Embed Size (px)
Citation preview

RSC Advances
PAPER
aCentre for Nano Materials, International
Metallurgy and New Materials, Hyderabad,bDepartment of Chemical Engineering, Ind
A.P., India. E-mail: [email protected] Diagnostics and Biomarkers La
A.P., India
† Electronic supplementary informa10.1039/c3ra44170a
Cite this: RSC Adv., 2014, 4, 12188
Received 5th August 2013Accepted 13th January 2014
DOI: 10.1039/c3ra44170a
www.rsc.org/advances
12188 | RSC Adv., 2014, 4, 12188–1219
Fabrication and surface functionalization ofelectrospun polystyrene submicron fibers withcontrollable surface roughness†
Anulekha K. Haridas,ab Chandra S. Sharma,*b V. Sritharanc and Tata N. Rao*a
Polystyrene (PS) submicron fibers of 14 wt% concentration were fabricated by electrospinning using
dimethylformamide (DMF)–tetrahydrofuran (THF) solvent system. The surface morphology of PS fibers
was modified from smooth to rough by changing the mixing ratio of DMF and THF in the spinning
solution. The electrospun PS fibers with smooth and rough surfaces were then amidomethylated by
treating with N-hydroxymethyl-2-chloroacetamide. PS fibers with higher roughness incorporated more
amidomethyl functional groups on their surface, as confirmed by XPS analysis. This observation was
further supported by BET adsorption isotherm results which showed a significant increase in specific
surface area of rough PS electrospun fibers. Interestingly, amidomethylation has altered rough
electrospun PS submicron fibers from extremely hydrophobic to hydrophilic. These chemically modified
electrospun PS fibers with controllable surface roughness and wettability may be utilized as a carrier for
proteins, mainly enzymes and antibodies, by covalent linkage through amino groups attached to their
surface.
1. Introduction
Polystyrene (PS) is one of the widely used polymers for numerousapplications in the form ofmicrospheres, thin lms and bers dueto its excellent mechanical properties and low cost. Conversely, italso shows properties like hydrophobicity and low chemicalresistance which restrict its use.Many surfacemodication studiessuch as UV and ozone treatments,1 plasma treatments,2,3 ion beamsputtering,4 chemical substitution reactions5,6 and even blendingwith hydrophilic polymers7,8 have been reported in order toenhance its biocompatibility, wettability and surface adhesion bybiomolecules. Among the various surface modication methods,the chemical substitution reaction is an easy as well as costeffective method. Due to the presence of a long carbon chain withpendant benzene rings, PS can be easily substituted by FriedelCras aromatic substitution reactions at the ortho- and para-positions with carboxyl and amine groups. One such chemicalmodication done on polystyrene was amidomethylation whichenhances both the covalent and ionic attachment of biomoleculeson the polystyrene surfaces. Amidomethylation can be considered
Advanced Research Centre for Powder
A. P., India. E-mail: [email protected]
ian Institute of Technology, Hyderabad,
boratory, Global Hospitals, Hyderabad,
tion (ESI) available. See DOI:
7
as an electrophilic substitution reaction and has been done byTeramoto9 for modifying the surfaces of PS Petri dishes withoutlosing their transparency. Also the same group has investigated thereaction with substituted PS like polymethylstyrene, poly(4-tert-butylstyrene) etc.10 Yamamoto et al.11 have studied the functional-ization of PS based micron sized fabric immobilized with anantibody for burn wound dressings. When the ber diameter isreduced to submicron and nanoscale and its surface ismodied soas to enhance the surface area, the application can be much moremeaningful. In one such attempt, Jia et al.12 used PS nanobers forthe immobilization of chymotrypsin.
Although there are a number of methods for the production ofnanobers such as drawing, phase separation, template synthesisand self-assembly, electrospinning has become the most exten-sively usedmethod for the fabrication of nanobers.13This is due toits notable advantages such as continuity in the formation of bers,easiness to scale up the process and simple and robust method offabrication. The process of electrospinning involves the formationof nanobers from a viscous solution under the application of highvoltage which is usually in the order of kilovolts.14–18 The appliedvoltage charges the viscous solution and overcomes its surfacetension, resulting in the formation of a Taylor cone, which ulti-mately initiates the spinning jet. In the second stage, the Taylorcone further stretches due to the applied eld and inter-particlerepulsion enabling the jet to elongate. The stretching of the Taylorcone is proceeded by instabilities in the air such as bending andwhippingmotions which ultimately result in evaporation of solventfrom the polymer jet. The polymer in the spinning jet is thencollected on the target electrode as nanobers.16,17 Another reason
This journal is © The Royal Society of Chemistry 2014

Paper RSC Advances
to choose electrospinning as a method to produce nanobers isthe ability of electrospun nanobers to add surface micro struc-tural features (surface pores, roughness, hollow and core–shellstructure) directly during the formation process. Few studies havebeen reported recently to produce porous electrospun nanobersby combining electrospinning with phase separation at the bersurface.18–28 The driving force for the phase separation to take placemay be either temperature, presence of non-solvent, relativehumidity or relative volatility of solvent systems. Preparation ofporous polyacrylonitrile (PAN) bers from solvent mixture ofdimethylformamide (DMF)–water was reported by Yu et al.19
whereas Huang et al.20 fabricated PAN and polysulfone nanoberswith surface features by simply varying the humidity in the spin-ning environment. In the case of PS, a few researchers have alreadyreported functional surface morphologies such as surface poresand wrinkled structures and also investigated the effect ofmolecular weight on the surface morphology of electrospun PSbers.23 In recent studies, several researchers21,22,26 reported theelectrospinning of porous PS polystyrene nanobers using solventmixtures of DMF and tetrahydrofuran (THF). Use of a solventmixture with differences in relative volatility leads to phase sepa-ration and hence facilitates the pore formation. In another study,Karthik et al.27 have shown the formation of surface pores onelectrospun nanobers while collecting the bers directly in a non-solvent bath. Further, Demir et al.28 have investigated the forma-tions of three different types of pores on the electrospun PS bersby varying the weight fraction of polymer at constant relativehumidity. Whereas, Pai et al.29 studied wrinkled PS ber formationby varying the humidity of the environment.
