Embed Size (px)
Citation preview

The gatekeeper residue and beyond: homologous calcium-dependent protein kinases as drug development targets forveterinarian Apicomplexa parasites
KATELYN R. KEYLOUN1†, MOLLY C. REID1†, RYAN CHOI1, YIFAN SONG2,ANNA M. W. FOX1, HEIDI K. HILLESLAND1, ZHONGSHENG ZHANG2,RAMASUBBARAO VIDADALA3, ETHAN A. MERRITT2, AUDREY O. T. LAU4,DUSTIN J. MALY3, ERKANG FAN2, LYNN K. BARRETT1, WESLEY C. VAN VOORHIS1
and KAYODE K. OJO1*1Department of Medicine, Division of Allergy and Infectious Diseases, Center for Emerging and Re-emerging InfectiousDiseases (CERID), University of Washington, Seattle, Seattle, WA, USA2Department of Biochemistry, University of Washington, Seattle, Seattle, WA, USA3Department of Chemistry, University of Washington, Seattle, Seattle, WA, USA4Veterinary Microbiology and Pathology, College of Veterinary Medicine, Washington State University, Pullman,WA, USA
(Received 4 March 2014; revised 28 April 2014; accepted 29 April 2014; first published online 13 June 2014)
SUMMARY
Specific roles of individual CDPKs vary, but in general they mediate essential biological functions necessary for parasitesurvival. A comparative analysis of the structure-activity relationships (SAR) of Neospora caninum, Eimeria tenella andBabesia bovis calcium-dependent protein kinases (CDPKs) together with those of Plasmodium falciparum, Cryptosporidiumparvum and Toxoplasma gondii was performed by screening against 333 bumped kinase inhibitors (BKIs). Structuralmodelling and experimental data revealed that residues other than the gatekeeper influence compound–protein interactionsresulting in distinct sensitivity profiles. We subsequently defined potential amino-acid structural influences within theATP-binding cavity for each orthologue necessary for consideration in the development of broad-spectrum apicomplexanCDPK inhibitors. Although the BKI library was developed for specific inhibition of glycine gatekeeper CDPKs combinedwith low inhibition of threonine gatekeeper human SRC kinase, some library compounds exhibit activity against serine- orthreonine-containing CDPKs. Divergent BKI sensitivity of CDPK homologues could be explained on the basis ofdifferences in the size and orientation of the hydrophobic pocket and specific variation at other amino-acid positions withinthe ATP-binding cavity. In particular, BbCDPK4 and PfCDPK1 are sensitive to a larger fraction of compounds thanEtCDPK1 despite the presence of a threonine gatekeeper in all three CDPKs.
Key words: Apicomplexa, calcium-dependent protein kinases, bumped kinase inhibitors.
INTRODUCTION
The Apicomplexa are a diverse group of unicellularparasites that infect humans and animals.Plasmodiumfalciparum and Toxoplasma gondii are among thebest-known Apicomplexa being among the leadingcauses of morbidity andmortality worldwide (Tenteret al. 2000; Guinovart et al. 2006).Malaria is endemicin 104 countries putting an estimated 3·4 billionpeople at risk, with 207 million reported cases and627000 deaths globally in 2012 (WHO 2013). Otherpathogens of importance include Cryptosporidium,Babesia, Eimeria and Neospora caninum whichcollectively can cause devastating and debilitating
encephalitis, diarrhoea and foetal abortion in the host.Treatment options, which rely heavily on chemo-therapeutics, are limited and an issue of immediateimportance due to increasing resistance of manyapicomplexans to available therapeutic agents (Nairet al. 2011). Currently available therapies includeantifolate and macrolide antimicrobials such assulfadiazine and pyrimethamine or clindamycin,spiramycin and imidocarb. All of these treatmentspresent obstacles by exhibiting poor pharmacokineticproperties, toxicity, teratogenicity, tolerability andallergenicity (Norrby, 1978; Reeves and Wilkinson,1979; Luft et al. 1993; Katlama et al. 1996).Most apicomplexan parasites are obligate intra-
cellular pathogens thatmust invade a host cell to growand replicate (Morrissette and Sibley, 2002). Theyhave distinctive structural and phenotypic charac-teristics that can be targeted for the developmentof safe and effective antimicrobial therapies. Recentstudies of apicomplexan kinase biology have† Authors contributed equally to this manuscript.
* Corresponding author: Department of Medicine,Division of Allergy and Infectious Diseases, Center forEmerging and Re-emerging Infectious Diseases (CERID),University of Washington, Seattle, WA 98109, USA.E-mail: [email protected]
1499SPECIAL ISSUE ARTICLE
Parasitology (2014), 141, 1499–1509. © Cambridge University Press 2014doi:10.1017/S0031182014000857

described the transmission of calcium signals underthe control of calcium-dependent protein kinases(CDPKs) to inter-related pathways as essential infacilitating host cell invasion, namely gliding motilityand microneme secretion (Lovett et al. 2002).Each CDPK has defined regulatory functions atdistinct stages of the apicomplexan life cycle, whichare integrated into various key phosphorylation-associated signalling events (Ojo et al. 2013).Hence, expression levels and specific functions ineach organism may vary based on the specific lifecycle requirements, which are not completely under-stood at present. However, we have previously shownthat interruption of these enzymes’ ability to transmitsignals by the application of specific inhibitorsresulted in inhibition of parasite cell replication andgrowth in in vitro and in vivo models for T. gondii(Ojo et al. 2010; Johnson et al. 2012; Doggett et al.2014), Cryptosporidium parvum (Murphy et al. 2010;Castellanos-Gonzalez et al. 2013) and P. falciparum(Ojo et al. 2012, 2014; Vidadala et al. 2014) withoutharmful effects to the hosts. The inhibitors’ librarystructure-activity relationships (SARs) for theseparasites are similar. A structure-activity relationshipis an iterative study that defines the contribution ofeach molecular group within an inhibitor’s chemicalstructure to the overall biological activity associatedwith it (inhibitor). Specific inhibition of CDPKs wasachieved using an adenosine-like pyrazolopyrimidinescaffold which binds to the hinge region of theactive site, similar to the binding motif of ATP, butwith an added bulky aromatic R1-substituent at theC3-position – the so-called ‘bump’ of bumped kinaseinhibitors (BKIs) (Ojo et al. 2010). Topologically,the aromatic R1-group projects into a hydrophobicpocket adjacent to the gatekeeper position of theATP-binding site (the gatekeeper pocket), a featureexclusive to protein kinases with small gatekeepers,such as many apicomplexan CDPKs. Most mam-malian kinases typically have larger gatekeeperresidues that exclude the ‘bump’ of BKIs, althougha fewmammalian protein kinases like SRCdo containrelatively smaller gatekeeper residues (threonine).Therefore, great care must be taken to maintainselectivity towards apicomplexan CDPKs over suchhuman kinases. Fortunately, preliminary results alsosuggest that slight topographical deviations of theATP-binding region of apicomplexan CDPKs fromSRC, namely, the ribose binding pocket, also conferalternative selectivity to BKIs. Larson et al. (2012)previously demonstrated that mammalian kinasessuch as SRC have shallow ribose pockets as comparedwith apicomplexan CDPKs. Co-crystal structures ofBKIs in complex with TgCDPK1 have revealed ahydrogen bond between residue E135, a conservedglutamic acid found within the ribose bindingpocket and shared among other apicomplexanCDPKs, and inhibitors with a 4-piperidinylmethy-lene R2 group, an interaction which further enhances
selectivity. Together, the gatekeeper residue and thediffering topology of the ribose binding pocket allowselectivity for broad-spectrum anti-apicomplexaninhibitors with specificity for CDPKs over humankinases.
