Embed Size (px)
Citation preview

GENES, CHROMOSOMES & CANCER 50:82–94 (2011)
Common Pathogenetic Mechanism Involving HumanChromosome 18 in Familial and Sporadic IlealCarcinoid Tumors
Janet L. Cunningham,1 Teresita Dıaz de Stahl,2 Tobias Sjoblom,2 Gunnar Westin,3
Jan P. Dumanski,2 and Eva T. Janson1*
1Departmentof Medical Sciences,Section of Endocrine Oncology,Uppsala University,Uppsala,Sweden2Departmentof Genetics and Pathology,Uppsala University,Uppsala,Sweden3Departmentof Surgical Sciences,Section of Endocrine Surgery,Uppsala University,Uppsala,Sweden
Serotonin producing endocrine carcinoma of small intestine (ileal carcinoid) is a clinically distinct endocrine tumor. It is
generally considered as a sporadic disease and its molecular etiology is poorly understood. We report comprehensive clin-
ical and molecular studies of 55 sporadic and familial patients diagnosed with this condition. Nine pedigrees encompassing
23 affected subjects were established, consistent with autosomal dominant mode of inheritance. Familial and sporadic
patients demonstrated indistinguishable clinical pictures. Molecular analyses of 61 tumors from 45 individuals, including
eight familial and 37 sporadic patients, aimed at determination of global copy number aberrations using BAC and Illumina
SNP arrays and gene expression profiling by Affymetrix chips. Chromosome 18 aberrations were identified in both spo-
radic and in familial tumors; 100% vs. 38%, respectively. Other, less frequent aberrations were also common for both
groups. Global expression profiles revealed no differentially expressed genes. Frequent gain of chromosome 7 was exclu-
sively observed in metastases, when patient matched primary tumors and metastases were compared. Notably, the latter
aberration correlated with solid growth pattern morphology (P < 0.01), a histopathological feature that has previously
been related to worse prognosis. The clinical and molecular similarities identified between sporadic and familial cases sug-
gest a common pathogenetic mechanism involved in tumor initiation. The familial variant of ileal carcinoid represents a pre-
viously unrecognized autosomal dominant inherited tumor disease, which we propose to call Familial Ileal Endocrine
Carcinoma (FIEC). Our findings indicate the location of a FIEC tumor suppressor gene near the telomere of 18q, involved
in development of inherited and sporadic tumors. VVC 2010 Wiley-Liss, Inc.
INTRODUCTION
Well differentiated serotonin producing endo-
crine carcinomas that originate in the small intes-
tine and proximal colon (the midgut) are
commonly denoted ileal carcinoids (Modlin et al.,
2008). They are malignant and most patients
present with metastatic disease. The median age
at diagnosis is 61 years and the incidence is 1-2/
100 000 inhabitants per year. The tumor is usu-
ally slow growing and the Ki67 proliferation index
is below 1% in the majority of cases (Cunning-
ham et al., 2007). Many patients present with ab-
dominal pain due to intestinal obstruction caused
by tumor growth. Hormones produced by the tu-
mor cells give rise to the carcinoid syndrome,
including flush, diarrhea, the carcinoid heart dis-
ease and bronchial constriction. The only curative
treatment is surgery, but this can be accom-
plished only in a minority of cases. First-line
medical treatment includes alpha-interferon and
somatostatin analogs of which the latter are
potent in reducing hormone secretion and thus
symptoms of the carcinoid syndrome (Modlin
et al., 2008).
Ileal carcinoid is generally considered a spo-
radic tumor. However, during the past five deca-
des some case reports of inherited variants have
been published describing families with 2 or 3
affected members (Eschbach and Rinaldo, 1962;
Moertel and Dockerty, 1973; Wale et al., 1983;
Kinova et al., 2001; Pal et al., 2001; Jarhult et al.,
2010). In contrast to patients with ileal carcinoid,
Additional Supporting Information may be found in the onlineversion of his article.
Supported by: The Swedish Research Council, Lions founda-tion for Cancer research at Uppsala University Hospital, SelandersFoundation, the Swedish Children’s Cancer Foundation and theSwedish Cancer Society.
The first two authors contributed equally to this work.
*Correspondence to: Eva T. Janson, Department of MedicalSciences, Uppsala University, University Hospital entrance 40, 5thfloor, 751 85 Uppsala, Sweden.E-mail: [email protected]
Received 12 July 2010; Accepted 11 October 2010
DOI 10.1002/gcc.20834
Published online 22 November 2010 inWiley Online Library (wileyonlinelibrary.com).
VVC 2010 Wiley-Liss, Inc.

subjects affected with multiple endocrine neo-
plasia type 1 (MEN1) develop lesions in the
pituitary, parathyroid and pancreas. Well differen-
tiated endocrine carcinomas originating in the
lung, thymic, gastric and duodenal area, earlier
denoted foregut carcinoids, are also associated
with MEN1. Furthermore, a large epidemiologi-
cal study failed to demonstrate an association
between ileal carcinoids and endocrine tumors
typically associated with the MEN1 syndrome
(Hemminki and Li, 2001). Recent genomic
(Kytola et al., 2001; Lollgen et al., 2001; D’Adda
et al., 2002) and expression studies (Duerr et al.,
2006) indicate that malignant ileal carcinoids
have a different etiology than other neuroendo-
crine tumors and genetic screening for mutation
in the MEN1 gene in two of the previously
described ileal carcinoid families was negative
(Kinova et al., 2001; Jarhult et al., 2010).
The genetic events leading to development of
an ileal carcinoid tumor are largely unknown. Pre-
vious studies have shown that deletion of 18q21-
qter is a common event (Lollgen et al., 2001). This
was confirmed by another group, which narrowed
the region to 18q22qter (Kytola et al., 2001). Other
alterations described include loss on 11q22-23, loss
on 16q21 and gain on 4p14. In two recent studies
using genomic profiling of ileal carcinoids, loss of
chromosome 18 (61% and 74%, respectively) was
confirmed ( Kulke et al., 2008; Andersson et al.,
2009). Other common aberrations were loss of 9p,
11q and 16q, as well as gain of chromosome 14.
The latter abnormality was associated with poor
survival in one of the studies (Andersson et al.,
2009). These reports provided information about
the genetic changes involved in the sporadic dis-
ease. There is, however, no available data on
genetic aberrations in familial cases.
In this study, we have characterized nine fami-
lies with a history of ileal carcinoid. To evaluate
the degree to which molecular events in familial
tumors resembles that of sporadic cases, we per-
formed high-resolution genomic and gene expres-
sion profiling in a comprehensive series of tumors
derived from familial and sporadic patients.
MATERIALS AND METHODS
Patient Samples
The Department of Endocrine Oncology at Upp-
sala University Hospital, Sweden has been a national
referral center for neuroendocrine tumors since the
1980’s. For this study we have collected clinical
records for 55 patients with ileal carcinoid, diagnosed
at the Laboratory of Pathology and Cytology and
treated at our department. The diagnosis was based
on international recommendations for the classifica-
tion of endocrine tumors (Solcia et al., 2000; Kloppel
et al., 2009). For nine of these patients, diagnosed
between 1988 and 2005, a family history of carcinoid
disease was uncovered (our index patients). During
the past 2 decades, 9 further patients were diagnosed
at our clinic within these families. Five additional
patients belonging to these families were identified
at other hospitals as having a confirmed ileal carci-
noid, but they were not subject to clinical or molecu-
lar studies. For these additional patients pathology
reports were reviewed and tissue specimens re-eval-
uated when necessary to establish the diagnosis.
When an index patient was discovered the patient
was interviewed and a family pedigree established.
A trained research nurse contacted living family
members to collect relevant data.
Plasma chromogranin A (CgA), urinary 5-
hydroxyindole acetic acid (U-5HIAA) and radiol-
ogy was performed at diagnosis. Data were ana-
lyzed using the statistical program package SPSS.
