Embed Size (px)
Citation preview

(324), ra42. [DOI: 10.1126/scisignal.2005049] 7Science SignalingKallakury, Jeffrey Toretsky, Anton Wellstein and Chunling Yi (6 May 2014) Wadhwa, Mahandranauth Chetram, Mrinmayi Joshi, Fen Wang, BhaskarGarrett Graham, Swati Gupta, Eveline E. Vietsch, Sean Z. Laughlin, Mandheer Weiying Zhang, Nivedita Nandakumar, Yuhao Shi, Mark Manzano, Alias Smith,for Neoplastic Progression to Pancreatic Ductal Adenocarcinoma
Downstream of Mutant KRAS, the Transcription Regulator YAP Is Essential`
This information is current as of 6 May 2014. The following resources related to this article are available online at http://stke.sciencemag.org.
Article Tools http://stke.sciencemag.org/cgi/content/full/sigtrans;7/324/ra42
Visit the online version of this article to access the personalization and article tools:
MaterialsSupplemental
http://stke.sciencemag.org/cgi/content/full/sigtrans;7/324/ra42/DC1 "Supplementary Materials"
Related Content
http://stke.sciencemag.org/cgi/content/abstract/sigtrans;6/291/ra77 http://stke.sciencemag.org/cgi/content/abstract/sigtrans;6/292/ra81 http://stke.sciencemag.org/cgi/content/abstract/sigtrans;6/302/rs15 http://stke.sciencemag.org/cgi/content/abstract/sigtrans;6/302/pe36
's sites:ScienceThe editors suggest related resources on
References http://stke.sciencemag.org/cgi/content/full/sigtrans;7/324/ra42#BIBL
1 article(s) hosted by HighWire Press; see: cited byThis article has been
http://stke.sciencemag.org/cgi/content/full/sigtrans;7/324/ra42#otherarticlesThis article cites 129 articles, 46 of which can be accessed for free:
Glossary http://stke.sciencemag.org/glossary/
Look up definitions for abbreviations and terms found in this article:
Permissions http://www.sciencemag.org/about/permissions.dtl
Obtain information about reproducing this article:
the American Association for the Advancement of Science; all rights reserved. byAssociation for the Advancement of Science, 1200 New York Avenue, NW, Washington, DC 20005. Copyright 2008
(ISSN 1937-9145) is published weekly, except the last week in December, by the AmericanScience Signaling
on May 6, 2014
stke.sciencemag.org
Dow
nloaded from
on May 6, 2014
stke.sciencemag.org
Dow
nloaded from

R E S E A R C H A R T I C L E
C A N C E R
Downstream of Mutant KRAS, the TranscriptionRegulator YAP Is Essential for NeoplasticProgression to Pancreatic Ductal AdenocarcinomaWeiying Zhang, Nivedita Nandakumar, Yuhao Shi, Mark Manzano, Alias Smith,Garrett Graham, Swati Gupta, Eveline E. Vietsch, Sean Z. Laughlin, Mandheer Wadhwa,Mahandranauth Chetram, Mrinmayi Joshi, Fen Wang, Bhaskar Kallakury, Jeffrey Toretsky,Anton Wellstein, Chunling Yi*
stke.sciD
ownloaded from
Pancreatic ductal adenocarcinoma (PDAC) is an aggressive cancer with poor survival rates and frequentlycarries oncogenic KRAS mutation. However, KRAS has thus far not been a viable therapeutic target. Wefound that the abundance of YAPmRNA, which encodes Yes-associated protein (YAP), a protein regulatedby the Hippo pathway during tissue development and homeostasis, was increased in human PDAC tissuecompared with that in normal pancreatic epithelia. In genetically engineered KrasG12D and KrasG12D:Trp53R172H mouse models, pancreas-specific deletion of Yap halted the progression of early neoplastic le-sions toPDACwithout affecting normal pancreatic development and endocrine function. AlthoughYapwasdispensable for acinar to ductal metaplasia (ADM), an initial step in the progression to PDAC, Yap was crit-ically required for the proliferation ofmutantKrasorKras:Trp53neoplastic pancreatic ductal cells in cultureand for their growth and progression to invasive PDAC in mice. Yap functioned as a critical transcriptionalswitch downstream of the oncogenic KRAS–mitogen-activated protein kinase (MAPK) pathway, promotingthe expression of genes encoding secretory factors that cumulatively sustained neoplastic proliferation, atumorigenic stromal response in the tumor microenvironment, and PDAC progression in Kras and Kras:Trp53mutant pancreas tissue. Together, our findings identified Yap as a critical oncogenic KRAS effectorand a promising therapeutic target for PDAC and possibly other types of KRAS-mutant cancers.
e
on May 6, 2014 ncem
ag.org
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is one of the most lethal cancerswith a 5-year survival rate of less than 5% (1). PDAC tumors are thought tooriginate from mature acinar cells that transdifferentiate into ductal-likecells, a process known as acinar to ductal metaplasia (ADM) (2, 3).ADM can be induced by pancreatitis or oncogenic mutations. Only undersustained oncogenic insult will pancreatic cells that have undergone ADMprogress through a series of histopathological stages called pancreatic in-traepithelial neoplasias (PanINs) to invasive PDAC.
Nearly all PDAC and a high percentage of early PanIN lesions haveKRAS mutations (4). The human ADM-to-PanIN-to-PDAC progressionhas been recapitulated using genetically engineered mouse models(GEMMs) in which endogenous expression of oncogenic Kras is inducedin the developing pancreas, either alone or in conjunction with the in-activation of other commonly mutated tumor suppressor genes, such asp16, Trp53, or Smad4 (5–8). When oncogenic Kras is activated alone,ADM and early PanINs readily develop, but progression into late-stagePanINs and eventually PDAC is delayed (8). This process is substantiallyaccelerated by mutation of p16, Trp53, or Smad4, suggesting that the pro-teins encoded by these genes inhibit the proliferative signals mediated bymutant Kras (5–7).
Despite recent progress, oncogenic KRAS itself remains an extremelychallenging therapeutic target (9, 10). Thus, much effort is devoted to iden-tifying critical downstream effectors of oncogenic KRAS. Because the de-velopment and progression of PDAC are strongly influenced by themicroenvironment, GEMMs represent the most physiologically relevant
Lombardi Comprehensive Cancer Center, Georgetown University MedicalCenter, Washington, DC 20057, USA.*Corresponding author. E-mail: [email protected]
models. In the past few years, the aforementioned GEMMs have beencrossedwithmice harboring various loss-of-functionmutant alleles, leadingto the identification of genes required for the initiation (EGFR, ADM17,PDK1, Gli1, and Sox9) or progression (Rac1, STAT3, IL-6, NEMO, andIKK2) of PDAC in the context of Krasmutation, either alone or concurrentwith p16 deletion (11–22). However, introduction of Trp53 mutations intothese models (which mimic the TP53 mutations found in the majority ofhuman PDAC) mitigated the requirement for many of these genes duringPDAC initiation and progression (11, 17, 19, 20). In accord with these find-ings in GEMMs, clinical trials of inhibitors targeting the epidermal growthfactor receptor (EGFR), the RAF–mitogen-activated protein kinase(MAPK) pathway, phosphoinositide 3-kinase, or the Hedgehog pathwayhave been largely unsuccessful (23). Thus, there remains an urgent needto discover the “Achilles’ heel” that governs PDACdevelopment in the pres-ence of KRAS:TP53 mutations.
The Yes-associated protein (YAP), encoded by YAP1, referred to hereinsimply asYap, is a bona fide oncoprotein, and its abundance and activity arefrequently increased in many types of cancers (24–36). YAP is a transcrip-tional coactivator that partners with the TEAD family of transcriptionfactors to promote the expression of pro-proliferative and antiapoptoticgenes (37–46). Extensive genetic studies that focused primarily on organsize control in Drosophila and mice identified that the Hippo pathway isthe canonical regulator of YAP activity (39, 47–50). The Hippo pathwayis composed of a kinase cascade in which the MST1 and MST2 (MST1/2)Hippo kinases are facilitated by scaffold protein WW45 to phosphorylatetheLATS1 andLATS2 (LATS1/2) kinases and their adaptor protein,MOB1(43, 45, 51–53). Phosphorylated LATS1/2 kinases in turn phosphorylateYAP, inactivating YAP by causing it to be retained in the cytoplasm anddegraded (54, 55). A host of factors [including cell density, extracellularmatrix stiffness, G protein (heterotrimeric guanine nucleotide–binding
www.SCIENCESIGNALING.org 6 May 2014 Vol 7 Issue 324 ra42 1

R E S E A R C H A R T I C L E
Dow
n
protein)–coupled receptors, protease-activated receptors, EGFR, andleukemia inhibitory factor receptor] influence YAP activity by modulat-ing the Hippo pathway (56–60). Additionally, accumulating evidenceindicates that YAP activity can be regulated by noncanonical, Hippo-independent mechanisms (61–67).
GEMMs developed in recent years have revealed critical functions ofYAP and the Hippo pathway in the maintenance of tissue homeostasis,the organ size checkpoint, and tumorigenesis. Tissue-specific overexpres-sion of Yap or inactivation of Hippo signaling through the homozygous de-letion ofMst1/2 or genes encoding other components of the Hippo pathwayresulted in enlargement of the liver, heart, and intestine, as well as tumori-genesis in the liver (47, 49, 68, 69). In contrast, deletion ofMst1/2 or ectopicexpression of Yap in the developingmouse pancreas induces ADM and im-pairs differentiation of exocrine and endocrine compartments withoutincreasing the size of the pancreas or inducing pancreatic tumor develop-ment (70, 71). These studies demonstrate that activation of YAP alone isinsufficient to induce PDAC, but have not determined whether YAP is nec-essary for PDAC development. Here, we examined YAP abundance inprimary humanPDAC, its role inmutantKRAS– andKRAS:TP53-mediatedPDAC initiation and progression, and the molecular mechanismsunderlying oncogenic RAS–YAP crosstalk.
on May 6, 2014
stke.sciencemag.org
loaded from
RESULTS
YAP is expressed in normal and neoplasticpancreatic ductal cellsYAP abundance is increased in human PDAC (72, 73). Through a meta-analysis of published human PDAC microarray data sets (74–77), we con-firmed that overall YAP mRNA abundance was significantly increased inhuman PDAC when compared to normal pancreatic tissues (fig. S1A). Forreference, the genotypes of all GEMMs used in this study are listed in table S1.The KrasG12D/+:p48-Cre (KC) and KrasG12D/+:Trp53R172H/+:p48-Cre (KPC)GEMMs in which p48-Cre (also known as Ptf1a-Cre) induces the expres-sion of mutant Kras alone or together with mutant Trp53 in murine pancre-atic epithelium fully recapitulate the pathogenesis of human PDAC and aregenerally regarded as two of the best GEMMs for human PDAC (23). ByWestern blot analysis, we found that Yap protein abundancewas also mark-edly greater in pancreatic tissue from KPC mice that had developed PDACcompared with that from wild-type mice (fig. S1B).
To explore this association further, we performed immunohisto-chemistry (IHC) analysis for YAP on two human tissue microarrays(TMAs) containing 31 normal pancreata, 64 chronic pancreatitis or tumoradjacent tissues, and 140 PDAC tumor cores, as well as pancreatic sectionsfrom multiple wild-type, KC, and KPC mice (figs. S1, C to I, and S2A).Consistent with previous reports (72, 73), we found that YAP abundancewas restricted to ductal and terminal-duct centroacinar cells in normal hu-man and mouse pancreas tissue (figs. S1D and S2A and table S2). Variableamounts of cytoplasmic or nuclear YAP were also detected in a significantfraction of neoplastic ductal epithelial cells and stromal cells in almost all ofthe PDAC tumors and adjacent tissues containing benign lesions and chron-ic pancreatitis (fig. S1, C and E to G, and table S2). Similar patterns of Yapstaining were observed in pancreata from KC and KPC mice that had earlyPanINs or fully established PDAC (fig. S1, H and I). Although a fraction ofhuman PDAC tumors exhibited intense staining for YAP (particularly inlate-stage tumors), the majority of human PDAC tumors appeared to havean abundance of YAP within individual tumor cells that was comparable tothat in normal centroacinar and ductal cells (fig. S1, C to G, and table S2).Thus, the overall increase in YAP abundance in PDACmeasured by micro-array and Western blot analyses (fig. S1, A and B) likely resulted from the
expansion of neoplastic ductal and stromal cell populations, rather thanfrom a net increase in YAP transcript and abundance in individual PDACcells.
Yap is dispensable for normal pancreatic developmentand glucose metabolismBecause YAP is present in normal centroacinar and ductal cells, we inves-tigated whether the deletion of Yap might affect the normal development orfunction of the pancreas by generating Yap flox/flox:p48-Cre (YC) mice. YCmice were born at the Mendelian ratio and had normal weight, physical ap-pearance, and life expectancy. IHC analysis confirmed the complete loss ofYap expression in the pancreatic epithelium of YCmice (fig. S2A). Despitethe absence of Yap, YC pancreata were indistinguishable from wild-typepancreata in overall histology by hematoxylin and eosin (H&E), stainingpatterns for major pancreatic cell lineage markers [including amylase (inacinar cells), cytokeratin 19 (CK19; in ductal cells), insulin (in b cellswithinthe islets), and glucagon (in a cells within the islets)], or organization ofductal networks (fig. S2, B and C). A glucose tolerance test also revealedno apparent difference between YC and wild-type mice in their ability tomodulate blood glucose levels (fig. S2D). Collectively, these data suggestthat YAP deletion from the pancreatic epithelium is inconsequential tonormal pancreatic development, homeostasis, and glucose metabolism.
Inactivation of Yap blocks PDAC development in Krasand Kras:Trp53 compound mutant pancreasTo determine whether Yap is involved in PDAC oncogenesis, we crossedYap flox/flox with KC and KPC mice to generate KrasG12D/+:Yap flox/+:p48-Cre,KrasG12D/+:p53R172H/+:Yap flox/+:p48-Cre, KrasG12D/+:Yap flox/flox:p48-Cre(KYC), and KrasG12D/+:p53R172H/+:Yap flox/flox:p48-Cre (KPYC) mice.
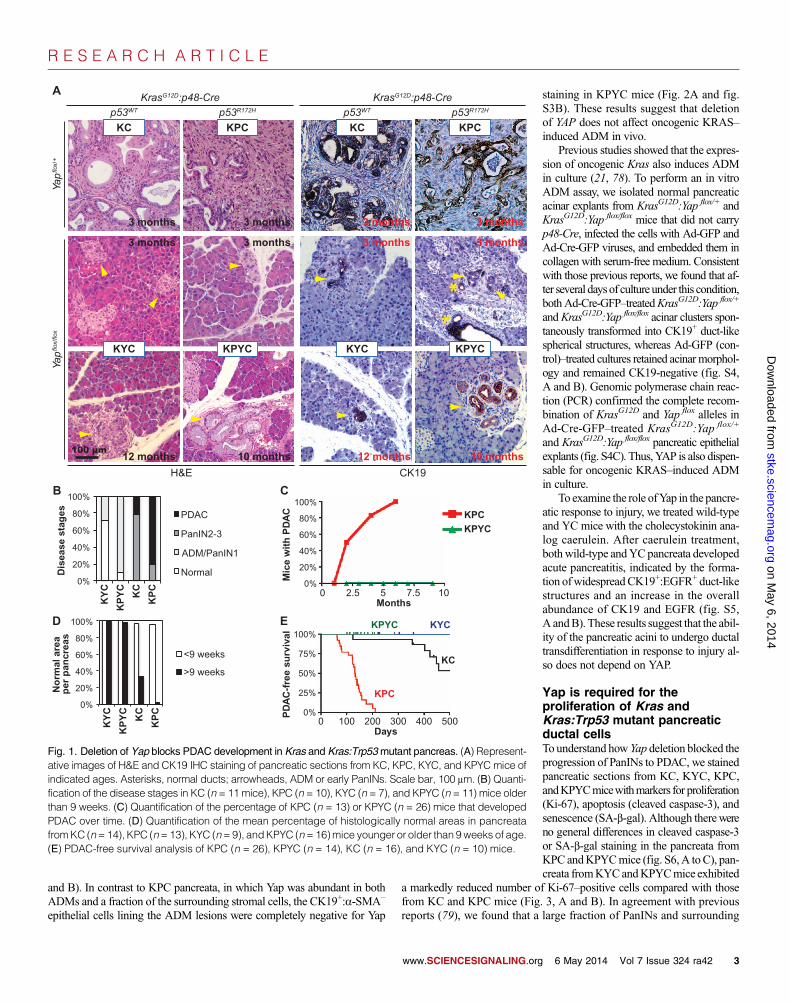
KC (includingKrasG12D/+:Yap+/+:p48-Cre andKrasG12D/+:Yap flox/+:p48-Cre)or KPC (including KrasG12D/+:p53R172H/+:Yap+/+:p48-Cre and KrasG12D/+:p53R172H/+:Yap flox/+:p48-Cre) mice with one or two intact Yap alleles devel-opedADMand early PanINs from4 to 8weeks of age, respectively (table S1and fig. S3). These ADM and early PanINs progressed through late-stagePanINs and eventually to invasive PDAC by 2 to 4 months in KPCmice, orfrom 6 months to 2 years in KC mice (Fig. 1, A to C, and fig. S3). In con-trast, all the KYC and KPYC mice with homozygous Yap deletions com-pletely lacked any late-stage PanINs or PDAC when analyzed at all thetested time points, even at the age of 12months (for KCmice) or 10months(for KPC mice) (Fig. 1, A to C, and fig. S3).
