Embed Size (px)
Citation preview

R
Ps
MI
a
AA
KICSPB
C
1aCta2p1p2sftt
0d
Journal of Chromatography A, 1216 (2009) 1861–1880
Contents lists available at ScienceDirect
Journal of Chromatography A
journa l homepage: www.e lsev ier .com/ locate /chroma
eview
artitioning behaviour of organic compounds between ionic liquids andupercritical fluids
ichal Roth ∗
nstitute of Analytical Chemistry of the ASCR, v. v. i., Veverí 97, 60200 Brno, Czech Republic
r t i c l e i n f o
rticle history:vailable online 15 October 2008
a b s t r a c t
Applications and prospects of two-phase, tuneable solvent systems composed of ionic liquids (ILs) andsupercritical fluids with an emphasis on supercritical carbon dioxide (scCO2) are reviewed. The IL–scCO2
eywords:onic liquidarbon dioxideupercritical fluid chromatographyartition coefficientiphasic solvent
biphasic systems have increasingly been used in diverse fields of chemistry and technology, and someexamples of these applications are mentioned here. Rational design of such applications can obviouslybenefit from pertinent data on phase equilibria including the partition coefficients of the prospectiveproducts and reactants between the two phases. Therefore, a reliable technique to measure the limitingpartition coefficients would be of value. Here, the pros and cons of supercritical fluid chromatography inthis respect are discussed. An overview of methods for predictive thermodynamic modelling of binary
(IL–scCO2) and ternary (solute–IL–scCO2) systems is also included.© 2008 Elsevier B.V. All rights reserved.
ontents
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18622. Ionic liquids and supercritical fluids. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1862
2.1. Biphasic solvent systems . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18622.2. Application fields of ionic liquid–supercritical carbon dioxide systems . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1863
2.2.1. Extractions from ionic liquids with supercritical carbon dioxide . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18632.2.2. Reactions in ionic liquid–supercritical carbon dioxide systems . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18642.2.3. Supercritical carbon dioxide as a phase separation switch . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1866
2.3. Phase behaviour in ionic liquid–supercritical carbon dioxide binary systems – measurement and modelling . . . . . . . . . . . . . . . . . . . . . . . . . . 1867
2.3.1. Solubility measurements of supercritical carbon dioxide2.3.2. Melting point depression in organic salts by compressed2.3.3. Thermodynamic models of ionic liquid–supercritical car3. Ternary organic nonelectrolyte–ionic liquid–carbon dioxide systems . . . .
Abbreviations: acac, acetylacetonate; APAM, N-acetyl-(S)-phenylalanine met-n-butyl-3-methylimidazolium; bmpy, 1-n-butyl-3-methylpyridinium; bupy, butylpyrlkyl; C6H4F9mim, 3-(3,3,4,4,5,5,6,6,6-nonafluorohexyl)-1-methylimidazolium; C8H4F13
andida antarctica lipase B; dca, dicyanoamide; dppe, 1,2-bis(diphenylphosphino)ethions of state; GC EoS, group contribution equation of state; HDS, hydrodesulfurimmonium acetate; HEAF, 2-(2-hydroxyethoxy) ammonium formate; HEAL, 2-(2-hydr-hydroxyethyl ammonium lactate; hmim, 1-n-hexyl-3-methylimidazolium; hmmim,oint; LSER, linear solvation energy relationship; MAAC, methyl-(Z)-�-acetamidocinnam-n-octyl-3-methylimidazolium; P[MATMA], poly([2-(methacryloyloxy)ethyl]trimethyl aoly(p-vinylbenzyltrimethyl ammonium); PCN, 2-(4-methyl-2-phenyl-1-piperazinyl)-3-,3-dimethylimidazolium; PVT, pressure–volume–temperature (behaviour); S–L, Sancheupercritical carbon dioxide; SILP, supported ionic liquid phase; soft-SAFT, soft statistical aonyl; Tf2N, bis(trifluoromethylsulfonyl)imide; TfO, trifluoromethanesulfonate (triflate); tri(2-hydroxy ethyl) ammonium lactate; thtdp, trihexyltetradecylphosphonium; tolBINAPruncated perturbed chain polar statistical associating fluid theory.∗ Tel.: +420 532290171; fax: +420 541212113.
E-mail address: [email protected].
021-9673/$ – see front matter © 2008 Elsevier B.V. All rights reserved.oi:10.1016/j.chroma.2008.10.032
in ionic liquids . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1867carbon dioxide . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1871
bon dioxide binary systems . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1871. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1873
hyl ester; ATR-IR, attenuated total reflectance infrared (spectroscopy); bmim,idinium; Cnmim, 1-n-alkyl-3-methylimidazolium with n carbon atoms in themim, 3-(3,3,4,4,5,5,6,6,7,7,8,8,8-tridecafluorooctyl)-1-methylimidazolium; CALB,
ane; emim, 1-ethyl-3-methylimidazolium; EoS, equation of state; EoSs, equa-sation; HEA, 2-hydroxyethyl ammonium acetate; HEAA, 2-(2-hydroxyethoxy)oxyethoxy) ammonium lactate; HEF, 2-hydroxyethyl ammonium formate; HEL,1-n-hexyl-2,3-dimethylimidazolium; IL, ionic liquid; LCEP, lower critical endate; MPhPz, 1-methyl-3-phenylpiperazine; NtN, 2-chloro-nicotinonitrile; omim,mmonium); P[VBMI], poly(1-(p-vinylbenzyl)-3-methylimidazolium); P[VBTMA],pyridinecarbonitrile; pmim, 1-propyl-3-methylimidazolium; pmmim, 1-propyl-z–Lacombe (equation of state); SAFT, statistical associating fluid theory; scCO2,ssociating fluid theory; SWCF, square well chain fluid (EoS); Tf, trifluoromethylsul-ha, tetrahexylammonium; THEAA, tri(2-hydroxyethyl) ammonium acetate; THEAL,, 2,2′-bis(di-p-tolylphosphino)-1,1′-binaphthyl; TON, turnover number; tPC-PSAFT,

1862 M. Roth / J. Chromatogr. A 1216 (2009) 1861–1880
4. Supercritical fluid chromatography niche in the studies of solute–ionic liquid–supercritical carbon dioxide systems . . . . . . . . . . . . . . . . . . . . . . . . . 18744.1. Limiting partition coefficients of solutes in ionic liquid–supercritical carbon dioxide systems . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1874
4.1.1. Experimental studies of solute partitioning . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18754.1.2. Modelling of solute partitioning . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18754.1.3. Pros and cons of supercritical fluid chromatography in solute–ionic liquid–supercritical carbon dioxide measurements . . . . . 1877
4.2. Supercritical fluid chromatography with ionic liquids in analytical separations? . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18785. Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1878
Acknowledgements . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1878. . . . . .
1
3irietpmit
pRmsmcpccphttavstr
i[osmnstesouStrylits
2
tsataotp
tWhica9csein
2
btrwratbnb[
tpfttcf
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. Introduction
Ionic liquids (ILs) are organic salts with melting points below73 K. After the discovery of the first IL, ethylammonium nitrate,n 1914 [1], this class of liquids composed solely of ions haveemained largely an academic curiosity for decades to come. Thentroduction of chloroaluminate ILs in 1982 [2] did not mark anssential change of the situation because the sensitivity of these ILso moisture prevented their widespread application. The change inerception of ILs probably came with the introduction of air- andoisture-stable ILs in 1992 [3]; recently, the year-to-year increase
n the number of publications regarding ILs has been exponen-ial.
Unlike molecular liquids, ILs usually have negligible vapourressures; recent reviews of physical properties of ILs include, e.g.,efs. [4–6]. ILs also often feature a wide liquidus range, high ther-al stability and wide electrochemical window. These desirable
olvent characteristics [7–10] make ILs worthwhile alternatives toolecular solvents for synthesis [11,12], catalysis [13,14], electro-
hemistry [15,16], or extractions [17,18]. Because of wide variety ofossible cation–anion combinations, the solvent properties of ILsan be “tailored” to fit a particular application; consequently, ILsan be considered “designer solvents” [19]. In some applications,urity of ILs can be a critical factor as the presence of impuritiesas been shown to modify the physical properties of ILs, notablyheir viscosity [20]. As the ILs do not evaporate, they do not con-ribute to pollution of atmosphere by volatile organic compounds,nd therefore, they have sometimes been referred to as “green sol-ents”. In general, however, the “green” label to ILs is not justified asome ILs are rather toxic to microbial and aquatic life [21–26], andhe frequently used ILs containing the PF6
− anion can hydrolyse toelease hydrogen fluoride [27].
Among their diverse applications, ILs have also often been usedn analytical chemistry, e.g., in chromatography and extraction28,29], electrophoresis [30,31], gas sensor development [32,33]r as matrices for matrix-assisted laser desorption/ionisation masspectrometry [34,35]. The relationship of ILs and gas–liquid chro-atography (GLC) has been two-sided. In analytical GLC, the dual
ature of ILs has been utilised, i.e., their ability to act as nonpolartationary liquids toward nonpolar analytes and as highly polar sta-ionary liquids toward polar analytes [36,37]. In turn, GLC has beenmployed as a thermodynamic measurement method in numeroustudies to determine limiting (infinite-dilution) activity coefficientsf solutes from the solute retention volumes measured with a col-mn containing the particular IL as the stationary liquid [38–44].ome of the GLC studies [45–47] also provided insight into con-ributions of different mechanisms of retention to the overall netetention volume of the solute. Retention measurements by GLC
ield “neat” information on solute–IL interactions because, at theow pressures employed, the carrier gas (usually helium) solubilityn the IL is negligible. At elevated pressures, however, gas solubili-ies in ILs are no longer negligible, especially in condensable gasesuch as CO2 or ethylene.ososA
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1878
. Ionic liquids and supercritical fluids
Among the diverse uses of ILs, a specific place belongs tohe applications combining the ILs with supercritical fluids. Thepecificity of IL–supercritical fluid biphasic systems follows fromvailability of several mechanisms for tuning the solvent proper-ies of such systems – apart from the wide selection of IL cationsnd IL anions to tailor the IL properties as mentioned above, theperating temperature and pressure are also available as variableso adjust the density and the solvent power of the supercritical fluidhase.
Historically, the first reports of IL–supercritical fluid interac-ions are probably those of Wightman and co-workers [48,49].
ithout referring specifically to ILs, these authors used tetra-exylammonium hexafluorophosphate ([tha][PF6]) as an additive
n voltammetry of ferrocene in water-containing supercriti-al carbon dioxide (scCO2) [48], and, later, [tha][NO3] as andditive in voltammetric studies of ferrocene, anthracene and,10-diphenylanthracene in water-containing scCO2 and in water-ontaining supercritical N2O [49]. They used IR spectroscopy tohow that [tha][PF6] was not soluble in scCO2 to any measurablextent [48], and they also noted large melting point depressionsnduced in [tha][PF6] [48] and in [tha][NO3] [49] by contact withear-critical fluids (see Section 2.3.2).
.1. Biphasic solvent systems
An important impulse for the current extent of application ofiphasic solvent systems has come from the development of phaseransfer catalysis in aqueous–organic systems [50–52] and fluo-ous biphasic catalysis in fluorous–organic systems [53]. By analogyith “aqueous”, the term “fluorous” [53] refers to highly fluo-
inated organic compounds (mostly alkanes, ethers and tertiarymines). The primary reason for introducing these techniques waso facilitate the separation and recovery of products and catalystsy employing widely different solubilities of the individual compo-ents in the two immiscible liquid phases. Accounts of history ofiphasic catalysis have been given, e.g., by Keim [54] and by Sheldon55].
Typically, a biphasic system for homogeneous catalysis containshe catalyst in the heavier (lower) phase solvent while the upperhase solvent carries the reactant(s) in and the products out. There-ore, the lower solvent should be able to dissolve the catalyst andhe reactant(s) whereas the upper solvent should be able to dissolvehe reactant(s) and products but neither the lower solvent nor theatalyst. In addition, the lower solvent should be easily separablerom the products and should be environmentally benign [56].
Compared with the classical aqueous–organic or fluorous–
rganic biphasic solvent systems, applications of the IL–upercritical fluid systems are more recent and still less numer-us but no less varied. The supercritical fluids involved includeupercritical carbon dioxide (scCO2) and hydrofluorocarbons.lthough a considerable effort has been invested in the study of
M. Roth / J. Chromatogr. A 1216 (2009) 1861–1880 1863
Fa1C
I[tApamIihcitceIooscb
2s
toK
2d
ttfTaIistuc
Fa1C
rpssertsspCif[essextractions were carried out at 313 K and 13.8 MPa. Fig. 3 indi-cates that the solute affinity toward the IL phase increased withthe dipole moment of the solute, and that solid solutes were muchmore difficult to extract as compared with liquid solutes.
ig. 1. Extraction of aromatic solutes from [bmim][PF6] with scCO2 at 313 Knd 13.8 MPa. Numbers in the legend indicate solute dipole moments (debye,D = 3.3356 × 10−30 C m). Reprinted with permission from [72]. © 2001 Americanhemical Society.
L–hydrofluorocarbon systems, especially as regards measurement57] and equation of state (EoS) modelling [58] of phase equilibria,he applications of IL–scCO2 biphasic systems remain dominant.pparently, the important reasons for that include affordablerice, ready availability and environmental compatibility of CO2s compared with hydrofluorocarbons. Another (and more funda-ental) reason may come from different phase behaviours of the
L–hydrofluorocarbon and IL–scCO2 binary systems: while mostmidazolium-based ILs are almost insoluble in scCO2 even at veryigh pressures [59], this is not necessarily the case in hydrofluoro-arbons as shown, e.g., by the phase equilibrium measurementsn [emim][PF6]–CHF3 system [60]. In most prospective applica-ions, an appreciable solubility of IL in supercritical fluid wouldomplicate the separation of the reaction products and the recov-ry of the IL, thus detracting from usefulness of the particularL–supercritical fluid system. Therefore, the practical insolubilityf most imidazolium-based ILs in scCO2 contributes to dominancef scCO2 in applications of IL–supercritical fluid biphasic solventystems. As shown by Wu et al. [61,62] and Zhang et al. [63], polarosolvents such as ethanol or acetone are needed to raise the solu-ilities of imidazolium ILs in scCO2 to readily measurable levels.
.2. Application fields of ionic liquid–supercritical carbon dioxideystems
The IL–scCO2 systems have predominantly been used in extrac-ions and reactions. Earlier reviews of applications and propertiesf IL–scCO2 systems include those of Dzyuba and Bartsch [64] andeskin et al. [65].
.2.1. Extractions from ionic liquids with supercritical carbonioxide
As mentioned above, ILs have increasingly been used as reac-ion media. After a reaction has been completed in an IL medium,he reaction products and residual reactants have to be separatedrom the IL. This can be done either by distillation or by extraction.he distillation route makes use of the effective nonvolatility of ILss opposed to non-ionic organic compounds (although even someLs can be distilled [66–70]). However, the applicability of distill-
ng the non-ionic components from a reaction mixture with an ILolvent may be limited. First, distillation is an energy-intensive andherefore costly process. Second, many prospective reaction prod-cts are thermally labile and would decompose on distilling. Theseonsiderations appear to favour the low-temperature extractionFaR
ig. 2. Extraction of aliphatic solutes from [bmim][PF6] with scCO2 at 313 Knd 13.8 MPa. Numbers in the legend indicate solute dipole moments (debye,D = 3.3356 × 10−30 C m). Reprinted with permission from [72]. © 2001 Americanhemical Society.
oute. Extractions with conventional liquid solvents can be com-licated because, when a particular non-ionic compound is to beeparated from a particular IL, finding a liquid solvent of sufficientelectivity is not easy, and the danger of cross-contamination of thextraction solvent with the IL is usually imminent. Besides, solventesidues in the extracted product(s) can be highly undesirable andheir removal costly. For the above reasons, Blanchard et al. [71]uggested the extraction of organic nonelectrolytes from ILs withcCO2 as a convenient procedure avoiding cross-contamination. Aarticularly attractive feature of this procedure is that both IL andO2 can be recycled. Shortly after, Blanchard and Brennecke [72]
nvestigated feasibility of using scCO2 to separate organic solutesrom ILs. They determined the solubilities of 20 organic solutes inbmim][PF6] and provided quantitative data on extraction recov-ry rates of aromatic and aliphatic solutes from [bmim][PF6] withcCO2. Figs. 1 and 2 show the recoveries of aromatic and aliphaticolutes, respectively, as functions of the CO2/solute mole ratio. The
ig. 3. Ease of extraction of solutes from [bmim][PF6] with scCO2 at 313 Knd 13.8 MPa versus the solute dipole moment (debye, 1D = 3.3356 × 10−30 C m).eprinted with permission from [72]. © 2001 American Chemical Society.

1 r. A 12
ata
rhr{ssws
T(tcert
ftwsc(g�t[ec
iatt
pistccttf
sr[mia1swsuamab
2s
erptdfrvSarnst
[CtppawtcN
w[bptost
otspsca
uaesdpIamddi3
864 M. Roth / J. Chromatog
Since the seminal papers by Blanchard et al. [71] and Blanchardnd Brenecke [72], diverse applications have been devised of extrac-ions of nonelectrolytes from ILs with scCO2, and some examplesre given below.
Brown et al. [56] used wet IL [bmim][PF6] for asymmet-ic hydrogenation of tiglic acid to 2-methylbutanoic acid withigh enantiomeric excess (>85%) and high conversion (>97%). Theeaction in the IL phase was catalysed by a rhodium chelateRu(O2CMe)2[(R)-tolBINAP]} and the product was extracted withcCO2 without cross-contamination by either IL or the catalyst. Theolution of the catalyst in [bmim][PF6] could be used repeatedlyithout significant losses of both enantioselectivity and conver-
ion.Bortolini et al. [73] employed several ILs [bmim][X] (X = BF4, PF6,
fO and Tf2N) for epoxidation of several electron-deficient olefins2-cyclohexen-1-one and its derivatives) with basic aqueous solu-ion of hydrogen peroxide at room temperature. In most cases, aomplete consumption of the substrate was observed in the pres-nce of a small excess of oxidant and in short reaction time. Theesultant epoxides could be almost quantitatively extracted fromhe reaction mixture with scCO2.
Supercritical CO2 can also be used to extract metal chelatesrom ILs. Mekki et al. [74] extracted Cu(II) from [bmim][Tf2N] inhe form of trifluoroacetonate or hexafluoroacetonate hydratesith a contingent addition of tributylphosphate as a CO2-philic
ynergist agent to stabilise the metal chelates. The extraction effi-iencies varied somewhat with the experimental arrangementstatic/dynamic extraction, with/without tributylphosphate) butenerally exceeded 95%. Mekki et al. [75] also used fluorinated-diketones, hexafluoroacetylacetonate and 4,4,4-trifluoro-1-(2-
hienyl)-1,3-butanedione, to extract Eu(III) and Ln(III) frombmim][Tf2N] or from aqueous solution via [bmim][Tf2N]. In somexperiments, tributylphosphate was added to stabilise the metalhelates, and extraction efficiencies exceeding 87% were achieved.
Zhou et al. [76] synthesised IL precursor [bmim][Cl] by conduct-ng the reaction of 1-methylimidazole with butylchloride in scCO2,nd used extraction with scCO2 to remove residual reactants afterhe reaction. Residual reactants could be completely removed andhe yield of [bmim][Cl] exceeded 90%.
