Embed Size (px)
Citation preview

Food, Medicine and Health CareAdministration and Control Authority
Strategies for Marketing Authorization ofpharmaceuticals


Executive summary
The government of Ethiopia has issued Proclamation 661/2009 in orderto protect the public health from unsafe, inefficacious and poorquality medicines and to promote healthy and productive community.Medicines safety, efficacy and quality are ensured throughstandardized premarketing evaluation of product information,manufacturing premises inspection, laboratory testing and postmarketing surveillance.The main objective of a pharmaceutical manufacturer is to producefinished products from a combination of materials including startingand packaging and labeling materials to ensure that Medicinal productsare consistently produced and controlled to the quality standardsappropriate to their intended therapeutic use and as required by themarketing authorization.
In the past few years of inspection of manufacturing facility for cGMPcompliance by the Authority, it was found that more than 60% offoreign manufacturers failed to comply for the cGMP and hence themarketing authorization. This is a remarkable risk reduction processto protect the public health from substandard and poor qualitymedicines.
The experience now a days in both developed and developing countriesis that the process of market authorization - dossier evaluation,cGMP inspection and pre-marketing quality control laboratory testingfocuses on risk based approach.
Thus in Ethiopia it means the market-authorization process-productdossier evaluation, manufacturers inspection and pre and post marketlaboratory testing should focus on risk based approach to ensure thatthe limited resources allocated are efficiently and effectivelyutilized to assess products of high risk and enhance public healthprotection and promotion in a timely and realistic manner.

The number of application for marketing authorization is increasingfrom time to time .This could be attributed to the economic growthcurrently happening in the country and the population size of thecountry as well as the attractive government investment policy.Meanwhile, the experience, skill ,qualification and educational levelof the human resource required in the medicine and food regulatoryauthorities is diversified and most of them are not readily availablein the market; moreover, the attrition rate of human resource inFMHACA is very high. Thus, using part time qualified and experiencedemployees is mandatory in order to cope up with the current demand forproduct registration and GMP inspection
Thus, this strategy points out how to deal with the current demand forthe marketing authorization, GMP inspection and laboratory testingon risk based approach and using outside experts so that safety,quality and efficacy of medicines will be ensured.
Table of Contents
Executive summary....................................................3Table of Contents....................................................4
Abbreviations........................................................5Acknowledgment.......................................................6
Definitions..........................................................7Introduction.........................................................8
Other Country Experience............................................10Marketing Authorization Strategies for FMHACA.......................11
Strategic direction.................................................11

Strategy for dossier evaluation.....................................12Dossier evaluation of low risk products..........................13
Rigorous evaluation for critical products based on the risks.....13Using Part time dossier assessors................................14
Re-registration of medicinal products............................15Strategy for Good Manufacturing Practice inspection................16
Strategy for pre-market Laboratory testing..........................17Products exempted from pre-market laboratory testing.............18
Products which require testing for selected critical parameter...18Rigorous and extensive testing...................................18
Annex I-The main elements of risk categorization for Product dossier assessment..........................................................19
Abbreviations
AMRP Abbreviated Medicine review process (AMPRP)
cGMP Current Good Manufacturing Practice
EFMHACA Ethiopian Food, Medicine & Health Care Administration
and Control Authority

GMP Good Manufacturing Practice
MCC Medicine Control Council
MOU Memorandum of Understanding
OTC Over the Counter
PQAD Product Quality Assessment Directorate
QA Quality Assurance
QC Quality Control
SADC South African Development Community
SRA Stringent Regulatory Authority
TGA Therapeutics Goods Administration of Australia
USFDA United States Food and Drug Administration
WHO World Health Organization
Acknowledgment
The Ethiopian Food, Medicine and Healthcare Administration and ControlAuthority (EFMHACA) would like to acknowledge the U.S Pharmacopeial convention’s Promoting the Quality Medicines program (USP/PQM) and U.S

Agency for International Development (USAID) for their technical and financial support in the preparation of this strategy for marketing authorization.
Definitions The definitions provided below apply to the words and phrases
used in these document.

