Embed Size (px)
Citation preview

Hydrodeoxygenation of Stearic Acid into Normal and Iso-Octadecane Biofuel with Zeolite Supported Palladium-OxalateCatalystO. B. Ayodele,*,† Hazzim F. Abbas,‡ and Wan Mohd Ashri Wan Daud*,†
†Department of Chemical Engineering, University of Malaya, Kuala Lumpur, Malaysia‡Department of Chemical Engineering, Nizwa University, Nizwa, Oman
ABSTRACT: This study reports the hydrodeoxygenation (HDO) of stearic acid (SA) into paraffinic biofuel with synthesizedpalladium-oxalate zeolite supported catalyst (PdOx/Zeol). The PdOx/Zeol was synthesized via the functionalization ofdihydrogen tetrachloropalladate (II) with aqueous oxalic acid (OxA) to form the polynuclear palladium(II) oxalate (PdOx),which was supported on zeolite. The SEM and XRD characterization results showed that the zeolite support is highly crystallinebut loss some degree of crystallinity in the PdOx/Zeol sample after PdOx incorporation. The activity of the PdOx/Zeol tested onthe HDO of SA showed that temperature, pressure, gas flow rate, and PdOx/Zeol loading have significant effects on the HDOprocess, and their best observed conditions were 360 °C, 20 bar, 100 mL/min, and 25 mg, respectively to achieve 92% biofuelproduction from 35 g SA. The biofuel product distribution showed 71% n-C18H38, 18% iso-C18H38, and 3% C17H36. The presenceof iso-C18H38, which is an excellent biofuel value-added-component due to its low freezing point, was ascribed to thefunctionalization of Pd with OxA, which increases PdOx/Zeol acidity. The results showed that PdOx/Zeol is a prospectivecatalyst toward further research and commercialization of HDO process of SA.
■ INTRODUCTION
The recent shift of attention on energy generation from fossilstoward renewable sources has led to different funded researchstudies, such as sourcing the most reliable renewable feedstocks and the most economical and safe operatingconditions.1−3 Other studies also focused on sustainable andefficient catalyst development, process equipment design, andengineering.4−6 The most fundamental in all these studies is thecatalyst development because it affects the rate of reaction,which is a major determining factor in reactor design andengineering. It also determines the types and qualities ofproduct(s) to be obtained, which in turn has a strong bearingon the product separation, purification, and standardization. Agood catalyst for the production of biofuel is expected toproduce both normal and iso-paraffin in order to have areasonably high cetane number as well as improve the biofuelcold flow properties. The presence of iso-paraffin enhances thecold flow properties of biofuel by lowering the freezing point ofthe fuel; for example, the freezing point of the C16−C18 n-paraffin lies between 18 and 28 °C, while with the presence of20% iso-paraffin it can be reduced to about 10−12 °C.7−9
Different catalysts have recently been researched andreported in the literature while concerted efforts at improvingboth the catalyst synthesis procedure and operating conditionsare still ongoing. Among the best reported active metals used inthe hydrodeoxygenation (HDO) process to remove oxygenmolecules from feed stocks is palladium. Madsen et al.5
reported the deoxygenation of dilute and concentrated stearicacid over 2% Pd/C beads in a continuous reactor at 300 °C, 20bar and achieved 95% conversion, but the production of iso-parafins were not reported. Similarly, in the work of Arend etal.,10 in a continuous gas phase deoxygenation of oleic acidusing granular 2 wt % Pd/C catalyst, a maximum selectivity of
28.5 mol % for the formation of heptadecane and heptadeceneswas found at a temperatures of 380 °C using 5 g 2 wt % Pd/Cin a 4 h reaction. Lestari et al.11 reported deoxygenation ofstearic acid into n-pentadecane and n-heptadecane withselectivity up to 67% after 5 h reaction using palladiumsupported on mesoporous silica SBA15 and MCM-41. Bernaset al.12 developed egg-shell type palladium catalyst supportedon mesoporous carbon and reported the production ofundecane and undecene from a feed stock of dodecanoic acidover temperature and pressure range of 300−360 °C and 5−20bar, respectively. Simakova et al.13 studied four synthesized Pd/C catalysts over the temperature range 260−300 °C on thecatalytic deoxygenation of palmitic and stearic acids in asemibatch reactor, their main liquid phase products were n-heptadecane and n-pentadecane. Other available reports14−16
on the use of supported Pd for the production of biofuel alsodo not report the presence of iso-paraffins.One of the promising methods of enhancing both the
catalytic and isomerization activity of a catalyst is to increase itsacidity; for example, Simacek et al.17 sulfided NiMo/Al2O3catalysts and obtained some quantities of isoparaffin over thetemperature range 260−360 °C and pressure of 70−150 barfrom rapeseed oil diluted with isooctane. Krar et al.18 andKubicka et al.19 also investigated the ability of sulfided NiMo/SiO2−Al2O3 and sulfided NiMo/Al2O3 catalysts in the HDO ofgas oil and rapeseed oil and reported increase in the iso-paraffincontents as temperature was increased and pressure decreased.In view of the environmental concerns on the use of sulfidefunctionalization, Kovacs et al.20 replaced the sulfide function-
Received: June 13, 2014Revised: August 4, 2014
Article
pubs.acs.org/EF
© XXXX American Chemical Society A dx.doi.org/10.1021/ef501325g | Energy Fuels XXXX, XXX, XXX−XXX

alization with fluoride and reported a reasonable level ofisomerization with the same operational variable trend earlierreported by Krar et al.18 and Kubicka et al.19 Results from arecent study21 on the catalytic deoxygenation of triglyceridesand fatty acids to hydrocarbons in a semibatch autoclave over20 wt % Ni/C and 5 wt % Pd/C catalysts showed that 20 wt %Ni/C yielded satisfactory blendable products range than 5 wt %Pd/C. According to the authors, the difference in theperformance of these two catalysts was attributed to the higheracidity of the Ni-based formulation. Another expedient methodof increasing catalyst acidity is via functionalization with oxalicacid (OxA) to develop metal−oxalate catalysts having organo-metallic abilities that have been reported to be more reactivethan the metal oxide counterparts22,23 due to the metal−oxalateligand’s ability to minimize the formation of intermediate orside products22−25 from reactions such as hydrocracking,methanization, cyclization, and water−gas-shift reaction.Consequently, they have the ability to maintain their catalyticactivity over a wide range of process conditions because of theactive metal stability after proper calcination.23−25
In view of the above and the fact that Pd is a very expensivemetal (for example, its cost is about 1000 times more expensivethan the cost of Ni),21 it is imperative to synthesize acidifiedsupported Pd catalyst that can achieve both HDO andisomerization in one single processing step to enhance theoverall process economics. Therefore, in this study, wefunctionalized palladium with OxA to develop a novelpalladium oxalate catalyst supported on zeolite A (PdOx/Zeol) with increased acidity. Zeolite A was selected because ofits thermal and structural stability, and in addition, it iscurrently being cheaply produced from coal fly ash (CFA),which hitherto is the waste product of combustion of coal incoal-fired power stations with about 800 million tons perannum CFA production.26 The catalyst was chacterized for thephysical and chemical properties and its HDO and isomer-ization activities were tested on stearic acid.
■ EXPERIMENTAL SECTIONMaterials. All chemicals and the zeolite support were purchased
from Sigma-Aldrich except oxalic acid (OxA), which was purchasedfrom Spectrum Chemicals.Catalyst Development. Prior to the synthesis of the palladium
oxalate zeolite supported catalyst (PdOx/Zeol), the zeolite (Zeol)support was oven-dried at 150 °C for 3 h to remove free and boundedmoisture as well as any partial occluded organic matter. The PdOx/Zeol was synthesized via functionalization of 2.36 g of H2PdCl4 withstoichiometric ratio of aqueous OxA and was ripened for 1 h todevelop the polynuclear palladium(II) oxalate complexes (PdOx) in analuminum foil wrapped 500 mL conical flask because of the metal−oxalate complex photosensitivity.23 The precursor was then added toalready prepared 50 g Zeol dispersion and left stirring for 4 h at 50 °Cfor the deposition of PdOx particles on the Zeol support and the pHwas observed to stabilize at 5.3 ± 0.3 as the stirring progressed. Thesynthesized PdOx/Zeol was filtered and washed to remove thechloride ions, followed by drying at 100 °C for 12 h, and then, it wascalcined at 400 °C for 3 h on the basis of a few preliminary studies.Catalyst Characterization. Thermal Gravimetric Analysis (TGA)
analysis was carried out with a SHIMADZU DTG-60/60H instrument.A known weight of the samples were heated in a silica crucible at aconstant heating rate of 10 °C/min operating in a stream of N2atmosphere with a flow rate of 40 mL/min from 25 to 700 °C andweight loss per time and temperature increment were recorded. X-rayfluorescence spectroscopy (XRF) analysis of the samples were done todetermine their chemical composition using a μXay μEDX 100Schmadzu, NY, and X-ray tube of rhodium anode and scintillation
detector operating on a 40 mA current and 40 mV voltage. Scanningelectron microscopy (SEM) was employed to study the surfacemorphology of both Zeol support and the PdOx/Zeol catalyst. Theanalysis was carried out using a scanning electron microscope (ModelEMJEOL- JSM6301-F) with an Oxford INCA/ENERGY-350 micro-analysis system. The samples were scanned at various magnifications.The X-ray diffraction (XRD) patterns of the PdOx/Zeol catalyst andthe Zeol support were measured with SIEMENS XRD D5000equipped with Cu Kα radiation and recorded in the range 5−90°with a scanning rate of 2°/min. Fourier Transformed InfraredSpectroscopy (FTIR) analysis was performed on the Zeol supportand the PdOx/Zeol catalyst using a PerkinElmer Spectrum GXInfrared Spectrometer with resolution of 4 cm−1, in the range 4000−400 cm−1 to determine their surface functional groups. The Ramanspectra for the Zeol support and the PdOx/Zeol catalyst were obtainedto study the Pd−zeolite interaction using a Spex Triplematespectrograph connected to a large area intensified diode array detector(Tracor Northern 1024) with a 488 nm line (Lexel Model No. 95 Ar+)laser as the excitation source with a grating monochromator forrejecting any spurious lines and background. Both Zeol support andthe PdOx/Zeol catalyst spectra were taken from 200 to 1600 cm−1
with resolution of 1 cm−1. Nitrogen adsorption−desorption measure-ments with Brunauer−Emmett−Teller (BET) method was performedat liquid nitrogen temperature (−196 °C) with an autosorb BETapparatus, Micromeritics ASAP 2020, to determine the samplestextural properties (surface area, pore volume and diameter).