Yoon et al. [77] employed extraction with scCO2 to separate theroducts of Heck reaction (Csp2–Csp2 coupling reaction) between
odobenzene and olefins from a reaction mixture in [bmim][PF6]olvent. The reaction was catalysed by PdCl2, and the reaction mix-ure also had to contain triethylamine to assist dissolution of theatalyst in [bmim][PF6]. Depending on temperature and on waterontent in the IL, the conversion of iodobenzene and styrene torans-stilbene exceeded 90% under most conditions. Extraction ofhe products with scCO2 made it possible to recycle the catalystour times without loss of activity.
Kroon et al. [78] demonstrated various applications of scCO2 ineparating organic solutes from ILs by experimental study of theecovery of N-acetyl-(S)-phenylalanine methyl ester (APAM) frombmim][BF4]. APAM is the product of asymmetric hydrogenation of
ethyl-(Z)-�-acetamidocinnamate (MAAC) that can be conductedn ILs. APAM extracted from [bmim][BF4] contained no detectablemount of the IL; the solubility of APAM in scCO2 at 323 K and2 MPa was 1.78 g kg−1 whereas the solubility of [bmim][BF4] incCO2 was negligible. The solubility of the reactant MAAC in scCO2as five times lower than the solubility of APAM so that sufficient
electivity toward extraction of APAM existed. In addition, the prod-
ct APAM could also be separated from the IL by using scCO2 as annti-solvent because the solubility of APAM in [bmim][BF4]–scCO2ixture was lower than the solubility of APAM in [bmim][BF4] atmbient conditions. After precipitation, the crystals of APAM coulde purified by washing with CO2.
pcapb
16 (2009) 1861–1880
.2.2. Reactions in ionic liquid–supercritical carbon dioxideystems
In addition to the examples discussed in the preceding sections,xtraction from IL with scCO2 has often been used in tandem with aeaction in IL medium as an integral part of the reactant → productathway. Flowing scCO2 can serve both to carry the substrate (reac-ant) to the IL and then extract the product(s) out of the IL. Afterecompression of the fluid mixture, the product(s) obtained areree from IL and from organic solvent residues whereas CO2 can beecompressed and recycled. Therefore, the process may result inery significant reduction of waste generation as measured, e.g., byheldon’s E factor [79,80] given by the ratio (mass of waste gener-ted)/(mass of product). Besides, if the reaction product does notequire any further refining and/or purification, the potential eco-omic benefit of the process can also be very important, as theeparation/purification operations in large-scale chemical plantsypically account for ∼50% of investment costs [81].
Liu et al. [82] demonstrated an application ofbmim][PF6]–scCO2 system for hydrogenation of olefins andO2. Hydrogenation of dec-1-ene proceeded in 98% conversiono n-decane in 1 h at 323 K and 4.8 MPa H2 pressure and a totalressure (H2 + CO2) of 20.7 MPa. Hydrogenation of cyclohexeneroceeded more slowly to dec-1-ene. 82% conversion after 2 hnd 96% conversion after 3 h under the same conditions asith Hydrogenation of CO2 in [bmim][PF6]–scCO2 system in
he presence of dialkylamines was catalysed by scCO2-insolubleomplex RuCl2(dppe)2 (dppe = Ph2PCH2CH2PPh2) and produced,N-dialkylformamides.
Sellin et al. [83] studied hydroformylation of linear alkenesith >5 carbon atoms (hex-1-ene, oct-1-ene and non-1-ene) in
bmim][PF6]–scCO2 biphasic system. The reaction was catalysedy a rhodium complex, and it was operated in a continuous flowrocess in which the substrate alkene, the gases (H2 and CO) andhe products were transported in and out of the reactor by a streamf scCO2. With oct-1-ene, the process could be operated at a con-tant rate for over 20 h and the collected product contained lesshan 1 ppm Rh.
Bösmann et al. [84] explored the possibility to immobiliserganometallic catalysts with transition metals in ILs for prospec-ive use in IL–scCO2 continuous flow systems. They employedeveral IL–scCO2 combinations to activate and immobilise a com-lex Ni-containing catalyst for the hydrovinylation of styrene. Theyhowed that the combination of a suitable IL and compressed CO2ould offer much more potential for process optimisation than justsimple protocol for batchwise catalyst recycling.
Another demonstration of how the presence of scCO2 can besed to feed the substrate to and remove the product from a cat-lytic reaction in IL medium was given by Ballivet-Tkatchenkot al. [85]. They used the [bmim][BF4]–scCO2 system for bipha-ic Pd(acac)2-catalysed dimerisation of methyl methacrylate toimethyl dihydromuconates. For comparison, the reaction was alsoerformed under monophasic conditions in the absence of scCO2.
n the biphasic system, the selectivity for the tail-to-tail dimers wass high as under monophasic reaction conditions (>98%). Whileethyl methacrylate was readily soluble in scCO2 throughout the
ensity range employed, an effective solubilisation of dimethylihydromuconates required the scCO2 density above 0.4 g cm−3,
.e., a pressure exceeding 15 MPa at the operating temperature of53 K.
In the study of enantioselective hydrogenation of N-(1-henylethylidene)aniline using cationic iridium complexes with
hiral phosphinooxazoline ligands, Solinas et al. [86] showed somedditional benefits of IL–CO2 biphasic reaction media. First, theresence of CO2 caused a dramatic increase in hydrogen solu-ility in the IL as indicated by high-pressure NMR. At 297 K and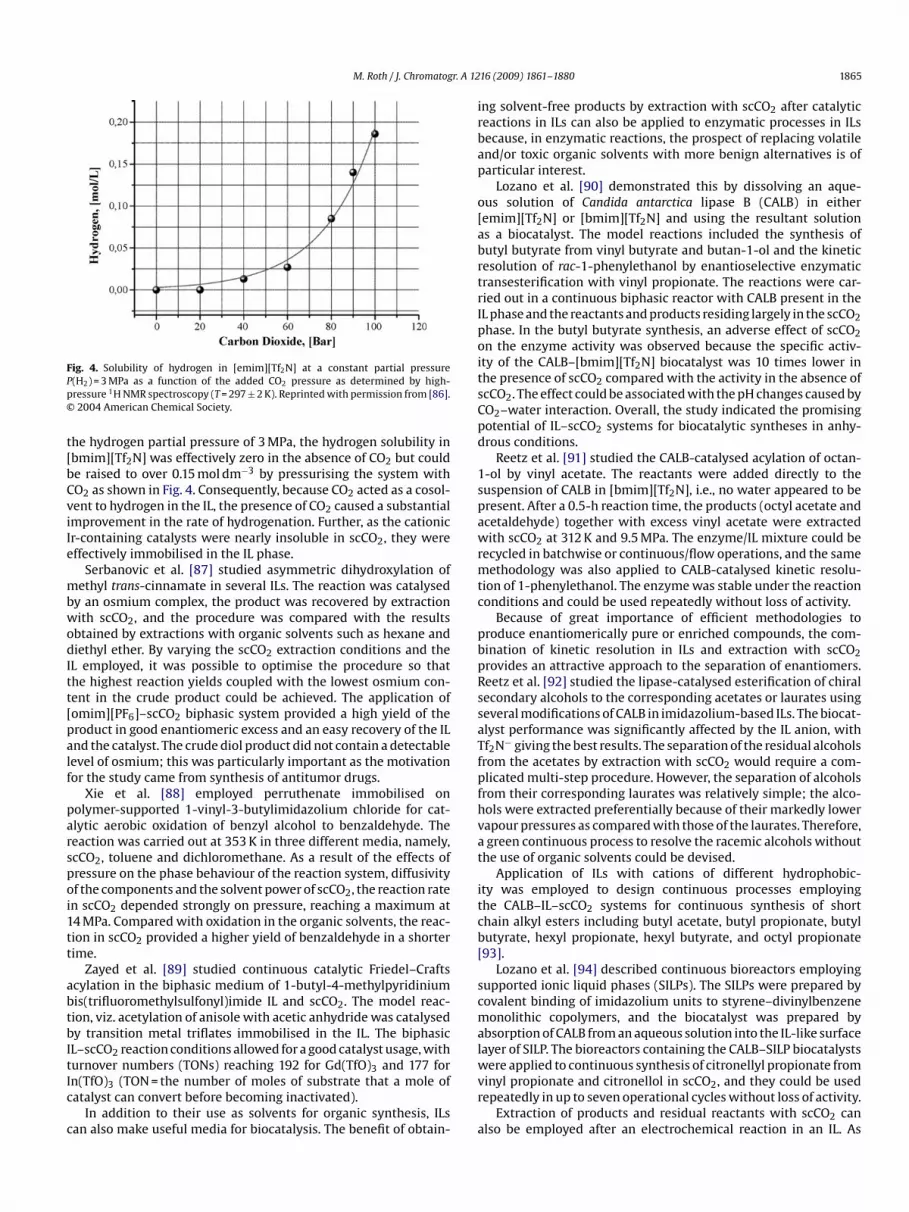
M. Roth / J. Chromatogr. A 12
FPp©
t[bCviIe
mbwodItt[palf
parspoi1tt
abtbItIc
c
irbap
o[abrtrIpoitsCpd
1spawrmtc
pbpRssaTfpfhvat
itcb[
scmal
ig. 4. Solubility of hydrogen in [emim][Tf2N] at a constant partial pressure(H2) = 3 MPa as a function of the added CO2 pressure as determined by high-ressure 1H NMR spectroscopy (T = 297 ± 2 K). Reprinted with permission from [86].2004 American Chemical Society.
he hydrogen partial pressure of 3 MPa, the hydrogen solubility inbmim][Tf2N] was effectively zero in the absence of CO2 but coulde raised to over 0.15 mol dm−3 by pressurising the system withO2 as shown in Fig. 4. Consequently, because CO2 acted as a cosol-ent to hydrogen in the IL, the presence of CO2 caused a substantialmprovement in the rate of hydrogenation. Further, as the cationicr-containing catalysts were nearly insoluble in scCO2, they wereffectively immobilised in the IL phase.
Serbanovic et al. [87] studied asymmetric dihydroxylation ofethyl trans-cinnamate in several ILs. The reaction was catalysed
y an osmium complex, the product was recovered by extractionith scCO2, and the procedure was compared with the results
btained by extractions with organic solvents such as hexane andiethyl ether. By varying the scCO2 extraction conditions and the
L employed, it was possible to optimise the procedure so thathe highest reaction yields coupled with the lowest osmium con-ent in the crude product could be achieved. The application ofomim][PF6]–scCO2 biphasic system provided a high yield of theroduct in good enantiomeric excess and an easy recovery of the ILnd the catalyst. The crude diol product did not contain a detectableevel of osmium; this was particularly important as the motivationor the study came from synthesis of antitumor drugs.
Xie et al. [88] employed perruthenate immobilised onolymer-supported 1-vinyl-3-butylimidazolium chloride for cat-lytic aerobic oxidation of benzyl alcohol to benzaldehyde. Theeaction was carried out at 353 K in three different media, namely,cCO2, toluene and dichloromethane. As a result of the effects ofressure on the phase behaviour of the reaction system, diffusivityf the components and the solvent power of scCO2, the reaction raten scCO2 depended strongly on pressure, reaching a maximum at4 MPa. Compared with oxidation in the organic solvents, the reac-ion in scCO2 provided a higher yield of benzaldehyde in a shorterime.
Zayed et al. [89] studied continuous catalytic Friedel–Craftscylation in the biphasic medium of 1-butyl-4-methylpyridiniumis(trifluoromethylsulfonyl)imide IL and scCO2. The model reac-ion, viz. acetylation of anisole with acetic anhydride was catalysedy transition metal triflates immobilised in the IL. The biphasicL–scCO2 reaction conditions allowed for a good catalyst usage, with
urnover numbers (TONs) reaching 192 for Gd(TfO)3 and 177 forn(TfO)3 (TON = the number of moles of substrate that a mole ofatalyst can convert before becoming inactivated).In addition to their use as solvents for organic synthesis, ILsan also make useful media for biocatalysis. The benefit of obtain-
wvr
a
16 (2009) 1861–1880 1865
ng solvent-free products by extraction with scCO2 after catalyticeactions in ILs can also be applied to enzymatic processes in ILsecause, in enzymatic reactions, the prospect of replacing volatilend/or toxic organic solvents with more benign alternatives is ofarticular interest.
Lozano et al. [90] demonstrated this by dissolving an aque-us solution of Candida antarctica lipase B (CALB) in eitheremim][Tf2N] or [bmim][Tf2N] and using the resultant solutions a biocatalyst. The model reactions included the synthesis ofutyl butyrate from vinyl butyrate and butan-1-ol and the kineticesolution of rac-1-phenylethanol by enantioselective enzymaticransesterification with vinyl propionate. The reactions were car-ied out in a continuous biphasic reactor with CALB present in theL phase and the reactants and products residing largely in the scCO2hase. In the butyl butyrate synthesis, an adverse effect of scCO2n the enzyme activity was observed because the specific activ-ty of the CALB–[bmim][Tf2N] biocatalyst was 10 times lower inhe presence of scCO2 compared with the activity in the absence ofcCO2. The effect could be associated with the pH changes caused byO2–water interaction. Overall, the study indicated the promisingotential of IL–scCO2 systems for biocatalytic syntheses in anhy-rous conditions.
Reetz et al. [91] studied the CALB-catalysed acylation of octan--ol by vinyl acetate. The reactants were added directly to theuspension of CALB in [bmim][Tf2N], i.e., no water appeared to beresent. After a 0.5-h reaction time, the products (octyl acetate andcetaldehyde) together with excess vinyl acetate were extractedith scCO2 at 312 K and 9.5 MPa. The enzyme/IL mixture could be
ecycled in batchwise or continuous/flow operations, and the sameethodology was also applied to CALB-catalysed kinetic resolu-
ion of 1-phenylethanol. The enzyme was stable under the reactiononditions and could be used repeatedly without loss of activity.
Because of great importance of efficient methodologies toroduce enantiomerically pure or enriched compounds, the com-ination of kinetic resolution in ILs and extraction with scCO2rovides an attractive approach to the separation of enantiomers.eetz et al. [92] studied the lipase-catalysed esterification of chiralecondary alcohols to the corresponding acetates or laurates usingeveral modifications of CALB in imidazolium-based ILs. The biocat-lyst performance was significantly affected by the IL anion, withf2N− giving the best results. The separation of the residual alcoholsrom the acetates by extraction with scCO2 would require a com-licated multi-step procedure. However, the separation of alcoholsrom their corresponding laurates was relatively simple; the alco-ols were extracted preferentially because of their markedly lowerapour pressures as compared with those of the laurates. Therefore,green continuous process to resolve the racemic alcohols without
he use of organic solvents could be devised.Application of ILs with cations of different hydrophobic-
ty was employed to design continuous processes employinghe CALB–IL–scCO2 systems for continuous synthesis of shorthain alkyl esters including butyl acetate, butyl propionate, butylutyrate, hexyl propionate, hexyl butyrate, and octyl propionate93].
Lozano et al. [94] described continuous bioreactors employingupported ionic liquid phases (SILPs). The SILPs were prepared byovalent binding of imidazolium units to styrene–divinylbenzeneonolithic copolymers, and the biocatalyst was prepared by
bsorption of CALB from an aqueous solution into the IL-like surfaceayer of SILP. The bioreactors containing the CALB–SILP biocatalysts
ere applied to continuous synthesis of citronellyl propionate frominyl propionate and citronellol in scCO2, and they could be usedepeatedly in up to seven operational cycles without loss of activity.
Extraction of products and residual reactants with scCO2 canlso be employed after an electrochemical reaction in an IL. As

1 r. A 1216 (2009) 1861–1880
atIIpcsw
itpat
Iehctcr
m
1rtiTafffwwhba
br0cptc
2
ta(pI
iTicapttp
FpS
aAKvct“paws[
haaomstii
be3Tptst
ifsma
cmiaI
866 M. Roth / J. Chromatog
n example, Zhao et al. [95] carried out electrochemical oxida-ion of benzyl alcohol to benzaldehyde in the liquid phase of anL–scCO2 biphasic system containing a small amount of water. TheLs employed were [bmim][PF6] and [bmim][BF4], with the latterroviding a more effective medium as measured by the Faraday effi-iency for benzaldehyde. The liquid products, benzaldehyde and amall percentage of benzyl ether, could be recovered by extractionith scCO2 and the IL could be reused.
To complete this section, a few applications follow of reactionsnvolving the use of ILs and CO2 where CO2 is not used as an extrac-ion agent but enters the reaction as a reactant. Such reactions canerhaps be useful in turning CO2 from a major greenhouse gas tovaluable C1 resource for organic synthesis. Most applications of
his kind involve synthesis of organic carbonates.Mun et al. [96] studied the performance of several imidazolium
Ls as catalysts for copolymerisation of CO2 and phenyl glycidylther to polycarbonate. The selection of ILs included emim+, bmim+,mim+ and omim+ cations and Cl−, BF4
− and PF6− anions. TON and
arbonate content increased as the temperature increased from 313o 353 K, but they decreased over 373 K. The highest TON and 100%arbonate content were obtained with [bmim][Cl] at 393 K with theeaction time of 6 h.
Sun et al. [97] reviewed the use of ILs as catalysts and/or reactionedia in the synthesis of cyclic carbonates from CO2.Zhang et al. [98] described an interesting application of
-(N,N-dimethylaminoethyl)-2,3-dimethylimidazolium trifluo-omethanesulfonate IL as a base to promote hydrogenation of CO2o formic acid in an aqueous medium. In the reaction, rutheniummmobilised on silica was employed as heterogeneous catalyst.he catalyst was first dispersed in the aqueous solution of the ILnd then H2 and CO2 were added. The partial pressure of H2 rangedrom 1 to 9 MPa, and CO2 was added to a total pressure rangingrom 4 to 18 MPa. After the reaction, the catalyst was separatedrom the mixture by filtration and could be reused directly. Wateras removed from the filtrate by heating to 383 K. The formic acidas separated from the resultant binary mixture with the IL byeating the mixture to 403 K under nitrogen flow, and the IL coulde reused directly. Therefore, the whole process did not produceny waste.
Zhang et al. [99] developed an electrosynthesis of organic car-onates from CO2 and alcohols in CO2-saturated [bmim][BF4]. Theeaction was carried out under mild conditions (CO2 pressure.1 MPa, 328 K) and did not involve the use of any toxic solvents,atalysts or additional supporting electrolytes. The conversion ofrimary and secondary alcohols proceeded with good yields whileertiary alcohols and phenol were unreactive. The IL could be recy-led.
