Embed Size (px)
Citation preview

& Supramolecular Chirality
Hierarchical Self-Assembly of Supramolecular Helical Fibres fromAmphiphilic C3-Symmetrical Functional Tris(tetrathiafulvalenes)
Flavia Pop,[a] Caroline Melan,[a] Ion Danila,[a] Mathieu Linares,[b] David Beljonne,[c]
David B. Amabilino,*[d] and Narcis Avarvari*[a]
Abstract: The preparation and self-assembly of the enantio-mers of a series of C3-symmetric compounds incorporatingthree tetrathiafulvalene (TTF) residues is reported. The chiralcitronellyl and dihydrocitronellyl alkyl chains lead to helicalone dimensional stacks in solution. Molecular mechanics anddynamics simulations combined with experimental and the-oretical circular dichroism support the observed helicity insolution. These stacks self-assemble to give fibres that havemorphologies that depend on the nature of the chiral alkylgroup and the medium in which the compounds aggregate.An inversion of macroscopic helical morphology of the citro-
nellyl compound is observed when compared to analogous2-methylbutyl chains, which is presumably a result of thestereogenic centre being further away from the core of themolecule. This composition still allows both morphologies tobe observed, whereas an achiral compound shows no helici-ty. The morphology of the fibres also depends on the flexi-bility at the chain ends of the amphiphilic components, asthere is not such an apparently persistent helical morpholo-gy for the dihydrocitronellyl derivative as for that preparedfrom citronellyl chains.
Introduction
The formation of chiral supramolecular aggregates is the resultof a self-assembly process during which the molecular buildingblocks engage in stereospecific non-covalent intermolecular in-teractions, such as p–p stacking and hydrogen bonding, themolecular chirality being transposed at the supramolecularlevel.[1–4] Whether this chirality is expressed or not also de-pends on weaker and less specific van der Waals interactions.[2]
Accordingly, various structural shapes, such as helical fibres,twisted wires or ribbons, helical tubes, or coiled ribbons, canbe observed in the solid state as expression of the supramolec-ular chirality at the nano- and mesoscale. Of particular interest
is the case when the molecular unit contains a functionalgroup with a specific property which can be then amplified inthe resulting chiral supramolecular assemblies.[5–8] Furthermore,collective properties such as electrical conductivity may be ob-served in the helical aggregates built up from p-conjugatedbuilding blocks.[9] These latter include oligothiophenes,[10, 11]
phenylene–vinylenes,[12–14] hexabenzocoronenes,[15, 16] and alsotetrathiafulvalene (TTF) units.[17] One of the main objectives ofour research is to use such supramolecular electroactive fibresin molecular electronics.[18, 19]
The interest in chiral conductors[20–22] has been recently high-lighted through the first experimental observation of the inter-play between chirality and electrical conductivity in a bulkcrystalline molecular conductor prepared from a chiral TTF de-rivative.[23] This phenomenon, referred to as electrical magneto-chiral anisotropy (eMChA) effect,[24] expresses the difference inthe electrical resistance between the two enantiomers ofa chiral conductor, measured in a magnetic field parallel to thedirection of the current.
C3-symmetric discotic platforms such as benzene-1,3,5-tricar-boxamides (BTA) substituted with different chiral units provedto be particularly efficient as triggers of the self-assembly ofmolecular units into helical aggregates; they have been thor-oughly investigated in solution by circular dichroism (CD) andin solid state by microscopy techniques.[25] Furthermore, evensymmetry breaking can occur in BTA derivatives with rigidachiral arms.[26] Meijer and co-workers have shown that thefunctionalisation of BTA by 3,3’-diamino-2,2’-bipyridinewedges,[27] which can be then appropriately substituted bychiral units, induces a propeller conformation to the molecularbuilding blocks, which is further transmitted to the supra-
[a] Dr. F. Pop, C. Melan, Dr. I. Danila, Dr. N. AvarvariUniversit� d’Angers, CNRSLaboratoire MOLTECH-Anjou, UMR 6200UFR Sciences, B�t. K, 2 Bd. Lavoisier, 49045 Angers (France)Fax: (+ 33) 02-41-73-54-05E-mail : [email protected]
[b] Dr. M. LinaresDepartment of Computational PhysicsIFM—Linkçpings Universitet, 581 83 Linkçping (Sweden)
[c] Dr. D. BeljonneLaboratory for Chemistry of Novel MaterialsUniversit� de Mons, Place du Parc 20, 7000 Mons (Belgium)
[d] Prof. D. B. AmabilinoInsitut de Ciencia de Materials de Barcelona (ICMAB-CSIC), 08193 Bellaterra(Spain)Present Address: School of Chemistry, The University of Nottingham,NG72RD (UK)E-mail : [email protected]
Supporting information for this article is available on the WWW underhttp ://dx.doi.org/10.1002/chem.201404753.
Chem. Eur. J. 2014, 20, 17443 – 17453 � 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim17443
Full PaperDOI: 10.1002/chem.201404753

molecular aggregates built up upon p–p stacking.[28, 29] Thechirality of the peripheral groups controls the helicity, M or P,of the primary fibres, and, moreover, non-linear effects, that is,“sergeants-and-soldiers”[30] and “majority rules”,[31] have beenevidenced by CD studies. Desymmetrisation of the C3 platformallowed the modulation of the number of chiral units, thenature of the appended groups, and, ultimately, the self-as-sembly properties.[32, 33] To prepare electroactive supramolecularaggregates, we have recently attached thioalkyl containing TTFunits to the 3,3’-bis(acylamino)-2,2’-bipyridine-substituted ben-zene-1,3,5-tricarboxamide (BiPy-BTA) moieties. Accordingly,organogels and conducting xerogel upon doping have beenobtained with three achiral bis(thioethyl)TTF fragments (com-pound A in Figure 1),[34] while by the use of non-amphiphilic
chiral 2-methyl-butyl groups (compound B in Figure 1), helicalfibres[35] or microscopic croissants[36] formed in the solid statedepending on the preparation conditions. Experimental CDstudies combined with theoretical CD calculations and molecu-lar mechanics and dynamics modelling clearly demonstratedthat for the (S) configuration of the alkyl chains the primaryfibres in solution were of left-handed (M) helicity, while inver-sion of the helical sense occurred from the solution to thesolid state.[35] Indeed, optical and electronic microscopy imageshave shown the presence of only right-handed (P) solid fibresor croissants for the configuration (S) of the alkyl substituent.Thus, inversion of helicity took place during the hierarchicalorganisation at the supramolecular level.
To illustrate the role of the chiral fragment on the self-assembly properties of the system, herein we investigate thereplacement of the short 2-methylbutyl groups (compound B)by the citronellyl and dihydrocitronellyl chains (compounds 1 aand 1 b, respectively, in Figure 1) that give a more classicalamphiphilic nature to the materials. When compared to com-pound B, in 1 a and 1 b the stereogenic centre has been shiftedby one carbon atom, which is expected to have some influ-ence on the helicity of the aggregates through the “odd–even”
effect.[37–41] Moreover, the dihydrocitronellyl unit hasdemonstrated its usefulness in the control of supramolecularorganisation of BTA derivatives[29–31, 42, 43] or other related C3-symmetric platforms.[44]
Results and Discussion
Synthesis and characterisation
The synthesis of the tris(TTF-citronellyl) derivatives follows thestrategy previously described for the ethyl and 2-methylbutylcompounds (A and B in Figure 1).[34, 35] First, the enantiopure(S,S) and (R,R) monoacylated derivatives 10 a,b have beenselectively obtained by reacting the acid chloride precursors8 a,b with the 3,3’-diamino-2,2’-bipyridine 9 at low tempera-ture to avoid the bis-acylation reaction. 8 a,b were classicallyprepared by a sequence involving first the reaction of thezinc(II) complex 2 with enantiopure citronellyl and dihydroci-tronellyl bromides affording the dithiole–thiones 3 a,b,[45]
which were then coupled with the diester dithiolone 4 toobtain the diester-TTFs 5 a,b. The latter were further mono-decarboxylated to the monoester-TTFs 6 a,b, hydrolysed to thecorresponding acids 7 a,b, and finally reacted with oxalylchloride to obtain 8 a,b (Scheme 1).
