Embed Size (px)
Citation preview

Computer Physics Communications 185 (2014) 908–916
Contents lists available at ScienceDirect
Computer Physics Communications
journal homepage: www.elsevier.com/locate/cpc
Generalized scalable multiple copy algorithms for moleculardynamics simulations in NAMDWei Jiang a,⇤,1, James C. Phillips d,1, Lei Huang c, Mikolai Fajer c, Yilin Mengc,James C. Gumbart b,f, Yun Luoa,2, Klaus Schultend,e,⇤⇤, Benoît Rouxb,c,⇤⇤a Argonne Leadership Computing Facility, Argonne National Laboratory, 9700 South Cass Avenue, Building 240, Argonne, IL 60439, United Statesb Biosciences Division, Argonne National Laboratory, 9700 South Cass Avenue, Building 202, Argonne, IL 60439, United Statesc Department of Biochemistry and Molecular Biology, Gordon Center for Integrative Science, University of Chicago, 929 57th Street, Chicago, IL 60637,United Statesd Beckman Institute, University of Illinois at Urbana–Champaign, Urbana, IL 61801, United Statese Department of Physics, University of Illinois at Urbana–Champaign, Urbana, IL 61801, United Statesf School of Physics, Georgia Institute of Technology, Atlanta, GA 30332, United States
a r t i c l e i n f o
Article history:Received 1 October 2013Received in revised form20 November 2013Accepted 8 December 2013Available online 18 December 2013
Keywords:MCANAMDTclCharm++
a b s t r a c t
Computationalmethodologies that couple the dynamical evolution of a set of replicated copies of a systemof interest offer powerful and flexible approaches to characterize complex molecular processes. Suchmultiple copy algorithms (MCAs) can be used to enhance sampling, compute reversible work and freeenergies, as well as refine transition pathways. Widely used examples of MCAs include temperature andHamiltonian-tempering replica-exchange molecular dynamics (T -REMD and H-REMD), alchemical freeenergy perturbation with lambda replica-exchange (FEP/�-REMD), umbrella sampling with Hamiltonianreplica exchange (US/H-REMD), and stringmethodwith swarms-of-trajectories conformational transitionpathways. Here, we report a robust and general implementation of MCAs for molecular dynamics (MD)simulations in the highly scalable program NAMD built upon the parallel programming system Charm++.Multiple concurrent NAMD instances are launched with internal partitions of Charm++ and locatedcontinuously within a single communication world. Messages between NAMD instances are passed bylow-level point-to-point communication functions, which are accessible through NAMD’s Tcl scriptinginterface. The communication-enabled Tcl scripting provides a sustainable application interface for endusers to realize generalized MCAs without modifying the source code. Illustrative applications of MCAswith fine-grained inter-copy communication structure, including global lambda exchange in FEP/�-REMD, window swapping US/H-REMD in multidimensional order parameter space, and string methodwith swarms-of-trajectories were carried out on IBM Blue Gene/Q to demonstrate the versatility andmassive scalability of the present implementation.
Published by Elsevier B.V.
1. Introduction
A long-standing challenge for molecular dynamics (MD) sim-ulations based on all-atom models is the difficulty to adequatelysample infrequent events, although what has routinely been at-tainable within typical MD simulations has evolved over the years.
⇤ Corresponding author. Tel.: +1 1016302528688.⇤⇤ Corresponding authors.E-mail addresses: [email protected] (W. Jiang), [email protected]
(K. Schulten), [email protected] (B. Roux).1 Co-first authors.2 Current address: Pharmaceutical Sciences Department, College of Pharmacy,
Western University of Health Sciences, Pomona, CA, United States.
With the advancement of novel accelerating computer chips andsupercomputers that consist of MD-specific integrated circuits, [1]simple brute-force MD simulations on the order of millisecondshave recently become possible. This is remarkablewhen comparedwith what was the state-of-the-art only barely one decade ago.However, the fact remains that a whole host of important biologi-cal processes take place on a timescale ranging from millisecondsto seconds, beyond what can be achieved with simple brute-forceMD. For this reason, it remains critical to continue to seek alterna-tive solutions to the sampling problem.
One broad computational strategy is offered by algorithmsbased on multiple copies. Rather than attempting to characterizethe system through a single and extremely long trajectory, suchmultiple copy algorithms (MCAs) adopt a ‘divide-and-conquer’strategy to carry out the desired computation using massive
0010-4655/$ – see front matter. Published by Elsevier B.V.http://dx.doi.org/10.1016/j.cpc.2013.12.014

W. Jiang et al. / Computer Physics Communications 185 (2014) 908–916 909
parallel resources. MCAs can be designed to calculate both equilib-rium and non-equilibrium properties [2–8]. For equilibrium prop-erties, the most widely used MCAs include parallel temperature[9–17] and Hamiltonian tempering with replica-exchange MD(REMD) simulations [18–25]. There are also several variants ofMCAs focused on non-equilibrium situations, such as the forward-flux sampling, [26] the stringmethodwith swarms-of-trajectories,[5] and time-dependent umbrella sampling [7,8]. MCAs offer someof the most extremely scalable MD methodologies, allowing oneto fully exploit the unprecedented computational power of lead-ership computers such as IBM Blue Gene/Q and Cray XT7. As themost popular applications of MCAs, REMD algorithms have beenextensively implemented in several MD engines such as AMBER,[27]. GROMACS [28] and Desmond [29] with special applicationinterface (API) as well as post-processing utility. Generally, theREMD algorithms are implemented as regular point-to-point dataexchange that is hard coded in source code, or driven by an ex-ternal master script working with specific machines. As a result,the existing REMD schemes are restricted to a number of plainapplications, such as parallel tempering or Hamiltonian exchangewith a single biasing parameter,while implementation of anymoreelaborate algorithm has to touch the low-level MPI programming.However, biological MD simulations often involve several slow de-grees of freedom that have to be addressed in multidimensionalspace of order parameters, [30–33] ormultiple thermodynamic co-ordinates [34–36] to accelerate the sampling efficiency of a reac-tion path. In addition to the demand of multidimensional REMDalgorithms, there can arise an irregular inter-copy communicationstructure for a complex biophysical problem, such as case of stringmethod with swarms-of-trajectories, [5,37] raising the necessityto develop a sustainable API to accommodate various novel MCAsolutions.