In this study, we used a xed 14 wt% PS solution in DMF–THFsolvent system to electrospin long, continuous and uniform PSsubmicron sized bers. We tuned the surface roughness andinternal porosity by varying the mixing ratios of DMF and THFsolvents in the PS electrospinning solution. Further we carried outamidomethylation functionalization by incorporating N-hydroxy-methyl-2-chloroacetamide (NMCA) on as-spun PS bers with leastand highest roughness. As mentioned previously, amidomethyla-tion is one of the preferred ways to enhance the attachment ofbiomolecules to a PS surface. Thus the main aim of this work is tostudy the effect of amidomethylation reaction on electrospun PSsubmicron bers that have enhanced surface area due to theirrough and porous morphology. Fourier Transform Infrared Spec-troscopy (FTIR), Brunauer–Emmett–Teller (BET) surface area andX-ray Photoelectron Spectroscopy (XPS) analyses were used toassist this analysis. Later we also studied the wettability charac-teristics of surface functionalized (amidomethylated) PS fabricwith least and highest surface roughness. PS submicron berswith hydrophilic nature may be utilised as a carrier for proteins,mainly enzymes and antibodies, and thus for various bio-medicalapplications such as selective removal of ligands, separationcolumns and even in antibacterial wound dressings.
2. Experimental section2.1. Materials
PS withmolecular weight (Mw) 350 000 gmol�1 and NMCA (99%pure) were purchased from Sigma Aldrich. DMF (99% purity)
This journal is © The Royal Society of Chemistry 2014
and THF (99% purity) were purchased from Merck India,whereas nitrobenzene, sulphuric acid and methanol werepurchased from Fischer Scientic, India. All the chemicals wereused without any modication.
2.2. Electrospinning of PS submicron sized bers
A large number of solution parameters affect the ability of apolymer to electrospin into continuous nano-sized bers. Thisincludes the dielectric constant and solubility parameter of thesolvents and therefore, the conductivity and viscosity of thepolymer solutions used in electrospinning.14–18 A mixture ofDMF and THF solution having dielectric constant values of 38and 7.5, respectively, was used as solvent for PS. Moreover thesolubility parameter values of DMF and THF are 12.1 and 9.5.
A 14 wt% PS solution was prepared by dissolving PS in DMF–THF solvent mixture at 50 �C using a magnetic stirrer. Themixing ratio of DMF and THF was varied as 100 : 0, 70 : 30,50 : 50, 30 : 70 and 0 : 100 while preparing the PS solution. PSsolutions with different DMF and THF solvent ratios were thenelectrospun vertically downward to yield submicron bers asshown in Fig. 1. The electrospinning set up was purchased fromE-spin Nanotech Pvt. Ltd (India). The size of the needle, appliedvoltage and the distance between the needle and the collector(Al foil) were some of the process parameters that were opti-mized in this study to obtain long, continuous and uniformbers. These parameters were as follows: 23 gauge syringeneedle, 18 kV applied voltage and 8 cm. All the experimentswere conducted at a temperature of 30 �C and 27% relativehumidity.
2.3. Surface modication of PS submicron bers byamidomethylation reaction
The electrospun PS bers with highest surface roughness andporosity were compared with the smooth morphological berswith respect to their ability to functionalize using amidome-thylation reaction in the presence of NMCA linker.9 Both typesof samples (smooth and rough surface morphology) were cutinto 5 � 5 cm2 mats with approximate weight of 40 mg. Ami-domethylating solution was prepared by dissolving different wt% of NMCA (7, 5, 2, and 1) in a mixture of sulphuric acid andnitropropane at an ice cold condition (0–5 �C). Aer obtaining aclear solution of NMCA in sulphuric acid–nitropropanemixture, the electrospun PS fabric was immersed in it for 1 and2 hours to allow the reaction. Aer the specied time periods,the fabric was taken out and washed thoroughly using ice coldmethanol, room temperature methanol followed by DI water,three times. All the samples were triplicated in this study. Aerwashing, the PS ber mats were observed to be a yellow colourand were then dried and characterized further.
2.4. Characterization of polymer solution and electrospunPS submicron bers
The viscosity of all PS solutions with varying DMF and THFmixing ratios was measured using an Anton Paar viscometerPhysica MCR 51 at 31 �C and at a constant shear rate of1000 s�1. The ionic conductivity/resistivity of the PS solutions at
RSC Adv., 2014, 4, 12188–12197 | 12189
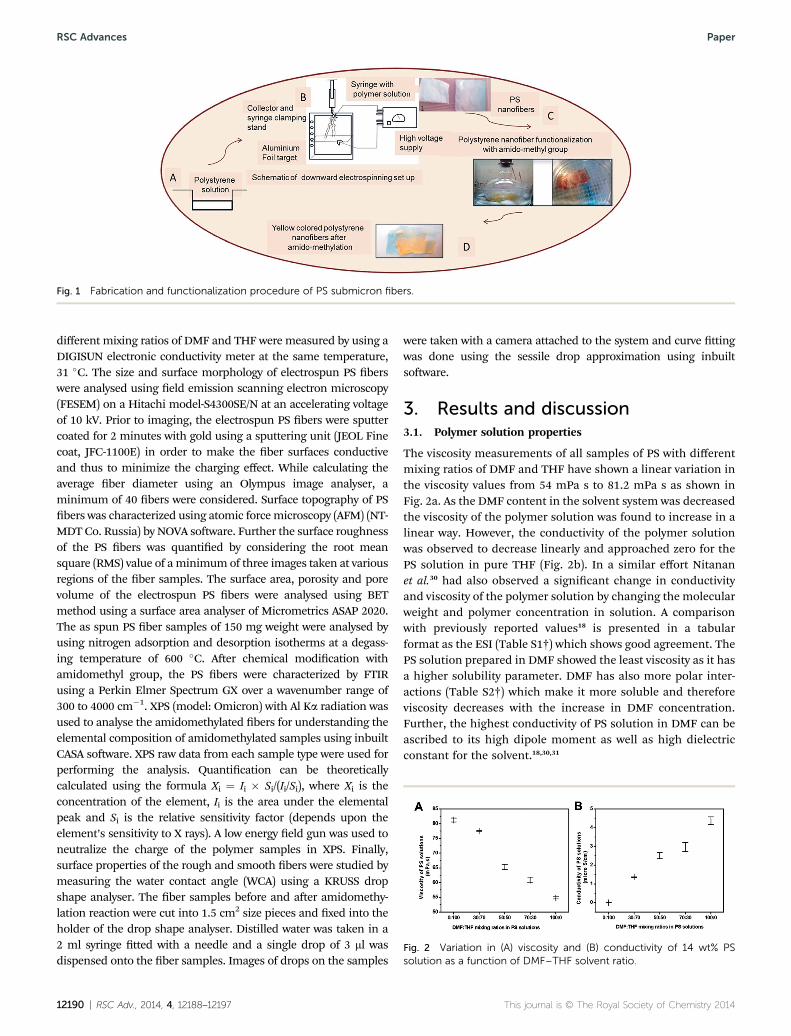
Fig. 1 Fabrication and functionalization procedure of PS submicron fibers.
Fig. 2 Variation in (A) viscosity and (B) conductivity of 14 wt% PSsolution as a function of DMF–THF solvent ratio.
