Embed Size (px)
Citation preview

Molecular and Cellular Neuroscience 39 (2008) 400–410
Contents lists available at ScienceDirect
Molecular and Cellular Neuroscience
j ourna l homepage: www.e lsev ie r.com/ locate /ymcne
Isoform-specific contribution of protein kinase C to prion processing
Moustapha Alfa Cissé a, Krystel Louis b, Uschi Braun c, Bernard Mari a, Michael Leitges c, Barbara E. Slack d,Abraham Fisher e, Patrick Auberger f, Frédéric Checler a,⁎, Bruno Vincent a,⁎a Institut de Pharmacologie Moléculaire et Cellulaire du CNRS, UMR6097, UNSA, Equipe labellisée Fondation pour la Recherche Médicale, 660 route des Lucioles,Sophia-Antipolis, 06560 Valbonne, Franceb Laboratoire de Biologie des Tumeurs et du Développement, CHU Sart Tilman, 4000, Liège, Belgiumc The Biotechnology Center of Oslo, University of Oslo, Oslo, Norwayd Boston University School of Medicine, Boston, MA 02118, USAe Israel Institute for Biological Research, 74100 Ness-Ziona, Israelf INSERM, U895, Faculté de Médecine, Equipe labellisée Ligue Nationale contre le Cancer, Nice, France
Abbreviations: PKC, protein kinase C; ADAM, A DisPDBu, Phorbol-12-13-dibutyrate; PAGE, polyacrylamenhanced chemiluminescence; HEK, human embryonic kCA, constitutively active; CJD, Creutzfeldt–Jakob disease⁎ Corresponding authors. Fax: +33 4 93 95 77 08.
E-mail addresses: [email protected] (F. Checler), v(B. Vincent).
1044-7431/$ – see front matter © 2008 Elsevier Inc. Aldoi:10.1016/j.mcn.2008.07.013
a b s t r a c t
a r t i c l e i n f oArticle history:
The cellular prion protein (P Received 26 May 2008Revised form 11 July 2008Accepted 16 July 2008Available online 29 July 2008Keywords:PKCPrPc
α-secretaseN1 fragmentRegulationDisintegrins
rPc) undergoes a physiological cleavage between amino acids 111 and 112, therebyleading to the secretion of an amino-terminal fragment referred to as N1. This proteolytic event is eitherconstitutive or regulated by protein kinase C (PKC) and is operated by the disintegrins ADAM9/ADAM10 orADAM17 respectively.We recently showed that the stimulation of theM1/M3muscarinic receptors potentiatesthis cleavage via the phosphorylation and activation of ADAM17.Wehave examined the contribution of variousPKC isoforms in the regulated processing of PrPc. First we show that the PDBu- and carbachol-stimulated N1secretions are blocked by the general PKC inhibitor GF109203X.We establish that HEK293 and human-derivedrhabdhomyosarcoma cells over-expressing constitutively active PKCα, PKCδ or PKCε, but not PKCζ, produceincreased amounts of N1 and harbor enhanced ability to hydrolyze the fluorimetric substrate of ADAM17,JMV2770. Conversely, over-expression of the corresponding dominant negative proteins abolishes PDBU-stimulated N1 secretion and restores N1 to levels comparable to constitutive production.Moreover, deletion ofPKCα lowers N1 recovery in primary cultured fibroblasts. Importantly, mutation of threonine 735 of ADAM17significantly lowers the PDBu-induced N1 formation while transient over-expression of constitutively activePKCα, PKCδ or PKCε, but not PKCζ, induced both the phosphorylation of ADAM17 on its threonine residues andN1 secretion. As a corollary, T735Amutation concomitantly reversed PKCα-, PKCδ- and PKCε-induced ADAM17phosphorylation and N1 recovery. Finally, we established that PKCε-dependent N1 production is fullyprevented by ADAM17 deficiency. Altogether, the present results provide strong evidence that the activation ofPKCα, δ and ε, but not ζ, isoforms leads to increased N1 secretion via the phosphorylation and activation ofADAM17, a process that likely accounts for M1/M3 muscarinic receptors-mediated control of N1 production.
© 2008 Elsevier Inc. All rights reserved.
Introduction
Prion diseases are fatal degenerative disorders of the centralnervous system. In humans, prion diseasesmay be of sporadic, familial,or iatrogenic origin and include Creutzfeldt-Jakob disease (CJD),Gertsmann-Sträussler-Scheinker disease, fatal familial insomnia andkuru (Aguzzi and Polymenidou, 2004). The common central event inthese pathologies involves the conversion of the host-encoded prionprotein (PrPc), into an insoluble and partially protease-resistantisoform (PrPsc) that accumulates in specific brain areas (Prusiner,1998).
integrin And Metalloprotease;ide gel electrophoresis; ECL,idney; DN, dominant negative;; RD, rhabdomyosarcome.
l rights reserved.
The cellular prion protein is physiologically cleaved in the middleof its 106-126 putative toxic core, at the 111/112 peptidyl bond, thusgenerating an amino-terminal fragment referred to as N1 and itscarboxy-terminal membrane tethered counterpart termed C1 (Chenet al., 1995). In CJD-affected brains, an additional cleavage occurs at the90/91 site, yielding fragments referred to as N2/C2 (Chen et al., 1995).Interestingly, this alternative cleavage preserves the 106-126 domain,which has been shown to be highly toxic in vitro (Forloni et al., 1993)and in vivo (Ettaiche et al., 2000). We established that the N1-generating physiological cleavage of PrPc could be either constitutiveor regulated by PKC (Vincent et al., 2000). We and others alsodemonstrated that the disintegrin ADAM10 was mainly responsiblefor the constitutive shedding of PrPc (Vincent et al., 2001; Laffont-Proust et al., 2005), whereas the PKC-mediated N1 release could befully mediated by ADAM17 (Vincent et al., 2001). Moreover, wedemonstrated that ADAM9 acted as an important upstream regulatorof ADAM10 activity (Alfa Cissé et al., 2005). We very recently showed
401M. Alfa Cissé et al. / Molecular and Cellular Neuroscience 39 (2008) 400–410
that stimulation of the M1/M3 muscarinic receptors with severalclassical or more specific agonists promote the physiological proces-sing of the cellular prion protein via ADAM17 phosphorylation on itsthreonine 735 and its subsequent catalytic activation (Alfa Cissé et al.,2007). Although the nature of the proteases involved as well as thereceptor-mediated regulation of PrPc processing at position 111/112are now clearly established, nothing is known concerning the identityof the PKC isoforms implicated and whether they are sufficient, per se,to elicit ADAM17 phosphorylation, thereby increasing N1 formation.
Protein kinase C (PKC) is a critical transducer of intracellularsignaling pathways (Mellor and Parker, 1998). The PKC family iscomposed of at least 12 members and is subdivided into three groupsaccording to their regulation by Ca2+ and diacylglycerol (DAG)(Nishizuka, 1988). The conventional PKC isoforms (cPKCs: α, βI/II, γ)are regulated by both DAG and calcium, while the novel isoforms(nPKCs: δ, ε,η, and θ) are only regulated byDAG.More recently, anothergroup referred to as atypical PKCs (aPKCs) including PKCτ and ζ wasidentified. The latter kinases are not regulated by DAG or calcium.
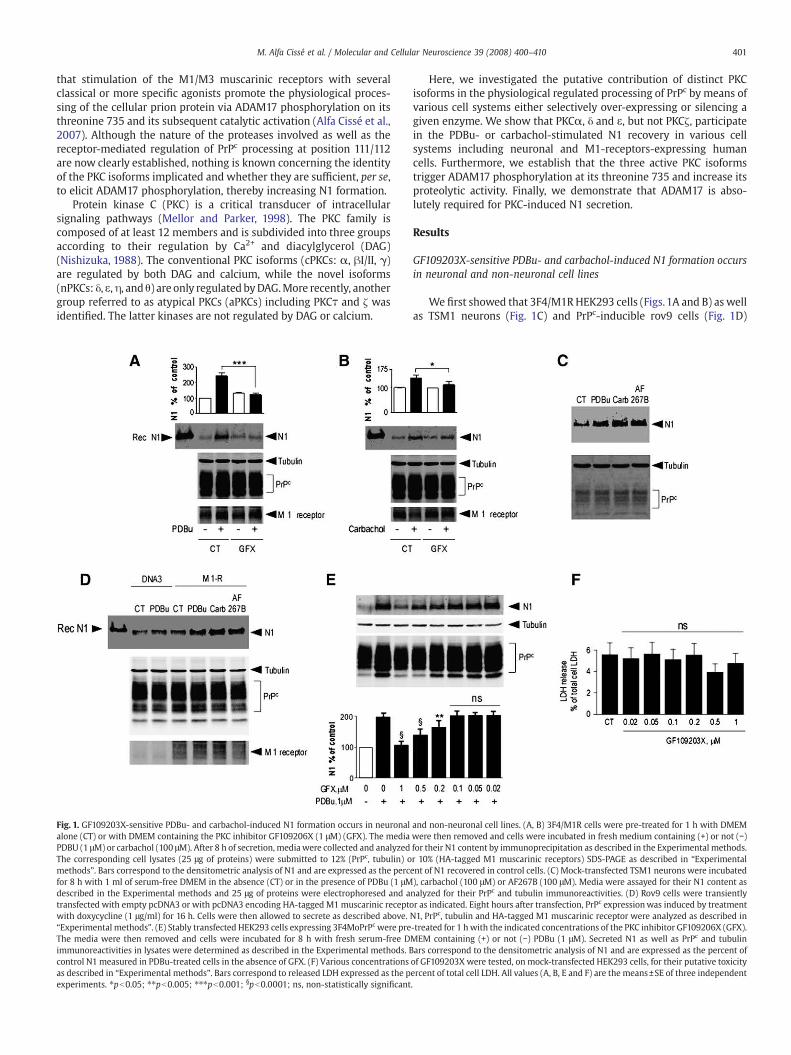
Fig. 1. GF109203X-sensitive PDBu- and carbachol-induced N1 formation occurs in neuronalalone (CT) or with DMEM containing the PKC inhibitor GF109206X (1 μM) (GFX). The mediaPDBU (1 μM) or carbachol (100 μM). After 8 h of secretion, mediawere collected and analyzedThe corresponding cell lysates (25 μg of proteins) were submitted to 12% (PrPc, tubulin) omethods”. Bars correspond to the densitometric analysis of N1 and are expressed as the percfor 8 h with 1 ml of serum-free DMEM in the absence (CT) or in the presence of PDBu (1 μMdescribed in the Experimental methods and 25 μg of proteins were electrophoresed and atransfected with empty pcDNA3 or with pcDNA3 encoding HA-tagged M1muscarinic receptowith doxycycline (1 μg/ml) for 16 h. Cells were then allowed to secrete as described above.“Experimental methods”. (E) Stably transfected HEK293 cells expressing 3F4MoPrPc were preThe media were then removed and cells were incubated for 8 h with fresh serum-free Dimmunoreactivities in lysates were determined as described in the Experimental methods. Bcontrol N1 measured in PDBu-treated cells in the absence of GFX. (F) Various concentrationsas described in “Experimental methods”. Bars correspond to released LDH expressed as the pexperiments. ⁎pb0.05; ⁎⁎pb0.005; ⁎⁎⁎pb0.001; §pb0.0001; ns, non-statistically significant
Here, we investigated the putative contribution of distinct PKCisoforms in the physiological regulated processing of PrPc by means ofvarious cell systems either selectively over-expressing or silencing agiven enzyme. We show that PKCα, δ and ε, but not PKCζ, participatein the PDBu- or carbachol-stimulated N1 recovery in various cellsystems including neuronal and M1-receptors-expressing humancells. Furthermore, we establish that the three active PKC isoformstrigger ADAM17 phosphorylation at its threonine 735 and increase itsproteolytic activity. Finally, we demonstrate that ADAM17 is abso-lutely required for PKC-induced N1 secretion.
