Embed Size (px)
Citation preview

1521-0103/350/2/412–424$25.00 http://dx.doi.org/10.1124/jpet.114.214221THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS J Pharmacol Exp Ther 350:412–424, August 2014Copyright ª 2014 by The American Society for Pharmacology and Experimental Therapeutics
Pharmacologic Profile of the Adnectin BMS-962476, a SmallProtein Biologic Alternative to PCSK9 Antibodies for Low-DensityLipoprotein Lowering
Tracy Mitchell, Ginger Chao, Doree Sitkoff, Fred Lo, Hossain Monshizadegan,Daniel Meyers, Simon Low, Katie Russo, Rose DiBella, Fabienne Denhez, Mian Gao,Joseph Myers, Gerald Duke, Mark Witmer, Bowman Miao, Siew P. Ho, Javed Khan,and Rex A. ParkerMolecular Discovery Technologies (T.M., G.C., D.S., S.L., K.R., R.D., F.D., M.G., J.M., G.D., M.W., J.K.), Applied Genomics(S.P.H.), and Cardiovascular Discovery Biology (F.L., H.M., D.M., B.M., R.A.P.), Bristol-Myers Squibb Research andDevelopment, Princeton, New Jersey
Received February 24, 2014; accepted June 9, 2014
ABSTRACTProprotein convertase subtilisin kexin-9 (PCSK9) is an importantpharmacological target for decreasing low-density lipoprotein(LDL) in cardiovascular disease, although seemingly inaccessibleto small molecule approaches. Compared with therapeutic IgGantibodies currently in development, targeting circulating PCSK9with smaller molecular scaffolds could offer different profiles andreduced dose burdens. This inspired genesis of PCSK9-bindingAdnectins, a protein family derived from human fibronectin-10th-type III–domain and engineered for high-affinity target binding.BMS-962476, an ∼11-kDa polypeptide conjugated to polyethyleneglycol to enhance pharmacokinetics, binds with subnanomolaraffinity to human. The X-ray cocrystal structure of PCSK9 with aprogenitor Adnectin shows ∼910 Å2 of PCSK9 surface coverednext to the LDL receptor binding site, largely by residues of a
single loop of the Adnectin. In hypercholesterolemic, over-expressing human PCSK9 transgenic mice, BMS-962476rapidly lowered cholesterol and free PCSK9 levels. In genomictransgenic mice, BMS-962476 potently reduced free humanPCSK9 (ED50 ∼0.01 mg/kg) followed by ∼2-fold increases in totalPCSK9 before return to baseline. Treatment of cynomolgusmonkeys with BMS-962476 rapidly suppressed free PCSK9.99% and LDL-cholesterol ∼55% with subsequent 6-fold in-crease in total PCSK9, suggesting reduced clearance of circulat-ing complex. Liver sterol response genes were consequentlydownregulated, following which LDL and total PCSK9 returned tobaseline. These studies highlight the rapid dynamics of PCSK9control over LDL and liver cholesterol metabolism and charac-terize BMS-962476 as a potent and efficacious PCSK9 inhibitor.
IntroductionProprotein convertase subtilisin/kexin type 9 (PCSK9) is
a 74-kDa protein secreted by liver that regulates hepatic low-density lipoprotein (LDL) receptor (LDLR) activity andcontrols plasma LDL cholesterol (LDL-C) levels. By virtue ofits genetics and biologic mechanism (Abifadel et al., 2003;Cohen et al., 2006), PCSK9 has emerged as an importanttherapeutic target for discovery and development of newdrugs for cardiovascular diseases (Steinberg and Witzum,
2009). PCSK9 binds to the LDLR within its well conservedepidermal growth factor precursor homology domain-A(EGFA) at the cell surface (Kwon et al., 2008). Upon binding,PCSK9 cointernalizes into endosomes and redirects receptorsorting into the lysosomal degradative pathway rather thanrecycling to the surface (Zhang et al., 2007). If PCSK9 bindingis inhibited, the biologically important cargo delivery functionof the receptor is increased, the liver intracellular cholesterolpool is augmented, and plasma LDL-C is decreased.Because small molecule approaches targeting PCSK9 have
proven difficult to date, unlike for some members of theproprotein convertase gene family (Seidah et al., 2008), drugdiscovery attention has focused upon antisense oligonucleo-tides (Graham et al., 2008; Fitzgerald et al., 2013) ormonoclonal antibody (mAb) based biologics (Chan et al.,2009; Ni et al., 2011; Liang et al., 2012; reviewed by Seidah
Use of the Advanced Photon Source was supported by the U S Department ofEnergy. Use of the IMCA-CAT beamline 17ID at the Advanced Photon Sourcewas supported by the companies of the Industrial Macromolecular Crystallog-raphy Association through a contract with the Center for Advanced RadiationSources at the University of Chicago.
dx.doi.org/10.1124/jpet.114.214221.
ABBREVIATIONS: apo, apolipoprotein; BMS-962476, PCSK9-specific Adnectin with 40-kDa branched PEG; cyno, cynomolgus monkey; DiI-LDL,3-39-dioctadecylindo carbocyanine labeled LDL; DSC, differential scanning calorimetry; EGFA, epidermal growth factor homology domain A of theLDLR; ELISA, enzyme linked immunosorbent assay; FACS, fluorescence-activated cell sorter; FRET, fluorescence resonance energy transfer; HDL,high-density lipoprotein; ITC, isothermal calorimetry; LDL-C, low-density lipoprotein-cholesterol; LDLR, low-density lipoprotein receptor; LPDS,lipoprotein-deficient serum; mAb, monoclonal antibody; PBS, phosphate-buffered saline; PCSK9, proprotein convertase substilisin kexin-9; PEG,poletheylene glycol; PK, pharmacokinetic; SPR, surface plasmon resonance; SREBP2, sterol response element binding protein 2.
412
at ASPE
T Journals on June 21, 2016
jpet.aspetjournals.orgD
ownloaded from

and Prat, 2012). Structure-function analysis suggests target-ing the EGFA-interacting region of PCSK9 for pharmacolog-ical inhibition (Zhang et al., 2007). The EGFA site involvesresidues 367–381 covering about 500 Å2 of PCSK9 surface(Kwon et al., 2008) and includes the D374Y locus, a gain-of-function mutation that increases activity 10-fold (Lagaceet al., 2006). Two high-affinity mAbs were found to covera somewhat larger region at or near the EGFA binding site(Chan et al., 2009; Liang et al., 2012). These and other PCSK9mAbs (Ni et al., 2011; Stein et al., 2012) lowered LDL inanimal studies and human clinical trials. At the other end ofthe size spectrum, Zhang et al. (2014) recently describeda linear peptide of 13 amino acids that contacted about 400 Å2
on the PCSK9 surface and inhibited EGFA binding in vitro;however, the binding affinity was too weak [KD ∼700 nM,compared with KD ∼200 nM for PCSK9:LDLR as shown byBottomley et al. (2009)] to support in vivo pharmacologystudies.The search for alternatives to immunoglobulin-based
inhibitors inspired the discovery of PCSK9-binding Adnec-tins, identified through selection from highly diverselibraries using mRNA display. Adnectins are based on the10th-type III–domain (10Fn3) of human fibronectin, whosevariable loops can be efficiently engineered to introducesurfaces that bind therapeutic targets with high affinity andspecificity (Koide et al., 1998; Wojcik et al., 2010). Adnectinsare small (#12 kDa), compact proteins without sequencehomology to immunoglobulins but possessing a b-sheet foldstructure with diversified loops analogous to antibody vari-able regions (Parker et al., 2005; Lipovsek, 2011). Adnectinshave no disulfides and are not glycosylated, exhibit highthermal stability and monomeric solution behavior, and areefficiently produced using bacterial expression systems. Bymodifying variable loop sequence and length while holdingscaffold residues constant, subnanomolar target bindingaffinity can be achieved while structural stability is main-tained. Given their size, Adnectins are rapidly filtered bythe kidney and therefore require pharmacokinetic (PK) en-hancement modification for in vivo applications. The highlystable Adnectin polypeptide scaffold allows incorporation ofvarious PK enhancement modalities through protein engi-neering. The present studies employ polyethylene glycolmodification as a convenient and established approach forPK enhancement.This report describes the discovery and characterization
of novel high-affinity PCSK9-binding Adnectins identifiedthrough screening for EGFA competitive interactions.The resulting optimized Adnectin exhibited subnanomolarbinding affinity to human PCSK9, displacement of EGFA andfull-length LDLR binding, and potent biologic effects in cells,transgenic mice, and nonhuman primates in vivo. The in vivoprofile exhibited remarkably fast pharmacodynamic effects oncholesterol metabolism and revealed changes in total circu-lating PCSK9 levels dependent on drug binding. Evaluationof sterol response gene expression in monkey liver led to aproposed model for regulation of key pathway genes includingPCSK9 itself. The cocrystal structure with a closely relatedprogenitor Adnectin revealed the binding mode and locus onPCSK9 adjacent to the EGFA interaction site and providedinsight into the marked species dependency of the PCSK9binding and inhibition profile observed.
Materials and MethodsPCSK9 Adnectin Selection, Expression, and Purification.
Generation of the Adnectins followed procedures described previously(Parker et al., 2005; Lipovsek, 2011). Briefly, an Adnectin DNAlibrary of high diversity (2.3 � 1012) produced by randomizing the 3variable loops (denoted BC, DE, and FG) of 10Fn3 was taken throughmultiple rounds of mRNA display. In each round, the library wastranscribed in vitro, translated using rabbit reticulolysate, and eachencoding mRNA was fused to its own Adnectin via a puromycinlinkage. Reverse transcription was used to generate a cDNA tail oneach Adnectin, and during selection, Adnectins that bound humanPCSK9 were amplified by polymerase chain reaction to serve astemplate for the next round. Cycles of selection and amplificationwere continued until significant target binding was observed byquantitative polymerase chain reaction, and binding populationswere cloned, sequenced, and assayed. Lead candidates were chosenbased on biophysical properties and inhibition potency in PCSK9:EGFA binding and cell-based assays. The progenitor Adnectin, clone1459D05, had the following amino acid sequence: GVSDVPRDLEVVAATPTSLLISWPPPSHGYGYYRITYGETGGNSPVQEFTVPPGKGTAT ISGLKPGVDYTITVYAVEYPYKHSGYYHRPISINYRTEIDKPSQHHHHHH.
To further enhance binding affinity, the 1459D05 sequence wasaffinity matured using an optimization library with repeated selec-tions of increasing stringency to favor clones with tighter affinity andslower off-rate. The PCSK9 Adnectin BMS-962476 is the optimizedvariant of 1459D05 with higher binding affinity and potency in func-tional assays. BMS-962476 has a protein molecular mass of 11.3 kDa,comprising 103 residues with the following sequence: GVSDVPR-DLEVVAATPTSLLISWVPPSDDYGYYRITYGETGGNSPVQEFTVP-IGKGTAT ISGLKPGVDYTITVYAVEFPWPHAGYYHRPISINYRTE-IEKPCQ.
BMS-962476 also contains a 40-kDa, 2-branched polyethyleneglycol moiety conjugated via standard maleimide chemistry at thepenultimate C-terminal cysteine residue (PEGylated Adnectin). TheC-terminal His6 purification-tagged form of the PEGylated proteinwas also studied (referred to as BMS-962476 his6). The non-PKenhanced, non-PEGylated form of this Adnectin (referred to as BMS-962476 his6, no PEG) exhibits very high thermal stability (Tmax 581°C) and solubility with monomeric behavior by gel filtrationchromatography.