Accredited Laboratory means pharmaceutical quality control laboratory
which has been accredited in accordance with all requirements of
current ISO 17025 for the required test and whose accreditation can be
accessed on the web.
Applicant means the person or entity who submits a registration
application of product to the Authority, and responsible for the
product information
Authority Means the “Ethiopian Food, Medicine and Health Care
Administration and Control Authority or the acronym “EFMHCACA”
established in accordance with Food, Medicine & Health Care
Administration & Control Regulation No 189/ 2010.
Manufacture means all operations of purchase of materials and
products, production, quality control, release, storage and
distribution of pharmaceutical products, and the related
controls.
Manufacturer means a company that carries out operations, such as
production, packaging, repackaging, labeling and relabeling of
pharmaceuticals.Marketing Authorization means an official document issued for the purpose
of marketing or free distribution of a product after evaluation of
safety, efficacy and quality of the product.
Pharmaceutical product means any material or product intended for
human use presented in its finished dosage form or as a starting
material for use in such a dosage form that is subject to control
by pharmaceutical legislation in the exporting state and/or the
importing state.

Risk analysis means method to assess and characterize the
critical parameters in the functionality of a process or an
equipment.
Stringent Regulatory Authority mean a regulatory authority of a
member of the International Conference on Harmonisation (ICH) (as
specified on www.ich.org); or an ICH observer, being the European
Free Trade Association (EFTA), as represented by Swiss Medic, and
Health Canada (as may be updated from time to time); or a
regulatory authority associated with an ICH member through a
legally-binding, mutual recognition agreement including
Australia, Iceland, Liechtenstein and Norway (as may be updated
from time to time) and WHO
pre-qualification program are considered to be products
registered with Stringent Regulatory Authority (SRA)
Introduction
The Authority Register human medicines, medical devices and other
health related products as per the Ethiopian Food, Medicine and
Health Care Administration and Control Proclamation No 661/2009.
As the economic development of the country is progressing
overseas and local manufacturing companies have shown increased
interest to register their products. However, the increasing
demand of companies for registration and the type of products
needed from the procurement agencies are not matched with the
capacity of the Authority to provide responsive actions for

registration application and procurement request effectively and
efficiently.
Root causes of the problems are the followings:
The product registration and licensing directorate has been
mandated with responsibilities which requires specialization
of staff and separate management system : Assessment of
application for medicine marketing authorization, Assessment
of application for medical Devices/Supplies marketing
authorization, GMP inspection which include receiving
applications for GMP inspection, organizing, participating
and approval of results of GMP inspection, Assessment of
application of Clinical trial protocol and authorization of
clinical trials , Inspection of clinical trials, Handling
Food registration and local food processing manufacturing
facilities inspection, Approval of Purchase orders for
Medicine, medical devices, raw materials for Pharmaceutical
Industries and Review and approval of promotional materials
and promoters. These responsibilities require their own
respective expertise, documentation and management system.
The Authority inadequate number of staff with appropriateknowledge, experience and skills to assess dossiers. Thereis also inadequate/poor professional mix particularly toassess safety, efficacy and quality of new chemical entities
Registration is not focused on risk reduction - extensiveevaluation should be on high risk products unlike the low

risk products (e.g. OTC) which could be registered withminimum requirements
The evaluators professional capability, skill and experience
coupled with inadequate professional mix on assessment of safety,
efficacy and quality of new chemical entities and, efficacy and
quality of multisource products is very limited which hampers the
efficiency of registration process.
As a result of the above not only the efficiency of registration
process is affected but also the reliability and credibility of
assessment results has been greatly questioned. Hence the call
for establishment of efficient and effective registration system
based on principles of risk management to address the nation’s
public health need is inevitable.
Over the past few years cGMP inspection was conducted on
significant number of foreign pharmaceuticals products
manufacturers located mainly in Asian and African countries as
part of the fulfillment of marketing authorization. The
inspection result revealed that about 60% of the manufacturers
were found to be non-compliant with the cGMP requirement.
In addition to premises GMP inspection requirement, the Authority
requests samples for quality control laboratory testing on
samples submitted by the applicant as an important element for
the issuance of marketing authorization after acceptance of the
dossier and the premises for GMP. There are a number of

