Embed Size (px)
Citation preview

Synergistic induction of theMUC4 mucin gene by interferon-c and retinoicacid in human pancreatic tumour cells involves a reprogramming
of signalling pathways
Mahefatiana Andrianifahanana1, Anshu Agrawal1, Ajay P Singh1, Nicolas Moniaux1,Isabelle van Seuningen2, Jean-Pierre Aubert2, Jane Meza3 and Surinder K Batra*,1
1Department of Biochemistry and Molecular Biology, Eppley Institute for Research in Cancer and Allied Diseases, University ofNebraska Medical Center, Omaha, NE 68198, USA; 2Department of Preventive and Societal Medicine, University of NebraskaMedical Center, Omaha, NE 68198, USA; 3INSERM, Place de Verdun, 59045 Lille Cedex, France
The transmembrane mucin, MUC4, is aberrantly ex-pressed with a high incidence in human pancreaticadenocarcinomas and plays an important role in thepathogenesis of the disease. Our recent studies have shownthat interferon-c (IFNc) and retinoic acid (RA) areimportant regulators of MUC4 in pancreatic tumourcells. Induction of MUC4 by IFNc occurs via a novelpathway involving upregulation of the signal transducerand activator of transcription 1 (STAT-1), whereas itsstimulation by RA requires mediation by the transforminggrowth factor b-2 (TGFb-2). In this study, we haveinvestigated the molecular mechanisms underlying theinteraction of IFNc and RA in MUC4 regulation in pan-creatic tumour cells. We demonstrate that these reagentsexert a synergistic induction of MUC4. Interestingly,while the upregulation of STAT-1 by IFNc is partiallyinhibited by RA, IFNc is shown to repress RA-drivenTGFb-2 induction, pointing to the involvement of alter-native mechanism(s) in IFNc–RA synergism. Moreover, adose-dependent and cooperative induction of MUC4promoter activity suggests a regulation at the transcrip-tional level, most likely by STAT-1 and RAR/RXR(RA receptor/retinoic X receptor) or other IFNc/RA-induced secondary intermediate effectors. Our findingsprovide potential mechanisms that may account for theaberrant expression of MUC4 in pancreatic tumour cellsand expose a novel molecular mechanism of geneinduction, whereby a reprogramming of signalling path-way through alternative route(s) operates during asynergistic interaction of biological modifiers.Oncogene (2005) 24, 6143–6154. doi:10.1038/sj.onc.1208756;published online 20 June 2005
Keywords: MUC4 mucin; IFNg; retinoic acid; syner-gism; gene regulation; pancreatic cancer
Introduction
Pancreatic cancer is a highly lethal malignancy char-acterized by an extremely poor prognosis (Warshaw andFernandez-del Castillo, 1992; Reber, 1998; Jemal et al.,2003). Efforts made to improve the treatment of thisneoplastic disorder have been hampered, in large part,by the elusive nature of its initiation and progression.Nonetheless, a wide variety of biochemical and/orgenetic aberrations commonly associated with pancrea-tic cancer have been identified, owing to the advent ofnewer and more efficient molecular biological andgenetic techniques (Reber, 1998; Sirivatanauksornet al., 1998). Among the series of genes whose expressionprofiles are frequently altered in pancreatic cancer aremucins (MUC), the members of an expanding class ofhigh-molecular-weight glycoproteins (Gendler and Spi-cer, 1995; Moniaux et al., 2001; Hollingsworth andSwanson, 2004). MUC4, a large transmembrane mucin,is aberrantly expressed in the majority (70–80%) ofpancreatic tumours and tumour cell lines while remain-ing undetectable in the normal pancreas (Andrianifaha-nana et al., 2001). In recent years, there has beenincreasing evidence supporting the role of this tumour-associated mucin in the pathogenesis of pancreaticcancer (Andrianifahanana et al., 2001; Swartz et al.,2002; Singh et al., 2004). Comparative expressionanalyses have revealed a positive correlation betweenMUC4 levels, the differentiation status of pancreatictumour cell lines (Andrianifahanana et al., 2001), andtumour grading in accordance with a model ofpancreatic tumour progression (Swartz et al., 2002).Moreover, functional studies using antisense- and/orshort-interfering RNA (siRNA) oligonucleotide-basedknockdown or ectopic expression of MUC4 haveprovided substantial evidence of its role in the promo-tion of pancreatic tumour cell growth and metastasis invivo (Singh et al., 2004). Taken together, these observa-tions suggest an intimate link between aberrant MUC4expression and the pathogenesis of pancreatic cancer.Therefore, an improved understanding of mechanism(s)underlying the regulation of this mucin gene may help toidentify key biochemical events and biologically relevant
Received 20 September 2004; revised 1 April 2005; accepted 1 April 2005;published online 20 June 2005
*Correspondence: SK Batra; E-mail: [email protected]
Oncogene (2005) 24, 6143–6154& 2005 Nature Publishing Group All rights reserved 0950-9232/05 $30.00
www.nature.com/onc

factors that may account for its aberrant upregulation inpancreatic tumour cells in vivo. This, in turn, maysignificantly facilitate the design of therapeutic strategiesusing MUC4 as a novel molecular target.
In our recent studies, we have used a pancreatictumour cell system to investigate the mechanismsregulating MUC4 expression. This model system iscomprised of the MUC4-producing parental cell line,CD18/HPAF, and its serum-free (SF)-adapted deriva-tive, CD18/HPAF-SF, which expresses low or undetect-able endogenous MUC4 (Choudhury et al., 2000). Wehave demonstrated that interferon-g (IFNg) (Andriani-fahanana et al., 2002) and retinoic acid (RA) (Choudh-ury et al., 2000) can potently induce MUC4 expressionin CD18/HPAF-SF cells. From a mechanistic stand-point, the proinflammatory cytokine, IFNg, canonicallyactivates the Janus kinase (JAK)/signal transducer andactivator of transcription 1 (STAT-1) pathway viabinding to its cell-surface receptor complex (IFNgreceptor (IFNGR) subunits 1 and 2). Activation ofreceptor-associated JAKs (JAK-1 and JAK-2) subse-quently ensues, leading to a cascade of molecular eventsthat culminate in the phosphorylation of latent cyto-plasmic STAT-1 protein at residues Tyr701 and Ser727.Activated STAT-1 molecules homodimerize and trans-locate to the nucleus to induce the transcription of targetgenes (Bach et al., 1997; Park and Schindler, 1998;Ramana et al., 2002). Stimulation of MUC4 expressionby IFNg in CD18/HPAF-SF cells was shown to engagea pathway, whereby upregulation of constitutivelySer727-phosphorylated STAT-1 promoted the nuclearaccumulation of this transcription factor and repre-sented a key regulatory step in MUC4 induction(Andrianifahanana et al., 2002). In contrast, theclassical STAT-1 Tyr701 phosphorylation (Bach et al.,1997; Park and Schindler, 1998; Ramana et al., 2002)did not appear to play an essential role in MUC4induction (Andrianifahanana et al., 2002). RA, on theother hand, is a differentiation factor commonlyencountered in the blood plasma (Hara et al., 2001)that exerts its effect via the nuclear RA receptors (RAR(isoforms a, b, and g)) and retinoic X receptors(RXR (isoforms a, b, and g)). Typically, heterodimersof RAR/RXR act as transcription factors to promotethe transcription of RA-induced genes (Leid et al., 1992;Hu et al., 2002). In CD18/HPAF-SF cells, MUC4induction by RA was a two-step process that involvedthe transforming growth factor b-2 (TGFb-2) as a keymediator. RA treatment elicited RARa-dependentTGFb-2 upregulation, which was necessary for MUC4induction via an as yet undefined pathway (Choudhuryet al., 2000). Interestingly, IFNg and RA are well knownfor their ability to evoke a synergistic effect, whichgenerally leads to an enhanced induction of targetgene(s) and an exacerbation of the associated biologicalresponse(s). The impact of this synergism has beenobserved in a wide range of malignant tumour cell types,including breast, oral, neuroblastoma, leukemic andrenal cancer cells (Ho, 1985; Windbichler et al., 1996;Widschwendter et al., 1997; Guzhova et al., 2001; Yanget al., 2001; Hu et al., 2002), but has remained largely
unexplored in pancreatic tumour cells and, therefore,raises important questions.