Results
GF109203X-sensitive PDBu- and carbachol-induced N1 formation occursin neuronal and non-neuronal cell lines
We first showed that 3F4/M1RHEK293 cells (Figs.1A and B) as wellas TSM1 neurons (Fig. 1C) and PrPc-inducible rov9 cells (Fig. 1D)
and non-neuronal cell lines. (A, B) 3F4/M1R cells were pre-treated for 1 h with DMEMwere then removed and cells were incubated in fresh medium containing (+) or not (−)for their N1 content by immunoprecipitation as described in the Experimental methods.r 10% (HA-tagged M1 muscarinic receptors) SDS-PAGE as described in “Experimentalent of N1 recovered in control cells. (C) Mock-transfected TSM1 neurons were incubated), carbachol (100 μM) or AF267B (100 μM). Media were assayed for their N1 content asnalyzed for their PrPc and tubulin immunoreactivities. (D) Rov9 cells were transientlyr as indicated. Eight hours after transfection, PrPc expressionwas induced by treatmentN1, PrPc, tubulin and HA-tagged M1 muscarinic receptor were analyzed as described in-treated for 1 hwith the indicated concentrations of the PKC inhibitor GF109206X (GFX).MEM containing (+) or not (−) PDBu (1 μM). Secreted N1 as well as PrPc and tubulinars correspond to the densitometric analysis of N1 and are expressed as the percent ofof GF109203X were tested, on mock-transfected HEK293 cells, for their putative toxicityercent of total cell LDH. All values (A, B, E and F) are the means±SE of three independent.
402 M. Alfa Cissé et al. / Molecular and Cellular Neuroscience 39 (2008) 400–410
responded to PDBu (Figs.1A, C and D), carbachol (Figs.1B, C and D) andAF267B (Figs. 1C and D) in regards with N1 production.
In 3F4/M1R HEK293 cells, the general PKC inhibitor GF109203X(which inhibits all PKC isozymes, except PKCμ) led to the inhibition ofboth PDBu- and carbachol-induced N1 recoveries without modifica-tion of the constitutive N1 formation or of PrPc expression levels (Figs.1A and B). Moreover, GF109203X affected N1 production in a dose-dependent manner with a half-maximal inhibition at about aconcentration of 0.5 μM of GF109203X in HEK293 cells (Fig. 1E). Thiseffect could not be explained by a putative toxicity associated withGF109203X treatment since we established by LDH assay that cellviability was not affected by the incubation of cells with concentra-tions of the inhibitor up to 1 μM (Fig. 1F). Note that recombinant N1used as a standard presents a slightly different migration patternwhen compared to secreted N1. This is due to a twelve amino acidssequence located upstream to the 111/112 site of PrPc that remainedafter cleavage of the pGEX-KG vector by thrombin.
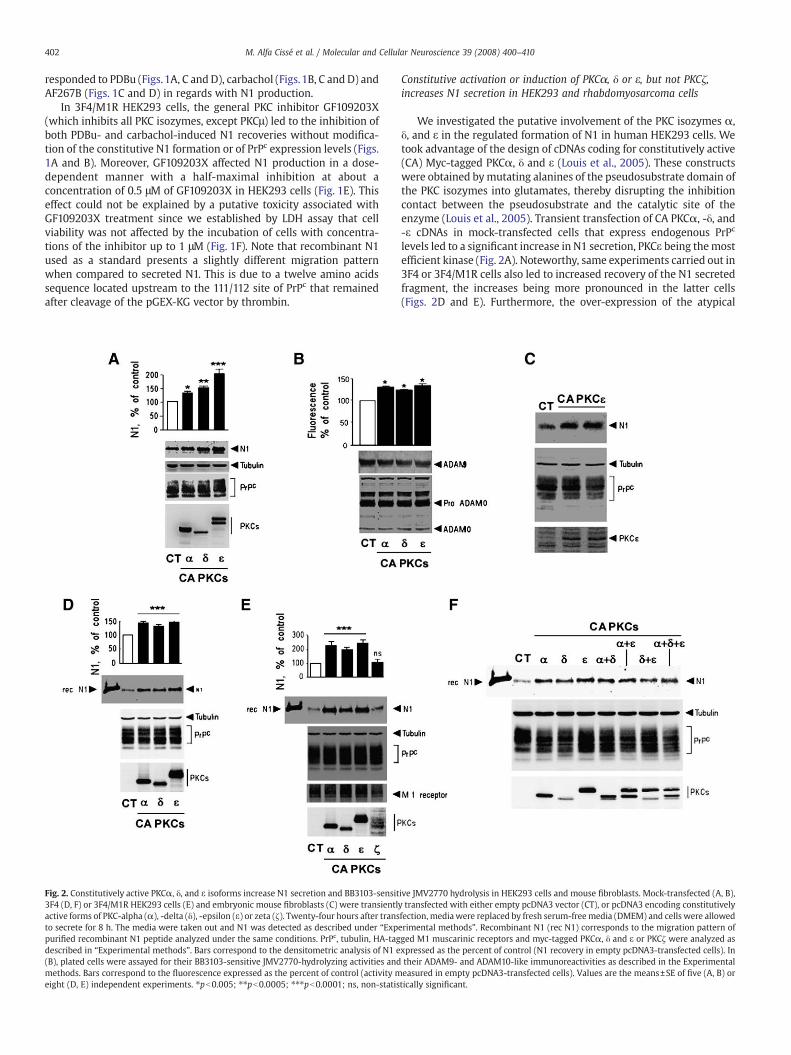
Fig. 2. Constitutively active PKCα, δ, and ε isoforms increase N1 secretion and BB3103-sensit3F4 (D, F) or 3F4/M1R HEK293 cells (E) and embryonic mouse fibroblasts (C) were transientlactive forms of PKC-alpha (α), -delta (δ), -epsilon (ε) or zeta (ζ). Twenty-four hours after transto secrete for 8 h. The media were taken out and N1 was detected as described under “Exppurified recombinant N1 peptide analyzed under the same conditions. PrPc, tubulin, HA-tagdescribed in “Experimental methods”. Bars correspond to the densitometric analysis of N1 e(B), plated cells were assayed for their BB3103-sensitive JMV2770-hydrolyzing activities andmethods. Bars correspond to the fluorescence expressed as the percent of control (activity meight (D, E) independent experiments. ⁎pb0.005; ⁎⁎pb0.0005; ⁎⁎⁎pb0.0001; ns, non-statis
Constitutive activation or induction of PKCα, δ or ε, but not PKCζ,increases N1 secretion in HEK293 and rhabdomyosarcoma cells
We investigated the putative involvement of the PKC isozymes α,δ, and ε in the regulated formation of N1 in human HEK293 cells. Wetook advantage of the design of cDNAs coding for constitutively active(CA) Myc-tagged PKCα, δ and ε (Louis et al., 2005). These constructswere obtained bymutating alanines of the pseudosubstrate domain ofthe PKC isozymes into glutamates, thereby disrupting the inhibitioncontact between the pseudosubstrate and the catalytic site of theenzyme (Louis et al., 2005). Transient transfection of CA PKCα, -δ, and-ε cDNAs in mock-transfected cells that express endogenous PrPc
levels led to a significant increase in N1 secretion, PKCε being themostefficient kinase (Fig. 2A). Noteworthy, same experiments carried out in3F4 or 3F4/M1R cells also led to increased recovery of the N1 secretedfragment, the increases being more pronounced in the latter cells(Figs. 2D and E). Furthermore, the over-expression of the atypical
ive JMV2770 hydrolysis in HEK293 cells and mouse fibroblasts. Mock-transfected (A, B),y transfected with either empty pcDNA3 vector (CT), or pcDNA3 encoding constitutivelyfection, mediawere replaced by fresh serum-freemedia (DMEM) and cells were allowederimental methods”. Recombinant N1 (rec N1) corresponds to the migration pattern ofged M1 muscarinic receptors and myc-tagged PKCα, δ and ε or PKCζ were analyzed asxpressed as the percent of control (N1 recovery in empty pcDNA3-transfected cells). Intheir ADAM9- and ADAM10-like immunoreactivities as described in the Experimentaleasured in empty pcDNA3-transfected cells). Values are the means±SE of five (A, B) ortically significant.
