Embed Size (px)
Citation preview

1.0 INTRODUCTION
Cancer is a broad group of various diseases characterized by
unregulated, uncontrolled abnormal cell proliferation. In a normal
healthy body, cells grow, die, and are replaced in a very controlled
manner. This process of replication is carefully initiated and
terminated to generate a specific amount of cells by a cascade of
signal rendering and silencing mechanisms mediated by growth
factors, inhibitors, and other associated molecules bearing in a
multi-step fashion signals to the nucleus with each step along the
way well monitored and controlled. This ordered signal transduction
sends to the nucleus the need to grow and divide or the need to stop
growing. Changes in the genetic material of cells (mutation) by
environment or internal factors sometimes result in a loss of
restraint on cell proliferation, culminating in cells that do not
die (loss of apoptosis), or that continue to multiply until a mass
of cancer cells or tumor developed. A portion of the mass of
cancerous cells can detach, enters the blood stream or the lymphatic
system, transport and start a new colony elsewhere and thereby
invade and destroy surrounding healthy tissues and organs via a
process called metastasis. Metastasis is a noxious character of
malignant cancers. Benign cancers on the other hand are less
offensive; they do not grow uncontrollably, do not invade
neighboring tissues, and do not spread throughout the body.
NORMAL CELL COMMUNITYWITH A TIGHT LEASH
CANCER CELLS FORM TUMORWHICH CONTINUES TO ANGIOGENESIS SUPPLIES
TUMOR CELLS WITH MORE
Figure 1: PICTORIAL DEPICTION OF HOW CANCER CELLS GROW AND INVADESURROUNDING TISSUES AND ORGANS

1.1 CANCER AS A MENACE
There are over 200 different known cancers that afflict humans. In
2007, cancer caused about 13% of all human deaths worldwide
(7.9 million). Rates are rising as more people age and as mass
lifestyle changes occur in the developing world. In 2009, three
hundred and twenty thousand, four hundred and sixty seven (320,467)
new cases of cancer were diagnosed in the UK (Jemal et al., 2011).
In the US, one in every four people dies of cancer. In the UK, more
than one in three people will develop some form of cancer during
their lifetime (NHS health apps Library).
CANCER CELLS MIGRATEVIA BLOODSTREAM CANCER CELLS INVADE
TISSUES AND ORGANSA HEALTHY BODY BREAKS
DOWN

To put an end to this menace therefore requires exploiting every
process known to be involved with Cancer. Interestingly many if not
all of the peculiar adaptive processes employ by cancer cells from
pre-malignant stage to metastatic and invasive stage sits on one key
metabolic figure; SUGAR. The metabolism of sugar plays a central
role in tumorigenesis. Exploiting sugar in cancer therapy is
therefore a reasonable target in the quest to end the scourge of
cancer.
Also targeted in cancer therapy include signal transduction in
cells, cell division, DNA replication, transcription, and
translation, metastasis, cell death (apoptosis), angiogenesis etc
all of which still have one or more connection to sugar as a major
participant in their metabolic pathway. While exploiting signal
transduction seeks to active and deploy speed breakers (e.g.
tyrosine kinase inhibitors) and brakes on cell proliferation by
finding a way of de-amplifying multiplying signals in the cell,
amplifying contact, some studies try to exploit mutations of proto
Oncogenes (genes liable to become mutated and cause cancer),
oncogenes, tumor suppressor genes etc.

However, the main quest here is find to seek a way of exploiting
sugar-the body primary fuel, in the diagnosis and treatment of
cancer.
CHAPTER TWO: KEY EVENTS WITHIN THE MASS OF CANCER CELLS
Having established the fact that a tumor is a mass of cancer cells,
researchers endeavor to expose the happenings within a tumor
community and seek to exploit these occurrences in cancer diagnosis
and treatment. Some of the usual happenings within a tumor community
include the followings;
2.1 ANGIOGENESIS
Angiogenesis is the physiological process through which new blood
vessels form from pre-existing vessels by dividing (the blood vessel
splitting into two wall to wall) or by growing branches called
sprouts, the latter is used by cancer cells in new blood vessels
formation with the new vessels shooting out from the existing ones
into the tumor. Angiogenesis is different from vasculogenesis in
that with vasculogenesis, entirely new i.e. de novo-formation of blood

vessels takes place especially where there are no pre-existing blood
vessels e.g. in the developing embryo (Flamme et al., 1997).
The development of blood vessels is an essential step in the growth
of a tumor. Key players in this process are growth factors capable
of stimulating blood vessels formation. Some notable growth factors
are fibroblast growth factor (FGF) (Ornitz DM, Itoh N., 2001) and
vascular endothelial growth factor (VEGF) tasked with the job of
inducing capillary growth into tumor, which in turn supply required
nutrients, allowing the tumor to continue expanding. Angiogenesis
also serves as a waste disposal, taking away the biological end
products secreted by rapidly dividing cancer cells. Whether
supplying nutrients or disposing waste, angiogenesis is a necessary
and required step for transition from a small harmless cluster of
cells (often said to be about the size of the metal ball at the end of a ball-point pen) to
a large tumor.
Angiogenesis is also required for the spread of a tumor, or
metastasis. Single cancer cells can break away from an established
solid tumor, enter the blood vessel, and be carried to a distant
site, where they can implant and begin the growth of a secondary
tumor. Evidence now suggests the blood vessel in a given solid tumor

