Embed Size (px)
Citation preview

1
Manganese binds to Clostrdium difficile Fbp68 and is essential for fibronectin binding Yi-Pin Lin1, Chih-Jung Kuo1, Xhelil Koleci1, Sean P. McDonough2, and Yung-Fu Chang1*
1Department of Population Medicine and Diagnostic Sciences, 2Department of
Biomedical Sciences, College of Veterinary Medicine, Cornell University, Ithaca, New York Running Title: Manganese binding to Fbp68 *Corresponding author: Department of Population Medicine and Diagnostic Sciences, College of Veterinary Medicine, Cornell University, Ithaca, NY 14853. Phone: (607) 253-2675. Fax: (607) 253-3943. E-mail: [email protected]
Clostridium difficile is an etiological agent of pseudomembranous colitis and antibiotic-associated diarrhea. Adhesion is the crucial first step in bacterial infection. Thus, in addition to toxins, the importance of colonization factors in C. diffcile-associated disease is recognized. In this study, we identified Fbp68, one of the colonization factors that bind to Fn, as a manganese binding protein (KD = 52.70 ±1.97nM). Further, the conformation of Fbp68 changed dramatically upon manganese binding. Manganese binding can also stabilize the structure of Fbp68 as evidenced by the increased Tm measured by thermo-denatured circular dichroism and differential scanning calorimetry (CD, Tm = 58oC to 65oC; DSC, Tm = 59oC to 66oC). In addition, enhanced tolerance to protease K also suggests greatly improved stability of Fbp68 through manganese binding. Fn binding activity was found to be dependent on manganese due to the lack of binding by manganese free Fbp68 to Fn. The C-terminal 194 amino acid residues of Fbp68 (Fbp68C) were discovered to bind to the N-terminal domain (NTD) of Fn (Fbp68C-NTD, KD = 233 ±10 nM, obtained from ITC). Moreover, adhesion of C. difficile to Caco-2 cells can be partially blocked if cells are
pretreated with Fbp68C, and the binding of Fbp68C on Fn siRNA transfected cells was significantly reduced. These results raise the possibility that Fbp68 plays a key role in C. difficile adherence on host cells to initiate infection. Introduction
Clostridium difficile is a gram-positive, spore-forming anaerobic bacterium that infects people and multiple animal species. Colonization of the gastrointestinal tract may be asymptomatic or cause a variety of disorders including mild diarrhea, pseudomembranous colitis and antibiotic-associated diarrhea (1). Recent outbreaks in North America and Europe document the seriousness of C. difficile infection (2). C. difficile toxins, including toxin A, toxin B and a recently identified toxin, binary ADP-ribosyltransferase toxin C. difficile transferase (CDT), are thought to be the primary virulence factors that mediate C. difficile-associated disease (3). Since C. difficile colonizes gastrointestinal tissues and enterocyte-like Caco-2 cells (4,5), colonization factors are also recognized as important virulence factors of C. difficile. A variety of colonization factors have been identified including the capsule (6), proteolytic enzymes (7,8), S-layer proteins P36 and P47 (9), adhesins such as SlpA,
http://www.jbc.org/cgi/doi/10.1074/jbc.M110.184523The latest version is at JBC Papers in Press. Published on November 9, 2010 as Manuscript M110.184523
Copyright 2010 by The American Society for Biochemistry and Molecular Biology, Inc.
by guest on June 26, 2016http://w
ww
.jbc.org/D
ownloaded from

2
Cwp66, Fbp68, 12kDa protein, 27kDa protein (4,9-14), flagellins such as FliC and FliD (15,16), and GroEL chaperones (17,18).
Adhesion is the first step in bacterial infection, and several adhesins known as Microbial Surface Components Recognizing Adhesive Matrix Molecules (MSCRAMMs) contribute to this step (19). MSCRAMMs located on bacterial surfaces mediate adhesion by binding to various extracellular matrices including fibronectin (Fn), laminin, collagen, elastin, and proteoglycan on host surfaces (19). Loss of some of these genes may attenuate virulence, indicating that MSCRAMMs are pivotal mediators of bacterial disease (20). Although a number of MSCRAMMs have been investigated in other bacterial pathogens, only a few clostridial adhesins, such as Fbp68, have been characterized, and the colonization mechanisms of C. difficile are still poorly understood (11).
Manganese binding proteins are important players in bacterial physiology by participating in cation homeostasis (21), carbon metabolism to promote nutrient acquisition (22), signal transduction (23), resistance to oxidative stress (24) and nutrient deprived stress (25). In addition, the interaction of some bacterial adhesins and ECM components can be modulated by metal ions such as calcium (26,27). To date, the ability of manganese to mediate bacterial adhesion has not been reported.
Previously, Fbp68 on the surface of C. difficile was shown to serve as an adhesin by binding to Fn, fibrinogen, and vitronectin (11). Interestingly, antibody to Fbp68 can be detected in sera from patients with C. difficile-associated disease, indicating that Fbp68 can induce a host immune response during C. difficile infection (28). Structural
analysis of Fbp68 indicates that it contains eight degenerated repeated sequences and a highly probable α helical region in amino acids 305 to 340 (11). In this study, we show that Fbp68 is a manganese binding protein, that manganese enhances the structural stability of Fbp68 and that manganese is required for Fbp68 binding to Fn. Furthermore, we localized the Fbp68 binding site on Fn to the N-terminal domain (NTD), whereas the fibronectin binding site on Fbp68 resides in the C-terminal 194 amino acids (Fbp68C). Finally, Fbp68C-NTD interaction was able to mediate the adhesion of C. difficile to Caco-2 cells, indicating that Fbp68 is an important colonization factor contributing to clostridial virulence. Materials and Methods Bacterial strains and cell culture- C. difficile 630 was used in this study (29). C. difficile was cultivated in prereduced anaerobically sterilized peptone yeast extract broth with glucose (Anaerobe Systems, Morgan Hill, CA). Escherichia coli strains were cultured in Luria-Bertani broth (LB) with appropriate antibiotics (Table 1). Caco2 cells were cultured in Dulbecco minimum essential medium (DMEM) containing 10% fetal bovine serum (GIBCO Laboratories, Grand Island, NY) and were grown at 37oC in a humidified atmosphere with 5% CO2 (30). Gene knock-out and characterization of the mutants- The fbp68 was an insertion knock-out using the ClosTron gene knockout system developed by Heap, et al. (31-33). The intron target sites within fbp68 recognized by L1.LtrB-derived introns were identified by using intron target tool, one of
by guest on June 26, 2016http://w
ww
.jbc.org/D
ownloaded from

3
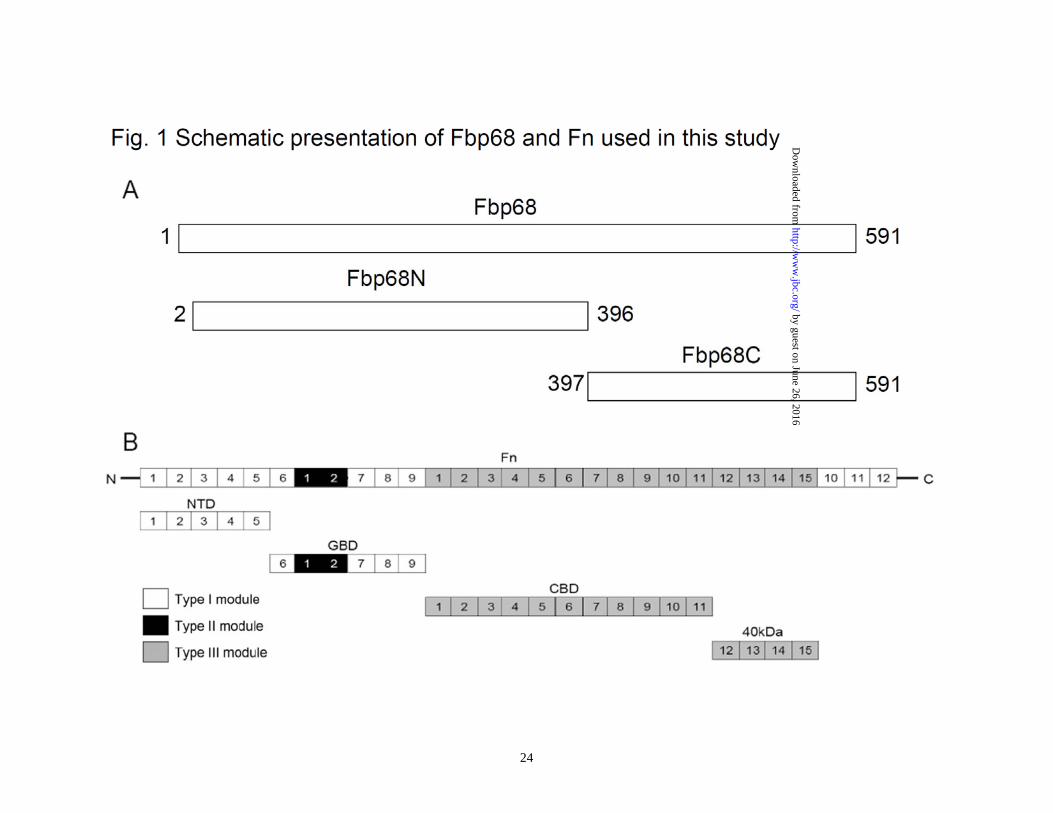
which, 102 bp from the start codon, was used to generate a mutant designated CD∆Fbp68102 (Table 1). The intron targeting region designated by the intron design tool was constructed synthetically by DNA 2.0 Inc. (Mento Park, CA). The synthetic construct was inserting into the ClosTron plasmid pMTL007C-E2 and the resulting plasmid pMTL007C-E2-CDI-Fbp68-102S (Table 1) was electroporated into the conjugative donor E. coli CA434 and then transferred via conjugation into C. difficile 630. Successful transconjugates were selected from a BHI plate supplemented with 250 µg/ml cycloserine (Sigma, St. Louis, Mo) and 8µg/ml of cefotaxine (Sigma, St. Louis, Mo) to select against the E. coli conjugal donor and 15 µg/ml thiamphenicol (Sigma, St. Louis, Mo) to select for the pMTL007C-E2-CDI-Fbp68-102S retargeted C. difficile. Subsequent integrants were selected on BHI agar supplemented with lincomycin (20 µg/ml)(Sigma, St. Louis, Mo). PCR using primers that flanked fbp68 and EBS universal primer was performed to demonstrate the integration of L1.LtrB-derived introns (Table 2). PCR using Thio-F1 and ErmB-R1 primers (Table 2) confirmed the Lincor phenotype was caused by the splicing of the group I intron from the group II intron following integration and not a spontaneous Lincor (32). Two primers, FliD-F and FliD-R, were used to demonstrate that the mutant is a C. difficile strain (Table 2). The PCR product of fbp68 using primers Fbp68F and Fbp68R (Table 2) was inserted into plasmid pMTL84151(Table 1), electroporated into the conjugative donor E. coli CA434 and then transferred via conjugation into CD∆Fbp68102 for complementary testing as previously described (33).
Reagents and antibodies– Fibronectin (human plasma fibronectin), NTD or GBD of Fn, Ethylenedaminetetraacetic acid (EDTA), sodium chloride, sodium phosphate monobasic, sodium phosphate dibasic, Tris, magnesium chloride, manganese chloride, zinc chloride, calcium chloride, protease K, and protein A-10nm gold particle colloidal suspension were purchased from Sigma-Aldrich (St. Louis, Mo). 120 kDa cell binding domain (CBD) or 40 kDa domain (40 kDa) of Fn, mouse anti-fibronectin, and mouse anti-α-actin were purchased from Chemicon International (Temecula, CA). Fbp68 antibody was prepared by vaccination of rabbits. Briefly, intramuscular inoculation with 100 µg of recombinant Fbp68 in Freund’s complete adjuvant (first vaccination) was followed by 100 µg intramuscularly in Freund’s incomplete adjuvant (booster) three weeks later. Three weeks after the second booster, the rabbit was sacrificed and serum was collected. Anti-C. difficile antibodies were previously prepared by vaccination of horses with whole killed cells of C. difficile. Mouse anti-histidine tag, Rabbit anti-GST antibody, Texas Red conjugated goat anti-rabbit IgG, FITC-conjugated goat anti-horse antibody, FITC-conjugated goat anti-mouse antibody, HRP-conjugated goat anti-mouse antibody, and HRP-conjugated goat anti-rabbit antibody were ordered from Invitrogen (Eugene, OR). HRP conjugated goat anti-horse antibody was purchased from KPL (Gaithersburg, MA). Plasmid construction and Protein purification– The construct for the expression of histidine-tag fused with Fbp68, GST and histidine-tag fused with Fbp68N
by guest on June 26, 2016http://w
ww
.jbc.org/D
ownloaded from

