Embed Size (px)
Citation preview

Bioorganic & Medicinal Chemistry xxx (2014) xxx–xxx
Contents lists available at ScienceDirect
Bioorganic & Medicinal Chemistry
journal homepage: www.elsevier .com/locate /bmc
Macrocyclic diterpenes resensitizing multidrug resistant phenotypes
http://dx.doi.org/10.1016/j.bmc.2014.05.0060968-0896/� 2014 Elsevier Ltd. All rights reserved.
⇑ Corresponding author. Tel.: +351 217946475; fax: +351 217946470.E-mail address: [email protected] (Maria-José U. Ferreira).
Please cite this article in press as: Reis, M. A.; et al. Bioorg. Med. Chem. (2014), http://dx.doi.org/10.1016/j.bmc.2014.05.006
Mariana A. Reis a, Angela Paterna a, Ricardo J. Ferreira a, Hermann Lage b, Maria-José U. Ferreira a,⇑a Instituto de Investigação do Medicamento (iMed.ULisboa), Faculdade de Farmácia, Universidade de Lisboa Av. Prof. Gama Pinto, 1649-003 Lisboa, Portugalb Charité Campus Mitte, Institute of Pathology, Berlin, Germany
a r t i c l e i n f o
Article history:Received 3 February 2014Revised 29 April 2014Accepted 5 May 2014Available online xxxx
Keywords:Collateral sensitivityMultidrug resistance in cancerMacrocyclic diterpenesAntiproliferative activityP-glycoprotein
a b s t r a c t
Herein, collateral sensitivity effect was exploited as a strategy to select effective compounds to over-come multidrug resistance in cancer. Thus, eleven macrocyclic diterpenes, namely jolkinol D (1), iso-lated from Euphorbia piscatoria, and its derivatives (2–11) were evaluated for their activity on threedifferent Human cancer entities: gastric (EPG85-257), pancreatic (EPP85-181) and colon (HT-29) eachwith a variant selected for resistance to mitoxantrone (EPG85-257RN; EPP85-181RN; HT-29RN) andone to daunorubicin (EPG85-257RD; EPP85-181RD; HT-29RD). Jolkinol D (1) and most of its derivatives(2–11) exhibited significant collateral sensitivity effect towards the cell lines EPG85-257RN (associatedwith P-glycoprotein overexpression) and HT-29RD (altered topoisomerase II expression). The benzoylderivative, jolkinoate L (8) demonstrated ability to target different cellular contexts with concomitanthigh antiproliferative activity. These compounds were previously assessed as P-glycoproteinmodulators, at non-cytotoxic doses, on MDR1-mouse lymphoma cells. A regression analysis betweenthe antiproliferative activity presented herein and the previously assessed P-glycoprotein modulatoryeffect showed a strong relation between the compounds that presented both high P-glycoprotein mod-ulation and cytotoxicity.
� 2014 Elsevier Ltd. All rights reserved.
1. Introduction
Drug resistance has been pointed as one of the main causes forthe failure of chemotherapy to treat cancer, affecting patientswith a variety of blood cancers and solid tumors, including breast,ovarian, lung, and lower gastrointestinal tract cancers.1 Resistanceto chemotherapy can be intrinsic or acquired; tumors with intrin-sic resistance fail to respond to the first chemotherapy given. Onthe other hand, in acquired resistance, tumors innately respond tochemotherapy but eventually there is a relapse in spite of the fol-lowing treatment. If resistance on cancer cells occurs to a broadspectrum of structurally and mechanistically diverse antitumoragents, it is designated as multidrug resistance (MDR). The phe-nomenon of MDR is considered to be a major hurdle for chemo-therapy success since multiple drugs of different classes are usedto treat most cancers.1–3 A better understanding of MDR natureand its molecular mechanisms is essential for overcoming MDRin tumor cells and is of the utmost importance for the successof future clinical treatments. Many different types of cellular
mechanisms in MDR have been described and are likely to occursimultaneously or on a cascade of events during the establish-ment of the MDR phenotype. In fact, clinical scenario MDR isoften a multifactorial phenomenon with more than one mecha-nism being present rather than a single mechanistic event.4
Among these mechanisms it can be found: alterations in the cellcycle checkpoints, failure of the apoptotic mechanisms (e.g., over-expression of Bcl-2 family proteins), repair of damaged cellulartargets, alterations in drug targets (e.g., decreased expression/and or activity of topoisomerase II), drug inactivation by glutathi-one-S-transferase and reduced drug accumulation through drugefflux or vesicular sequestration by ATP binding cassete (ABC)transporters.2,5–7
The three major types of MDR efflux pumps include membersof the ABCB (ABCB1/MDR1/P-glycoprotein), ABCC (ABCC1/MRP1,ABCC2/MRP2), and ABCG (ABCG2/MXR/BCRP) subfamilies. P-gly-coprotein (ABCB1, P-gp) is the most known and studied effluxpump associated with MDR.8 The overexpression of P-gp inmalignant cancer cells results in reduced intracellular concentra-tion of anticancer drugs to levels that lead to treatment failure,causing cross-resistance to several cytotoxic drugs such asVinca alkaloids, podophyllotoxins, anthracycline derivatives and

2 M. A. Reis et al. / Bioorg. Med. Chem. xxx (2014) xxx–xxx
taxanes.1,9 The MDR1 promoter activity can be up-regulated byvarious stimuli, such as anticancer drugs, DNA-damaging agents,heat shock, serum starvation and ultraviolet radiation. MDR1expression can also be up-regulated as a consequence of tumorprogression, as in cases where mutation in tumor suppressor genep53 and activation of ras oncogene are present.10 Research onselective and potent P-gp reversal agents that impair the effluxmechanism when co-administered with anticancer drugs hasbeen one of the most common approaches to avoid thechemotherapy failure. This pharmacological modulation has beenattempted since 1981 (with verapamil), however threedecades passed and despite several generations of P-gp modula-tors had gone into clinical trials, none has made it into theclinic.9,11
A valuable alternative strategy is the identification of com-pounds that selectively exert a more pronounced cytotoxicityon cells with MDR phenotype than in the parental nonresistantcells from which they are derived.9 On MDR phenotypes, resis-tance to one cytotoxic agent can, simultaneously, confer greatersensitivity to an alternate agent, being this effect greater thanin the parental cell line. This effect is termed ‘collateral sensitiv-ity’ and can be used to select compounds that are highly effectiveagainst drug-resistant phenotypes, since the geneticalteration that conferred resistance to a drug may sensitize it toother drugs. This effect is assessed in vitro by determining thecytotoxicity of a compound against parental line and its MDRsub-line.12,13
Macrocyclic diterpenes and ent-abietane lactones, isolatedfrom Euphorbia species, were identified as having a ‘collateralsensitivity’ effect on MDR cell variants with different MDR phe-notypes.14 Some of which, namely the macrocyclic diterpeneswith the lathyrane skeleton, were also recognized as potentP-gp modulators.15,16 The combination of drugs to target morethan one MDR mechanism simultaneously might be a solutionto control MDR in its complexity, which would notonly increase the cytotoxic effect but also decrease the occur-rence of drug resistance providing an optimal therapeuticoutcome.
