Embed Size (px)
Citation preview

Antidepressant pharmacotherapy in
epilepsy:
The effects of chronic fluoxetine and citalopram
treatments in a rat model of epileptogenesis
LISA CARDAMONE
Submitted in total fulfilment of the requirements of the
degree of Master of Philosophy (by Research)
December 2012
Department of Medicine
Royal Melbourne Hospital
Faculty of Medicine, Dentistry and Health Sciences
The University of Melbourne

ii

iii
Just keep swimming. Just keep swimming.
Just keep swimming, swimming, swimming.
Dory, Finding Nemo

iv

v
ABSTRACT
Introduction
In patients with epilepsy there is a high incidence of comorbid psychiatric illnesses, especially
mood and anxiety disorders, which have been associated with lower quality of life, impaired
function and an elevated risk of suicide. In fact, the occurrence of these illnesses in patients with
epilepsy has been reported to be a stronger predictor of quality of life than epilepsy variables
such as illness duration or seizure frequency. In addition, there is evidence that the depressed
state itself may predispose to seizures and epilepsy. For these reasons, effective management of
depressive and anxiety symptoms and syndromes in epilepsy is essential. Selective serotonin
reuptake inhibitors (SSRIs) are commonly used to treat depression in epilepsy, therefore it is
important to consider their impact on epilepsy. To date, many studies have suggested that SSRI
are safe for use in epilepsy, but the majority of these studies adminstered SSRIs only acutely or
for short periods, and investigated effects only on acute seizure endpoints. There is no
indication of the effect that chronic SSRI treatment may have on epileptogenesis and the
associated neurobiological changes that continue after seizures emerge. This thesis aimed to
investigate the effects of chronic SSRI treatment in a rat model of epileptogenesis, as well as
investigating common neurobiological substrates of SSRI treatment and epileptogenesis that
may also influence the disorder. It was hypothesised that chronic SSRI treatment would slow
the rate of kindling epileptogenesis, as well as mitigate effects on common neurobiological
substrates.
Methods
The amygdala kindling model was used to assess the effects of chronic SSRI treatment (with
fluoxetine or citalopram) on epileptogenesis. 9-11 week old male Wistar rats were surgical
implanted with a bipolar electrode into the left amygdala for electrical kindling and a
subcutaneously implanted osmotic pump filled with fluoxetine (10mg/kg/day, n=19) or vehicle
(50% DMSO, n=22) or citalopram (10mg/kg/day, n=26) or vehicle (50% DMSO, n=22),
comprising two separate cohorts. All rats were given 30 stimulations and then kindling rate,
seizure duration and seizure threshold before and after kindling were monitored. Effects on
anxiety- and depressive-like behaviours were also investigated after kindling using two well-
validated tests, the elevated plus maze and forced swim test respectively, as well as assessing
the corticosterone response to stress and dentate gyrus neurogenesis.

vi
Results
The key finding of this study was that rats chronically treated with SSRIs, either fluoxetine or
citalopram, demonstrated accelerated rates of kindling epileptogenesis, showing a more rapid
progression through the different stages of kindling compared to vehicle treated rats. The
increase in seizure duration was also accelerated in the early stages of kindling in both cohorts
of SSRI treated rats, however seizure threshold was not significantly different between vehicle
and fluoxetine or vehicle and citalopram treated rats, either before or after kindling. This
indicates that while epileptogenesis itself progressed at a faster rate during chronic SSRI
treatment, accelerating the increase in seizure severity and duration, the local excitability and
the threshold at which a seizure occurred was not affected by SSRI treatment. In order to
investigate potential mechanisms underlying this, neurobiological alterations common to
epileptogenesis and SSRI treatment were also investigated. Behavioural analyses found that
both fluoxetine and citalopram treatments did not affect anxiety- or depressive-like behaviours,
while kindling increased anxiety-like behaviour, but only in the fluoxetine treated cohort.
Dentate gyrus neurogenesis was not significantly affected by kindling or drug treatment while
stress-induced corticosterone levels were significantly reduced only by fluoxetine treatment.
These investigations do not suggest that these alterations are associated with accelerating
kindling rate during chronic SSRI treatment, however how these are affected during or
immediately after kindling was not investigated.
Conclusions
Chronic treatment with fluoxetine and citalopram, at clinically relevant doses, accelerated
kindling epileptogenesis in rodents. This highlights the need to investigate the effects of SSRI
treatment on epileptogenesis over time in both animal models and people with epilepsy, rather
than focusing solely on acute seizure time points. While the investigations in this study do not
suggest that alterations in behaviour, neurogenesis or neuroendocrine responses are associated
with accelerating kindling rate during chronic SSRI treatment, how these are affected during or
immediately after kindling was not investigated. Therefore, future studies should further
investigate the mechanisms underlying the effects of SSRIs on epileptogenesis at appropriate
time points, such as during kindling epileptogenesis and also in complementary animal models
of epilepsy, such as post-status epilepticus and post-traumatic models. It is essential to treat the
depressive symptoms that manifest in people with epilepsy; however whether these
medications affect the course of epilepsy and how they may do so should become priority areas
for future research.

vii
DECLARATION
This is to certify that:
i. the thesis comprises only my original work towards the Masters,
ii. due acknowledgement has been made in the text to all other material used,
iii. the thesis is no more than 50,000 words in length, exclusive of tables, maps,
bibliographies and appendices.
Signature ______________________________ Date ___________________

viii

ix
ACKNOWLEDGMENTS
To my supervisors, for being a great source of knowledge and advice throughout this journey as
well as for expertly guiding me on the road to becoming a scientifically-minded person.
Dr Nigel Jones, I would not only like to thank you for the sound experimental advice and
guidance you have given me, but also for being a friend over the years we have worked together.
The nicknames that you have given me will surely stick for years to come.
Associate Professor Michael Salzberg, I appreciate all the valuable knowledge you have shared
with me throughout, for listening to my ideas and for the many helpful conversations.
Professor Terence O’Brien, I greatly appreciate all the opportunities you have given me during
my time working in your laboratory, for teaching me how to see scientific concepts more clearly
and for being a wonderful advisor.
I would also like to thank:
Jenny Davis and Lara Mizhiritsky, for expertly managing the animal house and for providing care
to the rats used in this project. In particular, I would like to thank Jenny for always being flexible
with my many requests.
Amelia Koe, for teaching me many of the skills required to complete my experiments, reading
drafts and being excellent company. Meng Yang, for teaching me BrdU staining and providing
delicious foods. Lucy Vivash and Agi Swierczak, for reading drafts and always being only a phone
call away. Each of you has made this journey worth pursuing to the end. Thanks for the many
Friday night drinks, weird and wonderful conversations, gossip and shoulders to cry on (often!),
advice and laughter. I could not have chosen a better group of intelligent, entertaining and
supportive friends to spend this time with.
Gil Rind, for fixing many of the technical and experimental glitches along the way. You have been
invaluable provider of knowledge, puns and spaghetti songs.
Idrish Ali, for help with the kindling experiments, useful scientific discussions and for being a
great desk buddy.
Valentina Jovanoska, for passing on her expert cryostat skills to me.
Kim Powell, for the little but important things: a friend, advisor and go-to person.
Gabi Dezsi, Paul Anderson, Pablo Casillas-Espinosa, Caroline Ng, Thomas Zheng, Leena Van Raay,
Arun Gandrathi and all members of the O’Brien lab past and present who have been my second
family for many years, you have made it worth coming into work every day.
And finally, to my friends and family, for supporting my decisions and encouraging me with my
scientific and academic pursuits.

x

xi
ABBREVIATIONS
5HT Serotonin
8-OH-DPAT 8-hydroxy-2-(di-n-propylamino)tetralin
ACTH Adrenocorticotrophic hormone
ADT Afterdischarge threshold
AED Antiepileptic drug
BDNF Brain-derived neurotrophic factor
BrdU 5-bromo-2'-deoxyuridine
BUP Bupropion
CA Cornu ammonis
CIT Citalopram
CLV Clovoxamine
CRH Corticotrophin releasing hormone
DES Desipramine
Dex Dexamethsone
DMSO Dimethyl sulfoxide
DOI 2,5-dimethoxy-4-iodoamphetamine
DPX Dibutylphthalate polystyrene xylene
ECT Electroconvulsive therapy
EEG Electroencephalogram
EPM Elevated plus maze
FLV Fluvoxamine
FLX Fluoxetine
FST Forced swim test
GABA Gamma-amino-butyric acid
GEPR Genetically Epilepsy Prone Rats
GR Glucocorticoid receptor
HPA Hypothalamo-pituitary adrenal
ILAE International League Against Epilepsy
IL Interleukin
KA Kainic acid
LH Learned helplessness
MAOI Monoamine oxidase inhibitor
MAP Maprotiline

xii
MDD Major depressive disorder
MES Maximal electroshock
MIR Mirtazepine
MR Mineralocorticoid
PAR Paroxetine
PFA Paraformaldehyde
PTZ Pentylenetetrazol
SERT Serotonin transporter
SRS Spontaneous recurrent seizures
SRT Sertraline
SNRI Serotonin and noradrenaline reuptake inhibitor
SSRI Selective serotonin reuptake inhibitor
SUDEP Sudden unexpected death in epilepsy
REB Reboxetine
TBI Traumatic brain injury
TBS Tris-buffered saline
TCA Tricyclic antidepressant
TLE Temporal lobe epilepsy
VEN Venlafaxine
VGAT Vesicular GABA transporter

xiii
TABLE OF CONTENTS
Chapter 1: Literature Review
1.1. General introduction ........................................................................................................................................ 1
1.2. Epilepsy .................................................................................................................................................................. 2
1.2.1 Definitions and epidemiology ............................................................................................................. 2
1.2.2 Seizures ........................................................................................................................................................ 2
1.2.3 Temporal lobe epilepsy ......................................................................................................................... 3
1.2.3.1 Brief introduction to temporal lobe epilepsy ........................................................................... 3
1.2.3.2 Anatomical connectivity of the temporal lobe......................................................................... 4
1.2.3.3 Aetiology of temporal lobe epilepsy ............................................................................................ 6
1.2.3.4 Pathology of temporal lobe epilepsy ........................................................................................... 6
1.2.3.5 Treatment of temporal lobe epilepsy .......................................................................................... 7
1.2.4 Epileptogenesis ......................................................................................................................................... 7
1.2.5 Animal models of temporal lobe epilepsy ...................................................................................... 9
1.2.5.1 The post-status epilepticus model of temporal lobe epilepsy ....................................... 10
1.2.5.2 The kindling model of temporal lobe epilepsy ..................................................................... 10
1.3. The comorbidities of epilepsy ................................................................................................................... 12
1.3.1 Historical and current perspectives of the comorbidities of epilepsy ............................ 12
1.3.2 Prevalence and epidemiology of psychiatric disorders in epilepsy ................................. 14
1.3.3 Depression and anxiety in epilepsy ............................................................................................... 14
1.4. Antidepressant pharmacotherapy in epilepsy .................................................................................... 16
1.4.1 The discovery and use of first-generation antidepressants ................................................ 16
1.4.2 The discovery and use of selective serotonin reuptake inhibitors ................................... 18
1.4.3 Use of antidepressant pharmacotherapy in epilepsy ............................................................. 20
1.4.3.1 Human studies of SSRI pharmacotherapy in epilepsy ...................................................... 21
1.4.3.2 SSRI pharmacotherapy in animal models of epilepsy ....................................................... 25
1.5. Antidepressants and epileptogenesis: shared neurobiological substrates............................. 30

xiv
1.5.1 Hypothalamo-pituitary-adrenal axis and glucocorticoids ................................................... 30
1.5.2 Hippocampal neurogenesis ............................................................................................................... 33
1.5.3 Other intersecting intermediaries .................................................................................................. 36
1.5.3.1 Monoamines ...................................................................................................................................... 37
1.5.3.2 Brain-derived neurotrophic factor ............................................................................................ 37
1.5.3.3 Inflammation ...................................................................................................................................... 38
1.6. Study rationale ................................................................................................................................................. 40
1.7. Hypotheses and aims ..................................................................................................................................... 41
Chapter 2: Methods
2.1. Overall study design ..................................................................................................................................... 43
2.2. Experimental animals .................................................................................................................................. 43
2.3. Live animal experimental procedures .................................................................................................. 44
2.3.1 Osmotic pump preparation ................................................................................................................ 44
2.3.2 Osmotic pump implantation .............................................................................................................. 46
2.3.3 Bipolar and recording electrode implantation .......................................................................... 47
2.3.4 Seizure threshold testing .................................................................................................................... 48
2.3.5 Amygdala kindling ................................................................................................................................. 49
2.3.6 Osmotic pump re-implantation ........................................................................................................ 50
2.3.7 5-bromo-2-deoxyuridine injections ............................................................................................... 50
2.3.8 Elevated plus maze ................................................................................................................................ 50
2.3.9 Forced swim test and corticosterone stress response ........................................................... 51
2.3.10 Corticosterone radioimmunoassay ................................................................................................. 53
2.4. Post-mortem experimental procedures ............................................................................................... 53
2.4.1 Cardiac blood sampling, transcardial perfusion and tissue fixation ................................. 53
2.4.2 Cryosectioning ......................................................................................................................................... 54
2.4.3 Thionin staining and determination of electrode placement .............................................. 54
2.4.4 BrdU immunohistochemistry ............................................................................................................ 55
2.4.5 BrdU cell number quantification ..................................................................................................... 56

xv
2.4.7 Plasma sample analysis for fluoxetine levels ............................................................................. 57
Chapter 3: Pilot study - Tolerability of osmotic pumps and fluoxetine and the effects on behaviour, stress-induced corticosterone response and neurogenesis
3.1. Introduction ..................................................................................................................................................... 59
3.2. Materials and methods ................................................................................................................................ 61
3.2.1 Statistical analyses .................................................................................................................................. 62
3.3. Results ................................................................................................................................................................ 62
3.3.1 Fluoxetine administered by osmotic pumps does not affect weight gain ...................... 62
3.3.2 Rats treated with fluoxetine show serum levels within an appropriate therapeutic
range .............................................................................................................................................................................. 63
3.3.3 Fluoxetine-treated rats do not display an anxious phenotype in the elevated plus
maze ............................................................................................................................................................................... 63
3.3.4 Fluoxetine treated rats display a depressive-like phenotype ............................................... 65
3.3.5 Fluoxetine treated rats show a trend towards a suppressed stress response .............. 65
3.3.6 BrdU cell counts do not differ between fluoxetine and vehicle treatments .................... 66
3.4. Discussion ......................................................................................................................................................... 67
Chapter 4: Chronic SSRI treatment and its effects on kindling
epileptogenesis, behaviour, stress-induced corticosterone
response and neurogenesis
4.1. Introduction ..................................................................................................................................................... 71
4.2. Materials and methods ................................................................................................................................ 74
4.2.1 Statistical analyses ................................................................................................................................... 75
4.3. Results ................................................................................................................................................................ 76
4.3.1 Rats treated with fluoxetine had serum levels within an appropriate therapeutic range
.......................................................................................................................................................................................... 76
4.3.2 Seizure threshold is reduced by kindling, but is not affected by chronic SSRI treatment
.......................................................................................................................................................................................... 76

xvi
4.3.3 Chronic fluoxetine and citalopram treatments accelerate kindling epileptogenesis . 77
4.3.4 Chronic SSRI treatments accelerate the progression of increase in seizure duration
early in epileptogenesis ......................................................................................................................................... 77
4.3.5 Chronic fluoxetine and citalopram treatments differentially affect anxiety-like
behaviours ................................................................................................................................................................... 81
4.3.6 Kindling, fluoxetine or citalopram treatments do not affect depressive-like behaviour
.......................................................................................................................................................................................... 81
4.3.7 Chronic fluoxetine, but not citalopram, treatment reduces the corticosterone response
to swim stress ............................................................................................................................................................ 84
4.3.8 Neither kindling nor chronic fluoxetine treatment affect dentate gyrus neurogenesis85
4.4. Discussion ........................................................................................................................................................ 86
4.4.1 Kindling epileptogenesis is accelerated by chronic antidepressant treatment ............. 86
4.4.2 Chronic fluoxetine and citalopram treatments increase seizure duration in the early
stages of epileptogenesis ....................................................................................................................................... 89
4.4.3 Chronic fluoxetine and citalopram treatments do not affect seizure threshold ............ 90
4.4.4 Interim conclusion of kindling data .................................................................................................. 91
4.4.5 Chronic fluoxetine and citalopram treatment and effects on anxiety- and depressive-
like behaviours .......................................................................................................................................................... 91
4.4.6 Chronic fluoxetine, but not citalopram, treatment reduces the corticosterone response
to swim stress ............................................................................................................................................................ 92
4.4.7 Neurogenesis is not affected by kindling or chronic fluoxetine treatment ....................... 94
4.4.8 Final conclusions ...................................................................................................................................... 94
Chapter 5: General discussion
5.1. Key findings..................................................................................................................................................... 97
5.2. Potential mechanisms by which fluoxetine and citalopram accelerated kindling rate ... 99
5.2.1 Serotonin receptor alterations ...................................................................................................... 100
5.2.2 Alterations in other aspects of neuroplasticity ....................................................................... 101
5.2.3 Modulation of excitatory and inhibitory neurotransmission ........................................... 102
5.2.4 Overall conclusion of potential mechanisms ........................................................................... 102

xvii
5.3. The potential clinical implications of an accelerated rate of epileptogenesis ................... 103
5.4. Methodological considerations and limitations ............................................................................. 106
5.4.1 Validity of the kindling model for comparison to epileptogenesis in humans ........... 106
5.4.2 Normal rats versus the pathological state .................................................................................. 108
5.4.3 Time point of analysis of common neurobiological substrates ......................................... 108
5.4.4 Behavioural testing and comparison to human behaviours ............................................... 109
5.5. Future directions ........................................................................................................................................ 110
5.5.1 Investigating the mechanisms involved in accelerated kindling epileptogenesis during
chronic SSRI treatment ........................................................................................................................................ 110
5.5.2. Investigating the effects of SSRIs in a chronic epilepsy model ........................................... 111
5.5.3 Investigating the effects of SSRIs in a model of epilepsy and depression ..................... 111
5.5.4 Clinical directions .................................................................................................................................. 112
5.6. Final conclusions ........................................................................................................................................ 112
References………………………………………………………………………………………………………………………...114

xviii

xix
LIST OF FIGURES AND TABLES
Figure 1.1: Anatomy of the human temporal lobe. ............................................................................................. 5
Figure 1.2: Location and structure of the rodent hippocampus. .................................................................. 5
Figure 1.3: Anatomical connectivity of the rodent hippocampus. ............................................................... 6
Figure 1.4: A schematic representation of epileptogenesis. ........................................................................... 9
Figure 1.5: Schematic representation of epileptogenesis in animal models of temporal lobe
epilepsy. ............................................................................................................................................................................. 13
Figure 1.6: The time course of epileptogenesis and the typical stages at which antidepressants
may be clinically introduced. .................................................................................................................................... 17
Figure 1.7: The effects of SSRIs at the serotonergic synapse. ..................................................................... 19
Figure 1.8: The hypothalamo-pituitary-adrenal axis. ..................................................................................... 32
Figure 1.9: The process of adult hippocampal neurogenesis in the dentate gyrus. ........................... 34
Figure 2.1: Timeline of experimental procedures............................................................................................ 43
Figure 2.2: Internal structure of the osmotic pumps. ..................................................................................... 45
Figure 2.3: The sequence of procedures for osmotic pump implantation. ............................................ 47
Figure 2.4: Location of recording and bipolar electrodes............................................................................. 48
Figure 2.5: An example of a seizure induced by electrical stimulation of the amygdala. ................ 49
Figure 2.6: The elevated plus maze. ....................................................................................................................... 51
Figure 2.7: An example of the behaviours displayed in the forced swim test. ..................................... 52
Figure 2.8: Examples of sections cut on the cryostat. ..................................................................................... 54
Figure 2.9: An example of BrdU stained cells. ................................................................................................... 57
Figure 3.1: Timeline of experimental procedures............................................................................................ 61
Figure 3.2: Average weekly weight of fluoxetine- and vehicle-treated rats. ........................................ 62
Figure 3.3: Fluoxetine and norfluoxetine levels versus vehicle treatment as measured by mass
spectroscopy. ................................................................................................................................................................... 63
Figure 3.4: The effects of fluoxetine treatment on anxiety-like behaviours in the elevated plus
maze. ................................................................................................................................................................................... 64
Figure 3.6: Change in corticosterone concentration from pre-stress levels. ........................................ 66
Figure 3.7: BrdU cell counts in the dentate gyrus and the hilus. ............................................................... 66
Figure 4.1: Timeline of experimental procedures............................................................................................ 75
Figure 4.2: Fluoxetine and norfluoxetine levels as measured by mass spectroscopy. ..................... 76
Figure 4.3: The effects of kindling and chronic fluoxetine and citalopram treatment on seizure
threshold before and after kindling. ...................................................................................................................... 78
Figure 4.4: The effect of chronic fluoxetine and citalopram treatments on kindling
epileptogenesis. .............................................................................................................................................................. 79

xx
Figure 4.5: The effect of chronic fluoxetine and citalopram treatments on seizure duration. ...... 80
Figure 4.6: The effects of kindling and fluoxetine or citalopram treatment on anxiety-like
behaviours. ....................................................................................................................................................................... 82
Figure 4.7: The effects of kindling, fluoxetine and citalopram treatments on depressive-like
behaviours. ....................................................................................................................................................................... 83
Figure 4.8: The effects of kindling and fluoxetine or citalopram treatments on corticosterone
response following swim stress. ............................................................................................................................. 84
Figure 4.9: Total number of BrdU labelled cells in the subgranular zone of the dentate gyrus and
hilus. .................................................................................................................................................................................... 85
Table 1.1: Studies of antidepressant use in patients with epilepsy. ......................................................... 24
Table 1.2: Studies of antidepressant administration in animal models of seizures and
epilepsy. ............................................................................................................................................................................. 27
Table 2.1: Total sample size included in each experiment. .......................................................................... 44

1
CHAPTER 1: LITERATURE REVIEW
1.1. General introduction
There is a high incidence of psychiatric comorbidity in patients with epilepsy, the most
prevalent being depression (Gaitatzis et al., 2004, Kanner et al., 2012). Depression contributes
to a large burden of the disability in patients suffering from epilepsy, more so than other factors
such illness duration or seizure frequency (Boylan et al., 2004, Gilliam et al., 2004, Kanner et al.,
2010). Accordingly, considerable efforts have been made to improve the detection and
diagnosis of depression in epilepsy, resulting in many patients being treated with
antidepressant medication. Therefore, examining the effects of these medications in epilepsy is
essential. Older generations of antidepressants, such as tricyclic antidepressants (TCAs), have
been shown to induce seizures in some non-epileptic patients (Wroblewski et al., 1990,
Preskorn and Fast, 1992) and increase seizure frequency in patients with epilepsy (Pisani et al.,
1999). Therefore, although they are not contraindicated, TCAs need to be employed in patients
with epilepsy with caution. Currently, there is growing evidence to suggest that the newer
classes of antidepressants, in particular the selective serotonin reuptake inhibitors (SSRIs), have
markedly less effects on seizure susceptibility, and may in fact lead to improvements in the
severity of epilepsy itself. Studies in humans and in animal models of epilepsy suggest that SSRI
treatment does not adversely affect seizures (Harmant et al., 1990, Wada et al., 1999, Hovorka
et al., 2000, Kuhn et al., 2003, Borowicz et al., 2007), and in some cases may even improve
seizure outcomes (Favale et al., 1995, Favale et al., 2003, Specchio et al., 2004), with only a few
reports of proconvulsant effects (Gigli et al., 1994, Kanner et al., 2000, Thome-Souza et al.,
2007). However, in all of these studies, the only investigations have been to examine the effects
of SSRIs on short-term seizure outcomes. This includes assessing seizure threshold or seizure
frequency over a short time period, mostly following short periods of treatment or following
acute doses of SSRIs. To date, no studies have examined the effects of chronic SSRI exposure on
epileptogenesis, the underlying and progressive neurobiological alterations that lead to the
development of spontaneous seizures. This is essential to address, as SSRIs may be affecting the
neurobiological processes leading to epilepsy, rather than just affecting the occurrence of
seizures themselves.
This literature review first describes epilepsy and the pathological alterations occurring in
epileptogenesis, with a description of animal models of epilepsy. The comorbidities of epilepsy
are then discussed, examining their prevalence, epidemiology and theories of causation,

2
focusing on depressive disorders. Next, the use of antidepressant pharmacotherapy in epilepsy
is reviewed, describing the different antidepressants available, focusing on SSRIs, and reviewing
studies of SSRI use in patients and animal models of epilepsy. Finally, common neurobiological
substrates between antidepressant treatment and epileptogenesis are described, to give further
understanding of the effects antidepressants may have on epileptogenesis.
1.2. Epilepsy
1.2.1 Definitions and epidemiology
Epilepsy is a brain disorder that has been recognised from the early times of medical diagnosis.
It is highly prevalent, affecting approximately 50 million people worldwide (World Health
Organisation, 2006) with a bimodal distribution of seizure incidence, peaking in children and
the elderly, although it can occur at any age (Hauser et al., 1991, 1993, Kotsopoulos et al., 2002).
In fact, up to 10% of the population will experience a seizure during their lifetime, which can be
described as a period of abnormal and synchronous excitation of a population of neuronal cells.
Epilepsy itself is characterised by a history of a seizure that causes an enduring alteration in the
brain, increasing the likelihood of developing further seizures, with associated neurobiological,
cognitive and psychosocial conditions (Fisher et al., 2005).
The disorder poses a significant burden on quality of life, not only on the individual but also on
their families and society at large. While epilepsy is a widely recognised condition in the present
day, many issues still exist for patients with epilepsy. It can be a source of social discrimination
and stigma, results in an increased mortality rate, economically contributes to 0.5% of the
global burden of disease (Leonardi and Ustun, 2002), and importantly for this thesis, is
associated with a higher incidence of cognitive, social and psychiatric comorbidities, which will
be discussed in detail below.
1.2.2 Seizures
The definition of seizures has evolved throughout medical history with improvements in
neuroimaging, genomic and molecular biology technologies. John Hughlings Jackson proposed
one of the first definitions of a seizure in 1870 as “an occasional, an excessive and a disorderly
discharge of nerve tissue” (Fisher et al., 2005, Scharfman and Pedley, 2006). Presently, the
International League Against Epilepsy (ILAE) defines a seizure as “a transient occurrence of

3
signs and/or symptoms due to abnormal excessive or synchronous neuronal activity in the
brain” (Fisher et al., 2005). While all seizures arise from an imbalance of excitation and
inhibition in the brain, the initial causes vary, and therefore classification of seizures and
epilepsy types has evolved. Currently, the ILAE classifies seizures as either focal or generalised:
focal seizures originate in a specific region of the brain and are limited to a specific area and
only one hemisphere, while generalised seizures do not have a localised onset and rapidly
engage bilateral, and sometimes asymmetrically, cortical and subcortical networks (Berg and
Scheffer, 2011).
The underlying causes of seizures can be divided into their origins: genetic, where the epilepsy
is a direct result of a known or presumed genetic alteration from which seizures manifest;
structural or metabolic, where there is a distinct condition or disease that is associated with the
development of recurrent seizures, which may be acquired (e.g. stroke, brain injury) or genetic
(e.g. channelopathies); or unknown, where the origin of seizures is due to an unrecognised
genetic, structural or metabolic defect or a combination of these factors (Berg and Scheffer,
2011). Within these categories, many forms of epilepsies can be identified. The focus for this
thesis is temporal lobe epilepsy (TLE), which is classified as focal epilepsy with
structural/metabolic origins. It is one of the most common types of epilepsy in adults and is of
particular interest due to the high comorbidity of psychiatric conditions, and as such, treatment
of these conditions with pharmacotherapies.
1.2.3 Temporal lobe epilepsy
1.2.3.1 Brief introduction to temporal lobe epilepsy
Temporal lobe epilepsy, named so because seizures originate in temporal lobe structures (Van
Roost et al., 1998), accounts for approximately 80% of focal epilepsies in adults (Hauser et al.,
1991). It is frequently drug-resistant (Engel, 1996, 2001, Kwan et al., 2010) in that appropriate
pharmacological interventions fail to provide adequate seizure control (Cascino, 1994). Patients
typically experience focal seizures arising from the temporal lobe with or without impairment
of consciousness, evolving to bilateral convulsive seizures (Engel, 1996, Sharma et al., 2007).
Prior to discussing the pathophysiology of TLE, the normal structure and connectivity of the
temporal lobe will be described, as it is relevant to the understanding of TLE, antidepressant
action and their inter-relationship.

4
1.2.3.2 Anatomical connectivity of the temporal lobe
The temporal lobe is located bilaterally on the lateral sides of the brain, ventral to the Sylvian
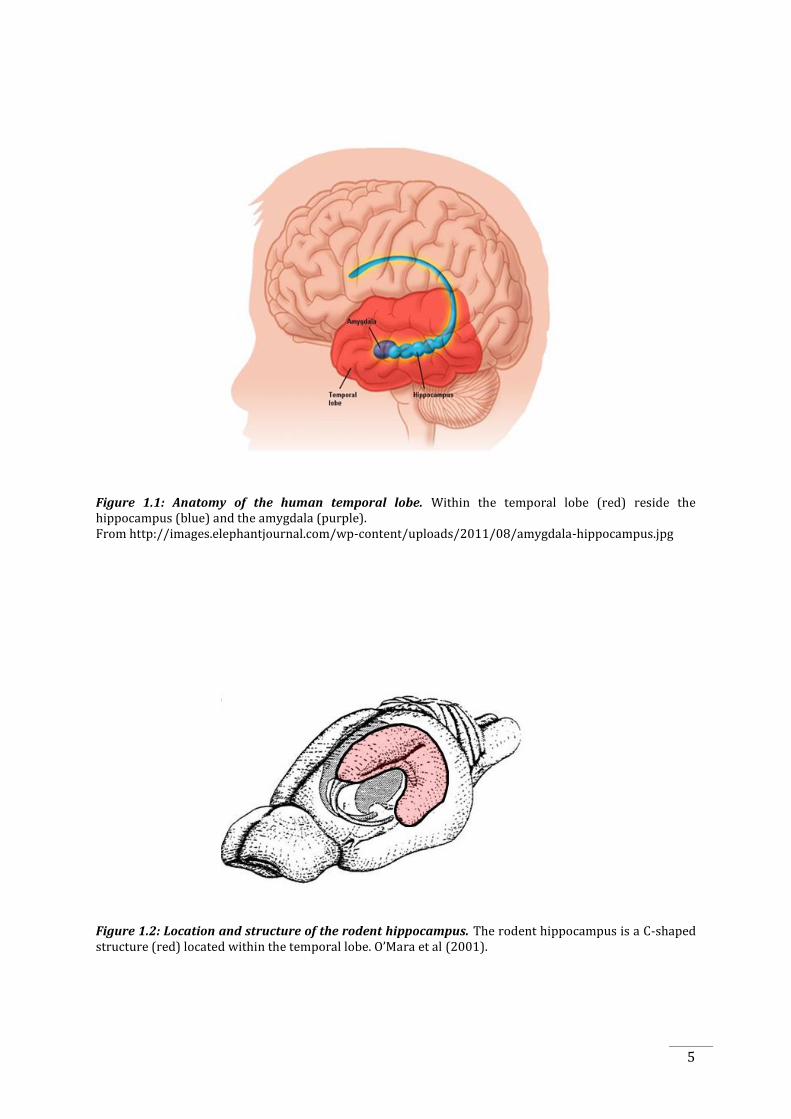
fissure and anterior to the parietal lobes (Figure 1.1). Its main structures, consisting of a
majority of structures found in the limbic system, include the parahippocampal gyrus (including
the parahippocampal, perirhinal and entorhinal cortices), the hippocampus and the amygdala.
The latter two structures will be further discussed due to their involvement in emotional
processing and in seizure development.
The hippocampus can be identified as a “C-shaped” structure in both the human and rodent
brains (Figure 1.2), containing the hippocampus proper, consisting of Cornu Ammonis (CA) 1, 2
and 3 and the dentate gyrus. While the structures anatomical location is different in the human
and rodent brains, the morphology and afferent and efferent connections are highly conserved.
Figure 1.3 pictorially displays hippocampal connectivity. The granule cells of the dentate gyrus
receive inputs from the entorhinal cortex via the perforant pathway. Mossy fibres, the axons of
the granule cells, project from the dentate gyrus and synapse onto the pyramidal neurons of
CA1, also synapsing onto CA3, from which the pyramidal neurons project via Schaeffer
collaterals onto CA1 or directly out of CA3 to the fornix. Pyramidal cells of the CA1 region then
project to the subiculum, which in turn projects out of the hippocampus to the hypothalamus via
the fornix or to the entorhinal cortex, the latter of which contains reciprocal connections with
the rest of the temporal lobe, serving as a gateway between the hippocampus and temporal lobe
(Amaral et al., 1984, Witter, 1993). The amygdalae are located rostral to the hippocampi (Figure
1.1), comprising of multiple nuclei, including the basolateral, medial and central nuclei. The
amygdala also has many reciprocal connections with the hippocampus (Amaral et al., 1987) and
various cortical areas.
5
Figure 1.1: Anatomy of the human temporal lobe. Within the temporal lobe (red) reside the hippocampus (blue) and the amygdala (purple). From http://images.elephantjournal.com/wp-content/uploads/2011/08/amygdala-hippocampus.jpg
Figure 1.2: Location and structure of the rodent hippocampus. The rodent hippocampus is a C-shaped structure (red) located within the temporal lobe. O’Mara et al (2001).

6
Figure 1.3: Anatomical connectivity of the rodent hippocampus. The hippocampus receives inputs from the entorhinal cortex via the perforant pathway (PP, lateral and medial), which synapse onto the pyramidal cells of the dentate gyrus (DG) and the CA3 pyramidal cells. The CA3 also receives inputs from the mossy fibres (MF) of the DG, which also synapse onto CA1 via the Schaffer Collateral (SC) pathway. CA1 also receives input from the contralateral hippocampus via the Associational Commissural (AC) pathway. CA1 neurons project to the subiculum (Sb), sending the output the entorhinal cortex (EC, lateral and medial) to form a reciprocal loop. From http://alfin2100.blogspot.com.au/2009/12/clues-on-short-term-memory-in.html
1.2.3.3 Aetiology of temporal lobe epilepsy
The causes of TLE vary greatly, and in many cases are unknown. It may occur as a result of
tumours, traumatic brain injuries or cortical and cerebrovascular malformations (Le Blanc and
Rasmussen, 1974), amongst other causes, all of which can be exacerbated by external influences
including stress (Haut et al., 2007, Koutsogiannopoulos et al., 2009), exercise (Dhanushkodi and
Shetty, 2008), drugs of abuse (Gordon and Devinsky, 2001) or pharmacotherapies (Preskorn
and Fast, 1992). However, the pathology of TLE remains largely similar regardless of the
causation.
1.2.3.4 Pathology of temporal lobe epilepsy
The hippocampal formation remains the main area of investigation of TLE pathogenesis. This
can be attributed to (1) electrographic recordings identifying the hippocampus as one of the
main structures where seizures originate (Van Roost et al., 1998), (2) in patients with drug-
resistant TLE, surgical removal of regions including the hippocampus ipsilateral to seizure onset
reduces and often abolishes seizures (Zentner et al., 1995) (3) the pathological alterations in
TLE patients largely involve the hippocampus. This includes hippocampal sclerosis, which is one
of the most commonly described neuropathologies (Armstrong, 1993, de Lanerolle et al., 2003,

7
Sharma et al., 2007, Bae et al., 2010, Zheng et al., 2011), defined as a selective loss of neurons in
the CA3 and CA1 regions, with relative sparing of CA2 and the granule cells of the dentate gyrus;
mossy fibre sprouting, which involves the granule cell axons that usually synapse onto CA3,
aberrantly projecting back onto granule cell apical dendrites and forming reciprocal excitatory
projections which can contribute to seizure generation (Houser et al., 1990, Mathern et al.,
1995, Buckmaster and Dudek, 1999, Gorter et al., 2001); gliosis, which is not entirely restricted
to the hippocampus having also been demonstrated in the amygdala (Feindel et al., 1952,
Feindel and Penfield, 1954, Pitkanen, 2002, Sharma et al., 2007, Shapiro et al., 2008). Whether
these neuropathological alterations are a cause or a consequence of epilepsy remains unclear.
1.2.3.5 Treatment of temporal lobe epilepsy
In all epilepsies, treatment relies upon seizure suppression with pharmacotherapy. However, as
mentioned above, up to 30% of patients become drug-resistant (Kwan and Brodie, 2000, Berg,
2008), while approximately 40% suffer significant drug-induced side effects (Kwan and Brodie,
2000). In drug-resistant patients, the only effective treatment option available to alleviate
seizures is surgical removal of the epileptogenic region. However, in patients who are drug-
responsive, treatment with antiepileptic drugs (AEDs) does not cure the condition. AEDs
merely suppress seizures affording a better quality of life, with many patients continuing to take
medication for the remainder of their lives. There is no alteration of the underlying pathology
causing TLE despite much investigation into the processes leading to and continuing after
seizures develop, that is, the process of epileptogenesis.
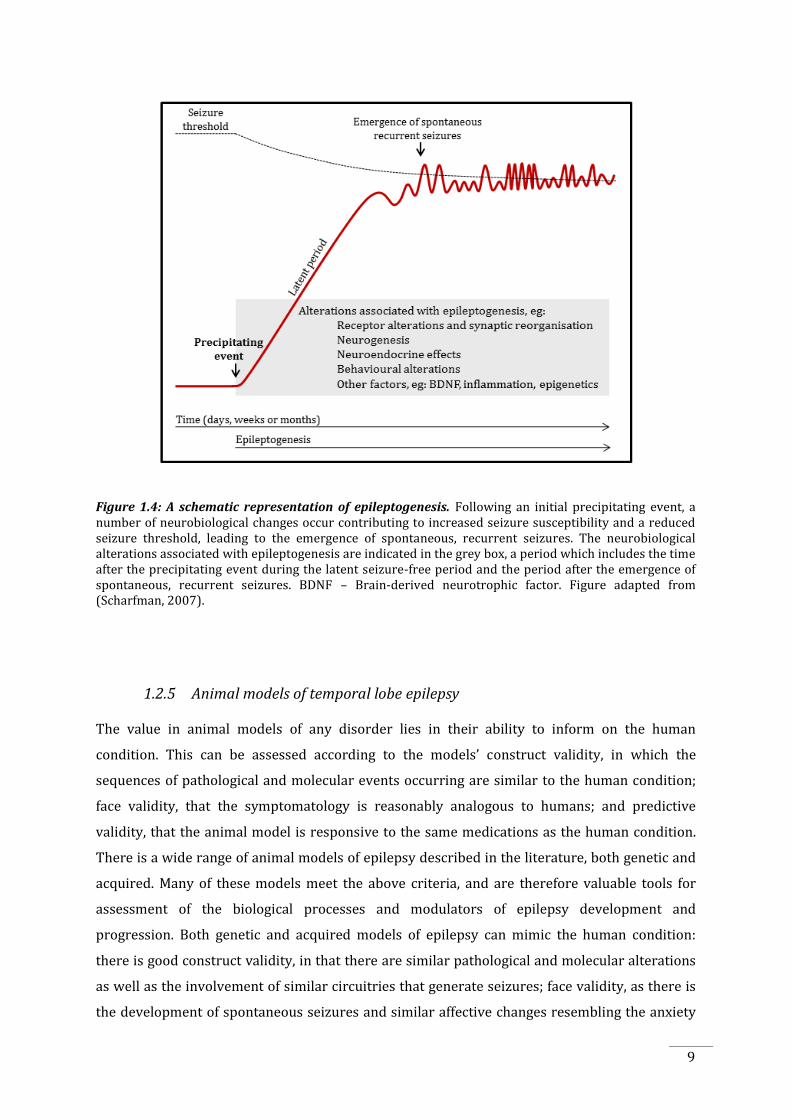
1.2.4 Epileptogenesis
Epileptogenesis is a dynamic process during which a variety of biochemical, anatomical and
physiological changes occur in the brain that alter neuronal excitability and transform a normal
brain into one that is capable of generating spontaneous seizures (Herman, 2002, Loscher,
2002, Pitkanen, 2002, Stables et al., 2002, Pitkanen et al., 2007, Engel and Pedley, 2008,
Pitkanen, 2010). While epileptogenesis is most often associated with focal epilepsies resulting
from structural lesions and focal seizures (Engel, 2001), it has also been suggested to occur in
generalised epilepsies, perhaps through alterations in gene expression over time that increase
seizure susceptibility (Zara and Bianchi, 2009). In the ‘three stage model’ of epileptogenesis,
following an initial, precipitating insult to the brain, for example head trauma (Stage 1), a
cascade of events are initiated that lead to altered excitability in the hippocampus and related
structures. This is followed by a seizure-free latent period (Stage 2) which can last from months
to years during which structural and biochemical changes occur, followed by the emergence of

8
spontaneous recurrent seizures (Stage 3) that can worsen epilepsy over time, further
contributing to the neurobiological changes and psychiatric comorbidities (Figure 1.4,
Scharfman, 2007). Additionally, the processes that underlie epileptogenesis do not cease once
seizures commence; the neurobiological alterations continue and may even contribute to the
progression of the disorder.
The progressive nature of epilepsy in humans is becoming more widely accepted, especially for
TLE (O'Brien et al., 1999, Tasch et al., 1999, Bouilleret et al., 2000, Cole, 2000, Pitkanen and
Sutula, 2002, Nearing et al., 2007, Cascino, 2009, Yang et al., 2010). This is due to evidence from
humans and animal studies showing the progression of changes associated with
epileptogenesis. As the epileptic disorder progresses, seizures may become more frequent and
severe (Sillanpaa et al., 1998, Kwan and Sander, 2004, Shorvon and Luciano, 2007). In the more
severe cases, as the disorder progresses, seizures may become resistant to conventional
medications, and more invasive procedures, such as surgery to remove the epileptic focus, may
need to be considered (Zentner et al., 1995). In association with this, psychiatric comorbidities
may manifest, which can include anxiety and depression, as well as psychoses, memory
impairments or cognitive decline (Kwan and Brodie, 2001, Motamedi and Meador, 2003,
Andersson-Roswall et al., 2010). The association of some of these alterations and the
implications of treating these comorbidities is a focus of this thesis, and will be discussed in
more detail below (Section 1.3 onwards).
Although there has been much study on the processes occurring during and associated with
epileptogenesis, it is still poorly understood, primarily due to the difficulties of studying
epileptogenesis in humans; for example, in the majority of cases, human brain tissue from TLE
patients is only available from those who are medically refractory, and at the most advanced
stages of their illness and therefore the epileptogenic process. Consequently, animal models
have provided a very valuable tool for gaining insights into the pathological changes occurring
in epileptogenesis, as the changes occur over a period of months after the initial insult (Tanaka
et al., 1992, Hellier et al., 1998, Kharatishvili et al., 2006) while in humans, similar processes can
take years (French et al., 1993, Mathern et al., 1995).
9
Figure 1.4: A schematic representation of epileptogenesis. Following an initial precipitating event, a number of neurobiological changes occur contributing to increased seizure susceptibility and a reduced seizure threshold, leading to the emergence of spontaneous, recurrent seizures. The neurobiological alterations associated with epileptogenesis are indicated in the grey box, a period which includes the time after the precipitating event during the latent seizure-free period and the period after the emergence of spontaneous, recurrent seizures. BDNF – Brain-derived neurotrophic factor. Figure adapted from (Scharfman, 2007).
1.2.5 Animal models of temporal lobe epilepsy
The value in animal models of any disorder lies in their ability to inform on the human
condition. This can be assessed according to the models’ construct validity, in which the
sequences of pathological and molecular events occurring are similar to the human condition;
face validity, that the symptomatology is reasonably analogous to humans; and predictive
validity, that the animal model is responsive to the same medications as the human condition.
There is a wide range of animal models of epilepsy described in the literature, both genetic and
acquired. Many of these models meet the above criteria, and are therefore valuable tools for
assessment of the biological processes and modulators of epilepsy development and
progression. Both genetic and acquired models of epilepsy can mimic the human condition:
there is good construct validity, in that there are similar pathological and molecular alterations
as well as the involvement of similar circuitries that generate seizures; face validity, as there is
the development of spontaneous seizures and similar affective changes resembling the anxiety

10
and depressive comorbidities of epilepsy and predictive validity, with similar responses to
pharmacological interventions. Genetic models of epilepsy will not be discussed here; instead
the focus will remain on models of acquired epilepsy, in which epileptogenesis is initiated by an
external brain insult. The two most commonly used models of acquired epilepsy in epilepsy
research are the post-status epilepticus model and the kindling model. The development of
epilepsy in these models is shown in Figure 1.5. Each model provides a good representation of
the human condition, as well as being complementary, giving insight into different features of
human TLE. However, each model has its own advantages and disadvantages, as will be
discussed below.
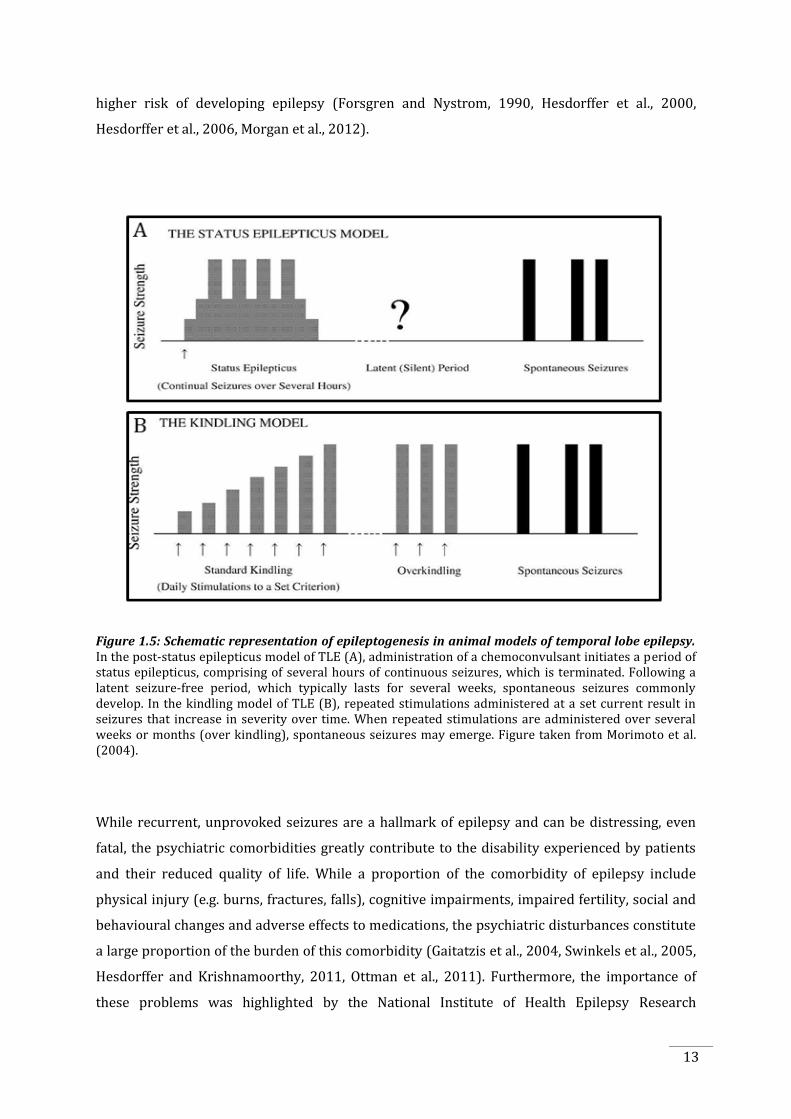
1.2.5.1 The post-status epilepticus model of temporal lobe epilepsy
The post-status epilepticus model of TLE is good model of the later stages of epilepsy, due to the
development of spontaneous seizures. In this model, animals are typically systemically injected
with a chemoconvulsant such as kainate or pilocarpine, which induce an acute period of
recurrent seizures (status epilepticus), which are then terminated with an anticonvulsant.
Following a latent period, seizures may spontaneously occur.
While the development of seizures in the post-status epilepticus model mimics the human
condition, there are some limitations. The expression of spontaneous seizures is variable
between different animals, a substantial portion of animals can die during status epilepticus and
there is extensive cell loss associated with the initial seizures following the chemoconvulsant, as
well as severe and widespread damage post-status epilepticus (Covolan and Mello, 2000,
Peredery et al., 2000). Thus, the kindling model was used instead for the work described in this
thesis, the advantages of which will be discussed below.
1.2.5.2 The kindling model of temporal lobe epilepsy
The kindling model has a number of advantages as a model of temporal lobe epileptogenesis
disease progression. Kindling itself is defined as the “progressive changes that result from
repeated electrical stimulation” (Goddard et al., 1969). Kindling was first demonstrated by
Alonso-DeFlorida and Delgado (1958) and thoroughly tested by Goddard (Goddard, 1967,
1969), primarily in rats, but has also been shown in other species (mice, dogs, cats and several
primate species) (Goddard et al., 1969, Racine, 1978). Various limbic structures can be kindled
to generate seizures, including the hippocampus and the amygdala (Delgado and Sevillano,
1961, Goddard et al., 1969, Racine, 1972b). The amygdala is the site most commonly kindled, as
comparatively few electrical stimulations are required to induce a kindling effect (McNamara,

11
1986). Additionally, studies by Adamec (1998) have shown that amygdala kindling is associated
with behavioural alterations including increases in anxiety-like behaviour (Adamec, 1998,
Adamec and Young, 2000, Adamec et al., 2004, Adamec et al., 2005) and cognitive deficits (Peele
and Gilbert, 1992). On the other hand, depression, although common, has not been consistently
shown to occur (Corcoran et al., 1992, Helfer et al., 1996, Adamec et al., 2004).
Amygdala kindling involves the repeated induction of focal seizures by an electrical current
delivered via an implanted electrode to a specific amygdala nucleus. This produces a
progressive increase in seizure susceptibility and the epileptic response, with a spread of
seizure activity from focal to extrafocal regions and the appearance of generalised convulsive
seizures, a condition that persists for months to years (Goddard et al., 1969, Racine, 1972b).
In conjunction with the increase in seizure susceptibility with subsequent stimulations,
increases in the severity of behavioural seizures are also observed. The progression of the
convulsive behaviours associated with amygdala kindling was extensively studied by Racine
(1972b), who published a standard grading of behavioural seizures, from class I to V, during
kindling. In the initial stages, when a focal seizure is elicited, an electrographic response occurs
without any behavioural changes (class 0). With subsequent stimulations, behavioural
responses begin to manifest which may include freezing and facial clonus (class I) followed by
ipsilateral eye twitching and closure and head nodding (class II). As stimulations continue more
distant structures from the initial seizure focus are recruited and forelimb clonus occurs (class
III). Eventually, seizures generalise to bilateral cortical regions, the motor cortices are recruited
and animals exhibit bilateral clonic seizures. This involves bilateral limb clonus, postural
alterations such as rearing and twisting (class IV), and loss of balance (class V). Additionally,
with subsequent electrical stimulations there is also a reduction in seizure threshold (Racine,
1972a, Tress and Herberg, 1972) and an increase in seizure duration, amplitude, spike
frequency and morphology (Racine, 1972a,b, 1975).
The kindling model reliably produces seizures of increasing severity with repeated stimulations
and is therefore an excellent model to assess the progression of epileptogenesis. Furthermore,
anecdotal reports suggested that kindling might occur in humans. For example, occasionally
electroconvulsive therapy with repeated stimulations unintentionally may result in
spontaneous seizures, increased seizure durations and seizure-associated behavioural
automatisms (Morrell, 1985, Sato et al., 1990, Coulter et al., 2002). However, a disadvantage of
the kindling model is that seizures must be induced, as there is rarely development of
spontaneous seizures (unless a large number of stimulations are given, termed “over kindling”,
in which approximately half the animals will experience spontaneous seizures (Pinel and

