Embed Size (px)
Citation preview

Published Ahead of Print 2 July 2008. 2008, 82(17):8579. DOI: 10.1128/JVI.01022-08. J. Virol.
Churchill and Tomasz I. MichalakClifford S. Guy, Patricia M. Mulrooney-Cousins, Norma D. Hepadnavirusduring Acute Infection with Woodchuck Responses Immediately after Invasion andwith Innate and Adaptive Immune Intrahepatic Expression of Genes Affiliated
http://jvi.asm.org/content/82/17/8579Updated information and services can be found at:
These include:
REFERENCEShttp://jvi.asm.org/content/82/17/8579#ref-list-1at:
This article cites 62 articles, 21 of which can be accessed free
CONTENT ALERTS more»articles cite this article),
Receive: RSS Feeds, eTOCs, free email alerts (when new
http://journals.asm.org/site/misc/reprints.xhtmlInformation about commercial reprint orders: http://journals.asm.org/site/subscriptions/To subscribe to to another ASM Journal go to:
on Decem
ber 11, 2012 by guesthttp://jvi.asm
.org/D
ownloaded from

JOURNAL OF VIROLOGY, Sept. 2008, p. 8579–8591 Vol. 82, No. 170022-538X/08/$08.00�0 doi:10.1128/JVI.01022-08Copyright © 2008, American Society for Microbiology. All Rights Reserved.
Intrahepatic Expression of Genes Affiliated with Innate and AdaptiveImmune Responses Immediately after Invasion and during Acute
Infection with Woodchuck Hepadnavirus�
Clifford S. Guy,1 Patricia M. Mulrooney-Cousins,1 Norma D. Churchill,1 and Tomasz I. Michalak1,2*Molecular Virology and Hepatology Research Group, Division of BioMedical Science,1 and Discipline of Laboratory Medicine,2
Faculty of Medicine, Health Science Centre, Memorial University, St. John’s, Newfoundland, Canada
Received 15 May 2008/Accepted 23 June 2008
The importance of effective immune responses in recovery from acute hepadnaviral hepatitis has beendemonstrated. However, there is no conclusive delineation of virological and immunological events occurringin the liver immediately after hepadnavirus invasion and during the preacute phase of infection. These veryearly events might be of primary importance in determining the recovery or progression to chronic hepatitisand the intrinsic hepadnaviral propensity to persist. In this study, applying the woodchuck model of acutehepatitis B, the hepatic kinetics of hepadnavirus replication and activation of genes encoding cytokines,cytotoxicity effectors, and immune cell markers were quantified in sequential liver biopsies collected from 1 hpostinoculation onward by sensitive real-time cDNA amplification assays. The results revealed that hepadna-virus replication is established in the liver as early as 1 hour after infection. In 3 to 6 h, significantly augmentedintrahepatic transcription of gamma interferon and interleukin-12 were evident, suggesting activation ofantigen-presenting cells. In 48 to 72 h, NK and NKT cells were activated and virus replication was transientlybut significantly reduced, implying that this early innate response is at least partially successful in limitingvirus propagation. Nonetheless, T cells were activated 4 to 5 weeks later when hepatitis became histologicallyevident. Collectively, our data demonstrate that virus replication is initiated and the innate response activatedin the liver soon after exposure to a liver-pathogenic dose of hepadnavirus. Nevertheless, this response isunable to prompt a timely adaptive T-cell response, in contrast to infections caused by other viral pathogens.
Hepatitis B virus (HBV) is the prototypic member of theHepadnaviridae family of small, enveloped, primarily hepato-tropic DNA viruses which cause acute and chronic hepatitisand hepatocellular carcinoma (reviewed in reference 9). Hep-adnaviruses display highly restricted host specificity. Thus,HBV infection is limited to humans and higher primates, whilewoodchuck hepatitis virus (WHV), despite sharing significantstructural, genomic, and antigenic similarities with HBV, in-fects the eastern North American woodchuck (Marmotamonax) (38). Notwithstanding their limited host ranges, HBVand WHV induce similar courses of hepatitis, which are pre-ceded by a long incubation period ranging from 6 weeks to 6months for hepatitis B and from 4 to 10 weeks for WHVhepatitis (9, 34). In contrast with the appearance of viremiaand adaptive T-cell immunity a few days after invasion withmost viral pathogens, a significant increase in HBV replicationis not observed until 3 to 5 weeks postinfection (p.i.) in chim-panzees, while activation of HBV-specific adaptive immunityoccurs several weeks later (4, 17, 59).
Despite the delay in activation of an HBV-specific T-cellresponse, a robust, multispecific T-cell reactivity seems to beessential for both induction of acute hepatitis (AH) and theclinical recovery (50, 51), although it is unable to clear the virusentirely (43, 55, 56). Experimental evidence acquired from the
chimpanzee and HBV-transgenic mouse systems, as well asthe woodchuck model of hepatitis B, has clearly indicated theimportance of virus-specific CD4� and CD8� T lymphocytesand antiviral cytokines, such as alpha interferon, beta inter-feron, gamma interferon (IFN-�), and tumor necrosis factoralpha (TNF-�), in the downregulation of virus replication (21,35, 36, 48), while bystander recruitment of virus-nonspecific Tcells and other immune cell subsets greatly contributes to liverinflammation and hepatocyte destruction (30, 58). In addition,the role of antiviral cytokines, particularly IFN-�, has beenascribed to the ability to inhibit replication of hepadnavirusin a noncytopathic manner (15, 17). In this regard, elevationsin the intrahepatic expression of IFN-� appeared sufficient inchimpanzees to reduce HBV viremia before the peak onset ofhepatitis (17). Also, an increased intrahepatic expression ofIFN-� was consistently detected for many years following re-covery from WHV-induced AH, implying its role in control ofresidual (occult) WHV replication continuing after resolutionof acute disease (21).
Natural killer (NK) and NK T (NKT) cells are componentsof the innate immune system. They are enriched among theresident lymphomononuclear cells in the liver and are capableof producing IFN-� and other cytokines within minutes follow-ing viral invasion (reviewed in reference 8). The results fromstudies with HBV transgenic mice suggest that activation ofNKT cells via CD1d-restricted �-galactosylceramide can down-regulate virus replication (22, 23), while nonclassical NKT cellsmay recognize HBV antigens expressed in the liver (1). It hasbeen suggested that NK- or NKT-derived IFN-� may be theprincipal mediator of HBV downregulation in acutely infected
* Corresponding author. Mailing address: Molecular Virology andHepatology Research Group, Faculty of Medicine, Health SciencesCentre, Memorial University, St. John’s, NL, Canada A1B 3V6.Phone: (709) 777 7301. Fax: (709) 777 8279. E-mail: [email protected].
� Published ahead of print on 2 July 2008.
8579
on Decem
ber 11, 2012 by guesthttp://jvi.asm
.org/D
ownloaded from
chimpanzees (17), while indirect evidence from the woodchuckmodel suggests that activated, cytotoxic NK or NKT cells maycontribute to hepatocyte killing and recovery from WHV in-fection (20).
Nonetheless, despite findings suggesting that hepadnavi-ruses may directly activate intrahepatic immune cells capableof producing cytokines promoting antiviral defense and favor-ing a T-helper-cell type 1 response (17, 59), comprehensivedata from a natural model of hepadnaviral infection are lack-ing. Furthermore, characterization of the liver immune re-sponse in the first hours and days following hepadnavirus in-vasion has not yet been accomplished. The aim of the currentstudy was to recognize, using the woodchuck model of hepatitisB, the nature and the kinetics of intrahepatic immune re-sponses occurring soon after exposure to WHV and during thepreacute and acute phases of hepadnaviral infection by quan-tifying hepatic transcriptional activities of genes encoding crit-ical proinflammatory and antiviral cytokines and markers spec-ifying individual immune cell subsets.
MATERIALS AND METHODS
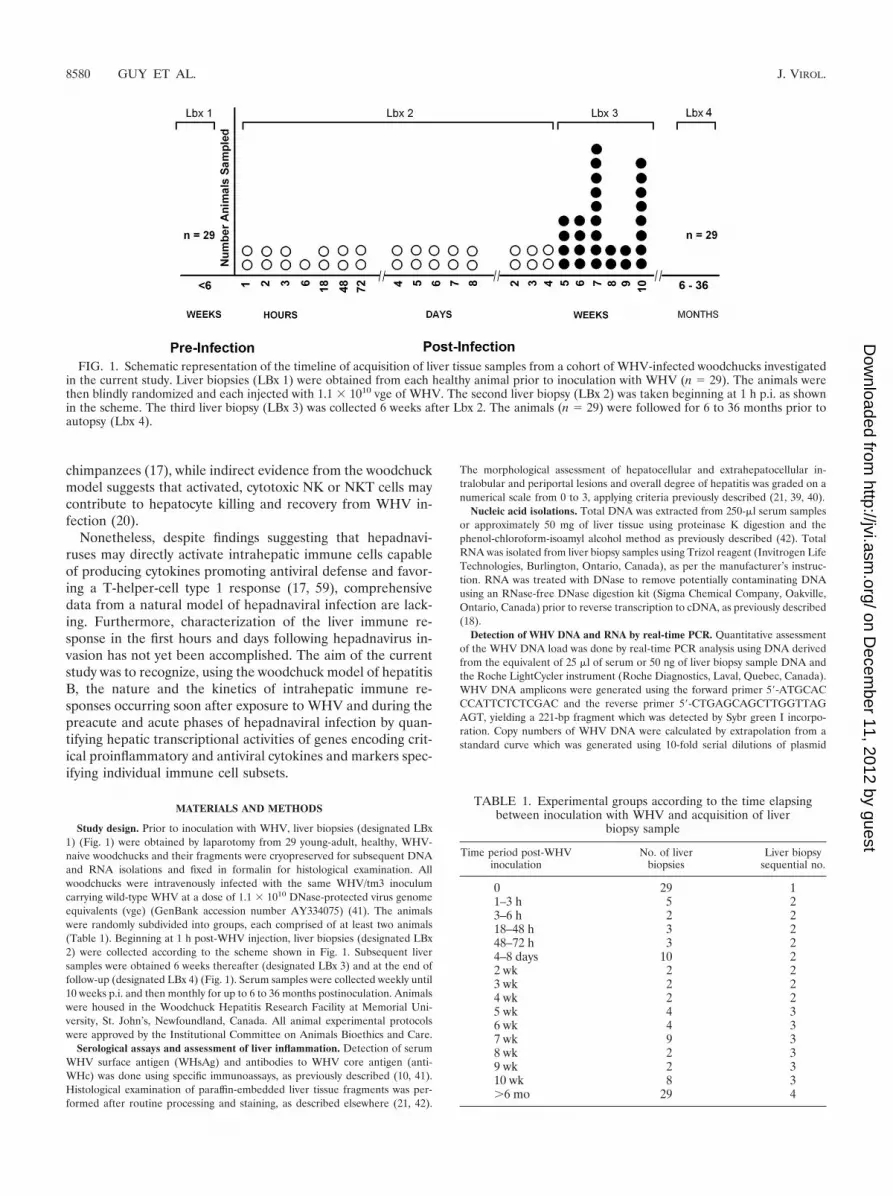
Study design. Prior to inoculation with WHV, liver biopsies (designated LBx1) (Fig. 1) were obtained by laparotomy from 29 young-adult, healthy, WHV-naive woodchucks and their fragments were cryopreserved for subsequent DNAand RNA isolations and fixed in formalin for histological examination. Allwoodchucks were intravenously infected with the same WHV/tm3 inoculumcarrying wild-type WHV at a dose of 1.1 � 1010 DNase-protected virus genomeequivalents (vge) (GenBank accession number AY334075) (41). The animalswere randomly subdivided into groups, each comprised of at least two animals(Table 1). Beginning at 1 h post-WHV injection, liver biopsies (designated LBx2) were collected according to the scheme shown in Fig. 1. Subsequent liversamples were obtained 6 weeks thereafter (designated LBx 3) and at the end offollow-up (designated LBx 4) (Fig. 1). Serum samples were collected weekly until10 weeks p.i. and then monthly for up to 6 to 36 months postinoculation. Animalswere housed in the Woodchuck Hepatitis Research Facility at Memorial Uni-versity, St. John’s, Newfoundland, Canada. All animal experimental protocolswere approved by the Institutional Committee on Animals Bioethics and Care.