Furthering our work with apicomplexan CDPKs,this study reports the analysis of sensitivity ofadditional apicomplexan CDPK orthologues to acompound library of 333 BKIs. Using purifiedCDPK orthologues of veterinary pathogens Eimeriatenella, N. caninum and Babesia bovis, we investigatethe impact of BKIs on several pathogens of greatrelevance to food security and veterinary medicineand further define potential structural constraints forconsideration in the development of broad-spectrumapicomplexan CDPK inhibitors.
MATERIALS AND METHODS
Compound library
Chemical synthesis of BKIs used in this study waspreviously described (Murphy et al. 2010; Johnsonet al. 2012; Zhang et al. 2012). The library iscomprised of 53 benzoylbenzimidazole; 14 imidazo[1, 5-a] pyrazine analogues, 259 pyrazolo [3,4-d]pyrimidine analogues, with 7 other bumped inhibi-tors. The purity of all compounds (>98%) wasconfirmed by reverse-phase HPLC and 1H-NMR.The <2% impurity should have an insignificanteffect on inhibitory activity at the final reactionconcentration of 3 μM.
Molecular cloning, protein expression and purificationof parasite CDPK enzymes
The complete coding region of wild-type apicom-plexan calcium-dependent protein kinase enzymesTgCDPK1 (T. gondiiME49),CpCDPK1 (C. parvumIowa II), NcCDPK1 (N. caninum Liverpool),PfCDPK1 (P. falciparum 3d7), PfCDPK4 (P.falciparum 3d7), EtCDPK1 (E. tenella) andBbCDPK4 (B. bovis T2Bo) were PCR amplifiedfrom the cDNA library of the respective parasitestrain in parentheses. Since the ATP binding sites ofapicomplexan CDPKs seem to be highly conservedeven between genera, we do not anticipate anystructural differences or response to inhibitorsbetween strains of the same species as shown byprevious studies inT. gondii (Ojo et al. 2010; Doggettet al. 2014). The amplicons were cloned into theligation-independent cloning (LIC) site of expressionvector AVA0421 or the maltose-binding proteinvariant (AVA0421_MBP) (Alexandrov et al. 2004;Mehlin et al. 2006). The inserts were sequenced andconfirmed to correspond with GenBank entries fortheir respective gene. Site-directed mutants ofTgCDPK1, in which the gatekeeper amino acid wassubstituted for a serine or threonine residue
1500Katelyn R. Keyloun and others

(TgG128SCDPK1 and TgG128TCDPK1 respect-ively), were also included to further our understand-ing of the gatekeeper residue influence. Recombinantexpression was in Rosetta® 2(DE3) competentEscherichia coli cells (Novagen EMD, Billerica,MA) using Studier auto-induction protocols at20 °C (Studier, 2005). Soluble enzymes were purifiedby immobilized metal-affinity chromatography(IMAC) in a Ni2+–NTA (Qiagen, Valencia, CA)column as earlier described (Ojo et al. 2011). Thebinding buffer is composed of 20mM HEPES pH7·25, 500mM NaCl, 5% glycerol, 30 mM imidazole,0·5% CHAPS and 1mM TCEP. Purified proteinswere eluted with the same buffer supplemented withimidazole up to 250mM. Human kinase SRC wasincluded as a control to examine off-target liabilitiesof threonine gatekeeper kinases. SRC was expressedand purified according to an established protocol(Seeliger et al. 2005).
Apicomplexan CDPK enzyme activity assay
Compound screening included 333 compounds at3 μM final concentration in the experimental reaction.Each screen for a given enzyme was performed inthree replicates. Inhibition concentration to give 50%reduction in enzyme activity (IC50) was determinedin eight serial dilutions of compounds from 2 μMto 0·0001 μM using the non-radioactive Kinaseglo©
luciferase assay (Promega,Madison,WI).Kinaseglo©
is a reagent containing the enzyme luciferase, whichutilizes ATP to give off light. Phosphorylation ofpeptide substrate (PLARTLSVAGLPGKK) (AmericanPeptide Company, Inc. Sunnyvale, CA) was in-directly assessed as previously described (Ojo et al.2010). Enzyme activity assays in the presence of20 μM peptide substrate; 2·11 nMTgCDPK1; 0·80 nM
NcCDPK1; 2·0 nM CpCDPK1; 4·06 nM EtCDPK1;1·84 nM TgG128SCDPK1; 146·5 nM PfCDPK4;8·80 nM PfCDPK1; 18·72 nM BbCDPK4; or7·32 nM TgG128TCDPK1 were performed in abuffered solution containing 1mM EGTA (pH 7·2),10 mM MgCl2, 20mM HEPES pH 7·5 (KOH), 0·1%BSA and enzyme activation reagent containing2mM CaCl2. The PfCDPK4 recombinant enzymeis marginally soluble even as a maltose-bindingprotein fusion, hence the necessity for a higherconcentration per reaction. The reaction was stoppedafter 90min incubation at 30 °C with the additionof 5mM EGTA. Each experiment included reactionswithout peptide substrate as controls and no ATPconsumption was noted without substrate.
SRC enzyme activity assay
Similarly, for the human SRC enzyme inhibitionassay, Kinaseglo© luciferase assay was used togenerate inhibition data with the compound library.Final concentration of components of assay buffer
included 40mM Tris-HCl (pH 7·5), 20mM MgCl2,1 mM MnCl2, 1 mM DTT and 0·1% BSA. SRCenzyme was used at a final concentration of 1·97 nM
(25 μL final reaction volume), while SRC sub-strate sequence Ac-EIYGEFKKK (GenScript,Piscataway, NJ) was used at a final concentration of61 μM. SRC substrate was purified on a C-18 reversephase prep column on a Varian Prostar® HPLC withan acetonitrile/water gradient containing 0·05%TFA. A final concentration of 10 μM ATP wasadded per well to initiate the reaction. The assaywas allowed to proceed for 90min at 30 °C. Enzymeinhibition was obtained indirectly by assessing thepercentage of unused ATP in the reaction vialuminescence with Kinaseglo® and comparing itwith SRC wells with no inhibitor (in the presenceof 1% DMSO to account for the DMSO in ex-perimental wells). This assay also included reactionswithout peptide substrate as controls. Luminescencewas read on a MicroBeta2® plate reader (PerkinElmer,Waltham, MA) after addition of Kinaseglo®.