.2.3. Supercritical carbon dioxide as a phase separation switchThe previous sections have illustrated some advantages of scCO2
o facilitate the reactant feed before and the product separationfter reactions in the IL media. However, there is yet another way forsc)CO2 to be useful in IL applications because the presence and/orressure of CO2 can serve to manipulate the phase behaviour in
L–organic or in IL–water systems.Scurto et al. [100] demonstrated that the addition of CO2 can
nduce formation of three phases in [bmim][PF6]–methanol system.he effect of rising pressure of CO2 on phase behaviour is shownn Fig. 5. At ambient conditions, [bmim][PF6] and methanol areompletely miscible. Addition of pressurised CO2 may induce the
ppearance of a three-phase system with two liquid phases; at thearticular temperature and the initial amounts of IL and methanol,he second liquid phase appears at the pressure corresponding tohe lower critical end point (LCEP). The bottom (most dense) liquidhase L1 is rich in IL, the middle liquid phase L2 is rich in methanol,[(tif
ig. 5. Schematic view of [bmim][PF6]–methanol phase behaviour with increasingressure of CO2. Reprinted with permission from [100]. © 2002 American Chemicalociety.
nd the upper (vapour) phase V is rich in CO2 and contains no IL.s the pressure of CO2 increases and reaches what is called the-point, the methanol-rich liquid phase merges with the CO2-richapour phase with the resultant CO2–methanol supercritical phaseontaining no detectable IL (the K-point transition only occurs ifhe temperature is above the critical temperature of CO2). Thesephase equilibrium switches” occur reversibly by changing the CO2ressure at a particular temperature and IL/methanol proportion,nd they can be used to separate ILs from organic solvents evenhen the IL was dilute in the original mixture with the organic
olvent. CO2 can also induce phase separation in IL–water systems101,102].
Najdanovic-Visak et al. [103] investigated the effect of addingigh-pressure CO2 to mixtures of [bmim][PF6], ethanol and water,nd presented some three-phase pressures at 313.15 K. Theylso demonstrated that the surprisingly varied phase behaviourf [bmim][PF6]–ethanol–water–CO2 mixtures, ranging from totaliscibility through partial miscibility to nearly complete phase
eparation, can be very useful in reaction/separation cycles. Whenhe epoxidation of isophorone to isophorone oxide was carried outn biphasic conditions, the product yield after 3 h was 27% whereasn single-phase conditions, the yield after 3 h was 79%.
Zhang et al. [104] studied the effect of CO2 on the phaseehaviour of the reaction system and equilibrium conversion forsterification of acetic acid and ethanol in [bmim][HSO4] (1-butyl--methylimidazolium hydrogen sulfate) at 333 K and up to 15 MPa.he reaction mixture was a single-phase system; with increasingressure of CO2, two-phase → three-phase → two-phase transi-ions occurred. The pressure of CO2 or the phase behaviour of theystem showed a marked effect on the equilibrium conversion ofhe reaction.
For an efficient utilisation of CO2 as a phase separation switcht is very important that the vapour or supercritical phase be freerom IL under all experimental conditions. Aki et al. [105] demon-trated that this is the case by vapour–liquid–liquid equilibriumeasurements on eight different IL–CO2–organic solvent systems
t 298 K and 313 K.Mellein and Brennecke [106] investigated the ability of
ompressed CO2 to induce liquid/liquid phase separation inixtures of ILs and organic solvents with different kinds of
ntermolecular interactions. The organic solvents used werecetonitrile, 2-butanone and 2,2,2-trifluoroethanol and theLs included [hmim][Tf2N], [hmim][TfO], [hmmim][Tf2N] andNMe2EtPr][Tf2N]. The ability of CO2 to induce phase separation
act as anti-solvent) dependent primarily on solubility of CO2 inhe particular IL–organic solvent mixture. Strong hydrogen bond-ng between the IL and the organic solvent made it more difficultor CO2 to induce phase separation.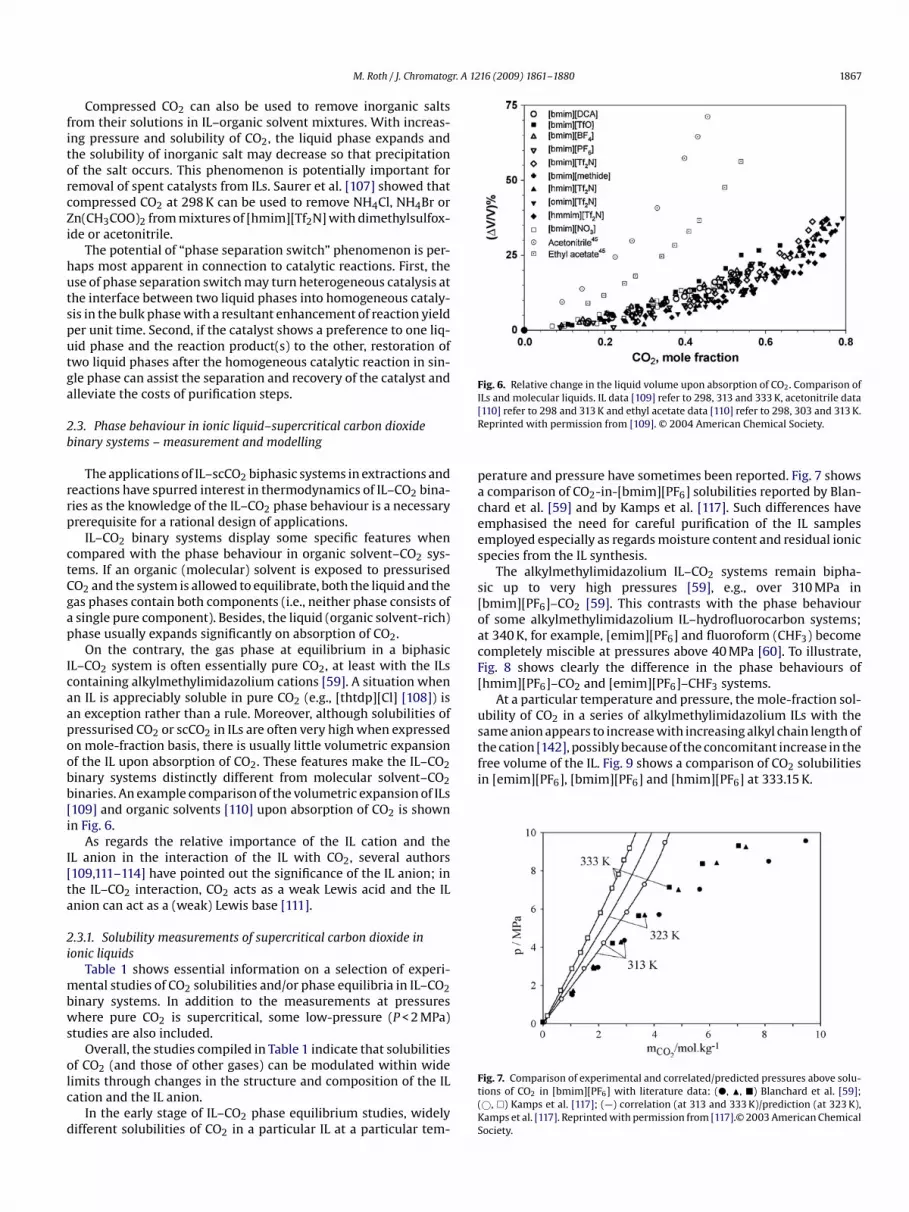
r. A 1216 (2009) 1861–1880 1867
fitorcZi
hutsputga
2b
rrp
ctCgap
Icaapoobb[i
I[ta
2i
mbws
olc
d
Fig. 6. Relative change in the liquid volume upon absorption of CO2. Comparison ofI[R
pacees
s[oacF[
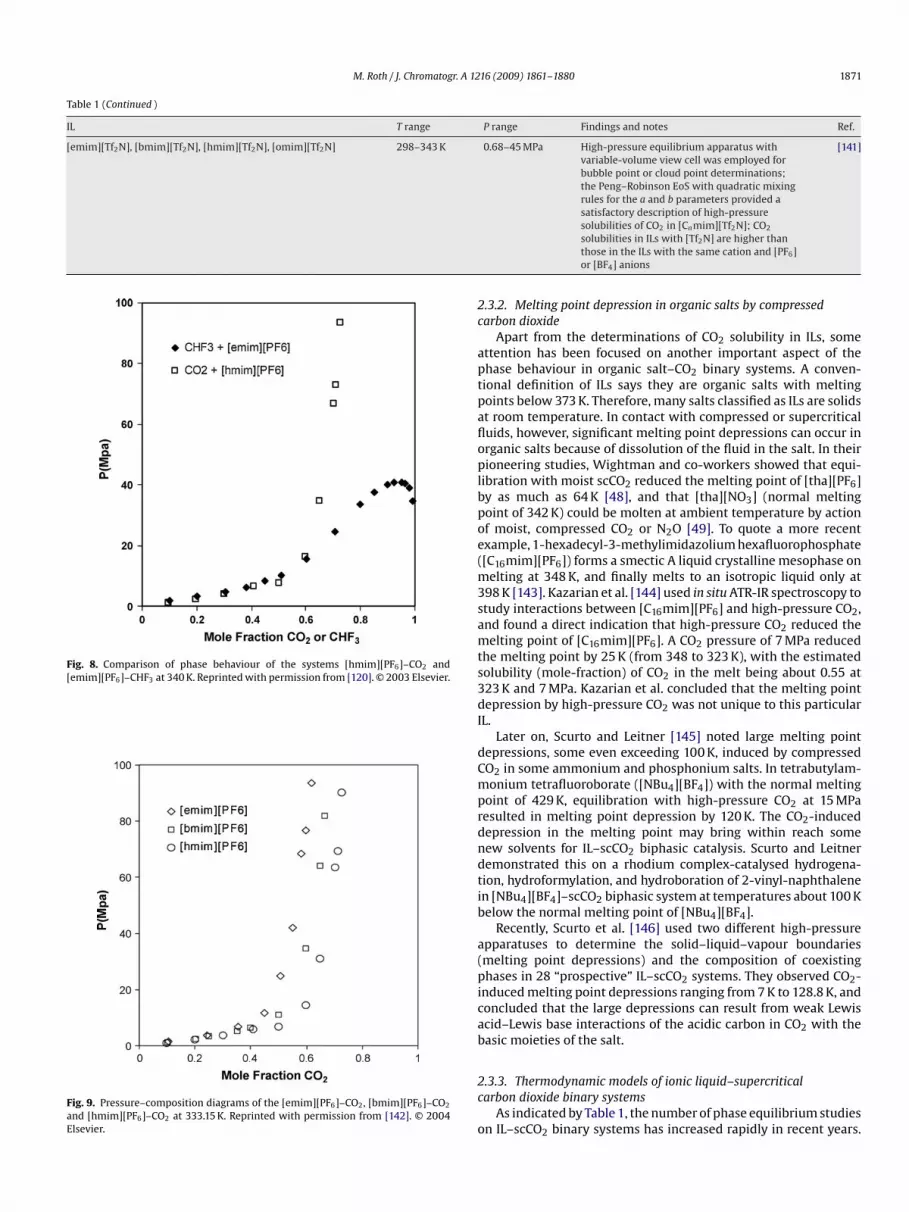
ustfree volume of the IL. Fig. 9 shows a comparison of CO2 solubilitiesin [emim][PF6], [bmim][PF6] and [hmim][PF6] at 333.15 K.
M. Roth / J. Chromatog
Compressed CO2 can also be used to remove inorganic saltsrom their solutions in IL–organic solvent mixtures. With increas-ng pressure and solubility of CO2, the liquid phase expands andhe solubility of inorganic salt may decrease so that precipitationf the salt occurs. This phenomenon is potentially important foremoval of spent catalysts from ILs. Saurer et al. [107] showed thatompressed CO2 at 298 K can be used to remove NH4Cl, NH4Br orn(CH3COO)2 from mixtures of [hmim][Tf2N] with dimethylsulfox-de or acetonitrile.
The potential of “phase separation switch” phenomenon is per-aps most apparent in connection to catalytic reactions. First, these of phase separation switch may turn heterogeneous catalysis athe interface between two liquid phases into homogeneous cataly-is in the bulk phase with a resultant enhancement of reaction yielder unit time. Second, if the catalyst shows a preference to one liq-id phase and the reaction product(s) to the other, restoration ofwo liquid phases after the homogeneous catalytic reaction in sin-le phase can assist the separation and recovery of the catalyst andlleviate the costs of purification steps.
.3. Phase behaviour in ionic liquid–supercritical carbon dioxideinary systems – measurement and modelling
The applications of IL–scCO2 biphasic systems in extractions andeactions have spurred interest in thermodynamics of IL–CO2 bina-ies as the knowledge of the IL–CO2 phase behaviour is a necessaryrerequisite for a rational design of applications.
IL–CO2 binary systems display some specific features whenompared with the phase behaviour in organic solvent–CO2 sys-ems. If an organic (molecular) solvent is exposed to pressurisedO2 and the system is allowed to equilibrate, both the liquid and theas phases contain both components (i.e., neither phase consists ofsingle pure component). Besides, the liquid (organic solvent-rich)hase usually expands significantly on absorption of CO2.
On the contrary, the gas phase at equilibrium in a biphasicL–CO2 system is often essentially pure CO2, at least with the ILsontaining alkylmethylimidazolium cations [59]. A situation whenn IL is appreciably soluble in pure CO2 (e.g., [thtdp][Cl] [108]) isn exception rather than a rule. Moreover, although solubilities ofressurised CO2 or scCO2 in ILs are often very high when expressedn mole-fraction basis, there is usually little volumetric expansionf the IL upon absorption of CO2. These features make the IL–CO2inary systems distinctly different from molecular solvent–CO2inaries. An example comparison of the volumetric expansion of ILs109] and organic solvents [110] upon absorption of CO2 is shownn Fig. 6.
As regards the relative importance of the IL cation and theL anion in the interaction of the IL with CO2, several authors109,111–114] have pointed out the significance of the IL anion; inhe IL–CO2 interaction, CO2 acts as a weak Lewis acid and the ILnion can act as a (weak) Lewis base [111].
.3.1. Solubility measurements of supercritical carbon dioxide inonic liquids
Table 1 shows essential information on a selection of experi-ental studies of CO2 solubilities and/or phase equilibria in IL–CO2
inary systems. In addition to the measurements at pressureshere pure CO2 is supercritical, some low-pressure (P < 2 MPa)
tudies are also included.Overall, the studies compiled in Table 1 indicate that solubilities
f CO2 (and those of other gases) can be modulated within wideimits through changes in the structure and composition of the ILation and the IL anion.
In the early stage of IL–CO2 phase equilibrium studies, widelyifferent solubilities of CO2 in a particular IL at a particular tem-
Ft(KS
Ls and molecular liquids. IL data [109] refer to 298, 313 and 333 K, acetonitrile data110] refer to 298 and 313 K and ethyl acetate data [110] refer to 298, 303 and 313 K.eprinted with permission from [109]. © 2004 American Chemical Society.
erature and pressure have sometimes been reported. Fig. 7 showscomparison of CO2-in-[bmim][PF6] solubilities reported by Blan-
hard et al. [59] and by Kamps et al. [117]. Such differences havemphasised the need for careful purification of the IL samplesmployed especially as regards moisture content and residual ionicpecies from the IL synthesis.
The alkylmethylimidazolium IL–CO2 systems remain bipha-ic up to very high pressures [59], e.g., over 310 MPa inbmim][PF6]–CO2 [59]. This contrasts with the phase behaviourf some alkylmethylimidazolium IL–hydrofluorocarbon systems;t 340 K, for example, [emim][PF6] and fluoroform (CHF3) becomeompletely miscible at pressures above 40 MPa [60]. To illustrate,ig. 8 shows clearly the difference in the phase behaviours ofhmim][PF6]–CO2 and [emim][PF6]–CHF3 systems.
At a particular temperature and pressure, the mole-fraction sol-bility of CO2 in a series of alkylmethylimidazolium ILs with theame anion appears to increase with increasing alkyl chain length ofhe cation [142], possibly because of the concomitant increase in the
ig. 7. Comparison of experimental and correlated/predicted pressures above solu-ions of CO2 in [bmim][PF6] with literature data: (�, �, �) Blanchard et al. [59];©, �) Kamps et al. [117]; (—) correlation (at 313 and 333 K)/prediction (at 323 K),amps et al. [117]. Reprinted with permission from [117].© 2003 American Chemicalociety.

1868 M. Roth / J. Chromatogr. A 1216 (2009) 1861–1880
Table 1CO2 solubility and phase equilibrium measurements in IL–CO2 binary systems.
IL T range P range Findings and notes Ref.
[bmim][PF6], [bmim][BF4] 313.15–333.15 K 6.8–20 MPa Molecular state of CO2 dissolved in ILs studiedby ATR-IR spectroscopy; in its interaction withCO2, BF4
− is a stronger Lewis base than PF6−;
CO2 solubilities estimated from absorbancedata
[111]
[bmim][PF6], [omim][PF6], [omim][BF4], [bmim][NO3],[emim][EtSO4], [N-butylpyridinium][BF4] ([N-bupy][BF4])
313.15–333.15 K 0–9.5 MPa Two different apparatuses (a static and adynamic flow); solubility trend[bmim][PF6]∼[omim][PF6] > [omim][BF4] > [N-bupy][BF4] > [bmim][NO3] > [emim][EtSO4]
[59]
[bmim][PF6] 283.15–323.15 K 0–1.3 MPa Other gases than CO2 (C2H4, C2H6, CH4, Ar, O2,CO, H2, N2); CO2 solubility at a constant P riseswith decreasing T; enthalpies and entropies ofgas dissolution reported
[115]
[bmim][BF4] 303.72–344.49 K Ambient (∼0.1 MPa) O2 solubilities also measured; thermodynamicfunctions of solvation reported
[116]
[bmim][PF6] 293.15–333.15 K 0–9.7 MPa CO2 solubilities markedly lower than thosereported by Blanchard et al. [59]; solubilitydata correlated with the extended Henry’s law;thermodynamic properties of solution reported
[117]
[pmim][Tf2N], [bmim][Tf2N], [hmim][Tf2N], [omim][Tf2N],[C8F13mim][Tf2N], [1,4-dibutyl-3-phenylimidazolium][Tf2N],[1-butyl-3-phenylimidazolium][Tf2N], [C3mim][PF6]
298.15 K 0.1 MPa or less Quartz crystal microbalance technique;Henry’s constants of CO2 in ILs reported; CO2
solubility in the IL with perfluorooctyl cation([C8F13mim][Tf2N]) enhanced (6.7 times) ascompared to that in [omim][Tf2N]; CO2
solubility in [pmim][PF6] lower than that in[pmim][Tf2N]
[118]
[emim][PF6] 308.14–366.03 K 1.49–97.10 MPa Vapour–liquid boundaries (bubble points)reported for CO2 concentrations from 10.4 to61.9 mol%; biphasic character of[emim][PF6] + CO2 system confirmed; differentphase behaviours of [emim][PF6] + CO2 and[emim][PF6] + CHF3 systems emphasised
[119]
[hmim][PF6] 298.31–363.58 K 0.64–94.60 MPa Vapour–liquid boundaries (bubble points)reported for CO2 concentrations from 9.8 to72.7 mol%; mole-fraction solubility of CO2 in[hmim][PF6] is much higher than in[emim][PF6] because of longer alkyl chain ofthe IL cation
[120]
[bmim][BF4], [bmim][methanesulfonate] ([bmim][CF3SO3]),[bmim][methide] ([bmim][(CF3SO2)3C]),[bmim][dicyanoamide] ([bmim][dca]), [bmim][PF6],[bmim][NO3], [bmim][PF6], [bmim][Tf2N], [hmim][Tf2N],[omim][Tf2N], [hmmim][Tf2N]
298.15–333.15 K 0–15 MPa Volumetric data also reported in addition toCO2 solubilities; in [bmim]-containing ILs, CO2
solubility does not correlate with hydrogenbond basicity of the anion, probably becausethe acid–base interaction is not the onlymechanism of IL–CO2 interaction; CO2
solubility trend in [bmim]-containing ILs variessomewhat with temperature: at 298.15 K:[NO3] < [dca] < [BF4] ∼ [PF6] < [CF3SO3] < [Tf2N]< [(CF3SO2)3C]; at 333.15 K:[NO3] < [BF4] < [dca] ∼ [PF6] ∼ [CF3SO3] < [Tf2N]< [(CF3SO2)3C]
[109]
[bmim][BF4], [bmim][Tf2N], [NBu3Me][Tf2N],[butylmethylpyrrolidinium][Tf2N],[triisobutylmethylphosphonium][p-toluenesulfonate]
283.15–323.15 K 0–1.3 MPa In [bmim][BF4] and [bmim][Tf2N], solubilitiesof other gases/vapours also reported includingO2, CO, C2H4, C2H6, N2O and C6H6; Henry’sconstants and entropies of absorption of thegases in ILs reported; IL anion appears to playthe most significant role in determining the gassolubilities; gases with large dipole orquadrupole moments have the highestsolubilities whereas the solubilities of nonpolargases correlate with the gas polarisability
[113]
[bmim][PF6] 298 K 4 and 15 MPa Intermolecular distribution functions obtainedfrom X-ray diffraction measurements at 298 Kin neat [bmim][PF6] and in [bmim][PF6]saturated with CO2 at 4 and 15 MPa; the resultsindicate preferential solvation of CO2 to PF6
−
because of specific interaction
[114]
[hmim][BF4] 293.18–368.16 K 0.54–86.60 MPa Vapour–liquid boundaries (bubble points)reported for CO2 concentrations from 10.4 to61.9 mol%; three-phase liquid–liquid–vapourequilibrium observed in the IL + CO2 mixture(xIL = 0.025) at T and P near the critical point ofCO2
[121]
M. Roth / J. Chromatogr. A 1216 (2009) 1861–1880 1869
Table 1 (Continued )
IL T range P range Findings and notes Ref.