Serological assays and assessment of liver inflammation. Detection of serumWHV surface antigen (WHsAg) and antibodies to WHV core antigen (anti-WHc) was done using specific immunoassays, as previously described (10, 41).Histological examination of paraffin-embedded liver tissue fragments was per-formed after routine processing and staining, as described elsewhere (21, 42).
The morphological assessment of hepatocellular and extrahepatocellular in-tralobular and periportal lesions and overall degree of hepatitis was graded on anumerical scale from 0 to 3, applying criteria previously described (21, 39, 40).
Nucleic acid isolations. Total DNA was extracted from 250-�l serum samplesor approximately 50 mg of liver tissue using proteinase K digestion and thephenol-chloroform-isoamyl alcohol method as previously described (42). TotalRNA was isolated from liver biopsy samples using Trizol reagent (Invitrogen LifeTechnologies, Burlington, Ontario, Canada), as per the manufacturer’s instruc-tion. RNA was treated with DNase to remove potentially contaminating DNAusing an RNase-free DNase digestion kit (Sigma Chemical Company, Oakville,Ontario, Canada) prior to reverse transcription to cDNA, as previously described(18).
Detection of WHV DNA and RNA by real-time PCR. Quantitative assessmentof the WHV DNA load was done by real-time PCR analysis using DNA derivedfrom the equivalent of 25 �l of serum or 50 ng of liver biopsy sample DNA andthe Roche LightCycler instrument (Roche Diagnostics, Laval, Quebec, Canada).WHV DNA amplicons were generated using the forward primer 5�-ATGCACCCATTCTCTCGAC and the reverse primer 5�-CTGAGCAGCTTGGTTAGAGT, yielding a 221-bp fragment which was detected by Sybr green I incorpo-ration. Copy numbers of WHV DNA were calculated by extrapolation from astandard curve which was generated using 10-fold serial dilutions of plasmid
FIG. 1. Schematic representation of the timeline of acquisition of liver tissue samples from a cohort of WHV-infected woodchucks investigatedin the current study. Liver biopsies (LBx 1) were obtained from each healthy animal prior to inoculation with WHV (n � 29). The animals werethen blindly randomized and each injected with 1.1 � 1010 vge of WHV. The second liver biopsy (LBx 2) was taken beginning at 1 h p.i. as shownin the scheme. The third liver biopsy (LBx 3) was collected 6 weeks after Lbx 2. The animals (n � 29) were followed for 6 to 36 months prior toautopsy (Lbx 4).
TABLE 1. Experimental groups according to the time elapsingbetween inoculation with WHV and acquisition of liver
biopsy sample
Time period post-WHVinoculation
No. of liverbiopsies
Liver biopsysequential no.
0 29 11–3 h 5 23–6 h 2 218–48 h 3 248–72 h 3 24–8 days 10 22 wk 2 23 wk 2 24 wk 2 25 wk 4 36 wk 4 37 wk 9 38 wk 2 39 wk 2 310 wk 8 3�6 mo 29 4
8580 GUY ET AL. J. VIROL.
on Decem
ber 11, 2012 by guesthttp://jvi.asm
.org/D
ownloaded from

containing known copy numbers of complete, recombinant WHV DNA. WHVRNA was similarly detected using 50 ng liver total RNA. The specificity ofreal-time PCR products was always confirmed by nucleic acid hybridization(NAH), i.e., Southern blot hybridization analysis, using complete recombinantWHV DNA as a probe and autoradiography, as previously described (10, 41).The sensitivity of the real-time PCR assay was 200 vge/�g DNA, 200 cop-ies/�g RNA, or 50 vge/ml serum.
Detection of WHV cccDNA. WHV covalently closed circular DNA (cccDNA),representing virus genome replicative intermediate, was detected in liver tissuesamples by PCR amplification with a sensitivity of 102 vge/�g total DNA, aspreviously described in detail (26, 41). Briefly, 4 �g of DNA was digested withmung bean nuclease prior to PCR amplification with oligonucleotide primersspanning the nicked region of the partially double-stranded WHV DNA genome.The specificity of PCR amplicons was routinely confirmed by NAH, as previouslydescribed (10, 42).
Identification of woodchuck cellular gene sequences. To facilitate analysis ofthe spectrum and the dynamics of intrahepatic immune response in WHV in-fection, a number of woodchuck gene sequences encoding markers specifyingdifferent immune cell subtypes, immune cell effector molecules, and cytokineswere determined, applying a strategy previously reported (18). In general, wood-chuck gene sequences were identified by reverse transcription-PCR (RT-PCR)using degenerate oligonucleotide primers whose sequences were deducedthrough interspecies comparison of the sequences available in GenBank. Theresulting amplicons were cloned into the PCRII TOPO TA cloning system(Invitrogen) and the excised fragments sequenced in both directions. Based onthe woodchuck sequences determined, the pairs of gene-specific primers weredesigned. Table 2 presents the list of woodchuck genes for which hepatic expres-sion was quantified in the current study and the primer pairs used for theirquantitative detection.
Analysis of woodchuck gene expression by real-time PCR. Real-time RT-PCRassays were developed for each cellular effector molecule and the cytokine geneanalyzed, using the Roche LightCycler instrument with Sybr green I detectionand PCR primer pairs specified in Table 2. Changes in gene expression levelswere determined by comparison to the baseline level of each gene’s transcriptiondetected in the liver biopsy sample obtained prior to infection with WHV foreach individual animal after normalization to expression of the housekeeping
gene -actin. Following measurement of a given gene’s expression in a particularliver sample from an individual animal, the mean expression level was deter-mined for all liver biopsy samples within the experimental groups showed inTable 1.
Inactivation of WHV inoculum. To ascertain that the observed changes in theintrahepatic gene expression levels were truly related to infection with WHV butnot to injection with serum components present in the WHV inoculum, a controlexperiment was performed. Thus, the WHV/tm3 inoculum was inactivated bytreatment with 50 �g/ml psoralen (Sigma) combined with exposure to 365 nmUV light for 90 min at 4°C. Animals were injected intravenously with 0.5 ml ofpsoralen-inactivated inoculum containing the equivalent of 1.1 � 1010 vge or with0.5 ml of similarly treated healthy woodchuck serum prior to euthanasia at 3 dayspostinjection.
Statistical analysis. A two-tailed, unpaired Student t test with 95% confidenceinterval was applied to compare the means of sample groups investigated, and Pvalues of �0.05 were considered statistically significant.
Accession numbers of woodchuck gene sequences identified. The accessionnumbers for the woodchuck gene sequences established in the course of thisstudy have been submitted to GenBank under the following accession numbers:for the CD4 gene, EF621765; for the CD8 gene, EF621766; for the CD40 ligand(CD40L), EF621170; for CD1d, EF621767; for NKp46, EF621768; and for in-terleukin-8 (IL-8), EF-126348.
RESULTS
Serologic and hepatic profiles of WHV infection. Inoculationof woodchucks with a WHV dose of 1.1 � 1010 vge resulted intransiently serum WHsAg-positive, self-limiting acute infec-tion in all 29 animals. The mean time of WHsAg appearancewas 18.5 days, while its average duration in the circulation was35.8 days. Anti-WHc remained detectable until the end of theobservation period (Fig. 2A). The pattern of serum WHsAgpositivity implied, based on the known profiles of serologicalmarkers of WHV infection (41, 42), that a self-limited episodeof AH had developed in all woodchucks investigated. This wasconfirmed by histological examination of serial liver biopsysamples collected.
Considering morphological alterations encountered in livertissue, their characteristics and the timing of their occurrenceclosely followed those previously reported for experimentalacute WHV infection (38, 42). In brief, the appearance ofscattered lymphocytes and neutrophils in sinusoids and peri-portal areas, accompanied by mild proliferations of sinusoidallining endothelium and bile ducts, was the first manifestation,which occurred at 4 to 5 weeks p.i. In the next few days,scattered lobular infiltrations, consisting mainly of lympho-cytes, degenerative or necrotic changes of single hepatocytes,progressing hyperplasia of Kupffer cells, and limited portalinfiltrations, occurred. Subsequently, more-severe lobular in-filtrations and hepatocyte degenerative and necrotic changesand the appearance of acidophilic bodies were observed. Atthe peak of AH, usually occurring between weeks 6 and 8 p.i.,both lobular and periportal inflammatory changes were evi-dent, the portal infiltrations became most intense and ex-panded occasionally through limiting plates, and multifocalnecroses of parenchyma with varying numbers of acidophilicbodies were found. However, the severity of AH varied, andwhile the changes in some woodchucks were intense, reachinghistological degree of hepatitis 2.5, in others they were mildand hepatitis did not exceed grade 1.5. Following resolution ofAH, intermittent minimal lobular and portal inflammatory al-terations and periods of normal or nearly normal liver mor-phology were observed, as previously reported (21, 41, 42).
