Embed Size (px)
Citation preview

ARTICLEdoi:10.1016/j.ymthe.2005.04.002
Long-Term Expression of Erythropoietin fromMyoblasts Immobilized in Biocompatible and
Neovascularized Microcapsules
G. Orive,1 M. De Castro,1 S. Ponce,1 R. M. Hernandez,1 A. R. Gascon,1 M. Bosch,2
J. Alberch,2 and Jose L. Pedraz1,*
1Laboratory of Pharmacy and Pharmaceutical Technology, Faculty of Pharmacy, University of the Basque Country, Vitoria-Gasteiz, Spain2Dept. Biologia Cellular i Anatomia Patologica, Facultat de Medicina, IDIBAPS, Universitat de Barcelona
*To whom correspondence and reprint requests should be addressed. Fax: +34 945 013040. E-mail: [email protected].
Available online 1 June 2005
MOLECULA
Copyright C
1525-0016/$
The present paper investigates the long-term functionality of an ex vivo gene therapy approachbased on cell microencapsulation for the continuous delivery of erythropoietin (EPO) withoutimplementation of immunosuppressive protocols. Polymer microcapsules (0.5 ml) loaded with EPO-secreting C2C12 myoblasts and releasing 15,490 F 600 IU EPO/24 h were implanted in theperitoneum and subcutaneous tissue of syngeneic and allogeneic mice. High and constanthematocrit levels were maintained for more than 100 days in all implanted mice. Capsules retrievedfrom the peritoneum were free-floating or forming small capsule clusters, and we detected only aweak fibroblast outgrowth in capsules adhered to organs, whereas capsules explanted from thesubcutaneous region appeared altogether as a richly vascularized structure with no signs of majorhost reaction. Interestingly, the functionality of capsules implanted in the allogeneic mice persisteduntil day 210 after implantation. These results highlight the feasibility of cell encapsulationtechnology for the long-term delivery of EPO independent of the method of administration and themouse strain.
R
Th
30
Key Words: cell encapsulation, alginate, cell therapy, erythropoietin, myoblasts,protein delivery, biocompatibility
INTRODUCTION
Somatic gene therapy is being considered in the treat-ment of a number of acquired chronic diseases. Oneinteresting approach is cell encapsulation, in whichengineered somatic cells are protected against immunecell- and antibody-mediated rejection by their immobili-zation in a polymer matrix surrounded by a semiperme-able membrane. The latter regulates the bidirectionaldiffusion of nutrients, oxygen, and waste, allowing thecontrolled and continuous delivery of therapeutic pro-teins in the absence of immunosuppression [1–3]. Inaddition to reducing sharply the frequency of admin-istration and thus improving patient comfort, cell encap-sulation strategy would improve the pharmacokinetics ofeasily degradable peptides and proteins, which oftenhave short half-lives in vivo [4].
In the past few years, much effort has been focused onstudying the biocompatibility of materials and capsules[5,6], designing new immunoisolation devices [7,8], or just
THERAPY Vol. 12, No. 2, August 2005
e American Society of Gene Therapy
.00
demonstrating the proof of principle of this cell-basedtechnology [9–11]. We have reported previously that care-ful selection and evaluation of purified alginates and celllines and fabrication of small and uniform microcapsulesare key requirements to ensure an optimal biocompatibil-ity, long-termfunctionality, andsuitablezero-orderkineticrelease of the therapeutic molecules [12–14]. However,little research has involved the study of parameters such asthe implantation site of the encapsulated cells, the feasi-bility of using the same approach for syngeneic or alloge-neic transplantation, or the application of a well-vascularized immobilization device to permit close contactbetween the encapsulated cells and the bloodstream andthus improve the long-term efficacy of the graft.
With the aim of addressing some of these issues, wehave immobilized genetically engineered C2C12 myoblastcells secreting erythropoietin (EPO) within optimizedpolymer microcapsules and we have tested their feasi-bility in two different murine models (C3H and Balb/c
283

FIG. 1. Photomicrographs showing capsules containing EPO-secreting C2C1
myoblasts before implantation in mice. Scale bar, 250 Am.
ARTICLE doi:10.1016/j.ymthe.2005.04.002
mice) after intraperitoneal and subcutaneous implanta-tion. EPO has been selected as a candidate molecule inpart because it is easy to monitor its expression andbioactivity in vivo by following the hematocrit level.Since EPO is widely used in the treatment of anemicpatients, the technology of cell encapsulation wouldavoid the repeated weekly injections currently practiced.In addition, EPO acts as an angiogenic factor enhancingmatrix metalloproteinase 2 production, Janus kinase-2phosphorylation, and endothelial cell proliferation [15].Thus, we hypothesized that in addition to its erythro-poietic effect, EPO will induce the formation of a vascularnetwork surrounding the microcapsule graft, facilitatingthe access of encapsulated cells to the blood supply andconsequently to oxygen and nutrients.
