agent showspromise moving into clinical applications inneuroblastoma.In summary, in this study, we report that the
small molecule (NSAID), tolfenamic acid inhibitsneuroblastoma cell growth even at low/therapeuticconcentrations. Tolfenamic acid induces cell apo-ptosis and causes cell cycle arrest potentiallythrough suppressing Sp1 and survivin and up-regu-lating critical regulators of apoptosis, includingcaspase-3/7 and c-PARP. The results presented inthis investigation suggest that targeting survivinthrough Sp1 degradation could be a promisingstrategy for the treatment of neuroblastoma. Usingsmall molecules, such as tolfenamic acid, to targetkey transcription factors associated with cancer is anovel approach and the results of these findingsprovide better understanding of genes and molecu-lar pathways that are susceptible to be targetedfor the treatment of this malignancy. These pre-clinical findings demonstrate the ability of TA as apotential anti-neoplastic agent for the treatment ofneuroblastoma.
ACKNOWLEDGMENTS
We thank MD Anderson Cancer Center Orlan-do’s Cancer Research Institute for providing finan-cial assistance. This work is partially supported bya Hyundai Hope on Wheels Scholar Award (DE)and Runway To Hope (DE).
REFERENCES
1. Bowen KA, Chung DH. Recent advances in neuroblasto-ma. Curr Opin Pediatr 2009;21:350–356.
2. Maris JM. Recent advances in neuroblastoma. N Engl JMed 2010;362:2202–2211.
3. Schwab M, Evans A. Neuroblastoma—Developmental andmolecular biology meet therapy design. Cancer Lett 2003;197:1.
4. Schwab M, Westermann F, Hero B, Berthold F. Neuroblas-toma: Biology and molecular and chromosomal patholo-gy. Lancet Oncol 2003;4:472–480.
5. van Ginkel PR, Sareen D, Subramanian L, et al. Resveratrolinhibits tumor growth of human neuroblastoma andmediates apoptosis by directly targeting mitochondria.Clin Cancer Res 2007;13:5162–5169.
6. Carosio R, Zuccari G, Orienti I, Mangraviti S, MontaldoPG. Sodium ascorbate induces apoptosis in neuroblastoma
cell lines by interfering with iron uptake. Mol Cancer2007;6:55.
7. Ebb DH, Green DM, Shamberger RC, Tarbell NJ. Solidtumors of childhood. In: DeVita VT, Hellman S, RosenbergSA, editors. Cancer: Principles and practice of oncology, 6thedition. Philadelphia: Williams andWilkins; 2001. p. 2169.
8. Abdelrahim M, Baker CH, Abbruzzese JL, Safe S. Tolfe-namic acid and pancreatic cancer growth, angiogenesis,and Sp protein degradation. J Natl Cancer Inst 2006;98:855–8868.
9. Basha R, Baker CH, Sankpal UT, et al. Therapeutic applica-tions of NSAIDS in cancer: Special emphasis on tolfenamicacid. Front Biosci (Schol Ed) 2011;3:797–805.
10. Chang WC, Chen BK. Transcription factor Sp1 functionsas an anchor protein in gene transcription of human12(S)-lipoxygenase. Biochem Biophys Res Commun 2005;338:117–121.
11. Lu S, Archer MC. Sp1 coordinately regulates de novo lipo-genesis and proliferation in cancer cells. Int J Cancer2009;2:416–425.
12. Basha R, Ingersoll SB, Sankpal UT, et al. Tolfenamic acidinhibits ovarian cancer cell growth and decreases the ex-pression of c-Met and survivin through suppressing speci-ficity protein transcription factors. Gynecol Oncol 2011;122:163–170.
13. Konduri S, Colon J, Baker CH, et al. Tolfenamic acidenhances pancreatic cancer cell and tumor response toradiation therapy by inhibiting survivin protein expression.Mol Cancer Ther 2009;8:533–542.
14. Maliakal P, Abdelrahim M, Sankpal UT, et al. Chemo-preventive effects of tolfenamic acid against esophagealtumorigenesis in rats. Invest New Drugs 2011; [Epubahead of print].
15. Eggert A, Grotzer MA, Ikegaki N, Liu XG, Evans AE,Brodeur GM. Expression of the neurotrophin receptorTrkA down-regulates expression and function of angio-genic stimulators in SH-SY5Y neuroblastoma cells. CancerRes 2002;62:1802–1808.
16. Look AT, Hayes FA, Shuster JJ, et al. Clinical relevance oftumor cell ploidy and N-myc gene amplification in child-hood neuroblastoma: A Pediatric Oncology Group study.J Clin Oncol 1991;9:581–591.