The amide protons of 10 a,b resonate at d= 14.85–14.87 ppm in 1H NMR, as a consequence of the withdrawingeffect of carbonyl and the intramolecular N�H···NPy hydrogenbond.[34] Further reaction of three equivalents of 10 a,b withone equivalent of trimesic acid chloride affords the tris(TTF) de-rivatives 1 a,b (Scheme 2) as red–brown powders that are verysoluble in chlorinated solvents and in toluene, insoluble in ace-tone and alcohols at room temperature, while in solvents likemethyethylketone or 1,4-dioxane they are partially soluble atroom temperature and become soluble upon heating. Interest-ingly, the dihydrocitronellyl derivative (1 b) is much moresoluble than the citronellyl species (1 a) in alkanes such asundecane or dodecane, which is probably because of themore rigid chains in the latter.
Molecular modelling of helical stacks
It is well-established that these chiral C3-symmetric precursorsself-assemble in solution to provide helical stacks. In the previ-ous study we have shown that when substituted with a (S)-2-methylbutyl group (compound B in Figure 1), the moleculesprefer to form a counterclockwise (CCW or M) helix,[35] which ismore stable by 2 kcal mol�1 per molecule than the clockwise(CW or P) helix. For the (S)-dihydrocitronellyl substituent ((S)-1 b), we have built two periodic stacks (CW and CCW) of 15molecules, which was sufficient to simulate an infinite stack, asthe twist from one unit to the next one is about 8.08 and themolecule is C3-symmetric. After a periodic optimisation of thestructure and the size of the box, we obtain intermoleculardistances of 3.67 � and 3.79 � for the CW and CCW helices,respectively (Figure 2).
We performed molecular dynamics (MD) simulations onthose two systems in the NVT ensemble, at a temperature of
Figure 1. C3-symmetrical tris(TTF) compounds.
Chem. Eur. J. 2014, 20, 17443 – 17453 www.chemeurj.org � 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim17444
Full Paper

300 K for 2 ns. When looking at the potential energy along thetrajectories, it does not allow us to determine which system isthe most stable because the fluctuations of energies are moreimportant than the difference of energy between the two sys-tems. To be able to differentiate them, we need to optimiseseveral points along the trajectories. Doing so on two hundredgeometries for each helix, the stabilisation energy per mole-cule converged to 131.3�0.7 kcal mol�1 for the CW helix and130.6�1.0 kcal mol�1 for the CCW helix, suggesting that theright-handed primary twist (P) is slightly more stable than theleft-handed twist. Thus, when changing the substituent from(S)-methylbutyl (compound B) to (S)-dihydrocitronellyl ((S)-1 b),we invert the chirality of the stack from CCW to CW, an inver-sion that is most likely related to the odd–even effect, as thestereogenic centre is shifted one carbon away from the TTFunit.[39] However, more importantly, the stability of one helicalsense over the opposite one (less than one kcal mol�1) is lesspronounced than for analogue B. For (S)-1 b, the CD spectrumhas been calculated based on one hundred structures extract-
ed from the MD trajectory (see computational details). Asexpected, we obtain a CD signal image of the one obtainedfor the precursor B (for a description of the main electronictransitions that involve contributions from both the TTF andbipyridine units, see Ref. [32]).
Electronic circular dichroism study
Circular dichroism (CD) spectroscopy has proved its efficiencyin the investigation of chiral supramolecular aggregates in so-lution,[46] and in particular in the case of precursor B (Figure 1),which shows strong CD signals in dioxane solutions at concen-trations of 2–5 � 10�5
m.[35] Accordingly, two intense negativebands centred at l= 387 and 368 nm, assigned to bipyridine-based transitions, and a shoulder at l= 319 nm, correspondingto a TTF transition, were observed for the (S) enantiomer of B,and the mirror image signals for the (R) enantiomer. These ex-perimental CD data combined with molecular mechanics/dy-namics simulation and theoretical CD calculations allowed to
Scheme 1. Synthesis of enantiopure tetrathiafulvalene(TTF)–amide–bipyridine precursors.
Scheme 2. Synthesis of enantiopure amphiphilic tris(TTFs).
Chem. Eur. J. 2014, 20, 17443 – 17453 www.chemeurj.org � 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim17445
Full Paper
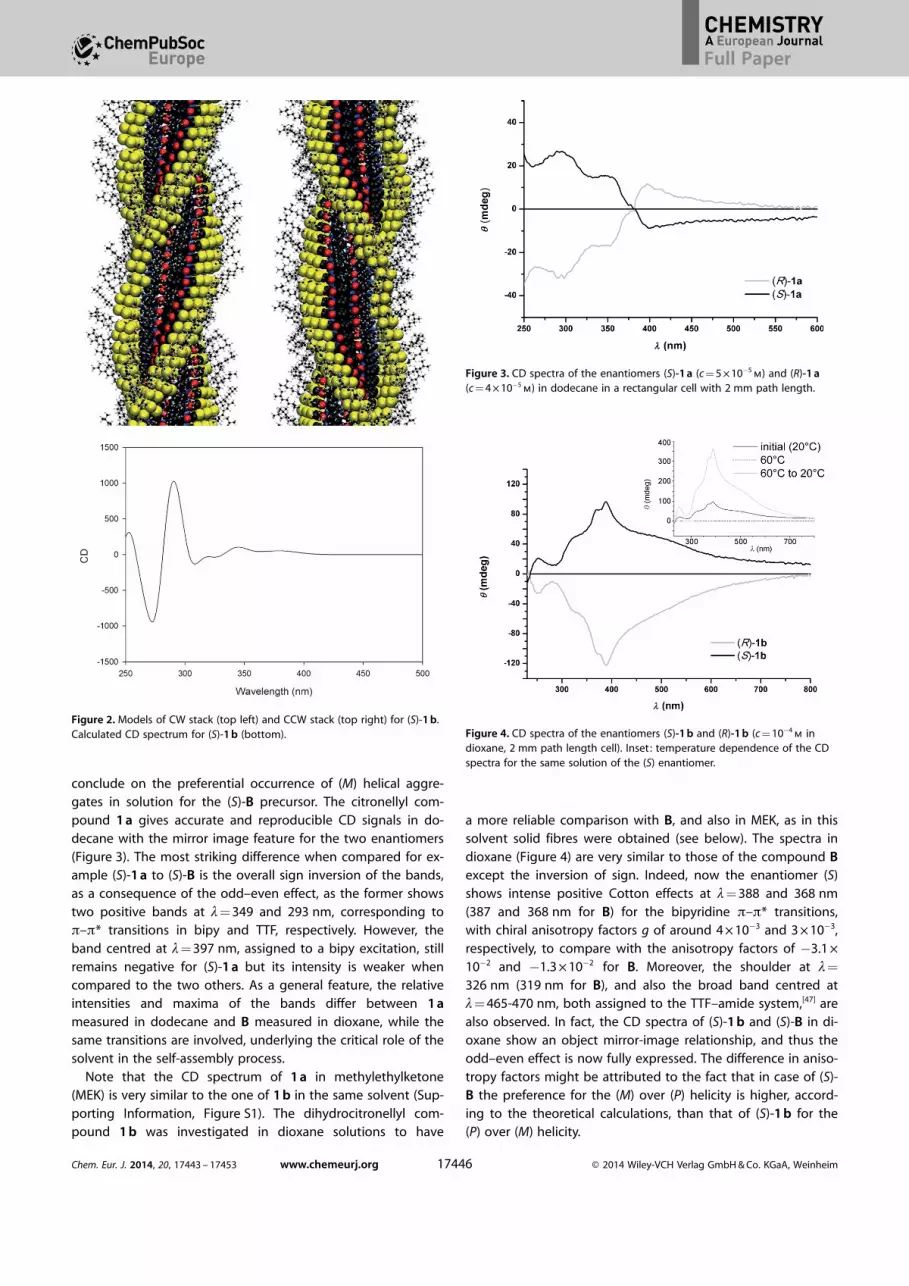
conclude on the preferential occurrence of (M) helical aggre-gates in solution for the (S)-B precursor. The citronellyl com-pound 1 a gives accurate and reproducible CD signals in do-decane with the mirror image feature for the two enantiomers(Figure 3). The most striking difference when compared for ex-ample (S)-1 a to (S)-B is the overall sign inversion of the bands,as a consequence of the odd–even effect, as the former showstwo positive bands at l= 349 and 293 nm, corresponding top–p* transitions in bipy and TTF, respectively. However, theband centred at l= 397 nm, assigned to a bipy excitation, stillremains negative for (S)-1 a but its intensity is weaker whencompared to the two others. As a general feature, the relativeintensities and maxima of the bands differ between 1 ameasured in dodecane and B measured in dioxane, while thesame transitions are involved, underlying the critical role of thesolvent in the self-assembly process.