In light of these observations, we have conducted thepresent study to investigate the interactions of IFNg- andRA-induced signalling pathways in the context of MUC4induction, and to characterize the molecular bases ofthese interactions. Using the CD18/HPAF-SF cellsystem, we demonstrate that IFNg and RA synergizepotently to induce MUC4 expression. RARa and RARgare upregulated by IFNg–RA combinations, pointing totheir potential implication in the synergism. Interestingly,examination of the signalling effectors associated withpathways activated by IFNg or RA indicates that theupregulation of STAT-1 by IFNg is partially inhibited byRA. Likewise, the TGFb-2-dependent pathway, which isnormally activated by RA (Choudhury et al., 2000), isrepressed by IFNg, suggesting the involvement of ad-ditional mechanism(s). Moreover, results from luciferasereporter gene assays suggest a cooperative activation ofthe MUC4 promoter by IFNg and RA. Together, thesedata reflect a reprogramming of signalling pathwaysthrough alternative routes during the synergistic inter-action of two distinct biological inducers.
Results
Synergistic induction of MUC4 expression by IFNg and RA
We have recently demonstrated that IFNg (Andrianifa-hanana et al., 2002) and RA (Choudhury et al., 2000)are potent inducers of MUC4 in pancreatic tumour cells.In various cell systems, both reagents have been shownto interact synergistically, thereby enhancing the expres-sion of specific sets of genes and the associatedbiological responses (Ho, 1985; Windbichler et al.,1996; Widschwendter et al., 1997; Guzhova et al.,2001; Hu et al., 2002). To evaluate the effect of acombined IFNg–RA treatment on MUC4 induction,CD18/HPAF-SF cells were stimulated with differentdoses of each reagent, which were applied either alone orin combination. Cells were harvested at 48 h poststi-mulation and subjected to RNA slot blot and Westernblot analyses. Our results revealed that cotreatment ofcells with IFNg and RA elicited a dramatic upregulationof MUC4 transcript and protein (Figure 1a and b,respectively). For all of the doses tested, cells concomi-tantly exposed to IFNg and RA exhibited enhancedMUC4-specific signals that were greater (up to three-fold; e.g. IFNg (25 ng/ml)/RA (10 nM); IFNg (50 ng/ml)/RA (10 nM)) than signals expected from an additiveeffect of both types of stimuli (Figure 1a). Theseobservations indicate that IFNg and RA interactsynergistically to induce MUC4 expression in CD18/HPAF-SF cells.
Involvement of alternative pathway(s) in the synergisticinduction of MUC4
We subsequently sought to delineate the molecularmechanism(s) underlying the synergistic interaction of
Regulation of MUC4 by IFNc and RAM Andrianifahanana et al
6144
Oncogene

IFNg and RA. Our recent studies have demonstratedthat MUC4 induction by IFNg in CD18-HPAF-SF cellscritically depends on STAT-1 upregulation (Andriani-fahanana et al., 2002). Moreover, various studies havereported the ability of RA to stimulate the production ofSTAT-1 and other signalling effectors in severalmalignant cell systems. Increased STAT-1 expressionwas shown to enhance the responsiveness of RA-treatedcells to IFNs (Kolla et al., 1996; Gianni et al., 1997;Chelbi-Alix and Pelicano, 1999; Niitsu et al., 2002).Given that STAT-1 upregulation is critical to MUC4induction by IFNg (Andrianifahanana et al., 2002), weinvestigated the expression profile of this transcriptionfactor in IFNg- and RA-stimulated CD18-HPAF-SF cells. Consistent with our previous observations(Andrianifahanana et al., 2002), Western blot analysesrevealed an augmented expression of STAT-1 in cellstreated with IFNg (Figure 2a, lanes 2–4). In contrast,RA treatment reduced the constitutive expression ofSTAT-1 (lanes 9 and 13). Furthermore, coadministra-tion of IFNg and RA resulted in a partial, albeitstatistically significant, inhibition of STAT-1 inductionby IFNg (lanes 6–8, 10–12, and 14–16). Thus, our data
point to two possibilities. First, the contribution ofSTAT-1 upregulation to the synergistic induction ofMUC4 by IFNg and RA may not be as critical as it is incells treated with IFNg alone. Alternatively, the extentto which this transcription factor is upregulated may besufficient to support its inducing effect during thesynergistic interaction.
A similar approach was taken to investigate the roleof TGFb-2, a key mediator of RA-induced MUC4expression (Choudhury et al., 2000), in IFNg–RAsynergism. Essentially, cells subjected to the sametreatments as above were examined for the expressionof this cytokine. In agreement with our previous findings(Choudhury et al., 2000), treatment of cells with RAelicited TGFb-2 upregulation in a dose-dependentmanner. However, IFNg exerted an inhibitory effecton both the basal and RA-induced expression of TGFb-2 (Figure 2b). Thus, TGFb-2 may not play a principalrole in mediating the synergistic induction of MUC4 asit is observed in cells treated with RA alone (Choudhuryet al., 2000). Taken together, these observations indicatethat the signalling pathway(s) involved in the synergisticinduction of MUC4 by IFNg and RA may differ from
a
IFNγ (ng/ml)
RA (nM)
0 12.5 25 50 0 0 0 12.5 12.5 12.5 25 25 25
0 0 0 0 1 10 100 1 10 100 1 10 100
50 50 50
11 0 100
MUC4
PGK
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16
b
0 12.5 25 50 0 0 0 12.5 12.5 12.5 25 25 25 50 50 50
0 0 0 0 1 10 100 1 10 100 1 10 100 1 10 100
0
5
10
15
20
MUC4
GAPDH
IFNγ (ng/ml)
RA (nM)
MU
C4
mR
NA
leve
l(a
rbit
rary
un
its)
Figure 1 Synergistic induction of MUC4 by IFNg and RA in CD18/HPAF-SF cells. (a) CD18/HPAF-SF cells were exposed to theindicated doses of IFNg and/or RA for 48 h. Cells were harvested and processed for RNA isolation. Total RNA was analysed by RNAslot blot using 32P-labelled cDNA probes specific for MUC4 or GAPDH (internal control). The bar graph represents the intensity ofMUC4-specific signal adjusted to GAPDH. Error bars indicate the standard deviation from triplicate values. (b) Samples of totalprotein from cell lysates were electrophoresed on 2% SDS–agarose gel and blotted onto PVDF membrane. Blots were subsequentlyprocessed according to the standard Western blotting protocol using a MUC4-specific monoclonal antibody. Aliquots from all sampleswere resolved on 10% Laemmli SDS–polyacrylamide gel. Blots were generated and probed with anti-PGK polyclonal antibody(internal control). Shown are representative data from at least three independent experiments
Regulation of MUC4 by IFNc and RAM Andrianifahanana et al
6145
Oncogene

those normally activated by either reagent, when appliedindividually.