Stearic Acid Hydrodeoxygenation Experiments. Hydrodeox-ygenation of stearic acid (SA) was conducted using a 100 mL highpressure batch reactor, the reaction temperature and pressure werevaried within 300−380 °C and 10−60 bar, respectively. The reactionpressure and flow of carrier gas inlet and outlet were controlled by apressure controller (Brooks 5866) and flow (Brooks 58505 S). In atypically experiment, 35 g (∼40 mL) of SA was placed in the reactorfollowed by 25 mg of PdOx/Zeol (except during the study of effect ofcatalyst loading) and the catalyst was reduced in situ under flowing H2at 200 °C for 1 h, after which the reactor was purged with He. Theoperating temperature was established and monitored by a type-KOmega thermocouple located within the reactor. Before the reactionstarted, 50 mL of 90 vol % N2 and 10 vol % H2 were passed throughthe reactor until the desired reaction pressure was reached and thereaction commences by turning on the stirrer. Based on preliminarystudies, all experiments were performed under 60 min and the reactorset up was cooled by forced air and dismantled for product analysis.Withdrawn liquid samples from the reactor were dissolved in pyridineand followed by silylation with (100 wt % excess of) N,O-bis(trimethyl)-trifluoroacetamide, BSTFA, in an oven at 60 °C for 1h prior to GC analysis. The internal standard eicosane (C20H42) wasadded for quantitative calculations. The analysis of the withdrawnsamples was achieved with a gas chromatograph (GC, HP 6890)equipped with DB-5 column (60 m × 0.32 mm × 0.5 mm) and a flameionization detector. A sample (1 mL) was injected into the GC withsplit ratio of 50:1, and helium was used as the carrier gas. Thechromatographic pressure program was carefully adjusted to obtain asatisfactory separation of the desired identified products which werevalidated with a gas chromatograph mass spectrometer (GCMS).Since there were technical limitations and difficulty in the onlinequantification and analysis of the evolved gases (Egas) during thestudy, they were calculated according to eq 1, and the liquid productdistribution was evaluated using eq 2.
= + −M M MEgas [ ]b H a2 (1)
ω =∑
nn
(%) 100ii
i (2)
where Mb is the mass of reactor with the OA and catalyst beforereaction, MH2
is the mass of total H2 gas required during theexperiment evaluated from the H2 flow rate and its density. Ma is themass of the reactor with the liquid product and used catalyst after
Energy & Fuels Article
dx.doi.org/10.1021/ef501325g | Energy Fuels XXXX, XXX, XXX−XXXB
reaction. Similarly, ωi(%) is the mass fraction of the components in theliquid product, and n refers to the total number of liquid components.Stearic Acid Hydrodeoxygenation Process Chemistry. Prior
to the studies of the factors that influence the hydro deoxygenation ofstearic acid (SA), a preliminary analysis was done to investigate thedeoxygenation process chemistry with reaction time. A high thermalresistant alumina crucible (HTRAC) located in the thermogravimetricanalyzer (STA 449 F1/F3 with Automatic Sample Changer, Netzsch,Germany) was used as the reactor chamber. The products exit portwas coupled to a FTIR spectroscopy instrument (OPUS v7.0, BrukerOptics) to concomitantly monitor the rate of SA disappearance (i.e.,rate of reaction) in the presence of PdOx/Zeol catalyst and thequalitative analysis of the evolved products of HDO of SA. In a typicalcatalytic experiment based on preliminary studies, 3.5 g SA and 2.5 mgof PdOx/Zeol were carefully premixed in the HTRAC. The mixturewas heated at the heating rate of 30 °C/min from 30 to 360 °C, with20 mL/min of nitrogen flow as protective gas for the HTRAC. Afterabout 10 min, when traces of products were noticed from the FTIRdisplay, a mixture of high purity H2/N2/ (5%) at a flow rate of 100mL/min was continuously bubbled into the reactor. The vapors of thereaction product were conducted into the FTIR instrument via atransfer tube which was kept at 200 °C to maintain the reactionproducts in the vapor phase for analysis. The analysis of the productsspectra was done every 7.03 s at a spectra resolution of 1 cm−1 in thewavenumber range 4000−600 cm−1. In order to verify that the massloss recorded by the TG analyzer was strictly due to the HDO of SAand not in conjuction with moisture and volatile matter from PdOx/Zeol, a blank experiment was earlier conducted solely for the thermalgravimetric behavior of PdOx/Zeol catalyst.
