Embed Size (px)
Citation preview

I N T E R N A T I O N A L A T O M I C E N E R G Y A G E N C Y , V I E N N A , 1 9 6 5


CHEMICAL EFFECTS OF NUCLEAR TRANSFORMATIONS
VOL. I


PROCEEDINGS SERIES
CHEMICAL EFFECTS OF NUCLEAR
TRANSFORMATIONS
PROCEEDINGS OF THE SYMPOSIUM ON CHEMICAL E F F E C T S ASSOCIATED WITH
NUCLEAR REACTIONS AND RADIOACTIVE TRANSFORMATIONS
HELD BY THE INTERNATIONAL ATOMIC ENERGY AGENCY
IN CO-OPERATION WITH THE JOINT COMMISSION ON APPLIED RADIOACTIVITY (ICSU)
IN VIENNA, 7-11 DECEMBER 1964
In two volumes
VOL.I
INTERNATIONAL ATOMIC ENERGY AGENCY VIENNA, 1965

Symposium on C hem ical E ffects A ssociated with N uclear R eactions and R adioactive T ra n sfo rm ations, Vienna 7 - 1 1 D ecem ber 1964.
P roceed in g s . . . held by the International Atom ic E n ergy Agency in co -op eratio n with the Joint C om m ission on Applied R adioactivity (ICSU) . . . Vienna, the Agency, 1965.
2 v o ls . (IAEA P roceed in g s se rie s )
5 4 1 . 2 8 5 4 1 . 1 5 6 2 1 . 0 3 9 . 8
CHEMICAL E F F E C T S OF NUCLEAR TRANSFORMATIONS, IAEA, VIENNA, 1965
ST I/PU B /91
Printed by the IAEA in Austria
April 1965

FOREWORD
The study of the chem ical changes consequent upon the nuclear tra n sformation of an atom that is linked with other atoms in a molecule and su rrounded by other sim ilar or d issim ilar m olecules has intrigued chem ists for a number of y e a rs . This interest is certainly not static but if anything is increasing. The main theme of this meeting was a discussion of the suggestions and theories that have been advanced to explain the wealth of ex perimental observations on the behaviour of atoms at energies and in situations not norm ally accessib le in the lab o ratory . Though the subject has some p ractica l im plications in the preparation of radioisotopes, this was not an im portant consideration at this Symposium.
The f irs t Symposium on hot-atom chem istry organized by the Agency was held in Prague in October 1960. Comparison of the past and the present state of the subject shows that a greater variety and sophistication of tech niques are now being applied as the sim pler approaches used in the past have been shown to be inadequate. P ro g re ss has been made in the understanding of the sim pler gas system , but in liquids and solids there is still much to clarify . It is also of interest that for the m ajority of the work r e ported in these Proceedings a re a c to r was the radiation source, and in this field much experim ental work still rem ains to be done.
The Symposium on Chemical Effects Associated with Nuclear Reactions and Radioactive T ransform ations was held from 7 to 11 Decem ber 1964 in Vienna, and was attended by 136 participants from 29 countries and 4 international organizations. It was organized by the International Atomic Energy Agency in co-operation with the Joint Commission on Applied Radioactivity. The publication of these Proceedings makes the content of the papers and discussion available to a wider audience than Was possible at the m eeting in Vienna.

EDITORIAL NOTE
The papers and discussions incorporated in the proceedings published by the International Atomic Energy Agency are edited by the Agency's editorial staff to the extent considered necessary for the reader's assistance. The views expressed and the general style adopted remain, however, the responsibility of the named authors or participants.
For the sake of speed of publication the present Proceedings have been printed by composition typing and photo-offset lithography. Within the limitations imposed by this method, every effort has been made to maintain a high editorial standard; in particular, the units and symbols employed are to the fullest practicable extent those standardized or recommended by the competent international scientific bodies.
The affiliations of authors are those given at the time of nomination.The use in these Proceedings of particular designations of countries or
territories does not imply any judgement by the Agency as to the legsd status of such countries or territories, of their authorities and institutions or of the delimitation of their boundaries.
The mention of specific companies or of their products or brand-names does not imply any endorsement or recommendation on the part of the International Atomic Energy Agency.

CONTENTS OF VOL. I
IONIZATION E FFE C T S AND TRITIUM HOT ATOM STUDIES IN THE GAS-PHASE (Sessions 1 and 2)
M ass spectrom etric studies of reactions of reco il ions withm olecules (SM-57 /69 ) ...................................................................... .............. 3S. Wexler (United States of America)
"Explosion" of multicharged m olecular ions: chem ical consequencesof inner shell vacancies in atoms (SM -57/88) .......................................... 23
.T. A : Carlson and R. M. White (United States of America)Ionization that follows a heavy-ion-induced nuclear reaction
(SM -5 7 / 2 8 ) .................................................................................................................... 35N. H. Steiger (Israel)
Discussion ................................................................. ....................................................... 53Kinetic isotope effects in recoil tritium reactions through m easure
ment of isotopic molecule yields (SM -57/85) ............................................ 55E. K. C. Lee, J. W. Root and F. S. Rowland (United States of America)
Quantitative studies of the reactions of hot tritium atoms withhydrocarbons and hydrocarbon m ixtures (S M -5 7 /3 9 ) ........................... 71D. S. Urch and M. J. Welch (United Kingdom)
Discussion ......................................................................................................................... 83Реакции горячих атомов водорода с этиленом. Роль возбужденных
этильных радикалов как промежуточных продуктов (SM-57 /61 ) . . . 87Б. Г. Дзантиев и А. П. Шведчиков (СССР)
Discussion ......................................................................................................................... 103
GASEOUS SYSTEMS (Session 2)
Reactions and mechanism s involving hot carbon atoms and N2 -H 2, N2-alkane and N2 -alkane-m oderator system s including theirrelationship to other simple system s (SM-5 7 / 6 7 ) .................................... 107H. Ache and A. P. Wolf (United States of America)
Competitive gas-phase reactions of C11 in binary oxygen-alkanesystem s (SM -57/50) .............................................................................................. 121G. Stocklin and A. P. Wolf (United States of America)
The effect of kinetic energy on the reactions of nucleogenic carbonatoms with hydrocarbons (S M -5 7 /7 0 ) ...................................................... 133J. Dubrin, H. Rosenberg, R. Wolfgang and C. MacKay (United States of America)
Discussion ......................................................................................................................... 145The reactions of hot fluorine-18 with gaseous carbon tetrafluoride
(SM -57/72) .................................................................................................................... 149N. Colebourne, J. F. J. Todd and R. Wolfgang (United States of America)
Discussion ......................................................................................................................... 175

Chemical reactions of N1 3 recoils from the C1 2 (d, n)N i 3 reaction(SM- 5 7 / 7 3 ) .................................................................................................................... 177W.S. Koski, D. Malinin and M. Berta (United States of America)
Gas-phase reactions of (n ;7 ) and isom eric transition-activated B r 8 0
with alkanes and haloalkanes (SM -57/79) ................................................... 185L. D. Spicer and A. A. Gordus (United States of America)
Chemical effects of the (n,p) reaction in gaseous system s: simple .alkanes and their chloroderivatives (SM -57/64) .................................... 195K. Pane к and K. Mudra (Czechoslovakia)
Discussion ....................................... . . ............................................................................. 205Разработка методов горячего синтеза меченных серой-35
биологически активных веществ (S M -5 7 /5 9 ).............................................. 209Б. Г. Дзантиев и А. В. Шишков (СССР)
Discussion ......................................................................................................................... 217
LIQUID SYSTEMS (Session 3) ■
Chemical effects of nuclear transform ations of halogens in organic ,media (SM -57/92) ................................................................................................... 221J. E. Willard (United States of America)
D iscussion............................................................................................................................ 236Химические эффекты ядерных превращений и процессы передачи
энергии возбуждения (SM -57/54) .................................................................... 239Ан.Н. Несмеянов и Э .С . Филатов (СССР )
Reactions of tritium recoil atoms in liquid organic m ixtures *(SM- 57/45a) ............................................................................................................... 255A. Soko/owska (Poland)
Hot phosphorus atom reactions in liquid organic m ixtures(SM -57/45b) .................................................................................................................. 265A. Siuda (Poland)
Discussion ......................................................................................................................... 273Реакции горячих атомов трития с алифатическими спиртами и
их смесями с бензолом и циклогексаном (SM -57/56) ........................ 277Э.С. Филатов, Ан.Н. Несмеянов и Цзян Тай-ван (СССР)
Discussion ......................................................................................................................... 299Reactions of hot Cl3 8 atoms in m ixtures of carbon tetrachloride
with aliphatic alcohols (SM -57/23) .............................................. .................. 301L. Vasar os (Hungary)
Chemical effects of nuclear recoil in organic halide system s: a new theoretical treatm ent and experim ental verification of the theory(SM- 5 7 / 3 6 ) ..................................................... .............................................................. 311S. S. Kontis, P. Sanitwongs and M. Weston (United Kingdom)
■ The stereoch em istry of the reactions of (n, y) halogen atoms withalkyl halides in the liquid phase (SM -57/76) ............................................ 333F. S. Rowland, C. M. Wai, С. T. Ting and G. Miller (United States of America)
Discussion ......................................................................................................................... 345

ORGANIC SYSTEMS (Session 4)
Effects of tem perature and p ressu re on hot-atom reactions in brom o-ethane (SM -57/93) ................................................................................ ................ 351A. J. Cole, M. D. Mia, G. E. Miller and P. F. D. Shaw (United Kingdom)
Discussion ......................................................................................................................... 371Взаимодействие атомов отдачи углерода-14 в бинарных системах,
содержащих гетероциклические соединения (S M -5 7 /5 2 )..................... 373Л,П . Фирсова, М.Ф. Баракат, М . Форысь иАн.Н. Несмеянов (СССР)
Chemical behaviour of С 1 1 in liquid hydrocarbons (SM -57/68) .......... 385A. F. Voigt, D. E. Clark and F. G. Mesich (United States of America)
Discussion ......................................................................................................................... 397Образование радиоактивных полимерных продуктов при реакциях
поливалентных атомов отдачи (SM -57/бО)..................................................... 399Б. Г. Дзантиев, Р. А. Стукан, А. П. Шведчиков и А. В. Шишков (СССР )
Discussion ..................................................................................... .................................... 407Реакции горячих атомов трития с аминокислотами (S M -5 7 /5 3 ) ......... 411
Е. Ф. Симонов и Ан.Н. Несмеянов (СССР)A triggering mechanism for the promotion of therm al annealing
in crystalline hexabromoethane by radiation-produced defects(SM- 57/4) ....................................................................................................................... 421
К. E. Collins (United States of America)Chemical effects of the nuclear isom eric transition of B r 80m in
glassy and polycrystalline alkyl bromides (SM -57/21) (Abstractonly) ................................................................................................................................ 433R. M. A. Hahne and J.E. Willard (United States of America)
Discussion .............................................................................................. .......................... 435


IONIZATION EFFECTS AND TRITIUM HOT ATOM STUDIES IN THE GAS PHASE
(Sessions 1 and 2)


MASS SPECTROMETRIC STUDIES OF REACTIONS OF RECOIL IONS WITH MOLECULES*
S. WEXLER
ARGONNE NATIONAL LABORATORY
ARGONNE, ILLINOIS, UNITED STA TES OF AMERICA
Abstract — Résumé — Аннотация — Resumen
MASS SPECTROMETRIC STUDIES OF REACTIONS OF RECOIL IONS W ITH MOLECULES. M echanism s
o f the reactions of re c o il species from n u clear transform ation h av e previously been inferred from th e d istri
bution of th e stable products. Two altern ative experim en tal approaches, w hich use mass sp ectrom etric te c h
niques to identify the tran sient io n ic in te rm e d ia te products o f r e c o il re a c tio n s , are d escrib ed . T h ese e x
periments provide ( l ) a d irect study of modes of energy loss by high-energy tritium recoils during slowing down
to the range of ch e m ica l reactio n energies and (2 ) evidence for the m echanism of the ch em ical reactio n with
C H 4 of the low -energy (TH e3 )+ daughter from 6 “ d ecay o f Тг.
Ionic products from interactions of 0 ,8 - 3 .7 5 MeV protons (as stand-ins for tritons) with several isolated noble gas atom s and hydrocarbon m olecu les (m eth an e, a cety len e , ethylene, ethan e, propane and n-b utane)
w ere observed in a portab le m ass sp ectro m eter that had sp ecial con stru ction al features. S pecies in various
ch arge states were produced from the noble gases. Partial ionization cross-sections determ ined for the in d i
vidual ch arge states varied between 1 0 " 1S and 1 0 "20 cm 2 m o lecu le"1. Collision of a proton with a p olyatom ic
hydrocarbon m o le cu le resulted in io n izatio n and exten siv e fragm entation . Only sin g ly -ch arg ed fragm ents
were found. Comparisons were m ade with the effects provoked by high energy electrons with the sam e velocity
as th at o f th e protons. Th e results show th a t e n e rg e tic tritiu m re co ils from th e H e 3 (n ,p )T 6 and Li6 (n , o$T
nuclear reactions must lose k in etic energy through in elastic ionizing and excitin g collisions with the m edium ,
and th at th e re co ils behave m uch lik e any ion izin g a g e n t. Consequences o f th ese processes in "h o t-a to m " ch em istry are discussed.
ÉTU D E, A L’AIDE D’ UN SPECTROM ÈTRE DE MASSE, DES RÉACTIONS ENTRE IONS DE RECUL ET
MOLECULES. Jusqu'à présent, on a d éterm in é les m écanism es des réactions provoquées par les espèces c h i
miques de recul provenant de transform ations nucléaires, en se fondant sur la distribution des produits stables.
L'auteur d écrit deux autres m éthodes expérim entales qui font appel à un spectrom ètre de m asse pour identifier
les produits ionisés interm édiaires des réactions de recul. Ces expériences p erm ettent: 1. d*étudier directem ent
les voies par lesquelles les tritons de recu l dotés d*une haute énergie perdent une partie de c e l l e - c i lorsqu'ils
sont ralentis au point d 'attein d re la g am m e des énergies des réactions chim iques; 2. d ’obtenir des indications
sur le m écanism e de la réactio n chim ique entre CH 4 e t (T?He)+ de faib le énergie , produit de la décroissance
S “ de T2 .
Les produits ionisés provenan t des in teractio n s en tre protons de 0 ,8 à 3 ,7 5 M eV (u tilisés à la p la c e de tritons) e t plusieurs atom es de g a z rares et m olécu les d ’hydrocarbures isolés (m éth an e , a cé ty lè n e , éth ylèn e,
éth ane, propane e t n -b u tan e) ont pu ê tre observés à l 'a id e d'un sp ectrom ètre de m asse p ortatif sp écialem en t
con çu à c e t effe t. Les g a z rares ont donné des espèces présentant différents é ta ts .d e ch a rg e . Les sections
efficaces partielles d ’ionisation, déterm inées pour les différents états de ch arge, varient de 1 0 ’ 15 à 1 0 ”20 cm 2
par m olécu le . La collision d'un proton av e c une m olécu le d ’hydrocarbure polyatom ique a provoqué une io n i
sation e t une fragm entation poussée. L’auteur n 'a trouvé que des fragm ents à ch arge unique. Il a com paré
ces effets av ec ceux qui sont provoqués par des électrons de haute énergie ayant la m êm e vitesse que les protons. Les résultats m ontrent que les tritons de recu l de haute énergie, qui proviennent des réactions nucléaires 3H e(n,p)T e t Ч л (п ,а )Т d oiven t perdre une p artie de leu r én erg ie cin étiqu e du fa it de chocs inélastiques ionisants et
e x cita n ts a v e c le m ilie u , e t que les a to m es d e re c u l ont un co m p o rte m e n t très sem b lab le à c e lu i de tous
les agents ionisants. L 'auteur exam in e T intérêt que c e phénomène présente pour la chim ie des atom es chauds.
il a utilisé des m éthodes d e spectrom étrie de m asse à « h a u te pression » pour prouver que l ’espèce p o ly
m ère СгН 4Т + constitue le principal in term édiaire ionique de longue période qui soit form é lors de la réactio n
* Based on work p erform ed under the auspices o f th e United S tates A to m ic Energy C om m ission .
3

4 S. WEXLER
entre le méthane et (T 3He)+, produit de la décroissance d de T2. Le précurseur C H iT * de C2H4T + semble avoir une période trop courte pour pouvoir être observé. Les données confirm ent la validité du m écanism e envisagé par Pratt e t Wolfgang pour exp liquer ré c h a n g e isotopique entre le tritium et le m éthane qui est provoqué par la décroissance de T 2. »
М А С С -С П Е К Т Р О М Е Т Р И Ч Е С К И Е И С С Л Е Д О В А Н И Я Р Е А К Ц И Й И О Н О В О Т Д А Ч И С М О Л Е К У Л А М И . М ехани зм ы реакций обр азц ов отдачи в р е з у л ь т а т е ядер ны х преобразований раньш е вы во д и л и сь и з р асп р ед ел ен и я у ст о й ч и в ы х п р о д у к то в . О п и сы ваю тся д в а д р у ги х э к с п е р и м ен тал ьн ы х м е т о д а , при к о то р ы х для р асп о зн аван и я п е р ехо д н ы х ионных п ром еж уточны х п р о дук то в реакций отдачи п ри м еняли сь м ето д ы м а с с -с п е к т р о м е т р и и . В р е з у л ь т а т е п р о ведения э ти х э к сп ери м ентов о б е сп е ч и в а ет ся 1 ) н еп о ср ед ствен н о е изучение форм потери энергии ат о м а м и отд ач и три ти я в ы со к о й э н ер ги и в о вр ем я за м е д л ен и я до д и ап азо н а э н ер ги й хи м и ч е с к и х реакций и 2 ) п од твер ж ден и е в отнош ении м е х а н и зм а х и м и ч еско й реакции д о ч е р н е го п р о д у к та ни зк о й э н ер ги и ( Т Н е 3 ) + , о б р а з о в а в ш е г о с я в р е з у л ь т а т е (3 -р а сп а д а Т ц с С Н 4 .
И онны е п р о д у к ты , о б р а з у е м ы е в р е з у л ь т а т е в за и м о д е й с т в и я п р о тон ов с э н ер ги ей 0 , 8 —3 ,7 5 М э в (н ап р и м ер в р е м е н н ы е з а м е н и т е л и т р и то н о в ) с н е к о то р ы м и и зол и р ованны м и атом ам и б л агор од н ы х г а з о в и м олекулам и у гл ев о д о р о д а (м е та н , ац ети л ен , эти лен, э тан , пропан и п -б у т а н ) , н абл ю дал и сь в п ер ен о сн о м м а с с -с п е к т р о м е т р е с о сп ец и ал ьны м и к о н с т р у к ционными о с о б е н н о с тя м и . О бразцы в р азн о м зар я д о во м состоя н и и производи ли сь из б л а го родн ы х г а з о в . П арц и альны е п о п ер ечн ы е се ч е н и я ионизации, о п р ед е л я е м ы е для о т д е л ь н ы х за р я д о в ы х со с то я н и й , м е н я л и сь в д и а п а зо н е м еж д у Ю ’^ и 1 0 -20с м 2 м о л е к у л а * ! . С т о л к н о вени е протона с м олекулой м н о го а т о м н о го у гл ев о д о р о д а приводило к ионизации и зн а ч и т ел ьной ф р агм ен тац и и . Б ы ли обнаруж ены т о л ь к о о скол ки с единичны м за р я д о м . П роводились ср авнен и я с э ф ф ек там и , вы зы ва е м ы м и эл ектр онам и вы сокой энергии с такой же ск о р о сть ю , к а к и п р о т о н ы . Р е з у л ь т а т ы п о к а з ы в а ю т , ч т о ат о м ы о тд ач и тр и ти я больш ой э н ер ги и , о б р а зу е м ы е в р е з у л ь т а т е я дер н ы х реакций Н е3 (п ,р )Т и Ы 6 ( п ,а ) Т , должны т е р я т ь к и н е т и ч е с кую энергию ч е р е з неупругую ионизацию и возбуж дени е стол кн овени й со средой и ч то атом ы отдачи в е д у т с е б я в зн ач и тел ьн о й ст еп ен и т а к ж е, к а к ионизирующий а г е н т . О б су ж д аю тся р е з у л ь т а т ы э ти х п р о ц ессо в в р а м к а х химии "го р я ч и х а т о м о в " .
М ето д ы м а сс -сп е к т р о м е т р и и "в ы с о к о г о д авл ен и я " прим енялись для п о к а з а , ч т о полим ерн ы е образцы C j Н4Т *п р е д с т а в л я ю т собой основной долгож ивущ ий ионный промеж уточный п р о дук т, об р азуем ы й при реакции д о ч е р н е го п родукта (Т Н е 3 )+ , о б р а з о ва в ш его ся в р е з у л ь т а т е б е т а -р а с п а д а Т г с м е та н о м . Ч асти ца СН4Т+ первичная в отношении C jH 4T , по-видимому, я в л я е т ся слиш ком короткож и вущ ей, чтобы е е можно было н а б л ю д ать . Д анны е подтверж даю т предлож енны й П р атто м и В о л ь ф г а н г о м м е х а н и зм н а в е д е н н о го р а сп а д а и зо т о п н о го о б м е н а тр и ти я с м е т а н о м . (Б о л ь ш а я ч а с т ь д а н н о го д о к л а д а п р е д с т а в л я е т соб ой н овы й м а т е р и а л , ч а с т ь же —о б су ж д ен и е н е д а в н о о п у б л и к о в ан н ы х р е з у л ь т а т о в ) . •
. ESTUDIO POR ESPECTROMETRIA DE MASAS DE LAS REACCIONES DE IONES DE RETROCESO CON MOLECULAS. Hasta ahora, e l m ecanism o de las reacciones de las especies de retroceso provenientes de transformaciones nucleares se ha deducido de la distribución de los productos estables. El autor describe otros dos procedimientos experim entales basados en la espectrometrfa de masas que sirven para identificar los productos iónicos intermedios de carácter transitorio de las reacciones de retroceso. Estos experimentos permiten:1 ) estudiar directam ente las modalidades de la pérdida de energfa* del tritio de retroceso de alta energía en e l transcurso de su m oderación hasta alcanzar los niveles de energía del orden de las reacciones quím icas;2) obtener pruebas del m ecanism o de la reacción quím ica del descendiente (T 3H e)+ de ba ja energía, proveniente de la desintegración 6 “ del T 2. con e l CH4.
En un espectrómetro de masas portátil de características especiales se has estudiado los productos iónicos provenientes de las interacciones de protones de 0 ,8 - 3 ,75 MeV (em pleadonen lugar de tritones) con varios átomos de gases nobles y m oléculas de hidrocarburos aislados (m etano, acetilen o , e tilen o , etano, propano y n-butano). Los gases nobles produjeron especies en estados diferentes de carga. Las secciones eficaces parciales de ionización determinadas para cada estado de carga oscilaron entre 1 0 "1* y 10“20 cm 2/m o lécu la . El choque de protones con moléculas poliatóm icas de hidrocarburo produjo com o resultado ionización y amplia fragmentación. Sólo se observaron fragmentos de una sola carga. Se efectuaron comparaciones con los efectos provocados por electrones de elevada energía animados de la misma velocidad que los protones. Los resultados muestran que e l tritio energético de retroceso proveniente de las reacciones nucleares sH e(n,p)T y ^ i(n , a )T debe perder energía cinética por choques inelásticos de ionización y excitación con el medio, y que las especies

REACTIONS OF RECOIL IONS WITH MOLECULES 5
de retroceso deben tener comportam iento muy parecido a l de cualquier agente ionizante. Se examinan las
repercusiones de estos procesos en la quím ica de los «áto m os calientes » .Se han aplicado técnicas de espectrometría de masas a «elev ad a presión»para demostrar que la especie
polim érica C 2H4T + constituye e l principal ion interm edio de periodo largo formado en la reacción del descendiente (T3He)+ , proveniente de la desintegración 6 del T 2, con e l CH *' El ion CH4T +, precursor del C 2H4T + , parece escapar a la observación por tener un período demasiado corto. Los datos confirman e l m ecanism o propuesto por Pratt y Wolfgang para e l intercam bio isotópico entre e l tritio y e l metano inducido por desintegración.
1. INTRODUCTION .
Experim ental studies of the chem istry of energetic atoms have almost exclusively involved the identification of the stable products formed by r e action with molecules of the radioactive recoils from spontaneous nuclear transform ation or nuclear reaction . M echanisms of these reactions have been inferred from the product distributions as affected by scavengers, molecu lar structure and state [1] . However, the information required for an adequate understanding of the mechanisms and kinetics of these often unique chemical interactions includes:
(a) The properties (charge, state of excitation, kinetic energy and molecular composition) of the recoil species at the instant of formation in the nuclear process;
(b) The modes of translational-and excitation-energy loss and charge change during the slowing down of the energetic recoils to the range of chemical reaction energies (< ~ 20 eV);
(c) The identity of the transient intermediate species (ionic, free radical or m olecular) taking part in the possible consecutive chemical reactions leading to the observed products; and
(d) The ways (e.g. dissociative charge neutralization, charge exchange, ion recombination and coílisional de-excitation) by which the m olecu lar species are stabilized as the observed products.
Much is known about the prim ary physical and chemical ch aracteristics of the nuclidic entities produced by radioactive decay, and some results have been obtained on the initial products of nuclear reactions [2] . Consequently, the next steps in this approach in hot atojn ch em istry should be the study of the m echanism s of energy loss during the slowing-down phase and the identification of transient interm ediates formed by reactions of the hot species with m olecules of the medium. F o r these types of investigations direct observation of the interactions of the recoils with isolated molecules is desirable and often required. This paper reports on a start that has been made on these lines. Two different m ass sp ectrorn etric techniques have been employed to study the transient ionic interm ediates formed by in teractions of recoil species with atoms and m olecules. One method has given direct evidence for modes of kinetic energy loss by high-energy tritium recoils during part of their slowing down to the range of chem ical reaction en ergies. The other has provided experim ental proof for a proposed m echanism of the sequence of chem ical reactions beginning with methane and the (THe3)+ daughter from beta decay of m olecular tritium .
Ionizing collisions of 0.8 to 3.75 MeV protons (as stand-ins for tritons) with the isolated noble gas atom s He, Ne, Ar andKr, and with the hydrocarbon

6 S. WEXLER
m olecules methane, acetylene, ethylene, ethane, propane and n-butane, were studied in a portable mass spectrom eter that had special constructional features. The slow positive fragments formed in these collisions were analysed as to chemical nature, charge state and abundance. Partial cross sections for form ation of several of the charge states of each of the noble gases were determined and compared with corresponding probabilities obtained by FEDORENKO et al. [3] for impact by 5 to 180 keV protons. The patterns of positively charged fragm ents resulting from bombardment by 2.25 MeV protons with the hydrocarbons were com pared with the sp ectra observed with 1225 eV electro n s. At these energies the two p ro jectiles possess the same velocity. This comparison provided an assessm ent of the behaviour of recoil tritons as ionizing agents.
The second experimental approach used the techniques of "high pressu re" m ass sp ectro m etry to establish the existence of the long-lived (> 1 0 ' 5 s) polym eric interm ediate involved in the decay-induced isotopic exchange of tritium with methane [41. The mechanism of tagging organic molecules with tritium by the widely used and simple technique of mixing T^ and the com pound, and allowing the two to stand for a time ("Wilzbach labelling" [5-7] ), has been investigated for several simple compounds by GANT and YANG [8 ] . Methane in p articular has been studied by PRATT and WOLFGANG [9] . Kinetic and scavenger procedures have led both groups of workers to conclude that there are two modes of labelling organic molecules with tritium : decay-induced labelling, which is initiated by reaction of the (THe3)+ daughter ion from radioactive transform ation of T2 ; and radiation-induced labelling, which is provoked by the excitation and ionization of the ta rg e t m olecule and T2 by the betas emitted in the nuclear decay and by secondary electrons. The m echanism s proposed for the two paths of isotopic tagging involve the reactions of positively-charged transient species.
Search for the ions suggested as intermediates in the decay-induced mechanism was made by introducing m ixtures of T2 and CH4 with and without added D2 into the source chamber of a "high-pressure" m ass spectrom eter [1 0 ] operated at a source pressure sufficiently high {—0 . 1 mm) so that consecutive ion-molecule reactions can occur. At this density of gas a primary ion m ay be expected to undergo sev eral collisions between form ation by nuclear decay and departure from the source chamber. As an illustration, in a gas at 0.1 mm an ion traversin g a length of 3.2 mm between point of origin and exit slit may collide with three molecules on the average. These experiments, then, serve to test the ionic mechanism proposed by Pratt and Wolfgang for the decay-induced isotopic exchange of tritium with methane. Investigation of the suggested radiation-induced m echanism s of tritiating CH4 has also been made [4] .
2. EXPER IM EN TA L
The object of this research necessitated the use of two completely different techniques of m ass spectrom etry. F o r the study of the behaviour ofhigh energy reco ils , a portable m ass sp ectrom eter was constructed on afram e provided with castors and jacks so that it could be readily moved into

REACTIONS OF RECOIL IONS WITH MOLECULES 7
position and aligned with the beam of high energy protons from the 4.5-M eV Van de Graaff electrostatic generator of this laboratory. F o r the investigation of the chem ical reactions following beta decay of m olecular tritium , a m ass sp ectrom eter ch aracterized by very large pumps, a vacuum-tight source chamber (except for the necessary tiny apertures) and a scintillation ion detector was designed. The spectrom eter could be operated with p ressures in the source as high as several tenths of a mm, and very low signals could be detected.
The design of the spectrom eter source section used in most of the high- energy projectile experiments is shown in Fig. 1. The energy-analysed and collim ated beam entered the source chamber through a knife-edge slit 2.0 mm high and 4.0 mm wide, and passed through the target gas on a path midway between the repeller plate and exit slit before being monitored in a deep air-cooled Faraday cup. It was stopped by a tantalum-coated iron plug that could be manoeuvered by an external magnet so as to perm it its observation through an end window. The position and profile of the beam could be judged from the fluorescence emitted when it struck a quartz plug adjacent to the window. A negative voltage on an adjacent electrode containing a slit 2 .4 mm high and 3.2 mm wide served to prevent secondary electro n s produced by the protons at the edges of the entrance slit from entering the cham ber. The Faraday cup was also maintained at a positive potential to suppress secondary electrons.
Slow positive ions formed in the ionizing collisions were repelled from the source chamber through a 6 . 3X18 . 8 mm slot covered by a grid of 60% open a re a . Two m ore electrodes containing slots of sim ilar dimensions, but covered with grid m aterial with 98% of its area unobstructed, accelerated and focused the ion beam. The ions then passed through the 0 .5X 15 .9 -m m object slit of the spectrom eter before being analysed by a 90°-secto r single directional focusing magnetic field of 9-in radius. They then entered the detector chamber through a 1 . 0 X1 5 . 9 - mm image slit, and their intensities w ere m easured by a plate surrounded by guard rings and an electrod e to suppress secondary electrons. A vibrating-reed electrom eter whose background was ~ 5 X 1 0 " 14 A measured the current to the detector. The spectrom eter could resolve fragments differing by one m ass unit at a m ass of 115. The pressure of the target gas in the source was approx. 10“ 4 mm. Generally, less than 1 0 ' 6 mm was observed for the pressures in the other com partments of the spectrom eter.
A high-energy electron gun designed by MONAHAN and STANTON [11] was attached to the source chamber for the studies of the fragmentation of polyatomic molecules by bombardment with electrons of the same velocity as that of the protons. The conditions of this part of the experim ent (direction and profile of the electron beam, the source pressure and the electr ic fields fo r extracting, focusing and accelerating the ions) were held as nearly the same as possible as those used in corresponding measurements with the proton beam. To minimize the deflection of the ionizing beams by the fringe field, the magnet was enclosed by a box of soft iron, and several layers of soft iron and aluminium were placed alternately between the magnet and the source of the sp ectrom eter. A m ore complete description of the experim ent has been published [ 1 2 ] .
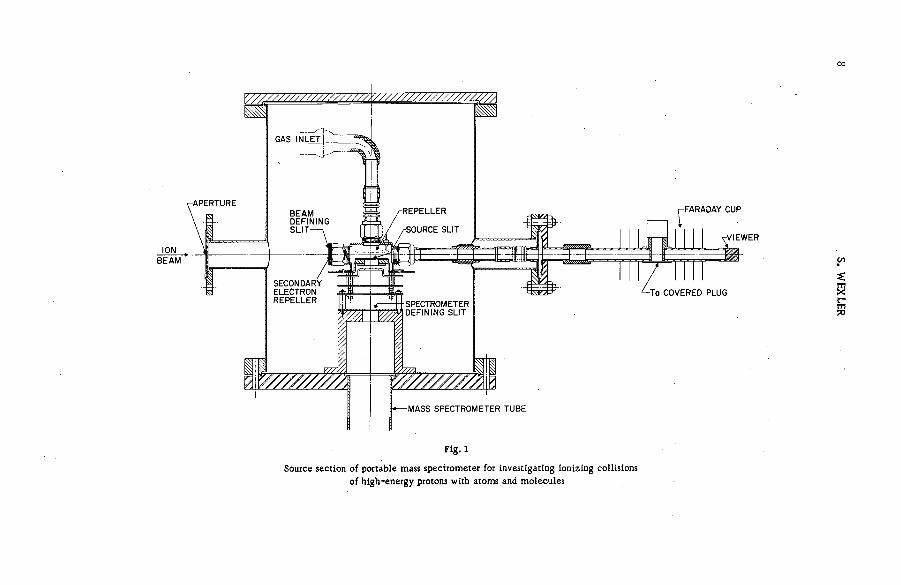
со
Fig. 1
Source section of portable mass spectrometer for investigating ionizing collisions
of high-energy protons with atoms and molecules
WE
XL
ER

REACTIONS OF RECOIL IONS WITH MOLECULES 9
T R A P S 4 5 0 1/ PUM P
A D J U S T A B L ES L IT
FOCUSING
ELEC TRO D
F IL A M E N
TO GAS
RESERVO IR
.SC INTILLATION ION
' DETECTOR
AD JU STABLE " S L IT
SOURCECH AM BER
60° SECTOR
'M AGNETIC
F IELD ( R = 12 in)
Fig. 2
High pressure" mass spectrometer
A sketch of the "h igh -p ressu re" m ass sp ectrom eter appears in F ig . 2. The magnetic deflection is conventional; a 60° sector of 12-in radius of cu rvature with single directional focusing analysed the positive species. But the spectrom eter has several distinguishing features which made the studies described here possible. Three 4 5 0 -1 /s m ercury-diffusion pumps, each exhausted through its own fore-pum p, were connected to the spectrom eter through short tubes of 5-in diam. ‘ The great pumping speed effected sharp decreases in pressure between the source chamber and other compartments of the instrument. Thus, when the pressure of a gas m ixture in the source was 0.15 mm, that in the chamber surrounding the source was 3X 10" 6 mm, while the pressure in the tube was 5X 10 ' 7 mm. A hem i-cylindrical repeller electrode established a field of 10 to 16 V/cm to impel positive ions produced in the chamber towards an exit slit 0 .025 mm high and 2 .0 mm wide. The only other aperture to the mass spectrom eter in the otherwise vacuum-tight chamber was a hole, 0.025 mm in d iam ., which, for testing purposes only, served to introduce an electron beam into the box. The low intensities (5 to 800 counts/m in above background were m easured) of the ionic species found were determined by pulse counting with a scintillation ion detector [13] extensively modified from the design of DALY [14] . 1
In the experiments designed to search for transient ionic intermediates of the decay-induced exchange of tritium with methane, m ixtures of T2, D2 and CH4 of known composition entered the source cham ber through la rg e - diam. tubes from an 11.4-1 re s e rv o ir . P ressu res in the source (of about0 . 1 mm) were calculated from the p ressu res in the re se rv o ir and the rate of flow in the inlet sy stem . Ionic species of positive charge w ere looked for by magnetic scanning. Only those ions resulting initially from the beta decay and from the accompanying internal radiation field in the source were observed. The electron gun was, of course, not used.
10 S. WËXLER
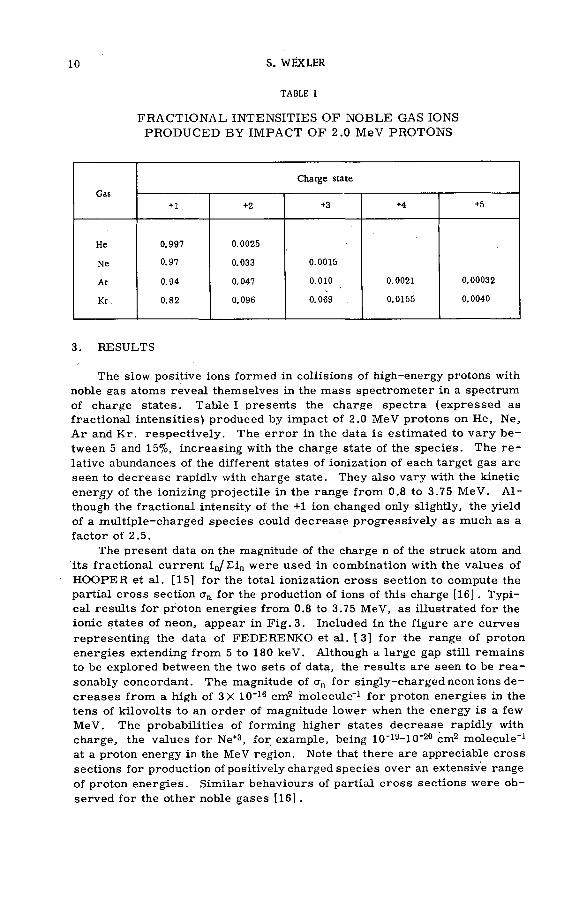
TABLE I
FRACTION AL INTENSITIES O F N O BLE GAS IONS PRODUCED B Y IM PACT O F 2 .0 MeV PROTONS
Gas
Charge state
+ 1 . +2 +3 +4 +5
He 0.997 0.0025
Ne 0.97 0.033 0.0015
Ar 0.94 0.047 0.010 0.0021 0.00032
Kr 0.82 0.096 0.069 . 0.0155 0.0040
3. RESULTS
The slow positive ions formed in collisions of high-energy protons with noble gas atoms reveal themselves in the m ass spectrom eter in a spectrum of charge sta tes. Table I presents the charge sp ectra (expressed as fractional intensities) produced by im pact of 2.0 MeV protons on He, Ne, A r and K r. respectiv ely . The e r r o r in the data is estim ated to v ary between 5 and 15%, increasing with the charge state of the species. The r e lative abundances of the different states of ionization of each target gas are seen to decrease rapidlv with charge state. They also vary with the kinetic energy of the ionizing p rojectile in the range from 0.8 to 3.75 MeV. A lthough the fractional intensity of the + 1 ion changed only slightly, the yield of a m ultiple-charged species could d ecrease p rogressively as much as a facto r of 2 .5 .
The present data on the magnitude of the charge n of the struck atom and its fractional cu rren t in/ E in w ere used in combination with the values of HOOPER et al. [15] for the total ionization c ro s s section to compute the partial cross section ста for the production of ions of this charge [16] . Typical results for proton energies from 0.8 to 3.75 MeV, as illustrated for the ionic states of neon, appear in F ig .3 . Included in the figure are curves representing the data of FEDERENKO et al. [3 ] for the range of proton energies extending from 5 to 180 keV. Although a large gap still rem ains to be explored between the two sets of data, the results are seen to be re a sonably concordant. The magnitude of an for singly-charged neon ions decre a s e s from a high of 3X 10 " 16 cm2 molecule" 1 fo r proton energies in the tens of kilovolts to an o rd er of magnitude low er when the energy is a few MeV. The probabilities of forming higher states d ecrease rapidly with charge, the values for Ne+3, for example, being 1 0 '19-10"2° cm2 molecule ' 1 at a proton energy in the MeV region. Note that there are appreciable cross sections for production of positively charged species over an extensive range of proton energies. Sim ilar behaviours of partial cro ss sections were observed for the other noble gases [16] .
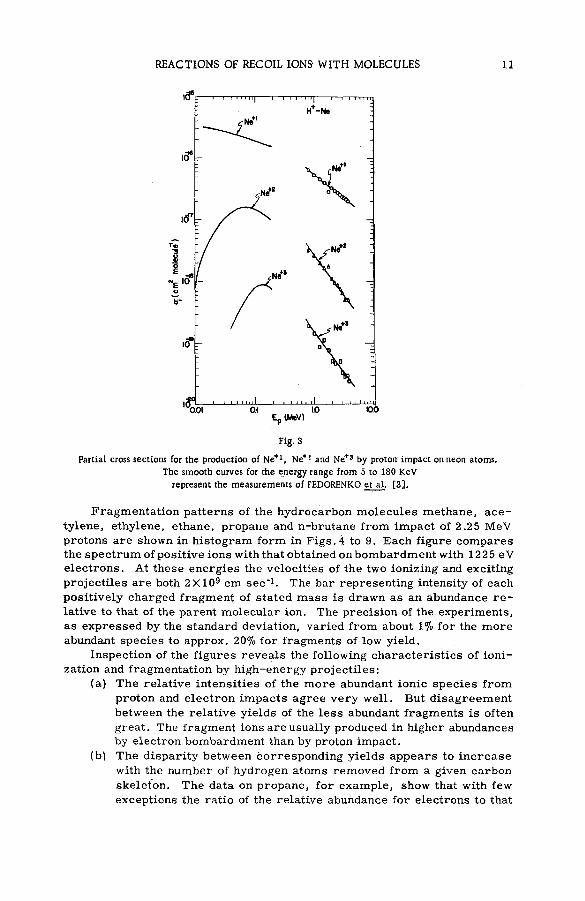
REACTIONS OF RECOIL IONS WITH MOLECULES 11
Fig. 3
Partia l cross sections for the production of Ne+1, Ne+! and Ne+S by proton: impact on neon atoms.
The smooth curves for the energy range from 5 to 180 KeV
represent the measurements of FEDORENKO et al. [3].
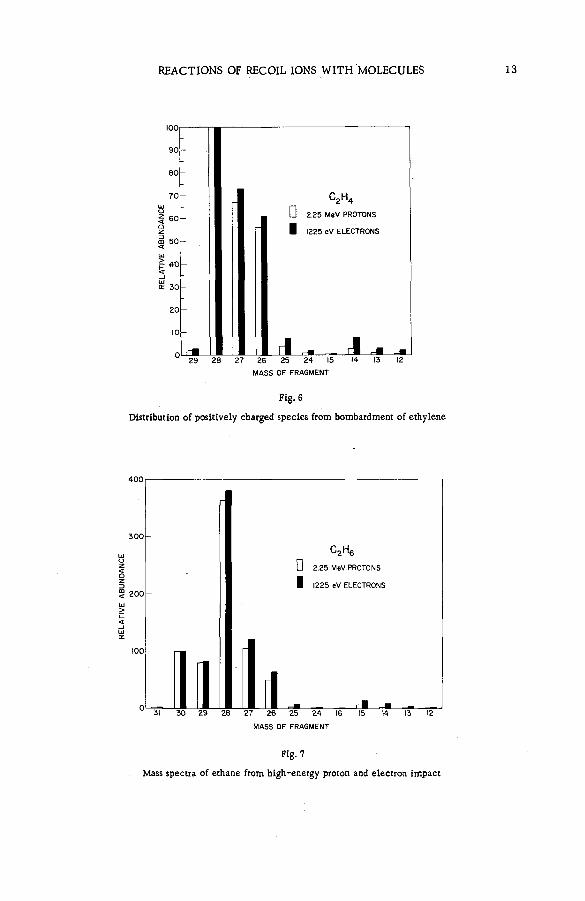
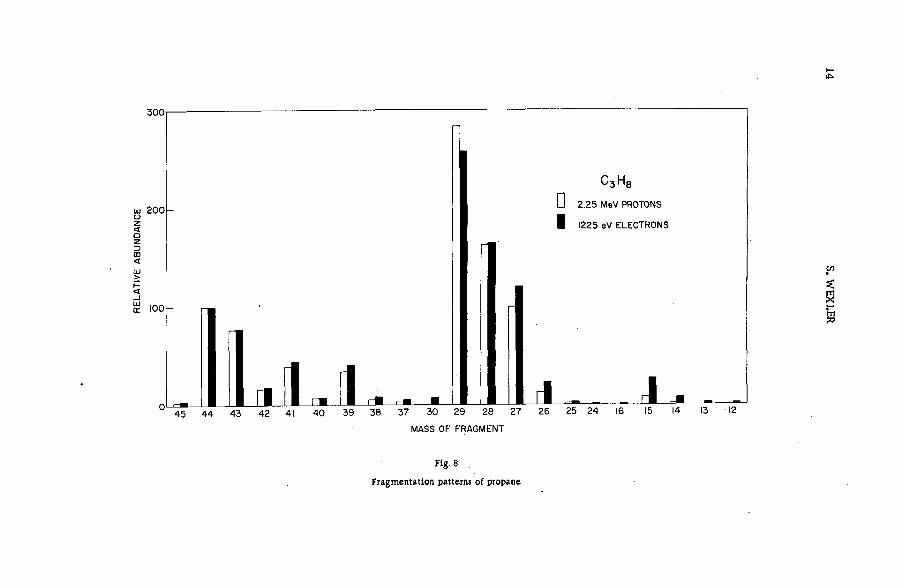
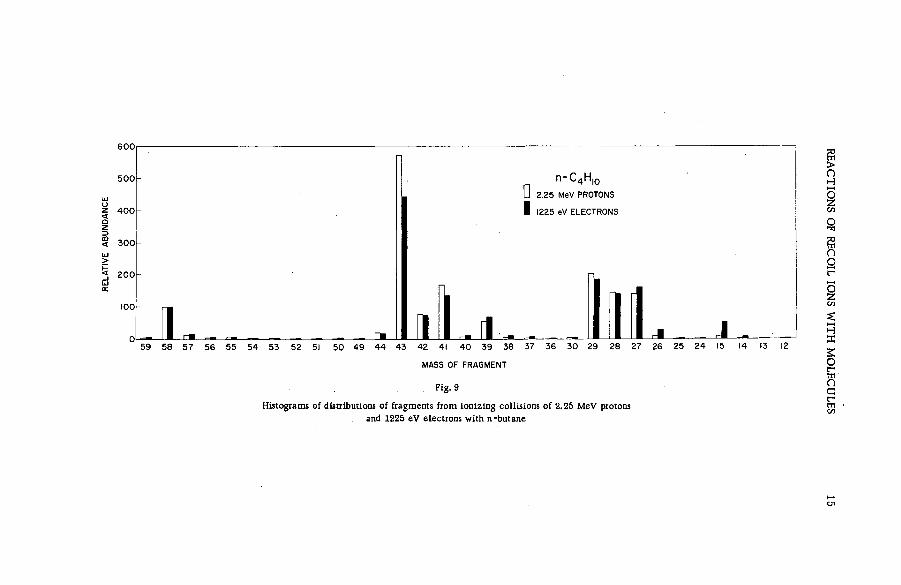
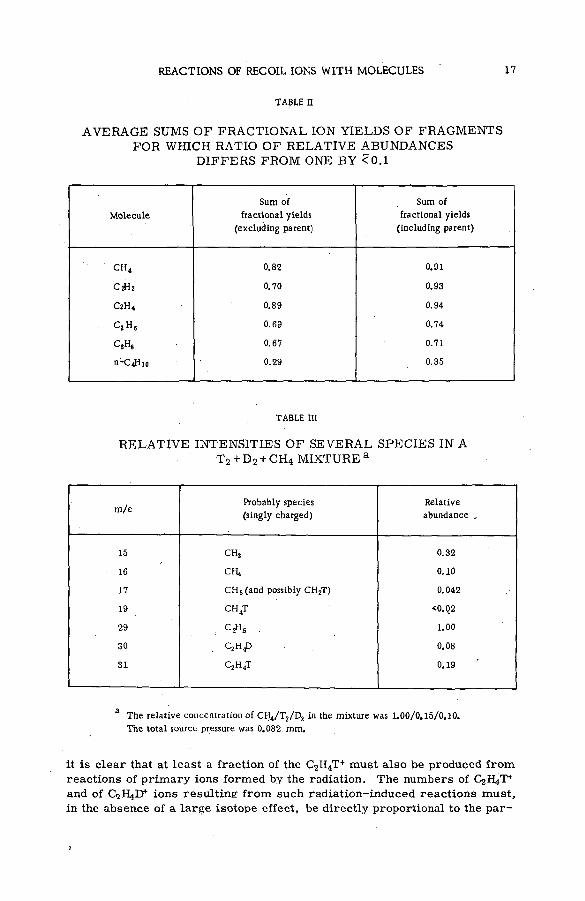
Fragm entation patterns of the hydrocarbon m olecules methane, a ce tylene, ethylene, ethane, propane and n-brutane from im pact of 2.25 MeV protons are shown in histogram form in Figs. 4 to 9. Each figure compares the spectrum of positive ions with that obtained on bombardment with 1225 eV electrons. At these energies the velocities of the two ionizing and exciting projectiles are both 2 X 10 9 cm sec"1. The bar representing intensity of each positively charged fragm ent of stated m ass is drawn as an abundance r e lative to that of the parent molecular ion. The precision of the experiments, as expressed by the standard deviation, varied from about 1 % for the more abundant species to approx. 2 0 % for fragments of low yield.
Inspection of the figures reveals the following ch aracteristics of ionization and fragmentation by high-energy projectiles:
(a) The relative intensities of the m ore abundant ionic species from proton and electron im pacts agree v ery well. But disagreem ent between the relative yields of the less abundant fragments is often great. The fragment ions are usually produced in higher abundances by electron bombardment than by proton impact.
(b) The disparity between corresponding yields appears to in crease with the number of hydrogen atoms removed from a given carbon skeleton. The data on propane, for exam ple, show that with few exceptions the ratio of the relative abundance for electrons to that
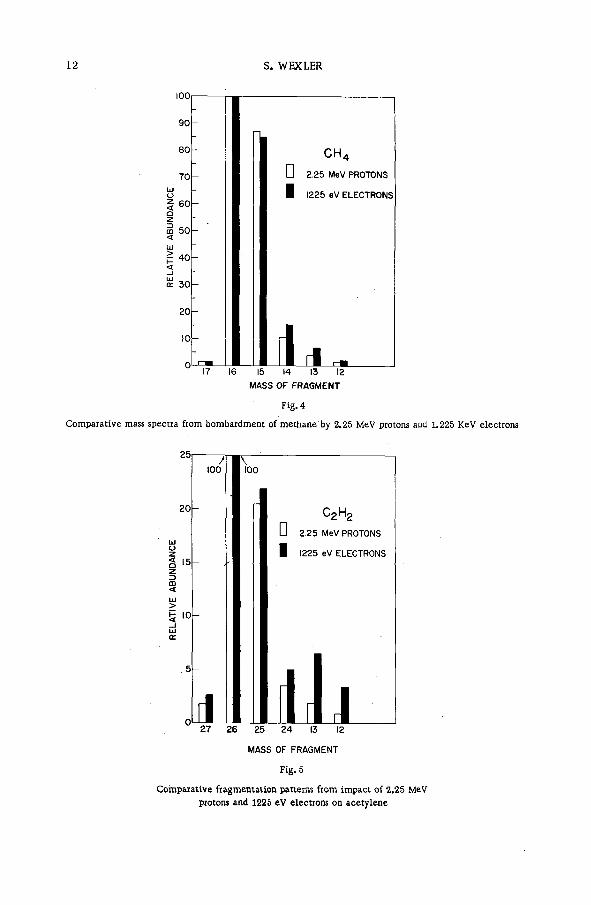
12 S. WEXLER
100
90
80
70
60
m 50<
<_i ш cc 30
20
10
CH4
G 2.25 MeV PROTONS
I 1225 eV ELECTRONS
1 I .17 16 15 14 13 12
MASS OF FRAGMENT
Fig. 4
Comparative mass spectra from bombardment of methane by 2.25 M eV protons and 1.225 K eV electrons
25
20
Шоz
Z)m<
10
/ \ 100 100
□I
C2H2
2.25 MeV PROTONS
1225 eV ELECTRONS
27 26 25 24 13 12
MASS OF FRAGMENT
F ig .5
Comparative fragmentation patterns from impact o f 2.25 MeV
protons and 1225 eV electrons on acetylene
RELATIV
E
ABU
ND
AN
CE
REACTIONS OF RECOIL IONS WITH MOLECULES
100
90-
с * 0 -
20 -
10-
с2н4D 2.25 MeV PROTONS
I 1225 eV ELECTRONS
29 28 27 26 25 24 15
MASS OF FRAGMENT
Fig. 6
Distribution of positive ly charged species from bombardment of ethylene
300
200
100
° 31 30 29 28 27 26 25 24 16 15 14 13 12
MASS OF FRAGMENT
Fig. 7
Mass spectra of ethane from high-energy proton and electron impact
Ii
C2H6
2.25 MeV PROTONS
1225 eV ELECTRONS
300
lu 2 0 0 - o z < o zD ffl <Ш
S loo
i ■__ ■45 44 43 42 41 40 39 38 37 30 29 28 27
MASS 0F FRAGMENT
Fig. 8
Fragmentation patterns of propane
2.25 MeV PROTONS
1225 eV ELECTRONS
RELATIV
E
AB
UN
DA
NC
E
600
500
400
300
200
100
n -C 4H|0D 2.25 MeV PROTONS
I 1225 eV ELECTRONS
59 58 57 56 55 54 53 52 51 50 49 44 43 42 41 40 39 38 37 36 30 29 28 27 26
MASS OF FRAGMENT
. F ig .9
Histograms of distributions of fragments from ion iz ing collisions of 2.25 M eV protons
and 1225 eV electrons w ith n-butane
_ - J ________ -25 24 15 14 13 12
Ul
REACTION
S OF
RECOIL IONS
WITH
MO
LECULES

16 S. WEXLER
for protons rise s progressively from 1.0 to 1.4 as the m ass of the ion d e creases from 4 3 (СзН7 ) to 37(СзН+). The behaviour is r e peated in the groups of species with two carbon atom s and again with one carbon atom in the molecular skeleton, although the magnitudes of the ratios change.
(c) There appears to be no relation between the ratio of relative yields and the number of carbon atoms in the skeleton of the ion. Although the disparity for all the multicarbon molecules studied is qualitatively g reater for the groups of ions than for groups of higher carbon content, there is no discernible difference between the C2, C3 and C4 groups.
(d) Only singly-charged species are observed.In a quantitative sense the ionization and fragmentation of a molecule
by collision of a high-energy proton is found to be v ery sim ilar to that by an electron of the same velocity. This may be seen in column 2 of Table II, which gives the averages from different experiments of the totsil fractional ion yield of those fragments for which the ratio of relative abundance from electron bombardment to that from proton impact deviates from unity by less than 10%. Because the abundances of each parent ion were given the same value, their yields w ere not included in the summations used to calculate the fractional yields of the fragm ents. Since each ionizing collision a c companied by excitation of the parent ion may lead to the appearance of a charged fragm ent, the data on methane, for example, show that in an average of 82% of such im pacts the behaviours of projectile and target are independent of the nature of the swiftly-moving p article . Only for n-butane is the percentage low, and this is undoubtedly the result of the relatively high ratio (1.23) found for the species C3H7 , an ion which accounts for 32 to 42% of the total ion yield in the fragment spectrum . Note that only collisions involving fragmentation of the target molecule were considered in the preparation of the data in column 2 of Table II. If the abundances of the parent species a re included, the fractions of collisions in which the behaviour of the molecule is the same for a high-energy proton as for an electron of the same velocity increase to the usually large values presented in column 3 of the table.
Tables III and IV summarize the results of the experiments that searched for transient intermediates of the decay-induced isotopic exchange of tritium with methane. The ionic species observed (Table III) when m ixtures of T 2 + D2 + CH4 w ere introduced at o v er-a ll p ressu res of about 0.1 mm into the source cham ber of the m ass sp ectrom eter included the p rim ary ions produced by self-rad iation of the gas m ixture by the betas from decay of T2 and by secondary electrons, and the secondary ions CH5 , C2 H5 , C2H4T+ and C2H4D+ resulting from ion-m olecule reactions of the (THe3)+ daughter of the nuclear transform ation and of the p rim a rie s . Deuterium gas was added to assess the effects of radiation-induced reaction of D2 (and thus T2 ) with CH4 , the assumption being made that T2 and D2 react in identical ways with electrons, CH¿, etc. One notes (Table IV) that the ratio of the intensity of m ass 31 (C2H4T +) to that of m ass 29 (C^Hj) is invariably g rea ter than the ratio of 30/29 (C2H4D+/C 2 H5 , corrected for C12C13H5 ). However, .nee the C2Ü4D+ ions observed can be formed only by radiation-induced processes,
REACTIONS OF RECOIL IONS WITH MOLECULES 17
TABLE II
AVERAGE SUMS OF FRACTIONAL ION YIELDS OF FRAGMENTS FOR WHICH RATIO OF RELATIVE ABUNDANCES
DIFFERS FROM ONE BY <0.1
M olecule
Sum of
fractional yields
(excluding parent)
Sum of
fractional yields
(including parent)
C H 4 0.82 0.91
CÜ2 0.70 0.93
CzH4 0.89 0.94
C2H 6 0.69 0.74
CSH8 0.67 0.71
П-С4Н 10 ' 0.29 0.35
TABLE I I I
RELATIVE INTENSITIES OF SEVERAL SPECIES IN A T 2 + D2 + CH4 MIXTURE a
m/eProbably species
(singly charged)
Relative
abundance ,
15 CH3 0.32
- 16 CH4 0.10
. 17 . C H 5(and possibly CH 2T) 0.042 .
19 C H 4T <0.02
29 . CPs ■ 1.00
30 с 2н р . . 0.08
31 C2H 4T ■ 0.19 '
a The re la tive concentration of CH4/ T 2/D 2 in the m ixtu re was 1.00/0.15/0.10.
The to ta l source pressure was 0.082 mm.
it is clear that at least a fraction of the C2H4T + must also be produced from reactions of prim ary ions formed by the radiation. The numbers of C^HjT* and of СгЩО* ions resulting from such radiation-induced reaction s m ust, in the absence of a large isotope effect, be directly proportional to the p ar-
18 S. WEXLER
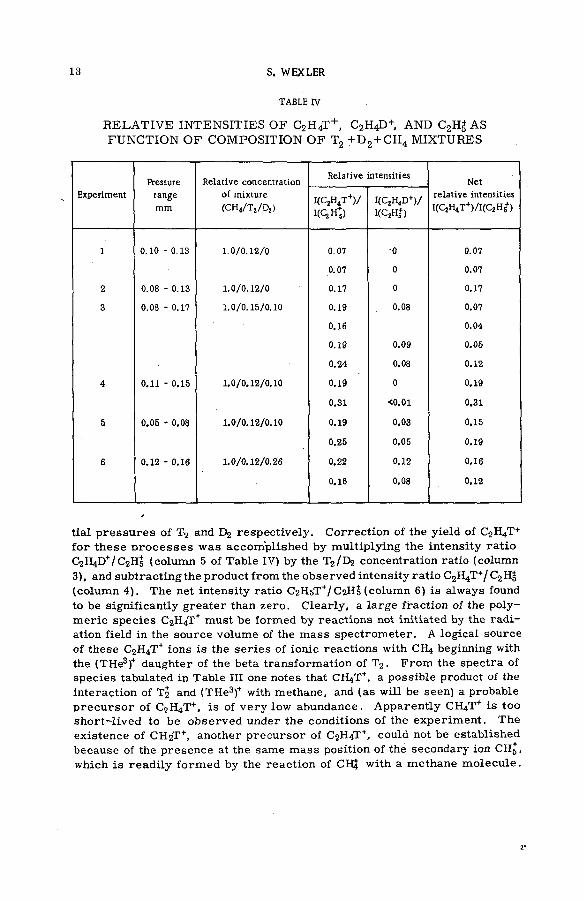
TABLE IV .
R ELA T IV E IN TEN SITIES O F C2 H4T + , C2H4D+, AND C2HJ AS FUNCTION O F COMPOSITION O F T2 + D 2 + CH4 M IXTURES
ExperimentPressure
rangemm
Relative concentration
of mixture
(C H 4/T2/D2)
Relative intensitiesNet
relative intensities
I(C2H4T +)/I(C2H s+)I(C 2H<T+)/I(C ,H +5)
I(C jH 4D +)/I(C 2H5+)
1 0.10 - 0.13 1 .0/0. 12 /0 0.07 0 0.07
0.07 0 0.07
2 0.08 - 0.13 1 .0/0. 12 /0 0.17 0 0.17
3 0.08 - 0.17 1.0/0.15/0.10 0.19 0.08 0.07
0 .16 0.04
0.19 0.09 0.05
0.24 0.08 0 .12
4 0.11 - 0.15 1 . 0/0. 1 2 /0.10 0.19 0 0.19
0.31 <0 .0 1 0.31
5 0.05 - 0.08 1 . 0/0. 1 2 /0.10 0.19 0.03 0.15
0.25 0.05 0.19
6 0 .12 - 0.16 1.0/0.12/0.26 0.22 0 .12 0.16
0.16 0.08 0 .12
tial p ressu res of T2 and respectively. Correction of the yield of C2 H4T+ for these p ro ce sse s was accom plished by multiplying the intensity ra tio C2 H4D+/C 2Hj (column 5 of Table IV) by the T2 /E>2 concentration ratio (column 3), and subtracting the product from the observed intensity ratio С2Н4Т+/ С2Щ (column 4). The net intensity ratio C2HsT+/C 2H5 (column 6 ) is always found to be significantly greater than zero. Clearly, a large fraction of the polym eric species C2H4T+ must be formed by reactions not initiated by the radiation field in the source volume of the m ass spectrom eter. A logical source of these C2H4T+ ions is the series of ionic reactions with CH4 beginning with the (THe3)+ daughter of the beta transformation of T2 . From the spectra of species tabulated in Table III one notes that CH4T+, a possible product of the interaction of T2 and (THe3)+ with methane, and (as will be seen) a probable p recu rso r of CqH4T+, is of very low abundance. Apparently СЩТ* is too short-lived to be observed under the conditions of the experim ent. The existence of CH2T +, another p recu rsor of C2H4T +, could not be established because of the presence at the same m ass position of the secondary ion CH5 , which is readily formed by the reaction of СЩ with a methane m olecule.

REACTIONS OF RECOIL IONS WITH MOLECULES 19
4 . DISCUSSION
PLATZMAN and HART [17] have pointed out that the chem istry of nuclear transform ations can be discussed in term s of the sam e three successive temporal stages invoked in the interpretation of events in customary radiation chem istry. Thus, the physical stage includes the prim ary effects of nuclear transition or reaction in the labelled or target molecule r e s pectively, and the slowing down of the reco il species with accom panying acceleratio n , excitation and ionization of the surroundings. A physicochem ical stage follows, and here the epithermal chemical behaviour of the recoiling nuclide is important. Finally the chemical stage appears, a time that is characterized by the reactions of species that have attained therm al energies. In this context, the studies reported here on the ionization and fragmentation of atoms and molecules by high-energy protons (and thus t r i tons) are concerned with aspect of the physical stage of nuclear-transform ation chem istry, while the "high-pressure" mass spectrom etric experiments that established the existence of an ionic intermediate taking part in the decay- induced tagging of methane with tritium may properly be included in the physico-chem ical stage.
(1) The mechanism of kinetic energy loss by energetic recoil tritons
An extensive literature has appeared on the chem ical reactions of hot tritium atom s with an organic m olecule [1] . The tritiu m atom s used in these studies are alm ost invariably produced by the nuclear reactions He3 (n, p)T and L i6 (n, a )T , in which the (probably) positive tritium ions are formed with translational energies of 192 and 2730 keV respectively. These energies are far in excess of the region for chem ical reactions, estimated to be in the range below 10 to 20 eV [18, 19]. The recoils must, therefore, be moderated considerably before they can form stable bonds with surrounding m olecular fragm ents. The experim ents with MeV protons d escribed here are consequently concerned with the mechanisms of energy loss by tritium ions during part of their moderation to energies of chemical interest.
A ccording to the theory of absorption of energetic projectiles in a medium [2 0 ], the retardation of the particle occurs through energy transfer, mainly as excitation and ionization, to atoms and molecules that are in the vicinity of the moving charged p rojectile . F ro m the early days of radioactivity it has been known that alphas, protons and other charged atom ic p artic les ionize and excite m olecules. However, the significance of the m ass sp ectrom etric data presented above is that the behaviour of a m olecule when struck by a high-energy proton is very sim ilar to that when hit by an electron of the sam e velocity . The m olecules a re ionized and e x cited by the collisions, and dissociate into a variety of ionic (and unobserved neutral) fragm ents. The distributions of the positively charged fragments from impacts of the two swiftly-moving projectiles are quite sim ilar. This result is in accordance with argum ents based on the Born approximation, as exp ressed in the Bethe equation [2 0 -2 5 ]. F u rth e r, the fragm entation patterns observed on bombardment with these high-energy p rojectiles are sim ilar to the sp ectra of ions produced in the m ass sp ectro m eter by

20 S. WEXLER
electrons of low ( ~ 7 0 eV) energy [25, 26]. In addition, the cro ss sections for ionization, as illustrated by the data for Ne (Fig. 3), are appreciable over alm ost the entire energy range of the triton during its moderation. From these observations, it may be concluded that an energetic triton acts much like any ionizing agent as it slows down.
A number of conclusions regarding the chem istry of recoil tritium may be derived from the foregoing paragraph. F irs tly , a fair fraction of the chem istry involved in the studies of tritium recoils is the radiation chemistry provoked by the tritons during their m oderation. F o r , from each He3 (n, p)T nuclear reaction 780 keV of energy is released . This amount accrues to the proton and triton as kinetic energy and is subsequently dissipated mainly as ionization and excitation of the medium. When tritium recoils are produced by the Li6 (n,a)T process, some 4.8 MeV is absorbed by the surroundings per tritium atom released. Taking 30 eV as the energy required to form an ion pair, one finds that about 26 0 0 0 molecules are ionized and fragm ented p er tritium and hydrogen ions slowed down after formation from a He3 target, and approx. 160 000 molecules are so affected when an alpha p article and a T* are stopped. Roughly another 26 000 and 160 0 0 0 m olecules respectively should be excited during the moderation of the two recoils in each case . The concentration of excited and ionized primary products so formed can be shown to be far higher (>25 times higher) than the density of activated molecules produced by the background radiation in the neutron re a c to rs in which the experim ents are perform ed. In fact, 1 h irrad iation of a compound mixed with 1% He3 o r L i 6 in a neutron flux of 1 0 12 cm ' 2 s e c * 1 will cause 1 % of the substance to be destroyed by e x citation o r ionization by the reco ils . The interm ediates and some products of the irrad iation will re a c t often m ore readily than the parent m olecule with both hot and th erm al tritium atom s.
Secondly, and m ore im portant, the distribution of the prim ary fra g ments, ionic and neutral, should be much like that resulting from the action of custom ary radiation fields, and consequently the interm ediate species and radiolytic products from both should be sim ilar. These sim ilarities would appear to justify the addition of scavengers (e .g . halogens, NO), originally employed for the purpose in radiation and photo-chem istry, to distinguish between hot and thermal processes in the chemistry of recoil âtoms. If the kind and -number of species form ed along the track of the energetic p article are like those produced by the usual radiation fields, the effects of the scavenger should be identical. The scavenger would be expected to re a c t efficiently with therm al radiolytic interm ediates and final products whether formed by recoils or by electrons and high-energy electromagnetic radiations.
Fu rth erm ore, tritium atom s that have survived chem ical reaction as hot species and have become thermalized should act like therm al hydrogen atoms produced by radiation, and therefore the distributions of stable products formed by the two should be the sam e. Although a definitive test of this conclusion has not appeared in the literatu re , the extensive studies of reactions of therm al tritium and of radiation effects by ROWLAND [19, 27, 28] and by WILLARD [291 indicate a qualitative similarity between the radioactive products from reactions of therm al tritium s and the compounds from radiolysis.

REACTIONS OF RECOIL IONS WITH MOLECULES 21
(2) The mechanism of the decay-induced isotopic exchange of tritium with methane
The ionic m echanism proposed by PRATT and WOLFGANG [9] to account for that part of the tritium labelling of CH4 initiated by reaction of the (THe3)+ daughter of Тг is composed of the following consecutive reactions:
T2 - (THe3)+ +j3~ (1)(THe3)+ +CH4^ (СЩТ+^ + Не3 (2)(CH4T+)*-» CH2T+ + H2 (3)С Н ^ + С ^ - G jH ^ + Ha (4)
+ rCH2T + C H 2 СгЩТ + e [ C H 3 + CHT (5)
CH2T +C H 4 - CH3T + CH3 (6 )
It is seen that, following beta decay of T2, the (THe3f daughter reacts with methane to form an excited СЩТ* ion, which dissociates quickly (in ~ KTn s [9 ]) to CH2T+ . The la tte r then re a c ts with CH4 to give the tritia ted ethyl ion СгЩТ*. Since it is proposed that this ionic species is neutralized by electron capture, it must be very long lived, for STEVENSON [30] has calculated that the lifetime of an ion against electron capture is about 1 0 ® times as long as the lifetim e against an ion-m olecule reaction in a gas at a p re - sure near one atmosphere, when the dose rate is 300 r sec"1. Under P ratt and W olfgang's and the presen t experim ental conditions the fa c to r would be even g re a te r . . •
The observations (Tables III and IV) from the study of CH4 + T2 + D2 m ixtures support the mechanism suggested by Pratt and Wolfgang. For example the ion СЩТ* was considered by them to be formed in an excited state and to dissociate quickly into CH2Tf . It should, therefore, be too short lived to be observed in the m ass sp ectrom eter, where ion life-tim es g re a te r than about 10"5 s are required for detection. The data (Table III) show this species (m /e = 19) to be absent from the spectra of ion species. Again, an ion of m ass 17 was found with reasonable yield, but it is not clear that it is the proposed dissociation product CH2T^ from (СЩ Т*)*, because CHS, a secondary ion formed readily by reaction of CH4 with methane, p ossesses the sam e m ass. However, the polym eric species Q K jT + was shown to be partially formed by ion-molecule reactions initiated by a prim ary precursor not produced by the radiation field. The most likely prim ary species that is not caused by radiation is the m olecular-ion (THe3)+, the daughter of T2 by beta decay. This ionic species has a lifetime greater than 10's s in collision-free space, for it has been shown by m ass spectrom etric techniques to be formed in 94.5% of the beta transitions of m olecular tritium [31] . Further, the condensation reaction of СЩ -type ions with CH4 to form C2 H$ - type secondaries proceeds v ery efficiently [32] . Also, the stability of C2H4T + against ion-m olecule reaction s with methane, implied in the m echanism of P ratt and Wolfgang, is in agreement with previous observation that ions of the C2H5-type do not re a c t with methane [10] . In conclusion, though the present experiments do not unequivocally establish the existence of the intermediate СН2Т* because of interference from CH5, the absence of СЩТ+ and the presence of CjHjT* in the ion pattern a re evidence for the co rre c tn e ss of the m echanism proposed by P ra tt and Wolfgang.

22 S. WEXLER
R E F E R E N C E S
[1 ] See, e .g . the reviews of CAMPBELL, L G . , Adv. Inorg. and Radiochem. 5^(1963) 135 and of WOLFGANG, R. (to be published).
[2 ] WEXLER, S . , in Actions Chimiques et Biologiques des Radiations 8_ (Haissinsky, M ., Ed. ) Masson and
Cie, Paris (1964).
[3 ] SOLOVEV, E.S. , ILIN, R. N. , OPARIN, V. A. and FEDORENKO, N. V . , Soviet Phys. - JETP 15 (1962) 459; 2. eksp. teor. Fiz. 4£ (1962) 659.
[4 ] WEXLER, S. , J. Amer. chem. Soc. 85 (1963) 272.[5 ] WILZBACH, K. E. , J. Amer. chem. Soc. 79 (1957) 1013.[ 6] WILZBACH, K. E ., and RIESZ, P., J. phys. Chem. 62 (1958) 6.[7 ] AHRENS, W. R., SA'UER, M. C. Jr. and WILLARD, J. E ., J. Amer. chem. Soc. 79 (1957) 3284.
[8 ] GANT, P. L. and YANG, K. , J. chem. Phys. 32 (1960) 1757< 31 (1959) 1589; 30 (1959) 1108; J. phys. Chem. 66 (1962) 1619.
[9 ] PRATT, T. H. and WOLFGANG, R. . J. Amer. chem. Soc. 83 (1961) 10.[10] WEXLER, S. andJESSE, N .. J. Amer. chem. Soc. 84 (1962) 3425.[11 ] MONAHAN, I.E . and STANTON, H. E ., J. chem. Phys. 37 (1962) 2654.[12] WEXLER, S . , I. chem. Phys. (in press).[13] WEXLER, S. , Argonne National Laboratory Report, ANL-6288, January-February (1961).[14] DALY, N. R ., Rev. Sci. Instrum. 31 (1960) 264.
[15 ] HOOPER, J. W . . MCDANIEL, E. W . , MARTIN, D. W. and HARMER, D. S . , Phys. Rev. 121 (1961) 1123; 125 (1962) 2000; Proc. 2nd Int. Conf. on the Physics of Electronic and Atomic Collisions, W. A. Benjamin, New York (1962) 67.
[16] WEXLER, S . , J. Chem. Phys. 41 (1964) 1714.[17] HART, E.J. and PLATZMAN, R. L ., "Radiation Chemistry", Mechanisms in Radiobiology I, Academic
Press (1961) 166.[18] CROSS, R.J. and WOLFGANG, R ., J. chem. Phys. 35 (1961) 2002.[19] ROWLAND, F. S ., LEE, J. K . , MUSGRAVE, B. and WHITE, R. M ., Chemical Effects of Nuclear Trans
formations Ц IAEA, Vienna (1961) 67.[20] BETHE, H ., Ann. Phys. 5 (1930) 325.[21] MOTT, N. F. and MASSEY, H. S. W . , The Theory of Atomic Collisions, Oxford University Press, London
(1949) 247, 271. •[22] BATES, D.R. and GRIFFING, G . . Proc. phys. S oc ., Lond., A66 (1953) 961; A67 (1954) 663; A 68
(1955) 90.[23] MAPLETON, R. A . , Phys. Rev. 109 (1958) 1166.
[24 ] MASSEY, H. S. W. and BURHOP, E. H. S ., Electron and Ionic Impact Phenomena, Oxford University
Press, London (1952), Chapter 3.
[25] KEBARLE, P. and GODBOLE, E. W . , J. chem. Phys. 36 (1962) 302.[26] MELTON, С. E. , J. chem. Phys. 32 (1962) 562.[27] LEE, J .K ., MUSGRAVE, B. and ROWLAND, F .S . , J. Amer. chem. Soc. 82 (1960) 3545; 81 (1959)
3803; Cañad. J. Chem. 38 (1960) 1756. .[28] WHITE, R. M. and ROWLAND, F. S. , J. Amer. chem. Soc. 82 (1960) 4713, 5345.[29] SAUER, М. C. and WILLARD, I .E ., J. phys. Chem. 64 (1960) 359.[30] STEVENSON, D. P. , J. phys. Chem. 61 (1957) 1453.[31 ] WEXLER, S ., J. inorg. nucl. Chem. 10 (1958) 8 ; see also SNELL, A. H. , PLEASONTON, F. and
LEMING, H. E. , ibid 5 (1957) 112.[32] SCHISSLER, D. O. and STEVENSON, D. P. , J. chem. Phys. 24 (1956) 926.

"EXPLOSION" OF MULTICHARGED MOLECULAR IONS: CHEMICAL CONSEQUENCES OF INNER SHELL
VACANCIES IN ATOMS*
T .A . CARLSON AND R. MILFORD WHITE**OAK RIDGE NATIONAL LABORATORY, OAK RIDGE,
TENNESSEE, USA
Abstract — Résumé — Аннотация — Resumen
"EXPLOSION” OF MULTICHARGED MOLECULAR IONS; CHEMICAL CONSEQUENCES OF INNER SHELL
VACANCIES IN ATOMS. Molecules containing atoms that undergo internal conversion or electron capture
are subject to extensive decomposition. This decomposition results from the large number of electrons that an atom loses as it adjusts to a vacancy in one of its inner shells. Electrons are pulled from the rest of the
molecule to the region of high positive charge, and the whole molecule literally explodes from Coulombic
repulsion.In this paper a short review is first made of the past work on the molecular consequences to inner shell
vacancies with particular emphasis on gases examined with a charge spectrometer. This previous work, however, has been limited to qualitative observations. A description is then given of a new charge spectrometer thât utilizes X-rays to initiate inner shell vacancies. With this spectrometer it is possible to measure the relative abundances of all the fragment ions formed in the decomposition of the parent molecule without the
errors that arose earlier from a dependence of collection efficiency on recoil energy. Furthermore, it has
been possible to measure the recoil spectrum for each of the ions.As an example, some recently acquired data are given on the decomposition of CH3I following vacancies
formed primarily in the L shell of iodine by X-rays. The decomposition is violent, with the molecule decomposing almost entirely into H+, Cn+ and In+, The relative abundance of molecular ions is very small.
The sums of the carbon, iodine and hydrogen ions are in the approximate ratios of 1 :1 :3 suggesting that
the quantity of neutral species is also small. The most abundant carbon ion is C2+, which possesses an average
recoil energy of about 50 eV. The most abundant iodine species is 1*+, which contrasts with an average charge
of eight from an analysis of Xe ions produced with X-rays of the same energy. These and other data on the
recoil and charge spectra from CH3I are compared with calculations using a model of a multiple-ion Coulomb
explosion.
«E X P LO S IO N » D'IONS MOLÉCULAIRES A CHARGES MULTIPLES: CONSEQUENCES CHIMIQUES DE
LA PRÉSENCE DE LACUNES DANS LES COUCHES INTERNES DES ATOMES. Les molécules contenant des
atomes qui sont le siège d'une conversion interne ou d-'une capture électronique, sont sujettes a une décomposition étendue. Cette décomposition résulte des nombreuses pertes d'électrons que subit un atome quand il s'adapte à une lacune dans une de ses couches internes. De partout dans la molécule, les électrons sont attirés
vers la région a charge positive élevée et la molécule tout entière explose littéralement sous l'action des
forces coulombiennes de répulsion. .. L’auteur commence par passer brièvement en revue les travaux antérieurs sur les conséquences, pour
la molécule, de lacunes dans les couches internes, dans lesquels une attention particulière a été accordée
aux gaz examinés avec un spectromètre de charge. Toutefois, ces premiers travaux avaient été limités à
des observations qualitatives. L’auteur décrit ensuite un nouveau spectromètre de charge, dans lequel on se
sert des rayons X pour provoquer les lacunes dans les couches internes. Avec ce spectromètre il est possible
de mesurer l'abondance relative de tous les ions fragmentaires qui sont formés lors de la décomposition de
la molécule mère, sans s'exposer aux erreurs imputables précédemment au fait que l'efficacité de la collection dépendait de l ’énergie de recul. De plus, il a été possible de mesurer le spectre de recul pour chacun
des ions.
* Research sponsored by the US Atom ic Energy Commission under contract w ith the Union Carbide
Corporation.
* * Summer Participant, present address: Baker University, Baldwin, Kansas.
23

24 T , A. CARLSON and R. MILFORD WHITE
L'auteur donne à titre d’exemple quelques renseignements obtenus récemment sur la décomposition
de CH3I consécutive à la formation de lacunes provoquées par les rayons X, principalement dans la couche L
de l'iode. La décomposition est brutale et la molécule se décompose presque entièrement en H+ , Cn+ et In+. La quantité relative d'ions moléculaires est très faible. Les nombres totaux d'ions carbone, iode et
hydrogène sont plus ou moins dans le rapport 1 :1 :3 , ce qui donne a penser que la quantité d'espèces neutres est faible elle aussi. L'ion carbone le plus abondant est C 2+; il possède une énergie moyenne de recul de l'ordre
de 50 eV. L’espèce iode la plus abondante est I5+, ce qui contraste avec une charge moyenne de 8 révélée par
une analyse des ions Xe produits par des rayons X de même énergie. L'auteur comparé ces données et d'autres
données relatives aux spectres de recul et de charge pour CH3I avec les résultats de calculs effectués au moyen
d'un modèle d'explosion coulombienne à ions multiples.
"В З Р Ы В ” М Н О Г О З А Р Я Ж Е Н Н Ы Х М О Л Е К У Л Я Р Н Ы Х И О Н О В : Х И М И Ч Е С К И Е П О С Л Е Д С Т В И Я ВА К А Н С И Й В Н У Т Р Е Н Н И Х О БО Л О Ч Е К В А Т О М А Х . Молекулы, содержащие атомы, которые претерпевают внутренние превращения или электронный захват , подвержены экстенсивному разложению. Это разложение является результатом потери атомом большого числа электронов, когда атом приспосабливается к вакансии в одной из своих внутренних оболочек . Электроны выталкиваются из остальной части молекулы в область с высоким позитивным зарядом , и вся молекула буквально взры вается, благодаря кулоновскому о т талкиванию .
Доклад содержит краткий обзор работ по молекулярным последствиям для вакансий внутренней оболочки с особым упором на газы , изучавшиеся с зарядным спектрометром. Эти работы , однако, ограничиваются качественными наблюдениями. Д ается описание нового зарядного спектрометра, где используются рентгеновские лучи для создания вакансий в н у тренней оболочки. С этим спектрометром можно измерить сравнительное количество всех фрагментных ионов, образованных при разложении родительской молекулы без погрешностей, которые возникали ранее и з -з а зависимости эффективности сбора от энергии отдачи. Далее можно измерить спектр отдачи для каждого из ионов.
В качестве примера приводятся недавно полученные данные по следующему разложению C H 3J вакансий, образованных главным образом в Ьгоболочке йода рентгеновскими лучами. Разлож ение является произвольным, причем молекула разлагается почти полностью на Нт , С п+ и J n+ . Относительный избыток м олекулярны х ионов является сравнительно м алы м . Суммы ионов углерода, йода и водорода находятся в приблизительных соотношениях, равных
1 :1 :3 , говоря о том, что количество нейтральных ионов также м ало. В наибольшем изобилии находится ион углерода С 2+ , который обладает средней энергией отдачи около 50 э в . В наибольшем изобилии среди ионов йода находится J5+ , который контрастирует со средним зарядом восемь из анализа ионов Х е , произведенных с помощью рентгеновских лучей такой Же энергии. Эти и другие данные по спектрам отдачи и заряда из С Н 3 J сравниваются с
расчетам и, используя модели многоионного кулоновского взры ва.
■ «E X P LO S IO N » DE IONES MOLECULARES PORTADORES DE CARGAS MULTIPLES; EFECTOS QUIMICOS
DE LAS VACANTES EN LAS CAPAS INTERNAS DE LOS ÁTOMOS. U s moléculas que contienen átomos en
los que se desarrollan procesos de conversión interna o de captura electrónica suelen experimentar una descomposición profunda. Dicha descomposición es consecuencia de la pérdida de gran número de electrones
por el átomo cuando la estructura de éste se reajusta para adaptarse a una vacante en una de sus capas interiores. Los electrones afluyen del resto de la molécula a la región de carga positiva elevada, y la molécula entera
explota literalmente a causa de la repulsión de Coulomb,En la primera parte de la memoria se reseñan los trabajos precedentes sobre los efectos moleculares
ejercidos por las. vacantes en capas atómicas interiores, prestando particular atención al examen de gases por espectrometría de cargas. Pero estos trabajos se han limitado a observaciones de carácter cualitativo. Seguidamente se describe un nuevo espectrómetro de cargas, que utiliza rayos X para crear vacantes en las capas
interiores. Este espectrómetro permite medir la abundancia relativa de todos los fragmentos iónicos formados
por descomposición de la molécula precursora, sin los errores a que antes daba origen la variación de la eficacia
de captación con la energía de retroceso. Asimismo, se ha podido medir el espectro de retroceso de cada
uno de los iones.A título de ejemplo, se presentan algunos datos recientemente obtenidos sobre la descomposición de
СНэ1 consecutiva a la aparición de vacantes creadas por los rayos X principalmente, en la capa L del yodo. La descomposición es violenta, desintegrándose la molécula casi enteramente en H+, Cn+ e In+. La abundancia
relativa de iones moleculares es muy reducida. Las sumas de los iones carbono, yodo e hidrógeno guardan

"EXPLOSION” OF MULTICHARGED MOLECULAR IONS 25
aproximadamente la relación 1 :1 :3 , lo que indica que la cantidad de especies neutras es también escasa.
El ion carbono más abundante es el C2+, que posee una energía media de retroceso de unos 50 eV. En cuanto
al yodo, la especie más abundante es el I5+, lo que contrasta con la carga media de 8 deducida de un análisis
de iones Xe obtenidos con ayuda de rayos X de la misma energía. Estos y otros datos relativos a los espectros
de retroceso y de carga del CH 3I se comparan con los resultados de cálculos efectuados sobre la base de un
modelo que supone una explosión de Coulomb en que intervienen iones de cargas, múltiples.
1. INTRODUCTION
Not long after the discovery of the Szilard-Chalmers effect, it was found that an atom undergoing internal conversion was separated from its parent m olecule, in spite of the p ro ce ss usually not im parting sufficient re co il energy to cause severan ce of the chem ical bond [1, 2] . In fact, HAMILL and YOUNG [3] dem onstrated that essentially all the Br^om transitions in methyl bromide were effective in bond rupture . At that time it was correctly realized [4, 5] that the decom position following internal conversion was caused by an inner shell vacancy in which an atom readjusts itself by a séries of radiative and non-radiative transitions. Each of the non-radiative transitions (Auger p ro ce sse s) resu lts in the loss of an e lectron , and the atom becomes highly charged. Such a series of Auger processes has been called a vacancy cascade [6 ] . If the atom is part of a molecule, the whole molecule decomposes violently.
The average charge ca rrie d by the ions resulting from internal conversion o r electron capture was measured for a number of radioactive atoms and m olecules [7, 8 ] . L a te r , m ass sp ectrom etric analysis was employed by KOFOED-HANSEN [9] and m ore extensively by SNELL and PLEASONTON [6 , 1 0 , 1 1 , 1 2 ] in m easuring the relative abundances of ions resulting from internal conversion and e lectro n capture of various r a r e g a s e s . These studies were extended to m olecular system s by W EXLER et a l. [13, 14] and by the author of this paper [151 . The results indicate that the atom undergoing internal conversion o r electron capture not only broke away from the parént m olecule, but that the parent m olecule itse lf underwent extensive decomposition. The studies on molecules w ere, however, only qualitative since the fragment ions received considerable recoil energy which strongly influenced their collection efficiencies. A new experim ental approach has been made, however, to the problem of m olecules undergoing violent decomposition as the result of inner shell vacancies, in which X -ray s are used to create the initial vacancy [16] . This new approach will be the main topic of this paper.
Since the chem ical consequences of internal conversion are caused by the formation of vacancies in the inner shells of an atom, it follows that the sam e phenomenon can-be obtained by producing these holes by other means such as photo-electron em ission. The use of X -ra y s gives the advantage of ( 1 ) not being restricted to certain radioactive isotopes and (2 ) being able to select by the proper choice Of X -ra y energy the shell where most of the vacancies will be produced. An extensive program m e is already being carried out to m easure the charge spectra of ra re gas ions following inner shell vacancies as produced by X -ra y s [17, 18] . As expected, the data from

26 T . A. CARLSON and R. MILFORD WHITE
radioactive studies and X -ra y studies a re nearly identical in ca.ses where the initial vacancy distributions are the sam e.
In this paper we shall present some recent results on the fragment ions resulting from the X -ra y bombardment of CH3I. The chemical consequences are essentially the same as if the iodine had undergone internal conversion. There is a large improvement, however, over radioactive studies in that the use of X -ray s has allowed us to operate our spectrom eter in such a way that the uncertainties of the relative abundances due to collection efficiencies are removed. In addition, we have also made measurements on the recoil spectra for m ost of the fragment ions. With this type of data it is now possible to have a m ore quantitative understanding of the violent m olecular decomposition that accom panies extensive ionization. In the case of CH3I it will be shown that the description of the decomposition most consistent with the data is an "explosion" of ions propelled by Coulombic force.
ANALYSERADJUSTABLE
LEAK
F i g . l
Source vo lum e for studying the charge d istribution of ions produced by X-rays
2. EXPERIMENTAL
The charge sp ectrom eter used in our study of methyl iodide has been described previously [19] . Ions born in the source volume (Fig. 1) are extracted and magnetically analysed. The tim es for atomic readjustment are very short, ~ 1 0 '14 s, compared to the time it takes an ion to leave the source volume, ~ 1 0 ‘ 5 s , so the phenomena we are investigating are completed before analysis. The X -ra y source is operated at 40 keV and is a M achlett A EG -50 tube with a tungsten target. When the irradiated gas is CH,I, this source of X -ra y s will produce initial vacancies m ostly in the L shell of iodine. The photoelectric cro ss-se c tio n s for carbon and hydrogen are negligible.
The experim ents reported in this paper w ere generally carried out at p ressu res of 2 X 10~5 to r r in the sou rce volume and 4 X 10 " 6 to r r in the analyser. All studies w ere repeated at higher p ressu res, and the results w ere extrapolated to zero p ressu re .

"EXPLOSION” OF MULTICHARGED MOLECULAR IONS 27
Experim ents on CH3I were carried out by two different methods of analysis, which will be described briefly.
Analysis No. 1: In this.analysis an extraction voltage, which is 4% of the total voltage, Vs , is applied between plates (a) and (b) as shown in Fig. 1. When an ion has a negligible recoil energy, as in the case of rare gas atoms, the collection efficiency is independent of the choice of V? . In the study of the fragment ions from molecules the recoil energy, however, is not negligible, and the collection efficiency is a function of Vsn /E r w here n is the charge of the ion and E r is its reco il energy. By m easuring the relative counting rates for each of the ions as a function of Vs, it is possible to extrapolate these intensities to 1 0 0 % counting efficiency, and even to evaluate the relative reco il energies of the different ions.
Analysis No. 2: In the second method for analysis, plates (a) and (b) are shorted so that the ions a re formed in a field -free region and are not extracted , but em erge from the second grid of plates (b) with only their initial recoil energy. Additional kinetic energy is then obtained as the ions pass through the potential field between plates (b) and ground. When the spectrum of fragment ions is measured, V, is set at a sufficiently high voltage to ensure that all the ions of a given species a re analysed with nearly the same total energy. By reducing Vs so that the recoil energy is a measurable fraction of the acceleratin g voltage, we can evaluate the reco il energy spectrum for each species.
The two analyses complement each other. The counting rates for analysis No. 2 are an order of magnitude lower than analysis No. 1, but the data are m ore dependable, particu larly for ions of low charge and high reco il energy; and it is possible to make direct measurements of the recoil spectra. The f irs t analysis is useful for those ions possessing high charge and low reco il energy, such as the iodine ions, which w ere collected with nearly 1 0 0 % efficiency when the maximum voltage V¡ was applied.
3. RESULTS AND DISCUSSIONS
Table I lis ts the weighted average of resu lts obtained from analyses No. 1 and No. 2 (see above) for the relative abundances of the fragm ent ions form ed following the X -rad iation of CH3I. Also listed a re the peak values of the recoil spectra for each of the ions examined. We note first the very low yield for molecule ions. Nearly all the observed fragm ents are either H+, C n+or In+. This is in sharp contrast to electron im pact studies and to decomposition following 0" decay [20] . The evidence in the present case is of a violent decomposition. It should also be noted from Table I that the sum of In+, Cn+, and H+ ions are in the ratios of 1 .0 :1 .0 :3 .0 , suggesting that all the atoms originally making up CH3I are ionized, and that very few neutral species occur in the decomposition.
In F ig . 2 are plotted (1) the spectrum of iodine ions from CHgl; (2) the spectrum of I ions from HI [21] ; (3) a spectrum of Xe ions [21]; and (4) a spectrum of Xe131 ions [6 ] . In the first three studies the initial atomic vacancies were produced by the sam e X -ra y source. In the fourth study ionization a rise s from internal conversion. The X en ions are com pared with the In+1 ions from HI. and the In+3 ions from CH3I. By plotting the charge
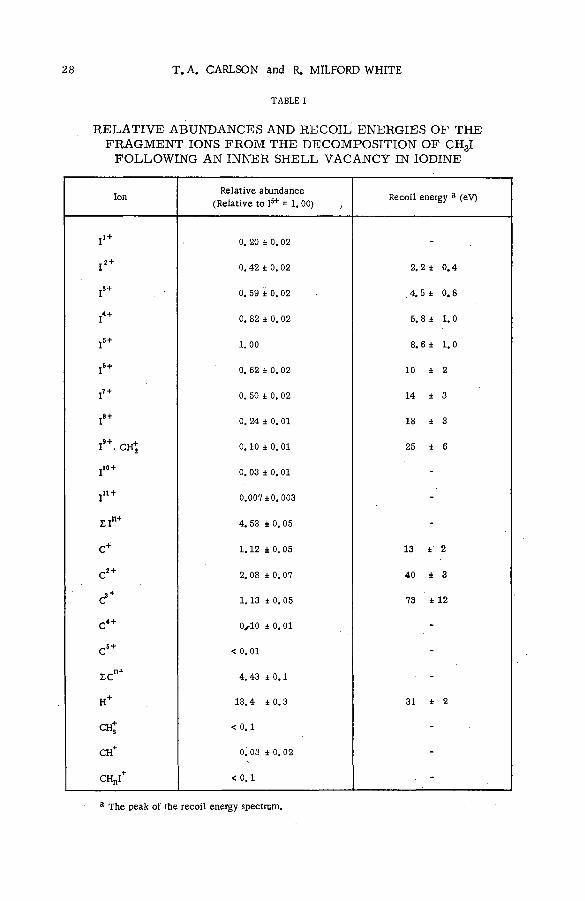
28 T . A. CARLSON and R. MILFORD WHITE
TABLE I
R E L A T IV E ABUNDANCES AND RECO IL EN ERG IES O F THE FRAG M ENT IONS FROM TH E DECOMPOSITION O F CH3I
FOLLOWING AN INNER SH ELL VACANCY IN IODINE
IonRelative abundance
(Re la tive to I st = 1. 00) ¡ Recoil energy a (eV)
j l + 0. 20 ± 0 .0 2 -
I 2 +0. 42 ± 0.02 2. 2 ± 0 .4
i 3+ ■ 0.59 ±0.02 . 4. 5 ± 0. 8
4 + 0. 82 ± 0. 02 ■ 5. 8 ± 1.0
5 + 1 .00 8. 6 ± 1 .0
6 +0.62 ± 0 .0 2 10 ± 2
_7 +0. 50 ± 0. 02 14 ± 3
8 +0 .24 ±0.01 18 ± 3
I 9+. C H t 0 .1 0 ± 0 .01 25 ± 6
10 +0. 03 ± 0. 01 -
-11 +0.007 ±0.003 -
£ I n+ 4. 53 ± 0. 05 -
C+ 1.12 ±0.05 13 ± 2
C2 + 2. 08 ± 0. 07 40 ± 3
C? + 1.13 ± 0. 05 73 ± 12
c4+ OylO ± 0. 01 -
c 5+ < 0. 01 . -
. 2 C n+ 4. 43 ±0.1 . -
H + 13.4 ± 0.3 31 ± 2
CH+ < 0 .1 - ■
CH+ 0.03 ± 0.02 -
CHnI + < 0 .1 . -
a The peak of the reco il energy spectrum.
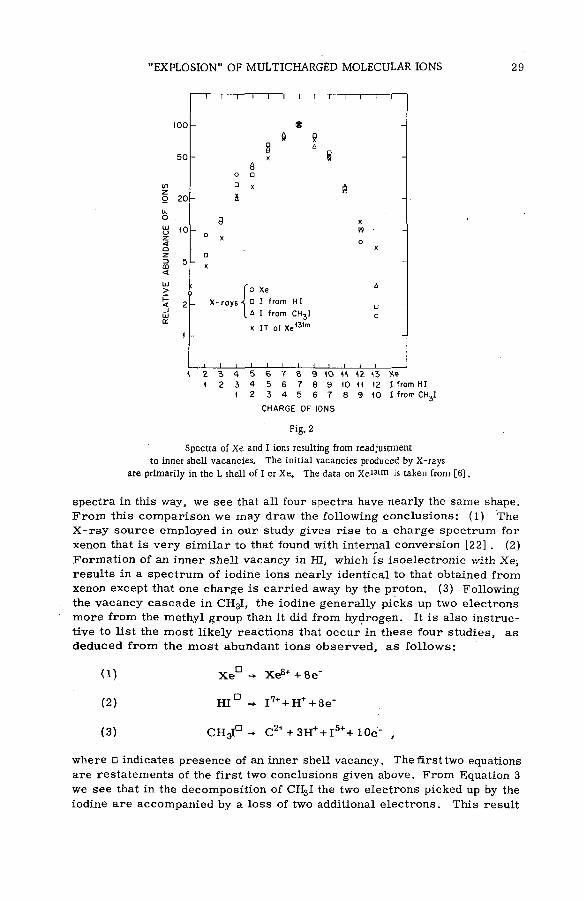
"EXPLOSION” OF MULTICHARGED MOLECULAR IONS 29
1 2 3 4 5 6 7 8 9 10 <1 12 I from HI1 2 3 4 5 6 7 8 9 10 1 from CHjI
CHARGE OF IONS
Fig. 2 .
Spectra of Xe and I ions resulting from readjustment
to inner shell vacancies. The initial vacancies produced by X-rays
are primarily in the L shell of I or Xe. The data on X e13lm is taken from [ 6] .
spectra in this way, we see that all four spectra have nearly the same shape. Fro m this com parison we may draw the following conclusions: (1) The X -ra y source employed in our study gives rise to a charge spectrum for xenon that is very sim ilar to that found with internal conversion [2 2 ] . (2 )Form ation of an inner shell vacancy in Ш, which is isoelectronic with Xe, results in a spectrum of iodine ions nearly identical to that obtained from xenon except that one charge is carried away by the proton. (3) Following the vacancy cascad e in CH3I, the iodine generally picks up two electrons m ore from the methyl group than it did from hydrogen. It is also instructive to list the m ost likely reactions that o ccu r in these four studies, as deduced from the m o st abundant ions o b served , as follow s:
<1} XeD -* Xe**+ + 8e‘
(2) H ID - I7++H + + 8e'
(3) СНэ1а C2+ + 3H++ I 5++10e- ,
where □ indicates presence of an inner shell vacancy. The first two equations are restatem ents of the first two conclusions given above. From Equation 3 we see that in the decomposition of CH3I the two electrons picked up by the iodine are accompanied by a loss of two additional electron s. This result
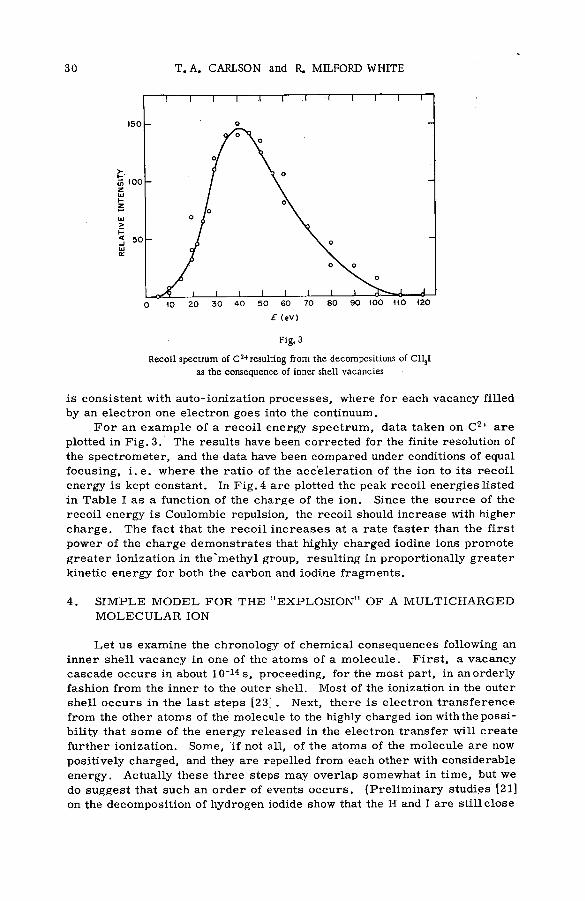
30 T . A. CARLSON and R. MILFORD WHITE
Fig.3
Recoil spectrum of C 2+resulting from the decompositions of CH3I as the consequence of inner shell vacancies
is consistent with auto-ionization processes, where for each vacancy filled by an electron one electron goes into the continuum.
F o r an exam ple of a re co il energy sp ectrum , data taken on C2+ a re plotted in Fig . 3. The results have been corrected for the finite resolution of the spectrom eter, and the data have been compared under conditions of equal focusing, i .e . where the ratio of the acceleratio n of the ion to its reco il energy is kept constant. In F ig . 4 are plotted the peak recoil energies listed in Table I as a function of the charge of the ion. Since the so u rce of the recoil energy is Coulombic repulsion, the recoil should increase with higher ch arg e. The fact that the reco il in cre a se s at a rate fa s te r than the first power of the charge dem onstrates that highly charged iodine ions promote g reater ionization in the'methyl group, resulting in proportionally greater kinetic energy for both the carbon and iodine fragm ents.
4 . SIM PLE MODEL FO R THE "EXPLO SIO N " OF A MULTICHARGEDM OLECULAR ION
L et us examine the chronology of chem ical consequences following an inner shell vacancy in one of the atom s of a m olecule. F ir s t , a vacancy cascade occurs in about 1 0 “1 4 s, proceeding, for the most part, in an orderly fashion from the inner to the outer shell. Most of the ionization in the outer shell o ccu rs in the last steps [23] . Next, there is electron tran sferen ce from the other atoms of the molecule to the highly charged ion with the possibility that some of the energy released in the electron tran sfer will create further ionization. Some, if not all, of the atoms of the molecule are now positively charged, and they are repelled from each other with considerable energy. Actually these three steps may overlap somewhat in tim e, but we do suggest that such an ord er of events o ccu rs . (Prelim inary studies [21] on the decomposition of hydrogen iodide show that the H and I are still close
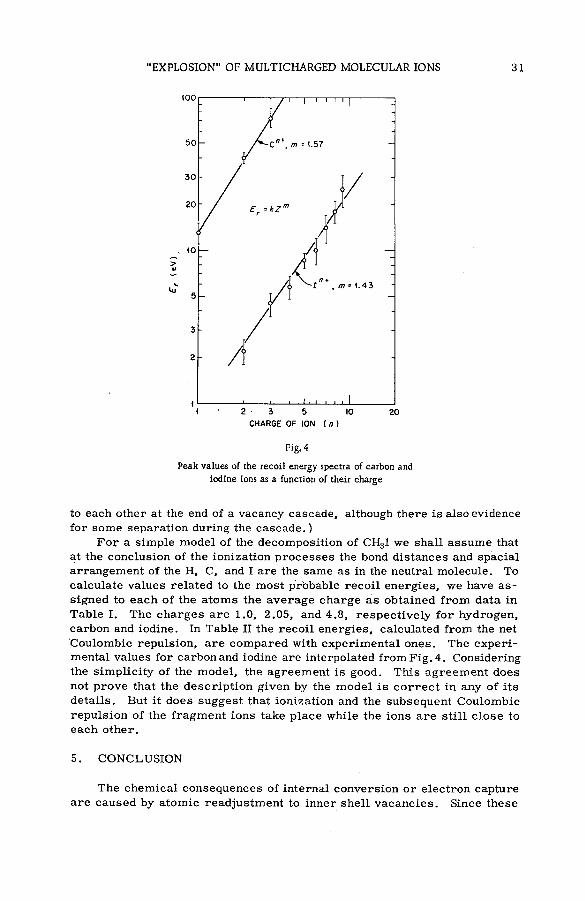
"EXPLOSION" OF MULTICHARGED MOLECULAR IONS 31
CHARGE OF ION ( n )
F ig . 4
Peak values of the recoil energy spectra of carbon and
iodine ions as a function of their charge
to each other at the end of a vacancy cascade, although there is also evidence for some separation during the cascade. )
F o r a simple model of the decomposition of CH3I we shall assum e that at the conclusion of the ionization p rocesses the bond distances and spacial arrangement of the H, C, and I are the same as in the neutral molecule. To calculate values related to the most probable recoil energies, we have a s signed to each of the atom s the average ch arge as obtained from data in Table I. The ch arges a re 1.0, 2 .05 , and 4 .8 , respectively for hydrogen, carbon and iodine. In Table II the recoil energies, calculated from the net Coulombic repulsion, a re com pared with experim ental ones. The experimental values for carbon and iodine are interpolated from Fig. 4 . Considering the sim plicity of the model, the agreem ent is good. This agreem ent does not prove that the description given by the model is c o r r e c t in any of its details. But it does suggest that ionization and the subsequent Coulombic repulsion of the fragm ent ions take place while the ions a re still clo se to each other.
5. CONCLUSION
The chem ical consequences of internal conversion o r electron capture are caused by atom ic readjustment to inner shell vacan cies. Since these

32 T . A. CARLSON and R. MILFORD WHITE
TABLE I I
COMPARISON OF EXPERIMENTAL RECOIL ENERGIES WITH CALCULATIONS BASED ON A COULOMB "EXPLOSION" MODEL
Recoil Energy (eV)
Iona Experiment b - Calculated '
+ 4 .8 .. I . 8 i 1 6
c + 2 .0541 i 3 59
H+ 31 ±2 . 50
a The charge in the average obtained from data in Tab le I
b Most probable reco il energy corresponding to the average charge from Tab le I
and Fig. 4
vacancies may also be produced by photoelectron em ission, X -ra y s can be used to study the sam e phenomena. This has been done for CH3I, and a new sp ectrom eter has been described in which quantitative information is obtained regarding the relative abundances and recoil energy spectra of the fragment ions formed as the result of X -irradiation . The data on CH3I giveevidence of violent decomposition and have been correlated with a model thatexplains the results as a Coulombic "explosion" of a multicharged molecular ion.
A C K N O W L E D G E M E N T
The assistance of the Petroleum Research Fund in providing travel funds for the presentation of this paper is gratefully acknowledged.
R E F E R E N C E S
[1 ] SEGRE, E. . HALFORD, R. S. and SEABORG, G. T . . Phys. Rev. 55 (1939) 321.[2 ] DE VAULT. D .C . and LIBBY. W. F . . Phys. Rev. 55 (1939) 322.[3 ] HAMILL, W. H. and YOUNG. J. A . . I. chem. Phys. 17 ( 1949) 215.[4 ] DE VAULT, D. and LIBBY. W. F . , J. Amer. chem. Soc. 63 (1941) 3216.[5 ] COOPER, E. P .. Phys. Rev. 61 (1942) 1.[ 6] PLEASONTON, F. and SNELL, A. H . , Proc. roy. Soc. 241A (1957) 141.[7 ] PERLMAN, M. L. and MISKEL, J. A . , Phys. Rev. 91 (1953) 899.[ 8] WEXLER, S ., Phys. Rev. 93 (1954) 182.[9 ] KOFOED-HANSEN, O . . Phys. Rev. 96(1954) 1045.
[10] SNELL, A. H. and PLEASONTON. F . , Phys. Rev. 100 (1955) 1396.[11] SNELL, A. H. and PLEASONTON, F . , Phys. Rev. 107 (1957) 740.[12] SNELL, A . H. and PLEASONTON, F . , Phys. Rev. I l l (1958) 1338.[13] WEXLER. S. and ANDERSON, G. R ., J. chem. Phys. 33 (1960) 850.[14] WEXLER, S ., J. chem. Phys. 36 (1962) 1992.[15] CARLSON, T. A . and WHITE. R .M ., ;. chem. Phys. 38 (1963) 2930.[16] Some earlier studies of fragment ions following X-radiation of molecules were carried out with a time-
of-flight spectrometer employing coincidence measurements between the charged fragments and ejected
electrons. The data, because of collection efficiency problems, are, however, only qua).lative.