In previous studies, WHV DNA in the circulation and the
TABLE 2. Primer sequences used in this study for real-timeRT-PCR quantifications of expression of woodchuck
genes encoding immune cell markers, cytokines,and cytotoxicity effector molecules
Gene Primer sequencea
Expectedsize of
amplicon(bp)
GenBankaccession
no.
CD3 F-CTGGGACTCTGCCTCTTATC 536 AF232727R-GCTGCCCTTTCCGGATGGGCTC
CD4b F-GGAGAATAAGAAGATAGAGG 560 EF621765R-TCAAGAGTCACAGTCAGG
CD8b F-AACGAGGGCTACTATTTCTGCTC 309 EF621766R-GTTTCCGGTGGTGACAGATGA
CD40 ligandb F-AGCATGTGTGCTACAGT 214 EF621770R-CCGCCCTGAGTAAGATT
CD1db F-TCCTAGATTAGGGAAGTCAGAAC 222 EF621767R-GCTCGGAGATACCACG
NKp46b F-TTGCCACCTAGTGACAG 200 EF621768R-CACCAGGAGCATCACC
CD95 ligandb F-CCATTTAACAGGTAAGCCC 250 AF152368R-TCATCATCTTGCCCTCC
Perforinb F-GCATCAACAATGACTGGCGGG 302 AF298158R-TGAAGTGGGTGCCGTAGTTGTGG
IL-4 F-TTTGCTGTCCCCAAGAAC 200 AF333965R-CCTGGATTCACTCACGG
IL-8b F-GGTAACCTGCCTACTTTC 207 EF216348R-GTTCAGGCAAAGCTCT
IL12p40 F-tTGGATTGGCACCCTGACAC 198 X97019R-GCATCTGGCTCAGAACTTCAC
IFN-� F-AGGAGCATGGACACCATCA 215 AF232728R-cCGACCCCGAATCGAAG
TNF-� F-TGAGCACTGAAAGTATGATCC 283 AF333967R-TGCTACAACATGGGCTACAG
2�,5�-OAS F-TCAGGCAAAGGCACTACCC 150 AF082498R-aCTTCTCTTTCGGACATGCT
a F, forward primer; R, reverse primer.b Woodchuck gene sequence identified in the current study.
VOL. 82, 2008 EARLY INTRAHEPATIC IMMUNITY IN HEPADNAVIRUS INFECTION 8581
on Decem
ber 11, 2012 by guesthttp://jvi.asm
.org/D
ownloaded from
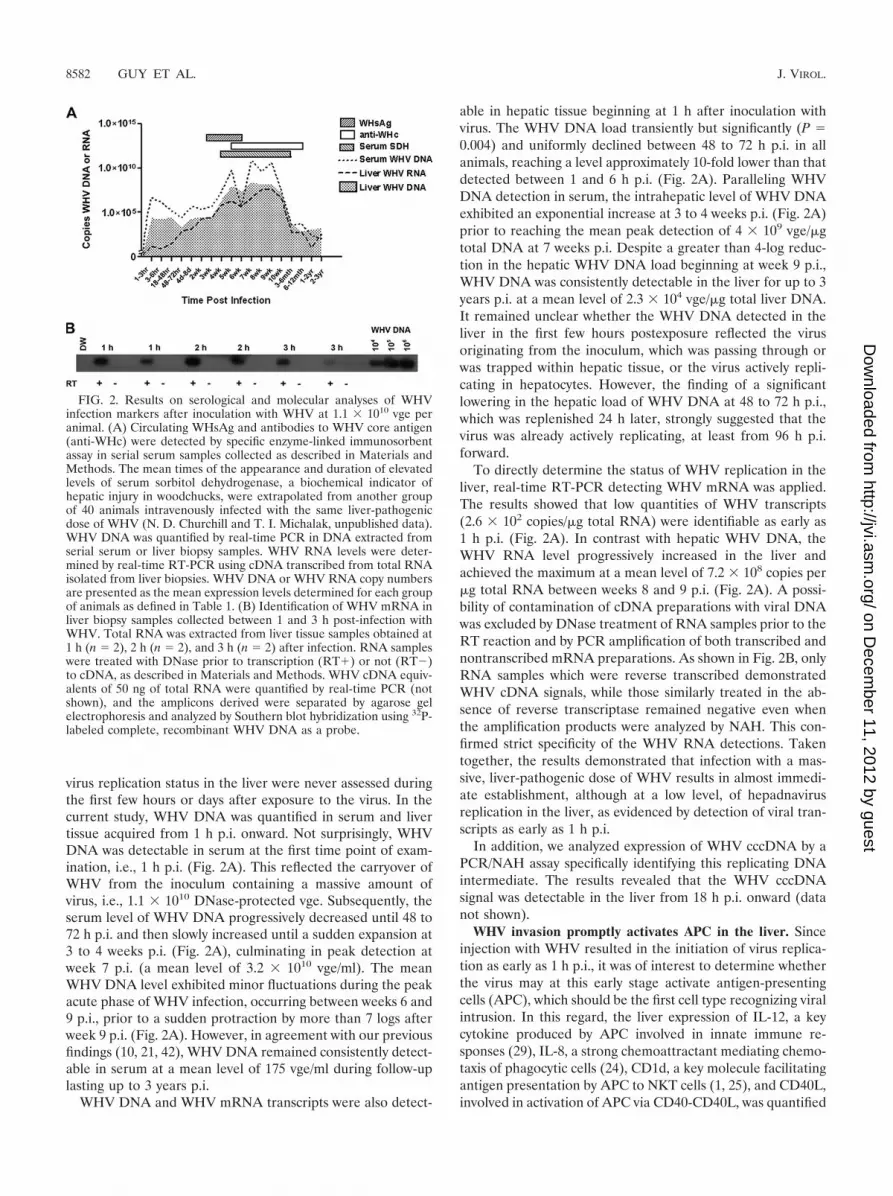
virus replication status in the liver were never assessed duringthe first few hours or days after exposure to the virus. In thecurrent study, WHV DNA was quantified in serum and livertissue acquired from 1 h p.i. onward. Not surprisingly, WHVDNA was detectable in serum at the first time point of exam-ination, i.e., 1 h p.i. (Fig. 2A). This reflected the carryover ofWHV from the inoculum containing a massive amount ofvirus, i.e., 1.1 � 1010 DNase-protected vge. Subsequently, theserum level of WHV DNA progressively decreased until 48 to72 h p.i. and then slowly increased until a sudden expansion at3 to 4 weeks p.i. (Fig. 2A), culminating in peak detection atweek 7 p.i. (a mean level of 3.2 � 1010 vge/ml). The meanWHV DNA level exhibited minor fluctuations during the peakacute phase of WHV infection, occurring between weeks 6 and9 p.i., prior to a sudden protraction by more than 7 logs afterweek 9 p.i. (Fig. 2A). However, in agreement with our previousfindings (10, 21, 42), WHV DNA remained consistently detect-able in serum at a mean level of 175 vge/ml during follow-uplasting up to 3 years p.i.
WHV DNA and WHV mRNA transcripts were also detect-
able in hepatic tissue beginning at 1 h after inoculation withvirus. The WHV DNA load transiently but significantly (P �0.004) and uniformly declined between 48 to 72 h p.i. in allanimals, reaching a level approximately 10-fold lower than thatdetected between 1 and 6 h p.i. (Fig. 2A). Paralleling WHVDNA detection in serum, the intrahepatic level of WHV DNAexhibited an exponential increase at 3 to 4 weeks p.i. (Fig. 2A)prior to reaching the mean peak detection of 4 � 109 vge/�gtotal DNA at 7 weeks p.i. Despite a greater than 4-log reduc-tion in the hepatic WHV DNA load beginning at week 9 p.i.,WHV DNA was consistently detectable in the liver for up to 3years p.i. at a mean level of 2.3 � 104 vge/�g total liver DNA.It remained unclear whether the WHV DNA detected in theliver in the first few hours postexposure reflected the virusoriginating from the inoculum, which was passing through orwas trapped within hepatic tissue, or the virus actively repli-cating in hepatocytes. However, the finding of a significantlowering in the hepatic load of WHV DNA at 48 to 72 h p.i.,which was replenished 24 h later, strongly suggested that thevirus was already actively replicating, at least from 96 h p.i.forward.
To directly determine the status of WHV replication in theliver, real-time RT-PCR detecting WHV mRNA was applied.The results showed that low quantities of WHV transcripts(2.6 � 102 copies/�g total RNA) were identifiable as early as1 h p.i. (Fig. 2A). In contrast with hepatic WHV DNA, theWHV RNA level progressively increased in the liver andachieved the maximum at a mean level of 7.2 � 108 copies per�g total RNA between weeks 8 and 9 p.i. (Fig. 2A). A possi-bility of contamination of cDNA preparations with viral DNAwas excluded by DNase treatment of RNA samples prior to theRT reaction and by PCR amplification of both transcribed andnontranscribed mRNA preparations. As shown in Fig. 2B, onlyRNA samples which were reverse transcribed demonstratedWHV cDNA signals, while those similarly treated in the ab-sence of reverse transcriptase remained negative even whenthe amplification products were analyzed by NAH. This con-firmed strict specificity of the WHV RNA detections. Takentogether, the results demonstrated that infection with a mas-sive, liver-pathogenic dose of WHV results in almost immedi-ate establishment, although at a low level, of hepadnavirusreplication in the liver, as evidenced by detection of viral tran-scripts as early as 1 h p.i.
In addition, we analyzed expression of WHV cccDNA by aPCR/NAH assay specifically identifying this replicating DNAintermediate. The results revealed that the WHV cccDNAsignal was detectable in the liver from 18 h p.i. onward (datanot shown).
WHV invasion promptly activates APC in the liver. Sinceinjection with WHV resulted in the initiation of virus replica-tion as early as 1 h p.i., it was of interest to determine whetherthe virus may at this early stage activate antigen-presentingcells (APC), which should be the first cell type recognizing viralintrusion. In this regard, the liver expression of IL-12, a keycytokine produced by APC involved in innate immune re-sponses (29), IL-8, a strong chemoattractant mediating chemo-taxis of phagocytic cells (24), CD1d, a key molecule facilitatingantigen presentation by APC to NKT cells (1, 25), and CD40L,involved in activation of APC via CD40-CD40L, was quantified
FIG. 2. Results on serological and molecular analyses of WHVinfection markers after inoculation with WHV at 1.1 � 1010 vge peranimal. (A) Circulating WHsAg and antibodies to WHV core antigen(anti-WHc) were detected by specific enzyme-linked immunosorbentassay in serial serum samples collected as described in Materials andMethods. The mean times of the appearance and duration of elevatedlevels of serum sorbitol dehydrogenase, a biochemical indicator ofhepatic injury in woodchucks, were extrapolated from another groupof 40 animals intravenously infected with the same liver-pathogenicdose of WHV (N. D. Churchill and T. I. Michalak, unpublished data).WHV DNA was quantified by real-time PCR in DNA extracted fromserial serum or liver biopsy samples. WHV RNA levels were deter-mined by real-time RT-PCR using cDNA transcribed from total RNAisolated from liver biopsies. WHV DNA or WHV RNA copy numbersare presented as the mean expression levels determined for each groupof animals as defined in Table 1. (B) Identification of WHV mRNA inliver biopsy samples collected between 1 and 3 h post-infection withWHV. Total RNA was extracted from liver tissue samples obtained at1 h (n � 2), 2 h (n � 2), and 3 h (n � 2) after infection. RNA sampleswere treated with DNase prior to transcription (RT�) or not (RT�)to cDNA, as described in Materials and Methods. WHV cDNA equiv-alents of 50 ng of total RNA were quantified by real-time PCR (notshown), and the amplicons derived were separated by agarose gelelectrophoresis and analyzed by Southern blot hybridization using 32P-labeled complete, recombinant WHV DNA as a probe.