may, in fact, be mosaic vessels, composed of endothelial cells and
tumor cells. This allows for substantial shedding of tumor cells
into the vasculature, possibly contributing to the appearance of
circulating tumor cells in the peripheral blood of patients with
malignancies. The subsequent growth of such metastases will also
require a supply of nutrients and oxygen and a waste disposal
pathway, therefore the chaotic process continues (W. Jeffrey Allard
et al. 2004).
Adaptation of cancerous cells causes mutations that destroy or
snuff out processes that are against angiogenesis, for instance the
inhibition of the anti-VEGF enzyme PKG. In normal cells PKG
apparently limits beta-catenin-a pro-angiogenetic substance (Bagri,
A; et al 2010).
The blood vessels created in this way are not exactly the same as
normal blood vessels. It is believed tumor blood vessels are not
smooth like normal tissues, and are not ordered sufficiently to give
oxygen effectively to all of the tissues (Risau, W; Flamme, I.,
1995).
2.1.1 EXPLOITING ANGIOGENESIS IN CANCER TREATMENT

Hindering the supply of nutrients e.g. glucose and oxygen to the
growing mass of cancer cells by the use of anti-angiogenic
substances is a naturally reasonable way of limiting cancer growth.
Since glucose and it’s metabolic processes seem to be central to the
very nature of cancer. The first line of defense against cancer may
be cutting off the supply of sugar to the cancer cells. This may be
achieved by inhibiting angiogenesis in cancer cells or by inhibiting
glucose transporters needed to transport glucose into the rebel
cells e.g. GLUT1.
On the prospect of cutting off the supply by targeting angiogenesis,
research has shown that endothelial cells are genetically more
stable than cancer cells-the latter assume the defensive position of
adapting to all assault-doing this requires a high level of
instability. This genomic stability of endothelial cells gives an
advantage of targeting endothelial cells using anti-angiogenic
therapy, which may be more effective in comparison to chemotherapy
directed at cancer cells as cancer cells rapidly mutate and acquire
'drug resistance' to treatment, endothelial cells are more stable
making them an ideal target for therapies directed against them
(Bagri, A.; et al 2010).

Angiogenesis research is therefore a cutting-edge field in cancer
research. Another application of endothelial cells genetic stability
is the use of radiation therapy on endothelial cell compartment,
rather than on tumor cell compartment. This may be effective as new
blood vessel formation is a relatively fragile process, subject to
disruptive interference at several levels. Again, since tumor cells
evolve resistance and adapt to an otherwise lethal environment
rapidly due to their rapid generation time (days) and genomic
instability (variation), endothelial cells are better targets of
radiation therapy as they possess long generation time (months),
genomic stability (low variation), and thus cannot quickly adapt to
hazardous environment to resist the killing agents (Brown JM,
Giaccia AJ, 1998).
Interestingly, these ideas perfectly depict natural selection in
action at the cellular level, using a selection pressure to target
and differentiate between varying populations of cells. The end
result is the extinction of one species or population of cells
(endothelial cells), followed by the collapse of the ecosystem (the
tumor) due either to nutrient deprivation or self-pollution from the
destruction of necessary waste pathways.

Angiogenesis-based tumor therapy presently relies on natural and
synthetic angiogenesis inhibitors like angiostatin, endostatin and
tumstatin. These are proteins that mainly originate as specific
fragments of pre-existing structural proteins like collagen or
plasminogen.
FDA-approved therapy targeted at angiogenesis in cancer is on the
market in the US. This is a monoclonal antibody directed against an
isoform of VEGF. The commercial name of this antibody is Avastin,
and the therapy approved for use in colorectal cancer in combination
with established chemotherapy.
2.2 HYPOXIA-AN IMPORTANT FEATURE OF CANCER
Control of cellular oxygen concentration under normal physiological
condition is very strict as the body tries to keep the oxygen
concentration within a normal physiological range termed normoxia,
and putting mechanisms in place to forestall oxygen concentration
from getting too high; hyperoxia or from getting too low; hypoxia.
Hyperoxia causes damage secondary to reactive oxygen species while
hypoxia leads to activation of a diverse array of downstream
transcriptional pathways including angiogenesis, glucose metabolism
and apoptosis (C Thirlwell, et al, 2011).

Despite the strict regulation of cellular oxygen concentration, the
unrestrained cell multiplication of cancer cells generates more
cells to be “fed” oxygen and glucose; this necessitates an urgent
angiogenesis which generates impaired microvasculature for
micronutrient supply and oxygenation. As this vasculature develops
in a chaotic way with structural malformations, regions of hypoxia
are present within all solid tumors. Normal oxygen tension in
healthy tissue is 7% (53 mmHg), levels of oxygenation in tumors may
vary from physiological levels (7%) to severe hypoxia (< 1%) which
is usually found in areas adjacent to necrotic tissue (Heddleston
JM, et al, 2010).
Within the same region of a given tumor, levels of oxygenation may
cycle due to poor vasculature and limited oxygen diffusion,
resulting in intermittent periods of hypoxia (Heddleston JM, et al,
2010). These cyclical episodes of hypoxia lead to increased
metastatic potential of cancer cells (Cairns RA, et al, 2001).
Hypoxia is therefore a promoter of cancer progression, invasion and
metastasis (Pennacchietti S; et al 2003).
Hypoxia may also be responsible for the voracious appetite of cancer
cells for glucose because intra-tumoral hypoxia causes the hypoxia-