"EXPLOSION" OF MULTICHARGED MOLECULAR IONS 33
KRAUSE, М ., VESTAL, M ., WAHRHAFTIG, A .L ., LAMPLE, F.W . and JOHNSTON, W .H ., Technical Report ASD-TDR-62-10 (1962) (unpublished).
[17] KRAUSE, M. O . , VESTAL, M. L . , JOHNSTON, W .H . and CARLSON, T .A . , Phys. Rev. 133 (1964) A385.
[18] CARLSON, T .A . and KRAUSE, M .O ., Bull. Amer. Phys. Soc. 9(1964) 51;KRAUSE. M .O . and CARLSON, T .A . , Bull. Am et Phys. Soc. 9(1964) 51.
[19] CARLSON, T .A . and KRAUSE, M. O. , (to be published in Phys. Rev).[20] CARLSON, T .A . and WHITE, R .M ., J. chem. Phys. 36(1962) 2883; 36(1963) 2075.[21] CARLSON, T .A . and WHITE, R.M , (unpublished results).[22] This is true despite the fact that most of the initial vacancies in our study are in the L shell, while
Il31m is mostly converted in the К shell. However, it should be noted that in 87% of the cases a К vacancyin Xe is transferred without ionization to a higher shell, usually L, by means of a radiative transition.
[23] For the justification of these statements examine the extensive calculations done on Auger transition
rates for several atoms: R.A. Rubenstein, Ph.D. Thesis, University of Illinois (1955) (unpublished).


IONIZATION THAT FOLLOWS A HEAVY-ION- INDUCED NUCLEAR REACTION
N. H. STEIGERDEPARTMENT OF NUCLEAR SCIENCE, ISRAEL INSTITUTE OF TECHNOLOGY,
. . HAIFA, ISRAEL
Abstract — Résumé — Аннотация — Resumen
IONIZATION THAT FOLLOWS A HEAVY-ION-INDUCED NUCLEAR REACTION. lonizaiion following
different modes of radioactive decay has previously been studied extensively. However, at present there
appears to be no experimental data on internal ionization obtained in products of induced nuclear reactions.
In this paper initial results are given of an experimental study on the internal ionization effect that follows a heavy-ion-induced nuclear reaction.
Charge spectrometry by means of magnetic analysis of the reaction products was applied. The effect
was investigated fot the teaction
Pri4i ( o 16, 8n)Ho149.
The 016 ions were obtained from the Berkeley heavy-ion linear accelerator.
The bombarding energy was 10.4 ± 0.2 MeV. Pr141 target thicknesses from 26.2 fig/cm2 down to
9.7 /ig/cm2 were used. For target thicknesses lower than about 25 Mg/cm2, non-equilibrium ionic-charge
spectra are obtained that tend towards lower probabilities for the charge states lower than the equi
librium mean charge, and tend towards higher probabilities for the charge states higher than the equilibrium mean charge. As a result, the mean ionic charge increases from 17.6 ± 0.5 for the equilibrium case
to 19.4 ± 1.0 for a target thickness of 9.7 Mg/cm2. By extrapolation to target thickness zero, an ionic-charge
spectrum is obtained that is considered to illustrate the "instantaneous internal ionization" obtained for the
interaction between two complex nuclei. "Instantaneous internal ionization" is defined as the ionization that takes place before the resulting species of a nuclear reaction starts moving inside the target material and is given the chance of charge-changing collisions.
The shape of the resulting extrapolated charge spectrum can be explained as being mainly composed
of the two processes responsible for the internal ionization:(a ) the non-diabatic part o f the transition following the large change in nuclear charge (Z = +8) ;(b ) the development of vacancy cascades as a result o f internal conversion.Based on the experimental results, the part of the ionization due to process (a ) was estimated to be less
than 800 eV. v • 4 ‘
IONISATION CONSÉCUTIVE A UNE RÉACTION NUCLÉAIRE PROVOQUÉE PAR DES IONS LOURDS, l'io n isation qui fait suite à différents modes de décroissance radioactive a déjà été abondamment étudiée. Il semble
toutefois qu’on ne dispose actuellement d'aucune donnée expérimentale concernant l ’ionisation interne obtenue
dans des produits de réactions nucléaires provoquées artificiellement.Dans ce mémoire, les auteurs communiquent les résultats initiaux d'une étude expérimentale sur l'ioni
sation interne consécutive à une réaction nucléaire provoquée par les ions lourds.L'auteur a utilisé comme méthode la spectrométrie de charge par analyse magnétique des produits de
la réaction. Les effets étudiés ont porté sur la réaction
w lPr(l60 ,8 n )U9Ho.
Les ions 160 ont été obtenus à i ’aide de l'accélérateur linéaire à ions lourds de Berkeley.
Pour cette expérience, i ’éneïgie de bombaîdemenî était de 10,4 ± 0,2 MeV et l ’on a utilisé des cibles
de 141Pr d’une épaisseur allant de 26,2 Mg/cm2 à 9,7 Mg/cm2. .A vec les épaisseurs inférieures à 25 Mg/cm2 environ, on a obtenu des spectres de chargé ionique de non-équilibre, dont la tendance est Í une probabilité
35

36 N. H. STEIGER
moindre pour les états de charge inférieurs à la charge moyenne d'équilibre et une probabilité plus forte pour
les états de charge supérieurs à la charge moyenne d'équilibre. Il en résulte que la charge ionique moyenne
passe de 17,6 ± 0,5 pour le cas de l'équilibre, à 19,4 ± 1,0 lorsque la cible a une épaisseur de 9,7 |ig/cm2.
En extrapolant ces résultats pour une cible d’épaisseur nulle, on obtient un spectre de charge ionique qui est censé représenter l ' « ionisation interne instantanée»obtenue lorsqu'il y a interaction entre deux noyaux complexes. Par «ionisation interne instantanée», on entend l'ionisation qui apparaît avant que l’espèce produite
dans une réaction nucléaire ne commence à se déplacer à l'intérieur de la cible et que ce mouvement ne
puisse donner lieu à des chocs entraînant un changement de la charge.On peut considérer que le spectre de charge obtenu par extrapolation résulte essentiellement de deux
processus qui déterminent l'ionisation interne:a) La partie non adiabatique de la transition consécutive au changement important de la charge nuclé
aire (Z = +8);b) La formation de lacunes en cascades résultant de la conversion interne.D'après les résultats expérimentaux obtenus, les auteurs estiment que la part d'ionisation due au processus
a) est inférieure à 800 eV.
И О Н И ЗАЦ И Я В Р Е З У Л Ь Т А Т Е Я Д Е РН Ы Х Р Е А К Ц И Й , Н А В Е Д Е Н Н Ы Х Т Я Ж Е Л Ы М И И О Н А М И . В прошлом была тщательно изучена ионизация, наступающая в результате разнообразны х радиоактивных распадов. Однако до настоящ его времени, по-видимому, не с у ществует экспериментальных данных относительно внутренней ионизации, происходящей в
продуктах ядерных реакций, наведенных тяжелыми ионами. .В настоящем докладе сообщаются предварительные результаты экспериментального
изучения эффекта внутренней ионизации, наступающей вслед за ядерными реакциями, наведенными тяжелыми ионами.
Для этого применялась спектрометрия заряда при помощи магнитного анализа продуктов реакции. Это явление было изучено для реакции:
Р г 141 (О 16, 8п) Н о 149 .
Ионы кислорода-16 были получены на линейном ускорителе тяжелых ионов лаборатории
в Баркли.Энергия бомбардирующих ионов составляла 1 0 ,4 ±0 ,2 М э в . Применялись мишени из
празеодима-141 толщиной 9,7 — 26,2 мкг/см 2 . Для мишеней толщиной менее приблизительно 25 м кг/см 2 получаются неравновесные спектры ионного заряда, дающие меньшую вероятность получения состояний заряда ниже среднего равновесного и большую вероятность получения состояний заряда, превышающих среднее равновесное. В конечном результате средний иойный заряд увеличивается с 17,6 ±0 ,5 для равновесного случая до 19,4 ±1 ,0 для мишени с толщиной в 9,7 мкг/см 2 . Путем экстраполяции до нулевой толщины мишени получается спектр ионного заряда, который можно принять за показатель "мгновенной внутренней ионизации" , получающейся в результате взаимодействия между двумя комплексными ядрами. Под "мгновенной внутренней ионизацией" подразумевается ионизация, наступающая до того, как продукты, образующиеся в результате ядерных реакций, начинают продвигаться внутри м атериала мишени и открывают путь для столкновений,сопровождаемых обменом заряда .
Форма получающегося экстраполированного спектра заряда может быть объяснена, как
состоящ ая главн ы м об р азо м из д в ух п ро ц ессов , вы зы ваю щ их внутреннюю ионизацию :а) недиабатическая часть перехода вслед за значительным изменением ядерного заряда
( Z = + 8);б) образование каскадов вакансий в результате внутренней конверсии.На основании экспериментальных результатов было определено, что часть ионизации,
вызываемая процессом а ), составляет менее 800 электрон -вольт.
IONIZACION CONSECUTIVA A UNA REACCION NUCLEAR INDUCIDA POR IONES PESADOS. La ionización consecutiva a diferentes modos de desintegración radiactiva ha sido estudiada extensamente. Sin em bargo, no se dispone aún de datos experimentales sobre la ionización interna de productos procedentes de
reacciones nucleares inducidas.En la presente memoria, el autor presenta los primeros resultados experimentales sobre el efecto de
ionización interna consecutivo a una reacción nuclear inducida por iones pesados.

IONIZATION FOLLOWING HEAVY-ION-INDUCED REACTION 37
Aplicó la espectrometría de cargas sobre la base del análisis magnético de los productos de la reacción
y estudió el efecto para la reacción
141Pr( 160 , 8 n) 149Ho.
Los iones 160 se obtuvieron en el acelerador lineal de iones pesados de Berkeley.
La energía de bombardeo era de 10,4 ± 0,2 MeV. El espesor del blanco de M1Pr estaba comprendido
entre 26,2 Mg/cm2 y 9,7 Mg/cm2. Cuando el espesor del blanco es inferior a uftos 25 Mg/cm2, se obtienen
espectros de cargas iónicas que no están en equilibrio y en los que tiende a disminuir la probabilidad de que
la carga sea menor que el valor medio de la carga de equilibrio y viceversa. Como resultado, la carga media
iónica aumentó de 17,6 ± 0,5, en el caso de equilibrio, a 19,4 ± 1,0, cuando el espesor del blanco es de9,7 Mg/cm2. Extrapolando estos resultados para un espesor del blanco igual a cero, se obtuvo un espectro
de cargas iónicas que sirvió para ilustrar la «ionización interna instantánea»correspondiente a la interacción
de dos núcleos complejos. La «ion ización interna instantánea » se define como la ionización que tiene
lugar antes de que la especie resultante de una reacción nuclear empiece a desplazarse en el seno del material del blanco y tenga la posibilidad de experimentar colisiones que cambien la carga.
La forma del espectro de cargas extrapolado resultante tiene su origen principalmente en los dos procesos que determinan la ionización interna: .
a) la parte adiabática de la transición consecutiva al gran cambio de la carga nuclear (Z- +8);b) la aparición de cascadas de vacantes como resultado de la conversión interna.Sobre la base de los resultados experimentales, el autor estima que la parte de la ionización causada
por el proceso a) es inferior a 800 eV.
I. INTRODUCTION .
Several m echanism s can lead to internal excitation and ionization in atoms that result from a nuclear transform ation. The most important are (1) the development of vacancy cascades by successive Auger events; (2) an electrostatic "shake-off" process caused by a change in nuclear charge; and(3) a non-nuclear "shake-off"produced by the change in e le ctric field following the sudden loss of an atom ic electron . One of these effects or a combination of them may occur as a result of various kinds of spontaneous or non-spontaneous nuclear transform ations.
These effects have been studied experimentally and theoretically for different modes of radioactive decay. A short compilation of these studies made prior to 1958, including a summary of references, has been published by BAULCH and DUNCAN [1]. More recent work has been published by se veral authors [2 - 1 0 ].
It has been shown that the ion ic-ch arge sp ectra of the product atom s resulting from nuclear transform ations reflect a sensitivity to the nature of the nuclear change that caused the ionization, as well as representing the sta tistica l outcome of the complex atom ic rearran gem en ts that follow electron loss from any of the sev eral electron shells of the atom s. Ionization following beta decay, internal conversion, orbital electron capture and various combinations of these p ro cesses have been studied extensively by applying charge sp ectrom etry to the product atom s [3 -1 4 ] . However, at present there appear to be no experim ental data on internal ionization obtained in products of induced nuclear reactio n s.
In two previous papers we reported on a detailed study of ionic-charge distributions of products of heavy-ion-induced nuclear reactions [15, 16].

38 N. H. STEIGER
These distributions were for an equilibrium state achieved by the reaction products within the target m aterial. Heavy particles passing through matter can lose or capture electrons in collisions with stationary atoms of the medium traversed. This very complicated process results in the establishment of an equilibrium distribution of charges. This distribution relates to an element of the path of the penetrating p a rtic le s , which is long enough to include a large number of charge-exchange collisions but too short to appreciably slow down the p articles. F o r a reaction product of certain nuclear charge, these charge distributions depend mainly on the velocity of the product, and are independent of the kind and ch aracteristics of the nuclear transformation. Target thicknesses used in these experiments were of the order of 100 n g ¡cm 2. It was found that equilibrium -charge d is tributions were practically obtained for target thicknesses larger than 25 /ug/cm2.
In the present report we present initial results on ionic-charge spectra of Ho149 p articles obtained as products of a heavy-ion-induced nuclear reaction when targ et thicknesses of le ss than 25 /ug/.cm2 w ere used. We hope that an extrapolation to zero target thickness will reflect (at least qualitatively) the "instantaneous internal ionization" obtained as a result of the interaction between two complex nuclei. By "instantaneous internal ionization" we mean the ionization that occurs before the resulting species of a nuclear reaction starts to move inside the target m aterial and is given the chance of charge-changing collisions. We have mentioned some p re lim inary results of these experim ents in a recent report [15].
II. EXPERIM EN TAL PROCEDURE
The experim ental method chosen was sim ilar to the one used for the studies of equilibrium-charge distributions of products of heavy-ion-induced nuclear reactions in the rare -earth region [15, 16].
A schematic representation of the experimental set-up is shown in Fig. 1. The heavy-ions w ere obtained from the Berkeley heavy-ion lin ear a c ce le ra to r (H ilac), which acce lera tes heavy-ions to an energy of 10.4 ± 0 .2 MeV/amu [171. Magnetic deflection was used as the charge-analysing method. The accelerated ion beam was deflected through 30° by a bending magnet before reaching the deflection cham ber. The heavy-ion beam entered the chamber through a small oval collimator (collim ator No. 1), pa|sed a narrow slit collim ator (collim ator No. 2) and hit the target ^after penetrating its backing m aterial. The resulting reaction products, together with the heavy- ion beam emerging from the target, passed through an additional slit collim ator (collim ator No. 3). Entering the gap of a permanent magnet of 3150 G field strength, the charged reaction products as well as the beam ions were horizontally deflected according to their momenta and effective charge states. The reaction products were finally collected on a thin Al catcher foil. The Faraday cup behind the catch er foil served to monitor the heavy-ion beam throughout the experim ent.
The experiments were performed at a pressure of lO 4 mm Hg or less. The horizontal distribution of the collected reaction products, which is proportional to the ionic-charge state of the particle , was recorded by taking

IONIZATION FOLLOWING HEAVY-ION-INDUCED REACTION 39
' Fig. 1
Schematic d iagram o f expérimentât arrangement
an autoradiograph of the catcher foil. The distribution was then determined by counting on the nuclear emulsion under a m icroscope the particle tracks originating from the alpha disintegration of a decay product of the prim ary reaction product.
The points below have been taken into account in choosing a suitable nuclear reaction for these experim ents. '
(a) A large difference in nuclear charge between reaction product and targetatom.
This would increase the internal ionization effect, which is expected to originate mainly from the change in nuclear charge. Because it is p ractically impossible to perform experim ents with targ ets below a certain thickn ess, a crude method of extrapolation to ta rg e t thickness zero had to be used. As the difference between the investigated internal ionization and the ionization obtained as a resu lt of charge-changing collisions m ay be relatively sm all, it was desirable that the internal ionization effect should be as high as possible. .
(b) High reaction cro ss section
Because of the very thin target thicknesses to be used, the low reaction yield thus obtained would create a demand for very highly integrated beam levels of the bombarding heavy-ions. This introduces a serious facto r of in terferen ce because of the alpha activity induced in heavy-elem ent im purities in the catcher-foil m aterial. As this induced alpha activity is proportional to the integrated beam level, a high reaction cro ss section would minimize this interference; however, as can be seen from the resu lts, it was impossible to eliminate this interference com pletely.

40 N. H. STEIGER
(c) Bombarding energies that could be obtained without the use of degrading foils.
Because of the low reaction yield obtained when using very thin targets, high beam levels of heavy—ions had to be used. These high beam levels would eventually burn the degrading foils. •
(d) A targ e t m ateria l that could be obtained in extrem ely thin la y e rs by evaporating on to a suitable backing m ateria l.
After taking into account all these considerations, we chose the reaction P r 141 (016, 8 n)Ho149. F o r this reaction, the difference in nuclear charge is equal to A Z = +8 . F o r the full energy beam, which after a slight energy degradation by the backing m aterial of the target has a lab. energy of 161.0 MeV, the reaction cro ss-se c tio n has the acceptable value of 2 0 0 mb [18].
Vacuum evaporation of natural praseodym ium on to thin Ni-backing foils was used to prepare various thicknesses of the РгШ targets to be bombarded. The thickness of the targ ets was determined by are a and weight determ inations.
The decay ch a ra c te ris tics of Ho149 a re as follows:
«7Н0 149 1 т *п-> „„Dy149 15 m *n } „Tbl49g -------Ü Ï 1 ----------) . . . .67 EC 66 y EC 65 0 .1 o (3 .9 5 MeV)
As the detecting method we used the technique of alpha-particle track counting of the decayed ТЫ49в described previously [15 ,16].
III. EXPERIM ENTAL ANALYSIS
On the basis of the fundamental equation for magnetic deflection of charged particles and from pure geom etrical considerations of the experimental set-up, we find that the horizontal deflection D on the catch er foil, with respect to the beam axis of a particle of m ass Av velocity v and ionic charge state z, is given by
Hez, i . XHez1 - cos a rc sin ------Ajcv
+l a J ^ --------- , (1)
A >CV / AHez\arcsin Aicv J
where H is the m agnetic field strength, с the velocity of light, e the elem entary charge, A the length of the magnetic field, and L the distance between the end of the magnetic field and the catcher foil. This relation was tested and calibrated by making use of the fully ionized O16 beam from the a cce lera to r [16]. The identification of the charge sp ectra obtained for the nuclear reaction products was made by use of E q .( l ) , introducing for Ai the m ass of the final nuclear reaction product, and for v the mean velocity < v > of these products (see Eq. (2)).

IONIZATION FOLLOWING HEAVY-ION-INDUCED REACTION 41
In order to calculate the velocity of the reaction products we have to consider the mechanism of the reaction by which they are produced. Experim ental investigations of the reco il properties of the final products of r e actions of the type under investigation have provided a test for the validity of the s ta tis tica l assumption and have been found to favour a "compound nucleus" reaction m echanism [19 -21 ] . The excitation energies obtained in these reactions v ary between about 35 and 125 MeV. At such high energies of excitation a very large number of overlapping levels will exist. If we assume that in the outgoing channels the random-phase approximation still applies, the angular distribution of the evaporated neutrons should be sym m etric about 7г /2 in the cen tre-of-m ass system . Let us therefore assume that, an incident beam p article of lab energy Еь is absorbed to form an excited state of a compound nucleus. Because in any model for a nuclear reaction linear momentum is conserved, the momentum of the incident beam p article must be equal to the momentum of the compound nucleus. With VçN denoting the velocity of the compound nucleus, which is identical with the velocity of the cen tre-o f-m ass, we have
where Ab and Ax denote the m asses of bombarding particle and target atom respectively . As the compound nucleus decays, the velocities of the r e sulting reaction products a re affected by this decay. However, when we re ca ll that we assum ed sym m etric angular distribution of the evaporated neutrons, we get the result that the mean velocity <v> of the reaction products is equal, to vCN and is therefore given by Eq. (2). .
In order to estim ate the velocity spread caused by nucleon evaporation, let us consider somewhat further the decay of the compound nucleus. Let V denote the velocity given to the reaction products in the cen tre-of-m ass system as a result of evaporation of all neutrons .Let us for further simplification assum e that the neutrons are emitted in random directions in the ce n tre - o f-m ass system . We are then able to find an expression for the mean square value of V for this random walk. The resulting momentum of a re coiling "mean m ass" of the evaporation chain is given by
during an evaporation p rocess of x neutrons, Tn is the average total energy removed by the evaporated neutrons, and mn is the m ass of the neutron.Tn is given by
where Q is the m ass difference between reactan ts and final products (theО values for both the target and heavy product nuclei have been calculated
(3)
(4)
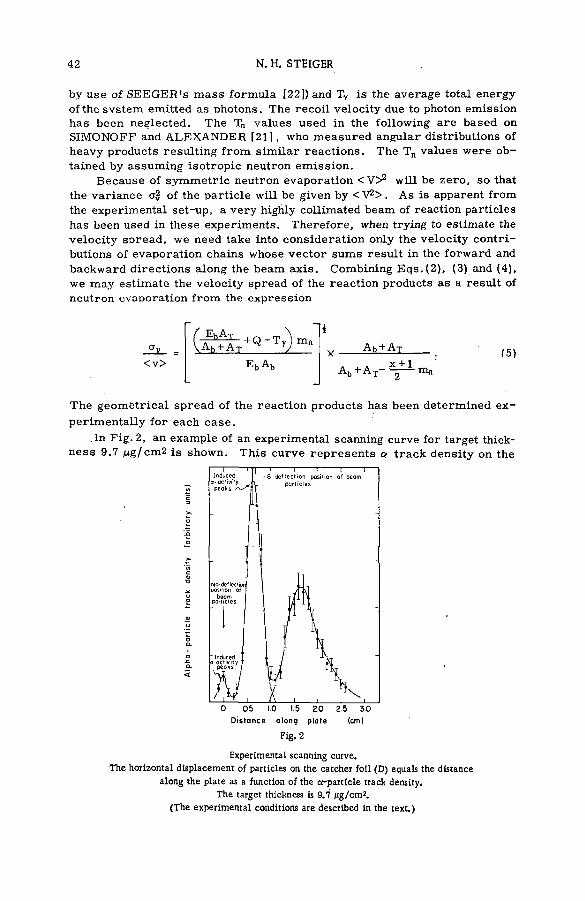
42 N. H. STEIGER
by use of SEEG ER 's m ass formula [22]) and Ty is the average total energy of the svstem emitted as ohotons. The recoil velocity due to photon emission has been neglected. The Tn values used in the following are based on SIMONOFF and ALEXANDER [211 . who m easured angular distributions of heavy products resulting from sim ilar reactions. The Tn values were obtained by assuming isotropic neutron em ission.
Because of sym m etric neutron evaporation < V>2 will be zero, so that the variance of the particle will be given by <V2> . As is apparent from the experimental set-up, a very highly collimated beam of reaction particles has been used in these experiments. Therefore, when trying to estimate the velocity spread, we need take into consideration only the velocity contributions of evaporation chains whose vector sums result in the forward and backward directions along the beam axis. Combining E q s .(2 ), (3) and (4), we may estim ate the velocity spread of the reaction products as a result of neutron evaporation from the expression
The geom etrical spread of the reaction products has been determined experim entally for each case .
In Fig. 2, an example of an experimental scanning curve for target thickness 9.7 цg/cm 2 is shown. This curve rep resents a track density on the
Ab + A T 15)< v > E bAb x 1Аь + AT- mn
Induced а -activity i peaks
+ 8 deflection position of beam porfíeles
0 0.5 1.0 1.5 2.0 2.5 3.0
D i s t a n c e a l o n g p l a t e (cm)
Fig. 2
Experimenta l scanning curve.
The ho rizonta l displacement of particles on the catcher fo il (D) equals the distance
along the p la te as a function of the a-partic le track density.
The target thickness is 9.7 Mg/cm2.
(The experim enta l conditions are described in the text.)
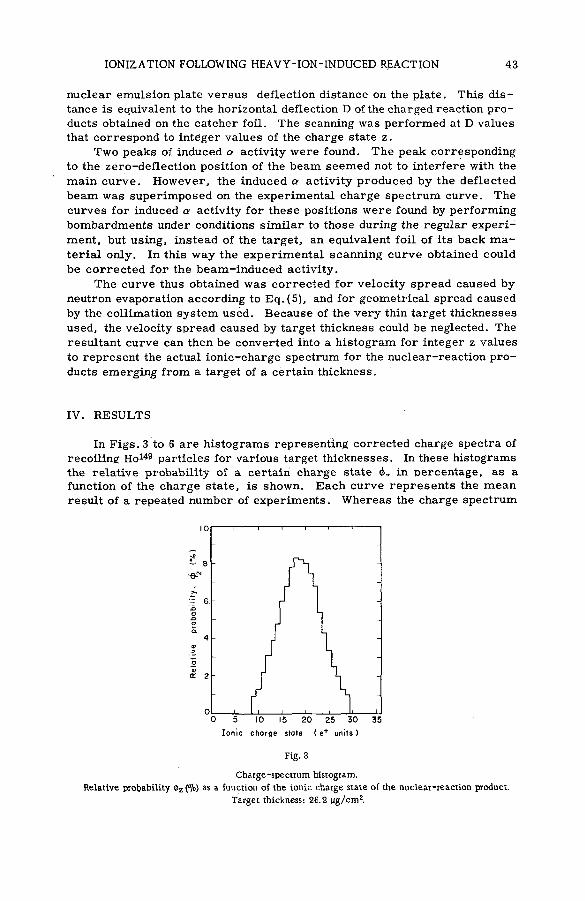
IONIZATION FOLLOWING HEAVY-ION-INDUCED REACTION 43
nuclear emulsion plate versus deflection distance on the plate. This distance is equivalent to the horizontal deflection D of the charged reaction products obtained on the catcher foil. The scanning was performed at D values that correspond to integer values of the charge state z.
Two peaks of induced a activity were found. The peak corresponding to the zero-deflection position of the beam seemed not to interfere with the main cu rv e . However, the induced a activity produced by the deflected beam was superimposed on the experimental charge spectrum curve. The curves for induced a activity for these positions were found by performing bombardments under conditions sim ilar to those during the regular experim ent, but using, instead of the ta rg e t, an equivalent foil of its back m ate ria l only. In this way the experim ental scanning curve obtained could be co rre c te d fo r the beam -induced activity .
The curve thus obtained was co rrected for velocity spread caused by neutron evaporation according to E q .(5 ), and for geom etrical spread caused by the collimation system used. Because of the very thin target thicknesses used, the velocity spread caused by target thickness could be neglected. The resultant curve can then be converted into a histogram for integer z values to represent the actual ionic-charge spectrum for the nuclear-reaction products emerging from a target of a certain thickness.
IV. RESULTS
In F igs. 3 to 6 are histograms representing corrected charge spectra of recoiling Ho149 particles for various target thicknesses. In these histograms the relative probability of a certain charge state in percentage, as a function of the charge state , is shown. E ach curve rep resen ts the mean result of a repeated number of experim ents. Whereas the charge spectrum
Fig.3
Charge-spectrum histogram.Relative probability ®z (<fo) as a function of the ionic charge state of the nuclear-reaction product.
Target thickness: 26.2 (ig/cm2.
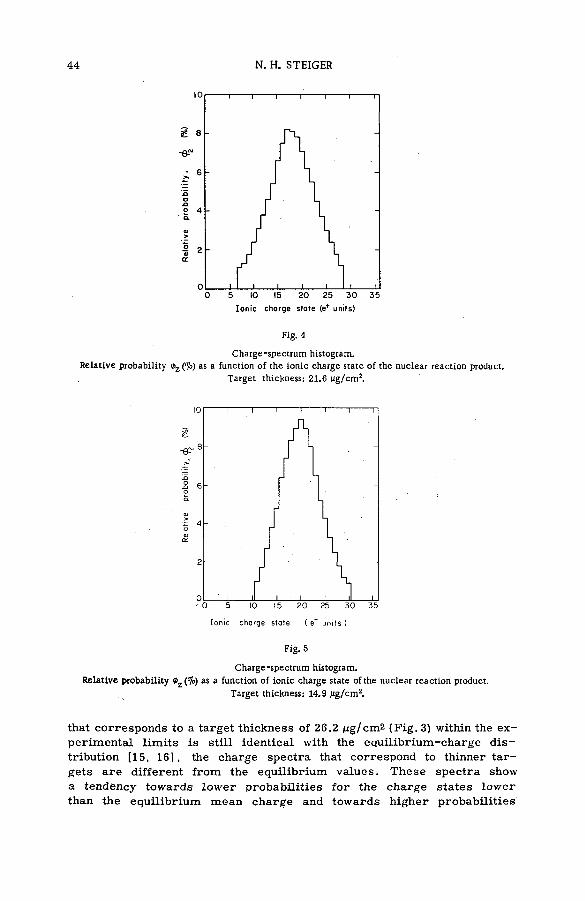
44 N. H. STEIGER
I o n i c c h a r g e s t a t e (ef u n i t s )
Fig. 4
Charge-spectrum histogram.Relative probability 0Z (%) as a function o f the ionic charge state of the nuclear reaction product. . Target thickness: 21.6 fig/cm2.
Io n ic charge state ( e+ un its )
Fig. 5
Charge-spectrum histogram.Relative probability 9z 0o) as a function of ionic charge state of the nuclear reaction product.
Target thickness: 14.9 (Jg/cm2.
that corresponds to a target thickness of 26.2 ;ug/cm2 (Fig . 3) within the experim ental lim its is still identical with the equilibrium -charge distribution [15, 161 , the charge spectra that correspond to thinner ta r gets are different from the equilibrium values. These sp ectra show a tendency towards low er probabilities for the charge states low er than the equilibrium mean charge and tow ards higher probabilities
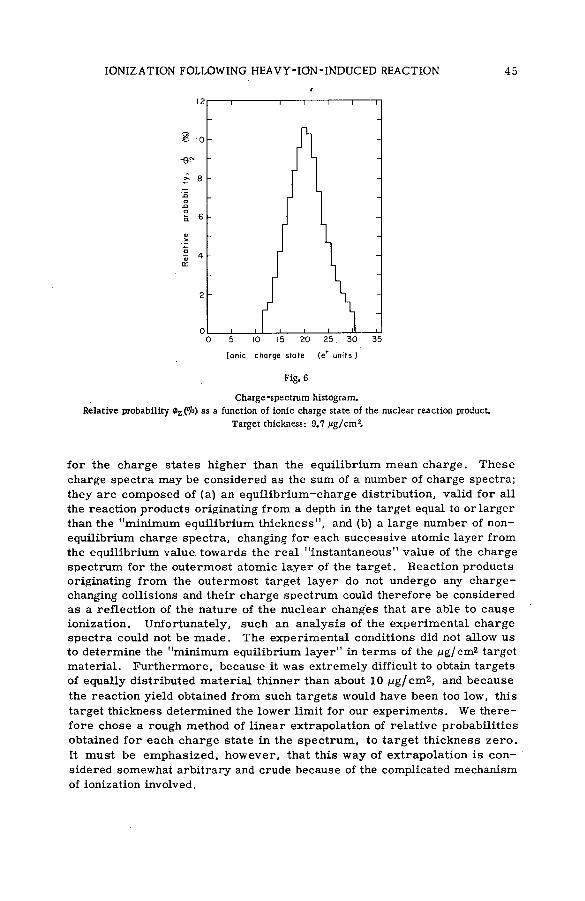
IONIZATION FOLLOWING HEAVY-ION-INDUCED REACTION 45
I o n i c c h a r g e s t a t e ( e + u n i t s )
. Fig. 6
Charge-spectrum histogram.Relative probability as a function of ionic charge state of the nuclear reaction product.
Target thickness: 9.7 jig/cm2.
for the charge states higher than the equilibrium mean charge. These charge spectra may be considered as the sum of a number of charge spectra; they are composed of (a) an equilibrium -charge distribution, valid for all the reaction products originating from a depth in the target equal to or larger than the "minimum equilibrium thickness", and (b) a large number of nonequilibrium charge spectra, changing for each successive atomic layer from the equilibrium value towards the real "instantaneous" value of the charge spectrum for the outerm ost atomic lay er of the targ et. Reaction products originating from the outerm ost target layer do not undergo any ch arge- changing collisions and their charge spectrum could therefore be considered as a reflection of the nature of the nuclear changes that are able to cause ionization. Unfortunately, such an analysis of the experim ental charge sp ectra could not be m ade. The experim ental conditions did not allow us to determine the "minimum equilibrium layer" in term s of the ¡ug/cnfi target m aterial. Furtherm ore, because it was extremely difficult to obtain targets of equally distributed m aterial thinner than about 1 0 ng/ cm 2 , and because the reaction yield obtained from such targets would have been too low, this target thickness determined the lower limit for our experiments. We therefore chose a rough method of linear extrapolation of relative probabilities obtained for each charge state in the spectrum , to targ et thickness zero . It must be em phasized, however, that this way of extrapolation is considered somewhat arbitrary and crude because of the complicated mechanism of ionization involved.
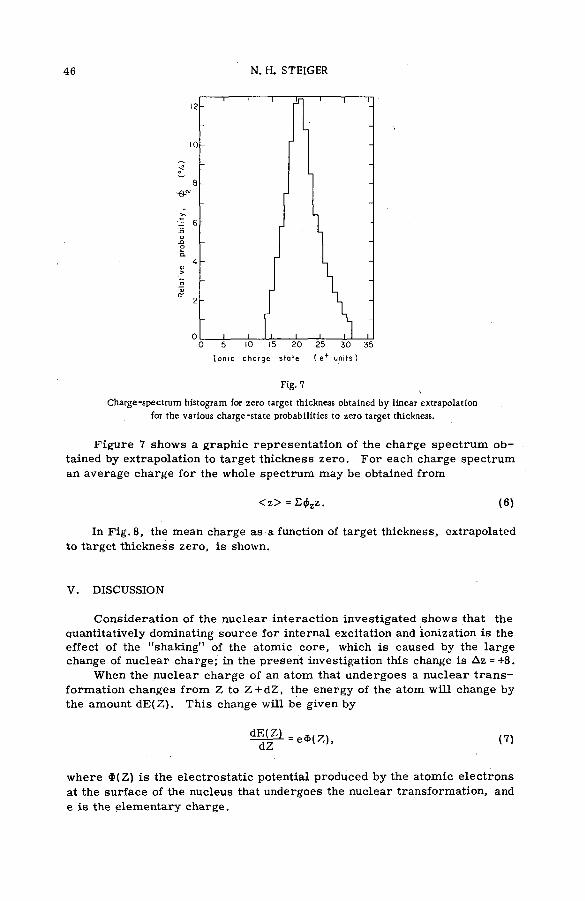
46 N. H. STEIGER
I o n ic c h a rg e s ta te ( e + u n its )
Fig. 7 NCharge-spectrum histogram for zero target thickness obtained by linear extrapolation
for the various charge-state probabilities to zero target thickness.
Figure 7 shows a graphic representation of the charge spectrum obtained by extrapolation to target thickness zero. F o r each charge spectrum an average charge for the whole spectrum may be obtained from
< z > = E 0 Z z. (6 )
In Fig. 8 , the mean charge as a function of target thickness, extrapolated to target thickness zero, is shown.
V. DISCUSSION
Consideration of the nuclear interaction investigated shows that the quantitatively dominating source for internal excitation and ionization is the effect of the "shaking" of the atomic core, which is caused by the large change of nuclear charge; in the present investigation this change is Az = +8 .
When the nuclear charge of an atom that undergoes a nuclear tra n sform ation changes from Z to Z + d Z , the energy of the atom will change by the amount dE( Z ). This change will be given by
^ ^ - = e * (Z ) , (7)
where $(Z ) is the e lectro static potential produced by the atomic electrons at the surface of the nucleus that undergoes the nuclear transformation, and e is the elementary charge.

IONIZATION FOLLOWING HEAVY-ION-INDUCED REACTION 47
T a r g e t t h i c k n e s s ( f ig / c m 2 )
Fig. 8
Mean ionic charge o f the product atom as a function of target thickness.The equilibrium mean charge is 17.6 ± 0.5 [15 ,16 ].
If in any nuclear reaction the charge p articles entering o r leaving the nucleus have velocities considerably greater than the velocities of the orbital electrons of the target atom, the electronic cloud cannot adjust adiabatically to the sudden change in nuclear charge. Thus a certain amount of electronic energy is available for atomic excitation, which in an adiabatic process would be absorbed by the incoming or emerging charged p articles. Following an approach which has been outlined by SERBER and SNYDER [23], it can be shown that this energy ДЕ is given by the difference between the energy absorbed by the charged particle in a reorientation of the electronic cloud in an adiabatic transition, and the energy absorbed in the actual non-adiábatic p ro cess. We then have
ДЕ = - [E (Z ') -E (Z )] + e (Z ' - Z) Ф (Z), (8 )
where E (Z ) is the total atom ic energy of the ta rg et atom and E (Z ') is thetotal atomic energy of the nuclear reaction product. The energy difference ДЕ appears as excitation and ionization energy of the final reaction product. A ccording to FO LD Y [24], the best E (Z ) values fo r heavy elem ents can be obtained when using the atom ic model of H artree , which gives
E(Z) = -R Z 12/ 5, (9)
where R is Rydberg's constant in energy units. Differentiating E q .(9 ), and introducing this result into E q .(7 ), we obtain
еФ(г) = -3 2 .6 4 Z 7/ s (eV). (10)
When E ( Z ') - E ( Z ) in E q .( 8 ) is expanded in a T ay lor se r ie s in Z ' - Z , and combined with E q .(10 ), we obtain an expression for the average energy of

48 N. H. STEIGER
excitation of a heavy nuclear reaction product, following a change in nuclear charge from Z to Z ':
ЛЕ = 22.85 Z2/ 5 ( Z '- Z )2 (eV). (11)
This value is an average taken over the probability distribution for the transition from the ground state of the target atom to the various final states of the reaction product. Applying E q .( l l ) to the heavy-ion induced nuclear reaction studied, and stating that Z 1- Z = 8 and Z = 59, we obtain ДЕ = 7473 eV. However, for two reasons, the actual value of ДЕ for this case will be considerably sm aller.
F irstly , Eq.(lO ), considered as an interpolation formula between integral values of Z, corresponds to the charge on the electrons being kept equal to the charge on the nucleus as the atomic number varies from Z to Z '; however, this is actually not so. The number of electrons involved in this process will be equal to the nuclear charge of the target atom (59) whereas the energy difference obtained from E q .(9 ) takes into account the difference in energy between the neutral atom of the reaction product and the neutral target atom.
Secondly, a comparative estimate of the velocity of the bombarding O16
ion and the velocities of the atomic electrons of the P r 149 target atoms shows that, in the case considered here, the transition is largely adiabatic and to a le ss e r degree non-adiabatic. The bombarding energy of the O16 ion co rresponds to a velocity of 4 .5 • 109 c m /s . This particle velocity is sm aller than the velocity of the К electrons of the target atom and comparable with the velocity of its L electron s. F o r the change in binding energy of these electrons as the result of the change in nuclear charge of the atom, the transition is thus adiabatic. F o r the change in binding energy of the r e maining electrons which occupy the outerm ost shells the transition will be non-adiabatic, and this energy difference will partly be available for intern al excitation and ionization of the reaction product. '
Consequently, the actual mean excitation energy of the final atom will only be a sm all fraction of the energy difference of ДЕ = 7473 eV as e stimated before from E q .( l l ) for the case of a com plete non-adiabatic transition .
As mentioned in the introduction, another main source for internal ionization is the development of vacancy cascad es. The main transitions r e s ponsible for this p rocess are internal conversion and orbital electron capture. To discuss the possible contribution of internal conversion to the ionization effect we need data concerning the gam m a-ray spectra of heavy-ion- induced compound nucleus reactions in the ra re earth region. M easu rements of gamma sp ectra of this kind give results of mean energies of em itted gam m a ray s of about 1.2 MeV, with low er side energies down to le ss than 0.5 MeV [25]. Furtherm ore, theoretical considerations seem to indicate a probability for the occurrence of even low er-energy gamma c a s cades of about 100 to 200 keV. F o r such low gamma energies, a high probability for internal conversion will exist, and the development of vacancy cascad es will o ccu r. Unfortunately, there do not exist any experim ental data on these low-energy gamma cascades and consequently no quantitative statement can be made about the internal-conversion probabilities and their

IONIZATION FOLLOWING HEAVY-ION-INDUCED REACTION 49
relative importance in the measured charge spectrum. Quálitatively however, it can be stated that the shape of the charge spectrum, which would have been obtained from vacancy cascades alone, is determined by the probabilities of internal conversion, the fluorescence yields associated with the various electron shells, the relative probabilities of competing X-ray transitions, and the nature of the competing radiationless transitions [13].
The electron -cap tu re decay of Hoi49 is certain ly slow in com parison with the tim e of flight of the final reaction product (of the ord er of 1 0 '8 s) p rio r to its m agnetic analysis. This p ro cess m ay therefore be excluded com pletely.
The third main source for ionization that has been mentioned in the introduction is the non-nuclear "shake-off", which has been suggested by WOLFSBERG and PERLMAN [261. According to these authors, the departure of Auger electrons will cause electrostatic perturbations in a sim ila r way as would a change in nuclear charge. In this way excitation and ionization can a rise . At present there seem s to be no way to m easure this effect experimentally. Nevertheless, in general this effect can be expected to be present to a certain extent when vacancy cascades occur.
Finally, two additional but minor sources for ionization maybe mentioned. F irs tly , a second o rd er "shake-off" as a resu lt of the sudden change in nuclear velocity can be expected. The mean recoil velocity of the r e action products in the heavy-ion reaction under investigation may be ca lculated by use of E q .(2 ) , and is found to be appreciable (4 .5 X 1 0 8 c m /s ) . However, an estim ate of some of the ionization probabilities as a result of recoil shaking shows that this effect will be of m inor im portance [2 7 ]. Secondly, as a result of collisions between the bombarding heavy-ion and the atomic electrons of the target atom some of these electrons may be "knocked out". The probability for this p ro cess (P(jc ) com pared with the probability for ejection due to the effect of "nuclear shaking" (Ps) has been estim ated by FEINBERG [28] for the case of beta decay in light atoms and found to be about 1 /1 0 0 0 . In heavier atoms and for collisions with heavy- ions, P(jc and Ps may become of the same order of magnitude only for electrons in the inner shells. As Ps is sm all for these electrons [29] the contribution of P<tc to the total ionization probability will always be negligible. Since, however, the rem oval of one of the inner electrons will to a certain extent give rise to a cascade of Auger electrons, the d irect collision may become important for that sm all fraction of the reaction products that are highly ionized.
We may now conclude that two p ro cesses a re mainly responsible for the internal ionization that o ccu rs in the product atoms of the investigated reaction and that is reflected in the charge spectrum of the Ho149 particles: ( a) the non-adiabatic part of the transition following a change in nuclear charge;(b) the development of vacancy cascades as a result of internal conversion.
Before considering the charge spectrum obtained as a result of the experim ents, it may be interesting to speculate what approxim ate shapes of charge spectra could be expected for each of the two mechanisms separately.
Even in the case of a completely adiabatic transition following the change in nuclear charge, the minimum state of ionization for* the reaction product is expected to be +8 . This charge state results from the fact that the bombarding O16 ion enters target nucleus in a completely ionized state [30]. Any

50 N. H. STEIGER
additional effect of ionization caused by the non-adiabatic part of the transition will therefore result in still higher charge sta te s . As the highest probability for ionization as a result of "shaking" will be for the existing outer electrons of the atom , alm ost no vacancy cascad es can be expected from this p ro cess. As a result of the first process alone we would therefore expect a charge spectrum starting from z = + 8 and dropping tow ards higher ch a rg e s . As a resu lt of the second m echanism alone we would expect a start of low intensity for the lower charge states, rising to a maximum and then decreasing again for high charge values. The low ch arg e-state side of the spectrum may be explained by two types of events. F irstly , the o ccurrence of radiative transitions when prim ary vacancies are filled mainly by X -ra y transitions. Secondly, conversion occurring in the outer part of the atom so that even for the radiationless part of the transitions no significant cascades can develop. The high charge side of the spectrum may be explained by the occurrence of multiplying radiationless transitions developing as a result of an original vacancy created in the inner part of the atom. The position and height of the maximum of the spectrum will depend on the character and relative probabilities of the various processes involved. Spectra of this kind have been obtained experimentally for isom eric transitions, where internal conversion alone was responsible for the ionization effect [13].
The resultant spectrum obtained in our case by a combination of both p ro cesses can be expected to s ta rt at the low er ch arg e-sta te side with a certain probability for charge z = + 8 , rising to a maximum and then decreasing again.
The charge spectrum obtained for target thickness zero, by crude extrapolation of our experimented charge spectra for various target thicknesses (see F ig .7), may be considered as approximately following this expectation.
If the entire internal ionization p ro cess were caused by prim ary ionization effects only, an estim ate of the expended ionization energy could be made, based on the charge spectrum given in F ig . 7. This energy will be the sum of the binding energies of all the electron s involved in the ionization effect. We have this estim ate, starting with a [Ho1491*8 ion, re p re senting the instantaneous product of the nuclear reaction after the evaporation of all the eight neutrons has been completed, and before any internal ionization could have happened. We have further assumed that the electronic configuration of the hypothetical [Ho149]+8 ion is equal to the electronic configuration of the neutral P r 14i target atom, only with different electron binding energies. All additional ionization as represented by the charge spectrum was calculated taking into account the binding energies of the outer electrons involved, and proceeding su ccessively into inner shells for the higher ch a rg e -s ta te probabilities. This estim ate gives approx. 800 eV.
However, as discussed, "vacancy cascad es" caused by internal conversion , as well as "p rim ary ionization" caused directly by the "non- adiabatic" part of the transition following "shake-off", are responsible for the internal ionization. Because the relative importance of these.two processes is unknown it can be stated only that the part of the ionization caused by the "shaking" effect will be less than 800 eV.
This investigation should be regarded as a first experimental approach to study the internal ionization effect following an induced nuclear reaction.

IONIZATION FOLLOWING HEAVY-ION-INDUCED REACTION 51
Because of the partly unknown basic information, such as the gamma c a s cades following the nuclear reaction , and the considerable experim ental difficulties in eliminating the influence of target thickness, the results should at this stage be regarded as rather qualitative. More detailed calculations of the expected absolute magnitude of the non-adiabatic transitions of the shaking effect a re in p ro g re ss . '
A C K N O W L E D G M E N T S
Experim ental part of this work was carried out under the auspices of the United States Atom ic E nergy Com m ission at the L aw ren ce Radiation L ab orato ry , U niversity of C alifornia, Berkeley, C a lif . , United States of A m erica .
The author wishes to thank D r. Torbj^rn Sikkeland for many helpful discussions concerning this work. He is greatly indebted to Dr. Albert Ghiorso for his encouraging interest during all its phases. He appreciates the assistan ce of the Hilac crew fo r many hours of operating tim e . The author exp resses his thanks to P ro fesso r Isadore Perlm an for the pleasant hospitality of-^the N uclear C hem istry Division of the Law ren ce Radiation L ab o rato ry .
R E F E R E N C E S
[1 ] BAULCH, D, L and DUNCAN, J. F, , Quart. Rev. (London), 12 (1958) 133.[2 ] BAULCH. D. L., DUNCAN, J. F. and THOMAS, F. G ,, Chemical Effects of Nuclear Transformations,
JIIA EA , Vienna (1961) 169-81.
[3 ]. SNELL, H. A. and PLEASONTON, F ., Phys. Rev. 11Г(1958) 1338.[4 ] SNELL, H. A . , PLEASONTON, F. and NEED, J. L. , Phys. Rev. 116 (1959) 1548.[5 ] SNELL, H. A . , PLEASONTON, F. and CARLSON, T. A . , Chemical Effects of Nuclear Transformations,
I. IAEA, Vienna (1961) 147-53.16] CARLSON, T. A. et al. . Chemical Effects of Nuclear Transformations. I IAEA, Vienna (1961) 155.[7 ] CARLSON, T. A. , PLEASONTON, f. and JOHNSON. С. H . , Phys. Rev. 129 (1963) 2220.[ 8 ] CARLSON, T. A . , Phys. Rev. 130 (1963) 2361.[9 ] CARLSON, T. A . , Phys. Rev. 131 (1963) 676.
[10 ] KRAUSE, M. O . , Phys. Rev. 133 (1964) A385. '[11] SNELL, H .A . and PLEASONTON, F . . Phys. Rev. 100 (1955) 1396.[12] SNELL, H. A. and PLEÁSONTON, F . , Phys. Rev. 102 (1956) 1419.[13] SNELL, H. A. and PLEASONTON, F . , Proc. rOy. Soc. A241 (1957) 141.
[14 ] SNELL, H .A . and PLEASONTON, F . , Phys. Rev. 101 (1957) 740.[15] STEIGER, N. H ., Proc. 3rd Conf. on Reactions Between Complex Nuclei, Asilomar, C a lif ., University
of California Press, Berkeley (1963),
[16] STEIGER, N. H ., Equilibrium Distributions of Effective Charges of Products of Heavy-ion Induced Spallation, UCRL 10806 (1963).
[17] HUBBARD, E. L et a l . , Rev. sci. Instrum. 32 (1961) 621.
[ 18 ] ALEXANDER, J. M ., (Lawrence Radiation Laboratory, Berkeley, Calif.), private communication.[19] WINSBERG, L. and ALEXANDER, J. M . , Phys. Rev. Ш (1961) 518 and 529.[20] ALEXANDER, J. M. and SISSON, D. H ., Phys. Rev. 128 (1962) 2288. .
[21] SIMONOFF, G. N. and ALEXANDER, J. M ., Angular Momentum Effects on Neutron Emission by Dy and Ту
Compound Nuclei, UCRL- 10099 Rev. (1962).

52 N. H. STEIGER
[22] SEEGER, P. A . , Nucl. Phys. 25 (1961) 1.[23] SERBER, R. and SNYDER, H. S. , Phys. Rev. 8 (1952) 152.[24] FOLDY, L. L. , Phys. Rev. 83 (1951) 397.[25] MOLLENAUER, J. F. , Effects of Angular Momentum on Gamma Ray Production in Compound Nucleus
Reactions (thesis), UCRL-9724 (1960).
[26] WOLFSBERG, M. and PERLMAN, M. L. , Phys. Rev. 99 (1955) 1833.[27] LEVINGER, J. S. , Phys. Rev. 90 (1953) 11.[28] FEINBERG, E. L. , J. Phys. USSR 4 (1941) 423.[29] MIGDAL, A . , J. Phys. USSR 4 (1941) 449.[30] HECKMAN, a H ., HUBBARD, E. L. and SIMON. W. G . , Phys. Rev. 129 (1963) 1240.

IONIZATION FOLLOWING HEAVY-ION-INDUCED REACTION
D IS C U S S IO N
(on the foregoing three papers)
F .S . ROWLAND: In connection with Dr. W exler's rem ark s on the r e lationship between radiation ch em istry and hot-atom ch em istry in re co il tritium system s, I should like to point out that this relationship fundamentally amounts to the fact that two processes occur simultaneously in the sam e system - the radiation effects of the high-energy triton and proton, and the bond-forming reactions of the recoil tritium atoms at the end of the range - but that otherwise they are unrelated to each other. The first process, induced by high-energy ions, includes ionization and excitation effects resulting in the formation of unlabelled products throughout the system . The second p ro ce ss , the reaction of the recoil tritium atom, crea tes a radioactively labelled compound. In reco il tritium studies, one is concerned only with the radioactivity and is not bothered by the radiation ch em istry unless the sam ple rem ains in the re a c to r too long. A number of studies have been made of the possible radiation effects on these radioactivity distributions, which indicate that, at the usual radiation times and doses (e .g .1 h at 1 0 u n/cm 2 .s ) , radiation perturbation of the recoil tritium atom distribution is usually not a problem.
S. W EX LER : I agree . I was, however, m erely trying to present evidence in support of the assumptions that constitute the basis for the use of scavengers distinguishing between hot processes and therm al p ro cesses. These scavengers are taken from radiation chem istry and it is assumed that they distinguish between hot and therm al species in our hot-atom studies. If this assumption is c o rre c t, then the types of reactan ts with which the scavengers re a c t must be the sam e in the two studies.
F .S . ROWLAND: I would add that there can be two reasons for using scavengers in recoil tritium system s. One is for the purpose of d istinguishing between the hot and thermal reactions of the recoil tritium atom, as indicated by Dr. W exler. The second reason may be to suppress radiation effect perturbation of the reco il tritium product spectrum . In some system s, especially if th ^p aren t molecule is insensitive to attack by the species formed by ionizing radiation, a particular radioactive product may re a c t in a highly preferential m anner. An extrem e example of this is СрНзТ formed by recoil T reactions with cyclopropane; the radiolytic hydrogen atom s form ed by ionizing radiation reactions rem ove this ethylene-t very efficiently. This system is very hard to investigate unless a scavenger is present that is capable of reacting with hydrogen atom s. Most system s, however, do not exhibit such a wide range in reactiv ity toward radiolytic species as ethylene and cyclopropane, and can thus be studied at more convenient total neutron dosages. The scavenger experim ents a re still r e quired to distinguish hot p ro ce sse s from therm al p ro ce ss e s .
A .G . MADDOCK: I entirely agree with what Dr. Rowland has just said as fa r as tritium work is concerned. On the other hand, the paper p re sented by C arlson and White shows that in some other system s there is a very close connection between the radiation ch em istry and, for exam ple, the effects of iso m eric change, o r - to be ra th er m ore p recise - between
53

54 N. H. STEIGER
the effects of isom eric change and a particular part of the radiolysis of the system . I found Dr. White's paper particularly pleasing because he refers to a p art of the radiolytic p ro cess that I postulated at a F arad ay Society meeting in 1951, and I v ery much hope he will be able to pursue his c a lculations, and ascertain the quantitative proportion between events of the kind he has described for the X -r a y irrad iation of methyl iodide and the general radiolysis of methyl iodide. He has naturally adjusted the experiment to emphasize photo-absorption in the К and L shells, which of course normally does not occur in ordinary radiolysis. I suspect, however, that the kind of event dealt with in this paper is responsible for what I would group together as the complex products of the rad iolysis of organic m ate ria ls . In other words, the kind of process involved here produces what I think P ro fesso r Willard has described as a radical nest, and gives rise to a m ore o r le ss random selection of com plicated products. Now we know that this is not the dominant kind of event; from experimental data, it would seem to amount usually to about 5%. It would be v ery satisfacto ry if this could be justified by a calculation based on the results of White and Carlson.

KINETIC ISOTOPE EFFECTS IN RECOIL TRITIUM REACTIONS THROUGH MEASUREMENT OF
ISOTOPIC MOLECULE YIELDS*
E. K.C. LEE, J.W . ROOT** AND F. S. ROWLAND*** DEPARTMENTS OF CHEMISTRY, UNIVERSITY OF KANSAS,
LAWRENCE. KANSAS, AND UNIVERSITY OF CALIFORNIA IRVINE,IRVINE, CALIFORNIA, UNITED STATES OF AMERICA
Abstract — Résumé — Аннотация — Resumen
KINETIC ISOTOPE EFFECTS IN RECOIL TRITIUM REACTIONS THROUGH MEASUREMENT.OF ISOTOPIC
MOLECULE YIELDS. Direct gas chromatographic separation of isotopic tritiated molecules has made it possible to obtain new data on both the intermolecular and intramolecular kinetic isotope effects on the hot displacement reaction. These data provide results for several new systems, as well as confirming some of the
experimental isotope effects determined previously by indirect competition methods.The intermolecular kinetic isotope effect in atom displacement reactions lh and Id
T * + C H 4------>CH,T + H lh
T * + CD4 ----- »C D 3T + D Id
has been shown to be 1.33 ± 0.04 for the ratio (CH3T/CH4)/(CD3T/CD4) in experiments carried out in CH4 -CD 4 mixtures. The intramolecular kinetic isotope effect in the replacement of an alkyl group by an energetic
tritium atom has been shown to be 1.25 ± 0.04 for the ratio C H 3T/CD3T from C H 3CD 3.
The four isotopic isomer molecules of propylene-t have been separated into C H 2TCH =CH 2 and a com
bined olefinic-t group by gas chromatography. A similar separation has been carried out for C H ^С = CH2
CH2T/ 'and (СНз)гС = С НТ, and for the other tritiated butenes. Application of this analytical method to the olefinic
radioactive products from recoil tritium reactions with gaseous propane, isobutane, n-butane and neo-pentance
has provided information about the origin of these products. The propylene n formed from reactions of tritium
with propane is about half CH2T C H -C H 2, indicating that the primary mechanism of its formation is by secondary decomposition of an excited molecule, rather than by a simultaneous double displacement of 2H atoms.
The separation of the products C2H3T, C 2H2DT, C 2HD2T and C 2D3T from recoil tritium reactions
with CHsCDs has shown the major yields to be only of the two molecules, C 2H2DT and C 2HD2T. The ratio
of C2HD2T/C2H2DT is about 1. 6. Taken with the other experiments concerning the origin of tritiated olefins
from alkanes. these results show an isotope effect in the elimination of HD from CH^rDCj* versus CH^CDgT. The probable explanation-for this effect is related to the energy deposition in the replacement of H versus
D in very high energy reactions.
DÉTERMINATION DES EFFETS ISOTOPIQUES CINÉTIQUES DANS LES RÉACTIONS AVEC LE TRITIUM
DE 'r e c u l , PAR MESURE DES RENDEMENTS EN MOLÉCULES ISOTOPIQUES. La séparation directe, par
chromatographie gazeuse, de molécules isotopiques tritiées a permis d'obtenir des données nouvelles concernant les effets isotopiques cinétiques intramoléculaires et intermoléculaires sur la réaction de déplacement
par atomes chauds. Ces données indiquent les résultats obtenus avec plusieurs nouveaux systèmes; elles con-
* A i l experimenta l work was carried out at the University of Kansas and was supported by the
D irectorate of Chem ical Sciences, A ir Force Office of Scientific Research, Grant No. 534-64 and Contract
No. A T - ( ll- l)- 4 07 w ith the United States Atom ic Energy Commission.
* * Present address : Un iversity of Ca lifo rn ia Los Angeles.
Present address : Un iversity of Ca lifo rn ia Irv ine.
55

56 E. К. C. LEE et a l.
firment eii outre certains des effets isotopiques qui avaient été déterminés expérimentalement par des méthodes
indirectes de réaction compétitive.
U a été établi que l'effet isotopique cinétique intermoléculaire dans les réactions de déplacement
d'atomes suivantes:
T * + C H 4----- »C H 3T + H
T * + CD 4----- »CD3T + D
est égal à 1,33 i 0,04 pour le rapport (C H 3T/CH4)/ (CD 3T/CD4) lorsque les expériences s'effectuent dans
des mélanges C H 4-C D 4. On a également établi que l ’effet isotopique cinétique intramoléculaire lors du
remplacement d'un groupe alkyle par un atome de tritium de haute énergie est égal à 1,25 ± 0,04 pour le
rapport CH3T/CD3T à partir de CH3CD3.Les quatre molécules isotopiques isomères de propylène tritié ont été séparées par chromatographie ga
zeuse pour donner CH2T C H =C H 2 et un groupe d'oléfines triciées combinées. Une séparation analogue a été effectuée pour
‘ C = CH2 CH2T ^
et (CH3)2C = CHT et pour les autres butènes tritiés. L’application de cette méthode d’analyse aux produits
radioactifs oléfiniques lors des réactions du tritium de recul avec les gaz propane, isobutane, n-butane et tétraméthylméthane a permis d'obtenir des renseignements sur l ’origine de ces produits. Le propylène tritié, formé lors des réactions entre le tritium et le propane, est constitué pour moitié environ de CH2TCH =CH 2, ce
qui montre que le mécanisme primaire de sa formation est une décomposition secondaire d'une molécule
excitée et non un déplacement simultané de deux atomes H.La séparation des produits C 2H3T. C 2H2DT, Q HD¡ T et Q D ST lors des réactions entre le tritium de
recul et C H 3CDs a montré que les rendements principaux ne portent que sur deux molécules: C 2H 2DT et C 2HD2T. Le rapport СгНйгТ/СгНгОТ est d’environ 1,6. Si on les considère avec les autres expériences
sur l ’origine des oléfines tritiées provenant des alcanes, ces résultats indiquent un effet isotopique dans l'é lim ination de HD dans CH 2TCE^ par rapport à CH3CD2T. L'explication de cet effet est probablement liée au dépôt d'énergie lors du remplacement de H par D dans les réactions de très haute énergie.
К И Н Е Т И Ч Е С К И Е И ЗО Т О П Н Ы Е ЭФ Ф ЕК ТЫ В Р Е А К Ц И Я Х О Т Д А Ч И Т Р И Т И Я , О П Р Е Д Е Л Е Н Н Ы Е П У Т Е М И З М Е Р Е Н И Я В Ы Х О Д О В И ЗО ТО П Н О Й М О Л Е К У Л Ы . Прям ое г а з о хроматографическое разделение насыщенных тритием изотопных молекул дало возможность получить новые данные как по интермолекулярным, так и по интрамолекулярным кинетическим изотопным эффектам по реакции горячего замещ ения. Эти данные обеспечиваю т р е зультаты для нескольких новых систем, а также подтверждают некоторые экспериментальные изотопны е эф ф екты , определенны е ран ее с помощью м етод а косвенной кон курен ц и и .
Интермолекулярный кинетический изотопный эффект в реакциях 1 h и Id замещения атом ов,
Т * + С Н 4 ------- ► С Н 3Т + Н lh
T * + C D 4 ------- ► C D 3T + D Id
как было показано, составляет 1,33 ±0 ,0 4 для соотношения
(С Н ,Т / С Н 4 )/fCD3T / C D 4 )
в экспериментах, выполненных в см есях СН 4 — CD 4 . Интрамолекулярный кинетический изотопный эффект при замене алкильной группы энергетическим атомом трития, как было показано, составляет 1,25 ±0 ,0 4 для соотношения C H 3T / C D 3T от C H 3C D 3 .
Четыре изотопных молекулы изомера пропилен-t были разделены на С Н 2Т С Н = СН 2 и объединенную олефиновую-t группу газовой хроматографией. Подобное же разделение было
осуществлено для
1 h
1 j
сщ т ^С = С Н2

KINETIC ISOTOPE EFFECTS IN RECOIL TRITIUM REACTIONS 57
й (С Н з)г С = C H T , а также для других насыщенных тритием бутенов. Применение этого аналитического метода к олефиновым радиоактивным продуктам реакций отдачи трития с га зо образным пропаном, изобутаном, n -бутаном и нео-пентаном обеспечило информацию относительно происхождения этих продуктов. Пропилен- t , образовавш ийся из реакций трития с пропаном, составляет примерно половину СН 2Т СН = СН 2 , указы вая, что первичный м еханизм его образования создается вторичным разложением возбужденной молекулы, а не одновременным двойным смещением атомов 2 Н.
Разделен и е продуктов С ?Н ЯТ , C 2 H2D T , C 2 HD2T h C 2D 3T и з реакции отдачи трития с
C H 3 C D 3 показало, что основными выходами являются лишь две молекулы: C 2H2DT и C2 HD2T . Отношение C 2 H D 2T / C 2 H2D T равняется 1,6. Сравнивая эти результаты с другими экспериментами, касающимися происхождения насыщенных тритием олефинов от алканов, можно видеть изотопный эффект при элинации HD из CH 2 TCD$* по сравнению с C H 3 C D 2T . В о з можное объяснение этого эффекта связано с энергией отложения при замещении Н на D в
реакциях с очень высокой энергией .
ESTUDIO DE LOS EFECTOS ISOTOPICOS CINETICOS EN LAS REACCIONES DEL TRITIO DÉ RETROCESO
POR MEDICION DE LOS RENDIMIENTOS MOLECULARES ISOTOPICOS. La separación directa por cromatografía
en fase gaseosa de moléculas isotópicas tritiadas ha permitido reunir nuevos datos sobre los efectos cinéticos
isotópicos, tanto intermoleculares como intramoleculares, que se producen en las'reacciones de desplazamiento por átomos calientes. Estos datos proporcionan información sobre varios sistemas nuevos, al tiempo
que confirman algunos de los efectos isotópicos ya determinados experimentalmente por métodos competitivos
indirectos.
Mediante experimentos realizados con mezclas de CH4 -C D 4 , se ha demostrado que el efecto isotópico
cinético de carácter intermolecular en las siguientes reacciones de desplazamiento atómico
T * + CH 4------* CH3T + H lh
T * + C D 4 ------»C D 3T + D ld
es de 1,33 ± 0,04 para la razón (СН 3Т)/(СН 4)/ (С 0 3Т )/ (С 0 4). Se ha demostrado asimismo que el efecto isotópico cinético de carácter intramolecular en la sustitución de un grupo albullo por un átomo energético de
tritio es de 1,25 ¿ 0,04 para la razón CHST/CD3T derivada de CH3CD3.Las cuatro moléculas isómeras isotópicas de propilenon se han separado, por cromatografía en fase
gaseosa, en CH2TCH = CH2 y un grupo pleffnico-^t combinado. En el caso del CH3,C = CH,/ *CH 2T
y del (C H j)2C = CHT, asi como de los restantes butenos tritiados, se ha efectuado una separación análoga. La
aplicación de este método analítico a los productos oleffnicos radiactivos provenientes de las reacciones del tritio de retroceso con propano, isobutano, n-butano y neopentano (tetrametílmetano) gaseosos ha permitido
obtener información sobre el origen de estos productos. El propileno-ч formado en virtud de las reacciones del tritio con el propano se compone, en un 50 о aproximadamente, de CH2TCH = CH2, lo que indica que el mecanismo primario de su formación es la descomposición secundaria de una molécula excitada, y no un desplazamiento doble y simultáneo de 2 átomos de H.
La separación de los productos C 2H3T, C 2H2DT, C 2HD2T, C 2D 3T. provenientes de las reacciones
del tritio de retroceso con CH3CD 3 muestra que los rendimientos principales corresponden únicamente a las
dos moléculas C 2H2DT y C 2HD2T. La razón C 2HD2T/C2H2DT es del orden de 1,6. Considerados juntamente
con los demás experimentos relativos al origen de las olefinas tritiadas provenientes de alcanos, estos resultados muestran un efecto isotópico en la eliminación de HD del CH2TCDjf referidos al CH3CD2T. Probablemente, la explicación de este efecto está relacionada con la energía acumulada en la sustitución de H por D en reacciones muy energéticas.
INTRODUCTION
Much of the recen t re s e a rch involving energetic tritium atom s from nuclear recoil has been concerned with the understanding of the mechanism

58 E. К. C. LEE et al.
of hot reaction [1, 2]. The most important of these reactions in hydrocarbon and halocarbon system s are summarized in equations (1) to (4):
T* + R H ----------------------> H T + R (1)
T* + RH ----------------------> RT + H (2)
• T* + RX -----------------------> RT + X (3)
T* + 7r-bond ----------------► Excited radical (4)
Each of these general reaction types has been established as being initiated by energetic tritium atom s through studies in the presence and absence of scavengers for therm al species, of m oderator m olecules, and through simultaneous m easurem ents of two reactions in the sam e system . The determination of those factors that are important in controlling the m echanism s of each of these reactions has been ca rrie d out largely through simultaneous m easurem ents of two reactio n s in the sam e system . The initial approaches by this method involved yield comparisons of reaction ( 1 ) with (2 ) for each of a se t of m olecules, with general inferences drawn from the variation of this H T/R T ratio with the nature of R [1].
Uncertainties in interpretation arise with this approach because a suitable standard for comparison is lacking; since all yields are hot reactions with m echanism s potentially different from th erm al reactio n s , none can be utilized as a fixed standard on an a p rio ri basis. Determinations of absolute yields are of little help in fixing a standard because these yields are dependent upon the energy loss mechanism in each individual sample. These energy lo sse s are them selves not n ecessarily constant from m olecule to molecule, even in a homologous series . Improvements in the competitive technique have been obtained through m easurem ents with two parent m olecules RH and R 1 H, permitting the direct comparison of (2) and (2 • ) [3 - 5 ]; through intram olecular determination of the tritium distribution, which also perm its comparison of ( 2 ) and (2 ' ) [6 ]; and by m easurem ent of the absolute yields from RH in the presence of an excess of an additional molecule. The latter approach has been applied for the yields of HT from various molecules RH in an excess of C2 D4 [7].
When reactions (1), (2) and (3) are investigated by these m ore precise competitive techniques, variations with the nature of the group R a re observed for each . The yields of the abstraction reaction (1) from hydrocarbons can be closely correlated with the bond dissociation energy of the RH bond involved [7, 8 ].
Kinetic isotope effects
The substitution of deuterium for hydrogen in the m olecules RH or RX offers the opportunity for observation of isotope effects in these hot reactions, and has been regularly used as a means of testing various hypotheses about the mechanism of hot reaction . The experim ental approaches to m easurement of isotope effects between RH and RD have been developed in a manner

KINETIC ISOTOPE EFFECTS IN RECOIL TRITIUM REACTIONS 59
sim ilar to the comparison of equation (2) for RH and R' H. The ideal m easurem ent of a kinetic isotope effect involves the direct m easurem ent of the relative yields in reaction with a protonated position and with the equivalent deuterated position.' This approach was first used to investigate the hydrogen abstraction reaction (1) by obtaining the HT/DT ratio from the molecule CH2E>2 and from m ixtures of CH4 and CD4 [9, 1 0 ]. The determination of the intram olecular location of tritium had been used e a rlie r for the investigation of the relative, yields of reactions (2) and (4) with both protonated and p a rtia lly deuterated isopropyl benzoates as the ta rg e t m o lecu les [ 1 1 ].
D irect m easurem ents of the relative yields of (1), (2) and (3) can be made for a protonated molecule and then for its deuterated counterpart [ 1 2 , 13]. Relative m easurem ents cam also be carried out readily for com petitions involving one m olecule RH, and the deuterated form , R ' D, of an entirely different molecule. The possible existence of several kinds of iso topic variation (e. g. different energy loss for non-reactive collisions with RH than RD, different average energy of the tritium atom at reaction with isotopic m olecules, different probability of reaction per collision with each one e tc . ) usually means that sev eral m easurem ents are n ecessary to d istinguish the contribution from each [14-16].
The d irect investigation of kinetic isotope effects has been hindered by the lack of suitable methods for the separation (with the exception of HT from DT) of tra c e r amounts of isotopic molecules. Recently, however, advances have been made in gas chromatographic techniques that perm it such separations with both alkanes and alkenes [17, 18, 8 ]. With these procedures investigation's have been carried out in several system s containing isotopic varian ts. These have included the direct comparison of reactions (2H) and (2D) with the molecules CH4 and CD4 for confirmation of the recent indirect m easurem ents of this ratio . The sam e separation of CH3T from CD3T has also been used for the m easurem ent of their relative yields from CH3CD3 in a test of kinetic isotope effects in reaction (3). The separation of all the different e th y len e-t's has been used to m easure the isotopic behaviour in reaction (2) of T* with СНэСОз, and in the subsequent loss of HD from the excited molecules CH3 CD2T* and CH2TCD3*. Finally, additional intram olecular measurements of HT versus DT have been performed.
EXPERIM ENTAL
Irradiations
R ecoil tritiu m atom s w çre produced from the He3(n, p)T reactio n in samples prepared according to the usual techniques [1, 2]. He3 was obtained from the Mound Laboratory, Monsato Research Corporation, and was purified from tritiated contaminants p rior to use. The deuterated hydrocarbons w ere obtained from M erck, Sharp and Dohme of Canada, and other gases were Phillips research grade used without further purification. The isotopic purities of the deuterated m olecules are given in deuterium atom per cent as follows: CD4 (99. 1%), CHaCD3 (98. 7%).
The methane samples were irradiated in a nominal flux of 1.0 X 1011 n /cm 2 • s for 90 min in the ro tary rack of the Omaha VA Hospital TRIGA re a c to r at

60 E. К. C . LEE et a l.
20°C ambient temp. The other sam ples w ere irradiated at a flux of l X 1 0 u n/c:m2 -s for 2 h in the University of Kansas Bendix re a c to r . The actual fluxes within the bulbs were reduced to about one half of the nominal flux by neutron absorption in the boron of the P yrex 1720 glass.
The gaseous tritium and deuterium labelled products w ere separated and m easured by radiogas chromatography using helium as the eluent gas and propane as the counting gas [19]. The separations w ere achieved by three different colum ns, one for each group:
(a) CH3T /C D 3T by an activated charcoal column. 100 ft, 3 /16 in O. D. copper tubing plus 50 ft, 1 /4 in O. D. copper tubing, with 3 0 -4 0 m esh a c tivated cocoanut-shell charcoal, operated at 25 lb/in2 inlet pressure, atmospheric outlet pressure, 52°C, and 0. 31 m l/s flow rate.
(b) HT/DT by a modified alumina column. 1 3 ft, 1/4 in, O. D. copper tubing with 50-60 mesh firebrick plus 7 ft, 1/4 in O. D. copper tubing, with 4 0 -5 0 m esh, 7 -alum ina coated with 3. 5% by weight of Fe^O^, operated at 2 0 lb /in 2 inlet p ressu re , atm ospheric outlet p ressu re , at liquid-nitrogen tem perature, and 0. 48 m l/s flow ra te .
(c) Isotopic position isom ers of ethylene-t and propylene-t by a silver nitrate-ethylene glycol column. 250 ft, 0. 138 in I. D. nylon tubing with 1000 m l of 3 0 -4 0 m esh chrom osorb P (HM DS-treated) coated with 40 ml of 83% saturated AgN03-ethylene glycol at room tem perature, operated at 26. 5 ib /in 2 inlet p ressu re , atm ospheric outlet p ressu re , at 0°C and 0. 280. 32 m l/s flow ra te .
A flow proportional counter with .85 m l active vol. was used for the CH3T /C D 3T and H T/D T analyses, and a 45 m l counter was used for the analysis of tritium -labelled olefins. The sm aller counter provided improved peak resolution while sacrificing some sensitivity of detection (by the ratio of the active volumes) [19].
RESULTS AND DISCUSSION4 .
Substituent effects in the T -f o r -X reaction
■ The substitution of reco il tritium atoms for various alkyl groups has been reported for a large number of hydrocarbons, and appears to be e s sentially universal in occurrence [1, 2]. Systematic comparison of relative probabilities for replacing different R groups has shown a strong preference for the formation of CH3T over higher labelled alkanes in all hydrocarbon system s [20]. Halocarbon experiments showed a preference (per C -F bond) for the formation of CH3T from CH3F versus CH2TFfrom CH2F2, with consistently lower yields as the number of F atoms in the molecule was increased [21]. The rotational inertia hypothesis is consistent with all these resu lts , and describes the critica l bond-formation step after the replacement of substituent X as being dependent upon the ability of the remnant radical to rotate quickly, and offering a bonding orbital to the T atom before it can escape. Methyl radical rotation would be fastest, and therefore CH3T yields highest, because of the low m ass of the H atom s, while heavier substituents (e. g. F o r CH3 for H) slow the rotation because of the g reater rotational inertia of the heavier substituent, and thus substantially reduce the probability of

KINETIC ISOTOPE EFFECTS IN RECOIL TRITIUM REACTIONS 61
bond-formation to the reacting T atom. While later experiments have shown that quantitative measurements require very careful evaluation of the effects of excitation energy in causing decomposition of particular labelled products, the general semi-quantitative outline of the yield ratios has been confirmed [2 2 ].
Secondary isotope effects in recoil tritium reactions
The substitution of a hot tritium atom for a substituent group X has been compared with the two molecules CH3F and CD3F , as in reactions (5) and (6 ):
T* + CH3X ------------------► CH3T + X (5)
T* + CD3X ------------------» CD3T + X . (6 )
The absolute yield of CH3T from CH3F was shown to be about 30% g rea ter than that of CD3T from CD3F in several experimental circum stances, offering clear evidence of a "secondary" isotope effect in recoil tritium replacement of F , favouring bond-forming reaction with CH3 versus CD3 [13-15].
An intram olecular measurement of a sim ilar isotope effect in the T -fo r- alkyl group reaction has been obtained with the ta rg e t m olecule CH3CD3 .The replacement of a methyl group by the hot tritium atom leads to the fo rmation of either CH3T or CD3T. The chromatographic separation of the two isotopic m ethanes indicates a preferen ce for the form ation of CH3T over CD3T by 1. 26 ± 0 . 04. Such an intramolecular measurement permits greater accuracy in the ratio measurement, while introducing the ambiguity that the X groups are not entirely identical, being CD3 and CH3 respectively, in equations (5) ànd (6 ). Nevertheless, the agreement with the methyl fluoride resu lts is very close¿ and the experim ent confirm s that bonds to CH3 a re 25-30% easier to form than bonds to CD3 during replacement of a substituent group X. The direction of this isotope effect is consistent with the effects of much heavier R groups, as expressed in the rotation al-in ertia concept. However, as discussed later, the magnitude of this isotope effect seems too large in comparison with actual changes in the moments of inertia for various groups.
M easurem ents have also been made of the relative yields of ethane-t and propane-t from various isotopic propanes, as shown in Table I. The quantitative interpretation of these experiments involves a problem in standardization. N evertheless, it is clear that the ethane-t_ yield is higher when the molecule formed is R-CH2T than when it is R-CD2T.
The T -fo r -H and T -fo r -D reactions in methane
The relative yields of substitution reactions for H versus D have been p reviously m easured by indirect methods [2, 12-16]. F o r exam ple, the yield of CHaT F from CH3 F has been com pared by m eans of cyclobutane com petition experim ents with the yield of CD,TF from CD3F , and found to be1. 3 3 ± 0. 04 tim es la rg e r [14]. However, no direct com parisons within the sam e sample have previously been made for isotopic m olecules.
The relative yields of CH3T from CH4 and CD3T from CD4 have been directly m easured in four CH4 -CD4 m ixtures covering a wide mole fraction
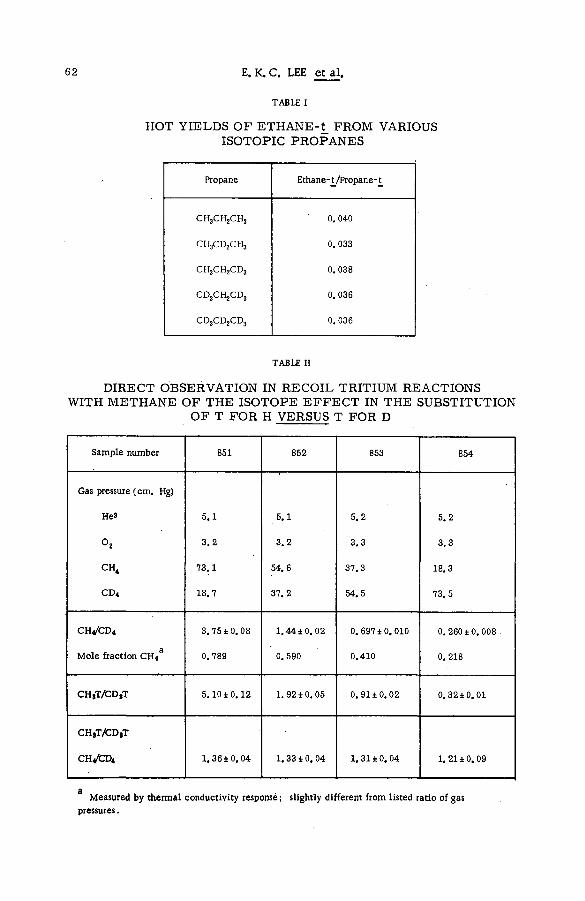
62 E. К. C . LEE et a l.
TABLE I
HOT YIELD S O F ETH A N E-t_ FROM VARIOUS ISOTOPIC PRO PANES
Propane Ethane- 1 /Propane-1
CH3CH2CH3 0. 040
c i i3c d 2c h 3 0.033
CH3CH2CD3 0. 038
c d 3c h 2c d 3 0. 036
c d 3c d 2c d 3 0.036
TABLE I I
D IRECT OBSERVATION IN R EC O IL TRITIUM REACTIONS WITH M ETHANE O F THE ISOTOPE E F F E C T IN TH E SUBSTITUTION
O F T FO R H VERSUS T FO R D
Sample number 851 852 853 854
Gas pressure (cm. Hg)
HeS 5.1 5. 1 5.2 5.2
o 2 3 .2 3 .2 3 .3 3 .3
CH 4 73.1 54.6 37.3 18.3
c d 4 18.7 37.2 54.5 73. 5
CH 4/CD4 3.75 ±0.08 1. 44± 0. 02 0. 697 ±0.010 0.260 ±0.008 ■
M o le fraction 0.789 0. 590 0.410 0. 218
C H jT/CDjT 5.10 ±0.12 1. 92 ±0. 05 0.91 ±0 .0 2 0.32 ±0 .01
C H jT/CDjT
CH /CD 4 1.36±0. 04 1. 33 ± 0. 04 1 .3 1 *0 . 04 1. 21 ±0.09
aMeasured by the rm a l conductiv ity response; s ligh tly d ifferent from listed ra tio of gas
pressures.

KINETIC ISOTOPE EFFECTS IN RECOIL TRITIUM REACTIONS 63
(ACTIVATED CHARCOAL)
(a) (b)
Fig* 1
Radio gas chromatogram showing the separation of isotopic methane molecules on a charcoal column.
The rad ioactiv ity measurement is shown by the solid Une, and the mass measurement
of the same sample is shown by the dotted line.
(a) Sample No. 583 (b) Sample No. 851.
range. The results obtained from these samples are summarized in Table П and illustrated in Fig . 1. The sample filling procedure requires the su ccessive introduction of four very low boiling gases into each sam ple bulb, with possible attendant e rro rs in the estimation of composition from back- diffusion etc . [15]. Accordingly, all m acroscopic compositions w ere obtained from the therm al conductivity responses recorded for each sample, as illustrated in Fig . 1 by the dotted lines. The observed areas under each m ass peak have been co rre cte d for the slightly different therm al conductivity responses of CH4 and CD4 [8 ].
These d irect m easurem ents of the competition between T -fo r-H and T -fo r-D reactions all show a kinetic isotope effect [23] favouring the former by a facto r of 1. 3 1 ± 0. 03. This facto r is approxim ately the sam e as the factor observed for the same competition for CH3F versus CD3F , and for various alkanes v ersu s deuterated alkanes [8 ], and also by indirect com petition.
Measurement of the yields of HT and DT from these sam e sam ples, as shown in Fig . 2, gave, abstraction/substitution ratio s of 0. 8 for each m olecule for all mole fractions. These abstraction yields are consistent with p rev io u sly observed co rre la tio n s with bond d isso cia te d en e rg y [7 , 8 ].
Prim ary versus secondary isotope effects
Two completely different approaches cam be made to explain the experimental comparison of the two reactions (7) and (8 ):
64 E. К. C. LEE et al.
T* + CD4 -------------------+ CD3T + D. (8 )
These two approaches can be summ arized in the following question: Is the isotope effect observed between reaction s with CH4 and CD4 a "p rim ary " isotope effect (i.e. replacem ent by tritium atom of H versus D), or a "secondary" isotope effect (formation of bond by tritium to CH3versus CD3)? All the previous discussions of the isotope effects in the replacem ent of H or D have emphasized the isotopic differences between the replaced atom s, although each experiment with a fully protonated or fully deuterated molecule simultaneously is subject to any isotopic variations arising from the differences between the radicals to which the tritium atom becomes bonded. The observed secondary isotope effect in the T -fo r -X reactions certainly indicates that its possible existence cannot be ignored in the T -fo r-H reaction .
T * + C H 4 --------------------» -С Нз Т+Н (7)
(T -A ljO j WITH FegOj )
Fig. 2Radio gas chromatogram showing the separation of isotopic HT and DT molecules
on a modified a lum ina column.
(a) Sample Ko, 854 (b) Sample No, 851.
A critica l test of the differences in these two approaches could be made through experim ents with the molecule CH2D^, as in reactions (9) and (10):
T * + C H 2D2 --------------- >CH TD 2+ H (9)
T ^ + C H ^ --------------- >C H 2D T +D . (10)
If the isotope effect observed between CH4 and CD4 w ere entirely caused by a prim ary isotope effect in the replacem ent of H or D, then the y : Id of

KINETIC ISOTOPE EFFECTS IN RECOIL TRITIUM REACTIONS 65
(9) would be expected to be about 30% greater than that of (10). On the other hand, if the CH4 -CD4 results are attributable entirely to a secondary isotopic difference between CH3 and CD3, then CH2D and CHDg would probably show about 2 0 % and 1 0 % effects, and reaction ( 1 0 ) would give a 1 0 % higher yield than reaction (9). A d irect measurerrient of the relative yields of CHTD2 and CH2DT would distinguish the contributions from the prim ary and secondary effects. Unfortunately, the gas chrom atographic separation of these two m olecules has not yet been achieved with sufficient resolution for this experim ent.
We believe that the present experim ental evidence is best accounted for through the hypothesis that the secondary isotopic effects of CH^versus CD4
are much m ore significant in reactions (7) and (8 ) than the prim ary isotope difference between the replacement of H or D. In the over-all consideration of the replacem ent of X in RX by T, the yield of RT is dependent upon the identity and nature of both X and R. However, all experim ents to date are consistent with the assumption that the substitution of D for H in R causes much larg er isotopic variations in the yield of RT than substitution of D for H in X . ,
The hypothesis that the replacem ent of H or D by tritium is simply another example of the T -fo r -X reaction offers as a corollary a consistent explanation of the results obtained with CH3F and CDsF, for which the yields of both T -f o r -F and T -fo r-H -o r-D , have been m easured. In I2-scavenged experim ents, the ratio s of CH3T /C H 2T F and CD3T/C D 2T F have been m easured as 0 .337± 0.002 and 0 .324± 0.002 respectively [13] . The comparable 0 2-scavenged figures are 0. 329±0. 003 and 0 .3 1 3 ± 0 .0 0 4 [15]. Both sets of data agree that the yield ra tio s are nearly the sam e, but that the ra tio is about 5 ± 1% larg er for reactions with the protonated species - - a small difference, but outside the statistical lim its of e rro r. This result can be adequately accounted for by secondary isotope effects: in the replacem ent of H or D, only two hydrogen atoms remain in the CH2F or CD2F entity to which the T atom becom es bonded. However, in the replacem ent of the F atom, three hydrogen atoms rem ain, and the secondary isotope effect is c o r r e s pondingly la rg er.
Origin of secondary isotope effects in recoil tritium reactions
The explanation for this secondary isotope effect in hot tritium reactions probably lies in the necessity for the remnant of the struck molecule to r e lax its stru ctu re sufficiently to perm it bond formation to the tritium atom. The concept of "rotational inertia" involves one possible mode of consideration for this relaxation p ro ce ss . Another possible concept is that som e atom ic relaxation is n e ce ssa ry and that the lighter atom s a re quicker to relax into a structure in which an electronic orbital is sufficiently available to cause bond-formation. This explanation implies that H atoms move more rapidly than D, and that heavier atoms hardly move at all.
This view of the relaxation p ro cess does not involve the consideration of the rotational motion of an entire radical, with comparatively fixed geom etry for the atomic components, as implied in the rotational inertia concept. Instead, the atom s bonded to the carbon atom must shift into a new

66 E. К. C . LEE et a l.
spatial arrangem ent perm itting som e orbital overlap tow ards the tritium atom . The resulting sp ecies m ay v ery well be in a spatial arrangem ent corresponding to extrem e vibrational excitation, but possessing a reasonable stable С-T bond. The high excitation energies observed for such products [4, 5] are consistent with this picture.
The replacem ent of Cl by T in C2HSC1 has a yield of about 6 8 relative to the sam e reaction in CH3CI as 100 [22]. This decrease is approximately the sam e as the magnitude of the observed secondary isotope effect in the substitution of T for F in methyl fluorides. The rotational inertia of the CH3-CH 2- radical is very much larg er than that of either СОз- or CH3- , yet the observed d ecrease in yields is com parable for CH3CH2- and CD3- . A relaxation of the positions of H and D atoms (but not Cl) could give a consistent picture of the magnitude of the observed effects.
Tritiated olefins from hot tritium reactions with alkanes
Intram olecular tritium distributions can be obtained directly through chromatographic methods based on the Ag+ ion-olefin complexes, for which substantial equilibrium hydrogen isotope effects are observed. F ig s . 3a and 3b illu strate the peaks observed after reco il tritium reactio n s with butene- 1 and tran s-b u ten e-2 , indicating resp ectiv ely CH2TCH = CH2 and CH3CH = CHT. Separate measurement with CH3CT = CH2 showed a peak coinciding with the la tte r . The propylene-t from both n-C 4 H10 and iso -C 4Hio gives two peaks each, as shown in (c) and (d) of F ig . 3. In each case , the stoichiom etry corresponds to equations ( 1 1 ) and ( 1 2 ), and the
T * + C 4 H10 -----------------v C4Hç,T* (11)
C4 HgT* -----------------» CH4 + СзН5Т ( lá)
reactions probably go as w ritten. Certainly, however, the initial hot step is not the simultaneous double displacement of two groups, for such a re a c tion would lead only to olefinic tritium with either parent m olecule, while the observed distributions include substantial contributions of alkyl-t [24]. The propylene-t from neo- C5H12 shows alm ost entirely o lefin ic-t, consistent with the expectation of CH3 CT = CH2 from the decom position of (CH3)3CT* form ed by the replacem en t of CH3. by T.
By analogy with the above reactions and with the formation of propylene-t from C3Hg, the expected m echanism for the form ation of ethylene-t from ethane would be through reactions (7) and (8 ):
T* + QiHfi -----------------> C2H5T* + H (13)
C2H5T* ---------- — > C 2H3T + H 2 . (14)
The analysis of the corresponding eth ylene-t's formed from CH3CD3 shows that C2HD2T is the largest peak, as in Fig. 4, and that ethylidene-type peaks(CH3CH: ------ » C2H4) are only a sm all fraction of the total. The observedratios for C2H3T : C2H2DT : C2HD2T : C2Dk¡T are 5 : 34 : 55 : 6 (± 2 for the larger
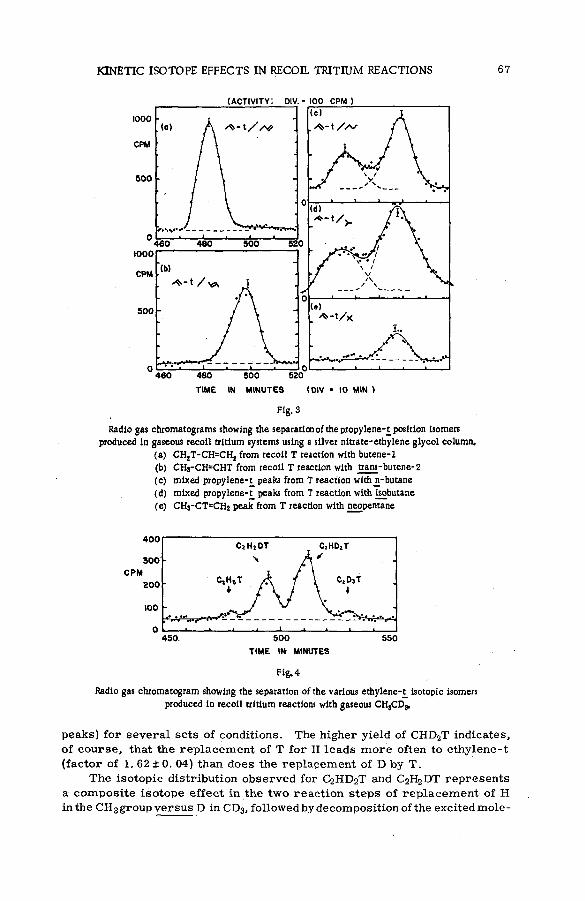
KINETIC ISOTOPE EFFECTS IN RECOIL TRITIUM REACTIONS 67
(a c t iv it y : DIV. = 100 CPM)
TIME IN MINUTES (OIV • 10 MIN >
Fig. 3
Radio gas chromatograms showing the separationofthepropylene-t position isomers
produced in gaseous reco il tr it iu m systems using a silver nitrate-ethylene g lyco l column.
(a) CH,T-CH=CHj from reco il T reaction w ith butene-1
(b) CHj-CH=CHT from reco il T reaction w ith trans-butene-2
(c) m ixed propylene-t peaks from T reaction with_n-butane
(d) m ixed propylene-t peaks from T reaction w ith isobutane
(e) CHj-CT=CH2 peak from T reaction w ith neopentane
TIME IN MINUTES
Fig. 4
Radio gas chromatogram showing the separation of the various ethylene-t isotopic isomers
produced in recoil t r it iu m reactions w ith gaseous CH jCDj,
peaks) for several sets of conditions. The higher yield of CHD2T indicates, of cou rse, that the replacem ent of T for H leads m ore often to ethylene-t (factor of 1 . 62 ± 0 . 04) than does the replacem ent of D by T.
The isotopic distribution observed for C2 HD2T and C2H2 DT represents a com posite isotope effect in the two reaction steps of replacem ent of H in the CH3group versus D in CD3, followed by decomposition of the excitedm ole-

68 E. К. C. LEE et a l.
cule by elim ination of HD. The o v e r-a ll replacem ent of H and D should show an isotope effect of about 1. 3; the enhancement to 1. 6 represents an additional facto r favouring the decomposition of CH2TCD3* over that of CH3 CD2T *. The m ost probable explanation for this resu lt is that the r e placement of H by T apparently leaves a higher excitation energy than does the replacem ent of D by T. This condition could arise from the exit of the D atoms at a slightly higher average energy than the corresponding H atoms. This observation would be consistent with the relative ease of tran sfer of energy from m ass 3 to m ass 2 versus m ass 1, but very much sm aller than ex pected on a "b illiard-ball" collision mechanism.
This m easurem ent involves only the behaviour of the upper end of the excitation range, since the total yield of ethylene-t is approxim ately 0. 05 X the observed total yield of ethane-t; most of the molecules in which T has replaced either H o r D are not sufficiently excited to undergo uni- m olecular elimination of HD. The rates of decomposition of isotopic m olecules are not identical for equivalent excitation energies, but the difference for CH2TCD3* and CH3 CD2T* should be quite sm all. One additional caution should be observed in interpretation - - the average energy of the exciting H or D atom might also be more a function of the remnant radical than of the atom itself.
R E F E R E N C E S
[1 ] Several articles, at least partial reviews, appeared in Chemical Effects of Nuclear Transformations П
IAEA, Vienna (1961). See [2 ].[2 ] A long review by R. Wolfgang, "The Hot-Atom Chemistry of Gas Phase Systems", has appeared as an
A .E .C. progress report, NYO-1957-50, and will appear as a Chapter in Vol. Ш of "Progress in Reaction
Kinetics". A short review by F. Schmidt-Bleek and F. S. Rowland has appeared in Angew. Chem.[3 ] ROOT, J. W. and ROWLAND, F. S ., J. Amer. chem. Soc. 84 (1962) 3027.[4 ] LEE, E. К. C. and ROWLAND, F. S . , J. Amer. chem. Soc. 85 (1963) 897.[5 ] TANG, T. N ., LEE, E. К. C. and ROWLAND, F. S ., J. Amer. chem. Soc.. 86(1964) 1280.[ 6] ODELL, A . , ROSENBERG. A . , FINK. R, and WOLFGANG, R .. J. chem. Phys. 40(1964 ) 3730.[7 ] BRECKENRTOGE, W . , ROOT. ]. W . and ROWLAND, F. S . , J. chem. Phys. 39 (1963) 2374.[ 8] ROOT, J..W ., Ph. D. Thesis, University of Kansas, 1964. '[9 ] LEE. J. K .. MUSGRAVE, B. and ROWLAND. F. S . , J. phys. Chem . 64 (1960) 1950.
[ 10 ] Neither of these examples is really "ideal" since the radical remaining behind is not identical for the
two competing reactions. A comparison of the abstraction of H from CH^D with D from CH¡D¡ would
involve reactions in which the remnant radical is СНгО in both cases. Strictly speaking, no ideal experiment exists since isotopic substitution in one position will always have at least small effects upon the
remainder of the molecule.[11] BROWN, W. G. and GARNETT, J. L . , Int. J. appl. Rad. Isotopes 5JJL959) 114.[12] CROSS, R.J. Jr. and WOLFGANG, R ., J. chem. Phys. 35_( 1961) 2002.[13] JURGELEIT, a C . and WOLFGANG, R., J. Amer. chem. Soc. 85(1963) 1057.[14] LEE, E. K.C. and ROWLAND, F .S ., J. Amer. chem. Soc. 85(1963) 2907.[15] LEE, E. K .C ., MILLER, G. and ROWLAND, F. S ., J. Amer. chem. Soc. (in press).[16] ROOT. J. W. and ROWLAND, F .S .. J. chem. Phys. 38(1962) 2030.[17] ROOT, J.W ., LEE, E. K.C. and ROWLAND, F .S ., Science 143_( 1964) 676.[18] LEE, E. K.C . and ROWLAND, F. S ., Analyt. Chem. 36(1964) 2181. See also the earlier reports by
CVETANOVIC, R. et a l . . Cañad. J. Chem.. 41(1963) 2095 and DUBRIN, J. et al. , J. Amer. chem.Soc. 86(1964) 959.
[19] LEE, J.K. et a l. , Analyt. Chem. 34 (1962) 741.

KINETIC ISOTOPE EFFECTS IN RECOIL TRITIUM REACTIONS 69
[20] URCH, D. and WOLFGANG, R.. J. Amer. chem. Soc. 83J1961) 2982.[21] ODUM, R. and WOLFGANG, R., J. Amer. chem. Soc. 85 (1963) 1050.[22] TANG. Y. N ., Ph. D. Thesis, University of Kansas, 1964.[23] The statistical errors do not permit the conclusion that the isotope effect varies with mole fraction.
More accurate experiments are needed to check the apparent trend indicated by the data,[24] LEE, E. K .C . , TANG, Y . N. and ROWLAND, F. S .. Amer, chem, Soc. 86 (1964) 5038.


QUANTITATIVE STUDIES OF THE REACTIONS' OF HOT TRITIUM ATOMS WITH HYDROCARBONS
AND HYDROCARBON MIXTURES
D.S.URCH AND M.J. WELCH DEPARTMENT OF CHEMISTRY, QUEEN MARY COLLEGE. UNIVERSITY OF LONDON, LONDON, UNITED KINGDOM
Abstract — Résumé — Аннотация — Resumen
QUANTITATIVE STUDIES OF THE REACTIONS OF HOT TRITIUM ATOMS WITH HYDROCARBONS AND
HYDROCARBON MIXTURES. The kinetic theory of hot-atom reactions is expanded to cover the general system
of two reactive species and an inert moderator. From results with a single reactant and moderator, two reactants without moderator, and two reactants with moderator, values of, or ratios of, the reactivity integral I and the average logarithmic energy loss per collision a can be calculated.
New results are presented from the reaction of hot tritium produced by the He3(n, p)H* reaction withthe following systems (a ll contain oxygen scavenger):
. ethane with helium moderatorbutane with helium moderator neopentane with helium moderator ’ethane and butane with and without helium moderator ethane and neopentane with and without helium moderator
Values of a and I for the three reactants are calculated from the three types of system and the agreement between the results is discussed in the terms of the accuracy of the model. The results obtained from the d ifferent systems agree to within the limits of the experimental error, and this shows that the kinetic theory model
can be applied to hot-atom systems with some certainty and that the assumptions made when considering mixtures are reasonable ones.
The ratios of the reactivity of the hydrocarbons in mixtures with regard to substitution reactions are not the same as the ratios of the number of hydrogen atoms in the hydrocarbons. The values obtained for theseratios are explained in terms of the values of I and S (the collision cross-section) for the hydrocarbons;
ETUDES QUANTITATIVES DES RÉACTIONS DES ATOMES CHAUDS DE TRITIUM AVEC LES HYDROCARBURES ET LES MÉLANGES D'HYDROCARBURES. Les auteurs étendent la théorie cinétique des réactions
des atomes chauds au système général formé par deux espèces réactives et un ralentisseur inerte. A partir des résultats obtenus au moyen d'un seul corps en réaction et d‘un ralentisseur, de deux corps en réaction sansralentisseur et de deux corps en réaction avec ralentisseur, il est possible de calculer les valeurs ou les rapportsde l'intégrale de réactivíté I, et de la perte d ’énergie logarithmique moyenne par collision a.
Les auteurs présentent les résultats obtenus en faisant réagir le tritium chaud produit par la réaction
3H e (n ,p )3H avec les systèmes suivants - qui contiennent tous de l ’oxygène agissant comme agent de balayage;
de l ’éthane avec de l ’hélium comme ralentisseur;du butane avec de l ’hélium comme ralentisseur;du tétraméthylméthane avec de l ’hélium comme ralentisseur;de l'éthane et du butane avec et sans hélium comme ralentisseur;de l ’éthane et du tétraméthylméthane avec et sans hélium comme ralentisseur.
Les auteurs calculent les valeurs de a et I obtenues pour les trois corps en réaction d’après ces trois types
de système, et ils discutent la concordance entre les résultats en fonction de l'exactitude du modèle. Les
résultats obtenus à l'aide des différents systèmes concordent aux erreurs d'expérimentation près, ce qui démontre
71

72 D .S. URCH and M .J. WELCH
la possibilité d'appliquer avec une certaine certitude le modèle de la théorie cinétique aux systèmes d'atomes
chauds et le bien-fondé des hypothèses admises pour l'étude des mélanges.
Les rapports exprimant la réactivité des hydrocarbures contenus dans des mélanges, en ce qui concerne les réactions de substitution, ne sont pas les mêmes que les rapports du nombre d'atomes d'hydrogène dans les
hydrocarbures. Les valeurs obtenues pour ces rapports sont analysées en fonction des valeurs de l et S (section
efficace de choc) pour les hydrocarbures.
К О Л И Ч Е С Т В Е Н Н О Е И С С Л Е Д О В А Н И Е РЕ А К Ц И Й Г О Р Я Ч И Х А Т О М О В Т Р И Т И Я С Г И Д Р О У Г Л Е Р О Д А М И И С М Е С Я М И Г И Д Р О У Г Л Е Р О Д О В . К и н ети ч еск ая теори я реакций горячи х ато м о в р асп р о стр ан яется на общую си ст ем у д в у х реакционно способных видов и инертный з а м е д л и т е л ь . Из р е з у л ь т а т о в с одним реагирую щ им в ещ ес тв о м и зам е д л и те л ем , двум я реагирующ ими в ещ ес тв а м и б е з зам ед л и тел я и двум я реагирующ ими вещ ествам и с зам ед л и тел ем можно вы чи сл и ть величины или коэфф ициенты и н тегр ал а I реакти вн ости и среднюю л о га риф м ическую потери энерги и на стол кн овен и е а . .
П р ед с та в л я ю тс я н о вы е р е з у л ь т а т ы , п ол ученн ы е в р е з у л ь т а т е реакции го р я ч е го трития в реакции Н е3 ( п .р . ) Н3 со следую щ ими си ст ем а м и (в с е сод ер ж ат акцептор радикалов ки сл орода):
этан с ге л и е вы м за м е д л и те л е м ;бутан с ге л и е вы м за м е д л и те л ем :т е т р а м ет и л м ет а н с ге л и е вы м за м е д л и те л ем ;э тан и б утан с ге л и е вы м зам е д л и те л ем и б е з н е го ;э тан и т е т р а м ет и л м ет а н с ге л и е вы м зам ед л и тел ем и б е з н е г о .
В ы ч и сл яю тся величины а и I для т р е х реагирую щ их в ещ ес тв из этих т р е х типов си стем ы и о б с у ж д а е т ся с о гл а с и е м еж ду р е зу л ь т а т а м и с точки зрени я точн ости м одел и . Р е з у л ь т а т ы , п олученны е из р азл и чн ы х с и с т е м , со гл а с у ю т ся в р ам ках пределов эксперим ентальной ошибки, и э то п о к а з ы в а е т , ч т о м одел ь к и н ети ческой теории м ож ет п р и м еняться к си ст е м а м гор я чи х ато м о в с некоторой оп р едел ен н остью и что сдел ан н ы е предположения при р ассм отр ени и с м е сей я вл я ю тся р азу м н ы м и .
К оэф ф ициенты р е а к т и в н о ст и ги д р о угл ер о д о в в с м е с я х в отнош ении реакций зам ещ ени я не я в л я ю т ся т ем и же с а м ы м и , к а к соо тн ош ен и я ч и сл а а т о м о в во д о р о д а в ги д р о у г л е р о д а х . П ол учен н ы е вели чи ны для э т и х к оэф ф и ц и ен тов о б ъ я сн я ю т ся вел и чи нам и I и S (п оп ер ечн ое се ч е н и е ст о л к н о в ен и я ) для ги д р о у г л е р о д о в .
ESTUDIO CUANTITATIVO DE LAS REACCIONES DE ATOMOS DE TRITIO CALIENTES CON HIDROCARBUROS Y MEZCLAS DE HIDROCARBUROS. Los autores amplían la teoría cinética de las reacciones de
los átomos calientes a fin de abarcar el sistema general de dos especies reactivas y un moderador inerte. A
partir de los resultados correspondientes a una sola especie reactiva y moderador, dos especies reactivas sin
moderador y dos especies reactivas con moderador, se pueden calcular valores y razones de la integral de reactividad I, y del valor medio del decremento logarítmico de energía por colisión a.
Se presentan nuevos resultados correspondientes a la reacción de tritio caliente, producido por la reacción
3H e(n ,p )3H, con los siguientes sistemas (todos los cuales contienen oxigeno como depurador);
etano con helio como moderador,butano con helio como moderador,neopentano con helio como moderador,etano y butano con y sin helio como moderador,
etano y neopentano con y sin helio como moderador..
Los autores calculan los valores de a y de I para las tres especies reactivas, a partir de los tres tipos
de sistemas, y discuten la concordancia entre los resultados, en relación con la exactitud del modelo. Los
resultados obtenidos para los diferentes sistemas son compatibles dentro de los límites del error experimental,lo que indica que el modelo de la teoría cinética puede aplicarse a los sistemas de átomos calientes con cierta
confianza y que las hipótesis adoptadas al considerar las mezclas son razonables.