8582 GUY ET AL. J. VIROL.
on Decem
ber 11, 2012 by guesthttp://jvi.asm
.org/D
ownloaded from

by using real-time RT-PCR assays specifically developed forthe purpose of this study.
Figure 3 shows that IL-12 achieved a maximum hepaticexpression (20-fold increase over the preinfection level; P �0.02) between 3 and 6 h p.i. (Fig. 3A, left panel). Interestingly,in contrast with this early increase, the remaining course ofWHV infection, including the period of histologically evidentAH with peaking histologically evident liver injury occurringbetween weeks 6 and 8 p.i. (Fig. 3A, right panel), was withoutany noticeable increase in expression of this cytokine.
The transcription level of IL-8 tended to rise (a 12.5-foldincrease) within 1 to 3 h p.i. above that detected in the periodprior to infection; however, this increase did not reach a sta-tistically significant value (P � 0.1) (Fig. 3A, left panel). Themaximum expression of the cytokine (a 35-fold increase overthe preinfection level; P � 0.05) occurred at week 7 p.i., duringthe phase of acute liver inflammation (Fig. 3A, right panel).
Since a strong induction of IL-12 expression detected at 3 to6 h p.i. could be directly associated with activation of APC(29), the intrahepatic expression of CD1d, which facilitatesantigen presentation to NKT cells, was also evaluated. Theresults showed that the level of CD1d mRNA reached a peak(a 3-fold increase over the preinfection level; P � 0.03) by 48to 72 h p.i. (Fig. 3B, left panel). Then, the level subsided untilWHV replication was drastically augmented at 3 to 4 weeks p.i.
(Fig. 3B, right panel). Thus, the upregulated transcription ofCD1d at 48 to 72 h was associated with a significant one-logdecrease in the hepatic WHV DNA load and a decline ofWHV DNA in circulation. Furthermore, the concomitant in-crease (P � 0.02) in CD40L expression (Fig. 3B, left panel)suggested activation of APC via CD40-CD40L interaction.
Activation of hepatic NK and NKT cells follows infectionwith WHV. WHV infection induced two distinct phases of veryearly immune activation in the liver, one between 3 and 6 h anda second at 48 to 72 h p.i. Since experimental evidence fromother viral infections indicates the ability of NK and NKT cellsto respond within minutes to hours to virus by secretion ofIFN-� or by acquisition of cytotoxic function (8), expression ofthe gene encoding the NK receptor NKp46 was investigated.Furthermore, since IL-12 is recognized as a key cytokine whichinfluences IFN-� secretion by NK cells and also increasesNK-mediated cytotoxicity, which is primarily facilitated by theperforin pathway (44), perforin mRNA levels were also quan-tified. The results showed that the earliest increase in tran-scription of IL-12, occurring between 3 to 6 h p.i. (Fig. 3A, leftpanel), coincided with significantly augmented transcription ofIFN-� at the same time (3.3-fold; P � 0.018) (Fig. 4B, leftpanel). However, apparent increases in the expression ofNKp46 (a 2.5-fold induction) and perforin (a 3.4-fold induc-
FIG. 3. WHV infection upregulates intrahepatic genes indicative of activation of APC. Profiles of expression of IL-12 and IL-8 (A) and CD1dand CD40L (B) are shown. Gene mRNA levels were quantified by real-time RT-PCR in liver biopsy samples. Data shown represent the meanexpression levels for each group of liver biopsy samples analyzed at the time points indicated and are presented relative to the maximum leveldetected for each gene analyzed. Mean levels of intrahepatic WHV DNA were determined as described in Materials and Methods and in thelegend to Fig. 2A. WHV DNA profiles are shown for the time periods between 0 and 2 weeks p.i. (left panel) and between 2 and 10 weeks p.i.(right panel) as a reference. Abbreviations for times post-inoculation with WHV: hr, hours; d, days; wk, weeks.
VOL. 82, 2008 EARLY INTRAHEPATIC IMMUNITY IN HEPADNAVIRUS INFECTION 8583
on Decem
ber 11, 2012 by guesthttp://jvi.asm
.org/D
ownloaded from
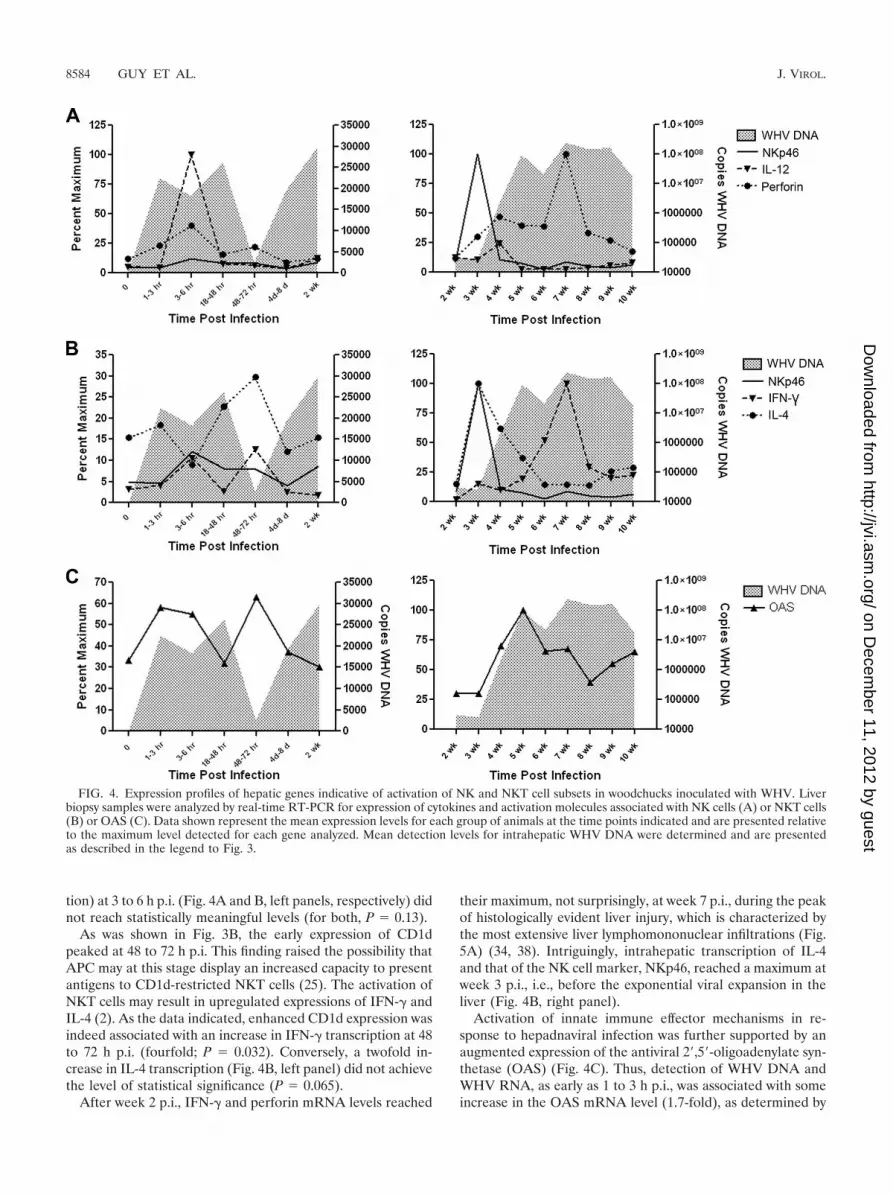
tion) at 3 to 6 h p.i. (Fig. 4A and B, left panels, respectively) didnot reach statistically meaningful levels (for both, P � 0.13).
As was shown in Fig. 3B, the early expression of CD1dpeaked at 48 to 72 h p.i. This finding raised the possibility thatAPC may at this stage display an increased capacity to presentantigens to CD1d-restricted NKT cells (25). The activation ofNKT cells may result in upregulated expressions of IFN-� andIL-4 (2). As the data indicated, enhanced CD1d expression wasindeed associated with an increase in IFN-� transcription at 48to 72 h p.i. (fourfold; P � 0.032). Conversely, a twofold in-crease in IL-4 transcription (Fig. 4B, left panel) did not achievethe level of statistical significance (P � 0.065).
After week 2 p.i., IFN-� and perforin mRNA levels reached
their maximum, not surprisingly, at week 7 p.i., during the peakof histologically evident liver injury, which is characterized bythe most extensive liver lymphomononuclear infiltrations (Fig.5A) (34, 38). Intriguingly, intrahepatic transcription of IL-4and that of the NK cell marker, NKp46, reached a maximum atweek 3 p.i., i.e., before the exponential viral expansion in theliver (Fig. 4B, right panel).
Activation of innate immune effector mechanisms in re-sponse to hepadnaviral infection was further supported by anaugmented expression of the antiviral 2�,5�-oligoadenylate syn-thetase (OAS) (Fig. 4C). Thus, detection of WHV DNA andWHV RNA, as early as 1 to 3 h p.i., was associated with someincrease in the OAS mRNA level (1.7-fold), as determined by
FIG. 4. Expression profiles of hepatic genes indicative of activation of NK and NKT cell subsets in woodchucks inoculated with WHV. Liverbiopsy samples were analyzed by real-time RT-PCR for expression of cytokines and activation molecules associated with NK cells (A) or NKT cells(B) or OAS (C). Data shown represent the mean expression levels for each group of animals at the time points indicated and are presented relativeto the maximum level detected for each gene analyzed. Mean detection levels for intrahepatic WHV DNA were determined and are presentedas described in the legend to Fig. 3.
