Embed Size (px)
Citation preview

471
Review
www.expert-reviews.com ISSN 1751-2433© 2010 Expert Reviews Ltd10.1586/ECP.10.35
Major depressive disorder (MDD), which is among the leading causes of disease bur-den, remains undertreated as well as under- recognized [1]. Despite the clear efficacy of modern anti depressants, response rates are still unsatisfy-ing [2–4] and successful treatment is accompanied by clear disadvantages, such as a delayed time of onset as well as a variety of minor side effects. Moreover, many patients only show a limited response or fail to respond at all and, unfortu-nately, a high risk of relapse persists even when remission is achieved [5,6].
Besides the well-studied effects of age, weight, comorbidity, gender, nutrition and coadministered drugs [7], individual genetic make-up is being increasingly appreciated as a profound factor which impacts on drug response [8,9]. This recent shift of attention is reflected in the growing number of publications over the past few years (Table 1) [10–12]. However, small effect sizes and a lack of replica-tions limit the clinical applicability of those studies [10–12]. Nevertheless, expectations of the capabili-ties of psycho pharmacogenetics to guide antide-pressant treatment remain high, even though the predictive utility of pharmacogenetic tests is still not ready for widespread clinical use [13,14].
Similar to pharmacogenetic studies, neuro-imaging studies investigating gene effects (imag-ing genetics) and drug response have become
tremendously popular over the last couple of years (Table 2). Those studies have highlighted the importance of brain circuitries of emotion processing, which are modulated by risk genes of depression as well as antidepressive drug effects (Figure 1) [15,16]. Since both the effects of antide-pressants and genes at risk of causing depression converge on almost identical neural substrates, it is tempting to amalgamate these findings with results of pharmacogenetic-association stud-ies in order to provide a better understanding of pharmacogenetic actions on a neurobiologi-cal level and to discuss possible implications for future research, as with the scope of this article. Publications have been selected by the authors from Pubmed according to the following inclu-sion criteria: the genetic variants chosen have been associated with depression and have been implicated in drug response or side effect rates. In addition, functional or anatomical neuro imaging studies have been performed in vivo, investigat-ing those genetic variants as well as antidepressive drug action in adult samples of healthy subjects or mood disorder patients. Although the section ‘Drug effects on regions of emotion processing’ contains several PET studies, the discussion of genetic impact only includes MRI studies, since MRI has become the standard technique used in imaging genetics.
Ulrich Rabl1, Christian Scharinger1, Markus Müller2 andLukas Pezawas†1
1Division of Biological Psychiatry, Department of Psychiatry and Psychotherapy, Medical University of Vienna, Waehringer Guertel 18–20, A-1090 Vienna, Austria2Department of Clinical Pharmacology, Medical University of Vienna, Austria †Author for correspondence:Tel.: +43 140 400 3568 Fax: +43 140 400 3099 [email protected]
Genetic variation of SLC6A4, HTR1A, MAOA, COMT and BDNF has been associated with depression, variable antidepressant drug responses as well as impacts on brain regions of emotion processing that are modulated by antidepressants. Pharmacogenetic studies are using psychometric outcome measures of drug response and are hampered by small effect sizes that might be overcome by the use of intermediate endophenotypes of drug response, which are suggested by imaging studies. Such an approach will not only tighten the relationship between genes and drug response, but also yield new insights into the neurobiology of depression and individual drug responses. This article provides a comprehensive overview of pharmacogenetic, imaging genetics and drug response studies, utilizing imaging techniques within the context of antidepressive drug therapy.
Keywords: amygdala • anterior cingulate cortex • antidepressants • hippocampus • imaging genetics • major depressive disorder • MRI • orbitofrontal cortex • pharmacogenetics
Imaging genetics: implications for research on variable antidepressant drug responseExpert Rev. Clin. Pharmacol. 3(4), 471–489 (2010)
Expert Rev. Clin. Pharmacol. 3(4), (2010)472
Review Rabl, Scharinger, Müller & Pezawas
This article attempts to provide a comprehensive overview of the rapidly growing fields of imaging genetics and pharmaco-genetic studies. Owing to the large body of available literature and space limitations, we will only sporadically discuss the design of, or analytical differences between, studies. In order to facilitate the recognition of study limitations, we will emphasize important issues in the discussion of this article. Since this article aims to amalgamate available evidence regarding the impact of genetic variants and antidepressant drugs on brain systems of emo-tion processing that have been implicated in the neurobiology of depression, we have restricted the number of possible brain regions to those for which overwhelming evidence exists for play-ing a prominent role in antidepressant drug response as well as depression neurobiology [15–17].
Neuroimaging methods Since numerous neuroimaging studies are at the core of this arti-cle, this section aims to enhance readability by providing a brief description of the neuroimaging techniques mentioned in the course of this article.
Functional MRI (fMRI) is currently the most popular in vivo imaging technique and takes advantage of the magnetic proper-ties of hemoglobin [18]. In more detail, neural activity leads to increased oxygen extraction by neurons, which is accompanied by an increase of blood flow resulting in an increased fraction of oxygenated hemoglobin. Since oxygenated and deoxygenated hemoglobin have different magnetic susceptibilities, an increased blood oxygen level-dependent signal can be detected in areas of the brain that become active [18]. Activation in brain regions of
interest is typically induced by specific neuropsychological tasks or stimuli, which are known to be accompanied by neural activity within these specific neural networks, while baseline (resting-state) activations are recorded without the need for any specific task [19,20].
Another method capable of measuring neural activity is PET, a nuclear imaging technique, that utilizes the application of radioactive tracers in order to indirectly assess neural activity, such as regional cerebral blood flow or glucose metabolism as well as fractional receptor binding, reflecting regional receptor distribution [21].
Computational neuroanatomy provides the possibility to detect regional morphometric alterations with MRI. To date, several methods have been developed such as voxel-based morphometry, which allows for a voxel-wise comparison of local tissue volumes (specifically gray matter) in the spatially normalized anatomical images [22,23]. Furthermore, diffusion tensor imaging (DTI) utilizes differences in the directional mobility of water molecules (frac-tional anisotropy) in order to study the orientation and integrity of white matter tracts [24].
Finally, this article also includes studies that have applied advanced MRI techniques such as functional or structural connectivity, which provide information on the inter-regional connection strengths of brain systems [25].
Drug effects on regions of emotion processingAmygdalaThe amygdala is considered to be a critical neural hub involved in both normal human behavior [26] as well as mental illness [27]. Increased amygdala activation has been related to fear condi-tioning and reaction [26,28] as well as to acute selective serotonin-reuptake inhibitor (SSRI) treatment [29–31], and is suggested to be an important endophenotype of acute MDD [17]. Moreover, specifically in children and adolescents, acute SSRI administration has been associated with a temporary increase of anxious [32,33] or even suicidal behavior [34]. Consequently, animal studies have reported an increase of fear following acute SSRI administration, but subsequent normalization under chronic treatment [35,36]. Similarly, studies in healthy humans reported decreases of fear [37] or decreases of amygdala reactivity in response to aversive stim-uli [38–40], and reduced basal glucose metabolism [41] under chronic SSRI treatment. Such an attenuation of amygdala reactivity is not unique to SSRIs, but has also been shown in response to other anxiolytic drugs such as benzodiazepines [42]. In MDD patients, fluoxetine [43], sertraline [44,45] and bupropion [46] have further been reported to normalize exaggerated amygdala activity under chronic treatment conditions, which appears to be tightly related to drug response.
Studies investigating the putative role of amygdala reactiv-ity as an imaging predictor of treatment response in mood and anxiety disorder samples are still limited. However, pretreatment amygdala reactivity has been found to positively correlate with fluoxetine treatment response [47] in pediatric anxiety disorder patients as well as long-term remission [48] and hospitalization [49] in MDD patients.
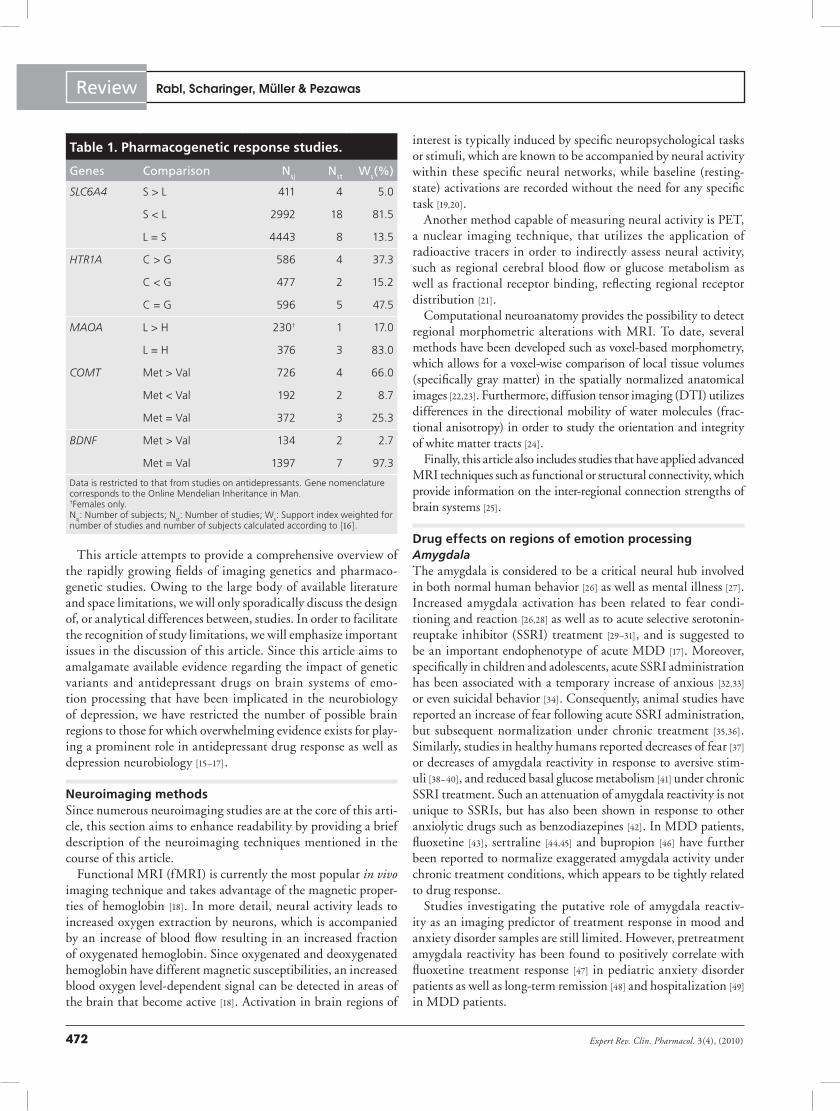
Table 1. Pharmacogenetic response studies.
Genes Comparison Nsj Nst Ws(%)
SLC6A4 S > L 411 4 5.0
S < L 2992 18 81.5
L = S 4443 8 13.5
HTR1A C > G 586 4 37.3
C < G 477 2 15.2
C = G 596 5 47.5
MAOA L > H 230† 1 17.0
L = H 376 3 83.0
COMT Met > Val 726 4 66.0
Met < Val 192 2 8.7
Met = Val 372 3 25.3
BDNF Met > Val 134 2 2.7
Met = Val 1397 7 97.3
Data is restricted to that from studies on antidepressants. Gene nomenclature corresponds to the Online Mendelian Inheritance in Man. †Females only.N
sj: Number of subjects; N
st: Number of studies; W
s: Support index weighted for
number of studies and number of subjects calculated according to [16].

www.expert-reviews.com 473
Review
Anterior cingulate cortex
Orbitofrontal cortex
AMY–ACC
Amygdala
SLC6A4, COMT
MAOA, COMT
SLC6A4, MAOA
SLC6A4, MAOA, COMT Hippocampus
MAOA, COMT, BDNF
A C P
A C
C
A C P
A C PBOLD signal change in response to acute treatment
Regional effects associated with chronic treatment
Regional alterations predict remission
A
C
P
BOLD response
Volume
Functional connectivity
Imaging genetics: implications for research on variable antidepressant drug response
Anterior cingulate cortexThe anterior cingulate cortex (ACC) is an area of critical impor-tance for emotion processing comprising of the perigenual ACC, implicated in emotional and autonomic integration, and the subgenual ACC (sACC), which is engaged in the expres-sion of autonomic states [50]. With regard to MDD, increased metabolism [51] and decreased tissue volume [52] have been found in the sACC, a cortical region that exhibits high densities of serotonin transporter (5-HTT) and serotonin receptor 1A (5-HT
1A) [53,54].
Similar to findings for the amygdala, an increase in ACC activa-tion has been reported for acute [29] or subchronic administration of SSRIs [55]. An attenuation under chronic treatment in healthy subjects has also been observed [38], which is in accordance with studies demonstrating a decrease of ACC activation or metabolism under antidepressant treatment in patient samples [46,56–58].
Furthermore, ACC activation or metabolism is thought to be an encouraging imaging indicator of individual treatment response – ACC hypermetabolism at baseline has been found to indicate treatment responders [59], whereas successful treatment with fluoxetine [43,60], buproprion [46] and venlafaxine [61,62] has been accompanied by a decrease of ACC activity or metabolism. In addition, a greater gray matter volume and density as well as greater functional activation in the ACC have been associated with a rapid symptom decrease in patients undergoing fluoxetine treatment [63,64]. However, contradictory results exist [65].
Orbitofrontal cortexAnother key structure in emotion process-ing is the orbitofrontal cortex (OFC) [66], which receives inputs from the ventral striatum and amygdala [67]. It is implicated in reinforcement-guided decision-mak-ing, emotion and social behavior, sharing mant of the similarities with the ACC [67]. Similar to findings in the ACC, loss of tissue volume and metabolic increases in MDD patients have also been reported for the OFC [68].
In healthy participants, acute citalo-pram administration [31] as well as short-term citalopram or reboxetine treatment [69] have been associated with attenuated activation of the OFC. Chronic treatment in bupropion-treated MDD patients also resulted in decreased OFC activation [46].
With regard to response prediction in MDD patients, decreased OFC metabo-lism has been found to reciprocally corre-late with responses to venlafaxine [61] and paroxetine [70]. Similarly, decreased OFC activation in MDD patients undergoing chronic bupropion treatment was highly
correlated with symptom improvement [46]. Finally, OFC gray matter density has been implicated in the prediction of residual symptoms after fluoxetine treatment [64].
Hippocampus The hippocampus is thought to be another key region of emo-tion processing [71,72] and, along with amygdala hyperactivation, hippocampal volume loss is the best studied endophenotype of MDD [16,17,73,74]. Since the hippocampus is among the brain regions that are most vulnerable to the deleterious effects of chronic stress [75], such volume reductions have been related to stress exposure. This finding is supported by animal stress models showing a reversal of stress-induced hippocampal atrophy by antidepressants [76]. Furthermore, antidepressants are thought to directly impact on hippocampal function. In studies of healthy subjects, acute citalo-pram administration translated into increased hippocampal activa-tion [29], whereas chronic treatment was accompanied by a dimin-ished hippocampal response [38,40,55]. Similar findings have also been reported for chronically treated MDD patients [46,57,60,77,78].
With regard to drug response prediction, increased pre treatment hippocampal activation has been demonstrated to proceed clini-cal improvement and to normalize during treatment with bupro-prion [46] or fluoxetine [79]. Furthermore, lower pretreatment
Figure 1. Genetic variants that affect drug response as well as developmental and functional properties of brain regions of emotion processing that are modulated by antidepressive action. Gene nomenclature corresponds to the Online Mendelian Inheritance in Man. AMY–ACC: Amygdala–anterior cingulate cortex; BOLD: Blood oxygen level dependent; BDNF: Brain-derived neurotrophic factor; COMT: Catechol-O-methyltransferase.