AlthoughADMand some PanIN-1 lesions that were CK19-positive andYap-negative (CK19+:Yap−) still developed in KYC and KPYC pancreata(Fig. 1, A and B, and fig. S3, A and B), nearly all pancreatic tissue areas inthesemicewere histologically normal across all age groups (Fig. 1,A andD,and fig. S3A). In contrast, from the age of 9 weeks, the number of lesions atvarious stages of progression increased in the pancreata from KC and KPCmice (Fig. 1, A and D, and fig. S3A).
Whereas nearly all KPC and half of KC mice died from PDAC by 6months or 1.5 years of age, respectively, not a single KPYC or KYCmousein our experimental cohort succumbed to PDAC within the same periods(Fig. 1E). These results clearly demonstrate that YAP is essential in mutantKRAS– and KRAS:TP53-mediated PDAC development.
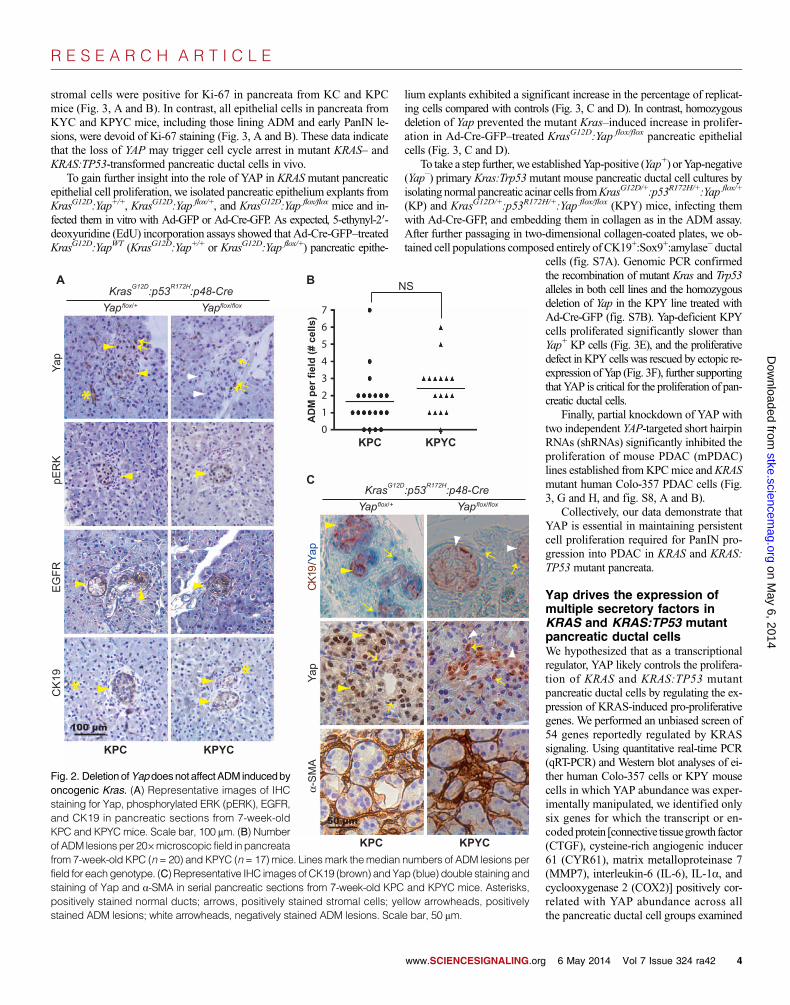
Deletion of Yap does not affect ADM induced byoncogenic Kras or pancreatitisADM caused by KRAS mutation or pancreatic injury is thought to be theinitiating step in PDAC development. Examination of pancreatic tissuesfrom young KPC and KPYC mice (4 to 8 weeks old) showed that pERK+
(phosphorylated extracellular signal–regulated kinase):EGFR+:CK19+
ADM initiated at a similar rate in KPC and KPYC littermates (Fig. 2, A
www.SCIENCESIGNALING.org 6 May 2014 Vol 7 Issue 324 ra42 2
R E S E A R C H A R T I C L E
on May 6, 2014
stke.sciencemag.org
Dow
nloaded from
and B). In contrast to KPC pancreata, in which Yap was abundant in bothADMs and a fraction of the surrounding stromal cells, the CK19+:a-SMA−
epithelial cells lining the ADM lesions were completely negative for Yap
www.SCIENCESIGNALING.org
staining in KPYC mice (Fig. 2A and fig.S3B). These results suggest that deletionof YAP does not affect oncogenic KRAS–induced ADM in vivo.
Previous studies showed that the expres-sion of oncogenic Kras also induces ADMin culture (21, 78). To perform an in vitroADM assay, we isolated normal pancreaticacinar explants from KrasG12D:Yap flox/+ andKrasG12D:Yap flox/flox mice that did not carryp48-Cre, infected the cells with Ad-GFP andAd-Cre-GFP viruses, and embedded them incollagen with serum-free medium. Consistentwith those previous reports, we found that af-ter several daysof culture under this condition,bothAd-Cre-GFP–treatedKrasG12D:Yap flox/+
andKrasG12D:Yap flox/flox acinar clusters spon-taneously transformed into CK19+ duct-likespherical structures, whereas Ad-GFP (con-trol)–treated cultures retained acinar morphol-ogy and remained CK19-negative (fig. S4,A and B). Genomic polymerase chain reac-tion (PCR) confirmed the complete recom-bination of KrasG12D and Yap flox alleles inAd-Cre-GFP–treated KrasG12D:Yap flox/+
and KrasG12D:Yap flox/flox pancreatic epithelialexplants (fig. S4C). Thus,YAP is also dispen-sable for oncogenic KRAS–induced ADMin culture.
To examine the role ofYap in the pancre-atic response to injury, we treated wild-typeand YC mice with the cholecystokinin ana-log caerulein. After caerulein treatment,bothwild-type andYCpancreata developedacute pancreatitis, indicated by the forma-tion ofwidespreadCK19+:EGFR+ duct-likestructures and an increase in the overallabundance of CK19 and EGFR (fig. S5,A andB). These results suggest that the abil-ity of the pancreatic acini to undergo ductaltransdifferentiation in response to injury al-so does not depend on YAP.
Yap is required for theproliferation of Kras andKras:Trp53 mutant pancreaticductal cellsTounderstand howYap deletion blocked theprogression of PanINs to PDAC, we stainedpancreatic sections from KC, KYC, KPC,andKPYCmicewithmarkers for proliferation(Ki-67), apoptosis (cleaved caspase-3), andsenescence (SA-b-gal). Although therewereno general differences in cleaved caspase-3or SA-b-gal staining in the pancreata fromKPCandKPYCmice (fig. S6,A toC), pan-creata fromKYCandKPYCmice exhibited
a markedly reduced number of Ki-67–positive cells compared with thosefrom KC and KPC mice (Fig. 3, A and B). In agreement with previousreports (79), we found that a large fraction of PanINs and surrounding
KP
YC
3 months 3 months
3 months 3 months
12 months 10 months
Yap
flox/
flox
Yap
flox/
+
KrasG12D:p48-Crep53R172Hp53WT
H&E
KrasG12D:p48-Crep53R172Hp53WT
3 months 3 months
3 months 3 months
CK19
10 months12 months
A
*
*
KPC
KYC
KC
KPYC
0%
25%
50%
75%
100%
PD
AC
-fre
e su
rviv
al
0 100 200 300 400 500Days
E
C
Mic
e w
ith
PD
AC
100%
80%
60%
40%
20%
0%
KPCKPYC
Months0 2.5 5 7.5 10
D
No
rmal
are
a p
er p
ancr
eas
B
Dis
ease
sta
ges
KC
KY
C
KP
C
KC KCKPC KPC
KYC KYCKPYC KPYC
0%
20%
40%
60%
80%
100%
KY
C
KP
YC
KC
KP
C
PDAC
PanIN2-3
ADM/PanIN1
Normal
0%
20%
40%
60%
80%
100%
<9 weeks
>9 weeks
Fig. 1. Deletion of Yap blocks PDAC development in Kras and Kras:Trp53mutant pancreas. (A) Represent-
ative images of H&E and CK19 IHC staining of pancreatic sections from KC, KPC, KYC, and KPYC mice ofindicated ages. Asterisks, normal ducts; arrowheads, ADM or early PanINs. Scale bar, 100 mm. (B) Quanti-fication of the disease stages in KC (n = 11mice), KPC (n = 10), KYC (n = 7), and KPYC (n = 11) mice olderthan 9 weeks. (C) Quantification of the percentage of KPC (n = 13) or KPYC (n = 26) mice that developedPDAC over time. (D) Quantification of the mean percentage of histologically normal areas in pancreatafromKC (n=14), KPC (n=13), KYC (n=9), andKPYC (n=16)mice younger or older than 9weeks of age.(E) PDAC-free survival analysis of KPC (n = 26), KPYC (n = 14), KC (n = 16), and KYC (n = 10) mice.6 May 2014 Vol 7 Issue 324 ra42 3
R E S E A R C H A R T I C L E
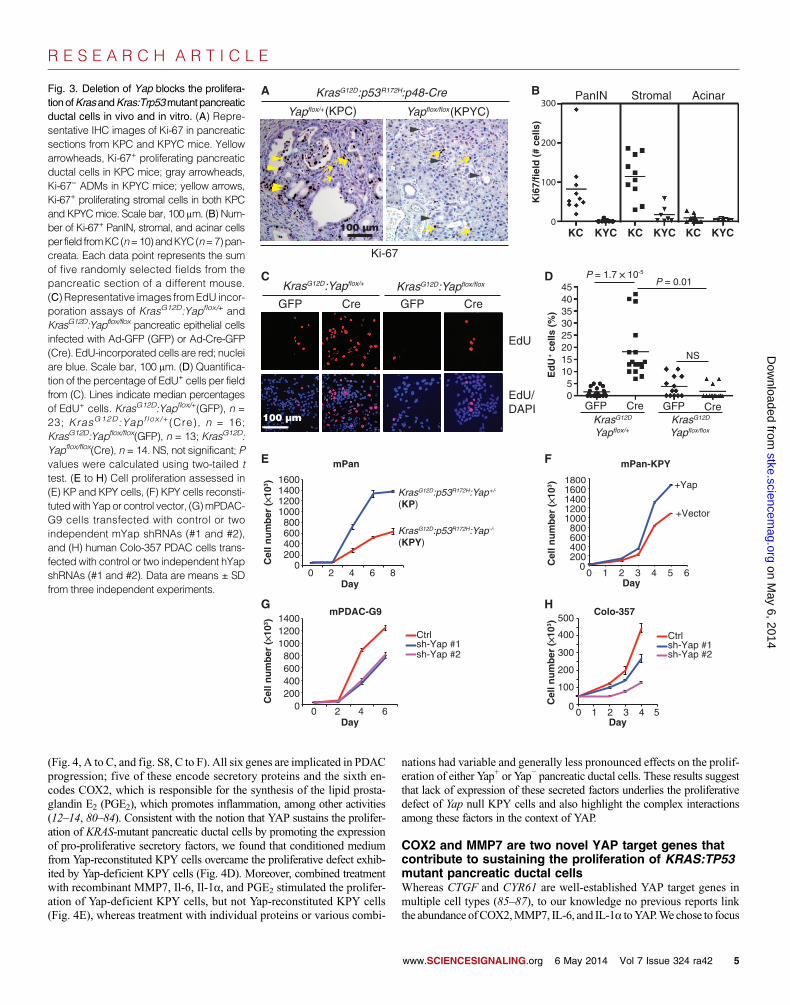
stromal cells were positive for Ki-67 in pancreata from KC and KPCmice (Fig. 3, A and B). In contrast, all epithelial cells in pancreata fromKYC and KPYC mice, including those lining ADM and early PanIN le-sions, were devoid of Ki-67 staining (Fig. 3, A and B). These data indicatethat the loss of YAP may trigger cell cycle arrest in mutant KRAS– andKRAS:TP53-transformed pancreatic ductal cells in vivo.
To gain further insight into the role of YAP in KRASmutant pancreaticepithelial cell proliferation, we isolated pancreatic epithelium explants fromKrasG12D:Yap+/+, KrasG12D:Yap flox/+, and KrasG12D:Yap flox/flox mice and in-fected them in vitro with Ad-GFP or Ad-Cre-GFP. As expected, 5-ethynyl-2′-deoxyuridine (EdU) incorporation assays showed that Ad-Cre-GFP–treatedKrasG12D:YapWT (KrasG12D:Yap+/+ or KrasG12D:Yap flox/+) pancreatic epithe-
lium explants exhibited a significant increase in the percentage of replicat-ing cells compared with controls (Fig. 3, C and D). In contrast, homozygousdeletion of Yap prevented the mutant Kras–induced increase in prolifer-ation in Ad-Cre-GFP–treated KrasG12D:Yap flox/flox pancreatic epithelialcells (Fig. 3, C and D).
To take a step further,we establishedYap-positive (Yap+) orYap-negative(Yap−) primary Kras:Trp53mutant mouse pancreatic ductal cell cultures byisolatingnormal pancreatic acinar cells fromKrasG12D/+:p53R172H/+:Yap flox/+
(KP) and KrasG12D/+:p53R172H/+:Yap flox/flox (KPY) mice, infecting themwith Ad-Cre-GFP, and embedding them in collagen as in the ADM assay.After further passaging in two-dimensional collagen-coated plates, we ob-tained cell populations composed entirely of CK19+:Sox9+:amylase− ductal
www.SCIENCESIGNALING.org
on May 6, 2014
stke.sciencemag.org
Dow
nloaded from
cells (fig. S7A). Genomic PCR confirmedthe recombination of mutant Kras and Trp53alleles in both cell lines and the homozygousdeletion of Yap in the KPY line treated withAd-Cre-GFP (fig. S7B). Yap-deficient KPYcells proliferated significantly slower thanYap+ KP cells (Fig. 3E), and the proliferativedefect inKPY cellswas rescued by ectopic re-expression ofYap (Fig. 3F), further supportingthat YAP is critical for the proliferation of pan-creatic ductal cells.
Finally, partial knockdown of YAP withtwo independent YAP-targeted short hairpinRNAs (shRNAs) significantly inhibited theproliferation of mouse PDAC (mPDAC)lines established fromKPCmice andKRASmutant human Colo-357 PDAC cells (Fig.3, G and H, and fig. S8, A and B).
Collectively, our data demonstrate thatYAP is essential in maintaining persistentcell proliferation required for PanIN pro-gression into PDAC in KRAS and KRAS:TP53 mutant pancreata.