■ RESULTS AND DISCUSSIONCatalyst Characterization. Thermal Gravimetric Analysis
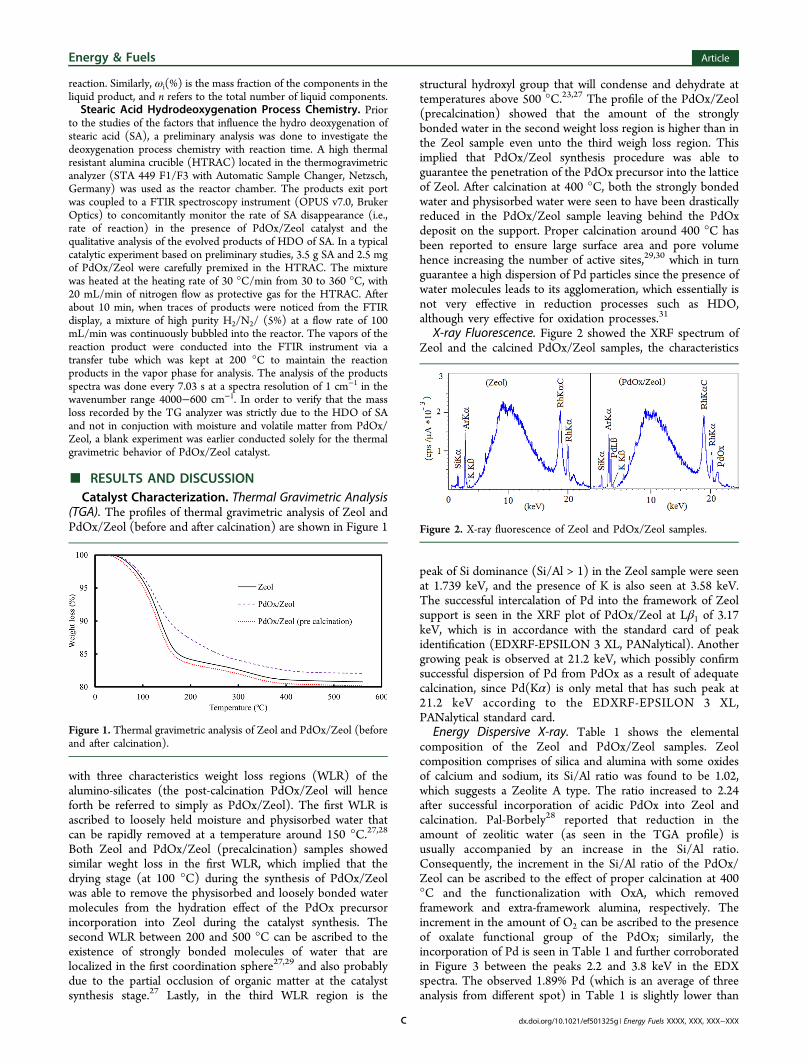
(TGA). The profiles of thermal gravimetric analysis of Zeol andPdOx/Zeol (before and after calcination) are shown in Figure 1
with three characteristics weight loss regions (WLR) of thealumino-silicates (the post-calcination PdOx/Zeol will henceforth be referred to simply as PdOx/Zeol). The first WLR isascribed to loosely held moisture and physisorbed water thatcan be rapidly removed at a temperature around 150 °C.27,28
Both Zeol and PdOx/Zeol (precalcination) samples showedsimilar weght loss in the first WLR, which implied that thedrying stage (at 100 °C) during the synthesis of PdOx/Zeolwas able to remove the physisorbed and loosely bonded watermolecules from the hydration effect of the PdOx precursorincorporation into Zeol during the catalyst synthesis. Thesecond WLR between 200 and 500 °C can be ascribed to theexistence of strongly bonded molecules of water that arelocalized in the first coordination sphere27,29 and also probablydue to the partial occlusion of organic matter at the catalystsynthesis stage.27 Lastly, in the third WLR region is the
structural hydroxyl group that will condense and dehydrate attemperatures above 500 °C.23,27 The profile of the PdOx/Zeol(precalcination) showed that the amount of the stronglybonded water in the second weight loss region is higher than inthe Zeol sample even unto the third weigh loss region. Thisimplied that PdOx/Zeol synthesis procedure was able toguarantee the penetration of the PdOx precursor into the latticeof Zeol. After calcination at 400 °C, both the strongly bondedwater and physisorbed water were seen to have been drasticallyreduced in the PdOx/Zeol sample leaving behind the PdOxdeposit on the support. Proper calcination around 400 °C hasbeen reported to ensure large surface area and pore volumehence increasing the number of active sites,29,30 which in turnguarantee a high dispersion of Pd particles since the presence ofwater molecules leads to its agglomeration, which essentially isnot very effective in reduction processes such as HDO,although very effective for oxidation processes.31
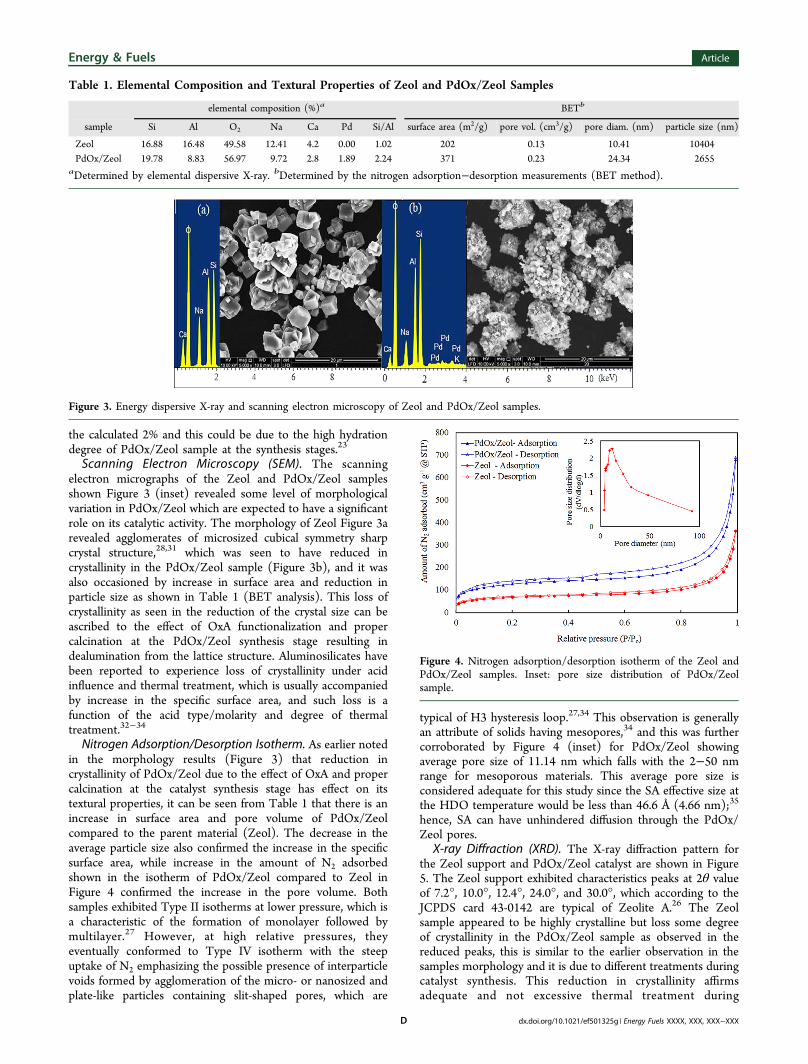
X-ray Fluorescence. Figure 2 showed the XRF spectrum ofZeol and the calcined PdOx/Zeol samples, the characteristics
peak of Si dominance (Si/Al > 1) in the Zeol sample were seenat 1.739 keV, and the presence of K is also seen at 3.58 keV.The successful intercalation of Pd into the framework of Zeolsupport is seen in the XRF plot of PdOx/Zeol at Lβ1 of 3.17keV, which is in accordance with the standard card of peakidentification (EDXRF-EPSILON 3 XL, PANalytical). Anothergrowing peak is observed at 21.2 keV, which possibly confirmsuccessful dispersion of Pd from PdOx as a result of adequatecalcination, since Pd(Kα) is only metal that has such peak at21.2 keV according to the EDXRF-EPSILON 3 XL,PANalytical standard card.
Energy Dispersive X-ray. Table 1 shows the elementalcomposition of the Zeol and PdOx/Zeol samples. Zeolcomposition comprises of silica and alumina with some oxidesof calcium and sodium, its Si/Al ratio was found to be 1.02,which suggests a Zeolite A type. The ratio increased to 2.24after successful incorporation of acidic PdOx into Zeol andcalcination. Pal-Borbely28 reported that reduction in theamount of zeolitic water (as seen in the TGA profile) isusually accompanied by an increase in the Si/Al ratio.Consequently, the increment in the Si/Al ratio of the PdOx/Zeol can be ascribed to the effect of proper calcination at 400°C and the functionalization with OxA, which removedframework and extra-framework alumina, respectively. Theincrement in the amount of O2 can be ascribed to the presenceof oxalate functional group of the PdOx; similarly, theincorporation of Pd is seen in Table 1 and further corroboratedin Figure 3 between the peaks 2.2 and 3.8 keV in the EDXspectra. The observed 1.89% Pd (which is an average of threeanalysis from different spot) in Table 1 is slightly lower than
Figure 1. Thermal gravimetric analysis of Zeol and PdOx/Zeol (beforeand after calcination).
Figure 2. X-ray fluorescence of Zeol and PdOx/Zeol samples.