REACTIONS OF HOT TRITIUM ATOMS WITH HYDROCARBONS 73
Las razones de reactividad de los hidrocarburos en mezclas, en lo que se refiere a las reacciones de sustitución, no coinciden con las razones del número de átomos de hidrógeno de los hidrocarburos. Los valores
obtenidos para dichos cocientes se explican en función de los valores de I y S (la sección eficaz de colisión)
correspondientes a los hidrocarburos.
INTRODUCTION
Hot tritium atoms produced by He3 (n, p)H3 were reacted with ethane, n-butane, neopentane and mixtures of ethane with the other two hydrocarbons. From the results, values of the reactivity integral I and the average logarithmic energy loss per collision a have been calculated.
EXPERIMENTAL
The hydrocarbons, together with about 1 cm Hg of He3 (purified using a zirconium "getter" [4]), about 2-4 cm of oxygen to act as a radical scavenger, and sometimes He4 moderator, were sealed into quartz tubes of internal diameter 2 cm and length 6±0.4 cm, at a total pressure of just less than 1 atm. The tubes were irradiated by a total neutron flux of 10i5n/cm2; this was done either in two weeks at a flux of 109n/cm2- s in the United Kingdom Atomic Energy Research Establishment (U. K. A . E. R. E. ) BEPO reactor, or in a day at a flux of low in the United Kingdom Atomic Weapons Research Establishment (U. K. A . W. R. E. ) Herald reactor. The total neutron flux was measured by inserting cobalt monitor wires with each set of samples.
The products were analysed by gas-phase chromatography and proportional flow counting [5, 6 ]. Electronic equipment of the I.D .L . "1800" series was used, and it was found that the scaler would not accept pulses of less than 3 V amplitude. The pulse spectrum of the pulses produced by tritium in the counter, and amplified by a head amplifier, was photographed using a Laben 512 channel analyser (Fig. 1). It was found that under normal
F ig . 1
Energy distribution of pulses, obtained using a Laben 512 channel pulse analyser
working conditions (with gain 500) the spectrum peak occurred when the pulses had 15 V amplitude and the maximum pulse height produced was over 50 V. Under these conditions counting efficiency was (82±3)%.
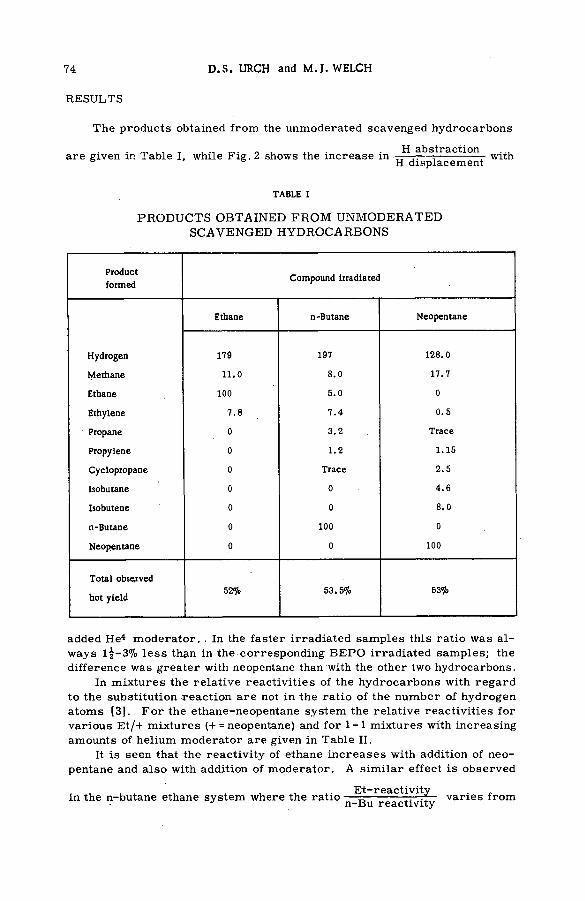
74 D .S. URCH and M .J. WELCH
The products obtained from the unmoderated scavenged hydrocarbons
• • гг, !_■, t t-, т-r о v. il. ■ ■ H abstractionare given m Table I, while Fig. 2 shows the increase in — ;--r with“ H displacement
RESULTS
TABLE I
PRODUCTS OBTAINED FROM UNMODERATED SCAVENGED HYDROCARBONS
Product
form edCompound irradiated
Ethane n-Butane Neopentane
Hydrogen 179 197 128.0
M ethane 1 1 . 0 8 . 0 17.7
Ethane 1 0 0 5 .0 0
Ethylene 7 .8 7 .4 0 .5
Propane 0 - 3 .2 . T race
Propylene 0 1 . 2 1.15
Cyclopropane 0 T race 2 .5
Isobutane 0 0 4 .6
Isobutene 0 0 8 . 0
n-Butane 0 1 0 0 0 .Neopentane 0 0 1 0 0
T o ta l obseived
hot y ie ld52% 53.5% 53%
added He4 moderator. . In the faster irradiated samples this ratio was always 1^-3% less th*m in the corresponding BEPO irradiated samples; the difference was greater with neopentane than with the other two hydrocarbons.
In mixtures the relative reactivities of the hydrocarbons with regard to the substitution reaction are not in the ratio of the number of hydrogen atoms [3]. For the ethane-neopentane system the relative reactivities for various Et/+ mixtures (+ = neopentane) and for 1-1 mixtures with increasing amounts of helium moderator are given in Table II.
It is seen that the reactivity of ethane increases with addition of neopentane and also with addition of moderator. A similar effect is observed
Et-reactivity . „in the n-butane ethane system where the ratio— =--------- -r.— varies from■ J n-Bu reactivity
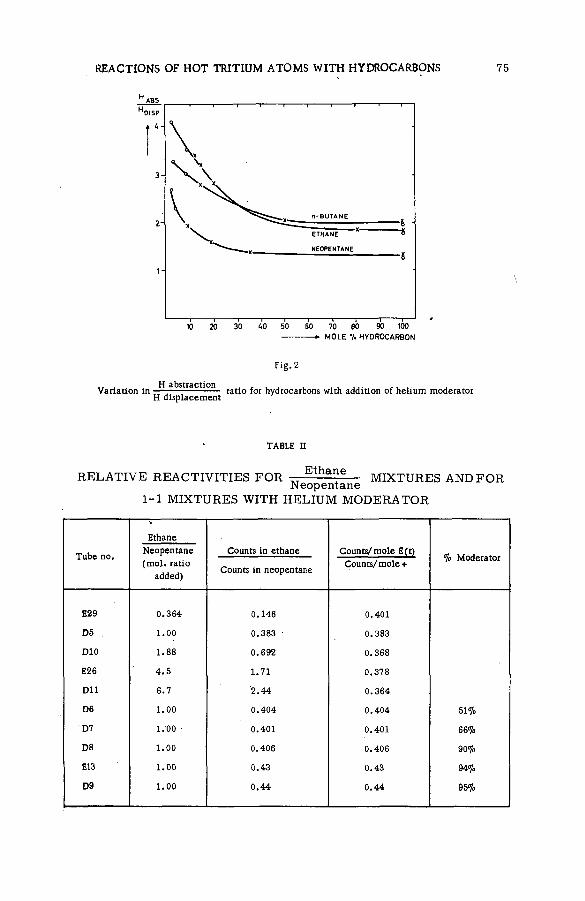
REACTIONS OF HOT TRITIUM ATOMS WITH HYDROCARBONS 75
------- ► MOLE •/. HYDROCARBON
F ig . 2
V ariation in t j ? e stracIion r a t ¡ 0 for hydrocarbons w ith addition o f helium moderator H disp lacem ent
• TABLE II
RELATIVE REACTIVITIES FOR Etha^e MIXTURES AND FOR Neopentane
1-1 MIXTURES WITH HELIUM MODERATOR
Tube no.
Ethane
Neopentane
(m o l. ratio
added)
Counts in ethane Counts/m ole E ft)% M oderator
Counts in neopentaneCounts/m ole +
E29 0.364 0.146 0.401
D5 , 1.00 0.383 0.383
DIO 1 . 8 8 0.692 0.368
E26 4 .5 1.71 0.378
D l l 6 .7 2 .44 0.364
D6 1.00 0.404 0.404 51%
D7 1.00 0.401 0.401 6 6 %
D 8 1.00 0.406 0.406 90%
E13 1.00 0.43 0.43 94%
D9 1.00 0.44 0.44 95%

76 D .S. URCH and M .J. WELCH
0.450 to 0.482 when the n-butane concentration is increased in unmoderated mixtures. In 1-1 mixtures the ratio increases from 0.475 in unmoderated to 0. 525 in highly moderated samples.
Kinetic theory of hot atom reactions
Wolfgang's application of the kinetic theory [2] gives for a single reactant with an inert moderator the following relation:
1 а react ^ a mod ^ react
ln (l-P ) I I freact
where P = probability of a particle undergoing a hot reaction.In the following fi = relative probability of collision with component i:
f i - X iSi X = mole fraction~ EjXjSj S = collision cross section.
1 ^ f react ^ r e a c tA plot of against —j------- has intercept — ----- and gradientl n i l - i r 1) I react 1
а , and also a plot of against ^ has intercept Ij .
On expanding the theory to two reactants y and z and an inert moderator we arrive at the two relations:
p ay fz az fl-(^y ^mod- + T - - 7 - + ------------- ------------- ------Pyln (l-P ) Iy fy Iy r fy Iy
_p_____ + _ aj_ | [1 - (fy + fz )1 mod
P .ln (l-P ) Iz fz Iz fz Iz
fzFor a series of samples with — =Rtv. 1У
P 1 . „ . a mod ^mod— [a y + R az)+ -Pyln (l-P ) ' I y V“ y ly fy •
Therefore a plot of - 1 against — should give a straight lineГ у 1 П I J X y
•yof gradient —:— and intercept (orv + R a z).
i y
P fFor non-moderated mixtures a plot of ~ p in (l -P ) aSa -nst should
cx &give a straight line with intercept and gradient -y2- . There are, ofl y l y
course, similar plots derivable from the P z relation.
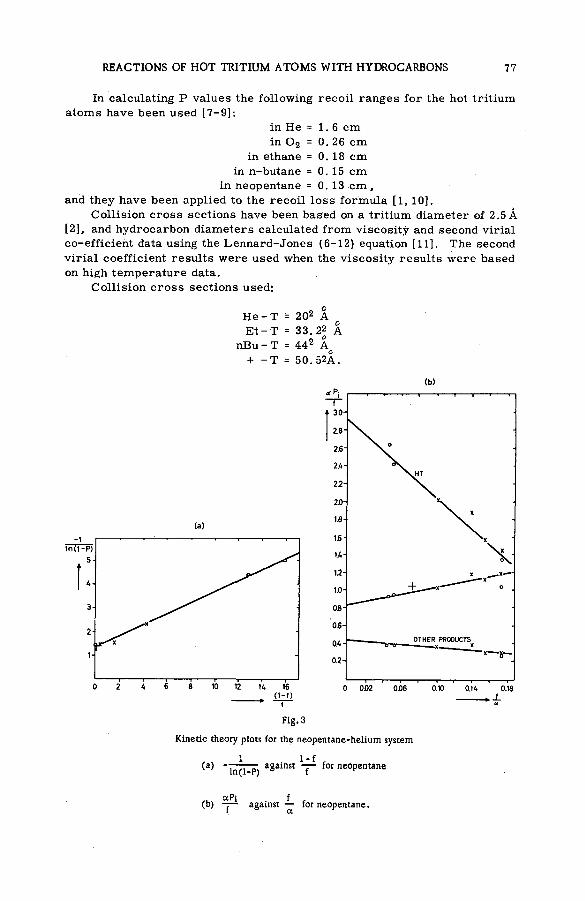
REACTIONS OF HOT TRITIUM ATOMS WITH HYDROCARBONS 77
In calculating P values the following recoil ranges for the hot tritium atoms have been used [7-9]:
in He =1 .6 cm in O2 = 0 . 26 cm
in ethane = 0 . 18 cm in n-butane = 0. 15 cm
in neopentane = 0. 13 cm, and they have been applied to the recoil loss formula [ 1 , 1 0 ].
Collision cross sections have been based on a tritium diameter of 2.5À [ 2 ], and hydrocarbon diameters calculated from viscosity and second virial co-efficient data using the Lennard-Jones (6-12) equation [11]. The second virial coefficient results were used when the viscosity results were based on high temperature data.
Collision cross sections used:
H e -T = 202 A.E t -T = 33. 22 A
n B u -T = 442 Ao + - T = 50. 52A.
(Ы
F ig .3
K in etic theory plots for the neopentane-helium system
, , 1 . 1 - f ,Tn(l-P) a8alnst ~ ‘or n“pentane
. . . ocPj . f(b ) - — against — for neopentane.
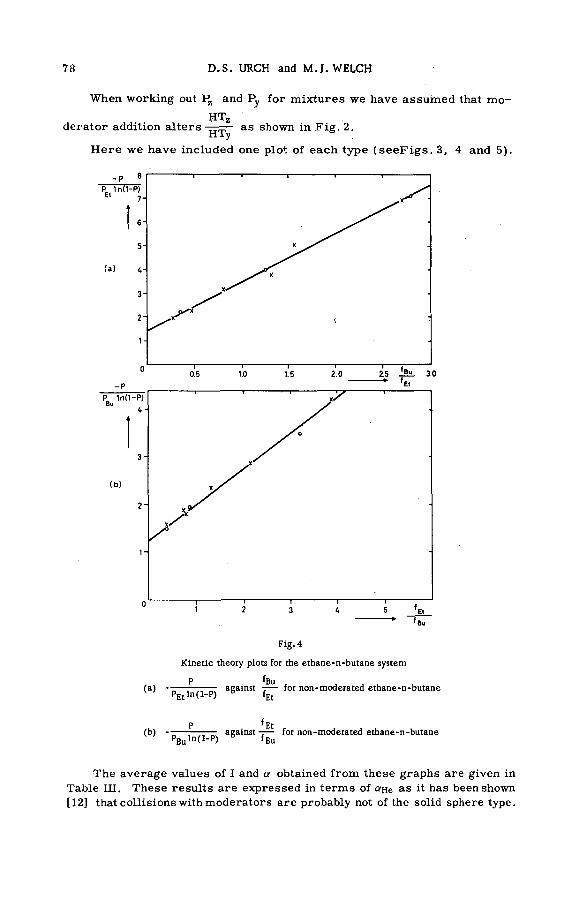
When working out FJ, and Py for mixtures we have assumed that mo- HTZ
derator addition alters TT_ as shown in Fig. 2.l l l y .
Here we have included one plot of each type (seeF igs. 3, 4 and 5).
78 D .S. URCH and M .J. WELCH
F ig . 4
K in e tic th eory plots for th e e th a n e -n -b u ta n e system
( a ) — :— :----- a g a in st - — for n o n -m o d e ra te d e th a n e -n -b u ta n e' ' PE t l n ( l - P ) fEt
(b ) ----------------- ag ain st —— for n o n -m o d e ra te d e th a n e -n -b u ta n eP B u ln (l -P ) Б f Bu
The average values of I and a obtained from these graphs are given in Table III. These results are expressed in terms of <*не as it has been shown [ 1 2 ] that collisions with moderators are probably not of the solid sphere type.

REACTIONS OF HOT TRITIUM ATOMS WITH HYDROCARBONS 79
(a )
(b)
F i g .5
K in e t ic th eory plots for th e e th a n e -n e o p e n ta n e -h e liu m system
P fm od( a ) - - — ^ a g a in st —------ for m od erated e th a n e -n e o p e n ta n e
^mod(b ) - - — — — a g a in st - — for m od erated e th a n e -n e o p e n ta n e .
DISCUSSION
The results from various plots agree to within 5% for I values and 10% for a values. This is the sort of error we can expect as there are many assumptions made in the theory and we are using values of recoil ranges and collision cross sections that are not accurately known.
f Pi aThe negative gradient in the — against —— plot for the substitution
reaction of neopentane is probably due to the difference between the samples irradiated for different times; the graph could be made up of two straight lines with gradient ~0. A finite L value in the expansion [2]

80 D .S. URCH and M .J. WELCH
p i = — I i - - V K i + - ^ 41 a 1 a2 1 cr3 1
would also give the plot a decreased gradient (i. e. К value).
f P¡ a j2For the — against - j— plot to be straight lines EKi should equal
for our plots we have found this relation is true to ± 10%.
TABLE III
AVERAGE VALUES OF I AND a IN TERMS OF aH e
H ydrocarbon I T o ta l a ГН Т I H ydrocarbonI M inor
products
E th an e ( 2 . 7 ± 0 .1 8 ) a H e ( 3 . 6 4 ¿ 0 .4 5 ) a H e 2 , l a H e 0 . 55<xH e 0 . 1 2 a H e
n -B u ta n e ( 3 . 8 8 ± 0 . 1 5 ) < * H e ( 5 . 0 5 i 0 . 4 ) o t H e 2 . 7 8 < ,He 0 Л 9 “ Н е 0 . 2 8 a He
N eop en tan e ( 4 . 2 2 ± 0 . 2 0 ) a H e ( 5 . 8 i 0 . 5 ) a He 2 . 9 3 a H e 0 .8 3 S o tHe 0 . 4 3 a H e
In a hydrocarbon mixture at high moderator concentration the ratio of reactivities should depend only on the reactivity integral and on the collision cross Section:
Reactivity of A _£a_^_£a Reactivity of B Ig X SB '
For ethane and n-butane the ratio of reactivities tends to 0.525 and
rEt v *gEt = O' 538; for ethane and neopentane the reactivity ratio is 0. 44 and Ib u x b Bu
IEt V o Et = 0.437.1+ X b +
The fact that the theory gives results from three types of system that agree within the limits of experimental error, and predicts the ratios of reactivities almost exactly, leads us to assume that the kinetic theory can be applied to hot atom systems with some certainty and that the assumption made [2] when considering mixtures is a reasonable one. The disagreement between our results and those of ROSENBERG and WOLFGANG [13], can probably be explained by different methods of neutron flux determination and the use of different tritium ranges and hydrocarbon cross sections.
The variation in reactivities of ethane and the higher hydrocarbons (Table II) may be caused by slightly different reaction probability curves. The ethane is more reactive when the ethane content of non-moderated mix-

REACTIONS OF HOT TRITIUM ATOMS WITH HYDROCARBONS 81
tures is decreased and when moderator is added. Both of these effects lead to less collisions in the high energy range, and therefore the trends can be explained if the heavier hydrocarbons are more reactive at'high energies.
This trend is contrary to the results obtained in similar liquid systems [16, 17], where the mixtures are non-ideal and the radiochemical yield is a linear function of concentration.
T, . .. • H abstraction . .. ;Variation m ——-— ;------------ ratiosH displacement
It has been suggested that for a single non-moderated scavenged hydro. H abstraction n .carbon L14J — ------------ =— —— г— ----—— -----—— — where n is theH displacement nj( 1 -ÇI) + пг(1 -£2)2 + пз( 1 — íí)3
number of hydrogen atoms; nj,n2 and П3 the numbers of primary, secondary and tertiary hydrogens; and £2 = 0.45. One would expect ethane and neo
* . , . . . H abstraction .. -, .pentane to have similar — :------------ ratios as both contain only primaryr H displacement r ■ •!hydrogen; the ratios however differ by 33%. This cannot be explained purely
by an inertial effect [181 as methane has an traction ratio less thanJ H displacement
neopentane. An inertial effect would give a variation proportional to mass;considering methane, ethane and neopentane, the ratios are 1, 1.8 and 1.3respectively.
If the hot atom does not react until it has slowed down to a velocity similar to that of a gaseous hydrocarbon molecule the difference can be explained by differences in molecular symmetry. In symmetrical neopentane and methane translational movement through the gas is accompanied by rotation. If the velocity of the hot atom and hydrocarbon are comparable, most collisions will be on the front face of the hydrocarbon. These collisions would normally be along the -C -H bond and lead to abstraction, but because of the accompanying rotation present in the more symmetrical molecules these collisions occur at angles to the bond and lead to substitution. If the theory is true, raising the energy of the system either by heat or by using greater
neutron fluxes should reduce the H abstraction ratj0 Qur results andH displacement
those of other workers [15] seem to show this trend, and we have observed a greater effect with the symmetrical neopentane than with the other hydrocarbons studied.
A C K N O W L E D G E M E N T S
One of us (M. J. W. ) would like to thank Queen Mary College for a maintenance grant. We would also like to thank Dr.A. Ashmore and M r.T . Pritchard for discussion concerning counter efficiencies and recoil ranges; the staffs of the two reactors used for their friendly co-operation; and Dr. A.G. Maddock for helpful comments on the preliminary draft of this paper.

82 D .S. URCH and M .J. WELCH
R E F E R E N C E S
[ 1 ] ESTRUP, P .J . and WOLFGANG, R ., J . A m er. c h e m . S o c . 82 (1 9 6 0 ) 2 6 5 5 .
[ 2 ] WOLFGANG, R . , J . ch e m . Phys. 39 (1 9 6 3 ) 2 9 8 3 .
[ 3 ] ROOT, J .W . and ROWLAND, F .S . , J . A m er. ch em . S o c . 84 (1 9 6 2 ) 3 0 2 7 . ■
[ 4 ] PRITCHARD, J . , URCH, D .S . and WELCH, M . J . , J . inorg. n u cl. C hem . 2 6 (1 9 6 4 ) 1121.
[ 5 ] WOLFGANG, R. and M A CK AY. C . F . , Nucleonics 16 (1 9 5 8 ) 6 9 .
[ 6 ] WOLFGANG, R. and ROWLAND, F .S . A nalyt. C h em . 30 (1 9 5 8 ) 9 0 3 .
[ 7 ] WHALING, WARD, Handbuch der Physik 3 4 , 2 0 8 -9 .
[ 8 ] ZAIMIDOROGA, O .A . e t a l . , Dubna ( U .S .S .R .) Reports (1 9 6 3 ) .
[ 9 ] PARK, J .T . and ZIMMERMAN, E . J . , USAEC T ID -15127 (1 9 6 2 )
[ 1 0 ] ESTRUP, P . J . , P h .D . Thesis, Y a le U niversity, 1 9 5 9 .[ U ] HIRZCHFELDER, J . O . , CURTISS, C .F . and BIRD, R .B ., M olecular Theory of Gases and Liquids, W iley,
New York (1 9 5 4 ) 1 1 1 0 .
[ 1 2 ] ESTRUP, P . J . , J . c h e m . Phys. 41 (1 9 6 4 ) 1 6 4 .
[ 1 3 ] ROSENBERG, H . and WOLFGANG. R . , J . ch em . Phys. 41 2 1 5 9 -6 7 (1 9 6 4 ) .
[ 1 4 ] HENCHMAN, М . , URCH, D .S . and WOLFGANG, R . , C añ ad . J . C h em . 3 8 ( 1 9 6 0 ) 1722.
[ 1 5 ] EL-SA YED , M .A . , ESTRUP, P .J . and WOLFGANG, R . . J . ch em . Phys. 62 (1 9 5 8 ) 1 3 5 6 .
[ 1 6 ] NESMEYANOV, A n .N . and FILATOV, E . S . , Radiohim ija 3 ( 1 9 6 1 ) 6 1 4 .
[ 1 7 ] AVDONINA, E .N . and NESMEYONOV, A n .N ., Radiohim ija 5 (1 9 6 3 ) 5 1 4 .
[ 1 8 ] ODUM, R. A . and WOLFGANG, R . . J . A m er. c h e m . S o c . 8 5 ( 1 9 6 3 ) 1 0 5 0 .

REACTIONS OF HOT TRITIUM ATOMS WITH HYDROCARBONS 83
D I S C U S S I O N
(on the foregoing two papers)
A. GORDUS: I gather that, in the work described in the paper presented by Mr. Lee, the tritiated isotopic ethanes were not analysed because of the lack of a suitable gas chromatographic separation procedure. I might note that we have been able to separate cm3 -atm amounts of isotopic methanes and ethanes in about 1 - 2 h. .
In connection with the use of neutron-moderation theory for the analysis of hot-atom data, I would suggest that it is inadvisable to place too much reliance on straight-line plots and internal correlations to prove the validity of the theory. Unless proper caution is exercised, one is liable to be unjustifiably confident. Recently we synthesized hot-atom data using a computer. Mixtures of a reactive and a non-reactive molecule were considered to undergo asymmetric scattering in collisions with the hot atoms. Data for the yields of any desired number of products as well as the total hot yield were calculated as a function of the mole fraction of the reactive gas. A typical set of data with a total yield of 73% for the pure system resulted in a perfectly straight line when plotted as -l/ ln (l-P ) versus (l-fi)/ fi. From the slope and intercept, the value of a^ja^ was calculated as 2.77, compared to the value of 3.72 for the same ratio that had been used in synthesizing the data. Thus use of the theory would result in a 34% error in the a ratio. From the plot, l/a2 was found to be equal to 3.70. Plots of Pia/fj versus i l /а were also straight lines for the individual yields and Lli/az = 3.72, which would suggest, incorrectly, that the apparent correlation between I values from the two plots proves the validity of using the theory. Other computer data show, in some cases, much larger discrepancies between a i j a % derived from such plots and aja<¿ used in creating the data, and in these cases the plots of the data were also linear and Eli = 1 .
S. WEXLER: How do your findings reflect on the validity of the Wolfgang-Estrup model? .
A. GORDUS: Even if eventually extremely accurate hot-atom experimental data are obtained, and even if these data should result in perfect straight-line plots and perfect internal correlation in terms of the Estrup- Wolfgang formulation, I would still be reluctant to accept, with 100% confidence, the various values of I and a derived from these plots since these derived values may not represent I and a defined in the original formulation.
F.S. ROWLAND: I would like to comment on Mr. Welch's paper, reply to Professor Gordus's comment, and then answer the question raised by Dr. Wexler.
Dr. Edward Lee and I published a figure in our paper on cyclobutane (J. Amer. chem. Soc. 85 (1963) 897) that showed that the yield of cyclo-CiH'/T rose from about 0.46 in arbitrary units at a pressure of 5 cm to 0.60 at a pressure of 80 cm, and to 0.82 in liquid-phase experiments. The accompanying figure shows a similar increase in yield with increasing gas pressure, and with the change from gas to liquid for C2H4TCI from recoil-tritium reactions with C2H5C1.
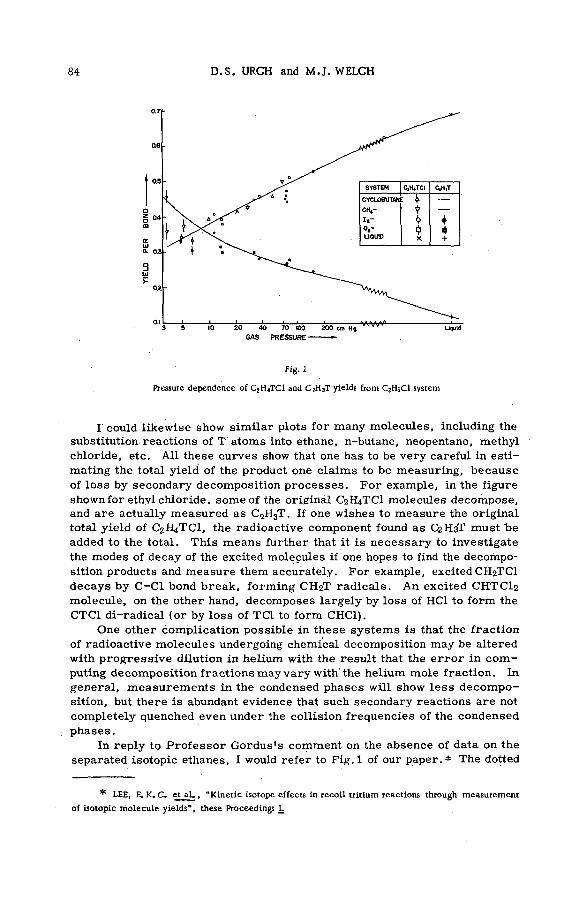
84 D .S. URCH and M .J. WELCH
Fig. 1Pressure dependence of C2 ЩТС1 and C 2H3T yields from CjHsCl system
I could likewise show similar plots for many molecules, including the substitution reactions of T atoms into ethane, n-butane, neopentane, methyl chloride, etc. All these curves show that one has to be very careful in estimating the total yield of the product one claims to be measuring, because of loss by secondary decomposition processes. For example, in the figure shown for ethyl chloride, some of the original C2H4TCI molecules decompose, and are actually measured as C2HgT. If one wishes to measure the original total yield of C2H4TCI, the radioactive component found as C2H3T must be added to the total. This means further that it is necessary to investigate the modes of decay of the excited molecules if one hopes to find the decomposition products and measure them accurately. For example, excited CH2TCI decays by C -C l bond break, forming CH2T radicals. An excited CHTCI2 molecule, on the other hand, decomposes largely by loss of HC1 to form the CTC1 di-radical (or by loss of TCI to form CHC1).
One other complication possible in these systems is that the fraction of radioactive molecules undergoing chemical decomposition may be altered with progressive dilution in helium with the result that the error in computing decomposition fractions may vary with the helium mole fraction. In general, measurements in the condensed phases will show less decomposition, but there is abundant evidence that such secondary reactions are not completely quenched even under the collision frequencies of the condensed phases.
In reply to Professor Gordus's comment on the absence of data on the separated isotopic ethanes, I would refer to F ig .l of our paper.* The dotted
* LEE, E. К . C . e t a i . , "K in e tic isotop e e ffe cts in r e c o il tritiu m re a ctio n s through m e a su re m e n t
o f iso to p ic m o le cu le y ie ld s" , th ese Proceedings L

REACTIONS OF HOT TRITIUM ATOMS WITH HYDROCARBONS 85
lines show the separation obtained with CH4 and CDi, also in cm3-atm amounts - which differ by four H versus fourD atoms. The solid lines show the separation of CH3T and CD3 T - differing by three H versus three D atoms. The separations he reports are of С2 Нб, CH3CD3 andC2D6, which also differ by three H versus D atoms - the same separations can be readily carried out on the columns used in this work. The ability to separate these species is of course not related to the problem of the ethanes formed in our experiment, since the species involved in this case are CH2TCD3 and CH3CD2T, or CHTD2
and CH2DT, which differ by only one H versus D pair, and are therefore much more difficult to separate. No one has yet reported such a separation with sufficient resolution to permit accurate measurements of relative ratios in experiments such as ours that involve cm3 -atm amounts of gas.
Finally, I should like to answer Dr. Wexler's question about the general validity of the Estrup-Wolfgang theory. I prefer to do this in general terms rather than to consider particular methods of application. If one measures the total-product yields from recoil tritium reactions with a particular molecule in its pure form, and then in the presence of another molecule, for example helium or another hydrocarbon, it is possible to obtain data that will give numerical values for two parameters for each molecule. One of these parameters is related to the reactivity integral. I, in Estrup- Wolfgang terminology, and the other is related to the values of a, the logarithmic energy decrement for non-reacting collisions between the recoil tritium atom and the molecule. Such experiments will produce measurements of two valid parameters in connection with these systems. The discussions and the arguments about such measurements are concerned with the accuracy with which the measured parameters correspond to the definitions given for them in the Estrup-Wolfgang approach. It is quite certain that such parameters can be obtained, that their relative values can be measured with reasonable accuracy and are of significance for the circumstances in which they are measured, and that there is as yet no inconsistency with the general approach used. The technique of moderator experiments is a very productive one in recoil tritium chemistry at the present time, and should soon make it possible to check the validity of parameters obtained for one molecule in one set of experiments against others involving the same molecule. As we shall hear later, moderator experiments also offer considerable promise in studies of the reactions of F18 and other re coil atoms.


РЕАКЦИЯ ГОРЯЧИХ АТОМОВ ВОДОРОДА С ЭТИЛЕНОМ. РОЛЬ ВОЗБУЖДЕННЫХ ЭТИЛЬНЫХ РАДИКАЛОВ КАК ПРОМЕЖУТОЧНЫХ ПРОДУКТОВ
Б .Г . ДЗАНТИЕВ и А.П. ШВЕДЧИКОВ ИНСТИТУТ ХИМИЧЕСКОЙ ФИЗИКИ АН СССР
СССР
Abstract — Résumé — Аннотация — Resumen
REACTIONS OF HOT HYDROGEN ATOMS WITH ETHYLENE. THE ROLE OF EXCITED ETHYL RADICALS
AS INTERMEDIATE PRODUCTS. It is well known that the nuclear reactions L i6(n , a) T and He3(n , p) T result
in the form ation o f hot atom s o f rad io activ e hydrogen. The ch a ra c te ris tics o f the c h e m ic a l consequences o f
nuclear transformations are largely determined by the high ch em ical activ ity of such atom s. However, hydrogen
atom s ca n play an essential role not only in nuclear chem istry but also in o th er branches o f high-energy ch em istry , such as radiolysis and photolysis.
In th e present re se a rch we attem p ted to m ak e a co m p a ra tiv e study o f th e behaviour o f hot hydrogen
atom s obtained in various w ays: by the Li6 (n , a ) T re a c tio n (re co il energy = 2 .7 M eV ); by radiolysis ( E ~ a few
e V ); by photolysis ( E ~ 1 - 1 ,5 eV ; hot hydrogen is obtained from HI by photolysis a t \ = 2 5 3 7 Â ) .
In order to obtain tritiu m atom s, we used crystals and film s of L i2C o s, L ieF , L i6OH, .Li6B 0 2* 8H 2 as 1
targ ets . Gaseous e th y len e (P = 5 - 1 0 a tm ) and its m ixtures with am m o n ia , h elium and inhibitors, w ere sub
je c te d to rad iatio n . R adiation was carried out on an IRT-100Q re a c to r w ith a th e rm a l neutron flu x o f 10U -
1012 a / c m 2 • s. T ritiu m -la b e lle d com pounds and products o f radiation ch em istry reactio n s w ere identified
by gas chrom ato graph y involving th e use o f tw o m onitoring units co n n ected in series (a k a th aro m eter and a G -M flow coun ter).
It is shown that the spectrum of labelled and unlabelled products in irradiated ethylene is very d istinct.
The m ain labelled products are C 2 H2 , C 2 H6 , C4 H10 and hydrocarbons > C4 ; the H2 yield is sm all. The m ain
unlabelled products are H T, C 2 H3 T and C4 H9 T ; th e yield of C 2H 5 T and esp ecially of C 2HT is insignificant.
Th e lab elled -co m p o u n d yield s a re depen dent on the introd uction o f additives (a m m o n ia , h e liu m , io d in e)
in to the e th y le n e , on th e ty p e o f l i th iu m -c o n ta in in g com poun d and on th e s tru c tu re o f th e ta r g e t .
Lowering the tem p eratu re from 5 0 * С to - 7 8 * С has little effe ct on the distribution of lab elled products;
how ever, in the transition to h eavy e th y len e ( - 1 9 6 eC ) a phase e ffe c t is observed; the HT and C 4H *T yield
decreases and the a c tiv ity of the parent com pound C 2 H3 T increases, ’In th e radiolysis o f C 2 H4 and its m ixtures w ith am m onia the ra tio C 2 H6 / C 4 H 10 is eq u al to 1 .0 ± 0 .3 ,
w hich does not agree with th e standard schem e for disproportionation and recom bin ation reaction s o f th erm al ethylene radicals (C 2 H6 / C 4 H10 = 0 .2 ± 0 .1 ).
On the basis o f d ata on the e ffe ct of heliu m addition and tem p eratu re on C 2 H6 and C4 Hi0 , yield in the
radiolysis of C 2 H 4, and in the photolysis of m ixtures o f C2 H4 and HI, we were able to g et an idea of the for
m ation of e x c ite d ethylene rad icals of C 2 H * These are obtained as a result of the addition of hot hydrogen
atom s (generated ra d io ly tica lly or p h o to ly tically ) to e th ylen e: H * + C 2 H4 = C 2 H ?. It is shown th at the C2 H f
rad ical easily enters into the stripping reaction C 2 H^ + R H =C 2 H6 + R, which accounts for the "e x c e s s ” quantity of ethane formed in the radiolysis of ethylene.
REACTIONS DES ATOMES CHAUDS D'HYDROGÈNE AVEC L ’ÉTHYLÈNE. LE RÔLE DES RADICAUX
ETH YLE EXCITÉS COMME PRODUITS INTERMÉDIAIRES. On sait qu’à la suite des réaction s n ucléaires
6L i ( n ,a ) T e t 3H e (n ,p )T se form ent des atom es chauds de tritiu m . La forte ré a c tiv ité chim ique de ces
atom es d é term in e à m ain ts égards les p articu larités chim iques des effets dus au x transform ations n u cléaires .
O r, les atom es chauds de tritium peuvent jouer un rôle im p ortant, non seulem ent en ch im ie n u clé a ire , m ais
aussi dans d 'autres dom aines de la ch im ie des hautes énergies, notam m ent dans la radiolyse, la photolyse, e tc .
Dans le m ém o ire , on essaie de com parer le com portem ent des atom es chauds de tritium obtenus par les
moyens suivants: ré a c tio n «Li(n, a ) T ( E sep = 2 ,7 M eV ); radiolyse (E ~ plusieurs e V ); photolyse (E ~ 1 à
1 ,5 eV ; le tritiu m se form e par photolyse à partir de H1 pour \ = 2 5 3 7 . ) .
87

88 Б.Г. ДЗАНТИЕВ и А.П. ШВЕДЧИКОВ
Pour obtenir des atom es de tritiu m , on a .u tilisé com m e cibles des cristaux et des films de Li2 C 0 3 , 6 LiF,
6LiOH e t ^iBCfe * 8 H2 O. On a irradié de l'é th y lèn e en phase gazeuse (sous une pression p de 5 à 10 atm ) et
des m élanges de c e corps a v e c l 'a m m o n ia c , l'h éliu m et des inhibiteurs. L 'irradiation a eu lieu à l'intérieur
d'un ré a cte u r du type IR T -1 0 0 0 dans un flux de neutrons therm iques de 1011 à Ю 12 n / c m 2* s. Les com posés
marqués au tritium e t les produits des réactions radiochim iques ont été déterminés par voie de chrom atographie
gazeuse a l ’aide de deux détecteurs m ontés en série (un « cath aro m ètre » et un com pteur G eiger à balayage
continu). .
Les auteurs m ontrent que dans l ’éthylène irradié, la gam m e des produits marqués se distingue sensible
m ent d e c e lle des produits non marqués, Les principaux produits non m arqués sont; C 2 H2 , C 2 HÔ, С 4 Ню e t
des carbures à plus de quatre atom es d ’hydrogène, le rendem ent en H2 étant faib le. Les principaux produits
m arqués sont; H T, C 2 HST e t C 4 H9 T , le rendem ent en C 2 H5T e t surtout e n C 2HT étan t n égligeab le . Le
rendem ent en com posés m arqués dépend des substances ajoutées à l ’éthylène (a m m o n ia c , h éliu m , iod e), du
com posé con tenant du lith iu m et de la structure de la cib le .
En ram en an t la te m p ératu re d e '50 à " 7 8 * , on ne m od ifie que fa ib lem en t la rép artitio n des produits
m arqués; cep en d an t, lors du passage de l 'é th y lè n e à l 'é ta t solide (-1 9 6 *C ), on observe un e ffe t d é p h ase;
la production d e H T e t C 4 H9 T d im in u e, alors que l 'a c t i v i t é d e l'é th y lè n e C 2 H3 T au g m en te . '
Les auteurs m ontrent que lors de la radiolyse de C 2 H4 e t des m élan ges de c e carbu re e t d 'a m m o n ia c ,
le rapport C 2 H6 / C 4 H l 0 est é g al à 1 ,0 ± 0 ,3 , c e qui ne co n co rd e pas a v e c le sch ém a classique dés réactions
de dism utation e t d e recom b in aison des rad icau x éthyle therm iques (С 2 Н 6 / С 4Ню = 0 , 2 ± 0 , 1 ).
En se fondant sur les données relativ es aux effets que les additions d 'h éliu m e t la tem p ératu re exercen t
sur le rendem ent en С гЩ e t C 4 H10 lors de la radiolyse de C 2 H4 e t de la photolyse des m élanges С гЩ -Н Г ; les
auteurs ont é ta b li une th é o rie expliq uant l 'ap p aritio n de rad icau x d 'é th y le e x c ité s C 2HgV Ces derniers se
forment lorsque des atom es chauds d'hydrogène (produits par la radiolyse ou la photolyse) se fixent sur l ’éthylène
suivant la form ule: H ^ C 2H4= С гН ^. On m ontre que le rad ical C 2H f subit facilem en t la réactio n G2Hjf+RH
= С2Нб+ R, c e qui expliq ue pourquoi la radiolyse de l'é th y lè n e donne lieu à la production d 'un « e x c é d e n t »
d 'é th a n e . • ■
Р Е А К Ц И Я Г О Р Я Ч И Х А Т О М О В В О Д О Р О Д А С Э Т И Л Е Н О М . Р О Л Ь В О З Б У Ж Д Е Н Н Ы Х Э Т И Л Ь Н Ы Х Р А Д И К А Л О В К А К П Р О М Е Ж У Т О Ч Н Ы Х П Р О Д У К Т О В . И з в е с т н о , ч то в р е з у л ь т а т е я д е р н ы х р еак ц и й L i 6 ( и ,а ) Т и Н е 3 ( п ,р )Т о б р а з у ю т с я го р я ч и е а т о м ы р а д и о а к т и в н о го в о д о р о д а . В ы с о к а я х и м и ч е с к а я а к т и в н о ст ь т а к и х а т о м о в в о м н о го м о п р е д е ля е т о с о б е н н о ст и х и м и ч е с к и х п о с л е д с т в и й я д е р н ы х п р евр а щ е н и й . О д н ак о го р я ч и е а т о м ы в о д о р о д а м о г у т и г р а т ь с у щ е с т в е н н у ю р о л ь не т о л ь к о в ядер ной х и м и и , но и в д р у г и х о б л а с т я х хи м и и в ы с о к и х э н е р г и й : при р а д и о л и з е , ф о т о л и зе и т . д .
В н аст о я щ ей р а б о т е с д е л а н а п оп ы тка с о п о ст а в и т ь п о веден и е го р я ч и х а т о м о в во д о р о д а , п о л у ч а е м ы х р а зл и ч н ы м п у т е м : при р еак ц и и L i 6 (п,ог) Т (Е о т д в 2 , 7 М э в ) , р а д и о л и т и ч е с к и (Е ~ н е с к о л ь к о оэ в ) , ф отоли тически ( Е —1 — 1 ,5 э в ; горячий водород п о л у ч а ет ся и з H J при ф ото
л и зе с Х 2 5 3 7 А ) .В к а ч е с т в е м иш еней для п олуч ен и я а т о м о в трития и с п о л ь зо в а л и с ь к р и с т а л л ы и пленки
L i 2 С О 3 , 1Д6 F , L i6 О Н , L i 6 В О г * 8 Н г О . О б лу ч ен и ю п о д в е р г а л и г а з о о б р а з н ы й э т и л е н ( Р = 5 — 1 0 а т м .) и е г о с м е с и с а м м и а к о м , г е л и е м и и н ги б и т о р а м и . О б лу ч ен и е п р о во д и ло сь н а р ё а к т о р е ти п а И Р Т - 1 0 0 0 при п о т о к е т е п л о в ы х н ей тр о н о в 1 0 11 — 1 0 12 н / с м 2 с е к . М е ч е н ные по тритию соеди н ен и я и п родукты р ади ац и он н о -хи м и ч еск и х реакций оп р еделяли г а з о х р о
м а т о гр а ф и ч е ск и с помощ ью д в у х п о с л е д о в а т е л ь н о со ед и н ен н ы х .д а т ч и к о в (к а т а р о м е т р ) и про
точный сч е тч и к Г е й г е р а ) .П о к а з а н о , ч т о с п е к т р м е ч е н ы х и н е м е ч е н ы х п р о д у к то в в о б л у ч а е м о м э т и л е н е з н а ч и
т е л ь н о о т л и ч а е т с я . О сн о в н ы м и н е м е ч е н ы м и п р о д у к т а м и я в л я ю т с я С 2 Н 2 , с 2 н е , С 4 Н 10 и у гл е в о д о р о д ы > С 4 ; в ы х о д Н2 м а л . О сн овн ы м и нем ечены м и п р одуктам и я в л я ю т ся H T , С 2 Н 3Т и . С 4 Н 9 Т , в ы х о д С 2 Н 5 Т и о со б ен н о С 2 Н Т н е з н а ч и т е л е н . В ы х о д ы м е ч е н ы х со еди н ен и й з а в и с я т от в в е д е н и я д о б а в о к в эт и л е н (а м м и а к , г е л и й , й о д ), в и д а л и т и й с о д е р ж а щ е го с о е д и н е н и я , ст р у к т у р ы м и ш ен и .
Пониж ение тем п ер атур ы с 5 0 до - 7 8 ° м а л о вли я ет на р асп р еделен и е м еч ен ы х п р одукто в, одн ако , при п ер еход е к т вер д о м у эти лен у ( -1 9 6 ° С ) н аб лю дается ф азо вы й эф ф ек т: у м ен ьш ается вы ход H T и С 4 Н 9 Т и у в е л и ч и в а е т с я акт и вн о ст ь м ат ер и н ск о го соединения С 2 Н зТ .
П о к а за н о , что при р ади о ли зе С 9 Н4 и е г о с м е с е й с ам м и ако м отнош ение С 2 Н 6 / С 4 Н 10
р авн о l , 0 t 0 , 3 , ч то не с о г л а с у е т с я с канон и ческой схе м о й для реакций диспропорционирования и рекомбинации т е п л о в ы х эт и ль н ы х р ади к ало в (С 2 Н6 / С 4 Н1 0 — 0 ,2 ± 0 , 1 ) .

РЕАКЦИЯ ГОРЯЧИХ АТОМОВ ВОДОРОДА С ЭТИЛЕНОМ 89
На основании данных по влиянию добавок гелия и температуры на выход С 2 Н6 и С4 Ню
при радиолизе С 2 Н4 и при фотолизе смесей С 2 Н4 —H J сформулировано представление об образовании возбужденных этильных радикалов С 2 Н5 * . Последние получаются за счет присоединения горячих атомов водорода (генерируемых радиолитически или фотолитически) к этилену: Н * + СгН4 =СгН 5 * .
П оказано, что С 2Н 5*-радикал легко вступает в реакцию отрыва C 2 H 5 * + R H ^ С з Н е + R , что объясняет "и збы точн ое" количество этан а , образую щ егося при рад и оли зе эт и л е н а .
REACCIONES DE ATOMOS CALIENTES DE HIDROGENO CON ETILENO. PAPEL DE LOS RADICALES
ETILICOS EXCITADOS COMO PRODUCTOS INTERMEDIOS. C o m o resultado de las reaccio n es nucleares
®Li(n, a ) T y 3 H e (n ,p )T se form an átom os calien tes de tritio . La elev ad a actividad qu ím ica de dichos
átom os deteim ina en gran parte las características de los efectos quím icos de las transform aciones nucleares
a so cia d a s. Sin e m b a rg o , los átom o s c a lie n te s d e tri t io pueden d ese m p e ñ a r un p a p e l e s e n c ia l no sólo
en la quím ica nuclear, sino tam bién en otras esferas de la quím ica de las altas energías: radiólisis, fotólísis, e tc .
En la p resente m e m o ria se co m p ara e l co m p o rtam ien to de los átom os c a lie n te s de tri t io , obtenidos
de diversos m odos: por la re a c c ió n 6Li(n , ot)T (E = 2 ,7 M eV ); por rad iólisis ( E ~ algunos e V ); por fotólisis
(E ~ 1 a 1 ,5 eV ; e l h id ró g en o c a l ie n te se o b tie n e a p artir d e l H I p or fo tó lis is , s ien d o \ = 2 5 3 7 Â ).
Com o blancos para obtener átom os de tritio los autores utilizaron cristales y películas de Li2 C 0 8 , 6LiF,
6LiOH, 6L iB 0 2 * 8 H 2 0 . Irradiaron e tile n o gaseoso ( P = 5 a 10 a tm ) y sus m e z c la s co n a m o n ia c o , h e lio e
inhibidores. La irradiación se efectu ó en un re a c to r del tipo 1R T -1000, con un flujo dé 1 0 11 a 1 0 12 neutrones
té rm ic o s /c m 2, s. Los com puestos tritiados y los productos de las reaccio n es inducidas por radiacion es se d e
term inaron por cro m ato g rafía de gases con ayuda d e dos d etecto res m ontados en s e rie : un c a ta ró m e tro y un contador G eiger de co rrien te gaseosa. .
Los autores dem uestran que e l esp ectro de los productos m arcados y sin m a rca r en e l e tilen o irradiado
presenta considerables diferencias. Los principales productos no m arcados son e l C 2 H2 , C 2 H6, C 4 H10 e hidro
carburos > C 4 ; e l ren dim iento de H2 es pequeño. Los principales productos m arcad os son e l H T, C 2 H3T y C 4 H9 T , elren dim ien to de C 2 H 5T y en especial de C 2H T es insignificante. Los rendimientos de los compuestos
m arcados dependen de la introducción d e aditivos (a m o n ia co , h e lio , yodo) en e l e tilen o del tipo d el c o m
puesto que co n tien e litio y d e la estructura del b lan co .
La dism inución de la tem p eratu ra desde 5 0 *C a -78®C influye esca sa m e n te en la distribución d e los
productos m arcad os; sin em bargo, a l pasar al e tilen o sólido, a -1 9 6 * C , se observa un e fe c to de fases: d is
m in u y e e l re n d im ie n to d e H T y d e C 4 H9 T y a u m e n ta la a c tiv id a d d e l c o m p u e s to o r ig in a l С 2 Нз T .
Los autores dem uestran que en la radiólisis d el C 2 H4 y de sus m e z cla s con am o n iaco , la relació n
C 2 H 6 /C 4 H1 0 es igual a 1 ,0 ± 0 ,3 , lo que no concuerda con e l esquem a c lá s ic o re la tiv o a las re a ccio n e s de
dism u tación y reco m b in ación té rm ica de los rad ica le s etileno (C 2 H 6 /C 4 Hi0 - 0 , 2 ± 0 , 1 ).
Basándose en los datos relativos a la influen cia ejercid a a l añadir h elio y al aum entar la tem p eratura
sobre e l rendim iento de C2 He y C4 H 10 por radiólisis del C2 H4 y fotólisis de m ezclas de C 2 H4 -H I, los autores
estab lecen una hipótesis sobre la form ación de rad icales etílicos excitad o s (C jH ^ ). Estos últim os se obtienen
co m o resultado de la co m b in ació n de átom os de hidrógeno ca lie n te s generados por radiólisis o por fotólisis, con e l e tile n o : Н *+ С 2 H 4= С 2H j.
Demuestran que e l rad ical C 2H f interviene fácilm ente en la reacció n C2 H*g+RH= C2 H 6+ R, que exp lica
e l « e x c e s o » de etano form ado durante la radiólisis del etileno.
fПри изучении химических последствий ядерных преобразований имеют
дело с частицами промежуточных энергий, спектр которых лежит между энергетическими областями, рассматриваемыми в ядерной физике и химической кинетике. Естественно, что это обстоятельство во многом определяет методы экспериментального исследования и теоретического анализа опытных данных. Некоторая ясность в понимании природы сложных химических процессов, следующих за ядерными превращениями, вносится разделением конечных меченых веществ на продукты '‘горячих" и "тепловых" реакций; это достигается обычными в химической кинетике методами, основанными на исследовании зависимости выходов от температуры и введения ингибиторов радикальных реакций. Заметный успех в изучении первичных процессов с участием нейтральных горячих атомов достигнут за

90 Б.Г . ДЗАНТИЕВ и А.П. ШВЕДЧИКОВ
счет применения методов нейтронной физики; наиболее последовательным развитием этого направления является "кинетический" метод Вольфганга. К сожалению, ряд обстоятельств, связанных с относительно низкой энергией горячих атомов и их малой концентрацией, затрудняет пока применение при исследовании химического поведения атомов отдачи наиболее эффективных методик ядерной физики (пучок монохроматических частиц) и химической кинетики (новые методы исследования радикалов). Однако и на современном методическом уровне в<?зможно получение новых результатов в области химии горячих атомов на основе более широкого привлечения методов и данных смежных областей физики и химии и рассмотрения их под новым углом зрения (хорошим примером здесь может служить "кинетический" метод).
Нам представляется, что определенный прогресс в понимании механизма химических процессов, следующих за ядерными превращениями, может быть достигнут в результате более полного и последовательного привлечения идей и результатов из области химической кинетики, в частности кинетики радикальных реакций, конечно, с учетом энергетических различий.
Вряд ли можно рассчитывать — даже для простейшего случая горячего атома водорода-свести все многообразие химических процессов, инициированных ядерными превращениями, к двум группам: первичным "горячим" реакциям отрыва и замещения быстрым атомом атомов и атомных групп и "тепловым" процессам с участием замедлившихся атомов, избежавших горячих реакций в ходе термализации. По мере уменьшения энергии нейтрального горячего атома, движущегося в среде, происходит не только изменение скорости потери энергии Э е / Э х , н о и соотношения упругих и неупругих потерь, степени изотропности рассеяния, сечений процессов отрыва и замещения (а в более сложных случаях процессов внедрения по С —С и С —Н-связям, присоединения по кратной связи). Вместе с тем изменяется стабильность и химическая реакционоспособность молекулярных и радикальных продуктов первичных элементарных реакций горячих атомов. Эти продукты должны обладать избытком энергии и поэтому быть способными участвовать в эндометрических или обладающих высоким активационным барьером реакциях.
Возбужденные меченые молекулы и радикалы могут претерпевать как мономолекулярные (изомеризация, распад), так и биомолекулярные (взаимодействие с молекулами среды и радикалами, образующимися при сопутствующем радиолизе) реакции, в том числе и неизвестные в тепловой химии или встречающиеся лишь при очень высоких температурах.
Логично предполагать, что конечное распределение активности по меченым продуктам в ряде случаев в заметной, если не в решающей степени, определяется вторичными химическими процессами с участием возбужденных частиц, образующихся при первичных элементарных реакциях горячих атомов.
Было бы несколько прямолинейно считать, что химическая стабилизация горячего атома в молекуле всегда происходит "в один акт", и полученный продукт сразу приобретает свойства стабильной молекулы, хотя это, повидимому, все же имеет место в специальных условиях (конденсированная фаза, высокое давление), когда обеспечен быстрый отвод энергии возбуждения. Законно ожидать, что химизм процесса с участием атомов отдачи в ряде случаев может быть понят на основе представления о цепочке

РЕАКЦИЯ ГОРЯЧИХ АТОМОВ ВОДОРОДА С ЭТИЛЕНОМ 91
высокоэнергетических реакций, начинающейся первичным элементарным актом X* + RH и продолжающейся в силу повышенной реакционоспособности возбужденных продуктов предшествующей реакции. Такие своеобразные "энергетические" цепочки, повидимому, должны состоять из небольшого числа (2 —4) быстро следующих друг за другом звеньев. В результате происходит эстафетная передача меченого атома с распределением его энергии по все увеличивающемуся числу степеней свободы j что приводит к возрастанию времени жизни промежуточных меченых возбужденных частиц и образованию в конце цепочки стабильной меченой молекулы. Закономерности химического поведения продуктов ядерного превращения не являются узкой спецификой только данной области.
Особенности химической стабилизации атомов отдачи в какой-то мере должны проявляться во всех областях химии, где могут генерироваться "горячие" атомы, обладающие высокой неравновесной энергией: в фотохимии [1], в радиационной химии [2 — 4], космохимии, возможно, также и в тепловой химии при высоких температурах (взрыв).
Таким образом, если, с одной стороны, для уточнения наших знанийо механизме реакций горячих радиоактивных атомов необходимо более тесное привлечение идей и методов химической кинетики, то, с другой стороны, своеобразные результаты, полученные при исследовании химизма процессов, связанных с ядерными преобразованиями, могут помочь в понимании опытных данных в более традиционных областях химической кинетики, не поддающихся объяснению в пределах известных закономерностей тепловой химии.
В настоящей работе проводилось экспериментальное исследование реакций горячих атомов водорода с этиленом с целью выяснения роли возбужденных этильных радикалов, как промежуточных продуктов. Исследовавшаяся система является достаточно простой,а поэтому имеет модельный характер.
Горячие атомы водорода — в отличие от поливалентных атомов — могут, в принципе, взаимодействовать с этиленом относительно небольшим числом способов. Возможны, в основном, три типа первичных элементарных процессов: отрыв водорода, замещение водорода и присоединение по двойной связи:
Н* +С 2 Н4 ------ -НН* + С2 Н3 (1)
Н * +С 2 Н4 ------ - С 2Н3Н* + Н (2)
Н * +С 2 Н4 ------ ► (С 2Н4Н*) (3)
Выбор в качестве вещества-мишени этилена обусловлен,в основном, тем, что, как известно из кинетики, он является эффективным скевенже- ром атомарного водорода. Образующиеся по реакции (3) этильные радикалы могут, в принципе,быть возбужденными и служить моделью "горячего" радикала. Свойства тепловых этильных радикалов хорошо известны в результате многочисленных исследований [5—7]. Кроме того, изучение реакций горячих атомов водорода с этиленом — простейшим олефином — представляет самостоятельный интерес, так как реакции Н* с предельными молекулами исследованы значительно лучше, чем с непредельными. Между

92 Б.Г . ДЗАНТИЕВ и А.П. ШВЕДЧИКОВ
тем, возможный спектр реакций Н*+олефин богаче, чем в случае Н*+пара- фин. Так как олефины всегда возникают в результате радиолиза предельных соединений под действием ядерных излучений (параллельно осуществлению ядерного процесса), то взаимодействие их с горячими атомами водорода (трития) должно накладывать отпечаток на спектр меченых продуктов также и в случае реакций Н * + парафин. ■
Проводилось сопоставление поведения горячих атомов водорода различных энергетических групп и различной природы. С этой целью горячие атомы водорода генерировались а) в результате ядерной реакции L i B(n,ar)T (Е0 =2,7 Мэв), б) в результате радиолиза этилена и аммиака под действием излучения ядерного реактора и продуктов (п,а) реакций (Е~несколько эв), в) путем фотолиза HJв ультрафиолетовой области (Е = 0,8 -1,8 эб). В последнем случае производилось облучение смесей С 2Н4 —HJ и С2Н4 —HJ —Не светом ртутной лампы А = 2537 А с последующим хроматографическим анализом.
Два первых варианта осуществлялись одновременно путем облучения в ядерном реакторе этилена в присутствии соединений лития и бора: LÍ2CO3 Li6F , L i6 ВО2 • 8Н2О, L i6 ОН. Облучению подвергался газообразный этилен (чистота 99,8%) при повышенном давлении (3 — 10 атм), а также смеси этилена с аммиаком, гелием, ингибиторами. Исследовавшиеся системы запаивались в предварительно эвакуированные кварцевые ампулы объемом 1 — 10 см3 и облучались в канале реактора в течение 0,5 — 25 час в потоке тепловых нейтронов Фтн = 1011 — 10 12 н/см2 сек. Поток острых нейтронов составлял ~10% от Фтн , 7 - Д °з а Dy равна (1 — 2) • 1014 эв/см Зсек, общая доза (D = Dy +Da +Dn) достигала ~ 1 0 15 эв/см3сек (при нормальных условиях). Измерения потоков нейтронов проводились методом радиоактивных индикаторов, дозиметрические измерения-ионизационным методом и с помощью химической дозиметрии. Температура при облучении изменялась от +50°С до —196°С. При низкотемпературных облучениях ампулы помещались в дюары, заполненные льдом (0°С) , смесью твердой углекислоты с ацетатом (—78°С) и жидким азотом (-196°С ). *
После вскрытия облученных ампул продукты химических реакций с участием горячих атомов трития и продукты радиолиза (с участием Н и Н*) подвергались анализу методом газовой хроматографии и радиохроматографии. Использовались два последовательно соединенных датчика: катаро- метр и проточный счетчик Гейгера. Хроматографические колонки снабжались программным обогревом. Для разделения легких углеводородов и водорода использовалась колонка, заполненная силикагелем, для разделения более тяжелых продуктов (> С4) применялась колонка, заполненная детергентом; в качестве жидкой фазы использовалась смесь скволан- трикрезилфосфат (1:4). В ряде случаев для разделения HT и СН3Т применялись молекулярные сита типа 5А.
Исследован спектр и установлены выходы меченых и немеченых продуктов. Показано, что соотношение выходов меченных тритием (реакции Т*+СгН 4 ) и не меченных (прямой радиолиз и реакции (Н *(Н ) +С2 Н4 ) продуктов различно. Основными немечеными продуктами являются ацетилен, этан и бутан . Заметный выход дают более тяжелые углеводороды > С4 . Выход молекулярного водорода невелик. Среди продуктов присутствуют в незначительном количестве метан, пропан, пропилен, изобутан.

РЕАКЦИЯ ГОРЯЧИХ АТОМОВ ВОДОРОДА С ЭТИЛЕНОМ 93
Относительные выходы основных продуктов радиолиза этилена: С2 Н2 , С2 Не, С4Н10, Н2 составляют в среднем 1:1:1:0,6.
Образование ацетилена в основном обусловлено прямым распадом возбужденной в первичном акте молекулы этилена:
(4а)
(46)
Из фотохимических данных известно, что с ростом энергии возбуждения вероятность реакции (46) увеличивается. Сравнительно малый выход молекулярного водорода указывает на то, что уровни возбуждения молекулы С2 Н4 при действии ионизирующего излучения таковы, что путь (46) предпочтительнее. Помимо реакции (46), атомарный водород может образовываться в процессе (5):
С2 Н4 ------ - (С2 Н4) * ------ - С2Нз +Н (5)
или при радиолизе водородсодержащих компонентов смесей, например:
NH3 ------► (NH3) * ------>NH2 +H ( 6 )
Некоторая часть ацетилена может получаться за счет диспропорциониро- вания винильных радикалов, возникающих по реакции (5). Возникновение остальных продуктов связано с реакциями (1—3) атомарного водорода; некоторая часть последнего, как будет показано ниже, образуется при радиолизе в "горячем" состоянии. В образовании продуктов С2, С4 и > С4 важную роль играют реакции радикалов: винильного и,особенно,этильного,появляющегося при присоединении атома Н к этилену по реакции (3).
Основными мечеными продуктами являются молекулярный водород HT, этилен С2Н3 Т и н-бутан С4 Н9 Т. Выходы этана С2Н5Т и,особенно,ацетилена С2НТ незначительны. В числе продуктов реакции Т * + С 2 Н4 наблюдаются бутен С4 Н7 Т и гексан СбНгзТ с небольшими выходами. При сопоставлении выходов меченых и немеченых продуктов бросается в глаза существенное различие в относительных весах молекулярного водорода, ацетилена и этана.
В табл.1 представлены результаты двух серий опытов по облучению тритонами смесей этилена с аммиаком. Серии различаются химическим составом и структурой датчика горячих атомов трития: в первом случае (I) брали тонкую пленку (толщина' ~ 1 мг/см2 ) L,i6B 0 2 - 8H2 0 на внутренней поверхности кварцевой ампулы, во втором случае (II) — кристаллы L Í2 CO3 . Выходы активных продуктов выражены в процентах от суммарной активности газофазных производных этилена (RT) и водорода (HT). Последний образуется при взаимодействии Т как с этиленом, так и с аммиаком. Условия облучения в известной мере сказываются на выходах активного бутана С4НдТ. Зависимость выходов HT, С2 Н3 Т и С2 Н5 Т в обеих сериях опытов согласуется достаточно хорошо. В чистом этилене относительные выходы основных меченых продуктов составляют: HT — 40 — 50%, С2 Н3Т — 25—30%, С 2Н 5Т — 5 — 6%, С4 Н9 Т — 10 —257о. С ростом концентрации аммиака (v) в системе С2 Н4 —NH3 относительные выходы (и) молекулярного водорода HT
С,Н2 П 4
С2 Н2 + 2Н
94 Б.Г. ДЗАНТИЕВ и А.П. ШВЕДЧИКОВ
Т аб ли ц а 1
ОТНОСИТЕЛЬНЫЕ ВЫХОДЫ АКТИВНЫХ ПРОДУКТОВ ПРИ ОБЛУЧЕНИИ СИСТЕМЫ ЭТИЛЕН-АММИАК
(Условия облучения: I. Внутренняя стенка ампулы покрыта пленкой Li6B 02 '8H 20 т о л щ и н о й ~1 м г/см 2. Объем ампулы
5 — 8 см3 , р = 5 — 8 атм. Ф = 10 11 н/см 2сек, время облучения —25 часов. II. Ампула заполнена
кристаллами LÍ2 CO3 (50 — 100 мг). Объем ампулы1 — 2 см 3, р = 8 —10 атм. Ф = 3,5-1011 н/см 2 сек,
время облучения 7—10 часов).
У с л о в и яоблучения
с 2н 4 ,м ол %
В ы х о д м еч ен ы х соеди н ен и й , %
H T С 2 Н 5 Т С 2 Н 3 Т С 3 Н 7 Т + С 2 Н Т С 4 Н 9 Т C 4 H 7 T + C 6 H i 3 T
100 45 4,5 32 2,7 8 7,8
80 47 3,0 28,5 2,2 12 7 ,1
75 36 4,5 38 - 12 9,0
60 3 7 5 ,7 28 2,8 1 6 1 0 ,4
I 50 3 4 3,6 3 4 1 .4 1 6 12,2
25 5 7 2 ,9 22 0,8 1 3 4,4 .
10 4 7 11,4 6 ,4 1 ,9 2 4 8,6
5 5 6 10,8 2,4 4 ,2 21 4,9
2 87 6,2 3 ,5 - 3 ,3 -
100 42 6,0 22 4 2 6 -
9 0 4 5 2,8 28 2,4 22 -
65 3 8 2 ,5 4 0 - 20 -
II 50 3 5 3 ,8 3 0 1 ,3 27 3 ,0
45 3 8 4 ,3 29 0 ,4 2 3 6,2
40 4 6 5 ,5 2 6 - 20 2,4
20 5 6 5 ,0 16 - 20 3 ,0
10 7 2 1 0 ,5 6 ,5 0 ,5 10 0 ,5
и этилена С2 Н3Т в области v = 0,4 — 0,5 проходят соответственно через нерезкие минимум и максимум; в этой точке их выходы равны и составляют 35%. С дальнейшим ростом концентрации аммиака выход HT довольно резко растет, а С 2 Н зТ — падает ; при и —* 1 и (HT) —*100, а /л(СгНзТ) —>0. Относительный выход этана С2 Н5 Т в широком интервале концентраций сохраняет практически постоянное значение ^(СгНаТ) =3 — 5%, но в области малых концентраций этилена (5 — 107») /^(C^HsT) увеличивается в 2—3 раза, а затем резко падает. В области концентраций аммиака i/ = 0,8—0,9 наблюдается резкое уменьшение относительного выхода и меченого бутана; при v—>• 1 м(С4 H 9 Т) *0 .
Заметим, что полученные в настоящей работе данные по распределению активности в продуктах реакций трития с чистым этиленом, согласуясь с результатами Сауэра и Вилларда [15] в отношении выходов С2 Н5 Т и С4 Н9 Т,

РЕАКЦИЯ ГОРЯЧИХ АТОМОВ ВОДОРОДА С ЭТИЛЕНОМ 95
заметно расходятся с ними при сравнении выходов молекулярного водорода и этилена. Согласно [15] относительные выходы HT и С2 Н3 Т составляют соответственно 16 и 53%, т.е. /и(НТ) в 2,5—3 раза меньше, а й(СгН3Т) в2 раза большврчем в данной работе. Возможно, отчасти это расхождение связано с различием источников тритонов, хотя этим обстоятельством вряд ли можно объяснить изменение вероятностей соотношения реакций отрыва и замещения водорода в несколько раз.
В табл.2 представлены данные, характеризующие зависимость относительных выходов меченых продуктов от температуры во время облучения. Понижение температуры этилена от 50° до -7 8° С не оказывает заметного влияния на относительный выход ни одного из наблюдаемых соединений. При -196°р выход молекулярного водорода HT уменьшается вдвое, а выходы меченых этилена и этана возрастают: С 2 Н3Т ~в 2 раза, а С2 Н5 Т ~в 3 раза. Однако—этот эффект скорее связан с изменением фазового состояния и, соответственно, условий стабилизации.
Табл.З характеризует зависимость относительного выхода активных продуктов от состава системы С2 Н4 — Не. Добавки гелия снижают относительный выход меченого материнского соединения С 2 Н3 Т, несколько повышают ц(НТ), не сказываясь заметно на относительных выходах остальных продуктов. Для понимания этих результатов существенно знать, как меняются абсолютные значения выходов. В отдельных опытах показано, что абсолютный выход HT (приведенный к одному количеству L i6 и одному потоку) падает при введении гелия в систему; при концентрациях гелия выше 20 — 25 мол % абсолютный выход HT меняется мало. В табл.4 приведены данные по влиянию добавок иода (ингибитор радикальных реакций) на соотношение выходов продуктов реакций трития с этиленом. С ростом концентрации J2 увеличиваются относительные выходы меченых этана и этилена за счет падения доли HT в суммарной активности газовой фазы. Относительный выход бутана С4Н 9Т практически не меняется.
Выяснение механизма образования меченых соединений по реакции Т* + С2 Н4—► продукты только на основании приведенных закономерностей затруднительно. Известно, что исследование механизма даже наиболее простых реакций в области тепловой химии представляет всегда весьма сложную задачу и требует комплексного подхода. Тем более это относится к рассматриваемому случаю, осложненному наличием значительного числа меченых продуктов, влиянием радиолиза, конкуренцией реакций Горячих и тепловых атомов Т, возможным, в принципе, в условиях опытов вкладом ионно-молекулярных реакций.
Одним из основных условий изучения механизма сложного химического процесса является выяснение характера и реакционной способности промежуточных продуктов. Одним из возможных промежуточных продуктов в реакциях Т * + С г Н 4 является возбужденный этильный радикал, который, в npHHunnejможет образоваться по реакции (3) при присоединении горячего атома Т к этилену по двойной связи. Для выяснения роли возбужденных этильных радикалов как промежуточных продуктов в реакциях атомов отдачи трития с этиленом была предпринята попытка получить C 2HJ и исследовать их реакцион'оспособность в более "чистых" условиях.
С этой целью проводился фотолиз смесей HJ +С 2 Н4 , заключенных в термостатируемую кювету, у/ф светом при Х = 2483—2804Â . В этой об-
со05
Т аб ли ц а 2
ЗАВИСИМОСТЬ ОТНОСИТЕЛЬНЫХ ВЫХОДОВ МЕЧЕНЫХ СОЕДИНЕНИЙ ОТ ТЕМ П ЕРАТУРЫ И ОБЛУЧЕНИЯ
(Облучаемая система —С 2 Н4. Объем ампул 2 — 3 см3. Ф = 2,7-1012 н/см2сек. ЛК — L Í2CO3 кристаллический, ЛБ — L i 6B 0 2 -ЭНгО кристаллический)
Т е м п е р а т у а ,° С
Д атчи ктритонов
В р ем яоб луч ен и я,
ч асы
Д а в л е н и е ,ат м
В ы х о д м е ч е н ы х со еди н ен и й , %
H T с 2н 5т с 2н 3т с 2 н т + с 3 н 7т C 4 H 9 T с 6н 13т
50 ЛК 2 10 38 8 25 5 18 6
50 ЛБ 10 2 35 4,5 29 6,5 22,5 2,5
50 ЛБ 10 4 41, 5 3, 5 25 4,5 21 4,5
50 ЛБ 0,5 10 40 7,5 33, 5 4,5 10 4,5
0 ЛК 2 8 3 5 7 2 7 5 ,5 1 6 8,5
* 7 8 Л К I 3 3 8 10 2 7 5 20 -
- 1 9 6 Л К 0 ,5 ТВ. 16 21 4 9 \ _ 14 -
-196 Л Б 0 ,5 Т В . 2 4 1 8 4 8 - 10 -
Б.Г
. Д
ЗАНТИ
ЕВ и
А.П
. Ш
ВЕД
ЧИ
КО
В

РЕАКЦИЯ ГОРЯЧИХ АТОМОВ ВОДОРОДА С ЭТИЛЕНОМ 97
Т а б л и ц а 3
ЗАВИСИМОСТЬ ОТНОСИТЕЛЬНОГО ВЫХОДА АКТИВНЫХ ПРОДУКТОВ РЕАКЦИИ Т + С 2Н4 ОТ СОСТАВА СИСТЕМЫ С2Н4-Н е
(Датчик трития L i6F(2 —5 мг). Время облучения 10 часов. Ф=3,5*1011 н/см2сек).
С 2 Н 4
м ол %Д а в л е н и е ,
а т м
. В ы х о д м е ч е н ы х со еди н ен и й , %
H T С 2 Н 5 Т С 2 Н3 Т С 2 Н Т + С 3 Н 7 Т С 4 Н 9 Т
1 0 0 5 4 2 6 2 2 4 2 6
9 5 5 4 4 4 ,2 ■ 2 4 4 ,3 2 4
9 0 3 3 9 9 , 4 1 8 7 ,6 2 6
7 5 2 - 4 ,8 17 4 , 8 -
5 7 2 5 1 ,5 8 , 8 1 1 ,5 - 2 7 ,5
5 0 2 5 4 7 ,2 1 0 ,5 6 ,5 2 2
15 1 ,5 5 8 7 ,8 7 ,8 - 2 7
Т аб ли ц а 4
ВЛИЯНИЕ ДОБАВОК J2 НА ОТНОСИТЕЛЬНЫЙ ВЫХОД АКТИВНЫХ ПРОДУКТОВ ПРИ ВЗАИМОДЕЙСТВИИ ТРИТИЯ С ЭТИЛЕНОМ
(Датчик тритонов—L i2 СОз (—100 мг). Время облучения 15 часов.Ф =3,5* 1011 н/см2сек).
В е с J 2 , м г
Д а в л е н и е ,а т м
В ы х о д м е ч е н ы х со еди н ен и й ,%
H T С 2 Н 5 Т С 2 Н 3 Т С 2 Н Т + С 3 Н 7 Т С 4 Н9 Т
0 ,5 5 5 0 8 ,5 2 2 4 ,5 15
1 > 0 3 ,5 4 8 1 3 ,5 2 2 2 ,5 1 4
2 , 0 6 . 4 0 19 2 8 2 ,5 1 1
3 , 0 6 3 6 17 2 5 2 2 0
4 ,0 6 3 4 19 3 2 2 13
5 ,0 5 ,5 3 0 19 4 0 . - 1 1
ласти спектра этилен не подвергается фотолизу, a HJ разлагается согласно схеме [1 , 8]:
HJ — ► Н + J*, ,
H + H J ------ >H2+J,

98 Б.Г. ДЗАНТИЕВ и А.П. ШВЕДЧИКОВ
Квантовый выход Ф = 2 и не зависит от температуры и добавок С2 Н4 . Атомы Н образуются при фотолизе HJ с повышенной неравновесной энергией
Е(Н*) = [Еу -D H j - e (J * ) ]^ j§ | -
При X = 2537Â (Еу = 4,9 эв = 112 ккал/моль) и энергии связи Dhj = 71 ккал/моль кинетическая энергия Н-атомов Е(Н *) = 19 ккал/моль (0,8'эв), если.1 образуется в состоянии PJ(E(J*) = 22 ккал/моль) и достигает 41 ккал/моль (1,8эв) в случае J (Р|2).
Горячие и термализованные атомы Н, присоединяясь к этилену, генерируют возбужденные и тепловые этильные радикалы:
Н*+'С2 Н4 ------ " с 2н * (3<)
н +С 2 Н4 ------ ► C2 H¿ (3 " )
В среде из тяжелых молекул (HJ, С2Н4 ) время жизни Н* достаточно велико, что обеспечивает эффективное образование С2н£ даже при малых отношениях C 2H4 /HJ. Так как для тепловых С2 Н5 возникающий при фотолизе йод является эффективным скевенджером, то образование органических продуктов фотолиза должно быть в основном связано с реакциями горячих радикалов С2Н 5 . Действительно, из кинетики хорошо известно, что основными реакциями тепловых этильных радикалов при комнатной температуре являются реакции рекомбинации и дйспропорционирования, приводящие к образованию бутана и этана:
C 2 H s + C 2 H 5
(7а)
(76)
Отношение констант К7а/К7б таково, что выход этана составляет 10 — 20% от выхода бутана [ 6 ,7]. Однако хроматографический анализ, продуктов фотолиза смеси С2 Н4 — HJ (10% С2 Н4 ) показывает отсутствие бутана при наличии значительных количеств С2 Нб.
Как видно из рис .la выход этана в широком температурном интервале 0 — 250° совершенно не зависит от температуры.
Введение в систему различных инертных газов различным образом влияет на выход этана. Эффективность инертного газа зависит от степени близости массы его атома и массы атома водорода. Так^скорость образования этана лишь незначительно изменяется при введении в систему HJ —С2 Н4 даже больших (до 80%) количеств аргона (рис.2а), но резко падает при добавлении гелия (рис.26).
Если смесь С2 Н4 — HJ — Не (~75% Не), где выход СгНб при комнатной температуре мал, нагревать в процессе фотолиза, что в отличие от чистой системы С2 Н4 —HJ, в этом случае для выхода этана наблюдается аррени- усовская зависимость (рис. 16). Эффективная энергия активации согласуется с величиной, вычисленной по правилу Семенова-Поляни [9] для тепловой реакции С2 H¿ + H J —► С2 Н6 + J .

РЕАКЦИЯ ГОРЯЧИХ АТОМОВ ВОДОРОДА С ЭТИЛЕНОМ 99
Р и с . 1
З а в и с и м о с т ь ск о р о ст и о б р азо ва н и я С 2 Н6 ( с м 3 /м и н ) от тем п е р а ту р ы при ф о т о л и зе :
а ) с и с т е м а H J — Û 2 H 4 = 9 : 1 ( J ) ;б) с и с т е м а J : Не = 2 : 8 .
Р и с . 2
З а в и с и м о с т ь в ы х о д а C 2 Hg от д о б а в о к ар го н а и г е л и я при ф о т о л и зе :
а ) с и с т е м а S — А г ;б) с и с т е м а J — Н е.
Рассмотренные результаты указывают на возникновение возбужденных этильных радикалов при присоединении горячих (обладающих повышенной кинетической энергией) атомов Н по тг-связи к этилену (реакция 3). Образование этана связано с отрывом возбужденным этильным радикалом Н-атома от молекулы среды:
C2 H¡ +HJ(C2H4) ------► С2Н6 +J(C2 H¿) (8)

100 Б.Г . ДЗАНТИЕВ и А.П. ШВЕДЧИКОВ
Необходимая для осуществления процесса энергия активации обеспечивается за счет энергии возбуждения радикалов. Реакционная способность С2Н| определяется степенью его возбуждения, зависящей от средней энергии горячих Н-атомов:
E (C 2H*) = E(H*)+q,
где q = 1,65 эв — тепловой эффект реакции присоединения атома Н к этилену. Природа энергии возбуждения этильного радикала обсуждается в [10].
Образование возбужденных радикалов, обладающих повышенной реакционной способностью, должно быть общим свойством систем, содержащих непредельные соединения, в которых тем или иным путем генерируются горячие ¿Томы водорода. В частности, рассмотренные выше эффекты должны наблюдаться при реакциях,атомов отдачи трития и при радиолизе.
Ряд авторов (Воеводский В.В. [2 — 4] Хардвик [11], Медведев [12,13]) высказывали мысль, что при радиолизе углеводородов образуются горячие атомы водорода, появление которых связано с распадом возбужденных излучением молекул. Однако вопрос о возникновении при радиолизе RH не тепловых Н-атомов считается дискуссионным.
Своеобразным индикатором наличия в любой системе горячих атомов водорода может служить этилен. Последний может содержаться в системе, специально в нее добавляться или образовываться под действием излучения. Критерием является отклонение экспериментально наблюдаемого отношения выходов этана и бутана г = СгНб/С4 Ню от "канонического" для тепловых этильных радикалов значения г 0 = 0 , 1 —0 , 2 , равного К ДИспр /К р е к . Если г> го, то добавочное количество этана определяется реакцией возбужденных этильных радикалов:
C2 H* + RH ------ - C 2H6+ R (81)
и, следовательно, наличием горячих атомов Н*, ответственных за появление СгН| согласно реакции (3).
При радиолизе этилена отношение выходов этана и бутана г = 1, т.е. г/г0 = 5 —10. При облучении смесей этилена и аммиака с большим содержанием NH 3 величина г достигает 5, а г/го = 25 — 50. Эти данные свидетельствуют об образовании при радиолизе этилена и аммиака, наряду с тепловыми Н-атомами,и горячих атомов водорода в результате процессов:
С2 Н | ------>C 2H¿+H* (9)
NH3 ------► NH¿ + Н* (10)
Это заключение находит ряд дополнительных и независимых экспериментальных подтверждений. Они получены в результате опытов с введением термализующих добавок, ингибиторов радикальных реакций, путем исследования радиолиза при пониженных температурах, облучения бинарных и тройных систем.
Реакционная способность возбужденных этильных радикалов, образующихся в результате последовательных реакций (9 — 10), (З1) и (81), должна зависеть от энергии возбуждения C2 HJ и,следовательно, от кинетичес
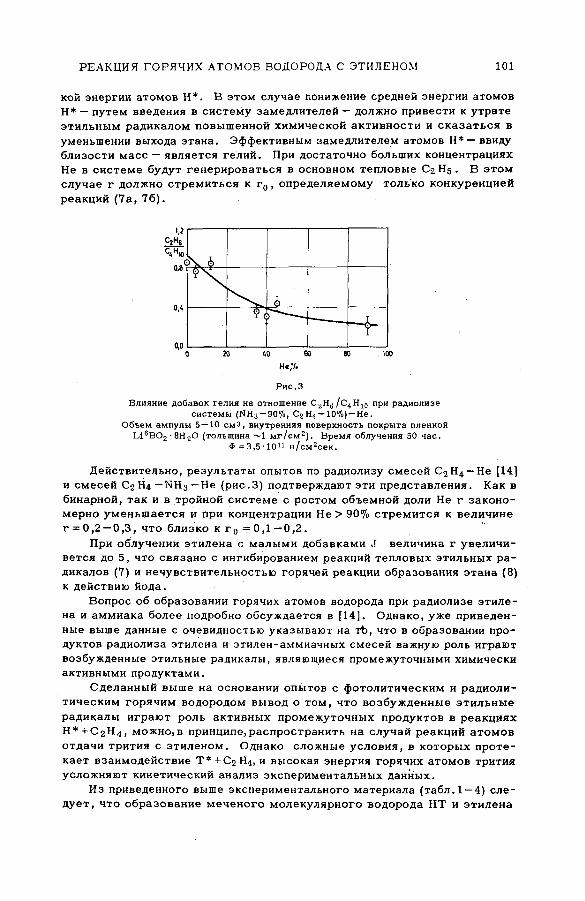
РЕАКЦИЯ ГОРЯЧИХ АТОМОВ ВОДОРОДА С ЭТИЛЕНОМ 101
кой энергии атомов Н*. В этом случае понижение средней энергии атомов Н* — путем введения в систему замедлителей — должно привести к утрате этильным радикалом повышенной химической активности и сказаться в уменьшении выхода этана. Эффективным замедлителем атомов Н* —ввиду близости масс — является гелий. При достаточно больших концентрациях Не в системе будут генерироваться в основном тепловые С2 Н5 . В этом случае г должно стремиться к г0, определяемому только конкуренцией реакций (7а, 76).
Р и с .З
В ли я н и е д о б а в о к ге л и я на отнош ение С 2 Не / С 4 Н 1 0 при р а д и о л и зе с и с т е м ы (N H 3 —9 0 % , С 2 Н4 — 10% ) — Н е .
О бъ ем ам п у лы 5 — 1 0 с м З , вн утр енн яя п о ве р хн о сть п окр ы та п ленкой L i s B 0 2 ' 8 H 20 (то льщ ин а ~ 1 м г / с м 2) . В р е м я облучен ия 5 0 ч а с .
Ф = 3 , 5 * 1 0 1 1 н / с м 2 с е к .
Действительно, результаты опытов по радиолизу смесей С2 Н4 —Не [14] и смесей С2 Н4 —NH3 —Не (рис.З) подтверждают эти представления. Как в бинарной, так и в тройной системе с ростом объемной доли Не г закономерно уменьшается и при концентрации Н е> 90% стремится к величине г = 0,2 —0,3, что близко к г 0 = 0,1—0,2 .
При облучении этилена с малыми добавками J величина г увеличи- вется до 5, что связано с ингибированием реакций тепловых этильных радикалов (7) и нечувствительностью горячей реакции образования этана ( 8 ) к действию йода.
Вопрос об образовании горячих атомов водорода при радиолизе этилена и аммиака более подробно обсуждается в [14]. Однако, уже приведенные выше данные с очевидностью указывают на тЬ, что в образовании продуктов радиолиза этилена и этилен-аммиачных смесей важную роль играют возбужденные этильные радикалы, являющиеся промежуточными химически активными продуктами.
Сделанный выше на основании опытов с фотолитическим и радиоли- тическим горячим водородом вывод о том, что возбужденные этильные радикалы играют роль активных промежуточных продуктов в реакциях Н * + С 2Н4, можно,в принципе,распространить на случай реакций атомов отдачи трития с этиленом. Однако сложные условия, в которых протекает взаимодействие Т * + С 2 Н4, и высокая энергия горячих атомов трития усложняют кинетический анализ экспериментальных данных.
Из приведенного выше экспериментального материала (табл. 1—4) следует, что образование меченого молекулярного водорода HT и этилена

102 Б.Г . ДЗАНТИЕВ и А.П. ШВЕДЧИКОВ
С2 Н3Т по крайней мере отчасти обусловлено прямыми горячими реакциями отрыва и замещения:
Т * + С 2 Н4 ------► HT +C 2H¿
- С2 Н3 T + H
( И )
( 12 )
Образование части HT, повидимому, связано с реакциями тепловых атомов трития. Нам представляется, что некоторая доля HT и С2 Н3Т образуется в результате распада возбужденного этильного радикала:
Т * * + С 2 Н4 (С2 Н4 Т)*HT +С 2 Н3
RH < 1 3 >н 2 + с 2н 2т ------► С2 Н3Т
и возбужденной молекулы этана.С последним процессом может быть связано то обстоятельство, что
отношение выходов меченых этана и бутана С 2Н5 Т/С4НдТ хотя и выше, чем Го, но ниже, чем в случае продуктов радиолиза. Относительное уменьшение выхода С 2 Н б Т и увеличение С 4 Н э Т может быть связано с высокими уровнями возбуждения горячего этильного радикала. Последнее обстоятельство может привести к цепочке процессов с участием горячих частиц с постепенной их стабилизацией в результате моно- и бимолекулярных процессов:
с 2н 4 ) УТ * * + С 2Н4 ---- ► (С2Н4Т) * * +
NH,HT
(С2Н5Т)*,НТ+С 2Н4
Н2 +с2н3т
ЧС4Н8Т) * +С 2Н4
NH 3
(14)
•С4Н9Т
Эта схема согласуется с результатами опытов по облучению этилен + Li в присутствии Не, J2 и аммиака.
Л И Т Е Р А Т У Р А
[1 ] B E N S O N S, T h e F ou n d ation s o f C h e m ic a l K in e t ic s . 369, 1960.[2 ] В О Е В О Д С К И Й В . В . , М О Л И Н Ю .Н . R a d ia t io n R e s e a rc h . 17, 3, 366 (1962 ).
[3 ] М О Л И Н Ю . Н . , Ч Х Е И Д З Е И . И . , К А П Л А Н Е . П . Б У Б Е Н И. Я. и В О Е В О Д С К И Й В . В . "К и н ети к а и к а т а л и з " , 3 , 5 (1 9 6 2 ) 674.
[4 ] М О Л И Н Ю .Н . , "К и н е т и к а и к а т а л и з " , ^ , 4 (1 9 6 0 ) 490 .[5 ] S T E A C IE E .W . R . " A t o m ic and F r e e R a d ic a l R e a c t io n s " N . Y . 1954.[ 6 ] S H E P P A . , K U T S C H K E K . J .C h e m . P h y s . 26 1020 (1 9 5 7 ).[7 ] IV IN K . J . , S T E A C IE E . W . R . P r o c . R o y . S oc . A 2 0 8 25 (1 9 5 1 ).[ 8 ] К О Н Д Р А Т Ь Е В B . H . , К О Н Д Р А Т Ь Е В А E . H . и Л А У Р И С А . Ж Ф Х , 5 , (1 9 3 4 ) 1411.

РЕАКЦИЯ ГОРЯЧИХ АТОМОВ ВОДОРОДА С ЭТИЛЕНОМ 103
[9 ] С Е М Е Н О В Н . Н . "О некоторых проблемах химической кинетики и реакционоспособности" М . , 1958.
[10] Д З А Н Т И Е В Б . Г . Ш ВЕ Д Ч И К О В А .П . ,и Б О РЩ А ГО В С К И Й Б . В . Д АН С С С Р , 157(1964) 653.
[11] H A R D W IC K T .J . J . Phys. Chem . 66, 9 (1962) 1611.[12] П Р А В Е Д Н И К О В А .Н . ,и М Е Д В Е Д Е В С .С . "Труды I Всесою зного совещания по радиа
ционной химии", М . , 1958, 269.[13] П РА В Е Д Н И К О В А . Н . , ИН ШЕН КАН,и М Е Д В ЕД Е В С .С . "Труды II Международной кон
ференции по мирному использованию атомной энергии. Женева, 1958". т .4, 241, М . 1959.[14] Д ЗА Н ТИ ЕВ Б . Г . и Ш ВЕДЧИКОВ А .П . , ЖФХ, 38, 11 (1964) 2745.[15] S A U E R М. С . , W IL L A R D J .E . J. Phys. Chem. 64 (1960) 359.
D I S C U S S I O N
F.S. ROWLAND: I have one comment concerning the addition of hydrogen atoms to ethylene. Mrs.Umezawa and I carried out some experiments with hydrogen atoms formed in the radiolysis of cyclopropane, allowing them to add to C2 H3T to form Q H 4T radicals. When these radicals subsequently reacted with C^Hg radicals, the products C^HgT and n-C4HgT were observed in the ratio 0.15 ±0.02, the usual value obtained for thermalized ethyl radicals. We did not,therefore, see any evidence for the higher ratio expected for excited ethyl radicals with this particular source of radiolytic hydrogen atoms.


GASEOUS SYSTEMS (Session 2)


REACTIONS AND MECHANISMS INVOLVING HOT CARBON ATOMS
AND Ñ2 -H 2, N2 -ALKANE AND N2 -ALKANE-MODERATOR SYSTEMS INCLUDING THEIR RELATIONSHIP
TO OTHER SIMPLE SYSTEMS*
H. ACH E AND A. P . W O L F
C H EM ISTRY D EPARTM ENT, BROOKHAVEN NATIONAL LABORATORY,
U PTO N , NEW YO RK ,UNITED STA TES OF AMERICA
Abstract — Résumé — Аннотация — Resumen
REACTIONS AND MECHANISMS INVOLVING HOT CARBON ATOMS AND N2 -H 2, N2 -ALKANE AND
N 2 -ALKANE-MODERATOR SYSTEMS INCLUDING THEIR RELATIONSHIP TO OTHER SIMPLE SYSTEMS, Carbon-11 produced by a variety of nuclear reactions, e .g . C 12(n ,2n)C11, C 12(p ,pn )Cu , N l4(p ,-a )C 11, Neip.iXJC11, Bu (p ,n )C u and Be9(H e3, n )Cn , has been used by us in studying the chemistry of hot carbon
atoms. The Ni*(p, a )C n reaction is particularly advantageous because the radiation dose to the system can be
readily controlled and high levels of activity can be produced. Furthermore the use of systems with multiple
substrates affords a convenient method for obtaining experimental "reaction cross-sections".The dominant reaction of hot carbon atoms with nitrogen in hydrogen-containing systems is the for
mation of HCn N, presumably through the intermediary of the C n N radical. Only HCn N and C n H4 are
formed in N 2 -H 2. The usual product spectrum characteristic of carbon-atom alkane interaction is found in
addition in N 2 -alkane systems. The addition of oxygen and/or moderator (He, Ne) to the N2 -alkane mixture
reveals the fact that little or no HCN is formed in thermal reactions. Experimental "reaction cross-sections” (probability of product formation factors) for particular products are
° C “ 0 Hot °H C H N Hot
° H CU N Hot “ 9 and ° H C H = C H H o t- 0 -22
obtained from reactions involving oxygen and nitrogen, and nitrogen and alkane respectively, competing for the hot carbon atom.
No amines are formed in N2“containing systems, suggesting that ‘ C n N cannot reduce itself to C U H3NH2. High radiation damage results in the production of nitriles in addition to HCU N. This is in contrast to NH3 or RNH2 systems where no HCn N is formed.
Evidence for the mechanism of formation of acetylene-C11 in hot carbon-alkane systems can be garnered
from studies in N2-alkane systems where N2 now serves only as a source of carbon-11. The following alkanes
were studied in oxygen-scavenged systems; methane-H|, ethane-l-H ^, ethane-1, 2-H§, propane-l-H§, propane-2-H2l propane-1, 3 -H 62, c -p ropan e-l-H 22, methane-methane-Hj, ethane-ethane-H62, propane- propane - H i , с -propane - с -propon e - H I*
The yields of HC 11 = CH, НС 11 г CD and DC 11 = CD from attack at primary and secondary carbon atoms
indicated a pronounced selectivity for primary C -H bonds (i.e , attack at methyl groups), the ratio of yields
per C -H bond ( l eC-H/2*C-H ) being about 1. 3. This is in excellent agreement with the structural dependence
of acetylene formation found by Stocklin and Wolf for acyclic hydrocarbons.
The data gives support to the excited intermediate or complex hypothesis, and indicates that the intermediate has a lifetime longer than'several bond vibrations:
[ :C * ]Í + RH------ * [excited intermediate]------ 1 | fragmentation. *-----* stabilization. '
* Research performed under the auspices of the United States Atomic Energy Commission.
107

108 H. ACHE and A. P. WOLF
RÉACTIONS ET MÉCANISMES FAISAN T INTERVENIR DES ATOM ES CHAUDS DE CARBONE ET DES
SYSTÈMES N 2-H 2, Nz -A LC A N E ET N 2-ALCANE-RALÈNTISSEUR; RELATIONS AVEC D'AUTRES SYSTÈMES
SIMPLES. Les auteurs ont étudié la ch im ie des atom es chauds de carbone en utilisant le c a rb o n e -1 1 produit
par diverses réactions nucléaires, telles que 12C (n, 2 n )u C , 12C (p, pn)u C , 14Ы (р ,а )и С , N e (p ,X )u C , u B (p ,n )n C e t 9B e (3He, n ) llC . La réactio n 14N(p, a ) n C est particu lièrem ent intéressante du fait qu’ il est fa cile de c o n
trô ler la dose de rayonnem en ts re çu e par le systèm e e t que l'o n peut obtenir des n iveaux d 'a c t iv i té é lev és .
En ou tre, l ’utilisation de systèm es à substrats m ultiples constitue une m éthode pratique pour d éterm iner e x
p érim en talem en t les « s e c tio n s e ff ica ce s de r é a c t i o n » .
La réaction dom inante des atom es chauds de carbone av e c l ’ azote dans les systèmes contenant de l'hydro
gèn e est c e lle qui donne lieu à la form ation de H n CN , prob ab lem ent par l 'in te rm é d ia ire du ra d ica l U CN.
Le systèm e N2 - H 2 ne donne lieu qu’ à la form ation de H llCN e t l lC H 4. La série h ab ituelle de produits qui
c a ra cté rise l ’ in teractio n a to m e de ca rb o n e -a lc a n e se retrouve ég a le m e n t dans les systèm es N2 -a lc a n e . En
ajoutant de i ’oxygène ou un ralentisseur (Не, Ne) au m élan ge N2 -a lc a n e , on constate qu’au cours des réactions
therm iques HCN n 'est produit qu’en p e tite quantité ou pas du tout. Les « s e c tio n s e ff ic a c e s d e r é a c t i o n »
e x p é rim e n ta le s (fa c te u rs e x p rim a n t la p ro b a b ilité d e fo rm a tio n d e prod u its) pour c e rta in s produ its sont
Q11C O Ch a Ud ° H 11C N c h au(j .
° H llC N c h a u d ' 9 61 ОН и С = С Н сЬа11с1е 0 ’2 2 *
On a obtenu ces valeurs à partir de réaction s dans lesquelles l ’oxygèn e e t l 'a z o te , e t l 'a z o te e t l 'a lc a n e resp e ctiv e m e n t, se font con cu rren ce pour l 'a to m e chaud de carbone. ,
11 ne se form e pas d 'am ines dans les systèm es contenant N2 , c e qui laisse supposer que le ra d ica l n CN
ne peu t ê tre réduit pour donner 11CH 3 NH2 . Lorsque le d om m age radioinduit est im portant, il se m an ifeste
par la production de nitriles venant s'ajou ter à c e lle de H n CN. En revan che, dans les systèmes NH3 ou RNH2 ,
H !1CN ne se forme pas. .
Pour obtenir des renseignem ents sur le m écan ism e de la form ation d 'a c é ty lè n e m arqué par И С dans
les systèm es a to m e chaud d e c a rb o n e -a lca n e , on peut étudier les systèm es N2 -a lca n e où N2 ne jo u e plus que
le rô le d 'un e source de c a rb o n e -1 1 . A l 'a id e de systèm es épurés par l 'o x y g è n e , les auteurs ont étu d ié les
alcan es suivants: m éth an e- Ш 2, é th a n e -2H3 - l , é th a n e -2H2 "1 , 2 , p rop an e-2H3 - l , propane-2H2 "2 , propane-
Î î e - 1 , 3 , c y c lo p ro p a n e -2H2 - l , m é th a n e -m é th a n e -2H4 , é th a n e -é th a n e -2H 6, p ro p a n e -p ro p a n e -2 H 8 ,
cy c lo p ro p a n e -c y c lo p ro p a n e -2Hg.
Les rendem ents en H l l C = C H , H U C = CD e t D u C s C D résultant d 'un e attaque au niveau des atom es
de carbone primaires et secondaires ont révélé nettem ent que les liaisons primaires C-H sont attaquées sé lectiv e
m ent (attaque au niveau des groupes m éth yle), le rapport entre les rendements par liaison C -H (C -H p rim aire/
C -H seco n d aire) é ta n t de l'o rd re de 1 ,3 . C e phén om ène est absolum ent co n fo rm e au fa it que la stru cture
du com posé prim aire e x e rc e une influence sur la form ation d 'a c é ty lè n e , co m m e Stocklin et W olf l'ont cons
ta té pour les hydrocarbures acycliq ues.
Ces données confirm ent l'hypothèse de l'e x iste n ce d 'interm édiaires ou de com plexes excités et indiquent
que la durée de v ie de l ’ interm édiaire est supérieure à plusieurs périodes de vibration des liaisons. .
[ : C - ] * + R H ------J [interm édiaire e x c ité ]-------- Г I *raB™entationL '------- ? stabilisation .
Р Е А К Ц И И И М Е Х А Н И З М Ы , С В Я З А Н Н Ы Е С Г О Р Я Ч И М И А Т О М А М И У Г Л Е Р О Д А И
С И С Т Е М А М И N2 - H 2 , N 2- А Л К А Н И N 2 - А Л К А Н -З А М Е Д Л И Т Е Л Ь , В К Л Ю Ч А Я И Х С В Я З И С Д Р У Г И М И П Р О С Т Ы М И С И С Т Е М А М И . У г л е р о д - 1 1 , о б р а з у е м ы й в р е з у л ь т а т е р а зн ы х я д е р н ы х р е ак ц и й , н ап р и м ер С 12 (п ,2 п ) С 11 С 12 (р ,р п ) С 11 , N 14 ( р ,а ) С 11 , N e ( р ,Х ) С 11 ,В 11 (р ,п ) С 1! и B e 9 (Н е З ,п ) С И , п р и м ен я лся при и зу ч ен и и хи м и и го р я ч и х а т о м о в у г л е р о д а . Р е а к ц и я N *4 ( р ,» ) С 11 особ ен н о п р и го д н а , п о ск о л ь к у м ож но л е г к о р е гу л и р о в а т ь д о зу и зл у ч е ния в с и ст е м е и п олучать акти вн ости в ы с о к о го у р о в н я . Д а л е е , и спо льзо ван и е си ст е м с м н огок р а т н ы м и с у б с т р а т а м и п р е д с т а в л я е т со б о й у д о б н ы й м е т о д п о лу ч е н и я э к с п е р и м е н т а л ь н ы х
"п о п е р е ч н ы х с е ч е н и й р е а к ц и й " . 'О сн овн ой р еак ц и ей "г о р я ч и х " а т о м о в у г л е р о д а с а з о т о м в во д о р о д о со д ер ж ащ и х с и с т е
м а х я в л я е т с я о б р а зо в а н и е Н С u N, в е р о я т н о , ч е р е з п р ом еж уто чн ы й р ад и к ал С 11 N. Т о л ь к о H C 1 j N h С 11 Н 4 о б р а з у ю т с я в N2 — Нг . Д оп о л н и тельн о в с и с т е м а х Ы г-а л к а н о б н а р у ж и в а е т с я с п е к т р об ы ч н ого п р о д у к т а , х а р а к т е р и зу е м ы й в за и м о д е й с т в и е м а т о м а у г л е р о д а и а л к а н а . В

REACTION OF HOT CARBON ATOMS WITH NITROGEN 109
результате добавления кислорода и/или замедлителя (Н е , N e ) в смесь Ыг-алкан выявляется, что м ало образуется или совсем не образуется H C N в результате реакций на тепловйх нейтронах. Экспериментальны е "поперечные сечения реакций" (вероятность коэффициентов образования продуктов) для конкретных продуктов составляю т:
ffr l l O СТН С 11С гор s 9 и гор s 0,22 .
СТН С “ СТН С Ч = СНгор гор
Они получаются в результате проведения реакций, связанных с применением соответственно кислорода и азота , и азота и алкана, с целью получения "горячих" атомов углерода.
В системах, содержащих N 2 , не образуется никаких аминов, что наталкивает на вывод о том, что * C H N не может само по себе восстановиться в C U H 3 N H 2 • Больш ие радиацион~
ные повреждения приводят к образованию нитрилов, помимо H C H N . В этом отличие от систем с NH3 или RNH2 , где не образуется никакого H C ^ N .
Д оказательство относительно механизма образования ацетилена-С 11 в системах "горячий" углеро д -алк ан может быть получено в результате исследований систем Ы г-алкан , в
которых N 2 в настоящее время используется только в качестве источника углеро да -11. С ледующие алканы изучались в системах, из которых удаляется кислород: метан-Нг, этан-1-Нз , этан-1,2 —Н2 , пропан-1 —Нз, пропан-2 — н\, пропан-1,3 —Не , с-пропан-1 — Н2 , м етан -м етан -н !, этан-этан —Не , пропан-пропан — Н | , с-пропан-с-пропан —н| .
Выходы Н С 11* CH , Н С 11 = C D и D C 11 * C D b результате воздействия на первичные и вторичные атомы углерода указывали на значительную селективность в отношении первичных связей С —Н (т .е . воздействие на метиловые группы), при этом величина выхода на связь
С —Н (1 °С —Н /2 °С —Н) составляла приблизительно 1,3. Этот результат очень хорошо согла суется с зависимостью структуры образования ацетилена, обнаруженной Стеклином и В оль фом у ациклических углеводородов.
Эти данные соответствуют гипотезе возбужденного промежуточного продукта или комплекса и свидетельствуют о том, что период полураспада промежуточного продукта меньше, чем некоторые колебания связи.
[ С . ] Î+- R H ----- - [возбужденное промежуточное соединение] — I * расщепление------ * стабилизация
REACCIONES DE ATOMOS CALIENTES DE CARBONO EN SISTEMAS N2 -H 2 , N2-ALCANO, N2‘ ALCANO-
MODERADOR, Y SUS RELACIONES CON OTROS SISTEMAS SIMPLES. Los autores han estudiado la qu ím ica
de los átom os ca lien tes de carbono con ayuda del carb o n o-11 producido en una serie de reaccion es nucleares,
a saber, 12C (n , 2n )1]C , lzC (p , pn)u C , 14N (p ,a ) 1JC , N e (p ,X )u C , 11B (p ,n )11C y % е(*Н е. n )n c . La re a cció n
14N(p, a ) 1JC se presta p a rticu la rm e n te para e llo , pues p erm ite obten er a ctiv id ad es e lev ad as y com p rob ar
fá c ilm e n te la dosis de ra d ia ció n recib id a por e l s is te m a . Por o tra p a rte , e l em p leo de sustratos m u ltiples
con stitu y e un m éto d o ad ecu ad o p ara d e te rm in a r las « s e c c i o n e s e f ic a c e s d e r e a c c i ó n » e x p e r i m e n t a le s .
La re a cció n predom inante de los átom os calien tes de carbono con nitrógeno en sistem as que contienen
hidrógeno es la fo rm ació n de H U C N , p rob ab lem en te por m ed iació n d el ra d ica l n CN . En N 2 -H 2 s ó l o íe
form an H llCN y LlC H 4 . En los sistem as N2 -a lc a n o se observa adem ás e l h ab itu al esp ectro de productos,
c a ra c te r ís tico d e la in te ra cc ió n á to m o de ca rb o n o -a lca n o . Añadiendo o xígen o y m oderador (H e , N e) a la
m e z c la N2 _a lca n o , se pone de r e l ie v e e lh e c h o d e que en las re a ccio n e s té rm ica s se form an can tidad es p e
queñas o nulas de HCN. Las « s e c c io n e s e f ic a c e s de r e a c c i ó n » ex p e rim e n ta le s (.factores de probabilidad
p ara la fo rm ació n de produ ctos) correspondientes a ca d a uno de éstos son
011C O ca lie n te ° H 11CN c a iie n te1 1 ^ Q _ — 1 11 n 1 is/ A f>fl
° H U C N c a l i e n t e ° H 1l C = C H c a i¿e n t e
deducidas de re a ccio n e s en que p articipan oxígeno y nitrógeno, y nitrógeno y a lca n o , resp ectiv am en te , que
com piten por el á tom o c a lie n te de carbono.En sistem as que contien en N2 no se form an am inas, lo que sugiere que el ra d ica l. U CN no puede r e
ducirse para dar origen a 11C H 3NH2 . Cuando los daños provocados por las radiacion es son considerables, se

110 H. ACHE and A. P. WOLF
forman nitrilos adem ás de H n CN , lo que contrasta con los sistem as que contienen NH3 o RNH2, en los que no
se form a H ^C N .
Los estudios sobre sistem as N2 -a lc a n o , en los que e l N2 d esem peñ a m era m e n te la función d e fuente
de ca rb o n o -1 1 , p erm iten aco p iar pruebas que corroboran e l m ecan ism o d e fo rm ación de a c e t i l e n o - 1^ en
sistem as carbono ca lie n te -a lc a n o . Se han estudiado los siguientes aléanos en sistemas depurados con oxígeno:
m e ta n o -гН2 , e t a n o - l - 2H3 , e t a n o - 1 ,2 - 2H2, propano- 1 - 2H3 , p ro p a n o -2 -2Ha , p ro p a n o -1 ,3 -2H 6, c-p ropano-
1 - 2 H2 , m e ta n o -m e ta n o -2H4 , e ta n o -e ta n o - ?H6 , propano-propano-2 H8, с -propano-c -propano - 2 H6 .
El rendim iento en HHC = CH , H 1IC = CD y D11C = CD , formados por ataque en átom os prim arios y s e
cundarios de carbono, indica una m arcad a selectividad por los enlaces C -H primarios (esto es, ataque en los
grupos m e tilo ), siendo la razón de los rendim ientos por e n la c e C -H (C -H p rim a rio /C -H secundario) de 1 ,3 ,
aproxim adam ente. Este dato concuerda m uy satisfacto riam en te con la variació n de la cantidad de a cetilen o
obtenida en función de la estructura, observada por Stô'cklin y W olf en los hidrocarburos a c íc lico s .
Los datos apoyan la hipótesis según la cu a l se form an productos in term ed ios o co m p lejo s e x c ita d o s e
ind ican que la duración d el p rodu cto in term ed io es superior a varias vib racio n es del e n la c e .
[ :C « ] * * R H ------- f [p rod ucto in term ed io e x c ita d o ] --------1 | fragm en tación------- 7 e s ta b iliz a ció n .
INTRODUCTION
The hot atom chemistry of carbon has been discussed in the recent reviews of WOLF [ l j and WOLFGANG [2J. Carbon-11 was first applied to studying inorganic salts by SHARMAN and M cCALLUM [3] . Organic systems were then reported on by SURYANARAYANA and WOLF 14] . Since that time, a large number of papers have appeared involving carbon- 1 1 in recoil studies. The C12 (n, 2n)Cn,' N^fp.aJC 11 and C 12(-y,n)Cn are the most frequently used nuclear reactions. Only one of these, the Ni4(p, aJC11 re action, allows control of the radiation damage to the system [5] . While the damage from the n, 2n and the y, n can be made quite low, the damage from these reactions cannot be readily varied or controlled. The NW(p>a)c n also allows the attainment of a high level of activity; however, in this respect the C 12(7 ,n )C U is probably preferable.
The mechanistic approach to the hot atom chemistry of carbon has been amply documented [1] [2] . Degradative evidence led W OLF et a l. [6 ] to postulate an insertion reaction, i .e. by methylene [СНг: ] , as a possible pathway for the formation of certain recoil labelled molecules. MacKAY et a l. [7] postulated insertion by carbon atoms in order to account for the labelled acetylene and ethylene arising from reaction between carbon - 1 1
and low molecular weight hydrocarbons. Evidence that insertion by methyne [CH:] is mainly responsible for the production of ethylene from the hydrocarbons was adduced from structure dependence studies by W OLF and STÔCKLIN [8 j . It has been suggested 11, 2, 9] that the insertion reactions lead to short-lived intermediates which have numerous alternate modes of decay. Evidence for this has also been presented by RACK et a l. [10]. The lifetime and detailed nature of this intermediate is still in some question. There is also conflicting evidence in the literature on phase dependence and structure dependence. The authors take the view that the intermediate is highly excited but has a lifetime long enough in most cases to allow intermolecular energy transfer (cf. [10]). Evidence w ill be presented in this