17. Nakagawara A, Arima-Nakagawara M, Scavarda NJ, AzarCG, Cantor AB, Brodeur GM. Association between highlevels of expression of the TRK gene and favorable out-come in human neuroblastoma. N Engl J Med 1993; 328:847–854.
18. Seeger RC, Brodeur GM, Sather H, et al. Associationof multiple copies of the N-myc oncogene with rapidprogression of neuroblastomas. N Engl J Med 1985;313:1111–1116.
19. Tang XX, Zhao H, Kung B, et al. The MYCN enigma: Sig-nificance of MYCN expression in neuroblastoma. CancerRes 2006;66:2826–2833.
20. Gately S, Li WW. Multiple roles of COX-2 in tumor angio-genesis: A target for antiangiogenic therapy. Semin Oncol2004;31:2–11.
21. Jacoby RF, Seibert K, Cole CE, Kelloff G, Lubet RA. Thecyclooxygenase-2 inhibitor celecoxib is a potent preventiveand therapeutic agent in the min mouse model of adeno-matous polyposis. Cancer Res 2000;60:5040–5044.
22. Tarnawski AS, Jones MK. Inhibition of angiogenesis byNSAIDs: Molecular mechanisms and clinical implications.J Mol Med 2003;81:627–636.
23. Colon J, Basha MR, Madero-Visbal R, et al. Tolfenamicacid decreases c-Met expression through Sp proteins deg-radation and inhibits lung cancer cells growth and tumorformation in orthotopic mice. Invest New Drugs 2011;29:41–51.
24. Yao JC, Wang L, Wei D, et al. Association between ex-pression of transcription factor Sp1 and increased vascular
TOLFENAMIC ACID INHIBITS NEUROBLASTOMA 9
Molecular Carcinogenesis

endothelial growth factor expression, advanced stage,and poor survival in patients with resected gastric cancer.Clin Cancer Res 2004;10:4109–4117.
25. Adwan LI, Basha R, Abdelrahim M, Subaiea GM, ZawiaNH. Tolfenamic acid interrupts the de novo synthesis ofthe beta-amyloid precursor protein and lowers amyloidbeta via a transcriptional pathway. Curr Alzheimer Res2011;8:385–392.
26. Subaiea GM, Alansi BH, Serra DA, Alwan M, Zawia NH.The ability of tolfenamic acid to penetrate the brain: Amodel for testing the brain disposition of candidate Alz-heimer’s drugs using multiple platforms. Curr AlzheimerRes 2011;8:860–867.
27. Deveraux QL, Reed JC. IAP family proteins—Suppressorsof apoptosis. Genes Dev 1999;13:239–252.
28. Doolittle H, Morel A, Talbot D. Survivin-directed antican-cer therapies. A review of pre-clinical data and early-phaseclinical trials. Eur Oncol 2010;6:10–14.
29. Mita AC, Mita MM, Nawrocki ST, Giles FJ. Survivin: Keyregulator of mitosis and apoptosis and novel target forcancer therapeutics. Clin Cancer Res 2008;14:5000–5005.
30. Altieri DC. The case for survivin as a regulator of microtu-bule dynamics and cell-death decisions. Curr Opin CellBiol 2006;18:609–615.
31. Adida C, Berrebi D, Peuchmaur M, Reyes-Mugica M,Altieri DC. Anti-apoptosis gene, survivin, and prognosis ofneuroblastoma. Lancet 1998;351:882–883.
32. Islam A, Kageyama H, Takada N, et al. High expressionof Survivin, mapped to 17q25, is significantly associatedwith poor prognostic factors and promotes cell survivalin human neuroblastoma. Oncogene 2000;19:617–623.
33. Obexer P, Hagenbuchner J, Unterkircher T, et al. Repres-sion of BIRC5/survivin by FOXO3/FKHRL1 sensitizes humanneuroblastoma cells to DNA damage-induced apoptosis.Mol Biol Cell 2009;20:2041–2048.
34. Chen Y, Wang X, Li W, et al. Sp1 upregulates survivinexpression in adenocarcinoma of lung cell line A549. AnatRec (Hoboken) 2011;294:774–780.
35. Mityaev MV, Kopantzev EP, Buzdin AA, Vinogradova TV,Sverdlov ED. Functional significance of a putative sp1transcription factor binding site in the survivin gene pro-moter. Biochemistry (Mosc) 2008;73:1183–1191.
36. Xu R, Zhang P, Huang J, Ge S, Lu J, Qian G. Sp1 and Sp3regulate basal transcription of the survivin gene. BiochemBiophys Res Commun 2007;356:286–292.
37. Aravindan N, Madhusoodhanan R, Ahmad S, Johnson D,Herman TS. Curcumin inhibits NFkappaB mediated radio-protection and modulate apoptosis related genes in hu-man neuroblastoma cells. Cancer Biol Ther 2008;7:569–576.
