Embed Size (px)
Citation preview

Real-Time Monitoring and Parameter Estimationof the Emulsion Polymerization of CarboxylatedStyrene/Butadiene Latexes
Matheus Soares,1 Fabricio Machado,2 Alessandro Guimaraes,3 Marcelo M. Amaral,4 Jose Carlos Pinto1
1 Programa de Engenharia Quımica/COPPE, Universidade Federal do Rio de Janeiro, Cidade Universitaria,CP 68502, Rio de Janeiro 21941-972 RJ, Brazil
2 Instituto de Quımica, Universidade de Brasılia, Campus Universitario Darcy Ribeiro, CP 04478,Brasılia 70910-900 DF, Brazil
3 IQT – Industrias Quımicas Taubate, Rua Irmaos Albernaz 300, Taubate 12050-190 SP, Brazil
4 Accenture – Av. Republica do Chile, 500/188 andar, Rio de Janeiro 20031-170 RJ, Brazil
This work presents a mathematical model for semibatchcarboxylated styrene/butadiene (XSBR) emulsion poly-merizations, intended for the online and real-time datareconciliation and monitoring of the polymerization re-actor. Proposed procedures assume that some parame-ters must be estimated in real time, for accommodationof the unavoidable fluctuations of industrial operationconditions. Pressure, temperature, and feed rate profilesare used for parameter estimation, allowing for real-timeprediction of important properties of XSBR latexes,including monomer conversions, solids contents, andcopolymer compositions. The proposed scheme wasvalidated with actual data obtained in Pilot-plant andfull-scale reactors. In all cases, estimated n valuesranged from 0.1 to 1.7, which agree with results reportedpreviously. As observed at plant, heat transfer coeffi-cients can change from batch-to-batch and experiencelarge changes during the batch due to modification ofthe reactor volume and accumulation of polymer mate-rial on the heat transfer areas. POLYM. ENG. SCI., 51:1919–1932, 2011. ª 2011 Society of Plastics Engineers
INTRODUCTION
The carboxylated styrene-butadiene (XSBR) industry
has suffered drastic changes in the last 20 years, due to
increasing demand for new products with enhanced per-
formance, the fierce negotiation between customers and
suppliers and the strong competition among producers.
These driving forces obliged XSBR latex producers to reply
rapidly to new demands from the market, through the offer
of high quality products at competitive prices [1].
Nowadays, emulsion polymers are extensively used in
several technological applications, although standard for-
mulations and reaction strategies used to perform emul-
sion polymerizations are still largely based on scientific
and technological developments carried out in the early
1950s [2]. However, it is also true that recent fundamental
discoveries and industrial applications are enabling the
development of the next generation of consumer and
industrial products [3]. Particularly, despite the wide-
spread use of XSBR latexes, there is an enormous gap
between the open scientific information and the industrial
practice in this field [4].
Polymer latexes produced by free-radical emulsion po-
lymerization are applied in multiple applications, includ-
ing paper and paperboard coating, adhesives, carpet back-
ing, textiles, among others [4, 5]. One of the main fields
of application of XSBR latexes is the paper and paper-
board industry. Copolymers of styrene-butadiene are
modified by the incorporation of a,b-unsaturated carbox-
ylic acids, whose presence contribute to change the opti-
cal properties and surface characteristics of the particle,
to enhance the colloidal stability, to increase the compati-
bility with inorganic fillers, and to produce films with
improved mechanical properties [6].
Because of the inherent characteristics of the polymeriza-
tion mechanism, emulsion polymerization processes are very
sensitive to small changes of the operation conditions, such
Correspondence to: Jose Carlos Pinto; e-mail: [email protected]
Contract grant sponsor: IQT – Industrias Quımicas Taubate (Sao Paulo,
Brazil); contract grant sponsor: CNPQ – Conselho Nacional de Desen-
volvimento Cientıfico e Tecnologico (Brazil).
DOI 10.1002/pen.22002
Published online in Wiley Online Library (wileyonlinelibrary.com).
VVC 2011 Society of Plastics Engineers
POLYMER ENGINEERING AND SCIENCE—-2011

as the presence of contaminants in the feed streams and mod-
ification of heat transfer coefficients due to fouling [7, 8].
Consequently, undesired fluctuations of the final products
properties are observed at plant site very frequently, impos-
ing the modification of production and operation policies
and leading to frequent plant intervention. Quite often, these
interventions are performed based solely on previous experi-
ence or tacit knowledge of plant engineers and operators,
making difficult the implementation of rational operation
and control strategies.
To control the final polymer quality, possibly compen-
sate for unknown perturbations of the process operation,
and assure the product specification, the on-line monitor-
ing of some key process variables, such as the monomer
conversion and the copolymer composition, can be of par-
amount importance [9]. In fact, it is well known that the
implementation of on-line monitoring and control strat-
egies leads to reduction of process variability and of pro-
duction costs, improvement of process consistency and
faster development of novel products, and responding to
client demands [10, 11].
Given the strong effect that reaction temperature exerts
on the chemical kinetics and final product properties, on
its importance for development and implementation of
safety procedures and on the easy and cheap measurement
of temperature signals at plant site, temperature profiles
are usually available in most polymerization processes.
For this reason, calorimetric techniques have been widely
used for the on-line monitoring and controlling of poly-
merization reactors in batch, semibatch, and continuous
operations [12–21]. Calorimetry can be defined as the
monitoring of heat balances in a reacting system, which
allows inferring of the rates of exothermic (or endother-
mic) reactions and of some additional correlated variables,
such as compositions. The technique is characterized by
its simplicity, as it depends almost exclusively on the
availability of temperature and flow-rate measurements of
the main process streams, which can be accomplished in
real time with the help of versatile, robust, and cheap
plant equipments.
Despite the many potential uses of calorimetry in a
real production environment, calorimetric techniques have
been normally used to monitor small lab-scale reactors,
usually in off-line mode [13, 14]. Besides, calorimetric
techniques have never been combined with first-principles
fundamental models to provide useful information about
the states of real full-scale industrial reactors, including
monomer conversions, copolymer composition, and solids
content.
Based on the previous paragraphs, the main objective
of this work is the development of a first-principles math-
ematical model to describe semibatch XSBR emulsion
polymerizations, intended for the online and real-time
data reconciliation and monitoring of polymerization reac-
tors. The proposed procedure assumes that some parame-
ters must be estimated in real time, for accommodation
of the unavoidable fluctuation of operation conditions, as
observed at plant site. A direct search complex algorithm
was used for estimation of model parameters based on
available pressure, temperature, and feed flow rate pro-
files, allowing for prediction of important properties of
XSBR latexes in real-time, such as the monomer conver-
sion, the solids content, and the copolymer composition.
The proposed model and monitoring strategy were vali-
dated with actual data obtained in a pilot plant reactor
and in a full-scale industrial process for the first time.
NUMERICAL PROCEDURES
Modeling
The kinetic mechanism used to describe the emulsion
copolymerization of XSBR latexes is based on the classi-
cal free radical polymerization model, comprising the
usual reaction steps: initiation, propagation, chain transfer
to monomer, chain transfer to a chain transfer agent,
chain transfer to dead polymer chains, and termination by
disproportionation and combination [22]. The terminal po-
lymerization model was used to describe the copolymer-
ization reactions [23, 24]. The basic reaction steps are
presented in Eqs. 1–25.