Note that the CD spectrum of 1 a in methylethylketone(MEK) is very similar to the one of 1 b in the same solvent (Sup-porting Information, Figure S1). The dihydrocitronellyl com-pound 1 b was investigated in dioxane solutions to have
a more reliable comparison with B, and also in MEK, as in thissolvent solid fibres were obtained (see below). The spectra indioxane (Figure 4) are very similar to those of the compound Bexcept the inversion of sign. Indeed, now the enantiomer (S)shows intense positive Cotton effects at l= 388 and 368 nm(387 and 368 nm for B) for the bipyridine p–p* transitions,with chiral anisotropy factors g of around 4 � 10�3 and 3 � 10�3,respectively, to compare with the anisotropy factors of �3.1 �10�2 and �1.3 � 10�2 for B. Moreover, the shoulder at l=
326 nm (319 nm for B), and also the broad band centred atl= 465-470 nm, both assigned to the TTF–amide system,[47] arealso observed. In fact, the CD spectra of (S)-1 b and (S)-B in di-oxane show an object mirror-image relationship, and thus theodd–even effect is now fully expressed. The difference in aniso-tropy factors might be attributed to the fact that in case of (S)-B the preference for the (M) over (P) helicity is higher, accord-ing to the theoretical calculations, than that of (S)-1 b for the(P) over (M) helicity.
Figure 2. Models of CW stack (top left) and CCW stack (top right) for (S)-1 b.Calculated CD spectrum for (S)-1 b (bottom).
Figure 3. CD spectra of the enantiomers (S)-1 a (c = 5 � 10�5m) and (R)-1 a
(c = 4 � 10�5m) in dodecane in a rectangular cell with 2 mm path length.
Figure 4. CD spectra of the enantiomers (S)-1 b and (R)-1 b (c = 10�4m in
dioxane, 2 mm path length cell). Inset: temperature dependence of the CDspectra for the same solution of the (S) enantiomer.
Chem. Eur. J. 2014, 20, 17443 – 17453 www.chemeurj.org � 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim17446
Full Paper

Upon heating a dioxane solution of (S)-1 b or (R)-1 b at 60 8Cfor a couple of minutes, the CD signal completely disappears(inset in Figure 4). Nevertheless, the cooled solution showsmuch more intense bands, suggesting that the final aggre-gates are better organised than in the initial solution, a similarbehaviour to that of compound B.[35] For example, the band atl= 388 nm has now an anisotropy factor of 1.4 � 10�2, whichrepresents a circa 3.5-fold increase with respect to the initialsolution, and approaches the value shown by (S)-B. Very likely,the helical aggregates disassemble upon heating, and at 60 8Conly short optically inactive aggregates remain. When coolingback the solution, these aggregates serve as nucleation seedsfor the self-assembly process of the helical fibres, which growin a more ordered fashion and in which the chiral preferencefor the (P) helicity is more clearly expressed. Moreover, whena solution of (S)-1 b (2 � 10�5
m) is heated at 50 8C for 30–40 min, the CD signal disappears and it is not then recoveredby cooling (Supporting Information, Figure S2), showing thata critical concentration in nucleation seeds is needed to ob-serve the self-assembly process. However, the addition ofa small volume (10 mL) of fresh solution of (S)-1 b in the initialsolution (0.2 mL), showing no more signal after heating, trig-gers the aggregation processes of the dispersed molecules, asdemonstrated by the CD signals of the new solution, whichare eventually more intense than those of the initial solutionbefore heating (Supporting Information, Figure S1).
The CD spectra of 1 b in MEK show basically the same char-acteristics as those in dioxane, with positive Cotton effects forthe (S) enantiomer and negative for the (R) enantiomer(Figure 5). The maximum of the two p–p* bipyridine transi-tions slightly change to l= 383 and 364 nm. As the differencein aggregation between the room temperature and 50 8C isvery weak in MEK only, a solution of MEK/CHCl3 9:1 was usedfor the temperature dependence studies (inset in Figure 5).
As observed, the CD signal disappears upon heating thesample at 50 8C for a couple of minutes, and then it is recov-
ered upon cooling back to 20 8C, reaching practically the sameintensity as before heating. The difference with the dioxanesolution probably comes from the fact that, as the moleculesdo not self-assemble in CHCl3, in the MEK/CHCl3 9:1 mixturepart of the molecules 1 b still remain dispersed in solution. Thisparticular mixture of solvents was chosen because solid helicalaggregates formed out of it upon re-precipitation (see below).No evidence for the “majority rules” effect could be observedfor either 1 a or 1 b, thus emphasising the role of kinetic effectssuch as nucleation and growth in the self-assembly process.
Microscopic investigation of macroscopic aggregates
The derivative 1 a with the six citronellyl chains forms longfibres from methylethylketone (MEK) upon cooling a hot solu-tion of the compound at a concentration of 1 mg per millilitreon the bench. For the (S) enantiomer these fibres have a clearlyleft-handed (M helical) morphology. In accord with the odd–even effect for chiral compounds, this is the opposite handed-ness for the fibres formed by same enantiomer of thecompound B with a 2-methylbutyl chain, in that case formedfrom dioxane.[35] In the present citronellyl-substituted case, nofibrous material was obtained from dioxane, presumablybecause of the very different solubility of the compound. Thefibres from MEK are very long (several tens of micrometres)and are between 600 nm and 1 mm wide. Scanning electronmicroscope (SEM) images (Figure 6) show that the fibres areleft-handed, although the texture is quite smooth. The ends ofthe fibres are tapered, and the fibres occasionally turn at rightangles. The fibres also show helical morphology in the opticalmicroscope using reflected light (Figure 6) where long regionsof helical order can be discerned. In transmission, no helicity isseen, but crossing polarisers shows that the fibres arebirefringent and as such they have a relatively high degree ofsupramolecular order within them.
When the same hot solution in MEK at the same concentra-tion is cooled slowly to room temperature in a bath overa period of one hour, very short “chiral croissants” are formed(Figure 7). The particles are between one and two micrometresin width and between four and seven micrometres in length,the aspect ratio being similar in all cases. Often, the objectsappear to be homochiral (see the optical microscope image inFigure 7), as they are clearly in the case of the chiral croissant-shaped objects formed under similar conditions in dioxane forthe compound B with the 2-methyl-butyl chain.[36] However,the SEM images show that there is a non-infrequent inversionof helicity. The main left-handed spiral is usually seen for the(S) enantiomer of the citronellyl-substituted species, but the in-version at the midpoint to give a right-handed helix is also ob-served (one can be seen right of centre in the SEM image inFigure 7) or simply a poorly defined structure. The material isalso birefringent, as witnessed by their brightness betweencrossed polarising filters in the optical microscope.
The citronellyl derivative 1 a is soluble in a wide range of sol-vents, and also shows aggregate formation in them, unlike thecompound B with 2-methylbutyl chains.[35, 36] When MEK ismixed with THF in a 9:1 ratio and the compound was dis-
Figure 5. CD spectra of the enantiomers (S)-1 b and (R)-1 b (c = 10�5m in
methylethylketone (MEK), 2 mm path length cell). Inset : temperature de-pendence of the CD spectra for the (S) enantiomer in MEK/CHCl3 9:1 solu-tion (c = 2 � 10�5
m).
