Embed Size (px)
Citation preview

lable at ScienceDirect
Biomaterials 31 (2010) 2371–2379
Contents lists avai
Biomaterials
journal homepage: www.elsevier .com/locate/biomateria ls
The effects of mixed MPEG–PLA/Pluronic� copolymer micelles on thebioavailability and multidrug resistance of docetaxel
Chao-Feng Mu a,b, Prabagar Balakrishnan b, Fu-De Cui a, Yong-Mei Yin b,c, Yong-Bok Lee d, Han-Gon Choi e,Chul Soon Yong e, Suk-Jae Chung b, Chang-Koo Shim b, Dae-Duk Kim b,*
a College of Pharmacy, Shenyang Pharmaceutical University, Shenyang 110016, Chinab College of Pharmacy and Research Institute of Pharmaceutical Sciences, Seoul National University, Seoul 151-742, South Koreac College of Pharmacy, Nankai University, Tianjin 300071, Chinad College of Pharmacy and Institute of Bioequivalence and Bridging Study, Chonnam National University, Gwangju 500-757, South Koreae College of Pharmacy, Yeungnam University, Gyongsan 712-749, South Korea
a r t i c l e i n f o
Article history:Received 24 September 2009Accepted 26 November 2009Available online 19 January 2010
Keywords:DocetaxelMixed micellesSolubilizationEnhanced bioavailabilityMultidrug resistance
* Corresponding author. Tel.: þ82 2 880 7870; fax:E-mail address: [email protected] (D.-D. Kim).
0142-9612/$ – see front matter � 2009 Elsevier Ltd.doi:10.1016/j.biomaterials.2009.11.102
a b s t r a c t
A mixed micelle that comprised of MPEG–PLA (MPP) and Pluronic� copolymers was developed forenhanced bioavailability and to overcome multidrug resistance of docetaxel in cancer therapy. The mixedmicelles that sufficiently solubilized docetaxel were evaluated for the effect of Pluronic� copolymersweight ratio on the mixed micelles with respect to drug loading and drug release. In vitro, cell viabilityand cytotoxicity studies in KB and KBv cells revealed that the mixed micellar formulations were morepotent than the commercial docetaxel formulation (Taxotere�). In vivo pharmacokinetics study in ratsshowed that the mixed micelles significantly enhanced the bioavailability of docetaxel (3.6 fold) thanTaxotere�. Moreover, antitumor activity assessed in KBv cancer xenograft BALB/C nude mice modelsshowed that the mixed micelles significantly reduced the tumor size than the control (Taxotere�). Cleardifferences in the intracellular uptake of docetaxel between MPP and mixed micelles were observedusing confocal laser scanning microscopy. This study presents not only a new micelle structure fora diblock–triblock copolymer system, but also a method for enhanced bioavailability of docetaxel and toovercome some of the limitations on its multidrug resistance in cancer therapy.
� 2009 Elsevier Ltd. All rights reserved.
1. Introduction
Docetaxel is of the chemotherapy drug class taxane, and isa semi-synthetic analogue of paclitaxel (Taxol�), an extract from therare Pacific yew tree Taxus brevifolia [1]. In the past decade, doce-taxel (Taxotere�, Sanofi–Aventis) has emerged as one of the mostimportant cytotoxic agents, with proven clinical efficacy againstmany cancers including advanced and metastatic breast cancer,non-small cell lung cancer, advanced gastric cancer, etc., [2,3].However, side effects caused by docetaxel have considerably over-shadowed its clinical use. Firstly, like most other classic chemo-therapeutic agents, docetaxel distributes throughout the body ina nonspecific manner; secondly, as docetaxel is poorly soluble inwater, non-ionic surfactant Tween 80 (polysorbate 80) and 13%ethanol in saline solution are being used to dissolve the agent in itscurrent commercial formulation (Taxotere�). The use of docetaxel inthe present formulation leads to the well-known adverse drug
þ82 2 873 9177.
All rights reserved.
reactions due to either the agent itself (e.g., fluid retention, neuro-toxicity, musculoskeletal toxicity and neutropenia) or to the solventsystem (e.g., hypersensitivity, fluid retention) [4]. Furthermore,many patients with breast cancer showed an inherent or acquiredresistance to docetaxel in the disease progression. Multidrugresistance (MDR) has been already a major clinical problem, whichlimits the effectiveness of docetaxel and other anticancer agents[5,6]. Thus, development of new approaches is required to make thechemotherapeutics achieve adequate local concentration at thetumor site and, at the same time, reduce side effects caused bynonspecific delivery.
In the past decade, polymeric micelles have been extensivelystudied for its prominent superiorities among the emerging nano-scopic carrier systems. It provides several advantages includingdrug solubilization, controlled drug release, escaping from reticu-loendothelial system (RES) uptake, tumor targeting by enhancedpermeability and retention (EPR) effect [7]. More recently, a largenumber of studies on mixed polymeric micelles have appeared dueto the prominent advantages of different types of copolymersconcentrated on a single polymeric micellar system. Di/multi-functional copolymer micelles can be realized by preparing mixed

C.-F. Mu et al. / Biomaterials 31 (2010) 2371–23792372
micelles [8,9]. The loading content and stability of drug in mixedmicelles can be improved greatly with different kinds of copolymerscompared with single copolymer micelles [10–13]. The release andfunction of micelles can be modified to be desirable by formingmixed micelles [14,15].
In this study, the objective was to develop a new mixed polymericmicellar formulation comprised of methoxy poly(ethylene glycol)–poly (lactide) polymer (MPEG–PLA, here after will be mentioned asMPP) and Pluronic� triblock polymers for enhanced bioavailabilityand to overcome MDR of docetaxel. The amphiphilic block polymerMPP was synthesized and used in this study for its excellent micelleformation, drug loading capability and release behavior [16–18].Pluronic� block copolymers consist of hydrophilic ethylene oxide(EO) and hydrophobic propylene oxide (PO) blocks arranged ina basic EO–PO–EO, were used in the study for its commercial avail-ability, biocompatibility and safety [19]. Moreover, recent studieshave reported that Pluronic� block copolymers sensitize the resis-tant cancer cell lines, resulting in an increase in the cytotoxic activityof the drug by 2–3 orders of magnitude [20]. However, the majorlimitations of Pluronic copolymers are its low micellization andsolubilization capacity to lots of hydrophobic drugs, and the lowstability of its self-assembled micelles upon dilution in the blood-stream because of its high apparent critical micelle concentration(CMC) [20]. Thus, the mixed micelles using MPP polymer and Plur-onic� copolymers (L61, L62 and P85) were prepared by co-solventevaporation method in order to enhance the bioavailability andovercome MDR of docetaxel. The prepared mixed micelles werecharacterized by drug loading, particle size and in vitro, releaseprofile, cytotoxicity, cellular uptake and intracellular distribution ofdocetaxel. Moreover, the optimized formulation was evaluated invivo by pharmacokinetics and antitumor efficacy studies.
2. Materials and methods
2.1. Materials
Docetaxel was purchased from Taihua Co. (Xi’an, China). Poly(ethylene glycol)methyl ether (MPEG, Mw: 2000), 3, 6-dimethyl-1, 4-dioxane-2, 5-dione (D,L-lactide)and stannous octoate were supplied by Aldrich (WI, USA). MPEG was dried by anazeptropic distillation in toluene, and D,L-lactide was recrystallized from ethylacetate for several times prior to use. Pluronic� copolymers L61, L62 and P85 wereobtained as a gift from BASF Corp. (Seoul, Korea). Tween 80 was purchased fromKanto Chemical Co. Inc. (Tokyo, Japan). Coumarin 6 and 3-(4, 5-dimethyltiazol-2-ly)-2, 5-diphenyltetrazolium bromide (MTT) were obtained from Sigma Chemical Co.(MO, USA). Acetonitrile (HPLC grade) was supplied by Fisher Scientific Korea Ltd.(Seoul, Korea). All other reagents were of analytical grade obtained from commercialsources and used without further purification.