Initiation
I �!kD 2R (1)
RþM1 �!k1 P�1 (2)
RþM2 �!k2 Q�1 (3)
Propagation
P�i þM1 �!
kP11P�iþ1 (4)
P�i þM2 �!
kP12Q�
iþ1 (5)
Q�i þM1 �!
kP21P�iþ1 (6)
Q�i þM2 �!
kP22Q�
iþ1 (7)
Chain Transfer to Monomer
P�i þM1 �!
ktrM11 Ci þ P�1 (8)
P�i þM2 �!
ktrM12 Ci þ Q�1 (9)
Q�i þM1 �!
ktrM21 Ci þ P�1 (10)
Q�i þM2 �!
ktrM22 Ci þ Q�1 (11)
1920 POLYMER ENGINEERING AND SCIENCE—-2011 DOI 10.1002/pen

Chain Transfer to Polymer
P�i þ Gj �!
ktrP11 Gi þ P�j (12)
P�i þ Gj �!
ktrP12 Gi þ Q�j (13)
Q�i þ Gj �!
ktrP21 Gi þ P�j (14)
Q�i þ Gj �!
ktrP22 Gi þ Q�j (15)
Incorporation of Pendant Double Bonds
P�i þ Gj �!
ktR1Q�
iþj (16)
Q�i þ Gj �!
ktR2Q�
iþj (17)
Transfer to Chain Transfer Agent
P�i þ CTA �!
ktrCTA1 Gi þ P�1 (18)
Q�i þ CTA �!
ktrCTA2 Gi þ Q�1 (19)
Termination by Disproportionation
P�i þ P�
j �!kTD11 Gi þ Gj (20)
P�i þ Q�
j �!kTD12 Gi þ Gj (21)
Q�i þ Q�
j �!kTD22 Gi þ Gj (22)
Termination by Combination
P�i þ P�
j �!kTC11 Giþj (23)
P�i þ Q�
j �!kTC12 Giþj (24)
Q�i þ Q�
j �!kTC22 Giþj (25)
In Eqs. 1–25 I, R, M1, M2, P�i , and Q�
i represent the ini-
tiator, primary radicals, monomers 1 (styrene) and 2 (buta-
diene), and living chains of size i containing mers 1 and 2
at the active site, respectively. kd, k1, and k2 correspond to
the rate constants for initiator decomposition and initiation
of monomers 1 and 2, respectively. Gi is a dead polymer
chain of size i, while CTA represents a chain transfer agent.
kPi,j, kTCi,j, kTDi,j, ktrMi,j, ktrATCi, ktrPij, and ktRi are, respec-tively, the kinetic rate constants for propagation of radical iwith monomer j, for termination by combination of radicals
i and j, for termination by disproportionation of radicals iand j, for chain transfer of radical i to monomer j, for chain
transfer of radical i to the chain transfer agent, for chain
transfer of radical i to incorporate monomer j in the dead
polymer chain, and for incorporation of pendant double
bonds in the dead polymer chain by radical i.Besides the kinetic mechanism, the following hypothe-
ses were also assumed to be valid during model develop-
ment: (a) living radical chains are sufficiently long (long-
chain assumption); (b) rates of production and consumption
of living radical chains are equal (quasi-steady state
assumption); (c) the initiator is soluble in water, where it
decomposes and form radicals; (d) polymerization takes
place predominantly within the polymer particles, without
relevant nucleation of new particles (polymerizations are
seeded and performed in starved conditions, in absence of
monomer droplets); (e) the mass of the gas phase is negligi-
ble; (f) reactor phases are in thermal equilibrium; (g) the
jacket dynamics can be described as the dynamics of a
stirred tank; (h) volumes are additive and the copolymer
densities are computed as composition-averages of the
homopolymer densities; (i) the XSBR polymerization reac-
tion can be properly parameterized in terms of the styrene
and butadiene monomers.
For the purposes of this work and according to
assumption i, the XSBR reactor can be represented as a
binary copolymerization system and the addition of a,b-unsaturated carboxylic acids to the reacting system can be
disregarded. It is important to emphasize that this is not
equivalent to saying that the carboxylic acids are not im-
portant for description of some of the final properties of
the latexes, but that carboxylic acids are added in small
quantities and do not affect the dynamic trajectory of
most reaction variables very significantly. This is very im-
portant to reduce the dimension of the reactor model and
allow for real-time implementation of the process estima-
tor. Besides, as discussed in the following paragraphs,
some process parameters are estimated on-line and in real
time, which can compensate for perturbations of the pro-
cess operation conditions due to modification of the car-
boxylic acids content.
Based on these hypotheses, the mass balance equations
can be derived for the XSBR emulsion polymerization
process performed in batch or semibatch mode, as
described below.
dI
dt¼ �kDI þ FI; Ið0Þ ¼ I0 (26)
where FI is the molar feed rate of initiator.
dCTA
dt¼ � �nNT
NAktrCTA1X1 þ ktrCTA2X2
� �CTA
Voþ FCTA;
CTAð0Þ ¼ CTA0: ð27Þ
where n represents the average number of radicals per
polymer particle, NT represents the total number of poly-
mer particles, FCTA is the molar feed rate of chain trans-
fer agent and
DOI 10.1002/pen POLYMER ENGINEERING AND SCIENCE—-2011 1921

Xi ¼kPjiMi
kPjiMi þ kPijMjand Xj ¼ 1� Xi; i ¼ 1; 2 (28)
dM1
dt¼ � �nNT
NA
kP11X1 þ kP21X2ð ÞM1
Vo
� �nNT
NA
ktrM11X1 þ ktrM21
X2
� �M1
VO
þ F1; M1ð0Þ¼M10 ð29Þ
where F1 is the molar feed rate of monomer 1.
dM2
dt¼ � �nNT
NA
kP12X1 þ kP22X2ð ÞM2
Vo
� �nNT
NA
ktrM12X1 þ ktrM22
X2
� �M2
VO
þ F2;
M2ð0Þ ¼ M20
(30)
where F2 is the molar feed rate of monomer 2.
d}1
dt¼ �nNT
NA
kP11X1 þ kP21X2ð ÞM1
Vo
þ �nNT
NA
ktrM11X1 þ ktrM21
X2
� �M1
VO
; }1 0ð Þ ¼ }10 ð31Þ
d}2
dt¼ �nNT
NA
kP12X1 þ kP22X2ð ÞM2
VO
þ �nNT
NA
ktrM12X1 þ ktrM22
X2
� �M2
VO
; }2 0ð Þ ¼ }20 ð32Þ
where }i represents the moles of monomer i that were
incorporated into polymer chains.
dMw
dt¼ Fw; Mwð0Þ ¼ Mw0 (33)
where Fw is the molar feed rate of water.
The total volumes of the organic, aqueous, and liquid
phases are:
VO ¼ M1
r1MW1 þM2
r2MW2 þ }1
r}1
MW1 þ }2
r}2
MW2 (34)
VW ¼ Mw
rwMWw (35)
VT ¼ VO þ VW (36)
where MWi and ri represent the molecular weights and
densities of species i.The vapor pressures can be calculated as
ln PSATi
� � ¼ Ai � Bi
T þ Ci(37)
where PSATi corresponds to the vapor pressure of compo-
nent i, and Ai, Bi, and Ci are the constants of the Antoine
Equation. Therefore, the total pressure of the gas phase
can be calculated as
P ¼Xi
pi þ PSATw þ PInert i ¼ 1; 2 (38)
where pi is the partial pressure of monomer i, calculatedas
lnpi
PSATi
� �¼ lnfi þ 1� 1
m
� �fP þ wf2
P i ¼ 1; 2 (39)
where
fi ¼wi=riPNCj¼1 wj
.rj
i ¼ 1; 2 (40)
where wi represents the mass fraction of monomer i in the
organic phase.
fP ¼ 1� f1 þ f2ð Þ (41)
where w is the Flory–Huggins interaction parameter and mrepresents the molar volume ratio of polymer and mono-
mer (n2/n1).It assumed that one of the components in the gas phase
is an inert, which accumulates during the reaction process.
The presence of the inert originates from impurities in the
butadiene stream and standard limitations of the vacuum
system. The mass balance of the inert component is given
by:
dMInert
dt¼ wInertF2; MInert ¼ MInert0 (42)
where wInert is the molar fraction of volatile impurities in
the butadiene stream, and MInert0 is the initial volatile
mass in the reactor. Both MInert and MInert0 can be related
to the reactor pressure as:
PInert ¼ MInertRT
VR � VT
(43)
where VR is the total reactor volume and ideal gas
behavior was assumed. Before the addition of the feed
streams, MInert0 can be given by:
MInert0 ¼ P0 � VR
R � T (44)
where P0 is the total reactor pressure when reactor load-
ing is initiated.
The overall energy balance equation can be written in
the form:
Xni¼1
riCpiVTdT
dt¼Xni¼1
FiCpiðTei � TÞþQR þ QA � QT�QP
(45)
1922 POLYMER ENGINEERING AND SCIENCE—-2011 DOI 10.1002/pen

The energy generated by the agitation shaft, QA, can
be monitored during the process. In most cases, QA can
be neglected without compromising the overall heat bal-
ance. The rate of energy loss to the surroundings can be
estimated as QP ¼ (UA)a(T 2 Ta). The overall heat trans-
fer coefficient (UA)a can be estimated with available
operation data. Generally, for large insulated industrial
reactors, as in the case considered here, this term can be
neglected. Consequently, Eq. 45 can be rewritten as:
Xni¼1
riCpiVTdT
dt¼
Xni¼1
FiCpiðTei � TÞ þ QR � QT;
Tð0Þ ¼ T0 ð46Þwhere Cpi is the specific heat capacity of chemical spe-
cies i, and the remaining heat terms are defined below.