Yap drives the expression ofmultiple secretory factors inKRAS and KRAS:TP53 mutantpancreatic ductal cellsWe hypothesized that as a transcriptionalregulator, YAP likely controls the prolifera-tion of KRAS and KRAS:TP53 mutantpancreatic ductal cells by regulating the ex-pression of KRAS-induced pro-proliferativegenes. We performed an unbiased screen of54 genes reportedly regulated by KRASsignaling. Using quantitative real-time PCR(qRT-PCR) and Western blot analyses of ei-ther human Colo-357 cells or KPY mousecells in which YAP abundance was exper-imentally manipulated, we identified onlysix genes for which the transcript or en-codedprotein [connective tissuegrowth factor(CTGF), cysteine-rich angiogenic inducer61 (CYR61), matrix metalloproteinase 7(MMP7), interleukin-6 (IL-6), IL-1a, andcyclooxygenase 2 (COX2)] positively cor-related with YAP abundance across allthe pancreatic ductal cell groups examined
*
*
A
flox/flox
KPC KPYC
*
NSB
AD
M p
er f
ield
(#
cells
)
0
1
2
3
4
5
6
7
KPC KPYC
pER
KC
K19
Yap
EG
FR
:p48-CreYapflox/floxYapflox/+
KrasG12D:p53R172H
KPC KPYC
C
CK
19/Y
apY
apα-
SM
AYapYapflox/+
:p48-CreKrasG12D:p53R172H
Fig. 2. Deletion ofYapdoesnot affectADM inducedbyoncogenic Kras. (A) Representative images of IHCstaining for Yap, phosphorylated ERK (pERK), EGFR,and CK19 in pancreatic sections from 7-week-oldKPC and KPYC mice. Scale bar, 100 mm. (B) Numberof ADM lesions per 20×microscopic field inpancreatafrom 7-week-old KPC (n = 20) and KPYC (n = 17) mice. Lines mark themedian numbers of ADM lesions per
field for eachgenotype. (C) Representative IHC images of CK19 (brown) andYap (blue) double staining andstaining of Yap and a-SMA in serial pancreatic sections from 7-week-old KPC and KPYC mice. Asterisks,positively stained normal ducts; arrows, positively stained stromal cells; yellow arrowheads, positivelystained ADM lesions; white arrowheads, negatively stained ADM lesions. Scale bar, 50 mm.6 May 2014 Vol 7 Issue 324 ra42 4
R E S E A R C H A R T I C L E
on May 6, 2014
stke.sciencemag.org
Dow
nloaded from
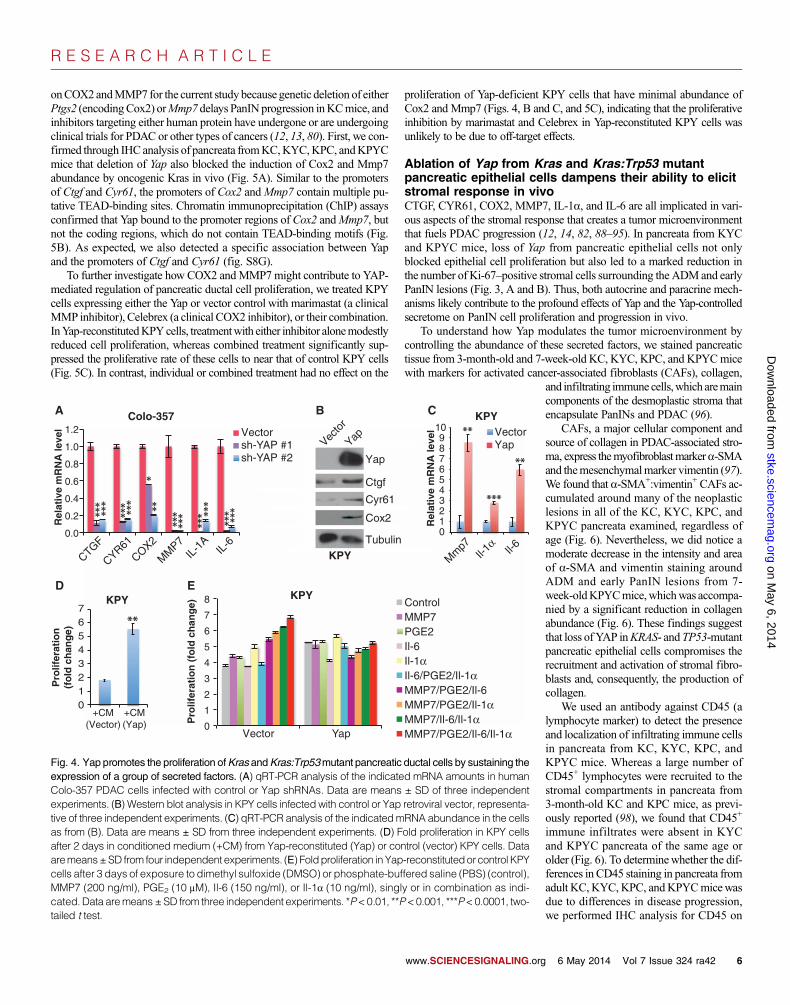
(Fig. 4, A to C, and fig. S8, C to F). All six genes are implicated in PDACprogression; five of these encode secretory proteins and the sixth en-codes COX2, which is responsible for the synthesis of the lipid prosta-glandin E2 (PGE2), which promotes inflammation, among other activities(12–14, 80–84). Consistent with the notion that YAP sustains the prolifer-ation of KRAS-mutant pancreatic ductal cells by promoting the expressionof pro-proliferative secretory factors, we found that conditioned mediumfrom Yap-reconstituted KPY cells overcame the proliferative defect exhib-ited by Yap-deficient KPY cells (Fig. 4D). Moreover, combined treatmentwith recombinant MMP7, Il-6, Il-1a, and PGE2 stimulated the prolifer-ation of Yap-deficient KPY cells, but not Yap-reconstituted KPY cells(Fig. 4E), whereas treatment with individual proteins or various combi-
nations had variable and generally less pronounced effects on the prolif-eration of either Yap+ or Yap− pancreatic ductal cells. These results suggestthat lack of expression of these secreted factors underlies the proliferativedefect of Yap null KPY cells and also highlight the complex interactionsamong these factors in the context of YAP.
COX2 and MMP7 are two novel YAP target genes thatcontribute to sustaining the proliferation of KRAS:TP53mutant pancreatic ductal cellsWhereas CTGF and CYR61 are well-established YAP target genes inmultiple cell types (85–87), to our knowledge no previous reports linkthe abundance ofCOX2,MMP7, IL-6, and IL-1a toYAP.We chose to focus
Yapflox/flox
KrasG12D:p53R172H:p48-Cre
Ki-67
Ki6
7/fi
eld
(#
cells
)
0 200 400 600 800
1000 1200 1400 1600
0 2 4 6 8
KrasG12D:p53R172H:Yap+/-
-/-
(KP)
KrasG12D:p53R172H:Yap(KPY)
Cel
l nu
mb
er (¥1
03 )
GFP Cre 0
10
KrasG12D
Yapflox/+KrasG12D
Yapflox/flox
Ed
U+
cells
(%
)
A B
C D
E
GFP Cre GFP Cre
KrasG12D KrasG12D:Yapflox/flox
EdU
EdU/DAPI
F
G H
0 2 4 6
Ctrlsh-Yap #1sh-Yap #2
0 200 400 600 800
1000 1200 1400
Cel
l nu
mb
er (¥1
03 )
Day
Day0 1 2 3 4
Ctrlsh-Yap #1sh-Yap #2
0
100
200
300
400
500
Cel
l nu
mb
er (¥1
03 )
Day
mPDAC-G9
mPan
Colo-357
0 1 2 3 4 5
+Yap
+Vector
mPan-KPY
0 200 400 600 800
1000 1200 1400 1600 1800
Cel
l nu
mb
er (¥1
03 )
Day
5
15202530354045
GFP Cre
P = 1.7 ¥ 10-5
NS
P = 0.01
0
100
200
300PanIN Stromal Acinar
KC KC KC KYCKYCKYC
6
5
Yapflox/+(KPC) (KPYC)
:Yapflox/+
100 µm
100 µm
Fig. 3. Deletion of Yap blocks the prolifera-tionofKrasandKras:Trp53mutant pancreaticductal cells in vivo and in vitro. (A) Repre-sentative IHC images of Ki-67 in pancreaticsections from KPC and KPYC mice. Yellowarrowheads, Ki-67+ proliferating pancreaticductal cells in KPC mice; gray arrowheads,Ki-67− ADMs in KPYC mice; yellow arrows,Ki-67+ proliferating stromal cells in both KPCand KPYCmice. Scale bar, 100 mm. (B) Num-ber of Ki-67+ PanIN, stromal, and acinar cellsper field fromKC(n=10)andKYC(n=7)pan-creata. Each data point represents the sumof five randomly selected fields from thepancreatic section of a different mouse.(C) Representative images fromEdU incor-poration assays of KrasG12D:Yapflox/+ andKrasG12D:Yapflox/flox pancreatic epithelial cellsinfected with Ad-GFP (GFP) or Ad-Cre-GFP(Cre). EdU-incorporated cells are red; nucleiare blue. Scale bar, 100 mm. (D) Quantifica-tion of the percentage of EdU+ cells per fieldfrom (C). Lines indicate median percentagesof EdU+ cells. KrasG12D:Yapflox/+(GFP), n =23; KrasG12D:Yapf lox /+(Cre), n = 16;KrasG12D:Yapflox/flox(GFP), n = 13; KrasG12D:Yapflox/flox(Cre), n = 14. NS, not significant; Pvalues were calculated using two-tailed ttest. (E to H) Cell proliferation assessed in(E) KP and KPY cells, (F) KPY cells reconsti-tutedwith Yapor control vector, (G)mPDAC-G9 cells transfected with control or twoindependent mYap shRNAs (#1 and #2),and (H) human Colo-357 PDAC cells trans-fected with control or two independent hYapshRNAs (#1 and #2). Data are means ± SDfrom three independent experiments.
www.SCIENCESIGNALING.org 6 May 2014 Vol 7 Issue 324 ra42 5
R E S E A R C H A R T I C L E
Dow
n
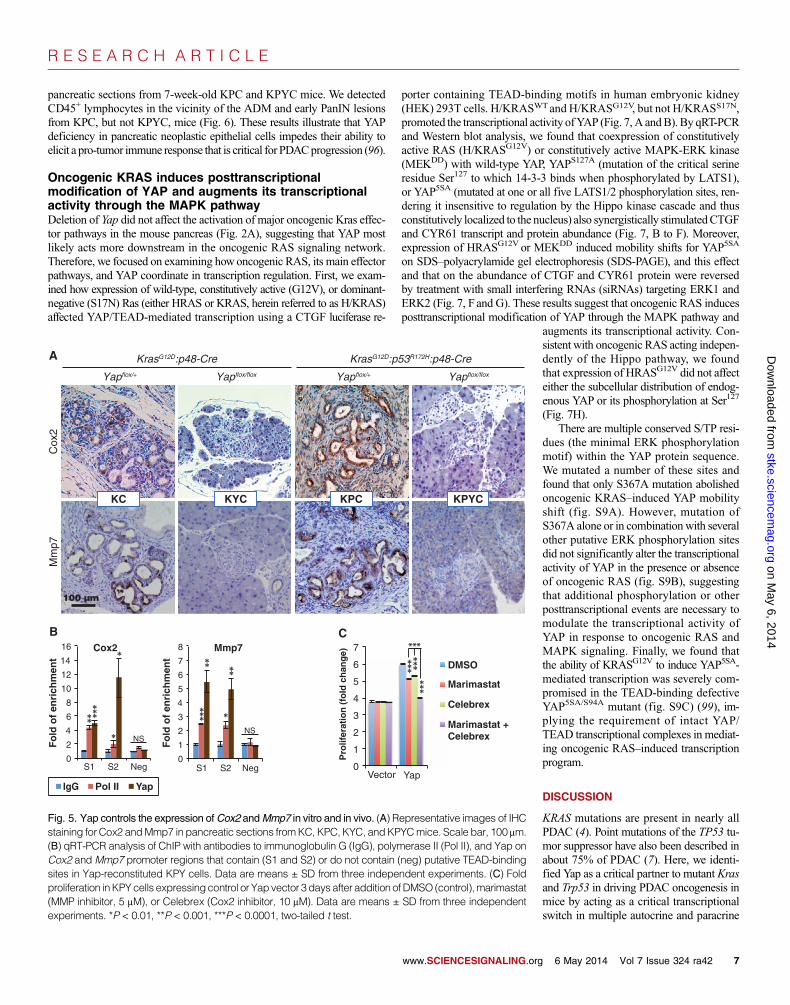
onCOX2andMMP7 for the current study because genetic deletion of eitherPtgs2 (encodingCox2) orMmp7delays PanINprogression inKCmice, andinhibitors targeting either human protein have undergone or are undergoingclinical trials for PDAC or other types of cancers (12, 13, 80). First, we con-firmed through IHCanalysis of pancreata fromKC,KYC,KPC, andKPYCmice that deletion of Yap also blocked the induction of Cox2 and Mmp7abundance by oncogenic Kras in vivo (Fig. 5A). Similar to the promotersof Ctgf and Cyr61, the promoters of Cox2 andMmp7 contain multiple pu-tative TEAD-binding sites. Chromatin immunoprecipitation (ChIP) assaysconfirmed that Yap bound to the promoter regions of Cox2 andMmp7, butnot the coding regions, which do not contain TEAD-binding motifs (Fig.5B). As expected, we also detected a specific association between Yapand the promoters of Ctgf and Cyr61 (fig. S8G).
To further investigate how COX2 andMMP7might contribute to YAP-mediated regulation of pancreatic ductal cell proliferation, we treated KPYcells expressing either the Yap or vector control with marimastat (a clinicalMMP inhibitor), Celebrex (a clinical COX2 inhibitor), or their combination.InYap-reconstitutedKPYcells, treatmentwith either inhibitor alonemodestlyreduced cell proliferation, whereas combined treatment significantly sup-pressed the proliferative rate of these cells to near that of control KPY cells(Fig. 5C). In contrast, individual or combined treatment had no effect on the
proliferation of Yap-deficient KPY cells that have minimal abundance ofCox2 and Mmp7 (Figs. 4, B and C, and 5C), indicating that the proliferativeinhibition by marimastat and Celebrex in Yap-reconstituted KPY cells wasunlikely to be due to off-target effects.
Ablation of Yap from Kras and Kras:Trp53 mutantpancreatic epithelial cells dampens their ability to elicitstromal response in vivoCTGF, CYR61, COX2, MMP7, IL-1a, and IL-6 are all implicated in vari-ous aspects of the stromal response that creates a tumor microenvironmentthat fuels PDAC progression (12, 14, 82, 88–95). In pancreata from KYCand KPYC mice, loss of Yap from pancreatic epithelial cells not onlyblocked epithelial cell proliferation but also led to a marked reduction inthe number of Ki-67–positive stromal cells surrounding the ADMand earlyPanIN lesions (Fig. 3, A and B). Thus, both autocrine and paracrine mech-anisms likely contribute to the profound effects of Yap and the Yap-controlledsecretome on PanIN cell proliferation and progression in vivo.
