Embed Size (px)
Citation preview

Retinal Abnormalities Characteristic of Inherited RenalDisease
Judy Savige, Sujiva Ratnaike, and Deb Colville
Department of Medicine, Northern Health, The University of Melbourne, Epping, Victoria, Australia
Inherited disease accounts for half ofall children and 1 in 5 adults with end-stage renal failure (Table 1).1 The prev-alence of individual diseases varies from 1in 5 to 10,000 for Alport syndrome andmembranoproliferative glomerulonephri-tis type 2, to 1 in 50,000 for the rarer con-ditions such as Fabry disease.2 The chal-lenge is to identify inherited renal diseasewhen the phenotype is mild or atypical, orwhen there is no family history, which oc-curs with de novo mutations, recessive in-heritance, and dominant disease with vari-able expression or penetrance.
Inherited renal disease is suspectedwhen there is a family history of similarfeatures, or when abnormalities affectmultiple systems without another ob-vious explanation.3 Databases such asORPHANET (http://www.orpha.net) andOMIM (http://www.ncbi.nlm.nih.gov/omim) suggest patterns of organ involve-ment. Many inherited renal diseases havehearing loss (Table 2), and ocular, espe-
cially retinal, abnormalities. Notable ex-ceptions, based on current knowledge, arethe autosomal dominant and recessiveforms of polycystic kidney disease, medul-lary cystic kidney disease, and thin base-ment membrane nephropathy.
WHY THE KIDNEY AND RETINASHARE INVOLVEMENT ININHERITED DISEASES
Inherited renal disease often also in-volves the retina because the kidneyand retina develop at the same embry-onic stage and share developmentalpathways;4 the glomerular filtrationbarrier and retinochoroidal junctionare structurally similar;5 the glomeru-lus and chorioretina are both large cap-illary beds, and the renal epithelial(podocyte) and retinal pigment epithe-lial (RPE) cells are critically dependenton cilia to function.
The Kidney and Eye ShareDevelopmental PathwaysBoth the PAX and WT1 pathways are im-portant in the embryogenesis of the kid-ney and retina.4 The PAX genes encodenuclear transcription factors that controldevelopment of the kidney, eye, ear,brain, vertebral column and limb mus-cles.6 PAX2 is required for developmentof the urogenital tract, eye, ear, andbrain. The WT1 gene is necessary for ure-teric bud formation and retinal ganglioncell differentiation.7,8 Mutations in PAX2result in the renal-coloboma syndromewith vesicoureteric reflux, and WT1 mu-tations produce Wilm’s tumor, and theWAGR, Frasier, and Denys-Drash syn-dromes.9 –11
The Retinochoroidal JunctionResembles the GlomerularFiltration BarrierRPE cells, Bruch’s membrane, and the fe-nestrated choriocapillaries of the retinaresemble the podocytes, glomerular base-ment membrane (GBM), and fenes-trated capillaries of the glomerular tuft,respectively. The epithelial cells have or-gan-specific functions, the basementmembranes support the adjacent struc-tures and form a barrier that prevents the
Published online ahead of print. Publication dateavailable at www.jasn.org.
Correspondence: Dr. Judy Savige, Department ofMedicine, Northern Health, The University of Mel-bourne, The Northern Hospital, Epping, Victoria3076, Australia. Phone: �613 8405 8823; Fax: �6138405 8724; E-mail: [email protected]
Copyright © 2011 by the American Society ofNephrology
ABSTRACTMany inherited renal diseases have retinal features that are helpful diagnosti-cally. These include coloboma, drusen, atrophy and pigmentation (retinitispigmentosa), hamartoma, vascular anomalies, and crystals. Retinal abnormali-ties occur because the kidney and retina share developmental pathways andstructural features including basement membrane collagen IV protomer com-position and their vascularity, and because both the kidney and retina arefunctionally dependent on ciliated cells. Diagnosis of inherited renal disease isimportant because of the risks of further renal and systemic complications, theimplications for other family members, the predictability of the clinical course,and the possibility of treatment. Furthermore, retinal abnormalities may helpexplain the pathogenesis of the renal disease, and can sometimes be used tomonitor its course.
J Am Soc Nephrol 22: 1403–1415, 2011. doi: 10.1681/ASN.2010090965
BRIEF REVIEW www.jasn.org
J Am Soc Nephrol 22: 1403–1415, 2011 ISSN : 1046-6673/2208-1403 1403

passage of macromolecules, and the cap-illaries provide nutrition and removewaste from nearby metabolically activecells. After infancy, the GBM, and the in-ternal limiting and Bruch’s membranes inthe retina comprise the same specializedcollagen IV �3�4�5 protomers.12,13 Muta-tions in the collagen IV genes (COL4A1and COL4A3 through COL4A5) result inHereditary Angiopathy with retinal tortu-osities, Nephropathy, Aneurysms, andmuscular Cramps (HANAC), and inX-linked and the autosomal recessiveforms of Alport syndrome, respectively.
Ciliated CellsCilia transmit mechanosensory, visual,and osmotic stimuli, and both podo-cytes and RPE cells depend on theirprimary cilia for specialized cell func-tions. Mutations affecting proteins in
the podocyte cilia or related structuresresult in cystic kidney disease14 includ-ing nephronophthisis and its variants(the nephrocystins) and Bardet-Biedlsyndrome (BBS1 through BBS12 etc.).The retina is commonly affected,15 andother clinical features include hearingloss, abnormal limb and digit develop-ment, developmental delay, situs inver-sus, liver and respiratory disease, andinfertility.16
RETINAL ABNORMALITIES ININHERITED RENAL DISEASE
Retinal abnormalities in inherited renaldisease include coloboma, drusen, atro-phy and pigmentation (retinitis pigmen-tosa), hamartoma, vascular anomalies,and crystals (Table 3; Figure 1). Thesechanges may also be accompanied by theretinal associations of renal failure suchas hemorrhage,17 arteriolar narrowing,exudates, infarcts,18 calcification,19,20 andmacular degeneration.21
COLOBOMA
Coloboma result from defective clo-sure of the embryonic fissure of the op-tic cup. These defects are typically lo-cated in the lower part of the iris,chorioretina, or optic disc. Optic disccoloboma occur in vesicoureteric re-flux and other structural urinary tractdisease.22 Optic disc and chorioretinaland iris coloboma are found in theCHARGE and COACH syndromes,and possibly in nephronophthisis andtuberous sclerosis.23–26 Coloboma areidentified by careful ophthalmoscopicexamination and photography of bothfundi of the presenting individual andtheir family members. Coloboma aretypically asymmetrical with, for exam-ple, a normal eye or mild defect in oneeye, and a severe central abnormality inthe other.27 Likewise, the severity of thedefect varies in individual members ofa family. There is no treatment andthey may be complicated by glaucoma,retinal detachment, and central serousretinopathy.28
Renal-Coloboma Syndrome(Papillorenal Syndrome, OMIM120330)Coloboma are found in �5% of patientswith reflux nephropathy or other renalstructural defects but many are probablyunrecognized. Inheritance is autosomaldominant, but only 50% of patients withthe renal-coloboma syndrome have PAX2mutations.22,27 The other genes are notknown. PAX2 mutations are different ineach family and have their effect throughinterfering with the vascular supply of theurinary tract and eye. Some mutationscause reflux only without coloboma29 butnone causes isolated coloboma. The renalabnormalities in renal-coloboma syn-drome include vesicoureteric reflux, renalhypoplasia, multicystic or dysplastic kid-neys, and renal failure.30,31 The age at pre-sentation and rate of progression to renalfailure vary even within families. Ocularfeatures vary from optic disc pits to largechorioretinal coloboma, and abnormali-ties are usually asymmetrical.27 Pits aresubtle and may be overlooked on ophthal-moscopy. In more severe disease the retinalvessels emerge from the periphery of theoptic disc.32 The surrounding retina appearsatrophic. Vision varies from normal to se-verely impaired.33 Fewer than 20% of pa-tients have associated sensorineural hearingloss, seizures, Arnold-Chiari malformations,or skin and joint laxity.30 Patients with papil-lorenal syndrome should be monitored forophthalmic complications and other familymembersscreenedcarefully foropticdiscde-fects. PAX2 mutations do not occur in theCHARGE or COACH syndromes and donot cause iris coloboma.
CHARGE Syndrome (OMIM214800)This comprises Coloboma of the iris, Heartanomalies, Atresia of the nasal choanae,Retardation of growth and/or develop-ment, Genital and/or urinary abnormali-ties (hypogonadism), and Ear abnormali-ties with deafness.24 This condition affects1 in 10,000 individuals and demonstratesautosomal dominant inheritance. Sixtypercent of patients have a mutation in theCHD7 gene34 which codes for a DNA-binding protein involved in early embry-onic development. Ninety percent of chil-
Table 1. Inherited renal diseases thatpresent in adults as well as children
Autosomal recessive polycystic kidneydisease140
Alport syndrome46
Membranoproliferative glomerulonephritisNephronophthisis141
Bardet-Biedl syndrome142
Alagille syndrome143,77
MELAS syndrome80,81
Kearns-Sayre syndrome82
LCAT deficiency115
Cystinosis129
Oxalosis121
Fabry disease144,145
Table 2. Inherited renal diseasesassociated with hearing loss
Renal coloboma syndrome146
CHARGE syndrome24
Alport syndrome47
MELAS syndrome80,81
Kearns-Sayre syndrome82
Leber AmaurosisAlstrom syndrome72
Fabry disease94
Charcot-Marie-Tooth diseaseBartter syndrome147
Wolfram syndromeHurler syndromeNephronophthisis63
Bardet-Biedl syndrome142
BRIEF REVIEW www.jasn.org
1404 Journal of the American Society of Nephrology J Am Soc Nephrol 22: 1403–1415, 2011
dren have coloboma of the optic disc,chorioretina or iris, 20% to 40% have astructural urinary tract anomaly includingsolitary kidney, renal hypoplasia, duplexkidney, or vesicoureteric reflux, and 60%have heart defects (often Fallot’s tetralogy).Kidney problems are often associated withipsilateral facial palsy.35
WAGR Syndrome (OMIM 194072)This is a rare genetic syndrome withWilm’s tumor, Aniridia, Genitourinaryanomalies including gonadal tumors, men-tal Retardation, and obesity.36,37 It resultsfrom a deletion involving the contiguousgenes, PAX6 and WT1, on chromosome11.38 PAX6 regulates neuronal migration
in the cerebral cortex.39 Loss of thesegenes produces ocular and genitourinaryanomalies, respectively. The iris is defi-cient, and the optic nerve and fovea arehypoplastic. These abnormalities may becomplicated by a fragile cornea, glaucoma,and retinal detachment.40 Fifty percent ofpatients develop renal cancer.