2.2. Cell line and cell culture
Human carcinoma cell line KB was purchased from the American Type CultureCollection (Rockville, MD, USA). Its multidrug resistant KBv cell line was kindly giftedby Dr. Dong Kwon Rhee (College of pharmacy, Sungkyunkwan University, Seoul,Korea). Cell culture medium (RPMI 1640, Waymouth), penicillin, streptomycin andheat-inactivated fetal bovine serum (FBS) were obtained from Gibco Life Technolo-gies, Inc. (NY, USA). Culture plates and dishes were purchased from Corning Inc.(NY, USA). The KB and KBv cells were maintained in RPMI 1640 medium supple-mented with 10% FBS, 2 mM L-glutamine, 10 mM HEPES, 24 mM NaHCO3, 1% antibiotics(100 U/mL penicillin G and 0.1 mg/mL streptomycin) and 20 mM vinblastine (KBv cellsonly). Cells were maintained at 37 �C in a humidifier with 5% CO2 atmosphere.
2.3. Synthesis and characterization of MPP block copolymers
MPP diblock polymer was synthesized by the anionic ring opening polymeri-zation. D,L-lactide, MPEG with Mw 2000 and toluene were added to a two-neckedround-bottle flask with a magnetic stirrer. The diblock polymer was synthesized asdescribed elsewhere using stannous octoate as catalyst [21]. The molecular weightdistribution of the polymers was determined by the relative elution time usingpolystyrene monodisperse as standard in gel permeation chromatography (GPC).The composition and the average molecular weight of each copolymer weredetermined using 400 MHz 1H NMR in CDCl3. The Mn and Mw/Mn were 4534 and1.42, respectively. The 1H NMR and GPC data were given as Supplemental data.
2.4. Preparation and characterization of empty and docetaxel-loaded mixed micelles
A co-solvent evaporation method was used for the self-assembly of MPP/Plur-onic� copolymers (L61, L62 and P85) and drug encapsulation [22]. The ratio oforganic to the aqueous phase and the order of addition of the phases in the co-solvent evaporation method were changed to optimize the preparation method interms of carrier size and encapsulation efficiency. MPP (30 mg), Pluronic� copoly-mers at different weight ratio and drug were dissolved in 0.2 mL acetone. Thissolution was added drop-wise to 4 mL of water corresponding to a final 1:20organic:aqueous phase ratio. The mixture was then stirred at room temperature for2 h. Vacuum was applied to remove the remainder of the organic solvent. At the endof encapsulation process, the micelle solutions were filtered through a 0.45 mmMinisart syringe filter (Sartorius, Germany) to remove free docetaxel and subse-quently lyophilized. Fluorescent coumarin 6 (10 mg/mL) loaded mixed micelleformulations were also prepared in the same way.
Mean diameter of the prepared empty and drug-loaded micellar solutions wasdefined by dynamic light scattering (NICOMP 370 Submicron Particle Sizer, CA, USA)at a polymer concentration of 10 mg/mL. Transmission electron microscopy (TEM)(JEM-1010, Tokyo, Japan) of polymeric micellar solution negatively stained by 2%aqueous solution of uranyl acetate was used to assess the morphology of preparedpolymeric carriers.
An aliquot of the micellar solution was diluted with acetonitrile to disrupt theself-assembled structures. The drug loading content (DLC) and entrapmentefficiency (EE) of micelles were measured using high performance liquid chroma-tography (HPLC). The HPLC instrument consisted of a dual pump (Waters 515),auto-sampler (Waters 717 plus), Capcell Pak C18 MG column (4.6 mm � 250 mm,5 mm, Shiseido, Japan) and UV detector (Waters 2487) set at 230 nm. Mobile phasewas a mixture of acetonitrile:water (65:35, v/v), filtered through 0.45 mm membranefilter and eluted at a flow rate of 1.0 mL/min. Docetaxel concentrations weredetermined using 20 mL of injection volume at room temperature. The inter- andintra-day variance of this HPLC method was within the acceptable range.
DLC and EE were calculated from the following equations:
Drug loading content ð%Þ ¼ weight of docetaxel in micellesweight of micelles
�100
Entrapment efficiency ð%Þ ¼ weight of docetaxel in micellesweight of docetaxel fed initially
�100
2.5. In vitro release studies
The release profile of docetaxel from polymeric micelles was evaluatedaccording to an earlier report with slight modification [23]. Docetaxel-loadedpolymeric micelles and control (equivalent to 50 mg docetaxel) were placed in a minidialysis kit (molecular cut-off: 6–8 kDa, Kfar–Hanagid, Israel) and placed intoa centrifugal tube containing 30 mL of phosphate buffer saline (PBS, pH 7.4) asrelease media. The system was maintained at 37.0 � 0.5 �C with stirring rate of100 rpm. The release media (0.2 mL) was sampled at predetermined time intervalsand replaced with equal volume of fresh medium. The samples were analyzed fordocetaxel content by HPLC.
2.6. Cellular uptake study
For the cellular uptake study, 1 � 105 cells (KB and KBv) were seeded on a 24-well cell culture plate for 24 h. The cell culture medium was then replaced with newmedium containing 1 mg/mL of docetaxel and incubated for different time intervals1, 2 and 4 h at 37 �C. Cells were then washed twice with cold PBS to remove cellsurface-bound docetaxel and incubated with 0.1 mL of 2% sodium dodecyl sulfatesolution for 30 min at 37 �C. An aliquot (10 mL) of cell lysate was used for cell proteincontent determination using BCA kit according to the manufacturer protocol. Theremaining portion was extracted for docetaxel with equal volume of acetonitrile.The suspension was centrifuged at 13,000 rpm for 5 min and the supernatant wasanalyzed by HPLC.
Coumarin 6 accumulated in KB and KBv cells from mixed micelles and controlwas localized using a Leica TCS SP (Germany) confocal laser scanning microscope(CLSM). The KB and KBv cells were seeded on chambered cover glass system (LAB-TEK�, Nagle Nunc, IL) at a density of 2.5 � 104 cells/well for 24 h at 37 �C ina humidifier with 5% CO2 atmosphere. The cells were washed twice with PBS thentreated with 0.4 mL of coumarin 6 loaded mixed micelle media solution and incu-bated for 4 h at 37 �C. Cells were then washed 3 times with cold PBS and mounted ona slide with 2% formaldehyde for CLSM observation.
2.7. In vitro cytotoxicity assay
For in vitro study, two different tumor cell lines KB and KBv were used. The invitro cytotoxicity of docetaxel-loaded micelles, commercial docetaxel formulationand empty vehicle (Tween 80, 13% (w/w) ethanol) were determined by standardMTT assays [24]. Cells were seeded in a 96-well bottom plate at a density of5000 cells per well and incubated at 37 �C in a humidified atmosphere with 5% CO2

MPEG-PPLA/Pluronic®
(w/w)
6:0 6:1 6:2 6:4 6:6 0:6
Mean
d
iam
eter (n
m)
0
50
100
150
200
1000
1100
1200
MPP/L61
MPP/L62
MPP/P85
Fig. 1. The influence of Pluronic� copolymers (L61, L61 and P85) weight ratio on themean volume diameter size of the empty mixed micelles (MPP/Pluronic�).