12
Rovner, 1978, Michalakis et al., 1998, Coulter et al., 2002, Sayin et al., 2003). The kindling model
is also associated with only minor pathological changes such as mild cell loss and gliosis,
dentate gyrus neurogenesis (Bengzon et al., 1997, Parent et al., 1998, Scott et al., 1998) and
mossy fibre sprouting (Sutula, 1990, Ebert and Loscher, 1995). This damage is not as extensive
as occurs following the post-status epilepticus models or in human TLE. However, this is
beneficial, as it allows for the study of the functional changes in epileptogenesis independent of
the confounding effects of cell loss and pathological alterations. For these reasons, the kindling
model was chosen for this study. Furthermore, if the effects of antidepressants were to be seen
with kindling in this study, this would justify further experiments employing the post-status
epilepticus model, which are technically more difficult and require greater numbers of animals.
1.3. The comorbidities of epilepsy
1.3.1 Historical and current perspectives of the comorbidities of epilepsy
The co-occurrence of epilepsy and psychiatric disorders has been recognised since antiquity, as
observed by Hippocrates, around 400 B.C., (Lewis, 1934) that “melancholics ordinarily become
epileptics, and epileptics melancholics: what determines the preference is the direction the
malady takes; if it bears upon the body, epilepsy, if upon the intelligence, melancholy.”
Hippocrates observed that epilepsy was a result of natural and not sacred causes, eventually
leading to medical investigations of the causes of epilepsy during the Renaissance, and the first
understanding of seizure origins by John Hughlings Jackson in 1870 (Fisher et al., 2005,
Scharfman and Pedley, 2006). From this, further investigations by Briquet in 1859 (Swinkels et
al., 2005) and Morel in 1860 (Swinkels et al., 2005) recognised that psychological disturbances,
behavioural and cognitive alterations occur between seizures as well as during the seizures
themselves (Swinkels et al., 2005). The introduction of electroencephalography (EEG), the
discovery of a temporal lobe origin of some seizures, and the association of the limbic structures
within the temporal lobe with emotional processing further added support to ideas about how
and why psychiatric disorders may be linked to epilepsy, especially epilepsy of temporal lobe
origin. Currently, the understanding of the link between epilepsy and psychiatric disturbances
recognises that this relationship is bidirectional, with several population-based studies finding
that patients with epilepsy have a 5 to 20 fold higher risk of developing affective disorders
(Tellez-Zenteno et al., 2007) and patients with a history of affective disorders have a 3 to 7 fold
13
higher risk of developing epilepsy (Forsgren and Nystrom, 1990, Hesdorffer et al., 2000,
Hesdorffer et al., 2006, Morgan et al., 2012).
Figure 1.5: Schematic representation of epileptogenesis in animal models of temporal lobe epilepsy. In the post-status epilepticus model of TLE (A), administration of a chemoconvulsant initiates a period of status epilepticus, comprising of several hours of continuous seizures, which is terminated. Following a latent seizure-free period, which typically lasts for several weeks, spontaneous seizures commonly develop. In the kindling model of TLE (B), repeated stimulations administered at a set current result in seizures that increase in severity over time. When repeated stimulations are administered over several weeks or months (over kindling), spontaneous seizures may emerge. Figure taken from Morimoto et al. (2004).
While recurrent, unprovoked seizures are a hallmark of epilepsy and can be distressing, even
fatal, the psychiatric comorbidities greatly contribute to the disability experienced by patients
and their reduced quality of life. While a proportion of the comorbidity of epilepsy include
physical injury (e.g. burns, fractures, falls), cognitive impairments, impaired fertility, social and
behavioural changes and adverse effects to medications, the psychiatric disturbances constitute
a large proportion of the burden of this comorbidity (Gaitatzis et al., 2004, Swinkels et al., 2005,
Hesdorffer and Krishnamoorthy, 2011, Ottman et al., 2011). Furthermore, the importance of
these problems was highlighted by the National Institute of Health Epilepsy Research

14
Benchmarks which nominated the comorbidities of epilepsy, including predominantly
psychiatric comorbidities, as priority areas of research (Kelley et al., 2009).
1.3.2 Prevalence and epidemiology of psychiatric disorders in epilepsy
Patients with epilepsy have been reported to have higher rates of psychiatric comorbidities
compared to the general population (Hesdorffer and Krishnamoorthy, 2011). These disorders
are still highly prevalent in patients with epilepsy even when adjusting for socioeconomic
disadvantage and other health issues (Rai et al., 2012). Psychiatric disorders in epilepsy can be
classified according to their temporal relationship with seizures: peri-ictal – related to the
seizure, or inter-ictal – between seizures and unrelated to the seizures themselves. Peri-ictal
symptoms occurring in relation to the seizure will not be discussed; instead the focus will be
upon the psychiatric alterations occurring independent of seizures themselves, during the inter-
ictal period.
1.3.3 Depression and anxiety in epilepsy
In patients with TLE, both depression and anxiety are the most prevalent of the psychiatric
disorders, with 17-44% of patients being diagnosed with depression and 7-35% of patients with
anxiety (Ettinger et al., 2004, Mensah et al., 2007, Tellez-Zenteno et al., 2007, Kanner, 2011b).
Community- and clinically-based studies have shown that the rates of occurrence of depressive
(McLaughlin et al., 2008, Fuller-Thomson and Brennenstuhl, 2009) and anxiety (Gaitatzis et al.,
2004, Kobau et al., 2006, Mensah et al., 2007, Kanner, 2011a) disorders in epilepsy are clearly
elevated above the rates observed in the general population (Tellez-Zenteno et al., 2007). Other
common disturbances, though less prevalent, include attention-deficit hyperactivity disorder
and psychoses (Gaitatzis et al., 2004, Hesdorffer and Krishnamoorthy, 2011).
Depression in epilepsy can take several forms, including major depressive disorder, dysthymic
disorder, adjustment disorder and the depressive phases of bipolar disorder. Studies have
shown that depression is associated with impaired quality of life, greater cognitive complaints
and deficits, and greater health care utilisation (Lacey et al., 2009), with some studies indicating
that depression is a greater predictor of quality of life over other epilepsy-related variables,
such as illness duration or seizure frequency (Boylan et al., 2004, Gilliam, 2005, Kanner, 2009,
Kanner et al., 2010). Depression has also been shown to be a strong risk factor for suicide and a
possible risk factor for Sudden Unexpected Death in Epilepsy (SUDEP) (Ridsdale et al., 2011).
Depression has been widely recognised as a common comorbidity of epilepsy while anxiety
disorders, although also highly prevalent in people with epilepsy, have been somewhat

15
neglected. In fact, some studies suggest that anxiety disorders may even be more prevalent than
depression and could be related to many of the same adverse consequences as depression (Li et
al., 2002, Shimizu et al., 2002, Kanner, 2011a). Anxiety disorders in epilepsy can include
generalised anxiety and panic disorders, which may also constitute risk factors for the
development of depression in epilepsy, similar to the greater likelihood of developing
depression in non-epileptic people who experience anxiety disorders (Kanner, 2011a).
Various epileptic factors have been examined with regard to the development of anxiety or
depression. These include factors such as age of onset of epileptic disorder, seizure frequency or
severity (Robertson et al., 1987), type of epilepsy syndrome and anatomical location of seizure
focus, all of which have been examined with largely inconclusive results (Adams et al., 2008,
Filho et al., 2008, Asmussen et al., 2009, Babu et al., 2009, Desai et al., 2010). In contrast to the
neurological factors, psychosocial factors such as life stress, coping mechanisms, social support,
perceived stigma and personality have shown to be more consistent predictors of the
development of a comorbid psychiatric condition in epilepsy (Ormel et al., 1993, Hermann et al.,
2000). Various studies suggest that TLE is more greatly associated with psychiatric disorders
(Torta and Keller, 1999, Hermann et al., 2000). However this was due, in part, to a
preponderance of studies involving TLE patients in tertiary epilepsy centres, which mostly
include patients who are the most severely affected by seizures and other comorbidities.
However, other studies suggest that rates of psychiatric comorbidities are similar across all
epilepsies, as these studies have shown that psychiatric comorbidities also occur in generalised
and extra-temporal epilepsies (Victoroff et al., 1994, Lambert and Robertson, 1999, Christensen
et al., 2007, Adams et al., 2008, Hermann et al., 2008).
The link between epilepsy and psychiatric comorbidities has also been demonstrated in a wide
variety of animal models of epilepsy. Both genetic and acquired models have been found to be
associated with behavioural features relevant to the psychiatric co-morbidities that are
commonly present in patients with epilepsy. This includes depressive- and anxiety-like
behaviours observed in models of acquired epilepsy such as kindling (Adamec and Young, 2000,
Kalynchuk, 2000, Post, 2002, Mazarati et al., 2007), post-status epilepticus (Groticke et al., 2007,
Koh et al., 2007, Groticke et al., 2008, Mazarati et al., 2008, Muller et al., 2009a, Muller et al.,
2009b), posttraumatic epilepsy (Jones et al., 2008a) and febrile seizure (Mesquita et al., 2006)
models, as well as in genetic models of epilepsy (Jobe and Browning, 2007, Jones et al., 2008b,
Bouilleret et al., 2009, Sarkisova and van Luijtelaar, 2011). Furthermore, various early life
psychosocial exposures have also been shown increase susceptibility to epilepsy development,

16
such as maternal separation (Salzberg et al., 2007, Jones et al., 2009, Kumar et al., 2011, Koe,
2012) and cross-fostering (Gilby, 2009).
As depressive and anxiety disorders are highly prevalent in epilepsy and may have adverse
consequences, treatment and management of these symptoms in patients with epilepsy should
be considered as an essential component of treatment (Barry et al., 2008).
1.4. Antidepressant pharmacotherapy in epilepsy
As highlighted above, the psychiatric comorbidities of epilepsy, particularly depression,
constitutes a large proportion of the burden of comorbidity (Gaitatzis et al., 2004, Hesdorffer
and Krishnamoorthy, 2011). As such, treatment of these depressive symptoms is essential.
When these psychopathologies are recognised, a common approach is to treat with
antidepressant pharmacotherapy. However, the way in which these drugs interact with the
disease processes of epilepsy have not been thoroughly investigated, and may not only be
treating the comorbid disorder, but may also be impacting on the epilepsy. Antidepressants may
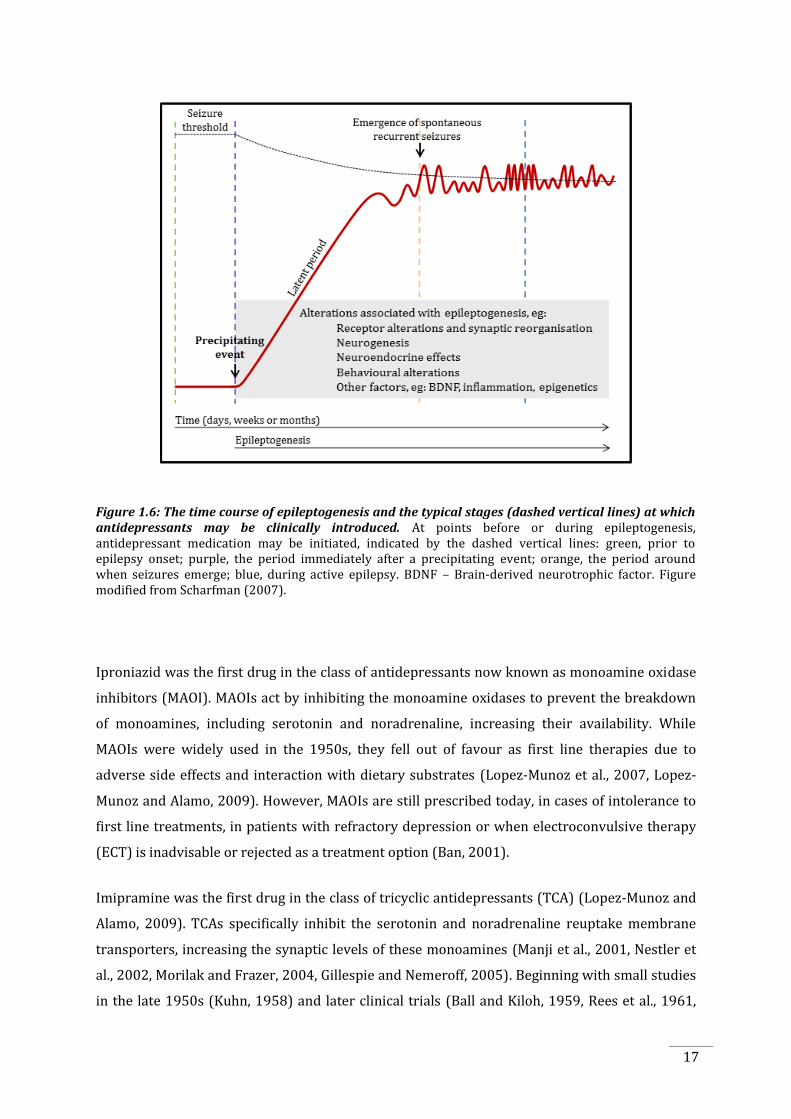
be introduced at various stages of epilepsy, shown by the dashed lines in Figure 1.6. Currently,
selective serotonin reuptake inhibitors (SSRIs) or serotonin-noradrenaline reuptake inhibitors
(SNRIs) are most commonly prescribed to patients with epilepsy. The following section will
briefly discuss antidepressant medications, before discussing studies addressing the effect of
SSRIs in epilepsy.
1.4.1 The discovery and use of first-generation antidepressants
Prior to the mid-20th century, pharmacological intervention to treat depression was not
commonly practiced. Early studies suggested that monoaminergic deficits caused depressive
symptoms and drugs that reversed this alleviated depressive symptoms. However, it was not
until the opportune discovery in the 1950s of drugs that unintentionally treated depressive
symptoms that there emerged the hypothesis of a monoaminergic deficit of depression. These
two drugs included iproniazid, used to effectively treat tuberculosis, and imipramine, used to
treat patients with schizophrenia. While these drugs effectively treated these disorders, they
were also found to alleviate depressive symptoms in the patients taking these drugs (Smith,
1953, Crane, 1957, Loomer et al., 1957, Kuhn, 1958, Sandler, 1990, Lopez-Munoz et al., 2007),
drugs later being discovered to act via monoaminergic mechanisms.
17
Figure 1.6: The time course of epileptogenesis and the typical stages (dashed vertical lines) at which antidepressants may be clinically introduced. At points before or during epileptogenesis, antidepressant medication may be initiated, indicated by the dashed vertical lines: green, prior to epilepsy onset; purple, the period immediately after a precipitating event; orange, the period around when seizures emerge; blue, during active epilepsy. BDNF – Brain-derived neurotrophic factor. Figure modified from Scharfman (2007).
Iproniazid was the first drug in the class of antidepressants now known as monoamine oxidase
inhibitors (MAOI). MAOIs act by inhibiting the monoamine oxidases to prevent the breakdown
of monoamines, including serotonin and noradrenaline, increasing their availability. While
MAOIs were widely used in the 1950s, they fell out of favour as first line therapies due to
adverse side effects and interaction with dietary substrates (Lopez-Munoz et al., 2007, Lopez-
Munoz and Alamo, 2009). However, MAOIs are still prescribed today, in cases of intolerance to
first line treatments, in patients with refractory depression or when electroconvulsive therapy
(ECT) is inadvisable or rejected as a treatment option (Ban, 2001).
Imipramine was the first drug in the class of tricyclic antidepressants (TCA) (Lopez-Munoz and
Alamo, 2009). TCAs specifically inhibit the serotonin and noradrenaline reuptake membrane
transporters, increasing the synaptic levels of these monoamines (Manji et al., 2001, Nestler et
al., 2002, Morilak and Frazer, 2004, Gillespie and Nemeroff, 2005). Beginning with small studies
in the late 1950s (Kuhn, 1958) and later clinical trials (Ball and Kiloh, 1959, Rees et al., 1961,

18
Klerman and Cole, 1965), their effectiveness in treating depressive symptoms was clearly
evident. However, many undesirable side effects were also reported, including lethargy, weight
gain and lethality in overdose (Ball and Kiloh, 1959, Rees et al., 1961, Klerman and Cole, 1965).
TCAs are still broadly used today for the treatment of disorders other than depression such as
obsessive-compulsive disorder and chronic pain.
The discovery of MAOIs and TCAs defined a turning point for psychopharmacological research
into depression. The use of these drugs to treat depressive disorders initiated the idea that
depression was caused by impairments and chemical alterations within the brain, challenging
the long-held belief that patients with psychiatric disorders, especially depression, were
“deranged individuals with moral defects, in need of moral therapy” (Lopez-Munoz and Alamo,
2009). This not only removed a considerable amount of burden from patients and their families,
but also provided an understanding for the neurobiological basis of depressive disorders, as
well as how antidepressant drugs worked.
1.4.2 The discovery and use of selective serotonin reuptake inhibitors
During this period in the 1950s and 1960s, as analytical techniques improved, it became
understood that one mechanism by which antidepressants exerted their effects was by actions
on the serotonergic system (Carlsson et al., 1968). For the first time, antidepressant drugs were
being developed with a clear rationale and design to target specific deficits in depression. This
paved the way for the development of drugs to specifically target the serotonin reuptake
transporter to increase serotonin availability, avoiding other target receptors to reduce adverse
side effects. This led to the discovery and introduction into popular use of the selective
serotonin reuptake inhibitors (SSRIs), and also selective noradrenaline reuptake inhibitors and
serotonin and noradrenergic reuptake inhibitors (SNRIs): drugs that specifically target the
serotonergic and/or noradrenergic systems.
Through the development and testing of a variety of compounds, fluoxetine hydrochloride was
found to potently inhibit serotonin reuptake 300 times more than noradrenaline (Richelson,
2001) and to less potently affect α1, histamine or muscarinic cholinergic receptors (Stahl,
1996). Reports of positive outcomes in clinical trials on depressive symptoms, fewer side effects
and better safety in overdose led to approval from the United States Food and Drug
Administration and the introduction of the first SSRI, fluoxetine hydrochloride (Prozac), into the
market in 1987. With further research into SSRIs, a range of successors followed, including
citalopram hydrobromide (Celexa) by Lundbeck in 1989. Citalopram is the most selective in its
serotonin reuptake action in both in vitro and in vivo studies, showing 3500 times more

19
potentcy in inhibiting serotonin reuptake than it is with noradrenaline reuptake (Richelson,
2001). While many other selective serotonergic and/or noradrenergic compounds have been
discovered and are used today, for relevance to this thesis the subsequent discussion will focus
upon fluoxetine and citalopram.
It is now widely established that one of the mechanisms of action of SSRIs, including both
fluoxetine and citalopram, is inhibition of the serotonin transporter (SERT, Barker and Blakely,
1995, Stahl, 1996, Stahl, 1998) resulting in increasing extracellular levels of serotonin in the
synaptic cleft of many brain regions (Fuller and Snoddy, 1990; Figure 1.7). With an increase in
serotonin levels, the somatodendritic autoreceptors on the presynaptic neuron become
desensitised. These autoreceptors normally act to inhibit further serotonin release but
desensitisation contributes to increased release of serotonin into the synapse (Stahl, 1996).
While both fluoxetine and citalopram are highly selective in their actions of increasing
serotonin, fluoxetine has also shown to inhibit the P450 liver breakdown enzyme (CYP2D6)
(Stahl, 1996), potentially resulting in adverse effects when combined with other medications as
it may interfere with their metabolism. Fluoxetine also has a mean half-life of 1-3 days and its
active metabolite norfluoxetine, a half-life of 7-15 days (Hyttel, 1994), which increases the
likelihood of adverse interaction with other drugs administered at the same time.
Figure 1.7: The effects of SSRIs at the serotonergic synapse. Serotonin is released from the presynaptic nerve ending from synaptic vesicles into the synapse to act on receptor sites at the postsynaptic nerve ending. Excess serotonin is taken up by the serotonin reuptake transporter and recycled. SSRIs act by blocking the reuptake transporter, increasing the availability of serotonin in the synapse to act on postsynaptic receptor sites. From http://www.bluelight.ru/vb/threads/532617-How-does-an-SSRI-work

20
Although fluoxetine was widely used as an antidepressant in earlier days, and is still commonly
used in the experimental literature, it is currently not regularly prescribed to patients with
depression, due to the availability of more selective and safe SSRIs, such as citalopram.
Citalopram is now one of the first line medications for clinical use in patients with depression,
due to its high specificity and low side effect profile. It has low drug-drug interactions as it is
less strongly protein bound, and its metabolites are not as biologically active as that of
fluoxetine (the metabolites of citalopram are 4-11 times less potent than citalopram itself
(Roseboom and Kalin, 2011). Furthermore, its 36 hour half-life results in rapid elimination prior
to trying another drug and citalopram does not inhibit the P450 liver isozymes, like fluoxetine
(Mula and Trimble, 2003, Roseboom and Kalin, 2011).
When SSRIs are administered, increases in serotonin occur very quickly, however, several
weeks of treatment are required for clinical benefits to manifest in patients (Goodnick and
Goldstein, 1998, Rosebook and Kalin, 2011), often resulting in poor compliance. In early studies,
this delayed time of action was thought to be due to effects on receptor adaptations and to the
delayed time of desensitisation of the somatodendritic and terminal serotonin receptors that act
to regulate serotonin release (Lopez-Munoz et al., 2007). Studies now suggest that SSRIs have
other targets of action in addition to reuptake inhibition, which may also explain the delayed
time of therapeutic effect. There are a number of mechanisms by which the delayed time of
response may occur, some of which will be discussed below in Section 1.5. Briefly, this may be
due to the delayed time to affect regulation of intracellular signal transduction pathways
(Nestler et al., 1989, Perez et al., 1991) and alterations in gene expression (Nibuya et al., 1996,
Frechilla et al., 1998), as well as alterations in neurotrophic mechanisms, especially BDNF
(Nibuya et al., 1995, Chen et al., 2001) and potentially through delayed effects on
neuroplasticity (Nestler et al., 1989, Duman et al., 1997, Fabel et al., 2003).
1.4.3 Use of antidepressant pharmacotherapy in epilepsy
For some time, antidepressants have been known to influence seizure susceptibility. The first-
generation antidepressants, the TCAs, were noted to have the side effect of triggering seizures in
non-epileptic patients (Wroblewski et al., 1990, Edwards and Wheal, 1992, Preskorn and Fast,
1992) as well as aggravating pre-existing epilepsy (Pisani et al., 1999). Furthermore,
experimental electrophysiological studies corroborated this ability of TCAs to aggravate
seizures in vivo (Trimble, 1978, Koella et al., 1979, Ago et al., 2006, Ago et al., 2007) and in vitro
(Luchins et al., 1984). As a consequence, antidepressant medications were more carefully
prescribed to patients with epilepsy, with an increasing reluctance from physicians to treat the
depressive symptoms in patients with epilepsy with antidepressants (Cotterman-Hart, 2010).

21
Consequently, many depressed patients with epilepsy remained, and in fact, still remain
undertreated or completely untreated for their mood disorder. However, as the newer, second-
generation antidepressants, the SSRIs came into popular use, being known for their reduced
side effects and relative safety in overdose compared to TCAs, their use in epilepsy also
increased and are considered a first-line medication for the treatment of depression in epilepsy.
One point to consider is also the concurrent use of AEDs with antidepressants and the
interactions that may result. Patients with epilepsy are commonly treated with AEDs, and as
such considering the effect of antidepressants on AEDs is also essential. Some AEDs have been
shown to increase clearance of antidepressants (e.g., carbamazepine) while some
antidepressants can inhibit clearance of AEDs through interaction with the CYP-450 hepatic
enzyme system (Barry et al., 2008). Additionally, various AEDs are also used to treat psychiatric
disorders such as mania and bipolar depression, and act as mood stabilisers in bipolar and
schizoaffective disorders (Schmitz, 2011). However, AEDs have also shown to possibly be a
cause of depressive symptoms in some patients (Mula and Monaco, 2009; Schmitz, 2011).
Therefore considering the interaction of antidepressants and AEDs is essential given the
common targets and indications of both drugs.
Various studies have attempted to address the issue of AED interaction with antidepressants
and the effect this may have on seizures (Borowicz et al., 2006, Borowicz et al., 2007, Borowicz
et al., 2011) while other studies have also investigated the way in which antidepressants
themselves may affect seizures, in both patients and animal models. However, while various
studies in humans and animals have investigated the effects of antidepressants on seizures, a
common theme is the lack of investigation of the effects of antidepressants on epileptogenesis.
Epileptogenesis is an important aspect to investigate, rather than just investigating the effects
on the symptoms of the disorder, the seizures. This is because there is evidence to suggest that
epilepsy is a progressive disorder that evolves over years in humans (O'Brien et al., 1999, Tasch
et al., 1999, Bouilleret et al., 2000, Cole, 2000, Cascino, 2009, Yang et al., 2010, Ono and
Galanopoulou, 2012) and while seizures may be controlled by AEDs, the underlying disorder
itself may continue to progress.
1.4.3.1 Human studies of SSRI pharmacotherapy in epilepsy
Currently, ten studies have reported on the effect of SSRI or SNRI treatment in patients with
epilepsy (Table 1.1). These studies involved both children and adult patients with either focal or
generalised epilepsies either with (Harmant et al., 1990, Hovorka et al., 2000, Kanner et al.,
2000, Kuhn et al., 2003, Specchio et al., 2004, Thome-Souza et al., 2007, Okazaki et al., 2011) or

22
without (Gigli et al., 1994, Favale et al., 1995, Favale et al., 2003) affective disorders. All patients
in the study groups were receiving AED therapy concurrently with antidepressant medications.
The antidepressants that were administered to the patients included various SSRIs and SNRIs as
well as TCAs in one study (Okazaki et al., 2011). In all studies, the baseline seizure incidence
(from 1-12 months prior to antidepressant administration) was obtained from medical records
or seizure diaries, while during treatment seizure incidence was monitored at regular intervals
for a time period of 1 to 15 months.
Of these ten studies, seven studies reported no change in seizure frequency (Harmant et al.,
1990; Gigli et al., 1994; Hovorka et al., 2000; Kanner et al., 2000; Kuhn et al., 2003; Thome-Souza
et al., 2007; Okazaki et al., 2011) and three studies reported an improvement in seizure
frequency, with some patients reporting complete seizure freedom during antidepressant
treatment (Favale et al., 1995; Favale et al., 2003; Specchio et al., 2004). While in some studies
patients reported an increase in seizure frequency (Gigli et al., 1994; Kanner et al., 2000;
Specchio et al., 2004, Thome-Souza 2007). However, these were rare occurrences and seizures
were returned to pre-antidepressant treatment rates by increasing AED medications or
removing the antidepressants.
Overall, these studies suggest that antidepressant medication administered to patients with
epilepsy does not adversely affect seizure frequency, which in itself is an important outcome.
Additionally, in the studies in which depressive symptoms were also monitored (Harmant et al.,
1990, Hovorka et al., 2000, Kanner et al., 2000, Kuhn et al., 2003, Specchio et al., 2004, Thome-
Souza et al., 2007, Okazaki et al., 2011), improvements were largely observed, which is
important, given the impact of depression on quality of life (Boylan et al., 2004, Gilliam et al.,
2004).
However, as with many clinical studies, various limitations exist when interpreting the results.
Firstly, no studies were performed with a double blind, randomised-control design. Secondly,
assessing the effects of the antidepressants on seizures may have been confounded by other
factors such as medication compliance, stress, insomnia, brain damage or cognitive
impairments. Thirdly, the magnitude of the effects may have been over- or underestimated due
to fluctuations in seizure frequency, common throughout the course of the illness. Most studies
were performed using small sample sizes resulting in low statistical power. Finally, in some
studies, patients were chosen from highly selected population samples, such as patients from
epilepsy clinics or patients being considered for epilepsy surgery.

23
Although there have been no placebo-controlled studies to assess the effect of antidepressants
on epilepsy itself, one important and influential study assessed the seizure-inducing effects of
antidepressant medications in non-epileptic patients (Alper et al., 2007). In this study, the
effects of second generation antidepressants (mostly SSRIs) on seizures in over 75000 patients
from phase II and III randomly controlled clinical trials for treatment of depression were
assessed. The results of this study suggested that in depressed (non-epileptic) placebo-treated
controls, the rates of spontaneous seizures were considerably higher than known population
rates and further suggested that this rate of spontaneous seizures was significantly reduced by
antidepressant treatment. Although patients with epilepsy were excluded from analysis in this
study, it does suggest an overall antiseizure effect for antidepressants when administered at
therapeutic doses, which follows from the ten studies described above that investigated
antidepressant medication and its effects on seizures in patients with epilepsy.
To date, there have been no clinical studies addressing the potential effects of antidepressant
treatment on disease progression or disease-modification in epilepsy, as opposed to the anti-
seizure effects. While it has been suggested in recent literature that such studies would be of
value (Danzer, 2011, Igelstrom, 2012, Payandemehr et al., 2012, Vermoesen et al., 2012), the
technical and logistic aspects make this a difficult undertaking. Such studies would require long-
term treatment and follow up, large sample sizes, the need for appropriately matched placebo-
treated controls, and the need to address the various manifestations of epilepsy progression
that may be affected by antidepressant treatment. This includes not only seizure variables such
as frequency, severity and duration but also other related factors such as type of AED
administered and AED resistance, neuropsychiatric and neurocognitive deficits and associated
structural brain changes. These factors are more readily addressed in animal models of epilepsy
in which there is greater control of experimental settings, comparisons can be made before and
after treatment, long-term follow up and assessment is more straight-forward and achievable in
a shorter time frame, and other aspects of the condition such as behavioural and neurochemical
alterations can be more easily addressed.
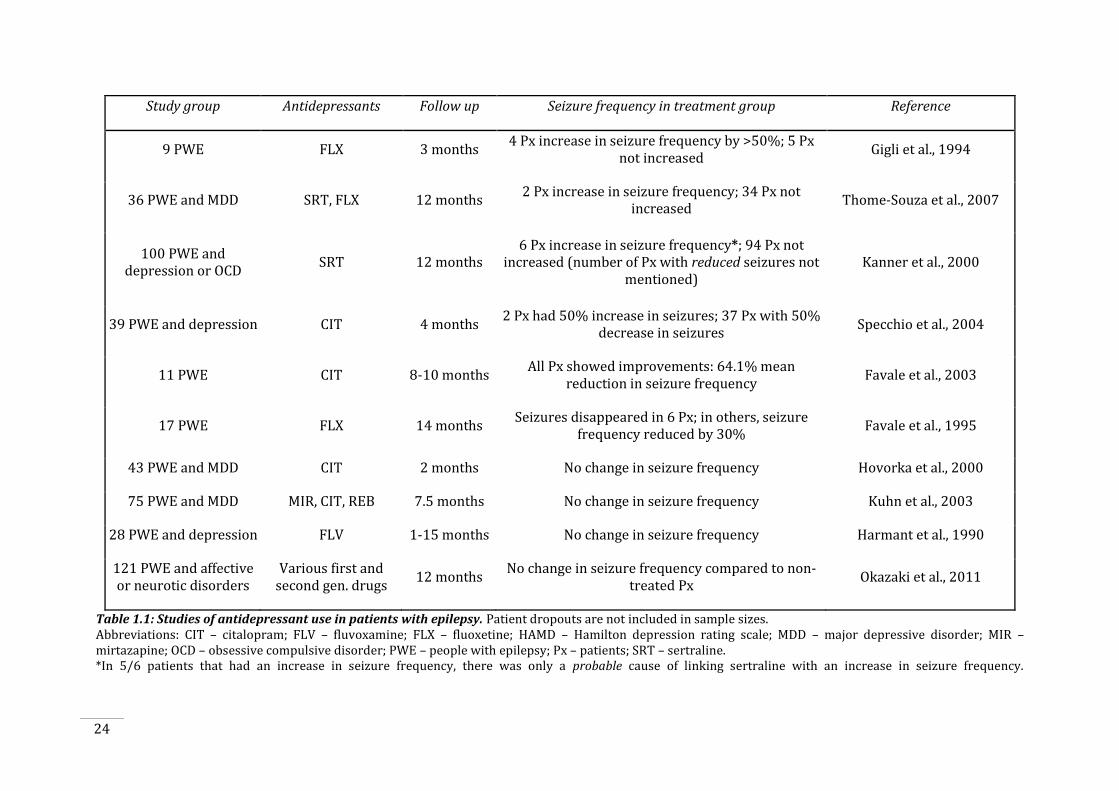
24
Study group Antidepressants Follow up Seizure frequency in treatment group Reference
9 PWE FLX 3 months 4 Px increase in seizure frequency by >50%; 5 Px
not increased Gigli et al., 1994
36 PWE and MDD SRT, FLX 12 months 2 Px increase in seizure frequency; 34 Px not
increased Thome-Souza et al., 2007
100 PWE and depression or OCD
SRT 12 months 6 Px increase in seizure frequency*; 94 Px not
increased (number of Px with reduced seizures not mentioned)
Kanner et al., 2000
39 PWE and depression CIT 4 months 2 Px had 50% increase in seizures; 37 Px with 50%
decrease in seizures Specchio et al., 2004
11 PWE CIT 8-10 months All Px showed improvements: 64.1% mean
reduction in seizure frequency Favale et al., 2003
17 PWE FLX 14 months Seizures disappeared in 6 Px; in others, seizure
frequency reduced by 30% Favale et al., 1995
43 PWE and MDD CIT 2 months No change in seizure frequency Hovorka et al., 2000
75 PWE and MDD MIR, CIT, REB 7.5 months No change in seizure frequency Kuhn et al., 2003
28 PWE and depression FLV 1-15 months No change in seizure frequency Harmant et al., 1990
121 PWE and affective or neurotic disorders
Various first and second gen. drugs
12 months No change in seizure frequency compared to non-
treated Px Okazaki et al., 2011
Table 1.1: Studies of antidepressant use in patients with epilepsy. Patient dropouts are not included in sample sizes. Abbreviations: CIT – citalopram; FLV – fluvoxamine; FLX – fluoxetine; HAMD – Hamilton depression rating scale; MDD – major depressive disorder; MIR – mirtazapine; OCD – obsessive compulsive disorder; PWE – people with epilepsy; Px – patients; SRT – sertraline. *In 5/6 patients that had an increase in seizure frequency, there was only a probable cause of linking sertraline with an increase in seizure frequency.

25
1.4.3.2 SSRI pharmacotherapy in animal models of epilepsy
Various animal studies have investigated the effects of antidepressants in animal models of
epilepsy. A recent review by Igelstrom (2012) which focused on determining the effects of
antidepressants on animal models of seizures and epilepsy, found that antidepressants exerted
an overall anticonvulsant effect. This follows the patient data in that antidepressants are not
adversely affecting seizures. However, similarly to the human studies, most of the animal
studies only address effects on short-term seizure outcomes (e.g., seizure threshold) and no
longitudinal studies have been carried out.
The animal studies that have investigated the effects on SSRIs and SNRIs on seizures are
summarised in Table 1.2. While human studies solely investigated seizure frequency, other
measures were assessed in the animal models including number of spontaneous seizures,
seizure frequency or duration, threshold to behavioural or electrical seizure, seizure severity,
latency to first seizure and survival (see Table 1.2).
In summary, a majority of the studies found that SSRIs and SNRIs exert beneficial effects on
seizures (anticonvulsant) or are without effect, indicated by reduced, or no change in, seizure
frequency, duration, threshold, severity and increased latency to seizure between
antidepressant-treated and vehicle-treated animals. However, a minority of studies did report a
proconvulsant effect of antidepressants, indicated by reduced threshold to behavioural seizures,
greater seizure severity, shorter latency and more frequent seizures. However this was a small
number of studies (7 out of 38 studies), and in some cases, seizures were occurring at doses of
antidepressants above the reported therapeutic range for animal models (Santos et al., 2002,
Ahern et al., 2006, Payandemehr et al., 2012, Vermoesen et al., 2012). In fact, studies have
reported a biphasic effect of serotonin (Clinckers et al., 2004a) and SSRI (Loscher, 2009,
Payandemehr et al., 2012) concentrations on seizures, whereby low doses are anticonvulsant
and higher doses are proconvulsant.
A strength of this collective data, suggesting that antidepressants have no adverse effect on
seizure outcomes or may even be anticonvulsant, is that the evidence has been derived from a
wide variety of different animal models including both chemoconvulsant- and electrically-
induced seizure models as well as genetically predisposed models of epilepsy. However, there
are some limitations to consider when interpreting this data. Firstly, in many of the models
there were no behavioural abnormalities that were indicative of the comorbid psychiatric
conditions that occur in patients with epilepsy. Secondly, many of the models employed were
models of acute seizures, such as pentylenetetrazol or maximal electroshock, in which seizure

26
susceptibility can be tested in response to administration of antidepressants. However, the
animals in these models are not epileptic and there would have been no associated pathological
alterations, which are found in epileptic brains of humans and animals (described in section
1.2.3.4), and therefore the drugs may be having different effects. Thirdly, although the variety of
animal models used is advantageous, it is also problematic to aggregate such data due to the
wide variety of experimental designs used, particularly variations in the dose and timing of
antidepressant administration relative to the seizure testing. Finally, in many of the studies,
antidepressants were only administered as a single dose. This does not mimic the clinical
situation, where several weeks of antidepressant treatment are required for clinically beneficial
effects to manifest in patients and exposure may continue for months or years.
Absent from the current literature are any studies investigating the effects of chronic
antidepressant treatment on epileptogenesis. Some studies have administered antidepressants
for three weeks or greater (Dailey et al., 1992, Wada et al., 1995, Raju et al., 1999, Ferrero et al.,
2005, Ahern et al., 2006). There have been three studies that investigated the effects of the
chronic antidepressant treatment on long-term seizure outcomes (Hernandez et al., 2002,
Mazarati et al., 2008, Vermoesen et al., 2012), all of which used a chemoconvulsant-induced
model and investigated the occurrence of spontaneous seizures following SSRI treatment for 4-
10 days. However, no studies have combined these measures to determine how chronic
treatment with antidepressant drugs may impact epileptogenesis.
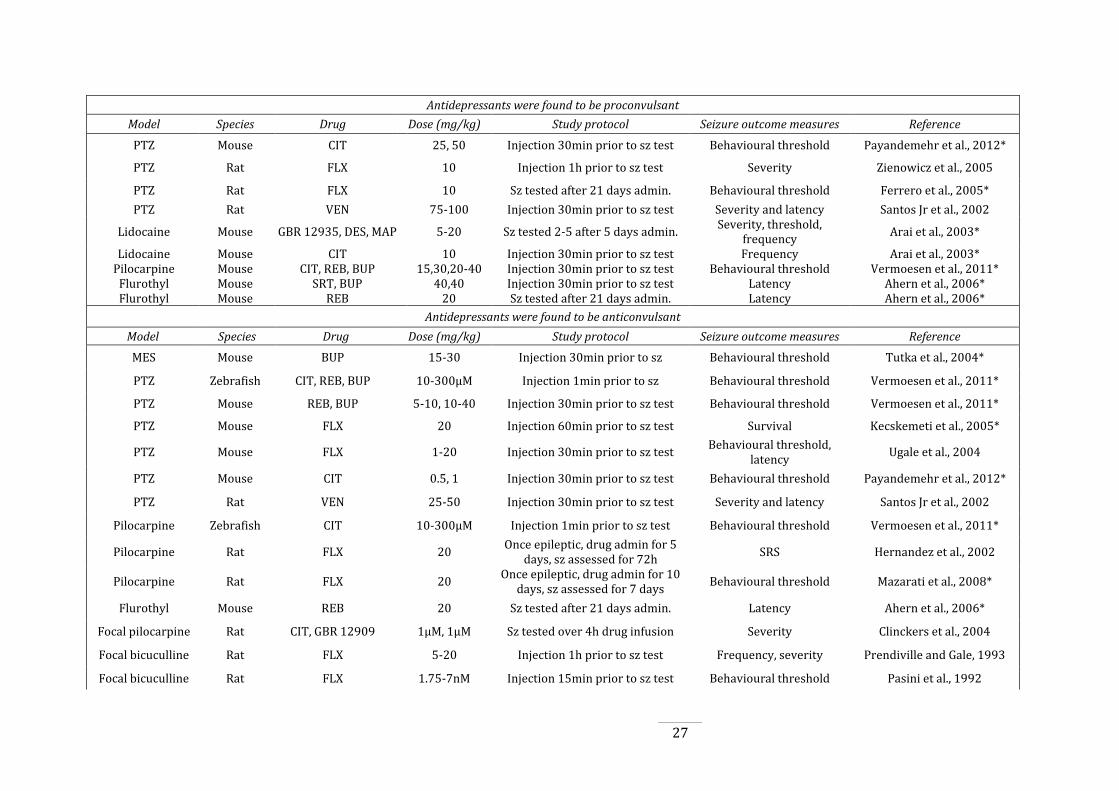
27
Antidepressants were found to be proconvulsant
Model Species Drug Dose (mg/kg) Study protocol Seizure outcome measures Reference
PTZ Mouse CIT 25, 50 Injection 30min prior to sz test Behavioural threshold Payandemehr et al., 2012*
PTZ Rat FLX 10 Injection 1h prior to sz test Severity Zienowicz et al., 2005
PTZ Rat FLX 10 Sz tested after 21 days admin. Behavioural threshold Ferrero et al., 2005*
PTZ Rat VEN 75-100 Injection 30min prior to sz test Severity and latency Santos Jr et al., 2002
Lidocaine Mouse GBR 12935, DES, MAP 5-20 Sz tested 2-5 after 5 days admin. Severity, threshold,
frequency Arai et al., 2003*
Lidocaine Mouse CIT 10 Injection 30min prior to sz test Frequency Arai et al., 2003* Pilocarpine Mouse CIT, REB, BUP 15,30,20-40 Injection 30min prior to sz test Behavioural threshold Vermoesen et al., 2011*
Flurothyl Mouse SRT, BUP 40,40 Injection 30min prior to sz test Latency Ahern et al., 2006* Flurothyl Mouse REB 20 Sz tested after 21 days admin. Latency Ahern et al., 2006*
Antidepressants were found to be anticonvulsant
Model Species Drug Dose (mg/kg) Study protocol Seizure outcome measures Reference
MES Mouse BUP 15-30 Injection 30min prior to sz Behavioural threshold Tutka et al., 2004*
PTZ Zebrafish CIT, REB, BUP 10-300µM Injection 1min prior to sz Behavioural threshold Vermoesen et al., 2011*
PTZ Mouse REB, BUP 5-10, 10-40 Injection 30min prior to sz test Behavioural threshold Vermoesen et al., 2011*
PTZ Mouse FLX 20 Injection 60min prior to sz test Survival Kecskemeti et al., 2005*
PTZ Mouse FLX 1-20 Injection 30min prior to sz test Behavioural threshold,
latency Ugale et al., 2004
PTZ Mouse CIT 0.5, 1 Injection 30min prior to sz test Behavioural threshold Payandemehr et al., 2012*
PTZ Rat VEN 25-50 Injection 30min prior to sz test Severity and latency Santos Jr et al., 2002
Pilocarpine Zebrafish CIT 10-300µM Injection 1min prior to sz test Behavioural threshold Vermoesen et al., 2011*
Pilocarpine Rat FLX 20 Once epileptic, drug admin for 5
days, sz assessed for 72h SRS Hernandez et al., 2002
Pilocarpine Rat FLX 20 Once epileptic, drug admin for 10
days, sz assessed for 7 days Behavioural threshold Mazarati et al., 2008*
Flurothyl Mouse REB 20 Sz tested after 21 days admin. Latency Ahern et al., 2006*
Focal pilocarpine Rat CIT, GBR 12909 1µM, 1µM Sz tested over 4h drug infusion Severity Clinckers et al., 2004
Focal bicuculline Rat FLX 5-20 Injection 1h prior to sz test Frequency, severity Prendiville and Gale, 1993
Focal bicuculline Rat FLX 1.75-7nM Injection 15min prior to sz test Behavioural threshold Pasini et al., 1992
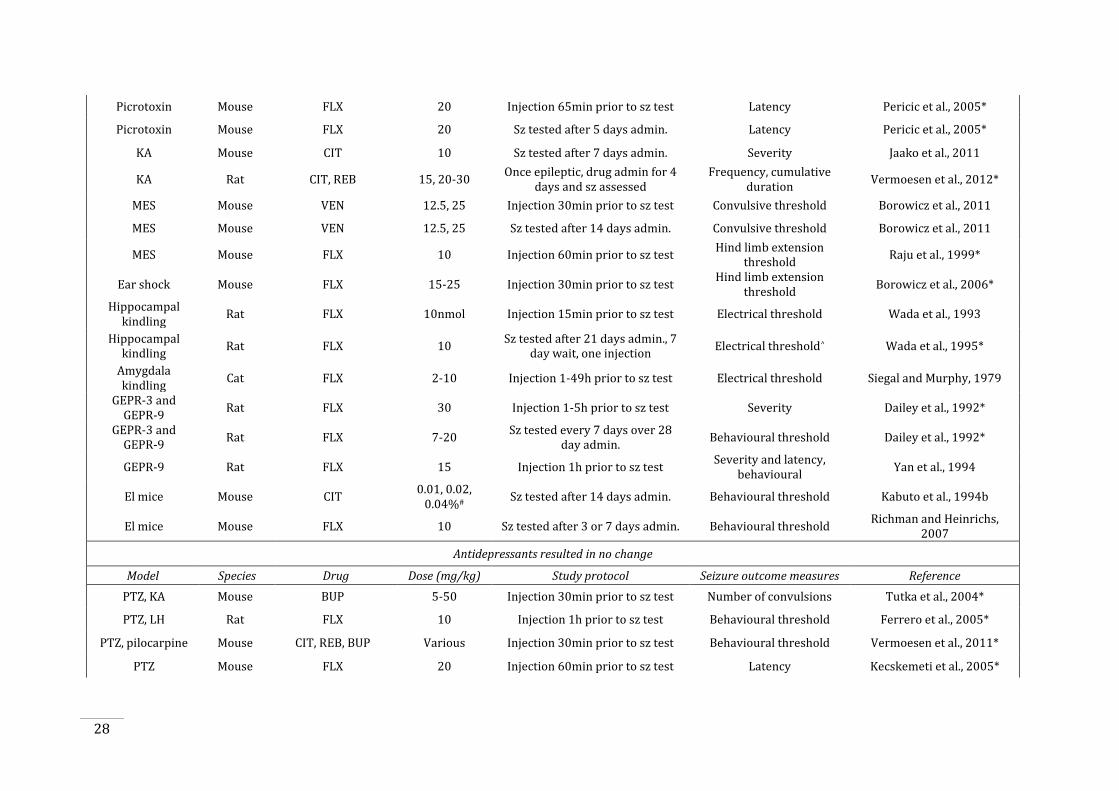
28
Picrotoxin Mouse FLX 20 Injection 65min prior to sz test Latency Pericic et al., 2005*
Picrotoxin Mouse FLX 20 Sz tested after 5 days admin. Latency Pericic et al., 2005*
KA Mouse CIT 10 Sz tested after 7 days admin. Severity Jaako et al., 2011
KA Rat CIT, REB 15, 20-30 Once epileptic, drug admin for 4
days and sz assessed
Frequency, cumulative duration
Vermoesen et al., 2012*
MES Mouse VEN 12.5, 25 Injection 30min prior to sz test Convulsive threshold Borowicz et al., 2011
MES Mouse VEN 12.5, 25 Sz tested after 14 days admin. Convulsive threshold Borowicz et al., 2011
MES Mouse FLX 10 Injection 60min prior to sz test Hind limb extension
threshold Raju et al., 1999*
Ear shock Mouse FLX 15-25 Injection 30min prior to sz test Hind limb extension
threshold Borowicz et al., 2006*
Hippocampal kindling
Rat FLX 10nmol Injection 15min prior to sz test Electrical threshold Wada et al., 1993
Hippocampal kindling
Rat FLX 10 Sz tested after 21 days admin., 7
day wait, one injection Electrical threshold^ Wada et al., 1995*
Amygdala kindling
Cat FLX 2-10 Injection 1-49h prior to sz test Electrical threshold Siegal and Murphy, 1979
GEPR-3 and GEPR-9
Rat FLX 30 Injection 1-5h prior to sz test Severity Dailey et al., 1992*
GEPR-3 and GEPR-9
Rat FLX 7-20 Sz tested every 7 days over 28
day admin. Behavioural threshold Dailey et al., 1992*
GEPR-9 Rat FLX 15 Injection 1h prior to sz test Severity and latency,
behavioural Yan et al., 1994
El mice Mouse CIT 0.01, 0.02,
0.04%# Sz tested after 14 days admin. Behavioural threshold Kabuto et al., 1994b
El mice Mouse FLX 10 Sz tested after 3 or 7 days admin. Behavioural threshold Richman and Heinrichs,
2007
Antidepressants resulted in no change
Model Species Drug Dose (mg/kg) Study protocol Seizure outcome measures Reference
PTZ, KA Mouse BUP 5-50 Injection 30min prior to sz test Number of convulsions Tutka et al., 2004*
PTZ, LH Rat FLX 10 Injection 1h prior to sz test Behavioural threshold Ferrero et al., 2005*
PTZ, pilocarpine Mouse CIT, REB, BUP Various Injection 30min prior to sz test Behavioural threshold Vermoesen et al., 2011*
PTZ Mouse FLX 20 Injection 60min prior to sz test Latency Kecskemeti et al., 2005*
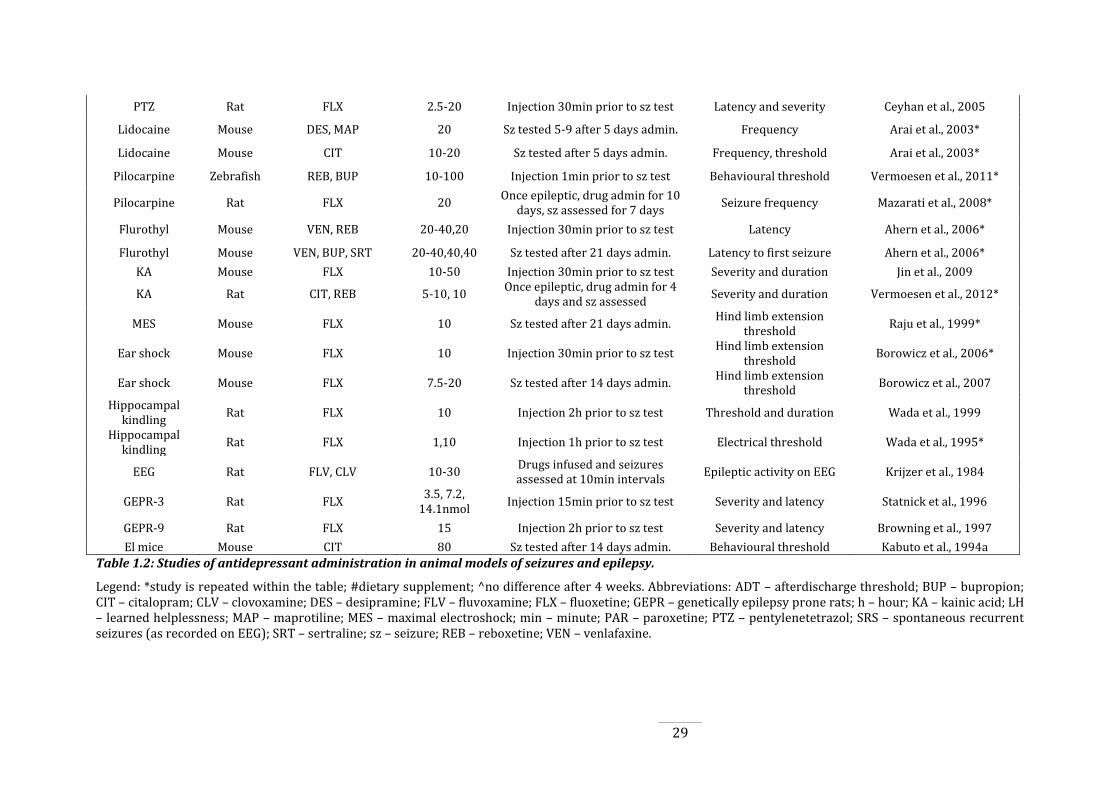
29
PTZ Rat FLX 2.5-20 Injection 30min prior to sz test Latency and severity Ceyhan et al., 2005
Lidocaine Mouse DES, MAP 20 Sz tested 5-9 after 5 days admin. Frequency Arai et al., 2003*
Lidocaine Mouse CIT 10-20 Sz tested after 5 days admin. Frequency, threshold Arai et al., 2003*
Pilocarpine Zebrafish REB, BUP 10-100 Injection 1min prior to sz test Behavioural threshold Vermoesen et al., 2011*
Pilocarpine Rat FLX 20 Once epileptic, drug admin for 10
days, sz assessed for 7 days Seizure frequency Mazarati et al., 2008*
Flurothyl Mouse VEN, REB 20-40,20 Injection 30min prior to sz test Latency Ahern et al., 2006*
Flurothyl Mouse VEN, BUP, SRT 20-40,40,40 Sz tested after 21 days admin. Latency to first seizure Ahern et al., 2006*
KA Mouse FLX 10-50 Injection 30min prior to sz test Severity and duration Jin et al., 2009
KA Rat CIT, REB 5-10, 10 Once epileptic, drug admin for 4
days and sz assessed Severity and duration Vermoesen et al., 2012*
MES Mouse FLX 10 Sz tested after 21 days admin. Hind limb extension
threshold Raju et al., 1999*
Ear shock Mouse FLX 10 Injection 30min prior to sz test Hind limb extension
threshold Borowicz et al., 2006*
Ear shock Mouse FLX 7.5-20 Sz tested after 14 days admin. Hind limb extension
threshold Borowicz et al., 2007
Hippocampal kindling
Rat FLX 10 Injection 2h prior to sz test Threshold and duration Wada et al., 1999
Hippocampal kindling
Rat FLX 1,10 Injection 1h prior to sz test Electrical threshold Wada et al., 1995*
EEG Rat FLV, CLV 10-30 Drugs infused and seizures assessed at 10min intervals
Epileptic activity on EEG Krijzer et al., 1984
GEPR-3 Rat FLX 3.5, 7.2,
14.1nmol Injection 15min prior to sz test Severity and latency Statnick et al., 1996
GEPR-9 Rat FLX 15 Injection 2h prior to sz test Severity and latency Browning et al., 1997
El mice Mouse CIT 80 Sz tested after 14 days admin. Behavioural threshold Kabuto et al., 1994a
Table 1.2: Studies of antidepressant administration in animal models of seizures and epilepsy.
Legend: *study is repeated within the table; #dietary supplement; ^no difference after 4 weeks. Abbreviations: ADT – afterdischarge threshold; BUP – bupropion; CIT – citalopram; CLV – clovoxamine; DES – desipramine; FLV – fluvoxamine; FLX – fluoxetine; GEPR – genetically epilepsy prone rats; h – hour; KA – kainic acid; LH – learned helplessness; MAP – maprotiline; MES – maximal electroshock; min – minute; PAR – paroxetine; PTZ – pentylenetetrazol; SRS – spontaneous recurrent seizures (as recorded on EEG); SRT – sertraline; sz – seizure; REB – reboxetine; VEN – venlafaxine.