A prototypical example of a MCA as a high-dimensional Hamil-tonian exchange algorithm was recently developed and imple-mented in the Distributed Replica (REPDSTR) module of CHARMMto calculate the absolute binding free energy of a ligand to a pro-tein [3,4]. With the 2-dimensional Hamiltonian replica-exchangescheme, an unprecedented global replica exchange through theentire reaction pathwas performed and accurate free energy infor-mation was obtained. Within REPDSTR, coordinates instead of bi-asing/scaling parameter were swapped during exchange attempts,relying on the legacy parallel structure of CHARMM based on astatic task distribution and atomic decomposition [38]. Followinga similar strategy, a variety of MCAs could be implemented withinthe CHARMM/REPDSTR code. However, due to the limitations ofthe internal parallel structure of CHARMM, this initial implemen-tation of MCAs has been limited to small and moderate size bio-logical systems. To allow scalable simulation of increasingly largesystems, the development of contemporary MD software has beendirected towards spatial decomposition and dynamic load balance[28,39]. In spite of its limitations, CHARMM/REPDSTR provides aprototype REMD scheme independent of specific potential termsor parameters and, therefore, can serve to guide further extensionin high performance MD software such as NAMD.
NAMD is a highly scalable program built onto a hybrid spa-tial/force decomposition managed by the asynchronous parallelprogramming systemCharm++ [39]. An attractive feature of NAMDis the flexible Tcl interface that provides a user-friendly scriptingplatform, where one can control simulation parameters on-the-fly.Furthermore, Tcl communication commands can be built on top oflow-level communication functions, while the Charm++ program-ming system can support concurrent multiple NAMD instances byremapping processing elements. With these advanced program-ming features, NAMD provides an ideal platform to implement ahighly flexible API for generalized MCAs.
In this article, we report a systematic implementation ofMCAs in NAMD’s Tcl interface and Charm++. Implementation
details are reported and four representative applications, paralleltempering, free energy perturbation (FEP)/REMD, US/H-REMDwith irregular order parameter locations in 2-dimensions, as wellas the string method with swarms-of-trajectories are carried outon the IBM Blue Gene/Q computer at Argonne National Laboratory.The superior scalability of the present MCA implementation isdemonstrated and discussed in detail.
2. Implementation details
2.1. MCAs implementation with Charm++ and NAMD Tcl interface
AprimitiveMCA can be realizedwithmultipleMPI_subcommu-nicators. In the MPI machine layer of Charm++, the MPI_Comm_split function splits the default MPI_COMM_WORLD intomultiple local subcommunicators, each of which runs an inde-pendent Charm++ and NAMD instance. A secondary set of MPIcommunicators, each spanning like rankswithin the local subcom-municators, allow exchanges between the independent NAMD in-stances. This exchange communication is implemented throughnew APIs in both the Charm++ Converse layer and the NAMDTcl scripting interface to provide a user-friendly interface. Aquick route towards REMD, MPI_subcommunicator is extensivelyadopted by many MD software, however such a plain applicationof many MPI_subcommunicators could lose significant networktopology information and, therefore, induce further network con-tention or latency. However, within the Charm++ Converse layerinstead ofMPI_COMM_WORLD, each Charm++ processing elementcan be logically mapped onto a designated local partition. WhenCharm++/NAMD enters the inter-copy communication phase, alllocalized processing elements are mapped back to global state.In addition, on leadership supercomputers, such internal parti-tions can obtain further performance gain benefiting from thelow-level machine specific communication library, such as ParallelActiveMessaging Interface (PAMI) [40] on IBMBlue Gene/Q or userGeneric Network Interface (uGNI) [41] on Cray XK7. Tcl interfaceof NAMD intends to provide maximum flexibility for a high-enduser to realize its special needs without touching the source code.For example, by Tcl scripting, variables and expressions used in ini-tially defining options can be changed during a running simulation.After user-friendly Tcl communication commands are built on topof the low-level point-to-point communication functions (eitherCharm++ Converse or MPI), a user can further design generic MCAswithout modifying or adding a single line of C++ (Fig. 1). Withina MCA Tcl script, inter-copy communications are executed by TclreplicaSend/Recv/Sendrecv functions on top of Converse commu-nication layer of Charm++, and a user needs to designate communi-cation partners for each copy. Appendix A exhibits an excerpt of Tclscripting code that controls temperature swaps in a parallel tem-pering MD simulation. A significant advantage of the present Tcl-based MCA scheme is that a user can realize any type MCA withinTcl scripting, as long as the energy terms or parameters to be biasedare registered in the Tcl scripting interface of NAMD. For example,in an absolute free energy calculation, nonbond scaling parametersof different alchemical types can be wrapped into a single parame-ter unit to be exchanged along the entire alchemical reaction path,or multiple orthogonal order parameters can be alternatively ex-changed to form a multidimensional umbrella sampling Hamilto-nian exchange (see Section 3).
2.2. Performance on IBM Blue Gene/Q
The string method with swarms-of-trajectories [5,37,42–44]represents a general, but challengingMCAdue to themassive num-ber of concurrent trajectories involved and themulti-level commu-nication relations between them. Previously such a sophisticated
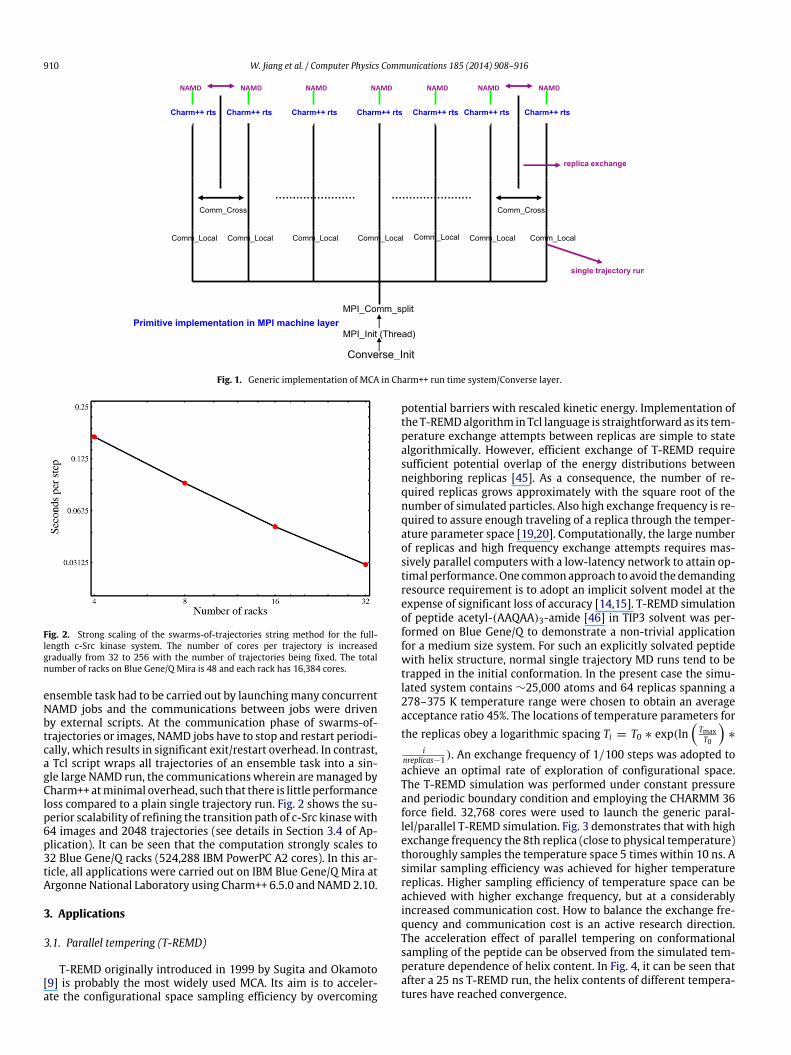
910 W. Jiang et al. / Computer Physics Communications 185 (2014) 908–916
Fig. 1. Generic implementation of MCA in Charm++ run time system/Converse layer.