The present study investigates the long-term function-ality, local angiogenesis induction, and biocompatibilityof an EPO continuous drug delivery system based onmicroencapsulated C2C12 cells. To test the feasibility ofthe approach, we have placed the graft in the absence ofimmunosuppression both in the peritoneum and insubcutaneous tissue of immunocompetent syngeneicand allogeneic animal models.
RESULTS
Microencapsulation of C2C12 Myoblasts and EpoExpression from the Immobilized CellsWe examined the in vitro EPO production by a murineEPO-secreting C2C12 cell line. We employed an ELISAspecific for detection of human EPO to measure murineEPO as has been previously reported by others [17,18].The clone selected for immobilization released 46,264 F1451 IU EPO/106 cells/24 h. Afterward, we fabricatedalginate–poly-l-lysine (PLL)–alginate microcapsulesloaded with 2 � 106 C2C12 cells/ml using purifiedalginates and an electrostatic droplet generator, whichenabled the production of uniform devices with a verynarrow size dispersion (485 F 15 Am) (Fig. 1).
We measured EPO secretion from 100 cell-loadedmicrocapsules at 93 F 34 IU EPO/24 h. Assuming theprevious data, we estimated that implantation of 0.5 ml ofcell-loaded microcapsules (approximately 106 immobi-lized cells) might result in a significant and constantincrease in mouse hematocrit level over time. The rate ofEPO release of the 0.5 ml of cell-loaded microcapsulesbefore implantation was 15,490 F 600 IU EPO/24 h.
Long-Term Hematocrit of C3H Mice ImplantedIntraperitoneally and Subcutaneously withEPO-Secreting Encapsulated CellsWe implanted 0.5 ml of polymer microcapsules loadedwith EPO-releasing C2C12 myoblasts (2 � 106 cells/mlalginate) in syngeneic C3H recipients both in theperitoneum (n = 5) and in the subcutaneous tissue (n =5). The hematocrit level of implanted recipients was
MOLECULAR THERAPY Vol. 12, No. 2, August 2005284Copyright C The American Society of Gene Therap
2
significantly increased compared to that of the controlmice (Fig. 2) (**P b 0.001; *P b 0.0005). In the case ofmice receiving capsules subcutaneously, plasma EPOhematocrit increased from a basal 52.5% before implan-tation to 81.3%, as early as 14 days postimplantation,peaking at 28 days (87.1%) and maintaining a constanthematocrit level until day 100 (83%) (Fig. 2A). Weobserved a similar behavior in mice implanted intra-peritoneally. In fact, hematocrit levels rose until day 21(83.8%) and then they maintained an asymptotic leveluntil day 100 (85%) (Fig. 2B). Control mice showed aconstant baseline during the study.
At explantation, the majority of the capsules orcapsule aggregates secreted detectable amounts of EPO(data not shown), confirming the long-term viability andfunctionality of the enclosed cells. However, we observedsome interesting differences between capsules placedintraperitoneally and those placed subcutaneously. Theformer appeared as small capsule clusters together withfree capsules, some of which were partially deformed(Figs. 3A and B), while the latter were altogether formingan irregular and neovascularized structure (Fig. 3C). Inaddition, capsules implanted in the subcutaneous tissueremained intact within the structure fully surrounded bya vascular network, which will provide the necessary
y
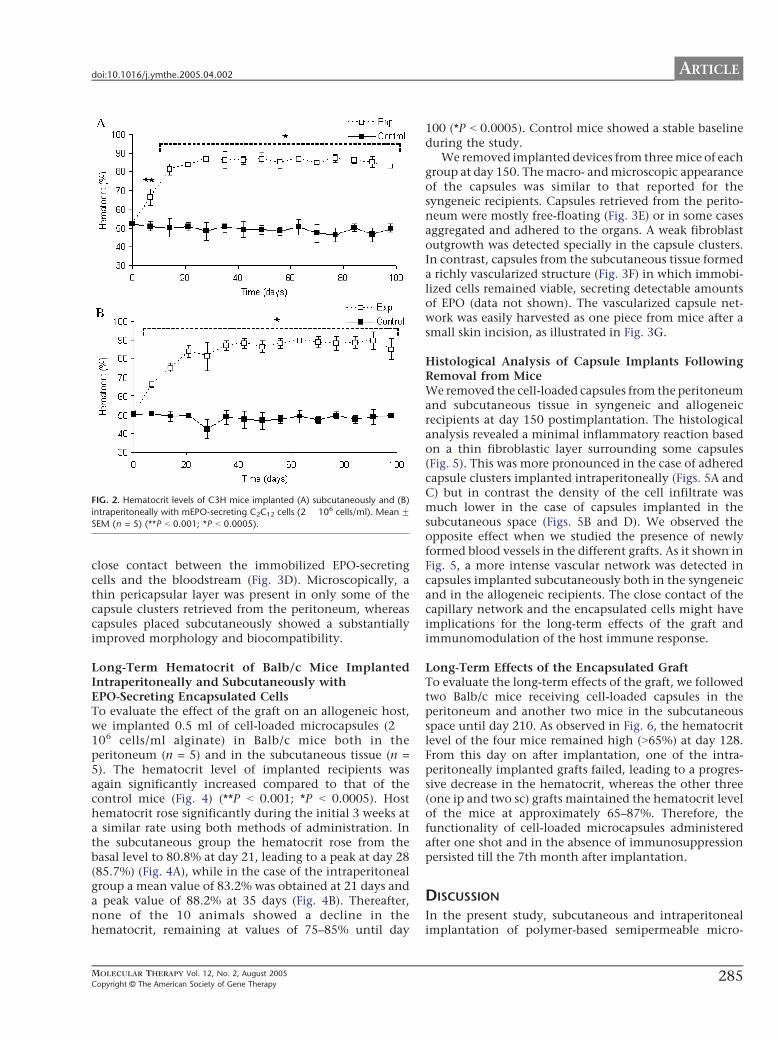
FIG. 2. Hematocrit levels of C3H mice implanted (A) subcutaneously and (B)
intraperitoneally with mEPO-secreting C2C12 cells (2 � 106 cells/ml). Mean FSEM (n = 5) (**P b 0.001; *P b 0.0005).