30
1.5. Antidepressants and epileptogenesis: shared neurobiological
substrates
Antidepressants affect a number of neurobiological processes in ways that could well affect
epileptogenesis. This has only recently begun to be appreciated (Mazarati et al., 2010, Danzer,
2011, Jaako et al., 2011, Payandemehr et al., 2012) and as yet these processes have not been
thoroughly investigated from this perspective. While determining how epileptogenesis may be
affected by antidepressant exposure can be investigated to a limited extent by analysing the
seizure outcomes that result from epileptogenesis, it is also essential to investigate the
mechanisms of action common to the two disorders. As such, it is important to determine the
common neurobiological mechanisms that are both affected by antidepressant treatment and
implicated in epileptogenesis, as these factors may also be contributing to the effects of
antidepressants in epilepsy. From the literature, it is clear that both epileptogenesis and
antidepressant treatment share some common mechanisms of action, providing pathways by
which antidepressants may potentially affect epileptogenesis. The most prominent of these are
the effects on neuroplasticity and neurogenesis and alterations in the hypothalamo-pituitary-
adrenal (HPA) axis, which are discussed below. There are also other factors and mechanisms
common to the two, including monoamines, brain-derived neurotrophic factor and
inflammatory processes, all of which will be briefly discussed but were not investigated in the
experiments reported in this thesis.
1.5.1 Hypothalamo-pituitary-adrenal axis and glucocorticoids
Glucocorticoids, the end product of the hypothalamo-pituitary adrenal (HPA) axis, are steroid
hormones that are released in a circadian and pulsatile rhythm, which become increasingly
released in times of stress. Both circulating and stress-induced release of glucocorticoids,
predominantly cortisol in humans and corticosterone in rodents, are homeostatically regulated
by positive and negative feedback loops involving the hippocampus, amygdala, hypothalamus,
frontal cortex and pituitary (Figure 1.8, Lupien et al., 2009). This occurs via activation of
glucocorticoid receptors (GR) and mineralocorticoid receptors (MR) (Sapolsky et al., 1984,
Jacobson and Sapolsky, 1991, Herman and Cullinan, 1997, Herman et al., 2003). The
hippocampus and amygdala are also structures highly implicated in epileptogenesis, depression
and antidepressant action. Therefore, considering the involvement of the HPA system in
epilepsy and during antidepressant treatment may shed some light on possible mechanisms of
their interaction.

31
Hyper-reactivity of the HPA axis has been reported to occur in greater than 50% of depressed
patients (Holsboer, 2001, Pariante and Lightman, 2008), which is presumably due to
dysfunctional negative feedback regulation of the HPA axis. This dysfunction has been shown to
be alleviated by antidepressant treatment, particularly by SSRIs (Holsboer, 2001, Pariante,
2006, Himmerich et al., 2007). However, in a systematic review of studies investigating changes
in cortisol levels in unipolar depression pre- and post-treatment, it was found that
approximately 56% of patients had similar cortisol levels before and after treatment, regardless
of depressive symptoms (McKay and Zakzanis, 2010). The ability of antidepressants to affect the
HPA axis has also been investigated in animal studies, with a suppression of HPA axis function
following treatment with fluoxetine or citalopram reported (Jensen et al., 1999, Jongsma et al.,
2005, Mazarati et al., 2008). The mechanism by which antidepressants alleviate HPA
dysfunction has been suggested to occur by increasing GR translocation in the hippocampus
(Molteni et al., 2009) and regulation of CRH mRNA production in the paraventricular nucleus
(Brady et al., 1991).
While hyper-reactivity of the HPA axis is a widely reported alteration in depressed patients
(Pariante and Lightman, 2008), fewer, but increasing, reports suggest that HPA axis dysfunction
also occurs in epilepsy. Various human and rodent studies have reported elevations in
adrenocorticotrophic hormone (ACTH) and cortisol/corticosterone following seizures
(Gallagher et al., 1984, Pritchard et al., 1985, Takeshita et al., 1986, Gallagher, 1987, Rao et al.,
1989, Kumar et al., 2011). Humans with epilepsy, without comorbid depression or anxiety, were
found to have a greater elevation in cortisol following the dexamethasone/corticotrophin-
releasing hormone (Dex/CRH) test compared to controls, indicative of a hyper-reactive HPA
axis (Zobel et al., 2004). In addition to this, animal studies suggest a cause/effect relationship
between HPA axis dysfunction and epilepsy. For example, amygdala kindling epileptogenesis is
accelerated with corticosterone supplementation (Karst et al., 1999, Taher et al., 2005, Kumar et
al., 2007), and conversely, retarded kindling rates have been demonstrated in surgically
adrenalectomised rats compared to sham-adrenalectomised rats (Cottrell et al., 1984, Weiss et
al., 1993, Edwards et al., 1999). These findings are also consistent with a study using a rat model
of epilepsy, in which HPA axis hyper-reactivity, assessed with the Dex/CRH test, was associated
with the development of spontaneous seizures, and therefore epilepsy, which also correlated
with the severity of depressive-like symptoms (Mazarati et al., 2009). Interestingly, in this
study, HPA axis hyper-reactivity was also observed in rats that were not spontaneously seizing,
indicating that the underlying epilepsy-associated pathological changes that contribute to the
HPA axis changes may be unrelated to the seizures themselves.
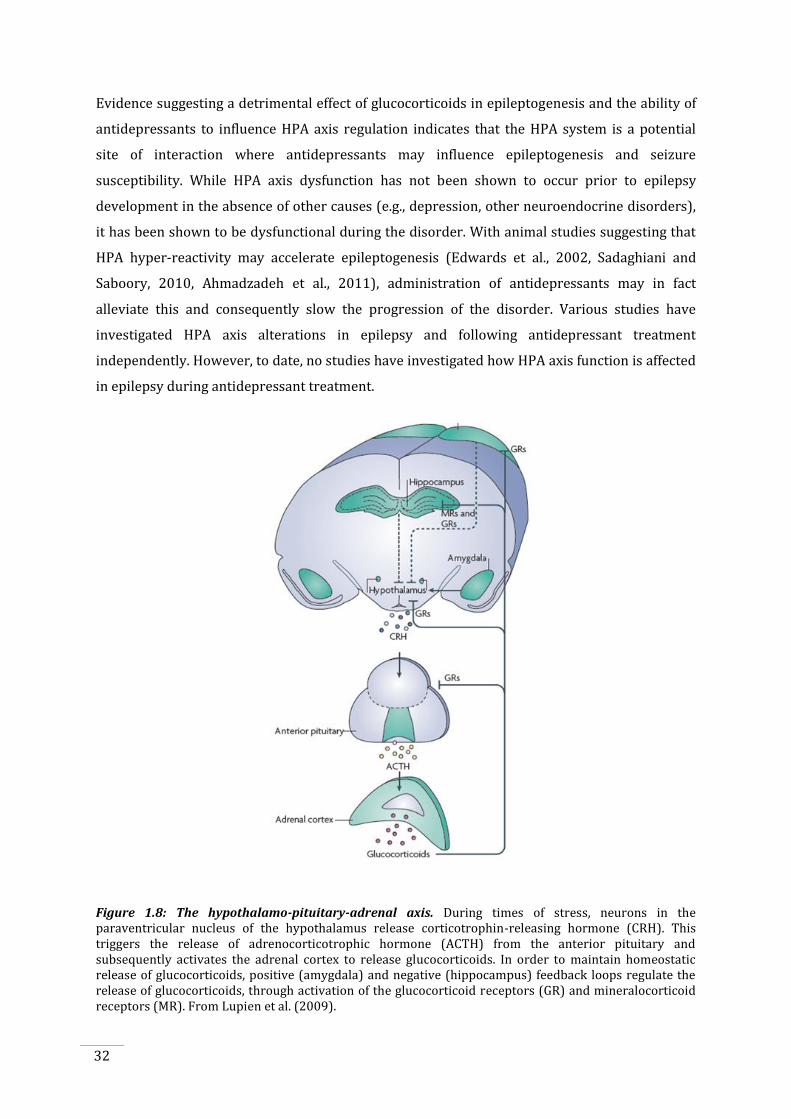
32
Evidence suggesting a detrimental effect of glucocorticoids in epileptogenesis and the ability of
antidepressants to influence HPA axis regulation indicates that the HPA system is a potential
site of interaction where antidepressants may influence epileptogenesis and seizure
susceptibility. While HPA axis dysfunction has not been shown to occur prior to epilepsy
development in the absence of other causes (e.g., depression, other neuroendocrine disorders),
it has been shown to be dysfunctional during the disorder. With animal studies suggesting that
HPA hyper-reactivity may accelerate epileptogenesis (Edwards et al., 2002, Sadaghiani and
Saboory, 2010, Ahmadzadeh et al., 2011), administration of antidepressants may in fact
alleviate this and consequently slow the progression of the disorder. Various studies have
investigated HPA axis alterations in epilepsy and following antidepressant treatment
independently. However, to date, no studies have investigated how HPA axis function is affected
in epilepsy during antidepressant treatment.
Figure 1.8: The hypothalamo-pituitary-adrenal axis. During times of stress, neurons in the paraventricular nucleus of the hypothalamus release corticotrophin-releasing hormone (CRH). This triggers the release of adrenocorticotrophic hormone (ACTH) from the anterior pituitary and subsequently activates the adrenal cortex to release glucocorticoids. In order to maintain homeostatic release of glucocorticoids, positive (amygdala) and negative (hippocampus) feedback loops regulate the release of glucocorticoids, through activation of the glucocorticoid receptors (GR) and mineralocorticoid receptors (MR). From Lupien et al. (2009).

33
1.5.2 Hippocampal neurogenesis
As discussed in Section 1.2.3.2, the hippocampus is an essential structure in TLE, being a
common region of seizure initiation (Babb, 2001), as well as showing significant pathological
alterations. This includes aberrant hippocampal plasticity, which is one of the key features of
acquired epilepsy models and in human epilepsy, which may contribute to the generation of the
hyperexcitable circuitry in the epileptic brain. The alteration of hippocampal neurogenesis may
therefore potentially affect epilepsy development and progression.
In recent years, one of the most widely reported aspects of hippocampal plasticity has been
hippocampal neurogenesis – the generation of newborn granule cells in the mammalian dentate
gyrus, and their subsequent integration into the existing hippocampal circuitry (Altman and
Das, 1965, Kaplan and Hinds, 1977, Cameron et al., 1993). Adult neurogenesis has been shown
to occur in all mammalian species examined to date (humans, Eriksson et al., 1998; other
species, Altman, 1962, Altman and Das, 1965, Gould et al., 1998, Kornack and Rakic, 1999)
under normal conditions. Figure 1.9 shows the normal process of adult hippocampal
neurogenesis that begins with neural progenitor cells giving rise to a pool of precursors. These
precursor cells adopt a neuronal fate, and over several weeks undergo differentiation,
maturation, migration and integration to become fully functional dentate granule cells (Eisch et
al., 2008). Throughout this period, the newborn cells develop all the features of granule cells
including the characteristic dendritic structure, axonal projections and afferent and efferent
connections (Kaplan and Hinds, 1977, Markakis and Gage, 1999, Ambrogini et al., 2004,
Overstreet et al., 2004, Laplagne et al., 2006, Ribak and Shapiro, 2007), becoming remarkably
similar to cells produced earlier in development (Esposito et al., 2005, Zhao et al., 2006, Rahimi
and Claiborne, 2007).

34
Figure 1.9: The process of adult hippocampal neurogenesis in the dentate gyrus. Progenitor cells (green) divide and multiply to give rise to a pool of precursor cells (blue) that adopt a neuronal fate (lavender) to become newborn neurons (purple). These immature neurons will develop over several weeks (pink) to eventually become fully integrated mature granule cells (orange). Figure adapted from Eisch et al (2008).
This process normally occurs in the brain continuously however insults to the brain may affect
its progress. Studies have shown that seizures and epilepsy can be associated with both
increases and decreases in neurogenesis. Acute seizures have been shown to stimulate
hippocampal neurogenesis (Bengzon et al., 1997, Parent et al., 1997, Gray and Sundstrom, 1998,
Parent et al., 1998), while chronic epilepsy in rodents resulted in a dramatic reduction in
differentiation (Hattiangady et al., 2004, Hattiangady and Shetty, 2010). Integration of these
cells into the hippocampal circuitry may also be affected by seizures. Following experimentally-
induced status epilepticus, which over time often results in epilepsy, it has been found that
newly generated neurons migrate ectopically into the dentate hilar region rather than into the
normal hippocampal circuitry (Scharfman et al., 2000, McCloskey et al., 2006, Parent et al., 2006,
Jessberger et al., 2007, Parent et al., 2007, Scharfman and Gray, 2007, Walter et al., 2007,
Murphy et al., 2011). These cells have also shown to develop aberrant basal dendrites
(Jessberger et al., 2007, Ribak and Shapiro, 2007, Shapiro et al., 2008) and contribute to mossy
fibre sprouting (Parent et al., 1997, Danzer et al., 2008, Kron et al., 2010). This can create
aberrant dendritic connectivity within the structure and potentially contribute to the
generation of the epileptic circuitry (Parent et al., 1997, Scharfman et al., 2007, Kron et al.,

35
2010). Several studies have also shown that blocking seizure-induced neurogenesis normalised
the behavioural and electrophysiological effects of seizures (Jung et al., 2004, Jessberger et al.,
2007, Raedt et al., 2007). However, other studies suggest that only a subset of newborn cells are
affected (Jessberger et al., 2007, Walter et al., 2007) and that newly generated neurons may in
fact protect the injured hippocampus from excessive excitability. These studies suggested that
newborn cells receive less excitatory input than age-matched cells from control, non-epileptic
animals (Jakubs et al., 2006, Murphy et al., 2011). Although it remains unclear, and
controversial, how the neurogenic response to seizures may impact epileptogenesis, it is an
important feature of epileptic pathology that is consistently observed and continues to be the
focus of active research.
Adult neurogenesis is also sensitive to pharmacological interventions, as studies have shown
that SSRIs, particularly fluoxetine but also citalopram and escitalopram, increase hippocampal
neurogenesis in both humans (Boldrini et al., 2009) and animals (Malberg et al., 2000). In many
studies neurogenesis was only observed after more than 14 days of SSRI treatment (Malberg et
al., 2000, Kodama et al., 2005, Huang and Herbert, 2006, Marcussen et al., 2008), although a
delayed time of action for SSRIs to increase neurogenesis is not observed in all studies
(Himmerich et al., 2007, David et al., 2009, Hanson et al., 2011). In the study by Santarelli et al.
(2003), it was found that neurogenesis was critical in mediating the positive behavioural
response to antidepressants in animal models, however, this is still subject to debate (Miller et
al., 2008, Zhao et al., 2008, Bessa et al., 2009). While neurogenesis may be a potential critical
component required for the response to SSRIs, the mechanisms by which this occurs are not
entirely clear. Studies have indicated that the enhancement of dentate gyrus neurogenesis
following chronic antidepressant treatment may be mediated by increased serotonin and
noradrenaline concentrations (Duman et al., 1997, Carlezon et al., 2005), an increased cAMP-
CREB cascade (Nestler et al., 1989, Nibuya et al., 1996, Thome et al., 2000) or increased
neurotrophic factors (Fabel et al., 2003, Khawaja et al., 2004).
There have been no studies specifically investigating the effect of long-term antidepressant
treatment on neurogenesis in epilepsy. Nevertheless, a few studies have reported findings that
may shed some light on this issue. For example, fluoxetine was able to reverse the maturation of
a significant proportion of matured dentate granule cells into cells with immature neuronal
properties (Kobayashi et al., 2010, Karpova et al., 2011). This may result in reduced synaptic
connectivity with these immature cells and their subsequent projections into CA3, which may be
relevant to the spread of seizure activity in the hippocampus. Other studies have investigated
neuroplastic alterations following chemoconvulsant-induced status epilepticus during chronic

36
treatment with citalopram. It was found that citalopram treatment reduced seizure-induced
levels of reactive gliosis and aberrant cell proliferation (Jaako et al., 2009) as well as cell death
and axonal sprouting (Jaako et al., 2011). This suggests that the pathological hippocampal
circuit remodelling occurring in response to seizures can be alleviated by chronic SSRI
treatment, which could potentially prevent the onset of epilepsy following an epileptic insult.
The importance of adult neurogenesis has also been debated. Although there is a high rate of
neurogenesis occurring in the adult hippocampus, of approximately 9000 cells per day in a
young adult rat, this is only 0.5% of the total mature granule cell population (Cameron and
McKay, 2001). Therefore, the relative importance of these cells is questionable. However, these
newborn cells possess greater levels of structural plasticity, especially in their immature stage
(Hastings and Gould, 1999, Zhao et al., 2006, Toni et al., 2007), suggesting that they may be able
to contribute to functionality more so than mature cells. As well has having greater plasticity,
these cells also exhibit enhanced excitability and reduced threshold for long-term potentiation
compared to mature granule cells (Snyder et al., 2001, Schmidt-Hieber et al., 2004, Saxe et al.,
2006, Ge et al., 2007). Furthermore, with a greater level of structural plasticity comes the
possibility of more robust changes in response to external influences, such as antidepressant
treatment or brain insults such as epilepsy. However, no studies to date have addressed
whether antidepressant treatment in epilepsy increases the production of normal or abnormal
granule cells, and whether these alterations in neurogenesis contribute to epileptogenesis
during antidepressant treatment. While increases in neurogenesis following antidepressant
treatment appear to be beneficial, whether such increases in the epileptic brain are also
beneficial is unknown. Addressing the alterations in neurogenesis in epilepsy and following
antidepressant treatment may provide insight into how antidepressants affect the epileptic
brain.
1.5.3 Other intersecting intermediaries
Alterations in the HPA axis and neuroplasticity, especially neurogenesis, are the focus in this
thesis and are investigated as possible substrates by which antidepressants may affect
epileptogenesis. However, there are other common substrates between antidepressant action
and epileptogenesis that should also be highlighted such as alterations in monoamines, brain-
derived neurotrophic factor (BDNF) and inflammation.

37
1.5.3.1 Monoamines
A broad literature describes monoamine abnormalities in both epilepsy and depression. With
many of the newer-generation antidepressants targeting monoaminergic neurotransmission,
this may be a possible candidate of interaction between epilepsy and antidepressant treatment.
Studies have shown that serotonergic and noradrenergic alterations contribute to
epileptogenesis. Wada et al. (1997) investigated kindling epileptogenesis following
administration of serotonin receptor agonists and found that a 5HT1A receptor agonist, 8-
hydroxy-2-(di-n-propylamino)tetralin (8-OH-DPAT) inhibited kindling rate while a 5HT2
receptor agonist, 2,5-dimethoxy-4-iodoamphetamine (DOI), facilitated kindling. The effect of
DOI was blocked by the addition of a 5HT2 receptor antagonist, ketanserin. This suggests a
pivotal role of serotonergic neurotransmission in epileptogenesis. The importance of serotonin
in epilepsy has also been highlighted in the Genetically Epilepsy Prone Rats (GEPR), a model of
generalised seizures, in which widespread reductions in serotonin concentrations and uptake,
as well as alterations in serotonergic receptor distributions have been reported (Statnick et al.,
1996a). Other studies have also highlighted the involvement of serotonergic neurotransmission
in modulating seizure susceptibility (Kilian and Frey, 1973, Buterbaugh, 1978, Hiramatsu et al.,
1987, Dailey et al., 1992, Przegalinski et al., 1994, Gerber et al., 1998, Filakovszky et al., 2001)
while other studies have investigated the effects of fluoxetine on the serotonergic system and its
relationship to seizures. In GEPRs fluoxetine reduced the severity of sound-induced convulsions,
the time course of which was paralleled by a fluoxetine-induced rise in serotonin levels (Dailey
et al., 1992). Additionally, in the pilocarpine model of status epilepticus, chronic fluoxetine
restored epilepsy-induced serotonergic deficits (Mazarati et al., 2008).
Noradrenergic alterations have also been shown to contribute to seizure susceptibility and
epileptogenesis. In animals, lesioning noradrenergic inputs from the locus coeruleus accelerated
amygdala kindling, implying an antiepileptogenic role for noradrenaline. Further, the GEPR-9
rats display seizure susceptibility concurrent with noradrenergic deficits (Jobe et al., 1995,
Statnick et al., 1996b) while reversing these neurotransmitter deficits reduces the seizure
susceptibility (Yan et al., 1993). Together, these studies suggest a role of monoamines in
mediating the effects of antidepressants on epileptogenesis.
1.5.3.2 Brain-derived neurotrophic factor
BDNF is another factor that has been shown to be involved in epilepsy and in the actions of
antidepressants. Studies have shown that BDNF is elevated in hippocampal tissue obtained from
patients with epilepsy (Mathern et al., 1997, Takahashi et al., 1999, Murray et al., 2000), and its

38
synthesis is increased in response to seizures (Ernfors et al., 1991, Rudge et al., 1998). However,
both proepileptogenic (Croll et al., 1999, Asztely et al., 2000, Zhu and Roper, 2001, Scharfman et
al., 2002, Koyama et al., 2004) and antiepileptogenic effects (Larmet et al., 1995, Osehobo et al.,
1999, Reibel et al., 2000a, Reibel et al., 2000b) of BDNF have been reported. As BDNF is a
potentially an essential component to epileptogenesis, alterations in this factor may affect
epilepsy.
SSRIs have also been shown to affect BDNF, with studies showing that BDNF and its receptor
TrkB are required for the action of antidepressants (D'Sa and Duman, 2002, Nestler et al., 2002,
Popoli et al., 2002). Clinical studies report increases in serum BDNF following antidepressant
treatment in depressed patients, correlating with improvements in mood (Karege et al., 2002,
Aydemir et al., 2005, Gervasoni et al., 2005, Gonul et al., 2005, Aydemir et al., 2006). Animal
studies have also found increases in BDNF mRNA (Nibuya et al., 1995, Russo-Neustadt et al.,
2000, Dias et al., 2003) and protein (Chen et al., 2001, Altar et al., 2003, Xu et al., 2003), as well
as TrkB expression (Nibuya et al., 1995) and activation (Saarelainen et al., 2003) in the
hippocampus and prefrontal cortex following antidepressant treatment, although other studies
have failed to demonstrate such elevations (Miro et al., 2002, Coppell et al., 2003, Altieri et al.,
2004). While there is a clear overlap of BDNF alterations following antidepressant treatment
and in epileptogenesis, no studies to date have investigated the effects of antidepressants on
BDNF during epileptogenesis.
1.5.3.3 Inflammation
Inflammation is another process implicated in epilepsy (Vezzani et al., 2012), depression (Miller
et al., 2009) and in antidepressant action (Janssen et al., 2010). In epilepsy, increases in
inflammatory mediators have been identified in resected brain tissue of TLE patients (Crespel et
al., 2002, Zattoni et al., 2011, Hirvonen et al., 2012), and in experimental seizure models
(Minami et al., 1991, Vezzani et al., 2000, Turrin and Rivest, 2004, Gorter et al., 2006, Aronica et
al., 2007). In depression, increases in inflammatory mediators (Maes et al., 1995, Lanquillon et
al., 2000, Mikova et al., 2001, Owen et al., 2001, Tiemeier et al., 2003, Alesci et al., 2005, Brietzke
et al., 2009) have also been shown to be alleviated by antidepressant treatment (Kenis and
Maes, 2002, Sutcigil et al., 2007), although other studies have failed to replicate this (Hannestad
et al., 2011). Furthermore, pro-inflammatory effects of antidepressants have also been
demonstrated (Piletz et al., 2009, Fornaro et al., 2011). As such, antidepressants may play an
anti-inflammatory role in epilepsy, but whether this is the case and if it is beneficial or
detrimental has not been thoroughly investigated.

39
Two studies have investigated the interaction of fluoxetine with IL-1β in the pilocarpine-
induced post-status epilepticus model. In these studies, rats rendered epileptic displayed
depressive-like behaviours, dysregulation of the HPA axis and abnormal serotonin signalling. It
was found that intrahippocampal infusion of an IL-1 receptor antagonist (IL-1ra) ameliorated
depressive-like behaviours, with no effect on spontaneous seizures (Mazarati et al., 2010). In a
follow up study, combined treatment of fluoxetine and IL-1ra improved the depressive-like
behaviours and abnormalities in serotonin signalling, but only partially alleviated HPA axis
hyper-reactivity while fluoxetine treatment in isolation had no effect (Pineda et al., 2012). These
findings indicate that co-treatment of antidepressants with anti-inflammatory agents could
reverse some of the pathological processes associated with epilepsy development.

40
1.6. Study rationale
As psychiatric disorders contribute to a major burden of disease in epilepsy, management of
these disorders is of great importance. As such, many patients with epilepsy may be prescribed
antidepressants at various stages of their disorder to treat comorbid psychiatric issues. This is
an important area to investigate, as treatment with antidepressants may impact the progression
of the epileptic disorder. To date, much of the literature has focused on the effects of
antidepressants on short-term seizure endpoints, with most studies only investigating seizures
(the occurrence, frequency or threshold to a seizure) rather than epilepsy (alterations in the
pathological and biochemical changes that lead to seizures and the occurrence of spontaneous
seizures). When taken together, these studies largely suggest that antidepressants are beneficial
in epilepsy and are therefore considered safe for use in patients with epilepsy. However, many
studies only administer antidepressants acutely or for short periods of time, which does not
follow the clinical situation in which antidepressants are administered for several weeks to
ensure a beneficial therapeutic effect. As such, there is no indication of the effect that chronic
antidepressant treatment has on epileptogenesis and the associated neurobiological changes
that continue even after seizures emerge. Investigation of long-term endpoints is needed to
determine the long-term effects of antidepressant treatment in epilepsy. This includes
investigation of seizure frequency over time and changes in the severity of the disorder during
antidepressant treatment, as well as exploring the effects on common substrates. As highlighted,
undertaking such studies in patient populations is technically and logistically difficult and time
consuming. Investigating this in animal models provides a much more straightforward and
achievable approach. If animal studies were to show effects of antidepressants on
epileptogenesis, this would then be a strong argument for conducting human studies to further
investigate this phenomenon.

41
1.7. Hypotheses and aims
The main research questions of this thesis are:
1. Does chronic treatment with the SSRIs fluoxetine and citalopram have effects on
amygdala kindling epileptogenesis?
2. Does chronic treatment with fluoxetine and citalopram influence the severity of anxiety-
and depressive-like behaviours, suppress the corticosterone response to stress and
reduce aberrant neurogenesis associated with the kindling model of epileptogenesis?
Study 1 – Pilot study: tolerability of osmotic pumps and fluoxetine and the effects on behaviour,
stress-induced corticosterone and neurogenesis (Chapter 3).
Prior to investigating the effects on epileptogenesis, the chosen method of drug delivery – SSRIs
delivered by osmotic pumps – was piloted. Additionally, the behavioural, neuroendocrine and
neuroplastic effects of fluoxetine were investigated.
Hypotheses:
1. Fluoxetine delivered by osmotic pumps will be an effective method of drug delivery to
rodents.
2. Chronic treatment with fluoxetine will affect behaviour, suppress the corticosterone
response to stress and increase hippocampal neurogenesis.
Aims:
1. To validate the use of osmotic pumps as an effective method to chronic deliver fluoxetine
treatment in rats.
2. To investigate the changes in depressive- and anxiety-like behaviours, stress-induced
corticosterone levels and hippocampal neurogenesis during treatment with fluoxetine.

42
Study 2 – The effects of chronic fluoxetine and citalopram treatment on kindling epileptogenesis
and related behavioural and physiological alterations (Chapter 4).
Hypotheses:
1. Compared to vehicle-treated rats, fluoxetine- and citalopram-treated rats would have a
slower rate of kindling epileptogenesis.
2. Kindling would increase anxiety and depressive-like behaviours and this would be
alleviated with fluoxetine and citalopram treatments.
3. Kindling would increase the corticosterone response to stress, and this would be
alleviated with fluoxetine and citalopram treatments.
Aims:
1. To investigate the effects of fluoxetine or citalopram on epileptogenesis using the
amygdala kindling model.
2. To investigate the kindling-induced changes in depressive- and anxiety-like behaviours,
stress-induced corticosterone levels and hippocampal neurogenesis and how these are
affected by fluoxetine and citalopram treatment.

43
CHAPTER 2: METHODS
2.1. Overall study design
This study was designed to evaluate effects of chronic antidepressant treatment on the
progression of epileptogenesis. A general timeline of experimental procedures is shown in
Figure 2.1. To ensure continuous delivery of drugs throughout the experiment, all rats were
implanted with osmotic pumps (each of four week delivery duration) at the beginning of the
experiment (week 0) and at week 4. A pilot study (Chapter 3) was designed to validate the
osmotic pumps as a method of drug delivery and to determine the tolerability of the osmotic
pumps for the experimental time period. Therefore, this cohort of rats went through identical
experimental procedures as set out in the timeline (Figure 2.1) excluding electrode
implantations, seizure threshold testing and kindling stimulations. Chapter 4 describes the
kindling studies that followed the timeline in Figure 2.1, with rats being treated with either
fluoxetine or citalopram.
Figure 2.1: Timeline of experimental procedures
2.2. Experimental animals
9-11 week old male Wistar rats were used (n=10 for pilot study, n=63 for fluoxetine kindling
study, n=69 for citalopram kindling study). Rats were bred and housed in the Department of

44
Medicine Biological Research Facility and maintained on a 12 hour light/dark cycle (lights on at
6am) at 19-24°C with ad libitum access to food and water.
Table 2.1: Total sample size included in each experiment. Reasons for exclusion: incorrect electrode
placement, non-detectable levels of drug in plasma, culled due to loss of electrode cap, culled due to
infection from osmotic pump, died during/post electrode surgery, kindling procedure incomplete (did not
receive thirty stimulations).
2.3. Live animal experimental procedures
All procedures were performed according to the guidelines set by the Australian Code of
Practice for the Care and Use of Animals for Scientific Purposes (2004) and approved by the
University of Melbourne Animal Ethics Committee (ethics number: 0911543).
2.3.1 Osmotic pump preparation
Osmotic pumps allow for the continuous systemic infusion of solutions into unrestrained rats.
The internal structure of a pump is displayed in Figure 2.2. To prepare the pumps, the drug is
injected into the impermeable reservoir. Upon implantation, water from the surrounding
environment permeates the semi-permeable membrane and expands the osmotic layer (which
contains a high osmolality solution, causing water to flux in). This displaces the solution from
the impermeable reservoir at a predetermined rate via the flow moderator, which controls the
release of the agent over the required time period (Theeuwes, 1975). This process begins

45
approximately 6 hours after implantation and continues uninterrupted until removal. The drug
is then taken up systemically, according to the site of implantation.
Figure 2.2: Internal structure of the osmotic pumps. Image taken from http://www.alzet.com/products/ALZET_Pumps/howdoesitwork.html
The pumps used in this study lasted between 32 and 35 days depending on the lot number
(2ML4, Alzet Osmotic Minipump, Alza, USA). Weight gain over the experimental period was
taken into account when calculating drug concentrations. The concentration of fluoxetine
hydrochloride (Aurobindo Pharma, India) or citalopram hydrobromide (Biotrend Chemicals,
Switzerland) to fill the pump was based on the predicted weight of the rat four weeks from date
of implantation to ensure the target dose of 10mg/kg was delivered by the end of the
experimental period. The average weight gain was predicted from a large inventory of data on
weight gain of rats over time from several different experiments within the laboratory. The dose
of 10mg/kg was chosen for both drugs on the basis of previous studies that have administered
fluoxetine and citalopram to rats using pumps with therapeutic plasma levels being detected in
rats that had received this dose (Czachura and Rasmussen, 2000, Castro et al., 2003, Hasegawa
et al., 2005, Hesketh et al., 2005, Hesketh et al., 2008, Riad et al., 2008).
To prepare the solutions of fluoxetine and citalopram, the appropriate concentration was
dissolved in a sterile 50% dimethyl sulfoxide (DMSO) and distilled water vehicle. In order to
ensure pumps were correctly filled, the pump with its flow moderator was weighed, the
solution injected into the pump under sterile conditions (performed in a laminar flow hood),
and the filled pump was weighed again. If the filled weight was >90% of the mean fill volume
(as specified from manufacturer’s instructions) the pump was considered filled and ready for
implantation.

46
2.3.2 Osmotic pump implantation
Pumps were prepared prior to commencement of surgical procedures. All rats were implanted
with osmotic pumps with the appropriate drug or vehicle. Rats were anaesthetised by
inhalation of isofluorane (5% induction, 1.5-2.5% maintenance, in 1:1 medical air:oxygen).
Heart rate and blood oxygenation were monitored with a pulse oximeter throughout surgery.
Once eye blink and footpad reflexes were absent, the site of incision was shaved and swabbed
with a topical antiseptic (10% w/v povidone-iodine, Betadine) from the nape of the neck to 1cm
below the shoulder blades along the midline and 2cm lateral to the spine on either side (Figure
2.3A).
The sequence of images in Figure 2.3 shows where the pump was implanted. A 2cm incision was
made at the shoulder blades through the skin, avoiding the underlying muscle and connective
tissue. Using haemostats, an area of subcutaneous tissue, 1cm lateral and parallel to the spine
was made above the left flank, 6-7cm long for the pump to fit lengthways. This is an area slightly
larger than the pump to allow the pump to move freely and not be of discomfort to the rat. The
pump was then inserted (Figure 2.3B), with the flow moderator end pointed caudally, until it
was at least 1cm away from the incision site (Figure 2.3C). This ensures the pump does not
adhere to tissue under the incision site, break the skin and become exposed. The incision was
closed with buried sutures using absorbable suture (Biovek 4/0, Dynek, Australia).
Rats in the kindling studies of Chapter 4 then underwent electrode implantation (Section 2.3.3).
For rats in the pilot study (Chapter 3), the incision area was swabbed with topical antiseptic and
the rat injected with 1ml saline (0.9%, subcutaneously) and an analgesic, 4mg/kg carprofen
(intraperitoneally, Rimadyl, Pfizer, Australia). Rats were recovered on a heat mat and weight
was monitored for 5 days or until pre-surgery weight was obtained. At the end of the
experimental period, pumps were removed and the remaining volume aspirated to ensure that
over 85% of the solution had been released as per manufacturer’s instructions.
Rats were also monitored regularly (at least once per week) for any signs of inflammation or
infection around the pump. A common post-surgery occurrence was seromas, an accumulation
of clear, sterile fluid around area of tissue trauma. This was observed in 15/73 rats, but drained
away naturally over time. Two rats were culled due to signs of infection, as the seroma did not
reduce. Other issues that arose were scratching around the site of incision. This was due to
irritation caused by the sutures and was treated with a fusidic acid/betamethasone gel
(5mg/1mg, Fuciderm, Lyppard, Australia). These mild reactions to the pumps did not have an
effect on the well-being of the rats, and were therefore not considered as a confounder to the
results.

47
Figure 2.3: The sequence of procedures for osmotic pump implantation. (A) A 2cm incision is made and underlying tissue dissected from surrounding tissue. (B) The pump is initially inserted diagonally then parallel to the spine (C) to ensure it is comfortable for the rat. (D) At 4 weeks, the pump is removed and replaced with another on the opposite side, filled with the same solution as the first pump (E).
2.3.3 Bipolar and recording electrode implantation
Bipolar and recording electrode implantations were only performed in kindling cohorts
(Chapter 4). Following osmotic pump implantation, while still under anaesthesia, the head was
shaved and cleaned with topical antiseptic and the rat placed into a stereotactic frame (David
Kopf Instruments, USA). A single midline incision was made along the skin, from between the
eyes to between the ears to reveal the surface of the skull, with bleeding stopped using diluted
hydrogen peroxide. Using an engraving drill (300 series variable rotary tool, Dremel, Australia)
with a 0.9cm drill bit attached (Australian Jewellers Supplies, Australia), five holes were made
through the skull and just above the dura – three for extradural electroencephalogram (EEG)
recordings and two for anchoring side screws (1.4x3mm, Mr Specs, Australia) to hold the head
cap in place (Figure 2.4A). Extradural electrodes were custom-made by soldering a ‘male’ pin to
a screw.
Once the anchoring screws and extradural electrodes were attached, a hole was drilled for
stereotactic insertion of a bipolar stimulating electrode (MS303/1, Plastics One, USA) targeting
the lateral amygdaloid nuclei (Figure 2.4B). Co-ordinates were 3mm caudal and 5mm lateral
from bregma, and 6.5mm ventral from the dura (Paxinos and Watson, 1998). The lateral
position was adjusted to 5.25mm for the citalopram cohort after evaluation of electrode
placement in the fluoxetine cohort. This assembly was held in place by dental acrylic (Vertex
Dental, The Netherlands) then the skin sutured (Dysilk 3/0, Dynek, Australia) around the
headpiece and cleaned with topical antiseptic. Rats were then injected with 1ml saline (0.9%,
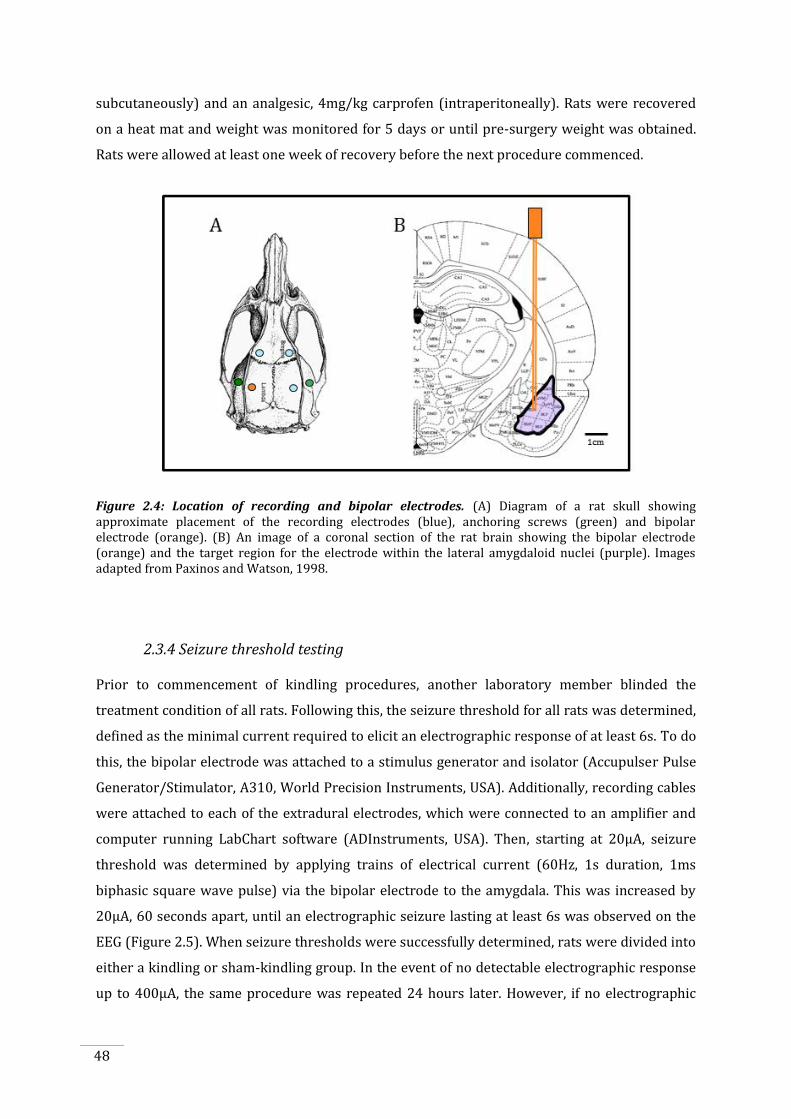
48
subcutaneously) and an analgesic, 4mg/kg carprofen (intraperitoneally). Rats were recovered
on a heat mat and weight was monitored for 5 days or until pre-surgery weight was obtained.
Rats were allowed at least one week of recovery before the next procedure commenced.
Figure 2.4: Location of recording and bipolar electrodes. (A) Diagram of a rat skull showing approximate placement of the recording electrodes (blue), anchoring screws (green) and bipolar electrode (orange). (B) An image of a coronal section of the rat brain showing the bipolar electrode (orange) and the target region for the electrode within the lateral amygdaloid nuclei (purple). Images adapted from Paxinos and Watson, 1998.
2.3.4 Seizure threshold testing
Prior to commencement of kindling procedures, another laboratory member blinded the
treatment condition of all rats. Following this, the seizure threshold for all rats was determined,
defined as the minimal current required to elicit an electrographic response of at least 6s. To do
this, the bipolar electrode was attached to a stimulus generator and isolator (Accupulser Pulse
Generator/Stimulator, A310, World Precision Instruments, USA). Additionally, recording cables
were attached to each of the extradural electrodes, which were connected to an amplifier and
computer running LabChart software (ADInstruments, USA). Then, starting at 20μA, seizure
threshold was determined by applying trains of electrical current (60Hz, 1s duration, 1ms
biphasic square wave pulse) via the bipolar electrode to the amygdala. This was increased by
20μA, 60 seconds apart, until an electrographic seizure lasting at least 6s was observed on the
EEG (Figure 2.5). When seizure thresholds were successfully determined, rats were divided into
either a kindling or sham-kindling group. In the event of no detectable electrographic response
up to 400μA, the same procedure was repeated 24 hours later. However, if no electrographic
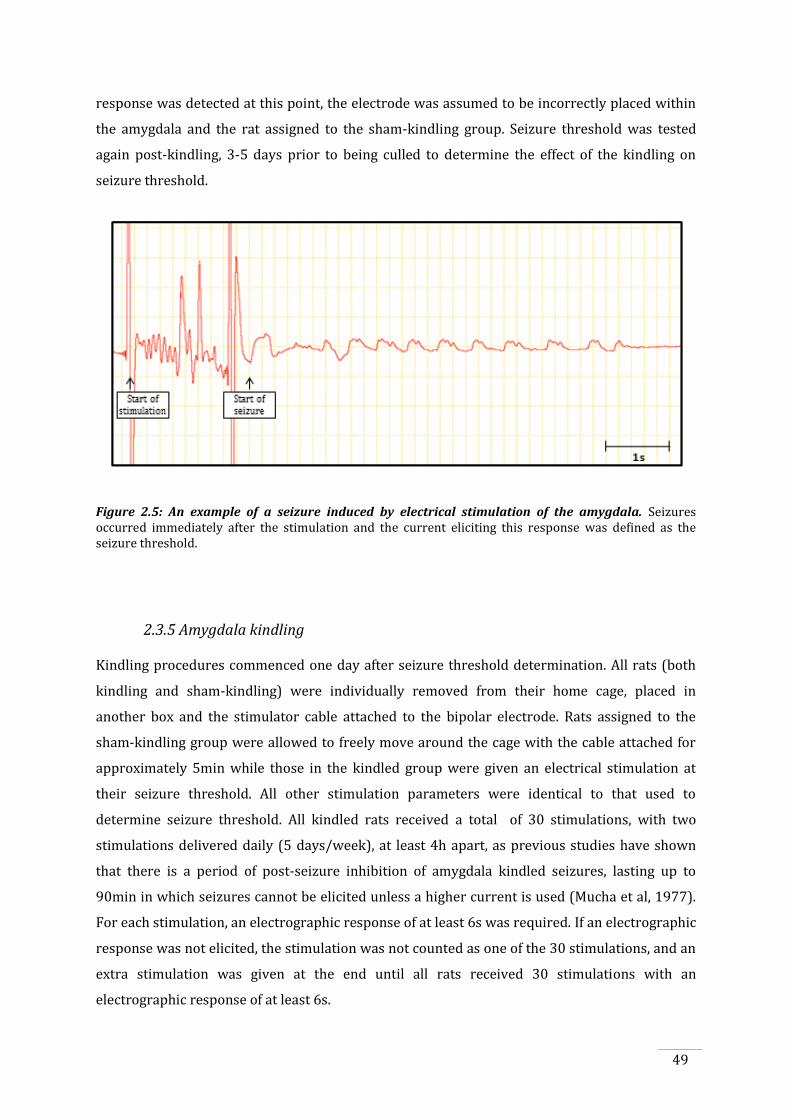
49
response was detected at this point, the electrode was assumed to be incorrectly placed within
the amygdala and the rat assigned to the sham-kindling group. Seizure threshold was tested
again post-kindling, 3-5 days prior to being culled to determine the effect of the kindling on
seizure threshold.
Figure 2.5: An example of a seizure induced by electrical stimulation of the amygdala. Seizures occurred immediately after the stimulation and the current eliciting this response was defined as the seizure threshold.
2.3.5 Amygdala kindling
Kindling procedures commenced one day after seizure threshold determination. All rats (both
kindling and sham-kindling) were individually removed from their home cage, placed in
another box and the stimulator cable attached to the bipolar electrode. Rats assigned to the
sham-kindling group were allowed to freely move around the cage with the cable attached for
approximately 5min while those in the kindled group were given an electrical stimulation at
their seizure threshold. All other stimulation parameters were identical to that used to
determine seizure threshold. All kindled rats received a total of 30 stimulations, with two
stimulations delivered daily (5 days/week), at least 4h apart, as previous studies have shown
that there is a period of post-seizure inhibition of amygdala kindled seizures, lasting up to
90min in which seizures cannot be elicited unless a higher current is used (Mucha et al, 1977).
For each stimulation, an electrographic response of at least 6s was required. If an electrographic
response was not elicited, the stimulation was not counted as one of the 30 stimulations, and an
extra stimulation was given at the end until all rats received 30 stimulations with an
electrographic response of at least 6s.

50
During kindling, behavioural seizures were graded by direct observation according to the
Racine (1972) scale: Class I – facial clonus, class II – head nodding, class III – unilateral forelimb
clonus, class IV – bilateral forelimb clonus and/or rearing, class V – rearing and falling. All
seizures were video recorded for blind observation by a second reviewer if required. Total
seizure duration was also recorded, defined as the period of the seizure from the end of the
stimulation to the end of the total electrographic response. If the seizure exceeded 300s, a total
seizure time of 300s was recorded.
2.3.6 Osmotic pump re-implantation
After the 29th stimulation, the osmotic pump was removed and replaced with another pump
containing the same solution (Figure 2.3D and E), to ensure continuous delivery for the
remainder of the experimental period. The reimplantation was performed after the 29th
stimulation to allow the rats to recover for 2 days, and then receive the 30th stimulation
followed immediately by BrdU injections (fluoxetine cohort only, Section 2.3.7). This was to
ensure post-surgical recovery did not interfere with BrdU uptake. Although citalopram-treated
rats did not receive BrdU injections, the same procedure for pump reimplantation was followed.
Briefly, under anaesthesia, the first pump was removed via an incision made at the caudal end
overlying the pump and closed with buried sutures. Then another incision was made 5mm
below the incision for the first pump, and the second pump was implanted over the right flank
following the same procedures as outlined in Section 2.3.2.
2.3.7 5-bromo-2-deoxyuridine injections
5-bromo-2-deoxyuridine (BrdU) is a chemical used to label dividing cells. It allows for post
mortem quantification of newborn cells that can be detected immunohistochemically (Section
2.4.4). The BrdU solution was prepared by dissolving in 0.9% saline at a concentration of
10mg/ml. This solution was covered with aluminium foil to avoid exposure to light, stored at
4°C and checked daily to ensure the solution had not precipitated. Beginning 5min after the 30th
stimulation, all rats received an injection of BrdU (100mg/kg, intraperitoneally) for 7 days, 22-
26h apart.
2.3.8 Elevated plus maze
The elevated plus maze was used to assess anxiety-like behaviour, taking advantage of two
opposing instincts in the rat: the fear of open spaces versus the desire to explore a new area.
The ability of this test to assess anxiety-like behaviours is based on studies by Handley and

51
Mithani (1984) that showed that anxiogenic agents decreased open arm entries while anxiolytic
agents increase entries. The maze consists of 1m x 1m plus-shaped platforms elevated 60cm off
the floor with two opposite arms being enclosed and the other two arms open (Figure 2.7A). On
the day of the test (performed between 10am and 12pm), the rat was brought into the testing
room and allowed to habituate for 30min. Lighting in the room was adjusted to 90-100 lux in
the open arms and centre area. Each rat was then placed individually into the centre of the maze
and allowed to explore the arena for 5min, with its movement tracked from above (Figure 2.7B)
by Ethovision tracking software (Noldus, The Netherlands). This software allows the objective
assessment of the number of entries into and time spent in the open arms. An avoidance of the
open arms is considered to be indicative of a heightened anxiety state (Rodgers and Dalvi,
1997).
Figure 2.6: The elevated plus maze. (A) Photograph of the elevated plus maze. (B) An image taken from Ethovision during an elevated plus maze acquisition period, with the red lines indicating the rat’s movement throughout the maze. Image A taken from http://www.mpipsykl.mpg.de/en/institute/services/emolab/index.html.
2.3.9 Forced swim test and corticosterone stress response
The forced swim test (FST) was used to assess depressive-like behaviour. The test consists of a
clear Perspex cylinder, diameter 30cm and height 40cm, filled with 25˚C water to a height of
30cm. The rat was brought into the testing room and allowed to habituate for 30min (test
performed between 10am and 12pm). The rat was then placed in the cylinder for 5min, during
which its movements were video-recorded from a horizontal angle for off-line analysis of
behaviours, examples of which are schematically shown in Figure 2.8. The time spent displaying
each of the behaviours was summed for the trial: (i) immobility (the primary outcome) –

52
defined as passive floating with only slight movements to keep its head above water with a lack
of movement of at least 3 paws, (ii) climbing – defined as vigorous active movements with all
four paws while parallel to the wall and its head and shoulders above the water, (iii) swimming
– all remaining time was considered swimming, with the rat making more vigorous movements
than necessary to keep its head above water and usually in a horizontal position and (iv) first
time immobile. At the end of the trial, rats were removed from the cylinder and towel-dried.
Behaviours that persisted for 2s or greater were counted, and each trial was blindly assessed
three times and the times averaged for each rat.
Figure 2.7: An example of the behaviours displayed in the forced swim test. Adapted from Cryan, 2005
According to established protocols (Porsolt et al., 1977), the FST is used to assess the efficacy of
antidepressants. In the traditional protocol, the test is conducted over two days, where on day 1
the rat is allowed to swim for 15min, a habituation trial, followed 24h later by administration of
the antidepressant and a 5min swim trial. It is during the 5min trial where the behaviours and
therefore efficacy of the antidepressant are assessed – the time immobile, indicative of
depressive-like behaviour, is compared between antidepressant and vehicle treatments.
However, as this two-day protocol is moderately stressful to rats, affecting outcomes of this
study such as neurogenesis, a one day, 5min trial was used for this study instead.
The corticosterone response to swim stress was measured by taking advantage of the mild
stress response induced by swimming in an inescapable tank for 5min (Duncan et al., 1998).
Prior to being placed into the FST, the rat was placed into a restraint tube with its tail
protruding at one end. A small incision was made at the end of the tail and a baseline blood
sample was collected (approximately 0.05ml into a heparinised syringe). This procedure took
between 30s and 1min. At the end of the 5min swimming trial, the rat was removed from the
FST, towel-dried for 1min and placed back into the restraint tube to take a “post-stress” sample

53
within 2min of completing the FST. All blood samples were transferred from the syringe into
individual 3ml Eppendorf tubes and stored on ice. Samples were then centrifuged at 4°C for
10min at 10,000rpm, after which plasma was collected, placed into a clean Eppendorf tube and
stored at -20°C for later analysis.
2.3.10 Corticosterone radioimmunoassay
Plasma samples obtained from the stress test were analysed for levels of corticosterone, both at
baseline and post-stress. This was performed with a rat specific corticosterone double antibody
125I radioimmunoassay kit (MP Biomedicals, Australia). Plasma samples and a set of samples
provided by the manufacturer to create a standard curve were prepared to manufacturer’s
instructions. Once complete, corticosterone levels were quantified using a Packard Cobra II
Auto-Gamma counter (Perkin Elmer, USA). Counts per minute were averaged for duplicate
samples, and using the values from the standard curve, corticosterone levels were calculated.
2.4. Post-mortem experimental procedures
2.4.1 Cardiac blood sampling, transcardial perfusion and tissue fixation
Prior to being culled, rats were given a 2000μA, 1s continuous electrical stimulation via the
bipolar electrode to induce the release of ferrous ions at the electrode tip. This allowed for
determination of electrode placement, as the ferrous ions will interact with the cyanide in the
fixative solution, resulting in a mark of ferrocyanide in the exact location of the electrode tip.
Immediately after, rats were given a lethal injection of phenobarbital (Lethabarb, 1ml/kg
intraperitoneally) and once eye-blink and footpad reflexes were absent, the sternum and rib
cage were cut open to expose the heart. For the determination of plasma fluoxetine and
citalopram levels, a 3-5ml blood sample was taken via the left ventricle into a heparinised
syringe (stored on ice, centrifuged at 4°C for 10min at 10,000rpm, plasma collected and stored
at -20°C for later analysis (Section 2.4.7). Immediately following this, rats were transcardially
perfused using a blunt 21-gauge needle placed into the aorta via the left ventricle using a
peristaltic pump (Cole Parmer Instruments, USA). This began with 200ml 0.1M phosphate-
buffered saline (37°C, pH 7.4) then 450ml 4% paraformaldehyde (PFA, dissolved in 0.1M PBS,
4°C, pH 7.4) containing 0.05% each of potassium ferrocyanide and potassium ferricyanide.
Brains were removed and post-fixed in 4% PFA overnight followed by 30% sucrose for 48h

54
(both at 4°C). Brains were wrapped in aluminium foil and frozen over dry ice for 10min and
stored at -80°C until cryosectioning.
2.4.2 Cryosectioning
Using a cryostat, 20μm thick coronal sections were cut. Sections of the dorsal and ventral
hippocampus were collected, beginning at bregma -1.60mm to -6.72mm ((Paxinos and Watson,
1998), Figure 2.9). In this hippocampal region, every alternating section was collected on
Superfrost Plus slides (Menzel-Glaser, Germany) culminating in 10 series, with 4 slides per
series and 3 sections per slide. In this arrangement, each slide held every 20th section. For
electrode placement and immunohistochemistry, the same series from each rat was allocated to
each staining procedure.
Due to the high rate of neurogenesis in the subventricular zone (Zheng et al., 2004), sections
from this region (between bregma +1.20mm and -0.40mm) were collected for positive controls
for BrdU immunohistochemistry. All slides were air-dried and stored at -80°C.
Figure 2.8: Examples of sections cut on the cryostat. (A) Rat brain atlas representation of the first hippocampal section collected and (B) the last hippocampal section collected. From Paxinos and Watson (1998).
2.4.3 Thionin staining and determination of electrode placement
In order to determine electrode placement, one series from each brain was stained for thionin.
Thionin staining allowed for visualisation of the ferrocyanide marking at the site of the bipolar
electrode tip. By staining the surrounding structures, this made them identifiable under the
microscope so that the exact location of the electrode could be determined.