Fig. 2. Strong scaling of the swarms-of-trajectories string method for the full-length c-Src kinase system. The number of cores per trajectory is increasedgradually from 32 to 256 with the number of trajectories being fixed. The totalnumber of racks on Blue Gene/Q Mira is 48 and each rack has 16,384 cores.
ensemble task had to be carried out by launchingmany concurrentNAMD jobs and the communications between jobs were drivenby external scripts. At the communication phase of swarms-of-trajectories or images, NAMD jobs have to stop and restart periodi-cally, which results in significant exit/restart overhead. In contrast,a Tcl script wraps all trajectories of an ensemble task into a sin-gle large NAMD run, the communications wherein aremanaged byCharm++ atminimal overhead, such that there is little performanceloss compared to a plain single trajectory run. Fig. 2 shows the su-perior scalability of refining the transition path of c-Src kinasewith64 images and 2048 trajectories (see details in Section 3.4 of Ap-plication). It can be seen that the computation strongly scales to32 Blue Gene/Q racks (524,288 IBM PowerPC A2 cores). In this ar-ticle, all applications were carried out on IBM Blue Gene/Q Mira atArgonne National Laboratory using Charm++ 6.5.0 and NAMD 2.10.
3. Applications
3.1. Parallel tempering (T-REMD)
T-REMD originally introduced in 1999 by Sugita and Okamoto[9] is probably the most widely used MCA. Its aim is to acceler-ate the configurational space sampling efficiency by overcoming
potential barriers with rescaled kinetic energy. Implementation ofthe T-REMDalgorithm in Tcl language is straightforward as its tem-perature exchange attempts between replicas are simple to statealgorithmically. However, efficient exchange of T-REMD requiresufficient potential overlap of the energy distributions betweenneighboring replicas [45]. As a consequence, the number of re-quired replicas grows approximately with the square root of thenumber of simulated particles. Also high exchange frequency is re-quired to assure enough traveling of a replica through the temper-ature parameter space [19,20]. Computationally, the large numberof replicas and high frequency exchange attempts requires mas-sively parallel computers with a low-latency network to attain op-timal performance. One commonapproach to avoid the demandingresource requirement is to adopt an implicit solvent model at theexpense of significant loss of accuracy [14,15]. T-REMD simulationof peptide acetyl-(AAQAA)3-amide [46] in TIP3 solvent was per-formed on Blue Gene/Q to demonstrate a non-trivial applicationfor a medium size system. For such an explicitly solvated peptidewith helix structure, normal single trajectory MD runs tend to betrapped in the initial conformation. In the present case the simu-lated system contains ⇠25,000 atoms and 64 replicas spanning a278–375 K temperature range were chosen to obtain an averageacceptance ratio 45%. The locations of temperature parameters forthe replicas obey a logarithmic spacing Ti = T0 ⇤ exp(ln
⇣TmaxT0
⌘⇤
inreplicas�1 ). An exchange frequency of 1/100 steps was adopted toachieve an optimal rate of exploration of configurational space.The T-REMD simulation was performed under constant pressureand periodic boundary condition and employing the CHARMM 36force field. 32,768 cores were used to launch the generic paral-lel/parallel T-REMD simulation. Fig. 3 demonstrates that with highexchange frequency the 8th replica (close to physical temperature)thoroughly samples the temperature space 5 times within 10 ns. Asimilar sampling efficiency was achieved for higher temperaturereplicas. Higher sampling efficiency of temperature space can beachieved with higher exchange frequency, but at a considerablyincreased communication cost. How to balance the exchange fre-quency and communication cost is an active research direction.The acceleration effect of parallel tempering on conformationalsampling of the peptide can be observed from the simulated tem-perature dependence of helix content. In Fig. 4, it can be seen thatafter a 25 ns T-REMD run, the helix contents of different tempera-tures have reached convergence.
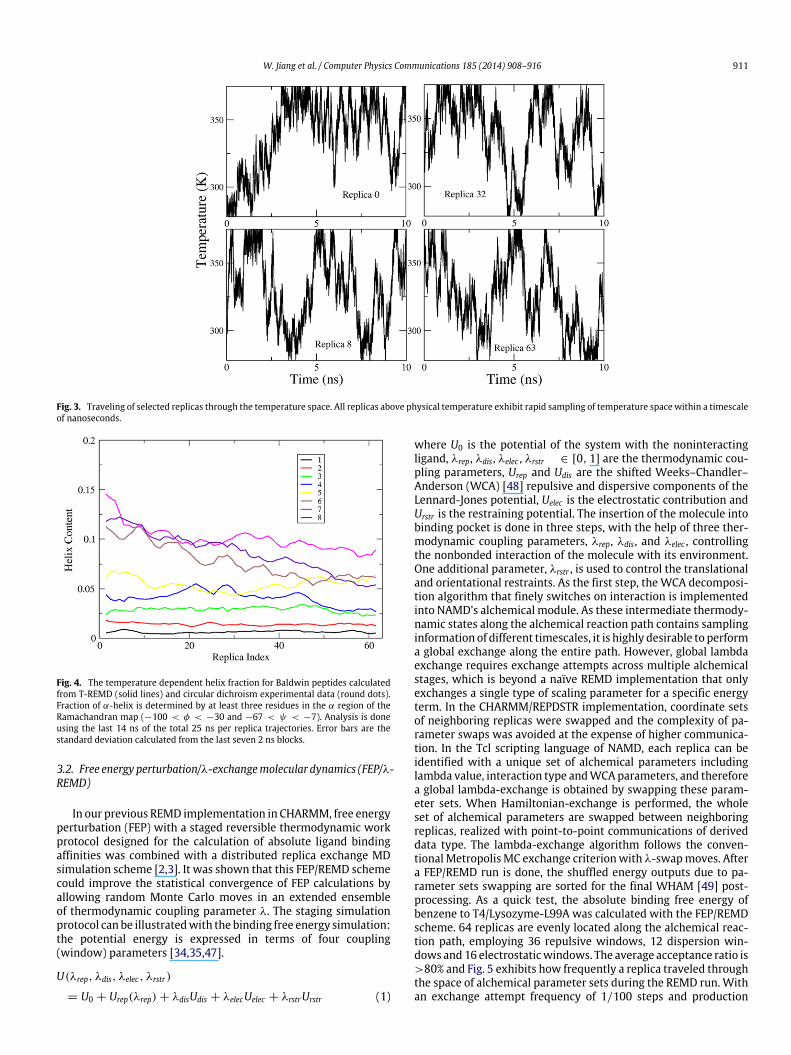
W. Jiang et al. / Computer Physics Communications 185 (2014) 908–916 911
Fig. 3. Traveling of selected replicas through the temperature space. All replicas above physical temperature exhibit rapid sampling of temperature space within a timescaleof nanoseconds.