To understand how Yap modulates the tumor microenvironment bycontrolling the abundance of these secreted factors, we stained pancreatictissue from 3-month-old and 7-week-old KC, KYC, KPC, and KPYCmicewith markers for activated cancer-associated fibroblasts (CAFs), collagen,
www.SCIENCESIGNALING.org
on May 6, 2014
stke.sciencemag.org
loaded from
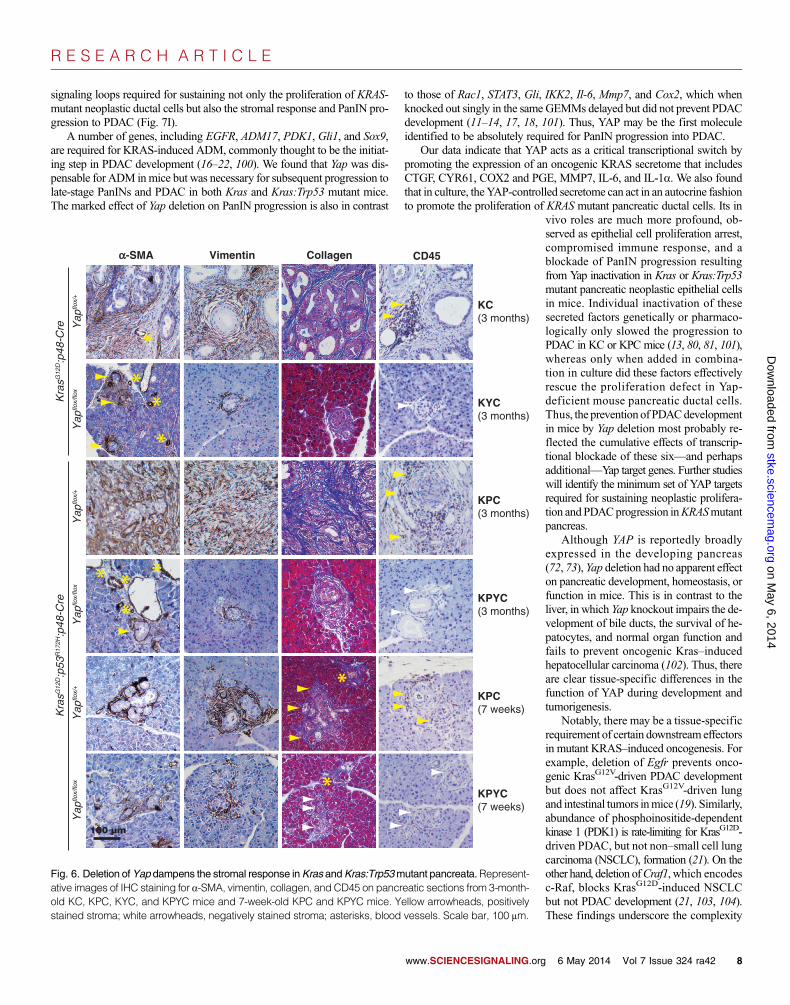
and infiltrating immune cells,which aremaincomponents of the desmoplastic stroma thatencapsulate PanINs and PDAC (96).
CAFs, a major cellular component andsource of collagen in PDAC-associated stro-ma, express themyofibroblastmarkera-SMAand themesenchymalmarker vimentin (97).We found that a-SMA+:vimentin+ CAFs ac-cumulated around many of the neoplasticlesions in all of the KC, KYC, KPC, andKPYC pancreata examined, regardless ofage (Fig. 6). Nevertheless, we did notice amoderate decrease in the intensity and areaof a-SMA and vimentin staining aroundADM and early PanIN lesions from 7-week-oldKPYCmice,whichwas accompa-nied by a significant reduction in collagenabundance (Fig. 6). These findings suggestthat loss ofYAP inKRAS- andTP53-mutantpancreatic epithelial cells compromises therecruitment and activation of stromal fibro-blasts and, consequently, the production ofcollagen.
We used an antibody against CD45 (alymphocyte marker) to detect the presenceand localization of infiltrating immune cellsin pancreata from KC, KYC, KPC, andKPYC mice. Whereas a large number ofCD45+ lymphocytes were recruited to thestromal compartments in pancreata from3-month-old KC and KPC mice, as previ-ously reported (98), we found that CD45+
immune infiltrates were absent in KYCand KPYC pancreata of the same age orolder (Fig. 6). To determinewhether the dif-ferences in CD45 staining in pancreata fromadult KC, KYC,KPC, andKPYCmicewasdue to differences in disease progression,we performed IHC analysis for CD45 on
+CM(Vector)
+CM(Yap)
0 1 2 3 4 5 6 7
DKPY
**
A
0.0
0.2
0.4
0.6
0.8
1.0
1.2
Rel
ativ
e m
RN
A le
vel
CTGF
CYR61
COX2 IL
-1A
IL-6
MM
P7
Colo-357
***
***
***
***
**
***
***
*** **
*
***
***
*
**
Vecto
r
Yap
0 1 2 3 4 5 6 7 8 9
10
Mm
p7
Il-1α
Il-6
Vector Yap
Rel
ativ
e m
RN
A le
vel
B C
KPY
KPY**
***
Vector sh-YAP #1 sh-YAP #2
1
2
3
4
5
6
7
Vector Yap
Control MMP7 PGE2 Il-6 Il-1α Il-6/PGE2/Il-1α MMP7/PGE2/Il-6 MMP7/PGE2/Il-1α MMP7/Il-6/Il-1α MMP7/PGE2/Il-6/Il-1α
KPYE
Pro
lifer
atio
n (
fold
ch
ang
e) 8
0
Pro
lifer
atio
n(f
old
ch
ang
e)
Tubulin
Yap
Cyr61
Ctgf
Cox2
Fig. 4. Yap promotes the proliferation ofKras andKras:Trp53mutant pancreatic ductal cells by sustaining theexpression of a group of secreted factors. (A) qRT-PCR analysis of the indicated mRNA amounts in humanColo-357 PDAC cells infected with control or Yap shRNAs. Data are means ± SD of three independentexperiments. (B) Western blot analysis in KPY cells infected with control or Yap retroviral vector, representa-tive of three independent experiments. (C) qRT-PCR analysis of the indicatedmRNA abundance in the cellsas from (B). Data are means ± SD from three independent experiments. (D) Fold proliferation in KPY cellsafter 2 days in conditioned medium (+CM) from Yap-reconstituted (Yap) or control (vector) KPY cells. Dataaremeans±SD from four independent experiments. (E) Foldproliferation inYap-reconstitutedorcontrol KPYcells after 3 days of exposure to dimethyl sulfoxide (DMSO) or phosphate-buffered saline (PBS) (control),MMP7 (200 ng/ml), PGE2 (10 mM), Il-6 (150 ng/ml), or Il-1a (10 ng/ml), singly or in combination as indi-cated. Data aremeans±SD from three independent experiments. *P<0.01, **P<0.001, ***P<0.0001, two-tailed t test.
6 May 2014 Vol 7 Issue 324 ra42 6
R E S E A R C H A R T I C L E
pancreatic sections from 7-week-old KPC and KPYC mice. We detectedCD45+ lymphocytes in the vicinity of the ADM and early PanIN lesionsfrom KPC, but not KPYC, mice (Fig. 6). These results illustrate that YAPdeficiency in pancreatic neoplastic epithelial cells impedes their ability toelicit a pro-tumor immune response that is critical for PDACprogression (96).
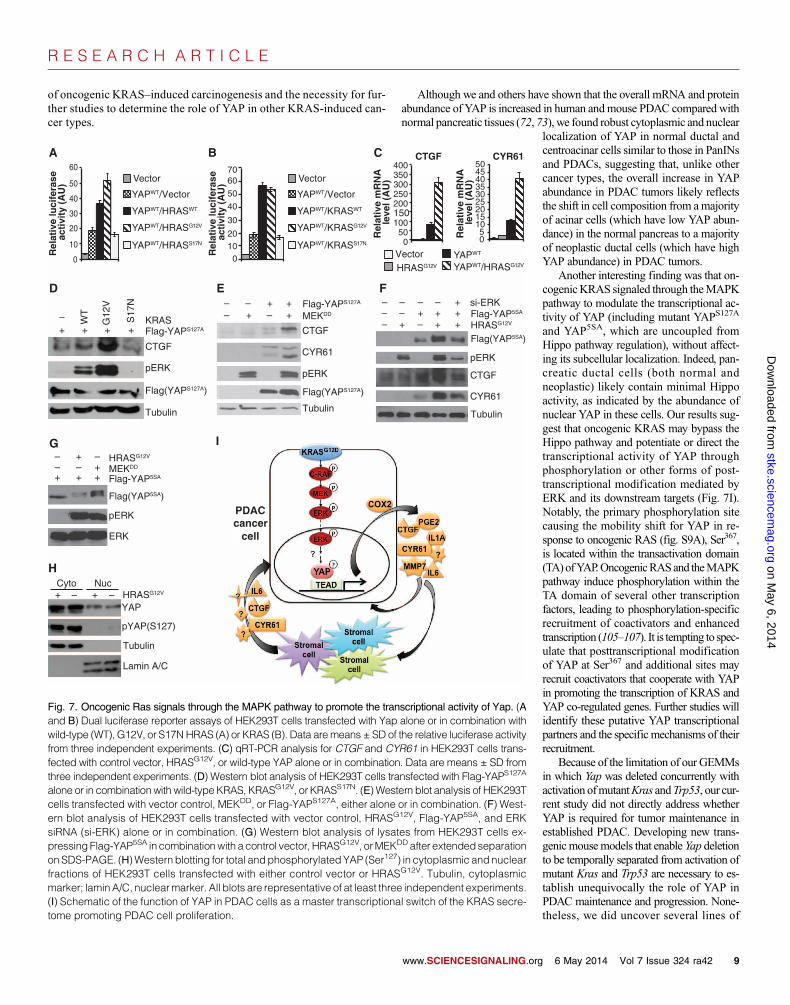
Oncogenic KRAS induces posttranscriptionalmodification of YAP and augments its transcriptionalactivity through the MAPK pathwayDeletion of Yap did not affect the activation of major oncogenic Kras effec-tor pathways in the mouse pancreas (Fig. 2A), suggesting that YAP mostlikely acts more downstream in the oncogenic RAS signaling network.Therefore, we focused on examining howoncogenic RAS, its main effectorpathways, and YAP coordinate in transcription regulation. First, we exam-ined how expression of wild-type, constitutively active (G12V), or dominant-negative (S17N) Ras (either HRAS or KRAS, herein referred to as H/KRAS)affected YAP/TEAD-mediated transcription using a CTGF luciferase re-
porter containing TEAD-binding motifs in human embryonic kidney(HEK) 293T cells. H/KRASWTand H/KRASG12V, but not H/KRASS17N,promoted the transcriptional activity ofYAP (Fig. 7, A andB). By qRT-PCRand Western blot analysis, we found that coexpression of constitutivelyactive RAS (H/KRASG12V) or constitutively active MAPK-ERK kinase(MEKDD) with wild-type YAP, YAPS127A (mutation of the critical serineresidue Ser127 to which 14-3-3 binds when phosphorylated by LATS1),or YAP5SA (mutated at one or all five LATS1/2 phosphorylation sites, ren-dering it insensitive to regulation by the Hippo kinase cascade and thusconstitutively localized to the nucleus) also synergistically stimulatedCTGFand CYR61 transcript and protein abundance (Fig. 7, B to F). Moreover,expression of HRASG12V or MEKDD induced mobility shifts for YAP5SA
on SDS–polyacrylamide gel electrophoresis (SDS-PAGE), and this effectand that on the abundance of CTGF and CYR61 protein were reversedby treatment with small interfering RNAs (siRNAs) targeting ERK1 andERK2 (Fig. 7, F and G). These results suggest that oncogenic RAS inducesposttranscriptional modification of YAP through the MAPK pathway and
www.SCIENCESIGNALING.org
on May 6, 2014
stke.sciencemag.org
Dow
nloaded from
augments its transcriptional activity. Con-sistent with oncogenic RAS acting indepen-dently of the Hippo pathway, we foundthat expression of HRASG12V did not affecteither the subcellular distribution of endog-enous YAP or its phosphorylation at Ser127
(Fig. 7H).There are multiple conserved S/TP resi-
dues (the minimal ERK phosphorylationmotif) within the YAP protein sequence.We mutated a number of these sites andfound that only S367A mutation abolishedoncogenic KRAS–induced YAP mobilityshift (fig. S9A). However, mutation ofS367A alone or in combinationwith severalother putative ERK phosphorylation sitesdid not significantly alter the transcriptionalactivity of YAP in the presence or absenceof oncogenic RAS (fig. S9B), suggestingthat additional phosphorylation or otherposttranscriptional events are necessary tomodulate the transcriptional activity ofYAP in response to oncogenic RAS andMAPK signaling. Finally, we found thatthe ability of KRASG12V to induce YAP5SA-mediated transcription was severely com-promised in the TEAD-binding defectiveYAP5SA/S94A mutant (fig. S9C) (99), im-plying the requirement of intact YAP/TEAD transcriptional complexes in mediat-ing oncogenic RAS–induced transcriptionprogram.
DISCUSSION
KRAS mutations are present in nearly allPDAC (4). Point mutations of the TP53 tu-mor suppressor have also been described inabout 75% of PDAC (7). Here, we identi-fied Yap as a critical partner to mutant Krasand Trp53 in driving PDAC oncogenesis inmice by acting as a critical transcriptionalswitch in multiple autocrine and paracrine
Yapflox/floxYapflox/+ Yapflox/floxYapflox/+
KrasG12D:p48-Cre KrasG12D:p53R172H:p48-Cre
Cox
2M
mp7
A
KC KYC KPC KPYC
DMSO
Marimastat
Celebrex
Marimastat +Celebrex
B
Yap Vector
C
***
***
***
***
0
1
2
3
4
5
6
7
0
1
2
3
4
5
6
7
8
0
2
4
6
8
10
12
14
16 Cox2
S1 S2 Neg
Mmp7
S1 S2 Neg
Fo
ld o
f en
rich
men
t
Fo
ld o
f en
rich
men
t
IgG Pol II Yap
** ***
*
NSNS
***
**
***
*
Pro
lifer
atio
n (
fold
ch
ang
e)
100 µm
Fig. 5. Yap controls the expression ofCox2 andMmp7 in vitro and in vivo. (A) Representative images of IHCstaining for Cox2 andMmp7 in pancreatic sections fromKC, KPC, KYC, andKPYCmice. Scale bar, 100 mm.(B) qRT-PCR analysis of ChIP with antibodies to immunoglobulin G (IgG), polymerase II (Pol II), and Yap onCox2 andMmp7 promoter regions that contain (S1 and S2) or do not contain (neg) putative TEAD-bindingsites in Yap-reconstituted KPY cells. Data are means ± SD from three independent experiments. (C) Foldproliferation inKPYcells expressingcontrol or Yapvector 3daysafter addition ofDMSO (control),marimastat(MMP inhibitor, 5 mM), or Celebrex (Cox2 inhibitor, 10 mM). Data are means ± SD from three independentexperiments. *P < 0.01, **P < 0.001, ***P < 0.0001, two-tailed t test.
6 May 2014 Vol 7 Issue 324 ra42 7
R E S E A R C H A R T I C L E
signaling loops required for sustaining not only the proliferation of KRAS-mutant neoplastic ductal cells but also the stromal response and PanIN pro-gression to PDAC (Fig. 7I).
A number of genes, including EGFR, ADM17, PDK1, Gli1, and Sox9,are required for KRAS-induced ADM, commonly thought to be the initiat-ing step in PDAC development (16–22, 100). We found that Yap was dis-pensable for ADM inmice but was necessary for subsequent progression tolate-stage PanINs and PDAC in both Kras and Kras:Trp53 mutant mice.The marked effect of Yap deletion on PanIN progression is also in contrast
to those of Rac1, STAT3, Gli, IKK2, Il-6, Mmp7, and Cox2, which whenknocked out singly in the same GEMMs delayed but did not prevent PDACdevelopment (11–14, 17, 18, 101). Thus, YAP may be the first moleculeidentified to be absolutely required for PanIN progression into PDAC.
