Embed Size (px)
Citation preview

Biotechnol. Appl. Biochem. (2007) 48, 167–178 (Printed in Great Britain) doi:10.1042/BA20060223 167
Small-scale immunopurification of cytochrome c oxidase for ahigh-throughput multiplexing analysis of enzyme activity andamount
James Murray*, Birgit Schilling†, Richard H. Row†, Chris B. Yoo†, Bradford W. Gibson†‡,Michael F. Marusich*§ and Roderick A. Capaldi*‖1
*Mitosciences Inc., Eugene, OR 97403-2095, U.S.A., †The Buck Institute for Age Research, Novato, CA 94945-1400, U.S.A.,‡Department of Pharmaceutical Chemistry, University of California, San Francisco, CA 94143-0446, U.S.A., §Institute ofNeuroscience, University of Oregon, Eugene, OR 97403-1254, U.S.A., and ‖Institute of Molecular Biology, University ofOregon, Eugene, OR 97403-1229, U.S.A.
COX (cytochrome c oxidase) deficiency is one ofthe main causes of genetic mitochondrial diseaseand presents with multiple phenotypes, depending onwhether the causative mutation exists in a mitochon-drial or nuclear gene and on whether it involvesan altered catalytic or structural component or anassembly factor for this membrane-embedded 13-subunit enzyme complex. COX deficiency is routinelyobserved in AD (Alzheimer’s disease), although thereis continuing debate about whether this is a causativeor a secondary consequence of the condition. Alteredlevels of COX and reduced oxidative phosphorylationcapacity have been reported in other common diseases,including cancer, and are seen as unwanted side effectsin a number of drug treatments, particularly withantiretroviral and antibiotic treatments. Here, we in-troduce a simple, rapid, high-throughput 96-well plateprotocol that uses a multiplex approach to determinethe amount and activity of COX, which should findwidespread use in evaluating the above diseases andin drug safety studies. Importantly, the method usesvery small amounts of cell material or tissue and doesnot require the isolation of mitochondria. We show theutility of this approach by example of the analysis offibroblasts from patients with COX activity deficiencyand the effect of the antiretroviral drug ddC (2′,3′-dideoxycytidine) on the biogenesis of the enzyme.
Introduction
Genetic defects in complexes of the OXPHOS (oxidativephosphorylation) machinery are the cause of a broad classof so-called mitochondrial disorders (for case examples see[1,2] and for reviews see [3,4]). Increasingly, deficits in thesesame complexes are being observed in common neurode-generative disorders such as PD (Parkinson’s disease) [5–7]and AD (Alzheimer’s disease) [8–11], as well as in the overall
process of aging [12]. In PD, there is strong evidence for aprimary involvement of Complex I, the NADH:ubiquinoneoxidoreductase, in neuronal degeneration [5,13]. In AD,the focus of recent research has been the involvement ofa COX (cytochrome c oxidase) deficiency, since severalstudies have reported a specific reduction of enzymaticactivity in AD patient tissues [8–11]. For example, Maureret al. [9] reported an approx. 50% reduction of activityspecifically in the hippocampus and temporal cortex of ADbrain, while the activity in the cerebellum and frontal cortexremains similar to controls [9]. It is unclear at this timewhether the observed reduction in COX activity is a resultof mitochondria-generated oxidative damage to mtDNA(mitochondrial DNA) with a consequent reduction in geneexpression, as proposed by several researchers [14,15], oris due to damage or inhibition of COX directly. A simple andefficient method of isolating the active enzyme from localizedbrain regions of mouse model or human post mortem isnecessary to answer this question.
In cancer cells, it is well accepted that a switch fromaerobic respiration to glycolysis occurs. This switch maybe due to p53 mutation, leading to reduced synthesis ofthe COX assembly factor SCO2 (synthesis of cytochromeoxidase 2) [16]. Additionally, an increased ratio of nuclearto mitochondrially encoded subunits has also been detectedin cancer cells [17,18]. Altered levels of COX between
Key words: Alzheimer’s disease (AD), cytochrome c oxidase (COX),mitochondrion, multiplexing, oxidative phosphorylation (OXPHOS),Parkinson’s disease (PD).
Abbreviations used: AD, Alzheimer’s disease; AP, alkaline phosphatase; BCA,bicinchoninic acid; CID, collision-induced dissociation; COX, cytochrome coxidase; ddC, 2′ ,3′-dideoxycytidine; ESI–MS/MS, electrospray-ionizationtandem MS;LC, liquid chromatography; LIT, linear ion trap; mAb,monoclonal antibody; MALDI–TOF, matrix-assisted laser-desorptionionization–time-of-flight; mtDNA, mitochondrial DNA; NRTI, nucleosideanalogue reverse transcriptase inhibitor; OXPHOS, oxidativephosphorylation; PD, Parkinson’s disease; PMF, peptide mass fingerprint;vMALDI, vacuum MALDI.
1 To whom correspondence should be addressed ([email protected]).
C© 2007 Portland Press Ltd

168 J. Murray and others
cancerous and normal surrounding tissue has been used toidentify tumour growth and stage before and after treatment[19–21]. A rapid, sensitive assay of assembly and activity ofCOX could be very useful in monitoring cancer, particularlyin confirming complete excision of a tumour by surgery.
Another common cause of mitochondrial dysfunctionis the adverse reaction of drugs. Inhibition of OXPHOShas been documented for several classes of drugs, includingdiabetes and anticancer drugs, as well as many antivirals andantibiotics [22–27]. Identifying mitochondrial toxicityand delineating the mechanism of this toxicity remain achallenge early in the drug development process. High-throughput, sensitive and specific assays of OXPHOS alter-ation are required.
We are developing mAb (monoclonal antibody)-basedimmunoprecipitation procedures for each of the OXPHOScomplexes from small amounts of tissue that isolate thesehydrophobic multisubunit complexes in an intact and en-zymatically active form, a method we term ‘immunocapture’[28–31]. Here, we describe a high-yield immunocaptureprotocol for the terminal enzyme of the respiratory chain,Complex IV (COX). We have characterized the isolatedenzyme by an extensive MS analysis using both MALDI–TOF (matrix-assisted laser-desorption ionization–time-of-flight) MS and nano-LC (liquid chromatography)–ESI–MS/MS(electrospray-ionization tandem MS) to confirm full subunitcomposition and to prepare for future studies on oxidativestress-induced post-translational modifications that may bea cause, or a consequence, of various diseases. In addition,we investigated proteolytic digests of Complex IV using a LIT(linear ion trap) equipped with a vMALDI (vacuum MALDI)source for fast protein identification. We also show that theenzyme can be immunocaptured by the antibody in the wellsof a microplate from the lysate of whole cells. The enzyme isactive and sensitive to cyanide, a specific inhibitor. We showthat such COX-immunocapture microplates can be usedto determine the relative amounts of the enzyme betweensamples and the enzyme activity, thereby giving a measureof specific activity. We provide examples of the use of theseplates in studying genetic defects of COX in human cell linesand in the evaluation of the effect of an antiviral drug onmtDNA replication as represented by altered COX levels.
Materials and methods
Preparation of cultured cells and heartmitochondriaNormal human fibroblasts (MRC5, a diploid strain derivedfrom fetal human lung) were obtained from the A.T.C.C.and used between doublings 30 and 45. MRC5 Rho0cells were derived by culturing cells for 12–14 passagesin ethidium bromide (50 ng/ml). For drug toxicity studies,the MRC5 cells were also cultured for 2, 4, 6, 8 and
10 population doublings in ddC (2′,3′-dideoxycytidine)(4 µM in 1% DMSO). Bovine heart mitochondria wereprepared as described by Smith [32]. Briefly, ventricleswere homogenized and particulate material was removedby centrifugation at 1000 g for 10 min. Mitochondriawere collected from the supernatant by spinning down at12 000 g for 15 min and resuspending in iso-osmotic buffer[10 mM Tris/HCl (pH 7.8), 0.25 M sucrose, 0.2 mM EDTAand 0.5 mM PMSF]. Kidney mitochondria were preparedin a similar fashion from bovine kidney cortex. Humanmitochondria from the heart of a 47-year-old man who diedof brain cancer were obtained, with the permission of thenext-of-kin, from Analytical Biological Services (Wilmington,DE, U.S.A.). Protein concentration was determined by theBCA (bicinchoninic acid) method (Pierce).