Fig. 4. The temperature dependent helix fraction for Baldwin peptides calculatedfrom T-REMD (solid lines) and circular dichroism experimental data (round dots).Fraction of ↵-helix is determined by at least three residues in the ↵ region of theRamachandran map (�100 < � < �30 and �67 < < �7). Analysis is doneusing the last 14 ns of the total 25 ns per replica trajectories. Error bars are thestandard deviation calculated from the last seven 2 ns blocks.
3.2. Free energy perturbation/�-exchangemolecular dynamics (FEP/�-REMD)
In our previous REMD implementation in CHARMM, free energyperturbation (FEP) with a staged reversible thermodynamic workprotocol designed for the calculation of absolute ligand bindingaffinities was combined with a distributed replica exchange MDsimulation scheme [2,3]. It was shown that this FEP/REMD schemecould improve the statistical convergence of FEP calculations byallowing random Monte Carlo moves in an extended ensembleof thermodynamic coupling parameter �. The staging simulationprotocol can be illustratedwith the binding free energy simulation:the potential energy is expressed in terms of four coupling(window) parameters [34,35,47].
U(�rep, �dis, �elec, �rstr)
= U0 + Urep(�rep) + �disUdis + �elecUelec + �rstrUrstr (1)
where U0 is the potential of the system with the noninteractingligand, �rep, �dis, �elec , �rstr 2 [0, 1] are the thermodynamic cou-pling parameters, Urep and Udis are the shifted Weeks–Chandler–Anderson (WCA) [48] repulsive and dispersive components of theLennard-Jones potential, Uelec is the electrostatic contribution andUrstr is the restraining potential. The insertion of the molecule intobinding pocket is done in three steps, with the help of three ther-modynamic coupling parameters, �rep, �dis, and �elec , controllingthe nonbonded interaction of the molecule with its environment.One additional parameter, �rstr , is used to control the translationaland orientational restraints. As the first step, theWCA decomposi-tion algorithm that finely switches on interaction is implementedinto NAMD’s alchemical module. As these intermediate thermody-namic states along the alchemical reaction path contains samplinginformation of different timescales, it is highly desirable to performa global exchange along the entire path. However, global lambdaexchange requires exchange attempts across multiple alchemicalstages, which is beyond a naïve REMD implementation that onlyexchanges a single type of scaling parameter for a specific energyterm. In the CHARMM/REPDSTR implementation, coordinate setsof neighboring replicas were swapped and the complexity of pa-rameter swaps was avoided at the expense of higher communica-tion. In the Tcl scripting language of NAMD, each replica can beidentified with a unique set of alchemical parameters includinglambda value, interaction type andWCAparameters, and thereforea global lambda-exchange is obtained by swapping these param-eter sets. When Hamiltonian-exchange is performed, the wholeset of alchemical parameters are swapped between neighboringreplicas, realized with point-to-point communications of deriveddata type. The lambda-exchange algorithm follows the conven-tionalMetropolisMC exchange criterionwith �-swapmoves. Aftera FEP/REMD run is done, the shuffled energy outputs due to pa-rameter sets swapping are sorted for the final WHAM [49] post-processing. As a quick test, the absolute binding free energy ofbenzene to T4/Lysozyme-L99A was calculated with the FEP/REMDscheme. 64 replicas are evenly located along the alchemical reac-tion path, employing 36 repulsive windows, 12 dispersion win-dows and16 electrostaticwindows. The average acceptance ratio is>80% and Fig. 5 exhibits how frequently a replica traveled throughthe space of alchemical parameter sets during the REMD run. Withan exchange attempt frequency of 1/100 steps and production
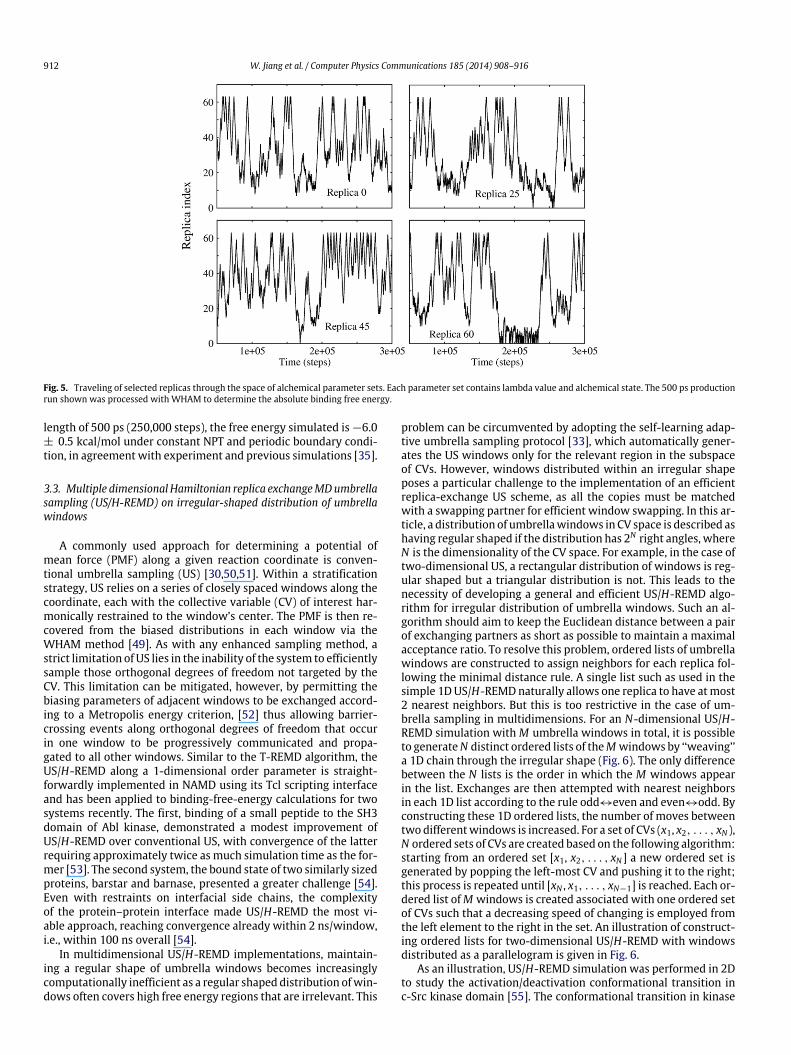
912 W. Jiang et al. / Computer Physics Communications 185 (2014) 908–916
Fig. 5. Traveling of selected replicas through the space of alchemical parameter sets. Each parameter set contains lambda value and alchemical state. The 500 ps productionrun shown was processed with WHAM to determine the absolute binding free energy.