Role of modulated receptor expression in IFNg–RAsynergism
Previous studies have established the role of RAreceptors as a basis for the synergistic interaction ofIFNg and RA in various cell types. Upregulation of
RARb was shown to enhance the responsiveness of cellsto treatment with IFNg (Niitsu et al., 2002). Moreover,our own studies have demonstrated the implication ofRARa in RA-induced MUC4 expression (Choudhuryet al., 2000). To evaluate the relevance of RARs toIFNg/RA synergism, we examined the expressionprofiles of transcripts for RARa, -b, and -g in CD18/HPAF-SF cells following treatment with various dosesof IFNg and RA, either alone or in combination. Total
a
b
0 12.5 25 50
0 1 10 100
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16
0 12.5 25 50 0 12.5 25 50 0 12.5 25 50IFNγ (ng/ml)
RA (nM)
STAT-1(91 kDa)
PGK(41 kDa)
ST
AT
-1 p
rote
in le
vel
(arb
itra
ry u
nit
)
0
20
40
60
IFNγ (ng/ml)
RA (nM)
0 12.5 50
100
1 2 3 4 5 6 7 8 9
RPL13A(319 bp)
TGF� -2(600 bp)
0 12.5 50
10
0 12.5 50
0
10
0
0
CD18/HPAF-SF
CD
18/H
PA
F
TG
F�
-2 m
RN
A le
vel
(arb
itra
ry u
nit
)
0
10
20
30
Figure 2 Role of STAT-1 and TGFb-2 in IFNg–RA synergism. CD18/HPAF-SF cells were treated with IFNg and/or RA at theindicated concentrations and harvested at 48 h after treatment. (a) Total protein was analysed by Western blotting using anti-STAT-1ap91 monoclonal or anti-PGK polyclonal (internal control) antibodies. The bar graphs (mean7standard deviation) represent theintensity of STAT-1-specific signals adjusted to PGK. Shown are data from three independent experiments. Analysis with one-wayANOVA indicated a statistically significant difference among the treatment groups (n¼ 3; Po0.001). Relevant comparisons by t-testthat revealed a statistically significant difference (Pp0.05) include lane 1 vs 9, 1 vs 13, 3 vs 7, and 7 vs 11. (b) Total RNA was analysedby RT–PCR using primers specific for TGFb-2 or GAPDH (internal control). PCR products were resolved on 2% agarose gel stainedwith ethidium bromide. Photographs were taken under UV light. The bar graphs (mean7standard deviation) represent the intensity ofTGFb-2-specific signals adjusted to RPL13A. Shown are representative data from at least three independent experiments. Analysis withone-way ANOVA indicated a statistically significant difference among the treatment groups (n¼ 3; Po0.001). Relevant comparisonsby t-test that revealed a statistically significant difference (Pp0.05) include lane 1 vs 2, 1 vs 3, 4 vs 6, 7 vs 8, 7 vs 9, and 8 vs 9. Lane 10shows the expression of TGFb-2 mRNA in the CD18/HPAF parental cells, grown in serum-containing medium
Regulation of MUC4 by IFNc and RAM Andrianifahanana et al
6146
Oncogene

RNAs from cells subjected to the indicated treatmentswere initially tested by semiquantitative reverse tran-scription (RT)–PCR at low cycle (linear range). Thiswas to determine whether any of the receptors understudy showed any changes in their respective expressionpatterns that paralleled that of MUC4. The receptorsthat fit this criterion were further analysed by quanti-tative RNA slot blotting, and one-way analysis ofvariance (ANOVA) was performed to reveal anystatistical differences among the treatments tested. Ourresults revealed a dose-dependent upregulation of RARafollowing treatment with IFNg (Po0.05) or with RAat 100 nM (P¼ 0.003) (Figure 3a). Interestingly, RARaexpression in cells cotreated with IFNg and RA wasfurther enhanced compared to cells treated with eitherreagent alone (Figure 3a). Likewise, RARg was dose-dependently upregulated both by IFNg and RA(Figure 3c), although the expression of this receptorisoform was further upregulated only in cells subjectedto a combined treatment with lower doses of thereagents (i.e., Figure 3c: (IFNg 12.5 ng/mlþRA 10 nM)vs (IFNg 12.5 ng/ml) or (RA 10 nM)). In contrast, underall concentrations of IFNg and RA tested, RARbexpression did not display a pattern consistent withthat of MUC4 (Figure 3b). Despite the slight upregula-tion of this transcript by some of the individualtreatments (e.g., IFNg 50 ng/ml, RAX10 nM), most ofthe combined treatments resulted in a reduced expres-sion of the receptor (e.g., Figure 3c; (IFNg 50 ng/ml)vs (IFNg 50 ng/mlþRA 100 nM)).
To further explore the role of RARa in the synergisticinduction of MUC4, cells were treated with IFNg, RA,or IFNgþRA, in the presence or absence of the RARa-specific inhibitor, Ro41-5253. Results from Western blotanalyses revealed that neither Ro41-5253 nor DMSO(solvent) affected the constitutive expression of MUC4(Figure 4a). Moreover, Ro41-5253 dose-dependentlyinhibited the RA-dependent MUC4 induction(Figure 4c), which was consistent with our previousobservations (Choudhury et al., 2000). Likewise, thesynergistic induction of MUC4 by a combination ofIFNg and RA was also inhibited by Ro41-5253(Figure 4d), indicating that RARa was required forthe IFNg/RA synergism. However, to our surprise, thestimulation of MUC4 by IFNg was also negated bytreatment with the RARa-specific inhibitor (Figure 4b).To determine the mechanistic basis of this inhibitoryeffect, we examined the expression profile of STAT-1,a transcription factor whose upregulation by IFNg iscritical to the induction of MUC4 by this cytokine(Andrianifahanana et al., 2002). Results from Westernblot analysis demonstrated that STAT-1 was down-regulated by Ro41-5253 in a dose-dependent manner,and that no further upregulation of this transcriptionfactor was observed in response to treatment with IFNg(Figure 4b, middle row). Thus, these data suggest thatRARa is required for both the constitutive expression ofSTAT-1 and the synergistic induction of MUC4 byIFNg and RA.
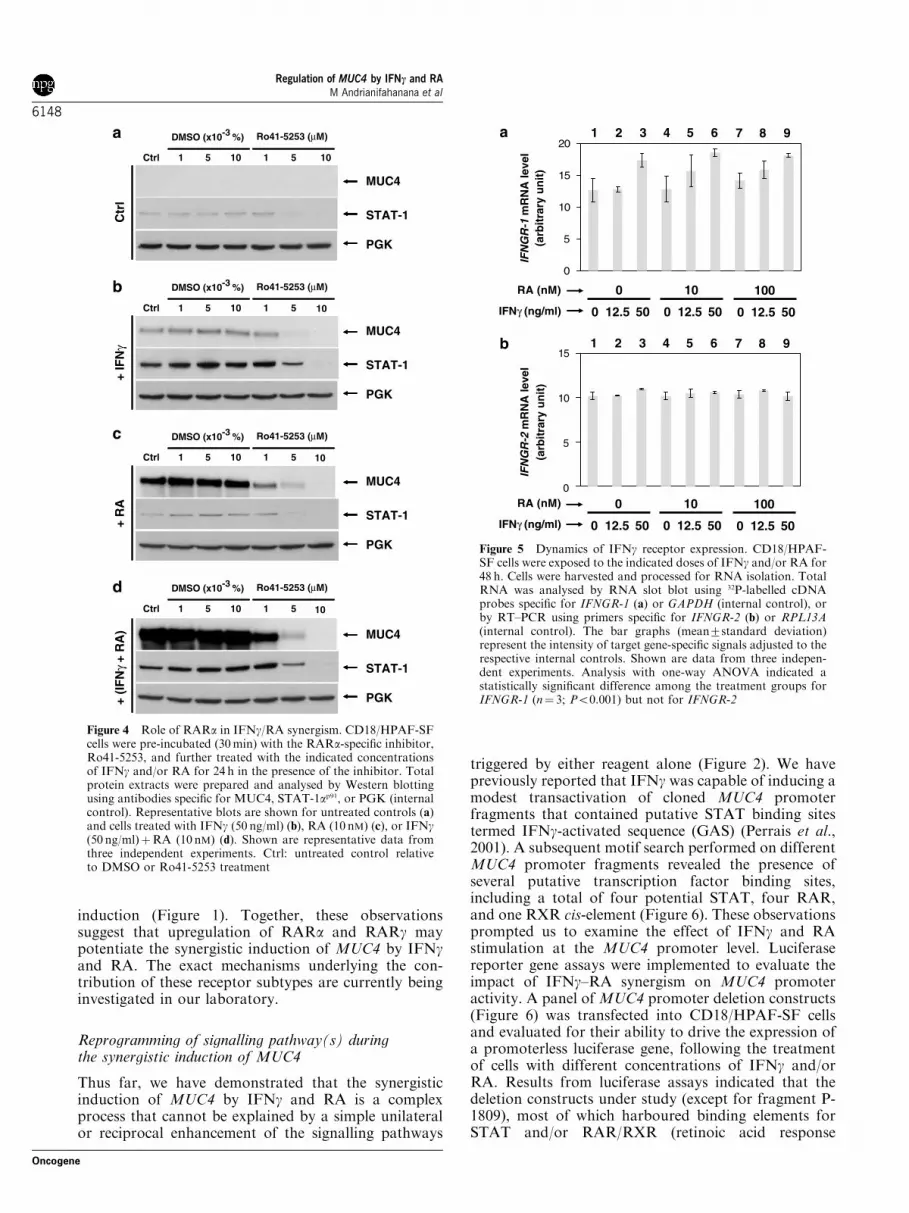
We next investigated the expression profiles of bothsubunits of the IFNg receptor. As evaluated by RNA
slot blot analysis, IFNg upregulated the expression ofIFNGR-1 mRNA at 50 ng/ml (P¼ 0.02), while RA didnot significantly affect the levels of this receptor(Figure 5a). Although certain IFNg/RA combinationsappeared to potentiate the stimulation of IFNGR-1mRNA expression by either reagent (e.g., (IFNg 12.5 ng/mlþRA 10 nM) vs (IFNg 12.5 ng/ml) or (RA 10 nM)),no statistically significant difference was observedamong the effects of these treatments (Figure 5a). Incontrast, the levels of IFNGR-2 mRNA did not exhibitany significant fluctuation in response to the treatmentstested, as revealed by one-way ANOVA (Figure 5b).Moreover, the overall pattern of expression for thisreceptor subunit was not consistent with MUC4
a
b
c
0 12.5 50
0
IFNγ (ng/ml)
RA (nM)
0 12.5 50
10
0 12.5 50
100
RA
R- α
mR
NA
leve
l(a
rbit
rary
un
it)
0
5
10
1531 2 4 5 6 7 8 9
0 12.5 50
0
IFNγ (ng/ml)
RA (nM)
0 12.5 50
10
0 12.5 50
100
0
5
10
0 12.5 50
0
0 12.5 50
10
0 12.5 50
100
0
20
40
RA
R- �
mR
NA
leve
l(a
rbit
rary