RSC Advances Paper
different mixing ratios of DMF and THF were measured by using aDIGISUN electronic conductivity meter at the same temperature,31 �C. The size and surface morphology of electrospun PS berswere analysed using eld emission scanning electron microscopy(FESEM) on a Hitachi model-S4300SE/N at an accelerating voltageof 10 kV. Prior to imaging, the electrospun PS bers were sputtercoated for 2 minutes with gold using a sputtering unit (JEOL Finecoat, JFC-1100E) in order to make the ber surfaces conductiveand thus to minimize the charging effect. While calculating theaverage ber diameter using an Olympus image analyser, aminimum of 40 bers were considered. Surface topography of PSbers was characterized using atomic forcemicroscopy (AFM) (NT-MDT Co. Russia) by NOVA soware. Further the surface roughnessof the PS bers was quantied by considering the root meansquare (RMS) value of aminimum of three images taken at variousregions of the ber samples. The surface area, porosity and porevolume of the electrospun PS bers were analysed using BETmethod using a surface area analyser of Micrometrics ASAP 2020.The as spun PS ber samples of 150 mg weight were analysed byusing nitrogen adsorption and desorption isotherms at a degass-ing temperature of 600 �C. Aer chemical modication withamidomethyl group, the PS bers were characterized by FTIRusing a Perkin Elmer Spectrum GX over a wavenumber range of300 to 4000 cm�1. XPS (model: Omicron) with Al Ka radiation wasused to analyse the amidomethylated bers for understanding theelemental composition of amidomethylated samples using inbuiltCASA soware. XPS raw data from each sample type were used forperforming the analysis. Quantication can be theoreticallycalculated using the formula Xi ¼ Ii � Si/(Ii/Si), where Xi is theconcentration of the element, Ii is the area under the elementalpeak and Si is the relative sensitivity factor (depends upon theelement’s sensitivity to X rays). A low energy eld gun was used toneutralize the charge of the polymer samples in XPS. Finally,surface properties of the rough and smooth bers were studied bymeasuring the water contact angle (WCA) using a KRUSS dropshape analyser. The ber samples before and aer amidomethy-lation reaction were cut into 1.5 cm2 size pieces and xed into theholder of the drop shape analyser. Distilled water was taken in a2 ml syringe tted with a needle and a single drop of 3 ml wasdispensed onto the ber samples. Images of drops on the samples
12190 | RSC Adv., 2014, 4, 12188–12197
were taken with a camera attached to the system and curve ttingwas done using the sessile drop approximation using inbuiltsoware.
3. Results and discussion3.1. Polymer solution properties
The viscosity measurements of all samples of PS with differentmixing ratios of DMF and THF have shown a linear variation inthe viscosity values from 54 mPa s to 81.2 mPa s as shown inFig. 2a. As the DMF content in the solvent system was decreasedthe viscosity of the polymer solution was found to increase in alinear way. However, the conductivity of the polymer solutionwas observed to decrease linearly and approached zero for thePS solution in pure THF (Fig. 2b). In a similar effort Nitananet al.30 had also observed a signicant change in conductivityand viscosity of the polymer solution by changing the molecularweight and polymer concentration in solution. A comparisonwith previously reported values18 is presented in a tabularformat as the ESI (Table S1†) which shows good agreement. ThePS solution prepared in DMF showed the least viscosity as it hasa higher solubility parameter. DMF has also more polar inter-actions (Table S2†) which make it more soluble and thereforeviscosity decreases with the increase in DMF concentration.Further, the highest conductivity of PS solution in DMF can beascribed to its high dipole moment as well as high dielectricconstant for the solvent.18,30,31
This journal is © The Royal Society of Chemistry 2014

Paper RSC Advances
3.2. Surface morphology analysis of electrospun PSsubmicron bers
The effect of varying DMF and THF ratio in PS solution on themorphology of electrospun bers is summarized in Fig. 3. When aPS solution with DMF–THF ratio 0 : 100 (when only THF was usedas solvent) was electrospun, bers with micron sized beadsmorphology were formed as observed in Fig. 3A1. However, a PSsolution with 100 : 0 mixing ratio of the solvents (only DMF) yiel-ded non-uniform bers (Fig. 3E1). The bead formation in PS0 : 100 samples can be attributed to the insufficient conductivityand high viscosity of polymer solution whereas low viscosity mightbe the reason for bead on stringmorphology in PS 100 : 0 samples.In PS 0 : 100 samples, charging of the polymer solution becameless due to less penetration of THF into the PS chains and hence itcould not stretch and form a Taylor cone. The inability of Taylorcone formation resulted in deposition of PS as beads of biggersizes. Whereas some drops of the PS solution which were able toform a Taylor cone have stretched and formed bers. In addition,the low relative humidity (27%) can also be the reason for theformation of beaded structures. In PS 100 : 0 sample, insufficientviscosity of the PS solution resulted in incomplete stretching of thepolymer solution creating bead on string morphology. All othersamples of electrospun PS bermats (Fig. 3B1, C1 and D1) showedne ber morphology without any formation of beads. This was inagreement with the studies of Zhang et al.32 where solvent ratios ofDMF and THF were varied for electrospinning PS. But there werenoticeable variations in the surface morphology for PS submicronbers when they were analysed at a high magnication of 40k.Appearance of surface pores on the bers and the presence of
Fig. 3 FESEM images of PS 14 wt% electrospun at varying solventratios of DMF and THF. Selected samples of fibers in varying DMF–THFratios are depicted where solvent ratios are (A) 0 : 100; (B) 30 : 70; (C)50 : 50; (D) 70 : 30; (E) 100 : 0.
This journal is © The Royal Society of Chemistry 2014
porous beads were observed in the FESEM image of bers spunfrom DMF–THF ratio 0 : 100 (Fig. 3A2). The formation of pores onthe beads and breath gures on bers in the sample can be due tothe high volatility of THF solvent used. Due to the fast phaseseparation that happened during electrospinning, some regions inthe spinning jet got separated into solventless and polymer richphases, creating porous surface features or breath gures.However, bymixing the DMFwith THF and thus allowing a changein relative volatility of the solvent mixture and therefore phaseseparation, surface protrusions occurred on the PS bers surface toyield rough bers.24–27 Among all the different solvent ratios tried,the bers in the solvent mixing ratio of DMF–THF: 70 : 30(Fig. 3D2) were seen as more rough. For PS 100 : 0 samples (onlyDMF), we observe a relatively smooth ber surface (Fig. 3E2)besides non-uniformity in the ber size distribution. This obser-vation is also in agreement with what has been reported earlier forthe formation of smooth PS electrospun bres in only DMF athigher relative humidity.29 The presence of surface features likewrinkles and roughness can also be described by the phase sepa-ration occurring at the ber surfaces during the electrospinningprocess. As also explained by Jinyou et al.,24 the volatility difference(vapour pressures) in the solvents of DMF and THF caused fastevaporation of THF during the spinning process giving rise tosolventless and solvent rich areas on the spinning jet. This resultsin different surface patterns like protrusions and wrinkles yieldingthe roughness on the ber surfaces (Fig. 3B2, C2 and D2). Hencevapour induced phase separation may be the reason for the vari-ations in surface morphologies of PS bers as the solvent ratioswere varied during electrospinning. The average diameter of theelectrospun bers as measured by an Olympus image analyser wasfound to be 780 � 185 nm and 650 � 58 nm for the PS bersamples with 100 : 0 and 70 : 30 solvent ratios, respectively.