limitations on these procedures. Testing of samples submitted by
a manufacturer has limitation in that:
It is very unlikely that the applicant will submit samples
which will fail to pass the quality specification.
Samples submitted by the applicant may not be representative
of commercial batches and does not represent the actual user
situation. This has been reflected through trend analysis
carried out by PQAD for samples submitted for registration
from the year 2007-2011. The result shows that most of
failures of samples submitted for PMS was higher (9.5%-15.5%)
than samples submitted for the purpose of pre-marketing
authorization (4.7% - 10.7%). This implies that the focus of
laboratory testing should be focused on samples withdrawn from
commercial batches as it appears in the market and/or from
consignment at the port of entry rather than samples submitted
by the applicant for the purpose of marketing authorization.
The requirement for testing and compliance should be in line with
specification described in the submission dossier and when tested
the product should meet the specification irrespective of whether
it is at pre-marketing and/or post marketing sampling levels.
The current revised registration guideline in CTD format demands
rigorous and thorough data.

Other Country Experience
Other Countries experience including South African, Australian
(TGA), Malaysian, US FDA-, Ugandan, German and Thailand market
authorization processes have been reviewed during development of
these strategies of dossier evaluation, cGMP inspection and pre-
market laboratory testing for market authorization.
According to a multi country study by WHO in 2010, assessment and
registration are not the same for all categories of products. How
extensive the assessment should depend on a number of factors,
for example in Australia the extensiveness of the assessment
depends mainly on two factors; the first one being the potential
risks of the product and the other the availability of human
resource for assessment. These factors are taken into account in
setting priorities & deciding the depth of the review, i.e. for
prescription drugs, some medical devices, some alternative
products with better efficacy and safety and these products are
subjected to extensive pre marketing evaluation & registration.
Low risk products are only evaluated for safety. For some product
groups, manufacturer’s declaration of safety is accepted and the
product is then subjected to more intensive post-marketing
surveillance.
In addition to exemptions based on the type of product (product
category) products may also be exempted from registration on the
basis of their source (country of origin).

Based on analysis of national current situations and
international experience the Authority has come out with the
strategic options outlined below for improvement of efficiency,
effectiveness, transparency and accountability of market
authorization of pharmaceuticals.
Marketing Authorization Strategies for FMHACA
General objectives
To improve pharmaceuticals Market Authorization in order to
promote public health focusing on National health priorities
and protect the public from substandard, unsafe, ineffective
pharmaceuticals
Specific objective
To improve the effectiveness and efficiency of
pharmaceuticals dossier evaluation, cGMP inspection and
premarket laboratory testing
To facilitate market authorization process through
implementation of risk based marketing authorization and
fast track registration
To effectively utilize limited resources
To effectively utilize skilled manpower outside the
authority

Strategic direction1. Marketing authorization of pharmaceutical should be
supported with cGMP compliance and should focus on products
essential for the promotion of key health problems of the
country and to protect the public from unsafe, ineffective &
unacceptable quality products.
2. Graduated system of implementation for product safety,
efficacy and quality assessment based on priority products
and focusing on risk of products
3. Proactive risk identification, management and control
4. Market authorization to be supported by regular testing of
actual consignment and samples collected through post market
surveillance.
The minimum necessary activities of marketing authorization are
as follows:
Establishing and maintaining an inventory of the products
available on the local market;
Premarket evaluation of new products:
Ensuring that a complete data-set on quality is available;
Evaluating data on quality
Ensuring that newly authorized products containing well
established drugs are
interchangeable with locally marketed products, and that the
approved product information is accurate and locally useful;