In a previous work, fractionation of the methanol extract ofthe aerial parts of Euphorbia piscatoria led to the isolation ofjolkinol D (1) in large amount that allowed to build a smalllibrary of derivatives, which were evaluated as MDR reversers.17
On the present work, jolkinol D and the derivatives 2–12 wereevaluated for their potential collateral sensitivity effect ongastric, pancreatic and colon cancer cells models (drugsensitive and drug resistant sublines) well characterized forMDR.18–26
2. Results and discussion
2.1. Antiproliferative activity and the collateral sensitivity effect
Compounds with the macrocyclic lathyrane-type scaffold havebeen isolated from several Euphorbia species and their potentialas P-gp modulators is being subject of investigation.16,27–29 Frac-tionation of the methanol extract of the aerial parts of Euphorbiapiscatoria led to the isolation of jolkinol D (1) in large amountthat allowed to build a small library of derivatives.17 Jolkinol Dderivatives (2–11) were designed in order to establish struc-ture-activity relationships with P-gp modulation. It was con-cluded that the presence of an aromatic moiety improvedsignificantly the inhibition of this efflux pump, at non-cytotoxicdoses.
Please cite this article in press as: Reis, M. A.; et al. Bioorg. Med. Chem.
3
OO
O
H3C
CH3
H3C
RO HCH3
21
16 45 6 7 8
91112
13
1415
1'2'
CH3
CH3
2019
18
10
3
OHO
CH3
H3C
HO HCH3
21
16 45 6 7 8
911
12
13
1415
17
CH3
CH3
2019
18
10
H
H
H
H
17Jolkinodiol (11)
R R
Jolkinol D (1) H Jolkinoate D (6) CH3
O
9 1''2''
12''
Jolkinoate A (2) CH3
O
1''2''Jolkinoate I (7)
O
3''
1''2''3''
4''
4''5''
Jolkinoate C (3)
O
CH3 1''4''
3''2'' Jolkinoate L (8)
O
CF3
1''2''
5'' 3''4''
6''
7''
Jolkinoate E (4)
O
CH3
CH3
1''2''3''
4''5'' Jolkinoate K (9)
O
3''
1''2''3''
4''
4''5''MeO
Jolkinoate G (5)
O
CH3
CH3 1''4''
3''6''
2''5''
Jolkinoate M (10)SO
O2''
2''
1''
3''
3''
4''
Pursuing our research on MDR in cancer, compounds 1–11 wereinvestigated for their potential collateral sensitivity effect on threedifferent cancer entities, using the SRB assay. For each human can-cer cell line EPP85-181 (pancreatic), EPG85-257 (gastric) and HT-29 (colon) there is a variant selected for resistance to mitoxantrone(RN) and one to daunorubicin (RD), which accounts for a specificmultidrug resistant phenotype (Table 1). In order to find selectiveantiproliferative compounds, similar studies have been reportedusing these cancer cell models.30–34 The relative resistance ratio(RR, determined by dividing the IC50 against a resistant line bythe IC50 against a parental line) was used to explore if the com-pounds could confer a greater sensitivity in the drug-resistantsub-lines than in the parental cell line. If this effect is observed,it is called as collateral sensitivity. In such cases, the RR value islower than 0.5, meaning that there is selectivity towards MDR phe-notypes. However, other cases can be verified, for instance thedevelopment of resistance to a drug and simultaneous cross-resis-tance to various cytotoxic agents may result in loss of sensitivity,which is reflected by a RR value higher than 2. If a compoundpresents the same response for both parental and resistant lines(2< RR >0.5), it cannot be considered as cross-resistance nor collat-eral sensitivity; instead it can be assumed that there is a lack ofresistance mechanisms for these compounds.13 And thus, thisapproach can lead to the selection of compounds that are highlyeffective against drug-resistant phenotypes. The cytotoxic agentsetoposide and cisplatin were used as positive controls. The antipro-liferative activity and collateral sensitivity effect of jolkinol D (1)and its derivatives (2–11) are presented on Tables 2–4.
In order to find out structure activity relationships between thetested compounds (1–11) and the different MDR phenotypes,the non-parametric Kruskal–Wallis rank test was performed.
(2014), http://dx.doi.org/10.1016/j.bmc.2014.05.006

Tabl
e1
Char
acte
rist
ics
ofca
ncer
cell
lines
and
drug
-res
ista
ntsu
blin
es
Ori
gin
Gas
tric
carc
inom
aPa
ncr
eati
cca
rcin
oma
Col
onca
rcin
oma
Cel
lli
ne
EPG
85-
257P
EPG
85-2
57R
NEP
G85
-257
RD
EPP8
5-18
1PEP
P85-
181R
NEP
P85-
181R
DH
T-29
PH
T-29
RN
HT-
29R
D
Sele
ctio
nag
ent*
�1
8M
itox
antr
one
(0.2
lg/
ml)
18
Dau
nor
ubi
cin
(2.5
lg/
ml)
18
�2
4M
itox
antr
one
(0.0
2l
g/m
l)2
4D
aun
oru
bici
n(2
.5l
g/m
l)2
4�
26
Mit
oxan
tron
e(0
.2l
g/m
l)2
6D
aun
oru
bici
n(0
.25
lg/
ml)
26
mR
NA
expr
essi
ngle
vel
mR
NA
expr
essi
ngle
vel
mR
NA
expr
essi
ngle
vel
Ref
Ref
Ref
MR
P1+
++
14+
++
14+
++
14M
RP2
++"
+"14
++"
+"14
++"
+"14
MR
P3�
��
14+;
+"+"
14+
+"+
14M
RP4
++"
+"14
++"
+"14
++"
+"14
MR
P5+
+"+"
14+
+"+"
14+
+"+"
14M
RP6
��
�14
+�
�14
+�
+"14
MR
P7+
++
14+
++
14+
++
14M
RP8
+;�
�14
��
�14
��
�14
MR
P9�
��
14�
��
14�
��
14M
DR
1�
�+"
14�
�+"
14,2
4�
��
14B
CR
P+;
+"+;
14,1
9�
��
14,2
4+;
+;+;
14G
PC3
+;+"
20,2
1TA
P+
+"+
23TO
POII
++;
22+
+;+;
25
MR
P=
mu
ltid
rug
resi
stan
cepr
otei
n;
MD
R1
=P-
gp;
BC
RP
=br
east
can
cer
resi
stan
cepr
otei
n;
GPC
3=
gtyp
ican
-3;
TAP
=tr
ansp
orte
ras
soci
ated
prot
ein
;TO
PO=
DN
Ato
pois
omer
ase;
+=
mR
NA
expr
esse
d;+;
=lo
wle
vels
ofm
RN
Aex
pres
sed;
+"=
hig
hle
vels
ofm
RN
Aex
pres
sed;�
=n
oex
pres
sion
.*
Toen
sure
the
MD
Rph
enot
ype
the
grow
thm
ediu
mof
each
cell
lin
eis
supp
lem
ente
dw
ith
ade
term
ined
con
cen
trat
ion
ofse
lect
ion
agen
t.