C.-F. Mu et al. / Biomaterials 31 (2010) 2371–2379 2373
for 24 h. The medium was then replaced with 200 mL of medium containing eitherdocetaxel-loaded micelles or conventional docetaxel formulation (docetaxelequivalent to 5 ng/mL and 50 ng/mL for KB and KBv cell lines, respectively). Controlwells were treated with equivalent volume of drug free media. The cells wereincubated at 37 �C for 72 h. After incubation, wells were rinsed with PBS, and thenMTT (0.5 mg/ml) solution was added to each well and the plate was incubated for4 h, allowing the viable cells to reduce the yellow MTT into dark-blue formazancrystals, which were dissolved in 200 mL of dimethyl sulphoxide (DMSO). Theabsorbance of individual wells was measured at 560 nm by a microplate reader(Molecular Devices Corporation, Sunnyvale, CA). All the results obtained from bothcytotoxicity assays were confirmed by repeating the experiment on three inde-pendent occasions and testing in triplicate each time.
2.8. Pharmacokinetics studies
Pharmacokinetic studies were performed in male Sprague–Dawley rats(weighing about 250 g). All rats were maintained in a light-controlled room kept ata temperature of 22 � 2 �C and a relative humidity of 55 � 5% (Animal Center forPharmaceutical Research, College of Pharmacy, Seoul National University, Korea).The experimental protocols involving animal study were approved by the AnimalCare and Use Committee of the College of Pharmacy, Seoul National University.
Pharmacokinetic studies were performed as described elsewhere [25]. Femoralarteries and veins of the rats were catheterized with polyethylene tubing (PE-50,Clay Adams, Parsippany, NJ, USA) filled with 50 IU/mL of heparin in saline underanesthesia by ketamine at supine position. Polymeric micelles and commercialformulation Taxotere� were administered intravenously at a dose of docetaxel 8 mg/kg. Blood samples (w0.25 mL) were withdrawn from the femoral artery at pre-determined time intervals for 8 h. Plasma samples were obtained by immediatelycentrifuging the blood samples at 7000g for 5 min, after which 100 mL of plasmasamples were transferred to new glass tubes and stored at�20 �C until analyzed byHPLC. The pharmacokinetic parameters of each formulation were attained using theWinNonlin� program (Version 3.1, Pharsight Co., Mountainview, CA, USA).
Plasma samples were extracted twice with diethyl ether before injection, aspreviously reported with minor modification [26]. Briefly, 100 mL of plasma wasspiked with 10 mL of Titrisol buffer (pH 5.0) and 5 mL of paclitaxel (5 mg/mL) as theinternal standard. Docetaxel was extracted with 1.0 mL of diethyl ether by vigorousmixing for 5 min. After centrifugation at 13,200 rpm for 5 min, the organic phasewas collected. The organic phases were combined after repeating the aboveextraction procedure two times then it was dried under nitrogen gas stream at 40 �C.The residue was then reconstituted with 40 mL of methanol and vortex for 5 min. Thesolution was centrifuged for 5 min at 13,200 rpm and 20 mL of the supernatant wasinjected into the HPLC system.
2.9. In vivo antitumor efficacy
The antitumor efficacy was investigated in KBv (drug resistant cancer cell line)cells bearing BALB/C nude mice (Charles River, Korea). For in vivo implantation, KBvcells were injected subcutaneously at 1 �106 cells in 0.1 mL in the right rear flank of4-weeks old male BALB/C nude mice (19 � 1 g). Tumor dimensions were measuredas the method described in tumor distribution studies. When tumors were 50–100 mm3 in volume, treatments were started and this day was designated as day 0.On day 0, mice were randomly divided into groups of 4. They were injected controland test formulations intravenously through the tail vein at a dose of docetaxel10 mg/kg for four times on days 0, 3, 6 and 9. Mice injected with saline andcommercial formulation Taxotere� were set up as control groups. Tumor size andthe body weight of the mice were measured once in 3 days. The antitumor efficacy ofdocetaxel-loaded micelles was compared with that of conventional docetaxelformulation by observing tumor volume and measuring total body weight.
2.10. Statistical analysis
All the experiments in the study were performed at least three times and thedata were expressed as the mean � standard deviation (S.D.). A two-tailed unpairedStudent’s t-test was performed at p < 0.05.
3. Results and discussion
3.1. Characterization of empty and docetaxel-loaded mixed micellarformulation
3.1.1. Preparation methodEncapsulation process has shown to affect loading efficiency of
a number of hydrophobic drugs in polymeric micelles depending onthe physicochemical properties of the block copolymers [7]. Forinstance, switching the encapsulation method from dialysis tosolvent evaporation is shown to increase the encapsulated levels of
amphotericin B in MePEO-block-poly (N-hexyl stearate L-asparta-mide) (MePEO-b-PHSA) micelles [27]. Assembly of MPP blockcopolymers and drug loading in the assembled structures haspreviously been accomplished through solution casting method[16,28], which employs heating process and is unfavorable to heatlabile drugs. In this study we employed co-solvent evaporationmethod to prepare MPP/Pluronic� copolymer self-assemblemicelles. In the co-solvent evaporation method, the block copolymerand the drug are dissolved in a volatile, water miscible organicsolvent (selective co-solvent for the core-forming block). Self-assembly and drug entrapment is then triggered by the addition ofthe organic phase to water (non-solvent for the core-forming block)(or vice versa) followed by the evaporation of the organic co-solvent.The co-solvent evaporation method bears several advantages overother methods including more feasibility for scale up and less chancefor drug loss during dialysis in the encapsulation process [23].Moreover, it has been reported that the type of organic co-solvent,the volume fraction of organic to aqueous phase during the assemblyprocess would affect the particle size and distribution of polymericmicelles [23]. Each factor was individually manipulated and theeffect of each change on carrier size, morphology and/or docetaxel-loaded levels was assessed (data not shown). Acetone was selected asorganic co-solvent and the volume fraction of organic to aqueousphase was optimized at 1:20 v/v as it produced the smallest meanparticle size and narrow size distribution of mixed micelles. Upon thedrop-wise addition of highly miscible acetone, with dissolved drugand unimers, to a large excess of mixing water, the unimers and drugare in a state of high supersaturation resulting in rapid and simul-taneous nucleation of micelle structures incorporating the drugmolecule into the hydrophobic core [29].
3.1.2. Size and morphologyAs shown in the Fig. 1, the size of mixed micelles did not vary
significantly from MPP micelles (w21.1 nm) when the ratios ofMPP/Pluronic� copolymer changed from 6:1 to 6:6. However, thesize of pure Pluronic� micelles was above 100 nm. This could bedue to its high tendency to aggregate in aqueous solution followedby liquid phase separation at relatively low concentration. As L61micelles showed much larger particle size compared to other purePluronic� micelles (>1100 nm) thus further studies on it was notperformed. It appeared in this study that the size of the mixedmicelles is dominated by the higher molecular weight diblockcopolymer. This is in good accordance with an earlier report where

C.-F. Mu et al. / Biomaterials 31 (2010) 2371–23792374
a higher molecular weight triblock copolymer dominated theparticle size [30]. Fig. 2A and B shows the representative sizedistribution and TEM micrograph of the empty MPP/L62 (6:1 w/w)mixed micelles, respectively. The narrow size distribution of themicelles (Fig. 2A) implied the co-micellization of the two copoly-mers. The di/triblock copolymers formed a uniform structure withhomogenous core. The TEM image showed that the morphology ofthe mixed polymeric micelle was spherical in shape (Fig. 2B andSupplemental data). The size of micelle is one of the properties,which largely influence the circulation time and organ distributionof the vehicle. Particles which are less than 200 nm are said to beless susceptible to RES clearance, and those less than 5 mm haveaccess to small capillaries [7]. Hence, the fabrication of mixedmicelle with Pluronic� copolymer is necessary to exploit the MDRinhibition effect of Pluronic� copolymers as they alone producelarger particle size micelles with broader size distribution (Fig. 1).