Issuing a written marketing authorization (or rejection) on
completion of the
assessment process.
Evaluating applications to make changes to product
information and to pharmaceutical aspects of existing
marketing authorizations;
The market authorization process also includes manufacturing premise inspection for cGMP compliance and laboratory testing where applicable.
The following strategies for improving efficiency and
effectiveness of Dossier Evaluation, cGMP Inspection and Pre-
market Authorization Laboratory Testing will be implemented
during evaluation of products submitted for marketing
authorization.
The strategies for market authorization are as shown below:-
The purpose of this guidance is neither to eliminate therequirement of dossier submission nor to limit the Authority forfull assessment of the product whenever deemed to be necessary.The main purpose of this guidance is to introduce a procedurewhich will facilitate the registration and hence enhancement ofavailability of the medicines to the public.
Strategy for dossier evaluation
Dossier evaluation of medicinal products is huge task by
itself .Thus, it requires its own management or functional unit

with its own specialized , skilled and experienced
staff/assessors. Thus, segregating/or restructuring the dossier
evaluation task/activities of the authority in to separate
department/unit/directorate will solve many of the problems
currently being encountered. Furthermore, within the dossier
evaluation department/unit/directorate there should be specializedunits/teams such as ;
1. units/team for pharmaceutical products
2. units teams for biological products
3. units/teams for medical device
4. units /teams for diagnostic /reagents
After reorganizing the current product registration and licensing
directorate in the above way, the following strategies should be
applied to cope up with ever increasing demand for market
authorization.
Dossier evaluation of low risk products Included in the low risk categories are products which are
used by small group of patients and which don’t have high
market (e.g. Certain category of OTC, multivitamin, class I
(low risk), II (Low-moderate risk) medical devices, and Class
A (no-public health risk or low personal risk & Class B (low
public health or moderate personal risk ) IVD, and
cosmetics). For further reference and information, please see
annex I of this document.

The requirements for registration of the product are as
described in their respective guideline.
Low risk products are characterized by their property as
described in annex I of this guideline
Low risk product will be assessed by one assessor only
( could permanent or part time assessor)
Rigorous evaluation for critical products based on the risks
Much time of the assessors will be spent on rigorous and
extensive evaluation products with high risk category. The
general approach for categorization of high risk products are
described in annex I of this document. Products considered as
high risk class during assessment are;
New products (not marketed in the country)
Biologicals and immunological;
Generic products with poor bioavailability, complicated
products etc(e.g. sterile product);
Medicine for major public health problems of the country
o ARV, Anti- TB, Anti-Malaria etc
Medicines with narrow therapeutic index;
Invasive medical devices categorized as class III and IV as
described in the registration guideline for medical devices
In-vitro devices that requires special expertise classified
as class C and D as described in the registration guideline
for medical devices

Using Part time dossier assessors
As number of application for marketing authorization is
increasing from time to time.This could be attributed to the
economic growth currently happening in the country and the
population size of the country( it is the second populous country
in Africa which makes the it huge market for
pharmaceuticals).Meanwhile, the experience, skill ,qualification
and educational level of the human resource required in the
medicine and food regulatory authorities is diversified and most
of them are not readily available in the market; moreover, the
attrition rate of human resource in regulatory authorities is
very high.
There are only few countries (e.g. Cyprus) whose public
employees are better paid than their private counterparts. For
instance, Drug regulatory authority personnel in the Netherlands
and Australia receive remuneration comparable with that which
they would receive in the private sector. However, medical
practitioners working with the TGA are paid less than their peers
in clinical practice. The lower level of remuneration generally
leads to two problems in personnel management: difficulty in
recruiting qualified people and difficulty in retaining them
(WHO,2009).
Because of the above mentioned reasons, most of the national
regulatory authorities use skilled and qualified human resource
outside their organization. This helps them to get skilled and