COACH Syndrome (OMIM 216360)This condition is characterized by Cerebellarvermis hypo/aplasia, Oligophrenia, congeni-tal Ataxia, ocular Coloboma, and Hepatic fi-brosis. This affects 1 in 200,000 individualsand inheritance is autosomal recessive. Itprobably represents a subtype of Joubertsyndrome with liver disease.41 Mutations
affect one of the MKS3, CC2D2A, orRPGRIP1L genes.42 MSK3 mutations ac-count for nearly 60% of cases.43 Pa-tients may have the molar tooth sign, amidbrain-hindbrain malformation alsoseen in Joubert syndrome and congenitalhepatic fibrosis. Although they havemedullary renal cysts, they do not neces-sarily develop renal failure. Optic nervecoloboma,44,41 mental retardation, nys-tagmus, and congenital ataxia occur.
DRUSEN
Drusen are yellowish-white depositsof cellular and inflammatory debris
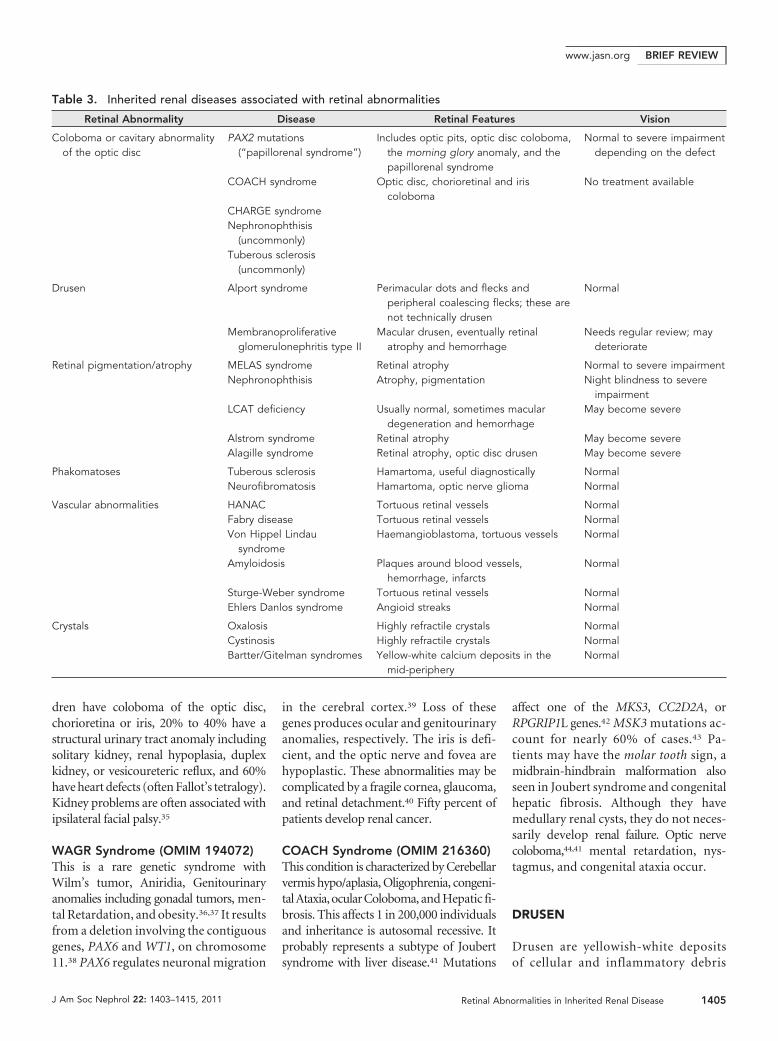
Table 3. Inherited renal diseases associated with retinal abnormalities
Retinal Abnormality Disease Retinal Features Vision
Coloboma or cavitary abnormalityof the optic disc
PAX2 mutations(“papillorenal syndrome”)
Includes optic pits, optic disc coloboma,the morning glory anomaly, and thepapillorenal syndrome
Normal to severe impairmentdepending on the defect
COACH syndrome Optic disc, chorioretinal and iriscoloboma
No treatment available
CHARGE syndromeNephronophthisis
(uncommonly)Tuberous sclerosis
(uncommonly)
Drusen Alport syndrome Perimacular dots and flecks andperipheral coalescing flecks; these arenot technically drusen
Normal
Membranoproliferativeglomerulonephritis type II
Macular drusen, eventually retinalatrophy and hemorrhage
Needs regular review; maydeteriorate
Retinal pigmentation/atrophy MELAS syndrome Retinal atrophy Normal to severe impairmentNephronophthisis Atrophy, pigmentation Night blindness to severe
impairmentLCAT deficiency Usually normal, sometimes macular
degeneration and hemorrhageMay become severe
Alstrom syndrome Retinal atrophy May become severeAlagille syndrome Retinal atrophy, optic disc drusen May become severe
Phakomatoses Tuberous sclerosis Hamartoma, useful diagnostically NormalNeurofibromatosis Hamartoma, optic nerve glioma Normal
Vascular abnormalities HANAC Tortuous retinal vessels NormalFabry disease Tortuous retinal vessels NormalVon Hippel Lindau
syndromeHaemangioblastoma, tortuous vessels Normal
Amyloidosis Plaques around blood vessels,hemorrhage, infarcts
Normal
Sturge-Weber syndrome Tortuous retinal vessels NormalEhlers Danlos syndrome Angioid streaks Normal
Crystals Oxalosis Highly refractile crystals NormalCystinosis Highly refractile crystals NormalBartter/Gitelman syndromes Yellow-white calcium deposits in the
mid-peripheryNormal
BRIEF REVIEWwww.jasn.org
J Am Soc Nephrol 22: 1403–1415, 2011 Retinal Abnormalities in Inherited Renal Disease 1405

located beneath the RPE. They are ob-vious on fundus ophthalmoscopy andphotographs especially red-free images.Occasional drusen occur normallywith aging and increased numbers are
found with some forms of glomerulo-nephritis.45 The dots and flecks in Al-port syndrome are not technicallydrusen because they affect the internallimiting membrane rather than the
RPE.46 They are smaller than thedrusen in membranoproliferative glo-merulonephritis type 2 and do not in-volve the macula. The drusen in densedeposit disease are large and soft and
a BA
C D
E F
HG
ji JI
LkK
nM nN
PO
p
Figure 1. Retinal abnormalities in inherited renal disease. (A) Optic pit. This is a mild form of the optic disc coloboma that occurswith reflux nephropathy due to PAX2 mutations. It may also be found in the CHARGE and COACH syndromes. Vision was normalin this eye but coloboma are typically asymmetrical. (B) Morning glory anomaly. Vision was impaired. (C) Normal central retina.The central retina was also normal in the fellow eye. This individual had a child with an iris coloboma and single kidney. (D) Thisis the peripheral retina of the individual whose fundus is shown in (C). Retinal coloboma were present in the periphery bilaterally.(E) Dot and fleck retinopathy in Alport syndrome. The retinopathy spares the macula and is bilateral. Vision was not affected;(F) Peripheral retinopathy in Alport syndrome. This is more common than the central retinopathy and comprises confluent dotsand flecks �2 disc diameters from the foveola. Again vision was not affected. (G) Widespread large soft macular drusen inmembranoproliferative glomerulonephritis (dense deposit disease). These are identical to the drusen in macular degeneration butare present from early adulthood. (H) Late form of dense deposit disease with extensive retinal drusen, hemorrhage, and atrophy.These individuals should be assessed and monitored by an ophthalmologist because of the risk of complications and thepossibility of treatment. (I) Retinal drusen and atrophy in Jeune syndrome, a form of nephronophthisis. (J) Red-free images ofJeune syndrome in which the abnormalities are more obvious. (K) Retinal atrophy in Joubert syndrome, another form ofnephronophthisis, demonstrated by optical coherence tomography. The black line indicates peripapillary thickness is 1% normal.(L) Retinal atrophy in MELAS syndrome. The patient initially had poor vision that progressed. (M) Chorio-retinal atrophy in KearnsSayre syndrome. The retina is thinned and depigmented. Vision was affected. (N) Hamartoma in tuberous sclerosis. A white tuberwith fine linear hemorrhage was present unilaterally. This abnormality is found in the majority of patients with tuberous sclerosisand must be distinguished from retinal infarcts due to hypertension, diabetes, or lupus. (O) Corkscrew vessels in Fabry syndrome.These are asymptomatic but sudden visual loss may occur with central artery occlusion. These must be distinguished from thechanges in hypertension. (P) Von Hippel Lindau syndrome. Tortuous vessels are seen leading to a hemangioblastoma just outsidethe field. The obvious macular lipid exudates are due to leakage from abnormal vessels. Vision was impaired.
BRIEF REVIEW www.jasn.org
1406 Journal of the American Society of Nephrology J Am Soc Nephrol 22: 1403–1415, 2011

located at the macula. They resemblethe drusen in macular degenerationbut occur in early adulthood and areaccompanied by renal manifestations.
Alport Syndrome (X-Linked OMIM301050; Autosomal RecessiveOMIM 203780)Alport syndrome affects 1 in 10,000 indi-viduals47 and accounts for 2% of adultswith ESRD. At least 80% of patients haveX-linked disease with COL4A5 muta-tions and the others have autosomal re-cessive or dominant inheritance withmutations in the COL4A3 or COL4A4genes. These genes code for the chainsthat comprise the collagen IV �3�4�5protomer which is found in the GBM,the stria vascularis of the cochlea, cornea,lens capsule, and internal limiting mem-brane, and Bruch’s membrane of the ret-ina.48 Individuals with Alport syndromehave hematuria and develop kidney fail-ure, hearing loss, corneal dystrophy, len-ticonus, and retinopathy. Retinopathy ispresent in half the males and 1 in 5 fe-males with X-linked disease, and proba-bly the majority of those with recessivedisease.47 Central perimacular dots andflecks and retinal thinning are typicaland, rarely, a macular hole occurs espe-cially in the temporal macula.49 –51,46 Insevere disease the flecks outline a peri-macular lozenge.52,53 Peripheral flecksare more common than the perimacularretinopathy. Retinal abnormalities arebest visualized using red-free photo-graphs and the peripheral retinopathyrequires multiple images. Vision is notaffected. The central and peripheral reti-nopathies are diagnostic for Alport syn-drome, but the diagnosis may also beconfirmed with a positive family history,GBM lamellation, and absence of the�3�4�5 protomers from the kidney orskin, or genetic testing. Many mutationshave been described in X-linked Alportsyndrome, and they are generally differ-ent in each family. Certain variants, suchas large deletions and rearrangements,nonsense mutations, and carboxy termi-nus missense mutations, result in earlyonset renal failure and retinopathy inmales.54 –56 Many fewer mutations have
been described in recessive and the raredominant forms, and their effect on phe-notype is not clear.