3.1.3. Drug loading capacity of micellar formulationsMany studies have reported that the major factor influencing
both the loading capacity and efficiency of polymeric micelles is the
Fig. 2. Representative NICOMP size distribution analysis of MPP/L62 (6:1 w/w) mixedmicelles (A) and its transmission electron micrograph (B).
compatibility between the solubilizate and core-forming block [31].To investigate the impact of different proportion of Pluronic�
copolymers on the loading capacity of docetaxel in mixed micelles,the drug loading content was determined in the mixed micelleswith varied weight ratio of MPP to L62 (L62 taken as a model for allPluronic� copolymers due to its higher DLC) at 5% theoreticalloading rate (Fig. 3). It was found that the drug loading did notaffect the mixed micelles particle size (Table 1). The results indi-cated that the drug loading capacity diminished remarkably withincreased weight ratio of L62 from 6:1 to 6:4 in the mixed micelles(MPP/L62). The drug loading capacity of MPP micelles was about10-fold higher compared to that of L62 micelles. This could beattributed to the fact that the higher affinity of drug to the relativelymore hydrophobic core PLA of MPP copolymer than PPO of Plur-onic� copolymer micelles. In addition to that the ‘‘liquid-like’’ stateof the core of Pluronic� micelles, which was demonstrated by NMRrelaxation study [32], also explains the low loading capacity ofPluronic� copolymer micelles. This explains the decreasing drugloading of mixed micelles with increasing Pluronic� copolymersratio in it. Thus, we fixed the ratio between MPP and Pluronic�
copolymers at 6:1 to obtain higher drug-loaded mixed micelles.The effect of amount of docetaxel used to prepare mixed
micelles consisting of MPP and Pluronic� copolymers (L61, L62 andP85) at 6:1 w/w ratio in the range of 5–20% theoretical loading onthe entrapment efficiency of docetaxel is profiled in Table 1. It wasobserved that the higher the drug used lower the entrapmentefficiency of MPP and mixed micelles. This could be due to the drugsaturation of the inner core of the micelles might be reached at 5%of drug incorporation. There was obvious drug precipitation, whenhigher amount of drug was used. This leads to the assumption thatmicelle formulations could enhance the solubility of certain poorlysoluble drugs but to a maximum limit after which any increase inthe drug concentration leads to drug precipitation [33]. Among thetested formulations, MPP/P85 micelles showed relatively low EE.This could be due to more hydrophilic nature of P85 polymer(HLB ¼ 16) than L61 and L62 polymers. In this study the maximumsolubilization capacity of docetaxel by the prepared mixed micelles(MPP/Pluronic�) at the ratio of 6:1 was 3.71% (MPP/L61) and theminimum was 3.17% (MPP/P85), which was in good accordancewith earlier reported poly(ethylene oxide)-block-poly (stereneoxide) (PEO-b-PSO) [34] and poly (N-vinylpyrrolidone)-block-poly(D,L-lactide) micelles [35].
MPEG-PLA/L62 (w/w)
6:0 6:1 6:2 6:4 0:6
Dru
g lo
ad
in
g co
nten
t (%
)
0
1
2
3
4
5
Fig. 3. The effect of Pluronic� L62 copolymer ratio on the drug loading content (%) ofmixed micelles (n ¼ 3, theoretical loading: 5%).
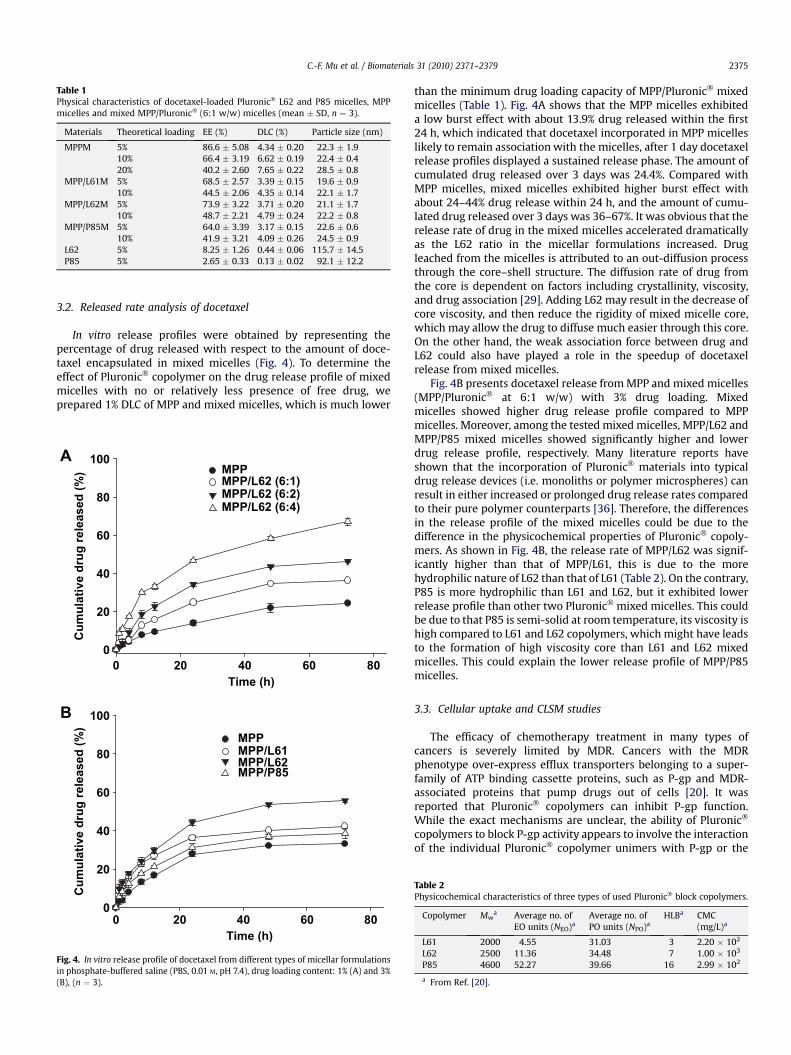
Table 1Physical characteristics of docetaxel-loaded Pluronic� L62 and P85 micelles, MPPmicelles and mixed MPP/Pluronic� (6:1 w/w) micelles (mean � SD, n ¼ 3).
Materials Theoretical loading EE (%) DLC (%) Particle size (nm)
MPPM 5% 86.6 � 5.08 4.34 � 0.20 22.3 � 1.910% 66.4 � 3.19 6.62 � 0.19 22.4 � 0.420% 40.2 � 2.60 7.65 � 0.22 28.5 � 0.8
MPP/L61M 5% 68.5 � 2.57 3.39 � 0.15 19.6 � 0.910% 44.5 � 2.06 4.35 � 0.14 22.1 � 1.7
MPP/L62M 5% 73.9 � 3.22 3.71 � 0.20 21.1 � 1.710% 48.7 � 2.21 4.79 � 0.24 22.2 � 0.8
MPP/P85M 5% 64.0 � 3.39 3.17 � 0.15 22.6 � 0.610% 41.9 � 3.21 4.09 � 0.26 24.5 � 0.9
L62 5% 8.25 � 1.26 0.44 � 0.06 115.7 � 14.5P85 5% 2.65 � 0.33 0.13 � 0.02 92.1 � 12.2
C.-F. Mu et al. / Biomaterials 31 (2010) 2371–2379 2375
3.2. Released rate analysis of docetaxel
In vitro release profiles were obtained by representing thepercentage of drug released with respect to the amount of doce-taxel encapsulated in mixed micelles (Fig. 4). To determine theeffect of Pluronic� copolymer on the drug release profile of mixedmicelles with no or relatively less presence of free drug, weprepared 1% DLC of MPP and mixed micelles, which is much lower
Time (h)
0 20 40 60 80
Cu
mu
lative d
ru
g released
(%
)
0
20
40
60
80
100A
B
MPP
MPP/L62 (6:1)
MPP/L62 (6:2)
MPP/L62 (6:4)
Time (h)
0 20 40 60 80
Cu
mu
lative d
ru
g released
(%
)
0
20
40
60
80
100
MPP
MPP/L61
MPP/L62
MPP/P85
Fig. 4. In vitro release profile of docetaxel from different types of micellar formulationsin phosphate-buffered saline (PBS, 0.01 M, pH 7.4), drug loading content: 1% (A) and 3%(B), (n ¼ 3).