[bmim][BF4] 278.47–368.22 K 0.58–67.62 MPa Vapour–liquid boundaries (bubble points)reported for CO2 concentrations from 10.22 to60.17 mol%; two-phase region of the systemextends to very high pressures
[122]
[trihexyltetradecylphosphonium][dodecylbenzenesulfonate]([thtdp][C12H25PhSO3]),[trihexyltetradecylphosphonium][methanesulfonate],([thtdp][MeSO3])
305–325 K 4–9 MPa When expressed as (moles of CO2/1 kg IL), theCO2 solubility in [thtdp][MeSO3] is higher thanthat in [thtdp][C12H25PhSO3]; data correlatedwith the extended Henry’s law
[123]
[bmim][PF6], [1,1,3,3-tetramethylguanidinium][lactate] (TMGL) 297–328 K 0–11 MPa CO2 solubility (moles of CO2/1 kg IL) in TGML isslightly (by about 12%) higher than that in[bmim][PF6]; data correlated with theextended Henry’s law
[124]
[bmim][PF6], [bmim][BF4] 283.15–348.15 K 0.01–2 MPa Time-dependent gravimetric microbalancemeasurements were used to obtain bothsolubilities and effective diffusivities of CO2 inILs; solubility data were correlated with aRedlich–Kwong type cubic EoS with a specificform of temperature dependence of purecomponent a parameters and modified van derWaals–Berthelot mixing rules; the observeddiffusion coefficients were about 10–100 timeslower than those in organic liquids and about10 times larger than those of ions in ILs
[125]
[bmim][PF6] 293.29–363.54 K 0.59–73.50 MPa Vapour–liquid boundaries (bubble points)reported for CO2 concentrations from 10 to65 mol%; the P–T projection of theliquid–liquid–vapour boundary determined upto the critical endpoint of the[bmim][PF6] + CO2 system (0.04 K above thecritical temperature of pure CO2); phasebehaviours compared of the [bmim][PF6] + CO2
and [bmim][PF6] + CHF3 systems
[126]
[omim][BF4] 303–363 K 0.1–100 MPa Vapour–liquid boundaries (bubble points)reported for CO2 concentrations from 10 to75 mol%; vapour–liquid equilibrium pressureincreases sharply for CO2 concentrations above60 mol%
[127]
[hmim][Tf2N] 293.15–413.2 K 0.6–10 MPa Solubility data correlated with the extendedHenry’s law; Henry’s constants of CO2 reportedand compared with other measurements on[hmim][Tf2N] + system (mostly agreementwithin uncertainty limits)
[128]
[bmim][MeSO4] 293.2–413.2 K 0.9–9.8 MPa Solubility data correlated with the extendedHenry’s law; Henry’s constants of CO2 reportedand fitted with a simple function oftemperature
[129]
[bmim][PF6] 313.2–333.2 K 1–25 MPa Effect of moisture content investigated withwater mass fraction in [bmim][PF6] rangingfrom 0.000067 to 0.016; at a particular T and P,the average variation of CO2 solubility withwater content of the IL was 6.7% (15%maximum); effect of IL water content onvolumetric properties of [bmim][PF6] + CO2
mixture relatively minor
[130]
[bmim][BF4], [hmim][BF4], [omim][BF4] 305–325 K 1–9 MPa CO2 solubilities decrease with increasing T andincrease with increasing P; solubility datacorrelated with the extended Henry’s law;Henry’s constants follow the order[bmim][BF4] > [hmim][BF4] > [omim][BF4], i.e.,the reverse order of solubilities
[131]
Poly(p-vinylbenzyltrimethyl ammonium tetrafluoroborate)(P[VBTMA][BF4]), poly([2-(methacryloyloxy)ethyl]trimethylammonium tetrafluoroborate) (P[MATMA][BF4])
298–348 K 0–1.5 MPa Magnetic suspension balance study;polymerised ammonium ILs; solubility datacorrelated with dual-mode sorption model;CO2 solubility in P[VBTMA][BF4] is higher thanthat in P[MATMA][BF4]
[132]
P[VBTMA][BF4], poly(1-(p-vinylbenzyl)-3-methylimidazoliumtetrafluoroborate) (P[VBMI][BF4])
298–348 K 0–18 MPa Magnetic suspension balance study; solubilitydata at low pressures (<1.5 MPa) correlatedwith dual-mode sorption model; CO2 solubilityin P[VBTMA][BF4] is higher than that inP[VBMI][BF4]
[133]

1870 M. Roth / J. Chromatogr. A 1216 (2009) 1861–1880
Table 1 (Continued )
IL T range P range Findings and notes Ref.
2-hydroxyethyl ammonium formate (HEF), 2-hydroxyethylammonium acetate (HEA), 2-hydroxyethyl ammoniumlactate (HEL), tri-(2-hydroxyethyl) ammonium acetate(THEAA), tri-(2-hydroxy ethyl) ammonium lactate (THEAL),2-(2-hydroxyethoxy) ammonium formate (HEAF),2-(2-hydroxyethoxy) ammonium acetate (HEAA),2-(2-hydroxyethoxy) ammonium lactate (HEAL)
303–323 K 0–11 MPa Solubility data correlated withKrichevsky–Kasarnovsky equation; Henry’sconstants and partial molar volumes of CO2 inILs reported; CO2 solubilities follow thesequenceTHEAL > HEAA > HEA > HEF > HEAL > THEAA ∼ HEL > HEAF
[134]
[emim][Tf2N] 310–450 K 0–15 MPa Vapour–liquid boundaries (bubble points)reported for CO2 concentrations from 12.3 to59.3 mol%; at 333 K and a particular P, the CO2
solubility in [emim][Tf2N] is much higher thanthat in [emim][PF6]
[135]
[1-(2-methoxyethyl)-3-methylimidazolium][Tf2N], [1-(2-(2-methoxyethoxy)ethyl)-3-methylimidazolium][Tf2N],[1-(2-(2-(2-methoxyethoxy)ethoxy)ethyl)-3-methylimidazolium][Tf2N],[C7mim][Tf2N]
313 K Ambient (∼0.1 MPa) Oligo(ethylene glycol) functionalised ILs; N2
and CH4 solubilities also measured in additionto those of CO2; Henry’s constants reported;oligoethylene-functionalised ILs show betterCO2/N2 and CO2/CH4 ideal solubilityselectivities as compared to those of thecorresponding alkyl-substituted ILs
[136]
[bmim][Tf2N], [1-propyl-2,3-dimethylimidazolium][Tf2N]([pmmim][Tf2N]), [1-butyl-3-methylpyridinium][Tf2N]([bmpy][Tf2N]),[1-(3,4,5,6-perfluorohexyl)-3-methylimidazolium][Tf2N]bis(trifluoromethyl ([perfluoro-hmim][Tf2N])), [bmim][BF4]
283–323 K 0.1–0.2 MPa Transient thin liquid film method enabled asimultaneous determination of Henry’sconstant and diffusivity of CO2; Henry’sconstants in the range of 2.55–8.4 MPa; in theILs with Tf2N− anion CO2 solubilities follow thetrend [pmmim] < [bmim] < [bmpy] < [perfluoro-hmim]
[137]
[hmim][Tf2N] 282–348 K 0.009–1.98 MPa IL samples from 2 different sources wereindistinguishable within experimentaluncertainties; solubility data were correlatedwith a Redlich–Kwong type cubic EoS with aspecific form of temperature dependence ofpure component a parameters and modifiedvan der Waals–Berthelot mixing rules;although based solely on vapour–liquidequilibrium information, the EoS model hascorrectly predicted the liquid–liquidimmiscibility gap in the [hmim][Tf2N]–CO2
system at temperatures below the critical ofCO2
[138]
[hmim][Tf2N], [hmmim][Tf2N], [bmim][trifluoroacetate],[bmim][pentadecafluorooctanoate], [1-butylnicotinic acidbutyl ester][Tf2N], [1-hexyl-3-methylpyridinium][Tf2N],[hmim][tris(pentafluoroethyl) trifluorophosphate],[hmim][tris(heptafluoropropyl) trifluorophosphate],[1-pentyl-3-methylimidazolium][tris(nonafluorobutyl)trifluorophosphate], [tetrabutylammonium][docusate],[3-(3,3,4,4,5,5,6,6,6-nonafluorohexyl)-1-methylimidazolium][Tf2N] ([C6H4F9mim][Tf2N]),[3-(3,3,4,4,5,5,6,6,7,7,8,8,8-tridecafluorooctyl)-1-methylimidazolium][Tf2N] ([C8H4F13mim][Tf2N]), PEG-5cocomonium methylsulfate (Ecoeng 500),[bmim][2-(2-methoxyethoxy) ethylsulfate] (Ecoeng 41 M),[hmim][saccharinate], [hmim][acesulfamate],[(1-methylimidazole)(triethylamine) boronium][Tf2N],[choline][Tf2N], [NBuMe3][Tf2N], [hmim][PF6],[bmim][Tf2N], [bmim][methide] ([bmim][(CF3SO2)3C])
283–333 K 0–9.1 MPa CO2 solubility measurements in a range of ILswith CO2-philic substituents with the aim toimprove the solubility of CO2 in ILs;low-pressure measurements (<1.3 MPa) withgravimetric microbalance, high-pressuremeasurements (>1.3 MPa) with statichigh-pressure apparatus; Henry’s constantswere obtained from low-pressure data;fluorination of the IL anion increased solubilityof CO2–solubility increased with increasinglength of the fluoroalkyl chain on the anion;fluorination of the IL cation also increasedsolubility of CO2 but to a lesser extentcompared with anion fluorination–the CO2
solubility was higher in [C8H4F13mim][Tf2N]than in [C6H4F9mim][Tf2N]; overall, theimprovement in CO2 solubility achieved withnon-fluorous CO2-philic ILs was not as good aswith fluorous CO2-philic ILs; nevertheless,non-fluorinated ILs-containing ether linkagesand flexible alkyl chains to increase freevolume can be designed to have a high affinityfor CO2
[139]
[bmim][acetate] 283–348 K 0.01–2.0 MPa Vapour–liquid equilibria (CO2 solubilities)were investigated with a gravimetricmicrobalance; solubility data were correlatedwith a Redlich–Kwong type cubic EoS with aspecific form of temperature dependence ofpure component a parameters and modifiedvan der Waals–Berthelot mixing rules;vapour–liquid–liquid equilibria (liquid–liquidimmiscibility) at temperatures below thecritical of CO2 were predicted by the EoS modeland confirmed experimentally
[140]
M. Roth / J. Chromatogr. A 1216 (2009) 1861–1880 1871
Table 1 (Continued )
IL T range P range Findings and notes Ref.
[emim][Tf2N], [bmim][Tf2N], [hmim][Tf2N], [omim][Tf2N] 298–343 K 0.68–45 MPa High-pressure equilibrium apparatus withvariable-volume view cell was employed forbubble point or cloud point determinations;the Peng–Robinson EoS with quadratic mixingrules for the a and b parameters provided asatisfactory description of high-pressure
[141]
Fig. 8. Comparison of phase behaviour of the systems [hmim][PF6]–CO2 and[emim][PF6]–CHF3 at 340 K. Reprinted with permission from [120]. © 2003 Elsevier.
Fig. 9. Pressure–composition diagrams of the [emim][PF6]–CO2, [bmim][PF6]–CO2
and [hmim][PF6]–CO2 at 333.15 K. Reprinted with permission from [142]. © 2004Elsevier.
2c
aptpafloplbpoe(m3samts3dI
dCmprdndtib
a(picab
2c
o
solubilities of CO2 in [Cnmim][Tf2N]; CO2
solubilities in ILs with [Tf2N] are higher thanthose in the ILs with the same cation and [PF6]or [BF4] anions
.3.2. Melting point depression in organic salts by compressedarbon dioxide
Apart from the determinations of CO2 solubility in ILs, somettention has been focused on another important aspect of thehase behaviour in organic salt–CO2 binary systems. A conven-ional definition of ILs says they are organic salts with meltingoints below 373 K. Therefore, many salts classified as ILs are solidst room temperature. In contact with compressed or supercriticaluids, however, significant melting point depressions can occur inrganic salts because of dissolution of the fluid in the salt. In theirioneering studies, Wightman and co-workers showed that equi-
ibration with moist scCO2 reduced the melting point of [tha][PF6]y as much as 64 K [48], and that [tha][NO3] (normal meltingoint of 342 K) could be molten at ambient temperature by actionf moist, compressed CO2 or N2O [49]. To quote a more recentxample, 1-hexadecyl-3-methylimidazolium hexafluorophosphate[C16mim][PF6]) forms a smectic A liquid crystalline mesophase on
elting at 348 K, and finally melts to an isotropic liquid only at98 K [143]. Kazarian et al. [144] used in situ ATR-IR spectroscopy totudy interactions between [C16mim][PF6] and high-pressure CO2,nd found a direct indication that high-pressure CO2 reduced theelting point of [C16mim][PF6]. A CO2 pressure of 7 MPa reduced
he melting point by 25 K (from 348 to 323 K), with the estimatedolubility (mole-fraction) of CO2 in the melt being about 0.55 at23 K and 7 MPa. Kazarian et al. concluded that the melting pointepression by high-pressure CO2 was not unique to this particular
L.Later on, Scurto and Leitner [145] noted large melting point
epressions, some even exceeding 100 K, induced by compressedO2 in some ammonium and phosphonium salts. In tetrabutylam-onium tetrafluoroborate ([NBu4][BF4]) with the normal melting
oint of 429 K, equilibration with high-pressure CO2 at 15 MPaesulted in melting point depression by 120 K. The CO2-inducedepression in the melting point may bring within reach someew solvents for IL–scCO2 biphasic catalysis. Scurto and Leitneremonstrated this on a rhodium complex-catalysed hydrogena-ion, hydroformylation, and hydroboration of 2-vinyl-naphthalenen [NBu4][BF4]–scCO2 biphasic system at temperatures about 100 Kelow the normal melting point of [NBu4][BF4].
Recently, Scurto et al. [146] used two different high-pressurepparatuses to determine the solid–liquid–vapour boundariesmelting point depressions) and the composition of coexistinghases in 28 “prospective” IL–scCO2 systems. They observed CO2-
nduced melting point depressions ranging from 7 K to 128.8 K, andoncluded that the large depressions can result from weak Lewiscid–Lewis base interactions of the acidic carbon in CO2 with theasic moieties of the salt.
.3.3. Thermodynamic models of ionic liquid–supercriticalarbon dioxide binary systems
As indicated by Table 1, the number of phase equilibrium studiesn IL–scCO2 binary systems has increased rapidly in recent years.

1 r. A 12
Ihioee
mopCspsda
m[mImrltmptmv[
t[p(cue
pounnsifwtcv[s
ailacatiEe
iAme
nv
pwdats
sctsitdtit
scHbdptttbeatLttpafttrvo
i[oTiedip
872 M. Roth / J. Chromatog
n line with the increase, a large variety of thermodynamic modelsave been proposed to correlate, interpret and generalise the exper-
mental data on phase equilibria. In most thermodynamic modelsf phase equilibrium in an IL–scCO2 system, the IL has been consid-red a single molecular species rather than being treated as a truelectrolyte consisting of a cation and an anion.
Ally et al. [147] employed a semiempirical irregular ionic latticeodel to correlate and predict CO2 solubilities in ILs. A limited set
f CO2 solubilities in a particular IL were used to regress the twoarameters of the model, and these were then employed to predictO2 solubilities in the particular IL at the temperature and pres-ure conditions where no experimental data were available. Therocedure was applied to [bmim][PF6]–CO2 and [omim][PF6]–CO2ystems with some success although deviations of the model pre-ictions from the experimental data were noted in the latter systemt high solubilities of CO2 (>60 mol%).
Gas solubilities in organic (molecular) solvents have often beenodelled with the regular solution theory [148]. Camper et al.
149] employed the regular solution theory to model the experi-ental data [59,116] on low-pressure solubilities of CO2 in several
Ls. With empirical constants, regular solution theory was able toodel low-pressure solubilities of CO2 and C2H4 in several ILs. This
esult may appear somewhat surprising because ILs certainly vio-ate one of the basic assumptions underlying the regular solutionheory, namely, the dominance of dispersion forces in the inter-
olecular interactions. It should also be noted that the IL solubilityarameters reported by Camper et al. are markedly lower thanhe square roots of IL cohesive energy densities obtained by other
ethods including molecular dynamics simulations [150,151], sol-ent effects on reaction rates [152] and viscosity measurements153].
In a follow-up study, Scovazzo et al. [154] empirically estimatedhe cohesive energies of several ILs ([bmim][PF6], [emim][Tf2N],emim][TfO], [emim][dca] and [thtdp][Cl]) from their meltingoints, and used the solubility parameters to model low-pressure<0.1 MPa) solubilities of CO2 in the ILs. They found that the ILs’ohesive energies and molar volumes influenced the relative sol-bilities of CO2 and that the IL–CO2 complexation was not thexclusive factor in determining the relative solubilities of CO2.
In applications of the regular solution concept to correlate low-ressure solubilities of gases in ILs, there are multiple possibilitiesf how to estimate the IL’s solubility parameter. Kilaru et al. [155]sed surface tension data to estimate solubility parameters of aumber of ILs containing imidazolium, ammonium and phospho-ium cations, and applied the results to correlate low pressureolubilities of CO2, ethylene, propylene, 1-butene and 1,3-butadienen the ILs. Except for 1,3-butadiene in ammonium ILs, the sur-ace tension-based correlations predicted the gas-in-IL solubilitiesithin an error of ±15% as measured by deviation from the respec-
ive experimental value. Alternatively, solubility parameter of an ILan be estimated from the activation energy of viscosity, and theiscosity-based correlation of gas-in-IL solubilities at low pressures156] provided a more flexible description as compared with theurface tension-based correlation.