The end point for survival analyses was event-
free survival time calculated from diagnosis to
carcinoid related death or, for censored observa-
tions, until last follow-up. A possible correlation
between number of genetic aberrations and tu-
mor morphology was tested using a Spearman
Rank test. Independent effects of specific chro-
mosome aberrations on tumor growth pattern
were tested using stepwise logistic regression
analysis. All patients developed metastatic dis-
ease; 4 cases having WHO stage IIIb and the
remaining 51 with stage IV.
Sixty-one tumor samples derived from 45 of
these patients (8 familial and 37 sporadic cases)
were subjected to genetic analysis. Twenty-seven
samples represented primary tumors and 34 me-
tastases (30 mesenterial and 4 hepatic). For 14
patients, patient-matched primary and metastatic
samples were profiled. Tumor material was
sampled during operation and stored at �80�C.Tumor tissue for DNA and RNA extraction was
carefully selected to obtain >70% tumor cells
content. Sections from frozen tissue were taken
before and after material collection, stained for
CgA and examined to ensure high tumor content.
Evaluation of tumor morphology was performed
as previously described (Cunningham et al.,
2007). The study was approved by the Uppsala
University Hospital ethics committees and all
patients gave informed consent.
FAMILIAL AND SPORADIC ILEAL CARCINOIDS 83
Genes, Chromosomes & Cancer DOI 10.1002/gcc

Array-Based Comparative Genomic
Hybridization: 32K Array
The 32K array was previously established (de
Stahl et al., 2008). DNA labeling, hybridization,
washing and scanning of arrays were performed
as earlier described (Buckley et al., 2002; de Stahl
et al., 2008). Blood DNA from a healthy female
(F1) was used as reference in all experiments.
The raw data were uploaded to a laboratory infor-
mation management system database (LCB;
http://base.lcb.uu.se) and filters were applied to
remove oversaturated spots (>5%), spots with
low signal-to-noise-ratio (<3) and spots flagged as
bad or absent (de Stahl et al., 2008). The data
was normalized using print-tip locally weighted
scatter-plot smoothing (Otsuka et al., 2008). Log2
ratio was exported into Nexus Copy Number 4.0
copy number analysis program (BioDiscovery,
Inc., El Segundo, CA). BioDiscovery’s Rank seg-
mentation algorithm was used for calling of copy
number alterations (CNAs). Copy number data
were used for statistical analysis using Fisher’s
Exact Test.
High-Density SNP Genotyping Arrays:
Illumina 610Q Chips
Profiling with SNP arrays was done using the
Illumina InfiniumII assay (Steemers et al., 2006)
on Illumina Human610Q beadchips (Illumina,
San Diego, CA). Normalization and calling was
done in BeadStudio v3.3 (Illumina). The median
call rate was 94.38% (range: 90.62-95.10%). Log
R ratio (LRR) and B-allele frequency (BAF)
were then exported into Nexus Copy Number
4.0 copy number analysis program (BioDiscovery,
Inc.). CNAs were called using BioDiscovery’s
SNPRank segmentation algorithm. The raw data
from 32K BAC array and Illumina beadchips are
being submitted to Gene Expression Omnibus
database.
Microarray Expression Analysis
Total RNA was prepared by RNeasy Mini Kit
(Qiagen, Hilden, Germany). RNA concentration
and quality was assessed as previously described
(Nord et al., 2010). 100 ng of total RNA were
used to prepare biotinylated fragmented cRNA
using a two-cycle amplification step, according to
the GeneChipVR
Expression Analysis Technical
Manual (Rev. 5, Affymetrix Inc., Santa Clara,
CA). Affymetrix GeneChipVR
expression arrays
(GeneChipVR
Human Genome U133 Plus 2.0
Array) were hybridized, washed and stained and
data analyzed as described in Nord et al. (2010).
To search for differentially expressed genes an
empirical Bayes moderated t-test was then
applied (Smyth, 2004), using the ‘limma’ package
(Smyth, 2005) and P-values were adjusted accord-
ing to Benjamini and Hochberg (Benjamini et al.,
2001). Hierarchical clustering of the samples was
made in Genesis (Sturn et al., 2002) using all
genes, Euclidian distance and average linkage.
Sequencing of Candidate Genes
RefSeq gene and transcript coordinates, human
genome sequence, and single nucleotide poly-
morphisms were obtained from the UCSC data-
base (http://genome.ucsc.edu). For each gene,
protein-encoding exons as well as flanking
intronic sequences and 5’ UTR and 3’ UTR
sequences were extracted. Primers used for PCR
amplification and sequencing have previously
been published (Sjoblom et al., 2006; Wood
et al., 2007). A sequencing primer (M13 forward,
5’-GTAAAACGACGGCCAGT-3’) was appended
to the 5’ end of one of the primers in each primer
pair. Whole genome amplification (WGA) was
used to increase quantities of DNA from tumor
and normal samples, according to published pro-
tocol (Sjoblom et al., 2006). For each sample, a
minimum of 5 independent WGA reactions were
pooled to reduce the effects of any allelic or locus
bias that may have occurred during amplification.
PCR amplification, dye terminator sequencing
and somatic mutation analysis was performed as
previously described (Sjoblom et al., 2006).
RESULTS
Familial and Sporadic Ileal Carcinoids Have
Similar Clinical Characteristics
Twenty-three patients were identified with a
family history of ileal carcinoids (Fig. 1) and com-
plete clinical data were available for 18 of them.
In two families, ileal carcinoids were seen in two
branches linked by an unaffected individual sug-
gesting an incomplete penetrance of the disease.
The clinical details of familial cases compared
with the sporadic ones are summarized in Table
1. No significant differences in age at diagnosis,
hormone levels or proliferation index were
observed between the two groups. A higher fre-
quency of other non-malignant endocrine condi-
tions (hyperparathyroidism and thyrotoxicosis)
was found in patients with a family history of
84 CUNNINGHAM ETAL.
Genes, Chromosomes & Cancer DOI 10.1002/gcc

ileal carcinoids, when compared with sporadic
cases. One family stands out (family E, Fig. 1)
where five out of six siblings as well as their
mother presented with thyrotoxicosis and three of
the siblings also developed ileal carcinoid tumors.
Similar Genomic Profiles of Familial and
Sporadic Ileal Carcinoids
Genomic aberrations in 61 tumor samples were
evaluated using a 32K BAC array and confirmed
with Illumina 610Q beadchips. Overall, 252
CNAs were identified in the 27 primary tumors
and 34 metastases (Table 2). The frequency of
copy number changes was calculated for all tumor
samples as well as for familial and sporadic
groups separately (Fig. 2A). Aberration of chro-
mosome 18 was the dominating finding and was
observed in 88.8% of tumors, including all 37
sporadic and three (out of eight) familial cases.
Monosomy 18 was often found in combination
with other changes, but was also observed as the
only detectable CNA in the tumors from 12
patients (Table 2, Figs. 2 and 3 and Supporting
Information Fig. 1). The two affected individuals
in family G displayed different CNAs on chromo-
some 18, one presented with monosomy 18 while
the other patient showed copy number neutral
loss of heterozygozity (LOH) of the whole chro-
mosome, without copy number reduction (ID6
and ID7, respectively, Fig. 3 and Table 2). The
data from tumor ID7 are consistent with loss of
one copy of chromosome 18 and reduplication of
the remaining one. The results from this family
show that two different second hits affected the
putative tumor suppressor gene located on 18q.
The comparison of genomic profiles between all
familial and all sporadic tumors revealed that
most of the genetic changes were in common
between the groups. As for differences, we
observed a tendency for higher number of chro-
mosomes affected by CNAs in familial vs. spo-
radic cases, average 5.6 vs. 3.4 /sample, (t-testP-value: 0.028). The identification of more aber-
rations in individuals with familial disease may,
however, be a consequence of over-representation
of liver metastases in this group (three liver
metastases out of eight familial cases vs. one out
of 37 in sporadic cases). Interestingly and in con-
tradiction to the above, one familial case (ID8)
with a thoroughly verified tumor content (>70%,
displayed no CNAs in the samples taken from
both primary tumor and metastasis.