4
(amino acids 2-396) or Fbp68C (amino acids 397-591) was generated using vectors pQE30 (Qiagen, Alencia, CA) and pGEX4T2 (GE Healthcare) (Fig. 1A). To perform the PCR reactions, the primers shown in Table 2 were utilized based on the Fbp68 sequence (11). Primers were engineered to introduce a BamHI site at the 5’ end of each fragment and a stop codon followed by a SalI site at the 3’ end of each fragment. PCR products were sequentially digested with BamHI and SalI and then ligated into pQE30 or pGEX4T2 cut with BamHI and SalI, respectively. The soluble forms of all of recombinant proteins were purified from E. coli as previously described (34,35). Isothermal Titration Calorimetry (ITC)– The experiments were carried out with a CSC 5300 microcalorimeter (Calorimetry Science Corp. Lindon, UT, USA) at 25°C as previously described (26). Before the ITC experiment, traces of metal ions were removed from Fbp68 solution through incubation with EDTA and subsequent dialysis in Tris buffer (25mM Tris, 150mM sodium chloride at pH 7.0). The cell contained 1 ml of a solution of Fbp68 and the syringe contained 250 μL of a solution of various metal ions at different concentrations, as indicated in Table 3. For the NTD binding experiments, 1mL of NTD (35 µM) was in the cell and 250 μL of Fbp68C (325 µM) with 100μM of MnCl2 were in the syringe. All solutions were in Tris buffer (pH 7.0). The concentration of each species is presented in Table 3. The titration was performed as follows: 25 injections of 10 μl with a stirring speed of 250 rpm with a delay time between injections of 5 minutes. Data were analyzed
using BindWorks software (Model CSC 5300, Calorimetry Science Corp. Lindon, UT, USA) fitting them to an independent binding model. Fluorescence Spectrometry–Fbp68 was treated with 50μM of EDTA and then extensively dialyzed in Tris buffer at pH 7.0 to remove metal ions. Fluorescence emission spectra were measured on a Hitachi F7500 spectrofluorometer (Hitachi. San Jose, CA). All spectra were recorded in correct spectrum mode of the instrument using excitation and emission band passes of 5 nm. The intrinsic Trp fluorescence of protein was recorded by exciting the solution at 295 nm and measuring the emission in the region from 305-400 nm. For manganese titration, 0, 25, 50, 100, 200, and 400 nM of manganese chloride were mixed with 1 μM of Fbp68 and spectra were recorded after 3 minutes. For the ANS fluorescence experiment, ANS binding was checked by adding 100 μM ANS solution (10 mM stock in 100% methanol) to a protein solution (1 µM), incubated for two minutes and spectra were recorded between 400 and 600 nm. Next, Fbp68 was treated with different concentrations of MnCl2 (0, 25, 50, 100, 200, 400 nM) for 5 minutes and then spectra were measured at an excitation wavelength of 295 nm. All spectra were recorded in the correct spectrum mode with excitation and emission band passes of 5 nm each and corrected for volume changes before further analysis. All measurements were performed at 25ºC. Circular dichroism (CD) spectrometry- CD spectra were recorded on a Jasco J-815 spectropolarimeter under N2 atmosphere at room temperature (25°C) in a 0.02 and 0.5
by guest on June 26, 2016http://w
ww
.jbc.org/D
ownloaded from

5
cm path length quartz cell for far- and near-UV respectively with 8 accumulations. The Fbp68 was treated with 50μM of EDTA and then extensively dialyzed in Tris buffer at pH 7.0 to remove metal ions. Aliquots of manganese chloride solution (0, 25, 50, 100, 200, 400 nM) were added to 1μM Fbp68 protein solution and incubated for 5 minutes. All spectra were recorded in Tris buffer, pH 7.0. In a melting temperature experiment, 10 μM of Fbp68 in the absence or presence of 100 μM MnCl2 was subjected to thermal unfolding, and data were collected at 1°C/minute increments from 25°C to 100°C recording the ellipticity at 205 nm, with 30 second temperature equilibrations, followed by 30 second data averaging. In order to measure the melting point, a first order derivative was applied to the results from the melting experiment. In all CD experiments, the background spectrum of Tris buffer (pH 7.0) alone was subtracted from the protein containing spectra. Differential Scanning Calorimetry (DSC)- Excess heat capacity Cp(T) of Fbp68C with or without MnCl2 was measured using a DSC Q1000 microcalorimeter (Waters, New Castle, DE). The Fbp68 was treated with 50μM of EDTA and then extensively dialyzed in Tris buffer at pH 7.0 to remove metal ions. Degassed sample containing 30 µM of Fbp68 with or without 100 μM of MnCl2 in Tris buffer were heated at 0.1 K/min scan rate. Heat Capacity, Cp(T) data were recorded, corrected for buffer baseline, and normalized to the amount of the sample. The TA Universal Analysis software (Waters, New Castle, DE) was used for the data analysis and display. All calorimetric experiments in this study were repeated 3 times to ensure reproducibility.
Protease K resistance experiment- The recombinant histidine tagged Fbp68 used in this study was treated with 50μM of EDTA, and subsequently dialyzed in Tris buffer at pH 7.0 to remove trace metal ions. One μM of Fbp68 was mixed with or without 100μM of MnCl2 then dialyzed in Tris buffer to remove the unbound MnCl2 in order to prevent unbound MnCl2 from interfering with the activity of protease K. Then, Fbp68 samples with and without MnCl2 were analyzed for the sensitivity of protease K by treating with 0-60 ng/μL Protease K (PK) at 37oC for 1hour. The reaction was stopped by adding 1μL of protease inhibitor without EDTA (Thermoscientific, Logan, UT), and then mixing with Laemmli sample loading buffer consisting of 50 mM Tris-HCl (pH6.8), 100 mM dithiothreitol, 2% sodium dodecyl sulfate, 0.25 mM PMSF, and 0.1% bromophenol blue in 20% glycerol. The digested Fbp68 was subjected to 10% SDS-PAGE and electroblotted onto polyvinylidene difluoride membranes. The membranes were incubated in 5% skim milk in PBS/T overnight and then incubated with mouse anti-histdine tag antibody (1:1,000x). The immunocomplexes were detected with an HRP-conjugated goat anti-mouse IgG antibody (1:5,000x). Binding assays by ELISA- To determine the binding of untreated or manganese treated Fbp68 to Fn (Fig. 4A), 100μL of 1μM Fn or BSA (negative control and data not shown) were coated on microtiter plate wells and blocked subsequently as previously described (36). Various concentrations of 100μM MnCl2 treated histidine tagged LigBCen2 (a known Fn binding protein from Leptospira interrogans to serve as
by guest on June 26, 2016http://w
ww
.jbc.org/D
ownloaded from

6
positive control) (36), untreated or 100 μM MnCl2 treated histidine tagged Fbp68, or LigBCon4 (a protein from L. interrogans lacking Fn binding activity to serve as negative reference) (37) in 100 μl Tris buffer (pH 7.0) were added into microtiter plate wells and incubated for 1 hour at 37°C. To map the binding sites of Fn on Fbp68 (Fig. 5A), different concentrations of 100 μM MnCl2 treated GST fused Fbp68 (positive control), LigBCon (a protein from L. interrogans lacking Fn binding activity to serve as negative control) (37), Fbp68N, or Fbp68C in 100 μl Tris buffer (pH 7.0) were added into microtiter plate wells and incubated for 1 hour at 37°C. To localize the Fbp68C binding sites on Fn (Fig. 5B), 100μL of 1μM Fn (positive control), NTD, GBD, CBD, 40kDa, or BSA (negative control) were coated on microtiter plate wells and blocked subsequently as previously described above. Serial concentrations of 100μM of MnCl2 treated GST fused Fbp68C or LigBCon (negative reference and data not shown) in 100 μl Tris buffer (pH 7.0) were added into microtiter plate wells and incubated for 1 hour at 37°C. To detect the binding of Fbp68C to Caco-2 cells (Fig. 7A), 100μL of 10μM GST fused Fbp68C or GST (negative control) was added to microtiter plate wells cultured with 105 Caco-2 cells for 1 hour. To determine the inhibitory effect on C. difficile adhesion caused by pretreatment of Fbp68C (Fig. 7B), 105 Caco-2 cells were treated with 100μL of 10μM GST fused Fbp68C or GST (negative reference) at 37℃ for 1 hour prior to incubation with 107 C. difficile at 37℃ for 1 hour. To detect the binding of histidine tagged Fbp68, LigBCen2, LigBCon, mouse anti-histidine tagged (1:200x) and HRP-
conjugated goat anti-mouse IgG (1:1,000x) were used as primary and secondary antibodies, respectively (34,35). To measure the binding of GST fused LigBCon, Fbp68, Fbp68N, Fbp68C, rabbit anti-GST (1:200x) and HRP-conjugated goat anti-rabbit IgG (1:1,000x) were used as primary and secondary antibodies, respectively. To determine the adhesion of C. difficile, horse anti-GST (1:200x) and HRP-conjugated goat anti-horse IgG (1:1,000x) were used as primary and secondary antibodies, respectively. After washing the plates thrice with TBST (0.05% Tween-20 in Tris buffer), 100 μl of TMB (KPL, Gaithersburg, MD) was added to each well and incubated for 5 min. The reaction was stopped by adding 100 μl of 0.5% hydrofluoric acid to each well. Each plate was read at 630 nm using an ELISA plate reader (Bioteck EL-312, Winoski, VT). Each value represents the mean ± SEM of three trials in triplicate samples. Statistically significant (P<0.05) differences are indicated by *. Bacterial attachment assay – To study the adhesion of C. difficile 630 (wild type), CD∆Fbp68102(mutant) and CD∆Fbp68102/pMTL84151-fbp68 (complemented mutant) to Caco-2 cells (Fig. 7E), an attachment assay was slightly modified from the method reported previously (38). Basically, ice-cold PBS buffer was used to wash the cells prior to the assay to prevent C. difficile invasion. A total of 108 CD630WT, CD∆Fbp68102, or CD∆Fbp68102/pMTL84151-fbp68 bacteria suspended in ice-cold medium were then incubated with 106 Caco-2 cells at 4oC for 1 hour. The wells incubated without Caco-2 cells served as negative controls. Unattached bacteria were removed by five washes with
by guest on June 26, 2016http://w
ww
.jbc.org/D
ownloaded from

7
PBS. PBS containing 1% (v/v) Triton X-100 was used to resuspend adherent bacteria, and the 104-fold dilution of the adherent bacterial suspensions were spread on C. difficile agar plates (CSDA, BD, Sparks, MD) to determine the number of cell-associated bacteria per well. Surface Plasmon Resonance (SPR) – The Fbp68-Fn or Fbp68C-NTD interaction was analyzed by a SPR technique using a Biacore 2000 instrument (GE Healthcare, Piscataway, NJ). Briefly, Fbp68 and Fbp68C were treated with 50μM of EDTA and subsequently dialyzed in Tris buffer at pH 7.0 to remove trace metal ions. To determine the Fn binding activity of Fbp68 in the presence or absence of manganese (Fig. 4B), 100μM of manganese chloride in Tris buffer (pH 7.0) was mixed with 1.5μM of histidine tagged Fbp68 when immobilized on an NTA chip (GE Healthcare). Then, 10μL of 1,000nM Fn was injected into the flow cell at 10μL/min and 25oC. To measure the binding affinity of Fbp68C and Fn (Fig. 5E), 1.5μM of histidine tagged Fbp68C was incubated with Tris buffer with or without 100μM of manganese chloride when immobilized on an NTA chip, and the NTA chip was conjugated to 500μM of nickel sulfate prior to the immobilization of Fbp68 or Fbp68C. A control flow cell was injected with Tris buffer without Fbp68 or Fbp68C. Then, 10μL of a serial concentration (0, 62.5, 125, 250, 500, 1,000nM) of NTD was injected into the flow cell at 10μL/min and 25℃. All sensogram data were subtracted from the negative control flow cell. To obtain the kinetic parameters of the interaction, the data of the sensograms were fitted by BIAevaluation software version 3.0 using the one step biomolecular association
reaction model (1:1 Langmuir model), which resulted in optimum mathematical fits with the lowest Chi values. Binding assays by confocal laser-scanning microscopy (CLSM)- To determine the binding inhibition of C. difficile to Caco-2 cells by Fbp68C by CLSM, 106 Caco-2 cells were preincubated with 50 μM of GST-Fbp68C or GST (negative control) in 100 μL of PBS for 1 h at 37 oC. Then, 108 C. difficile 630 were added to each well and incubated for 1 h at 37 oC (Fig. 7C). To measure the adhesion of C. difficile to Fn (Fig. 7D), 108 wild type C. difficile (CD630WT), fbp68 knockout mutant (CD∆Fbp68102), or fbp68 knockout mutant complemented with intact fbp68 (CD∆Fbp68102/pMTL84151-fbp68) were added to glass slides in 24-well plates coated with 1μM of Fn. BSA coated slides served as negative controls. To determine the attachment of C.difficile and the binding of GST-Fbp68C or GST to Caco-2 cells, (Fig. 7C and D) rabbit anti-GST (1:250x) and horse anti-C. difficile antibodies (1:100x) served as primary antibodies, and Texas Red conjugated goat anti-rabbit IgG (1:250x) and FITC-conjugated goat anti-horse IgG (1:250x) were used as secondary antibodies. Fixation and immunofluorescence staining were performed as previously described (36) with slight modifications. Briefly, C. difficile and Caco-2 cells were fixed in 2% paraformaldehyde for 60 min at RT. For antibody labeling, fixed bacteria were incubated in PBS containing 0.3% BSA for 10 min at RT. The primary and secondary antibodies in PBS containing 0.3% BSA were incubated sequentially for 60min at RT. After incubation with primary and
by guest on June 26, 2016http://w
ww
.jbc.org/D
ownloaded from