8584 GUY ET AL. J. VIROL.
on Decem
ber 11, 2012 by guesthttp://jvi.asm
.org/D
ownloaded from

real-time RT-PCR, but this was not significant (P � 0.14) (Fig.4C, left panel). However, at 48 to 72 h p.i., a significant in-crease (P � 0.05) in OAS (Fig. 4C, left panel) correlated witha significant (P � 0.03) elevation in expression of IFN-� (Fig.4B, left panel), a known inducer of OAS (45, 60). This oc-curred at the time when the WHV load significantly decreasedin the liver. Taken together, the coincidence of these eventssupports the notion that the transient upregulation of hepaticIFN-� occurring in the first 72 h following hepadnaviral expo-sure was biologically relevant and exerted a strong antiviraleffect.
Intrahepatic CD4� and CD8� T cells are quiescent forweeks after WHV infection. The prominent reduction in hep-adnavirus replication and resolution of AH appear reliantupon a strong and multispecific antiviral T-cell response, whichis characterized by secretion of IFN-� and TNF-� (15, 16). Toassess whether the observed initial elevations in intrahepaticexpression of IFN-� (Fig. 4B, left panel) could be due to thepresence of activated T cells, hepatic transcription of genesencoding CD4, CD8, and CD3 T-cell markers was quantified.It was found that CD4 and CD8 mRNA levels tended toincrease (by 20 to 25%) in the first 3 h post-WHV exposure,but these increases did not reach a statistically significant dif-ference (P � 0.18 and 0.12, respectively) over the preinfectionlevels. The CD4 and CD8 mRNA levels remained consistentlylow (10 to 15% of the preinfection levels; P � 0.3 and 0.2,respectively) until week 4 p.i. (Fig. 5A, left panel). However,not surprisingly, very prominent increases in the mRNA levelsof CD4 (8.3-fold; P � 0.017) and CD8 (6.7-fold; P � 0.03) weredetected during the peak of histologically evident liver inflam-
mation, which also correlated with detection of the maximallevel of IFN-� mRNA (a 31.3-fold increase; P � 0.003). On theother hand, the accompanied elevation (a 17-fold increase) inTNF-� mRNA did not achieve a statistically different level(P � 0.07) (Fig. 5A, right panel).
In contrast to CD4 and CD8, the level of CD3 mRNAshowed a distinctive peak (a 2.9-fold increase; P � 0.03) at 48to 72 h p.i. (Fig. 5A, left panel). Subsequently, the CD3 ex-pression level became again augmented beginning at week3 p.i., preceding the rise in CD4 and CD8 transcription byapproximately 1 week. Then, the expression profiles of CD3,CD4, and CD8 paralleled each other both during and after theacute phase of hepatitis (Fig. 5A, right panel). These resultssuggested that upregulated intrahepatic expression of IFN-�(Fig. 4B, left panel) detected at 48 to 72 h p.i. reflected acti-vation of cells of the innate immune system rather than con-ventional T-cell subsets; however, a contribution of the lattercannot be completely excluded. In the same time period, atendency to elevate transcription of TNF-� (a 2.2-fold increaseover the preinfection level) was also observed, but this increasedid not achieve a statistically meaningful difference (P � 0.08)(Fig. 5A, left panel). The augmented coincident expression ofCD3, CD1d (Fig. 3B, left panel), and IFN-� (Fig. 4B, leftpanel) at 48 to 72 h p.i. implied that NKT cells also becameactivated soon after exposure to pathogenic hepadnavirus.
In addition, the enhanced expression of CD3, preceding therise in CD4 and CD8 expression during the acute phase ofWHV hepatitis (Fig. 5A, right panel) and coinciding with theaugmented transcription of NKp46, may suggest that the acti-vation of both NKp46-positive NK cells and CD3-positive NKT
FIG. 5. Expression of T-cell-affiliated genes in sequential liver samples collected from animals infected with a liver-pathogenic dose of WHV.Profiles of expression of CD3, CD4, and CD8 T-cell markers and TNF-� (A) and cytotoxic effector molecules CD95L mRNA and perforin mRNA(B) are shown. Data shown represent mean expression levels and are presented relative to the maximum level detected for each gene analyzed.Mean levels of intrahepatic WHV DNA were determined and are shown for reference (see the legend to Fig. 3).
VOL. 82, 2008 EARLY INTRAHEPATIC IMMUNITY IN HEPADNAVIRUS INFECTION 8585
on Decem
ber 11, 2012 by guesthttp://jvi.asm
.org/D
ownloaded from

cells was taking place before major infiltration of the liver withCD4� and CD8� T lymphocytes (Fig. 5A, right panel). Inaddition, it appears that CD3-positive NKT cells at this rela-tively late stage of infection, i.e., week 3 p.i., were respondingby producing IL-4 (Fig. 4B, right panel).
Upregulated transcription of CD95L and perforin corre-lates with early immune response to WHV. The intrahepaticexpression of perforin was significantly augmented (1.9-fold;P � 0.05) at 48 to 72 h p.i. (Fig. 5B, left panel). To ascertainwhether an increase in the local cytotoxic activity in the livermight be responsible for the transient decrease in hepaticWHV DNA, expression of CD95L, an effector molecule capa-ble of inducing death of hepatocytes, which are naturally en-dowed with CD95 (49), was quantified by real-time RT-PCR.As shown in Fig. 5B (left panel), CD95L was significantly (P �0.03) upregulated (2.6-fold) at 48 to 72 h p.i., suggesting thatindeed augmented hepatic cytotoxicity, possibly mediated byboth perforin and CD95L, might contribute to the transientdepletion of WHV DNA seen at 48 to 72 h p.i. In the timeperiod after week 2 p.i., CD95L expression in the liver becamenoticeably enhanced, reaching a maximum (a 4.8-fold increase;P � 0.01) at week 4 p.i. (Fig. 5B, right panel). Surprisingly, thisCD95L mRNA peak preceded both the expansion of virus inhepatic tissue and the rise in CD4 and CD8 mRNA levelscoinciding with the appearance of histologically evident AH(Fig. 5A, right panel). On the other hand, this peak of CD95Lexpression occurred together with the upregulated transcrip-tion of CD3, suggesting a possible contribution of NKT cells tothe increased intrahepatic detection of CD95L. However, wehave previously reported that hepatocytes also constitutivelyexpress CD95L (18). Thus, it cannot be completely excludedthat an increase in the intrahepatic CD95L mRNA level wasdue at least in part to the elevated transcription of CD95L inhepatocytes. It is potentially possible, since IFN-� upregulatesCD95L transcription in hepatocytes and intrahepatic expres-sion of this cytokine was augmented (P � 0.032) between 3 and6 h p.i. (Fig. 4B, left panel).
Liver immune activation is reproducibly induced soon afterinvasion with WHV. To further ascertain that WHV inducedactivation of an intrahepatic immune response as early as 72 hp.i., two additional healthy woodchucks were intravenouslyinjected with 1.1 � 1010 vge of the WHV/tm3 inoculum andanalyzed at 3 days p.i. In agreement with the first set of data,significant elevations in CD3 (P � 0.0001), NKp46 (P � 0.03),and CD1d (P � 0.0001) expression levels were detected 72 hafter injection with virus, suggesting again an accumulationand/or local activation of NK and NKT cells (Fig. 6A). How-ever, transcription of CD4 and CD8 was also meaningfullyupregulated (by 47% and 21%; P � 0.001 and 0.003, respec-tively) compared with the hepatic expression of the genes priorto WHV infection (Fig. 6A). Despite these increases, we didnot notice any morphological evidence of lymphomononuclearinfiltrations, suggesting that this augmented expression couldbe due to the activation of the cells already residing within theliver.
Quantification of the cytokine expression at 72 h p.i. alsoconfirmed the data obtained from the first experiment demon-strating significant elevation of IFN-� and a trend toward aug-mented expression of IL-4, possibly reflecting activation ofNKT cells (Fig. 6B). In this supplementary experiment, both
IFN-� (P � 0.004) and IL-4 (P � 0.0001) transcription wassignificantly augmented over the preinfection levels. In addi-tion, a meaningful (P � 0.0001) increase in TNF-� may reflectactivation of intrahepatic macrophages, as was previously sug-gested in regard to a transient elevation in IL-12 mRNA (Fig.3A, left panel) at 3 to 6 h p.i., which had subsided by 72 h p.i.(Fig. 3A, left panel, and 6B). Similarly, as shown in Fig. 4C (leftpanel), detection of OAS mRNA was significantly (P � 0.0017)elevated at 72 h (Fig. 6B), confirming that the invading WHVis recognized by effector immune cells. Finally, expression ofboth the cytotoxic effector molecules CD95L (P � 0.0001) andperforin (P � 0.047) was significantly elevated at 72 h p.i. (Fig.6C), raising again the possibility that antiviral cytotoxic mech-anisms also are activated in the liver at this very early stage ofhepadnavirus infection.
Infectious WHV is required to activate early hepatic im-mune response. To exclude the possibility that exposure tocomponents of serum carrying the virus but not to WHV itselfmight be responsible for activating the intrahepatic immuneresponse, additional control experiments were performed us-ing an inactivated WHV inoculum or healthy woodchuck se-rum. For this purpose, a sample of woodchuck serum servingas WHV/tm3 inoculum and containing 1.1 � 1010 WHV vgeand serum from a healthy woodchuck were treated with pso-ralen under UV light. As shown in Fig. 6D, inactivated WHVinoculum or similarly treated control normal woodchuck se-rum was unable to upregulate expression of the hepatic genes,which have been previously found to be significantly aug-mented at 72 h p.i. Specifically, there were no increases inIFN-� or CD3 mRNA levels and no induction of OAS (Fig.6D). These results confirmed that productive WHV infectionbut not exposure to serum components or circulating viralantigens was responsible for activation of intrahepatic immuneresponses as found in our study.
DISCUSSION
A unique feature of hepadnaviral hepatitis is a prolongedincubation period where no apparent clinical symptoms orbiochemical manifestations of liver injury are evident. Previousstudies, using both the woodchuck model of hepatitis B (11, 34,35) and HBV-infected chimpanzees (47, 62), have suggestedthat viral replication in the liver remains largely undetectableuntil 3 to 4 weeks p.i., at which time exponential virus expan-sion leads to infection of all or almost all hepatocytes. Thesestudies have also demonstrated that antiviral immunity, medi-ated predominantly by virus-specific CD8� cytotoxic T lym-phocytes via both noncytolytic and cytotoxic mechanisms, isfinally responsible for downregulation of hepadnaviral replica-tion and clinical recovery from hepatitis. Furthermore, manip-ulations of the antiviral immune response in HBV-transgenicmice have suggested an involvement of innate immune cellsubsets in inhibition of viral replication (22, 23). Previous ob-servations with HBV-infected chimpanzees (62) and WHV-infected woodchucks (20) have also suggested an involvementof the innate immune system in controlling hepadnavirus in-fection. However, these studies commenced evaluation of virusreplication and intrahepatic immune responses not earlier than1 week postinfection (62).