MembranoproliferativeGlomerulonephritis Type 2 (DenseDeposit Disease, orMesangiocapillaryGlomerulonephritis, Associatedwith Factor H Deficiency, OMIM609814)This accounts for 2% of all glomerulardisease.57 Patients have hematuria andproteinuria and develop renal impair-ment by early adulthood. Facial andshoulder girdle lipodystrophy, and C3nephritic factor, and low C3 levels mayoccur.5,58 At least some forms of this dis-ease are inherited, and mutations anddisease haplotypes have been identifiedin the complement Factor H (CFH)gene.59,60 The intramembranous GBMdeposits and retinal drusen have an iden-tical composition.61 Vision is affected,and patients should be assessed and re-viewed regularly by an ophthalmologistbecause of the risk of retinal complica-tions such as neovascular membranes.62
RETINAL ATROPHY ANDPIGMENTATION (RETINITISPIGMENTOSA)
Individuals with renal disease and retinalatrophy first complain of poor night vi-sion and even tunnel vision. Initially, theabnormality is only demonstrated withan electroretinogram but subsequentlythe retinal appearance is abnormal withpallor, pigmentation and mottling, at-tenuated arterioles and venules, and ob-vious choroidal vessels.
NephronophthisisThese diseases affect 1 in 50,000 individualsand account for 10% to 25% of all childrenwith inherited renal failure.63 Inheritance isautosomal recessive. Mutations affect theNPHP1 through NPHP10 (nephrocystin)genes. Homozygous NPHP1 mutations ac-count for 25% of patients, and mutations intheothergenes, eachfor�3%.64 Thesegenescode for proteins found in the primary ciliaor centrosome, and result in defective signal-
ing and abnormal tissue differentiation andmaintenance. Additional genes still have tobe identified. Patients present from 1 year ofage through to the fourth decade with poly-uria and polydipsia but not hematuria, pro-teinuria, or hypertension. Their kidneys aresmall- or normal-sized with corticomedul-lary cysts. The ciliary theory explains the pat-tern of organs involved with retinal dystro-phy/retinitis pigmentosa, ataxia, situsinversus, liver fibrosis, and mental retar-dation. Hearing loss may be present.65
Retinitis pigmentosa (Senior Loken syn-drome) occurs with 10% to 30% NPHP1through NPHP4 mutations, and allNPHP5 and NPHP6 mutations.16,64 Theretinal defects vary from abnormal reti-nal function tests to blindness. Theremay also be tunnel vision early,16 nystag-mus and poor pupillary reflexes, andfundus abnormalities. Vision deterio-rates in parallel with worsening renalfunction.
Rare related disorders result from reces-sive mutations in genes coding for otherproteins in the cilia, basal bodies, or cen-trosomes of renal and retinal epithelialcells. Most of these disorders also featurerenal medullary cysts and retinal atrophy/pigmentation. Bardet-Biedl syndrome (BBS,OMIM 209900) affects 1 in 50,000 in-dividuals. Mutations affect the 14 BBSgenes, one of which accounts for 50%of all mutations (BBS1, BBS10). Some-times mutations affect two differentgenes, and disease severity may also de-pend on a modifier gene.66 This syn-drome is characterized clinically by fo-cal and segmental glomerular sclerosis(FSGS), obesity, impaired cognition,and polydactyly. The phenotype varieswithin families and individuals maynot present until the fifth decade. Theretina is normal early, but atrophywithout much pigmentation occurslater. The visual abnormalities includeabnormal electroretinogram, nightblindness, peripheral field defects, andblindness in early adulthood.67 Carri-ers probably have subtle defects of ret-inal function.68
Joubert syndrome and related disor-ders (OMIM 213300) are due to muta-tions in the NPHS1, NPHS6, AHI1,CEP290, and additional unidentified
BRIEF REVIEWwww.jasn.org
J Am Soc Nephrol 22: 1403–1415, 2011 Retinal Abnormalities in Inherited Renal Disease 1407

genes.69,70 The kidneys are often cystic.Other clinical features include hepaticfibrosis, polydactyly, cerebellar ataxia(with cerebellar vermis hypoplasia andthe molar tooth sign on MRI), and ab-normal eye movements including ro-tary nystagmus, psychomotor retarda-tion, and developmental delay. Retinalabnormalities include retinitis pig-mentosa and possibly chorioretinalcoloboma.16 As with the Senior Lokensyndrome, the retinitis pigmentosa isoften severe and the electroretinogramis abnormal early.16
Cogan syndrome (oculomotor apraxia;OMIM 257550) is also due to NPHSmutations and may be a mild form ofJoubert syndrome. Renal failure, poorhearing, and reduced muscle tone oc-cur but there are no retinal abnormal-ities.71
Meckel-Gruber syndrome (OMIM249000) may be caused by mutations inthe same genes as Joubert syndrome butis fatal in early life. It is characterized bycystic kidney dysplasia, liver fibrosis, oc-cipital encephalocoele, and polydactyly.
Jeune syndrome (asphyxiating tho-racic dystrophy; OMIM 208500) is char-acterized by cystic kidney disease, a longnarrow thorax, short limb dwarfism,brachydactyly, and other skeletal defects.Respiratory distress may be severe.72 Ret-inal atrophy occurs, but is mild.
Alstrom disease (OMIM 203800) isdue to mutations in the ALMS1 geneand clinically resembles the Bardet-Biedl syndrome. Affected individualshave renal failure with FSGS and tubu-lointerstitial fibrosis.73 They also havehearing loss, liver dysfunction, dilatedcardiomyopathy, delayed puberty, obe-sity, and diabetes without mental retar-dation, polydactyly, or hypogonadism.Retinal atrophy occurs with large pig-ment clumps,73 vascular attenuation,and optic disc pallor. Vision is poor.
Nephronophthisis and its variantsmust be distinguished from Medullarycystic kidney disease (OMIM 191845),which presents in adulthood, and dem-onstrates autosomal dominant inheri-tance. It is often caused by mutationsin the UMOD gene, which codes foruromodulin, or Tamm Horsfall pro-
tein.74 –76 This disease typically resultsin renal failure in middle age, some-times with a high serum urate, butthere are no known retinal associa-tions.
Alagille Syndrome (ArteriohepaticDysplasia, OMIM 118450)This is very rare and �50 families havebeen described worldwide. Inheritance isautosomal dominant, and the diagnosisis usually made in children with choles-tasis or their adult relatives, but some-times in isolated adults.77 It results frommutations in the JAG1 gene, which en-codes a ligand for Notch1. This stimu-lates a signaling pathway that determinescell fate in embryogenesis.78 Clinical fea-tures include renal arterial stenosis, hy-pertension, and renal failure but the kid-neys may not be involved. There arereduced intrahepatic bile ducts and liverfailure that typically presents with neo-natal jaundice. In addition, peripheralpulmonary artery hypoplasia results inpulmonary hypertension, and various car-diac anomalies, vertebral arch defects, anda peculiar facies are present. The retina iscommonly diffusely hypopigmented orspeckled, and optic disc anomalies, espe-cially drusen, are present in nearly all af-fected individuals.79
MELAS (Myopathy,Encephalopathy, Lactic Acidosis,Stroke-like Episodes Syndrome,OMIM 540000)This condition affects 1 in 5 to 10,000individuals and is due to an A3243Gmutation in the mitochondrial DNAcoding for tRNA (Leu).80,81 Inheri-tance is mitochondrial with transmis-sion from mother to child. Presenta-tion occurs from childhood throughearly adulthood. In addition to theclassical features, patients usually haveFSGS and steroid-resistant nephroticsyndrome that progresses to end-stagerenal failure before middle age. Hear-ing loss, cardiomyopathy, and diabetesare also common but clinical featuresvary between families. Retinal atrophyoccurs in at least half the patients80 butthe electroretinogram is abnormal in90%. The diagnosis is confirmed with a
muscle biopsy demonstrating ragged-red fibers, or on mitochondrial DNAsequencing.
Kearns-Sayre Syndrome (OMIM530000)Kearns-Sayre is a rare mitochondrial dis-ease affecting 1 in 30,000 to 100,000 in-dividuals. It typically results from a4.9-kb deletion in mitochondrial DNA.Many cases occur de novo. The onset isusually before the age of 20 but some-times later.82 Kearns-Sayre syndrome isassociated with a Bartter-like tubular de-fect,83 together with chronic progressiveexternal ophthalmoplegia, ptosis, hear-ing loss, cardiac conduction abnormali-ties, cerebellar ataxia, and diabetes. De-spite retinal atrophy and pigmentation,visual loss is usually mild. Again the di-agnosis is confirmed with ragged-red fi-bers on muscle biopsy, or with mito-chondrial DNA sequencing.
PHAKOMATOSES(NEUROCUTANEOUSSYNDROMES)
These disorders include neurofibromato-sis, tuberous sclerosis, von Hippel Lindaudisease, and Sturge-Weber syndrome. VonHippel Lindau and Sturge-Weber syn-dromes also cause vascular anomalies andare considered later.
Neurofibromatosis (NF) Types I and2 (OMIM 162200 and 101000)Neurofibromatosis 1 and 2 affect 1 in3000 and 1 in 100,000 live births and aredue to mutations in the NF1 or NF2genes, which code for neurofibromin, aGTP-ase-activating enzyme, or merlin, acytoskeletal protein, respectively. Inher-itance is autosomal dominant but abouthalf of all mutations occur de novo. NF1and NF2 mutations are characterized bypigmented skin lesions, neurofibromas,and acoustic neuroma (NF2). Hyperten-sion occurs because of proximal renal ar-tery stenosis, coarctation of the aorta, orphaemochromocytoma.84 Retinal abnor-malities include ischemia,85 hamartomaincluding Combined Hamartoma of theRetina and Retinal Pigment Epithelium
BRIEF REVIEW www.jasn.org
1408 Journal of the American Society of Nephrology J Am Soc Nephrol 22: 1403–1415, 2011

(CHRRPE, both NF1 and NF286), opticglioma (NF1), and optic atrophy second-ary to pressure on the optic nerve.
Tuberous Sclerosis (TSC) Types 1and 2 (OMIM 191100 and 191092)TSC affects 1 in 10,000 individuals, andinheritance is autosomal dominant.Mutations occur in the TSC1 gene ormore commonly the TSC2 gene, whichcode for hamartin and tuberin, respec-tively.87 Twenty percent of patientshave no mutation identified. BothTSC1 and TSC2 are tumor suppressorgenes that require a second hit beforetumors develop. TSC mutations arehighly penetrant but have variable ex-pression. TSC2 is contiguous with thePKD1 gene for polycystic kidney dis-ease, and patients with large deletionshave both TSC and renal cysts.88 Sixtyto 80% of patients with TSC have mul-tiple bilateral angiomyolipoma in thekidney.89 These result in hematuria, andsometimes cancer. Thirty percent of pa-tients have renal cysts. Facial adenomasebaceum, cardiac rhabodomyoma,and pulmonary cysts are common. Oc-ular coloboma and eyelid tumors arerare. Most patients have at least oneretinal or optic nerve hamartoma.90
These are highly vascular and eventu-ally calcify, and must be differentiatedfrom hypertensive infarcts. Vision re-mains normal. Retinal hamartoma arehelpful diagnostically, but the diagno-sis of TSC is also confirmed with ge-netic testing.