Another intriguing aberration of chromosome
18 was noted in one sporadic tumor ID11 (Sup-
porting Information Fig. 1 and Table 2). In this
sample, the Log R ratio (LRR) values reflecting
fluorescent intensity and B-allele frequency
Figure 1. Pedigrees of nine families (A–I) showing patients affectedwith ileal carcinoid are in agreement with autosomal dominant inheri-tance pattern and incomplete penetrance. Individuals with ileal carci-noids are represented as filled symbols. Patients studied molecularlyare denoted by Arabic numerals, which correspond to patient ID inTable 2. Generations are shown using Roman numerals on the leftand diagonal lines indicate deceased patients. Other diagnoses are:thyrotoxicosis (TT), hyperparathyroidism (HP), hypernephroma (HN)and colon adenocarcinoma (CA).
TABLE 1. Clinical Data From Patients with Familiar andSporadic Ileal Carcinoids
ParameterFamilial ilealcarcinoid
Sporadic ilealcarcinoid
Number of patients 18 37Age at diagnosis (years) 62 [24–73] 61 [37–85]Sex (M:F) 7:11 19:18Survival (months) 79 [27–310] 75 [7–399]Status (DWD:AWD)a 11:7 8:29Ki67 (%) <1 [<1–10] <1 [<1–4]U-5HIAA (lmol/24 h) 141 [28–2720] 300 [16–2112]CgA (nmol/L) 46 [4–1280] 22 [4–382]Other malignancy 1b 5c
Other endocrine disease 6d 2e
M, Male; F, Female; DWD, dead with disease; AWD, alive with
disease; Survival is calculated as months from diagnosis to death;
U-5HIAA, Urinary 5-hydroxyindoleacetic acid (serotonin metabolite)
normal reference is 50 lmol/24 h; CgA, Chromogranin A, normal
reference is <4 nmol/L; Ki67 (%), proliferation index in tumor areas
with highest proliferation.aA higher number of patients died in the familial group than among
sporadic cases. This is likely due to the longer duration of follow up
in familial group of patients as there is no difference in duration of
survival between the two groups.bColon adenocarcinoma (1).cBreast cancer (1), colon adenocarcinoma (1), folicular thyroid cancer
(1), uterus adenocarcinoma (1), prostate cancer (1).dThyrotoxicosis (4), hyperparathyroidism (2).eHypothyroidism (1), thyrotoxicosis (1).
FAMILIAL AND SPORADIC ILEAL CARCINOIDS 85
Genes, Chromosomes & Cancer DOI 10.1002/gcc
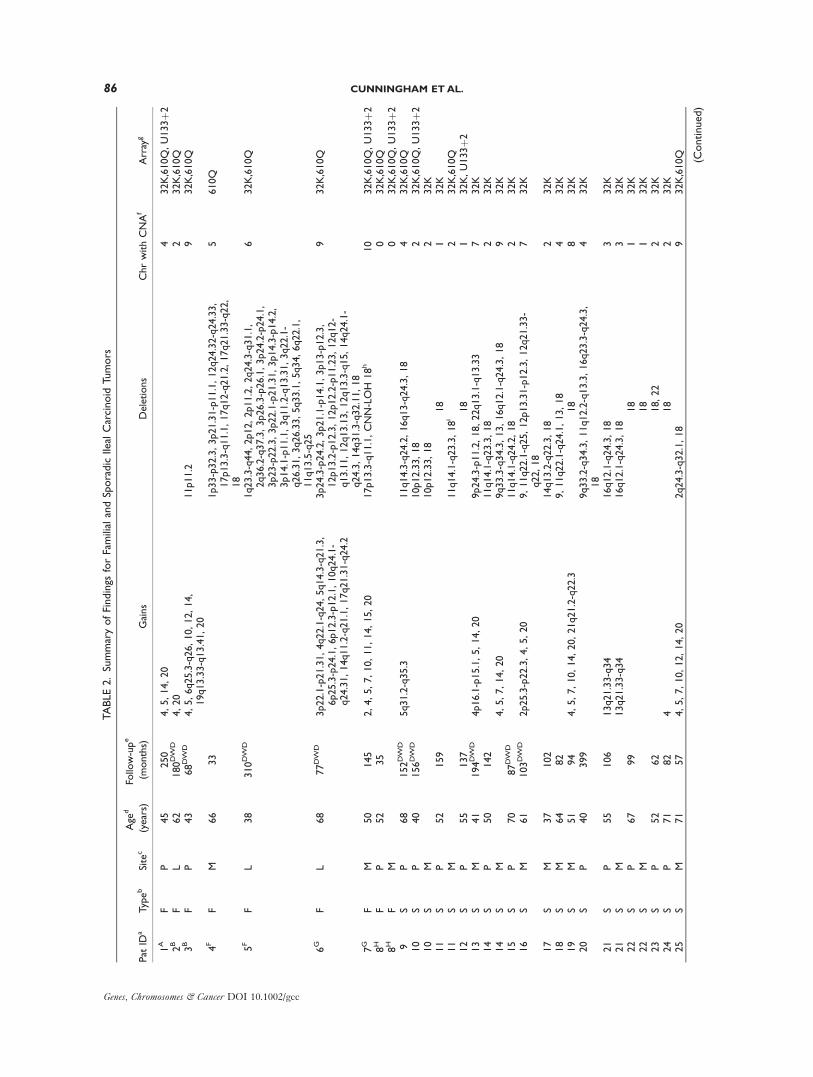
TABLE2.SummaryofFindings
forFamilialandSporadicIlealCarcinoid
Tumors
Pat
IDa
Typeb
Site
c
Age
d
(years)
Follow-upe
(months)
Gains
Deletions
ChrwithCNAf
Array
g
1A
FP
45
250
4,5,14,20
432K,610Q,U133þ2
2B
FL
62
180DW
D4,20
232K,610Q
3B
FP
43
68DW
D4,5,6q25.3-q26,10,12,14,
19q13.33-q13.41,20
11p11.2
932K,610Q
4F
FM
66
33
1p33-p32.3,3p21.31-p11.1,12q24.32-q24.33,
17p13.3-q11.1,17q12-q21.2,17q21.33-q22,
18
5610Q
5F
FL
38
310DW
D1q23.3-q44,2p12,2p11.2,2q24.3-q31.1,
2q36.2-q37.3,3p26.3-p26.1,3p24.2-p24.1,
3p23-p22.3,3p22.1-p21.31,3p14.3-p14.2,
3p14.1-p11.1,3q11.2-q13.31,3q22.1-
q26.31,3q26.33,5q33.1,5q34,6q22.1,
11q13.5-q25
632K,610Q
6G
FL
68
77DW
D3p22.1-p21.31,4q22.1-q24,5q14.3-q21.3,
6p25.3-p24.1,6p12.3-p12.1,10q24.1-
q24.31,14q11.2-q21.1,17q21.31-q24.2
3p24.3-p24.2,3p21.1-p14.1,3p13-p12.3,
12p13.2-p12.3,12p12.2-p11.23,12q12-
q13.11,12q13.13,12q13.3-q15,14q24.1-
q24.3,14q31.3-q32.11,18
932K,610Q
7G
FM
50
145
2,4,5,7,10,11,14,15,20
17p13.3-q11.1,CNN-LOH
18h
10
32K,610Q,U133þ2
8H
FP
52
35
032K,610Q
8H
FM
032K,610Q,U133þ2
9S
P68
152DW
D5q31.2-q35.3
11q14.3-q24.2,16q13-q24.3,18
432K,610Q
10
SP
40
156DW
D10p12.33,18
232K,610Q,U133þ2
10
SM
10p12.33,18
232K
11
SP
52
159
18
132K
11
SM
11q14.1-q23.3,18i
232K,610Q
12
SP
55
137
18
132K,U133þ2
13
SM
41
194DW
D4p16.1-p15.1,5,14,20
9p24.3-p11.2,18,22q13.1-q13.33
732K
14
SP
50
142
11q14.1-q23.3,18
232K
14
SM
4,5,7,14,20
9q33.3-q34.3,13,16q12.1-q24.3,18
932K
15
SP
70
87DW
D11q14.1-q24.2,18
232K
16
SM
61
103DW
D2p25.3-p22.3,4,5,20
9,11q22.1-q25,12p13.31-p12.3,12q21.33-
q22,18
732K
17
SM
37
102
14q13.2-q22.3,18
232K
18
SM
64
82
9,11q22.1-q24.1,13,18
432K
19
SM
51
94
4,5,7,10,14,20,21q21.2-q22.3
18
832K
20
SP
40
399
9q33.2-q34.3,11q12.2-q13.3,16q23.3-q24.3,
18
432K
21
SP
55
106
13q21.33-q34
16q12.1-q24.3,18
332K
21
SM
13q21.33-q34
16q12.1-q24.3,18
332K
22
SP
67
99
18
132K
22
SM
18
132K
23
SP
52
62
18,22
232K
24
SP
71
82
418
232K
25
SM
71
57
4,5,7,10,12,14,20
2q24.3-q32.1,18
932K,610Q (Continued)