Thermodynamic models applied to IL–(sc)CO2 binary systemslso include those based on cubic equations of state (EoSs). Thiss probably because cubic EoSs have often been used to corre-ate/predict phase equilibria in the binary mixtures of CO2 andn organic nonelectrolyte [157,158], and so applications to IL-ontaining systems are a natural and logical extension of the
pplication domain of cubic EoSs. Some applications of cubic EoSso model IL–(sc)CO2 binary systems [125,138,140,141] have beenncluded in Table 1. The pure component parameters of most cubicoSs can readily be obtained from the component’s critical prop-rties. Therefore, the critical properties of ILs would be very usefulbPLa
16 (2009) 1861–1880
n the development of cubic EoS models of IL-containing mixtures.lthough the critical properties of ILs cannot be measured experi-entally because of thermal decomposition, several approaches to
stimate them have been worked out.Rebelo et al. [159] attempted to estimate critical temperatures,
ormal boiling points and vapour pressures of ILs from measurablealues of surface tensions and molar volumes.
Valderrama et al. [160,161] attempted an estimation of criticalroperties, normal boiling temperatures and acentric factors of ILsith a group contribution method. Since no definite experimentalata on these properties of ILs are available, one should allow formple uncertainty in the estimated values. Because of the impor-ance of these properties of ILs, their estimated values have beenubject to some discussion [162,163].
Another class of thermodynamic models for IL–CO2 binaryystems have made use of several versions of the statistical asso-iating fluid theory (SAFT) [164,165]. Compared with cubic EoSs,he key advantage of the SAFT models is their much more solidtatistical-mechanics basis and flexibility as regards the kinds ofntermolecular interactions to be taken into account. In applica-ions to IL–CO2 systems, several SAFT-based models were employediffering in the selection of intermolecular interactions included inhe expression for the residual Helmholtz free energy of the fluid,.e., for the difference between the real Helmholtz free energy ofhe fluid and the Helmholtz free energy for an ideal gas.
Kroon et al. [166] modelled the phase behaviour of IL–CO2ystems with the truncated perturbed chain polar statistical asso-iating fluid theory (tPC-PSAFT) EoS. In tPC-PSAFT, the residualelmholtz free energy of the fluid is expressed as a sum of contri-utions arising from hard-sphere, chain formation, association andispersion interactions. To account for all these interactions, severalarameters per pure component are needed, and the parame-ers are obtained by fitting a selection of the pure component’shermodynamic and physicochemical properties. In addition, oneemperature-dependent binary interaction parameter per IL–CO2inary is needed, and this has been adjusted by fitting thexperimental vapour–liquid equilibrium data. Because of properccounting for the dipolar interactions between the IL molecules,he quadrupolar interactions between the CO2 molecules, and theewis acid–Lewis base association between the IL molecules andhe CO2 molecules, the tPC-PSAFT EoS could accurately describehe CO2 solubility in alkylmethylimidazolium ILs up to very highressure (∼100 MPa). The best agreement with experimental datand the lowest binary interaction parameter values were observedor IL–CO2 mixtures with the longest alkyl chains and at the lowestemperatures. Subsequently, the tPC-PSAFT EoS was reparame-erised employing density data of pure ILs over a wide temperatureange [167]. The reparameterised model predicted more realisticapour pressures of pure ILs, and it provided improved predictionsf gas solubilities in IL–CO2, IL–CO, IL–O2 and IL–CHF3 systems.
A square well chain fluid (SWCF) EoS used before in describ-ng the solubility of CO2 in polymers was employed by Wang et al.168] to model the PVT behaviour of 21 pure ILs and the solubilitiesf several gases (CO2, C3H6, C3H8 and C4H10) in some of the ILs.he SWCF EoS did not take into account the contributions of polarnteractions and specific interactions to the residual Helmholtz freenergy. This was probably the reason why the EoS did not succeed inescribing the CO2 solubilities at high pressures (>15 MPa) although
t performed well up to 50 mol% CO2 concentration in the liquidhase.
Andreu and Vega [169] used soft-SAFT EoS to model CO2 solu-ilities in several alkylmethylimidazolium ILs containing BF4
− andF6
− anions. Although their model did not account for any specificewis acid–Lewis base interaction between IL and CO2, it has beenble to reproduce the experimentally observed sharp increase in
M. Roth / J. Chromatogr. A 1216 (2009) 1861–1880 1873
F[
ep
wmmv
e[1opiIspfgl
mgdcaspimaGsap1
Ioctamp
iraCiaoeo
Fig. 11. Solubility isotherm of CO2 in [hmim][Tf2N] at 333 K from various sources:(tnC
csfibFll
3d
hetpaIpcb
saMocC[igtAst
si(NtN), 1-methyl-3-phenylpiperazine (MPhPz) and 2-(4-methyl-
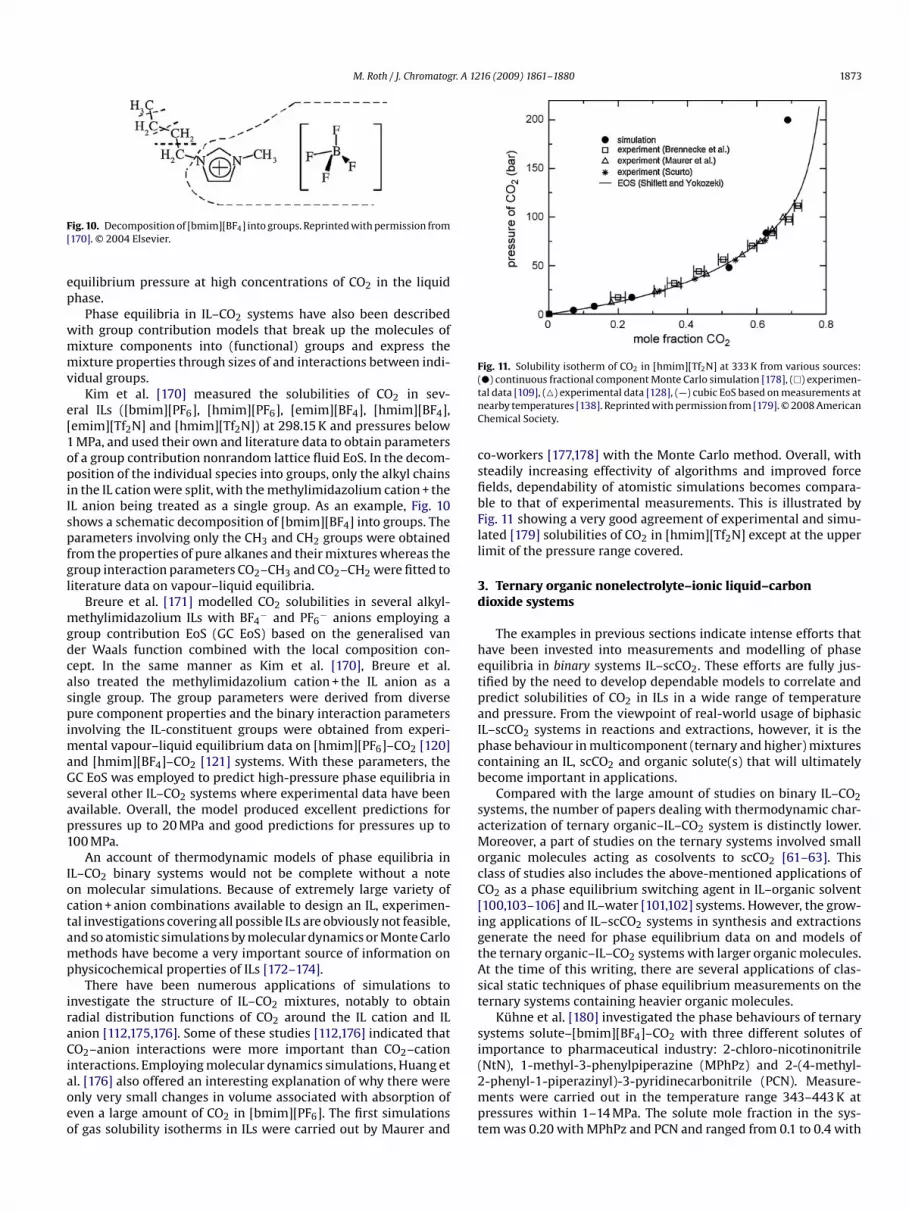
ig. 10. Decomposition of [bmim][BF4] into groups. Reprinted with permission from170]. © 2004 Elsevier.
quilibrium pressure at high concentrations of CO2 in the liquidhase.
Phase equilibria in IL–CO2 systems have also been describedith group contribution models that break up the molecules ofixture components into (functional) groups and express theixture properties through sizes of and interactions between indi-
idual groups.Kim et al. [170] measured the solubilities of CO2 in sev-
ral ILs ([bmim][PF6], [hmim][PF6], [emim][BF4], [hmim][BF4],emim][Tf2N] and [hmim][Tf2N]) at 298.15 K and pressures belowMPa, and used their own and literature data to obtain parametersf a group contribution nonrandom lattice fluid EoS. In the decom-osition of the individual species into groups, only the alkyl chains
n the IL cation were split, with the methylimidazolium cation + theL anion being treated as a single group. As an example, Fig. 10hows a schematic decomposition of [bmim][BF4] into groups. Thearameters involving only the CH3 and CH2 groups were obtainedrom the properties of pure alkanes and their mixtures whereas theroup interaction parameters CO2–CH3 and CO2–CH2 were fitted toiterature data on vapour–liquid equilibria.
Breure et al. [171] modelled CO2 solubilities in several alkyl-ethylimidazolium ILs with BF4
− and PF6− anions employing a
roup contribution EoS (GC EoS) based on the generalised vaner Waals function combined with the local composition con-ept. In the same manner as Kim et al. [170], Breure et al.lso treated the methylimidazolium cation + the IL anion as aingle group. The group parameters were derived from diverseure component properties and the binary interaction parameters
nvolving the IL-constituent groups were obtained from experi-ental vapour–liquid equilibrium data on [hmim][PF6]–CO2 [120]
nd [hmim][BF4]–CO2 [121] systems. With these parameters, theC EoS was employed to predict high-pressure phase equilibria ineveral other IL–CO2 systems where experimental data have beenvailable. Overall, the model produced excellent predictions forressures up to 20 MPa and good predictions for pressures up to00 MPa.
An account of thermodynamic models of phase equilibria inL–CO2 binary systems would not be complete without a noten molecular simulations. Because of extremely large variety ofation + anion combinations available to design an IL, experimen-al investigations covering all possible ILs are obviously not feasible,nd so atomistic simulations by molecular dynamics or Monte Carloethods have become a very important source of information on
hysicochemical properties of ILs [172–174].There have been numerous applications of simulations to
nvestigate the structure of IL–CO2 mixtures, notably to obtainadial distribution functions of CO2 around the IL cation and ILnion [112,175,176]. Some of these studies [112,176] indicated thatO2–anion interactions were more important than CO2–cation
nteractions. Employing molecular dynamics simulations, Huang et
l. [176] also offered an interesting explanation of why there werenly very small changes in volume associated with absorption ofven a large amount of CO2 in [bmim][PF6]. The first simulationsf gas solubility isotherms in ILs were carried out by Maurer and2mpt
�) continuous fractional component Monte Carlo simulation [178], (�) experimen-al data [109], (�) experimental data [128], (—) cubic EoS based on measurements atearby temperatures [138]. Reprinted with permission from [179]. © 2008 Americanhemical Society.
o-workers [177,178] with the Monte Carlo method. Overall, withteadily increasing effectivity of algorithms and improved forceelds, dependability of atomistic simulations becomes compara-le to that of experimental measurements. This is illustrated byig. 11 showing a very good agreement of experimental and simu-ated [179] solubilities of CO2 in [hmim][Tf2N] except at the upperimit of the pressure range covered.
. Ternary organic nonelectrolyte–ionic liquid–carbonioxide systems
The examples in previous sections indicate intense efforts thatave been invested into measurements and modelling of phasequilibria in binary systems IL–scCO2. These efforts are fully jus-ified by the need to develop dependable models to correlate andredict solubilities of CO2 in ILs in a wide range of temperaturend pressure. From the viewpoint of real-world usage of biphasicL–scCO2 systems in reactions and extractions, however, it is thehase behaviour in multicomponent (ternary and higher) mixturesontaining an IL, scCO2 and organic solute(s) that will ultimatelyecome important in applications.
Compared with the large amount of studies on binary IL–CO2ystems, the number of papers dealing with thermodynamic char-cterization of ternary organic–IL–CO2 system is distinctly lower.oreover, a part of studies on the ternary systems involved small
rganic molecules acting as cosolvents to scCO2 [61–63]. Thislass of studies also includes the above-mentioned applications ofO2 as a phase equilibrium switching agent in IL–organic solvent100,103–106] and IL–water [101,102] systems. However, the grow-ng applications of IL–scCO2 systems in synthesis and extractionsenerate the need for phase equilibrium data on and models ofhe ternary organic–IL–CO2 systems with larger organic molecules.t the time of this writing, there are several applications of clas-ical static techniques of phase equilibrium measurements on theernary systems containing heavier organic molecules.
Kühne et al. [180] investigated the phase behaviours of ternaryystems solute–[bmim][BF4]–CO2 with three different solutes ofmportance to pharmaceutical industry: 2-chloro-nicotinonitrile
-phenyl-1-piperazinyl)-3-pyridinecarbonitrile (PCN). Measure-ents were carried out in the temperature range 343–443 K at
ressures within 1–14 MPa. The solute mole fraction in the sys-em was 0.20 with MPhPz and PCN and ranged from 0.1 to 0.4 with
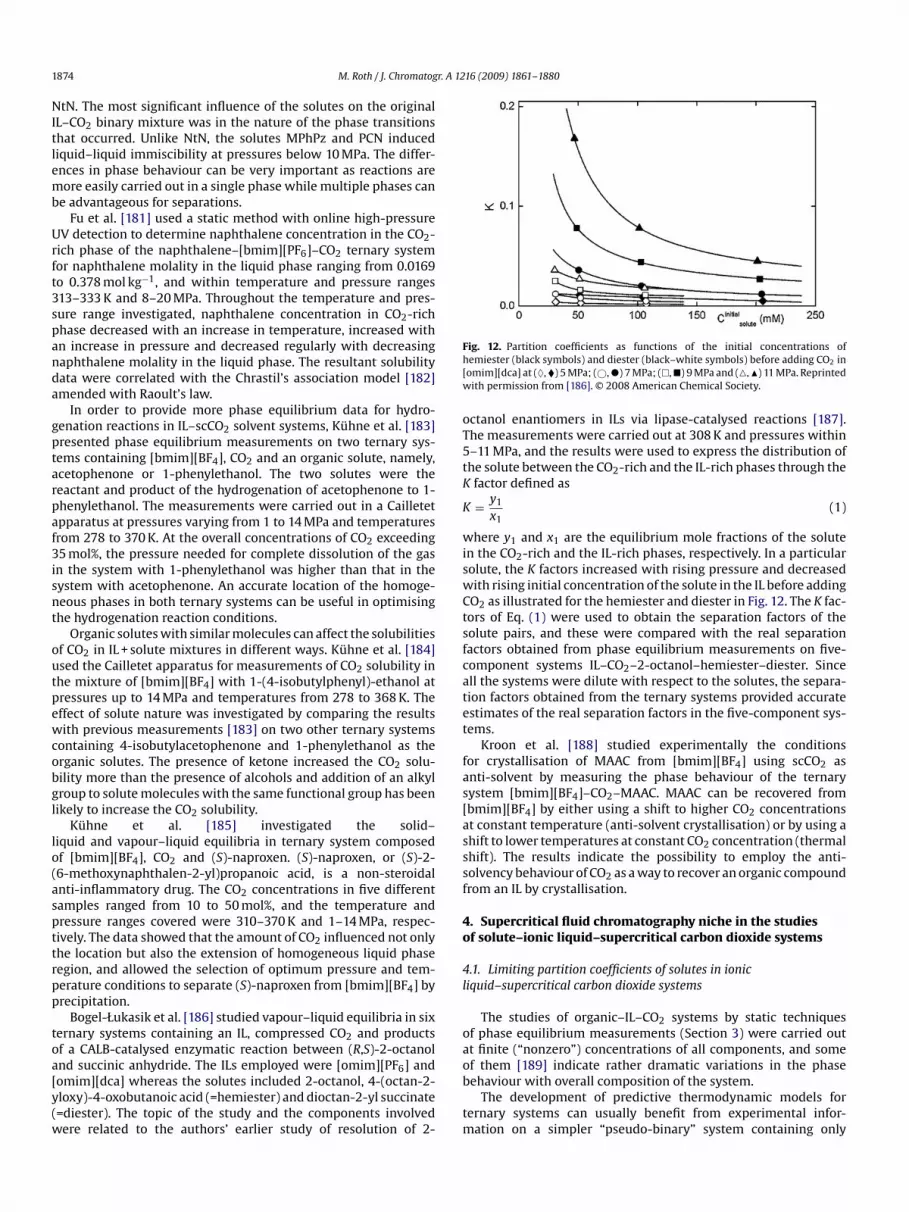
1 r. A 1216 (2009) 1861–1880
NItlemb
Urft3spanda
gptarpaf3isnt
outpewcobgl
lo(aspttrpp
toa[y(w
Fh[w
oT5tK
K
wiswCtsfcatet
fas[asssf
4o
4l
oa
874 M. Roth / J. Chromatog
tN. The most significant influence of the solutes on the originalL–CO2 binary mixture was in the nature of the phase transitionshat occurred. Unlike NtN, the solutes MPhPz and PCN inducediquid–liquid immiscibility at pressures below 10 MPa. The differ-nces in phase behaviour can be very important as reactions areore easily carried out in a single phase while multiple phases can
e advantageous for separations.Fu et al. [181] used a static method with online high-pressure
V detection to determine naphthalene concentration in the CO2-ich phase of the naphthalene–[bmim][PF6]–CO2 ternary systemor naphthalene molality in the liquid phase ranging from 0.0169o 0.378 mol kg−1, and within temperature and pressure ranges13–333 K and 8–20 MPa. Throughout the temperature and pres-ure range investigated, naphthalene concentration in CO2-richhase decreased with an increase in temperature, increased withn increase in pressure and decreased regularly with decreasingaphthalene molality in the liquid phase. The resultant solubilityata were correlated with the Chrastil’s association model [182]mended with Raoult’s law.
In order to provide more phase equilibrium data for hydro-enation reactions in IL–scCO2 solvent systems, Kühne et al. [183]resented phase equilibrium measurements on two ternary sys-ems containing [bmim][BF4], CO2 and an organic solute, namely,cetophenone or 1-phenylethanol. The two solutes were theeactant and product of the hydrogenation of acetophenone to 1-henylethanol. The measurements were carried out in a Cailletetpparatus at pressures varying from 1 to 14 MPa and temperaturesrom 278 to 370 K. At the overall concentrations of CO2 exceeding5 mol%, the pressure needed for complete dissolution of the gasn the system with 1-phenylethanol was higher than that in theystem with acetophenone. An accurate location of the homoge-eous phases in both ternary systems can be useful in optimisinghe hydrogenation reaction conditions.