un
it)
RA
R- �
mR
NA
leve
l(a
rbit
rary
un
it)
IFNγ (ng/ml)
RA (nM)
31 2 4 5 6 7 8 9
31 2 4 5 6 7 8 9
Figure 3 Dynamics of RA receptor expression. CD18/HPAF-SFcells were exposed to the indicated doses of IFNg and/or RA for48 h. Cells were harvested and processed for RNA isolation. TotalRNA was analysed by RNA slot blot using 32P-labelled cDNAprobes specific for RARa (a), RARg (c), or GAPDH (internalcontrol), or by RT–PCR using primers specific for RARb (b) orRPL13A (internal control). The bar graphs (mean7standarddeviation) represent the intensity of target gene-specific signalsadjusted to the respective internal controls. Shown are data fromthree independent experiments. Analysis with one-way ANOVAindicated a statistically significant difference among the treatmentgroups for all experiments (n¼ 3; Po0.001). Relevant comparisonsby t-test that revealed a statistically significant difference (Pp0.05)include the following: Panel a: lane 1 vs 7, 2 vs 8, 3 vs 6, 3 vs 9, and 7vs 9; Panel b: lane 1 vs 4 and 3 vs 9; Panel c: lane 2 vs 5, 2 vs 8, and4 vs 5
Regulation of MUC4 by IFNc and RAM Andrianifahanana et al
6147
Oncogene
induction (Figure 1). Together, these observationssuggest that upregulation of RARa and RARg maypotentiate the synergistic induction of MUC4 by IFNgand RA. The exact mechanisms underlying the con-tribution of these receptor subtypes are currently beinginvestigated in our laboratory.
Reprogramming of signalling pathway(s) duringthe synergistic induction of MUC4
Thus far, we have demonstrated that the synergisticinduction of MUC4 by IFNg and RA is a complexprocess that cannot be explained by a simple unilateralor reciprocal enhancement of the signalling pathways
triggered by either reagent alone (Figure 2). We havepreviously reported that IFNg was capable of inducing amodest transactivation of cloned MUC4 promoterfragments that contained putative STAT binding sitestermed IFNg-activated sequence (GAS) (Perrais et al.,2001). A subsequent motif search performed on differentMUC4 promoter fragments revealed the presence ofseveral putative transcription factor binding sites,including a total of four potential STAT, four RAR,and one RXR cis-element (Figure 6). These observationsprompted us to examine the effect of IFNg and RAstimulation at the MUC4 promoter level. Luciferasereporter gene assays were implemented to evaluate theimpact of IFNg–RA synergism on MUC4 promoteractivity. A panel of MUC4 promoter deletion constructs(Figure 6) was transfected into CD18/HPAF-SF cellsand evaluated for their ability to drive the expression ofa promoterless luciferase gene, following the treatmentof cells with different concentrations of IFNg and/orRA. Results from luciferase assays indicated that thedeletion constructs under study (except for fragment P-1809), most of which harboured binding elements forSTAT and/or RAR/RXR (retinoic acid response
Ctr
l
Ctrl
MUC4
STAT-1
PGK
MUC4
STAT-1
PGK
MUC4
STAT-1
PGK
MUC4
STAT-1
PGK
1 5 10 1 5 10
10
10
10
DMSO (x10-3 %) Ro41-5253 (µM)
Ctrl 1 5 10 1 5
DMSO (x10-3 %) Ro41-5253 (µM)
Ctrl 1 5 10 1 5
DMSO (x10-3 %) Ro41-5253 (µM)
Ctrl 1 5 10 1 5
DMSO (x10-3 %) Ro41-5253 (µM)
+ IF
Nγ
+ R
A+
(IF
Nγ
+ R
A)
a
b
c
d
Figure 4 Role of RARa in IFNg/RA synergism. CD18/HPAF-SFcells were pre-incubated (30 min) with the RARa-specific inhibitor,Ro41-5253, and further treated with the indicated concentrationsof IFNg and/or RA for 24 h in the presence of the inhibitor. Totalprotein extracts were prepared and analysed by Western blottingusing antibodies specific for MUC4, STAT-1ap91, or PGK (internalcontrol). Representative blots are shown for untreated controls (a)and cells treated with IFNg (50 ng/ml) (b), RA (10 nM) (c), or IFNg(50 ng/ml)þRA (10 nM) (d). Shown are representative data fromthree independent experiments. Ctrl: untreated control relativeto DMSO or Ro41-5253 treatment
0 12.5 50
0
IFNγ (ng/ml)
RA (nM)
IFNγ (ng/ml)
RA (nM)
0 12.5 50
10
0 12.5 50
100
IFN
GR
-1 m
RN
A le
vel
(arb
itra
ry u
nit
)
0
5
10
15
201 2 3 4 5 6 7 8 9
0 12.5 50
0
0 12.5 50
10
0 12.5 50
100
IFN
GR
-2 m
RN
A le
vel
(arb
itra
ry u
nit
)
0
5
10
151 2 3 4 5 6 7 8 9
a
b
Figure 5 Dynamics of IFNg receptor expression. CD18/HPAF-SF cells were exposed to the indicated doses of IFNg and/or RA for48 h. Cells were harvested and processed for RNA isolation. TotalRNA was analysed by RNA slot blot using 32P-labelled cDNAprobes specific for IFNGR-1 (a) or GAPDH (internal control), orby RT–PCR using primers specific for IFNGR-2 (b) or RPL13A(internal control). The bar graphs (mean7standard deviation)represent the intensity of target gene-specific signals adjusted to therespective internal controls. Shown are data from three indepen-dent experiments. Analysis with one-way ANOVA indicated astatistically significant difference among the treatment groups forIFNGR-1 (n¼ 3; Po0.001) but not for IFNGR-2
Regulation of MUC4 by IFNc and RAM Andrianifahanana et al
6148
Oncogene

element (RARE)), were responsive to stimulation byIFNg or RA in a dose-dependent manner (Figure 6). Ofparticular interest was the deletion construct P-2150,which evidenced a cooperative effect of IFNg and RAon promoter activity. In cells transfected with thisconstruct, transcriptional activity was induced 1.7- and1.2-fold in the presence of IFNg (50 ng/ml) and RA(100 nM), respectively, and further enhanced (2.5-fold)in the simultaneous presence of both reagents (Figure 6and Table 1). Interestingly, this fragment carried alarger number of potential STAT binding sites ascompared with other fragments and highlighted thepresence of an RXR cis-element in addition to the RARbinding sites (Figure 6). A similar pattern of inductionwas also observed with the P-1959 and P-1641 frag-ments, which, in turn, were located within the TATA-containing region (Figure 6 and Table 1). An enhancedtranscriptional activity (P-1959 (2.4-fold); P-1641 (3.2-fold)) was also recorded in cells cotreated with IFNg orRA, as compared with those individually stimulatedwith IFNg at 50 ng/ml (P-1959 (1.7-fold); P-1641 (2.3-fold)) or RA at 100 nM (P-1959 (1.1-fold); P-1641 (1.3-fold)) (Table 1). Importantly, these observations areconsistent with findings from studies that identifiedseveral active transcription start sites both withinTATA-containing and TATA-less regions of theMUC4 promoter (Perrais et al., 2001). Overall, our dataindicate that multiple regions within the MUC4
promoter constitute potential zones of convergence fortranscription factors activated by IFNg and RA andmay partly account for the synergistic effect on MUC4gene induction. In consideration of the respectivepathways activated by IFNg and RA in the context ofMUC4 regulation, these findings suggest a rerouting ofsignalling via alternative pathway(s) during the syner-gistic interaction of these MUC4 inducers.