ARTICLEdoi:10.1016/j.ymthe.2005.04.002
close contact between the immobilized EPO-secretingcells and the bloodstream (Fig. 3D). Microscopically, athin pericapsular layer was present in only some of thecapsule clusters retrieved from the peritoneum, whereascapsules placed subcutaneously showed a substantiallyimproved morphology and biocompatibility.
Long-Term Hematocrit of Balb/c Mice ImplantedIntraperitoneally and Subcutaneously withEPO-Secreting Encapsulated CellsTo evaluate the effect of the graft on an allogeneic host,we implanted 0.5 ml of cell-loaded microcapsules (2 �106 cells/ml alginate) in Balb/c mice both in theperitoneum (n = 5) and in the subcutaneous tissue (n =5). The hematocrit level of implanted recipients wasagain significantly increased compared to that of thecontrol mice (Fig. 4) (**P b 0.001; *P b 0.0005). Hosthematocrit rose significantly during the initial 3 weeks ata similar rate using both methods of administration. Inthe subcutaneous group the hematocrit rose from thebasal level to 80.8% at day 21, leading to a peak at day 28(85.7%) (Fig. 4A), while in the case of the intraperitonealgroup a mean value of 83.2% was obtained at 21 days anda peak value of 88.2% at 35 days (Fig. 4B). Thereafter,none of the 10 animals showed a decline in thehematocrit, remaining at values of 75–85% until day
MOLECULAR THERAPY Vol. 12, No. 2, August 2005
Copyright C The American Society of Gene Therapy
100 (*P b 0.0005). Control mice showed a stable baselineduring the study.
We removed implanted devices from three mice of eachgroup at day 150. The macro- and microscopic appearanceof the capsules was similar to that reported for thesyngeneic recipients. Capsules retrieved from the perito-neum were mostly free-floating (Fig. 3E) or in some casesaggregated and adhered to the organs. A weak fibroblastoutgrowth was detected specially in the capsule clusters.In contrast, capsules from the subcutaneous tissue formeda richly vascularized structure (Fig. 3F) in which immobi-lized cells remained viable, secreting detectable amountsof EPO (data not shown). The vascularized capsule net-work was easily harvested as one piece from mice after asmall skin incision, as illustrated in Fig. 3G.
Histological Analysis of Capsule Implants FollowingRemoval from MiceWe removed the cell-loaded capsules from the peritoneumand subcutaneous tissue in syngeneic and allogeneicrecipients at day 150 postimplantation. The histologicalanalysis revealed a minimal inflammatory reaction basedon a thin fibroblastic layer surrounding some capsules(Fig. 5). This was more pronounced in the case of adheredcapsule clusters implanted intraperitoneally (Figs. 5A andC) but in contrast the density of the cell infiltrate wasmuch lower in the case of capsules implanted in thesubcutaneous space (Figs. 5B and D). We observed theopposite effect when we studied the presence of newlyformed blood vessels in the different grafts. As it shown inFig. 5, a more intense vascular network was detected incapsules implanted subcutaneously both in the syngeneicand in the allogeneic recipients. The close contact of thecapillary network and the encapsulated cells might haveimplications for the long-term effects of the graft andimmunomodulation of the host immune response.
Long-Term Effects of the Encapsulated GraftTo evaluate the long-term effects of the graft, we followedtwo Balb/c mice receiving cell-loaded capsules in theperitoneum and another two mice in the subcutaneousspace until day 210. As observed in Fig. 6, the hematocritlevel of the four mice remained high (N65%) at day 128.From this day on after implantation, one of the intra-peritoneally implanted grafts failed, leading to a progres-sive decrease in the hematocrit, whereas the other three(one ip and two sc) grafts maintained the hematocrit levelof the mice at approximately 65–87%. Therefore, thefunctionality of cell-loaded microcapsules administeredafter one shot and in the absence of immunosuppressionpersisted till the 7th month after implantation.