PCSK9 Protein Expression and Purification. Recombinantfull-length human PCSK9 (31–692) and truncated human PCSK9(53–451, for crystallography) and full-length PCSK9 from cynomolgusmonkey (cyno), Guinea pig, and mouse cDNA sequences were producedand purified from baculovirus expression or stably transfectedmammalian cell systems by standard molecular biology and chroma-tography techniques, essentially as described previously (Benjannetet al., 2010).
Biophysical Assessment of Adnectins. Size exclusion chroma-tography was used to profile solution monomeric behavior. Sizeexclusion chromatography runs were performed on an Agilent 1200system (Agilent Technologies, Santa Clara, CA) using Superdex-7510/300 column (GE Healthcare, Life Sciences, Piscataway, NJ).Mobile phase for all SEC runs consisted of 100 mM NaPO4, 100 mMNaSO4, 150mMNaCl, pH 6.8, and data analysis was performed usingChemstation software package provided by Agilent Technologies.Differential scanning calorimetry (DSC) was performed on samplesdialyzed into phosphate-buffered saline (PBS) and normalized to0.5 mg/ml before run. DSC sample runs were conducted at a scan rateof 60°C/hour from 15 to 95°C on a MicroCal VP-DSC (GE Healthcare,Life Sciences). Data analysis was performed using Origin softwarepackage (GE Healthcare, Life Sciences).
PCSK9/Adnectin Complex Crystallization and StructureDetermination. Purified human PCSK9 (53–451) and Adnectin1459D05 were mixed in a 1:3 molar ratio and incubated on ice for5 hours with gentle mixing every 30 minutes. To remove excess
Structure and Pharmacodynamics of an Adnectin PCSK9 Inhibitor 413
at ASPE
T Journals on June 21, 2016
jpet.aspetjournals.orgD
ownloaded from

unbound Adnectin, the complex was run over Superdex-75 (16/60)column equilibrated in 25 mM HEPES pH 7.5, 0.2 M NaCl, and 5%glycerol at flow rate 1 ml/min; peak fractions containing 1:1 Adnectin:PCSK9 complex were pooled and concentrated. The sample wasclarified by centrifugation and concentrated to 10 mg/ml for use incrystallization experiments. Crystals of PCSK9/1459D05 were ob-tained by hanging drop vapor diffusion method at 22°C by equil-ibrating the complex against a reservoir solution containing 22% (v/v)PEG 200, 1% (v/v) ethylene glycol, and 0.1 M MES pH 6.5. Crystalswere harvested by flash-cooling in liquid nitrogen using 30% (v/v)PEG200 and 0.1 M MES pH 6.5 as a cryoprotectant.
X-ray data for the cocrystal complex were collected at beamline17ID at IMCA-CAT of the Advanced Photon Source at ArgonneNational Laboratory (Lemont, IL). The wavelength used was 1.00 Åand the detector was a MAR-165 CCD (Marresearch GmbH,Norderstedt, Germany). The diffraction images were indexed, in-tegrated, and scaled with D*TREK (Pflugrath, 1999). The structurewas determined by molecular replacement using the programPHASER (McCoy et al., 2007). Two different PDBs served as searchmodels for molecular replacement: 1) PDB ID 2W2M (Bottomley et al.,2009) for PCSK9 and 2) PDB ID 1FNF (Leahy et al., 1996) as searchmodel for the Adnectin. From the latter, the variable loops and N andC termini were removed before running molecular replacement.Model building was done for one monomer of the PCSK9-Adnectincomplex, which was then used as search model to find the secondcomplex in the asymmetric unit. The model was built using theprogram COOT (Emsley and Cowtan, 2004). The structure wasinitially refined using Refmac5 (CCP4 Suite) and the final cyclesusing autoBUSTER (Global Phasing LTD, Cambridge, UK). Figureswere generated using pymol (PyMOL Molecular Graphics System,v1.5.0.5; Schrödinger, LLC, New York, NY). The final structure hasa resolution of 2.69 Å, an R factor 5 19.9% and Rfree 5 23.08%.Coordinates and structure amplitudes are deposited as PDB ID 4OV6.
Measurement of Binding Affinity and Kinetics. Bindingstudies were conducted in PBS buffer at pH 7.4 unless otherwisenoted. Adnectin binding to immobilized PCSK9 was measured bybiolayer interferometry (Octet Red 96, using Superstreptavidinsensor tips; ForteBio, Menlo Park, CA). Association and dissociationevents were measured in real time for a series of Adnectin concen-trations with biotinylated full-length PCSK9 captured on sensor tips.Binding curves were globally fit to produce values for KD, kon, and koff.With the use of an alternative solid phase format, Adnectin bindingaffinity for immobilized PCSK9 was assessed by surface plasmonresonance (SPR) in PBS 1 0.05% Tween-20 using a ProteOn-XPR(Bio-Rad, San Francisco, CA), with data reduction using the Langmuir1:1 interaction model and constant parameter fitting with ProteOnManager software. Affinity was also measured by solution phasemethods using the kinetic exclusion assay essentially as described byDrake et al. (2004), which is particularly well suited to measuringsubnanomolar affinities. As a final test, affinity was measured usingsolution-phase isothermal calorimetry (ITC) with a VP-ITC calorimeter(MicroCal/GE Healthcare Life Sciences). ITC, which measures thethermodynamics and stoichiometry of protein:protein binding byquantifying the amount of heat released or absorbed, is in some casesthe most accurate method, although systems with subnanomolaraffinities at ambient temperature require measurements at highertemperatures with extrapolation to ambient (Doyle, 1997). Althoughthe PEG moiety is positioned away from the presumed target bindingregion of the Adnectin, in some assay platforms with various proteinsthe PEG moiety can interfere with binding measurements (Kubetzkoet al., 2005). Therefore it was relevant to study the non-PEGylated formof the Adnectin and to test multiple different assay platforms.
LDLR-EGFA Competitive Inhibition Fluorescence Reso-nance Energy Transfer Assay. A competitive binding assay basedon homogeneous time-resolved fluorescence resonance energy trans-fer (FRET) was used to measure displacement of the EGFA peptide toPCSK9 protein. EGFA peptide synthesis by solid-phase chemistryand the assay conditions were essentially as described previously
(Benjannet et al., 2010). Briefly, the FRET assay buffer was 150 mMNaCl, 5 mM CaCl2, 10 mMHEPES, 0.05% bovine serum albumin, pH7.4, and the assay in 384-well plates contained 10 nM PCSK9 (or asspecified in data), 0.5 nM europium chelate-labeled anti-PCSK9mAb-4H5, 400 nM EGFA (-biotinyl), 40 nM streptavidin-allophycocyanin,and specified levels of Adnectins. mAb4H5 is a BMS custom antibodyproduced by Lampire Biologic Laboratories (Pipersville, PA) thatrecognizes the C-terminal domain of PCSK9. Plates were incubated at20°C for 16 hours, followed by measurement of time-resolved fluores-cence using a PerkinElmer (Waltham, MA) Envision instrument withlaser excitation (337/615/665 nm). Data were fitted as nonlinearregression sigmoidal dose response variable slope using logarithm ofconcentration with GraphPad Prism software (GraphPad Software,La Jolla, CA).
Cell-Based PCSK9 Uptake and LDLR Functional and Pro-tein Level Assays. To assay LDLR-dependent PCSK9 uptake intocells, human hepatomaHepG2 cells (HB8065; American Type CultureCollection, Manassas, VA) were incubated for 18 hours in RPMI1640 media (11835; Invitrogen, Grand Island, NY) containing 5%lipoprotein-deficient serum (LPDS; Intracel, Frederick, MD; RP054),mevalonate (50 mM), and the statin BMS-423526 (100 nM) toupregulate LDLR expression. Adnectins diluted in media were addedat concentrations ranging typically from 0.03 to 300 nM together withfluorescently labeled PCSK9-AF647 at 25 nM. PCSK9 covalentlycoupled with AF647 fluorophore was prepared using the protocolprovided by Molecular Probes (A20173; Invitrogen, Carlsbad, CA).The cell plates were incubated at 37°C for 4 hours and washed andfixed with 2% formaldehyde in the presence of 4 mg/ml of HoechstDNA stain. The plates were read for intracellular (endosomal) AF647fluorescence using a Cellomics ArrayScan, and image data werequantified as the mean total intensity of fluorescence per cell fromquadruplicate wells with an average read of 600 cells/well. Data wereanalyzed in GraphPad Prism 4 using nonlinear regression sigmoidaldose response variable slope curvefits for IC50 and EC50 values.
LDLR functional activity was assayed using HepG2 cells pre-treated as described above. After 18 hours, purified PCSK9 (135 nM)was added with Adnectins as indicated in the data (note, higherPCSK9 levels are required to observe functional activity on LDLR bythis method). After 2-hour incubation of cells at 37°C, DiI-LEL (3-39-dioctadecylindo carbocyanine labeled LDL) (BT-904; BiomedicalTechnologies, Stoughton, MA) at 5 mg/ml was microfiltered andmixed with phospholipid vesicles (40 mg/ml) and added to the cellmedia. Cells were returned to tissue culture incubators for anadditional 2 hours for DiI-LDL uptake. Cells were then fixed andquantified by Cellomics (Halethorpe, MD) Array-Scan essentially asdescribed for the PCSK9-AF647 assay above.
LDLR protein levels were immunoassayed by fluorescence-activatedcell sorter (FACS) after an overnight incubation of HepG2 cells inLPDS media, after which cells were removed from plates with trypsin(0.05%) and resuspended in 5% LPDS-containing RPMI 1640 media.The cells in suspension received Adnectins in media in the presenceof 10 nM PCSK9 and were incubated overnight at 37°C still insuspension. On the following day, the level of LDLR protein on theHepG2 cell surface was assessed by FACS analysis using biotinylatedanti-LDLRpolyclonal antibody and streptavidin-PE conjugated detec-tion antibody. Curvefits were as described above.
Enzyme-Linked Immunosorbent Assay and FRET Assaysfor Free and Total PCSK9 in Blood Plasma. Enzyme-linkedimmunosorbent assays (ELISAs) specific for human and cynomolgusPCSK9 that do not detect mouse PCSK9 were developed. The free(unbound) PCSK9 assay employed streptavidin-pretreated 96-wellplates coated with 2 mg/ml of biotinylated PCSK9-Adnectin ascapturing reagent. Plasma samples frozen once only were diluted asappropriate in ELISA buffer (25 mM Tris, 150 mM NaCl, pH 7.2 with0.05% Tween-20 and 0.1% bovine serum albumin), added to wells andincubated for 1 hour at 20°C. Wells were then washed and incubatedwith 5 mg/ml of rabbit polyclonal anti-human PCSK9 IgG (BMScustom antibody produced by Lampire Biologic Laboratories) for
414 Mitchell et al.
at ASPE
T Journals on June 21, 2016
jpet.aspetjournals.orgD
ownloaded from

1 hour, followed by processing for horseradish peroxidase–labeledanti-rabbit IgG with 3,39,5,59-tetramethylbenzidine by standardELISA methods. The total PCSK9 ELISA assay was conductedsimilarly as described above except mAb-4H5 (BMS custom antibodyproduced by Lampire Biologic Laboratories) was incorporated as thecapture antibody. The mAb-4H5 binds the C-terminal domain ofPCSK9 and when bound to the 96-well plates efficiently captures totalPCSK9 (both Adnectin-PCSK9 complex plus free PCSK9). Standardcurves were generated using purified recombinant human or cyno-molgus PCSK9.