86 CUNNINGHAM ETAL.
Genes, Chromosomes & Cancer DOI 10.1002/gcc

TABLE2.SummaryofFindings
forFamilialandSporadicIlealCarcinoid
Tumors
(Continued)
Pat
IDa
Typeb
Site
c
Age
d
(years)
Follow-upe
(months)
Gains
Deletions
ChrwithCNAf
Array
g
26
SM
85
25DW
D12p13.33-p13.32,17q21.32-q24.1
16q22.1-q24.3,18
432K
27
SP
52
142
18
132K
27
SM
16q12.2-q24.1,18
232K
28
SL
45
44
4,5,8,10,12,14,17,20,21
9p24.3-p11.2,17q11.2,18,22
13
32K
29
SM
67
34DW
D18
132K,610Q,U133þ2
30
SM1
70
48
18q22.1-q22.2
132K,610Q
30
SM2
18
132K
31
SP
78
41
16q12.2-q24.1,18
232K
31
SM
4,20q11.23-q13.33
16q12.2-q24.3,18
432K
32
SP
74
7DW
D9,18
232K
33
SM
47
133
18
132K,610Q,U133þ2
34
SP
51
37
18
132K
34
SM
18
132K
35
SP
65
35
18
132K
35
SM
18
132K
36
SP
76
39
16q12.2-q24.3,18
232K
37
SP
71
34
4,5q11.2-q35.3,14,20
2q36.1-q37.3,9p24.3-p12,18q21.1-q21.31
732K,610Q,U133þ2
37
SM
4,5q11.2-q35.3,14,20
2q36.1-q37.3,9p24.3-p12,18q21.1-q21.31
732K
38
SM1
40
75
18
132K
38
SM2
14
9p24.3-p12,18
332K
39
SM
68
32
2p25.3-p25.1,17p13.3-p11.2,18
332K
40
SP
71
34
10q26.13-q26.3,20q13.13-q13.33
12p13.31-p12.3,18q21.1-q21.32,18q22.3-
qter
432K,610Q,U133þ2
41
SP
41
37
18
132K
41
SM
18
132K
42
SP
66
30
18
132K
42
SM
2p25.3-p25.1,2p21-p16.3,2q24.1-q24.3,
4p16.2-p15.33,7p21.1-p15.1,
8q21.3,8q24.12-q24.22,14q32.2-q32.33,
17p12-q11.1,20q13.33
3p21.31,11q22.3-q25,12q24.31-q24.33,18
11
32K,610Q
43
SP
43
130
4,10,14,15,20
16q13-q24.3,18,22
832K
44
SP
65
67
6q23.3,13q12.3-q13.2,13q13.3-q21.2,18
332K
44
SM
5,7,10,14,20
18
632K
45
SP
57
72
4,5,20
18
432K
Copy
numberalterations(C
NAs)
identifiedin
61tumorsamplesderivedfrom
45patients
withfamilialandsporadicilealcarcinoid.
aThefamily
towhichindividualsbelongisindicatedbyacapitalsuperscriptletter(A,B,F,G
orH),seeFig.1.
bS,
Sporadic;F,Familialilealcarcinoid.
cP,Primarytumor;
M,Mesenterial
metastasis;L,Livermetastasis.Fo
r14patients
matchedprimaryandmetastatic
tumorwere
profiled.In
6ofthematchedpairs
(ID:11,14,27,31,42,and44),additional
alterationswere
observedin
metastasis.Fo
rID:30and38tw
ometastasesderivedfrom
thesamepatientwere
profiled(M
1andM2),whichrevealedtumorheterogeneity.
dAge
atdiagnosis.
ePatients
DW
D,dead
withdiseaseareindicated.
f NumberofchromosomesaffectedbyCNA.
gThearrayusedto
profile
thetumorsamplesisindicated:32K,Illumina610Q
orAffym
etrix
U133þ2
chips.
hCNN-LOH
18,copy
numberneutralLOH
ofchromosome18.
i Loss
ofchromosome18withoutallelic
imbalance,seetextfordetails.
FAMILIAL AND SPORADIC ILEAL CARCINOIDS 87
Genes, Chromosomes & Cancer DOI 10.1002/gcc
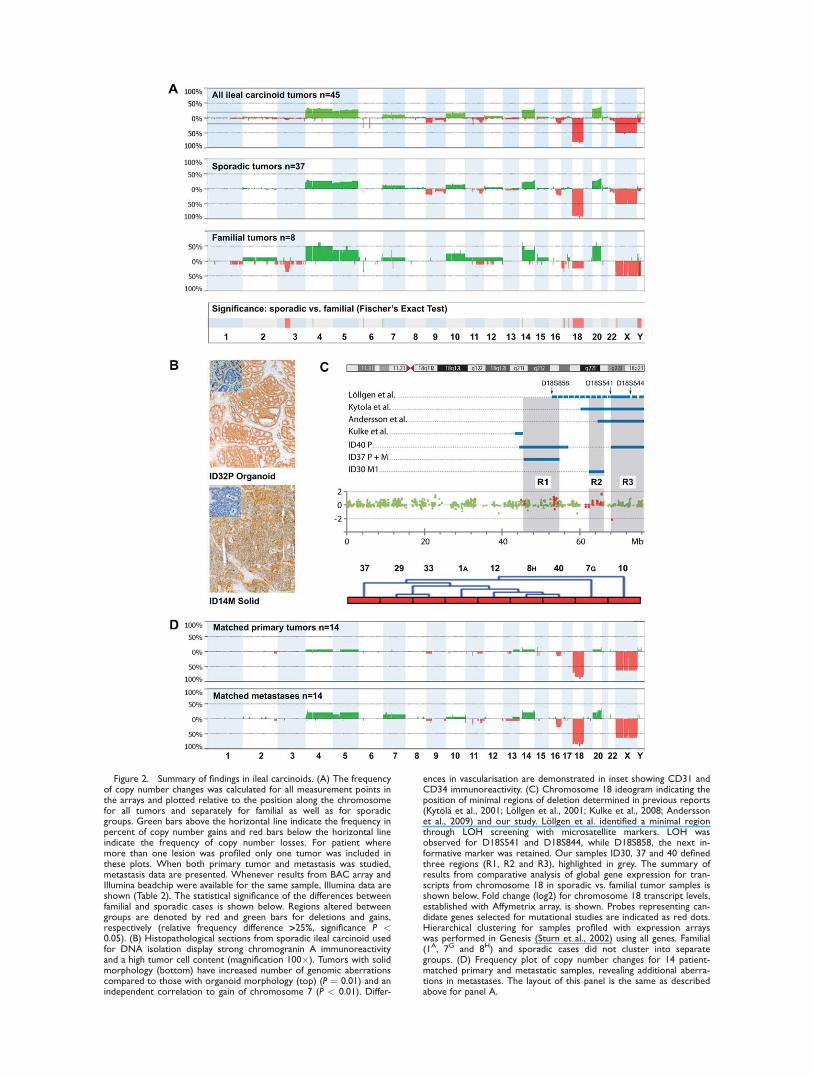
Figure 2. Summary of findings in ileal carcinoids. (A) The frequencyof copy number changes was calculated for all measurement points inthe arrays and plotted relative to the position along the chromosomefor all tumors and separately for familial as well as for sporadicgroups. Green bars above the horizontal line indicate the frequency inpercent of copy number gains and red bars below the horizontal lineindicate the frequency of copy number losses. For patient wheremore than one lesion was profiled only one tumor was included inthese plots. When both primary tumor and metastasis was studied,metastasis data are presented. Whenever results from BAC array andIllumina beadchip were available for the same sample, Illumina data areshown (Table 2). The statistical significance of the differences betweenfamilial and sporadic cases is shown below. Regions altered betweengroups are denoted by red and green bars for deletions and gains,respectively (relative frequency difference >25%, significance P <0.05). (B) Histopathological sections from sporadic ileal carcinoid usedfor DNA isolation display strong chromogranin A immunoreactivityand a high tumor cell content (magnification 100�). Tumors with solidmorphology (bottom) have increased number of genomic aberrationscompared to those with organoid morphology (top) (P ¼ 0.01) and anindependent correlation to gain of chromosome 7 (P < 0.01). Differ-
ences in vascularisation are demonstrated in inset showing CD31 andCD34 immunoreactivity. (C) Chromosome 18 ideogram indicating theposition of minimal regions of deletion determined in previous reports(Kytola et al., 2001; Lollgen et al., 2001; Kulke et al., 2008; Anderssonet al., 2009) and our study. Lollgen et al. identified a minimal regionthrough LOH screening with microsatellite markers. LOH wasobserved for D18S541 and D18S844, while D18S858, the next in-formative marker was retained. Our samples ID30, 37 and 40 definedthree regions (R1, R2 and R3), highlighted in grey. The summary ofresults from comparative analysis of global gene expression for tran-scripts from chromosome 18 in sporadic vs. familial tumor samples isshown below. Fold change (log2) for chromosome 18 transcript levels,established with Affymetrix array, is shown. Probes representing can-didate genes selected for mutational studies are indicated as red dots.Hierarchical clustering for samples profiled with expression arrayswas performed in Genesis (Sturn et al., 2002) using all genes. Familial(1A, 7G and 8H) and sporadic cases did not cluster into separategroups. (D) Frequency plot of copy number changes for 14 patient-matched primary and metastatic samples, revealing additional aberra-tions in metastases. The layout of this panel is the same as describedabove for panel A.