Expert Rev. Clin. Pharmacol. 3(4), (2010)474
Review Rabl, Scharinger, Müller & Pezawas
hippocampal volumes and gray matter densities have been related to a reduced likelihood of remission [80–84] and an increased risk of residual symptoms in MDD patients [64]. Finally, hippo campal growth has been linked to antidepressant drug treatment in patients, probably reflecting neurotrophic effects [81].
Amygdala–ACC circuitry The limbic system is highly interconnected and complex, mak-ing research in the field of neuroimaging a challenge. In order to address this problem, it has been suggested to dissect this com-plex system into smaller neural circuitries [25]. The most studied subsystem today is the amygdala–ACC circuitry [85], which has
frequently been implicated in depressive illness [86,87] and comprises feed-forward projections from the amygdala to the sACC (unci-natus bundle), as well as feedback projections from the perigenual ACC/anterior midcingulate cortex back to the amygdala (cingulum bundle) [88].
A reduction of amygdala–ACC connectivity has been found in unmedicated MDD patients compared with healthy sub-jects [87]. Under chronic treatment with sertraline [45] and fluox-etine [89], amygdala–ACC connectivity has proven to normal-ize, likely reflecting neuroplastic remodeling of a dysfunctional neural circuitry [45]. It should be noted that the functional con-nectivity [88,90] and pathway strength [91] of this neural circuitry,
Table 2. Imaging genetics studies.
Genes Measure Comparison Amygdala ACC OFC Hippocampus
Nst Nsj Nst Nsj Nst Nsj Nst Nsj
SLC6A4 Function S > L 14 622 5 166 – – 1 48
S < L 1 54† 1 48 – – – –
L = S 1 29 – – – – – –
Volume S > L 1 37 – – – – – –
S < L 3 302 3 232 1 41 1 77
L = S 1 45 – – – – 3 129
HTR1A Function C > G 1 89 – – – – – –
C = G 1 54† – – – – – –
MAOA Function L > H 2 189 1 47† – – 2 189
L < H – – 2 177 2 177 – –
Volume L > H – – – – 2 101† – –
L < H 1 97 1 97 – – – –
L = H – – 1 59‡ 1 55† – –
COMT
Function Met > Val 3 129 2 94 1 27 4 268
Met < Val 1 74 1 27 2 40 – –
Met = Val 1 101 – – – – – –
Volume Met > Val 3 203 – – – – 4 353
Met < Val – – – – 1 53 – –
Met = Val – – – – – – 1 76
BDNF Function Met > Val 2 107 – – – – 2 58
Met < Val 1 29 1 70 1 29 2 95
Met = Val 4 153 – – – – – –
Volume Met < Val 1 87 1 42 – – 9 649
Met = Val 5 483 – – – – 2 167
Restricted to studies on brain function and morphology. Gene nomenclature corresponds to the Online Mendelian Inheritance in Man. †Females only.‡Males only.ACC: Anterior cingulate cortex; BDNF: Brain-derived neurotrophic factor; COMT: Catechol-O-methyltransferase; N
sj: Number of subjects; N
st: Number of studies;
OFC: Orbitofrontal cortex.

www.expert-reviews.com 475
ReviewImaging genetics: implications for research on variable antidepressant drug response
as highlighted by fMRI and DTI studies, negatively correlate with trait anxiety, which is thought to be a vulnerable marker for depression.
Depression risk genes & drug responseFunctional polymorphisms of genes expressed in the brain, blood–brain barrier and liver have been linked to variable treat-ment responses to anti depressants [11,92]. Whereas genetic vari-ants associated with pharmacodynamic effects might also reflect disease-specific individual differences, polymorphisms affecting pharmacokinetics are agnostic against the variable neuro biology of depression. Pharmacokinetic genetic variability, such as in MDR1, affecting the penetration of drugs into brain tissue [11], has not been the focus of neuroimaging studies since it is unlikely to affect brain function or structure. Pharmacokinetic genetic variability in other genes, such as the cytochrome P450 (CYP450) genes involved in drug metabolism [11], may also impact on the brain since they are putatively involved in both drug and neurotransmitter metabo-lism [93]. By contrast, genes involved in pharmaco dynamic vari-ability have been investigated by both pharmacogenetic as well as imaging genetics studies and are probably implicated in the neuro biology of depression and variable treatment response, which will be the focus of this section of the article.
Serotonin transporter geneSerotonin-reuptake inhibitors are the first-line treatment for MDD [94] and target 5-HTT, which is expressed by the serotonin transporter gene (SLC6A4). A variable number of tandem repeats (VNTR) polymorphism (5-HTTLPR) within the promotor region of SLC6A4 resulting in variable 5-HTT expression [95], has been the primary target of research over the last couple of years since it has been associated with anxiety-related temperamental traits as well as depression in the context of environmental adversity [96,97]. Approximately two thirds of Caucasians are carriers of the short (S) allele of 5-HTTLPR, whereas allele frequencies in other ethnicities differ [98]. The S allele of 5-HTTLPR is associated with a twofold decreased expression and transport activity in human cell lines in vitro [96]. Although 5-HTT concentrations in the synaptic cleft cannot be measured directly, it is assumed that the S allele results in higher extracellular concentrations of serotonin (5-HT) in the synaptic cleft [99]. Several PET studies measuring 5-HTT binding have reported increased serotonin transporter binding in individu-als homozygous for the long (L) allele [100–102]. However, others have been unable to show the effects of 5-HTTLPR on 5-HTT binding in vivo [103–106] or postmortem [107]. Such inconsistencies have been explained by time-specific effects such as seasonality [108] and advances in tracer development [109], as well as the neurodevel-opmental effects of serotonin [53,110,111] underpinned by an animal study demonstrating that transient inhibition of 5-HTT during early development results in anxious behavior in adults [112].
Since blockade of 5-HTT is an important mode of action of anti-depressants, 5-HTTLPR has also been investigated with regard to drug response. Available evidence from pharmacogenetic studies in Caucasians who were homozygous for the S allele suggests lower
remission rates of MDD under SSRI treatment [113]. Similarly, the majority of available studies reported that the S allele was associated with lower response rates to antidepressant drugs [10]. In detail, studies reported a better or faster treatment outcome in those with the L/L genotype undergoing fluvoxamine [114–118], paroxetine [115–117,119–122], fluoxetine [123–126], citalopram [127], escitalopram [128], sertraline [129] and antidepressant treatment in general [130,131]. However, studies in Asian samples suggest the opposite [132–135], which might be caused by differences in genetic background as well as study lengths. In addition, several negative studies exist for milnacipran [136], sertraline [137], fluoxetine [138], citalopram [139,140], escitalopram [141] and antidepressant treatment in general [142,143].
In addition to treatment response, pharmacogenetic studies also investigate the effects on treatment tolerability – the L allele has been reported to be associated with a lower side effect rate and to be a possible predictor of treatment tolerance [10] for drugs such as fluoxetine [144] and citalopram [139], and antidepressant drug treat-ment in general [131,145,146]. Similar to studies on drug response, conflicting data exist, for example, some studies indicate fewer side effects in S-allele carriers undergoing mirtazapine treatment [121] and other studies report a lack of association between 5-HTTLPR and observed side effect frequencies [117,147–149].
The majority of studies have consistently demonstrated an S allele attenuated treatment response; however, several negative studies exist, which is not suprising, since effect sizes are probably to be smaller than originally believed. Today, it is estimated that 5-HTTLPR only accounts for a 2–3% explained variance in treat-ment response [113]. This may be explained, at least partly, by less studied genetic variability in SLC6A4 [150] such as a single nucleotide polymorphism (A>G substitution, rs25531) within the L allele of 5-HTTLPR [101,141,151] and a VNTR in the second intron [152,153]. Moreover, other promising genetic locations [154] and contributing factors [155] affecting antidepressive drug binding to 5-HTT have been found in vitro. However, as of yet none of those preclinical findings have been transferred to humans.
By contrast, results of fMRI studies investigating the neural impact of 5-HTTLPR on amygdala reactivity in healthy subjects are highly consistent [156,157]. They suggest that S-allele carriers show an exaggerated amygdala reactivity in the presence of fearful or negatively valenced stimuli [49,88,156,158–168] as well as under base-line conditions [161,166] in comparison with those with a L/L geno-type. Similar results have been obtained in a limited number of studies in MDD patients [49,162,169].
With regard to ACC function, increases in ACC basal metabo-lism [170] as well as blood flow [166,171] in healthy subjects have been associated with the S allele. Accordingly, S-allele-driven increases have been reported by activation studies [49,162,167,172,173]. Only one study reported opposing results [174]. Structural MRI studies in healthy subjects revealed S-allele-moderated gray matter volume loss in the ACC, or sACC [88,160,175], which is similar to a primate study with orthologous 5-HTTLPR (rh5-HTTLPR) [176]. Patient studies have been inconclusive with authors reporting S-allele-driven increases [49,177] or decreases of ACC activation [174], and results are lacking [162,175].

Expert Rev. Clin. Pharmacol. 3(4), (2010)476
Review Rabl, Scharinger, Müller & Pezawas
Finally, 5-HTTLPR effects have also been demonstrated on a brain systems level, demonstrating that the S allele reduces structural and functional connectivity between the amygdala and sACC [88]. These measurements suggest a relative uncoupling of those struc-tures resulting in a disinhibition of this feedback loop and hence, an increased amygdala activity [88]. This finding is in accordance with other fMRI [162,165,169,173] and DTI studies [178] of patients or healthy subjects reporting S-allele effects on amygdala–ACC coupling.
Serotonin receptor 1A geneThe G allele of C(-1019)G (rs6295), a single nucleotide polymor-phism within the promotor region of the 5-HT
1A gene (HTR1A),
can be found in 50% of Caucasians and 21% of Asians [148] and has been associated with impaired transcriptional repression mecha-nisms leading to increased 5-HT
1A density [179]. It is assumed to
be a risk allele for MDD, since it has been reported to be over-represented in MDD patients and increases of post synaptic 5-HT
1A
signaling have been implicated in the mode of action of several anti-depressant treatments [180,181]. Similar to SLC6A4, it has been sug-gested that HTR1A specifically impacts early brain development, since transient gene knockout in newborn mice leads to anxiety-like behavior, whereas its absence during adult life has no effect [182]. Furthermore, it has been found that the anxiety phenotype of HTR1A-knockout mice can be moderated by early environmental factors, suggesting gene–environment interactions [183].
With regard to drug response studies, homozygous G(-1019) allele carriers have been reported to show a diminished response to flibanserin [184], which has failed to show antidepressive effects, as well as fluoxetine [126,148] and several other anti depressants compared with subjects with the C/C genotype [185]. By con-trast, other authors reported the G allele to be associated with favorable response rates in patients undergoing fluvoxamine, par-oxetine or milnacipran treatment [186] along with several other antidepressants [187].
Some of this conf licting evidence might be explained by gene–gene interactions (epistasis) between C(-1019)G HTR1A and 5-HTTLPR in SLC6A4 with respect to citalopram treatment [188], or between C(-1019)G HTR1A and Val66Met BNDF [189], which has been linked to treatment resistance.
However, evidence for the pharmacogenetic effects of C(-1019)G HTR1A is very limited since negative studies exist for fluoxetine [184,190], fluvoxamine [191] and SSRIs in general [192,193], and a recent meta-analysis was not able to detect an overall impact on treatment efficacy [10].
Similarly discouraging are findings of imaging genetics stud-ies reporting a G allele-associated amygdala activation decrease in healthy subjects [194], whereas another study in patients by Dannlowski et al found the opposite [169]. Furthermore, a replication study failed in a group of healthy Asian patients [195].
Monoamine oxidase A geneThe X-chromosome-linked MAOA gene encodes monoamine oxidase A, a key enzyme in the degradation of 5-HT, which is expressed in the outer mitochondrial membrane of monoaminer-gic neurons. The promoter region of the MAOA gene comprises
a functional VNTR polymorphism resulting in a highly active MAOA-H and less active MAOA-L allele depending on gene expression differences [196]. Since MAOA inhibitors are highly effective drugs in MDD treatment, associations of the MAOA VNTR with antidepressant drug response have been investigated. However, few studies are available so far and the majority of stud-ies lack results. Accordingly, no asssociations of MAOA VNTR genotype with drug response to fluvoxamine [197], paroxetine [197], moclobemide [198] and fluvoxamine [199] have been found. To our knowledge, only a single study reported a favorable treatment response to fluoxetine in females who were homozygous for the MAO-L allele compared with MAO-H allele carriers [200].
With regard to imaging genetics studies, genetic variation in MAOA has repeatedly been associated with functional and structural alterations in depression-related brain circuitries. Functional studies reported increases of brain activation in the amygdala and hippocampus [201,202], as well as decreases in the OFC and ACC [172,202] of healthy MAOA-L allele carriers. With regard to brain structure, increases of OFC [202,203] and decreases of ACC volume [202] have been found in healthy MAOA-L car-riers. However, negative findings exist with regard to amygdala and ACC volume in an underpowered sample [203]. Moreover, in a non-Caucasian and gender-restricted sample, a lack of impact on OFC and inverse effects on ACC function have been reported [201].
Finally, on a brain systems level, the effects of MAOA on the amygdala–ACC circuitry have been reported in healthy subjects demonstrating MAOA-L allele-induced increases in functional connectivity that correlate positively with harm avoidance and negatively with reward dependence [204]. In addition, a study in MDD patients demonstrated decreases of amygdala–ACC cou-pling, which have been linked to a more severe clinical course of depression, in carriers of the MAO-H allele [205].
Catechol-O-methyltransferase geneCatechol-O-methyltransferase (COMT) is a monoamine-degrading, extracellularly located enzyme encoded by the COMT gene [206], which profoundly impacts on dopamine catabolism in the prefrontal cortex, a region in which dopamine transport-ers are rare [207]. The higher active and phylogenetically ancestral Val allele of Val158Met COMT (rs4680) has been demonstrated to increase cognitive flexibility [208]. Moreover, it also has been reported to impact on the human reward center, which is involved in anhedonia, a key symptom of depression, as well as euphoria, which can be induced by dopaminergic drug abuse [209].
Pharmacogenetic studies of Val158Met COMT are contra-dictory. Some authors found a diminished treatment response in homozygous Val allele carriers [210] for paroxetine [211], milnacip-ran [212] and fluoxetine [213]. However, opposing effects have been found for mirtazapine [214] and citalopram [215], as well as lack-ing effects for paroxetine [214,215] and duloxetine [216]. In addition, a reduced risk of antidepressant-induced weight gain has been reported for Val158Met heterozygotes [217].
By contrast, results of imaging genetics studies on COMT por-tray a more distinct picture. The Met allele has been associated with increased amygdala volume [218–220] and activation [167,221–222].