DISCUSSION
In the present study, subcutaneous and intraperitonealimplantation of polymer-based semipermeable micro-
285
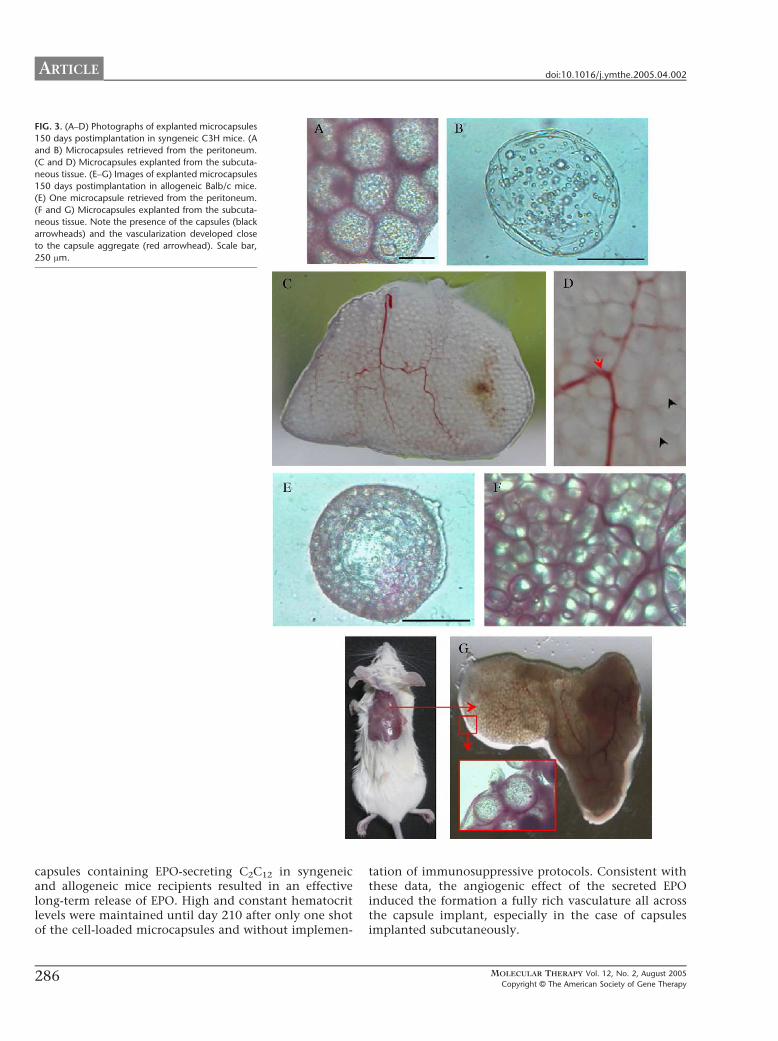
FIG. 3. (A–D) Photographs of explanted microcapsules
150 days postimplantation in syngeneic C3H mice. (A
and B) Microcapsules retrieved from the peritoneum.
(C and D) Microcapsules explanted from the subcuta-
neous tissue. (E–G) Images of explanted microcapsules
150 days postimplantation in allogeneic Balb/c mice.
(E) One microcapsule retrieved from the peritoneum.
(F and G) Microcapsules explanted from the subcuta-
neous tissue. Note the presence of the capsules (black
arrowheads) and the vascularization developed close
to the capsule aggregate (red arrowhead). Scale bar,
250 Am.
ARTICLE doi:10.1016/j.ymthe.2005.04.002
capsules containing EPO-secreting C2C12 in syngeneicand allogeneic mice recipients resulted in an effectivelong-term release of EPO. High and constant hematocritlevels were maintained until day 210 after only one shotof the cell-loaded microcapsules and without implemen-
286
tation of immunosuppressive protocols. Consistent withthese data, the angiogenic effect of the secreted EPOinduced the formation a fully rich vasculature all acrossthe capsule implant, especially in the case of capsulesimplanted subcutaneously.