The spinnability–solubility map for the 14 wt% PS solutionwith varying solvent ratio is also prepared and shown as Fig. 4.The fractional solubility parameters for solvent mixture werecalculated using fractional parameters for the solvent compo-nents, following the lever rule21 and are summarized in TableS2.† This map also suggests that spinnability of PS solution isbetter with increased DMF concentration. This behaviour is inagreement with what has been reported by Jarusuwannapoomet al.18 This is mainly attributed to the relatively high dipolemoment and high conductivity for DMF. In the ternary diagram(Fig. 4), this can be endorsed to the fact that a moderate value ofpolar forces is pivotal for electrospinning.
The cross section of the PS ber samples with breath gures(Fig. 3A2), highest surface roughness (Fig. 3D2) and least surfaceroughness (Fig. 3E2) were also analysed using FESEM. As the DMFcontent in the solvent mixture increases from 0 to 100, the interiorporosity in the bers also changes. The bers electrospun withDMF and THF ratios of 0 : 100 and 100 : 0, respectively (Fig. 5Aand C), can be viewed as having solid cross sections. However thePS bers with highest surface roughness (70 : 30) show internalporosity with platelet like structures as perceived in the cross-sectional view (Fig. 5B). It can be understood from these resultsthat maximum phase separation occurred in the case of PS berswith 70 : 30 solvent ratio as compared to the other two samples,and the least phase separation in the case of 100 : 0 solvent ratio.
RSC Adv., 2014, 4, 12188–12197 | 12191

Fig. 4 Spinnability–solubility map for 14 wt% PS solutions prepared forvarying DMF–THF solvent ratios.
Fig. 6 2-D surface imaging of PS spun fiber samples with (A) 100 : 0and (C) 70 : 30 DMF–THF solvent ratio. (B) and (D) show the 3-Dprofiles with fitted value of roughness, respectively. Image in inset of(B) and (D) shows the histogram for surface roughness.
RSC Advances Paper
Further, in order to analyse and quantify the surfaceroughness of PS smooth and rough bers (100 : 0 and 70 : 30),the samples were scanned by AFM in non-contact mode. Asshown in the 2-D and 3-D surface proles (Fig. 6), the root meansquare value of roughness for PS 100 : 0 ber (Fig. 6A and B) wasfound to be 31.79 nm, which increased to 42.73 nm for PS70 : 30 ber sample (Fig. 6C and D). A histogram plot of thesurface roughness was also provided as an inset in Fig. 6B andD. Thus a 34% increase in surface roughness has been observeddue to vapour phase separation when the THF content in the PSsolution was increased from 0 to 30%.
BET adsorption isotherms further characterized the PS smoothand rough ber samples for their surface area, pore volume andporosity. These results are summarized in Table 1.We observed thatBET surface area increased signicantly (�57%) from 52.11 m2 g�1
to 82.2 m2 g�1, while total pore volume also showed a similarincrease for the rough PS bers (70 : 30) as compared to relativelysmooth bers (100 : 0). Average pore diameter of rough PS berswas reduced to 23.26 nm from 32.15 nm for smooth bers. Thehigher surface area value for the PS bers 70 : 30 can be attributedto the high internal porosity (as shown in Fig. 4b) and surfaceroughness, as also illustrated by AFM scanning results (Fig. 6).
3.3. Surface modication of electrospun PS submicronbers by amidomethylation reaction
The amidomethylation reaction can be considered as an elec-trophilic substitution reaction on the benzene ring of PS by the
Fig. 5 FESEM images showing the cross section of PS fibers with (A) surfaand (C) least surface roughness (DMF–THF: 100 : 0).
12192 | RSC Adv., 2014, 4, 12188–12197
carbonium ion – RONHCH from NMCA,9,10 as shown below ineqn (1).
(1)
Eqn (1): electrophilic substitution reaction of PS usingNMCA.9,10
When electrospun PS submicron bers were immersed inamidomethylation solution, a reddish gel was formed. This canbe attributed to the cross linking reaction by formaldehyde,present in NMCA, on polystyrene as reported by Termato et al.9
The amidomethylation reaction is expected to occur at the para-position of the benzene ring, due to steric hindrance.9 Theamidomethylated polystyrene (AmPS) bers aer washing withcold methanol followed by methanol at room temperature anddistilled water, were observed as yellow colour ber mats. Ter-mato et al.9 used �7 wt% NMCA in complete amidomethylationreaction for 2 hours. However, in the case of electrospun PSsubmicron bers, 7 wt% NMCA concentration seems to be veryhigh as the fabric mat became too fragile to handle. To over-come this, we varied the concentration from 7 wt% to 1 wt%.
ce pores (DMF–THF: 0 : 100) (B) maximum surface roughness (70 : 30)
This journal is © The Royal Society of Chemistry 2014

Table 1 BET surface area of the as spun PS submicron fibers
S. no. Nanober sampleBET surfacearea (m2 g�1)
BJH pore volume(cm3 g�1)
Average poresize (nm)
1 Smooth surface (DMF–THF: 100 : 0) 52.11 0.4121 32.152 Rough surface (DMF–THF: 70 : 30) 82.20 0.4722 23.26
Paper RSC Advances
Further, we carried out the reaction at two different temper-atures, room temperature (30 �C) and at low temperature(�5 �C). At room temperature, all the samples showed orangecoloration with brittleness, which eventually resulted inpowdering of the samples. However at low temperature(�5 �C), by reducing the concentration of NMCA from 7 to 1 wt%, AmPS samples showed pale yellow coloration while theber morphology also remains intact. We further examinedthe effect of reaction time on integrity of fabric matrix byconsidering the two different PS ber samples with lowestconcentration of (1 wt%) NMCA (Fig. 7). 1 hour reaction timeyielded AmPS bers without losing their morphology (Fig. 7A1and B1). The individual bers in 100 : 0 and 70 : 30 solventratio samples of AmPS have swollen a little bit and have comemuch closer compared to the as spun bers. Moreover, for2 hours reaction time, the individual bers started fusingtogether, losing their identity and formed a layer type struc-ture (Fig. 7A2 and B2). Based on these observations, we mayconclude the optimized parameters for amidomethylationreaction onto electrospun PS bers, which are summarized asfollows: 1 wt% NMCA solution, reaction temperature: �5 �C,reaction time: 1 hour. This tuning of reaction parameters wasonly based on intact PS ber morphology, however, FTIRand XPS analysis as discussed below further conrmedthese observations.