Organic solutes with similar molecules can affect the solubilitiesf CO2 in IL + solute mixtures in different ways. Kühne et al. [184]sed the Cailletet apparatus for measurements of CO2 solubility inhe mixture of [bmim][BF4] with 1-(4-isobutylphenyl)-ethanol atressures up to 14 MPa and temperatures from 278 to 368 K. Theffect of solute nature was investigated by comparing the resultsith previous measurements [183] on two other ternary systems
ontaining 4-isobutylacetophenone and 1-phenylethanol as therganic solutes. The presence of ketone increased the CO2 solu-ility more than the presence of alcohols and addition of an alkylroup to solute molecules with the same functional group has beenikely to increase the CO2 solubility.
Kühne et al. [185] investigated the solid–iquid and vapour–liquid equilibria in ternary system composedf [bmim][BF4], CO2 and (S)-naproxen. (S)-naproxen, or (S)-2-6-methoxynaphthalen-2-yl)propanoic acid, is a non-steroidalnti-inflammatory drug. The CO2 concentrations in five differentamples ranged from 10 to 50 mol%, and the temperature andressure ranges covered were 310–370 K and 1–14 MPa, respec-ively. The data showed that the amount of CO2 influenced not onlyhe location but also the extension of homogeneous liquid phaseegion, and allowed the selection of optimum pressure and tem-erature conditions to separate (S)-naproxen from [bmim][BF4] byrecipitation.
Bogel-Łukasik et al. [186] studied vapour–liquid equilibria in sixernary systems containing an IL, compressed CO2 and productsf a CALB-catalysed enzymatic reaction between (R,S)-2-octanol
nd succinic anhydride. The ILs employed were [omim][PF6] andomim][dca] whereas the solutes included 2-octanol, 4-(octan-2-loxy)-4-oxobutanoic acid (=hemiester) and dioctan-2-yl succinate=diester). The topic of the study and the components involvedere related to the authors’ earlier study of resolution of 2-ob
tm
ig. 12. Partition coefficients as functions of the initial concentrations ofemiester (black symbols) and diester (black–white symbols) before adding CO2 inomim][dca] at (♦,�) 5 MPa; (©, �) 7 MPa; (�,�) 9 MPa and (�,�) 11 MPa. Reprintedith permission from [186]. © 2008 American Chemical Society.
ctanol enantiomers in ILs via lipase-catalysed reactions [187].he measurements were carried out at 308 K and pressures within–11 MPa, and the results were used to express the distribution ofhe solute between the CO2-rich and the IL-rich phases through thefactor defined as
= y1
x1(1)
here y1 and x1 are the equilibrium mole fractions of the soluten the CO2-rich and the IL-rich phases, respectively. In a particularolute, the K factors increased with rising pressure and decreasedith rising initial concentration of the solute in the IL before adding
O2 as illustrated for the hemiester and diester in Fig. 12. The K fac-ors of Eq. (1) were used to obtain the separation factors of theolute pairs, and these were compared with the real separationactors obtained from phase equilibrium measurements on five-omponent systems IL–CO2–2-octanol–hemiester–diester. Sincell the systems were dilute with respect to the solutes, the separa-ion factors obtained from the ternary systems provided accuratestimates of the real separation factors in the five-component sys-ems.
Kroon et al. [188] studied experimentally the conditionsor crystallisation of MAAC from [bmim][BF4] using scCO2 asnti-solvent by measuring the phase behaviour of the ternaryystem [bmim][BF4]–CO2–MAAC. MAAC can be recovered frombmim][BF4] by either using a shift to higher CO2 concentrationst constant temperature (anti-solvent crystallisation) or by using ahift to lower temperatures at constant CO2 concentration (thermalhift). The results indicate the possibility to employ the anti-olvency behaviour of CO2 as a way to recover an organic compoundrom an IL by crystallisation.
. Supercritical fluid chromatography niche in the studiesf solute–ionic liquid–supercritical carbon dioxide systems
.1. Limiting partition coefficients of solutes in ioniciquid–supercritical carbon dioxide systems
The studies of organic–IL–CO2 systems by static techniquesf phase equilibrium measurements (Section 3) were carried outt finite (“nonzero”) concentrations of all components, and some
f them [189] indicate rather dramatic variations in the phaseehaviour with overall composition of the system.The development of predictive thermodynamic models forernary systems can usually benefit from experimental infor-
ation on a simpler “pseudo-binary” system containing only

r. A 12
aoifisrswildcmbad
titsae
K
ws
K
wpk
K
a
K
w(oeir
4
l[ecaaratruTsotI
tS
ss[ttTtficebva
fi[aAUtad
sm4btdufpsrmwapdiIrawopasooowrta
M. Roth / J. Chromatog
trace amount of the third component. In the particular casef organic–IL–CO2 ternary systems, valuable complementarynformation comes from limiting (infinite-dilution) partition coef-cients of the organic solute between both phases in IL–(sc)CO2ystem. To minimise disturbing the binary IL–CO2 phase equilib-ium by the presence of the solute, the amount of solute presenthould obviously be extremely low. This is precisely the situationhere the power of chromatography can be employed, and the
mportant developments of the experimental equipment for ana-ytical chromatography put to work for a purpose outside the veryomain of analytical chemistry itself. Further, it is just at the con-entration limits where the predictions of mixture properties areost sensitive to correct description of intermolecular interactions
etween unlike species. That’s why the infinite-dilution propertiesre particularly valuable in the development of molecular thermo-ynamic models of mixtures.
In an analogy with applications of GLC to low-pressure solutionhermodynamics, limiting partition coefficients of organic solutesn biphasic IL–CO2 systems can be obtained from solute reten-ion factors in a column with the IL as the stationary liquid andcCO2 as the carrier fluid. Solute distribution between the station-ry (IL + CO2) and mobile (scCO2) phase is usually quantified usingither partition coefficient,
c = c1s
c1m(2)
here c1s and c1m are the molar concentration of the solute in thetationary and the mobile phases, respectively, or K-factor,
= x1m
x1s(3)
here x1s and x1m are the mole fractions of the solute in the twohases. The relationships of Kc and K to the solute retention factor,1, are
c = k1Vm1 − x3s
n2svs(4)
nd
= M3n2s
k1Vm�m (1 − x3s)(5)
here Vm is the void volume of the column, x3s is the solubilityequilibrium mole-fraction) of CO2 in the IL, n2s is the numberf moles of the IL in the column, vs is the molar volume of CO2-xpanded IL in the column, M3 is the molar mass of CO2, and �m
s the density (mass/volume) of CO2. The quantities x3s, vs and �m
efer to the temperature and mean pressure in the column.
.1.1. Experimental studies of solute partitioningPlaneta et al. measured retention factors for a number of
ow-to-medium volatility solutes in [bmim][PF6]–scCO2 [190],bmim][BF4]–scCO2 [191] and [thtdp][Cl]–scCO2 [192] systemsmploying SFC with open tubular, wall-coated fused silica capillaryolumns. The open tubular column format was chosen to obtainfavourably low surface-to-volume ratio of the IL phase, with an
dded benefit of minimising the column pressure drop. The soluteetention factors were measured at temperatures from 313 to 353 Knd pressures ranging within 8.1–23.2 MPa. The resultant parti-ion coefficients were expressed in a relative way, i.e., they wereeferred to the partition coefficient of naphthalene in the partic-lar IL–scCO2 system at the particular temperature and pressure.
he relative partition coefficients are convenient for several rea-ons. First, they are not sensitive to the uncertainty in the amountf IL in the column (see Section 4.1.3). Second, they also minimisehe effect of uncertainty in the solubility of scCO2 in the particularL. Third, they are also convenient for modelling and correlation via4
etc
16 (2009) 1861–1880 1875
he Abraham’s linear solvation energy relationships (LSERs) (seeection 4.1.2).
Sakellarios and Kazarian [193] employed the ATR-IR spectro-copic technique in tandem with transmission spectroscopy totudy the partitioning of a model solute between both phases inbmim][PF6]–scCO2 system. The applicability of IR spectroscopy tohe solute–IL–scCO2 systems appears to be somewhat limited byhe overlap of the absorption bands of the individual components.herefore, benzil (1,2-diphenylethane-1,2-dione) was chosen ashe model solute because it provided clearly separated bandsor the spectroscopic study. Assuming a constant (i.e., medium-ndependent) value of the molar absorptivity of benzil, the partitionoefficient could be readily obtained from the absorbances andffective path lengths in the two phases. The initial concentration ofenzil in [bmim][PF6] was 0.05 (mole-fraction). The spectroscopicalues of Kc for benzil were consistent with the results of Planetand Roth [190].
Machida et al. [194] obtained infinite-dilution partition coef-cients of benzene, toluene, chlorobenzene and naphthalene inbmim][PF6]–scCO2 system employing packed column SFC withlab-assembled apparatus. The IL was coated on Chromosorb WW DMCS 60–80 mesh, and the coated support was packed into a-shaped, 1/4-in. tube column (40 cm long, 4 mm I.D.). The parti-
ion coefficients of naphthalene at 313.2 and 333.2 K were in goodgreement with the data of Planeta and Roth [190] whereas someeviations were noted at 353.2 K.
In a more application-oriented study, Planeta et al. [195] mea-ured the partition coefficients of several sulfur-containing aro-atic solutes (thianaphthene, benzothiazole, dibenzothiophene,
,6-dimethyldibenzothiophene, phenoxathiin and thianthrene) iniphasic [hmim][Tf2N]–scCO2 system. The purpose of this inves-igation was related to prospective applications of ILs in deepesulfurisation of oil refinery streams to meet the stringent reg-latory limits on the maximum content of sulfur in transportationuels. Currently, the prevailing method is high-temperature, high-ressure catalytic hydrodesulfurisation (HDS). As a result of HDS,ulfur is converted to hydrogen sulfide which is relatively easy toemove from the fuel stream. However, some sulfur-containing aro-atics, e.g., 4,6-dimethyldibenzothiophene and other compoundshere the sulfur atom is sterically hindered by alkyl substituents,
re highly resistant to HDS. Besides, HDS is an energy-intensiverocess. Therefore, alternative or complementary methods ofesulfurisation have been sought for, and one of the suggestions
nvolves extraction of sulfur compounds from the oil streams withLs [196]. In such applications, the IL would obviously have to beecycled for both economic and environmental reasons. Esser etl. [197] noted that re-extraction of sulfur compounds from ILsith scCO2 could be a possible way of recycling and purification
f the IL before the IL is repeatedly used to extract sulfur com-ounds from the oil streams. Esser et al. also expressed doubtsbout the applicability of the re-extraction process on a multiton-cale because of energy expenses. The partition coefficient databtained by Planeta et al. [195] suggest that the solvent powerf scCO2 is not high enough to secure an efficient re-extractionf the sulfur-containing aromatics even from [hmim][Tf2N], an ILith relatively low cohesive energy density. Overall, therefore, the
esults suggest that multiton-scale application of re-extraction ofhe sulfur compounds from ILs with scCO2 would be questionablet best.
.1.2. Modelling of solute partitioningAs opposed to a wide selection of thermodynamic models
mployed to describe the phase equilibria in binary IL–scCO2 sys-ems (see Section 2.3.3), there have been only a few attempts yet toorrelate/predict the limiting partition coefficients of organic non-

1 r. A 12
ewf
c[tr
l
waiaasts
dc[nfopaIona
bcflLafip
Fa[
ap1
tbnhtuipi[mfqoowtiatttcetam
pt[
�
876 M. Roth / J. Chromatog
lectrolyte solutes in biphasic IL–scCO2 systems. At the time of thisriting, only two different approaches appear to have been applied
or the purpose.Planeta et al. attempted to correlate the relative partition
oefficients of solutes in [bmim][BF4]–scCO2 systems [191] andthtdp][Cl]–scCO2 systems [192] via the Abraham’s LSER correla-ions. The partition coefficient of a solute A relative to that of theeference solute B (naphthalene) can be expressed by
og(
KcA
KcB
)= eEA + sSA + aAA + bBA + vVA (6)
here the descriptors of solute A [198,199] EA, SA, AA, BAnd VA describe the solute’s excess molar refractivity, dipolar-ty/polarisability, hydrogen bond acidity (hydrogen bond donatingbility), hydrogen bond basicity (hydrogen bond accepting ability)nd the McGowan’s characteristic volume [200], respectively. In aet of experimental data comprising a sufficient number of solutes,he coefficients e, s, a, b and v can be determined by linear regres-ion.
There is a vast number of LSER applications to data sets oniverse properties related to a change in the Gibbs energy, includinghromatographic retention data [8,201–203] and IL–gas systems8,203,204]. The success of LSERs is tied to their partly empiricalature in breaking a Gibbs energy-related property to contributions
rom the individual kinds of intermolecular interactions employingnly pure-component parameters for the individual solutes. Thisarticular feature is essential as it makes LSERs readily applicable tony solute for which the molecular descriptors are available [198].n the particular case of LSER applications to ILs, the overall numberf the equation coefficients needed to cover all ILs can even be sig-ificantly reduced by dissecting the coefficients into cation-specificnd anion-specific contributions [205,206].
Generally, one should bear in mind that LSERs have originallyeen designed for applications in incompressible media. Therefore,are is needed when applying LSERs to systems with supercriticaluids of wide-ranging densities. Planeta et al. [191,192] applied theSER-based correlations to data sets obtained at a fixed temperature
nd pressure (and CO2 density). The results were relatively satis-actory, and the accuracy of the predictions seems sufficient for annitial engineering design of real-world processes employing thearticular IL–scCO2 biphasic system. As an example, Fig. 13 showsig. 13. LSER fit of relative partition coefficients in [bmim][BF4]–CO2 system at 333 Knd 12.1 MPa. Calculated versus experimental data. Reprinted with permission from191].© 2005 American Chemical Society.
w
T
wPcIbctbi
P
w�
b
�
w
odp
16 (2009) 1861–1880
comparison of the LSER-fitted and experimental values of relativeartition coefficients in [bmim][BF4]–scCO2 systems at 333 K and2.1 MPa.
Expectedly, the deviations of the LSER-predicted values fromhe experimental data are most prominent in the solutes capa-le of hydrogen bonding; the reason for this, however, need notecessarily come from possible difficulties of LSERs in describingydrogen-bonded systems (see Section 4.1.3). The LSER correla-ions are obtained by multiparameter linear regression so that thenderlying mathematics is very simple and no specialised software
s needed since common spreadsheet programs are adequate. Therincipal strength of the LSER correlations comes from availabil-
ty of the molecular descriptors for large selections of solutes, e.g.,198]; this provides the LSERs with significant predictive power. The
ain disadvantage of LSERs in this particular application comesrom the necessity to handle compressible media, and conse-uently, to accommodate the effects of temperature and pressuren solvent characteristics of the phases involved. This applies notnly to the scCO2 mobile phase but also to the stationary phase asell because the solubilities of CO2 in ILs vary widely within the
emperature and pressure range concerned. As indicated by approx-mate calculations of Planeta et al. [191,192], dissolution of CO2 inn IL tends to decrease the effective cohesive energy density (andhe solubility parameter) of the IL–CO2 mixture as compared withhe pure IL, probably because of “dilution” of the ion–ion interac-ions by the molecules of CO2. A possible way to overcome theseomplications could be to obtain LSER correlations at several differ-nt pairs of temperature and pressure conditions, and then attempto correlate the resultant LSER coefficients as functions of temper-ture and pressure. Unfortunately, this procedure requires ratherassive amount of experimental data as the input.In another approach, Machida et al. [194] modelled the limiting
artition coefficients of organic solutes in biphasic IL–scCO2 sys-ems with the Sanchez–Lacombe (S–L) mean-field lattice fluid EoS207,208]. The S–L EoS has the form
˜ 2 + P + T[
ln(1 − �) +(
1 − 1r
)�]
= 0 (7)
ith
˜ = T
T∗ P = P
P∗ � = �
�∗ r = MP∗
kBT∗�∗ (8)
here T is the temperature, P is the pressure, � is the density and T*,* and �* are the respective characteristic parameters of the pureomponent with molar mass M, and kB is the Boltzmann constant.n pure components, the characteristic parameters T*, P* and �* cane obtained from the pure component properties. In mixtures, theharacteristic parameters depend on the pure-component charac-eristic parameters through mixing and combining rules describedy Sanchez and Lacombe [208]. The mixing rule for the character-stic pressure P* is
∗ =∑
i
�iP∗i − 1
2
∑i
∑j
�i�j�P∗ij (9)
here �i is the volume fraction of component i in the mixture andP∗
ijis related to the characteristic pressures of components i and j
y the combining rule
P∗ij = P∗
i + P∗j − 2
(1 − �ij
)√P∗
iP∗
j(10)
here �ij is an adjustable binary interaction parameter.Application of the S–L EoS to the solute–IL–CO2 system is based
n the assumption that the presence of a trace amount of soluteoes not disturb the solubility of CO2 in the IL, and that the chemicalotentials of the solute in both phases are the same at equilibrium.

M. Roth / J. Chromatogr. A 12
F[S[
TSfipItostlpcLc�otw
mbEsksit
4s
cbttfspswiti[e
dst
hdw
pemIwcpIwapiamtaaeheibbn
aacalrgabasamoptt
sotcetsca
ig. 14. Infinite-dilution partition coefficients of naphthalene in naphthalene–bmim][PF6]–CO2 system. Experimental data: (�) 313.2 K, (�) 333.2 K, (♦) 353.2 K.–L EoS: (- - - -) 313.2 K, (-·-·-) 333.2 K, (—) 353.2 K. Reprinted with permission from194].© 2007 Elsevier.
he expression for the chemical potential can be derived from the–L EoS so that, finally, the relationship for the solute partition coef-cient can be deduced. To use it, however, the unlike interactionarameter �ij values (Eq. (10)) must be known for the respective
L–CO2, solute–CO2 and solute–IL pairs. Machida et al. obtainedhe [bmim][PF6]–CO2 parameters from the phase equilibrium dataf Shariati et al. [126] and the parameters for the individualolute–CO2 pairs from the literature data on equilibrium proper-ies of the respective solute–CO2 binary system. Because of theack of the solute–[bmim][PF6] binary data, the respective binaryarameters had to be obtained by fitting the measured partitionoefficients of the respective solute. It has turned out that the S–EoS provided a good representation of the solute partition coeffi-ients as indicated by Fig. 14. However, as the interaction parameterij value is unique to the particular i–j pair, the S–L EoS descriptionf one system is difficult to generalise to other systems. Thereby,he ability of the S–L EoS to serve as a predictive tool for systemshere no experimental data are available is rather limited.
To some extent, the LSER and the S–L EoS models are comple-entary because the strengths and weaknesses differ markedly
etween the two models. Compared with the LSER model, the S–LoS is more rigorous but also more difficult to generalise to a wideelection of prospective solute–IL–scCO2 systems. This particularind of systems combine an organic nonelectrolyte, an ionic sub-tance and a supercritical fluid, making dependable modelling anntricate task. Consequently, a real progress would probably requirehe use of multiple modelling approaches.