Human cell toxicity assay
Cell viability assays were performed to determinepotential toxicity of compounds against mammaliancells. CRL-8155 human lymphocytic cells (ATCC,WIL2-NS) cultured in RPMI-1640 growth mediumsupplemented with 10% heat inactivated foetalbovine serum (FBS), 10mM HEPES, 1mM sodiumpyruvate and 1mM L-glutamine, were incubatedin the presence of compounds. The Alamar Blue®
assay (Life Technologies, Grand Island, NY), whichmeasures general cellular metabolism, was usedto quantify cell growth. Mid-log cells were seededin 96-well flat-bottom plates at a density of3×105 cells mL−1 containing compounds (20 mM
stock solutions dissolved in DMSO) at six concentra-tions (40, 20, 10, 5, 2·5 and 1·25 μM) in quadruplicateand grown for 48 h at 37 °C in a 5% CO2 humidifiedincubator. 1/10th volume of Alamar Blue® develop-ing reagent was added to each well and incubated foran additional 3 h and fluorescence was measured atthe respective excitation and emission wavelengths of560 and 590 nm in a FLx800 microplate reader(Biotek, Winooski, VT). Per cent growth inhibitionsby test compounds were calculated based on DMSOvehicle and negative (50 μM Quinacrine) controls,which represent 0 and 100% growth inhibition,respectively. Additionally, Quinacrine was used asa positive control in each assay for quality assurance.
Data analysis
Luminescence data were analysed and equated to percent ATP usage. The results were then converted toper cent inhibition produced by each compound,subsequent IC50 analysis was performed usingGraphpad® Prism software (GraphPad Software,
1501Homologous Apicomplexa CDPKs as drug targets

San Diego, CA). Statistical analysis of data usingPearson’s Correlation test was performed usingMicrosoft® Excel analysis package. For each en-zyme’s reaction plate, the Z′-factor, a coefficient thatdefines assay signal dynamic range and data variationassociated with signal measurements were deter-mined as earlier described (Zhang et al. 1999). TheZ′-factor was calculated as an intra-assay assessmentof the quality of the assay using the equation below(Zhang et al. 1999).
RESULTS
Overall inhibition of CDPKs and SRC from theBKI library
In order to establish the structure activity relation-ships (SAR) of BKIs against the CDPKs, we assayedthe entire BKI library for inhibition of the recombi-nant apicomplexan CDPKs at a concentration of3 μM. Sensitivity to a compound at this final reactionconcentration was defined as the inhibition of enzymekinase activity by greater than 70% relative to the noinhibitor full reaction (positive control). Resistancewas defined as the inhibition of an enzyme kinaseactivity by less than 30% relative to the positivecontrol. The average Z′-factor of the compoundreaction plate for enzymes screened ranged from0·7112 for BbCDPK4 to 0·8949 for CpCDPK1(Table 1). A high Z′-factor reflects two characteristicsof the assay. It reflects a wide separation between themaximum signals of the assay (in this case, highestamount of luminescence in zero ATP consumptionfound in negative controls) and the lowest signal(lowest amount of luminescence found in positiveenzyme reactions with no inhibitors causing a
reduction of the initial ATP concentration). It alsoshows that standard deviations are low when asses-sing positive and negative controls. A Z′-factor valueof greater than 0·5 implies a quality assay, and aperfect assay will approach 1·0 (Zhang et al. 1999).The most sensitive CDPK orthologues to our BKIcompound library were wild-type NcCDPK1,TgCDPK1 and CpCDPK1, where 90% (301 com-pounds), 89% (296) and 86% (287) of inhibitorsexhibited over 70% to absolute inhibition of kinase
activity, respectively (Table 1). These three enzymesshare a glycine gatekeeper residue. These results werefollowed by PfCDPK4 with 55% (183 compounds),PfCDPK1 with 44% (145), BbCDPK4 with 39%(129), EtCDPK1 with 21% (71) and SRC with 10%(33) effective inhibitors. PfCDPK4 has a serine gate-keeper, whilePfCDPK1,BbCDPK4,EtCDPK1 andthe human enzyme SRC all share a threoninegatekeeper. The level of resistance to BKIs wasmuch higher for EtCDPK1 and SRC compared withother kinases (Table 1).
The selectivity of the BKIs’ inhibition towardsthe different protein kinases tested was variable.Fourteen of the 333 compounds have greater than90% inhibition of kinase activity in all of thewild-type apicomplexan CDPK enzymes tested. Ofthese 14 compounds, five were completely non-selective as they also inhibited the human kinaseSRC activity by greater than 90%. Forty compoundshave >80% inhibition of all CDPKs with 11 of these40 compounds also inhibiting SRC by greaterthan 80%. Sixty of the 333 compounds caused>70% inhibition of CDPKs where 17 also inhibitedSRC by greater than 70%. Of these 60 compounds,