403M. Alfa Cissé et al. / Molecular and Cellular Neuroscience 39 (2008) 400–410
constitutively active isoform PKCζ in 3F4/M1R cells failed to modifyN1 recovery (Fig. 2E) thus showing that PKC-related effects wereisoform-specific. Transient transfection of constitutively active PKCε inmouse embryonic fibroblasts also significantly increased N1 produc-tionwithout any modification of PrPc levels (Fig. 2C). Importantly, PrPc
immunoreactivities remained unaffectedwhatever the conditions andthe cell lines considered (Figs. 2A, C, D and E). In addition, intact mock-transfected HEK293 cells over-expressing PKCα, δ or ε also displayedan increased ability to cleave the fluorimetric JMV2770 substrate ofADAM proteases mimicking the 111/112 cleavage site on PrPc (AlfaCissé et al., 2006) without modification of ADAM9- and ADAM10-likeimmunoreactivities (Fig. 2B). Finally, the PKCα-, δ- and ε-associatedeffects on N1 recoverywere clearly not additive as illustrated in Fig. 2F.
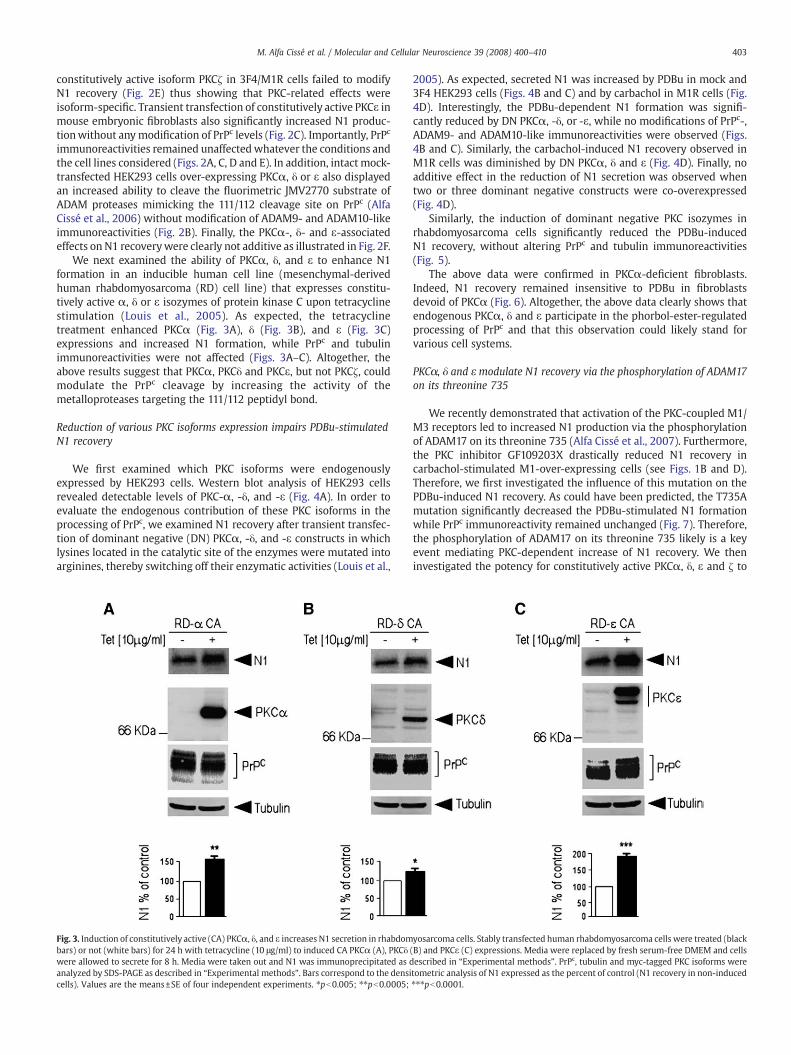
We next examined the ability of PKCα, δ, and ε to enhance N1formation in an inducible human cell line (mesenchymal-derivedhuman rhabdomyosarcoma (RD) cell line) that expresses constitu-tively active α, δ or ε isozymes of protein kinase C upon tetracyclinestimulation (Louis et al., 2005). As expected, the tetracyclinetreatment enhanced PKCα (Fig. 3A), δ (Fig. 3B), and ε (Fig. 3C)expressions and increased N1 formation, while PrPc and tubulinimmunoreactivities were not affected (Figs. 3A–C). Altogether, theabove results suggest that PKCα, PKCδ and PKCε, but not PKCζ, couldmodulate the PrPc cleavage by increasing the activity of themetalloproteases targeting the 111/112 peptidyl bond.
Reduction of various PKC isoforms expression impairs PDBu-stimulatedN1 recovery
We first examined which PKC isoforms were endogenouslyexpressed by HEK293 cells. Western blot analysis of HEK293 cellsrevealed detectable levels of PKC-α, -δ, and -ε (Fig. 4A). In order toevaluate the endogenous contribution of these PKC isoforms in theprocessing of PrPc, we examined N1 recovery after transient transfec-tion of dominant negative (DN) PKCα, -δ, and -ε constructs in whichlysines located in the catalytic site of the enzymes were mutated intoarginines, thereby switching off their enzymatic activities (Louis et al.,
Fig. 3. Induction of constitutively active (CA) PKCα, δ, and ε increases N1 secretion in rhabdombars) or not (white bars) for 24 h with tetracycline (10 μg/ml) to induced CA PKCα (A), PKCδwere allowed to secrete for 8 h. Media were taken out and N1 was immunoprecipitated as danalyzed by SDS-PAGE as described in “Experimental methods”. Bars correspond to the densicells). Values are the means±SE of four independent experiments. ⁎pb0.005; ⁎⁎pb0.0005;
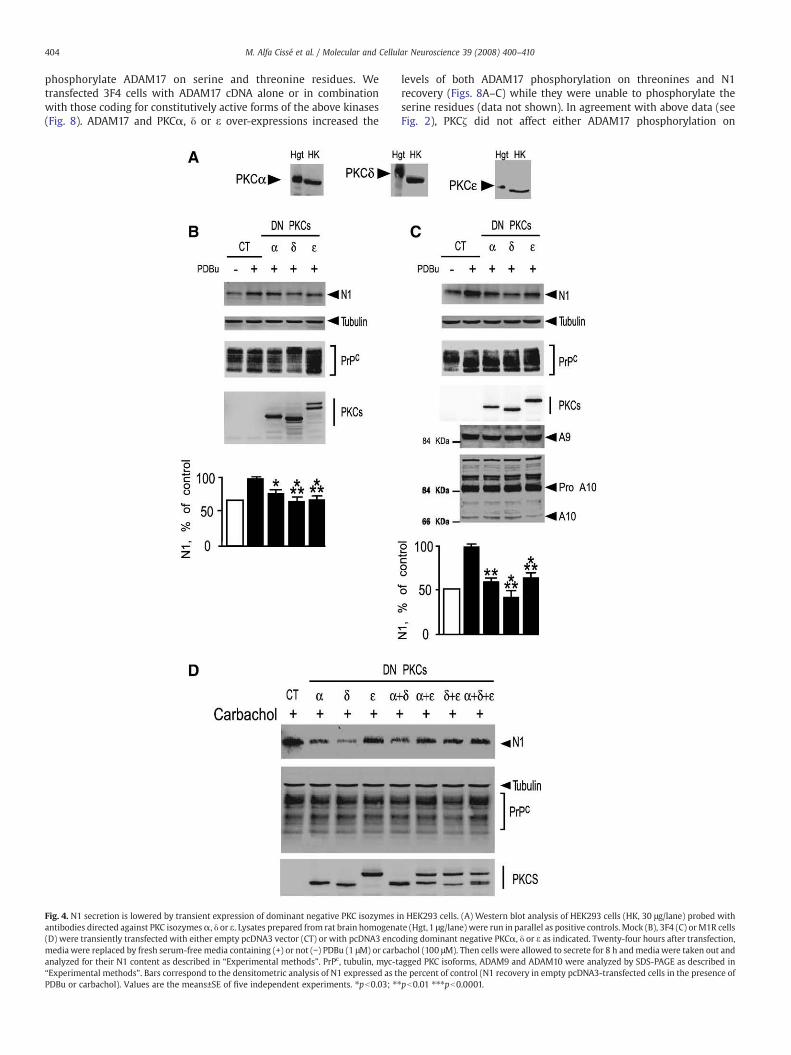
2005). As expected, secreted N1 was increased by PDBu in mock and3F4 HEK293 cells (Figs. 4B and C) and by carbachol in M1R cells (Fig.4D). Interestingly, the PDBu-dependent N1 formation was signifi-cantly reduced by DN PKCα, -δ, or -ε, while no modifications of PrPc-,ADAM9- and ADAM10-like immunoreactivities were observed (Figs.4B and C). Similarly, the carbachol-induced N1 recovery observed inM1R cells was diminished by DN PKCα, δ and ε (Fig. 4D). Finally, noadditive effect in the reduction of N1 secretion was observed whentwo or three dominant negative constructs were co-overexpressed(Fig. 4D).
Similarly, the induction of dominant negative PKC isozymes inrhabdomyosarcoma cells significantly reduced the PDBu-inducedN1 recovery, without altering PrPc and tubulin immunoreactivities(Fig. 5).
The above data were confirmed in PKCα-deficient fibroblasts.Indeed, N1 recovery remained insensitive to PDBu in fibroblastsdevoid of PKCα (Fig. 6). Altogether, the above data clearly shows thatendogenous PKCα, δ and ε participate in the phorbol-ester-regulatedprocessing of PrPc and that this observation could likely stand forvarious cell systems.