.1.3. Pros and cons of supercritical fluid chromatography inolute–ionic liquid–supercritical carbon dioxide measurements
Some of the “pros” to be mentioned here are common to allhromatographic studies of solution thermodynamics rather thaneing limited to systems with ILs. These common pros includehe relative rate of data generation as a large number of parti-ioning data can be obtained in a short time. This ability resultsrom separation power of chromatography – because of that, severalolutes can be handled in a single experimental run and the soluteurity is not a primary concern. Further, the required amounts ofolute(s) and the stationary phase (IL) are very low as comparedith those needed for static techniques of phase equilibrium stud-
es. As the ILs are capable of a large variety of interactions withhe solutes, it is possible to use a single column (=a single load-ng with IL) to handle solutes of widely different volatilities. Inbmim][BF4]–CO2 system at 313 K and a fixed density of CO2, forxample, the solute retention factors [191] ranged well within three
s
pir
16 (2009) 1861–1880 1877
ecadic orders of magnitude whereas the vapour pressures of theolutes spanned across more than five decadic orders of magni-ude.
Also, the physicochemical and solvent properties of IL samplesave been known to vary with water content in the IL. If sufficientlyry CO2 is used in SFC, the flow of CO2 through the column can stripater off the stationary IL.
The above pros, or at least a part of them, are certainly accom-anied by several “cons”. Compared with static phase equilibriumxperiments employing bulk ILs, the chromatographic measure-ents can suffer from much larger surface-to-volume ratio of the
L phase. The ratio is generally higher in packed columns comparedith wall-coated open tubular (WCOT) columns but even in WCOT
olumns it is still higher than that in the static systems. Moreover,reparation of a stable and reasonably efficient WCOT column with
L may not be easy as the non-ionic surface of silica is not readilyetted with ILs. This problem can partly be alleviated by preparingmicrocrystalline layer of NaCl [209] in the fused-silica capillaryrior to coating the capillary with the IL [190]. Then, however, there
s a danger of ion exchange reaction (“metathesis”) between the ILnd the underlying layer of NaCl, particularly if the system is notoisture-free. Alternatively, therefore, it may be useful to increase
he inner surface area of fused silica tubing by making it rough,nd thereby increase somewhat the stability of the IL coating inWCOT column. To this end, Planeta et al. [192,195] used 24-h
tching of the inner surface with saturated solution of ammoniumydrogen difluoride ([NH4][HF2]) in anhydrous methanol; surfacetching with [NH4][HF2] was originally employed for applicationsn electrochromatography [210,211]. Fused silica capillaries can alsoe treated with 2-chloro-1,1,2-trifluoroethylmethylether that haseen used to prepare nanowires to enlarge the inner surface ofarrow bore tubing [212,213].
The large surface-to-volume ratio of IL in the column bringsbout uncertainty in the retention mechanism as the solutedsorption on the IL surface can become significant. In GLC, theontributions of partitioning and adsorption to solute retentionre usually separated by employing several columns with differentoadings of the stationary phase and extrapolating the resultant netetention volumes to infinite volume of the stationary phase as sug-ested long ago by Conder et al. [214]. This procedure has also beendopted in some recent measurements of activity coefficients in ILsy GLC [45–47]. Application of this procedure obviously requiresprecise control of column temperature so that the variations in
olute retention with temperature could not interfere with the vari-tions caused by solute adsorption. In SFC, however, the situation isore complicated as the solute retention depends strongly not only
n temperature but, unlike GLC, also on pressure. This effectivelyrevents the application of Conder et al.’s [214] procedure in SFC ashe column pressure is generally more difficult to control preciselyhan is the column temperature.
Calculation of limiting partition coefficients of solutes in bipha-ic IL–scCO2 systems requires an accurate knowledge of the amountf IL in the column. Obviously, the amount of IL is more easily con-rolled in a column packed with IL-coated support than in a WCOTolumn coated with IL. On the other hand, packed columns gen-rally require a higher pressure drop than WCOT columns, and inhermodynamic measurements by SFC the column pressure drophould be minimised. Therefore, in this particular field of SFC appli-ations, both packed and WCOT columns each have relative meritsnd drawbacks of their own, and neither format of the column
hould be indiscriminately preferred in all situations.Some of the phenomena discussed above can certainly com-romise the accuracy of the partitioning data obtained by SFC,
n particular with highly polar solutes. However, even with thiseservation, SFC still provides the easiest way to measure the

1 r. A 12
ls
4a
ssdbdwm
ibCiuTmpS
tb[Iia
aokGmctc
5
lbpmadustrsi
A
a
dAP
R
878 M. Roth / J. Chromatog
imiting partition coefficients of solutes in IL–scCO2 biphasicystems.
.2. Supercritical fluid chromatography with ionic liquids innalytical separations?
To date, all applications of SFC with ILs as stationary phaseserved an extra-analytical purpose, namely, determination of theolute partition coefficients to be used for process design or forevelopment of predictive thermodynamic models of the phaseehaviour in solute–IL–scCO2 systems. Thus, the situation in SFC isifferent from that in GLC where the IL stationary phases have beenidely used for both analytical separations and thermodynamiceasurements.Apart from an overall decrease of interest in SFC as an analyt-
cal separation technique, another major reason why ILs have noteen used in analytical SFC comes probably from high solubility ofO2 in most ILs. The high solubility of CO2 is likely to bring a higher
mportance of dispersion forces and other non-specific intermolec-lar interactions in IL–CO2 mixtures as compared with pure ILs.herefore, the dissolved CO2 tends to make the IL less selective andore “nonpolar”. Consequently, the case for utilising the specific
roperties of ILs (their dual nature [36]) as stationary phases forFC is certainly weaker as compared with GLC.
Besides, in spite of very low solubilities of most ILs in scCO2,he amount of IL in a chromatographic column decreases slowlyut definitely when the column is being used in SFC with scCO2192,195]. Therefore, preparation of reasonably stable columns withLs for analytical SFC would almost certainly require the use ofmmobilised/cross-linked ILs such as those employed by Andersonnd Armstrong [215] in high-temperature GC.
Alternatively, poly(ionic liquid)s could be used, i.e., the materi-ls prepared by polymerisation of imidazolium ILs containing vinylr other polymerisable substituents on the imidazolium ring. Thisind of poly-ILs has recently been used as stationary phases forC [216]. However, some of these materials tend to absorb evenore CO2 than “ordinary” ILs [217,218]. Again, therefore, any spe-
ific selectivity of poly-ILs in SFC would probably be “diluted” byhe absorbed CO2 and would manifest itself to a lesser extent asompared with poly-IL stationary phases in GC.
. Conclusion
As of the time of this writing, several studies have been pub-ished of limiting partition coefficients of organic solutes betweenoth phases of IL–scCO2 systems by SFC at different operating tem-eratures and pressures. Although there is still a lack of test dataeasured by independent experimental methods, SFC emerges aspowerful technique to study the interphase distribution of highlyilute solutes in IL–scCO2 systems. In this sense, SFC can be a veryseful complement to other methods of phase equilibrium mea-urements in systems with ILs since it provides the easiest wayo measure the limiting partition coefficients. A major issue to beesolved in the future is the reliability of SFC data, namely, the role ofpurious retention mechanisms and the ensuing systematic errorsn the resultant partition coefficients.
cknowledgements
I am indebted to an anonymous reviewer for bringing Refs. [48]
nd [49] to my attention.This contribution was supported by the Czech Science Foun-ation (Projects GA203/08/1465 and GA203/08/1536) and by thecademy of Sciences of the Czech Republic (Institutional Researchlan No. AV0Z40310501).
16 (2009) 1861–1880
eferences
[1] P. Walden, Bull. Acad. Imper. Sci. St. Peters. 8 (1914) 405.[2] J.S. Wilkes, J.A. Levisky, R.A. Wilson, C.L. Hussey, Inorg. Chem. 21 (1982) 1263.[3] J.S. Wilkes, M.J. Zaworotko, J. Chem. Soc. Chem. Commun. (1992) 965.[4] K.N. Marsh, J.A. Boxall, R. Lichtenthaler, Fluid Phase Equilib. 219 (2004) 93.[5] C.A. Angell, N. Byrne, J.-P. Belieres, Acc. Chem. Res. 40 (2007) 1228.[6] H. Weingärtner, Angew. Chem. Int. Ed. 47 (2008) 654.[7] K.N. Marsh, A. Deev, A.C.-T. Wu, E. Tran, A. Klamt, Korean J. Chem. Eng. 19
(2002) 357.[8] C.F. Poole, J. Chromatogr. A 1037 (2004) 49.[9] L.P.N. Rebelo, J.N.C. Lopes, J.M.S.S. Esperanca, H.J.R. Guedes, J. Łachwa, V.
Najdanovic-Visak, Z.P. Visak, Acc. Chem. Res. 40 (2007) 1114.[10] C. Hardacre, J.D. Holbrey, M. Nieuwenhuyzen, T.G.A. Youngs, Acc. Chem. Res.
40 (2007) 1146.[11] T. Welton, Chem. Rev. 99 (1999) 2071.[12] P. Wasserscheid, T. Welton (Eds.), Ionic Liquids in Synthesis, Wiley-VCH, Wein-
heim, 2003.[13] Z.C. Zhang, Adv. Catal. 49 (2006) 153.[14] V.I. Pârvulescu, C. Hardacre, Chem. Rev. 107 (2007) 2615.[15] D.R. MacFarlane, M. Forsyth, P.C. Howlett, J.M. Pringle, J. Sun, G. Annat, W. Neil,
E.I. Izgorodina, Acc. Chem. Res. 40 (2007) 1165.[16] P. Hapiot, C. Lagrost, Chem. Rev. 108 (2008) 2238.[17] J.G. Huddleston, H.D. Willauer, R.P. Swatloski, A.E. Visser, R.D. Rogers, Chem.
Commun. (1998) 1765.[18] H. Zhao, S. Xia, P. Ma, J. Chem. Technol. Biotechnol. 80 (2005) 1089.[19] M. Freemantle, Chem. Eng. News 76 (1998) 32.[20] K.R. Seddon, A. Stark, M.-J. Torres, Pure Appl. Chem. 72 (2000) 2275.[21] J. Pernak, K. Sobaszkiewicz, I. Mirska, Green Chem. 5 (2003) 52.[22] R.P. Swatloski, J.D. Holbrey, S.B. Memon, G.A. Caldwell, K.A. Caldwell, R.D.
Rogers, Chem. Commun. (2004) 668.[23] B. Jastorff, K. Mölter, P. Behrend, U. Bottin-Weber, J. Filser, A. Heimers, B. Ondr-
uschka, J. Ranke, M. Schaefer, H. Schröder, A. Stark, P. Stepnowski, F. Stock, R.Störmann, S. Stolte, U. Welz-Biermann, S. Ziegert, J. Thöming, Green Chem. 7(2005) 362.
[24] D.J. Couling, R.J. Bernot, K.M. Docherty, J.K. Dixon, E.J. Maginn, Green Chem. 8(2006) 82.
[25] S. Stolte, M. Matzke, J. Arning, A. Böschen, W.-R. Pitner, U. Welz-Biermann, B.Jastorff, J. Ranke, Green Chem. 9 (2007) 1170.
[26] K.J. Kulacki, G.A. Lamberti, Green Chem. 10 (2008) 104.[27] R.P. Swatloski, J.D. Holbrey, R.D. Rogers, Green Chem. 5 (2003) 361.[28] S. Carda-Broch, A. Berthod, D.W. Armstrong, Anal. Bioanal. Chem. 375 (2003)
191.[29] X. Han, D.W. Armstrong, Acc. Chem. Res. 40 (2007) 1079.[30] M. Vaher, M. Koel, M. Kaljurand, Electrophoresis 23 (2002) 426.[31] W. Qin, S.F.Y. Li, Electrophoresis 23 (2002) 4110.[32] X. Jin, L. Yu, D. Garcia, R.X. Ren, X. Zeng, Anal. Chem. 78 (2006) 6980.[33] A. Saheb, M. Josowicz, J. Janata, Anal. Chem. 80 (2008) 4214.[34] D.W. Armstrong, L.-K. Zhang, L. He, M.L. Gross, Anal. Chem. 73 (2001) 3679.[35] A. Tholey, E. Heinzle, Anal. Bioanal. Chem. 386 (2006) 24.[36] D.W. Armstrong, L. He, Y.-S. Liu, Anal. Chem. 71 (1999) 3873.[37] J.L. Anderson, D.W. Armstrong, Anal. Chem. 75 (2003) 4851.[38] A. Heintz, D.V. Kulikov, S.P. Verevkin, J. Chem. Eng. Data 46 (2001) 1526.[39] T.M. Letcher, B. Soko, D. Ramjugernath, N. Deenadayalu, A. Nevines, P.K.
Naicker, J. Chem. Eng. Data 48 (2003) 708.[40] T.M. Letcher, U. Domanska, M. Marciniak, A. Marciniak, J. Chem. Thermodyn.
37 (2005) 587.[41] F. Mutelet, J.-N. Jaubert, M. Rogalski, M. Boukherissa, A. Dicko, J. Chem. Eng.
Data 51 (2006) 1274.[42] M.-L. Ge, L.-S. Wang, M.-Y. Li, J.-S. Wu, J. Chem. Eng. Data 52 (2007) 2257.[43] A. Heintz, S.P. Verevkin, J.K. Lehmann, T.V. Vasiltsova, D. Ondo, J. Chem. Ther-
modyn. 39 (2007) 268.[44] U. Domanska, A. Marciniak, J. Phys. Chem. B 111 (2007) 11984.[45] F. Mutelet, J.-N. Jaubert, J. Chromatogr. A 1102 (2006) 256.[46] F. Mutelet, J.-N. Jaubert, M. Rogalski, J. Harmand, M. Sindt, J.-L. Mieloszynski,
J. Phys. Chem. B 112 (2008) 3773.[47] M. Sobota, Experimental determination of infinite dilution activity coeffi-
cients of volatile organic compounds in ionic liquids, Diploma Thesis, Instituteof Chemical Technology, Prague, 2008.
[48] D. Niehaus, M. Philips, A. Michael, R.M. Wightman, J. Phys. Chem. 93 (1989)6232.
[49] D.E. Niehaus, R.M. Wightman, P.A. Flowers, Anal. Chem. 63 (1991) 1728.[50] M. Makosza, M. Wawrzyniewicz, Tetrahedron Lett. 10 (1969) 4659.[51] C.M. Starks, J. Am. Chem. Soc. 93 (1971) 195.[52] C.M. Starks, C.L. Liotta, M. Halpern, Phase-Transfer Catalysis—Fundamentals,
Applications, and Industrial Perspectives, Chapman & Hall, New York, 1994.[53] I.T. Horváth, J. Rábai, Science 266 (1994) 72.[54] W. Keim, Green Chem. 5 (2003) 105.
[55] R.A. Sheldon, Green Chem. 7 (2005) 267.[56] R.A. Brown, P. Pollet, E. McKoon, C.A. Eckert, C.L. Liotta, P.G. Jessop, J. Am. Chem.Soc. 123 (2001) 1254.[57] M.B. Shiflett, A. Yokozeki, AIChE J. 52 (2006) 1205.[58] M.B. Shiflett, A. Yokozeki, J. Chem. Eng. Data 51 (2006) 1931.[59] L.A. Blanchard, Z. Gu, J.F. Brennecke, J. Phys. Chem. B 105 (2001) 2437.

r. A 12
[[[
[
[
[[[[
[
[
[[
[
[
[[[[[
[
[[
[[
[[[
[[[
[
[[[
[[[
[
[
[[[[
[[
[[[
[
[
[[[
M. Roth / J. Chromatog
[60] A. Shariati, C.J. Peters, J. Supercrit. Fluids 25 (2003) 109.[61] W. Wu, J. Zhang, B. Han, J. Chen, Z. Liu, T. Jiang, J. He, W. Li, Chem. Commun.
(2003) 1412.[62] W. Wu, W. Li, B. Han, T. Jiang, D. Shen, Z. Zhang, D. Sun, B. Wang, J. Chem. Eng.
Data 49 (2004) 1597.[63] Z. Zhang, W. Wu, B. Wang, J. Chen, D. Shen, B. Han, J. Supercrit. Fluids 40 (2007)
1.[64] S.V. Dzyuba, R.A. Bartsch, Angew. Chem. Int. Ed. 42 (2003) 148.[65] S. Keskin, D. Kayrak-Talay, U. Akman, Ö. Hortacsu, J. Supercrit. Fluids 43 (2007)
150.[66] M.J. Earle, J.M.S.S. Esperanca, M.A. Gilea, J.N. Canongia Lopes, L.P.N. Rebelo,
J.W. Magee, K.R. Seddon, J.A. Widegren, Nature 439 (2006) 831.[67] D.H. Zaitsau, G.J. Kabo, A.A. Strechan, Y.U. Paulechka, A. Tschersich, S.P.
Verevkin, A. Heintz, J. Phys. Chem. A 110 (2006) 7303.[68] B.A. DaSilveira Neto, L.S. Santos, F.M. Nachtigall, M.N. Eberlin, J. Dupont,
Angew. Chem. Int. Ed. 45 (2006) 7251.[69] J.P. Armstrong, C. Hurst, R.G. Jones, P. Licence, K.R.J. Lovelock, C.J. Satterley, I.J.
Villar-Garcia, Phys. Chem. Chem. Phys. 9 (2007) 982.[70] J.A. Widegren, Y.-M. Wang, W.A. Henderson, J.W. Magee, J. Phys. Chem. B 111
(2007) 8959.[71] L.A. Blanchard, D. Hancu, E.J. Beckman, J.F. Brennecke, Nature 399 (1999) 28.[72] L.A. Blanchard, J.F. Brennecke, Ind. Eng. Chem. Res. 40 (2001) 287.[73] O. Bortolini, S. Campestrini, V. Conte, G. Fantin, M. Fogagnolo, S. Maietti, Eur.
J. Org. Chem. (2003) 4804.[74] S. Mekki, C.M. Wai, I. Billard, G. Moutiers, C.H. Yen, J.S. Wang, A. Ouadi, C.
Gaillard, P. Hesemann, Green Chem. 7 (2005) 421.[75] S. Mekki, C.M. Wai, I. Billard, G. Moutiers, J. Burt, B. Yoon, J.S. Wang, C. Gaillard,
A. Ouadi, P. Hesemann, Chem. Eur. J. 12 (2006) 1760.[76] Z. Zhou, T. Wang, H. Xing, Ind. Eng. Chem. Res. 45 (2006) 525.[77] B. Yoon, C.H. Yen, S. Mekki, S. Wherland, C.M. Wai, Ind. Eng. Chem. Res. 45
(2006) 4433.[78] M.C. Kroon, J. van Spronsen, C.J. Peters, R.A. Sheldon, G.-J. Witkamp, Green
Chem. 8 (2006) 246.[79] R.A. Sheldon, Chem. Ind. (London) 23 (1992) 903.[80] R.A. Sheldon, Green Chem. 9 (2007) 1273.[81] J.M. Prausnitz, R.N. Lichtenthaler, E.G. de Azevedo, Molecular Thermodynam-
ics of Fluid-Phase Equilibria, 2nd ed., Prentice-Hall, Englewood Cliffs, NJ, 1986,p. 2.
[82] F. Liu, M.B. Abrams, R.T. Baker, W. Tumas, Chem. Commun. (2001) 433.[83] M.F. Sellin, P.B. Webb, D.J. Cole-Hamilton, Chem. Commun. (2001) 781.[84] A. Bösmann, G. Franciò, E. Janssen, M. Solinas, W. Leitner, P. Wasserscheid,
Angew. Chem. Int. Ed. 40 (2001) 2697.[85] D. Ballivet-Tkatchenko, M. Picquet, M. Solinas, G. Franciò, P. Wasserscheid, W.