Temporal aspect of IFNg/RA-triggered MUC4 induction
In a parallel study, we have consistently observed theexpression of IFNg transcript in pancreatic tumours,which contrasted with a lack of detection in normalpancreatic tissues (Andrianifahanana et al., 2005).
Figure 6 Mapping and transcriptional activity of MUC4 pGL3 deletion constructs in CD18/HPAF-SF cells. A series of MUC4promoter deletion constructs subcloned into the promoterless pGL3 Basic vector were transfected into CD18/HPAF-SF cells, whichwere subsequently treated with the indicated concentrations of IFNg, RA, or their combination. Transcriptional activity was assessedby luciferase reporter gene assay as described in Materials and methods. Locations of the deletion constructs are indicated to the leftand their respective transcriptional activities to the right of the figure. Position (�1) represents the first nucleotide upstream of theATG start codon. Asterisk indicates the position of the first active TATA box (�2672/�2668) (Perrais et al., 2001). Relative positionsof putative binding sites: STAT: �3062/�3054, �964/�956, �415/�407, �123/�115; RAR: �2872/�2867, �2751/�2745, �318/�306,�150/�142; RXR: �801/�791. Analysis with one-way ANOVA indicated a statistically significant difference (n¼ 3; Po0.001) amongthe treatment groups for P-1641, P-1959, and P-2150, but not for P-1809 or pGL3 basic. Pairwise comparisons by t-test revealed astatistically significant difference (Pp0.05) among all treatments within each group except for the following: P-1959: (untreated) vs(RA (100 nM)); and P-1641: (IFNg (50 ng/ml)) vs (IFNg (50 ng/ml)þRA (100 nM)). Comparisons did not include (IFNg (50 ng/ml)) vs(RA (100 nM))
Table 1 Analysis of MUC4 promoter deletion constructs byluciferase reporter gene assay
pGL3 deletionconstructs
Fold inductiona
IFNg (50) RA (100) IFNg (50)+RA(100)
P-1641 2.3 1.3 3.2P-1959 1.7 1.1 2.4P-2150 1.7 1.2 2.5P-1809 1.1 1.1 1.3
aAverage fold induction relative to untreated cells IFNg, in ng/ml; RA,in nM
Regulation of MUC4 by IFNc and RAM Andrianifahanana et al
6149
Oncogene

Together with the common occurrence of RA in theblood circulation (Hara et al., 2001), these observationspointed to a potential role of both IFNg and RA in theregulation of MUC4 in pancreatic tumour cells in vivo.However, given that the local tumour microenvironmentmay not always favour a simultaneous exposure oftumour cells to both compounds, we sought todetermine whether a concomitant and continuousstimulation was required to yield a synergistic inductionof MUC4. To this end, we carried out experimentswhereby CD18/HPAF-SF cells were subjected to prim-ing (preincubation) with different concentrations ofIFNg or RA for 8 or 16 h, followed by a duplicated(i.e., IFNg-IFNg or RA-RA) or reciprocal (i.e.,IFNg-RA or RA-IFNg) treatment with eitherreagent. Results from Western blot analyses revealedthat duplicated treatments with IFNg (Figure 7, lanes 3and 6) but not RA (lanes 10 and 13) were more potent atinducing MUC4 than the corresponding individualtreatments (IFNg: lanes 2 and 5; RA: lanes 8 and 10,respectively). Moreover, when cells were primed withIFNg, the extent of MUC4 induction in cells that hadreceived reciprocal treatments (IFNg-RA: lanes 4 and7) appeared to exceed those in cells subjected toduplicated treatments (lanes 3 and 6). Most interestingly,the levels of MUC4 in these reciprocally treated cellswere comparable to those detected in cells exposed to theIFNg–RA control treatment (lane 16). Taken together,these observations indicate that the continuous presenceof IFNg or RA is not required for a synergistic inductionof MUC4, and that priming with either reagent for aperiod of time as short as 8 h is sufficient to potentiateeach other’s effect. In consideration of the functionalproperties of MUC4 (see Discussion), these findings mayhave important biological implications with respectto the development of pancreatic tumours in vivo.
Discussion
In this study, we have elucidated important aspects ofthe molecular events associated with the synergistic
induction of the MUC4 gene by IFNg and RA inpancreatic tumour cells. We demonstrate that themechanism(s) underlying the concerted regulation ofMUC4 by these biological modifiers diverge to someextent from those operated by cells subjected tostimulation with each reagent alone. Moreover, theprofiles of MUC4 promoter activity support a coopera-tive activation by both reagents at the transcriptionallevel. Together, these observations indicate a reroutingof signalling during the synergistic interaction of IFNgand RA, and reflect an extensive flexibility of thesignalling pathways activated by each or both biologicalstimuli within a single cell system.