www.expert-reviews.com 477
ReviewImaging genetics: implications for research on variable antidepressant drug response
So far, only one study has been unable to confirm COMT impact on amygdala activation [223], whereas another reported opposing effects, particularly in females [224]. Analogous to findings in the amygdala, increased activation has also been detected in the ACC of healthy Met allele carriers [167,221,225].
With regard to the findings in OFC, the majority of studies have demonstrated a Met allele-associated decrease of activation [225–227] and volume [219] in healthy controls. Similar decreased activa-tion [167,222,223,228] and volume [218–220,229] have been reported for the hippocampus. Only a single study has been unable to detect COMT volume effects in a healthy control group [230], which is probably related to ethnicity [230].
Finally, on a brain system level, reduced functional coupling of the amygdala–OFC circuitry has been linked to Val allele carriers [223].
Brain-derived neurotrophic factor gene The neurotrophin brain-derived neurotrophic factor (BDNF) is a key protein involved in long-term potentiation and memory encoding. Its precursor (pro-BDNF) binds to the p75 neuro-trophin receptor (p75NTR), eliciting apoptosis, whereas its mature form predominantly activates the tropomyosin-related kinase B receptor, promoting neurotrophic and antiapoptotic effects [231].
Over the last few years, BDNF has been implicated in various neurologic and psychiatric phenotypes including mood disor-ders [232]. BDNF signaling interacts with the monoaminergic as well as the excitatory and inhibitory neurotransmitter system [233], which are all supposed to contribute to the complex neurobiology of depression. Further support stems from animal studies linking stress exposure and glucocorticoid administration to reduced hip-pocampal BDNF mRNA expression, as well as from studies show-ing antidepressant efficacy of intracerebral BDNF infusions [234].
These observations have led to the so-called neurotrophin hypothesis of depression, suggesting a loss of BDNF effects to be the underlying cause of depression due to introducing dysfunc-tional anatomical mood circuitries [235]. However, contradictory results have been obtained from animal models of depression, where BDNF mRNA is increased within the reward system and local BDNF perfusion promotes depressive-like behavior [236]. Therefore, the exact role of BDNF is currently far from being understood and complex molecular mechanisms, such as tran-scription through multiple promoters, trafficking, cleavage and secretion, have been suggested to contribute to the observed inconsistencies [237]. In addition, BDNF expression seems to be subjected to profound epigenetic control [238].
The BDNF gene contains several polymorphic sites [239]; how-ever, most attention has been paid to Val66Met BDNF (rs6265), which has been shown to impair intracellular trafficking and the activity-dependent secretion of BDNF [240].
In overall meta-analyses, no significant association between Val66Met and MDD has been found [241–243], although, taking gender into account, one study reported a significant association of the Met allele with MDD in males [242]. However, with regard to personality traits, the Met allele has been associated with lower neuroticism scores [244].
Whereas the distinct role of BDNF in mood disorder etiology remains unclear, its involvement in antidepressant action has been more consistently demonstrated in a variety of studies [236], for example, it has been observed that heterozygous BDNF-knockout mice are unresponsive to antidepressants [245]. Consequently, sev-eral pharmacogenetic studies addressing BDNF have been pub-lished. Effects of BDNF polymorphisms on treatment response have been studied for antidepressive drug therapy [239,246–252] in MDD as well as lithium treatment in bipolar disorder [253]. A favorable response to antidepressive treatment has been reported for Met allele carriers of Val66Met BNDF in Asian samples, including drugs such as citalopram [251], fluvoxamine and mil-nacipran [247]. Opposing results have been found in Caucasians, reporting higher response rates in Val allele carriers undergoing citalopram treatment, but this was not replicated within a large-scale study [250]. Furthermore, a lack of results have been reported for antidepressant treatments in general [246,248], and specifically for nortriptyline [249], escitalopram [249] and despiramine [239]. Finally, an improved response to fluoxetine in heterozygous patients (Val/Met) compared with the homozygous groups has been reported [252]. However, recent meta-analyses [10,254] found evidence for increased response rates in Met allele carriers, spe-cifically in Asian populations [247,251,252,254]. Counterintuitively, preclinical evidence obtained from genetically engineered mice suggested the Met allele to be unresponsive to fluoxetine [255].
With regard to brain structure, healthy carriers of the Met allele of Val66Met BDNF have been overwhelmingly asso-ciated with smaller hippocampal volume [256–263]. However, Jessen & Koolschijn failed to detect volumetric effects [264,265] and a co-dependence on neuroticism has been reported [266]. Functional studies are as yet less conclusive, associating the Val allele with increased hippocampal activation in healthy subjects [267,268], in contrast to others favoring the Met allele [240,259]. With respect to patient studies, reduced hippocampal volume has also been reported for Met carriers independent of clinical diagnosis [256,261], although one author was not able to replicate those findings [264]. Further support stems from animal studies confirming the assumption of BDNF-mediated effects on the hippocampus: BDNF has been demonstrated to be crucial for hippocampal functioning and integrity [269,270]. Moreover, selec-tive loss of BDNF in the dentate gyrus has been demonstrated to attenuate antidepressant efficacy [271]. Less evidence exists for BDNF-related effects on the amygdala. Met allele-associated amygdalar volume reduction has recently been reported [272]
although earlier studies have been unable to show such an effect [256–259,273]. Similarly, most studies failed to detect the effects of Val66Met on amyg dala function [240,259,267,268]. However, a single study found increased activity in Val homo zygotes [274], whereas studies two reported the Met allele to be associated with increased amygdala response [275,276]. The latter observation, however, is vaguely supported by animal studies in which Met homozygous mice exhibited increased anxiety [255] and impaired extinc-tion learning [275] – features tightly associated with amygdala functioning. Similarly, limited evidence is available for BDNF effects on the ACC, where reduced volume and function in Met

Expert Rev. Clin. Pharmacol. 3(4), (2010)478
Review Rabl, Scharinger, Müller & Pezawas
carriers have been reported in two studies [257,275]. Finally, one study reported an increase in OFC activation in Val homozygous individuals [274].
DiscussionSince neuroimaging and pharmacogenetic studies have both merits and demerits, we will discuss some major issues in the course of this section. More indepth information can be found elsewhere [20,92,277].
When tools for gene identification, such as linkage studies, became available, these methods were enthusiastically adopted for psychiatric research [278]. However, in contrast to Mendelian disorders, results have been discouraging, which is understandable in the face of obvious differences in the disorders’ genetic het-erogeneity, risk gene effect size and the mode of inheritance [279]. Association studies comparing the prevalence of candidate gene markers derived from a priori hypotheses in affected and unaf-fected individuals have been considered to be a more powerful approach [280] and, within a short period of time, have also been utilized in clinical pharmacology within pharmacogenetic asso-ciation studies of variable drug response [92]. However, although meta-analyses indicate that such studies are capable of risk variant identification [281], a lack of replication appears to be more the rule rather than the exception, since effect sizes of such risk variants are very small [282]. One typically applied exit strategy is to dra-matically increase sample sizes. As compared with initial studies including not more than 100 participants [114], recent trials such as the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study enrolled up to 2000 patients [283] in order to com-pensate for small effect sizes. Given the neuro biological complexity of psychiatric disorders, the small effect sizes typically found in association studies are no surprise. The same argument can be applied to antidepressive treatment response, which is also highly complex, and the gene–gene as well as gene–environment inter-actions impacting on it are currently poorly understood [284,285]. However, association studies are agnostic to the biological under-pinnings of such complex traits and, therefore, are only capable of a crude risk variant identification.
In order to overcome the missing link between genes and behav-ioral traits, the concept of intermediate endophenotypes has been introduced [286,287]. Intermediate endophenotypes are quantita-tive traits that are thought to more directly index the biology of a complex behavioral phenotype [287]. This framework attempts to bypass the uncertainty of psychiatric diagnoses using more objective measures and promises to inform more deeply regarding the underlying biological processes [287].
However, this concept has been criticized as well, arguing that the biological basis of some endophenotypes might be less com-plicated than the phenotype itself [288]. Nevertheless, although this might be the case for biologically complex traits such as Wisconsin Card Sorting Task performance, an endophenotype used in schizophrenia research [288], meta-analyses of imaging genetics studies reveal a different picture [157,289]. For instance, in contrast to the 5-HTTLPR polymorphism accounting for only 2–3% of the estimated variance of treatment response [113], it has
been shown to account for up to 10% of the variance in amygdala activation [157]. Evidence from single studies promise even stronger effect sizes for connectivity measurements [25,88,204,223].
Owing to their obvious advantages, neuroimaging techniques have been widely accepted in the study of mental disorders and gained tremendous popularity, partially because they provide seem-ingly simplistic explanations of complex behavioral phenotypes [18]. However, despite this superficial simplicity, neuro imaging methods suffer from important limitations and are potentially error-prone, since they are fairly sophisticated and the measured signals are rather small [277]. Briefly, the following limitations have to be considered: the underlying mechanism of the blood oxygen level-dependent signal used in fMRI is still not fully understood and it only rep-resents a surrogate measure for neuronal mass activity [277]. Low task-related signals are compromised by large levels of non-task-related noise caused by thermal noise, system variability in scanner hardware and physiological variability or head motion requiring sophisticated preprocessing procedures [290]. Another important concern is how to deal with multiple comparisons, especially in voxel-wise fMRI analy ses, but empirical evidence suggests that cur-rently used methods are overly strict [291]. Furthermore, there is no genuine experimental design that fits for all fMRI studies, and a design that is not properly adjusted to the hypothesis may lead to the wrong conclusions being drawn, since irrelevant brain activation can be elicited. However, several well-estalished tasks are available today and have been sufficiently used in MDD, such as a paradigm employing fearful faces [164]. Small effect sizes of genetic variants require a power analysis in order to choose the appropriate sample size for pharmacogenetic as well as for imaging genetics studies. While the appropriate sample size for a given genetic variant still needs to be determined for pharmacogenetic imaging studies, it has been estimated that for imaging genetic studies investigating SLC6A4, the minimal sample size is 70 subjects [157], which is widely acknowledged by recent studies. It should be noted that this number is significantly smaller than the number of subjects needed for asso-ciation studies, which supports the intermediate endophenotype concept. Another important issue is the impact of demographic data, such as age and gender data, as well as clinical specifiers, which can profoundly affect neuroimaging results. This underlines the neces-sity for thoughtfully collected samples and statistical tests correcting for confounding variables in order to control neural effects between genotype and age [272,273], disease course [292,293] or treatment [294].
In summary, neuroimaging techniques require several caveats, since they are fairly sophisticated. Nevertheless, when properly applied, they offer intruiging insights into the neurobiology of mental disorders.
Conclusion & future directionsGenetic variants in SLC6A4, HTR1A, MAOA, COMT and BDNF have been linked to depression as well as antidepressant drug response and impact on brain regions of emotion processing that are modulated by antidepressants.
Both depression and antidepressant drug response are geneti-cally complex and probably result from interactions of multiple genetic variants (epistasis) with small effect sizes, and they are

www.expert-reviews.com 479
ReviewImaging genetics: implications for research on variable antidepressant drug response
further affected by gene–environment interactions [285]. Hence, single genetic variants only exhibit subtle biological effects and do not encode for behavioral phenotypes [287]. Similarly, effect sizes of association and pharmacogenetic studies have been, at most, mod-est or even undetectable [113,287,295]. Behavioral measures of depres-sion or drug response add further inaccuracy to the highly variable neurobiology of depression. Recently, further criticism has been raised that the use of symptom-based anti depressive response scales (e.g., Hamilton Depression Rating Scale) as clinical end points does not sufficently account for the multi dimensional and widely subjective nature of depression symptoms [296]. Such problems may be, at least, partially overcome by studies assessing intermediate endophenotypes that more directly impact the neurobiological underpinnings of symptom-derived psychiatric diagnoses [287]. Several intermediate phenotypes have been demonstrated to be closely related to MDD with most evidence available for amygdala hyper-reactivity and hippocampal volume reduction [16,17]. In addi-tion, other regions of emotion processing, particulary the ACC and OFC, appear to be further valid intermediate phenotypes, since they have been found to be structurally and functionally affected in acute depression; connectivity measurements encom-passing the ACC–amygdala circuitry may be even more powerful [16,17]. Imaging genetics studies highlight that such intermediate endophenotypes are under heavy genetic control. Moreover, phar-macological fMRI and pharmacological MRI studies have iden-tified intermediate endophenotypes of antidepressant treatment response, which are intriguingly similar to those of depression. Since both depression risk genes and variable treatment response have been linked to several identical neural targets, it is tempting to propose that genetic variants impact on antidepressant treat-ment response via neurocircuitries that have been identified as intermediate endophenotypes of depression. Such an approach indicates the notion that individually varying drug effects can-not simply be explained by mechanistic pharmacogenetic models of variable protein expression, but implicate more complex proc-esses. Among those are likely to be developmental genetic effects, which have been demonstrated by structural imaging studies (e.g., smaller sACC volume and amygdala–ACC connectivity in S-allele carriers of 5-HTTLPR) and probably result in altered individual responsiveness [49,88,160,162,167,172,173,175]. Hence, lower response rates in S-allele carriers might be, at least partly, explained by develop-mental deficits of the amygdala–ACC circuitry [88,162,165,169,173,178], which has proven to be regulated by SSRI treatment [45,89]. While there is overwhelming evidence of an important role during brain development for SLC6A4 [53] and BDNF [297], there is also some evidence for the remaining genes, which have been the focus of this article [53,112,204,223].
Whereas risk variants are indistinguishable in their contri-bution to clinical treatment response, they seem to affect char-acteristic patterns of brain regions on a brain systems level. Following this assumption, the variable antidepressant drug response related to SLC6A4 might be mediated over the amy-gdala [49,88,156,158–169], sACC [49,88,160,162,167,172,173,175] and amy-gdala–sACC circuitry [88,162,165,169,173,178], whereas MAOA affects a broader variety of brain systems such as the OFC [172,202–203],
ACC [202], amygdala [201,202], hippocampus [195,202] and amyg-dala–ACC circuitry [204,205]. Similar to MAOA, COMT affects the OFC [219,225–227], ACC [167,221,225], amygdala [167,218–222], hippo-campus [167,218–220,222,223,228,229] and amygdala–OFC circuitry [223]. While genetic variation in HTR1A has not been demonstrated to impact significantly on drug response or emotion processing in humans, genetic variability in BDNF has been clearly proven to affect the hippocampus [240,256–263,267,268].
Following this chain of arguments, we propose that the applica-tion of imaging endophenotypes within the framework of variable antidepressant treatment response might provide several advantages: first, studies investigating the impact of different genotypes on neu-ral correlates of antidepressant drug action might reveal information regarding subtypes of antidepressant treatment response that oth-erwise would be indiscriminable in clinical behavioral assessments and, therefore, are capable of bridging the gap between varying genetic make-up and variable drug response rates. This would prob-ably be an important step forward towards personalized treatment strategies, since genetic testing on its own provides too little infor-mation to be translated from bench to bedside (e.g., 5-HTTLPR accounting for only 2–3% of the variance of treatment response)[113]. Accordingly, it has been suggested that personalized treatment can only be achieved by a combination of different strategies, such as imaging, genetics, proteomics or neuroendocrinology [14].
Growing numbers of studies have begun to address more com-plex models, such as haplotypes, epistasis and gene–environment interactions [16]. An imaging genetics approach might be specifi-cally suitable for the investigation of more complex mechanisms including gene–gene or gene–environment interactions as well as haplotypes, since imaging endophenotypes may dramatically decrease the required sample size. Several recent publications underline the feasability of such an approach [161,167,169,229,298–301].
Recently, the focus of pharmacogenetic studies has moved from a likely biased candidate gene approach based on a priori hypoth-eses to an ‘agnostic’ genome-wide association (GWA) approach. New and unexpected candidate genes associated with drug response [302,303] and depression [172,304–307] have been reported. Interestingly, such studies only partly support the findings of can-didate gene studies and suggest that multiple loci of small effect sizes may determine treatment response [302,303]. Although several open questions remain regarding the analysis of genome-wide association studies (GWAS), such as the appropriate correction for multiple comparisons, they will likely stimulate research in the field of variable treatment responses [308]. Imaging genetics studies have been demonstrated to be a valuable tool to further evaluate risk variant candidates derived from GWAS [309,310]. Moreover, the first voxel-wise GWAS are on the horizon [311].
Variable antidepressant treatment response is genetically deter-mined; however, multiple loci of small effect sizes hamper the applicability of genetic testing for personalized treatment regimes. Therefore, more translational-oriented research is urgently needed to disentangle the underlying neurobiological mechanisms. Among other techniques, such as genetics, proteomics and animal models, neuroimaging will likely provide a powerful armamentarium of tools towards the goal of personalized treatment regimes.

