Embed Size (px)
Citation preview

Free Radical Biology & Medicine 49 (2010) 757–769
Contents lists available at ScienceDirect
Free Radical Biology & Medicine
j ourna l homepage: www.e lsev ie r.com/ locate / f reeradb iomed
Original Contribution
Long-lasting inhibition of presynaptic metabolism and neurotransmitter release byprotein S-nitrosylation
Alena Rudkouskaya a,1, Vasiliy Sim a,2, Aabha A. Shah a, Paul J. Feustel a,David Jourd'heuil b, Alexander A. Mongin a,⁎a Center for Neuropharmacology and Neuroscience, Albany Medical College, Albany, NY 12208, USAb Center for Cardiovascular Sciences, Albany Medical College, Albany, NY 12208, USA
Abbreviations: BCA, bicinchoninic acid; CysNO, S-nthreitol; GA-3-P, glyceraldehyde 3-phosphate; GAPDHdehydrogenase; NAC,N-acetylcysteine; NEM, N-ethylmalesensitive fusion factor; RNS, reactive nitrogen species; RSNNONOate; TBOA, DL-threo-β-benzyloxyaspartic acid.⁎ Corresponding author. Fax: +1 518 262 5799.
E-mail address: [email protected] (A.A. Mong1 Present address: Department of Physiology and
Western Ontario, London, ON, Canada.2 Present address: Department of Surgery, Brookdale U
Center, Brooklyn, NY, USA.3 The term “S-nitrosation” is defined in chemical
nitrosonium (NO+) equivalent to a sulfhydryl group toThe term “S-nitrosylation” is commonly used tomodification of proteins through the formation of RSterm “S-nitrosylation” is used throughout this paper.
0891-5849/$ – see front matter © 2010 Elsevier Inc. Adoi:10.1016/j.freeradbiomed.2010.05.032
a b s t r a c t
a r t i c l e i n f oArticle history:Received 12 January 2010Revised 21 May 2010Accepted 28 May 2010Available online 8 June 2010
Keywords:Nitric oxideS-nitroso-L-cysteineS-nitrosylationS-nitrosationNeurotransmitter releaseEnergetic metabolismBrainFree radicals
Nitric oxide (NO) and related reactive nitrogen species (RNS) play a major role in the pathophysiology ofstroke and other neurodegenerative diseases. One of the poorly understood consequences of stroke is a long-lasting inhibition of synaptic transmission. In this study, we tested the hypothesis that RNS can producelong-term inhibition of neurotransmitter release via S-nitrosylation of proteins in presynaptic nerve endings.We examined the effects of exogenous sources of RNS on the vesicular and nonvesicular L-[3H]glutamaterelease from rat brain synaptosomes. NO/RNS donors, such as spermine NONOate, MAHMA NONOate,S-nitroso-L-cysteine, and SIN-1, inhibited only the vesicular component of glutamate release with an orderof potency that closely matched levels of protein S-nitrosylation. Inhibition of glutamate release persistedfor N1 h after RNS donor decomposition and washout and strongly correlated with decreases in theintrasynaptosomal ATP levels. Post-NO treatment of synaptosomes with thiol-reducing reagents decreasedthe total content of S-nitrosylated proteins but had little effect on glutamate release and ATP levels. Incontrast, post-NO application of the end-product of glycolysis, pyruvate, partially rescued neurotransmitterrelease and ATP production. These data suggest that RNS suppress presynaptic metabolism andneurotransmitter release via poorly reversible modifications of glycolytic and mitochondrial enzymes,one of which was identified as glyceraldehyde-3-phosphate dehydrogenase. A similar mechanism maycontribute to the long-term suppression of neuronal communication during nitrosative stress in vivo.
itroso-L-cysteine; DTT, dithio-, glyceraldehyde-3-phosphateimide; NSF, N-ethylmaleimide-O, nitrosothiol; SpNO, spermine
in).Pharmacology, University of
niversity Hospital andMedical
terms as the addition of aform an S-nitrosothiol (RSNO).denote the posttranslationalNOs at cysteine residues. The
ll rights reserved.
© 2010 Elsevier Inc. All rights reserved.
The biologically active molecule nitric oxide (NO) and relatedreactive nitrogen species (RNS) regulate cellular functions via interac-tions with metalloproteins, such as guanylyl cyclase and cytochromec oxidase, but also via a variety of chemical modifications ofbiomolecules, such as oxidation, nitration, and S-nitrosylation3 [1–4].NO and RNS can be harmful if produced in excessive quantities or ifnormal intracellular redox homeostasis is impaired. In the context of
brain pathophysiology, NO and RNS have been implicated in a varietyof neural disorders, such as stroke, brain, trauma, Alzheimer andParkinson diseases, multiple sclerosis, and others [5–7].
The pathological significance of NO and RNS is particularly wellstudied in cerebral ischemia (stroke). Transient or permanentdisruption of the blood supply triggers rapid damage and death ofneuronal cells, which is initially restricted to the ischemic core, theregion where blood flow is reduced by ∼80% or more [8]. However,over a few hours to 1–3 days, the initial infarction spreads toneighboring tissue regions of the ischemic penumbra, where bloodflow remains partially preserved because of collateral circulation [8–10]. NO and RNS are major players in both acute and delayed formsof ischemic brain damage. In ischemia and reperfusion, cytoplasmicCa2+ overload leads to uncontrolled activation of neuronal nitricoxide synthase (nNOS), which is followed by delayed inflammatoryupregulation of the inducible NOS (iNOS) activity. There is a goodevidence that both nNOS and iNOS mediate ischemic tissue damagebecause their genetic deletion or pharmacological inhibition stronglyreduces infarction volume in animal models of cerebral ischemia[5,7,11–13].
Unlike the much-studied mechanisms of cell death, the potentialpathological impact of NO and RNS on neuronal communication is

758 A. Rudkouskaya et al. / Free Radical Biology & Medicine 49 (2010) 757–769
poorly understood. Long-term changes in neurotransmission are seeneven in the remote periinfarct areas where cellular viability is notaffected. Chronic modifications of synaptic transmission are associatedeither with reductions in the release of the inhibitory neurotransmitterGABA [14,15] or with suppression of the release of the excitatoryneurotransmitter glutamate [16–18]. The in vivo mechanisms contrib-uting to the defects in synaptic communication are not well defined butmay involve impaired neurotransmitter release [16], downregulation ofpostsynaptic receptors [14], and/or degeneration of neuronal processes[19]. Importantly, synaptic transmission may be particularly vulnerableto ischemia. In one recent study, neuroprotective treatments thatprevented neuronal death did not prevent the loss of synaptic functionor the structural loss of neuronal projections [20].
In this work, we explore the idea that NO and RNS can producelasting changes in neurotransmitter release via protein S-nitrosyla-tion, the covalent attachment of NO to the thiol groups of proteincysteine residues. S-nitrosylation has recently emerged as a wide-spread and important mechanism regulating protein and cellularfunctions in many tissues including the central nervous system [4,21–24]. Levels of S-nitrosothiols (RSNOs) are strongly elevated undervarious pathological conditions inside and outside of the brain andseem to contribute to the etiology of diseases [25–31], although this ispoorly defined mechanistically. Nevertheless, several in vitro studiesestablished that NO-generating compounds depolarize presynapticnerve endings and potently inhibit the vesicular neurotransmitterrelease in a manner that is dependent on modification (includingS-nitrosylation) of intrasynaptosomal sulfhydryl groups [32–34]. Innonneuronal tissue, physiological levels of S-nitrosylation have beenshown to suppress exocytosis via inhibition of the N-ethylmaleimidefusion protein (NSF) [35]. Therefore, our initial hypothesis was thatvesicular neurotransmitter release can be suppressed via an identicalmechanism [36]. However, our findings in isolated nerve endings(synaptosomes) strongly suggest that protein S-nitrosylation potentlyand persistently inhibits neurotransmitter release primarily viainhibition of energetic metabolism. These observations may be ofrelevance to the pathophysiology of stroke and other neurodegener-ative disorders that are associated with nitrosative stress.
Experimental procedures
Reagents
Spermine–nitric oxide complex (spermine NONOate), 6-(2-hydroxy-1-methyl-2-nitrosohydrazino)-N-methyl-1-hexanamine (MAHMANONOate), lactate dehydrogenase, pyruvate kinase, glyceraldehyde3-phosphate, 2-phosphoglycerate, and ATP luciferin–luciferase assaykit were purchased from Sigma–Aldrich (St. Louis, MO, USA). 3-MorpholinylsydnoneimineCl (SIN-1) andDL-threo-β-benzyloxyasparticacid (TBOA)were fromTocris Bioscience (Ellisville,MO,USA). The thiol-labeling reagent Nα-(3-maleimidylpropionyl)biocytin was from Invi-trogen (Carlsbad, CA, USA). L-[3H]glutamate (51 Ci/mmol) wasobtained from Amersham–GE Healthcare (Piscataway, NJ, USA).45CaCl2 (0.91 Ci/mmol) was purchased from PerkinElmer (Boston,MA, USA). And all other salts and reagentswere purchased from Sigma–Aldrich and were of the highest purity available.
Preparation of rat forebrain synaptosomes
Presynaptic nerve endings (synaptosomes) were isolated fromforebrains of male Sprague–Dawley rats (150–200 g), according to themethod of Hajos [37] with modifications as outlined below. All animalprocedures were approved by the Institutional Animal Use and CareCommittee. Animals were euthanized by rapid decapitation; wholebrains were rapidly removed and transferred into ice-cold sucrosemedium. Cerebellum and brain stem were dissected, and remainingtissuewas homogenized in 0.32 M sucrose/5 mMHepes (pH7.4) using a
Teflon–glass homogenizer. The homogenate was centrifuged for 10 minat 900 g (2 °C) to remove nuclei and cell debris. The first pellet wasdiscarded and the supernatant was further centrifuged for 20 min at9000 g (2 °C). The resulting pellet (P2) was resuspended in 0.32 Msucrose, layered over 0.8 M sucrose, and additionally centrifuged for20 min at 9000 g (2 °C). The myelin-containing layer at the 0.32–0.8 Msucrose interface was aspirated, the 0.8 M sucrose supernatant contain-ing synaptosomes was removed, and the 0.8 M sucrose pellet containingpredominantly mitochondria was discarded. The synaptosomal suspen-sion in 0.8 M sucrose was slowly diluted with an equal volume of thephosphate-buffered basal medium containing (in mM) 125 NaCl, 5 KCl,1.2MgSO4, 1CaCl2, 2.5NaH2PO4, 7.5Na2HPO4, and10D-glucose (pH7.4).The resulting suspensionwas centrifuged for 20 min at 9000 g (2 °C). Thefinal synaptosomal pellet was resuspended in the same phosphate-buffered basal solution and used in the subsequent experiments. In eachexperiment the synaptosomal suspension (∼1.5–2 mg protein/ml) wasinitially preincubated for 1 h at 37 °C with constant agitation to restorenormal metabolic status and transmembrane ion gradients.
Treatment with exogenous RNS donors
Synaptosomal suspensions were allowed to recover metabolicallyas described above and then were divided into several aliquots andtreated for 30 min at 37 °C in the dark with one of the NO donors,spermine NONOate (half-life at 37 °C ∼39 min) or MAHMA NONOate(half-life at 37 °C ∼1.5 min); the peroxynitrite donor SIN-1 (half life at37 °C ∼90–230 min); or the nitrosothiol compound S-nitroso-L-cysteine. All agents were added from freshly prepared stock solutionsto a final concentration of 100 μM, except for SIN-1, which was used atboth 100 and 500 μM. Spermine NONOate (100 mM) and MAHMANONOate (100 mM) were prepared in 10 mM NaOH. SIN-1 stocksolution (500 mM) was prepared immediately before experiments inH2O and stored on ice. S-Nitroso-L-cysteine (200 mM) was freshlysynthesized for each experiment from L-cysteine and NaNO2 underacidic conditions as previously described [34]. All stock solutionswerediluted 1000-fold and tested for potential changes in pH (nonefound). Vehicles were routinely added to the control samples. In ourprevious work [34] we performed control experiments with light-decomposed S-nitroso-L-cysteine and found no significant effects onvesicular neurotransmitter release up to the concentration of 1 mM.
After the initial 30-min treatment with NO donors, we used twodifferent experimental designs for studying the functional impact of NOon synaptosomal functions. In the first type of experiment, after NOtreatment, synaptosomes were pelleted by 2-min centrifugation at10,000 g in an excess of basal phosphate medium; washed twice with2 ml of Hepes-buffered basal medium containing (in mM) 135 NaCl,3.8 KCl, 1.2 MgSO4, 1.3 CaCl2, 1.2 KH2PO4, 10 D-glucose, and 10 Hepes(pH 7.4); and used for the subsequent assays (see below). These ex-periments were conducted to reveal the lasting functional impact of NOafter its removal. In the second type of experiment, synaptosomes werewashed of extracellular NO donors as described above and then wereallowed to recover for one additional hour in the basal Hepes-bufferedmedium. The recovered synaptosomes were washed three additionaltimes and then used for functional assays. This second experimentaldesign was employed to study the reversibility of NO effects uponmetabolic recovery. To study if functional recovery after the NOtreatment can be accelerated, in some experiments we supplementedthe recovery medium with thiol-reducing reagents or metabolicsubstrates as indicated in the text and figure legends. The concentrationof synaptosomal protein during all treatments was maintained between0.9 and 1.4 mg/ml.
