Embed Size (px)
Citation preview

INFECTION AND IMMUNITY, Apr. 2002, p. 1936–1948 Vol. 70, No. 40019-9567/02/$04.00�0 DOI: 10.1128/IAI.70.4.1936–1948.2002Copyright © 2002, American Society for Microbiology. All Rights Reserved.
In Vivo Clearance of an Intracellular Bacterium, Francisella tularensisLVS, Is Dependent on the p40 Subunit of Interleukin-12 (IL-12)
but Not on IL-12 p70Karen L. Elkins,* Allison Cooper, Susan M. Colombini, Siobhán C. Cowley, and Tara L. Kieffer
Laboratory of Mycobacteria, Division of Bacterial, Parasitic, and Allergenic Products, CBER/FDA, Rockville, Maryland 20852
Received 26 September 2001/Returned for modification 20 December 2001/Accepted 10 January 2002
To determine the role of interleukin-12 (IL-12) in primary and secondary immunity to a model intracellularbacterium, we have comprehensively evaluated infection with Francisella tularensis LVS in three murine modelsof IL-12 deficiency. Mice lacking the p40 protein of IL-12 (p40 knockout [KO] mice) and mice treated in vivowith neutralizing anti-IL-12 antibodies survived large doses of primary and secondary LVS infection but nevercleared bacteria and exhibited a chronic infection. In dramatic contrast, mice lacking the p35 protein (p35 KOmice) of heterodimeric IL-12 readily survived large doses of primary sublethal LVS infection as well asmaximal secondary lethal challenge, with only a slight delay in clearance of bacteria. LVS-immune wild-type(WT) lymphocytes produced large amounts of gamma interferon (IFN-�), but p35 KO and p40 KO lympho-cytes produced much less; nonetheless, similar amounts of NO were found in all cultures containing immunelymphocytes, and all immune lymphocytes were equally capable of controlling intracellular growth of LVS invitro. Purified CD4� and CD8� T cells from both WT and p40 KO mice controlled intracellular growth, eventhough T cells from WT mice produced much more IFN-� than those from p40 KO mice, and p40 KO T cellsdid not adopt a Th2 phenotype. Thus, while IL-12 p70 stimulation of IFN-� production may be important forbacteriostasis, IL-12 p70 is not necessary for appropriate development of LVS-immune T cells that are capableof controlling intracellular bacterial growth and for clearance of primary or secondary LVS infection. Instead,an additional mechanism dependent on the IL-12 p40 protein, either alone or in another complex such as thenewly discovered heterodimer IL-23, appears to be responsible for actual clearance of this intracellularbacterium.
People with defects in interleukin-12 (IL-12) production orIL-12 receptor expression, as well as in expression of gammainterferon (IFN-�) receptors, appear to be unusually suscepti-ble to Mycobacteria and Salmonella infections (9, 24, 33). How-ever, the specific contributions of each cytokine to susceptibil-ity remain incompletely understood. IFN-� is clearly a keycytokine in responses to intracellular pathogens such as Myco-
bacteria and Salmonella, in part through activation of macro-phages that results in nitric oxide (NO) production (at least inmice) and killing of intracellular parasites (reviewed in refer-ence 39). IL-12 is a 70-kDa cytokine comprised of two disul-fide-linked proteins (p35, which is constitutively expressed, andp40, which is inducible). IL-12 has a variety of biological func-tions, including stimulation of natural killer cell activity andinduction of Th1 T-cell development (reviewed in reference16). IL-12 production by antigen-presenting cells (includingmacrophages, dendritic cells, and B cells) greatly increasesIFN-� production by natural killer cells and T cells. IL-12 alsoupregulates expression of its own receptor on activated T cells,NK cells, and activated B cells, resulting in further IFN-�production (41). Thus, this positive feedback loop alone mightreadily explain the importance of IL-12 in intracellular infec-tions, through regulation of IFN-� production.
Experimental studies on the role of IL-12 in intracellular
infections have been performed using several different modelsof IL-12 deficiency, including in vivo depletion of IL-12 fromnormal mice by treatment with neutralizing anti-IL-12 antibod-ies (49); targeted mutation of the p35 chain of the IL-12 mol-ecule in the mouse genome, resulting in p35 knockout (KO)mice (30); and targeted mutation of the p40 chain (p40 KOmice) (26). In some cases, infection of any IL-12-deficientmouse with a pathogen that stimulates cell-mediated immunityresulted in death. These include Leishmania major infection inp35 KO, p40 KO, and anti-IL-12-treated mice (28, 30, 40) andToxoplasma gondii infection in p40 and anti-IL-12-treated mice(17, 50). In other cases, there are discrepancies in the pheno-type of infection between p35 KO and p40 KO mice. Forexample, p40 KO mice infected intravenously with the fungalpathogen C. neoformans exhibited higher infection burdens,poorer granuloma formation in lungs, and earlier death thanp35 KO mice, although both died more rapidly than wild-type(WT) mice (8). Of note, IFN-� production was deficient butcomparable in both types of KO mice (8). These results sug-gested a more complex picture of IL-12 dependence thanmight be expected based only on IL-12’s role in Th1 T-celldevelopment and IFN-� production.
To further study the contributions of IL-12 and its subunitsto protective immunity to intracellular bacteria, we have char-acterized murine infection with Francisella tularensis LVS in allthree models of IL-12 deficiency. To our knowledge, this is thefirst such comprehensive evaluation of primary and secondaryintracellular bacterial infection using the same infections in all
* Corresponding author. Mailing address: LOM/DBPAP/CBER/FDA, 1401 Rockville Pike, HFM 431, Rockville, MD 20852. Phone:(301) 496-0544. Fax: (301) 402-2776. E-mail: [email protected].
1936

three circumstances. LVS is a well-characterized intracellularbacterium that replicates in macrophages and disseminates toorgans of the reticuloendothelial system (primarily the spleen,liver, lung, and lymph nodes [for a review, see reference 45]).Murine LVS infection initiated intradermally (i.d.) or subcu-taneously is sublethal, while infection initiated intraperitone-ally (i.p.) is lethal (13, 15). Similar to virtually all other intra-cellular pathogens, innate resistance to primary sublethal (i.d.)infection with LVS is clearly dependent on early production ofIFN-� and tumor necrosis factor alpha (TNF-�) (1, 11, 12, 25),and IL-12 is produced within a day after primary or secondaryLVS infection (43); specific long-term protective immunity isdependent on Th1 T cells (51). Here we show that anti-IL-12-treated or p40 KO mice infected with LVS exhibit a chronicinfection despite development of normal T-cell function, whileresolution of LVS infection in p35 KO mice is nearly normal.Thus, clearance of this intracellular bacterial infection is notdependent on IL-12 p70 but on an unrelated function of p40.
MATERIALS AND METHODS
Mice. Specific-pathogen-free male BALB/cByJ and C57BL/6J mice were pur-
chased from Jackson Laboratories (Bar Harbor, Maine). Male IL-12a� (p35)
and IL-12b� (p40) KO mice in a C57BL/6J background were purchased from the
Induced Mutant Resource of Jackson Laboratories; male IL-12b� (p40) KO
mice in a BALB/c background were a generous gift from Dorothy Scott, Center
for Biologics Research and Evaluation (CBER), Food and Drug Administration
(FDA). All mice were housed in sterile microisolator cages in a barrier environ-
ment at the CBER, fed autoclaved food and water ad libitum, and routinely
tested for common murine pathogens by a diagnostic service provided by the
Division of Veterinary Services, CBER. Within an experiment, all WT and KO
mice were carefully age matched (� 1 week), as some age-related differences in
responses to F. tularensis LVS have been previously noted. In conducting the
research described in this report, the investigators adhered to a protocol ap-
proved by the Animal Care and Use Committee of CBER.
Bacteria and growth conditions. F. tularensis LVS (ATCC 29684; American
Type Culture Collection, Rockville, Md.) was cultured on modified Mueller-
Hinton (MH) agar plates or in modified MH broth (Difco Laboratories, Detroit,
Mich.) supplemented with ferric pyrophosphate and IsoVitalex as previously
described (Becton Dickinson, Cockeysville, Md.) (3, 15). One-milliliter aliquots
of bacteria were frozen in broth alone at �70°C and periodically thawed for use,
and viable bacteria were quantified by plating serial dilutions on MH agar plates.
The number of CFU after thawing varied less than 10% over a 12-month period.
In vivo bacterial infections. Mice were given 0.5 ml i.p. or 0.1 ml i.d. of the
indicated dilution of LVS; actual doses of inoculated bacteria were simulta-
neously determined by plate count. All materials, including bacteria, were di-
luted in phosphate-buffered saline (PBS) (BioWhittaker, Walkersville, Md.) con-
taining �0.01 ng of endotoxin per ml. In vivo organ burden and serum studies
used groups of three to five mice, as indicated. Experiments enumerating num-
bers of CFU in organs of various mice were performed as previously described
(15); briefly, spleens, livers, and lungs were removed aseptically and emulsified in
a Stomacher (Tekmar, Cincinnati, Ohio) in 5 to 10 ml of sterile PBS, and
appropriate dilutions were plated on MH plates.