MOLECULAR THERAPY Vol. 12, No. 2, August 2005
Copyright C The American Society of Gene Therapy
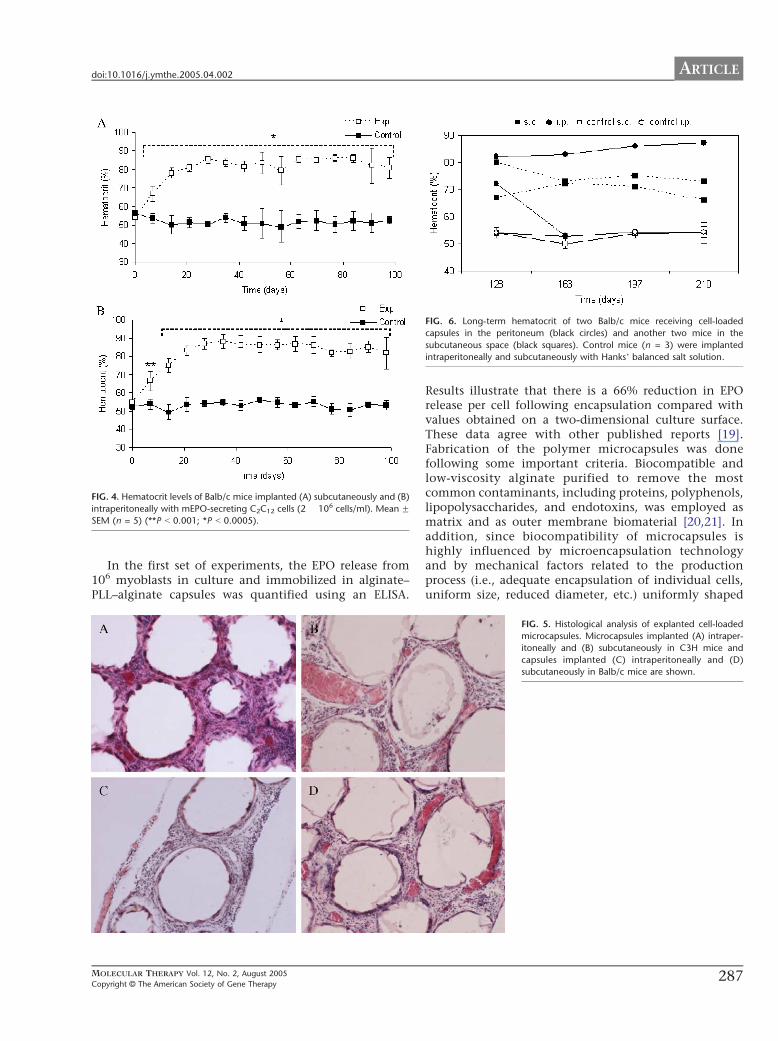
FIG. 4. Hematocrit levels of Balb/c mice implanted (A) subcutaneously and (B)
intraperitoneally with mEPO-secreting C2C12 cells (2 � 106 cells/ml). Mean FSEM (n = 5) (**P b 0.001; *P b 0.0005).
IG. 6. Long-term hematocrit of two Balb/c mice receiving cell-loaded
apsules in the peritoneum (black circles) and another two mice in the
bcutaneous space (black squares). Control mice (n = 3) were implanted
traperitoneally and subcutaneously with HanksT balanced salt solution.
ARTICLEdoi:10.1016/j.ymthe.2005.04.002
In the first set of experiments, the EPO release from106 myoblasts in culture and immobilized in alginate–PLL–alginate capsules was quantified using an ELISA.
MOLECULAR THERAPY Vol. 12, No. 2, August 2005
Copyright C The American Society of Gene Therapy
F
c
su
in
Results illustrate that there is a 66% reduction in EPOrelease per cell following encapsulation compared withvalues obtained on a two-dimensional culture surface.These data agree with other published reports [19].Fabrication of the polymer microcapsules was donefollowing some important criteria. Biocompatible andlow-viscosity alginate purified to remove the mostcommon contaminants, including proteins, polyphenols,lipopolysaccharides, and endotoxins, was employed asmatrix and as outer membrane biomaterial [20,21]. Inaddition, since biocompatibility of microcapsules ishighly influenced by microencapsulation technologyand by mechanical factors related to the productionprocess (i.e., adequate encapsulation of individual cells,uniform size, reduced diameter, etc.) uniformly shaped
FIG. 5. Histological analysis of explanted cell-loaded
microcapsules. Microcapsules implanted (A) intraper-
itoneally and (B) subcutaneously in C3H mice and
capsules implanted (C) intraperitoneally and (D)
subcutaneously in Balb/c mice are shown.
287

ARTICLE doi:10.1016/j.ymthe.2005.04.002
capsules with a diameter of 450 Am were prepared for thisstudy. The small and uniform microcapsules obtainedoffer many advantages, including a higher degree ofbiocompatibility [22,23], a reduced total implant volume,better cell-product kinetics [24], improved cell oxygen-ation [25], and potential access to different implantationsites.
In vivo, the hematocrit levels of all recipients implantedwith cell-loaded microcapsules increased during the first 3or 4 weeks postimplantation, until they reached a plateaulevel of approximately 80% that lasted to the end of thestudy. These results were obtained independent of themouse strain and the method of administration. This isparticularly interesting in the case of subcutaneouslyimplanted grafts, since previous reports testing thismethod resulted in poor implant viability and inconsis-tency in the hematocrit response to EPO secretion, even insyngeneic recipients [26]. We hypothesize that the opti-mized volume–surface relation of the microcapsules addedto the angiogenic effects of the EPO molecule couldhave provided the necessary supply of nutrition, oxy-gen, and growth factors to the immobilized graft [27].The subcutaneous administration of EPO and the con-tinuous versus bolus administration of the moleculeshould be taken into account for future potentialclinical application [28]. In fact, patients receivingconstant subcutaneous release of EPO require lowerdoses and less frequent injections than patients treatedwith intravenous injections; therefore the former is amore effective treatment [29,30].