L-[3H]Glutamate release assay
To measure vesicular and nonvesicular glutamate release weemployed a radiotracer assay. Synaptosomes were loaded with L-[3H]

759A. Rudkouskaya et al. / Free Radical Biology & Medicine 49 (2010) 757–769
glutamate (10 μCi/ml) at 37 °C for 30 minwith constant agitation. Thehigh specific activity of 3H tracer was critical to achieve significantlabeling of the vesicular L-glutamate pool. To terminate loading andremove the extracellular isotope, 10 volumes of ice-cold sucrose stopsolution (medium S) was added to the suspension, and synaptosomeswere centrifuged for 20 min at 9000 g (2 °C). Medium S contained(mM) 243 sucrose (iso-osmotic replacement for 135 mM NaCl), 3.8KCl, 1.2 MgSO4, 1.2 KH2PO4, 10 Hepes, 10 D-glucose (pH 7.4). Theresulting pellets were stored on ice and resuspended in 8 ml ice-coldmedium S immediately before neurotransmitter release measure-ments. This procedure prevents spontaneous synaptosome depolar-ization and neurotransmitter release during storage on ice [34].
To start the efflux assay, 400-μl aliquots of L-[3H]glutamate-loadedsynaptosomes were dispensed into tubes containing 4.5 ml of warmbasal medium or high-K+ medium or Ca2+-free high-K+ medium. Inhigh-K+ medium [K+]o was elevated to 43 mM (final [K+]o 40 mM) byequimolar replacement of Na+, leaving all other components the sameas in basal medium. The composition of Ca2+-free high-K+ mediumwas similar, except 1 mM CaCl2 was replaced by 1 mM MgCl2 and50 μM EGTA was added. After 5 min incubation at 37 °C, the glutamateefflux was terminated by a rapid vacuum filtration through the GF/Bfilters (Whatman–GE Healthcare, Florham Park, NJ, USA). The filterswere placed overnight into scintillation vials with 4 ml Ecoscint Ascintillation cocktail (National Diagnostics, Atlanta, GA, USA) andcounted for radioactivity that remained in synaptosomes. Fractionalrelease of L-[3H]glutamate was calculated relative to total isotopecontent in synaptosomes at time 0. In control experiments wedetermined that at the beginning of the experiment N85% of L-[3H]glutamate was contained inside synaptosomes and that nonspecificbinding of L-[3H]glutamate to the filters was negligible (b1–2% of themeasured values).
Measurements of intrasynaptosomal ATP content
Intrasynaptosomal levels of ATP were measured using a commer-cially available luciferin–luciferase ATP assay kit (Sigma). Synapto-somes were treated with NO donors and subjected to wash proceduressimilar to those in the neurotransmitter release experiments. Onehundredmicroliters of solution containing 100 mMperchloric acid plus50 mM EDTA was added to 1-ml synaptosomal aliquots, and they wereimmediately boiled for 30 s, followed by sedimentation of thedenatured proteins (10,000 g at room temperature, 22 °C). Super-natants were neutralized by adding 25-μl aliquots of 3 M KOH plus 1 MTris. Twenty-five microliters of lysate samples or the freshly preparedATP standards was added to 2 ml of luciferin–luciferase mix diluted1:200 with the ATP assay dilution buffer (Sigma). ATP levels werequantified as light production in a TriCarb TR1900 scintillation counter(PerkinElmer, Waltham, MA, USA) and compared to standards withknown ATP content. ATP levels were then normalized to proteincontent in each sample as determined using a standard bicinchoninicacid (BCA) assay kit and bovine serum albumin as a standard (Pierce-Thermo Scientific, Rockford, IL, USA).
Chemiluminescence assay of intrasynaptosomal S-nitrosothiols
Total amount of protein and lipid-bound intrasynaptosomal NO andintrasynaptosomal RSNO levels were measured using triiodide-depen-dent reductive release of bound NO followed by detection of ozone-based chemiluminescence as described elsewhere [25,38]. Briefly,synaptosomal samples were washed with phosphate-buffered basalmedium, sedimented at 10,000 g, and then lysed in 2 ml of 4 mMphosphate buffer (pH 7.5) containing 100 μM EDTA and 10 mM NEM.Samples were then pretreated in the dark with acidified sulfanilamide(final concentration 10 mM, 100 mM sulfanilamide stock prepared in1 N HCl) for 15 min to remove nitrite with or without HgCl2 (finalconcentration 4.9 mM) to evaluate the presence of RSNOs. Hg2+
selectively decomposes RSNOs [25] such that RSNO concentrationsmaybe determined from the difference in NO signal with or withoutpretreatment of paired sampleswith HgCl2. Four hundredmicroliters ofeach sample was injected in a purge vessel that contained 4.5 ml ofglacial acetic acid and 500 μl of an aqueous mixture of 450 mMpotassium iodide and 100 mM iodine. The vessel was kept at 70 °C, andthe solution was constantly purged with nitrogen and changed everyfour injections. The amounts of NO released from the purge vessel werequantified by gas-phase chemiluminescence (NOA 280; Sievers Instru-ments, Boulder, CO, USA). Peak integration was performed, and theresults were converted to NO concentrations using authentic NO as astandard and then normalized to the protein content in each sample.
Biotin-switch assay of intrasynaptosomal nitrosothiols
To visualize the extent of protein S-nitrosylation we employed abiotin-switch technique developed by Jaffrey et al. [23], with severalmodifications as described below. Twomain changes were as follows:(1) We used NEM (final concentration ∼23 mM) instead of theoriginally proposed MMTS. This modification allowed for moreconsistent blocking at room temperature (22 °C) and stronglyreduced nonspecific background biotinylation of the masked freethiols. (2) We used a different biotinylating agent, Nα-(3-maleimi-dylpropionyl)biocytin, which in our hands worked more consistentlyand produced lower background signal than the biotin–HPDPproposed in the original method. Synaptosomes were washed of NOdonors three times with Hepes-buffered basal medium and thenincubated for 1 h at 25 °C in the dark in 1 ml of blocking solutioncontaining 1 volume of 0.25 M NEM, 9 volumes of HEN buffer, pH 7.7,and 1 volume of 25% SDS. HEN buffer was composed of (mM) 250Hepes, 1 EDTA, 0.1 neocuproine. To remove NEM, the proteins wereprecipitated by adding 2 ml of prechilled acetone for 15 min at−20 °C, followed by sedimentation. After the acetone was removed,the RSNOs were labeled for 1 h at room temperature (22 °C) in thedark with 0.5 ml of reducing/labeling solution with final concentra-tions of 3 mM ascorbate and 1 mM Nα-(3-maleimidylpropionyl)biocytin in HEN buffer, pH 7.0. The samples were desalted withacetone one more time, pelleted, and resuspended in the basalmedium. Small aliquots were used for determination of proteinconcentration by a colorimetric BCA assay kit (Pierce–ThermoScientific), the rest was diluted with 2× Laemmli reducing buffer(Bio-Rad, Hercules, CA, USA) and used for Western blot analysis. Theproteins were resolved by SDS–polyacrylamide gel electrophoresis(10%) and transferred onto an Immun-Blot polyvinylidene difluoridemembrane (Bio-Rad). After being blocked with 5% milk in phosphate-buffered saline buffer containing 0.05% Tween 20 (PBST), themembrane was incubated with polyclonal anti-biotin antibody(1:50,000 dilution; Bethyl Laboratories, Montgomery, TX, USA) for1 h at 25 °C, followed by five washes for 5 min with 1% milk PBST. Themembrane was incubated for 1 h with secondary anti-rabbit horse-radish peroxidase-conjugated antibodies (Amersham–GE Healthcare;1:20,000 dilution), followed by four PBST washes. Chemilumines-cence was detected using an ECLplus kit (Amersham–GE Healthcare)and CL-Xposure film (Pierce).
GAPDH activity assay
The activity of GAPDHwas measured by monitoring the enzymaticreduction of NAD+ to NADH in the presence of the GAPDH substrateglyceraldehyde 3-phosphate (GA-3-P). Washed and pelleted synap-tosomes were lysed in 0.5 ml of ice-cold lysis buffer (4 mM phosphatebuffer, 500 μM EDTA, Roche protease inhibitor cocktail, pH 7.5) andhomogenized using a hypodermic syringe. The lysates were clarifiedby 10 min centrifugation at 10,000 g (2 °C). Fifty microliters ofclarified lysates were added to 950 μl of the GAPDH reaction mixcontaining (inmM) 100 glycine, 100 KH2PO4, 5 EDTA, 1 NAD+, and 1.5
760 A. Rudkouskaya et al. / Free Radical Biology & Medicine 49 (2010) 757–769
GA-3-P (pH 8.9, adjusted with NaOH). NAD+ and GA-3-P were addedto the mix immediately before the assay. The GAPDH reaction wasinitiated by addition of synaptosomal lysates and carried out for 5 minat 25 °C. Enzymatic production of NADH was measured as increase inthe optical density at 340 nm using an ELx800 plate reader (Bio-TekInstruments, Winooski, VT, USA) and calculated using the NADHmolar extinction coefficient of 6300 cm−1 M−1. Total protein con-centration in the samples was determined using a BCA assay. Resultswere expressed as nmol NADH produced/mg total protein/5 min.
Enolase activity assay
The neuron-specific enolase (NSE) activity was assayed in acoupled enzymatic assay by monitoring the conversion of NADH toNAD+ resulting in a decrease in absorbance at 340 nm. The lysateswere prepared in the same manner as for GAPDH assay (see above).Fifty microliters of clarified lysates was added to 950 μl of the NSEreaction mix containing 100 mM Hepes, 25 mM MgSO4, 100 mM KCl,0.2 mM NADH, 1.3 mM ADP, 2 mM 2-phosphoglycerate, 10 U lactatedehydrogenase, 7 U pyruvate kinase (pH 7.4, adjusted with NaOH).Reaction was carried out for 5 min at 25 °C. Enzymatic production ofNAD+ was measured as a decrease in the optical density at 340 nmusing an ELx800 plate reader and calculated using the NADH molarextinction coefficient of 6300 cm−1 M−1. Total protein concentrationin the samples was determined using a BCA assay. Results wereexpressed as μmol NADH consumed/mg total protein/5 min.
Ca2+ uptake via the voltage-gated Ca2+ channels
Depolarization-induced Ca2+ uptake was measured using 45Ca2+ asa radiotracer [39]. Synaptosomes were incubated with NO donors andsubjected to the same procedures as in all other assays and thenpelleted in an excess of ice-cold sucrose stop solution S (for com-position see above) and transferred onto ice. Before measurements thepellets were resuspended in the same sucrose solution (proteinconcentration 3–4 mg/ml). One-hundred-microliter aliquots of synap-tosomes were injected into 900 μl of warm basal Hepes-buffered
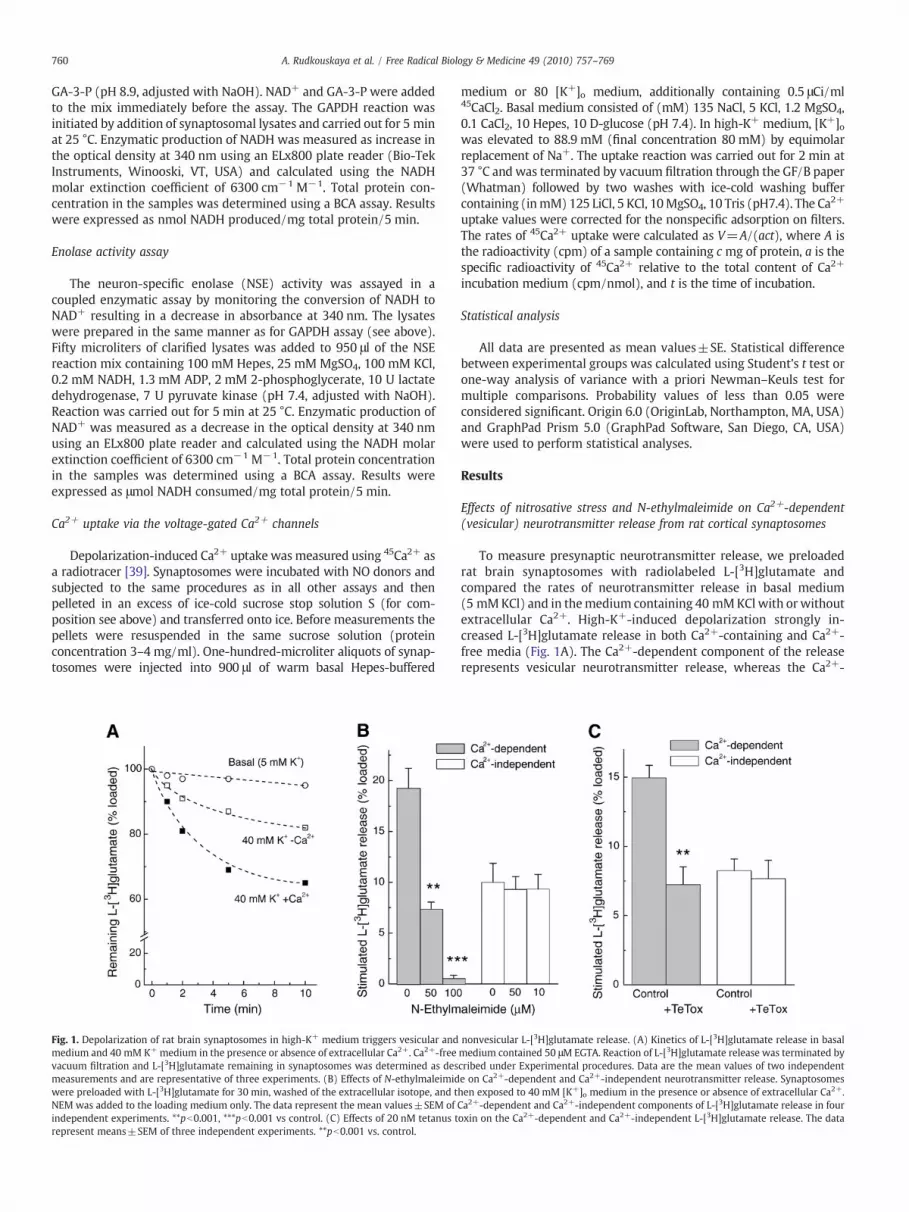
Fig. 1. Depolarization of rat brain synaptosomes in high-K+ medium triggers vesicular andmedium and 40 mM K+ medium in the presence or absence of extracellular Ca2+. Ca2+-freevacuum filtration and L-[3H]glutamate remaining in synaptosomes was determined as desmeasurements and are representative of three experiments. (B) Effects of N-ethylmaleimidwere preloaded with L-[3H]glutamate for 30 min, washed of the extracellular isotope, and tNEM was added to the loading medium only. The data represent the mean values±SEM ofindependent experiments. **pb0.001, ***pb0.001 vs control. (C) Effects of 20 nM tetanus trepresent means±SEM of three independent experiments. **pb0.001 vs. control.