Similarly, the heat balance for the jacket can be given by:
rCCpCVC
dTCdt
¼ FCCpCðTeC � TCÞ þ QT; TCð0Þ ¼ TC0
(47)where:
QR ¼X2i¼1
Rið�DHiÞ (48)
andQT ¼ UAðT � TCÞ (49)
Finally, the overall monomer conversion, the solids
content, and the average copolymer compositions can be
calculated, respectively, in the form:
xM ¼ MW1}1 þMW2}2
MW1 }1 þM1ð Þ þMW2 }2 þM2ð Þ (50)
xS ¼ MW1}1 þMW2}2
MW1 }1 þM1ð Þ þMW2 }2 þM2ð Þ þMWwMw
(51)
= ¼ }1
}1 þ }2
(52)
Model equations were implemented in FORTRAN and
solved numerically with the integration package DDASSL
[25]. It is very important to emphasize that there is very little
information in the literature regarding the XSBR latex poly-
merization process, when polymerization is performed at high
temperature (above 60 8C). Whenever possible, kinetic pa-
rameters available in the open literature were used for simula-
tions [26–30]. When parameters were unavailable, estimation
was performed off-line with experimental data obtained from
the industrial polymerization process. All parameters required
for model simulations are presented in Table 1.
On-Line Data Reconciliation and Parameter Estimation
During real time applications, model parameters were
estimated on-line with the Monte Carlo method [31].
(Other methods, including the Complex algorithm [32],
the Gradient technique [33, 34], and the Newton method
[35], were also tested, but the performance of stochastic
optimization procedures were always much better both in
terms of computation speed and rate of failure, as also
described in the literature [36].) Direct search methods
are very useful for real-time applications, because they do
not require the calculation of the derivatives, the inversion
of matrices or definition of continuous objective functions
and constraints. As good initial guesses are always avail-
able in real-time applications, direct search methods are
usually faster and more robust [37].
The optimization problem can be defined as
minf x1; x2; :::; xnð Þ (53)
subject to
li � xi � ui i ¼ 1; 2; :::;m (54)
where x1,x2,. . .,xn, are the manipulated variables, and f isthe objective function that must be minimized. In this ar-
ticle, the objective function was defined as:
f x1; x2; :::; xnð Þ ¼XNYi¼1
yci � yei� �2
s2i(55)
where y is a model output (superscript c) that can be
compared to an available process measurement (reactor
pressure, reactor temperature, and jacket temperature, as
indicated by superscript e). s2i is an estimate of the var-
iance of the measurement error. The upper (ui) and lower
(li) limits of the constraints can be constant or functions
of the manipulated variables.
As the obtained optimum solution must satisfy the pro-
cess model, as described in Eqs. 1–52, the sequential optimi-
zation procedure was implemented as recommended in the
literature and presented in Fig. 1 [38]. According to the se-
quential optimization procedure, the first stage corresponded
to the optimization task, when optimum candidates were
generated. According to the Monte Carlo procedure, the can-
didate optimum solutions of the estimation problem must be
obtained by generating random numbers in the form
xjð Þ
i;k ¼ xj�1ð Þi;k þ r
ðjÞi;kD
ðjÞi i ¼ 1; :::;m k ¼ 1; :::; n ð56Þ
where rðjÞi;k are pseudorandom numbers uniformly distributed
in the interval (�1, 1) and DðjÞi defines a search interval for
variable i at iteration j. Convergence can be achieved by
reducing the search interval along the iterative procedure in
the form
DðjÞi ¼ Dðj�1Þ
i
li ¼ 1; :::;m (57)
where l is a real number greater than 1. The numerical pa-
rameters that must be defined for implementation of the
DOI 10.1002/pen POLYMER ENGINEERING AND SCIENCE—-2011 1923

optimization tasks are the number of candidate optimum
solutions (n, equal to 10, unless stated otherwise), the
search interval reduction factor (l, equal to 1.5, unless
stated otherwise) and the initial search intervals (Dð0Þi , equal
to 3si, unless stated otherwise). Initial guesses for measured
variables xð0Þi;k were always equal to the available values,
whereas initial values for estimated process parameters are
described in the following sections.
After generation of the candidate optimum solutions at
iteration j, the second stage comprised the numerical
integration of the algebraic-differential equations for each
optimum candidate and the posterior computation of
the objective function that must be minimized. Dynamic
trajectories were computed with minimum relative preci-
sion of 1.0 3 1026.
The numerical procedure was implemented in a desk-
top computer, equipped with an Intel1 Pentium1 4 proc-
essor, with CPU clock of 3 GHz, and 1 GB of RAM
memory. The operation data were imported from a propri-
etary spreadsheet, provided by the supplier of the dataFIG. 1. Proposed numerical scheme for the in-line parameter estimation.
TABLE 1. Model parameters used in the styrene/butadiene copolymerization model.
Parameters Units References
kd ¼ 1.0 � 1022 � cxp(2205000/R � T) s21 Brandrup et al. [26]
kp11 ¼ 2.16 � 107 � exp(232500/R � T) L mol21 s21 Brandrup et al. [26]
kp22 ¼ 1.2 � 107 � exp(239000/R � T) L mol21 s21 Brandrup et al. [26]
r1 ¼ 0.78 Dimensionless Hamielec et al. [27]
r2 ¼ 1.39 Dimensionless Hamielec et al. [27]
CtrM11¼ 0.65 � 1024 Dimensionless Brandrup et al. [26]
CtrM22¼ 2.32 � 1024 Dimensionless Brandrup et al. [26]
CtrATC¼ 0.66 Dimensionless Brandrup et al. [26]
n Dimensionless Estimated
NT Dimensionless Estimated
R J mol21 K21 Admitted
Cp1 ¼ 26.747 þ 1471 � 1021 � T29.609 � 1025 � T2 – 2.373 � 1028 � T3
cal g21 K21 Kalfas et al. [28]
Cp2 ¼ 123.60 J Kg21 K21 Brandrup et al. [26]
Cp}1¼ 0.283 2 0.966 � 1023 � T cal g21 K21 Kalfas et al. [28]
Cp}2¼ 1600 J Kg21 K21 Brandrup et al. [26]
CpH2O¼ �203:606þ 1523:290 � h
� 3196:413 � h2 þ 2474:455 � h3
þ 3:855532
h2
Com y ¼ 1023 � T
J mol21 K21 Chase [29]
(2DH1) ¼ 68500.0 J mol21 Brandrup et al. [26]
(2DH2) ¼ 73000.0 J mol21 Brandrup et al. [26]
q1 ¼ 0.9193 2 6.65�1024 � (T 2 273.15) g cm3 Kalfas et al. [28]
q2 ¼ 0.650 g cm3 Brandrup et al. [26]
q}1¼ 0.9926 2 2.65�1024 � (T 2 273.15) g cm3 Kalfas et al. [28]
q}2¼ 0.890 g cm3 Brandrup et al. [26]
qInic ¼ 2.5 g cm3 Brandrup et al. [26]
qCTA ¼ 2.1 g cm3 Brandrup et al. [26]
qw ¼ 1.000663 2 7.35�1025 � (T 2 273.15)
23.5 � 1026 � (T 2 273.15)2g cm3 Perry e Green [30]
1924 POLYMER ENGINEERING AND SCIENCE—-2011 DOI 10.1002/pen
acquisition system, and stored as text input files for poste-
rior manipulation of the FORTRAN code. Similarly,
estimated monomer conversion, solids content, and copol-
ymer composition were exported to the data acquisition
spreadsheet for posterior analysis and manipulation.
Unless stated otherwise, sampling times of 5 min were
used for estimation of model parameters and states, taking
less than 2 min for the optimization task to be finished.
RESULTS AND DISCUSSION
Model Validation
First, the process model was validated with experimen-
tal datasets obtained from polymerization reactions carried
out in a small Pilot-plant reactor (3 l) and in a full-scale
industrial plant. Detailed description of reactor geometry
and of polymerization recipe is not provided for proprie-
tary reasons.