PKCα, δ and ε modulate N1 recovery via the phosphorylation of ADAM17on its threonine 735
We recently demonstrated that activation of the PKC-coupled M1/M3 receptors led to increased N1 production via the phosphorylationof ADAM17 on its threonine 735 (Alfa Cissé et al., 2007). Furthermore,the PKC inhibitor GF109203X drastically reduced N1 recovery incarbachol-stimulated M1-over-expressing cells (see Figs. 1B and D).Therefore, we first investigated the influence of this mutation on thePDBu-induced N1 recovery. As could have been predicted, the T735Amutation significantly decreased the PDBu-stimulated N1 formationwhile PrPc immunoreactivity remained unchanged (Fig. 7). Therefore,the phosphorylation of ADAM17 on its threonine 735 likely is a keyevent mediating PKC-dependent increase of N1 recovery. We theninvestigated the potency for constitutively active PKCα, δ, ε and ζ to
yosarcoma cells. Stably transfected human rhabdomyosarcoma cells were treated (black(B) and PKCε (C) expressions. Media were replaced by fresh serum-free DMEM and cellsescribed in “Experimental methods”. PrPc, tubulin and myc-tagged PKC isoforms weretometric analysis of N1 expressed as the percent of control (N1 recovery in non-induced⁎⁎⁎pb0.0001.
404 M. Alfa Cissé et al. / Molecular and Cellular Neuroscience 39 (2008) 400–410
phosphorylate ADAM17 on serine and threonine residues. Wetransfected 3F4 cells with ADAM17 cDNA alone or in combinationwith those coding for constitutively active forms of the above kinases(Fig. 8). ADAM17 and PKCα, δ or ε over-expressions increased the
Fig. 4. N1 secretion is lowered by transient expression of dominant negative PKC isozymes iantibodies directed against PKC isozymesα, δ or ε. Lysates prepared from rat brain homogena(D) were transiently transfected with either empty pcDNA3 vector (CT) or with pcDNA3 encomedia were replaced by fresh serum-free media containing (+) or not (−) PDBu (1 μM) or carbanalyzed for their N1 content as described in “Experimental methods”. PrPc, tubulin, myc-t“Experimental methods”. Bars correspond to the densitometric analysis of N1 expressed as thPDBu or carbachol). Values are the means±SE of five independent experiments. ⁎pb0.03; ⁎⁎
levels of both ADAM17 phosphorylation on threonines and N1recovery (Figs. 8A–C) while they were unable to phosphorylate theserine residues (data not shown). In agreement with above data (seeFig. 2), PKCζ did not affect either ADAM17 phosphorylation on
n HEK293 cells. (A) Western blot analysis of HEK293 cells (HK, 30 μg/lane) probed withte (Hgt,1 μg/lane) were run in parallel as positive controls. Mock (B), 3F4 (C) orM1R cellsding dominant negative PKCα, δ or ε as indicated. Twenty-four hours after transfection,achol (100 μM). Then cells were allowed to secrete for 8 h andmedia were taken out andagged PKC isoforms, ADAM9 and ADAM10 were analyzed by SDS-PAGE as described ine percent of control (N1 recovery in empty pcDNA3-transfected cells in the presence ofpb0.01 ⁎⁎⁎pb0.0001.
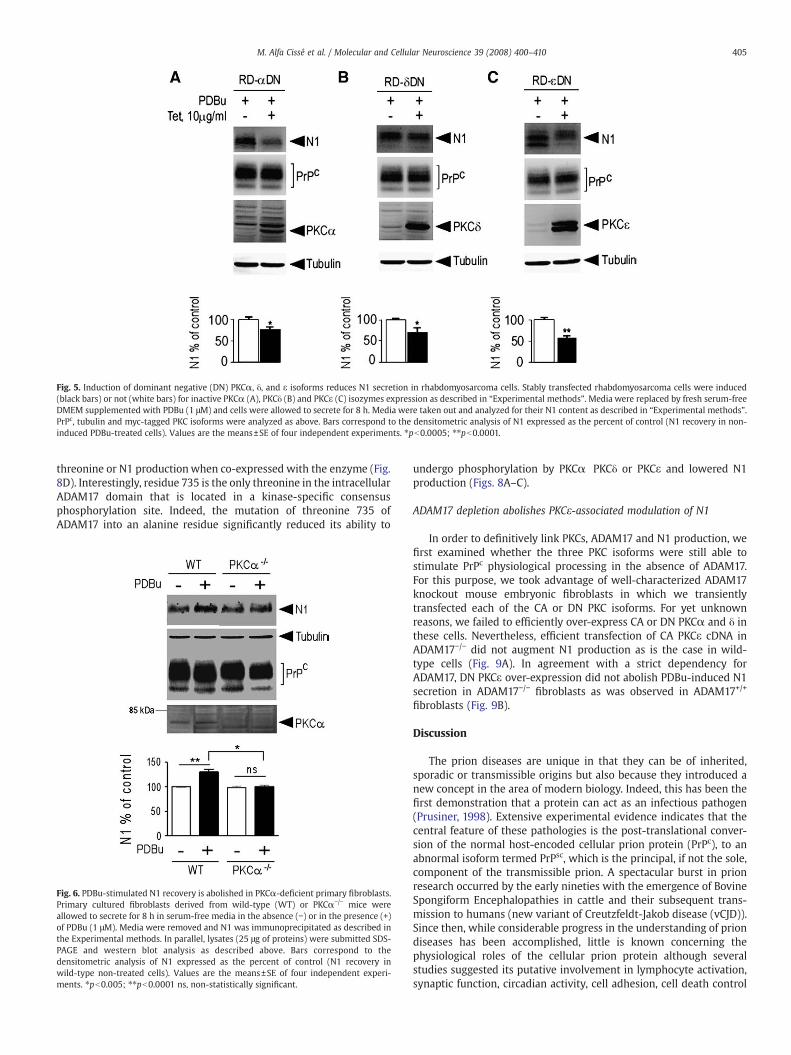
Fig. 5. Induction of dominant negative (DN) PKCα, δ, and ε isoforms reduces N1 secretion in rhabdomyosarcoma cells. Stably transfected rhabdomyosarcoma cells were induced(black bars) or not (white bars) for inactive PKCα (A), PKCδ (B) and PKCε (C) isozymes expression as described in “Experimental methods”. Media were replaced by fresh serum-freeDMEM supplemented with PDBu (1 μM) and cells were allowed to secrete for 8 h. Media were taken out and analyzed for their N1 content as described in “Experimental methods”.PrPc, tubulin and myc-tagged PKC isoforms were analyzed as above. Bars correspond to the densitometric analysis of N1 expressed as the percent of control (N1 recovery in non-induced PDBu-treated cells). Values are the means±SE of four independent experiments. ⁎pb0.0005; ⁎⁎pb0.0001.
405M. Alfa Cissé et al. / Molecular and Cellular Neuroscience 39 (2008) 400–410
threonine or N1 productionwhen co-expressed with the enzyme (Fig.8D). Interestingly, residue 735 is the only threonine in the intracellularADAM17 domain that is located in a kinase-specific consensusphosphorylation site. Indeed, the mutation of threonine 735 ofADAM17 into an alanine residue significantly reduced its ability to
Fig. 6. PDBu-stimulated N1 recovery is abolished in PKCα-deficient primary fibroblasts.Primary cultured fibroblasts derived from wild-type (WT) or PKCα−/− mice wereallowed to secrete for 8 h in serum-free media in the absence (−) or in the presence (+)of PDBu (1 μM). Media were removed and N1 was immunoprecipitated as described inthe Experimental methods. In parallel, lysates (25 μg of proteins) were submitted SDS-PAGE and western blot analysis as described above. Bars correspond to thedensitometric analysis of N1 expressed as the percent of control (N1 recovery inwild-type non-treated cells). Values are the means±SE of four independent experi-ments. ⁎pb0.005; ⁎⁎pb0.0001 ns, non-statistically significant.
undergo phosphorylation by PKCα PKCδ or PKCε and lowered N1production (Figs. 8A–C).
ADAM17 depletion abolishes PKCε-associated modulation of N1
In order to definitively link PKCs, ADAM17 and N1 production, wefirst examined whether the three PKC isoforms were still able tostimulate PrPc physiological processing in the absence of ADAM17.For this purpose, we took advantage of well-characterized ADAM17knockout mouse embryonic fibroblasts in which we transientlytransfected each of the CA or DN PKC isoforms. For yet unknownreasons, we failed to efficiently over-express CA or DN PKCα and δ inthese cells. Nevertheless, efficient transfection of CA PKCε cDNA inADAM17−/− did not augment N1 production as is the case in wild-type cells (Fig. 9A). In agreement with a strict dependency forADAM17, DN PKCε over-expression did not abolish PDBu-induced N1secretion in ADAM17−/− fibroblasts as was observed in ADAM17+/+
fibroblasts (Fig. 9B).