Antibodies and in vivo antibody treatments. The following neutralizing anti-
bodies, purified and in azide-free and low-endotoxin format, used for blocking
studies were purchased from BD PharMingen (San Diego, Calif.): anti-IFN-�
(clone R4-6A2), anti-IL-12 (clone C17.8, rat immunoglobulin G2a [IgG2a]), and
anti-IL-4 (clone 11B11, rat IgG1). Antibodies for in vivo depletion of IL-12
(clones C15.6 and C15.1, both rat IgG1 [49]) were produced as ascites in BALB/c
nu/nu mice, precipitated from clarified ascites with 50% ammonium sulfate,
quantified using a capture enzyme-linked immunosorbent assay (ELISA) specific
for rat IgG1 using monoclonal rat IgG1 as a standard (PharMingen), and tested
for endotoxin levels using a kit from BioWhittaker; all ascites preparations
contained �10 EU of endotoxin per ml. Hybridoma cells producing both anti-
IL-12 antibodies were a generous gift of Maria Wysocka and Giorgio Trincheri
(Wistar Institute, Philadelphia, Pa.). Anti-IL-4 antibody (clone 11B11) used for
in vivo treatments was a generous gift from Jerko Barbic and Mary Leef and was
also produced as ascites and tested as described above. To deplete mice of IL-12,
mice were treated i.p. with 500 �g each of C15.6 and C15.1 monoclonal anti-
bodies or control anti-IL-4 antibodies on days �1 and 0 relative to the day of
infection with LVS and then weekly thereafter. The treatment regimen used,
employing a combination of two anti-IL-12 monoclonal antibodies on days �1
and 0 relative to infection and weekly thereafter, has previously been shown to
effectively neutralize in vivo IL-12 bioactivity from peripheral sites in mice (40,
49).
Characterization of antibody response. Sera were obtained via the lateral tail
vein and pooled from the indicated groups of mice both before (prebleed) and
after LVS infection at the indicated time points. Titers of specific anti-LVS
serum antibodies were determined by ELISA using Immulon 1 plates coated
with whole LVS bacteria and isotype-specific detection antibodies as previously
described (37). End-point titer was defined as the lowest dilution of immune
serum that had a mean optical density at 405 nm that was greater than the mean
value of the matched dilution of normal prebleed mouse serum plus 3 standard
deviations of the mean and was also greater than 0.050 (37).
In vitro assessment of control of intracellular bacterial growth in bone mar-
row-derived macrophages (BMM�) and T-cell enrichment. The in vitro culture
system used to analyze the ability of immune lymphocytes to control intracellular
replication of LVS bacteria and validation of the culture system’s reflection of
known parameters of in vivo control of bacterial growth have been described in
detail elsewhere (4). Briefly, BMM derived from femurs of the indicated mice
were cultured in 24-well plates (Costar, Corning, N.Y.) in complete medium
(Dulbecco’s modified Eagle’s medium, 10% fetal bovine serum, glutamine, and
nonessential amino acids) containing 10% L929 supernatant for 7 to 8 days, until
a confluent monolayer was obtained. Macrophage monolayers were infected with
F. tularensis LVS at a multiplicity of infection of 1:20 (bacteria to BMM) for 2 h
and then treated for 45 min with medium containing 50 �g of gentamicin per ml
to eliminate extracellular bacteria. Single-cell suspensions of lymphocytes (5
106/well; approximately one lymphocyte to two BMM) obtained by standard
techniques from either naive or LVS-immune mice (mice previously infected
with a sublethal dose of LVS i.d., as indicated) were then added to duplicate or
triplicate infected macrophage cultures. CD4� and CD8� T cells were enriched
from LVS-immune splenic lymphocyte preparations using a MACS Midi system
and the appropriate magnetic beads according to the manufacturer’s instructions
(Miltenyi Biotec, Auburn, Calif.); the purity of the resulting T-cell preparations
was assessed by flow cytometry as previously described (51) using the appropriate
fluorochrome-labeled antibodies (BD PharMingen) in one- and two-color stain-
ing protocols. Following incubation of the infected macrophage-lymphocyte cul-
tures, supernatants were harvested after 72 h and stored at �70°C until assay
(see below). Growth of LVS in BMM was monitored as previously described
(4) by lysing cultures with water at the indicated time points and plating serial
dilutions of the lysate on MH agar plates.
Quantification of cytokines and NO in BMM� culture supernatants. Culture
supernatants were assayed for IFN-�, IL-12, IL-18, TNF-�, IL-4, and IL-10 by
standard sandwich ELISAs. All antibody pairs and recombinant standards were
purchased from BD PharMingen, and ELISAs were performed according to the
manufacturer’s instructions. Cytokines were quantified by comparison to recom-
binant standards using four-parameter fit regression in the SoftMax Pro ELISA
analysis software (Molecular Devices, Sunnyvale, Calif.).
NO was detected in culture supernatants by the Griess reaction as previously
described (20). Briefly, equal volumes of culture supernatants, diluted as neces-
sary, were incubated with commercial Griess reagent (Sigma, St. Louis, Mo.) for
5 min at room temperature, and absorbance at 490 nm was measured. NO2 was
quantified by comparison to serially diluted NaNO2 as a standard using four-
parameter fit regression as described above.
RESULTS
Characterization of primary i.d. F. tularensis LVS infection
in anti-IL-12-treated normal mice. Following in vivo depletionof IL-12 from normal mice by anti-IL-12 treatment, mice wereinfected with sublethal (104 or 105) i.d. doses of F. tularensis
LVS or a dose (106) that approaches the i.d. 50% lethal dose(LD50) (4, 10, 13, 15). Serum samples were obtained bothbefore and after LVS infection, and survival was monitored;other mice were sacrificed at various time intervals to deter-mine organ burdens. Overall survival results are shown in Ta-ble 1. Mice treated with either PBS or anti-IL-4 antibodies andinfected with LVS exhibited a typical dose-dependent survivalpattern, and their survival rates at the highest dose were not
VOL. 70, 2002 F. TULARENSIS LVS CLEARANCE DEPENDS ONLY ON IL-12 p40 1937

significantly different from each other (by Fisher’s exact test;two-sided P value of �0.05). Anti-IL-12-treated mice exhibiteddecreased overall survival of the highest dose studied, 106 LVSi.d., compared to either PBS-treated mice, anti-IL-4-treatedmice, or both control groups combined (P � 0.05 in all threecases). However, there were no differences in survival betweencontrol or anti-IL-12-treated mice at the lower two doses ofLVS i.d. studied (P � 0.05 in all cases).
Impacts on both organ burdens and circulating cytokinelevels in anti-IL-12-treated mice were also detected. Bacterialburdens in the spleens of mice treated with anti-IL-12 andinfected with 106 LVS i.d. were not different from those ofcontrol mice at day 3 (Fig. 1). However, on day 18, numbers ofbacteria in spleens of anti-IL-12-treated mice had declined butwere significantly higher (by Student’s t test; P � 0.05) thannumbers in either PBS-treated or anti-IL-4-treated mice (Fig.1A). Similar trends were evident in the other major sites ofLVS infection, livers and lungs, of anti-IL-12-treated mice(data not shown). Remaining mice sacrificed on days 24 to 28,after the last available treatment with anti-IL-12, still con-tained substantial numbers of bacteria in all organs (data notshown).
Circulating levels of IL-12 and IFN-� in treated and infectedmice were also studied. Large amounts of IL-12 were readilydetected in serum from PBS- and anti-IL-4-treated mice both2 and 7 days after LVS infection; in contrast, no IL-12 wasdetected in anti-IL-12-treated mice at any time point, in this orother similar experiments (data not shown), confirming theeffectiveness and maintenance of depletion (Fig. 1B). On day2, large amounts of IFN-� were present in the sera of PBS- oranti-IL-4-treated mice, but barely detectable amounts thatwere significantly different from those of either control group(by Student’s t test; P � 0.05) were found in the sera ofanti-IL-12-treated mice. By day 7, serum levels of IFN-� hadgreatly declined in PBS- or anti-IL-4-treated mice and wereless than the very low levels still detected in anti-IL-12-treatedmice (Fig. 1C). In more limited studies, similar differences inbacterial burdens and serum cytokine levels were seen in micetreated with anti-IL-12 and infected with a 104 dose of LVS i.d.
(data not shown), despite the lack of impact of anti-IL-12treatment on overall survival of this lower dose of LVS (Table1).
Characterization of primary i.d. and secondary i.p. F. tula-
rensis LVS infection and specific anti-LVS antibody responses
in p35 and p40 IL-12 KO mice. The course of LVS infection inanti-IL-12-treated mice was compared to that in IL-12 KOmice, using mice in which IL-12 production and normal bio-
TABLE 1. Survival of primary LVS infection by BALB/cByJ micetreated with anti-IL-12 antibodies
Treatmenta i.d. dose of LVS (CFU) No. of deaths/total
PBS 104 0/5105 0/5106 6/29
Anti-IL-4 antibody 105 0/5106 7/16
Anti-IL-12 antibodies 104 2/10105 1/5106 20/29
a BALB/cByJ mice were treated i.p. with either 0.5 ml of PBS, 1 mg ofanti-IL-4 antibody or a combination of 500�g of C15.G anti-IL-12 antibody plus500 �g of C15.1 anti-IL-12 antibody on days �1, 0, 7, 14, and 21. On day 0, micewere infected i.d. with the indicated dose of LVS; actual doses were confirmedby plate count at the time of infection. Since deaths of normal mice from i.d. LVSinfection occur between days 4 and 7, survival was monitored through at least day11, at which time selected mice were sacrificed to assess bacterial burdens; mostmice were monitored through day 24 before they were sacrificed. The combinedresults of five independent experiments, all with five or more mice per group, areshown.