inducible factor (HIF)1α pathway to be activated in many tumors
cells, resulting in the direct up-regulation of lactate
dehydrogenase (LDH) and increased glucose consumption (Cairns RA, et
al 2011.).
Another important effect of hypoxia is autophagy or self-eating-
process by which cells degrade own cellular components to survive
during starvation or to eliminate damaged organelles after oxidative
stress. Mitophagy, or mitochondrial-autophagy, is particularly
important to remove damaged or reactive oxygen species (ROS)-
generating mitochondria or a result of cancer genes shutting down
the mitochondria because they are involved in the cell's apoptosis
program which would otherwise kill cancerous cells. An autophagy or
mitophagy program can be triggered by hypoxia (Azad MB, Gibson SB;
2010).
Degrading the mitochondria in this way may explain the lack of
apoptosis in cancer cells. The hypoxia inducible factor can be said
to be the mediator and key promoter of many characteristics of
cancer cells, being involved in angiogenesis, up regulation of
glycolytic enzymes, cancer’s genetic instability making cancer cells
evolve and remain adamant to therapy and the loss of apoptosis.
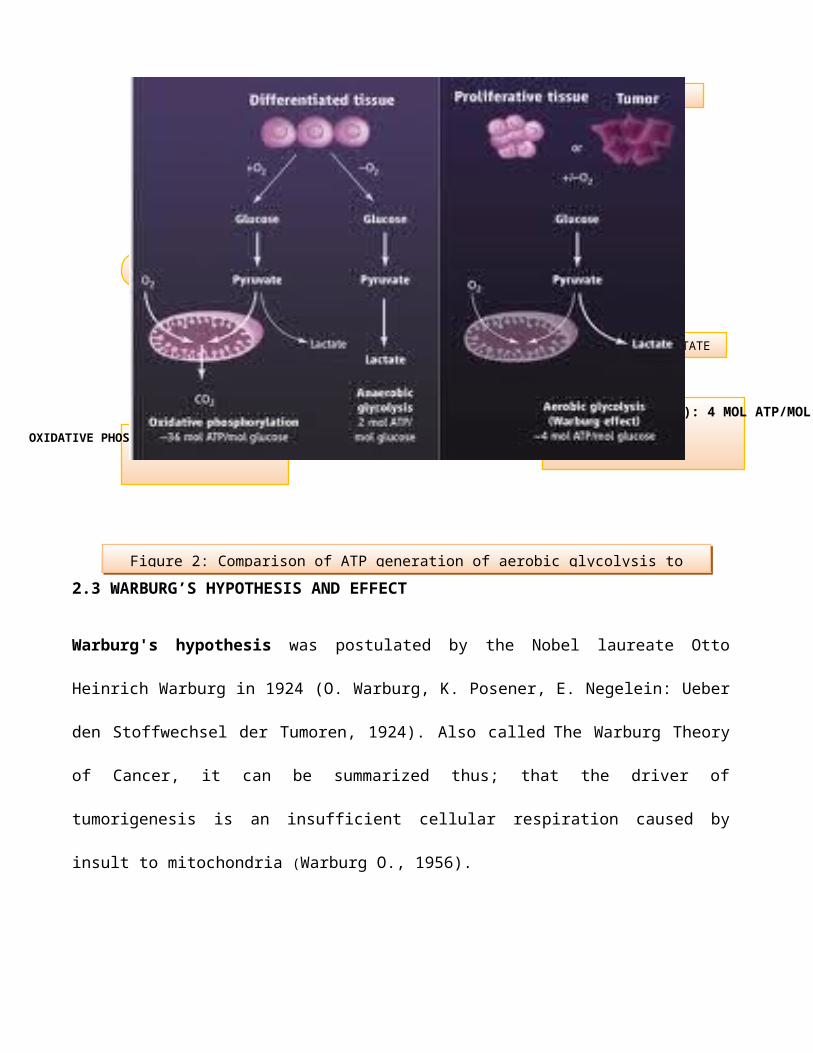
DIFFERENTIATED TISSUE
GLUCOSE
PYRUVATE PYRUVATE PYRUVATE
LACTATELACTATE
OXIDATIVE PHOSPHORYLATION: 36 MOL ATP/MOL GLUCOSE
ANAEROBIC GLYCOLYSIS: 2 MOL ATP/MOL GLUCOSEAEROBIC GLYCOLYSIS (WARBURG EFFECT): 4 MOL ATP/MOL GLUCOSE
-O2+O2
GLUCOSE
TUMOR
+/-O2
CO2
O2 O2
2.3 WARBURG’S HYPOTHESIS AND EFFECT
Warburg's hypothesis was postulated by the Nobel laureate Otto
Heinrich Warburg in 1924 (O. Warburg, K. Posener, E. Negelein: Ueber
den Stoffwechsel der Tumoren, 1924). Also called The Warburg Theory
of Cancer, it can be summarized thus; that the driver of
tumorigenesis is an insufficient cellular respiration caused by
insult to mitochondria (Warburg O., 1956).
PROLIFERATIVE
Figure 2: Comparison of ATP generation of aerobic glycolysis to
Oxidative phosphorylation36mol ATP/mol glucose
Anaerobic glycolysis 2 mol ATP/mol glucose
Aerobic glycolysis (Warburg effect)4mol ATP/mol glucose
Warburg hypothesized that cancer, malignant growth, and tumor growth
are caused by the fact that tumor cells mainly generate energy (as
e.g. adenosine triphosphate ATP) by non-oxidative breakdown of
glucose (glycolysis). This is in contrast to "healthy" cells which
mainly generate energy from oxidative breakdown of pyruvate.
Pyruvate is an end-product of glycolysis, and is oxidized within the
mitochondria. Hence, according to Warburg, the driver of cancer
cells should be interpreted as stemming from a lowering of
mitochondrial respiration. Warburg reported a fundamental difference
between normal and cancerous cells to be the ratio of glycolysis to
respiration; this observation is also known as the Warburg effect.
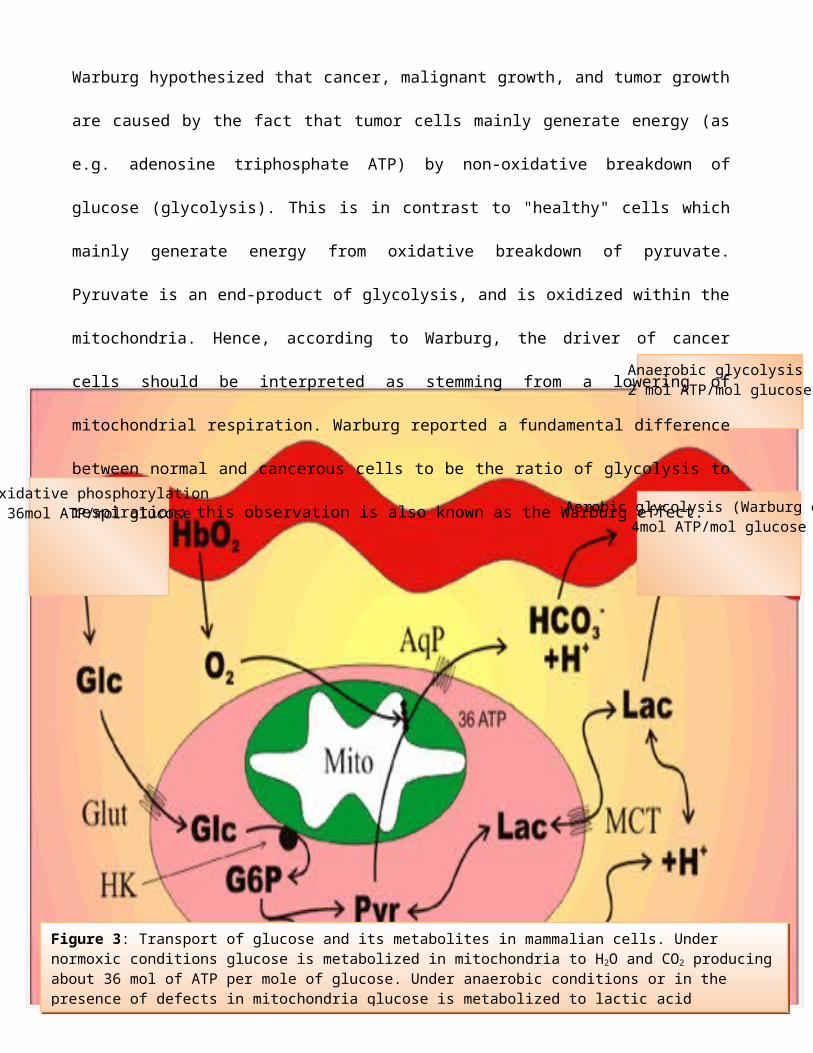
Figure 3: Transport of glucose and its metabolites in mammalian cells. Under normoxic conditions glucose is metabolized in mitochondria to H2O and CO2 producingabout 36 mol of ATP per mole of glucose. Under anaerobic conditions or in the presence of defects in mitochondria glucose is metabolized to lactic acid