Temperature and Pressure Profiles in the Industrial Plant
For validation of model performance in the industrial
process, available jacket temperatures and polymerization
recipes were used as inputs to calculate pressure and tem-
perature profiles, which were then compared to available
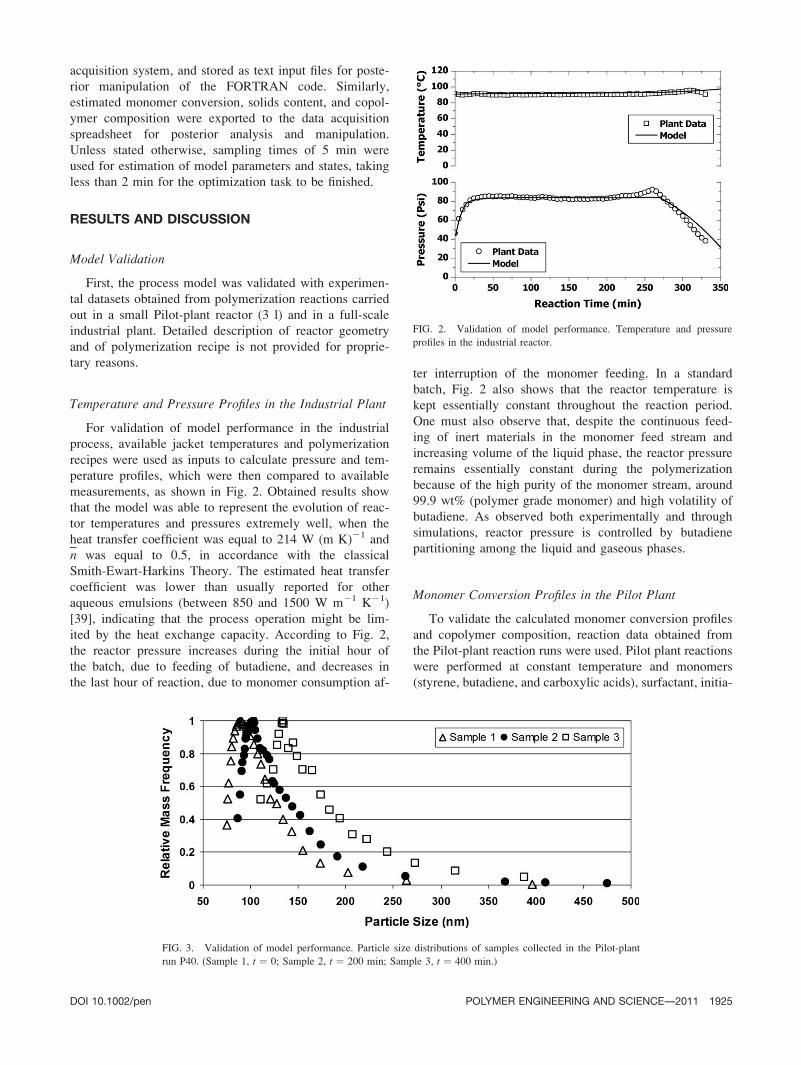
measurements, as shown in Fig. 2. Obtained results show
that the model was able to represent the evolution of reac-
tor temperatures and pressures extremely well, when the
heat transfer coefficient was equal to 214 W (m K)21 and
n was equal to 0.5, in accordance with the classical
Smith-Ewart-Harkins Theory. The estimated heat transfer
coefficient was lower than usually reported for other
aqueous emulsions (between 850 and 1500 W m21 K21)
[39], indicating that the process operation might be lim-
ited by the heat exchange capacity. According to Fig. 2,
the reactor pressure increases during the initial hour of
the batch, due to feeding of butadiene, and decreases in
the last hour of reaction, due to monomer consumption af-
ter interruption of the monomer feeding. In a standard
batch, Fig. 2 also shows that the reactor temperature is
kept essentially constant throughout the reaction period.
One must also observe that, despite the continuous feed-
ing of inert materials in the monomer feed stream and
increasing volume of the liquid phase, the reactor pressure
remains essentially constant during the polymerization
because of the high purity of the monomer stream, around
99.9 wt% (polymer grade monomer) and high volatility of
butadiene. As observed both experimentally and through
simulations, reactor pressure is controlled by butadiene
partitioning among the liquid and gaseous phases.
Monomer Conversion Profiles in the Pilot Plant
To validate the calculated monomer conversion profiles
and copolymer composition, reaction data obtained from
the Pilot-plant reaction runs were used. Pilot plant reactions
were performed at constant temperature and monomers
(styrene, butadiene, and carboxylic acids), surfactant, initia-
FIG. 2. Validation of model performance. Temperature and pressure
profiles in the industrial reactor.
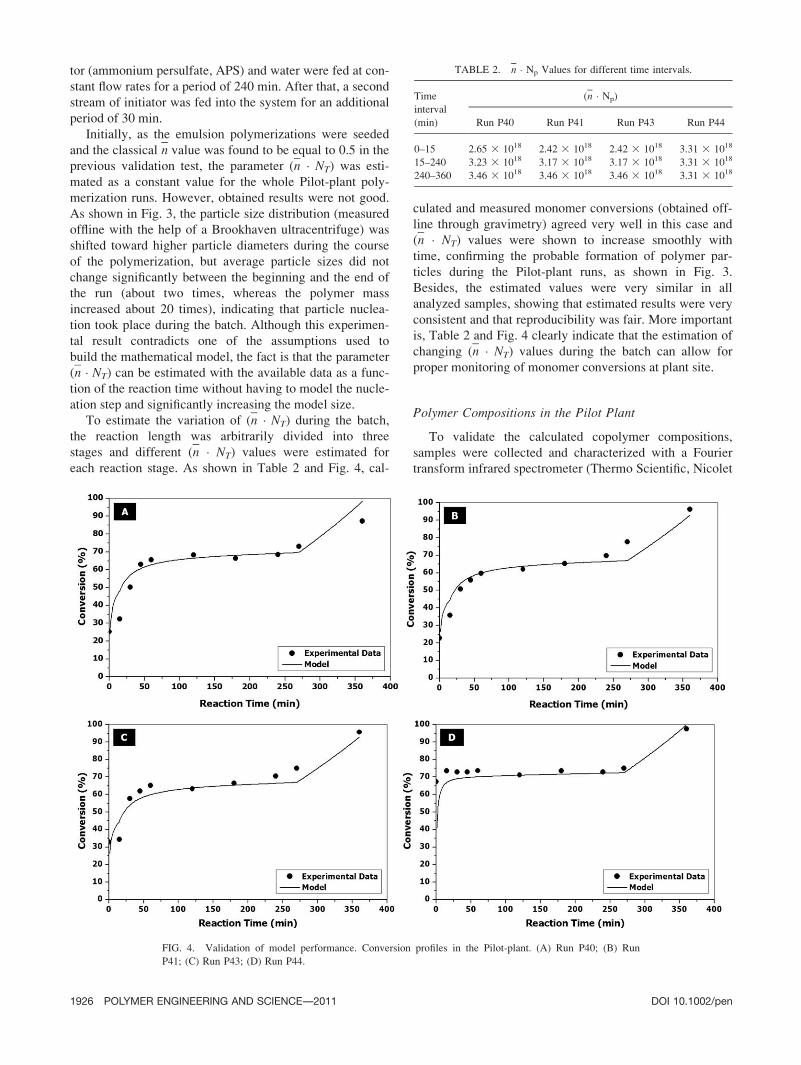
FIG. 3. Validation of model performance. Particle size distributions of samples collected in the Pilot-plant
run P40. (Sample 1, t ¼ 0; Sample 2, t ¼ 200 min; Sample 3, t ¼ 400 min.)
DOI 10.1002/pen POLYMER ENGINEERING AND SCIENCE—-2011 1925
tor (ammonium persulfate, APS) and water were fed at con-
stant flow rates for a period of 240 min. After that, a second
stream of initiator was fed into the system for an additional
period of 30 min.
Initially, as the emulsion polymerizations were seeded
and the classical n value was found to be equal to 0.5 in the
previous validation test, the parameter (n � NT) was esti-
mated as a constant value for the whole Pilot-plant poly-
merization runs. However, obtained results were not good.