55
Slides were removed from the -80°C freezer and allowed to thaw for 45min or until all
condensation had evaporated. Sections were then post-fixed in 10% potassium-buffered
formalin (10min), then taken through the following solutions: distilled water (3x5min), 0.03%
thionin working solution (5min), distilled water (2 dips in 3 changes of water), 96% ethanol (6
dips), 100% ethanol (2-4min, depending in darkness of stain), histolene (2x5min). Slides were
then coverslipped with DPX (dibutylphthalate polystyrene xylene, BDH Chemicals, United
Kingdom) and dried in a fumehood. Once dried, excess DPX was scraped off and slides cleaned
with ethanol.
Images were taken of one slide per rat, in which the blue dot at the electrode tip or electrode
track was visible. From these images, electrode placement was confirmed by comparing the
image of the section to the rat atlas (Paxinos and Watson, 1998), using landmarks such as the 3rd
ventricle, piriform cortex, rhinal fissure and shape of the section to mark the electrode’s
placement on the appropriate section. With treatment blinded, two observers decided on
inclusion according to set criteria (a standardised outline of the amygdala from sections in the
rat atlas in which the marking should be located). An example of the target region is shown in
Figure 2.4B. Incorrect electrode placement resulted in exclusion of that rat from all kindling,
behavioural, corticosterone and histological analyses.
2.4.4 BrdU immunohistochemistry
Sections were stained to detect any newborn cells that were labelled with BrdU (i.e., cells born
after kindling). For this protocol, one series was used for BrdU staining. In addition, a negative
control slide (3 sections, omission of primary antibody) and positive control (one slide, three
sections from the SVZ) were included in each batch.
All washes were performed in 0.1M Tris-buffered saline (TBS, pH 7.4, 4x5min each). Slides were
removed from the -80°C freezer and thawed for 45min. Once thawed, slides were placed in 10%
phosphate-buffered formalin (3min), washed and incubated in a 1% hydrogen peroxide/50%
methanol solution to remove endogenous peroxidases (30min). Slides were washed again and
incubated in 2M hydrochloric acid (37°C, 30min). This was performed to denature the DNA and
expose the sites for recognition by the primary antibody (Wojtowicz, 2006). Following
neutralisation in 0.1M sodium tetraborate (pH 8.5) and washing, slides were incubated for 2h in
10% normal goat serum to block non-specific binding sites of the secondary antibody. As
previous studies in the laboratory have shown, BrdU staining produces a lot of non-specific
background staining, making it difficult to distinguish BrdU-labelled cells. Therefore, an
additional blocking step was included using CAS-block (10min, Invitrogen, Australia), a
universal blocking agent for non-specific background staining. Slides were then incubated in the

56
primary antibody overnight (at least 16h, 4°C). This consisted of 2% NGS with 0.3% Triton X-
100 containing the monoclonal rat anti-BrdU antibody (Accurate, USA) at a 1:400 dilution.
The next day, slides were washed to remove unbound primary antibody and incubated in
secondary biotinylated antibody for 90min (rabbit anti-rat, 1:500, Vector Laboratories, USA).
Following another wash, sections were incubated in avidin-biotin peroxidase for 90min
(Vectastain ABC kit, Vector Laboratories, USA). After washing, staining was visualised by
developing the peroxidase with a diaminobenzidine (DAB) substrate, activated with 3μl
hydrogen peroxide. Each section was covered with DAB solution until a colorimetric reaction
occurred, after which DAB was washed off with TBS into bleach (to deactivate DAB solution).
Finally, sections were washed, counterstained in thionin (20-30s), dehydrated in ethanol (70%,
96% and 100%), cleared in histolene and coverslipped.
2.4.5 BrdU cell number quantification
Using the Cavalieri principles to estimate cell number, as described in Miki et al (2005), the
number of BrdU-positive cells were quantified in the left and right granule cell layer and
subgranular zone (defined as being within two cell bodies of the granule cell layer) of the
dentate gyrus and the hilus (ectopic cells). Inclusion criteria for positive cells were those with
diffuse, dark brown staining throughout the nucleus or punctuate staining (multiple distinct
spots of staining within the nucleus) and a rounded shape (Figure 2.10). Elongated, diffusely
dark brown stained endothelial-like cells, and small (<5μm diameter) BrdU-positive nuclei,
indicative of glial cells were not counted.
Per brain, every twentieth section throughout the left and right hippocampus, were counted on
an Olympus BX51 (Olympus, USA) microscope at x20 magnification, using StereoInvestigator
software (MBF Bioscience, USA). In both the validation study (Chapter 3) and fluoxetine and
kindling epileptogenesis study (Chapter 4), no significant difference in the total number of cells
between the left and right dentate gyrus or hilus were found, therefore cell counts were
combined from both sides to give a total dentate gyrus cell count and hilar cell count.

57
Figure 2.9: An example of BrdU stained cells. (A) The regions quantified for BrdU-positive cells, dentate gyrus outlined in green and the hilus located within (the end of the region marked by the dashed lines) and (B) examples of BrdU-positive cells (red circles).
2.4.7 Plasma sample analysis for fluoxetine levels
Plasma samples obtained at the end of the experiment were sent to Sydney Southwest
Pathology where levels of fluoxetine and its metabolite, norfluoxetine, were measured using gas
chromatography-mass spectroscopy with selected ion monitoring. The results of the analysis
were provided as levels of fluoxetine and norfluoxetine in each rat in ng/ml. Those with plasma
levels below the detectable limit were excluded from all analyses. For citalopram-treated rats,
plasma citalopram levels were not analysed. This was due to two reasons: (i) there was no
company within Australia that was able to perform this procedure for rodents and (ii) as only
3/63 rats in the fluoxetine-treated group had non-detectable plasma levels, the pumps were
considered to be an effective method to deliver the drugs, and it was reasonable to assume that
citalopram was also effectively delivered by the pumps to the large majority of rats.

58

59
CHAPTER 3: PILOT STUDY
Tolerability of osmotic pumps and fluoxetine and the effects on
behaviour, stress-induced corticosterone response and neurogenesis
3.1. Introduction
There have been a number of studies that have investigated the effects of SSRIs in animal
models of epilepsy (section 1.4.3.2); however in most studies SSRIs were only acutely
administered. This is a limitation in terms of clinical relevance, as patients are administered
SSRIs chronically rather than as a single dose, and several weeks of SSRI administration are
required before therapeutic benefits manifest (Goodnick and Goldstein, 1998, Rosebook and
Kalin, 2011). Throughout this period of delay, several biochemical and physiological changes
occur that contribute to the observed therapeutic effects of SSRIs. This includes receptor
adaptations such as desensitisation of the somatodendritic and terminal serotonin receptors
that normally act to regulate serotonin release (Stahl, 1998), which results in an increase in
serotonin (Fuller and Snoddy, 1990). While such effects of SSRIs occur soon after
administration, other downstream alterations have been shown to occur that may also explain
the delayed time for manifestation of therapeutic effects. This includes effects on intracellular
signalling pathways (Nestler et al., 1989, Perez et al., 1991, Tardito et al., 2006), gene expression
(Nibuya et al., 1996, Frechilla et al., 1998, Tardito et al., 2006), neurotrophic mechanisms
(Nibuya et al., 1995, Chen et al., 2001) and potentially effects on neuroplasticity (Nestler et al.,
1989, Duman et al., 1997, Fabel et al., 2003). In acute dosing paradigms, such effects would not
occur rendering these results less clinically relevant when investigating the effects of SSRIs on
epilepsy. Therefore, the effects of chronic SSRI treatment in animal models of epilepsy should be
investigated however there have been few studies that have done so.
In this thesis, the next chapter (Chapter 4) describes a study in which chronic SSRI treatment
was used in an animal model of epilepsy. However prior to this, the chosen method of chronic
drug delivery, by osmotic pumps, was piloted and is the focus of this chapter (Chapter 3).
Osmotic pumps allow for the continuous systemic infusion of solutions into unrestrained rats
and are advantageous for two main reasons:
1. The pumps are implanted at one time point, after which the drug is infused
continuously, eliminating the need for repeated daily injection and stressful handling
procedures. This is important for this study, as factors that are affected by stress such as

60
kindling epileptogenesis, behavioural outcomes, corticosterone levels and neurogenesis
are outcome measures.
2. Daily injections result in fluctuating drug plasma concentrations (Cremers et al., 2000).
Infusion with osmotic pumps maintains the drug at constant therapeutic levels. While
bolus injection does mimic human administration (i.e., one pill per day), the faster drug
metabolism (Martignoni et al., 2006) and shorter half-life of drugs (Lemberger, 1972,
Caccia et al., 1990) in rats compared to humans produces fluctuating levels rather than
the chronically high doses seen in patients.
There are also some disadvantages to using osmotic pumps. However the negative attributes
can be minimised:
Plasma levels of the drug can only be assessed at the end and not throughout the
experimental period unless a cannula is permanently implanted which would allow for
regular blood sampling to monitor drug levels. However, it is sufficient to inspect the
pump at the end of the experimental period to ensure it has emptied and therefore the
solution has been absorbed systemically, and cardiac blood taken at euthanasia is a
practical method for assessing the blood concentrations of the drug achieved.
Drug concentration needs to be calculated at implantation. Although it is possible to take
into account the rat’s weight to work out drug concentration (refer section 2.3.1), this is
only an average predicted weight over the chronic treatment period. Therefore,
individual rats may receive slightly higher or lower doses depending upon individual
weight gain during the study.
The SSRI chosen for this pilot study was fluoxetine. This drug has been widely used for research
in animal models of depression, and many of the studies investigating the effects of
antidepressants in animal models of seizures have also used fluoxetine (Dailey et al., 1992,
Prendiville and Gale, 1993, Raju et al., 1999, Ugale et al., 2004, Ferrero et al., 2005, Zienowicz et
al., 2005, Mazarati et al., 2008).
In addition to assessing the tolerability of the osmotic pumps for the experimental time period,
behavioural and physiological markers of the effects of fluoxetine were also assessed. As
outlined in Chapter 1, both SSRI treatment and epilepsy have similar neurobiological targets
and alterations (section 1.5). Chronic fluoxetine treatment has been shown to have effects on
behaviour (Santarelli et al., 2003, Cryan et al., 2005, Mazarati et al., 2008), HPA axis function
(Mazarati et al, 2009) and dentate gyrus neurogenesis (Malberg et al., 2000) all of which have
also shown to be altered during epileptogenesis (Parent et al., 1997, Zobel et al., 2004, Jones et
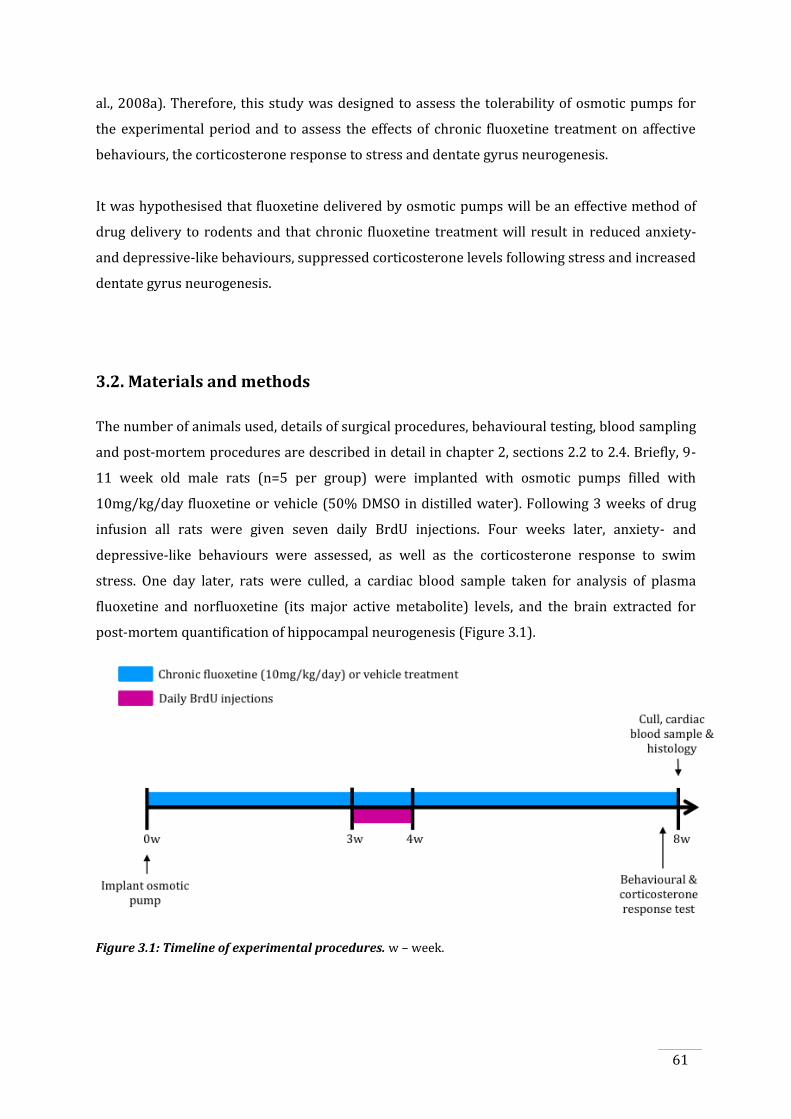
61
al., 2008a). Therefore, this study was designed to assess the tolerability of osmotic pumps for
the experimental period and to assess the effects of chronic fluoxetine treatment on affective
behaviours, the corticosterone response to stress and dentate gyrus neurogenesis.
It was hypothesised that fluoxetine delivered by osmotic pumps will be an effective method of
drug delivery to rodents and that chronic fluoxetine treatment will result in reduced anxiety-
and depressive-like behaviours, suppressed corticosterone levels following stress and increased
dentate gyrus neurogenesis.
3.2. Materials and methods
The number of animals used, details of surgical procedures, behavioural testing, blood sampling
and post-mortem procedures are described in detail in chapter 2, sections 2.2 to 2.4. Briefly, 9-
11 week old male rats (n=5 per group) were implanted with osmotic pumps filled with
10mg/kg/day fluoxetine or vehicle (50% DMSO in distilled water). Following 3 weeks of drug
infusion all rats were given seven daily BrdU injections. Four weeks later, anxiety- and
depressive-like behaviours were assessed, as well as the corticosterone response to swim
stress. One day later, rats were culled, a cardiac blood sample taken for analysis of plasma
fluoxetine and norfluoxetine (its major active metabolite) levels, and the brain extracted for
post-mortem quantification of hippocampal neurogenesis (Figure 3.1).
Figure 3.1: Timeline of experimental procedures. w – week.
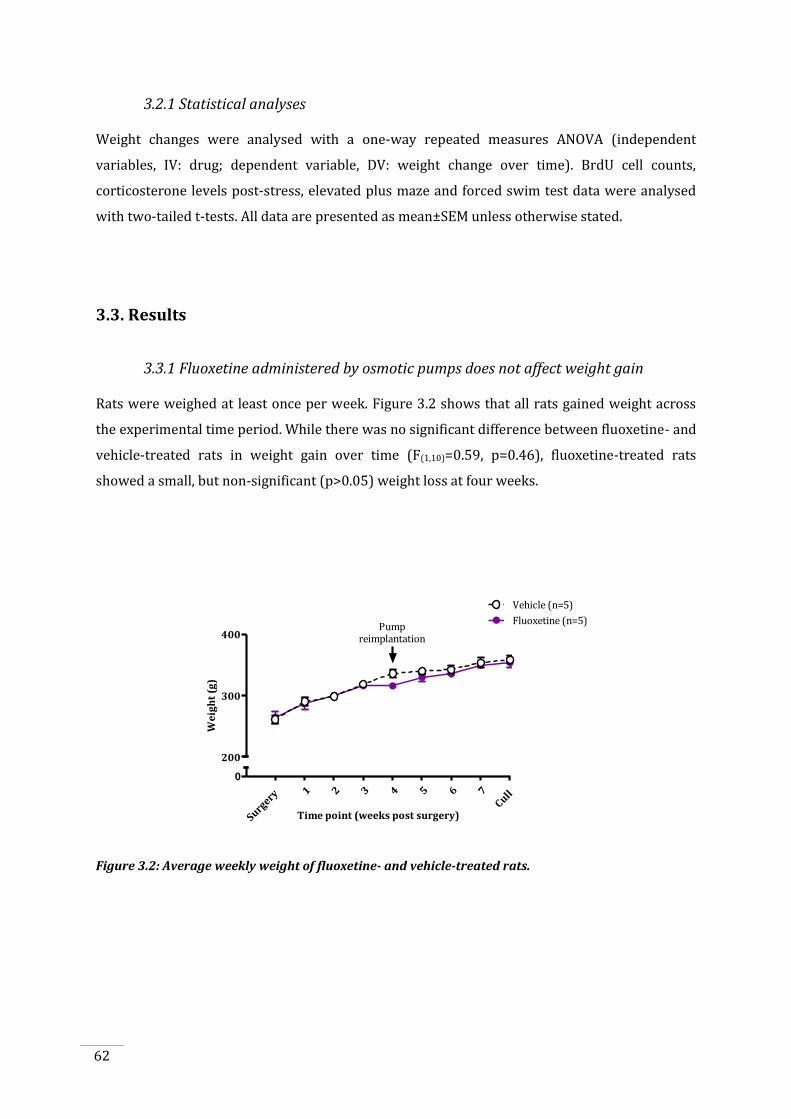
62
3.2.1 Statistical analyses
Weight changes were analysed with a one-way repeated measures ANOVA (independent
variables, IV: drug; dependent variable, DV: weight change over time). BrdU cell counts,
corticosterone levels post-stress, elevated plus maze and forced swim test data were analysed
with two-tailed t-tests. All data are presented as mean±SEM unless otherwise stated.
3.3. Results
3.3.1 Fluoxetine administered by osmotic pumps does not affect weight gain
Rats were weighed at least once per week. Figure 3.2 shows that all rats gained weight across
the experimental time period. While there was no significant difference between fluoxetine- and
vehicle-treated rats in weight gain over time (F(1,10)=0.59, p=0.46), fluoxetine-treated rats
showed a small, but non-significant (p>0.05) weight loss at four weeks.
0
200
300
400
Fluoxetine (n=5)
Vehicle (n=5)
Pumpreimplantation
Time point (weeks post surgery)
We
igh
t (g
)
Surgery
1 2 3 4 5 6 7Cull
Figure 3.2: Average weekly weight of fluoxetine- and vehicle-treated rats.
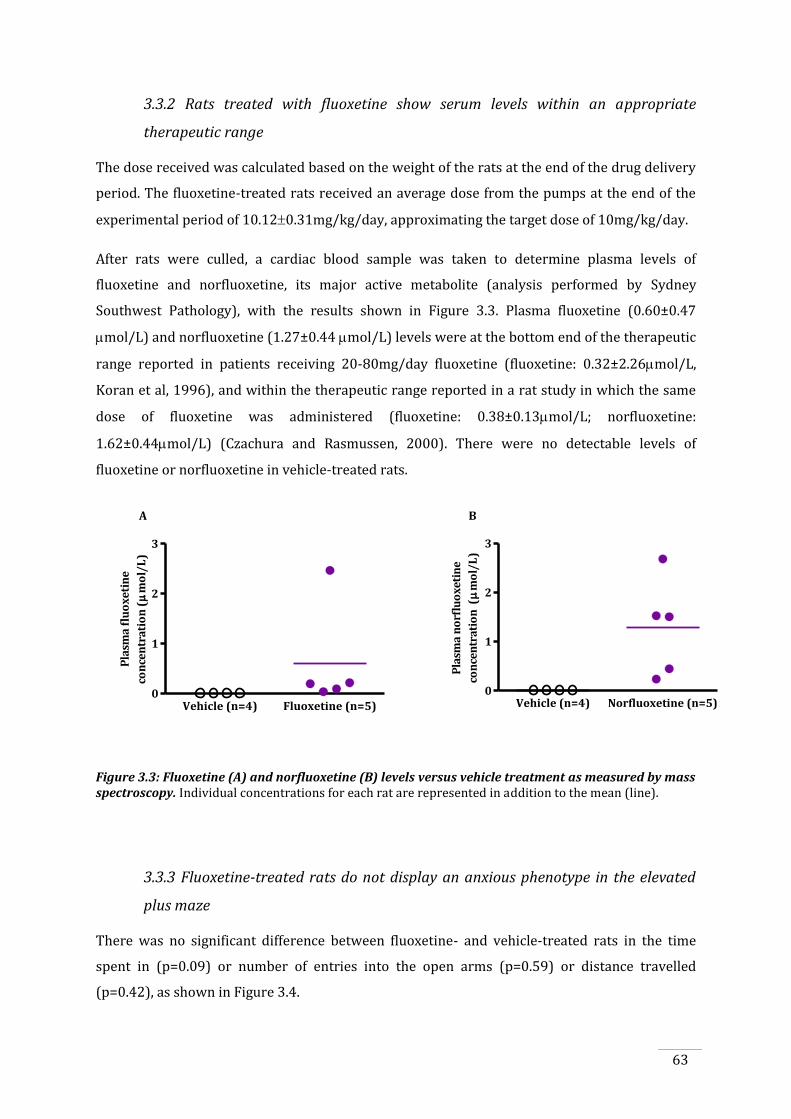
63
3.3.2 Rats treated with fluoxetine show serum levels within an appropriate
therapeutic range
The dose received was calculated based on the weight of the rats at the end of the drug delivery
period. The fluoxetine-treated rats received an average dose from the pumps at the end of the
experimental period of 10.120.31mg/kg/day, approximating the target dose of 10mg/kg/day.
After rats were culled, a cardiac blood sample was taken to determine plasma levels of
fluoxetine and norfluoxetine, its major active metabolite (analysis performed by Sydney
Southwest Pathology), with the results shown in Figure 3.3. Plasma fluoxetine (0.60±0.47
mol/L) and norfluoxetine (1.27±0.44 mol/L) levels were at the bottom end of the therapeutic
range reported in patients receiving 20-80mg/day fluoxetine (fluoxetine: 0.32±2.26mol/L,
Koran et al, 1996), and within the therapeutic range reported in a rat study in which the same
dose of fluoxetine was administered (fluoxetine: 0.38±0.13mol/L; norfluoxetine:
1.62±0.44mol/L) (Czachura and Rasmussen, 2000). There were no detectable levels of
fluoxetine or norfluoxetine in vehicle-treated rats.
Vehicle (n=4) Fluoxetine (n=5)0
1
2
3
Pla
sma
flu
ox
eti
ne
con
cen
tra
tio
n (
mo
l/L
)
Vehicle (n=4) Norfluoxetine (n=5)0
1
2
3
Pla
sma
no
rflu
ox
eti
ne
con
cen
tra
tio
n (
mo
l/L
)
A B
Figure 3.3: Fluoxetine (A) and norfluoxetine (B) levels versus vehicle treatment as measured by mass spectroscopy. Individual concentrations for each rat are represented in addition to the mean (line).
3.3.3 Fluoxetine-treated rats do not display an anxious phenotype in the elevated
plus maze
There was no significant difference between fluoxetine- and vehicle-treated rats in the time
spent in (p=0.09) or number of entries into the open arms (p=0.59) or distance travelled
(p=0.42), as shown in Figure 3.4.
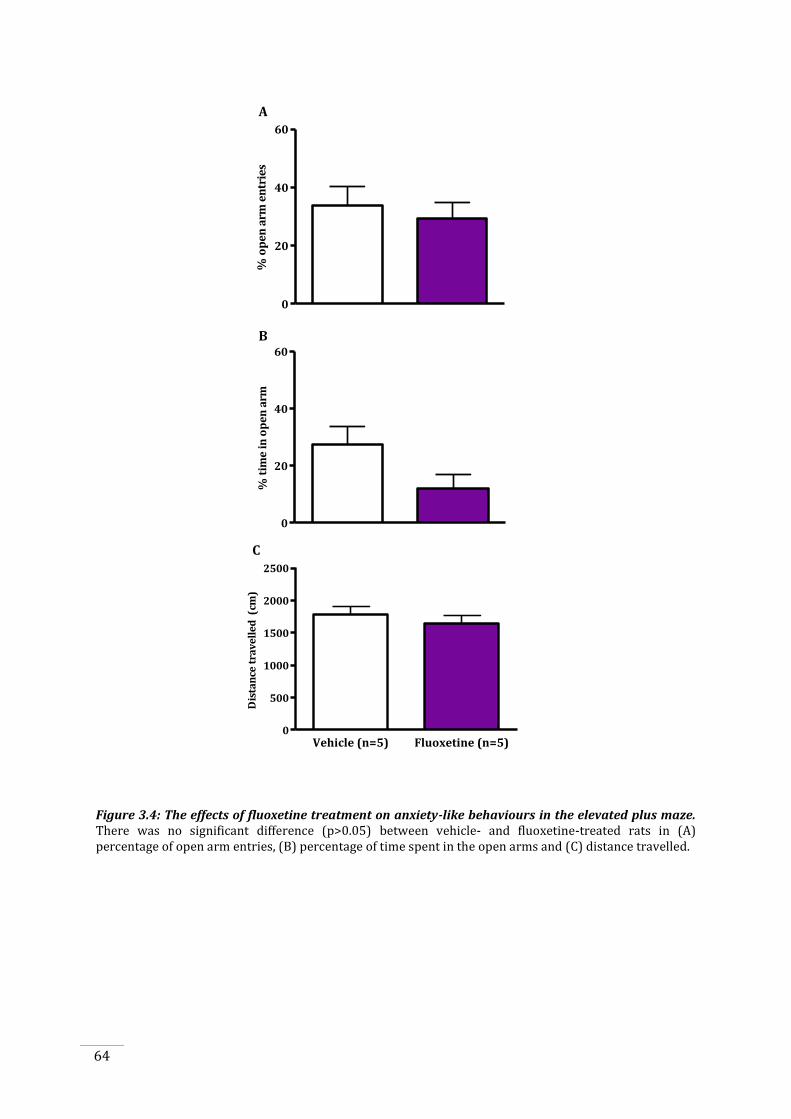
64
0
20
40
60
% o
pe
n a
rm e
ntr
ies
0
20
40
60
% t
ime
in
op
en
arm
Vehicle (n=5) Fluoxetine (n=5)0
500
1000
1500
2000
2500
Dis
tan
ce t
rav
ell
ed
(cm
)
A
B
C
Figure 3.4: The effects of fluoxetine treatment on anxiety-like behaviours in the elevated plus maze. There was no significant difference (p>0.05) between vehicle- and fluoxetine-treated rats in (A) percentage of open arm entries, (B) percentage of time spent in the open arms and (C) distance travelled.
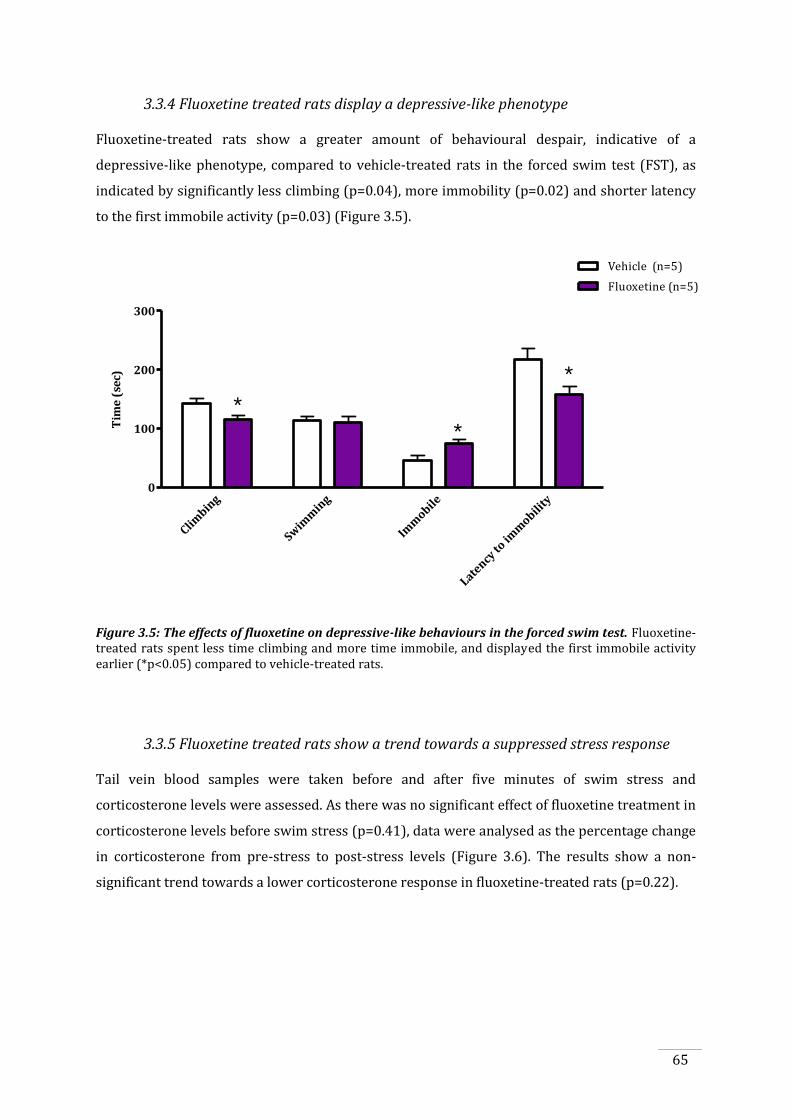
65
3.3.4 Fluoxetine treated rats display a depressive-like phenotype
Fluoxetine-treated rats show a greater amount of behavioural despair, indicative of a
depressive-like phenotype, compared to vehicle-treated rats in the forced swim test (FST), as
indicated by significantly less climbing (p=0.04), more immobility (p=0.02) and shorter latency
to the first immobile activity (p=0.03) (Figure 3.5).
Climbin
g
Swim
min
g
Imm
obile
Latency
to im
mobili
ty0
100
200
300
Vehicle (n=5)
Fluoxetine (n=5)
*
*
*
Tim
e (
sec)
Figure 3.5: The effects of fluoxetine on depressive-like behaviours in the forced swim test. Fluoxetine-treated rats spent less time climbing and more time immobile, and displayed the first immobile activity earlier (*p<0.05) compared to vehicle-treated rats.
3.3.5 Fluoxetine treated rats show a trend towards a suppressed stress response
Tail vein blood samples were taken before and after five minutes of swim stress and
corticosterone levels were assessed. As there was no significant effect of fluoxetine treatment in
corticosterone levels before swim stress (p=0.41), data were analysed as the percentage change
in corticosterone from pre-stress to post-stress levels (Figure 3.6). The results show a non-
significant trend towards a lower corticosterone response in fluoxetine-treated rats (p=0.22).
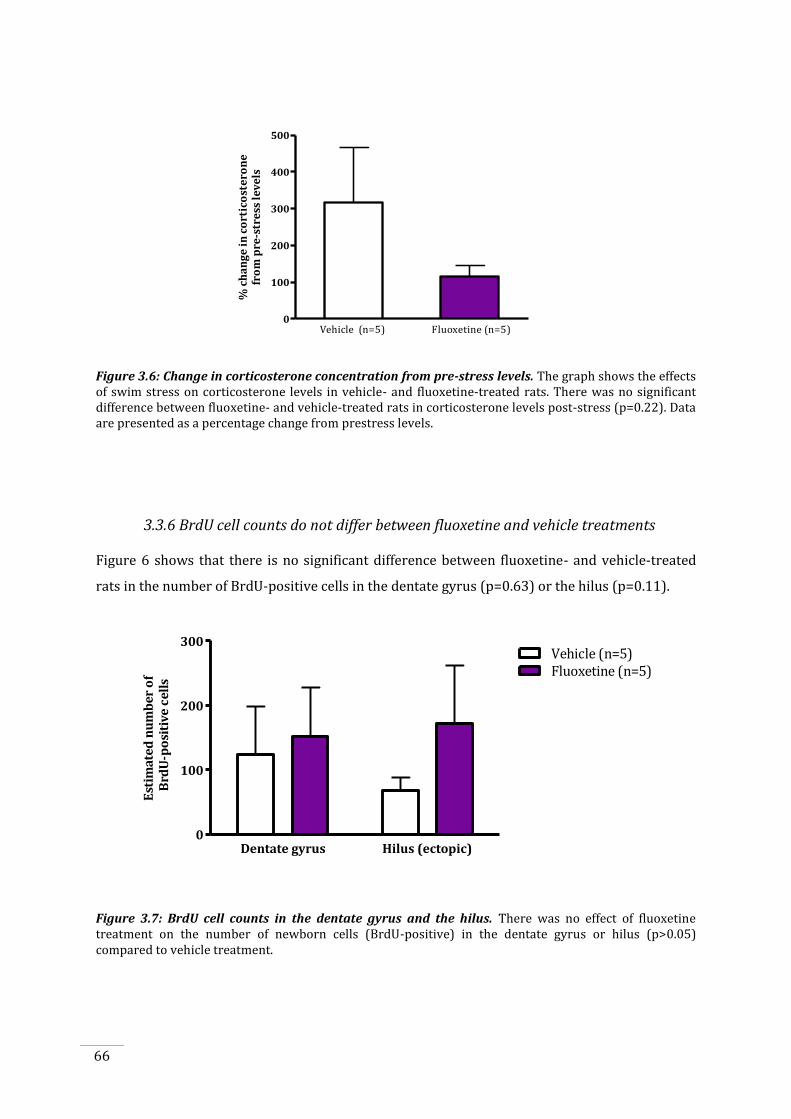
66
10
100
200
300
400
500
% c
ha
ng
e i
n c
ort
ico
ste
ron
efr
om
pre
-str
ess
le
ve
ls
Vehicle (n=5) Fluoxetine (n=5)
Figure 3.6: Change in corticosterone concentration from pre-stress levels. The graph shows the effects of swim stress on corticosterone levels in vehicle- and fluoxetine-treated rats. There was no significant difference between fluoxetine- and vehicle-treated rats in corticosterone levels post-stress (p=0.22). Data are presented as a percentage change from prestress levels.
3.3.6 BrdU cell counts do not differ between fluoxetine and vehicle treatments
Figure 6 shows that there is no significant difference between fluoxetine- and vehicle-treated
rats in the number of BrdU-positive cells in the dentate gyrus (p=0.63) or the hilus (p=0.11).
Est
ima
ted
nu
mb
er
of
Brd
U-p
osi
tiv
e c
ell
s
Dentate gyrus Hilus (ectopic)0
100
200
300
Fluoxetine (n=5)Vehicle (n=5)
Figure 3.7: BrdU cell counts in the dentate gyrus and the hilus. There was no effect of fluoxetine treatment on the number of newborn cells (BrdU-positive) in the dentate gyrus or hilus (p>0.05) compared to vehicle treatment.

67
3.4. Discussion
This chapter reports on the validation pilot study for the use of osmotic pumps as a method to
chronically deliver fluoxetine to rats. It was found that the osmotic pumps were well-tolerated
for the experimental time period and that this method of delivery was able to achieve
therapeutically relevant plasma concentrations. While fluoxetine treatment resulted in
behavioural alterations contrary to the hypothesis, the effects of fluoxetine on the
neuroendocrine system and on neurogenesis, albeit not statistically significant, trended to be
consistent with the prior hypotheses, with the inability to demonstrate a significant effect
potentially due to small sample sizes.
There was a wide range of plasma levels of fluoxetine found in this study, however these fell
within the therapeutic range reported in a previous study in which the same dose of fluoxetine
was administered, and therapeutically relevant plasma and cerebrospinal fluid fluoxetine and
norfluoxetine concentrations were detected (Czachura and Rasmussen, 2000) (fluoxetine:
0.38±0.13mol/L compared to 0.60±0.47mol/L in this thesis; norfluoxetine: 1.62±0.44mol/L
compared to 1.27±0.44 mol/L in this thesis). The variability range may be due to the small
sample size used, variability in individual drug clearance or variability in the assay detection.
However, there was no correlation found between plasma concentrations and any outcome
measure including behaviour, corticosterone levels following stress or number of newborn cells
(data not shown). This is also reported to be the case in humans, where one study has shown
clinical response to be independent of plasma antidepressant concentrations (Kelly et al., 1989).
Additionally, plasma fluoxetine concentrations were not related to health of the rats (assessed
by weight gain over time).
Assessment of anxiety- and depressive-like behaviours both showed unexpected outcomes. It
was shown that treatment with fluoxetine, while not significant, had a trend to increase anxiety
and also resulted in significantly more time spent immobile during the FST, indicative of
behavioural despair and a depressed-like state in fluoxetine-treated rats. While these results
may be due to the small sample size used in this study, other factors may have had an effect. One
point to consider is that normal rats were used in this study, rather than those already
exhibiting a depressed-like phenotype. Previous studies suggest that fluoxetine does not reduce
anxiety in non-anxious rats (Durand et al., 1999, File et al., 1999, Griebel et al., 1999, Silva and
Brandao, 2000) while other studies suggest that chronic fluoxetine treatment can increase
anxiety-like behaviour in mice (Oh et al., 2009, Kobayashi et al., 2011) and rats (Silva et al.,
1999) in the elevated plus maze. In terms of depression, studies have also shown that fluoxetine
does not act as a mood elevator or reduce depressive-like behaviours in otherwise normal rats,

68
as was the case in this study, or healthy patient controls (Gelfin et al., 1998, Geyer and Markou,
2002). Behavioural testing of subtle alterations in mood should be assessed by a battery of tests,
and not just one test for each endophenotype as was performed in this study (i.e., EPM only for
anxiety, FST only for depression). As behavioural changes were not the main outcome of this
study, only one test was used to assess each of the behaviours. As these are subtle alterations,
future studies should use a broader array of behavioural tests that will give a better indicator of
the effects of fluoxetine in this study.
Measurements of changes in corticosterone levels following a stressor are another way to
assess the biological effects of fluoxetine. In this study, it was found that following a brief
stressor, fluoxetine-treated rats showed a trend for suppressed levels of corticosterone
compared to vehicle-treated rats. This is consistent with data from previous studies in animals
(Pariante, 2006) and humans (Linkowski et al., 1987, Souetre et al., 1989, Steiger et al., 1989)
showing that fluoxetine normalises the HPA dysfunction together with recovery of depressive
symptoms. However, there a several reasons that may explain why there was no significant
difference in the studies reported in this thesis, such as the small sample size, the use of non-
depressed rats, which do not exhibit a hyper-reactive HPA axis response, and the mild intensity
of the stressor (swim stress). While swim stress is generally considered to be sufficiently
stressful, studies have previously found that corticosterone levels post-stress are increased to
300 to 400ng/ml (Duncan et al., 1998, Bouilleret et al., 2011) while the highest level of
corticosterone observed in this study was 288.58ng/ml (mean±SEM: 132.29±29.43ng/ml).
While this may be due to the protocol itself, in that swim stress was not sufficiently stressful, it
may also be due to the time point of analysis post-stress. Although previous studies have shown
that peak corticosterone concentrations are found 30 minutes after stress (Joels et al., 2007, de
Kloet et al., 2008), other studies contradict this, finding no change (Duncan et al., 1998, Mathews
et al., 2008, Norcross et al., 2008) or increases (Gozen et al., 2007, Droste et al., 2008, Steiner et
al., 2008) in corticosterone levels after swim stress during chronic fluoxetine treatment.
BrdU cell counts were also assessed post mortem to provide an indicator of the effects of
fluoxetine on hippocampal neurogenesis. The results suggest that there was a large variability
in the number of BrdU-positive cells, although there does appear to be a trend for fluoxetine-
treated rats to have more BrdU-positive cells in the hilus. High cell counts in the hilus indicate
that cells have ectopically migrated from the dentate gyrus. The reasons for this trend occurring
in this study are not clear, as in other studies cells migrate ectopically only following a brain
insult, for example following status epilepticus (Scharfman and Pedley, 2006, Jessberger et al.,
2007, Walter et al., 2007, Kron et al., 2010) or stroke (Niv et al., 2012). However this was not the
case here. Ultimately, the results suggest that there was no significant effect of fluoxetine on

69
dentate gyrus neurogenesis. While the small sample size may explain why there was no
difference, methodological limitations should also be considered such as inadequate detection
of BrdU-positive cells with the immunohistochemical methods used or inadequate uptake of
BrdU from the intraperitoneal injection. Overall, conclusions regarding the effects of fluoxetine
on newborn granule cell number cannot be drawn from this study.
Taken together, the results of this study show that osmotic pumps are a reliable method of drug
delivery and that rats are able to tolerate the pumps for the experimental time period and
receive the targeted dose of fluoxetine. Furthermore, although not significantly different, it is
likely fluoxetine is having a biological effect in the rats, as it was shown to have effects on
depressive-like behaviours and trends for effects on anxiety-like behaviours and suppression of
the corticosterone response to stress.
Following the validation of osmotic pumps as a reliable method of fluoxetine delivery and
indications of biological and behavioural effects, the next investigation will focus on the effects
of fluoxetine, and a more clinically relevant SSRI, citalopram, on epileptogenesis.

70

71
CHAPTER 4
Chronic SSRI treatment and its effects on kindling epileptogenesis,
behaviour, stress-induced corticosterone response and neurogenesis
4.1. Introduction
The high incidence of comorbid psychiatric illnesses, especially mood and anxiety disorders, in
patients with epilepsy (Gaitatzis et al., 2004) can have a significant impact on quality of life and
cognitive functioning as well as increasing the likelihood of suicide (Hesdorffer et al., 2006) and
possibly Sudden Unexpected Death in Epilepsy (SUDEP) (Ridsdale et al., 2011). In fact, the
manifestation of these illnesses in patients with epilepsy has been reported to be a greater
predictor of quality of life compared to illness duration or seizure frequency (Boylan et al., 2004,
Gilliam et al., 2004, Kanner et al., 2010). Therefore, management of these depressive and anxiety
symptoms and syndromes is an essential part of comprehensive treatment of people with
epilepsy.
Presently, the use of selective serotonin reuptake inhibitors (SSRIs) in patients with epilepsy is
considered to be acceptably safe (Jobe and Browning, 2005, Bagdy et al., 2007, Kanner, 2009,
Kondziella and Asztely, 2009), however antidepressants have not always been considered safe
for use in epilepsy. This is mainly due to reports of seizures as a side effect of many ‘first
generation’ antidepressants, particularly tricyclic antidepressants (TCAs) (Preskorn and Fast,
1992, Rosenstein et al., 1993, Salzberg and Vajda, 2001). The seizure aggravating effects of TCAs
have been demonstrated in both in vitro (Luchins and Ananth, 1976) and in vivo (Trimble, 1978)
experimental models. Furthermore, TCAs were recognised to have the capacity to induce
seizures in non-epileptic patients (Wroblewski et al., 1990, Preskorn and Fast, 1992) and to
aggravate seizures in pre-existing epilepsy (Pisani et al., 1999). Consequently, clinicians became
hesitant to prescribe antidepressants to patients with epilepsy (Cotterman-Hart, 2010).
Currently, there is still uncertainty amongst physicians regarding the use of antidepressant
medications in epilepsy and, as such, many depressed patients with epilepsy remain either
undertreated or completely untreated for their mood disorders.
However, there is a growing body of evidence to suggest that the newer, or ‘second generation’,
antidepressants have less effects on brain excitability, and may in fact reduce it. The newer
antidepressants, notably the SSRIs and serotonin and noradrenergic reuptake inhibitors

72
(SNRIs), have been shown to have a better safety profile in terms of seizure occurrence during
treatment in patients with epilepsy. There have been many studies that have investigated the
effect of SSRIs on seizures in both people with epilepsy and in animal models (reviewed in
Cardamone et al., 2012). Studies conducted in patients with epilepsy have found that chronic
treatment with SSRIs or SNRIs did not affect seizure frequency and, in some cases, even reduced
seizure frequency (no effect: Harmant et al., 1990, Gigli et al., 1994, Hovorka et al., 2000, Kanner
et al., 2000, Kuhn et al., 2003, Thome-Souza et al., 2007, Okazaki et al., 2011; reduction: Favale
et al., 1995, Favale et al., 2003, Specchio et al., 2004). Similar patterns are found in animal
models of epilepsy. Many of these studies report an anticonvulsant effect (Dailey et al., 1992,
Kabuto et al., 1994a, Wada et al., 1995, Hernandez et al., 2002, Pericic et al., 2005, Ahern et al.,
2006, Richman and Heinrichs, 2007, Mazarati et al., 2008, Borowicz et al., 2011, Jaako et al.,
2011, Vermoesen et al., 2012) or no effect (Raju et al., 1999, Ahern et al., 2006, Borowicz et al.,
2007, Mazarati et al., 2008, Choi et al., 2010, Vermoesen et al., 2012) of chronic treatment with
SSRIs or SNRIs. While reports of a proconvulsant effect of SSRIs in patients with epilepsy are
few, and occur primarily in those who have taken overdoses (Judge and Rentmeester, 2011), a
proconvulsant effect has been observed in some animal studies after chronic administration of
SSRIs or SNRIs at therapeutic doses (Arai et al., 2003, Ferrero et al., 2005, Ahern et al., 2006).
However, in all of these studies, seizures were only assessed at one time point after SSRI/SNRI
administration, rather than investigating the effects on longer-term epileptic outcomes such as
changes in seizure frequency over time.
In both human and animal studies, much of the focus has been upon short-term outcomes, such
as changes in seizure frequency and threshold in response to short-term, or even single-dose,
antidepressant treatment. Together, the data from both human and animal studies largely
suggest that SSRIs do not adversely affect seizure frequency and threshold, and as they alleviate
depressive symptoms in patients with epilepsy, are considered safe to use in this context.
However, absent from the literature are studies investigating the effects of chronic
antidepressant treatment on epileptogenesis, the underlying neurobiological changes that are
associated with the development of epilepsy that also continue even after seizures emerge.
In the human studies, the long-term effects on epilepsy beyond the period of assessment (1 to
15 months) have not been examined. Furthermore, only three animal studies have investigated
the effects of chronic SSRI treatment on long-term seizure outcomes using post-status
epilepticus models of epilepsy. Using the post-pilocarpine status epilepticus model, in which
rats develop spontaneous recurrent seizures therefore allowing for long-term monitoring of
seizure outcomes, Hernandez et al. (2002) found that five days of fluoxetine treatment inhibited

73
spontaneous recurrent seizures, while in the same model Mazarati et al. (2008) found no
differences in spontaneous recurrent seizures following ten days of fluoxetine treatment. In the
post-kainic acid induced status epilepticus model, Vermoesen et al. (2012) investigated the
effects of four days of citalopram treatment on seizure frequency, severity and duration. In this
study, it was found that when administered at therapeutically relevant doses, citalopram
reduced seizure frequency and cumulative seizure duration, without affecting seizure severity.
While these three studies provide some insight on the effects of chronic SSRI treatment on
seizures, epileptogenesis was not investigated as antidepressants were administered after the
emergence of spontaneous seizures. In addition, the periods of treatment used in these studies,
between four and ten days, are relatively short and cannot be considered as chronic treatment.
As such, investigation of the effects of chronic SSRI administration on epileptogenesis is needed.
Long-term studies investigating changes in seizure frequency over time and severity of the
disorder during antidepressant treatment would aid in understanding the impact of
antidepressant treatment during epilepsy.
Investigating this critical issue in patient populations would be technically and logistically
difficult and require very long-term studies. In contrast, using animal models to examine the
effects of chronic SSRI treatment on epileptogenesis provides a much more practical approach.
Investigating epileptogenesis in animals can be accomplished with the amygdala kindling model
of epileptogenesis. In this model, a bipolar electrode is implanted into the amygdala through
which an electrical current is delivered twice daily to induce focal seizures (Goddard, 1967,
Goddard et al., 1969, Racine, 1978). This produces a progressive increase in seizure
susceptibility and the epileptic response, with a spread of seizure activity from focal to
extrafocal regions and the appearance of generalised convulsive seizures (Goddard et al., 1969,
Racine, 1972b). During this kindling period, various aspects of seizures and epileptogenesis can
be monitored, including seizure threshold before and after kindling, seizure duration with
subsequent stimulations, and importantly for this study, the progression of seizure severity
with repeated electrical stimulations. This will allow for close monitoring of the progression of
epileptogenesis, and other kindling parameters, during chronic SSRI treatment.
The SSRIs used in the experiments discussed in this chapter were fluoxetine and citalopram,
administered by osmotic pumps. The use of fluoxetine to affect behaviour and investigate its
effects on acute seizure outcomes has been well-established in various animal models, allowing
for relevant comparison of this study with the literature. Citalopram was chosen for this study
as it is one of the first-line medications currently used in clinical practice for the treatment of
depression in epilepsy, due to its low side effect profile and low levels of interaction with AEDs,

74
and has also been previously used in the experimental literature. As such, citalopram is
considered safe for use in epilepsy with studies showing that is does not adversely influence
seizure frequency (Hovorka et al., 2000, Favale et al., 2003, Specchio et al., 2004). The study
presented in this chapter investigated the effects of chronic antidepressant treatment on
epileptogenesis. Changes in behaviour, hypothalamo-pituitary-adrenal axis function and
neurogenesis were also investigated, as these factors have been shown to be involved in
epileptogenesis and are also affected by SSRI treatment.
It was hypothesised that:
1. Compared to vehicle-treated rats, fluoxetine- and citalopram-treated rats would have a
slower rate of kindling epileptogenesis.
2. Kindling would increase anxiety- and depressive-like behaviours and this would be
mitigated with fluoxetine and citalopram treatments.
3. Kindling would increase the corticosterone response to stress, and this would be
mitigated with fluoxetine and citalopram treatments.
4.2. Materials and methods
The number of animals used, details of electrode implantation, surgical, kindling, behavioural
testing, blood sampling and post-mortem procedures are described in detail in Chapter 2,
Sections 2.2 to 2.4. Throughout this chapter, results are presented separately for the fluoxetine-
treated cohort and their vehicle-treated counterparts, and for the citalopram-treated cohort and
their vehicle-treated counterparts. As such, two separate vehicle-treated cohorts (treated with
the same vehicle) were used for comparison with the respective SSRI treatment.
In this study, seizure threshold was assessed prior to and at the end of the kindling procedures:
prior to kindling to determine the current at which to stimulate during kindling, as well as to
determine any effects of SSRI treatment on seizure threshold at this time; at the end of kindling
to assess the effects of kindling and as well as chronic SSRI treatment on seizure threshold.
Kindling procedures commenced one day after seizure threshold determination during which
all rats assigned to the kindling group received 30 stimulations, twice daily (5 days/week).
Following kindling, rats in the fluoxetine cohort received seven daily BrdU injections. Three