Discussion
The prion diseases are unique in that they can be of inherited,sporadic or transmissible origins but also because they introduced anew concept in the area of modern biology. Indeed, this has been thefirst demonstration that a protein can act as an infectious pathogen(Prusiner, 1998). Extensive experimental evidence indicates that thecentral feature of these pathologies is the post-translational conver-sion of the normal host-encoded cellular prion protein (PrPc), to anabnormal isoform termed PrPsc, which is the principal, if not the sole,component of the transmissible prion. A spectacular burst in prionresearch occurred by the early nineties with the emergence of BovineSpongiform Encephalopathies in cattle and their subsequent trans-mission to humans (new variant of Creutzfeldt-Jakob disease (vCJD)).Since then, while considerable progress in the understanding of priondiseases has been accomplished, little is known concerning thephysiological roles of the cellular prion protein although severalstudies suggested its putative involvement in lymphocyte activation,synaptic function, circadian activity, cell adhesion, cell death control
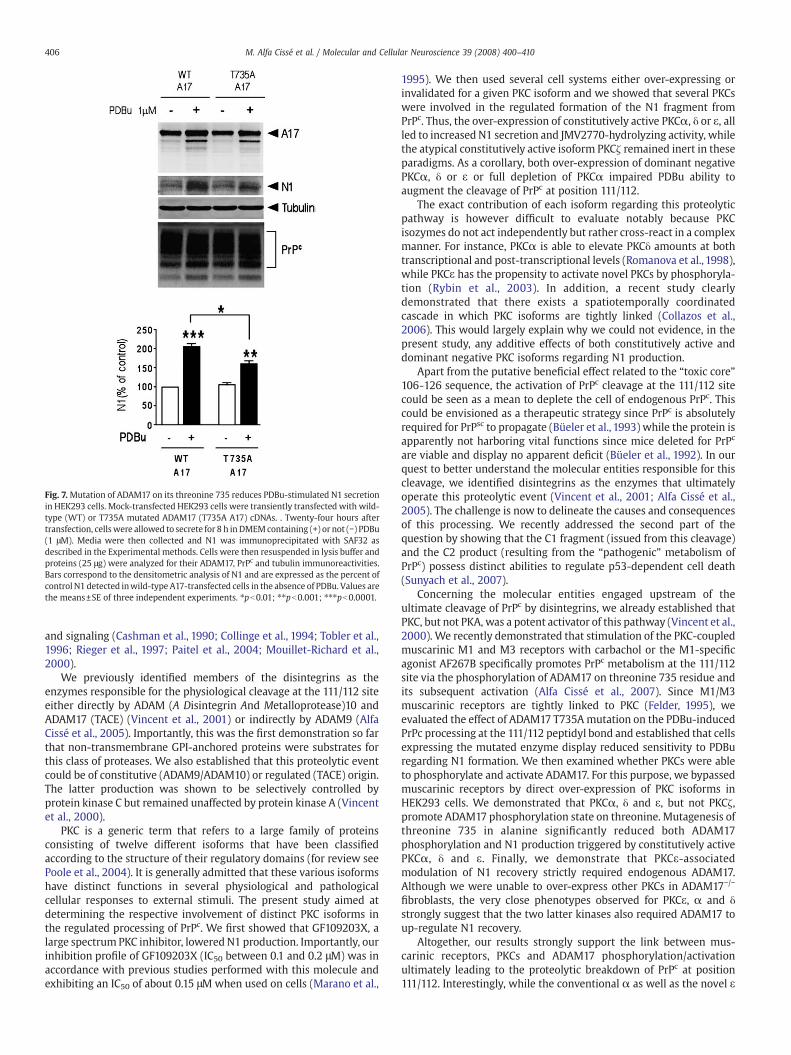
Fig. 7. Mutation of ADAM17 on its threonine 735 reduces PDBu-stimulated N1 secretionin HEK293 cells. Mock-transfected HEK293 cells were transiently transfected with wild-type (WT) or T735A mutated ADAM17 (T735A A17) cDNAs. . Twenty-four hours aftertransfection, cells were allowed to secrete for 8 h inDMEMcontaining (+) or not (−) PDBu(1 μM). Media were then collected and N1 was immunoprecipitated with SAF32 asdescribed in the Experimental methods. Cells were then resuspended in lysis buffer andproteins (25 μg) were analyzed for their ADAM17, PrPc and tubulin immunoreactivities.Bars correspond to the densitometric analysis of N1 and are expressed as the percent ofcontrol N1 detected inwild-type A17-transfected cells in the absence of PDBu. Values arethe means±SE of three independent experiments. ⁎pb0.01; ⁎⁎pb0.001; ⁎⁎⁎pb0.0001.
406 M. Alfa Cissé et al. / Molecular and Cellular Neuroscience 39 (2008) 400–410
and signaling (Cashman et al., 1990; Collinge et al., 1994; Tobler et al.,1996; Rieger et al., 1997; Paitel et al., 2004; Mouillet-Richard et al.,2000).
We previously identified members of the disintegrins as theenzymes responsible for the physiological cleavage at the 111/112 siteeither directly by ADAM (A Disintegrin And Metalloprotease)10 andADAM17 (TACE) (Vincent et al., 2001) or indirectly by ADAM9 (AlfaCissé et al., 2005). Importantly, this was the first demonstration so farthat non-transmembrane GPI-anchored proteins were substrates forthis class of proteases. We also established that this proteolytic eventcould be of constitutive (ADAM9/ADAM10) or regulated (TACE) origin.The latter production was shown to be selectively controlled byprotein kinase C but remained unaffected by protein kinase A (Vincentet al., 2000).
PKC is a generic term that refers to a large family of proteinsconsisting of twelve different isoforms that have been classifiedaccording to the structure of their regulatory domains (for review seePoole et al., 2004). It is generally admitted that these various isoformshave distinct functions in several physiological and pathologicalcellular responses to external stimuli. The present study aimed atdetermining the respective involvement of distinct PKC isoforms inthe regulated processing of PrPc. We first showed that GF109203X, alarge spectrum PKC inhibitor, lowered N1 production. Importantly, ourinhibition profile of GF109203X (IC50 between 0.1 and 0.2 μM) was inaccordance with previous studies performed with this molecule andexhibiting an IC50 of about 0.15 μM when used on cells (Marano et al.,
1995). We then used several cell systems either over-expressing orinvalidated for a given PKC isoform and we showed that several PKCswere involved in the regulated formation of the N1 fragment fromPrPc. Thus, the over-expression of constitutively active PKCα, δ or ε, allled to increased N1 secretion and JMV2770-hydrolyzing activity, whilethe atypical constitutively active isoform PKCζ remained inert in theseparadigms. As a corollary, both over-expression of dominant negativePKCα, δ or ε or full depletion of PKCα impaired PDBu ability toaugment the cleavage of PrPc at position 111/112.
The exact contribution of each isoform regarding this proteolyticpathway is however difficult to evaluate notably because PKCisozymes do not act independently but rather cross-react in a complexmanner. For instance, PKCα is able to elevate PKCδ amounts at bothtranscriptional and post-transcriptional levels (Romanova et al., 1998),while PKCε has the propensity to activate novel PKCs by phosphoryla-tion (Rybin et al., 2003). In addition, a recent study clearlydemonstrated that there exists a spatiotemporally coordinatedcascade in which PKC isoforms are tightly linked (Collazos et al.,2006). This would largely explain why we could not evidence, in thepresent study, any additive effects of both constitutively active anddominant negative PKC isoforms regarding N1 production.
Apart from the putative beneficial effect related to the “toxic core”106-126 sequence, the activation of PrPc cleavage at the 111/112 sitecould be seen as a mean to deplete the cell of endogenous PrPc. Thiscould be envisioned as a therapeutic strategy since PrPc is absolutelyrequired for PrPsc to propagate (Büeler et al., 1993) while the protein isapparently not harboring vital functions since mice deleted for PrPc
are viable and display no apparent deficit (Büeler et al., 1992). In ourquest to better understand the molecular entities responsible for thiscleavage, we identified disintegrins as the enzymes that ultimatelyoperate this proteolytic event (Vincent et al., 2001; Alfa Cissé et al.,2005). The challenge is now to delineate the causes and consequencesof this processing. We recently addressed the second part of thequestion by showing that the C1 fragment (issued from this cleavage)and the C2 product (resulting from the “pathogenic” metabolism ofPrPc) possess distinct abilities to regulate p53-dependent cell death(Sunyach et al., 2007).
Concerning the molecular entities engaged upstream of theultimate cleavage of PrPc by disintegrins, we already established thatPKC, but not PKA, was a potent activator of this pathway (Vincent et al.,2000). We recently demonstrated that stimulation of the PKC-coupledmuscarinic M1 and M3 receptors with carbachol or the M1-specificagonist AF267B specifically promotes PrPc metabolism at the 111/112site via the phosphorylation of ADAM17 on threonine 735 residue andits subsequent activation (Alfa Cissé et al., 2007). Since M1/M3muscarinic receptors are tightly linked to PKC (Felder, 1995), weevaluated the effect of ADAM17 T735Amutation on the PDBu-inducedPrPc processing at the 111/112 peptidyl bond and established that cellsexpressing the mutated enzyme display reduced sensitivity to PDBuregarding N1 formation. We then examined whether PKCs were ableto phosphorylate and activate ADAM17. For this purpose, we bypassedmuscarinic receptors by direct over-expression of PKC isoforms inHEK293 cells. We demonstrated that PKCα, δ and ε, but not PKCζ,promote ADAM17 phosphorylation state on threonine. Mutagenesis ofthreonine 735 in alanine significantly reduced both ADAM17phosphorylation and N1 production triggered by constitutively activePKCα, δ and ε. Finally, we demonstrate that PKCε-associatedmodulation of N1 recovery strictly required endogenous ADAM17.Although we were unable to over-express other PKCs in ADAM17−/−
fibroblasts, the very close phenotypes observed for PKCε, α and δstrongly suggest that the two latter kinases also required ADAM17 toup-regulate N1 recovery.