As shown in Fig. 3, the particle size distribution (measured
offline with the help of a Brookhaven ultracentrifuge) was
shifted toward higher particle diameters during the course
of the polymerization, but average particle sizes did not
change significantly between the beginning and the end of
the run (about two times, whereas the polymer mass
increased about 20 times), indicating that particle nuclea-
tion took place during the batch. Although this experimen-
tal result contradicts one of the assumptions used to
build the mathematical model, the fact is that the parameter
(n � NT) can be estimated with the available data as a func-
tion of the reaction time without having to model the nucle-
ation step and significantly increasing the model size.
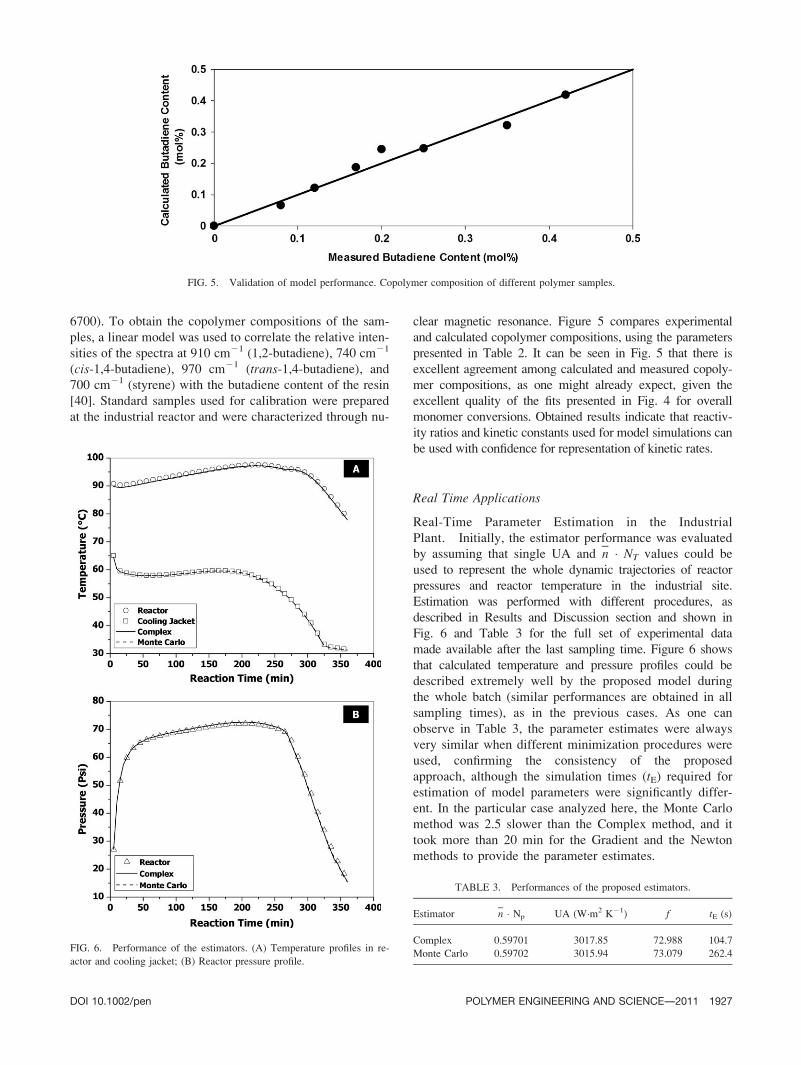
To estimate the variation of (n � NT) during the batch,
the reaction length was arbitrarily divided into three
stages and different (n � NT) values were estimated for
each reaction stage. As shown in Table 2 and Fig. 4, cal-
culated and measured monomer conversions (obtained off-
line through gravimetry) agreed very well in this case and
(n � NT) values were shown to increase smoothly with
time, confirming the probable formation of polymer par-
ticles during the Pilot-plant runs, as shown in Fig. 3.
Besides, the estimated values were very similar in all
analyzed samples, showing that estimated results were very
consistent and that reproducibility was fair. More important
is, Table 2 and Fig. 4 clearly indicate that the estimation of
changing (n � NT) values during the batch can allow for
proper monitoring of monomer conversions at plant site.
Polymer Compositions in the Pilot Plant
To validate the calculated copolymer compositions,
samples were collected and characterized with a Fourier
transform infrared spectrometer (Thermo Scientific, Nicolet
TABLE 2. n � Np Values for different time intervals.
Time
interval
(min)
(n � Np)
Run P40 Run P41 Run P43 Run P44
0–15 2.65 3 1018 2.42 3 1018 2.42 3 1018 3.31 3 1018
15–240 3.23 3 1018 3.17 3 1018 3.17 3 1018 3.31 3 1018
240–360 3.46 3 1018 3.46 3 1018 3.46 3 1018 3.31 3 1018
FIG. 4. Validation of model performance. Conversion profiles in the Pilot-plant. (A) Run P40; (B) Run
P41; (C) Run P43; (D) Run P44.
1926 POLYMER ENGINEERING AND SCIENCE—-2011 DOI 10.1002/pen
6700). To obtain the copolymer compositions of the sam-
ples, a linear model was used to correlate the relative inten-
sities of the spectra at 910 cm21 (1,2-butadiene), 740 cm21
(cis-1,4-butadiene), 970 cm21 (trans-1,4-butadiene), and
700 cm21 (styrene) with the butadiene content of the resin
[40]. Standard samples used for calibration were prepared
at the industrial reactor and were characterized through nu-
clear magnetic resonance. Figure 5 compares experimental
and calculated copolymer compositions, using the parameters
presented in Table 2. It can be seen in Fig. 5 that there is
excellent agreement among calculated and measured copoly-
mer compositions, as one might already expect, given the
excellent quality of the fits presented in Fig. 4 for overall
monomer conversions. Obtained results indicate that reactiv-
ity ratios and kinetic constants used for model simulations can
be used with confidence for representation of kinetic rates.
Real Time Applications
Real-Time Parameter Estimation in the Industrial
Plant. Initially, the estimator performance was evaluated
by assuming that single UA and n � NT values could be
used to represent the whole dynamic trajectories of reactor
pressures and reactor temperature in the industrial site.
Estimation was performed with different procedures, as
described in Results and Discussion section and shown in
Fig. 6 and Table 3 for the full set of experimental data
made available after the last sampling time. Figure 6 shows
that calculated temperature and pressure profiles could be
described extremely well by the proposed model during
the whole batch (similar performances are obtained in all
sampling times), as in the previous cases. As one can
observe in Table 3, the parameter estimates were always
very similar when different minimization procedures were
used, confirming the consistency of the proposed
approach, although the simulation times (tE) required for
estimation of model parameters were significantly differ-
ent. In the particular case analyzed here, the Monte Carlo
method was 2.5 slower than the Complex method, and it
took more than 20 min for the Gradient and the Newton
methods to provide the parameter estimates.
FIG. 6. Performance of the estimators. (A) Temperature profiles in re-
actor and cooling jacket; (B) Reactor pressure profile.
FIG. 5. Validation of model performance. Copolymer composition of different polymer samples.
TABLE 3. Performances of the proposed estimators.