Since the half-life of HBV in serum is estimated to be as
8586 GUY ET AL. J. VIROL.
on Decem
ber 11, 2012 by guesthttp://jvi.asm
.org/D
ownloaded from

FIG. 6. Challenge with WHV but not with inactivated virus or normal woodchuck serum upregulates genes indicative of activation of hepaticimmune response. For panels A to C, two adult healthy woodchucks were injected with WHV 72 h prior to euthanasia. Intrahepatic expressionof genes encoding immune cell subsets (A), antiviral and proinflammatory cytokines and OAS (B), or cytotoxic effector molecules (C) werequantified by real-time RT-PCR. Data represent the mean gene expression levels determined for both woodchucks, with each cDNA sampleanalyzed for expression of a particular gene in triplicate. In control experiments (D), woodchucks were inoculated with 0.5 ml healthy woodchuckserum or with 0.5 ml of serum used as WHV inoculum containing 1 � 1010 vge which were treated with psoralen and UV as described in Materialsand Methods. Animals were sacrificed 72 h p.i., and hepatic transcription levels of the genes indicated were quantified by real-time RT-PCR. Geneexpression levels shown in panels A to D are presented relative to those determined in liver biopsy samples obtained from the respective animalsprior to injection with the infectious inoculum, inactivated inoculum, or inactivated normal woodchuck serum. Differences marked with an asteriskwere significant at P values indicated in Results.
VOL. 82, 2008 EARLY INTRAHEPATIC IMMUNITY IN HEPADNAVIRUS INFECTION 8587
on Decem
ber 11, 2012 by guesthttp://jvi.asm
.org/D
ownloaded from

short as 4 h (46), although it might even be shorter (13), itcould be expected that infection of hepatocytes may occurpromptly after exposure to virus, at least in situations when thehost is exposed to liver-pathogenic doses of virions (41). Fur-thermore, it is acknowledged that cells of the innate immunesystem are activated within minutes to hours following invasionwith viral pathogens (8). Taken together, we hypothesized thatinoculation with WHV doses known to induce serologicallyand histologically evident AH, i.e., exceeding 103 to 104 virions(41), should result in activation of the hepatic innate immunity,although one pertinent study using three chimpanzees infectedwith HBV, in which expression of the innate immune response-affiliated genes was assessed by cDNA microarray analysis(61), suggested otherwise. To investigate these issues, a largecohort of WHV-naive woodchucks was infected with a well-characterized WHV inoculum containing 1.1 � 1010 DNase-protected vge per dose. This large cohort of animals permittedreliable determination of viral kinetics and the status of intra-hepatic immune activation starting as early as 1 h p.i.
Our quantitative analysis, applying a real-time PCR assay,demonstrated WHV DNA in serum (4 � 106 vge/ml) andliver tissue (6 � 105 vge/�g total DNA) at 1 h after injectionwith virus. However, these high levels of the WHV genomesdetected almost immediately after inoculation almost certainlyoriginated to a large degree, if not entirely, from carryover ofthe inoculum injected. On the other hand, quantification ofWHV RNA provided a direct insight into the status of virusreplication in hepatic tissue. At 1 h p.i., WHV transcripts werefound at levels approximating 200 to 400 copies per �g totalliver RNA. This indicated that the virus was able to enterhepatocytes, repair its partially double-stranded DNA, andtranscribe DNA to mRNA within 1 h after injection. In thisregard, our study is the first where the evidence of such earlyreplication of hepadnavirus in vivo has been documented. Inour work, viral mRNA was detected by sensitive real-timeRT-PCR (sensitivity of �200 vge/�g RNA). In one pertinentbut not fully compatible study, in which HBV RNA was eval-uated by an RNase protection assay with HBV-infected chim-panzees, HBV pregenomic RNA transcripts were detected inthe liver beginning at 3 to 4 weeks p.i., with subsequent expo-nential expansion between weeks 4 and 6 p.i. (61). The differ-ence between ours and the study mentioned is almost certainlydue to the greater sensitivity of the PCR-based WHV mRNAdetection. Our finding of WHV replication in the liver as earlyas 1 h p.i. is not unique and appears to be compatible withreplication kinetics delineated for some other viruses. For ex-ample, evidence of de novo synthesis of measles virus RNA inHeLa cells was found as early as 2 h p.i. when analyzed byreal-time RT-PCR; however, the earlier time points postinocu-lation were not examined in this study (52). Also, transcriptionof the nucleopolyhedrovirus in its host was detected at 1 h p.i.by RT-PCR (14).
Our attempt to detect WHV cccDNA in the liver in the firstfew hours after inoculation was not successful. This replicativeintermediate of the WHV genome, which constitutes an obli-gate prerequisite for the generation of hepadnaviral mRNAtranscripts (32), was for the first time identified at 18 h p.i. Thediscrepancy between the time of the earliest detection of WHVmRNA and WHV cccDNA was not surprising and was mostcertainly related to a greater sensitivity of the WHV mRNA
RT-PCR assay than the PCR assay available for WHVcccDNA detection (at least 5- to 10-fold) and to naturallyoccurring lower copy numbers of WHV cccDNA than WHVmRNA in infected cells. In one related study, in which earlykinetics of duck HBV replication were investigated using invitro-infected primary duck hepatocytes examined by classicalSouthern blot hybridization methods, virus cccDNA and sin-gle-stranded DNA, both indicative of active replication, weredetected at 48 h p.i. (53).
While the hepatic load of WHV mRNA transcripts progres-sively increased starting from 1 h p.i., there was only a slightparallel increase in the WHV DNA level until 3 weeks p.i.compared with that detected at 48 to 72 h p.i. From week 3 to6 weeks p.i., a strong coordinated expansion of levels of WHVDNA and mRNA was apparent, suggesting exponential viralreplication. This result is in agreement with previous findingsfrom HBV infection showing that exponential viral replicationincludes proportional increases in expression of both hepad-naviral genomes and replicative intermediates (47, 62). Subse-quently, a parallel increase in viral RNA transcripts and DNA,although of a lower magnitude, continued until 8 to 9 weeksp.i. However, there was a noticeable transient, but not statis-tically meaningful, decrease in the detection of both nucleicacid forms in the liver and WHV DNA in serum around week6 p.i. From week 10 p.i. forward, a progressive decline inhepatic loads of WHV RNA and DNA occurred, lasting until30 weeks p.i. Nonetheless, traces of WHV DNA and RNAremained detectable in hepatic tissue until the end of theobservation period, which was as long as 3 years p.i. in someanimals. This finding was consistent with the results of previousstudies where the life-long persistence of low-level replicationof infectious WHV after seemingly complete serological andbiochemical resolution of AH was documented (10, 21, 42;reviewed in reference 37).
The microenvironment of the liver displays unique immu-nological properties which have been ascribed to hepatic APC,including liver sinusoidal endothelial cells and Kupffer cells,and to the disproportionate occurrence of NK and NKT cells(reviewed in reference 54). Furthermore, recruitment of NKand NKT cells into the liver from the splenic compartment hasbeen observed following viral infection (57). Our results sug-gested that WHV infection resulted in apparent sequentialactivation of APC and NK or NKT cells within the liver, lead-ing to a temporary decrease in the viral load. Specifically, asignificant (P � 0.004) one-log decrease in hepatic WHV DNAcontent was detected in all animals whose livers were sampledbetween 48 to 72 h after infection. The hepatic WHV DNAlevel returned to that seen prior to this decrease approximately24 h later, implying that the rebound was due to active WHVreplication. This temporary reduction of WHV DNA was as-sociated with significantly augmented hepatic transcription ofantiviral mediators, such as IFN-� and OAS, cellular markersCD1d and CD3, and cytolytic effector molecules CD95L andperforin. This strongly argues that an activation of intrahepaticinnate immunity caused this transient but significant decline inthe viral load, although without apparent modification in thelevel of virus transcription. These results are the first of thiskind, and they do not conform with those reported for exper-imental HBV infection in chimpanzees, which suggested thatinnate immunity is not activated by hepadnaviral infection
8588 GUY ET AL. J. VIROL.
on Decem
ber 11, 2012 by guesthttp://jvi.asm
.org/D
ownloaded from

(61). However, the aforementioned study began examinationof the gene expression in serial liver biopsies from 1 week p.i.and applied less-sensitive microarray analysis, while evaluationof the host response in our study was commenced within 1 hafter WHV inoculation and applied 5- to 10-fold more-sensi-tive real-time RT-PCR assays.
Experimental evidence accumulated from infections withother viral pathogens clearly indicates that NK and NKT cellshave the ability to respond to virus by production of IFN-� orby acquisition of a cytotoxic function within minutes or hoursafter infection (8). In this regard, recognition of viral antigensby the activating NK receptor NKp46 has been implicated as akey stimulus during influenza virus infection (31). Our datademonstrated that as early as 3 to 6 h after exposure to WHV,there was a significant increase in intrahepatic expression ofIFN-�. This was accompanied by trends, although withoutreaching statistically significant difference, toward elevated ex-pression of NKp46 (a 2.5-fold induction) and perforin (a 3.4-fold induction), a key effector molecule mediating NK cellcytotoxicity. This may suggest that NK cells are activated al-most immediately following WHV infection, although the cur-rent data are not conclusive in this regard. These events oc-curred in parallel with a significant upregulated expression ofIL-12, implying that the initial production of IFN-� may haveaugmented the activation of APC.
Based on detection of WHV transcription shortly after ex-posure and the fact that hepadnaviral envelope proteins mayactivate NKT cells (1), it is reasonable to suggest that earlyactivation of NK cells could lead to enhanced presentation ofWHV antigens to CD1d-restricted NKT cells, culminating inelevations of intrahepatic IFN-� and IL-4 at 48 to 72 h p.i. Insupport of this possibility, CD3 expression was also found toexhibit a distinct peak (a 2.9-fold increase comparing to thepreinfection level) at 48 to 72 h p.i. (see Fig. 5A, left panel).Since CD1d-restricted NKT cells express a T-cell receptor(TCR) comprised of TCR�/ chains in combination with theCD3 receptor complex (25), significant elevations in CD3 butnot CD4 or CD8 T-cell markers could be interpreted in sup-port of the idea that WHV infection also activated intrahepaticNKT cells, resulting in the increased expression of IFN-�. Ithas been shown that activation of CD1d-restricted NKT cellsinhibits viral replication in HBV-transgenic mice via noncyto-pathic mechanisms mediated by IFN-� (22). Overall, the re-sults obtained suggest that WHV infection shortly after inva-sion may first activate NK cells and subsequently NKT cells,with the latter possibly contributing to a transient decrease inviral DNA. The activation of the intrahepatic innate immunitywas transient, waning by 72 h p.i. Therefore, it might not besurprising that this innate immune response was undetected inthe liver in previous studies which commenced evaluation ofexpression of the genes affiliated with this response at 1 weekafter inoculation of chimpanzees with HBV (61).