(BAF) data showing allelic status demonstrated
inconsistent results for chromosome 18. LRR val-
ues indicate monosomy 18 but BAF values did
not show the expected allelic imbalance. It
should be stressed that this tumor also showed in-
terstitial deletion of chromosome 11 with the
expected appearance of both LRR and BAF val-
ues. Several explanations can be proposed to
these unexpected results. For instance, this tumor
sample might be bi-clonal with two tumor cell
populations co-existing at a similar relative fre-
quency and each cell population having lost
either the maternal or paternal chromosome.
Another possible explanation is the presence of
tetraploid cancer cells in this tumor, exhibiting
loss of two copies (one maternal and one pater-
nal) of chromosome 18.
The high resolution of the applied analysis
platforms and a considerable number of studied
tumors permitted us to identify four partial dele-
tions on 18q in sporadic cases, which defined
three candidate intervals: 18q21.1-q21.31 (45.80-
54.36Mb), 18q22.1-q22.2 (62.68-65.99Mb) and
18q22.3-q23 (68.38-76.12Mb) indicated as R1, R2
and R3 (Fig. 2C and Table 2). These regions
were 8.56, 3.31 and 7.73 Mb in size and encom-
passed 30, 6, and 29 genes, respectively. A sec-
ond major region affected by deletions in six
sporadic and one familial tumors mapped to
11q22.3-23.3 (Fig. 2). Genes mapped to this locus
include several members of the cysteine-aspartic
acid protease (caspase) family: CASP4, CASP1and CASP5 as well as MLL and SDHD. Restrictedregions of deletion were also observed at 3p12.3-
p14.3, in 3 familial tumors, two of these being
liver metastasis (Fig. 2 and Table 2). The pres-
ence of deletions at 11q22.3-23.3 and 3p12.3-
p14.3 and loss of chromosome 18 were not mutu-
ally exclusive indicating that these deletions may
have a synergistic effect on tumor progression.
Copy number gains frequently encompassed
whole chromosomes 4, 5, 14 and 20 and were
observed in more than 20% of cases (Fig. 2). The
above mentioned array results are best compatible
with trisomy for these chromosomes in the tumor
cells and these aberrations were often present con-
comitantly. Since gain of chromosome 14 has been
suggested to be a predictor of poor outcome in ileal
carcinoids (Andersson et al., 2009), survival was com-
pared in patients with and without this alteration. In
this study, 14 patients displayed partial or complete
gain. The shortest observation time within the group
with chromosome 14 gain was 34 months and the
shortest survival time was 68 months (Table 3).
Kaplan-Meier survival analysis showed no difference
in survival between patients with and without gain of
chromosome 14 (not shown). Eleven cases (10 spo-
radic and 1 familial), revealed gain of chromosome 14
together with loss of chromosome 18, another familial
case displayed gain of chromosome 14 along with
copy number neutral LOH of chromosome 18, while
2 familial cases displayed gain of chromosome 14
without deletions on chromosome 18. Segmental
gains on chromosome 14 were observed in 2 patients,
at position chr14:22,093,320-37,596,612 in case ID6
and at position chr14:97,434,551-106,368,585 in case
ID4 (Table 1). Interestingly, a locus on chromosome
14 at position chr14:19,704,670-22,817,404 showed
the highest copy number gain consistent with four
copies, which was present on top of trisomy 14 (case
ID3, band 14q11.2). The overlap between the seg-
mental gain in case ID6 and the high copy gain in
case ID3 defines a candidate region at chr14:
22,093,320-22,817,404.
Familial and Sporadic Ileal Carcinoids Reveal
Similar Expression Profiles
To identify gene expression differences leading
to the familial or sporadic variants of ileal carcinoids,
global gene expression profiles were established
using U133Plus2.0 arrays with RNA extracted from
9 tumors (6 sporadic and 3 familial, Table 2). We
first searched for differentially expressed genes
along chromosome 18. Mean expression levels for
transcripts along the whole chromosome were com-
parable in both groups of samples. Only four probes
representing the SYT4, SERPINB5, DOK6 and
CBLN2 genes presented with the absolute mean
fold change in log2 ratio > 1 (Fig. 2C). However,
none of these transcripts were significantly differen-
tially expressed (P-value > 0.7) between the familial
and sporadic tumors. At the genome wide level, only
one probe (212143_s_at), representing IGFBP3 (in-
sulin-like growth factor binding protein 3), was
found differentially expressed between the groups
(P-value ¼ 0.040, log2 fold change �1.93, sporadic
vs. familial). However, the second probe represent-
ing this gene (210095_s_at) was not expressed differ-
entially (P-value ¼ 0.631, log2 fold change �1.70).
Clustering analysis showed that familial and sporadic
cases did not cluster into separate groups (Fig. 2C).