medium or 80 [K+]o medium, additionally containing 0.5 μCi/ml45CaCl2. Basal medium consisted of (mM) 135 NaCl, 5 KCl, 1.2 MgSO4,0.1 CaCl2, 10 Hepes, 10 D-glucose (pH 7.4). In high-K+ medium, [K+]owas elevated to 88.9 mM (final concentration 80 mM) by equimolarreplacement of Na+. The uptake reaction was carried out for 2 min at37 °C and was terminated by vacuum filtration through the GF/B paper(Whatman) followed by two washes with ice-cold washing buffercontaining (inmM) 125 LiCl, 5 KCl, 10MgSO4, 10 Tris (pH7.4). The Ca2+
uptake values were corrected for the nonspecific adsorption on filters.The rates of 45Ca2+ uptake were calculated as V=A/(act), where A isthe radioactivity (cpm) of a sample containing c mg of protein, a is thespecific radioactivity of 45Ca2+ relative to the total content of Ca2+
incubation medium (cpm/nmol), and t is the time of incubation.
Statistical analysis
All data are presented as mean values±SE. Statistical differencebetween experimental groups was calculated using Student's t test orone-way analysis of variance with a priori Newman–Keuls test formultiple comparisons. Probability values of less than 0.05 wereconsidered significant. Origin 6.0 (OriginLab, Northampton, MA, USA)and GraphPad Prism 5.0 (GraphPad Software, San Diego, CA, USA)were used to perform statistical analyses.
Results
Effects of nitrosative stress and N-ethylmaleimide on Ca2+-dependent(vesicular) neurotransmitter release from rat cortical synaptosomes
To measure presynaptic neurotransmitter release, we preloadedrat brain synaptosomes with radiolabeled L-[3H]glutamate andcompared the rates of neurotransmitter release in basal medium(5 mMKCl) and in themedium containing 40 mMKClwith orwithoutextracellular Ca2+. High-K+-induced depolarization strongly in-creased L-[3H]glutamate release in both Ca2+-containing and Ca2+-free media (Fig. 1A). The Ca2+-dependent component of the releaserepresents vesicular neurotransmitter release, whereas the Ca2+-
nonvesicular L-[3H]glutamate release. (A) Kinetics of L-[3H]glutamate release in basalmedium contained 50 μM EGTA. Reaction of L-[3H]glutamate release was terminated bycribed under Experimental procedures. Data are the mean values of two independente on Ca2+-dependent and Ca2+-independent neurotransmitter release. Synaptosomeshen exposed to 40 mM [K+]o medium in the presence or absence of extracellular Ca2+.Ca2+-dependent and Ca2+-independent components of L-[3H]glutamate release in fouroxin on the Ca2+-dependent and Ca2+-independent L-[3H]glutamate release. The data
761A. Rudkouskaya et al. / Free Radical Biology & Medicine 49 (2010) 757–769
independent release is most likely due to the reversal of membraneglutamate transporters [40]. To verify the vesicular nature of Ca2+-dependent release in our preparation, we performed two types ofcontrols. We first treated synaptosomes with the sulfhydryl-modify-ing reagent NEM,which blocks exocytosis [41]. As seen in Fig. 1B, NEMsuppressed the Ca2+-dependent (vesicular) L-[3H]glutamate releasein a dose-dependent fashion with a complete inhibition seen at∼100 μM, but was totally ineffective against Ca2+-independent(transporter-mediated) neurotransmitter release. We next pretreatedsynaptosomes for 1 h with 20 nM tetanus toxin, a bacterial toxin thatenzymatically cleaves the vesicle-associated SNARE protein synapto-brevin and thereby prevents exocytosis [42]. Tetanus toxin inhibitedthe Ca2+-dependent neurotransmitter release by∼55–60%without anyeffect on the Ca2+-independent component of the release (Fig. 1C).Incomplete inhibition of the Ca2+-dependent L-[3H]glutamate releasewas probably due to partial cleavage of the intrasynaptosomalsynaptobrevin. Taken together, these data validate the use of Ca2+-dependent L-[3H]glutamate efflux as a measure of vesicular neuro-transmitter release.
We next exposed synaptosomes to several NO-generating com-pounds during the whole 30-min duration of the L-[3H]glutamateloading. After 30-min incubation, NO donors were washed from theextracellular medium together with extracellular isotope. This exper-imental design emphasized long-lasting rather than acute effects of NOtreatment, because the short-lived NO donors completely decomposeduring incubation and the long-lived donors are removed duringwashes. The levels of L-[3H]glutamate loading were not significantlyaffected by treatments with “pure” NO donors and SIN-1 (loadingranged between 85 and 115% of controls in various preparations),but were increased in a number experiments after treatment withS-nitroso-L-cysteine (range 100–160%of control values). Because L-[3H]glutamate release values were calculated as percentage of the totalloading (see Experimental procedures), this allowed us to negate anypotential impact of variations in loading levels.
To explore whether there is a difference in actions betweendifferent RNS, we compared the effects of the short-lived NO donorMAHMANONOate (half-life ∼1.5 min), the more slowly decomposingNO donor spermine NONOate (SpNO; half life ∼39 min), the NO/peroxynitrite generator SIN-1 (half life ∼90–230 min), and the
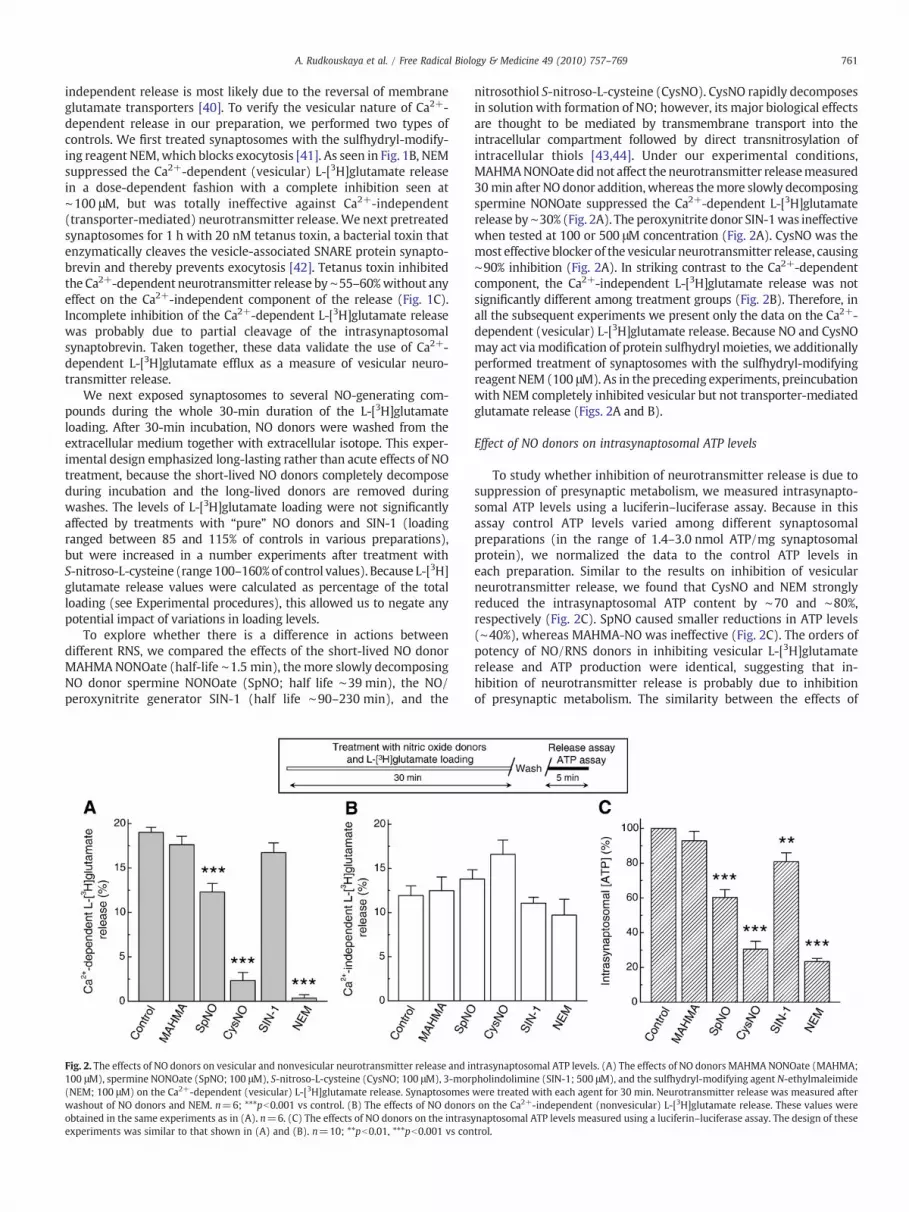
Fig. 2. The effects of NO donors on vesicular and nonvesicular neurotransmitter release and in100 μM), spermine NONOate (SpNO; 100 μM), S-nitroso-L-cysteine (CysNO; 100 μM), 3-mor(NEM; 100 μM) on the Ca2+-dependent (vesicular) L-[3H]glutamate release. Synaptosomeswashout of NO donors and NEM. n=6; ***pb0.001 vs control. (B) The effects of NO donorobtained in the same experiments as in (A). n=6. (C) The effects of NO donors on the intrasexperiments was similar to that shown in (A) and (B). n=10; **pb0.01, ***pb0.001 vs con
nitrosothiol S-nitroso-L-cysteine (CysNO). CysNO rapidly decomposesin solution with formation of NO; however, its major biological effectsare thought to be mediated by transmembrane transport into theintracellular compartment followed by direct transnitrosylation ofintracellular thiols [43,44]. Under our experimental conditions,MAHMANONOate did not affect the neurotransmitter releasemeasured30min after NO donor addition,whereas themore slowly decomposingspermine NONOate suppressed the Ca2+-dependent L-[3H]glutamaterelease by∼30% (Fig. 2A). The peroxynitrite donor SIN-1was ineffectivewhen tested at 100 or 500 μM concentration (Fig. 2A). CysNO was themost effective blocker of the vesicular neurotransmitter release, causing∼90% inhibition (Fig. 2A). In striking contrast to the Ca2+-dependentcomponent, the Ca2+-independent L-[3H]glutamate release was notsignificantly different among treatment groups (Fig. 2B). Therefore, inall the subsequent experiments we present only the data on the Ca2+-dependent (vesicular) L-[3H]glutamate release. Because NO and CysNOmay act viamodification of protein sulfhydryl moieties, we additionallyperformed treatment of synaptosomes with the sulfhydryl-modifyingreagent NEM (100 μM). As in the preceding experiments, preincubationwith NEM completely inhibited vesicular but not transporter-mediatedglutamate release (Figs. 2A and B).
Effect of NO donors on intrasynaptosomal ATP levels
To study whether inhibition of neurotransmitter release is due tosuppression of presynaptic metabolism, we measured intrasynapto-somal ATP levels using a luciferin–luciferase assay. Because in thisassay control ATP levels varied among different synaptosomalpreparations (in the range of 1.4–3.0 nmol ATP/mg synaptosomalprotein), we normalized the data to the control ATP levels ineach preparation. Similar to the results on inhibition of vesicularneurotransmitter release, we found that CysNO and NEM stronglyreduced the intrasynaptosomal ATP content by ∼70 and ∼80%,respectively (Fig. 2C). SpNO caused smaller reductions in ATP levels(∼40%), whereas MAHMA-NO was ineffective (Fig. 2C). The orders ofpotency of NO/RNS donors in inhibiting vesicular L-[3H]glutamaterelease and ATP production were identical, suggesting that in-hibition of neurotransmitter release is probably due to inhibitionof presynaptic metabolism. The similarity between the effects of
trasynaptosomal ATP levels. (A) The effects of NO donors MAHMANONOate (MAHMA;pholindolimine (SIN-1; 500 μM), and the sulfhydryl-modifying agent N-ethylmaleimidewere treated with each agent for 30 min. Neurotransmitter release was measured afters on the Ca2+-independent (nonvesicular) L-[3H]glutamate release. These values wereynaptosomal ATP levels measured using a luciferin–luciferase assay. The design of thesetrol.