Energy & Fuels Article
dx.doi.org/10.1021/ef501325g | Energy Fuels XXXX, XXX, XXX−XXXC
the calculated 2% and this could be due to the high hydrationdegree of PdOx/Zeol sample at the synthesis stages.23
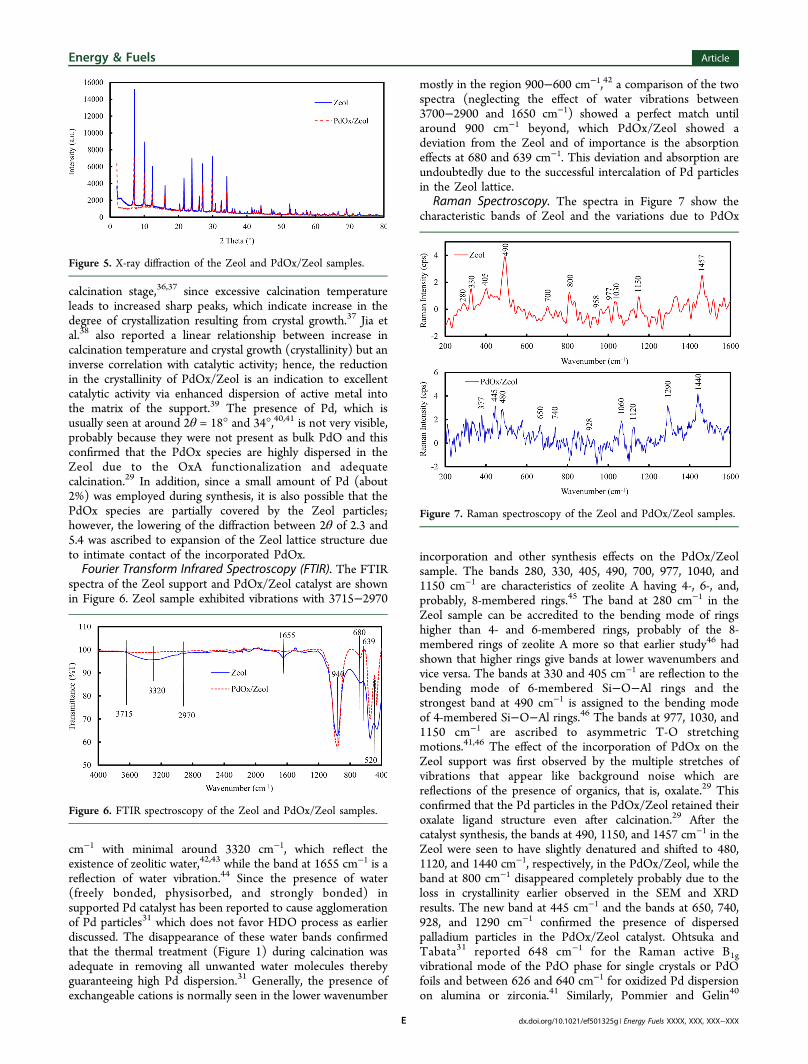
Scanning Electron Microscopy (SEM). The scanningelectron micrographs of the Zeol and PdOx/Zeol samplesshown Figure 3 (inset) revealed some level of morphologicalvariation in PdOx/Zeol which are expected to have a significantrole on its catalytic activity. The morphology of Zeol Figure 3arevealed agglomerates of microsized cubical symmetry sharpcrystal structure,28,31 which was seen to have reduced incrystallinity in the PdOx/Zeol sample (Figure 3b), and it wasalso occasioned by increase in surface area and reduction inparticle size as shown in Table 1 (BET analysis). This loss ofcrystallinity as seen in the reduction of the crystal size can beascribed to the effect of OxA functionalization and propercalcination at the PdOx/Zeol synthesis stage resulting indealumination from the lattice structure. Aluminosilicates havebeen reported to experience loss of crystallinity under acidinfluence and thermal treatment, which is usually accompaniedby increase in the specific surface area, and such loss is afunction of the acid type/molarity and degree of thermaltreatment.32−34
Nitrogen Adsorption/Desorption Isotherm. As earlier notedin the morphology results (Figure 3) that reduction incrystallinity of PdOx/Zeol due to the effect of OxA and propercalcination at the catalyst synthesis stage has effect on itstextural properties, it can be seen from Table 1 that there is anincrease in surface area and pore volume of PdOx/Zeolcompared to the parent material (Zeol). The decrease in theaverage particle size also confirmed the increase in the specificsurface area, while increase in the amount of N2 adsorbedshown in the isotherm of PdOx/Zeol compared to Zeol inFigure 4 confirmed the increase in the pore volume. Bothsamples exhibited Type II isotherms at lower pressure, which isa characteristic of the formation of monolayer followed bymultilayer.27 However, at high relative pressures, theyeventually conformed to Type IV isotherm with the steepuptake of N2 emphasizing the possible presence of interparticlevoids formed by agglomeration of the micro- or nanosized andplate-like particles containing slit-shaped pores, which are
typical of H3 hysteresis loop.27,34 This observation is generallyan attribute of solids having mesopores,34 and this was furthercorroborated by Figure 4 (inset) for PdOx/Zeol showingaverage pore size of 11.14 nm which falls with the 2−50 nmrange for mesoporous materials. This average pore size isconsidered adequate for this study since the SA effective size atthe HDO temperature would be less than 46.6 Å (4.66 nm);35
hence, SA can have unhindered diffusion through the PdOx/Zeol pores.
X-ray Diffraction (XRD). The X-ray diffraction pattern forthe Zeol support and PdOx/Zeol catalyst are shown in Figure5. The Zeol support exhibited characteristics peaks at 2θ valueof 7.2°, 10.0°, 12.4°, 24.0°, and 30.0°, which according to theJCPDS card 43-0142 are typical of Zeolite A.26 The Zeolsample appeared to be highly crystalline but loss some degreeof crystallinity in the PdOx/Zeol sample as observed in thereduced peaks, this is similar to the earlier observation in thesamples morphology and it is due to different treatments duringcatalyst synthesis. This reduction in crystallinity affirmsadequate and not excessive thermal treatment during
Table 1. Elemental Composition and Textural Properties of Zeol and PdOx/Zeol Samples
elemental composition (%)a BETb
sample Si Al O2 Na Ca Pd Si/Al surface area (m2/g) pore vol. (cm3/g) pore diam. (nm) particle size (nm)
Zeol 16.88 16.48 49.58 12.41 4.2 0.00 1.02 202 0.13 10.41 10404PdOx/Zeol 19.78 8.83 56.97 9.72 2.8 1.89 2.24 371 0.23 24.34 2655
aDetermined by elemental dispersive X-ray. bDetermined by the nitrogen adsorption−desorption measurements (BET method).
Figure 3. Energy dispersive X-ray and scanning electron microscopy of Zeol and PdOx/Zeol samples.
Figure 4. Nitrogen adsorption/desorption isotherm of the Zeol andPdOx/Zeol samples. Inset: pore size distribution of PdOx/Zeolsample.
Energy & Fuels Article
dx.doi.org/10.1021/ef501325g | Energy Fuels XXXX, XXX, XXX−XXXD
calcination stage,36,37 since excessive calcination temperatureleads to increased sharp peaks, which indicate increase in thedegree of crystallization resulting from crystal growth.37 Jia etal.38 also reported a linear relationship between increase incalcination temperature and crystal growth (crystallinity) but aninverse correlation with catalytic activity; hence, the reductionin the crystallinity of PdOx/Zeol is an indication to excellentcatalytic activity via enhanced dispersion of active metal intothe matrix of the support.39 The presence of Pd, which isusually seen at around 2θ = 18° and 34°,40,41 is not very visible,probably because they were not present as bulk PdO and thisconfirmed that the PdOx species are highly dispersed in theZeol due to the OxA functionalization and adequatecalcination.29 In addition, since a small amount of Pd (about2%) was employed during synthesis, it is also possible that thePdOx species are partially covered by the Zeol particles;however, the lowering of the diffraction between 2θ of 2.3 and5.4 was ascribed to expansion of the Zeol lattice structure dueto intimate contact of the incorporated PdOx.Fourier Transform Infrared Spectroscopy (FTIR). The FTIR
spectra of the Zeol support and PdOx/Zeol catalyst are shownin Figure 6. Zeol sample exhibited vibrations with 3715−2970
cm−1 with minimal around 3320 cm−1, which reflect theexistence of zeolitic water,42,43 while the band at 1655 cm−1 is areflection of water vibration.44 Since the presence of water(freely bonded, physisorbed, and strongly bonded) insupported Pd catalyst has been reported to cause agglomerationof Pd particles31 which does not favor HDO process as earlierdiscussed. The disappearance of these water bands confirmedthat the thermal treatment (Figure 1) during calcination wasadequate in removing all unwanted water molecules therebyguaranteeing high Pd dispersion.31 Generally, the presence ofexchangeable cations is normally seen in the lower wavenumber
mostly in the region 900−600 cm−1,42 a comparison of the twospectra (neglecting the effect of water vibrations between3700−2900 and 1650 cm−1) showed a perfect match untilaround 900 cm−1 beyond, which PdOx/Zeol showed adeviation from the Zeol and of importance is the absorptioneffects at 680 and 639 cm−1. This deviation and absorption areundoubtedly due to the successful intercalation of Pd particlesin the Zeol lattice.
Raman Spectroscopy. The spectra in Figure 7 show thecharacteristic bands of Zeol and the variations due to PdOx
incorporation and other synthesis effects on the PdOx/Zeolsample. The bands 280, 330, 405, 490, 700, 977, 1040, and1150 cm−1 are characteristics of zeolite A having 4-, 6-, and,probably, 8-membered rings.45 The band at 280 cm−1 in theZeol sample can be accredited to the bending mode of ringshigher than 4- and 6-membered rings, probably of the 8-membered rings of zeolite A more so that earlier study46 hadshown that higher rings give bands at lower wavenumbers andvice versa. The bands at 330 and 405 cm−1 are reflection to thebending mode of 6-membered Si−O−Al rings and thestrongest band at 490 cm−1 is assigned to the bending modeof 4-membered Si−O−Al rings.46 The bands at 977, 1030, and1150 cm−1 are ascribed to asymmetric T-O stretchingmotions.41,46 The effect of the incorporation of PdOx on theZeol support was first observed by the multiple stretches ofvibrations that appear like background noise which arereflections of the presence of organics, that is, oxalate.29 Thisconfirmed that the Pd particles in the PdOx/Zeol retained theiroxalate ligand structure even after calcination.29 After thecatalyst synthesis, the bands at 490, 1150, and 1457 cm−1 in theZeol were seen to have slightly denatured and shifted to 480,1120, and 1440 cm−1, respectively, in the PdOx/Zeol, while theband at 800 cm−1 disappeared completely probably due to theloss in crystallinity earlier observed in the SEM and XRDresults. The new band at 445 cm−1 and the bands at 650, 740,928, and 1290 cm−1 confirmed the presence of dispersedpalladium particles in the PdOx/Zeol catalyst. Ohtsuka andTabata31 reported 648 cm−1 for the Raman active B1gvibrational mode of the PdO phase for single crystals or PdOfoils and between 626 and 640 cm−1 for oxidized Pd dispersionon alumina or zirconia.41 Similarly, Pommier and Gelin40
Figure 5. X-ray diffraction of the Zeol and PdOx/Zeol samples.
Figure 6. FTIR spectroscopy of the Zeol and PdOx/Zeol samples.