Z′ − Factor = 1− 3×(Standard deviation of negative control) + 3×(Standard deviation of positive control)Mean of negative control−Mean of postive control
Table 1. Recombinant enzymes tested, gatekeeper residues, evaluation of reproducibility of assays(Z′-factor) and % BKIs that effectively inhibited the various enzymes
EnzymeGatekeeperresidue
AverageZ′-factor
Sensitive (>70%) Resistant (<30%)
Count % Count %
TgCDPK1 Glycine 0·880 296 89 15 5NcCDPK1 Glycine 0·869 301 90 15 5CpCDPK1 Glycine 0·895 287 86 22 7PfCDPK4 Serine 0·844 183 55 59 18TgG128SCDPK1 Serine 0·809 241 72 36 11PfCDPK1 Threonine 0·716 145 44 104 31BbCDPK4 Threonine 0·711 129 39 142 43EtCDPK1 Threonine 0·898 71 21 192 58TgG128TCDPK1 Threonine 0·854 73 22 187 56SRC Threonine 0·808 33 10 241 72
1502Katelyn R. Keyloun and others
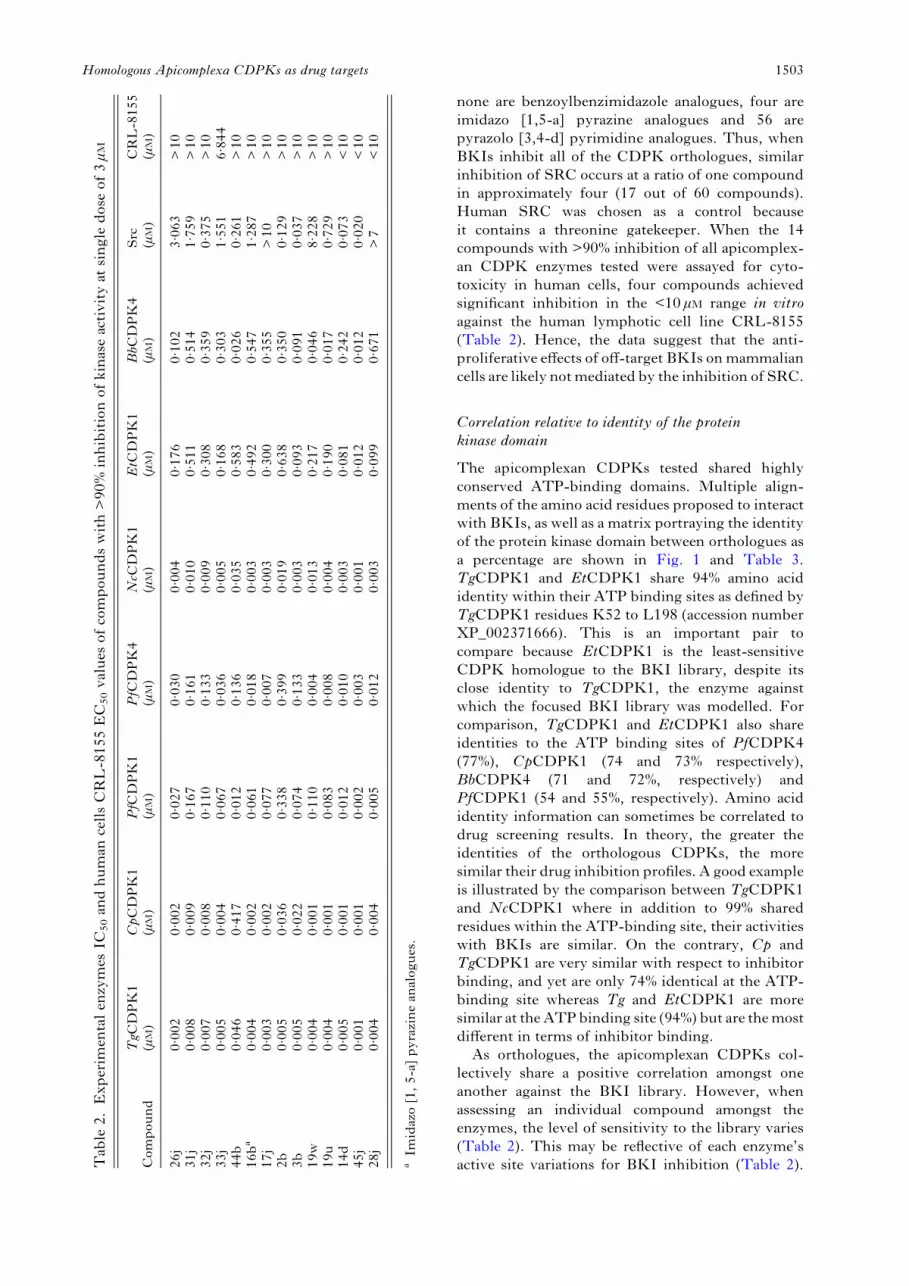
none are benzoylbenzimidazole analogues, four areimidazo [1,5-a] pyrazine analogues and 56 arepyrazolo [3,4-d] pyrimidine analogues. Thus, whenBKIs inhibit all of the CDPK orthologues, similarinhibition of SRC occurs at a ratio of one compoundin approximately four (17 out of 60 compounds).Human SRC was chosen as a control becauseit contains a threonine gatekeeper. When the 14compounds with >90% inhibition of all apicomplex-an CDPK enzymes tested were assayed for cyto-toxicity in human cells, four compounds achievedsignificant inhibition in the <10 μM range in vitroagainst the human lymphotic cell line CRL-8155(Table 2). Hence, the data suggest that the anti-proliferative effects of off-target BKIs onmammaliancells are likely notmediated by the inhibition of SRC.
Correlation relative to identity of the proteinkinase domain
The apicomplexan CDPKs tested shared highlyconserved ATP-binding domains. Multiple align-ments of the amino acid residues proposed to interactwith BKIs, as well as a matrix portraying the identityof the protein kinase domain between orthologues asa percentage are shown in Fig. 1 and Table 3.TgCDPK1 and EtCDPK1 share 94% amino acididentity within their ATP binding sites as defined byTgCDPK1 residues K52 to L198 (accession numberXP_002371666). This is an important pair tocompare because EtCDPK1 is the least-sensitiveCDPK homologue to the BKI library, despite itsclose identity to TgCDPK1, the enzyme againstwhich the focused BKI library was modelled. Forcomparison, TgCDPK1 and EtCDPK1 also shareidentities to the ATP binding sites of PfCDPK4(77%), CpCDPK1 (74 and 73% respectively),BbCDPK4 (71 and 72%, respectively) andPfCDPK1 (54 and 55%, respectively). Amino acididentity information can sometimes be correlated todrug screening results. In theory, the greater theidentities of the orthologous CDPKs, the moresimilar their drug inhibition profiles. A good exampleis illustrated by the comparison between TgCDPK1and NcCDPK1 where in addition to 99% sharedresidues within the ATP-binding site, their activitieswith BKIs are similar. On the contrary, Cp andTgCDPK1 are very similar with respect to inhibitorbinding, and yet are only 74% identical at the ATP-binding site whereas Tg and EtCDPK1 are moresimilar at the ATPbinding site (94%) but are themostdifferent in terms of inhibitor binding.As orthologues, the apicomplexan CDPKs col-
lectively share a positive correlation amongst oneanother against the BKI library. However, whenassessing an individual compound amongst theenzymes, the level of sensitivity to the library varies(Table 2). This may be reflective of each enzyme’sactive site variations for BKI inhibition (Table 2).T
able
2.Exp
erim
entalen
zymes
IC50an
dhuman
cellsCRL-815
5EC50values
ofcompou
ndswith>90
%inhibitionof
kinaseactivity
atsingledoseof
3μM
Com
pou
nd
TgC
DPK1
(μM)
CpC
DPK1
(μM)
PfC
DPK1
(μM)
PfC
DPK4
(μM)
NcC
DPK1
(μM)
EtC
DPK1
(μM)
BbC
DPK4
(μM)
Src
(μM)
CRL-815
5(μ
M)
26j
0·00
20·00
20·02
70·03
00·00
40·17
60·10
23·06
3>10
31j
0·00
80·00
90·16
70·16
10·01
00·51
10·51
41·75
9>10
32j
0·00
70·00
80·11
00·13
30·00
90·30
80·35
90·37
5>10
33j
0·00
50·00
40·06
70·03
60·00
50·16
80·30
31·55
16·84
444
b0·04
60·41
70·01
20·13
60·03
50·58
30·02
60·26
1>10
16ba
0·00
40·00
20·06
10·01
80·00
30·49
20·54
71·28
7>10
17j
0·00
30·00
20·07
70·00
70·00
30·30
00·35
5>10
>10
2b0·00
50·03
60·33
80·39
90·01
90·63
80·35
00·12
9>10
3b0·00
50·02
20·07
40·13
30·00
30·09
30·09
10·03
7>10
19w
0·00
40·00
10·11
00·00
40·01
30·21
70·04
68·22
8>10
19u
0·00
40·00
10·08
30·00
80·00
40·19
00·01
70·72
9>10
14d
0·00
50·00
10·01
20·01
00·00
30·08
10·24
20·07
3<10
45j
0·00
10·00
10·00
20·00
30·00
10·01
20·01
20·02
0<10
28j
0·00
40·00
40·00
50·01
20·00
30·09
90·67
1>7
<10
aIm
idazo[1,5-a]
pyrazinean
alog
ues.