FIG. 1. Bacterial burdens and serum cytokine levels in anti-IL-12-treated mice following primary sublethal F. tularensis LVS infection.BALB/cByJ mice were treated with PBS or anti-cytokine antibodies asdescribed in Table 1 and infected with 106 LVS i.d. At the indicatedtimes after infection, groups of four mice were sacrificed for determi-nation of bacterial burdens in spleens (A) and to obtain serum forassessment of IL-12 p40 protein (B) or IFN-� (C) by ELISA. No IL-12p40 or IFN-� was detected in prebleed serum samples from any mice;the limit of detection was approximately 150 pg/ml. Results from onerepresentative experiment of four independent experiments of similardesign are shown.
1938 ELKINS ET AL. INFECT. IMMUN.

activity were eliminated by targeted mutation of either the p35chain of IL-12 (p35 KO mice) or the p40 chain of IL-12 (p40KO mice). Use of KO mice also permitted the study of sec-ondary infection, which was greatly limited in anti-IL-12-treated mice by the difficulty and expense of maintaininganti-IL-12 treatment in vivo. Since C57BL/6J mice are approx-imately 1 log more resistant to both primary and secondaryLVS infection than the BALB/cByJ mice described above (10,13, 15, 25), correspondingly higher doses of LVS were studiedin the C57-p35 KO mice. As shown in Table 2, C57-p35 KOmice readily survived primary i.d. LVS infection at rates com-parable to those of WT C57BL6/J mice, with the exception ofthe highest dose studied, 107 LVS i.d. (significantly different byFisher’s exact test; P � 0.05). Selected mice were sacrificedbetween days 24 and 28, and no bacteria were detected inspleens, livers, and lungs of any C57BL/6J WT or C57-p35 KO
mice (�50 CFU/organ). Further, virtually all C57BL/6J WT aswell as C57-p35 KO mice survived maximal secondary lethalchallenge of 5 106 LVS i.p. (equivalent to 500,000 LD50s)given on day 30 (Table 2), indicating the development of pro-tective immunity in both cases. Importantly, bacteria were alsocleared from all organs of both C57BL/6J WT and C57-p35KO mice within 2 months after secondary lethal challenge (seefootnotes to Table 2). To examine bacterial burdens in moredetail, other mice were infected with 106 LVS i.d. and sacri-ficed at earlier time points. As illustrated in Fig. 2A, C57BL/6JWT and C57-p35 KO mice had comparable levels of bacteriain spleens at day 3. By day 14 C57BL/6J WT mice had nodetectable bacteria in spleens, while C57-p35 KO mice exhib-ited declining numbers of bacteria that were eliminated by day24; similar trends were evident in livers and lungs (data notshown). Sera from both C57BL/6J WT and C57-p35 LVS-
TABLE 2. Survival of primary and secondary LVS infection by IL-12 p35 KO micea
Mouse straini.d. dose of LVS on day 0(primary infection) (CFU)
No. ofdeaths/total
i.p. dose of LVS on day 30(secondary challenge) (CFU)
No. ofdeaths/total
Bacterial clearanceby day 90
C57BL/6J 0 (PBS only) 0/5 5 106 5/5 NA105 2/5 5 106 0/3 Yes106 0/5 5 106 1/5 Yes
3 106 2/8 5 106 0/4b Yes107 1/5 5 106 0/4 Yes
C57-p35 KO 105 0/5 5 106 1/5 Yes106 1/5 5 106 0/4 Yes
3 106 2/8 5 106 0/4b Yes107 5/5 5 106 NA NA
a The indicated mice were infected on day 0 i.d. with the indicated dose of LVS; actual doses were confirmed by plate count at the time of infection. On day 30 afterprimary infection, surviving mice were lethally challenged with 5 106 (500,000 LD50s) CFU of LVS i.p. On day 90 (60 days after secondary challenge), surviving micewere sacrificed and assessed for bacterial burdens. The limit of detection was 50 CFU/organ (1.7 logs). NA, not applicable. The results of two independent experiments,with either five or eight mice per group, are shown.
b Because some mice were sacrificed for use in other experiments, only the total shown were challenged.
FIG. 2. Bacterial burdens and serum cytokine levels in p35 KO mice following primary sublethal F. tularensis LVS infection. C57BL/6J WT orC57-p35 KO mice were infected with 106 LVS i.d. At the indicated times after infection, groups of four mice were sacrificed for determination ofbacterial burdens in spleens (A) and to obtain serum for assessment of IL-12 p40 protein or IFN-� (B) by ELISA; the limit of detection wasapproximately 150 pg/ml. Results from one representative experiment of four independent experiments of similar design are shown.
VOL. 70, 2002 F. TULARENSIS LVS CLEARANCE DEPENDS ONLY ON IL-12 p40 1939

infected KO mice had large amounts of IL-12 p40 protein atday 3 after i.d. LVS infection, but very low levels by day 28(Fig. 2B); as expected, C57-p35 KO mice had no detectablecomplete IL-12 p70 (�50 pg/ml in all cases; data not shown).While sera from LVS-infected C57BL/6J WT mice had largeamounts of serum IFN-� on day 3 that declined below detect-able levels by day 28, sera from LVS-infected C57-p35 KOmice had no detectable IFN-� at either time point (Fig. 2B).
Similar studies were conducted in p40 KO mice, which wereavailable in both the BALB/c and C57BL/6J background. Asshown in Table 3, p40 KO mice survived primary sublethal i.d.LVS infection at rates comparable to those of either strain ofWT mice. However, when selected mice were sacrificed be-tween days 24 and 28, bacteria were detected in spleens, livers,and lungs of p40 KO but not WT mice (see below). Virtually allWT as well as p40 KO mice survived maximal lethal secondaryLVS i.p. challenge given on day 28 for about 2 months afterlethal challenge (Table 3). However, 50 days after secondarylethal challenge some p40 KO mice in the BALB/c backgroundexhibited obvious morbidity, and according to protocol, werehumanely sacrificed (when their ability to obtain food andwater was judged to be compromised). Consistent levels ofbacteria were found in spleens (Fig. 3A) as well as livers andlungs (data not shown) of i.d. LVS-infected BALB/c-p40 KOmice, while no bacteria were detected in comparable BALB/cByJ WT mice (randomly chosen from the matched infectiongroup). Visible signs of illness were not observed in LVS-infected p40 KO mice on the C57BL/6J background, likelyreflecting the slightly greater resistance of mice of that inbredstrain.
To examine the bacterial burdens in p40 KO mice in moredetail, other mice were infected with a sublethal dose of 106
LVS i.d. and sacrificed at various time points after both pri-mary infection and secondary lethal i.p. challenge. As illus-trated in Fig. 3B, i.d. LVS-infected C57BL/6J WT and p40 KOmice had comparable levels of bacteria in spleens on days 3and 7; by day 14, however, C57BL/6J WT mice had no detect-able bacteria in spleens, while C57-p40 mice exhibited detect-able burdens that were maintained through day 24. All remain-ing infected mice survived and were given a maximal secondarylethal challenge on day 30 of 106 CFU i.p. (equivalent to100,000 LD50s in nonimmune mice). C57BL/6J WT micecleared secondary LVS challenge by day 42 (12 days after
challenge), but C57-p40 mice contained large numbers of bac-teria in spleens through day 78 (Fig. 3B). Similar results wereobtained in livers and lungs (data not shown). Sera from LVS-infected C57BL/6J WT mice had large amounts of IL-12 p40protein on day 3 after i.d. LVS infection, but very low levels byday 22. As expected, sera from LVS-infected C57-p40 KO micehad no detectable p40 protein (Fig. 3C) or complete IL-12 p70(�50 pg/ml in all cases; data not shown). However, while serafrom LVS-infected C57BL/6J WT mice had large amounts ofserum IFN-� on day 3 that declined below detectable levels byday 22, sera from LVS-infected C57-p40 KO mice had nodetectable IFN-� at either time point (Fig. 3B).
The quantity and isotype of specific anti-LVS antibodieswere also measured in all three types of mice. Three weeksafter sublethal i.d. LVS infection of C57BL/6J WT mice, seracontained low amounts of IgM (mean end-point titer in serafrom five mice of 1:640)- and IgG2a (1:80)-specific anti-LVSantibodies (but not specific antibodies of other isotypes [36]).Sera from LVS-infected C57-p35 KO mice had somewhathigher anti-LVS IgM levels of 1:1,280, IgG2a titers of 1:1,280,and IgG1 titers of 1:20. In the sera of LVS-infected C57-p40KO mice, anti-LVS IgM titers of 1:2,560, IgG2a titers of1:1,280, and IgG1 titers of 1:640 (but no titers for IgG2b orIgG3) were found.
Role of T cells, cytokines, and NO in control of intracellular
LVS infection in IL-12 KO mice. IL-12 is considered necessaryfor appropriate development of Th1 cells and production ofIFN-� (see reference 16). To examine T-cell development andthe contribution of IFN-� in KO mice more directly, we useda recently developed in vitro model system that assesses theability of immune lymphocytes to control intracellular bacterialgrowth (4). To date, this in vitro culture system has reflectedknown immunological parameters of in vivo immunity to LVS,particularly dependence on CD4� or CD8� T cells in second-ary (memory) responses (4).