Figure 7. Raman spectroscopy of the Zeol and PdOx/Zeol samples.
Energy & Fuels Article
dx.doi.org/10.1021/ef501325g | Energy Fuels XXXX, XXX, XXX−XXXE
confirmed successful incorporation of palladium particles intozeolite support at 928 cm−1 forming stretching vibrations withthe T−O bonds.Hydrodeoxygenation of Stearic AcidEffect Of
Operational Variables. A systematic approach was adoptedfor the HDO of SA, first effect of temperature was studied as athermodynamic state function by conducting the HDO atdifferent temperatures while keeping other variables constant.Second, the kinetics of the process was studied by varing thePdOx/Zeol loading over the tested temperature range. Lastly,the effect of pressure and gas flow were investigated at the besttemperature and PdOx/Zeol loading, and all studies wereconducted within 60 min reaction time.Effect of Temperature and PdOX/Zeol Loading on the
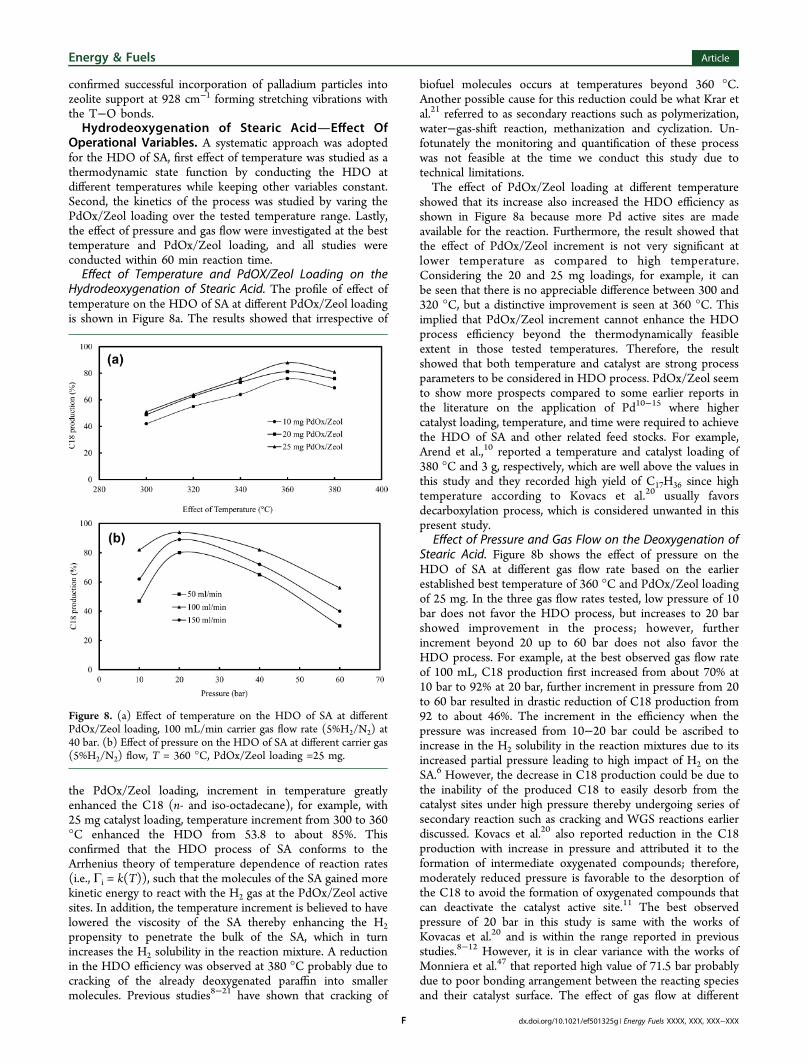
Hydrodeoxygenation of Stearic Acid. The profile of effect oftemperature on the HDO of SA at different PdOx/Zeol loadingis shown in Figure 8a. The results showed that irrespective of
the PdOx/Zeol loading, increment in temperature greatlyenhanced the C18 (n- and iso-octadecane), for example, with25 mg catalyst loading, temperature increment from 300 to 360°C enhanced the HDO from 53.8 to about 85%. Thisconfirmed that the HDO process of SA conforms to theArrhenius theory of temperature dependence of reaction rates(i.e., Γi = k(T)), such that the molecules of the SA gained morekinetic energy to react with the H2 gas at the PdOx/Zeol activesites. In addition, the temperature increment is believed to havelowered the viscosity of the SA thereby enhancing the H2propensity to penetrate the bulk of the SA, which in turnincreases the H2 solubility in the reaction mixture. A reductionin the HDO efficiency was observed at 380 °C probably due tocracking of the already deoxygenated paraffin into smallermolecules. Previous studies8−21 have shown that cracking of
biofuel molecules occurs at temperatures beyond 360 °C.Another possible cause for this reduction could be what Krar etal.21 referred to as secondary reactions such as polymerization,water−gas-shift reaction, methanization and cyclization. Un-fotunately the monitoring and quantification of these processwas not feasible at the time we conduct this study due totechnical limitations.The effect of PdOx/Zeol loading at different temperature
showed that its increase also increased the HDO efficiency asshown in Figure 8a because more Pd active sites are madeavailable for the reaction. Furthermore, the result showed thatthe effect of PdOx/Zeol increment is not very significant atlower temperature as compared to high temperature.Considering the 20 and 25 mg loadings, for example, it canbe seen that there is no appreciable difference between 300 and320 °C, but a distinctive improvement is seen at 360 °C. Thisimplied that PdOx/Zeol increment cannot enhance the HDOprocess efficiency beyond the thermodynamically feasibleextent in those tested temperatures. Therefore, the resultshowed that both temperature and catalyst are strong processparameters to be considered in HDO process. PdOx/Zeol seemto show more prospects compared to some earlier reports inthe literature on the application of Pd10−15 where highercatalyst loading, temperature, and time were required to achievethe HDO of SA and other related feed stocks. For example,Arend et al.,10 reported a temperature and catalyst loading of380 °C and 3 g, respectively, which are well above the values inthis study and they recorded high yield of C17H36 since hightemperature according to Kovacs et al.20 usually favorsdecarboxylation process, which is considered unwanted in thispresent study.
Effect of Pressure and Gas Flow on the Deoxygenation ofStearic Acid. Figure 8b shows the effect of pressure on theHDO of SA at different gas flow rate based on the earlierestablished best temperature of 360 °C and PdOx/Zeol loadingof 25 mg. In the three gas flow rates tested, low pressure of 10bar does not favor the HDO process, but increases to 20 barshowed improvement in the process; however, furtherincrement beyond 20 up to 60 bar does not also favor theHDO process. For example, at the best observed gas flow rateof 100 mL, C18 production first increased from about 70% at10 bar to 92% at 20 bar, further increment in pressure from 20to 60 bar resulted in drastic reduction of C18 production from92 to about 46%. The increment in the efficiency when thepressure was increased from 10−20 bar could be ascribed toincrease in the H2 solubility in the reaction mixtures due to itsincreased partial pressure leading to high impact of H2 on theSA.6 However, the decrease in C18 production could be due tothe inability of the produced C18 to easily desorb from thecatalyst sites under high pressure thereby undergoing series ofsecondary reaction such as cracking and WGS reactions earlierdiscussed. Kovacs et al.20 also reported reduction in the C18production with increase in pressure and attributed it to theformation of intermediate oxygenated compounds; therefore,moderately reduced pressure is favorable to the desorption ofthe C18 to avoid the formation of oxygenated compounds thatcan deactivate the catalyst active site.11 The best observedpressure of 20 bar in this study is same with the works ofKovacas et al.20 and is within the range reported in previousstudies.8−12 However, it is in clear variance with the works ofMonniera et al.47 that reported high value of 71.5 bar probablydue to poor bonding arrangement between the reacting speciesand their catalyst surface. The effect of gas flow at different
Figure 8. (a) Effect of temperature on the HDO of SA at differentPdOx/Zeol loading, 100 mL/min carrier gas flow rate (5%H2/N2) at40 bar. (b) Effect of pressure on the HDO of SA at different carrier gas(5%H2/N2) flow, T = 360 °C, PdOx/Zeol loading =25 mg.