Our recent studies have shown that upregulation ofMUC4 by IFNg in CD18/HPAF-SF cells depends onelevated expression of STAT-1 transcription factor(Andrianifahanana et al., 2002), and that its stimulationby RA implicates TGFb-2 as a critical mediator(Choudhury et al., 2000). Thus, one may predict that asynergistic induction of this mucin gene by IFNg andRA would occur via amplification of the molecularsignals instigated by each type of stimulus. Alterna-tively, modulation of receptor expression may impartenhanced cell responsiveness to either ligand, therebyleading to a synergistic effect, as observed with othersystems (Niitsu et al., 2002). However, as demonstratedin this study, treatment of cells with RA repressed theconstitutive expression of STAT-1 and partially inhi-bited its induction by IFNg (Figure 2a). Furthermore,expression of the RA signalling mediator, TGFb-2, wasrepressed by IFNg (Figure 2b). In contrast, RARa(Figure 3a) and RARg (Figure 3c) were induced by RAand/or IFNg, and these receptor isoforms were furtherupregulated by certain IFNg–RA combinations. There-fore, one of the most plausible explanations to thesynergistic induction of MUC4 is a rerouting of thesignalling pathway(s) engaged in the simultaneouspresence of IFNg and RA, whereby upregulation ofRARa and/or RARg may play a role in IFNg–RAsynergism. Indeed, analysis of the transcriptionalactivity of various MUC4 promoter deletion constructsevidenced a cooperative effect of IFNg and RA at the
MUC4(> 1MDa)
IFNγ ( 25ng/ml)Treatment
RA (10 nM)
PGK(41 kDa)
- + + + + ++ + + + +-
- - - - - - - - -- - - -- - - - -
++
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16
IFNγ (25 ng/ml)
RA (10 nM)
Priming(hr)
0
0
8 16
0 0 8 16 0
00 0
Figure 7 Effect of priming by IFNg or RA on MUC4 induction. CD18/HPAF-SF cells were primed (preincubated) with the indicatedconcentrations of IFNg or RA for 8 or 16 h and subjected to duplicated (i.e., IFNg-IFNg or RA-RA) or reciprocal (i.e., IFNg-RAor RA-IFNg) treatments for 48 h. Positive controls consisted of cells stimulated with IFNg (lane 14), RA (lane 15), or IFNg-RA (lane16), for 48 h without any prior treatment. Total protein was analysed by Western blotting using antibodies specific for MUC4 or PGK(internal control). Shown are representative data from three (two full and one partial) independent experiments
Regulation of MUC4 by IFNc and RAM Andrianifahanana et al
6150
Oncogene

promoter level (Figure 6), suggesting a potentialinteraction of the transcription factors, STAT-1 andRAR/RXR, with the MUC4 promoter. These argu-ments are corroborated by the presence of severalpotential STAT and RAR/RXR binding sites withinthe MUC4 promoter region (Figure 6). In that line ofreasoning, these observations indicate that cotreatmentof cells with IFNg and RA may activate alternativepathway(s) that bypass those engaged by each reagentalone, specifically the TGFb pathway in the presentcase. At this point, the exact mechanism(s) involved isstill unclear and is the subject of our ongoing investiga-tions. One of the hypotheses being tested is that, in thepresence of RA alone, the native chromosomal contextof the MUC4 promoter may prohibit the access ofRAR/RXR to their cognate cis-elements, which wouldfavour the prevalence of TGFb signalling as the majorpathway for RA-driven MUC4 gene regulation. Induc-tion of STAT-1 following IFNg treatment may alleviatethe constraints imposed by the chromosomal structures,thereby increasing the accessibility of RAR/RXRbinding sites and allowing for the synergistic inductionto take place. Under these circumstances, the repressiveeffect of IFNg on RA-induced TGFb expression wouldhave little or no impact on MUC4 induction. Here,inhibition of TGFb-2 expression may have resulted fromupregulation of SMAD7 (our unpublished data), aninhibitory SMAD that functions as a negative regulatorof the TGFb signalling pathway (Ulloa et al., 1999). Inthis respect, a recent study has described the interferenceof IFNg-induced SMAD7 with TGFb-dependent
regulation of the sialomucin complex (SMC), theMUC4 homologue in rat (Soto et al., 2003). Overall, asimplified model is proposed to summarize our findingsregarding MUC4 regulation in pancreatic tumour cells(Figure 8). This model depicts the signalling pathwaysimplicated in MUC4 upregulation by IFNg and RA,and highlights the complex interplay between pathwaysactivated during the synergism.
On the other hand, additional signalling pathway(s)may likewise be involved in this synergistic interaction.As illustrated by the case of the promoter fragment, P-1959, a dose-dependent induction of luciferase geneexpression had taken place although no STAT bindingsite was identified in this fragment (Figure 6). In thisregard, we have recently shown that the STAT-1-mediated elicitation of MUC4 expression is dependenton JAK(s) (Andrianifahanana et al., 2002), a tyrosinekinase that serves as a platform for the activation ofseveral distinct IFNg-triggered pathways (Ramanaet al., 2002). Interestingly, the upregulation of IFNGR-1 mRNA by IFNg (Figure 5a) may be of particularrelevance to this point, although the exact role of thisreceptor subunit in this context still remains unclear.Moreover, the influence of secondary responses inIFNg–RA synergism cannot be ruled out. Intermediateeffectors whose expression is induced by factorsactivated by the primary stimuli (IFNg or RA) maycome into play during the synergistic interaction.
The importance of these findings lies with theirimplications in the pathobiology of pancreatic cancer,particularly with respect to two sets of observations. On
Figure 8 Model of MUC4 regulation by IFNg and RA. This simplified model describes the molecular mechanisms involved in MUC4gene regulation by IFNg and RA, either individually or in synergism. The pathways activated are colour-coded (blue: IFNg; green: RA;red: IFNg/RA synergism). Arrows indicate the flow of signalling. Circled arrows indicate upregulation of the targeted signallingcomponents/genes. Left panel: regulation of the MUC4 gene by IFNg alone. Induction of MUC4 occurs via upregulation of STAT-1,whose constitutive expression may involve an RARa-dependent pathway(s) (not shown). Middle panel: regulation of MUC4 by RAalone. Induction of MUC4 results primarily from upregulation of TGFb-2 in an RARa-dependent manner (Choudhury et al., 2000),although the regulatory process may also involve a direct activation of MUC4 by an RARa-containing complex (dotted green arrow).Right panel: synergistic regulation of MUC4 by IFNg and RA. Induction of MUC4 involves a reprogramming of signallingpathway(s). While STAT-1 induction by IFNg is partially repressed by RA (red dotted line), the upregulation of TGFb-2 by RA isinhibited by IFNg. The dotted blue line depicts a presumptive pathway (Ulloa et al., 1999) (our unpublished data) that may account forthe inhibition of TGFb-2 expression, which leads to the attenuation of the TGFb-2-dependent pathway (dotted green arrow)
Regulation of MUC4 by IFNc and RAM Andrianifahanana et al
6151
Oncogene

the one hand, IFNg and RA are naturally occurringbiological agents with proven cell growth inhibitoryand/or differentiating properties (Ho, 1985; Windbichleret al., 1996; Widschwendter et al., 1997; Chelbi-Alix andPelicano, 1999; Guzhova et al., 2001; Yang et al., 2001;Hu et al., 2002). Moreover, our recent studies haveindicated a differential expression of IFNg in pancreatictumours, as opposed to the lack of detection in thenormal pancreas (Andrianifahanana et al., 2005). Onthe other hand, both compounds are presently shown topromote the synergistic induction of MUC4, a tumour-associated molecule that is known to promote tumourcell growth and/or metastasis in several experimentalsystems (Komatsu et al., 2000; Singh et al., 2004). Theseobservations raise important questions regarding theultimate behaviour of pancreatic tumour cells inresponse to IFNg and RA in vivo. A legitimate avenueto addressing this situation is to draw on informationabout the functional properties of MUC4. Although theexact biological functions of human MUC4 have onlybegun to be understood (Singh et al., 2004), the highdegree of structural similarity between this mucin and itsorthologue, rat Muc4/SMC, strongly suggests similarfunctions (Sheng et al., 1992; Wu et al., 1994; Moniauxet al., 1999). Rat Muc4/SMC has been implicated in thepromotion of tumour cell metastasis in vivo (Komatsuet al., 2000). Its overexpression on the cell surfaceenables human A375 melanoma cells to escape immunesurveillance, presumably via masking of key recognitionmolecules that are necessary for tumour cell targeting byimmune effector cells (Komatsu et al., 1999). Likewise,ectopic expression of MUC1, another member of thetransmembrane mucin family, has been shown topotentiate the immune evasion capabilities of A375cells. Overexpression of this mucin provides augmentedprotection of the tumour cells against killing byactivated cytotoxic immune effector cells (van de Wiel-van Kemenade et al., 1993). MUC1 has also been shownto inhibit cell–cell interaction mediated by the celladhesion molecule, E-cadherin (Wesseling et al., 1996;Kondo et al., 1998). In view of the structural analogybetween MUC4 and its rat counterpart (Sheng et al.,1992; Wu et al., 1994; Moniaux et al., 1999), thefunctional roles of MUC1 (van de Wiel-van Kemenadeet al., 1993; Wesseling et al., 1996; Kondo et al., 1998),and the predicted large size and extended structure ofMUC4 (Moniaux et al., 1999), the aberrant expressionof MUC4 on the surface of pancreatic tumour cells maypotentially provide protection against the host’s acti-vated immune system while conferring antiadhesive/metastatic properties upon the cells. These statementsare further supported by information regarding thestructural features of MUC4, whose ectodomain ispredicted to extend B2 mm (vs 200–500 nm for MUC1;Wesseling et al., 1996) above the cell surface (Moniauxet al., 1999). Furthermore, our observations from recentfunctional studies suggest a role of MUC4 in promotingthe growth and metastasis of pancreatic tumour cells invivo (Singh et al., 2004). On the other hand, the presentresults from priming experiments indicated that induc-tion of MUC4 did not require the continuous presence
of either IFNg or RA (Figure 7). Although IFNg hasbeen previously shown to inhibit pancreatic tumour cellgrowth in vitro (Matsubara et al., 1991; Detjen et al.,2001), details about the temporal aspect of this processhave remained unexplored. In addition, we haveobserved that RA does not affect the proliferation ofCD18/HPAF-SF cells while promoting the growth ofthe parental cell line, CD18/HPAF (AP Singh and SKBatra, unpublished observations). Evaluation of therequirements (e.g., time and duration of exposure) thatdefine the respective physiological effects of IFNg andRA would therefore be of particular interest in order toidentify their impact on pancreatic tumour cell beha-viour. Overall, the current data on MUC4 generegulation provide a potential mechanism underlyingits aberrant expression in pancreatic tumour cellsin vivo, and illustrate how it may interfere withthe proper functioning of the host’s defence system,thereby contributing to the pathogenesis of pancreaticcancer.