Fig. 7 The figure illustrates change in morphology of AmPS fibers as afunction of the amidomethylation reaction times. A1, A3, A5 and B1, B3,B5 show low magnification FESEM images of PS (100 : 0) and PS(70 : 30) sub-micron fibers while A2, A4, A6 and B2, B4, B6 are highmagnification FESEM images of PS (100 : 0) and PS (70 : 30) sub-micron fibers after 0, 1 and 2 hours of amidomethylation reactiontimes, respectively.
3.4. FTIR analysis before and aer amidomethylation ofelectrospun PS submicron bers
The FTIR analysis of PS submicron bers before and aeramidomethylation is shown in Fig. 8. The peaks at 1260 and1280 cm�1 show symmetric methylene stretching while bendingvibrations of out-of-plane C–H bonds of aromatic benzene ringwere observed at 698 and 756 cm�1 (Fig. 8a). A few other peaksat 601, 1493 and 1452 cm�1 showed aromatic ring breathingmodes of benzene ring in PS. Also 3082, 3061 and 3027 cm�1
were observed for the aromatic C–H stretches of benzene ring(Fig. 8a). However aer amidomethylation reaction with 1 wt%NMCA for 1 and 2 h (Fig. 8b and c) the AmPS bers showedcharacteristic peaks of amide I, amide II, N–H and C–Clbending vibrations at 1640, 1545, 3300 and 764 cm�1, respec-tively. A peak broadening in the aromatic breathing modes of PSbenzene ring was also observed at 1601, 1493 and 1452 cm�1.Moreover, the presence of peaks at 800–850 cm�1 (Fig. 8b and c)conrms the substitution (amidomethylation) taking placeat the para position of the benzene ring. Also an intensestretching peak of N–H bond was observed at 3300 cm�1, whichwas later conrmed by XPS. A –CH2 stretching vibration wasobserved at 2920 cm�1. This also conrms the presence of
This journal is © The Royal Society of Chemistry 2014
–CH2NHCOCH2Cl group at para position of the benzene ringof PS.
3.5. Surface analysis of amidomethylated PS submicronbers by XPS
PS submicron bers with rough and smooth surfacemorphology (DMF–THF: 70 : 30 and 100 : 0, respectively) aertreating with 1 wt% NMCA in cold condition for 1 and 2 hourswere analysed using XPS in order to check the chemical modi-cation both qualitatively and quantitatively. PS submicronbers have shown sharp characteristic aliphatic and aromaticcarbon peak at 284.6 eV (Fig. 9a). A p–p* shake up which isusually present in aromatic rings was also observed in the PSber sample at a binding energy (B.E.) of 291.4 eV (ref. 33)
RSC Adv., 2014, 4, 12188–12197 | 12193
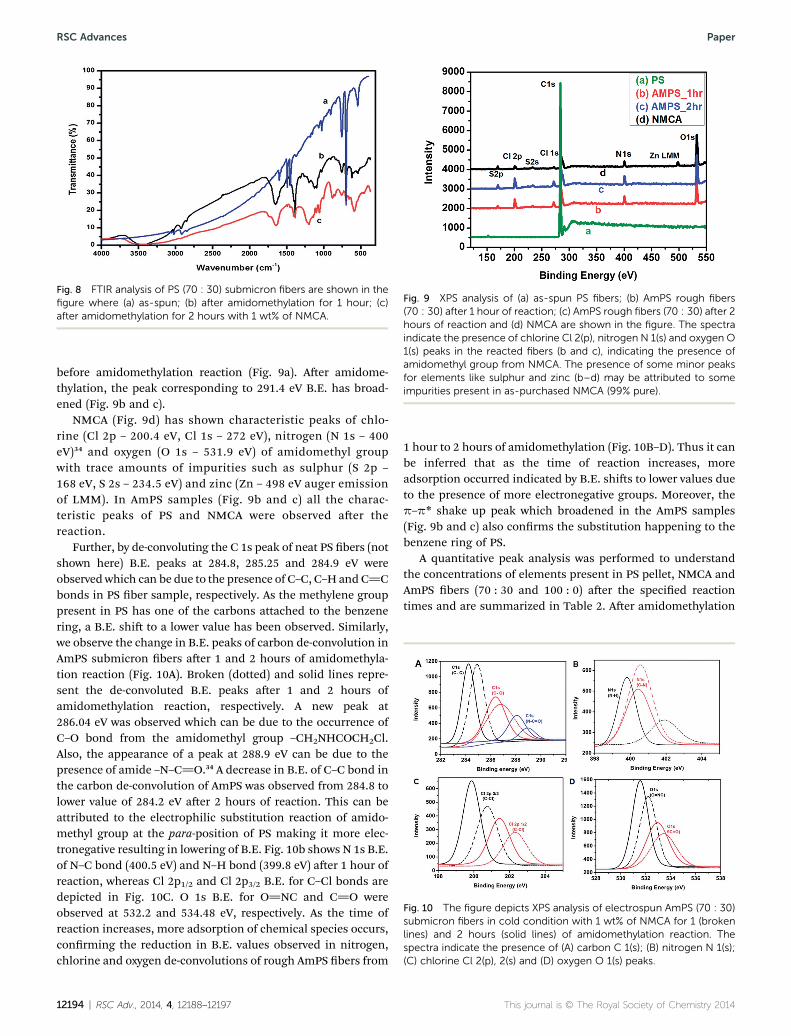
Fig. 8 FTIR analysis of PS (70 : 30) submicron fibers are shown in thefigure where (a) as-spun; (b) after amidomethylation for 1 hour; (c)after amidomethylation for 2 hours with 1 wt% of NMCA.
Fig. 9 XPS analysis of (a) as-spun PS fibers; (b) AmPS rough fibers(70 : 30) after 1 hour of reaction; (c) AmPS rough fibers (70 : 30) after 2hours of reaction and (d) NMCA are shown in the figure. The spectraindicate the presence of chlorine Cl 2(p), nitrogen N 1(s) and oxygen O1(s) peaks in the reacted fibers (b and c), indicating the presence ofamidomethyl group from NMCA. The presence of some minor peaksfor elements like sulphur and zinc (b–d) may be attributed to someimpurities present in as-purchased NMCA (99% pure).