1503Homologous Apicomplexa CDPKs as drug targets

The shared concordance could be correlated byPearson’sR value (Table 4). Relative to other enzymepairings, TgCDPK1, with glycine gatekeeper resi-due, andEtCDPK1, with threonine as the gatekeeperresidue, have only fair correlation (Table 4), despitehigh protein kinase active site identity (Fig. 1 andTable 3). CpCDPK1’s inhibition spectrum towardsBKIs is very similar to TgCDPK1 and NcCDPK1(Table 4), and distinct from the BKIs that inhibitedEtCDPK1 (Pearson R value = 0·4252). PfCDPK4,the only CDPK studied with a serine gatekeeper,had fair to poor correlation when compared withother enzymes, achieving the most correlationwith TgCDPK1 (Pearson R= 0·6028), CpCDPK1(Pearson R = 0·6921) and NcCDPK1 (PearsonR = 0·5851 (Table 4).
Effects of a gatekeeper residue mutation on sensitivityand characteristics of inhibitory compounds
Mutants were generated to explore the effect ofthe gatekeeper residue on the inhibition pattern.A TgCDPK1 threonine mutant, TgG128TCDPK1,was shown to have an almost perfect correlationand BKI compound sensitivity compared withEtCDPK1 (Tables 1 and 4). The compellingsimilarity between EtCDPK1 and the mutantTgG128TCDPK1 under the same conditions, aswell as their relative resistance to compoundscompared with the other orthologues with smallergatekeeper residues further supports the hypo-thesis that gatekeeper residues are key drivers ofcompound binding. However, when comparingTgG128TCDPK1’s SAR to BbCDPK4 andPfCDPK1, sensitivities to the compound libraryshowed discordance (37% for BbCDPK4 and 44%for PfCDPK1 versus 21% for TgG128TCDPK1 orEtCDPK1, respectively). Thus, EtCDPK1 andTgG128TCDPK1 are more resistant to the librarythan BbCDPK4 and PfCDPK1.
Weighted identity scores ([WS] in Fig. 1) ofconserved differences in amino acids proposed to
directly interact with BKIs were derived by assigninga unitless value of 1 for every property difference,based on well-established properties of charge, po-larity and size as previously described (Livingstoneand Barton, CABIOS, 1993). The gatekeepers wereexcluded from this analysis since the influence ofthese residues on BKI inhibition of CDPKs is welldefined. By this measurement, PfCDPK1 andCpCDPK1 have the highest divergence from theTgCDPK1 ATP binding site with a score of 15 forPfCDPK1 and 9 for CpCDPK1 (Fig. 1). However,CpCDPK1 showed identical SAR profile and withhigh sensitivity towards the library, much likeTgCDPK1, probably due to the higher influence ofthe shared glycine gatekeeper residue. Conversely,the SAR profiles of Nc/TgCDPK1 versus those ofPfCDPK1, BbCDPK4 and EtCDPK1 make itchallenging to attribute all the inhibitor libraryobserved characteristics to the gatekeeper size alone.In the latter case, we can partially attribute highersensitivity to increased divergence from the model ofTgCDPK1 on which compound design was based.However, it is likely that a combinatory effectresulting from differences in types and chemicalproperties of amino acids within the active sites ofapicomplexa CDPKs can produce a variety of BKIinhibition SARs. Analysis of amino acid differencesbetween PfCDPK1 and TgCDPK1 demonstrateddeviations capable of promoting the differentialsensitivity observed. Through modelling basedon previous crystal structures of TgCDPK1 andCpCDPK1 co-crystallized with BKI inhibitors,probable structural models were created to simulatebinding motifs of other CDPK enzymes. PfCDPK1was compared to this effect with TgCDPK1 (Fig. 2).Three possible differences from the sequences ofPfCDPK1 and TgCDPK1 were noted that mayinfluence the binding of BKIs to PfCDPK1. Theseinclude TgCDPK1 F134V (Residue 117 inPfCDPK1), M112I (Residue 129 in PfCDPK1)and I194V (Residue 221 in PfCDPK1). Perhaps themost influential variation is at the position ofmethionine 112 in TgCDPK1 (isoleucine 129 in
Fig. 1. Multiple alignments of the amino acid residues proposed to interact with BKIs. Regions contributing tothe active site as seen in crystal structures of several TgCDPK1 orthologues are shown. Numbering was based onTgCDPK1 protein database accession number XP_002371666. Specific residues that may confer differential sensitivityto some BKI are shaded yellow. Residues that are shaded grey are conserved between all the enzymes. The gatekeeperresidues are shown in red. Weighted identity score (WS) of differences in amino acids, compared with Tg/NcCDPK1(note, Tg identical to Nc in the active site) proposed to directly interact with BKIs in each enzyme is shown on the farright. The gatekeepers were excluded from this analysis since the influence of these residues on BKI inhibition ofCDPKs is well defined.
1504Katelyn R. Keyloun and others

Table 3. Comparison of amino acid identities (%) of CDPK orthologues’ ATP-binding sites as defined by TgCDPK1 residues K52 to L198 (accession numberXP_002371666). The smallest sum probability by BLASTP search is shown in parentheses
Enzyme
Similarity (identity)
PfCDPK1 EtCDPK1 BbCDPK4 TgCDPK1 CpCDPK1 NcCDPK1
PfCDPK4 53 (5·0×10−42) 77 (5·0×10−72) 66 (4·0×10−60) 77 (8·0×10−72) 68 (4·0×10−57) 78 (2·0×10−72)PfCDPK1 55 (3·0×10−49) 53 (2·0×10−45) 54 (2·0×10−47) 53 (4·0×10−46) 54 (2·0×10−46)EtCDPK1 71 (4·0×10−66) 94 (5·0×10−89) 73 (6·0×10−75) 93 (1·0×10−88)BbCDPK4 72 (2·0×10−65) 68 (3·0×10−59) 72 (5·0×10−66)TgCDPK1 74 (6·0×10−65) 99 (5·0×10−95)CpCDPK1 73 (4·0×10−64)
Table 4. Pearson correlation R values: Statistical comparative analysis of inhibition data for each enzyme relative to the other using Pearson’s correlation test.Higher correlations among CDPKs with similar gatekeeper residues support their influence on BKI inhibition. The data showed relatively lower inhibitioncorrelation of most CDPK enzymes with human SRC kinases
Enzyme PfCDPK1 EtCDPK1 BbCDPK4 TgCDPK1 TgG128SCDPK1 TgG128TCDPK1 CpCDPK1 NcCDPK1 SRC
PfCDPK4 0·607 0·597 0·544 0·603 0·771 0·593 0·692 0·585 0·261PfCDPK1 0·854 0·865 0·578 0·719 0·846 0·534 0·563 0·483EtCDPK1 0·866 0·447 0·639 0·962 0·425 0·433 0·456BbCDPK4 0·454 0·632 0·858 0·401 0·438 0·369TgCDPK1 0·855 0·449 0·911 0·989 0·310TgG128SCDPK1 0·658 0·794 0·847 0·348TgG128TCDPK1 0·421 0·431 0·495CpCDPK1 0·901 0·294NcCDPK1 0·303
1505Hom
ologousApicom
plexaCDPKsas
drugtargets

PfCDPK1). The larger (glycine->threonine) gate-keeper residue on one side of the binding pocket ispaired with a complementary smaller residue(methionine-> isoleucine) on the opposite side ofthe binding pocket (Fig. 2). This allows the BKIbinding pose to shift slightly away from the gate-keeper, something that is otherwise sterically restric-ted in TgCDPK1 and EtCDPK1. Thus the effect ofthe larger threonine gatekeeper in PfCDPK1 ispartially offset by the presence of a complementaryamino acid variation elsewhere in the active site,which may explain its increased BKI sensitivityrelative to EtCDPK1 (Fig. 2).