Infection with lymphocytic choriomeningitis virus has beenshown to induce NKT-cell activation, resulting in IFN-� andIL-4 expression, which is immediately followed by a decrease intheir levels and a subsequent increase several weeks later (19,28). Interestingly, peak expression of the NK marker, NKp46,occurred in our study at 3 weeks p.i., coinciding with significantelevations in IL-4 expression, in the absence of upregulatedtranscription of IFN-�. Although we cannot determine the
cellular site of augmented expression of IL-4, the histologicalanalysis showed a lack of lymphomononuclear inflammatoryinfiltrations in the liver at that time. Thus, NKT cells mayrepresent the predominant cell type responsible for the in-creased hepatic expression of IL-4 in our study. Since specificantibodies for detection of woodchuck NKT cells or IL-4 arecurrently lacking, the explanation of this possibility will requirefurther investigation when such reagents become available.Furthermore, WHV infection skewing toward the T-helper-cell type 2 response, characterized by IL-4 and the absence ofIFN-� expression, is another enticing possibility.
Despite activation of innate immune cell subsets in the liverduring days following WHV infection, coinciding with signifi-cant reductions in viral load, this initial antiviral responsewaned and failed to promptly induce a CD4� and CD8� T-cellresponse until 5 to 6 weeks later. These findings are in contrastwith those encountered during infections with other viralpathogens, which tend to induce timely sequential activation ofinnate and adaptive immune cell subsets, leading normally tothe self-resolution of acute infection. The prolonged period ofantigen-specific immunological ignorance to hepadnaviral in-fection, as depicted in our study by a lack of hepatic expressionof CD4 and CD8 T-cell marker genes and an absence of liverinflammation following activation of the innate immune re-sponse or in the studies by others as a lack of hepadnavirus-specific T-cell responses (4, 35), may be partially explained bythe tolerance-inducing effect of the liver. The capacity of theliver to induce tolerance to oral or allogeneic antigens is nowwell recognized (3, 5, 27). It is thought that this occurs viaseveral mechanisms, including suboptimal T-cell primingand induction of T-cell anergy (reviewed in references 6 and12). In our study, the expression of CD4 and CD8 wastransiently elevated immediately following infection. In ad-dition, inflammatory mediators, including IFN-�, have beenshown to influence the expression of adhesion molecules onendothelial cells, which have been implicated in mediatingT-cell trapping in the liver (33). Furthermore, transient ac-tivation of antigen-specific T cells has been observed inTCR-transgenic mouse models, wherein cognate antigenpresentation was restricted to hepatocytes, leading to dys-functional priming of naive T cells (5, 7). Thus, initial trap-ping of CD4 and CD8 T cells, followed by suboptimal prim-ing or deletion, may potentially facilitate hepadnaviralsubversion of the adaptive immune response.
In summary, our findings provide new insights into the fea-tures of the immune responses associated with hepadnaviralinfection. The data obtained indicate that infection induced bya liver-pathogenic dose of hepadnavirus rapidly initiates viralreplication in hepatic tissue and activates the local immunesystem. The infection appears to almost immediately stimulateintrahepatic innate immune cells, including APC, NK, andNKT cells, which coincides with a transient but profound sup-pression of the hepatic virus load. However, in contrast withother viral infections, this very early immune activity does notprecipitate a swift adaptive T-cell immune response. The rea-son behind this is unknown, but it could be due to as yetunidentified viral factors or a consequence of the liver’s abilityto induce immune tolerance.
VOL. 82, 2008 EARLY INTRAHEPATIC IMMUNITY IN HEPADNAVIRUS INFECTION 8589
on Decem
ber 11, 2012 by guesthttp://jvi.asm
.org/D
ownloaded from

ACKNOWLEDGMENTS
We thank Colleen L. Trelegan for her expert technical assistance,Luke Grenning for assistance during woodchuck laparotomies, andJudy Foote for histological processing of liver biopsies. We also thankJulia Pohling for her contribution to identification of woodchuck IL-8gene sequence.
This research was supported by grant MOP-14818 from the Cana-dian Institutes of Health Research (to T.I.M.). C.S.G. was supported inpart by a Canadian Liver Foundation doctoral fellowship. T.I.M. is theCanada Research Chair (Tier 1) in Viral Hepatitis/Immunology, sup-ported by the Canadian Institutes of Health Research and the CanadaFoundation for Innovation.
REFERENCES
1. Baron, J. L., L. Gardiner, S. Nishimura, K. Shinkai, R. Locksley, and D.Ganem. 2002. Activation of a nonclassical NKT cell subset in a transgenicmouse model of hepatitis B virus infection. Immunity 16:583–594.
2. Bendelac, A., P. B. Savage, and L. Teyton. 2007. The biology of NKT cells.Annu. Rev. Immunol. 25:297–336.
3. Berg, M., G. Wingender, D. Djandji, S. Hegenbarth, F. Momburg, G. Ham-merling, A. Limmer, and P. Knolle. 2006. Cross-presentation of antigensfrom apoptotic tumor cells by liver sinusoidal endothelial cells leads totumor-specific CD8� T cell tolerance. Eur. J. Immunol. 36:2960–2970.
4. Bertoletti, A., and C. Ferrari. 2003. Kinetics of the immune response duringHBV and HCV infection. Hepatology 38:4–13.
5. Bertolino, P., D. G. Bowen, G. W. McCaughan, and B. Fazekas de St Groth.2001. Antigen-specific primary activation of CD8� T cells within the liver.J. Immunol. 166:5430–5438.
6. Bertolino, P., G. W. McCaughan, and D. G. Bowen. 2002. Role of primaryintrahepatic T-cell activation in the ‘liver tolerance effect.’ Immunol. CellBiol. 80:84–92.
7. Bertolino, P., M. C. Trescol-Biemont, and C. Rabourdin-Combe. 1998.Hepatocytes induce functional activation of naive CD8� T lymphocytes butfail to promote survival. Eur. J. Immunol. 28:221–236.
8. Biron, C. A., K. B. Nguyen, G. C. Pien, L. P. Cousens, and T. P. Salazar-Mather. 1999. Natural killer cells in antiviral defense: function and regula-tion by innate cytokines. Annu. Rev. Immunol. 17:189–220.
9. Chisari, F. V., and C. Ferrari. 1995. Hepatitis B virus immunopathogenesis.Annu. Rev. Immunol. 13:29–60.
10. Coffin, C. S., T. N. Pham, P. M. Mulrooney, N. D. Churchill, and T. I.Michalak. 2004. Persistence of isolated antibodies to woodchuck hepatitisvirus core antigen is indicative of occult infection. Hepatology 40:1053–1061.
11. Cote, P. J., B. E. Korba, R. H. Miller, J. R. Jacob, B. H. Baldwin, W. E.Hornbuckle, R. H. Purcell, B. C. Tennant, and J. L. Gerin. 2000. Effects ofage and viral determinants on chronicity as an outcome of experimentalwoodchuck hepatitis virus infection. Hepatology 31:190–200.
12. Crispe, I. N. 2003. Hepatic T cells and liver tolerance. Nat. Rev. Immunol.3:51–62.
13. Dandri, M., J. M. Murray, M. Luetgehetmann, T. Volz, A. Lohse, and J.Petersen. 2008. Virion half-life in chronic hepatitis B infection is stronglycorrelated with viral load. J. Hepatol. 48(Suppl.):S30–S31.
14. Duffy, S. P., E. M. Becker, B. H. Whittome, C. J. Lucarotti, and D. B. Levin.2007. In vivo replication kinetics and transcription patterns of the nucle-opolyhedrovirus (NeabNPV) of the balsam fir sawfly, Neodiprion abietis.J. Gen. Virol. 88:1945–1951.
15. Guidotti, L. G., K. Ando, M. V. Hobbs, T. Ishikawa, L. Runkel, R. D.Schreiber, and F. V. Chisari. 1994. Cytotoxic T lymphocytes inhibit hepatitisB virus gene expression by a noncytolytic mechanism in transgenic mice.Proc. Natl. Acad. Sci. USA 91:3764–3768.
16. Guidotti, L. G., and F. V. Chisari. 2001. Noncytolytic control of viral infec-tions by the innate and adaptive immune response. Annu. Rev. Immunol.19:65–91.
17. Guidotti, L. G., R. Rochford, J. Chung, M. Shapiro, R. Purcell, and F. V.Chisari. 1999. Viral clearance without destruction of infected cells duringacute HBV infection. Science 284:825–829.
18. Guy, C. S., J. Wang, and T. I. Michalak. 2006. Hepatocytes as cytotoxiceffector cells can induce cell death by CD95 ligand-mediated pathway. Hepa-tology 43:1231–1240.
19. Hobbs, J. A., S. Cho, T. J. Roberts, V. Sriram, J. Zhang, M. Xu, and R. R.Brutkiewicz. 2001. Selective loss of natural killer T cells by apoptosis follow-ing infection with lymphocytic choriomeningitis virus. J. Virol. 75:10746–10754.
20. Hodgson, P. D., M. D. Grant, and T. I. Michalak. 1999. Perforin and Fas/Fasligand-mediated cytotoxicity in acute and chronic woodchuck viral hepatitis.Clin. Exp. Immunol. 118:63–70.
21. Hodgson, P. D., and T. I. Michalak. 2001. Augmented hepatic interferongamma expression and T-cell influx characterize acute hepatitis progressingto recovery and residual lifelong virus persistence in experimental adultwoodchuck hepatitis virus infection. Hepatology 34:1049–1059.
22. Kakimi, K., L. G. Guidotti, Y. Koezuka, and F. V. Chisari. 2000. Natural
killer T cell activation inhibits hepatitis B virus replication in vivo. J. Exp.Med. 192:921–930.
23. Kakimi, K., T. E. Lane, F. V. Chisari, and L. G. Guidotti. 2001. Cutting edge:inhibition of hepatitis B virus replication by activated NK T cells does notrequire inflammatory cell recruitment to the liver. J. Immunol. 167:6701–6705.