REACTION OF HOT CARBON ATOMS WITH NITROGEN 111
paper to show that the common phenomenon of labile hydrogen migration also takes place during the lifetime of this intermediate.
The use of nitrogen as a carbon-11 source has allowed us to investigate the reactions of nitrogen with carbon- 1 1 and the reactions of carbon- 1 1 with organic compounds used in admixture. The reactions of carbon atoms with inorganic molecules, among them nitrogen and its oxides, has been reported on by DUBRIN et a l. [13].
Recently DUBRIN et al. 111 j have reported on the use of deuterium labelled hydrocarbons in studying the mechanism of the formation of ethylene. The double-label (C 11 and H2 in the same compound) technique has also been applied by these authors [9b] in studying acetylene formation. The method of CVETATTOVIC [12] was used to separate the various partially and fully deuterated olefinic compounds produced. As its second part, this paper will present our work along these lines and the implications of the results for the detailed mechanism of reaction.
EXPERIMENTAL -
The technique of irradiation was essentially that of CACACE and WOLF [5] . A ll hydrocarbons used were Phillips Research Grade, purified in all cases by repeated crystallization. Nitrogen, hydrogen, oxygen and the rare gases (Aireo Research Grade) were all assayed at less than 4 ppm of other impurities and were used as such. The deuterated compounds were mainly obtained from Merck Sharpe and Dohme, Canada. Some were synthesized at Brookhaven National Laboratory. Deuterium analyses were provided by Merck and were also carried out at Brookhaven National Laboratory.
The 10 MeV proton beam ofthe Brookhaven 60-incyclotronwasusedfor all irradiations. The irradiation of nitrogen-hydrogen and nitrogen- hydrocarbon mixtures was carried out with the gases in a quartz tank having a 10 mil1 window. A beam intensity of 0.5/uA for 10 s was used in most cases. This corresponds to a radiation dose of about 2X 10' 3 eV/mol. Oxygen was used as scavenger where necessary.
The irradiation of the deuterated and partially deuterated hydrocarbons in the double-label work was carried out with the gases contained in an aluminium vessel with a 3 mil aluminium window.
A ll specifically deuterated hydrocarbons and all deuterated-non- deuterated hydrocarbon mixtures were irradiated with a 5 дА beam for 30 s. In each case the gas composition was 50% hydrocarbon or hydrocarbon mixture, 45.5% N 2 and 4.5% Ог- The gas pressure was 760 mm. Concentrations of oxygen as high as 20% did not alter the product ratios. Analysis of products was done by gas liquid chromatography, references for which can be found in the cited reviews [l , 2 ] .
The doubly labelled acetylenes were isolated from the bulk irradiated gases using a 40-60 mesh silica gel filled column. (No exchange of acety- lenic hydrogens occur when the deuterated and partially deuterated acetylene pass through this column.) The acetylene was collected and reduced [14]
1 mil = 0.001 in

112 H. ACHE and A. P. WOLF
using chromous chloride. No exchange occurs during this process. The reduction is 98% complete in 1 0 min. The three possible ethylenes, C 11H2= CH2, CHH2= C(n )HD 'and C1! DH= CDH, were then separated by the method of CVETANOVIC [12] using the recycling method of ROOT et a l. [15]. An effective column length of 200 ft was used.
THE NITROGEN-ALKANE SYSTEM
In addition to the normal spectrum of products observed from the re action between carbon-11 atoms and alkanes, good yields of HCn = N are also obtained. When nitrogen is present, interestingly enough no amines could be detected, in marked contrast to what is observed in ammonia [5] and in alkyl amines.
ThatHC1:1 = N is not produced by thermal reactions is supported by two facts:
1. A series of runs were made on mixtures made up of 98.7% N 2 and1. 3% ethane in the presence of amounts of oxygen varying from 0% to 20% of the total gas volume. The results indicated a typical scavenger [1] effect for acetylene and ethylene yields but only a very slow decrease in H C^ = N yield with increasing oxygen concentration. When plotting the
tH C 1 1 ^ N 1 n o 0 2 - t H C 1 1 ^ ] obs. [ t 0 2 ]
[ H C U 5 N ] ob,[+02]
ratio versus the [O 2] [ N2] ratio, a straight line is obtained going through the origin (Fig. l ) . This strongly suggests that 0 2 is having no scavenging effect on the formation of HCn = N. Since it is known that thermal carbon atoms react very rapidly with 02, this would support the thesis that HCU = N is formed only in hot reactions. Furthermore, the numerator of the ratio used as ordinate (Fig. 1) is essentially equal to the hot Cu O yield, and the slope of this line then gives us the ratio of a quantity which is in essence a reaction cross section, i .e. ctCO i h o t / o' h c n , h o t = 9 . This means to say that oxygen is about nine times as reactive as nitrogen towards "hot" carbon atoms.
2. The yield of HCu s N is sharply reduced when an increasing amount of moderator (He or Ne) is present in a mixture of 89.5% N2 and 10.5%СгНв during irradiation with protons (Fig. 2). Consistent with theory, neon is more effective than helium as a moderator in this system. The HCU 5 N must therefore be formed in major fraction in non-thermal reactions.
These results for hydrocarbon systems essentially agree with those of DUBRIN et al. [13], who investigated the neon moderation of carbon-11 using ethylene as a scavenger for ’C s N radicals (НС 11 = N and CH2 = CHC11 = N are the products).
In order to determine the experimental "reaction cross section" ratio for HCu s N and НС11 = CH, a plot similar to that in Fig. 1 was made using the [HCn = N ]H0T/[HC1:1 s CHJH0T ratio as ordinate and the [N 2] / [Substrate] ratio as abscissa (F ig.3 ). Here we find that c t h c h N h o t / ctc , h , h o t =0.22.
[0 2] / [n 2] x I 0 '1
Fig . 1
The N w( p . « ) C 1 1 reaction in the system N 2 -C 2H 6.
Ratio o f ДНС 1 1 a N -H O » s N. Д Н С " a N = C “ О HOT-
% Y
IEL
D
HC
MN
Fig . 2
The N 1 4 (p , a )C lt reaction in the system N 2 - C 2 H 6 - Ne or He.
The HC<*=N y ie ld versus moderator concentration.
REAC
TION
OF
HOT CARBO
N
ATO
MS
WITH
NITRO
GEN
113
114 H. ACHE and A. P. WOLF
J M[ s i
F ig .3
T h e N14 ( p ,a ) C n reaction in th e system N2-C 2 H6 .
R atio o f th e HC11 = N - HC“ = C H y ield s.
Using these two experimentally determined ratios, it can be seen that CTco h o t / c t C » h , h o t = 2, a result which is in reasonable agreement with the CTCO h o t /ffc H h o t = 2 . 5 found for non-nitrogen-containing hydrocarbon systems by STÔ’CKLÎN and WOLF [16] .
THE NITROGEN-HYDROGEN SYSTEM
The only products detectable by us resulting from the irradiation of the N2 -H2 system were C 1:1H4 and HCu sN. At high nitrogen concentrations (> 95%), the yield of HCU = N is larger than that of Cn H 4. How much of the Cn H4 (75%) found in a system composed of 82.5% H2 and 17.5% N 2 was due to hot reaction, and how much to the radiolytic reduction of thermal carbon atoms was not determined.
THE INSERTION RATIOS FOR ENERGETIC CARBON ATOMS
The indiscriminate action of singlet methylene (cf. [ 1J ) in its reactions with carbon-hydrogen bonds in alkanes is well known. It was therefore of

REACTION OF HOT CARBON ATOMS WITH NITROGEN 115
interest, in view of the postulated insertion by carbon atoms [7] , to see whether or not the carbon atom itself shows any discrimination in a system involving only carbon-hydrogen bonds. It has been suggested by RACK et al. [10] that carbon-11 insertion is largely statistical. MARSHALL et a l. [9a] have found that both thermal and "hot" carbon atoms prefer attack on v bonds rather than a bonds although the selectivity of attack is less pronounced for the "hot" carbons.
The structural dependence observed by STÔCK.LIN and WOLF 116] (cf. data in [ 1 ]) indicated that the energetic carbon atoms had a marked preference for attack at methyl groups in acyclic hydrocarbons. This preference is born out by our studies on specifically deuterated propanes. The results obtained from propane-l-H|, propane-2-H| and propane-1, 3-Hg are given in Table I.
TABLE I
AC ETYLE N E YIELDS FROM S PE C IF IC A LLY DEUTERATED PROPANES
Compound A cety len e yie lds in % a
H - C “ s C - H b H - C u s C ^ - D b D - C 1 1 a C - D b
C D 3 - C H 2 - C H 3 56.5 1 2 . 1 31 .4
c h 3 - c d 2 - c h 3 69.9 1 2 . 6 17.5
CDj - C H 2 - C D 3 1 7 .4 18.0 64.6
a Sum o f a ll a c t iv ity in a l l three acetylenes = 100%.
b These y ie ld s have been corrected for the actual isotopic com position o f the deuterated
compound. It was c lea r ly not possible to prepare 100% iso top ica lly pure compounds.
The [prim. C-H/s С -Н ]с2нг insertion ratio for "hot" carbon atoms calculated from the results in Table I is = 1. 26. This ratio is only valid for acetylene production. The true prim. C-H/s C-H insertion ratio for "hot" carbon atoms could be obtained only if we knew the complete "hot" product spectrum resulting from carbon insertion in the C-H bonds of propane. The ratio must also ultimately be corrected for the isotope effect (vide infra) . This w ill not alter the value appreciably. The uncertainty in exact isotope composition of the propanes and the uncertainty in the values of the isotope effect make this correction difficult at the present time. It is further interesting to note that the ratio [prim. C-H/s С -Н ]с,н 2 obtained from the structure dependence curves of Stocklin and Wolf is = 1.22. This is remarkable agreement in view of the magnitude of the experimental errors involved in studies using carbon-11. These results suggest that the "hot" carbon atom is a voracious electrophile but also that the nature of the hot process involves a structural dependence which may have to do with the collision cross section of methyl groups and methylene groups. The fact that

116 H. ACHE and A. P. WOLF
a hot process is involved during the insertion is bolstered by the observed distinct lack of selectivity for the weaker secondary carbon-hydrogen band. It is hoped that the detailed energetics and geometry of the insertion process may ultimately result from these studies.
THE ISOTOPE EFFECT
At least three distinct isotope effects may arise from the attack on the hydrocarbon and subsequent decomposition of the intermediate.
These are (relative to the fully protonated compound):1. Insertion isotope effect; a Io isotope effect.
(l) denotes excess kinetic energy; (2) denotes highly excited complex.2. Carbon-carbon homolytic bond scission isotope effect; a 2°e-effect.
3. Carbon-hydrogen homolytic'bond scission isotope effect; a Io isotope effect.
to imply the necessary order of their occurrence.The results for a series of hydrocarbon mixtures are listed in Table II.The HCH= CH/DC11 s CD ratios, corrected for isotopic composition of
the deuterated hydrocarbon, are listed in Table III.Assuming the value for methyl groups (from methane and ethane
CH3 '/ CD3" is = 1.26 and the value for methylene groups (from cyclopropane and cyclohexane) - СНг"/- CD2 * is = 1.08, the values for propane and butane agree rather well with what would be predicted from strict proportionality.
The results do indicate that there are at least two distinct isotope effects operative, and there may be three. The alicyclic compounds can in principle only exhibit effects one and two and the acyclic compounds can exhibit all three. It can be pointed out that the maximum effect would probably involve C-D bond rupture (effect No.3) and this is qualitatively supported by the data.
[ • C11: ] + 1 +D — CD2 - CD3 -» [D - C11- CD2 - CD3] * 2
*+C D 3.
D
The order in which these effects (i.e. 2 and 3) were written is not meant
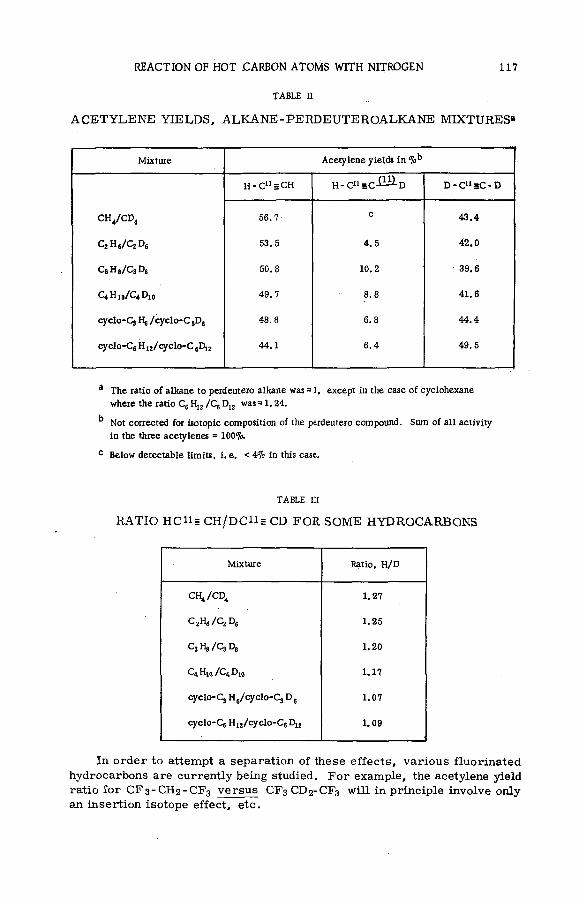
REACTION OF HOT CARBON ATOMS WITH NITROGEN 117
TABLE II
AC E TYLE N E YIELDS, ALK ANE-PERDEUTERO ALK ANE MIXTURES»
Mixture Acetylene yields in
H - C U =C H H - C n = C - ^ D D - C l l a C - D
c h 4/c d 4 56.7 с 43.4
C2 H б/С г Df; . 53.5 4 .5 42.0
C3 Hg/Cg Dg 50.8 10.2 39.6
C4H10/C4DK1 49.7 8.8 41.6
cyc lo -C j Hj / cy c lo -C 3D6 48.8 6.8 44.4
cy c lo -C 6 H 12/cycIo-C 6Di2 44.1 6.4 49.5
a The ratio o f a lkane to perdeutero a lkane was = 1, excep t in the case o f cyclohexane
where the ratio C6 H12 /C6 Dlz was = 1.24.
k Not corrected for isotopic com position o f the perdeutero compound. Sum o f a ll a c tiv ity
in the three acetylenes = ЮС)"?».
c Below d etec tab le lim its , i . e . < 4% in this case.
TABLE III
RATIO HCH= CH/DCH= CD FOR SOME HYDROCARBONS
M ixture Ratio, H/D
c h 4 / c d 4 1.27
1.25
C 3 H, /С 3 D8 1 . 2 0
C tH w / Q D io 1.17
c y c lo -C 3 H 6/ c y c lo -C 3 D 6 1.07
c y c lo -C 6 Н 1 2 /сус1о-С 6 0 1г 1.09
In order to attempt a separation of these effects, various fluorinated hydrocarbons are currently being studied. For example, the acetylene yield ratio for CF 3 -CH 2 -CF 3 versus CF3 CD2-CF3 will in principle involve only an insertion isotope effect, etc.
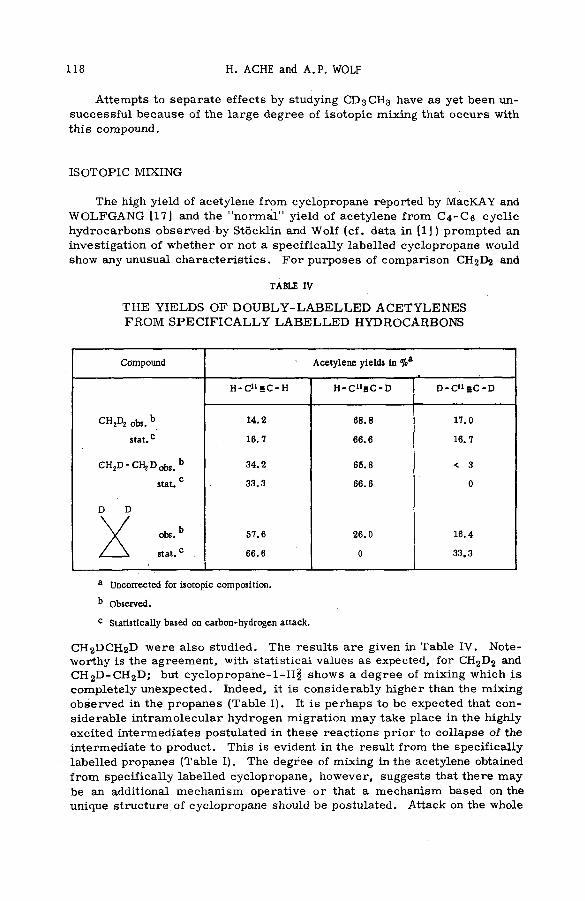
118 H. ACHE and A. P. WOLF
Attempts to separate effects by studying CD3 CH3 have as yet been unsuccessful because of the large degree of isotopic mixing that occurs with this compound.
ISOTOPIC MIXING
The high yield of acetylene from cyclopropane reported by MacKAY and WOLFGANG L17J and the "normal" yield of acetylene from C4-C6 cyclic hydrocarbons observed by Stôcklin and Wolf (cf. data in [1J ) prompted an investigation of whether or not a specifically labelled cyclopropane would show any unusual characteristics. For purposes of comparison CH2D2 and
TABLE IV
THE YIELDS OF D O U B L Y -L A B E L LE D ACETYLENES FROM SPEC IF ICALLY LA B E LLE D HYDROCARBONS
Compound A cety len e yields in °!oa
H - C U = C - H H - C “ b C - D D - C “ = C - D
C H 2D2 0bs. ^ 1 4 .2 6 8 .8 1 7 .0
s t a t .c 1 6 .7 6 6 .6 1 6 .7
e H 2D - C H D obs. b 3 4 .2 6 5 .8 < 3
s ta t. c 3 3 .3 6 6 .6 0
D D
Y obs- b5 7 .6 2 6 .0 1 6 .4
/_ __Л s ta t. c 6 6 .6 0 3 3 .3
a Uncorrected for isotop ic com position,
b Observed.
c S ta tis tica lly based on carbon-hydrogen a tta c k .
CH2DCH2D were also studied. The results are given in Table IV. Noteworthy is the agreement, with statistical values as expected, for CH2D2 and CH 2D -C H 2D; but cyclopropane-1-H§ shows a degree of mixing which is completely unexpected. Indeed, it is considerably higher than the mixing observed in the propanes (Table I). It is perhaps to be expected that considerable intramolecular hydrogen migration may take place in the highly excited intermediates postulated in these reactions prior to collapse of the intermediate to product. This is evident in the result from the specifically labelled propanes (Table I). The degree of mixing in the acetylene obtained from specifically labelled cyclopropane, however, suggests that there may be an additional mechanism operative or that a mechanism based on the unique structure of cyclopropane should be postulated. Attack on the whole

REACTION OF HOT CARBON ATOMS WITH NITROGEN 119
molecule by an initial collision perpendicular to the cyclopropane ring can be suggested. Mixing would then take place prior to reorientation of the appropriate bonds to give the intermediate leading to acetylene.
Mixing because of radiation damage is not probable, since all compounds involved in these double-label studies were irradiated under identical conditions. Thus, the alkane-perdeutero alkane mixtures should have given approximately the same degree of mixing as was observed in the acetylene from the specifically labelled tri- and hexadeutero propanes.
CONCLUSIONS
1. "Hot" carbon atoms will react with nitrogen to give H C s N . Thepresumed intermediate, the cyano radical [ - CN ] , can only be. formed by a hot reaction. :
2. Relative reactivity ratios have been obtained for various substrates in reaction with "hot" carbon atoms. These are сгсо(НОТ)/сгнсн{НОТ) = 9 and о h c n (нот) / a с ,нг (нот) = 0. 22. A "calculated" value of а со (НОТ)/а"сгн2 (нот)=2 is in agreement with the experimental value of STOCKLIN and WOLF 116] .
3. The insertion ratio for CH + primary C-H bonds versus Cn +secón- dary C-H bonds to give acetylene [ГС-Н/20 С-Н]с2нг is =1.26. The preference for primary C-H bonds is in agreement with the structured dependence of acetylene yields.
4. The isotope effect in the over-all reaction giving acetylene is small, ranging from 1.07 to 1.27, and probably related to the so-called "high temperature limit" effects. The detailed nature of these effects requires further study.
5. Isotopic mixing seen in doubly labelled acetylene produced by "hot" reaction on specifically labelled alkanes reflects both the intramolecular nature of the process (since 1 / 1 mixtures of alkane-perdeutero alkane do. not show the same degree of mixing) and the large amount of excitation energy available in the reaction complex. Evidence for mechanistic structural dependence was adduced from the cyclopropane- 1 -Hi^ results.
6 . Detailed studies of this nature using the double-label technique should allow us to determine both the energy and geometry of the excited complex and thus give a better understanding of a specific Hhot"-atom process and high energy reactions (not only those brought about by nuclear transformation) in general.
A C K N O W L E D G E M E N T
The authors wish to acknowledge the invaluable assistance of Dr. Christman of these laboratories who prepared some of the specificallylabelled compounds used in this work.and also his assistance during our carbon - 1 1 "runs" when time was of major importance.

120 H. ACHE and A. P. WOLF
R E F E R E N C E S
[ 1 ] W O LF, A . P . , A d van ces in P h y sical O rganic C h em istry 2 (GO LD, V . , E d .) A c a d e m ic P ress, London
( 1 9 6 4 ) 2 0 1 .
[2 ] WOLFGANG, R . , A d van ces in N u clear C h em istry , (in press).
[ 3 ] SHARM AN, L .J . and M cC A LLU M , K . J . , I. A m e r. c h e m . S o c . T7 (1 9 5 5 ) 2 9 8 9 .
[ 4 ] SU RYAN ARATAN A. B. and W OLF, A . P. , J. phys. C h e m . 6 2 (1 9 5 8 ) 1 3 6 9 .
[5 ] C A C A C E , F . and W O LF. A . P . , J . A m e r. c h e m . S o c . 8 4 ( 1 9 6 2 ) 3 2 0 2 .
[ 6 ] W OLF, A . P . , GORDON. B . and ANDERSON. R .C . , J . A m e r. c h e m . S o c . 78 (1 9 5 6 ) 2 6 5 7 .
[7 ] M acK A Y , C . , PANDO W, М ., POLAK, P . and WOLFGANG, R . , C h e m ica l Effects o f N uclear Transfor
m ations П IAEA, V ienna (1 9 6 1 ) 1 7 .
[8 ] WOLF, A . P. and STÔCKLIN, G. . A bstracts of 146th A .C .S . M eetin g . D en ver. C o lo . (1 9 6 4 ) 32C .
[9 ] C f. also (a ) MARSHALL, M . , M acK A Y , C . and WOLFGANG, R . , J. A m er. c h e m . S o c . (in press);
(b ) DUBRIN, J . , M acK A Y , C . and WOLFGANG, R . , J . A m er. ch e m . S o c . (in press).
[1 0 ] RACK. E. P . . LANG. C .E . and V O IG T, A .F . ; J . c h e m . Phys. 3 8 (1 9 6 3 ) 1 2 1 1 .
[ 1 1 ] DUBRIN, J . . M acK A Y . C . and WOLFGANG, R . , J . A m er. ch e m . S o c . 8 6 ( 1 9 6 4 ) 9 5 9 .
[1 2 ] CV ETANO VIC, R . J . , DUNCAN, F . J . and FALCONER, W . E . , C añ ad . J . C h em . 4 1 ( 1 9 6 3 ) 2 0 9 5 .
[1 3 ] DUBRIN, J . , M acK A Y , C . , PANDOW, M . and WOLFGANG, R . , J . A m er. ch e m . S o c . (in press).
[1 4 ] CROMPTON. C .E . and WOODRUFF. N . . N ucleonics 7 . 3 (1 9 5 0 ) 4 9 . 5 3 .
[1 5 ] ROOT, J. W . . LEE, E .K .C . and ROWLAND, F . S . . S c ie n ce 143 (1 9 6 4 ) 6 7 6 .
[1 6 ] STÔ CKLIN , G . and WOLF, A .P . these"proceedings.
[1 7 ] M acK A Y . C . and WOLFGANG, R. , J . A m er. c h e m . S o c . 83 (1 9 6 1 ) 2 3 9 9 .

COMPETITIVE GAS-PHASE REACTIONS OF C 11 IN BINARY OXYGEN-ALKANE SYSTEMS*
G. S T Ô C K U N * * AND A. P. WOLF
BROOKHAVEN N ATIO N AL LABORATORY, UPTON, LONG ISLAND, NEW YORK,
U N ITED ST A T ES OF AMERICA
Abstract — Résumé — Аннотация — Resumen
COM PETITIVE GAS-PHASE REACTIONS OF C 11 IN BINARY OXYGEN-ALKANE SYSTEM S. C om petition
exp erim en ts h a v e b een ca rrie d out in order to study th e k in etics o f e n e rg e tic r e c o il c a r b o n -1 1 form ed ^
th e C 12(n , 2 n )C 11 and th e C ^ p .p n J C 11 -p ro cess in a lk a n e -o x y g e n m ixtu res .
Th e o x y g e n -a lk a n e system is e sp e cia lly w ell suited for co m p e titio n experim en ts s in ce both substrates
yield very sp e cific produ cts: the re a ctio n of re c o il carbon with oxy g en only (v ia the 0 16(p , pn a ) C u -p rocess)
leads to 98°}o o f th e to ta l induced a c tiv i ty in CO 11, w hile th e m ajo r products in alkanes a re a c e ty le n e and
eth y le n e .
The yields of the representative products have been determ ined in dependence of the oxygen /alkan e ratio
for th e system CH4 / 0 2 , C 2 H6 / 0 2 and C 3 H8 / 0 2 . Th e plot of the e x p e rim e n ta l values for [C O ]/ [C 2H2] and
[C O j/E C g H j versus [O 2J/[su b stra te ] ; is in good agreem ent with the k in etic prediction based on the assumption
of carbon and m ethyne species yielding a ce ty le n e and eth ylen e resp ectiv ely , and the carbon m onoxide being
exclu siv ely form ed v ia carbon atom s. It can be shown that different values for ° c c / 0 C2 H2 anc* ° C o / ° C 2 H4
for d ifferent alkanes are due to a structure d epen den ce of these "re a c tio n cross-sections”. From ' the structure
dependence of the m ajor products in a v a rie ty o f alkanes (C t - C 5) a t constant oxygen co n cen tra tion structure term s h av e b een d eterm in ed . This leads to a unified expression for the yield ratios allow ing th e p red iction
o f the m ajor produ ct yields for sim p le alkanes a t a g iven oxygen co n cen tratio n .
Fu rth erm o re, our results suggest th at the r e a c tiv i ty of a n o n -th e rm a l carb on w ith o xy g en is about s ix
tim es g re a te r than th a t w ith C -H bonds.
REACTIONS COM PÉTITIVES DE UC EN PHASE GAZEUSE DANS DES SYSTEMES BINAIRES O XYGENE-
ALCANE. Les auteurs ont provoqué des réactio n s co m p é titiv e s pour étu d ier, dans des m élan ges d ’ a lca n e e t d 'o xy g èn e , la cin étiq ue d ’atom es chauds de ca rb o n e -1 1 de recu l produits par les réactions n u cléaires 12C (n , 2n) UC e t i2C(p, pn) “ C .
Le systèm e o x y g è n e -a lc a n e se prête p articu lièrem en t bien aux réactions com p étitives du fait que ces
deux corps donnent des produits bien spécifiques: la ré a ctio n du carb o n e d e recu l a v e c l 'o x y g è n e seul (par
la ré a c tio n 160 (р , p n a ) *1C) donne d e l 'o x y d e de carb o n e m arqué (u CO ) dans lequ el se retrou ven t 98% de
l 'a c t i v i t é to ta le in d u ite ; a v e c les a lc a n e s , les p rin cip a u x p rod u its sont l 'a c é t y l è n e e t l 'é th y lè n e .
On a d é te rm in é les rend em ents des produits ca ra cté ristiq u e s en fo n ctio n du rapport o x y g è n e -a lc a n e
pour les systèm es CH 4 / 0 2 , C 2H 6/ 0 2 e t C 3H8 / 0 2 . La courbe des valeurs exp érim en tales pour [C O ]/[C 2 H2]
e t [ C O ] / [ C 2H4 ] en fon ction d e [ 0 ¿ ) /[c o rp s en e x p é r ie n c e ] co n co rd e bien a v e c la c in étiq u e que l 'o n a v a it
prévue en supposant que les espèces carbone et m éthyne produisent respectivem ent de l 'a cé ty lè n e et de l 'é th y -
lè n e , l 'o x y d e d e carbon e provenan t e x clu siv e m e n t des atom es de carb o n e. Il est possible de dém ontrer que
si les valeurs des rapports ° ç o / ° c H e t °C O ^ °C H sont différentes P °ur différents a lca n e s , c 'e s t que les
« s e c tio n s e ffica ce s de ré a ctio n varien t selon la structure. On a d éterm in é les term es de structure à partir
des variations, selon la structure, des principaux produits dans divers alcan es ( C j - C s ) , pour une concentration
en oxygèn e constante. C e c i a perm is ensuite de form uler une définition généralisée des rapports de rendem ent
qui perm et de prévoir les rendem ents en produits principaux pour les alcan es sim ples e t pour une concentration en oxygène donnée.
* Work p erform ed under th e au sp ices of the U nited S tates A to m ic E n ergy C o m m issio n .
* * P resen t address; In stitu t für R a d io ch e m ie , K e rn fo rsch u n g san lag e JD lich , F e d e ra l R ep u b lic o f
G erm an y .
121

122 G. STOCKLIN and A. P. WOLF
En outre, les résultats obtenus par les auteurs donnent à penser que la ré a c tiv ité d'un carbone non ther
m ique a v e c l'oxygèn e est environ le sextuple de la ré a c tiv ité a v e c des liaisons C -H .
К О Н К У Р И Р У Ю Щ И Е Р Е А К Ц И И У Г Л Е Р О Д А - U В Г А З О В О Й Ф А ЗЕ В Б И Н А Р Н Ы Х С И С Т Е М А Х К И С Л О Р О Д —П Р Е Д Е Л Ь Н Ы Е У Г Л Е В О Д О Р О Д Ы . П р о во д и ли сь эк сп е р и м ен т ы по и зуч ени ю ки нетики конкурирую щ их реакций э н е р ге т и ч е с к и х а т о м о в отдачи у г л е р о д а - 1 1 , о б р а зу ю щ и х ся в р е з у л ь т а т е я д е р н ы х реакций С 1 2 ( п , 2 п ) С 11 и С*2 ( p ^ n J C 1* в с м е с я х к и с л о - р о д -п р е д е л ь н ы е у г л е в о д о р о д ы . Э т а с и с т е м а осо б ен н о п одхо ди т дл я и зуч ения конкурирую щих п р о ц е с с о в , п о с к о л ь к у об а к о м п о н ен т а д а ю т в р е з у л ь т а т е р еакц и й сп ец и ф и ч ески е п р о
д у к т ы . Р е а к ц и я а т о м о в отдач и у г л е р о д а с ки слор одо м т о ль к о ( з а с ч е т п р о ц есса О1 6 { р ,р п а) С 11 д а е т 9 8 % общ ей индуцйрованной а к т и в н о ст и в в и д е С О 1 1 в т о в р е м я , ка к гл а в н ы м и прод у к т а м и реакц и и с п р ед ел ь н ы м и у г л е в о д о р о д а м и я в л я ю т ся ац ети лен и э т и л е н .
В ы х о д ы х а р а к т е р н ы х п р о д у к то в о п р е д е л я л и сь в з а в и с и м о с т и от отнош ения к и сл о р о д -
у гл е во д о р о д для с и ст е м С Н 4 /О 2 , С 2 Н6 /О 2 и С3 Н 8 /О 2 • Гр аф и к зави си м ости эксп ер и м ен тальн ы х зн ачен и й дл я отнош ений Í C O 3 / C 2 H 2 ] и [ С О ] / С г Н 4 ] от и зм ен ен и я соотнош ения [ Ог ] / [ с у б с т р а т ] хорош о с о г л а с у е т с я с п р ед ск а за н и я м и ки нетики р еакц и и , основанн ы м и на п р едполож ении, что у гл е р о д с р адикалам и метильной группы д а е т ацетилен и этилен с о о т в е т ст в е н но, в то вр е м я к а к м он ооки сь у г л е р о д а о б р а з у е т с я и склю ч и тельн о з а сч е т а т о м ар н о го у г л е
р о д а . М ож но п о к а з а т ь , ч то р азл и ч н ы е зн ачен и я отношений O co /ac*H t и a c o / ac tH4 для Р а з _ ли чн ы х у гл е в о д о р о д о в об ъ я сн я ю т ся стр уктур н о й за в и с и м о с т ь ю эт и х "сеч ен и й р е а к ц и й ". И з стр уктур ной за в и с и м о с т и осн о вн ы х продуктов в эк сп е р и м ен т а х с различны м и углеводо р одам и ( C j — С 5 ) при постоянной концентрации ки слор ода бы ли оп ределены стр уктур ны е т е р м ы . Э г о
п р и води т к уни ф и ц и р ованн ом у вы р аж ен и ю дл я соотнош ений в ы х о д о в , п о зво л я ю щ е м у п р е д с к а з ы в а т ь вы ходы о сн о вн ы х п р одукто в для п р о ст ы х у гл е в о д о р о д о в при заданн ой ко нц ентр ации к и сл о р о д а .
К р ом е т о г о , п олуч ен н ы е н ам и р е зу л ь т а т ы даю т о сн о ван и я п о л а г а т ь , что реакционная сп о со б н о ст ь н е т е р м а л и зо в а н н о го у гл е р о д а по отнош ению к ки слор оду примерно в ш есть р а з вы ш е реакционной сп о со б н о ст и по отношению к С - Н - с в я з и .
REACCIONES COMPETITIVAS EN FASE GASEOSA DEL n C EN SISTEMAS BINARIOS OXIGENO-ALCANO.
Se han re a liz a d o exp erim en tos co m p etitivo s para estudiar la c in é tic a del á to m o en e rg é tico c a rb o n o -1 1 de
re tr o c e s o , form ad o p o r las r e a c c io n e s ^ ( n , 2 n )n C y 1 2 C (p , p n )n C en m e z c la s d e a l c a n o -o x íg e n o .
El sistem a o x íg e n o -a lca n o se presta particu larm en te para experim en tos de esa índole, dado que ambos
substratos dan productos muy específicos: la reacción d el carbono de retroceso con el oxígeno solam ente (según
e l proceso 1 6 0 (p , p n a )n C ) da origen a 1 1 CO , que representa e l 98^0 de la activid ad to ta l inducida, m ientras
que los principales productos formados en los aléanos son e l a ce tile n o y el etilen o.El rendim iento de los productos representativos se ha determ inado en función de la razón o xíg en o /
alca n o para los sistem as C H 4 /O 2 , C 2 H 6 / 0 2 y C 3 H 8 / 0 2 . La representación g ráfica de los valores e x
perim en tales correspondientes a [ C 0 ] / [ C 2 H 2] y [C O ]/[C 2 H 4 ] en función de [Ог ]/[su b stra to j concuerda
s a tis fa c to ria m e n te co n las previsiones d e c a r á c te r c in é tic o basadas en la hipótesis d e que e l carbono
y e l m etin o produ cen a c e tile n o y e tile n o , re sp e ctiv a m e n te , y de que ¿1 СО p ro vien e e x c lu s iv a
m ente de átomos de carbono. Puede demostrarse que las diferencias en los valores de ° ç o ^ ° C У
correspondientes a distintos a lcan o s se deben a una v a ria ció n de estas « s e c c io n e s e fica ce s de r e a c c i ó n » e n
función de la estructu ra. Basándose en dicha v a ria ció n se han determ in ado los térm inos estru ctu rales en e l
caso de los productos principales de una serie de alcanos (C x - C 5) para una concentración constante de oxígeno.
De esta m anera se d ed u ce una expresión unificada de las razones de rendim ien to , que p erm ite p red ecir los
rendim ientos de los principales productos, en e l caso de alcan os sim ples y para una co n centración de oxígeno
determ inada.Asim ism o, los resultados obtenidos por los autores indican que la reactividad de un carbono no térm ico
con e l oxígeno es unas seis v eces m ayor que la del e n la ce C -H .
INTRODUCTION
Among hot atom studies the reactions of the recoil carbon plays a special role since the chemistry of atomic carbon is a subject of great interest

COMPETITIVE GAS-PHASE REACTIONS OF C 11 123
in physical organic chemistry. So far, its in situ production by means of nuclear processes seems to be the best method for producing this species.
The short-lived C11 offers the advantage of great flexibility in irradiation conditions and concomitant low radiation damage. Considerable work has been done during the past few years, mainly in hydrocarbons, using the 20-min recoil С 11 produced by different nuclear reactions (cf. review by A. P. W OLF [1] ).
Insertion reactions of carbon and its secondary species methyne and methylene seem to provide a reasonably consistent over-all picture [2 - 1 1 ] . Furthermore, the assumption of a collision complex formation and its subsequent de-excitation or decay is also consistent with what is known to date.
Competition experiments in binary oxygen-alkane systems seem to provide additional information on the relative reactivity of atomic carbon and on the postulated mechanisms. In order to study competitive reactions of recoil carbon it is necessary for the competing second substrate to give rise to a product that is different from those of the first substrate. Furthermore, the new product should be simple and stable. Oxygen, besides being an efficient radical- and thermal carbon atom scavenger, is suitable for this type of experiment since it gives rise to CO as the only non-thermal competition product.
EXPERIMENTAL
The nuclear reactions used in this study are the C12 (n, 2n)C1:1-reaction brought about by fast neutrons produced in a LiH target bombarded with 20 MeV deuterons from the Brookhaven 60" cyclotron, and the C12 (p, pn) С 11-reaction brought about by 2-3 GeV protons from the Brookhaven Cos- motron. Additional C11 was produced in the latter reaction (when oxygen was present) by the 0 I6 (p-pna)C1 1 -process.
Irradiations were carried out in quartz or 2 S aluminium vessels. The accompanying radiation dose was between 10-3 and 10~4 eV/mol. Phillips Research Grade hydrocarbons were used after purification by freezing and melting cycles on a vacuum line. Purification of methane has been described before [12] . Matheson oxygen (lecture bottle) was used without additional purification.
Conventional vacuum techniques were used to handle the gaseous samples before and after irradiation. Analysis was carried out by means of radiogas chromatography. Our specific technique has been described before [13] .
RESULTS AND DISCUSSION
In Fig. la - с we have plotted the yield of the major reaction products, carbon monoxide, acetylene and ethylene in percentage of the total induced C11 - activity versus the oxygen-concentration (in volume per cent) for the methane, ethane and propane system. Cn O was the only oxygenated product obtained in the oxygen concentration region studied. Other oxygenated C11- labelled products such as alcohols are present in negligible amounts and
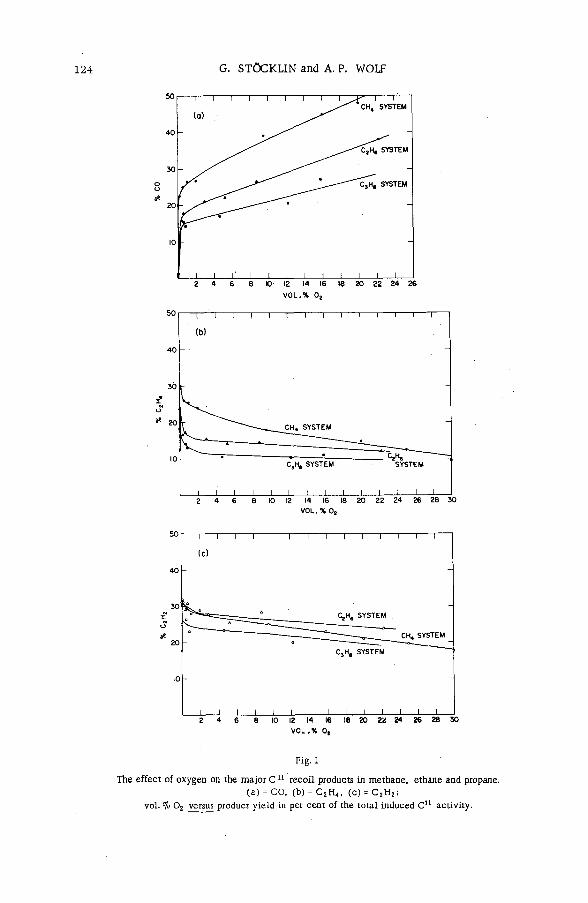
124 G. STOCKLIN and A. P. WOLF
VOL.% 02
F i g . l
T h e e f f e c t o f o x y g e n on th e m a jo r С 11 r e c o il produ cts in m e th a n e , e th a n e and propane,
( a ) = C O , (b ) = C 2 H 4 , ( c ) = C 2H 2 ;
v o l. °¡o 0 2 versus p rodu ct y ie ld in per c e n t o f th e to ta l ind uced C 11 a c tiv i ty .

COMPETITIVE GÁS-PHASE REACTIONS OF С 11 125
their yield does not increase with increasing oxygen concentration. This is consistent with the fact that the C11 - activity balance remains practically unchanged within the 0 2-concentration region studied.
The sharp increase in the carbon monoxide yield at low oxygen concentrations is accompanied by a complete elimination of saturated thermal products such-as methane and ethane (not shown in the figure), and indicates a fast thermal scavenging reaction reaching a saturation at about 1 vol.% oxygen. Actually this saturation is masked by a slow further increase (linear) in the carbon monoxide yield accompanied by a concomitant decrease in the major reaction products acetylene and ethylene. This suggests a competition reaction between oxygen and the substrate for the non-thermal carbon. When we use the term "non-thermal" carbon, we wish to distinguish between the thermal species which can easily be scavenged by small amounts of oxygen,and those species which either have kinetic energies above thermal or excited electronic states or both.
At low oxygen-concentrations the acetylene and ethylene yields suggest that these products also contain a fraction that is more sensitive to oxygen and probably produced by a thermal reaction. Above 3-4 vol.% O?., however, the curves are linear and only one reaction path seems to be predominant for each product. Thus, for the kinetic treatment of the non-thermal re actions we shall exclude the thermal part of the curves and only consider yields at oxygen-concentrations above 3 vol.%.
Implicit in our mechanistic hypothesis [10] is the assumption that the non-thermal acetylene is exclusively formed through the reaction of carbon atoms with the substrate by the insertion reaction postulated by MacKAY et al. [ 4-6] : ■ '.V
R R[С -11]* + HCH-> [ H - Cn -CH]* -> -R + -R' + HCn = CH. (1)
•• R' - R'
If the insertion step is the "rate-determining" reaction (the decay of the excited intermediate being fast), we may write for the rate of the acetylene production:
RC2H2 = ôC2h2[S ] [C] . (2)
It is further implicit in our second mechanistic hypothesis that ethylene is exclusively formed by the insertion reaction of methyne into primary C -H bonds of the hydrocarbon [1 0 ] .
[C UH]+ * + H3C-CH2R -» Ç11 -CH 2 íCH 2R] * -» • CH2R + H2Cn = CH2. (3)
The "rate" of the ethylene production is then:
RC2h4 6C2hJ SH C H ]. (4)

126 G. STOCKLIN and A. P. WOLF
The sigmas are structure-dependent "reaction cross-sections" rather than rate constants, and S stands for substrate.
For the "rate" of the CO-production we imply that this product is exclusively formed via the reaction of atomic carbon with oxygen:
[С-11]* + Оч -> [CH0 2I* - » C U O + -Ô-. (5)
This reaction has been postulated by MacKAY et al. f4 ] and we have also shown [141 that the reaction of recoil carbon with pure oxygen (using the O16 (p, pna)Cn -reaction) leads to С110 with 98% of the total induced activity, and that practically no phase effect can be observed when this reaction is carried out in liquid oxygen where a stabilization of the excited C 0 2 intermediate could be expected.
If the first step of Eq. (5) is the rate-determining step (the decay of the excited intermediate again being fast), we may write for the rate of the C o production:
R C O = W ° 2 H C ] . ( 6 )
In binary hydrocarbon-oxygen systems we have to assume that reactions of the secondary species CH and CH9 do not lead to CO. If this is true, the ratio [С] /[CH] should be a constant over the oxygen-concentration range studied.
The ratio [C ] /[CH] is connected with the yield ratio [C 2H2] / [C 2H4] by the expression:
R c 2h 2 6 c 2h 2 [ c ] t C 2 H 2 l
Rc2Hj = 5Сгн4 [CH] = [C2H4] ' ( >
This means that the ratio [C 2 Щ ] /[C 2 H4 ] should be a constant within the oxygen-concentration range studied when [C]/[CH] is constant. This is the case at least for the ethane and propane system. Within the experimental error the ratio [ C2HjJ /[C2H4] is constant for the ethane and propane system. In the methane system this ratio increases by a factor of about 2.8 between 0.5 and 50% 0 2, but only by a factor of 1.3 in the range from 9.5 to 30% 0 9, the latter range having been used. For the kinetic treatment we shall exclude the thermal part of the oxygen curves (Fig.la-c). Hence, we will only use values from the oxygen-concentration range above3 vol. %. From this concentration on the carbon monoxide as well as the acetylene and ethylene curves are straight lines and possible thermal contributions more sensitive to oxygen have already been eliminated. In case of the CO we have to substract the thermal contribution by extrapolating the linear part of the curve to the intercept with the ordinate.
For the ratio [COhot] /[C2H2 ] and [COhot] /[C 2H4] we obtain:
[COhoJ _ ¿со W IC 2H2] ¿C2H2[S] ’
(8)
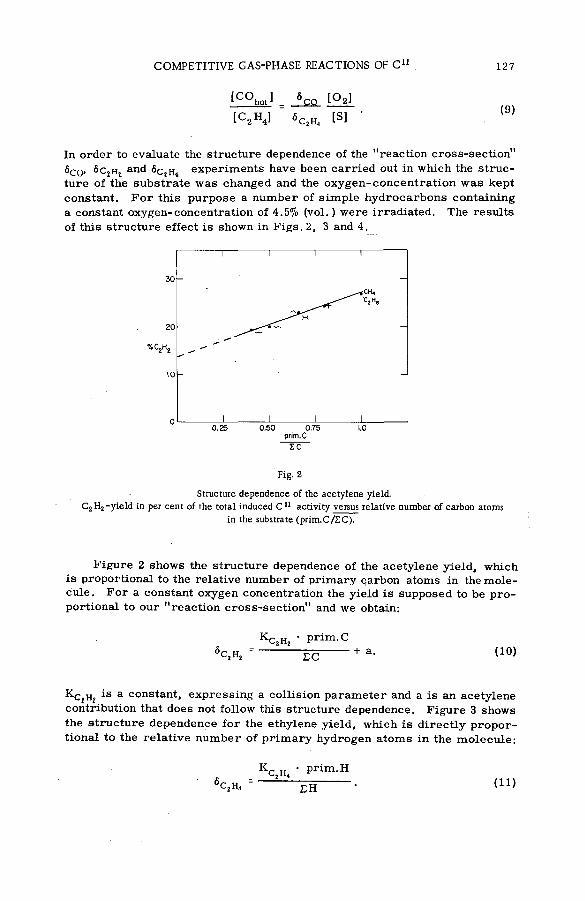
COMPETITIVE GAS-PHASE REACTIONS OF C 11 127
tCQhot] . _^£L Ш[C 2 H4] ôC2H4 [S] • (9)
In order to evaluate the structure dependence of the "reaction cross-section" ôco, 5c2Hj and óc¡Hl experiments have been carried out in which the structure of the substrate was changed and the oxygen-concentration was kept constant. For this purpose a number of simple hydrocarbons containing a constant oxygen-concentration of 4.5% (vol. ) were irradiated. The results of this structure effect is shown in Figs. 2, 3 and 4.
zc
F ig . 2
■ S tru ctu re d e p e n d e n ce o f the a c e ty le n e y ie ld .
C 2 H2 - y ie ld in p er c e n t o f th e to ta l in d u ced С 11 a c tiv i ty versus r e la t iv e nu m b er o f ca rb o n ato m s
in th e substrate (p r im .C /S C ) .
Figure 2 shows the structure dependence of the acetylene yield, which is proportional to the relative number of primary carbon atoms in the molecule. For a constant oxygen concentration the yield is supposed to be proportional to our "reaction cross-section" and we obtain:
k c 2h 2 ' prim. С
Ôc2h2 = LC + a' (10)
Ксгн2 is a constant, expressing a collision parameter and a is an acetylene contribution that does not follow this structure dependence. Figure 3 shows the structure dependence for the ethylene yield, which is directly proportional to the relative number of primary hydrogen atoms in the molecule:
K_ „ ■ prim.H £ - c t H<________________
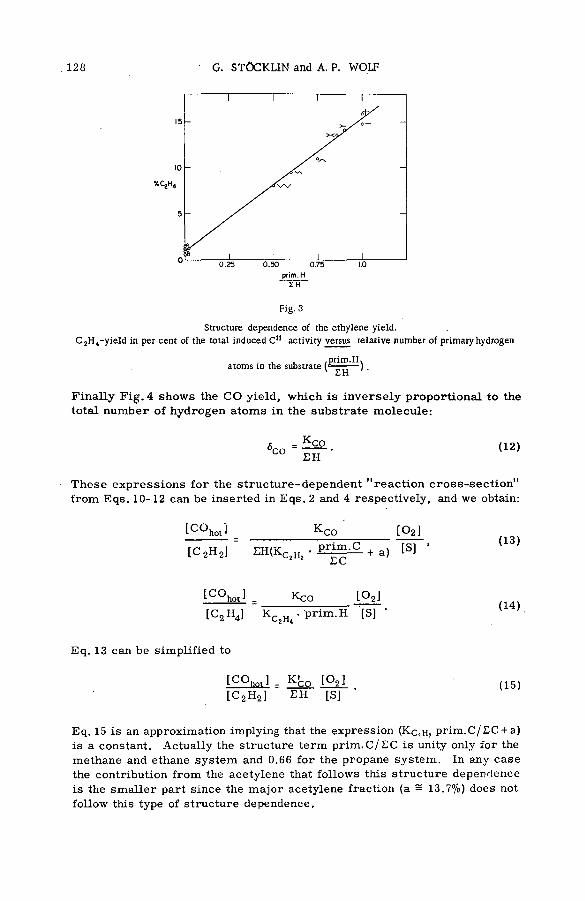
128 G. STOCKLIN and A. P. WOLF
Ï H
Fig- 3S tru ctu re d ep en d en ce of the e th y le n e y ie ld . .
C 2H 4-y ie ld in p er c e n t of th e to ta l in d u ced C 11 a c tiv i ty versus r e la t iv e n u m ber o f p rim a ry h ydrogen
ato m s in th e sub strate ) .E H
Finally Fig. 4 shows the CO yield, which is inversely proportional to the total number of hydrogen atoms in the substrate molecule:
6 = Î S ç oCO EH
(12)
These expressions for the structure-dependent "reaction cross-section" from Eqs. 10-12 can be inserted in Eqs. 2 and 4 respectively, and we obtain:
[ C O К CO ___ [OzJ_
[C 2H 2] EH(KciHi . pr“g — + a) [S] ’(13)
[CO hot1[C2 H4]
Kco [0 2]Kr „ • prim.H [S] (14)
Eq. 13 can be simplified to
[ C O j ^ ] = к CO [ o 2
[C 2H2] EH [S](15)
Eq. 15 is an approximation implying that the expression (Kc,h, prim.C/EC + a) is a constant. Actually the structure term prim.C/EC is unity only for the methane and ethane system and 0.66 for the propane system. In any case the contribution from the acetylene that follows this structure dependence is the smaller part since the major acetylene fraction (a = 13.7%) does not follow this type of structure dependence.
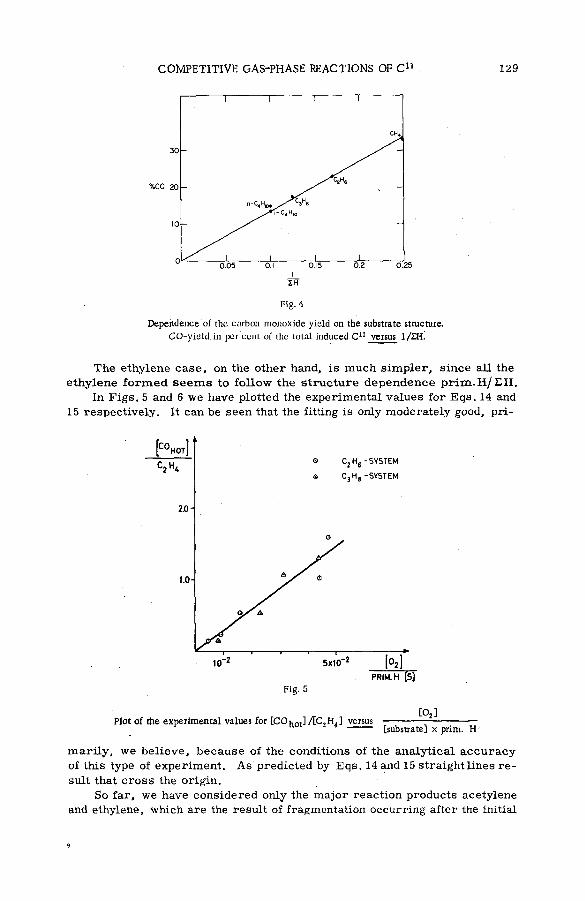
COMPETITIVE GAS-PHASE REACTIONS OF C 11 129
F ig . 4
D ep en d en ce o f th e carb o n m o n o xid e yield on th e su bstrate s tru ctu re .
C O -y ie ld in per c e n t of the to ta l induced C u versus 1 /2 H .
The ethylene case, on the other hand, is much simpler, since all the ethylene formed seems to follow the structure dependence prim -H/EH.
In Figs. 5 and 6 we have plotted the experimental values for Eqs. 14 and 15 respectively. It can be seen that the fitting is only moderately good, pri-
Fig . 5
t 0 2 ]P lo t o f th e e x p e r im e n ta l v a lu e s for [C O h n t] Д С .Н . ] versus ---------------------------------------
[su b strate] x p rim . H
marily, we believe, because of the conditions of the analytical accuracy of this type of experiment. As predicted by Eqs. 14 and 15 straightlines result that cross the origin.
So far, we have considered only the major reaction products acetylene and ethylene, which are the result of fragmentation occurring after the initial
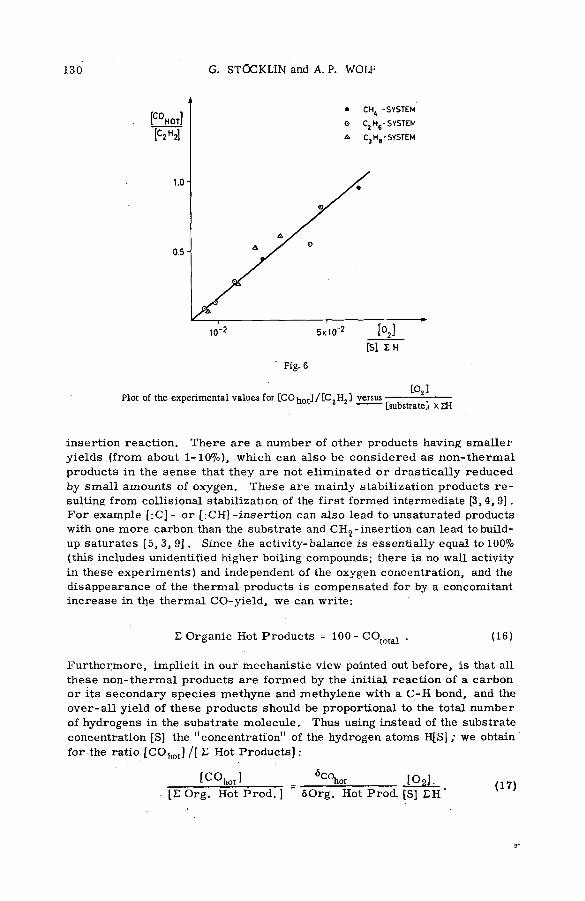
130 G. STOCKLIN and A. P. WOLF
' F ig . 6
[o2]P lo t o f th e e x p e r im e n ta l v a lu e s for [CO v,nr] / [ C , H , ] v e rs u s ----------------------------
uui z t ---------- [su bstrate] x f f i
insertion reaction. There are a number of other products having smaller yields (from about 1 - 1 0 %), which can also be considered as non-thermal products in the sense that they are not eliminated or drastically reduced by small amounts of oxygen. These are mainly stabilization products resulting from collisional stabilization of the first formed intermediate [3,4, 9]. For example [:C ] - or [:CH] -insertion can also lead to unsaturated products with one more carbon than the substrate and CH^-insertion can lead to buildup saturates [5, 3, 9]. Since the activity-balance is essentially equal to 100% (this includes unidentified higher boiling compounds; there is no wall activity in these experiments) and independent of the oxygen concentration, and the disappearance of the thermal products is compensated for by a concomitant increase in the thermal CO-yield, we can write:
E Organic Hot Products = 100- COtotal . (16)
Furthermore, implicit in our mechanistic view pointed out before, is that all these non-thermal products are formed by the initial reaction of a carbon or its secondary species methyne and methylene with a C-H bond, and the over-all yield of these products should be proportional to the total number of hydrogens in the substrate molecule. Thus using instead of the substrate concentration [S] the "concentration" of the hydrogen atoms H[S] ; we obtain for the ratio [COhot] /[ L Hot Products] :
[COhoJ _ 6c°hot [О 9].[E Org. Hot Prod. ] ôOrg. Hot Prod. [S] EH ’
(17)
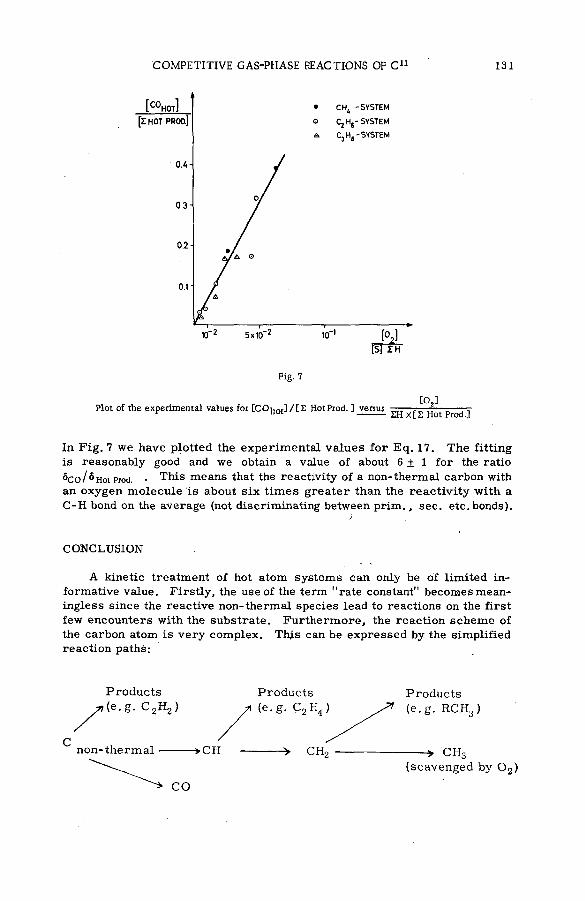
COMPETITIVE GAS-PHASE REACTIONS OF C 11 131
Plot of the experim enta l values for [CO jj0t]/[2; Hot Prod. ] versus д н x [2 Prod 3
In F ig . 7 we have plotted the experim ental values for Eq . 17. The fitting is reasonably good and we obtain a value of about 6 + 1 for the ratio 5Co/ 5 Hot Prod. • This means that the reactivity of a non-thermal carbon with an oxygen m olecule is about six tim es g re a te r than the reactiv ity with aC-H bond on the average (not discriminating between p rim ., sec. etc. bonds).
; .
CONCLUSION .
A kinetic treatm ent of hot atom system s can only be of limited informative value. F irstly , the use of the term "ra te constant" becomes meaningless since the reactive non-thermal species lead to reactions on the first few encounters with the substrate. Fu rth erm ore, the reaction schem e of the carbon atom is very complex. This can be expressed by the simplified reaction paths:
Prod u cts (e .g . RCH3 )
------> CH3(scavenged by 0 2)
P rod u cts f (e .g . C 2 H2 )
P rod u cts (e .g . C2 H4 ;
n on-therm al -»CH CH9
CO

132 G. STOCKLIN and A. P. WOLF
The o v e r-a ll reactiv ity of an alkane with re s p e c t to n on-therm al carbon and its secondary species is essentially due to the number of available C-H bonds in the substrate m olecule. The m ore detailed treatm ent of the acetylene and ethylene production and the reasonable good agreement between the experim ental results and the postulated "k in etics" provides additional evidence for the m echanistic view which we have exp ressed in these assum ptions.
R E F E R E N C E S
[1] WOLF, A. P. , Advances in Physical Organic Chemistry I I (GOLD, V . , Ed. ), Academic Press (1964)
201-77.
[2] WOLF, A. P ., GORDON, R. and ANDERSON, R .C ., J. Amer. chem. Soc. 78 (1956) 2657.
I3J YANG, J. Y. and WOLF, A. P. , J. Amer. chem. Soc. 82 (1960 ) 448.
[4] MacKAY, C. , PANDOW, M . , POLAK, P. and WOLFGANG, R. , Chemical Effects of Nuclear Trans
formations^. IAEA, Vienna (1961) 17-26.
[5] MacKAY, C. and WOLFGANG, R. , J. Amer. chem. Soc. 83 (1961) 2399.
[6] MacKAY, C. , POLAK, P., ROSENBERG, H. E. and WOLFGANG, R. L . , J. Amer. chem. Soc. 84(1962)
308. *
[7] MacKAY, C. and WOLFGANG, R , Radiochim. Acta 1_ (1962) 42. '
[8] RACK, E. P. and VOIGT, A. F. , J. phys. Chem. 67 (1963) 198.
[9] STÔCKLIN, G. and WOLF, A. P ., J. Amer. chem. Soc. 8£ (1963) 229.
[10] STÔCKLIN, G. and WOLF, A. P. , Methods of Preparing and Storing Marked Molecules, EURATOM,
Presses Académiques Européennes, Brussels (1964) and: Chem. and Ind. 42 4(1964)46.
[11] DUBRIN, J ., MacKAY, C. and WOLFGANG, R ., J. Amer. chem. Soc. 86 (1964) 959.
[12] STÔCKLIN, G. , WOLF, A. P. et al. , J. phys. Chem. 67 (1963).
[13] STÔCKLIN, G. , CACACE, F. and WOLF, A. P ., Z. anal. Chem. 194 (1963) 406.
[14] STÔCKLIN, G. and WOLF, A. P ., to be published.

THE E F F E C T OF KINETIC EN ERG Y ON THE REACTIONS OF NUCLEOGENIC CARBON ATOMS
WITH HYDROCARBONS
J . DUBRIN, H. ROSENBERG, R. WOLFGANG STERLING CHEMISTRY LABORATORY,
YALE UNIVERSITY, NEW HAVEN, CONN.,AND
C. MacKAY HAVERFORD COLLEGE, HAVERFORD, PA.,
UNITED STATES OF AMERICA
Abstract — Résumé — Аннотация — Resumen
THE EFFECT OF K INETIC ENERGY ON THE REACTIONS OF NUCLEOGENIC CARBON ATOMS W ITH
HYDROCARBONS. The basic technique for studying the effect of kinetic energy on reactions of С atoms pro
duced by nuclear transformation involves the well-known method of moderation w ith inert gases. However,
this can be combined with other techniques such as (1) the use of scavengers to detect processes involving long-
lived radicals, (2) degradative studies that serve to fix the position occupied by the labelled atom, (3) double
tracer studies in which a reactant is pa rtia lly labelled w ith deuterium and the isotopic composition of the
labelled products is determined in order to establish the origin of the hydrogen which they contain. In this
paper new results on product yields in two systems, neon-ethylene and neon-ethane, w il l be presented, and
the relation of these results to other work involving degradative studies and double tracer experiments w il l be
discussed.
The discussion of the neon-ethylene results is in terms of the two previously postulated insertion m e
chanisms, insertion of the С atom into the C = C and into the C-H bonds to give C-C2 H^adducts, As neon
concentration is increased no product is elim inated, but the relative yields of products are altered markedly.
Those products such as acetylene and v in y l acetylene which can be formed from the in it ia l C-C2H 4 adducts
v ia processes with a high energy requirement decrease in importance, and the yields of products formed in
low energy processes, such as C5 compounds, increase. The ethane pattern is sim ilar.
Degradative studies have already shown that the intramolecular C*1 distribution in aliene and methyl-
acetylene formed from ethylene is affected by neon moderation. These results imply both participation of the
C *C and C-H bonds in formation of these products, and a dependence of the ratio of attack at the two bond
types on the kinetic energy of the reacting С atom.
Other work involving double tracer studies on acetylene formation from single molecules such as CH2CD2,
CH3CH2D, CD9CDH2 and from various mixtures of other labelled alkanes and alkenes indicates that the re
moval of k ine tic energy from, the С atom has li t t le effect on the re la tive rate of attack at various types of
C-H bonds. ,
EFFET DE L’ENERGIE CINETIQUE SUR LES RÉACTIONS DES ATOMES DE CARBONE NUCLÉOGÉNIQUES
AVEC DES HYDROCARBURES. Le procédé fondamental permettant d'étudier l'e ffet de l'énergie cinétique
sur les réactions des atomes de carbone produits par transformation nucléaire se fonde sur la méthode bien
connue du ralentissement 5 l'aide de gaz inertes. Toutefois, i l peut être combiné I d'autres procédés: 1. em
ploi d'agents de balayage pour déceler les processus dans lesquels interviennent des radicaux de longue période;
2, études de dégradation qui servent à déterminer la position occupée par l'atome actif; 3, études à l'a ide
d'un double indicateur, dans lesquelles un coips en réaction est partiellement marqué par le deutérium et la
composition isotopique des produits marqués est déterminée en vue d’étab lir l'orig ine de l'hydrogène que ces
produits contiennent. Les auteurs présentent des résultats nouveaux sur les rendements dans les deux systèmes
néon-éthylène et néon-éthane, et discutent la relation entre ces résultats et ceux d’autres travaux fondés sur
des études de dégradation et des expériences à l ’aide d’un double indicateur.
133

134 J. DUBRIN et al.
Pour ce qui est des résultats relatifs au système néon-éthylène, la discussion se fonde sur les deux mé
canismes d’insertion antérieurement admis, à savoir insertion de l ’atome de carbone dans la double liaison
C = C et dans la liaison C-H, de manière à donner des produits d'addition C-C2H4. Lorsque la concentration
en néon augmente, on retrouve tous les produits, mais les rendements relatifs varient sensiblement. Des produits
tels que l ’acétylène proprement dit et le vinyl-acétylène, qui peuvent se former à partir des produits d’addition
initiaux C-C2H4par des processus exigeant une forte énergie, perdent de leur importance, tandis que le rende
ment en produits formés à basse énergie, tels que les composés d<; C5, accusent une augmentation. L'éthane
suit un processus sim ila ire. '
Les études de dégradation ont déjà montré que la distribution de i*C intramolécula ire dans l ’aliène
et le méthylacétylène formés à partir de l ’éthylène est influencée par le ralentissement dû au néon. Ces
résultats impliquent à la fois la participation de la double liaison C = C et de la liaison C-H à la formation
des produits et le fa it que le rapport d’attaque aux deux types de liaisons dépend de l ’énergie cinétique de
de l'atome de carbone en réaction.
D'autres travaux fondés sur des études à l ’aide d'un double indicateur, sur la formation d'acétylène
à partir de molécules isolées, telles que CH2CD2 , CH3CH2D, CD3CDH2, et à partir de divers mélanges
d’autres alcanes et alcènes marqués indiquent que la perte d'énergie cinétique par l'atome de carbone exerce
peu d’effet sur la vitesse d'attaque re lative aux diverses liaisons C-H.
Э Ф Ф Е К Т К И Н Е Т И Ч Е С К О Й Э Н Е Р Г И И П Р И Р Е А К Ц И Я Х А Т О М О В Я Д Е Р Н О Г Е Н И Ч Е С К О -
Г О У Г Л Е Р О Д А С У Г Л Е В О Д О Р О Д А М И . О с н о в н а я м е т о д и к а и з у ч е н и я э ф ф е к т а к и н е
т и ч е с к о й э н е р г и и п р и р е а к ц и я х а т о м о в у г л е р о д а , о б р а з у е м ы х в р е з у л ь т а т е я д е р н ы х п р е о б р а
з о в а н и й , с в я з а н а с и с п о л ь з о в а н и е м х о р о ш о и з в е с т н о г о м е т о д а з а м е д л е н и я с п о м о щ ь ю и н е р т
н ы х г а з о в . О д н а к о э т о т м е т о д м о ж е т с о ч е т а т ь с я с д р у г о й м е т о д и к о й , к а к , н а п р и м е р , 1 ) и с
п о л ь з о в а н и е а к ц е п т о р о в р а д и к а л о в д л я о б н а р у ж е н и я п р о ц е с с о в , с в я з а н н ы х с д о л г о ж и в у щ и м и
р а д и к а л а м и ; 2 ) и с с л е д о в а н и я я в л е н и й д е г р а д а ц и и д л я у с т а н о в л е н и я п о л о ж е н и я , з а н и м а е м о г о
м е ч е н ы м а т о м о м ; 3 ) и с с л е д о в а н и я с и с п о л ь з о в а н и е м д в о й н ы х и н д и к а т о р о в , в к о т о р ы х р е а г и р у
ю щ е е в е щ е с т в о ч а с т и ч н о м е т и т с я д е й т е р и е м , и и з о т о п н ы й с о с т а в м е ч е н ы х п р о д у к т о в о п р е д е
л я е т с я с ц е л ь ю у с т а н о в л е н и я п р о и с х о ж д е н и я в о д о р о д а , к о т о р ы й в н и х с о д е р ж и т с я . В д о к л а д е
п р е д с т а в л е н ы н о в ы е р е з у л ь т а т ы о т н о с и т е л ь н о в ы х о д о в п р о д у к т о в в д в у х с и с т е м а х , н е о н -
э т и л е н и н е о н — э т а н , и о б с у ж д а е т с я в о п р о с о в з а и м о с в я з и э т и х р е з у л ь т а т о в с д а н н ы м и д р у г о й
р а б о т ы , связанной с и с с л е д о в а н и я м и я в л е н и й д е г р а д а ц и и и п р о в е д е н и е м о п ы т о в с и с п о л ь з о
в а н и е м д в о й н ы х и н д и к а т о р о в .
О б с у ж д е н и е р е з у л ь т а т о в и з у ч е н и я с и с т е м ы н е о н — э т и л е н п р о и с х о д и т в р а м к а х д в у х р а
н е е о п р е д е л е н н ы х м е х а н и з м о в в в е д е н и я а т о м о в у г л е р о д а в с в я з и С — С и С — Н с ц е л ь ю п о
л у ч е н и я п р о и з в о д н ы х С — С 2 Н 4 . П о м е р е у в е л и ч е н и я к о н ц е н т р а ц и и н е о н а н е у с т р а н я е т с я н и
к а к о г о п о о л у к т а . о л н а к о з а м е т н о м е н я ю т с я о т н о с и т е л ь н ы е в ы х о д ы п р о д у к т о в . Т а к и е п р о
д у к т ы , к а к а ц е т и л е н и в и н и л а ц е т и л е н , к о т о р ы е м о г у т б ы т ь о б р а з о в а н ы и з п е р в о н а ч а л ь н ы х
п р о и з в о д н ы х С — С 2 Н 4 в р е з у л ь т а т е о с у щ е с т в л е н и я п р о ц е с с о в с б о л ь ш и м р а с х о д о м э н е р г и и ,
т е р я ю т с в о е з н а ч е н и е , в т о в р е м я к а к у в е л и ч и в а ю т с я в ы х о д ы п р о д у к т о в , о б р а з у е м ы х в х о д е
п р о ц е с с о в н и з к о й э н е р г и и , к а к н а п р и м е р , С 5 с о е д и н е н и я . С х е м а э т а н а я в л я е т с я а н а л о г и ч н о й .
И с с л е д о в а н и я я в л е н и й д е г р а д а ц и и у ж е п о к а з а л и , ч т о н а в н у т р и м о л е к у л я р н о е р а с п р е д е
л е н и е С 11 в а л л е н е и м е т и л а ц е т и л е н е , о б р а з у е м о м и з э т и л е н а , о к а з ы в а е т в л и я н и е з а м е д л е
н и е н е о н а . Э т и р е з у л ь т а т ы п о д р а з у м е в а ю т к а к у ч а с т и е с в я з е й С — С и С — Н в о б р а з о в а н и и
э т и х п р о д у к т о в , т а к и з а в и с и м о с т ь в е л и ч и н ы в о з д е й с т в и я н а о б а т и п а с в я з и о т к и н е т и ч е с к о й
э н е р г и и р е а г и р у ю щ е г о а т о м а у г л е р о д а .
Р е з у л ь т а т ы д р у г о й р а б о т ы , с в я з а н н о й с исследованиями и с п о л ь з о в а н и я д в о й н ы х и н д и
к а т о р о в , п о п р о б л е м а м о б р а з о в а н и я а ц е т и л е н а и з е д и н и ч н ы х м о л е к у л , к а к н а п р и м е р , C H 2 C D 2 ,
CH 3CH 2D, CD 3CDH 2 и и з р а з л и ч н ы х с м е с е й д р у г и х м е ч е н ы х а л к а н о в и а л к е н о в , п о к а з ы в а ю т ,
ч т о с н я т и е к и н е т и ч е с к о й э н е р г и и с а т о м а у г л е р о д а о к а з ы в а е т н е б о л ь ш о е в л и я н и е н а о т н о
с и т е л ь н у ю с к о р о с т ь в о з д е й с т в и я п р и р а з л и ч н ы х т и п а х с в я з е й С — Н .
EFECTO DE LA ENERGIA C INETICA EN LAS REACCIONES DE ATOMOS NUCLEOGENOS DE CARBONO
CON HIDROCARBUROS. La técnica básica para estudiar la influencia ejercida por la energía cinética en las
reacciones de átomos de carbono producidos por transformación nuclear recurre a l método bien conocido de
moderación con gases inertes. Ahora bien, éste puede combinarse con otras técnicas tales como; 1) empleo
de depuradores para detectar procesos en que intervengan radicales de período largo; 2) estudios por degrada
ción para determinar la posición ocupada por el átomo marcado; 3) estudios con indicadores dobles, en los
que un reactivo se marca parcialmente con deuterio y se determina la composición isotópica de los productos
marcados con miras a establecer e l origen del hidrógeno que contienen. En la memoria se exponen nuevos

% EFFECT OF KINETIC ENERGY ON CARBON ATOMS REACTIONS 135
resultados sobre e l rendimiento en productos de dos sistemas, neón-etileno y neôn-etano, y se estudia la re
lación de estos resultados con otros trabajos que comprenden estudios por degradación y experimentos
con indicadores dobles. .
Los resultados referentes al sistema neón-etileno se discuten en función de dos mecanismos de inserción
previamente propuestos, a saber, la inserción del átomo de carbono en los enlaces C = C y C-H para formar
compuestos de adición C-C2H4. A l aumentar la concentración de neón no desaparece ningún producto, pero
los rendimientos relativos sufren modificaciones considerables. Los productos tales como e l acetileno y el
vin il-acetileno que pueden formarse a partir de los compuestos in icia les de adición C-C2H4, en virtud de
procesos que requieren gran cantidad de energía, disminuyen en importancia, mientras que aumenta el rendi
miento de los productos formados por procesos que exigen poca energía, tales como los compuestos C5. Algo
semejante sucede con el etano. .
Los estudios por degradación han mostrado que la distribución intramolecular de UC en e l aleño y e l
m etilacetileno formados a partir de etileno es afectada por e l efecto moderador del neón. Ello im p lica la
participación de los enlaces C = C y C-H en la formación de estos productos, así como una relación de de
pendencia entre la razón de ataque en esos dos tipos de enlace y la energía cinética del átomo de carbono
que interviene en la reacción.
Otros estudios con indicadores dobles acerca de la formación de acetileno a partir de moléculas sen
c illas tales como CH2CD2, CH3CH2D, CD3CDH 2 y a partir de varias mezclas de otros alcanos y alquenos
marcados indican que la pérdida de energía cinética por el átomo de carbono ejerce escaso efecto en el índice
relativo de ataque en varios tipos de enlace C-H.
INTRODUCTION
Reaction mechanisms oí îree carbon atoms have been investigated using C11 produced by n u clear reactio n . These carbon atom s, because of the nuclear reco il associated with the formation p ro cess , will p ossess excess kinetic energy. Thus in the photonuclear form ation of C11,
C1 2 + y -> C11 +n ,
the C il will initially p ossess 105 eV o r m o re . This energy will be lost in successive collisions. When an energy of the o rd er of magnitude of 10 eV (still quite high by chem ical standards) is reached the atom m ay undergo a hot reaction . A lternatively, it m ay be furth er degraded to th erm al en ergies, norm ally 1 0 '1- 10-2 eV.
Two questions then a r is e : Do m ost carbon atom s re a c t while hot o r afte r therm alization? What is the effect of e x ce s s kinetic energy on the modes of reaction of the carbon atom? Are hot and therm al reactions basically sim ilar o r d issim ilar? The usual method of deciding such questions is by the use of a m oderator. In this paper we report on the consequences of adding neon as a m oderator to system s in which carbon atoms react with hydrocarbons. An e x ce ss of neon will reduce the probability of collision and reaction while hot, and will therefore favour th erm al p ro c e s s e s .
In sim pler hot atom system s, notably recoil tritium , the only important effect of adding noble gases appears to be to m oderate the kinetic energy of the hot atom . However, with carbon other effects must be considered:
(1) Carbon atom s can form excited interm ediates having a lifetim e of many m olecular vibrations. These may decompose in a variety of m odes, depending on th eir internal energy, to give sev era l products. Addition of

136 J. DUBRIN et al.
a m oderator will effect collisional deactivation of these interm ediates and therefore the final product spectrum . Dilution of a hydrocarbon by a noble gas at a constant total p ressu re will favour high energy modes of decom position, since the noble gas is le ss effective as a collisional d eactivato r than the hydrocarbon itself.
(2) Carbon atoms produced by high energy nuclear recoil are believed to reach the energy region where chem ical reaction can occur as a mixture of C (P 3), ground state , and C^D1) states [i ] . Addition of m oderator will change the nature and number of collisions that the carbon m akes before it reacts and can therefore effect the ratio of these states. A change in product yields will result since the two electronic states appear to differ in the nature of their reactions.
EXPERIM ENTAL
The C11 was produced through the use of either the Yale Heavy Ion or Electron A ccelerators. Detailed methods of production of the C11 and a description of the radiation vessels are presented elsewhere 11, 2, 3 ] . The total
eVradiation dose delivered to sam ples, at either a cce le ra to r , was 1 0 " 2 ------r-. m ol.o r less as determined using benzene production from acetylene as a crude d osim eter. A fter irradiation , analyses w ere perform ed by m eans of gas chrom atography using a window flow proportional counter in s e r ie s with a standard therm isto r d etector so that m ass and activity analyses could be perform ed simultaneously [4]. Identities and purities of reported species w ere confirmed by analysis on m ore than one column and by use of special trapping m a teria ls to distinguish certa in organic bond types [2]. Yields of various products can be computed as p er cent of total volatile activity o r p er cent of total expected activity (absolute yields) [3]. The fo rm er is arrived at by comparing the activity of a specific peak with the total volatile activity . The la tte r was computed for only heavy ion irradiations by com paring the activity of a specific peak with the total volatile activity of a standard sample (monitor) of known absolute recov ery . Gaseous oxygen is used as the absolute m onitor, since it has been shown that all the C11stopped in the gas phase re a c ts to form only Cn O and Cn 0 2 [3] . Thep ressu re of the oxygen m onitor is adjusted to the sam e stopping power as the sam ple. A fter appropriate normalization of the sample and monitor for differences in irradiation time and integrated beam intensities, the absolute yields are then calculated by dividing the total activity in the oxygen monitor (СПО+С^Ог) into the observed activity of any product.
RESULTS
Table I lists the yield of C5 products from the reaction of C11 with ethylene under various experim ental conditions. The yields are expressed as p er cent total volatile activity and rep resen t the average of two o r m ore independent determinations. Figures 1 and 2 illustrate the effect of the neon
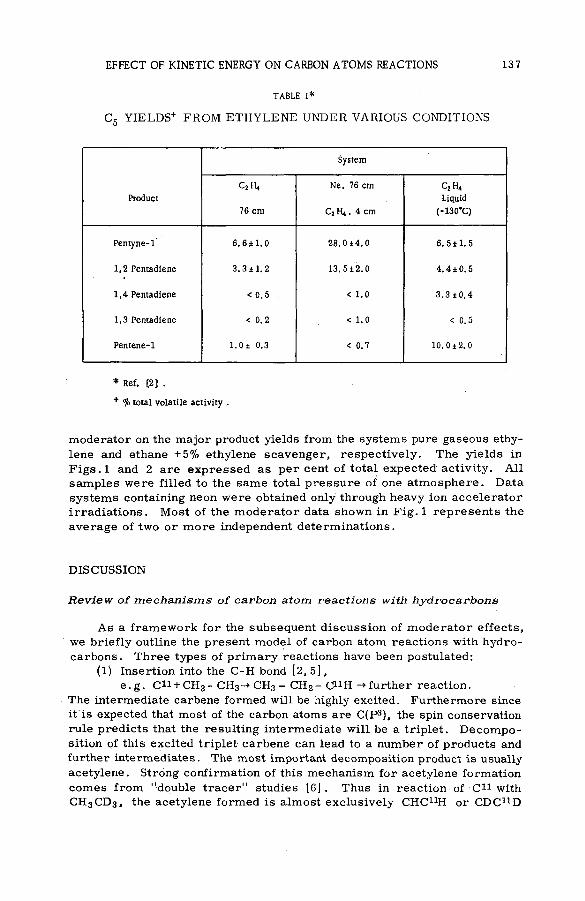
EFFECT OF KINETIC ENERGY ON CARBON ATOMS REACTIONS 137
TABLE I*
C5 YIELDS* FROM ETH YLEN E UNDER VARIOUS CONDITIONS
Product
System
C2H4
76 cm
Ne, 76 cm
C2 H i , 4 cm
C2H4Liquid
(-130’C)
Pentyne-1 6. 6 ±1.0 28.0 ±4.0 6. 5± 1. 5
1,2 Pentadiene 3.3 ± 1. 2 13. 5 ±2.0 4 .410 .5
1,4 Pentadiene < 0.5 < 1.0 3.3 ± 0.4
1,3 Pentadiene < 0 .2 . < 1.0 < 0 .5
Pentene-1 l . O i 0.3 < 0 .7 10. 0 ±2.0
* Ref. [2] .
+ % total volatile activity .
m oderator on the m ajor product yields from the system s pure gaseous ethylene and ethane +5% ethylene scaven ger, resp ectiv ely . The yields in F ig s . 1 and 2 a re exp ressed as p er cent of total expected activ ity . All sam ples w ere filled to the sam e total p ressu re of one atm osp here. Data system s containing neon w ere obtained only through heavy ion a cc e le ra to r irrad ia tio n s. Most of the m o d erato r data shown in F ig . 1 rep resen ts the average of two o r m ore independent determ inations.
DISCUSSION
R eview o f m echanism s o f carbon atom reactions with hydrocarbons
As a fram ew ork fo r the subsequent discussion of m o d erato r effects , we briefly outline the present model of carbon atom reactions with hydrocarbons. Three types of p rim ary reactions have been postulated:
(1) Insertion into the C-H bond [2, 5 ] ,e .g . CII + CH3 -C H 3-» OH3 - C H 2 - C ^ H -^further reaction .
The intermediate carbene formed will be highly excited. Furtherm ore since it is expected that most of the carbon atoms are CfP3), the spin conservation rule p redicts that the resulting interm ediate will be a trip le t. Decom position of this excited trip let carbene can lead to a number of products and further interm ediates. The most important decomposition product is usually acetylene. Strong confirmation of this mechanism for acetylene formation com es from "double t r a c e r " studies 16J . Thus in reaction of С 11 with CH3 CD3 , the acetylene formed is alm ost exclusively CHCnH or CDC1:1D

138 J. DUBRIN et al.
[V]. Double tra c e r , phase dependence and scavenger studies of most p roducts other than acetylene indicate that they also derive from reactions of an intermediate formed by prim ary insertion of a C11 atom into a C-H bond.
(2) Insertion into С = С bond [1, 5] .In alkenes there is a second important prim ary mechanism of C11 reaction;
Degradation studies confirm that m ost of the aliene and m ethylacetylene form ed from ethylene a re indeed cen tre labelled [1] . (The sm aller yield of end-labelled products presumably results from C-H bond insertion .) Double t r a c e r studies indicate that the final product incorp o rates all the hydrogen atom s of the ethylene molecule attacked [6] . If spin conservation is assum ed, this means that the original adduct is a singlet. This in turn implies that С = С insertion to yield aliene from ethylene is caused largely by the reactions of C(Di) atom s.
(3) H atom abstraction [8] .The С atom may "pick up" H atoms to form CH, CH2 and CH3 . Yields of ch aracteris tic products resulting from reaction of these species have been observed in yields of 5 - 10% of the carbon atom s available fo r reaction ;
General effects of neon moderation on С atom reactions with hydrocarbons
The most striking aspect of the results, shown in F ig s. 1 and 2, is that with the possible exception of vinylacetylene ( F i g .l ) all products form ed in pure ethylene o r ethane (scavenged) are still formed as dilution with neon approaches infinity. Thus the reaction properties of hot and thermal carbon atoms are qualitatively sim ilar, although there can be considerable quantitative differences. This situation results from two related facto rs: F irs tly , many reaction s of th erm al carbon atom s a re highly exo th erm ic, and, secondly, the in trin sic reactiv ity of the th érm al carbon atom appears to be so high that its threshold or activation energy for many processes is close to zero.
H2 C-CH2e .g . CH2 = CH2 + C n -> -»CH2= C n =CH|
СИ
e .g . CU H2 + C 2Hg -» CH3 CH2 CiiH3
СН2 - СН2 СИН2 +С 2 Н4 -> /
спна
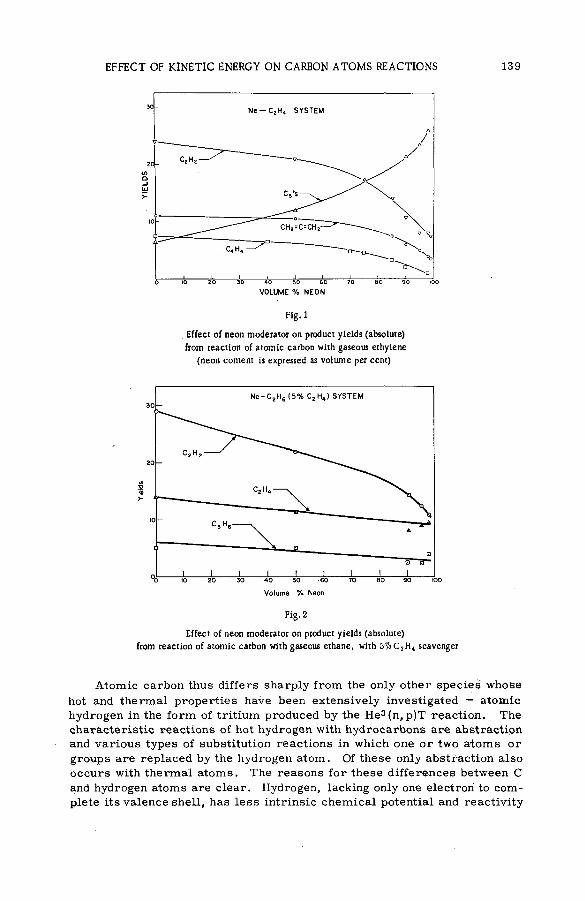
EFFECT OF KINETIC ENERGY ON CARBON ATOMS REACTIONS 139
Fig. 1
Effect of neon moderator on product yields (absolute)
from reaction of atomic carbon with gaseous ethylene
(neon content is expressed as volume per cent)
Fig. 2
Effect of neon moderator on product yields (absolute)
from reaction of atomic carbon with gaseous ethane, with scavenger
Atomic carbon thus differs sharply from the only other species whose hot and therm al properties have been extensively investigated — atomic hydrogen in the form of tritium produced by the He3 (n, p)T reaction . The ch aracteris tic reactions of hot hydrogen with hydrocarbons are abstraction and various types of substitution reactions in which one o r two atom s o r groups are replaced by the hydrogen atom . Of these only abstraction also o ccu rs with therm al atom s. The reasons for these differences between С and hydrogen atoms are clear. Hydrogen, lacking only one electron to complete its valence shell, has less intrinsic chem ical potential and reactivity

140 J. DUBRIN et al.
than carbon, which lacks four. Hence addition of kinetic energy cau ses a qualitative change in its p ro p erties.
The effects of moderation on the probability of hot hydrogen atom r e action have been successfully treated by a simple kinetic theory of hot reactions [9] . This theory only considers the effect of m o d erato r in r e moving kinetic energy from the hot atom. As mentioned in the introduction, a m oderator may have other effects in a hot carbon atom system : modifying the ratio of C (P 3) and C(Di) atom s and influencing the modes of decomposition of the relatively long-lived interm ediates that a re formed*. The simple kinetic theory is therefore not applicable to these sy stem s.
While a simple quantitative interpretation of the present results is not possible, a consideration of m oderator yield dependences of individual products (see F ig s .l and 2) does indicate that the kinetic energy of the carbon atom s changes th eir relative probability of forming the sev eral final p roducts. As a co ro llary it can be concluded that in an unmoderated gaseous system most carbon atoms react while they are still hot; were this not the case the yield ratios would show little dependence on the neon concentration.
Effect of moderator on individual products from C 11 reaction with ethylene
Acetylene and the С compounds produced in reaction of carbon atoms with ethylene are postulated to result from a common p recu rsor formed by insertion of C(P3 ) into ethylene [2 ] . As shown e a rlie r the m ost general mode of acetylene form ation resu lts from decom position of the adduct (I) form ed by insertion of C (P3) into a C-H bond.
However, in the case of ethylene we must also consider decom position of the adduct (II) form ed by carbon insertion into the 7r-bond.
ÇH + C2H4 ->H2C = C H - CnH <->H2 C - CH = C11H(I)
-» HC= Cn H + CH2 (1)
CH2 - CH2C 11 + C2 H4 -» \ / -»H2C = C 11- C H 2 (II)■ ¿ 1 1
CH2 = Cn - CH2-> C n = CH2 +CH 2
C n =CH 2 ->H C ll=C H . (2)
* Such long-lived intermediates do not appear to be formed in reaction of hot hydrogen atoms w ith
saturated systems [10].

EFFECT OF KINETIC ENERGY ON CARBON ATOMS REACTIONS 141
Depending on the energy content of the adducts (I and II) and the available tim e before collisional deactivation they m ay: (a) undergo very extensive decomposition to form some fragment (C2, СгН ог CCH2 ) that then reacts with another ethylene molecule to form vinylacetylene; (b) decompose less d ra stica lly to form acetylene; (c) p re se rv e th e ir skeletal s tru ctu re arid eventually re a c t with a second ethylene m olecule to yield C5 compounds.
In accordance with these mechanisms yields of acetylene, vinylacetylene and C 5 compounds show com plem entary m o d erato r dependences 12 J . In unmoderated sam ples the p rim ary adduct will usually be form ed by a hot atom and will therefore be more highly excited. This will then enhance ex tensive decomposition p ro cesses (I or II) with eventual production of vinylacetylene. Furth erm ore, formation of acetylene by means of (2), involving an energetically unfavourable step, will be enhanced. In thermalized (highly moderated) system s these adducts will be less excited, and these high activation energy fragmentation p rocesses to C2 entities will be less important. Instead, the adducts will eventually undergo reactions involving addition of ethylene to yield С 5 products. Even at infinite moderation acetylene is not eliminated. This is in accordance with the fact that some possible pathways to this product, in p articu lar reaction (1 ), a re not only exo ergic but also involves no highly endoergic step s.
Examination of the nature and yields of the C5 compounds formed under various conditions (see Table I) indicates certain subtle aspects of moderator action. Even with therm al carbon atoms the prim ary (triplet) C3 H4 adduct (I o r II) will have sufficient internal energy to elim inate a hydrogen atom . This should be p articu larly im portant in highly m oderated sam ples where collisional deactivation is inefficient. Indeed, it is not surprising to find in an excess of neon only those C5 products that are expected from reaction of CHC2 H3 with ethylene, namely pentyne- 1 and ethylallene [2 J .
НС = С - CH2 CH2 CH2 CH2 C sC H CH3 CH2 CH2C = CH
+ С 2н / +H and (3). * \ — *
HC = C = CH2 \ iC H 2=C = CH-CH2-CH2 CH3CH2CH = C = CH2
It is interesting to contrast highly moderated (gaseous) with condensed (unmoderated) ethylene system s L2j . In the la tte r the initial adducts are p re sumably formed largely by hot atoms and are therefore highly excited. But collisional deactivation is so rapid that decomposition to acetylene and vinylacetylene is relatively dim inished. F u rth erm ore decom position to C 3H3 would also be expected to be reduced. Although th ere is a larg e yield of C 5 products, they a re different from those form ed in the neon m oderated system and appear to a rise largely through C3 H4 (triplet) and C 3H5 (allyl ra d ica l).
Thus the trend of acetylene, vinylacetylene and С 5 product yields can be accounted for in te rm s of the effect of neon in moderating hot atom s and in being a poor third body for collisional deactivation.
F igu re 1 shows that aliene, like acetylene, is reduced in im portance with increasing neon concentration. However, comparison of the m oderator

142 J. DUBRIN et al.
curves suggests that the dependency of aliene (unlike acetylene) on increasing neon dilution m ay be larg ely caused by both the inefficiency of neon as a third body and the assumed decrease in the ratio of singlet to triplet carbon atoms [1 ], rather than the moderation of hot carbon atoms. These possibilities are discussed below in connection with the formation of aliene.
(1) The reaction of a C^D1) with ethylene to form aliene is highly exo ergic (175 kcal). Experim ental evidence indicates that the activation energy of formation of aliene by insertion reactions is very low, as expected[1]. This in turn implies that therm al carbon atoms should have a high probability of reaction with ethylene to form aliene.
(2) The highly excited C3 H4 interm ediate must be collisionally stabilized to aliene before isom erization o r possible fragmentation o ccu rs . Dilution of ethylene with neon will result in p oo rer collisional d e-excitation and will effectively reduce the aliene yield. This effect is witnessed by the increased degree of isomerization of aliene to methylacetylene with increased neon content [1 ] .
(3) Aliene has been postulated to result from reaction of a CiD1) with ethylene. In highly moderated system s, the ratio of CÍD1) to C(P3) may be reduced as a result of the extra collisions undergone in excess neon. Hence the aliene yield would also be expected to decrease.
The ratio of ce n tre -to end-labelled aliene changes from about 1 . 8 in pure gaseous ethylene to 3. 2 in system s containing 95% neon [1 ]. Since end labelling is associated with C-H insertion and cen tre labelling with C = C insertion , this resu lt im plies that therm al carbon atom s show m ore d iscrim ination in favour of double bond attack than do hot atom s.
SUMMARY
It has been shown that thermalization of carbon atoms changes the yield patterns with hydrocarbons quantitatively but not qualitatively. This finding, that hot and therm al carbon atom s re a c t sim ilarly , is consistent with the high, chem ical reactivity of the therm al carbon atom . In an unmoderated system m ost carbon atom s while they a re still hot undergo insertion r e actions to form highly excited interm ediates. This additional internal energy supplied to the intermediates by the hot atom, enhances certain fragmentation m odes of high activation energy. L e s s excited interm ediates formed by therm al carbon atoms, will reduce but not eliminate the yields of certain products. Instead products resulting from lower energy decomposition or addition modes will be increased in importance. In addition thermal carbon atoms show m ore discrimination in favour of double bond attack than do hot atom s. x
In addition to moderating hot carbon atom s, neon is inefficient in collisionally stabilizing long-lived interm ediates and may modify the ratio of C(P3) to C(D l); these additional effects can independently change the yields of products in highly m oderated system s. Hence the kinetic theory of hot atom reactions, successfully applied to monovalent hot atoms, is not applicable to these hydrocarbon system s.

EFFECT OF KINETIC ENERGY ON CARBON ATOMS REACTIONS 143
' R E F E R E N C E S
[1] MARSHALL, M . , MacKAY, C. and WOLFGANG, R ., J. Amer. chem. Soc. (in press).
[2] DUBRIN, J . , MacKAY, C. and WOLFGANG, R. , J . Amer. chem. Soc. (in press).
[3] DUBRIN, J . , MacKAY, C. and WOLFGANG, R. , J . inorg. nucl. Chem. (in press).
[4] WOLFGANG, R. and ROWLAND, F. S ., Analyt. Chem. 30 (1958) 903 ; WOLFGANG, R. and MacKAY, C . ,
Nucleonics 16 10 (1958) 69.
[5] MacKAY, C. and WOLFGANG, R ., J . Amer. chem. Soc. 83(1961) 2394; MacKAY, C . , POLAK, P..
ROSENBERG, H. E. and WOLFGANG, R ., I. Amer. chem. Soc. 84 (1962) 308.
[6] DUBRIN, J . , M acKAY, C. and WOLFGANG, R. . J . Amer. chem. Soc. 86 (1964) 959.
[7] DUBRIN, J . , M acKAY, C. and WOLFGANG, R. , J . chem. Phys. (in press).
[8] STÔCKLIN, G. and WOLF, A. P., J. Amer. chem. Soc. 85 (1963) 229; RACK, E. P ., LANG, C.E. and
VO IG T , A .F . , J. 'chem. Phys. 38 (1963) 1211; M acKAY, C . , PANDOW, М ., POLAK, P. and
WOLFGANG, R. , in Chem ical Effects of Nuclear Transformations П, IAEA, Vienna (1961) 17.
[9] ROSENBERG, A . H. and WOLFGANG, R . , J. chem. Phys. 41 (1964) 2159.
[10] WOLFGANG, R ., "The Hot Atom Chemistry of Gas Phase Systems", Progress in Reaction Kinetics I I I
( in press).


EFFECT OF KINETIC ENERGY ON CARBON ATOMS REACTIONS 145
D IS C U S S IO N
(on the foregoing three papers)
A. ATEÑ: I should like to make a rem ark of a qualitative nature in connection with the information given by Dr. Wolf on the ease of formation of the CN group in HCN from C11. In the course of investigations carried out recently at A m sterdam in collaboration with M r. Delvenne we found quite a high retention of СИ from the (n, 2n) reaction in solid NaCN - about 59% in fact. The retention of N13 was even higher (about 91%).
A. WOLF: Regarding the paper presented by M r. Dubrin I should liketo comment on the thorny problem of the validity of spin-state determ inations. We have obtained evidence for the spin-state of "hot methylene" by observing the ste reo -se lectiv e addition of methylene to ç is - and tra n s- butene-2. The reaction of hot 'C H ^: with cis-butene-2 results prim arilyin c i s - l , 2-dim ethylcyclopropane-3-C 11 and the reaction with tran s-butene-2 gives p rim arily the tra n s-1 , 2-dim ethylcyclopropane-3-C i1 . This is what one would expect in the case of an attack by singlet methylene on a double bond. Caution has to be exercised, however, since the kinetic energy of the attacking methylene o r the exotherm icity of the initial attack, o r both to gether, might be sufficient to bring about a spin change, e .g . in a case where the methylene attacking was a triplet and therefore needed to undergo spin inversion in order to form the final covalent bond after the f irs t bond had been form ed. The argum ents that are advanced on spin are certainly reasonable but I do not think they a re unequivocal.
B . DZANTIEV: I wonder whether the authors of these extrem ely interesting papers on hot carbon reactions would ca re to comment on the nature of the energy responsible for the reactivity of the hot carbon atoms. Does kinetic energy or some other type of energy play a decisive role? The papers submitted have presented a variety of experimental evidence on this subject. On the one hand, attention has been drawn to the part played by the therm alization of the atom s (additions of He, Ne). On the other hand, the view has been expressed that the reactivity is p ractically identical for hot and therm al atom ic carbon.
It might perhaps be useful to supplement work on C11 and C14 atom s with investigations on rad ical reactions with therm al and excited radicals of CH, CH^ and CH3, which are certainly intermediate products in hot carbon atom reactions. I would be interested to hear Dr. Wolf's views on whether one of the differences between hot carbon and therm al carbon might be that the thermal carbon atom is a bi-radical sim ilar to CH2 (the methylene radical) w hereas the hot atom can be a te tra ra d ica l. It is known that carbon in the ground state is a trip let and that it is a b i-rad ical. In order to find an additional pair of bond electrons, additional energy is required to bring it into an active valent state . Perhaps the hot carbon atoms obtain this additional energy from the energy of the nuclear reaction that produces them.
A. W OLF: The energy state of the carbon atom just p rior to the first reactive collision is difficult to determine at the present state of our knowledge of the reactions of carbon atom s. The question whether the reactivity , of the carbon atom is due to kinetic energy or to some other form of energy is rendered obscure by the fact that the carbon atom can give sim ilar prod-
t o '

146 J. DUBRIN et al.
ucts when it re a c ts as a hot atom and when it re a c ts as a therm al atom, as has been shown by both D r. Dubrin and ourselves. The product d istribution does a lte r as one goes from hot to therm al carbon atom s, and the p recise nature of the f irs t reactiv e collision in the hot region has not as yet been fully determined. The ground state of the carbon atom is a triplet and the first two excited states are low-lying singlets, being some 1-3 eV above the ground state. Interconversion between these states may be easily possible at the collision energies involved. As far as I am aware, no direct experim ental evidence is currently available. Physical organic chem ists have described the mechanism of the insertion reaction of methylene in considerable detail; we have taken th eir description and have form ulated an argument by analogy for the presumed formation of methylene in the hot- atom réactions. P rofessors MacKay and Wolfgang have used this insertion analogy to postulate insertion by carbon atom s. In our paper we have p resented further evidence that bears on this analogy. It nevertheless seem s premature to us to offer a detailed explanation of the nature of the spin state of carbon just p rior to the initial reactive collision in the hot range and furtherm ore to relate that spin state to the observed product distribution. What we are attempting to do is to provide an internally consistent picture based on what is known about organic reaction m echanism s. Arguments about energy states must of cou rse be consistent with both experim ental observation and the postulated m echanism s. S o la r at least insertion r e actions seem to offer a satisfactory hypothesis for explaining a large number of the products observed in simple alkanes. They do not, however, offer a particularly good explanation for "re -e n try " products.
J . DUBRIN: In the past few y ears, studies have been made at Yale of the reactions of С atoms with N2, N20 and NO. When either an unsaturated or a saturated scavenger has been added to these system s, products typical of cyanide (CUN] attack have been found in significant yield. Effective therm alization by neon moderation can remove the measured cyanide yield from the N2 but not the N20 system ; this finding and the products observed are consistent with the initial formation of C11 N by direct carbon-atom attack on either N2 or N20 . Our data suggest that the reduction in the cyanide yield with added oxygen is a result of the reaction of CN with the oxygen. In system s consisting of only N2 + 0 2 o r N20 + 0 2, a significant yield of СЗ-Юд is found in addition to Cn O; this may well be the.result of a reaction or series of reactions of CUN with oxygen.
The discussion of the effect of kinetic energy on carbon-atom reactions with hydrocarbons does not depend on the assumption of specific spin states of the carbon. A great number of analogies have been made to the reactions of methylene. It is difficult to say how many of these are justifiable; indeed in the simple system s studied thus far at Yale all the results obtained indicate that methylene is a relatively unimportant p recu rso r.
I should like to em phasize once again that in all the system s studied at Yale we have found that acetylene form ation does not for the m ost part involve any in te r- or in tra-m olecular mixing. This is consistent with the formation of acetylene by direct carbon-atom insertion. The cyclopropane system might well represent a rather intriguing but special case.
A. WOLF: I certainly agree that too much can be made of the analogies with m ethÿlene-insertion reactio n s . Indeed this is p recise ly the point I
Ю*

EFFECT OF KINETIC ENERGY ON CARBON ATOMS REACTIONS 147
have been trying to s tre s s . The arguments are based wholly on an analogy with what physical organic chemists have determined over the past ten years. Insertion as described by the physical organic chemist involves orthogonal attacks on a covalent bond in which both electronic and s te ric facto rs can be involved. There is a great deal known about methylene. The only knowledge we have of carbon-atom "insertion" is what has been suggested as a hypothesis to explain the occurrence of certain products in hot-atom reaction s.
Secondly, my own feeling as to what constitutes a m ajor product is that this is based solely on what one is trying to study at a particu lar tim e. It seem s to me the use of the word "m ajor" involves us in semantic difficulties. Is "m ajo r" to be defined as implying values greater than 50%; or is it to be defined in term s of any radiochem ical yield that is greater than any other, o r, again, is a "m ajo r" product to be defined as one that is derived "directly" from the carbon-atom "insertion" alone? In the case of propane, for example, the yield of acetylene presumably derived from carbon-atom insertion is about 26% in the gas phase, 18% in the liquid phase and 1 1 % in the solid phase. The methylene "in sertion " products are about 6 % in the gas phase and 12-13% in the liquid and solid phases. One could therefore say that the yield of the methylene insertion product is "m ajo r" in the solid phase. It is of course clear that the methylene itself comes from some sort of carbon-atom reaction with the alkane. In studying methylamine, one finds that the sum of the yield of dimethylamine and ethylamine (С -insertion) is 28% and the acetylene yield is 12.5%, in the liquid phase. Clearly, the carbon-atom -insertion product is not "m ajo r" here.
To sum up, the im portant thing in my view is to try to co rre la te the yields of products with different types of reactive pathways or mechanisms, if you will, in the hope that it will eventually be possible to predict product distributions in new system s rather than focus attention on the major yields alone. The whole system is of importance.
A .F . VOIGT: In connection with this problem of the relative importance of carbon and methylene reactio n s, I should like to draw attention to the finding reported in our paper on liquid hydrocarbons *, namely that the carbon-insertion reaction appears to be more important than the methylene reaction. In these system s the yields of acetylene and ethylene are e s sentially as high as they are in gaseous system s.
* VO IGT. A .F . et a l„ "Chem ical behaviour of Cn in liqu id hydrocarbons", these Proceedings J.