Within the TgG128TCDPK1 inhibitor SAR,patterns of compound characteristics were identified.In general, inhibition of TgG128TCDPK1 andits highly similar threonine partner, EtCDPK1,requires relatively small substituents at the C3position (R1 group), such as an alkoxybenzylgroup, 4-chlorophenyl, or naphthalen-2-yl (Fig. 3).Presumably, larger substituents exhibit steric clashwith the threonine gatekeeper, disfavouring binding.However, a few compounds with larger R1 groups,such as 23j, 24j, 31j, 32j, 33j, 26a, 24a, 25j, 30a, 39a,27a, 34a, 6j, did cause >70% inhibition of EtCDPK1kinase activity, irrespective of the R2 groups size.Such constraints on inhibitors were not as obviouswith the other threonine enzymes BbCDPK4and PfCDPK1. A wider range of compounds wereable to achieve inhibition for these orthologues
(BbCDPK4 and PfCDPK1) relative to EtCDPK1,including compounds with larger R1 moieties(6-alkoxy- and 6-benzyloxy-naphthol) without theneed of a smaller R2 group that may be associatedwith the spatial constraints of enzymes with largergatekeeper residues.
Contrary to the near-identical sensitivity and cor-relation seen withEtCDPK1 andTgG128TCDPK1,the serine mutant TgG128SCDPK1 was sensitive to241 (72%) of compounds while its serine gate-keeper partner PfCDPK4 was sensitive to 183(55%). Although this enzyme pair shared the samegatekeeper residue, there was a difference in sensi-tivity patterns to compounds and change in corre-lation (Table 4: Pearson R value 0·7714) that wasnot dependent on the gatekeeper. A series ofcompounds was isolated which differentially inhib-ited TgG128SCDPK1 over PfCDPK4. This seriesshifted inhibition towards TgG128SCDPK1 bygreater than 30% over PfCDPK4. Compounds withthis pattern include 15k, 4a, 13a, 31a, 32a, 36a, 35a,37a, 30a, 41b, 43b, 40b, 25b, 6b, 12b, 10b.
PfCDPK4 seems to accommodate R1 groups suchas a naphthyl ring connected in an ortho configura-tion, while the larger threonine gatekeeper residueenzyme, PfCDPK1, was able to accommodate quitelarge and extended groups at this position, such as6-alkoxy- and 6-benzyloxynaphthol substituents. Infact, a group of compounds with extended moietiesinhibited PfCDPK1, yet against PfCDPK4 with aserine gatekeeper residue, did not achieve the samelevel of inhibition. These compounds include 35a, 7a,8a, 9a, 13a, 36a, 35a, 11j, 10b (Fig. 3).
DISCUSSION
A comparative analysis of the structural activityrelationship of veterinary pathogens N. caninum,E. tenella andB. bovis; zoonotic pathogensC. parvumand T. gondii CDPKs and those of human pathogenP. falciparum CDPKs was determined by screeningagainst 333 BKI analogues. The degree of sensitivityor resistance of CDPK enzymes to BKIs was earlierpostulated to be due to the size and characteristic ofthe gatekeeper residue and the adjacent pocket.Indeed, correlation of screening data was bestamong enzymes with the same gatekeeper residue,which corroborates the gatekeeper hypothesis(Table 4). However, sensitivity to compoundsamong enzymes with the same gatekeeper residuedoes not correlate perfectly, suggesting alternativemodes of CDPK–drug interaction. We explored thisby constructing TgCDPK1 mutants that substitutedserine or threonine for the wild-type glycine atthe gatekeeper position, and comparing their BKIsensitivity profile with that of the homologues withthe corresponding wild-type gatekeeper residue; i.e.TgCDPK1G128S can be compared to PfCDPK4,and TgCDPK1G128T can be compared to
Fig. 2. BKI interactions in the ATP-binding domainsof PfCDPK1 and TgCDPK1. Model of PfCDPK1based on previous crystal structure of TgCDPK1 incomplex with inhibitor 19j (Larson et al. 2012; PDB3sx9). Gatekeeper residues and other residues postulatedto affect inhibition are shown as red (TgCDPK1) andyellow (PfCDPK1) sticks. The larger G128M (Residue145 in PfCDPK1) gatekeeper residue is paired with acomplementary smaller residue M112I (Residue 129 inPfCDPK1), and I194V (Residue 221 in PfCDPK1) inPfCDPK1. This partially offset the effect of the largergatekeeper in PfCDPK1 by the presence of acomplementary amino acid variation elsewhere in theactive site, which may explain its increased BKIsensitivity relative to EtCDPK1.
1506Katelyn R. Keyloun and others

PfCDPK1, EtCDPK1 andBbCDPK4 (Tables 3 and4). Although EtCDPK1 has the closest sequenceidentity to wild-type Tg/NcCDPK1 in the ATPbinding pocket (Table 3: 94%), it nonetheless provedto be the least sensitive enzyme to BKIs, with poorcorrelation to the sensitivity profile of wild-typeTgCDPK1. However, the sensitivity of EtCDPK1correlates very well with that of TgCDPK1G128T.The BKI sensitivity of two other enzymes witha threonine gatekeeper, BbCDPK4 and PfCDPK1,is also more similar to that of the T. gondiiG128T mutant than to that of the wild-typeTgCDPK1. However, these orthologues share lesssequence identity with the T. gondii enzyme andhave weaker correlation with the sensitivity ofthe TgCDPK1G128T mutant. TgCDPK1 andNcCDPK1 are also similar in their ATP bindingdomain identity (>99%) and predictably sharesimilar sensitivity to compounds. Yet, CpCDPK1 isnot as similar in identity to TgCDPK1 as EtCDPK1(94% identity versus 74%) or NcCDPK1 (>99%versus 74%) (Table 3). However, the fact thatTg/NcCDPK1 andCpCDPK1 are the most sensitiveenzymes to compounds, yet share less sequencesimilarity (74% identity) than that of EtCDPK1,returns us to the enormous role played by thesize of the gatekeeper residue in determining thelevel of sensitivity to BKIs. This assertion wassupported with experimental evidence showingexcellent correlation and characteristics of structureactivity relationship between EtCDPK1 andTgG128TCDPK1, completely reversing suscepti-bility of TgCDPK1.