Morphological and histological analysis of the ex-planted microcapsules 150 days after implantationrevealed some differences between capsules placed intra-peritoneally and those placed subcutaneously. In general,no signs of major inflammatory reaction were detected inthe explanted grafts. Capsules retrieved from the perito-neum were free-floating or forming small capsule clusters,with only a weak fibroblast outgrowth detected incapsules adhered to organs. In contrast, capsules ex-planted from the subcutaneous region appeared alto-gether as a richly vascularized structure with no signs ofmajor host reaction. In the past few years, many scientistshave investigated the factors limiting the long-termefficacy of microencapsulated cells. Lymphocytes andmacrophage-derived factors such as interleukin (IL)-1h,tumor necrosis factor (TNF)-a, IL-6, monocyte chemo-attractant protein 1 (MCP-1) and others can exert delete-rious effects and trigger graft failure [6,31]. EPO has beendemonstrated recently to prevent cell inflammationthrough pathways that involve phosphatidylserine andthe regulation of caspases [32,33]. More interestingly, EPOaddresses cellular inflammation directly by inhibitingseveral proinflammatory cytokines, including IL-6, TNF-a, and MCP-1 [34]. Thus, the ability of EPO to offercytoprotection and immunomodulation could haveplayed a role in the long-term efficacy and biocompati-
288
bility of the grafts implanted in syngeneic and allogeneicmice recipients after both administration methods.
Although several studies have reported EPO genedelivery using various plasmid and viral vectors [35,36]or enclosing EPO-secreting cells in hollow fibers [37], thelong-term expression of microencapsulated EPO-secretingcells in different recipients and after different methods ofadministration has not been reported. The data presentedin this study highlight the feasibility of cell encapsulationtechnology as a long-term drug delivery system independ-ent of the recipient and the administration method. Thecombination of the optimized microcapsules, the EPO-induced neovascularized network, and the low initial cellload, which could probably improve cell survival and theiradaptation to their microenvironment with minimal riskof metabolic failure [26], resulted in a continuous EPOrelease for more than 7 months. However, although yearsof clinical administration in patients with anemia haveshown EPO to be well tolerated and safe [38], a controlledEPO gene expression will be necessary to avoid expandedred cell mass (polycythemia). The microencapsulation ofcells with an oxygen-regulated EPO expression could shedlight on this issue, making EPO release by cell micro-encapsulation a more attractive and safer approach inclinical setting.
MATERIALS AND METHODS
Cell culture. C2C12 myoblasts derived from the skeletal muscle of a C3H
mouse and genetically engineered to secrete murine EPO were kindly
provided by the Institute des Neurosciences (Ecole Polytechnique Federale
de Lausanne, Lausanne, Switzerland). Cells were grown in DulbeccoTsmodified Eagle medium containing 10% fetal bovine serum, 2 mM l-
glutamine, 4.5 g/L glucose, 100 U/ml penicillin, and 100 U/ml strepto-
mycin. Cells were passed every 2–3 days and maintained at 378C in 5%
CO2. All the components of the culture medium were purchased from
Gibco BRL (Invitrogen S.A., Spain).
Before cell encapsulation EPO secretion from 1 � 106 cells/ml/24 h
was determined using a sandwich ELISA kit for human EPO (R&D
Systems, Minneapolis, MN, USA). Cross-reaction of the kit allowed
detection of mouse EPO in culture supernatants.
Cell encapsulation. We encapsulated engineered myoblasts into alginate–
poly-l-lysine–alginate microcapsules prepared using an electrostatic
droplet generator and according to a modification of Lim and SumTsprocedure [16]. Cells were suspended in 1.5% (w/v) low-viscosity high-
glucuronic acid alginate (FMC Biopolymer, Norway), obtaining a cell
density of 2 � 106 cells/ml alginate. This suspension was extruded into a
55 mM calcium chloride solution and the resulting alginate beads were
successively coated with poly-l-lysine 0.05% (w/v) for 5 min (MW
15,000–30,000; Sigma, St. Louis, MO, USA) and alginate 0.1% (w/v) for
5 min. Microcapsules were prepared at room temperature and under
sterile conditions and cultured in complete medium. The mean micro-
capsule diameter was 485 F 15 Am.
Microcapsule implantation. C3H and Balb/c mice (Charles River,
Germany) were used as syngeneic and allogeneic recipients, respectively.
Before implantation capsules were washed several times in HanksTbalanced salt solution (HBSS). For transplantation, animals were anes-
thetized by ether inhalation. Cell-loaded capsules (0.5 ml, 2 � 106 cells/
ml) suspended in 0.5 ml of HBSS were implanted subcutaneously or
intraperitoneally in both mouse strains using a catheter (Nipro 18 gauge;
MOLECULAR THERAPY Vol. 12, No. 2, August 2005
Copyright C The American Society of Gene Therapy

ARTICLEdoi:10.1016/j.ymthe.2005.04.002
Nissho Corp.). Control animals received 1 ml of HBSS by subcutaneous or
intraperitoneal route. Upon recovery, animals had access to food and
water ad libitum. Every week, we collected blood samples by retroorbital
plexus on anesthetized animals using heparinized capillary tubes. The
hematocrit was measured by a standard microhematocrit method.