Our data indicate that YAP acts as a critical transcriptional switch bypromoting the expression of an oncogenic KRAS secretome that includesCTGF, CYR61, COX2 and PGE, MMP7, IL-6, and IL-1a. We also foundthat in culture, theYAP-controlled secretome can act in an autocrine fashionto promote the proliferation of KRAS mutant pancreatic ductal cells. Its in
www.SCIENCESIGNALING.org
on May 6, 2014
stke.sciencemag.org
Dow
nloaded from
vivo roles are much more profound, ob-served as epithelial cell proliferation arrest,compromised immune response, and ablockade of PanIN progression resultingfrom Yap inactivation in Kras or Kras:Trp53mutant pancreatic neoplastic epithelial cellsin mice. Individual inactivation of thesesecreted factors genetically or pharmaco-logically only slowed the progression toPDAC in KC or KPC mice (13, 80, 81, 101),whereas only when added in combina-tion in culture did these factors effectivelyrescue the proliferation defect in Yap-deficient mouse pancreatic ductal cells.Thus, the prevention of PDACdevelopmentin mice by Yap deletion most probably re-flected the cumulative effects of transcrip-tional blockade of these six—and perhapsadditional—Yap target genes. Further studieswill identify the minimum set of YAP targetsrequired for sustaining neoplastic prolifera-tion andPDACprogression inKRASmutantpancreas.
Although YAP is reportedly broadlyexpressed in the developing pancreas(72, 73), Yap deletion had no apparent effecton pancreatic development, homeostasis, orfunction in mice. This is in contrast to theliver, in which Yap knockout impairs the de-velopment of bile ducts, the survival of he-patocytes, and normal organ function andfails to prevent oncogenic Kras–inducedhepatocellular carcinoma (102). Thus, thereare clear tissue-specific differences in thefunction of YAP during development andtumorigenesis.
Notably, there may be a tissue-specificrequirement of certain downstream effectorsin mutant KRAS–induced oncogenesis. Forexample, deletion of Egfr prevents onco-genic KrasG12V-driven PDAC developmentbut does not affect KrasG12V-driven lungand intestinal tumors inmice (19). Similarly,abundance of phosphoinositide-dependentkinase 1 (PDK1) is rate-limiting for KrasG12D-driven PDAC, but not non–small cell lungcarcinoma (NSCLC), formation (21). On theother hand, deletion ofCraf1, which encodesc-Raf, blocks KrasG12D-induced NSCLCbut not PDAC development (21, 103, 104).These findings underscore the complexity
Yap
flox/flo
xYap
flox/
+Yap
flox/flo
xYap
flox/
+
Collagen-SMAαα Vimentin CD45
KrasG
12D:p48
-Cre
KrasG
12D:p53
R17
2H:p48
-Cre
Yap
flox/flo
xYap
flox/
+
KC(3 months)
KYC(3 months)
KPYC(3 months)
KPYC(7 weeks)
KPC(3 months)
KPC(7 weeks)
*
*
*
*
**
* **
*
100 µm
Fig. 6. Deletion ofYap dampens the stromal response inKras andKras:Trp53mutant pancreata.Represent-ative images of IHC staining for a-SMA, vimentin, collagen, and CD45 on pancreatic sections from 3-month-old KC, KPC, KYC, and KPYC mice and 7-week-old KPC and KPYC mice. Yellow arrowheads, positivelystained stroma; white arrowheads, negatively stained stroma; asterisks, blood vessels. Scale bar, 100 mm.
6 May 2014 Vol 7 Issue 324 ra42 8
R E S E A R C H A R T I C L E
of oncogenic KRAS–induced carcinogenesis and the necessity for fur-ther studies to determine the role of YAP in other KRAS-induced can-cer types.
Although we and others have shown that the overall mRNA and proteinabundance of YAP is increased in human andmouse PDAC compared withnormal pancreatic tissues (72, 73), we found robust cytoplasmic and nuclear
www.SCIENCESIGNALING.org
on May 6, 2014
stke.sciencemag.org
Dow
nloaded from
localization of YAP in normal ductal andcentroacinar cells similar to those in PanINsand PDACs, suggesting that, unlike othercancer types, the overall increase in YAPabundance in PDAC tumors likely reflectsthe shift in cell composition from amajorityof acinar cells (which have low YAP abun-dance) in the normal pancreas to a majorityof neoplastic ductal cells (which have highYAP abundance) in PDAC tumors.
Another interesting finding was that on-cogenicKRAS signaled through theMAPKpathway to modulate the transcriptional ac-tivity of YAP (including mutant YAPS127A
and YAP5SA, which are uncoupled fromHippo pathway regulation), without affect-ing its subcellular localization. Indeed, pan-creatic ductal cells (both normal andneoplastic) likely contain minimal Hippoactivity, as indicated by the abundance ofnuclear YAP in these cells. Our results sug-gest that oncogenic KRAS may bypass theHippo pathway and potentiate or direct thetranscriptional activity of YAP throughphosphorylation or other forms of post-transcriptional modification mediated byERK and its downstream targets (Fig. 7I).Notably, the primary phosphorylation sitecausing the mobility shift for YAP in re-sponse to oncogenic RAS (fig. S9A), Ser367,is located within the transactivation domain(TA)ofYAP.OncogenicRASand theMAPKpathway induce phosphorylation within theTA domain of several other transcriptionfactors, leading to phosphorylation-specificrecruitment of coactivators and enhancedtranscription (105–107). It is tempting to spec-ulate that posttranscriptional modificationof YAP at Ser367 and additional sites mayrecruit coactivators that cooperate with YAPin promoting the transcription of KRAS andYAP co-regulated genes. Further studies willidentify these putative YAP transcriptionalpartners and the specific mechanisms of theirrecruitment.
Because of the limitation of our GEMMsin which Yap was deleted concurrently withactivation ofmutantKras andTrp53, our cur-rent study did not directly address whetherYAP is required for tumor maintenance inestablished PDAC. Developing new trans-genicmousemodels that enable Yap deletionto be temporally separated from activation ofmutant Kras and Trp53 are necessary to es-tablish unequivocally the role of YAP inPDAC maintenance and progression. None-theless, we did uncover several lines of
A B
0
10
20
30
40
50
60 Vector
YAPWT/Vector
YAPWT/HRASWT
YAPWT/HRASG12V
YAPWT/HRASS17N
Rel
ativ
e lu
cife
rase
act
ivit
y (A
U)
400
0 50
100 150 200 250 300 350
CTGF
Rel
ativ
e m
RN
A le
vel (
AU
)
0 5
10 15 20 25 30 35 40 45 50
CYR61
HRASG12V
YAPWT
YAPWT/HRASG12V Vector
+ ++ +
WT
G12
V
S17
N
CTGF
Tubulin
– ––
pERK
CYR61
CTGF
+–+++– Flag-YAPS127A
MEKDD
Tubulin
++–+++
––
+–
– – +– –
pERK
CTGF
CYR61
Tubulin
HRASG12V
si-ERKFlag-YAP5SA
Flag(YAP5SA)
Cyto Nuc
YAP
pYAP(S127)
Tubulin
Lamin A/C
HRASG12V– –++
––
+––+
pERK
ERK
MEKDD
Flag(YAP5SA)
HRASG12V
C
D E F
G
H
Vector
YAPWT/Vector
YAPWT/KRASWT
YAPWT/KRASG12V
YAPWT/KRASS17N 0
10 20
30
40
50
60 70
KRAS
Flag(YAPS127A)Flag(YAPS127A)
pERK
Flag-YAPS127A
Flag-YAP5SA+ ++
I
Rel
ativ
e lu
cife
rase
act
ivit
y (A
U)
Rel
ativ
e m
RN
A le
vel (
AU
)
PDACcancer
cell
Fig. 7. Oncogenic Ras signals through the MAPK pathway to promote the transcriptional activity of Yap. (Aand B) Dual luciferase reporter assays of HEK293T cells transfected with Yap alone or in combination withwild-type (WT), G12V, or S17NHRAS (A) or KRAS (B). Data aremeans ± SD of the relative luciferase activityfrom three independent experiments. (C) qRT-PCR analysis for CTGF and CYR61 in HEK293T cells trans-fected with control vector, HRASG12V, or wild-type YAP alone or in combination. Data are means ± SD fromthree independent experiments. (D) Western blot analysis of HEK293T cells transfected with Flag-YAPS127A
alone or in combinationwith wild-type KRAS, KRASG12V, or KRASS17N. (E) Western blot analysis of HEK293Tcells transfected with vector control, MEKDD, or Flag-YAPS127A, either alone or in combination. (F) West-ern blot analysis of HEK293T cells transfected with vector control, HRASG12V, Flag-YAP5SA, and ERKsiRNA (si-ERK) alone or in combination. (G) Western blot analysis of lysates from HEK293T cells ex-pressing Flag-YAP5SA in combinationwith a control vector, HRASG12V, orMEKDD after extendedseparationon SDS-PAGE. (H)Western blotting for total andphosphorylated YAP (Ser127) in cytoplasmic and nuclearfractions of HEK293T cells transfected with either control vector or HRASG12V. Tubulin, cytoplasmicmarker; laminA/C, nuclearmarker. All blots are representative of at least three independent experiments.(I) Schematic of the function of YAP in PDAC cells as a master transcriptional switch of the KRAS secre-tome promoting PDAC cell proliferation.
6 May 2014 Vol 7 Issue 324 ra42 9

R E S E A R C H A R T I C L E
Dow
nloa
evidence supporting the continued requirement for YAP in establishedPDAC. First, we demonstrated that YAP is widely expressed in primary hu-man PDAC. Second, we showed that YAP expression is necessary for main-taining the expression of the six noted secreted factors in KRAS mutanthuman and mouse PDAC lines. Last, we found that even a modest de-crease in YAP expression was sufficient to inhibit the proliferation of theaforementioned PDAC lines.
As a transcriptional regulatorwith no enzymatic pocket, YAPposes sub-stantial challenges as a drug target. Despite this, there has been some recentprogress in developing small-molecule YAP inhibitors (108–112). Analternative to directly targeting YAP to treat PDAC may be to combine acocktail of inhibitors or therapeutic antibodies against multiple componentsof the KRAS/YAP secretome. Clinical trials have been conducted withCOX2 and MMP7 inhibitors as single agents for PDAC, yielding mostlydisappointing results (113–115). A clinical trial is currently ongoing for aCTGF-targeted antibody in PDAC and other cancers (116). Clinical agentstargeting IL-6 and IL-1a are also available, although they have not beentested in PDAC (117). We found that combination of COX2 and MMP7inhibitorsmore efficiently inhibited the proliferation ofYap-positive pancre-atic ductal cells than either inhibitor alone, suggesting that combinationstrategies with available agents may achieve optimum clinical outcome inPDAC without causing unmanageable toxicity.
on May 6, 2014
stke.sciencemag.org
ded from
MATERIALS AND METHODS
Generation of mouse strainsGenetically engineeredmouse strainsYap flox/flox,KrasLSL-G12D,Trp53LSL-R172H,and p48-Cre (102, 118–120) were interbred to generate all experimentalcohorts (table S1). All animal experiments were conducted according toprotocol #11-055 approved by the Institutional Animal Care and Use Com-mittee at Georgetown University.
Cell culture, transfection, and infectionThe human pancreatic cancer cell lineColo-357was cultured inRPMI 1640(Sigma-Aldrich Corp.) supplemented with 10% fetal bovine serum (FBS).HEK293T, 293 Phoenix-A, andmouse pancreatic tumor cell line mPDAC-G9were cultured in Dulbecco’s modified Eagle’s medium (Sigma-Aldrich)supplemented with 10% FBS. The primary mouse pancreatic cells werecultured in Waymouth’s MB 752/1 (Sigma-Aldrich) supplemented with10% FBS and soybean trypsin inhibitor (STI; 0.1 mg/ml; AMRESCO,LLC) on collagen-coated plates.
pLKO-shRNA lentiviral constructs targeting human and mouse Yap(TTRCN0000107266: 5′-TTCTTTATCTAGCTTGGTGGC-3′,TRCN0000107268: AAAGGATCTGAGCTATTGGTC, andTRCN0000095864: 5′-TTAACAAAGGAATCTGTCTGC-3′) were pur-chased from Thermo Scientific Open Biosystems. Lentiviral and retroviralproductions were performed as previously described (121).
Mouse acinar explant preparation and in vitro ADM assayMouse pancreatic explant cultures were established bymodifying previous-ly published protocols (122, 123). Briefly, whole pancreata were harvestedand digested in collagenase type 4 (4000 U/ml, Worthington BiochemicalCorp.) for 50 min at 37°C. After multiple washes with Waymouth’s MB752/1 supplemented with 5% FBS, collagenase-digested pancreatic tissuewas sequentially filtered through 90-µmmetal mesh filters. The filtrate wasthen passed through a 30% FBS cushion by centrifugation for 5 min at1000g. The cell pellet was resuspended in Waymouth’s culture medium[Waymouth’sMB 752/1Medium supplemented with penicillin/streptomycinand STI (0.1 mg/ml)]. After incubation with adenoviruses carrying GFP
w
or Cre-GFP (Gene Transfer Vector Core of University of Iowa) at 37°C for1 hour, the cells were pelleted and resuspended in Waymouth’s culture me-dium. An equal volume of neutralized rat-tail collagen type I (RTC) (BDBiosciences) mixture was added to the cellular suspension. The cellular/RTCsuspension was pipetted into culture dish precoated with RTC. After solidi-fication, Waymouth’s culture medium was added. Cultures were maintainedat 37°C in a 5% CO2 humidified chamber for up to 3 days, then switched toWaymouth’s culture medium supplemented with 10% FBS.
Cell proliferation and EdU incorporation assaysFor cell proliferation assays, human or mouse pancreatic tumor cell lineswere seeded in triplicate onto 12-well plates, trypsinized, and counted atthe indicated times using Multisizer 3 Coulter Counter (Beckman CoulterInc.). Colo-357 and mPDAC-G9 cells expressing control or Yap shRNAswere plated at 5 × 104 cells per well. Primary mouse pancreatic cellsexpressing pBABE vector or pBABE-Yap were plated at 2 × 104 cellsper well. For treatment with secreted factors, recombinant human MMP7(200 ng/ml, Millipore), murine Il-6 (150 ng/ml, Invitrogen), murine Il-1a(10 ng/ml, Sino Biological Inc.), and PGE2 (10 mM, Enzo Life Sciences Inc.)were added singly or in combination to the medium, and cells were incu-bated for three additional days.
For EdU incorporation assays, freshly isolated pancreatic epithelial cellsinfected with Ad-GFP or Ad-Cre-GFP were plated onto collagen-coatedcoverslips inWaymouth’sMB 752/1Medium supplemented with penicillinG (1000 U/ml), streptomycin (100 mg/ml), STI (0.1 mg/ml), and 10% FBS.After 3 days in culture, EdU was added to the medium, and cells were in-cubated for an additional 14 hours. EdU-positive cells were detected usingthe Click-iT EdUAlexa Fluor 594 Imaging Kit according to the manufac-turer’s instructions (Invitrogen). Nuclei were counterstained with 4′,6-diamidino-2-phenylindole.
Glucose tolerance testThree-month-old YC mice with age-matched wild-type littermates weresubjected to fasting for 12 hours before the baseline blood glucose levelwas first measured for each mouse using OneTouch UItraMini Blood Glu-coseMonitoring System according to the manufacturer’s instructions (Life-Scan Inc.). Sterilized D-glucose (200 mg/ml) was then intraperitoneallyinjected into eachmouse (2mg/g in PBS), and blood glucosewasmeasuredagain at 30, 60, and 120 min after the injection.
Acute pancreatitis inductionTwo-month-old wild-type and YCmicewere intraperitoneally injected sev-en times with caerulein (50 mg/kg) at 1-hour intervals. Mice were eutha-nized 48 hours after the first injection, and the entire pancreas wasdissected and fixed in 10% formalin.