experienced human resource with limited financial resource
thereby increase the safety, efficacy and quality of medicines
available in their respective countries.
Thus using skilled and qualified human resources (pharmacist and
other appropriate professionals) dossier assessment available in
academia (teaching hospitals), regional regulatory
bureaus/authorities/agencies and EFMAHAC branch offices is
mandatory to EFMHACA in order to deal with increasing demand for
marketing authorization. This will benefit the authority in the
following ways
1. Increase the efficiency of registration marketingauthorization while enhancing the reliability and assessmentresults.
2. Cope up with high attrition rate of its employees
3. Give faster response to its clients and promote goodgovernance
4. Increase access and alternatives of safe, efficacious andquality medicines to the public
5. Creat of pool of expertise who can be used whenever thereis need
To be Part time employees should be free from conflict of
interest and should sign declaration for conflict of interest and
Part time employees should not be from pharmaceutical
manufacturers, Importers, distributors, hospitals and from
retail outlet of medicines as employees of such institutions are
prone to conflict of interest directly or indirectly.

Re-registration of medicinal products
As it is indicated in the Proclamation No. 661/2009, re-registration
of medicinal products shall be done very four years. Currently the
authority is treating re-registration application as new registration
application whether there is variation /change or not. This practice
has been contributing to accumulation dossiers (backlogs).
This strategy recommends that if the manufacturer or the company
responsible for the re-registration of the medicinal product declare
that there is no change/variation from the previously registered
products and presents valid GMP compliance certificate from FMHACA or
stringent regulatory authority, the marketing authorization will be
renewed without further requirements of documents and evaluation. The
manufacturer or company will submit confirmatory letter indicating
there has been no change from the previous registration condition.
If variation is declared during the re-registration application, the
re-registration process will be treated by the variation handling
guideline of the authority.
Strategy for Good Manufacturing Practice inspection
GMP is recognized as a vital component of the control of
pharmaceuticals. All sites of
manufacture for new marketing authorizations, and new sites for
existing products, should be

cleared with respect to GMP by the FMHACA own inspectorate or by
means of valid GMP certificate from stringent regulatory agency.
Whether local sites are given a general inspection, or whether a
separate inspection is conducted for each new marketing
authorization, is a matter for local legislation or policy. At
least finished-product manufacturing sites should be certified.
Manufacturing of pharmaceutical product requires in built quality
control and quality assurance system to produce products meeting
its marketing authorization. In other words quality of product
should be built in the process rather than testing on the end
products. Moreover, there are several quality requirements that
can’t be tested in the product such as processing conditions,
systems and manufacturing premises. Thus, inspection of
manufacturing premises to assure consistent production and avoid
mix ups and contamination, on site audit of the manufacturing
premises to augment marketing authorization is indispensable.
This has been proven in the past few years inspection where it
was found that more than 60% of overseas manufacturers failed to
meet the requirements for cGMP which implies the risk to be posed
to the public health and scarce resource wastage on the
procurement of unsafe and substandard medicines.
FMHACA has the responsibility to inspect foreign medicine
manufacturers at least once in every four years based on the
Proclamation No. 661/2009.The number of GMP inspection
application by foreign manufacturers are increasing too. Thus,

FMHACA has no longer has the resource to fulfil this statutory
requirements;hence ,EFMHACA should have adequate manpower and
should able to use external skilled manpower in this area.
The inspection of manufacturing premises requires skilled
manpower and adequate financial resources. An audit of one
particular manufacturing premise requires a minimum of three
expertise and sufficient days for audit and report writing.
Therefore, in order to use the scarce resource appropriately
strategic alternatives for the acceptance of a facility for cGMP
is mandatory.
Conducting GMP inspection using FMHACA staff only will not solve
the questions raised y
the stakeholder in timely manner as well as will compromise other
directorates responsibilities as they have to contribute
personnel to GMP inspection.EFMHACA will not able to respond
teimely to applicants for GMP inspection using its own human
resource. In addition ,theaddition, the attrition rate is also
high and makes it difficult to get experienced and qualified GMP
inspectors as required.
Thus, due to the factors mentioned above , using qualified and
experienced GMP inspectors outside FMHACA will be mandatory to
the authority in order to cope up with the current demand of
qualified GMP inspectors. Moreover, it increases the reliability
and credibility of the GMP inspection as GMP inspectors are
assigned based on merit .