The Warburg effect or aerobic glycolysis can be defined as the
propensity of cancer cells to take up high levels of glucose and to
secrete lactate in the presence of oxygen (Warburg O., 1956; Dang
CV., 2009).
The Warburg Effect describes the observation that cancer cells, and
many cells grown in-vitro, exhibit glucose fermentation even when
enough oxygen is present to properly respire. In other words,
instead of fully respiring in the presence of adequate oxygen,
cancer cells ferment. The current popular opinion is that cancer
cells ferment glucose while keeping up the same level of respiration
that was present before the process of carcinogenesis (Vazquez, A.,
et al, 2010).
That cancer cells consistently use more glucose and produce more
lactic acid than normal tissue may represents adaptation to intra-
tumoral hypoxia due to disordered angiogenesis and blood flow
(explained above), with recent researches it is revealed that much
seems to be due to the curious phenomenon of “aerobic glycolysis”—
i.e. continued use of the glycolytic metabolic pathway even in the
presence of adequate oxygen. Warburg explained his observation with
his hypothesis that proposed aerobic glycolysis was a manifestation

of a deficiency in respiration. However, extensive research clearly
demonstrated this to be untrue.
From a casual and hasty perception, aerobic glycolysis should not
contribute positively to cell’s fitness and survival, in fact it
should do more damage than good to the survival of cancer cells
because; it is much less efficient in energy production than is
aerobic metabolism—specifically glycolysis produces only 2 mol
ATP/mole of glucose while oxidative metabolism of glucose results in
about 36 molATP/mole of glucose; also glycolysis increases acid
production resulting in a highly acidic extra-cellular environment.
This should result in local toxicity including cell death and extra-
cellular matrix degradation due to release of proteolytic enzymes
(Williams, A. C., Collard, T. J., & Paraskeva, C., 1999; Park, H.
J., et al, 1990; Rohzin, J., et al, 1994; Cuvier, C., Jang,
A.,&Hill, R. P., 1997).
How then do cancer cells survive if increase glycolysis is a shot in
the leg for them? A deeper study of cancer cells sugar metabolism
reveal how cancer cells beat the odds through sugar metabolism;
2.4 SUGAR METABOLISM AND CANCER CELLS SURVIVAL