VASCULAR ABNORMALITIES
Some inherited renal diseases are asso-ciated with tortuous retinal vesselswith hemorrhages and infarcts (cottonwool spots). These are evident on ophthal-moscopy and in retinal photographs. Themajor differential diagnoses are hyper-tension and diabetes, which are usuallyobvious on history. However, retinalabnormalities can persist for monthsafter a single episode of severe hyper-tension, and the diagnosis of diabetesmay not be obvious if glucose control
improves as renal function deterio-rates.
HANAC (Hereditary Angiopathywith Retinal Tortuosities,Nephropathy (Manifesting asHematuria or Cysts), Aneurysms,and Muscular Cramps Syndrome,OMIM 611773)To date, only a few families have beendescribed with this disease.91–93 It re-sults from mutations in the COL4A1gene. Affected individuals have hema-turia, renal cysts, and GBM defects.Small and large arteries are tortuous.Other features include headache, mus-cle cramps with elevated levels of crea-tine kinase, supraventricular cardiac ar-rhythmia, Raynaud’s phenomenon, andleukoencephaly. The retina has tortu-ous vessels, and repeated retinal hem-orrhage results in transient visual im-pairment that resolves spontaneously.
Fabry Disease (OMIM 301500)This lysosomal storage disorder affectsat least 1 in 50,000 males but milderforms are probably more common.94
Inheritance is X-linked and disease re-sults from mutations in the GLA geneencoding �-galactosidase. Mutationsresult in the accumulation of the glyco-lipid, ceramide, in blood vessels andother tissues.95 Mutations are differentin each family but missense variants aremost common (76%). Mutation typecorrelates with age at onset of renalfailure. Symptoms appear in childhoodbut become particularly troublesomein the fourth decade. Fifty percent ofpatients have proteinuria by 35 years ofage, and renal failure by 42.96 The renalbiopsy demonstrates FSGS with lamel-lated zebra bodies. Other features in-clude angiokeratoma on the lower ab-domen and thighs, cardiomyopathy,mitral valve prolapse, painful periph-eral neuropathy, hearing loss, stroke,and lack of sweating. Patients withatypical disease (cardiac or visceral fea-tures without angiokeratoma, renaldisease, or corneal keratopathy) arerecognized increasingly.97,98 Featuresin females vary from asymptomatic tosevere.99 Fabry disease is also found in a
series of patients with renal failure ondialysis, with cardiomyopathy, and inyoung patients with stroke.100 –102 Theretinal vessels are tortuous. Other ocu-lar abnormalities include corneal verti-cillata or horse tails, and tortuous con-junctival vessels. Vision is normal. Thediagnosis is confirmed on renal biopsy,with low enzyme levels (which may benormal in affected symptomatic fe-males), or with genetic testing.
Von Hippel Lindau Disease (OMIM193300)This condition affects 1 in 36,000 indi-viduals. It results from mutations inthe VHL gene, which codes for the pro-tein that regulates hypoxia-induciblefactor activity.103 Mutations result inupregulation of vascular endothelialgrowth factor and increased endothe-lial cell growth and migration.104 In-heritance is autosomal dominant, but20% of mutations occur de novo. How-ever, two mutations are required fordisease, one germline and one somatic.Germline mutations are different ineach family. Nonsense mutations anddeletions result in more severe dis-ease.105 The timing of the somatic mu-tation probably determines the age atonset of disease, the organs affected,and severity. Thus, phenotypes varyeven within families but penetrance isnearly 100% by age 60. Affected indi-viduals develop renal cysts, and some-times clear cell renal cancer. They alsohave cafe au lait spots, nervous systemhemangioblastomas, and sometimes,phemochromocytoma and pancreaticislet cell tumors. Fifty percent ofpatients have retinal hemangioblas-toma.106 These are usually multiple,vary in size, and are located in the cen-tral and mid-peripheral retina. Theyhave a globular reddish appearance(sugar-coated raspberries) and adjoin-ing vessels are tortuous. They may beasymptomatic for years and regressspontaneously. However, they may becomplicated by retinal hemorrhage,retinal tears, and detachment. Evensmall asymptomatic lesions should betreated. Early detection and treatmenthelps preserve vision,107 and in the fu-
BRIEF REVIEWwww.jasn.org
J Am Soc Nephrol 22: 1403–1415, 2011 Retinal Abnormalities in Inherited Renal Disease 1409

ture, antiangiogenic factors may provehelpful. The diagnosis of von HippelLindau syndrome is usually made clini-cally, but may be confirmed also with ge-netic testing.
Sturge-Weber Syndrome (OMIM185300)One in 50,000 individuals are affectedand many cases are sporadic. No mutantgene has been identified. The conditionis characterized by facial nevi, cerebralhemangioma, and intracranial calcifica-tion. Vascular malformations occur inthe kidney, within the renal pelvis, pa-pilla, or urinary bladder.108 These aretypically solitary and result in hematuria.They can be treated with laser. Vascularmalformations of the retina and choroidmay be complicated by retinal detach-ment or optic neuropathy.109,110
AmyloidosisAmyloid deposits are rigid, linear non-branching aggregated fibrils with an an-tiparallel �-pleated sheet configuration.The inherited forms (AA) include thosedue to transthyretin mutations, familialamyloidotic polyneuropathy, and famil-ial Mediterranean fever. Inheritance isautosomal recessive or dominant de-pending on the type. Inherited amyloid-osis affects many tissues including thekidney and eye.111,112 It commonly resultsin kidney failure, but other features includecardiomyopathy, polyneuropathy, and au-tonomic dysfunction. Retinal changes in-clude tortuous vessels, vessel cuffing, hem-orrhage, and infarcts.112–114 The diagnosisis confirmed with the demonstration ofCongo red deposits with apple green bire-fringence under polarized light in a con-junctival biopsy, or with genetic testing.
Lecithin-Cholesterol Acyltransferase(LCAT) Deficiency (OMIM 606967)This condition appears to be very rare,affecting �1 in 1,000,000 individuals,but is also probably under-recognized.115
Inheritance is autosomal recessive, andmore than 40 mutations have been de-scribed. LCAT catalyzes the formation ofcholesterol esters and its deficiency re-sults in lipid deposition in the kidney andother tissues. Clinical features vary in dif-
ferent members of a family with the samemutation, and carriers are usuallyasymptomatic.116 LCAT deficiency pro-duces steroid-resistant FSGS and ESRDby the fourth decade, but sometimes thekidneys are not affected. Other featuresinclude dyslipidemia and xanthelasma,corneal opacities, and stomatocytes withincreased membrane phospholipid, re-sulting in hemolytic anemia. Retinalmanifestations include macular degener-ation and hemorrhage from involvementand rupture of Bruch’s membrane.117
LCAT deficiency is diagnosed whenLCAT levels are negligible, unesterifiedcholesterol and cholesterol ester levelsare low, and LDL and triglyceride levelsare increased. The diagnosis is also madewith genetic testing.
RETINAL CRYSTALS
Retinal crystals are small and highly re-fractile, often with associated pigmenta-tion (cystinosis, oxalosis) or large anddull (ectopic calcium deposits in chronicrenal failure, Bartter and Gitelman syn-drome). Vascular deposits may result inischemia. The crystals must be distin-guished from the normal youthful retinalsheen. Other causes of crystals are usu-ally obvious on history (cholesterol em-boli, tamoxifen, talc in intravenous drugusers, and calcified drusen).118
Oxalosis (OMIM 260000)Oxalosis affects 1 in 100,000 individu-als.119 Inheritance is autosomal recessiveand due to mutations in the alanineglyoxylate aminotransferase (AGT) orthe glyoxylate reductase/hydroxypyru-vate reductase (GRHPR) genes.120 AGTdeficiency is more common and causesmore serious disease. Mutations result inincreased synthesis of oxalate and subse-quent urinary excretion and depositionof insoluble calcium oxalate in the kid-ney. As renal function deteriorates withprogressive involvement, oxalate accu-mulates in other organs. Oxalosis resultsin nephrolithiasis, nephrocalcinosis, andrenal failure. Renal failure occurs frominfancy through to the sixth decade.121
The characteristic retinal findings are
highly refractile crystals often in the arte-rioles, producing a flecked retinopa-thy,122 and crystals in the RPE layer, pro-ducingahyperpigmentedcentersurroundedby hypopigmentation (ringlets).123 Optic at-rophy occurs with severe disease. Visionis usually normal, but may be impaired.The diagnosis of oxalosis is suspected inpatients with stones or nephrocalcinosisin childhood, recurrent calcium oxalatestones in adulthood, or renal insuffi-ciency associated with stones or nephro-calcinosis. The diagnosis is confirmedwith urinary oxalate levels and genetictesting.
Cystinosis (OMIM 219800)This condition also affects about 1 in100,000 individuals124 and accounts for5% of children with renal failure.125 In-heritance is autosomal recessive, andmutations occur in the CTNS gene forcystinosin.126 Many Caucasians have thesame mutation consistent with a foundereffect.127 Compound heterozygotes havemilder, usually adult-onset, disease andcarriers are asymptomatic. Affected chil-dren typically present in infancy withpolyuria and dehydration from damagedproximal tubular cells. Untreated, theydevelop renal failure before the teenageyears.124 However, retinal (and cornealand iris) crystals occur even without sys-temic features. Affected individuals com-plain of gritty eyes and, if severely af-fected, have poor night and color visionand reduced visual acuity. They also haveretinal crystals with hypopigmentation,and pigmentary stippling.128 Adult-onsetdisease was formerly considered primar-ily ocular but probably all patients haveat least some renal involvement.129 Thediagnosis is based on clinical features, in-creased leukocyte cystine levels, and ge-netic testing.124,125
Tubular Defects (Bartter Syndrome,OMIM 607364 and GitelmanSyndrome, OMIM 263800)These each affect about 1 in 50,000 indi-viduals and inheritance is autosomal re-cessive.130 Bartter syndrome130 is due tomutations in genes coding for proteinsfound in the thick ascending limb of theloop of Henle and is characterized by hy-
BRIEF REVIEW www.jasn.org
1410 Journal of the American Society of Nephrology J Am Soc Nephrol 22: 1403–1415, 2011

pokalemia, alkalosis, and normal or lowblood pressure. Patients present in uteroor before school age. They typically havepolyuria, polydipsia, and a tendency fordehydration, features also seen with loopdiuretics such as furosemide. Mutationsaffect the NKCC2, ROMK, CLCNKB,BSND, and CASR genes. Patients also of-ten have a magnesium deficiency and de-velop widespread calcium deposits. Gitel-man syndrome results from mutations inthe SLC12A3 gene, which codes for thethiazide-sensitive Na-Cl cotransporter inthe distal convoluted tubule.131 This dis-ease is mild and patients present in ado-lescence or later. They are asymptomaticor have fatigue, muscle cramps, andrarely seizures or cardiac arrhythmias.Symptoms are due mainly to hypokale-mia. BP is normal. Carriers may havemild features. Patients with either condi-tion sometimes have large dullish yel-low-white deposits in the retinal midpe-riphery,132,133 sometimes with pigmentdeposition. Visual fields and acuity arenormal.