Expert Rev. Clin. Pharmacol. 3(4), (2010)480
Review Rabl, Scharinger, Müller & Pezawas
Expert commentaryGenetic variation implicated in the pharmacodynamic action of antidepressants is frequently associated with variable drug response. However, since drug response is exclusively evaluated by psychometric scales, low associations with gene candidates have been found. The use of intermediate endophenotypes for drug response research, which are already available in the field of neuroimaging, will likely tighten the relationship between these genotypes and drug response. It will further facilitate the biological understanding of a complex behavioral trait such as antidepressant drug response.
Five-year viewAlthough the neurobiological understanding of depression is still in its infancy, there is reason to believe that growing insights from human neuroimaging studies will doubtlessly yield a more thor-ough understanding of the relationship between variable antide-pressive treatment response and underlying differences in brain architecture and function. Several intermediate endophenotypes of depression derived from neuroimaging studies are already avail-able today and are ready to serve as biological indicators in the context of drug response studies, which might eliminate problems
inherent in the current psychometric assessment of drug response. Psychopharmacogenetic effect sizes of single disease-related genes are small and, therefore, of limited clinical use. This could be over-come by the use of more sophisticated psychopharmocogentic mod-els appreciating the complex neuro biology of depression and drug response such as gene–gene or gene–environment interactions, epi-genetics and imprinting, some of which have already demonstrated neurobiological relevance in imaging genetics studies.
AcknowledgementsThis article is part of the educational efforts of the Special Research Project SFB-35 (Project No. F3514-B11 and F3506-B11), funded by the Austrian Science Fund (FWF).
Financial & competing interests disclosureThe authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
Key issues
• Shortcomings of antidepressant treatment such as unsatisfying drug response, undesirable side effects and high recurrence rates have been promoting the hunt for personalized treatment regimes.
• Genetics is appreciated as an important factor that impacts on drug response; however, the genes implicated in the pharmacodynamics are still controversial.
• Effect sizes of genetic associations with clinical measures such as drug response are small and can be overcome by the use of intermediate endophenotypes.
• Pharmacological imaging studies have identified the neural targets of drug response.
• Imaging genetics studies have identified the neural targets of genes that are related to pharmacodynamic action.
• Imaging genetics and pharmacological imaging studies suggest that simple mechanistic pharmacogenetic models cannot be translated on a brain systems level in a simplistic fashion.
• A comprehensive perspective on drug effects obtained from interdisciplinary research including imaging genetics will probably stimulate the development of personalized treatment regimes.
ReferencesPapers of special note have been highlighted as:• of interest
1 Wong ML, Licinio J. Research and treatment approaches to depression. Nat. Rev. Neurosci. 2(5), 343–351 (2001).
2 Melander H, Salmonson T, Abadie E, Van Zwieten-Boot B. A regulatory apologia – a review of placebo-controlled studies in regulatory submissions of new-generation antidepressants. Eur. Neuropsychopharmacol. 18(9), 623–627 (2008).
3 Cipriani A, Furukawa TA, Salanti G et al. Comparative efficacy and acceptability of 12 new-generation antidepressants: a multiple-treatments meta-analysis. Lancet 373(9665), 746–758 (2009).
4 Kirsch I, Deacon BJ, Huedo-Medina TB, Scoboria A, Moore TJ, Johnson BT. Initial
severity and antidepressant benefits: a meta-analysis of data submitted to the food and drug administration. PLoS Med. 5(2), e45 (2008).
5 Trivedi MH, Rush AJ, Wisniewski SR et al. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am. J. Psychiatry 163(1), 28–40 (2006).
6 Moncrieff J, Kirsch I. Efficacy of antidepressants in adults. BMJ 331(7509), 155–157 (2005).
7 Seeringer A, Kirchheiner J. Pharmacogenetics-guided dose modifications of antidepressants. Clin. Lab. Med. 28(4), 619–626 (2008).
8 Weinshilboum R. Inheritance and drug response. N. Engl. J. Med. 348(6), 529–537 (2003).
9 Roden DM, George AL Jr. The genetic basis of variability in drug responses. Nat. Rev. Drug Discov. 1(1), 37–44 (2002).
10 Kato M, Serretti A. Review and meta-analysis of antidepressant pharmacogenetic findings in major depressive disorder. Mol. Psychiatry 15(5), 473–500 (2008).
• Comprehensivereviewandmeta-analysisonpharmacogeneticfindingsonvariableantidepressantdrugresponse.
11 Schosser A, Kasper S. The role of pharmacogenetics in the treatment of depression and anxiety disorders. Int. Clin. Psychopharmacol. 24(6), 277–288 (2009).
12 Horstmann S, Binder EB. Pharmacogenomics of antidepressant drugs. Pharmacol. Ther. 124(1), 57–73 (2009).

www.expert-reviews.com 481
ReviewImaging genetics: implications for research on variable antidepressant drug response
13 Arranz MJ, Kapur S. Pharmacogenetics in psychiatry: are we ready for widespread clinical use? Schizophr. Bull. 34(6), 1130–1144 (2008).
14 Holsboer F. How can we realize the promise of personalized antidepressant medicines? Nat. Rev. Neurosci. 9(8), 638–646 (2008).
15 Anderson IM, Mckie S, Elliott R, Williams SR, Deakin JF. Assessing human 5-ht function in vivo with pharmacoMRI. Neuropharmacology 55(6), 1029–1037 (2008).
16 Scharinger C, Rabl U, Sitte HH, Pezawas L. Imaging genetics of mood disorders. Neuroimage DOI: doi:10.1016/j.neuroimage.2010.02.019 (2010) (Epub ahead of print).
17 Savitz JB, Drevets WC. Imaging phenotypes of major depressive disorder: genetic correlates. Neuroscience 164(1), 300–330 (2009).
18 Logothetis NK. The ins and outs of fMRI signals. Nat. Neurosci. 10(10), 1230–1232 (2007).
19 Raichle ME, Snyder AZ. A default mode of brain function: a brief history of an evolving idea. Neuroimage 37(4), 1083–1090 (2007).
20 Poldrack RA, Fletcher PC, Henson RN, Worsley KJ, Brett M, Nichols TE. Guidelines for reporting an fMRI study. Neuroimage 40(2), 409–414 (2008).
21 Bandettini PA. What’s new in neuroimaging methods? Ann. N. Y. Acad. Sci. 1156, 260–293 (2009).
22 Ashburner J, Friston KJ. Voxel-based morphometry – the methods. Neuroimage 11(6 Pt 1), 805–821 (2000).
23 Ridgway GR, Henley SM, Rohrer JD, Scahill RI, Warren JD, Fox NC. Ten simple rules for reporting voxel-based morphometry studies. Neuroimage 40(4), 1429–1435 (2008).
24 Sexton CE, Mackay CE, Ebmeier KP. A systematic review of diffusion tensor imaging studies in affective disorders. Biol. Psychiatry 66(9), 814–823 (2009).
25 Meyer-Lindenberg A. Neural connectivity as an intermediate phenotype: brain networks under genetic control. Hum. Brain Mapp. 30(7), 1938–1946 (2009).
26 Ledoux J. The amygdala. Curr. Biol. 17(20), R868–R874 (2007).
27 Anand A, Shekhar A. Brain imaging studies in mood and anxiety disorders: special emphasis on the amygdala. Ann. N. Y. Acad. Sci. 985, 370–388 (2003).
28 Sehlmeyer C, Schoning S, Zwitserlood P et al. Human fear conditioning and extinction in neuroimaging: a systematic review. PLoS ONE 4(6), E5865 (2009).
29 Mckie S, Del-Ben C, Elliott R et al. Neuronal effects of acute citalopram detected by pharmacoMRI. Psychopharmacology (Berl.) 180(4), 680–686 (2005).
30 Bigos KL, Pollock BG, Aizenstein HJ, Fisher PM, Bies RR, Hariri AR. Acute 5-HT reuptake blockade potentiates human amygdala reactivity. Neuropsychopharmacology 33(13), 3221–3225 (2008).
31 Del-Ben CM, Deakin JF, Mckie S et al. The effect of citalopram pretreatment on neuronal responses to neuropsychological tasks in normal volunteers: an fMRI study. Neuropsychopharmacology 30(9), 1724–1734 (2005).
32 Bhagwagar Z, Cowen PJ, Goodwin GM, Harmer CJ. Normalization of enhanced fear recognition by acute ssri treatment in subjects with a previous history of depression. Am. J. Psychiatry 161(1), 166–168 (2004).
33 Browning M, Reid C, Cowen PJ, Goodwin GM, Harmer CJ. A single dose of citalopram increases fear recognition in healthy subjects. J. Psychopharmacol. 21(7), 684–690 (2007).
34 Hetrick S, Merry S, Mckenzie J, Sindahl P, Proctor M. Selective serotonin reuptake inhibitors (ssris) for depressive disorders in children and adolescents. Cochrane Database Syst. Rev. (3), CD004851 (2007).
35 Burghardt NS, Sullivan GM, Mcewen BS, Gorman JM, Ledoux JE. The selective serotonin reuptake inhibitor citalopram increases fear after acute treatment but reduces fear with chronic treatment: a comparison with tianeptine. Biol. Psychiatry 55(12), 1171–1178 (2004).
36 Burghardt NS, Bush DE, Mcewen BS, Ledoux JE. Acute selective serotonin reuptake inhibitors increase conditioned fear expression: blockade with a 5-HT(2c) receptor antagonist. Biol. Psychiatry 62(10), 1111–1118 (2007).
37 Harmer CJ, Shelley NC, Cowen PJ, Goodwin GM. Increased positive versus negative affective perception and memory in healthy volunteers following selective serotonin and norepinephrine reuptake inhibition. Am. J. Psychiatry 161(7), 1256–1263 (2004).
38 Harmer CJ, Mackay CE, Reid CB, Cowen PJ, Goodwin GM. Antidepressant drug treatment modifies the neural processing of nonconscious threat cues. Biol. Psychiatry 59(9), 816–820 (2006).
39 Arce E, Simmons AN, Lovero KL, Stein MB, Paulus MP. Escitalopram effects on insula and amygdala BOLD activation during emotional processing. Psychopharmacology (Berl.) 196(4), 661–672 (2008).
40 Windischberger C, Lanzenberger R, Holik A et al. Area-specific modulation of neural activation comparing escitalopram and citalopram revealed by pharmaco-fMRI: a randomized cross-over study. Neuroimage 49(2), 1161–1170 (2010).
41 Drevets WC, Bogers W, Raichle ME. Functional anatomical correlates of antidepressant drug treatment assessed using pet measures of regional glucose metabolism. Eur. Neuropsychopharmacol. 12(6), 527–544 (2002).
42 Paulus MP, Feinstein JS, Castillo G, Simmons AN, Stein MB. Dose-dependent decrease of activation in bilateral amygdala and insula by lorazepam during emotion processing. Arch. Gen. Psychiatry 62(3), 282–288 (2005).
43 Fu CH, Williams SC, Cleare AJ et al. Attenuation of the neural response to sad faces in major depression by antidepressant treatment: a prospective, event-related functional magnetic resonance imaging study. Arch. Gen. Psychiatry 61(9), 877–889 (2004).
44 Sheline YI, Barch DM, Donnelly JM, Ollinger JM, Snyder AZ, Mintun MA. Increased amygdala response to masked emotional faces in depressed subjects resolves with antidepressant treatment: an fMRI study. Biol. Psychiatry 50(9), 651–658 (2001).
45 Anand A, Li Y, Wang Y, Gardner K, Lowe MJ. Reciprocal effects of antidepressant treatment on activity and connectivity of the mood regulating circuit: an fMRI study. J. Neuropsychiatry Clin. Neurosci. 19(3), 274–282 (2007).
46 Robertson B, Wang L, Diaz MT et al. Effect of bupropion extended release on negative emotion processing in major depressive disorder: a pilot functional magnetic resonance imaging study. J. Clin. Psychiatry 68(2), 261–267 (2007).
47 Mcclure EB, Adler A, Monk CS et al. FMRI predictors of treatment outcome in pediatric anxiety disorders. Psychopharmacology (Berl.) 191(1), 97–105 (2007).
48 Canli T, Cooney RE, Goldin P et al. Amygdala reactivity to emotional faces predicts improvement in major depression. Neuroreport 16(12), 1267–1270 (2005).