length of 500 ps (250,000 steps), the free energy simulated is �6.0± 0.5 kcal/mol under constant NPT and periodic boundary condi-tion, in agreement with experiment and previous simulations [35].
3.3. Multiple dimensional Hamiltonian replica exchangeMDumbrellasampling (US/H-REMD) on irregular-shaped distribution of umbrellawindows
A commonly used approach for determining a potential ofmean force (PMF) along a given reaction coordinate is conven-tional umbrella sampling (US) [30,50,51]. Within a stratificationstrategy, US relies on a series of closely spaced windows along thecoordinate, each with the collective variable (CV) of interest har-monically restrained to the window’s center. The PMF is then re-covered from the biased distributions in each window via theWHAM method [49]. As with any enhanced sampling method, astrict limitation of US lies in the inability of the system to efficientlysample those orthogonal degrees of freedom not targeted by theCV. This limitation can be mitigated, however, by permitting thebiasing parameters of adjacent windows to be exchanged accord-ing to a Metropolis energy criterion, [52] thus allowing barrier-crossing events along orthogonal degrees of freedom that occurin one window to be progressively communicated and propa-gated to all other windows. Similar to the T-REMD algorithm, theUS/H-REMD along a 1-dimensional order parameter is straight-forwardly implemented in NAMD using its Tcl scripting interfaceand has been applied to binding-free-energy calculations for twosystems recently. The first, binding of a small peptide to the SH3domain of Abl kinase, demonstrated a modest improvement ofUS/H-REMD over conventional US, with convergence of the latterrequiring approximately twice as much simulation time as the for-mer [53]. The second system, the bound state of two similarly sizedproteins, barstar and barnase, presented a greater challenge [54].Even with restraints on interfacial side chains, the complexityof the protein–protein interface made US/H-REMD the most vi-able approach, reaching convergence already within 2 ns/window,i.e., within 100 ns overall [54].
In multidimensional US/H-REMD implementations, maintain-ing a regular shape of umbrella windows becomes increasinglycomputationally inefficient as a regular shaped distribution ofwin-dows often covers high free energy regions that are irrelevant. This
problem can be circumvented by adopting the self-learning adap-tive umbrella sampling protocol [33], which automatically gener-ates the US windows only for the relevant region in the subspaceof CVs. However, windows distributed within an irregular shapeposes a particular challenge to the implementation of an efficientreplica-exchange US scheme, as all the copies must be matchedwith a swapping partner for efficient window swapping. In this ar-ticle, a distribution of umbrellawindows in CV space is described ashaving regular shaped if the distribution has 2N right angles, whereN is the dimensionality of the CV space. For example, in the case oftwo-dimensional US, a rectangular distribution of windows is reg-ular shaped but a triangular distribution is not. This leads to thenecessity of developing a general and efficient US/H-REMD algo-rithm for irregular distribution of umbrella windows. Such an al-gorithm should aim to keep the Euclidean distance between a pairof exchanging partners as short as possible to maintain a maximalacceptance ratio. To resolve this problem, ordered lists of umbrellawindows are constructed to assign neighbors for each replica fol-lowing the minimal distance rule. A single list such as used in thesimple 1DUS/H-REMDnaturally allows one replica to have atmost2 nearest neighbors. But this is too restrictive in the case of um-brella sampling in multidimensions. For an N-dimensional US/H-REMD simulation with M umbrella windows in total, it is possibleto generateN distinct ordered lists of theM windows by ‘‘weaving’’a 1D chain through the irregular shape (Fig. 6). The only differencebetween the N lists is the order in which the M windows appearin the list. Exchanges are then attempted with nearest neighborsin each 1D list according to the rule odd$even and even$odd. Byconstructing these 1D ordered lists, the number of moves betweentwo differentwindows is increased. For a set of CVs (x1, x2, . . . , xN ),N ordered sets of CVs are created based on the following algorithm:starting from an ordered set [x1, x2, . . . , xN ] a new ordered set isgenerated by popping the left-most CV and pushing it to the right;this process is repeated until [xN , x1, . . . , xN�1] is reached. Each or-dered list ofM windows is created associated with one ordered setof CVs such that a decreasing speed of changing is employed fromthe left element to the right in the set. An illustration of construct-ing ordered lists for two-dimensional US/H-REMD with windowsdistributed as a parallelogram is given in Fig. 6.
As an illustration, US/H-REMD simulation was performed in 2Dto study the activation/deactivation conformational transition inc-Src kinase domain [55]. The conformational transition in kinase

W. Jiang et al. / Computer Physics Communications 185 (2014) 908–916 913
–9
–10
–11
2 3 4 5 6 2 3 4 5 6
A B
Fig. 6. A graphic demonstration of the two order lists that could be used in 2D US/H-REMD. (A) Ordered list when x-axis is the fast-changing coordinate. (B) Ordered listwhen y-axis is the fast-changing coordinate.
A B
Fig. 7. Conformational changes investigated by US/H-REMD calculations. (A) Structure of inactive c-Src kinase domain (adapted from PDB 2SRC). (B) Structure of active-likec-Src kinase domain (adapted from PDB 1Y57). Only the catalytic domain (residue 260 to 521) of c-Src kinase is employed in this study. The ↵C helix and the A-loop arecolored in magenta and cyan, respectively. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
domain primarily involves the ↵C helix and the activation loop(Fig. 7) and, two order parameters that characterize the confor-mational transition were identified based on the relative motionsbetween relevant atom groups. Two-dimensional self-learningadaptive umbrella sampling [33] calculations are carried out be-fore US/H-REMD calculations in order to define the landscape to beexplored by US/H-REMD. 1097 umbrella windows are determinedto be essential to characterize the landscape and are further uti-lized in the US/H-REMD simulation. The two ordered lists utilizedin the two-dimensional US/H-REMD calculations are illustrated inFig. 8. Each umbrella window is propagated for 5 ns under con-stant NPT and periodic boundary condition, and an exchange is at-tempted every 1 ps. The probability distribution of acceptance ratioin the calculation of c-Src kinase domain conformational transitionis given in Fig. 9A. The average acceptance ratio is 0.49 and the twoextremes are 0.36 and 0.68 for all neighboring pairs. Fig. 9B showsthe trajectory of replica 435 in the replica space against exchangeattempts (time). With the large acceptance ratio, a wide range ofreplicas (a span of >350 replicas) have been traversed multipletimes, however the entire replica space still is not fully covered.This indicates that either a sampling time of 5 ns per window isnot long enough or the exchange frequency is too low, and the PMFcould still be refined.