Although the similarity of a homologue toTgCDPK1’s ATP-binding site sequence is anattractive marker for determining sensitivity toBKIs for enzymes whose full binding pocket isuncharacterized, the binding pocket size, charge andhydrophobicity are all factors that ultimately deter-mine sensitivity to BKIs. A combination of thesefactors contributes directly or indirectly to thesensitivity of CDPKs to BKIs. Further research isnecessary to elucidate the relative impact of eachfactor to CDPK sensitivity to BKIs. Crystal struc-tures of these new homologues may help understandthe complex nature of the binding pocket. With thisnew information, we conclude that a large gatekeeperresidue does not necessarily mean complete resistanceto BKIs. In the example of our threonine gatekeeperenzymes, low nanomolar inhibition was achieved,even by BKIs designed to target small gatekeepers.It is important that future drug development
focuses on exploiting other characteristics of thedrug-binding pocket in addition to the gatekeeper. Inthis respect, compounds could be optimized to targetthe conserved apicomplexan ribose-binding pocket.Similarly, targeting the calcium-binding regulatorydomain could prevent activation of CDPK enzymeswith a resultant disruption of downstream process.Compounds that inhibit EtCDPK1 but not SRC areimportant as lead compounds for the developmentof broad-spectrum antimicrobials. Because of itsthreonine gatekeeper and unaccommodating bindingpocket towards BKIs, if potent compounds to targetEtCDPK1 are synthesized, then clinically, thesecompounds can be useful as monotherapy with low
Fig. 3. R1 and R2 substructures of compounds.
1507Homologous Apicomplexa CDPKs as drug targets

likelihood of easily conferring resistance acrossCDPKs of the apicomplexa. Within our BKI library,34 compounds effectively inhibited EtCDPK1 by>70% and inhibit SRC kinase activity by <30%. Thestructure and characteristics of these compounds willbe the focus of future studies. These compoundsinclude 19a, 19j, 22j, 23j, 26j, 24j, 21j, 31j, 33j, 25j, 3j,38a, 30a, 42b, 19b, 10j, 17j, 6j, 25l, 19e, 16j, 19q, 19r,19t, 19v, 19w, 20j, 5j, 5l, 19s, 20a, 28j.
CONCLUSION
It is apparent that the highly conserved nature ofthe ATP-binding domain shared by apicomplexanCDPK homologues could be exploited to someextent in development of potential broad-spectruminhibitors if potential structural constraints are welldefined and understood. A combination of theseconstraints concerning the gatekeeper residue, thestructure and characteristic of the binding pocket,and differences among compounds, all contributesubtle distinctions in sensitivity to compoundsamong these highly conserved CDPKs. Furtherresearch into the structural constraints for thedevelopment of broad-spectrum, potent inhibitorsof apicomplexan CDPKs is warranted.
ACKNOWLEDGEMENTS
The authors wish to express our gratitude to SteveM. Johnson, Ryan C. Murphy and Edward J. Dale fortechnical and valuable support.
FINANCIAL SUPPORT
This work is supported by theNational Institutes ofHealthgrants R01AI089441 (E.A.M. and W.C.V.V.) and 5 R01AI080625 (W.C.V.V.). The Maly Lab was supported byNIH grant R01GM086858. K.R.K. was supported by atraining scholarship from the University of WashingtonPlein Endowment for Geriatric Pharmacy Research.
REFERENCES
Alexandrov, A., Vignali, M., LaCount, D. J., Quartley, E., de Vries, C.,De Rosa, D., Babulski, J., Mitchell, S. F., Schoenfeld, L.W., Fields, S.,Hol,W. G., Dumont,M. E., Phizicky, E.M. andGrayhack, E. J. (2004).A facile method for high-throughput co-expression of protein pairs.Molecular and Cellular Proteomics 3, 934–938. doi: 10.1074/mcp.T400008-MCP200.Castellanos-Gonzalez, A., White, A. C., Ojo, K. K., Vidadala, R. S.,Zhang, Z., Reid,M. C., Fox, A.M., Keyloun, K. R., Rivas, K., Irani, A.,Dann, S.M., Fan, E., Maly, D. J. and Van Voorhis, W. C. (2013). A novelcalcium-dependent protein kinase inhibitor as a lead compound for treatingcryptosporidiosis. Journal of Infectious Diseases 208, 1342–1348. doi:10.1093/infdis/jit327.Doggett, J. S., Ojo, K. K., Fan, E., Maly, D. J. and Van Voorhis, W. C.(2014). Bumped kinase inhibitor 1294 treats established Toxoplasma gondiiinfection. Antimicrobial Agents and Chemotherapy 58, 3547–3549. AAC01823–13.Guinovart, C., Navia, M.M., Tanner, M. and Alonso, P. L. (2006).Malaria: burden of disease. Current Molecular Medicine 6, 137–140.Johnson, S.M., Murphy, R. C., Geiger, J. A., DeRocher, A. E.,Zhang, Z., Ojo, K. K., Larson, E. T., Perera, B. G., Dale, E. J., He, P.,Reid, M. C., Fox, A.M., Mueller, N. R., Merritt, E. A., Fan, E.,Parsons, M., Van Voorhis, W. C. and Maly, D. J. (2012). Development
of Toxoplasma gondii calcium-dependent protein kinase 1 (TgCDPK1)inhibitors with potent anti-toxoplasma activity. Journal of MedicinalChemistry 55, 2416–2426. doi: 10.1021/jm201713h.Katlama, C., De Wit, S., O’Doherty, E., Van Glabeke, M. andClumeck, N. (1996). Pyrimethamine-clindamycin vs. pyrimethamine-sulfadiazine as acute and long-term therapy for toxoplasmic encephalitis inpatients with AIDS. Clinical Infectious Diseases 22, 268–275.Larson, E. T., Ojo, K. K., Murphy, R. C., Johnson, S.M., Zhang, Z.,Kim, J. E., Leibly, D. J., Fox, A.M., Reid, M. C., Dale, E. J.,Perera, B. G., Kim, J., Hewitt, S. N., Hol, W. G., Verlinde, C. L.,Fan, E., Van Voorhis, W. C., Maly, D. J. and Merritt, E. A. (2012).Multiple determinants for selective inhibition of apicomplexan calcium-dependent protein kinase CDPK1. Journal ofMedicinal Chemistry 55, 2803–2810. doi: 10.1021/jm201725v.Livingstone, C. D. and Barton, G. J. (1993). Protein sequence alignments:a strategy for the hierarchical analysis of residue conservation. ComputerApplications in the Biosciences 9, 745–756.Lovett, J. L., Marchesini, N., Moreno, S. N. and Sibley, L. D.