M. A. Reis et al. / Bioorg. Med. Chem. xxx (2014) xxx–xxx 3
Please cite this article in press as: Reis, M. A.; et al. Bioorg. Med. Chem. (2
The significant statistical differences were explored by compar-ing the antiproliferative activity on the pairs of parental anddrug-resistant cell lines. Through this approach, significant statis-tical differences were only found between the parental cell lineEPG85-257 and EPG85-257RD (p value = 0.001) and betweenEPG85-257RD and EPG85-257RN (p value = 0.017). The observeddifferences are due to the lower IC50 values found onEPG85-257RD cell line (Table 2). Jolkinol D (1) and all derivatives(except 6), were highly effective towards this cell line, exhibitingan antiproliferative activity comparable to the positive controlscisplatin (IC50 = 4.0 ± 0.3 lM; RR = 1) and etoposide (IC50 = 6.2 ±0.3 lM; RR = 59). These results suggest that compounds 1 andderivatives were more selective towards the gastric cancer cellline, in which the MDR phenotype is characterized by P-gp over-expression. This fact is also supported by RR values (Fig. 1), sincejolkinol D (1) and all derivatives (2–11) exhibited a collateralsensitivity effect on EPG85-257RD cell line. For the other MDRcounterpart EPG85-257RN, only substituted aroyl derivativesjolkinoate L (8) (IC50 = 3.58 ± 1.93 lM; RR = 0.39) and jolkinoateK (9) (IC50 = 9.47 ± 3.04 lM; RR = 0.42) displayed both highantiproliferative activity and collateral sensitivity effect(Table 2, Fig. 1). The other derivatives displayed an ‘indifferenteffect’ (2< RR >0.5); nevertheless compound 7 (IC50 = 5.40 ± 1.50lM; RR = 0.54) displayed an antiproliferative activity similar tocompounds 8 and 9 and comparable to cisplatin (IC50 =2.6 ± 0.2 lM).
For the pancreatic cancer cell lines no significant statisticaldifferences were detected between their IC50 (p values >0.05).This was verified because the compounds presented a similarantiproliferative profile for both parental and resistant lines,independently from their MDR phenotype (Table 3). This is in linewith the RR values found for EPP85-181RD and EPP85-181RN(Fig. 1), in which, all the compounds (except 1 and 2) presentedan ‘insensitive’ effect (2< RR >0.5). This effect can be regardedas lack of resistance mechanisms in these cells towards thetested compounds. A suitable example is jolkinoate L (8) thatexhibited the highest antiproliferative activity on pancreaticcancer cell lines: 17.76 ± 5.65 lM (EPP85-181P); 10.11 ± 6.65 lM(EPP85-181RN; RR = 0.57) and 11.34 ± 4.07 lM (EPP85-181RD;RR = 0.64). It should be noted that, besides the lower IC50 foundfor etoposide and cisplatin, both agents present cross-resistancein the MDR cell lines (Table 3).
Regarding colon cancer cell lines, no significant statistical dif-ferences could be found for the antiproliferative activity of thecompounds 1–11 (p values >0.05). However, there seems to bea collateral sensitivity effect towards the MDR phenotype charac-terized by an altered topoisomerase II expression (HT-29RN),since all the compounds (except 1, 2 and 6) presented a RR<0.5 (Fig. 1). Nevertheless, in overall the antiproliferative activityof the compounds in these cell lines was considered to bemoderate (Table 4). Compound 8 stands out from this set,as it showed to have an effective antiproliferative activity onHT-29RN (IC50 = 3.82 ± 1.08 lM; RR = 0.41) and on HT-29RD(IC50 = 9.67 ± 2.77 lM; RR = 1.04).