Cancer cells strategy to overcoming consistent environmental
pressures is the up-regulated glycolytic metabolic pathways (Gatenby
& Gillies, 2004).
Under hazardous conditions (hypoxia and acidosis), cellular traits
that promote constitutive up-regulation of glycolysis and resistance
to acid-induced toxicity occur in cancer cells. Mathematical models
and empirical observation suggest the growth advantage conferred by
this combination of phenotypic traits is substantial because the
cells create an acidic local environment (due to up-regulated
glycolysis) that is toxic to other cells (Gatenby & Gawlinski,
1996).
There is some evidence that this advantage along with acid-induced
degradation of extracellular matrix is critical for tumor invasion
into normal host tissue (Gatenby et al., 2006).
Studying extensively the multi-faceted manipulative use of sugar
metabolism by cancer cells opens our eyes to the elusive nature of
this menace greatly. A very interesting idea suggested that
increased sugar metabolism including glycolysis (which has cropped
up as a chief culprit in cancer survival plot) and also pentose
phosphate metabolism may have a way of conferring other measures of

adaptive stone walling power to cancer cells, if it allows excess
pyruvate to be available for lipid synthesis or providing essential
anabolic substrates, such as ribose for nucleic acid synthesis
(Homem de Bittencourt, Peres, Yano, Hiratea, & Curi, 1993).
Glucose consumption through the pentose pathway may also provide
essential reducing equivalents (NADPH) to reduce the toxicity of
reactive oxygen species conferring resistance (Kondoh, Lieonart,
Bernard, & Gil, 2007; Kondoh, Lieonart, Gil, Beach, & Peters, 2005).
These evolutionary advantages can explain the remarkable prevalence
of the glycolytic phenotype in human cancers and the otherwise
puzzling observation that malignant cells remain glycolytic even in
the presence of normoxia. This conceptual model is supported by
empirical studies that demonstrate constitutive upregulation of
glycolysis is consistently observed during the transition from
premalignant lesions and invasive cancer (Gambhir, 2002; Abbey et
al, 2006; Yasuda et al., 2001; Abbey et al., 2004).
The molecular basis for evolution of the glycolytic phenotype has
been clarified by recent advances in understanding the hypoxia-
inducible factor, HIF-1 system (Ryan, Lo, & Johnson, 1998).

It is clear that up-regulation of the HIF system elicits a multi-
phasic response that includes increased expression of components of
the glycolytic pathways including membrane glucose transporters
(Greijer et al., 2005). At present over 70 genes directly regulated
by HIF have been identified and several hundred are directly or
indirectly influenced by HIF. HIF stabilization occurs in response
to diminished oxygen concentrations.
Interestingly, increased levels of pyruvate (a glycolytic product)
also stabilize HIF providing a potential feed forward loop which may
be critical to aerobic glycolysis (Vangellur, Phillips, Bogenesch, &
LaPres, 2005; Kim, Tchernyshyov, Semenza, & Dang, 2006). HIF
activity can also be stabilized in the presence of oxygen by growth
Figure 4: A summary of the HIF system demonstrating several factors whichgovern the level of HIF-1 and some of its protean effects on cell
metabolism, survival, proliferation and their microenvironment which

factors that also participate in carcinogenesis, including ras, HSP
90, Cox 2, HER, and the AKT/mTOR pathway (Sang, Stiehl, et al,
2003).
Cells with loss of HIF activity demonstrate reduced (but not
completely absent) expression of glucose transporter and glycolytic
enzymes in response to hypoxia when compared to normal (Vengellur,
Woods, Ryan, Johnson, & LaPres, 2003).
Interestingly, HIF1 activation can also lead to expression of cell
death factors so that full activation by severe hypoxia can also
lead to apoptosis and necrosis (Burke et al., 2003; Cramer et al.,
2003). Clearly, much additional work will be required to fully
understand the complex mechanisms involved in hypoxic response,
metabolic controls, and adaptation to acidosis in cancer
progression. Despite gaps in understanding, the complex pathways
that result in increased glycolytic metabolism in the vast majority
of human cancers provide multiple potential targets for treatment
strategies designed to alter tumor glucose metabolism (Semenza,
2003).
Since this phenotype emerges during carcinogenesis, it may represent
a possible target in cancer prevention. In advanced metastatic

cancers, understanding of the molecular and physiological causes and
consequences of upregulated glycolysis may lead to targeted
therapies.
CHAPTER THREE: EXPLOITING SUGAR IN CANCER DIAGNOSIS AND THERAPY
3.1 EXPLOITING SUGAR IN CANCER DIAGNOSIS
The Warburg effect has important medical applications, as high
aerobic glycolysis by malignant tumors is exploited clinically to
diagnose and monitor treatment responses of cancers by imaging
uptake of 2-18F-2-deoxyglucose (FDG) (a radioactive modified
hexokinase substrate) with positron emission tomography (PET).
Positron Emission Tomography (PET) is a nuclear imaging technique
that takes the advantage of the fact that cancer cells absorb
glucose more than normal cells because of their dependence on low