than the minimum drug loading capacity of MPP/Pluronic� mixedmicelles (Table 1). Fig. 4A shows that the MPP micelles exhibiteda low burst effect with about 13.9% drug released within the first24 h, which indicated that docetaxel incorporated in MPP micelleslikely to remain association with the micelles, after 1 day docetaxelrelease profiles displayed a sustained release phase. The amount ofcumulated drug released over 3 days was 24.4%. Compared withMPP micelles, mixed micelles exhibited higher burst effect withabout 24–44% drug release within 24 h, and the amount of cumu-lated drug released over 3 days was 36–67%. It was obvious that therelease rate of drug in the mixed micelles accelerated dramaticallyas the L62 ratio in the micellar formulations increased. Drugleached from the micelles is attributed to an out-diffusion processthrough the core–shell structure. The diffusion rate of drug fromthe core is dependent on factors including crystallinity, viscosity,and drug association [29]. Adding L62 may result in the decrease ofcore viscosity, and then reduce the rigidity of mixed micelle core,which may allow the drug to diffuse much easier through this core.On the other hand, the weak association force between drug andL62 could also have played a role in the speedup of docetaxelrelease from mixed micelles.
Fig. 4B presents docetaxel release from MPP and mixed micelles(MPP/Pluronic� at 6:1 w/w) with 3% drug loading. Mixedmicelles showed higher drug release profile compared to MPPmicelles. Moreover, among the tested mixed micelles, MPP/L62 andMPP/P85 mixed micelles showed significantly higher and lowerdrug release profile, respectively. Many literature reports haveshown that the incorporation of Pluronic� materials into typicaldrug release devices (i.e. monoliths or polymer microspheres) canresult in either increased or prolonged drug release rates comparedto their pure polymer counterparts [36]. Therefore, the differencesin the release profile of the mixed micelles could be due to thedifference in the physicochemical properties of Pluronic� copoly-mers. As shown in Fig. 4B, the release rate of MPP/L62 was signif-icantly higher than that of MPP/L61, this is due to the morehydrophilic nature of L62 than that of L61 (Table 2). On the contrary,P85 is more hydrophilic than L61 and L62, but it exhibited lowerrelease profile than other two Pluronic� mixed micelles. This couldbe due to that P85 is semi-solid at room temperature, its viscosity ishigh compared to L61 and L62 copolymers, which might have leadsto the formation of high viscosity core than L61 and L62 mixedmicelles. This could explain the lower release profile of MPP/P85micelles.
3.3. Cellular uptake and CLSM studies
The efficacy of chemotherapy treatment in many types ofcancers is severely limited by MDR. Cancers with the MDRphenotype over-express efflux transporters belonging to a super-family of ATP binding cassette proteins, such as P-gp and MDR-associated proteins that pump drugs out of cells [20]. It wasreported that Pluronic� copolymers can inhibit P-gp function.While the exact mechanisms are unclear, the ability of Pluronic�
copolymers to block P-gp activity appears to involve the interactionof the individual Pluronic� copolymer unimers with P-gp or the
Table 2Physicochemical characteristics of three types of used Pluronic� block copolymers.
Copolymer Mwa Average no. of
EO units (NEO)aAverage no. ofPO units (NPO)a
HLBa CMC(mg/L)a
L61 2000 4.55 31.03 3 2.20 � 102
L62 2500 11.36 34.48 7 1.00 � 103
P85 4600 52.27 39.66 16 2.99 � 102
a From Ref. [20].

C.-F. Mu et al. / Biomaterials 31 (2010) 2371–23792376
lipid microenvironment surrounding of P-gp. Additionally, ATPdepletion induced by Pluronic� copolymers in MDR cells isconsidered as one potential reason for chemosensitization of thesecells [37]. Moreover, it has been observed that block copolymerswith longer PO and shorter EO segments are more likely to interactwith cell membranes, particularly, the most efficient block copol-ymers are those with intermediate length of the hydrophobic blockranging from 30 to 60 repeating units like L61, L62 and P85copolymers [20,37].
The cellular uptake and CLSM study were performed in KB andKBv cell lines. The results of the cellular uptake of docetaxel fromcommercial formulation, MPP and mixed micelles by KB and KBvcell lines at 37 �C for distinct durations are shown in Fig. 5A and B,respectively. As shown in Fig. 5A, there was no significant differ-ence in the cellular uptake of docetaxel from different formulations.Free docetaxel may transport across the cell membrane throughdiffusion in KB cells, a pathway that is affected in MDR KBv cells byP-gp [38,39]. As shown in Fig. 5B, after exposure of KBv cells to
Taxote
re®
MPP
MPP/L
61
MPP/L
62
MPP/P
85
Do
cetaxel co
nten
t (n
g/m
g p
ro
tein
)
0
20
40
60
80
1 h
2 h
4 h
Taxote
re®
MPP
MPP/L
61
MPP/L
62
MPP/P
85
Do
cetaxel co
nten
t (n
g/m
g p
ro
tein
)
0
10
20
30
40
501 h
2 h
4 h ** *
A
B
Fig. 5. Cellular uptake of docetaxel in KB (A) and KBv (B) cells with Taxotere�, differentdocetaxel-loaded micellar formulations (docetaxel equivalent to 1 mg/mL) followingincubation at 37 �C for 1, 2 and 4 h. (n ¼ 3) *p < 0.05 compared with Taxotere� andMPP micelles.
Texotere� and MPP micelles, the accumulation of docetaxel in cellswas limited throughout the entire course of the study. This obser-vation suggested that the uptake of docetaxel was extruded out ofKBv cells by the P-gp efflux pump. On the contrary, for the doce-taxel-containing mixed micelles, the accumulation of docetaxel inKBv cells increased with increased duration of incubation, indi-cating that Pluronic� copolymers in mixed micelles were able toeffectively inhibit P-gp function. This may be attributed to theinteraction of the individual copolymer unimers (L61, L62 and P85)with the P-gp in KBv cells [20].
Fig. 6 presents the cellular association and localization of fluo-rescent coumarin 6 loaded MPP and mixed micellar formulations inKB and KBv cells at 37 �C by CLSM. It can be observed that the KBcells at 4 h showed stronger fluorescent signals for all the test andcontrol formulations than the KBv cells (Fig. 6). Stronger fluorescentsignals indicated higher internalization of coumarin 6. In KB cellline there was no obvious difference in the fluorescent signals at4 h, however, in KBv cell lines, a distinct difference between themixed and MPP micelles was observed. There was no significantinternalization of coumarin 6 inside the cells treated with MPPmicelles, but in mixed micelles the internalization was significantcompared to MPP micelles. These results supported our cellularuptake studies in these cell lines.