Chem. Eur. J. 2014, 20, 17443 – 17453 www.chemeurj.org � 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim17447
Full Paper

solved in the hot mixture at a concentration of 2.45 mg mL�1,cooling on the bench resulted in fibres that show a muchclearer helical morphology than those formed in MEK alone(Figure 8). The fibres, that have clearly tapered ends, are ap-
proximately one-and-a-half micrometres wide and are of theorder of hundreds of micrometres long. Shorter fibres are alsoobserved with lengths in the region of 50 mm. Interestingly, inthese fibres both M and P morphologies are observed clearlyin all the material. The M morphology is still the dominantone, but the P morphology is present in many fibres. Helical in-version points of these macroscopic objects can be seen clear-ly in the SEM images. The inversion points are also present inthe shorter fibres that are also formed in this precipitation pro-cess (Figure 9). They can also be inferred in the optical micro-scope images of these shorter fibres (Figure 9) that are in sizebetween the chiral croissant-like objects and the longer fibres.In all of the samples, very short (1–2 mm long) and narrowfibres (hundreds of nanometres) are also observed, similar tothose seen in the emergent chiral structures in the 2-methyl-butyl derivative B.[36]
The careful inspection of more than 150 aggregates presenton 21 SEM images allows for a more quantitative estimation ofthe helical preference of the fibres formed out of (S)-1 a inMEK/THF. Accordingly, 66 % of the fibres show M morphology,8 % P, 18 % show both M and P with a centre of inversion,while about 8 % of the fibres do not show any clear helicity.The observation of an inversion of fibre chirality indicates thedelicate balance in the twisting of the aggregates one way orthe other. It is only in the micrometre-sized aggregates ofthese molecules that the helical morphology can be observed;the smaller structures show a smooth surface. Despite this fact,chirality is clearly present in the assemblies because they pres-ent very significant circular dichroic signals. The inversion offibre morphology was seen by us in micrometre-sized fibresformed by a racemic mixture of self-assembling C3 compoundsB,[35] but the observation of the phenomenon here in an enan-tiopure sample is unprecedented insofar as we are aware.Moreover, in the enantiopure B inversion of helicity in the solidfibres has not been observed, which is in agreement with thehigher degree of helical twist discrimination, suggested by thetheoretical calculations, in B than in 1 a,b. As in the latter the
Figure 6. Scanning electron microscope image (top) of the fibres of the (S)enantiomer of 1 a formed by cooling of a hot solution in MEK on the bench,and (below) a reflection optical microscope image (left) of fibres in the samesample and a transmission optical image with crossed polarisers (bottom).Scale bars: 10 mm.
Figure 7. Scanning electron microscope image (top) of the “chiral croissants”of (S)-1 a formed by slow cooling of a hot solution in MEK in a bath, and(below) a reflection optical microscope image of the objects. Scale bars:5 mm.
Figure 8. Scanning electron microscope image of the fibres of the (S) enan-tiomer of the citronellyl-substituted compound formed by cooling of a hotsolution in MEK/THF 9:1 on the bench. The morphological helicity of thefibres is indicated next to them, and a clear inversion point is indicated.Scale bar: 10 mm.
Chem. Eur. J. 2014, 20, 17443 – 17453 www.chemeurj.org � 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim17448
Full Paper

difference in energy between the M and P primary twists forthe same enantiomer is much weaker (see above), helicalinversion might occur with higher probability than in the caseof B.
The dihydrocitronellyl compounds 1 b form fibres from MEK,and mixtures of this solvent with 10 % of either chloroform orTHF. In all cases the morphologies are similar. The fibres are rel-atively short, from several tens to a few hundreds of microme-tres long (Supporting Information, Figure S3). The helicity isnot always evident in their morphology, but when it is thefibres show a clearly P helical morphology for the (R)enantiomer (Figure 10), in the same way as the citronellylderivative does. The width of the fibres is quite uniform, and isapproximately 700 nm.
Therefore in the assembly of dihydrocitronellyl compounds1 b the expression of the chirality from the stereogenic centresto the morphology of the macroscopic fibres is not as clear asin the case of the citronellyl compounds, in that smooth mor-phology is observed frequently. On the other hand, no oppo-site handedness are seen in the fibres. This subtle situation,that in a way is a contradiction in that the compound withenantiomorphic fibres shows greater tendency for helical mor-phology, is probably a result of the different conformationsand relative rigidity of the chain ends where alkyl or alkenegroups are present.
On the other hand, slower cooling of the dihydrocitronellylcompounds also leads to fibres that show helical morphologyon occasion (Figure 11). The length of the individual objects isbetween ten and twenty micrometres, somewhat shorter thanthose formed under faster cooling conditions. The width of thefibres is similar in both cases (approximately half a micrometre).
The SEM images shown in Figure 11 show areas of particularlywell-resolved fibres, where it can be observed that the helicalfibres are formed by intertwining smaller ones that sometimesseparate at the ends of the larger objects. In any case, themorphology is completely different to the helical objects with
Figure 9. Scanning electron microscope image (left) and reflection opticalmicroscope image (right) of the fibres of the (S) enantiomer of the citronell-yl-substituted compound formed by cooling of a hot solution in MEK/THF9:1 on the bench. The inversion of the morphological helicity of the fibrescan be seen at their thickest part and also in the SEM image in the lowersection. Scale bars: 10 mm.
Figure 10. Optical micrographs of the fibres formed by the (R) enantiomerof 1 b upon precipitation from MEK. Transmission (top), transmission withcrossed polarisers (middle), and reflection (bottom). Scale bars : 5 mm.
Chem. Eur. J. 2014, 20, 17443 – 17453 www.chemeurj.org � 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim17449
Full Paper

a smaller aspect ratio formed by either 1 a or the analogouscompounds bearing the 2-methylbutyl group.[36] Fibres are alsoformed by the enantiomers of 1 b in cyclohexanol, although inthis case a totally smooth morphology is observed (SupportingInformation, Figures S4, S5), and no hint of helicity wasobserved in either optical or electronic microscopes.
Conclusion
The use of chiral citronellyl derivatives in C3-symmetric com-pounds incorporating three TTF units gives the moleculesa preference for one handedness in their self-assembly intoone-dimensional stacks. The odd–even effect in the assembly(compared with a previous 2-methylbutyl substituted com-pound) is followed. Great differences are observed in thehigher-order fibres that assemble upon precipitation of thecompounds. A fast cooling of the saturated solutions leads tothe formation of fibres. For the citronellyl substituted com-pound, the transmission of chirality from the single stacks tothe macroscopic fibres is not as selective as in the case of 2-methylbutyl chains. Thus, despite the fact that a clear prefer-ence for single handedness is seen in one dimensional stacksin solution, the macroscopic objects have morphologies thatdisplay both helical senses despite the presence of a singleenantiomer.
One the other hand, a degree of structural rigidity apparent-ly favours the emergence of helical morphology in certaintypes of aggregate. In the samples that precipitate on relative-ly slow cooling, chirality is very evident for citronellyl and 2-methylbutyl substituted compounds in the croissant-like shortfibres that they form. On the other hand, the dihydrocitronellylcompound forms somewhat longer fibres in which the helicalmorphology is not so evident and where the componentsmaller fibres tend in some instances to unwind near the fibreends, perhaps pointing to a weaker interaction as a result ofthe greater flexibility at the chain end compared with theother compounds. These new series of electroactive C3 com-pounds demonstrate the flexibility of our approach towardsa control of the helical sense, morphologies, and dimensionsof the supramolecular aggregates. Although so far only hydro-phobic chains have been attached at the periphery of the disk-shaped platform, the possibility of using hydrophilic polyetherchains is also envisaged. Moreover, desymmetrisation of the C3
core through the well-established strategy[32, 33] would possiblyallow for the modulation of the helical pitch of the mesoscopicsize fibres.
Experimental Section
General
Reactions were carried out under argon and using toluene HPLC.Nuclear magnetic resonance spectra were recorded on a BrukerAvance DRX 500 spectrometer (operating at 500.04 MHz for 1Hand 125 MHz for 13C) and a Bruker Avance DRX 300 automaticspectrometer (operating at 300 MHz for 1 H, and 75 MHz for 13C).Chemical shifts are expressed in parts per million (ppm) downfieldfrom external TMS. The following abbreviations are used: s singlet,d doublet, m multiplet, br broad. MALDI-TOF MS spectra were re-corded on Bruker Biflex-IIITM apparatus, equipped with a 337 nmN2 laser. Elemental analyses were recorded using Flash 2000 FisherScientific Thermo Electron analyser. The compound 3,3’-diamino-2,2’-bipyridine 9 has been synthesised according to a previouslyreported procedure.[48]
Synthesis
General method for 6 a or 6 b: 5 a or 5 b (see the SupportingInformation; 1 equiv) and LiBr (18 equiv) were mixed in dimethyl-formamide. The solution was stirred at 85 8C for 5 h, the formationof the product being monitored by TLC. Brine was added to the re-action mixture, the product was extracted with ethyl acetate andthe organic phase was washed with brine and water and thendried over MgSO4. The solvent was removed under vacuum andthe product was purified by chromatography on a silica columnusing petroleum spirit with 5–10 % of ethyl acetate.