THE REACTIONS OF HOT FLUO RIN E-18 WITH GASEOUS CARBON TETRAFLU O RID E*
N. COLEBOURNE, J. F.J. TODD AND R. WOLFGANG 'YALE UNIVERSITY, NEW HAVEN, CONN.,
UNITED STATES OF AMERICA
Abstract — Résumé — Аннотация — Resumen
THE REACTIONS OF HOT FLUORINE-18 W ITH GASEOUS CARBON TETRAFLUORIDE. Studies on the
reactions of hot Fie atoms with carbon tetrafluoride are reported. Gaseous samples were exposed to the
40-60 MeV (maximum) bremsstrahlung beam of the Yale University Electron Accelerator. The F19(y,n)F18 process produces F18 w ith a kinetic energy of the order of 105-106 eV. These species lose energy by collision
and are expected to reach the "chem ical" energy range (< 100 eV) as ground state atoms. Ethylene was found
to be a good scavenger for thermal F18 atoms. Analysis of products was made using standard radio-gas chroma
tography techniques. The system was found to be quite sensitive to extraneous radiation damage effects and
appropriate precautions were taken.
Hot displacement reactions, sim ila r to those observed for hot hydrogen, but much less effic ient, were
found:
f 18+ c f4 -» CF3 F18+F ,
F» +CF4 -* CF2F «+ (F + F ),
It was impossible to study the abstraction reaction
FU +CF4 -* CF3 +FF18
directly. However, indirect evidence suggests that it also has a low efficiency. ’
Detailed studies of the effect of moderator on the F18 +CF4 system have been made. The data obtained
were analysed by means of the k inetic theory of hot reactions. The system was found to be in accord w ith
this formalism, providing quantitative confirmation of the present interpretation of the results.
The carbon tetrafluoride and methane systems provide a basis for some tentative conclusions on the
mechanisms of hot fluorine atom reactions. A t present it appears that w ith certain important, but natural,
modifications the model first developed for hot hydrogen atoms is applicable.
RÉACTIONS DES ATOMES “ F CHAUDS AVEC LE TE'tRAFLUORURE DE CARBONE EN PHASE GAZEUSE.
Le mémoire est consacré à des études sur les réactions des atomes 18 F chauds avec le tétrafluorure de carbone.
Des échantillons gazeux ont été exposés à un faisceau de rayonnements de freinage de 40 à 60 MeV (maximum)
émis par l'accélérateur d’électrons de l'Université Yale. Le processus l 9F(y, n)18F donne des atomes de 18F
d'une énergie cinétique de l ’ordre de 105 à 10® eV. Ces espèces perdent de l'énergie par choc et doivent
atteindre la gamme des énergies «chim iques » (< 100 eV) en tant qu'atomes à l'é ta t fondamental. Les
auteurs ont constaté que Véthylène était un excellent agent de balayage pour les atomes 18F thermiques. Ils
ont analysé les produits à l'aide de méthodes courantes de radiochromatographie en phase gazeuse. Le système
s'est révélé très sensible aux effets de rayonnements étrangers; aussi les mesures de précaution appropriées
ont-elles été prises.
Des réactions de déplacement, analbgues à celles qui ont été observées pour l'hydrogène chaud mais
beaucoup moins efficaces, ont été relevées; ce sont les réactions suivantes:
* Studies supported by the United States A tom ic Energy Commission under Contract SAR/AT(30-1)
-1957.
149

150 N. COLEBOURNE, et al.
«F + C F j -» CF3 18F + F, ■ .
1! F+CF4 -f CF2 18F + (F + F).
I l n'a pas été possible d'étudier directement la réaction d'enlèvement .
18F + CF. C F + F 18F,4 ? .
Cependant, certains indices donnent à penser que cette réaction a également une faib le efficacité.
Les auteurs ont étudié en déta il l'effet de ralentissement sur le système 18F + CF4 . Ils ont analysé les
données obtenues en se fondant sur la théorie cinétique des réactions chaudes. Ils ont constaté que le système
était conforme à cette théorie, ce qui a fourni une confirmation quantitative de l'interprétation actuelle des
résultats.
Les systèmes méthane et tétrafluorure de carbone fournissent une base à certaines conclusions provisoires
sur les mécanismes des réactions des atomes de fluor chauds. I l semble à présent que, moyennant certaines
modifications importantes mais naturelles, le modèle qui avait tout d'abord été mis au point pour les atomes
d'hydrogène chauds soit applicable aux atomes de fluor chauds.
Р Е А К Ц И И Г О Р Я Ч И Х А Т О М О В Ф Т О Р А - 1 8 С Г А З О В О Й Ф А З О Й Т Е Т Р А Ф Т О Р М Е Т А Н А .
С о о б щ а е т с я о р е а к ц и я х г о р я ч и х а т о м о в F 1S с т е т р а ф т о р о м е т а н о м . Г а з о о б р а з н ы е о б р а з ц ы
о б л у ч а л и с ь т о р м о з н ы м и з л у ч е н и е м с э н е р г и е й 4 0 — 6 0 М э в ( м а к с и м у м ) н а э л е к т р о н н о м у с к о
р и т е л е Й е л ь с к о г о у н и в е р с и т е т а . П р о ц е с с F 19 ( 7 > n ) F l 8 f l a e T F 18 с к и н е т и ч е с к о й э н е р г и е й
п о р я д к а 1 0 s — 1 0 6 э в . Э т и о б р а з ц ы т е р я ю т э н е р г и ю в р е з у л ь т а т е с т о л к н о в е н и й и , к а к п р е д
п о л а г а е т с я , д о с т и г н у т в е л и ч и н ы " х и м и ч е с к о й " э н е р г и и ( < 1 0 0 э в ) к а к о с н о в н о г о с о с т о я н и я
а т о м о в . Б ы л о н а й д е н о , ч т о э т и л е н я в л я е т с я х о р о ш и м а к ц е п т о р о м т е п л о в ы х а т о м о в F 1 8 .
Б ы л с д е л а н а н а л и з п р о д у к т о в п р и и с п о л ь з о в а н и и с т а н д а р т н ы х м е т о д о в р а д и о - г а з о х р о м а т о -
г р а ф и и . С и с т е м а , к а к б ы л о н а й д е н о , я в л я е т с я в п о л н е ч у в с т в и т е л ь н о й к в н е ш н и м э ф ф е к т а м
р а д и а ц и о н н о г о п о в р е ж д е н и я , и п о э т о м у б ы л о п р и н я т ы с о о т в е т с т в у ю щ и е м е р ы п р е д о с т о р о ж
н о с т и .
Б ы л и н а й д е н ы с л е д у ю щ и е р е а к ц и и г о р я ч е г о з а м е щ е н и я , п о д о б н ы е р е а к ц и я м , н а б л ю д а е
м ы м д л я г о р я ч е г о в о д о р о д а , н о г о р а з д о м е н е е э ф ф е к т и в н ы е :
' F 18 + C F 4 — > C F 3 F 1 8 + F ,
F 18 - t - C F 4 — ► C F 2 F 1 8 + ( F + F ) .
О к а з а л о с ь н е в о з м о ж н ы м и з у ч и т ь р е а к ц и ю о т д е л е н и я п р я м о : :
F le +CF4 — > C F 3 + F F l b .
О д н а к о к о с в е н н ы е д о к а з а т е л ь с т в а г о в о р я т о т о м , ч т о о н а т а к ж е и м е е т н и з к у ю э ф ф е к т и в
н о с т ь .
П о д р о б н о и з у ч а л и с ь э ф ф е к т ы з а м е д л и т е л я н а с и с т е м у F 10 + C F 4 . П о л у ч е н н ы е д а н н ы е
а н а л и з и р о в а л и с ь с п о м о щ ь ю к и н е т и ч е с к о й т е о р и и г о р я ч и х р е а к ц и й . Б ы л о н а й д е н о , ч т о э т а
с и с т е м а с о г л а с у е т с я с д а н н ы м о б ъ я с н е н и е м п р и у с л о в и и к о л и ч е с т в е н н о г о п о д т в е р ж д е н и я
т а к о г о т о л к о в а н и я р е з у л ь т а т о в .
С и с т е м ы т е т р а ф т о р м е т а н а и м е т а н а о б е с п е ч и в а ю т о с н б в у д л я н е к о т о р ы х п р е д в а р и т е л ь
н ы х в ы в о д о в о т н о с и т е л ь н о м е х а н и з м а р е а к ц и й г о р я ч и х а т о м о в ф т о р а . В н а с т о я щ е е в р е м я
п р е д с т а в л я е т с я , ч т о с н е к о т о р ы м и в а ж н ы м и , н о е с т е с т в е н н ы м и и з м е н е н и я м и , м о д е л ь , в п е р
в ы е р а з р а б о т а н н а я д л я г о р я ч и х а т о м о в в о д о р о д а , п р и м е н и м а и з д е с ь .
REACCIONES DE ATOMOS CALIENTES DE FLÚOR-18 CON TETRAFLUORURO DE CARBONO GASEOSO.
La memoria describe estudios de las reacciones de átomos calientes de 18F con tetrafluoruro de carbono. Se
expusieron muestras gaseosas a l haz de 40 a 60 MeV (máximo) de radiaciones de frenado del acelerador de
electrones de la Universidad de Yale. En virtud de la reacción l 9F(y,n)*8F se produce 18F con una energía
cinética del orden de 105 a 10® eV. Estas especies pierden energía por choque y se espera que alcancen el

REACTIONS OF HOT FLUORINE-18 151
in tervalo de energías «qu ím icas» (< 100 eV) como átomos en e l estado fundamental. Se comprobó que el
etileno constituye un buen depurador para los átomos de 18F térmicos. El análisis de los productos se efectuó
por técnicas corrientes de radiocromatografía en fase gaseosa. Se encontró que el sistema es bastante sensible
a los daños producidos por radiaciones externas, por lo que se adoptaron precauciones para evitar la penetración
de éstas.
Se observaron reacciones de desplazamiento calientes análogas a las que se producen con hidrógeno
caliente, pero de rendim iento mucho menos elevado,
1SF + CF4 -» CF3 l8F + F .
18F + CF4 -> CF218F + (F + F).
No fue posible estudiar directamente la reacción de abstracción '
- 18F+CF4 -) CF3 + F 18F ; ■
pero pruebas indirectas sugieren que su rendim iento es también reducido. . , ..
Se ha estudiado detalladamente el efecto de un moderador sobre e l sistema 18F + CF4. Los datos e x
perimentales se analizaron con ayuda de la teoría cinética de las reacciones calientes. Se encontró que el
sistema se ajusta a este formalismo, lo cual confirma cuantitativamente la interpretación actual de los
resultados.
Los sistemas tetrafluoruro de carbono y metano han servido de.base para formular algunas hipótesis sobre
los mecanismos de reacción de los átomos calientes de flúor. A ju ic io de los autores, e l modelo establecido
para los átomos calientes de hidrógeno puede, con ciertas modificaciones lógicas, aplicarse a dichos sistemas.
1. INTRODUCTION
The recent studies in the field of hot-atom chem istry have been largely concerned with hydrogen [1]. A sa result most current ideas on the mechanism of reactions of atoms of high translational energy are based on this species[2] . The presen t work on hot fluorine atom reaction s is p art of an effort to extend our knowledge of the field [3, 4] .
V ery high energy F 1S atom s are produced by reco il from a nuclear r e action, F 19( y, n )F18, o ccurring in CF4 . The fast atom s (> 1 0 4eV) obtained lose energy by moderating collisions in the medium. Below a certain energy (<100 eV) such a collision with a CF4 molecule may result in a hot reaction with ch em ical combination of the fluorine. Atom s that a re m oderated to th erm al en ergies without having become, combined a re taken up by an appropriate scav en g er. P roducts of these reactio n s a re sep arated by gas chrom atography 15, 6 J and assayed by m easuring the radioactivity of the contained F 18(112 m in).
The resulting data on probabilities of reaction to form Tjarious products are analysed using the kinetic theory of hot reactions [7, 8 ] . This analysis provides a further indication [9] of the validity of. this theory. The results thus obtained for the system F + СБ4 are then compared with sim ilar results, obtained previously, on H + CH4 . This serves as a basis for a discussion of com parative mechanism. An attempt is made to decide which aspects of the m echanism s previously proposed for hot hydrogen atom reactions are valid for hot species in general, and which aspects differ according to the nature of the hot atom involved.

152 N. COLEBOURNE, et al.
The reaction was carried out in cylindrical P yrex tubes of about 60 ml capacity , fitted with long necks of capillary tubing and a vacuum stopcock. The vessels were filled on a vacuum line with the appropriate reactant (CF4 ), m oderator (He, Ne o r A r)and scavengers (C2H4 and I2). The reagents used had been shown by gas chrom atography to contain <0 .1% im p u rities . In most experiments the tota.1 pressure was 1 atm at the experimental tem perature of 25°C.
The filled vessels were exposed to the bremsstrahlung beam of the Yale 4 0 -6 0 MeV electron a cce le ra to r, ca re being taken to shield the stopcocks. Such irradiation produced of the order of 108 atoms F 18. Most samples were irrad iated in groups, the v e sse ls being mounted on a turntable so as to receiv e identical radiation doses.
Radiation damage was minimized by using the high-energy bremsstrahlung sp ectra obtained by im pact of a 40 to 60 MeV electron beam on a relatively thin (10 g /cm 2) targ et. This minimized the fractions of low-energy gamma ra y s that cause radiation damage without contributing to F 18 production. Irradiation of an acetylene dosim eter under sim ilar conditions yielded no detectable amount of benzene, indicating that the total radiation absorption by the system is <0 .0 1 eV/m ol [10] . In agreement with this, no m acroscopic ( > 0 .1 %) decomposition of samples was observed in most cases and for properly scavenged sam ples and irrad iation tim es of approxim ately one hour there was no indication of any radiation effect on the product yields. However, prolonged exposure to the beam caused gross variations in the product distribution. This was attributed either to direct decomposition of the p rim ary products of the reactions of F ls or to failure of the scavenging system to remove all the therm al F 18 atoms in the presence of an excess of radicals produced by the radiation.
2 . 2 . Product identification and analysis
The analytical technique used was identical to that employed in this laboratory for studies of the chem istry of atomic carbon [2] . This involves separation of the products (present in trace amounts) by gas chromatography, followed by their radioassay in a flow counter in series with the usual thermal conductivity device of the chrom atograph. This technique [5] and the high- efficiency thin-window flow counter [6 ] have been described elsew h ere .
After allowing three hours for the induced C11 (20.4 min) activity to decay, an irradiated sample was divided into several aliquots. Two aliquots were analysed using 8 -m and 1-m silicone (General E le ctric SF 96, 40 g /100 g fireb rick ) colum ns at 2 5 ° С and a 2 -m dim ethylform am ide column (DMF) (approx. 20 g /100 g firebrick) at 0°C. Helium was used as the flow gas and " P - 1 0 " (Matheson C o .) as the counter g as . Two other aliquots w ere a s sayed, without separation, for total volatile activity. This procedure provides a comparison between the sum of the activities of the individual volatile products and the total volatile activity.
2. EXPERIM ENTAL
2.1. Irradiations

REACTIONS OF HOT FLUORINE-18 153
Initial identifications of the products w ere made using the silicone colum ns, a 1 -ft dim ethylform am ide DMF (20%DM F on fireb rick ) column and 1-ft and 10-ft de-activated alumina columns. Relative retention volumes (Rv) w ere determ ined by injection of m acro amounts of non-labelled form s of the suspected products. Results of this calibration procedure are given in Table I.
TABLE I
RETENTION VOLUMES
Column
and temperaturePeak 1 Rv( m l) Peak 2 Rv(ml) Peak 3 Rv(ml)
8-m silicone
(25°C)
c f4 0 < w203 CF3I 1014
1-m silicone
(25°C)
CF.. CjHgF,
CF3I
0 C f t F I 824
1-m DMF(0* q
c f4 0 СЛ Р238
2-m DMF
(O'C)
CF4, CfyF 0 CF3I 836
10-m alumina
(de-activated)
(25“q
c f4 55 С Д 265< W
1300
2-ft alumina
(de-activated)
(25°Q
cf4. c2^ f 0 CF3I 880
2. 3. Activity balance and estimation of absolute yields
The yields of products w ere expressed as fractions of the total volatile activity as determined by injection of aliquots directly into the counter 1 1 1 ] . The resu lts of the gas chrom atographic assay s showed that the identified products accounted for substantially all (90 ± 10%) of the volatile yield. The total volatile activity was always found to be d irectly proportional to the CP content of a sample indicating that it constituted the same fraction of the total F 18 formed at all levels of moderation. However, it was possible that a substantial fraction of the activity might be in a non-volatile form . Since it is desirable to know product yields on an absolute basis, a separate series of experiments was performed to check on this possibility.
Small (approx. 1 cm diam. ) quartz ampoules were filled with representative CF4 -sca v e n g e r-m o d e ra to r m ixtu res, sealed and irrad iated in the Hbrmal m anner. A fter irrad iation the unopened ampoule was counted in a w ell-scin tillation counter fo r a sufficient tim e to establish the 1 1 2 min half-life. It was then opened and a known aliquot of its contents allowed to expand into á sim ilar glass ampoule that was sealed and counted under

154 N. COLEBOURNE, et al.
sim ilar geom etry to establish the amount of volatile activity. In the meantime the quartz ampoule was pumped out and washed with acetone and 10~ - M NaF solution to rem ove any activity which might have settled on the walls of the ampoule. The ampoule was then counted again. A certain residual activity was found that could not readily be removed by repeated washing. This residual activity rep resents activation of im purities in the quartz wail and F 18 that has recoiled into the wall (and is therefore unavailable for hot r e action in the gas phase). The total amount of F 18 available for hot reaction was thus taken as the total F 18 content of the ampoule, c o rre c te d fo r the residual activity trapped in the wall. As is shown in Table II, the volatile activity accounts for nearly all this available F 18.
We conclude that <20% of the total F 18 is in form s which are not com pletely volatile at room tem p eratu re . As a consequence product yields determined as a fraction of the total activity in the form of identified products a re within a possible sy stem atic e r r o r of 15% rep resentativ e of absolute fractional y ie ld s .
2.4. Experim ental e rro rs
Standard deviations due to sta tistica l fluctuations in counting w ere of the following magnitudes: (1 ) for major products (those arising from therma- lized F 18 i .e . C2H3F 18 and C2H4F 18I): ±1 - 3%; (2) since hot products (CF3 F 18 and CF2F 18I) represented a relatively small fraction of the total F 1S, standard deviations on th eir assay s w ere as high as ± 1 0 % for som e unm oderated sam ples. F o r sam ples containing 95% m oderator these e r r o r s averaged ±30% . (This was due to the decrease in F 18 produced in moderated samples, as well as the diminished fraction that reacted to give hot products. ) The limits of e rro r may be somewhat larger in runs where argon was used as the m oderator because of the indirect method employed for determining the total volatile activity (see [ 1 1 ] ).
Possible system atic e r r o r s due to scavenger action a re discussed in the section on resu lts . E r r o r s involved in taking yields, determined as a fraction of the total identified yield, as absolute fractional yields a re d iscussed above. No other large sou rces of possible system atic e r r o r w ere obvious to the exp erim en ters.
F u rth er discussion of e rro rs in the derived quantities I and a is given in the section on kinetic analysis. E r ro rs quoted in the text are estim ated standard deviations (unless otherwise specified).
3. RESULTS '
3. 1. Scavenging of thermal fluorine atoms
In addition to CF4 , sm aller amounts of ethylene and I2 were present in the system to serve as a scavenger for those F 1E atoms that reached thermal energies without reaction. The development of this scavenger has been de-

TABLE II
V O LA TILE ACTIVITY AS A FRACTION O F TO TA L A CTIV ITY
Sample composition
To ta l a c tiv ity
of ampoule
(counts/in in
re la tive)
Residual a c tiv ity
before washing
(counts/tain)
Residual a c tiv ity
after washing
(counts/min)
A va ilab le
ac tiv ity
(counts/min)
V o la t ile
a c tiv ity
(counts/min)
V o la t ile a c tiv ity
A va ila b le a c tiv ity
9 4 .4 e/oCF1, 5.6%C2H 4
I2, 0% Ne27 700 1400 640 27 060 21922 •81. 0°!o
47. 2% CF4, 5.0%
I j , 47. 2% Ne48 200' . 2480 1830 46370 46100 99.4%
REACTION
S OF
HOT FLUO
RINE-18
155

156 N. COLEBOURNE, et al.
scribed in a prelim inary communication [4] . It acts as follows: Therm al fluorine atom s add to ethylene
C2H4 + F 18 ~ C2H4F 1S* . (1)
The excited ethyl radical formed can decompose.
C2H4F 18* - OjHaF18* + H. (2)
Alternatively it may lose energy by collision
C2H4F 18* + M - C ^ F 18 + M, (3)
and pick up an iodine atom
C2H4F 18 + I2 -* C2H4F 18I + I. (4)
Therm alized F 18 thus appears either as vinyl fluoride C2H3F i 8 o r as fluoro- iodoethane C2H4F 18I. Detailed confirmation of this mechanism of scavenger action is presented in the Appendix.
In the absence of scavenger the therm alized F 18 appears largely in the form of C^FjF18 and additional CF3F 18. These products are presumably formed by reaction of the thermalized fluorine with sm all concentrations of radicals o r ions produced by the radiation field. Figure 1 shows the quantity of ethylene required to suppress these processes to a level where essentially all the CF3F 18 is produced by hot reaction. (The p ressure of iodine vapour used was in a ll c a s e s the saturation p re ssu re at 25°C as ensured by the p resen ce of a sm all c ry s ta l . )
It is noted from F ig . 1 that an appreciable p ressu re of ethylene is r e quired to suppress sufficiently CF3 F l a of therm al origin . This is due to the sm all ratio of fluorine atom s that react hot relative to those that re a c t therm ally . In runs containing up to 80% m oderator the ratio of CF4 :C 2H4 used was 18:1 . Scavenger curves of the type shown in F ig . 1 w ere also run for a moderated system containing 90% neon. These showed that, as expected because of the sm aller fraction of F 18 atoms reacting hot, a larg er ratio of scavenger was required at high m oderator concentrations. A ccordingly, in samples containing more than 80% moderator the ratio of CF4 :C2H4w as4 : l .
In all ca s e s the use of different scavenger ratio s (within the ranges indicated) at a given m o d erato r concentration did not affect the yield of CF3F 18 outside the experimental uncertainty. This shows that although these amounts of ethylene are relatively high in term s of usual levels of scavenger concentrations, they are not high enough to remove enough hot fluorine atoms to appreciably affect the yield of the hot reaction with CF4 . However, a slight decreasing trend in the yield of C F 2F 18I with СгЩ concentration (see F ig . 1) may be real. This product is believed to arise from the combination with I2 of CF2 F 18 as formed by hot reaction. It is plausible that ethylene at
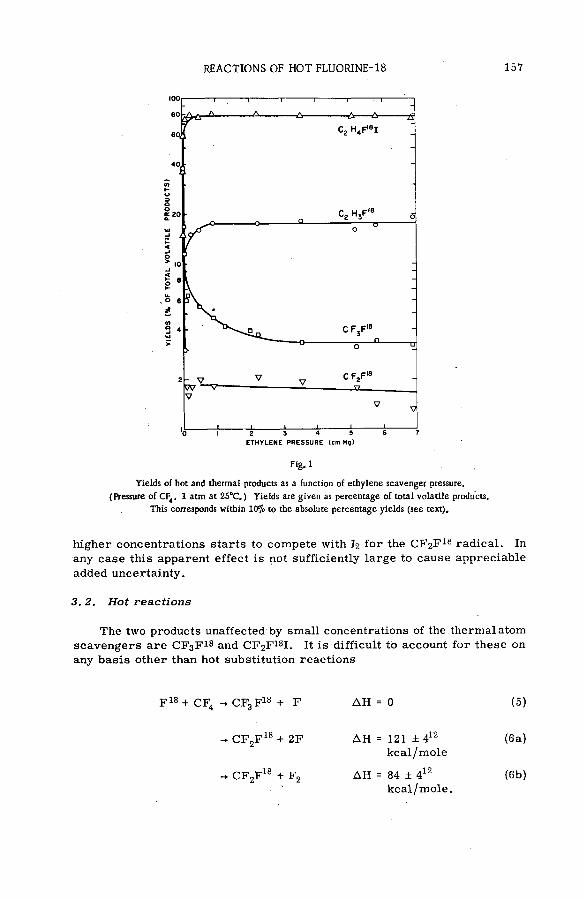
REACTIONS OF HOT FLUORINE-18 157
Fig- 1
Yields of hot and thermal products as a function of ethylene scavenger pressure.
(Pressure of CF,, 1 atm at 25'C.) Yields are given as percentage of total vo la tile products.
This corresponds within 10% to the absolute percentage yields (see text).
higher concentrations sta rts to compete with I2 for the C F 2F i s rad ical. In any case this apparent effect is not sufficiently large to cause appreciable added uncertainty.
3. 2. Hot reactions
The two products unaffected by small concentrations of the thermal atom scavengers a re CF3 F 18 and C F 2F 18I. It is difficult to account for these on any basis other than hot substitution reactions
F 18 + CF4 - CF3 F 18 + F ДН = 0 (5)
-» C FgF18 + 2F ДН = 121 ± 412 (6 a)kcal/m ole
- C F2F 18 + F 2 ДН = 84 ± 412 (6 b)kcal/m ole.

158 N. COLEBOURNE, et al.
(The C F2F 18 will subsequently combine with I2 to form CF3I. ) The yields of these reactions in unmoderated CF4 are : (5) 2.9 ± 0.3% and (6 ) 2.3 ±0.3% of the total fluorine atoms.
These substitution reactions are analogous to reactions of hot hydrogen atoms with methane, although their yields are considerably sm aller. The abstraction reaction observed in the H + CH4 system cannot be observed in the present case because of the reactivity of the product F 2
F 18 + CF4 - CF3 + F F 18 ДН = 84 ± 4 (7)kcal/m ole.
However, we can place an upper limit on this p ro cess. F 2 will either (1) go to the wall and become bound there; o r (2 ) re a c t with the ethylene to form С гН ^г. The la tte r m olecule has not been found and rep resen ts < 0.2% of the total F 18. However, since it would have been formed in a highly excited state it might reasonably be expected to decompose largely to C2H3 F and HF; and C2H3F 18 thus formed would not be detectable because of the large yield of this product form ed by scavenging of th erm al F 18. However, the conjugate product HF18 would probably co llect on the w all. .
F ro m Table II we see that the fraction of the total F 18 activity in unmoderated sam ples that rem ains on the ampoule wall is less than about 5%. Furth erm ore, the activity that could be removed from the walls by washing amounted to only 2.5% of the total. Therefore we may conclude that the yield of reaction (7) is well under 10% but may be about 2.5%.
The finding that the total reactivity of F 18 (P) is low is of im portance in both the kinetic analysis and the discussion of m echanism .
3. 3. E ffect o f p ressu re .
Figure 2 dem onstrates that, within experim ental e r ro r , the fractional yields of CF3F 18 and CF2 F 18I are independent of pressure over a range from 200 to 1400 total pressure (to rr). This shows that these products are formed with insufficient excitation energy to cause appreciable unim olecular decom position. In p articu lar it dem onstrates that the reaction
C F 3F 18* - C F 2F 18 + F (8 )
is not im portant in this system . .
3. 4. Effect o f moderator
F igu re 3 shows the dependence of the yields of C F 3F 18 and CF2 F 18I on the mole fraction of Ne and He m oderators. A few points obtained with argon a re also included. As expected for hot products these yields fall with increasing m oderator concentrations. Furtherm ore the relative efficiency for m oderating the F 18 is qualitatively in the o rd e r expected : Ne > A r > He, although the difference between the m oderators is not as apparent in the case of C F2F 18I production as in that of CF3F 18. A contrast can be drawn with the
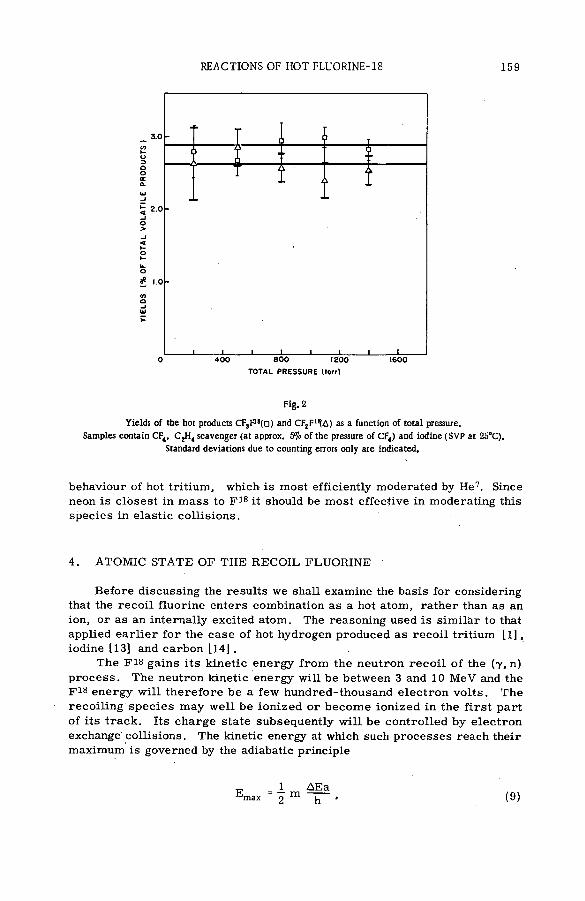
REACTIONS OF HOT FLUORINE-18 159
_l______ I_______I______ I_______1______ 1______ I______ L.0 400 800 1200 1600
TOTAL P R E S S U R E (torr)
Fig. 2
Yields of the hot products CFsP 8(d ) and CF2F*^A) as a function of total pressure.
Samples contain CF4, CjH4 scavenger (at approx. 5% of the pressure of CF4) and iodine (SVP at 25°C).
Standard deviations due to counting errors only are indicated.
behaviour of hot tritium , which is most efficiently moderated by He7. Since neon is closest in m ass to F 18 it should be most effective in moderating this species in elastic collisions.
4 . ATOMIC STATE OF THE RECOIL FLUORINE
Before discussing the results we shall examine the basis for considering that the recoil fluorine enters combination as a hot atom, rath er than as an ion, or as an internally excited atom. The reasoning used is sim ilar to that applied e a rlie r for the case of hot hydrogen produced as recoil tritium [1 ] , iodine [.13] and carbon [14] . .
The F is gains its kinetic energy from the neutron re co il of the (7 , n) p ro cess . The neutron kinetic energy will be between 3 and 10 MeV and the F * 8 energy will therefore be a few hundred-thousand electron vo lts . The recoiling sp ecies may well be ionized o r becom e ionized in the f irs t part of its tra ck . Its charge state subsequently will be controlled by electron exchange collisions. The kinetic energy at which such processes reach their maximum is governed by the adiabatic principle
„ _ I ДЕа"шах 2 m h ’
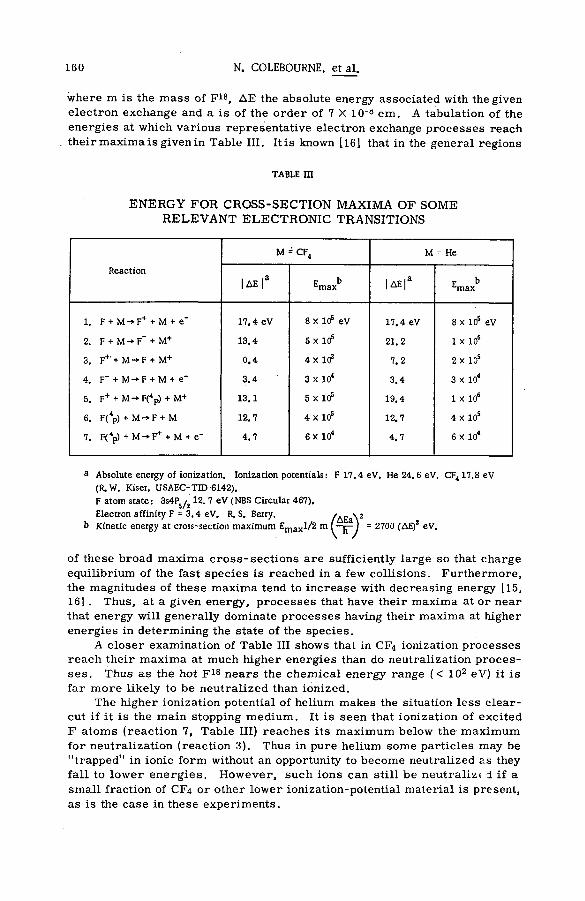
160 N. COLEBOURNE, et al.
where m is the m ass of F ls, ДЕ the absolute energy associated with the given electron exchange and a is of the ord er of 7 X 10" 8 cm . A tabulation of the energies at which various representative electron exchange processes reach their m axim ais given in Table III. Itis known [16] that in the general regions
TABLE ш
E N E R G Y F O R C R O S S - S E C T I O N M A X IM A O F S O M E R E L E V A N T E L E C T R O N I C T R A N S IT IO N S
M = c f 4 M = He
Reaction
1 AE I3 ^max*5 |ЛЕ|3 E ^ max
1. F + M->F+ + M + e“ 17.4 eV 8 X 10s eV 17.4 eV 8 X 10s eV
2. F + M ->F‘ + M + 13.4 5 X 10s 2 1 .2 1 X 106
3. F+'+ M - *F + M + 0 .4 4 X 102 7.2 2 X 10s
4. F" + M-+F + M + e" 3 .4 3 x 104 3 .4 3 X 104
5. F+ + M -» F ( 4p) + M + 1 3 .1 5 X 105 19.4 1 X 106
6. F(4p) + M -* F + M 12.7 4 X 10s 12.7 4 X 10s
7 . F (4p) + M - * F + + M + e " 4.7 6 X 104 4.7 6 X 104
a Absolute energy of ionization. Ionization potentials : F 17.4 eV, He 24.6 eV, CF4 17.8 eV
(R.W. Kiser, USAEC-ТШ 6142).
F atom state: 3s4P5/ 2 12. 7 eV (NBS Circular 467).
Electron affinity F = 3 .4 eV. R. S. Berry. /ДЕа\2
b Kinetic energy at cross-section maximum Emax1/ 2 m H r ) = 2700 (ЛЕ>2 eV-
of these broad m axim a cro ss-sectio n s are sufficiently large so that charge equilibrium of the fast species is reached in a few collisions. Furtherm ore, the magnitudes of these maxima tend to increase with decreasing energy [15, 16] . Thus, at a given energy, processes that have their maxima at or near that energy will generally dominate processes having their maxima at higher energies in determining the state of the species.
A clo ser examination of Table III shows that in CF4 ionization processes reach their m axim a at much higher energies than do neutralization p ro cess e s . Thus as the hot F i8 n ears the chem ical energy range ( < 102 eV) it is far m ore likely to be neutralized than ionized.
The higher ionization potential of helium makes the situation less clear- cut if it is the main stopping medium. It is seen that ionization of excited F atom s (reaction 7, Table III) reach es its maximum below the maximum for neutralization (reaction 3). Thus in pure helium some particles may be "trapped" in ionic form without an opportunity to become neutralized as they fall to low er en erg ies. However, such ions can still be neutralizí i if a sm all fraction of CF4 or other lower ionization-potential m aterial is present, as is the case in these experim ents.

REACTIONS OF HOT FLUORINE-18 161
We m ust further consider the possibility of re-io n ization subsequent to the reco il by delayed nuclear p ro cesses . The reco il has a duration of the magnitude of 10"lu- 0~a s at 1 atm pressu re. If the (7 ,n ) reaction populates nuclear levels with a longer lifetim e than this, the F 18 may be p e r turbed a fte rith a s com eto re s t. Levels that might be relevant in this respect are the 5+, 1.13 MeV and 3+, 0.94 MeV states [17] . Any F 18 in these levels would undergo E2 gamma transitions which can have a half-life of the magnitude of the recoil time [18] . These transitions would be relatively unlikely to cause re-ionization by internal conversion [19] although the gamma recoil may again make the F 18 a hot atom, this time with an initial energy of about 20 eV. The expectation that such delayed p ro cesses a re of negligible im portance is confirmed by the finding (see section on results) that variation of p re ssu re , and hence re c o il tim e o ver an o rd er of magnitude, has no appreciable effect.
The conclusion that we are indeed dealing with ground state hot atom s [2 0 ] is further confirmed by the data. If the reactions w ere due to ions, a dependence on additives of different ionization potential would be expected. None was found. If the reactions w ere due to internal excitation energy the observed m oderator dependence would be totally unexplained. As it is , the behaviour with m oderator quantitatively confirms (see Discussion) that the F 18 reacts by virtue of its excess kinetic energy.
5. DISCUSSION
5. 1, Kinetic analysis o f results
The kinetic theory of hot reactions [7, 8 ] provides a basis for expressing data from experim ents such as these in term s of two types of p aram eters: "R eactivity In tegrals", I, probabilities of reaction on collision integrated over all energies (on a logarithmic scale), and " а -values", m easures of the average logarithm ic energy loss p er collision. We use here a form of the theory, with assumptions and development as outlined in a recent publication [8 ] . Definitions of symbols are given in Table IV.
5 . 1 . 1. Average logarithmic energy decrement, а
We determine the average energy loss p er collision, using the equation
_ 1 ' ln (l - P)
Here P is the total probability that a hot atom becomes combined in any hot product and I is the corresponding total reactivity integral. On plotting the left side of Equation (10) against 1 - freact/freact a straight line should result, with slope Q-mod/I and in tercep t areact /I . Knowing th eir ra tio , a mod M e a c t ,
we can then determ ine ctteact.
а а , (1 - f )react mod K react1 / т т f * \*-V)

162 N. COLEBOURNE, et al.
TABLE IV ;
G L O S S A R Y
a Average logarithmic energy loss per collision of hot atom.
otXi j Average logarithmic energy loss per collision of hot atom x with component j.
E r Lower energy limit for hot atom chemical combination reaction.
E2 Upper energy limit for hot atom chemical combination reaction,
fj Relative probability of collision with component j.
1 _ j 2® Reactiv ity integral. Refers to p robab ility for a ll hot reactions.
Ei ■b -
= / 1 dE: Reactivity integral. Refers to probability of reaction to form product i.E, E . *
m Mass of hot atom.
Mj Mass of component j.
p(E): Probability of chemical combination on collision.
Pi(E) : Probability of reaction on collision to give product i.
P Total probability of chemical combination reaction of a hot atom before
thermalization.
Pi Total probability of reaction to give product i.
SiCollision cross-section for hot atom and component j.
X iMole fraction of component j. _
Now P is the sum of probabilities of form ing individual products
. P " P CF,f'' + P CF,F“ + P FFJi •
While we have determined Pcf3f18 + Рсяг f 18 at various collision fractions (f)
of C F 4 m ixed with m oderator (see F ig . 3), we only have an upper lim it on P FF18 . However, as pointed out in [8 , (Appendix II)] if the reactivity is small, as in this case , P for only part of the products may be used in Equation (10) without appreciably affecting the ratio of slope to intercept.
Figure 4 shows plots according to Equation (10) for Ne, Ar and He modera to r se r ie s . The P ’s used a re the sum of P c f 3 f u and P c f z f 18 (F ig .3 ) and
Pppis is taken as zero (in accordan ce with the previous statem ent). The collision fraction f react was calculated using
' ' X : . S. f = ____________ CF« 4______________ / 1?\
• .. . . X CF4 : SCF4 + (l ' X CF4 ) ^Moderator
(where collisions with ethylene are neglected). The evaluation of Equation
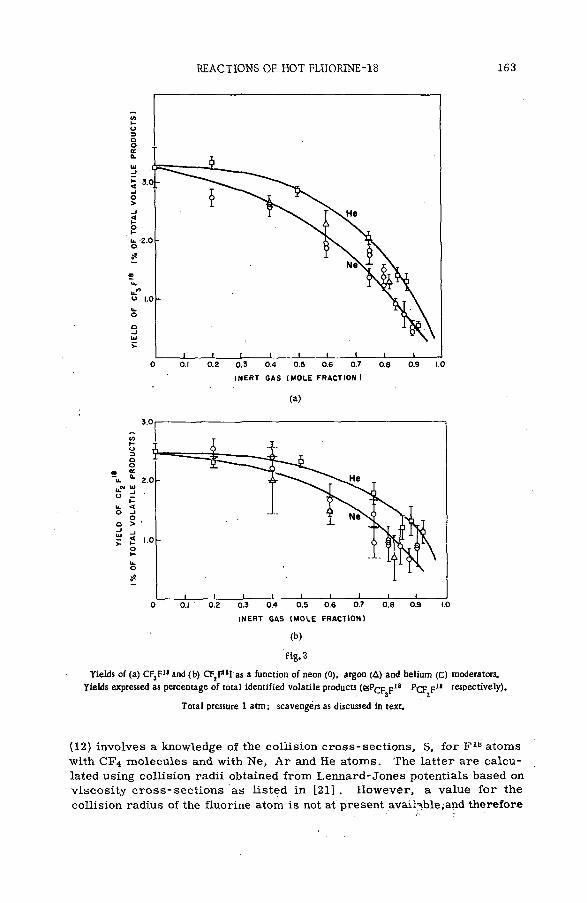
REACTIONS OF HOT FLUORINE-18 163
I N E R T G A S (M O LE F R A C T I O N )
(a)
I N E R T G A S ( M O L E FRA C TI O N )
(b)
Fig.3
Yields of (a) CF3F1S and (b) CF F^I'as a function of neon (0), argon (Д) and helium (□) moderators.
Yields expressed as percentage of total identified volatile products (sPçp p18 Pq : f 18 resPectively).
Total pressure 1 atm; scavengers as discussed in text.
(12) involves a knowledge of the collision cro ss-se ctio n s , S, for F ly atoms with C F4 m olecules and with Ne, A r and He atom s. The la tte r are calcu lated using collision radii obtained from Lennard-Jones potentials based on visco sity c ro s s -s e c tio n s as listed in [21] . How ever, a value for the collision radius of the fluorine atom is not at present available,and therefore

•Ín
(l-P
)
(a)
0 - V / 4 о- Ч ’/ Ч ‘ ' - v /ЧFig. 4
Plots of total probabilities of reaction according to Equation (10).
Data for (a) Ne, (b) Ar and (c) He moderators are represented.
p = PCFjF1* + pCFjF** ; Pff1* has been taken as zero in this plot (see text).
164 N.
COLEBO
URNE,
et al,

REACTIONS OF HOT FLUORINE-18 165
it was taken as being equal to that of neon. The following collision c r o s s sections w ere used:
SF. CF = 4 .4 9 X 1 O' 15 cm2, Sp_He = 2.33 X 10 ' 15 cm 2
SF. Ne = 2.57 X 10 ' 15 cm 2, SF_Ar = 3.14 X 10 ‘ 15 cm 2.
As required by Equation(10) straight lines a re obtained. Ratios of slopes to intercepts «mod/0, react are given in Table V [22] .
In order to deduce absolute values of <*react and I it is n ecessary to evaluate <zmod- An estim ate of this param eter may be readily obtained a s suming hard sphere e lastic collisions. However, E stru p 123] has shown that a m ore rea lis tic model for this type of collision may involve "so fte r" interm olecular potentials. Since it is the interpretation of the kinetic data ra th e r than the kinetic analysis itse lf that depends upon the knowledge of «mod» it is preferable to express values for the param eters a¿F and I in term s4of a mod. F u rth erm o re , it is c le a r that the absolute values of ap.CF4 and
reactivity integrals determined using different m oderators should be independent of the m oderator. This allows us also to exp ress values for «f, Ar and <*FtHe in term s of q,Fi Ne . A sum m ary of a and I values calculated from the values in Table V is listed in Table VI.
5 .1 .2 . Reactivity integrals
The p artial reactivity integrals, I c f 3 f 18 and I c f 2f 18 » the sum of which
should equal I a, may be determined using
^CF4 f C F<K i f CF4 L iP = ------------- ------5— + ----- g— •••. (13)
1 a ot¿ aJ
H ere,
a - fCF «F.CF, + (1 "fcF, ) a F. mod- (14)4 4 4
If, as expected in view of the sm all P ’ s andl’ s in this system , all term s but the first two are negligible, plots of (aV^cF*)1 v ersu s içp ja : should give straight lines with in tercep ts I ¡ and elopes K ¡. Such plots a re shown inF ig .5 . Values of P Cp3pi8 and Pçf f1* are taken from F ig . 3 ; the values ofa, exp ressed in units of <*F Ne, a re calculated from equation (14) and the relationships in Table VI.
Figu re 5 shows plots of CF3 F 18 and C F 2F 18I according to Equation (13). Because of low activities in this system e rro rs are large. N evertheless, it can be seen that points for different m oderators do fall on the sam e line as
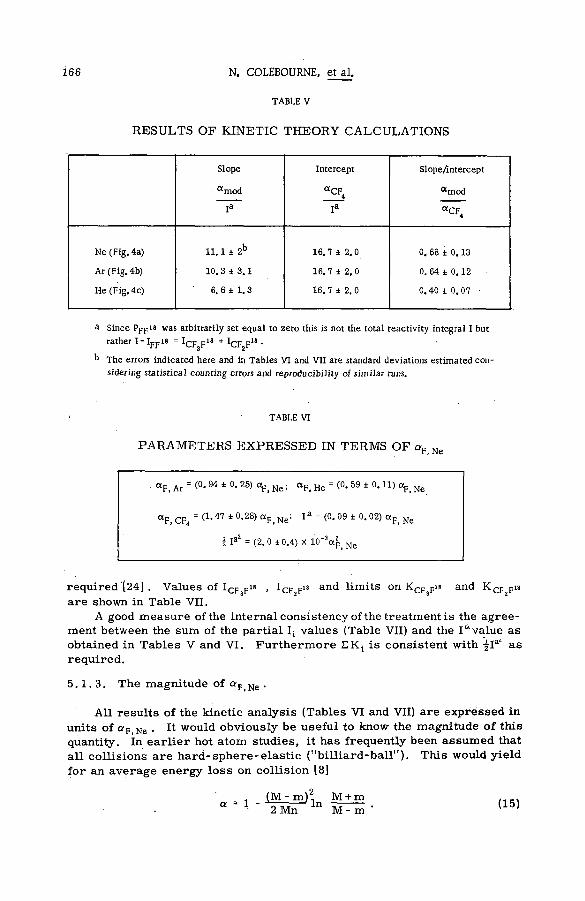
TABLE V
166 N. COLEBOURNE, et al.
RESULTS OF KINETIC TH EO RY CALCULATIONS
Slope Intercept SlopeAntercept
a mod “ c f4 “ mod
I a I a “ c f 4
Ne (Fig. 4a) 11 .1 ± 2b 16. 7 ± 2. 0 0. 68 ± 0.13
A r (Fig, 4b) 10. 3 i 3.1 16.7 ± 2 .0 0.64 ± 0.12
He (Fig.4c) 6. 6 ± 1. 3 16. 7 ± 2. 0 0.40 ± 0.07 ■
a Since Ppp 18 was a rb itra rily set equal to zero this is not the to ta l reac tiv ity in tegral I but
rather I- Ip p i8 Iq f pi® + ^CF F*8 *
b The errors indicated here and in Tables V I and V I I are standard deviations estimated con
sidering statistica l counting errors and rep roduc ib ility of s im ila r runs.
TABLE V I
PA RA M ETERS EX P R ESSED IN TERMS OF aFNe
, . a F A r - (0.94 è 0.25) OpiN e ; otF_ He = (0. 59 è 0.11) Ng
a F, CF¿ ~ ^ * 0*28) I а = (0. 09 ± 0, 02) otp
I I a2 = (2.0 ±0.4) X 10-34 iN e
required [24] . Values of ICFjFis , IcFjF18 anc* lim its on K ^pis and K Cp2pi8 are shown in Table VII.
A good m easure of the internal consistency of the treatm ent is the agreement between the sum of the partial I¡ values (Table VII) and the Ia value as obtained in Tables V and VI. F u rth erm o re EK ¡ is consistent with ^Ia‘ as required.
5 . 1 .3 . The magnitude of Ne .
AH results of the kinetic analysis (Tables VI and VII) are expressed in units of <*FiNe . It would obviously be useful to know the magnitude of this quantity. In e a rlie r hot atom studies, it has frequently been assum ed that aH collisions a re h a rd -sp h e re -e la stic ("b illiard -b all'1). This would yield for an average energy loss on collision [8 ]
REACTIONS OF HOT FLUORINE-18 167
■ I1 l\ li Ï 1jfi; fi1 1
■ i i
Í rr^
i i i . j ----------O.l 0.2 0.3 0.4 0.5 0.6 0.7
f/ „ (units of a ; 'N<)
f/a (units of a "'Nt) '
(b)
Fig. 5
Determination of partial reactivity integrals (a) Içp p‘« and (b) Iq : pie
according to Equation (13) for reactions moderated by neon (0), argon (Д) and helium (d),
On the basis of an examination of known atom -m olecule potentials, Estrup has concluded that collisions of hot hydrogen atoms are better described as soft sphere and that a values may be as much as an order of magnitude less than those calculated on the billiard-ball model.
TABLE v i l
168 N. COLEBOURNE, et al.
PA RTIA L REA CTIV ITY IN TEG RALSa
i l i K i O j
CF jF18 0. 050 ± 0. 01° - c r
0. 092 ± 0. 03 o . o i s 4 * ' 024"0.013
CF^ 8 0. 042 ± 0. 01 0 . 0 1 3 « : «
a Expressed in units of a p
The present results provide experimental evidence that collision of hot fluorine atom s not only with CF4 but with m oderator deviate from the billiard- ball c h a ra c te r . Table VIII com p ares ra tio s of a values calculated from
t a b l e v n i
COMPARISON BETW EEN CALCULATED HARD SPHERE AND ACTUAL a - VALUE RATIOS
“ F.He a F, Ar “ f . c f 4
“ F.Ne “ F. Ne “ F.Ne
B illiard-ball
Experimental
0.40
0. 59 ± 0.11
0. 69
0. 94 ± 0. 25
0.36
1.47 i 0.28
Equation (15) and those experim entally obtained. The fact that CF4 is a b etter m o d erator than suggested by the b illiard -b all hypothesis was to be expected because of the internal degrees of freedom available. The deviation of ratio of orp,не to <*F,Ne from h ard -sp h ere values is perhaps m ore s u rprising and indicates that this model seem s to have little validity even for hot atom -atom collisions.
At present F -N e potentials are not available and a F Ne is therefore unknown. It will certainly be less than 0.98, the billiard-ball value, but it is difficult to conceive that the F -N e potential will be so soft that it would be under 0 .1 . This corresponds to the following lim its on the absolute values
: 1-4 - 0-14
I a n 0.09 - 0.009

REACTIONS OF HOT FLUORINE-18 169
It appears that the kinetic theory in the form used is completely adequate as a basis for interpreting data in this system . It successfully co llates a la rg e amount of data in te rm s of two fundamental quantities, and does so without the use of arb itrary o r adjustable param eters. However, it must be pointed out that the assumptions of this form of the kinetic theory would be expected to hold best in just such a low reactivity system as this.
5.2. Conclusions as to mechanism o f hot fluorine reaction
5 .2 .1 . Interesting aspects
Three aspects of the results a re of p articular in terest:(a) A most striking feature of the data is the low total probability of reaction (P) as reflected in the sm all reactivity integrals (I). These values are about an o rd er of magnitude low er than those obtained fo r analogous reactio n s of hot hydrogen with methane and other m olecules 17,25] . Some difference in com parative reactiv ities in these system s might be expected because of the g rea ter strength of the С - F versus the C-H bond [12] . But, because of the excess energy available, hot atom reactions should not have the sharp energy dependences found in th erm al p ro ce sse s [26] . A difference of an ord er of magnitude in reactiv ities can therefore not be explained by a halfe lectro n volt spread in bond en erg ies . F u rth e rm o re , frag m en tary data [3, 13] on gas-phase reaction s of hot B r and I indicate that these halogens also tend to have low reactivities, regardless of the bond strengths involved.(b) A second feature of these results is the relatively high energy loss per collision of F with C F 4 in the "chem ical" energy range. The most efficient energy tran sfer in elastic collision of a fluorine atom would be with an entity of sim ilar m ass, i. e. Ne. Y et the resu lts show that energy loss with the much heavier CF4 molecule is g rea ter. Thus it is obvious that collisions of hot F 18 with CF4 a re highly in elastic whether o r not reaction takes p lace .
Certainly the b illiard -b all model for hot-atom reaction , as suggested by Libby [28] for liquid-phase hot halogen p ro cesses, is not operative here. Such a m echanism would p redict a high reactivity integral for substitution of F by F , and this is just what is not found. This result further confirm s e a rlie r experim ental and theo retical findings [27] that indicate that the b illiard -b all model is not applicable to gas-phase hot-atom p ro c e s s e s .(c) The probability of F 18 replacing two fluorine atoms to form CF2F 18
(reaction 6 ) is about as great as that of replacing one atom to form CF3F 18
(reaction 5 ) . The ratio of reactivity integrals is I c f 2f 18 / I c f 3f 18 s 1 • The
corresponding ratio for reaction of hot hydrogen (tritium ) with methane toform CH2T and CH3T respectively is ICH t / I ch t = ° - 1- This is completelyt- 3[7] con trary to what would be expected on the basis of simple bond-energy consideration. The formation of CF2 F !8 requires at least 4 eV (reaction 6 b) m ore than form ation of C F 2F 1S, while the analogous reaction to give CH2T requires no m ore energy than that yielding CH3T . This situation provides
5 .1 .4 . Summary

170 N. COLEBOURNE, et al.
an illustration of the principle that considerations of energy economy do not prim arily determine the path of hot-atom reactions 129] .
5 .2 .2 . Models
Two facto rs m ust be considered in attempting to extend the model of hot-atom reaction developed for hydrogen to hot fluorine:(a) Substitution reactions of hot hydrogen a re postulated to involve a fast, localized collision in which one o r two bonds are broken at the point of impact [29] . Because of the la rg er size and, at a given energy, the lower velocity of a hot fluorine atom , the collisions that it m akes cannot be regarded as either fast o r localized. There will therefore be m ore opportunity for the translational energy of the incident atom to diffuse throughout the molecule struck within the tim e of the interaction . This delocalization of available energy makes it less likely that any one bond in the a re a of impact will be broken during the collision . The probability of substitution reaction may thus be reduced. In higher energy collisions bonds will of course be broken and combination of the hot atom can o ccu r. However the p ro cess is then liable to involve so much energy that, because of the energy delocalization, m ore than one bond breaks and m olecular fragments result.(b) An alternative approach is to com pare the s te ric hindrance which the fluorine atom experiences when interacting with the C -F bond in CF4 with the ease of attack on a C -H bond by a tritium atom . It is evident that in the form er case the possible angles of approach of the fluorine atom to the C -F bond are very much more restricted than in the tritium-methane interaction, thereby reducing the relative probability of the F lb undergoing a substitution reactio n . F u rth erm o re , because of its g re a te r size com pared to that of hydrogen, when the hot F 18 atom does meet the steric requirements for interaction with the C -F bond there is a relatively greater chance of simultaneous interaction with a second, neighbouring C -F bond, thereby favouring the double displacem ent reaction to yield C F 2F 18.
A possible m eans of evaluating the relative im portance of these two factors is to investigate the reaction of hot fluorine with hydrocarbons. B ecause the C-H stretching frequency is greater than that of C -F , energy flow should be m ore rapid in hydrocarbons. If this factor is dominant, even lower total reactivity and higher fragmentation would be expected in F + hydrocarbon sy stem . On the other hand s te ric fa cto rs would p redict an enhanced r e activity and relatively diminished fragmentation, with the F 18 + CH4 system falling somewhere between H3 +C H 4 and F 18 + C F4.
P relim in ary resu lts on F í8+ CH4 indicate that it is the second s te ric factor that appears to be most important in causing the differences between the reaction s of hot fluorine and hydrogen atom s. In any ca se the basic model of hot-atom reactions as developed for hydrogen and modified by these two facto rs appears to provide a natural b asis fo r accounting for the low reactivity of the fluorine atom, the high relative yield of the fragment CF2F 18 and, of course, the inelastic nature of collisions in which the hot atom escapes combination. A sim ilar pattern for substitution reaction of other heavy hot atoms in other gas-phase system s is expected [13] . The model will, however, probably require modification when applied to liquid-phase system s. It seem s

REACTIONS OF HOT FLUORINE-18 171
quite likely that the stabilizing effect of the solvent cage will increase yields while diminishing the relative importance of fragm entary products.
A C K N O W L E D G E M E N T S
The authors wish to thank the staff of the Yale University Linear Electron A ccelerator for their helpful co-operation.
APPENDIX
UNIMOLECULAR DECOMPOSITION OF EXCITED FLUOROETHYL RADICAL
The scavenging of th erm al F 1B atom s by ethylene was confirm ed by studying the unimolecular decomposition of the resulting fluoroethyl radical. Such decomposition to vinyl fluoride
C2H4F 18* r* C2H3F 18 + H (2)
is competitive with third body de-excitation
C2H4F 18* + CF4 - C2H4F ^8 + C F4* (3a)
. ., , C2H4F 18* + Ne - C2H4F 18 + Ne*. (3b)
The de-excited radical then combines with iodine to form C2H4F 18I.
Pressu re dependence
The dependence on total pressure of the ratio C2H3F /C 2H2FI in unmoderated CF4 system s is shown in Fig . 6 . As expected, this ratio increases with decreasing pressure.
Moderator dependence
Figu re 7 shows the dependence of the relative yields C2H3F /C 2H3F I on dilution with neon m oderator at constant pressure. The decreasing trend of this ratio shows that Ne is a poorer m oderator than CF4 .
The form ation of the two th erm al products is governed by the usual equations
d(C2H4F I) ’ ■-------- ----------‘ = k3J C2H4F )(c f 4 ) + ^ „ ( C ^ F 18 )(Ne)
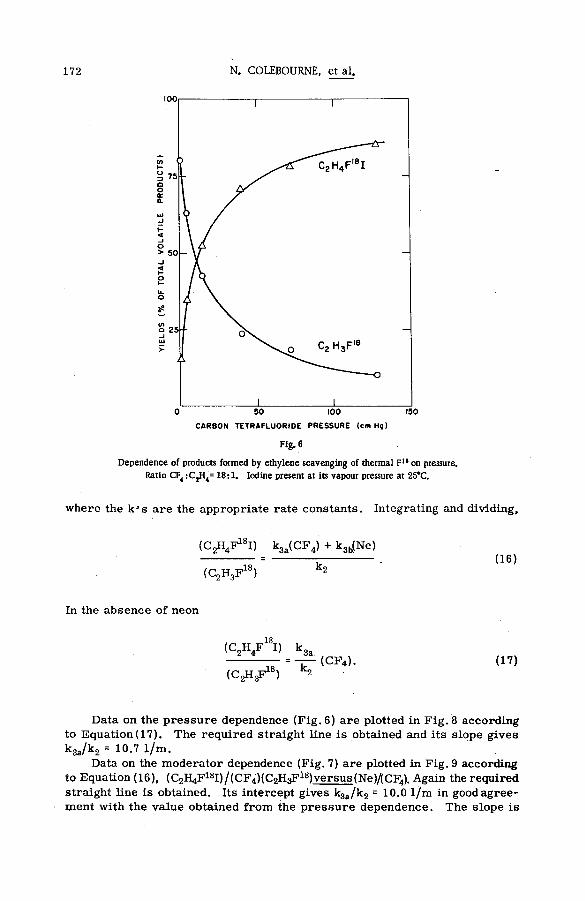
172 N. COLEBOURNE, et al.
CA R BO N T E T R A F L U O R I D E P R E S S U R E (cm Hg)
Fig.6Dependence of products formed by ethylene scavenging of thermal Fu on pressure.
Ratio CF4 :C jH 4= 18:1. Iodine present at its vapour pressure at 25°C.
where the k ’ s are the appropriate rate constants. Integrating and dividing,
( C ^ F 18!) k3a(C F4) + k3b(Ne)
(C ,H ,F18)(16)
In the absence of neon
18( C 2H 4F ^ k 3a
(C ^ a F 18) k 2(CF4). (17)
Data on the p ressu re dependence (Fig . 6 ) are plotted in F ig . 8 according to Equation (17). The required straight line is obtained and its slope givesk 3 a /k 2 = 1 ° - 7 l / m
Data on the m oderator dependence (Fig. 7) are plotted in Fig . 9 according to Equation (16), (C2H4F 18I)/(C F 4 )(C2H3F lii)versus(Ne)/(CF4). Again the required straight line is obtained. Its intercept gives k3a/k 2 = 10.0 l/m in good agreement with the value obtained from the p ressu re dependence. The slope is

REACTIONS OF HOT FLUORINE-18 173
NEON ( M O L E FRACTION)
Fig. 7
Dependence of products formed by ethylene scavenging of thermal F18 on the concentration of
Ne at a to ta l pressure of 1 atm. Ratio CF4 :C jH 4 as discussed in text.
Iodine present at its vapour pressure at 25°C.
. Fig . 8
Plot of pressure dependence according to Equation (16)
k3b/k 2 = 1.3 1 / m. The ra tio of intercept to slope кза/кзь = 7. 5. This r e presents the relative efficiencies of CF4 and Ne in removing energy from the excited C2H4F18.
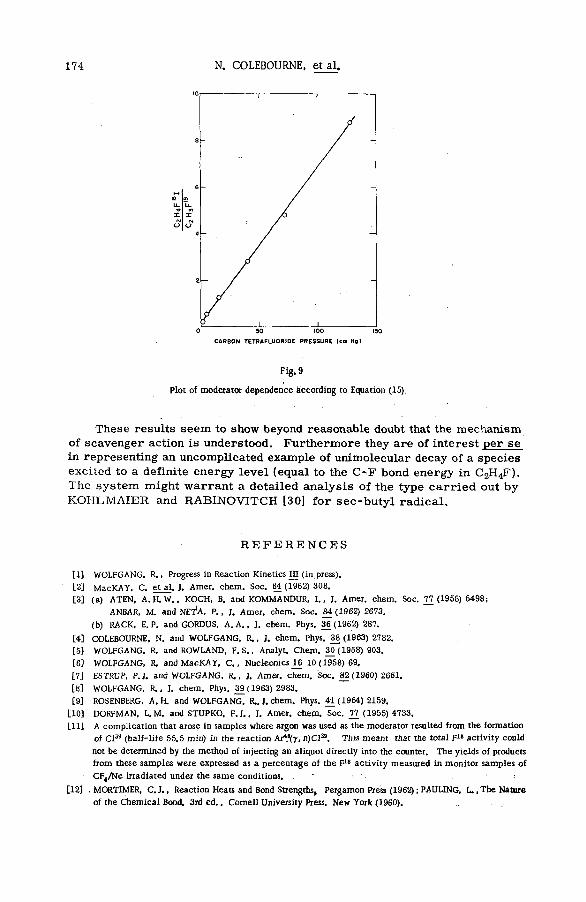
174 N. COLEBOURNE, et al.
Fig. 9
Plot of moderator dependence According to Equation (15)
These results seem to show beyond reasonable doubt that the mechanism of scavenger action is understood. Furtherm ore they are of interest per se in representing an uncomplicated example of unimolecular decay of a species excited to a definite energy level (equal to the C -F bond energy in C2H4F ). The system might w arran t a detailed analysis of the type c a r r ie d out by KOHLMAIER and RABINOVITCH [30] fo r sec-b u ty l rad ica l.
R E F E R E N C E S
[1] WOLFGANG. R., Progress in Reaction Kinetics Ш (in press).
L2J Mac KAY, C. et a l. J. Amer. chem. Soc. 84 (1962) 308.
[3] (a) ATEN. A .H .W .. KOCH. B. and KOMMANDUR, I. , J. Amer. chem. Soc. 77 (1955) 5498;
ANBAR. M. and NETA. P .. J. Amer. chem. Soc. 84 (1962) 2673.
(b) RACK. E. P. and GORDUS, A. A . . J. chem. Phys. 36 (1962) 287.
[4] COLEBOURNE. N. and WOLFGANG, R ., J. chem. Phys. 38 (1963) 2782.
[5] WOLFGANG, R. and ROWLAND, F. S ., Analyt. Chem. 30 (1958) 903.
[6] WOLFGANG, R. and MacKAY, C . , Nucleonics 1_6 10(1958) 69.
L7J ESTRUP, P.J. and WOLFGANG, R .. J. Amer. chem. Soc. 82(1960) 2661.
[8] WOLFGANG, R ., J. chem. Phys. 39(1963) 2983.
[9] ROSENBERG, A. a and WOLFGANG, R., J. chem. Phys. 41(1964) 2159.
CIO] DORFMAN, L. M. and STUPKO, F .J .. J. Amer. chem. Soc. 77 (1955) 4733.
L11J A complication that arose in samples where argon was used as the moderator resulted from the formation
of C lw (half- life 55.5 min) in the reaction Ai^û(y, n)C lS9. This meant that the total F1B ac tiv ity could
not be determined by the method of injecting an aliquot directly into the counter. The yields of products
from these samples were expressed as a percentage of the F*“ a c tiv ity measured in monitor samples of
CF4/Nc irradiated under the same conditions. . . •
[12] , MORTIMER, C. J ., Reaction Heats and Bond Strengths, Pergamon Press (1962) ; PAULING, L . . The Nature
of the Chemical Bond, 3rd ed.. Cornell University Press, New York (1960).

REACTIONS OF HOT FLUORINE-18 1-75
[13] CROSS, R.J. and WOLFGANG. R ., Radiochim. Acta 2 (1964) 112.
[14] MARSHALL, М ., MacKAY, C. and WOLFGANG, R..-J. Amer. chem. Soc. (in press).
[15] M O TT , N. and MASSEY, H .. The Theory of A tom ic Collisions. Oxford ( 1949) ; MASSEY, a and
. BURHOP, E. H. S .. Electronic and Ionic Impact Phenomena, Oxford (1952).
[16] HASTED, J.B .. ALLISON, S. K. and GARCIA-MUNOZ, М .. Atomic and Molecular Processes (D. R. Bates.
Ed.) Academic Press (1962); HASTED. J. B.. Adv. Electron. 13 (1960) 1.
[17] Nuclear Data Sheets V Set 2 (Dec. 1962) Nat. Acad. Sci., Nat. Res. Council.
[18] EVANS, R .D ., The Atom ic Nucleus, McGraw-Hill (1955) 215.
[19] EVANS, R .D ., The Atomic Nucleus, McGraw-Hill (1955) 222.
[20] By "ground state" we mean both states of the ground state doublet (separated by 0.05 eV).
[21] HIRSCHFELDER. J. O ., CURTISS, C. F. and BIRD, R ., Molecular theory of Gases and Liquids, John W iley
and Sons, In c ., New York (1954) 1110.
[22] It is readily verified that if, instead of taking Ppp18 = 0, it is set equal to some small m u ltip le of Pq; p18
(see section on results) there is no appreciable change in this ratio. This is illustrated by Fig. 1 in [8] in which
Pppis is taken as equal to PCF p ie. .
[23] ESTRUP, P .J ., J. chem. Phys. 41(1964) 164.
[24] No significance is attached to upward and downward trends for the yields of CFjF18 and CF3F18 respectively
at high moderation since there are appreciable sources of systematic error as we ll as the statistical errors
indicated when determ ining the yields of hot products at activ ities that were sometimes as low as
50 counts.
[25] The I values in [7] are actua lly low (see [8], footnote 15). I t appears that I- values for hot hydrogen
substitution in hydrocarbons are p 0.5 (see [9]).
[26] WOLFGANG. R ., J. Amer. chem. Soc. 84 (1962) 4586.
[27] CROSS, R.J. and WOLFGANG, R ., J. chem. Phys. 35 (1961) 2002.
[28] LIBBY, W .F . , J. Amer. chem. Soc. 69(1947) 2523.
[29] HENCHMAN, M . e t.a l. , Cañad. J. chem. 38 (1960) 1722.
[30] KOHLMAIER, G. H. and RABINOVITCH, B. S ., J. chem. Phys. 38 (1963) 1692.
D I S C U S S I O N
B. DZANTIEV: Did you observe any products of interaction between the excited radical C2 H4 F l8 and the CF4 m olecule, i . e . the product C 2H4F18 F19 ?
J . DUBRIN: We did not observe this product and this is not unreasonable in view of the fact that the bond strength of the CF4 is approximately 118kcal, whereas that of the iodine is approximately 35 kcal. Unless the intermediate, the fluoroethyl radical, were highly excited, one would expect its reaction with CF4 leading to the extraction of fluorine to be a relatively unimportant p ro cess. The product has actually been looked for.
F . S . ROWLAND: As the results given in this paper show, the problem of F 18 reco ils in gaseous system s prom ises to be a very interesting field of study. Com parisons of the reaction s of F18 with CE4 and those of T with CH4 immediately point to important differences in behaviour. The observation that the "double-substitution" product CF2 Fi8 has a yield about as large as that of CF3F 18 is interesting, especially in view of the fact that in our recent work in recoil-tritium systems we have failed to find any evidence for a direct double-substitution process in the prim ary hot reaction. In this work, essentially all of the "double-substitution" product observed in each system has been shown to be formed from secondary decomposition

176 N. COLEBOURNE, et al.
reactions following the initial prim ary single displacement reaction. This work has just been reported in the Journal of the A m erican Chemical Society*.
*J. Amer. chem. Soc. 86 (1964) 5038.

CH EM IC A L REACTIO N S OF N13 R E C O ILS FRO M TH E C12(d, n)N13 REACTIO N
W.S. KOSKI, D. MALININ AND M. BERTA THE JOHNS HOPKINS UNIVERSITY, DEPARTMENT OF CHEMISTRY,
BALTIMORE, MD., UNITED STATES OF' AMERICA
. Abstract — Résumé — Аннотация — Resumen
CHEMICAL REACTIONS OF N13 RECOILS FROM C12(d. n)Nls REACTION. Earlier studies of N « recoils
produced by the nuclear reaction Clz(d, n)Nw in CH4, CH8OH, CC14, etc. showed that the fina l radioactive
gaseous products were en tire ly cyanides such as HCN, CHSCN and C1CN. No ammonia or amines were
detected. In this study the investigation has been extended to benzene and CF4. In addition reactions of
N* ions w ith CC14 and CF4 have been examined in a tandem mass spectrometer. In the case of Nls recoils
reacting with benzene HCN was the main product and small amounts of benzonitrile were formed. No aniline
or pyridine were produced. This w i l l be contrasted with reported studies in which active nitrogen produced
by electrical discharge reacted with benzene. In the case of CF4 , the only radioactive product detected was
FCN. In both cases polymeric materials were produced on the walls of the reaction vessels. No other products
such as NF3 were detected. Studies of the effect of rare gas additives in the case of methanol indicated that
ion-molecule reactions were involved a t least in part. For this reason, the reactions of N+ ions w ith CC I4 and CF4 were studied in a tandem mass spectrometer in the bombarding ion energy range from 2 eV to. 200 eV.
In this study the re la tive cross-sections for various ion production were investigated as a function of energy.
In addition to ions composed of carbon and chlorine, various nitrogen-containing ions such as NC1+. CNC1+
and CN+ were detected. The shapes of the cross-section curves were such as to indicate complex formation
possibly (CC14N)+, which decomposed to give the product ions. The above complex was not detected directly.
REACTIONS CHIMIQUES DES ATOMES « n DE RECUL PRODUITS PAR LA REACTION 12Q d, n)»N. Des
études antérieures sur les réactions des atomes i*N de recul produits par la réaction nucléaire 12C(d, n)13N avec
CH4, CH3OH, C C I* e tc ., avaient montré que les gaz radioactifs finals sont constitués exclusivement par
des cyanures tels que HCN, CH3CN et CÏCN. On n’avait décelé aucune trace d'ammoniac ou d'amines. Dans
l ’étude qui fa it l'ob jet du mémoire, les auteurs ont étendu les recherches au benzène et au CF4. En outre,
ils ont étudié les réactions des ions N+ avec CC14 et CF4 dans un spectromètre de masse « tandem ». En ce
qui concerne la réaction des atomes 18N de recul avec le benzène, le produit p rincipal é ta it HCN et i l y a
eu formation de petites quantités de benzonitrile. I l ne s’est formé ni aniline ni pyridine. Ces résultats sont
comparés à ceux d'autres études dont i l a été fa it état et dans lesquelles l'azote ac tif produit par décharge
électrique avait réagi avec le benzène. Dans le cas de CF4, le seul produit radioactif qui a it pu être décelé
était FCN. Dans les deux cas, des polymères se sont formés sur les parois du récipient. On n’a pu déceler
aucun autre produit, par exemple du NF8. Des études sur l'effet de gaz rares ajoutés aux corps en réaction
dans le cas du méthanol ont montré que des réactions ion-molécule intervenaient au moins en partie. Pour
cette raison, les auteurs ont étudié les réactions d’ions N+ avec CC14 et CF4, dans un spectromètre de masse
« ta n d e m » pour une gamme d’énergies des ions projectiles a llan t de 2 à 200 eV. Dans ce trava il, ils ont
fa it des recherches sur les variations des sections efficaces relatives de production de divers ions selon l ’énergie.
En plus d’ions"composés de carbone et de chlore, ils ont décelé divers ions contenant de l ’azote, tels que
NC1+, CNC1+ et CN+. Si l'on en juge d'après l'a llu re des courbes de section efficace, i l se forme un com
plexe, peut-être (CC14N)+, qui se décompose pour donner les ions décelés, mais le complexe lui-même n’a
pas été décelé directement.
Х И М И Ч Е С К И Е Р Е А К Ц И И А Т О М О В О Т Д А ЧИ А 3 0 Т А - 1 3 ИЗ Р Е А К Ц И И УГЛ Е Р О Д -12
(d,n) А 3 0 Т - 1 3 . Предыдущие исследования атомов отдачи аэота-13, произведенных ядерной
реакцией углерод-12 (d,n) азот-13 в СН4 , С Н 3ОН, СС14 и т . д . , показали, что конечными
радиоактивными газообразными продуктами были только цианиды, такие как H CN , C H 3CN
и C 1C N . Аммиака или аминов обнаружено не было. В данном исследовании работа была
распространена на бензол и C F 4 . Кроме того, были изучены реакции ионов Н+ с ССЦ и
C F 4 в тандем мас-спектрометре. В случае атомов отдачи азота-13, реагирующих с бензо
17712

178 W .S . KQSKI et al.
лом, HCN был основным продуктом, а также были образованы небольшие количества бензо
нитрила. Анилина или пиридина образовано не было. Это противоречит исследованиям, в
которых активный азот, получающийся в результате электрического разряда, реагировал
с бензолом. В случае C F 4 единственным обнаруженным радиоактивным продуктом был FCN.
В обоих случаях полимерные материалы отлагались на стенках реактивных сосудов. Других
продуктов, таких как N F 3 , обнаружено не было. Изучение эффекта добавок редких газов в
случае метанола показало, что по крайней мере отчасти сюда были вовлечены ионо-моле-
кулярные реакции. По этой причине изучались реакции ионов IV^c СС14 и C F 4 в тандем
мас-спектрометре при бомбардировке ионами с энергией от 2 до 200 электронвольт. В этой
работе исследовались относительные сечения для производства различных ионов в функции
энергии. Помимо ионов, состоящих из углерода и хлора, были обнаружены различные азото
содержащие ионы, такие как NCI4 , CNC1+ и C N +. Формы кривых сечений были такими, что
указали на возможность комплексного образования (C C l4N)+ , которое при разложении дает
продукт ионов. Вышеуказанный комплекс не был прямо обнаружен.
REACCIONES QUIMICAS DEL WN DE RETROCESO RESULTANTE DE LA REACCION lzC(d,n)i3N. Anteriores
estudios del 13N de retroceso resultante de la reacción nuclear 12C(d, n)I3N en CH* CH3OH, CCI* etc., habfan
demostrado que los productos finales de carácter radiactivo y gaseoso consistían fntegramente en cianuros ta
les como CHN, CH3CN y C1CN, sin que se detectara la presencia de amoniaco n i de aminas. En el presente
estudio las investigaciones se extendieron a l benceno y a l CF4. Asimismo, se han investigado mediante un
espectrómetro de masas en tándem las reacciones de iones N* con CC14 y CF4. En la reacción del 1SN de
retroceso con el benceno, e l producto principal consistió en HCN, formándose también pequeñas cantidades
de benzonitrilo. No se observó la presencia de anilina ni piridina. Estos resultados se confrontan con los de
otros estudios publicados, en que el nitrógeno activo producido por descarga eléctrica reaccionó con el benceno.
En el caso del CF4, e l tínico producto radiactivo detectado fue el FCN. En ambos casos se produjeron sustancias
poliméricas en las paredes de los recipientes de reacción. No se detectaron otros productos, tales como el
N F j. Los estudios sobre e l efecto de la adición d¿ gases nobles en e l caso del metanol indicaron que
intervienen, al menos en parte, reacciones ion-molécula. Por esta razón, se han estudiado con un espectró
metro de masas en tándem las reacciones de iones N+ con CC14 y CF4, para una energía de los iones incidentes
comprendida entre 2 y 200 eV. En este estudio se investigaron las secciones eficaces relativas correspondientes
a la formación de diversos iones en función de la energía. Además de los iones compuestos de carbono y
cloro, se detectaron otros varios que contenían nitrógeno, tales como NC1+, CNCl'1' y CN+. El perfil de las
curvas de sección eficaz sugirió la formación de complejos, posiblemente (CC l^ í)+, que se descomponían
para dar origen a los iones indicados. E l complejo mencionado no se detectó directamente.
INTRODUCTION
When carbon-containing compounds are bombarded with deuterons of appropriate energy, the nuclear reaction C12(d, n)N13 o ccu rs. The nitrogen recoils as a positively charged ion with a considerable amount of translational and electronic energy. It dissipates this energy by collision with the walls of the container or with molecules in the gas phase. The charge is reduced by charge tran sfer processes and, finally, when the nitrogen has approached energies not very much higher than bond energies, it reacts with the molecules as a singly charged ion or as a neutral nitrogen atom. When the i r radiated gas was CH3B r [1] or CHgCl, the main product was HCN. In the cases of chloroform . C1CN was found in addition to HCN. This work has also been extendedlo methyl [21 alcohol, ethyl alcohol and methane. HCN and CHgCN are the gaseous activities found for CH4 and ÇH3OH. These activities plus CgH^CN were found in the ethanol bombardments. Experiments with ra re gas additives and B r? suggested that cyano ions o r radicals are the N13 c a r r ie rs and that the HCN was formed by hydrogen abstraction with

CHEMICAL REACTIONS OF N13 RECOILS 179
m aterials on the wall of the reaction vessel, whereas CH3CN was formed by reaction with ta rg et gas m olecules in the gas phase. No ammonia or amines w ere detected in any of these experim ents indicating that probably no abstraction of hydrogen by nitrogen was taking place. In the study r e ported here, the N13 recoil work was extended to CF4 and benzene. In addition some studies have been made on the reaction of N-1- ions with CCI4 and CF4 in a tandem m ass spectrom eter over an ion energy range of 3 to 200 eV.
EXPERIMENTAL
The bombardments were made with 2 MeV deuterons from an electro static generator in our laboratory . Beam current and bombardment times w ere kept as low as possible to minimize complications from reactions involving radiation dam age p roducts. C urrents as low as О.ОЗуи A and i r radiation tim es of 2 min can be taken as low er lim its used for these p aram eters. The irradiations were made through a 0.0001-inch nickel foil. The irradiation cells were made of b rass and of glass and ranged from 2-7 cm in diam. and w ere about 16 cm long. The gas chromatograph was an allglass instrum ent. It contained a 12—ft column of "C elite" coated with silicone oil. Helium was used as a c a r r ie r gas and a flow of 40 cm 3/m in was maintained by a p ressu re regulator. Detection of m acro amounts of m aterial was realized with a therm al conductivity cell. The radioactive gases were detected with 2ж methane proportional counters viewing diam etrically opposite Mylar windows of a glass cell through which the gas passed on exit from the chromatograph. The response of the detectors were recorded with a two-pen re c o rd e r . Identification of the compounds was made through a comparison of their retention tim es with those of the reference compounds.
All the chem icals used here w ere obtained from com m ercial sources and were subjected to further purification by trap -to -trap distillation, gas chrom atography o r, in the case of benzene, by repeated fractional crystallization .
The m ass sp ectro m eter used in this study was a tandem instrum ent. The prim ary ion beam is generated by a 180° magnetic m ass spectrom eter having a 1 cm radius of curvature. This ion beam passes between deflection plates and enters an ion lens which focuses it into a reaction chamber. This lens also adjusts the ion acceleration to any value between 2 and 200 V. The p rim ary ion beam c ro s se s the reaction chamber at right angles to the direction in which secondary ions are extracted through the exit slit. The slit is oriented so that its long dimension runs parallel to the prim ary beam direction,
A fter extraction from the reaction cham ber, secondary ions are a c celerated to 2 o r 3 keV and m ass analysed in an 8 -in radius, 60° magnetic sector m ass spectrom eter. In order to facilitate the detection of secondary ions from momentum tran sfer reactions, Z-direction deflectors have been added to the secondary m ass sp ectrom eter ion acceleratin g lens. These deflectors can be adjusted to counteract the Z -direction momentum tran sferred to the secondary ions permitting the secondary ions to reach the electron m ultiplier d etector. The ability of the Z defleótors to cancel the Z -d irection momentum of the secondary ions is dem onstrated by the fact
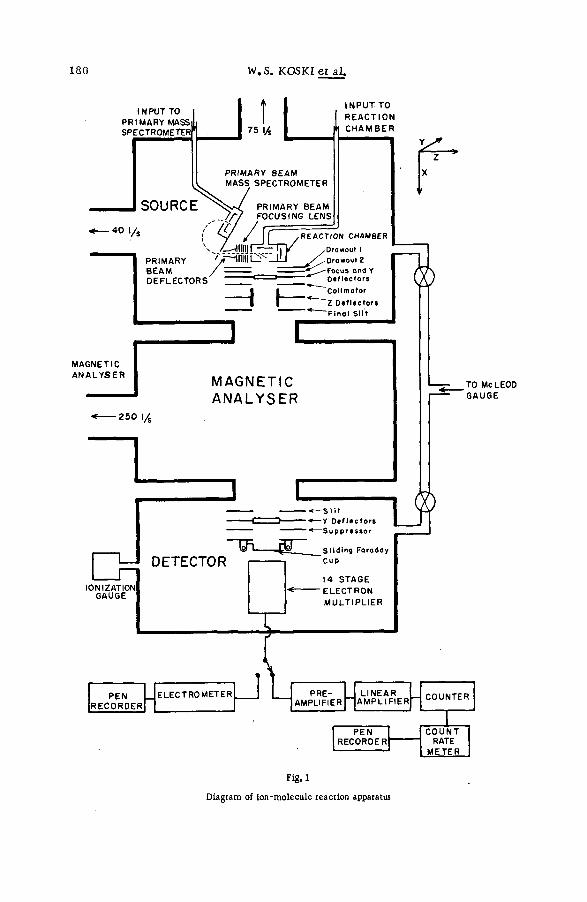
180 W .S . KOSKI et a l.
INPUT TO PRIMARY MASS
MAGNETICANALYSER
E L E C T R O M E T E R 1 1 PRE LINEAR COUNTERAMPLIFIER AMPLIFIER
PENRECORDER
COUNTRATE
METER
Fig. 1
Diagram of ion-molecule reaction apparatus

CHEMICAL REACTIONS OF №» RECOILS 181
that the prim ary beam itself can be focused on electron multiplier detector in the secondary m ass spectrom eter for energies up to about 40 eV by using the Z d eflectors with higher than usual rep eller voltages in the reaction cham ber. The resulting prim ary ion peaks are flat-topped and undistorted. A m ore detailed description of this instrument is given elsew here [3]. A schem atic diagram is. given in F ig . 1.
RESULTS AND DISCUSSION
Irradiation o í C Cl 4 and CF¿
Gas chrom atographic analysis of radioactive products produced by the deuteron irradiation of ССЦ showed that the dominant activity was due to CICNI3. The only other peak that appeared in the chrom atogram s was a sm all peak due to N14N13, which is believed to be produced by the reaction of N13 with N? absorbed on the walls of the reaction vessel. In studying the reaction s of N+ with CCU in the tandem m ass sp ectrom eter, presence of ions such as NC1+ was observed. It therefore may not be unreasonable to expect a product such as NCI3 in the deuteron irradiation of CC14. However, no peak corresponding to such a compound was observed in the gas chromatogram . In view of the low stability and high reactivity of NCI3, it might be expected that, if it were initially present in the radiation products, reaction o r decomposition might result either on certain m etal components of the apparatus o r in the packing of the chrom atographic column.
In o rd er to explore this feature further, we made some deuteron i r radiations of CF4, since if any N -F bonded compound such as NF3 was formed it would probably be readily detected because of its much greater stability com oared to NCI3 . Gas chrom atographic analysis of the radioactive products from the bombardment of CF4 ffave onlv one peak. At the time of writing of this report we have not made a completely unambiguous identification of this peak since no FCN was available to determine the retention tim e of this compound. This uncertainty will be removed as soon as some FCN is synthesized. However, in comparing the retention times of cyanides such as C1CN, BrCN , e t c . , it appears v ery probable that the peak in question is due to FC N . No other peaks w ere observed and no peak was found in the region that NF3 would be expected. We have tentatively concluded that no NF3 was form ed in this reaction .
Irradiation o f benzene
The chief product of the deuteron bombardments of benzene was identified as hydrogen cyanide. In addition, a sm all amount of benzonitrile was detected, the product ratio being about 10:1. Accumulation of a non-volatile polym eric m aterial on the walls of the target chamber indicated the occurrence of some radiation damage when gaseous benzene was irradiated. The amounts of this polymer were decreased and the gaseous products c o rre s pondingly increased when a solid benzene target was used. In this case, the benzene vapour was frozen out on the bottom of the glass cell at liquid nitrogen tem peratures before bombardment, then warmed to room temperature

182 W. S. KOSKI et al.
after irradiation for analysis of the products. Increasing the pressure of the benzene vapour in the target chamber from 15 to 60 mm gave increasing yields of HCN and slightly decreasing yields of benzonitrile.
P relim in ary studies involving the irradiation of m ixtures of benzene vapour and argon show that the yield of HCN increases with the addition of increasing amounts of argon as a m oderator. This result agrees with earlier work [21 on the alcohols where it was concluded that the HCN formed resulted from a therm al ra th er than a hot atom p ro ce ss . The sam e conclusions lead us to believe on the other hand that the benzonitrile is formed by a reaction with benzene of an N13 c a rrie r of greater than thermal energy.
It is of interest to compare our results with those obtained by ARONOVICH et a l . [4, 5] in a study of the action of active nitrogen on benzene in a discharge tube. These investigators found the two chief products of the 50-h reaction to be HCN and a polymer sim ilar to that described in the present study. In addition, tra ce amounts of benzonitrile, phenylisonitrile and pyridine were identified spectrographically. Form ation of the nitrile and isonitrile had been predicted as the result of the reaction of phenyl and cyano radicals. The unexpected presence of pyridine among the reaction products was attributed to a cleavage of the benzene ring and subsequent reaction of this fragm ent with nitrogen.
In the present study, neither phenylisonitrile nor pyridine was detected as a product of the deuteron bombardments of benzene. Although the p resence of the form er is perhaps to be expected in the course of the reaction, its detection as a product is alm ost certainly precluded by the relative instability of the isonitrile compared with benzonitrile. The occurrence of pyridine as a reaction product is considered improbable under our experimental conditions. The extrem ely low concentrations of our N13 ca rrie rs , which are believed to be cyano rad icals, suggest that the HCN and the benzonitrile a r is e s as a result of hydrogen abstraction and replacem ent, respectively , ra th e r than from ra d ica l-ra d ica l reaction s.
Reactions of N + with CC1¿
In the chem ical èffects of nuclear transform ation the question of the role of ion molecules frequently arises since the newly formed species re sulting from the transform ation may be ionic. F o r this reason we have investigated the reaction s of N+ ions with CCI4, CF4 and CF3C1. Typical resu lts are indicated in F ig . 2, where the relative cro ss-se ctio n s are given as a function of N+ ion beam energy. The curves for CF4 and CF3 C1 are sim ilar except in these la tte r two cases no ionic products involving nitrogen were observed. In the ССЦ case, in addition to the ionic reactions indicated in the figure, sm all amounts of CN+ were formed. The presence of the secondary ions such as NC1+ , NCC1+ and CN+ indicated that collision complex is formed between the incident ion and the target molecule at low energies in the CCI4 case . As the relative energy increases, the flattening of the cro ss-se ctio n s for the CCI4 fragment ions and the disappearance of the nitrogen-containing secondary ions shows that reactions become dominated by head-on collisions in which complex formation is increasingly im probable. These observations suggest that if a high energy nitrogen ion resulting from a nuclear transformation can survive the thermalization pro-
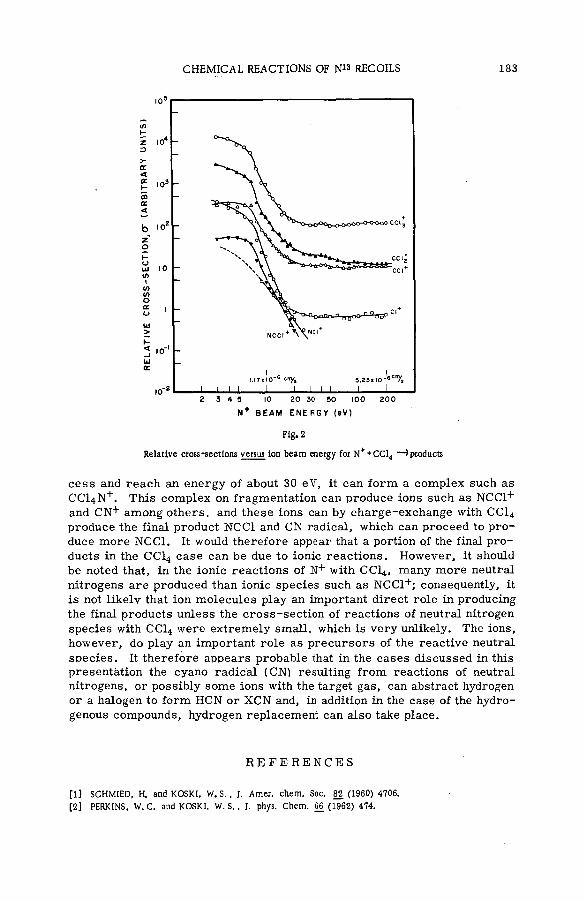
CHEMICAL REACTIONS OF №3 RECOILS 183
Fig. 2
Relative cross-sections versus ion beam energy for N+ + CC l4 —* products
ce s s and reach an energy of about 30 eV, it can form a com plex such as CCl4 N+ . This complex on fragmentation can produce ions such as NCC1+ and CN+ among others, and these ions can by charge-exchange with CC14 produce the final product NCC1 and CN radical, which can proceed to produce more NCC1. It would therefore appear that a portion of the final products in the CC14 case can be due to ionic reactio n s. However, it should be noted that, in the ionic reaction s of N+ with CCLt, many m ore neutral nitrogens are produced than ionic species such as NCC1+ ; consequently, it is not likelv that ion m olecules play an important d irect role in producing the final products unless the cro ss-sectio n of reactions of neutral nitrogen species with CC14 were extrem ely sm all, which is very unlikely. The ions, however, do play an important role as p recu rso rs of the reactive neutral species. It therefore appears probable that in the cases discussed in this presentation the cyano rad ical (CN) resulting from reactions of neutral nitrogens, or possibly some ions with the target gas, can abstract hydrogen or a halogen to form HCN or XCN and, in addition in the case of the hydrogenous compounds, hydrogen replacement can also take place.
R E F E R E N C E S
[1] SCHMIED, H. and KOSKI, W. S ., J. Amer. chem. Soc. 82 (1960) 4706.
[2] PERKINS. W.C. and KOSKI, W. S ., J. phys. Chem. 66 (1962) 474.

184 W. S. KOSKI et al.
[3] WEINER, E. R., HERTEL, G. R. and KOSKI, W. S . , J.'Amer. chem. Soc. 86 (1964) 788.
[4] ARONOVICH, P.M. and MIKHAILOV, В. М ., Izv. Akad. Nauk SSSR, Otdel. him. Nauk (1956) Г>44.
[5] ARONOVICH, P .M ., BEL'SKIl, N. K. and MIKHAILOV, В. M . , Izv. Akad. Nauk SSSR, Otdel. him.
Nauk (1956) 696.

G A S-P H A SE REA CTIO N S OF (11,7) AND ISO M ERIC T R A N SIT IO N -A C T IV A T ED B r 80
WITH A L K A N E S AND H A LO A LK A N ES
L.D. SPICER* AND A. A. GORDUS DEPARTMENT OF CHEMISTRY, UNIVERSITY OF MICHIGAN,
ANN ARBOR, MICH. , UNITED STATES OF AMERICA
Abstract — Résumé — Аннотация — Resumen
GAS-PHASE REACTIONS OF (n,y) AND ISOMERIC TRANSITION-ACTIVATED &M W ITH ALKANES AND HALO
ALKANES. Experimental data are presented on the gas-phase reactions of alkanes and haloalkanes with bromine atoms
and ions activated by nuclear transformations. The target molecules include CH* CD*, C2He, C2D6, CHSC1,
CH2C l2, CHCI3, CC14, CH2F2, CHFg, CF4, C2F6, CF3Br, and CH3Br. The nuclear reactions and transformations
used in producing the energetic recoil atoms and ions were Br8°m (isomeric transition), Br80, and Br79(n,y)Br80.
The percentage of the radioactivity found in organic combination (the organic yield, O. Y .) was determined
as a /unction of the concentration of the target molecule in the mole-fraction range of about 0.95 to 1.00.
E lemental Br2 served both as a source of hot atoms and as a scavenger. Usually 20-50 separate samples of
each reaction system were examined and the data of O. Y. as a function of the concentration o f scavenger
were plotted and extrapolated to un it mole fraction of target molecule. In a ll cases, die O. Y. decreased
w ith increasing halogen concentration.
Data on the (n,y) activated reactions of Br80 w ith isotopic alkanes suggest a .comparable extrapolated
O. Y. for CgHg and CgDg, but an O. Y. for CD* about half of the O. Y. with CH* Gas chromatographic analysis
of the organic products indicates that about 90% of the O. Y. in CH4 is caused by CHsBr and 10% by CHaBr*
For CD4 as the target the distribution of organic ac tiv ity is approx. 75% CDsBr and 25% CDjBrj.
These various data are discussed in terms of possible mechanisms involving hot halogen atoms and ions.
RÉACTIONS EN PHASE GAZEUSE DE еовг, PRODUIT PAR TRANSFORMATION NUCLÉAIRE ET PAR TRAN
SITION ISOMËRIQUE, AVEC DES ALCANES ET DES HALOGÉNURES D’ALCANES. Dans ce mémoire sont pré
sentées des données expérimentales sur les réactions en phase gazeuse d’alcanes et d'halogénures d'alcanes
avec des atomes et ions de brome activé par des transformations nucléaires. Les molécules cibles comprennent
CH* CD4, СгН& C2D6, СН,£1, СН£1г CHC13, CCI* CH ^Z, CHFS, CF4, C2F6, CF3Br et CH3Br, Les réaction
et transformation nucléaires utilisées pour la production d’atomes et d’ions de recul de haute énergie étaient
80mBr (transition isomérique) 80Br et 79Bi(n, y)®0Br. Les auteurs ont déterminé le pourcentage de radioactivité
en combinaison organique (rendement organique, RO) en fonction de la concentration de la molécule cible
dans la bande des fractions molaires a llant d’environ 0,95 à 1,00. Le gaz Br2 a joué le double rôle de source
d’atomes chauds et d’agent de balayage. De 20 à 50 échantillons provenant de chacun des systèmes en réaction
ont été étudiés, et on a porté sur un graphique les données relatives au RO en fonction de la concentration
en agent de balayage, puis on les a extrapolées pour la fraction de molécule cible égale à une mole. Dans
tous les cas, RO a diminué quand la concentration en brome a augmenté.
Les données relatives aux réactions du brome activé par 79Br(n,y)8CBr avec des alcanes isotopiques font
penser que les valeurs extrapolées de RO sont comparables pour C2H6 et C2DÔ, mais que la valeur de RO pour
CD4est environ la moitié de sa valeur pour CH4. L’analyse par chromatographie gazeuse des produits orga
niques indique que 90% environ de RO dans CH4 sont dus à CH3Br et 10% à CH2Br2. Avec CD4 pour cible,
la répartition de l'a c t iv ité organique est plus ou moins la suivante: 75% de CD jB r e t 25% de CD jB r2.
Les auteurs discutent ces données en fonction de mécanismes possibles dans lesquels interviendraient
des ions et des atomes d'halogène chauds.
РЕАКЦ И И БРО М А-80, АКТИ В И РО В АН Н О ГО ПРОЦЕССОМ (n,y) И ИЗОМЕРНЫ М П Е Р Е
ХОД ОМ, С А Л К А Н А М И И ГА Л О - А Л К А Н А М И В ГАЗОВОЙ Ф АЗЕ, Сообщаются эксперимен
тальные данные о газофазных реакциях алканов и гало-алканов с атомами и ионами брома,
* Undergraduate Honours Research Participant 4
185

186 L .D . SPICER and A . A . GORDUS
активированными ядерными превращениями. В молекулы мишеней входили: СН4 , C D 4 ,
С2 Н6 , C 2 D 6 ,CH 3 C1, СН 2 С1 2 , СНС13 , СС14 , C H 2 F 2( C H F 3 , c r i , C 2 F 6 , C F 3B r , и C H 3B r .
Ядерными реакциями и превращениями для получения атомов и ионов отдачи с большой энер
гией послужили реакции: Вг80т(изомерный переход), В г 80 и В г 79 (п ,7 ) В г 80 . Процентную
ная долю радиоактивности, обнаруженную в органических комплексах (т .н . органический вы
ход "O B '*), определяли в зависимости от концентрации молекул мишени в диапазоне доли
грамм-молекулы приблизительно от 0,95 до 1,00. Элементарный В г 2 служил как источни
ком горячих атомов, так и акцептором. Обычно рассматривалось от 20 до 50 отдельных
проб для каждой группы реакций и строились диаграммы, дающие О В в зависимости от
концентрации акцептора; полученная кривая экстраполировалась до целого значения доли
грамм-молекулы мишени. Во всех случаях О В уменьшался по мере увеличения концентра
ции галогена.
На основании данных по реакциям брома-80, активированного процессом (11,7 ), с изо
топными алканами можно вывести сравнимую экстраполированную величину О В для C 2Hg
и C2D6 ; однако О В для CD4 составляет всего лишь приблизительно половину О В, получен
ного для СН4 . Газовый хроматографический анализ органических продуктов показывает,
что приблизительно 90% О В для СН4 приходится на долю СН3Вг и 10%—на долю СН2В г2 . Для
мишени из CD 4 органическая активность распределяется приблизительно в пропорции 75%
на долю C D 3B r и 25% на долю C D 2B r 2 .Эти различные данные обсуждаются в связи с возможным механизмом участия горячих
атомов и ионов галогенов в этих реакциях.
REACCIONES EN FASE GASEOSA DE 8<fer OBTENIDO POR REACCION (n.y) Y TRANSICION ISOMERICA
CON ALCANOS Y HALOALCANOS. Se facilitan datos experimentales acerca de las reacciones en fase ga
seosa de alcanos y haloalcanos con átomos e iones bromuro activados por transformaciones nucleares. Como
blanco se han utilizado moléculas de CH4, CD4, C2Hç, CH3CI, CH2C12, CHC13, CCI*, C H ^ , CHF3, CF4,
CgFg, CF3Br y CHgBr, Las reacciones y transformaciones nucleares utilizadas para producir los átomos e iones
energéticos de retroceso fueron вотвг (transición isomérica), *<Br y 75Br(n, y)8(Br. La radiactividad porcentual
del compuesto orgánico (rendim iento orgánico, R.O. ) se determinó en función de la concentración de la
molécula blanco en el intervalo de fracciones molares 0, 95 a 1, 00, aproximadamente. Se u tilizó Br2 e le
mental como fuente de átomos calientes y en calidad de depurador. Por lo general se examinaron de 20 a
50 muestras de cada sistema de reacción y los datos relativos a l RO en función de la concentración molar 1
de molécula blanco. En todos los casos, e l RO disminuyó a l aumentar la concentración de l halógeno.
Los datos referentes a las reacciones activadas por (n.y) del 8®Br con alcanos isotópicos indican un valor
extrapolado del RO comparable en el caso del Q H gy del QDe, pero en el caso del CD4 dicho valor es apro
ximadamente la mitad del correspondiente a l CH^, El análisis por cromatografía en fase gaseosa de los pro
ductos orgánicos indica que aproximadamente e l 90°Jo del RO en e l CH4se debe a l CH^r, y el 10°¡o a l CH£r2.
Cuando se u tiliza CD4 como blanco, la distribución de los productos orgánicos de carácter radiactivo es, apro
ximadamente, 75°}o de CD$Br y 25°¡o de CD2Br2.
Se estudian estos datos con relación a los mecanismos probables en que intervienen átomos e iones
calientes de halógenos.
INTRODUCTION
Reactions of (n, 7 ) and isom eric transition-activated B r 80 and (n, 7 ) a c tivated I128 with gaseous CH4 have been studied extensively [1-6] . The I128 appears to re a c t with CH4 to yield CH3I principally via reactions involving ionic and electronically excited 1128 of high translational kinetic energy [5, 6 ]. However, the reaction of (n, 7 ) -activated B r 80 to yield CH3B r appears to occur as a result of the recoil kinetic energy acquired by the B r80 [4]. Sim ilarly , B r 80 activated by the B r80m isom eric transition appears to re a c t with CH4 to yield СН3ВГ in a manner that uses the sm all amount of B r 8<> kinetic energy acquired by Coulombic repulsion [3].
Data to be used in comparing the extents of reaction of hydrocarbons and alkyl-halides with B r80 activated by the two methods should a s s is t in

GAS-PHASE REACTIONS OF ( n , y ) AND I.T .-A C T IV A T E D Br80 187
determining the mechanisms of hot B r 80 gas-phase reactions. In this paper is presented the p er cent B r 80 stabilized as organic activity for reaction s of B r 80 with isotopic m ethanes and ethanes, m eth y l-h alid es and C2 F 6 .
E X P E R IM E N T A L
Phillips re se a rch grade CH4 and C2H6, M erck of Canada CD4 and C2Dg, Matheson CF4, CHF3, CH3B r, CH3C1 and CF3B r, and du Pont re se a rch sample CH2F2 and C2F,; were all used without purification. CHC13 and CH2C12 were stirred for 1-2 h with concentrated H2S04; CC14 was photobrominated. Each was then washed with Na2 C 03, dried with CaCl2, and fractionated, taking the centre cut.
Samples prepared in 4 -5 ml silica (for n, у experiments) or pyrex (for I. T . experim ents) bulblets usually contained about 2- 10 mm of B r, (or B rB r80m). The hydrocarbon or alkyl halide pressure was about 600 except when its boiling point was less than room tem perature. In such cases the p ressu re was never m ore than about 80% of the vapour p ressu re at room tem perature.
The B rB r80® was form ed by irradiating liquid B r 2 with neutrons for about 10 min and was used in the I. T. experiments 1-5 h after irradiation. Under these conditions the percentage Br82 ( 36 h) activity fo r which c o r rection must be made was only about 5%. The B rB r 80 reaction mixture was allowed to stand 1. 5 to 2 h to perm it B r80m - B r 80 p aren t-d au gh ter equilibrium to be established [3]. After this period of tim e the sam ples w ere broken in a separatory funnel beneath a two-phase m ixture of CHC13 - aqueous 0 . 5 M Na2S 03. The organic phase contained activity of which at least 97% was B r8<> (18 min), thus indicating that a proper chemical separation was achieved. The inorganic phase was allowed to stand 1. 5-2 h to r e establish parent-daughter equilibrium. It was then counted to determ ine the amount of 4 . 4 - h B r 80m activity (corrected for B r82). After applying density and decay corrections, the (per cent) organic yield, O. Y. = 100 X organic activity/(organic + inorganic activity), was calculated.
Energetic (n, y ) activated B r 80 was formed by irradiating the sam ples for a few seconds in The University of Michigan-Ford Nuclear Reactor. The re a c to r power level was usually about 1 or 2 MeV. The therm al neutron flux at 2 MeV was about 3X1012 n /cm 2- s and the gamma radiation dosage was approx. 1 . 6 X 1 0 4 r /m in .
Separation into organic and inorganic fractions was achieved by the same method used in the I. T . experim ents. Counting was usually started about 10 min after irradiation at which tim e the (4. 4 h) B r 80m represented < 5% of the total observed activity. F o r the irradiation periods used, the (36 h) B r 82 activity was negligible. The organic yield was defined as given above.
Experim ents using B r82-tagged CH3B r and B r2 suggest that the solvent extraction procedure used is not 100% efficient as was found in the I. T. runs. When tagged CH3B r dissolved in CHC13 was shaken with an equal volume of aqueous 0. 5M Na2S03 about 2-3% of the B r82 activity was "extracted" by the aqueous phase. This could be repeated using the organic phase from the first "extraction" and a fresh sample of aqueous sulphite. When tagged Br2
was dissolved in CHC13 and shaken with an equal volume of aqueous 0. 5 M

188 L. D. SPICER and A . A . GORDUS
NagSC^, less than 1% of the B r 82 was found in the organic phase. This "o r ganic" activity could not be extracted by a fresh sample of aqueous sulphite. Sim ilarly, the shaking of the inorganic phase with a fresh sample of CHC13 did not result in tran sfer of any B r 82 to the organic phase.
Although these inefficiencies in the solvent extraction method are small, they could influence the experimental data, especially for samples exhibiting low O. Y . We would estim ate therefore that the O. Y . values reported in this paper may be high by probably not m ore than 1 (%) unit. This is undoubtedly an upper lim it since some of the system s, e. g. I. T. B r 80 + CF4 , exhibit reasonably consistent O. Y. values less than 1. 0%.
An ideal experim ental procedure for the determination of the O. Y. for system s of the type reported in this paper would be a method that allowed determ ination of both organic and inorganic activity without the need for solvent extraction. It would appear that gas-chrom atography should be an ideal technique. However, we were not successful in devising a gas chromatography arrangem ent that could be used to separate quantitatively B r 2
(or H Br) from organic brom ides. An alternative procedure, reaction of Br2 with C2 H4 p rio r to gas-chrom atographic separation, appears feasible, but has as yet not been perfected to the point of being quantitative.
B r 80 ACTIVATION PROCESSES
The isom eric transition of B r80m is followed by Auger electron emission and can result in a highly-charged m olecule. F o r exam ple, using m a ss- spectrom etric techniques, W EXLER and ANDERSON [7] have observed B r+n (n = 1 to 13) following isom eric transition of B r 80m in С Н 3В Г . The B r 80 daughter ion could acquire appreciable kinetic energy due to coulombic r e pulsion if charge distribution over the molecule occurs before dissociation due to loss of binding electrons takes place. For example, a B r 80+4 - B r +1 molecule, upon dissociation, results in bromine ions having 12. 5 eV of kinetic energy [3]. Using different m olecular form s of B r 80m it was shown that the relative organic yields of B r80 with CH4 appeared to parallel the B r 80 energies if calculated on the assumption that Coulombic repulsion resu lts following I. T . [3].
The (n, y) activation p ro cess, on the other hand, results in B r 80 atoms and ions of a maximum kinetic energy equal to 358 eV when the Вг^э is a free atom. In m olecular form , the maximum Br80 energy will depend principally on the m ass of the radical to which the Br^9 is bonded [8 , 9]. When B r2 is used as a neutron target, B r 8<> atoms of 178 eV maximum energy are formed. However, partial cancellation of prompt gam m a-cascade momenta could resu lt in a distribution of B r80 energies from zero to the maximum[8 ] although m ost of the B r 80 will probably be formed with energies in e x cess of 10- 20 eV [9]. At least 18% of the B r 80 is positively charged [10], but the reaction with CH4 appears to o ccu r principally as a resu lt of the B r8o re c o il kinetic energy [4].
Values of the O. Y . for various B r80 reaction s w ere determ ined as a function of the concentration of B r2 in the range zero to 0. 1 m ole-fraction Br^. F o r each ta rg e t m olecule the data w ere extrapolated to zero m ole-
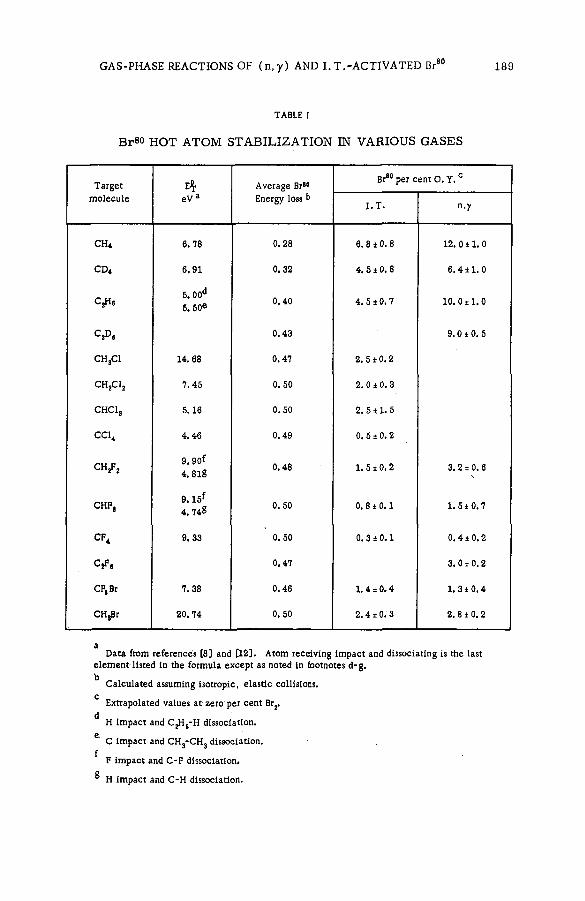
GAS-PHASE REACTIONS OF ( n , y ) AND I.T .-A C T IV A T E D Br80 189
TABLE I
B r 80 HOT ATOM STABILIZATION IN VARIOUS GASES
TargetEÎ- Average Br8°
Br80 per cent O. Y. c
molecule e V a Energy loss bI . T . n.y
CH4 6. 78 0.28 6. 8 ± 0 .8 12. 0 ± 1. 0
CD* 6.91 0.32 4. 5 ± 0. 8 6.4 ±1 .0
C jH í5. 00d
5. 50e0.40 4. 5 ±0.7 1 0 .0 ±1 .0
c 2d , 0.43 9.0 ±0.5
CH jC I 14. 68 0.47 2. 5 ±0.2
CH 2C12 7.45 0. 50 2. 0 ± 0.3
CHC1, 5.16 0. 50 2. 5 ± 1. 5
CC14 4.46 0.49 0. 5 ± 0. 2
C H ^9 .90 Í
4. 8 lE0.48 1.5 ±0 .2 3.2 ± 0. 6
\
CHF,9 .15 f
4. 7480. 50 0. 8 ± 0 . 1 1.5 ±0.7
c f 4 9. 33 0. 50 0. 3 ± 0. 1 0 .4 ±0 .2
C f , 0.47 3. 0 ± 0.2
CFjBr 7. 38 0.46 1. 4 t 0.4 1.3 ±0.4
CHjBr 20.74 0. 50 2 .4 ±0.3 2. 8 ± 0 .2
Data from references [8] and [12]. Atom receiving impact and dissociating is the last
elem ent listed in the formula except as noted in footnotes d-g.
bCalculated assuming isotropic, elastic collisions.
сExtrapolated values at zero per cent Br2.
dH impact and C jH j-H dissociation.
gС im pact and C H 3*CH8 dissociation. . .
F impact and C-F dissociation,
gp H impact and C-H dissociation.