Immunocapture of COX on Protein G–agarosebeadsCOX immunocapture beads are available from Mitosciencesas product MS401. To prepare the beads, 5 mg of mouseanti-COX mAb 31E9B82G9 (or 7E5BA4) was incubatedby constant turning overnight at 4 ◦C with 1 ml of swollenProtein G–agarose beads (Sigma). Beads were washed inPBS [1.4 mM KH2PO4, 8 mM NaH2PO4, 140 mM NaCl and2.7 mM KCl (pH 7.3)]. The antibody–bead conjugate wascross-linked for 30 min with 20 mM dimethylpimelimidate(Sigma) in 0.2 M sodium borate (pH 9.0). The reaction wasstopped by incubation with 0.1 M ethanolamine (pH 8.0) for3 h at room temperature. Beads were resuspended in PBS.For the immunocapture of COX, mitochondria or culturedcells were suspended at 5.5 mg/ml in PBS. Membraneswere solubilized by adding 0.1 vol. of 0.2 M dodecylβ-D-maltoside (Calbiochem) to a final concentration of20 mM and incubation on ice for 30 min. Insoluble materialwas removed by centrifugation at 70000 g for 30 min.The recovered supernatant was incubated with 10 µl ofantibody-conjugated beads overnight at 4 ◦C while turning.Beads were washed three times in 1 ml of PBS, 0.3 mMdodecyl β-D-maltoside. Immunocaptured protein was elutedby the addition of 1% SDS. The protein concentration ofa small portion of the eluate was established by the BCAmethod (Pierce). It was determined that beads, prepared inthis way, can capture 1 µg of COX per µl of beads.
Electrophoresis of bovine heart and kidney COXCOX (10 µg) isolated by the above immunocapture methodfrom bovine heart and kidney mitochondria in SDS/PAGEsample buffer [50 mM Tris/HCl, pH 6.8, 5 % (v/v) glycerol,2 % (w/v) SDS and 0.02 mg/ml Bromophenol Blue] wasresolved by BisTris/4–12%-(w/v)-polyacrylamide NuPAGEgels using Mes buffer (Invitrogen). Gels were stained withCoomassie Brilliant Blue (Sigma). Beads coated with COXcapture mAb 31E9B82G9 or 7E5BA4 yielded an identicalCOX protein gel pattern (see Supplementary Figure S5).
C© 2007 Portland Press Ltd

Small-scale immunopurification of cytochrome c oxidase 169
In-gel proteolytic digestion of proteinsFor proteolysis, sequencing grade modified porcine trypsin(Promega) and bovine chymotrypsin (Roche) were used.HPLC solvents such as acetonitrile and water were obtainedfrom Burdick & Jackson. Protein spots of interest weremanually excised out of the gel and processed with the auto-matic in-gel digester robot ProGest (Genomic Solutions).The gel spots were destained and dehydrated with aceto-nitrile. Subsequently, they were reduced with 10 mM di-thiothreitol at 60 ◦C for 30 min and alkylated with 100 mMiodoacetamide (37 ◦C, 45 min). All samples were thenincubated with 125 ng of sequencing-grade trypsin at 37 ◦Cfor 4 h. The resultant tryptic peptides were extracted fromgel slices by 10% (v/v) formic acid extraction and analysedby MS. In addition, gel bands from bovine heart COX weredigested with chymotrypsin as well. After reduction andalkylation as described above, samples were incubated withchymotrypsin at room temperature for 4 h. The result-ant chymotryptic peptides were extracted with 50 % (v/v)acetonitrile/5% formic acid, concentrated and analysed.
MSMass spectra of digested gel spots were obtained byMALDI–TOF MS on a Voyager DE STR plus instrument(Applied Biosystems). All mass spectra were acquiredin positive-ionization mode with reflectron optics. Theinstrument was equipped with a 337 nm nitrogen laser andoperated under delayed extraction conditions; delay time190 ns, grid voltage 66–70% of full acceleration voltage (20–25 kV). All peptide samples were prepared using a matrixsolution consisting of 33 mM α-cyano-4-hydroxycinnamate(Agilent Technologies) in acetonitrile/methanol (1:1, v/v);1 µl of analyte (0.1–1 pmol of material) was mixed with 1 µlof matrix solution and then air-dried at room temperatureon a stainless-steel target. Typically, 50–100 laser shots wereused to record each spectrum. The mass spectra obtainedwere externally calibrated with an equimolar mixture ofangiotensin I, corticotropin-, 1–17, -18–39 and -7–38.
In all cases, the proteolytic peptide mixtures derivedfrom immunocaptured COX were analysed by reverse-phase nano-HPLC–ESI–MS/MS. Briefly, peptides wereseparated on an Ultimate nanocapillary HPLC systemequipped with a PepMapTM C18 nanocolumn (75 µm internaldiameter × 15 cm long; Dionex) and a CapTrap Micro guardcolumn of 0.5 µl bed volume (Michrom). Peptide mixtureswere loaded on to the guard column and washed with theloading solvent (0.05% formic acid; flow rate: 20 µl/min) for5 min, then transferred on to the analytical C18 nanocapil-lary HPLC column and eluted at a flow rate of 300 nl/minby using the following gradient: 2% solvent B in A (from 0to 5 min) and 2–70% solvent B in A (from 5 to 55 min).Solvent A consisted of 0.05% formic acid in 98% water/2%acetonitrile and solvent B consisted of 0.05% formic acid in
98% acetontrile/2% water. The column eluent was directlycoupled with a Q3STAR Pulsar i quadrupole orthogonal TOFmass spectrometer (MDS SCIEX) equipped with a Protanananospray ion source (ProXeon Biosystems). The nanosprayneedle voltage was typically 2300 V in the HPLC–MS mode.ESI–MS and ESI–MS/MS were recorded in positive-ion modewith a resolution of 12000–15000 full-width half-maximum.For CID (collision-induced dissociation) MS/MS, the masswindow for precursor ion selection of the quadrupolemass analyser was set to +− 1 m/z. The precursor ionswere fragmented in a collision cell using nitrogen as thecollision gas. Spectra were calibrated in static nanospraymode using MS/MS fragment-ions of a renin peptide standard(His immonium-ion with m/z at 110.0713 and b8-ion with m/zat 1028.5312) providing a mass accuracy of � 50 p.p.m.
In some cases, data were also acquired on a FinniganLTQTM LIT instrument coupled with a vMALDI ion source(Thermo Fisher Scientific, San Jose, CA, U.S.A.). The peptidesamples obtained from tryptic proteolysis of the one-dimen-sional SDS/PAGE gel spots were prepared without priorchromatographic separation. Additional experimentaldetails for vMALDI-MS and -MS/MS analyses are containedin the Supplementary material (http://www.babonline.org/bab/480167add.htm).
Database searches for protein identificationMS data [both PMF (peptide mass fingerprint) and MS/MSdata] were analysed with the bioinformatics search engineMascot (Matrix Sciences) [33]. Routinely, MALDI-MS datawere analysed using the Mascot Wizard tool (MatrixSciences) for PMFs matching against peptides from knownprotein sequences entered in publicly available proteindatabases (e.g. Swiss-Prot) or in-house custom databasesusing the following parameters: internal calibration usingtrypsin autolysis masses (m/z 842.5100 and 2211.1046),100 p.p.m. mass accuracy, two missed proteolytic cleavagesallowed. In all cases, tryptic and chymotryptic digestionextracts of proteins were analysed by HPLC–ESI–MS/MS,these results were then submitted to the search engineMascot (using Mascot Daemon, Matrix Sciences) whichanalyses peptide sequence information from tandem massspectra (100 p.p.m. mass accuracy). For vMALDI-MS/MSdatabase searches, see the Supplementary material. Toimprove the efficiency of identifying peptides derived fromComplex IV, we incorporated a custom-designed database(‘bovine Complex IV’) into our in-house licensed searchengine Mascot. The search engine Mascot uses a probability-based ‘Mowse Score’ to evaluate data obtained from tandemmass spectra (significance threshold P < 0.05) [33].
COX activity in mitochondrial membranes andafter isolation in a 96-well microtitre plateKCN-sensitive COX activity was measured in bovine andhuman heart mitochondria and human fibroblast cell lysate
C© 2007 Portland Press Ltd

170 J. Murray and others
by a novel microtitre plate immunocapture method availableas MS423 from Mitosciences. The substrate of the reaction,reduced ferrocytochrome c, was generated by dissolving250 mg of bovine heart cytochrome c (Sigma) in 1 mlof 50 mM KH2PO4 (pH 7.2) and reduced by addition ofL-(+)-ascorbic acid (MCB Reagents). Reduced bovine heartcytochrome c was purified at 4 ◦C over 20 ml of SephadexG-25 (Amersham Biosciences) in a column equilibrated with50 mM KH2PO4 (pH 7.2) and collected in 1 ml fractionsbefore concentration measurement at 550 nm in a BeckmanDU7 [molar absorption coefficient (ε) 19.1 · mM−1 · cm−1].To prepare the immunocapture plate, each well of a NuncMaxisorp 96-well plate was loaded with 1 µg of COXcapture mAb in 200 µl of 50 mM KH2PO4 (pH 7.2) andstored overnight at 4 ◦C. Wells were then blocked with300 µl of 50 mg/ml milk solution in 50 mM KH2PO4 (pH 7.2)for 2 h at room temperature. Wells were then rinsed in50 mM KH2PO4 (pH 7.2) to remove milk residue. The COXenzyme was captured by adding 200 µl of solubilized sample(solubilization described above) at the appropriate proteinconcentration in 20 mM KH2PO4 (pH 7.2) and 0.3 mMdodecyl β-D-maltoside. The plate was incubated at roomtemperature for 3 h. Wells were then rinsed three times in20 mM KH2PO4 (pH 7.2) and 0.3 mM dodecyl β-D-maltosideto remove unbound material. COX activity could then bemeasured by adding reaction buffer (20 mM KH2PO4, pH 7.2,2 mM dodecyl β-D-maltoside and 30 µM ferrocytochromec) and following the oxidation of ferrocytochrome c at 30 ◦Cby using a Victor2 1460 multiplate reader (PerkinElmer)with a narrow-bandpass 550 nm filter. Whereas theoxidation of ferrocytochrome c oxidation was monitoredfor 105 min, the activity rates were calculated from a linearslope of the initial rates at time points less than 11 min.The rates determined using mAbs 31E9B82G9 and 7E5BA4were very similar for bovine heart mitochondria, whereas31E9B82G9 gave higher rates with human fibroblast celllysate and was therefore chosen for all further experiments.