8
secondary antibodies, the glass slides were mounted with coverslips using Prolong Antifade (Molecular Probe, Eugene, OR) and viewed with a 60x objective by CLSM (Olympus, America, Inc., Melville, NY). An Olympus Fluoview 500 confocal laser-scanning imaging system equipped with krypton, argon, and He-Ne lasers on an Olympus IX70 inverted microscope with a PLAPO 60X objective was used. The settings were identical for all captured images. Images were processed using Adobe Photoshop CS2. For counting the attachment of C. difficile to Fn coated wells, three fields (40X objective) were selected at random to count the number of bound organisms. All studies were repeated three times and the attachment of C. difficile to Caco-2 cells was scored by an operator who was blinded to the treatment group.
Small interfering RNA (siRNA) inhibition of Fbp68C binding- siRNA duplexes directed against human Fn (Ambion ID AM121357) and negative siRNA duplex (Ambion ID AM4611) were purchased from Ambion (Foster City, CA). RNA duplexes were introduced into Caco-2 cells by the method of lipofection (37), and 8 × 105 cells were transfected with 0.4 μg negative siRNA and Fn-siRNA. Adhesion assays were performed 72 h after lipofection (37). The knock down efficiency of endogeneous Fn expression was determined as previously described (37) with slight modification. The total protein content of 106 Caco-2 cells was analyzed using Western immunoblotting as described under ‘Protease K resistance experiments’. The protein bands of actin derived from Caco-2 cells were measured as a control using a mouse anti-actin antibody (1:5,000x). The band intensity was measured by
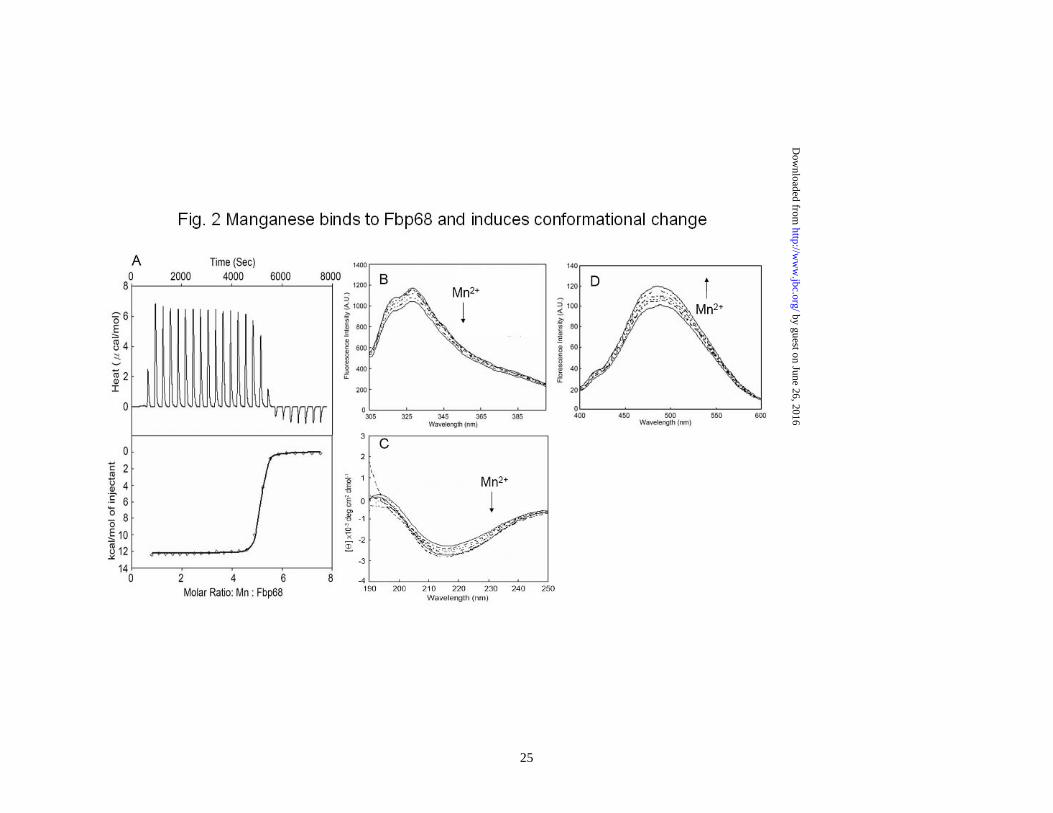
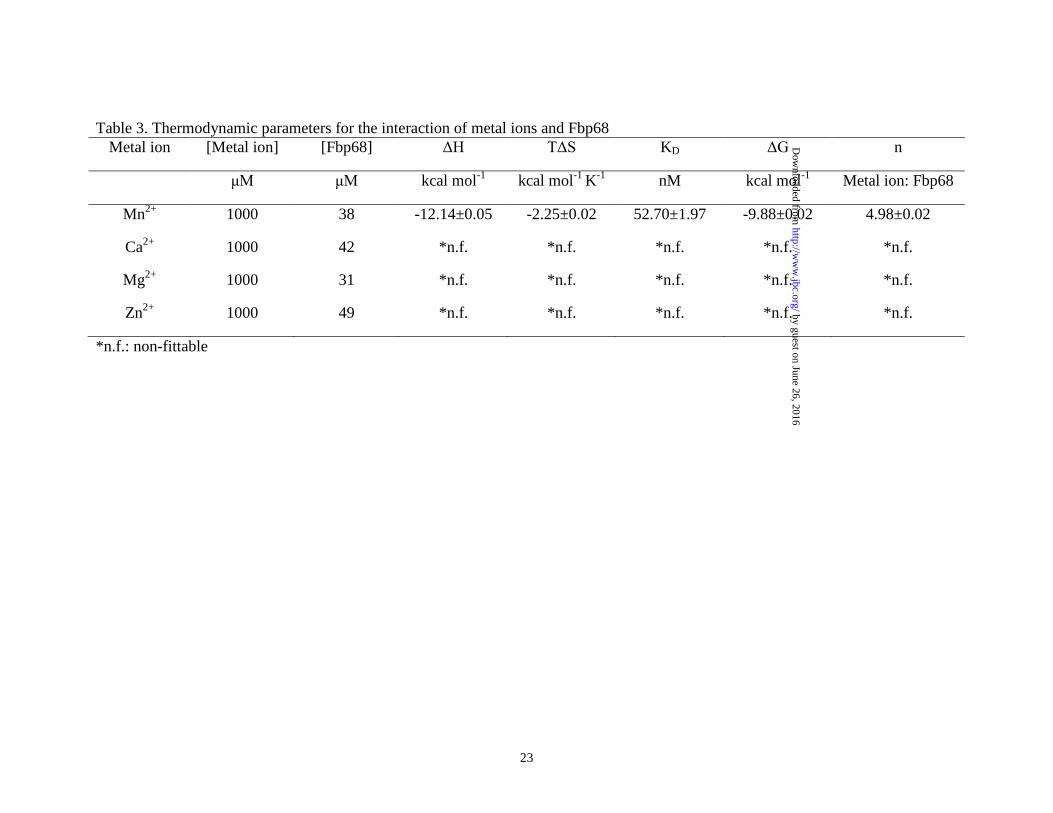
densitometry using Image J software (National Institutes of Health, Bethesda, MD, USA) (39). Fbp68C binding assay was performed 72 h after lipofection. To determine the binding of Fbp68C to Fn, 100μL of 50μM GST-Fbp68C or GST was added to 106 Caco-2 cells transfected with Fn or negative siRNA. To determine the binding of Fbp68C and the expression of Fn on Caco-2 cells, rabbit anti-GST (1:250x) and mouse anti-Fn (1:250x) served as the primary antibodies, and FITC-conjugated goat anti-mouse IgG (1:250x) and Texas Red-conjugated goat anti-rabbit IgG (1:250x) were used as secondary antibodies. Fixation, immunofluorescence staining, image detection and processing were as described in the previous sections. All the experiments were performed in triplicate. Statistical analysis- Each data point represents the mean ± standard error of the mean (SEM) for each sample tested in triplicate. Data were analyzed by Student’s t test and statistically significant differences were claimed at p values < 0.05. Results Manganese binds to Fbp68 and induces conformational change- Previously, Fbp68 was identified as a Fn binding protein (11). Since metal ions can modulate the ECM binding activities of some ECM binding proteins (26,27,40-42), the binding activity of Fbp68 to Ca2+, Mg2+, Zn2+ and Mn2+ was examined in this study through ITC. As shown in Fig. 1A and Table 3, Fbp68 bound to Mn2+ (KD = 52 ±1.97 nM) but not to Ca2+, Mg2+, or Zn2+, and the stoichiometric values indicated that 5 manganese ions were able to bind to 1 Fbp68 (Table 3). Furthermore, a significant quenching of tryptophan
by guest on June 26, 2016http://w
ww
.jbc.org/D
ownloaded from