190 L. D. SPICER and A . A . GORDUS
fraction Bi^. The extrapolated values of the О. У. with their visually estim ated uncertainties are listed in Table I. Usually between 20 and 50 individual yields were used in determining these extrapolated values.
As noted in Table I B r 80 produced by (n, y ) re a c ts to a g re a te r extent than I. T . activated B r 80. This is consistent with the fact that the (n, 7 )- activated B r8<> is formed not only with larg er initial energies but with initial energies which generally exceed the energies required for reaction . As a result, the (n, 7 )-activated В г8°, unlike the I. T. -activated B r 80, has available the full reaction energy range [ 1 1 ] in which to undergo reactiv e collisions.
There is no direct, simple method for calculating the reaction energy range. However, it may be possible to use as an approximation of the lower reaction energy threshold values of E^. calculated for m om entum -transfer to an atom in a m olecule [8 , 12]. These Eif values, given in Table I, r e present the average minimum energy that must be tran sferred to an atom in a molecule in order that the atom receiving the energy dissociates from the m olecule. Since the calculations of E^- take into account the m ass and spatial configuration of the radical attached to the activated atom, inertial effects of the type discussed by ODUM and WOLFGANG [13] a re at le a st p artially included.
Also given in Table I a re values of the average B r80 energy loss a s suming isotrop ic, e lastic collisions. These values a re only the crud est of approximations since the average collisional energy losses cannot be made because of lack of inelastic scattering asymmetry data [11]. For B r80, isotrop ic, elastic scatterin g, the average energy loss would be 160/(M +80)2[11] where M is the m olecular weight of the targ et m olecule. As seen in Table I, these values a re all approx. 0. 5 except for CH4 , CD4 , С2Нб and C2D6 . In addition to moderation efficiencies and reaction energy rahges, the probability of reactin g per collision would also affect the extent of r e action to form inorganic products [11]. No information is available concerning the reaction probabilities.
I. T.-A CTIV A TED B r80 REACTIONS
The yields in the two series : CH3 C1, CH2C12, CHClg, CCI4 and CH2F 2, CHF3, CF4 , both d ecrease with the decreasing number of hydrogen atoms per m olecule. The Br*® moderation efficiencies are probably sim ilar for these molecules and the variation in yields would appear to be caused principally by the com position of the m olecule. As noted in Table I, ES}, is 4.81 eV for C-H rupture in CH2F2 but 9.90 eV for C - F rupture. Similarly, the C-H values of E j should be less than the C-Cl values for CH3 CI, CH2C12
and CHCI3 listed in Table I.G as-chrom atographic analysis of the products of these reactions has
not been attempted, but it is expected that both H and Cl or F displacement by Br80 should be possible since non-zero organic yields are observed for CH4 , СБ4 and CCI4 . The values of E^., th erefo re , appear to be a useful guide in that they indicate higher energy requirem ents for C -F com pared with C-Cl bond rupture. As a result, the yield, for example, of B r 80 and

GAS-PHASE REACTIONS OF ( n , y ) AND I.T .-A C T IV A T E D Br80 191
CH2C12 would be predicted to be higher than the yield of B r 80 + CH2F2 as found experimentally. ,
These values of Ejj. could also partially help to explain the B r80 + CH4, CD4 and C2Hg yields. Although values are generally lower for these r e actions, the average collisional energy losses may also be less thus allowing the B r 8o a g rea ter number of collisions in which to re a c t . However, the reactions of B rso with CF3B r and CH3B r may proceed principally via an exchange p ro cess that perhaps involves an ion-m olecule interm ediate [14].
None of the above possible explanations a re , of co u rse , com pletely satisfactory since it is possible that reaction probability effects could be the main factor that determines the organic yields.
n, -/-ACTIVATED B r80 REACTIONS
Analysis of the data for the gaseous reactions of (n, y )-activated B r80 is limited by lack of knowledge of reaction param eters as noted in the p receding section . A few observations, how ever, m e rit fu rth er com m ent.
Reactions of B r 80 with C2Hg and C2D6, within experimental e rro r, result in the same organic yield, the data being consistent with published values [3]. Gas-chromatographic analyses indicate four major organic products, CH3B r, CH2B r2, C2H5B r and C2H4Br2, (or their deuterated analogues) present re la tive to CH3B r in an amount 1. 0, 0. 17, 1. 6 and 0. 3 respectively. As noted in Table I, values of E .; are alm ost equal for C -C and C-H bond rupture. Similarly, the CH3B r +.СН2ВГ£ yield is almost equal to the C2H5B r + yield.
Fig. 1
Organic yields as a function of Br2 concentration for Br80+CH4, O , and Br80+CD4, • , reactions.
Yields for the other isotopic pair, CH4-CD4, are most interesting in that the reaction of B r 80 with СЩ resu lts in alm ost twice as much organic a c

192 L.D . SPICER and A . A . GORDUS
tivity as the reaction with CD4. These data are given in Fig. 1. The limiting yield for the CH4 system is in agreem ent with the value, 13. 3 ± 0. 5%, determ ined previously [4]. Two organic brom ides are detected by gas- chromatographic analysis: CH3B r and CH2 Br2, or their deuterated analogues. In the Br80 + CH4 reaction , 89% of the organic activity is due to CH gBr80. F o r the Br80 + CD4 reaction , 76% of the organic activity is due to СОзВгво. Thus, the lim iting organic yields are 10. 7% СН3 ВГ8 О and 1. 3% CH2 B r B r 8 0
compared with 4. 9% CD3Br80 and 1. 5% СОгВгВгво. We know of no simple explanation for these differences1.
SUMMARY '
Low organic yields for the reaction of hot B r 8 0 with alkanes and halo- alkanes are found suggesting that the average reaction probability [ 1 1 ] to form organic compounds is relatively sm all. The data do not, how ever, provide information about the inorganic reaction probabilities since no experimental means are available for separating inorganic B r8o resulting from hot and therm al reactions.
The yields from I. T . activated Br80 are low er than the yields from (n, 7 )-activated B r80, as would be expected since the (n, 7 ) activation results in Br80 of greater initial energies.
Further experiments using gas chromatographic analysis would be useful. In p articu lar, product yield dependence on the total p ressu re of the system may provide valuable information which can be used in evaluating the m echanism s. ^
A C K N O W L E D G E M E N T S
This work was supported by the United States Atomic Energy Commission, Division of R esearch , and The University of M ichigan-M em orial Phoenix P ro je c t. L . D. S. wishes to exp ress appreciation to the National Science Foundation for an undergraduate re s e a rch fellowship. The assistan ce of W. G. Rado and W. D. B latter in determining the isom eric transition yields is gratefully acknowledged.
R E F E R E N C E S
[1 ] HORNIG, J .F ., LEVEY. G. and WILLARD, J.E. , J. chem. Phys. 20 (1952) 1556.[2] LEVEY, G and WILLARD, I .E . , J. chem. Phys. 25 (1956) 904.[3] GORDUS, A. A. and WILLARD, J.E. , J. Amer. chem. Soc. 76 (1957) 4609.[4] RACK, E.P. and GORDUS, A .A ., J. phys. chem. 65 (1961) 944.
1 The possible importance of a slight difference between the ionization potential o f CH3Br (10. 53 eV)and CDjBr was investigated because the Br2 ionization potential is also about 10.53 eV. It seemed possiblethat CHsBr+ may be stabilized by charge exchange with Вгг whereas CD3Bi+ may hot easily undergo such chargetransfer. Organic yields were determined for CH4 containing a few mole per cent CH3Br or CD3Br and for CD4containing a few mole per cent of СОзВг or CH3Br. The yields in these systems were consistent with theC H 4- C D 4 data in Table I, indicating that small differences in ionization potentials did not influence th e O. Y.

GAS-PHASE REACTIONS OF (n , y ) AND I.T.-ACTIVATED Br80 193
[5] RACK, E.P. and GORDUS, A .A . , J. chem. Phys, 34 (1961) 1855,[6] RACK, E.P. and GORDUS, A .A . , J. chem, Phys. 36 (1962) 287.
[7] WEXLER, S. and ANDERSON, G .R ., J. chem. Phys. 33 (1960) 850.[8] HSIUNG, C. and GORDUS, A. A . , J. chem. Phys. 36 (1962) 947.[9] GORDUS, A. A. and HSIUNG, C. , J. chem. Phys. 36 (1962) 954.
[10] WEXLER, S. and DAVIES, H. , J. chem. Phys. 20 (1952) 1688.[11] HSIUNG, C. and GORDUS, A .A . , I. Amet. chem. Soc. 86(1964)2782.[12] HSIUNG, C . , Ph.D. Dissertation, The University o f Michigan, Ann Arbor (1962).[13] ODUM, R. and WOLFGANG, R ., I. Amer. chem. Soc. 83 (1961) 4668.[14] For example, an analogotu species CHjlJ has been reported: POTTIE, R. F. , BARKER, R. and
■ HAMILL, W.H. Rad. Res. 10 (1959) 644.


CHEMICAL EFFECTS OF THE (n, p) REACTION IN GASEOUS SYSTEMS:
SIMPLE ALKANES AND THEIR CHLORODERIVATIVES
K. PÁNEK AND K. MUDRA NUCLEAR RESEARCH INSTITUTE OF THE
CZECHOSLOVAK ACADEMY OF SCIENCES, &E¿, PRAGUE, CZECHOSLOVAK SOCIALIST REPUBLIC
Abstract — Résumé — Аннотация — Resumen
CHEMICAL EFFECTS OF THE (n, p) REACTION IN GASEOUS SYSTEMS: SIMPLE ALKANES AND THEIR
CHLORODERIVATIVES. The chemical effects accompanying the reaction C l35(n, p)S35 in organic system*, have been studied in this paper.
Chemical species in which the hot S s atoms are stabilized were studied in the first part of this article. Methyl chloride, ethyl chloride and methane-hydrogen chloride mixture in gaseous state were used as target
materials. A ll samples were prepared by vacuum technique and irradiated in a horizontal channel o f the VVR-S reactor. The irradiated samples were analysed by radio-gas chromatography.
The pressure dependence of yields of individual species has been also investigated in methyl chloride and ethyl chloride with no additives. Only a slight effect o f pressure (in the range of 200-500 mm Hg) was found for irradiated methyl chloride, while the apparent increase o f the yield o f some simple S35-labelled compounds appeared in ethyl chloride under the same conditions.
The reactions of recoil S35 atoms with gaseous methane have been examined in detail. For this purpose,
the methane-hydrogen chloride mixture with additives were irradiated under constant conditions. Argon and nitric oxide were used as moderator and scavenger respectively. The composition of irradiated samples varied in the range o f 20-260 mm Hg o f methane and argon, 0.02-12 mm Hg o f nitric oxide, the partial pressure of hydrogen chloride keeping constant (100 mm Hg).
The S35-labelled hydrogen sulphide and methyl mercaptane were found as main products in irradiated samples. The results obtained show that the yield of hydrogen sulphide is mostly affected by the concentration of nitric oxide. On the other hand the amount o f methyl mercaptane formed is determined mainly by the
concentration of argon.
EFFETS CHIMIQUES DE LA RÉACTION (n,p) DANS DES SYSTEMES GAZEUX, DES ALCANES SIMPLES ET LEURS DERIVES CHLORES. Le mémoire traite des effets chimiques qui accompagnent dans des systèmes organiques la réaction 35Cl(n, p)3sS.
Sa première partie porte sur les espèces chimiques dans lesquelles les atomes S5S chauds sont stabilisés. Le chlorure de méthyle, le chlorure d’ éthyle et le mélange méthane-acide chlorhydrique à l'état gazeux ont été utilisés comme cibles. Tous les échantillons ont été préparés suivant la technique du vide et irradiés dans un canal horizontal du réacteur VVR-S, Les échantillons irradiés ont été analysés par radiochromatographie gazeuse. •
La mesure dans laquelle les rendements des diverses espèces dépendent de la pression a également été étudiée dans les cas du chlorure de méthyle et du chlorure d'éthyle exempts d'additifs. On n'a noté qu'une faible incidence de la pression (de l'ordre de 200 à 500 mm Hg) pour le chlorure de méthyle irradié mais il y a eu un accroissement apparent du rendement de certains composés simples marqués avec 3ss «pour le chlorure d'éthyle dans les mêmes conditions. .
Les réactions des atomes 3 S de recul avec le méthane gazeux ont fait l'objet d*un examen détaillé.
Pour cela, le mélange méthane-acide chlorhydrique avec additifs a été irradié dans des conditions maintenues constantes. L’argon et le bioxyde d'azote ont été utilisés, l'un comme ralentisseur, l ’ autre comme agent de balayage. La composition des échantillons irradiés a varié de 20 à 260 mm Hg pour le méthane et l'argon, de 0, 02 à 12 mm Hg pour le bioxyde d'azote, la pression partielle de l'acide chlorhydrique étant maintenue constante (100 mm Hg).
195

196 K. PÀNEK and K. MUDRA
L’ acide sulfhydrique et le thioalcool méthylique marqués avec 85S sont les principaux produits dont la présence a été constatée dans les échantillons irradiés. Les résultats obtenus montrent que le rendement en acide sulfhydrique dépend principalement de la concentration en bioxyde d'azote. En revanche, la quantité de thioalcool méthylique est principalement fohction de la concentration en argon.
ХИ М И ЧЕ С К О Е Д Е Й С Т В И Е РЕ АК Ц И И (n,p) В ГА ЗО В Ы Х С И С Т Е М А Х : П Р О С Т Ы Е А ЛК АН Ы И ИХ ХЛОРГГРОИЗВОДНЫ Е. В этом докладе изучаются химические эффекты реакции С 35 (n,p) S35 в органических систем ах .
В первой части этой статьи изучаются химические образцы, в которых стабилизируются горячие а том ы .S35 . В качестве материалов мишени использовались хлористый этил и см есь хлористого метана и хлористого водорода. Все образцы изготовлялись вакуумными методами и облучались в горизонтальном канале реактора В В Р -С . Облученные образцы анализировались методами радиогазохром атограф ии.
В хлористом метиле и хлористом этиле без добавок изучается также влияние давления на выход отдельны х образцов. Д ля облученного хлористого м етила установлено только слабое влияние давления (в диапазоне 200—500 мм р т .с т . ) , в то время как в хлористом этиле при тех же условиях имело м есто явное увеличение выхода некоторых простых соединений, меченных S35.
Подробно изучались реакции атомов отдачи S35 с газообразны м м ета н ом . Для этой цели см есь хлористого метана и хлористого водорода с добавками облучалась при постоянных условиях. Аргон и окись азота соответственно использовались в качестве замедлителя и аксептора радикалов. В состав облученных образцов в диапазоне давления 20 — 260 мм рт. столба входили метан и аргон, в диапазоне 0,02 — 12 мм р т . с т .—окись азота, парциальное давление хлористого водорода поддерживалось постоянным (100 мм р т .с т . ) .
Бы ло установлено, что меченный S ’*0 сероводород и метилмеркаптан являются о с новными продуктами в облученных образцах. Достигнуты е результаты свидетельствую т о том, что на выход сероводорода очень большое влияние оказывает концентрация окиси азота . С другой стороны, количество образующегося метилмеркаптана определяется главным образом степенью концентрации аргона.
EFECTOS QUIMICOS DE LA REACCION (n, p) EN SISTEMAS GASEOSOS: ALCANOS SIMPLES Y SUS CLORODERIVADOS. En la memoria se estudian los efectos químicos asociados a la reacción 85Cl(n, p)85S en sistemas orgánicos.
En la primera parte se examinan las especies químicas en que los átomos calientes de 85S se convierten
por estabilización. Como blancos se han utilizado cloruro de metilo, cloruro de etilo y una mezcla de metano y cloruro de hidrógeno, en estado gaseoso. Todas las muestras se han preparado al vacfo y se han irradiado en un canal horizontal del reactor VVR-S. El análisis de las muestras irradiadas se ha efectuado por radio- cromatografía en fase gaseosa. .
También se ha investigado en cloruro de metilo y cloruro de etilo sin aditivos e l efecto ejercido por la presión sobre el rendimiento de cada especie. Sólo se ha observado un ligero efecto de la presión (en el intervalo 200-500 mm. de Hg) en el caso del cloruro de metilo; en cambio, el cloruro de etilo presenta en las m is m a s condiciones un manifiesto incremento del rendimiento de algunos compuestos poco complejos marcados>con 35S.
Se han examinado en detalle las reacciones de los átomos de retroceso’de 3ss con metano en estado gaseoso. A tal efecto, se ha irradiado en condiciones constantes la mezcla de metano y cloruro de hidrógeno con aditivos. Como moderador y depurador se han utilizado argón y óxido nítrico, respectivamente. La composición de las muestras irradiadas osciló entre 20-260 mm de Hg de metano y argón, y 0,02-12 mm de Hg de óxido nítrico, permaneciendo constante la presión parcial del cloruro de hidrógeno (100 mm de Hg).
Los principales productos hallados en las muestras irradiadas fueron el sulfuro de hidrógeno marcado con S5S y el metil-mercaptano. Los resultados obtenidos muestran que la cantidad de sulfuro de hidrógeno depende en gran medida de la concentración de óxido nítrico. En cambio, la cantidad de metil-mercaptano formado varfa en primer lugar con la concentración de argón.

CHEMICAL EFFECTS OF THE (n,p) REACTION
INTRODUCTION
197
The highly energetic sulphur atom s resulting from nuclear p ro cesses have previously been studied in both solid [1, 2] and liquid phases [3] , but scarcely any gas-phase experim ents have been carried out. The gas-phase reactions of sulphur atom s created by the S3 4 (n, y)S3 5 nuclear reaction with m olecules containing sulphur have been investigated [4] . The S35-labelled carbonyl sulphide has been reported as a result of the Cl3 5 (n, p)S3 5 nuclear reaction in gaseous m ixtures of halocarbons with carbon monoxide and dioxide [5] .
N evertheless there a re not yet enough experim ental resu lts to com pare the behaviour of recoil sulphur atoms with other nuclear recoil nuclides created by the (n, p) process in simple organic systems.
The presen t paper rep o rts on a study of the reco il sulphur atom s in such sim ple sy s te m s .
E X P E R I M E N T A L
Sample preparation
Technical methyl chloride was purified by repeated low -tem perature vacuum distillation. The gas-chrom atographic analysis showed less than0. 2% im purities in the product. Ethyl chloride was prepared and purified in the sam e way. L e ss than 0 . 5% impurities were found by gas liquid chrom atography. Methane and argon were used directly from the storage tanks. The stated purity was 99.5%formethane and 99.8% for argon. Hydrogen chloride and nitric oxide were freshly prepared for each group of sam ples, purified by low tem perature vacuum distillation and filled directly into the storage glass cylinders of the vacuum apparatus. The purity was not determ ined.
All sam ples w ere prepared by the common vacuum technique. Since the p ressu re was m easured with the accu racy of ± 0 . 5 mm Hg the sm all amounts of n itric oxide w ere added already mixed with methane.
The irradiation bulbs used were made from Sial glass and equipped with two b reak-seals. The volume was 10. 5 ml.
Irradiation
The samples were irradiated in a horizontal channel of the VVR-S r e a c to r . The irrad iation tim e was 2 h in a neutron flux of З Х Ю И п / cm2, s .
Analysis
The irradiated sam ples were stored for three months to allow all the short-lived isotopes, including P 32 , to decay. Just before the analysis each bulb was cooled down in liquid a ir , one seal was broken off and a m ixture of c a r r ie r s was added. Immediately afterw ards the bulb was attached to

198 К. PÁNEK and K. MUDRA
the by-pass sampling valve of the gas-chrom atographic apparatus, heated, and the whole contents w ere tran sferred into the chrom atographic column.
The column used was made of 3 m by 4 mm internal diameter aluminium tubing containing 4. 3 g packing p er m eter of 0 . 2 5 - 0 . 3 0 mm Celite 545 im pregnated with 15 wt . % tricresylph osp h ate. Methane was used as the c a r r ie r gas at a flow rate of 35 m l/m in . Column inlet p ressu re was 1 . 01 atm , the column tem perature was 36. 2°C. The te rm is to r d etector was used. The /3 activity of S3 5 was m easured by an internal proportional flow -counter attached d irectly to the outlet of the heat conductivity ce ll.
Since the activity of S3 5 trapped on the wall of the bulb was assum ed to be not negligible, each bulb was treated after the chrom atographic analysis with n itric acid , the ex tract was evaporated to dryness and counted under the end window G-M tube. All n ecessary corrections were applied.
Total activity of the sample
The sum of activities measured, i . e . the activity of each isolated peak and activity trapped on the w alls, was com pared with the activity ca lcu lated from irradiation conditions (cro ss-sectio n of 0. 3 b was used). Since the values obtained varied within the range of neutron flux changes, the average sum of activity was used for evaluation of experimental results. Butyl - m ercaptane-S 3 5 of known activity was used as a standard fo r com parison of a ctiv itie s m easu red by proportional co u n ter o r by window co u n te r .
Identification of radioactivity as S35
Although no other activity was expected, the separation of some radioactive fractions from the c a r r ie r gas stream was carried out. The liquid a ir trap was used. The content of the trap was counted by window counter after chemical treatm ent and the absorption analysis showed that the activity of particular peaks was due to S3® only.
Separation and identification of individually labelled compounds
The gas chromatography of sulphur compounds has been the subject of sev eral recent studies [6 , 7 ] . The method reported by RYCE and BRYCE [8 ] was used under the conditions described above for these experim ents. The effect of all compounds used on the efficiency of the proportional counting was also investigated [9] . All c a r r ie r s used were prepared and checked on purity by gas liquid chromatography analysis. Mercaptans were prepared by means of cleavage of corresponding isothiuronium salts [1 0 ] . The sym m etrical dialkyl sulphides were obtained by the reaction of sodium sulphide with alkyl brom ides [11] . The unsym m etrical dialkyl sulphides w ere prepared by alkylation of the m ercaptides [12] . The chlorosulphides were synthetized according to BÓHME [13]. The retention data under various conditions were determined for all used c a rr ie rs .
The separation of p articu lar labelled compounds was perform ed both with and without the c a r r ie r s . The analysis without c a r r ie r s showed that the corrected retention data of the particular radioactive peaks corresponded
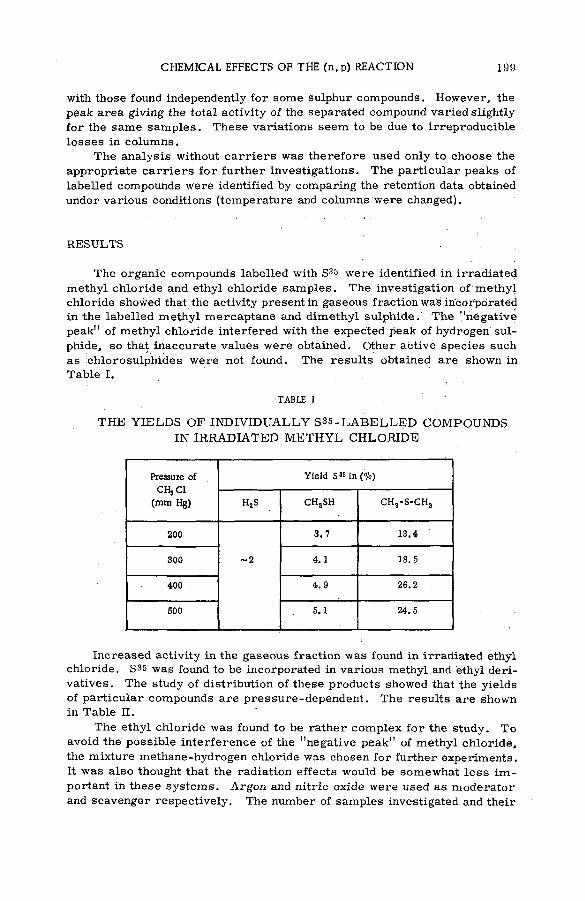
CHEMICAL EFFECTS OF THE (n,p) REACTION 199
with those found independently for some sulphur compounds. However, the peak area giving the total activity of the separated compound varied slightly for the sam e sam ples. These variations seem to be due to irreproducible losses in columns.
The analysis without c a r r ie r s was therefore used only to choose the appropriate c a r r ie r s for further investigations. The p articu lar peaks of labelled compounds were identified by comparing the retention data obtained under various conditions (temperature and columns were changed).
RESULTS .
The organic compounds labelled with S3 5 were identified in irradiated methyl chloride and ethyl chloride sam ples. The investigation of'm ethyl chloride showed that the activity present in gaseous fraction was incorpóratéd in the labelled m ethyl m ercaptane and dimethyl sulphide. The "negative peak" of methyl chloride interfered with the expected peak of hydrogen sulphide, so that inaccurate values were obtained. Other active species such as chlorosulphides were not found. The results obtained a re shown in Table I.
TABLE I
TH E YIELD S OF INDIVIDUALLY S3 5 -L A B E L L E D COMPOUNDS IN IRRADIATED M ETH YL CHLORIDE
Pressure o f C H jC l
(mm Hg)
Y ield S35 in (% )
H2S CH3SH CHj-S -CH j
200 3.7 13.4
300 ~2 4.1 18.5
400 4.9 26.2
500 5.1 24.5
Increased activity in the gaseous fraction was found in irradiated ethyl chloride. S35 was found to be incorporated in various methyl and ethyl derivatives. The study of distribution of these products showed that the yields of p articu lar compounds are pressure-dependent. The results are shown in Table II. ’
The ethyl chloride was found to be rath er com plex for the study. To avoid the possible interference of the "negative peak" of methyl chloride, the mixture methane-hydrogen chloride was chosen for further experiments. It was also thought that the radiation effects would be somewhat le ss im portant in these system s. Argon and nitric oxide were used as m oderator and scavenger respectively. The number of samples investigated and their
2 0 0 К. PÁNEK and K. MUDRA
TABLE II
TH E YIELD S OF INDIVIDUALLY S 35-L A B E L L E D COMPOUNDS IN IRRADIATED E T H Y L CHLORIDE
Pressure o f
C 2HBC1 (mm Hg)
Y ie ld S * ' in (% )
H2S CH,SH C ^ S H (CH s)2S C H j-S -C 2H5 (C ZH5) 2S
200 3.1 . 31.0 7.4 - 4.2 9.1
300 4.2 39.6 6.5 3.0 3.8 12.2
400 5.5 49.5 / 5.3 2 .5 4.2 14.4
485 6.9 57.0 4.2 1.5 3.9 16.2
composition w ere chosen in such a way as to make sta tis tica l evaluation possible. The G reek-Latiii square combination was used. Some sam ples were also irradiated in addition to the statistical system,and the results obtained were compared with those evaluated from the statistical system. The composition of samples and experimental results found are shown in Table III. The values presented a re the average of at least two experim ents unless otherw ise stated .
Experim ental e rro r in the range of ± 10% was found for methyl m ercap- tane yields and of ± 1.5% for hydrogen sulphide yields. Such erro rs might be due to the interactions or to the large difference in the counting efficiencies of the proportional and the window counter.
The following effects were to be caused by the presence of the respective additives: (1) Effect of scavenger. The large influence of very small amounts of scavenger is c le a r from Fig . 1, where the yield of hydrogen sulphide is plotted against the partial pressure of nitric oxide in the sample. The yield of methyl mercaptane seem s to be unaffected within the experimental e rro r . (2) Effect of methane. The yield of hydrogen sulphide is unaffected by the methane p ressu re within the experim ental e r r o r . On the other hand, only very slight dependence (as compared with the e rro r) of the yield of methyl m ercaptane on methane p ressure was found. (3) Effect of argon. The presence of argon seem s to be without any effect on hydrogen sulphide yield, while a large influence on the yield of methyl m ercaptane is evident from F ig . 2, which shows the plot of the yield as a function of the p re ssu re of argon.
DISCUSSION
It is interesting to com pare the results obtained with those reported by HYDER and MARKOWITZ [4] . Although S35 atom s created in the (n, y) reaction p ossess considerably lower energy (760 eV) than those from the reaction (n, p) ( 31 . 2 keV) [2] , some sim ilar results have been found. The
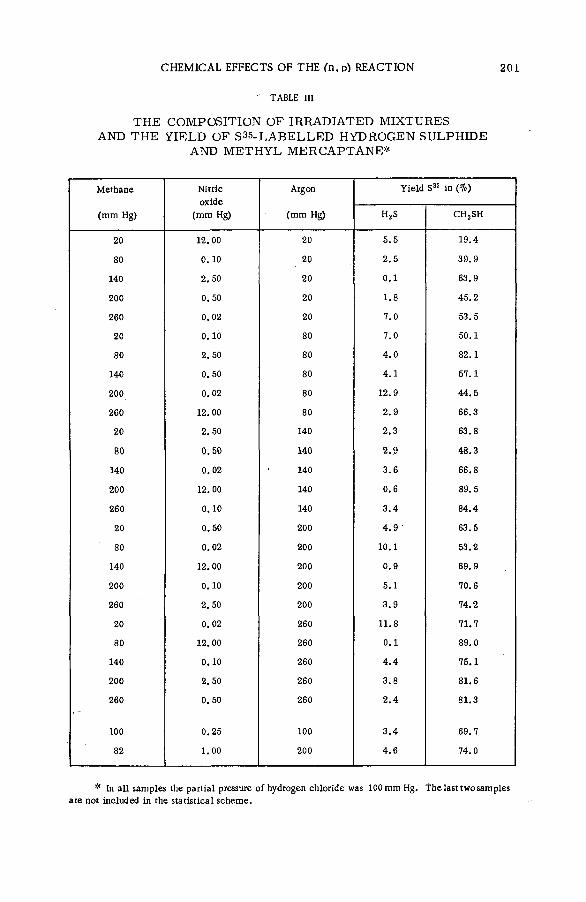
CHEMICAL EFFECTS OF THE fn.D) REACTION 2 0 1
' TABLE ill
TH E COMPOSITION OF IRRADIATED M IXTURES AND TH E YIELD OF S35-LA B ELLED HYDROGEN SULPHIDE
AND M ETH YL M ERCA PTA N E*
Methane
(mm Hg)
Nitric oxide
(mm Hg)
Argon
(mm Hg)
Y ield S35 in (% )
H2S CHjSH
20 12.00 20 5.5 19.4
80 0.10 20 2.5 39.9
140 2.50 20 0.1 63.9
200 0.50 20 1.8 45.2
260 0.02 20 7.0 53.5
20 0.10 80 7.0 50.1
80 2.50 80 4.0 82.1
140 0.50 80 4.1 57.1
200 0.02 80 12.9 44.5
260 12.00 80 2.9 66.3
20 2. 50 140 2.3 63.8
80 0.50 140 2.9 48.3
140 0.02 ■ 140 3.6 66.8
200 12.00 140 0.6 89.5
260 0.10 140 3.4 84.4
20 0.50 200 4.9 63.5
80 0.02 200 10.1 53.2
140 12. 00 200 0.9 69.9 ,
200 0.10 200 5.1 70.6
260 2. 50 200 3.9 74.2
20 0.02 260 11.8 71.7
80 12. 00 260 0.1 89.0
140 0.10 260 4.4 75.1
200 2. 50 260 3.8 81.6
260 0. 50 260 2.4 81.3
100 0.25 100 3.4 69.7
82 1.00 200 4.6 74.0
* In a ll samples the partial pressure o f hydrogen chloride was 100 mm Hg. Thelasttwosamples are not included in the statistical scheme.
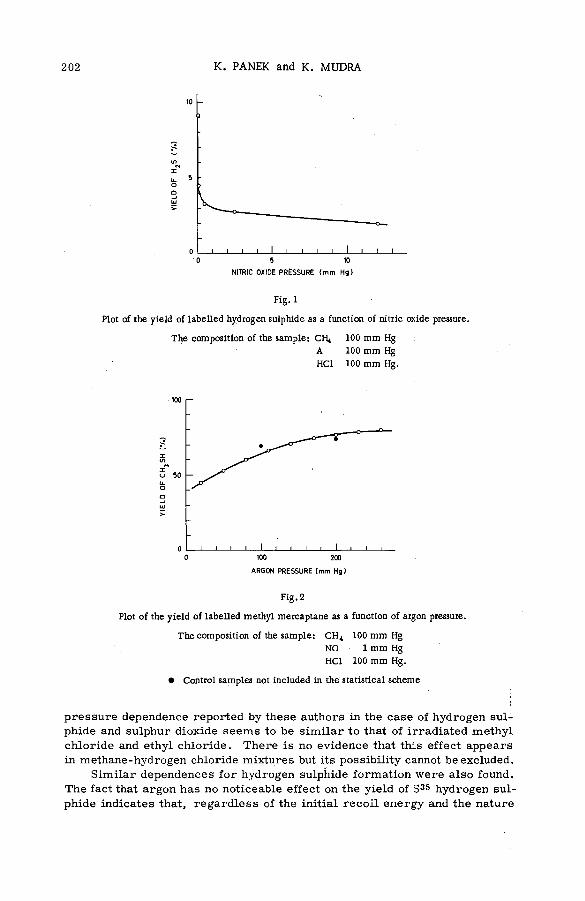
2 0 2 K. PANEK and K. MUDRA
NITRIC OXIDE PRESSURE (m m H g )
. Fig- 1Plot o f the yield o f labelled hydrogen sulphide as a function o f nitric oxide pressure.
The composition o f the sample: СЦ, 100 mm Hg :' A 100 mm Hg
HC1 100 mm Hg.
ARGON PRESSURE (m m H g )
Fig. 2
Plot o f the yield o f labelled methyl mercaptane as a function o f argon pressure.
The composition o f the sample: CH4 100 mm HgNO 1 mm Hg HC1 100 mm Hg.
• Control samples not included in the statistical scheme
' i p ressure dependence reported by these authors in the case of hydrogen sulphide and sulphur dioxide seem s to be sim ilar to that of irradiated methyl chloride and ethyl chloride. There is no evidence that this effect appears in methane-hydrogen chloride mixtures but its possibility cannot be excluded.
Sim ilar dependences for hydrogen sulphide formation were also found. The fact that argon has no noticeable effect on the yield of S3S hydrogen sulphide indicates that, reg ard less of the initial reco il energy and the nature

CHEMICAL EFFECTS OF THE (n,p) REACTION 203
of surroundings, hot reactions are not involved in the formation of hydrogen sulphide. Furtherm ore, it is evident from the shape of the curve of hydrogen sulphide yield in dependence on nitric oxide concentration ( F i g . l ) that the reactions involving radical therm al recombination m echanism should be dominant for the incorporation of the activity into hydrogen sulphide.
The reactions responsible for the production of labelled methyl m ercaptane appear to be rather complex. It has been found that the yield is fairly sensitive to the presence of the moderator, but in contrast with the behaviour of the typical hot reaction [14] the yield of m ercaptane has risen as the pressure of argon has increased.
Such behaviour could be understood if the m ercaptane were not formed by a hot reaction of S3 5 species. Furtherm ore, these reactions very likely take place in the gas phase as the activity on the walls appears to decrease with an increase in the pressure of the m oderator.
- A sim ilar m oderation effect occurred in irrad iated gaseous sulphur dioxide [4], but the increase in the yield of sulphur dioxide was not so obvious in the presence of argon. The investigation of the possible role of the m oderator in the course of the formation of any product shows that it is reasonable to consider the potential moderation ability of other constituents present in the sam ple. One could expect, for instance, the moderation ability of sulphur dioxide o r hydrogen chloride to be not negligible so that the effect of argon added would be the more evident the sm aller the amount of constituents with comparable m ass present in the sample.
This might be illustrated by the hydrogen chloride - methane system . Supposing that the moderation ability of hydrogen chloride would approach that of argofi (the effect of hydrogen is not considered although its influence could not be excluded), the yield of any form of activity would then depend on the sum of p artial p ressu res of hydrogen chloride and argon. In F i g . 3 the yield of methyl m ercaptane is plotted against the sum of the mole fra c tions of "m o d erato rs", x , in the sam ple. The dotted line was obtained by graphical extrapolation.
That the yield of methyl m ercaptane was not related to the presence of nitric oxide was a little surprising. This can be explained by postulating the reaction s to be of an ion-m olecule ch a ra c te r, as it is known that such reactions are not related to the presence of nitrogen oxide [15] . It is hoped that further investigations will be made into this possibility.
It is believed in general that the m echanism of the form ation of S35- labelled compounds is ra th er com plex, as the probability of the creation of any stable compound from only one collision is v ery sm all. The m ore probable mechanism would therefore involve at least two steps, corresponding to the formation of two chemical bonds. This might be written
S 3 5 *-> [X S 34 * - »X S X 3 5
where X stands for hydrogen, alkyl group o r any other species. Following this schem e it is probable that only an interm ediate product might result from the hot p ro cess , if this o ccurs at all. Although it is possible that the interm ediate, if any, would be in som e excited state , its consecutive r e actions would probably be of a therm al nature. Unfortunately very few experimental results are available at present to support any such mechanism.
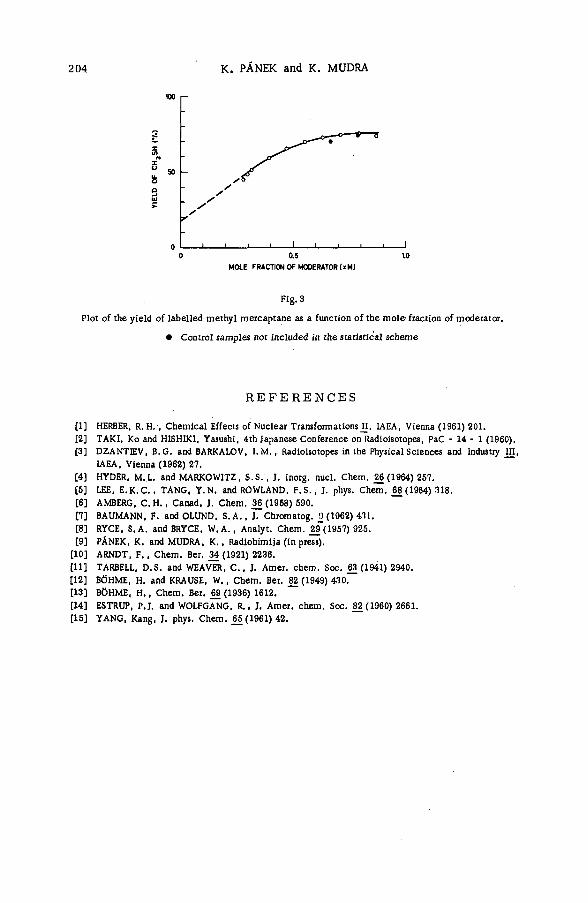
204 К. PÁNEK and K. MUDRA
MOLE FRACTION OF MODERATOR (x M )
Fig. 3
Plot o f the yield o f labelled methyl mercaptane as a function o f the mole-fraction o f moderator.
• Control samples not included in the statistical scheme
R E F E R E N C E S
[1] HERBER, R. H. , Chemical Effects o f Nuclear Transformations H, IAEA, Vienna (1961) 201.[2] TA K I, Ко and HISHIKI, Yasushi, 4th Japanese Conference on Radioisotopes, PaC - 14 - 1 (1960).[3] DZANTŒV, B.G. and BARKALOV, I. M . , Radioisotopes in the Physical Sciences and Industry Ш.
IAEA, Vienna (1962) 27.[4] HYDER, M. L. and MARKOWITZ, S.S. , J. inorg. nucl. Chem. 26 (1964) 257.[5] LEE, E .K .C ., TANG, Y .N. and ROWLAND, F .S . , J. phys. Chem. 68 (1964) 318.[6] AMBERG, С. H ., Cañad. J. Chem. 36 (1958) 590.[7] BAUMANN, F. and OLUND, S. A . , J. Chromatog. j) (1962) 411.[8] RYCE, S .A . and BRYCE, W .A . , Analyt. Chem. 29(1957) 925.
[9 ] PÁNEK, K. and MUDRA, K . , Radiohimija (in press).[10] ARNDT, F . , Chem. Ber. 34 (1921) 2236.[11] TARBELL, D.S. and WEAVER, C . , J. Amer. chem. Soc. 63 (1941) 2940.[12] ВбНМЕ, H. and KRAUSE, W ., Chem. Ber. 82 (1949) 430.[13] BÜHME, H . , Chem. Ber. 69 (1936) 1612.[14] ESTRUP, P.J. and WOLFGANG, R ., J. Amer. chem. Soc. 82 (1960) 2661.[15] YANG, Kang, I. phys. Chem. 65(1961) 42.

CHEMICAL EFFECTS OF THE fn.p) REACTION
D I S C U S S I O N
(on the foregoing three papers)
R. H. HERBER: From experiments we have carried out on the chemical fate of S3 5 form ed by the (n, p) reaction in CC14, as well as from K oski's early results on ordinary and outgassed KC1, it seem s likely that sulphur chlorides are formed as p rim ary chemical products in irradiations of this so rt. This postulate is based on the likelihood of the halogen acting as an electron donor to the positively charged sulphur atom undergoing re co il. The observation that the addition of NO does not influence the CH3 S3 5H yield suggests that, in the presence of nitric oxide, sulphur oxides ( e . g . , SO and/ o r SO2 ) are form ed that a re not recovered in the m ercaptan fraction . To shed some light on this possibility, perhaps D r. Pánek could comment on the extent to which he can account for the total S3 5 activity in those experiments in which no scavenger was added. I imagine that experim ents with an excess of halogen m olecules in the target phase would help to elucidate these questions further.
K. PÁNEK: Of the expected compounds, only H2 S3 5 and CH¿S35H were found. N either SO nor SQ w ere present in the m ixture of c a r r ie r s used for the chrom atographic analysis so that the presence of these compounds cannot be excluded even when they were not found. N evertheless, SO or SO2 could only be present in v ery sm all amounts, if at all.
Furtherm ore, the sum of activity of all the separated fractions together with the activity trapped on the walls of the bulb was compared with the a c tivity calculated from the irradiation conditions (a cross-section of 0.3 b was used). It was found that all values varied in the range of neutron-flux variation .
F . S . ROWLAND: The problem of the reactions of recoil S3 5 includes the question of the electronic state of the atom at the time of reaction. Spectroscopic investigations.of atom ic sulphur show that the ground state is 3 P ( 3 P2, O; 3Pi , 1130 cal/m ole ; 3 FJj, 1640 cal/m ole) with an excited 1 D2
state at 26 390 ca l/m o le , and an excited iSo state at 63 370 ca l/m o le . To study how S atoms react, one needs to know the electronic state involved - whether it is singlet or trip let, o r even !D 2 or iSo. Gunning's studies on the photolysis of 0=C=S are interpreted as involving singlet sulphur atoms, and the observed reaction s are closely analogous to those observed with methylene rad icals . The sulphur atoms insert d irectly into the C-H bond of hydrocarbons to give the corresponding m ercaptan - with methane, the yield of methyl mercaptan depends on the gas pressure at which the experiments are conducted. If the p ressure is not high enough, the methyl m ercaptan can undergo unimolecular decomposition to give a complex m ixture of products. T herefore, if recoil S3 5 atoms w ere in one of the excited singlet sta tes , the form ation of CH3 S35H from a d irect reaction with CH4
is a logical expectation.If a m oderator such as argon is introduced into the system , more than
one effect can result. F irs t , it can remove excess kinetic energy from the recoil atom. Secondly, these collisions can shift the electronic energy state of the S3 5 atom, as is well known from methylene experim ents. Since the
205

206 К. PÁNEK and К. MUDRA
triplet S atoms probably do not insert into C-H bonds, and the effects of the differences between !S and 45 are unknown, it is not yet possible to predict the over-all effect of a moderating species on the reactions of recoil S3S.
Furthermore, since free radical scavengers, such as NO, have little or no effect on singlet reactions, one would not expect the S35 reaction forming CH3S35H to be affected by the presence of NO. Without being able to say much about the kinetic energy of these atomic species at the time of reaction, I should have thought that the experiments reported by Dr. Pánek were quite consistent with singlet-sulphur-atom reactions.
R. BETTS (Chairman): Would Dr. Rowland care to expand on the relevance of ground-state or low-lying atomic levels of S35 to the chemical processes involved in the light of the fact that the new-born S35 may possess many hundreds of eV of excitation? .
F.S. ROWLAND: If a recoil atom undergoes an interaction with a molecule while it still possesses tens of electron-volts of energy, there is very little probability of a bond capable >of survival being formed - the extra energy is so large that the recoil atom breaks loose again, and is free for further reaction. Since these systems are examined by radio-gas chromatography, only molecules containing the radioactive species are observed. Only when the recoil atom gets into a range of energies comparable to chemical bond energies,(approx. in the 10 eV range) can the bonds formed by these atoms survive for subsequent product analysis. This means that initial energies of 100 eV or 105 eV are not verv important for the hot-atom chemistry of the system - almost all this energy will be lost before a chemical reaction forming a stable bond fot- the radioactive atom occurs.
The electronic energies of the S atom singlet states are of course of a magnitude similar to chemical bond energies.
A. WOLF: The matter of excitation energy, which has been raised on a number of occasions at this meeting, is of fundamental importance in hot- atom chemistry. The questions that are asked in this connection are, I believe, straightforward. What is the excitation state of the atom immediately prior to the first reactive collision? How much kinetic energy does the atom have? Is the atom charged? What electronic state is it in? When the atom undergoes a reactive collision, i. e. one in which at least one covalent bond to it is formed but not necessarily one in which a stable product is formed, one would like to know how the excitation of the incoming atom affects the first excited intermediate formed. Is there enough energy available from the colliding atom or from the exothermicity of the reaction to make the spin state of the incoming atom of minor or no importance, i.e . does the energy of thé complex allow easy spin interconversion? Is the structure of the excited intermediate well defined or are some of the atoms very much perturbed in relation to their normal geometrical configuration? How rapidly does energy transfer - primarily vibrational - take place in the intermediate before subsequent steps take place? .
There are many other questions one can ask. In working on organic systems, the answers are obtained by inference, primarily from the product distribution. The work of Lee and White and their collaborators and other work presented at this meeting is beginning to give us quantitative

CHEMICAL EFFECTS OF THE (n,p) REACTION 207
answers to some of these questions. It is to be hoped that work in carbon systems is also progressing along similar lines. It may be that hot-atom chemistry can begin to provide some of the answers to the much broader problem of the chemical reactions that occur well above the thermal range.
S. WEXLER: Professor Gordus, you report a rather large isotope effect in the bromine-plus-methane/deuterated-m ethane experiments. It is known that substantial isotope effects occur in ion-molecule-reactions - the lighter species always being the more reactive - but the isotope effect is considerably smaller (perhaps 20%). In consecutive ion-molecule reactions, however, the isotope effect can build up to a factor of 2 , again with the lighter species being more reactive. I remember that in an earlier paper you provided evidence of ion-molecule reactions forming iodomethane in the iodine(n, y) reaction on methane. ■ '
At that time I think you were inclined to believe that there were no ion- molecule reactions in the case of bromine. I should be interested to hear your views on this.
A. GORDUS: I do not think that was exactly our interpretation in the case of bromine. Our conclusion was simply that kinetic energy appears to be a requirement for the reaction, and it is not clear from the data whether or not the bromine is ionized or neutral or excited. In one of our papers some data were given in a footnote that indicated that for the (n, y) reaction in iodine+CD4 the organic yield was 52% compared with 54% in the iodine+ CH 4 reaction mixture. In the case of the (n, y)-produced iodine, there is, I think, quite good evidence that ionic iodine - ionic excited iodine, in'fact - is indeed involved in the reaction. I agree that there is a possibility of ion-molecule or secondary-ion-molecule reactions occurring in the case of bromine-methane, but I unfortunately do not know of any very simple way of testing this.
G. HARBOTTLE: In connection with Dr. Gordus's work, it seems to me that the reaction (Br80)n+ + C H 4 , CD4 is somewhat analogous to the "explosion" of CH3 I that occurs following irradiation with X-rays and the production of К-vacancies, i .e . to a multi-centre charged Coulomb repulsion as described by Dr. White*. If the isotopic effects in the experiment CH3Br, CD3Br (X-rays) were studied by charge spectrometry one might obtain a useful correlation with Dr. Gordus's results.
A. GORDUS: This would certainly be a most interesting study, but it would probably not provide definitive information about the Br80 + C H 4 or CD4 reaction since the Br^o is probably reduced in charge to +1 before it reaches the energy range of 10-20 eV, below which most reactions leading to stable products occur.
F.S. ROWLAND: At the hot-atom meeting at Amsterdam in May 1963, Peter Shaw reported large isotope effects in (n, y) reactions in liquid C2H5Br and QI^Br, comparable in magnitude to those reported by Dr. Gordus. Would Dr. Shaw care to comment on the origin of these isotope effects?
P. SHAW: About two years ago B .C . Patterson found that substitution of Br80m in (liquid) bromoethane was greater in QH5Br that in C2D^Br. This
CARLSON, T. A. and WHITE, R.M ., "Explosion o f multicharged molecular ions: chemical con
sequences o f inner shell vacancies in atoms", these Proceedings I. ,

208 К. PÁNEK and K. MUDRA
was tentatively attributed to differences in the bond dissociation energies of C-H and C-D bonds caused by differences in zero point energy. However, in the liquid state the reactions will probably be very different to those found in the gas phase.
J.E. LEDUC: Considerable attention has been devoted at this meeting to concepts such as reaction cross-sections and organic yields. It seems as though rapid progress is being made in the interpretation of the fundamental mechanisms involved and that, in some cases at least, it is now possible to predict the results of the induced reactions. What practical implications do you think this work might have in the field of chemistry?
A. GORDUS: I can give you some of my personal views on a few of the uses of gas-phase hot-atom experiments. The kind of information needed to interpret gas-phase reactions is also of basic interest for all studies of chemical kinetics. These include scattering asymmetry, collisional inelasticity and reaction probabilities, about which virtually nothing is known. Up to now, scientists working with atomic and molecular beams have had difficulty in obtaining beams of atoms with energies in the range of chemical interest, i. e. 1-20 eV, so that they have not been able to carry out such studies. Work on gas-phase hot-atom reactions may help us to understand these parameters. These studies are also a useful aid to an understanding of the chemical reactions that occur in the upper atmospheres not only in that of the earth but also in those of other planets.
B. DZANTIEV: On the subject of the practical singificance of hot-atom chemistry, I can see two possible applications. One is the "hot" synthesis of labelled compounds. In this field a multi-stage, lengthy and laborious process of chemical synthesis can be replaced by a rapid, single-stage process. In one of our papers*, we examine the scientific basis of a method for the hot synthesis of complex active biological compounds labelled with sulphur-35. Another possible application is the control of radiochemical - s.ynthesis processes in organic and aqueous systems, in which hot atoms of hydrogen and excited radicals are produced as a result of the radiation- induced disintegration of molecules + .
In connection with papers SM-57/64 and 73, I should like to make the point that investigations into the reactions of hot atoms of S and N - an important field of study per se - are particularly interesting if one considers the reactions of hot atoms of different valencies - H(T) - S35 - N 13 - C 14 - with identical molecules. A few years ago at the Chemical Physics Institute in Moscow we did some work in which we obtained hot atoms of № 3 through the nuclear reaction №4 (n, 2n) № 3 (irradiation with (T-D) neutrons) and a new type of double nuclear reaction, n - a - n : Li6 + n -i o, +t, о' + В10-* № 3 +n (in pile irradiation of Li6B10O2 salt). The reaction of hot atoms of N13 with benzene and urea produced hydrocyanic acid HCN13 and pyridine C5H5NI3. Parallel with the reaction n - t -n : Li6 + n -» or+t, O16 +t -> F18 +n, hot atoms of radioactive F18 were obtained.
sjcДзантиев Б .Г . и Шишков A . В "Р а з р а б о т к а м етодов гор ячего синтеза меченных
серой-35 биологически активных вещ еств", these P ro c ee d in g s I.t See e .g . 2. fiz . Him. 38 11 (1964) 2139. .

РАЗРАБОТКА МЕТОДОВ ГОРЯЧЕГО СИНТЕЗА МЕЧЕННЫХ СЕРОЙ-35 БИОЛОГИЧЕСКИ
АКТИВНЫХ ВЕЩЕСТВ
Б .Г . ДЗАНТИЕВ и А .В . ШИШКОВ ИНСТИТУТ ХИМИЧЕСКОЙ ФИЗИКИ АН СССР, МОСКВА
СССР
Abstract — Résumé — Аннотация — Resumen
DEVELOPMENT OF METHODS FOR THE HOT SYNTHESIS OF SS5-LABELLED BIOLOGICALLY ACTIVE SUBSTANCES. It was found in investigations with model systems that sulphur-35 recoil atoms are capable
of entering atom and atom-group substitution reactions through interaction with cyclic and heterocyclic compounds, as well as by way o f the C-C bond. We therefore considered that it would be interesting to use the specific properties of hot sulphur atoms for the synthesis of labelled biologically active compounds. We selected 4-methyl-50-hydroxyethyl thiazole (an intermediate product of vitamin B* synthesis), triethylenimine thiophosphoramide and amino acids (methionine, norvaline and norleucine) for investigation. Binary systems containing the compounds enumerated above, as well as CCI4 or HC1 (donors of hot S3s atoms) were investigated.
Irradiation was carried out in an IRT-1000 reactor channel at a thermal neutron flux o f 1011-1012 n/cm2*s. The S35 recoil atoms, formed by the reaction Cl 35(n, p)S3S with a recoil energy of 16 keV, interact with the thiazole giving thiazole-S*5. The yield of labelled product is highly dependent on the composition o f the system, reaching a maximum of 20-25%. The addition of benzene (acceptor of the excitation energy)'increases the yield of-product, the maximum yield being reached when the ratio o f CC14, thiazole and benzene
is 1:1 si. When the mixture of triethylenimine thiophosphoramide and CCI4 is irradiated, an initial product labelled with S35 and P32 is obtained, the P32 being formed by the reactions С135(п,с£)Р32, S32(n, p)P32 and P*Hn,y)P«.
Methionine-S35 is obtained by irradiating methionine and HC1 in an aqueous solution. The labelled product formed is usually diluted by a carrier, although it is possible to obtain compounds without a carrier by selecting the initial compounds appropriately. Thus, by irradiating systems o f norvaline-HCl and norleucine-HCl one obtains methionine-S®5 without a carrier, due to the entry o f sss by way o f the C-C bond into the norvaline molecule or the substitution o f the CH2 group in the norleucine» In both cases the yield of m e th io n in e - S s5 amounts to 7*12%. The products in question were separated and identified both chemically and by paper chromatography. '
In the case o f triethylenimine thiophosphoramide biological tests were carried out on the labelled products. . . .
The "hot synthesis" method thus enables one to obtain complex, biologically active compounds by means of a single-stage process, in many cases without a carrier. 4
ÉLABORATION DE MÉTHODES POUR LA‘«SYNTHESE CHAUDE» DE SUBSTANCES BIOLOGIQUEMENT ACTIVES MARQUÉES AU SOUFRE-35. En étudiant des systèmes modèles, on a montré que lors d'une interaction avec des hydrocarburès 'cycliques" et hétéro-cycliques, les atomes de recul de WS entrent dans des ré-' actions de substitution d’ atomes et groupes d'atomes ou s’ intercalent dans les liaisons C-C. Il a semblé intéressant de tirer parti des propriétés spécifiques des atomes chauds de soufre pour réaliser la synthèse de composés marqués biologiquement actifs. Pour procéder à ces recherches, on avait choisi les produits suivants: méthyl-
4-oxyéthyl-5Ô-thiazole (soüs-produit de'la synthèse de la vitamine-B,), triéthylaminothiophosphoramide, aminoacides (méthionine, nôrvalihe et norleucine). L'étude a porté sur des systèmes binaires contenant les composés mentionnés ci-dessus, ainsi que du ССЦ ou du НС1 (générateurs d'atomes chauds 35S). L’ irradiation'
avait lieu dans le canal du réacteur IRT-1000. à l'aidè d'un flux de neutrons thermiques de 10*1 à 10l2n/cm2»s. Les atomes de recul de 35S, qui se forment dans la réaction nucléaire 55Cl(n, p)S5S oü Erec = 16 keV, réagissent avec le thiazole en donnant du thiazole-35S. Le rèndement en produit marqué dépend de la composition du système et atteint au maximum 20 à 25Pjo» Une addition de benzole (accepteur d’ énergie d’excitation) augmente le rendement qui atteint le maximum lorsque le rapport entre les composantes CC14, thiazole et benzole est
20914

2 1 0 Б . Г . ДЗАНТИЕВ и А . В . ШИШКОВ
de 1ДД. En irradiant un mélange de triéthylaminothiophosphoramide et de ССЦ, on obtient un produit final marqué par 3ss et par S2p ; celui-ci se forme dans les réactions nucléaires: 35С1(п,а)32р, 32S(n,P)82p*31P(n,y)32P.
Lorsque de la méthionine et du HCl sont irradiés en solution aqueuse, il se forme de la méthionine-35S. Le produit marqué obtenu est habituellement dilué dans un entraîneur, mais en choisissant des composés initiaux appropriés on parvient à obtenir des préparations sans entraîneur. Ainsi, lors de l'irradiation des systèmes norvaüne-HCl et norleucine-HCl, il y a formation de méthionine-35s libre d'entraîneur aux dépens de l'in corporation de 35S à la liaison C-C de la molécule de norvaline ou aux dépens de la substitution du groupe CH2 dans la norleucine. Dans les deux cas, le rendement en méthionine-35S varie de 7 à 12с/о. Les produits finals ont été séparés et identifiés par des méthodes chimiques et par chromatographie sur papier.
Dans le cas de la triéthylaminothiophosphoramide, on a soumis le produit marqué à des essais biologiques.On peut conclure que la «synthèse chaude» permet d'obtenir par un processus à une seule étape des
préparations complexes biologiquement actives et, dans certains cas, libres d'entraîneur.
Р А З Р А Б О Т К А М Е ТО Д О В Г О Р Я Ч Е Г О СИ Н ТЕЗА М ЕЧЕННЫ Х СЕРО Й -35 Б И О ЛО ГИ Ч Е С К И А К ТИ В Н Ы Х В Е Щ Е С ТВ . В исследованиях на модельных систем ах было показано, что атомы отдачи серы-35 способны при взаимодействии с циклическими и гетероциклическими углеводородами вступать в реакции замещения атомов и атомны х групп, а также внедряться по С —С связи . П редставляло интерес использовать специфические свойства горячих атомов серы для синтеза меченых, биологически активных соединений. В качестве объектов исследования были выбраны 4 -м ети л-5 -8 -ок си эти д ти а зол (полупродукт синтеза витамина В1), триэтилениминотиофосфорамид (тиоТЭ Ф ), аминокислоты (метионин, норвалин и норлейцин). И сследовались бинарные системы , содержащие перечисленные выше соединения и C C I4 или НС1 (датчики горячих атомов серы -35 ). Облучение проводилось в канале реактора И РТ (1000 при потоке тепловы х нейтронов 1011 —1012 н/см2 с е к . А том ы отдачи серы-35, образующиеся по ядерной реакции C l35 (n ,p ) S35 с энергией отдачи 16 кэв, втупают во взаимодействие с тиазолом, давая тиазол, меченный серой-35. Выход меченого продукта зависит экстремально от состава системы, достигая в максимуме 20 — 25% . Добавка бензола (акцептор возбуждения) повышает выход продукта, причем максимальный выход достигается при соотношении компонентов C C I4 тиозол и бензол, равном 1 :1 :1 . При облучении смеси тиоТЭФ'а с C C I4 получается исходный продукт, меченный серой-35 и фосфором-32 (n ,p ) Р 32 ;Р31 (п ,7) Р 32 •
При облучении метионина и НС1 в водном растворе получается метионин-S 35 . Обра
зующийся меченый продукт обычно разбавлен носителем , однако при соответствующем подборе исходных соединений оказывается возможным получить препараты без носителя. Так, при облучении систем норвалин-H C l и норлейцин-H C l образуется метионин-S 36 без н о с и т е л я
за счет внедрения S35 по С -С -с в я з и в м олекулу норвалина или замещения СН2 -группы в норлейцине. В обоих случаях выход метионина-S 35 составляет 7 — 12% . Выделение и идентификация целевых продуктов осущ ествлялись химически и методом хроматографии на бумаге.
В случае тиоТЭФ 'а проводились биологические испытания меченого продукта.Таким образом , метод "горячего синтеза" позволяет получать сложные биологически
активные препараты п утем одноактного процесса, причем в ряде случаев б е з н оси теля .
ELABORACION DE METODOS PARA LA SINTESIS CALIENTE DE SUSTANCIAS BIOLOGICAMENTE AC
TIVAS MARCADAS CON AZUFRE-35. El estudio de sistemas modelos ha demostrado que por reacción con hidrocarburos cfclicos y heterocfclicos los átomos de retroceso de 3sS pueden sustituir a átomos o a grupos de átomos, o incorporarse en los enlaces C-C. Por ello se consideró de interés aprovechar las propiedades específicas de los átomos de azufre caliente para lograr la síntesis de compuestos marcados biológicamente activos. Para proceder a esas investigaciones los autores emplearon los siguientes compuestos: 4-metil-5S- oxietiltiazol (producto intermedio en la sfntesis de la vitamina Bi), trietilaminotiofosforamida (tioTEF), y aminoácidos (metionina, norvalina y norleucina). Los autores estudiaron sistemas binarios, que contenían los compuestos enumerados más arriba, así como CCI4 o HCl (generadores de átomos de 35S calientes). La irradiación se efectuó en un canal del reactor IRT-1000, con un flujo de 10n -1012 neutrones térmicos/cm2. s. Los átomos de retroceso de 95S que se forman en virtud de la reacción nuclear 3SCl(n, p )3 S, cuya energía de retroceso es de 16 keV, reaccionan con el tiazol, dando tiazo l-3SS. El rendimiento del producto marcado
depende de la composición del sistema y alcanza un máximo de 20 a 25°Jo. La adición de benceno (aceptor de la energía de excitación) aumenta el rendimiento del producto, alcanzándose un máximo cuando la rela
ción entre CCI4» tiazol y benceno es de 1:1:1. A l irradiar mezclas de trietilaminotiofosforamida y de CCI4, se obtiene un producto final marcado con 35S о 3Ф; este último se forma de acuerdo con las reacciones nucleares В Д ( П , а ) м Р , « S (n ,p )3 2 p , 3 Jp (n ,y )3 2 p ,

РАЗРАБОТКА МЕТОДОВ ГОРЯЧЕГО СИНТЕЗА 2 1 1
Рог irradiación de metionina у НС1 en solución acuosa, se forma metionina-S5S, El producto marcado que se obtiene suele estar diluido en el portador; sin embargo, por elección adecuada de los compuestos iniciales, se pueden obtener compuestos libres de portador. Asf, al irradiar el sistema norvalina-HCl y norleucina-HCl se forma metionina-SSS sin portador a expensas de la incorporación del 35S en el enlace C-C de la molécula de norvalina o de la sustitución del grupo CH en la norleucina. En ambos casos, el rendimiento de metionina-35S varfa entre 7 y 12%. La separación e identificación de los productos se efectuó por métodos qufmicos y por cromatografía sobre papel.
En el caso del trietilaminotiofosforamida, se efectuaron ensayos biológicos del producto marcado.La «sfntesis ca lien te » permite por tanto obtener, por un proceso de una sola etapa, preparados com
plejos biológicamente activos y, en algunos casos, libres de portador.
В настоящее время в биологии и медицине ширится интерес к изотопным методам исследования. Действительно, только применение изотопов и меченых соединений дает возможность глубже понять такие биохимические процессы, как белковой обмен, развитие опухолевых заболеваний, понять механизм действия лекарственных средств. В связи с этим значительно расширяется номенклатура необходимых меченых соединений, причем одно из важнейших мест среди них занимают препараты, меченые по сере-35. К классу серусодержащих соединений относится большая часть природных биологически активных веществ, например аминокислоты (метионин), витамины, сложные эфиры и многие другие.
Однако обычно используемый для получения меченых по сере-35 сложных органических препаратов метод химического синтеза часто оказывается слишком трудоемким и дорогостоящим. Отсюда возникает необходимость изыскания новых более простых и быстрых путей получения меченых соединений. Наиболее эффективные результаты в этом смысле дает метод изотопного обмена, однако в случае серусодержащих соединений этим методом могут быть получены лишь немногие вещества. Так, например, атомы серы, входящие в состав сульфидных групп типа R —S —R, в обмен ни при каких условиях не вступают.
Метод горячего синтеза, которому посвящена настоящая работа, позволяет в ряде случаев ввести радиоактивный изотоп в молекулу целевого соединения в результате "одноактного" физического процесса. Метод основан на использовании в синтетических целях энергии отдачи радиоактивных атомов, возникающих в результате ядерных превращений. Величина сечения ядерной реакции Cl35(n, p)S35, идущей как на тепловых, так и на быстрых нейтронах, а также период полураспада радиоактивной серы-35 делают метод горячего синтеза перспективным для получения меченых серусодержащих препаратов.
Практическое осуществление метода сводится к облучению системы, содержащей вещество, являющееся датчиком горячих атомов^и вещество- мишень, в молекулу которого вводится радиоактивная метка, на ядерном реакторе с последующим разделением облученной смеси и выделением целевого продукта теми или иными методами. Именно с последней стадией связаны значительные трудности метода, так как, как правило, при облучении образуется целая гамма меченых продуктов. Однако, как показывают исследование,на простых модельных системах (С6 Н12 —СС14, С6Н6 — СС14 и др.) оказывается возможным, изменяя состав и'замедляющие и стабилизирующие свойства системы, создать условия, при которых выход изотопного продукта оказывается оптимальным.

2 1 2 Б . Г . ДЗАНТИЕВ и А . В . ШИШКОВ.
В настоящей работе исследовалась возможность использования в целях синтеза меченых биологически активных веществ специфических процессов, протекающих с участием горячих атомов серы-35. Замещение атомов и атомных групп и внедрение по С —С связи:
S3s +R -C H 2- R ------ - R -S 3&-R +СН2 ; (1)
S35+ R - S - R ------ R — S 35 — R + S ; (2)
S3s+ R -R ------ > R -S 3s- R . (3)
Особый интерес представляют, по-видимому, процессы (1) и (3), так как в этом случае образующийся продукт не содержит носителя и может быть выделен со 1 0 0 -процентной удельной активностью.
В качестве объектов исследования были выбраны 4-метил-5-6-оксиэтил- тиазол (являющийся полупродуктом синтеза витамина Bi), аминокислоты (метионин, норвалин, норлейцин) и триэтилениминотиофосфорамид (тиоТЭФ).
Облучению подвергались бинарные системы, представляющие собой смеси перечисленных веществ с CCI4 или НС1. Последние являются датчиком горячих атомов серы, образующейся по реакции Cl35(n,p)S35.
В табл.1 представлена зависимость выхода меченого 4-метил-5-|3- оксиэтилтиазола от соотношения компонентов в системе.
Таблица 1
ЗАВИСИМОСТИ ВЫХОДА МЕЧЕННОГО СЕРОЙ-35 4-МЕТИЛ-5Д-ОКСИЭТИЛТИАЗОЛА
ОТ СОСТАВА ОБЛУЧАЕМОЙ СИСТЕМЫ
М олярная доля СС14
1 В ы ход м еч ен ого продукта, %
М олярная доля б ен зола при соотношении
С С 14/тиаэол l / l
В ы ход м еч ен ого продукта, %
0,18 „ 8 0 100,21 , ¿.i 40 20 15
0,24 - 18,3 30 „ 210,28 , 20,5 . 39 25,3
0,35 24,5 48 12,60,37 24 60 80,42 19 65 80,44 15,7 68 7,9
0,62 10,5
Меченый тиазол образуется за счет замещения атомом отдачи серыв тиазольном цикле: . ,
. ' - ' t ■
N -------------СН3 / N -------- —Г— £ Н 3
s 35+ II - JLcHaCHíOH ^ СН2СН2ОН +s •N'4 s ^ ■ ■ * ■ s35 - -
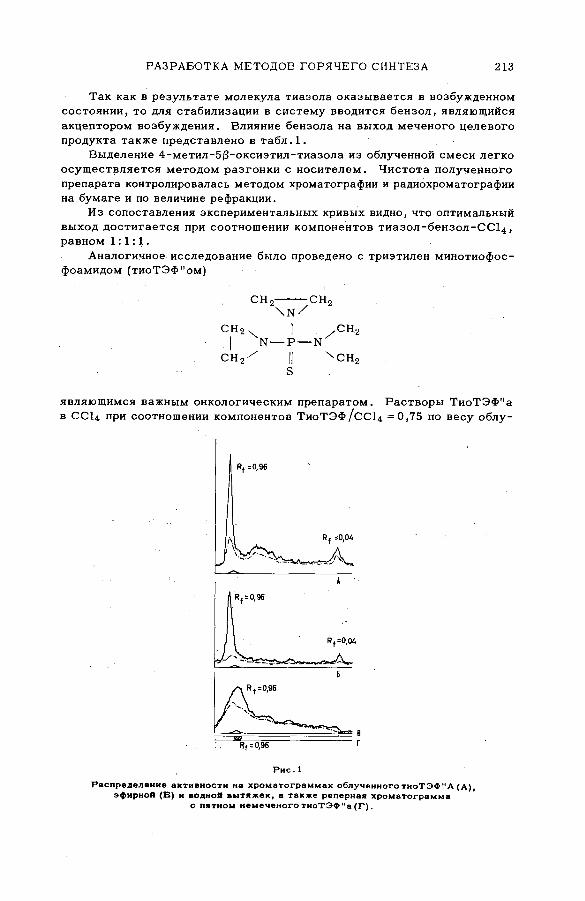
РАЗРАБОТКА МЕТОДОВ ГОРЯЧЕГО СИНТЕЗА 213
Так как в результате молекула тиазола оказывается в возбужденном состоянии, то для стабилизации в систему вводится бензол, являющийся акцептором возбуждения. Влияние бензола на выход меченого целевого продукта также представлено в табл. 1 .
Выделение 4-метил-5|3-оксиэтил-тиазола из облученной смеси легко осуществляется методом разгонки с носителем. Чистота полученного препарата контролировалась методом хроматографии и радиохроматографии на бумаге и по величине рефракции.
Из сопоставления экспериментальных кривых видно, что оптимальный выход достигается при соотношении компонентов тиазол-бензол-СС14, равном 1 : 1 : 1 .
Аналогичное исследование было проведено с триэтилен минотиофос- фоамидом (тиоТЭФ"ом)
СН2-------- СН2\ n / .
с н 2 ч I С Н 2. I N — Р — N
. СН2^ II \ с н 2S .
являющимся важным онкологическим препаратом. Растворы ТиоТЭФ"а в CCI4 при соотношении компонентов ТиоТЭф/сС14 =0,75 по весу облу-
Rf =0,96 Г
Р и с .1
Расп р еделен и е активности на хр ом атогр ам м ах о б луч е н н о го т и о Т Э Ф мА (А ) , эфирной (Б ) и водной вы тяж ек, а также реперная хр ом атогр ам м а
с пятном н е м е ч е н о г о т и о Т Э Ф "а (Г ).

214 Б . Г . ДЗАНТИЕВ и А . В . ШИШКОВ
чались смесью тепловых и быстрых нейтронов в канале реактора. При этом, наряду с серой-35, индуцируемой по реакции на хлоре, имеет место образование значительных количеств радиофосфора Р32 по реакциям C l3 5 ( n ,a ) P 32, S3 2 ( n ,p ) P 32 и P 3 1 ( n , 7 ) P 32j в результате чего целевой продукт получается с двойной меткой по S35 и Р32.
Анализ облученной мишени проводился методом хроматографии и радиохроматографии на бумаге. На рис.1 представлена типичная хроматограмма облученной смеси (А), а также реперная хроматограмма с пятном чистого немеченного тиоТЭФ"а. Из рисунка видно, что основная доля активности падает на целевой продукт, причем около 50% активности тиоТЭФ "а приходится на долю фосфора-32. Хорошие результаты дает также метод очистки тиоТЭФ "а путем последовательных перекристаллизаций из эфирного и водного раствора попеременно с осаждением РО4 и SO4 -ионов из водного раствора обычными химическими методами. На рис.1 представлены хроматограммы эфирной (Б) и водной (В) вытяжек, из которых видно, что таким путем могут быть получены достаточно чистые препараты. Меченный серой-35 и фосфором-32 тиоТЭФ был использован при биологических исследованиях, результаты которых явились дополнительным подтверждением химической чистоты синтезированного горячим методом соединения.
Чрезвычайно важную роль при различного рода биохимических исследованиях играет метионин CH3 SCH2CH2 CH(NH2)COOH, который в настоящее время производится в больших количествах изотопной промышленностью.
Меченный серой метионин получался по методу горячего синтеза при облучении метионина в водном растворе хлористого водорода. Протекающий при этом процесс может быть представлен схемой:
S 35 + C H 3 S C H 2 C H 2 C H (N H 2 ) C 0 0 H --------► C H 3 S 3 5 C H 2 C H 2 C H (N H 2 ) C 0 0 H + s
На примере процесса горячего синтеза меченого метионина было интересно проверить возможность получения меченых препаратов, не содержащих носителя, т .е . использовать в синтетических целях способность атомов отдачи серы замещать в молекулах вещества мишени СН2 -группы, а также внедряться в молекулу по С —С связи. Указанные процессы были обнаружены ранее [1 ] в системах, содержащих циклические углеводороды, как,например, циклопентан. Представлялось также интересным и с теоретической точки зрения проверить, реализируются ли указанные процессы в случае углеводородов с открытой цепью.
Для этого облучению подвергались норвалин [1] -аминокислота с тем же числом углеродных атомов, что и в молекуле метионина, и норлейцин [2 ] - аминокислота, в молекуле которой на одну СН2-группу больше, чем в метионине, в солянокислых растворах. При этом за счет реакций
S35 +CH3CH2 CH2CH2 CH(NH2 )СООН------ ► CH3S35CH2 CH2CH(NH2 )C00H +
S35 + СН3СН2 CH2CH(NH2 )СООН------► CH3S35CH2 CH2CH(NH2)COOH+CH2
должен был бы получаться меченный серой-35 метионин. Для анализа облученных систем использовался также метод хроматографии и радиохроматографии на бумаге.

РАЗРАБОТКА МЕТОДОВ ГОРЯЧЕГО СИНТЕЗА 215
Р и с . 2
Р а сп р еделен и е активности на хр ом атогр ам м ах : а — о б л у ч е н н ы й раствор метионина;6 — р а с т в о р м е т и о н и н а , о б р а б о т а н н ы й 3 0 % Н 2 О 2 .
На рис. 2а, За, 4а представлен спектр меченых продуктов, образующихся при облучении солянокислых растворов метионина, норвалина и нор- лейцина, соответственно. На всех трех хроматограммах четко выражен пик с Rf метионина (0,57), причем на долю его приходится около 10% от общей активности. С целью химической индентификации метионина производилось окисление последнего перегидролем до метионин-сульфона. На рисунках 26, 36, 46 видно, что в результате окисления пик активности, соответствующий метионину, исчезает и возникает пик активности с величиной Rf, соответствующей сульфону. Наряду с метионином-S35 при облучении образуются также в незначительных количествах сульфон, сульфо- окись метионина и гомоцистеин (1%) и до30% активности серы-35 содержится в форме сульфат-иона.
Рассмотренные исследования показывают, что в случае изотопа серы-35, фосфора-32, а также,очевидно^ для целого ряда изотопов, особенно короткоживущих, применение метода горячего синтеза позволяет простым быстрым и экономичным путем синтезировать достаточно сложные меченые вещества, в том числе и биологически активные препараты, причем в ряде случаев возможно получение препаратов с очень высокой удельной активностью без носителя.
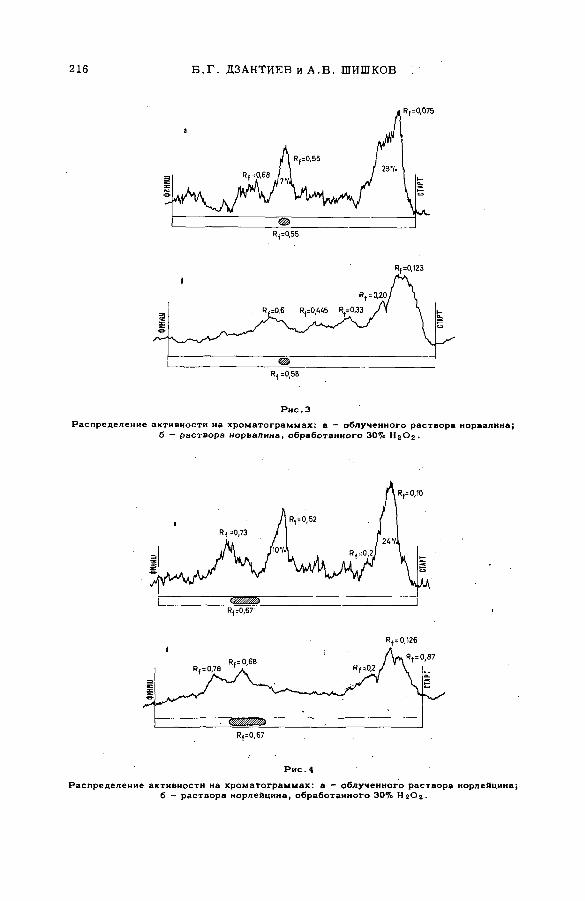
216 Б . Г . ДЗАНТИЕВ и А . В . ШИШКОВ
Р и с .З
Р а сп р еделен и е активности на хр ом атогр ам м а х : а — облуч ен н ого раствора норвалина; б - раствора норвалина, обр аботан н ого 30% Н20 2 .
R f = 0,126
Р и с . 4
Расп р ед елен и е активности на хр ом атогр ам м а х : а - облуч ен н ого раствора норлейцина; б — раствора норлейцина, обр аботан н ого 30% Н гО г .

РАЗРАБОТКА МЕТОДОВ ГОРЯЧЕГО СИНТЕЗА 217
Л И Т Е Р А Т У Р А
[1 ] Д З А Н Т И Е В Б . Г . и Б А Р К А Л О В И . М .
[2 ] Т р у д ы М е ж д у н а р о д н о й к о н ф е р е н ц и и п о п р и м е н е н и ю и з о т о п о в , К о п е н г а г е н , 1 9 6 0 .
D I S C U S S I O N
Y . G H O O S: D id y o u fin d a n y in d ic a t io n o f a m in o - a c id p o ly m e r iz a t io n in th is w o r k ?
B . D Z A N T IE V : W e w e r e n ot c o n c e r n e d in th is p a p e r w ith the p r o b le m o f th e p r o d u c t - f o r m a t io n m e c h a n is m a lth ou g h it d o e s l o o k a s th ou g h , a s in th e c a s e o f s im p le r m o l e c u l e s , th e p r o d u c t f o r m a t io n c o u ld b e th e r e s u l t o f th e s u b s t itu t io n o f a CH 2 g r o u p b y a s u lp h u r a to m . H o w e v e r , r a d ia t io n e f f e c t s c o u ld a ls o b e in v o lv e d . W e d id not in v e s t ig a t e th e p r o b le m o f p o ly m e r i z a t i o n b e c a u s e o u r a im w a s m e r e l y to o b s e r v e th e c h r o m a t o g r a p h ic p e a k s o f p a r t ic u la r p r o d u c t s . T h e p e r c e n t a g e s sh ow n on the c h r o m a t o g r a m a r e n ot p e r c e n t a g e s o f to ta l su lp h u r a c t iv ity but p e r c e n t a g e s o f th e a c t iv i t y o n th e c h r o m a t o g r a m . T h e c h r o m a t o g r a m p e a k s m a y in c lu d e p o l y m e r i c p r o d u c t s b u t w e d id n ot id e n t i fy th e m a s s u c h .
N . G E T O F F : W e h a v e s tu d ie d th e c a r b o x y la t io n o f d i f f e r e n t o r g a n i c c o m p o u n d s in a q u e o u s s o lu t io n f o l lo w in g e x p o s u r e to g a m m a - r a y s an d U V lig h t (X = 1 8 4 .9 m m ), and w e h a v e fo u n d that u n d e r th e s e c o n d it io n s a m in e s ca n b e t r a n s fo r m e d in to a m in o a c i d s .
W ith C 1 4 0& on e ca n o b ta in a m in o a c id s la b e l le d in th e C O O H g r o u p . F u r t h e r m o r e , S35, P 32, e t c . ca n a ct as s c a v e n g e r s o f the d if fe re n t f r e e r a d i c a ls f o r m e d b y r a d io ly s is o r p h o to ly s is o f th e a m in e in th e p r e s e n c e o f CO^ g a s and th e y ca n b e in c o r p o r a t e d in th e r e s u lt in g a m in o a c id s .
W e h ave a ls o foun d that th e lo w e r a m in o a c id s that h ave on e o r tw o c a r b o n a to m s a r e m u c h m o r e s e n s i t iv e to r a d ia t io n th an th e h ig h e r o n e s that h a v e m o r e . I f i r r a d ia t io n i s p r o lo n g e d , th e r e i s a c o n c e n t r a t io n o f a m in o a c id s o f th e la t t e r ty p e .
Amino acids such as glycine and alanine are formed directly in aqueous solutions from NH3, C02 and water by UV light. The yield is, however, very low.


LIQUID SYSTEMS (Session 3)


CHEMICAL EFFECTS OF NUCLEAR TRANSFORMATIONS
OF HALOGENS IN ORGANIC MEDIA
J.E. WILLARD DEPARTMENT OF CHEMISTRY,
UNIVERSITY OF WISCONSIN, MADISON, WIS.,' UNITED STATES OF AMERICA
Abstract — Résumé — Аннотация — Resumen
C H E M I C A L E F F E C T S O F N U C L E A R T R A N S F O R M A T IO N S O F H A L O G E N S IN O R G A N IC M E D I A . T h is
r e v i e w is c o n c e r n e d w ith r e c e n t w o r k a n d s o m e u n s o lv e d p r o b le m s o f t h e c h e m i c a l e f f e c t s o f n u c le a r a c t i
v a t io n o f h a lo g e n s in g a s e o u s , l iq u id a n d s o l id o r g a n ic m e d i a . T h e p r o b a b i l i t y o f b o n d ru p tu r e , th e p r im a r y
p h y s ic a l an d c h e m ic a l p ro c e s se s fo l lo w in g n e u tro n c a p tu r e , an d c e r t a in m e c h a n is m s fo r r e c o i l a to m r e c o m b in
a t io n a r e d isc u sse d in so m e d e t a i l .
E F F E T S C H IM IQ U E S D E S T R A N S F O R M A T I O N S N U C L É A IR E S D E S H A L O G È N E S D A N S D E S M I L I E U X
O R G A N IQ U E S . C e t t e é t u d e d 'e n s e m b le p o r t e su r d e s t r a v a u x r é c e n t s e t su r c e r t a in s p r o b lè m e s n o n r é s o lu s
q u e p o se n t l e s e f f e t s c h im iq u e s d e l 'a c t i v a t i o n n u c l é a i r e d es h a lo g è n e s d a n s d e s m i l i e u x o r g a n iq u e s s o l id e s ,
l iq u id e s e t g a z e u x . L 'a u t e u r e x a m in e e n d é t a i l l a p r o b a b il it é d 'u n e ru p tu re d e l ia i s o n , le s p ro ce ssu s p h y s iq u e s
e t c h im iq u e s p r i m a i r e s c o n s é c u t i f s à l a c a p t u r e d 'u n n e u tro n , e t c e r t a in s m é c a n is m e s d e r e c o m b in a i s o n d e s
a t o m e s d e r e c u l .
ХИМИЧЕСКИЕ ЭФФЕКТЫ -ЯДЕРНЫ Х ПРЕВРАЩ ЕНИЙ ГАЛО ГЕ Н О В В ОРГАНИЧЕСКОЙ СРЕ Д Е . В этом обзоре затрагиваются проводившаяся в последнее время работа и некоторые нерешенные проблемы химического действия ядерной активации галогенов в газообразны х, жидких и твердых органических средах. Довольно подробно обсуждаются вероятность разрыва связи, первичные физические и химические процессы, следующие после захвата нейтронов, и некоторые механизмы рекомбинации атомов отдачи. . ,
E F E C T O S Q U IM IC O S D E L A S T R A N S F O R M A C IO N E S N U C L E A R E S D E L O S H A L O G E N O S E N M E D IO S
O R G A N IC O S . E l e s t u d io t r a t a d e lo s t r a b a jo s r e c i e n t e m e n t e e f e c t u a d o s , y d e lo s p r o b le m a s p e n d ie n t e s
d e s o lu c ió n , e n m a t e r ia d e e f e c t o s q u í m i c o s d e l a a c t i v a c i ó n n u c le a r d e lo s h a ló g e n o s e n m e d i o s o r g á n ic o s
g a s e o s o s , lfq u id o s y s ó l id o s . S e e x a m in a n c o n b a s t a n t e d e t a l l e l a p r o b a b i l id a d d e l a ru p tu ra d e e n la c e s , lo s
p r o c e s o s p r im a r io s d e c a r á c t e r f f s i c o y q u f m ic o c o n s e c u t iv o s a l a c a p t u r a n e u t r ó n ic a y a lg u n o s m e c a n is m o s
d e r e c o m b in a c i ó n d e á t o m o s d e r e t r o c e s o .
INTRODUCTION
At the IAEA Symposium on Chemical Effects of Nuclear Transformations held in Prague in 1960 a summary was given [1] of work on nuclear transformations of halogens in organic media. It is the purpose of the presént paper to review investigations since that time and to identify some of the unsolved problems in this field.
No attempt will be made to review the literature completely. References to the earlier work are included in previous reviews [1, 2]. SIUDA [3] has compiled a comprehensive bibliography of all work on chemical effects of
2 2 1

2 2 2 J. E. WILLARD
nuclear transformations from 1914 to the end of 1962. Review articles since 1960 dealing in part with chemical effects of nuclear activation of halogens in organic media include a thorough critical review by CAMPBELL [4], a treatment of primary physical and chemical effects by WEXLER [5] and a short summary of the field [6 ] . WOLFGANG [7] has discussed halogens briefly as part of a review of all gas-phase hot atom reactions.
Investigations during the last four years have included studies designed to find out more about both the primary physical and chemical processes, and the mechanisms of chemical stabilization following these processes. Predominant among the problems to which answers are being sought by workers in the field are: (1 ) the relative importance of kinetic energy and of charge in determining the chemical fate of the atoms studied; and (2 ) the effect of phase on the mechanisms that lead to chemical stabilization. Among the subjects of interest on which there have been publications are: the charge spectra of ion fragments from the isomeric transition of СС1зВг80т [8 ], from nuclear electron capture in C2H5 1125 an(j CH3 I125 [9] and from the Э, T decays of RI131 and R B r 8 2 compounds [8 ,10]; further evidence that atoms produced by the (n, y) process may remain in metastable states that then charge the atom by internal conversion after it has broken its parent chemical bonds[1 1 ]; extensive evidence on the extent of failure of bond rupture following the (n, y) process both in the gas phase [12,13] and liquid phase [14, 15, 16, 17a, 18]; definitive evidence that the gas phase yield of the reaction of recoil 1 28 atoms with CH4 requires electronically excited positive ions as well as atoms or ions with kinetic energy, whereas the analogous Br80- CH4 reaction apparently requires only kinetic energy [19]; studies of both solution [18,20] and gas-phase [21] F18 recoil chemistry; a procedure for distinguishing hot displacement reactions from those radical combination reactions that occur within the "hot spot," by extrapolation to 100%scavenger[2 2 ]; an attempt to determine the number of radicals present in the liquid volume within which an 1128 recoil atom undergoes diffusive combination [17a] ; a model of liquid phase retention based on the possibility that all radical formation occurs by hot (epithermal) bimolecular displacement reactions[23]; consideration of the application of the Estrup-Wolfgang kinetic theory to condensed systems [24]; evidence that the fate of recoil atoms in condensed systems may be determined by the fragments produced around them by their own conversion electrons and vacancy cascade electrons [25]; evidence for an isotope effect when bromine is activated by the (n, y) process in polyhalogenated methanes [26] . Publications concerned with the solid state have included: correlations of organic yields'of the B r 7 9 ( n , 7 ) B r 8 0 process with heat capacity and dielectric constant changes at phase transitions [27 ] ; studies of the organic yields resulting from the B r 8 0 m isomeric transition in alkyl bromide solutions as a function of state (glassy or polycrystalline), temperature, bromine concentration, and parent compound [28]; studies of the kinetics by which defects produced in С2ВГ6 by gamma irradiation either before or after neutron irradiation of СгВге crystals can interact with the metastable B r 8 2 sites to change the form of chemical combination in which the Br82 is found when the С2ВГ6 is dissolved after warming 129, 30].

NUCLEAR TRANSFORMATIONS OF HALOGENS 223
PRIMARY PHYSICAL AND CHEMICAL PROCESSES
Radiative neutron capture has been the mode of activation in the majority of studies of chemical activation of halogens by nuclear processes. Current knowledge of the gamma-ray cascades from (n, 7 ) processes makes it seem probable that a halogen atom that captures a neutron usually receives from the first and most energetic gamma emitted a recoil energy much in excess of its chemical bond energy, and that the probability of sufficient cancellation of momentum to lead to failure of bond rupture is low even if all gammas are emitted simultaneously. In the cases studied 111,31, 32] at least a substantial fraction of all atoms produced by the (n, 7 ) process becomes charged by internal conversion and emission of Auger electrons in vacancy cascades. In those cases studied [11, 32] such charging occurs long enough after the initial (n, 7 ) event so that the parent chemical bonds of the recoiling atom have been broken. Since metastable state lifetimes may be as long as 10-6 - 10-5 s, the charging process may occur after the atom has been stabilized in new chemical combination following recoil. If a charge of +2 or greater is acquired while the atom is chemically bound, it is usually capable of producing disintegration of the molecule by Coulombic forces even in the absence of recoil energy 18, 9] . A discussion of much of the work dealing with these activation steps has been given in earlier reviews [1, 2, 4, 5), with the physical aspects receiving particularly complete discussion in the review by WEXLER [5] .
A very important type of information thus far not available is knowledge of the fraction of (n, y) events which results in charging of the atom for each of the halogen nuclides used in hot atom studies. The clear cut evidence that charging occurs with Br and I sets only lower limits on the fraction charged. The actual fraction may be higher because either some charge transfer may occur even in the low pressure gases used 131 J; or neutral atoms may be collected in a time too short for charging to have occurred. No similar measurements have been made for Cl and F. It is known that Cl36 from the Cl35 (n, 7 )C136 process is stabilized by gamma cascades 133] . If a К electron were removed by internal conversion, vacancy cascades would occur 134]. The probability of internal conversion would, however, be expected to be less for Cl and F than for Br and I because of their lower atomic numbers.
Of great interest is the discovery [35, 36] that the initial product of about 90% of the radiative neutron capture events by B r81 is BrS2™, which decays by isomeric transition with a half-life of about 6 min to Br82 (36 h). The decay goes through a 46 keV transition with an internal conversion coefficient of ak = 268 [36] so that essentially all the atoms are charged. This means that the organic yields and individual product yields that have been attributed to (n, 7 ) activation in the many studies made with Br82have, in fact, been due almost exclusively to activation occurring by isomeric transition of the Вг82ш, since its lifetime is short compared to the usual irradiation times used in most Br82 studies, and compared to the times of standing following irradiation before analysis.
GORDUS and co-workers have evaluated the probability of failure of bond rupture following gamma-ray cascades [37], and the partition of recoil

224 J. E. WILLARD
energy between the fragments of a variety of parent molecules [12]. They have determined experimentally for 18 gaseous organic halides the fraction of radiative captures not leading to bond rupture [13]. The fraction is in all cases 0 .0 1 or below, as expected both from the calculations and earlier experimental work on relatively few compounds.
Recent experiments by THOMPSON and MILLER [11] have shown that recoil atoms produced by the (n, 7 ) process on In, 1п2Оз, InF3 , Mn, КМПО4 and Оу2Оз coated on a graphite rod in an evacuated cóntainer may be collected on aluminized Mylar films surrounding the rod. By determinations with applied voltages it was found that the proportion of positively charged recoils, which ranged from 40 to 60%, was, within experimental error, independent of the chemical form of the atom before recoil. This supports the earlier evidence [32], obtained from the recoil of Au and In atoms from gold and indium foils, that charging of these atoms by internal conversion follows recoil from the initial gamma ray by a time sufficiently long for the atom to have escaped from its molecular bonding.
Since the Prague Symposium, further mass spectrometric evidence on the fate of alkyl halides undergoing beta decay, isomeric transition and electron capture in the gas phase has been published. These results are of interest as primary chemical effects of nuclear transformations, and also because, in the cases of isomeric transition and electron capture, they simulate processes that must occur when a halogen atom that has undergone the (n,7 ) process is charged by the loss of an internal conversion electron. Among the interesting results are the ion yields from the /3, У decay of СН3ЦЗ1 that produces 69% СН3ХеШ+ and 2% CHJ, of CH3I130 that yields 34% CH3Xe13ü+and 39% CHJ and of C2H5I13i that gives 1.4% C2H5Xei3i+ and 89% C2H5 . The authors, CARLSON and WHITE [10], attribute the greater stability of the CH3Xei3i+ comparéd with the CH3Xei30+to lower average recoil energy from the 0 ray and accompanying y rays of I131 (0.22 eV as compared to 0.78 eV). The results suggest a bond energy of 0 .5 -1 eV for the C-Xe bond. The much lower stability of the C2H5Xe131+ is attributed to higher electron density on the central carbon favouring transfer of an electron to the Xe and bond rupture. The same authors 110] have found ion abundances for the ¡3 decay of CH3 Br82, 1, 2- C2Щ ВгВг82 and CCl3Br82 to be 89% CHJ, 70% C2 H4Br+ and 64% CClJ respectively. For the СС1зВг82 |3 decay, Wexler reports ratios of CCÍJ, CC1+ , Cl+ and C+ to CC13 of 0.20,0 .19, 0.12 and 0.045.
Recent mass spectrometric determinations of the charged fragments produced as a consequence of isomeric transition and nuclear electron capture processes in gaseous molecules confirm earlier examples showing that removal of an electron from an inner shell leads to build-up of multiple positive charge that causes the molecules to explode into a variety of fragments as a result of internal Coulombic repulsion. Thus the isomeric transition of Br80m in СС13Вг80т produces a spectrum of positive Br80 ions peaking at Br+8 and extending to Br+14 18]. On a relative abundance scale where (Br 80)+5 = 1, some other values are (Br80)+8 = 1. 3, Cl+1 = 1. 4, Cl',6 = 0 .3 , C*=1.4, C+2 =0 .4 , CC13 Br+1 = 0. 02, CClf = 1. 2, С С Г =1.6 . Electron capture by 1125 in CH3 Ii25 and C2 HSI125 produces spectra of Те ions peaking with values of about 12% abundance for all ions from Te+7 through Te+11.