Direct structure activity relationships between this set ofcompounds (1–11) and antiproliferative activity was not clear,conversely to what was found for P-gp modulation. As mentionabove, the aroyl derivatives (7–10) were particularly able tomodulate P-gp in comparison with the remaining compounds.17
On a recent work,17 the lathyranes (1–11) were evaluated asP-glycoprotein modulators (at non-cytotoxic doses) on L5178mouse T lymphoma cells transfected with the human MDR1 geneand on the parental ones. These cells are a good model for thispurpose, because no other efflux pump is being expressed. P-gpmodulation was measured through the rhodamine-123 exclusionassay. This fluorescent dye is a substrate of P-gp and the extent of
014), http://dx.doi.org/10.1016/j.bmc.2014.05.006
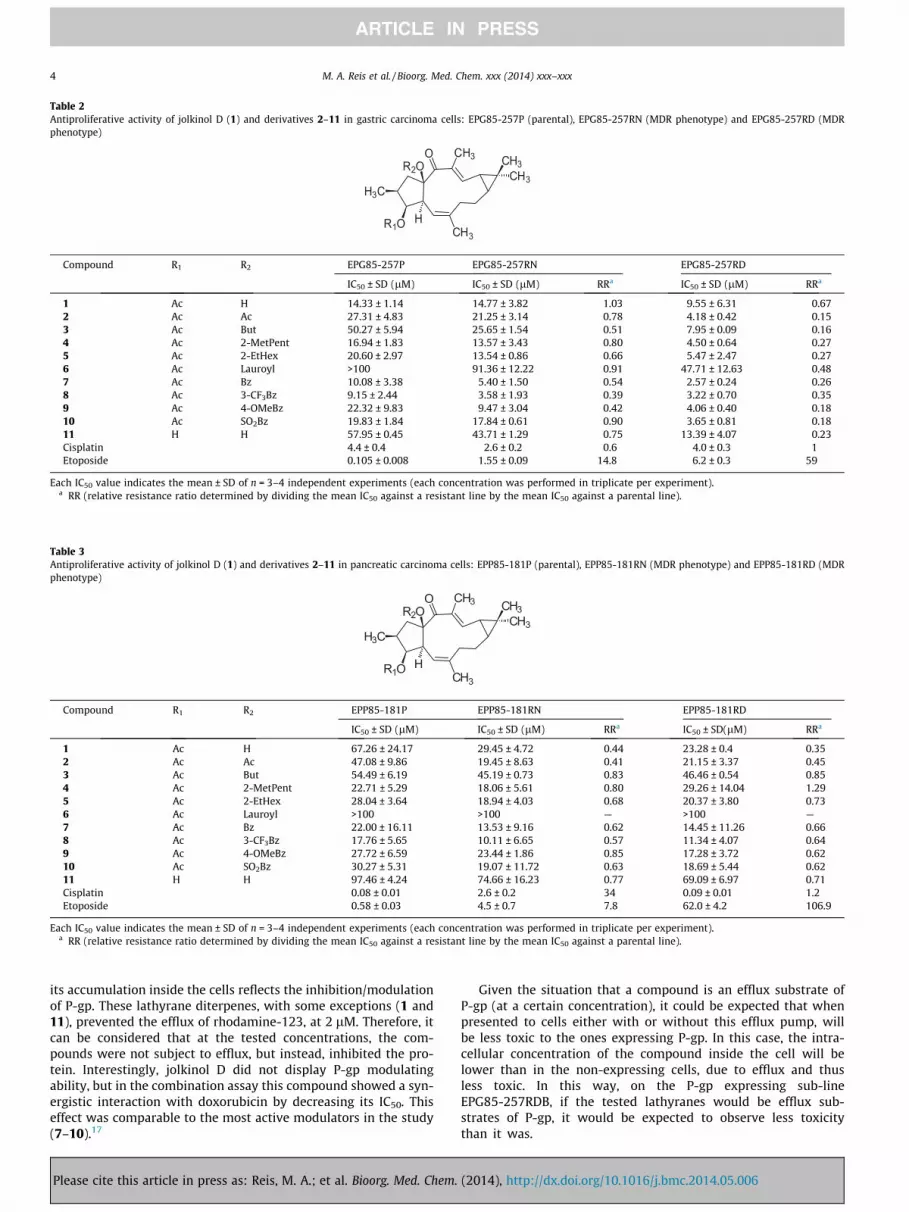
Table 2Antiproliferative activity of jolkinol D (1) and derivatives 2–11 in gastric carcinoma cells: EPG85-257P (parental), EPG85-257RN (MDR phenotype) and EPG85-257RD (MDRphenotype)
OR2O
CH3
H3C
R1O HCH3
CH3CH3
Compound R1 R2 EPG85-257P EPG85-257RN EPG85-257RD
IC50 ± SD (lM) IC50 ± SD (lM) RRa IC50 ± SD (lM) RRa
1 Ac H 14.33 ± 1.14 14.77 ± 3.82 1.03 9.55 ± 6.31 0.672 Ac Ac 27.31 ± 4.83 21.25 ± 3.14 0.78 4.18 ± 0.42 0.153 Ac But 50.27 ± 5.94 25.65 ± 1.54 0.51 7.95 ± 0.09 0.164 Ac 2-MetPent 16.94 ± 1.83 13.57 ± 3.43 0.80 4.50 ± 0.64 0.275 Ac 2-EtHex 20.60 ± 2.97 13.54 ± 0.86 0.66 5.47 ± 2.47 0.276 Ac Lauroyl >100 91.36 ± 12.22 0.91 47.71 ± 12.63 0.487 Ac Bz 10.08 ± 3.38 5.40 ± 1.50 0.54 2.57 ± 0.24 0.268 Ac 3-CF3Bz 9.15 ± 2.44 3.58 ± 1.93 0.39 3.22 ± 0.70 0.359 Ac 4-OMeBz 22.32 ± 9.83 9.47 ± 3.04 0.42 4.06 ± 0.40 0.1810 Ac SO2Bz 19.83 ± 1.84 17.84 ± 0.61 0.90 3.65 ± 0.81 0.1811 H H 57.95 ± 0.45 43.71 ± 1.29 0.75 13.39 ± 4.07 0.23Cisplatin 4.4 ± 0.4 2.6 ± 0.2 0.6 4.0 ± 0.3 1Etoposide 0.105 ± 0.008 1.55 ± 0.09 14.8 6.2 ± 0.3 59
Each IC50 value indicates the mean ± SD of n = 3–4 independent experiments (each concentration was performed in triplicate per experiment).a RR (relative resistance ratio determined by dividing the mean IC50 against a resistant line by the mean IC50 against a parental line).