9
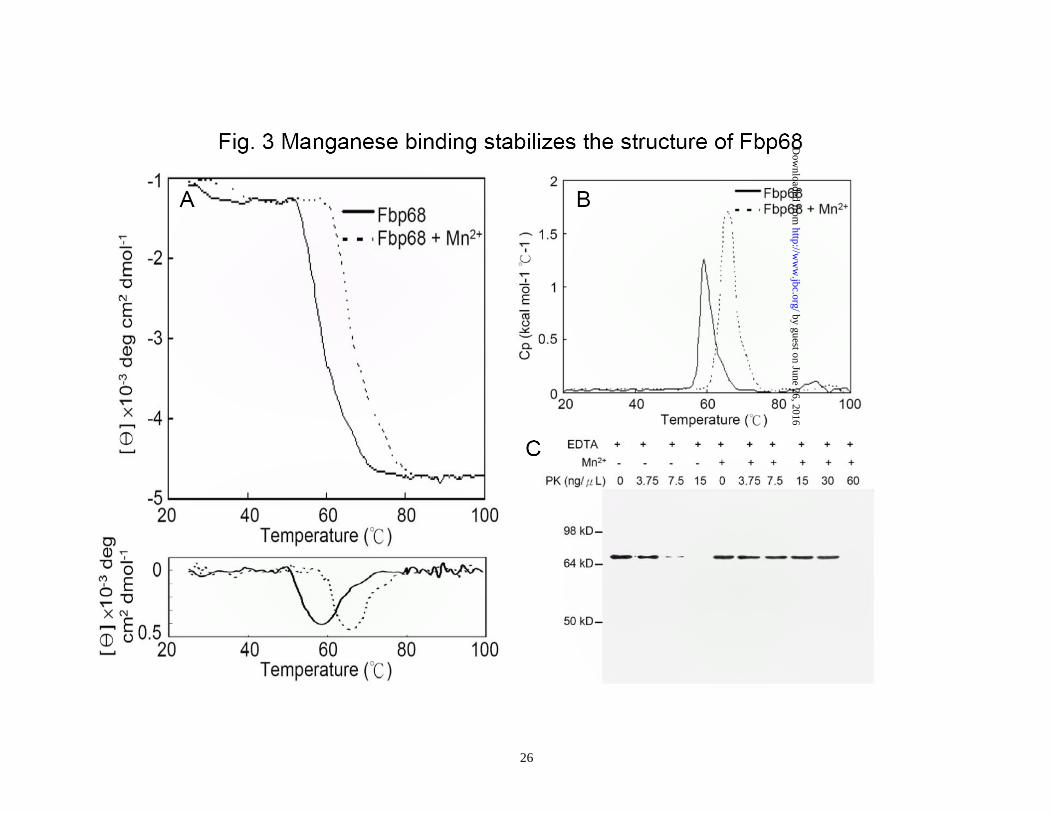
fluorescence spectra for Fbp68 (12%) upon Mn2+ binding indicateed manganese binding could also alter the conformation of Fbp68 (Fig. 2B). Interestingly, the Fbp68 conformational changes upon Mn2+ binding were mainly in the random coiled region since the proportion of α-helix increased while that of random coil was reduced when Fbp68 titrated with Mn2+ was measured through far-UV CD spectrometry (α-helix from 56% to 69%, β-strand remaining 25%, random coil from 19% to 6%.)(Fig. 2C). In addition, the conformational change occured in a hydrophobic area because the fluorescence of ANS-Fbp68 increased dramatically (20%) upon Mn2+ binding (Fig. 2D). Manganese binding enhances the stability of Fbp68-The function of Mn2+ binding is generally recognized as maintaining protein stability (43,44). To gain more insight about the function of manganese binding to Fbp68, DSC thermo-unfolding and thermo-denatured CD was performed. As shown on Fig. 3A, the CD profiles of heat induced folding to unfolding transition in Mn2+ bound Fbp68 shifted to a significantly higher temperature compared to the apo-Fbp68, and the Tm value of Mn2+ bound Fbp68 also dramatically increased (Apo-Fbp68, Tm = 58.0 ±2.1oC; Mn2+ bound Fbp68, Tm = 65.9 ±1.3oC). A similar result was also observed in the DSC experiment (Apo-Fbp68, Tm =59.3 ±2.1oC; Mn2+ bound Fbp68, Tm =66.1 ±3.2oC) (Fig. 3B). It has also been reported that Mn2+ bound proteins can better resist protease digestion compared to apo-proteins. Therefore, protease K resistance assays were applied to Mn2+ bound or apo-Fbp68 to test their stability. As indicated in Fig. 3C, Mn2+ bound Fbp68 was
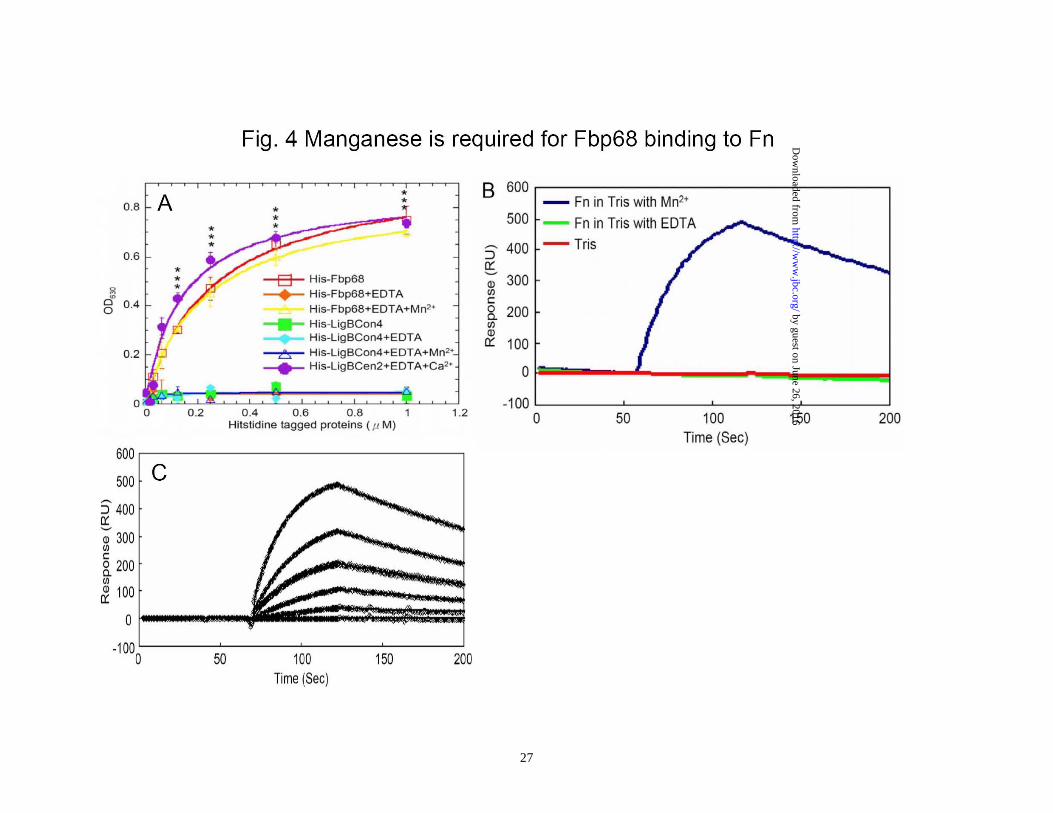
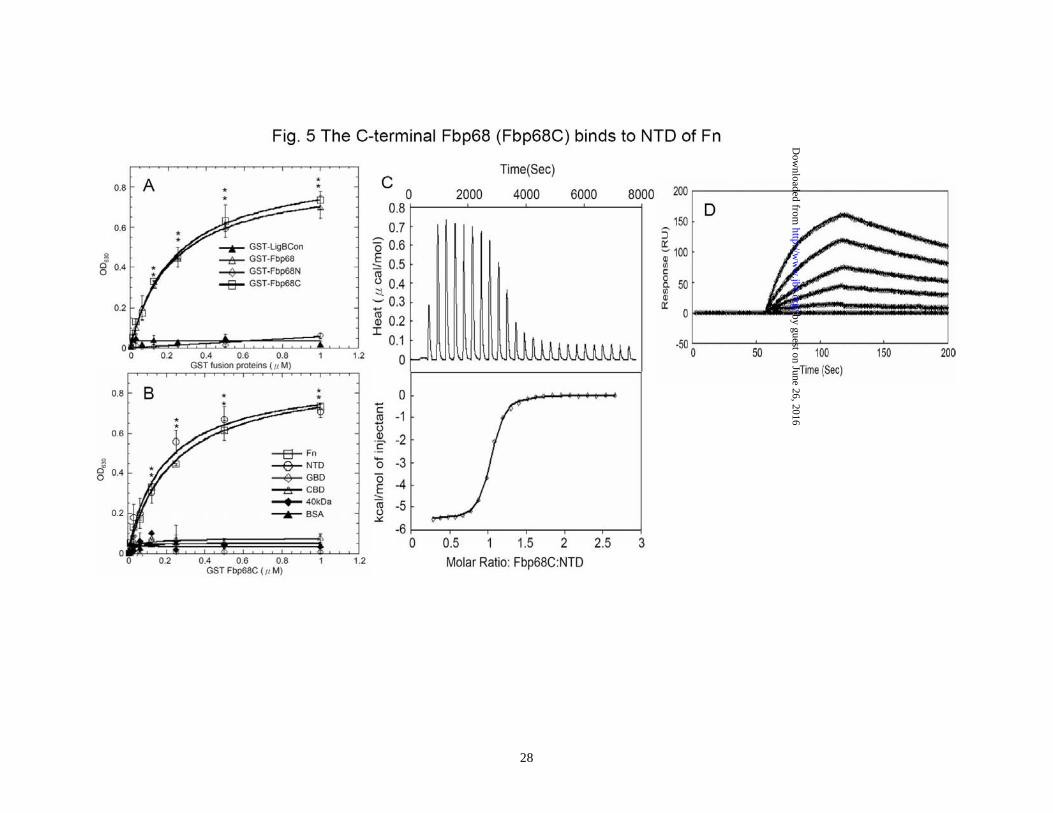
able to tolerate a higher concentration of protease K since the decomposition of Mn2+ bound Fbp68 could not be observed until the addition of 60ng/μL of protease K. However, apo-Fbp68 was vulnerable to protease K digestion at a comparatively lower concentration of protease K (7.5 ng/μL) (Fig. 3C). The interaction of Fbp68 and Fn requires manganese- Fbp68 is a Fn binding protein. In order to elucidate the effect of manganese on Fbp68-Fn interaction, histidine tagged Fbp68 was treated with EDTA and dialyzed in PBS buffer. Then, the binding of Fn to untreated Fbp68 or EDTA treated Fbp68 with or without Mn2+ was measured by ELISA. As shown on Fig. 4A, both untreated Fbp68 and Mn2+ bound Fbp68 could associate with Fn with similar affinities (Fbp68, KD = 253 ±23 nM; Mn2+ bound Fbp68, KD = 221 ±10 nM). Strikingly, EDTA treated Fbp68 completely lost Fn binding activity on removal of divalent cation (Fig. 4A). These results clearly indicate that manganese is essential for Fbp68 binding to Fn. The SPR results (Fig. 4B and 4C) support the same conclusion since Mn2+ bound Fbp68 was bound tightly to Fn while EDTA treated Fbp68 failed to bind to Fn. Mapping the Fbp68 and NTD binding sites-To better define the Fn binding site of Fbp68, Fbp68 was truncated into two fragments, Fbp68N (Residues 2-396) and Fbp68C (Residues 397-591) (Fig. 1A). ELISA was performed to determine the interaction of Fn with Fbp68N or Fbp68C. As presented in Fig. 5A, GST fused Fbp68C was strongly bound to immobilized Fn (KD = 234 ±22 nM) whereas Fbp68N could not bind. In addition,
by guest on June 26, 2016http://w
ww
.jbc.org/D
ownloaded from