Fig. 10 The figure depicts XPS analysis of electrospun AmPS (70 : 30)submicron fibers in cold condition with 1 wt% of NMCA for 1 (brokenlines) and 2 hours (solid lines) of amidomethylation reaction. Thespectra indicate the presence of (A) carbon C 1(s); (B) nitrogen N 1(s);(C) chlorine Cl 2(p), 2(s) and (D) oxygen O 1(s) peaks.
RSC Advances Paper
before amidomethylation reaction (Fig. 9a). Aer amidome-thylation, the peak corresponding to 291.4 eV B.E. has broad-ened (Fig. 9b and c).
NMCA (Fig. 9d) has shown characteristic peaks of chlo-rine (Cl 2p – 200.4 eV, Cl 1s – 272 eV), nitrogen (N 1s – 400eV)34 and oxygen (O 1s – 531.9 eV) of amidomethyl groupwith trace amounts of impurities such as sulphur (S 2p –
168 eV, S 2s – 234.5 eV) and zinc (Zn – 498 eV auger emissionof LMM). In AmPS samples (Fig. 9b and c) all the charac-teristic peaks of PS and NMCA were observed aer thereaction.
Further, by de-convoluting the C 1s peak of neat PS bers (notshown here) B.E. peaks at 284.8, 285.25 and 284.9 eV wereobserved which can be due to the presence of C–C, C–H andC]Cbonds in PS ber sample, respectively. As the methylene grouppresent in PS has one of the carbons attached to the benzenering, a B.E. shi to a lower value has been observed. Similarly,we observe the change in B.E. peaks of carbon de-convolution inAmPS submicron bers aer 1 and 2 hours of amidomethyla-tion reaction (Fig. 10A). Broken (dotted) and solid lines repre-sent the de-convoluted B.E. peaks aer 1 and 2 hours ofamidomethylation reaction, respectively. A new peak at286.04 eV was observed which can be due to the occurrence ofC–O bond from the amidomethyl group –CH2NHCOCH2Cl.Also, the appearance of a peak at 288.9 eV can be due to thepresence of amide –N–C]O.34 A decrease in B.E. of C–C bond inthe carbon de-convolution of AmPS was observed from 284.8 tolower value of 284.2 eV aer 2 hours of reaction. This can beattributed to the electrophilic substitution reaction of amido-methyl group at the para-position of PS making it more elec-tronegative resulting in lowering of B.E. Fig. 10b shows N 1s B.E.of N–C bond (400.5 eV) and N–H bond (399.8 eV) aer 1 hour ofreaction, whereas Cl 2p1/2 and Cl 2p3/2 B.E. for C–Cl bonds aredepicted in Fig. 10C. O 1s B.E. for O]NC and C]O wereobserved at 532.2 and 534.48 eV, respectively. As the time ofreaction increases, more adsorption of chemical species occurs,conrming the reduction in B.E. values observed in nitrogen,chlorine and oxygen de-convolutions of rough AmPS bers from
12194 | RSC Adv., 2014, 4, 12188–12197
1 hour to 2 hours of amidomethylation (Fig. 10B–D). Thus it canbe inferred that as the time of reaction increases, moreadsorption occurred indicated by B.E. shis to lower values dueto the presence of more electronegative groups. Moreover, thep–p* shake up peak which broadened in the AmPS samples(Fig. 9b and c) also conrms the substitution happening to thebenzene ring of PS.
A quantitative peak analysis was performed to understandthe concentrations of elements present in PS pellet, NMCA andAmPS bers (70 : 30 and 100 : 0) aer the specied reactiontimes and are summarized in Table 2. Aer amidomethylation
This journal is © The Royal Society of Chemistry 2014

Paper RSC Advances
reaction, the presence of elements such as nitrogen, oxygen andchlorine was observed in both types of AmPS samples (roughand smooth). However, we observed more adsorption of thechloroacetamidomethyl group in rough PS bers (70 : 30)than their smooth counterpart (100 : 0) in terms of theiratomic wt% of elements adsorbed aer the reaction. As thereaction time was increased from 1 to 2 hours, nitrogen,oxygen and chlorine contents were found to be increasedfrom 9.08, 19.40, and 9.07 for 1 hour to 11.03, 21.71, and 9.53for 2 hours of reaction, respectively. Further, as the numberof nitrogen atoms is equal to the chloroacetamidomethylgroup introduced during the reaction, the degree of substi-tution on PS bers was calculated using the percentagenitrogen content from XPS data using the formula DS ¼ (nM)/(1401 � 105.5n),9,10 where n is the percentage nitrogencontent; M is the molecular weight of intact polystyrene.Rough PS (70 : 30) bers have been observed to be moresubstituted with chloroacetamidomethyl group than smoothPS (100 : 0) ber samples aer one (DS ¼ 2.13) and two (DS ¼4.83) hours of reaction (Table 2). This can be attributed tohigher specic surface area, large pore volume and increasedsurface roughness for PS bers (70 : 30) as compared to thesmooth PS bers spun with only DMF as solvent. A schematicillustrating amidomethylation reaction of PS bers is shownin Fig. 11.
Fig. 11 Schematic illustrating amidomethylation reaction on PS(100 : 0) and (70 : 30) fibres.
3.6. Wettability analysis of amidomethylated PS submicronbers with different surface morphology
As-spun PS smooth bers (100 : 0) show hydrophobic naturewith WCA of 124 � 6� (Fig. 12A1) whereas rough PS bers(70 : 30) show WCA of 140 � 4� (Fig. 12B1) approachingtowards superhydrophobicity. The results obtained can becorrelated to the ndings of Zheng et al.35 and Ma et al.36
regarding electrospinning of PS bers in DMF and THF. Thesignicant change in WCA values of PS bers may beattributed to extra roughness generated by ber surfacemorphology as compared to smooth thin lms.37–39 Cassieand Baxter's ndings corroborates that the heterogeneoussurfaces enable air entrapment leading to superhydrophobicbehaviour.39 Aer the amidomethylation reaction thesmooth PS bers show a slow decrease in the contact anglestarting from 116 � 4� to 109 � 2� aer 1 minute of obser-vation (Fig. 12A2). Whereas a rapid decrease in the contact
Table 2 Estimation of degree of substitution (DS) from XPS elementalNMCA
S. no. Sample name
A
C
1 Polystyrene pellet 12 NMCA3 AmPS bers (70 : 30) Aer 1 hour of reaction4 Aer 2 hours of reaction5 AmPS bers (100 : 0) Aer 1 hour of reaction6 Aer 2 hours of reaction
This journal is © The Royal Society of Chemistry 2014
angle was observed for rough PS bers from an initial valueof 66� to 15� within a minute of placing the water drop(Fig. 12B2). This rapid decrease in contact angle in rough PSbers can be attributed to their high porosity which enabledthe coverage of amidomethyl groups (which have the pres-ence of nitrogen, oxygen and chlorine responsible for H-bonding) over a greater surface area making their surfaceextremely hydrophilic. Further, it may be due to the reactioninside the pores, which is supported by swelling that hasoccurred during amidomethylation. The amidomethylationreaction occurring inside the pores of the rough PS bers assupported by their swelling behavior may also be attributedto the observed rapid decrease in contact angle. It is illus-trated with these results that by controlling the surfacemorphology from thin lms to smooth to rough bers,wettability of electrospun PS submicron bers may becontrolled over a wide range of contact angles from hydro-philic to nearly superhydrophobic. Further chemical modi-cation (amidomethylation in this case) of these PSelectrospun bers makes them extremely hydrophilic. Thiscontrolled evolution of PS electrospun submicron bersfrom nearly superhydrophobic to extremely hydrophilicshows a polynomial trend line of order three (y ¼ �1.77x3 +12.94x2 � 39.72x + 167.67), as shown in Fig. 13. This broadrange of wettability characteristics opens a wide variety ofpotential applications for these electrospun AmPS submi-cron bers. Using these fabrics as bio-sensing devices byimmobilizing various biomolecules on them is currentlyunder investigation in our group.