Bis(citronellylthio)methoxycarbonyltetrathiafulvalene (S,S)-6 a :General procedure, starting from (S,S)-5 a (2.42 g, 3.66 mmol) andLiBr (5.72 g, 65.9 mmol). The product was obtained as an orangeoil. Yield 1.70 g (2.82 mmol, 77 %).
Bis(citronellylthio)methoxycarbonyltetrathiafulvalene (R,R)-6 a :General procedure, starting from (R,R)-5 a (1.85 g, 2.80 mmol) andLiBr (4.37 g, 50.4 mmol). The product was obtained as an orangeoil. Yield 1.60 g (2.60 mmol, 95 %). 1H NMR (300 MHz, CDCl3): d
(ppm) 7.35 (s, 1 H, HTTF), 5.09 (t, 2 H, 3J = 6.9 Hz, C=CH), 3.82 (s, 3 H,CO2CH3), 2.83 (m, 4 H, SCH2), 1.98 (m, 4 H), 1.69 (s, 6 H, C(CH3)2
Figure 11. SEM micrographs of the fibres formed by the (S) enantiomer of1 b upon precipitation from a mixture of MEK and chloroform (9:1 v/v). Scalebars : 10 mm.
Chem. Eur. J. 2014, 20, 17443 – 17453 www.chemeurj.org � 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim17450
Full Paper

trans), 1.61 (s, 6 H, C(CH3)2 cis), 1.43–1.70 (m, 6 H), 1.15–1.38 (m, 4 H,SCH2CH2), 0.91 (d, 6 H, 3J = 6.2 Hz, CHCH3). 13C NMR (75 MHz, CDCl3):d 159.7 (CO2CH3), 131.9 (CCO2CH3), 131.4 (HC=C(CH3)2), 128.3, 128.0,127.3, 124.5 (HC=C(CH3)2), 52.7 (CO2CH3), 36.8 (CH2), 36.6 (CH2), 34.2(SCH2), 31.7 (CHCH3), 25.7 (C=CHCH2), 25.4 (CH3 trans), 19.2(CHCH3), 17.7 (CH3 cis). MS (MALDI-TOF) m/z : 601.6 [M+] ; theor.602.15.
Bis((3,7-methyloct-6-octyl)thio)methoxycarbonyltetrathiafulva-lene (S,S)-6 b : General procedure, starting from (S,S)-5 b (4.25 g,6.4 mmol) and LiBr (10 g, 115.2 mmol). The product was obtainedas an orange–red oil. Yield 3.84 g (6.3 mmol, 99 %).
Bis((3,7-methyloct-6-octyl)thio)methoxycarbonyltetrathiafulva-lene (R,R)-6 b : General procedure, starting from (R,R)-5 b (3 g,4.5 mmol) and LiBr (7 g, 81 mmol). The product was obtained as anorange-red oil. Yield 2.6 g (4.3 mmol, 95 %). 1H NMR (300 MHz,CDCl3): d 7.34 (s, 1 H, HTTF), 3.84 (s, 3 H, CO2CH3), 2.78–2.96 (m, 4 H,SCH2), 1.40–1.69 (m, 8 H), 1.10–1.33 (m, 12 H), 0.87 (d, 6 H, 3J =6.9 Hz, *CHCH3), 0.86 (d, 12 H, 3J = 6.6 Hz, CH(CH3)2). 13C NMR(75 MHz, CDCl3): d 159.8 (CO2CH3), 132.0 (CCO2CH3), 128.3, 128.1,127.3 (CH2SC), 111.9, 109.9, 52.7 (CO2CH3), 39.2, 36.8, 36.7, 34.2,32.0, 27.9, 24.6, 22.7, 22.6, 19.2. MS (MALDI-TOF) m/z : 605.9 [M+] ;theor. 606.18.
General method for 7 a and 7 b: A mixture of LiOH·H2O (5 equiv)was degassed for 15 min and was added under argon to a solutionof 6 a or 6 b (1 equiv) in dioxane. After a night at room tempera-ture, the mixture was neutralised with 5 m HCl (2 mL) and thecolour turned to red. After 15 min. at room temperature diethylether and several mL of water were added in order to form twophases. 5 m HCl was added until the pH of the mixture became 1;the two phases were separated and the aqueous phase was ex-tracted with diethyl ether. The combined organic phases weredried over MgSO4 and the solvent was removed by evaporation.
Bis((citronellylthio)tetrathiafulvalenylcarboxylic acid (S,S)-7 a :General procedure, starting from (S,S)-6 a (1.61 g, 2.67 mmol) andLiOH·H2O (0.56 g, 13.35 mmol). The product was obtained as anorange–red oily solid. Yield 1.44 g (2.37 mmol, 89 %).
Bis((citronellylthio)tetrathiafulvalenylcarboxylic acid (R,R)-7 a :General procedure, starting from (R,R)-6 a (1.60 g, 2.65 mmol) andLiOH·H2O (0.55 g, 13.25 mmol). The product was obtained as anorange–red oily solid. Yield 1.53 g (2.6 mmol, 99 %). 1H NMR(300 MHz, CDCl3, selected signals): d (ppm) 6.93 (s, 1 H, HTTF), 5.09(t, 2 H, 3J = 7.1 Hz, C=CH), 2.74–2.91 (m, 4 H, SCH2), 1.98 (m, 4 H),1.69 (s, 6 H, C(CH3)2 trans), 1.60 (s, 6 H, C(CH3)2 cis), 1.20–1.60 (m,10 H), 0.90 (d, 6 H, 3J = 6.0 Hz, CHCH3). 13C NMR (75 MHz, CDCl3): d
(ppm) 131.4–131.4 (HC=C(CH3)2), 124.6–124.5 (HC=C(CH3)2), 36.7(CH2), 31.7 (CHCH3), 25.7 (C=CHCH2), 25.4 (CH3 trans), 19.2 (CHCH3),17.7 (CH3 cis). MS (MALDI-TOF) m/z : 587.4 [M+] ; theor. 588.14.
Bis((3,7-methyloct-6-octyl)thio)tetrathiafulvalenylcarboxylic acid(S,S)-7 b : General procedure, starting from (S,S)-6 b (3.8 g,6.26 mmol) and LiOH·H2O (1.31 g, 31.3 mmol). The product was ob-tained as an orange–red oily solid. Yield 3.71 g (6.25 mmol, 99 %).
Bis((3,7-methyloct-6-octyl)thio)tetrathiafulvalenylcarboxylic acid(R,R)-7 b : General procedure, starting from (R,R)-6 b (2.5 g,4.11 mmol) and LiOH·H2O (0.86 g, 20.6 mmol). The product was ob-tained as an orange–red oily solid. Yield 2.45 g (4.11 mmol, 100 %).1H NMR (300 MHz, CDCl3): d (ppm) 6.91 (s, 1 H, HTTF), 2.79–3.15 (m,4 H, SCH2), 1.42–1.69 (m, 8 H, CH2), 1.25–1.31 (m, 12 H, CH,CH2), 0.85(d, 6 H, 3J = 6.0 Hz, *CHCH3), 0.86 (d, 12 H, 3J = 6.6 Hz, CH(CH3)2). MS(MALDI-TOF) m/z : 592.2 [M+] ; theor. 592.17.
General method for 8 a and 8 b : 7 a or 7 b (1 equiv) was dissolvedin THF under argon and the solution was heated at 45 8C. Oxalylchloride (3 equiv) and pyridine 10 % v/v in THF were immediately
added dropwise. After 3 h at 45 8C the colour of the solutionturned to dark violet. The solvent and the excess oxalyl chloridewere removed under vacuum. The resulting dark violet oil,obtained in quantitative yield, was used in the next stepwithout further purification.
Bis((citronellylthio)chloroformyltetrathiafulvalene (S,S)-8 a: Gen-eral procedure, starting from (S,S)-7 a (1.44 g, 2.37 mmol) andoxalyl chloride (0.88 g, 7.11 mmol). The product was obtained asa dark violet oil.
Bis((citronellylthio)chloroformyltetrathiafulvalene (R,R)-8 a: Gen-eral procedure, starting from (R,R)-7 a (1.5 g, 2.54 mmol) and oxalylchloride (0.94 g, 7.62 mmol). The product was obtained as a darkviolet oil.