Sequencing-Based Mutation Analysis of
Candidate Genes
In an attempt to identify a gene involved in
the development of familial and sporadic ileal
FAMILIAL AND SPORADIC ILEAL CARCINOIDS 89
Genes, Chromosomes & Cancer DOI 10.1002/gcc

carcinoids, we performed mutation analysis of 18
genes from three candidate regions (Fig. 2C).
The protein encoding sequences of SMAD7,ACAA2, ST8SIA3, ONECUT2, FECH, NARS,ATP8B1, NEDD4L, ALPK2, CDH7, CDH19,DSEL, TXNDC10, CCDC102B, DOK6, CD226,SOCS6, and CBLN2 were studied in 23 sporadic
tumors as well as in patient-matched blood DNA.
The sequencing encompassed � 37 kb target
sequence (175 exons including at least four flak-
ing bases in the splice donor- and acceptor sites),
generating a total of 888 kb of Sanger sequence.
The sequence coverage for the studied genes is
shown in Supporting Information Table 1. Exons
with putative mutations were re-sequenced in tu-
mor and matched normal tissue. However, no tu-
mor-specific mutations were confirmed in these
genes.
Comparison of Patient-Matched Primary Tumors
and Metastases
Sixteen pairs of tumor samples, derived from
the same individual, were profiled. For 14
patients, DNA was isolated from matched pri-
mary and metastatic tumor, and for the remaining
two from two different metastases. Loss of chro-
mosome 18 was seen in all primary tumors and
Figure 3. Familial ileal carcinoid tumors derived from two individ-uals in family G presented with different second hit events. Wholegenome results from samples ID6 and ID7, profiled with Illumina610Q chips are shown. Red arrows indicate chromosome 18. Panels
A and C show Log R ratio (LRR) data from the tumors, while panelsB and D represent B-allele frequency (BAF) values. Sample ID6 dis-played monosomy 18, while ID7 presented with copy number neutralLOH for the entire chromosome 18, without copy number change.
90 CUNNINGHAM ETAL.
Genes, Chromosomes & Cancer DOI 10.1002/gcc

matched metastases. A metastasis from case ID30
displayed a regional loss at 18q22.1-q22.2, which
defines a new region on chromosome 18, while
another metastasis from the same patient dis-
played total loss of chromosome 18. The remain-
ing eight pairs showed indistinguishable profiles
in primary and metastatic tumors. In four cases,
both primary tumors and metastases displayed
loss of chromosome 18, as the only detectable
change, further confirming that this aberration is
an early event in the pathogenesis of ileal
carcinoids.
Aberrations additional to those identified in pri-
mary tumors were observed in the metastases of
eight matched pairs. These included mostly gains
of entire chromosomes 4, 5, 7, 14 and/or 20,
which could indicate a tendency towards increas-
ing genomic instability in the metastatic tumors
(Fig. 2D). Interestingly, gain of chromosome 7
was exclusively observed in metastases of the
matched samples. Furthermore, an examination
of the whole tumor series shows that five out of
totally 32 metastases, but none of the 27 primary
tumors, presented with gain of entire chromo-
some 7 (Table 2).
Gain of Chromosome 7 was Related to a Distinct
Morphological Feature
Thirty-two molecularly studied tumors were
morphologically categorized as organoid (small tu-
mor nests surrounded by a highly vascularised
stroma), 11 as solid (large tumor areas with pene-
trating vascular network) and 18 as mixed, with
both patterns present (Fig. 2B). Tumors with
solid morphology presented with higher number
of genomic aberrations. This was found in both
primary tumors (rs 0.6, P < 0.01) and metastases
(rs ¼ 0.8, P < 0.01). Gain of chromosome 7 was
the only aberration with an independent associa-
tion to ‘‘solid’’ tumor morphology (P<0.01).
DISCUSSION
We have investigated a comprehensive series of
sporadic and familial well differentiated serotonin
producing endocrine carcinomas originating in the
small intestine and proximal colon, commonly
known as ileal carcinoids. Nine families were char-
acterized encompassing 23 patients with an inher-
ited disease, making this the largest collection of
hereditary cases described to date. The histopatho-
logical characteristics such as immunoreactivity for
relevant markers and proliferation index are similar
for the familial and sporadic tumors. Furthermore,
the clinical features of familial cases, including age
at disease onset, symptoms, hormone production
and survival are indistinguishable from sporadic
patients. The pattern of inheritance suggests an
autosomal dominant two-hit inherited susceptibility
to ileal carcinoid, first elucidated for retinoblastoma
and further shown as operative for numerous other
inherited tumor diseases (Vogelstein and Kinzler,
2002). For familial ileal carcinoids, the disease pen-
etrance is difficult to evaluate as the disease usu-
ally is diagnosed late in life and the tumors are
seldom recognized before symptoms due to meta-
static disease occur. It is further very likely that
some individuals in the families are carrying clini-
cally silent tumors, as suggested by autopsy studies
(Berge and Linell, 1976). There are previous
reports of families with two or more members
affected with ileal carcinoid tumors (Eschbach and
Rinaldo, 1962; Moertel and Dockerty, 1973; Wale
et al., 1983; Kinova et al., 2001; Pal et al., 2001;
Jarhult et al., 2010). The most recent one describes
a three generation family from Sweden which is
not included in our report (Jarhult et al., 2010).
There are also epidemiological studies supporting
the notion of a familial variant of this disease
(Babovic-Vuksanovic et al., 1999; Hemminki and
Li, 2001). Based on these results and on the results
presented here, we propose that the Familial IlealEndocrine Carcinoma (FIEC) represent a so far
unrecognized inherited tumor disease.
The patients included in this study represent a
carefully selected cohort. All met the WHO crite-
ria for diagnosis and patients with other gastroin-
testinal endocrine tumors (i.e. appendix
carcinoids) were excluded, making this a homoge-
nous group. We have included tumor tissue from
both sporadic and familial cases in all assays; the
results show that there is no obvious difference
in genetic aberrations or gene expression between
the two groups. Chromosome 18 aberrations
detected in both sporadic and familial forms of
TABLE 3. Median Time from Diagnosis to Death or LatestFollow-Up for Patients with and Without Gain of
Chromosome 14
Statusa
No gain ofchromosome 14
Gain ofchromosome 14
NMedian survival
(range in months) NMedian survival
(range in months)
AWD 22 65 (30–159) 11 94 (34–250)DWD 9 103 (7–310) 3 77 (68–194)
N, number of patients; AWD, alive with disease; DWD, dead with
disease.
FAMILIAL AND SPORADIC ILEAL CARCINOIDS 91
Genes, Chromosomes & Cancer DOI 10.1002/gcc

ileal carcinoids (100% vs. 38%, respectively) indi-
cates that these tumor variants share a common
mechanism and strengthens the notion of a gene
involved in tumor development located to this
chromosome. The frequency of chromosome 18
aberration in sporadic tumors was considerably
higher in our study compared with previous
reports (43%-88%) (Kytola et al., 2001; Lollgen
et al., 2001; 2002; Zikusoka et al., 2005; Kulke
et al., 2008). This might be due to strict patient
inclusion criteria and careful dissection of the tis-
sue. To ensure high tumor cell content, we
examined CgA stained 4 lm tissue sections,
taken both before and after dissection of the tu-
mor tissue.
The data regarding chromosome 18 reinforce the
notion that this chromosome contains the primary
ileal carcinoid gene. Our results identified, how-
ever, three minimal overlapping regions of dele-
tions in sporadic cases, mapping to 18q21.1-q21.31,
18q22.1-q22.2 and 18q22.3-q23 (Fig. 2C, Table 2).
Two of these loci are novel and the third one,
mapping to the telomeric end of the q-arm, overlap
with that previously reported by Lollgen et al.