Quantitative detection of COX subunits bymultiplexing microplate assayCOX quantification was performed by the antibody‘sandwich’ method. Briefly, after the activity of theimmunocaptured COX was measured in each well, 200 µlof anti-(COX III) mAb (clone DA5BC4 isotype IgG 2a) wasadded to each well at 0.5 µg/ml in 20 mM KH2PO4 (pH 7.2),0.3 mM dodecyl β-D-maltoside and 1 µg/ml BSA. The platewas incubated for 1 h and then the wells were washed threetimes with 300 µl of 20 mM KH2PO4 (pH 7.2) and 0.3 mMdodecyl β-D-maltoside to remove unbound antibody.Next, 200 µl of AP (alkaline phosphatase)-conjugated goatanti-mouse IgG 2a antibody was added 1:10000 (SouthernBiotechnology) to each well in 20 mM KH2PO4 (pH 7.2),
0.3 mM dodecyl β-D-maltoside and 1 µg/ml BSA. The platewas incubated for 1 h and then the wells were washedthree times with 300 µl of 20 mM Tris (pH 7.2) and 0.3 mMdodecyl β-D-maltoside to remove unbound antibody. BoundAP-conjugated antibody was measured by phosphatasereaction using 15 mM p-nitrophenyl phosphate (Sigma) in0.1 M diethanolamine and 1 mM MgCl2 (pH 9.8).
Results
Quantitative immunocapture of COX by usingantibody bound to beadsMice were immunized with purified bovine COX. The mAbs7E5BA4 and 31E9B82G9 were generated, which, as shownbelow, specifically immunocapture COX from bovine andhuman mitochondria samples. A typical immunocapture,as described in the Materials and methods section, uses10 µl of antibody–bead conjugate and yields 10 µg of COXfrom as little as 250 µg of mitochondria. These amounts ofmitochondria are obtainable from platelets, needle biopsy orcultured cells. Two consecutive immunocapture steps with10 µl of beads effectively bound all available COX from thebovine sample, a total yield of 20 µg from 250 µg of mito-chondria. From this result, we estimate that 1 mg of bovineheart mitochondrial membranes contains a total of 80 µg ofCOX, i.e. 0.4 nmol/mg. These results are in excellent agree-ment with results from previous studies quantifying the levelsof the OXPHOS enzymes within mitochondria [34,35].
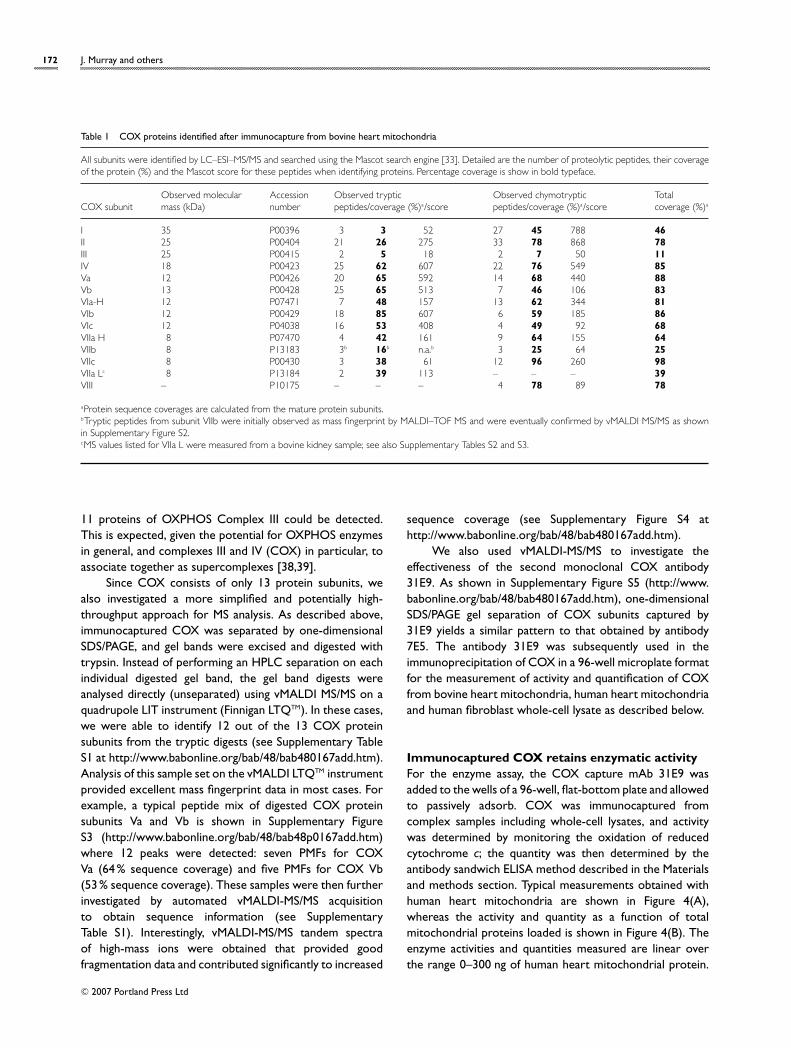
Immunocaptured COX contains all 13 subunits ofthe complexFigure 1 shows a typical subunit profile of COXimmunocaptured from bovine heart mitochondria andhuman heart mitochondria when resolved using the SDS/4–12% polyacrylamide Bis-Tris NuPAGE gel system. Other gelsystems exist that would separate the low-molecular-masssubunits better. However, this gel system was chosen be-cause it is commercially available and therefore more easilyreproduced between laboratories. The subunits of bovineCOX were identified by peptide mass fingerprinting andMS/MS (nano-HPLC–MS/MS). Protein bands were excisedfrom the gel, digested with trypsin and/or chymotrypsin, andanalysed by MALDI–TOF MS prior to subjecting the extractsto nano-HPLC–MS/MS to obtain peptide sequence data. Asshown in Figure 2, MALDI-MS PMF data are often sufficientto rapidly identify proteins from gel bands. As an example,when the band annotated as bovine heart mitochondriaCOX subunit IV (see Figure 1) was excised and fully analysed,13 subunit IV tryptic peptides were observed by MALDIMS. These PMFs provided coverage of 51% of the mature(processed) protein sequence (Mascot score 139). Similarly,
C© 2007 Portland Press Ltd
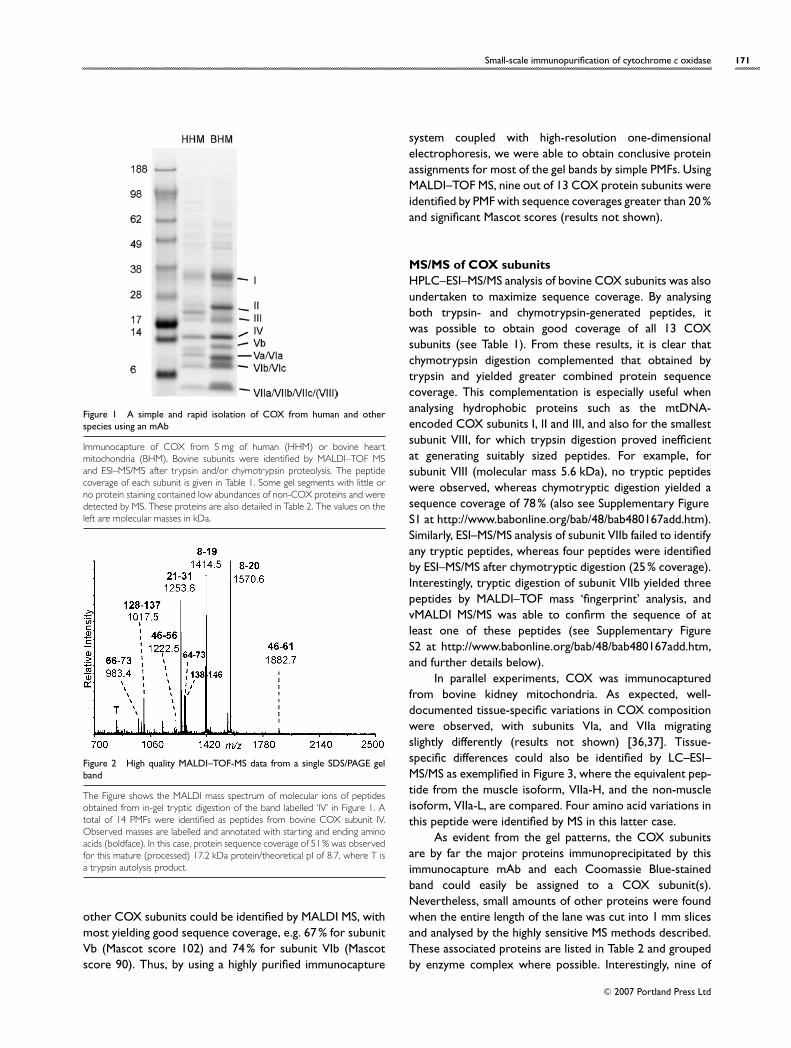
Small-scale immunopurification of cytochrome c oxidase 171
Figure 1 A simple and rapid isolation of COX from human and otherspecies using an mAb
Immunocapture of COX from 5 mg of human (HHM) or bovine heartmitochondria (BHM). Bovine subunits were identified by MALDI–TOF MSand ESI–MS/MS after trypsin and/or chymotrypsin proteolysis. The peptidecoverage of each subunit is given in Table 1. Some gel segments with little orno protein staining contained low abundances of non-COX proteins and weredetected by MS. These proteins are also detailed in Table 2. The values on theleft are molecular masses in kDa.