INCIDENTAL RETINAL FINDINGS
Nail Patella Syndrome (HereditaryOsteo-onychodysplasis, OMIM161200)This affects about 1 in 50,000 individ-uals134 and inheritance is autosomaldominant. It is due to mutations in theLMX1B gene, which encodes the tran-scription factor that regulates expres-sion of collagen IV, and determinesdorsal limb and eye development in theembryo.135 However, LMX1B is alsoexpressed after birth in the glomerularpodocyte, suggesting it has a regulatoryrole in that cell.136 Mutations affect acritical region of the gene (the LIM orhomeodomain) and result in haploin-sufficiency. Most patients have thumbnail hypoplasia, absent patellae, radialhead dislocation, and iliac horns. Thepresence of renal disease (hematuriaand proteinuria in 40% to 60%) de-pends on the underlying mutation and15% of patients develop renal failureby middle age.134 The renal biopsydemonstrates variable amounts of glo-
merulosclerosis, with GBM thickening,rarefaction, and collagen III fibrils.
About one third of patients developprimary open-angle glaucoma by theage of 40, and sometimes optic atro-phy.137 Other ocular associations areptosis, epicanthal folds, microcornea,keratoconus, cataracts, and possiblycloverleaf iris pigmentation.138 Indi-viduals with the nail and bone manifes-tations of the nail-patella syndromeshould be screened for hematuria andproteinuria, and reviewed annually forincreased intraocular pressure andglaucoma.139
CONCLUSIONS
Many inherited renal diseases are diag-nosed with expensive and labor-inten-sive laboratory techniques. However,retinal abnormalities in inherited renaldisease are sufficiently common andcharacteristic to help with the diagnosis.Most of these features are obvious onophthalmoscopy or in retinal photo-graphs. Some are easier to identify withred-free images or peripheral retinalviews. Retinal abnormalities may affectvision, and most patients with inheritedrenal disease require an ophthalmologi-cal assessment and sometimes furthertesting, monitoring, and treatment. Thediagnosis of inherited renal disease is im-portant because of the risk of complica-tions, the implications for family mem-bers, and the possibility of treatment.Retinal abnormalities may also assistwith assessing disease progression andthe response to treatment, and in ex-plaining the pathogenesis of the renal le-sions. Inherited forms of renal diseasewhere, currently, there are no known ret-inal associations, warrant further oph-thalmic investigation.
ACKNOWLEDGMENT
This project was supported by the National
Health and Research Council of Australia
and Kidney Health Australia. We would
like to thank the many patients who took
part in these studies and their clinicians,
especially Prof. David Power, A/Prof. James
Elder, A/Prof. Justin O’Day, and Dr. Kathy
Nicholls.
DISCLOSURESNone.
REFERENCES
1. Grunfeld, J-P: Congenital/inherited kidneydiseases: How to identify them early andhow to manage them. Clin Exp Nephrol 9:192–194, 2005
2. Levy M, Feingold J: Estimating prevalencein single-gene kidney diseases progressingto renal failure. Kidney Int 58: 925–943,2000
3. Grunfeld JP, Chauveau D, Levy M: Ander-son-Fabry disease: Its place among othergenetic causes of renal disease. J Am SocNephrol 13: S126–S129, 2002
4. Izzedine H, Bodaghi B, Launay-Vacher V,Deray G: Eye and kidney: From clinicalfindings to genetic explanations. J Am SocNephrol 14: 516–529, 2003
5. Appel GB, Cook HT, Hageman G, JennetteJC, Kashgarian M, Kirschfink M, LambrisJD, Lanning L, Lutz HU, Meri S, Rose NR,Salant DJ, Sethi S, Smith RJH, Smoyer W,Tully HF, Tully SP, Walker P, Welsh M,Wurzner R, Zipfel PF: Membranoprolifera-tive glomerulonephritis type II (dense de-posit disease): An update. J Am Soc Neph-rol 16: 1392–1403, 2005
6. Dahl E, Koseki H, Balling R: Pax genes andorganogenesis. Bioessays 19: 755–765,1997
7. Mrowka C, Schedl A: Wilms’ tumor sup-pressor gene WT1: From structure to renalpathophysiologic features. J Am SocNephrol 11: S106–S115, 2000
8. Wagner N, Wagner KD, Schley G, Coup-land SE, Heimann H, Grantyn R, Scholz H:The Wilms’ tumor suppressor Wt1 is asso-ciated with the differentiation of retinoblas-toma cells. Cell Growth Differ 13: 297–305,2002
9. Pelletier J, Bruening W, Li FP, Haber DA,Glaser T, Housman DE: WT1 mutationscontribute to abnormal genital system-de-velopment and hereditary Wilms-tumor.Nature 353: 431–434, 1991
10. Pelletier J, Bruening W, Kashtan CE, MauerSM, Manivel JC, Striegel JE, Houghton DC,Junien C, Habib R, Fouser L, Fine RN, Sil-verman BL, Haber DA, Housman D: Germ-line mutations in the Wilms-tumour sup-pressor gene are associated with abnormalurogenital development in Denys-Drashsyndrome. Cell 67: 437–447, 1991
11. Barbaux S, Niaudet P, Gubler MC, Grun-
BRIEF REVIEWwww.jasn.org
J Am Soc Nephrol 22: 1403–1415, 2011 Retinal Abnormalities in Inherited Renal Disease 1411

feld JP, Jaubert F, Kuttenn F, Fekete CN,Souleyreau-Therville N, Thibaud E, Fel-lous M, McElreavey K: Donor splice-sitemutations in WT1 are responsible forFrasier syndrome. Nat Genet 17: 467–470, 1997
12. Harvey SJ, Zheng KQ, Sado Y, Naito I,Ninomiya Y, Jacobs RM, Hudson BG,Thorner PS: Role of distinct type IV colla-gen networks in glomerular developmentand function. Kidney Int 54: 1857–1866,1998
13. Ljubimov AV, Burgeson RE, Butkowski RJ,Couchman JR, Zardi L, Ninomiya Y, Sado Y,Huang ZS, Nesburn AB, Kenney MC: Base-ment membrane abnormalities in humaneyes with diabetic retinopathy. J HistochemCytochem 44: 1469–1479, 1996
14. Watnick T, Germino G: From cilia to cyst.Nat Genet 34: 355–356, 2003
15. Hildebrandt F, Otto E: Cilia and centro-somes: A unifying pathogenic concept forcystic kidney disease? Nat Rev Genet 6:928–940, 2005
16. Adams NA, Awadein A, Toma HS: The ret-inal ciliopathies. Ophthalmol Genet 28:113–125, 2007
17. Evans RD, Rosner M: Ocular abnormalitiesassociated with advanced kidney diseaseand hemodialysis. Semin Dial 18: 252–257,2005
18. Bajracharya L, Shah D, Raut K, Koirala S:Ocular evaluation in patients with chronicrenal failure–a hospital based study. NepalMed Coll J 10: 209–214, 2008
19. Haddad R, Witzmann K, Braun O: Meta-static calcification to the peripheral fundusin chronic renal failure. Ophthalmologica179: 178–183, 1979
20. Patel DV, Snead MP, Satchi K: Retinal arte-riolar calcification in a patient with chronicrenal failure. Br J Ophthalmol 86: 1063,2002
21. Liew G, Mitchell P, Wong TY, Iyengar SK,Wang JJ: CKD increases the risk of age-related macular degeneration. J Am SocNephrol 19: 806–811, 2008
22. Sanyanusin P, Schimmenti LA, McNoe LA,Ward TA, Pierpont MEM, Sullivan MJ,Dobyns WB, Eccles MR: Mutation of thePAX2 gene in a family with optic-nervecolobomas, renal anomalies and vesi-coureteral reflux. Nat Genet 9: 358–364,1995
23. Alsing A, Christensen C: Atypical macularcoloboma (dysplasia) associated with famil-ial juvenile nephronophthisis and skeletalabnormality. Ophthalmol Pediatr Genet 9:149–155, 1988
24. Pagon RA, Graham JM, Zonana J, Yong SL:Coloboma, congenital heart disease, andchoanal atresia with multiple anomalies:CHARGE association. J Pediatr 99: 223–227, 1981
25. Verloes A, Lambotte C: Further delineationof a syndrome of cerebellar vermis hypo/
aplasia, oligophrenia, congenital ataxia,coloboma, and hepatic fibrosis. Am J MedGenet 32: 227–232, 1989
26. Mullaney PB, Jacquemin C, Abboud E, Kar-cioglu ZA: Tuberous sclerosis in infancy.J Pediatr Ophthalmol Strabismus 34: 372–375, 1997
27. Dureau P, Attie-Bitach T, Salomon R, Bet-tembourg O, Amiel J, Uteza Y, Dufier JL:Renal coloboma syndrome. Ophthalmol-ogy 108: 1912–1916, 2001
28. Bron AJ, Burgess SEP, Awdry PN, Oliver D,Arden G: Papillo-renal syndrome. An inher-ited association of optic disk dysplasia andrenal-disease. Report and review of the lit-erature. Ophthalmol Pediatr Genet 10:185–198, 1989
29. Nishimoto K, Iijima K, Shirakawa T,Kitagawa K, Satomura K, Nakamura H, Yo-shikawa N: PAX2 gene mutation in a familywith isolated renal hypoplasia. J Am SocNephrol 12: 1769–1772, 2001
30. Fletcher J, Hu M, Berman Y, Collins F,Grigg J, McIver M, Juppner H, AlexanderSI: Multicystic dysplastic kidney and vari-able phenotype in a family with a noveldeletion mutation of PAX2. J Am SocNephrol 16: 2754–2761, 2005