10
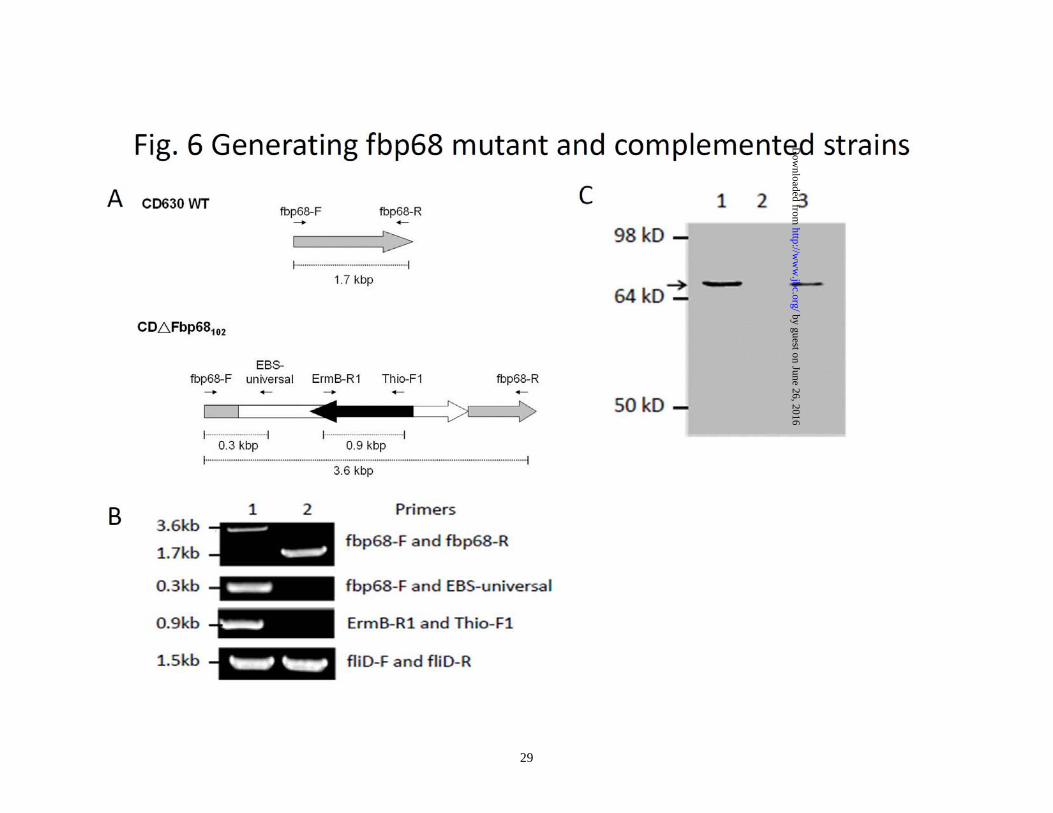
the Fbp68 binding site on Fn was also mapped via ELISA, and only NTD of Fn was able to interact with GST fused Fbp68C (KD = 220±93 nM) (Fig. 1B and 5B). ITC and SPR studies yielded similar affinities (ITC, KD = 233 ±10 nM; SPR, KD = 216 ±56 nM) (Fig. 5. C and D), providing further support to the Fbp68C-NTD interaction determined by ELISA Generating fbp68 mutant and complemented strains- In order to elucidate physiological roles of the Fbp68-Fn interaction, fbp68 mutant, and fbp68 complemented strains were generated. To obtain fbp68 mutant, the lincomycin resistant gene was inserted between 102nd and 103rd base pair (Fig. 6A). The transconjugants of C. difficile 630 with the inserted intron would confer a lincomycin resistant phenotype. In order to select the fbp68 knockout mutant, PCR analyses with different primer pairs was performed. As shown in Fig. 6B, 3.6kb of the DNA fragment could be amplified from the PCR reaction with the primer pairs, Fbp68-F and Fbp68-R, in the selected fbp68 knockout mutant, but only 1.7kb PCR amplificon was obtained with the same primer pairs in wild type C. difficile 630 due to the lack of the inserted intron (Fig. 6B). In addition, the 0.9kb lincomycin resistant gene was amplified by primer pairs, ErmB-R1 and Thio-F1, and 0.3kb of 5’ adjacent fragment of inserted intron amplified by Fbp68-F and EBS-Universal primer was observed in this fbp68 knockout mutant (Fig. 6B). This mutation was complemented in trans with plasmid pMTL84151-fbp68 (Table 1). The immunoblot analysis was also used to demonstrate that Fbp68 was absent in the mutant but restored in the complemented strain (Fig. 6C). This
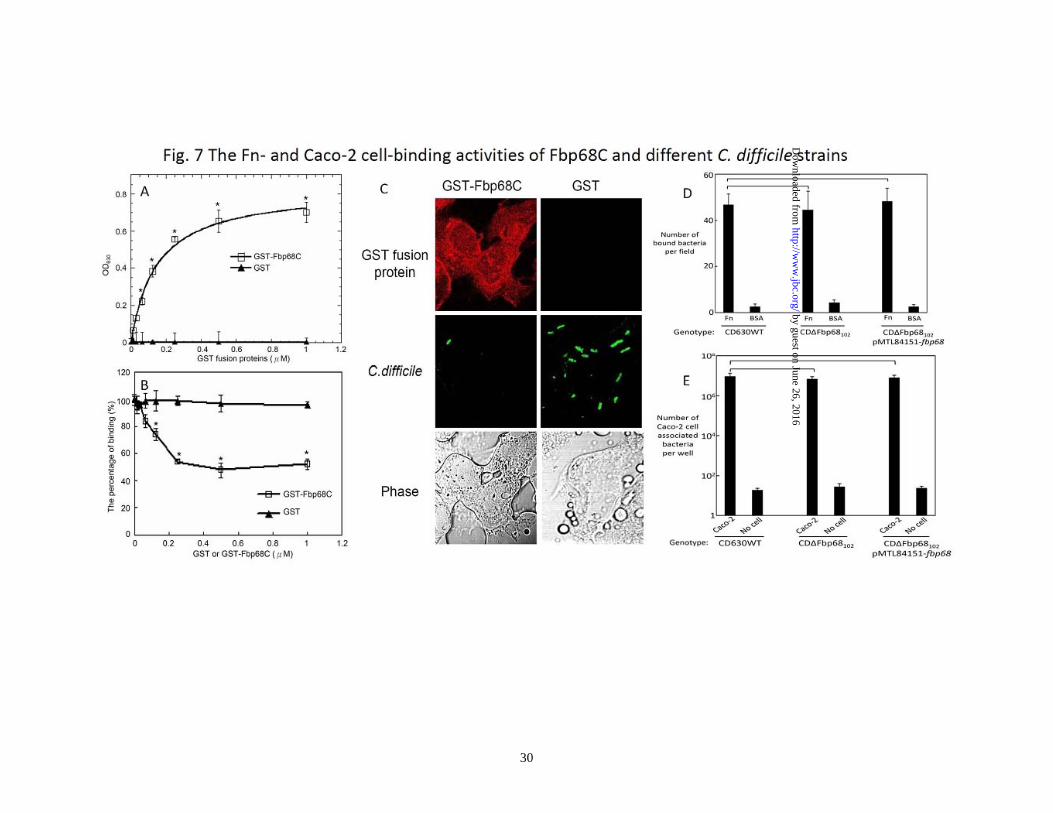
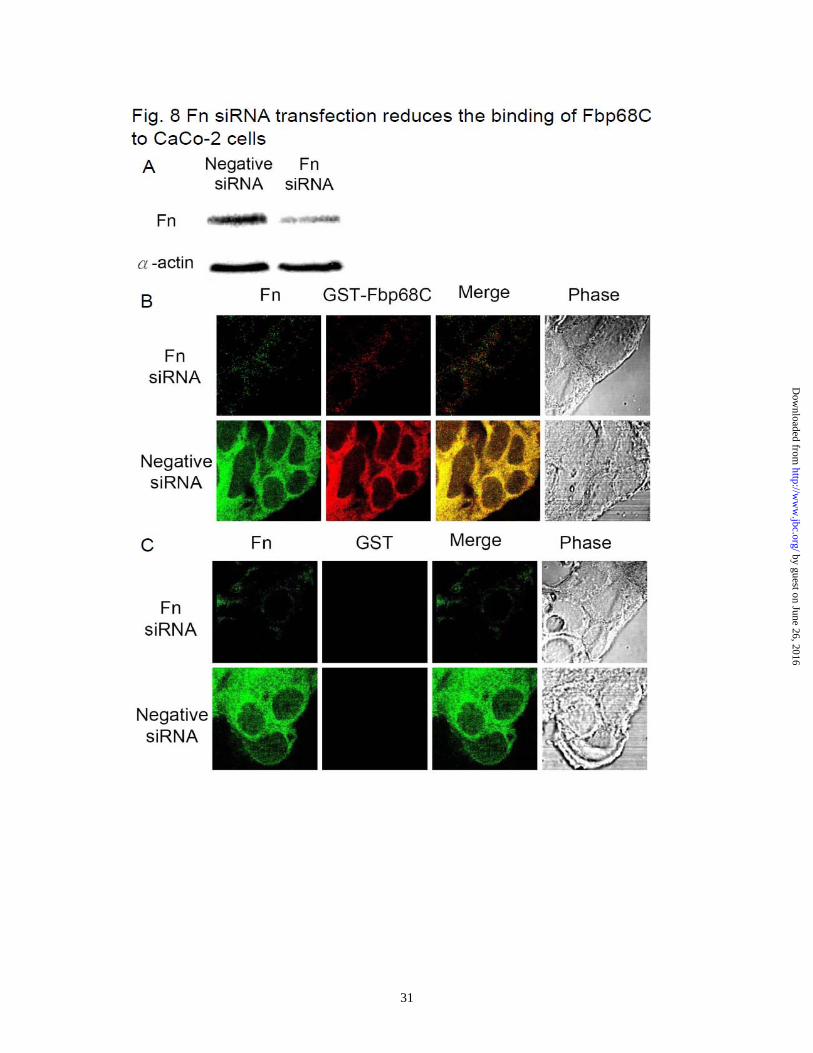
mutation was complemented in trans with plasmid pMTL84151-fbp68 (Table 1). The treatment of Fbp68C and the transfection of Fn siRNA inhibit C. difficile binding on the mammalian cells - Bacterial Fn binding proteins contribute to host cell adhesion (36,37,45). Since Fbp68 is located on the surface of C. difficile (Supplemental Fig. 1, 21), it is reasonable to hypothesize that Fbp68 might be one of the adhesins mediating C. difficile adhesion. To gain more understanding of the physiological relevance of Fbp68-Fn interaction, Caco-2 cells, a human epithelial colorectal cell line, were used to test the binding activity of Fbp68C. Compared to GST (negative control), GST-Fbp68C was able to bind to Caco-2 cells as shown in Fig. 7A. In addition, pretreatment of Caco-2 cells with Fbp68C decreased C. difficile adhesion by 51% (Fig. 7B and C), in agreement with the binding affinity assay results. However, the indistinguishable Fn- and cell-binding activity of wild type C. difficile 630, fbp68 knockout mutant, or fbp68 complemented strains suggest redundant adhesins appearing on C. difficile contribute to adhesion (Fig. 7D and E). To further elucidate the receptor role of Fn on Caco-2 cells for putative binding partners such as Fbp68 of C. difficile, the binding of GST-Fbp68C or C. difficile to Fn or negative siRNA transfected cells was examined. As shown in Fig. 8A, Fn siRNA duplex specifically reduced the expression of Fn. Moreover, the decreased binding of GST-Fbp68C and a 35% reduction in adhesion of C. difficile to Fn siRNA transfected cells (compared to the negative siRNA transfected cells) validated the conclusion that Fn plays a pivotal role in
by guest on June 26, 2016http://w
ww
.jbc.org/D
ownloaded from

11
the adherence of C. difficile (Fig. 8B and data not shown). Discussion
Two high-molecular-weight toxins (toxins A and B) of C. difficile play major roles in the pathogenesis of pseudomembranous colitis and antibiotic-associated diarrhea (1). However, not all C. difficile-associated disease is caused by toxigenic strains of C. difficile (5,8,14,46,47). Thus, other virulence factors, such as adhesins, are likely important in the pathogenesis of C. difficile-associated disease. Adhesion is a crucial first step that allows pathogenic bacteria, including C. difficile, to infect host cells. A group of virulence factors, termed Microbial Surface Components Recognizing Adhesive Matrix Molecules (MSCRAMMs), mediates adhesion of a wide variety of pathogenic bacteria including Staphylococcus, Streptococcus, Enterococcus, Borrelia, Leptospira, and others. (19,26,34,35,37,48,49). Generally, MSCRAMMs are located on the outer surface of bacteria thereby mediating attachment to host cells by interacting with fibrinogen or various ECM components such as Fn, laminin, collagen, elastin, and proteoglycan (19). A number of C. difficile MSCRAMMs that bind to either host cells or various ECM components have been described and include Fbp68 (11), SlpA (4), Cwp66(14), and 27kDa protein (50). These adhesins may be significant factors in the virulence of different C. difficile strains.
Numerous studies indicate that at least some MSCRAMMs, such as LipL32 and Lig proteins of L. interrogans and ClfA of S. aureus, are metalloproteins (26,27,41,51) In this study, we demonstrated that Fbp68 is a
manganese binding protein and also showed that it binds to manganese with high affinity and specificity (KD = 52 ±1.97nM). This is the first study to identify a bacterial manganese binding MSCRAMM. Since a higher concentration of free manganese would be an oxidative stress in the cells (52), the concentration of free manganese in vivo is extremely low (10-7M) (53). In addition, manganese is a trace element and is present at very low levels in the environment (10-8M) (52). In order to acquire environmental manganese, the binding affinity of most manganese binding proteins must be high (e.g. E. coli manganese superoxide dismutase, KD = 3.12nM)(44). Thus, the high affinity of Fbp68 for manganese would help overcome the low concentration of manganese both in vivo and in the environment. Most metalloproteins undergo conformational changes upon metal ion binding. In certain extreme cases, apo-metalloproteins lose their conformation since metal ions stabilize the structure (54). In other cases, the geometry of the metal ion binding sites is dominated by metalloproteins so the structure of apo-proteins is able to be maintained while partial alteration of the conformation is still observed upon metal ion binding (55). Manganese binding changed the global conformation of Fbp68, but the structure of apo-Fbp68 can still be discerned in Far-UV spectra. Thus, our results suggest that Fbp68 dominates the geometry of the manganese binding region.
Manganese usually enhances protein structural stability. For example, when manganese binds to manganese superoxide dismutase (SOD) of E. coli, the protein structure is stabilized as evidenced by the increased Tm of manganese bound proteins
by guest on June 26, 2016http://w
ww
.jbc.org/D
ownloaded from