Figure 2 High quality MALDI–TOF-MS data from a single SDS/PAGE gelband
The Figure shows the MALDI mass spectrum of molecular ions of peptidesobtained from in-gel tryptic digestion of the band labelled ‘IV’ in Figure 1. Atotal of 14 PMFs were identified as peptides from bovine COX subunit IV.Observed masses are labelled and annotated with starting and ending aminoacids (boldface). In this case, protein sequence coverage of 51% was observedfor this mature (processed) 17.2 kDa protein/theoretical pI of 8.7, where T isa trypsin autolysis product.
other COX subunits could be identified by MALDI MS, withmost yielding good sequence coverage, e.g. 67% for subunitVb (Mascot score 102) and 74% for subunit VIb (Mascotscore 90). Thus, by using a highly purified immunocapture
system coupled with high-resolution one-dimensionalelectrophoresis, we were able to obtain conclusive proteinassignments for most of the gel bands by simple PMFs. UsingMALDI–TOF MS, nine out of 13 COX protein subunits wereidentified by PMF with sequence coverages greater than 20%and significant Mascot scores (results not shown).
MS/MS of COX subunitsHPLC–ESI–MS/MS analysis of bovine COX subunits was alsoundertaken to maximize sequence coverage. By analysingboth trypsin- and chymotrypsin-generated peptides, itwas possible to obtain good coverage of all 13 COXsubunits (see Table 1). From these results, it is clear thatchymotrypsin digestion complemented that obtained bytrypsin and yielded greater combined protein sequencecoverage. This complementation is especially useful whenanalysing hydrophobic proteins such as the mtDNA-encoded COX subunits I, II and III, and also for the smallestsubunit VIII, for which trypsin digestion proved inefficientat generating suitably sized peptides. For example, forsubunit VIII (molecular mass 5.6 kDa), no tryptic peptideswere observed, whereas chymotryptic digestion yielded asequence coverage of 78 % (also see Supplementary FigureS1 at http://www.babonline.org/bab/48/bab480167add.htm).Similarly, ESI–MS/MS analysis of subunit VIIb failed to identifyany tryptic peptides, whereas four peptides were identifiedby ESI–MS/MS after chymotryptic digestion (25% coverage).Interestingly, tryptic digestion of subunit VIIb yielded threepeptides by MALDI–TOF mass ‘fingerprint’ analysis, andvMALDI MS/MS was able to confirm the sequence of atleast one of these peptides (see Supplementary FigureS2 at http://www.babonline.org/bab/48/bab480167add.htm,and further details below).
In parallel experiments, COX was immunocapturedfrom bovine kidney mitochondria. As expected, well-documented tissue-specific variations in COX compositionwere observed, with subunits VIa, and VIIa migratingslightly differently (results not shown) [36,37]. Tissue-specific differences could also be identified by LC–ESI–MS/MS as exemplified in Figure 3, where the equivalent pep-tide from the muscle isoform, VIIa-H, and the non-muscleisoform, VIIa-L, are compared. Four amino acid variations inthis peptide were identified by MS in this latter case.
As evident from the gel patterns, the COX subunitsare by far the major proteins immunoprecipitated by thisimmunocapture mAb and each Coomassie Blue-stainedband could easily be assigned to a COX subunit(s).Nevertheless, small amounts of other proteins were foundwhen the entire length of the lane was cut into 1 mm slicesand analysed by the highly sensitive MS methods described.These associated proteins are listed in Table 2 and groupedby enzyme complex where possible. Interestingly, nine of
C© 2007 Portland Press Ltd
172 J. Murray and others
Table 1 COX proteins identified after immunocapture from bovine heart mitochondria
All subunits were identified by LC–ESI–MS/MS and searched using the Mascot search engine [33]. Detailed are the number of proteolytic peptides, their coverageof the protein (%) and the Mascot score for these peptides when identifying proteins. Percentage coverage is show in bold typeface.
Observed molecular Accession Observed tryptic Observed chymotryptic TotalCOX subunit mass (kDa) number peptides/coverage (%)a/score peptides/coverage (%)a/score coverage (%)a
I 35 P00396 3 3 52 27 45 788 46II 25 P00404 21 26 275 33 78 868 78III 25 P00415 2 5 18 2 7 50 11IV 18 P00423 25 62 607 22 76 549 85Va 12 P00426 20 65 592 14 68 440 88Vb 13 P00428 25 65 513 7 46 106 83VIa-H 12 P07471 7 48 157 13 62 344 81VIb 12 P00429 18 85 607 6 59 185 86VIc 12 P04038 16 53 408 4 49 92 68VIIa H 8 P07470 4 42 161 9 64 155 64VIIb 8 P13183 3b 16b n.a.b 3 25 64 25VIIc 8 P00430 3 38 61 12 96 260 98VIIa Lc 8 P13184 2 39 113 – – – 39VIII – P10175 – – – 4 78 89 78
aProtein sequence coverages are calculated from the mature protein subunits.bTryptic peptides from subunit VIIb were initially observed as mass fingerprint by MALDI–TOF MS and were eventually confirmed by vMALDI MS/MS as shownin Supplementary Figure S2.cMS values listed for VIIa L were measured from a bovine kidney sample; see also Supplementary Tables S2 and S3.
11 proteins of OXPHOS Complex III could be detected.This is expected, given the potential for OXPHOS enzymesin general, and complexes III and IV (COX) in particular, toassociate together as supercomplexes [38,39].
Since COX consists of only 13 protein subunits, wealso investigated a more simplified and potentially high-throughput approach for MS analysis. As described above,immunocaptured COX was separated by one-dimensionalSDS/PAGE, and gel bands were excised and digested withtrypsin. Instead of performing an HPLC separation on eachindividual digested gel band, the gel band digests wereanalysed directly (unseparated) using vMALDI MS/MS on aquadrupole LIT instrument (Finnigan LTQTM). In these cases,we were able to identify 12 out of the 13 COX proteinsubunits from the tryptic digests (see Supplementary TableS1 at http://www.babonline.org/bab/48/bab480167add.htm).Analysis of this sample set on the vMALDI LTQTM instrumentprovided excellent mass fingerprint data in most cases. Forexample, a typical peptide mix of digested COX proteinsubunits Va and Vb is shown in Supplementary FigureS3 (http://www.babonline.org/bab/48/bab48p0167add.htm)where 12 peaks were detected: seven PMFs for COXVa (64 % sequence coverage) and five PMFs for COX Vb(53 % sequence coverage). These samples were then furtherinvestigated by automated vMALDI-MS/MS acquisitionto obtain sequence information (see SupplementaryTable S1). Interestingly, vMALDI-MS/MS tandem spectraof high-mass ions were obtained that provided goodfragmentation data and contributed significantly to increased
sequence coverage (see Supplementary Figure S4 athttp://www.babonline.org/bab/48/bab480167add.htm).
We also used vMALDI-MS/MS to investigate theeffectiveness of the second monoclonal COX antibody31E9. As shown in Supplementary Figure S5 (http://www.babonline.org/bab/48/bab480167add.htm), one-dimensionalSDS/PAGE gel separation of COX subunits captured by31E9 yields a similar pattern to that obtained by antibody7E5. The antibody 31E9 was subsequently used in theimmunoprecipitation of COX in a 96-well microplate formatfor the measurement of activity and quantification of COXfrom bovine heart mitochondria, human heart mitochondriaand human fibroblast whole-cell lysate as described below.