Table 3Antiproliferative activity of jolkinol D (1) and derivatives 2–11 in pancreatic carcinoma cells: EPP85-181P (parental), EPP85-181RN (MDR phenotype) and EPP85-181RD (MDRphenotype)
OR2O
CH3
H3C
R1O HCH3
CH3CH3
Compound R1 R2 EPP85-181P EPP85-181RN EPP85-181RD
IC50 ± SD (lM) IC50 ± SD (lM) RRa IC50 ± SD(lM) RRa
1 Ac H 67.26 ± 24.17 29.45 ± 4.72 0.44 23.28 ± 0.4 0.352 Ac Ac 47.08 ± 9.86 19.45 ± 8.63 0.41 21.15 ± 3.37 0.453 Ac But 54.49 ± 6.19 45.19 ± 0.73 0.83 46.46 ± 0.54 0.854 Ac 2-MetPent 22.71 ± 5.29 18.06 ± 5.61 0.80 29.26 ± 14.04 1.295 Ac 2-EtHex 28.04 ± 3.64 18.94 ± 4.03 0.68 20.37 ± 3.80 0.736 Ac Lauroyl >100 >100 — >100 —7 Ac Bz 22.00 ± 16.11 13.53 ± 9.16 0.62 14.45 ± 11.26 0.668 Ac 3-CF3Bz 17.76 ± 5.65 10.11 ± 6.65 0.57 11.34 ± 4.07 0.649 Ac 4-OMeBz 27.72 ± 6.59 23.44 ± 1.86 0.85 17.28 ± 3.72 0.6210 Ac SO2Bz 30.27 ± 5.31 19.07 ± 11.72 0.63 18.69 ± 5.44 0.6211 H H 97.46 ± 4.24 74.66 ± 16.23 0.77 69.09 ± 6.97 0.71Cisplatin 0.08 ± 0.01 2.6 ± 0.2 34 0.09 ± 0.01 1.2Etoposide 0.58 ± 0.03 4.5 ± 0.7 7.8 62.0 ± 4.2 106.9
Each IC50 value indicates the mean ± SD of n = 3–4 independent experiments (each concentration was performed in triplicate per experiment).a RR (relative resistance ratio determined by dividing the mean IC50 against a resistant line by the mean IC50 against a parental line).
4 M. A. Reis et al. / Bioorg. Med. Chem. xxx (2014) xxx–xxx
its accumulation inside the cells reflects the inhibition/modulationof P-gp. These lathyrane diterpenes, with some exceptions (1 and11), prevented the efflux of rhodamine-123, at 2 lM. Therefore, itcan be considered that at the tested concentrations, the com-pounds were not subject to efflux, but instead, inhibited the pro-tein. Interestingly, jolkinol D did not display P-gp modulatingability, but in the combination assay this compound showed a syn-ergistic interaction with doxorubicin by decreasing its IC50. Thiseffect was comparable to the most active modulators in the study(7–10).17
Please cite this article in press as: Reis, M. A.; et al. Bioorg. Med. Chem.
Given the situation that a compound is an efflux substrate ofP-gp (at a certain concentration), it could be expected that whenpresented to cells either with or without this efflux pump, willbe less toxic to the ones expressing P-gp. In this case, the intra-cellular concentration of the compound inside the cell will belower than in the non-expressing cells, due to efflux and thusless toxic. In this way, on the P-gp expressing sub-lineEPG85-257RDB, if the tested lathyranes would be efflux sub-strates of P-gp, it would be expected to observe less toxicitythan it was.
(2014), http://dx.doi.org/10.1016/j.bmc.2014.05.006
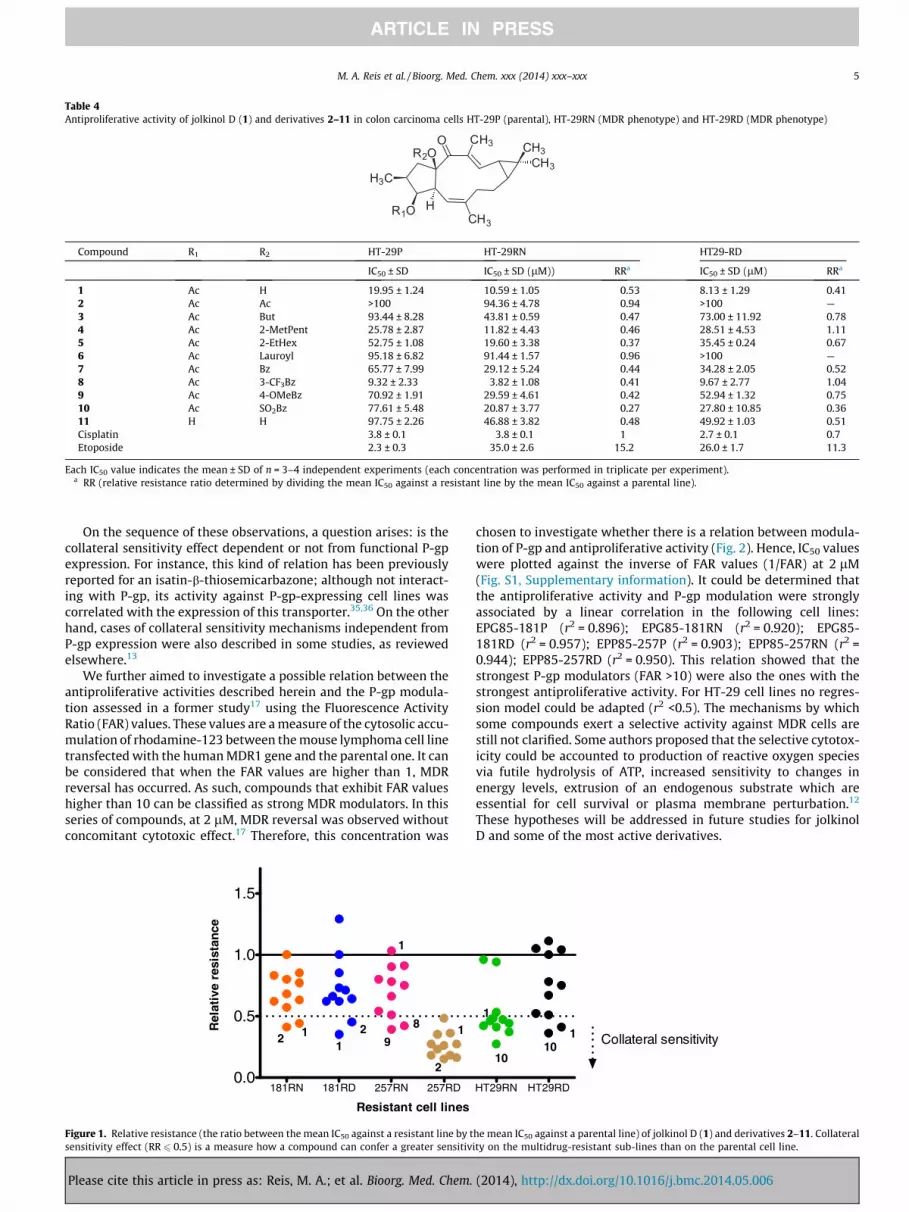
Table 4Antiproliferative activity of jolkinol D (1) and derivatives 2–11 in colon carcinoma cells HT-29P (parental), HT-29RN (MDR phenotype) and HT-29RD (MDR phenotype)
OR2O
CH3
H3C
R1O HCH3
CH3CH3
Compound R1 R2 HT-29P HT-29RN HT29-RD
IC50 ± SD IC50 ± SD (lM)) RRa IC50 ± SD (lM) RRa
1 Ac H 19.95 ± 1.24 10.59 ± 1.05 0.53 8.13 ± 1.29 0.412 Ac Ac >100 94.36 ± 4.78 0.94 >100 —3 Ac But 93.44 ± 8.28 43.81 ± 0.59 0.47 73.00 ± 11.92 0.784 Ac 2-MetPent 25.78 ± 2.87 11.82 ± 4.43 0.46 28.51 ± 4.53 1.115 Ac 2-EtHex 52.75 ± 1.08 19.60 ± 3.38 0.37 35.45 ± 0.24 0.676 Ac Lauroyl 95.18 ± 6.82 91.44 ± 1.57 0.96 >100 —7 Ac Bz 65.77 ± 7.99 29.12 ± 5.24 0.44 34.28 ± 2.05 0.528 Ac 3-CF3Bz 9.32 ± 2.33 3.82 ± 1.08 0.41 9.67 ± 2.77 1.049 Ac 4-OMeBz 70.92 ± 1.91 29.59 ± 4.61 0.42 52.94 ± 1.32 0.7510 Ac SO2Bz 77.61 ± 5.48 20.87 ± 3.77 0.27 27.80 ± 10.85 0.3611 H H 97.75 ± 2.26 46.88 ± 3.82 0.48 49.92 ± 1.03 0.51Cisplatin 3.8 ± 0.1 3.8 ± 0.1 1 2.7 ± 0.1 0.7Etoposide 2.3 ± 0.3 35.0 ± 2.6 15.2 26.0 ± 1.7 11.3
Each IC50 value indicates the mean ± SD of n = 3–4 independent experiments (each concentration was performed in triplicate per experiment).a RR (relative resistance ratio determined by dividing the mean IC50 against a resistant line by the mean IC50 against a parental line).