(2002). Toxoplasma gondii microneme secretion involves intracellularCa(2+) release from inositol 1,4,5-triphosphate (IP(3))/ryanodine-sensitivestores. Journal of Biological Chemistry 277, 25870–25876. doi: 10.1074/jbc.M202553200.Luft, B. J., Hafner, R., Korzun, A. H., Leport, C., Antoniskis, D.,Bosler, E.M., Bourland, D. D., Uttamchandani, R., Fuhrer, J. andJacobson, J. (1993). Toxoplasmic encephalitis in patients with the acquiredimmunodeficiency syndrome. Members of the ACTG 077p/ANRS 009Study Team.NewEngland Journal ofMedicine 329, 995–1000. doi: 10.1056/NEJM199309303291403.Mehlin, C., Boni, E., Buckner, F. S., Engel, L., Feist, T., Gelb, M.H.,Haji, L., Kim, D., Liu, C., Mueller, N., Myler, P. J., Reddy, J. T.,Sampson, J. N., Subramanian, E., Van Voorhis, W. C., Worthey, E.,Zucker, F. and Hol, W. G. (2006). Heterologous expression of proteinsfrom Plasmodium falciparum: results from 1000 genes. Molecular andBiochemical Parasitology 148, 144–160. doi: 10.1016/j.molbio-para.2006.03.011.Morrissette, N. S. and Sibley, L. D. (2002). Cytoskeleton of apicomplex-an parasites. Microbiology and Molecular Biology Reviews 66, 21–38.Murphy, R. C., Ojo, K. K., Larson, E. T., Castellanos-Gonzalez, A.,Perera, B. G., Keyloun, K. R., Kim, J. E., Bhandari, J. G.,Muller, N. R., Verlinde, C. L., White, A. C., Merritt, E. A., VanVoorhis, W. C. and Maly, D. J. (2010). Discovery of potent and selectiveinhibitors of calcium-dependent protein kinase 1 (CDPK1) fromC. parvumand T. gondii. ACS Medicinal Chemistry Letters 1, 331–335. doi: 10.1021/ml100096t.Nair, S. C., Brooks, C. F., Goodman, C. D., Strurm, A.,McFadden, G. I., Sundriyal, S., Anglin, J. L., Song, Y.,Moreno, S. N. J. and Striepen, B. (2011). Apicoplast isoprenoid precursorsynthesis and the molecular basis of fosmidomycin resistance inToxoplasmagondii. Journal of Experimental Medicine 208, 1547–1559. doi: 10.1084/jem.20110039.Norrby, R. (1978). A review of the penetration of antibiotics into CSFand its clinical significance. Scandinavian Journal of Infectious Diseases(Suppl. 4), 296–309.Ojo, K. K., Larson, E. T., Keyloun, K. R., Castaneda, L. J.,Derocher, A. E., Inampudi, K. K., Kim, J. E., Arakaki, T. L.,Murphy, R. C., Zhang, L., Napuli, A. J., Maly, D. J., Verlinde, C. L.,Buckner, F. S., Parsons, M., Hol, W. G., Merritt, E. A. and VanVoorhis, W. C. (2010). Toxoplasma gondii calcium-dependent proteinkinase 1 is a target for selective kinase inhibitors. Nature Structural andMolecular Biology 17, 602–607. doi: 10.1038/nsmb.1818.Ojo, K. K., Arakaki, T. L., Napuli, A. J., Inampudi, K. K.,Keyloun, K. R., Zhang, L., Hol, W. G., Verlinde, C. L., Merritt, E. A.and Van Voorhis, W. C. (2011). Structure determination of glycogensynthase kinase-3 from Leishmania major and comparative inhibitorstructure-activity relationships with Trypanosoma brucei GSK-3.Molecular and Biochemical Parasitology 176, 98–108. doi: 10.1016/j.molbiopara.2010.12.009.Ojo, K. K., Pfander, C., Mueller, N. R., Burstroem, C., Larson, E. T.,Bryan, C.M., Fox, A.M., Reid, M. C., Johnson, S.M., Murphy, R. C.,Kennedy, M., Mann, H., Leibly, D. J., Hewitt, S. N., Verlinde, C. L.,Kappe, S., Merritt, E. A., Maly, D. J., Billker, O. and VanVoorhis, W. C. (2012). Transmission of malaria to mosquitoes blocked bybumped kinase inhibitors. Journal of Clinical Investigation 122, 2301–2305.doi: 10.1172/JCI61822.Ojo, K. K., Merritt, E. A., Maly, D. J. and Van Voorhis, W. C. (2013).Calcium-dependent protein kinases of Apicomplexan parasites as drugtargets. In Protein Phosphorylation in Parasites: Novel Targets forAntiparasitic Intervention (ed. Doerig, C., Späth, G. and Wiese, M.),
1508Katelyn R. Keyloun and others

pp. 293–316. Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim,Germany. doi: 10.1002/9783527675401.ch14.Ojo, K. K., Eastman, R. T., Vidadala, R., Zhang, Z.,Rivas, K. L., Choi, R., Lutz, J. D., Reid, M. C., Fox, A.M.,Hulverson, M. A., Kennedy, M., Isoherranen, N., Kim, L.M.,Comess, K.M., Kempf, D. J., Verlinde, C. L., Su, X. Z.,Kappe, S. H., Maly, D. J., Fan, E. and Van Voorhis, W. C. (2014). Aspecific inhibitor of PfCDPK4 blocks malaria transmission: chemical-genetic validation. Journal of Infectious Diseases 209, 275–284. doi: 10.1093/infdis/jit522.Reeves, D. S. and Wilkinson, P. J. (1979). The pharmacokinetics oftrimethoprim and trimethoprim/sulphonamide combinations, includingpenetration into body tissues. Infection 7 (Suppl. 4), S330–S341.Seeliger, M. A., Young, M., Henderson, M.N., Pellicena, P.,King, D. S., Falick, A.M. and Kuriyan, J. (2005). High yield bacterialexpression of active c-Abl and c-Src tyrosine kinases. Protein Science 14,3135–3139. doi: 10.1110/ps.051750905.Studier, F.W. (2005). Protein production by auto-induction in highdensity shaking cultures. Protein Expression and Purification 41, 207–234.Tenter, A.M., Heckeroth, A. R. and Weiss, L.M. (2000). Toxoplasmagondii: from animals to humans. International Journal for Parasitology 30,1217–1258.
Vidadala, R. S., Ojo, K. K., Johnson, S.M., Zhang, Z., Leonard, S. E.,Mitra, A., Choi, R., Reid, M. C., Keyloun, K. R., Fox, A.M.,Kennedy, M., Silver-Brace, T., Hume, J. C., Kappe, S.,Verlinde, C. L., Fan, E., Merritt, E. A., Van Voorhis, W. C. andMaly, D. J. (2014). Development of potent and selective Plasmodiumfalciparum calcium-dependent protein kinase 4 (PfCDPK4) inhibitors thatblock the transmission of malaria to mosquitoes. European Journal ofMedicinal Chemistry 74C, 562–573.World Health Organization (2013). World Malaria Report2013. 1–2. Publications of the World Health Organization, Geneva,Switzerland.Zhang, J. H., Chung, T. D. and Oldenburg, K. R. (1999).A simple statistical parameter for use in evaluation and validation ofhigh throughput screening assays. Journal of Biomolecular Screening 4,67–73.Zhang, Z., Ojo, K. K., Johnson, S.M., Larson, E. T., He, P.,Geiger, J. A., Castellanos-Gonzalez, A., White, A. C., Parsons, M.,Merritt, E. A., Maly, D. J., Verlinde, C. L., Van Voorhis, W. C. andFan, E. (2012). Benzoylbenzimidazole-based selective inhibitors targetingCryptosporidium parvum and Toxoplasma gondii calcium-dependent proteinkinase-1. Bioorganic and Medicinal Chemistry Letters 22, 5264–5267.doi: 10.1016/j.bmcl.2012.06.050.
1509Homologous Apicomplexa CDPKs as drug targets





