3.4. String method using swarms-of-trajectories
A putative transition path between conformational states canbe refined by the swarms-of-trajectories approach [5,37]. Thestring is a set of discrete conformations, or images, along thepath. Aset of collective variables is typically used to reduce the complexityof the system. The free energy gradient at each image can be com-puted and the images propagated along the component orthogonalto the path, iterating until the string has converged to a local mini-mum transition path. The swarms-of-trajectories approach utilizesmany molecular dynamics trajectories started from a single imagethat can be combined to yield an average drift. This requires com-munication between the N copies of a specific image, illustratedby Fig. 10. The average drifts for each image compose an updatestring, and the images are redistributed along this string to removeany drift tangential to the path. The string update process requirescommunicationbetween theM images that compose the string. Af-ter the reparameterization with the current average drifts the newimagesmust be prepared before the next cycle of drifts can be per-formed, achieved by performing molecular dynamics simulationswith constraints that slowly move the CVs to their reparameter-ized positions. The multiple copy implementation of this method

914 W. Jiang et al. / Computer Physics Communications 185 (2014) 908–916
!C H
elix
Mov
emen
t
Activation Loop Opening!C
Hel
ix M
ovem
ent
Activation Loop Opening
!C H
elix
Mov
emen
t
Activation Loop Opening
kcal/mol
–12
15
12
9
–9
6
–6
3
–3
0
2 4 6 8 10 12
15
12
–12
9
–9
6
–6
3
–3
0
2 4 6 8 10 12
15
12
–12
9
–9
6
–6
3
–3
0
2 4 6 8 10 12
00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.0
A B
C
Fig. 8. (A) Ordered list when the x-axis is the fast-changing coordinate. (B) Ordered list when the y-axis is the fast-changing coordinate. (C) PMF determined from US/H-REMD simulations using the two ordered lists. The ↵C helix movement is characterized by the difference between two salt-bridges (d1 � d2), and the A-loop opening isrepresented by the average of three distances listed in the text. Both coordinates have the unit of Å.
Acceptance Ratio
Prob
abili
ty D
ensi
ty
Exchange Attempt Index
Rep
lica
Inde
x
0.20
0.15
0.10
0.05
0.000.35 0.40 0.45 0.50 0.55 0.60 0.65 0.70
700
600
500
400
3000 1000 2000 3000 4000 5000
A B
Fig. 9. Diagnostics of US/H-REMD using ordered lists of neighboring windows. (A) Probability distribution of acceptance ratio for all exchanging pairs. (B) The trajectory ofreplica 435 in the space of replicas.
utilizes MN independent copies that must intercommunicate onceper iteration.
In this article, the string method with swarms-of-trajectorieswas used to refine the activation/deactivation conformationaltransition in c-Src kinase domain (identical with the Section 3.3)in the presence of the regulatory SH2/SH3 domains. The full-lengthc-Src structure is threaded along the initial string taken from pre-vious work of the kinase domain only. This threading is composedof restraining the CVs of the full-length to the string values andslowly moving those restraints along the string. A total of 64 im-ages are selected from the initial string. Each swarm composes of
32 5-ps trajectories, and the equilibration following the reparam-eterization is another 5-ps of restrained molecular dynamics.
The discrete Fréchet distance is an order-preserving measureof the similarity between two strings. The distance between eachstring iteration to the initial string and the distance to the finalstrings is shown in Fig. 11 and are either monotonically increas-ing or decreasing, respectively. Themonotonicity indicates that theswarms-of-trajectories method is performing a steepest descentminimization of the transition path. Furthermore the flatteningof the discrete Fréchet distance to the final string around itera-tion 250 suggests that the transition path is nearly converged. The

W. Jiang et al. / Computer Physics Communications 185 (2014) 908–916 915
A B C
Fig. 10. Multi-level communications are involved in the swarms-of-trajectories stringmethod (Panel A). Each image between the two end states owns a swarmof trajectoriesand during the refinement iteration of reaction path each trajectory has to talk to each other trajectory in a swarm to compute the position of the next swarm’s center (PanelC). Then every swarm must communicate every other swarm to reparameterize the string overall (Panel B).
String iteration
Frec
het d
ista
nce
(Å)
Initial stringFinal string
Fig. 11. The discrete Fréchet distance between each string iteration and either theinitial string (blue) or the final string (green). (For interpretation of the references tocolour in this figure legend, the reader is referred to the web version of this article.)
converged transition path can be studied directly, but is most use-ful as a starting point for free energy calculation like the US/H-REMD discussed above.
4. Conclusion
We have developed and implemented generalized multiplecopy algorithms (MCAs) in the highly scalable programNAMDbuiltupon the parallel programming system Charm++. Inter-copy com-munication is realized by low-level point-to-point communicationfunctions between independent NAMD instances. At the applica-tion level the MCAs are implemented via NAMD’s Tcl scripting in-terface, which provides great flexibility for a user to design novelMCA solutions for complex biophysical problems without modify-ing the source code. The application of MCAs could be extendedto multiple thermodynamic parameters (or reaction coordinates),multidimension and irregular communication between copies.Wehave demonstrated the versatility of the present implementationwith three novel MCA applications, global lambda exchange alongan entire reaction path in an absolute binding free energy calcula-tion, high-dimensional umbrella sampling with irregular reactioncoordinates, and stringmethodwith swarms-of-trajectories. Novelapplications to complex biological problems are currently under-way.
Acknowledgments
We would like to acknowledge the Parallel ProgrammingLaboratory, University of Illinois at Urbana–Champaign, for the
implementation of Charm++ on IBM Blue Gene/Q. This researchis supported by the Early Science Program of Argonne LeadershipComputing Facility, Department of Energy Office of Science andused resources of the Argonne Leadership Computing Facility atArgonne National Laboratory, which is supported by the Officeof Science of the U.S. Department of Energy under contract DE-AC02-06CH11357. This work also is supported by the NationalInstitutes of Health through grants 9P41GM104601 (K.S. and J.P.),U54GM087519 (K.S. and B.R.), and K22-AI100927 (J.C.G.) and byMCB-0920261 (B.R.) from the National Science Foundation (NSF).