(2001) and Andersson et al. (2009). The fact that
these regions are not overlapping is intriguing and
renders identification of a suppressor gene involved
in ileal carcinoid more difficult. It also points to
the possible involvement of more than one 18q-
located gene in the tumorigenesis. We performed a
comprehensive somatic mutation analysis of 18
genes from three candidate regions but were
unable to observe any tumor-specific sequence var-
iants upon comparison with the patient-matched
blood DNA, which suggest that these 18 genes are
unlikely tumor suppressor candidates. It is note-
worthy in this context that 18q deletions have
been proposed as a molecular predictor of hepatic
metastasis in e.g. colorectal cancer (Tanaka et al.,
2009), which is a very common feature of ileal car-
cinoids. It is, therefore, plausible that chromosome
18 contains more than one gene involved in the
course of the disease of ileal carcinoids; allowing
for an initiating tumor suppressor and a metastasis-
related gene(s). The MEN1 gene on 11q13.1 maps
outside the minimal candidate interval and only
one sporadic tumor in our series (sample ID25)
presented with a deletion encompassing MEN1.This is in agreement with previous studies showing
that MEN1 alterations are rare in ileal carcinoids
(Debelenko et al., 1997; Lollgen et al., 2001).
We have applied two platforms (BAC array and
Illumina SNP beadchips) for characterization of tu-
mor-specific genomic aberrations. This permits an
important validation step of results obtained from
one platform by another methodologically distinct
approach. Furthermore, Illumina beadchips allow
two levels of analysis. In addition to the LRR val-
ues representing fluorescent intensity of probes on
the array, the BAF data displays allelic status of
genotyped SNPs. The BAF-derived results allowed
us to uncover copy number neutral LOH, of an
affected chromosomal segment. This type of aber-
ration is consistent with loss of one parental allele
followed by reduplication of the remaining one.
The findings from family G are particularly impor-
tant as two family members displayed two differ-
ent second hits affecting the putative tumor
suppressor on 18q, one of them being a copy num-
ber neutral LOH. This result is consistent with
segregation of a ‘‘first hit’’ inherited mutation in
the FIEC predisposition gene.
A recent publication suggested that gain of chro-
mosome 14 was associated with short survival for
ileal carcinoid patients; 7 of 8 patients died within
20 months from diagnosis (Andersson et al., 2009).
Since the survival of ileal carcinoid patients has
been reported as considerably longer [median sur-
vival 92 months (Janson et al., 1997)], the gain of
chromosome 14 (Andersson et al., 2009) was sug-
gested as an indicator of poor prognosis and FISH
analysis was proposed to be performed in all ileal
carcinoid tumors. We were, however, not able to
confirm this result. We observed only three death
events in patients with tumor-specific gain of chro-
mosome 14, after 68, 77 and 194 months (Table 2).
The median duration of follow-up in our patients
with chromosome 14 gain was 92 months (range
34-250). Furthermore, we could not demonstrate
difference in overall survival time between patients
with and without chromosome 14 gain. The differ-
ence between the two studies regarding the possi-
ble effect of chromosome 14 gain on survival
merits further investigation. However, we currently
see no indication to incorporate FISH analysis in
the general work-up of ileal carcinoid patients.
In a previous study of ileal carcinoid tumor
morphology a defined morphologic feature, the
solid growth pattern, was shown to be an inde-
pendent risk factor for shorter survival (Cunning-
ham et al., 2007). In the present study this
morphologic feature is correlated to genomic
instability and demonstrates an independent cor-
relation to trisomy 7, which was only present in
metastases. ‘‘Solid’’ morphology is distinguished
from ‘‘organiod’’ by larger tumor cell aggrega-
tions, intratumoral vascular structures and the ab-
sence of palisade cell arrangement. Future
92 CUNNINGHAM ETAL.
Genes, Chromosomes & Cancer DOI 10.1002/gcc

understanding of the role of trisomy 7 in the reg-
ulation of tumor growth pattern may yield impor-
tant insights of prognostic and therapeutic
significance.
In conclusion, similarities between sporadic
and familial cases suggest that FIEC represent a
new autosomal dominant inherited tumor disease.
One major point of our report is raising the
awareness about the existence of a familial vari-
ant of ileal carcinoid disease. Loss of chromosome
18 is, without doubt, a very early event in the de-
velopment of both familial and sporadic tumors
and the presented evidence suggests a shared
pathogenetic mechanism, while gain of chromo-
some 7 seems to correlate to a change in growth
pattern. However, the actual mutated gene(s)
that initiates tumor development still remains to
be identified. Based on our clinical findings we
propose that patients presenting with abdominal
pain or hormone-related symptoms and a positive
family history of ileal carcinoid should be investi-
gated on a wide indication for biochemical and
radiological signs of such tumors.
ACKNOWLEDGMENTS
The authors thank Dr. Carl Bruder and Dr.
Johanna Sandgren for producing the BAC arrays
and Monica Lindman and Daniel Moreno Bergg-
ren for performing the sequence analysis. The
Illumina genotyping was performed at the SNP
Technology Platform, Uppsala, Sweden (www.ge-
notyping.se), supported by Uppsala University
and the Knut and Alice Wallenberg foundation.
The Affymetrix expression analysis was per-
formed at the Uppsala Array Platform
(www.medsci.uu.se/klinfarm/arrayplatform/).
REFERENCES
Andersson E, Sward C, Stenman G, Ahlman H, Nilsson O. 2009.High-resolution genomic profiling reveals gain of chromosome14 as a predictor of poor outcome in ileal carcinoids. EndocrRelat Cancer 16:953–966.
Babovic-Vuksanovic D, Constantinou CL, Rubin J, Rowland CM,Schaid DJ, Karnes PS. 1999. Familial occurrence of carcinoidtumors and association with other malignant neoplasms. CancerEpidemiol Biomarkers Prev 8:715–719.
Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I. 2001. Con-trolling the false discovery rate in behavior genetics research.Behav Brain Res 125:279–284.
Berge T, Linell F. 1976. Carcinoid tumours. Frequency in adefined population during a 12-year period. Acta Pathol Micro-biol Scand A 84:322–330.
Buckley PG, Mantripragada KK, Benetkiewicz M, Tapia-Paez I,Diaz De Stahl T, Rosenquist M, Ali H, Jarbo C, De Bustos C,Hirvela C, Sinder Wilen B, Fransson I, Thyr C, Johnsson BI,Bruder CE, Menzel U, Hergersberg M, Mandajl N, Blennow E,Wedell A, Beare DM, Collins JE, Dunham I, Albertson D, Pin-kel D, Bastian BC, Faruqi AF, Lasken RS, Ichimura K, CollinsVP, Dumanski JP. 2002. A full-coverage, high-resolution human
chromosome 22 genomic microarray for clinical and researchapplications. Hum Mol Genet 11:3221–3229.
Cunningham JL, Grimelius L, Sundin A, Agarwal S, Janson ET.2007. Malignant ileocaecal serotonin-producing carcinoidtumours: the presence of a solid growth pattern and/or Ki67index above 1% identifies patients with a poorer prognosis.Acta Oncol 46:747–756.
D’Adda T, Pizzi S, Azzoni C, Bottarelli L, Crafa P, Pasquali C,Davoli C, Corleto VD, Delle Fave G, Bordi C. 2002. Differentpatterns of 11q allelic losses in digestive endocrine tumors.Hum Pathol 33:322–329.
de Stahl TD, Sandgren J, Piotrowski A, Nord H, Andersson R,Menzel U, Bogdan A, Thuresson AC, Poplawski A, von Tell D,Hansson CM, Elshafie AI, Elghazali G, Imreh S, NordenskjoldM, Upadhyaya M, Komorowski J, Bruder CE, Dumanski JP.2008. Profiling of copy number variations (CNVs) in healthyindividuals from three ethnic groups using a human genome 32K BAC-clone-based array. Hum Mutat 29:398–408.