24. Kobayashi, Y. 2008. The role of chemokines in neutrophil biology. Front.Biosci. 13:2400–2407.
25. Kronenberg, M., and L. Gapin. 2002. The unconventional lifestyle of NKTcells. Nat. Rev. Immunol. 2:557–568.
26. Lew, Y. Y., and T. I. Michalak. 2001. In vitro and in vivo infectivity andpathogenicity of the lymphoid cell-derived woodchuck hepatitis virus. J. Vi-rol. 75:1770–1782.
27. Limmer, A., J. Ohl, G. Wingender, M. Berg, F. Jungerkes, B. Schumak, D.Djandji, K. Scholz, A. Klevenz, S. Hegenbarth, F. Momburg, G. J. Hammer-ling, B. Arnold, and P. A. Knolle. 2005. Cross-presentation of oral antigensby liver sinusoidal endothelial cells leads to CD8 T cell tolerance. Eur.J. Immunol. 35:2970–2981.
28. Lin, Y., T. J. Roberts, C. R. Wang, S. Cho, and R. R. Brutkiewicz. 2005.Long-term loss of canonical NKT cells following an acute virus infection.Eur. J. Immunol. 35:879–889.
29. Ma, X., and G. Trinchieri. 2001. Regulation of interleukin-12 production inantigen-presenting cells. Adv. Immunol. 79:55–92.
30. Maini, M. K., C. Boni, C. K. Lee, J. R. Larrubia, S. Reignat, G. S. Ogg, A. S.King, J. Herberg, R. Gilson, A. Alisa, R. Williams, D. Vergani, N. V. Naou-mov, C. Ferrari, and A. Bertoletti. 2000. The role of virus-specific CD8(�)cells in liver damage and viral control during persistent hepatitis B virusinfection. J. Exp. Med. 191:1269–1280.
31. Mandelboim, O., N. Lieberman, M. Lev, L. Paul, T. I. Arnon, Y. Bushkin,D. M. Davis, J. L. Strominger, J. W. Yewdell, and A. Porgador. 2001.Recognition of haemagglutinins on virus-infected cells by NKp46 activateslysis by human NK cells. Nature 409:1055–1060.
32. Mason, W. S., M. S. Halpern, J. M. England, G. Seal, J. Egan, L. Coates, C.Aldrich, and J. Summers. 1983. Experimental transmission of duck hepatitisB virus. Virology 131:375–384.
33. Mehal, W. Z., F. Azzaroli, and I. N. Crispe. 2001. Antigen presentation byliver cells controls intrahepatic T cell trapping, whereas bone marrow-de-rived cells preferentially promote intrahepatic T cell apoptosis. J. Immunol.167:667–673.
34. Menne, S., and P. J. Cote. 2007. The woodchuck as an animal model forpathogenesis and therapy of chronic hepatitis B virus infection. World J.Gastroenterol. 13:104–124.
35. Menne, S., J. Maschke, M. Lu, H. Grosse-Wilde, and M. Roggendorf. 1998.T-cell response to woodchuck hepatitis virus (WHV) antigens during acuteself-limited WHV infection and convalescence and after viral challenge.J. Virol. 72:6083–6091.
36. Menne, S., C. A. Roneker, M. Roggendorf, J. L. Gerin, P. J. Cote, and B. C.Tennant. 2002. Deficiencies in the acute-phase cell-mediated immune re-sponse to viral antigens are associated with development of chronic wood-chuck hepatitis virus infection following neonatal inoculation. J. Virol. 76:1769–1780.
37. Michalak, T. I. 2007. Characteristics and consequences of experimentaloccult hepatitis B virus infection in the woodchuck model of hepatitis B.Curr. Topics Virol. 6:1–13.
38. Michalak, T. I. 1998. The woodchuck animal model of hepatitis B. ViralHepat. Rev. 4:139–165.
39. Michalak, T. I., and B. Lin. 1994. Molecular species of hepadnavirus coreand envelope polypeptides in hepatocyte plasma membrane of woodchuckswith acute and chronic viral hepatitis. Hepatology 20:275–286.
40. Michalak, T. I., B. Lin, N. D. Churchill, P. Dzwonkowski, and J. R. Desousa.1990. Hepadna virus nucleocapsid and surface antigens and the antigen-specific antibodies associated with hepatocyte plasma membranes in exper-imental woodchuck acute hepatitis. Lab. Investig. 62:680–689.
41. Michalak, T. I., P. M. Mulrooney, and C. S. Coffin. 2004. Low doses ofhepadnavirus induce infection of the lymphatic system that does not engagethe liver. J. Virol. 78:1730–1738.
42. Michalak, T. I., I. U. Pardoe, C. S. Coffin, N. D. Churchill, D. S. Freake, P.Smith, and C. L. Trelegan. 1999. Occult lifelong persistence of infectioushepadnavirus and residual liver inflammation in woodchucks convalescentfrom acute viral hepatitis. Hepatology 29:928–938.
43. Michalak, T. I., C. Pasquinelli, S. Guilhot, and F. V. Chisari. 1994. HepatitisB virus persistence after recovery from acute viral hepatitis. J. Clin. Investig.93:230–239.
44. Moretta, A., E. Marcenaro, S. Parolini, G. Ferlazzo, and L. Moretta. 2008.NK cells at the interface between innate and adaptive immunity. Cell DeathDiffer. 15:226–233.
45. Mullan, P. B., A. M. Hosey, N. E. Buckley, J. E. Quinn, R. D. Kennedy, P. G.Johnston, and D. P. Harkin. 2005. The 2�,5�-oligoadenylate synthetase/RNaseLpathway is a novel effector of BRCA1- and interferon-gamma-mediatedapoptosis. Oncogene 24:5492–5501.
46. Murray, J. M., R. H. Purcell, and S. F. Wieland. 2006. The half-life ofhepatitis B virions. Hepatology 44:1117–1121.
8590 GUY ET AL. J. VIROL.
on Decem
ber 11, 2012 by guesthttp://jvi.asm
.org/D
ownloaded from

47. Murray, J. M., S. F. Wieland, R. H. Purcell, and F. V. Chisari. 2005.Dynamics of hepatitis B virus clearance in chimpanzees. Proc. Natl. Acad.Sci. USA 102:17780–17785.
48. Nakamura, I., J. T. Nupp, M. Cowlen, W. C. Hall, B. C. Tennant, J. L. Casey,J. L. Gerin, and P. J. Cote. 2001. Pathogenesis of experimental neonatalwoodchuck hepatitis virus infection: chronicity as an outcome of infection isassociated with a diminished acute hepatitis that is temporally deficient forthe expression of interferon gamma and tumor necrosis factor-alpha mes-senger RNAs. Hepatology 33:439–447.
49. Ogasawara, J., R. Watanabe-Fukunaga, M. Adachi, A. Matsuzawa, T. Ka-sugai, Y. Kitamura, N. Itoh, T. Suda, and S. Nagata. 1993. Lethal effect ofthe anti-Fas antibody in mice. Nature 364:806–809.
50. Penna, A., M. Artini, A. Cavalli, M. Levrero, A. Bertoletti, M. Pilli, F. V.Chisari, B. Rehermann, G. Del Prete, F. Fiaccadori, and C. Ferrari. 1996.Long-lasting memory T cell responses following self-limited acute hepatitisB. J. Clin. Investig. 98:1185–1194.
51. Penna, A., G. Del Prete, A. Cavalli, A. Bertoletti, M. M. D’Elios, R. Sor-rentino, M. D’Amato, C. Boni, M. Pilli, F. Fiaccadori, and C. Ferrari. 1997.Predominant T-helper 1 cytokine profile of hepatitis B virus nucleocapsid-specific T cells in acute self-limited hepatitis B. Hepatology 25:1022–1027.
52. Plumet, S., W. P. Duprex, and D. Gerlier. 2005. Dynamics of viral RNAsynthesis during measles virus infection. J. Virol. 79:6900–6908.
53. Qiao, M., C. A. Scougall, A. Duszynski, and C. J. Burrell. 1999. Kinetics ofearly events in duck hepatitis B virus replication in primary duck hepato-cytes. J. Gen. Virol. 80:2127–2135.
54. Racanelli, V., and B. Rehermann. 2006. The liver as an immunological organ.Hepatology 43:S54–S62.
55. Rehermann, B., C. Ferrari, C. Pasquinelli, and F. V. Chisari. 1996. Thehepatitis B virus persists for decades after patients’ recovery from acute viral
hepatitis despite active maintenance of a cytotoxic T-lymphocyte response.Nat. Med. 2:1104–1108.
56. Rehermann, B., P. Fowler, J. Sidney, J. Person, A. Redeker, M. Brown, B.Moss, A. Sette, and F. V. Chisari. 1995. The cytotoxic T lymphocyte responseto multiple hepatitis B virus polymerase epitopes during and after acute viralhepatitis. J. Exp. Med. 181:1047–1058.
57. Salazar-Mather, T. P., and K. L. Hokeness. 2003. Calling in the troops:regulation of inflammatory cell trafficking through innate cytokine/chemo-kine networks. Viral Immunol. 16:291–306.
58. Sitia, G., M. Isogawa, K. Kakimi, S. F. Wieland, F. V. Chisari, and L. G.Guidotti. 2002. Depletion of neutrophils blocks the recruitment of antigen-nonspecific cells into the liver without affecting the antiviral activity ofhepatitis B virus-specific cytotoxic T lymphocytes. Proc. Natl. Acad. Sci. USA99:13717–13722.
59. Thimme, R., S. Wieland, C. Steiger, J. Ghrayeb, K. A. Reimann, R. H.Purcell, and F. V. Chisari. 2003. CD8(�) T cells mediate viral clearance anddisease pathogenesis during acute hepatitis B virus infection. J. Virol. 77:68–76.
60. Wang, J., S. A. Gujar, L. Cova, and T. I. Michalak. 2007. Bicistronic wood-chuck hepatitis virus core and gamma interferon DNA vaccine can protectfrom hepatitis but does not elicit sterilizing antiviral immunity. J. Virol.81:903–916.
61. Wieland, S., R. Thimme, R. H. Purcell, and F. V. Chisari. 2004. Genomicanalysis of the host response to hepatitis B virus infection. Proc. Natl. Acad.Sci. USA 101:6669–6674.
62. Wieland, S. F., H. C. Spangenberg, R. Thimme, R. H. Purcell, and F. V.Chisari. 2004. Expansion and contraction of the hepatitis B virus transcrip-tional template in infected chimpanzees. Proc. Natl. Acad. Sci. USA 101:2129–2134.
VOL. 82, 2008 EARLY INTRAHEPATIC IMMUNITY IN HEPADNAVIRUS INFECTION 8591
on Decem
ber 11, 2012 by guesthttp://jvi.asm
.org/D
ownloaded from





