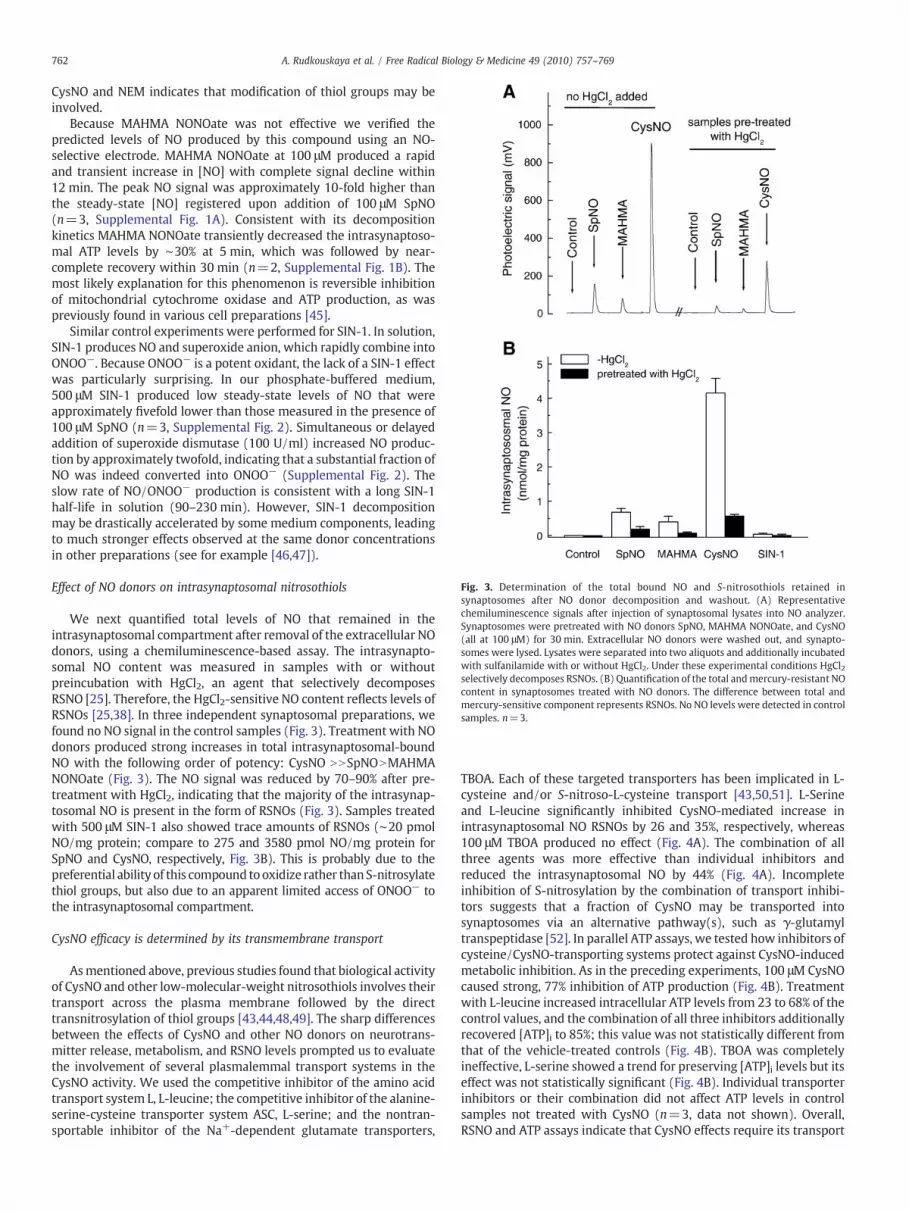
Fig. 3. Determination of the total bound NO and S-nitrosothiols retained insynaptosomes after NO donor decomposition and washout. (A) Representativechemiluminescence signals after injection of synaptosomal lysates into NO analyzer.Synaptosomes were pretreated with NO donors SpNO, MAHMA NONOate, and CysNO(all at 100 μM) for 30 min. Extracellular NO donors were washed out, and synapto-somes were lysed. Lysates were separated into two aliquots and additionally incubatedwith sulfanilamide with or without HgCl2. Under these experimental conditions HgCl2selectively decomposes RSNOs. (B) Quantification of the total andmercury-resistant NOcontent in synaptosomes treated with NO donors. The difference between total andmercury-sensitive component represents RSNOs. No NO levels were detected in controlsamples. n=3.
762 A. Rudkouskaya et al. / Free Radical Biology & Medicine 49 (2010) 757–769
CysNO and NEM indicates that modification of thiol groups may beinvolved.
Because MAHMA NONOate was not effective we verified thepredicted levels of NO produced by this compound using an NO-selective electrode. MAHMA NONOate at 100 μM produced a rapidand transient increase in [NO] with complete signal decline within12 min. The peak NO signal was approximately 10-fold higher thanthe steady-state [NO] registered upon addition of 100 μM SpNO(n=3, Supplemental Fig. 1A). Consistent with its decompositionkinetics MAHMA NONOate transiently decreased the intrasynaptoso-mal ATP levels by ∼30% at 5 min, which was followed by near-complete recovery within 30 min (n=2, Supplemental Fig. 1B). Themost likely explanation for this phenomenon is reversible inhibitionof mitochondrial cytochrome oxidase and ATP production, as waspreviously found in various cell preparations [45].
Similar control experiments were performed for SIN-1. In solution,SIN-1 produces NO and superoxide anion, which rapidly combine intoONOO−. Because ONOO− is a potent oxidant, the lack of a SIN-1 effectwas particularly surprising. In our phosphate-buffered medium,500 μM SIN-1 produced low steady-state levels of NO that wereapproximately fivefold lower than those measured in the presence of100 μM SpNO (n=3, Supplemental Fig. 2). Simultaneous or delayedaddition of superoxide dismutase (100 U/ml) increased NO produc-tion by approximately twofold, indicating that a substantial fraction ofNO was indeed converted into ONOO− (Supplemental Fig. 2). Theslow rate of NO/ONOO− production is consistent with a long SIN-1half-life in solution (90–230 min). However, SIN-1 decompositionmay be drastically accelerated by some medium components, leadingto much stronger effects observed at the same donor concentrationsin other preparations (see for example [46,47]).
Effect of NO donors on intrasynaptosomal nitrosothiols
We next quantified total levels of NO that remained in theintrasynaptosomal compartment after removal of the extracellular NOdonors, using a chemiluminescence-based assay. The intrasynapto-somal NO content was measured in samples with or withoutpreincubation with HgCl2, an agent that selectively decomposesRSNO [25]. Therefore, the HgCl2-sensitive NO content reflects levels ofRSNOs [25,38]. In three independent synaptosomal preparations, wefound no NO signal in the control samples (Fig. 3). Treatment with NOdonors produced strong increases in total intrasynaptosomal-boundNO with the following order of potency: CysNO NNSpNONMAHMANONOate (Fig. 3). The NO signal was reduced by 70–90% after pre-treatment with HgCl2, indicating that the majority of the intrasynap-tosomal NO is present in the form of RSNOs (Fig. 3). Samples treatedwith 500 μM SIN-1 also showed trace amounts of RSNOs (∼20 pmolNO/mg protein; compare to 275 and 3580 pmol NO/mg protein forSpNO and CysNO, respectively, Fig. 3B). This is probably due to thepreferential ability of this compound tooxidize rather thanS-nitrosylatethiol groups, but also due to an apparent limited access of ONOO− tothe intrasynaptosomal compartment.
CysNO efficacy is determined by its transmembrane transport
Asmentioned above, previous studies found that biological activityof CysNO and other low-molecular-weight nitrosothiols involves theirtransport across the plasma membrane followed by the directtransnitrosylation of thiol groups [43,44,48,49]. The sharp differencesbetween the effects of CysNO and other NO donors on neurotrans-mitter release, metabolism, and RSNO levels prompted us to evaluatethe involvement of several plasmalemmal transport systems in theCysNO activity. We used the competitive inhibitor of the amino acidtransport system L, L-leucine; the competitive inhibitor of the alanine-serine-cysteine transporter system ASC, L-serine; and the nontran-sportable inhibitor of the Na+-dependent glutamate transporters,
TBOA. Each of these targeted transporters has been implicated in L-cysteine and/or S-nitroso-L-cysteine transport [43,50,51]. L-Serineand L-leucine significantly inhibited CysNO-mediated increase inintrasynaptosomal NO RSNOs by 26 and 35%, respectively, whereas100 μM TBOA produced no effect (Fig. 4A). The combination of allthree agents was more effective than individual inhibitors andreduced the intrasynaptosomal NO by 44% (Fig. 4A). Incompleteinhibition of S-nitrosylation by the combination of transport inhibi-tors suggests that a fraction of CysNO may be transported intosynaptosomes via an alternative pathway(s), such as γ-glutamyltranspeptidase [52]. In parallel ATP assays, we tested how inhibitors ofcysteine/CysNO-transporting systems protect against CysNO-inducedmetabolic inhibition. As in the preceding experiments, 100 μM CysNOcaused strong, 77% inhibition of ATP production (Fig. 4B). Treatmentwith L-leucine increased intracellular ATP levels from 23 to 68% of thecontrol values, and the combination of all three inhibitors additionallyrecovered [ATP]i to 85%; this value was not statistically different fromthat of the vehicle-treated controls (Fig. 4B). TBOA was completelyineffective, L-serine showed a trend for preserving [ATP]i levels but itseffect was not statistically significant (Fig. 4B). Individual transporterinhibitors or their combination did not affect ATP levels in controlsamples not treated with CysNO (n=3, data not shown). Overall,RSNO and ATP assays indicate that CysNO effects require its transport
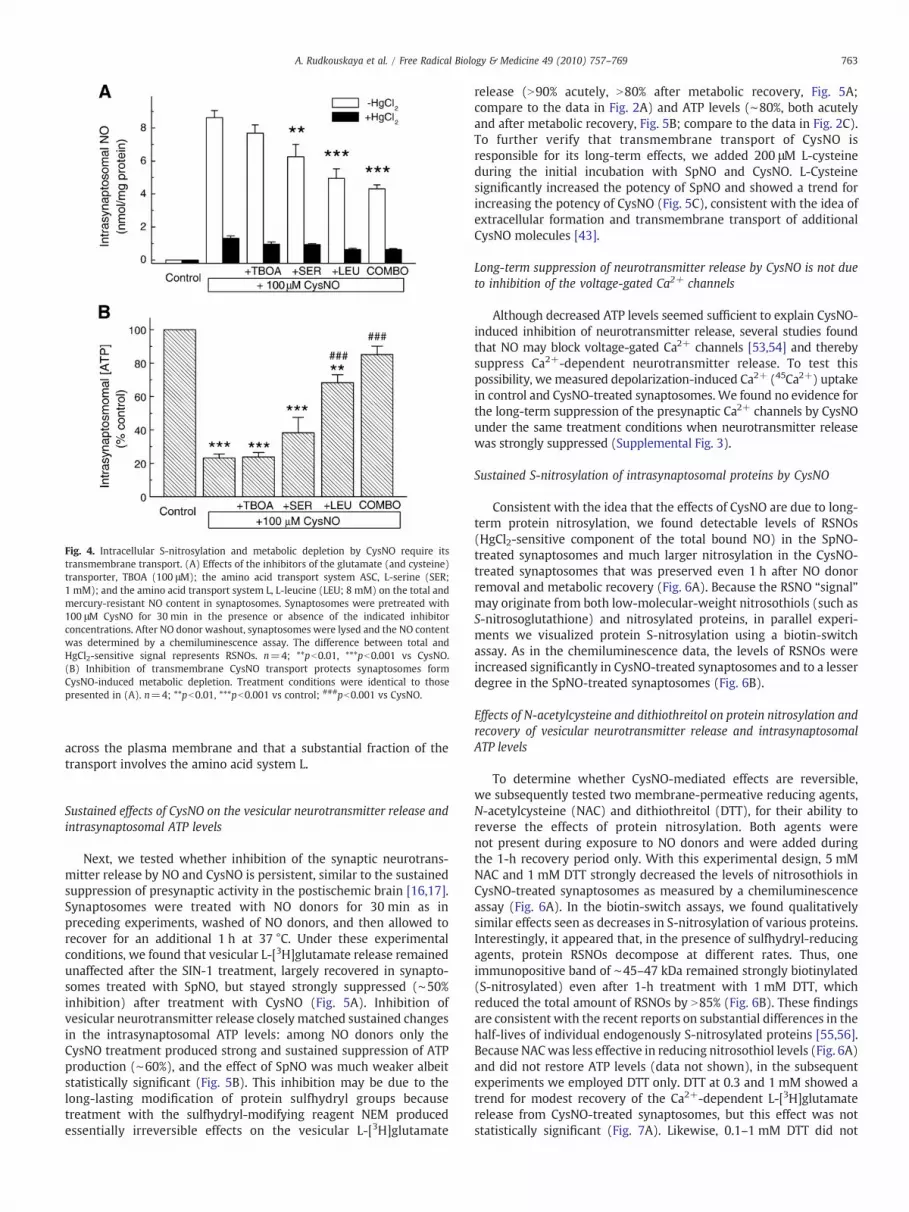
Fig. 4. Intracellular S-nitrosylation and metabolic depletion by CysNO require itstransmembrane transport. (A) Effects of the inhibitors of the glutamate (and cysteine)transporter, TBOA (100 μM); the amino acid transport system ASC, L-serine (SER;1 mM); and the amino acid transport system L, L-leucine (LEU; 8 mM) on the total andmercury-resistant NO content in synaptosomes. Synaptosomes were pretreated with100 μM CysNO for 30 min in the presence or absence of the indicated inhibitorconcentrations. After NO donor washout, synaptosomes were lysed and the NO contentwas determined by a chemiluminescence assay. The difference between total andHgCl2-sensitive signal represents RSNOs. n=4; **pb0.01, ***pb0.001 vs CysNO.(B) Inhibition of transmembrane CysNO transport protects synaptosomes formCysNO-induced metabolic depletion. Treatment conditions were identical to thosepresented in (A). n=4; **pb0.01, ***pb0.001 vs control; ###pb0.001 vs CysNO.