energy yielding aerobic glycolysis, which extract only about 5% of
the available energy in the glucose, calling for more glucose
absorption. This voracious appetite for glucose is taken advantage
of in the use of radioactive tracers that allows delving deeper into
the physiologic processes occurring intracellularly. PET primarily
looks at one specific tracer the 18F-labeled glucose analog 2-
deoxyglucose (2DG) (Zhang et al., 2006), which can be administered
through a vein and observed with the PET scanner after a while. The
PET scanner is used to take pictures that show the use of the
radioactively label glucose by different organs and tissues in the
body. Cancer cells use more glucose than normal cells, and the
ability to detect that on an image is a way of diagnosing the
presence of cancer-and evaluating tumor size and a very effective
way of doing that.
One of the hurdles FDG-PET has helped to scale is the pre-operative
staging of the carcinoma of the stomach which has a poor prognosis
because many patients have advanced disease at the time of diagnosis
(Siewert et al, 1998). Therefore, pretreatment assessment and
staging of disease is essential for managing gastric carcinoma. The
tumor stage provides the basis for selecting the most appropriate

therapeutic strategy (Fleming et al., 1997). Preoperative staging
currently relies on a standard noninvasive imaging modality of
spiral-computed tomography (CT) of the abdomen and pelvis. However,
CT is an anatomy-based diagnostic technique with certain drawbacks,
including limited sensitivity from falsenegative findings due to
non-enlarged invaded lymph nodes and limited specificity from false-
positive findings due to enlarged inflammatory lymph nodes.
Therefore, a better preoperative evaluation strategy would greatly
aid the preparation of treatment plans for patients with gastric
carcinoma. Positron emission tomography (PET) using the radiolabeled
glucose analogue, 18 F-fluorodeoxyglucose (FDG), as a tracer is a well
established imaging technique that offers new perspectives in
staging malignant diseases. FDG-PET scanning enables observation of
altered glucose metabolism in neoplastic cells (Tschmelitsch et al,
2000). Images from PET scanners are complementary to traditional
morphologic images, such as those produced by CT, and may be more
sensitive because functional changes often precede anatomic changes.
Jian Chen et al in a research report on the application of FDG-PET
in the pre-operative staging of gastric adenocarcinoma that proved
that FDG-PET improves the preoperative TNM staging of gastric

adenocarcinoma. Based on its superior specificity, FDG-PET can
facilitate the selection of patients for a curative resection by
confirming a nodal status identified by CT (Jian Chen et al, 2005).
Based on the work of Jian Chen et al, PET may play a complementary
role in pretreatment evaluation, as results of this research showed
FDG-PET upstaged 6% patients from the false-negative CT findings,
and downstaged 9% patients from CT false-positive findings.Also, PET
provided important information for making decisions regarding
treatment; splenectomy was performed in patient(s) confirmed to have
a spleen metastasis, and lymphadenectomy was performed in another
patient. Therefore, those patients who benefited from the FDG-PET
detection method were treated with a timely curative resection,
without the need for any extra neoadjuvant chemotherapy.
3.2 EXPLOITING SUGAR IN CANCER TREATMENT
Cancer cells with increased aerobic glycolysis due to activated
oncogenes (including Ras, Her-2, and Akt) or loss of tumor
suppressor function (including TCS1/2, p53, LKB1) has been shown to

undergo rapid apoptosis when placed in culture conditions with low
glucose concentrations (Inoki, Zhu, & Guan, 2003; Xu et al., 2005;
Jones et al., 2005). If such low glucose concentration environment
can therefore be created through glucose deprivation, it is expected
that that will lead to cancer cells apoptosis. In human patients,
these culture conditions could be mimicked in vivo by transiently
reducing blood and interstitial glucose concentrations through
administration of insulin, diet alteration and lifestyle adjustment.
3.2.1 ANTI-GLYCOLYTIC MEASURES
Since cancer cells rely on anaerobic metabolism to produce a
variable but generally significant portion of their energy
requirements, inhibition of the glycolytic pathway is an obvious
approach that may exploit the high glucose consumption by cancer
cells. There are evidences that support this approach; that
inhibition of glycolysis can result in cancer cell’s death
particularly in hypoxic environment due to ATP depletion (Jones et
al., 2005).
3.2.1.1 BROMOPYRUVATE: HEXOKINASE AND G-3-P DEHYDROGENASE INHIBITOR

An obvious therapeutic target in the glycolytic pathways is
hexokinase which catalyzes phosphorylation of glucose to glucose-6-
phosphate, the first and rate-limiting step in glucose metabolism.
Bromopyruvic acid is a synthetic brominated derivative of pyruvic acid. It
is being studied as a potential treatment for certain types of cancer.
Initial studies in laboratory animals’ researchers at Johns Hopkins showed
that bromopyruvic acid is effective at eliminating aggressive liver tumors
(Ko et al, 2001).
A study stated that 3-Bromopyruvate markedly reduce ATP
concentrations leading to cytotoxicicity in tumor cells that are
hypoxic or possess mitochondrial defects by inhibiting hexokinase
(Gerschwind et al, 2002).
Another study observed that the mechanism of action of bromopyruvic
acid involves interruption of the glycolytic process by the
inhibition of the enzyme Glyceraldehyde 3-phosphate-dehydrogenase
(Ganapathy-Kanniappan, S; Geschwind, JF 2012).
The research group of J.F. Geschwind reported that intra-arterial
delivery of bromopyruvic acid directly to the site of a tumor
represents a new strategy for stopping the growth of liver cancer
while minimizing toxic side-effects (Geschwind JF, et al. 2002). The