Energy & Fuels Article
dx.doi.org/10.1021/ef501325g | Energy Fuels XXXX, XXX, XXX−XXXF
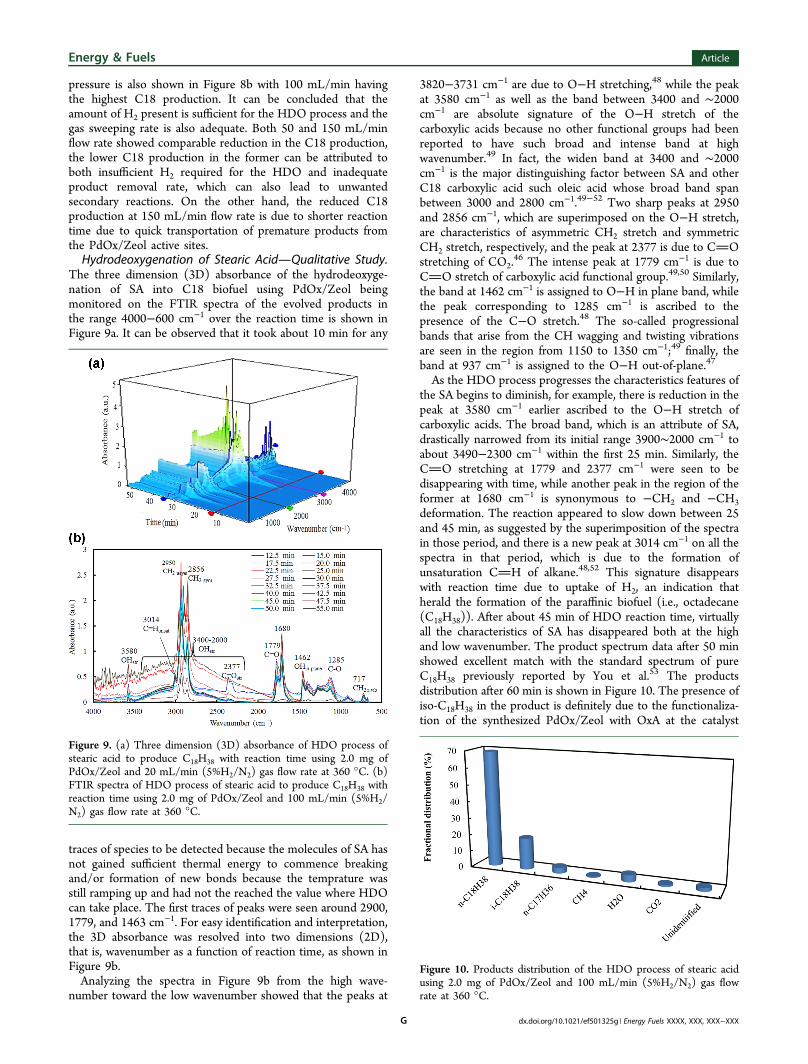
pressure is also shown in Figure 8b with 100 mL/min havingthe highest C18 production. It can be concluded that theamount of H2 present is sufficient for the HDO process and thegas sweeping rate is also adequate. Both 50 and 150 mL/minflow rate showed comparable reduction in the C18 production,the lower C18 production in the former can be attributed toboth insufficient H2 required for the HDO and inadequateproduct removal rate, which can also lead to unwantedsecondary reactions. On the other hand, the reduced C18production at 150 mL/min flow rate is due to shorter reactiontime due to quick transportation of premature products fromthe PdOx/Zeol active sites.Hydrodeoxygenation of Stearic AcidQualitative Study.
The three dimension (3D) absorbance of the hydrodeoxyge-nation of SA into C18 biofuel using PdOx/Zeol beingmonitored on the FTIR spectra of the evolved products inthe range 4000−600 cm−1 over the reaction time is shown inFigure 9a. It can be observed that it took about 10 min for any
traces of species to be detected because the molecules of SA hasnot gained sufficient thermal energy to commence breakingand/or formation of new bonds because the temprature wasstill ramping up and had not the reached the value where HDOcan take place. The first traces of peaks were seen around 2900,1779, and 1463 cm−1. For easy identification and interpretation,the 3D absorbance was resolved into two dimensions (2D),that is, wavenumber as a function of reaction time, as shown inFigure 9b.Analyzing the spectra in Figure 9b from the high wave-
number toward the low wavenumber showed that the peaks at
3820−3731 cm−1 are due to O−H stretching,48 while the peakat 3580 cm−1 as well as the band between 3400 and ∼2000cm−1 are absolute signature of the O−H stretch of thecarboxylic acids because no other functional groups had beenreported to have such broad and intense band at highwavenumber.49 In fact, the widen band at 3400 and ∼2000cm−1 is the major distinguishing factor between SA and otherC18 carboxylic acid such oleic acid whose broad band spanbetween 3000 and 2800 cm−1.49−52 Two sharp peaks at 2950and 2856 cm−1, which are superimposed on the O−H stretch,are characteristics of asymmetric CH2 stretch and symmetricCH2 stretch, respectively, and the peak at 2377 is due to COstretching of CO2.
46 The intense peak at 1779 cm−1 is due toCO stretch of carboxylic acid functional group.49,50 Similarly,the band at 1462 cm−1 is assigned to O−H in plane band, whilethe peak corresponding to 1285 cm−1 is ascribed to thepresence of the C−O stretch.48 The so-called progressionalbands that arise from the CH wagging and twisting vibrationsare seen in the region from 1150 to 1350 cm−1;49 finally, theband at 937 cm−1 is assigned to the O−H out-of-plane.47
As the HDO process progresses the characteristics features ofthe SA begins to diminish, for example, there is reduction in thepeak at 3580 cm−1 earlier ascribed to the O−H stretch ofcarboxylic acids. The broad band, which is an attribute of SA,drastically narrowed from its initial range 3900∼2000 cm−1 toabout 3490−2300 cm−1 within the first 25 min. Similarly, theCO stretching at 1779 and 2377 cm−1 were seen to bedisappearing with time, while another peak in the region of theformer at 1680 cm−1 is synonymous to −CH2 and −CH3deformation. The reaction appeared to slow down between 25and 45 min, as suggested by the superimposition of the spectrain those period, and there is a new peak at 3014 cm−1 on all thespectra in that period, which is due to the formation ofunsaturation CH of alkane.48,52 This signature disappearswith reaction time due to uptake of H2, an indication thatherald the formation of the paraffinic biofuel (i.e., octadecane(C18H38)). After about 45 min of HDO reaction time, virtuallyall the characteristics of SA has disappeared both at the highand low wavenumber. The product spectrum data after 50 minshowed excellent match with the standard spectrum of pureC18H38 previously reported by You et al.53 The productsdistribution after 60 min is shown in Figure 10. The presence ofiso-C18H38 in the product is definitely due to the functionaliza-tion of the synthesized PdOx/Zeol with OxA at the catalyst
Figure 9. (a) Three dimension (3D) absorbance of HDO process ofstearic acid to produce C18H38 with reaction time using 2.0 mg ofPdOx/Zeol and 20 mL/min (5%H2/N2) gas flow rate at 360 °C. (b)FTIR spectra of HDO process of stearic acid to produce C18H38 withreaction time using 2.0 mg of PdOx/Zeol and 100 mL/min (5%H2/N2) gas flow rate at 360 °C.
Figure 10. Products distribution of the HDO process of stearic acidusing 2.0 mg of PdOx/Zeol and 100 mL/min (5%H2/N2) gas flowrate at 360 °C.
Energy & Fuels Article
dx.doi.org/10.1021/ef501325g | Energy Fuels XXXX, XXX, XXX−XXXG

development stage, which increased the acidity. Previousstudies2,18−20 have shown that acidic catalysts are promisingtoward the production of iso-paraffins, which are known to begood biofuel additive due to their low freezing points. Thepresence of water confirmed that the O2 extraction proceeds viaHDO process (since CO was not observed) while that of CO2is likely due to decarboxylation process which justifies the tracesof C17H36 observed. Part of the CO2 perhaps had undergonemethanization reaction with the H2 in the carrier gas stream toform the traces of CH4 observed. However, since no otherlower and higher hydrocarbons were observed, it implied thatthe operating conditions were not at extremes that could favorcracking and polymerization reactions and also the PdOx/Zeolhas the ability to minimize the formation of other sideproduct(s) due to the presence of the metal−oxalateligands.23−26 In view of the foregoing, a comparison of thePdOx/Zeol activity with other related studies4−6,9−15 showedthat PdOx/Zeol is comparably highly promising especiallyconsidering their feed/catalyst dosage ratio, higher reactiontemperature, longer reaction time, and product purity. Forexample, Arend et al.10 used 3 g of 2% Pd/C and could onlyachieve deoxygenation at 380−450 °C; hence, the catalystsuffered deactivation due to carbon deposition. Similarly,Lestari et al.11 achieved catalytic deoxygenation of SA overPd supported on acid modified mesoporous silica SBA15 andMCM-41 with 67% liquid n-pentadecane selectivity after 5 hreaction compared to over 90% combined n- and iso-octadecane recorded in this study in 1 h HDO time. In allthose studies,4−6,9−15 the authors did not claim any formationof isomerized products, which are value added components forbiofuel to enhanced its cold flow properties.Catalyst Reusability. The catalyst reusability was studied at
360 °C, 100 mL/min gas flow rate and 20 bar in 60 minreaction time. After three consecutive experiments, thereusability results were consistent with 92% biofuel productioncomprising 71% n-C18H38, 18% iso-C18H38, and 3% C17H36from 35 g SA. This ability was ascribed to the PdOx/Zeolsynthesis protocol that employs the functionalization ofpalladium with OxA to develop organometallic palladium IIoxalate complex catalyst precursor with increased acidity.Generally metal−oxalate catalysts have been reported to bemore reactive than the metal oxide catalysts and are highlyresistance to leaching of the active metal due to the presence ofthe strong Mn+−oxalate ligand, which also minimizes thetendencies of multiple side reactions.22,24,54 However, about 2%reduction in isomerization efficiency was observed probablydue to slight reduction in the PdOx/Zeol acidity. Kovacs et al.20
in their study on hydrotreating of triglycerides using fluorinatedNiMo/Al2O3 catalyst also reported some degree of acidity losswhich resulted into reduced iso-paraffin production.