763A. Rudkouskaya et al. / Free Radical Biology & Medicine 49 (2010) 757–769
across the plasma membrane and that a substantial fraction of thetransport involves the amino acid system L.
Sustained effects of CysNO on the vesicular neurotransmitter release andintrasynaptosomal ATP levels
Next, we tested whether inhibition of the synaptic neurotrans-mitter release by NO and CysNO is persistent, similar to the sustainedsuppression of presynaptic activity in the postischemic brain [16,17].Synaptosomes were treated with NO donors for 30 min as inpreceding experiments, washed of NO donors, and then allowed torecover for an additional 1 h at 37 °C. Under these experimentalconditions, we found that vesicular L-[3H]glutamate release remainedunaffected after the SIN-1 treatment, largely recovered in synapto-somes treated with SpNO, but stayed strongly suppressed (∼50%inhibition) after treatment with CysNO (Fig. 5A). Inhibition ofvesicular neurotransmitter release closely matched sustained changesin the intrasynaptosomal ATP levels: among NO donors only theCysNO treatment produced strong and sustained suppression of ATPproduction (∼60%), and the effect of SpNO was much weaker albeitstatistically significant (Fig. 5B). This inhibition may be due to thelong-lasting modification of protein sulfhydryl groups becausetreatment with the sulfhydryl-modifying reagent NEM producedessentially irreversible effects on the vesicular L-[3H]glutamate
release (N90% acutely, N80% after metabolic recovery, Fig. 5A;compare to the data in Fig. 2A) and ATP levels (∼80%, both acutelyand after metabolic recovery, Fig. 5B; compare to the data in Fig. 2C).To further verify that transmembrane transport of CysNO isresponsible for its long-term effects, we added 200 μM L-cysteineduring the initial incubation with SpNO and CysNO. L-Cysteinesignificantly increased the potency of SpNO and showed a trend forincreasing the potency of CysNO (Fig. 5C), consistent with the idea ofextracellular formation and transmembrane transport of additionalCysNO molecules [43].
Long-term suppression of neurotransmitter release by CysNO is not dueto inhibition of the voltage-gated Ca2+ channels
Although decreased ATP levels seemed sufficient to explain CysNO-induced inhibition of neurotransmitter release, several studies foundthat NO may block voltage-gated Ca2+ channels [53,54] and therebysuppress Ca2+-dependent neurotransmitter release. To test thispossibility, we measured depolarization-induced Ca2+ (45Ca2+) uptakein control and CysNO-treated synaptosomes. We found no evidence forthe long-term suppression of the presynaptic Ca2+ channels by CysNOunder the same treatment conditions when neurotransmitter releasewas strongly suppressed (Supplemental Fig. 3).
Sustained S-nitrosylation of intrasynaptosomal proteins by CysNO
Consistent with the idea that the effects of CysNO are due to long-term protein nitrosylation, we found detectable levels of RSNOs(HgCl2-sensitive component of the total bound NO) in the SpNO-treated synaptosomes and much larger nitrosylation in the CysNO-treated synaptosomes that was preserved even 1 h after NO donorremoval and metabolic recovery (Fig. 6A). Because the RSNO “signal”may originate from both low-molecular-weight nitrosothiols (such asS-nitrosoglutathione) and nitrosylated proteins, in parallel experi-ments we visualized protein S-nitrosylation using a biotin-switchassay. As in the chemiluminescence data, the levels of RSNOs wereincreased significantly in CysNO-treated synaptosomes and to a lesserdegree in the SpNO-treated synaptosomes (Fig. 6B).
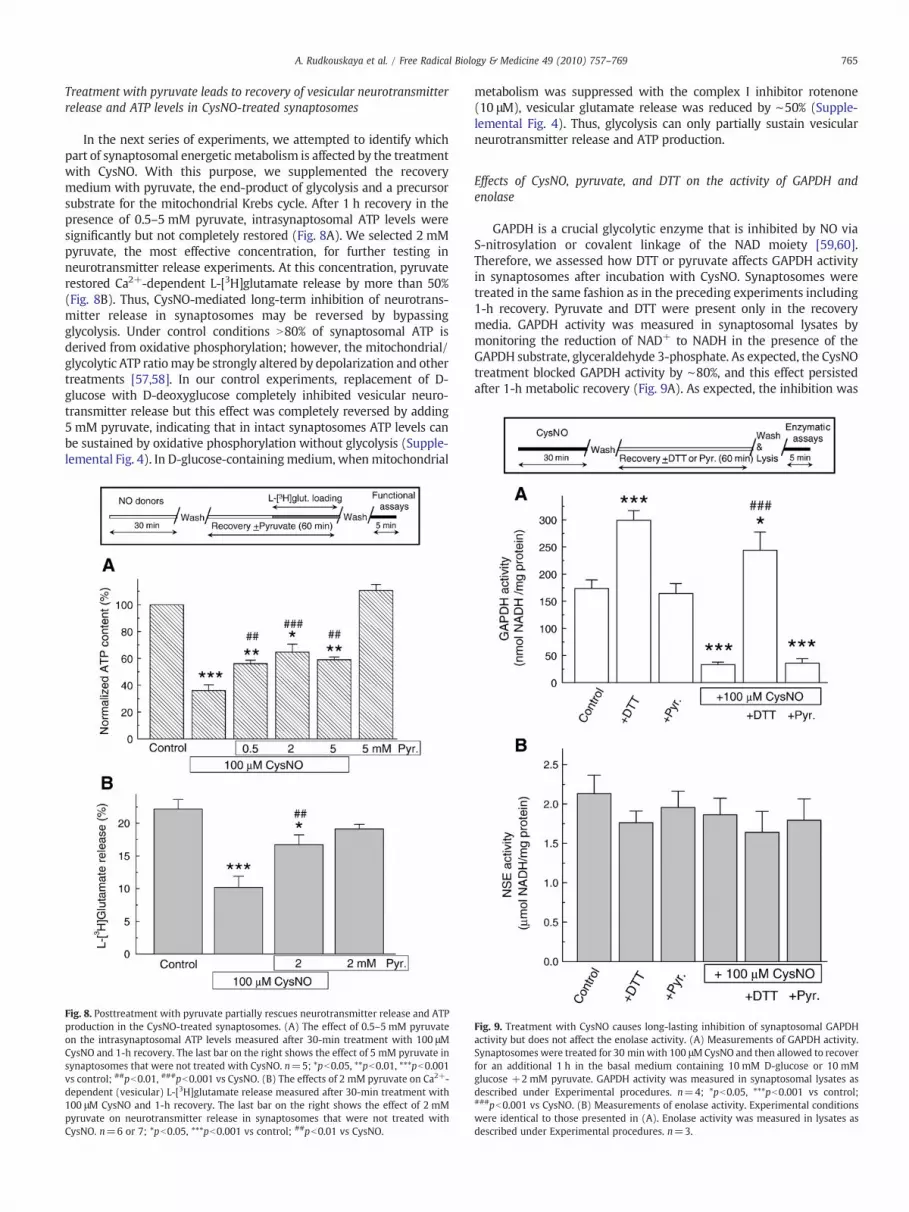
Effects of N-acetylcysteine and dithiothreitol on protein nitrosylation andrecovery of vesicular neurotransmitter release and intrasynaptosomalATP levels
To determine whether CysNO-mediated effects are reversible,we subsequently tested two membrane-permeative reducing agents,N-acetylcysteine (NAC) and dithiothreitol (DTT), for their ability toreverse the effects of protein nitrosylation. Both agents werenot present during exposure to NO donors and were added duringthe 1-h recovery period only. With this experimental design, 5 mMNAC and 1 mM DTT strongly decreased the levels of nitrosothiols inCysNO-treated synaptosomes as measured by a chemiluminescenceassay (Fig. 6A). In the biotin-switch assays, we found qualitativelysimilar effects seen as decreases in S-nitrosylation of various proteins.Interestingly, it appeared that, in the presence of sulfhydryl-reducingagents, protein RSNOs decompose at different rates. Thus, oneimmunopositive band of ∼45–47 kDa remained strongly biotinylated(S-nitrosylated) even after 1-h treatment with 1 mM DTT, whichreduced the total amount of RSNOs by N85% (Fig. 6B). These findingsare consistent with the recent reports on substantial differences in thehalf-lives of individual endogenously S-nitrosylated proteins [55,56].Because NACwas less effective in reducing nitrosothiol levels (Fig. 6A)and did not restore ATP levels (data not shown), in the subsequentexperiments we employed DTT only. DTT at 0.3 and 1 mM showed atrend for modest recovery of the Ca2+-dependent L-[3H]glutamaterelease from CysNO-treated synaptosomes, but this effect was notstatistically significant (Fig. 7A). Likewise, 0.1–1 mM DTT did not
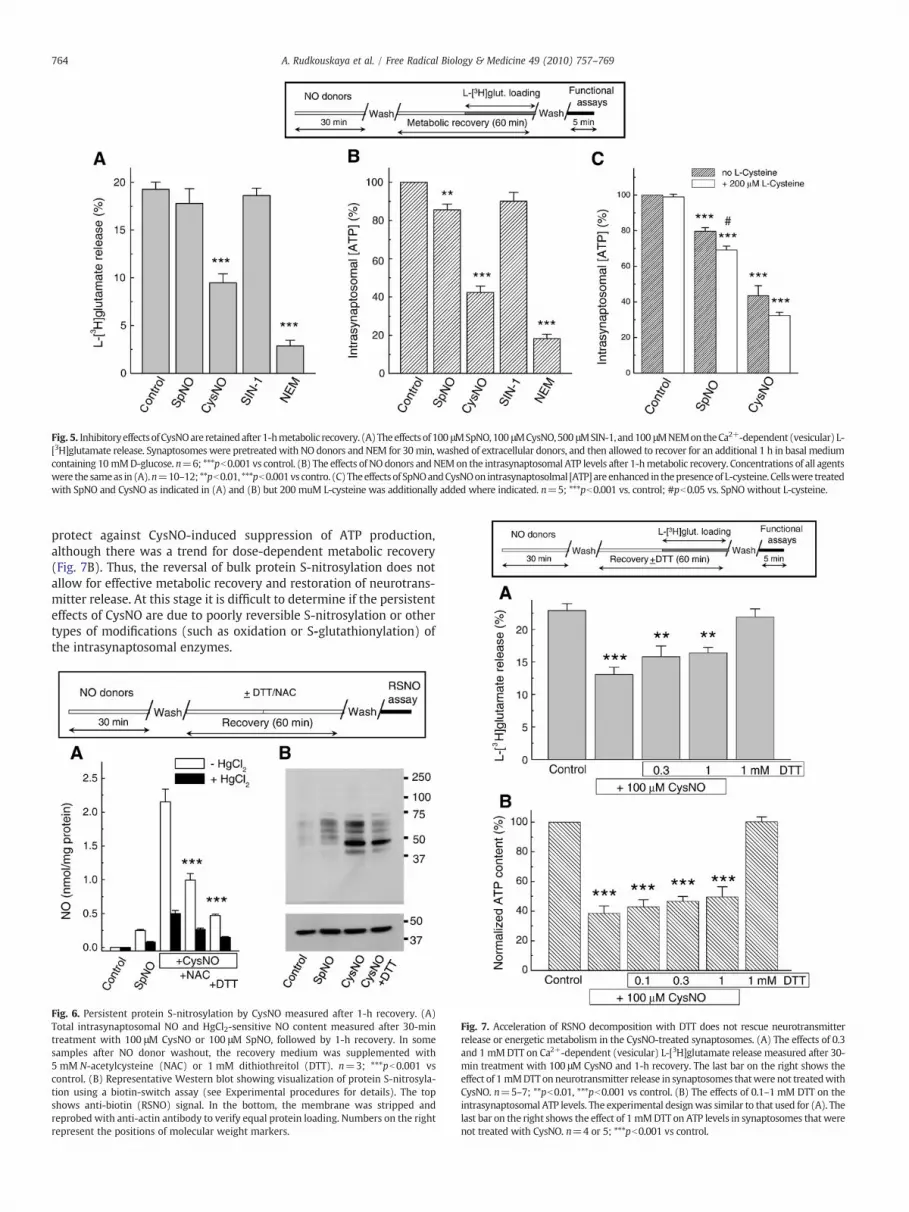
Fig. 5. InhibitoryeffectsofCysNOare retainedafter1-hmetabolic recovery. (A)Theeffectsof 100 μMSpNO,100 μMCysNO,500 μMSIN-1, and100 μMNEMontheCa2+-dependent (vesicular) L-[3H]glutamate release. Synaptosomes were pretreatedwith NO donors and NEM for 30min, washed of extracellular donors, and then allowed to recover for an additional 1 h in basal mediumcontaining 10mMD-glucose. n=6; ***pb0.001 vs control. (B) The effects of NOdonors andNEMon the intrasynaptosomal ATP levels after 1-hmetabolic recovery. Concentrations of all agentswere the sameas in (A).n=10–12; **pb0.01, ***pb0.001vs contro. (C)Theeffectsof SpNOandCysNOon intrasynaptosolmal [ATP]areenhanced in thepresenceof L-cysteine. Cellswere treatedwith SpNO and CysNO as indicated in (A) and (B) but 200 muM L-cysteine was additionally added where indicated. n=5; ***pb0.001 vs. control; #pb0.05 vs. SpNO without L-cysteine.