Estimator n � Np UA (W�m2 K21) f tE (s)
Complex 0.59701 3017.85 72.988 104.7
Monte Carlo 0.59702 3015.94 73.079 262.4
DOI 10.1002/pen POLYMER ENGINEERING AND SCIENCE—-2011 1927
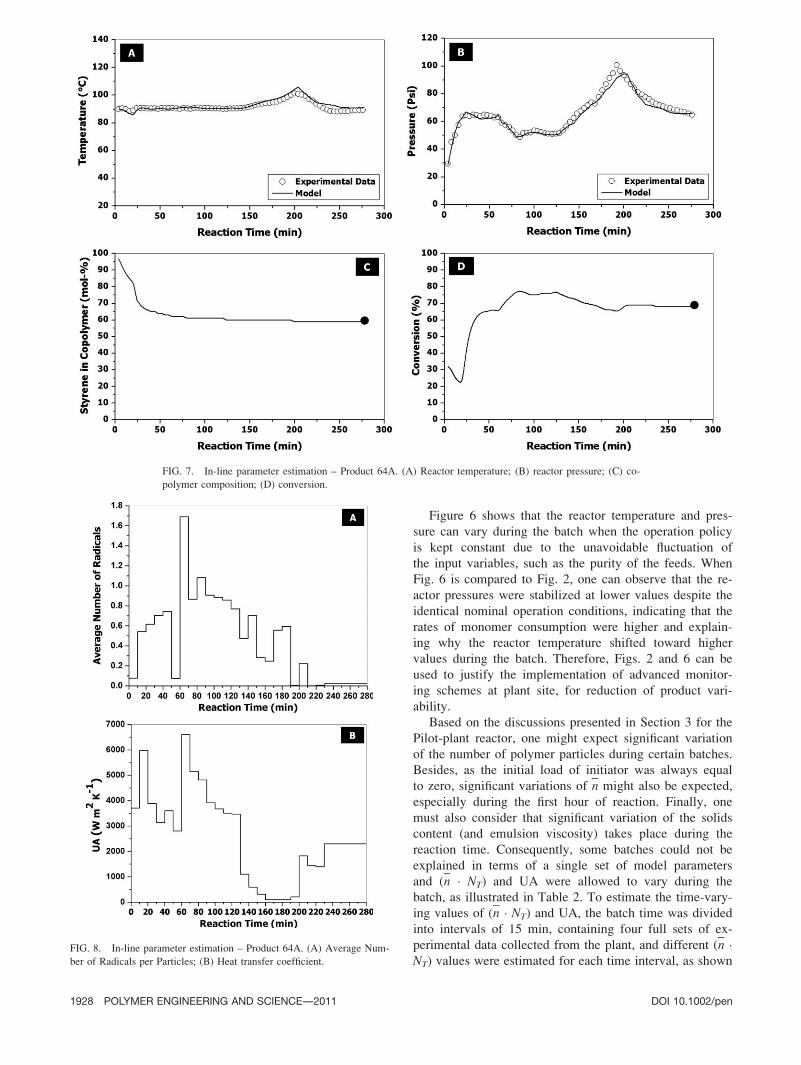
Figure 6 shows that the reactor temperature and pres-
sure can vary during the batch when the operation policy
is kept constant due to the unavoidable fluctuation of
the input variables, such as the purity of the feeds. When
Fig. 6 is compared to Fig. 2, one can observe that the re-
actor pressures were stabilized at lower values despite the
identical nominal operation conditions, indicating that the
rates of monomer consumption were higher and explain-
ing why the reactor temperature shifted toward higher
values during the batch. Therefore, Figs. 2 and 6 can be
used to justify the implementation of advanced monitor-
ing schemes at plant site, for reduction of product vari-
ability.
Based on the discussions presented in Section 3 for the
Pilot-plant reactor, one might expect significant variation
of the number of polymer particles during certain batches.
Besides, as the initial load of initiator was always equal
to zero, significant variations of n might also be expected,
especially during the first hour of reaction. Finally, one
must also consider that significant variation of the solids
content (and emulsion viscosity) takes place during the
reaction time. Consequently, some batches could not be
explained in terms of a single set of model parameters
and (n � NT) and UA were allowed to vary during the
batch, as illustrated in Table 2. To estimate the time-vary-
ing values of (n � NT) and UA, the batch time was divided
into intervals of 15 min, containing four full sets of ex-
perimental data collected from the plant, and different (n �NT) values were estimated for each time interval, as shown
FIG. 7. In-line parameter estimation – Product 64A. (A) Reactor temperature; (B) reactor pressure; (C) co-
polymer composition; (D) conversion.
FIG. 8. In-line parameter estimation – Product 64A. (A) Average Num-
ber of Radicals per Particles; (B) Heat transfer coefficient.
1928 POLYMER ENGINEERING AND SCIENCE—-2011 DOI 10.1002/pen
in Figs. 7 and 8. (The use of Kalman filters was avoided to
avoid the increase of the system dimension and the manipu-
lation and inversion of matrixes of derivatives.)
Figures 7 and 8 show that the parameter estimates can
change quite significantly during some batches, as already
explained, allowing for proper representation of reactor
temperature and pressure trajectories. Particularly, Fig. 7A
and B show that the reactor operation was driven to run-
away operation conditions, probably due to the uncon-
trolled increase of particle nucleation, followed afterward
by particle coagulation. In the analyzed case, feeding of
organic and aqueous streams was kept constant during the
first 200 min of operation and was halted after runaway
for safety reasons (therefore, normal operation was inter-
rupted after 200 min). Particle nucleation can justify the
continuous increase of (n � NT) during the first part of the
batch (represented in terms of n, when NT is assumed
constant and equal to the initial particle seed concentra-
tion), whereas particle coagulation can justify the steep
decrease of n � NT and UA during the second part of the
run. Despite that, it is important to note that the n values
shown in Fig. 8 are also consistent with experimental
results reported by Abdollahi and Sharifpour [41], as
these authors showed that n can be significantly influ-
enced by the nature of the carboxylic acid used in XSBR
emulsion polymerizations and can range from 0.3 to 1.7
in presence of acrylic, methacrylic, and itaconic acids.
Figure 7C and D illustrate how important the imple-
mentation of soft sensors can be at plant site, as there
were clear indications of dangerous monomer accumula-
tion inside the reactor during the first hour of reaction,
given the very low estimated monomer conversions in the
first part of the run, before the sudden increase of n � NT.
If estimated conversion data are compared to the mono-
mer solubility in the polymer particles (ranging from 39
to 40 wt%), it can clearly be concluded that formation of
monomer droplets takes place, as predicted by the pro-
posed polymerization model. Figure 7C also illustrates the
very significant drift of the copolymer composition during
the first reaction stage, as butadiene was accumulated pri-
marily in the gas phase. Both pieces of information could
be used for posterior optimization of the operation proce-
dure.
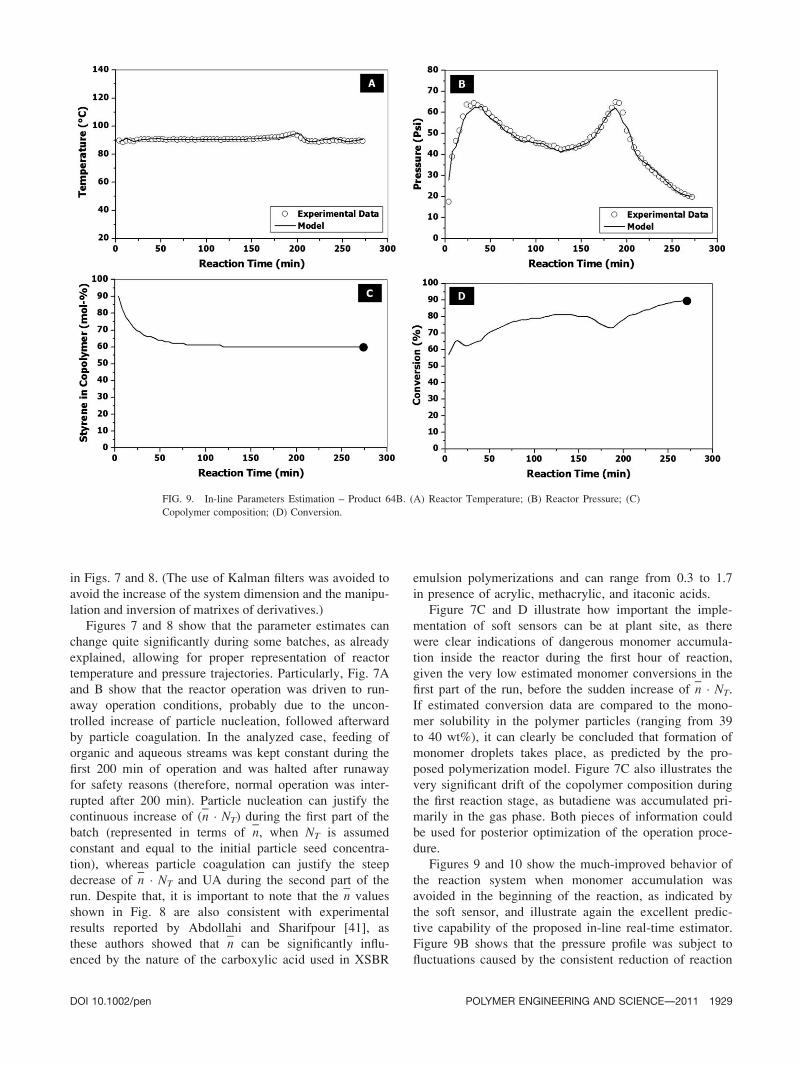
Figures 9 and 10 show the much-improved behavior of
the reaction system when monomer accumulation was
avoided in the beginning of the reaction, as indicated by
the soft sensor, and illustrate again the excellent predic-
tive capability of the proposed in-line real-time estimator.