3.4. In vitro cytotoxicity of different docetaxel-loaded formulations
As a next step, we investigated whether the increased cellulardocetaxel accumulation by mixed micelle formulations increasedthe cytotoxicity in cancer cells. To determine the cytotoxic activitiesof docetaxel in mixed micelles, the formulations were testedagainst KB and its MDR KBv cell line, which have been previouslyused in studying the anticancer activity and reversal of MDR. [40].For better understanding of the effect of different mixed micelleformulations and their corresponding blank vehicles on KB and KBvcell lines, the IC50 of docetaxel for these cancer cell lines weredetermined separately and the cytotoxicity studies were performedat IC50 concentrations (5 ng/mL of docetaxel for KB and 50 ng/mL ofdocetaxel for KBv cells). The cytotoxicity of tested formulations inKB and KBv cells were profiled in Fig. 7. The cell viability of KB cellsfor the controls and test formulations (at 5 ng/mL of docetaxel) hadno obvious differences between them (Fig. 7A). However, the mixedmicelles (at 50 ng/mL of docetaxel) showed significant cytotoxicityin KBv cells, the cell viability was much lower (8–15%) compared toTaxotere� and MPP micelles (Fig. 7B). These results were in goodagreement with an earlier reported study [38] as well as ourprevious studies mentioned in Section 3.3, quantitative detection ofdocetaxel uptake and CLSM internalization studies in these celllines.
3.5. Pharmacokinetics studies
Linearity in the standard curves was demonstrated for plasmaover the concentration range studied, and chromatograms werefree of interference from endogenous components. The plasmadocetaxel concentration vs. time profiles observed for Taxotere�,MPP and mixed micellar formulations were shown in Fig. 8. A rapiddecline in concentrations, representing a distribution phase, wasobserved for the first 2 h after dosing. This was followed by anelimination phase with t1/2 of 1–2 h. However, after dose normal-ization, docetaxel in MPP and mixed micelles yielded higherplasma concentrations than did Taxotere�. Non-compartmentalanalysis of the plasma concentrations showed a significant changein pharmacokinetic parameters of docetaxel in mixed micellescompared to that of commercial formulation (Table 3). Mixedmicelles provided significantly higher (3.5 fold) AUC compared to
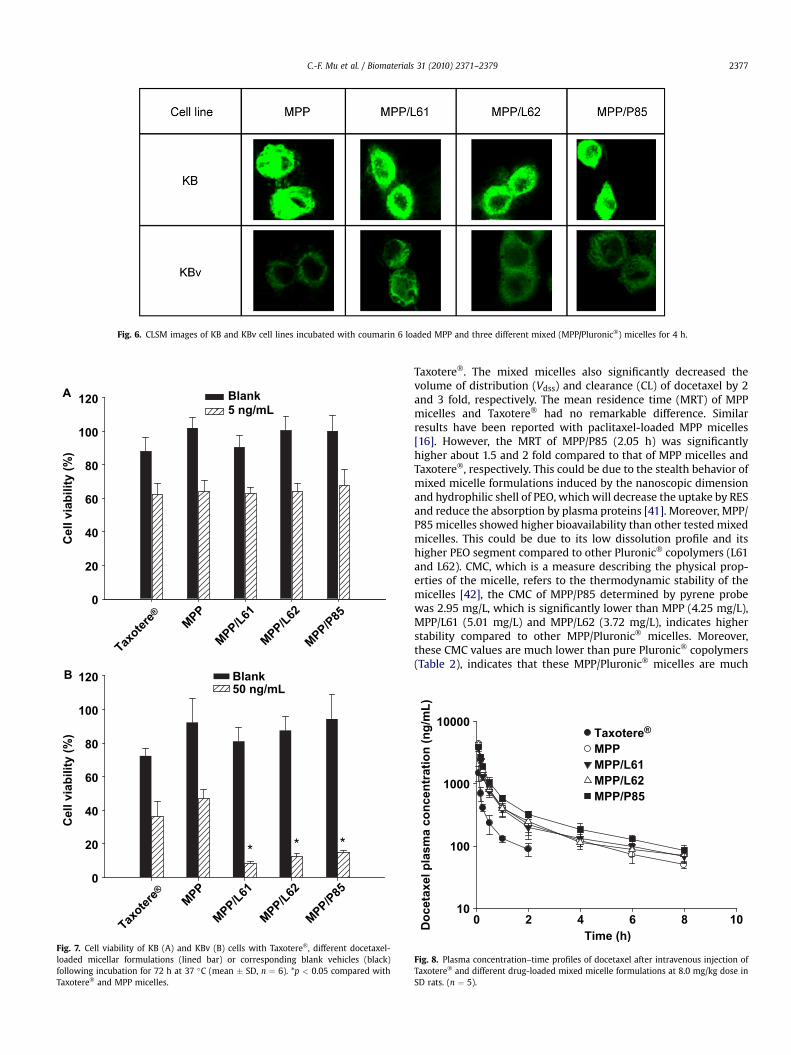
Fig. 6. CLSM images of KB and KBv cell lines incubated with coumarin 6 loaded MPP and three different mixed (MPP/Pluronic�) micelles for 4 h.
Taxote
re®
MPP
MPP/L
61
MPP/L
62
MPP/P
85
Cell viab
ility (%
)C
ell viab
ility (%
)
0
20
40
60
80
100
120A
B
Blank
5 ng/mL
Taxote
re®
MPP
MPP/L
61
MPP/L
62
MPP/P
85
0
20
40
60
80
100
120 Blank
50 ng/mL
* * *
Fig. 7. Cell viability of KB (A) and KBv (B) cells with Taxotere�, different docetaxel-loaded micellar formulations (lined bar) or corresponding blank vehicles (black)following incubation for 72 h at 37 �C (mean � SD, n ¼ 6). *p < 0.05 compared withTaxotere� and MPP micelles.
C.-F. Mu et al. / Biomaterials 31 (2010) 2371–2379 2377
Taxotere�. The mixed micelles also significantly decreased thevolume of distribution (Vdss) and clearance (CL) of docetaxel by 2and 3 fold, respectively. The mean residence time (MRT) of MPPmicelles and Taxotere� had no remarkable difference. Similarresults have been reported with paclitaxel-loaded MPP micelles[16]. However, the MRT of MPP/P85 (2.05 h) was significantlyhigher about 1.5 and 2 fold compared to that of MPP micelles andTaxotere�, respectively. This could be due to the stealth behavior ofmixed micelle formulations induced by the nanoscopic dimensionand hydrophilic shell of PEO, which will decrease the uptake by RESand reduce the absorption by plasma proteins [41]. Moreover, MPP/P85 micelles showed higher bioavailability than other tested mixedmicelles. This could be due to its low dissolution profile and itshigher PEO segment compared to other Pluronic� copolymers (L61and L62). CMC, which is a measure describing the physical prop-erties of the micelle, refers to the thermodynamic stability of themicelles [42], the CMC of MPP/P85 determined by pyrene probewas 2.95 mg/L, which is significantly lower than MPP (4.25 mg/L),MPP/L61 (5.01 mg/L) and MPP/L62 (3.72 mg/L), indicates higherstability compared to other MPP/Pluronic� micelles. Moreover,these CMC values are much lower than pure Pluronic� copolymers(Table 2), indicates that these MPP/Pluronic� micelles are much
Time (h)
0 2 4 6 8 10
Do
cetaxel p
lasm
a co
ncen
tratio
n (n
g/m
L)
10
100
1000
10000
Taxotere®
MPP
MPP/L61
MPP/L62
MPP/P85
Fig. 8. Plasma concentration–time profiles of docetaxel after intravenous injection ofTaxotere� and different drug-loaded mixed micelle formulations at 8.0 mg/kg dose inSD rats. (n ¼ 5).

Table 3Pharmacokinetic parameters of docetaxel after intravenous administration of Tax-otere�, MPP micelles and three MPP/Pluronic� (6:1) mixed micellar formulations inrats (8 mg/kg of docetaxel, mean � SD, n ¼ 3–5).