Leitner, Green Chem. 5 (2003) 232.[86] M. Solinas, A. Pfaltz, P.G. Cozzi, W. Leitner, J. Am. Chem. Soc. 126 (2004) 16142.[87] A. Serbanovic, L.C. Branco, M. Nunes da Ponte, C.A.M. Afonso, J. Organometal.
Chem. 690 (2005) 3600.[88] Y. Xie, Z. Zhang, S. Hu, J. Song, W. Li, B. Han, Green Chem. 10 (2008) 278.[89] F. Zayed, L. Greiner, P.S. Schulz, A. Lapkin, W. Leitner, Chem. Commun. (2008)
79.[90] P. Lozano, T. de Diego, D. Carrié, M. Vaultier, J.L. Iborra, Chem. Commun. (2002)
692.[91] M.T. Reetz, W. Wiesenhöfer, G. Franciò, W. Leitner, Chem. Commun. (2002)
992.[92] M.T. Reetz, W. Wiesenhöfer, G. Franciò, W. Leitner, Adv. Synth. Catal. 345
(2003) 1221.[93] P. Lozano, T. de Diego, S. Gmouh, M. Vaultier, J.L. Iborra, Biotechnol. Prog. 20
(2004) 661.[94] P. Lozano, E. García-Verdugo, R. Piamtongkam, N. Karbass, T. De Diego, M.I.
Burguete, S.V. Luis, J.L. Iborra, Adv. Synth. Catal. 349 (2007) 1077.[95] G. Zhao, T. Jiang, W. Wu, B. Han, Z. Liu, H. Gao, J. Phys. Chem. B 108 (2004)
13052.[96] N.-Y. Mun, K.-H. Kim, D.-W. Park, Y. Choe, I. Kim, Korean J. Chem. Eng. 22 (2005)
556.[97] J. Sun, S. Fujita, M. Arai, J. Organometal. Chem. 690 (2005) 3490.[98] Z. Zhang, Y. Xie, W. Li, S. Hu, J. Song, T. Jiang, B. Han, Angew. Chem. Int. Ed. 47
(2008) 1127.[99] L. Zhang, D. Niu, K. Zhang, G. Zhang, Y. Luo, J. Lu, Green Chem. 10 (2008) 202.100] A.M. Scurto, S.N.V.K. Aki, J.F. Brennecke, J. Am. Chem. Soc. 124 (2002) 10276.101] A.M. Scurto, S.N.V.K. Aki, J.F. Brennecke, Chem. Commun. (2003) 572.102] Z. Zhang, W. Wu, H. Gao, B. Han, B. Wang, Y. Huang, Phys. Chem. Chem. Phys.
6 (2004) 5051.103] V. Najdanovic-Visak, A. Serbanovic, J.M.S.S. Esperanca, H.J.R. Guedes, L.P.N.
Rebelo, M. Nunes da Ponte, ChemPhysChem 4 (2003) 520.104] Z. Zhang, W. Wu, B. Han, T. Jiang, B. Wang, Z. Liu, J. Phys. Chem. B 109 (2005)
16176.105] S.N.V.K. Aki, A.M. Scurto, J.F. Brennecke, Ind. Eng. Chem. Res. 45 (2006) 5574.106] B.R. Mellein, J.F. Brennecke, J. Phys. Chem. B 111 (2007) 4837.107] E.M. Saurer, S.N.V.K. Aki, J.F. Brennecke, Green Chem. 8 (2006) 141.
108] J.W. Hutchings, K.L. Fuller, M.P. Heitz, M.M. Hoffmann, Green. Chem. 7 (2005)475.109] S.N.V.K. Aki, B.R. Mellein, E.M. Saurer, J.F. Brennecke, J. Phys. Chem. B 108
(2004) 20355.[110] A. Kordikowski, A.P. Schenk, R.M. Van Nielen, C.J. Peters, J. Supercrit. Fluids 8
(1995) 205.
[[
[[
16 (2009) 1861–1880 1879
[111] S.G. Kazarian, B.J. Briscoe, T. Welton, Chem. Commun. (2000) 2047.112] C. Cadena, J.L. Anthony, J.K. Shah, T.I. Morrow, J.F. Brennecke, E.J. Maginn, J.
Am. Chem. Soc. 126 (2004) 5300.[113] J.L. Anthony, J.L. Anderson, E.J. Maginn, J.F. Brennecke, J. Phys. Chem. B 109
(2005) 6366.[114] M. Kanakubo, T. Umecky, Y. Hiejima, T. Aizawa, H. Nanjo, Y. Kameda, J. Phys.
Chem. B 109 (2005) 13847.[115] J.L. Anthony, E.J. Maginn, J.F. Brennecke, J. Phys. Chem. B 106 (2002) 7315.[116] P. Husson-Borg, V. Majer, M.F. Costa Gomes, J. Chem. Eng. Data 48 (2003)
480.[117] A.P.-S. Kamps, D. Tuma, J. Xia, G. Maurer, J. Chem. Eng. Data 48 (2003) 746.[118] R.E. Baltus, B.H. Culbertson, S. Dai, H. Luo, D.W. DePaoli, J. Phys. Chem. B 108
(2004) 721.[119] A. Shariati, C.J. Peters, J. Supercrit. Fluids 29 (2004) 43.120] A. Shariati, C.J. Peters, J. Supercrit. Fluids 30 (2004) 139.121] M. Costantini, V.A. Toussaint, A. Shariati, C.J. Peters, I. Kikic, J. Chem. Eng. Data
50 (2005) 52.122] M.C. Kroon, A. Shariati, M. Costantini, J. van Spronsen, G.-J. Witkamp, R.A.
Sheldon, C.J. Peters, J. Chem. Eng. Data 50 (2005) 173.123] S. Zhang, Y. Chen, R.X.-F. Ren, Y. Zhang, J. Zhang, X. Zhang, J. Chem. Eng. Data
50 (2005) 230.124] S. Zhang, X. Yuan, Y. Chen, X. Zhang, J. Chem. Eng. Data 50 (2005) 1582.125] M.B. Shiflett, A. Yokozeki, Ind. Eng. Chem. Res. 44 (2005) 4453.126] A. Shariati, K. Gutkowski, C.J. Peters, AIChE J. 51 (2005) 1532.127] K.I. Gutkowski, A. Shariati, C.J. Peters, J. Supercrit. Fluids 39 (2006) 187.128] J. Kumełan, A.P.-S. Kamps, D. Tuma, G. Maurer, J. Chem. Thermodyn. 38 (2006)
1396.129] J. Kumełan, A.P.-S. Kamps, D. Tuma, G. Maurer, J. Chem. Eng. Data 51 (2006)
1802.130] D. Fu, X. Sun, J. Pu, S. Zhao, J. Chem. Eng. Data 51 (2006) 371.131] Y. Chen, S. Zhang, X. Yuan, Y. Zhang, X. Zhang, W. Dai, R. Mori, Thermochim.
Acta 441 (2006) 42.132] A. Blasig, J. Tang, X. Hu, Y. Shen, M. Radosz, Fluid Phase Equilib. 256 (2007) 75.133] A. Blasig, J. Tang, X. Hu, S.P. Tan, Y. Shen, M. Radosz, Ind. Eng. Chem. Res. 46
(2007) 5542.134] X. Yuan, S. Zhang, J. Liu, X. Lu, Fluid Phase Equilib. 257 (2007) 195.135] A.M. Schilderman, S. Raeissi, C.J. Peters, Fluid Phase Equilib. 260 (2007) 19.136] J.E. Bara, C.J. Gabriel, S. Lessmann, T.K. Carlisle, A. Finotello, D.L. Gin, R.D. Noble,
Ind. Eng. Chem. Res. 46 (2007) 5380.137] Y. Hou, R.E. Baltus, Ind. Eng. Chem. Res. 46 (2007) 8166.138] M.B. Shiflett, A. Yokozeki, J. Phys. Chem. B 111 (2007) 2070.139] M.J. Muldoon, S.N.V.K. Aki, J.L. Anderson, J.K. Dixon, J.F. Brennecke, J. Phys.
Chem. B 111 (2007) 9001.140] M.B. Shiflett, D.J. Kasprzak, C.P. Junk, A. Yokozeki, J. Chem. Thermodyn. 40
(2008) 25.141] E.-K. Shin, B.-C. Lee, J.S. Lim, J. Supercrit. Fluids 45 (2008) 282.142] A. Shariati, C.J. Peters, J. Supercrit. Fluids 34 (2005) 171.143] C.M. Gordon, J.D. Holbrey, A.R. Kennedy, K.R. Seddon, J. Mater. Chem. 8 (1998)
2627.144] S.G. Kazarian, N. Sakellarios, C.M. Gordon, Chem. Commun. (2002) 1314.145] A.M. Scurto, W. Leitner, Chem. Commun. (2006) 3681.146] A.M. Scurto, E. Newton, R.R. Weikel, L. Draucker, J. Hallett, C.L. Liotta, W. Leitner,
C.A. Eckert, Ind. Eng. Chem. Res. 47 (2008) 493.147] M.R. Ally, J. Braunstein, R.E. Baltus, S. Dai, D.W. DePaoli, J.M. Simonson, Ind.
Eng. Chem. Res. 43 (2004) 1296.148] J.M. Prausnitz, R.N. Lichtenthaler, E. Gomes de Azevedo, Molecular Thermo-
dynamics of Fluid-Phase Equilibria, 2nd ed., Prentice-Hall, Englewood Cliffs,NJ, 1986, Ch. 8, p. 371.
149] D. Camper, P. Scovazzo, C. Koval, R. Noble, Ind. Eng. Chem. Res. 43 (2004) 3049.150] T.I. Morrow, E.J. Maginn, J. Phys. Chem. B 106 (2002) 12807.151] Z. Liu, S. Huang, W. Wang, J. Phys. Chem. B 108 (2004) 12978.152] K. Swiderski, A. McLean, C.M. Gordon, D.H. Vaughan, Chem. Commun. (2004)
2178.153] S.H. Lee, S.B. Lee, Chem. Commun. (2005) 3469.154] P. Scovazzo, D. Camper, J. Kieft, J. Poshusta, C. Koval, R. Noble, Ind. Eng. Chem.
Res. 43 (2004) 6855.155] P.K. Kilaru, R.A. Condemarin, P. Scovazzo, Ind. Eng. Chem. Res. 47 (2008) 900.156] P.K. Kilaru, P. Scovazzo, Ind. Eng. Chem. Res. 47 (2008) 910.157] H. Orbey, S.I. Sandler, Modelling Vapor–Liquid Equilibria. Cubic Equations of
State and Their Mixing Rules, Cambridge University Press, New York, 1998.158] A. Anderko, in: J.V. Sengers, R.F. Kayser, C.J. Peters, H.J. White Jr. (Eds.), Equa-
tions of State for Fluids and Fluid Mixtures, Experimental ThermodynamicsSeries, Vol. V, Elsevier, Amsterdam, 2000, p. 75.
159] L.P.N. Rebelo, J.N. Canongia Lopes, J.M.S.S. Esperanca, E. Filipe, J. Phys. Chem.B 109 (2005) 6040.
160] J.O. Valderrama, P.A. Robles, Ind. Eng. Chem. Res. 46 (2007) 1338.161] J.O. Valderrama, W.W. Sanga, J.A. Lazzús, Ind. Eng. Chem. Res. 47 (2008) 1318.162] R.G. Jones, P. Licence, K.R.J. Lovelock, I.J. Villar-Garcia, Ind. Eng. Chem. Res. 46
(2007) 6061.
163] J.O. Valderrama, P.A. Robles, Ind. Eng. Chem. Res. 46 (2007) 6063.164] W.G. Chapman, K.E. Gubbins, G. Jackson, M. Radosz, Fluid Phase Equilib. 52(1989) 31.165] S.H. Huang, M. Radosz, Ind. Eng. Chem. Res. 29 (1990) 2284.166] M.C. Kroon, E.K. Karakatsani, I.G. Economou, G.-J. Witkamp, C.J. Peters, J. Phys.
Chem. B 110 (2006) 9262.

1 r. A 12
[
[[[
[[
[
[
[[[
[[[
[
[[
[
[
[[
[[[[[[
[[
[
[[[[[[
[
[[[[
[
[[
880 M. Roth / J. Chromatog
167] E.K. Karakatsani, I.G. Economou, M.C. Kroon, C.J. Peters, G.-J. Witkamp, J. Phys.Chem. C 111 (2007) 15487.
168] T. Wang, C. Peng, H. Liu, Y. Hu, Fluid Phase Equilib. 250 (2006) 150.169] J.S. Andreu, L.F. Vega, J. Phys. Chem. C 111 (2007) 16028.170] Y.S. Kim, W.Y. Choi, J.H. Jang, K.-P. Yoo, C.S. Lee, Fluid Phase Equilib. 228–229
(2005) 439.[171] B. Breure, S.B. Bottini, G.-J. Witkamp, C.J. Peters, J. Phys. Chem. B 111 (2007)
14265.172] E.J. Maginn, Acc. Chem. Res. 40 (2007) 1200.173] A.A.H. Pádua, M.F. Costa Gomes, J.J.A. Canongia Lopes, Acc. Chem. Res. 40
(2007) 1087.[174] R.M. Lynden-Bell, M.G. del Pópolo, T.G.A. Youngs, J. Kohanoff, C.G. Hanke, J.B.
Harper, C.C. Pinilla, Acc. Chem. Res. 40 (2007) 1138.175] J. Deschamps, M.F. Costa Gomes, A.A.H. Pádua, ChemPhysChem 5 (2004) 1049.
[176] X. Huang, C.J. Margulis, Y. Li, B.J. Berne, J. Am. Chem. Soc. 127 (2005) 17842.177] J. Kumełan, Á.P.-S. Kamps, I. Urukova, D. Tuma, G. Maurer, J. Chem. Thermodyn.
37 (2005) 595.178] I. Urukova, J. Vorholz, G. Maurer, J. Phys. Chem. B 109 (2005) 12154.179] W. Shi, E.J. Maginn, J. Phys. Chem. B 112 (2008) 2045.180] E. Kühne, C.J. Peters, J. van Spronsen, G.-J. Witkamp, Green Chem. 8 (2006)
287.181] D. Fu, X. Sun, Y. Qiu, X. Jiang, S. Zhao, Fluid Phase Equilib. 251 (2007) 114.182] J. Chrastil, J. Phys. Chem. 86 (1982) 3016.183] E. Kühne, S. Santarossa, E. Perez, G.J. Witkamp, C.J. Peters, J. Supercrit. Fluids
46 (2008) 93.184] E. Kühne, E. Saez Calvo, G.J. Witkamp, C.J. Peters, J. Supercrit. Fluids 45 (2008)
293.185] E. Kühne, S. Santarossa, G.J. Witkamp, C.J. Peters, Green Chem. 10 (2008) 762.186] R. Bogel-Łukasik, V. Najdanovic-Visak, S. Barreiros, M. Nunes da Ponte, Ind.
Eng. Chem. Res. 47 (2008) 4473.
187] R. Bogel-Łukasik, N.M.T. Lourenco, P. Vidinha, M.D.R. Gomes da Silva, C.A.M.Afonso, M. Nunes da Ponte, S. Barreiros, Green Chem. 10 (2008) 243.188] M.C. Kroon, V.A. Toussaint, A. Shariati, L.J. Florusse, J. van Spronsen, G.-J.
Witkamp, C.J. Peters, Green Chem. 10 (2008) 333.189] E. Kühne, E. Perez, G.J. Witkamp, C.J. Peters, J. Supercrit. Fluids 45 (2008) 27.190] J. Planeta, M. Roth, J. Phys. Chem. B 108 (2004) 11244.
[[
[
16 (2009) 1861–1880
191] J. Planeta, M. Roth, J. Phys. Chem. B 109 (2005) 15165.192] J. Planeta, P. Karásek, M. Roth, J. Phys. Chem. B 111 (2007) 7620.193] N.I. Sakellarios, S.G. Kazarian, J. Chem. Thermodyn. 37 (2005) 621.194] H. Machida, Y. Sato, R.L. Smith Jr., J. Supercrit. Fluids 43 (2008) 430.195] J. Planeta, P. Karásek, M. Roth, Green Chem. 8 (2006) 70.196] A. Bösmann, L. Datsevich, A. Jess, A. Lauter, C. Schmitz, P. Wasserscheid, Chem.
Commun. (2001) 2494.197] J. Eßer, P. Wasserscheid, A. Jess, Green Chem. 6 (2004) 316.198] M.H. Abraham, H.S. Chadha, G.S. Whiting, R.C. Mitchell, J. Pharm. Sci. 83 (1994)
1085.199] M.H. Abraham, N. Benjelloun-Dakhama, J.M.R. Gola, W.E. Acree, W.S. Cain, J.E.
Cometto-Muniz, New J. Chem. 24 (2000) 825.200] M.H. Abraham, J.C. McGowan, Chromatographia 23 (1987) 243.201] M.H. Abraham, A. Ibrahim, A.M. Zissimos, J. Chromatogr. A 1037 (2004) 29.202] M. Vitha, P.W. Carr, J. Chromatogr. A 1126 (2006) 143.203] Z.S. Breitbach, D.W. Armstrong, Anal. Bioanal. Chem. 390 (2008) 1605.204] M.H. Abraham, W.E. Acree, Green Chem. 8 (2006) 906.205] L. Sprunger, M. Clark, W.E. Acree Jr., M.H. Abraham, J. Chem. Inf. Model. 47
(2007) 1123.206] L.M. Sprunger, A. Proctor, W.E. Acree Jr., M.H. Abraham, Fluid Phase Equilib.
265 (2008) 104.207] I.C. Sanchez, R.H. Lacombe, J. Phys. Chem. 80 (1976) 2352.208] I.C. Sanchez, R.H. Lacombe, Macromolecules 11 (1978) 1145.209] S.C. Dhanesar, M.E. Coddens, C.F. Poole, J. Chromatogr. Sci. 23 (1985) 320.210] J.J. Pesek, M.T. Matyska, J. Chromatogr. A 736 (1996) 255.
[211] J.J. Pesek, M.T. Matyska, S. Velpula, J. Chromatogr. A 1126 (2006) 298.212] A. Woldegiorgis, K. Jansson, M. Curcio, J. Roeraade, Electrophoresis 25 (2004)
3660.213] A. Woldegiorgis, K. Jansson, J. Roeraade, J. Mater. Sci. 40 (2005) 583.214] J.R. Conder, D.C. Locke, J.H. Purnell, J. Phys. Chem. 73 (1969) 700.
215] J.L. Anderson, D.W. Armstrong, Anal. Chem. 77 (2005) 6453.216] Y.-N. Hsieh, R.S. Horng, W.-Y. Ho, P.-C. Huang, C.-Y. Hsu, T.-J. Whang, C.-H. Kuei,Chromatographia 67 (2008) 413.[217] J. Tang, H. Tang, W. Sun, M. Radosz, Y. Shen, J. Polym. Sci. A: Polym. Chem. 43
(2005) 5477.218] J. Tang, W. Sun, H. Tang, M. Radosz, Y. Shen, Macromolecules 38 (2005) 2037.





