Altogether, our results strongly support the link between mus-carinic receptors, PKCs and ADAM17 phosphorylation/activationultimately leading to the proteolytic breakdown of PrPc at position111/112. Interestingly, while the conventional α as well as the novel ε
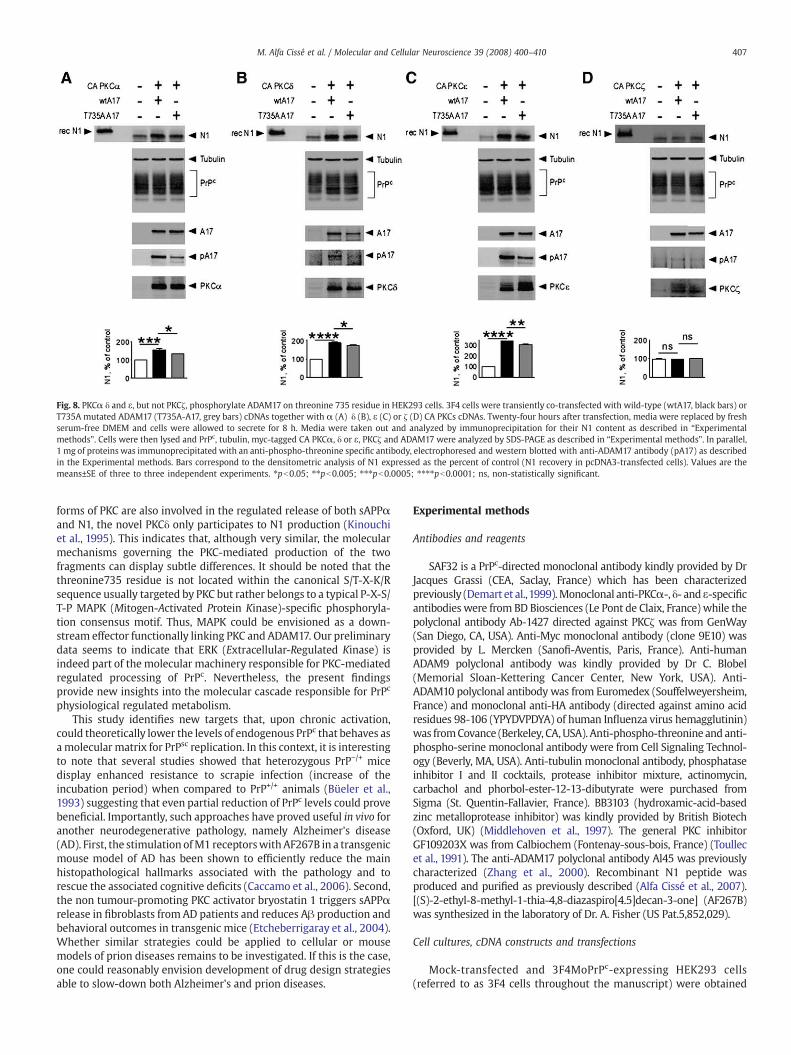
Fig. 8. PKCα δ and ε, but not PKCζ, phosphorylate ADAM17 on threonine 735 residue in HEK293 cells. 3F4 cells were transiently co-transfected with wild-type (wtA17, black bars) orT735A mutated ADAM17 (T735A-A17, grey bars) cDNAs together with α (A) δ (B), ε (C) or ζ (D) CA PKCs cDNAs. Twenty-four hours after transfection, media were replaced by freshserum-free DMEM and cells were allowed to secrete for 8 h. Media were taken out and analyzed by immunoprecipitation for their N1 content as described in “Experimentalmethods”. Cells were then lysed and PrPc, tubulin, myc-tagged CA PKCα, δ or ε, PKCζ and ADAM17 were analyzed by SDS-PAGE as described in “Experimental methods”. In parallel,1 mg of proteins was immunoprecipitated with an anti-phospho-threonine specific antibody, electrophoresed and western blotted with anti-ADAM17 antibody (pA17) as describedin the Experimental methods. Bars correspond to the densitometric analysis of N1 expressed as the percent of control (N1 recovery in pcDNA3-transfected cells). Values are themeans±SE of three to three independent experiments. ⁎pb0.05; ⁎⁎pb0.005; ⁎⁎⁎pb0.0005; ⁎⁎⁎⁎pb0.0001; ns, non-statistically significant.
407M. Alfa Cissé et al. / Molecular and Cellular Neuroscience 39 (2008) 400–410
forms of PKC are also involved in the regulated release of both sAPPαand N1, the novel PKCδ only participates to N1 production (Kinouchiet al., 1995). This indicates that, although very similar, the molecularmechanisms governing the PKC-mediated production of the twofragments can display subtle differences. It should be noted that thethreonine735 residue is not located within the canonical S/T-X-K/Rsequence usually targeted by PKC but rather belongs to a typical P-X-S/T-P MAPK (Mitogen-Activated Protein Kinase)-specific phosphoryla-tion consensus motif. Thus, MAPK could be envisioned as a down-stream effector functionally linking PKC and ADAM17. Our preliminarydata seems to indicate that ERK (Extracellular-Regulated Kinase) isindeed part of the molecular machinery responsible for PKC-mediatedregulated processing of PrPc. Nevertheless, the present findingsprovide new insights into the molecular cascade responsible for PrPc
physiological regulated metabolism.This study identifies new targets that, upon chronic activation,
could theoretically lower the levels of endogenous PrPc that behaves asamolecular matrix for PrPsc replication. In this context, it is interestingto note that several studies showed that heterozygous PrP−/+ micedisplay enhanced resistance to scrapie infection (increase of theincubation period) when compared to PrP+/+ animals (Büeler et al.,1993) suggesting that even partial reduction of PrPc levels could provebeneficial. Importantly, such approaches have proved useful in vivo foranother neurodegenerative pathology, namely Alzheimer's disease(AD). First, the stimulation ofM1 receptorswith AF267B in a transgenicmouse model of AD has been shown to efficiently reduce the mainhistopathological hallmarks associated with the pathology and torescue the associated cognitive deficits (Caccamo et al., 2006). Second,the non tumour-promoting PKC activator bryostatin 1 triggers sAPPαrelease in fibroblasts from AD patients and reduces Aβ production andbehavioral outcomes in transgenic mice (Etcheberrigaray et al., 2004).Whether similar strategies could be applied to cellular or mousemodels of prion diseases remains to be investigated. If this is the case,one could reasonably envision development of drug design strategiesable to slow-down both Alzheimer's and prion diseases.
Experimental methods
Antibodies and reagents
SAF32 is a PrPc-directed monoclonal antibody kindly provided by DrJacques Grassi (CEA, Saclay, France) which has been characterizedpreviously (Demart et al.,1999).Monoclonal anti-PKCα-, δ- and ε-specificantibodies were from BD Biosciences (Le Pont de Claix, France) while thepolyclonal antibody Ab-1427 directed against PKCζ was from GenWay(San Diego, CA, USA). Anti-Myc monoclonal antibody (clone 9E10) wasprovided by L. Mercken (Sanofi-Aventis, Paris, France). Anti-humanADAM9 polyclonal antibody was kindly provided by Dr C. Blobel(Memorial Sloan-Kettering Cancer Center, New York, USA). Anti-ADAM10 polyclonal antibody was from Euromedex (Souffelweyersheim,France) and monoclonal anti-HA antibody (directed against amino acidresidues 98-106 (YPYDVPDYA) of human Influenza virus hemagglutinin)was fromCovance (Berkeley, CA,USA). Anti-phospho-threonine and anti-phospho-serine monoclonal antibody were from Cell Signaling Technol-ogy (Beverly, MA, USA). Anti-tubulin monoclonal antibody, phosphataseinhibitor I and II cocktails, protease inhibitor mixture, actinomycin,carbachol and phorbol-ester-12-13-dibutyrate were purchased fromSigma (St. Quentin-Fallavier, France). BB3103 (hydroxamic-acid-basedzinc metalloprotease inhibitor) was kindly provided by British Biotech(Oxford, UK) (Middlehoven et al., 1997). The general PKC inhibitorGF109203X was from Calbiochem (Fontenay-sous-bois, France) (Toullecet al., 1991). The anti-ADAM17 polyclonal antibody Al45 was previouslycharacterized (Zhang et al., 2000). Recombinant N1 peptide wasproduced and purified as previously described (Alfa Cissé et al., 2007).[(S)-2-ethyl-8-methyl-1-thia-4,8-diazaspiro[4.5]decan-3-one] (AF267B)was synthesized in the laboratory of Dr. A. Fisher (US Pat.5,852,029).
Cell cultures, cDNA constructs and transfections
Mock-transfected and 3F4MoPrPc-expressing HEK293 cells(referred to as 3F4 cells throughout the manuscript) were obtained
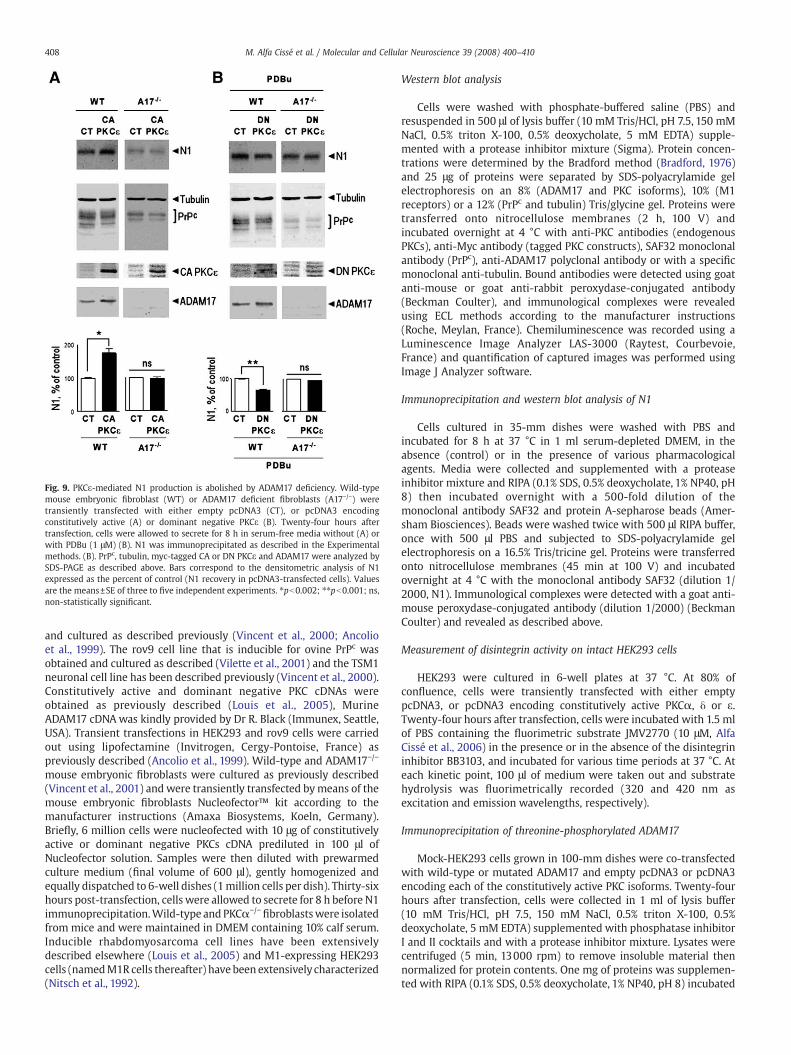
Fig. 9. PKCε-mediated N1 production is abolished by ADAM17 deficiency. Wild-typemouse embryonic fibroblast (WT) or ADAM17 deficient fibroblasts (A17−/−) weretransiently transfected with either empty pcDNA3 (CT), or pcDNA3 encodingconstitutively active (A) or dominant negative PKCε (B). Twenty-four hours aftertransfection, cells were allowed to secrete for 8 h in serum-free media without (A) orwith PDBu (1 μM) (B). N1 was immunoprecipitated as described in the Experimentalmethods. (B). PrPc, tubulin, myc-tagged CA or DN PKCε and ADAM17 were analyzed bySDS-PAGE as described above. Bars correspond to the densitometric analysis of N1expressed as the percent of control (N1 recovery in pcDNA3-transfected cells). Valuesare the means±SE of three to five independent experiments. ⁎pb0.002; ⁎⁎pb0.001; ns,non-statistically significant.