75
weeks later, anxiety- and depressive-like behaviours were assessed in all cohorts, as well as the
corticosterone response to swim stress. One day later, rats were culled, a cardiac blood sample
was taken for analysis of levels of plasma fluoxetine and norfluoxetine (its major active
metabolite; not performed for citalopram cohort), and the brain extracted for determination of
electrode placement (both cohorts) or post-mortem quantification of hippocampal
neurogenesis (fluoxetine cohort only) (Figure 4.1).
4.2.1 Statistical analyses
Seizure threshold and kindling rate were each analysed with a one-way repeated measures
ANOVA (IV: drug; DV: threshold current, number of stimulations to reach seizure class).
Individual data points for seizure duration were analysed with a Student’s t-test (seizure
duration at each stimulation). Data for the elevated plus maze, forced swim test, corticosterone
concentrations and BrdU cell counts were each analysed with a two-way repeated measures
ANOVA (IV: drug, kindling; DV: time spent in open arms/number entries to open arms/distance
travelled, time spent climbing/immobile/time first immobile, plasma corticosterone
concentrations and cell counts respectively). All data is presented as mean±SEM unless
otherwise stated.
Figure 4.1: Timeline of experimental procedures. w – week.
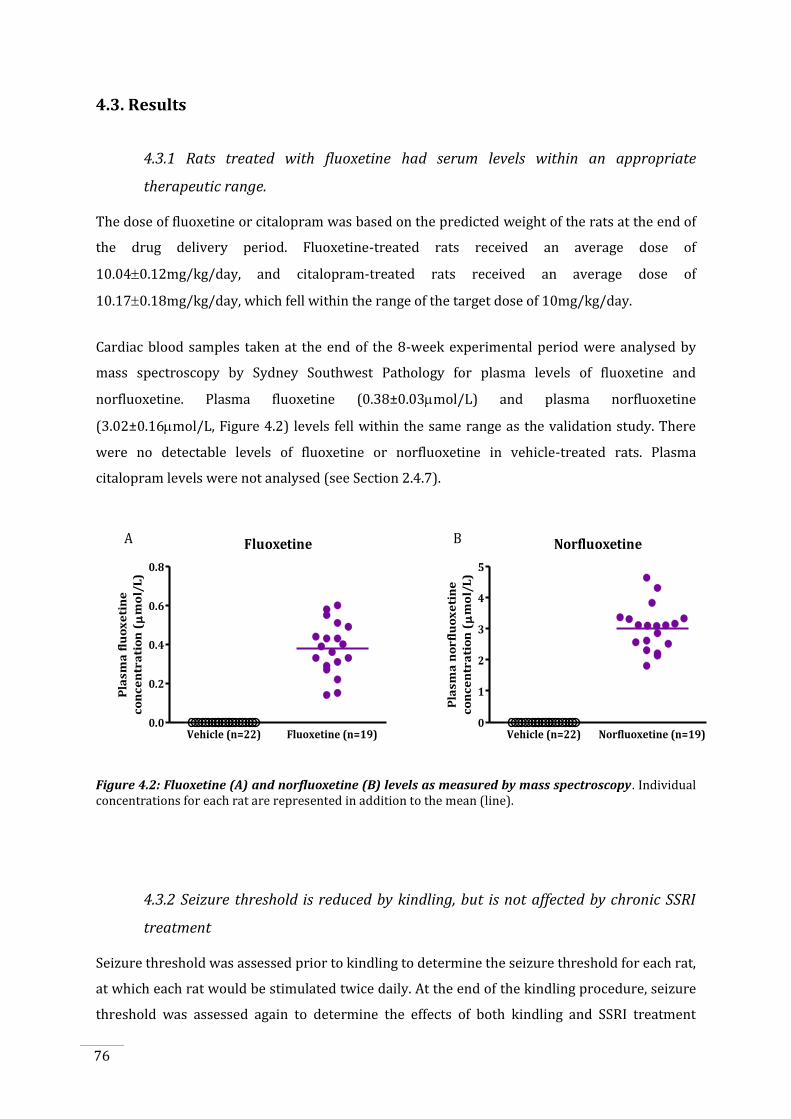
76
4.3. Results
4.3.1 Rats treated with fluoxetine had serum levels within an appropriate
therapeutic range.
The dose of fluoxetine or citalopram was based on the predicted weight of the rats at the end of
the drug delivery period. Fluoxetine-treated rats received an average dose of
10.040.12mg/kg/day, and citalopram-treated rats received an average dose of
10.170.18mg/kg/day, which fell within the range of the target dose of 10mg/kg/day.
Cardiac blood samples taken at the end of the 8-week experimental period were analysed by
mass spectroscopy by Sydney Southwest Pathology for plasma levels of fluoxetine and
norfluoxetine. Plasma fluoxetine (0.38±0.03mol/L) and plasma norfluoxetine
(3.02±0.16mol/L, Figure 4.2) levels fell within the same range as the validation study. There
were no detectable levels of fluoxetine or norfluoxetine in vehicle-treated rats. Plasma
citalopram levels were not analysed (see Section 2.4.7).
Fluoxetine
Vehicle (n=22) Fluoxetine (n=19)0.0
0.2
0.4
0.6
0.8
Pla
sm
a f
luo
xe
tin
e
co
nc
en
tra
tio
n (
mo
l/L
)
Norfluoxetine
Vehicle (n=22) Norfluoxetine (n=19)0
1
2
3
4
5
Pla
sm
a n
or
flu
ox
eti
ne
co
nc
en
tra
tio
n (
mo
l/L
)
A B
Figure 4.2: Fluoxetine (A) and norfluoxetine (B) levels as measured by mass spectroscopy . Individual concentrations for each rat are represented in addition to the mean (line).
4.3.2 Seizure threshold is reduced by kindling, but is not affected by chronic SSRI
treatment
Seizure threshold was assessed prior to kindling to determine the seizure threshold for each rat,
at which each rat would be stimulated twice daily. At the end of the kindling procedure, seizure
threshold was assessed again to determine the effects of both kindling and SSRI treatment

77
(Figure 4.3). As expected, repeated electrical stimulations resulted in a significantly reduced
seizure threshold (fluoxetine cohort: F(1,17)=30.62, p<0.0001; citalopram cohort: F(1,33)=91.84,
p<0.0001) however neither fluoxetine (F(1,17)=0.007, p=0.933) nor citalopram (F(1,33)=3.321,
p=0.078) treatments significantly affected seizure threshold before or after kindling.
4.3.3 Chronic fluoxetine and citalopram treatments accelerate kindling
epileptogenesis
During the kindling procedure, seizure severity was recorded for each stimulation, from class 1
(least severe) to class 5 (most severe). Significant differences were observed in the progression
of kindling and in response to drug treatment (Figure 4.4). As expected, all treatment groups
showed an increase in seizure severity with progressive stimulations (fluoxetine cohort:
F(1,17)=46.71, p<0.0001; citalopram cohort: F(1,33)=159.36, p<0.0001). This effect was accelerated
in both fluoxetine- and citalopram-treated rats, with rats both groups showing a more rapid
progression through the different stages of kindling (fluoxetine: F(1,17)=5.23, p=0.040;
citalopram: F(1,33)=16.18, p=0.0004). These results suggest that chronic treatment with
fluoxetine and citalopram accelerates the rate of kindling epileptogenesis.
4.3.4 Chronic SSRI treatments accelerate the progression of increase in seizure
duration early in epileptogenesis
Total seizure duration was recorded at each stimulation, from the end of the stimulation to the
end of the elicited electrographic seizure, to a maximum length of 300s (Figure 4.5). Usually
during kindling, seizure duration increases and plateaus once maximal seizure duration is
reached. In this study, in order to appropriately compare the differences in seizure duration as
epileptogenesis progressed, data for seizure duration was analysed with t-tests at each
subsequent group of stimulations, comparing the seizure duration between fluoxetine- or
citalopram-treated groups with their respective vehicle-treated counterparts (as depicted in the
x-axis of Figure 4.5A and B). Analysis showed that the increase in seizure duration was
accelerated in the early stages of kindling, occurring significantly quicker in both fluoxetine- and
citalopram-treated rats. This increase in seizure duration was significant at stimulations 1 to 3
(fluoxetine: p=0.018; citalopram: p=0.001), 4 to 6 (fluoxetine: p=0.044; citalopram: p=0.004)
and 7 to 9 (fluoxetine: p=0.018; citalopram: p=0.020). From stimulation 10 onwards, seizure
duration in all treatment groups reached a plateau, with no significant differences in seizure
duration from stimulations 10 to 30 (p>0.05).
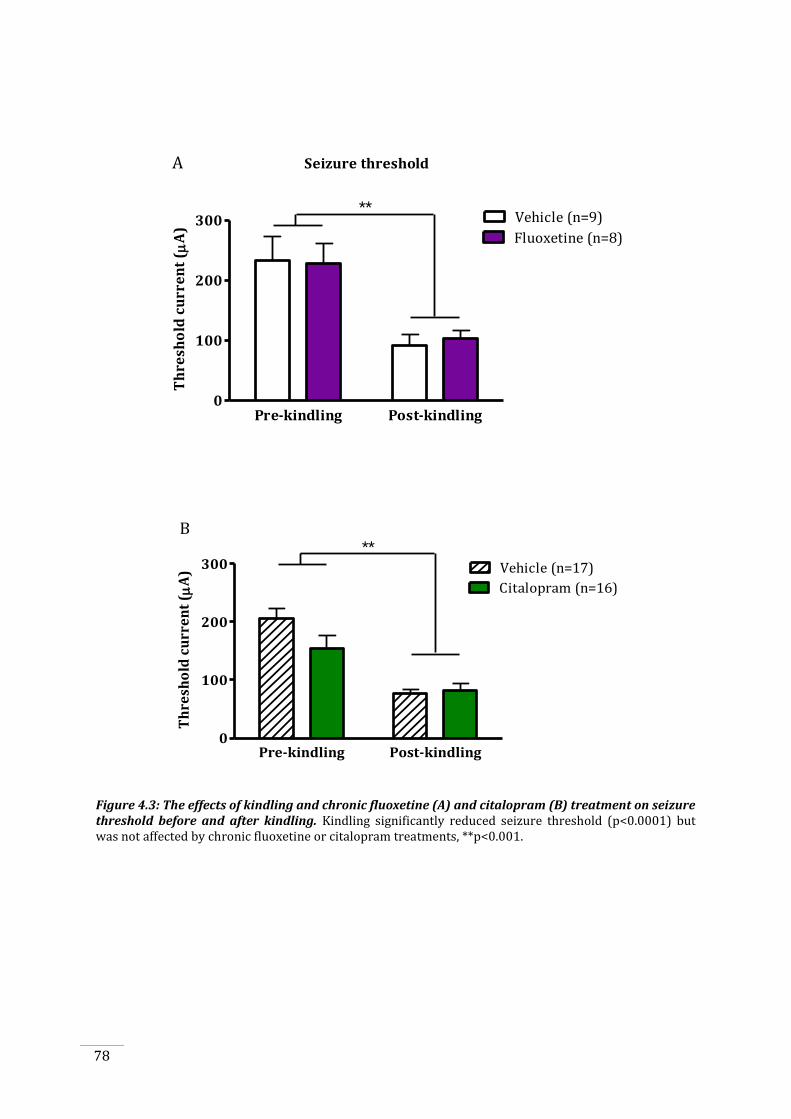
78
Seizure threshold
Pre-kindling Post-kindling0
100
200
300Fluoxetine (n=8)
Vehicle (n=9)
Th
resh
old
cu
rre
nt
( A
)
**
A
Pre-kindling Post-kindling0
100
200
300
Citalopram (n=16)
Vehicle (n=17)
Th
resh
old
cu
rre
nt
( A
)
**B
Figure 4.3: The effects of kindling and chronic fluoxetine (A) and citalopram (B) treatment on seizure threshold before and after kindling. Kindling significantly reduced seizure threshold (p<0.0001) but was not affected by chronic fluoxetine or citalopram treatments, **p<0.001.
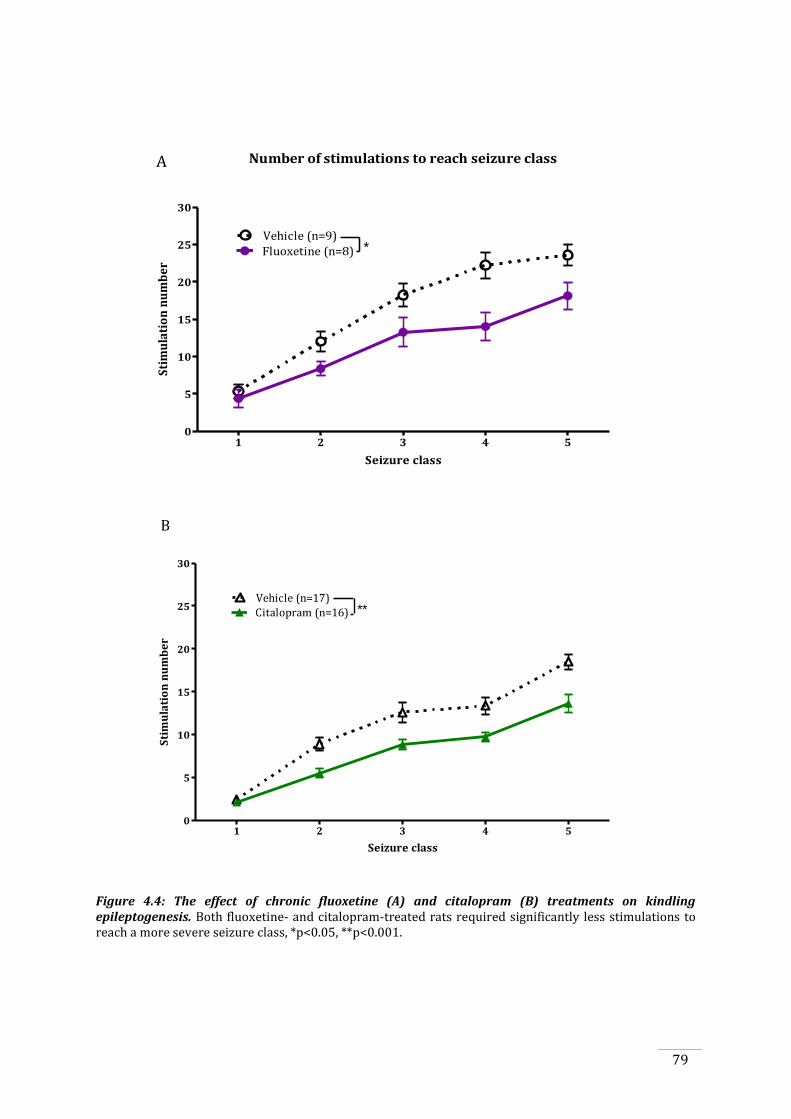
79
Number of stimulations to reach seizure class
1 2 3 4 50
5
10
15
20
25
30
Fluoxetine (n=8)
Vehicle (n=9)*
Seizure class
Sti
mu
lati
on
nu
mb
er
A
1 2 3 4 50
5
10
15
20
25
30
Citalopram (n=16)
Vehicle (n=17)
Seizure class
Sti
mu
lati
on
nu
mb
er
**
B
Figure 4.4: The effect of chronic fluoxetine (A) and citalopram (B) treatments on kindling epileptogenesis. Both fluoxetine- and citalopram-treated rats required significantly less stimulations to reach a more severe seizure class, *p<0.05, **p<0.001.
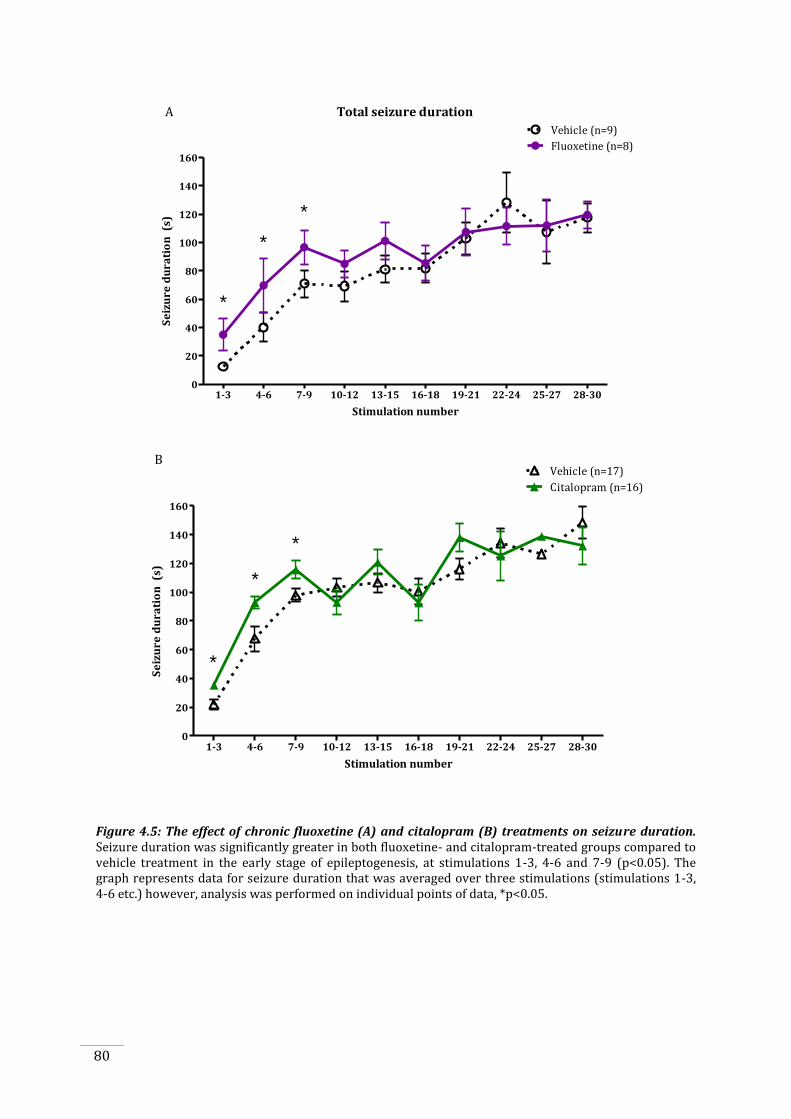
80
Total seizure duration
1-3 4-6 7-9 10-12 13-15 16-18 19-21 22-24 25-27 28-300
20
40
60
80
100
120
140
160Fluoxetine (n=8)
Vehicle (n=9)
Stimulation number
Se
izu
re d
ura
tio
n (
s)
A
*
*
*
1-3 4-6 7-9 10-12 13-15 16-18 19-21 22-24 25-27 28-300
20
40
60
80
100
120
140
160
Citalopram (n=16)
Vehicle (n=17)
Stimulation number
Se
izu
re d
ura
tio
n (
s)
B
*
*
*
Figure 4.5: The effect of chronic fluoxetine (A) and citalopram (B) treatments on seizure duration. Seizure duration was significantly greater in both fluoxetine- and citalopram-treated groups compared to vehicle treatment in the early stage of epileptogenesis, at stimulations 1-3, 4-6 and 7-9 (p<0.05). The graph represents data for seizure duration that was averaged over three stimulations (stimulations 1-3, 4-6 etc.) however, analysis was performed on individual points of data, *p<0.05.

81
4.3.5 Chronic fluoxetine and citalopram treatments differentially affect anxiety-like
behaviours
Anxiety-like behaviours were assessed approximately three weeks after the end of the kindling
procedures. Firstly, in the fluoxetine cohort, kindled rats made significantly fewer entries
(F(3,40)=5.19, p=0.03, Figure 4.6A) and spent significantly less time (F(3, 40)=4.58, p=0.04, Figure
4.6B) in the open arms of the plus maze, indicating an anxiogenic effect of kindling. However, in
the citalopram cohort, there was no significant effect of kindling on anxiety-like behaviours,
with no significant differences in the percentage of open arm entries (F(3,48)=2.93, p=0.09, Figure
4.6D) or percentage of time spent in the open arms (F(3,48)=1.77, p=0.19, Figure 4.6E) of the plus
maze.
Analysis of the effect of drug treatment revealed no significant effect of either fluoxetine or
citalopram treatments in the percentage of open arm entries (fluoxetine: F(3,40)=2.60, p=0.12;
citalopram: F(3,48)=2.67, p=0.11) or percentage of time spent (fluoxetine: F(3,40)=3.14, p=0.08;
citalopram: F(3,48)=2.35, p=0.13) in the open arms of the plus maze.
Locomotor activity was assessed by quantifying the total distance travelled during the testing
session. For rats in the fluoxetine cohort, neither kindling (F(3,40)=2.91, p=0.09, Figure 4.6C) nor
drug treatment (F(3,40)=0.01, p=0.91) affected locomotor activity, while kindling (F(3,48)=5.46,
p=0.02) but not SSRI treatment (F(3,48)=0.51, p=0.48) significantly reduced locomotor activity in
the citalopram cohort (Figure 4.6F).
4.3.6 Kindling, fluoxetine or citalopram treatments do not affect depressive-like
behaviour
Depressive-like behaviours were assessed using the forced swim test. It was found that there
was no effect of kindling, fluoxetine or citalopram treatments on time spent climbing (fluoxetine
cohort: drug, F(3,40)=0.0002, p=0.98; kindling, F(3,40)=0.10, p=0.75; citalopram cohort: drug,
F(3,46)=1.52, p=0.22; kindling, F(3,46)=0.82, p=0.37), time spent immobile (fluoxetine cohort: drug,
F(3,40)=0.94, p=0.33; kindling, F(3,40)=0.33, p=0.56; citalopram cohort: drug, F(3,46)=0.39, p=0.54;
kindling, F(3,46)=0.90, p=0.35) or in the latency to the first immobile activity (fluoxetine cohort:
drug, F(3,40)=0.33, p=0.57; kindling, F(3,40)=1.58, p=0.22; citalopram cohort: drug, F(3,46)=0.99,
p=0.33; kindling, F(3,46)=0.11, p=0.74) (Figure 4.7).
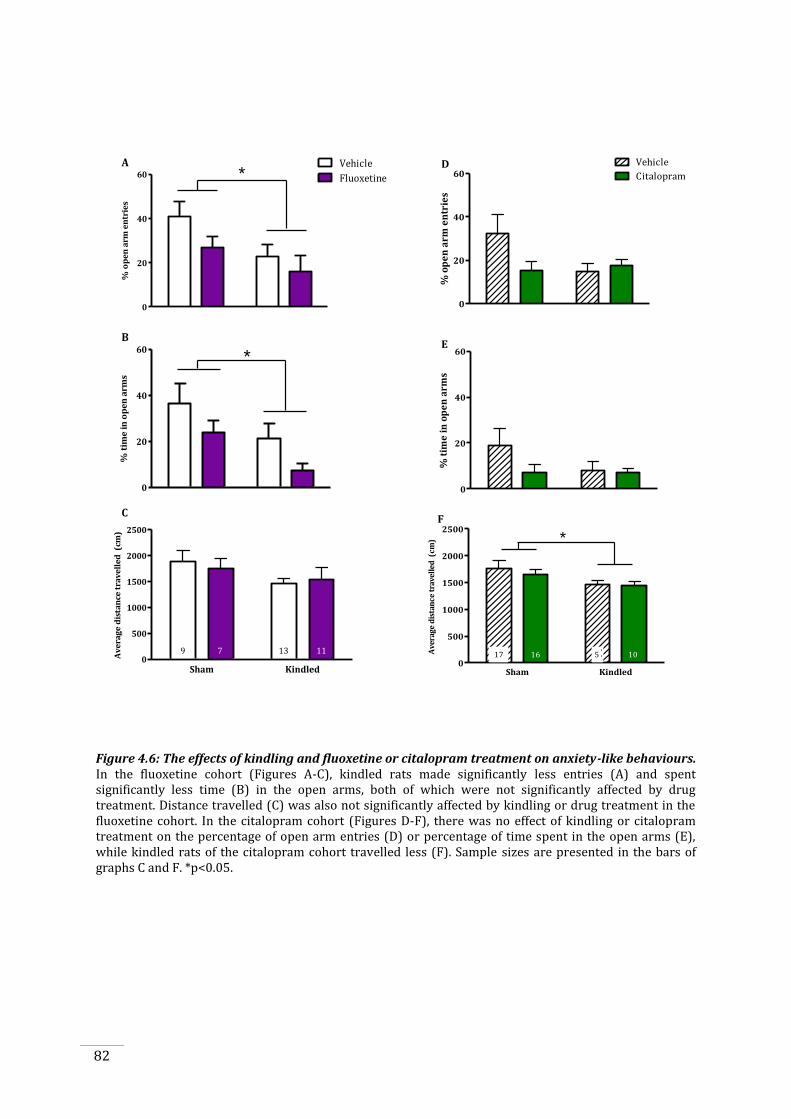
82
0
20
40
60 Fluoxetine
Vehicle*
% o
pe
n a
rm e
ntr
ies
0
20
40
60
% t
ime
in
op
en
arm
s
*
Sham Kindled0
500
1000
1500
2000
2500
Av
era
ge
dis
tan
ce t
rav
ell
ed
(cm
)
9 7 13 11
A
C
B
Sham Kindled0
20
40
60 Citalopram
Vehicle
% o
pe
n a
rm e
ntr
ies
Sham Kindled0
20
40
60
% t
ime
in
op
en
arm
s
Sham Kindled0
500
1000
1500
2000
2500
Ave
rage
dis
tan
ce t
rave
lled
(cm
)
17 516 10
*
D
E
F
Figure 4.6: The effects of kindling and fluoxetine or citalopram treatment on anxiety-like behaviours. In the fluoxetine cohort (Figures A-C), kindled rats made significantly less entries (A) and spent significantly less time (B) in the open arms, both of which were not significantly affected by drug treatment. Distance travelled (C) was also not significantly affected by kindling or drug treatment in the fluoxetine cohort. In the citalopram cohort (Figures D-F), there was no effect of kindling or citalopram treatment on the percentage of open arm entries (D) or percentage of time spent in the open arms (E), while kindled rats of the citalopram cohort travelled less (F). Sample sizes are presented in the bars of graphs C and F. *p<0.05.
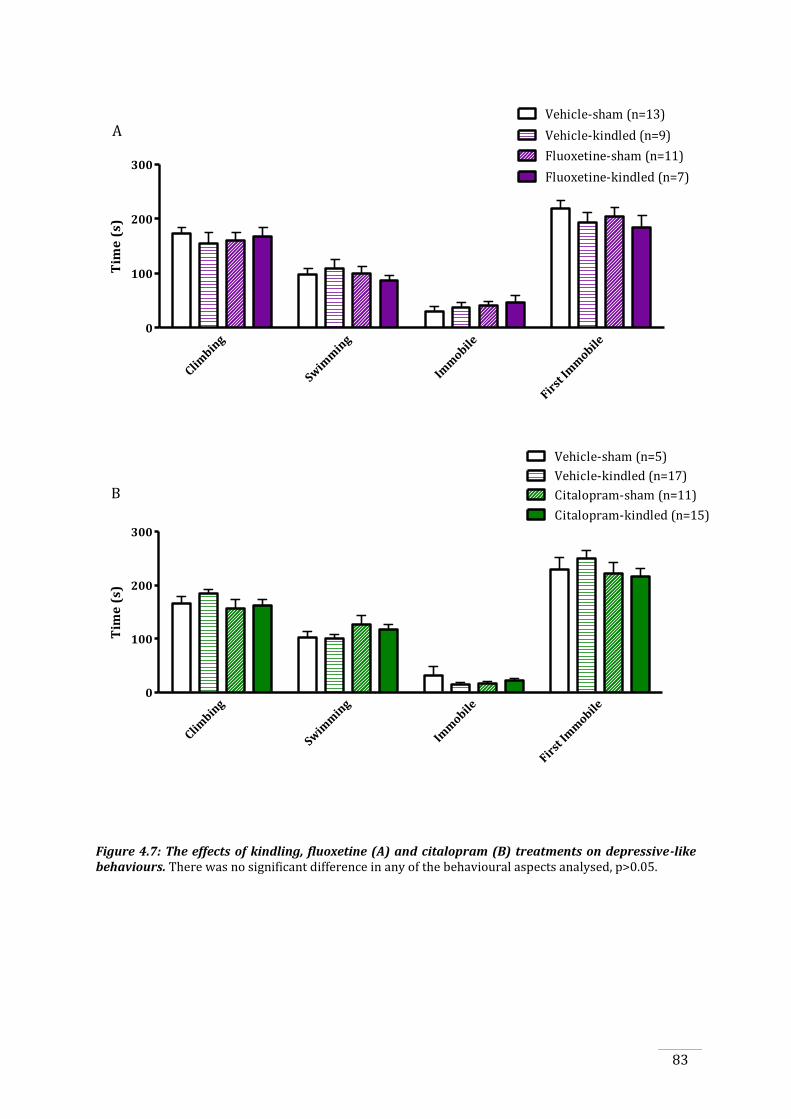
83
Climbin
g
Swim
min
g
Imm
obile
First
Imm
obile0
100
200
300Fluoxetine-kindled (n=7)
Fluoxetine-sham (n=11)
Vehicle-kindled (n=9)
Vehicle-sham (n=13)T
ime
(s)
A
Climbin
g
Swim
min
g
Imm
obile
First
Imm
obile0
100
200
300
Citalopram-kindled (n=15)
Citalopram-sham (n=11)
Vehicle-kindled (n=17)
Vehicle-sham (n=5)
Tim
e (
s)
B
Figure 4.7: The effects of kindling, fluoxetine (A) and citalopram (B) treatments on depressive-like behaviours. There was no significant difference in any of the behavioural aspects analysed, p>0.05.
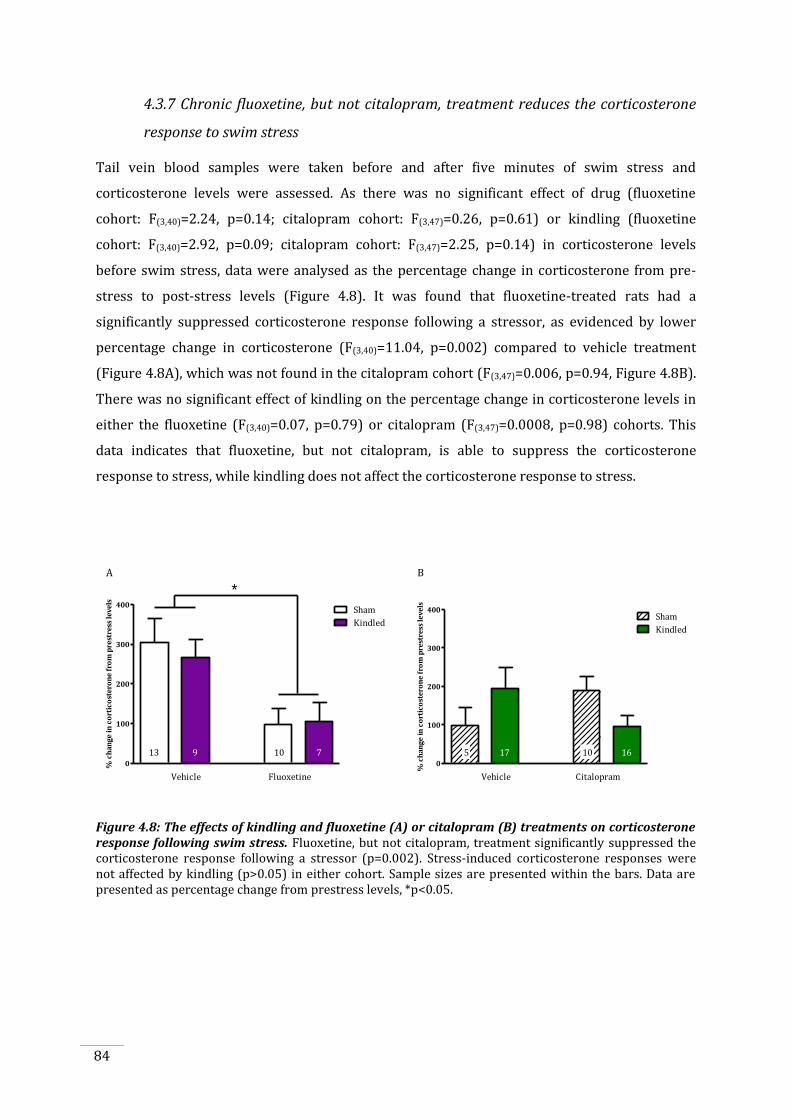
84
4.3.7 Chronic fluoxetine, but not citalopram, treatment reduces the corticosterone
response to swim stress
Tail vein blood samples were taken before and after five minutes of swim stress and
corticosterone levels were assessed. As there was no significant effect of drug (fluoxetine
cohort: F(3,40)=2.24, p=0.14; citalopram cohort: F(3,47)=0.26, p=0.61) or kindling (fluoxetine
cohort: F(3,40)=2.92, p=0.09; citalopram cohort: F(3,47)=2.25, p=0.14) in corticosterone levels
before swim stress, data were analysed as the percentage change in corticosterone from pre-
stress to post-stress levels (Figure 4.8). It was found that fluoxetine-treated rats had a
significantly suppressed corticosterone response following a stressor, as evidenced by lower
percentage change in corticosterone (F(3,40)=11.04, p=0.002) compared to vehicle treatment
(Figure 4.8A), which was not found in the citalopram cohort (F(3,47)=0.006, p=0.94, Figure 4.8B).
There was no significant effect of kindling on the percentage change in corticosterone levels in
either the fluoxetine (F(3,40)=0.07, p=0.79) or citalopram (F(3,47)=0.0008, p=0.98) cohorts. This
data indicates that fluoxetine, but not citalopram, is able to suppress the corticosterone
response to stress, while kindling does not affect the corticosterone response to stress.
10
100
200
300
400
% c
han
ge in
co
rtic
ost
ero
ne
fro
m p
rest
ress
le
vels
Vehicle Fluoxetine
13 10
Sham
Kindled
*
A
9 7
10
100
200
300
400
% c
ha
ng
e i
n c
ort
ico
ste
ron
e f
rom
pre
stre
ss l
ev
els
Vehicle Citalopram
5 10
Sham
Kindled
B
17 16
Figure 4.8: The effects of kindling and fluoxetine (A) or citalopram (B) treatments on corticosterone response following swim stress. Fluoxetine, but not citalopram, treatment significantly suppressed the corticosterone response following a stressor (p=0.002). Stress-induced corticosterone responses were not affected by kindling (p>0.05) in either cohort. Sample sizes are presented within the bars. Data are presented as percentage change from prestress levels, *p<0.05.
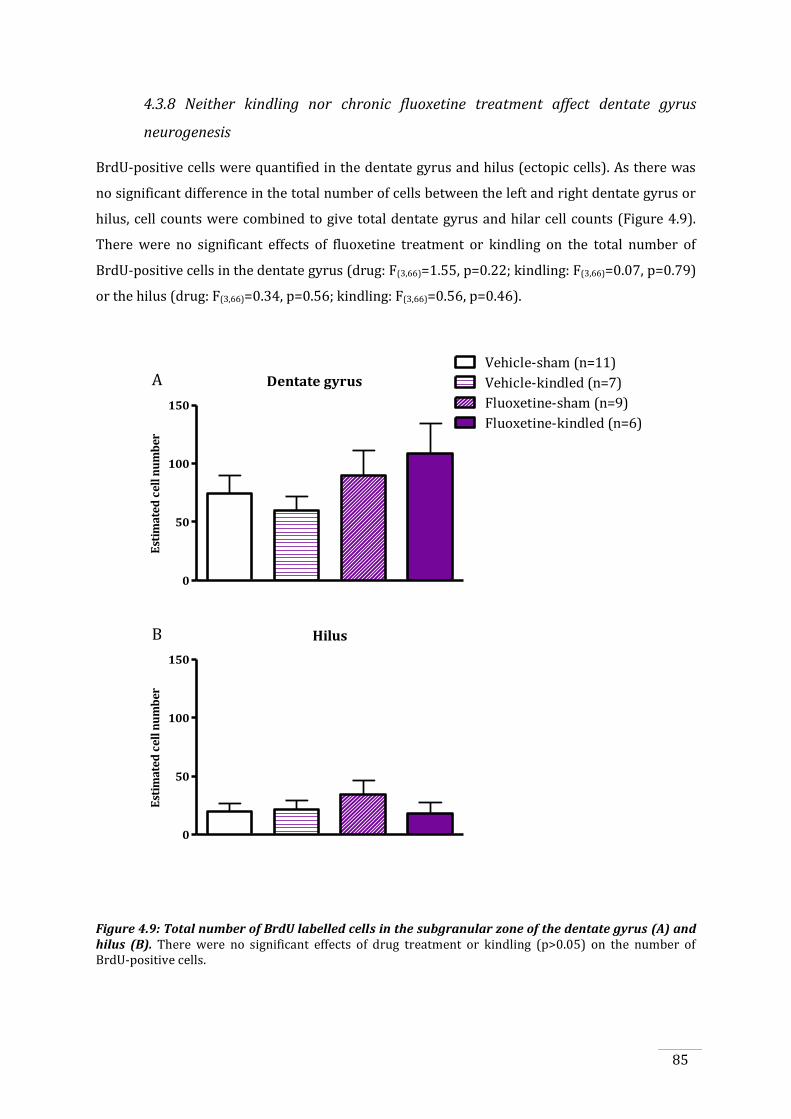
85
4.3.8 Neither kindling nor chronic fluoxetine treatment affect dentate gyrus
neurogenesis
BrdU-positive cells were quantified in the dentate gyrus and hilus (ectopic cells). As there was
no significant difference in the total number of cells between the left and right dentate gyrus or
hilus, cell counts were combined to give total dentate gyrus and hilar cell counts (Figure 4.9).
There were no significant effects of fluoxetine treatment or kindling on the total number of
BrdU-positive cells in the dentate gyrus (drug: F(3,66)=1.55, p=0.22; kindling: F(3,66)=0.07, p=0.79)
or the hilus (drug: F(3,66)=0.34, p=0.56; kindling: F(3,66)=0.56, p=0.46).
Dentate gyrus
Est
imat
ed c
ell n
um
ber
Dentate
gyru
s0
50
100
150
Vehicle-sham (n=11)
Vehicle-kindled (n=7)
Fluoxetine-sham (n=9)
Fluoxetine-kindled (n=6)
A
Hilus
Est
imat
ed c
ell n
um
ber
Hilus 0
50
100
150
B
Figure 4.9: Total number of BrdU labelled cells in the subgranular zone of the dentate gyrus (A) and hilus (B). There were no significant effects of drug treatment or kindling (p>0.05) on the number of BrdU-positive cells.

86
4.4. Discussion
In the majority of studies that report on the effects of antidepressant treatment in epilepsy,
acute dosing paradigms and acute seizure models have been used, with few studies employing
chronic treatment paradigms or performing any long-term follow up of epilepsy progression
(Cardamone et al., 2012). Furthermore, reports of the proconvulsant effects of SSRIs mostly
derive from animal studies in which acute seizure models were used or from patient reports of
SSRIs used in overdose. This is the first study to report specifically on the effects of chronic
SSRIs treatment at therapeutically relevant levels on progression of the epileptic disorder, by
investigating the effect on the progression of kindling epileptogenesis. The key finding of this
study was that rats chronically treated with SSRIs, either fluoxetine or citalopram,
demonstrated an accelerated rate of kindling epileptogenesis, in which rats developed more
severe seizures with less stimulations, compared to vehicle-treated rats. Seizure duration was
also significantly greater in both fluoxetine- and citalopram-treated rats in the early stage of
epileptogenesis, but not in the later stages and there was no significant effect of fluoxetine or
citalopram treatment on seizure threshold. Anxiety-like behaviours were differentially affected
by kindling and drug treatments but depressive-like behaviours were not significantly affected.
Stress-induced corticosterone responses were significantly reduced by fluoxetine but not
citalopram treatment and it was also found that neurogenesis was not significantly affected by
fluoxetine treatment or kindling.
4.4.1 Kindling epileptogenesis is accelerated by chronic antidepressant treatment
The kindling model of epileptogenesis is a useful one to investigate epileptogenesis, as it allows
for the close monitoring of the progressive increase in seizure susceptibility and the epileptic
response with subsequent electrical stimulations, by assessing behavioural severity of seizures
and quantifying electrographic seizure activity. In this study, chronic fluoxetine and citalopram
treatments accelerated the progression of kindling epileptogenesis, with fluoxetine- and
citalopram-treated rats displaying more severe seizures after less electrical stimulation. This
indicates that both chronic fluoxetine and citalopram treatments have a proepileptogenic effect.
This is neither consistent nor inconsistent with previous studies, as there have been none to
investigate epileptogenesis during chronic antidepressant treatment, and the effect was not due
to alterations in the acute excitability of the brain as seizure threshold was the same between
SSRIs and vehicle treatments. However, this is an unexpected finding as much of the previous
literature in humans and animals suggests that antidepressant treatment has no effect on
seizures, or even an anticonvulsant effect.

87
In the human literature, many studies have focused on more patient-relevant outcomes that
affect a person with epilepsy’s daily life, such as seizure frequency. Of these studies, it was found
that chronic SSRI treatment (treatment periods of 1 to 15 months) in epilepsy had no overall
effect on seizure frequency (Harmant et al., 1990, Gigli et al., 1994, Hovorka et al., 2000, Kanner
et al., 2000, Kuhn et al., 2003, Thome-Souza et al., 2007, Okazaki et al., 2011), with some
reporting an improvement in seizure frequency (Favale et al., 1995, Favale et al., 2003, Specchio
et al., 2004). Specifically, studies with fluoxetine in patients with epilepsy have shown that
treatment for three to fourteen months resulted in an overall reduction in seizure frequency
when compared to baseline seizure frequency, with some patients even reporting complete
seizure freedom. There were also reports of four patients having an increase in seizure
frequency (Gigli et al., 1994, Favale et al., 1995). Other studies reported that there were no
changes in seizure frequency with citalopram treatment (Hovorka et al., 2000, Kuhn et al., 2003)
or overall reductions in seizure frequency (Hovorka et al., 2000, Favale et al., 2003, Specchio et
al., 2004), while high doses of citalopram (in overdose) have been associated with increases in
seizure occurrence (Favale et al., 2003, Isbister et al., 2004, Kelly et al., 2004).
While the reported improvements or no effects of SSRIs on seizure frequency are an important
outcome, interpretation of the results of these studies is limited. This is because a majority of
these studies used a relatively short time-frame of SSRI treatment and follow up, and no studies
were performed with untreated or placebo-treated controls. Therefore, none of these studies
made comparisons with appropriate controls of the changes in seizure outcomes, such as
seizure frequency or severity, over time. This is an important outcome given that there is some
evidence that epilepsy may be a progressive disorder (Pitkanen and Sutula, 2002, Nearing et al.,
2007, Cascino, 2009), and therefore it is important to investigate the effects of SSRIs on longer
term outcomes, rather than just the effects on seizures at discreet time points of investigation.
Of the few animal studies that have reported proconvulsant effects of SSRI treatment, a majority
have only administered SSRIs acutely, with only two studies treating for up to 21 days. In the
studies that found proconvulsant effects of fluoxetine, Zienowicz et al. (2005) found that acute
treatment with 10mg/kg fluoxetine increased seizure severity and Ferrero et al. (2005) found
that 21-day treatment with 10mg/kg/day fluoxetine reduced seizure threshold. Studies with
citalopram that found proconvulsant effects have utilised only acute doses, finding a reduced
seizure threshold (Payandemehr et al., 2012, Vermoesen et al., 2011) or an increase in seizure
frequency (Arai et al., 2003). Overwhelmingly, animal studies investigating SSRI treatment in
animal models of epilepsy find that there is an anticonvulsant, or no effect of SSRI treatment on
seizures.

88
Overall, data from the previous studies would suggest that chronic treatment with fluoxetine or
citalopram, at therapeutically relevant doses, should not adversely affect seizure frequency. The
kindling model does not specifically investigate seizure frequency, as seizures are induced by
electrical stimulation and rats do not experience spontaneous seizures. However, this model is
advantageous in that it allows for the monitoring of changes in seizure susceptibility overtime,
allowing for monitoring of the progression of the disorder, which in this study has shown that
seizure susceptibility increases and consequently the rate of epileptogenesis is accelerated
during chronic SSRI treatment. Furthermore, the results of this study were replicated across
two separate cohorts using two different SSRIs, in experiments conducted independently of
each other, adding support to the notion that the results were not merely a false-positive.
Another advantage of this study is the use of a chronic treatment paradigm. In the majority of
animal studies of epilepsy, acute treatments are administered, and if SSRIs are administered
chronically, the period of treatment, in a majority of studies, is relatively short (2-14 days)
compared to the current study, with few studies treating for longer periods (21-28 days). SSRIs
are known to have a delayed onset of action, in which at least 3 weeks of treatment are required
for the therapeutic effect to manifest (Goodnick and Goldstein, 1998, Rosebook and Kalin,
2011). During this period, adaptive changes to biological systems that exert the effects of
antidepressants are taking place, which can only occur with chronic treatment paradigms. As
such, chronic treatment with these agents would display different effects than acute or even
shorter-term (less than 3 weeks) chronic treatment regimes.
One example of the adaptive changes occurring during chronic treatment, which may be
relevant to kindling, is alterations in serotonergic receptors (Eison and Mullins, 1996, Stahl,
1998, Keltner et al., 2002). Various studies have investigated the effects of serotonin receptor
agonists and antagonists on kindling epileptogenesis. These studies are important to consider,
given that one mechanism by which SSRIs exert their effects is by increasing serotonin levels,
which has subsequent effects on serotonergic receptors. There has been only one study which
investigated amygdala kindling epileptogenesis following administration of serotonin (5HT)
agonists and antagonists (Wada et al., 1997). In this study, it was found that a 5HT1A receptor
agonist, 8-hydroxy-2-(di-n-propylamino)tetralin (8-OH-DPAT) inhibited kindling rate while a
5HT2 receptor agonist, 2,5-dimethoxy-4-iodoamphetamine (DOI) facilitated kindling. The effect
of DOI was blocked by the addition of a 5HT2 receptor antagonist, ketanserin. Other studies
have investigated the effects of serotonin receptor agonists and antagonists on seizure
susceptibility, overall finding that 5HT1A receptor agonists have antiseizure effects (Wada et al.,
1992, Wada et al., 1993, Gariboldi et al., 1996a, Lu and Gean, 1998, Pericic et al., 2005) while

89
agonists of 5HT2 and 5HT3 receptors have proconvulsant effects (Wada et al., 1992, Wada et al.,
1999). However, Watanabe et al. (2000) found that antagonists of 5HT2A, 5HT3, 5HT2B and 5HT2C
do not change seizure threshold and Watanabe et al. (1998) found that the administration of
agonists of 5HT1A, 5HT2A, 5HT2C and 5HT3 receptors had no anticonvulsant effect in the kindling
model. The inconsistencies between these studies may be explained by disparity in the routes of
administration or by the seizure models employed.
Other studies have specifically examined which serotonergic receptors are involved in the
effects of SSRIs on seizures. For example, Payandemehr et al. (2012) found that citalopram dose
dependently exerted anticonvulsant (0.5 and 1mg/kg) and proconvulsant (28 and 50mg/kg)
effects on seizures in the pentylenetetrazol model of seizures. A dose dependant effect has also
been shown by Clinckers et al. (2004a). Although the anticonvulsant actions of citalopram at
low doses are not completely understood yet, it was verified that citalopram mediated its
anticonvulsant effect through the 5HT3 receptor, and its proconvulsant effect through a
modulatory role of nitric oxide. The proconvulsant effect of nitric oxide has also been reported
in previous studies (De Sarro et al., 1991, Osonoe et al., 1994, Gholipour et al., 2010, Bahremand
et al., 2011). Overall, these studies indicate that the proconvulsant effects of SSRIs may be
mediated through activation of 5HT2 receptors and this may also be mediated through effects on
nitric oxide. Whether such effects on these targets occurred in the present study cannot be
confirmed, and furthermore, the effects of fluoxetine and citalopram are more wide-ranging
than solely effects on specific serotonergic receptors, as discussed in Section 1.4.2. Investigating
these alterations in future studies should be considered.
4.4.2 Chronic fluoxetine and citalopram treatments increase seizure duration in the
early stages of epileptogenesis
In this study, it was found that both fluoxetine and citalopram treatments significantly increased
seizure durations in the early stages of kindling epileptogenesis, from stimulations 1 to 9. In this
period, seizure lengths increased differently between SSRI-treated and vehicle-treated rats,
with fluoxetine- and citalopram-treated rats having longer seizures. Seizure duration reached a
plateau in all treatment groups from stimulation 10 onwards: at this stage rats were beginning
to experience class 3 seizures, which corresponds to later stages of epileptogenesis when
seizures are beginning to generalise and become more severe, indicating a ceiling effect on
seizure duration. As such, all treatment groups had similar seizure durations at the end of
kindling however the fluoxetine- and citalopram-treated rats reached this stage faster.

90
This data indicates that, as expected, repeated electrical stimulation increase seizure duration,
and in fluoxetine- and citalopram-treated rats, this increase in seizure duration progressed at a
faster rate. This is consistent with a proepileptogenic effect of fluoxetine and citalopram
treatments, in that the epileptic disorder is progressing to a more severe stage at a quicker rate.
The results of this study conflict with other studies in which treatment with fluoxetine or
citalopram reduced or had no effect on seizure duration. For example, it has been reported that
2mg/kg fluoxetine but not 6 or 20mg/kg reduced primary after discharge duration (a measure
of seizure activity after a stimulation) (Watanabe et al., 1998), that four days of 15mg/kg
citalopram treatment but not 5 or 10mg/kg reduced cumulative seizure duration (Vermoesen et
al., 2012) while other studies found no effect of acute fluoxetine treatment on seizure duration
at doses of 10-50mg/kg (Wada et al., 1999, Jin et al., 2009). These studies are not consistent
with the current study, which may be explained by the use of different epilepsy models, as these
studies used post-status epilepticus models, in which rats experience spontaneous seizures, as
well as using a shorter period of drug administration. Furthermore, these studies only
investigated seizure duration at a single time point, or for short periods of analysis (maximum
of 3 days), and did not investigate the change in seizure duration as the epileptic disorder
progressed. There have been no studies to investigate the changes in seizure duration over time
during longer-term SSRI treatment.
4.4.3 Chronic fluoxetine and citalopram treatments do not affect seizure threshold
There was no effect of fluoxetine or citalopram treatments on seizure threshold before or after
kindling. As already discussed, most other studies focused on seizure threshold, as it is the key
indicator of neural excitability. Most such studies found that chronic SSRI treatment does not
affect seizure threshold. For example, following 14-21 days of fluoxetine treatment, it was found
that there was no difference in seizure threshold in models of maximal electroshock (Raju et al.,
1999) and ear shock (Borowicz et al., 2007). Additionally, no difference in seizure threshold was
found following chronic citalopram treatment in El mice, a genetic model of sensory-
precipitated seizures (Kabuto et al., 1994a) or following lidocaine-induced seizures (Arai et al.,
2003). Conversely, a number of studies found that chronic fluoxetine (Dailey et al., 1992, Wada
et al., 1995, Richman and Heinrichs, 2007, Mazarati et al., 2008) and citalopram (Kabuto et al.,
1994b) treatments increased seizure threshold while only one study found that chronic
fluoxetine treatment reduced seizure threshold (Ferrero et al., 2005).

91
4.4.4 Interim conclusion of kindling data
Overall, this data indicate that chronic fluoxetine and citalopram treatments accelerate the
progression of kindling epileptogenesis, as demonstrated by an accelerated kindling rate and
greater seizure duration in the early stages of epileptogenesis, however chronic SSRI treatment
did not affect seizure threshold and therefore local excitability within the brain. Potentially,
these results could have implications for the treatment of depressive symptoms in epilepsy,
which will be discussed in detail in Chapter 5.
4.4.5 Chronic fluoxetine and citalopram treatment and effects on anxiety- and
depressive-like behaviours
Psychiatric disorders are highly prevalent in patients with epilepsy, particularly in TLE
(Altshuler et al., 1999, Gaitatzis et al., 2004). Similarly, anxiety- and depressive-like behaviours
have been reported in animal models of epilepsy (Adamec and Young, 2000, Groticke et al.,
2007, Jones et al., 2008b). However when specifically investigating the kindling model, reports
of the effects of kindling on anxiety-like behaviour are mixed. Studies have reported both
anxiogenic (Adamec, 1990, Helfer et al., 1996) and anxiolytic (Adamec and Morgan, 1994, Jones
et al., 2009) effects of kindling, the results of which depend on various factors such as kindled
hemisphere (Adamec and Morgan, 1994), bipolar electrode location (Adamec et al., 2004) and
baseline state of anxiety (Adamec et al., 2005). For depressive-like behaviours, other studies
have reported no effect of kindling (Corcoran et al., 1992, Helfer et al., 1996, Adamec et al.,
2004), which was also found in this study. This supports the notion that the behavioural
alterations associated with kindling, in many studies, specifically affect anxiety-like behaviours.
In this study, kindled rats in the fluoxetine cohort displayed increased anxiety-like behaviours.
This study involved kindling of the left basolateral amygdala, and therefore is contrary to
previous studies that have found that left basolateral amygdala kindling has anxiolytic effects
(Adamec and Morgan, 1994, Jones et al., 2009). However, this was only found in the fluoxetine
but not citalopram cohort, the reasons for which are unclear. In the citalopram cohort, kindled
rats moved less, demonstrated by a reduced distance travelled in the EPM, however there was
no significant difference in mobility in the FST, with no significant differences between any
treatment groups in any of the active behaviours such as swimming or climbing. This difference
for distance travelled in the EPM may be related to the small sample size in the vehicle-sham
group, and furthermore, determining effects on locomotor activity would need to be confirmed
using a battery of tests for locomotion for which analysis of anxiety-like behaviours is not the
main outcome.