Expert Rev. Clin. Pharmacol. 3(4), (2010)482
Review Rabl, Scharinger, Müller & Pezawas
49 Dannlowski U, Ohrmann P, Bauer J et al. 5-HTTLPR biases amygdala activity in response to masked facial expressions in major depression. Neuropsychopharmacology 33(2), 418–424 (2008).
50 Vogt BA. Cingulate Neurobiology and Disease. Oxford University Press, London, UK (2009).
51 Drevets WC, Price JL, Simpson JR Jr. et al. Subgenual prefrontal cortex abnormalities in mood disorders. Nature 386(6627), 824–827 (1997).
52 Ongur D, Drevets WC, Price JL. Glial reduction in the subgenual prefrontal cortex in mood disorders. Proc. Natl Acad. Sci. USA 95(22), 13290–13295 (1998).
53 Gaspar P, Cases O, Maroteaux L. The developmental role of serotonin: news from mouse molecular genetics. Nat. Rev. Neurosci. 4(12), 1002–1012 (2003).
• Comprehensivereviewpointingoutthedevelopmentalroleofserotonininthebrain,whichfacilitatestheunderstandingofthebiologicalconsequencesofgeneticvariationwithintheserotonintransporter.
54 Kranz GS, Kasper S, Lanzenberger R. Reward and the serotonergic system. Neuroscience 166(4), 1023–1035 (2010).
55 Rose EJ, Simonotto E, Spencer EP, Ebmeier KP. The effects of escitalopram on working memory and brain activity in healthy adults during performance of the n-back task. Psychopharmacology (Berl.) 185(3), 339–347 (2006).
56 Brody AL, Saxena S, Stoessel P et al. Regional brain metabolic changes in patients with major depression treated with either paroxetine or interpersonal therapy: preliminary findings. Arch. Gen. Psychiatry 58(7), 631–640 (2001).
57 Goldapple K, Segal Z, Garson C et al. Modulation of cortical-limbic pathways in major depression: treatment-specific effects of cognitive behavior therapy. Arch. Gen. Psychiatry 61(1), 34–41 (2004).
58 Bauer M, London ED, Rasgon N et al. Supraphysiological doses of levothyroxine alter regional cerebral metabolism and improve mood in bipolar depression. Mol. Psychiatry 10(5), 456–469 (2005).
59 Mayberg HS, Brannan SK, Mahurin RK et al. Cingulate function in depression: a potential predictor of treatment response. Neuroreport 8(4), 1057–1061 (1997).
60 Mayberg HS, Brannan SK, Tekell JL et al. Regional metabolic effects of fluoxetine in major depression: serial changes and relationship to clinical response. Biol. Psychiatry 48(8), 830–843 (2000).
61 Kennedy SH, Konarski JZ, Segal ZV et al. Differences in brain glucose metabolism between responders to cbt and venlafaxine in a 16-week randomized controlled trial. Am. J. Psychiatry 164(5), 778–788 (2007).
62 Davidson RJ, Irwin W, Anderle MJ, Kalin NH. The neural substrates of affective processing in depressed patients treated with venlafaxine. Am. J. Psychiatry 160(1), 64–75 (2003).
63 Chen CH, Ridler K, Suckling J et al. Brain imaging correlates of depressive symptom severity and predictors of symptom improvement after antidepressant treatment. Biol. Psychiatry 62(5), 407–414 (2007).
64 Costafreda SG, Chu C, Ashburner J, Fu CH. Prognostic and diagnostic potential of the structural neuroanatomy of depression. PLoS ONE 4(7), E6353 (2009).
65 Walsh ND, Williams SC, Brammer MJ et al. A longitudinal functional magnetic resonance imaging study of verbal working memory in depression after antidepressant therapy. Biol. Psychiatry 62(11), 1236–1243 (2007).
66 Rolls ET, Grabenhorst F. The orbitofrontal cortex and beyond: from affect to decision-making. Prog. Neurobiol. 86(3), 216–244 (2008).
67 Rushworth MF, Behrens TE, Rudebeck PH, Walton ME. Contrasting roles for cingulate and orbitofrontal cortex in decisions and social behaviour. Trends Cogn. Sci. 11(4), 168–176 (2007).
68 Drevets WC, Price JL, Furey ML. Brain structural and functional abnormalities in mood disorders: implications for neurocircuitry models of depression. Brain Struct. Funct. 213(1–2), 93–118 (2008).
69 Mccabe C, Mishor Z, Cowen PJ, Harmer CJ. Diminished neural processing of aversive and rewarding stimuli during selective serotonin reuptake inhibitor treatment. Biol. Psychiatry 67(5), 439–445
70 Brody AL, Saxena S, Silverman DH et al. Brain metabolic changes in major depressive disorder from pre- to post-treatment with paroxetine. Psychiatry Res. 91(3), 127–139 (1999).
71 Phillips ML, Drevets WC, Rauch SL, Lane R. Neurobiology of emotion perception I: the neural basis of normal emotion perception. Biol. Psychiatry 54(5), 504–514 (2003).
72 Phillips ML, Drevets WC, Rauch SL, Lane R. Neurobiology of emotion perception II: implications for major psychiatric disorders. Biol. Psychiatry 54(5), 515–528 (2003).
73 Koolschijn PC, Van Haren NE, Lensvelt-Mulders GJ, Hulshoff Pol HE, Kahn RS. Brain volume abnormalities in major depressive disorder: a meta-analysis of magnetic resonance imaging studies. Hum. Brain Mapp. 30(11), 3719–3735 (2009).
74 Videbech P, Ravnkilde B. Hippocampal volume and depression: a meta-analysis of MRI studies. Am. J. Psychiatry 161(11), 1957–1966 (2004).
75 Mcewen BS. Plasticity of the hippocampus: adaptation to chronic stress and allostatic load. Ann. N. Y. Acad. Sci. 933, 265–277 (2001).
76 Kasper S, Mcewen BS. Neurobiological and clinical effects of the antidepressant tianeptine. CNS Drugs 22(1), 15–26 (2008).
77 Kennedy SH, Evans KR, Kruger S et al. Changes in regional brain glucose metabolism measured with positron emission tomography after paroxetine treatment of major depression. Am. J. Psychiatry 158(6), 899–905 (2001).
78 Aihara M, Ida I, Yuuki N et al. HPA axis dysfunction in unmedicated major depressive disorder and its normalization by pharmacotherapy correlates with alteration of neural activity in prefrontal cortex and limbic/paralimbic regions. Psychiatry Res. 155(3), 245–256 (2007).
79 Fu CH, Williams SC, Brammer MJ et al. Neural responses to happy facial expressions in major depression following antidepressant treatment. Am. J. Psychiatry 164(4), 599–607 (2007).
80 Macqueen GM, Yucel K, Taylor VH, Macdonald K, Joffe R. Posterior hippocampal volumes are associated with remission rates in patients with major depressive disorder. Biol. Psychiatry 64(10), 880–883 (2008).
81 Frodl T, Jager M, Smajstrlova I et al. Effect of hippocampal and amygdala volumes on clinical outcomes in major depression: a 3-year prospective magnetic resonance imaging study. J. Psychiatry Neurosci. 33(5), 423–430 (2008).
82 Kronmuller KT, Pantel J, Kohler S et al. Hippocampal volume and 2-year outcome in depression. Br. J. Psychiatry 192(6), 472–473 (2008).
83 Vakili K, Pillay SS, Lafer B et al. Hippocampal volume in primary unipolar major depression: a magnetic resonance imaging study. Biol. Psychiatry 47(12), 1087–1090 (2000).

www.expert-reviews.com 483
ReviewImaging genetics: implications for research on variable antidepressant drug response
84 Frodl TS, Koutsouleris N, Bottlender R et al. Depression-related variation in brain morphology over 3 years: effects of stress? Arch. Gen. Psychiatry 65(10), 1156–1165 (2008).
85 Ressler KJ, Mayberg HS. Targeting abnormal neural circuits in mood and anxiety disorders: from the laboratory to the clinic. Nat. Neurosci. 10(9), 1116–1124 (2007).
86 Wang F, Kalmar JH, He Y et al. Functional and structural connectivity between the perigenual anterior cingulate and amygdala in bipolar disorder. Biol. Psychiatry 66(5), 516–521 (2009).
87 Anand A, Li Y, Wang Y et al. Activity and connectivity of brain mood regulating circuit in depression: a functional magnetic resonance study. Biol. Psychiatry 57(10), 1079–1088 (2005).
88 Pezawas L, Meyer-Lindenberg A, Drabant EM et al. 5-HTTLPR polymorphism impacts human cingulate-amygdala interactions: a genetic susceptibility mechanism for depression. Nat. Neurosci. 8(6), 828–834 (2005).
• Importantimaginggeneticsstudy,whichdemonstratesthedevelopmentalandfunctionaleffectsof5-HTTLPRonabrainsystemslevel.Itfurtherhighlightstheimportanceofabrainsystemsviewwithregardtotemperamentalriskfactorsofdepression.
89 Chen CH, Suckling J, Ooi C et al. Functional coupling of the amygdala in depressed patients treated with antidepressant medication. Neuropsychopharmacology 33(8), 1909–1918 (2008).
90 Cremers HR, Demenescu LR, Aleman A et al. Neuroticism modulates amygdala–prefrontal connectivity in response to negative emotional facial expressions. Neuroimage 49(1), 963–970 (2010).
91 Kim MJ, Whalen PJ. The structural integrity of an amygdala–prefrontal pathway predicts trait anxiety. J. Neurosci. 29(37), 11614–11618 (2009).
92 Nebert DW, Zhang G, Vesell ES. From human genetics and genomics to pharmacogenetics and pharmacogenomics: past lessons, future directions. Drug Metab. Rev. 40(2), 187–224 (2008).
93 Kirchheiner J, Seeringer A, Godoy AL et al. CYP2D6 in the brain: genotype effects on resting brain perfusion. Mol. Psychiatry DOI: 10.1038/mp.2010.42 (2010) (Epub ahead of print).
94 Bauer M, Whybrow PC, Angst J, Versiani M, Moller HJ. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of unipolar depressive disorders, part 1: acute and continuation treatment of major depressive disorder. World J. Biol. Psychiatry 3(1), 5–43 (2002).
95 Heils A, Teufel A, Petri S et al. Allelic variation of human serotonin transporter gene expression. J. Neurochem. 66(6), 2621–2624 (1996).
96 Lesch KP, Bengel D, Heils A et al. Association of anxiety-related traits with a polymorphism in the serotonin transporter gene regulatory region. Science 274(5292), 1527–1531 (1996).
97 Caspi A, Sugden K, Moffitt TE et al. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science 301(5631), 386–389 (2003).
• Seminalstudyreportingstress-relatedgene–environmentinteractioninthe5-HTTLPRpromoterpolymorphismofSLC6A4.
98 Kunugi H, Hattori M, Kato T et al. Serotonin transporter gene polymorphisms: ethnic difference and possible association with bipolar affective disorder. Mol. Psychiatry 2(6), 457–462 (1997).
99 Canli T, Lesch KP. Long story short: the serotonin transporter in emotion regulation and social cognition. Nat. Neurosci. 10(9), 1103–1109 (2007).
100 Kalbitzer J, Frokjaer VG, Erritzoe D et al. The personality trait openness is related to cerebral 5-HTT levels. Neuroimage 45(2), 280–285 (2009).
101 Praschak-Rieder N, Kennedy J, Wilson AA et al. Novel 5-HTTLPR allele associates with higher serotonin transporter binding in putamen: a [(11)c] DASB positron emission tomography study. Biol. Psychiatry 62(4), 327–331 (2007).
102 Reimold M, Smolka MN, Schumann G et al. Midbrain serotonin transporter binding potential measured with [11c]DASB is affected by serotonin transporter genotype. J. Neural. Transm. 114(5), 635–639 (2007).
103 Parsey RV, Hastings RS, Oquendo MA et al. Effect of a triallelic functional polymorphism of the serotonin-transporter-linked promoter region on expression of serotonin transporter in the human brain. Am. J. Psychiatry 163(1), 48–51 (2006).
104 Murthy NV, Selvaraj S, Cowen PJ et al. Serotonin transporter polymorphisms (SLC6A4 insertion/deletion and rs25531)
do not affect the availability of 5-HTT to [(11)c] DASB binding in the living human brain. Neuroimage 52(1), 50–54 (2010).
105 Van Dyck CH, Malison RT, Staley JK et al. Central serotonin transporter availability measured with [123i]b-CIT SPECT in relation to serotonin transporter genotype. Am. J. Psychiatry 161(3), 525–531 (2004).
106 Shioe K, Ichimiya T, Suhara T et al. No association between genotype of the promoter region of serotonin transporter gene and serotonin transporter binding in human brain measured by pet. Synapse 48(4), 184–188 (2003).
107 Mann JJ, Huang YY, Underwood MD et al. A serotonin transporter gene promoter polymorphism (5-HTTLPR) and prefrontal cortical binding in major depression and suicide. Arch. Gen. Psychiatry 57(8), 729–738 (2000).
108 Kalbitzer J, Erritzoe D, Holst KK et al. Seasonal changes in brain serotonin transporter binding in short serotonin transporter linked polymorphic region-allele carriers but not in long-allele homozygotes. Biol. Psychiatry 67(11), 1033–1039 (2010).
109 Meyer JH. Imaging the serotonin transporter during major depressive disorder and antidepressant treatment. J. Psychiatry Neurosci. 32(2), 86–102 (2007).
110 Sibille E, Lewis DA. Sert-ainly involved in depression, but when? Am. J. Psychiatry 163(1), 8–11 (2006).
111 Homberg JR, Schubert D, Gaspar P. New perspectives on the neurodevelopmental effects of ssris. Trends Pharmacol. Sci. 31(2), 60–65 (2010).
112 Ansorge MS, Zhou M, Lira A, Hen R, Gingrich JA. Early-life blockade of the 5-HT transporter alters emotional behavior in adult mice. Science 306(5697), 879–881 (2004).
113 Serretti A, Kato M, De Ronchi D, Kinoshita T. Meta-analysis of serotonin transporter gene promoter polymorphism (5-HTTLPR) association with selective serotonin reuptake inhibitor efficacy in depressed patients. Mol. Psychiatry 12(3), 247–257 (2007).
114 Smeraldi E, Zanardi R, Benedetti F, Di Bella D, Perez J, Catalano M. Polymorphism within the promoter of the serotonin transporter gene and antidepressant efficacy of fluvoxamine. Mol. Psychiatry 3(6), 508–511 (1998).
115 Serretti A, Cusin C, Rossini D, Artioli P, Dotoli D, Zanardi R. Further evidence of a combined effect of SERTPR and TPH on