M. A. Reis et al. / Bioorg. Med. Chem. xxx (2014) xxx–xxx 5
On the sequence of these observations, a question arises: is thecollateral sensitivity effect dependent or not from functional P-gpexpression. For instance, this kind of relation has been previouslyreported for an isatin-b-thiosemicarbazone; although not interact-ing with P-gp, its activity against P-gp-expressing cell lines wascorrelated with the expression of this transporter.35,36 On the otherhand, cases of collateral sensitivity mechanisms independent fromP-gp expression were also described in some studies, as reviewedelsewhere.13
We further aimed to investigate a possible relation between theantiproliferative activities described herein and the P-gp modula-tion assessed in a former study17 using the Fluorescence ActivityRatio (FAR) values. These values are a measure of the cytosolic accu-mulation of rhodamine-123 between the mouse lymphoma cell linetransfected with the human MDR1 gene and the parental one. It canbe considered that when the FAR values are higher than 1, MDRreversal has occurred. As such, compounds that exhibit FAR valueshigher than 10 can be classified as strong MDR modulators. In thisseries of compounds, at 2 lM, MDR reversal was observed withoutconcomitant cytotoxic effect.17 Therefore, this concentration was
181RN 181RD 257RN 257RD0.0
0.5
1.0
1.5
2 1 21 9
8
1
1
2
Resistant cell lines
Rel
ativ
e re
sist
ance
Figure 1. Relative resistance (the ratio between the mean IC50 against a resistant line by tsensitivity effect (RR 6 0.5) is a measure how a compound can confer a greater sensitiv
Please cite this article in press as: Reis, M. A.; et al. Bioorg. Med. Chem.
chosen to investigate whether there is a relation between modula-tion of P-gp and antiproliferative activity (Fig. 2). Hence, IC50 valueswere plotted against the inverse of FAR values (1/FAR) at 2 lM(Fig. S1, Supplementary information). It could be determined thatthe antiproliferative activity and P-gp modulation were stronglyassociated by a linear correlation in the following cell lines:EPG85-181P (r2 = 0.896); EPG85-181RN (r2 = 0.920); EPG85-181RD (r2 = 0.957); EPP85-257P (r2 = 0.903); EPP85-257RN (r2 =0.944); EPP85-257RD (r2 = 0.950). This relation showed that thestrongest P-gp modulators (FAR >10) were also the ones with thestrongest antiproliferative activity. For HT-29 cell lines no regres-sion model could be adapted (r2 <0.5). The mechanisms by whichsome compounds exert a selective activity against MDR cells arestill not clarified. Some authors proposed that the selective cytotox-icity could be accounted to production of reactive oxygen speciesvia futile hydrolysis of ATP, increased sensitivity to changes inenergy levels, extrusion of an endogenous substrate which areessential for cell survival or plasma membrane perturbation.12
These hypotheses will be addressed in future studies for jolkinolD and some of the most active derivatives.
HT29RN HT29RD
1010
1
1
Collateral sensitivity
he mean IC50 against a parental line) of jolkinol D (1) and derivatives 2–11. Collaterality on the multidrug-resistant sub-lines than on the parental cell line.