Characterization of implanted capsules. Before implantation micro-
capsules were characterized in terms of morphology and EPO production.
The former was tested by microscopic observation using an inverted
optical microscope (Nikon TMS) equipped with a camera (Sony CCD-Iris).
The latter was measured using the ELISA mentioned above. EPO secretion
from 100 cell-loaded microcapsules and from 0.5 ml of capsules in 24 h
was determined.
Histological analysis. At day 150, capsules were explanted and fixed in
4% paraformaldehyde solution in 0.1 M sodium phosphate, pH 7.2, for a
minimum of 2 days. Thereafter, tissues were placed in increasing 10 to
30% sucrose/PBS and frozen in dry-ice-cooled isopentane. Serial hori-
zontal cryostat sections (14 Am) were processed for hematoxylin–eosin
staining.
Statistical analysis. Data are presented as means F SEM. We analyzed
data for statistical significance between control and experimental groups
using a Student t test according to the results of the Levene test of
homogeneity of variances. A P value b 0.001 was considered statistically
significant. All statistical computations were performed using SPSS11.0
(SPSS, Inc., Chicago, IL, USA).
ACKNOWLEDGMENTS
This project was partially supported by University of the Basque Country
(UPV/EHU) (9/UPV 00101.125-13496/2001). We also address our thanks to
the Department of Cellular Biology and Pathological Anatomy of the
Universidad Autonoma de Barcelona (IDIBAPS, Barcelona, Spain) for technical
assistance with the histological analysis.
RECEIVED FOR PUBLICATION MARCH 23, 2005; ACCEPTED APRIL 11, 2005.
REFERENCES1. Orive, G., et al. (2003). Cell encapsulation: promise and progress. Nat. Med. 9:
104 – 107.
2. Uludag, H., De Vos, P., and Tresco, P. A. (2000). Technology for mammalian cell
encapsulation. Adv. Drug Delivery Rev. 42: 29 – 64.
3. Chang, T. M. S. (2005). Therapeutic applications of polymeric artificial cells. Nat. Rev.
Drug Discovery 4: 221 – 235.
4. Dove, A. (2002). Cell-based therapies go live. Nat. Biotechnol. 20: 339 – 343.
5. Leinfelder, U., et al. (2003). A highly sensitive cell assay for validation of purification
regimes of alginates. Biomaterials 24: 4161 – 4172.
6. Juste, S., Lessard, M., Henley, N., Menard, M., and Halle, J. P. (2005). Effect of poly-L-
lysine coating on macrophage activation by alginate-based microcapsules: assessment
using a new in vivo method. J. Biomed. Mater. Res. 72: 389 – 398.
7. Wang, T., et al. (1997). An encapsulation system for the immunoisolation of pancreatic
islets. Nat. Biotechnol. 15: 358 – 362.
8. Dusseault, J., et al. (2005). Microencapsulation of living cells in semi-permeable
membranes with covalently cross-linked layers. Biomaterials 26: 1515 – 1522.
9. Prakash, S., and Chang, T. M. S. (1996). Microencapsulated genetically engineered live
E. coli DH5 cells administered orally to maintain normal plasma urea level in uremic
rats. Nat. Med. 2: 883 – 887.
10. Joki, T., Machluf, M., Atala, A., Zhu, J., Seyfried, N. T., and Dunn, I. F. (2001).
Continuous release of endostatin from microencapsulated engineered cells for tumor
therapy. Nat. Biotechnol. 19: 35 – 39.
11. Koo, J., and Chang, T. M. (1993). Secretion of erythropoietin from microencapsulated
rat kidney cells: preliminary results. Int. J. Artif. Organs 16: 557 – 560.
12. Orive, G., Ponce, S., Hernandez, R. M., Gascon, A. R., Igartua, M., and Pedraz, J. L.
(2002). Biocompatibility of microcapsules for cell immobilization elaborated with
different type of alginates. Biomaterials 23: 3825 – 3831.
MOLECULAR THERAPY Vol. 12, No. 2, August 2005
Copyright C The American Society of Gene Therapy
13. Orive, G., Hernandez, R. M., Gascon, A. R., Igartua, M., and Pedraz, J. L. (2003). Survival
of different cell lines in alginate–agarose microcapsules. Eur. J. Pharm. Sci. 18: 23 – 30.
14. Orive, G., Carcaboso, A. M., Gascon, A. R., Hernandez, R. M., and Pedraz, J. L. (2005).
Biocompatibility studies of alginate and alginate microcapsules. Biomacromolecules 6:
927 – 931.
15. Ribatti, D., et al. (1999). Human erythropoietin induces a pro-angiogenic phenotype
in cultured endothelial cells and stimulated neovascularization in vivo. Blood 93:
2627 – 2636.