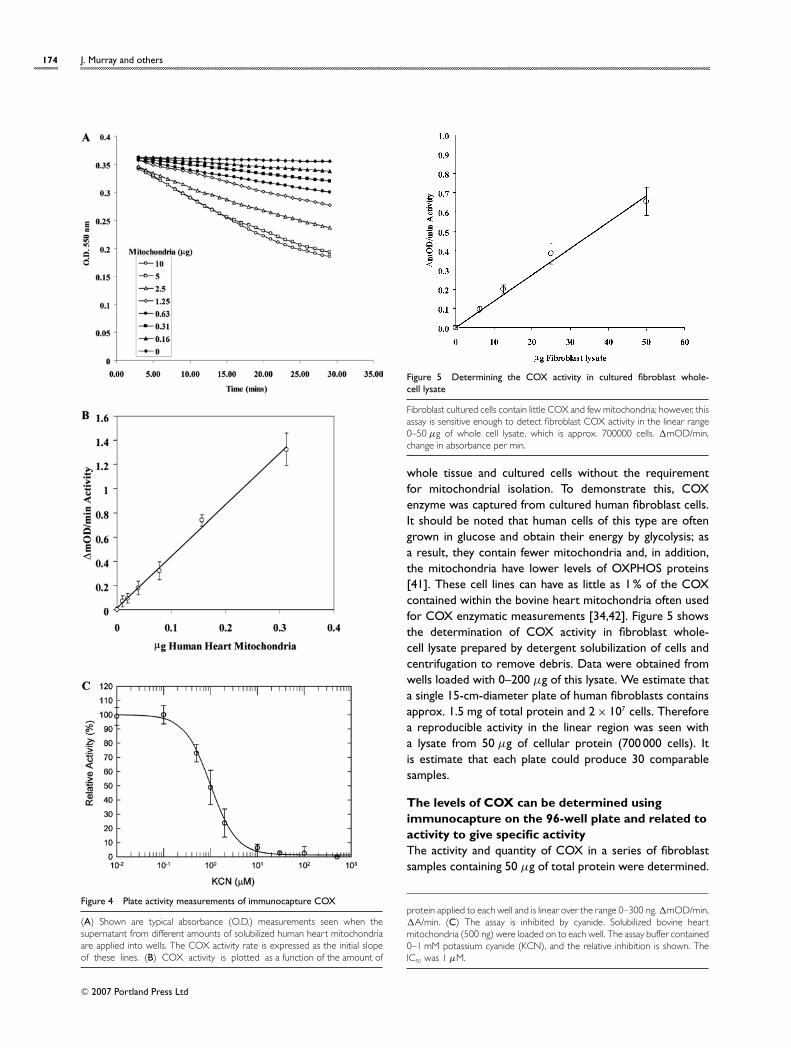
Immunocaptured COX retains enzymatic activityFor the enzyme assay, the COX capture mAb 31E9 wasadded to the wells of a 96-well, flat-bottom plate and allowedto passively adsorb. COX was immunocaptured fromcomplex samples including whole-cell lysates, and activitywas determined by monitoring the oxidation of reducedcytochrome c; the quantity was then determined by theantibody sandwich ELISA method described in the Materialsand methods section. Typical measurements obtained withhuman heart mitochondria are shown in Figure 4(A),whereas the activity and quantity as a function of totalmitochondrial proteins loaded is shown in Figure 4(B). Theenzyme activities and quantities measured are linear overthe range 0–300 ng of human heart mitochondrial protein.
C© 2007 Portland Press Ltd
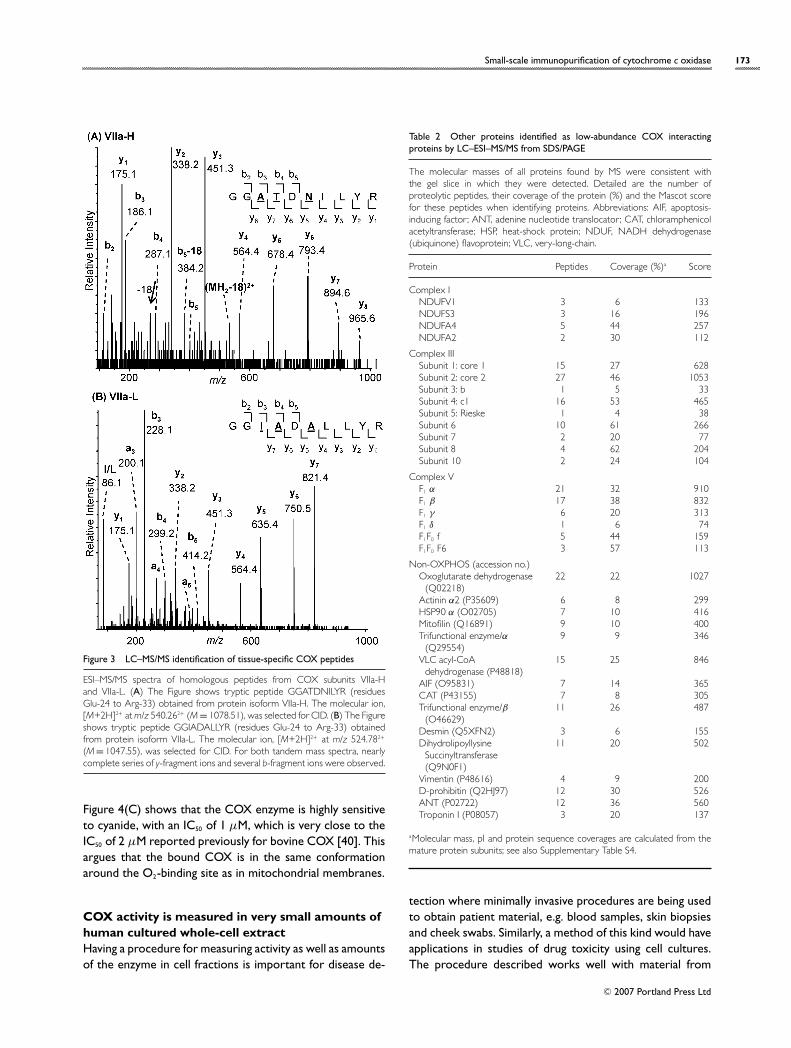
Small-scale immunopurification of cytochrome c oxidase 173
Figure 3 LC–MS/MS identification of tissue-specific COX peptides
ESI–MS/MS spectra of homologous peptides from COX subunits VIIa-Hand VIIa-L. (A) The Figure shows tryptic peptide GGATDNILYR (residuesGlu-24 to Arg-33) obtained from protein isoform VIIa-H. The molecular ion,[M+2H]2+ at m/z 540.262+ (M = 1078.51), was selected for CID. (B) The Figureshows tryptic peptide GGIADALLYR (residues Glu-24 to Arg-33) obtainedfrom protein isoform VIIa-L. The molecular ion, [M+2H]2+ at m/z 524.782+
(M = 1047.55), was selected for CID. For both tandem mass spectra, nearlycomplete series of y-fragment ions and several b-fragment ions were observed.
Figure 4(C) shows that the COX enzyme is highly sensitiveto cyanide, with an IC50 of 1 µM, which is very close to theIC50 of 2 µM reported previously for bovine COX [40]. Thisargues that the bound COX is in the same conformationaround the O2-binding site as in mitochondrial membranes.
COX activity is measured in very small amounts ofhuman cultured whole-cell extractHaving a procedure for measuring activity as well as amountsof the enzyme in cell fractions is important for disease de-
Table 2 Other proteins identified as low-abundance COX interactingproteins by LC–ESI–MS/MS from SDS/PAGE
The molecular masses of all proteins found by MS were consistent withthe gel slice in which they were detected. Detailed are the number ofproteolytic peptides, their coverage of the protein (%) and the Mascot scorefor these peptides when identifying proteins. Abbreviations: AIF, apoptosis-inducing factor ; ANT, adenine nucleotide translocator; CAT, chloramphenicolacetyltransferase; HSP, heat-shock protein; NDUF, NADH dehydrogenase(ubiquinone) flavoprotein; VLC, very-long-chain.