(2014), http://dx.doi.org/10.1016/j.bmc.2014.05.006
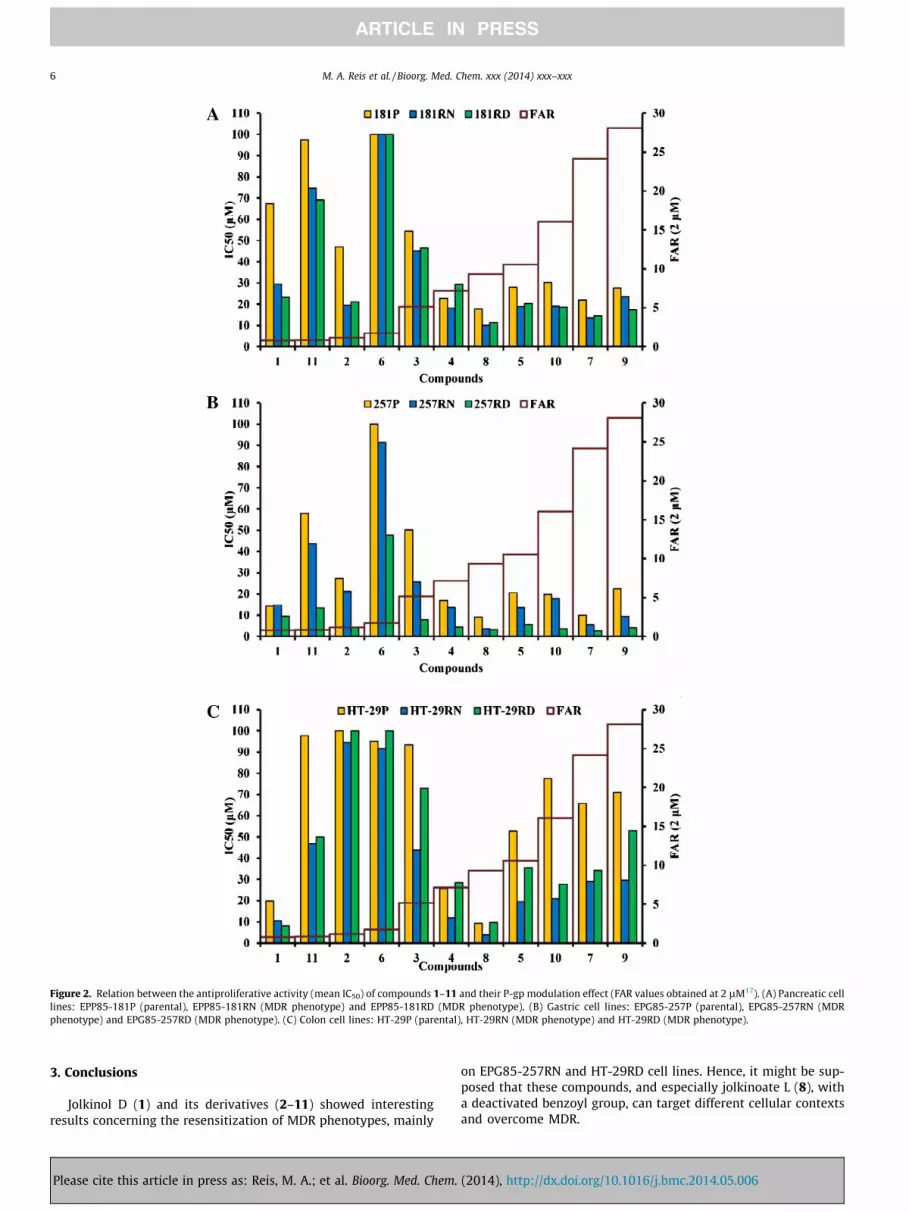
Figure 2. Relation between the antiproliferative activity (mean IC50) of compounds 1–11 and their P-gp modulation effect (FAR values obtained at 2 lM17). (A) Pancreatic celllines: EPP85-181P (parental), EPP85-181RN (MDR phenotype) and EPP85-181RD (MDR phenotype). (B) Gastric cell lines: EPG85-257P (parental), EPG85-257RN (MDRphenotype) and EPG85-257RD (MDR phenotype). (C) Colon cell lines: HT-29P (parental), HT-29RN (MDR phenotype) and HT-29RD (MDR phenotype).
6 M. A. Reis et al. / Bioorg. Med. Chem. xxx (2014) xxx–xxx
3. Conclusions
Jolkinol D (1) and its derivatives (2–11) showed interestingresults concerning the resensitization of MDR phenotypes, mainly
Please cite this article in press as: Reis, M. A.; et al. Bioorg. Med. Chem.
on EPG85-257RN and HT-29RD cell lines. Hence, it might be sup-posed that these compounds, and especially jolkinoate L (8), witha deactivated benzoyl group, can target different cellular contextsand overcome MDR.
(2014), http://dx.doi.org/10.1016/j.bmc.2014.05.006

M. A. Reis et al. / Bioorg. Med. Chem. xxx (2014) xxx–xxx 7
4. Experimental section
4.1. Compounds tested
Compound 1 was previously isolated in a large amount from theplant Euphorbia piscatoria;17 its derivatization gave rise to thederivatives 2–11.17
4.2. Cell lines, cell culture and cell proliferation assay
The establishment and characterization of the Human carci-noma cell lines and drug-resistant sublines have been describedpreviously (Table 1). The EPP85-181 (pancreatic) and EPG85-257(gastric) were grown in Leibovitz L-15 medium (Biowhittak-er,Walkersville,MD). HT-29 (colon) was grown in DMEM medium(Biowhittaker,Walkersville,MD). All were supplemented with10% fetal calf serum (FCS) (GIBCO/BRL,GrandIsland,NY), 1 mML-glutamine, 6.25 mg/L fetuin, 80 IE/L insulin, 2.5 mg/mL, transfer-rin, 0.5 g/L glucose, 1.1 g/L NaHCO3, 1% minimal essential vitaminsand 20,000 kIE/L trasylol. The cultures were maintained in ahumidified atmosphere of 5% CO2 at 37 �C. Drug-resistant cell lineswere established from parental cell lines by continuous exposureof the cells to step wise increasing concentrations of antineoplasticagents. For maintenance of drug-resistant phenotypes, the mediumof drug-resistant sublines was supplemented respectively with theselection agent mitoxantrone and daunorubicin (Table 1).
The antiproliferative assay was based on sulforhodamine B(SRB) staining. All compounds were dissolved in DMSO. Briefly,5 � 103/mL (EPG85-257P and EPP85-181P) and 7.5 � 103/mL(EPG85-257RN, EPG85-257RD, EPP85-181RN, EPP85-181RD, HT-29P, HT-29RN and HT-29RD) were seeded in 96-well plates in trip-licates. After 48 h attachment, a particular compound was added indilution series for 5 days incubation. Cells were fixed with 10% coldtrichloroacetic acid for 1 h at 4 �C and were washed five times withtap water. Staining was performed with 0.4% SRB in 1% acetic acidfor 10 min at room temperature. Then the plates were washed with1% acetic acid, dried overnight and resolubilized in 20 mM Tris–HCl(pH = 10) for one hour at room temperature. Cell growth wasmeasured at 562 nm against the reference wavelength of 690 nm.Mean IC50 values were obtained by best fitting the dose-dependentinhibition curves in GraphPadPrism5 program, from three to fourindependent experiments in triplicate for each cell line. Relativeresistance (RR) values were determined as:
RR ¼Mean IC50 MDR resistant cell lineMean IC50 parental cell line
4.3. Statistical analysis
The non-parametric Kruskal–Wallis rank test (probability valuep <0.05 was considered statistically significant) and regressionanalysis were performed using SPSS 16.0 statistical package.