Figure 9B shows that the pressure profile was subject to
fluctuations caused by the consistent reduction of reaction
FIG. 9. In-line Parameters Estimation – Product 64B. (A) Reactor Temperature; (B) Reactor Pressure; (C)
Copolymer composition; (D) Conversion.
DOI 10.1002/pen POLYMER ENGINEERING AND SCIENCE—-2011 1929
activity during the second part of the batch, as illustrated
in Fig. 10A, but the maximum attained pressure value
was significant lower than in the previous cases. For this
reason, reaction was not halted when monomer feeding
was interrupted after about 3 h of reaction. When Fig. 9
is compared to Fig. 7, one can clearly observe that mono-
mer conversion continued to increase after interruption of
monomer feeding, as reaction was continued in batch
mode.
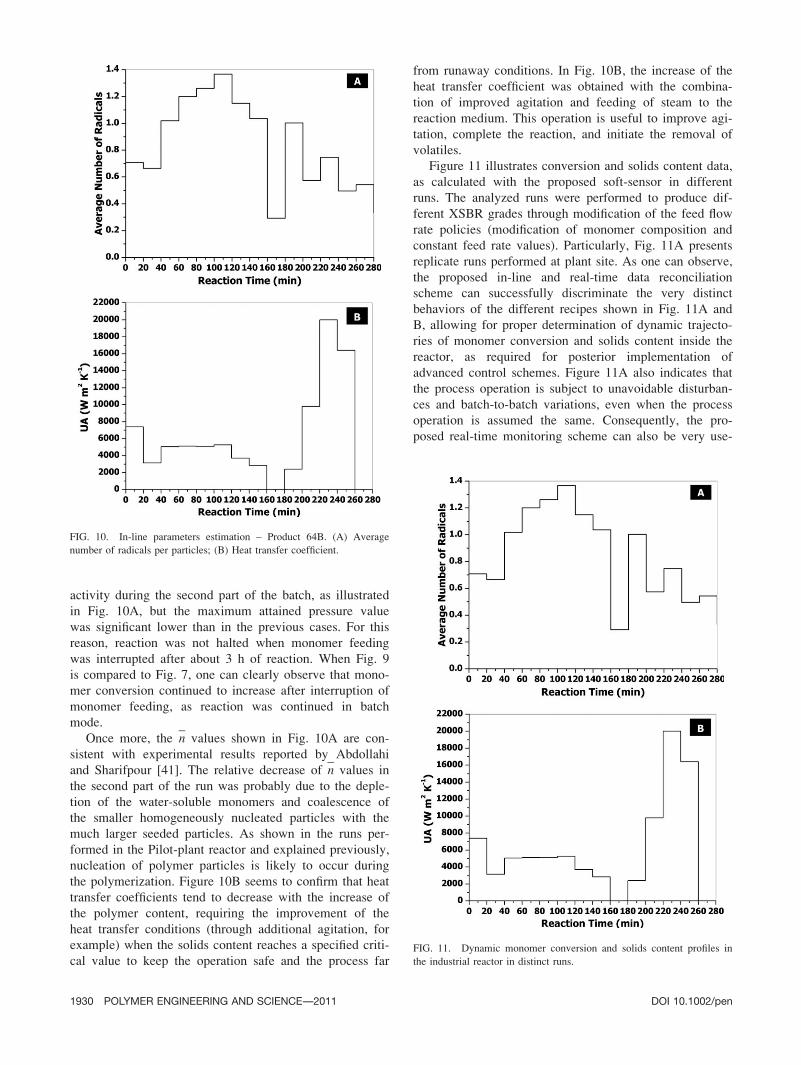
Once more, the n values shown in Fig. 10A are con-
sistent with experimental results reported by Abdollahi
and Sharifpour [41]. The relative decrease of n values in
the second part of the run was probably due to the deple-
tion of the water-soluble monomers and coalescence of
the smaller homogeneously nucleated particles with the
much larger seeded particles. As shown in the runs per-
formed in the Pilot-plant reactor and explained previously,
nucleation of polymer particles is likely to occur during
the polymerization. Figure 10B seems to confirm that heat
transfer coefficients tend to decrease with the increase of
the polymer content, requiring the improvement of the
heat transfer conditions (through additional agitation, for
example) when the solids content reaches a specified criti-
cal value to keep the operation safe and the process far
from runaway conditions. In Fig. 10B, the increase of the
heat transfer coefficient was obtained with the combina-
tion of improved agitation and feeding of steam to the
reaction medium. This operation is useful to improve agi-
tation, complete the reaction, and initiate the removal of
volatiles.
Figure 11 illustrates conversion and solids content data,
as calculated with the proposed soft-sensor in different
runs. The analyzed runs were performed to produce dif-
ferent XSBR grades through modification of the feed flow
rate policies (modification of monomer composition and
constant feed rate values). Particularly, Fig. 11A presents
replicate runs performed at plant site. As one can observe,
the proposed in-line and real-time data reconciliation
scheme can successfully discriminate the very distinct
behaviors of the different recipes shown in Fig. 11A and
B, allowing for proper determination of dynamic trajecto-
ries of monomer conversion and solids content inside the
reactor, as required for posterior implementation of
advanced control schemes. Figure 11A also indicates that
the process operation is subject to unavoidable disturban-
ces and batch-to-batch variations, even when the process
operation is assumed the same. Consequently, the pro-
posed real-time monitoring scheme can also be very use-
FIG. 11. Dynamic monomer conversion and solids content profiles in
the industrial reactor in distinct runs.
FIG. 10. In-line parameters estimation – Product 64B. (A) Average
number of radicals per particles; (B) Heat transfer coefficient.
1930 POLYMER ENGINEERING AND SCIENCE—-2011 DOI 10.1002/pen

ful for reduction of process variability in posterior
advanced control procedures.
CONCLUSIONS
A first-principles mathematical model was developed
and implemented to represent semibatch XSBR emulsion
polymerizations. As the model was intended for the
online and real-time data reconciliation and monitoring of
polymerization reactors, the proposed modeling procedure
assumed that some parameters should be estimated in real
time, for accommodation of the unavoidable fluctuation of
operation conditions, as observed at plant site. A direct
search complex algorithm was employed for estimation of
model parameters (the heat transfer coefficient and the
number of active radicals) based on available pressure,
temperature, and feed flow rate profiles, allowing for pre-
diction of important properties of XSBR latexes in real-
time, such as the monomer conversion, the solids content,
and the copolymer composition. The proposed model and
monitoring strategy were validated with actual data
obtained in a Pilot-plant reactor and in a full-scale indus-
trial process, showing very good agreement with the ex-
perimental data obtained in both the laboratory and indus-
trial scales. As illustrated with real operation data, the
proposed estimation strategy allowed for the successful
monitoring of process and polymer properties in real time,
constituting an important tool for posterior implementa-
tion of advanced control procedures. Particularly, the esti-
mated n values ranged from 0.1 to 1.7 in all cases, which
are in agreement with experimental results reported in the
open literature. It was also observed that heat transfer
coefficients can change significantly from batch-to-batch
and can experience large changes during the batch due to
modification of the reactor volume and accumulation of
polymer material on the heat transfer areas.