NUCLEAR TRANSFORMATIONS OF HALOGENS 225
O th er s p e c ie s from C 2H 5H25 in c lu d e 1.2% C 2 H 5T e +, 1.3% C 2H j, 0.3% C2H$,0.8% CJ, 3.2% C+ + C+2 [9J.
GAS-PHASE REACTIONS
Following rupture of its parent bond, the atom or ion formed as a result of nuclear transformation loses energy, or charge, or both in collisions with other molecules of the medium until it has been reduced to an energy of tens of eV or below and a charge of +1 or zero, at which time it may enter permanent stable combination if an appropriate reaction path is available. The early investigations of such processes in the gas phase (summarized in [2, 6 ] ) have now been extended by the definitive evidence of RACK and GORDUS 119J on the nature of the reactive species in the (n, 7 ) activated reactions of I and Br with CH4. Using additives of different ionization potentials, extrapolated to 10 0% concentration, and a consideration of available spectroscopic information, they conclude that of the normal 54% yield of CH3I128 from activation of iodine in gaseous methane 18% must result from atoms or ions with excess kinetic energy, 25% from translationally thermalized but electronically excited I+('D 2) ions and 1 1 % from I+ ions in lower states than I+('D2) 119aJ. Other experiments by these authors 119bJ strongly indicate that the reaction of Br® from the Br79(n, v)Br80 process with CH4 is probably not dependent upon excited states or charge.
In the first work to be published on gas-phase fluorine recoil chemistry, COLEBOURNE and WOLFGANG 121 ] studied the reactions of F 18 produced by the Fi9 (7 , n)Fi8 process from CF4 containing up to 10 mole % СгН2, and low pressures of I2. The yield of CF3 FI8 is reduced from ~ 40% to 5% of the F18 by less than 1 mole % СгНг-Ь- In the scavenged system, 80% of the Fis atoms are found as C2H4FI8I. CROSS AND WOLFGANG [38 J have reported a yield of ~ 4% for the production of CH3I126 from iodine produced by the 1127(7, n)Ii26 process in gaseous CH3I scavenged with I2. This is similar to, though slightly higher than, the organic yield of 1% found for iodine activated by the 1127(n, 7)1128 process in C2H5I [39] .
Two groups of workers have observed the organic iodine activity formed when fission product recoil atoms are stopped in methane [40, 41] or methyl iodide [41] . The results for the ratios of the yields of 1135to I133 are interpreted to indicate that reaction occurs more efficiently when the I atom is a primary fission recoil fragment than when it is formed by beta decay of a precursor.
Facts now established about gas-phase reactions of halogens activated by nuclear processes include the following. All halogens, F, Cl, Br andI, when activated by nuclear processes can enter stable organic combination by bimolecular displacement reactions in the gas phase. These reactions are in contrast to the abstraction reactions that at present appear to be the exclusive mode of attack of thermal halogen atoms. Reactions of CH4 with1128 from the l^^n, 7)1128 process occur by three types of processes, one requiring kinetic energy of the atom or ion, one electronic excitation and charge, and the other at least charge. The analogous reaction of Br with methane seems to require only kinetic energy. The bimolecular replace-

226 J. E. WILLARD
ment processes observed for Cl with n-PrCl and other organic chlorides tlj suggest that, depending on its energy and/or charge and direction of attack, a Cl atom can enter stable combination by displacing any atom or group in the molecule, and that the excess energy sometimes leads subsequently to olefin formation by elimination of HC1. Multipath reaction probabilities with formation of a variety of products have also been shown for I and Br gas-phase reactions 142] . Notable are the great differences in reaction probability for reactions with rather similar molecules. Thus the yield of CH3I128 from recoil I128 in CH4 (54%) is much higher than that of the analogous reaction of 1Ш with С2Нв (2%) or C2H5I (1%). The data do not enable a decision to be made as to whether the differences are due to competition between insertion and abstraction or between insertion and thermalisation. In seeking explanations the properties of the target molecule which must be considered include its ionization potential, ability to deactivate excited states of the attacking atom, mass, steric shielding of the carbon atoms, angular momentum. Extensive consideration has been given to the role of steric effects and angular momentum in reactions of recoil T atoms [7 ] .
LIQUID PHASE
Probability of bond rupture following radiative neutron capture
IYER and MARTIN 114] have carried out neutron irradiations of mixtures of CH3 I and Cglifl in which the dilute component was synthesized from 1129 while the major component contained only I121. I130 was formed by theIi29(n, 7 )I130 process. By determining its yield in the form of the parent molecule in excess of the yield when I2 was used as the source of I12**, a value of about 4% was obtained for the fraction of (n, 7 ) events in which the recoil atom either failed to rupture the parent bond or recombined with the parent. We [15] have extended this technique to solutions of 0 .1, 1 and 10 mole % Í-C3 H7I, and C2H5I in CqH\4 containing a few tenths mole % IJ29. The results indicate that: (1) recoil I130 and I128 activated by the (n, 7 ) process give essentially the same total organic yield and the same distribution of organic products if they originate from the same chemical parent; (2 ) the yields are essentially independent of whether the recoil I atom originates from RI or I2; and (3) failure of bond rupture occurs in less than 1% of the n, 7 events. STURM [43] has reported the interesting observation that aqueous solutions of 0.01 mole fraction CH3I with variable I2 scavenger concentrations in the range of 10-5 mole fraction give an organic yield from 1121(п, 7 )P28 activation of about 12%. This organic yield in an inorganic solvent has been attributed to failure of bond rupture. To test this conclusion we 116] have carried out the same experiments using I 29 rather than I^ as the scavenger. We obtain nearly the same organic yield (~ 10%) from the I130 atoms originating from the Ii29(n, 7)1130 process in I2 as for the I128 atoms originating from the СНз1127 in the same system. Thus the observed yield is not the result of failure of bond rupture. Preliminary results from work in progress indicate that the organic yield of 1130 is within 2 % of that for X128 when all the

NUCLEAR TRANSFORMATIONS OF HALOGENS 227
1129 is maintained in the form of I" by the presence of excess SO2. Use of1129 as iodide ion precludes the possibility that the I129 is held in contact with the CH3 I by a CH31-12 complex at the time of neutron capture. A plausible explanation of the result is that the thermalized П30 atoms combine with CH3 radicals formed by the CH3I+e' -» CH3 +I' process as postulated in the Auger electron reaction hypothesis [25] .
In work with solutions of I2 and C6H5I in benzene SHAW [17] has reasoned that if escape from the parent partner always occurs following (n, y) activation, an iodine atom originally combined as iodobenzene should give the same organic yield as one combined as I2. On comparing the yields from benzene solutions containing 10-5 mole fraction I2 with similar solutions containing also 10-5 mole fraction CgHsI (and correcting for the fact that only one-third of the iodine originates from CeH5I in the latter case) he concludes that the organic yield is 2.7% higher for СбН51 than for I2, indicating 2.7% failure of bond rupture. By similar reasoning, values of 2 .4 and 1.6% are obtained for CH3I and C2H5I in СбН6. Shaw notes that these values are only slightly higher than the organic yield from C2H5 I in the gas phase [19] indicating that relatively few molecules are stabilized inthe parent form by caging or energy removal by the solvent. ANBAR and NETA [18] have obtained several types of evidence that indicate about 3% failure of carbon-fluorine bond rupture by F 1S produced in a variety of compounds by the Fi9(n, 2n)Fi8 process. This is unexpected because of the very high recoil energy likely to come from the neutron emission. Fast neutron irradiations of aqueous fluorobenzoic acid at concentrations as low as 1 mole % show organic labelling by about 3% of the F18. The labelling yields of an aliphatic side chain by fluorine generated from an aromatic ring and the labelling of the ring by fluorine generated from the side chain are reported to be similar to the yields obtained for fluorine originating from "extra- molecular" sources whereas the organic radical to which the fluorine was originally bound is labelled with much higher yield. Aqueous solutions of 3 mole % o-FCgHjOH gave 2.6% organic yield with most of the activity as o-FC6H4OH while for solutions of p-FCgH4OH the total yield was similar but was predominantly p-FCg H4 OH. The retention of F18 produced by the Oi6(H3 ,n)Fl8 process on О in 1 mole % P-FC6 H4 OOLÍ6 in water was 0.3% while that of the fluorine produced by the F19 (n, 2n)Fi8 process in the same system was 3. 5%. The authors suggest that the failure of bond rupture may result from (n, 2n) events in which the entering neutron knocks out a neutron in an act which does not contribute momentum to the nucleus as a whole.
Mechanism of re-entry
Following bond rupture in the liquid phase the recoil halogen atom must, as in the gas phase, lose kinetic energy or charge or both before chemical stabilization is possible. In the scavenged gas phase the chances that the recoil atom will have a second encounter with a molecule to which it has lost energy or charge are negligible. Inthe liquid phase caging effects, slowness of diffusion, and the close proximity of solvent molecules exposed to radiolytic decomposition by any Auger electrons from the recoil atom

228 J. E. WILLARD
make it necessary to consider the possibility that the atom will react with a radical or ion resulting from its own energy transfer processes. Because of the variety and complexity of these processes and because of lack of knowledge of actual recoil energies, of the fraction of atoms charged and of the physics and chemistry of charge and energy transfer in condensed organic materials it is impossible to decide from a priori reasoning how recoil atoms enter stable combination in such media.
Certain features of the typical process are, however, known from experiment (for references see [Í, 2, 3, 4, 6] ). Thus it is reasonable to suppose that the hot replacement reactions which have been shown to occur by bimolecular collisions in the gas phase will also occur in solution. They may possibly be more efficient because of the ability of adjacent solvent molecules to remove energy from the energy-rich product molecules and thus lessen the probability of decomposition. It is known that a significant fraction of bromine or iodine atoms activated by nuclear processes in organic liquids enters organic combination by diffusive combination with a radical it has formed in losing its excess energy. This is shown by the effect of low concentrations of scavengers. (Similar scavengeable diffusive reactions have been demonstrated for recoil chlorine atoms in a solvent like CC14 that does not itself act as a scavenger for Cl atoms.) The current focus of attention of work in the liquid state is on the relative roles of displacement and radical formation mechanisms for those combination events which are not sensitive to low concentrations of scavengers, and on the specific mechanisms by which the radicals are formed. A paramount question reflected in the current literature is: does radical formation occur by impact of neutral atoms on molecules of the medium, or by ion molecule reactions, or by neutralization of a solvent envelope that has been charged by charge transfer to the recoil atom following á vacancy cascade in the latter, or by the radiolytic effects of conversion electrons and Auger electrons, or by the billiard ball mechanism of impact with individual atoms? How many radicals are there with which the recoil atom has a chance to react; are these only radicals formed from the last molecule with which the atom collides before stabilization, or is the atom surrounded by many radicals as a result of one or more of the energy loss mechanisms enumerated above? These specific questions have been raised and answers to them actively sought during the last four years in a number of laboratories. Progress has been made both conceptually and experimentally in defining the areas of uncertainty. Conclusive answers to most of the questions still iie in the future.
HARRIS [22] has shown that if the yields of the several organic products produced by the (n, 7 ) activation of bromomethanes in bromine are plotted against Br2 concentration, after normalization by dividing by the mole fraction of CHxBry present, certain products extrapolate to zero yield at 100 mole % Br2 and others show a positive intercept. He reasons that the value at the intercept must indicate a yield of the product formed by hot displacement processes. Thus he is able to divide the processes that occur within the hot spot, and that were previously considered to be indistinguishable, into what he terms "hot" reactions and "hot-spot diffusive" reactions. The yields of the latter decrease linearly to zero with increasing scavenger

NUCLEAR TRANSFORMATIONS OF HALOGENS 229
concentration. The "thermal" category of product formation remains as that portion sensitive to very low scavenger concentrations of the parent product. For CH3 Br, CH2Br2, CHBr3 and С2Н5 ВГ Harris obtains "hot" yields of the parent product of 5. 4, 9. 4, 14. 3 and 3% respectively and "hot spot diffusive" yields of 16, 21, 20 and 9%. For non-parent products the yields from the first three compounds in the series were: "hot", 13, 13 and 14%, and "hot spot diffusive", 2 .5 , 6 and 9%. Other investigations using a series of progressively more highly brominated methanes [26] and ethanes [44] have shown that the organic yields in scavenged solutions increase with increasing bromination of the solvent. Plotting organic yields (normalized on the basis of the fraction of the total bromine atoms in the system that are in the organic bromide) against bromine concentration SHAW and coworkers [44] have found that the yields of the parent molecule in Br2- scavenged solutions are consistent with a billiard-ball hypothesis. This hypothesis assumes that the yield of organically bound recoil atoms is proportional to the fraction of the total bromine atoms in the system that are organically bound. They note that other hypotheses could also explain the results. In the same paper they have investigated the use of HBr as a scavenger in place of Br2 .
In addition to their consideration of experimental results in terms of the billiard-ball hypotheses, as noted above, SHAW and co-workers have analysed the applicability of "thermal spike" theory to liquid systems [45], have considered evidence that the hot-spot reactions in ethyl bromide involve attack on a single molecule 146], and have sought to determine the number ánd source of radicals available for combination with recoil P28 atoms in solutions of organic iodides in organic solvents by correlating product yields with postulated competitive reactions in the diffusive zone 117]. The latter reasoning leads to the conclusion that there are half a dozen solvent radicals present in the "diffusive zone1'. At 10-3 mole fraction of CH3I or C2H5I and 10*5 mole fraction I2 the probability of an activated I128 combining with its parent radical is estimated to be about 2% in QH6, 5% in СбН5 ШЭ2 and 6 % in C6H14. * .
MILMAN [23] has considered whether the observed facts of liquid phase Szilard-Chalmers chemistry can be rationalized by a model that assumes that all (n, y) recoil atoms react as neutral species and that the only collision important in determining their fate is the final reactive collision by which the atom becomes incorporated in a stable molecule or forms a free radical with which it Subsequently combines. Is it, she asks, unnecessary to postulate the possibility of the atom reacting with radicals that it has formed by fragmentation collisions prior to the final thermalizing collision in the course of reducing its energy? She has tabulated the displacement-type radical-formation processes that might be expected, and has analysed the changes in product spectrum that might be expected with increasing scavenger concentration competing for these radicals. Comparing these predictions with published data on C2H5Br [46] and other experimental results, she shows that, within the scope of the evidence considered, the fate of the recoil atoms can be explained without the necessity for reaction of radicals other than those produced by displacement or abstraction.
In other work designed to test the applicability of different hypotheses to liquid phase systems, MILMAN [24a] has determined the organic yield of

230 J. E. WILLARD
bromine for a wide range of concentrations of bromine in benzene, hexane, cyclohexane, tetradecane, and bromoform, and has analysed the data by applying the Estrup-Wolf gang theory developed for gas phase reactions. The predicted organic yield versus scavenger concentration curves for Br2 in benzene, cyclohexane, and tetradecane fit the experimental values well when the energy loss per collision is calculated on the basis of molecular masses rather than atomic masses. In earlier experiments the effect of benzene as a moderator for recoil bromine atoms reacting with С2Н5 ВГ in both liquid and solid systems was investigated [24b] .
The similarity between the relative yields of RI131 products from the gamma-ray radiolysis of I2(Ii3i)-C H 3 I -C 5H12 solutions [47] and those of the R I 1 2 8 products resulting from neutron irradiation of similar solutions [25J has led to the hypothesis that the fate of the I128 atom is usually determined by its reaction with radicals formed in its own solvent envelope by radiolysis by its own internal conversion and Auger electrons. This would explain, for example, the fact that the I128 product spectrum is much the same as that for radiolysis even at I2 scavenger concentrations too high to permit interaction of the I128 with radicals produced by the gamma-ray background radiation. The hypothesis has been called the "Auger electron reaction hypothesis" or "autoradiation hypothesis" 125]. SHAW [17] observed for the (n,7 ) process a rapid increase in labelled RI with increasing RI concentration at low RI concentrations in hydrocarbons. Similar increases have been discussed in terms of the Auger reaction hypothesis [25J. He has also noted [17a] the possibility that neutralization of charged I128 atoms may produce fragments simulating those found in radiation chemistry.
WALTON [48] has discussed the relative merits of different hypotheses for the mechanism of recoil atom reactions and has suggested that the factor determining reaction in many cases may be electronic excitation energy.
WAI, TING and ROWLAND 149] have carried out the first investigation of the steriochemical effects of recoil halogen atom substitution. Following neutron irradiation of the dl form of 2, 3-dichlorobutane they have found the Cl3®-meso/Cl38-dl ratio in the liquid state to be 0 .4 at 20°С, and 0.65 at -56°C, while that in the solid was unity at both -78°C and -100°C, with or without scavenger present. Irradiation of the meso form yielded Cl38-dl/Cl38- meso ratios in the liquid of about 0 .4 at 20°C and 0.5 at -78°C, the ratios in the solid at -114°C and -190°C being about 0 .65 . In the gas phase the 2, 3-dichlorobutane yields were all in the form of the parent diasteriomer but were so low that they may have been due wholly to failure of bond rupture. The results show that, unlike recoil tritium substitution for H in hydrocarbons, recoil Cl38 atom substitution for Cl does not occur with complete retention of configuration. Racemization by inversion in the substitution act or by radical formation and racemization of the radical is favoured by decrease in temperature and perhaps also by conversion to the solid phase. The authors indicate that no change in the sum of the yields of the two diasteriomers occurs with change in temperature and suggest that the change in ratios may be controlled by differences in the activation energy of racemization of radicals formed by the C138 atom and the process of diffusion of such atoms to the radicals.
Other investigations in the liquid state of interest include extensive tests (NESMEYANOV, FILATOV and co-workers) of the influence of substituents

NUCLEAR TRANSFORMATIONS OF HALOGENS 231
on organic yields of Br82, Br80m and Br80 produced by the (n, 7 ) process in compounds in the series CHBr3, CH2Br2, СНгСШг, CHCl2Br, СС1зВг, CCl2 Br2, ChClBra, CH2 BrI, CBr3F and CHBr2F [26]. As noted in a later section, evidence has been found for isotope effects in some of these reactions . The effects of mass and type of substituent on yield have also been analysed and discussed. Other work from the same^laboratory 150J has studied the yields of Br82 in benzene derivatives as a function of their substituents and in acetic acid and haloacetic acids [51, 52J, and has investigated the influence of aniline on the yields of labelled products from (n, y) activated Br in C3H7 Br, CH2Br2 and CHBr3 [53J . Additional discussion of these results is given in [54] .
Also of importance are the investigations of STOCKLIN, SCHMIDT- BLEEK and HERR on the individual product yields of recoil Cl, Br and I from several alkyl iodides with particular reference to the effects of amine scavengers [55].
The first paper [20] published on the chemical reactions of F18 produced by the 0 16(H3, n)F!8 process, using tritium produced in situ by the Li6 (n,a)H3 process, reports organic labelling yields in a series of both aliphatic and aromatic solid lithium salts, and in solutions as a function concentration. Total yields were in the range of 0.1 to 6%, these being increased to as much as 30% in the solid salts when these were annealed before dissolving for analysis.
SVOBODA 156] has found that when CH3I is stirred with 10‘2 M Na2 S03 while irradiating with neutrons at very low y flux the organic yield 1128 from the (n, 7 ) process is 50% whereas similar samples irradiated in the pure state without stirring give yields of 99%. He attributes this to rapid exchange between radioiodine and methyl iodide at very low I2 concentrations, which is precluded when the I2 is continuously extracted during irradiation. Evidence for exchange of iodine between I2 and alkyl iodides at a rate, in moles 1*1 h*1, independent of total iodine concentration, which leads to rapid transfer of radioactivity at very low I2 concentrations, and to a negligible rate of transfer at higher concentrations, has also been given by other authors [57] . .
SOLID PHASE
Studies of the Szilard-Chalmers chemistry of halogens in solid organic compounds have in general suffered from the limitation that compounds with melting points below room temperature have usually been chosen for investigation in order to allow comparison of the results with those obtained for the same substance in earlier liquid state studies. Consequently it has been difficult to carry out annealing studies or to dissolve the crystals at temperatures below the melting point. The studies have shown, (for references see [1, 2, 3] ) that the total organic yields and specific product yields in the splid state are higher than in the liquid in some cases and lower in others, but no systematic understanding of the phenomena has been achieved.

232 J.E. WILLARD
A s te p in th e d ir e c t io n o f a m o r e d e ta ile d r e c o g n it io n and u n d e rsta n d in g o f th e c o m p le x p r o c e s s e s that c o n t r o l o r g a n ic s o l id s ta te hot a to m c h e m is t r y h a s b e e n m a d e in t h r e e p a p e r s 129, 30 J o n th e fa te o f Br82 f r o m th e B r 8 i(n , 7)B r82 p r o c e s s . T h e s e in v e s t ig a t io n s s h o w th a t th e a q u e o u s y i e ld i s a fu n c t io n o f th e d o s e and d o s e r a t e o f g a m m a r a y s to w h ich th e c r y s t a l s a r e e x p o s e d e i t h e r b e f o r e o r a f t e r n e u tr o n ir r a d ia t io n ,a n d to th e t e m p e r a t u r e and t im e o f t h e r m a l a n n e a lin g f o l l o w in g i r r a d ia t io n p r i o r t o a n a ly s i s . A n a ly s is o f a n n e a lin g c u r v e s at d i f f e r e n t t e m p e r a t u r e s f o r d i f f e r e n t s a m p le s e x p o s e d o v e r a w id e ra n g e o f g a m m a d o s e s r e v e a ls a c o m p le x p a tte rn , w h ich i s d i s c u s s e d in t e r m s o f t r a p p e d d e f e c t s h a v in g a v a r i e t y o f a c t iv a t io n e n e r g ie s f o r r e l e a s e , and th e in te r a c t io n o f th e r e le a s e d d e le c t s w ith th e r e c o i l a to m s . T h e r e s u l t s in d ic a te th at f o l lo w in g th e n u c le a r e v e n t b y w h ich th e y a r e p r o d u c e d a s u b s ta n tia l f r a c t io n o f th e Br82 a to m s a r e le f t in a c h e m ic a l ly m e t a s t a b le s t a t e . If th e s o l i d i s d i s s o l v e d in an o r g a n i c s o lv e n t w ith o u t p r i o r a n n e a lin g a t r o o m t e m p e r a t u r e , and e x t r a c t e d w ith a q u e o u s r e d u c in g a g e n t, t h e s e a t o m s a p p e a r in th e a q u e o u s p h a s e . If, h o w e v e r , th e c r y s t a l s a r e t h e r m a l ly a n n e a le d p r i o r t o d is s o lu t io n , e v e n ts o c c u r th a t r e t u r n th e Br82 t o s ta b le o r g a n ic c o m b in a t io n . M a n y o f th e s e e v e n ts r e q u ir e th a t g a m m a - p r o d u c e d d e f e c t s in f lu e n c e th e fa te o f Br82 a to m s s itu a te d m a n y m o le c u la r d ia m e t e r s a w a y (p r e s u m a b ly b y e n e r g y , c h a r g e o r a to m t r a n s f e r ) . O th e rs s h o w an in d e p e n d e n c e o f g a m m a d o s e in d ic a t in g th a t th e s t a b i l i z a t io n in to o r g a n i c c o m b in a t io n o c c u r s a s an i n t r a - s i t e p r o c e s s . In v ie w o f th e d i s c o v e r y th a t r a d ia t iv e n e u tr o n c a p t u r e p r o d u c e s Br822 p r e d o m in a n t ly in th e Br82m e x c i t e d s ta te th a t d e c a y s to B r82 b y i s o m e r i c t r a n s i t io n [35, 36] th e m e t a s ta b le B r82 a to m s o b s e r v e d in th e a b o v e С г В г е s tu d ie s m u st h av e b e e n p r o d u c e d b y i s o m e r i c t r a n s i t io n r a t h e r th a n d i r e c t l y b y th e (n , 7 ) p r o c e s s . S u ch a to m s m a y , h o w e v e r , r e s id e in , o r c l o s e t o , a d e fe c t t r a c k p r o d u c e d w h en th e a to m l o s t it s e n e r g y and a n y c h a r g e r e s u l t in g f r o m th e (n , 7 ) p r o c e s s .
I n c id e n t a l t o t h e i r p u r p o s e o f u n d e r s t a n d in g s o l id s ta t e h a lo g e n a to m r e c o i l c h e m is t r y th e C 2 B rg in v e s t ig a t io n s h a v e s u g g e s t e d a n ew m e th o d o f s tu d y in g g a m m a -in d u c e d d e fe c t s in o r g a n ic h a lid e s . T h u s , i f С гВ г6 c r y s t a ls th a t h a v e b e e n s u b je c t e d to g a m m a i r r a d ia t i o n a r e t h e r m a l ly a n n e a le d at d i f f e r e n t t e m p e r a t u r e s and f o r d i f f e r e n t t i m e s , fo l lo w e d b y n e u tro n i r r a d i a t io n an d a s ta n d a r d t h e r m a l a n n e a lin g , th e a q u e o u s y i e l d s s e r v e a s a m e a s u r e o f th e ex ten t to w h ich th e g a m m a d e fe c t s in tro d u ce d p r i o r to n eu tron i r r a d i a t i o n w e r e r e m o v e d b y th e a n n e a lin g p r i o r t o n e u t r o n i r r a d i a t i o n .
Studies of the chemical fate of B r 8 0 atonis formed from the B r 8 0 m isomeric transition in glassy and poly crystalline П-С3 Н7ВГ and П-С4Н9ВГ [28] give organic yields that are essentially independent of homogeneously dispersed Br2 scavenger concentration, indicating that the fate of the B r 8 0 is determined within a few molecular diameters of the site of formation. When the B r 8 0 m is in B r B r 8 0 m the organic yields are higher in the glassy than in the polycrystalline state. In both the glassy and polycrystalline states the organic yields are much higher when the B r 8 0 m is present in С 4 Н 9 В Г 8ОП) than when it is in B r B r 8 0 m . These results suggest caging with the parent fragment although earlier [58j work showing that the "hot" organic yields from B r B r 8 0 m in the liquid state increase with increasing chain length of pure hydrocarbon solvent cannot be rationalized by caging with the parent fragment.

NUCLEAR TRANSFORMATIONS OF HALOGENS 233
Two laboratories have observed a striking increase in organic yield of bromine in a solid hydrocarbon as a result of addition of a low concentration of alkyl bromide. In one case [24b] low mole fractions of С2Н5ВГ in benzene containing 3X IO-3 mole fraction Вгг at 77°K raised the organic yield of the Br79(n,y)Br80 process sharply. In the other [28] it was observed that the organic yield of the isomeric transition from BrBr80m was ~ 5% in n-CgHi4 containing 10"'¿ mole fraction Br2 and 10‘ 3 mole fraction П-С3Н7ВГ, but rose to 20% when the concentrations were 10"2 mole fraction Br2 and 10-2 mole fraction П-С3 Н 7ВГ, both at 77°K and 150°K. Product analysis showed that the increase in yield was not predominantly as С3Н7ВГ but spread over many products. The effect has not been satisfactorily explained.
MILMAN [27] has noted a correspondence between abrupt changes in the organic yield of the Br79(n, Y)Br80 process in 1, 2 -С 2Н4 ВГ5, which occur at the melting and at a solid state transition, and changes in heat capacity that occur at the same temperatures. She notes similar abrupt changes in organic yield at the melting point and each of two solid state transitions in СС1зВг, and a correspondence of these to changes in the dielectric constant at the same temperatures.
Other significant recent observations on solid state halogen recoil reactions include the increase in organic yield of F1® from the 0 16(H3, n)F18 process on annealing solid neutron irradiated lithium salts of organic compounds 120], and the fact that enhanced racemization of the Cl38 products of neutron irradiated meso and dl 2 , ЗС4 Н 8С1г is observed in the solid state [49]..
It appears clear that a sound understanding of the mechanisms of solid state Szilard-Chalmers reactions requires an understanding of the mechanisms of solid state radiation chemistry. Important advantages are currently being made in knowledge' of the differences in behaviour between glassy and crystalline systems, of exciton and electron transfer, and of the annealing behaviour of the variety of trapped species that may be observed in gamma- irradiated solid alkyl halides by their optical and esr spectra. References to some of these have been given [28].
ISOTOPE EFFECTS
Nuclides of a given element activated by drastically different nuclear processes usually (though not always) give both the same total organic yield and same distribution of products (for a summary of such studies see [1 ]). An interesting exception to this pattern has been reported by NESMEYANOV et al. 126] who find higher organic yields for Br82than for B r 8 0 m , over a wide range of scavenger concentrations, for reactions with liquid halomethanes containing two different halogens in the same molecule. The effect is not significant for compounds containing only one species of halogen or in the solid state. In view of the discovery that radiative neutron capture by B r 8 i
produces predominantly Br82m (6 'min) that decays by internal conversion to B r 8 2 it seems probable that the process responsible for the fact that higher organic yields are observed for Br«n than for Br»u™ in the mixed halomethanes is the B r 8 2 m isomeric transition rather than the B r 8 l ( n , y ) B r 8 2 process. If

234 J.E. WILLARD
this is the case, it is consistent with the fact that higher organic yields have been observed for the B r 8 0 m - > B r 8 0 isomeric transition than for the Br79(n, ■yjBrBOm process in СС1зВг and CCI4 [59, 61] . Differences in yields for B r 8 2 and B r 8 0 m have also been reported for inorganic systems [60] .
Very recently GILROY, MILLER and SHAW [62] have reported the ratio of Br80m to Br82 yields for each of seven products formed as a consequence of neutron irradiation of C2H5 Br. The ratios determined at each of five bromine concentrations from 10-3 to 0.75 mole fraction show isotope effects that vary from 0.45 to 3.05 depending on the chemical species and the bromine concentration, although the ratios of the total organic yields for the two isotopes are close to unity at all the bromine concentrations tested.
The experiments of ANBAR and NETA [18] comparing the organic yields of the F19 (n, 2n)F!8 process with those of the 0 16(H3 ,n)Fi8 process indicate that when F18 is produced by the (n, 2n) process in CH2FCOOLÍ, the organic yield is 1 0% while if produced by the H3 reaction it is about 2%.
R E FE R E N C E S
[1] WILLARD, J .E ., in Chemical Effects o f Nuclear Transformations H IAEA, Vienna (1961) 216-27.[2 ] WILLARD, J .E ., Annu. Rev. nucl. Sci. Ш (1953) 193; Annu. Rev. phys. Chem. 6 (1955) 141.[3] SIUDA, A . , Chemical Effects o f Nuclear Transformations, Polish Atomic Energy Commission, Warsaw
(1963).[4] CAMPBELL, I. G . , in Advances in Inorganic and Radiochemistry V Academic Press, New York (1963)
135-214.[5] WEXLER, S . , in Actions Chimiques et Biologiques des Radiations VIII (HA 1SSINSKSY, M .. Ed. )MASSON
et Cie, Paris (in press).[6] WILLARD, J.E .. Nucleonics 19(1961) 61.[7] WOLFGANG, R ., in Progress in Reaction Kinetics (G. PORTER, Ed.) Pergamon Press, New York(in press) L8J WEXLER, S., J. chem. Phys. 36(1962) 1991!.[9] CARLSON, T .A . and WHITE. R. M .. J. chem. Phys. 38 (1963) 2930. .
[10] CARLSON, T .A . and WHITE. R. M ., J. Chem. Phys. 36 (1962) 2883 ; 38 (1963) 2075.[11] THOMPSON, J.L. and MILLER, W. W. , J. chem. Phys. 38 (1963) 2477.[12] HSUING, C. and GORDUS, A .A ., J. chem. Phys. 36 (1962) 947.[13] GORDUS, A. A. and HSUING, C . , J. chem. Phys. 36 (1962) 954.[14] IYER, R.M. and MARTIN, G . , in Chemical Effects of Nuclear Transformations И IAEA, Vienna(1960)
281.[15] CHANG, H .M . and WILLARD, J.E. , unpublished.[16] BRENS IKE, J .F ., W1LKEY, D.D. and WILLARD. J .E ., unpublished.[17] (a ) SHAW, P .F .D .. Radiochim. Acta£(1963) 76;
(b) McCRAE, J.E.C. and SHAW, P.F.D . , J. inorg. nucl. Chem. 24(1962) 1337.[18] ANBAR, M. and NETA, P ., J. chem. Phys. £7(1962) 2557.[19] RACK. E.P. and GORDUS, A .A . , J. chem. Phys. (a) 34 (1961) 1855; (b) 36 (1962) 287; J. phys. Chem.
65 (1961) 944.[20] ANBAR, M. and NETA, P . . J. Amer. chem. Soc. 84 (1962) 2673.[21] COLEBOURNE, N. and WOLFGANG, R. , J. chem. Phys. 38 (1963) 2782.[22] HARRIS, W.E. (a) in Chemical Effects of Nuclear Transformations H IAEA, Vienna (1960) 229;
(b) Cañad. J. Chem. 39 (1961) 121.[23] MILMAN, М ., Radiochim. Acta 2 (1962) 15.[24] MILMAN, M. (a)Radiochim. Acta2(1964) 181; (b) J. phys. Chem. 67 (1963) 537.
[25] GEISSLER, P.R. and WILLARD, J.E ., J. phys. Chem. 67 (1963) 1675.

NUCLEAR TRANSFORMATIONS OF HALOGENS 235
[26] NESMEYANOV. A .N . , FILATOV. E .S .. BORISOV. E. A. and SHUKLA, B. M . . in Chemical Effects of Nuclear Transformations И IAEA. Vienna (1960) 259.
[27] MILMAN, M . , J. Amer. chem. Soc. 86 (1964) 3567.
[28] HAHNE. R. M .A . and WILLARD. I .E .. J. phys. Chem. 68 (1964) 2582.129) COLLINS. K.E. and WILLARD. J .E .. J. chem. Phys. 37 (1962) 1908.[30] COLLINS. K.E. and HARBOTTLE. G. (a) Radiochim. Acta 3 (1964) 21; (b) Radiochim. Acta3(1964)29.[31] WEXLER. S. and DAVIES. Т .Н . . J. chem. Phys. 20 (1952) 1688.[32] YOSIM, S. and DAVIES, Т .Н ., I. phys. Chem. 56 (1952) 599.
[33] GROSHEV. L .V . , LUTSENKO. V .N . . DEMIDOV, A .V . and PELEKOV, V. I . . Atlas of Gam m a-Ray Spectra ftom Radiative Capture o f Thermal Neutrons, Pergamon Press, London (1959).
[34] SNELL. A. H. and PLEASANTON. F . . Phys. Rev. 100 (1955) 1396.[35] ANDERS, O .U ., Paper 25, Division o f Nuclear Chemistry and Technology, 148th National Meeting
of American Chemical Society, Chicago (Sept. 1964).136] EMERY, J. F . , Paper 26, Division o f Nuclear Chemistry and Technology, 148th National Meeting of
the American Chemical Society, Chicago (Sept. 1964).[37] HSIUNG, C „ HSIUNG, H. and GORDUS. A . A., J. chem. Phys. 34 (1961) 535.[38] CROSS. R.J. and WOLFGANG, R ., Radiochim. Acta J. (1962) 42.[39] HORNIG, J.F. , LEVEY, G. and WILLARD, I.E . . I. chero. Phys. 20 (1952) 1556.[40] DENSCHLAG, H .O .. HENZEL. N. and HERRMANN, G . , Radiochim. Acta j. (1963) 172.[41] PARSS, Y . and AMIEL, S . . J. Amer. chem. Soc. 86 (1964) 233.[42] GORDUS, A . A . and WILLARD, J.E. , J. Amer. chem. Soc. 47 (1957) 4609.[43] STURM, J.E. and DAVIS, D .G . , Paper 106, Division of Physical Chemistry, 140th National Meeting
o f the American Chemical Society (Sept. 1961).
[44] MALLINSON, J. H .. MILLER, G.E. and SHAW. P .F .D .. Radiochim. Acta 1 (1963) 137.[45] SHAW, P .F .D .. Radiochim. Acta 1 (1963) 177.[46] KNIGHT, B .. MILLER, G.E. and SHAW, P .F .D .. J. inorg. nucl. Chem. 23 (1961) 15.
[47] GEISSLER, P.R. and WILLARD, J.E. . J. Amer. chem. Soc. 84 (1962) 4627.[48] WALTON. G. N ., Radiochim. Acta 2 (1964) 108.[49] WAI, C .M ., TING. С. T . and ROWLAND. F .S .. J. Amer. chem. Soc. 86 (1964) 2525.[50] NESMEYANOV. A .N . , FILATOV. E.S. and MANSFIELD. A . , Radiohimija 3 (1961) 610.[51] NESMEYANOV, A .N . . FILATOV. E.S. and GUSAKOVSKAYA, J .G .. Radiohimija 4 (1962)462.[52] NESMEYANOV. A .N . and FILATOV, G .E .. Radiohimija 4 (1962) 613.[53] NESMEYANOV, A .N . and FILATOV. G .E .. Radiohimija 5 (1963) 378.[54] FILATOV. G .E ., Uspehi Him 31 (1962) 752.[55] STÜCKLIN, G . . SCHMIDT-BLEEK and HERR, W ., in Chemical Effects of Nuclear Transformations
П IAEA, Vienna (1961) 245.[56] SVOBODA, K . , Nature. Lond. 198 (1963) 986.[57] (a ) BEHRENS. H. and MADDOCK. A .G ., J. chem. Phys. 22 (1954) 1139; (b ) COHEN, E. and
TRUMBORE, C . , Paper 97, Division o f Physical Chemistry, 148th National Meeting of the American Chemical Society, Chicago (Sept. 1964); (c) KLASSEN, H ., private communication.
[58] AD ITYA , S. and WILLARD, J .E ., J. Amer. chem. Soc. 79 (1957) 3367.[59] (a ) GOLDHABER, S . , CHIANG, R.S.H. and WILLARD, J.E. , J. Amer. chem. Soc. 73 (1951) 2271;
(b ) HORNIG, J. and WILLARD. J.E. , J. Amer. chem. Soc. J75 (1953 ) 461.[60] HARBOTTLE, G. and SUTIN, N ., Advances in Inorganic Chemistry and Radiochemistry I(EMELEUS, H.J.
and SHARP, A .G ., Eds.) Academic Press, New York(1959) 268.[61] HORNIG. J.F. and WILLARD, J.E ., J. Amer. chem. Soc. 75 (1963) 461.[62] GILROY. T .E ., MILLER, G. and SHAW, P .F .D ., J. Amer. chem. Soc., 86 (1964) 5033.

236 J. E. WILLARD
D I S C U S S I O N *
G . N . W A L T O N : A t H a r w e ll w e h a v e m e a s u r e d th e o r g a n ic r e te n t io n o f io d in e in the th e r m a l n e u tro n ir r a d ia t io n o f p h en y l io d id e b y c a r r y in g out the r e a c t io n in the p r e s e n c e o f io d in e la b e lle d w ith I131, s o that the b eh a v iou r o f the I 131 and I 128 w a s o b s e r v e d s im u lta n e o u s ly . T o s e p a r a te the r a d io ly t ic e f fe c t s , w e o b s e r v e d the d e c r e a s e in the re te n t io n w hen the s y s te m w as s u r ro u n d e d w ith a c a d m iu m b o x . U n d e r th e s e c o n d it io n s th e r e i s n o (n, 7 ) r e a c t io n in the p h e n y l io d id e , but th e s y s te m is s u b je c t to g a m m a ir r a d ia t io n f r o m th e (n, 7 ) p r o c e s s e s in th e s u r r o u n d in g c a d m iu m . T h e g a m m a -e f f e c t h a s a ls o b e e n m e a s u r e d b y i r r a d ia t in g th e s y s t e m w ith g a m m a r a y s f r o m c o b a l t - 6 0 .
T h e r e te n t io n on n e u tro n ir r a d ia t io n w a s found to b e o f the o r d e r o f 10 5
a to m s o f io d in e r e ta in e d f o r e v e r y (n, 7 ) e v e n t (I128 a to m f o r m e d ) a ft e r a l lo w a n c e h ad b e e n m a d e f o r th e r a d io ly t i c r e t e n t io n . T h is r e t e n t io n i s t o o g r e a t to b e e a s i l y a ttr ib u te d to th e r e c o i l e n e r g y o f th e I 128 a to m , w h ich is a b ou t 10 0 e V . W e c o n s i d e r th a t th is e v id e n c e s t r o n g ly s u p p o r t s th e v ie w that a la r g e fr a c t io n o f the 6 M eV tr a n s fo r m a t io n e n e r g y m u st g o , as it w e re , in b u r s t in g the b r e a c h , i . e . in e le c t r o n ic e x c ita tio n o f the bonds o f su rro u n d in g m o le c u le s by , f o r in s ta n ce , in te rn a l c o n v e r s io n and C o u lo m b ic r e p u ls io n c o m p a r a b le to that d e s c r ib e d b y C a r ls o n and W h ite * * .
R . H . H E R B E R : P r o f e s s o r W i l l a r d 's c o m m e n t s c o n c e r n i n g th e i m p o r ta n c e o f in te r v a l c o n v e r s io n in d e c id in g the f in a l c h e m ic a l fate o f an (n, 7 ) p r o d u c t a r e , I th in k , v e r y im p o r ta n t and s u c h p r o c e s s e s a r e e x p e c t e d to h a v e a m a jo r in f lu e n c e on th e s p e c t r u m o f th e c h e m ic a l p r o d u c t s th at a r e o b s e r v e d . In th is c o n n e c t io n I s h o u ld l ik e to a sk i f t h e r e i s e v id e n c e th at in te r n a l c o n v e r s io n o c c u r s a ft e r r e c o i l , s in c e n u c le a r r e la x a t io n t im e s a r e t y p ic a l ly 1 0 -1 7 to 1 0 ' 23 s , e l e c t r o n i c r e la x a t io n t im e s ~ 1 0 ‘ 12 s and t r a n s p o r t (m o le c u la r r e la x a t io n t im e s ) ~ 1 0 " 6 s . I sh ou ld a ls o l ik e to k n ow w hat i s th e s p e c i f i c r o l e o f in te r n a l c o n v e r s io n in th e d a ta w e h e a r d in w h ich the r e a c t io n C H 3 I -» C H 3. + Г w a s f o l lo w e d b y I131 - s c a v e n g in g o f th e m e th y l r a d i c a l s .
I s h o u ld a ls o b e g r a t e fu l i f P r o f e s s o r W il la r d w o u ld c o m m e n t o n th e in te r n a l c o n v e r s i o n c o e f f i c i e n t o f th e 6 - m in B r 82m th at h a s r e c e n t ly b e e n found, o r p e rh a p s g iv e u s s o m e id ea o f the d e c a y s ch e m e ( i . e . sp in and p a r i ty as w e ll a s e n e r g y ch a n g e s ) in th is tra n s it io n .
J . W IL L A R D : In a w id e v a r ie t y o f n u c l id e s s tu d ie d it h a s b e e n sh ow n that the e x c ita t io n e n e r g y g iv e n to th e n u c le u s b y the r a d ia t iv e n eu tron c a p t u r e p r o c e s s i s n o r m a l ly l o s t b y g a m m a - r a y c a s c a d e s , e a c h in v o lv in g a n u m b e r o f g a m m a r a y s , in c lu d in g s o m e in th e e n e r g y ra n g e o f 1 to l OOke V. R e la t iv e ly l o n g - l iv e d m e ta s ta b le s ta te s a re c o m m o n in the c a s e o f tra n s ition s in th is e n e r g y r a n g e , an d t h e i r d e c a y i s v e r y c o m m o n ly a c c o m p a n ie d b y in te r n a l c o n v e r s io n and a v a c a n c y c a s c a d e o f A u g e r e le c t r o n s . I f s u ch an even t la s ts as lo n g a s , sa y , 1 0 " 9 s a ft e r r e c o i l f r o m the in itia l, h igh ly e n e r g e t ic g a m m a r a y , the la b e l le d a to m w il l h ave had t im e to e n te r in to a s ta b le
* See also these Proceedings, discussion following paper SM-57/21. . .* * CARLSON. T. A. and WHITE, R. M ., "Explosion" o f multicharged molecular ions: chemical
consequences o f inner shell vacancies in atoms” , these Proceedings I.

NUCLEAR TRANSFORMATIONS OF HALOGENS 237
combination prior to the charging process. Whether or not it has done so, it will be immediately surrounded by a relatively high concentration of ions, electrons, free radicals and excited molecules with a spectrum of species similar to that which would be produced by gamma irradiation of the bulk of the solution. Since the low-energy Auger electrons have a high linear energy transfer, the concentration of such species in the solvent envelope immediately round the labelled atom will be much higher than that produced in the bulk of the solution by the background gamma radiation. As a result, the labelled atom will have an opportunity to react with one of these species without appreciable interference from normal concentrations of scavenger. As observed, the distribution of labelled products formed would be expected to be similar to the distribution of I131 -labelled products when the same system (say 1 mole % CH3I, 0. 25 mole % I2 (I131)) in hexane is irradiated with gamma rays. In the recent experiments cited it was observed that neutron irradiation of aqueous solutions of 10"2 mole fraction CH3I127 with lower concentrations of I129, present as I 29, (IJ29)- or (I129)- , gave organic yields of about 10% for the H30 formed by the I129 (n, y)I130 process. The autoradiation hypothesis is a plausible explanation of the ability of the recoil atom to enter into an organic combination with the dilute organic solute in such a scavenged non-organic system. Thus either the electrons or the H atoms produced by the radiolysis of the solvent envelope round the atom may react with the CH3I to produce methyl radicals with which the I13U may combine before it encounters scavenger from the bulk of the medium ¡
Experimental evidence that charging of recoil atoms from the (n, y) process occurs after recoil in indium, gold, manganese and dysprosium has been presented in the work of Yosim and Davies and of Thompson and Miller, who have surrounded cylindrical layers of these elements or their compounds with catcher foils while neutron-irradiating them. By studying the effect of applied fields they showed that at least 40 - 60% of the recoiling atoms were charged. They reasoned that the charging must occur after escape of the recoil atom from the surface since, if it occurred before, neutralization by the electrons in the surface would be faster than the movement of the atom away from the surface. '
Your last point is dealt with in Reference [36] to my paper; Emery found в к for Br82m to be 268, indicating internal conversion in nearly every event.
R.H. HERBER: Professor Willard's comments were most enlightening, but I should like to return briefly to the first part of the question since I think the lifetime of the Auger process is intimately connected with the recoil atom chemical product spectrum. The initial neutron capture leaves the compound nucleus with ~7 MeV of excitation. This excitation energy is lost in a cascade involving a variety of intermediate nuclear states with vastly differing lifetimes, spin and parity changes, and I therefore think it dangerous to extrapolate the results of the foil experiments in heavy atoms to a general case for the halogens. My feeling is that the competition between electron shake-off due to the Auger sequence, and translation of the recoiling atom through the medium must be examined for each nuclide in question in terms of the information that is known about the accessible nuclear levels. Moreover, the study of the relationship between the sequence

238 J.E. WILLARD
of the above events and the observation of positively charged ions in the foil experiments requires a detailed knowledge of the mechanism of recoil from the lattice, which I think is not very well understood. Clearly, however, Professor Willard's comments have postulated an interesting and most important mechanism in these processes.
J. WILLARD: It has been demonstrated experimentally that both bromine and iodine atoms*, as well as the elements used in the catcher experiments referred to, may be charged as a consequence of radiative neutron capture, that gamma cascades involving low-lying levels are a common mode of deexcitation following radiative neutron capturet, and that metastable states with lifetimes longer than that required for the recoil atom to lose its kinetic energy in condensed media frequently occur+. Therefore the evidence seems to require consideration of the post-recoil charging phenomena as a factor that may often play a part in determining the fate of the recoil atom.
J.I. VARGAS: The formation of F centres in KBr has been investigated in our laboratory by letting the Br80m activity decay in pre-annealed crystals. The observed F-centre formation proceeds with 4.4 h half-growth time. Although self-irradiation may contribute to the formation of the observed F-centres, the use of thin crystals might perhaps permit the observation of the B r8 2 m isomeric transitions.
Could Professor Willard explain in greater detail the annealing experiments done by Claridge in his laboratory?
J. WILLARD: The optical spectrum of gamma-irradiated C2 H5I glass at 77 °K and its change with time, as well as the ESR spectra of gamma- irradiated glassy and polycrystalline C2 H5I and the curves demonstrating that the rate of disappearance of gamma-induced radicals is accelerated by gamma irradiation, were all shown to illustrate the variety of processes that must be considered in seeking to explain the phenomena of the radiation chemistry - and, hence, the "hot-atom" chemistry - of organic solids. The annealing observed occurred in the dark and was accelerated by illumination.
J.I. VARGAS: Am I correct in assuming that no monochromatic light annealing of recoil atoms was observed?
J. WILLARD: We have not done any annealing of recoil atoms with monochromatic light. Such annealing of the optical and ESR peaks of gamma- irradiated alkyl iodides was done with a view to its application in recoil atom studies.
* Reference [31] in SM-57/92 t Reference [5] in SM-57/92
* Ibid.

ХИМИЧЕСКИЕ ЭФФЕКТЫ ЯДЕРНЫХ ПРЕВРАЩЕНИЙ И ПРОЦЕССЫ ПЕРЕДАЧИ ЭНЕРГИИ ВОЗБУЖДЕНИЯ
Ан.Н. НЕСМЕЯНОВ и Э .С . ФИЛАТОВ МОСКОВСКИЙ ГОСУДАРСТВЕННЫЙ УНИВЕРСИТЕТ, МОСКВА
СССР
Abstract — Résumé — Аннотация — Resumen
CHEMICAL EFFECTS OF NUCLEAR TRANSFORMATIONS AND ENERGY TRANSFER PROCESSES. As a result o f the interaction of hot atoms with molecules, excited molecules containing a hot atom are formed. The molecules become stabilized through a dissipation of energy resulting from a transfer of energy from the site of the collision of the hot atom with the molecule, either intramolecularly or intermolecularly. In certain
cases the excited molecule enters into chemical reactions with surrounding molecules.Radiation chemical transformations have shown the possibility of the dissipation of excitation energy
in binary mixtures. Excitation-energy transfer phenomena of this kind were found by the authors in a number of binary systems of organic compounds, on the example of the reactions of recoil atoms of tritium and bromine.
The problem of an unambiguous interpretation of the results obtained is discussed in the light of the lack
of knowledge concerning any o f the parameters for the process of interaction of hot atoms with molecules in a binasy m\xt\ne that has different ïeaction cross-sections for the components, the probability values for impact reactions, the change in the logarithmic decrement of extinction and the energy o f the liquid cage in response to a change in the composition of the system.
The paper also discusses the intramolecular transfer o f excitation energy in the reactions o f tritium recoil atoms with a number o f organic compounds.
EFFETS CHIMIQUES DES TRANSFORMATIONS NUCLÉAIRES ET PROCESSUS-DE TRANSFERT D'ÉNERGIE. L'interaction entre atomes chauds et molécules entraîne la formation de molécules excitées comportant un atome chaud. Les molécules se stabilisent par suite d’un transfert d’ énergie intramoléculaire ou intermoléculaire à partir du point d'impact. Dans certains cas, la molécule excitée entre en réaction chimique avec les molécules environnantes.
Les transformations chimiques radioinduites ont révélé la possibilité d'une diffusion de l ’énergie d’ excitation dans les mélanges binaires. L'auteur a observé dans plusieurs mélanges binaires de composés organiques des phénomènes de transfert de l'énergie d'excitation analogues à ceux qui se produisent dans les réactions entre des tritons de recul et du brome.
И examine dans quelle mesure on donne une interprétation sûre aux résultats obtenus, étant donné que l'on ne connaît pas tous les paramètres du processus d'interaction des àtomés chauds et des molécules dans un mélange binaire dont les composants ont des sections efficaces de réaction différentes, et que l'on ignore la probabilité d’une réaction par choc ainsi que les modifications que subissent le décrément logarithmique d’ amortissement et l ’ énergie de la cellule liquide lorsqu'on modifie la composition du mélange.
L’auteur examine également le transfert intramoléculaire de l'énergie d’ excitation lors des réactions provoquées par les atomes chauds de tritium avec plusieurs composés organiques.
ХИМ ИЧЕСКИЕ ЭФФЕКТЫ ЯДЕРНЫ Х ПРЕВРАЩ ЕНИЙ И ПРОЦЕССЫ ПЕРЕДАЧИ ВОЗБУЖ ДЕНИЯ ЭН ЕРГИ И . В результате взаимодействия горячих атомов с молекулами образуются возбужденные молекулы, включающие горячий атом.
Стабилизация молекул происходит путем рассеяния энергии в результате передачи энергии от лок альн ого м еста соударения гор ячего атома с м олекулой внутримолекулярно или межмОЛекулярно. В отдельных случаях возбужденная молекула вступает в химические реакции с окружающими молекулами. .
Радиационно химические превращения показали возможность рассеяния энергии возбуждения в бинарных см есях .
Подобные явления передачи энергии возбуждения обнаружены автором в ряде бинарных си стем ор ганических соединений на прим ере реакций а том о в отдачи трития и б р о м а .
239

240 A h . H. НЕСМЕЯНОВ и Э . С . ФИЛАТОВ
Обсуждается вопрос однозначности трактовки полученных результатов в связи с отсутствием знания всех параметров процесса взаимодействия горячих атомов с м олекулами в бинарной см еси , имеющей различные сечения реакции компонентов, величины вероятности реакции на удар, изменение логарифмического декремента затухания и энергии жидкой клетки с изменением состава системы.
В докладе рассматривается также внутримолекулярная передача энергии возбуждения при реакциях атомов отдачи трития с рядом органических соединений.
EFECTOS QUIMICOS DE LAS TRANSFORMACIONES NUCLEARES Y PROCESOS DE TRANSFERENCIA DE ENERGIA. Como resultado de la interacción de átomos calientes con moléculas, se forman moléculas excitadas que contienen un átomo caliente. Estas moléculas se estabilizan por transferencia intra o intermolecular
de la energía desde el lugar de impacto. En ciertos casos, la molécula excitada reacciona químicamente con las moléculas circundantes.
Las transformaciones químicas radioinducidas han demostrado la posibilidad de gue se produzca una disipación de la energía de excitación en mezclas binarias. El autor ha observado en. diversas mezclas binarias de compuestos orgánicos fenómenos de transferencia de energía de excitación análogos a los que se registran en las reacciones entre tritones de retroceso y bromo.
En la memoria, estudia en qué medida pueden interpretarse con seguridad los resultados obtenidos, dado que no se conocen todos los parámetros del proceso de interacción de los átomos calientes y las moléculas en una mezcla binaria, cuyos componentes poseen diferentes secciones eficaces de reacción, y que se ignora la probabilidad de que se produzca una reacción por choque, así como las modificaciones que experimentan el decremento logarítmico de amortiguamiento y la energía de la celda líquida cuando se altera la composición de la mezcla.
En lá memoria se estudia asimismo la transferencia intramolecular de la energía de excitación en el caso de reacciones provocadas por átomos calientes de tritio con toda una serie de compuestos orgánicos.
Горячие атомы, получающиеся в результате большинства ядерных превращений, имеют столь высокую кинетическую энергию, что в начале пути химически не взаимодействуют с окружающими молекулами, а их действие, подобно ядерным излучениям, сводится к ионизации атомов и молекул среды. В результате этого процесса энергия горячих атомов снижается до величин, при которых начинают устанавливаться химические связи — идут процессы замещения, присоединения и т.п . — химические реакции. Энергия атомов отдачи после (п, 7 ) реакции лежит в области прямого химического взаимодействия.
Как было установлено в ряде работ, взаимодействие тяжелых атомов отдачи с атомами близких масс удовлетворительно описывается механизмом упругих [1—7] и неупругих [7 — 10] соударений, а для действия горячих атомов трития хорошее согласие опыта с теорией получается при применении модели ударов Вольфганга [11,12].
Даже при применении механизма упругих соударений трудно себе представить, что после замещения какого-либо атома или группы атомов на атом отдачи вновь образованная молекула будет находиться в нормальном энергетическом состоянии. Несомненно, что в большинстве случаев молекулы, включающие горячий атом, будут возбуждены. Энергия возбуждения может затрачиваться на химические процессы, в результате которых изменяется состав продуктов, включающих атомы отдачи, удержание в форме материнской молекулы уменьшается, или энергия рассеивается при взаимодействии с молекулами среды и внутримолекулярно.

ХИМИЧЕСКИЕ ЭФФЕКТЫ ЯДЕРНЫХ ПРЕВРАЩЕНИЙ 241
Такая концепция приводит к выводу о том, что удельная активность должна увеличиваться в присутствии веществ или атомных групп в молекулах, способных рассеивать энергии возбуждения.
Радиационно-химические исследования показали, что радиолиз веществ под действием ядерных излучений в присутствии бензола,, имеющего сопряженные связи, резко снижается [13]. Наблюдается отклонение изменения выхода продуктов радиолиза (водорода) от линейного с изменением состава смеси.
Для объяснения этого явления были высказаны несколько точек зрения. Первая точка зрения, Бартона [14,15], связывает снижение водорода с меж- молекулярной передачей энергии от молекулы, подвергшейся воздействию излучения, к молекулам окружающей среды. Для передачи энергии необходимо вещество,"ароматичное" по природе [16]. В противоположность первой точке зрения, модель Своллоу-Инокути [17] предполагает, что отклонение от аддитивности связано не с передачей энергии, а с различием в сечении возбуждения молекул компонентов смеси ионизирующим излучением, т .е . избирательным поглощением энергии. Радиолиз бинарной смеси в газовой фазе показал отличие в закономерностях, обнаруженных для конденсированных систем. В связи с этим было высказано сомнение в справедливости гипотезы Своллоу-Инокути [18].
Третья точка зрения [19] объясняет изменение выхода водорода при радиолизе поглощением атомов водорода молекулами бензола. Однако ей противоречит нелинейный выход других продуктов радиолиза. Поэтому более целесообразным является подход, учитывающий как поглощение атомов водорода при малых добавках акцептора, так и передачу энергии, которая становится заметной при больших концентрациях (>5 молярных процентов) [20]. Поскольку передача энергии предполагает существование долгоживущих возбужденных состояний, что спектроскопически не доказано, некоторые авторы проводят мысль об образовании при радиолизе горячих атомов водорода, способных вступать в реакции отщепления водорода [21—25] или галоида [26]. Из работ по радиолизу бинарных смесей веществ следует вывод о том, что передача энергии, видимо, играет существенную роль в процессе стабилизации возбужденных молекул. Предложено несколько схем такого процесса [27]. Большие отклонения от аддитивности продуктов радиолиза (заметные уже при добавках акцептора меньше 0,01 М) были обнаружены в тех случаях, когда донор и акцептор энергии — ароматические соединения, и объяснены радиационной сенсибилизацией — безызлучатель- ным электромагнитным переходом энергии на расстояние порядка 50 —100к [28]. Альтернативным положением последней точке зрения является гипотеза о существовании в конденсированных системах ассоциатов, столкновение с которыми равносильно столкновению с возбужденной молекулой [27].
В некоторых случаях заметное отклонение от аддитивности выходов продуктов радиолиза наблюдается лишь при значительных добавках акцептора (более 5 —ЮМ). Такие "малые отклонения" от аддитивности могут быть объяснены образованием донорно-акцепторных комплексов [28]. Поскольку при столкновении горячих атомов с молекулами возможно значительное возбуждение молекул, а вероятность образования донорно-акцеп- торного комплекса пропорциональна величине возбуждения [28], то идея
16
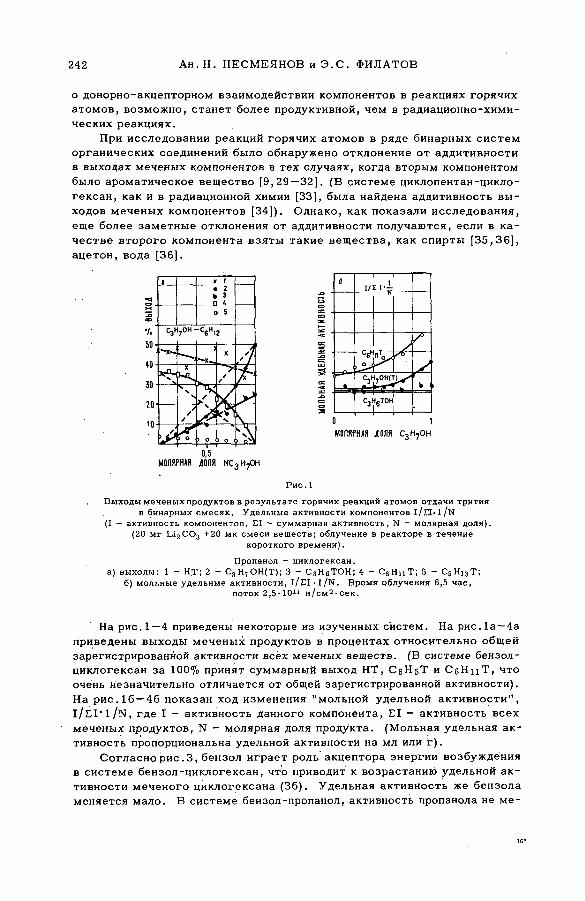
242 Ан. Н. НЕСМЕЯНОВ и Э . С . ФИЛАТОВ
о донорно-акцепторном взаимодействии компонентов в реакциях горячих атомов, возможно, станет более продуктивной, чем в радиационно-химических реакциях.
При исследовании реакций горячих атомов в ряде бинарных систем органических соединений было обнаружено отклонение от аддитивности в выходах меченых компонентов в тех случаях, когда вторым компонентом было ароматическое вещество [9,29—32]. (В системе циклопентан-цикло- гексан, как и в радиационной химии [33], была найдена аддитивность выходов меченых компонентов [34]). Однако, как показали исследования, еще более заметные отклонения от аддитивности получаются, если в качестве второго компонента взяты такие вещества, как спирты [35,36], ацетон, вода [36].
МОЛЯРНАЯ Л О Л А С 3Н 7 О Н
МОЛЯРНАЯ Д О ЛЯ N C j H y O H
Рис.1
Выходы меченых продуктов в результате горячих реакций атомов отдачи трития в бинарных см есях. Удельные активности компонентов I/EI’ I/ n
(I — активность компонентов, EÏ - суммарная активность, N — молярная доля). (20 м г L i2C 0 3 +20 мк Смеси веществ; облучение в реакторе в течение
короткого времени).
Пропанол - циклогексан.а) выходы: 1 - H T ; 2 - С3Н7О Н (Т ); 3 - С 3Н6ТОН; 4 - С6НП Т ; 5 - С5Н13Т ;
б) мольные удельные активности, l/ £ I * l/ N . Время облучения 6,5 час, поток 2,5-ХО11 н/см2-сек.
На рис. 1—4 приведены некоторые из изученных систем. На рис. 1а —4а приведены выходы меченых продуктов в процентах относительно общей зарегистрированной активности всех меченых веществ. (В системе бензол- циклогексан за 100% принят суммарный выход HT, С6Н5Т и СбНцТ, что очень незначительно отличается от общей зарегистрированной активности). На рис.16 —46 показан ход изменения "мольной удельной активности", I/E I’ I /N , где I - активность данного компонента, £1 - активность всех меченых продуктов, N - молярная доля продукта. (Мольная удельная активность пропорциональна удельной активности на мл или г ) .
Согласно рис.3 , бензол играет роль акцептора энергии возбуждения в системе бензол-циклогексан, что приводит к возрастанию удельной активности меченого циклогексана (36). Удельная активность же бензола меняется мало. В системе бензол-пропанол, активность пропанола не ме
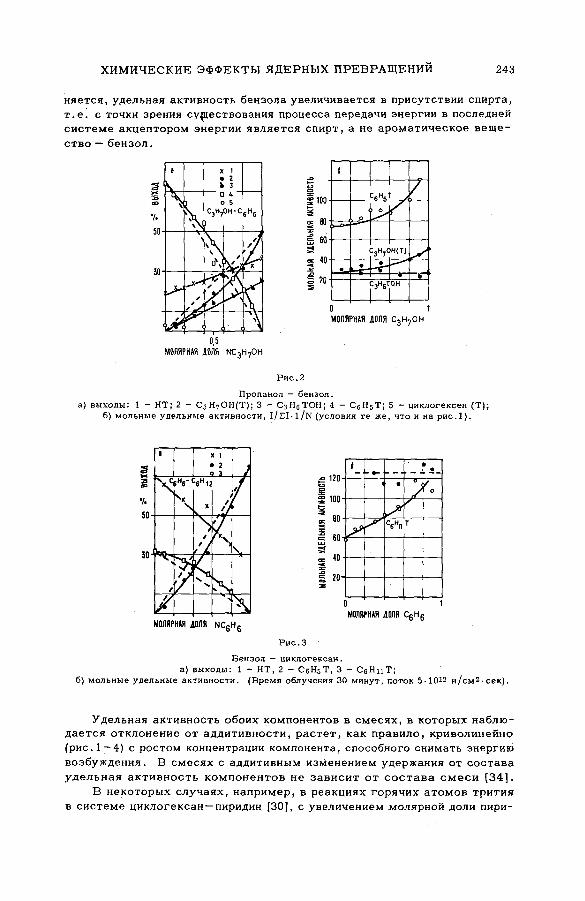
ХИМИЧЕСКИЕ ЭФФЕКТЫ ЯДЕРНЫХ ПРЕВРАЩЕНИЙ 243
няется, удельная активность бензола увеличивается в присутствии спирта, т .е . с точки зрения существования процесса передачи энергии в последней системе акцептором энергии является спирт, а не ароматическое вещество — бензол.
МОЛЯРНАЯ доля n c 3 h 7o h
МОЛЯРНАЯ доля С3Н7ОН
Ри с . 2
Пропанол - бензол.а) выходы: 1 - H T; 2 - С зН 7О Н (Т ); 3 - С зН6ТОН; 4 - СбН5Т ; 5 - циклогексен (Т ) ;
б ) мольные удельные активности, I/ 131 » 1 /N (условия те же, что и на ри с .1 ).
Р и с . 3
Бензол - циклогексан.а) выходы: 1 - H T, 2 - С 6Н5Т , 3 - С еН ц Т ;
б) мольные удельные активности. (Время облучения 30 минут, поток 5-1012 н/см2*сек ).
Удельная активность обоих компонентов в смесях, в которых наблюдается отклонение от аддитивности, растет, как правило, криволинейно (рис. 1—4) с ростом концентрации компонента, способного снимать энергию возбуждения. В смесях с аддитивным изменением удержания от состава удельная активность компонентов не зависит от состава смеси [34].
В некоторых случаях, например, в реакциях горячих атомов трития в системе циклогексан —пиридин [30], с увеличением молярной доли пири
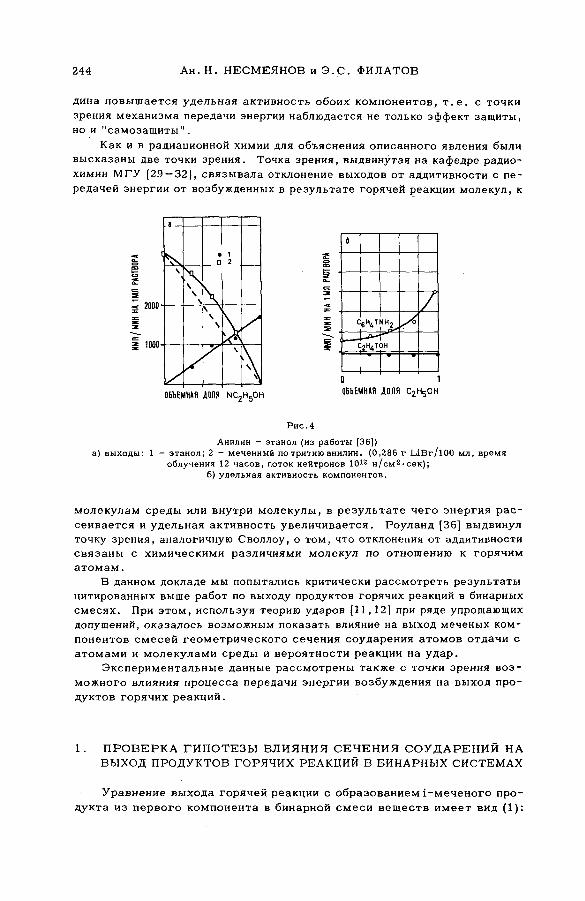
244 Ан. Н. НЕСМЕЯНОВ и Э . С . ФИЛАТОВ
дина повышается удельная активность обоих компонентов, т .е . с точки зрения механизма передачи энергии наблюдается не только эффект защиты, но и "самозащиты".
Как и в радиационной химии для объяснения описанного явления были высказаны две точки зрения. Точка зрения, выдвинутая на кафедре радиохимии МГУ [29—32], связывала отклонение выходов от аддитивности с передачей энергии от возбужденных в результате горячей реакции молекул, к
Рис. 4
Анилин — эганол (из работы [36]) а) выходы: 1 - этанол; 2 - меченный по тритию анилин. (0,286 г LiBr/lOO м л, время
облучения 12 часов, поток нейтронов 1012 н/см2-сек );б) удельная активность компонентов.
молекулам среды или внутри молекулы, в результате чего энергия рассеивается и удельная активность увеличивается. Роуланд [36] выдвинул точку зрения, аналогичную Своллоу, о том, что отклонения от аддитивности связаны с химическими различиями молекул по отношению к горячим атомам.
В данном докладе мы попытались критически рассмотреть результаты цитированных выше работ по выходу продуктов горячих реакций в бинарных смесях. При этом, используя теорию ударов [11,12] при ряде упрощающих допущений, оказалось возможным показать влияние на выход меченых компонентов смесей геометрического сечения соударения атомов отдачи с атомами и молекулами среды и вероятности реакции на удар.
Экспериментальные данные рассмотрены также с точки зрения возможного влияния процесса передачи энергии возбуждения на выход продуктов горячих реакций.
1 . ПРОВЕРКА ГИПОТЕЗЫ ВЛИЯНИЯ СЕЧЕНИЯ СОУДАРЕНИЙ НА ВЫХОД ПРОДУКТОВ ГОРЯЧИХ РЕАКЦИЙ В БИНАРНЫХ СИСТЕМАХ
Уравнение выхода горячей реакции с образованием i-меченого продукта из первого компонента в бинарной смеси веществ имеет вид (1 ):

ХИМИЧЕСКИЕ ЭФФЕКТЫ ЯДЕРНЫХ ПРЕВРАЩЕНИЙ 245
R i i = | 1 P l i № ) - ^ P i i ( E ) [ f i ( p 1 ( E ) - p 2 ( E ) ) + p 2( E ) ] , (1)
где lu — активность i -меченого продукта из первого компонента;2Л - активность всех меченых продуктов в системе, Ri¡ = Ií/EI;fi — геометрическая вероятность соударения с первым компонентом
_____ смеси;P j i ( E ) - средняя вероятность реакции на удар с образованием i-меченого
продукта из первого компонента;P i ( E ) - средняя вероятность реакции на удар с образованием всех мече
ных продуктов из первого компонента; р2(Е) - из второго;
Ç — логарифмический декремент энергии.Геометрическая вероятность соударения определяется через отноше
ние (2 ):fl = ____ Nl ginlT3______ = ____ N^zj______(
1 N i C T i n j Y ! + N 2 CT2 n 2 T 2 N 1 ( z 1 - z 2 ) + z 2
z l = CTl n l ' ) ' i . z 2 = c r 2 n 2 T 2 ,
где Ni и N2 — мол. доли первого и второго компонентов бинарной смеси;a i и сг2 - микросечения соударения (удар по связи);ni и п2 - число реакционно-способных "мест" в молекуле, при соуда
рении с которыми возможны химические реакции;Ti и 72 — коэффициенты стерического затруднения при атаке.Уравнение (1) получено на основе закономерностей, найденных при
исследовании термализации нейтронов при наличии захвата [11,12,37] и справедливо при выполнении следующих условий:
1 ) горячие атомы замедляются либо на атомах близких масс, либо Е < § 3Ео, где Eq — начальная энергия отдачи;
2) "функция возбуждения" химических реакций, р(Е), носит резонансный характер;
3) при столкновении атома с молекулой происходит или химическая реакция или рассеяние;
4) средняя потеря энергии на столкновение не зависит от энергии;5) микросечение соударения (от) и коэффициент стерического затрудне
ния (у) не зависит от энергии.Первые три условия, очевидно, выполняются в реакциях горячих ато
мов. Четвертое условие о независимости f от Е может быть принято лишь как первое весьма грубое приближение, т.к. было найдено [38], что потеря энергии горячими атомами происходит благодаря химическому взаимодействию при столкновении, не приводящему при больших Е к образованию связи с горячими атомами. Пятое условие является грубым приближением: в общем случае, сг и у являются функциями энергии. Однако для анализа уравнения (1 ) эти условия необходимо принять.
Введем обозначения:Rii = fi А - fi A [fi С + В], (3)
где A = £ 1L Í!); c = £ l M | £ 2ÍE). В = ^

246 Ан. Н. НЕСМЕЯНОВ и Э . С . ФИЛАТОВ
Определим по знаку второй производной от R по N вид зависимости R от N. Оказывается, что знак d2R/dN2 зависит от знака выражения (4):- 2AC(df/dN)2, т .е . от знака
“ P l i №) • [pîTË) - р ^ ( Ё ) ] / € 2 - (4 )
Знак выражения (4) в общем виде определить нельзя. Проведем его определение в следующих частных случаях.1. В случае p! (Е) > рг(Е), d2R/dN 2 < О и зависимость R от N имеет выпуклость вверх.2. В случае pi (Е) <р 2(Е), d2R/dN2> 0 и зависимость R от N отклонена от линейной к оси N.
Производная d2 R/dN2 не меняет знак во всем интервале концентраций N от 0 до 1, т .е . зависимость R от N при d2 R/dN2 #0 отклонена от линейной либо вверх, либо вниз в зависимости от соотношения между P i ( E ) и Рг(Е). __ __
Из приведенного анализа следует, что при р1(Ё) £ р2(Е) выходы меченых продуктов горячих реакций в бинарной смеси зависят от концентрации нелинейно и лишь при pi(È) - р2(Е) зависимость может стать линейной. Поэтому объяснение отклонений выходов меченых продуктов от аддитивности с точки зрения влияния процесса межмолекулярной передачи не является однозначным.
Рассмотрим с изложенных позиций имеющиеся данные для некоторых бинарных систем.1. Реакция Вг81(п,-у)Вг82 в системе С Н 2 В г 2 + С е Н 6 [7,32] (аналогичные данные получены для системы С 2 Н 5 В г + С б Н б [9]). Выход С Н 2 В г В г 82 меняется от NCH2 Br2 по S-образной кривой, т .е . d2R/dN2 меняет знак: при малых N d2R/dN2 <0 при N> 0,5 М. d2R/dN2 > 0 . Если d2R/dN2 > 0 при больших концентрациях бром-метилена, то pi (Е) <р2(Е). Но в области малых концентраций CH2Br2 d2 R/dN2 <0, следовательно, рг(Е)> р2(В). Значит, атомы отдачи брома "избирательно" реагируют с компонентами бинарной смеси С Н 2В г 2 +С$Нб: выход горячих реакций с образованием всех меченых продуктов больше для того компонента, чья концентрация меньше.2. Реакция L i6 (n,o')T в системах С 6 Н б(1) + С з Н 7 0 Н ( 2 ) и С б Н 1 2 ( 1 ) + + С 3 Н 7 О Щ 2 ) . Здесь найдены для выхода C g H s T и С б Н ц Т положительные отклонения от аддитивности во всем интервале концентраций, d2R/dN2 <0 (рис.1 и 2). Согласно проведенному анализу в этом случае должно быть Pi(E) > р2(Ё). Таким образом, тритий, видимо, "избирательно" реагирует с компонентами бинарных смесей и тем предпочтительнее, чем меньше концентрация компонента. Анализ данных [29] по реакциям горячих атомов трития в системах циклогексан (1)— бензол(2) и анилин(1) — спирт(2) [36] приводит к выводу о том, что pi(E)> р2(Е).
Для дальнейшего анализа экспериментальных данных есть необходимость рассмотреть зависимость R/N от N, т .е . "молярной удельной активности" от концентрации. (Зависимость удельной активности на 1 мл от объемных долей имеет тот же вид). Перепишем формулу (1) в виде (5).

ХИМИЧЕСКИЕ ЭФФЕКТЫ ЯДЕРНЫХ ПРЕВРАЩЕНИЙ 247
' Rii _ Z!Ph (E)___________ zipii(E)Nl 5[Ni (zi - 2 2) + z2] f 2[N! (zi - z2)+ z 2]
x { n , ( z " - Z¿ , Za 1Д 5 -) - M g ) l + i ^ g ) | ■ (5 )
Отсюда следует, что зависимость Rü /N i должна быть нелинейной. Наклонкривой R j i /Ni в области малых Nj равен ■
-Pii(E)[Pi(E)- Р 2(Е)] (6 )
в области больших:
- £Ji 2 [pi (E) - р2 (Е)]
т .е . наклоны функций, R/Nj = F(Ni) для малых и больших Ni отличаются в (z 1 /z 2 )2 раз. Если в бинарных смесях пропанола (2) с бензолом (1) и циклогексаном (1 ) Zj> z 2, то "молярная удельная" активность бензола и циклогексана, Rj; /N j , должна непрерывно возрастать по мере разбавления спиртом, что и подтверждается экспериментальными данными (рис. 16 и 26). Аналогично обстоит дело с реакциями горячих атомов трития в системе бензол —циклогексан, спирт —анилин, где было найдено увеличение удельных активностей циклогексана и анилина по мере разбавления в первом случае бензолом и во втором спиртом (рис.36, 46). Значит, кроме соотношения между вероятностями горячих реакций pi(E) и р2(Е), на выходы меченых продуктов оказывают существенное влияние такие параметры, как микросечение соударения, число реакционно-способных мест в молекулах и коэффициенты стерического затруднения при атаке горячего атома (z = any).
Решения уравнения (1) и для малых Nj и при Nj = 1 имеют вид:
и (7)
где Rn и R j — выходы меченого первого компонента при Ni в области. малых концентраций и при Nj = 1 соответственно;и | 2 - средние логарифмические декременты энергии для пер
вого и второго компонентов. Если полагать что Ej» pj(E) и £2 » р2(Е) для первого компонента, если он разбавлен,

248 Ан. Н. НЕСМЕЯНОВ и Э . С . ФИЛАТОВ
и для второго компонента, если он присутствует в малой концентрации:
Rk = í*_I, те z i ^ R k . í a -Л .Ш. о)« S i ? ! f 2 Z 2 U g i ?1 V l - N 2 (9)
где RÍ>¡ и ^ 2í ~ ВЫХ°ДЫ меченого второго компонента при малых концентрациях и при N2 =1 соответственно.
Отсюда
Rii . R2í r f .R°. R®. 1 2 ’Il 2i
Или
IÍl .Il Х .^ к .1 ^ _ Л ___ Niüa____=1 no).R î i ?2 / V R 2î S i V d - N ^ d - N a ) ( 1 0 )
(Здесь N2 #1 — Ni , Nj и N2 —малые величины концентраций). При N: и N2 ---- ► 0 имеем из (8 ) и (9):
Д ц туг - I 2. . Z 2. и R 2i , т _ ? 2 , z ¿ . , , , *R °íN i" € i Zl И R2i ~ р - ( *
Отсюда
Ri. R1 (12)
а при Nj = N2 = N ■
f f - f f - N 2- ! . (13)•Klí K 2i .
Уравнение (13) можно проверить экспериментально. Однако мы не располагаем данными по выходам меченых соединений в разбавленных растворах (при N<0,1 молярной доли). Поэтому оценку правильности закона(13) проведем лишь приближенно.
Для раствора бензола (1) в пиридине (2) при молярной доле бензола 0,27 отношение R iС6 Н5 T/H 0. 27 СбН5Т-0,27 = 1,1, а из данных по выходу меченых соединений в разбавленном растворе пиридина в бензоле, получаем R1C5 Н4 TN/R0-23 С5 Н4 TN.0,23 = 0,77. Произведение отношений выходов согласно (13) равно 0,85, т .е . близко к 1. Аналогичные расчеты для других систем приведены в табл.1. По-видимому, отклонения от закона (13) связаны с тем, что взятие концентрации слишком велики.
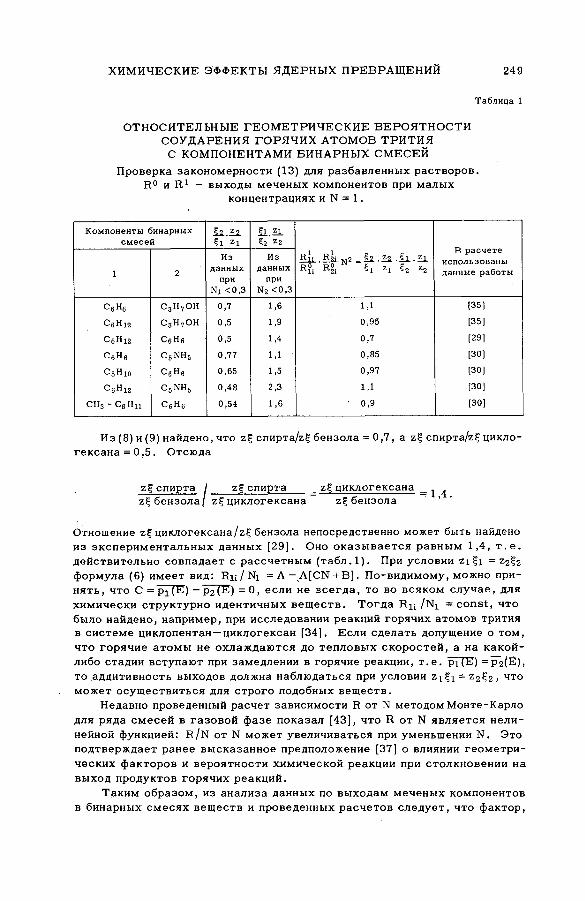
ХИМИЧЕСКИЕ ЭФФЕКТЫ ЯДЕРНЫХ ПРЕВРАЩЕНИЙ 249
Таблица 1
ОТНОСИТЕЛЬНЫЕ ГЕОМЕТРИЧЕСКИЕ ВЕРОЯТНОСТИ СОУДАРЕНИЯ ГОРЯЧИХ АТОМОВ ТРИТИЯ
С КОМПОНЕНТАМИ БИНАРНЫХ СМЕСЕЙ Проверка закономерности (13) для разбавленных растворов.
R0 и R1 - выходы меченых компонентов при малых концентрациях и N = 1.
Компоненты бинарных смесей
I2 . . Í2Il Z1
l l . i l .?2 z2
S i l . Ü2i_ „ 2 = i i . . i l • í i Rîi R2i ?1 Z1 2 z2
В расчете использованы данные работы1 2
Из данных
при N i <0,3
Из данных
при N 2 <0,3
с 6н6 С 3Н 7ОН 0,7 1,6 i , i [35]
С ен а2 С 3Н,ОН 0,5 1,9 0,95 [35]
C 6Hl2 С6н 6 0,5 1.4 0,7 [29]
с 6н 6 c 5n h 5 0,77 1,1 0,85 [30]
С 5Ню С 6Н6 0,65 1,5 0,97 [30]
с 6н 12 C 6NH6 0,48 2,3 1,1 [30]
СН3 - С 6НП С 6Н6 0,54 1,6 • 0,9 [30]
Из (8 ) и (9) найдено, что zg спирта/z! бензола = 0,7, a zg спирта/zg цикло- гексана = 0,5. Отсюда
zg спирта I zg спирта_____zg циклогексана _ ^zgбензола/ zgциклогексана zgбензола ’ '
Отношение zg циклогексана/zg бензола непосредственно может быть найдено из экспериментальных данных [29]. Оно оказывается равным 1,4, т .е . действительно совпадает с рассчетным (табл.1). При условии zifi = z2g2 формула (6) имеет вид: Ri¡ / Ni =А —A[CN+B]. По-видимому, можно принять, что С - pi (К) - р2 (Е) = 0 , если не всегда, то во всяком случае, для химически структурно идентичных веществ. Тогда Ri¡ /Ni = const, что было найдено, например, при исследовании реакций горячих атомов трития в системе циклопентан —циклогексан [34]. Если сделать допущение о том, что горячие атомы не охлаждаются до тепловых скоростей, а на какой- либо стадии вступают при замедлении в горячие реакции, т .е . P i ( E ) =р 2(Ё), то аддитивность выходов должна наблюдаться при условии Zigi = z2g2, что может осуществиться для строго подобных веществ.
Недавно проведенный расчет зависимости Е от N методом Монте-Карло для ряда смесей в газовой фазе показал [43], что R от N является нелинейной функцией: R/N от N может увеличиваться при уменьшении N. Это подтверждает ранее высказанное предположение [37] о влиянии геометрических факторов и вероятности химической реакции при столкновении на выход продуктов горячих реакций.
Таким образом, из анализа данных по выходам меченых компонентов в бинарных смесях веществ и проведенных расчетов следует, что фактор,

250 Ан. Н. НЕСМЕЯНОВ и Э . С . ФИЛАТОВ
включающий в себя вероятность столкновения, стерический параметр, число реакционных мест в молекуле и величину средней потери энергии может определять кинетику горячей реакции.
2. ПРОВЕРКА КОНЦЕПЦИИ О ВЛИЯНИИ ПРОЦЕССА МЕЖМОЛЕКУ- ЛЯРНОЙ ПЕРЕДАЧИ ЭНЕРГИИ НА ВЫХОД ПРОДУКТОВ ГОРЯЧИХ РЕАКЦИЙ
Концепция межмолекулярной передачи энергии предполагает наличие межмолекулярного взаимодействия между компонентами. По-видимому, оно устанавливается после процесса "пометки" в результате того, что молекула одного из компонентов становится возбужденной. В радиационной химии образованием таких "донорно-акцепторных комплексов" пытаются трактовать отклонения от аддитивности радиационно-химических выходов [28]. Возможно, что в химии горячих атомов образование донорно- акцепторного комплекса после "пометки" более предпочтительно, т.к . вероятность смещения валентных электронов увеличивается по мере увеличения энергии возбуждения. Снятие возбуждения идет по схеме:
Энергия возбуждения комплекса переходит в кинетическую энергию А и В, т .е . комплекс распадается на невозбужденные частицы (В системе цикло- гексан-бензол в радиационно-химических реакциях это твердо установлено).
Если бы передача энергии осуществлялась при простом столкновении возбужденной молекулы с молекулой акцептора энергии, то снижение радиационно-химических выходов или увеличение удельных активностей продуктов горячих реакций было бы пропорционально молярной доле акцептора энергии. Ни в радиационной химии, ни в химии горячих атомов этого не наблюдается. Как правило, R/N зависит от N нелинейно. Это еще раз указывает на возможность существования в растворе образований типа донорно-акцепторного комплекса.
Вероятность ингибирования радиационного разложения равна [39]:
где А =р(и )/и ; p(w) — плотность уровней ингибитора;« 1 и « 2 “ const; •и — энергия возбуждения.Формула (12) справедлива при следующих условиях. При радиолизе
образуются молекулярные ионы, уровень возбуждения которых ниже первого возбужденного уровня основного компонента. Ингибитор имеет непрерывный спектр уровней, т .е . среда между возбужденной молекулой и ингибитором "прозрачна". Закон 2/3 для малых концентраций ингибитора хорошо выполняется. В ряде систем закон (12) выполняется в широком интервале концентраций ("Закон 4 /3 " и "6 /3 ") . В химии горячих атомов не образуются молекулярные ионы, по крайней мере, в области, где идут
А* + В ---- ►[А+ В' ] * ---- ►А +В.

ХИМИЧЕСКИЕ ЭФФЕКТЫ ЯДЕРНЫХ ПРЕВРАЩЕНИЙ 251
горячие реакции. Поэтому выражение (12) непосредственно не может быть применено в химии горячих атомов.
При анализе данных изменения выхода продуктов горячих реакций ДИ от концентрации акцептора возбуждения, N, линейная зависимость получается только в координатах ДИ—№ . Полученные данные приведены на
z 5а: : I * 1,5
0,5
1//
//
/ ///
j
X1 // f
/ /к г/ /
/ { / ' •У
V/
уИ/ / Ч/ S<W°
Io
о"
N - КУ Б. МОЛЯРНЫЕ ДОЛИ АКЦЕПТОРА ЭНЕРГИИ
Р и с . 5
Зависимость относительного изменения выхода меченого компонента (1) бинарной смеси от среднего обратного расстояния 1 /г = N3 между
молекулами акцептора энергии возбуждения (компонент 2) для систем (N - молярная доля акцептора):
*СН2В г2( 1 ) - СбН б(2) - реакция горячих атомов брома-82;
С 6Н :2(1) — С зН 70Н (2) выход С б Н ц Т ;с бн 12( 1 ) ” СбН б(2 ), выход С б Н ц Т ; Реакция горячих атомов трития С 6Н 6<1) - С3 Н7ОН(2), выход С 6Н5 Т .
(Максимальный разброс данных показан для N3 = 0,51 при уменьшении N разброс уменьш ается).
рис.5. На оси ординат отложено относительное изменение радиохимического выхода
»‘г R/( 1 — N) - R (при N = 0 )R (при N = 0)
по оси абсцисс — N3 (N — молярная доля акцептора энергии возбуждения).Из рис.5 видно, что для всех рассмотренных систем (CgHg —С3 Н7 ОН,
С6Н12- С 3Н7ОН, Се Hj2 “ CgHg, СН2ВГ2 —CgHg) зависимость AR от N3 при N3 > 0,2 или N >0,6 линейна.
В то же время при концентрации акцептора возбуждения ниже 0,6 наблюдается резкое отклонение от линейности.

252 А н . Н . НЕСМЕЯНОВ и Э . С . ФИЛАТОВ
Из молекулярной физики известно, что линейная зависимость от l / r 3 (г — среднее расстояние между молекулами добавки) говорит о передаче энергии при столкновении, линейная зависимость от l / r 6 указывает на передачу энергии при дипольном взаимодействии.
Найденная зависимость AR от N указывет на существование донорно- акцепторного механизма передачи энергии возбуждения. Так как N3 = l /r и N> 0,6 (для того, чтобы AR = const-N3), то можно заключить, что возбужденная молекула должна быть окружена не менее, чем шестью молекулами акцептора энергии, чтобы механизм отвода энергии стал эффективным и линейно зависимым от l /r .
Проанализируем с изложенных позиций данные по выходу горячей реакции атомов отдачи брома в системе СН2ВГ2 +СбНб .
Выше было показано, что гипотеза о влиянии геометрической вероятности соударения применительно к системе СН2 Вгг (1 ) +СеНб(2 ) согласуется с экспериментальными данными только в области больших концентраций СН2Вг2.
Действительно, расчет отношения гг /z i по формуле (8 ) показывает что отношение z2 /zi быстро меняется при N ~0,4 —0,5 (Ç — в расчете не учитывалось). Микросечение соударения, коэффициент стерического затруднения или замедляющая способность среды меняются от состава плавно. (Образование других продуктов в реакциях горячих атомов брома-82 в системе СНгВгг +СбНб, кроме СНгВг82Вг, например, продуктов замещения водорода, не конкурирует с реакцией замещения брома [7], поэтому рг(Ё)~0 , и изменение отношения z2 /г 1; можно проследить во всем интервале концентраций). Следовательно, в области малых концентраций на выход СН2 Вг82Вг оказывает влияние какой-то другой фактор, кроме геометрической вероятности соударения. Можно допустить, что играет роль передача энергии возбуждения от свежеобразованных меченых молекул метиленбромида к молекулам бензола, причем эффективная передача энергии начинает осуществляться при концентрации бензола, больше 0,6. (Это следует из графика R/N2 о т 1 — N2 [32], N2 - молярная доля СЯ2Вг2. Это согласуется со скачкообразным изменением отношения, о чем было указано выше, и объясняет изменение знака второй производной.
3. ПРОЦЕССЫ ВНУТРИМОЛЕКУЛЯРНОЙ ПЕРЕДАЧИ ЭНЕРГИИ
На этом вопросе остановимся кратко.В работе [40] было установлено, что разрыв связей в алкилбензолах,
одинаково отстоящих от бензола кольца, идет в одинаковой степени для алкилбензолов с различной длиной цепи и строения заместителя (табл. 2 ), т . е . , по-видимому, осуществляется защита типа "губки", когда энергия возбуждения трансформируется из боковой цепи в кольцо.
Существование процесса эффективной внутримолекулярной передачи энергии было подтверждено также при изучении реакций атомов отдачи трития в алифатических спиртах [35]. Видимо, отмеченное ранее декарбо- ксилирование возбужденных молекул карбоновых кислот [41] также может быть объяснено внутримолекулярной миграцией энергии возбуждения от места удара к группе СООН.

Х И М И Ч Е С К И Е Э Ф Ф Е К Т Ы Я Д Е Р Н Ы Х П Р Е В Р А Щ Е Н И Й 253
. Таблица 2
О Т Н О С И Т Е Л Ь Н А Я В Е Р О Я Т Н О С Т Ь Р А З Р Ы В А С В Я З Е Й С - С В А Л К И Л Б Е Н З О Л А Х , %
Меченые продукты
Толуол Этилбензол Н-пропилбензол Изо-пропилбензол
Этилбензол 75 ± 8
н-пронилбензол 75 ± 1 66±9
н-бутилбензол 75 ± 4 65 ±4 47 ± 6
Вторичный бутилбензол 0 35 ± 3 35 22 ±3
Трет-бутилбензол 0 0 0 - 42 ± 3
В к р и ст а л л и ч е с к и х в е щ е с т в а х э т о т п р о ц есс п е р е с т а е т и г р а т ь р о л ь , ч т о б ы ло п р о д ем о н стр и р о ва н о на прим ере ам и н о к и сл о т [42 ] .
ЗА К Л Ю Ч ЕН И Е
И з п р овед ен н о го ан ал и за с л е д у е т , что имею щ иеся эксп ер и м ен тал ьн ы е данн ы е н е д о ста т о ч н ы для о д н о зн ачн о го реш ения воп р о са о роли м еж м о л е- кулярной п ередачи энерги и при реакц и ях а то м о в отдачи и роли сеч ен и я их в за и м о д е й ст ви я с м о л екул ам и с р е д ы . Н еобходим ы б о л ее то чн ы е д анн ы е для м а л ы х концентраций о д н о го ко м п о н ен та в д р у го м . С наш ей т о ч ки зр ен и я , и м е е т м е с т о вли яни е на вы хо д п р о д у к то в реакци й го р я ч и х а т о м о в п ар ал л ел ьн о об о и х ф а к т о р о в , к о то р ы е д е й с т в у ю т в р я д е с л у ч а е в в одном , а в н ек о то р ы х с л у ч а я х в противополож ных нап р авлени ях, приводя к S -о б р азн ы м кривы м за ви си м о сти удельной акти вн о сти от с о с т а в а с м е си . При больших концентрациях вто р о го ком понента (акцептора энергии) о сн о вную р оль и гр а е т п е р е д а ч а э н е р ги и , а при м а л ы х е г о к о н ц ен тр ац и ях п р ева л и р у е т м е х а н и зм и зб и р а т е л ь н о го д е й ст ви я го р я ч и х а т о м о в .
У ч е т влияния и зм ен ен и я с п е к т р а го р я ч и х а т о м о в на в ы х о д м е ч е н ы х п р одуктов в за ви си м о сти о т с о с т а в а бинарной см е си услож нит предлож енный "ко м п л ексн ы й " м еха н и зм го р я чи х реакций.
Л И Т Е Р А Т У Р А
[1 ) L IB B Y W . F ., J. A m . Chem . S o c ., 69 (1947) 2523.[2J F R IE D M A N L . , L IB B Y W . F . , J . Chem . P h y s ., 17 (1949) 647.[3 ] F O X M .S . , L IB B Y W . F . , J . Chem . P h y s ., 20 (1952) 487.[41 N IL L E R I. M . , D O D SO N R .W . , J. Chem . P h y s ., 18 (1950) 579.[5] C A P R O N P . C . , O SH IM A Y . . J. Chem . P h y s .. 20 (1952) 1403.[ 6] Н Е С М Е Я Н О В Ан . H . , Ф И Л А Т О В Э . С . , М А Н С Ф Е Л ЬД А . , Радиохимия, 3, 5 (1961) 610. [71 Ф И Л А Т О В Э . С . , Н Е С М Е Я Н О В А н .Н . , Ч Е П Ы Ж Е В Ю .Б . , Радиохимия, 6 , 5 (1964) 595. [81 Н Е С М Е Я Н О В А н .Н . , Б О Р И С О В Е . А . , З В А Р А И . , Р ад и охи м и я , 1, 3 (1 9 5 9 ) 3 25 . [91 Н Е С М Е Я Н О В А н . Н . , Ф И Л А Т О В Э . С . , Р ади охи м и я , 3 , 5 (1 96 1 ) 614.
[101 Н Е С М Е Я Н О В А н . Н . , Ф И Л А Т О В Э . С . , Р ад и охи м и я , 3 , 5 (1961 ) 601.[11 ] W O L F G A N G R . , E S T R U P P . , J . A m . C h e m . S o c . , 82 (1960 ) 2665.

254 А н . Н . Н Е С М Е Я Н О В и Э . С . Ф И Л А Т О В
[12 ] W O L F G A N G R . , J . C h em . P h y s . , 39 (1963 ) 2983.[131 С В О Л Л О У А . , Радиационная химия органических соединений. И Л 1963, с т р .156— 164. fl4| M A N IO N I. Р . , B U R T O N М . , J. Phys. C h em ., 56 (1952) 560.[15] B U R T O N М . , L IP S K Y S. , J. Phy s. C h e m ., 61 (1957) 1461.[16] D R E E S K A M P H . . B U R T O N М . , D isc . F a ra d . S o c ., 27 (1959) 64.[17] L A M B O R N J . , S W A L L O W A . J . , J. Phys. C h em ., 65 6 (1960) 920.[18] B L A C H F O R D J . , D Y N E P . , Can. J. C h em ., 42, 5 (1964) 1165. .[19] F R E E M A N G . , J. Chem . P h y s ., 36, 6 (1962) 1534.[2 0 ] H A R D W IC K T . , J. Phys. C h e m ., 66, ; ; (1962) 2132.[2 1 ] B A X E N D A L E J . , M E L L O W S F . , J . A m . Chem . S o c ., 83, 23 (1961) 4720.[2 2 ] H A R D W IC K T . , J . P hys. C h e m ., 66, 9 (1962) 1611.[23] H A R D W IC K T . , J . P h y s. C h e m ., 64, 11 (1960) 1623,[24 ] H A R D W IC K T . , J . Phy s. C h em ., 66, 1 (1962) 117.[25] M c D O N E L L W . , G O R D O N S . , J . Chem . P h y s ., 23, 1 (1955) 208.[26] H A R D W IC K T . , J . P h y s. C h em ., 66, 11 (1962) 2246.[27] К Р О Н Г А У З B . A . , Успехи химии, 31, вып. 2 (1962) 222.[28] Б А Г Д А С А Р Я Н Х . С . , Труды II Всесоюзного совещания по радиационной химии АН С С С Р ,
1962, с т р .52.[29] АВД О Н ИН А Е .Н . , Радиохим ия,^ (1962) 617.[30] А В Д О Н И Н А Е . Н . , Н Е С М Е Я Н О В А н .Н . , У Н -Х А О -М И Н , Радиохимия, _6 , 3 (1963 ) 323.[31 ] А В Д О Н И Н А Е . Н . , М У Д Р А К . , Н Е С М Е Я Н О В А н . Н . , Рад и охи м и я , j>, 5 (1963 ) 633 .[32] Ф И Л А ТО В Э . С . , Н Е С М Е Я Н О В А н .Н . , ЧЕПЫ Ж ЕВ Ю .Б . , Вестник Московского Универ
ситета, серия химии, 6 , (1963) 45.[33] M U C C IN I G . , S H U LE R R ., J. Phys. Chem ., 64 (1960) 1436.[34] АВД О Н И Н А Е . Н . , Н Е С М Е Я Н О В А н .Н . , Радиохимия, 5, 4 (1963) 514.[35] Ф И Л А Т О В Э . С . , ЦЗЯН Т А Й -В А Н , Н Е С М Е Я Н О В А н .Н . , Сообщение на настоящем сим
позиуме.[36] S O K O LO W S K A А . , H A S K IN L . , R O W L A N D F . , J . A m . Chem . S o c ., 84 (1962) 2469.[37] Ф И Л А ТО В Э .С . , Н Е С М Е Я Н О В А н .Н . , Вестник Московского Университета, 4 (1964) 13.[38] R O O T J . , R O W L A N D F . S ., J . Chem . P h y s ., 38 (1963) 2030.[39] Радиолиз углеводородов, И АН С С С Р , под ред . А . В . Топчиева и Л . С . П олака, М . , 1962,
стр.158 .[40] ПОЗДЕЕВ В . В . , ДЗАН ТИ ЕВ Б . Г . , Н ЕСМ ЕЯ Н О В А н .Н . , Кинетика и катализ, 3 (1962) 613.[41] E L A T R A S H A . , JO H N SE N R . , W O L F G A N G R . , J. A m . Chem . S o c ., J. Phy s. C h em .,
64 (1960) 785.[42] Н ЕСМ ЕЯ Н О В А н .Н . , СИ М ОНОВ Е . Ф . , Реакции горячих атомов трития с аминокислотами.
См . в этом томе.[43] R O W L A N D F . , C O U L T E R P . , Rad. Acta, b. 2, h 4 (1964) 163.

REACTIONS OF TRITIUM RECOIL ATOMS IN LIQUID ORGANIC MIXTURES
A. SOKOÍOWSKA DEPARTMENT OF RADIOCHEMISTRY,
INSTITUTE OF NUCLEAR RESEARCH, WARSAW, POLAND
Abstract — Résumé — Аннотация — Resumen
REACTIONS OF TRITIUM RECOIL ATOMS IN LIQUID ORGANIC MIXTURES. Studies on reactions of recoil tritium atoms in organic liquids were carried out in the mixtures of cyclohexane and benzene.
It was observed that the activities found in the form of tritiated benzene and cyclohexane are linearly
proportional to their mole fraction in the mixture. It was concluded that the reactions of T for H substitution
in cyclohexane and benzene molecules in the studied system do not depend on the composition of a mixture.
RÉACTIONS DES ATOMES DE RECUL DE TRITIUM DANS LES SYSTÈMES LIQUIDES. On a examiné
les réactions des atomes de recul de tritium dans les mélanges liquides de cyclohexane et de benzène.Il a été observé que les activités trouvées sous la forme.de benzène tritié et de cyclohexane tritié sont
proportionnelles à la fraction molaire des substances de départ. On en a conclu que les réactions de substitution de H par T dans les molécules de cyclohexane et de benzène ne dépendent pas, dans les systèmes examinés, de la composition des mélanges. .
РЕАК Ц И И А Т О М О В О Т Д А Ч И Т Р И Т И Я В ЖИДКОЙ О РГА Н И Ч Е С К О Й С И С Т Е М Е . И сследована реакция атомов отдачи трития в жидкой бинарной системе циклогексан — б ен зол .
П оказано, что активности тритобензола и тритоциклогексана линейно зёвисят от их молярной доли в см еси.
Было заключено, что реакции замещения водорода тритием в молекулах циклогексана и бензола не зависят от состава смесей. .
REACCIONES DE LOS ATOMOS DE RETROCESO DE TRITIO EN LAS MEZCLAS DE LIQUIDOS ORGANICOS. Se han estudiado las reacciones de los átomos de retroceso de tritio en mezclas lfquidas de ciclohexano y
benceno.Se ha observado que las actividades halladas bajo la forma de benceno y de ciclohexano tritiados son
proporcionales a la fracción molar de esas sustancias en la mezcla. Se ha llegado a la conclusión de que las reacciones de sustitución de H por T en las moléculas de ciclohexano y de benceno no dependen, en los sistemas examinados, de la composición de la mezcla.
INTRODUCTION
One of the essential problems still unsolved in studies on tritium recoil reactions in liquids is whether or not the model assumed in the gas phase for substitution and abstraction reactions (i.e. reactions leading tó formation of labelled parent compound and HT) can accomodate all the results observed in the liquid phase. Due to close packing of molecules their interaction is much stronger than in the gas phase and can influence the yield of labelled stable products. Some additional processes can contribute also, at least partially, to the formation of tritium labelled parent molecules and HT or interfere with it.
255

256 A. SOKOÏOWSKA
Studies on the systems containing two different kinds of molecules at various concentrations can reveal some information concerning the role of the medium upon the reactions of recoiling tritium atoms. When 4he composition of a mixture is varied gradually over the full range of mole fraction of the two reactants, the moderating or stabilizing properties of the mixture are also varied. This could influence the yield of these products whose formation is affected by surrounding medium.
In this paper will be presented some results of studies on tritium recoil reactions in the benzene-cyclohexane mixtures.
Reactions of recoil tritium atoms in this mixture were investigated previously by AVDONINA [1] . The gas-chromatography method was used for separation and analysis of the products. The yield of the three main products,i.e . HT, tritiated cyclohexane and tritiated benzene was calculated as the ratio of radioactivity eluted in a corresponding peak to the sum of activity eluted from the column in a given run. On the basis of the results obtained the author concluded that the presence of benzene stimulates a higher yield of tritiated cyclohexane than could be expected if it depended only on the probability of a recoiling tritium atom encountering a cyclohexane molecule.
In previous studies of reactions of recoiling tritium in the mixtures aniline-ethanol and aniline-acetone [2] it has been found that the yield of tritiated acetone and ethanol is proportional to their mole fractions, and the yield of tritiated aniline in the samples in which the aniline concentration is low exceeds the expected values.
The discrepancy of results obtained in the two papers mentioned above might be explained in the following ways:(1) In both cases aromatic-aliphatic mixtures were examined; one case
used a mixture of pure hydrocarbons, and the other case a mixture of amine and alcohol or ketone.
(2) The methods of separation and analysis were different: in one case gas-chromatography was used and in the other chemical methods.
EXPER IM ENTAL
The recoil tritium atoms were formed by thermal neutron irradiation of Li6 present as L i2C03. Due to insolubility of any lithium salt in hydrocarbons the two-phase system had to be used.
The mixtures of cyclohexane and benzene containing the known amount of lithium carbonate (about 10% by weight) were sealed in quartz capillaries. Some of the samples were evacuated and some saturated with oxygen. The range of diameter of lithium carbonate particles varied from 100 - 110 цт . Samples were irradiated for 1 h at a flux of 1 X 1012n/crr£* s or for 5 h at a flux of 3 X 10n n/crrf.s at the reactor "EW A" in Swierk in several series. All the samples for a given series received a common irradiation exposure except for the flux variations between capillaries. Each series covered the full range of concentration ratios and consisted of no more than 30 capillaries.
Samples were analysed by the gas-liquid chromatography method at a temperature of 80°C with /3, /З'-oxydipropionitryle used as the stationary phase and propane as the mobile phase. The counting measurements were made by means of a gas-proportional counter.
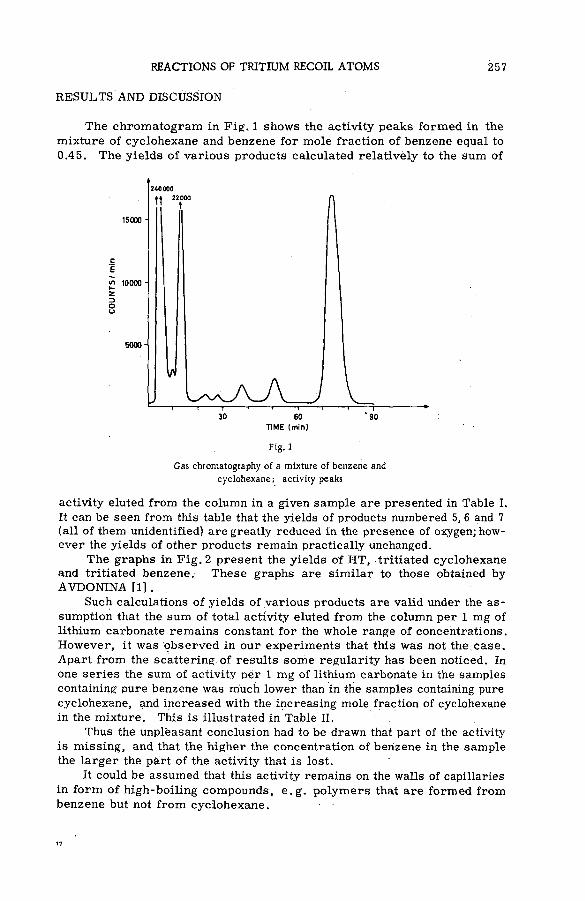
REACTIONS OF TRITIUM RECOIL ATOMS 257
RESULTS AND DISCUSSION
The chromatogram in Fig. 1 shows the activity peaks formed in the mixture of cyclohexane and benzene for mole fraction of benzene equal to0.45. The yields of various products calculated relatively to the sum of
TIME (min)
Fig. 1
Gas chromatography of a mixture of benzene and
cyclohexane; activity peaks
activity eluted from the column in a given sample are presented in Table I. It can be seen from this table that the yields of products numbered 5, 6 and 7 (all of them unidentified) are greatly reduced in the presence of oxygen; however the yields of other products remain practically unchanged.
The graphs in F ig. 2 present the yields of HT, tritiated cyclohexane and tritiated benzene. These graphs are similar to those obtained by AVDONINA [1 ]. ,
Such calculations of yields of various products are valid under the assumption that the sum of total activity eluted from the column per 1 mg of lithium carbonate remains constant for the whole range of concentrations. However, it was observed in our experiments that this was not the case. Apart from the scattering of results some regularity has been noticed. In one series the sum of activity per 1 mg of lithium carbonate in the samples containing pure benzene was much lower than in the samples containing pure cyclohexane, and increased with the increasing mole fraction of cyclohexane in the mixture. This is illustrated in Table II. .
Thus the unpleasant conclusion had to be drawn that part of the activity is missing, and that the higher the concentration of benzene in the sample the larger the part of the activity that is lost. '
It could be assumed that this activity remains on the walls of capillaries in form of high-boiling compounds, e .g . polymérs that are formêd from benzene but not from cyclohexane. .
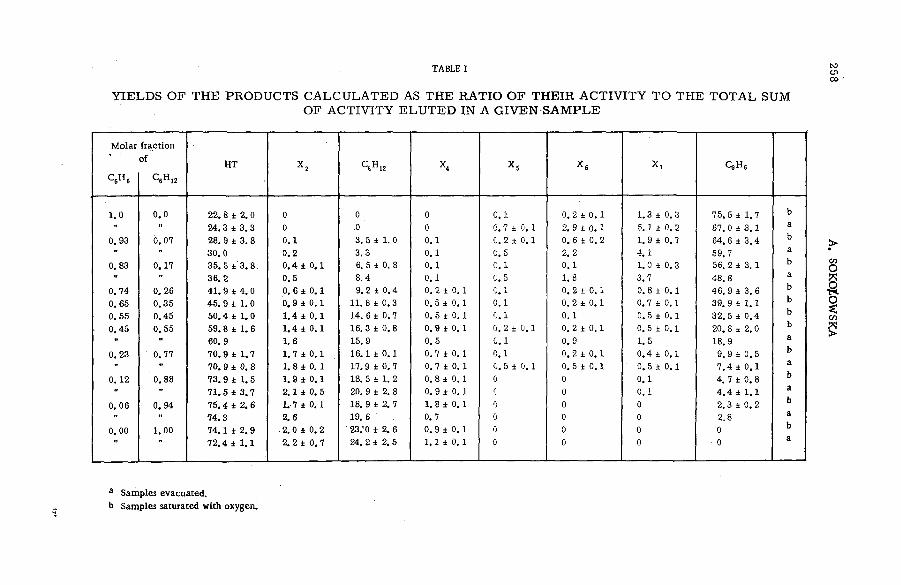
TABLE I
YIELDS OF THE PRODUCTS C A LC U LA TE D AS THE RATIO OF THEIR AC T IV IT Y TO THE TO TA L SUM OF ACTIV ITY E LU T E D IN A G IVEN -SAM PLE
Molar
c 6h 6
fraction
of
c 6h 12HT х г C6H 12 X4 *5 * 6 X , Q H 6
1 . 0 0.0 22. 8 ± 2 . 0 0 0 0 0 . 1 0 . 2 ± 0 . 1 1.3 è 0.3 75.5 ± 1 .7 b
" " 24. 3 ± 3. 3 0 -0 0 0. 7 ± 0.1 2. 9 ± 0.1 5.1 ± 0. 2 67. 0 ± 3.1 a
0. 93 0. 07 28. 9 ± 3. 8 0 . 1 3. 5 ± 1. 0 0 . 1 0. 2 ± 0 . 1 0. 6 ± 0. 2 1.9 ± 0.7 64. 6 ± 3 .4 b
" " 30.0 0 . 2 3.3 0 . 1 0.5 2 . 2 4.1 59.7 a
0. 83 0.17 35.5 ± 3 .8 . 0 .4 ± 0 .1 6. 5 ± 0. 8 0 . 1 0 . 1 0 . 1 1. 0 ± 0. 3 56. 2 ± 3.1 b
" " 36.2 0.5 8.4 0 . 1 0.5 1 . 8 3.7 48.8 a
0. 74 0. 26 41. 9 ± 4. 0 0. 6 ± 0 . 1 9. 2 ± 0.4 0. 2 ± 0 . 1 0 . 1 0. 2 ± 0. 1 0. 8 è 0 . 1 46. 9 ± 3. 6 b
0. 65 0.35 45. 9 * 1 . 0 0. 9 ± 0.1 1 1 . 8 ± 0.3 0.5 ± 0 . 1 0 . 1 0. 2 ± 0 . 1 0. 7 ± 0.1 39. 9 ± 1.1 b
0. 55 0.45 50.4 ± 1 .0 1.4 ± 0.1 14. 6 ± 0. 7 0. 5 ± 0.1 0 . 1 0 . 1 0.5 ± 0 . 1 32.5 ± 0.4 b
0.45 0. 55 59. 8 ± 1. 6 1.4 ± 0 .1 16. 3 ± 0. 8 0.9 ± 0 . 1 0. 2 ± 0 . 1 0. 2 ± 0 . 1 0. 5 ± 0.1 20. 8 ± 2 . 0 b
" " 60. 9 1 . 6 15.9 0.5 0 . 1 0.9 1.5 18.9 a
0.23 0.77 70.9 ± 1.7 1. 7 ± 0. 1 16.1 ± 0 . 1 0. 7 ± 0.1 0 . 1 0. 2 è 0 . 1 0.4 ± 0 .1 9. 9 ± 0.5 b
” ” 70. 9 ± 0. 8 1 . 8 ± 0 . 1 17.9 ± 0 .7 0. 7 ± 0.1 . 0. 5 ± 0. 1 0. 5 ± 0.1 0. 5 ± 0.1 7 .4 ± 0 .1 a
0 . 1 2 0. 88 73. 9 ± 1. 5 1. 9 ± 0.1 18. 5 ± 1. 2 0. 8 ± 0 . 1 0 0 0 . 1 4. 7 ± 0. 8 b
'• ” 71. 5 ± 3.7 2.1 ± 0. 5 20. 9 ± 2. 8 0. 9 ± 0.1 0 0 0 . 1 4 .4 ± 1 .1 a
0.06 0.94 75.4 ± 2 .6 1.-7 ± 0 . 1 18. 9 ± 2. 7 1. 8 ± 0.1 0 0 0 2.3 ± 0 . 2 b
" " 74.3 2 . 6 19. 6 ' . 0.7 0 0 0 2.8 a
0. 00 1.00 74.1 ± 2. 9 -2 . 0 ± 0 . 2 23;0 ± 2. 6 0. 9 ± 0.1 0 0 0 0 b
"72 .4 ± 1.1 2. 2 ± 0. 7 24. 2 ± 2. 5 1 . 1 ± 0 . 1 0 0 0 0 a
a Samples evacuated,
b Samples saturated w ith oxygen.
258 A.
SOK
OÍO
WSK
A
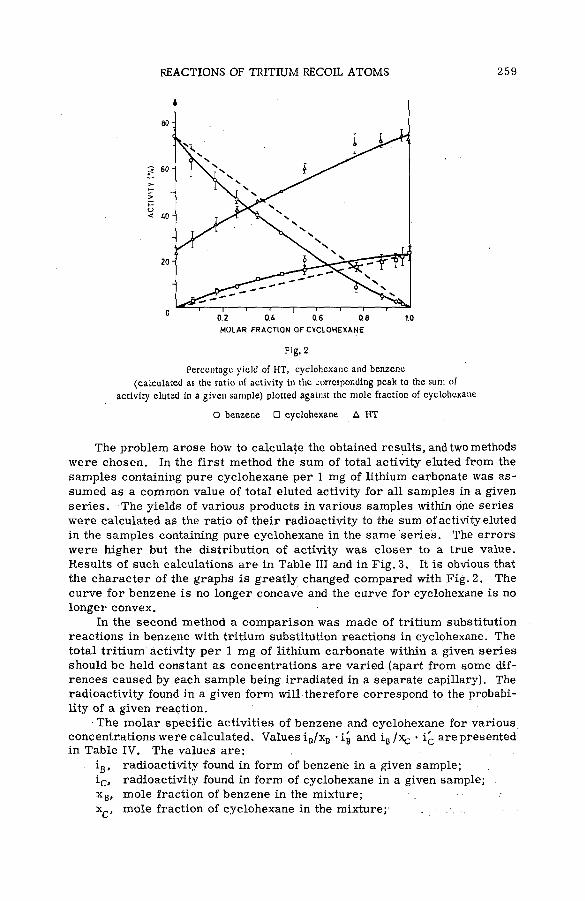
REACTIONS OF TRITIUM RECOIL ATOMS 259
Fig. 2
Percentage yield of HT, cyclohexane and benzene
(calculated as the ratio of activity in the corresponding peak to the sum of activity eluted in a given sample) plotted against the mole fraction of cyclohexane
О benzene □ cyclohexane Д HT
The problem arose how to calculate the obtained results, and two methods were chosen. In the first method thé sum of total activity eluted from the samples containing pure cyclohexane per 1 mg of lithium carbonate was assumed as a common value of total eluted activity for all samples in a given series. The yields of various products in various samples within one series were calculated as the ratio of their radioactivity to the sum of activity eluted in the samples containing pure cyclohexane in the same series. The errors were higher but the distribution of activity was closer to a true value.Results of such calculations are in Table III and in Fig. 3. It is obvious thatthe character of the graphs is greatly changed compared with Fig. 2. The curve for benzene is no longer concave and the curve for cyclohexane is no longer convex.
In the second method a comparison was made of tritium substitution reactions in benzene with tritium substitution reactions in cyclohexane. The total tritium activity per 1 mg of lithium carbonate within a given series should be held constant as concentrations are varied (apart from some dif- rences caused by each sample being irradiated in a separate capillary). The radioactivity found in a given form will therefore correspond to the probability of a given reaction.
The molar specific activities of benzene and cyclohexane for various concentrations were calculated. Values iB/xB • ig and iB /■Xq • i¿. arepresentedin Table IV. The values are: ,
ig, radioactivity found in form of benzene in a given sample;ic, radioactivity found in form of cyclohexane in a given sample;x£, mole fraction of benzene in the mixture; xc , mole fraction of cyclohexane in the mixture; . . .