Expert Rev. Clin. Pharmacol. 3(4), (2010)484
Review Rabl, Scharinger, Müller & Pezawas
SSRIS response in mood disorders. Am. J. Med. Genet. B Neuropsychiatr. Genet. 129B(1), 36–40 (2004).
116 Kato M, Ikenaga Y, Wakeno M et al. Controlled clinical comparison of paroxetine and fluvoxamine considering the serotonin transporter promoter polymorphism. Int. Clin. Psychopharmacol. 20(3), 151–156 (2005).
117 Kato M, Fukuda T, Wakeno M et al. Effects of the serotonin type 2a, 3a and 3b receptor and the serotonin transporter genes on paroxetine and fluvoxamine efficacy and adverse drug reactions in depressed japanese patients. Neuropsychobiology 53(4), 186–195 (2006).
118 Zanardi R, Serretti A, Rossini D et al. Factors affecting fluvoxamine antidepressant activity: influence of pindolol and 5-HTTLPR in delusional and nondelusional depression. Biol. Psychiatry 50(5), 323–330 (2001).
119 Pollock BG, Ferrell RE, Mulsant BH et al. Allelic variation in the serotonin transporter promoter affects onset of paroxetine treatment response in late-life depression. Neuropsychopharmacology 23(5), 587–590 (2000).
120 Zanardi R, Benedetti F, Di Bella D, Catalano M, Smeraldi E. Efficacy of paroxetine in depression is influenced by a functional polymorphism within the promoter of the serotonin transporter gene. J. Clin. Psychopharmacol. 20(1), 105–107 (2000).
121 Murphy GM Jr, Hollander SB, Rodrigues HE, Kremer C, Schatzberg AF. Effects of the serotonin transporter gene promoter polymorphism on mirtazapine and paroxetine efficacy and adverse events in geriatric major depression. Arch. Gen. Psychiatry 61(11), 1163–1169 (2004).
122 Bozina N, Peles AM, Sagud M, Bilusic H, Jakovljevic M. Association study of paroxetine therapeutic response with SERT gene polymorphisms in patients with major depressive disorder. World J. Biol. Psychiatry 9(3), 190–197 (2008).
123 Rausch JL, Johnson ME, Fei YJ et al. Initial conditions of serotonin transporter kinetics and genotype: influence on ssri treatment trial outcome. Biol. Psychiatry 51(9), 723–732 (2002).
124 Yu YW, Tsai SJ, Chen TJ, Lin CH, Hong CJ. Association study of the serotonin transporter promoter polymorphism and symptomatology and antidepressant response in major depressive disorders. Mol. Psychiatry 7(10), 1115–1119 (2002).
125 Joyce PR, Mulder RT, Luty SE et al. Age-dependent antidepressant pharmacogenomics: polymorphisms of the serotonin transporter and g protein b3 subunit as predictors of response to fluoxetine and nortriptyline. Int. J. Neuropsychopharmacol. 6(4), 339–346 (2003).
126 Hong CJ, Chen TJ, Yu YW, Tsai SJ. Response to fluoxetine and serotonin 1A receptor (c-1019g) polymorphism in Taiwan Chinese major depressive disorder. Pharmacogenomics J. 6(1), 27–33 (2006).
127 Arias B, Catalan R, Gasto C, Gutierrez B, Fananas L: 5-HTTLPR polymorphism of the serotonin transporter gene predicts non-remission in major depression patients treated with citalopram in a 12-weeks follow up study. J. Clin. Psychopharmacol. 23(6), 563–567 (2003).
128 Huezo-Diaz P, Uher R, Smith R et al. Moderation of antidepressant response by the serotonin transporter gene. Br. J. Psychiatry 195(1), 30–38 (2009).
129 Durham LK, Webb SM, Milos PM, Clary CM, Seymour AB. The serotonin transporter polymorphism, 5HTTLPR, is associated with a faster response time to sertraline in an elderly population with major depressive disorder. Psychopharmacology (Berl.) 174(4), 525–529 (2004).
130 Lee MS, Lee HY, Lee HJ, Ryu SH. Serotonin transporter promoter gene polymorphism and long-term outcome of antidepressant treatment. Psychiatr. Genet. 14(2), 111–115 (2004).
131 Wilkie MJ, Smith G, Day RK et al. Polymorphisms in the SLC6A4 and HTR2A genes influence treatment outcome following antidepressant therapy. Pharmacogenomics J. 9(1), 61–70 (2009).
132 Kim DK, Lim SW, Lee S et al. Serotonin transporter gene polymorphism and antidepressant response. Neuroreport 11(1), 215–219 (2000).
133 Yoshida K, Ito K, Sato K et al. Influence of the serotonin transporter gene-linked polymorphic region on the antidepressant response to fluvoxamine in japanese depressed patients. Prog. Neuropsychopharmacol. Biol. Psychiatry 26(2), 383–386 (2002).
134 Kang RH, Wong ML, Choi MJ, Paik JW, Lee MS. Association study of the serotonin transporter promoter polymorphism and mirtazapine antidepressant response in major depressive disorder. Prog. Neuropsychopharmacol. Biol. Psychiatry 31(6), 1317–1321 (2007).
135 Kim H, Lim SW, Kim S et al. Monoamine transporter gene polymorphisms and antidepressant response in koreans with late-life depression. JAMA 296(13), 1609–1618 (2006).
136 Yoshida K, Takahashi H, Higuchi H et al. Prediction of antidepressant response to milnacipran by norepinephrine transporter gene polymorphisms. Am. J. Psychiatry 161(9), 1575–1580 (2004).
137 Dogan O, Yuksel N, Ergun MA et al. Serotonin transporter gene polymorphisms and sertraline response in major depression patients. Genet. Test 12(2), 225–231 (2008).
138 Kraft JB, Slager SL, Mcgrath PJ, Hamilton SP. Sequence analysis of the serotonin transporter and associations with antidepressant response. Biol. Psychiatry 58(5), 374–381 (2005).
139 Hu XZ, Rush AJ, Charney D et al. Association between a functional serotonin transporter promoter polymorphism and citalopram treatment in adult outpatients with major depression. Arch. Gen. Psychiatry 64(7), 783–792 (2007).
140 Mrazek DA, Rush AJ, Biernacka JM et al. Slc6a4 variation and citalopram response. Am. J. Med. Genet. B Neuropsychiatr. Genet. 150B(3), 341–351 (2009).
141 Maron E, Tammiste A, Kallassalu K et al. Serotonin transporter promoter region polymorphisms do not influence treatment response to escitalopram in patients with major depression. Eur. Neuropsychopharmacol. 19(6), 451–456 (2009).
142 Minov C, Baghai TC, Schule C et al. Serotonin-2a-receptor and -transporter polymorphisms: lack of association in patients with major depression. Neurosci. Lett. 303(2), 119–122 (2001).
143 Kirchheiner J, Nickchen K, Sasse J, Bauer M, Roots I, Brockmoller J. A 40-basepair vntr polymorphism in the dopamine transporter (dat1) gene and the rapid response to antidepressant treatment. Pharmacogenomics J. 7(1), 48–55 (2007).
144 Perlis RH, Mischoulon D, Smoller JW et al. Serotonin transporter polymorphisms and adverse effects with fluoxetine treatment. Biol. Psychiatry 54(9), 879–883 (2003).
145 Popp J, Leucht S, Heres S, Steimer W. Serotonin transporter polymorphisms and side effects in antidepressant therapy – a pilot study. Pharmacogenomics 7(2), 159–166 (2006).
146 Smits K, Smits L, Peeters F et al. Serotonin transporter polymorphisms and the occurrence of adverse events during

www.expert-reviews.com 485
ReviewImaging genetics: implications for research on variable antidepressant drug response
treatment with selective serotonin reuptake inhibitors. Int. Clin. Psychopharmacol. 22(3), 137–143 (2007).
147 Takahashi H, Yoshida K, Ito K et al. No association between the serotonergic polymorphisms and incidence of nausea induced by fluvoxamine treatment. Eur. Neuropsychopharmacol. 12(5), 477–481 (2002).
148 Yu YW, Tsai SJ, Liou YJ, Hong CJ, Chen TJ. Association study of two serotonin 1a receptor gene polymorphisms and fluoxetine treatment response in chinese major depressive disorders. Eur. Neuropsychopharmacol. 16(7), 498–503 (2006).
149 Tanaka M, Kobayashi D, Murakami Y et al. Genetic polymorphisms in the 5-hydroxytryptamine type 3b receptor gene and paroxetine-induced nausea. Int. J. Neuropsychopharmacol. 11(2), 261–267 (2008).
150 Ogilvie AD, Battersby S, Bubb VJ et al. Polymorphism in serotonin transporter gene associated with susceptibility to major depression. Lancet 347(9003), 731–733 (1996).
151 Hu XZ, Lipsky RH, Zhu G et al. Serotonin transporter promoter gain-of-function genotypes are linked to obsessive-compulsive disorder. Am. J. Hum. Genet. 78(5), 815–826 (2006).
152 Hranilovic D, Stefulj J, Schwab S et al. Serotonin transporter promoter and intron 2 polymorphisms: relationship between allelic variants and gene expression. Biol. Psychiatry 55(11), 1090–1094 (2004).
153 Smits KM, Smits LJ, Schouten JS, Stelma FF, Nelemans P, Prins MH. Influence of SERTPR and STin2 in the serotonin transporter gene on the effect of selective serotonin reuptake inhibitors in depression: a systematic review. Mol. Psychiatry 9(5), 433–441 (2004).
154 Andersen J, Taboureau O, Hansen KB et al. Location of the antidepressant binding site in the serotonin transporter: importance of ser-438 in recognition of citalopram and tricyclic antidepressants. J. Biol. Chem. 284(15), 10276–10284 (2009).
155 Tavoulari S, Forrest LR, Rudnick G. Fluoxetine (prozac) binding to serotonin transporter is modulated by chloride and conformational changes. J. Neurosci. 29(30), 9635–9643 (2009).
156 Brown SM, Hariri AR. Neuroimaging studies of serotonin gene polymorphisms: exploring the interplay of genes, brain, and behavior. Cogn. Affect. Behav. Neurosci. 6(1), 44–52 (2006).
157 Munafo MR, Brown SM, Hariri AR. Serotonin transporter (5-HTTLPR) genotype and amygdala activation: a meta-analysis. Biol. Psychiatry 63(9), 852–857 (2008).
158 Hariri AR, Drabant EM, Munoz KE et al. A susceptibility gene for affective disorders and the response of the human amygdala. Arch. Gen. Psychiatry 62(2), 146–152 (2005).
159 Bertolino A, Arciero G, Rubino V et al. Variation of human amygdala response during threatening stimuli as a function of 5́ HTTLPR genotype and personality style. Biol. Psychiatry 57(12), 1517–1525 (2005).
160 Canli T, Omura K, Haas BW, Fallgatter A, Constable RT, Lesch KP. Beyond affect: a role for genetic variation of the serotonin transporter in neural activation during a cognitive attention task. Proc. Natl Acad. Sci. USA 102(34), 12224–12229 (2005).
161 Canli T, Qiu M, Omura K et al. Neural correlates of epigenesis. Proc. Natl Acad. Sci. USA 103(43), 16033–16038 (2006).
162 Friedel E, Schlagenhauf F, Sterzer P et al. 5-HTT genotype effect on prefrontal-amygdala coupling differs between major depression and controls. Psychopharmacology (Berl.) 205(2), 261–271 (2009).
163 Dannlowski U, Konrad C, Kugel H et al. Emotion specific modulation of automatic amygdala responses by 5-HTTLPR genotype. Neuroimage DOI: 10.1016 (2009).
164 Hariri AR, Mattay VS, Tessitore A et al. Serotonin transporter genetic variation and the response of the human amygdala. Science 297(5580), 400–403 (2002).
• ImportantpioneerimaginggeneticsstudyreportingonamygdalahyperreactivityinhealthyS-allelecarriers.
165 Heinz A, Braus DF, Smolka MN et al. Amygdala-prefrontal coupling depends on a genetic variation of the serotonin transporter. Nat. Neurosci. 8(1), 20–21 (2005).
166 Rao H, Gillihan SJ, Wang J et al. Genetic variation in serotonin transporter alters resting brain function in healthy individuals. Biol. Psychiatry 62(6), 600–606 (2007).
167 Smolka MN, Buhler M, Schumann G et al. Gene–gene effects on central processing of aversive stimuli. Mol. Psychiatry 12(3), 307–317 (2007).
168 Williams LM, Gatt JM, Schofield PR, Olivieri G, Peduto A, Gordon E. ‘Negativity bias’ in risk for depression and
anxiety: brain-body fear circuitry correlates, 5-HTT-LPR and early life stress. Neuroimage 47(3), 804–814 (2009).
169 Dannlowski U, Ohrmann P, Bauer J et al. Serotonergic genes modulate amygdala activity in major depression. Genes Brain Behav. 6(7), 672–676 (2007).
170 Graff-Guerrero A, De La Fuente-Sandoval C, Camarena B et al. Frontal and limbic metabolic differences in subjects selected according to genetic variation of the SLC6A4 gene polymorphism. Neuroimage 25(4), 1197–1204 (2005).
171 Fukudo S, Kanazawa M, Mizuno T et al. Impact of serotonin transporter gene polymorphism on brain activation by colorectal distention. Neuroimage 47(3), 946–951 (2009).
172 Passamonti L, Cerasa A, Gioia MC et al. Genetically dependent modulation of serotonergic inactivation in the human prefrontal cortex. Neuroimage 40(3), 1264–1273 (2008).
173 Roiser JP, De Martino B, Tan GC et al. A genetically mediated bias in decision making driven by failure of amygdala control. J. Neurosci. 29(18), 5985–5991 (2009).
174 Shah MP, Wang F, Kalmar JH et al. Role of variation in the serotonin transporter protein gene (SLC6A4) in trait disturbances in the ventral anterior cingulate in bipolar disorder. Neuropsychopharmacology 34(5), 1301–1310 (2009).
175 Frodl T, Koutsouleris N, Bottlender R et al. Reduced gray matter brain volumes are associated with variants of the serotonin transporter gene in major depression. Mol. Psychiatry 13(12), 1093–1101 (2008).
176 Jedema HP, Gianaros PJ, Greer PJ et al. Cognitive impact of genetic variation of the serotonin transporter in primates is associated with differences in brain morphology rather than serotonin neurotransmission. Mol. Psychiatry 15(5), 512–122 (2009).
177 Benedetti F, Bernasconi A, Blasi V et al. Neural and genetic correlates of antidepressant response to sleep deprivation: a functional magnetic resonance imaging study of moral valence decision in bipolar depression. Arch. Gen. Psychiatry 64(2), 179–187 (2007).
178 Pacheco J, Beevers CG, Benavides C, Mcgeary J, Stice E, Schnyer DM. Frontal-limbic white matter pathway associations with the serotonin transporter gene promoter region (5-HTTLPR) polymorphism. J. Neurosci. 29(19), 6229–6233 (2009).