Because the Adnectin binds cynomolgus PCSK9 more weakly thanhuman, dissociation of the complex in cyno plasma was observedusing ELISA assay format, therefore a homogeneous FRET-basedassay was established to measure free PCSK9 in cyno monkeyplasma. Samples were assayed with minimal dilution (typically 1:4final) to minimize complex dissociation during the homogeneousassay, which was adapted from the procedure described above for thePCSK9:EGFA FRET assay. A similar assay was developed for usewith human and transgenic mouse plasma as described in the results.
Human PCSK9 Transgenic Mouse Models. Human PCSK9overexpresser transgenic mice originally described by Lagace et al.(2006) were obtained from Dr. Jay Horton (University of Texas SouthWestern Medical School, Dallas, TX) and cohorts were bred andgenotyped at BMS. Hemizygous transgenic mice were maintained asmixed strain SJL � C57BL6/J and fed a normal chow diet. This isa human PCSK9 cDNA-driven overexpression model that avidlysecretes human PCSK9 to achieve high circulating plasma levels(∼500 mg/ml, ∼7 mM in male hemizygous mice), more than 500-foldgreater than normal human levels. The very high levels of humanPCSK9 strongly reduce mouse liver LDLR protein leading tohypercholesterolemia.
A second transgenic mouse model was developed that exhibitsnormal levels of human PCSK9. These mice express full-lengthhuman PCSK9 driven from a chromosomal insertion of a human DNAfragment (∼180,000 base pairs) surrounding the PCSK9 gene (22,000base pairs). Fluorescent in situ hybridization analysis showedinsertion in chromosome 5, distinct from endogenous mouse PCSK9on chromosome 4, which continues to be expressed in thesemice. BothmRNAs are expressed in liver of the transgenic mice and undergoregulation by feeding/fasting similarly (data not shown). Plasmalevels of human PCSK9 in homozygous male transgenic mice weretypically ∼3–5 nM (similar to endogenous mouse protein). The modelexhibited no obvious cholesterol phenotype compared with wild type.
Studies used male mice, 28–35 g weight, for both transgenicstrains. For dosing, Adnectins were formulated in sterile PBS solutionfor intraperitoneal injection.
Cynomolgus Monkey Studies. Vivarium management and thein vivo dosing and sampling protocols for the cyno studies wereconducted at Shin Nippon Biomedical Laboratories, Ltd (Everett,WA) in compliance with guidelines from the Association forAssessment and Accreditation of Laboratory Animal Care. Femalecynomolgus monkeys, body weight 3.0–4.6 kg, age 4–7 years wereused. Blood plasma (K2EDTA) and serum for PCSK9 assays, PK, andclinical chemistry assays were taken at the indicated times and quick
frozen for analysis with only a single freeze-thaw cycle. BMS-962476was formulated in sterile PBS solution for single dose intravenousinjection into monkeys.
Liver mRNA Expression Levels. Extraction and assay byreverse transcription-polymerase chain reaction of liver mRNAs forkey sterol pathway genes was conducted as described by Parker et al.(2013).
Lipoprotein Profile, Serum LDL-C, and Clinical ChemistryAssays. The plasma lipoprotein profile from transgenic mice wasanalyzed by size exclusion chromatography using a Superose 6column essentially as described (Ha and Barter, 1985) with assay oftotal cholesterol in the resolved fractions. Serum analytes wereassayed on a Siemens Advia 1800 Clinical Chemistry System usingstandardized enzymatic procedures. LDL-cholesterol was assayed bythe direct LDL method (Roche Diagnostics, Indianapolis, IN). Otheranalytes tested were aspartate aminotransferase, alanine amino-transferase, alkaline phosphatase, gamma glutamyltransferase, totalbilirubin, blood urea nitrogen, creatinine, total cholesterol, trigylcer-ide, high-density lipoprotein, low-density lipoprotein, glucose, totalprotein, albumin, globulins, albumin/globulin ratio, calcium, inorganicphosphorus, sodium; potassium, and chloride.
Pharmacokinetic Measurements. BMS-962476 and relatedprotein drug levelsweremeasured in cyno plasmausing theMesoscaleDiscovery technology platform. Biotinylated human PCSK9 was usedto capture the Adnectin and detection was via a rabbit antibodydirected against PEG (Epitomics, Burlingame, CA) and a sulfo-taggedgoat anti-rabbit polyclonal antibody. Noncompartmental analyseswere performed using Phoenix WinNonlin 6.3 (Pharsight Corpora-tion, Mountain View, CA) using a plasma model and linear up/logdown calculation method.
ResultsPCSK9 Binding Affinity and Kinetics. Binding of the
Adnectin BMS-962476 to human PCSK9, by both immobilizedtarget and solution phase techniques, exhibited subnanomo-lar KD values (Table 1). With the use of biolayer interferom-etry and surface plasmon resonance methods, BMS-962476(PEGylated form) exhibited only slightly lower affinity andslower on-rate relative to the unconjugated form, suggestingminimal interference from the PEG chains. With the use ofthe equilibrium solution method (kinetic exclusion assay),subnanomolar binding to human PCSK9 was also seen(Table 1). A progression to ∼10-fold tighter binding over theprogenitor Adnectin, clone 1459D05, was seen (Tables 1 and2). Although the progenitor Adnectin did not detectably bindcyno PCSK9, BMS-962476 bound to cyno PCSK9 withmoderate affinity, attributed mainly to faster intrinsic off-rates compared with human PCSK9 (Table 2). Furthermore,no appreciable specific binding of the Adnectin to mouserecombinant PCSK9 was observed by SPR, suggesting that
TABLE 1Affinity of binding to human PCSK9 protein assessed by three different biophysical methods at 25°C, pH 7.4Data are mean 6 S.D., for n replicates.
Form of Adnectin Biolayer Interferometry(Octet Red) KD
Surface Plasmon Resonance(ProteOn) KD
Equilibrium Solution Affinity(KinExA) KD
nM
1459D05 (progenitor) 3.6 (n = 6) 1.58 6 0.18 (n = 3) N.D.BMS-962476, his6, no PEG 0.36 (n = 6) 0.29 6 0.01 (n = 2) 0.07 (n = 2)BMS-962476 his6, PEGylated N.D. 0.65 N.D.BMS-962476 N.D. 0.85 6 0.3 (n = 3) 0.22 (n = 2)
KinExA, kinetic exclusion assay; N.D., not determined,
Structure and Pharmacodynamics of an Adnectin PCSK9 Inhibitor 415
at ASPE
T Journals on June 21, 2016
jpet.aspetjournals.orgD
ownloaded from
rodent models could not be employed for functional studies.However the moderate affinity for cyno PCSK9 was sufficientto support target engagement and pharmacodynamic studiesin nonhuman primates.Using ITC to study the thermodynamics and stoichiometry
of the interaction of BMS-962476 with human PCSK9 showedthat the equilibrium binding constant (KD) was too tight tomeasure directly at 25°C (temperature used in the methodsemployed in Table 1). Measuring ITC at 37°C gave an averageKD of 1.36 0.2 nM, and a van’t Hoff–extrapolated KD value of0.24 nM human PCSK9 at 25°C was obtained, agreeing wellwith the methods above. The change in enthalpy observed(DH 5 229.9 6 0.1 kcal/mol) suggested structural stabiliza-tion of the complex via specific bond formation at the expenseof entropic losses (–TDS 5 117.4 kcal/mol at 37°C). Further-more, the ITC results indicated unimolecular binding withstoichiometry approaching 1. Similar ITC results were alsoobserved for the non-PEGylated version of the Adnectin.The ability of BMS-962476 to bind PCSK9 in undiluted
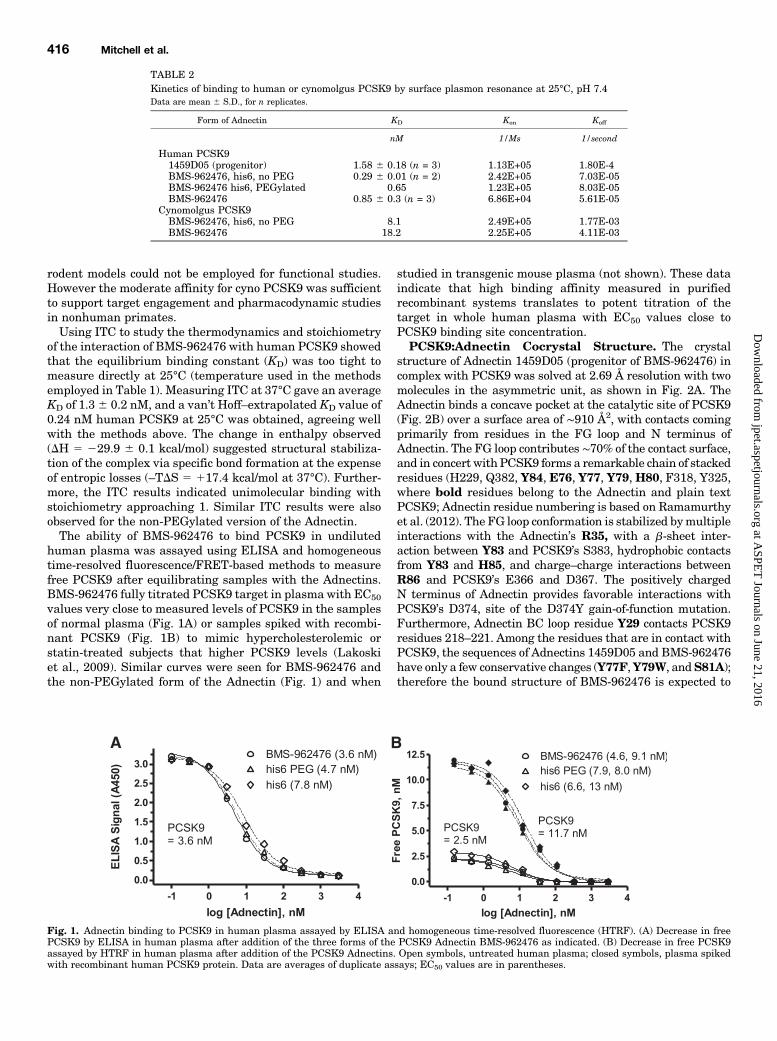
human plasma was assayed using ELISA and homogeneoustime-resolved fluorescence/FRET-based methods to measurefree PCSK9 after equilibrating samples with the Adnectins.BMS-962476 fully titrated PCSK9 target in plasma with EC50
values very close to measured levels of PCSK9 in the samplesof normal plasma (Fig. 1A) or samples spiked with recombi-nant PCSK9 (Fig. 1B) to mimic hypercholesterolemic orstatin-treated subjects that higher PCSK9 levels (Lakoskiet al., 2009). Similar curves were seen for BMS-962476 andthe non-PEGylated form of the Adnectin (Fig. 1) and when
studied in transgenic mouse plasma (not shown). These dataindicate that high binding affinity measured in purifiedrecombinant systems translates to potent titration of thetarget in whole human plasma with EC50 values close toPCSK9 binding site concentration.PCSK9:Adnectin Cocrystal Structure. The crystal
structure of Adnectin 1459D05 (progenitor of BMS-962476) incomplex with PCSK9 was solved at 2.69 Å resolution with twomolecules in the asymmetric unit, as shown in Fig. 2A. TheAdnectin binds a concave pocket at the catalytic site of PCSK9(Fig. 2B) over a surface area of ∼910 Å2, with contacts comingprimarily from residues in the FG loop and N terminus ofAdnectin. The FG loop contributes ∼70% of the contact surface,and in concert with PCSK9 forms a remarkable chain of stackedresidues (H229, Q382, Y84, E76, Y77, Y79, H80, F318, Y325,where bold residues belong to the Adnectin and plain textPCSK9; Adnectin residue numbering is based on Ramamurthyet al. (2012). The FG loop conformation is stabilized bymultipleinteractions with the Adnectin’s R35, with a b-sheet inter-action between Y83 and PCSK9’s S383, hydrophobic contactsfrom Y83 and H85, and charge–charge interactions betweenR86 and PCSK9’s E366 and D367. The positively chargedN terminus of Adnectin provides favorable interactions withPCSK9’s D374, site of the D374Y gain-of-function mutation.Furthermore, Adnectin BC loop residue Y29 contacts PCSK9residues 218–221. Among the residues that are in contact withPCSK9, the sequences of Adnectins 1459D05 and BMS-962476have only a few conservative changes (Y77F,Y79W, andS81A);therefore the bound structure of BMS-962476 is expected to
Fig. 1. Adnectin binding to PCSK9 in human plasma assayed by ELISA and homogeneous time-resolved fluorescence (HTRF). (A) Decrease in freePCSK9 by ELISA in human plasma after addition of the three forms of the PCSK9 Adnectin BMS-962476 as indicated. (B) Decrease in free PCSK9assayed by HTRF in human plasma after addition of the PCSK9 Adnectins. Open symbols, untreated human plasma; closed symbols, plasma spikedwith recombinant human PCSK9 protein. Data are averages of duplicate assays; EC50 values are in parentheses.