Appendix A
A.1. Excerpt of Tcl scripting code for parallel tempering
if {$replica(index) < $replica(index.$swap)} {# Exchangeattempt is performed by the replica that has smaller index
set temp $replica(temperature)set temp2 $replica(temperature.$swap) ; # Temperature pa-
rameters of a replica pair before an exchange attemptset dbeta [expr ((1.0/$temp) - (1.0/$temp2)) / $BOLTZMAN]set pot $POTENTIALset pot2 [replicaRecv $replica(loc.$swap)] ; # Receive instant
potential value from exchange partnerset delta [expr $dbeta * ($pot2 - $pot)]set doswap [expr $delta < 0. || exp(-1. * $delta) > rand()] ; #
Metropolis Monte Carlo algorithmreplicaSend $doswap $replica(loc.$swap) ; # Send swap
decision to exchange partner}if {$replica(index)> $replica(index.$swap)} {# Replica that has
larger index sends instant potential value to exchange partner forexchange attempt and receives swap decision
replicaSend $POTENTIAL $replica(loc.$swap)set doswap [replicaRecv $replica(loc.$swap)]}set newloc $rif {$doswap} {# If an exchange attempt is accepted, update the
physical location of a replicaset newloc $replica(loc.$swap)set replica(loc.$swap) $r}set replica(loc.$other) [replicaSendrecv $newloc $replica
(loc.$other) $replica(loc.$other)] ; # Exchange updated physical lo-cation with next round exchange partner
if {$doswap} {

916 W. Jiang et al. / Computer Physics Communications 185 (2014) 908–916
set OLDTEMP $replica(temperature)array set replica [replicaSendrecv [array get replica] $newloc
$newloc] ; # Update all local attributes (temperature, exchangepartner) for my replica if an exchange attempt is accepted
set NEWTEMP $replica(temperature) ; # Set new temperatureparameter for the local MD run
rescalevels [expr sqrt(1.0*$NEWTEMP/$OLDTEMP)]langevinTemp $NEWTEMP ; # After temperature parameter
is updated, velocities are rescaled and Langevin temperature isupdated as well
}
Appendix B
The Tcl scripts for the MCA applications can be downloadedalong with NAMD version 2.10 or nightly build source codevia http://www.ks.uiuc.edu/Development/Download/download.cgi?PackageName=NAMD. In the NAMD source tree, the directorylib/replica contains the relevant MCA scripts and test examples.Replica utility scripts, including replica sorting, visualization andWHAM post-processing, are also provided.
References
[1] D.E. Shaw, et al., Anton, a special-purpose machine for molecular dynamicssimulation, Commun. ACM 51 (2008) 91–97.
[2] W. Jiang, M. Hodoscek, B. Roux, Computation of absolute hydration andbinding free energy with free energy perturbation distributed replica-exchange molecular dynamics, J. Chem. Theory Comput. 5 (2009) 2583–2588.
[3] W. Jiang, B. Roux, Free energy perturbation Hamiltonian replica-exchangemolecular dynamics (FEP/H-REMD) for absolute ligand binding free energycalculations, J. Chem. Theory Comput. 6 (9) (2010) 2559–2565.
[4] W. Jiang, et al., Calculation of free energy landscape in multi-dimensionswith Hamiltonian-exchange umbrella sampling on petascale supercomputer,J. Chem. Theory Comput. 8 (2012) 4672–4680.
[5] A.C. Pan, D. Sezer, B. Roux, Finding the transition pathways using the stringmethod with swarms of trajectories, J. Phys. Chem. B 112 (2008) 3432–3440.
[6] W.P. Bhimalapuram, A.R. Dinner, Nonequilibriumumbrella sampling in spacesof many order parameters, J. Chem. Phys. 130 (2009) 074104.
[7] A. Dickson, A.R. Dinner, Enhanced sampling of nonequilibrium steady states,Annu. Rev. Phys. Chem. 61 (2010) 441–459.
[8] A. Dickson, et al., Flow-dependent unfolding and refolding of an RNAby nonequilibrium umbrella sampling, J. Chem. Theory Comput. 7 (2011)2710–2720.
[9] Y. Sugita, Y. Okamoto, Replica-exchange molecular dynamics method forprotein folding, Chem. Phys. Lett. 314 (1999) 141–151.
[10] Y. Sugita, A. Kitao, Y. Okamoto,Multidimensional replica-exchangemethod forfree-energy calculations, J. Chem. Phys. 113 (15) (2000) 6042–6051.
[11] A. Mitsutake, Y. Sugita, Y. Okamoto, Generalized-ensemble algorithms formolecular simulations of biopolymers, Biopolymers 60 (2001) 96–123.
[12] A.Mitsutake, Y. Okamoto, Frommultidimentional replica-exchangemethod tomultidimensional multicanonical algorithm and simulated tempering, Phys.Rev. E 79 (2009) 047701.
[13] K. Sanbonmatsu, A. Garcia, Structure of Met-enkephalin in explicit aqueoussolution using replica exchange molecular dynamics, Proteins: Struct., Funct.,Bioinf. 46 (2002) 225–234.
[14] R. Zhou, B. Berne, Can a continuum solvent model reproduce the free energylandscape of a beta-hairpin folding in water? Proc. Natl. Acad. Sci. USA 99(2002) 12777–12782.
[15] R. Zhou, Free energy landscape of protein folding in water:explicit vs. implicitsolvent, Proteins 53 (2003) 148–161.
[16] K. Ostermeir, M. Zacharis, Advanced replica-exchange sampling to study theflexibility and plasticity of peptides and proteins, Biochim. BioPhys. Acta 1834(5) (2013) 847–853.
[17] S. Kannan, M. Zacharis, Simulation of DNA double-strand dissociation andformation during replica-exchange molecular dynamics simulations, Phys.Chem. Chem. Phys. 11 (45) (2009) 10589–10595.
[18] C.J. Woods, J.W. Essex, M.A. King, Enhanced configurational sampling inbinding free-energy calculations, J. Phys. Chem. B 107 (2003) 13711–13718.
[19] D. Sindhikara, Y. Meng, A. Roitberg, Exchange frequency in replica exchangemolecular dynamics, J. Chem. Phys. 128 (2008) 024103.
[20] D.J. Sindhikara, D.J. Emerson, A.E. Roitberg, Exchange often and properly inreplica exchange molecular dynamics, J. Chem. Theory Comput. 6 (2010)2804–2808.
[21] S. Moors, S. Michielssens, A. Ceulemans, Improved replica exchange methodfor native-state protein sampling, J. Chem. Theory Comput. 7 (2010) 231–237.