In recognition of the possibility that nonclassical lympho-cytes, including ��� T cells and even B cells, might participatein control of intracellular bacterial growth, initially all immunelymphocytes were studied. LVS-immune lymphocytes were ob-tained from spleens of WT and KO mice that survived bothprimary and secondary LVS infections. All in vitro experimentsshown here used cells from C57BL/6J WT and p35 or p40 KOmice in the C57 background, although similar results were
TABLE 3. Survival of primary and secondary LVS infection by IL-12 p40 KO micea
Mouse straini.d. dose of LVS on day 0(primary infection) (CFU)
No. ofdeaths/total
i.p. dose of LVS on day 28(secondary challenge) (CFU)
No. of deaths/total(through day 50)
Bacterial clearanceafter day 50
BALB/cByJ 0 (PBS only) 0/5 106 5/5 NA104 0/8 106 1/8 Yes105 1/8 106 1/7 Yes
C57BL/6J 106 0/8 106 0/4b YesBALB/c-p40 KO 104 0/6 106 2/6 No
105 2/5 106 0/3 No106 0/5 106 3/5 No
C57-p40 KO 106 2/8 106 1/3b No
a The indicated mice were infected i.d. with various doses of LVS on day 0. On day 28 after primary infection, surviving mice were lethally challenged i.p. with 106
LVS, and survival through day 50 was recorded. Visibly ill mice were sacrificed at various time points thereafter (ranging from days 56 to 70), and spleens, livers, andlungs were assessed for bacterial burdens (see Fig. 5). The limit of detection was 50 CFU/organ (1.7 logs). NA, not applicable. The results of two independentexperiments, with either five or eight mice per group, are shown.
b Because some mice were sacrificed for use in other experiments, only the total shown were challenged.
1940 ELKINS ET AL. INFECT. IMMUN.

obtained using cells from BALB/cByJ WT mice and BALB/c-p40 KO mice (data not shown; BALB/c-p35 KO mice were notavailable). Macrophages derived from bone marrow (BMM)of either WT mice or p40 KO mice were infected in vitro withLVS; macrophages from p35 KO mice were not studied, sincethe phenotype of LVS infection in p35 KO mice was similar tothat of WT mice. Immediately after LVS infection, spleniclymphocytes from nonimmune and LVS-immune mice wereadded to infected macrophages. Infected macrophages werelysed to determine the extent of bacterial replication and sur-vival over time, and culture supernatants were collected after72 h. As shown in Fig. 4, LVS replicated exponentially in bothC57BL/6J WT and C57-p40 KO BMM over 3 days, andbacterial growth was essentially unaffected by the addition ofnonimmune WT lymphocytes. However, the addition of im-mune lymphocytes from either WT, p35 KO, or p40 KO miceto either infected WT or p40 KO macrophages controlled
bacterial growth dramatically by 72 h to a comparable degree.Of note, spleens obtained from p40 KO mice themselves con-tained LVS bacteria, due to chronic infection, at the time ofaddition to culture; no effort was made to remove this addi-tional bacterial burden before the addition of cells.
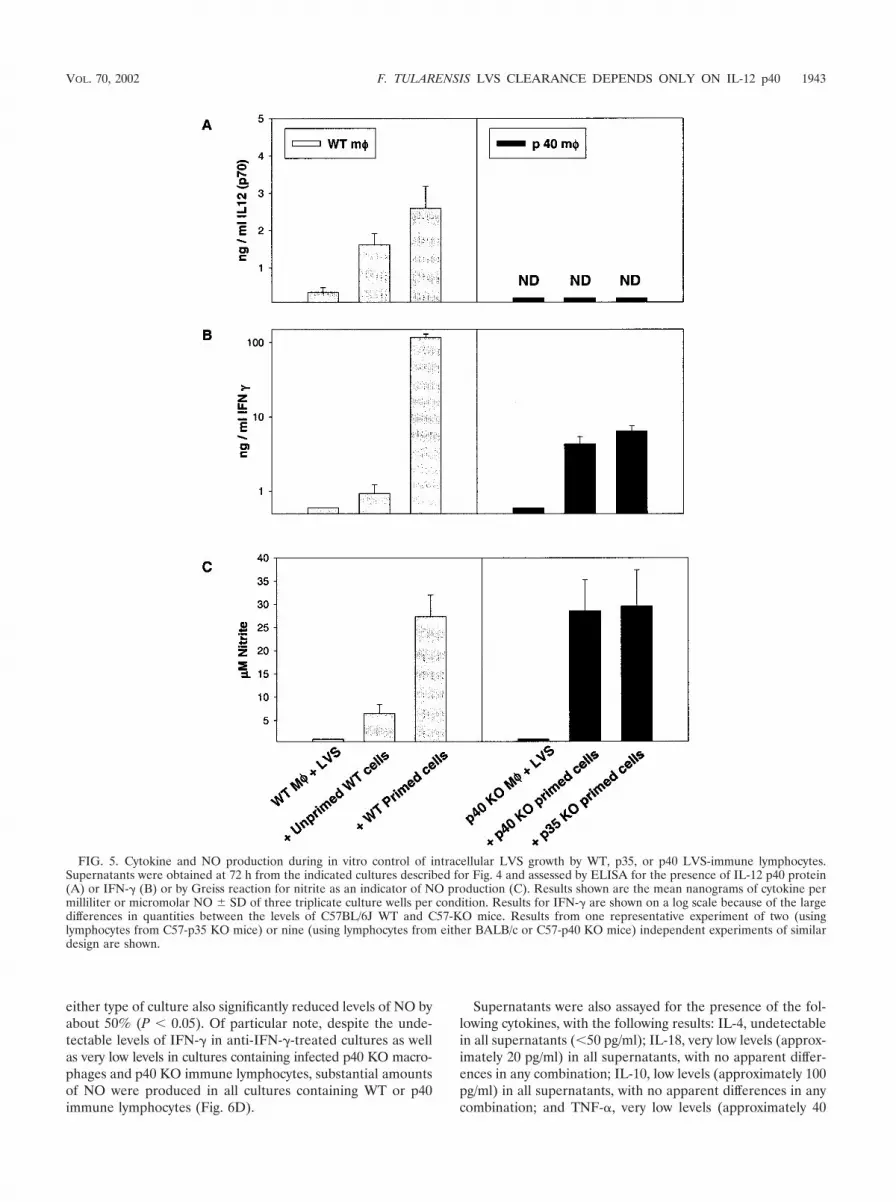
Supernatants obtained after 72 h from these cultures wereassayed for the presence of a panel of cytokines as well as forNO. For clarity, only results from cultures containing LVS-infected WT macrophages with WT lymphocytes or LVS-in-fected p40 KO macrophages with either type of KO lympho-cyte are shown (Fig. 5), although other combinations weretested and similar to the homologous cultures (data notshown). As seen previously (4), infected macrophages fromWT mice produced small amounts of IL-12 that were increasedupon addition of either unprimed or primed cells; as expected,no IL-12 p70 was detected in cultures containing KO macro-phages and lymphocytes (Fig. 5A). The addition of primed, but
FIG. 3. Bacterial burdens and serum cytokine levels in p40 KO mice following primary and secondary F. tularensis LVS infection. BALB/cByJWT or BALB/c-p40 KO mice were infected with 106 LVS i.d. and challenged on day 30 (arrow) with 106 LVS i.p. (1,000,000 LD50s for BALB/cByJmice). At the indicated times after secondary lethal challenge, individual mice were sacrificed for determination of bacterial burdens in spleens(A). Other C57BL/6J WT or C57-p40 KO mice, in groups of three, were sacrificed at the indicated time points after primary or secondary (arrow)LVS infection for determination of bacterial burdens in spleens (B), or serum was obtained after primary LVS infection for assessment of IL-12p40 protein (C, left panel) or IFN-� (C, right panel) by ELISA; the limit of detection was approximately 150 pg/ml. Results from one representativeexperiment of two (B) or three (C) independent experiments of similar design are shown.
VOL. 70, 2002 F. TULARENSIS LVS CLEARANCE DEPENDS ONLY ON IL-12 p40 1941

not unprimed, WT cells to infected WT macrophages resultedin production of large amounts of IFN-� (Fig. 5B) and NO(Fig. 5C). In contrast, much smaller amounts of IFN-� wereproduced in cultures containing KO macrophages and eitherp35 or p40 primed lymphocytes, and the quantities producedwere very similar to each other (Fig. 5B; note the logarithmicscale). Interestingly, despite large differences in the levels ofIFN-� produced, levels of NO produced were comparable incultures containing either WT, p35, or p40 primed lymphocytes(Fig. 5C).