408 M. Alfa Cissé et al. / Molecular and Cellular Neuroscience 39 (2008) 400–410
and cultured as described previously (Vincent et al., 2000; Ancolioet al., 1999). The rov9 cell line that is inducible for ovine PrPc wasobtained and cultured as described (Vilette et al., 2001) and the TSM1neuronal cell line has been described previously (Vincent et al., 2000).Constitutively active and dominant negative PKC cDNAs wereobtained as previously described (Louis et al., 2005), MurineADAM17 cDNAwas kindly provided by Dr R. Black (Immunex, Seattle,USA). Transient transfections in HEK293 and rov9 cells were carriedout using lipofectamine (Invitrogen, Cergy-Pontoise, France) aspreviously described (Ancolio et al., 1999). Wild-type and ADAM17−/−
mouse embryonic fibroblasts were cultured as previously described(Vincent et al., 2001) and were transiently transfected bymeans of themouse embryonic fibroblasts Nucleofector™ kit according to themanufacturer instructions (Amaxa Biosystems, Koeln, Germany).Briefly, 6 million cells were nucleofected with 10 μg of constitutivelyactive or dominant negative PKCs cDNA prediluted in 100 μl ofNucleofector solution. Samples were then diluted with prewarmedculture medium (final volume of 600 μl), gently homogenized andequally dispatched to 6-well dishes (1million cells per dish). Thirty-sixhours post-transfection, cells were allowed to secrete for 8 h before N1immunoprecipitation.Wild-type and PKCα−/−
fibroblastswere isolatedfrom mice and were maintained in DMEM containing 10% calf serum.Inducible rhabdomyosarcoma cell lines have been extensivelydescribed elsewhere (Louis et al., 2005) and M1-expressing HEK293cells (namedM1R cells thereafter) have been extensively characterized(Nitsch et al., 1992).
Western blot analysis
Cells were washed with phosphate-buffered saline (PBS) andresuspended in 500 μl of lysis buffer (10 mM Tris/HCl, pH 7.5, 150 mMNaCl, 0.5% triton X-100, 0.5% deoxycholate, 5 mM EDTA) supple-mented with a protease inhibitor mixture (Sigma). Protein concen-trations were determined by the Bradford method (Bradford, 1976)and 25 μg of proteins were separated by SDS-polyacrylamide gelelectrophoresis on an 8% (ADAM17 and PKC isoforms), 10% (M1receptors) or a 12% (PrPc and tubulin) Tris/glycine gel. Proteins weretransferred onto nitrocellulose membranes (2 h, 100 V) andincubated overnight at 4 °C with anti-PKC antibodies (endogenousPKCs), anti-Myc antibody (tagged PKC constructs), SAF32 monoclonalantibody (PrPc), anti-ADAM17 polyclonal antibody or with a specificmonoclonal anti-tubulin. Bound antibodies were detected using goatanti-mouse or goat anti-rabbit peroxydase-conjugated antibody(Beckman Coulter), and immunological complexes were revealedusing ECL methods according to the manufacturer instructions(Roche, Meylan, France). Chemiluminescence was recorded using aLuminescence Image Analyzer LAS-3000 (Raytest, Courbevoie,France) and quantification of captured images was performed usingImage J Analyzer software.
Immunoprecipitation and western blot analysis of N1
Cells cultured in 35-mm dishes were washed with PBS andincubated for 8 h at 37 °C in 1 ml serum-depleted DMEM, in theabsence (control) or in the presence of various pharmacologicalagents. Media were collected and supplemented with a proteaseinhibitor mixture and RIPA (0.1% SDS, 0.5% deoxycholate, 1% NP40, pH8) then incubated overnight with a 500-fold dilution of themonoclonal antibody SAF32 and protein A-sepharose beads (Amer-sham Biosciences). Beads were washed twice with 500 μl RIPA buffer,once with 500 μl PBS and subjected to SDS-polyacrylamide gelelectrophoresis on a 16.5% Tris/tricine gel. Proteins were transferredonto nitrocellulose membranes (45 min at 100 V) and incubatedovernight at 4 °C with the monoclonal antibody SAF32 (dilution 1/2000, N1). Immunological complexes were detected with a goat anti-mouse peroxydase-conjugated antibody (dilution 1/2000) (BeckmanCoulter) and revealed as described above.
Measurement of disintegrin activity on intact HEK293 cells
HEK293 were cultured in 6-well plates at 37 °C. At 80% ofconfluence, cells were transiently transfected with either emptypcDNA3, or pcDNA3 encoding constitutively active PKCα, δ or ε.Twenty-four hours after transfection, cells were incubated with 1.5 mlof PBS containing the fluorimetric substrate JMV2770 (10 μM, AlfaCissé et al., 2006) in the presence or in the absence of the disintegrininhibitor BB3103, and incubated for various time periods at 37 °C. Ateach kinetic point, 100 μl of medium were taken out and substratehydrolysis was fluorimetrically recorded (320 and 420 nm asexcitation and emission wavelengths, respectively).
Immunoprecipitation of threonine-phosphorylated ADAM17
Mock-HEK293 cells grown in 100-mm dishes were co-transfectedwith wild-type or mutated ADAM17 and empty pcDNA3 or pcDNA3encoding each of the constitutively active PKC isoforms. Twenty-fourhours after transfection, cells were collected in 1 ml of lysis buffer(10 mM Tris/HCl, pH 7.5, 150 mM NaCl, 0.5% triton X-100, 0.5%deoxycholate, 5 mM EDTA) supplemented with phosphatase inhibitorI and II cocktails and with a protease inhibitor mixture. Lysates werecentrifuged (5 min, 13000 rpm) to remove insoluble material thennormalized for protein contents. One mg of proteins was supplemen-ted with RIPA (0.1% SDS, 0.5% deoxycholate, 1% NP40, pH 8) incubated

409M. Alfa Cissé et al. / Molecular and Cellular Neuroscience 39 (2008) 400–410
overnight with immunoprecipitating anti-phospho-threonine anti-body and protein A-sepharose beads (Amersham Biosciences) (4 μg/1 mg of proteins), then incubated overnight. Beads werewashed twicewith 500 μl RIPA buffer, then once with 500 μl PBS and submitted toSDS-polyacrylamide gel electrophoresis for ADAM17 expressionanalysis, as described above.
Site-directed mutagenesis of ADAM17 on its threonine 735
Mouse ADAM17 inwhich threonine 735was replaced by an alanineresidue (T735A-A17) was obtained by oligonucleotide-directed muta-genesis from mouse wild-type ADAM17 cDNA by means of aquikChange™ site-directed mutagenesis kit (Stratagene). The twoprimers 5′-CCTGCACCCCAGGCTCCAGGTCGTC-TGC-3′ and 5′-GCAGAC-GACCTGGAGC-CTGGGGTGC-AGG-3′ containing the T735A mutationwere designed according to the manufacturer's conditions. Final cDNAconstructions were sequenced in their entirety.
Lactate dehydrogenase (LDH) assays
Cells grown in 24-well plates were incubated for 1 h without (CT)or with various GFX109203X concentrations and allowed to secreteduring 8 h in 1ml of DMEM containing PDBu (1 μM). Forty μl of culturemedium were then analyzed for the release of LDH using CytoTox-ONE™ homogeneous membrane integrity assay (Promega, Charbon-nières, France) according to the manufacturer's recommendations.
Statistical analysis
Statistical analyses were performed with the Prism software(Graphpad software, San Diego, CA) using the unpaired t-test forpairwise comparisons.
Acknowledgments
Wewould like to thank Dr C. Sunyach for the preparation and supplyof the recombinant N1 fragment, Dr R. Black for his kind gift of ADAM17cDNA, anti-ADAM17 antibody and ADAM17−/− fibroblasts and Dr D.Vilette for his purchase of rov9 cells.We are grateful to Dr L.Mercken forproviding us with anti-myc 9E10 antibody. We wish to thank Drs J.F.Hernandez and J. Martinez for providing JMV2770. This work wassupported by the Centre National de la Recherche Scientifique (CNRS)and The Fondation pour la Recherche Médicale (FRM).
References
Aguzzi, A., Polymenidou, M., 2004. Mammalian prion biology: one century of evolvingconcepts. Cell 116, 1–20.
Alfa Cissé, M., Sunyach, C., Lefranc-Jullien, S., Postina, R., Vincent, B., Checler, F., 2005.The disintegrin ADAM9 indirectly contributes to the physiological processing ofcellular prion by modulating ADAM10 activity. J. Biol. Chem. 281, 40624–40631.
Alfa Cissé, M., Gandreuil, C., Hernandez, J.F., Martinez, J., Checler, F., Vincent, B., 2006.Design and characterization of a novel cellular prion-derived quenched fluorimetricsubstrate of α-secretase. Biochem. Biophys. Res. Commun. 347, 254–260.
Alfa Cissé, M., Sunyach, C., Slack, B.E., Fisher, A., Vincent, B., Checler, F., 2007. M1 and M3muscarinic receptors control physiological processing of cellular prion protein bymodulating ADAM17 phosphorylation and activity. J. Neurosci 27, 4083–4092.
Ancolio, K., Dumanchin, C., Barelli, H., Warter, J.M., Brice, A., Campion, D., Frébourg, T.,Checler, F., 1999. Unusual phenotypic alteration of β amyloid precursor protein(βAPP) maturation by a new Val-715/Met βAPP-770 mutation responsible forprobable early-onset Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 96, 4119–4124.