764 A. Rudkouskaya et al. / Free Radical Biology & Medicine 49 (2010) 757–769
protect against CysNO-induced suppression of ATP production,although there was a trend for dose-dependent metabolic recovery(Fig. 7B). Thus, the reversal of bulk protein S-nitrosylation does notallow for effective metabolic recovery and restoration of neurotrans-mitter release. At this stage it is difficult to determine if the persistenteffects of CysNO are due to poorly reversible S-nitrosylation or othertypes of modifications (such as oxidation or S-glutathionylation) ofthe intrasynaptosomal enzymes.
Fig. 6. Persistent protein S-nitrosylation by CysNO measured after 1-h recovery. (A)Total intrasynaptosomal NO and HgCl2-sensitive NO content measured after 30-mintreatment with 100 μM CysNO or 100 μM SpNO, followed by 1-h recovery. In somesamples after NO donor washout, the recovery medium was supplemented with5 mM N-acetylcysteine (NAC) or 1 mM dithiothreitol (DTT). n=3; ***pb0.001 vscontrol. (B) Representative Western blot showing visualization of protein S-nitrosyla-tion using a biotin-switch assay (see Experimental procedures for details). The topshows anti-biotin (RSNO) signal. In the bottom, the membrane was stripped andreprobed with anti-actin antibody to verify equal protein loading. Numbers on the rightrepresent the positions of molecular weight markers.
Fig. 7. Acceleration of RSNO decomposition with DTT does not rescue neurotransmitterrelease or energetic metabolism in the CysNO-treated synaptosomes. (A) The effects of 0.3and 1 mMDTT on Ca2+-dependent (vesicular) L-[3H]glutamate release measured after 30-min treatment with 100 μM CysNO and 1-h recovery. The last bar on the right shows theeffect of 1 mMDTTonneurotransmitter release in synaptosomes thatwere not treatedwithCysNO. n=5–7; **pb0.01, ***pb0.001 vs control. (B) The effects of 0.1–1 mM DTT on theintrasynaptosomal ATP levels. The experimental designwas similar to that used for (A). Thelast bar on the right shows the effect of 1 mMDTT on ATP levels in synaptosomes that werenot treated with CysNO. n=4 or 5; ***pb0.001 vs control.
765A. Rudkouskaya et al. / Free Radical Biology & Medicine 49 (2010) 757–769
Treatment with pyruvate leads to recovery of vesicular neurotransmitterrelease and ATP levels in CysNO-treated synaptosomes
In the next series of experiments, we attempted to identify whichpart of synaptosomal energetic metabolism is affected by the treatmentwith CysNO. With this purpose, we supplemented the recoverymedium with pyruvate, the end-product of glycolysis and a precursorsubstrate for the mitochondrial Krebs cycle. After 1 h recovery in thepresence of 0.5–5 mM pyruvate, intrasynaptosomal ATP levels weresignificantly but not completely restored (Fig. 8A). We selected 2 mMpyruvate, the most effective concentration, for further testing inneurotransmitter release experiments. At this concentration, pyruvaterestored Ca2+-dependent L-[3H]glutamate release by more than 50%(Fig. 8B). Thus, CysNO-mediated long-term inhibition of neurotrans-mitter release in synaptosomes may be reversed by bypassingglycolysis. Under control conditions N80% of synaptosomal ATP isderived from oxidative phosphorylation; however, the mitochondrial/glycolytic ATP ratiomay be strongly altered by depolarization and othertreatments [57,58]. In our control experiments, replacement of D-glucose with D-deoxyglucose completely inhibited vesicular neuro-transmitter release but this effect was completely reversed by adding5 mM pyruvate, indicating that in intact synaptosomes ATP levels canbe sustained by oxidative phosphorylation without glycolysis (Supple-lemental Fig. 4). In D-glucose-containingmedium, whenmitochondrial
Fig. 8. Posttreatment with pyruvate partially rescues neurotransmitter release and ATPproduction in the CysNO-treated synaptosomes. (A) The effect of 0.5–5 mM pyruvateon the intrasynaptosomal ATP levels measured after 30-min treatment with 100 μMCysNO and 1-h recovery. The last bar on the right shows the effect of 5 mM pyruvate insynaptosomes that were not treated with CysNO. n=5; *pb0.05, **pb0.01, ***pb0.001vs control; ##pb0.01, ###pb0.001 vs CysNO. (B) The effects of 2 mM pyruvate on Ca2+-dependent (vesicular) L-[3H]glutamate release measured after 30-min treatment with100 μM CysNO and 1-h recovery. The last bar on the right shows the effect of 2 mMpyruvate on neurotransmitter release in synaptosomes that were not treated withCysNO. n=6 or 7; *pb0.05, ***pb0.001 vs control; ##pb0.01 vs CysNO.
metabolism was suppressed with the complex I inhibitor rotenone(10 μM), vesicular glutamate release was reduced by ∼50% (Supple-lemental Fig. 4). Thus, glycolysis can only partially sustain vesicularneurotransmitter release and ATP production.
Effects of CysNO, pyruvate, and DTT on the activity of GAPDH andenolase
GAPDH is a crucial glycolytic enzyme that is inhibited by NO viaS-nitrosylation or covalent linkage of the NAD moiety [59,60].Therefore, we assessed how DTT or pyruvate affects GAPDH activityin synaptosomes after incubation with CysNO. Synaptosomes weretreated in the same fashion as in the preceding experiments including1-h recovery. Pyruvate and DTT were present only in the recoverymedia. GAPDH activity was measured in synaptosomal lysates bymonitoring the reduction of NAD+ to NADH in the presence of theGAPDH substrate, glyceraldehyde 3-phosphate. As expected, the CysNOtreatment blocked GAPDH activity by ∼80%, and this effect persistedafter 1-h metabolic recovery (Fig. 9A). As expected, the inhibition was
Fig. 9. Treatment with CysNO causes long-lasting inhibition of synaptosomal GAPDHactivity but does not affect the enolase activity. (A) Measurements of GAPDH activity.Synaptosomes were treated for 30 minwith 100 μMCysNO and then allowed to recoverfor an additional 1 h in the basal medium containing 10 mM D-glucose or 10 mMglucose +2 mM pyruvate. GAPDH activity was measured in synaptosomal lysates asdescribed under Experimental procedures. n=4; *pb0.05, ***pb0.001 vs control;###pb0.001 vs CysNO. (B) Measurements of enolase activity. Experimental conditionswere identical to those presented in (A). Enolase activity was measured in lysates asdescribed under Experimental procedures. n=3.

766 A. Rudkouskaya et al. / Free Radical Biology & Medicine 49 (2010) 757–769
not reversed by post-CysNO treatment with pyruvate (Fig. 9A).However, treatment with 1 mM DTT completely reversed the CysNOeffect and even increasedGAPDHactivity to levels higher than in controlsamples. Additionally, DTT strongly elevated GAPDH activity on its own(Fig. 9A). This finding is consistent with the literature report of apartially oxidized state of GAPDH even in the absence of oxidative/nitrosylative stress [61]. However, in contrast to restoration of GAPDHactivity, DTT did not reverse the CysNO-induced suppression ofneurotransmitter release and ATP synthesis (see Fig. 7). Thus, thereduction of SH groups in GAPDH is not sufficient for restoration ofcellular functions in CysNO-treated synaptosomes and additionalglycolytic enzymes are likely to be irreversibly blocked. Recently, ithas been found that neuron-specific enolase is one of the prominentlynitrosylated enzymes in an animal model of multiple sclerosis [31].Therefore, we measured enolase activity in the lysates of CysNO-, DTT-,and pyruvate-treated synaptosomes. Unlike GAPDHactivity in the samesamples, the enolase activity was unchanged regardless of type oftreatment (Fig. 9B).
Discussion
It is well established that NO and RNS originating from nNOS andiNOS participate in the acute and delayed forms of brain damage instroke and other neuropathologies [5,7,13,62]. In this study weexploredmolecularmechanisms thatmay link nitrosative stress to thelong-term inhibition of synaptic communication. Such inhibition hasbeen found in animal models of stroke [16–18] but its mechanismsremain poorly understood. We established that in synaptosomalpreparations NO donors potently suppress neurotransmitter releasevia inhibition of energetic metabolism, with the order of potencycorrelating with S-nitrosylation of intrasynaptosomal proteins. Incontrast, the total amount of released NO was not related to theinhibition of synaptic metabolism. Global changes in ATP levels didnot allow us to test our original hypothesis on nitrosative inhibition ofthe NSF protein, the activity of which is sustained by ATP hydrolysis[36]. Overall, our findings are in agreement with early observations byBrorson et al., who have found long-term inhibition of energeticmetabolism by authentic NO and NO donors in cultured hippocampalneurons [63]. However, in synaptosomes, energetic metabolism andneurotransmitter release seem to be more sensitive to inhibition ofglycolysis than in intact neuronal preparations and could be rescuedby addition of the end-product of glycolysis, pyruvate.
Protein S-nitrosylation causes long-term inhibition of vesicularneurotransmitter release
The major finding of our study is that S-nitrosylation ofintrasynaptosomal proteins produces potent inhibition of the vesic-ular neurotransmitter release that persisted after washout ofextracellular RNS donors and additional metabolic recovery. Theefficacy of RNS-generating compounds was not related to the totalamount of released NO or the kinetics of NO donor decomposition.Instead, we found a very close correlation between the inhibitorypotency of individual RNS-generating compounds, the decreasedlevels of intrasynaptosomal ATP, and the increased levels ofintrasynaptosomal RSNOs. NO itself possesses very limited reactivitytoward SH groups compared to other NO metabolites such as •NO2,N2O3, RSNOs, or peroxynitrite, all of which oxidize and/or nitrosylatethiols much more readily than NO itself [38,64–66]. This may explainthe weak potency of the “pure” NO donors spermine NONOate andMAHMA NONOate in our experiments.
S-nitroso-L-cysteine was the most effective among the testedcompounds. Our analysis of bound intrasynaptosomal NO indicatedthat among possible modifications more than 85% of the CysNO-mediated increase in the intrasynaptosomal NO was represented byRSNOs. Thus, protein S-nitrosylation, or related RSNO-driven mod-
ifications such as S-glutathionylation, produces a long-term impact onpresynaptic metabolism and ATP-dependent vesicular neurotrans-mitter release. Persistent ATP depletion was not related to thereversible inhibition of mitochondrial cytochrome oxidase by NO[67,68], which in our experiments has been seen only during acutetreatment with NO donors. Instead, inhibition of the ATP productioninvolved more complex mechanisms.
Consistent with previous studies [43,44,48,69], the high efficacy ofCysNO was related to its transmembrane transport, which wasprobably followed by direct protein transnitrosylation. Similar tothe results obtained in nonneuronal preparations (see for example[43]), extracellularly added L-cysteine increased the potency of the“pure” NO donor SpNO, probably by producing extracellular CysNO.Previous pharmacological analysis and siRNA-knockdown studiesestablished an essential role for the amino acid transport system L forCysNO uptake and its biological activity in several cell types [44,48]. Inour experiments, L-leucine, a competitive inhibitor of the amino acidtransport system L, strongly suppressed formation of the intrasynap-tosomal thiols. Additional effects of L-serine, which blocks the aminoacid transport system ASC, indicate that this system may alsocontribute to CysNO transfer into synaptosomes. When both the Land the ASC transporter inhibitors were combined, intracellular RSNOformation was only partially prevented, suggesting that othertransport mechanisms may additionally contribute. Based on thepharmacological evidence, it has been proposed that γ-glutamyltranspeptidase may serve as an alternative route for transmembraneS-nitrosothiol transfer [52].
Mechanisms and reversibility of nitrosative inhibition of energeticmetabolism
Although the effects of SpNO and CysNO on synaptic function werelong-lasting, we did observe slow spontaneous recovery of neurotrans-mitter release and ATP levels after removal of RNS donors. Weattempted to hasten the recovery from nitrosative stress by includingin the recoverymediumthe thiol-reducing agentDTTor themembrane-permeative glutathione precursor N-acetylcysteine. Although bothagents strongly accelerated RSNO decomposition, seen as a reductionin the total RSNO signal and decreased biotinylation of individualprotein bands in the biotin-switch assays, they were paradoxicallyineffective at restoring ATP levels or neurotransmitter release. Onepotential explanation is relative resistance of some CysNO-modifiedthiol groups to the reducing agents. Consistent with this idea, weobserved theDTT-resistantnitrosylation of anunidentifiedproteinbandwith an apparent molecular weight of ∼45–48 kDa. In the brain, manyenzymes belonging to the glycolytic pathway and mitochondrialelectron transfer chain may be modified and inhibited by nitrosativestress [7,68]. Paige et al. identified in brain cytosolic lysates 10intracellular proteins, the S-nitrosylation of which was highly resistantto reducing reagents; these included two metabolic enzymes—GAPDHand pyruvate kinase [56]. In our experiments, supplementation of therecoverymediumwith the end-product of glycolysis, pyruvate, stronglyrestored both ATP levels and neurotransmitter release, pointing to theimportant role of glycolysis in the observed metabolic inhibition. Theglycolytic enzyme GAPDH is the known intracellular target fornitrosative inhibition [55,59,70]. NO and RSNOs may reversibly inhibitGAPDH via S-nitrosylation or irreversibly block its activity via covalentNAD attachment [59,60,71,72]. In our study, strong and persistentreductionofGAPDHactivitywas reversedafter treatmentwithDTT. Thisfinding probably points to S-nitrosylation, but inhibition via S-glutathionylation has also been reported in both cellular and cell-freeassay systems [73]. Strong suppressionof theGAPDHactivity seen in ourexperiments is quantitatively important for the CysNO-mediatedinhibition of synaptosomal ATP production. However, because theactivity of GAPDH was restored by the thiol-reducing agents withoutconcurrent recoveryofATP levels, ourfindings suggest that activity of an

767A. Rudkouskaya et al. / Free Radical Biology & Medicine 49 (2010) 757–769
additional unidentified glycolytic enzyme(s) should be persistentlysuppressed in a DTT-resistant manner. Neuron-specific enolase wasrecently found to be prominently nitrosylated in vivo in an animalmodel of multiple sclerosis [31] but its activity was not affected in theCysNO-treated samples in our experiments.