Formulations AUC (mg/ml h) MRT (h) Vss (L/kg) CL (L/h/kg) BA (%)
Taxotere� 0.82 � 0.06 1.04 � 0.27 10.25 � 1.78 9.07 � 0.45 100MPP 2.32 � 0.35* 1.34 � 0.35 4.47 � 0.38* 3.50 � 0.46* 283MPP/L61 2.28 � 0.36* 1.47 � 0.20 5.21 � 0.65* 3.56 � 0.55* 278MPP/L62 2.61 � 0.33* 1.53 � 0.22 4.66 � 0.16* 3.10 � 0.42* 318MPP/P85 2.92 � 0.21* 2.05 � 0.33*# 5.46 � 0.62* 2.75 � 0.19* 356
*p < 0.05 compared with Taxotere�, #p < 0.05 compared with MPP micelles.
C.-F. Mu et al. / Biomaterials 31 (2010) 2371–23792378
more stable than pure Pluronic� micelles at diluted condition [42].This also explains the obtained pharmacokinetic profile from MPP/Pluronic� micelles.
3.6. In vivo antitumor efficacy
To evaluate therapeutic efficacy of MPP/P85 micelle in vivo, weintravenously administered Taxotere�, MPP/P85 micelles, MPPmicelles (10 mg/kg of docetaxel) and saline as the control into thetail vein of tumor-bearing mice. Fig. 9 profiled the antitumor effi-cacy of the test and the control formulations. In Fig. 9A, it could be
Time (day)
0 5 10 15 20 25
#
30 35
Tu
mo
r vo
lu
me (m
m3)
0
500
1000
1500
2000
2500
3000A
B
Saline
Taxotere®
MPP
MPP/P85
Time (day)
0 5 10 15 20 25 30 35
Bo
dy
w
eig
ht (g
)
16
18
20
22
24
26
28
30 Saline
Taxotere®
MPP
MPP/P85
*
*
Fig. 9. (A) Antitumor effect of control and micellar docetaxel formulations on BALB/ctumor-bearing nude mice. (B) Body weight change of BALB/c tumor-bearing nude miceafter intravenous injection according to a dose schedule regimen of four injections at 3days intervals. *p < 0.05 compared with Taxotere�, #p < 0.05 compared with MPPmicelles. (n ¼ 4).
observed that the volume of tumor treated with MPP/P85 micelleswas 1.7-fold and 3.4-fold smaller than those treated with MPPmicelles and Taxotere� at the end of 24 days, respectively. Thetumor growth data of different mice showed a substantial evidenceof tumor-targeting specificity of MPP/P85 micelles in tumor-bearing mice (Fig. 9A). The MPP/P85 mixed micelles inhibitedtumor growth most efficiently, followed by MPP micelle and Tax-otere� (p < 0.05). This could be due to the longer circulation timeobserved in pharmacokinetic study, higher dissolution profilecompared to MPP micelles and overcoming MDR observed incellular uptake and cytotoxicity studies (in vitro) by the MPP/P85mixed micellar formulation. Fig. 9B shows the body weight changeof the tested mice groups. The results displayed that there was noserious body weight loss treated with MPP/P85 mixed micellarformulations compared to that of Taxotere�, moreover, bodyweight have gradually recovered over a period of treatment. Theseresults indicated that the fabricated mixed micellar formulationsinduced less systemic toxicity, probably due to high solubilizationcapability of micelles prevented the random drug release in thebody and the smaller size (about 20 nm) enhanced the tumorspecificity and therapeutic efficacy of docetaxel.
4. Conclusions
Mixed micelles consist of MPP/Pluronic� copolymers at 6:1 w/wratio with high solubilization of docetaxel (>3% w/w) and smallerparticle size (20–25 nm) to enhance the bioavailability and over-come MDR were successfully developed. The physicochemicalproperties of Pluronic� polymers played pivotal role with respect todrug loading, drug release, pharmacokinetic profile and eventuallyovercoming MDR in vivo of mixed micelles. Thus, preparing mixedmicelles with desired pharmacokinetic profile depends on thephysicochemical properties of the copolymers used to fabricatemixed micelles. The pharmacokinetic and antitumor evaluation ofthe mixed micelles prepared in this study showed that theseformulations could be an alternative to the conventional formula-tions in cancer chemotherapy to fight MDR effectively.
Acknowledgements
This study was financially supported by the Ministry of Educa-tion, Science and Technology (F104AA010008-08A0101-00810) inKorea. The authors thank Dr. Dong Kwon Rhee at SungkyunkwanUniversity for generous donation of KBv cell line.
Appendix
Figure with essential colour discrimination. Fig. 6 of this articlemay be difficult to interpret in black and white. The full colourimages can be found in the online version, at doi:10.1016/j.biomaterials.2009.11.102.
Appendix. Supplementary data
Note: the supplementary material associated with this articlecan be viewed at doi:10.1016/j.biomaterials.2009.11.102.
References
[1] Bissery MC. Preclinical pharmacology of docetaxel. Eur J Cancer1995;31A(Suppl. 4):S1–6.
[2] Baker J, Ajani J, Scotte F, Winther D, Martin M, Aapro MS, et al. Docetaxel –related side effects and their management. Eur J Oncol Nurs 2009;13:49–59.
[3] Kintzel PE, Michaud LB, Lange MK. Doetaxel-associated epiphora. Pharmaco-therapy 2006;26:853–67.

C.-F. Mu et al. / Biomaterials 31 (2010) 2371–2379 2379
[4] Persohn E, Canta A, Schoepfer S, Traebert M, Mueller L, Gilardini A, et al.Morphological and morphometric analysis of paclitaxel and docetaxel-induced peripheral neuropathy in rats. Eur J Cancer 2005;41:1460–6.
[5] Brown I, Shalli K, McDonald SL, Moir SE, Hutcheon AW, Heys SD, et al. Reducedexpression of p27 is a novel mechanism of docetaxel resistance in breastcancer cells. Breast Cancer Res 2004;6:R601–7.
[6] McDonald SL, Stevenson DA, Moir SE, Hutcheon AW, Haites NE, Heys SD, et al.Genomic changes identified by comparative genomic hybridisation in doce-taxel-resistant breast cancer cell lines. Eur J Cancer 2005;41:1086–94.
[7] Gaucher G, Dufresne MH, Sant VP, Kang N, Maysinger D, Leroux JC. Blockcopolymer micelles: preparation, characterization and application in drugdelivery. J Control Release 2005;109:169–88.
[8] Wang Y, Yu L, Han L, Sha X, Fang X. Difunctional Pluronic copolymer micellesfor paclitaxel delivery: synergistic effect of folate-mediated targeting andPluronic-mediated overcoming multidrug resistance in tumor cell lines. Int JPharm 2007;337:63–73.
[9] Lee ES, Gao Z, Kim D, Park K, Kwon IC, Bae YH. Super pH-sensitive multi-functional polymeric micelle for tumor pHe specific TAT exposure and multi-drug resistance. J Control Release 2008;129:228–36.
[10] Gao Z, Lukyanov AN, Chakilam AR, Torchilin VP. PEG–PE/phosphatidylcholinemixed immunomicelles specifically deliver encapsulated Taxol to tumor cellsof different origin and promote their efficient killing. J Drug Target2003;11:87–92.
[11] Sawant RR, Sawant RM, Torchilin VP. Mixed PEG–PE/vitamin E tumor-targetedimmunomicelles as carriers for poorly soluble anti-cancer drugs Improveddrug solubilization and enhanced in vitro cytotoxicity. Eur J Pharm Biopharm2008;70:51–7.
[12] Mu L, Elbayoumi TA, Torchilin VP. Mixed micelles made of poly(ethyleneglycol)–phosphatidylethanolamine conjugate and d-a-tocopheryl poly-ethylene glycol 1000 succinate as pharmaceutical nanocarriers for campto-thecin. Int J Pharm 2005;306:142–9.
[13] Li L, Tan YB. Preparation and properties of mixed micelles made of pluronicpolymer and PEG–PE. J Colloid Interface Sci 2008;31:326–31.