31. Cheong HI, Cho HY, Kim JH, Yu YS, Ha IS,Choi Y: A clinico-genetic study of renalcoloboma syndrome in children. PediatrNephrol 22: 1283–1289, 2007
32. Eccles MR, Schimmenti LA: Renal-colobomasyndrome: A multi-system developmentaldisorder caused by PAX2 mutations. ClinGenet 56: 1–9, 1999
33. Parsa CF, Silva ED, Sundin OH, GoldbergMF, De Jong MR, Sunness JS, Zimer R,Hunter DG: Redefining papillorenal syn-drome: An underdiagnosed cause of ocularand renal morbidity. Ophthalmology 108:738–749, 2001
34. Vissers L, van Ravenswaaij CMA, AdmiraalR, Hurst JA, de Vries BBA, Janssen IM, vander Vliet WA, Huys E, de Jong PJ, HamelBCJ, Schoenmakers E, Brunner HG, Velt-man JA, van Kessel AG: Mutations in a newmember of the chromodomain gene familycause CHARGE syndrome. Nat Genet 36:955–957, 2004
35. Ragan DC, Casale AJ, Rink RC, Cain MP,Weaver DD: Genitourinary anomalies in theCHARGE association. J Urol 161: 622–625,1999
36. Fischbach BV, Trout KL, Lewis J, Luis CA,Sika M: WAGR syndrome: A clinical reviewof 54 cases. Pediatrics 116: 984–988, 2005
37. Clericuzio, C: WAGR syndrome. In: Man-agement of Genetic Syndromes. 2nd ed.edited by Cassidy S, Allanson J, New York,:John Wiley & Sons, 2004
38. Glaser T, Jepeal L, Edwards JG, Young SR,Favor J, Maas RL: PAX6 gene dosage effectin a family with congenital cataracts, aniridia,anophthalmia and centra-nervous-systemdefects. Nat Genet 7: 463–471, 1994
39. Talamillo A, Quinn JC, Collinson JM, CaricD, Price DJ, West JD, Hill RE: PAX6 regu-lates regional development and neuronalmigration in the cerebral cortex. Dev Biol255: 151–163, 2003
40. Lee H, Khan R, O’Keefe M: Aniridia: Cur-rent pathology and management. ActaOphthalmol 86: 708–715, 2008
41. Satran D, Pierpont MEM, Dobyns WB: Cer-ebello-oculo-renal syndromes includingArima, Senior-Loken and COACH syn-dromes: More than just variants of Joubertsyndrome. Am J Med Genet 86: 459–469,1999
42. Doherty D, Parisi M, Finn L, Gunay-AygunM, Al-Mateen M, Bates D, Clericuzio C,Demir H, Dorschner M, van Essen A, GahlWA, Gentile M, Gorden N, Hikida A, Knut-zen D, Ozyurek H, Phelps I, Rosenthal P,Verloes A, Weigand H, Chance P, DobynsWB, Glass I: Mutations in 3 genes (MKS3,CC2D2A, RPGRIP1L) cause COACH syn-drome (Joubert syndrome with congenitalhepatic fibrosis). J Med Genet 47: 8–21,2010
43. Brancati F, Iannicelli M, Travaglini L, Maz-zotta A, Bertini E, Boltshauser E, D’ArrigoS, Emma F, Fazzi E, Gallizzi R, Gentile M,Loncarevic D, Mejaski-Bosnjak V, Panta-leoni C, Rigoli L, Salpietro DC, Signorini S,Stringini G, Verloes A, Zabloka D, the In-ternational JSRD Study Group, Dallapi-ccola B, Gleeson J, Valente EM: MKS3/TMEM67 mutations are a major cause ofCOACH Syndrome, a Joubert Syndromerelated disorder with liver involvement.Hum Mutat 30: E432–E442, 2009
44. Kumar S, Rankin R: Renal insufficiency is acomponent of COACH syndrome. Am JMed Genet 61: 122–126, 1996
45. Mullins RF, Aptsiauri N, Hageman GS:Structure and composition of drusen asso-ciated with glomerulonephritis: Implica-tions for the role of complement activationin drusen biogenesis. Eye 15: 390–395,2001
46. Savige J, Liu J, Cabrera DeBuc D, HandaJT, Hageman GS, Wang YY, Parkin JD,Vote B, Fassett R, Sarks S, Colville, D: Ret-inal basement membrane abnormalitiesand the retinopathy of Alport syndrome.Invest Ophthalmol Vis Sci 51: 1621–1627,2010
47. Savige J, Colville D: Ocular features aid thediagnosis of Alport syndrome. Nat RevNephrol 5: 356–360, 2009
48. Chen L, Miyamura N, Ninomiya Y, HandaJT: Distribution of the collagen IV isoformsin human Bruch’s membrane. Br J Ophthal-mol 87: 212–215, 2003
49. Shaw EA, Colville D, Wang YY, Zhang KW,Dagher H, Fassett R, Guymer R, Savige J:Characterization of the peripheral retinop-athy in X-linked and autosomal recessiveAlport syndrome. Nephrol Dial Transplant22: 104–108, 2007
BRIEF REVIEW www.jasn.org
1412 Journal of the American Society of Nephrology J Am Soc Nephrol 22: 1403–1415, 2011

50. Usui T, Ichibe M, Hasegawa S, Miki A, BabaE, Tanimoto N, Abe H: Symmetrical re-duced retinal thickness in a patient withAlport syndrome. Retina 24: 977–979,2004
51. Rahman W, Banerjee S: Giant macular holein Alport syndrome. Can J Ophthalmol 42:314–315, 2007
52. Cervantes-Coste G, Fuentes-Paez G, Ye-shurun I, Jimenez-Sierra JM: Tapetal-likesheen associated with fleck retinopathy inAlport syndrome. Retina 23: 245–247,2003
53. Colville D, Wang YY, Tan R, Savige J: Theretinal “lozenge’’ or ”dull macular reflex’’in Alport syndrome may be associatedwith a severe retinopathy and early-onsetrenal failure. Br J Ophthalmol 93: 383–386, 2009
54. Jais JP, Knebelmann B, Giatras I, De MarchiM, Rizzoni G, Renieri A, Weber M, Gross O,Netzer KO, Flinter F, Pirson Y, Verellen C,Wieslander J, Persson U, Tryggvason K, Mar-tin P, Hertz JM, Schroder C, Sanak M, Krej-cova S, Carvalho MF, Saus J, Antignac C,Smeets H, Gubler MC: X-linked Alport syn-drome: Natural history in 195 families andgenotype-phenotype correlations in males.J Am Soc Nephrol 11: 649–657, 2000
55. Gross O, Netzer KO, Lambrecht R, Sei-bold S, Weber M: Meta-analysis of geno-type-phenotype correlation in X-linked Al-port syndrome: Impact on clinical counselling.Nephrol Dial Transplant 17: 1218 –1227,2002
56. Tan R, Colville D, Wang YY, Rigby L,Savige, J: Alport retinopathy results from“severe” COL4A5 mutations and predictsearly renal failure. Clin J Am Soc Nephrol 5:34–38, 2010
57. Simon P, Ramee MP, Autuly V, Laruelle E,Charasse C, Cam G, Ang KS: Epidemiologyof primary glomerular-diseases in a Frenchregion. Variations according to period andage. Kidney Int 46: 1192–1198, 1994
58. Smith RJH, Alexander J, Barlow PN, BottoM, Cassavant TL, Cook HT, de Cordoba SR,Hageman GS, Jokiranta TS, KimberlingWJ, Lambris JD, Lanning LD, Levidiotis V,Licht C, Lutz HU, Meri S, Pickering MC,Quigg RJ, Rops AL, Salant DJ, Sethi S,Thurman JM, Tully HF, Tully SP, van derVlag J, Walker PD, Wurzner R, Zipfel PF:New approaches to the treatment of densedeposit disease. J Am Soc Nephrol 18:2447–2456, 2007
59. Licht C, Heinen S, Jozsi M, Loschmann I,Saunders RE, Perkins SJ, Waldherr R,Skerka C, Kirschfink M, Hoppe B, Zipfel PF:Deletion of Lys224 in regulatory domain 4of Factor H reveals a novel pathomecha-nism for dense deposit disease (MPGN II).Kidney Int 70: 42–50, 2006
60. Abrera-Abeleda MA, Nishimura C, SmithJLH, Sethi S, McRae JL, Murphy BF, Silves-tri G, Skerka C, Jozsi M, Zipfel PF, Hage-
man GS, Smith RJH: Variations in the com-plement regulatory genes factor H (CFH)and factor H related 5 (CFHR5) are associ-ated with membranoproliferative glomeru-lonephritis type II (dense deposit disease).J Med Gen 43: 582–589, 2006
61. Duvall-Young J, Short CD, Raines MF,Gokal R, Lawler W: Fundus changes in me-sangiocapillary glomerulonephritis type II:Clinical and fluorescein angiographic find-ings. Br J Ophthalmol 73: 900–906, 1989
62. Colville D, Guymer R, Sinclair RA, Savige,J: Visual impairment caused by retinalabnormalities in mesangiocapillary(membranoproliferative) glomerulone-phritis type II (“dense deposit disease”).Am J Kidney Dis 42: E2–E5, 2003
63. Hildebrandt F, Zhou WB: Nephronophthi-sis-associated ciliopathies. J Am Soc Neph-rol 18: 1855–1871, 2007
64. Hildebrandt F, Attanasio M, Otto E:Nephronophthisis: Disease mechanisms of aciliopathy. J Am Soc Nephrol 20: 23–35,2009
65. Clarke MP, Sullivan TJ, Francis C, BaumalR, Fenton T, Pearce WG: Senior-Loken syn-drome. Case reports of two siblings andassociation with sensorineural deafness.Br J Ophthalmol 76: 171–172, 1992
66. Badano JL, Leitch CC, Ansley SJ, May-Simera H, Lawson S, Lewis RA, Beales PL,Dietz HC, Fisher S, Katsanis N: Dissectionof epistasis in oligogenic Bardet-Biedl syn-drome. Nature 439: 326–330, 2006
67. Beales PL, Elcioglu N, Woolf AS, Parker D,Flinter FA: New criteria for improved diag-nosis of Bardet-Biedl syndrome: Results ofa population survey. J Med Genet 36: 437–446, 1999
68. Kim LS, Fishman GA, Seiple WH, Szlyk JP,Stone EM: Retinal dysfunction in carriers ofBardet-Biedl syndrome. Ophthalmic Genet28: 163–168, 2007