Bradford, M.M., 1976. A rapid and sensitive method for the quantification of microgramquantities of protein utilizing the principle of protein-dye binding. Anal. Biochem.72, 248–259.
Büeler, H., Fischer, M., Lang, Y., Bluethmann, H., Lipp, H., DeArmond, S., Prusiner, S.B.,Aguet, M., Weissmann, C., 1992. Normal development and behaviour of micelacking the neuronal cell-surface PrP protein. Nature 356, 577–582.
Büeler, H., Aguzzi, A., Sailer, A., Greiner, R., Autenried, P., Aguet, M., Weissmann, C., 1993.Mice devoid of PrP are resistant to scrapie. Cell 73, 1339–1347.
Caccamo, A., Oddo, S., Billings, L.M., Green, K.N., Martinez-Coria, H., Fisher, A., La Ferla,F.M., 2006. M1 receptors play a central role in modulating AD-like pathology intransgenic mice. Neuron 49, 671–682.
Cashman, N.R., Loertscher, R., Nalbantoglu, J., Shaw, I., Kascsak, R.J., Bolton, D.C.,Bendheim, P.E., 1990. Cellular isoform of the scrapie agent protein participates inlymphocyte activation. Cell 61, 185–192.
Chen, S.G., Teplow, D.B., Parchi, P., Teller, J.K., Gambetti, P., Autilio-Gambetti, L., 1995.Truncated forms of the human prion protein in normal brain and prion diseases. J.Biol. Chem. 270, 19173–19180.
Collazos, A., Diouf, B., Guérineau, N., Quittau-Prévostel, C., Peter, M., Coudane, F.,Hollande, F., Joubert, D., 2006. A spatiotemporally coordinated cascade of proteinkinase C activation controls isoform-selective translocation. Mol. Cell. Biol. 26,2247–2261.
Collinge, J., Whittington, M.A., Sidle, K.C.L., Smith, C.J., Palmer, M.S., Clarke, A.R., Jefferys,J.G.R., 1994. Prion protein is necessary for normal synaptic function. Nature 370,295–297.
Demart, S., Fournier, J.G., Cremignon, C., Frobert, J., Lamoury, F., Marce, D., Lasmézas, C.,Dormont, D., Grassi, J., Deslys, J.P., 1999. New insight into abnormal prion proteinusing monoclonal antibodies. Biochem. Cell Biol. 265, 652–657.
Etcheberrigaray, R., Tan, M., Dewachter, I., Kuipéri, C., Van der Auwera, I., Wera, S., Qiao,L., Bank, B., Nelson, T.J., Kozikowski, A.P., Van Leuven, F., Alkon, D.L., 2004.Therapeutic effects of PKC activators in Alzheimer's disease transgenic mice. Proc.Natl. Acad. Sci. U.S.A. 101, 11141–11146.
Ettaiche, M., Pichot, R., Vincent, J.P., Chabry, J., 2000. In vivo cytotoxicity of the prionprotein fragment 106-126. J. Biol. Chem. 275, 36487–36490.
Felder, C.C., 1995. Muscarinic acetylcholine receptors: signal transduction throughmultiple effectors. FASEB J. 9, 619–625.
Forloni, G., Angeretti, N., Chiesa, R., Monzani, E., Salmona, M., Bugiani, O., Tagliavini, F.,1993. Neurotoxicity of a prion protein fragment. Nature 362, 543–546.
Kinouchi, T., Sorimachi, H., Maruyama, K., Mizuno, K., Ohno, S., Ishiura, S., Suzuki, K.,1995. Conventional protein kinase C (PKC)-α and novel PKCε, but not δ, increasethe secretion of an N-terminal fragment of Alzheimer's disease amyloidprecursor protein from PKC cDNA transfected 3Y1 cells. FEBS Lett. 364,203–206.
Laffont-Proust, I., Faucheux, B.A., Hässig, R., Sazdovitch, V., Simon, S., Grassi, J., Hauw, J.J.,Moya, K.L., Haïk, S., 2005. The N-terminal cleavage of cellular prion protein in thehuman brain. FEBS Lett. 579, 6333–6337.
Louis, K., Guérineau, N., Fromigue, O., Defamie, V., Collazos, A., Anglard, P., Shipp, M.A.,Auberger, P., Joubert, D., Mari, B., 2005. Tumor cell-mediated induction of thestromal factor stromelysin-3 requires heterotypic cell contact-dependent activationof specific protein kinase C isoforms. J. Biol. Chem. 280, 1272–1283.
Marano, C.W., Laughlin, K.V., Russo, L.M., Mullin, J.M., 1995. The protein kinase Cinhibitor, bisindolylmaleimide, inhibits the TPA-induced but not the TNF-inducedincrease in LLC-PK1 transepithelial permeability. Biochem. Biophys. Res. Commun.209, 669–676.
Mellor, H., Parker, P.J., 1998. The extended protein kinase C superfamily. Biochem. J. 332,281–292.
Middlehoven, P.J., Ager, A., Roos, D., Verhoeven, A.J., 1997. Involvement of ametalloprotease in the shedding of human neutrophil FcgRIIIB. FEBS Lett. 414,14–18.
Mouillet-Richard, S., Ermonval, M., Chebassier, C., Laplanche, J.L., Lehmann, S., Launay,J.M., Kellermann, O., 2000. Signal transduction through prion protein. Science 289,1925–1928.
Nishizuka, Y., 1988. Themolecular heterogeneity of protein kinase C and its implicationsfor cellular regulation. Nature 334, 661–665.
Nitsch, R.M., Slack, B.E., Wurtman, R.J., Growdon, J.H., 1992. Release of Alzheimeramyloid precursor derivatives stimulated by activation of muscarinic acetylcholinereceptors. Science 258, 304–307.
Paitel, E., Sunyach, C., Alves da Costa, C., Bourdon, J.C., Vincent, B., Checler, F., 2004.Primary cultured neurons devoid of cellular prion display lower responsiveness tostaurosporine through the control of p53 at both transcriptional and post-transcriptional levels. J. Biol. Chem. 279, 612–618.
Poole, A.W., Pula, G., Hers, I., Crosby, D., Jones, M.L., 2004. PKC-interacting proteins: fromfunction to pharmacology. Trends in Pharm. Sci. 25, 529–535.
Prusiner, S.B., 1998. Prions. Proc. Natl. Acad. Sci. U.S.A. 95, 13363–13383.Rieger, R., Edenhofer, F., Lasmezas, C.I., Weiss, S., 1997. The human 37-kDa laminin
receptor precursor interacts with the prion protein in eukacyotic cells. Nat. Med. 3,1383–1388.
Romanova, L.Y., Alexandrov, I.A., Nordan, R.P., Blagosklonny, M.V., Mushinski, J.F., 1998.Cross-talk between protein kinase C-alpha (PKC-alpha) and -delta (PKC-delta):PKC-alpha elevates the PKC-delta protein level, altering its mRNA transcription anddegradation. Biochemistry U.S.A. 37, 5558–5565.
Rybin, V.O., Sabri, A., Short, J., Braz, J.C., Molkentin, J.D., Steinberg, S.F., 2003. Cross-regulation of novel protein kinase C (PKC) isoform function in cardiomyocytes. Roleof PKC epsilon in activation loop phosphorylations and PKC delta in hydrophobicmotif phosphorylations. J. Biol. Chem. 278, 14555–14564.
Sunyach, C., Alfa Cissé, M., Alves da Costa, C., Vincent, B., Checler, F., 2007. The C-terminalproducts of cellular prion protein processing, C1 and C2, exert distinct influence onp53-dependent staurosporine-induced caspase-3 activation. J. Biol. Chem. 282,1956–1963.
Tobler, I., Gaus, S.E., Deboer, T., Achermann, P., Fisher, M., Rülicke, T., Moser, M., Oesch, B.,McBride, P.A., Manson, J.C., 1996. Altered circadian activity rhythms and sleep inmice devoid of prion protein. Nature 380, 639–642.
Toullec, D., Pianetti, P., Coste, H., Bellevergue, P., Grand-Perret, T., Ajakane, M., Baudet,V., Boissin, P., Boursier, E., Loriolle, F., 1991. The bisindolethylmaleimide GF109203X is a potent and selective inhibitor of protein kinase C. J. Biol. Chem. 266,15771–15781.
Vilette, D., Andreoletti, O., Archer, F., Madeleine, M.F., Vilotte, J.L., Lehmann, S., Laude, H.,2001. Ex vivo propagation of infectious sheep scrapie agent in heterologous

410 M. Alfa Cissé et al. / Molecular and Cellular Neuroscience 39 (2008) 400–410
epithelial cells expressing ovine prion protein. Proc. Natl. Acad. Sci. U.S.A. 98,4055–4059.
Vincent, B., Paitel, E., Frobert, Y., Lehmann, S., Grassi, J., Checler, F., 2000. Phorbol esters-regulated cleavage of normal prion protein in HEK293 human cells and murineneurons. J. Biol. Chem. 275, 35612–35616.
Vincent, B., Paitel, E., Saftig, P., Frobert, Y., Hartmann, D., de Strooper, B., Grassi, J., Lopez-Perez, E., Checler, F., 2001. The disintegrins ADAM10 and TACE contribute to the
constitutive and phorbol ester-regulated normal cleavage of the cellular prionprotein. J. Biol. Chem. 276, 37743–37746.
Zhang, Y., Jiang, J., Black, R.A., Baumann, G., Franck, S.J., 2000. Tumor necrosisfactor-α converting enzyme (TACE) is a growth hormone binding protein(GHBP) sheddase: the metalloprotease TACE/ADM17 is critical for (PMA-induced) GH receptor proteolysis and GHBP generation. Endocrinology 141,4342–4348.


![A Non-Q/N-Rich Prion Domain of a Foreign Prion, [Het-s], Can Propagate as a Prion in Yeast](https://img.dokumen.tips/doc/110x75/63498a64d6804e97060ada39/a-non-qn-rich-prion-domain-of-a-foreign-prion-het-s-can-propagate-as-a-prion.jpg)


