first clinical use of 3-bromopyruvate to treat patients with late
stage liver cancer was reported to have taken place in 2010 under a
compassionate use protocol, where a total of two patients received
3-bromopyruvate intra-arterially. The patients were treated using a
special patented formulation of 3-bromopyruvate invented by Ko,
Pederson and Geschwind.
3.2.1.2 LONIDAMINE
Lonidamine, a derivative of indazole-3-carboxylinc acid decreases
glycolysis in vitro and in vivo probably through inhibition of
mitochondrial bound hexokinase (Floridi et al., 1981). Lonidamine
has been shown to decrease intra-cellular ATP and lactate production
in cancer cells but, interestingly, appears to enhance aerobic
glycolysis in non-transformed cells. Although lonidamine is
Figure 5: The structure of 3-bromopyruvic acid: a potent inhibitor ofG-3-P dehydrogenase and Hexokinase
IUPAC name: 3-Bromo-2-oxopropanoic acid

currently under investigation as a primary therapy for benign
prostatic hypertrophy, its main role in malignant therapy is in
combination with cytotoxic drugs.
3.2.1.3 MANNOHEPTULOSE
Mannoheptulose- a heptose, a monosaccharide with seven carbon atoms
is a hexokinase inhibitor. By occupying the active site of
glucokinase, it prevents glucose phosphorylation thus inhibiting
glycolysis. It is naturally found as D-mannoheptulose in avocado
plants (Persea americana) (Dai et al., 1999). A purified avocado extract
of D-mannoheptulose was added to a number of tumor cell lines tested in
vitro by researchers in the Department of Biochemistry at Oxford University
in Britain, their findings showed that it inhibited tumor cell glucose
uptake by 25 to 75 percent, their findings also show it has a inhibitory
effect on the enzyme glucokinase and on the growth rate of the cultured
tumor cell lines. Lab animals were administered 1.7 mg/g body weight dose
of mannoheptulose for five days and it reduced tumors by 65 to 79 percent
(Board et al., 1995) Based on these studies, there is good reason to
believe that avocado extract could help cancer patients by limiting glucose
use of the cancer cells.
Figure 6: D-Mannoheptulose-a heptosewith glucokinase inhibitory activity.
IUPAC Name: D-Manno-hept-2-ulose

3.2.2 INHIBITING THE ALTERNATIVE SUGAR SOURCE: GLUCONEOGENESIS
Glucose can be made by cells via gluconeogenesis. Joseph Gold, M.D.,
director of the Syracuse (N.Y.) Cancer Research Institute and former
U.S. Air Force research physician, surmised that a chemical called
hydrazine sulfate, used in rocket fuel, could inhibit the excessive
gluconeogenesis (making sugar from amino acids) that occurs in
cachectic cancer patients. Gold's work demonstrated hydrazine
sulfate's ability to slow and reverse cachexia in advanced cancer
patients. A placebo-controlled trial followed 101 cancer patients
taking either 6 mg hydrazine sulfate three times/day or placebo.
After one month, 83 percent of hydrazine sulfate patients increased
their weight, compared to 53 percent on placebo (Chlebowski et al.,
1987). A similar study by the same principal researchers backed by
the National Cancer Institute, Bethesda, followed 65 patients;
hydrazine sulfate was shown to potentiate the lives of those it was
administered to by 17 weeks (Chlebowski et al., 1990).
3.2.2 USING GLUCOSE ANALOGS

2-deoxy glucose has been shown to compete with glucose for trans-
membrane transport, making it useful not just in imaging techniques
but also in limiting glucose absorption. 2DG will not inhibit
glucokinase, infact it is bound by the enzyme and phosphorylated
just like normal glucose-to 2-deoxy glucose phosphate which cannot
be further metabolized. This is employed in the imaging technique
using the 18F labeled 2DG, as the accumulation of this metabolite in
the tumor cells is an obvious signal of the massive appetite that
cancer cells display for glucose or its derivatives. Several studies
have confirmed that 2DG depletes intra-cellular ATP leading to cell
death in vitro (Zhang et al., 2006). it is currently being explored
as an agent to enhance the therapeutic effects of radiation therapy
or cytotoxic drugs such as adriamycin and paclitaxel (Maschek et
al., 2004).
3.2.2 EXPLOITING HIF1 AND OTHER GENES
HIF1 is an essential regulator of adaptation to low oxygen levels.
It is a heterodimer composed of an oxygen regulated α-subunit and a
constitutively expressed β-subunit. The abundance of α subunit is
primarily regulated by a family of prolyl hydroxylases. In normoxia,
prolyl hydroxylase is activated and it directs the degradation of

the α subunit by ubiquiting proteasome pathway (McNeil et al.,
2002). Under hypoxia, porlyl hydroxylase activity decreases which
leads to the stabilization and translocation to the nucleus α
subunit which then heterodimerize with the β-subunit. HIF1 acitivity
is primarily regulated by the abundance of the HIF1- α subunit. HIF1
has also been shown to regulated many pro inflammatory genes such as
tumor necrosis factor- α (TNF- α), interleukin-8 (IL-8) and
vascular endothelial growth factor A (VEGF-A)-which is important to
angiogenesis. HIF1 promotes he expression of phosphofructokinase,
lactose dehydrogenase etc (Semenza GL, 2000).
Having establish the critical role of the HIF system in upregulating
glycolysis as an adaptation to hypoxia and non-adaptively in cancer
(producing aerobic glycolysis) has resulted in considerable interest
in developing inhibiting strategies. Exploiting HIF1 can be achieved
by direct influence; altering transcription or degradation, or by
indirect alterations of HIF by targeting control proteins and also
by alteration of effector proteins (Lee et al., 2006).
Probably the most direct of these is simply inhibiting the
transcription of HIF. PX-478, a recently developed drug that