Acknowledgments
This study was financially supported by Fundação para a Ciên-cia e a Tecnologia (FCT), Portugal (Projects: PTDC/QUI-QUI/099815/
Please cite this article in press as: Reis, M. A.; et al. Bioorg. Med. Chem.
2008; Pest-OE/SAU/UI4013/2011; PTDC/QEQ-MED/0905/2012.PhD Grant SFRH/BD/72915/2010).
Supplementary data
Supplementary data associated with this article can be found, inthe online version, at http://dx.doi.org/10.1016/j.bmc.2014.05.006.
References and notes
1. Gottesman, M. M.; Fojo, T.; Bates, S. E. Nat. Rev. Cancer 2002, 2, 48.2. Lehnert, M. Eur. J. Cancer 1996, 32A, 912.3. Baird, R. Eur. J. Cancer 2003, 39, 2450.4. Lage, H. Cell. Mol. Life Sci. 2008, 65, 3145.5. Baguley, B. C. Mol. Biotechnol. 2010, 46, 308.6. Longley, D. B.; Johnston, P. G. J. Pathol. 2005, 205, 275.7. O’Connor, R. Curr. Cancer Drug Targets 2009, 9, 273.8. Sarkadi, B.; Homolya, L.; Szakács, G.; Váradi, A. Physiol. Rev. 2006, 86, 1179.9. Szakács, G.; Paterson, J. K.; Ludwig, J. A.; Booth-Genthe, C.; Gottesman, M. M.
Nat. Rev. Drug Discovery 2006, 5, 219.10. Tsuruo, T.; Naito, M.; Tomida, A.; Fujita, N.; Mashima, T.; Sakamoto, H.; Haga,
N. Cancer Sci. 2003, 94, 15.11. Shukla, S.; Ohnuma, S.; Ambudkar, S. V. Curr. Drug Targets 2011, 12, 621.12. Pluchino, K. M.; Hall, M. D.; Goldsborough, A. S.; Callaghan, R.; Gottesman, M.
M. Drug Resist. Updat. 2012, 15, 98.13. Hall, M. D.; Handley, M. D.; Gottesman, M. M. Trends Pharmacol. Sci. 2009, 30,
546.14. Lage, H.; Duarte, N.; Coburger, C.; Hilgeroth; Ferreira, M. J. U. Phytomedicine
2010, 17, 441.15. Duarte, N.; Gyémánt, N.; Abreu, P. M.; Molnár, J.; Ferreira, M. J. U. Planta Med.
2006, 72, 162.16. Duarte, N.; Varga, A.; Cherepnev, G.; Radics, R.; Molnár, J.; Ferreira, M. J. U.
Bioorg. Med. Chem. 2007, 15, 546.17. Reis, M.; Ferreira, R. J.; Santos, M. M. M.; dos Santos, D. J. V.; Molnár, J.; Ferreira,
M. J. U. J. Med. Chem. 2013, 56, 748.18. Dietel, M.; Arps, H.; Lage, H.; Niendorf, A. Cancer Res. 1990, 6100.19. Ross, D. D.; Yang, W.; Abruzzo, L. V.; Dalton, W. S.; Schneider, E.; Lage, H.;
Dietel, M.; Greenberger, L.; Cole, S. P. C.; Doyle, L. A. J. Natl. Cancer I 1990, 429.20. Lage, H.; Dietel, M. Gene 1997, 188, 151.21. Wichert, A.; Stege, A.; Midorikawa, Y.; Holm, P. S.; Lage, H. Oncogene 2004, 23,
945.22. Kellner, U.; Hutchinson, L.; Seidel, A.; Lage, H.; Danks, M. K.; Dietel, M.;
Kaufmann, S. H. Int. J. Cancer 1997, 71, 817.23. Lage, H.; Perlitz, C.; Abele, R.; Tampé, R.; Dietel, M.; Schadendorf, D.; Sinha, P.
FEBS Lett. 2001, 503, 179.24. Lage, H.; Jordan, A.; Scholz, R.; Dietel, M. Int. J. Hyperthermia 2000, 16, 291.25. Lage, H.; Dietel, M. J. Cancer Res. Clin. Oncol. 2002, 128, 349.26. Sinha, P.; Dietel, M.; Schadendorf, D.; Lage, H.; Kooperations-, K.; Mannheim, H.
Electrophoresis 1990, 20, 2961.27. Jiao, W.; Dong, W.; Li, Z.; Deng, M.; Lu, R. Bioorg. Med. Chem. 2009, 17, 4786.28. Ferreira, R. J.; dos Santos, D. J. V.; Ferreira, M. J. U.; Guedes, R. C. J. Chem. Inf.
Model. 2011, 51, 1315.29. Sousa, I. J.; Ferreira, M. J. U.; Molnár, J.; Fernandes, M. X. Eur. J. Pharm. Sci. 2012,
48, 542.30. Borska, S.; Sopel, M.; Chmielewska, M.; Zabel, M.; Dziegiel, P. Molecules 2010,
15, 857.31. Coburger, C.; Lage, H.; Molnár, J.; Langner, A.; Hilgeroth, A. J. Pharm. Pharmacol.
2010, 62, 1704.32. Drag, M.; Surowiak, P.; Drag-Zalesinska, M.; Dietel, M.; Lage, H.; Oleksyszyn, J.
Molecules 2009, 14, 1639.33. Duarte, N.; Lage, H.; Abrantes, M.; Ferreira, M. J. U. Planta Med. 2010, 76, 975.34. Hilgeroth, A.; Baumert, C.; Coburger, C.; Seifert, M.; Krawczyk, S.; Hempel, C.;
Neubauer, F.; Krug, M.; Molnár, J.; Lage, H. Med. Chem. 2013, 9, 487.35. Hall, M. D.; Brimacombe, K. R.; Varonka, M. S.; Pluchino, K. M.; Monda, J. K.; Li,
J.; Walsh, M. J.; Boxer, M. B.; Warren, T. H.; Fales, H. M.; Gottesman, M. M. J.Med. Chem. 2011, 5878.
36. Ludwig, J.; Szakács, G.; Martin, S. E.; Chu, B. F.; Cardarelli, C.; Sauna, Z. E.;Caplen, N. J.; Fales, H. M.; Ambudkar, S. V.; Weinstein, J. N.; Gottesman, M. M.Cancer Res. 2006, 66, 4808.
(2014), http://dx.doi.org/10.1016/j.bmc.2014.05.006





