TABLE 2Kinetics of binding to human or cynomolgus PCSK9 by surface plasmon resonance at 25°C, pH 7.4Data are mean 6 S.D., for n replicates.
Form of Adnectin KD Kon Koff
nM 1/Ms 1/second
Human PCSK91459D05 (progenitor) 1.58 6 0.18 (n = 3) 1.13E+05 1.80E-4BMS-962476, his6, no PEG 0.29 6 0.01 (n = 2) 2.42E+05 7.03E-05BMS-962476 his6, PEGylated 0.65 1.23E+05 8.03E-05BMS-962476 0.85 6 0.3 (n = 3) 6.86E+04 5.61E-05
Cynomolgus PCSK9BMS-962476, his6, no PEG 8.1 2.49E+05 1.77E-03BMS-962476 18.2 2.25E+05 4.11E-03
416 Mitchell et al.
at ASPE
T Journals on June 21, 2016
jpet.aspetjournals.orgD
ownloaded from
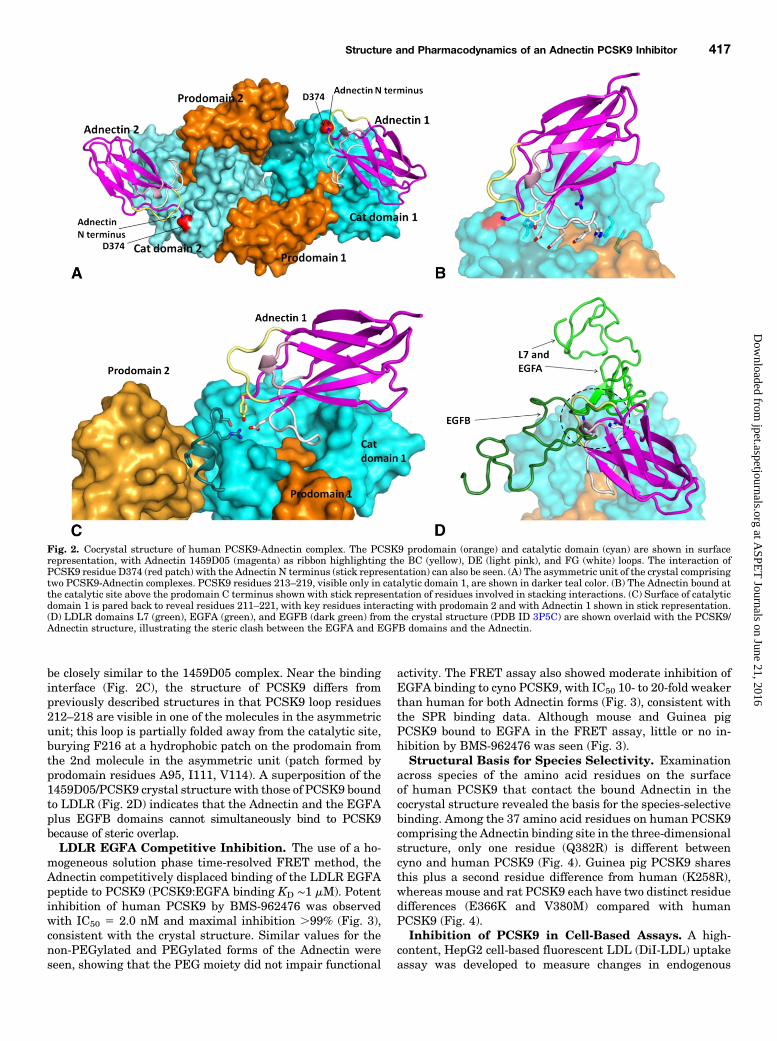
be closely similar to the 1459D05 complex. Near the bindinginterface (Fig. 2C), the structure of PCSK9 differs frompreviously described structures in that PCSK9 loop residues212–218 are visible in one of the molecules in the asymmetricunit; this loop is partially folded away from the catalytic site,burying F216 at a hydrophobic patch on the prodomain fromthe 2nd molecule in the asymmetric unit (patch formed byprodomain residues A95, I111, V114). A superposition of the1459D05/PCSK9 crystal structure with those of PCSK9 boundto LDLR (Fig. 2D) indicates that the Adnectin and the EGFAplus EGFB domains cannot simultaneously bind to PCSK9because of steric overlap.LDLR EGFA Competitive Inhibition. The use of a ho-
mogeneous solution phase time-resolved FRET method, theAdnectin competitively displaced binding of the LDLR EGFApeptide to PCSK9 (PCSK9:EGFA binding KD ∼1 mM). Potentinhibition of human PCSK9 by BMS-962476 was observedwith IC50 5 2.0 nM and maximal inhibition .99% (Fig. 3),consistent with the crystal structure. Similar values for thenon-PEGylated and PEGylated forms of the Adnectin wereseen, showing that the PEG moiety did not impair functional
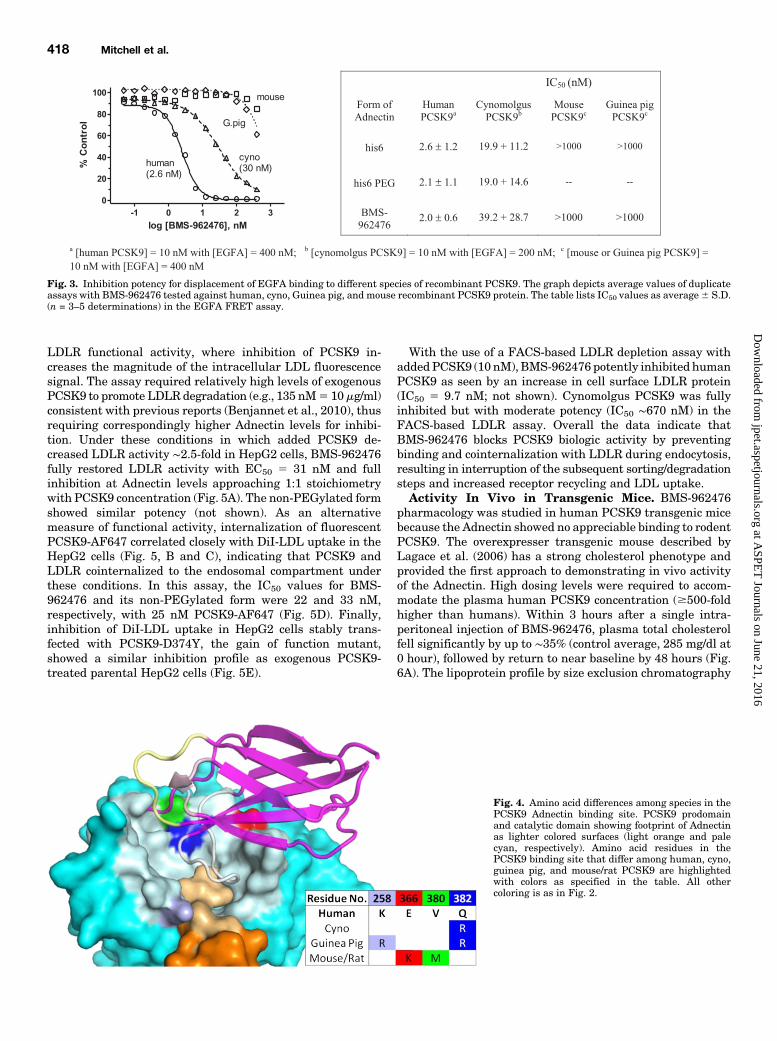
activity. The FRET assay also showed moderate inhibition ofEGFA binding to cyno PCSK9, with IC50 10- to 20-fold weakerthan human for both Adnectin forms (Fig. 3), consistent withthe SPR binding data. Although mouse and Guinea pigPCSK9 bound to EGFA in the FRET assay, little or no in-hibition by BMS-962476 was seen (Fig. 3).Structural Basis for Species Selectivity. Examination
across species of the amino acid residues on the surfaceof human PCSK9 that contact the bound Adnectin in thecocrystal structure revealed the basis for the species-selectivebinding. Among the 37 amino acid residues on human PCSK9comprising the Adnectin binding site in the three-dimensionalstructure, only one residue (Q382R) is different betweencyno and human PCSK9 (Fig. 4). Guinea pig PCSK9 sharesthis plus a second residue difference from human (K258R),whereas mouse and rat PCSK9 each have two distinct residuedifferences (E366K and V380M) compared with humanPCSK9 (Fig. 4).Inhibition of PCSK9 in Cell-Based Assays. A high-
content, HepG2 cell-based fluorescent LDL (DiI-LDL) uptakeassay was developed to measure changes in endogenous
Fig. 2. Cocrystal structure of human PCSK9-Adnectin complex. The PCSK9 prodomain (orange) and catalytic domain (cyan) are shown in surfacerepresentation, with Adnectin 1459D05 (magenta) as ribbon highlighting the BC (yellow), DE (light pink), and FG (white) loops. The interaction ofPCSK9 residue D374 (red patch) with the Adnectin N terminus (stick representation) can also be seen. (A) The asymmetric unit of the crystal comprisingtwo PCSK9-Adnectin complexes. PCSK9 residues 213–219, visible only in catalytic domain 1, are shown in darker teal color. (B) The Adnectin bound atthe catalytic site above the prodomain C terminus shown with stick representation of residues involved in stacking interactions. (C) Surface of catalyticdomain 1 is pared back to reveal residues 211–221, with key residues interacting with prodomain 2 and with Adnectin 1 shown in stick representation.(D) LDLR domains L7 (green), EGFA (green), and EGFB (dark green) from the crystal structure (PDB ID 3P5C) are shown overlaid with the PCSK9/Adnectin structure, illustrating the steric clash between the EGFA and EGFB domains and the Adnectin.