analysis after amidomethylation reaction of PS samples with 1 wt% of
tomic weight percentage of elements present (%)DS (%) ¼ (nM)/(1401 � 105.5n)1(s) O 1(s) N 1(s) Cl 2(p)
00 — — — —56.04 31.65 7.97 4.33 —62.45 19.40 9.08 9.07 2.3157.73 21.71 11.03 9.53 4.8365.13 17.37 8.82 8.68 1.9463.09 21.72 7.65 7.54 1.33
RSC Adv., 2014, 4, 12188–12197 | 12195
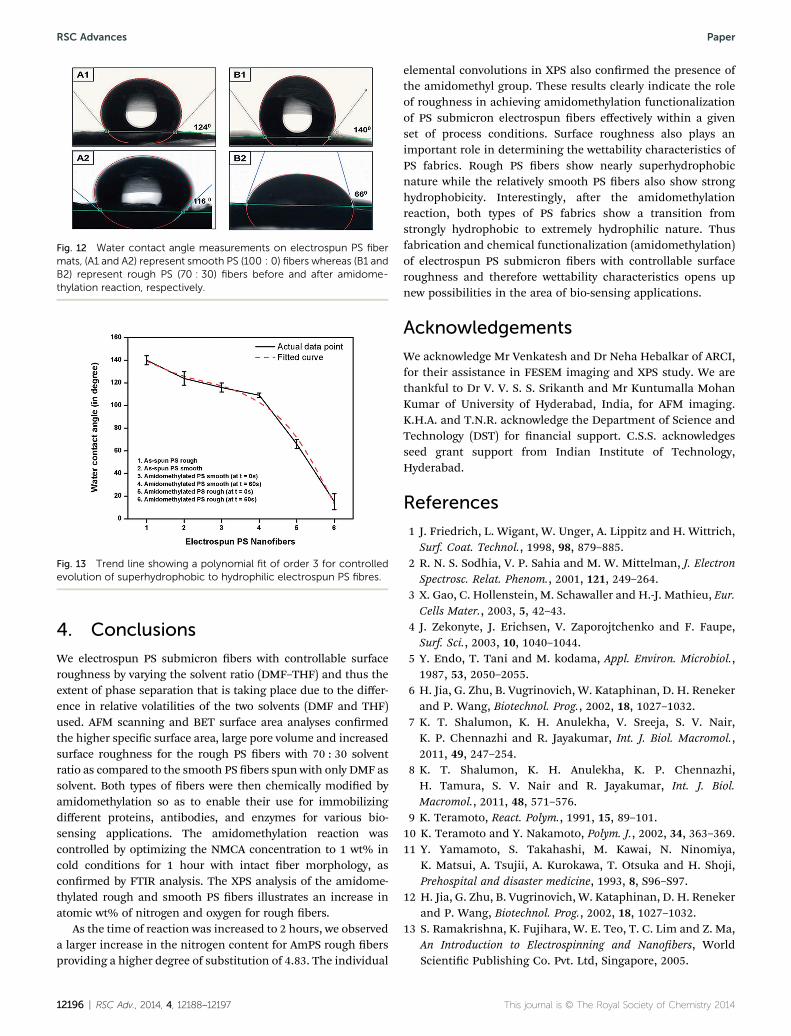
Fig. 12 Water contact angle measurements on electrospun PS fibermats, (A1 and A2) represent smooth PS (100 : 0) fibers whereas (B1 andB2) represent rough PS (70 : 30) fibers before and after amidome-thylation reaction, respectively.
Fig. 13 Trend line showing a polynomial fit of order 3 for controlledevolution of superhydrophobic to hydrophilic electrospun PS fibres.
RSC Advances Paper
4. Conclusions
We electrospun PS submicron bers with controllable surfaceroughness by varying the solvent ratio (DMF–THF) and thus theextent of phase separation that is taking place due to the differ-ence in relative volatilities of the two solvents (DMF and THF)used. AFM scanning and BET surface area analyses conrmedthe higher specic surface area, large pore volume and increasedsurface roughness for the rough PS bers with 70 : 30 solventratio as compared to the smooth PS bers spun with only DMF assolvent. Both types of bers were then chemically modied byamidomethylation so as to enable their use for immobilizingdifferent proteins, antibodies, and enzymes for various bio-sensing applications. The amidomethylation reaction wascontrolled by optimizing the NMCA concentration to 1 wt% incold conditions for 1 hour with intact ber morphology, asconrmed by FTIR analysis. The XPS analysis of the amidome-thylated rough and smooth PS bers illustrates an increase inatomic wt% of nitrogen and oxygen for rough bers.
As the time of reaction was increased to 2 hours, we observeda larger increase in the nitrogen content for AmPS rough bersproviding a higher degree of substitution of 4.83. The individual
12196 | RSC Adv., 2014, 4, 12188–12197
elemental convolutions in XPS also conrmed the presence ofthe amidomethyl group. These results clearly indicate the roleof roughness in achieving amidomethylation functionalizationof PS submicron electrospun bers effectively within a givenset of process conditions. Surface roughness also plays animportant role in determining the wettability characteristics ofPS fabrics. Rough PS bers show nearly superhydrophobicnature while the relatively smooth PS bers also show stronghydrophobicity. Interestingly, aer the amidomethylationreaction, both types of PS fabrics show a transition fromstrongly hydrophobic to extremely hydrophilic nature. Thusfabrication and chemical functionalization (amidomethylation)of electrospun PS submicron bers with controllable surfaceroughness and therefore wettability characteristics opens upnew possibilities in the area of bio-sensing applications.