Protein Peptides Coverage (%)a Score
Complex INDUFV1 3 6 133NDUFS3 3 16 196NDUFA4 5 44 257NDUFA2 2 30 112
Complex IIISubunit 1: core 1 15 27 628Subunit 2: core 2 27 46 1053Subunit 3: b 1 5 33Subunit 4: c1 16 53 465Subunit 5: Rieske 1 4 38Subunit 6 10 61 266Subunit 7 2 20 77Subunit 8 4 62 204Subunit 10 2 24 104
Complex VF1 α 21 32 910F1 β 17 38 832F1 γ 6 20 313F1 δ 1 6 74F1F0 f 5 44 159F1F0 F6 3 57 113
Non-OXPHOS (accession no.)Oxoglutarate dehydrogenase
(Q02218)22 22 1027
Actinin α2 (P35609) 6 8 299HSP90 α (O02705) 7 10 416Mitofilin (Q16891) 9 10 400Trifunctional enzyme/α
(Q29554)9 9 346
VLC acyl-CoAdehydrogenase (P48818)
15 25 846
AIF (O95831) 7 14 365CAT (P43155) 7 8 305Trifunctional enzyme/β
(O46629)11 26 487
Desmin (Q5XFN2) 3 6 155Dihydrolipoyllysine
Succinyltransferase(Q9N0F1)
11 20 502
Vimentin (P48616) 4 9 200D-prohibitin (Q2HJ97) 12 30 526ANT (P02722) 12 36 560Troponin I (P08057) 3 20 137
aMolecular mass, pI and protein sequence coverages are calculated from themature protein subunits; see also Supplementary Table S4.
tection where minimally invasive procedures are being usedto obtain patient material, e.g. blood samples, skin biopsiesand cheek swabs. Similarly, a method of this kind would haveapplications in studies of drug toxicity using cell cultures.The procedure described works well with material from
C© 2007 Portland Press Ltd
174 J. Murray and others
Figure 4 Plate activity measurements of immunocapture COX
(A) Shown are typical absorbance (O.D.) measurements seen when thesupernatant from different amounts of solubilized human heart mitochondriaare applied into wells. The COX activity rate is expressed as the initial slopeof these lines. (B) COX activity is plotted as a function of the amount of
Figure 5 Determining the COX activity in cultured fibroblast whole-cell lysate
Fibroblast cultured cells contain little COX and few mitochondria; however, thisassay is sensitive enough to detect fibroblast COX activity in the linear range0–50 µg of whole cell lysate, which is approx. 700000 cells. �mOD/min,change in absorbance per min.
whole tissue and cultured cells without the requirementfor mitochondrial isolation. To demonstrate this, COXenzyme was captured from cultured human fibroblast cells.It should be noted that human cells of this type are oftengrown in glucose and obtain their energy by glycolysis; asa result, they contain fewer mitochondria and, in addition,the mitochondria have lower levels of OXPHOS proteins[41]. These cell lines can have as little as 1 % of the COXcontained within the bovine heart mitochondria often usedfor COX enzymatic measurements [34,42]. Figure 5 showsthe determination of COX activity in fibroblast whole-cell lysate prepared by detergent solubilization of cells andcentrifugation to remove debris. Data were obtained fromwells loaded with 0–200 µg of this lysate. We estimate thata single 15-cm-diameter plate of human fibroblasts containsapprox. 1.5 mg of total protein and 2 × 107 cells. Thereforea reproducible activity in the linear region was seen witha lysate from 50 µg of cellular protein (700 000 cells). Itis estimate that each plate could produce 30 comparablesamples.
The levels of COX can be determined usingimmunocapture on the 96-well plate and related toactivity to give specific activityThe activity and quantity of COX in a series of fibroblastsamples containing 50 µg of total protein were determined.
protein applied to each well and is linear over the range 0–300 ng. �mOD/min,�A/min. (C) The assay is inhibited by cyanide. Solubilized bovine heartmitochondria (500 ng) were loaded on to each well. The assay buffer contained0–1 mM potassium cyanide (KCN), and the relative inhibition is shown. TheIC50 was 1 µM.
C© 2007 Portland Press Ltd
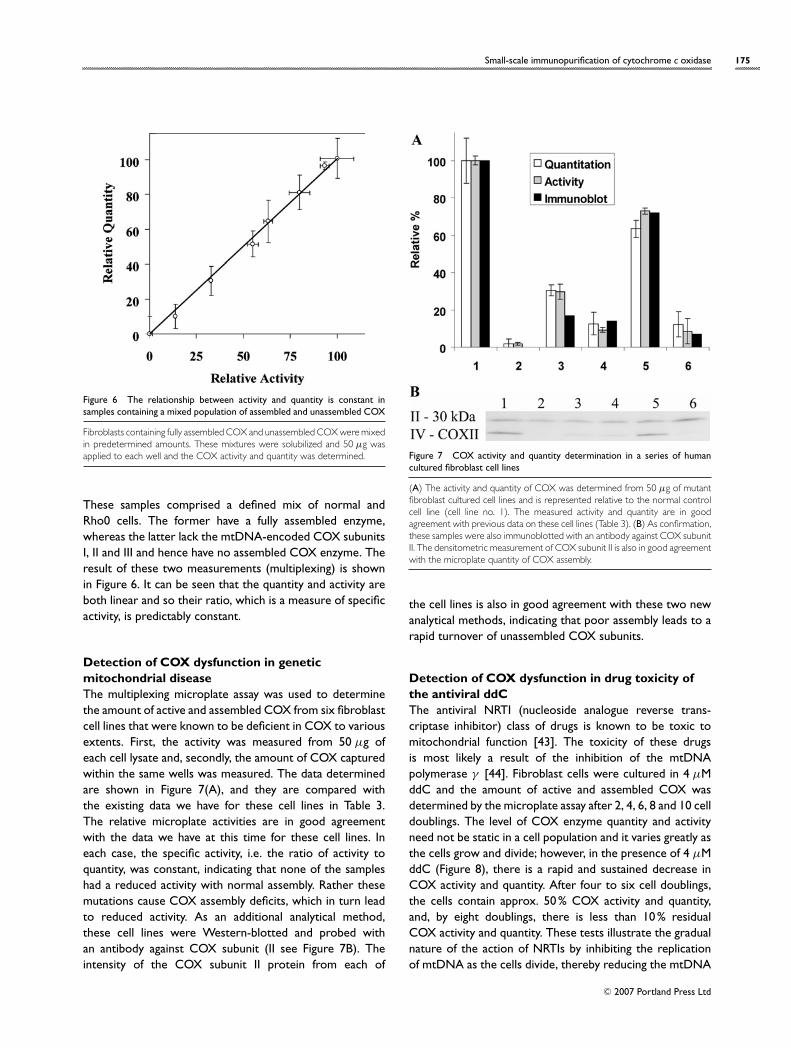
Small-scale immunopurification of cytochrome c oxidase 175
Figure 6 The relationship between activity and quantity is constant insamples containing a mixed population of assembled and unassembled COX
Fibroblasts containing fully assembled COX and unassembled COX were mixedin predetermined amounts. These mixtures were solubilized and 50 µg wasapplied to each well and the COX activity and quantity was determined.
These samples comprised a defined mix of normal andRho0 cells. The former have a fully assembled enzyme,whereas the latter lack the mtDNA-encoded COX subunitsI, II and III and hence have no assembled COX enzyme. Theresult of these two measurements (multiplexing) is shownin Figure 6. It can be seen that the quantity and activity areboth linear and so their ratio, which is a measure of specificactivity, is predictably constant.
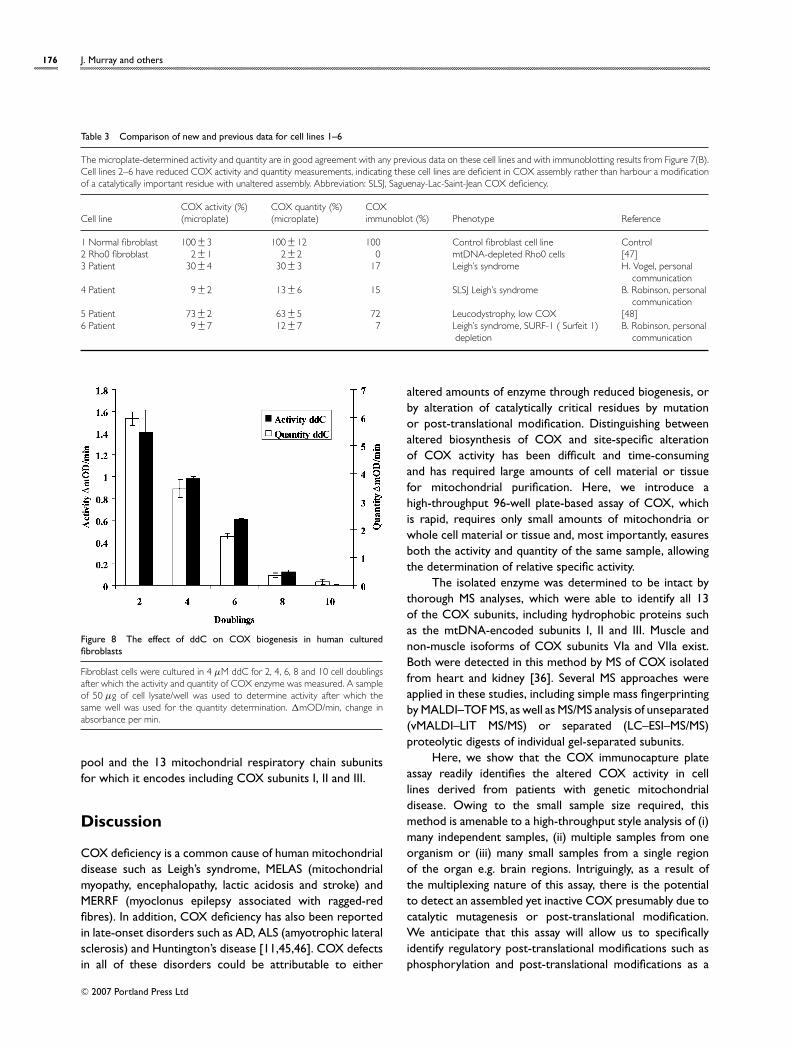
Detection of COX dysfunction in geneticmitochondrial diseaseThe multiplexing microplate assay was used to determinethe amount of active and assembled COX from six fibroblastcell lines that were known to be deficient in COX to variousextents. First, the activity was measured from 50 µg ofeach cell lysate and, secondly, the amount of COX capturedwithin the same wells was measured. The data determinedare shown in Figure 7(A), and they are compared withthe existing data we have for these cell lines in Table 3.The relative microplate activities are in good agreementwith the data we have at this time for these cell lines. Ineach case, the specific activity, i.e. the ratio of activity toquantity, was constant, indicating that none of the sampleshad a reduced activity with normal assembly. Rather thesemutations cause COX assembly deficits, which in turn leadto reduced activity. As an additional analytical method,these cell lines were Western-blotted and probed withan antibody against COX subunit (II see Figure 7B). Theintensity of the COX subunit II protein from each of
Figure 7 COX activity and quantity determination in a series of humancultured fibroblast cell lines
(A) The activity and quantity of COX was determined from 50 µg of mutantfibroblast cultured cell lines and is represented relative to the normal controlcell line (cell line no. 1). The measured activity and quantity are in goodagreement with previous data on these cell lines (Table 3). (B) As confirmation,these samples were also immunoblotted with an antibody against COX subunitII. The densitometric measurement of COX subunit II is also in good agreementwith the microplate quantity of COX assembly.