To be Part time GMP inspector for the authority one should be
free from conflict of interest and should sign declaration for
conflict of interest and TOR prepared by FMHACA for this purpose.
Part time GMP inspectors should not be from pharmaceutical
manufacturers, Importers and distributors as employees of such
institutions are prone to conflict of interest directly or
indirectly.
Both fulltime and part time GMP inspectors shall fulfil the
requirement set by the guidance on requirements for GMP inspector
qualification, training and experience requirements.
Strategy for pre-market Laboratory testing Medicine Quality Monitoring
The samples to be tested as well as the methods to be used for
analysis should rely on the risks identified during assessment.
Thus, in order to realize the proposed strategic dossier
assessment and registration process discussed above the current
sample testing based on ‘sample requisition’ from the applicant
should be shifted to representative sampling from commercial
batches withdrawn from consignment and/or market. There is low
tendency that an applicant will submit samples which may not
comply with the specification.
Moreover, experiences of other regulatory authorities shows that
marketing authorization is mainly based on dossier assessment and

GMP evaluation whereas the national laboratories are mainly
focused on actual consignment, PMS and addressing complaints from
public and users of the product. The current proposed strategy
does not eliminate testing when found to be necessary. It is
mandatory that when the samples tested at any time it should
comply with the approved specification as described in the
submission dossier.
The following are strategies for laboratory testing for the
purpose of marketing authorization;
Products exempted from pre-market laboratory testing Product registered by stringent regulatory Authorities in
SRA region,
Commercial batch Products tested by accredited Quality
Control Lab in SRA region
Low risk products
Products which require testing for selected critical parameter
Products registered by SRA but manufacturer located in
non- SRA region
Product that are not registered by SRA with moderate risk
and complexity
Biological and immunological products that are not
registered by SRA
Rigorous and extensive testing