Quantitative real-time RT-PCRTotal mRNA was isolated using TRIzol reagent (Invitrogen). Reversetranscription was performed using iScript cDNA Synthesis Kit ac-cording to the manufacturer’s instructions (Bio-Rad LaboratoriesHeadquarters). The resulting complementary DNA (cDNA) productswere amplified with iTaq Universal SYBR Green Supermix (Bio-RadLaboratories). All reactions were performed in triplicate. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (for human) or hypoxanthine-guanine phosphoribosyltransferase (HPRT) (for mouse) was used fornormalization. Relative gene expression was calculated as unit valueof 2−DCt = 2−[Ct (GAPDH or HPRT) − Ct (gene of interest)], where Ct is thethreshold cycle value defined as the fractional cycle number at whichthe target fluorescent signal passes a fixed threshold above baseline.The sequences of the primers used in the study are listed in table S3.
ww.SCIENCESIGNALING.org 6 May 2014 Vol 7 Issue 324 ra42 10

R E S E A R C H A R T I C L E
on May 6, 2014
stke.sciencemag.org
Dow
nloaded from
Chromatin immunoprecipitationChIP analysis was performed as previously described using normal rabbitIgG (Santa Cruz Biotechnology, sc-2027X) and antibodies against Yap(Santa Cruz Biotechnology, sc-15407X) and Pol II (Santa Cruz Bio-technology, sc-899X) (121). Precipitated DNA was eluted and amplifiedusing qRT-PCR with primer pairs flanking different regions of the mouseCyr61, Ctgf, Cox2, and Mmp7 promoters that contain putative TEAD-binding sites. The primer sequences used in the study are listed in table S4.
Western blot, IHC, and immunofluorescenceWestern blot analysis was performed as previously described (124). Humanpancreatic TMAs (#PA1001 and #PA207)were purchased fromUSBiomaxInc. Mouse pancreas was fixed in 10% buffered formalin and embedded inparaffin. For IHC, unstained pancreatic slides were deparaffinized andheated in standard citrate or tris-EDTA retrieval buffer for 30 min at 95°C.After incubation overnight with the primary antibodies at 4°C, the slideswereincubated with biotinylated secondary antibodies (Vector Laboratories Ltd.)for 1 hour at room temperature. Antibody labeling was visualized using theVECTASTAINABCkit (Vector Laboratories Ltd.) followed by stainingwith3,3′-diaminobenzidine tetrahydrochloride plus (DAB+) according to themanufacturer’s instructions (Thermo Scientific) or using the Vector BlueAlkaline Phosphatase Substrate Kit (Vector Laboratories Ltd.). The anti-bodies used for Western blot and IHC analyses are listed in table S5. Collagenstaining was performed using theMasson Trichrome Stain Kit according tothe manufacturer’s instructions (Sigma).
Subcellular fractionationFor subcellular fractionation experiments, the cytoplasmic fraction wasextracted with hypotonic buffer [10 mM Hepes (pH 7.9), 1.5 mM MgCl2,10mMKCl, and 0.5mM fresh dithiothreitol], and pellets containing nucleiwere washed twice with hypotonic buffer and subsequently lysed in radio-immunoprecipitation assay buffer.
Luciferase reporter assayThe CTGF luciferase reporter was a gift from K. Lyons [University ofCalifornia, Los Angeles (UCLA)] (125). Renilla luciferase vector was pur-chased from Promega. Luciferase reporter activities were determined withthe Dual Luciferase Assay Kit (Promega). The reporter’s firefly luciferaseactivity was normalized to that of the internal control Renilla luciferasebefore statistical analysis. The annotated relative luciferase activity is theratio between firefly and Renilla luciferase activities.
Microarray data mining and statistical analysisGene Expression Omnibus data sets GSE15471, GSE18670, GSE19650, andGSE16515 containing either normal pancreatic tissue or PDAC tumor cDNAsamples hybridized to Human Genome U133 Plus 2.0 GeneChips were in-cluded in our analysis. Array data were normalized, and samples having anSEgreater than 1.1were excluded.GC-RMAwas used for probe-level normal-ization of array intensities (126). Batch effects caused by multiple data sourceswere corrected using ComBat (127). Probes from each probe set with thegreatest interquartile range were retained for gene expression analysis. TheLinearModels forMicroarray (LIMMA) packagewas used for expression cal-culations (128). Bonferroni-corrected Student’s t testswere used to calculateP values. All analyseswere done in theR programming and language softwareenvironment using packages available through Bioconductor (129).
SUPPLEMENTARY MATERIALSwww.sciencesignaling.org/cgi/content/full/7/324/ra42/DC1Fig. S1. YAP is expressed in PDAC.Fig. S2. Deletion of Yap does not affect normal pancreatic development and glucose metabolism.
w
Fig. S3. Ablation of Yap blocks PDAC development in Kras and Kras:Trp53mutant pancreas.Fig. S4. Yap is not required for oncogenic Kras–induced ADM in vitro.Fig. S5. Yap is not required for induction of ADM by acute pancreatitis in vivo.Fig. S6. Deletion of Yap does not cause significant changes in apoptosis or senescence.Fig. S7. Characterization of Yap+ and Yap− primary Kras:Trp53 mutant pancreatic ductalcells.Fig. S8. Yap selectively promotes the expression of a group of genes encoding oncogenicKras–induced secretory factors.Fig. S9. Ser367 is the primary phosphorylation site responsible for the oncogenic RAS–induced mobility shift of YAP.Table S1. Abbreviations of all the mouse genotypes used in this study.Table S2. IHC analysis of YAP in human pancreatic TMAs.Table S3. Mouse and human primers used in qRT-PCR assay.Table S4. Mouse primers used in ChIP assay.Table S5. Antibodies used in IHC and Western blot analyses.
REFERENCES AND NOTES1. A. L. Warshaw, C. Fernández-del Castillo, Pancreatic carcinoma. N. Engl. J. Med.
326, 455–465 (1992).2. A. Maitra, S. D. Leach, Disputed paternity: The uncertain ancestry of pancreatic duc-
tal neoplasia. Cancer Cell 22, 701–703 (2012).3. R. H. Hruban, R. E. Wilentz, S. E. Kern, Genetic progression in the pancreatic ducts.
Am. J. Pathol. 156, 1821–1825 (2000).4. M. Löhr, G. Klöppel, P. Maisonneuve, A. B. Lowenfels, J. Lüttges, Frequency of K-ras
mutations in pancreatic intraductal neoplasias associated with pancreatic ductal adeno-carcinoma and chronic pancreatitis: A meta-analysis. Neoplasia 7, 17–23 (2005).
5. A. J. Aguirre, N. Bardeesy, M. Sinha, L. Lopez, D. A. Tuveson, J. Horner, M. S. Redston,R. A. DePinho, Activated Kras and Ink4a/Arf deficiency cooperate to produce metastaticpancreatic ductal adenocarcinoma. Genes Dev. 17, 3112–3126 (2003).
6. N. Bardeesy, K. H. Cheng, J. H. Berger, G. C. Chu, J. Pahler, P. Olson, A. F. Hezel,J. Horner, G. Y. Lauwers, D. Hanahan, R. A. DePinho, Smad4 is dispensable fornormal pancreas development yet critical in progression and tumor biology of pan-creas cancer. Genes Dev. 20, 3130–3146 (2006).
7. S. R. Hingorani, L. Wang, A. S. Multani, C. Combs, T. B. Deramaudt, R. H. Hruban,A. K. Rustgi, S. Chang, D. A. Tuveson, Trp53R172H and KrasG12D cooperate to pro-mote chromosomal instability and widely metastatic pancreatic ductal adenocarcino-ma in mice. Cancer Cell 7, 469–483 (2005).
8. S. R. Hingorani, E. F. Petricoin, A. Maitra, V. Rajapakse, C. King, M. A. Jacobetz, S. Ross,T. P. Conrads, T. D. Veenstra, B. A. Hitt, Y. Kawaguchi, D. Johann, L. A. Liotta,H. C. Crawford, M. E. Putt, T. Jacks, C. V.Wright, R. H. Hruban, A. M. Lowy, D. A. Tuveson,Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse.Cancer Cell 4, 437–450 (2003).
9. Y. Wang, C. E. Kaiser, B. Frett, H. Y. Li, Targeting mutant KRAS for anticancer ther-apeutics: A review of novel small molecule modulators. J. Med. Chem. 56, 5219–5230(2013).
10. H. Thompson, US National Cancer Institute’s new Ras project targets an old foe.Nat. Med. 19, 949–950 (2013).
11. I. Heid, C. Lubeseder-Martellato, B. Sipos, P. K. Mazur, M. Lesina, R. M. Schmid,J. T. Siveke, Early requirement of Rac1 in a mouse model of pancreatic cancer. Gas-troenterology 141, 719–730.e7 (2011).
12. M. Lesina, M. U. Kurkowski, K. Ludes, S. Rose-John, M. Treiber, G. Klöppel, A. Yoshimura,W. Reindl, B. Sipos, S. Akira, R. M. Schmid, H. Algül, Stat3/Socs3 activation by IL-6transsignaling promotes progression of pancreatic intraepithelial neoplasia and devel-opment of pancreatic cancer. Cancer Cell 19, 456–469 (2011).
13. A. Fukuda, S. C. Wang, J. P. Morris IV, A. E. Folias, A. Liou, G. E. Kim, S. Akira,K. M. Boucher, M. A. Firpo, S. J. Mulvihill, M. Hebrok, Stat3 and MMP7 contribute to pan-creatic ductal adenocarcinoma initiation and progression. Cancer Cell 19, 441–455 (2011).
14. J. Ling, Y. Kang, R. Zhao, Q. Xia, D. F. Lee, Z. Chang, J. Li, B. Peng, J. B. Fleming,H. Wang, J. Liu, I. R. Lemischka, M. C. Hung, P. J. Chiao, KrasG12D-induced IKK2/b/NF-kBactivation by IL-1a and p62 feedforward loops is required for development of pancreaticductal adenocarcinoma. Cancer Cell 21, 105–120 (2012).
15. H. J. Maier, M. Wagner, T. G. Schips, H. H. Salem, B. Baumann, T. Wirth, Require-ment of NEMO/IKKg for effective expansion of KRAS-induced precancerous lesionsin the pancreas. Oncogene 32, 2690–2695 (2013).
16. E. Maniati, M. Bossard, N. Cook, J. B. Candido, N. Emami-Shahri, S. A. Nedospasov,F. R. Balkwill, D. A. Tuveson, T. Hagemann, Crosstalk between the canonical NF-kBand Notch signaling pathways inhibits Pparg expression and promotes pancreaticcancer progression in mice. J. Clin. Invest. 121, 4685–4699 (2011).
17. M. Rajurkar, W. E. De Jesus-Monge, D. R. Driscoll, V. A. Appleman, H. Huang, J. L. Cotton,D. S. Klimstra, L. J. Zhu, K. Simin, L. Xu, A. P. McMahon, B. C. Lewis, J. Mao, The activityof Gli transcription factors is essential for Kras-induced pancreatic tumorigenesis.Proc. Natl.Acad. Sci. U.S.A. 109, E1038–E1047 (2012).
18. L. D. Mills, Y. Zhang, R. J. Marler, M. Herreros-Villanueva, L. Zhang, L. L. Almada, F. Couch,C. Wetmore, M. Pasca di Magliano, M. E. Fernandez-Zapico, Loss of the transcription
ww.SCIENCESIGNALING.org 6 May 2014 Vol 7 Issue 324 ra42 11

R E S E A R C H A R T I C L E
on May 6, 2014
stke.sciencemag.org
Dow
nloaded from
factor GLI1 identifies a signaling network in the tumor microenvironment mediatingKRAS oncogene–induced transformation. J. Biol. Chem. 288, 11786–11794 (2013).
19. C. Navas, I. Hernández-Porras, A. J. Schuhmacher, M. Sibilia, C. Guerra, M. Barbacid,EGF receptor signaling is essential for K-Ras oncogene-driven pancreatic ductal ade-nocarcinoma. Cancer Cell 22, 318–330 (2012).
20. C. M. Ardito, B. M. Grüner, K. K. Takeuchi, C. Lubeseder-Martellato, N. Teichmann,P. K. Mazur, K. E. Delgiorno, E. S. Carpenter, C. J. Halbrook, J. C. Hall, D. Pal, T. Briel,A. Herner, M. Trajkovic-Arsic, B. Sipos, G. Y. Liou, P. Storz, N. R. Murray, D. W. Threadgill,M. Sibilia, M. K. Washington, C. L. Wilson, R. M. Schmid, E. W. Raines, H. C. Crawford,J. T. Siveke, EGF receptor is required for KRAS-induced pancreatic tumorigenesis.Cancer Cell 22, 304–317 (2012).
21. S. Eser, N. Reiff, M. Messer, B. Seidler, K. Gottschalk, M. Dobler, M. Hieber, A. Arbeiter,S. Klein, B. Kong, C. W. Michalski, A. M. Schlitter, I. Esposito, A. J. Kind, L. Rad,A. E. Schnieke, M. Baccarini, D. R. Alessi, R. Rad, R. M. Schmid, G. Schneider,D. Saur, Selective requirement of PI3K/PDK1 signaling for Kras oncogene-drivenpancreatic cell plasticity and cancer. Cancer Cell 23, 406–420 (2013).
22. J. L. Kopp, G. von Figura, E. Mayes, F. F. Liu, C. L. Dubois, J. P. Morris 4th, F. C. Pan,H. Akiyama, C. V. Wright, K. Jensen, M. Hebrok, M. Sander, Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation ofpancreatic ductal adenocarcinoma. Cancer Cell 22, 737–750 (2012).
23. P. A. Pérez-Mancera, C. Guerra, M. Barbacid, D. A. Tuveson, What we have learnedabout pancreatic cancer from mouse models. Gastroenterology 142, 1079–1092(2012).
24. Y. Wang, Q. Dong, Q. Zhang, Z. Li, E. Wang, X. Qiu, Overexpression of yes-associatedprotein contributes to progression and poor prognosis of non-small-cell lung cancer.Cancer Sci. 101, 1279–1285 (2010).
25. C. A. Hall, R. Wang, J. Miao, E. Oliva, X. Shen, T. Wheeler, S. G. Hilsenbeck, S. Orsulic,S. Goode, Hippo pathway effector Yap is an ovarian cancer oncogene. Cancer Res. 70,8517–8525 (2010).
26. X. Zhang, J. George, S. Deb, J. L. Degoutin, E. A. Takano, S. B. Fox, AOCS Studygroup, D. D. Bowtell, K. F. Harvey, The Hippo pathway transcriptional co-activator,YAP, is an ovarian cancer oncogene. Oncogene 30, 2810–2822 (2011).
27. W. Kang, J. H. Tong, A. W. Chan, T. L. Lee, R. W. Lung, P. P. Leung, K. K. So, K. Wu,D. Fan, J. Yu, J. J. Sung, K. F. To, Yes-associated protein 1 exhibits oncogenic propertyin gastric cancer and its nuclear accumulation associates with poor prognosis. Clin.Cancer Res. 17, 2130–2139 (2011).
28. L. Zhang, D. X. Ye, H. Y. Pan, K. J. Wei, L. Z. Wang, X. D. Wang, G. F. Shen, Z. Y. Zhang,Yes-associated protein promotes cell proliferation by activating Fos Related Activator-1 in oral squamous cell carcinoma. Oral Oncol. 47, 693–697 (2011).
29. Z. Hélias-Rodzewicz, G. Pérot, F. Chibon, C. Ferreira, P. Lagarde, P. Terrier, J. M. Coindre,A. Aurias, YAP1 and VGLL3, encoding two cofactors of TEAD transcription factors, areamplified and overexpressed in a subset of soft tissue sarcomas.Genes ChromosomesCancer 49, 1161–1171 (2010).
30. B. A. Orr, H. Bai, Y. Odia, D. Jain, R. A. Anders, C. G. Eberhart, Yes-associatedprotein 1 is widely expressed in human brain tumors and promotes glioblastomagrowth. J. Neuropathol. Exp. Neurol. 70, 568–577 (2011).
31. T. Yokoyama, H. Osada, H. Murakami, Y. Tatematsu, T. Taniguchi, Y. Kondo, Y. Yatabe,Y. Hasegawa, K. Shimokata, Y. Horio, T. Hida, Y. Sekido, YAP1 is involved in mesotheli-oma development and negatively regulated byMerlin through phosphorylation.Carcinogen-esis 29, 2139–2146 (2008).
32. T. Muramatsu, I. Imoto, T. Matsui, K. Kozaki, S. Haruki, M. Sudol, Y. Shimada, H. Tsuda,T. Kawano, J. Inazawa, YAP is a candidate oncogene for esophageal squamous cellcarcinoma. Carcinogenesis 32, 389–398 (2011).