To assess the contribution of IL-12 and IFN-� to the controlof intracellular growth in vitro, neutralizing antibodies to eachcytokine were added at the initiation of cultures, and the im-pact on bacterial growth, cytokine production, and NO pro-duction was assessed. In these studies, we focused on compar-ison of control of intracellular bacterial growth by immunelymphocytes obtained following primary LVS infection of WTand p40 KO mice (since the in vivo phenotype of p40 mice wasdefective, but the phenotype of p35 KO mice was similar tothat of WT mice). For clarity, only results from cultures withLVS-infected C57BL/6J WT macrophages and WT immunelymphocytes or LVS-infected C57-p40 KO macrophages withp40 KO immune lymphocytes at the 72-h time point are shown(Fig. 6). As seen above, addition of both WT and p40 KOprimed lymphocytes to either type of infected macrophageeffectively and comparably controlled the intracellular growthof LVS bacteria (Fig. 6A). Addition of neutralizing anti-IL-12antibodies or isotype-matched control anti-IL-4 antibodies hadno significant impact (P � 0.05 for all combinations) on thecontrol of intracellular bacterial growth by either type of lym-phocytes. In contrast, the addition of anti-IFN-� partially butnot completely reversed the control of intracellular bacterialgrowth. Control of intracellular bacterial growth in the pres-
ence of anti-IFN-� was significantly less than that seen in thepresence of either type of lymphocytes alone (P � 0.05 for allcombinations, by Student’s t test), but bacterial growth in anti-IFN-�-treated cultures was still significantly less than that seenin infected macrophages alone (P � 0.05 for all combinations).Importantly, the relative impact of anti-IFN-� in reversingcontrol of growth was significantly greater in the cultures con-taining LVS-infected p40 KO macrophages and p40 KO im-mune lymphocytes than in the corresponding WT cultures (P
� 0.05 in five of six repeated experiments).When the corresponding production of soluble mediators
was examined, supernatants from WT cultures treated withanti-IL-12 antibodies contained no detectable IL-12, confirm-ing the effectiveness of neutralization, and all cultures contain-ing p40 KO cells produced no p40 (Fig. 6B) or p70 (data notshown), as expected. Throughout, the addition of isotype-matched control anti-IL-4 antibodies had no significant effecton levels of any of the cytokines tested or on NO levels (Fig. 6;also, see below). In WT cultures, the addition of anti-IL-12antibodies greatly reduced the amount of IFN-� produced(Fig. 6C) but not the amount of NO produced (Fig. 6D).However, the addition of anti-IL-12 antibodies to cultures con-taining p40 KO primed cells, in which the amounts of IFN-�produced were very low initially (Fig. 6C), had no significantimpact on production of either IFN-� (Fig. 6C) or NO (Fig.6D).
Supernatants from both WT and p40 cultures treated withanti-IFN-� antibodies contained no detectable IFN-� (Fig.6C), confirming the effectiveness of neutralization (despite thelarge amounts of IFN-� produced in WT cultures). Addition ofanti-IFN-� antibodies significantly reduced the amount ofIL-12 produced in cultures containing WT primed cells, byabout 50% (P � 0.05) (Fig. 6B). Addition of anti-IFN-� to
FIG. 4. LVS-immune lymphocytes from WT, p35, or p40 KO mice control intracellular LVS growth in vitro. BMM were derived from eitherWT C57BL/6J mice (left) or from C57-p40 KO mice (right) and infected with LVS. Splenic lymphocytes from the indicated mice (unprimedC57BL/6J mice or LVS-primed mice [mice that were infected with 106 LVS i.d. on day 0 and then challenged on day 35 with 5 106 LVS i.p.,with spleens removed 4 weeks later]) were added immediately after infection, and growth of bacteria in macrophages was monitored over time.Results shown are the mean CFU/well � standard deviations (SD) of three triplicate culture wells per condition. Results from one representativeexperiment of two (using lymphocytes from C57-p35 KO mice) or nine (using lymphocytes from either BALB/c or C57-p40 KO mice) independentexperiments of similar design are shown.
1942 ELKINS ET AL. INFECT. IMMUN.
either type of culture also significantly reduced levels of NO byabout 50% (P � 0.05). Of particular note, despite the unde-tectable levels of IFN-� in anti-IFN-�-treated cultures as wellas very low levels in cultures containing infected p40 KO macro-phages and p40 KO immune lymphocytes, substantial amountsof NO were produced in all cultures containing WT or p40immune lymphocytes (Fig. 6D).
Supernatants were also assayed for the presence of the fol-lowing cytokines, with the following results: IL-4, undetectablein all supernatants (�50 pg/ml); IL-18, very low levels (approx-imately 20 pg/ml) in all supernatants, with no apparent differ-ences in any combination; IL-10, low levels (approximately 100pg/ml) in all supernatants, with no apparent differences in anycombination; and TNF-�, very low levels (approximately 40
FIG. 5. Cytokine and NO production during in vitro control of intracellular LVS growth by WT, p35, or p40 LVS-immune lymphocytes.Supernatants were obtained at 72 h from the indicated cultures described for Fig. 4 and assessed by ELISA for the presence of IL-12 p40 protein(A) or IFN-� (B) or by Greiss reaction for nitrite as an indicator of NO production (C). Results shown are the mean nanograms of cytokine permilliliter or micromolar NO � SD of three triplicate culture wells per condition. Results for IFN-� are shown on a log scale because of the largedifferences in quantities between the levels of C57BL/6J WT and C57-KO mice. Results from one representative experiment of two (usinglymphocytes from C57-p35 KO mice) or nine (using lymphocytes from either BALB/c or C57-p40 KO mice) independent experiments of similardesign are shown.
VOL. 70, 2002 F. TULARENSIS LVS CLEARANCE DEPENDS ONLY ON IL-12 p40 1943

pg/ml) in all supernatants, with no apparent differences in anycombination.
Thus, LVS-immune splenic lymphocytes from WT, p35 KO,and p40 KO mice were comparable in their ability to controlintracellular bacterial growth (Fig. 4 to 6), even though p40 KOmice were clearly defective in bacterial clearance in vivo whileWT and p35 KO mice were not (Tables 2 and 3; Fig. 2 and 3).Previous data demonstrated that CD4� and CD8� T cells, butnot B cells, were effector cells in this in vitro system when WTcells were studied (4). Here we compared T cells in particularfrom LVS-infected (defective) p40 KO mice to those fromLVS-immune WT (normal) mice. Purified CD4� and CD8� Tcells were prepared from both types of LVS-immune primedspleen cells at 4 to 8 weeks after LVS sublethal infection(priming) and assessed using the homologous LVS-infectedmacrophages (Fig. 7). As seen in Fig. 7A, both WT and p40
KO CD4� and CD8� primed T cells were readily able tocontrol intracellular bacterial growth to a degree comparableto that of unseparated primed spleen cells and were not sig-nificantly different from each other (P � 0.05 in all cases).Levels of IL-12 were moderate in WT T-cell cultures andundetectable, as expected, in all p40 KO cultures (Fig. 7B).Both CD4� and CD8� WT primed T cells produced largeamounts of IFN-�, compared to much lower amounts in p40KO primed lymphocyte or CD4�-T-cell cultures (note logscale); in repeated experiments, both CD4� and CD8� T cellsfrom LVS-immune p40 KO mice produced very low levels ofIFN-� that were near the limit of detection (here, below thelimit of detection for CD8� T cells). IL-4 was undetectablefrom all cultures, whether they contained WT or p40 KOsplenic lymphocytes or separated T-cell subpopulations (�50pg/ml). Finally, levels of NO produced were high and compa-
FIG. 6. Effect of anticytokine treatment on in vitro control of intracellular LVS growth and production of mediators by WT or p40 LVS-immune lymphocytes. BMM were derived from either WT C57BL/6J mice or from C57-p40 KO mice and infected with LVS. Splenic lymphocytesfrom either C57BL/6J WT or C57-p40 KO immune mice (that had received a primary sublethal i.d. LVS infection of 105 CFU 35 days earlier) wereadded immediately after infection. Results using only LVS-infected WT macrophages with WT immune lymphocytes (black bars) or LVS-infectedp40 KO macrophages with p40 KO immune lymphocytes (hatched bars) are shown. Growth of bacteria in macrophages was assessed at 72 h afterinfection (A); results shown are the mean CFU/well � SD of three triplicate culture wells per condition. Supernatants were obtained at 72 h fromthe same cultures and assessed by ELISA for the presence of IL-12 p40 protein (B) or IFN-� (C) or by Greiss reaction for nitrite as an indicatorof NO production (D). Results shown are the mean nanograms of cytokine per milliliter or micromolar NO � SD of three triplicate culture wellsper condition. Results for IFN-� are shown on a log scale because of the large differences in quantities between the levels of WT and KO mice.Results from one representative experiment of six independent experiments of similar design are shown.
1944 ELKINS ET AL. INFECT. IMMUN.

rable in cultures containing unseparated spleen cells from bothWT and p40 KO mice, as well as those containing both types ofCD4� T cells (Fig. 7D); levels of NO were lower but compa-rable in cultures containing CD8� T cells from both types ofmice. Overall, levels of NO were not correlated with IFN-�production from these cultures (Fig. 7C). Although we havebeen unable to obtain sufficient age-matched WT, p35, and p40KO mice to simultaneously prepare purified T-cell subpopula-tions from all three types of mice, more limited studies usingpurified CD4� and CD8� T cells from LVS-immune p35 KOmice with LVS-infected p35 KO macrophages demonstratedthat, as expected, purified p35 KO T cells controlled intracel-lular bacterial growth, comparable to T cells from WT KO
mice, but produced reduced levels of IFN-� compared to cul-tures from WT mice (data not shown).