Because the effect of pyruvate on synaptosomal ATP levels wasincomplete, this finding points to impaired mitochondrial function.Previously, Erecinska et al. showed that CysNO strongly inhibitsmitochondrial O2 consumption and decreases synaptosomal ATPlevels with additional moderate inhibition of glycolysis [74], but N25,80, and 70% decreases in the activities of mitochondrial complexes I,III, and IV, respectively, had to be reached before any changes insynaptosomal ATP production were observed [75]. In mitochondria,complexes I, II, and IV and the tricarboxylic acid cycle enzymeaconitase are potently inhibited by RNS, particularly by peroxynitrite(reviewed in [68]). Modification via S-nitrosylation strongly sup-presses the activity of complex I [76,77] and inhibits aconitase [78].
Pathological and translational relevance of the present findings
Although chronic inhibition of synaptic transmission has beenreported in both focal and global animal ischemia models, itsmechanisms remain poorly understood. The defects in synaptictransmission may last for several weeks and are seen despitecompletely recovered membrane potentials and metabolism inneuronal bodies [17,79]. It is plausible that because of the length ofneuronal projections, the “remote” metabolic and functional proper-ties, particularly at the presynaptic sites, are disproportionally affectedin the postischemic tissue. Under this scenario, local nitrosative andoxidative inhibition of metabolic enzymes and disruption of mito-chondrial function in synaptic endings (and dendrites) would requirede novo protein synthesis and long-distance axonal delivery ofmitochondria. One recent study found chronic degeneration ofneuronal processes and disruption of synaptic communication in theCA1 hippocampal region 10 weeks after brief global ischemia [20].Importantly, in that work a combinatorial pharmacological treatmentthat afforded nearly complete protection of neuronal bodies did notpreserve neuronal projections and synaptic function.
Our study proposes one potential mechanism responsible forfunctional and metabolic deficits at the presynaptic sites. The in vitroresults presented here have to be considered and interpreted withcaution. Our model offers very limited representation of themultifaceted processes in the ischemic brain and does not reflectthe complete spectrum of pathologically produced reactive oxygenspecies and RNS. Although in our work and in cultured neurons [63]the peroxynitrite donor SIN-1 showed very low potency, this may bedue to low steady-state ONOO− levels and limited permeability ofneuronalmembranes to ONOO−, perhaps due to the low expression ofthe anion exchanger that is necessary for peroxynitrite transport [80].Another important question that requires additional work is whetherpathological levels of nitrosylation reach those attained in ourexperiments. As mentioned in the introduction, measuring RSNOlevels in vivo is a challenging task. Nevertheless, numerous brainproteins are nitrosylated even under physiological conditions by thenNOS-derived RNS [23], and a strong increase in protein nitrosylationhas been identified in the CNS in various animal models of neuro-degenerative diseases and in human patients [27–29,31]. Theassociated in vivo chemistry and the identity of S-nitrosylatingspecies are still being debated. However, decreased O2 levels, asexpected in the ischemic tissue, seem to enhance the RSNO formation[38,81].
Despite all the above-mentioned limitations our work providessome important clues for developing novel therapeutic approaches forpreservation and restoration of the synaptic function in variousneuropathologies that are associated with nitrosative stress. Applica-tion of pyruvate or its derivatives may allow bypassing of the per-
sistently inhibited steps in the glycolytic pathway. As seen in ourexperiments, if mitochondrial function is partially preserved, pyru-vate, when added after nitrosative stress, restores intrasynaptosomalATP levels and synaptic neurotransmitter release. Incidentally,pyruvate and its derivative ethyl pyruvate, which is more stable insolution, have been already tested in animal ischemia models.Systemic delivery of pyruvate or ethyl pyruvate affords potentneuroprotection in rodent models of global and focal ischemia withan extended therapeutic window [82–84], as well as againstischemia–reperfusion injury in several other tissues (reviewed in[85]). It remains to be tested whether pyruvate may effectivelyprotect against loss of synaptic activity or be used for restoration ofsynaptic communication in the postischemic brain in vivo.
Acknowledgments
We thank Aniqa Anwar for measuring the activity of the voltage-gated Ca2+ channels, Katharine E. Halligan and Frances L. Jourd'heuilfor help with chemiluminescence RSNO assays, and Preeti Dohare andNicole Bowens for experimental help. This work was supported byGrant NS061953 from the National Institutes of Health (A.A.M.) andAmerican Heart Association Student Scholarships in CerebrovascularDiseases and Stroke (Vasiliy Sim and Aniqa Anwar).
Appendix A. Supplementary data
Supplementary data associated with this article can be found, inthe online version, at doi:10.1016/j.freeradbiomed.2010.05.032.
References
[1] Stamler, J. S. Redox signaling: nitrosylation and related target interactions of nitricoxide. Cell 78:931–936; 1994.
[2] Beckman, J. S.; Koppenol, W. H. Nitric oxide, superoxide, and peroxynitrite: thegood, the bad, and ugly. Am. J. Physiol. 271:C1424–C1437; 1996.
[3] Wink, D. A.; Mitchell, J. B. Chemical biology of nitric oxide: insights intoregulatory, cytotoxic, and cytoprotective mechanisms of nitric oxide. Free Radic.Biol. Med. 25:434–456; 1998.
[4] Hess, D. T.; Matsumoto, A.; Kim, S. O.; Marshall, H. E.; Stamler, J. S. Protein S-nitrosylation: purview and parameters. Nat. Rev. Mol. Cell Biol. 6:150–166; 2005.
[5] Samdani, A. F.; Dawson, T. M.; Dawson, V. L. Nitric oxide synthase in models offocal ischemia. Stroke 28:1283–1288; 1997.
[6] Moncada, S.; Bolanos, J. P. Nitric oxide, cell bioenergetics and neurodegeneration.J. Neurochem. 97:1676–1689; 2006.
[7] Pacher, P.; Beckman, J. S.; Liaudet, L. Nitric oxide and peroxynitrite in health anddisease. Physiol. Rev. 87:315–424; 2007.
[8] Hossmann, K. A. Viability thresholds and the penumbra of focal ischemia. Ann.Neurol. 36:557–565; 1994.
[9] Astrup, J.; Siesjo, B. K.; Symon, L. Thresholds in cerebral ischemia—the ischemicpenumbra. Stroke 12:723–725; 1981.
[10] Dirnagl, U.; Iadecola, C.; Moskowitz, M. A. Pathobiology of ischaemic stroke: anintegrated view. Trends Neurosci. 22:391–397; 1999.
[11] Nishikawa, T.; Kirsch, J. R.; Koehler, R. C.; Bredt, D. S.; Snyder, S. H.; Traystman, R. J.Effect of nitric oxide synthase inhibition on cerebral blood flow and injury volumeduring focal ischemia in cats. Stroke 24:1717–1724; 1993.
[12] Huang, Z.; Huang, P. L.; Panahian, N.; Dalkara, T.; Fishman, M. C.; Moskowitz, M. A.Effects of cerebral ischemia in mice deficient in neuronal nitric oxide synthase.Science 265:1883–1885; 1994.
[13] Lo, E. H.; Dalkara, T.; Moskowitz, M. A. Mechanisms, challenges and opportunitiesin stroke. Nat. Rev. Neurosci. 4:399–415; 2003.
[14] Schiene, K.; Bruehl, C.; Zilles, K.; Qu, M.; Hagemann, G.; Kraemer, M.; Witte, O. W.Neuronal hyperexcitability and reduction of GABAA-receptor expression in thesurround of cerebral photothrombosis. J. Cereb. Blood Flow Metab. 16:906–914;1996.
[15] Qu, M.; Mittmann, T.; Luhmann, H. J.; Schleicher, A.; Zilles, K. Long-term changesof ionotropic glutamate and GABA receptors after unilateral permanent focalcerebral ischemia in the mouse brain. Neuroscience 85:29–43; 1998.
[16] Bolay, H.; Dalkara, T. Mechanisms of motor dysfunction after transient MCAocclusion: persistent transmission failure in cortical synapses is a majordeterminant. Stroke 29:1988–1993; 1998.
[17] Neumann-Haefelin, T.; Witte, O. W. Periinfarct and remote excitability changesafter transient middle cerebral artery occlusion. J. Cereb. Blood Flow Metab. 20:45–52; 2000.
[18] Bolay, H.; Gursoy-Ozdemir, Y.; Sara, Y.; Onur, R.; Can, A.; Dalkara, T. Persistentdefect in transmitter release and synapsin phosphorylation in cerebral cortexafter transient moderate ischemic injury. Stroke 33:1369–1375; 2002.

768 A. Rudkouskaya et al. / Free Radical Biology & Medicine 49 (2010) 757–769
[19] Kovalenko, T.; Osadchenko, I.; Nikonenko, A.; Lushnikova, I.; Voronin, K.;Nikonenko, I.; Muller, D.; Skibo, G. Ischemia-induced modifications in hippocam-pal CA1 stratum radiatum excitatory synapses. Hippocampus 16:814–825; 2006.
[20] Iyirhiaro, G. O.; Brust, T. B.; Rashidian, J.; Galehdar, Z.; Osman, A.; Phillips, M.;Slack, R. S.; MacVicar, B. A.; Park, D. S. Delayed combinatorial treatment withflavopiridol and minocycline provides longer term protection for neuronal somabut not dendrites following global ischemia. J. Neurochem. 105:703–713; 2008.
[21] Stamler, J. S.; Toone, E. J.; Lipton, S. A.; Sucher, N. J. (S)NO signals: translocation,regulation, and a consensus motif. Neuron 18:691–696; 1997.
[22] Stamler, J. S.; Lamas, S.; Fang, F. C. Nitrosylation: the prototypic redox-basedsignaling mechanism. Cell 106:675–683; 2001.
[23] Jaffrey, S. R.; Erdjument-Bromage, H.; Ferris, C. D.; Tempst, P.; Snyder, S. H. ProteinS-nitrosylation: a physiological signal for neuronal nitric oxide. Nat. Cell Biol. 3:193–197; 2001.
[24] Lipton, S. A.; Choi, Y. B.; Takahashi, H.; Zhang, D.; Li, W.; Godzik, A.; Bankston, L. A.Cysteine regulation of protein function—as exemplified by NMDA-receptormodulation. Trends Neurosci. 25:474–480; 2002.
[25] Feelisch, M.; Rassaf, T.; Mnaimneh, S.; Singh, N.; Bryan, N. S.; Jourd'heuil, D.; Kelm,M. Concomitant S-, N-, and heme-nitros(yl)ation in biological tissues and fluids:implications for the fate of NO in vivo. FASEB J. 16:1775–1785; 2002.
[26] Liu, L.; Yan, Y.; Zeng, M.; Zhang, J.; Hanes, M. A.; Ahearn, G.; McMahon, T. J.;Dickfeld, T.; Marshall, H. E.; Que, L. G.; Stamler, J. S. Essential roles of S-nitrosothiols in vascular homeostasis and endotoxic shock. Cell 116:617–628;2004.
[27] Bizzozero, O. A.; DeJesus, G.; Bixler, H. A.; Pastuszyn, A. Evidence of nitrosativedamage in the brain white matter of patients with multiple sclerosis. Neurochem.Res. 30:139–149; 2005.
[28] Uehara, T.; Nakamura, T.; Yao, D.; Shi, Z. Q.; Gu, Z.; Ma, Y.; Masliah, E.; Nomura, Y.;Lipton, S. A. S-nitrosylated protein-disulphide isomerase links protein misfoldingto neurodegeneration. Nature 441:513–517; 2006.
[29] Fang, J.; Nakamura, T.; Cho, D. H.; Gu, Z.; Lipton, S. A. S-nitrosylation ofperoxiredoxin 2 promotes oxidative stress-induced neuronal cell death inParkinson's disease. Proc. Natl Acad. Sci. USA 104:18742–18747; 2007.
[30] Nakamura, T.; Lipton, S. A. Emerging roles of S-nitrosylation in protein misfoldingand neurodegenerative diseases. Antioxid. Redox Signal. 10:87–101; 2008.
[31] Bizzozero, O. A.; Zheng, J. Identification of major S-nitrosylated proteins in murineexperimental autoimmune encephalomyelitis. J. Neurosci. Res. 87:2881–2889;2009.
[32] Mongin, A. A.; Nedvetsky, P. I.; Fedorovich, S. V. Depolarization of isolated brainnerve endings by nitric oxide donors: membrane mechanisms. Biochemistry(Moscow) 63:662–670; 1998.