the cell lines is also in good agreement with these two newanalytical methods, indicating that poor assembly leads to arapid turnover of unassembled COX subunits.
Detection of COX dysfunction in drug toxicity ofthe antiviral ddCThe antiviral NRTI (nucleoside analogue reverse trans-criptase inhibitor) class of drugs is known to be toxic tomitochondrial function [43]. The toxicity of these drugsis most likely a result of the inhibition of the mtDNApolymerase γ [44]. Fibroblast cells were cultured in 4 µMddC and the amount of active and assembled COX wasdetermined by the microplate assay after 2, 4, 6, 8 and 10 celldoublings. The level of COX enzyme quantity and activityneed not be static in a cell population and it varies greatly asthe cells grow and divide; however, in the presence of 4 µMddC (Figure 8), there is a rapid and sustained decrease inCOX activity and quantity. After four to six cell doublings,the cells contain approx. 50% COX activity and quantity,and, by eight doublings, there is less than 10% residualCOX activity and quantity. These tests illustrate the gradualnature of the action of NRTIs by inhibiting the replicationof mtDNA as the cells divide, thereby reducing the mtDNA
C© 2007 Portland Press Ltd
176 J. Murray and others
Table 3 Comparison of new and previous data for cell lines 1–6
The microplate-determined activity and quantity are in good agreement with any previous data on these cell lines and with immunoblotting results from Figure 7(B).Cell lines 2–6 have reduced COX activity and quantity measurements, indicating these cell lines are deficient in COX assembly rather than harbour a modificationof a catalytically important residue with unaltered assembly. Abbreviation: SLSJ, Saguenay-Lac-Saint-Jean COX deficiency.
COX activity (%) COX quantity (%) COXCell line (microplate) (microplate) immunoblot (%) Phenotype Reference
1 Normal fibroblast 100 +− 3 100 +− 12 100 Control fibroblast cell line Control2 Rho0 fibroblast 2 +− 1 2 +− 2 0 mtDNA-depleted Rho0 cells [47]3 Patient 30 +− 4 30 +− 3 17 Leigh’s syndrome H. Vogel, personal
communication4 Patient 9 +− 2 13 +− 6 15 SLSJ Leigh’s syndrome B. Robinson, personal
communication5 Patient 73 +− 2 63 +− 5 72 Leucodystrophy, low COX [48]6 Patient 9 +− 7 12 +− 7 7 Leigh’s syndrome, SURF-1 ( Surfeit 1) B. Robinson, personal
depletion communication
Figure 8 The effect of ddC on COX biogenesis in human culturedfibroblasts
Fibroblast cells were cultured in 4 µM ddC for 2, 4, 6, 8 and 10 cell doublingsafter which the activity and quantity of COX enzyme was measured. A sampleof 50 µg of cell lysate/well was used to determine activity after which thesame well was used for the quantity determination. �mOD/min, change inabsorbance per min.
pool and the 13 mitochondrial respiratory chain subunitsfor which it encodes including COX subunits I, II and III.
Discussion
COX deficiency is a common cause of human mitochondrialdisease such as Leigh’s syndrome, MELAS (mitochondrialmyopathy, encephalopathy, lactic acidosis and stroke) andMERRF (myoclonus epilepsy associated with ragged-redfibres). In addition, COX deficiency has also been reportedin late-onset disorders such as AD, ALS (amyotrophic lateralsclerosis) and Huntington’s disease [11,45,46]. COX defectsin all of these disorders could be attributable to either
altered amounts of enzyme through reduced biogenesis, orby alteration of catalytically critical residues by mutationor post-translational modification. Distinguishing betweenaltered biosynthesis of COX and site-specific alterationof COX activity has been difficult and time-consumingand has required large amounts of cell material or tissuefor mitochondrial purification. Here, we introduce ahigh-throughput 96-well plate-based assay of COX, whichis rapid, requires only small amounts of mitochondria orwhole cell material or tissue and, most importantly, easuresboth the activity and quantity of the same sample, allowingthe determination of relative specific activity.
The isolated enzyme was determined to be intact bythorough MS analyses, which were able to identify all 13of the COX subunits, including hydrophobic proteins suchas the mtDNA-encoded subunits I, II and III. Muscle andnon-muscle isoforms of COX subunits VIa and VIIa exist.Both were detected in this method by MS of COX isolatedfrom heart and kidney [36]. Several MS approaches wereapplied in these studies, including simple mass fingerprintingby MALDI–TOF MS, as well as MS/MS analysis of unseparated(vMALDI–LIT MS/MS) or separated (LC–ESI–MS/MS)proteolytic digests of individual gel-separated subunits.
Here, we show that the COX immunocapture plateassay readily identifies the altered COX activity in celllines derived from patients with genetic mitochondrialdisease. Owing to the small sample size required, thismethod is amenable to a high-throughput style analysis of (i)many independent samples, (ii) multiple samples from oneorganism or (iii) many small samples from a single regionof the organ e.g. brain regions. Intriguingly, as a result ofthe multiplexing nature of this assay, there is the potentialto detect an assembled yet inactive COX presumably due tocatalytic mutagenesis or post-translational modification.We anticipate that this assay will allow us to specificallyidentify regulatory post-translational modifications such asphosphorylation and post-translational modifications as a
C© 2007 Portland Press Ltd

Small-scale immunopurification of cytochrome c oxidase 177
result of oxidative stress such as nitration and carbonylation;this work is ongoing.
Finally, we show that the COX multiplexing assay can beused in drug toxicity studies where a compound is predictedto alter mitochondrial biogenesis, either by reducingmtDNA copy number and/or by reducing mitochondrialprotein synthesis through reaction with the mito-chondrial ribosome. We show the parallel decline in boththe quantity and activity of COX in response to 4 µMddC treatment. Also, the effect was proportional to theconcentration of ddC in the media (results not shown).
In summary, we describe a novel high-throughputmultiplexed assay for COX activity and the correlation ofthis activity with the levels of the enzyme complex. Weanticipate that this assay will find widespread use in basicscience studies of mitochondrial diseases, both geneticand late-onset, induced by or involving modification ofthe enzyme by reaction with oxygen species. Ultimately,it should be possible to generate a more comprehensiveimmunocapture screen of OXPHOS defects in all five of thecomplexes of the OXPHOS chain [28–31].
Acknowledgements
We thank Thermo Fisher Scientific for providing a LTQTM
mass spectrometer for evaluation equipped with a v-MALDI(OPTON 30021) ion source. B. S. thanks Mr Chip Wittand Mr Michael Cusack for bioinformatics support. Thiswork was supported in part by NIH (National Institutesof Health) Small Business Technology Transfer grant2R42GM071052-02 to M. F.M. M. F.M., R.A.C., and J.M.acknowledge a financial interest in these studies. We thankProfessor Hannes Vogel (Department of Neuropathology,Stanford University Medical Center, Palo Alto, CA, U.S.A.)and Professor Brian Robinson (Genetics and GenomeResearch Program, The Hospital for Sick Children, Toronto,ON, Canada) for supplying cell lines.