High risk Products not registered by SRA
Generic products with poor bioavailability, complicated
products etc(e.g. sterile product);
Medicine for major public health problems of the country
o ARV, Anti- TB, Anti-Malaria etc
Medicines with narrow therapeutic index;
Invasive medical devices categorized as class III and IV as
described in the registration guideline for medical devices
In-vitro devices that requires special expertise classified
as class C and D as described in the registration guideline
for medical devices
Annex I-The main elements of risk categorization for Product dossier assessment Each product will be categorized and in of two risk categories for the subsequent dossier assessment, laboratory testing and premises requirement for cGMP.
Parameters Low Risk High Risk
Over the counter product
Products which have the following characteristics are in general considered aslow risk
The potential for
Products containing problematic API such asbioavailability, solubility, polymorphism
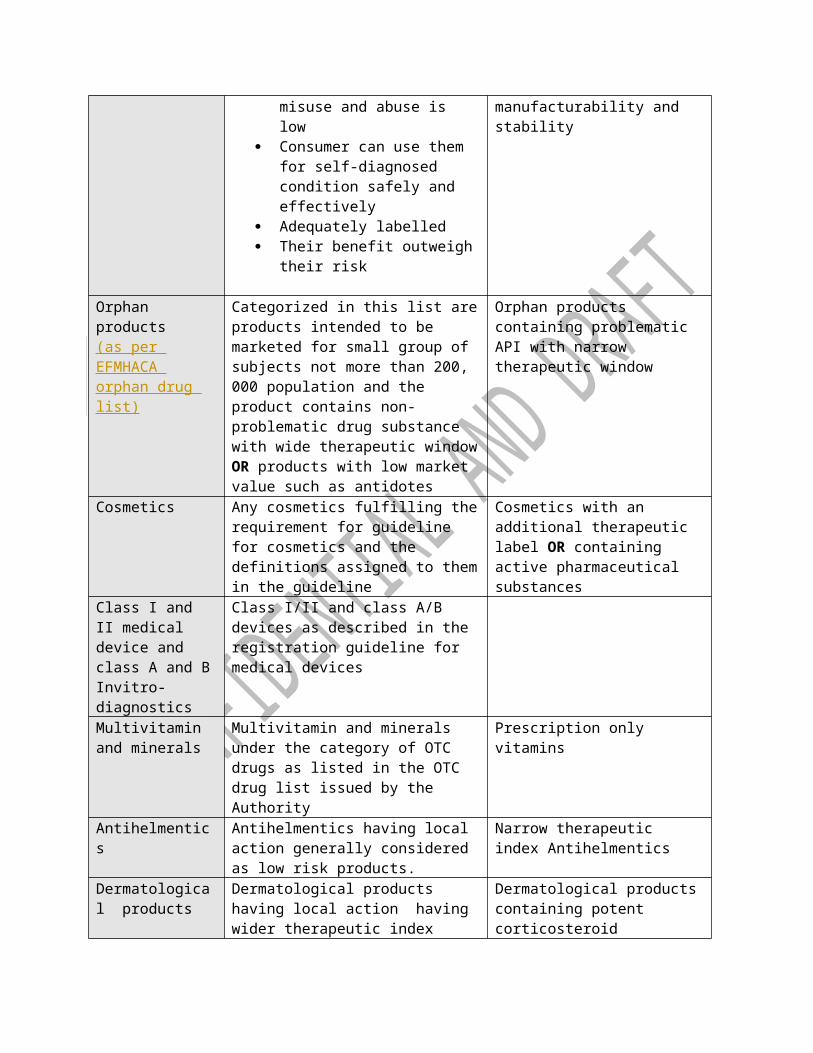
misuse and abuse is low
Consumer can use them for self-diagnosed condition safely and effectively
Adequately labelled Their benefit outweigh
their risk
manufacturability and stability
Orphan products(as per EFMHACA orphan drug list)
Categorized in this list areproducts intended to be marketed for small group of subjects not more than 200, 000 population and the product contains non-problematic drug substance with wide therapeutic windowOR products with low market value such as antidotes
Orphan products containing problematic API with narrow therapeutic window
Cosmetics Any cosmetics fulfilling therequirement for guideline for cosmetics and the definitions assigned to themin the guideline
Cosmetics with an additional therapeutic label OR containing active pharmaceutical substances
Class I and II medical device and class A and BInvitro-diagnostics
Class I/II and class A/B devices as described in the registration guideline for medical devices
Multivitamin and minerals
Multivitamin and minerals under the category of OTC drugs as listed in the OTC drug list issued by the Authority
Prescription only vitamins
Antihelmentics
Antihelmentics having local action generally considered as low risk products.
Narrow therapeutic index Antihelmentics
Dermatological products
Dermatological products having local action having wider therapeutic index
Dermatological productscontaining potent corticosteroid

considered as low risk considered as high riskAnti-inflammatory /ant allergicmedicine
Non steroidal and antihistaminic having wide therapeutic index
Narrow therapeutic product and product having potential for causing dependence are considered a high risk product.
Other Products
Products containing drug
substances with the
property such aso High solubility and
permeability
o Wide therapeutic index
o Non-significant
effect in the event
of treatment failure
o Products full filling
SRA requirement
Products containing problematic API such as bioavailability, solubility, polymorphism manufacturability andstability
New products to the public
Products with narrow therapeutic window
Based on case by case situation and current knowledge and
understanding products categorized as low risk will be exempted
from sample testing for pre-marketing authorization. Although, in
general products categorized as low risk can be eligible for
waiver for GMP requirement this depends on the product nature. As
for example, cosmetics for the mere beauty purpose are exempted
from GMP inspection at the time of registration.





