■ CONCLUSIONZeolite supported palladium oxalate (PdOx/Zeol) catalyst wassynthesized via functionalization of palladium with aqueousoxalic acid (OxA) to form the polynuclear palladium(II) oxalatecomplex, which was supported on zeolite support (Zeol). ThePdOx/Zeol characterization showed enhancement in thetextural properties due to the catalyst synthesis protocol. Thepresence of highly dispersed Pd particle was confirmed by EDX,XRF, FTIR, and Raman spectroscopy. The activity of PdOx/Zeol and effect of process variables were studied onhydrodeoxygenation (HDO) of stearic acid (SA). The effectof temperature, pressure and gas flow rate on the HDO process
showed that increase in their initial values enhanced the HDOprocess efficiency up to a certain extent beyond which furtherincrement leads to lower HDO efficiency. The best observedoperating condition was 360 °C, 25 mg of PdOx/Zeol, 100mL/min of gas flow under a reaction pressure of 20 bar toachieve about 92% biofuel production from 35 g SA. Theprocess chemistry showed the composition of the biofuel to be71% n-C18H38, 18% iso-C18H38, and 3% C17H36. The presenceof the isomerized product was ascribed to the functionalizationof the catalyst with OxA which increases its acidity. This studyis very promising toward the advancement of biofuel researchand commercialization.
■ AUTHOR INFORMATIONCorresponding Authors*Tel +60164955453. Email: [email protected].*Email: [email protected].
NotesThe authors declare no competing financial interest.
■ ACKNOWLEDGMENTSThe authors sincerely acknowledge the financial support fromHigher Impact Research Ministry of Higher Education projectno. D000011-16001, Faculty of Engineering, University ofMalaya.
■ REFERENCES(1) Ong, H. C.; Masjuki, H. H.; Mahlia, T. M. I.; Silitonga, A. S.;Chong, W. T.; Leong, K. Y. Optimization of Biodiesel Production andEngine Performance from High Free Fatty Acid CalophyllumInophyllum Oil in CI Diesel Engine. Energy Convers. Manage. 2014,81, 30−40.(2) Ayodele, O. B.; Abbas, H. F.; Daud, W. M. A. W. CatalyticUpgrading of Oleic Acid into Biofuel Using Mo Modified ZeoliteSupported Ni Oxalate Catalyst Functionalized with Fluoride Ion.Energy Convers . Manage. 2014 , DOI: 10.1016/j .encon-man.2014.02.014.(3) An, H.; Yang; Maghbouli, W. M. A.; Li, J.; Chua, K. J. A SkeletalMechanism for Biodiesel Blend Surrogates Combustion. EnergyConvers. Manage. 2014, 81, 51−59.(4) Somnuk, K.; Niseng, S.; Prateepchaikul, G. Optimization of HighFree Fatty Acid Reduction in Mixed Crude Palm Oils UsingCirculation Process through Static Mixer Reactor and Pilot-Scale ofTwo-Step Process. Energy Convers. Manage. 2014, 80, 374−381.(5) Takase, M.; Zhang, M.; Feng, W.; Chen, Y.; Zhao, T.; Cobbina, S.J.; Yang, L.; Wu, X. Application of Zirconia Modified with KOH AsHeterogeneous Solid Base Catalyst to New Nonedible Oil forBiodiesel. Energy Convers. Manage. 2014, 80, 117−125.(6) Kwon, K. C.; Mayfield, H.; Marolla, T.; Nichols, B.; Mashburn,M. Catalytic Deoxygenation of Liquid Biomass for Hydrocarbon Fuels.Renewable Energy 2011, 36, 907−915.(7) Lapuerta, M.; Villajos, M.; Agudelo, J. R.; Boehman, A. L. KeyProperties and Blending Strategies of Hydrotreated Vegetable Oil AsBiofuel for Diesel Engines. Fuel Process. Technol. 2011, 92, 2406−2411.(8) Hancsok, J.; Krar, M.; Magyar, Sz.; Boda, L.; Hollo, A.; Kallo, D.Investigation of the Production of High Quality Bio Gas Oil from Pre-Hydrogenated Vegetable Oils over Pt/SAPO-11/Al2O3. Stud. Surf. Sci.Catal. 2007, 170 (B), 1605−1610.(9) Krar, M.; Kovacs, S.; Kallo, D.; Hancsok, J. Fuel PurposeHydrotreating of Sunflower Oil on CoMo/Al2O3 Catalyst. Bioresour.Technol. 2010, 101, 9287−9293.(10) Arend, M.; Nonnen, T.; Hoelderich, W. F.; Fischer, J.; Groos, J.Catalytic Deoxygenation of Oleic Acid in Continuous Gas Flow for theProduction of Diesel-Like Hydrocarbons. Appl. Catal., A 2011, 399,198−204.
Energy & Fuels Article
dx.doi.org/10.1021/ef501325g | Energy Fuels XXXX, XXX, XXX−XXXH

(11) Lestari, S.; Beltramini, J.; Max Lu, G. Q. CatalyticDeoxygenation of Stearic Acid over Palladium Supported on AcidModified Mesoporous Silica. Stud Surf. Sci. Catal. B 2008, 174, 1339−1342.(12) Bernas, H.; Eranen, K.; Simakova, I.; Leino, A.-R.; Kordas, K.;Myllyoja, J.; Maki-Arvela, P.; Salmi, T.; Murzin, D. Y. Deoxygenationof Dodecanoic Acid under Inert Atmosphere. Fuel 2010, 89, 2033−2039.(13) Simakova, I.; Simakova, O.; Maki-Arvela, P.; Simakov, A.;Estrada, M.; Murzin, D. Y. Deoxygenation of Palmitic and Stearic Acidover Supported Pd Catalysts: Effect of Metal Dispersion. Appl. Catal.,A 2009, 355, 100−108.(14) Ping, E. W.; Wallace, R.; Pierson, J.; Fuller, T. F.; Jones, C. W.Highly Dispersed Palladium Nanoparticles on Ultra-Porous SilicaMesocellular Foam for the Catalytic Decarboxylation of Stearic Acid.Microporous Mesoporous Mater. 2010, 132, 174−180.(15) Na, J.-G.; Yi, B. E.; Han, J. K.; Oh, Y.-K.; Park, J. H.; Jung, T. S.;Han, S. S.; Yoon, H. C.; Kim, J. N.; Lee, H.; Ko, C. H. Deoxygenationof Microalgal Oil into Hydrocarbon with Precious Metal Catalysts:Optimization of Reaction Conditions and Supports. Energy 2012, 47,25−30.(16) Snare, M.; Kubickova, I.; Maki-Arvela, P.; Chichova, D.; Eranen,K.; Murzin, D. Y. Catalytic Deoxygenation of Unsaturated RenewableFeedstocks for Production of Diesel Fuel Hydrocarbons. Fuel 2008,87, 933−945.(17) Simacek, P.; Kubicka, D.; Sebor, G.; Pospisil, M. Hydro-processed Rapeseed Oil As a Source of Hydrocarbon-Based Biodiesel.Fuel 2009, 88, 456−460.(18) Krar, M.; Thernesz, A.; Toth, Cs.; Kasza, T.; Hancsok, J.Investigation of Catalytic Conversion of Vegetable Oil/Gas OilMixtures. In Silica and Silicates in Modern Catalysis; Halasz, I., Ed.;Transworld Research Network: India, Kerala, 2010; pp 435−455,ISBN 978-81-7895-455-4.(19) Kubicka, D.; Chudoba, J.; Simacek, P. Catalytic Conversion ofVegetable Oils into Transportation Fuels. In Energetische Nutzung vonBiomassen -Velen VIII DGMK-; Fachbereichstagung: Germany, Velen,2008; pp 101−106.(20) Kovacs, S.; Kasza, T.; Thernesz, A.; Horvath, I. W.; Hancsok, J.Fuel Production by Hydrotreating of Triglycerides on NiMo/Al2O3/FCatalyst. Chem. Eng. J. 2011, 176−177, 237−243.(21) Santillan-Jimenez, E.; Morgan, T.; Lacny, J.; Mohapatra, S.;Crocker, M. Catalytic Deoxygenation of Triglycerides and Fatty Acidsto Hydrocarbons over Carbon-Supported Nickel. Fuel 2013, 103,1010−1017.(22) Tanev, P. T., Lange De Oliveira, A. Methane aromatizationcatalyst, method of making and method of using the catalyst. UnitedStates Patent No. US 2012/0123176 A1.(23) Ayodele, O. B.; Hameed, B. H. Synthesis of Copper PillaredBentonite Ferrioxalate Catalyst for Degradation of 4-Nitrophenol inVisible Light Assisted Fenton Process. J. Ind. Eng. Chem. 2012, 19 (3),966−974.(24) Ng, K. Y. S.; Zhou, X.; Gulari, E. Spectroscopic Characterizationof Molybdenum Oxalate in Solution and on Alumina. J. Phys. Chem.1985, 89, 2411−2481.(25) Li, J.; Xia, Z.; Lai, W.; Zheng, J.; Chen, B.; Yi, X.; Fang, W.Hydrodemetallation (HDM) of Nickel-5,10,15,20-tetraphenylporphyr-in Ni-TPP over NiMo/γ-Al2O3 Catalyst Prepared by One-Pot Methodwith Controlled Precipitation of the Components. Fuel 2012, 97,504−511.(26) Hui, K. S.; Hui, K. N.; Lee, S. K. A Novel and Green Approachto Produce Nano-Porous Materials Zeolite A and MCM-41 from CoalFly Ash and their Applications in Environmental Protection. Int. J.Chem.Biol. Eng. 2009, 2, 165−175.(27) Xue, T.; Wang, Y. M.; He, M.-Y. Facile Synthesis of Nano-SizedNH4-ZSM-5 Zeolites. Microporous Mesoporous Mater. 2012, 156, 29−35.(28) Pal-Borbely, G. Thermal Analysis of Zeolites. Mol. Sieves 2007,5, 67−101.