References
1 Coenen, M. J., Smeitink, J. A., Pots, J. M., van Kaauwen, E.,Trijbels, F. J., Hol, F. A. and van den Heuvel, L. P. (2006) J. ChildNeurol. 21, 508–511
2 Benit, P., Chretien, D., Kadhom, N., de Lonlay-Debeney, P.,Cormier-Daire, V., Cabral, A., Peudenier, S., Rustin, P., Munnich,A. and Rotig, A. (2001) Am. J. Hum. Genet. 68, 1344–1352
3 Finsterer, J. (2004) Eur. J. Neurol. 11, 163–1864 von Kleist-Retzow, J. C., Schauseil-Zipf, U., Michalk, D. V. and
Kunz, W. S. (2003) Exp. Physiol. 88, 155–1665 Schapira, A. H., Cooper, J. M., Dexter, D., Clark, J. B., Jenner, P.
and Marsden, C. D. (1990) J. Neurochem. 54, 823–827
6 Betarbet, R., Sherer, T. B., MacKenzie, G., Garcia-Osuna, M.,Panov, A. V. and Greenamyre, J. T. (2000) Nat. Neurosci. 3,1301–1306
7 Mizuno, Y., Ohta, S., Tanaka, M., Takamiya, S., Suzuki, K., Sato, T.,Oya, H., Ozawa, T. and Kagawa, Y. (1989) Biochem. Biophys.Res. Commun. 163, 1450–1455
8 Bosetti, F., Brizzi, F., Barogi, S., Mancuso, M., Siciliano, G., Tendi,E. A., Murri, L., Rapoport, S. I. and Solaini, G. (2002) Neurobiol.Aging 23, 371–376
9 Maurer, I., Zierz, S. and Moller, H. J. (2000) Neurobiol. Aging21, 455–462
10 Mancuso, M., Filosto, M., Bosetti, F., Ceravolo, R., Rocchi, A.,Tognoni, G., Manca, M. L., Solaini, G., Siciliano, G. and Murri, L.(2003) Exp. Neurol. 182, 421–426
11 Cardoso, S. M., Proenca, M. T., Santos, S., Santana, I. andOliveira, C. R. (2004) Neurobiol. Aging 25, 105–110
12 Genova, M. L., Pich, M. M., Bernacchia, A., Bianchi, C., Biondi,A., Bovina, C., Falasca, A. I., Formiggini, G., Castelli, G. P. andLenaz, G. (2004) Ann. N.Y. Acad. Sci. 1011, 86–100
13 van der Walt, J. M., Nicodemus, K. K., Martin, E. R., Scott,W. K., Nance, M. A., Watts, R. L., Hubble, J. P., Haines, J. L.,Koller, W. C., Lyons, K. et al. (2003) Am. J. Hum. Genet. 72,804–811
14 Simonian, N. A. and Hyman, B. T. (1994) J. Neuropathol.Exp. Neurol. 53, 508–512
15 Chandrasekaran, K., Hatanpaa, K., Brady, D. R. and Rapoport,S. I. (1996) Exp. Neurol. 142, 80–88
16 Matoba, S., Kang, J. G., Patino, W. D., Wragg, A., Boehm, M.,Gavrilova, O., Hurley, P. J., Bunz, F. and Hwang, P. M. (2006)Science 312, 1650–1653
17 Herrmann, P. C., Gillespie, J. W., Charboneau, L., Bichsel, V. E.,Paweletz, C. P., Calvert, V. S., Kohn, E. C., Emmert-Buck,M. R., Liotta, L. A. and Petricoin, III, E. F. (2003) Proteomics 3,1801–1810
18 Krieg, R. C., Knuechel, R., Schiffmann, E., Liotta, L. A., Petricoin,III, E. F. and Herrmann, P. C. (2004) Proteomics 4, 2789–2795
19 Jiang, W. W., Rosenbaum, E., Mambo, E., Zahurak, M.,Masayesva, B., Carvalho, A. L., Zhou, S., Westra, W. H., Alberg,A. J. and Sidransky, D. et al. (2006) Clin. Cancer Res. 12,1564–1569
20 Jiang, W. W., Masayesva, B., Zahurak, M., Carvalho,A. L., Rosenbaum, E., Mambo, E., Zhou, S., Minhas, K.and Benoit, N. et al. (2005) Clin. Cancer Res. 11,2486–2491
21 Payne, C. M., Holubec, H., Bernstein, C., Bernstein, H., Dvorak,K., Green, S. B., Wilson, M., Dall’Agnol, M., Dvorakova,B., Warneke, J. and Garewal, H. (2005) Cancer Epidemiol.Biomarkers Prev. 14, 2066–2075
22 Jove, M., Salla, J., Planavila, A., Cabrero, A., Michalik, L.,Wahli, W., Laguna, J. C. and Vazquez-Carrera, M. (2004)J. Lipid Res. 45, 113–123
23 Grandjean, F., Bremaud, L., Robert, J. and Ratinaud, M. H.(2002) Biochem. Pharmacol. 63, 823–831
C© 2007 Portland Press Ltd

178 J. Murray and others
24 Merten, K. E., Feng, W., Zhang, L., Pierce, W., Cai, J., Klein, J. B.and Kang, Y. J. (2005) J. Pharmacol. Exp. Ther. 315, 1314–1319
25 Yerroum, M., Pham-Dang, C., Authier, F. J., Monnet, I., Gherardi,R. and Chariot, P. (2000) Acta Neuropathol. (Berlin) 100,82–86
26 Chariot, P., Monnet, I. and Gherardi, R. (1993) Ann. Neurol.34, 561–565
27 McKee, E. E., Ferguson, M., Bentley, A. T. and Marks, T. A.(2006) Antimicrob. Agents Chemother. 50, 2042–2049
28 Murray, J., Zhang, B., Taylor, S. W., Oglesbee, D., Fahy, E.,Marusich, M. F., Ghosh, S. S. and Capaldi, R. A. (2003) J. Biol.Chem. 278, 13619–13622
29 Schilling, B., Murray, J., Yoo, C. B., Row, R. H., Cusack, M. P.,Capaldi, R. A. and Gibson, B. W. (2006) Biochim. Biophys. Acta1762, 213–222
30 Aggeler, R., Coons, J., Taylor, S. W., Ghosh, S. S., Garcia, J. J.,Capaldi, R. A. and Marusich, M. F. (2002) J. Biol. Chem. 277,33906–33912
31 Lib, M., Rodriguez-Mari, A., Marusich, M. F. and Capaldi, R. A.(2003) Anal. Biochem. 314, 121–127
32 Smith, A. L. (1967) Methods Enzymol. 10, 81–8633 Perkins, D. N., Pappin, D. J., Creasy, D. M. and Cottrell, J. S.
(1999) Electrophoresis 20, 3551–356734 Murray, J., Gilkerson, R. and Capaldi, R. A. (2002) FEBS Lett.
529, 173–17835 Capaldi, R. A., Halphen, D. G., Zhang, Y. Z. and Yanamura, W.
(1988) J. Bioenerg. Biomembr. 20, 291–31136 Taanman, J. W., Hall, R. E., Tang, C., Marusich, M. F., Kennaway,
N. G. and Capaldi, R. A. (1993) Biochim. Biophys. Acta 1225,95–100
37 Anthony, G., Stroh, A., Lottspeich, F. and Kadenbach, B. (1990)FEBS Lett. 277, 97–100
38 Cruciat, C. M., Brunner, S., Baumann, F., Neupert, W. andStuart, R. A. (2000) J. Biol. Chem. 275, 18093–18098
39 Schagger, H. and Pfeiffer, K. (2000) EMBO J. 19, 1777–1783
40 Tschischka, K., Abele, D. and Portner, H. O. (2000) J. Exp. Biol.203, 3355–3368
41 Rossignol, R., Gilkerson, R., Aggeler, R., Yamagata, K.,Remington, S. J. and Capaldi, R. A. (2004) Cancer Res. 64,985–993
42 Musatov, A., Carroll, C. A., Liu, Y. C., Henderson, G. I.,Weintraub, S. T. and Robinson, N. C. (2002) Biochemistry41, 8212–8220
43 Brinkman, K., Smeitink, J. A., Romijn, J. A. and Reiss, P. (1999)Lancet 354, 1112–1115
44 Collins, M. L., Sondel, N., Cesar, D. and Hellerstein, M. K. (2004)J. Acquir. Immune Defic. Syndr. 37, 1132–1139
45 Schapira, A. H. (2002) J. Inherit. Metab. Dis. 25, 207–21446 Shoubridge, E. A. (2001) Am. J. Med. Genet. 106, 46–5247 Marusich, M. F., Robinson, B. H., Taanman, J. W., Kim, S. J.,
Schillace, R., Smith, J. L. and Capaldi, R. A. (1997) Biochim.Biophys. Acta 1362, 145–159
48 Hanson, B. J., Carrozzo, R., Piemonte, F., Tessa, A., Robinson,B. H. and Capaldi, R. A. (2001) J. Biol. Chem. 276,16296–16301
Received 6 November 2006/12 February 2007; accepted 18 May 2007Published as Immediate Publication 18 May 2007, doi:10.1042/BA20060223
C© 2007 Portland Press Ltd





