DISCUSSION
Unlike mice treated with anti-IFN-� or anti-TNF-�, whichdie within 5 to 7 days after primary sublethal LVS infection (1,11, 12, 25), both IL-12 p40 KO mice and mice treated withneutralizing anti-IL-12 antibodies survived and initially con-trolled primary LVS infection; they then exhibited a chronicinfection even following secondary challenge that resulted intolerable, and nearly homeostatic, numbers of bacteria in or-gans (Tables 1 and 3; Fig. 1 and 3). In contrast, p35 KO mice,
FIG. 7. In vitro control of intracellular LVS growth, cytokine production, and NO production by WT or p40 LVS-immune T-cell subpopula-tions. BMM were derived from either WT C57BL/6J mice or C57-p40 KO mice and infected with LVS. Splenic lymphocytes or purified CD4�
or CD8� T cells from either C57BL/6J WT or C57-p40 KO immune mice (that had received a primary sublethal i.d. LVS infection of 105 CFU45 days earlier) were added to the homologous LVS-infected macrophages immediately after infection. By flow cytometry, WT CD4� T cells were94.3% CD4�, WT CD8� T cells were 90.8% CD8�, p40 KO CD4� T cells were 90.4% CD4�, and p40 KO CD8� T cells were 87.3% CD8�. Resultsusing only LVS-infected WT macrophages with WT immune lymphocytes (black bars) or LVS-infected p40 KO macrophages with p40 KO immunelymphocytes (hatched bars) are shown. Growth of bacteria in macrophages was assessed at 72 h after infection (A); results shown are the meanCFU/well � SD of three triplicate culture wells per condition. Supernatants were obtained at 72 h from the same cultures and assessed by ELISAfor the presence of IL-12 p40 protein (B) or IFN-� (C) or by Greiss reaction for nitrite as an indicator of NO production (D). Results shown arethe mean nanograms of cytokine per milliliter or micromolar NO � SD of three triplicate culture wells per condition. Results for IFN-� are shownon a log scale because of the large differences in quantities between the levels of WT and KO mice. Results from one representative experimentof three independent experiments of similar design using WT and p40 KO cultures are shown.
VOL. 70, 2002 F. TULARENSIS LVS CLEARANCE DEPENDS ONLY ON IL-12 p40 1945

which also lack the ability to produce complete IL-12 p70protein, readily survived and cleared both primary and second-ary LVS infection, with only a slight delay in bacterial clear-ance (Table 2; Fig. 2). These results clearly lead to the surpris-ing conclusion that resolution of a classic intracellular bacterialinfection does not depend on IL-12 p70 but instead is depen-dent on a separate positive, not antagonistic (29), activity ofthe p40 protein itself.
Immunity to F. tularensis LVS is considered to be dependenton Th1 T-cell responses and IFN-� production, similar to manyother intracellular pathogens, and IL-12 can clearly be impor-tant in directing optimal Th1 T-cell development (16). How-ever, results using the in vitro culture system demonstratedthat the inability of p40 KO mice to resolve LVS infection wasnot due solely to poor T-cell function or memory, as primed Tcells from p40 KO mice were functionally comparable to thosefrom WT and p35 KO mice in controlling intracellular bacte-rial growth (Fig. 5 to 7). Previous results using this culturesystem demonstrated a close correlation between in vivo and invitro effector mechanisms, but here no difference in the effec-tiveness of WT or p40 T cells could be detected, despite obvi-ous differences in the course of in vivo infection (Table 3, Fig.2 and 3). Of note, not all bacteria are eliminated in this in vitroculture system, even when WT cells are studied and culturesare continued beyond 3 days (see reference 4). To date, wecannot determine whether this is due to inherent limitations inthe efficiency of the culture system or to factors that render itimpossible for this culture system to detect the final mecha-nism(s) that is responsible for full in vivo clearance. As mightbe expected, IFN-� production from both p40 KO mice orcultures was greatly reduced, but there were minimal or nochanges in the magnitude of production of other cytokines(Fig. 3 and 5 to 7) and no obvious Th2 switch in the absence ofIL-12. In other infection models, it is also becoming clear thatabsence of IL-12 does not necessarily result in a switch towardTh2 development. Leishmania infection in mice is indeed as-sociated with increased IL-4 production in IL-12-deficient an-imals (28, 30). On the other hand, there were no dramaticincreases in IL-4, IL-5, and/or IL-13 production in IL-12-defi-cient animals infected with Mycobacterium tuberculosis (7), My-
cobacterium bovis BCG (48), T. gondii (50), Schistosoma man-
soni (22), or viruses (34). Our data indicate that developmentof T cells capable of clearing LVS infection (arguably Th1 cellsalthough they are deficient in IFN-� production) and mainte-nance of LVS T-cell memory does not necessarily requireIL-12 p70; this is in contrast to results obtained using L. major
(44) and T. gondii (50), which suggested that Th1 developmentand maintenance of memory required IL-12.
The lack of change to a Th2 phenotype may also be reflectedin the continued production of larger amounts of IgG2a anti-LVS antibodies in both p35 and p40 LVS-infected KO mice(see text). We previously assumed that IgG2a anti-LVS anti-bodies were related to the large amounts of IFN-� producedafter LVS infection, but these studies suggest instead thateither small amounts of IFN-� are sufficient or that IgG2a is aninherently dominant isotype of specific antibody produced inresponse to chronic stimulation by this pathogen. Of note,other studies in viral and parasitic infection models also dem-onstrate that IgG2a production can be IFN-� independent(27).
There is substantial literature to date on the outcome ofintracellular bacterial and parasitic infections in various mod-els of IL-12 deficiency (see reference 16), with results rangingfrom dramatic to intermediate to minimal impact. In somecases, infection of any IL-12-deficient mouse resulted in death.These include L. major infection in p35 KO, p40 KO, andanti-IL-12-treated mice (28, 30, 40) and T. gondii infection inp40 and anti-IL-12-treated mice (17, 50). Interestingly, infec-tions with obligate intracellular pathogens (including viruses[34]) have been much less affected by IL-12 deficiency. Anti-IL-12-treated mice survived and cleared both Chlamydia pneu-
moniae (18) and Chlamydia trachomatis (35), with only minordifferences from WT mice in magnitude of organ burdens orlength of shedding. Some discrepancies in the phenotype ofinfection between p35 KO and p40 KO mice have also beenreported. Although both types died more rapidly than WTmice, C. neoformans-infected p35 KO mice died more slowly,with fewer obvious pathological differences and lower infectionburdens, than p40 KO mice despite comparable deficiencies inIFN-� production in both types of KO mice (8). Infections withthe classic intracellular bacterium Listeria monocytogenes havenot been comprehensively characterized in all three types ofIL-12 deficiency, but some results suggest that the outcome ofListeria infection may be different between the KO mice and/ormay be highly dose dependent. Anti-IL-12 treatment beforeinfection with a large primary sublethal dose of Listeria (ap-proaching the LD50) resulted in death, while secondary lethalListeria challenge was only minimally affected by anti-IL-12treatment (46, 47). A recent study demonstrated that p35 KOmice died following large sublethal Listeria doses but survivedand cleared lower primary and secondary doses, despite poorIFN-� production (5). Further, p40 KO mice also survived andcleared, with some delay, a low primary dose of Listeria (34).Of particular note, while intravenous M. tuberculosis infectionof p40 KO mice resulted in rapid death (7), very recent resultsindicate that intravenous M. bovis BCG infection was con-trolled well by WT and p35 KO mice but was chronic in p35-p40 double KO mice; further, WT and p35 KO mice given alow-dose aerosol infection with M. tuberculosis exhibitedgreater survival times, stronger delayed-type hypersensitivityresponses, and improved granuloma formation compared toM. tuberculosis-infected p35-p40 double KO mice (23). Al-though p40 single-KO mice were not included in that study, therelative difference in outcome after Mycobacteria infection ap-pears to be quite similar to that seen here with LVS.
While it may appear difficult to generalize about the role ofIL-12 p70 in intracellular infections, many studies of facultativeintracellular parasites are consistent with the central notionthat p40 protein may be necessary for clearance of maximaldoses. Direct comparison of LVS infection in all three modelsof IL-12 deficiency clearly indicates that long-term clearance isdependent on p40 but not on IL-12 p70. In other infectionssuch as Leishmania and Mycobacteria, the role of p40 may beobscured by the absolute dependence on complete IL-12 p70for control of initial primary infection. There are two obviouspossibilities concerning the means by which the IL-12 p40protein, which is present in mouse serum in excess of the p70heterodimer (16), may effect its role. As has been previouslysuggested (19), p40 protein as either a monomer or a ho-modimer may have positive biological activity in vivo that is not
1946 ELKINS ET AL. INFECT. IMMUN.

evident from in vitro studies indicating antagonistic activities(29). Alternatively, p40 may be present in complex with otherproteins. In particular, recent studies have described the com-bination of p40 with a 19-kDa protein to form a new cytokine,designated IL-23 (32). IL-23 is apparently produced by mac-rophages and dendritic cells, and in vitro this molecule sup-ported proliferation of memory T cells; this is therefore anattractive candidate for having a role in T-cell-mediated clear-ance of bacteria. Thus, future studies testing the ability of theclearance-promoting activities of either p40 alone or the p19-p40 complex are in progress.