In conclusion, we have demonstrated that IFNg andRA can synergize potently to effect MUC4 inductionin pancreatic tumour cells. An improved understandingof the mechanism(s) underlying the regulation of thismucin gene may provide useful insights into themolecular events underlying the pathogenesis of pan-creatic cancer, and may significantly impact the deve-lopment of therapeutic strategies using MUC4 as anovel target.
Materials and methods
Cell lines and reagents
The pancreatic tumour cell line, CD18/HPAF, and itsderivative, CD18/HPAF-SF, were used in this study. CD18/HPAF cells were maintained in a 1 : 1 mixture of Dulbecco’smodified Eagle’s medium (DMEM) : Ham’s F-12 mediumsupplemented with 10% fetal bovine serum (FBS). CD18/HPAF-SF cells were grown in the same culture mediumwithout serum. All reagents for cell culture were purchasedfrom Invitrogen. In stimulation experiments, CD18/HPAF-SFcells were seeded in six-well plates or in 10-cm Petri plates andallowed to reach B70% confluency prior to treatment. Cellswere treated with the indicated doses of IFNg (Peprotech)and/or all-trans retinoic acid (RA) (Sigma), which was usedeither individually or in combination. For priming experi-ments, cells were treated with the indicated concentrations ofIFNg or RA for 8 or 16 h, rinsed twice in culture medium, andsubsequently restimulated with either reagent for 48 h accord-ing to the described experimental layout. Cells were harvestedand processed for RNA or protein extraction as describedbelow.
In inhibition experiments, cells were preincubated with theRARa-specific inhibitor, Ro41-5253 (a gift from Hoffman/LaRoche), for 30 min prior to the indicated treatments.
RNA isolation and reverse transcription–PCR analysis
Total RNA was isolated from cells using the RNeasys MiniKit (Qiagen). Reverse transcription was performed on 2mg ofRNA per sample using the SuperScripttII RNase� ReverseTranscriptase system (Invitrogen). The cDNA samples were
Regulation of MUC4 by IFNc and RAM Andrianifahanana et al
6152
Oncogene

subjected to PCR amplification using the parameters providedin Table 2 (Choudhury et al., 2000; Andrianifahanana et al.,2001; Jesnowski et al., 2002). RPL13A was used as an internalcontrol. PCR products were electrophoretically resolved on2% agarose gels stained with ethidium bromide. Photographswere taken under UV light, and densitometric analyses ofDNA bands were carried out using the Gel Expert softwaresystem (Nucleotech).
RNA slot blot analysis
Total RNA (10 mg per sample) was denatured in a buffercontaining formamide/formaldehyde and blotted onto nylonmembranes by vacuum suction, using a 48-well slot filtrationmanifold system (Invitrogen). All samples were analysedin triplicate. Membranes were prehybridized at 421C for 12 hin 6� saline/sodium phosphate/ethylene diamine tetra acetate,5� Denhardt’s reagent, 0.5% sodium dodecyl sulphate, 50%formamide, and 100 mg/ml of sheared salmon sperm DNA.Hybridization was conducted under the same conditions in thepresence of 32P-labelled cDNA probes specific for the indicatedtarget genes. Membranes were exposed to a PhosphorImagerscreen, and subsequent analyses were carried out using theImageQuaNTt software (Molecular Dynamics). Membraneswere further stripped and rehybridized with a 32P-labelled,glyceraldehyde-3-phosphate dehydrogenase (GAPDH)-specificcDNA probe (internal control).
Western blot analysis
Cells were lysed in modified RIPA buffer (50mM Tris-HCl, pH7.4; 0.25% Na-deoxycholate; 150mM NaCl; 1% NP-40; 1 mM
EDTA), supplemented with 1 mg/ml aprotinin, 1 mg/mlleupeptin, 5mM NaF, 5 mM Na3VO4, and 1mM phenylmethyl-sulphonyl fluoride (PMSF). Proteins were extracted on ice for30min and further cleared by centrifugation at 16 000 g for10min. Supernatants (protein extracts) were collected andstored at �801C until further use.
Total protein was electrophoretically fractionated on 10%Laemmli SDS–polyacrylamide gels and electroblotted ontoPVDF membranes. Blotted membranes were blocked in 5%blotto (nonfat dry milk in phosphate-buffered saline) andsubsequently exposed to primary antibodies specific for STAT-1p91 (Santa Cruz Biotechnology) diluted in 1% blotto.Following incubation in the appropriate secondary antibody,membranes were treated with ECL reagents (AmershamBiosciences) and exposed to autoradiographic films. Mem-branes were stripped and reprobed with antibodies specific forphosphoglycerate kinase (PGK) (internal control; kindlyprovided by Dr JK Vishwanatha, UNMC, Omaha, NE, USA).
Due to the predicted large size of the mature MUC4glycoprotein (in the megadalton range), Western blot analysisof this mucin was carried out on 2% SDS–agarose gels.Subsequent experimental procedures were essentially as statedabove with the exception that protein transfer was achievedby capillary blotting. The primary antibody consisted of anMUC4-specific mouse monoclonal antibody produced in ourlaboratory (clone 8G7) (Swartz et al., 2002; Moniaux et al.,2004).
MUC4 promoter deletion constructs, transfection, and luciferasereporter gene assay
A total of four promoter deletion constructs carrying DNAfragments covering various regions of the MUC4 gene 50-flanking sequence were used in this study (Figure 6; Perraiset al., 2001). DNA inserts consisted of two fragments locatedwithin a TATA-less region of MUC4 50-flanking sequence (P-1809 (�219/�1) and P-2150 (�1187/�1)) and two fragmentswithin a TATA-containing region (P-1959 (�2781/�2572)and P-1641 (�3135/�2572)). All inserts were subcloned intothe promoterless pGL3 Basic vector (Promega) in the senseorientation. Constructs (2 mg) were cotransfected with thepSV-bGal vector (0.2 mg) (Promega) into CD18/HPAF-SFcells 24 h after seeding using the FuGENE 6 transfectionreagent (Roche). Cells were treated with the indicatedconcentrations of IFNg and/or RA at 24 h post-transfectionand allowed to incubate in the presence or absence of thereagents for another 40 h prior to harvesting. Cell lysates wereprepared in triplicate, and luciferase activity was assessed andcorrected with b-galactosidase activity for transfection effi-ciency.