Acknowledgements
We acknowledge Mr Venkatesh and Dr Neha Hebalkar of ARCI,for their assistance in FESEM imaging and XPS study. We arethankful to Dr V. V. S. S. Srikanth and Mr Kuntumalla MohanKumar of University of Hyderabad, India, for AFM imaging.K.H.A. and T.N.R. acknowledge the Department of Science andTechnology (DST) for nancial support. C.S.S. acknowledgesseed grant support from Indian Institute of Technology,Hyderabad.
References
1 J. Friedrich, L. Wigant, W. Unger, A. Lippitz and H. Wittrich,Surf. Coat. Technol., 1998, 98, 879–885.
2 R. N. S. Sodhia, V. P. Sahia and M. W. Mittelman, J. ElectronSpectrosc. Relat. Phenom., 2001, 121, 249–264.
3 X. Gao, C. Hollenstein, M. Schawaller and H.-J. Mathieu, Eur.Cells Mater., 2003, 5, 42–43.
4 J. Zekonyte, J. Erichsen, V. Zaporojtchenko and F. Faupe,Surf. Sci., 2003, 10, 1040–1044.
5 Y. Endo, T. Tani and M. kodama, Appl. Environ. Microbiol.,1987, 53, 2050–2055.
6 H. Jia, G. Zhu, B. Vugrinovich, W. Kataphinan, D. H. Renekerand P. Wang, Biotechnol. Prog., 2002, 18, 1027–1032.
7 K. T. Shalumon, K. H. Anulekha, V. Sreeja, S. V. Nair,K. P. Chennazhi and R. Jayakumar, Int. J. Biol. Macromol.,2011, 49, 247–254.
8 K. T. Shalumon, K. H. Anulekha, K. P. Chennazhi,H. Tamura, S. V. Nair and R. Jayakumar, Int. J. Biol.Macromol., 2011, 48, 571–576.
9 K. Teramoto, React. Polym., 1991, 15, 89–101.10 K. Teramoto and Y. Nakamoto, Polym. J., 2002, 34, 363–369.11 Y. Yamamoto, S. Takahashi, M. Kawai, N. Ninomiya,
K. Matsui, A. Tsujii, A. Kurokawa, T. Otsuka and H. Shoji,Prehospital and disaster medicine, 1993, 8, S96–S97.
12 H. Jia, G. Zhu, B. Vugrinovich, W. Kataphinan, D. H. Renekerand P. Wang, Biotechnol. Prog., 2002, 18, 1027–1032.
13 S. Ramakrishna, K. Fujihara, W. E. Teo, T. C. Lim and Z. Ma,An Introduction to Electrospinning and Nanobers, WorldScientic Publishing Co. Pvt. Ltd, Singapore, 2005.
This journal is © The Royal Society of Chemistry 2014

Paper RSC Advances
14 J. Doshi and D. H. Reneker, J. Electrost., 1995, 35, 151–160.
15 D. H. Reneker and I. Chun, Nanotechnology, 1996, 7, 216.16 D. H. Reneker, A. L. Yarin, E. Zussman and H. Xu, Adv. Appl.
Mech., 2007, 41, 44.17 D. H. Reneker and A. L. Yarin, Polymer, 2008, 49, 2387–2425.18 T. Jarusuwannapoom, W. Hongrojjanawiwat, S. Jitjaicham,
L. Wannatong, M. Nithitanakul, C. Pattamaprom,P. Koombhongse, R. Rangkupan and P. Supaphol, Eur.Polym. J., 2005, 41, 409–421.
19 X. Yu, H. Xiang, Y. Long, N. Zhao, X. Zhang and J. Xu,Mater.Lett., 2010, 64, 2407–2409.
20 L. Huang, N.-N. Bui, S. M. Seetha and R. McCutcheon Jeffrey,J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 1734–1744.
21 C. J. Luo, M. Nangrejo andM. Edirisinghe, Polymer, 2010, 51,1654–1662.
22 W. Zhong, F. Li, L. Chen, Y. Chen and Y. Wei, J. Mater.Chem.,2012, 22, 5523–5530.
23 C. L. Casper, J. S. Stephens, N. G. Tassi, D. B. Chase andJ. F. Rabolt, Macromolecules, 2004, 37, 573–578.
24 J. Lin, B. Ding, J. Yu and Y. Hsieh, ACS Appl. Mater. Interfaces,2010, 2, 521–528.
25 J. Lin, B. Ding, J. Yang, J. Yu and S. Gang, Nanoscale, 2012, 4,176–182.
26 B. Ding, J. Lin, X. Wang, J. Yu, J. Yang and Y. Cai, SoMatter,2011, 7, 8376–8383.
This journal is © The Royal Society of Chemistry 2014
27 K. Nayani, H. Katepalli, C. S. Sharma, A. Sharma, S. Patil andR. Venkataraghavan, Ind. Eng. Chem. Res., 2012, 51, 1761–1766.
28 M. M. Demir, eXPRESS Polym. Lett., 2010, 4, 2–8.29 C.-L. Pai, M. C. Boyce and G. C. Rutledge, Macromolecules,
2009, 42, 2102–2114.30 T. Nitanan, O. Praneet, A. Prasert, R. Theerasak, N. Tanasait
and S. Pitt, Korean J. Chem. Eng., 2012, 29, 173–181.31 T. Uyar and F. Besenbacher, Polymer, 2008, 49, 5336–5343.32 Y. Z. Zhang, Y. Feng, Z.-M. Huang, S. Ramakrishna and
C. T. Lim, Nanotechnology, 2006, 17, 901–908.33 M. M. Browne, G. V. Lubarsky, M. R. Davidson and
R. H. Bradley, Surf. Sci., 2004, 553, 155–167.34 M. Mohorc, I. Jerman, M. Zorko, L. Butinar, B. Orel, R. Jerala
and J. Friedrich, J. Mater. Sci.: Mater. Med., 2010, 21, 2775–2782.
35 J. Zheng, A. He, J. Li, J. Xu and C. C. Han, Polymer, 2006, 47,7095–7102.
36 M. Ma, R. M. Hill, J. L. Lowery, S. V. Fridrikh andG. C. Rutledge, Langmuir, 2005, 21, 5549–5554.
37 M. Ma, M. Gupta, Z. Li, L. Zhai, G. K. Karen, C. E. Robert,M. F. Rubner and G. C. Rutledge, Adv. Mater., 2007, 19,255–259.
38 Y. Li, J. Q. Pham, K. P. Johnston and P. F. Green, Langmuir,2007, 23, 9785–9793.
39 A. B. D. Cassie and S. Baxter, Trans. Faraday Soc., 1944, 40,546.
RSC Adv., 2014, 4, 12188–12197 | 12197





