33. L. Wang, S. Shi, Z. Guo, X. Zhang, S. Han, A. Yang, W. Wen, Q. Zhu, Overexpres-sion of YAP and TAZ is an independent predictor of prognosis in colorectal cancerand related to the proliferation and metastasis of colon cancer cells. PLOS One 8,e65539 (2013).
34. T. Liu, Y. Liu, H. Gao, F. Meng, S. Yang, G. Lou, Clinical significance of Yes-associatedprotein overexpression in cervical carcinoma: The differential effects based on histo-types. Int. J. Gynecol. Cancer 23, 735–742 (2013).
35. L. L. Su, W. X. Ma, J. F. Yuan, Y. Shao, W. Xiao, S. J. Jiang, Expression of Yes-associated protein in non-small cell lung cancer and its relationship with clinicalpathological factors. Chin. Med. J. 125, 4003–4008 (2012).
36. J. Zhang, Z. P. Xu, Y. C. Yang, J. S. Zhu, Z. Zhou, W. X. Chen, Expression of Yes-associated protein in gastric adenocarcinoma and inhibitory effects of its knockdownon gastric cancer cell proliferation and metastasis. Int. J. Immunopathol. Pharmacol.25, 583–590 (2012).
37. M. Sudol, P. Bork, A. Einbond, K. Kastury, T. Druck, M. Negrini, K. Huebner, D. Lehman,Characterization of the mammalian YAP (Yes-associated protein) gene and its role indefining a novel protein module, the WW domain. J. Biol. Chem. 270, 14733–14741(1995).
38. M. Sudol, Yes-associated protein (YAP65) is a proline-rich phosphoprotein that bindsto the SH3 domain of the Yes proto-oncogene product. Oncogene 9, 2145–2152(1994).
w
39. F. Kanai, P. A. Marignani, D. Sarbassova, R. Yagi, R. A. Hall, M. Donowitz, A. Hisaminato,T. Fujiwara, Y. Ito, L. C. Cantley, M. B. Yaffe, TAZ: A novel transcriptional co-activatorregulated by interactions with 14-3-3 and PDZ domain proteins. EMBO J. 19, 6778–6791(2000).
40. R. Yagi, L. F. Chen, K. Shigesada, Y. Murakami, Y. Ito, A WW domain-containingyes-associated protein (YAP) is a novel transcriptional co-activator. EMBO J. 18,2551–2562 (1999).
41. A. Vassilev, K. J. Kaneko, H. Shu, Y. Zhao, M. L. DePamphilis, TEAD/TEFtranscription factors utilize the activation domain of YAP65, a Src/Yes-associatedprotein localized in the cytoplasm. Genes Dev. 15, 1229–1241 (2001).
42. R. W. Justice, O. Zilian, D. F. Woods, M. Noll, P. J. Bryant, The Drosophila tumorsuppressor gene warts encodes a homolog of human myotonic dystrophy kinaseand is required for the control of cell shape and proliferation. Genes Dev. 9, 534–546(1995).
43. K. F. Harvey, C. M. Pfleger, I. K. Hariharan, The Drosophila Mst ortholog, hippo,restricts growth and cell proliferation and promotes apoptosis. Cell 114, 457–467(2003).
44. S. Wu, J. Huang, J. Dong, D. Pan, hippo encodes a Ste-20 family protein kinase thatrestricts cell proliferation and promotes apoptosis in conjunction with salvador andwarts. Cell 114, 445–456 (2003).
45. N. Tapon, K. F. Harvey, D. W. Bell, D. C. Wahrer, T. A. Schiripo, D. Haber, I. K. Hariharan,salvador promotes both cell cycle exit and apoptosis in Drosophila and is mutated inhuman cancer cell lines. Cell 110, 467–478 (2002).
46. Z. C. Lai, X. Wei, T. Shimizu, E. Ramos, M. Rohrbaugh, N. Nikolaidis, L. L. Ho, Y. Li,Control of cell proliferation and apoptosis by Mob as tumor suppressor, Mats. Cell120, 675–685 (2005).
47. J. Dong, G. Feldmann, J. Huang, S. Wu, N. Zhang, S. A. Comerford, M. F. Gayyed,R. A. Anders, A. Maitra, D. Pan, Elucidation of a universal size-control mechanism inDrosophila and mammals. Cell 130, 1120–1133 (2007).
48. B. Zhao, X. Wei, W. Li, R. S. Udan, Q. Yang, J. Kim, J. Xie, T. Ikenoue, J. Yu, L. Li,P. Zheng, K. Ye, A. Chinnaiyan, G. Halder, Z. C. Lai, K. L. Guan, Inactivation of YAPoncoprotein by the Hippo pathway is involved in cell contact inhibition and tissuegrowth control. Genes Dev. 21, 2747–2761 (2007).
49. F.D.Camargo,S.Gokhale,J.B.Johnnidis,D.Fu,G.W.Bell,R.Jaenisch,T.R.Brummelkamp,YAP1 increases organ size and expands undifferentiated progenitor cells. Curr. Biol. 17,2054–2060 (2007).
50. Q. Zeng, W. Hong, The emerging role of the Hippo pathway in cell contact inhibition,organ size control, and cancer development in mammals. Cancer Cell 13, 188–192(2008).
51. K. Harvey, N. Tapon, The Salvador–Warts–Hippo pathway—An emerging tumour-suppressor network. Nat. Rev. Cancer 7, 182–191 (2007).
52. R. S. Udan, M. Kango-Singh, R. Nolo, C. Tao, G. Halder, Hippo promotes prolifer-ation arrest and apoptosis in the Salvador/Warts pathway. Nat. Cell Biol. 5, 914–920(2003).
53. J. Huang, S. Wu, J. Barrera, K. Matthews, D. Pan, The Hippo signaling pathwaycoordinately regulates cell proliferation and apoptosis by inactivating Yorkie, theDrosophila homolog of YAP. Cell 122, 421–434 (2005).
54. Y. Hao, A. Chun, K. Cheung, B. Rashidi, X. Yang, Tumor suppressor LATS1 is anegative regulator of oncogene YAP. J. Biol. Chem. 283, 5496–5509 (2008).
55. B. Zhao, L. Li, K. Tumaneng, C. Y. Wang, K. L. Guan, A coordinated phosphorylationby Lats and CK1 regulates YAP stability through SCFb-TRCP. Genes Dev. 24, 72–85(2010).
56. B. V. Reddy, K. D. Irvine, Regulation of Hippo signaling by EGFR-MAPK signalingthrough Ajuba family proteins. Dev. Cell 24, 459–471 (2013).
57. R. Fan, N. G. Kim, B. M. Gumbiner, Regulation of Hippo pathway by mitogenic growthfactors via phosphoinositide 3-kinase and phosphoinositide-dependent kinase-1.Proc. Natl. Acad. Sci. U.S.A. 110, 2569–2574 (2013).
58. J. S. Mo, F. X. Yu, R. Gong, J. H. Brown, K. L. Guan, Regulation of the Hippo-YAPpathway by protease-activated receptors (PARs). Genes Dev. 26, 2138–2143(2012).
59. S. Piccolo, M. Cordenonsi, S. Dupont, Molecular PATHWAYS: YAP and TAZ takecenter stage in organ growth and tumorigenesis. Clin. Cancer Res. 19, 4925–4930(2013).
60. D. Chen, Y. Sun, Y. Wei, P. Zhang, A. H. Rezaeian, J. Teruya-Feldstein, S. Gupta,H. Liang, H. K. Lin, M. C. Hung, L. Ma, LIFR is a breast cancer metastasis suppres-sor upstream of the Hippo-YAP pathway and a prognostic marker. Nat. Med. 18,1511–1517 (2012).
61. M. J. Oudhoff, S. A. Freeman, A. L. Couzens, F. Antignano, E. Kuznetsova, P. H. Min,J. P. Northrop, B. Lehnertz, D. Barsyte-Lovejoy, M. Vedadi, C. H. Arrowsmith, H. Nishina,M. R. Gold, F. M. Rossi, A. C. Gingras, C. Zaph, Control of the Hippo pathway by Set7-dependent methylation of Yap. Dev. Cell 26, 188–194 (2013).
62. H. B. Nguyen, J. T. Babcock, C. D. Wells, L. A. Quilliam, LKB1 tumor suppressorregulates AMP kinase/mTOR-independent cell growth and proliferation via the phos-phorylation of Yap. Oncogene 32, 4100–4109 (2013).
ww.SCIENCESIGNALING.org 6 May 2014 Vol 7 Issue 324 ra42 12

R E S E A R C H A R T I C L E
on May 6, 2014
stke.sciencemag.org
Dow
nloaded from
63. J. Wang, J. S. Park, Y. Wei, M. Rajurkar, J. L. Cotton, Q. Fan, B. C. Lewis, H. Ji, J. Mao,TRIB2 acts downstream of Wnt/TCF in liver cancer cells to regulate YAP and C/EBPafunction. Mol. Cell 51, 211–225 (2013).
64. W. M. Konsavage Jr., S. L. Kyler, S. A. Rennoll, G. Jin, G. S. Yochum, Wnt/b-cateninsignaling regulates Yes-associated protein (YAP) gene expression in colorectal car-cinoma cells. J. Biol. Chem. 287, 11730–11739 (2012).
65. V. Tomlinson, K. Gudmundsdottir, P. Luong, K. Y. Leung, A. Knebel, S. Basu, JNKphosphorylates Yes-associated protein (YAP) to regulate apoptosis. Cell Death Dis.1, e29 (2010).
66. K. K. Lee, S. Yonehara, Identification of mechanism that couples multisite phospho-rylation of Yes-associated protein (YAP) with transcriptional coactivation and regula-tion of apoptosis. J. Biol. Chem. 287, 9568–9578 (2012).
67. S. Hata, J. Hirayama, H. Kajiho, K. Nakagawa, Y. Hata, T. Katada, M. Furutani-Seiki,H. Nishina, A novel acetylation cycle of transcription co-activator Yes-associatedprotein that is downstream of Hippo pathway is triggered in response to SN2 alkylat-ing agents. J. Biol. Chem. 287, 22089–22098 (2012).
68. L. Lu, Y. Li, S. M. Kim, W. Bossuyt, P. Liu, Q. Qiu, Y. Wang, G. Halder, M. J. Finegold,J. S. Lee, R. L. Johnson, Hippo signaling is a potent in vivo growth and tumor sup-pressor pathway in the mammalian liver. Proc. Natl. Acad. Sci. U.S.A. 107, 1437–1442(2010).
69. D. Zhou, C. Conrad, F. Xia, J. S. Park, B. Payer, Y. Yin, G. Y. Lauwers, W. Thasler,J. T. Lee, J. Avruch, N. Bardeesy, Mst1 and Mst2 maintain hepatocyte quiescenceand suppress hepatocellular carcinoma development through inactivation of theYap1 oncogene. Cancer Cell 16, 425–438 (2009).
70. T. Gao, D. Zhou, C. Yang, T. Singh, A. Penzo-Méndez, R. Maddipati, A. Tzatsos, N. Bardeesy,J. Avruch, B. Z. Stanger, Hippo signaling regulates differentiation and maintenance inthe exocrine pancreas. Gastroenterology 144, 1543–1553.e1 (2013).
71. N. M. George, C. E. Day, B. P. Boerner, R. L. Johnson, N. E. Sarvetnick, Hippo sig-naling regulates pancreas development through inactivation of Yap. Mol. Cell. Biol.32, 5116–5128 (2012).
72. J. Guo, J. Kleeff, Y. Zhao, J. Li, T. Giese, I. Esposito, M. W. Büchler, M. Korc, H. Friess,Yes-associated protein (YAP65) in relation to Smad7 expression in human pancreaticductal adenocarcinoma. Int. J. Mol. Med. 17, 761–767 (2006).
73. C. H. Diep, K. M. Zucker, G. Hostetter, A. Watanabe, C. Hu, R. M. Munoz, D. D. Von Hoff,H. Han, Down-regulation of Yes Associated Protein 1 expression reduces cell proliferationand clonogenicity of pancreatic cancer cells. PLOS One 7, e32783 (2012).
74. L. Badea, V. Herlea, S. O. Dima, T. Dumitrascu, I. Popescu, Combined gene expres-sion analysis of whole-tissue and microdissected pancreatic ductal adenocarcinomaidentifies genes specifically overexpressed in tumor epithelia. Hepatogastroenterology55, 2016–2027 (2008).
75. H. Pei, L. Li, B. L. Fridley, G. D. Jenkins, K. R. Kalari, W. Lingle, G. Petersen, Z. Lou,L. Wang, FKBP51 affects cancer cell response to chemotherapy by negatively reg-ulating Akt. Cancer Cell 16, 259–266 (2009).
76. N. Hiraoka, R. Yamazaki-Itoh, Y. Ino, Y. Mizuguchi, T. Yamada, S. Hirohashi, Y. Kanai,CXCL17 and ICAM2 are associated with a potential anti-tumor immune response inearly intraepithelial stages of human pancreatic carcinogenesis. Gastroenterology140, 310–321 (2011).
77. G. Sergeant, R. van Eijsden, T. Roskams, V. Van Duppen, B. Topal, Pancreaticcancer circulating tumour cells express a cell motility gene signature that predictssurvival after surgery. BMC Cancer 12, 527 (2012).
78. C. Guerra, A. J. Schuhmacher, M. Cañamero, P. J. Grippo, L. Verdaguer, L. Pérez-Gallego,P. Dubus, E. P. Sandgren, M. Barbacid, Chronic pancreatitis is essential for induction ofpancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 11,291–302 (2007).
79. D. A. Tuveson, A. T. Shaw, N. A.Willis, D. P. Silver, E. L. Jackson, S. Chang, K. L.Mercer,R. Grochow, H. Hock, D. Crowley, S. R. Hingorani, T. Zaks, C. King, M. A. Jacobetz,L. Wang, R. T. Bronson, S. H. Orkin, R. A. DePinho, T. Jacks, Endogenous onco-genic K-rasG12D stimulates proliferation and widespread neoplastic and develop-mental defects. Cancer Cell 5, 375–387 (2004).
80. R. Hill, Y. Li, L. M. Tran, S. Dry, J. H. Calvopina, A. Garcia, C. Kim, Y. Wang, T. R. Donahue,H. R. Herschman, H. Wu, Cell intrinsic role of COX-2 in pancreatic cancer development.Mol. Cancer Ther. 11, 2127–2137 (2012).
81. A. Neesse, K. K. Frese, T. E. Bapiro, T. Nakagawa, M. D. Sternlicht, T. W. Seeley,C. Pilarsky, D. I. Jodrell, S. M. Spong, D. A. Tuveson, CTGF antagonism with mAbFG-3019 enhances chemotherapy response without increasing drug delivery in mu-rine ductal pancreas cancer. Proc. Natl. Acad. Sci. U.S.A. 110, 12325–12330(2013).
82. I. Haque, S. Mehta, M. Majumder, K. Dhar, A. De, D. McGregor, P. J. Van Veldhuizen,S. K. Banerjee, S. Banerjee, Cyr61/CCN1 signaling is critical for epithelial-mesenchymaltransition and stemness and promotes pancreatic carcinogenesis. Mol. Cancer 10, 8(2011).
83. I. Haque, A. De, M. Majumder, S. Mehta, D. McGregor, S. K. Banerjee, P. Van Veldhuizen,S. Banerjee, The matricellular protein CCN1/Cyr61 is a critical regulator of SonicHedgehog in pancreatic carcinogenesis. J. Biol. Chem. 287, 38569–38579 (2012).
w
84. A. Kirane, J. E. Toombs, K. Ostapoff, J. G. Carbon, S. Zaknoen, J. Braunfeld,R. E. Schwarz, F. J. Burrows, R. A. Brekken, Apricoxib, a novel inhibitor of COX-2,markedly improves standard therapy response in molecularly defined models of pancre-atic cancer. Clin. Cancer Res. 18, 5031–5042 (2012).