12
in thermo-unfolding experiments (apo-SOD, Tm = 52.5oC; Mn-SOD, Tm = 68.6oC)(44). Likewise, manganese bound prion protein resists higher concentrations of protease K, which also indicates the protein structure is stabilized by manganese (apo-prion, protease K = 2μg/mL; Mn-prion, protease K > 25μg/mL) (43). In this study, the dramatically increased Tm and the significantly decreased susceptibly of manganese bound Fbp68 to protease K suggest that manganese binding stabilizes the structure of Fbp68, similar to other manganese binding proteins. Enhanced Fbp68 stability upon manganese binding may aid C. difficile survival by preventing protease digestion, thereby providing a competitive advantage over other bacteria in maintaining a foothold in a highly competitive environment such as the gut.
Several recent studies show that metal ions modulate the functions of bacterial adhesins. Reportedly, calcium binding enhanced but was not essential for the Fn binding activities of LipL32 and Lig proteins from L. interrogans (26,27,41). However, ClfA binding to calcium reduces its fibrinogen binding affinity (51). Interestingly, we discovered that manganese not only improved Fbp68 binding to Fn but was required for Fn binding to Fbp68. Our results suggest that the mechanism by which manganese promotes the binding of clostridial Fbp68 to Fn differs from that of calcium bound to leptospiral LipL32 or Lig proteins. It is believed that metal ion binding dominates protein-protein interactions through several different mechanisms. In some cases, metal ion binding causes a conformational change and the binding partner is able to selectively and specifically bind to holo-proteins instead of apo-proteins.
A global structural transition was observed in Fbp68 upon manganese binding, which suggests a conformational change mediated the ability of Fbp68 to bind to Fn in the presence of manganese. Alternatively, metal ions can serve as a bridge to link a protein to its binding partner. For example, the general metal ion-dependent adhesion site (MIDAS) on the α-subunit of integrin CR3 coordinates magnesium or manganese binding to its binding partners (56). However, Fn binding is entirely dependent upon the manganese within Fbp68 and Fbp68 lacks affinity for magnesium. Thus, manganese might form a cross-link between Fbp68 and Fn, but the binding motif should be different from the MIDAS of the α-subunit of integrin. As shown in Fig. 2 and Table 3, the stoichiometry of manganese binding to Fbp68 is five. Since Fbp68 possesses 8 degenerated repeated sequences (11), it is possible that the manganese binding motif of Fbp68 is located in these degenerated repeated sequences. However, an attempt to correlate these repeated amino acid sequences of Fbp68 with other known manganese binding motifs was unsuccessful Thus, Fbp68 may utilize a novel manganese binding motif. C. difficile Fbp68 possesses high sequence similarity with other known Fn binding proteins including Fbp54 of Streptococcus pyogenes (39% identity), PavA of S. pneumoniae (38% identity), FbpA of S. gordonii (30% identity), and Fbp of Bacillus subtitlis (44% identity) (11,57-59). The Fn binding region was mapped to the C-terminal 189 amino acids of PavA based on binding inhibition of full-length PavA or S. pneumoniae to immobilized Fn by truncated PavA without the C-terminal 189 amino acids (59). Similarly, Fn can only
by guest on June 26, 2016http://w
ww
.jbc.org/D
ownloaded from

13
bind to Fbp68C, the C-terminal 194 amino acids, which show 42 % sequence homology to the Fn binding region of PavA. Furthermore, the binding site for Fbp68 on Fn was localized to NTD, the heparin binding domain of Fn, consistent with PavA and Fbp54 (59). Taken together, it is likely that the mechanism of binding of PavA, Fbp54, and Fbp68 to Fn is similar. Recently, it was proposed that a group of similar NTD binding motifs on diversified bacterial proteins such as FnbpA and FnbpB of S. aureus, FnBB of S. dysgalactiae, SfbI of S. pyogenes, and BBK32 of B. burgdorferi possess a general binding mechanism called tandem β-zipper for bacterial MSCRAMMs binding to NTD (49,60-63). On the other hand, two NTD binding regions, unique Fn binding domain (UFbD) and repeated Fn binding domain (FbRD) were identified in Streptococcus PrtF1 and PrtF2 proteins (64). Neither of these Fn binding domains has sequence similarity with Fbp68, Fbp54, or PavA (data not shown). Thus, it is highly probable that Fbp68, Fbp54 and PavA possess a conserved but novel NTD binding motif that utilizes an as yet undetermined Fn binding mechanism. ECM binding, including Fn binding, is regularly elicited by pathogens in order to adhere to host cells (19,48,65). Moreover, Fn can also serve as a mediator to induce endocytosis and initiate the entry of bacteria when it binds to bacterial Fn binding proteins (65,66). Thus, the Caco-2 cell binding activity of Fbp68 and the reduced binding effects of C. difficile in Fbp68 or Fn siRNA treated Caco-2 cells suggest an adhesive role for Fbp68. However, the fbp68 knockout mutant showed similar cell- and Fn-binding activities compared to wild type
C. difficile 630, strongly suggesting multiple adhesins are present on the surface of C. difficle with redundant adhesive properties (5, 10-11, 13-15). Furthermore, since C. difficile is not an intracellular pathogen, the rationale for C. difficile to bind to Fn is unlikely to be related to invasion as is the case for other bacteria. It was reported that the toxin of C. difficile could be digested by certain proteases in the cecum of mice (67) so adhesion of C. diffficile may be able to target the toxin to the cell, thereby increasing toxin concentration at the cell surface and avoiding proteolysis, which would enhance toxin efficacy. On the other hand, another adhesin of C. difficile, SlpA, binds to ECM components and causes further epithelial damage following toxin-induced destruction of tissue (4). It has also been observed that the adherence of C. difficile to Caco-2 cells is enhanced through the addition of clostridial CDT toxin (68). It is reasonable to hypothesize that Fbp68 and other adhesins of C. difficile might bind to ECM receptors that have been unmasked by tissue injury, resulting in more severe damage in the intestine and colon. Thus, clostridial toxins and adhesins may act synergistically in the pathogenesis of C. difficile-associated disease. In conclusion, we have demonstrated that Fbp68 is a manganese binding protein, manganese binding stabilizes the structure of Fbp68, and that Fbp68 binds to the NTD of Fn. In addition, this is the first study to identify a manganese binding adhesin for Fn. Further studies to identify the binding motifs and define the mechanisms of Fn and manganese interaction with Fbp68 are ongoing in our laboratory.
by guest on June 26, 2016http://w
ww
.jbc.org/D
ownloaded from

14
References: 1. Kelly, C. P., and LaMont, J. T. (1998) Annu. Rev. Med. 49, 375-390 2. McDonald, L. C., Killgore, G. E., Thompson, A., Owens, R. C., Jr., Kazakova, S. V.,
Sambol, S. P., Johnson, S., and Gerding, D. N. (2005) N. Engl. J. Med. 353, 2433-2441 3. Giesemann, T., Egerer, M., Jank, T., and Aktories, K. (2008) J. Med. Microbiol. 57, 690-
696 4. Calabi, E., Calabi, F., Phillips, A. D., and Fairweather, N. F. (2002) Infect. Immun. 70,
5770-5778 5. Eveillard, M., Fourel, V., Barc, M. C., Kerneis, S., Coconnier, M. H., Karjalainen, T.,
Bourlioux, P., and Servin, A. L. (1993) Mol. Microbiol. 7, 371-381 6. Davies, H. A., and Borriello, S. P. (1990) Microb. Pathog. 9, 141-146 7. Poilane, I., Karjalainen, T., Barc, M. C., Bourlioux, P., and Collignon, A. (1998) Can. J.
Microbiol. 44, 157-161 8. Seddon, S. V., and Borriello, S. P. (1992) J. Med. Microbiol. 36, 307-311 9. Calabi, E., Ward, S., Wren, B., Paxton, T., Panico, M., Morris, H., Dell, A., Dougan, G.,
and Fairweather, N. (2001) Mol. Microbiol. 40, 1187-1199 10. Cerquetti, M., Molinari, A., Sebastianelli, A., Diociaiuti, M., Petruzzelli, R., Capo, C.,
and Mastrantonio, P. (2000) Microb. Pathog. 28, 363-372 11. Hennequin, C., Janoir, C., Barc, M. C., Collignon, A., and Karjalainen, T. (2003)
Microbiology 149, 2779-2787 12. Karjalainen, T., Waligora-Dupriet, A. J., Cerquetti, M., Spigaglia, P., Maggioni, A.,
Mauri, P., and Mastrantonio, P. (2001) Infect. Immun. 69, 3442-3446 13. Karjalainen, T., Saumier, N., Barc, M. C., Delmee, M., and Collignon, A. (2002) J. Clin.
Microbiol. 40, 2452-2458 14. Waligora, A. J., Hennequin, C., Mullany, P., Bourlioux, P., Collignon, A., and
Karjalainen, T. (2001) Infect. Immun. 69, 2144-2153 15. Tasteyre, A., Karjalainen, T., Avesani, V., Delmee, M., Collignon, A., Bourlioux, P., and
Barc, M. C. (2001) J. Clin. Microbiol. 39, 1178-1183 16. Tasteyre, A., Barc, M. C., Collignon, A., Boureau, H., and Karjalainen, T. (2001) Infect.
Immun. 69, 7937-7940 17. Hennequin, C., Porcheray, F., Waligora-Dupriet, A., Collignon, A., Barc, M., Bourlioux,
P., and Karjalainen, T. (2001) Microbiology 147, 87-96 18. Hennequin, C., Collignon, A., and Karjalainen, T. (2001) Microb. Pathog. 31, 255-260 19. Patti, J. M., Allen, B. L., McGavin, M. J., and Hook, M. (1994) Annu. Rev. Microbiol. 48,
585-617 20. Seshu, J., Esteve-Gassent, M. D., Labandeira-Rey, M., Kim, J. H., Trzeciakowski, J. P.,
Hook, M., and Skare, J. T. (2006) Mol. Microbiol. 59, 1591-1601 21. Papp-Wallace, K. M., and Maguire, M. E. (2006) Annu. Rev. Microbiol. 60, 187-209 22. Ainsworth, S., and Macfarlane, N. (1975) Biochem. J. 145, 63-71 23. Shi, L., Kehres, D. G., and Maguire, M. E. (2001) J Bacteriol 183, 7053-7057
by guest on June 26, 2016http://w
ww
.jbc.org/D
ownloaded from

15
24. Santos, R., Franza, T., Laporte, M. L., Sauvage, C., Touati, D., and Expert, D. (2001) Mol. Plant Microbe Interact. 14, 758-767
25. Johnson, G. S., Adler, C. R., Collins, J. J., and Court, D. (1979) J. Biol. Chem. 254, 5483-5487
26. Lin, Y. P., Raman, R., Sharma, Y., and Chang, Y. F. (2008) J. Biol. Chem. 283, 25140-25149
27. Tung, J. Y., Yang, C. W., Chou, S. W., Lin, C. C., and Sun, Y. J. (2010) J. Biol. Chem. 285, 3245-3252.
28. Pechine, S., Gleizes, A., Janoir, C., Gorges-Kergot, R., Barc, M. C., Delmee, M., and Collignon, A. (2005) J. Med. Microbiol. 54, 193-196
29. Janvilisri, T., Scaria, J., Thompson, A. D., Nicholson, A., Limbago, B. M., Arroyo, L. G., Songer, J. G., Grohn, Y. T., and Chang, Y. F. (2009) J Bacteriol 191, 3881-3891.
30. Janvilisri, T., Scaria, J., and Chang, Y. F. (2010) J. Infect. Dis. 202, 282-290. 31. Heap, J. T., Kuehne, S. A., Ehsaan, M., Cartman, S. T., Cooksley, C. M., Scott, J. C., and
Minton, N. P. (2010) J. Microbiol. Methods 80, 49-55 32. Heap, J. T., Pennington, O. J., Cartman, S. T., Carter, G. P., and Minton, N. P. (2007) J.
Microbiol. Methods 70, 452-464 33. Heap, J. T., Pennington, O. J., Cartman, S. T., and Minton, N. P. (2009) J. Microbiol.
Methods 78, 79-85 34. Lin, Y. P., Lee, D. W., McDonough, S. P., Nicholson, L., Sharma, Y., and Chang, Y. F.
(2009) J. Biol. Chem. 284, 19380-19391. 35. Lin, Y. P., Greenwood, A., Nicholson, L. K., Sharma, Y., McDonough, S. P., and Chang,
Y. F. (2009) J. Biol. Chem. 284, 23547-23557. 36. Lin, Y. P., and Chang, Y. F. (2007) Biochem. Biophys. Res. Commun. 362, 443-448 37. Lin, Y. P., and Chang, Y. F. (2008) J. Vet. Sci. 9, 133-144. 38. Chessa, D., Winter, M. G., Nuccio, S. P., Tukel, C., and Baumler, A. J. (2008) Mol.
Microbiol. 68, 573-587 39. Vendrame, F., Segni, M., Grassetti, D., Tellone, V., Augello, G., Trischitta, V.,
Torlontano, M., and Dotta, F. (2006) J. Clin. Endocrinol. Metabol. 91, 5064-5068 40. Dugan, T. A., Yang, V. W., McQuillan, D. J., and Hook, M. (2003) J. Biol. Chem. 278,
13655-13662 41. Hauk, P., Guzzo, C. R., Ramos, H. R., Ho, P. L., and Farah, C. S. (2009) J. Mol. Biol.
390, 722-736. 42. Mould, A. P., Akiyama, S. K., and Humphries, M. J. (1995) J. Biol. Chem. 270, 26270-
26277 43. Brown, D. R., Hafiz, F., Glasssmith, L. L., Wong, B. S., Jones, I. M., Clive, C., and
Haswell, S. J. (2000) Embo J. 19, 1180-1186 44. Mizuno, K., Whittaker, M. M., Bachinger, H. P., and Whittaker, J. W. (2004) J. Biol.
Chem. 279, 27339-27344 45. Coburn, J., Fischer, J. R., and Leong, J. M. (2005) Mol. Microbiol. 57, 1182-1195 46. Borriello, S. P., Davies, H. A., Kamiya, S., Reed, P. J., and Seddon, S. (1990) Rev. Infect.
Dis. 12 Suppl 2, S185-191
by guest on June 26, 2016http://w
ww
.jbc.org/D
ownloaded from