[22] Y. Meng, D. Sabri, A. Roitberg, Computing alchemical free energy differenceswith Hamiltonian replica exchange molecular dynamics (H-REMD) simula-tions, J. Chem. Theory Comput. 7 (9) (2011) 2721–2727.
[23] A.W. Matthew, V.P. Rohit, Satisfying the fluctuatio theorem in free-energycalculations with Hamiltonian replica exchange, Phys. Rev. E 77 (2008)026104.
[24] S. Kannan, M. Zacharis, Enhanced sampling of peptide and protein conforma-tions using replica exchange simulations with a peptide backbone biasing-potential, Proteins: Struct., Funct., Bioinf. 66 (2007) 697–706.
[25] M. Fajer, D. Hamelberg, J.A. McCammon, Replica-exchange acceleratedmolecular dynamics (REXAMD) applied to thermodynamic integration, J.Chem. Theory Comput. 4 (2008) 1565–1569.
[26] R. Allen, P. Warren, P. Wolde, Sampling rare switching events in biochemicalnetworks, Phys. Rev. Lett. 94 (2005) 018104.
[27] R. Salomon-Ferrer, D. Case, R.C. Walker, An overview of the Amberbiomolecular simulation package, WIREs Comput. Mol. Sci. 3 (2013) 198–210.
[28] B. Hess, et al., GROMACS 4: algorithms for highly efficient, load-balanced, andscalable molecular simulation, J. Chem. Theory Comput. 4 (2008) 435–447.
[29] K. Bowers, et al. Scalable algorithms for molecular dynamics simulationson commodity clusters, in: Proceedings of the ACM/IEEE Conference onSupercomputing, SC06, 2006: p. Tampa, Florida, November 11–17.
[30] J. Kastner, Umbrella sampling, Wiley Interdisciplinary Reviews: Computa-tional Molecular Science 1 (6) (2011) 932.
[31] T.W. Allen, O.S. Anderson, B. Roux, Molecular dynamics — potential of meanforce calculations as a tool for understanding ion permeation and selectivityin narrow channels, Biophys. Chem. 124 (2006) 251–267.
[32] A. Laio, F. Gervasio, Metadynamics: a method to simulate rare events andreconstruct the free energy in biophysics, chemistry andmaterial science, Rep.Progr. Phys. 71 (2008).
[33] W. Wojtas-Niziurski, et al., Self-learning adaptive umbrella sampling methodfor the determination of free energy landscapes in multiple dimensions, J.Chem. Theory Comput. 9 (2013) 1885–1895.
[34] Y. Deng, B. Roux, Hydration of amino acid side chains: nonpolar andelectrostatic contributions calculated from staged molecular dynamics freeenergy simulations with explicit water molecules, J. Phys. Chem. 108 (2004)16567–16576.
[35] Y. Deng, B. Roux, Calculation of standard binding free energies: aromaticmolecules in the T4 lysozyme L99Amutant, J. Chem. Theory Comput. 2 (2006)1255–1273.
[36] Y. Deng, B. Roux, Computation of binding free energywithmolecular dynamicsand grand canonical monte carlo simulations, J. Chem. Phys. 128 (2008)115103.
[37] W. Gan, S. Yang, B. Roux, Atomistic view of the conformational activation ofSrc kinase using the string method with swarms-of-trajectories, Biophys. J. 97(2009) L8–L10.
[38] B.R. Brooks, et al., CHARMM: a program for macromolecular energy,minimization, and dynamics calculations, J. Comput. Chem. 4 (1983) 187–217.
[39] J. Phillips, et al., Scalable molecular dynamics with NAMD, J. Comput. Chem.26 (2005) 1781–1802.
[40] S. Kumar, Y. Sun, L. Kale, Acceleration of an asynchronous message drivenprogramming paradigm on IBM blue Gene/Q, in: IEEE 27th InternationalSymposium on Parallel & Distributed Processing, 2013. pp. 689–699.
[41] Y. Sun, et al. Optimizing Fine-grained communication in a biomolecularsimulation application on cray XK6, in: SC12 Proceedings of the InternationalConference on High Performance Computing, Networking, Storage andAnalysis, 2012.
[42] W. E, W. Ren, E. Vanden-Eijnden, Transition pathways in complex systems:reaction coordinates, isocommittor surfaces, and transition tubes, Chem. Phys.Lett. 413 (2005) 242–247.
[43] W. E, W. Ren, E. Vanden-Eijnden, Finite temperature string method for thestudy of rare events, J. Phys. Chem. B 109 (2005) 6688–6693.
[44] W. Ren, et al., Transition pathways in complex systems: application of thefinite-temperature string method to the alanine dipeptide, J. Chem. Phys. 123(2005) 134109.
[45] H. Fukunishi, O. Watanabe, S. Takada, On the Hamiltonian replica exchangemethod for efficient sampling of biomolecular systems: application to proteinstructure prediction, J. Chem. Phys. 116 (2002) 9058–9067.
[46] W. Shalongo, L. Dugad, E. Stellwagen, Distribution of helicity within themodelpeptide. Acetyl(AAQAA)3amide, J. Am. Chem. Soc. 116 (1994) 8288–8293.
[47] J. Wang, Y. Deng, B. Roux, Absolute binding free energy calculations usingmolecular dynamics simulations with restraining potentials, Biophys. J. 91(2006) 2798–2814.
[48] J.D. Weeks, D. Chandler, H.C. Anderson, Role of repulsive forces in forming theequilibrium structure of simple liquids, J. Chem. Phys. 54 (1971) 5237–5247.
[49] S. Kumar, et al., THE weighted histogram analysis method for free-energycalculations on biomolecules. I. The method, J. Comput. Chem. 13 (1992)1011–1021.
[50] J. Kirkwood, Statistical mechanics of fluid mixtures, J. Chem. Phys. 3 (1935)300.
[51] G. Torrier, J. Valleau, Monte Carlo free energy estimates using non-Boltzmannsampling: application to the sub-critical Lennard-Jones fluid, Chem. Phys. Lett.28 (1974) 578.
[52] K. Murata, Y. Sugita, Y. Okamoto, Free energy calculations for DNA basestacking by replica-exchange umbrella sampling, Chem. Phys. Lett. 385 (2004)1–7.
[53] J.C. Gumbart, B. Roux, C. Chipot, Standard binding free energies from computersimulations: what is the best strategy? J. Chem. Theory Comput. 9 (2013)794–802.
[54] J. Gumbart, B. Roux, C. Chipot, Efficient determination of protein–proteinstandard binding free energies from first principles, J. Chem. Theory Comput.9 (2013) 3789–3798.
[55] W. Xu, Crystal structures of c-Src reveal features of its autoinhibitorymechanism, Molecular Cell 3 (1999) 629–638.





