(29) Ghule, A. V.; Ghule, K.; Punde, T.; Liu, J.; Tzing, S.; Chang, J.;Chang, H.; Ling, Y. In Situ Monitoring of NiO−Al2O3 NanoparticlesSynthesis by Thermo-Raman Spectroscopy. Mater. Chem. Phys. 2010,119, 86−92.(30) Liu, F.; Asakura, K.; He, H.; Liu, Y.; Shan, W.; Shi, X.; Zhang, C.Influence of Calcination Temperature on Iron Titanate Catalyst forthe Selective Catalytic Reduction of NOx with NH3. Catal. Today2011, 164, 520−527.(31) Ohtsuka, H.; Tabata, T. Effect of Water Vapor on theDeactivation of Pd-Zeolite Catalysts for Selective Catalytic Reductionof Nitrogen Monoxide by Methane. Appl. Catal., B 1999, 21, 133−139.(32) Xiaoling, L.; Yan, W.; Xujin, W.; Yafei, Z.; Yanjun, G.; Qinghu,X.; Jun, X.; Feng, D.; Tao, D. Characterization and CatalyticPerformance in n-Hexane Cracking of HEU-1 Zeolites DealuminatedUsing Hydrochloric Acid and Hydrothermal Treatments. Chin. J.Catal. 2012, 33, 1889−1900.(33) Ayodele, O. B.; Togunwa, O. S. Catalytic Activity of SynthesizedBentonite Supported Cuprospinel Oxalate Catalyst on the Degrada-tion and Mineralization Kinetics of Direct Blue 71, Acid Green 25, andReactive Blue 4 Pollutants in Photo-Fenton Process. Appl. Catal., A2014, 470, 285−293.(34) Ayodele, O. B.; Lim, J. K.; Hameed, B. H. Development ofKaolin Supported Ferric Oxalate Heterogeneous Catalyst forDegradation of 4-Nitrophenol in Photo Fenton Process. Appl. ClaySci. 2013, 83−84, 171−181.(35) Moniruzzaman, M.; Sundararajan, P. R. Morphology of Blendsof Self-Assembling Long-Chain Carbamate and Stearic Acid. Pure Appl.Chem. 2004, 76, 1353−1363.(36) Ayodele, O. B.; Abbas, H. F.; Daud, W. M. A. W.Hydrodeoxygenation of Shea Butter to Produce Diesel-Like FuelUsing Acidified and Basic Al2O3 Supported Molybdenum OxalateCatalyst Based on Aspen Hysys Simulation Study with Aspen HysysSimulation Study. Energy Educ. Sci. Technol. Part A 2014, 32, 447−460.(37) Oh, S. W.; Bang, H. J.; Bae, Y. C.; Sun, Y.-K. Effect ofCalcination Temperature on Morphology, Crystallinity, and Electro-chemical Properties of Nano-Crystalline Metal Oxides (Co3O4, CuO,and NiO) Prepared via Ultrasonic Spray Pyrolysis. J. Power Sources2007, 173, 502−509.(38) Jia, H.; Stark, J.; Zhou, L. Q.; Ling, C.; Sekito, T.; Markin, Z.Different Catalytic Behavior of Amorphous and Crystalline CobaltTungstate for Electrochemical Water Oxidation. RSC Adv. 2012, 2,10874−10881.(39) Li, S.; Chen, J.; Hao, T.; Lianga, W.; Liu, X.; Sun, M.; Ma, X.Pyrolysis of Huang Tu Miao Coal over Faujasite Zeolite andSupported Transition Metal Catalysts. J. Anal. Appl. Pyrol 2012, 161DOI: 10.1016/j.jaap.2012.12.029.(40) Pommier, B.; Gelin, P. On the Nature of Pd Species Formedupon Exchange of H-ZSM-5 with Pd((NH3)4)
2+ and Calcination inO2. Phys. Chem. Chem. Phys. 1999, 1, 1665−1672.(41) Gannouni, A.; Rozanska, X.; Albela, B.; Zina, M. S.; Delbecq, F.;Bonneviot, L.; Ghorbel, A. Theoretical and Experimental Inves-tigations on Site Occupancy for Palladium Oxidation States inMesoporous Al-MCM-41 Materials. J. Catal. 2012, 289, 227−237.(42) Breck, D. W. Zeolite Molcular Sieves; Wiley, New York, 1974; pp45−50.(43) Zhang, Q.; Wang, T.; Xu, Y.; Zhang, Q.; Ma, L. Production ofLiquid Alkanes by Controlling Reactivity of Sorbitol Hydrogenationwith a Ni/HZSM-5 Catalyst in Water. Energy Convers. Manage. 2014,77, 262−268.(44) Xie, W.; Zhao, L. Production of Biodiesel by Transesterificationof Soybean Oil Using Calcium Supported Tin Oxides AsHeterogeneous Catalysts. Energy Convers. Manage. 2013, 76, 55−62.(45) Yu, Y.; Xiong, G.; Li, C.; Xiao, F. S. Characterization ofAluminosilicate Zeolites by UV Raman Spectroscopy. MicroporousMesoporous Mater. 2001, 46, 23−34.(46) Elaiopoulos, K.; Perraki, Th.; Grigoropoulou, E. Monitoring theEffect of Hydrothermal Treatments on the Structure of a NaturalZeolite through a Combined XRD, FTIR, XRF, SEM, and N2-
Energy & Fuels Article
dx.doi.org/10.1021/ef501325g | Energy Fuels XXXX, XXX, XXX−XXXI

Porosimetry Analysis. Microporous Mesoporous Mater. 2010, 134, 29−43.(47) Monniera, J.; Sulimma, H.; Dalai, A.; Caravaggio, G.Hydrodeoxygenation of Oleic Acid and Canola Oil over Alumina-Supported Metal Nitrides. Appl. Catal., A 2010, 382, 176−180.(48) Wang, D.; Xiao, R.; Zhang, H.; He, G. Comparison of CatalyticPyrolysis of Biomass with MCM-41 and CaO Catalysts by UsingTGA-FTIR Analysis. J. Anal. Appl. Pyrolysis 2010, 89, 171−177.(49) Wu, N.; Fu, L.; Su, M.; Aslam, M.; Wong, K. C.; Dravid, V. P.Interaction of Fatty Acid Monolayers with Cobalt Nanoparticles. NanoLett. 2004, 4, 383−386.(50) Silverstein, R. M.; Webster, F. X.; Kiemle, D. J. SpectrometricIdentification of Organic Compounds, 7th ed.; John Wiley & Sons, Inc.:Hoboken, NJ, 2005; pp 72−126.(51) Lee, S. J.; Kim, K. Diffuse Reflectance Infrared Spectra of StearicAcid Self-Assembled on Fine Silver Particles. Vib. Spectrosc. 1998, 18,187−201.(52) http://www2.ups.edu/faculty/hanson/Spectroscopy/IR/IRfrequencies.html (accessed May 2013).(53) You, M.; Wang, X.; Zhang, X.; Zhang, L.; Wang, J.Microencapsulated n-Octadecane with Styrene-Divinybenzene Co-Polymer Shells. J. Polym. Res. 2011, 18, 49−58.(54) Ayodele, O. B.; Togunwa, O. S.; Abbas, H. F.; Daud, W. M. A.W. Preparation and characterization of alumina supported nickel-oxalate catalyst for the hydrodeoxygenation of oleic acid into normaland iso-octadecane biofuel. Energy Conversion and Management, http://dx.doi.org/10.1016/j.enconman.2014.05.099.
Energy & Fuels Article
dx.doi.org/10.1021/ef501325g | Energy Fuels XXXX, XXX, XXX−XXXJ





