Structure and Pharmacodynamics of an Adnectin PCSK9 Inhibitor 417
at ASPE
T Journals on June 21, 2016
jpet.aspetjournals.orgD
ownloaded from
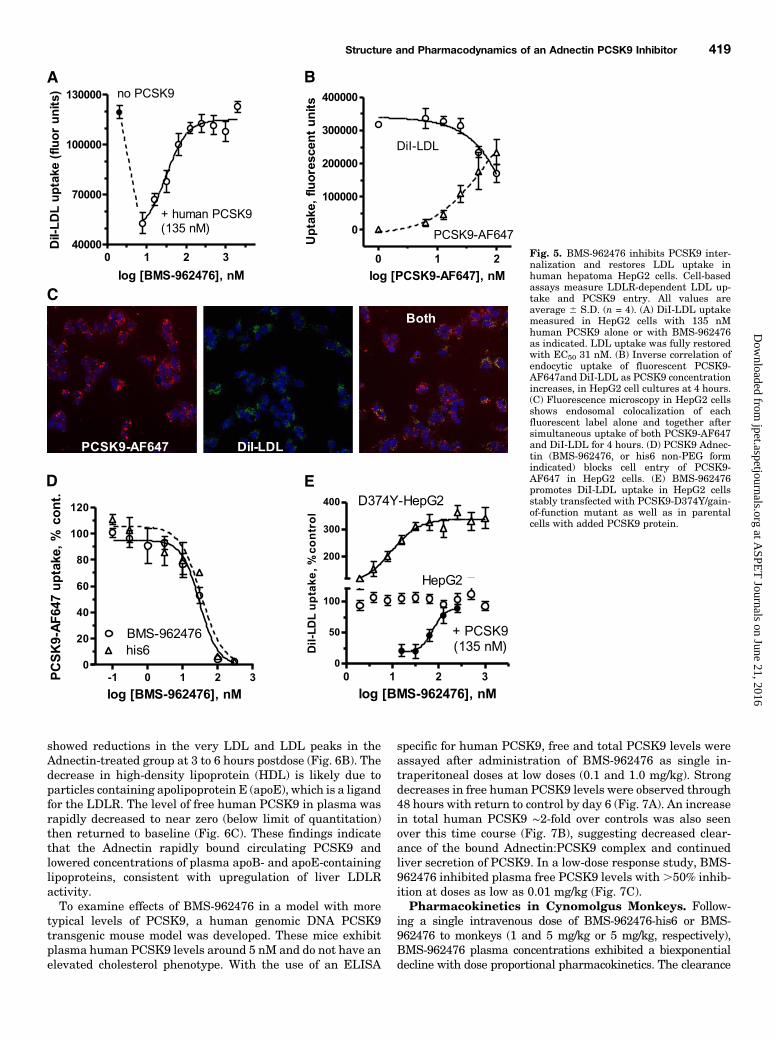
LDLR functional activity, where inhibition of PCSK9 in-creases the magnitude of the intracellular LDL fluorescencesignal. The assay required relatively high levels of exogenousPCSK9 to promote LDLR degradation (e.g., 135 nM5 10mg/ml)consistent with previous reports (Benjannet et al., 2010), thusrequiring correspondingly higher Adnectin levels for inhibi-tion. Under these conditions in which added PCSK9 de-creased LDLR activity ∼2.5-fold in HepG2 cells, BMS-962476fully restored LDLR activity with EC50 5 31 nM and fullinhibition at Adnectin levels approaching 1:1 stoichiometrywith PCSK9 concentration (Fig. 5A). The non-PEGylated formshowed similar potency (not shown). As an alternativemeasure of functional activity, internalization of fluorescentPCSK9-AF647 correlated closely with DiI-LDL uptake in theHepG2 cells (Fig. 5, B and C), indicating that PCSK9 andLDLR cointernalized to the endosomal compartment underthese conditions. In this assay, the IC50 values for BMS-962476 and its non-PEGylated form were 22 and 33 nM,respectively, with 25 nM PCSK9-AF647 (Fig. 5D). Finally,inhibition of DiI-LDL uptake in HepG2 cells stably trans-fected with PCSK9-D374Y, the gain of function mutant,showed a similar inhibition profile as exogenous PCSK9-treated parental HepG2 cells (Fig. 5E).
With the use of a FACS-based LDLR depletion assay withaddedPCSK9 (10 nM), BMS-962476 potently inhibited humanPCSK9 as seen by an increase in cell surface LDLR protein(IC50 5 9.7 nM; not shown). Cynomolgus PCSK9 was fullyinhibited but with moderate potency (IC50 ∼670 nM) in theFACS-based LDLR assay. Overall the data indicate thatBMS-962476 blocks PCSK9 biologic activity by preventingbinding and cointernalization with LDLR during endocytosis,resulting in interruption of the subsequent sorting/degradationsteps and increased receptor recycling and LDL uptake.Activity In Vivo in Transgenic Mice. BMS-962476
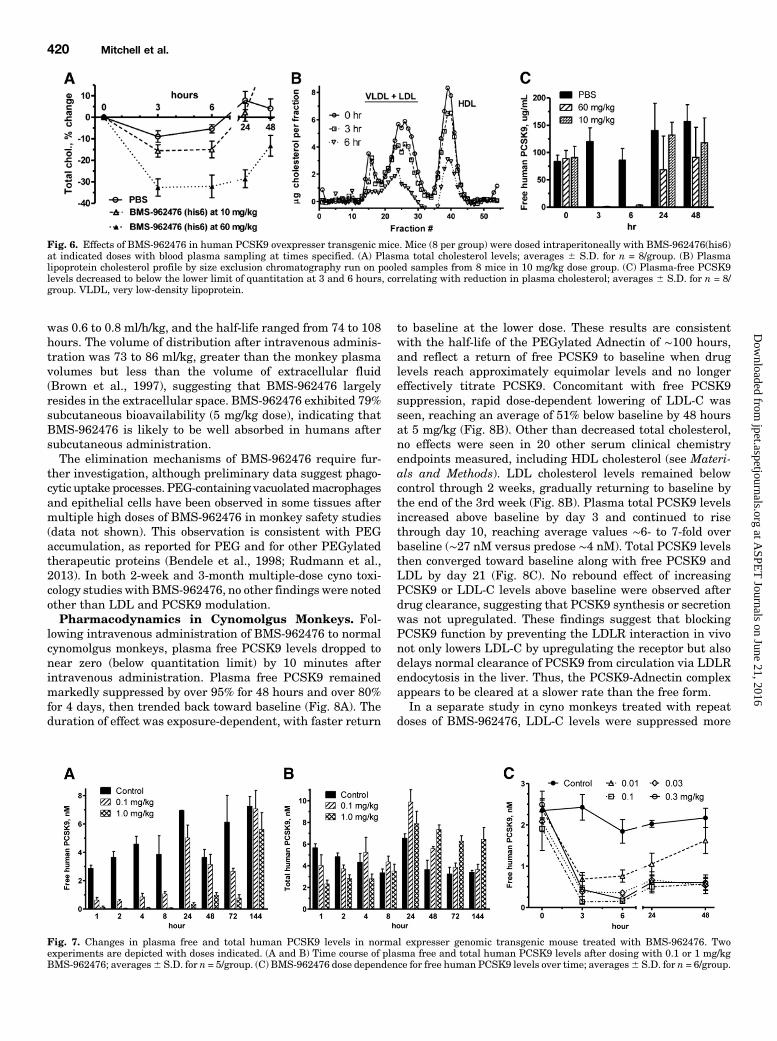
pharmacology was studied in human PCSK9 transgenic micebecause the Adnectin showed no appreciable binding to rodentPCSK9. The overexpresser transgenic mouse described byLagace et al. (2006) has a strong cholesterol phenotype andprovided the first approach to demonstrating in vivo activityof the Adnectin. High dosing levels were required to accom-modate the plasma human PCSK9 concentration ($500-foldhigher than humans). Within 3 hours after a single intra-peritoneal injection of BMS-962476, plasma total cholesterolfell significantly by up to ∼35% (control average, 285 mg/dl at0 hour), followed by return to near baseline by 48 hours (Fig.6A). The lipoprotein profile by size exclusion chromatography
Fig. 3. Inhibition potency for displacement of EGFA binding to different species of recombinant PCSK9. The graph depicts average values of duplicateassays with BMS-962476 tested against human, cyno, Guinea pig, and mouse recombinant PCSK9 protein. The table lists IC50 values as average6 S.D.(n = 3–5 determinations) in the EGFA FRET assay.
Fig. 4. Amino acid differences among species in thePCSK9 Adnectin binding site. PCSK9 prodomainand catalytic domain showing footprint of Adnectinas lighter colored surfaces (light orange and palecyan, respectively). Amino acid residues in thePCSK9 binding site that differ among human, cyno,guinea pig, and mouse/rat PCSK9 are highlightedwith colors as specified in the table. All othercoloring is as in Fig. 2.
418 Mitchell et al.
at ASPE
T Journals on June 21, 2016
jpet.aspetjournals.orgD
ownloaded from
showed reductions in the very LDL and LDL peaks in theAdnectin-treated group at 3 to 6 hours postdose (Fig. 6B). Thedecrease in high-density lipoprotein (HDL) is likely due toparticles containing apolipoprotein E (apoE), which is a ligandfor the LDLR. The level of free human PCSK9 in plasma wasrapidly decreased to near zero (below limit of quantitation)then returned to baseline (Fig. 6C). These findings indicatethat the Adnectin rapidly bound circulating PCSK9 andlowered concentrations of plasma apoB- and apoE-containinglipoproteins, consistent with upregulation of liver LDLRactivity.To examine effects of BMS-962476 in a model with more
typical levels of PCSK9, a human genomic DNA PCSK9transgenic mouse model was developed. These mice exhibitplasma human PCSK9 levels around 5 nM and do not have anelevated cholesterol phenotype. With the use of an ELISA
specific for human PCSK9, free and total PCSK9 levels wereassayed after administration of BMS-962476 as single in-traperitoneal doses at low doses (0.1 and 1.0 mg/kg). Strongdecreases in free human PCSK9 levels were observed through48 hours with return to control by day 6 (Fig. 7A). An increasein total human PCSK9 ∼2-fold over controls was also seenover this time course (Fig. 7B), suggesting decreased clear-ance of the bound Adnectin:PCSK9 complex and continuedliver secretion of PCSK9. In a low-dose response study, BMS-962476 inhibited plasma free PCSK9 levels with.50% inhib-ition at doses as low as 0.01 mg/kg (Fig. 7C).Pharmacokinetics in Cynomolgus Monkeys. Follow-
ing a single intravenous dose of BMS-962476-his6 or BMS-962476 to monkeys (1 and 5 mg/kg or 5 mg/kg, respectively),BMS-962476 plasma concentrations exhibited a biexponentialdecline with dose proportional pharmacokinetics. The clearance
Fig. 5. BMS-962476 inhibits PCSK9 inter-nalization and restores LDL uptake inhuman hepatoma HepG2 cells. Cell-basedassays measure LDLR-dependent LDL up-take and PCSK9 entry. All values areaverage 6 S.D. (n = 4). (A) DiI-LDL uptakemeasured in HepG2 cells with 135 nMhuman PCSK9 alone or with BMS-962476as indicated. LDL uptake was fully restoredwith EC50 31 nM. (B) Inverse correlation ofendocytic uptake of fluorescent PCSK9-AF647and DiI-LDL as PCSK9 concentrationincreases, in HepG2 cell cultures at 4 hours.(C) Fluorescence microscopy in HepG2 cellsshows endosomal colocalization of eachfluorescent label alone and together aftersimultaneous uptake of both PCSK9-AF647and DiI-LDL for 4 hours. (D) PCSK9 Adnec-tin (BMS-962476, or his6 non-PEG formindicated) blocks cell entry of PCSK9-AF647 in HepG2 cells. (E) BMS-962476promotes DiI-LDL uptake in HepG2 cellsstably transfected with PCSK9-D374Y/gain-of-function mutant as well as in parentalcells with added PCSK9 protein.