Bis((3,7-methyloct-6-octyl)thio)chloroformyltetrathiafulvalene(S,S)-8 b : General procedure, starting from (S,S)-7 b (2 g, 3.37 mmol)and oxalyl chloride (1.28 g, 10.11 mmol). The product was obtainedas a dark violet oil.
Bis((3,7-methyloct-6-octyl)thio)chloroformyltetrathiafulvalene(R,R)-8 b : General procedure, starting from (R,R)-7 b (1.5 g,2.5 mmol) and oxalyl chloride (1 g, 7.5 mmol). The product was ob-tained as a dark violet oil.
General method for 10 a and 10 b : A solution of 3,3’-diamino-2,2’-bipyridine 9 (1 equiv) and triethylamine (1 equiv) in dry CH2Cl2 wascooled at 0 8C, and then a solution of the acid chloride 8 a or 8 b(1 equiv) in dry CH2Cl2 was added dropwise. The mixture wasstirred at 0 8C for 2 h and overnight at room temperature, affordinga brown solution. The entire mixture was washed several timeswith water and once with brine. The organic phase was then sepa-rated and then dried over magnesium sulfate to afford a red–brown solid, which was further purified as follows for each type ofcompound.
3’-{[Bis(citronellylthio)tetrathiafulvalenyl]formylamino}2,2’-bipyr-idine-3-amine (S,S)-10 a: General procedure, starting from 3,3’-dia-mino-2,2’-bipyridine 9 (0.44 g, 2.4 mmol) and (S,S)-8 a (1.5 g,2.4 mmol). The obtained red–brown crude was purified by chroma-tography on a silica column with dichloromethane. The productwas obtained as an orange–red solid. Yield 1.39 g (1.8 mmol, 76 %).
3’-{[Bis[((R)-citronellyl)thio)]tetrathiafulvalenyl]formylamino}-2,2’-bipyridine-3-amine (R,R)-10 a: General procedure, starting from3,3’-diamino-2,2’-bipyridine 9 (0.47 g, 2.54 mmol) and (R,R)-8 a(1.54 g, 2.54 mmol). The obtained red–brown crude was purifiedby chromatography on a silica column with dichloromethane. Theproduct was obtained as an orange–red solid. Yield 1.85 g(2.4 mmol, 96 %). 1H NMR (300 MHz, CDCl3) d (ppm): 14.87 (s, 1 H,NH), 9.07 (dd, 1 H, 3J = 8.5 Hz, 4J = 1.5 Hz, Hpy), 8.34 (dd, 1 H, 3J =4.6 Hz, 4J = 1.6 Hz, Hpy), 8.11 (dd, 1 H, 3J = 4.0 Hz, 4J = 1.9 Hz, Hpy),7.29 (m, 1 H, HTTF), 7.27 (m, 1 H, Hpy), 7.13–7.20 (m, 2 H, Hpy), 6.69 (br,2 H, NH2), 5.09 (m, 2 H, C=CH), 2.86 (m, 4 H), 1.99 (m, 4 H, C=CHCH2),1.67 (d, 6 H, CH3 trans), 1.59 (d, 6 H, CH3 cis), 1.30–1.70 (m, 6 H),1.10–1.26 (m, 4 H), 0.94 (t, 6 H, 3J = 7.2 Hz, *CHCH3). 13C NMR(75 MHz, CDCl3) d (ppm): 157.8 (C=O), 145.3, 143.2, 141.2, 138.0,135.7, 134.8, 133.8, 128.4, 128.0, 126.5, 125.4, 124.3, 122.6, 112.2,109.9, 36.1, 31.8, 21.7, 13.6. MS (MALDI-TOF) m/z = 756.2 [M+] ;theor. 756.22.
3’-{[Bis[(S)-((3,7-methyloct-6-octyl)thio)]tetrathiafulvalenyl]for-mylamino}-2,2’-bipyridine-3-amine (S,S)-10 b: General procedure,starting from 3,3’-diamino-2,2’-bipyridine 9 (0.62 g, 3.3 mmol) and(S,S)-8 b (2 g, 3.3 mmol). The obtained red–brown crude was puri-fied by chromatography on a silica column with dichloromethane/petroleum spirits 2:1. The product was obtained as a red–brownpasty solid. Yield 1.56 g (2.04 mmol, 62 %).
Chem. Eur. J. 2014, 20, 17443 – 17453 www.chemeurj.org � 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim17451
Full Paper

3’-{[Bis[(R)-((3,7-methyloct-6-octyl)thio)]tetrathiafulvalenyl]for-mylamino}-2,2’-bipyridine-3-amine (R,R)-10 b: General procedure,starting from 3,3’-diamino-2,2’-bipyridine 9 (0.47 g, 2.54 mmol) and(R,R)-8 b (1.54 g, 2.54 mmol). The obtained red–brown crude waspurified by chromatography on a silica column with dichlorome-thane/petroleum spirits 2:1. The product was obtained as a red–brown pasty solid. Yield 1.15 g (2.4 mmol, 61 %). 1H NMR (300 MHz,CDCl3): d (ppm) 14.85 (br, 1 H, CONH), 9.06 (dd, 1 H, 3J = 8.4 Hz, 4J =1.8 Hz, Hpy), 8.32 (dd, 1 H, 3J = 4.5 Hz, 4J = 1.5 Hz, Hpy), 8.09 (dd, 1 H,3J = 4.3 Hz, 4J = 1.8 Hz, Hpy), 7.27 (m, 1 H, Hpy), 7.27 (1 H, HTTF), 7.13–7.16 (m, 2 H, Hpy), 6.67 (br, 2 H, NH2), 2.74–2.94 (m, 4 H, SCH2), 1.42–1.70 (m, 8 H), 1.21–1.31 (m, 6 H), 1.00–1.17 (m, 6 H), 0.89 (d, 6 H, 3J =6.3 Hz, *CHCH3), 1.02 (d, 12 H, 3J = 6.3 Hz, CH(CH3)2. 13C NMR(75 MHz, CDCl3): d (ppm) 157.7 (C=O), 145.2, 143.1, 141.1, 137.9,135.7, 134.7, 133.8, 128.3, 127.9, 127.8, 126.4, 125.4, 124.3, 122.6,112.2, 109.7, 39.2, 36.8, 3.3, 34.2, 32.1, 27.9, 24.6, 22.7, 22.6, 19.3.MS (MALDI-TOF) m/z : 759.9 [M+] ; theor. 760.25.
General method for 1 a and 1 b : A solution of 10 a or 10 b(1 equiv) and triethylamine (1 equiv) in dry CH2Cl2 was cooled at0 8C, and then a solution of trimesic chloride 11 (0.33 equiv) in dryCH2Cl2 was added dropwise. After 2 h stirring at 0 8C and one nightat room temperature water was added and the product was ex-tracted with dichloromethane. The organic phase was furtherwashed several times with water and the solvent was evaporated.The resulting solid was further purified as follows for each com-pound.
N,N’,N’’-Tris{3[3’-{[Bis(((S)-citronellyl)thio)]tetrathiafulvalenyl]for-mylamino]-2,2’-bipyridyl}benzene-1,3,5-tricarboxamide (S,S)-1 a :General procedure, starting from (S,S)-10 a (1.29 g, 1.70 mmol) andtrimesic chloride 11 (0.15 g, 0.56 mmol). The obtained solid waswashed several times with methanol, ethyl acetate, and acetone,and dried under vacuum. The product was obtained as an analyti-cally pure red–brown powder. Yield 1.02 g (0.42 mmol, 74 %). MS(MALDI-TOF) m/z : 2426.2; theor. 2424.73. Elemental analysiscalcd (%) for C120H144N12O6S18: C 59.37, H 5.98, N 6.92, O 3.93,S 23.77; found: C 59.22, H 6.02, N 6.73, S 23.09.
N,N’,N’’-Tris{3[3’-{[Bis(((R)-citronellyl)thio)]tetrathiafulvalenyl]for-mylamino]-2,2’-bipyridyl}benzene-1,3,5-tricarboxamide (R,R)-1 a :General procedure, starting from (R,R)-10 a (1.85 g, 2.44 mmol) andtrimesic chloride 11 (0.21 g, 0.81 mmol). The obtained solid waswashed several times with methanol, ethyl acetate, and acetone,and dried under vacuum. The product was obtained as an analyti-cally pure red–brown powder. Yield 1.6 g (0.37 mmol, 81 %). MS(MALDI-TOF) m/z : 2426.2; theor. 2424.73. Elemental analysiscalcd (%) for C120H144N12O6S18: C 59.37, H 5.98, N 6.92, O 3.93,S 23.77; found: C 58.08, H 5.83, N 6.18, S 23.37.