69. Ferland RJ, Eyaid W, Collura RV, Tully LD,Hill RS, Al-Nouri D, Al-Rumayyan A,Topcu M, Gascon G, Bodell A, ShugartYY, Ruvolo M, Walsh CA: Abnormal cer-ebellar development and axonal de-cussation due to mutations in AHI1 inJoubert syndrome. Nat Genet 36: 1126,2004
70. Valente EM, Brancat F, Silhavy JL, CastoriM, March SE, Barrano G, Bertini E, Bolt-shauser E, Zaki MS, Abdel-Aleem A, Abdel-Salam GMH, Bellacchlo E, Battini R, CruseRP, Dobyns WB, Krishnamoorthy KS,Lagier-Tourenne C, Magee A, Pascual-Cas-troviejo I, Salpietro CD, Sarco D, Dallapi-ccola B, Gleeson JG: AHI1 gene mutationscause specific forms of Joubert syndrome-related disorders. Ann Neurol 59: 527–534,2006
71. Mollet G, Salomon R, Gribouval O, Silber-mann F, Bacq D, Landthaler G, Milford D,Nayir A, Rizzoni G, Antignac C, Saunier S:The gene mutated in juvenile nephronoph-
thisis type 4 encodes a novel protein thatinteracts with nephrocystin. Nat Genet 32:459, 2002
72. Tuysuz B, Baris S, Aksoy F, Madazli R,Ungur S, Sever L: Clinical variability ofasphyxiating thoracic dystrophy (Jeune)syndrome: Evaluation and classificationof 13 patients. Am J Med Genet 149A:1727–1733, 2009
73. Marshall JD, Bronson RT, Collin GB, Nord-strom AD, Maffei P, Paisey RB, Carey C,MacDermott S, Russell-Eggitt I, Shea SE,Davis J, Beck S, Shatirishvili G, Mihai CM,Hoeltzenbein M, Pozzan GB, Hopkinson I,Sicolo N, Nagget JK, Nishina PM: New Al-strom syndrome phenotypes based on theevaluation of 182 cases. Arch Int Med 165:675–683, 2005
74. Kottgen A, Hwang SJ, Larson MG, Van EykJE, Fu Q, Benjamin EJ, Dehghan A, GlazerNL, Kao WH, Harris TB, Gudnason V, Sh-lipak MG, Yang Q, Coresh J, Levy D, FoxCS: Uromodulin levels associate with acommon UMOD variant and risk for inci-dent CKD. J Am Soc Nephrol 21: 337–344,2010
75. Bleyer AJ: Improving the recognition of he-reditary interstitial kidney disease. J AmSoc Nephrol 20: 11–13, 2009
76. Dahan K, Devuyst O, Smaers M, Vertom-men D, Loute G, Poux JM, Viron B, JacquotC, Gagnadoux MF, Chauveau D, BuchlerM, Cochat P, Cosyns JP, Mougenot B,Rider MH, Antignac C, Verellen-DumoulinC, Pirson Y: A cluster of mutations in theUMOD gene causes familial juvenile hype-ruricemic nephropathy with abnormal ex-pression of uromodulin. J Am Soc Nephrol14: 2883–2893, 2003
77. Jacquet A, Guiochon-Mantel A, Noel LH,Sqalli T, Bedossa P, Hadchouel M, Grun-feld JP, Fakhouri F: Alagille syndrome inadult patients: It is never too late. Am JKidney Dis 49: 705–709, 2007
78. Li LH, Krantz ID, Deng Y, Genin A, BantaAB, Collins CC, Qi M, Trask BJ, Kuo WL,Cochran J, Costa T, Pierpont MEM, RandEB, Piccoli DA, Hood L, Spinner NB:Alagille syndrome is caused by mutationsin human Jagged1, which encodes a li-gand for Notch1. Nat Genet 16: 243–251,1997
79. Hingorani M, Nischal KK, Davies A, BentleyC, Vivian A, Baker AJ, Mieli-Vergani G, BirdAC, Aclimandos WA: Ocular abnormalitiesin Alagille syndrome. Ophthalmology 106:330–337, 1999
80. Sue CM, Mitchell P, Crimmins DS, Moshe-gov C, Byrne E, Morris JGL: Pigmentaryretinopathy associated with the mitochon-drial DNA 3243 point mutation. Neurology49: 1013–1017, 1997
81. Jansen JJ, Maassen JA, van der Woude FJ,Lemmink HA, van den Ouweland JM, t’ HartLM, Smeets HJ, Bruijn JA, Lemkes HH: Mu-tation in mitochondrial tRNA(Leu(UUR)) gene
BRIEF REVIEWwww.jasn.org
J Am Soc Nephrol 22: 1403–1415, 2011 Retinal Abnormalities in Inherited Renal Disease 1413

associated with progressive kidney disease.J Am Soc Nephrol 8: 1118–1124, 1997
82. Yerdelen D, Koc F, Koc Z: Delayed diagno-sis of Kearns-Sayre syndrome in a 38-year-old male patient: A case report. Int J Neu-rosci 118: 267–275, 2008
83. Emma F, Pizzini C, Tessa A, Di Giando-menico S, Onetti-Muda A, Santorelli FM,Bertini E, Rizzoni G: “Bartter-like” pheno-type in Kearns-Sayre syndrome. PediatrNephrol 21: 355–360, 2006
84. Booth C, Preston R, Clark G, Reidy J:Management of renal vascular disease inneurofibromatosis type 1 and the role ofpercutaneous transluminal angioplasty.Nephrol Dial Transplant 17: 1235–1240,2002
85. Lecleire-Collet A, Cohen SY, Vignal C,Gaudric A, Quentel G: Retinal ischaemia intype 1 neurofibromatosis. Br J Ophthalmol90: 117, 2006
86. Vianna RNG, Pacheco DF, VasconcelosMM, de Laey, J-J: Combined hamartomaof the retina and retinal pigment epithe-lium associated with neurofibromatosistype-1. Int Ophthalmol 24: 63– 66, 2001
87. Crino PB, Nathanson KL, Henske EP: Thetuberous sclerosis complex. N Engl J Med355: 1345–1356, 2006
88. Brook-Carter PT, Peral B, Ward CJ, Thomp-son P, Hughes J, Maheshwar MM, NellistM, Gamble V, Harris PC, Sampson JR: De-letion of the TSC2 and PKD1 genes asso-ciated with severe infantile polycystic kid-ney disease: A contiguous gene syndrome.Nat Genet 8: 328–332, 1994
89. Rakowski SK, Winterkorn EB, Paul E, SteeleDJR, Halpern EF, Thiele EA: Renal manifes-tations of tuberous sclerosis complex: Inci-dence, prognosis, and predictive factors.Kidney Int 70: 1777–1782, 2006
90. Robertson D: Ophthalmic manifestations oftuberous sclerosis. Ann N Y Acad Sci 615:17–25, 1991
91. Gould DB, Marchant JK, Savinova OV,Smith RS, John SWM: COL4A1 mutationcauses endoplasmic reticulum stress andgenetically modifiable ocular dysgenesis.Hum Mol Genet 16: 798–807, 2007
92. Plaisier E, Alamowitch S, Gribouval O,Mougenot B, Gaudric A, Antignac C, Roul-let E, Ronco P: Autosomal-dominant famil-ial hematuria with retinal arteriolar tortuos-ity and contractures: A novel syndrome.Kidney Int 67: 2354–2360, 2005
93. Plaisier E, Gribouval O, Alamowitch S,Mougenot B, Prost C, Verpont MC, MarroB, Desmettre T, Cohen SY, Roullet E, Dra-con M, Fardeau M, Van Agtmael T, Kerj-aschki D, Antignac C, Ronco P: COL4A1mutations and hereditary angiopathy, ne-phropathy, aneurysms, and muscle cramps.N Engl J Med 357: 2687–2695, 2007
94. Clarke JTR: Narrative review: Fabry dis-ease. Ann Intern Med 146: 425– 433,2007
95. Nance CS, Klein CJ, Banikazemi M, DikmanSH, Phelps RG, McArthur JC, Rodriguez M,Desnick RJ: Later-onset Fabry disease: Anadult variant presenting with the cramp-fasciculation syndrome. Arch Neurol 63:453–457, 2006
96. Branton MH, Schiffmann R, Sabnis SG,Murray GJ, Quirk JM, Altarescu G, Gold-farb L, Brady RO, Balow JE, Austin HA,Kopp JB: Natural history of Fabry renal dis-ease: Influence of alpha-galactosidase Aactivity and genetic mutations on clinicalcourse. Medicine 81: 122–138, 2002
97. Flynn DM, Boothby CB, Lake BD, Young,EP: Gut lesions in Fabry’s disease without arash. Arch Dis Child 47: 26–33, 1972
98. Ko YH, Kim HJ, Roh YS, Park CK, Kwon CK,Park MH: Atypical Fabry’s disease: An oli-gosymptomatic variant. Arch Pathol LabMed 120: 86–89, 1996
99. Wang RY, Lelis A, Mirocha J, Wilcox WR:Heterozygous Fabry women are not justcarriers, but have a significant burden ofdisease and impaired quality of life. GenetMed 9: 34–45, 2007
100. Nakao S, Kodama C, Takenaka T, TanakaA, Yasumoto Y, Yoshida A, Kanzaki T, En-riquez ALD, Eng CM, Tanaka H, Tei C,Desnick RJ: Fabry disease: Detection of un-diagnosed hemodialysis patients and iden-tification of a “renal variant” phenotype.Kidney Int 64: 801–807, 2003
101. Nagao Y, Nakashima H, Fukuhara Y,Shimmoto M, Oshima A, Ikari Y, Mori Y,Sakuraba H, Suzuki Y: Hypertrophic car-diomyopathy in late-onset variant ofFabry disease with high residual activityof alpha-galactosidase-A. Clin Genet 39:233–237, 1991
102. Wozniak MA, Kittner SJ, Tuhrim S, ColeJW, Stern B, Dobbins M, Grace ME,Nazarenko I, Dobrovolny R, McDade E,Desnick RJ: Frequency of unrecognizedFabry disease among young European-American and African-American men withfirst ischemic stroke. Stroke 41: 78 – 81,2010
103. Latif F, Tory K, Gnarra J, Yao M, Duh FM,Orcutt ML, Stackhouse T, Kuzmin I, ModiW, Geil L, Schmidt L, Zhou FW, Li H, WeiMH, Chen F, Glenn G, Choyke P, WaltherMM, Weng YK, Duan DSR, Dean M, Gla-vac D, Richards FM, Crossey PA, Fergu-sonsmith MA, Lepaslier D, Chumakov I,Cohen D, Chinault AC, Maher ER, Line-han WM, Zbar B, Lerman MI: Identifica-tion of the von Hippel-Lindau disease tu-mor-suppressor gene. Science 260:1317–1320, 1993
104. Chan CC, Vortmeyer AO, Chew EY, GreenWR, Matteson DM, Shen DF, Linehan WM,Lubensky IA, Zhuang ZP: VHL gene dele-tion and enhanced VEGF gene expressiondetected in the stromal cells of retinal an-gioma. Arch Ophthalmol 117: 625–630,1999