16
47. Tasteyre, A., Barc, M. C., Karjalainen, T., Dodson, P., Hyde, S., Bourlioux, P., and Borriello, P. (2000) Microbiology 146 ( Pt 4), 957-966
48. Schwarz-Linek, U., Hook, M., and Potts, J. R. (2004) Mol. Microbiol. 52, 631-641 49. Schwarz-Linek, U., Pilka, E. S., Pickford, A. R., Kim, J. H., Hook, M., Campbell, I. D.,
and Potts, J. R. (2004) J. Biol. Chem. 279, 39017-39025 50. Karjalainen, T., Barc, M. C., Collignon, A., Trolle, S., Boureau, H., Cotte-Laffitte, J., and
Bourlioux, P. (1994) Infect. Immun. 62, 4347-4355 51. O'Connell, D. P., Nanavaty, T., McDevitt, D., Gurusiddappa, S., Hook, M., and Foster, T.
J. (1998) J. Biol. Chem. 273, 6821-6829 52. Silva, J. J. R. F., and Williams, R. J. F. (eds). (2001) The biological chemistry of the
elements: The inorganic chemistry of life, 2nd edition Ed., Oxford University Press 53. Hesketh, S., Sassoon, J., Knight, R., and Brown, D. R. (2008) Mol. Cell. Neurosci. 37,
590-598 54. Barondeau, D. P., and Getzoff, E. D. (2004) Curr. Opin. Struct. Biol. 14, 765-774 55. Tainer, J. A., Getzoff, E. D., Richardson, J. S., and Richardson, D. C. (1983) Nature 306,
284-287 56. Lee, J. O., Rieu, P., Arnaout, M. A., and Liddington, R. (1995) Cell 80, 631-638 57. Christie, J., McNab, R., and Jenkinson, H. F. (2002) Microbiology 148, 1615-1625 58. Courtney, H. S., Dale, J. B., and Hasty, D. I. (1996) Infect. Immun. 64, 2415-2419 59. Holmes, A. R., McNab, R., Millsap, K. W., Rohde, M., Hammerschmidt, S., Mawdsley, J.
L., and Jenkinson, H. F. (2001) Mol. Microbiol. 41, 1395-1408 60. Bingham, R. J., Rudino-Pinera, E., Meenan, N. A., Schwarz-Linek, U., Turkenburg, J. P.,
Hook, M., Garman, E. F., and Potts, J. R. (2008) Proc Natl Acad Sci U S A 105, 12254-12258
61. Meenan, N. A., Visai, L., Valtulina, V., Schwarz-Linek, U., Norris, N. C., Gurusiddappa, S., Hook, M., Speziale, P., and Potts, J. R. (2007) J. Biol. Chem. 282, 25893-25902
62. Raibaud, S., Schwarz-Linek, U., Kim, J. H., Jenkins, H. T., Baines, E. R., Gurusiddappa, S., Hook, M., and Potts, J. R. (2005) J. Biol. Chem. 280, 18803-18809
63. Schwarz-Linek, U., Werner, J. M., Pickford, A. R., Gurusiddappa, S., Kim, J. H., Pilka, E. S., Briggs, J. A., Gough, T. S., Hook, M., Campbell, I. D., and Potts, J. R. (2003) Nature 423, 177-181
64. Jaffe, J., Natanson-Yaron, S., Caparon, M. G., and Hanski, E. (1996) Mol. Microbiol. 21, 373-384
65. Peacock, S. J., Foster, T. J., Cameron, B. J., and Berendt, A. R. (1999) Microbiology 145 ( Pt 12), 3477-3486
66. Mempel, M., Schnopp, C., Hojka, M., Fesq, H., Weidinger, S., Schaller, M., Korting, H. C., Ring, J., and Abeck, D. (2002) Br. J. Dermatol. 146, 943-951
67. Corthier, G., Muller, M. C., Elmer, G. W., Lucas, F., and Dubos-Ramare, F. (1989) Infect. Immun. 57, 3922-3927
68. Schwan, C., Stecher, B., Tzivelekidis, T., van Ham, M., Rohde, M., Hardt, W. D., Wehland, J., and Aktories, K. (2009) PLoS pathogens 5, e1000626
by guest on June 26, 2016http://w
ww
.jbc.org/D
ownloaded from

17
FOOTNOTES
This work was supported with federal funds from the National Institute of Allergy and Infectious Disease, National Institute of Health, Department of Health and Human Services, under contract N01-AI-30054, project no. ZC005-06 and ZC008-09. Thanks are given to Dr Timothy J. Foster for his kind gift of Staphylococcal ClfAN2N3 clones, to Drs. Bhargavi Jayaraman and Charlene Mottler for help with ITC, to Dr. Richard Medville for assistance with Electron Microscopy, to Drs. Moonsoo Jin and Marci Scidmore for kindly allowing us to use their surface plasmon resonance and confocal laser scanning microscope, respectively, to Dr. John Heap for his advice for the construction of C. difficle mutants and Mr. Peter R. Harpending for his technical assistance. We also thank Dr. Nigel Minton for the gifts of plasmids (pMTL007-E2 and PMTL84151) and Dr. Mark McBride for the gift of E. coli CA434. Abbreviations: CD; circular dichroism; CLSM, confocal laser scanning microscopy; DSC, differential scanning calorimetry; ECM, extracellular matrix; ITC, isothermal titration calorimetry; MSCRAMMs, microbial surface component recognizing adhesive matrix molecules; NTD, N-terminal domain; siRNA, small interfering RNA; SPR, surface plasmon resonance
by guest on June 26, 2016http://w
ww
.jbc.org/D
ownloaded from

18
Figure Legend
Figure 1. Schematic presentation of Fbp68 and Fn used in this study. (A) A schematic diagram showing the structure of Fbp68 and the truncated Fbp68 proteins Fbp68N (residues 2 to 396) and Fnbp68C (residues 397 to 591) used in this study. (B) A chart representing fibronectin (Fn) and truncated Fn including N-terminal domain (NTD), gelatin binding domain (GBD), 70kDa domain (70kDa), cell binding domain (CBD), and heparin binding domain 2 (Hep-2) used in this study. Figure 2 Manganese binds to Fbp68 and induces conformational change. (A) Binding affinity determined by the ITC profile of Fbp68 with manganese chloride. The heat change obtained with ligand (Mn2+) titration is shown in the upper panel. In the lower panel, the solid lines represent the best fits to a single-site binding model after peak (◇) integration, subtraction of blank titration data (not shown), and concentration normalization. Molar heats of binding are plotted vs. the molar ratio of Mn2+ to protein. Binding potency of protein with ligand (KD) is 52.70±1.97 nM. The thermodynamic data that were obtained from ITC are shown in Table 3. (B) Intrinsic fluorescence spectrum of Fbp68 in the presence and absence of Mn2+. 1μM of Fbp68 in Tris buffer (pH 7.0) was excited at 295 nm. The figure shows Trp fluorescence in the presence of 0, 12.5, 25, 50, 100, or 200 nM of manganese chloride (Inner plot). (C) Fluorescence emission spectra of ANS-Fbp68 complex in the presence of 0, 12.5, 25, 50, 100, or 200 nM of manganese chloride. The excitation wavelength was set at 375 nm. The buffer consisted of 25 mM Tris (pH 7.0), 150 mM NaCl (Inner plot). The KD of Manganese and Fbp68 was determined by monitoring the increasing fluorescence intensities of ANS-Fbp68 titrated by manganese. (D) Far-UV CD spectra of Fbp68 were recorded in the presence of manganese. Protein concentration was 1μM in Tris buffer (pH 7.0). Spectra were recorded with a 0.02 cm path length cuvette. Aliquots of calcium chloride solution were added to a final concentration of 0, 12.5, 25, 50, 100, or 200 nM to the protein solution and CD spectra recorded. Manganese chloride was added to achieve final concentrations of 0, 12.5, 25, 50, 100, or 200 nM. In (B)-(D), direction of arrows indicates increasing order of manganese addition. Figure 3. Manganese binding stabilizes the structure of Fbp68. (A) Thermal unfolding transitions of Fbp68 with or without magnesium monitored by CD. Unfolding of 10 μM of Fbp68 either in the presence or absence of 100 μM -manganese chloride was followed by CD, measuring ellipticity at 205 nm from 25 to 70°C. A transition point was found in both manganese bound and free forms of protein. The melting temperatures were determined by the location of the peak in the plot of the derivative of ellipticity versus temperature. The midpoints of transitions for manganese bound or free form of the protein are 65.9±1.3°C and 58.0±2.1°C respectively. (B) Molar heat capacity [kcal/(mol.K)] was plotted against the temperature (°C ) for thermal denaturation of 30 μM of Fbp68 in the absence or in the presence of manganese chloride (100 μM) as monitored by DSC. Transitions occur with midpoint temperatures of 66.1±3.2°C and 59.3±2.1°C for manganese bound or free form of the protein, respectively. (C) Protease K resistance experiment. Dialyzed untreated or 100μM of manganese chloride treated Fbp68 were electrophoresed and blotted using Western blot.
by guest on June 26, 2016http://w
ww
.jbc.org/D
ownloaded from

19
Parallel samples were treated with increasing concentrations of PK (0, 3.75, 7.5, 15, 30, 60 ng/μL). Fbp68 was detected using a polyclonal antibody as described in Materials and Methods. The experiment was repeated three times. Figure 4. Manganese is required for Fbp68 binding to Fn. (A) Fn binding to Fbp68 assayed by ELISA. Binding of Fbp68, LigBCen2, or LigBCon4 in the absence of Mn2+ or in the presence of varying concentrations of Mn2+ (1.0, 0.5, 0.25, 0.125, 0.062, 0.037, 0.015, 0 μM) to immobilized NTD or BSA (negative control and data not shown). 10 nM of histidine tagged LigBCen2 (positive control), 100μM manganese bound or free forms of Fbp68 or LigBCon4 (negative control) were added to a serial dilution of NTD or BSA coated wells. Bound proteins were estimated by ELISA. Each value represents the mean± SEM of three trials in triplicate samples. Statistically significant differences (P<0.05) are indicted by *. (B) SPR analysis of Fbp68 interacting with Fn. 1.5 μM of recombinant histidine-tagged Fbp68 was immobilized on the surface of a Ni-NTA chip. 1 μM of Fn in Tris buffer in the presence or absence of 100 μM MnCl2 at pH 7.0 flowed through the chip. (C) The determination of KD of manganese bound Fbp68 and Fn by SPR analysis. 1.5 μM of recombinant histidine-tagged Fbp68 was immobilized on the surface of Ni-NTA chip. Fn was flowed through the chip in Tris buffer at pH 7.0, and the concentrations of Fn ranged from 1000 to 62.5nM (from top to bottom). The KD, kon, koff were obtained from the average of duplicate experiments (kon = 1.09 ± 0.35 × 104 s-1M-1, koff = 2.48 ± 0.52 × 10-3 s-1 , KD = 228±20 nM). Figure 5. Fbp68C binds to NTD of Fn. (A) Binding of serial concentrations of Fbp68, Fbp68N, or Fbp68C to immobilized Fn by ELISA. Serial concentrations of GST-Fbp68N, GST-Fbp68C, GST-Fbp68 (positive control), or GST-LigBCon (negative control) were added to wells coated with 1 μM of Fn or BSA (negative control, data not shown). (B) Binding of serial concentrations of Fbp68C to immobilized Fn by ELISA. Serial concentrations of GST-Fbp68C or GST-LigBCon (negative control, data not shown) were added to microtiter plate wells coated with 1 μM of Fn, NTD, GBD, CBD, 40kDa, or BSA (negative control). Bound proteins were measured by ELISA. (C) Determination of binding affinity by ITC. The cell contained 1 ml of NTD and the syringe contained 250μL of Fbp68C (upper panel). Heat differences were obtained from 25 injections of Fbp68C; (lower panel) Integrated curve with experimental data (◇) and the best fit (—). The thermodynamic parameters are shown in Table 3. The thermodynamic parameters are shown as the average of duplicate experiments (KD = 233 ± 10 nM, ΔH = -5.95 ± 0.38 kcal mol-1, TΔS = 3.05 ± 0.6 kcal mol-1 K-1, n= 0.98 ± 0.02). (D) SPR analysis of Fbp68C and NTD. 1.5 μM of recombinant histidine-tagged Fbp68C was immobilized on the surface of a Ni-NTA chip. NTD was flowed through the chip in Tris Buffer at pH 7.0, and the concentrations of NTD ranged from 1000 to 62.5nM (from top to bottom). The KD, kon, koff were obtained from the average of duplicate experiments (kon = 1.23 ± 0.25 × 104 s-1M-
1, koff = 2.66 ± 0.05 × 10-3 s-1 , KD = 216±56 nM).
by guest on June 26, 2016http://w
ww
.jbc.org/D
ownloaded from