N,N’,N’’-Tris{3[3’-{[Bis(S)-((3,7-methyloct-6-octyl)thio)]tetrathiaful-valenyl]formylamino]-2,2’-bipyridyl}benzene-1,3,5-tricarboxa-mide (S,S)-1 b : General procedure, starting from (S,S)-10 b (1.3 g,1.70 mmol) and trimesic chloride 11 (0.15 g, 0.56 mmol). The ob-tained solid was washed several times with methanol, pentane,and acetone, and dried under vacuum. The product was obtainedas an analytically pure red–brown powder. Yield 1.29 g (0.53 mmol,95 %). MS (MALDI-TOF) m/z: 2437.6; theor. 2436.72. Elemental anal-ysis : calcd for C120H156N12O6S18: C 59.07, H 6.44, N 6.89, O 3.93,S 23.66; found: C 57.48, H 6.30, N 6.51, S 22.63.
N,N’,N’’-Tris{3[3’-{[Bis(R)-((3,7-methyloct-6-octyl)thio)]tetrathiaful-valenyl]formylamino]-2,2’-bipyridyl}benzene-1,3,5-tricarboxa-mide (R,R)-1 b : General procedure, starting from (R,R)-10 b (1.1 g,1.45 mmol) and trimesic chloride 11 (0.13 g, 0.48 mmol). The ob-tained solid was washed several times with methanol, ethyl ace-tate, and acetone, and dried under vacuum. The product was ob-tained as an analytically pure red–brown powder. Yield 1.15 g
(0.47 mmol, 98 %). MS (MALDI-TOF) m/z : 2437.6; theor. 2436.72. El-emental analysis calcd (%) for C120H156N12O6S18: C 59.07, H 6.44,N 6.89, O 3.93, S 23.66); found: C 58.67, H 6.29, N 6.87, S 22.83.
Microscopy
Optical images were collected with a polarising optical microscopeOlympus BX51TRF. SEM images were acquired by scanning electronmicroscope (SEM) QUANTA FEI 200 FEG-ESEM system on samplesdeposited on gold on mica from the suspension. Contacts weremade between the gold surface and the sample holder withgraphite paste to avoid charging.
Computational details of molecular mechanics and molecu-lar dynamics
All calculations were performed using the Tinker package[49] andthe MM3 force-field.[50] MD simulations were carried out in the can-onical ensemble with an integration step of 1 fs at a temperatureof 300 K maintained using a Berendsen thermostat.[51] Simulationsof the single helices were performed using periodic boundary con-dition in order to simulate infinite stacks during 1 ns for equilibra-tion followed by 2 ns to collect data. Stabilisation energies werecalculated by optimising snapshots along the MD trajectories every10 ps, as follows:
Estab ¼ ðn Eisol�EstackÞ=n
Where n is the number of molecule in the stack, Eisol is the energyof an isolated molecule, and Estack is the energy of the stack.
Computational details of CD calculations
The calculation of the excitonic CD spectra proceeds in two steps.First, the lowest 50 excited states of the 15 molecules involved ina full turn of the helical structures are computed at the INDO/SCIlevel (using an active space of 60 occupied and 60 empty molecu-lar orbitals). Then, an excitonic Hamiltonian encompassing a totalof 15 � 60 basis functions (60 localised excitations per molecule) isbuilt on the basis of INDO/SCI[52] excitation energies and excitoncouplings. The latter are calculated as Coulomb interactions be-tween transition densities, thus going beyond the usual pointdipole model.[53] Diagonalisation of this Hamiltonian yields a set of900 exciton states a with energies �hwa and wavefunctions jya> ,for which the rotational strength Ra is computed as:[54]
Ra ¼�hwa
c
X
i;n
X
j;n0
yah jmi;n
Gj i � Gh jmj;n0
yaj i � ðrn � rn0 Þ
mi;n
������ m
j;n0
������
;
where c is the speed of light, mi,n the transition dipole momentfrom the ground state jg> to the excited state j i> of molecule nalong the stack, m
i;n¼ m
i;nð i; nj i gh j þ hcÞ the corresponding dipole
operator, jG> the ground state of the helical stack (product stateof all jg> ), and yaj i ¼
Pi;n
cai;n i; nj i the exciton state wavefunctions
expanded in terms of the cai;n eigenvectors. The CD response at
input frequency w is calculated on the basis of the rotationalstrengths as:
CDðwÞ ¼<X
a
Ra Gðw� waÞ > ;
Chem. Eur. J. 2014, 20, 17443 – 17453 www.chemeurj.org � 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim17452
Full Paper

where G(w�wa) is a Gaussian function centred around wa with var-iance s= 0.1 eV. The brackets denote a configurational averageover the positional and energetic disorder as explored during theMD simulations. Here, a total of 100 supramolecular helical struc-tures, each consisting of 15 molecules, were used; this approachwas found to yield CD spectra that are stable with respect toconfigurational averaging.
Acknowledgements
This work was supported in France by the Ministry of Educa-tion and Research (grants to I.D. and C.M.), the NationalAgency for Research (ANR, Project 09-BLAN-0045–01), theCNRS and the University of Angers (Ariane mobility program).I. Freuze and C. M�zi�re are thanked for performing the MSanalysis, while V. Bonnin is thanked for the elemental analyses.M.L. thanks SERC (Swedish e-Science Research Center) for fund-ing and SNIC for providing computer resources. D.B. is a FNRSResearch Director.
Keywords: C3 symmetry · circular dichroism · electronicmicroscopy · supramolecular chirality · tetrathiafulvalene
[1] T. Aida, E. W. Meijer, S. I. Stupp, Science 2012, 335, 813 – 817.[2] A. Brizard, R. Oda, I. Huc, Top. Curr. Chem. 2005, 256, 167 – 218.[3] M. Crego-Calama, D. N. Reinhoudt, Supramolecular Chirality. Topics in
Current Chemistry, Springer: Berlin, Heidelberg, New York, 2006.[4] D. B. Amabilino (Ed.), Chirality at the Nanoscale, Wiley-VCH, Weinheim,
2009.[5] C. C. Lee, C. Grenier, E. W. Meijer, A. P. H. J. Schenning, Chem. Soc. Rev.
2009, 38, 671 – 683.[6] V. K. Praveen, S. S. Babu, C. Vijayakumar, R. Varghese, A. Ajayaghosh,
Bull. Soc. Chem. Jpn. 2008, 81, 1196 – 1211.[7] D. Pijper, B. L. Feringa, Soft Matter 2008, 4, 1349 – 1372.[8] A. R. A. Palmans, E. W. Meijer, Angew. Chem. Int. Ed. 2007, 46, 8948 –
8968; Angew. Chem. 2007, 119, 9106 – 9126.[9] M. Hasegawa, M. Iyoda, Chem. Soc. Rev. 2010, 39, 2420 – 2427.
[10] F. S. Schoonbeek, J. H. van Esch, B. Wegewijs, D. B. A. Rep, M. P. de Haas,T. M. Klapwijk, R. M. Kellogg, B. L. Feringa, Angew. Chem. Int. Ed. 1999,38, 1393 – 1397; Angew. Chem. 1999, 111, 1486 – 1490.
[11] D. B. A. Rep, R. Roelfsema, J. H. van Esch, F. S. Schoonbeek, R. M. Kellogg,B. L. Feringa, T. T. M. Palstra, T. M. Klapwijk, Adv. Mater. 2000, 12, 563 –566.
[12] P. Jonkheijm, P. van der Schoot, A. P. H. J. Schenning, E. W. Meijer, Sci-ence 2006, 313, 80 – 83.
[13] A. P. H. J. Schenning, P. Jonkheijm, E. Peeters, E. W. Meijer, J. Am. Chem.Soc. 2001, 123, 409 – 416.
[14] R. P. Sijbesma, E. W. Meijer, Chem. Commun. 2003, 5 – 16.[15] W. Jin, Y. Yamamoto, T. Fukushima, N. Ishii, J. Kim, K. Kato, M. Takata, T.