Expert Rev. Clin. Pharmacol. 3(4), (2010)486
Review Rabl, Scharinger, Müller & Pezawas
179 Lemonde S, Turecki G, Bakish D et al. Impaired repression at a 5-hydroxytryptamine 1a receptor gene polymorphism associated with major depression and suicide. J. Neurosci. 23(25), 8788–8799 (2003).
180 Neff CD, Abkevich V, Packer JC et al. Evidence for HTRLA and LHPP as interacting genetic risk factors in major depression. Mol. Psychiatry 14(6), 621–630 (2009).
181 Savitz J, Lucki I, Drevets WC. 5-HT(1A) receptor function in major depressive disorder. Prog. Neurobiol. 88(1), 17–31 (2009).
182 Gross C, Zhuang X, Stark K et al. Serotonin 1a receptor acts during development to establish normal anxiety-like behaviour in the adult. Nature 416(6879), 396–400 (2002).
183 Zanettini C, Carola V, Lo Iacono L, Moles A, Gross C, D’amato FR. Postnatal handling reverses social anxiety in serotonin receptor 1a knockout mice. Genes Brain Behav. 9(1), 26–32 (2010).
184 Lemonde S, Du L, Bakish D, Hrdina P, Albert PR. Association of the C(-1019)G 5-HT1A functional promoter polymorphism with antidepressant response. Int. J. Neuropsychopharmacol. 7(4), 501–506 (2004).
185 Parsey RV, Olvet DM, Oquendo MA, Huang YY, Ogden RT, Mann JJ. Higher 5-HT
1A receptor binding potential during a
major depressive episode predicts poor treatment response: preliminary data from a naturalistic study. Neuropsychopharmacology 31(8), 1745–1749 (2006).
186 Kato M, Fukuda T, Wakeno M et al. Effect of 5-HT1A gene polymorphisms on antidepressant response in major depressive disorder. Am. J. Med. Genet. B Neuropsychiatr. Genet. 150B(1), 115–123 (2009).
187 Baune BT, Hohoff C, Roehrs T, Deckert J, Arolt V, Domschke K. Serotonin receptor 1A-1019C/G variant: impact on antidepressant pharmacoresponse in melancholic depression? Neurosci. Lett. 436(2), 111–115 (2008).
188 Arias B, Catalan R, Gasto C, Gutierrez B, Fananas L. Evidence for a combined genetic effect of the 5-HT(1A) receptor and serotonin transporter genes in the clinical outcome of major depressive patients treated with citalopram. J. Psychopharmacol. 19(2), 166–172 (2005).
189 Anttila S, Huuhka K, Huuhka M et al. Interaction between 5-HT1A and BDNF genotypes increases the risk of treatment-
resistant depression. J. Neural. Transm. 114(8), 1065–1068 (2007).
190 Peters EJ, Slager SL, Mcgrath PJ, Knowles JA, Hamilton SP. Investigation of serotonin-related genes in antidepressant response. Mol. Psychiatry 9(9), 879–889 (2004).
191 Serretti A, Artioli P, Lorenzi C, Pirovano A, Tubazio V, Zanardi R. The C(-1019)G polymorphism of the 5-HT1A gene promoter and antidepressant response in mood disorders: preliminary findings. Int. J. Neuropsychopharmacol. 7(4), 453–460 (2004).
192 Levin GM, Bowles TM, Ehret MJ et al. Assessment of human serotonin 1a receptor polymorphisms and SSRI responsiveness. Mol. Diagn. Ther. 11(3), 155–160 (2007).
193 Lin E, Chen PS, Chang HH et al. Interaction of serotonin-related genes affects short-term antidepressant response in major depressive disorder. Prog. Neuropsychopharmacol. Biol. Psychiatry 33(7), 1167–1172 (2009).
194 Fakra E, Hyde LW, Gorka A et al. Effects of HTR1A C(-1019)G on amygdala reactivity and trait anxiety. Arch. Gen. Psychiatry 66(1), 33–40 (2009).
195 Lee BT, Ham BJ. Serotonergic genes and amygdala activity in response to negative affective facial stimuli in korean women. Genes Brain Behav. 7(8), 899–905 (2008).
196 Sabol SZ, Hu S, Hamer D. A functional polymorphism in the monoamine oxidase A gene promoter. Hum. Genet. 103(3), 273–279 (1998).
197 Cusin C, Serretti A, Zanardi R et al. Influence of monoamine oxidase a and serotonin receptor 2a polymorphisms in SSRI antidepressant activity. Int. J. Neuropsychopharmacol. 5(1), 27–35 (2002).
198 Muller DJ, Schulze TG, Macciardi F et al. Moclobemide response in depressed patients: association study with a functional polymorphism in the monoamine oxidase a promoter. Pharmacopsychiatry 35(4), 157–158 (2002).
199 Yoshida K, Naito S, Takahashi H et al. Monoamine oxidase: a gene polymorphism, tryptophan hydroxylase gene polymorphism and antidepressant response to fluvoxamine in japanese patients with major depressive disorder. Prog. Neuropsychopharmacol. Biol. Psychiatry 26(7–8), 1279–1283 (2002).
200 Yu YW, Tsai SJ, Hong CJ, Chen TJ, Chen MC, Yang CW. Association study of a monoamine oxidase a gene promoter polymorphism with major depressive
disorder and antidepressant response. Neuropsychopharmacology 30(9), 1719–1723 (2005).
201 Lee BT, Ham BJ. Monoamine oxidase A-UVNTR genotype affects limbic brain activity in response to affective facial stimuli. Neuroreport 19(5), 515–519 (2008).
202 Meyer-Lindenberg A, Buckholtz JW, Kolachana B et al. Neural mechanisms of genetic risk for impulsivity and violence in humans. Proc. Natl Acad. Sci. USA 103(16), 6269–6274 (2006).
203 Cerasa A, Gioia MC, Labate A et al. Mao a vntr polymorphism and variation in human morphology: a VBM study. Neuroreport 19(11), 1107–1110 (2008).
204 Buckholtz JW, Callicott JH, Kolachana B et al. Genetic variation in MAOA modulates ventromedial prefrontal circuitry mediating individual differences in human personality. Mol. Psychiatry 13(3), 313–324 (2008).
205 Dannlowski U, Ohrmann P, Konrad C et al. Reduced amygdala–prefrontal coupling in major depression: association with maoa genotype and illness severity. Int. J. Neuropsychopharmacol. 12(1), 11–22 (2009).
206 Craddock N, Owen MJ, O’Donovan MC. The catechol-O-methyl transferase (COMT ) gene as a candidate for psychiatric phenotypes: evidence and lessons. Mol. Psychiatry 11(5), 446–458 (2006).
207 Tunbridge EM, Harrison PJ, Weinberger DR. Catechol-O-methyltransferase, cognition, and psychosis: Val158Met and beyond. Biol. Psychiatry 60(2), 141–151 (2006).
208 Krugel LK, Biele G, Mohr PN, Li SC, Heekeren HR. Genetic variation in dopaminergic neuromodulation influences the ability to rapidly and flexibly adapt decisions. Proc. Natl Acad. Sci. USA 106(42), 17951–17956 (2009).
209 Stein DJ. Depression, anhedonia, and psychomotor symptoms: the role of dopaminergic neurocircuitry. CNS Spectr. 13(7), 561–565 (2008).
210 Baune BT, Hohoff C, Berger K et al. Association of the COMT Val158Met variant with antidepressant treatment response in major depression. Neuropsychopharmacology 33(4), 924–932 (2008).
211 Benedetti F, Colombo C, Pirovano A, Marino E, Smeraldi E. The catechol-O-methyltransferase Val(108/158)Met polymorphism affects antidepressant response to paroxetine in a naturalistic setting. Psychopharmacology (Berl.) 203(1), 155–160 (2009).

www.expert-reviews.com 487
ReviewImaging genetics: implications for research on variable antidepressant drug response
212 Yoshida K, Higuchi H, Takahashi H et al. Influence of the tyrosine hydroxylase Val81Met polymorphism and catechol-O-methyltransferase Val158Met polymorphism on the antidepressant effect of milnacipran. Hum. Psychopharmacol. 23(2), 121–128 (2008).
213 Tsai SJ, Gau YT, Hong CJ, Liou YJ, Yu YW, Chen TJ. Sexually dimorphic effect of catechol-O-methyltransferase Val158Met polymorphism on clinical response to fluoxetine in major depressive patients. J. Affect. Disord. 113(1–2), 183–187 (2009).
214 Szegedi A, Rujescu D, Tadic A et al. The catechol-O-methyltransferase Val108/158Met polymorphism affects short-term treatment response to mirtazapine, but not to paroxetine in major depression. Pharmacogenomics J. 5(1), 49–53 (2005).
215 Arias B, Serretti A, Lorenzi C, Gasto C, Catalan R, Fananas L. Analysis of COMT gene (Val 158 Met polymorphism) in the clinical response to ssris in depressive patients of european origin. J. Affect. Disord. 90(2–3), 251–256 (2006).
216 Perlis RH, Fijal B, Adams DH, Sutton VK, Trivedi MH, Houston JP. Variation in catechol-O-methyltransferase is associated with duloxetine response in a clinical trial for major depressive disorder. Biol. Psychiatry 65(9), 785–791 (2009).
217 Secher A, Bukh J, Bock C et al. Antidepressive-drug-induced bodyweight gain is associated with polymorphisms in genes coding for COMT and TPH1. Int. Clin. Psychopharmacol. 24(4), 199–203 (2009).
218 Taylor WD, Zuchner S, Payne ME et al. The COMT Val158Met polymorphism and temporal lobe morphometry in healthy adults. Psychiatry Res. 155(2), 173–177 (2007).
219 Cerasa A, Gioia MC, Labate A, Liguori M, Lanza P, Quattrone A. Impact of catechol-O-methyltransferase Val(108/158)Met genotype on hippocampal and prefrontal gray matter volume. Neuroreport 19(4), 405–408 (2008).
220 Ehrlich S, Morrow EM, Roffman JL et al. The COMT Val108/158Met polymorphism and medial temporal lobe volumetry in patients with schizophrenia and healthy adults. Neuroimage DOI: 10.1016/j.neuroimage.2009.12.046 (2009) (Epub ahead of print).
221 Williams LM, Gatt JM, Grieve SM et al. COMT Val(108/158)Met polymorphism effects on emotional brain function and negativity bias. Neuroimage (2010).
222 Smolka MN, Schumann G, Wrase J et al. Catechol-O-methyltransferase Val158Met genotype affects processing of emotional stimuli in the amygdala and prefrontal cortex. J. Neurosci. 25(4), 836–842 (2005).
223 Drabant EM, Hariri AR, Meyer-Lindenberg A et al. Catechol O-methyltransferase Val158Met genotype and neural mechanisms related to affective arousal and regulation. Arch. Gen. Psychiatry 63(12), 1396–1406 (2006).
224 Kempton MJ, Haldane M, Jogia J et al. The effects of gender and COMT Val158Met polymorphism on fearful facial affect recognition: a fMRI study. Int. J. Neuropsychopharmacol. 12(3), 371–381 (2009).
225 Pomarol-Clotet E, Fatjo-Vilas M, Mckenna PJ et al. COMT Val158Met polymorphism in relation to activation and de-activation in the prefrontal cortex: a study in patients with schizophrenia and healthy subjects. Neuroimage DOI: 10.1016/j.neuroimage.2010.04.018 (2010) (Epub ahead of print).
226 Bishop SJ, Cohen JD, Fossella J, Casey BJ, Farah MJ. COMT genotype influences prefrontal response to emotional distraction. Cogn. Affect. Behav. Neurosci. 6(1), 62–70 (2006).
227 Dreher JC, Kohn P, Kolachana B, Weinberger DR, Berman KF. Variation in dopamine genes influences responsivity of the human reward system. Proc. Natl Acad. Sci. USA 106(2), 617–622 (2009).
228 Krach S, Jansen A, Krug A et al. COMT genotype and its role on hippocampal-prefrontal regions in declarative memory. Neuroimage DOI: 10.1016/j.neuroimage.2009.12.090 (2010).
229 Honea R, Verchinski BA, Pezawas L et al. Impact of interacting functional variants in COMT on regional gray matter volume in human brain. Neuroimage 45(1), 44–51 (2009).
230 Ohnishi T, Hashimoto R, Mori T et al. The association between the Val158Met polymorphism of the catechol-O-methyl transferase gene and morphological abnormalities of the brain in chronic schizophrenia. Brain 129(Pt 2), 399–410 (2006).
231 Lu B, Pang PT, Woo NH. The yin and yang of neurotrophin action. Nat. Rev. Neurosci. 6(8), 603–614 (2005).
232 Hu Y, Russek SJ. BDNF and the diseased nervous system: a delicate balance between adaptive and pathological processes of gene regulation. J. Neurochem. 105(1), 1–17 (2008).
233 Stein DJ, Daniels WM, Savitz J, Harvey BH. Brain-derived neurotrophic factor: the neurotrophin hypothesis of psychopathology. CNS Spectr. 13(11), 945–949 (2008).
234 Kozisek ME, Middlemas D, Bylund DB. Brain-derived neurotrophic factor and its receptor tropomyosin-related kinase B in the mechanism of action of antidepressant therapies. Pharmacol. Ther. 117(1), 30–51 (2008).
235 Castren E. Is mood chemistry? Nat. Rev. Neurosci. 6(3), 241–246 (2005).
236 Groves JO. Is it time to reassess the BDNF hypothesis of depression? Mol. Psychiatry 12(12), 1079–1088 (2007).
237 Martinowich K, Manji H, Lu B. New insights into BDNF function in depression and anxiety. Nat. Neurosci. 10(9), 1089–1093 (2007).
238 Tsankova NM, Berton O, Renthal W, Kumar A, Neve RL, Nestler EJ. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat. Neurosci. 9(4), 519–525 (2006).
239 Licinio J, Dong C, Wong ML. Novel sequence variations in the brain-derived neurotrophic factor gene and association with major depression and antidepressant treatment response. Arch. Gen. Psychiatry 66(5), 488–497 (2009).
• Recentstudyaddressingthepotentialofextensiveresequencingofkeycandidategenes,whichcanleadtothediscoveryofsubstantialnumbersofnewvariants.
240 Egan MF, Kojima M, Callicott JH et al. The BDNF Val66Met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell 112(2), 257–269 (2003).
241 Chen L, Lawlor DA, Lewis SJ et al. Genetic association study of BDNF in depression: finding from two cohort studies and a meta-analysis. Am. J. Med. Genet. B Neuropsychiatr. Genet. 147B(6), 814–821 (2008).
242 Verhagen M, Van Der Meij A, van Deurzen PA et al. Meta-analysis of the VDNF Val66Met polymorphism in major depressive disorder: effects of gender and ethnicity. Mol. Psychiatry 15(3), 260–271 (2010).
243 Lopez-Leon S, Janssens AC, Gonzalez-Zuloeta Ladd AM et al. Meta-analyses of genetic studies on major depressive disorder. Mol. Psychiatry 13(8), 772–785 (2008).
244 Frustaci A, Pozzi G, Gianfagna F, Manzoli L, Boccia S. Meta-analysis of the