92
Overall, SSRI treatment had no effect on anxiety-like behaviours. This is consistent with Borsini
et al. (2002), in which 107 studies analysing the effects of antidepressants in animal model of
anxiety were reviewed, finding that in a majority of studies, antidepressant treatment does not
affect anxiety-like behaviours. Specifically, there were studies that reported no effect (Durand et
al., 1999, File et al., 1999, Griebel et al., 1999, Silva and Brandao, 2000) of chronic fluoxetine or
citalopram treatment on anxiety-like behaviours in the EPM, but this was contrary to others in
which an anxiogenic-like effect was found, which has been reported only in studies using
fluoxetine (Durand et al., 1999, File et al., 1999, Silva et al., 1999, Kurt et al., 2000, Oh et al.,
2009, Kobayashi et al., 2011). Therefore, the results found in this study are consistent with
literature, in that SSRI treatment had no effect on anxiety-like behaviours.
4.4.6 Chronic fluoxetine, but not citalopram, treatment reduces the corticosterone
response to swim stress
It is widely reported that depression is associated with alterations in HPA axis functioning
(Pariante and Lightman, 2008) and that antidepressant treatment normalises these alterations,
returning cortisol (in humans; Holsboer, 2001, Himmerich et al., 2007) and corticosterone (in
rodents; Jensen et al., 1999, Jongsma et al., 2005, Mazarati et al., 2008) levels back to normal.
Studies have also investigated the role of corticosterone and HPA hyper-reactivity in epilepsy,
with studies in rats suggesting that stress accelerates kindling rate (Kumar et al., 2007) which is
associated with increased corticosterone levels (Jones et al., 2012) and maternal separation, a
model of early life stress, augments the corticosterone response to a seizure, which is reversed
by a corticosterone-synthesis antagonist, metyrapone (Koe, 2012). However, there have been
no studies to determine the effects of chronic SSRI treatment on HPA functionality in epilepsy.
This was explored in this study, in which it was found that kindling had no effect on stress-
induced corticosterone levels, but there was an effect of fluoxetine treatment, in which
fluoxetine-treated rats had a significantly reduced stress response compared to vehicle-treated
rats, consistent with previous studies showing that fluoxetine is able to normalise the increases
in corticosterone that occur after stress (Linkowski et al., 1987, Souetre et al., 1989, Steiger et
al., 1989, Pariante, 2006) however inconsistent with the notion that corticosterone may be
accelerating kindling rate in these rats during chronic SSRI treatment. Furthermore, there was
no effect of citalopram on stress-induced corticosterone levels. There have been no previous
studies to investigate the effects of chronic citalopram treatment on corticosterone levels post-
swim stress, however using other stress paradigms, studies have showed that chronic
citalopram treatment has no effect on corticosterone levels post-stress (Jensen et al., 1999,
Moncek et al., 2003, Hesketh et al., 2005), consistent with this study.

93
While the lack of difference between vehicle- and citalopram-treated groups could be due to
small sample size in the vehicle-treated group of the citalopram cohort, this factor also makes
interpretation of this data difficult. Pharmacological differences between fluoxetine and
citalopram may also explain the disparity of the results between the two SSRI treatments. While
both drugs have similar mechanisms of action, there are differences in their affinity for blocking
the serotonin transporter (Hyttel, 1994), thus having effects on serotonin concentrations and on
potential subsequent downstream effects on the HPA axis. Furthermore, the dose of citalopram
may not have been sufficient to induce alterations in the HPA axis. While doses of 10mg/kg/day,
as used in this study, have been used in previous studies and shown to increase brain
concentrations serotonin (Marsteller et al., 2007, Kanemaru et al., 2009) and to affect behaviour
(Kugelberg et al., 2002), other studies commonly use doses of 20-60mg/kg/day (Kugelberg et
al., 2001, Wegener et al., 2003, Payandemehr et al., 2012). However, due to solubility issue with
using the osmotic pumps, a dose of 10mg/kg/day was chosen. While 10mg/kg/day maybe have
been a dose sufficiently high enough to have effects on kindling, it may not have been effective
on other physiological parameters such as the HPA axis. Comparatively, the dose of fluoxetine at
10mg/kg/day was sufficiently high enough to affect kindling rate and the corticosterone
response to stress, the latter consistent with previous studies (Serra et al., 2001, Zhang et al.,
2010, Pawluski et al., 2012, Pineda et al., 2012).
In both cohorts, there was no effect of kindling on the corticosterone response to swim stress.
While previous studies implicate a role of corticosterone in epileptogenesis (Kumar et al., 2007,
Jones et al., 2012, Koe, 2012), this was not specifically investigated in this study. One previous
study found that kindling does not alter basal corticosterone levels (Adamec and McKay, 1993),
suggesting that it is unlikely that corticosterone levels were elevated throughout kindling in the
studies reported in this thesis. Therefore, it is unlikely that increases in corticosterone are a
mechanism by which SSRIs accelerate kindling rate here. Furthermore, the results of this study
suggest that fluoxetine was able to suppress the corticosterone response to stress four weeks
after kindling which could indicate that there may also have been suppression of corticosterone
throughout kindling.
One limitation of the analysis of the corticosterone response to stress in this study is the time
point of blood sampling post-stress. Blood samples were taken five minutes after swim stress,
which may not have been sufficient time for corticosterone levels to peak. Previous studies have
shown that corticosterone levels take up to 30 minutes to reach a peak post-stress (de Kloet et
al., 2008, Joels et al., 2007), and that at five minutes the difference between treatments,
particularly in the kindled versus sham-kindled rats, may not be large enough to be determined.

94
Nevertheless, analysis of corticosterone levels at 5 minutes post-stress would still be able to
give an initial indicator of any possible differences between treatment groups.
Overall, the results suggest that it is unlikely that corticosterone is mediating the accelerated
kindling rate observed in the SSRI-treated rats in this study. Future studies could investigate the
alterations in corticosterone receptors or other HPA intermediaries during kindling and SSRI
treatment, which would give a better understanding the effects of chronic SSRI treatment on the
HPA axis during kindling epileptogenesis.
4.4.7 Neurogenesis is not affected by kindling or chronic fluoxetine treatment
In this study, there were no differences in the number of BrdU-positive cells between any
treatment groups of the fluoxetine cohort. This is opposite to what was hypothesised, as it was
expected that there would be increases in neurogenesis in response to kindling and during
fluoxetine treatment, which has been shown to occur in previous studies (Boldrini et al., 2009,
Malberg et al, 2000, Parent et al, 1998). This may be because not all rats experienced the same
number of class 5 seizures, which could potentially mean that neurogenesis would not have
occurred to the same extent in all rats, creating greater variability in the data. However, there
was no correlation between the number of class 5 seizures and the number of BrdU-positive
cells (data not shown). Cell death may also account for the low levels of neurogenesis observed,
as cells were labelled with BrdU immediately after kindling, however rats were not culled until
3 weeks later. Any cells counted at 3 weeks post-kindling would indicate cell survival after
kindling and chronic SSRI administration, which may not be as pronounced as cell proliferation
immediately after kindling. Future studies should investigate the differences in cell proliferation
during kindling and chronic antidepressant treatment. Additionally, BrdU administration is only
able to provide information on the number of cells that survive, and not of the integration and
functionality of the surviving cells. Future experiments should also include an investigation of
cell phenotype, with double labelling for markers of mature neuronal cells, as well as
electrophysiological investigation of the functionality of BrdU-positive cells.
4.4.8 Final conclusions
In summary, this study demonstrated that chronic treatment with the SSRIs fluoxetine and
citalopram both accelerate the progression of kindling epileptogenesis. While this may have
implications for the way in which antidepressants are considered for use in people with
epilepsy, this effect should also be established in another model of epilepsy, such as the post-
status epilepticus model. The kindling model is advantageous as it provides comparatively

95
easily attainable data on disease progression however there is no development of spontaneous
seizures and little associated epileptic pathology. As such, replicating this study in a more
clinically-relevant epilepsy model is crucial. Furthermore, while biomarkers of kindling
epileptogenesis were investigated in this study, such as the effects on behaviour, HPA
functionality and neurogenesis, no clear associations were found, and mechanisms underlying
the SSRI-induced increase in kindling vulnerability remain to be established.

96

97
CHAPTER 5: GENERAL DISCUSSION
5.1. Key findings
The high prevalence of psychiatric symptoms and syndromes in patients with epilepsy
highlights the importance of treating these disorders within this patient group. Antidepressants,
in particular SSRIs, are widely prescribed to patients with epilepsy and are administered
chronically over months or years to alleviate depressive symptoms and improve quality of life.
In recent times, studies have further emphasised the increasing need for the screening and
treatment of mood disorders in epilepsy (Kanner, 2003, Cotterman-Hart, 2010). However, most
studies, in both humans and animals, have limited their investigations to short-term seizure
outcomes following short-term SSRI treatment periods; little data is available on the effects of
chronic SSRI treatment on epileptogenesis. These types of studies are essential for a
comprehensive investigation of the effects of antidepressants on epilepsy, as increasing
evidence suggests that epilepsy is a progressive disorder (Pitkanen and Sutula, 2002, Nearing et
al., 2007, Cascino, 2009) that evolves over several years (Ono and Galanopoulou, 2012). As such,
while the symptoms of the disorder, in particular the seizures, are generally controlled, the
underlying pathological mechanisms and the associated adverse consequences continue to
progress. Whether disease progression is affected by chronic SSRI treatment has not been
previously investigated. The studies in this thesis aimed to address this, by investigating the
effects of chronic fluoxetine and citalopram treatments on the progression of the epileptic
disorder in a rat kindling model of epileptogenesis.
The results presented in Chapter 3 showed that fluoxetine administered by osmotic pumps was
an effective method of drug delivery for the experimental time period and that fluoxetine
treatment had measurable effects in this model. Osmotic pumps were an advantageous method
of drug delivery as they negated the need for repeated daily handling and injections during the
kindling experiments, which in itself may have affected kindling epileptogenesis. These studies
also confirmed that fluoxetine was delivered at the appropriate concentration at the end of the
experimental time period and that appropriate plasma levels were attained. Contrary to
expectations, fluoxetine treatment also significantly increased depressive-like behaviours, and
there was a trend for an effect on anxiety-like behaviour and if sample sizes had been larger,
likely would have also significantly suppressed the corticosterone response to stress and
increased dentate gyrus neurogenesis. These results indicated that fluoxetine had biological
effects in rodents that have also been previously reported.

98
The effects of chronic SSRI treatment on epileptogenesis, using fluoxetine, and a more clinically
relevant SSRI, citalopram, were described in Chapter 4. The effects on some of the common
neurobiological substrates of SSRI treatment and epileptogenesis were also investigated. The
results of this study demonstrated that chronic treatment with fluoxetine or citalopram had a
proepileptogenic effect in the kindling model: fluoxetine- and citalopram-treated rats displayed
accelerated kindling rates, reaching a more severe stage of seizures earlier than those in the
vehicle-treated group. This was associated with an accelerated increase in seizure duration.
These results indicated that chronic fluoxetine and citalopram treatments accelerated the
progression of epileptogenesis. This is inconsistent with the few previous studies that employed
comparable paradigms to this study, with most studies reporting that antidepressants were
anticonvulsant, or had no effect on seizures. However, as discussed in Section 4.4.1, a majority
of these studies only administered antidepressants acutely. Importantly, seizure threshold (the
point at which the brain becomes susceptible to seizures) was not significantly affected by
chronic fluoxetine or citalopram treatments, which is consistent with previous studies as
discussed in Section 4.4.3, suggesting a truly disease-modifying intervention (as opposed to
changing excitability or being “proconvulsive” as has been reported for tricyclic
antidepressants). Overall, this data indicates that while excitability within the brain is not
affected by chronic SSRI treatment, the disorder itself progresses at a faster rate.
The original hypothesis of this study was that SSRIs would retard the rate of kindling
epileptogenesis, and in doing so, would also limit the behavioural and neuroendocrinological
consequences of kindling. After evaluating the proepileptogenic effects on kindling, one would
anticipate that the other consequences of kindling would likewise be exacerbated. However, in
investigating these associated alterations few conclusions could be drawn from this data to
explain the accelerated kindling rate during SSRI treatment. None of the common alterations
investigated showed an effect of kindling, likely due to the time point of analysis. These
behavioural and neuroendocrinological alterations were investigated after the completion of
kindling, and not during the kindling period. As such, it is only possible to speculate upon how
each of these factors may be affected by SSRI treatment in epileptogenesis.
The behavioural outcomes of this study found that, kindling itself had some effect on anxiety-
like behaviours but no effect on depressive-like behaviours. In the fluoxetine cohort, kindled
rats displayed an increase in anxiety-like behaviour compared to sham-kindled rats, with no
difference between kindled and sham-kindled rats in depressive-like behaviours. However, this
was not replicated in the citalopram cohort. Neither fluoxetine nor citalopram treatments
affected anxiety- or depressive-like behaviours. However, fluoxetine treatment was able to

99
suppress the corticosterone response to stress, with a trend towards such a suppression also
seen after citalopram treatment, while there was no effect of kindling in any treatment group.
Similar to the behavioural tests, these data were analysed after the completion of kindling, and
as such cannot implicate corticosterone in the accelerated kindling rate in this study. These
results suggest that even though kindling itself did not have long-term effects on corticosterone
release following a stressor, the stress response was still successfully suppressed by fluoxetine
treatment. Although an effect of fluoxetine was found in the corticosterone analysis, no effect of
either SSRI treatment or kindling was found in BrdU cell counts. However, the lack of effect is
likely due to methodological limitations, as discussed in Chapter 4.
In summary, the common neurobiological alterations of kindling and SSRI treatment
investigated in this study were not significantly affected by kindling at the time point analysed,
therefore their effects cannot be implicated as associative or causal factors to fluoxetine or
citalopram accelerating kindling epileptogenesis in this study. However, this does not exclude
the possibility that these alterations may still play a role in the effect of SSRIs on kindling
epileptogenesis, and will need to be investigated with further studies, as described below
(Section 5.5).
5.2. Potential mechanisms by which fluoxetine and citalopram accelerated
kindling rate
A number of possible mechanisms could explain the proepileptogenic effect of fluoxetine and
citalopram seen in this study. Particularly relevant to the SSRIs would be effects on serotonin
levels, serotonergic neurotransmission and receptor alterations during kindling. However,
increases in synaptic serotonin levels following SSRI administration are not the sole mechanism
by which SSRIs exert their effects and other downstream alterations may also be involved.
Furthermore, concentrations within the nanomolar range are required for blockade of the
serotonin transporter by SSRIs (Owens et al., 1997) while in humans, therapeutic
concentrations of SSRIs in the brain can range from 5-30uM (Karson et al., 1993, Bolo et al.,
2000). Therefore, at these much higher concentrations, fluoxetine and indeed other SSRIs are
unlikely to be specific for SERT and may have off-target effects (Bianchi and Botzolakis, 2010).
As reviewed in Chapter 1 (section 1.5), there are a number of common neurobiological
substrates between SSRI treatment and epileptogenesis, and any number of these may have
been contributing to the proepileptogenic effects observed here. Potential mechanisms by

100
which each of these factors may be contributing to the accelerated kindling rate are discussed
individually below however it is possible that the results found in this thesis are due to a
combination of factors.
5.2.1 Serotonin receptor alterations
Since serotonergic alterations have been implicated in epileptogenesis (Bonnycastle et al., 1957,
Lowenstein, 1996, Bagdy et al., 2007) and the primary pharmacological action of SSRIs is to
increase synaptic serotonin levels, it is reasonable to suggest a role for serotonin in the
proepileptogenic effects of SSRIs seen in this study. Previous studies have shown that 5HT1A
(Sarnyai et al., 2000), 5HT2C (Przegalinski et al., 1994, Tecott et al., 1995, Brennan et al., 1997,
Upton et al., 1998, Isaac, 2005) and 5HT3 (Wada et al., 1997, Payandemehr et al., 2012)
receptors are particularly relevant to epilepsy while these receptors are also affected by SSRI
treatment.
Previous studies have shown that 5HT1A receptor knockout mice have a reduced seizure
threshold (Sarnyai et al., 2000) and display increased mortality following administration of the
chemoconvulsant kainic acid (Parsons et al., 2001). Similarly, 5HT1A receptor antagonists have
been shown to increase seizure severity and duration (Watanabe et al., 2000; Pericic et al.,
2005), while agonists are protective against induced seizures (Hagan et al., 1995, Gariboldi et
al., 1996b). Autoradiographic analysis of serotonin receptors in the fully kindled rat brain
showed a selective increase in 5HT1A receptor binding in the dentate gyrus, indicating that
5HT1A receptors may increase in order to inhibit seizure activity, particularly in the
hippocampus (Clark et al., 1993). In addition to this, an established delayed effect of SSRIs is a
desensitisation of the presynaptic 5HT1A autoreceptors (Charney et al., 1981, Charney et al.,
1991, Stahl, 1992, Stahl, 1994, Leonard, 1996, Stahl, 1998). Therefore, if kindling leads to an
increase in 5HT1A receptor levels as a compensatory mechanism to inhibit and protect against
seizures, but chronic SSRI treatment is desensitising these 5HT1A receptors, this desensitisation
may be contributing to a proconvulsant effect of fluoxetine and citalopram.
Similar to 5HT1A receptors, 5HT2C receptor knockout mice are more susceptible to audiogenic
seizures and are prone to spontaneous death from seizures (Tecott et al., 1995, Brennan et al.,
1997, Applegate and Tecott, 1998). This suggests that 5HT2C receptors are necessary to
suppress neuronal excitability and seizure activity. Previous studies have also shown that
fluoxetine is a potent antagonist of the 5HT2C receptor, (Maj and Moryl, 1993, Kennett et al.,
1994, Palvimaki et al., 1996, Stahl, 1996, Ni and Miledi, 1997) therefore if fluoxetine is acting to
inhibit the 5HT2C receptor, this may be contributing to its proconvulsant effect, as found in this

101
study. This action has not been reported with citalopram treatment, and as such cannot explain
the proepileptogenic effect observed with citalopram. However, as discussed in Chapter 4,
citalopram may mediate a proconvulsant effect through other mechanisms. This has been
suggested by Payandemehr et al. (2012) where it was found that the proconvulsant activity of
citalopram was mediated through a modulatory role on nitric oxide, also shown in other studies
(De Sarro et al., 1991, Osonoe et al., 1994, Gholipour et al., 2010, Bahremand et al., 2011).
Therefore, although only speculative, it may be that functional or expression alterations in
5HT1A and 5HT2C receptors, as well as modulation of nitric oxide, may be involved in
accelerating kindling rate during chronic fluoxetine and citalopram treatments.
5.2.2 Alterations in other aspects of neuroplasticity
Although no alterations in neurogenesis were found in this study, it is possible that subtle
alterations in other neuroplastic mechanisms may be involved in the actions of SSRIs on the
kindled brain. There is considerable evidence implicating a role for aberrant hippocampal
plasticity, which also includes neurogenesis, in acquired epilepsies and epileptogenesis
(Bengzon et al., 1997, Parent et al., 1997, Hattiangady et al., 2004, Jakubs et al., 2006, Parent,
2007, Scharfman et al., 2007, Hattiangady and Shetty, 2008, Kron et al., 2010, Murphy et al.,
2011) however the results of this study do not suggest aberrant hippocampal plasticity
occurred following kindling. Furthermore, there was no effect of fluoxetine on neurogenesis,
which is contrary to reports from previous studies (Malberg et al., 2000, Santarelli et al., 2003,
Encinas et al., 2006, Boldrini et al., 2009, Kobayashi et al., 2010, Karpova et al., 2011). This
indicates that either there were methodological limitations in detection of BrdU-positive cells
(see Section 4.4.7 in Chapter 4), or that other, more subtle, alterations occurred that were not
detected in the experiments presented here.
Previous studies have found that SSRIs have effects on the functional characteristics of newborn
cells, rather than solely affecting newborn cell number. There have been two recent studies that
have shown SSRI treatment reversed the maturation of a large proportion of dentate granule
cells into cells with immature neuronal properties (Kobayashi et al., 2010; Karpova et al., 2011).
Reversing the maturational stage of dentate granule cells may result in reduced synaptic
facilitation from the dentate gyrus to the CA3 region. While this may potentially reduce seizures,
it was not specifically investigated in these studies and the function of these immature cells is
still speculative. It is possible that these immature cells may be rendered hyperexcitable, and as
such contribute to seizures rather than suppress them. While this was not specifically
investigated in the studies presented in this thesis, it may be possible that the functionality of
newborn cells following kindling and SSRI treatment was altered. However, in order to confirm

102
this, newborn cell function using electrophysiological techniques would need to be employed to
determine such an effect.
5.2.3 Modulation of excitatory and inhibitory neurotransmission
Alterations in excitatory and inhibitory neurotransmission are also potential candidates to
explain the effects of SSRIs on epileptogenesis. There are a small number of studies that suggest
antidepressants may influence the gamma-aminobutyric acid (GABA) system (Krystal et al.,
2002) and enhance GABA activity (Robinson et al., 2003, Ugale et al., 2004). Other studies have
also shown that SSRIs can increase (Sanacora et al., 2002) or decrease (Cuenca et al., 2004,
Isbister et al., 2004) the concentration of GABA in the brain, which could have potential effects
on synaptic transmission during a seizure. However, most studies investigating the effects of
SSRIs on GABAergic neurotransmission generally use fluoxetine and hence it is unclear whether
such results are common to all SSRIs.
A study by Choi et al. (2010) specifically investigated the effects of various SSRIs, including
fluoxetine and citalopram, on vesicular GABA transporter (VGAT) immunoreactivity in normal
and epileptic rats. It was found that overall VGAT immunoreactivity was increased in the
hippocampus of epileptic but not normal rats, indicating a compensatory mechanism in the
epileptic hippocampus to increase inhibitory neurotransmission. Then, following SSRI
treatment, VGAT immunoreactivity was decreased in both normal and epileptic hippocampi,
which suggests that chronic SSRI treatment reduces GABAergic inhibitory neurotransmission,
which could potentially increase seizure susceptibility in normal brains, and increase seizure
frequency in epileptic brains.
Other studies have shown that high levels of monoamines, in particular dopamine and
serotonin, promoted glutamate release which may potentially lead to an increase in neuronal
excitability and proconvulsant effects (Clinckers et al., 2004b, 2005). However, the same studies
have also shown that the sustained increase in monoamine levels also contributed to an
anticonvulsant effect. These studies suggest that other factors, besides the effects on
monoamine levels, also contribute to the effects that monoamines are having on
neurotransmission.
5.2.4 Overall conclusion of potential mechanisms
As previously stated, currently we can only speculate on possible mechanisms for the
accelerated kindling rate found in SSRI-treated rats in this study. The studies supporting a
proepileptogenic effect of various mechanisms are few and it is likely that a combination of

103
factors is involved. Further studies will be required to elucidate the mechanisms by which
chronic SSRI treatment accelerates kindling epileptogenesis.
5.3. The potential clinical implications of an accelerated rate of
epileptogenesis
An accelerated rate of epileptogenesis during SSRI treatment potentially has significant
implications for the treatment of patients with epilepsy. While these outcomes discussed below
are speculative, they are outcomes that have been reported to manifest in patients, and as such
are extrapolated from these data. The results of this study suggest that although the threshold at
which a seizure occurs may not be affected in a patient taking SSRIs, the underlying disorder
itself may develop or progress more rapidly. If this is the case, it is possible that the adverse
alterations that occur during epileptogenesis may manifest at an earlier stage as compared to
patients with epilepsy who are not taking SSRIs. This is discussed in more detail below.
In animal studies, the alterations that occur in association with epileptogenesis, as discussed in
Section 1.2.4, are changes which have been shown to develop in the months after the initial
insult (Tanaka et al., 1992, Hellier et al., 1998, Kharatishvili et al., 2006) while in humans this
can take years (French et al., 1993, Mathern et al., 1995). Current antiepileptic drugs (AED) are
only able to suppress the symptoms of the disorder, i.e. the seizures, and no medications are
currently able to prevent epileptogenesis in humans. While several studies in animals have
shown that AEDs can delay the process of kindling (Loscher 1998a, b, 1999), this has not been
shown to occur in people with epilepsy, whereby AEDs could slow the progression of
epileptogenesis. In fact, in many patients with epilepsy, while the seizures are being managed,
the underlying disorder itself may continue to progress. That epilepsy is a progressive disorder
is becoming more accepted, particularly for TLE (O'Brien et al., 1999, Tasch et al., 1999,
Bouilleret et al., 2000, Cole, 2000, Pitkanen and Sutula, 2002, Nearing et al., 2007, Cascino, 2009,
Yang et al., 2010) and various alterations have been shown to occur in association with
epileptogenesis, which are discussed below. While there is some data to support the association
of these changes with epileptogenesis, the way in which the underlying neurobiology is altered
during epileptogenesis is undiscovered, and therefore such alterations may be affected by
antidepressants in ways that are yet unclear. Additionally, it is unclear whether these changes
occur in all patients, and whether the severity of these alterations is increasing in patients.
Hence, the way in which SSRIs may affect these alterations is speculative and requires more
thorough investigation.

104
There are a number of potential clinical implications of an accelerated rate of epileptogenesis.
Firstly, treatment with SSRIs for a comorbid mood disorder may increase the risk that a person
develops epilepsy after a brain insult, such as traumatic brain injury (TBI). Secondly, an
increased severity and duration, as well as frequency, of seizures associated with accelerated
disease progression (Sillanpaa et al., 1998, Kwan and Sander, 2004, Shorvon and Luciano, 2007)
may increase the chance of physical injury from a seizure, while also leading to decreased
quality of life from a loss of independence, both of which can affect employment and family life.
In addition to this, as seizures become more frequent and severe, AEDs may need to be altered
or increased, which in themselves have side effects. With progression of the disorder, there may
also be the possibility of drug-resistance (Kwan and Brodie, 2000, Arts et al., 2004, Mohanraj
and Brodie, 2006, Berg et al., 2009, Kwan et al., 2010). Drug-resistance is also of great concern
to patients, as seizures cannot be managed with conventional medication. While there are other
options for seizure management such as dietary manipulations (Bergqvist et al., 2005, Neal et
al., 2008), more invasive measures may also need to be considered, such as surgical removal of
the epileptogenic region (Zentner et al., 1995). On the other hand, increased rates of anxiety and
a history of depression have been found to be associated with an increased risk of developing
seizure recurrence in newly treated epilepsy (Hitiris et al., 2007, Petrovski et al., 2010) and
following epilepsy surgery (Harden et al., 2007, Kanner, 2007). Therefore, it is possible that in
patients with mood disorders the pharmacological effects of SSRIs to enhance epileptogenesis
may be balanced by reducing the proepileptogenic effect of mood disorders.
Other potential consequences an accelerated progression of epileptogenesis includes an
increased severity of psychiatric comorbidities (Hermann et al., 2008), or the emergence of
comorbidities that previously were not present. This can include anxiety and depression, as well
as psychoses, memory impairments or cognitive decline (Kwan and Brodie, 2001, Motamedi and
Meador, 2003, Andersson-Roswall et al., 2010). In fact, previous studies have shown that
cognitive impairment correlated with seizure severity. Therefore if seizures become
progressively worse as epileptogenesis progresses this may greatly impact upon cognitive
functioning (Beume and Steinhoff, 2010, Ono and Galanopoulou, 2012). While treatment with
SSRIs may at the same time treat these psychiatric and cognitive symptoms, management of
these symptoms may also become more difficult as epileptogenesis progresses.
There may also be a progressive increase in neuropathological alterations, changes that have
been consistently observed in the brain tissue of patients with epilepsy post-mortem, and in
animal models of epilepsy. This can include neuronal cell loss (Armstrong, 1993, de Lanerolle et
al., 2003, Sharma et al., 2007, Bae et al., 2010, Zheng et al., 2011) which may result from

105
neuronal damage occurring due to excessive glutamate release during prolonged seizures
(Naegele, 2007); gliosis (Feindel et al., 1952, Feindel and Penfield, 1954, Pitkanen and Sutula,
2002, Sharma et al., 2007, Shapiro et al., 2008) which has been shown to affect neuronal
excitability (Pitkanen and Sutula, 2002, Vezzani et al., 2008) and potentially contribute to the
development of epilepsy; synaptic remodelling and formation of aberrant circuitry, possibly as a
result of mossy fibre sprouting which can lead to a hyperexcitable hippocampus (Houser et al.,
1990, Mathern et al., 1995, Buckmaster and Dudek, 1999, Gorter et al., 2001, Sutula, 2002). It is
possible that these neuropathological changes may also be affected by SSRI treatment, which
themselves have been shown to affect neurogenesis and synaptic plasticity (Malberg et al., 2000,
Kobayashi et al., 2010, Kron et al., 2010, Murphy et al., 2011). However, how these
neuropathological alterations are affected by SSRI treatment in epileptogenesis has not been
investigated.
The alterations discussed and the ways in which they progress with SSRI treatment in epilepsy
remain speculative. However, as the literature suggests that such candidate mechanisms that
occur in epileptogenesis (for example, neuroplasticity) may be affected by antidepressants, it is
reasonable to speculate upon how such changes may progress during epileptogenesis and
antidepressant treatment. It is possible that the benefits that SSRIs are providing to patients, in
terms of managing their depressive symptoms, may outweigh any longer-term adverse effects
of SSRIs on epileptogenesis. Epileptogenesis and the alterations associated with it may not
manifest in all patients and therefore treatment of the current depressive symptoms, along with
management of seizures, is currently the most important aspect to consider.
Future studies should still investigate the effects of SSRIs on epileptogenesis in epilepsy patient
populations, which would not only include monitoring of the depressive symptoms, but also
careful monitoring of changes in seizures over time. In addition to this, changes in various
biomarkers of disease progression could also be assessed and followed over time. The search
for biomarkers of epileptogenesis is an active field of research (Pitkanen, 2010, Pitkanen and
Lukasiuk, 2011, Galanopoulou et al., 2012a, Galanopoulou et al., 2012b), and ultimately, the goal
is to target these alterations and produce truly antiepileptogenic medications, rather than
antiseizure medications (Galanopoulou et al., 2012a). Various biomarkers of epileptogenesis
have been reported, and can be followed up over time in a patient with epilepsy taking SSRIs.
One such example is the alteration in brain structures identified with neuroimaging techniques.
In rat models of TBI, in which a percentage of animals develop seizures post-injury, or post-
status epilepticus, MRI scans have shown that increases in T2 signal (a marker of tissue
abnormality) in the piriform and entorhinal cortices can predict the development of seizures

106
(Roch et al., 2002a, b, Kharatishvili and Pitkanen, 2010, Pitkanen et al., 2011). In humans,
sporadic reports suggest that early changes in MRI that are indicative of hippocampal sclerosis
occur after prolonged seizures or even prior to seizure onset (Wieshmann et al., 1997,
VanLandingham et al., 1998, Kuster et al., 2007). Therefore, monitoring alterations in
epileptogenic structures such as the hippocampus with MRI scans over time may provide
insight into disease progression. Other more sensitive and advanced neuroimaging techniques
that can detect functional alterations may also be employed, such as magnetic resonance
spectroscopy, diffusion tensor imaging or positron emission tomography (Tokumitsu et al.,
1997, Rugg-Gunn et al., 2001, Mirrione et al., 2006). Additionally, assessment of memory,
cognitive and social changes, as well as changes in neuropsychiatric symptoms over time could
also be monitored (Motamedi and Meador, 2003, Pitkanen and Lukasiuk, 2011).
5.4. Methodological considerations and limitations
The findings of this thesis suggest that epileptogenesis and the associated consequences, as
discussed above, may become more pronounced or manifest more severely in patients who are
also taking SSRIs. However, as this was investigated in an animal model of epileptogenesis,
caution must be exercised when applying these results to the human epileptic condition. This
section outlines the methodological limitations of this study that should be accounted for when
translating the findings to human disease.
5.4.1 Validity of the kindling model for comparison to epileptogenesis in humans
As outlined in Chapter 1 (Section 1.2.5), animal models need to be able to reliably and
measurably model the human condition in order for comparisons and conclusions to be drawn.
However, it should always be acknowledged that animal models do not replicate the human
disorders and should only be considered as experimental systems by which certain aspects of
disorders can be investigated where it is not possible to do so in humans. Nevertheless, animal
models of epilepsy have shown to be valuable tools to investigate epilepsy. In particular, the
kindling model shows various elements of a good animal model for the human epileptic
condition: there is the potential for the development of spontaneous seizures (Pinel and Rovner,
1978, Michalakis et al., 1998, Coulter et al., 2002, Sayin et al, 2003); similar circuitry to the
human condition is involved; similar behavioural and neuropathological alterations are
observed (Peele and Gilbert, 1992, Ebert and Loscher, 1995, Helfer et al., 1996, Bengzon et al.,
1997, Adamec, 1998, Bertram, 2007); it is responsive to AEDs (Honack and Loscher, 1989,

107
Becker et al., 1995, Amano et al., 1998, Gu et al., 2004); and importantly for this study, the model
shows elements of disease progression which can be easily monitored during controlled
stimulations.
However, the kindling model does not always appropriately model the human epileptic
condition. As summarised in Bertram (2007), there are two common concepts of
epileptogenesis in humans. Firstly, there is the process whereby following an initial insult, a
sequence of events occurs during the seizure-free latent period that increases neuronal
excitability to the point where seizures begin to spontaneously occur (as shown in Figure 1.4).
The other concept of epileptogenesis is more in parallel with the kindling model. This process is
similar to the first, in that following the first spontaneous seizure (not occurring due to a known
precipitating event), there is an enhancement of seizure strength and an engagement of wider-
ranging neuronal networks. As this occurs, seizures begin to spread and become more severe
(in terms of frequency and duration). This is comparable to the kindling model, whereby
repeated stimulations, and consequently seizures, induce an increase in the epileptic response,
beginning with focal seizures that spread and engage ipsilateral and eventually bilateral
neuronal circuits to generate seizures. However, epilepsy does not always occur via this
pathway in humans, and therefore the kindling model does not always accurately model the
human condition. Whether it is that “seizures beget seizures” (Gowers, 1881) or that underlying
changes continue to develop which lead to more seizures, is not always clear.
Additionally, although aspects of disease progression are apparent in the kindling model, it is
limited in some areas. Seizures must be induced, and do not normally arise spontaneously
(unless “overkindled”), as in post-status epilepticus models or in people with epilepsy.
Furthermore, kindled rats only model certain aspects of TLE and the associated emotional
alterations. The neuropathology associated with kindling is not as extensive or defined as that
which occurs in models of spontaneous seizures and in the human condition however these
were not investigated in this thesis. Furthermore, behavioural alterations do not always
consistently manifest in kindling models, and in most studies, specifically affect anxiety-like
behaviours, similar to what was found in this study. Additionally, as kindling requires the long-
term implantation of an electrode into the temporal lobe, damage from this electrode may
contribute to the epileptogenic process. Previous studies have shown that merely having an
electrode implanted in the amygdala can produce proepileptogenic effects (Loscher et al., 1995,
Niespodziany et al., 1999).

108
5.4.2 Normal rats versus the pathological state
In this study, rats did not display aspects of depressive-like behaviours prior to or after kindling.
Therefore, the effects of SSRIs in this setting may not be entirely applicable to a person with
both epilepsy and depression. Divergence in the effects of SSRIs in normal rats versus those
displaying aspects of depressive-like behaviours has been shown. For example, Ferrero et al.
(2005) found that naïve rats chronically treated with fluoxetine had a reduced seizure threshold
following administration of the chemoconvulsant, pentylenetetrazol (PTZ), while there was no
effect of fluoxetine on seizure threshold observed in rats exposed to the learned helplessness
paradigm, a model of depression, when treated with PTZ. Furthermore, previous studies have
shown that SSRIs have little effect on elevating mood in normal animals or healthy controls
(Gelfin et al., 1998, Barr et al., 2002, Geyer and Markou, 2002). Therefore, in this study, SSRIs
were shown to accelerate epileptogenesis in rats that were not currently depressed. Whether
the effect on kindling epileptogenesis would remain the same or differ in a rat model displaying
depressive-like behaviours has not been investigated.
A further methodological limitation of this study was the timing of drug administration.
Fluoxetine and citalopram were administered prior to kindling, to rats that were normal and
subsequently had seizures induced. While it is important to consider that a small number of
patients may be depressed prior to epilepsy onset and therefore already taking SSRIs, the more
common clinical scenario is that patients with epilepsy are administered SSRIs after epilepsy
onset to treat depressive symptoms. While it may not be common for patients to be taking SSRIs
prior to the development of epilepsy, and as such difficult to investigate in a similar fashion to
the rat studies in this thesis, it would still be an important group of patients to follow. In
addition, there are studies to suggest that there may be a bidirectional relationship between
epilepsy and depression (Tellez-Zenteno et al., 2007, Forsgren and Nystrom, 1990, Hesdorffer et
al., 2000, Hesdorffer et al., 2006, Morgan et al., 2012) and the study by Alper et al (2007) found
that antidepressants significantly reduced the rate of spontaneous seizures in depressed (non-
epileptic) patients. As antidepressants have been shown to reduce seizure occurrence, future
studies following the potential development of seizures in patients with depression treated with
SSRIs could be investigated. In addition, whether epileptogenesis would still be accelerated
when SSRIs are first administered after seizures emerge, or during the epileptic period, should
also be investigated.

109
5.4.3 Time point of analysis of common neurobiological substrates
As discussed above, kindling did not appear to affect outcomes of the behavioural tests, the
corticosterone response to stress or neurogenesis. While it is feasible that such changes may not
have been detected at the time point investigated (four weeks after kindling), previous studies
do suggest that kindling is a permanent state (Racine, 1972b, Goddard, 1969) and if any
alterations in the factors investigated did occur, that they would persist four weeks after
kindling. In particular, behavioural and HPA axis changes would likely still be present four
weeks after kindling if they were also present immediately after kindling. However, alterations
in cell proliferation may have occurred immediately after kindling (which was not specifically
investigated), while cell survival, which was investigated in this study, was not altered. In fact,
previous studies have shown that some changes occurring soon after kindling (24-72h) are not
present in the weeks after kindling, including effects on CRH mRNA, neurogenesis and gene and
protein expression (Smith et al., 1991, Nakagawa et al., 2000, Sohl et al., 2000, Beheshti et al.,
2010). As such, it cannot be ruled out that such changes may have occurred immediately after
kindling.
5.4.4 Behavioural testing and comparison to human behaviours
Depression and anxiety are heterogeneous disorders with symptoms that occur not only as
behavioural manifestations, but also as psychological, emotional, cognitive and physiological
alterations. Clinically, these disorders are assessed with various tools, including assessment and
rating scales of depression, as well as self-reporting. Mimicking these in animals is difficult, and
many aspects of the disorder do not arise in animals, such as guilt or thoughts of suicide, and
only measurable aspects of the disorder can be investigated. This is achieved with the use of
various behavioural tests, such as those used here. However despite being validated for their
ability to assess certain aspects of behaviour, the behavioural tests used in this study also have
various limitations.
Firstly, the FST was initially designed to assess the effectiveness of antidepressant medications,
rather than the test itself being able to assess depressive-like behaviours, although it is
commonly used for this purpose (Bilge et al., 2008, Mazarati et al., 2008, Homberg et al., 2011,
Xing et al., 2011). Additionally, the FST only investigates behavioural despair, a single aspect of
the depressive disorder, and therefore other aspects associated with the depressive disorder,
such as social behaviours, cognition, memory, attention and anhedonia, should also be assessed.
These are all dysfunctions that have been shown to occur as part of depressive disorders and

110
are endophenotypes that can be easily assessed in rodents and are comparable to humans
(Cryan and Mombereau, 2004).
Secondly, the EPM was first designed to assess the effectiveness of benzodiazepine anxiolytic
drugs, rather than specifically to assess anxiety-like behaviours in other paradigms. Similar to
the FST, only certain aspects of the anxiety disorder are able to be determined with the EPM:
anxiety induced by exposure to a novel environment. The full spectrum of anxiety traits that are
present in an anxiety disorder are not assessed by the EPM, and a battery of tests that probe
different aspects should also be considered including, fear-conditioning, social interactions,
sustained attention and fear-associated memory (Haller and Alicki, 2012).
It is important to study as many aspects of the disorder as possible in animal models to gain a
more comprehensive understanding of behavioural changes in relation to epilepsy. However,
this was not performed in this study as the exposure of animals to multiple procedures, some of
which may act as stressors, was considered to be a potential confounder to the main
investigations of the study.
5.5. Future directions
5.5.1 Investigating the mechanisms involved in accelerated kindling epileptogenesis
during chronic SSRI treatment
The mechanisms by which SSRIs are accelerating kindling rate need to be further investigated.
Specifically, future studies should investigate the effects on synaptic serotonin levels as well as
alterations in serotonin receptors, particularly those implicated in epileptogenesis and
antidepressant action, such as the 5HT1A and 5HT2C receptors, with a focus on expression and
function.
Various neuroplastic endpoints, besides BrdU labelling, could also be investigated. While
quantifying cell numbers is an important first step, this does not given an indication of newborn
cell functionality or connectivity. Immunohistochemistry could be carried out to determine cell
phenotype, such as staining with NeuN for mature neurons. In addition to this, BrdU and other
thymidine analogues could be administered at different time points throughout kindling for
analysis by immunohistochemistry, with each thymidine analogue giving an indication of
neurogenesis at different time points.

111
The functionality and connectivity of newborn cells could also be investigated using
electrophysiological techniques. This will allow for determination of whether the cells are
functionally integrated or aberrantly connected, and whether this is contributing to the
proepileptogenic effect observed. This should also be investigated throughout kindling to
determine the changes occurring throughout epileptogenesis, in addition to the same time point
investigated in this study (four weeks after kindling) in order to give an indication of cell
survival. Investigating each of these intermediaries at the appropriate time points will give
some indication as to their effects and/or roles in epileptogenesis.
5.5.2. Investigating the effects of SSRIs in a chronic epilepsy model
The post-status epilepticus model could be used to confirm the result found in this study. This
model is advantageous as there is development of spontaneous seizures, which can be
monitored over a period of months to determine the effects of chronic SSRI treatment over time.
SSRIs could be administered immediately after status epilepticus, and treatment would need to
continue throughout the latent period, prior to the emergence of spontaneous seizures. In this
model, the latency to spontaneous seizure occurrence, as well as seizure severity and frequency
could be monitored. Another option may be to administer SSRIs at other time points that are
clinically relevant, such as after the emergence of spontaneous seizures, to investigate the
effects of chronic SSRI treatment on the seizure severity and the progression of epileptogenesis
in the established disorder. Behavioural alterations could also be investigated, both prior to the
emergence of spontaneous seizures and once seizures emerge, to better determine the effects of
SSRI treatment upon epilepsy-related behaviours.
5.5.3 Investigating the effects of SSRIs in a model of epilepsy and depression
As discussed above, the effects of SSRIs on a normal brain as compared to one in a disease state
can differ. Therefore, investigating the effects of SSRIs on epileptogenesis in a model of
depression would be more relevant. A model of social defeat or unpredictable chronic mild
stress, where the socio-environmental factors that contribute to depression will induce the
depressive-like behaviours, could be used. In these models, SSRI treatment and kindling could
also be completed in a similar fashion to what was done in this study.
Furthermore, as most previous clinical studies are limited to investigating the effects of SSRI on
seizures in patients with epilepsy already taking AED medication (which, ethically, cannot be
stopped) investigating the effects in an animal model in which AEDs are also administered

112
should also be considered. The rats in this study were not being administered AEDs and the
AEDs themselves may be interacting with SSRIs. This in itself may be having effects on
epileptogenesis. Whether the effect is the same in an animal model in which AEDs are
concurrently administered is unknown.
5.5.4 Clinical directions
Future clinical studies could follow patients with epilepsy being treated with SSRIs over time,
and monitor the progression of the epileptic symptoms. One possible study could be to
investigate this in a population of patients that are susceptible to developing epilepsy, such as in
patients following TBI. Studies have shown that up to 86% of patients may develop epilepsy in
the days or months after TBI (Haltiner et al., 1997, Annegers et al., 1998). If these patients could
be stratified based on treatment with SSRIs and compared to an untreated group, the delay to
which seizures manifest could be investigated. However, this may not be ethically acceptable
based on the results of this study, which suggested that SSRIs accelerated epileptogenesis.
Therefore, this could be investigated retrospectively, taking data from TBI patients who were
administered SSRIs and those who were not and comparing the development of epilepsy in
these patients. However, patient follow up, as discussed above in section 5.3, should be
monitored closely to determine disease progression. Similarly, retrospective analysis of data
indicative of disease progression could be performed, comparing SSRI and non-SSRI treated
patients with epilepsy, rather than initiating a new study which would not only be costly and
time consuming, but also raise various ethical concerns.
5.6. Final conclusions
The findings of this thesis challenge the belief that SSRIs can be safely used in patients with
epilepsy. It was found here that although SSRI treatments were not proconvulsant, chronic SSRI
treatment was associated with acceleration in the progression of epileptogenesis. This thesis
highlights the need to investigate the progression of the epileptic disorder over time, rather
than just examine symptomatic endpoints. In addition, chronic epilepsy models should be used
to determine the effects of SSRIs in epilepsy, rather than solely using acute seizure tests, given
that acute models do not exhibit the underlying alterations associated with epileptogenesis.
Although a chronic model of epileptogenesis was used in this study, none of the common
alterations investigated could account for the acceleration of kindling rate seen with fluoxetine
and citalopram treatment. This is possibly due to the time points chosen for analysis and

113
therefore investigation of these alterations at appropriate time points may give a better
indication of whether or not these factors (either alone, or in combination with each other) are
contributing to the effects of SSRIs in epileptogenesis. Clearly, it is essential to treat the
depressive symptoms that manifest in people with epilepsy; however whether these
medications affect the course of epilepsy and how they may do so should become priority areas
for future research.

114

115
REFERENCES
(2004). Australian code of practice for the care and use of animals for scientific purposes. National Health and Medical Research Council (NHMRC), Australia.
Adamec R (1998). Amygdala kindling and rodent anxiety. New York: Plenum.
Adamec R, Blundell J, Burton P (2004). Anxiolytic effects of kindling role of anatomical location of the kindling electrode in response to kindling of the right basolateral amygdala. Brain Res 1024:44-58.
Adamec R, Shallow T, Burton P (2005). Anxiolytic and anxiogenic effects of kindling--role of baseline anxiety and anatomical location of the kindling electrode in response to kindling of the right and left basolateral amygdala. Behav Brain Res 159:73-88.
Adamec R, Young B (2000). Neuroplasticity in specific limbic system circuits may mediate specific kindling induced changes in animal affect-implications for understanding anxiety associated with epilepsy. Neurosci Biobehav Rev 24:705-723.
Adamec RE (1990). Does kindling model anything clinically relevant? Biol Psychiatry 27:249-279.
Adamec RE, McKay D (1993). Amygdala kindling, anxiety, and corticotrophin releasing factor (CRF). Physiol Behav 54:423-431.
Adamec RE, Morgan HD (1994). The effect of kindling of different nuclei in the left and right amygdala on anxiety in the rat. Physiol Behav 55:1-12.
Adams SJ, O'Brien TJ, Lloyd J, Kilpatrick CJ, Salzberg MR, Velakoulis D (2008). Neuropsychiatric morbidity in focal epilepsy. Brit J Psychiat 192:464-469.
Ago J, Ishikawa T, Matsumoto N, Ashequr Rahman M, Kamei C (2006). Mechanism of imipramine-induced seizures in amygdala-kindled rats. Epilepsy Res 72:1-9.
Ago J, Ishikawa T, Matsumoto N, Rahman MA, Kamei C (2007). Epileptiformic activity induced by antidepressants in amygdala-kindled rats. Eur J Pharmacol 560:23-28.
Ahern TH, Javors MA, Eagles DA, Martillotti J, Mitchell HA, Liles LC, et al. (2006). The effects of chronic norepinephrine transporter inactivation on seizure susceptibility in mice. Neuropsychopharmacology 31:730-738.
Ahmadzadeh R, Saboory E, Roshan-Milani S, Pilehvarian AA (2011). Predator and restraint stress during gestation facilitates pilocarpine-induced seizures in prepubertal rats. Dev Psychobiol 53:806-812.
Alesci S, Martinez PE, Kelkar S, Ilias I, Ronsaville DS, Listwak SJ, et al. (2005). Major depression is associated with significant diurnal elevations in plasma interleukin-6 levels, a shift of its circadian rhythm, and loss of physiological complexity in its secretion: clinical implications. J Clin Endocrinol Metab 90:2522-2530.
Alonso-Deflorida F, Delgado JM (1958). Lasting behavioral and EEG changes in cats induced by prolonged stimulation of amygdala. Am J Physiol 193:223-229.

116
Alper K, Schwartz KA, Kolts RL, Khan A (2007). Seizure incidence in psychopharmacological clinical trials: an analysis of Food and Drug Administration (FDA) summary basis of approval reports. Biol Psychiatry 62:345-354.
Altar CA, Whitehead RE, Chen R, Wortwein G, Madsen TM (2003). Effects of electroconvulsive seizures and antidepressant drugs on brain-derived neurotrophic factor protein in rat brain. Biol Psychiatry 54:703-709.
Altieri M, Marini F, Arban R, Vitulli G, Jansson BO (2004). Expression analysis of brain-derived neurotrophic factor (BDNF) mRNA isoforms after chronic and acute antidepressant treatment. Brain Res 1000:148-155.
Altman J (1962). Are new neurons formed in the brains of adult mammals? Science 135:1127-1128.
Altman J, Das GD (1965). Autoradiographic and histological evidence of postnatal hippocampal neurogenesis in rats. The Journal of comparative neurology 124:319-335.
Altshuler L, Rausch R, Delrahim S, Kay J, Crandall P (1999). Temporal lobe epilepsy, temporal lobectomy, and major depression. J Neuropsychiatry Clin Neurosci 11:436-443.
Amano K, Hamada K, Yagi K, Seino M (1998). Antiepileptic effects of topiramate on amygdaloid kindling in rats. Epilepsy Res 31:123-128.
Amaral DG, Insausti R, Cowan WM (1984). The commissural connections of the monkey hippocampal formation. The Journal of comparative neurology 224:307-336.
Amaral DG, Insausti R, Cowan WM (1987). The entorhinal cortex of the monkey: I. Cytoarchitectonic organization. The Journal of comparative neurology 264:326-355.
Ambrogini P, Lattanzi D, Ciuffoli S, Agostini D, Bertini L, Stocchi V, et al. (2004). Morpho-functional characterization of neuronal cells at different stages of maturation in granule cell layer of adult rat dentate gyrus. Brain Res 1017:21-31.
Andersson-Roswall L, Engman E, Samuelsson H, Malmgren K (2010). Cognitive outcome 10 years after temporal lobe epilepsy surgery: a prospective controlled study. Neurology 74:1977-1985.
Annegers JF, Hauser WA, Coan SP, Rocca WA (1998). A population-based study of seizures after traumatic brain injuries. N Engl J Med 338:20-24.
Applegate CD, Tecott LH (1998). Global increases in seizure susceptibility in mice lacking 5-HT2C receptors: A behavioral analysis. Experimental Neurology 154:522-530.
Arai S, Morita K, Kitayama S, Kumagai K, Kumagai M, Kihira K, et al. (2003). Chronic inhibition of the norepinephrine transporter in the brain participates in seizure sensitization to cocaine and local anesthetics. Brain Res 964:83-90.
Armstrong DD (1993). The neuropathology of temporal lobe epilepsy. J Neuropathol Exp Neurol 52:433-443.
Aronica E, Boer K, van Vliet EA, Redeker S, Baayen JC, Spliet WG, et al. (2007). Complement activation in experimental and human temporal lobe epilepsy. Neurobiol Dis 26:497-511.