Aida, J. Am. Chem. Soc. 2008, 130, 9434 – 9440.[16] T. Yamamoto, T. Fukushima, A. Kosaka, W. Jin, Y. Yamamoto, N. Ishii, T.
Aida, Angew. Chem. Int. Ed. 2008, 47, 1672 – 1675; Angew. Chem. 2008,120, 1696 – 1699.
[17] Y. Tatewaki, T. Hatanaka, R. Tsunashima, t. Nakamura, M. Kimura, H.Shirai, Chem. Asian J. 2009, 4, 1474 – 1479.
[18] A. P. H. J. Schenning, E. W. Meijer, Chem. Commun. 2005, 3245 – 3258.[19] F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer, A. P. H. J. Schenning, Chem.
Rev. 2005, 105, 1491 – 1546.[20] N. Avarvari, J. D. Wallis, J. Mater. Chem. 2009, 19, 4061 – 4076.[21] A. M. Madalan, C. R�thor�, M. Fourmigu�, E. Canadell, E. B. Lopes, M. Al-
meida, P. Auban-Senzier, N. Avarvari, Chem. Eur. J. 2010, 16, 528 – 537.[22] F. Pop, P. Auban-Senzier, A. Frackowiak, K. Ptaszynski, I. Olejniczak, J. D.
Wallis, E. Canadell, N. Avarvari, J. Am. Chem. Soc. 2013, 135, 17176 –17186.
[23] F. Pop, P. Auban-Senzier, E. Canadell, G. L. J. A. Rikken, N. Avarvari, Nat.Commun. 2014, 5, 3757.
[24] G. L. J. A. Rikken, J. Fçlling, P. Wyder, Phys. Rev. Lett. 2001, 87, 236602 –1 – 4.
[25] S. Cantekin, T. F. A. de Greef, A. R. A. Palmans, Chem. Soc. Rev. 2012, 41,6125 – 6137.
[26] D. Wang, Y. Huang, J. Li, L. Xu, M. Chen, J. Tao, L. Li, Chem. Eur. J. 2013,19, 685 – 690.
[27] A. R. A. Palmans, J. A. J. M. Vekemans, H. Fischer, R. A. Hikmet, E. W.Meijer, Chem. Eur. J. 1997, 3, 300 – 307.
[28] L. Brunsveld, H. Zhang, M. Glasbeek, J. A. J. M. Vekemans, E. W. Meijer, J.Am. Chem. Soc. 2000, 122, 6175 – 6182.
[29] J. J. van Gorp, J. A. J. M. Vekemans, E. W. Meijer, J. Am. Chem. Soc. 2002,124, 14759 – 14769.
[30] A. R. A. Palmans, J. A. J. M. Vekemans, E. E. Havinga, E. W. Meijer, Angew.Chem. Int. Ed. Engl. 1997, 36, 2648 – 2651; Angew. Chem. 1997, 109,2763 – 2765.
[31] J. van Gestel, A. R. A. Palmans, B. Titulaer, J. A. J. M. Vekemans, E. W.Meijer, J. Am. Chem. Soc. 2005, 127, 5490 – 5494.
[32] M. H. C. J. van Houtem, R. Mart�n-Rapffln, J. A. J. M. Vekemans, E. W.Meijer, Chem. Eur. J. 2010, 16, 2258 – 2271.
[33] M. A. J. Gillissen, M. M. E. Koenigs, J. J. H. Spiering, J. A. J. M. Vekemans,A. R. A. Palmans, I. K. Voets, E. W. Meijer, J. Am. Chem. Soc. 2014, 136,336 – 343.
[34] I. Danila, F. Riob�, J. Puigmarti-Luis, . P�rez del Pino, J. D. Wallis, D. B.Amabilino, N. Avarvari, J. Mater. Chem. 2009, 19, 4495 – 4504.
[35] I. Danila, F. Riob�, F. Piron, J. Puigmart�-Luis, J. D. Wallis, M. Linares, H.�gren, D. Beljonne, D. B. Amabilino, N. Avarvari, J. Am. Chem. Soc. 2011,133, 8344 – 8353.
[36] I. Danila, F. Pop, C. Escudero, L. N. Feldborg, J. Puigmart�-Luis, F. Riob�,N. Avarvari, D. B. Amabilino, Chem. Commun. 2012, 48, 4552 – 4554.
[37] J. Tabei, R. Nomura, F. Sanda, T. Masuda, Macromolecules 2004, 37,1175 – 1179.
[38] P. J. M. Stals, M. M. J. Smulders, R. Mart�n-Rapffln, A. R. A. Palmans, E. W.Meijer, Chem. Eur. J. 2009, 15, 2071 – 2080.
[39] M. M. J. Smulders, P. J. M. Stals, T. Mes, T. F. E. Paffen, A. P. H. J. Schen-ning, A. R. A. Palmans, E. W. Meijer, J. Am. Chem. Soc. 2010, 132, 620 –626.
[40] J. Zhi, Z. Zhu, A. Liu, J. Cui, X. Wan, Q. Zhou, Macromolecules 2008, 41,1594 – 1597.
[41] D. B. Amabilino, J.-L. Serrano, T. Sierra, J. Veciana, J. Polym. Sci. Part A2006, 44, 3161 – 3174.
[42] P. J. M. Stals, J. F. Haveman, R. Mart�n-Rapffln, C. F. C. Fiti�, A. R. A. Pal-mans, E. W. Meijer, J. Mater. Chem. 2009, 19, 124 – 130.
[43] B. Narayan, C. Kulkarni, S. J. George, J. Mater. Chem. C 2013, 1, 626 –629.
[44] F. Garc�a, L. Snchez, J. Am. Chem. Soc. 2012, 134, 734 – 742.[45] E. Gomar-Nadal, C. Rovira, D. B. Amabilino, Tetrahedron 2006, 62, 3370 –
3379.[46] G. Gottarelli, S. Lena, S. Masiero, S. Pieraccini, G. P. Spada, Chirality 2008,
20, 471 – 485.[47] S. A. Baudron, N. Avarvari, E. Canadell, P. Auban-Senzier, P. Batail, Chem.
Eur. J. 2004, 10, 4498 – 4511.[48] C. R. Rice, S. Onions, N. Vidal, J. D. Wallis, M.-C. Senna, M. Pilkington, H.
Stoeckli-Evans, Eur. J. Inorg. Chem. 2002, 1985 – 1997.[49] J. W. Ponder, TINKER, Ver. 4.2 ; 2004 ; http://dasher.wustl.edu/tinker.[50] a) L. N. Allinger, Y. H. Yuh, J. H. Lii, J. Am. Chem. Soc. 1989, 111, 8551 –
8566; b) B. Ma, J. H. Lii, L. N. Allinger, J. Comput. Chem. 2000, 21, 813 –825.
[51] H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola, J. R.Haak, Chem. Phys. 1984, 81, 3684 – 3690.
[52] J. Ridley, M. Zerner, Theor. Chim. Acta 1973, 32, 111 – 134.[53] D. Beljonne, J. Cornil, R. Silbey, P. Millie, J. L. Bredas, J. Chem. Phys. 2000,
112, 4749 – 4758.[54] F. C. Spano, S. C. J. Meskers, E. Hennebicq, D. Beljonne, J. Am. Chem.
Soc. 2007, 129, 7044 – 7054.
Received: August 6, 2014Published online on October 30, 2014
Chem. Eur. J. 2014, 20, 17443 – 17453 www.chemeurj.org � 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim17453
Full Paper

![Buffers and Ionic Salts: Densities and Solubilities of Aqueous and Electrolyte Solutions of Tris(hydroxymethyl)aminomethane and N -Tris[hydroxymethyl]-4-amino-butanesulfonic Acid †](https://img.dokumen.tips/doc/110x75/63576ba53fe58b1a61066738/buffers-and-ionic-salts-densities-and-solubilities-of-aqueous-and-electrolyte-solutions.jpg)








![The basicity of [tris-(trimethylphosphine)(cyclooctadiene)iridium(I)]](https://img.dokumen.tips/doc/110x75/6348fd132cd4c1a3540d4821/the-basicity-of-tris-trimethylphosphinecyclooctadieneiridiumi.jpg)


