Statistical analyses
For each experiment, one-way ANOVA was used to determinewhether there was a statistically significant difference amongthe groups. Further pairwise comparisons to examine differ-ences between the treatments groups were performed usingt-tests.
Acknowledgements
We thank Erik Moore (UNMC) for technical support and theMolecular Biology Core Facility for oligonucleotide synthesisand DNA sequencing. We also thank Dr JK Vishwanatha(UNMC) for the generous gift of anti-PGK antibody. TheRARa-specific inhibitor, Ro41-5253, was a generous giftfrom Hoffman/LaRoche. This work was supported by agrant from the National Institutes of Health (CA 78590)and the Nebraska Department of Health LB506 Program(2002-04).
Table 2 Primer characteristics and PCR parameters
Gene Primer sequence AT (1C) Cycles
IFNGR-2 F: 50-GTGGCCCTGAGCAATAGCACGA-30 55 25R: 50-GGCCTTTGACCTGTTGGATTCCT-30
TGFb-2 F: 50-GCTTTTCTGATCCTGCATCTG-30 60 35R: 50-CAATACCTGCAAATCTTGCTTC-30
RARb F: 50-GGAACGCATTCGGAAGGCTT-30 55 30R: 50-GGAAGACGGACTCGCAGTGT-30
RPL13A F: 50-ATCGTGGCTAAACAGGTACTG-30 58 23R: 50-GCACGACCTTGAGGGCAGCC-30
F, forward; R, reverse; AT, annealing temperature
Regulation of MUC4 by IFNc and RAM Andrianifahanana et al
6153
Oncogene

References
Andrianifahanana M, Moniaux N and Batra SK. (2002). Proc.Am. Assoc. Cancer Res., 43, 607.
Andrianifahanana M, Moniaux N, Schmied BM, Ringel J,Friess H, Hollingsworth MA, Buchler MW, Aubert JP andBatra SK. (2001). Clin. Cancer Res., 7, 4033–4040.
Andrianifahanana M, Moniaux N, Singh AP, Varshney GCand Batra SK. (2002). Proc. Am. Assoc. Cancer Res., 43,1135.
Bach EA, Aguet M and Schreiber RD. (1997). Annu. Rev.Immunol., 15, 563–591.
Chelbi-Alix MK and Pelicano L. (1999). Leukemia, 13,1167–1174.
Choudhury A, Singh RK, Moniaux N, El Metwally TH,Aubert JP and Batra SK. (2000). J. Biol. Chem., 275,33929–33936.
Detjen KM, Farwig K, Welzel M, Wiedenmann B andRosewicz S. (2001). Gut, 49, 251–262.
Gendler SJ and Spicer AP. (1995). Annu. Rev. Physiol., 57,607–634.
Gianni M, Terao M, Fortino I, LiCalzi M, Viggiano V,Barbui T, Rambaldi A and Garattini E. (1997). Blood, 89,1001–1012.
Guzhova I, Hultquist A, Cetinkaya C, Nilsson K, Pahlman Sand Larsson LG. (2001). Int. J. Cancer, 94, 97–108.
Hara I, Taguchi I, Miyake H, Hara S, Gotoh A andKamidono S. (2001). Int. J. Oncol., 19, 959–962.
Ho CK. (1985). Cancer Res., 45, 5348–5351.Hollingsworth MA and Swanson BJ. (2004). Nat. Rev. Cancer,4, 45–60.
Hu W, Verschraegen CF, Wu WG, Nash M, Freedman RS,Kudelka A and Kavanagh JJ. (2002). Int. J. Gynecol.Cancer, 12, 202–207.
Jemal A, Murray T, Samuels A, Ghafoor A, Ward E and ThunMJ. (2003). CA Cancer J. Clin., 53, 5–26.
Jesnowski R, Backhaus C, Ringel J and Lohr M. (2002).Pancreatology, 2, 421–424.
Kolla V, Lindner DJ, Xiao W, Borden EC and KalvakolanuDV. (1996). J. Biol. Chem., 271, 10508–10514.
Komatsu M, Tatum L, Altman NH, Carothers Carraway CAand Carraway KL. (2000). Int. J. Cancer, 87, 480–486.
Komatsu M, Yee L and Carraway KL. (1999). Cancer Res.,59, 2229–2236.
Kondo K, Kohno N, Yokoyama A and Hiwada K. (1998).Cancer Res., 58, 2014–2019.
Leid M, Kastner P and Chambon P. (1992). Trends Biochem.Sci., 17, 427–433.
Matsubara N, Fuchimoto S and Orita K. (1991). Int. J.Pancreatol., 8, 235–243.
Moniaux N, Escande F, Porchet N, Aubert JP and Batra SK.(2001). Front. Biosci., 6, D1192–D1206.
Moniaux N, Nollet S, Porchet N, Degand P, Laine A andAubert JP. (1999). Biochem. J., 338, 325–333.
Moniaux N, Varshney GC, Chauhan SC, Copin MC, Jain M,Wittel UA, Andrianifahanana M, Aubert JP and Batra SK.(2004). J. Histochem. Cytochem., 52, 253–261.
Niitsu N, Higashihara M and Honma Y. (2002). Leuk. Res.,26, 745–755.
Park C and Schindler C. (1998). Methods, 15, 175–188.Perrais M, Pigny P, Ducourouble MP, Petitprez D, Porchet N,
Aubert JP and Van SI. (2001). J. Biol. Chem., 276,30923–30933.
Ramana CV, Gil MP, Schreiber RD and Stark GR. (2002).Trends Immunol., 23, 96–101.
Reber HA. (1998). Pancreatic Cancer: Pathogenesis, Diagnosis,and Treatment. Humana Press: Totowa, NJ.
Sheng Z, Wu K, Carraway KL and Fregien N. (1992). J. Biol.Chem., 267, 16341–16346.
Singh AP, Moniaux N, Chauhan SC, Meza JL and Batra SK.(2004). Cancer Res., 64, 622–630.
Sirivatanauksorn V, Sirivatanauksorn Y and Lemoine NR.(1998). Langenbecks Arch. Surg., 383, 105–115.
Soto P, Price-Schiavi SA and Carraway KL. (2003). J. Biol.Chem., 278, 20338–20344.
Swartz MJ, Batra SK, Varshney GC, Hollingsworth MA, YeoCJ, Cameron JL, Wilentz RE, Hruban RH and Argani P.(2002). Am. J. Clin. Pathol., 117, 791–796.
Ulloa L, Doody J and Massague J. (1999). Nature, 397,710–713.
van de Wiel-van Kemenade, Ligtenberg MJ, de Boer AJ, BuijsF, Vos HL, Melief CJ, Hilkens J and Figdor CG. (1993).J. Immunol., 151, 767–776.
Warshaw AL and Fernandez-del Castillo C. (1992). N. Engl. J.Med., 326, 455–465.
Wesseling J, van der Valk SW and Hilkens J. (1996). Mol. Biol.Cell, 7, 565–577.
Widschwendter M, Daxenbichler G, Culig Z, Michel S, ZeimetAG, Mortl MG, Widschwendter A and Marth C. (1997). Int.J. Cancer, 71, 497–504.
Windbichler GH, Hensler E, Widschwendter M, Posch A,Daxenbichler G, Fritsch E and Marth C. (1996). Gynecol.Oncol., 61, 387–394.
Wu K, Fregien N and Carraway KL. (1994). J. Biol. Chem.,269, 11950–11955.
Yang JB, Duan ZJ, Yao W, Lee O, Yang L, Yang XY, Sun X,Chang CC, Chang TY and Li BL. (2001). J. Biol. Chem.,276, 20989–20998.
Regulation of MUC4 by IFNc and RAM Andrianifahanana et al
6154
Oncogene





