Structure and Pharmacodynamics of an Adnectin PCSK9 Inhibitor 419
at ASPE
T Journals on June 21, 2016
jpet.aspetjournals.orgD
ownloaded from
was 0.6 to 0.8 ml/h/kg, and the half-life ranged from 74 to 108hours. The volume of distribution after intravenous adminis-tration was 73 to 86 ml/kg, greater than the monkey plasmavolumes but less than the volume of extracellular fluid(Brown et al., 1997), suggesting that BMS-962476 largelyresides in the extracellular space. BMS-962476 exhibited 79%subcutaneous bioavailability (5 mg/kg dose), indicating thatBMS-962476 is likely to be well absorbed in humans aftersubcutaneous administration.The elimination mechanisms of BMS-962476 require fur-
ther investigation, although preliminary data suggest phago-cytic uptake processes. PEG-containing vacuolatedmacrophagesand epithelial cells have been observed in some tissues aftermultiple high doses of BMS-962476 in monkey safety studies(data not shown). This observation is consistent with PEGaccumulation, as reported for PEG and for other PEGylatedtherapeutic proteins (Bendele et al., 1998; Rudmann et al.,2013). In both 2-week and 3-month multiple-dose cyno toxi-cology studies with BMS-962476, no other findings were notedother than LDL and PCSK9 modulation.Pharmacodynamics in Cynomolgus Monkeys. Fol-
lowing intravenous administration of BMS-962476 to normalcynomolgus monkeys, plasma free PCSK9 levels dropped tonear zero (below quantitation limit) by 10 minutes afterintravenous administration. Plasma free PCSK9 remainedmarkedly suppressed by over 95% for 48 hours and over 80%for 4 days, then trended back toward baseline (Fig. 8A). Theduration of effect was exposure-dependent, with faster return
to baseline at the lower dose. These results are consistentwith the half-life of the PEGylated Adnectin of ∼100 hours,and reflect a return of free PCSK9 to baseline when druglevels reach approximately equimolar levels and no longereffectively titrate PCSK9. Concomitant with free PCSK9suppression, rapid dose-dependent lowering of LDL-C wasseen, reaching an average of 51% below baseline by 48 hoursat 5 mg/kg (Fig. 8B). Other than decreased total cholesterol,no effects were seen in 20 other serum clinical chemistryendpoints measured, including HDL cholesterol (see Materi-als and Methods). LDL cholesterol levels remained belowcontrol through 2 weeks, gradually returning to baseline bythe end of the 3rd week (Fig. 8B). Plasma total PCSK9 levelsincreased above baseline by day 3 and continued to risethrough day 10, reaching average values ∼6- to 7-fold overbaseline (∼27 nM versus predose ∼4 nM). Total PCSK9 levelsthen converged toward baseline along with free PCSK9 andLDL by day 21 (Fig. 8C). No rebound effect of increasingPCSK9 or LDL-C levels above baseline were observed afterdrug clearance, suggesting that PCSK9 synthesis or secretionwas not upregulated. These findings suggest that blockingPCSK9 function by preventing the LDLR interaction in vivonot only lowers LDL-C by upregulating the receptor but alsodelays normal clearance of PCSK9 from circulation via LDLRendocytosis in the liver. Thus, the PCSK9-Adnectin complexappears to be cleared at a slower rate than the free form.In a separate study in cyno monkeys treated with repeat
doses of BMS-962476, LDL-C levels were suppressed more
Fig. 6. Effects of BMS-962476 in human PCSK9 ovexpresser transgenic mice. Mice (8 per group) were dosed intraperitoneally with BMS-962476(his6)at indicated doses with blood plasma sampling at times specified. (A) Plasma total cholesterol levels; averages 6 S.D. for n = 8/group. (B) Plasmalipoprotein cholesterol profile by size exclusion chromatography run on pooled samples from 8 mice in 10 mg/kg dose group. (C) Plasma-free PCSK9levels decreased to below the lower limit of quantitation at 3 and 6 hours, correlating with reduction in plasma cholesterol; averages 6 S.D. for n = 8/group. VLDL, very low-density lipoprotein.
Fig. 7. Changes in plasma free and total human PCSK9 levels in normal expresser genomic transgenic mouse treated with BMS-962476. Twoexperiments are depicted with doses indicated. (A and B) Time course of plasma free and total human PCSK9 levels after dosing with 0.1 or 1 mg/kgBMS-962476; averages6 S.D. for n = 5/group. (C) BMS-962476 dose dependence for free human PCSK9 levels over time; averages6 S.D. for n = 6/group.
420 Mitchell et al.
at ASPE
T Journals on June 21, 2016
jpet.aspetjournals.orgD
ownloaded from
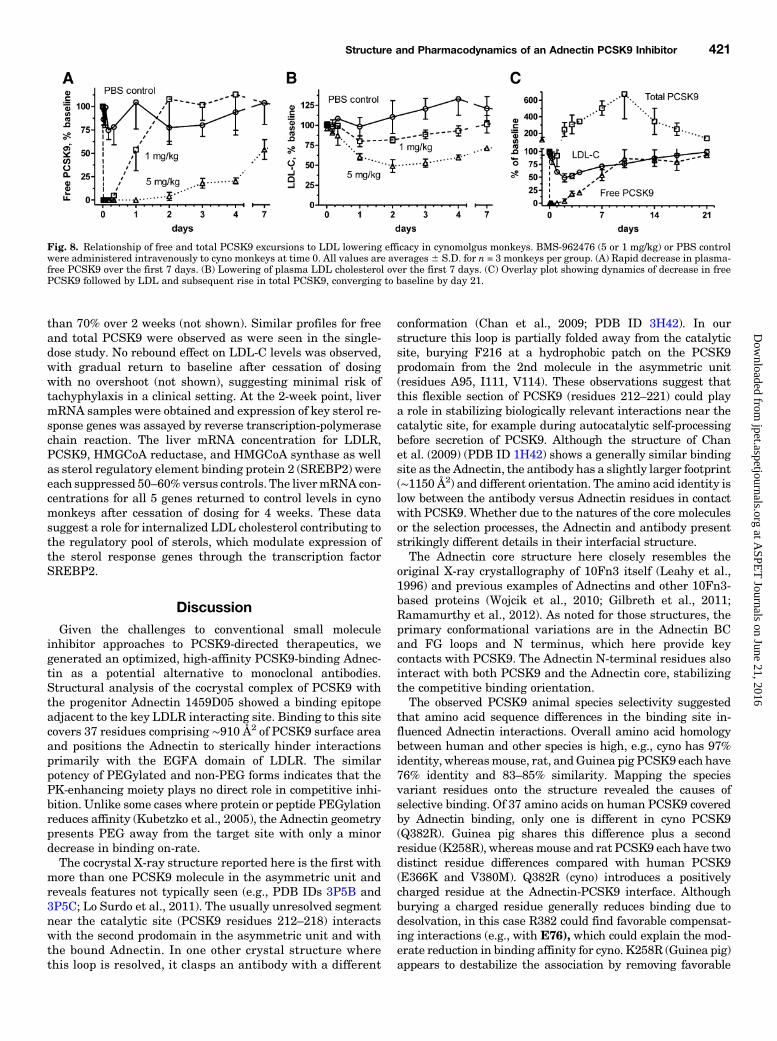
than 70% over 2 weeks (not shown). Similar profiles for freeand total PCSK9 were observed as were seen in the single-dose study. No rebound effect on LDL-C levels was observed,with gradual return to baseline after cessation of dosingwith no overshoot (not shown), suggesting minimal risk oftachyphylaxis in a clinical setting. At the 2-week point, livermRNA samples were obtained and expression of key sterol re-sponse genes was assayed by reverse transcription-polymerasechain reaction. The liver mRNA concentration for LDLR,PCSK9, HMGCoA reductase, and HMGCoA synthase as wellas sterol regulatory element binding protein 2 (SREBP2) wereeach suppressed 50–60%versus controls. The livermRNA con-centrations for all 5 genes returned to control levels in cynomonkeys after cessation of dosing for 4 weeks. These datasuggest a role for internalized LDL cholesterol contributing tothe regulatory pool of sterols, which modulate expression ofthe sterol response genes through the transcription factorSREBP2.