Expert Rev. Clin. Pharmacol. 3(4), (2010)488
Review Rabl, Scharinger, Müller & Pezawas
brain-derived neurotrophic factor gene (BDNF) Val66Met polymorphism in anxiety disorders and anxiety-related personality traits. Neuropsychobiology 58(3–4), 163–170 (2008).
245 Saarelainen T, Hendolin P, Lucas G et al. Activation of the TrkB neurotrophin receptor is induced by antidepressant drugs and is required for antidepressant-induced behavioral effects. J. Neurosci. 23(1), 349–357 (2003).
246 Wilkie MJ, Smith D, Reid IC et al. A splice site polymorphism in the g-protein b subunit influences antidepressant efficacy in depression. Pharmacogenet. Genomics 17(3), 207–215 (2007).
247 Yoshida K, Higuchi H, Kamata M et al. The G196A polymorphism of the brain-derived neurotrophic factor gene and the antidepressant effect of milnacipran and fluvoxamine. J. Psychopharmacol. 21(6), 650–656 (2007).
248 Gratacos M, Soria V, Urretavizcaya M et al. A brain-derived neurotrophic factor (BDNF) haplotype is associated with antidepressant treatment outcome in mood disorders. Pharmacogenomics J. 8(2), 101–112 (2008).
249 Rajewska-Rager A, Skibinska M, Szczepankiewicz A et al. [Association between polymorphisms of Val66Met in the BDNF gene and the response to escitalopram and nortriptyline treatment in the light of the neurodevelopmental hypothesis of depression]. Psychiatr. Pol. 42(6), 915–923 (2008).
250 Domschke K, Lawford B, Laje G et al. Brain-derived neurotrophic factor (BDNF) gene: no major impact on antidepressant treatment response. Int. J. Neuropsychopharmacol. 13(1), 93–101 (2010).
251 Choi MJ, Kang RH, Lim SW, Oh KS, Lee MS. Brain-derived neurotrophic factor gene polymorphism (Val66Met) and citalopram response in major depressive disorder. Brain Res. 1118(1), 176–182 (2006).
252 Tsai SJ, Cheng CY, Yu YW, Chen TJ, Hong CJ. Association study of a brain-derived neurotrophic-factor genetic polymorphism and major depressive disorders, symptomatology, and antidepressant response. Am. J. Med. Genet. B Neuropsychiatr. Genet. 123B(1), 19–22 (2003).
253 Masui T, Hashimoto R, Kusumi I et al. Lithium response and Val66Met polymorphism of the brain-derived neurotrophic factor gene in japanese patients with bipolar disorder. Psychiatr. Genet. 16(2), 49–50 (2006).
254 Zou YF, Ye DQ, Feng XL, Su H, Pan FM, Liao FF. Meta-analysis of BDNF Val66Met polymorphism association with treatment response in patients with major depressive disorder. Eur. Neuropsychopharmacol. DOI: 10.1016/j.euroneuro.2009.12.005 (2010) (Epub ahead of print).
255 Chen ZY, Jing D, Bath KG et al. Genetic variant BDNF (Val66Met) polymorphism alters anxiety-related behavior. Science 314(5796), 140–143 (2006).
256 Frodl T, Schule C, Schmitt G et al. Association of the brain-derived neurotrophic factor Val66Met polymorphism with reduced hippocampal volumes in major depression. Arch. Gen. Psychiatry 64(4), 410–416 (2007).
257 Matsuo K, Walss-Bass C, Nery FG et al. Neuronal correlates of brain-derived neurotrophic factor Val66Met polymorphism and morphometric abnormalities in bipolar disorder. Neuropsychopharmacology 34(8), 1904–1913 (2009).
258 Pezawas L, Verchinski BA, Mattay VS et al. The brain-derived neurotrophic factor Val66Met polymorphism and variation in human cortical morphology. J. Neurosci. 24(45), 10099–10102 (2004).
259 Schofield PR, Williams LM, Paul RH et al. Disturbances in selective information processing associated with the BDNF Val66Met polymorphism: evidence from cognition, the p300 and fronto-hippocampal systems. Biol. Psychol. 80(2), 176–188 (2009).
260 Montag C, Weber B, Fliessbach K, Elger C, Reuter M. The BDNF Val66Met polymorphism impacts parahippocampal and amygdala volume in healthy humans: incremental support for a genetic risk factor for depression. Psychol. Med. 39(11), 1831–1839 (2009).
261 Chepenik LG, Fredericks C, Papademetris X et al. Effects of the brain-derived neurotrophic growth factor Val66Met variation on hippocampus morphology in bipolar disorder. Neuropsychopharmacology 34(4), 944–951 (2009).
262 Bueller JA, Aftab M, Sen S, Gomez-Hassan D, Burmeister M, Zubieta JK. BDNF Val66Met allele is associated with reduced hippocampal volume in healthy subjects. Biol. Psychiatry 59(9), 812–815 (2006).
263 Szeszko PR, Lipsky R, Mentschel C et al. Brain-derived neurotrophic factor Val66Met polymorphism and volume of the hippocampal formation. Mol. Psychiatry 10(7), 631–636 (2005).
264 Jessen F, Schuhmacher A, Von Widdern O et al. No association of the Val66Met polymorphism of the brain-derived neurotrophic factor with hippocampal volume in major depression. Psychiatr. Genet. 19(2), 99–101 (2009).
265 Koolschijn PC, van Haren NE, Bakker SC, Hoogendoorn ML, Pol HE, Kahn RS. Effects of brain-derived neurotrophic factor Val66Met polymorphism on hippocampal volume change in schizophrenia. Hippocampus DOI: 10.1002/hipo.20699 (2009) (Epub ahead of print).
266 Joffe RT, Gatt JM, Kemp AH et al. Brain derived neurotrophic factor Val66Met polymorphism, the five factor model of personality and hippocampal volume: implications for depressive illness. Hum. Brain Mapp. 30(4), 1246–1256 (2009).
267 Hashimoto R, Moriguchi Y, Yamashita F et al. Dose-dependent effect of the Val66Met polymorphism of the brain-derived neurotrophic factor gene on memory-related hippocampal activity. Neurosci. Res. 61(4), 360–367 (2008).
268 Hariri AR, Goldberg TE, Mattay VS et al. Brain-derived neurotrophic factor Val66Met polymorphism affects human memory-related hippocampal activity and predicts memory performance. J. Neurosci. 23(17), 6690–6694 (2003).
269 Monteggia LM, Barrot M, Powell CM et al. Essential role of brain-derived neurotrophic factor in adult hippocampal function. Proc. Natl Acad. Sci. USA 101(29), 10827–10832 (2004).
270 Heldt SA, Stanek L, Chhatwal JP, Ressler KJ. Hippocampus-specific deletion of BDNF in adult mice impairs spatial memory and extinction of aversive memories. Mol. Psychiatry 12(7), 656–670 (2007).
271 Adachi M, Barrot M, Autry AE, Theobald D, Monteggia LM. Selective loss of brain-derived neurotrophic factor in the dentate gyrus attenuates antidepressant efficacy. Biol. Psychiatry 63(7), 642–649 (2008).
272 Sublette ME, Baca-Garcia E, Parsey RV et al. Effect of BDNF Val66Met polymorphism on age-related amygdala volume changes in healthy subjects. Prog. Neuropsychopharmacol. Biol. Psychiatry 32(7), 1652–1655 (2008).
273 Nemoto K, Ohnishi T, Mori T et al. The Val66Met polymorphism of the brain-derived neurotrophic factor gene affects age-related brain morphology. Neurosci. Lett. 397(1–2), 25–29 (2006).

www.expert-reviews.com 489
ReviewImaging genetics: implications for research on variable antidepressant drug response
274 Gasic GP, Smoller JW, Perlis RH et al. BDNF, relative preference, and reward circuitry responses to emotional communication. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 150B(6), 762–781 (2009).
275 Soliman F, Glatt CE, Bath KG et al. A genetic variant BDNF polymorphism alters extinction learning in both mouse and human. Science 327(5967), 863–866 (2010).
276 Montag C, Reuter M, Newport B, Elger C, Weber B. The BDNF Val66Met polymorphism affects amygdala activity in response to emotional stimuli: evidence from a genetic imaging study. Neuroimage 42(4), 1554–1559 (2008).
277 Logothetis NK. What we can do and what we cannot do with fMRI. Nature 453(7197), 869–878 (2008).
278 Riley BP, Mcguffin P. Linkage and associated studies of schizophrenia. Am. J. Med. Genet. 97(1), 23–44 (2000).
279 Menzel S. Genetic and molecular analyses of complex metabolic disorders: genetic linkage. Ann. N. Y. Acad. Sci. 967, 249–257 (2002).
280 Altshuler D, Daly MJ, Lander ES. Genetic mapping in human disease. Science 322(5903), 881–888 (2008).
281 Lohmueller KE, Pearce CL, Pike M, Lander ES, Hirschhorn JN. Meta-analysis of genetic association studies supports a contribution of common variants to susceptibility to common disease. Nat. Genet. 33(2), 177–182 (2003).
282 Hirschhorn JN, Lohmueller K, Byrne E, Hirschhorn K. A comprehensive review of genetic association studies. Genet. Med. 4(2), 45–61 (2002).
283 Garriock HA, Hamilton SP. Genetic studies of drug response and side effects in the STAR*D study, part 1. J. Clin. Psychiatry 70(8), 1186–1187 (2009).
284 Krishnan V, Nestler EJ. The molecular neurobiology of depression. Nature 455(7215), 894–902 (2008).
285 Lesch KP. Gene-environment interaction and the genetics of depression. J. Psychiatry Neurosci. 29(3), 174–184 (2004).
286 Gottesman, II, Gould TD. The endophenotype concept in psychiatry: etymology and strategic intentions. Am. J. Psychiatry 160(4), 636–645 (2003).
287 Meyer-Lindenberg A, Weinberger DR. Intermediate phenotypes and genetic mechanisms of psychiatric disorders. Nat. Rev. Neurosci. 7(10), 818–827 (2006).
288 Flint J, Munafo MR. The endophenotype concept in psychiatric genetics. Psychol. Med. 37(2), 163–180 (2007).
289 Mier D, Kirsch P, Meyer-Lindenberg A. Neural substrates of pleiotropic action of genetic variation in COMT: a meta-analysis. Mol. Psychiatry DOI: 0.1038/mp.2009.36 (2009) (Epub ahead of print).
290 Huettel SA, Song AW, Mccarthy G. Functional Magnetic Resoncance Imaging. Sinauer Associates, Inc., MA. USA (2009).
291 Meyer-Lindenberg A, Nicodemus KK, Egan MF, Callicott JH, Mattay V, Weinberger DR. False positives in imaging genetics. Neuroimage 40, 655–661 (2008).
292 Frodl T, Meisenzahl EM, Zetzsche T et al. Larger amygdala volumes in first depressive episode as compared to recurrent major depression and healthy control subjects. Biol. Psychiatry 53(4), 338–344 (2003).
293 Kronmuller KT, Schroder J, Kohler S et al. Hippocampal volume in first episode and recurrent depression. Psychiatry Res. 174(1), 62–66 (2009).
294 Neumeister A, Wood S, Bonne O et al. Reduced hippocampal volume in unmedicated, remitted patients with major depression versus control subjects. Biol. Psychiatry 57(8), 935–937 (2005).
295 Burmeister M, Mcinnis MG, Zollner S. Psychiatric genetics: progress amid controversy. Nat. Rev. Genet. 9(7), 527–540 (2008).
296 Della Pasqua O, Santen GW, Danhof M. The missing link between clinical endpoints and drug targets in depression. Trends Pharmacol. Sci. 31(4), 144–152 (2010).
297 Duman RS, Monteggia LM. A neurotrophic model for stress-related mood disorders. Biol. Psychiatry 59(12), 1116–1127 (2006).
298 Pezawas L, Meyer-Lindenberg A, Goldman AL et al. Evidence of biologic epistasis between BDNF and SLC6A4 and implications for depression. Mol. Psychiatry 13(7), 709–716 (2008).
• MorphologicalimaginggeneticsstudydealingwiththecomplexinteractionsofSLC6A4andBDNFanditseffectonmoodbraincircuitries.
299 Canli T, Congdon E, Todd Constable R, Lesch KP. Additive effects of serotonin transporter and tryptophan hydroxylase-2 gene variation on neural correlates of affective processing. Biol. Psychol. 79(1), 118–125 (2008).
300 Gatt JM, Nemeroff CB, Dobson-Stone C et al. Interactions between BDNF
Val66Met polymorphism and early life stress predict brain and arousal pathways to syndromal depression and anxiety. Mol. Psychiatry 14(7), 681–695 (2009).
301 Meyer-Lindenberg A, Nichols T, Callicott JH et al. Impact of complex genetic variation in COMT on human brain function. Mol. Psychiatry 11(9), 867–877, 797 (2006).
302 Ising M, Lucae S, Binder EB et al. A genomewide association study points to multiple loci that predict antidepressant drug treatment outcome in depression. Arch. Gen. Psychiatry 66(9), 966–975 (2009).
303 Garriock HA, Kraft JB, Shyn SI et al. A genomewide association study of citalopram response in major depressive disorder. Biol. Psychiatry 67(2), 133–138 (2010).
304 Mcmahon FJ, Akula N, Schulze TG et al. Meta-analysis of genome-wide association data identifies a risk locus for major mood disorders on 3p21.1. Nat. Genet. 42(2), 128–131 (2010).
305 Shyn SI, Shi J, Kraft JB et al. Novel loci for major depression identified by genome-wide association study of sequenced treatment alternatives to relieve depression and meta-analysis of three studies. Mol. Psychiatry DOI: 10.1038/mp.2009.125 (2009) (Epub ahead of print).
306 Shi J, Potash JB, Knowles JA et al. Genome-wide association study of recurrent early-onset major depressive disorder. Mol. Psychiatry DOI: 10.1038/mp.2009.124 (2010) (Epub ahead of print).
307 Muglia P, Tozzi F, Galwey NW et al. Genome-wide association study of recurrent major depressive disorder in two european case–control cohorts. Mol. Psychiatry 15(6), 589–601 (2010).
308 Mcmahon FJ. Pioneering first steps and cautious conclusions. Biol. Psychiatry 67(2), 99–100 (2010).
309 Esslinger C, Walter H, Kirsch P et al. Neural mechanisms of a genome-wide supported psychosis variant. Science 324(5927), 605 (2009).
310 Walter H, Schnell K, Erk S et al. Effects of a genome-wide supported psychosis risk variant on neural activation during a theory-of-mind task. Mol. Psychiatry DOI: 10.1038/mp.2010.18 (2010) (Epub ahead of print).
311 Stein JL, Hua X, Lee S et al. Voxelwise genome-wide association study (vGWAS). Neuroimage DOI: 10.1016/j.neuroimage.2010.02.032 (2010) (Epub ahead of print).





