[14] Lo CL, Huang CK, Lin KM, Hsiue GH. Mixed micelles formed from graft anddiblock copolymers for application in intracellular drug delivery. Biomaterials2007;28:1225–35.
[15] Yin H, Lee ES, Kim D, Lee KH, Oh KT. Physicochemical characteristics ofpH-sensitive poly (L-histidine)-b-poly(ethylene glycol)/poly (L-lactide)-b-poly(ethylene glycol) mixed micelles. J Control Release 2008;126:130–8.
[16] Burt HM, Zhang X, Toleikis P, Embree L, Hunter WL. Development of copoly-mers of poly(D,L-lactide) and methoxy polyethylene glycol as micellar carriersof paclitaxel. Colloids Surf B Biointerfaces 1999;16:161–71.
[17] Blanco E, Bey EA, Dong Y, Weinberg BD, Sutton DM, Boothman DA, et al.b-Lapachone-containing PEG–PLA polymer micelles as novel nanotherapeuticsagainst NQO1-overexpressing tumor cells. J Control Release 2007;122:365–74.
[18] Xiong MP, Yanez JA, Kwon GS, Davues NM, Forrest ML. A cremophor-freeformulation for tanespimycin (17-AAG) using PEO-b-PDLLA micelles: charac-terization and pharmacokinetics in rats. J Pharm Sci 2009;98:1577–86.
[19] Chiappetta DA, Sosnik A. Poly(ethylene oxide)-poly(propylene oxide) blockcopolymer micelles as drug delivery agents: improved hydrosolubility stabilityand bioavailability of drugs. Eur J Pharm Biopharm 2007;66:303–17.
[20] Kabanov AV, Batrakova EV, Alakhov VY. Pluronic� block copolymers forovercoming drug resistance in cancer. Adv Drug Deliv Rev 2002;54:759–79.
[21] Tobio M, Sanchez A, Vila A, Soriano I, Evora C, Vila-Jato JL, et al. The role of PEGon the stability in digestive fluids and in vivo fate of PEG–PLA nanoparticlesfollowing oral administration. Colloids Surf B Biointerfaces 2000;18:315–23.
[22] Aliabadi HM, Elhasi S, Mahmud A, Gulamhusein R, Mahdipoor P, Lavasanifar A.Encapsulation of hydrophobic drugs in polymeric micelles through co solventevaporation: the effect of solvent composition on micellar properties and drugloading. Int J Pharm 2007;329:158–65.
[23] Yang M, Ding Y, Zhang L, Qian X, Jiang X, Liu B. Novel thermosensitive poly-meric micelles for docetaxel delivery. J Biomed Mater Res A 2007;81:847–57.
[24] Twentyman PR, Luscombe M. A study of some variables in a tetrazolium dye(MTT) based assay for cell growth and chemosenitivity. Br J Cancer1987;56:279–85.
[25] Yin YM, Cui FD, Mu CF, Choi MK, Kim JS, Chung SJ, et al. Docetaxel micro-emulsion for enhanced oral bioavailability: preparation and in vitro and invivo evaluation. J Control Release 2009; doi:10.1016/j.jconrel.2009.08.01.
[26] Ciccolini J, Catalin J, Blachon MF, Durand A. Rapid high-performance liquidchromatographic determination of docetaxel (Taxotere) in plasma usingliquid–liquid extraction. J Chromatogr B Biomed Sci Appl 2001;759:299–306.
[27] Lavasanifar A, Samuel J, Kwon GS. Micelles self-assembled from poly(ethyleneoxide)-block-poly(N-hexyl stearate l-aspartamide) by a solvent evaporationmethod: effect on the solubilization and haemolytic activity of amphotericinB. J Control Release 2001;77:155–60.
[28] Zhang X, Jackson JK, Burt HM. Development of amphiphilic diblock copoly-mers as micellar carriers of taxol. Int J Pharm 1996;132:195–206.
[29] Forrest ML, Won CY, Malick AW, Kwon GS. In vitro release of the mTORinhibitor rapamycin from poly(ethylene glycol)-b-poly(3-caprolactone)micelles. J Control Release 2006;110:370–7.
[30] Dai S, Ravi P, Leong CY, Tam KC, Gan LH. Synthesis and aggregation behavior ofamphiphilic block copolymers in aqueous solution: di- and tri-block copoly-mers of poly (ethylene oxide) and poly (ethyl acrylate). Langmuir U S A2004;20:1597–604.
[31] Allen C, Maysinger D, Eisenberg A. Nano-engineering block copolymeraggregates for drug delivery. Colloids Surf B Biointerfaces 1999;16:3–27.
[32] Ma J, Guo C, Tang Y, Xiang J, Chen S, Wang J, et al. Micellization in aqueoussolution of an ethylene oxide–propylene oxide triblock copolymer, investi-gated with 1H NMR spectroscopy, pulsed-field gradient NMR and NMRrelaxation. J Colloid Interface Sci 2007;312:390–6.
[33] Balakrishnan P, Shanmugam S, Lee WS, Lee WM, Kim JO, Oh DH, et al.Formulation and in vitro assessment of minoxidil niosomes for enhanced skindelivery. Int J Pharm 2009;377(1–2):1–8.
[34] Elsabahy M, Perron ME, Bertrand N, Yu GE, Leroux JC. Leroux, solubilization ofdocetaxel in poly (ethylene oxide)-block-poly(butylenes/styrene oxide)micelles. Biomacromolecules 2007;8:2250–7.
[35] Le GD, Gori S, Luo L, Lessard D, Smith DC, Yessine MA, et al. Poly(N-vinyl-pyrrolidone)-block-poly(D, L-lactide) as a new polymeric solubilizer forhydrophobic anticancer drugs: in vitro and in vivo evaluation. J ControlRelease 2004;99:83–101.
[36] DesNoyer JR, McHugh AJ. The effect of Pluronic on the protein release kineticsof an injectable drug delivery system. J Control Release 2003;86:15–24.
[37] Batrakova E, Lee S, Li S, Venne A, Alakhov V, Kabanov A. Fundamental rela-tionships between the composition of pluronic block copolymers and theirhypersensitization effect in MDR cancer cells. Pharm Res 1999;16:1373–9.
[38] Mahmud A, Lavasanifar A. The effect of block copolymer structure on theinternalization of polymeric micelles by human breast cancer cells. ColloidsSurf B Biointerfaces 2005;45:82–9.
[39] Xiong XB, Uludag H, Lavasanifar A. Biodegradable amphiphilic poly(ethyleneoxide)-block-polyesters with grafted polyamines as supramolecular nano-carriers for efficient siRNA delivery. Biomaterials 2009;30:242–53.
[40] Yang TF, Chen CN, Chen MC, Lai CH, Liang HF, Sung HW. Shell-crosslinkedPluronic L121 micelles as a drug delivery vehicle. Biomaterials 2007;28:725–34.
[41] Aliabadi HM, Brocks DR, Lavasanifar A. Polymeric micelles for the solubiliza-tion and delivery of cyclosporine A: pharmacokinetics and biodistribution.Biomaterials 2005;26:7251–9.
[42] Vladimir PT. Polymeric micelles as pharmaceutical carriers. In: Uchegbu IF,Schatzlein AG, editors. Polymers in drug delivery. Florida: CRC Press; 2006. p.108–22.


![[Randomized clinical case-control trial for the comparison of docetaxel plus thiotepa versus docetaxel plus capecitabine in patients with metastatic breast cancer]](https://img.dokumen.tips/doc/110x75/63536a1d6ff1b55f420e545b/randomized-clinical-case-control-trial-for-the-comparison-of-docetaxel-plus-thiotepa.jpg)


