These studies also raise new questions about the role andrelative importance of IFN-� in intracellular infection. Sinceall three types of IL-12-deficient mice restrict initial bacterialorgan burdens to the same degree as WT mice but producelittle if any systemic IFN-� (Fig. 1 to 3), this initial control ofbacterial replication apparently requires relatively little IFN-�(perhaps locally produced). Thus, further study is necessary todistinguish between the possibilities that relatively low levels ofIFN-� are sufficient to clear LVS infection or that clearance oflate primary or secondary infection is actually independent ofIFN-�. Previous studies clearly demonstrated that initial sur-vival of WT mice was dependent on the availability of IFN-�within the first 2 days after LVS infection (1, 11, 12, 25), as isinitial survival of both p35 and p40 KO mice (S. C. Cowley andK. L. Elkins, manuscript in preparation). However, it is intrigu-ing to note that in LVS-infected WT mice, IFN-� was readilydetected in serum at very early time points only, within 1 to 3days after LVS infection, but not at later time points (Fig. 1 to3). Further, mice depleted of IFN-� immediately before sec-ondary LVS challenge died from higher challenge doses butnot from lower doses (42). Other evidence strongly indicatesthat while IFN-� may be necessary to control intracellularinfection, it is not likely to be sufficient. For example, CD4-deficient mice were clearly defective in their ability to controlprimary M. tuberculosis infection, despite having only minimalor transient differences in IFN-� production compared to WTmice (6, 38). Importantly, IFN-� KO mice vaccinated with anattenuated mutant of L. monocytogenes are nearly normal intheir ability to survive secondary lethal Listeria challenge (2,21). In fact, the relative importance of IFN-� production in lateprimary or secondary intracellular infection has not been wellstudied to date, due in large part to the acute death of IFN-�-deficient mice following primary intracellular infection.
The relative importance of activated macrophage produc-tion of NO in primary or secondary LVS infection also remainsto be revealed. In previous in vitro studies of LVS replicationin resident peritoneal or inflammatory murine macrophages,inhibition of NO partially but not completely reversed controlof intracellular bacterial growth (14). Further, control of LVSgrowth in IFN-�-stimulated alveolar macrophages was mini-mally affected by inhibition of NO (36). Here, there was acorrelation between production of NO and in vitro control ofintracellular LVS growth but not between IFN-� and NO pro-duction (see Fig. 5 to 7). In particular, large amounts of NOwere produced in cultures containing p40 KO immune lym-phocytes and LVS-infected p40 KO macrophages, where min-imal IFN-� was produced (Fig. 5 to 7). Thus, results to datesuggest the existence of significant IFN-�- and NO-indepen-
dent mechanisms in immunity to LVS, but more direct studiesusing inducible NO synthase KO mice are also planned.
Taken together, these data suggest that one mechanism ofcontrol of intracellular LVS growth is due to the well-knownIL-12–IFN-� macrophage activation–NO production cascadebut that another equally important mechanism is independentof these elements and dependent on p40. Thus, initial survivalof LVS infection is dependent on immediate production (with-in the first 2 days) of IFN-�; the cellular source of this earlyIFN-� remains to be directly identified, but is clearly not T cells(11, 12), and in other intracellular infections is proposed to benatural killer cells (39) or even CD8�� lymphoid dendriticcells, which are also an important source of IL-12 (31). Laterproduction of IFN-� is derived from T cells. We suggest thatthe activity of IFN-� is primarily bacteriostatic. A p40-depen-dent function is responsible for ultimate clearance of in vivoinfection, but clearance may be only partially IL-12 p70 and/orIFN-� dependent. This hypothesis is consistent with the obser-vation (Fig. 5 and 6; also see above) that anti-IFN-� antibodytreatment had a greater impact on control of intracellulargrowth by LVS-immune p40 KO lymphocytes (which lack theputative p40-dependent mechanism but have the IFN-�-de-pendent mechanism) than on that by WT lymphocytes (whichhave both mechanisms). Thus, future studies will examine thespecific role and form of p40 activity and the closely relatedquestion of the relative importance of IFN-� throughout LVSinfection.
ACKNOWLEDGMENTS
We are most grateful to Suzanne Epstein and Dorothy Scott forthoughtful and critical reviews of the manuscript; to Dorothy Scott forgenerously providing BALB/c-p40 KO mice; and to Catharine Bosiofor continued insightful discussions.
REFERENCES
1. Anthony, L. S. D., E. Ghadirian, F. P. Nestel, and P. A. L. Kongshavn. 1989.The requirement for gamma interferon in resistance of mice to experimentaltularemia. Microb. Pathog. 7:421–428.
2. Badovinac, V. P., and J. T. Harty. 2000. Adaptive immunity and enhancedCD8� T cell response to Listeria monocytogenes in the absence of perforinand IFN-gamma. J. Immunol. 164:6444–6452.
3. Baker, C. N., D. G. Hollis, and C. Thornsberry. 1985. Antimicrobial suscep-tibility testing of Francisella tularensis with a modified Mueller-Hinton broth.J. Clin. Microbiol. 22:212–215.
4. Bosio, C. M., and K. L. Elkins. 2001. Susceptibility to secondary Francisellatularensis LVS infection in B-cell-deficient mice is associated with neutro-philia but not with defects in specific T-cell-mediated immunity. Infect.Immun. 69:194–203.
5. Brombacher, F., A. Dorfmuller, J. Magram, W. J. Dai, G. Kohler, A.
Wunderlin, K. Palmer-Lehmann, M. K. Gately, and G. Alber. 1999. IL-12 isdispensable for innate and adaptive immunity against low doses of Listeriamonocytogenes. Int. Immunol. 11:325–332.
6. Caruso, A. M., N. Serbina, E. Klein, K. Triebold, B. R. Bloom, and J. L.
Flynn. 1999. Mice deficient in CD4 T cells have only transiently diminishedlevels of IFN-gamma, yet succumb to tuberculosis. J. Immunol. 162:5407–5416.
7. Cooper, A. M., J. Magram, J. Ferrante, and I. M. Orme. 1997. Interleukin 12(IL-12) is crucial to the development of protective immunity in mice intra-venously infected with Mycobacterium tuberculosis. J. Exp. Med. 186:39–45.
8. Decken, K., G. Kohler, K. Palmer-Lehmann, A. Wunderlin, F. Mattner, J.
Magram, M. K. Gately, and G. Alber. 1998. Interleukin-12 is essential for aprotective Th1 response in mice infected with Cryptococcus neoformans.Infect. Immun. 66:4994–5000.
9. Dorman, S. E., and S. M. Holland. 2000. Interferon-gamma and interleu-kin-12 pathway defects and human disease. Cytokine Growth Factor Rev.11:321–333.
10. Elkins, K. L., C. M. Bosio, and T. R. Rhinehart-Jones. 1999. Importance ofB cells, but not specific antibodies, in primary and secondary protectiveimmunity to the model intracellular bacterium Francisella tularensis livevaccine strain. Infect. Immun. 67:6002–6007.
VOL. 70, 2002 F. TULARENSIS LVS CLEARANCE DEPENDS ONLY ON IL-12 p40 1947

11. Elkins, K. L., T. R. Rhinehart-Jones, S. J. Culkin, D. Yee, and R. K. Win-
egar. 1996. Minimal requirements for murine resistance to infection withFrancisella tularensis LVS. Infect. Immun. 64:3288–3293.
12. Elkins, K. L., T. Rhinehart-Jones, C. A. Nacy, R. K. Winegar, and A. H.
Fortier. 1993. T-cell-independent resistance to infection and generation ofimmunity to Francisella tularensis. Infect. Immun. 61:823–829.
13. Elkins, K. L., R. K. Winegar, C. A. Nacy, and A. H. Fortier. 1992. Introduc-tion of Francisella tularensis at skin sites induces resistance to infection andgeneration of protective immunity. Microb. Pathog. 13:417–421.
14. Fortier, A. H., T. Polsinelli, S. J. Green, and C. A. Nacy. 1992. Activation ofmacrophages for destruction of Francisella tularensis: identification of cyto-kines, effector cells, and effector molecules. Infect. Immun. 60:817–825.
15. Fortier, A. H., M. V. Slayter, R. Ziemba, M. S. Meltzer, and C. A. Nacy. 1991.Live vaccine strain of Francisella tularensis: infection and immunity in mice.Infect. Immun. 59:2922–2928.
16. Gately, M. K., L. M. Renzetti, J. Magram, A. S. Stern, L. Adorini, U. Gubler,
and D. H. Presky. 1998. The interleukin-12/interleukin-12-receptor system:role in normal and pathologic immune responses. Annu. Rev. Immunol.16:495–521.
17. Gazzinelli, R. T., M. Wysocka, S. Hayashi, E. Y. Denkers, S. Hieny, P.
Caspar, G. Trinchieri, and A. Sher. 1994. Parasite-induced IL-12 stimulatesearly IFN-gamma synthesis and resistance during acute infection with Tox-oplasma gondii. J. Immunol. 153:2533–2543.
18. Geng, Y., K. Berencsi, A. Gyulai, T. Valy-Nagy, E. Gonczol, and G. Trinch-
ieri. 2000. Roles of interleukin-12 and gamma interferon in murine Chla-mydia pneumoniae infection. Infect. Immun. 68:2245–2253.
19. Germann, T., and E. Rüde. 1995. The IL-12 p40 homodimer as a specificantagonist of the IL-12 heterodimer. Immunol. Today 16:500–503.