117
Arts WF, Brouwer OF, Peters AC, Stroink H, Peeters EA, Schmitz PI, et al. (2004). Course and prognosis of childhood epilepsy: 5-year follow-up of the Dutch study of epilepsy in childhood. Brain 127:1774-1784.
Asmussen SB, Kirlin KA, Gale SD, Chung SS (2009). Differences in self-reported depressive symptoms between patients with epileptic and psychogenic nonepileptic seizures. Seizure 18:564-566.
Asztely F, Kokaia M, Olofsdotter K, Ortegren U, Lindvall O (2000). Afferent-specific modulation of short-term synaptic plasticity by neurotrophins in dentate gyrus. Eur J Neurosci 12:662-669.
Aydemir C, Yalcin ES, Aksaray S, Kisa C, Yildirim SG, Uzbay T, et al. (2006). Brain-derived neurotrophic factor (BDNF) changes in the serum of depressed women. Prog Neuropsychopharmacol Biol Psychiatry 30:1256-1260.
Aydemir O, Deveci A, Taneli F (2005). The effect of chronic antidepressant treatment on serum brain-derived neurotrophic factor levels in depressed patients: a preliminary study. Prog Neuropsychopharmacol Biol Psychiatry 29:261-265.
Babb TL (2001). Pathology of the temporal lobe: hippocampal sclerosis. (H.O., L. and G., C. Y., eds), pp 901-906 Philadelphia: Lippincott Williams & Wilkins.
Babu CS, Satishchandra P, Sinha S, Subbakrishna DK (2009). Co-morbidities in people living with epilepsy: hospital based case-control study from a resource-poor setting. Epilepsy Res 86:146-152.
Bae EK, Jung KH, Chu K, Lee ST, Kim JH, Park KI, et al. (2010). Neuropathologic and clinical features of human medial temporal lobe epilepsy. J Clin Neurol 6:73-80.
Bagdy G, Kecskemeti V, Riba P, Jakus R (2007). Serotonin and epilepsy. J Neurochem 100:857-873.
Bahremand A, Payandemehr B, Rahimian R, Ziai P, Pourmand N, Loloee S, et al. (2011). The role of 5-HT(3) receptors in the additive anticonvulsant effects of citalopram and morphine on pentylenetetrazole-induced clonic seizures in mice. Epilepsy Behav 21:122-127.
Ball JR, Kiloh LG (1959). A controlled trial of imipramine in treatment of depressive states. British medical journal 2:1052-1055.
Ban TA (2001). Pharmacotherapy of depression: a historical analysis. J Neural Transm 108:707-716.
Barker EL, Blakely RD (1995). Norepinephrine and serotonin transporters: molecular targets of antidepressant drugs. In: Psychopharmacology: The Fourth Generation of Progress(Bloom, F. E. and Kupfer, D. J., eds), pp 321–334 New York: Raven Press.
Barr AM, Markou A, Phillips AG (2002). A 'crash' course on psychostimulant withdrawal as a model of depression. Trends Pharmacol Sci 23:475-482.
Barry JJ, Ettinger AB, Friel P, Gilliam FG, Harden CL, Hermann B, et al. (2008). Consensus statement: the evaluation and treatment of people with epilepsy and affective disorders. Epilepsy Behav 13 Suppl 1:S1-29.

118
Becker A, Grecksch G, Brosz M (1995). Antiepileptic drugs--their effects on kindled seizures and kindling-induced learning impairments. Pharmacol Biochem Behav 52:453-459.
Beheshti S, Sayyah M, Golkar M, Sepehri H, Babaie J, Vaziri B (2010). Changes in hippocampal connexin 36 mRNA and protein levels during epileptogenesis in the kindling model of epilepsy. Prog Neuropsychopharmacol Biol Psychiatry 34:510-515.
Bengzon J, Kokaia Z, Elmer E, Nanobashvili A, Kokaia M, Lindvall O (1997). Apoptosis and proliferation of dentate gyrus neurons after single and intermittent limbic seizures. Proc Natl Acad Sci U S A 94:10432-10437.
Berg AT (2008). The natural history of mesial temporal lobe epilepsy. Curr Opin Neurol 21:173-178.
Berg AT, Levy SR, Testa FM, D'Souza R (2009). Remission of epilepsy after two drug failures in children: a prospective study. Ann Neurol 65:510-519.
Berg AT, Scheffer IE (2011). New concepts in classification of the epilepsies: entering the 21st century. Epilepsia 52:1058-1062.
Bergqvist AG, Schall JI, Gallagher PR, Cnaan A, Stallings VA (2005). Fasting versus gradual initiation of the ketogenic diet: a prospective, randomized clinical trial of efficacy. Epilepsia 46:1810-1819.
Bertram E (2007). The relevance of kindling for human epilepsy. Epilepsia 48 Suppl 2:65-74.
Bessa JM, Ferreira D, Melo I, Marques F, Cerqueira JJ, Palha JA, et al. (2009). The mood-improving actions of antidepressants do not depend on neurogenesis but are associated with neuronal remodeling. Mol Psychiatry 14:764-773, 739.
Beume LA, Steinhoff BJ (2010). Long-term outcome of difficult-to-treat epilepsy in childhood. Neuropediatrics 41:135-139.
Bianchi MT, Botzolakis EJ (2010). Targeting ligand-gated ion channels in neurology and psychiatry: is pharmacological promiscuity an obstacle or an opportunity? BMC Pharmacol 10:3.
Bilge S, Bozkurt A, Bas DB, Aksoz E, Savli E, Ilkaya F, et al. (2008). Chronic treatment with fluoxetine and sertraline prevents forced swimming test-induced hypercontractility of rat detrusor muscle. Pharmacol Rep 60:872-879.
Boldrini M, Underwood MD, Hen R, Rosoklija GB, Dwork AJ, John Mann J, et al. (2009). Antidepressants increase neural progenitor cells in the human hippocampus. Neuropsychopharmacology 34:2376-2389.
Bolo NR, Hode Y, Nedelec JF, Laine E, Wagner G, Macher JP (2000). Brain pharmacokinetics and tissue distribution in vivo of fluvoxamine and fluoxetine by fluorine magnetic resonance spectroscopy. Neuropsychopharmacology 23:428-438.
Bonnycastle DD, Giarman NJ, Paasonen MK (1957). Anticonvulsant compounds and 5-hydroxytryptamine in rat brain. British journal of pharmacology and chemotherapy 12:228-231.
Borowicz KK, Furmanek-Karwowska K, Sawicka K, Luszczki JJ, Czuczwar SJ (2007). Chronically administered fluoxetine enhances the anticonvulsant activity of conventional

119
antiepileptic drugs in the mouse maximal electroshock model. Eur J Pharmacol 567:77-82.
Borowicz KK, Golyska D, Luszczki JJ, Czuczwar SJ (2011). Effect of acutely and chronically administered venlafaxine on the anticonvulsant action of classical antiepileptic drugs in the mouse maximal electroshock model. Eur J Pharmacol 670:114-120.
Borowicz KK, Stepien K, Czuczwar SJ (2006). Fluoxetine enhances the anticonvulsant effects of conventional antiepileptic drugs in maximal electroshock seizures in mice. Pharmacol Rep 58:83-90.
Borsini F, Podhorna J, Marazziti D (2002). Do animal models of anxiety predict anxiolytic-like effects of antidepressants? Psychopharmacology (Berl) 163:121-141.
Bouilleret V, Cardamone L, Liu C, Koe AS, Fang K, Williams JP, et al. (2011). Confounding neurodegenerative effects of manganese for in vivo MR imaging in rat models of brain insults. J Magn Reson Imaging 34:774-784.
Bouilleret V, Hogan RE, Velakoulis D, Salzberg MR, Wang L, Egan GF, et al. (2009). Morphometric abnormalities and hyperanxiety in genetically epileptic rats: a model of psychiatric comorbidity? Neuroimage 45:267-274.
Bouilleret V, Nehlig A, Marescaux C, Namer IJ (2000). Magnetic resonance imaging follow-up of progressive hippocampal changes in a mouse model of mesial temporal lobe epilepsy. Epilepsia 41:642-650.
Boylan LS, Flint LA, Labovitz DL, Jackson SC, Starner K, Devinsky O (2004). Depression but not seizure frequency predicts quality of life in treatment-resistant epilepsy. Neurology 62:258-261.
Brady LS, Whitfield HJ, Jr., Fox RJ, Gold PW, Herkenham M (1991). Long-term antidepressant administration alters corticotropin-releasing hormone, tyrosine hydroxylase, and mineralocorticoid receptor gene expression in rat brain. Therapeutic implications. J Clin Invest 87:831-837.
Brennan TJ, Seeley WW, Kilgard M, Schreiner CE, Tecott LH (1997). Sound-induced seizures in serotonin 5-HT2c receptor mutant mice. Nat Genet 16:387-390.
Brietzke E, Stertz L, Fernandes BS, Kauer-Sant'anna M, Mascarenhas M, Escosteguy Vargas A, et al. (2009). Comparison of cytokine levels in depressed, manic and euthymic patients with bipolar disorder. J Affect Disord 116:214-217.
Buckmaster PS, Dudek FE (1999). In vivo intracellular analysis of granule cell axon reorganization in epileptic rats. J Neurophysiol 81:712-721.
Buterbaugh GG (1978). Effect of drugs modifying central serotonergic function on the response of extensor and nonextensor rats to maximal electroshock. Life Sci 23:2393-2404.
Caccia S, Cappi M, Fracasso C, Garattini S (1990). Influence of dose and route of administration on the kinetics of fluoxetine and its metabolite norfluoxetine in the rat. Psychopharmacology (Berl) 100:509-514.
Cameron HA, McKay RD (2001). Adult neurogenesis produces a large pool of new granule cells in the dentate gyrus. The Journal of comparative neurology 435:406-417.

120
Cameron HA, Woolley CS, McEwen BS, Gould E (1993). Differentiation of newly born neurons and glia in the dentate gyrus of the adult rat. Neuroscience 56:337-344.
Cardamone L, Salzberg MR, O'Brien TJ, Jones NC (2012). Antidepressant therapy in epilepsy - can treating the comorbidities affect the underlying disorder? Br J Pharmacol.
Carlezon WA, Jr., Duman RS, Nestler EJ (2005). The many faces of CREB. Trends Neurosci 28:436-445.
Carlsson A, Fuxe K, Ungerstedt U (1968). The effect of imipramine on central 5-hydroxytryptamine neurons. J Pharm Pharmacol 20:150-151.
Cascino GD (1994). Epilepsy: contemporary perspectives on evaluation and treatment. Mayo Clin Proc 69:1199-1211.
Cascino GD (2009). Temporal lobe epilepsy is a progressive neurologic disorder: Time means neurons! Neurology 72:1718-1719.
Castro ME, Diaz A, del Olmo E, Pazos A (2003). Chronic fluoxetine induces opposite changes in G protein coupling at pre and postsynaptic 5-HT1A receptors in rat brain. Neuropharmacology 44:93-101.
Charney DS, Delgade PL, Price LH, Heninger GR (1991). The receptor sensitivity hypothesis of antidepressant action: a review of antidepressant effects on serotonin function. In: The Role of Serotonin in Psychiatric Disorders(Brown, S. L. and van Praag, H. M., eds) New York: Brunner/Mazel.
Charney DS, Menkes DB, Heninger GR (1981). Receptor sensitivity and the mechanism of action of antidepressant treatment. Implications for the etiology and therapy of depression. Arch Gen Psychiatry 38:1160-1180.
Chen B, Dowlatshahi D, MacQueen GM, Wang JF, Young LT (2001). Increased hippocampal BDNF immunoreactivity in subjects treated with antidepressant medication. Biol Psychiatry 50:260-265.
Choi HC, Kim YI, Song HK, Kim JE, Kim DS, Kang TC (2010). Effects of selective serotonin reuptake inhibitors on GABAergic inhibition in the hippocampus of normal and pilocarpine induced epileptic rats. Brain Res 1357:131-141.
Christensen J, Vestergaard M, Mortensen PB, Sidenius P, Agerbo E (2007). Epilepsy and risk of suicide: a population-based case-control study. Lancet Neurol 6:693-698.
Clark M, Weiss SR, Post RM (1993). Autoradiographic analysis of serotonin receptors and transporter in kindled rat brain. Neurosci Lett 161:21-26.
Clinckers R, Smolders I, Meurs A, Ebinger G, Michotte Y (2004a). Anticonvulsant action of GBR-12909 and citalopram against acute experimentally induced limbic seizures. Neuropharmacology 47:1053-1061.
Clinckers R, Smolders I, Meurs A, Ebinger G, Michotte Y (2004b). Anticonvulsant action of hippocampal dopamine and serotonin is independently mediated by D and 5-HT receptors. J Neurochem 89:834-843.

121
Clinckers R, Smolders I, Meurs A, Ebinger G, Michotte Y (2005). Hippocampal dopamine and serotonin elevations as pharmacodynamic markers for the anticonvulsant efficacy of oxcarbazepine and 10,11-dihydro-10-hydroxycarbamazepine. Neurosci Lett 390:48-53.
Cole AJ (2000). Is epilepsy a progressive disease? The neurobiological consequences of epilepsy. Epilepsia 41 Suppl 2:S13-22.
Coppell AL, Pei Q, Zetterstrom TS (2003). Bi-phasic change in BDNF gene expression following antidepressant drug treatment. Neuropharmacology 44:903-910.
Corcoran ME, Lanius R, Duren A (1992). Reinforcing and punishing consequences of kindling. Epilepsy Res 13:179-186.
Cotterman-Hart S (2010). Depression in epilepsy: why aren't we treating? Epilepsy Behav 19:419-421.
Cottrell GA, Nyakas C, de Kloet ER, Bohus B (1984). Hippocampal kindling: corticosterone modulation of induced seizures. Brain Res 309:377-381.
Coulter DA, McIntyre DC, Loscher W (2002). Animal models of limbic epilepsies: what can they tell us? Brain Pathol 12:240-256.
Covolan L, Mello LE (2000). Temporal profile of neuronal injury following pilocarpine or kainic acid-induced status epilepticus. Epilepsy Res 39:133-152.
Crane GE (1957). Iproniazid (marsilid) phosphate, a therapeutic agent for mental disorders and debilitating diseases. Psychiatr Res Rep Am Psychiatr Assoc 8:142-152.
Cremers TI, de Boer P, Liao Y, Bosker FJ, den Boer JA, Westerink BH, et al. (2000). Augmentation with a 5-HT(1A), but not a 5-HT(1B) receptor antagonist critically depends on the dose of citalopram. Eur J Pharmacol 397:63-74.
Crespel A, Coubes P, Rousset MC, Brana C, Rougier A, Rondouin G, et al. (2002). Inflammatory reactions in human medial temporal lobe epilepsy with hippocampal sclerosis. Brain Res 952:159-169.
Croll SD, Suri C, Compton DL, Simmons MV, Yancopoulos GD, Lindsay RM, et al. (1999). Brain-derived neurotrophic factor transgenic mice exhibit passive avoidance deficits, increased seizure severity and in vitro hyperexcitability in the hippocampus and entorhinal cortex. Neuroscience 93:1491-1506.
Cryan JF, Mombereau C (2004). In search of a depressed mouse: utility of models for studying depression-related behavior in genetically modified mice. Mol Psychiatry 9:326-357.
Cryan JF, Page ME, Lucki I (2005). Differential behavioral effects of the antidepressants reboxetine, fluoxetine, and moclobemide in a modified forced swim test following chronic treatment. Psychopharmacology (Berl) 182:335-344.
Cuenca PJ, Holt KR, Hoefle JD (2004). Seizure secondary to citalopram overdose. J Emerg Med 26:177-181.
Czachura JF, Rasmussen K (2000). Effects of acute and chronic administration of fluoxetine on the activity of serotonergic neurons in the dorsal raphe nucleus of the rat. Naunyn Schmiedebergs Arch Pharmacol 362:266-275.

122
D'Sa C, Duman RS (2002). Antidepressants and neuroplasticity. Bipolar Disord 4:183-194.
Dailey JW, Yan QS, Mishra PK, Burger RL, Jobe PC (1992). Effects of fluoxetine on convulsions and on brain serotonin as detected by microdialysis in genetically epilepsy-prone rats. J Pharmacol Exp Ther 260:533-540.
Danzer SC (2011). Depression, stress, epilepsy and adult neurogenesis. Exp Neurol.
Danzer SC, Kotloski RJ, Walter C, Hughes M, McNamara JO (2008). Altered morphology of hippocampal dentate granule cell presynaptic and postsynaptic terminals following conditional deletion of TrkB. Hippocampus 18:668-678.
David DJ, Samuels BA, Rainer Q, Wang JW, Marsteller D, Mendez I, et al. (2009). Neurogenesis-dependent and -independent effects of fluoxetine in an animal model of anxiety/depression. Neuron 62:479-493.
de Kloet ER, Karst H, Joels M (2008). Corticosteroid hormones in the central stress response: quick-and-slow. Front Neuroendocrinol 29:268-272.
de Lanerolle NC, Kim JH, Williamson A, Spencer SS, Zaveri HP, Eid T, et al. (2003). A retrospective analysis of hippocampal pathology in human temporal lobe epilepsy: evidence for distinctive patient subcategories. Epilepsia 44:677-687.
De Sarro GB, Donato Di Paola E, De Sarro A, Vidal MJ (1991). Role of nitric oxide in the genesis of excitatory amino acid-induced seizures from the deep prepiriform cortex. Fundam Clin Pharmacol 5:503-511.
Delgado JM, Sevillano M (1961). Evolution of repeated hippocampal seizures in cat. Electroencephalography and Clinical Neurophysiology 13:722-733.
Desai SD, Shukla G, Goyal V, Singh S, Padma MV, Tripathi M, et al. (2010). Study of DSM-IV Axis I psychiatric disorders in patients with refractory complex partial seizures using a short structured clinical interview. Epilepsy Behav 19:301-305.
Dhanushkodi A, Shetty AK (2008). Is exposure to enriched environment beneficial for functional post-lesional recovery in temporal lobe epilepsy? Neurosci Biobehav Rev 32:657-674.
Dias BG, Banerjee SB, Duman RS, Vaidya VA (2003). Differential regulation of brain derived neurotrophic factor transcripts by antidepressant treatments in the adult rat brain. Neuropharmacology 45:553-563.
Droste SK, de Groote L, Atkinson HC, Lightman SL, Reul JM, Linthorst AC (2008). Corticosterone levels in the brain show a distinct ultradian rhythm but a delayed response to forced swim stress. Endocrinology 149:3244-3253.
Duman RS, Heninger GR, Nestler EJ (1997). A molecular and cellular theory of depression. Arch Gen Psychiatry 54:597-606.
Duncan GE, Knapp DJ, Carson SW, Breese GR (1998). Differential effects of chronic antidepressant treatment on swim stress- and fluoxetine-induced secretion of corticosterone and progesterone. J Pharmacol Exp Ther 285:579-587.
Durand M, Berton O, Aguerre S, Edno L, Combourieu I, Mormede P, et al. (1999). Effects of repeated fluoxetine on anxiety-related behaviours, central serotonergic systems, and the corticotropic axis axis in SHR and WKY rats. Neuropharmacology 38:893-907.

123
Ebert U, Loscher W (1995). Differences in mossy fibre sprouting during conventional and rapid amygdala kindling of the rat. Neurosci Lett 190:199-202.
Edwards HE, Burnham WM, Mendonca A, Bowlby DA, MacLusky NJ (1999). Steroid hormones affect limbic afterdischarge thresholds and kindling rates in adult female rats. Brain Res 838:136-150.
Edwards HE, Dortok D, Tam J, Won D, Burnham WM (2002). Prenatal stress alters seizure thresholds and the development of kindled seizures in infant and adult rats. Horm Behav 42:437-447.
Edwards JG, Wheal HV (1992). Assessment of epileptogenic potential: experimental, clinical and epidemiological approaches. J Psychopharmacol 6:204-213.
Eisch AJ, Cameron HA, Encinas JM, Meltzer LA, Ming GL, Overstreet-Wadiche LS (2008). Adult neurogenesis, mental health, and mental illness: hope or hype? J Neurosci 28:11785-11791.
Eison AS, Mullins UL (1996). Regulation of central 5-HT2A receptors: a review of in vivo studies. Behav Brain Res 73:177-181.
Encinas JM, Vaahtokari A, Enikolopov G (2006). Fluoxetine targets early progenitor cells in the adult brain. P Natl Acad Sci USA 103:8233-8238.
Engel J, Jr. (1996). Introduction to temporal lobe epilepsy. Epilepsy Res 26:141-150.
Engel J, Jr. (2001). Mesial temporal lobe epilepsy: what have we learned? Neuroscientist 7:340-352.
Engel J, Pedley T (eds.) (2008). What Is Epilepsy? Philidelphia: Lippincott Williams & Wilkins.
Eriksson PS, Perfilieva E, Bjork-Eriksson T, Alborn AM, Nordborg C, Peterson DA, et al. (1998). Neurogenesis in the adult human hippocampus. Nat Med 4:1313-1317.
Ernfors P, Bengzon J, Kokaia Z, Persson H, Lindvall O (1991). Increased levels of messenger RNAs for neurotrophic factors in the brain during kindling epileptogenesis. Neuron 7:165-176.
Esposito MS, Piatti VC, Laplagne DA, Morgenstern NA, Ferrari CC, Pitossi FJ, et al. (2005). Neuronal differentiation in the adult hippocampus recapitulates embryonic development. J Neurosci 25:10074-10086.
Ettinger A, Reed M, Cramer J (2004). Depression and comorbidity in community-based patients with epilepsy or asthma. Neurology 63:1008-1014.
Fabel K, Tam B, Kaufer D, Baiker A, Simmons N, Kuo CJ, et al. (2003). VEGF is necessary for exercise-induced adult hippocampal neurogenesis. Eur J Neurosci 18:2803-2812.
Favale E, Audenino D, Cocito L, Albano C (2003). The anticonvulsant effect of citalopram as an indirect evidence of serotonergic impairment in human epileptogenesis. Seizure 12:316-318.
Favale E, Rubino V, Mainardi P, Lunardi G, Albano C (1995). Anticonvulsant effect of fluoxetine in humans. Neurology 45:1926-1927.

124
Feindel W, Penfield W (1954). Localization of discharge in temporal lobe automatism. AMA Arch Neurol Psychiatry 72:603-630.
Feindel W, Penfield W, Jasper H (1952). Localization of epileptic discharge in temporal lobe automatism. Trans Am Neurol Assoc 56:14-17.
Ferrero AJ, Cereseto M, Reines A, Bonavita CD, Sifonios LL, Rubio MC, et al. (2005). Chronic treatment with fluoxetine decreases seizure threshold in naive but not in rats exposed to the learned helplessness paradigm: Correlation with the hippocampal glutamate release. Prog Neuropsychopharmacol Biol Psychiatry 29:678-686.
Filakovszky J, Kantor S, Halasz P, Bagdy G (2001). 8-OH-DPAT and MK-801 affect epileptic activity independently of vigilance. Neurochem Int 38:551-556.
File SE, Ouagazzal AM, Gonzalez LE, Overstreet DH (1999). Chronic fluoxetine in tests of anxiety in rat lines selectively bred for differential 5-HT1A receptor function. Pharmacol Biochem Behav 62:695-701.
Filho GM, Rosa VP, Lin K, Caboclo LO, Sakamoto AC, Yacubian EM (2008). Psychiatric comorbidity in epilepsy: a study comparing patients with mesial temporal sclerosis and juvenile myoclonic epilepsy. Epilepsy Behav 13:196-201.
Fisher RS, van Emde Boas W, Blume W, Elger C, Genton P, Lee P, et al. (2005). Epileptic seizures and epilepsy: definitions proposed by the International League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE). Epilepsia 46:470-472.
Fornaro M, Martino M, Battaglia F, Colicchio S, Perugi G (2011). Increase in IL-6 levels among major depressive disorder patients after a 6-week treatment with duloxetine 60 mg/day: a preliminary observation. Neuropsychiatr Dis Treat 7:51-56.
Forsgren L, Nystrom L (1990). An incident case-referent study of epileptic seizures in adults. Epilepsy Res 6:66-81.
Frechilla D, Otano A, Del Rio J (1998). Effect of chronic antidepressant treatment on transcription factor binding activity in rat hippocampus and frontal cortex. Prog Neuropsychopharmacol Biol Psychiatry 22:787-802.
French JA, Williamson PD, Thadani VM, Darcey TM, Mattson RH, Spencer SS, et al. (1993). Characteristics of medial temporal lobe epilepsy: I. Results of history and physical examination. Ann Neurol 34:774-780.
Fuller-Thomson E, Brennenstuhl S (2009). The association between depression and epilepsy in a nationally representative sample. Epilepsia 50:1051-1058.
Fuller RW, Snoddy HD (1990). Serotonin receptor subtypes involved in the elevation of serum corticosterone concentration in rats by direct- and indirect-acting serotonin agonists. Neuroendocrinology 52:206-211.
Gaitatzis A, Carroll K, Majeed A, J WS (2004). The epidemiology of the comorbidity of epilepsy in the general population. Epilepsia 45:1613-1622.
Galanopoulou AS, Buckmaster PS, Staley KJ, Moshe SL, Perucca E, Engel J, Jr., et al. (2012a). Identification of new epilepsy treatments: issues in preclinical methodology. Epilepsia 53:571-582.

125
Galanopoulou AS, Gorter JA, Cepeda C (2012b). Finding a better drug for epilepsy: the mTOR pathway as an antiepileptogenic target. Epilepsia 53:1119-1130.
Gallagher BB (1987). Endocrine abnormalities in human temporal lobe epilepsy. Yale J Biol Med 60:93-97.
Gallagher BB, Murvin A, Flanigin HF, King DW, Luney D (1984). Pituitary and adrenal function in epileptic patients. Epilepsia 25:683-689.
Gariboldi M, Tutka P, Samanin R, Vezzani A (1996a). Stimulation of 5-HT1A receptors in the dorsal hippocampus and inhibition of limbic seizures induced by kainic acid in rats. Br J Pharmacol 119:813-818.
Gariboldi M, Tutka P, Samanin R, Vezzani A (1996b). Stimulation of 5-HT1A receptors in the dorsal hippocampus and inhibition of limbic seizures induced by kainic acid in rats. Brit J Pharmacol 119:813-818.
Ge S, Yang CH, Hsu KS, Ming GL, Song H (2007). A critical period for enhanced synaptic plasticity in newly generated neurons of the adult brain. Neuron 54:559-566.
Gelfin Y, Gorfine M, Lerer B (1998). Effect of clinical doses of fluoxetine on psychological variables in healthy volunteers. Am J Psychiatry 155:290-292.
Gerber K, Filakovszky J, Halasz P, Bagdy G (1998). The 5-HT1A agonist 8-OH-DPAT increases the number of spike-wave discharges in a genetic rat model of absence epilepsy. Brain Res 807:243-245.
Gervasoni N, Aubry JM, Bondolfi G, Osiek C, Schwald M, Bertschy G, et al. (2005). Partial normalization of serum brain-derived neurotrophic factor in remitted patients after a major depressive episode. Neuropsychobiology 51:234-238.
Geyer M, Markou A (2002). The role of preclinical models in the development of psychotropic drugs. In: Neuropsychopharmacology: The Fifth Generation of Progress(Davis, K. L. et al., eds), pp 446-455 Philadelphia: Lippincott Williams & Wilkins.
Gholipour T, Ghasemi M, Riazi K, Ghaffarpour M, Dehpour AR (2010). Seizure susceptibility alteration through 5-HT(3) receptor: modulation by nitric oxide. Seizure 19:17-22.
Gigli GL, Diomedi M, Troisi A, Baldinetti F, Marciani MG, Girolami E, et al. (1994). Lack of potentiation of anticonvulsant effect by fluoxetine in drug-resistant epilepsy. Seizure 3:221-224.
Gilby KL (2009). Investigating epigenetic influences on seizure disposition. Can J Neurol Sci 36 Suppl 2:S78-81.
Gillespie CF, Nemeroff CB (2005). Hypercortisolemia and depression. Psychosom Med 67 Suppl 1:S26-28.
Gilliam FG (2005). Diagnosis and treatment of mood disorders in persons with epilepsy. Curr Opin Neurol 18:129-133.
Gilliam FG, Santos J, Vahle V, Carter J, Brown K, Hecimovic H (2004). Depression in epilepsy: ignoring clinical expression of neuronal network dysfunction? Epilepsia 45 Suppl 2:28-33.

126
Goddard GV (1967). Development of epileptic seizures through brain stimulation at low intensity. Nature 214:1020-1021.
Goddard GV, McIntyre DC, Leech CK (1969). A permanent change in brain function resulting from daily electrical stimulation. Exp Neurol 25:295-330.
Gonul AS, Akdeniz F, Taneli F, Donat O, Eker C, Vahip S (2005). Effect of treatment on serum brain-derived neurotrophic factor levels in depressed patients. European archives of psychiatry and clinical neuroscience 255:381-386.
Goodnick P, Goldstein B (1998). Selective serotonin reuptake inhibitors in affective disorders. II. Efficacy and quality of life. Psychopharmacol 3:S21–S54.
Gordon E, Devinsky O (2001). Alcohol and marijuana: effects on epilepsy and use by patients with epilepsy. Epilepsia 42:1266-1272.
Gorter JA, van Vliet EA, Aronica E, Breit T, Rauwerda H, Lopes da Silva FH, et al. (2006). Potential new antiepileptogenic targets indicated by microarray analysis in a rat model for temporal lobe epilepsy. J Neurosci 26:11083-11110.
Gorter JA, van Vliet EA, Aronica E, Lopes da Silva FH (2001). Progression of spontaneous seizures after status epilepticus is associated with mossy fibre sprouting and extensive bilateral loss of hilar parvalbumin and somatostatin-immunoreactive neurons. Eur J Neurosci 13:657-669.
Gould E, Tanapat P, McEwen BS, Flugge G, Fuchs E (1998). Proliferation of granule cell precursors in the dentate gyrus of adult monkeys is diminished by stress. Proc Natl Acad Sci U S A 95:3168-3171.
Gowers WR (1881). Epilepsy and other chronic convulsive disorders: their causes, symptoms and treatment. London: J&A Churchill.
Gozen O, Balkan B, Yararbas G, Koylu EO, Kuhar MJ, Pogun S (2007). Sex differences in the regulation of cocaine and amphetamine-regulated transcript expression in hypothalamic nuclei of rats by forced swim stress. Synapse 61:561-568.
Gray WP, Sundstrom LE (1998). Kainic acid increases the proliferation of granule cell progenitors in the dentate gyrus of the adult rat. Brain Res 790:52-59.
Griebel G, Cohen C, Perrault G, Sanger DJ (1999). Behavioral effects of acute and chronic fluoxetine in Wistar-Kyoto rats. Physiol Behav 67:315-320.
Groticke I, Hoffmann K, Loscher W (2007). Behavioral alterations in the pilocarpine model of temporal lobe epilepsy in mice. Exp Neurol 207:329-349.
Groticke I, Hoffmann K, Loscher W (2008). Behavioral alterations in a mouse model of temporal lobe epilepsy induced by intrahippocampal injection of kainate. Exp Neurol 213:71-83.
Gu J, Lynch BA, Anderson D, Klitgaard H, Lu S, Elashoff M, et al. (2004). The antiepileptic drug levetiracetam selectively modifies kindling-induced alterations in gene expression in the temporal lobe of rats. Eur J Neurosci 19:334-345.
Hagan JJ, Hatcher JP, Slade PD (1995). The role of 5-HT1D and 5-HT1A receptors in mediating 5-hydroxytryptophan induced myoclonic jerks in guinea pigs. European Journal of Pharmacology 294:743-751.

127
Haller J, Alicki M (2012). Current animal models of anxiety, anxiety disorders, and anxiolytic drugs. Curr Opin Psychiatry 25:59-64.
Haltiner AM, Temkin NR, Dikmen SS (1997). Risk of seizure recurrence after the first late posttraumatic seizure. Arch Phys Med Rehabil 78:835-840.
Handley SL, Mithani S (1984). Effects of alpha-adrenoceptor agonists and antagonists in a maze-exploration model of 'fear'-motivated behaviour. Naunyn Schmiedebergs Arch Pharmacol 327:1-5.
Hannestad J, DellaGioia N, Bloch M (2011). The effect of antidepressant medication treatment on serum levels of inflammatory cytokines: a meta-analysis. Neuropsychopharmacology 36:2452-2459.
Hanson ND, Owens MJ, Nemeroff CB (2011). Depression, Antidepressants, and Neurogenesis: A Critical Reappraisal. Neuropsychopharmacology 36:2589-2602.
Harden CL, Maroof DA, Nikolov B, Fowler K, Sperling M, Liporace J, et al. (2007). The effect of seizure severity on quality of life in epilepsy. Epilepsy Behav 11:208-211.
Harmant J, van Rijckevorsel-Harmant K, de Barsy T, Hendrickx B (1990). Fluvoxamine: an antidepressant with low (or no) epileptogenic effect. Lancet 336:386.
Hasegawa S, Watanabe A, Nguyen KQ, Debonnel G, Diksic M (2005). Chronic administration of citalopram in olfactory bulbectomy rats restores brain 5-HT synthesis rates: an autoradiographic study. Psychopharmacology (Berl) 179:781-790.
Hastings NB, Gould E (1999). Rapid extension of axons into the CA3 region by adult-generated granule cells. The Journal of comparative neurology 413:146-154.
Hattiangady B, Rao MS, Shetty AK (2004). Chronic temporal lobe epilepsy is associated with severely declined dentate neurogenesis in the adult hippocampus. Neurobiol Dis 17:473-490.
Hattiangady B, Shetty AK (2008). Implications of decreased hippocampal neurogenesis in chronic temporal lobe epilepsy. Epilepsia 49 Suppl 5:26-41.
Hattiangady B, Shetty AK (2010). Decreased neuronal differentiation of newly generated cells underlies reduced hippocampal neurogenesis in chronic temporal lobe epilepsy. Hippocampus 20:97-112.
Hauser WA, Annegers JF, Kurland LT (1991). Prevalence of epilepsy in Rochester, Minnesota: 1940-1980. Epilepsia 32:429-445.
Hauser WA, Annegers JF, Kurland LT (1993). Incidence of epilepsy and unprovoked seizures in Rochester, Minnesota: 1935-1984. Epilepsia 34:453-468.
Haut SR, Hall CB, Masur J, Lipton RB (2007). Seizure occurrence: precipitants and prediction. Neurology 69:1905-1910.
Helfer V, Deransart C, Marescaux C, Depaulis A (1996). Amygdala kindling in the rat: anxiogenic-like consequences. Neuroscience 73:971-978.

128
Hellier JL, Patrylo PR, Buckmaster PS, Dudek FE (1998). Recurrent spontaneous motor seizures after repeated low-dose systemic treatment with kainate: assessment of a rat model of temporal lobe epilepsy. Epilepsy Res 31:73-84.
Herman JP, Cullinan WE (1997). Neurocircuitry of stress: central control of the hypothalamo-pituitary-adrenocortical axis. Trends Neurosci 20:78-84.
Herman JP, Figueiredo H, Mueller NK, Ulrich-Lai Y, Ostrander MM, Choi DC, et al. (2003). Central mechanisms of stress integration: hierarchical circuitry controlling hypothalamo-pituitary-adrenocortical responsiveness. Front Neuroendocrinol 24:151-180.
Herman ST (2002). Epilepsy after brain insult: targeting epileptogenesis. Neurology 59:S21-26.
Hermann B, Seidenberg M, Jones J (2008). The neurobehavioural comorbidities of epilepsy: can a natural history be developed? Lancet Neurol 7:151-160.
Hermann BP, Seidenberg M, Bell B (2000). Psychiatric comorbidity in chronic epilepsy: identification, consequences, and treatment of major depression. Epilepsia 41 Suppl 2:S31-41.
Hernandez EJ, Williams PA, Dudek FE (2002). Effects of fluoxetine and TFMPP on spontaneous seizures in rats with pilocarpine-induced epilepsy. Epilepsia 43:1337-1345.
Hesdorffer DC, Hauser WA, Annegers JF, Cascino G (2000). Major depression is a risk factor for seizures in older adults. Ann Neurol 47:246-249.
Hesdorffer DC, Hauser WA, Olafsson E, Ludvigsson P, Kjartansson O (2006). Depression and suicide attempt as risk factors for incident unprovoked seizures. Ann Neurol 59:35-41.
Hesdorffer DC, Krishnamoorthy ES (2011). Neuropsychiatric disorders in epilepsy: epidemiology and classification. In: The neuropsychiatry of epilepsy(Trimble, M. and Schmitz, B., eds) Cambridge: Cambridge University Press.
Hesketh S, Jessop DS, Hogg S, Harbuz MS (2005). Differential actions of acute and chronic citalopram on the rodent hypothalamic-pituitary-adrenal axis response to acute restraint stress. J Endocrinol 185:373-382.
Hesketh SA, Brennan AK, Jessop DS, Finn DP (2008). Effects of chronic treatment with citalopram on cannabinoid and opioid receptor-mediated G-protein coupling in discrete rat brain regions. Psychopharmacology (Berl) 198:29-36.
Himmerich H, Zimmermann P, Ising M, Kloiber S, Lucae S, Kunzel HE, et al. (2007). Changes in the hypothalamic-pituitary-adrenal axis and leptin levels during antidepressant treatment. Neuropsychobiology 55:28-35.
Hiramatsu M, Ogawa K, Kabuto H, Mori A (1987). Reduced uptake and release of 5-hydroxytryptamine and taurine in the cerebral cortex of epileptic El mice. Epilepsy Res 1:40-45.
Hirvonen J, Kreisl WC, Fujita M, Dustin I, Khan O, Appel S, et al. (2012). Increased in vivo expression of an inflammatory marker in temporal lobe epilepsy. J Nucl Med 53:234-240.
Hitiris N, Mohanraj R, Norrie J, Sills GJ, Brodie MJ (2007). Predictors of pharmacoresistant epilepsy. Epilepsy Res 75:192-196.

129
Holsboer F (2001). Stress, hypercortisolism and corticosteroid receptors in depression: implications for therapy. J Affect Disord 62:77-91.
Homberg JR, Olivier JD, Blom T, Arentsen T, van Brunschot C, Schipper P, et al. (2011). Fluoxetine exerts age-dependent effects on behavior and amygdala neuroplasticity in the rat. PLoS One 6:e16646.
Honack D, Loscher W (1989). Amygdala-kindling as a model for chronic efficacy studies on antiepileptic drugs: experiments with carbamazepine. Neuropharmacology 28:599-610.
Houser CR, Miyashiro JE, Swartz BE, Walsh GO, Rich JR, Delgado-Escueta AV (1990). Altered patterns of dynorphin immunoreactivity suggest mossy fiber reorganization in human hippocampal epilepsy. J Neurosci 10:267-282.
Hovorka J, Herman E, Nemcova II (2000). Treatment of Interictal Depression with Citalopram in Patients with Epilepsy. Epilepsy Behav 1:444-447.
Huang GJ, Herbert J (2006). Stimulation of neurogenesis in the hippocampus of the adult rat by fluoxetine requires rhythmic change in corticosterone. Biol Psychiatry 59:619-624.
Hyttel J (1994). Pharmacological characterization of selective serotonin reuptake inhibitors (SSRIs). Int Clin Psychopharmacol 9 Suppl 1:19-26.
Igelstrom KM (2012). Preclinical antiepileptic actions of selective serotonin reuptake inhibitors-Implications for clinical trial design. Epilepsia 53:596-605.
Isaac M (2005). Serotonergic 5-HT2C receptors as a potential therapeutic target for the design antiepileptic drugs. Current Topics in Medicinal Chemistry 5:59-67.
Isbister GK, Bowe SJ, Dawson A, Whyte IM (2004). Relative toxicity of selective serotonin reuptake inhibitors (SSRIs) in overdose. J Toxicol Clin Toxicol 42:277-285.
Jaako K, Aonurm-Helm A, Kalda A, Anier K, Zharkovsky T, Shastin D, et al. (2011). Repeated citalopram administration counteracts kainic acid-induced spreading of PSA-NCAM-immunoreactive cells and loss of reelin in the adult mouse hippocampus. European Journal of Pharmacology 666:61-71.
Jaako K, Zharkovsky T, Zharkovsky A (2009). Effects of repeated citalopram treatment on kainic acid-induced neurogenesis in adult mouse hippocampus. Brain Research 1288:18-28.
Jacobson L, Sapolsky R (1991). The role of the hippocampus in feedback regulation of the hypothalamic-pituitary-adrenocortical axis. Endocr Rev 12:118-134.
Jakubs K, Nanobashvili A, Bonde S, Ekdahl CT, Kokaia Z, Kokaia M, et al. (2006). Environment matters: synaptic properties of neurons born in the epileptic adult brain develop to reduce excitability. Neuron 52:1047-1059.
Janssen DG, Caniato RN, Verster JC, Baune BT (2010). A psychoneuroimmunological review on cytokines involved in antidepressant treatment response. Hum Psychopharmacol 25:201-215.
Jensen JB, Jessop DS, Harbuz MS, Mork A, Sanchez C, Mikkelsen JD (1999). Acute and long-term treatments with the selective serotonin reuptake inhibitor citalopram modulate the HPA axis activity at different levels in male rats. J Neuroendocrinol 11:465-471.

130
Jessberger S, Zhao C, Toni N, Clemenson GD, Jr., Li Y, Gage FH (2007). Seizure-associated, aberrant neurogenesis in adult rats characterized with retrovirus-mediated cell labeling. J Neurosci 27:9400-9407.
Jin Y, Lim CM, Kim SW, Park JY, Seo JS, Han PL, et al. (2009). Fluoxetine attenuates kainic acid-induced neuronal cell death in the mouse hippocampus. Brain Res 1281:108-116.
Jobe P, Browning R (2007). Animal models of depression and epilepsy: the genetically epilepsy-prone rat. In: Psychiatric issues in epilepsy(Ettinger, A. B. and Kanner, A. M., eds): Lippencott W&W.
Jobe PC, Browning RA (2005). The serotonergic and noradrenergic effects of antidepressant drugs are anticonvulsant, not proconvulsant. Epilepsy Behav 7:602-619.
Jobe PC, Mishra PK, Adams-Curtis LE, Deoskar VU, Ko KH, Browning RA, et al. (1995). The genetically epilepsy-prone rat (GEPR). Ital J Neurol Sci 16:91-99.
Joels M, Karst H, Krugers HJ, Lucassen PJ (2007). Chronic stress: implications for neuronal morphology, function and neurogenesis. Front Neuroendocrinol 28:72-96.
Jones NC, Kumar G, O'Brien TJ, Morris MJ, Rees SM, Salzberg MR (2009). Anxiolytic effects of rapid amygdala kindling, and the influence of early life experience in rats. Behav Brain Res 203:81-87.
Jones NC, Lee HE, Yang M, Rees SM, Morris MJ, O'Brien TJ, et al. (2012). Repeatedly stressed rats have enhanced vulnerability to amygdala kindling epileptogenesis. Psychoneuroendocrinology.
Jones NC, Cardamone L, Williams JP, Salzberg MR, Myers D, O'Brien TJ (2008a). Experimental traumatic brain injury induces a pervasive hyperanxious phenotype in rats. J Neurotrauma 25:1367-1374.
Jones NC, Salzberg MR, Kumar G, Couper A, Morris MJ, O'Brien TJ (2008b). Elevated anxiety and depressive-like behavior in a rat model of genetic generalized epilepsy suggesting common causation. Exp Neurol 209:254-260.
Jongsma ME, Bosker FJ, Cremers TI, Westerink BH, den Boer JA (2005). The effect of chronic selective serotonin reuptake inhibitor treatment on serotonin 1B receptor sensitivity and HPA axis activity. Prog Neuropsychopharmacol Biol Psychiatry 29:738-744.
Judge BS, Rentmeester LL (2011). Antidepressant overdose-induced seizures. Neurol Clin 29:565-580.
Jung KH, Chu K, Kim M, Jeong SW, Song YM, Lee ST, et al. (2004). Continuous cytosine-b-D-arabinofuranoside infusion reduces ectopic granule cells in adult rat hippocampus with attenuation of spontaneous recurrent seizures following pilocarpine-induced status epilepticus. Eur J Neurosci 19:3219-3226.
Kabuto H, Yokoi I, Endo A, Takei M, Kurimoto T, Mori A (1994a). Chronic administration of citalopram inhibited El mouse convulsions and decreased monoamine oxidase-A activity. Acta Med Okayama 48:311-316.
Kabuto H, Yokoi I, Takei M, Kurimoto T, Mori A (1994b). The anticonvulsant effect of citalopram on El mice, and the levels of tryptophan and tyrosine and their metabolites in the brain. Neurochem Res 19:463-467.

131
Kalynchuk LE (2000). Long-term amygdala kindling in rats as a model for the study of interictal emotionality in temporal lobe epilepsy. Neurosci Biobehav Rev 24:691-704.
Kanemaru K, Nishi K, Hasegawa S, Diksic M (2009). Chronic citalopram treatment elevates serotonin synthesis in flinders sensitive and flinders resistant lines of rats, with no significant effect on Sprague-Dawley rats. Neurochem Int 54:363-371.
Kanner AM (2003). Depression in epilepsy: a frequently neglected multifaceted disorder. Epilepsy Behav 4 Suppl 4:11-19.
Kanner AM (2007). Epilepsy and mood disorders. Epilepsia 48 Suppl 9:20-22.
Kanner AM (2009). Psychiatric issues in epilepsy: the complex relation of mood, anxiety disorders, and epilepsy. Epilepsy Behav 15:83-87.
Kanner AM (2011a). Anxiety disorders in epilepsy: the forgotten psychiatric comorbidity. Epilepsy Curr 11:90-91.
Kanner AM (2011b). Depression and epilepsy: A bidirectional relation? Epilepsia 52 Suppl 1:21-27.
Kanner AM, Barry JJ, Gilliam F, Hermann B, Meador KJ (2010). Anxiety disorders, subsyndromic depressive episodes, and major depressive episodes: do they differ on their impact on the quality of life of patients with epilepsy? Epilepsia 51:1152-1158.
Kanner AM, Kozak AM, Frey M (2000). The Use of Sertraline in Patients with Epilepsy: Is It Safe? Epilepsy Behav 1:100-105.
Kanner AM, Schachter SC, Barry JJ, Hersdorffer DC, Mula M, Trimble M, et al. (2012). Depression and epilepsy: epidemiologic and neurobiologic perspectives that may explain their high comorbid occurrence. Epilepsy Behav 24:156-168.
Kaplan MS, Hinds JW (1977). Neurogenesis in the adult rat: electron microscopic analysis of light radioautographs. Science 197:1092-1094.
Karege F, Perret G, Bondolfi G, Schwald M, Bertschy G, Aubry JM (2002). Decreased serum brain-derived neurotrophic factor levels in major depressed patients. Psychiatry Res 109:143-148.
Karpova NN, Pickenhagen A, Lindholm J, Tiraboschi E, Kulesskaya N, Agustsdottir A, et al. (2011). Fear erasure in mice requires synergy between antidepressant drugs and extinction training. Science 334:1731-1734.
Karson CN, Newton JE, Livingston R, Jolly JB, Cooper TB, Sprigg J, et al. (1993). Human brain fluoxetine concentrations. J Neuropsychiatry Clin Neurosci 5:322-329.
Karst H, de Kloet ER, Joels M (1999). Episodic corticosterone treatment accelerates kindling epileptogenesis and triggers long-term changes in hippocampal CA1 cells, in the fully kindled state. Eur J Neurosci 11:889-898.
Kelley MS, Jacobs MP, Lowenstein DH (2009). The NINDS epilepsy research benchmarks. Epilepsia 50:579-582.

132
Kelly CA, Dhaun N, Laing WJ, Strachan FE, Good AM, Bateman DN (2004). Comparative toxicity of citalopram and the newer antidepressants after overdose. J Toxicol Clin Toxicol 42:67-71.
Kelly MW, Perry PJ, Holstad SG, Garvey MJ (1989). Serum fluoxetine and norfluoxetine concentrations and antidepressant response. Ther Drug Monit 11:165-170.
Keltner NL, McAfee KM, Taylor CL (2002). Mechanisms and treatments of SSRI-induced sexual dysfunction. Perspect Psychiatr Care 38:111-116.
Kenis G, Maes M (2002). Effects of antidepressants on the production of cytokines. Int J Neuropsychopharmacol 5:401-412.
Kennett GA, Lightowler S, Debiasi V, Stevens NC, Wood MD, Tulloch IF, et al. (1994). Effect of Chronic Administration of Selective 5-Hydroxytryptamine and Noradrenaline Uptake Inhibitors on a Putative Index of 5-Ht2c/2b Receptor Function. Neuropharmacology 33:1581-1588.
Kharatishvili I, Nissinen JP, McIntosh TK, Pitkanen A (2006). A model of posttraumatic epilepsy induced by lateral fluid-percussion brain injury in rats. Neuroscience 140:685-697.
Kharatishvili I, Pitkanen A (2010). Association of the severity of cortical damage with the occurrence of spontaneous seizures and hyperexcitability in an animal model of posttraumatic epilepsy. Epilepsy Res 90:47-59.
Khawaja X, Xu J, Liang JJ, Barrett JE (2004). Proteomic analysis of protein changes developing in rat hippocampus after chronic antidepressant treatment: Implications for depressive disorders and future therapies. J Neurosci Res 75:451-460.
Kilian M, Frey HH (1973). Central monoamines and convulsine thresholds in mice and rats. Neuropharmacology 12:681-692.
Klerman GL, Cole JO (1965). Clinical Pharmacology of Imipramine and Related Antidepressant Compounds. Pharmacol Rev 17:101-141.
Kobau R, Gilliam F, Thurman DJ (2006). Prevalence of self-reported epilepsy or seizure disorder and its associations with self-reported depression and anxiety: results from the 2004 HealthStyles Survey. Epilepsia 47:1915-1921.
Kobayashi K, Ikeda Y, Sakai A, Yamasaki N, Haneda E, Miyakawa T, et al. (2010). Reversal of hippocampal neuronal maturation by serotonergic antidepressants. P Natl Acad Sci USA 107:8434-8439.
Kobayashi K, Ikeda Y, Suzuki H (2011). Behavioral destabilization induced by the selective serotonin reuptake inhibitor fluoxetine. Mol Brain 4:12.
Kodama M, Fujioko T, Duman RS (2005). Chronic olanzapine or fluoxetine administration increases cell proliferation in hippocampus and prefrontal cortex of adult rats (vol 56, pg 570, 2004). Biol Psychiat 57:199-199.
Koe AS (2012). Early life stress and mesial temporal lobe epilepsy: the role of the hypothalamic-pituitary-adrenal axis. In: Medicine, Dentistry & Health Sciences - Medicine (RMH & WH), vol. PhD thesis Melbourne: University of Melbourne.

133
Koella WP, Glatt A, Klebs K, Durst T (1979). Epileptic phenomena induced in the cat by the antidepressants maprotiline, imipramine, clomipramine, and amitriptyline. Biol Psychiatry 14:485-497.
Koh S, Magid R, Chung H, Stine CD, Wilson DN (2007). Depressive behavior and selective down-regulation of serotonin receptor expression after early-life seizures: reversal by environmental enrichment. Epilepsy Behav 10:26-31.
Kondziella D, Asztely F (2009). Don't be afraid to treat depression in patients with epilepsy! Acta Neurol Scand 119:75-80.
Kornack DR, Rakic P (1999). Continuation of neurogenesis in the hippocampus of the adult macaque monkey. Proc Natl Acad Sci U S A 96:5768-5773.
Kotsopoulos IA, van Merode T, Kessels FG, de Krom MC, Knottnerus JA (2002). Systematic review and meta-analysis of incidence studies of epilepsy and unprovoked seizures. Epilepsia 43:1402-1409.
Koutsogiannopoulos S, Adelson F, Lee V, Andermann F (2009). Stressors at the onset of adult epilepsy: implications for practice. Epileptic Disord 11:42-47.
Koyama R, Yamada MK, Fujisawa S, Katoh-Semba R, Matsuki N, Ikegaya Y (2004). Brain-derived neurotrophic factor induces hyperexcitable reentrant circuits in the dentate gyrus. J Neurosci 24:7215-7224.
Kron MM, Zhang H, Parent JM (2010). The developmental stage of dentate granule cells dictates their contribution to seizure-induced plasticity. J Neurosci 30:2051-2059.
Krystal JH, Sanacora G, Blumberg H, Anand A, Charney DS, Marek G, et al. (2002). Glutamate and GABA systems as targets for novel antidepressant and mood-stabilizing treatments. Mol Psychiatry 7 Suppl 1:S71-80.
Kugelberg FC, Apelqvist G, Bengtsson F (2002). Effects of chronic citalopram treatment on central and peripheral spontaneous open-field behaviours in rats. Pharmacol Toxicol 90:303-310.
Kugelberg FC, Apelqvist G, Carlsson B, Ahlner J, Bengtsson F (2001). In vivo steady-state pharmacokinetic outcome following clinical and toxic doses of racemic citalopram to rats. Br J Pharmacol 132:1683-1690.
Kuhn KU, Quednow BB, Thiel M, Falkai P, Maier W, Elger CE (2003). Antidepressive treatment in patients with temporal lobe epilepsy and major depression: a prospective study with three different antidepressants. Epilepsy Behav 4:674-679.
Kuhn R (1958). The treatment of depressive states with G 22355 (imipramine hydrochloride). Am J Psychiatry 115:459-464.
Kumar G, Couper A, O'Brien TJ, Salzberg MR, Jones NC, Rees SM, et al. (2007). The acceleration of amygdala kindling epileptogenesis by chronic low-dose corticosterone involves both mineralocorticoid and glucocorticoid receptors. Psychoneuroendocrinology 32:834-842.
Kumar G, Jones NC, Morris MJ, Rees S, O'Brien TJ, Salzberg MR (2011). Early life stress enhancement of limbic epileptogenesis in adult rats: mechanistic insights. PLoS One 6:e24033.