Debelenko LV, Brambilla E, Agarwal SK, Swalwell JI, KesterMB, Lubensky IA, Zhuang Z, Guru SC, Manickam P, OlufemiSE, Chandrasekharappa SC, Crabtree JS, Kim YS, Heppner C,Burns AL, Spiegel AM, Marx SJ, Liotta LA, Collins FS, TravisWD, Emmert-Buck MR. 1997. Identification of MEN1 genemutations in sporadic carcinoid tumors of the lung. Hum MolGenet 6:2285–2290.
Duerr E, Mizukami Y, Warshaw A, Kulke M, Chung C. 2006. Geneexpression profiles of pancreatic neuroendocrine tumors and gas-trointestinal carcinoids. Gastroenterology 130(Suppl 2):S1887.
Eschbach J, Rinaldo J. 1962. Metastatic carcinoid, a familial occur-rence. Ann Intern Med 57:647–650.
Hemminki K, Li X. 2001. Familial carcinoid tumors and subse-quent cancers: A nation-wide epidemiologic study from Swe-den. Int J Cancer 94:444–448.
Janson ET, Holmberg L, Stridsberg M, Eriksson B, TheodorssonE, Wilander E, Oberg K. 1997. Carcinoid tumors: analysis ofprognostic factors and survival in 301 patients from a referralcenter. Ann Oncol 8:685–690.
Jarhult J, Landerholm K, Falkmer S, Nordenskjold M, Sundler F,Wierup N. 2010. First report on metastasizing small bowel car-cinoids in first-degree relatives in three generations. Neuroen-docrinology 91:318–323.
Kinova S, Duris I, Kovacova E, Stvrtina S, Galbavy S, Makaiova I. 2001.Malignant carcinoid in two brothers. Bratisl Lek Listy 102:231–234.
Kloppel G, Couvelard A, Perren A, Komminoth P, McNicol AM,Nilsson O, Scarpa A, Scoazec JY, Wiedenmann B, Papotti M,Rindi G, Plockinger U. 2009. ENETS consensus guidelines forthe standards of care in neuroendocrine tumors: Towards astandardized approach to the diagnosis of gastroenteropancreaticneuroendocrine tumors and their prognostic stratification. Neu-roendocrinology 90:162–166.
Kulke MH, Freed E, Chiang DY, Philips J, Zahrieh D, GlickmanJN, Shivdasani RA. 2008. High-resolution analysis of genetic alter-ations in small bowel carcinoid tumors reveals areas of recurrentamplification and loss. Genes Chromosomes Cancer 47:591–603.
Kytola S, Hoog A, Nord B, Cedermark B, Frisk T, Larsson C,Kjellman M. 2001. Comparative genomic hybridization identi-fies loss of 18q22-qter as an early and specific event in tumori-genesis of midgut carcinoids. Am J Pathol 158:1803–1808.
Kytola S, Nord B, Elder EE, Carling T, Kjellman M, CedermarkB, Juhlin C, Hoog A, Isola J, Larsson C. 2002. Alterations ofthe SDHD gene locus in midgut carcinoids, Merkel cell carci-nomas, pheochromocytomas, and abdominal paragangliomas.Genes Chromosomes Cancer 34:325–332.
Lollgen RM, Hessman O, Szabo E, Westin G, Akerstrom G. 2001.Chromosome 18 deletions are common events in classicalmidgut carcinoid tumors. Int J Cancer 92:812–815.
Modlin IM, Oberg K, Chung DC, Jensen RT, de Herder WW,Thakker RV, Caplin M, Delle Fave G, Kaltsas GA, Krenning EP,Moss SF, Nilsson O, Rindi G, Salazar R, Ruszniewski P, SundinA. 2008. Gastroenteropancreatic neuroendocrine tumours. LancetOncol 9:61–72.
Moertel CG, Dockerty MB. 1973. Familial occurrence of metasta-sizing carcinoid tumors. Ann Intern Med 78:389–390.
Nord H, Segersten U, Sandgren J, Wester K, Busch C, Menzel U,Komorowski J, Dumanski JP, Malmstrom PU, de Stahl TD.2010. Focal amplifications are associated with high grade andrecurrences in stage Ta bladder carcinoma. Int J Cancer 126:1390–1402.
Otsuka R, Tamakoshi K, Wada K, Matsushita K, Ouyang P, HottaY, Takefuji S, Mitsuhashi H, Toyoshima H, Shimokata H,
FAMILIAL AND SPORADIC ILEAL CARCINOIDS 93
Genes, Chromosomes & Cancer DOI 10.1002/gcc

Yatsuya H. 2008. Having more healthy practice was associatedwith low white blood cell counts in middle-aged Japanese maleand female workers. Ind Health 46:341–347.
Pal T, Liede A, Mitchell M, Calender A, Narod SA. 2001. Intesti-nal carcinoid tumours in a father and daughter. Can J Gastroen-terol 15:405–409.
Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD,Mandelker D, Leary RJ, Ptak J, Silliman N, Szabo S, Buck-haults P, Farrell C, Meeh P, Markowitz SD, Willis J, DawsonD, Willson JK, Gazdar AF, Hartigan J, Wu L, Liu C, Parmi-giani G, Park BH, Bachman KE, Papadopoulos N, VogelsteinB, Kinzler KW, Velculescu VE. 2006. The consensus codingsequences of human breast and colorectal cancers. Science314:268–274.
Smyth GK. 2004. Linear models and empirical bayes methods forassessing differential expression in microarray experiments. StatAppl Genet Mol Biol 3:Article3.
Smyth GK. 2005. Limma: Linear models for microarray data. In:Gentleman RVC, Dudoit S, Irizarry R, Huber W, editors. Bioin-formatics and Computational Biology Solutions using R andBioconductor. New York: Springer, pp.397–420.
Solcia E, Kloppel G, Sobin LH. 2000. Histological Typing of En-docrine Tumours. WHO International Histological Classificationof Tumours, 2nd ed. Berlin: Springer.
Steemers FJ, Chang W, Lee G, Barker DL, Shen R, GundersonKL. 2006. Whole-genome genotyping with the single-baseextension assay. Nat Methods 3:31–33.
Sturn A, Quackenbush J, Trajanoski Z. 2002. Genesis: clusteranalysis of microarray data. Bioinformatics 18:207–208.
Tanaka T, Watanabe T, Kitayama J, Kanazawa T, Kazama Y,Tanaka J, Kazama S, Nagawa H. 2009. Chromosome 18q dele-tion as a novel molecular predictor for colorectal cancer with si-multaneous hepatic metastasis. Diagn Mol Pathol 18:219–225.
Wale RJ, Williams JA, Beeley AH, Hughes ES. 1983. Familialoccurrence in carcinoid tumours. Aust N Z J Surg 53:325–328.
Vogelstein B, Kinzler KW. 2002. The genetic basis of human can-cer, 2nd ed. New York: McGrawh-Hill.
Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, Shen D,Boca SM, Barber T, Ptak J, Silliman N, Szabo S, Dezso Z, Ustyank-sky V, Nikolskaya T, Nikolsky Y, Karchin R, Wilson PA, KaminkerJS, Zhang Z, Croshaw R, Willis J, Dawson D, Shipitsin M, WillsonJK, Sukumar S, Polyak K, Park BH, Pethiyagoda CL, Pant PV, Bal-linger DG, Sparks AB, Hartigan J, Smith DR, Suh E, PapadopoulosN, Buckhaults P, Markowitz SD, Parmigiani G, Kinzler KW, Velcu-lescu VE, Vogelstein B. 2007. The genomic landscapes of humanbreast and colorectal cancers. Science 318:1108–1113.
Zikusoka MN, Kidd M, Eick G, Latich I, Modlin IM. 2005. Themolecular genetics of gastroenteropancreatic neuroendocrinetumors. Cancer 104:2292–2230.
94 CUNNINGHAM ETAL.
Genes, Chromosomes & Cancer DOI 10.1002/gcc




