DiscussionGiven the challenges to conventional small molecule
inhibitor approaches to PCSK9-directed therapeutics, wegenerated an optimized, high-affinity PCSK9-binding Adnec-tin as a potential alternative to monoclonal antibodies.Structural analysis of the cocrystal complex of PCSK9 withthe progenitor Adnectin 1459D05 showed a binding epitopeadjacent to the key LDLR interacting site. Binding to this sitecovers 37 residues comprising ∼910 Å2 of PCSK9 surface areaand positions the Adnectin to sterically hinder interactionsprimarily with the EGFA domain of LDLR. The similarpotency of PEGylated and non-PEG forms indicates that thePK-enhancing moiety plays no direct role in competitive inhi-bition. Unlike some cases where protein or peptide PEGylationreduces affinity (Kubetzko et al., 2005), the Adnectin geometrypresents PEG away from the target site with only a minordecrease in binding on-rate.The cocrystal X-ray structure reported here is the first with
more than one PCSK9 molecule in the asymmetric unit andreveals features not typically seen (e.g., PDB IDs 3P5B and3P5C; Lo Surdo et al., 2011). The usually unresolved segmentnear the catalytic site (PCSK9 residues 212–218) interactswith the second prodomain in the asymmetric unit and withthe bound Adnectin. In one other crystal structure wherethis loop is resolved, it clasps an antibody with a different
conformation (Chan et al., 2009; PDB ID 3H42). In ourstructure this loop is partially folded away from the catalyticsite, burying F216 at a hydrophobic patch on the PCSK9prodomain from the 2nd molecule in the asymmetric unit(residues A95, I111, V114). These observations suggest thatthis flexible section of PCSK9 (residues 212–221) could playa role in stabilizing biologically relevant interactions near thecatalytic site, for example during autocatalytic self-processingbefore secretion of PCSK9. Although the structure of Chanet al. (2009) (PDB ID 1H42) shows a generally similar bindingsite as the Adnectin, the antibody has a slightly larger footprint(∼1150 Å2) and different orientation. The amino acid identity islow between the antibody versus Adnectin residues in contactwith PCSK9. Whether due to the natures of the core moleculesor the selection processes, the Adnectin and antibody presentstrikingly different details in their interfacial structure.The Adnectin core structure here closely resembles the
original X-ray crystallography of 10Fn3 itself (Leahy et al.,1996) and previous examples of Adnectins and other 10Fn3-based proteins (Wojcik et al., 2010; Gilbreth et al., 2011;Ramamurthy et al., 2012). As noted for those structures, theprimary conformational variations are in the Adnectin BCand FG loops and N terminus, which here provide keycontacts with PCSK9. The Adnectin N-terminal residues alsointeract with both PCSK9 and the Adnectin core, stabilizingthe competitive binding orientation.The observed PCSK9 animal species selectivity suggested
that amino acid sequence differences in the binding site in-fluenced Adnectin interactions. Overall amino acid homologybetween human and other species is high, e.g., cyno has 97%identity, whereasmouse, rat, andGuinea pig PCSK9 each have76% identity and 83–85% similarity. Mapping the speciesvariant residues onto the structure revealed the causes ofselective binding. Of 37 amino acids on human PCSK9 coveredby Adnectin binding, only one is different in cyno PCSK9(Q382R). Guinea pig shares this difference plus a secondresidue (K258R), whereasmouse and rat PCSK9 each have twodistinct residue differences compared with human PCSK9(E366K and V380M). Q382R (cyno) introduces a positivelycharged residue at the Adnectin-PCSK9 interface. Althoughburying a charged residue generally reduces binding due todesolvation, in this case R382 could find favorable compensat-ing interactions (e.g., with E76), which could explain the mod-erate reduction in binding affinity for cyno. K258R (Guinea pig)appears to destabilize the association by removing favorable
Fig. 8. Relationship of free and total PCSK9 excursions to LDL lowering efficacy in cynomolgus monkeys. BMS-962476 (5 or 1 mg/kg) or PBS controlwere administered intravenously to cyno monkeys at time 0. All values are averages 6 S.D. for n = 3 monkeys per group. (A) Rapid decrease in plasma-free PCSK9 over the first 7 days. (B) Lowering of plasma LDL cholesterol over the first 7 days. (C) Overlay plot showing dynamics of decrease in freePCSK9 followed by LDL and subsequent rise in total PCSK9, converging to baseline by day 21.
Structure and Pharmacodynamics of an Adnectin PCSK9 Inhibitor 421
at ASPE
T Journals on June 21, 2016
jpet.aspetjournals.orgD
ownloaded from

packing of the aliphatic portion of K258 against Y79. E366K(mouse and rat) would eliminate the favorable charge-chargeinteraction with Adnectin’s R86, and V380M (mouse and rat)may diminish complementary with Y84. These explanationsare based on the crystal structure of 1459D05 with PCSK9;however, the overall binding mode for BMS-962476 is expectedto be similar with only three conservative changes in PCSK9-contacting residues (Y77F, Y79W, S81A in the Adnectin,which do not contact the species-variant residues).The species-dependent binding profile led us to study
human PCSK9 transgenic mouse models for in vivo pharma-cology. The overexpresser human PCSK9 transgenic mousedescribed by Lagace et al. (2006) provided a strongly hyper-cholesterolemic model driven by very high circulating levels ofhuman PCSK9. When administered at proportionately highdosages, the Adnectin quickly (3 hours) reduced plasma totalcholesterol in concert with free human PCSK9 in plasma, andboth returned to baseline 2–3 days later, further illustratingthe dynamic physiologic response to PCSK9 modulation.To study the pharmacodynamics in a preclinical model with
normal human PCSK9 levels, a genomic DNA human PCSK9transgenic mouse model was developed. These mice expressfull-length human PCSK9 driven from the insertion of a hu-man DNA fragment surrounding the PCSK9 gene, includingpromoter elements. In homozygous transgenic mice, this re-sulted in human PCSK9 plasma levels of ∼5 nM, insufficientto cause a cholesterol phenotype but allowing binding studies.Here BMS-962476 was very potent with ED50 ∼0.01mg/kg i.p.for suppression of plasma-free human PCSK9 levels. Therapid decreases in free PCSK9 were followed by 2-fold in-creases in total human PCSK9. Although increased synthesisof PCSK9 may play a role in the rise of total PCSK9, the lackof a rebound effect in both mouse and monkey studies sug-gests this is unlikely. Our data are more consistent with thepossibility of diminished clearance of the bound PCSK9complex.In cynomolgus monkeys dosed with BMS-962476, the
excursion in total PCSK9 levels reached ∼6- to 7-fold overbaseline before returning to control levels by 3 weeks, inparallel with return to baseline of LDL-C levels. The effect ontotal PCSK9 is consistent with the proposal that PCSK9 exitsthe circulation primarily through liver LDLR endocytosis.Previous studies inmice and humans have shown that PCSK9clearance is rapid and involves both LDLR-dependent and-independent pathways, but overall the receptor-dependentendocytic clearance process seems to dominate (Grefhorstet al., 2008; Stein and Raal, 2013; Tavori et al., 2013). Thetemporal pattern of decrease in free PCSK9, drop in LDL, andrise then fall in total PCSK9 concentration is likely coupled tothe formation of the PCSK9:Adnectin complex in circulation.This suggests that increased LDL uptake into liver may be onemechanism linked to return to baseline of total PCSK9. Con-sistent with the proposal that PCSK9 clearance is primarilythrough LDLR cointernalization, the rise in total PCSK9 levelsappears to reflect continued secretion coinciding with reducedclearance. Whether this pattern differs from mAbs, which mayinvolve Fc receptor-mediated mechanisms for clearance ofcomplexes, remains to be seen. In our studies, the return tobaseline in total PCSK9 appears to be the consequence of theAdnectin complex dissociating over time (drug levels falling),allowing PCSK9 to interact and internalize with the LDLR. Inmultiple-dose cyno studies (data not shown), cessation of
Adnectin dosing was associated with eventual return of LDLto predose levels without rebound to higher levels. The overallpattern of cholesterol homeostasis is achieved in this in vivomodel through the multiple levels of transcriptional and post-transcriptional feedback control affecting LDLR, cholesterolbiosynthetic pathway, and PCSK9 expression and function.Changes in liver secretion of new PCSK9 and in expression
of related genes may also play a role in the excursions of totalPCSK9 after treatment with the Adnectin or antibodies. Toaddress this, we measured the levels of expression for severalsterol response genes in liver samples of treated cynomonkeys. Under conditions where repeated dosing in cynomonkeys reduced LDL-C levels markedly over 2 weeks, livermRNA levels for LDLR, PCSK9, and HMGCoA reductase andHMGCoA synthase, as well as SREBP2, were each sup-pressed 50–60% at 2 weeks, followed by return to control4 weeks after dosing stopped (washout). These data suggesta role for internalized LDL-derived cholesterol contributingto the regulatory sterol pool in hepatocytes that modulatescoordinate expression of key sterol response genes throughSREBP2 (Horton et al., 2007; Parker et al., 2013). Althoughthe changes in circulating total PCSK9 levels after inhibitionwith protein-ligand approaches are not well understood, themodel depicted in Fig. 9 provides a possible integration of thepharmacodynamics after administration of a potent PCSK9inhibitor. The restoration of homeostasis, seen in return ofplasma LDL-C and total PCSK9 levels to baseline, reflects thehighly responsive dynamics of the PCSK9-LDLR pathway.The influence of cholesterol internalized into liver modifyingSREBP2-responsive gene expression would add considerablyto the changes seen in the in vivo studies.The decrease in HDL cholesterol levels observed in over-
expressing transgenic mice after treatment with BMS-962476is consistent with other reports showing reductions in HDLwith PCSK9 suppression in mouse models (Graham et al.,2008; Choi et al., 2013). It is well established that apoE isa ligand for the LDLR protein family and that some sub-classes of HDL particles contain apoE (Gordon et al., 1983),providing a likely mechanism for HDL cholesterol loweringthrough PCSK9 inhibition. Mice and rats, lacking expressionof cholesterol ester transfer protein, carry most of their totalplasma cholesterol within HDL, unlike nonhuman primatesand humans. HDL cholesterol reduction in PCSK9 antibody-treated monkeys has been noted in some studies (Chan et al.,2009), but not in others (Liang et al., 2012), and has not beenreported in human clinical trials with antibody or antisensedrugs (e.g., Stein et al., 2012; Fitzgerald et al., 2013). Anextensive analysis of plasma lipoproteins and cholesterol andbile acid metabolism in our PCSK9 Adnectin multiple-dosemonkey studies (Parker et al., 2013) suggests that significantHDL lowering in response to PCSK9 inhibition is mainly arodent physiologic phenomenon.In conclusion, BMS-962476 represents a new class of high-
affinity PCSK9 binding agents that exhibits many of thefunctional properties of inhibitory monoclonal antibodies toPCSK9 but fits between peptides and immunoglobulins inmolecular size range. The crystal structure of the PCSK9:Adnectin complex, the subnanomolar human PCSK9-specificbinding affinity, and the in vitro/in vivo pharmacologic profiledescribed in these studies highlights the biologically impor-tant and rapid dynamics of PCSK9 control over LDLmetabolism. The data characterize BMS-962476 preclinically
422 Mitchell et al.
at ASPE
T Journals on June 21, 2016
jpet.aspetjournals.orgD
ownloaded from

as a potent and efficacious PCSK9 inhibitor that may havetherapeutic potential for LDL lowering in patients withhypercholesterolemia in the treatment of cardiovasculardiseases.
Acknowledgments
Diffraction data were collected by Rick Walter and Gina Ranieri ofShamrock Structures, LLC, using beamlines at the Advanced PhotonSource. The transgenic mouse studies were made possible by theexcellent work of George Psaltis, Jian Chen, Carol Ryan, and RichardYang. The authors also thank Gabe Mintier, Dawn Mulligan andYaqun for cloning and protein production and purification, ClaudioMapelli for EGFA peptide synthesis, Vikas Malaiya for assistancewith mRNA display, Samantha Fong for protein chemistry analysis,Lumelle Schneweis for kinetic exclusion assays, Kevin O’Malley forOctet Red, Christine Cummings and Debra Wescott for clinicalchemistry assays, and Anjaneya Chimalakonda for PK/PD analysis.
Authorship Contributions
Participated in research design: Mitchell, Chao, Khan, Parker.Conducted experiments: Lo, Monshizadegan, Meyers, Low, Russo,
DiBella, Denhez, Myers, Ho, Miao, Khan.Contributed new reagents or analytic tools: Low, Gao, Myers, Duke,
Witmer, Miao.Performed data analysis: Mitchell, Chao, Sitkoff, Khan, Parker.Wrote or contributed to the writing of the manuscript: Mitchell,
Sitkoff, Parker.
References
Abifadel M, Varret M, Rabès JP, Allard D, Ouguerram K, Devillers M, Cruaud C,Benjannet S, Wickham L, and Erlich D, et al. (2003) Mutations in PCSK9 causeautosomal dominant hypercholesterolemia. Nat Genet 34:154–156.