134
Kurt M, Arik AC, Celik S (2000). The effects of sertraline and fluoxetine on anxiety in the elevated plus-maze test in mice. J Basic Clin Physiol Pharmacol 11:173-180.
Kuster GW, Braga-Neto P, Santos-Neto D, Garcia Santana MT, Maia Jr AC, Povoas Barsottini OG (2007). Hippocampal sclerosis and status epilepticus: cause or consequence? A MRI study. Arq Neuropsiquiatr 65:1101-1104.
Kwan P, Arzimanoglou A, Berg AT, Brodie MJ, Allen Hauser W, Mathern G, et al. (2010). Definition of drug resistant epilepsy: consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia 51:1069-1077.
Kwan P, Brodie MJ (2000). Epilepsy after the first drug fails: substitution or add-on? Seizure 9:464-468.
Kwan P, Brodie MJ (2001). Neuropsychological effects of epilepsy and antiepileptic drugs. Lancet 357:216-222.
Kwan P, Sander JW (2004). The natural history of epilepsy: an epidemiological view. J Neurol Neurosurg Psychiatry 75:1376-1381.
Lacey CJ, Salzberg MR, Roberts H, Trauer T, D'Souza WJ (2009). Psychiatric comorbidity and impact on health service utilization in a community sample of patients with epilepsy. Epilepsia 50:1991-1994.
Lambert MV, Robertson MM (1999). Depression in epilepsy: etiology, phenomenology, and treatment. Epilepsia 40 Suppl 10:S21-47.
Lanquillon S, Krieg JC, Bening-Abu-Shach U, Vedder H (2000). Cytokine production and treatment response in major depressive disorder. Neuropsychopharmacology 22:370-379.
Laplagne DA, Esposito MS, Piatti VC, Morgenstern NA, Zhao C, van Praag H, et al. (2006). Functional convergence of neurons generated in the developing and adult hippocampus. PLoS biology 4:e409.
Larmet Y, Reibel S, Carnahan J, Nawa H, Marescaux C, Depaulis A (1995). Protective effects of brain-derived neurotrophic factor on the development of hippocampal kindling in the rat. Neuroreport 6:1937-1941.
Le Blanc F, Rasmussen T (1974). Handbook of Clincal Neurology. Amsterdam: Elsevier.
Lemberger L (1972). Role of drug metabolism in drug research and development: importance of drug metabolism in clinical pharmacological evaluation of new drugs. J Pharm Sci 61:1690-1694.
Leonard BE (1996). The comparative pharmacological properties of selective serotonin reuptake inhibitors in animals. In: Selective Serotonin Reuptake Inhibitors, 2nd Edition: Advances in Basic Research and Clinical Practice(Feighner, J. P. and Boyer, W. F., eds) Chichester: Wiley.
Leonardi M, Ustun TB (2002). The global burden of epilepsy. Epilepsia 43 Suppl 6:21-25.
Lewis A (1934). Melancholia: a historical review. J Ment Sci 80:1-42.

135
Li JQ, Jia YX, Yamaya M, Arai H, Ohrui T, Sekizawa K, et al. (2002). Neurochemical regulation of cough response to capsaicin in guinea-pigs. Autonomic & autacoid pharmacology 22:57-63.
Linkowski P, Mendlewicz J, Kerkhofs M, Leclercq R, Golstein J, Brasseur M, et al. (1987). 24-hour profiles of adrenocorticotropin, cortisol, and growth hormone in major depressive illness: effect of antidepressant treatment. J Clin Endocrinol Metab 65:141-152.
Loomer HP, Saunders JC, Kline NS (1957). A clinical and pharmacodynamic evaluation of iproniazid as a psychic energizer. Psychiatr Res Rep Am Psychiatr Assoc 8:129-141.
Lopez-Munoz F, Alamo C (2009). Monoaminergic neurotransmission: the history of the discovery of antidepressants from 1950s until today. Curr Pharm Des 15:1563-1586.
Lopez-Munoz F, Alamo C, Juckel G, Assion HJ (2007). Half a century of antidepressant drugs: on the clinical introduction of monoamine oxidase inhibitors, tricyclics, and tetracyclics. Part I: monoamine oxidase inhibitors. J Clin Psychopharmacol 27:555-559.
Loscher, W. (1998a). New visions in the pharmacology of anticonvulsion. Eur . Pharmacol 342: 1–13 Loscher, W. (1998b) Pharmacology of glutamate receptor antagonists in the kindling model of epilepsy. Prog Neurobiol 54: 721–741 Loscher, W. (1999). Animal models of epilepsy and epileptic seizures. In: Antiepileptic Drugs. Handbook of Experimental Pharmacology (Eadie, M.J. and Vajda, F., eds) Berlin: Springer
Loscher W (2002). Current status and future directions in the pharmacotherapy of epilepsy. Trends Pharmacol Sci 23:113-118.
Loscher W (2009). Preclinical assessment of proconvulsant drug activity and its relevance for predicting adverse events in humans. Eur J Pharmacol 610:1-11.
Loscher W, Wahnschaffe U, Honack D, Rundfeldt C (1995). Does prolonged implantation of depth electrodes predispose the brain to kindling? Brain Res 697:197-204.
Lowenstein DH (1996). Recent advances related to basic mechanisms of epileptogenesis. Epilepsy Res Suppl 11:45-60.
Lu KT, Gean PW (1998). Endogenous serotonin inhibits epileptiform activity in rat hippocampal CA1 neurons via 5-hydroxytryptamine1A receptor activation. Neuroscience 86:729-737.
Luchins D, Ananth J (1976). Therapeutic implications of tricyclic antidepressant plasma levels. J Nerv Ment Dis 162:430-436.
Luchins DJ, Oliver AP, Wyatt RJ (1984). Seizures with antidepressants: an in vitro technique to assess relative risk. Epilepsia 25:25-32.
Lupien SJ, McEwen BS, Gunnar MR, Heim C (2009). Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nat Rev Neurosci 10:434-445.
Maes M, Meltzer HY, Bosmans E, Bergmans R, Vandoolaeghe E, Ranjan R, et al. (1995). Increased plasma concentrations of interleukin-6, soluble interleukin-6, soluble interleukin-2 and transferrin receptor in major depression. J Affect Disord 34:301-309.

136
Maj J, Moryl E (1993). Effects of Fluoxetine given chronically on the responsiveness of 5-HT receptor subpopulations to their agonist. European Neuropsychopharmacology 3:85-94.
Malberg JE, Eisch AJ, Nestler EJ, Duman RS (2000). Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J Neurosci 20:9104-9110.
Manji HK, Drevets WC, Charney DS (2001). The cellular neurobiology of depression. Nat Med 7:541-547.
Marcussen AB, Flagstad P, Kristjansen PE, Johansen FF, Englund U (2008). Increase in neurogenesis and behavioural benefit after chronic fluoxetine treatment in Wistar rats. Acta Neurol Scand 117:94-100.
Markakis EA, Gage FH (1999). Adult-generated neurons in the dentate gyrus send axonal projections to field CA3 and are surrounded by synaptic vesicles. The Journal of comparative neurology 406:449-460.
Marsteller DA, Barbarich-Marsteller NC, Patel VD, Dewey SL (2007). Brain metabolic changes following 4-week citalopram infusion: increased 18FDG uptake and gamma-amino butyric acid levels. Synapse 61:877-881.
Martignoni M, Groothuis GM, de Kanter R (2006). Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition and induction. Expert Opin Drug Metab Toxicol 2:875-894.
Mathern GW, Babb TL, Micevych PE, Blanco CE, Pretorius JK (1997). Granule cell mRNA levels for BDNF, NGF, and NT-3 correlate with neuron losses or supragranular mossy fiber sprouting in the chronically damaged and epileptic human hippocampus. Mol Chem Neuropathol 30:53-76.
Mathern GW, Babb TL, Vickrey BG, Melendez M, Pretorius JK (1995). The clinical-pathogenic mechanisms of hippocampal neuron loss and surgical outcomes in temporal lobe epilepsy. Brain 118 ( Pt 1):105-118.
Mathews IZ, Wilton A, Styles A, McCormick CM (2008). Increased depressive behaviour in females and heightened corticosterone release in males to swim stress after adolescent social stress in rats. Behav Brain Res 190:33-40.
Mazarati A, Shin D, Auvin S, Caplan R, Sankar R (2007). Kindling epileptogenesis in immature rats leads to persistent depressive behavior. Epilepsy Behav 10:377-383.
Mazarati A, Siddarth P, Baldwin RA, Shin D, Caplan R, Sankar R (2008). Depression after status epilepticus: behavioural and biochemical deficits and effects of fluoxetine. Brain 131:2071-2083.
Mazarati AM, Pineda E, Shin D, Tio D, Taylor AN, Sankar R (2010). Comorbidity between epilepsy and depression: role of hippocampal interleukin-1beta. Neurobiol Dis 37:461-467.
Mazarati AM, Shin D, Kwon YS, Bragin A, Pineda E, Tio D, et al. (2009). Elevated plasma corticosterone level and depressive behavior in experimental temporal lobe epilepsy. Neurobiol Dis 34:457-461.

137
McCloskey DP, Hintz TM, Pierce JP, Scharfman HE (2006). Stereological methods reveal the robust size and stability of ectopic hilar granule cells after pilocarpine-induced status epilepticus in the adult rat. Eur J Neurosci 24:2203-2210.
McKay MS, Zakzanis KK (2010). The impact of treatment on HPA axis activity in unipolar major depression. J Psychiatr Res 44:183-192.
McLaughlin DP, Pachana NA, McFarland K (2008). Depression in a community-dwelling sample of older adults with late-onset or lifetime epilepsy. Epilepsy Behav 12:281-285.
McNamara JO (1986). Kindling model of epilepsy. Adv Neurol 44:303-318.
Mensah SA, Beavis JM, Thapar AK, Kerr MP (2007). A community study of the presence of anxiety disorder in people with epilepsy. Epilepsy Behav 11:118-124.
Mesquita AR, Tavares HB, Silva R, Sousa N (2006). Febrile convulsions in developing rats induce a hyperanxious phenotype later in life. Epilepsy Behav 9:401-406.
Michalakis M, Holsinger D, Ikeda-Douglas C, Cammisuli S, Ferbinteanu J, DeSouza C, et al. (1998). Development of spontaneous seizures over extended electrical kindling. I. Electrographic, behavioral, and transfer kindling correlates. Brain Res 793:197-211.
Miki T, Satriotomo I, Li HP, Matsumoto Y, Gu H, Yokoyama T, et al. (2005). Application of the physical disector to the central nervous system: estimation of the total number of neurons in subdivisions of the rat hippocampus. Anatomical science international 80:153-162.
Mikova O, Yakimova R, Bosmans E, Kenis G, Maes M (2001). Increased serum tumor necrosis factor alpha concentrations in major depression and multiple sclerosis. Eur Neuropsychopharmacol 11:203-208.
Miller AH, Maletic V, Raison CL (2009). Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry 65:732-741.
Miller BH, Schultz LE, Gulati A, Cameron MD, Pletcher MT (2008). Genetic regulation of behavioral and neuronal responses to fluoxetine. Neuropsychopharmacology 33:1312-1322.
Minami M, Kuraishi Y, Satoh M (1991). Effects of kainic acid on messenger RNA levels of IL-1 beta, IL-6, TNF alpha and LIF in the rat brain. Biochem Biophys Res Commun 176:593-598.
Miro X, Perez-Torres S, Artigas F, Puigdomenech P, Palacios JM, Mengod G (2002). Regulation of cAMP phosphodiesterase mRNAs expression in rat brain by acute and chronic fluoxetine treatment. An in situ hybridization study. Neuropharmacology 43:1148-1157.
Mirrione MM, Schiffer WK, Siddiq M, Dewey SL, Tsirka SE (2006). PET imaging of glucose metabolism in a mouse model of temporal lobe epilepsy. Synapse 59:119-121.
Mohanraj R, Brodie MJ (2006). Diagnosing refractory epilepsy: response to sequential treatment schedules. Eur J Neurol 13:277-282.
Molteni R, Calabrese F, Cattaneo A, Mancini M, Gennarelli M, Racagni G, et al. (2009). Acute stress responsiveness of the neurotrophin BDNF in the rat hippocampus is modulated

138
by chronic treatment with the antidepressant duloxetine. Neuropsychopharmacology 34:1523-1532.
Moncek F, Duncko R, Jezova D (2003). Repeated citalopram treatment but not stress exposure attenuates hypothalamic-pituitary-adrenocortical axis response to acute citalopram injection. Life Sci 72:1353-1365.
Morgan VA, Croft ML, Valuri GM, Zubrick SR, Bower C, McNeil TF, et al. (2012). Intellectual disability and other neuropsychiatric outcomes in high-risk children of mothers with schizophrenia, bipolar disorder and unipolar major depression. Br J Psychiatry 200:282-289.
Morilak DA, Frazer A (2004). Antidepressants and brain monoaminergic systems: a dimensional approach to understanding their behavioural effects in depression and anxiety disorders. Int J Neuropsychopharmacol 7:193-218.
Morimoto K, Fahnestock M, Racine RJ (2004). Kindling and status epilepticus models of epilepsy: rewiring the brain. Prog Neurobiol 73:1-60.
Morrell F (1985). Secondary epileptogenesis in man. Arch Neurol 42:318-335.
Motamedi G, Meador K (2003). Epilepsy and cognition. Epilepsy Behav 4 Suppl 2:S25-38.
Mula M, Trimble M (2003). Pharmacokinetic Interactions between Antiepileptic and Antidepressant Drugs. World J Biol Psychiatry 4:21-24.
Muller CJ, Bankstahl M, Groticke I, Loscher W (2009a). Pilocarpine vs. lithium-pilocarpine for induction of status epilepticus in mice: development of spontaneous seizures, behavioral alterations and neuronal damage. Eur J Pharmacol 619:15-24.
Muller CJ, Groticke I, Bankstahl M, Loscher W (2009b). Behavioral and cognitive alterations, spontaneous seizures, and neuropathology developing after a pilocarpine-induced status epilepticus in C57BL/6 mice. Exp Neurol 219:284-297.
Murphy BL, Pun RY, Yin H, Faulkner CR, Loepke AW, Danzer SC (2011). Heterogeneous integration of adult-generated granule cells into the epileptic brain. J Neurosci 31:105-117.
Murray KD, Isackson PJ, Eskin TA, King MA, Montesinos SP, Abraham LA, et al. (2000). Altered mRNA expression for brain-derived neurotrophic factor and type II calcium/calmodulin-dependent protein kinase in the hippocampus of patients with intractable temporal lobe epilepsy. The Journal of comparative neurology 418:411-422.
Naegele JR (2007). Neuroprotective strategies to avert seizure-induced neurodegeneration in epilepsy. Epilepsia 48 Suppl 2:107-117.
Nakagawa E, Aimi Y, Yasuhara O, Tooyama I, Shimada M, McGeer PL, et al. (2000). Enhancement of progenitor cell division in the dentate gyrus triggered by initial limbic seizures in rat models of epilepsy. Epilepsia 41:10-18.
Neal EG, Chaffe H, Schwartz RH, Lawson MS, Edwards N, Fitzsimmons G, et al. (2008). The ketogenic diet for the treatment of childhood epilepsy: a randomised controlled trial. Lancet Neurol 7:500-506.

139
Nearing K, Madhavan D, Devinsky O (2007). Temporal lobe epilepsy: a progressive disorder? Rev Neurol Dis 4:122-127.
Nestler EJ, Barrot M, DiLeone RJ, Eisch AJ, Gold SJ, Monteggia LM (2002). Neurobiology of depression. Neuron 34:13-25.
Nestler EJ, Terwilliger RZ, Duman RS (1989). Chronic antidepressant administration alters the subcellular distribution of cyclic AMP-dependent protein kinase in rat frontal cortex. J Neurochem 53:1644-1647.
Ni YG, Miledi R (1997). Blockage of 5HT(2C) serotonin receptors by fluoxetine (Prozac). P Natl Acad Sci USA 94:2036-2040.
Nibuya M, Morinobu S, Duman RS (1995). Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J Neurosci 15:7539-7547.
Nibuya M, Nestler EJ, Duman RS (1996). Chronic antidepressant administration increases the expression of cAMP response element binding protein (CREB) in rat hippocampus. J Neurosci 16:2365-2372.
Niespodziany I, Klitgaard H, Margineanu DG (1999). Chronic electrode implantation entails epileptiform field potentials in rat hippocampal slices, similarly to amygdala kindling. Epilepsy Res 36:69-74.
Niv F, Keiner S, Krishna, Witte OW, Lie DC, Redecker C (2012). Aberrant neurogenesis after stroke: a retroviral cell labeling study. Stroke 43:2468-2475.
Norcross M, Mathur P, Enoch AJ, Karlsson RM, Brigman JL, Cameron HA, et al. (2008). Effects of adolescent fluoxetine treatment on fear-, anxiety- or stress-related behaviors in C57BL/6J or BALB/cJ mice. Psychopharmacology (Berl) 200:413-424.
O'Brien TJ, So EL, Meyer FB, Parisi JE, Jack CR (1999). Progressive hippocampal atrophy in chronic intractable temporal lobe epilepsy. Ann Neurol 45:526-529.
O'Mara SM, Commins S, Anderson M, Gigg J (2001). The subiculum: a review of form, physiology and function. Prog Neurobiol 64:129-155.
Oh JE, Zupan B, Gross S, Toth M (2009). Paradoxical anxiogenic response of juvenile mice to fluoxetine. Neuropsychopharmacology 34:2197-2207.
Okazaki M, Adachi N, Ito M, Watanabe M, Watanabe Y, Kato M, et al. (2011). One-year seizure prognosis in epilepsy patients treated with antidepressants. Epilepsy Behav 22:331-335.
Ono T, Galanopoulou AS (2012). Epilepsy and epileptic syndrome. Adv Exp Med Biol 724:99-113.
Ormel J, Von Korff M, Van den Brink W, Katon W, Brilman E, Oldehinkel T (1993). Depression, anxiety, and social disability show synchrony of change in primary care patients. Am J Public Health 83:385-390.
Osehobo P, Adams B, Sazgar M, Xu Y, Racine RJ, Fahnestock M (1999). Brain-derived neurotrophic factor infusion delays amygdala and perforant path kindling without affecting paired-pulse measures of neuronal inhibition in adult rats. Neuroscience 92:1367-1375.

140
Osonoe K, Mori N, Suzuki K, Osonoe M (1994). Antiepileptic effects of inhibitors of nitric oxide synthase examined in pentylenetetrazol-induced seizures in rats. Brain Res 663:338-340.
Ottman R, Lipton RB, Ettinger AB, Cramer JA, Reed ML, Morrison A, et al. (2011). Comorbidities of epilepsy: results from the Epilepsy Comorbidities and Health (EPIC) survey. Epilepsia 52:308-315.
Overstreet LS, Hentges ST, Bumaschny VF, de Souza FS, Smart JL, Santangelo AM, et al. (2004). A transgenic marker for newly born granule cells in dentate gyrus. J Neurosci 24:3251-3259.
Owen BM, Eccleston D, Ferrier IN, Young AH (2001). Raised levels of plasma interleukin-1beta in major and postviral depression. Acta Psychiatr Scand 103:226-228.
Owens MJ, Morgan WN, Plott SJ, Nemeroff CB (1997). Neurotransmitter receptor and transporter binding profile of antidepressants and their metabolites. J Pharmacol Exp Ther 283:1305-1322.
Palvimaki EP, Roth BL, Majasuo H, Laakso A, Kuoppamaki M, Syvalahti E, et al. (1996). Interactions of selective serotonin reuptake inhibitors with the serotonin 5-HT2c receptor. Psychopharmacology (Berl) 126:234-240.
Parent JM (2007). Adult neurogenesis in the intact and epileptic dentate gyrus. Prog Brain Res 163:529-540.
Parent JM, Elliott RC, Pleasure SJ, Barbaro NM, Lowenstein DH (2006). Aberrant seizure-induced neurogenesis in experimental temporal lobe epilepsy. Ann Neurol 59:81-91.
Parent JM, Janumpalli S, McNamara JO, Lowenstein DH (1998). Increased dentate granule cell neurogenesis following amygdala kindling in the adult rat. Neurosci Lett 247:9-12.
Parent JM, Jessberger S, Gage FH, Gong C (2007). Is neurogenesis reparative after status epilepticus? Epilepsia 48 Suppl 8:69-71.
Parent JM, Yu TW, Leibowitz RT, Geschwind DH, Sloviter RS, Lowenstein DH (1997). Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. J Neurosci 17:3727-3738.
Pariante CM (2006). The glucocorticoid receptor: part of the solution or part of the problem? J Psychopharmacol 20:79-84.
Pariante CM, Lightman SL (2008). The HPA axis in major depression: classical theories and new developments. Trends Neurosci 31:464-468.
Parsons LH, Kerr TM, Tecott LH (2001). 5-HT1A receptor mutant mice exhibit enhanced tonic, stress-induced and fluoxetine-induced serotonergic neurotransmission. Journal of Neurochemistry 77:607-617.
Pawluski JL, Charlier TD, Fillet M, Houbart V, Crispin HT, Steinbusch HW, et al. (2012). Chronic fluoxetine treatment and maternal adversity differentially alter neurobehavioral outcomes in the rat dam. Behav Brain Res 228:159-168.
Paxinos G, Watson C (1998). The rat brain in stereotaxic coordinates. San Diego: Academic Press.

141
Payandemehr B, Bahremand A, Rahimian R, Ziai P, Amouzegar A, Sharifzadeh M, et al. (2012). 5-HT(3) receptor mediates the dose-dependent effects of citalopram on pentylenetetrazole-induced clonic seizure in mice: Involvement of nitric oxide. Epilepsy Res.
Peele DB, Gilbert ME (1992). Functional dissociation of acute and persistent cognitive deficits accompanying amygdala-kindled seizures. Behav Brain Res 48:65-76.
Peredery O, Persinger MA, Parker G, Mastrosov L (2000). Temporal changes in neuronal dropout following inductions of lithium/pilocarpine seizures in the rat. Brain Res 881:9-17.
Perez J, Tinelli D, Bianchi E, Brunello N, Racagni G (1991). cAMP binding proteins in the rat cerebral cortex after administration of selective 5-HT and NE reuptake blockers with antidepressant activity. Neuropsychopharmacology 4:57-64.
Pericic D, Lazic J, Svob Strac D (2005). Anticonvulsant effects of acute and repeated fluoxetine treatment in unstressed and stressed mice. Brain Res 1033:90-95.
Petrovski S, Szoeke CE, Jones NC, Salzberg MR, Sheffield LJ, Huggins RM, et al. (2010). Neuropsychiatric symptomatology predicts seizure recurrence in newly treated patients. Neurology 75:1015-1021.
Piletz JE, Halaris A, Iqbal O, Hoppensteadt D, Fareed J, Zhu H, et al. (2009). Pro-inflammatory biomakers in depression: treatment with venlafaxine. World J Biol Psychiatry 10:313-323.
Pineda EA, Hensler JG, Sankar R, Shin D, Burke TF, Mazarati AM (2012). Interleukin-1beta causes fluoxetine resistance in an animal model of epilepsy-associated depression. Neurotherapeutics 9:477-485.
Pinel JP, Rovner LI (1978). Experimental epileptogenesis: kindling-induced epilepsy in rats. Exp Neurol 58:190-202.
Pisani F, Spina E, Oteri G (1999). Antidepressant drugs and seizure susceptibility: from in vitro data to clinical practice. Epilepsia 40 Suppl 10:S48-56.
Pitkanen A (2002). Drug-mediated neuroprotection and antiepileptogenesis: animal data. Neurology 59:S27-33.
Pitkanen A (2010). Therapeutic approaches to epileptogenesis--hope on the horizon. Epilepsia 51 Suppl 3:2-17.
Pitkanen A, Bolkvadze T, Immonen R (2011). Anti-epileptogenesis in rodent post-traumatic epilepsy models. Neurosci Lett 497:163-171.
Pitkanen A, Kharatishvili I, Karhunen H, Lukasiuk K, Immonen R, Nairismagi J, et al. (2007). Epileptogenesis in experimental models. Epilepsia 48 Suppl 2:13-20.
Pitkanen A, Lukasiuk K (2011). Mechanisms of epileptogenesis and potential treatment targets. Lancet Neurol 10:173-186.
Pitkanen A, Sutula TP (2002). Is epilepsy a progressive disorder? Prospects for new therapeutic approaches in temporal-lobe epilepsy. Lancet Neurol 1:173-181.

142
Popoli M, Gennarelli M, Racagni G (2002). Modulation of synaptic plasticity by stress and antidepressants. Bipolar Disord 4:166-182.
Porsolt RD, Le Pichon M, Jalfre M (1977). Depression: a new animal model sensitive to antidepressant treatments. Nature 266:730-732.
Post RM (2002). Do the epilepsies, pain syndromes, and affective disorders share common kindling-like mechanisms? Epilepsy Res 50:203-219.
Prendiville S, Gale K (1993). Anticonvulsant effect of fluoxetine on focally evoked limbic motor seizures in rats. Epilepsia 34:381-384.
Preskorn SH, Fast GA (1992). Tricyclic antidepressant-induced seizures and plasma drug concentration. J Clin Psychiatry 53:160-162.
Pritchard PB, 3rd, Wannamaker BB, Sagel J, Daniel CM (1985). Serum prolactin and cortisol levels in evaluation of pseudoepileptic seizures. Ann Neurol 18:87-89.
Przegalinski E, Baran L, Siwanowicz J (1994). Role of 5-Hydroxytryptamine Receptor Subtypes in the 1-[3-(Trifluoromethyl)Phenyl] Piperazine-Induced Increase in Threshold for Maximal Electroconvulsions in Mice. Epilepsia 35:889-894.
Racine R (1978). Kindling: the first decade. Neurosurgery 3:234-252.
Racine RJ (1972a). Modification of seizure activity by electrical stimulation. I. After-discharge threshold. Electroencephalogr Clin Neurophysiol 32:269-279.
Racine RJ (1972b). Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol 32:281-294.
Racine RJ (1975). Modification of seizure activity by electrical stimulation: cortical areas. Electroencephalogr Clin Neurophysiol 38:1-12.
Raedt R, Boon P, Persson A, Alborn AM, Boterberg T, Van Dycke A, et al. (2007). Radiation of the rat brain suppresses seizure-induced neurogenesis and transiently enhances excitability during kindling acquisition. Epilepsia 48:1952-1963.
Rahimi O, Claiborne BJ (2007). Morphological development and maturation of granule neuron dendrites in the rat dentate gyrus. Prog Brain Res 163:167-181.
Rai D, Kerr MP, McManus S, Jordanova V, Lewis G, Brugha TS (2012). Epilepsy and psychiatric comorbidity: a nationally representative population-based study. Epilepsia 53:1095-1103.
Raju SS, Noor AR, Gurthu S, Giriyappanavar CR, Acharya SB, Low HC, et al. (1999). Effect of fluoxetine on maximal electroshock seizures in mice: acute vs chronic administration. Pharmacol Res 39:451-454.
Rao ML, Stefan H, Bauer J (1989). Epileptic but not psychogenic seizures are accompanied by simultaneous elevation of serum pituitary hormones and cortisol levels. Neuroendocrinology 49:33-39.
Rees L, Brown AC, Benaim S (1961). A controlled trial of imipramine ("Tofranil") in the treatment of severe depressive states. J Ment Sci 107:552-559.

143
Reibel S, Larmet Y, Carnahan J, Marescaux C, Depaulis A (2000a). Endogenous control of hippocampal epileptogenesis: a molecular cascade involving brain-derived neurotrophic factor and neuropeptide Y. Epilepsia 41 Suppl 6:S127-133.
Reibel S, Larmet Y, Le BT, Carnahan J, Marescaux C, Depaulis A (2000b). Brain-derived neurotrophic factor delays hippocampal kindling in the rat. Neuroscience 100:777-788.
Riad M, Rbah L, Verdurand M, Aznavour N, Zimmer L, Descarries L (2008). Unchanged density of 5-HT(1A) autoreceptors on the plasma membrane of nucleus raphe dorsalis neurons in rats chronically treated with fluoxetine. Neuroscience 151:692-700.
Ribak CE, Shapiro LA (2007). Dendritic development of newly generated neurons in the adult brain. Brain Res Rev 55:390-394.
Richelson E (2001). Pharmacology of antidepressants. Mayo Clin Proc 76:511-527.
Richman A, Heinrichs SC (2007). Seizure prophylaxis in an animal model of epilepsy by dietary fluoxetine supplementation. Epilepsy Res 74:19-27.
Ridsdale L, Charlton J, Ashworth M, Richardson MP, Gulliford MC (2011). Epilepsy mortality and risk factors for death in epilepsy: a population-based study. Br J Gen Pract 61:e271-278.
Robertson MM, Trimble MR, Townsend HR (1987). Phenomenology of depression in epilepsy. Epilepsia 28:364-372.
Robinson RT, Drafts BC, Fisher JL (2003). Fluoxetine increases GABA(A) receptor activity through a novel modulatory site. J Pharmacol Exp Ther 304:978-984.
Roch C, Leroy C, Nehlig A, Namer IJ (2002a). Magnetic resonance imaging in the study of the lithium-pilocarpine model of temporal lobe epilepsy in adult rats. Epilepsia 43:325-335.
Roch C, Leroy C, Nehlig A, Namer IJ (2002b). Predictive value of cortical injury for the development of temporal lobe epilepsy in 21-day-old rats: an MRI approach using the lithium-pilocarpine model. Epilepsia 43:1129-1136.
Rodgers RJ, Dalvi A (1997). Anxiety, defence and the elevated plus-maze. Neurosci Biobehav Rev 21:801-810.
Rosebook P, Kalin N (2011). Citalopram and s-Citalopram. In: The American Psychiatric Publishing Textbook of Psychopharmacology(Schatzberg, A. and Nemeroff, C. B., eds): American Psychiatric Publishing.
Roseboom PH, Kalin NH (2011). Citalopram and s-citalopram. In: The American Psychiatric Publishing Textbook of Psyhcopharmacology(Schatzberg, A. F. and Nemeroff, C. B., eds): American Psychiatric Publishing.
Rosenstein DL, Nelson JC, Jacobs SC (1993). Seizures associated with antidepressants: a review. J Clin Psychiatry 54:289-299.
Rudge JS, Mather PE, Pasnikowski EM, Cai N, Corcoran T, Acheson A, et al. (1998). Endogenous BDNF protein is increased in adult rat hippocampus after a kainic acid induced excitotoxic insult but exogenous BDNF is not neuroprotective. Exp Neurol 149:398-410.
Rugg-Gunn FJ, Eriksson SH, Symms MR, Barker GJ, Duncan JS (2001). Diffusion tensor imaging of cryptogenic and acquired partial epilepsies. Brain 124:627-636.

144
Russo-Neustadt AA, Beard RC, Huang YM, Cotman CW (2000). Physical activity and antidepressant treatment potentiate the expression of specific brain-derived neurotrophic factor transcripts in the rat hippocampus. Neuroscience 101:305-312.
Saarelainen T, Hendolin P, Lucas G, Koponen E, Sairanen M, MacDonald E, et al. (2003). Activation of the TrkB neurotrophin receptor is induced by antidepressant drugs and is required for antidepressant-induced behavioral effects. J Neurosci 23:349-357.
Sadaghiani MM, Saboory E (2010). Prenatal stress potentiates pilocarpine-induced epileptic behaviors in infant rats both time and sex dependently. Epilepsy Behav 18:166-170.
Salzberg M, Kumar G, Supit L, Jones NC, Morris MJ, Rees S, et al. (2007). Early postnatal stress confers enduring vulnerability to limbic epileptogenesis. Epilepsia 48:2079-2085.
Salzberg MR, Vajda FJ (2001). Epilepsy, depression and antidepressant drugs. J Clin Neurosci 8:209-215.
Sanacora G, Mason GF, Rothman DL, Krystal JH (2002). Increased occipital cortex GABA concentrations in depressed patients after therapy with selective serotonin reuptake inhibitors. Am J Psychiatry 159:663-665.
Sandler M (1990). Monoamine oxidase inhibitors in depression: history and mythology. J Psychopharmacol 4:136-139.
Santarelli L, Saxe M, Gross C, Surget A, Battaglia F, Dulawa S, et al. (2003). Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 301:805-809.
Santos JG, Jr., Do Monte FH, Russi M, Agustine PE, Lanziotti VM (2002). Proconvulsant effects of high doses of venlafaxine in pentylenetetrazole-convulsive rats. Braz J Med Biol Res 35:469-472.
Sapolsky RM, Krey LC, McEwen BS (1984). Glucocorticoid-sensitive hippocampal neurons are involved in terminating the adrenocortical stress response. Proc Natl Acad Sci U S A 81:6174-6177.
Sarkisova K, van Luijtelaar G (2011). The WAG/Rij strain: a genetic animal model of absence epilepsy with comorbidity of depression [corrected]. Prog Neuropsychopharmacol Biol Psychiatry 35:854-876.
Sarnyai Z, Sibille EL, Pavlides C, Fenster RJ, McEwen BS, Toth M (2000). Impaired hippocampal-dependent learning and functional abnormalities in the hippocampus in mice lacking serotonin(1A) receptors. Proc Natl Acad Sci U S A 97:14731-14736.
Sato K, Morimoto K, Okamoto M, Nakamura Y, Otsuki S, Sato M (1990). An analysis of anticonvulsant actions of GABA agonists (progabide and baclofen) in the kindling model of epilepsy. Epilepsy Res 5:117-124.
Saxe MD, Battaglia F, Wang JW, Malleret G, David DJ, Monckton JE, et al. (2006). Ablation of hippocampal neurogenesis impairs contextual fear conditioning and synaptic plasticity in the dentate gyrus. Proc Natl Acad Sci U S A 103:17501-17506.
Sayin U, Osting S, Hagen J, Rutecki P, Sutula T (2003). Spontaneous seizures and loss of axo-axonic and axo-somatic inhibition induced by repeated brief seizures in kindled rats. J Neurosci 23:2759-2768.

145
Scharfman H, Goodman J, McCloskey D (2007). Ectopic granule cells of the rat dentate gyrus. Developmental neuroscience 29:14-27.
Scharfman HE (2007). The neurobiology of epilepsy. Curr Neurol Neurosci Rep 7:348-354.
Scharfman HE, Goodman JH, Sollas AL (2000). Granule-like neurons at the hilar/CA3 border after status epilepticus and their synchrony with area CA3 pyramidal cells: functional implications of seizure-induced neurogenesis. J Neurosci 20:6144-6158.
Scharfman HE, Goodman JH, Sollas AL, Croll SD (2002). Spontaneous limbic seizures after intrahippocampal infusion of brain-derived neurotrophic factor. Exp Neurol 174:201-214.
Scharfman HE, Gray WP (2007). Relevance of seizure-induced neurogenesis in animal models of epilepsy to the etiology of temporal lobe epilepsy. Epilepsia 48 Suppl 2:33-41.
Scharfman HE, Pedley TA (2006). Temporal lobe epilepsy. New York: Academic Press.
Schmidt-Hieber C, Jonas P, Bischofberger J (2004). Enhanced synaptic plasticity in newly generated granule cells of the adult hippocampus. Nature 429:184-187.
Schmitz B (2011). The effects of antiepileptic drugs on behavior. In: The effects of antiepileptic drugs on behaviour (Trimble, M. and Schmitz, B., eds), pp133-142 Cambridge: Cambridge University Press.
Scott BW, Wang S, Burnham WM, De Boni U, Wojtowicz JM (1998). Kindling-induced neurogenesis in the dentate gyrus of the rat. Neurosci Lett 248:73-76.
Serra M, Pisu MG, Muggironi M, Parodo V, Papi G, Sari R, et al. (2001). Opposite effects of short- versus long-term administration of fluoxetine on the concentrations of neuroactive steroids in rat plasma and brain. Psychopharmacology (Berl) 158:48-54.
Shapiro LA, Wang L, Ribak CE (2008). Rapid astrocyte and microglial activation following pilocarpine-induced seizures in rats. Epilepsia 49 Suppl 2:33-41.
Sharma AK, Reams RY, Jordan WH, Miller MA, Thacker HL, Snyder PW (2007). Mesial temporal lobe epilepsy: pathogenesis, induced rodent models and lesions. Toxicol Pathol 35:984-999.
Shimizu Y, Minatoguchi S, Hashimoto K, Uno Y, Arai M, Wang N, et al. (2002). The role of serotonin in ischemic cellular damage and the infarct size-reducing effect of sarpogrelate, a 5-hydroxytryptamine-2 receptor blocker, in rabbit hearts. J Am Coll Cardiol 40:1347-1355.
Shorvon S, Luciano AL (2007). Prognosis of chronic and newly diagnosed epilepsy: revisiting temporal aspects. Curr Opin Neurol 20:208-212.
Sillanpaa M, Jalava M, Kaleva O, Shinnar S (1998). Long-term prognosis of seizures with onset in childhood. N Engl J Med 338:1715-1722.
Silva MT, Alves CR, Santarem EM (1999). Anxiogenic-like effect of acute and chronic fluoxetine on rats tested on the elevated plus-maze. Braz J Med Biol Res 32:333-339.

146
Silva RC, Brandao ML (2000). Acute and chronic effects of gepirone and fluoxetine in rats tested in the elevated plus-maze: an ethological analysis. Pharmacol Biochem Behav 65:209-216.
Smith JA (1953). The use of the isopropyl derivative of isonicotinylhydrazine (marsilid) in the treatment of mental disease; a preliminary report. Am Pract Dig Treat 4:519-520.
Smith MA, Weiss SR, Abedin T, Kim H, Post RM, Gold PW (1991). Effects of amygdala kindling and electroconvulsive seizures on the expression of corticotropin-releasing hormone in the rat brain. Mol Cell Neurosci 2:103-116.
Snyder JS, Kee N, Wojtowicz JM (2001). Effects of adult neurogenesis on synaptic plasticity in the rat dentate gyrus. J Neurophysiol 85:2423-2431.
Sohl G, Guldenagel M, Beck H, Teubner B, Traub O, Gutierrez R, et al. (2000). Expression of connexin genes in hippocampus of kainate-treated and kindled rats under conditions of experimental epilepsy. Brain Res Mol Brain Res 83:44-51.
Souetre E, Salvati E, Belugou JL, Pringuey D, Candito M, Krebs B, et al. (1989). Circadian rhythms in depression and recovery: evidence for blunted amplitude as the main chronobiological abnormality. Psychiatry Res 28:263-278.
Specchio LM, Iudice A, Specchio N, La Neve A, Spinelli A, Galli R, et al. (2004). Citalopram as treatment of depression in patients with epilepsy. Clin Neuropharmacol 27:133-136.
Stables JP, Bertram EH, White HS, Coulter DA, Dichter MA, Jacobs MP, et al. (2002). Models for epilepsy and epileptogenesis: report from the NIH workshop, Bethesda, Maryland. Epilepsia 43:1410-1420.
Stahl S (1994). 5HT1A receptors and pharmacotherapy. Is serotonin receptor down-regulation linked to the mechanism of action of antidepressant drugs? Psychopharmacol Bull 30:39-43.
Stahl SM (1992). Serotonin 1A Receptors in Depression and Anxiety. New York: Raven Press.
Stahl SM (1996). Essential Pharmacology. New York: University Press.
Stahl SM (1998). Mechanism of action of serotonin selective reuptake inhibitors. Serotonin receptors and pathways mediate therapeutic effects and side effects. J Affect Disord 51:215-235.
Statnick M, Dailey J, Jobe P, Browning R (1996a). Neither intranigral fluoxetine nor 5,7-dihydroxytryptamine alter audiogenic seizures in genetically epilepsy-prone rats. Eur J Pharmacol 299:93-102.
Statnick MA, Dailey JW, Jobe PC, Browning RA (1996b). Abnormalities in 5-HT1A and 5-HT1B receptor binding in severe-seizure genetically epilepsy-prone rats (GEPR-9s). Neuropharmacology 35:111-118.
Steiger A, von Bardeleben U, Herth T, Holsboer F (1989). Sleep EEG and nocturnal secretion of cortisol and growth hormone in male patients with endogenous depression before treatment and after recovery. J Affect Disord 16:189-195.

147
Steiner MA, Marsicano G, Wotjak CT, Lutz B (2008). Conditional cannabinoid receptor type 1 mutants reveal neuron subpopulation-specific effects on behavioral and neuroendocrine stress responses. Psychoneuroendocrinology 33:1165-1170.
Sutcigil L, Oktenli C, Musabak U, Bozkurt A, Cansever A, Uzun O, et al. (2007). Pro- and anti-inflammatory cytokine balance in major depression: effect of sertraline therapy. Clin Dev Immunol 2007:76396.
Sutula T (2002). Seizure-Induced Axonal Sprouting: Assessing Connections Between Injury, Local Circuits, and Epileptogenesis. Epilepsy Curr 2:86-91.
Sutula TP (1990). Experimental models of temporal lobe epilepsy: new insights from the study of kindling and synaptic reorganization. Epilepsia 31 Suppl 3:S45-54.
Swinkels WA, Kuyk J, van Dyck R, Spinhoven P (2005). Psychiatric comorbidity in epilepsy. Epilepsy Behav 7:37-50.
Taher TR, Salzberg M, Morris MJ, Rees S, O'Brien TJ (2005). Chronic low-dose corticosterone supplementation enhances acquired epileptogenesis in the rat amygdala kindling model of TLE. Neuropsychopharmacology 30:1610-1616.
Takahashi M, Hayashi S, Kakita A, Wakabayashi K, Fukuda M, Kameyama S, et al. (1999). Patients with temporal lobe epilepsy show an increase in brain-derived neurotrophic factor protein and its correlation with neuropeptide Y. Brain Res 818:579-582.
Takeshita H, Kawahara R, Nagabuchi T, Mizukawa R, Hazama H (1986). Serum prolactin, cortisol and growth hormone concentrations after various epileptic seizures. Jpn J Psychiatry Neurol 40:617-623.
Tanaka T, Tanaka S, Fujita T, Takano K, Fukuda H, Sako K, et al. (1992). Experimental complex partial seizures induced by a microinjection of kainic acid into limbic structures. Prog Neurobiol 38:317-334.
Tardito D, Perez J, Tiraboschi E, Musazzi L, Racagni G, Popoli M (2006). Signaling pathways regulating gene expression, neuroplasticity, and neurotrophic mechanisms in the action of antidepressants: a critical overview. Pharmacol Rev 58:115-134.
Tasch E, Cendes F, Li LM, Dubeau F, Andermann F, Arnold DL (1999). Neuroimaging evidence of progressive neuronal loss and dysfunction in temporal lobe epilepsy. Ann Neurol 45:568-576.
Tecott LH, Sun LM, Akana SF, Strack AM, Lowenstein DH, Dallman MF, et al. (1995). Eating disorder and epilepsy in mice lacking 5-HT2c serotonin receptors. Nature 374:542-546.
Tellez-Zenteno JF, Patten SB, Jette N, Williams J, Wiebe S (2007). Psychiatric comorbidity in epilepsy: a population-based analysis. Epilepsia 48:2336-2344.
Theeuwes F (1975). Elementary osmotic pump. J Pharm Sci 64:1987-1991.
Thome-Souza MS, Kuczynski E, Valente KD (2007). Sertraline and fluoxetine: safe treatments for children and adolescents with epilepsy and depression. Epilepsy Behav 10:417-425.
Thome J, Sakai N, Shin K, Steffen C, Zhang YJ, Impey S, et al. (2000). cAMP response element-mediated gene transcription is upregulated by chronic antidepressant treatment. J Neurosci 20:4030-4036.

148
Tiemeier H, Hofman A, van Tuijl HR, Kiliaan AJ, Meijer J, Breteler MM (2003). Inflammatory proteins and depression in the elderly. Epidemiology 14:103-107.
Tokumitsu T, Mancuso A, Weinstein PR, Weiner MW, Naruse S, Maudsley AA (1997). Metabolic and pathological effects of temporal lobe epilepsy in rat brain detected by proton spectroscopy and imaging. Brain Res 744:57-67.
Toni N, Teng EM, Bushong EA, Aimone JB, Zhao C, Consiglio A, et al. (2007). Synapse formation on neurons born in the adult hippocampus. Nat Neurosci 10:727-734.
Torta R, Keller R (1999). Behavioral, psychotic, and anxiety disorders in epilepsy: etiology, clinical features, and therapeutic implications. Epilepsia 40 Suppl 10:S2-20.
Tress KH, Herberg LJ (1972). Permanent reduction in seizure threshold resulting from repeated electrical stimulation. Exp Neurol 37:347-359.
Trimble M (1978). Non-monoamine oxidase inhibitor antidepressants and epilepsy: a review. Epilepsia 19:241-250.
Turrin NP, Rivest S (2004). Innate immune reaction in response to seizures: implications for the neuropathology associated with epilepsy. Neurobiol Dis 16:321-334.
Ugale RR, Mittal N, Hirani K, Chopde CT (2004). Essentiality of central GABAergic neuroactive steroid allopregnanolone for anticonvulsant action of fluoxetine against pentylenetetrazole-induced seizures in mice. Brain Res 1023:102-111.
Upton N, Stean T, Middlemiss D, Blackburn T, Kennett G (1998). Studies on the role of 5-HT2C and 5-HT2B receptors in regulating generalised seizure threshold in rodents. European Journal of Pharmacology 359:33-40.
Van Roost D, Solymosi L, Schramm J, van Oosterwyck B, Elger CE (1998). Depth electrode implantation in the length axis of the hippocampus for the presurgical evaluation of medial temporal lobe epilepsy: a computed tomography-based stereotactic insertion technique and its accuracy. Neurosurgery 43:819-826; discussion 826-817.
VanLandingham KE, Heinz ER, Cavazos JE, Lewis DV (1998). Magnetic resonance imaging evidence of hippocampal injury after prolonged focal febrile convulsions. Ann Neurol 43:413-426.
Vermoesen K, Massie A, Smolders I, Clinckers R (2012). The antidepressants citalopram and reboxetine reduce seizure frequency in rats with chronic epilepsy. Epilepsia.
Vezzani A, Friedman A, Dingledine R (2012). The role of inflammation in epileptogenesis. Neuropharmacology.
Vezzani A, Moneta D, Conti M, Richichi C, Ravizza T, De Luigi A, et al. (2000). Powerful anticonvulsant action of IL-1 receptor antagonist on intracerebral injection and astrocytic overexpression in mice. Proc Natl Acad Sci U S A 97:11534-11539.
Vezzani A, Ravizza T, Balosso S, Aronica E (2008). Glia as a source of cytokines: implications for neuronal excitability and survival. Epilepsia 49 Suppl 2:24-32.
Victoroff JI, Benson F, Grafton ST, Engel J, Jr., Mazziotta JC (1994). Depression in complex partial seizures. Electroencephalography and cerebral metabolic correlates. Arch Neurol 51:155-163.

149
Wada Y, Hasegawa H, Nakamura M, Yamaguchi N (1992). Behavioral and electroencephalographic effects of a serotonin receptor agonist (5-methoxy-N,N-dimethyltryptamine) in a feline model of photosensitive epilepsy. Neurosci Lett 138:115-118.
Wada Y, Hirao N, Shiraishi J, Nakamura M, Koshino Y (1999). Pindolol potentiates the effect of fluoxetine on hippocampal seizures in rats. Neurosci Lett 267:61-64.
Wada Y, Nakamura M, Hasegawa H, Shiraishi J, Yamaguchi N (1993). Microinjection of the serotonin uptake inhibitor fluoxetine elevates hippocampal seizure threshold in rats. Neuroscience Research Communications 13:143-148.
Wada Y, Shiraishi J, Nakamura M, Hasegawa H (1995). Prolonged but not acute fluoxetine administration produces its inhibitory effect on hippocampal seizures in rats. Psychopharmacology (Berl) 118:305-309.
Wada Y, Shiraishi J, Nakamura M, Koshino Y (1997). Role of serotonin receptor subtypes in the development of amygdaloid kindling in rats. Brain Res 747:338-342.
Walter C, Murphy BL, Pun RY, Spieles-Engemann AL, Danzer SC (2007). Pilocarpine-induced seizures cause selective time-dependent changes to adult-generated hippocampal dentate granule cells. J Neurosci 27:7541-7552.
Watanabe K, Ashby CR, Jr., Katsumori H, Minabe Y (2000). The effect of the acute administration of various selective 5-HT receptor antagonists on focal hippocampal seizures in freely-moving rats. Eur J Pharmacol 398:239-246.
Wegener G, Bandpey Z, Heiberg IL, Mork A, Rosenberg R (2003). Increased extracellular serotonin level in rat hippocampus induced by chronic citalopram is augmented by subchronic lithium: neurochemical and behavioural studies in the rat. Psychopharmacology (Berl) 166:188-194.
Weiss G, Lucero K, Fernandez M, Karnaze D, Castillo N (1993). The effect of adrenalectomy on the circadian variation in the rate of kindled seizure development. Brain Res 612:354-356.
Wieshmann UC, Woermann FG, Lemieux L, Free SL, Bartlett PA, Smith SJ, et al. (1997). Development of hippocampal atrophy: a serial magnetic resonance imaging study in a patient who developed epilepsy after generalized status epilepticus. Epilepsia 38:1238-1241.
Witter MP (1993). Organization of the entorhinal-hippocampal system: a review of current anatomical data. Hippocampus 3 Spec No:33-44.
Wojtowicz JM (2006). Irradiation as an experimental tool in studies of adult neurogenesis. Hippocampus 16:261-266.
World Health Organisation (2006). Mental Health: Neurology and Public Health.
Wroblewski BA, McColgan K, Smith K, Whyte J, Singer WD (1990). The incidence of seizures during tricyclic antidepressant drug treatment in a brain-injured population. J Clin Psychopharmacol 10:124-128.

150
Xing B, Zhao Y, Zhang H, Dang Y, Chen T, Huang J, et al. (2011). Microinjection of valproic acid into the ventrolateral orbital cortex exerts an antidepressant-like effect in the rat forced swim test. Brain Res Bull 85:153-157.
Xu H, Steven Richardson J, Li XM (2003). Dose-related effects of chronic antidepressants on neuroprotective proteins BDNF, Bcl-2 and Cu/Zn-SOD in rat hippocampus. Neuropsychopharmacology 28:53-62.
Yan QS, Jobe PC, Dailey JW (1993). Thalamic deficiency in norepinephrine release detected via intracerebral microdialysis: a synaptic determinant of seizure predisposition in the genetically epilepsy-prone rat. Epilepsy Res 14:229-236.
Yang T, Zhou D, Stefan H (2010). Why mesial temporal lobe epilepsy with hippocampal sclerosis is progressive: uncontrolled inflammation drives disease progression? J Neurol Sci 296:1-6.
Zara F, Bianchi A (2009). The impact of genetics on the classification of epilepsy syndromes. Epilepsia 50 Suppl 5:11-14.
Zattoni M, Mura ML, Deprez F, Schwendener RA, Engelhardt B, Frei K, et al. (2011). Brain infiltration of leukocytes contributes to the pathophysiology of temporal lobe epilepsy. J Neurosci 31:4037-4050.
Zentner J, Hufnagel A, Wolf HK, Ostertun B, Behrens E, Campos MG, et al. (1995). Surgical treatment of temporal lobe epilepsy: clinical, radiological, and histopathological findings in 178 patients. J Neurol Neurosurg Psychiatry 58:666-673.
Zhang Y, Gu F, Chen J, Dong W (2010). Chronic antidepressant administration alleviates frontal and hippocampal BDNF deficits in CUMS rat. Brain Res 1366:141-148.
Zhao C, Deng W, Gage FH (2008). Mechanisms and functional implications of adult neurogenesis. Cell 132:645-660.
Zhao Z, Sun P, Chauhan N, Kaur J, Hill MD, Papadakis M, et al. (2006). Neuroprotection and neurogenesis: modulation of cornus ammonis 1 neuronal survival after transient forebrain ischemia by prior fimbria-fornix deafferentation. Neuroscience 140:219-226.
Zheng W, Nowakowski RS, Vaccarino FM (2004). Fibroblast growth factor 2 is required for maintaining the neural stem cell pool in the mouse brain subventricular zone. Developmental neuroscience 26:181-196.
Zheng XY, Zhang HL, Luo Q, Zhu J (2011). Kainic acid-induced neurodegenerative model: potentials and limitations. J Biomed Biotechnol 2011:457079.
Zhu WJ, Roper SN (2001). Brain-derived neurotrophic factor enhances fast excitatory synaptic transmission in human epileptic dentate gyrus. Ann Neurol 50:188-194.
Zienowicz M, Wislowska A, Lehner M, Taracha E, Skorzewska A, Maciejak P, et al. (2005). The effect of fluoxetine in a model of chemically induced seizures--behavioral and immunocytochemical study. Neurosci Lett 373:226-231.
Zobel A, Wellmer J, Schulze-Rauschenbach S, Pfeiffer U, Schnell S, Elger C, et al. (2004). Impairment of inhibitory control of the hypothalamic pituitary adrenocortical system in epilepsy. European archives of psychiatry and clinical neuroscience 254:303-311.

Minerva Access is the Institutional Repository of The University of Melbourne
Author/s:CARDAMONE, LISA
Title:Antidepressant pharmacotherapy in epilepsy: the effects of chronic fluoxetine andcitalopram treatments in a rat model of epileptogenesis
Date:2012
Citation:Cardamone, L. (2012). Antidepressant pharmacotherapy in epilepsy: the effects of chronicfluoxetine and citalopram treatments in a rat model of epileptogenesis. Masters Researchthesis, Department of Medicine (Royal Melbourne Hospital), Faculty of Medicine, Dentistryand Health Sciences, The University of Melbourne.
Persistent Link:http://hdl.handle.net/11343/38205
Terms and Conditions:Terms and Conditions: Copyright in works deposited in Minerva Access is retained by thecopyright owner. The work may not be altered without permission from the copyright owner.Readers may only download, print and save electronic copies of whole works for their ownpersonal non-commercial use. Any use that exceeds these limits requires permission fromthe copyright owner. Attribution is essential when quoting or paraphrasing from these works.





