105. Chen F, Kishida T, Yao M, Hustad T, GlavacD, Dean M, Gnarra JR, Orcutt ML, Duh FM,Glenn G, Green J, Hsia YE, Lamiell J, Li H,Wei MH, Schmidt L, Tory K, Kuzmin I,Stackhouse T, Latif F, Linehan WM, LermanM, Zbar B: Germline mutations in the vonHippel-Lindau disease tumor-suppressorgene: Correlations with phenotype. HumMutat 5: 66–75, 1995
106. Dollfus H, Massin P, Taupin P, Nemeth C,Amara S, Giraud S, Beroud C, Dureau P,Gaudric A, Landais P, Richard S: Retinalhemangioblastoma in von Hippel-Lindaudisease: A clinical and molecular study. In-vest Ophthalmol Vis Sci 43: 3067–3074,2002
107. Wittebol-Post D, Hes FJ, Lips CJM: Theeye in von Hippel-Lindau disease. Long-term follow-up of screening and treatment:Recommendations. J Intern Med 243: 555–561, 1998
108. Khan GA, Melman A, Bank N: Renal in-volvement in neurocutaneous syndromes.J Am Soc Nephrol 5: 1411–1417, 1995
109. Sullivan TJ, Clarke MP, Morin JD: The oc-ular manifestations of the Sturge-Webersyndrome. J Pediatr Ophthalmol Strabis-mus 29: 349–356, 1992
110. Sadda SR, Miller NR, Tamargo R, Wityk R:Bilateral optic neuropathy associated withdiffuse cerebral angiomatosis in Sturge-Weber syndrome. J Neuro-Ophthalmol 20:28–31, 2000
111. Said R, Hamzeh Y, Said S, Tarawneh M,al-Khateeb M: Spectrum of renal involve-ment in familial mediterranean fever. Kid-ney Int 41: 414–419, 1992
112. Ando E, Ando Y, Maruoka S, Sakai Y, Wa-tanabe S, Yamashita R, Okamura R, Araki S:Ocular microangiopathy in familial amy-loidotic polyneuropathy, type 1. GraefesArch Clin Exp Ophthalmol 230:1–5, 1992
113. Kawaji T, Ando Y, Nakamura M, Ya-mashita T, Wakita M, Ando E, Hirata A,Tanihara H: Ocular amyloid angiopathyassociated with familial amyloidotic poly-neuropathy caused by amyloidogenic tran-sthyretin Y114C. Ophthalmology 112: 2212–2218, 2005
114. Noble KG: Bilateral multifocal retinal arte-riolar sheathing as the only ocular finding inhereditary amyloidosis. Am J Ophthalmol125: 111–113, 1998
115. Frasca GM, Soverini L, Tampieri E, France-schini G, Calabresi L, Pisciotta L, Preda P,Vangelista A, Stefoni S, Bertolini S: A 33-year-old man with nephrotic syndrome andlecithin-cholesterol acyltransferase (LCAT)deficiency. Description of two new muta-tions in the LCAT gene. Nephrol DialTransplant 19: 1622–1624, 2004
116. Ayyobi AF, McGladdery SH, Chan S, Man-cini GBJ, Hill JS, Frohlich JJ: Lecithin: Cho-lesterol acyltransferase (LCAT) deficiencyand risk of vascular disease: 25 year follow-up. Atherosclerosis 177: 361–366, 2004
BRIEF REVIEW www.jasn.org
1414 Journal of the American Society of Nephrology J Am Soc Nephrol 22: 1403–1415, 2011

117. Hirano K, Kachi S, Ushida C, Naito M: Cor-neal and macular manifestations in a caseof deficient lecithin: cholesterol acyltrans-ferase. Jpn J Ophthalmol 48: 82–84, 2004
118. Nadim F, Walid H, Adib J: The differentialdiagnosis of crystals in the retina. Int Oph-thalmol 24: 113–121, 2001
119. Kopp N, Leumann E: Changing pattern ofprimary hyperoxaluria in Switzerland. Neph-rol Dial Transplant 10: 2224–2227, 1995
120. Bobrowski AE, Longman CB: The primaryhyperoxalurias. Semin Nephrol 28: 152–162, 2008
121. Blaschke S, Grupp C, Haase J, KleinoederT, Hallermann C, Troche I, Grone HJ,Muller, GA: A case of late-onset primaryhyperoxaluria type 1. Am J Kidney Dis 39:E11, 2002
122. Wells CG, Johnson RJ, Luo QL, BuntmilamAH, Kalina RE: Retinal oxalosis. A clinico-pathologic report. Arch Ophthalmol 107:1638–1643, 1989
123. Small KW, Letson R, Scheinman J: Ocularfindings in primary hyperoxaluria. ArchOphthalmol 108: 89–93, 1990
124. Gahl WA, Thoene JG, Schneider, JA: Cys-tinosis. N Engl J Med 347: 111–121, 2002
125. Nesterova G, Gahl W: Nephropathic cysti-nosis: Late complications of a multisys-temic disease. Pediatr Nephrol 23: 863–878, 2008
126. Town M, Jean G, Cherqui S, Attard M,Forestier L, Whitmore SA, Callen DF, Gri-bouval O, Broyer M, Bates GP, van’t HoffW, Antignac C: A novel gene encoding anintegral membrane protein is mutated innephropathic cystinosis. Nat Genet 18:319–324, 1998
127. Shotelersuk V, Larson D, Anikster Y, Mc-Dowell G, Lemons R, Bernardini I, Guo JR,Thoene J, Gahl WA: CTNS mutations in anAmerican-based population of cystinosispatients. Am J Hum Genet 63: 1352–1362,1998
128. Tsilou ET, Rubin BI, Reed G, Caruso RC,Iwata F, Balog J, Gahl WA, Kaiser-Kupfer MI:Nephropathic cystinosis: Posterior segmentmanifestations and effects of cysteamine
therapy. Ophthalmology 113: 1002–1009,2006
129. Servais A, Moriniere V, Grunfeld JP, NoelLH, Goujon JM, Chadefaux-Vekemans B,Antignac C: Late-onset nephropathic cysti-nosis: Clinical presentation, outcome, andgenotyping. Clin J Am Soc Nephrol 3: 27–35, 2008
130. Ji W, Foo JN, O’Roak B, Zhao H, Larson M,Simon D, Newton-Cheh C, State M, LevyD, Lifton R: Rare independent mutations inrenal salt handling genes contribute toblood pressure variation. Nat Genet 40:592–599, 2008
131. Gitelman HJ, Graham JB, Welt LG: A newfamilial disorder characterized by hypoka-lemia and hypomagnesemia. Trans AssocAm Physicians 79: 221–235, 1966
132. Marchini G, Tosi R, Parolini B, Castagna G,Zarbin M: Choroidal calcification in Bartter syn-drome. Am J Ophthalmol 126: 727–729, 1998
133. Honavar SG, Shields CL, Demirci H, ShieldsJA: Sclerochoroidal calcification: Clinicalmanifestations and systemic associations.Arch Ophthalmol 119: 833–840, 2001
134. Lemley KV: Kidney disease in nail-patellasyndrome. Pediatr Nephrol 24: 2345–2354,2009
135. Dreyer SD, Zhou G, Baldini A, WinterpachtA, Zabel B, Cole W, Johnson RL, Lee B:Mutations in LMX1B cause abnormal skel-etal patterning and renal dysplasia in nailpatella syndrome. Nat Genet 19: 47–50,1998
136. Chen H, Lun Y, Ovchinnikov D, Kokubo H,Oberg KC, Pepicelli CV, Gan L, Lee B,Johnson RL: Limb and kidney defects inLmx1b mutant mice suggest an involve-ment of LMX1B in human nail patella syn-drome. Nat Genet 19: 51–55, 1998
137. Mimiwati Z, Mackey DA, Craig JE, Mac-Kinnon JR, Rait JL, Liebelt JE, Ayala-LugoR, Vollrath D, Richards JE: Nail-patella syn-drome and its association with glaucoma: Areview of eight families. Br J Ophthalmol90: 1505–1511, 2006
138. Sweeney E, Fryer A, Mountford R, Green A,McIntosh I: Nail patella syndrome: A review
of the phenotype aided by developmentalbiology. J Med Genet 40: 153–162, 2003
139. Galloway G, Vivian A: An ophthalmicscreening protocol for nail-patella syn-drome. J Pediatr Ophthalmol Strabismus40: 51–53, 2003
140. Fonck C, Chauveau D, Gagnadoux MF, Pir-son Y, Grunfeld JP: Autosomal recessivepolycystic kidney disease in adulthood.Nephrol Dial Transplant 16: 1648–1652,2001
141. Bollee G, Fakhouri F, Karras A, Noel LH,Salomon R, Servais A, Lesavre P, MoriniereV, Antignac C, Hummel A: Nephronophthi-sis related to homozygous NPHP1 genedeletion as a cause of chronic renal failurein adults. Nephrol Dial Transplant 21:2660–2663, 2006
142. Azari AA, Aleman TS, Cideciyan AV,Schwartz SB, Windsor EAM, Sumaroka A,Cheung AY, Steinberg JD, Roman AJ,Stone EM, Sheffield VC, Jacobson SG: Ret-inal disease expression in Bardet-Biedl syn-drome-1 (BBS1) is a spectrum from macu-lopathy to retina-wide degeneration. InvestOphthalmol Vis Sci 47: 5004–5010, 2006
143. Schonck M, Hoorntje S, vanHooff J: Renaltransplantation in Alagille syndrome. Neph-rol Dial Transplant 13: 197–199, 1998
144. Meikle PJ, Hopwood JJ, Clague AE, CareyWF: Prevalence of lysosomal storage disor-ders. JAMA 281: 249–254, 1999
145. Hillsley RE, Hernandez E, Steenbergen C,Bashore TM, Harrison JK: Inherited restric-tive cardiomyopathy in a 74-year-old wom-an: A case of Fabry’s disease. Am Heart J129: 199–202, 1995
146. Devriendt K, Matthijs G, Van Damme B,Van Caesbroeck D, Eccles M, Vanrenter-ghem Y, Fryns JP, Leys A: Missense muta-tion and hexanucleotide duplication in thePAX2 gene in two unrelated families withrenal-coloboma syndrome (MIM 120330).Hum Genet 103: 149–153, 1998
147. Zaffanello M, Taranta A, Palma A, BettinelliA, Marseglia GL, Emma F: Type IV Barttersyndrome: Report of two new cases. Pedi-atr Nephrol 21: 766–770, 2006
BRIEF REVIEWwww.jasn.org
J Am Soc Nephrol 22: 1403–1415, 2011 Retinal Abnormalities in Inherited Renal Disease 1415





