38. Saulnier Sholler GL, Brard L, Straub JA, et al. Nifurtimoxinduces apoptosis of neuroblastoma cells in vitro and invivo. J Pediatr Hematol Oncol 2009;31:187–193.
39. Matthay KK. Neuroblastoma: A clinical challenge and bio-logic puzzle. CA Cancer J Clin 1995;45:179–192.
40. Abdelrahim M, Safe S, Baker C, Abudayyeh A. RNAi andcancer: Implications and applications. J RNAi Gene Silenc-ing 2006;2:136–145.
41. Papineni S, Chintharlapalli S, Abdelrahim M, et al. Tolfe-namic acid inhibits esophageal cancer through repressionof specificity proteins and c-Met. Carcinogenesis 2009;30:1193–1201.
42. Pennati M, Folini M, Zaffaroni N. Targeting survivin incancer therapy: Fulfilled promises and open questions.Carcinogenesis 2007;28:1133–1139.
43. Li Y, Xie M, Yang J, et al. The expression of antiapoptoticprotein survivin is transcriptionally upregulated by DEC1primarily through multiple sp1 binding sites in the proxi-mal promoter. Oncogene 2006;25:3296–3306.
44. Li Z, Zhang J, Liu Z, Woo CW, Thiele CJ. Downregulationof Bim by brain-derived neurotrophic factor activation ofTrkB protects neuroblastoma cells from paclitaxel but notetoposide or cisplatin-induced cell death. Cell Death Differ2007;14:318–326.
45. Grossman D, Kim PJ, Schechner JS, Altieri DC. Inhibitionof melanoma tumor growth in vivo by survivin targeting.Proc Natl Acad Sci USA 2001;98:635–640.
46. Shankar SL, Mani S, O’Guin KN, Kandimalla ER, AgrawalS, Shafit-Zagardo B. Survivin inhibition induces humanneural tumor cell death through caspase-independent and-dependent pathways. J Neurochem 2001;79:426–436.
47. Zhang XD, Gillespie SK, Hersey P. Staurosporine inducesapoptosis of melanoma by both caspase-dependent and -independent apoptotic pathways. Mol Cancer Ther 2004;3:187–197.
48. Collins I, Garrett MD. Targeting the cell division cycle incancer: CDK and cell cycle checkpoint kinase inhibitors.Curr Opin Pharmacol 2005;5:366–373.
49. Craig C, Wersto R, Kim M, et al. A recombinant adenovi-rus expressing p27Kip1 induces cell cycle arrest and lossof cyclin-Cdk activity in human breast cancer cells. Onco-gene 1997;14:2283–2289.
50. Lee YM, Lim do Y, Cho HJ, et al. Piceatannol, a naturalstilbene from grapes, induces G1 cell cycle arrest in andro-gen-insensitive DU145 human prostate cancer cells via theinhibition of CDK activity. Cancer Lett 2009;285:166–173.
51. MacLachlan TK, Sang N, Giordano A. Cyclins, cyclin-dependent kinases and cdk inhibitors: Implications in cellcycle control and cancer. Crit Rev Eukaryot Gene Expr1995;5:127–156.
52. Paternot S, Bockstaele L, Bisteau X, Kooken H, CoulonvalK, Roger PP. Rb inactivation in cell cycle and cancer: Thepuzzle of highly regulated activating phosphorylation ofCDK4 versus constitutively active CDK-activating kinase.Cell Cycle 2010;9:689–699.
53. Hendel L, Larsen E, Bonnevie O. A comparative study ofthe influence of tolfenamic acid (Clotam) and diclofenacsodium (Voltaren) on the gastrointestinal mucosa inpatients with a history of NSAID-related dyspeptic symp-toms. Pharmacol Toxicol 1994;75:49–50.
54. Khalifah RG, Hignite CE, Pentikainen PJ, Penttila A,Neuvonen PJ. Human metabolism of tolfenamic acid. II.Structure of metabolites and C-13 NMR assignments offenamates. Eur J Drug Metab Pharmacokinet 1982;7:269–276.
55. Vale JA, Meredith TJ. Acute poisoning due to non-steroi-dal anti-inflammatory drugs. Clinical features and man-agement. Med Toxicol 1986;1:12–31.
56. Eskerod O. Gastrointestinal tolerance studies on tolfe-namic acid in humans and animals. Pharmacol Toxicol1994;75:44–48.
57. Pentikainen PJ, Tokola O, Alhava E, Penttila A. Pharmaco-kinetics of tolfenamic acid: Disposition in bile, blood andurine after intravenous administration to man. Eur J ClinPharmacol 1984;27:349–354.
10 ESLIN ET AL.
Molecular Carcinogenesis





