20. Green, L. C., A. Wagner, J. Glogowski, P. L. Skipper, J. S. Wishnok, and
S. R. Tannenbaum. 1982. Analysis of nitrate, nitrite, and [15N]nitrate inbiological fluids. Anal. Biochem. 126:131–138.
21. Harty, J. T., and M. J. Bevan. 1995. Specific immunity to Listeria monocy-togenes in the absence of IFN-�. Immunity 3:109–117.
22. Hoffmann, K. F., S. L. James, A. W. Cheever, and T. A. Wynn. 1999. Studieswith double cytokine-deficient mice reveal that highly polarized Th1- andTh2-type cytokine and antibody responses contribute equally to vaccine-induced immunity to Schistosoma mansoni. J. Immunol. 163:927–938.
23. Hölscher, C., R. A. Atkinson, B. Arendse, N. Brown, E. Myburgh, G. Alber,
and F. Brombacher. 2001. A protective and agonistic function of IL-12 p40in mycobacterial infection. J. Immunol. 167:6957–6966.
24. Jouanguy, E., R. Doffinger, S. Dupuis, A. Pallier, F. Altare, and J. L.
Casanova. 1999. IL-12 and IFN-gamma in host defense against mycobacteriaand salmonella in mice and men. Curr. Opin. Immunol. 11:346–351.
25. Leiby, D. A., A. H. Fortier, R. M. Crawford, R. D. Schreiber, and C. A. Nacy.
1992. In vivo modulation of the murine immune response to Francisellatularensis LVS by administration of anticytokine antibodies. Infect. Immun.60:84–89.
26. Magram, J., S. E. Connaughton, R. R. Warrier, D. M. Carvajal, C. Y. Wu, J.
Ferrante, C. Stewart, U. Sarmiento, D. A. Faherty, and M. K. Gately. 1996.IL-12-deficient mice are defective in IFN-gamma production and type 1cytokine responses. Immunity 4:471–481.
27. Markine-Goriaynoff, D., J. T. van der Logt, C. Truyens, T. D. Nguyen, F. W.
Heessen, G. Bigaignon, Y. Carlier, and J. P. Coutelier. 2000. IFN-gamma-independent IgG2.a production in mice infected with viruses and parasites.Int. Immunol. 12:223–230.
28. Mattner, F., K. Di Padova, and G. Alber. 1997. Interleukin-12 is indispens-able for protective immunity against Leishmania major. Infect. Immun. 65:
4378–4383.29. Mattner, F., S. Fischer, S. Guckes, S. Jin, H. Kaulen, E. Schmitt, E. Rüde,
and T. Germann. 1993. The interleukin-12 subunit p40 specifically inhibitseffects of the interleukin-12 heterodimer. Eur. J. Immunol. 23:2202–2208.
30. Mattner, F., J. Magram, J. Ferrante, P. Launois, K. Di Padova, R. Behin,
M. K. Gately, J. A. Louis, and G. Alber. 1996. Genetically resistant micelacking interleukin-12 are susceptible to infection with Leishmania major andmount a polarized Th2 cell response. Eur. J. Immunol. 26:1553–1559.
31. Ohteki, T., T. Fukao, K. Suzue, C. Maki, M. Ito, M. Nakamura, and S.
Koyasu. 1999. Interleukin 12-dependent interferon g production by CD8��
lymphoid dendritic cells. J. Exp. Med. 189:1981–1986.32. Oppmann, B., R. Lesley, B. Blom, J. C. Timans, Y. Xu, B. Hunte, F. Vega, N.
Yu, J. Wang, K. Singh, F. Zonin, E. Vaisberg, T. Churakova, M. Liu, D.
Gorman, J. Wagner, S. Zurawski, Y.-J. Liu, J. S. Abrams, K. W. Moore, D.
Rennick, R. de Wall-Malefyt, C. Hannum, J. F. Bazan, and R. A. Kastelein.
2000. Novel p19 protein engages IL-12 p40 to form a cytokine, IL-23, withbiological activities similar to as well as distinct from IL-12. Immunity 13:
715–725.33. Ottenhoff, T. H., D. Kumararatne, and J. L. Casanova. 1998. Novel immu-
nodeficiencies reveal the essential role of type 1 cytokines in immunity tointracellular bacteria. Immunol. Today 19:491–494.
34. Oxenius, A., U. Karrer, R. M. Zinkernagel, and H. Hengartner. 1999. IL-12is not required for induction of type 1 cytokine responses in viral infections.Immunology 162:965–973.
35. Perry, L. L., K. Feilzer, and H. D. Caldwell. 1997. Immunity to Chlamydiatrachomatis is mediated by T helper 1 cells through IFN-gamma-dependentand -independent pathways. J. Immunol. 158:3344–3352.
36. Polsinelli, T., M. S. Meltzer, and A. H. Fortier. 1994. Nitric oxide-indepen-dent killing of Francisella tularensis by IFN-�-stimulated murine alveolarmacrophages. J. Immunol. 153:1238–1245.
37. Rhinehart-Jones, T. R., A. H. Fortier, and K. L. Elkins. 1994. Transfer ofimmunity against lethal murine Francisella infection by specific antibodydepends on host gamma interferon and T cells. Infect. Immun. 62:3129–3137.
38. Scanga, C. A., V. P. Mohan, K. Yu, H. Joseph, K. Tanaka, J. Chan, and J. L.
Flynn. 2000. Depletion of CD4� T cells causes reactivation of murine per-sistent tuberculosis despite continued expression of interferon gamma andnitric oxide synthase 2. J. Exp. Med. 192:347–358.
39. Schaible, U. E., H. L. Collins, and S. H. Kaufmann. 1999. Confrontationbetween intracellular bacteria and the immune system. Adv. Immunol. 71:
267–377.40. Scharton-Kersten, T., L. C. Afonso, M. Wysocka, G. Trinchieri, and P. Scott.
1995. IL-12 is required for natural killer cell activation and subsequent Thelper 1 cell development in experimental leishmaniasis. J. Immunol. 154:
5320–5330.41. Sinigaglia, F., D. D’Ambrosio, P. Panina-Bordignon, and L. Rogge. 1999.
Regulation of the IL-12/IL-12R axis: a critical step in T-helper cell differ-entiation and effector function. Immunol. Rev. 170:65–72.
42. Sjöstedt, A., R. J. North, and J. W. Conlan. 1996. The requirement of tumournecrosis factor-alpha and interferon-gamma for the expression of protectiveimmunity to secondary murine tularaemia depends on the size of the chal-lenge inoculum. Microbiology 142:1369–1374.
43. Stenmark, S., D. Sunnemark, A. Bucht, and A. Sjöstedt. 1999. Rapid localexpression of interleukin-12, tumor necrosis factor alpha, and gamma inter-feron after cutaneous Francisella tularensis infection in tularemia-immunemice. Infect. Immun. 67:1789–1797.
44. Stobie, L., S. Gurunathan, C. Prussin, D. L. Sacks, N. Glaichenhaus, C. Y.
Wu, and R. A. Seder. 2000. The role of antigen and IL-12 in sustaining Th1memory cells in vivo: IL-12 is required to maintain memory/effector Th1 cellssufficient to mediate protection to an infectious parasite challenge. Proc.Natl. Acad. Sci. USA 97:8427–8432.
45. Tärnvik, A. 1989. Nature of protective immunity to Francisella tularensis.Rev. Infect. Dis. 11:440–451.
46. Tripp, C. S., M. K. Gately, J. Hakimi, P. Ling, and E. R. Unanue. 1994.Neutralization of IL-12 decreases resistance to Listeria in SCID and C.B-17mice. J. Immunol. 152:1883–1888.
47. Tripp, C. S., O. Kanagawa, and E. R. Unanue. 1995. Secondary response toListeria infection requires IFN-� but is partially independent of IL-12. J. Im-munol. 155:3427–3432.
48. Wakeham, J., J. Wang, J. Magram, K. Croitoru, R. Harkness, P. Dunn, A.
Zganiacz, and Z. Xing. 1998. Lack of both type 1 and 2 cytokines, tissueinflammatory responses, and immune protection during pulmonary infectionby Mycobacterium bovis bacille Calmette-Guerin in IL-12-deficient mice.J. Immunol. 160:6101–6111.
49. Wysocka, M., M. Kubin, L. Q. Vieira, L. Ozmen, G. Garotta, P. Scott, and G.
Trinchieri. 1995. Interleukin-12 is required for interferon-gamma produc-tion and lethality in lipopolysaccharide-induced shock in mice. Eur. J. Im-munol. 25:672–676.
50. Yap, G., M. Pesin, and A. Sher. 2000. IL-12 is required for the maintenanceof IFN-gamma production in T cells mediating chronic resistance to theintracellular pathogen Toxoplasma gondii. J. Immunol. 165:628–631.
51. Yee, D., T. R. Rhinehart-Jones, and K. L. Elkins. 1996. Loss of either CD4�
or CD8� T cells does not affect the magnitude of protective immunity to anintracellular pathogen, Francisella tularensis strain LVS. J. Immunol. 157:
5042–5048.
Editor: R. N. Moore
1948 ELKINS ET AL. INFECT. IMMUN.





