20
Figure. 6. Generating fbp68 mutant and a complemented strain. (A) The structure of fbp68 wild type (CD630WT, top) and mutant (CD∆Fbp68102, bottom). Oligonucleotide binding sites used in constructing and screening fbp68 mutant are represented as arrows (fbp68-F, fbp68-R, EBS universal, EmB-R1, Thio-F1; see Table 2). (B) PCR analysis of the fbp68 deletion mutant. Lane 1, CD∆Fbp68102 (∆Fbp68::Lincor); Lane 2, CD630WT (parental strain). The primers used are indicated on the right (see panel A and Table 2). fliD gene amplified from fliD-F and fliD-R primer pairs served as a positive reference. (C) Western blot analysis to detect the expression of fbp68. Samples were immunoblotted using anti-Fbp68 polyclonal antibodies. Lane 1, CD630WT (parental strain); Lane2, CD∆Fbp68102 (∆Fbp68::Lincor); Lane 3, CD∆Fbp68102/pMTL84151-fbp68 (∆Fbp68::Lincor with a plasmid that contains an intact fbp68). All of the lanes contain the protein from 2 x 107 C. difficile 630 cells. The arrow indicates the location of Fbp68. Figure 7. The Fn- and Caco-2 cell-binding activities of Fbp68C and different C. difficile strains (A) Binding of Fbp68C to Caco-2 cells. A series concentration (0, 0.015, 0.03125, 0.0625, 0.125, 0.25, 0.5, or 1 μM) of GST-Fbp68C or GST (negative control) was added to Caco-2 cells (105). The binding of each of these proteins to Caco-2 cells was measured by ELISA. (B) Fbp68C inhibits the binding of C. difficile to Caco-2 cells. Caco-2 cells were incubated with a series concentration (0, 0.015, 0.03125, 0.0625, 0.125, 0.25, 0.5, or 1 μM) of GST-Fbp68C or GST (negative control) prior to the addition of C.difficile (107). The adhesion of C. difficile to Caco-2 cells (105) was detected by ELISA. The reduced percentage of attachment was determined relative to the attachment of C. difficile to untreated Caco-2 cells. (C). Fbp68C inhibits the binding of C. difficile to Caco-2 cells. Caco-2 cells (106) were pre-treated with 50μM of GST-Fbp68C or GST (negative control) prior to the addition of C. difficile (108). The adhesion of C. difficile or the binding of these proteins to Caco-2 cells were detected by CLSM. (D and E) Fn- and cell-binding activity of wild type C. difficile (CD630WT), fbp68 mutant (CD∆Fbp68102), or fbp68 complemented strain (CD∆Fbp68102/pMTL84151-fbp68). (D) A total of 108 CD630WT, CD∆Fbp68102, or CD∆Fbp68102/pMTL84151-fbp68 were added to Fn-coated wells (1µM/well). The wells coated with 1μM of BSA served as the negative control. Three CLSM fields were selected to count the number of bacteria that were bound to the Fn and BSA coated wells to determine the Fn-binding activity of various genotypes of C. difficile as described in Materials and Methods. (E) A total of 108 cells of CD630WT, CD∆Fbp68102, or CD∆Fbp68102/pMTL84151-fbp68 were incubated in wells cultured with 106 Caco-2 cells. The wells incubated without Caco-2 cells served as the negative control. Cell-binding activity was measured by the bacterial attachment assay as described in Materials and Methods; results are shown as the number of cell associated bacteria per well. In (A), (B), (D), and (E), each value represents the mean± SEM of three trials in triplicate samples. Statistically significant differences (P<0.05) are indicted by *. In (C), the CLSM settings were identical for all the captured images. Images were processed using Adobe Photoshop CS2. Figure 8. The binding of Fbp68C to Fn siRNA transfected Caco-2 cells was reduced. (A). Detection of the expression of Fn and actin in Caco-2 cells 72 hours after transfection with Fn or negative siRNA. Fn and α-actin were detected on immunoblots probed by actin
by guest on June 26, 2016http://w
ww
.jbc.org/D
ownloaded from
21
or Fn antibodies. (B) Binding of GST-Fbp68C was reduced in siRNA transfected cells. (C) Fifty μM of GST-Fbp68C or GST (negative control) was added to Fn or negative siRNA transfected Caco-2 cells. Expression of Fn and the binding of these proteins to Caco-2 cells were detected by CLSM. The CLSM settings were identical for all the captured images. Images were processed using Adobe Photoshop CS2. Table 1. Strains and plasmids used in this study Strain or plasmid Genotype or characteristic References or Sources C. difficile strains 630WT Wild type C. difficile strain (29) CD∆Fbp68102 630WT∆fbp68::Lincor This study CD∆Fbp68102/pMTL84151-fbp68 630WT∆fbp68::Lincor, complemented with fbp68 This study E. coli strains DH5α F�Φ80lacZ∆M15 ∆(lacZYA-argF)U169 recA1 endA1 hsdR17(rK�
mK+) supE44 thi-1 gyrA96 relA1λ� Invitrogen
BL21 F–, ompT, hsdSB (rB–, mB–), dcm, gal, λ(DE3) Promega M15[pREP4] nalS strS rifS thi- lac- ara- gal+ mtl- F- recA+ uvr+ lon+ [pREP4
KanR], Qiagen
CA434
F- mcrB mrr hsdS20(rB- mB-) recA13 leuB6 ara-14 proA2 lacY1 galK2 xyl-5 mtl-1 rpsL20(SmR) glnV44 λ- , R702
(33)
Plasmids pQE30 pGEX4T2 pQE30-fbp68 pQE30-fbp68N pQE30-fbp68C
Ampr, His-tag protein expression vector Ampr, GST-tag protein expression vector pQE30 carrying intact fbp68 pQE30 carrying fbp68N pQE30 carrying fbp68C
Qiagen GE This study This study This study
by guest on June 26, 2016http://w
ww
.jbc.org/D
ownloaded from

22
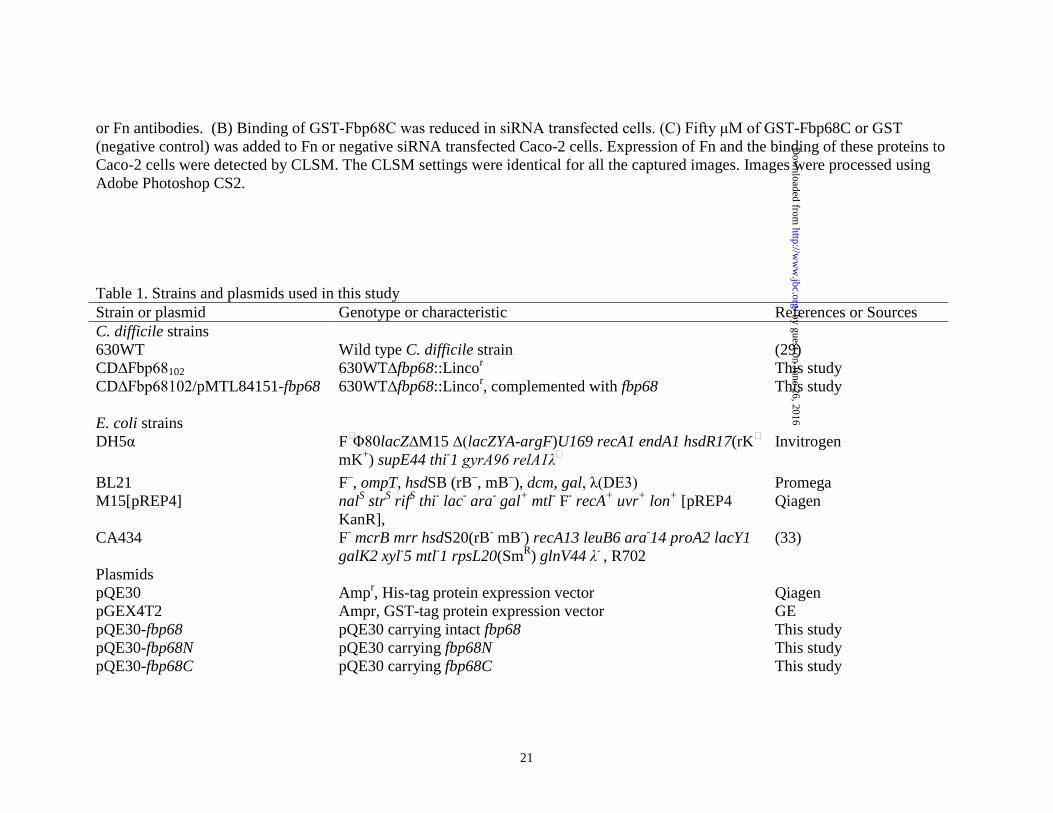
pQE30-ligBCon4 pQE30-ligBCen2 pGEX4T2-fbp68 pGEX4T2-fbp68N pGEX4T2-fbp68C pGEX4T2-ligBCon
pQE30 carrying ligBCon4 pQE30 carrying ligBCen2 pGEX4T2 carrying fbp68 pGEX4T2 carrying fbp68N pGEX4T2 carrying fbp68C pGEX4T2 carrying ligBCon
(37) (36) This study This study This study (37)
pMTL007C-E2 pMTL007C-E2-CDI-Fbp68-102S pMTL84151 pMTL84151-fbp68
Cycloseriner, Cefotaxiner; shuttle vector pMTL007C-E2 carrying the intron for target fbp68 102-103S site. Cefotoxinr, Thiamphenicolr, Cycloseriner; expression vector pMTL84151 carrying intact fbp68
(31) This study (33) This study
Table 2. Oligonucleotides used in this study Primer/Vector Sequence fbp68-F* CCGGAATTCATGGCATTAGATGGATTAGTTATAC fbp68-R* CGCGGATCCTTATTCAGATTTAACCTTAAGCTTGG EBS Universal* CGAAATTAGAAACTTGCGTTCAGTAAAC ErmB-R1* GAACGCGTGCGACTCATAGAATTATTTCCTCCCG Thio-F1* CTACTAGTACGCGTTATATTGATAAAAATAATAATAGTGGG fliD-F* CATATGATGTCAAGTATAAGTCCAGTAAG fliD-R* TTAATTACCTTGTGCTTGTGAG Fbp68fp CGGGATCCGCATTCGATGGA Fbp68rp CGGTCGACTTATTCAGATTTAACCTT Fbp68Nfp CGGGATCCGCATTCGATGGA Fbp68Nrp CGGTCGACCTTTTTAAAGTATTTTTG Fbp68Cfp CGGGATCCTATAACAAAATGAAACAT Fbp68Crp CGGTCGACTTATTCAGATTTAACCTT * Primers used for mutant generation.
by guest on June 26, 2016http://w
ww
.jbc.org/D
ownloaded from
23
Table 3. Thermodynamic parameters for the interaction of metal ions and Fbp68 Metal ion [Metal ion] [Fbp68] ΔH TΔS KD ΔG n
μM μM kcal mol-1 kcal mol-1 K-1 nM kcal mol-1 Metal ion: Fbp68
Mn2+ 1000 38 -12.14±0.05 -2.25±0.02 52.70±1.97 -9.88±0.02 4.98±0.02
Ca2+ 1000 42 *n.f. *n.f. *n.f. *n.f. *n.f.
Mg2+ 1000 31 *n.f. *n.f. *n.f. *n.f. *n.f.
Zn2+ 1000 49 *n.f. *n.f. *n.f. *n.f. *n.f.
*n.f.: non-fittable
by guest on June 26, 2016http://w
ww
.jbc.org/D
ownloaded from

Yi-Pin Lin, Chih-Jung Kuo, Xhelil Koleci, Sean P. McDonough and Yung-Fu ChangManganese binds to Clostrdium difficile Fbp68 and is essential for fibronectin binding
published online November 9, 2010J. Biol. Chem.
10.1074/jbc.M110.184523Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/early/2010/11/09/jbc.M110.184523.full.html#ref-list-1
This article cites 0 references, 0 of which can be accessed free at
by guest on June 26, 2016http://w
ww
.jbc.org/D
ownloaded from