[33] Sequeira, S. M.; Carvalho, A. P.; Carvalho, C. M. Both protein kinase G dependentand independent mechanisms are involved in the modulation of glutamaterelease by nitric oxide in rat hippocampal nerve terminals. Neurosci. Lett. 261:29–32; 1999.
[34] Nedvetsky, P. I.; Konev, S. V.; Rakovich, A. A.; Petrenko, S. V.; Mongin, A. A. Effectsof nitric oxide donors on Ca2+-dependent [14C]GABA release from brainsynaptosomes: the role of SH-groups. Biochemistry (Moscow) 65:1027–1035;2000.
[35] Matsushita, K.; Morrell, C. N.; Cambien, B.; Yang, S. X.; Yamakuchi, M.; Bao, C.;Hara, M. R.; Quick, R. A.; Cao, W.; O'Rourke, B.; Lowenstein, J. M.; Pevsner, J.;Wagner, D. D.; Lowenstein, C. J. Nitric oxide regulates exocytosis by S-nitrosylation of N-ethylmaleimide-sensitive factor. Cell 115:139–150; 2003.
[36] Mongin, A. A. Nitric oxidemay contribute to the long-term impairment of synaptictransmission after transient ischemia. Stroke 33:2348–2350; 2002.
[37] Hajos, F. An improved method for the preparation of synaptosomal fractions inhigh purity. Brain Res. 93:485–489; 1975.
[38] Jourd'heuil, D.; Jourd'heuil, F. L.; Feelisch, M. Oxidation and nitrosation of thiols atlow micromolar exposure to nitric oxide: evidence for a free radical mechanism.J. Biol. Chem. 278:15720–15726; 2003.
[39] Mongin, A. A.; Aksentsev, S. L.; Orlov, S. N.; Konev, S. V. Hypoosmotic shockactivates Ca2+ channels in isolated nerve terminals. Neurochem. Int. 31:835–843;1997.
[40] Nicholls, D. G. The glutamatergic nerve terminal. Eur. J. Biochem. 212:613–631;1993.
[41] Banerjee, A.; Barry, V. A.; DasGupta, B. R.; Martin, T. F. N-ethylmaleimide-sensitivefactor acts at a prefusion ATP-dependent step in Ca2+-activated exocytosis. J. Biol.Chem. 271:20223–20226; 1996.
[42] Schiavo, G.; Benfenati, F.; Poulain, B.; Rossetto, O.; Polverino, D. L.; DasGupta, B. R.;Montecucco, C. Tetanus and botulinum-B neurotoxins block neurotransmitterrelease by proteolytic cleavage of synaptobrevin. Nature 359:832–835; 1992.
[43] Zhang, Y.; Hogg, N. The mechanism of transmembrane S-nitrosothiol transport.Proc. Natl Acad. Sci. USA 101:7891–7896; 2004.
[44] Broniowska, K. A.; Zhang, Y.; Hogg, N. Requirement of transmembrane transportfor S-nitrosocysteine-dependent modification of intracellular thiols. J. Biol. Chem.281:33835–33841; 2006.
[45] Clementi, E.; Brown, G. C.; Foxwell, N.; Moncada, S. On the mechanism by whichvascular endothelial cells regulate their oxygen consumption. Proc. Natl Acad. Sci.USA 96:1559–1562; 1999.
[46] Martin-Romero, F. J.; Gutierrez-Martin, Y.; Henao, F.; Gutierrez-Merino, C.Fluorescence measurements of steady state peroxynitrite production upon SIN-1 decomposition: NADH versus dihydrodichlorofluorescein and dihydrorhoda-mine 123. J. Fluoresc. 14:17–23; 2004.
[47] Konishi, K.; Watanabe, N.; Arai, T. SIN-1 cytotoxicity to PC12 cells is mediated bythiol-sensitive short-lived substances generated through SIN-1 decomposition inculture medium. Nitric Oxide 20:270–278; 2009.
[48] Li, S.; Whorton, A. R. Identification of stereoselective transporters for S-nitroso-L-cysteine: role of LAT1 and LAT2 in biological activity of S-nitrosothiols. J. Biol.Chem. 280:20102–20110; 2005.
[49] Romero, J. M.; Bizzozero, O. A. Extracellular S-nitrosoglutathione, but not S-nitrosocysteine or N2O3, mediates protein S-nitrosation in rat spinal cord slices. J.Neurochem. 99:1299–1310; 2006.
[50] Shotwell, M. A.; Jayme, D. W.; Kilberg, M. S.; Oxender, D. L. Neutral amino acidtransport systems in Chinese hamster ovary cells. J. Biol. Chem. 256:5422–5427;1981.
[51] Chen, Y.; Swanson, R. A. The glutamate transporters EAAT2 and EAAT3 mediatecysteine uptake in cortical neuron cultures. J. Neurochem. 84:1332–1339; 2003.
[52] Lipton, A. J.; Johnson, M. A.; Macdonald, T.; Lieberman, M.W.; Gozal, D.; Gaston, B.S-nitrosothiols signal the ventilatory response to hypoxia. Nature 413:171–174;2001.
[53] Summers, B. A.; Overholt, J. L.; Prabhakar, N. R. Nitric oxide inhibits L-type Ca2+
current in glomus cells of the rabbit carotid body via a cGMP-independentmechanism. J. Neurophysiol. 81:1449–1457; 1999.
[54] D'Ascenzo, M.; Martinotti, G.; Azzena, G. B.; Grassi, C. cGMP/protein kinase G-dependent inhibition of N-type Ca2+ channels induced by nitric oxide in humanneuroblastoma IMR32 cells. J. Neurosci. 22:7485–7492; 2002.
[55] Romero, J. M.; Bizzozero, O. A. Intracellular glutathione mediates the denitrosyla-tion of protein nitrosothiols in the rat spinal cord. J. Neurosci. Res. 87:701–709;2009.
[56] Paige, J. S.; Xu, G.; Stancevic, B.; Jaffrey, S. R. Nitrosothiol reactivity profiling identifiesS-nitrosylated proteins with unexpected stability. Chem. Biol. 15:1307–1316; 2008.
[57] Erecinska, M.; Dagani, F. Relationships between the neuronal sodium/potassiumpump and energy metabolism: effects of K+, Na+, and adenosine triphosphate inisolated brain synaptosomes. J. Gen. Physiol. 95:591–616; 1990.
[58] Erecinska, M.; Dagani, F.; Nelson, D.; Deas, J.; Silver, I. A. Relations betweenintracellular ions and energy metabolism: a study with monensin in synapto-somes, neurons, and C6 glioma cells. J. Neurosci. 11:2410–2421; 1991.
[59] Vedia, L.; McDonald, B.; Reep, B.; Brune, B.; Di Silvio, M.; Billiar, T. R.; Lapetina, E. G.Nitric oxide-induced S-nitrosylation of glyceraldehyde-3-phosphate dehydroge-nase inhibits enzymatic activity and increases endogenous ADP-ribosylation.J. Biol. Chem. 267:24929–24932; 1992.
[60] McDonald, L. J.; Moss, J. Stimulation by nitric oxide of an NAD linkage toglyceraldehyde-3-phosphate dehydrogenase. Proc. Natl Acad. Sci. USA 90:6238–6241; 1993.
[61] Leichert, L. I.; Gehrke, F.; Gudiseva, H. V.; Blackwell, T.; Ilbert, M.; Walker, A. K.;Strahler, J. R.; Andrews, P. C.; Jakob, U. Quantifying changes in the thiol redoxproteome upon oxidative stress in vivo. Proc. Natl Acad. Sci. USA 105:8197–8202;2008.
[62] Guix, F. X.; Uribesalgo, I.; Coma, M.; Munoz, F. J. The physiology andpathophysiology of nitric oxide in the brain. Prog. Neurobiol. 76:126–152; 2005.
[63] Brorson, J. R.; Schumacker, P. T.; Zhang, H. Nitric oxide acutely inhibits neuronalenergy production. J. Neurosci. 19:147–158; 1999.
[64] Moro, M. A.; Darley-Usmar, V. M.; Goodwin, D. A.; Read, N. G.; Zamora-Pino, R.;Feelisch, M.; Radomski, M. W.; Moncada, S. Paradoxical fate and biological actionof peroxynitrite on human platelets. Proc. Natl Acad. Sci. USA 91:6702–6706; 1994.
[65] Arnelle, D. R.; Stamler, J. S. NO+, NO, and NO− donation by S-nitrosothiols:implications for regulation of physiological functions by S-nitrosylation andacceleration of disulfide formation. Arch. Biochem. Biophys. 318:279–285; 1995.
[66] Espey, M. G.; Miranda, K. M.; Thomas, D. D.; Wink, D. A. Distinction betweennitrosating mechanisms within human cells and aqueous solution. J. Biol. Chem.276:30085–30091; 2001.
[67] Brown, G. C.; Cooper, C. E. Nanomolar concentrations of nitric oxide reversiblyinhibit synaptosomal respiration by competing with oxygen at cytochromeoxidase. FEBS Lett. 356:295–298; 1994.
[68] Brown, G. C. Nitric oxide and mitochondrial respiration. Biochim. Biophys. Acta1411:351–369; 1999.
[69] Jia, L.; Bonaventura, C.; Bonaventura, J.; Stamler, J. S. S-nitrosohaemoglobin: adynamic activity of blood involved in vascular control. Nature 380:221–226; 1996.
[70] Dimmeler, S.; Lottspeich, F.; Brune, B. Nitric oxide causes ADP-ribosylation andinhibition of glyceraldehyde-3-phosphate dehydrogenase. J. Biol. Chem. 267:16771–16774; 1992.
[71] Padgett, C. M.; Whorton, A. R. S-nitrosoglutathione reversibly inhibits GAPDH byS-nitrosylation. Am. J. Physiol. 269:C739–C749; 1995.
[72] Mohr, S.; Stamler, J. S.; Brune, B. Posttranslational modification of glyceraldehyde-3-phosphate dehydrogenase by S-nitrosylation and subsequent NADH attach-ment. J. Biol. Chem. 271:4209–4214; 1996.
[73] Mohr, S.; Hallak, H.; de Boitte, A.; Lapetina, E. G.; Brune, B. Nitric oxide-induced S-glutathionylation and inactivation of glyceraldehyde-3-phosphate dehydroge-nase. J. Biol. Chem. 274:9427–9430; 1999.
[74] Erecinska, M.; Nelson, D.; Vanderkooi, J. M. Effects of NO-generating compoundson synaptosomal energy metabolism. J. Neurochem. 65:2699–2705; 1995.
[75] Davey, G. P.; Peuchen, S.; Clark, J. B. Energy thresholds in brain mitochondria:potential involvement in neurodegeneration. J. Biol. Chem. 273:12753–12757; 1998.
[76] Clementi, E.; Brown, G. C.; Feelisch, M.; Moncada, S. Persistent inhibition of cellrespiration by nitric oxide: crucial role of S-nitrosylation of mitochondrialcomplex I and protective action of glutathione. Proc. Natl Acad. Sci. USA 95:7631–7636; 1998.
[77] Borutaite, V.; Budriunaite, A.; Brown, G. C. Reversal of nitric oxide-, peroxynitrite-and S-nitrosothiol-induced inhibition of mitochondrial respiration or complex Iactivity by light and thiols. Biochim. Biophys. Acta 1459:405–412; 2000.
[78] Tortora, V.; Quijano, C.; Freeman, B.; Radi, R.; Castro, L. Mitochondrial aconitasereaction with nitric oxide, S-nitrosoglutathione, and peroxynitrite: mechanisms

769A. Rudkouskaya et al. / Free Radical Biology & Medicine 49 (2010) 757–769
and relative contributions to aconitase inactivation. Free Radic. Biol. Med. 42:1075–1088; 2007.
[79] Mittmann, T.; Qu, M.; Zilles, K.; Luhmann, H. J. Long-term cellular dysfunctionafter focal cerebral ischemia: in vitro analyses. Neuroscience 85:15–27; 1998.
[80] Denicola, A.; Souza, J. M.; Radi, R. Diffusion of peroxynitrite across erythrocytemembranes. Proc. Natl Acad. Sci. USA 95:3566–3571; 1998.
[81] Bosworth, C. A.; Toledo Jr., J. C.; Zmijewski, J. W.; Li, Q.; Lancaster Jr., J. R.Dinitrosyliron complexes and the mechanism(s) of cellular protein nitro-sothiol formation from nitric oxide. Proc. Natl Acad. Sci. USA 106:4671–4676;2009.
[82] Lee, J. Y.; Kim, Y. H.; Koh, J. Y. Protection by pyruvate against transient forebrainischemia in rats. J. Neurosci. 21:RC171; 2001.
[83] Yu, Y. M.; Kim, J. B.; Lee, K. W.; Kim, S. Y.; Han, P. L.; Lee, J. K. Inhibition of thecerebral ischemic injury by ethyl pyruvate with a wide therapeutic window.Stroke 36:2238–2243; 2005.
[84] Yi, J. S.; Kim, T. Y.; Kyu, K. D.; Koh, J. Y. Systemic pyruvate administrationmarkedlyreduces infarcts and motor deficits in rat models of transient and permanent focalcerebral ischemia. Neurobiol. Dis. 26:94–104; 2007.
[85] Fink, M. P. Ethyl pyruvate: a novel anti-inflammatory agent. J. Intern. Med. 261:349–362; 2007.





