specifically targets HIF expression, and has demonstrated anti-tumor
effects in animal models (Macpherson & Figg, 2004).
A number of treatment candidates have been developed to indirectly
influence HIF levels by targeting non-hypoxic controlling pathways.
This includes drugs such as CCI-779, which inhibits expression of
Mtor and consequently reduces HIF expression (Georger et al., 2001).
Similarly, drugs that alter expression of heat shock protein (Hsp90)
such as geldanamycin and apigenin also influence HIF1 expression.
Tumor suppressor genes are normal genes that slow down cell
division, repair DNA mistakes, or signal to the cells when to die (a
process known as apoptosis or programmed cell death). When tumor
suppressor genes don't work properly, cells can grow out of control,
which can lead to cancer. Treating problems in tumor suppressor
genes is more difficult. It would mean restoring normal
tumor suppressor gene functions, which researchers have not yet
figured out how to do effectively.
A major stumbling block lies in how to get new DNA into the cancer
cells. Another problem is that

most cancers have several mutations, so replacing one gene may not
be enough to stop the cancer
cells from growing and spreading.
One major breakthrough was achieved by inserting normal TP53 genes
into viruses and then trying to infect tumor cells with these
viruses. This worked well in the lab, but not in human studies. A
newer approach targets the weakness in the cell caused by the
abnormal tumor suppressor genes, rather than trying to restore
normal gene function. For example, some people inherit a mutation in
one of the BRCA genes (BRCA1 or BRCA2). If the second copy of this
gene is damaged, the gene no longer works and they may develop a
cancer. In cells where a BRCA gene no longer works (like cancer
cells), drugs called PARP inhibitors cause DNA damage that can lead
to cell death. Cells that have normally functioning BRCA genes can
repair this damage. This allows the PARP inhibitor to target the
cancer cells while doing little damage to the normal cells.
3.2.3 EXPLOITING ACIDOSIS
One of the consequences of increased glycolysis is acidification of
the extra-cellular space (Gillies et al., 2002). Acidosis in the

extracellular space brings about resistance to acid-mediated
toxicity, and that these enhance tumor growth and invasion. It also
represents, however, an opportunity for therapy because the tumor
microenvironment is typically extremely acidic which, in combination
with hypoxia, may produce significant cellular stress leading to
intratumoral regions of necrosis. Strategies to enhance this
toxicity and extend the regions of necrosis stem from recognition
that acid escapes from tumors by only two mechanisms: venous efflux
and local diffusion. Using mathematical models, it was demonstrated
that venous outflow can be significantly inhibited using even mild
systemic acidosis (Gatenby et al., 2002). This is due to the fact
that acid flow into intratumoral blood vessels is in part dependent
on the concentration gradient between the interstitial space and the
blood. If the acid concentration in the blood is increased, vascular
outflow of acid from the tumor dramatically decreases and the
intratumoral pH declines precipitously. As a result local acid
concentrations exceed the tolerance of even the tumor cells
triggering extensive tumor necrosis. Theoretical results have not
been explicitly tested but some indirect results are supportive.

Harguindey, Henderson, and Naeher (1979) demonstrated tumor-bearing
rats fed acidic chow had a substantial survival advantage when
compared to control (Harguindey et al., 1979).
Gatenby et al. (2002) found that patients who develop renal failure
(which is typically associated with metabolic acidosis) after
cytoreductive nephrectomy for metastatic renal cancer have a
substantial survival advantage compared to those who maintain normal
post operative renal function (survival of 17 months versus 4
months) (Gatenby et al., 2002).
Kelley, Manon, Buerk, Bauer, and Fraker, 2002) used isolate limb
perfusion in a rat model to examine simultaneous perfusion of acid
and melphalan for treating melanoma. They found the combination
resulted in consistent cure of the animals. However, in addition,
they showed that acid perfusion alone (pH of 6.8 for 10 min) induced
extensive apoptosis in the tumors and significant survival benefit
(Kelley et al., 2002).

CHAPTER FOUR: CONCLUSION
Tumorigenesis though beginning like a normal process in the body has
been shown to fascinatingly evolve abnormal but highly defensively
and offensively equipped cells. Finding a loophole to exploit in
cancer treatment must have been daunting over the years, as cancer
cells learn to adapt to different hazardous environment. However,
the evasiveness of cancer cells seems to solely rely on glucose and
glucose metabolism. The Warburg effect may have wide application
many of which still have gaps that need to be filled, but successful
inhibition of glycolytic enzymes especially hexokinase is given a
thumps up in the treatment of cancer. Tumor cells have been shown to
undergo apoptosis in the absence of the desired sugar. Also since a
lot of adaptive tendencies of cancer cells depend on HIF1
expression, which though emerges in response to hypoxia seems confer
on cancer cells the ability to survive. More research works need to
go into exploiting HIF1 then as it is central to tumorigenesis and
cancer development. Possible areas to exploit may be the up
regulation of the expression of von hippel lindau (VHL) protein-a

tumor suppressor capable of degrading HIF1. With HIF1 llimited,
tumorigenesis is highly restricted in terms of angiogenesis-HIF1 is
implicated in the expression of VEGF, expression of glycolytic
enzymes, and the downregulation of the tumor suppressor genes. Also
much of these works may be use in dietary adjustments for cancer
patients. Research works need to be devoted to exploiting sugar in
cancer metabolism as much prospects exist in future cancer therapy
and diagnosis from this perspective.





