16. Lim, F., and Sun, A. M. (1980). Microencapsulated islets as bioartificial endocrine
pancreas. Science 210: 908 – 909.
17. Regulier, E., et al. (1998). Continuous delivery of human and mouse erythropoietin
in mice genetically engineered polymer encapsulated myoblasts. Gene Ther. 5:
1014 – 1022.
18. Eliopoulos, N., Lejeune, L., Martineau, D., and Galipeau, J. (2004). Human-compatible
collagen matrix for prolonged and reversible systemic delivery of erythropoietin in mice
from gene-modified marrow stromal cells. Mol. Ther. 10: 741 – 748.
19. Sommer, B., et al. (2002). Long-term doxycycline-regulated secretion of erythropoietin
by encapsulated myoblasts. Mol. Ther. 6: 155 – 161.
20. Klfck, G., et al. (1994). Production of purified alginate suitable for use in
immunoisolated transplantation. Appl. Microbiol. Biotechnol. 40: 638 – 643.
21. Jork, A., et al. (2000). Biocompatible alginate from freshly collected Laminaria palida for
implantation. Appl. Microbiol. Biotechnol. 53: 224 – 229.
22. Robitaille, R., Pariseau, J. F., Leblond, F. A., Lamoureux, M., Lepage, Y., and Halle, J. P.
(1999). Studies on small (b350 Am) alginate–poly-L-lysine microcapsules: III.
Biocompatibility of smaller versus standard microcapsules. J. Biomed. Mater. Res. 44:
116 – 120.
23. Lum, Z. P., Krestow, M., Tai, I. T., Vacek, I., and Sun, A. M. (1992). Xenografts of rat
islets into diabetic mice: an evaluation of new smaller capsules. Transplantation 53:
1180 – 1183.
24. Chicheportiche, D., and Reach, G. (1988). In vitro kinetics of insulin release by
microencapsulated rat islets: effect of the size of the microcapsules. Diabetologia 31:
54 – 57.
25. Schrezenmeir, J., et al. (1992). The role of oxygen supply in islets transplantation.
Transplant. Proc. 24: 2925 – 2929.
26. Schneider, B. L., Schwenter, F., Pralong, W. F., and Aebischer, P. (2003). Prevention of
the initial host immuno-inflammatory response determines the long-term survival of
encapsulated myoblasts genetically engineered for erythropoietin delivery. Mol. Ther.
7: 506 – 514.
27. De Vos, P., Hamel, A. F., and Tatarkiewicz, K. (2002). Considerations for successful
transplantation of encapsulated pancreatic islets. Diabetologia 45: 159 – 173.
28. Schwenter, F., Deglon, N., and Aebischer, P. (2003). Optimization of human
erythropoietin secretion from MLV-infected human primary fibroblasts used for
encapsulated cell therapy. J. Gene Med. 5: 246 – 257.
29. Macdougall, I. C., et al. (1999). Pharmacokinetics of novel erythropoiesis stimulating
protein compared with epoetin alfa in dialysis patients. J. Am. Soc. Nephrol. 10:
2392 – 2395.
30. Sohmiya, M., Kakiba, T., and Kato, Y. (1998). Therapeutic use of continuous
subcutaneous infusion of recombinant human erythropoietin in malnourished predial-
ysis anemic patients with diabetic nephropathy. Eur. J. Endocrinol. 139: 367 – 370.
31. Martijin de Groot, M. S., Schuurs, T. A., Leuvenink, H. G. D., and van Schilfgaarde, M.
D. R. (2003). Macrophage overgrowth affects neighboring nonovergrown encapsu-
lated islets. J. Surg. Res. 115: 235 – 241.
32. Chong, Z. Z., et al. (2003). Erythropoietin prevents early and late neuronal demise
through modulation of Akt1 and induction of caspase 1, 3 and 8. J. Neurosci. Res. 71:
659 – 669.
33. Chong, Z. Z., et al. (2003). Erythropoietin fosters both intrinsic and extrinsic neuronal
protection through modulation of microglia, Akt1, Bad, and caspase-mediated
pathways. Br. J. Pharmacol. 138: 1107 – 1118.
34. Olsen, N. V. (2003). Central nervous system frontiers for the use of erythropoietin. Clin.
Infect. Dis. 37: S323 – S330.
35. Snyder, R. O., et al. (1997). Efficient and stable adeno-associated virus-mediated
transduction in the skeletal muscle of adult immunocompetent mice. Hum. Gene Ther.
8: 1891 – 1900.
36. Johnston, J., et al. (2003). Regulated expression of erythropoietin from an AAV vector
safely improves the anemia of h-thalassemia in a mouse model. Mol. Ther. 7: 493 – 497.
37. Dalle, B., et al. (1999). Improvement of mouse h-thalassemia upon erythropoietin
delivery by encapsulated myoblasts. Gene Ther. 6: 157 – 161.
38. Jelkman, W. (1992). Erythropoietin: structure, control of production, and function.
Physiol. Rev. 72: 449 – 489.
289





