85. D. Pan, The Hippo signaling pathway in development and cancer. Dev. Cell 19,491–505 (2010).
86. B. Zhao, X. Ye, J. Yu, L. Li, W. Li, S. Li, J. D. Lin, C. Y.Wang, A. M. Chinnaiyan, Z. C. Lai,K. L. Guan, TEAD mediates YAP-dependent gene induction and growth control. GenesDev. 22, 1962–1971 (2008).
87. D. Lai, K. C. Ho, Y. Hao, X. Yang, Taxol resistance in breast cancer cells is mediatedby the Hippo pathway component TAZ and its downstream transcriptional targetsCyr61 and CTGF. Cancer Res. 71, 2728–2738 (2011).
88. K. L. Bennewith, X. Huang, C. M. Ham, E. E. Graves, J. T. Erler, N. Kambham, J. Feazell,G. P. Yang, A. Koong, A. J. Giaccia, The role of tumor cell–derived connective tissuegrowth factor (CTGF/CCN2) in pancreatic tumor growth. Cancer Res. 69, 775–784(2009).
89. M. Hartel, F. F. Di Mola, A. Gardini, A. Zimmermann, P. Di Sebastiano, A. Guweidhi,P. Innocenti, T. Giese, N. Giese, M. W. Büchler, H. Friess, Desmoplastic reactioninfluences pancreatic cancer growth behavior. World J. Surg. 28, 818–825 (2004).
90. C. A. Iacobuzio-Donahue, B. Ryu, R. H. Hruban, S. E. Kern, Exploring the host des-moplastic response to pancreatic carcinoma: Gene expression of stromal and neo-plastic cells at the site of primary invasion. Am. J. Pathol. 160, 91–99 (2002).
91. J. K. Colby, R. D. Klein, M. J. McArthur, C. J. Conti, K. Kiguchi, T. Kawamoto, P. K. Riggs,A. I. Pavone, J. Sawicki, S. M. Fischer, Progressive metaplastic and dysplastic changes inmouse pancreas induced by cyclooxygenase-2 overexpression. Neoplasia 10, 782–796(2008).
92. K. Müller-Decker, G. Fürstenberger, N. Annan, D. Kucher, A. Pohl-Arnold, B. Steinbauer,I. Esposito, S. Chiblak, H. Friess, P. Schirmacher, I. Berger, Preinvasive duct-derivedneoplasms in pancreas of keratin 5–promoter cyclooxygenase-2 transgenic mice. Gas-troenterology 130, 2165–2178 (2006).
93. M. Groblewska, M. Siewko, B. Mroczko, M. Szmitkowski, The role of matrix metal-loproteinases (MMPs) and their inhibitors (TIMPs) in the development of esophagealcancer. Folia Histochem. Cytobiol. 50, 12–19 (2012).
94. V. Tjomsland, A. Spangeus, J. Valila, P. Sandstrom, K. Borch, H. Druid, S. Falkmer,U. Falkmer, D. Messmer, M. Larsson, Interleukin 1a sustains the expression of in-flammatory factors in human pancreatic cancer microenvironment by targetingcancer-associated fibroblasts. Neoplasia 13, 664–675 (2011).
95. L. W. Feurino, Y. Zhang, U. Bharadwaj, R. Zhang, F. Li, W. E. Fisher, F. C. Brunicardi,C. Chen, Q. Yao, L. Min, IL-6 stimulates Th2 type cytokine secretion and upregulatesVEGF and NRP-1 expression in pancreatic cancer cells. Cancer Biol. Ther. 6, 1096–1100(2007).
96. C. Feig, A. Gopinathan, A. Neesse, D. S. Chan, N. Cook, D. A. Tuveson, The pan-creas cancer microenvironment. Clin. Cancer Res. 18, 4266–4276 (2012).
97. B. Hinz, Formation and function of the myofibroblast during tissue repair. J. Invest.Dermatol. 127, 526–537 (2007).
98. C. E. Clark, S. R. Hingorani, R. Mick, C. Combs, D. A. Tuveson, R. H. Vonderheide,Dynamics of the immune reaction to pancreatic cancer from inception to invasion.Cancer Res. 67, 9518–9527 (2007).
99. Z. Li, B. Zhao, P. Wang, F. Chen, Z. Dong, H. Yang, K. L. Guan, Y. Xu, Structuralinsights into the YAP and TEAD complex. Genes Dev. 24, 235–240 (2010).
100. P. K. Mazur, H. Einwächter, M. Lee, B. Sipos, H. Nakhai, R. Rad, U. Zimber-Strobl,L. J. Strobl, F. Radtke, G. Klöppel, R. M. Schmid, J. T. Siveke, Notch2 is required forprogression of pancreatic intraepithelial neoplasia and development of pancreaticductal adenocarcinoma. Proc. Natl. Acad. Sci. U.S.A. 107, 13438–13443 (2010).
101. H. Funahashi, M. Satake, D. Dawson, N. A. Huynh, H. A. Reber, O. J. Hines, G. Eibl,Delayed progression of pancreatic intraepithelial neoplasia in a conditional KrasG12D
mouse model by a selective cyclooxygenase-2 inhibitor. Cancer Res. 67, 7068–7071(2007).
102. N. Zhang, H. Bai, K. K. David, J. Dong, Y. Zheng, J. Cai, M. Giovannini, P. Liu, R. A. Anders,D. Pan, The Merlin/NF2 tumor suppressor functions through the YAP oncoprotein toregulate tissue homeostasis in mammals. Dev. Cell 19, 27–38 (2010).
103. F. A. Karreth, K. K. Frese, G. M. DeNicola, M. Baccarini, D. A. Tuveson, C-Raf isrequired for the initiation of lung cancer by K-RasG12D. Cancer Discov. 1, 128–136(2011).
104. R. B. Blasco, S. Francoz, D. Santamaría, M. Cañamero, P. Dubus, J. Charron, M. Baccarini,M. Barbacid, c-Raf, but not B-Raf, is essential for development of K-Ras oncogene-driven non-small cell lung carcinoma. Cancer Cell 19, 652–663 (2011).
105. C. E. Foulds, M. L. Nelson, A. G. Blaszczak, B. J. Graves, Ras/mitogen-activatedprotein kinase signaling activates Ets-1 and Ets-2 by CBP/p300 recruitment. Mol.Cell. Biol. 24, 10954–10964 (2004).
106. G. Behre, S. M. Singh, H. Liu, L. T. Bortolin, M. Christopeit, H. S. Radomska, J. Rangatia,W.Hiddemann,A.D. Friedman,D.G. Tenen,Rassignalingenhances theactivity ofC/EBPato induce granulocytic differentiation by phosphorylation of serine 248. J. Biol. Chem. 277,26293–26299 (2002).
ww.SCIENCESIGNALING.org 6 May 2014 Vol 7 Issue 324 ra42 13

R E S E A R C H A R T I C L E
on May 6, 2014
stke.sciencemag.org
Dow
nloaded from
107. P. Monje, M. J. Marinissen, J. S. Gutkind, Phosphorylation of the carboxyl-terminaltransactivation domain of c-Fos by extracellular signal-regulated kinase mediatesthe transcriptional activation of AP-1 and cellular transformation induced by platelet-derived growth factor. Mol. Cell. Biol. 23, 7030–7043 (2003).
108. Y. Liu-Chittenden, B. Huang, J. S. Shim, Q. Chen, S. J. Lee, R. A. Anders, J. O. Liu,D. Pan, Genetic and pharmacological disruption of the TEAD–YAP complex sup-presses the oncogenic activity of YAP. Genes Dev. 26, 1300–1305 (2012).
109. M. Sudol, D. C. Shields, A. Farooq, Structures of YAP protein domains revealpromising targets for development of new cancer drugs. Semin. Cell Dev. Biol.23, 827–833 (2012).
110. Y. Bao, K. Nakagawa, Z. Yang, M. Ikeda, K. Withanage, M. Ishigami-Yuasa, Y. Okuno,S. Hata, H. Nishina, Y. Hata, A cell-based assay to screen stimulators of the Hippopathway reveals the inhibitory effect of dobutamine on the YAP-dependent genetranscription. J. Biochem. 150, 199–208 (2011).
111. H. W. Park, K. L. Guan, Regulation of the Hippo pathway and implications for anti-cancer drug development. Trends Pharmacol. Sci. 34, 581–589 (2013).
112. S. Jiao, H. Wang, Z. Shi, A. Dong, W. Zhang, X. Song, F. He, Y. Wang, Z. Zhang,W. Wang, X. Wang, T. Guo, P. Li, Y. Zhao, H. Ji, L. Zhang, Z. Zhou, A peptidemimicking VGLL4 function acts as a YAP antagonist therapy against gastric cancer.Cancer Cell 25, 166–180 (2014).
113. T. Dragovich, H. Burris III, P. Loehrer, D. D. Von Hoff, S. Chow, S. Stratton, S. Green,Y. Obregon, I. Alvarez, M. Gordon, Gemcitabine plus celecoxib in patients with ad-vanced or metastatic pancreatic adenocarcinoma: Results of a phase II trial. Am. J.Clin. Oncol. 31, 157–162 (2008).
114. L. M. Coussens, B. Fingleton, L. M. Matrisian, Matrix metalloproteinase inhibitorsand cancer: Trials and tribulations. Science 295, 2387–2392 (2002).
115. S. Zucker, M. Hymowitz, E. E. Rollo, R. Mann, C. E. Conner, J. Cao, H. D. Foda,D. C. Tompkins, B. P. Toole, Tumorigenic potential of extracellular matrix metalloprotei-nase inducer. Am. J. Pathol. 158, 1921–1928 (2001).
116. N. Dornhöfer, S. Spong, K. Bennewith, A. Salim, S. Klaus, N. Kambham, C. Wong,F. Kaper, P. Sutphin, R. Nacamuli, M. Höckel, Q. Le, M. Longaker, G. Yang, A. Koong,A. Giaccia, Connective tissue growth factor–specific monoclonal antibody therapy inhib-its pancreatic tumor growth and metastasis. Cancer Res. 66, 5816–5827 (2006).
117. S. A. Jones, J. Scheller, S. Rose-John, Therapeutic strategies for the clinical block-ade of IL-6/gp130 signaling. J. Clin. Invest. 121, 3375–3383 (2011).
118. L. Johnson, K. Mercer, D. Greenbaum, R. T. Bronson, D. Crowley, D. A. Tuveson, T. Jacks,Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature410, 1111–1116 (2001).
119. K. P. Olive, D. A. Tuveson, Z. C. Ruhe, B. Yin, N. A. Willis, R. T. Bronson, D. Crowley,T. Jacks, Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome.Cell 119, 847–860 (2004).
120. Y. Kawaguchi, B. Cooper, M. Gannon, M. Ray, R. J. MacDonald, C. V. Wright, Therole of the transcriptional regulator Ptf1a in converting intestinal to pancreatic pro-genitors. Nat. Genet. 32, 128–134 (2002).
121. C. Yi, Z. Shen, A. Stemmer-Rachamimov, N. Dawany, S. Troutman, L. C. Showe,Q. Liu, A. Shimono, M. Sudol, L. Holmgren, B. Z. Stanger, J. L. Kissil, The p130 isoformof angiomotin is required for Yap-mediated hepatic epithelial cell proliferation and tumor-igenesis. Sci. Signal. 6, ra77 (2013).
122. R. C. De Lisle, C. D. Logsdon, Pancreatic acinar cells in culture: Expression of ac-inar and ductal antigens in a growth-related manner. Eur. J. Cell Biol. 51, 64–75(1990).
123. S. Githens, C. L. Patke, J. A. Schexnayder, Isolation and culture of rhesus monkeypancreatic ductules and ductule-like epithelium. Pancreas 9, 20–31 (1994).
w
124. C. Yi, S. Troutman, D. Fera, A. Stemmer-Rachamimov, J. L. Avila, N. Christian,N. L. Persson, A. Shimono, D. W. Speicher, R. Marmorstein, L. Holmgren, J. L. Kissil,A tight junction-associated Merlin-angiomotin complex mediates Merlin’s regulation ofmitogenic signaling and tumor suppressive functions. Cancer Cell 19, 527–540 (2011).
125. B. L. Huang, S. M. Brugger, K. M. Lyons, Stage-specific control of connective tissuegrowth factor (CTGF/CCN2) expression in chondrocytes by Sox9 and b-catenin. J.Biol. Chem. 285, 27702–27712 (2010).
126. Z. J. Wu, R. A. Irizarry, R. Gentleman, F. Martinez-Murillo, F. Spencer, A model-basedbackground adjustment for oligonucleotide expression arrays. J. Am. Stat. Assoc. 99,909–917 (2004).
127. L. Barrera, C. Benner, Y. C. Tao, E. Winzeler, Y. Zhou, Leveraging two-way probe-level block design for identifying differential gene expression with high-density oligo-nucleotide arrays. BMC Bioinformatics 5, 42 (2004).
128. G. K. Smyth, Linear models and empirical Bayes methods for assessing differentialexpression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 3, Article3(2004).
129. R. C. Gentleman, V. J. Carey, D. M. Bates, B. Bolstad, M. Dettling, S. Dudoit, B. Ellis,L. Gautier, Y.Ge, J.Gentry,K.Hornik, T.Hothorn,W.Huber,S. Iacus,R. Irizarry, F. Leisch,C. Li, M.Maechler, A. J. Rossini, G. Sawitzki, C. Smith,G. Smyth, L. Tierney, J. Y. Yang,J. Zhang, Bioconductor: Open software development for computational biology and bio-informatics. Genome Biol. 5, R80 (2004).
Acknowledgments: We thank D. J. Pan (Johns Hopkins University) for providing us withYap conditional knockout mice and K. Lyons (UCLA) for providing us with the CTGF lu-ciferase reporter. We are thankful for the technical assistance from J. Garee in ChIP analysis,from D. L. Berry, E. Permaul, and S. Sen of Histopathology & Tissue Shared Resource(HTSR) in IHC analysis, and from P. Johnson of Lombardi Cancer Center Microscopy &Imaging Shared Resource (MISR) in microscope imaging. Funding: This work is supportedby a Georgetown Lombardi Cancer Center new faculty startup fund to C.Y., National CancerInstitute grant CA71508 to A.W., and the Burroughs Wellcome Clinical Scientist Award inTranslational Research and NIH grants R01CA133662 and R01CA138212 to J.T. TheLombardi Cancer Center Shared Resource is supported by a Cancer Center SupportGrant, CA051008. Author contributions: C.Y., W.Z., and N.N. designed and conductedthe bulk of the experiments. C.Y., W.Z., and G.G. co-wrote the manuscript. Y.S., S.Z.L.,and M.W. carried out the mouse studies. M.M., A.S., M.C., and S.G. contributed to some ofthe in vitro experiments. G.G. analyzed the microarray data and performed other statisticalanalysis. E.E.V. generated PDAC cell lines. M.J. and F.W. contributed to IHC analysis. B.K.performed pathological evaluations of all human and mouse pancreatic tissues. A.W. andJ.T. provided intellectual input into the design and presentation of the study. C.Y., A.W., andJ.T. obtained funding for the project. Competing interests: Georgetown University is filing apatent application related to technology described in this paper. One or more authors haveintellectual property interests in the technology related to this research.
Submitted 6 January 2014Accepted 11 April 2014Final Publication 6 May 201410.1126/scisignal.2005049Citation: W. Zhang, N. Nandakumar, Y. Shi, M. Manzano, A. Smith, G. Graham,S. Gupta, E. E. Vietsch, S. Z. Laughlin, M. Wadhwa, M. Chetram, M. Joshi, F. Wang,B. Kallakury, J. Toretsky, A. Wellstein, C. Yi, Downstream of mutant KRAS, thetranscription regulator YAP is essential for neoplastic progression to pancreatic ductaladenocarcinoma. Sci. Signal. 7, ra42 (2014).
ww.SCIENCESIGNALING.org 6 May 2014 Vol 7 Issue 324 ra42 14




















![Yap - Good Urban Governance in Southeast Asia [2008]](https://img.dokumen.tips/doc/110x75/631a1e57fd704e1d3909fcbd/yap-good-urban-governance-in-southeast-asia-2008.jpg)