NOMENCLATURE
Parameters
A heat exchange area of the jacket
APS ammonium persulfate
Ai constants of the Antoine equation
Bi constants of the Antoine equation
Ci constants of the Antoine equation
CTA chain transfer agent
CTA0 initial concentration of chain transfer agent
Cpi heat capacity of species iFi feed flow rate of species iI initiator concentration
I0 initial concentration of initiator
kD kinetic constant for initiator decomposition
kPi,j kinetic constant for propagation of the radical iwith the monomer j
kTCi,j kinetic constant for termination by combination
of radicals i and jkTDi,j kinetic constant for termination by dispropor-
tionation of radicals i and jktrATCi kinetic constant for transfer of radical i to
chain transfer agent
ktrMi,j kinetic constant for transfer of radical i to
monomer jktrPi,j kinetic constant for transfer of radical i to
incorporated monomer jktRi kinetic constant for incorporation of pendant
double bonds by radical ili lower limit of the restrictions
m molar volume ratio of polymer and monomer
Mi concentration of monomer iMi0 initial concentration of monomer iMInert0 initial concentration of inert
MWi molecular weight of species iMw0 initial concentration of water
n average number of radical per polymer particle
NA Avogadro’s number
NT total number of particles in the reaction me-
dium
P0 total reactor pressure at the beginning of the re-
actor loading
PSATi vapor pressure of pure component i
pi pressure of the component iPinert pressure of the inert component
PSATw vapor pressure of water
P�i radical chain containing i mers of the species 1
Q�i radical chain containing i mers of the species 2
QA energy generated by the agitator
QP energy lost to the surroundings
QR energy generated by the polymeric reaction
QT energy exchanged between the reactor medium
and the jacket
R primary radical of initiator (Eq. 1) and Univer-
sal Gas Constant (Eq. 43)Ri polymerization rate of species iri reactivity ratio of monomer i (polymerization
model) and pseudo-random numbers (Eq. 56).t reaction time
T reaction temperature
xM monomer conversion
Ta ambient Temperature
tE computational time
Tei feed flow temperature of species iT0 initial system temperature
TC0 initial jacket temperature
(UA)a overall heat transfer coefficient to the surroundings
U overall heat transfer coefficient
ui upper limit of the restrictions
V volume
Vo volume of organic the phase
VR total reactor volume
VT total reactor volume occupied by the liquid phase
Vt total volume of the system
DOI 10.1002/pen POLYMER ENGINEERING AND SCIENCE—-2011 1931

Vw volume of aqueous phase
xi independent variables
xM monomers conversion
xS solid content
wi mass fraction of component iwInert molar fraction of volatile impurities (inert) in
the butadiene stream
Greek Symbols
I copolymer composition
/i volume fraction of component iGk dead polymer chain containing k mers
}i concentration of polymer i}i0 initial concentration of polymer iRk radical of polymer containing k mers
Oi probability of radicals in the polymer particle
present a mer of type i at the end of the polymer
chain
mi molar volume of component ipi pure density of species iv Flory–Huggins interaction parameter
(2DHi) enthalpy of homopolymerization of species i
Subscripts
1 styrene
2 butadiene
REFERENCES
1. T. Cosse. ‘‘Structural Changes in the Global Carboxylated
Styrene-Butadiene Latex Industry,’’ in International LatexConference, Rubber & Plastics News, Akron, USA (2003).
2. D.C. Blackley, Emulsion Polymerization: Theory and Prac-tice, Springer, New York (1975).
3. R.B. Gilbert, Emulsion Polymerization: A MechanisticApproach, Academic Press, London (1995).
4. J.C. Daniel and C. Pichot, Les Latex Synthetiques, Tec &
Doc Lavoisier, Paris (2006).
5. P.A. Lovell and M.S. El-Aasser, Emulsion Polymerizationand Emulsion Polymers, Wiley, New York (1997).
6. L. Vorwerg and R.G. Gilbert,Macromolecules, 33, 6693 (2000).
7. J.Y.M. Chew, S.J. Tonneijk, W.R. Paterson, and D.I. Wil-
son, Ind. Eng. Chem. Res., 44, 4605 (2005).
8. J.S. Nettleton, M.J.G. Davidson, and H.L. Williams, Ind.Eng. Chem., 45, 1896 (1953).
9. J.R. Leiza and J.C. Pinto, ‘‘Control of Polymerization Reac-
tors,’’ in Polymer Reaction Engineering, J.M. Asua, Ed.
Blackwell Publishing, Oxford (2007).
10. J.M. de Faria Jr., F. Machado, E.L. Lima, and J.C. Pinto,
Macromol. React. Eng., 4, 11 (2010).
11. J.M. de Faria Jr., F. Machado, E.L. Lima, and J.C. Pinto,
Macromol. React. Eng., 4, 486 (2010).
12. I.S. De Buruaga, M. Arotcarena, P.D. Armitage, L.M. Gugliotta,
J.R. Leiza, and J.M. Asua, Chem. Eng. Sci., 51, 2781 (1996).
13. L.M. Gugliotta, M. Arotcarena, J.R. Leiza, and J.M. Asua,
Polymer, 36, 2019 (1995).
14. B. Alhamad, V.G. Gomes, and J.A. Romagnoli, Int. J.
Chem. Reac. Eng., 4, 1 (2006).
15. I.W. Cheong and J.H. Kimu, Coll. Surf. A: Physicochem.Eng. Asp., 153, 137 (1999).
16. M. Vicente, S. Benamor, L.M. Gugliotta, J.R. Leiza, and
J.M. Asua, Ind. Eng. Chem. Res., 40, 218 (2001).
17. M.F. Kemmere, J. Meuldijk, A.A.H. Drinkenburg, and A.L.
German, J. Appl. Polym. Sci., 79, 944 (2001).
18. F. Korber, K. Hauschild, and G. Fink, Macromol. Chem.
Phys., 202, 3329 (2001).
19. F. Korber, K. Hauschild, M. Winter, and G. Fink, Macro-
mol. Chem. Phys., 202, 3323 (2001).
20. G. Maschio, I. Ferrara, C. Bassani, and H. Nieman, Chem.
Eng. Sci., 54, 3273 (1999).
21. J.R. Vega, L.M. Gugliotta, and G.R. Meira, Polym. React.
Eng., 10, 59 (2002).
22. L.M. Gugliotta, M.C. Brandolini, J.R. Vega, E.O. Iturralde,
J.L. Azum, and G.R. Meira, Polym. React. Eng., 3, 201 (1995).
23. C. Sayer, E.L. Lima, and J.C. Pinto, Chem. Eng. Sci., 52,
341 (1997).
24. C.C. Chang, A.F. Halasa, J.W. Miller Jr., and W.L. Hsu,
Polym. Int., 33, 151 (1994).
25. IMSL STAT/LIBRARY User’s Manual, 1991, Version 2.0,
IMSL, Houston.
26. J. Brandup, E.H. Immergut, and E.A. Grulke, PolymerHandbook, 4 ed., Wiley, New York (1999).
27. A.E. Hamielec, J.F. Mcgregor, S. Webb, and T. Spychaj,
‘‘Thermal and Chemical Initiated Copolymerization of Styrene
Acrylic Acid at High Temperatures and Conversions in a Con-
tinuous Stirred Tank Reactor,’’ in Polymer Reaction Engineering,
K.H.G. Reichert, W., Eds., Huthig and Wepf: Basel, 185 (1986).
28. G. Kalfas, H. Yuan, and W.H. Ray, Ind. Eng. Chem. Res.,32, 1831 (1993).
29. M.W. Chase Jr., J. Phys. Chem. Ref. Data, 9, 1 (1998).
30. R.H. Perry and D.W. Green, Perry’s Chemical Engineers’
Handbook, 7th ed., McGraw-Hill, New York (1997).
31. B. Hesselbo and R.B. Stinchcombe, Phys. Rev. Lett., 74,2151 (1995).
32. N.J. Box, Comput. J., 8, 42 (1965).
33. E.S. Lee, Ind. Eng. Chem. Fund., 3, 373 (1964).
34. P.E. Gill, W. Murray, and M.H. Wright, Practical Optimiza-tion, Academic Press, New York (1982).
35. E.K.P. Chong and S.H. Zak, An Introduction to Optimiza-
tion, 2nd ed., Wiley, New York (2001).
36. D.M. Prata, M. Schwaab, E.L. Lima, and J.C. Pinto, Chem.
Eng. Sci., 64, 3953 (2009).
37. W.H. Swann, ‘‘Constrained Optimization by Direct Search,’’ in
Numerical Methods for Constrained Optimization, P.E. Gill and
W. Murray, Eds., Academic Press, New York, USA (1974).
38. F. Machado, E.L. Lima, and J.C. Pinto, Polym. Eng. Sci.,
50, 697 (2010).
39. F.P. Incropera and D.P. Dewitt, Fundamentos de Transferen-
cia de Calor e de Massa, LTC Editora, Rio de Janeiro (2003).
40. J.G. Ivaschenko, N.N. Karpova, and O.V. Fedotova, Chem.Comput. Simul. Butlerov Commun., 4, 21 (2001).
41. M. Abdollahi and M. Sharifpour, Polymer, 48, 2035 (2007).
1932 POLYMER ENGINEERING AND SCIENCE—-2011 DOI 10.1002/pen





