Benjannet S, Saavedra YG, Hamelin J, Asselin MC, Essalmani R, Pasquato A,Lemaire P, Duke G, Miao B, and Duclos F, et al. (2010) Effects of the prosegmentand pH on the activity of PCSK9: evidence for additional processing events. J BiolChem 285:40965–40978.
Bottomley MJ, Cirillo A, Orsatti L, Ruggeri L, Fisher TS, Santoro JC, Cummings RT,Cubbon RM, Lo Surdo P, and Calzetta A, et al. (2009) Structural and biochemicalcharacterization of the wild type PCSK9/EGF-AB complex and natural familialhypercholesterolemia mutants. J Biol Chem 284:1313–1323.
Brown RP, Delp MD, Lindstedt SL, Rhomberg LR, and Beliles RP (1997) Physio-logical parameter values for physiologically based pharmacokinetic models. ToxicolInd Health 13:407–484.
Bendele A, Seely J, Richey C, Sennello G, and Shopp G (1998) Short Communiction:Renal tubular vacuolation in animals treated with polyethelene-glycol-conjugatedproteins. Toxicol Sci 42:152–157.
Chan JC, Piper DE, Caoa Q, Liu D, King C, Wang W, Tang J, Liu Q, Higbee J,and Xia Z, et al. (2009) A proprotein convertase subtilisin/kexin type 9 neutralizingantibody reduces serum cholesterol in mice and nonhuman primates. Proc NatlAcad Sci USA 106:9820–9825.
Choi S, Aljakna A, Srivastava U, Peterson BR, Deng B, Prat A, and Korstanje R(2013) Decreased APOE-containing HDL subfractions and cholesterol efflux ca-pacity of serum in mice lacking Pcsk9. Lipids Health Dis 12:112–121.
Cohen JC, Boerwinkle E, Mosley TH, Jr, and Hobbs HH (2006) Sequence variationsin PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med354:1264–1272.
Doyle ML (1997) Characterization of binding interactions by isothermal titrationcalorimetry. Curr Opin Biotechnol 8:31–35.
Drake AW, Myszka DG, and Klakamp SL (2004) Characterizing high-affinityantigen/antibody complexes by kinetic- and equilibrium-based methods. AnalBiochem 328:35–43.
Emsley P and Cowtan K (2004) Coot: model-building tools for molecular graphics.Acta Crystallogr D Biol Crystallogr 60:2126–2132.
Fitzgerald K, Frank-Kamenetsky M, Shulga-Morskaya S, Liebow A, Bettencourt A,Sutherland J, Hutabarat R, Clausen V, Karsten V, and Cehelsky J, et al. (2013)Effect of an RNA interference drug on the synthesis of PCSK9 and concentration ofserum LDL cholesterol in healthy volunteers: phase 1 trial. Lancet 383:60–68.
Gilbreth RN, Truong K, Madu I, Koide A, Wojcik JB, Li N-S, Piccirilli JA, Chen Y,and Koide S (2011) Isoform-specific monobody inhibitors of small ubiquitin-relatedmodifiers engineered using structure-guided library design. Proc Natl Acad SciUSA 108:7751–7756.
Graham M, Lemonidis K, Whipple C, Subramaniam A, Monia B, Crooke S,and Crooke R (2008) Antisense inhibition of proprotein convertase subtilisin/kexintype 9 reduces serum LDL in hyperlipidemic mice. J Lipid Res 48:763–767.
Grefhorst A, McNutt MC, Lagace TA, and Horton JD (2008) Plasma PCSK9 prefer-entially reduces liver LDL receptors in mice. J Lipid Res 49:1303–1311.
Ha YC and Barter PJ (1985) Rapid separation of plasma lipoproteins by gel perme-ation chromatography on agarose gel Superose 6B. J Chromatogr A 341:154–159.
Horton JD, Cohen JC, and Hobbs HH (2007) Molecular biology of PCSK9: its role inLDL metabolism. Trends Biochem Sci 32:71–77.
Gordon V, Innerarity TL, and Mahley RW (1983) Formation of cholesterol- and apo-protein E-enriched high density lipoproteins in vitro. J Biol Chem 258:6202–6212.
Koide A, Bailey CW, Huang X, and Koide S (1998) The fibronectin type III domain asa scaffold for novel binding proteins. J Mol Biol 284:1141–1151.
Kubetzko S, Sarkar CA, and Plückthun A (2005) Protein PEGylation decreases ob-served target association rates via a dual blocking mechanism. Mol Pharmacol 68:1439–1454.
Kwon HJ, Lagace TA, McNutt MC, Horton JD, and Deisenhofer J (2008) Molecular basisfor LDL receptor recognition by PCSK9. Proc Natl Acad Sci USA 105:1820–1825.
Lagace TA, Curtis DE, Garuti R, McNutt MC, Park SW, Prather HB, Anderson NN,Ho YK, Hammer RE, and Horton JD (2006) Secreted PCSK9 decreases the numberof LDL receptors in hepatocytes and in livers of parabiotic mice. J Clin Invest 116:2995–3005.
Lakoski SG, Lagace TA, Cohen JC, Horton JD, and Hobbs HH (2009) Genetic andmetabolic determinants of plasma PCSK9 levels. J Clin Endocrinol Metab 94:2537–2543.
Leahy DJ, Aukhil I, and Erickson HP (1996) 2.0 A crystal structure of a four-domainsegment of human fibronectin encompassing the RGD loop and synergy region. Cell84:155–164.
Liang H, Chaparro-Riggers J, Strop P, Geng T, Sutton JE, Tsai D, Bai L, Abdiche Y,Dilley J, and Yu J, et al. (2012) Proprotein convertase substilisin/kexin type 9antagonism reduces low-density lipoprotein cholesterol in statin-treated hyper-cholesterolemic nonhuman primates. J Pharmacol Exp Ther 340:228–236.
Lipovsek D (2011) Adnectins: engineered target-binding protein therapeutics. ProteinEng Des Sel 24:3–9.
Lo Surdo P, Bottomley MJ, Calzetta A, Settembre EC, Cirillo A, Pandit S, Ni YG,Hubbard B, Sitlani A, and Carfí A (2011) Mechanistic implications for LDL re-ceptor degradation from the PCSK9/LDLR structure at neutral pH. EMBO Rep 12:1300–1305.
McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ(2007) Phaser crystallographic software. J Appl Cryst 40:658–674.
Ni YG, Di Marco S, Condra JH, Peterson LB, Wang W, Wang F, Pandit S, HammondHA, Rosa R, and Cummings RT, et al. (2011) A PCSK9-binding antibody thatstructurally mimics the EGF(A) domain of LDL-receptor reduces LDL cholesterolin vivo. J Lipid Res 52:78–86.
Parker MH, Chen Y, Danehy F, Dufu K, Ekstrom J, Getmanova E, Gokemeijer J, XuL, and Lipovsek D (2005) Antibody mimics based on humanfibronectin type-threedomain engineered for thermostability and high-affinity binding to vascular en-dothelial growth factor two. Protein Eng Des Sel 18:435–444.
Parker RA, Garcia R, Ryan CS, Liu X, Shipkova P, Livanov V, Patel P, and Ho SP(2013) Bile acid and sterol metabolism with combined HMG-CoA reductase andPCSK9 suppression. J Lipid Res 54:2400–2409.
Fig. 9. Model for the rise and fall in circulatingPCSK9 levels following dosing with a PCSK9 in-hibitor Adnectin. Integration of the dynamics ofchange in circulating PCSK9 through cholesterolfeedback control of liver gene expression followingadministration of a potent PCSK9 inhibitor. LDLparticles omitted for clarity.
Structure and Pharmacodynamics of an Adnectin PCSK9 Inhibitor 423
at ASPE
T Journals on June 21, 2016
jpet.aspetjournals.orgD
ownloaded from

Pflugrath JW (1999) The finer things in X-ray diffraction data collection. ActaCrystallogr D Biol Crystallogr 55:1718–1725.
Ramamurthy V, Krystek SR, Jr, Bush A, Wei A, Emanuel SL, Das Gupta R, JanjuaA, Cheng L, Murdock M, and Abramczyk B, et al. (2012) Structures of adnectin/protein complexes reveal an expanded binding footprint. Structure 20:259–269.
Rudmann DG, Alston JT, Hanson JC, and Heidel S (2013) High molecular weightpolyethylene glycol cellular distribution and PEG-associated cytoplasmic vacuo-lation is molecular weight dependent and does not require conjugation to proteins.Toxicol Pathol 41:970–983.
Seidah NG, Mayer G, Zaid A, Rousselet E, Nassoury N, Poirier S, Essalmani R,and Prat A (2008) The activation and physiological functions of the proproteinconvertases. Int J Biochem Cell Biol 40:1111–1125.
Seidah NG and Prat A (2012) The biology and therapeutic targeting of the proproteinconvertases. Nat Rev Drug Discov 11:367–383.
Stein EA, Mellis S, Yancopoulos GD, Stahl N, Logan D, Smith WB, Lisbon E,Gutierrez M, Webb C, and Wu R, et al. (2012) Effect of a monoclonal antibody toPCSK9 on LDL cholesterol. N Engl J Med 366:1108–1118.
Stein EA and Raal FJ (2013) Insights into PCSK9, low-density lipoprotein receptor,and low-density lipoprotein cholesterol metabolism: of mice and man. Circulation127:2372–2374.
Steinberg D and Witztum JL (2009) Inhibition of PCSK9: a powerful weapon forachieving ideal LDL cholesterol levels. Proc Natl Acad Sci USA 106:9546–9547.
Tavori H, Fan D, Blakemore JL, Yancey PG, Ding L, Linton MF, and Fazio S (2013)Serum proprotein convertase subtilisin/kexin type 9 and cell surface low-densitylipoprotein receptor: evidence for a reciprocal regulation. Circulation 127:2403–2413.
Wojcik J, Hantschel O, Grebien F, Kaupe I, Bennett KL, Barkinge J, Jones RB, KoideA, Superti-Furga G, and Koide S (2010) A potent and highly specific FN3 monobodyinhibitor of the Abl SH2 domain. Nat Struct Mol Biol 17:519–527.
Zhang DW, Lagace TA, Garuti R, Zhao Z, McDonald M, Horton JD, Cohen JC,and Hobbs HH (2007) Binding of proprotein convertase subtilisin/kexin type 9 toepidermal growth factor-like repeat A of low density lipoprotein receptor decreasesreceptor recycling and increases degradation. J Biol Chem 282:18602–18612.
Zhang Y, Eigenbrot C, Zhou L, Shia S, Li W, Quan C, Tom J, Moran P, Di Lello P,and Skelton NJ, et al. (2014) Identification of a small peptide that inhibits PCSK9protein binding to the low density lipoprotein receptor. J Biol Chem 289:942–955.
Address correspondence to: Dr. Rex A. Parker, Cardiovascular DiscoveryBiology, Bristol-Myers Squibb Research & Development, Princeton, NJ 08543-4000. E-mail: [email protected]; or Dr. Tracy Mitchell, MolecularDiscovery Technologies, Bristol-Myers Squibb Research & Development,Princeton, NJ 08543-4000. E-mail: [email protected]
424 Mitchell et al.
at ASPE
T Journals on June 21, 2016
jpet.aspetjournals.orgD
ownloaded from





