260 A. SOKOÍOWSKA
TABLE П
COM PARISON OF THE SUM OF THE TOTAL TRITIUM AC TIV ITY E LU TED DURING GAS-CHROM ATOGRAPHY PROCESSES
IN THE SAM PLES CONTAINING PURE BEN ZEN E + L i2C0 3 AND IN THE SAM PLES CONTAINING PURE CYCLOHEXANE + L i2C03
Series
No.
Total activity
eluted in benzene samples
counts
Total activity eluted in
cyclohexane samples
counts
Ratio
A Cmg of Li2CO j mg of Li2C 0 3
A C
3 20 700 40400 0. 51a
4 32000 44 600 0 .7 1 b
5 61 250 111300 0.55 b
7 69 300 111400 0. 62 b
10 73 040 150 950 0 .4 8 c
1 1 107 970 213 900 0. 50 c
1 1 145 500 260 600 . 0. 56a
1 2 357 500 461900 0 .7 7 a
1 2 248 000 530 500 0.47 c
14 118300 188 000 0. 63 c
a Samples evacuated. k Samples with air. c Samples saturated with oxygen.
ig, radioactivity found in form of benzene in the samples containing pure benzene in a given series;
iç, radioactivity found in form of cyclohexane in the samples containing pure cyclohexane in a given series.
The specific activities of benzene and cyclohexane relative to the radioactivity found in form of benzene (or cyclohexane respectively) inthe samples containing pure benzene (or pure cyclohexane) in a given series were calculated in order to compare results obtained from different series.
The values in Table IV are equal, within an experimental error, to 1. The constant specific activities of cyclohexane and benzene indicate that the probability of substitution reactions of H by T in benzene and cyclohexane molecules is linearly proportional to the mole fraction of each of the component or is independent of the composition of a mixture. .
The ratio of specific activities iB • xc/xB • ic shown in F ig .4. does not depend on the irradiation conditions. As can be seen this ratio remains constant (within an experimental error) over the whole range of concentrations and equals about 1 . 8.
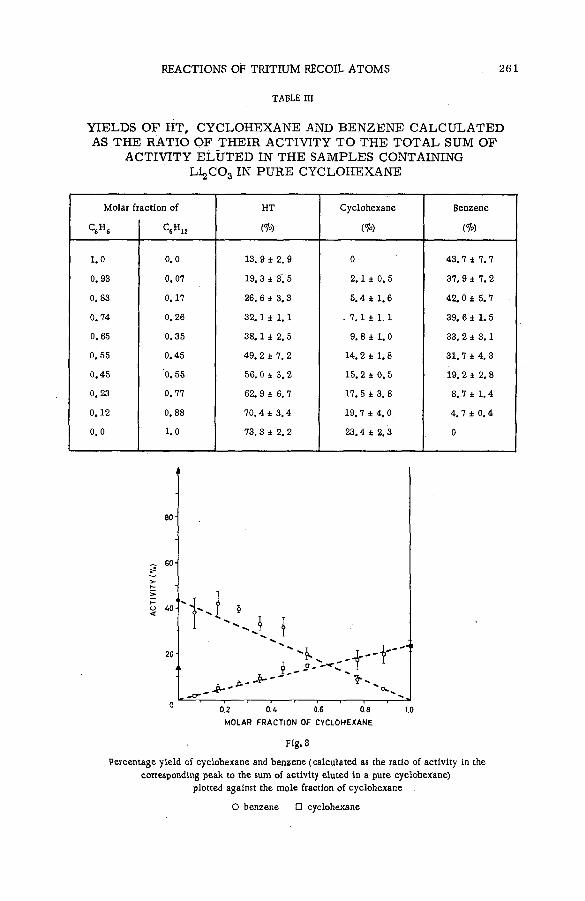
REACTIONS OF TRITIUM RECOIL ATOMS 261
TABLE III
YIELDS OF HT, CYCLOHEXANE AND BENZENE CALCULATED AS THE RATIO OF THEIR ACTIVITY TO THE TOTAL SUM OF
ACTIVITY ELUTED IN THE SAMPLES CONTAINING LLjCOg IN PURE CYCLOHEXANE
Molar fraction of HT Cyclohexane Benzene
9 ,H . c 6h 12 (%) (%) m
1 . 0 0.0 13. 9 è 2. 9 0 43.7 ± 7.7
0.93 0. 07 19. 3 è 3. 5 2 . 1 ± 0.5 37, 9 ± 7. 2
0. 83 0.17 26. 6 ± 3.3 5.4 i 1. 6 42. 0 ± 5. 7
0.74 0. 26 32.1 è 1.1 • 7 .1 è 1.1 39. 6 è 1. 5
0.65 0.35 38.1 è 2. 5 9. 8 ± 1. 0 33. 2 ± 3 .1
0.55 0.45 49. 2 i 7. 2 14. 2 ± 1. 8 31. 7 ± 4. 3
0,45 0.55 56. 0 ± 3. 2 15. 2 ± 0. 5 19. 2 i 2. 8
0.23 0.77 62. 9 i 6. 7 17. 5 ± 3. 8 8. 7 ± 1.4
0 . 1 2 0. 88 70.4 1 3 .4 19.7 ± 4 .0 4 .7 ± 0 .4
0.0 1 . 0 73. 3 à 2. 2 23.4 i 2. 3 0
■ Fig.3
Percentage yield of cyclohexane and benzene (calculated as the ratio of activity in the
corresponding peak to the sum of activity eluted in a pure cyclohexane) plotted against the mole fraction of cyclohexane
О benzene □ cyclohexane
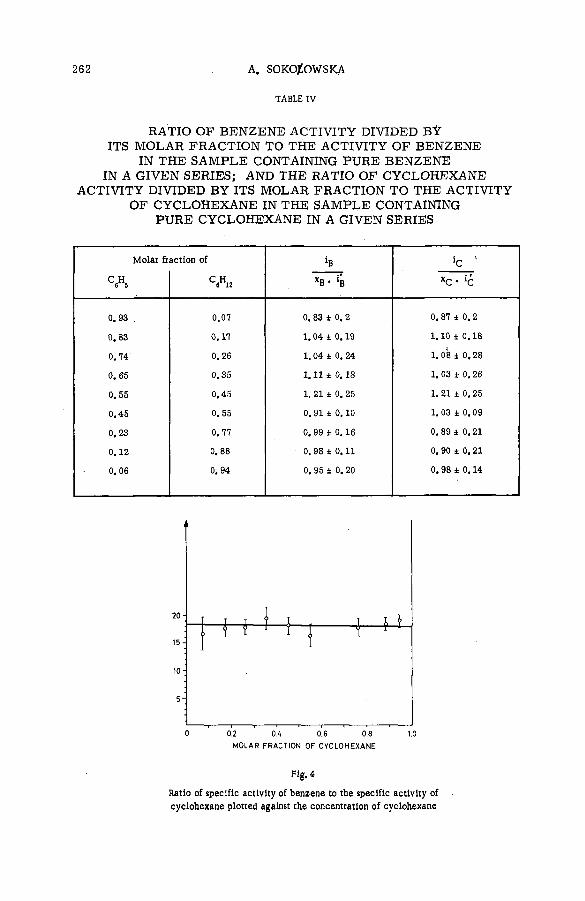
262 A, SOKOJtoWSKA
TABLE IV
RATIO OF BEN ZEN E AC TIV ITY DIVIDED BY ITS M OLAR FRACTION TO THE ACTIV ITY OF BENZENE
IN THE SAM PLE CONTAINING PURE BEN ZEN E IN A G IVEN SERIES; AND THE RATIO OF CYCLOHEXANE
AC TIV ITY DIVIDED BY ITS MOLAR FRACTION TO THE ACTIV ITY OF CYCLOHEXANE IN THE SAM PLE CONTAINING
PURE CYCLOHEXANE IN A GIVEN SERIES
Molar fraction of ‘в
C 6H5 с л г хв • ‘в хс • *С
0.93 0.07 0. 83 ± 0. 2 0. 87 ± 0. 2
0.83 0.17 1.04 ± 0.19 1 . 1 0 ± 0 .1 8
0. 74 0. 26 1. 04 ± 0. 24 1 . 08 è 0.28
0.65 0.35 1 . 1 1 ± 0.18 1. 03 ± 0. 26
0. 55 0.45 1. 21 à 0. 25 1. 21 ± 0.25
0.45 0. 55 0.91 ± 0 . 1 0 1. 03 ± 0.09
0.23 0.77 0. 99 ± 0.16 0. 89 ± 0.21
0 . 1 2 0. 88 0. 98 ± 0.11 0. 90 i 0.21
- 0.06 0.94 0. 95 ± 0. 20 0. 98 ± 0.14
10 - .
5 -
T----------r i---------- 1---------- 1 i----------г--------- 1---------- 1---------- 1 '
0 0.2 0.4 0.6 0.8 1.0
MOLAR FRACTION OF CYCLOHEXANE
Fig. 4
Ratio of specific activity of benzene to the specific activity of
cyclohexane plotted against the concentration of cyclohexane
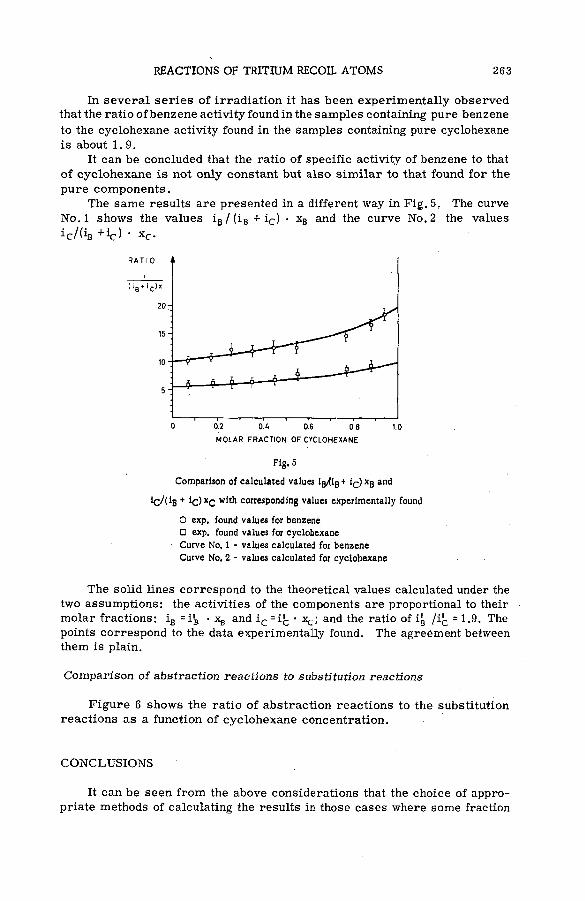
REACTIONS OF TRITIUM RECOIL ATOMS 263
In several series of irradiation it has been experimentally observed that the ratio of benzene activity found in the samples containing pure benzene to the cyclohexane activity found in the samples containing pure cyclohexane is about 1.9.
It can be concluded that the ratio of specific activity of benzene to that of cyclohexane is not only constant but also sim ilar to that found for the pure components.
The same results are presented in a different way in Fig. 5. The curve No. 1 shows the values iB/ (iB + ip) • xB and the curve No. 2 the values
*сА*в ‘ xc-
MOLAR FRACTION OF CYCLOHEXANE
Fig. 5
Comparison of calculated values lg/tlg + ic ) Xg and
ic/(ÍB + *c ) XC wlt*1 corresponding values experimentally found
О exp. found values for benzene
□ exp. found values for cyclohexane
Curve No. 1 - values calculated for benzene
Curve No. 2 - values calculated for cyclohexane
The solid lines correspond to the theoretical values calculated under the two assumptions: the activities of the components are proportional to their molar fractions: iB =i'B • xB and ic = i{- • and the ratio of ig /i¡~ =1.9. The points correspond to the data experimentally found. The agreement between them is plain.
Comparison of abstraction rea c tion s to substitution reactions
Figure 6 shows the ratio of abstraction reactions to the substitution reactions as a function of cyclohexane concentration.
CONCLUSIONS
It can be seen from the above considerations that the choice of appropriate methods of calculating the results in those cases where some fraction
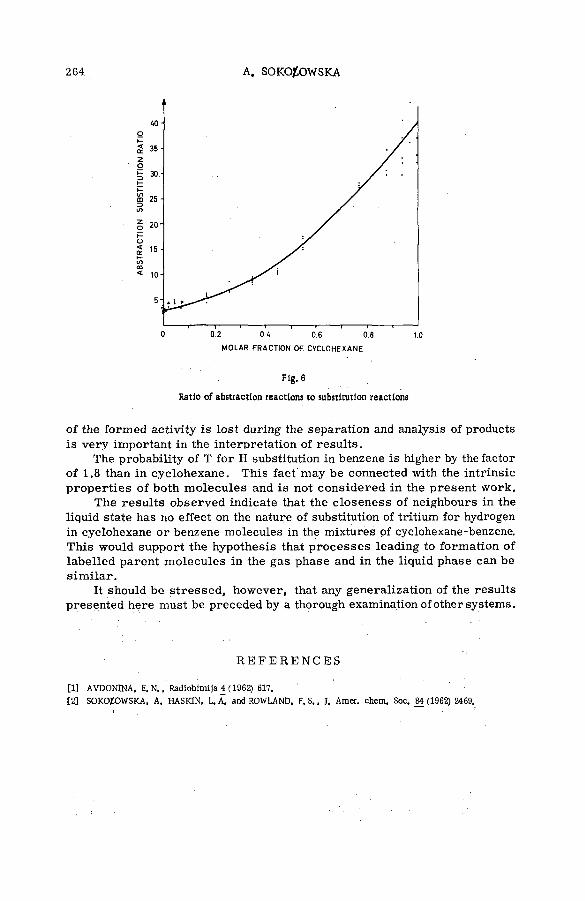
264 A. SOKOÍOWSKA
MOLAR FRACTION OF CYCLOHEXANE
Fig. 6
Ratio of abstraction reactions to substitution reactions
of the formed activity is lost during the separation and analysis of products is very important in the interpretation of results.
The probability of T for H substitution in benzene is higher by the factor of 1.8 than in cyclohexane. This fact may be connected with the intrinsic properties of both molecules and is hot considered in the present work.
The results observed indicate that the closeness of neighbours in the liquid state has no effect on the nature of substitution of tritium for hydrogen in cyclohexane or benzene molecules in the mixtures of cyclohexane-benzene. This would support the hypothesis that processes leading to formation of labelled parent molecules in the gas phase and in the liquid phase can be sim ilar.
It should be stressed, however, that any generalization of the results presented here must be preceded by a thorough examination of other systems.
R E F E R E N C E S
[1 ] AVDONINA, E .N .. Radiohimija 4 (1962) 617. ' ‘[2 ] SOKOXOWSKA, A. HASKIN, L. A. and ROWLAND, F. S .. J. Amer. chem. Soc. 84 (1962) 2469.

HOT PHOSPHORUS ATOM REACTIONS IN LIQUID ORGANIC MIXTURES
A, SIUDADEPARTMENT OF RADIOCHEMISTRY. INSTITUTE OF NUCLEAR RESEARCH,
WARSAW, POLAND
Abstract — Résumé — Аннотация — Resumen
HOT PHOSPHORUS ATOM REACTIONS IN LIQUID ORGANIC MIXTURES. The effect of the organic
diluents on the yield of P32-labelled phenylphosphonic acid formed in 1M benzene solutions of phosphorus
trichloride as a result of the P3Hn, y)P32 reaction has been investigated. The yield of phenylphosphonic acid
decreases linearly with the increase in cyclohexane mole fraction. On the other hand, with carbon tetrachloride dilution the yields of phenylphosphonic acid are greater than one could expect from the composition
of the mixtures. The results show that the reactions between the recoil phosphorus atoms and the benzene
molecules resulting in phenylphosphonic acid can be affected by surrounding molecules.
REACTIONS DES ATOMES CHAUDS DE PHOSPHORE DANS LES MELANGES LIQUIDES ORGANIQUES. On a examiné l ’ influence des diluants organiques sur le rendement en acide phénylphosphonique marqué avec
32p et formé par la réaction 31P(n,y)32P dans une solution molaire de trichlorure de phosphore dans du benzène. Le rendement de l'acide phénylphosphonique diminue d’une façon linéaire avec l ’augmentation de la fraction
molaire du cyclohexane. Par contre, le rendement de l'acide phénylphosphonique dans le tétrachlorure de
carbone est supérieur à celui auquel on aurait pu s’attendre sur la base de la composition chimique des mélanges. Les résultats indiquent que la formation de l’acide phénylphosphonique, due aux réactions des atomes
de recul de phosphore avec les molécules du benzène, dépend de la nature du milieu.
РЕАК Ц И И Г О Р Я Ч Е Г О А Т О М А Ф ОСФОРА В ЖИДКИХ О Р Г А Н И Ч Е С К И Х С М Е С Я Х . И с следовано влияние органических разбавителей на выход фенилфосфоновой кислоты, меченной P 3Î после реакции Р 31 (п ,-у )Р 32 в 1 М растворах трихлорида фосфора в бензоле . Выход фенилфосфоновой кислоты линейно уменьшается с увеличением молярной доли циклогексана. Однако при разбавлении четыреххлористым углеродом выход фенилфосфоновой кислоты больше ож идаемого из состава см есей.. Из полученных результатов следует, что на образование фенилфосфоновой кислоты вследствие реакции атомов отдачи фосфора с молекулами бензола воздействуют смежные молекулы.
REACCIONES DE LOS ATOMOS CALIENTES DE FOSFORO EN MEZCLAS DE LIQUIDOS ORGANICOS. Se ha estudiado la influencia de los diluentes orgánicos sobre el rendimiento de ácido fenilfosfónico.-32P y
formado en la reacción 31P(n,y)32P, en soluciones de tricloruro de fósforo 1 M en benceno. El rendimiento
de ácido fenilfosfónico es inversamente proporcional a la fracción molar del ciclohexano. Por el contrario, el rendimiento del ácido fenilfosfónico en el tetracloruro de carbono es superior al que cabfa esperar sobre
la base de la composición de las mezclas. Los resultados indican que la formación de ácido fenilfosfónico
en virtud de las reacciones entre los átomos de retroceso de fósforo y las moléculas de benceno depende de la
naturaleza de las moléculas del medio.
INTRODUCTION
Atoms created by nuclear transformations usually possess a high kinetic and potential energy, the latter being due to an electric charge caused by ionization and excitation of electronic orbitals. These atoms, because of
265

266 A. SIUDA
their energy, show a high reactivity towards a medium, which leads the formation of stable chemical combinations.
In general one can assume that the yield of a given product formed by the recoiling atom after it has been released from the parent molecule depends on the following factors:1. Moderation, or in other words the energy loss from the recoil atom.
This process can have a substantial influence on the fate of the hot atom, since it determines a number of atoms that are able to react in a given way as well as their energy, charge and degree of excitation. The moderation process itself should be dependent on the initial energy of the atom and moderating properties of the environment.
2. Collision of the recoiling atom with a molecule of the medium, leading to the formation of a chemical bond. It should be independent of the environmental properties, and the only factors determining this process are the structure and properties of the struck molecule on one hand, and the energy state of the hot atom on the other.
3. Stabilization of a product formed by the collision. Such a newly formed product can possibly possess some excitation energy and can be stabilized as a chemical compound only when the excess of its energy will be transferred to the vicinal molecules. This process ought to be dependent on the environmental properties, particularly regarding the ability of a given medium to receive the energy from the excited species.
The above model concerns only the reactions of atoms with an energy exceeding the thermal level; i. e. it can be applied only to the hot reactions.
The processes leading to the formation of the stable chemical combinations, especially in organic liquid systems, are very complex and can be influenced by many factors. One of the most important is the physicochemical properties of the medium. It seems that the investigation of re coil reactions in liquid organic mixtures at various molar fractions of the components can offer some information concerning the role of the environment. If in such a system the yield of a given product does not linearly depend on the mole fraction of reacting components one can conclude that the reaction is influenced by the composition of the medium. From the point of view of the moderation and stabilization mechanism a study of a given hot atom reaction in the mixtures of various organic solvents could be also interesting.
In this work the formation of phenylphosphonic acid by recoiling phosphorus-32 in the mixtures of benzene and cyclohexane as well as benzene and carbon tetrachloride has been investigated.
EXPÉRIMENTAL
The recoil phosphorus atoms were produced in the nuclear reaction P 31/ n , Y / P 3 2 . The samples of benzene-cyclohexane and benzene-carbon tetrachloride mixtures containing the same amounts of phosphorus trichloride corresponding to 1M solution sealed in glass ampoules in the presence of air were irradiated at a flux of 3X ion n/cm2, s in the thermal column of the reactor "EW A" for 2 h, the gamma dose rate being 4. 7X 106 rads per h.
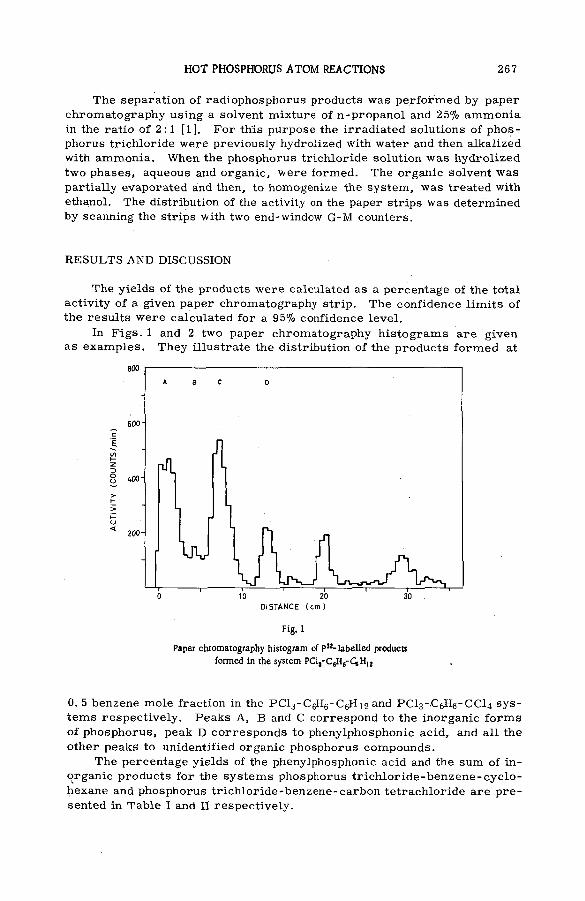
HOT PHOSPHORUS ATOM REACTIONS 267
The separation of radiophosphorus products was performed by paper chromatography using a solvent mixture of n-propanol and 25% ammonia in the ratio of 2 :1 [1]. For this purpose the irradiated solutions of phosphorus trichloride were previously hydrolized with water and then alkalized with ammonia. When the phosphorus trichloride solution was hydrolized two phases, aqueous and organic, were formed. The organic solvent was partially evaporated and then, to homogenize the system, was treated with ethanol. The distribution of the activity on the paper strips was determined by scanning the strips with two end-window G-M counters.
RESULTS AND DISCUSSION
The yields of the products were calculated as a percentage of the total activity of a given paper chromatography strip. The confidence limits of the results were calculated for a 95% confidence level.
In Figs. 1 and 2 two paper chromatography histograms are given as examples. They illustrate the distribution of the products formed at
Fig. 1
Paper chromatography histogram of PS2-labelled products
formed in the system PC1S-C6H6-QH12 .
0. 5 benzene mole fraction in the PC^-C^Hg-CgH^ and PCI3-C 6H6-CC I4 systems respectively. Peaks A, В and С correspond to the inorganic forms of phosphorus, peak D corresponds to phenylphosphonic acid, and all the other peaks to unidentified organic phosphorus compounds.
The percentage yields of the phenylphosphonic acid and the sum of inorganic products for the systems phosphorus trichloride-benzene-cyclohexane and phosphorus trichloride-benzene-carbon tetrachloride are presented in Table I and II respectively.

268 A. SIUDA
Fig. 2
Paper chromatography histogram of P32-labelled products
formed in the system PC13-C6H 6-CC14
It seems that the yield of phenylphosphonic acid decreases linearly (Fig. 3) within the experimental error with increasing cyclohexane concentration, but the yield of phenylphosphonic acid per molar fraction of benzene
TABLE I
YIELD OF PHENYLPHOSPHONIC ACID IN THE SYSTEM PHOSPHORUS TRICHLORIDE-BENZENE-CYCLOHEXANE
Mole fractionTotal P32 activity
C6H 6 C6H 12 P. . inorganic
PhPO/OH/2
1.0 0.0 77 ±4 18. 7 ± 1
0.9 0.1 70 ±1 16.0*0 ,4
0.7 0.3 69 ±4 12. 2±0.9
0.5 0.5 68 8. 6 *0 .9
0.4 0.6 70 5. 6* 0. 9
0.3 0.7 65. 5±0. 8 . 4. 2*0. 9
0.2 0.8 61 2. 9± 0. 9
0.1 0.9 58.4 ± 0.4 1 ±1
0.0 1.0 55 ±1 0 il
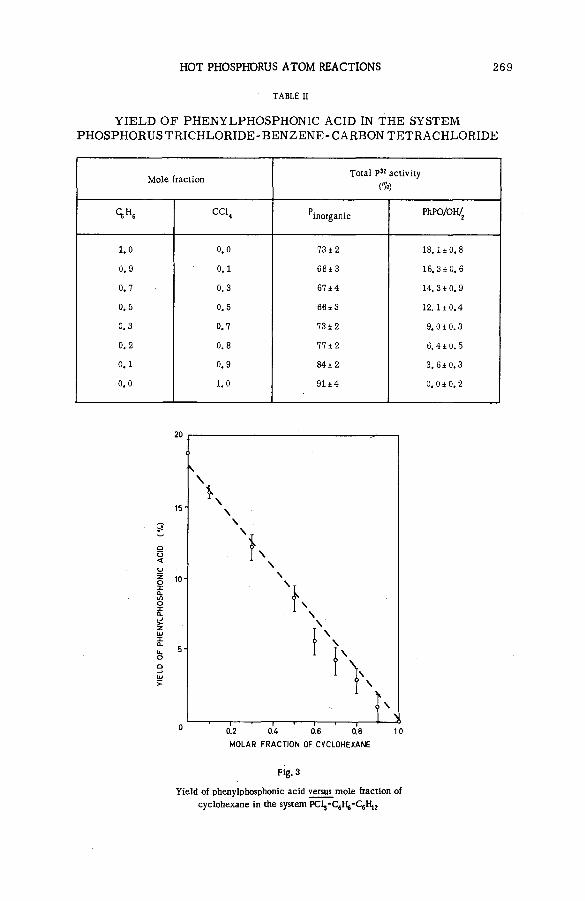
HOT PHOSPHORUS ATOM REACTIONS 269
TABLE II
YIELD OF PHENYLPHOSPHONIC ACID IN THE SYSTEM PHOSPHORUSTRICHLORIDE-BENZENE-CARBON TETRACHLORIDE
Mole fractionTotal P32 activity
ч н бcc i4 P- .
inorganicPhPO/OH/2
1.0 0.0 73 ±2 18. liO .8
0.9 ' 0.1 6813 16.31 0. 6
0.7 0.3 6714 14. 31 0. 9
0.5 0.5 6613 12. 1Ю .4
0.3 0.7 73 ±2 9. 01 0. 3
0.2 0.8 7712 6. 41 0.5
0.1 0.9 841 2 3. 6Ю .З
0.0 1.0 9114 0.0 Ю . 2
Fig.3
Yield of phenylphosphonic acid versus mole fraction of
cyclohexane in the system PClj-CgHj-CjHu
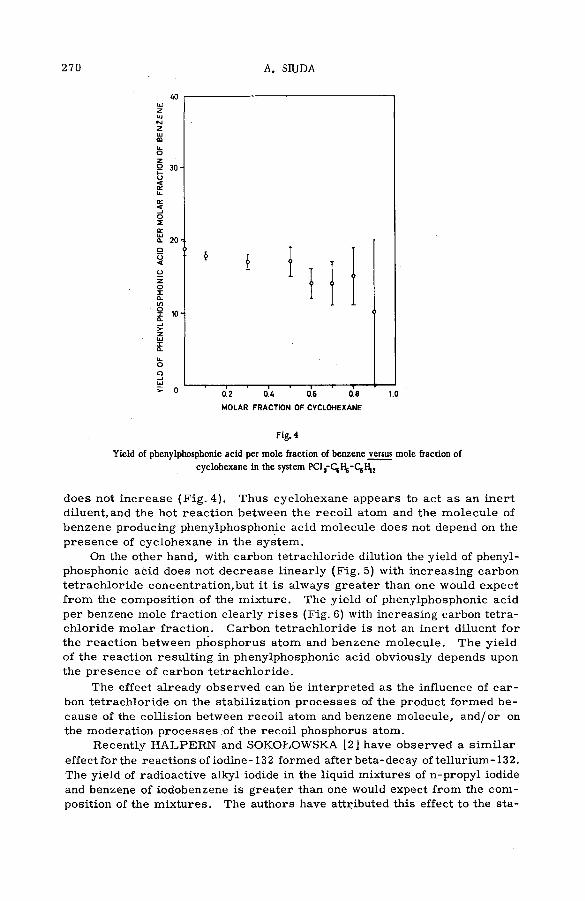
270 A. SIUDA
Fig. 4
Yield of phenylphosphonic acid per mole fraction of benzene versus mole fraction of
cyclohexane in the system PClj-C^-CjH,;
does not increase (Fig. 4). Thus cyclohexane appears to act as an inert diluent, and the hot reaction between the recoil atom and the molecule of benzene producing phenylphosphonic acid molecule does not depend on the presence of cyclohexane in the system.
On the other hand, with carbon tetrachloride dilution the yield of phenylphosphonic acid does not decrease linearly (Fig. 5) with increasing carbon tetrachloride с on centration, but it is always greater than one would expect from the composition of the mixture. The yield of phenylphosphonic acid per benzene mole fraction clearly rises (Fig. 6) with increasing carbon tetrachloride molar fraction. Carbon tetrachloride is not an inert diluent for the reaction between phosphorus atom and benzene molecule. The yield of the reaction resulting in phenylphosphonic acid obviously depends upon the presence of carbon tetrachloride.
The effect already observed can be interpreted as the influence of carbon tetrachloride on the stabilization processes of the product formed because of the collision between recoil atom and benzene molecule, and/or on the moderation processes of the recoil phosphorus atom.
Recently HALPERN and SOKOLOWSKA [2J have observed a sim ilar effect for the reactions of iodine-132 formed after beta-dec ay of tellurium-132. The yield of radioactive alkyl iodide in the liquid mixtures of n-propyl iodide and benzene of iodobenzene is greater than one would expect from the composition of the mixtures. The authors have attributed this effect to the sta-
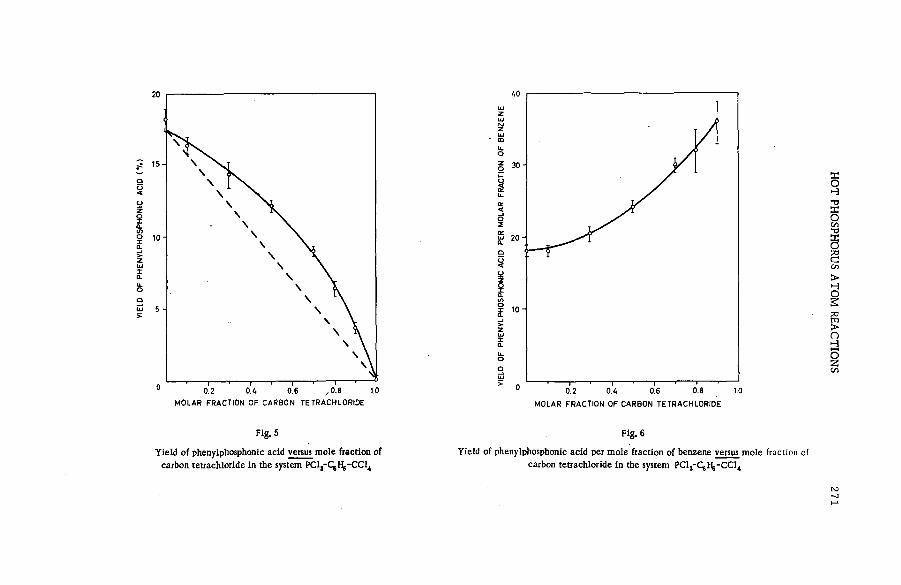
Fig. 5
Yield of phenylphosphonic acid versus mole fraction of
carbon tetrachloride in the system РС13-С^Ц-СС14
YIEL
D OF
PH
ENVL
PHO
SPHO
NJC
ACID
PE
R MO
LAR
FRAC
TION
OF
B
EN
ZEN
E
MOLAR FRACTION OF CARBON TETRACHLORIDE
Fig. 6
Yield of phenylphosphonic acid per mole fraction of benzene versus mole fraction of
carbon tetrachloride in the system РС18-С^Ц-СС14
HOT PH
OSPH
ORU
S ATOM
REA
CTIO
NS
271

272 A. SIUDA
bilization process of the created product. In the aromatic solvents the latter can be easily stabilized since the excitation energy can be transferred to benzene or iodobenzene molecules because the ionization potentials of aromatic compounds are lower than those of alkyl iodides.
However, in the phosphorus trichloride solutions the ionization potentials of both the diluents are higher than those of benzene. Under these conditions the energy transfer from the excited products to neighbouring molecules according to the mechanism suggested above seems unlikely. Even if the reaction of phenylphosphonic acid formation is associated with any unstable and excited product the mechanism of the stabilization of the latter is possibly different.
It is also possible that the presence of carbon tetrachloride in the system is important for the moderation of the phosphorus atom. It might be caused by the sim ilar atomic masses of phosphorus and chlorine. The slowing-down process would therefore be operated by another mechanism; and the energetic state of the atom colliding with the benzene molecule could be different, which could, of course, change the yield of phenylphosphonic acid.
On the basis of these results it is not possible to decide which of these two factors, the moderation or stabilization, is more important as far as the fate of the radiophosphorus atom in benzene is concerned. The experiments are being continued.
A C K N O W L E D G E M E N T S .
The author wishes to express his gratitude to S. Siekierski and A. Sokoiowska for helpful discussions.
R E F E R E N C E S
LU SIUDA, A . , to be published.[2 ] HALPERN, A. and SOKOfcOWSKA, A . , J. inorg. nucl. Chem. (in press).

HOT PHOSPHORUS ATOM REACTIONS 273
D IS C U S S IO N
(on the foregoing two papers)
O. JOVANOVKÍ: In connection with the paper presented by Dr. Siuda we should like to report briefly on the following work.
At the Institute for Nuclear Physics Research in Amsterdam, Netherlands, we determined, in co-operation with Professor Aten and Dr. Lindner, the distribution of P 3 2 recoils produced in solid triphenylphosphate (the triester of orthophosphoric acid). The retention was found to be strongly influenced by the irradiation conditions, ranging from 25% to 7% as a lower limit. P 32-labelled products different from the parent compound were analysed after extraction into an aqueous phase by high voltage electrophoresis (Fig. 1). This figure shows a typical electrophoresis histogram in which at least 15 activity peaks can be recognized. The majority of these peaks are believed to be identifiable as shown in Table I. I draw your attention to the first two columns listing the names of the species. Peaks K, L, M and N are inorganic species that are, respectively, orthophosphate, pyrophosphate, phosphite and hypophosphite, and together make up about 20% of the total activity. The remaining 11 peaks are organic species of two different types: first, 50% of the activity as esters with a P -O -C bond like R, triphenylphosphate (representing the retention), F, diphenylphosphate, G, monophenylphosphate, I, diphenylphosphite and H, the non-aromatic ester, monomethylphosphate; secondly, organophosphorus compounds with direct С-P bonds like J, monophenylphosphinic acid, D, diphenylphosphinic acid, C, monophenylphosphonic acid and again one non-aromatic species, E, which is dimethylphosphinic acid. Peaks A and В have not.yet been identified but are believed to be also of an aromatic nature.
We wpuld draw attention, in particular, to the main findings resulting from our work, which are: first, the much lower inorganic yield of less than 20% compared to 75% in pure benzene, as reported by Dr. Siuda; secondly, the small yield of aliphatic compounds, probably reflecting the relative stability of the aromatic ring towards recoils; and thirdly, the formation of С-P bonds, which are most reasonably explained by direct hot hydrogen displacement.
I wonder whether Dr. Siuda can say something about the nature of the inorganic : species determined in his system, and whether he has an explanation for the very large difference in the inorganic yield compared with our system. .
A. SIUDA: In answer to your questions I should like to draw attention to Figs. 1 and 2 in the paper, which illustrate the distribution of the products formed at 0. 5 benzene mole fraction in the P C I3-C 6 Нб -Ce H12 and РС13-СбН 6-C C I4 systems respectively. Peak A migrates with the same velocity as pyrophosphate ion, peak В corresponds to orthophosphate and hypophosphate ions, and peak С corresponds to phosphite ion. The sum of the percentage yields of peaks A, В and С is the inorganic yield given in Tables I and II.
With regard to the second question, it seems that to make a direct comparison between these two systems - solid triphenylphosphate and liquid benzene solutions of phosphorus trichloride - would be rather difficult. How-
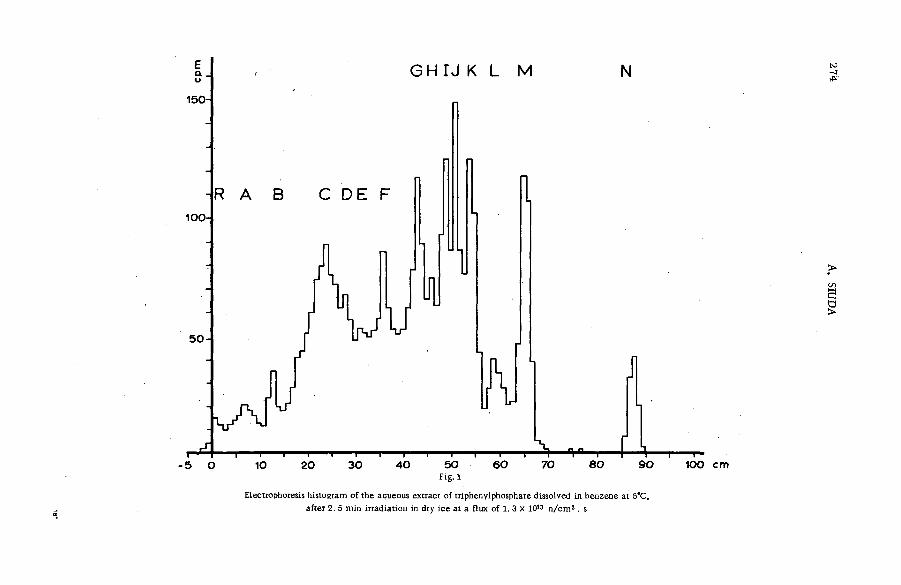
Fig-1
Electrophoresis histogram of the aqueous extract of triphenylphosphate dissolved in benzene at 5°C, after 2. 5 min irradiation in dry ice at a flux of 1.3 x 1013 n/cm2 . s
van
is
у

HOT PHOSPHORUS ATOM REACTIONS 275
TABLE I
AVERAGE DISTRIBUTION OF P32 RECOILS, SPECIES IN AQUEOUS SOLUTION
Special treatment none
(0O )SP5 R 25.5
? A 2 .4
? в 2 .9
0 lP 3 С 11.3
Î 0гР1 D 3.8
CL)О ( C H ^ P5 E 3.7a)eu
(00)2 P5 F 6.7
13(.00)1 P5 G 8.6
Л(C H jO hP5 H 2 .6
'a. (0O)¡¡ P3 I 6.8
<Zi P1 J 6.2
P 5 K 6.5
p50 p5 L 3.4
p3 M 6.8
P1 N 3.0
ever, in the case of tributylphosphate, which was investigated in our laboratory a few years ago, we found about 20% of P 32 activity in the inorganic forms and the primary retention in the form of tributylphosphate was low, about 5%. The inorganic yield was strongly dependent on the ir radiation conditions.
F .S . ROWLAND: As pointed out by Dr. Sokolowska, the discrepancy between her results and those obtained by Dr. Avdonina, referred to in the introduction to the paper, arises from a different basis of calculation of the total activity. Dr. Avdonina's results are calculated on the basis of total observed activity, while Dr. Sokotowska's results are calculated on an absolute yield basis, and show a decrease in observed activity as the mole fraction of benzene increases. Dr. Sokoîowska's interpretation suggests that a large amount of the tritium activity obtained from recoil tritium re actions with benzene does not pass through the usual gas chromatographic detector, and is therefore not taken into account in Dr. Avdonina’s estimate of the total activity.
Dr. John Garland and I have investigated the reactions of recoil tritium atoms with benzene in both the liquid and gas phases*. For liquid phase ir radiations, the experiments were performed in the following manner: The
* Ph.D. thesis, University of Kansas (1963).

276 A. SIUDA
ordinary gas chromatographic procedures were used for determining HT and benzene-T and other products (cyclohexadiene-T etc.). The gas-flow stream was then reversed and allowed to flow backwards for a long period. At the time corresponding to the total time of forward flow, a large broad radioactivity peak emerged from the column. This tritium activity represents volatile polymetric tritiated products. Dr. Garland also washed the container walls and found non-volatile high polymeric tritiated species. The total polymeric tritium activity thus found by Dr. Garland corresponds rather closely with the amount of activity suggested as "missing" by Dr. Sokoiowska's experiments, and confirms the validity of her method of calculation based on total yield.

РЕАКЦИИ ГОРЯЧИХ АТОМОВ ТРИТИЯ С АЛИФАТИЧЕСКИМИ СПИРТАМИ И ИХ
СМЕСЯМИ С БЕНЗОЛОМ И ЦИКЛОГЕКСАНОМ
Э .С . Ф ИЛАТОВ,Ан.Н . НЕСМЕЯНОВ и ЦЗЯН ТАЙ-ВАН МОСКОВСКИЙ ГОСУДАРСТВЕННЫЙ УНИВЕРСИТЕТ, МОСКВА
СССР
Abstract — Résumé — Аннотация — Resumen
REACTIONS OF HOT TRITIUM ATOMS WITH ALCOHOLS AND WITH SOME OF THEIR BINARY HYDROCARBON
MIXTURES. The reaction yields of hot tritium atoms were studied with the aim of investigating the influence
exerted by processes of intramolecular and intermolecular energy transfer in reactions of hot tritium atoms with, firstly, aliphatic alcohols, and, secondly, in binary mixtures of alcohols with benzene and cyclohexane,
as a function of concentration of components, and length of the alcohol molecule.Mixtures of lithium carbonate and the experimental substances were irradiated in a reactor at fluxes
varying between 1011 and 1013 n/cm2»s, over periods of between 15 and 6 h. Analysis was carried out on a chromatographic unit with interchangeable columns (molecular sieves, tricresylphosphate and liquid
petrolatum on kieselguhi, and cotton wool treated with methyl alcohol).• The data obtained can be explained in terms of the existence of a mechanism for the effective intra
molecular energy transfer from the collision site of the hot atom along the hydrocarbon chain of the alcohol molecules to the OH group capable of dissipating it. The influence of the intermolecular energy transfer process has not been unequivocally established. It is possible that a considerable part is played in this process
by the difference in microscopic cross-section of the primary event'in the interaction of the hot atom with
the molecule, and by the change in the moderating atom spectrum with the increase in concentration of one
of the components.
RÉACTIONS DES ATOMES CHAUDS DE TRITIUM AVEC DES ALCOOLS ET CERTAINS MELANGES BINAIRES D'ALCOOLS ET D'HYDROCARBURES. En vue d'étudier les effets qu'exercent les processus de transfert d'énergie intramoléculairès et intermoléculaires lors des réactions provoquées par les atomes chauds de tritium, les auteurs ont étudié les rendements des réactions entre ces atomes et des alcools aliphâtiques d*une part, des mélanges binaires alcool-benzol et alcool-cyclohexane d’autre part, en fonction de la concentration des
composants et de la longueur des molécules d'alcool.Des mélanges de carbonate de lithium et de substances à étudier ont été irradiés en pile dans un flux
de neutrons de 10*1 à Ш 9 n/cm2-s pendant 6 à 15 heures. L'analyse a été faite à l'a ide d'un appareil de
chromatographie sur colonne avec supports interchangeables (tamis moléculaires, phosphate de tricrésol et huile de vaseline sur terre d'infusoires, coton hydrophile traité à l'a lcool méthylique).
Les résultats obtenus peuvent s'expliquer par l'existence d'un mécanisme efficace de transfert d'énergie
intramoléculaire entre le point d'impact de l’atome chaud et le groupe OH, capable de dissiper cette énergie, en passant par la chaîne aliphatique des molécules d’alcool. L'influence du processus de transfert d'énergie
intermoléculaire n'a pu être établie avec précision. Il se peut que des influences tout aussi importantes soient celle de la différence qu'accusent les sections efficaces microscopiques d'interaction primaire atomes chauds- molécules, et celle de la modification que subit le spectre des atomes lors de leur ralentissement, à mesure
qu'augmente la concentration de l'un des éléments.
РЕАК Ц И И Г О Р Я Ч И Х А Т О М О В Т Р И Т И Я СО СП И РТ А М И И Н ЕК О ТО РЫ М И ИХ Б И Н А Р НЫ МИ С М Е С Я М И С У Г Л Е В О Д О Р О Д А М И . С целью исследования влияния процессов внутримолекулярной и межмолекулярной передачи энергии при реакциях горячих атомов трития были исследованы выходы реакции горячих атомов трития:
1 ) с алифатическими спиртами; 2 ) в бинарных смесях спиртов с бензолом и циклогекса- ном, в зависимости от концентрации компонентов и от длины спиртовой молекулы.
Смеси карбоната лития с исследуемыми веществами были облучены на реакторе в потоках от Ю Н до 1013 н/см 2 • сек в течение 15 — 6 часов. Анализ проведен на хроматографе
277

2 7 8 Э . С . Ф И Л А Т О В и д р .
со сменными колонками (молекулярные сита, трикреэилфосфат и вазелиновое масло на диатомите, вата, обработанная метиловым спиртом).
Полученные данные м огут быть объяснены с точки зрения существования механизма эффективной внутримолекулярной передачи энергии от места удара горячего атома по у гле водородной цепи молекул спиртов к группе ОН , способной ее рассеивать- Влияние процесса межмолекулярной передачи энергии не установлено однозначно. Возможно, не меньшую роль играют различие микросечений первичного акта взаимодействия горячего атома с молекулой и изменение спектра замедляющихся атомов по мере роста концентрации одного из компонентов.
REACCIONES DE ATOMOS DE TRITIO CALIENTES CON ALCOHOLES Y CON ALGUNAS DE SUS MEZCLAS
BINARIAS HIDROCARBURADAS. Con el fin de estudiar la influencia de los procesos de transferencia intramolecular de energía en reacciones de átomos de tritio calientes, los autores investigaron los rendimientos
de las reacciones de este tipo de átomos, primero, con alcoholes alifáticos, y segundo, en mezclas binarias
de alcoholes con benceno y ciclohexano, en función de la concentración de los componentes y la longitud
de la molécula de alcohol.Los autores irradiaron en un reactor con flujos de ion a 2013 neutrones /cm2*s, durante 15 a 6 h, mezclas
de carbonato de litio con las sustancias que se querían estudiar. Efectuaron el análisis por cromatografía con
columnas intercambiables (tamices moleculares, fosfato de tricresilo y parafina líquida en diatomeas, algodón
hidrófilo tiatado con metanol).Los datos obtenidos pueden explicarse suponiendo la existencia de un mecanismo eíicaz para la trans
ferencia intramolecular de energía del lugar del impacto del átomo caliente en la cadena carbonada de las
moléculas de los alcoholes hacia el grupo OH, que es capaz de disiparla. No se ha podido establecer con
certeza absoluta la influencia de la transmisión intermolecular de energía. Es posible que desempefle un papel igualmente importante la existencia de distintas secciones eficaces microscópicas para la interacción primaria
del átomo caliente con la molécula y la modificación del espectro de los átomos moderadores a medida que
aumenta la concentración de uno de los componentes.
Для изучения кинетики высокоэнергетических реакций в настоящее время используются горячие атомы, получающиеся при ядерных превращениях. В отличие от атомов отдачи галогенов, где существование реакций упругих столкновений показано с определенностью [1 —4], механизм первичного акта взаимодействия атомов отдачи трития с органическими молекулами не решен однозначно. Только ли пространственные факторы и вероятность реакции на удар по связи играют роль [5,6] или осуществляется переходный комплекс, вероятность образования которого определяется сечением взаимодействия, а вероятность pacnáfla энергией химической связи [7 ]? При значительных энергиях движения атомов время существования возбужденного комплекса атом —молекула очень мало и тем меньше, чем больше возбуждение. При времени жизни ~10-14 сек, т .е . порядка времени колебания связи, рассмотрение какого-либо комплекса в обычном понимании оказывается невозможным. Однако несомненно, что какое-то переходное состояние между начальной стадией (до столкновения) и стадией связанного атома должно осуществляться. Процесс локального удара горячего атома по молекуле с возбуждением уровней энергии у ближайших атомов при неизменном состоянии остальной части молекулы .затрудняет выяснение механизма реакции. '
Изучение изотопных эффектов в реакциях горячих атомов трития с простыми и дейтерированнымисоединениями [8 — 10 ] показали, что они совпадают с отношением частот нулевых колебаний связанных атомов водорода и дейтерия. В теории кинетики химических реакций это соответствует предельному случаю сохранения длины связи в переходном состоя

Р Е А К Ц И И Г О Р Я Ч И Х А Т О М О В Т Р И Т И Я 279
нии [ 1 2 ] и указывает на прямой, одноступенчатый характер реакции без образования комплекса с равномерным распределением энергии по степеням свободы.
Считается установленным, что образовавшаяся меченая молекула возбуждена. Естественно предположить, что энергия возбуждения, первоначально локализованная на одной—двух связях, мигрирует внутри-, а затем межмолекулярно. Предположено, что передача энергии происходит не при любых столкновениях: определяющую роль здесь играет природа окружения [ 12 — 14], а при внутримолекулярной передаче энергии —природа остатка молекулы, поглощающего избыточную энергию [16] .
Остается неизвестной относительная роль перечисленных процессов. В частности, не установлено однозначно, влияет ли процесс межмолеку- лярной передачи энергии на выход меченых молекул [12,13] или выход продуктов определяется только микросечениями соударения по связям [17 ], вероятностью химической реакции и изменением спектра замедляющихся атомов [18] . В данном случае имеется в виду не простая дезактивация возбужденных молекул, зависящая от числа столкновений, а специфическая передача энергии возбуждения непосредственно с электронных уровней молекулы . Специфическая передача энергии возбуждения связана с образованием донорно-акцепторного комплекса, внутри которого происходит перераспределение энергии.
Мы исследовали горячие реакции атомов отдачи трития с алифатическими спиртами и бинарными смесями спиртов с бензолом и циклогексаном. Главное внимание при исследовании было уделено выявлению роли процессов внутри- и межмолекулярного переноса энергии возбуждения.
ПРОВЕДЕНИЕ ЭКСПЕРИМЕНТА
В работе использовались спирты марки "х.ч", перегнанные на колонке. Бензол и циклогексан ("х .ч ." ) дополнительно очищали встряхиванием с kohu.H2S0 4 , сушили и перегоняли над металлическим натрием. По 20 мг исследуемого вещества в смеси с 20 мг углекислого лития (рамер зерен около 3 микрон) облучали в кварцевых ампулах (40 х 30 мм) потоком нейтронов от 1011 до 1013н.сек-1 см-2 при 40°С в ядерном реакторе ИРТ-1000.
Ампулы перед облучением откачивали до остаточного давления 20 мм воздуха. Анализ проводили на хроматографе со сменными колонками. В табл. Í приведены параметры колонок.
Для регистрации активности был использован проточный счетчик Гейгера-Мюллера, работающий на смеси гелий-метан (гелий-метан-спирт в случае измерения активности спирта). В случае разделения высококипя- щих продуктов счетчик подогревали до температуры 80“С. Результаты измерений приведены в таблицах 2—10. Из таблиц следует, что относительный выход продуктов замещения водорода на тритий мало зависит от времени вплоть до 10 часового облучения в потоке 1012н • см' 2 • сек- 1.Плотность потока нейтронов не влияет на распределение активности, если существенно не меняются интегральные потоки. Радиационная выживаемость спиртов составила 95 — 97% при облучении в потоке1013н-см~2 -сек-1 в течение 15 минут. В случае облучения бинарных смесей не было замечено изменения общей активности трития. Как было установлено ранее[19],
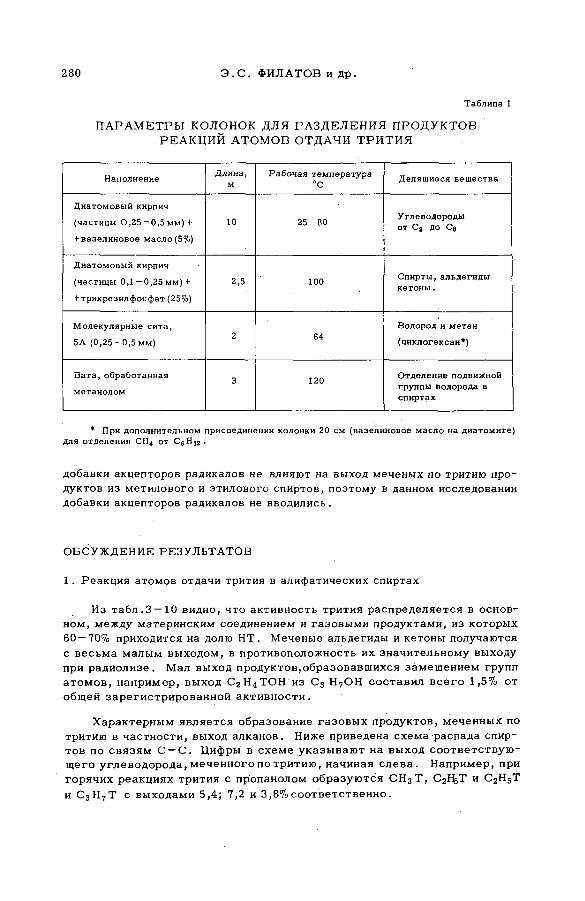
280 Э . С . Ф И Л А Т О В и д р .
ПАРАМ ЕТРЫ КОЛОНОК ДЛЯ РАЗДЕЛЕНИЯ ПРОДУКТОВ РЕАКЦИЙ АТОМОВ ОТДАЧИ ТРИТИЯ .
Таблица 1
НаполнениеДлина,
мРабочая температура
°СДелящиеся вещества
Диатомовый кирпич
(частицы 0 ,25 — 0,5 мм) +
+вазелиновое м асло (5%)
10 2 5 -8 0Углеводороды от С 2 до Се
Диатомовый кирпич
(частицы 0 ,1 — 0,25 мм) +
+ трикрезилфосфат (25%)
2,5 1 00 Спирты, альдегиды кетоны .
Молекулярные сита,
5А (0,25 — 0,5 мм)2 64
Водород и метан
(циклогексан*)
В ата , обработанная
метанолом3 120 Отделение подвижной
группы водорода в спиртах
* При дополнительном присоединении колонки 20 см (вазелиновое м асло на диатомите) для отделения СН 4 от С в Н ^ .
добавки акцепторов радикалов не влияют на выход меченых по тритию продуктов из метилового и этилового спиртов, поэтому в данном исследовании добавки акцепторов радикалов не вводились.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
1. Реакция атомов отдачи трития в алифатических спиртах
Из табл.3 — 10 видно, что активность трития распределяется в основном, между материнским соединением и газовыми продуктами, из которых 60—70% приходится на долю HT. Меченые альдегиды и кетоны получаются с весьма малым выходом, в противоположность их значительному выходу при радиолизе. Мал выход продуктов,образовавшихся замещением групп атомов, например, выход С2Н4ТОН из С 3 Н7ОН составил всего 1,5% от общей зарегистрированной активности.
Характерным является образование газовых продуктов, меченных по тритию в частности, выход алканов. Ниже приведена схема распада спиртов по связям С —С. Цифры в схеме указывают на выход соответствующего у г л е в о д о р о д а , меченного по тритию, начиная слева. Например, при горячих реакциях трития с пропанолом образуются СН3Т, С2Н5Т и С2Н5Т и СзН7Т с выходами 5,4; 7,2 и 3,8% соответственно.
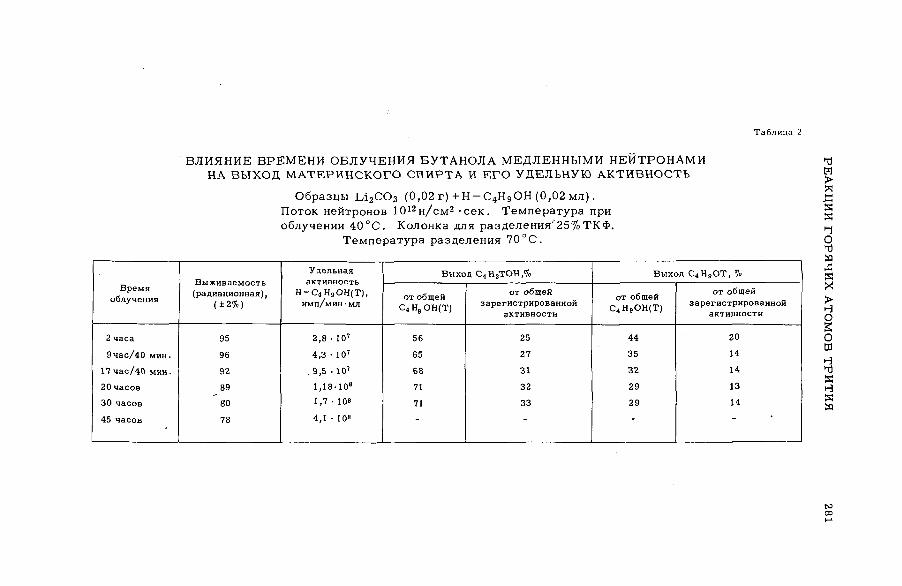
Таблица 2
ВЛИЯНИЕ ВРЕМЕНИ ОБЛУЧЕНИЯ БУТАНОЛА МЕДЛЕННЫМИ НЕЙТРОНАМИ НА ВЫХОД МАТЕРИНСКОГО СПИРТА И ЕГО УДЕЛЬНУЮ АКТИВНОСТЬ
Образцы 1л2СОз (0,02 г )+ Н —С4Н9ОН (0,02 м л ).Поток нейтронов 1012н/см2 - сек. Температура при облучении 40°С. Колонка для разделения'25% ТКФ.
Температура разделения 70°С .
Времяоблучения
Выживаемость (радиационная),
( ± 2 % )
Удельнаяактивность
Н - С 4Н 9О Н (Т ),имп/мин*мл
Выход С 4Н 3ТО Н ,% Выход С 4Н 9О Т , %
от общей С 4 Н9 О Н (Т )
от общей зарегистрированной
активности
от общей С 4Н 9О Н (Т )
от общей зарегистрированной
активности
2 часа 95 2,8 ■ 1 0 7 56 25 44 20
9 час/40 мин. 96 4,3 • 107 65 27 35 14
17 час/40 м ин. 92 9,5 • 107 68 31 32 14
20 часов 89 1,18-10® 71 32 29 13
30 часов 80 1,7 108 71 33 29 14
45 часов 78 4,1 - 108 - * ** "
РЕАКЦ
ИИ
ГО
РЯЧ
ИХ
АТО
МО
В ТРИ
ТИЯ
281
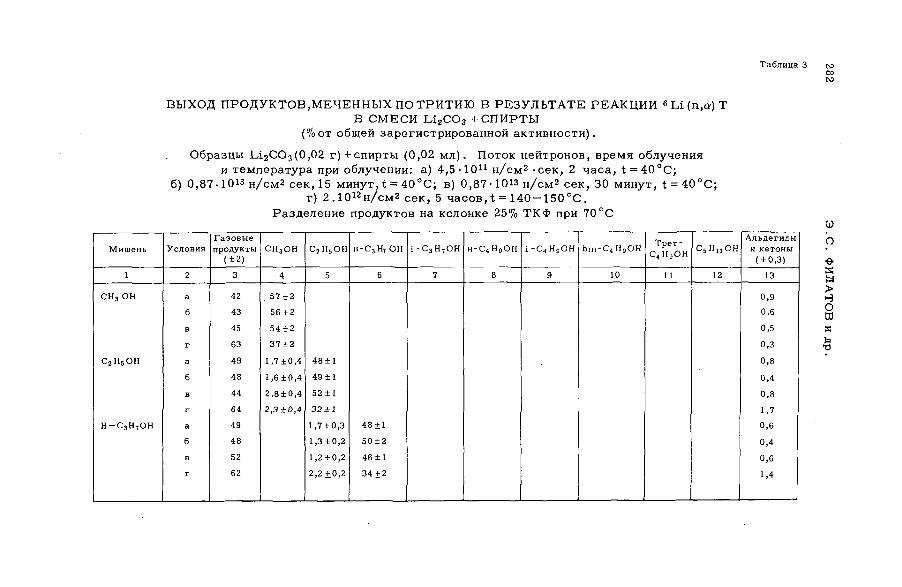
Таблица 3
ВЫХОД ПРОДУКТОВ,МЕЧЕННЫХ ПО ТРИТИЮ В РЕЗУЛЬТАТЕ РЕАКЦИИ 6L i (n ,a )TВ СМЕСИ 1Л2С03 + СПИРТЫ
(%от общей зарегистрированной активности).
. Образцы L i2C 0 3(0,02 г)+спирты (0,02 мл). Поток нейтронов, время облучения и температура при облучении: а) 4,5-1011 н/см2 - сек, 2 часа, t= 4 0 °C ;
б) 0,87 • 1013 н/см2 сек, 15 минут, t = 40°С ; в) 0,87 • 1013 н/см2 сек, 30 минут, t = 40°С; г) 2.10 1гн/см2 сек, 5 часов, t = 140 —150 °С .
Разделение продуктов на колонке 25% ТКФ при 70 °С
Мишень УсловияГазовыепродукты
( ± 2 )С Н 3ОН С 2 Н5ОН н - С з Н7 О Н i - С 3 Н 7ОН Н -С 4Н 9ОН i - C 4 H9OH b m -C 4 H9OH Т р ет -
С 4 Н 9ОН С 5 Н „ О НАльдегиды
и кетоны (± 0 ,3 )
1 2 3 4 5 6 7 8 9 10 1 1 12 13
С Н 3 ОН а 42 . 57 i 2 0,9
б 43 56 t2 0,6
в 45 54 ±2 0,5
Г 63 37 ± 2 0,3
с2н5он а 49 1,7 ±0 ,4 48 ± 1 0,8
б 48 1,6 ±0 ,4 49 ±1 0,4
в 44 2 ,8 ± 0,4 52 ± 1 0,8
г 64 2,3 ±0,4 32 ±1 1,7
н -с 3н7он а 49 1,7 ±0 ,3 48 ± 1 0,6
б 48 1,3 ± 0,2 50 ± 2 0,4
в 52 1 ,2 ± 0,2 46 ±1 0,6
Г 62 2,2 + 0,2 34 ±2 1,4
1
282 Э
. С. Ф
ИЛАТО
В и
др.
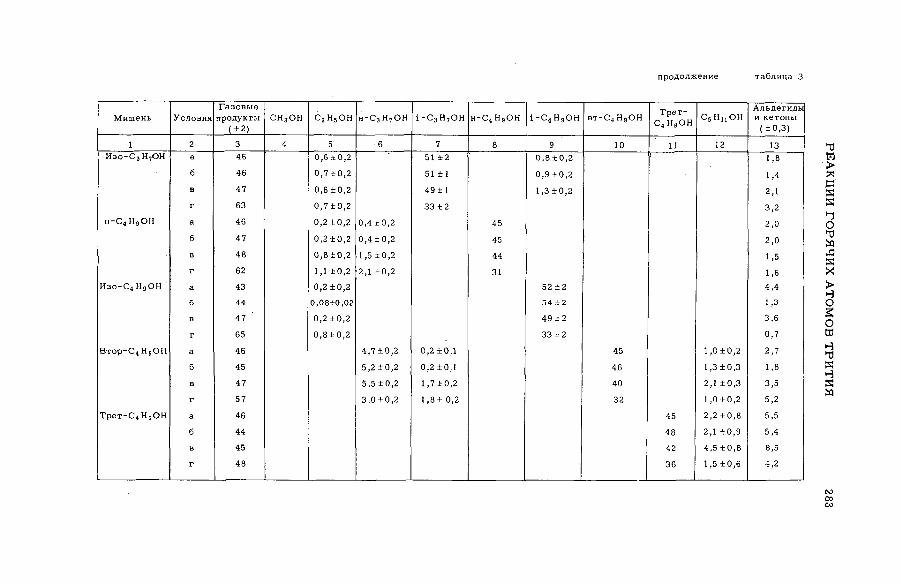
продолжение таблица 3
Мишень УсловияГазовы епродукты
( ± 2 )СН 3ОН С 2 Н5ОН н -С 3 Н7ОН i - С 3 Н 7ОН н -С 4Н 9ОН i - С 4Н 9ОН в т -С 4 НдОН
Т рет -с 4 н 9о н
С 5 Н и ОНАльдегиды и кетоны
(± 0 ,3 )
1 2 3 4 5 6 7 8 9 10 11 12 13И з о -С 3 Н 7ОН а 46 0,6 ± 0,2 51 ±2 0,8 ± 0,2 1,8
б 46 0,7 ±0,2 51 ±1 0,9 ±0,2 1,4
в 47 0,6 ± 0,2 49 ±1 1,3 ±0,2 2,1
г 63 0,7 ± 0,2 33 ±2 3,2
н -С 4Н 9ОН а 46 0,2 1 0,2 0,4 ±0,2 45 2,0
б 47 0,2 ± 0,2 0,4 ±0,2 45 2,0
в 48 0 ,8 ± 0,2 1,5 ±0,2 44 1,5
г 62 1,1 ± 0,2 2,1 ± 0,2 31 1,6
И з о -С 4 Н9ОН ' а 43 0,2 ± 0,2 52 ±2 4,4
б 44 0,08±0,02 54 ±2 1,3
в 47 0,2 ± 0,2 49 ± 2 3,6
Г 65 0,8 ± 0,2 33 ±2 0,7
В т о р -С 4 Н 9ОН а 46 4,7 ±0,2 0,2 ± 0,1 45 1,0 ± 0,2 2,7
б 45 5,2 ±0,2 0,2 ± 0,1 46 1,3 ±0,3 1,8
в 47 5,5 ±0,2 1,7 ± 0,2 40 2,1 ±0,3 3,5
г 57 3 ,0 ± 0,2 1,8 ± 0,2 32 1,0 ± 0,2 5,2
Т р е т -С 4Н эОН а 46 45 2,2 ± 0,8 5,5
б 44 48 2,1 ±0,9 5,4
в 45 42 4,5 ±0 ,8 8,5
г 48 36 1,5 ±0,6 4,2
РЕАКЦ
ИИ
ГО
РЯЧ
ИХ
АТО
МО
В ТРИ
ТИЯ
283

284 Э . С . Ф И Л А Т О В и д р .
- . 11,5 4,8. с н 3- с н 2 - о н
5,4 V, 8 3,8с н 3 - с н 2 - с н 2 - о н
4,9 3,8 5,5 2,0с н 3 - с н 2 - с н 2 - с н 2 - о н
3,2 3,7 2,6 4,0 1,6с н 3 - с н 2 - с н 2- с н 2- с н 2 - о н
2,6 2,5 1,5 1,1 2,6 1,0с н 3 - с н 2 - с н 2 - с н 2 - с н 2- с н 2 - о н
2,4 1,8 1,6 1.5 1,5 4,7 1,5с н 3- с н 2 — с н 2 — с н 2 — с н 2 - с н 2 - с н 2— о н
2,1 1,9 1,2 1,1 1,1 1,2 2,4 0,9с н 3 - с н 2 - с н 2 - с н 2 - с н 2 — с н 2 — с н 2 - с н 2 - о н
Ошибка ± 0, 3
Из приведенных данных можно сделать несколько выводов. Вероятность разрыва связи С —С определяется ее положением в молекуле. Предпочтительным является образование метана и этана. При увеличении длины цепи спиртовой молекулы выход углеводородов, образованных разрывом данного типа связи, уменьшается в согласии со статистическим уменьшением вероятности соударения по данной связи (рис.1). Отношение выходов НТ/СН3Т меняется линейно от параметра: отношение числа связей С —н/число связей С —СН3 (рис . 2). Это подтверждает модель ударов [5,6] и "инерционный механизм" [21] реакций горячих атомов.
В рамках модели ударов нет возможности объяснить предпочтительный разрыв а -связи с образованием меченого углеводорода, имеющего на один атом углерода меньше, чем исходная молекула. При изучении реакций горячих атомов трития [22] и брома [23] с карбоновыми кислотами эффект разрыва связи при o'-углеродном атоме удалось объяснить реакцией де- карбоксилирования "нагретых" меченых молекул. В случае спиртов такой подход не дает результатов: возбужденные молекулы спирта должны либо превращаться в альдегиды (кетоны), либо вступать в реакции с образованием эфиров. Однако ни того ни другого не наблюдается.
Можно полагать, что повышенный выход углеводорода, образованного разрывом а-связи обусловлен Меньшей электронной плотностью «-положения .
Возможны два механизма реакций. Первый —с образованием переходного состояния —комплекса соударения типа
19С Н 3 - О Н
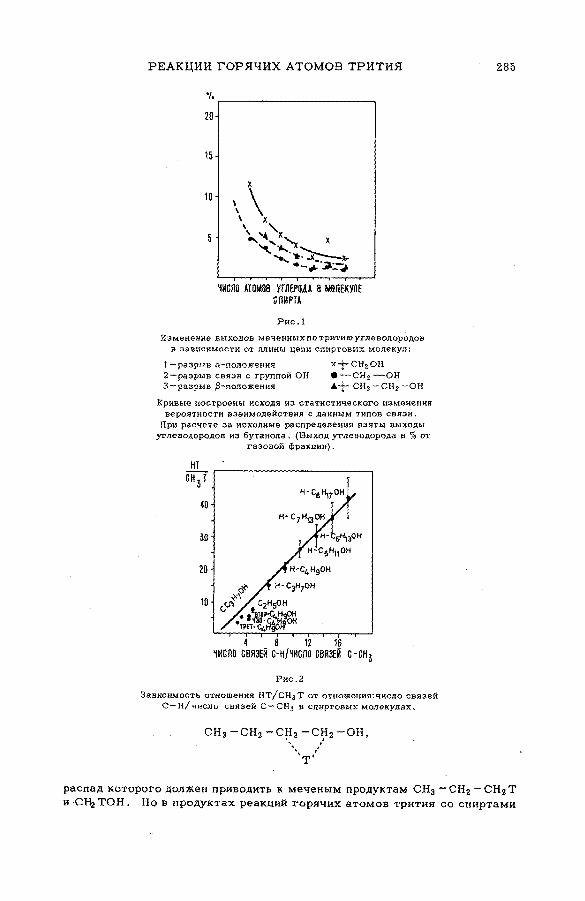
РЕАКЦИИ ГО Р Я Ч И Х АТОМОВ Т РИ Т И Я 285
V.
20
15
10
5
ЧИСЛО АТОМОВ УГЛЕРОДА В МОЛЕКУЛЕ СПИРТА
Р и с . 1
Изменение выходов меченных по тритию углеводородов , в зависимости от длины цепи спиртовых молекул:
^ р а з р ы в а -п о л о ж е н и я х -£ -С Н гО Н2 —разрыв связи с группой ОН • - — C H g— ОН3 —разрыв /3-положения А -J- СН 2 — СН 2 — ОН
Кривые построены исходя из статистического изменения вероятности взаимодействия с данным типов связи.
При расчете за исходные распределения взяты выходы углеводородов из бутанола. (Выход углеводорода в % от
газовой фракции).
HT
CV40
50
20
10
4 8 12 16ЧИСЛО СВЯЗЕЙ С-Н/ЧИСЛ0 СВЯЗЕЙ C -C H j
Рис .2
Зависимость отношения Н Т /С Н зТ от отношения:число связей С —Н /число связей С —СН 3 в спиртовых м олекулах.
с н 3 - с н 2 —с н а - с н 2 - о н ,
' т * '
распад которого должен приводить к меченым продуктам СН3 — СНг~СНгТ и СНгТОН. Но в продуктах реакций горячих атомов трития со спиртами
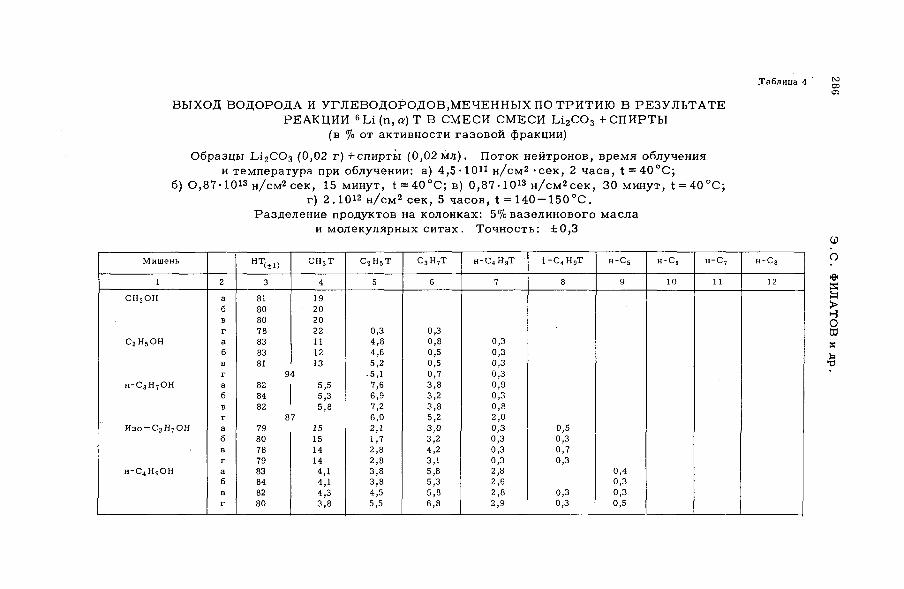
ВЫХОД ВОДОРОДА И УГЛЕВОДОРОДОВ,МЕЧЕННЫ Х ПО ТРИТИЮ В РЕЗУЛЬТАТЕ РЕАКЦИИ 6L i (n , » )T В СМЕСИ СМЕСИ L i2C 0 3 + СПИРТЫ
(в % от активности газовой фракции)
О бр а зц ы L 12CO3 (0,02 г) -f-спирты (0,02 мл). Поток нейтронов, время облучения и температура при облучении: а) 4,5 • 1011 н/см2 • сек, 2 часа, t = 40°С;
б) 0,87 • 1013 н/см2 сек, 15 минут, t = 40 °С; в) 0,87 • 1013 н/см2 сек, 30 минут, t = 40 °С; г) 2 . lo i2 н/см2 сек, 5 часов, t = 140—150 °С .
Разделение продуктов на колонках: 5% вазелинового масла и молекулярных ситах . Точность: ±0,3
.Таблица 4
Мишень HT(tl> СН 3Т С 2 Н5Т с 3н 7т H - О4 H дТ i - C 4H 9T н -С 5 н-Св н -С 7 н -С 8
1 2 3 4 5 6 7 8 9 10 И 12
С Н 3ОН а 81 19б 80 20в 80 20г 78 22 0,3 0,3
с 2н 5о н а 83 И 4,8 0,8 0,3б 83 12 4,6 0,5 0,3в 81 13 5,2 0,5 0,3г 94 -5,1 0,7 0,3
н -С 3Н 7ОН а 82 5,5 7,6 3,8 0,9б 84 5,3 6,9 3,2 0,3в 82 5,8 7,2 3,8 0,8г 87 6,0 5,2 2,0
И зо — С3 Н7ОН а 79 15 2, 1 3,0 0,3 0,5б 80 15 1 , 7 3,2 0,3 0,3в 78 14 2,8 4,2 0,3 0,7г 79 14 2,8 3,1 0,3 0,3
н -С 4Н 9ОН а 83 4,1 3,8 5,8 2,8 0,4б 84 4,1 3,8 5,3 2,6 0,3в 82 4,3 4,5 5,8 2,8 0,3 0,3г 80 3,8 5,5 6,8 2,9 0,3 0,5
286 Э
. С. Ф
ИЛАТО
В и
др.
РЕАКЦИИ Г О Р Я Ч И Х АТОМОВ ТРИ ТИ Я 287
юсон
Xо>Xчочоас
о "1X
12
1,0
и 1Я
X
О о о t» со«-1 *—1 «-Н
X
О со со со со о ю оэ CD <£> со со юа>
о о о о со го Гч1 *•4 гмX
h05
X о о о <о ем о
Vсм см со см с - ю со со
-
н05 ю ID с~ Е— со от-н СО оо tn -н ю т-<
г - о о о о -н гч см -н со -н
и
X
Н£— 1Л со eg со «—i LO со со LO СО см
X to со 00 Í - -Н -н -н СМ см ÍNJ -н —
и
нсо О) см о о СО о СМ о см «— со со
X ю о о со см со -н со СО со со со -н г -
Ó '
Е- см ем от *3*
з Г 10 со m to О) о со о со со со <N СМ см
осм
д.
о см ю со г - h- in in h-
яг - г~ |> со СП со со со со со 00 СО 00
см л ю 03 ь со ю n и СО ю 0 СО СО СО
Xщ Г)( )
X X ас Xх V 0 О О о
3 и £ 2 2 51 X X X X
> н оn
фа и и и О
5 н X X X X

288 Э . С . ФИЛАТОВ и др.
не обнаружено меченых молекул метилового спирта (несмотря на то, что "инерционный механизм" способствует его образованию). По-видимому, изложенный механизм не имеет места.
Второй возможный механизм реакции включает процесс миграции энергии возбуждения по углеводородному скелету от места удара к группе ОН, способной рассеивать энергию. Не исключено, что в этом процессе принимают участие водородные связи. Когда волна возмущения доходит до ослабленной »-связи, последняя разрывается с образованием лишь одного меченого продукта
—iAAP— ►СН3 -С Н 2 -С Н 2 -С Н 2 -О Н =Ф сн3-с н -с н 2-С Н 2ОН => СН3СНТСН3
Здесь возможен радикальный или мономолекулярный перенос водорода на концевой углеродный атом.
С изложенной точки зрения можно объяснить без привлечения "инерционного механизма" повышенные выходы метана и этана. При распространении энергии возбуждения в противоположном направлении разрывается связь с концевыми алкильными группами. Данные по изомерным, вторичным и третичному спиртам подтверждают предположение об определяющей роли процесса миграции энергии внутри молекулы после первичного акта завязывания связи и возбуждения локальных уровней энергии.
С точки зрения модели о влиянии процесса внутримолекулярной миграции энергии возбуждения следует ожидать увеличения в выходе меченых
Таблица 5
ОПРЕДЕЛЕНИЕ КОЛИЧЕСТВ ПОДВИЖНОГО И НЕПОДВИЖНОГО ТРИТИЯ В МОЛЕКУЛАХ МАТЕРИНСКИХ СПИРТОВ.
Образцы L Í2 CO3 0,02 г + спирты 0,02 мл.. Поток нейтронов 4,5 • 1011 н/см2 • сек.
Время облучения 2 часа. Температура при облучении 40°С. Разделение продуктвов на колонках 25% ТКФ (при 70°С)
и колонка с ватой (при 120°С).
Мишень
В ы х о д , % Вы ход,%
от общей активности
материнского спирта
от общей зарегистри
рованной активности
от общей активности
материнского спирта
от общей зарегистрированной
активности
С Н 3ОН 47 27 53 30
С 2 Н5ОН 52 25 48 23н -С 3 Н7ОН 50 24 50 24
И з о -С „ Н 9ОН 61 31 39 20
н -С 4Н 9ОН 56 25 44 20
И з о -С 4Н 9ОН 58 30 42 22
В тО р-С 4 НдОН 71 32 29 13
Т р е т -С 4 НдОН 69 31 31 14
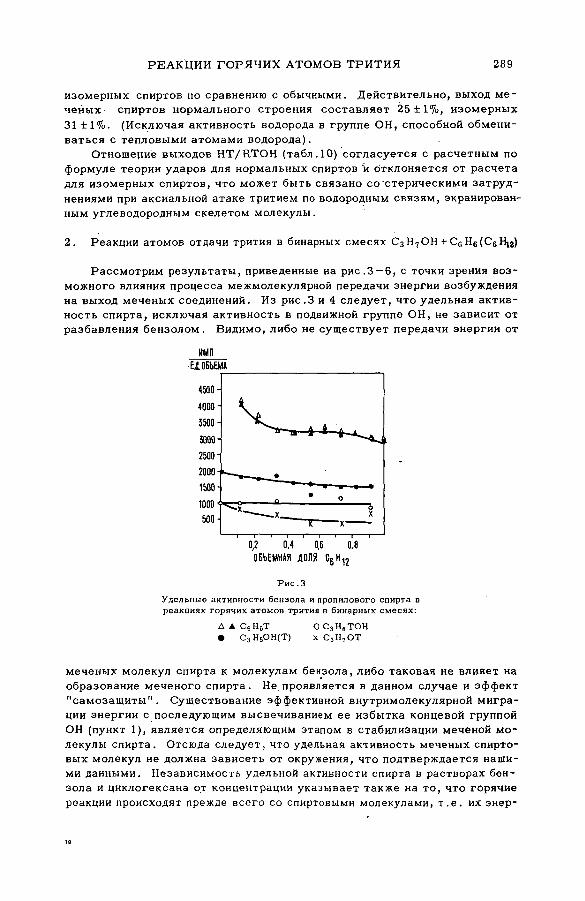
Р Е А К Ц И И Г О Р Я Ч И Х А Т О М О В Т Р И Т И Я 289
изомерных спиртов по сравнению с обычными. Действительно, выход меченых- спиртов нормального строения составляет 25 ±1%, изомерных 31 ±1%. (Исключая активность водорода в группе ОН, способной обмениваться с тепловыми атомами водорода).
Отношение выходов HT/RTOH (табл. 10) согласуется с расчетным по формуле теории ударов для нормальных спиртов ‘и отклоняется от расчета для изомерных спиртов, что может быть связано со стерическими затруднениями при аксиальной атаке тритием по водородным связям, экранированным углеводородным скелетом молекулы.
2. Реакции атомов отдачи трития в бинарных смесях С3Н7ОН+С6Н6(С6Н1г)
Рассмотрим результаты, приведенные на рис.З —6, с точки зрения возможного влияния процесса межмолекулярной передачи энергии возбуждения на выход меченых соединений. Из рис.З и 4 следует, что удельная активность спирта, исключая активность в подвижной группе ОН, не зависит от разбавления бензолом. Видимо, либо не существует передачи энергии от
имлE ¿ ОБЪЕМА
ОБЪЕМНАЯ ДОЛЯ С6 Н 12
Р и с .3
Удельные активности бензола и пропилового спирта в реакциях горячих атомов трития в бинарных смесях:
Д А С 6Н5Т О С 3 НвТОН• С 3 Н5О Н (Т ) х С 3Н7О Т
меченых молекул спирта к молекулам бензола, либо таковая не влияет на образование меченого спирта. Не проявляется в данном случае и эффект "самозащиты". Существование эффективной внутримолекулярной миграции энергии с последующим высвечиванием ее избытка концевой группой ОН (пункт 1), является определяющим этапом в стабилизации меченой молекулы спирта. Отсюда следует, что удельная активность меченых спиртовых молекул не должна зависеть от окружения, что подтверждается нашими данными. Независимость удельной активности спирта в растворах бензола и циклогексана от концентрации указывает также на то, что горячие реакции происходят прежде всего со спиртовыми молекулами, т.е. их энер-
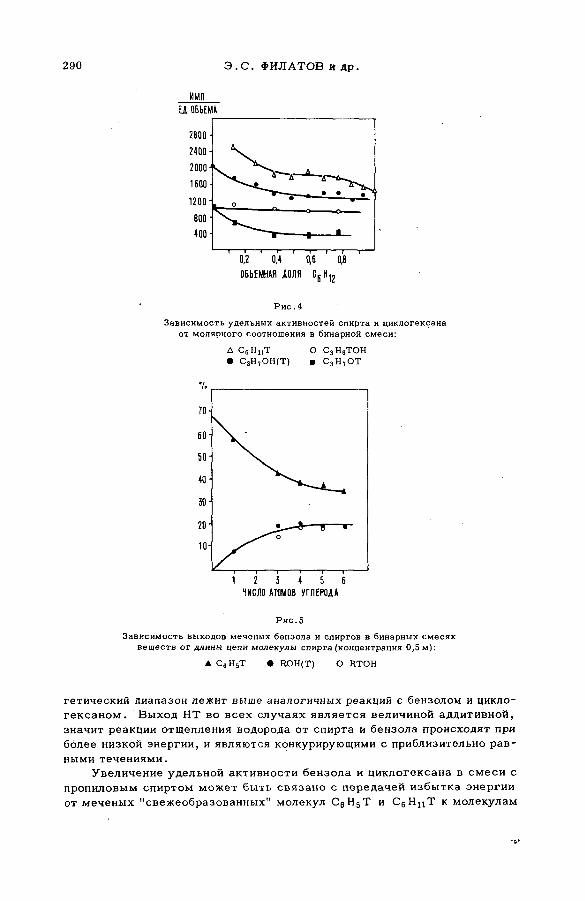
290 Э . С . ФИЛАТОВ и др.
импЕА ОБЬЕМА
Р и с .4
Зависимость удельных активностей спирта и циклогексана от молярного соотношения в бинарной смеси:
Д С 6Н цТ О С 3Н6ТОН• С 3Н ,О Н (Т ) ■ С 3Н ,О Т
ЧИСЛО АТОМОВ УГЛЕРОДА
Р и с . 5
Зависимость выходов меченых бензола и спиртов в бинарных смесях веществ от длины цепи молекулы спирта (концентрация 0,5 м ):
▲ С 6Н5Т • R O H (T ) О RTOH
гетический диапазон лежит выше аналогичных реакций с бензолом и цикло- гексаном. Выход HT во всех случаях является величиной аддитивной, значит реакции отщепления водорода от спирта и бензола происходят при более низкой энергии, и являются конкурирующими с приблизительно равными течениями.
Увеличение удельной активности бензола и циклогексана в смеси с пропиловым спиртом может быть связано с передачей избытка энергии от меченых "свежеобразованных" молекул С6Н5Т и С6НцТ к молекулам
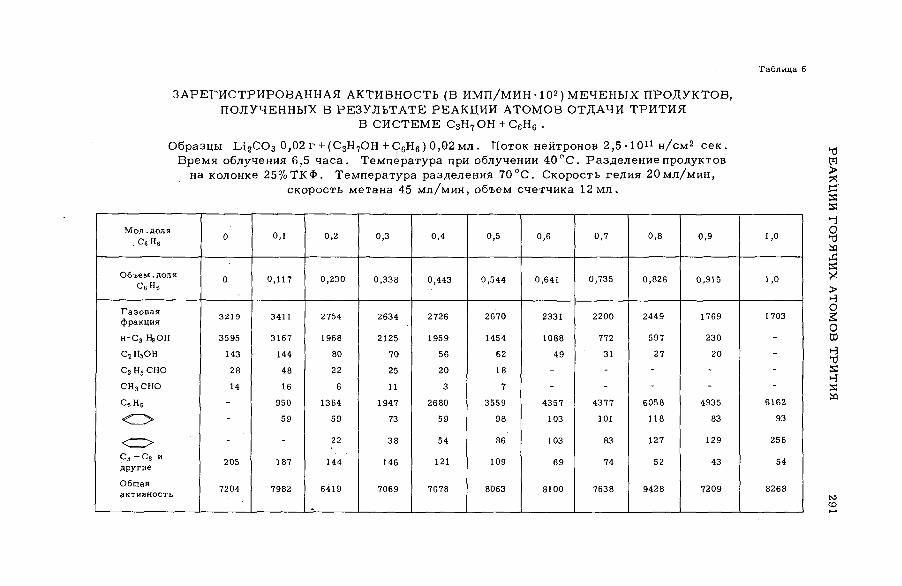
Таблица 6
ЗАРЕГИСТРИРОВАННАЯ АКТИВНОСТЬ (В ИМП/МИН • 102 ) МЕЧЕНЫХ ПРОДУКТОВ, ПОЛУЧЕННЫХ В РЕЗУЛЬТАТЕ РЕАКЦИИ АТОМ ОВ ОТДАЧИ ТРИТИЯ
В СИСТЕМ Е С3Н7ОН + С6Н6 .
Образцы 1л2СОз 0,02 г + (С 3Н7ОН + С6Н6 ) 0,02 мл . Поток нейтронов 2,5 • 1011 н/см2 сек. Время облучения 6,5 часа. Температура при облучении 40°С . Разделение продуктов
на колонке 25%ТКФ. Температура разделения 70 °С . Скорость гелия 20мл/мин, скорость метана 45 мл/мин, объем счетчика 12мл.
М ол доля. Се н6
0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9 1 , 0
О бъ ем . доляCç н 6
0 0,117 0,230 0,338 0,443 0,544 0,641 0,735 0,826 0,915 1 ,0
Газоваяфракция 3219 3411 2754 2634 2726 2670 2331 2200 2449 1769 1703
н -С 3 НдОН 3595 3167 1968 2125 1959 1454 1088 772 597 230 -
С 2 Н5ОН 143 144 80 70 56 62 49 31 27 20 -
С 2 Н5 СНО 28 48 22 25 20 18 - - - ' -
С Н 3 СНО 14 16 6 1 1 3 7 - - - - -
с 6н е - 950 1364 1947 2680 3559 4357 4377 6058 4935 6162
< г > - 59 59 73 59 98 103 1 0 1 118 83 93
< ^ > - ' - 22 38 54 86 103 83 127 129 256
С 5 ~ Cg и другие 205 187 144 146 1 2 1 109 69 74 52 43 54
Общаяактивность 7204 7982 6419 7069 7678 8063 8100 7638 9428 7209 8268
РЕАКЦ
ИИ
ГО
РЯЧ
ИХ
АТО
МО
В ТРИ
ТИЯ
291
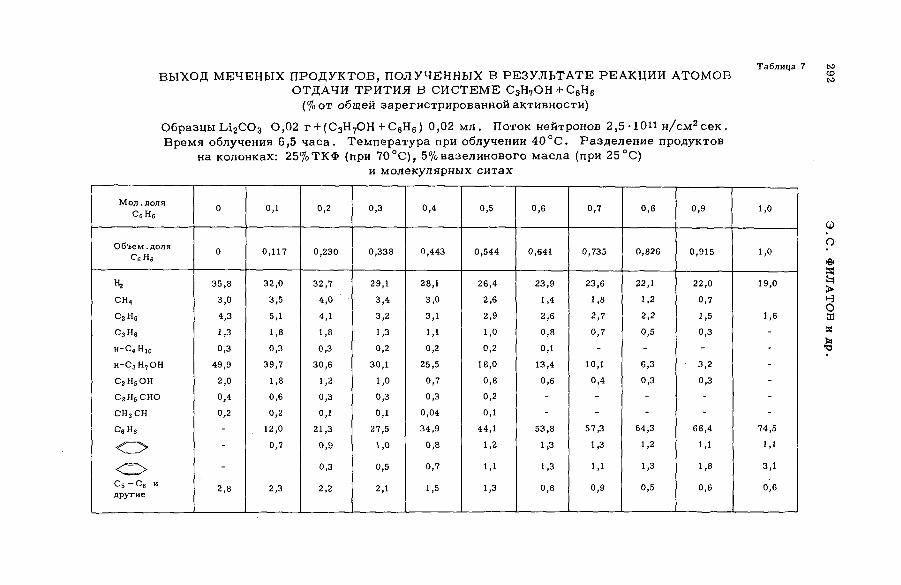
ВЫХОД МЕЧЕНЫХ ПРОДУКТОВ, ПОЛУЧЕННЫХ В РЕЗУЛЬТАТЕ РЕАКЦИИ АТОМ ОВ ОТДАЧИ ТРИТИЯ В СИСТЕМ Е С3Н7ОН + С6Н6
(%от общей зарегистрированной активности)
Образцы L i2C 0 3 0,02 г + (С 3Н7О Н + С 6Н6) 0,02 мл. Поток нейтронов 2,5-104 н/см2 сек.Время облучения 6,5 часа. Температура при облучении 40°С . Разделение продуктов
на колонках: 25%ТКФ (при 70°С ), 5%вазелинового масла (при 25 °С)и молекулярных ситах
Таблица 7
М ол .доляс 6н 6
0 ОД 0,2 0,3 0,4 0,5 0,6 0,7 0 ,8 0,9 1,0
Объем .доляс е н б
0 0,117 0,230 0,338 0,443 0,544 0,641 0,735 0,826 0,915 1,0
Ц, 35,8 32,0 32,7 29,1 28,1 26,4 23,9 23,6 22,1 22,0 19,0
с н 4 3,0 3,5 4,0 3,4 3,0 2,6 1,4 1,8 1,2 0,7
С 2Н6 4,3 5,1 4,1 3,2 3,1 2,9 2,6 2,7 2,2 1,5 1,6
С 3 н 8 1,3 1,8 1 , 8 1,3 1Д 1,0 0,8 0,7 0,5 0,3 -
н -С 4 Ню 0,3 0,3 0,3 0,2 0,2 0,2 0,1 - - - -
н -С 3 Н?ОН 49,9 39,7 30,6 30,1 25,5 18,0 13,4 10,1 6,3 3,2 -
с 2н 5о н 2,0 1,8 1,2 1,0 0,7 0,8 0,6 0,4 0,3 0,3 -
с 2н 5с н о 0,4 0,6 0,3 0,3 0,3 0,2 - - - - -
СН3СН 0,2 0,2 0,1 0,1 0,04 0,1 - - - - -
С 6н б - 12,0 21,3 27,5 34,9 44,1 53,8 57,3 64,3 68,4 74,5
< с ^ > - 0,7 0,9 1 , 0 0 ,8 1,2 1,3 1,3 1,2 1,1 1,1
- 0,3 0,5 0,7 1,1 1,3 1,1 1,3 1,8 3,1
С 5 — С 6 и другие
2,8 2,3 2,2 2,1 1,5 1,3 0,8 0,9 0,5 0,6 0,6
292 Э
.С.
ФИ
ЛАТОВ
и др.
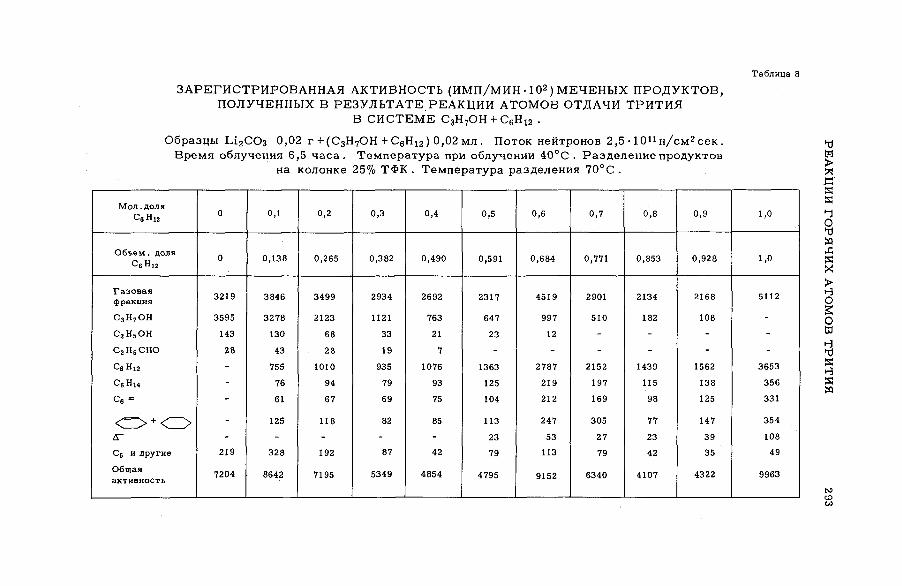
Таблица 8
ЗАРЕГИСТРИРОВАННАЯ АКТИВНОСТЬ (ИМП/МИН • 102 ) МЕЧЕНЫХ ПРОДУКТОВ, ПОЛУЧЕННЫХ В РЕЗУЛЬТАТЕ РЕАКЦИИ АТОМ ОВ ОТДАЧИ ТРИТИЯ
В СИСТЕМЕ С3Н7ОН + С6Н12 .
Образцы 1л.2СОз 0)02 г + (С 3Н7ОН + C6Hi2 ) 0,02 мл . Поток нейтронов 2,5 • 10и н/см2 сек . Время облучения 6,5 часа. Температура при облучении 40°С . Разделение продуктов
на колонке 25% ТФК . Температура разделения 70°С.
М ол .доляСе Н 12 0 0, 1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9 1 ,0
Объем , доля С 6 Н 12 0 0,138 0,265 0,382 0,490 0,591 0,684 0,771 0,853 0,928 1 ,0
Газоваяфракция 3219 3846 3499 2934 2692 2317 4519 2901 2134 2168 5112
С 3 Н 7ОН 3595 3278 2123 1 1 2 1 763 647 997 510 182 108 -
С 2 Н5ОН 143 130 68 33 21 23 12 - - - - '
С 2 Н 5СН О 28 43 28 19 7 - - - - - -
Се к 12 - 755 1 0 1 0 935 1076 1363 2787 2152 1439 1562 3653
С 6Н 14 - 76 94 79 93 125 219 197 115 138 356
С 6 = - 61 67 69 75 104 2 1 2 169 98 125 331
^ > + < z > - 125 118 82 85 113 247 305 77 147 354
/ г - - - ■ - - 23 53 27 23 39 108
С 5 и другие 219 328 192 87 42 79 113 79 42 35 49
Общаяактивность
7204 8642 7195 5349 4854 4795 9152 6340 4107 4322 9963
РЕАКЦ
ИИ
ГО
РЯЧ
ИХ
АТО
МО
В ТРИ
ТИЯ
293
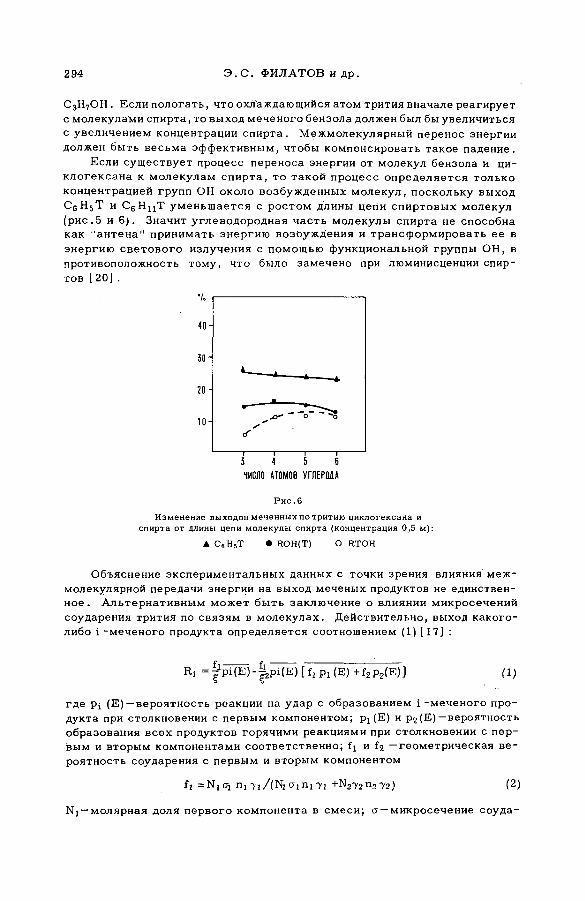
294 Э . С . Ф И Л А Т О В и д р .
С3Н7ОН . Если пологать, что охлаждающийся атом трития вначале реагирует с молекулами спирта, то выход меченого бензола должен был бы увеличиться с увеличением концентрации спирта. Межмолекулярный перенос энергии должен быть весьма эффективным, чтобы компенсировать такое падение.
Если существует процесс переноса энергии от молекул бензола и циклогексана к молекулам спирта, то такой процесс определяется только концентрацией групп ОН около возбужденных молекул, поскольку выход С6Н5Т и С6НиТ уменьшается с ростом длины цепи спиртовых молекул (рис.5 и 6). Значит углеводородная часть молекулы спирта не способна как "антена" принимать энергию возЬужд'ения и трансформировать ее в энергию светового излучения с помощью функциональной группы ОН, в противоположность тому, что было замечено при люминисценции спиртов I 20J .
Р и с . 6
Изменение выходов меченных по тритию циклогексана и спирта от длины цепи молекулы спирта (концентрация 0,5 м ):
▲ С 6Н5Т • R O H (T ) О RTOH
Объяснение экспериментальных данных с точки зрения влияния меж- молекулярной передачи энергии на выход меченых продуктов не единственное . Альтернативным может быть заключение о влиянии микросечений соударения трития по связям в молекулах. Действительно, выход какого- либо i -меченого продукта определяется соотношением (1) [17] :
Ri = |^Г(Ё )-| 2 (Ё ) [ f lP l (E )+ f2p2(E )] (1)
где Pi (Е) —вероятность реакции на удар с образованием i -меченого продукта при столкновении с первым компонентом; рх (Е) и рг(Е )—вероятность образования всех продуктов горячими реакциями при столкновении с первым и вторым компонентами соответственно; fi и i<¿ —геометрическая вероятность соударения с первым и вторым компонентом
fl =N iO i ni 7i/(Ni a ¡n i 7i +N272n272) (2)
Nj —молярная доля первого компонента в смеси; сг —микросечение соуда-

ВЫХОД МЕЧЕНЫХ ПРОДУКТОВ, ПОЛУЧЕННЫХ В РЕЗУЛЬТАТЕ РЕАКЦИИ АТОМОВ ОТДАЧИ ТРИТИЯ В СИСТЕМ Е С 3Н7ОН + С6Н12
(%от общей зарегистрированной активности)
Образцы L i2C 0 3 0,02 г + (С 3Н7ОН + С 6Н12) 0,02 мл. Поток нейтронов 2,5.10й н/см2 -сек.Время облучения 6,5 часа. Температура при облучении 40°С . Разделение продуктов
на колонках 25%ТКФ (при 70°С) 5% вазелинового масла (при 25°С ) и молекулярных сит с дополнительной колонкой для отделения циклогексана.
Таблица 9
М о л . ДОЛЯ
с 6н 120 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9 1,0
Объем .доляс 6н 12 0 0,138 0,265 0,382 0,490 0,591 0,684 0,771 0,853 0,928 1,0
и 2 34,8 33,1 38,5 45,6 48,1 42,0 42,6 38,5 47,7 46,0 47,1
с н 4 5,3 4,7 3,9 3,9 2,8 2,6 3,1 2,9 1,4 1,4 1,4
с 2н 6 5,3 4,0 3,9 3,3 2,7 2,4 2,0 2,1 1,6 1,2 1,3
с 3 н 8 1,9 2,1 1,7 1,4 1,2 1,1 1,5 1,1 1,0 1,0 1,0
Н-С4 н 10 0,4 0,6 0,6 0,6 0,6 0,2 0,1 0,2 0,2 0,5 0,5
с 3 н, он 49,9 37,9 29,5 21,0 15,7 13,5 10,9 8,0 4,4 2,5 -
с 2 Н5ОН 2,0 1,5 0,9 0,6 0,4 0,5 0,1 - - - -
С 2 Н5СНО 0,4 0,4 0,4 . 0,3 0,1 - - - - - -
С 6 н 12 - 8,7 14,0 17,5 22,2 28,2 30,4 33,9 35,0 36,1 36,3
С 6 Н ,4 - 0,9 1,3 1,5 1,9 2,6 . 2,4 3,1 2,8 3,2 3,6
С 6 = - 0,7 0,9 1,3 1,5 2,1 2,3 2,6 2,4 2,9 3,3
0 * 0"
- 1,4 1 , 6 1,5 1 , 8 2,3 2,7 4,8 1 ,8 3,4 3,6
д- -■ - - - 0,5 0,6 0,4 0,6 0,9 1 , 1С 5 и другие 3,0 3,8 2,6 1 , 6 0,9 1 , 8 1 , 2 1,3 1 , 1 * 0,8 0,5
РЕАКЦ
ИИ
ГО
РЯЧ
ИХ
АТО
МО
В ТРИ
ТИЯ
295

296 Э . С . ФИЛАТОВ и др.
р е н и я *; п —чи сло с в я з е й , д о сту п н ы х а т а к е ; 7 i и 72 коэф ф и ц и енты , у ч и т ы вающие сте р и ч е ск о е затр удн ен и е при а т а к е ; Ç —логариф м ический д екр ем ен т э нер ги и . В общем сл у ч а е | за ви си т от с о с т а в а бинарной с м е си .
П о ск о л ь к у у д е л ь н а я а к т и в н о с т ь сп и р та не м е н я е т с я при р азб авл ен и и бензол ом и ц и к л о ге к са н о м , можно п о л а г а т ь , что P i ( E ) б ен зо л а леж ит ниже р 2 (Е ) сп и р т а . Т о г д а в ы х о д сп и р та , м е ч е н н о го п о три ти ю , б у д ет п о д чи н яться ур авн ен и ю :
fe — 7£л I ÜRi (RTOH) = - j j Pi (E ) l a- f 2 Pi (E) p 2 (E ) ¡-c o n s t . (3 )спирта
По-видимому, в данном случае потеря энергии при столкновении не зависит от состава:
Ri (RTOH) = л Í2 p¡ (E ) - Í2 p¡ (E ) P 2 (E ) ^ c o n s t . (4)
Е с л и вы хо д о с т а л ь н ы х м еч ен ы х п р одуктов мал или реакции их о б р а зо вани я л еж а т в бол ее низкой э н ер гети ч еск о й о б л а сти , то
R¡ (R T O H ) = f 2 p i (E ) • c o n s t . (5)
Э т о , п о -в и д и м о м у , с п р а в е д л и в о , т а к к а к реакция отщ епления водор ода прои сх о д и т при б о л е е н и зко й э н е р ги и , ч е м за м ещ е н и е [ 24 J . В ы х о д д р у г и х п р о д у к то в м а л . Т о г д а
Í2 - N20-2 П2Т2 и R i (R T O H ) = c o n s t . N2 p¡ (E ) . (6)
О тсю д а
Ri / N2 • P i (E ) = c o n s t . (7)
ч т о и бы ло н ай дено в д е й с т в и т е л ь н о с т и : ради охи м и чески й в ы х о д , Rí/N2, м е ч е н о го спи рта не за в и си т от концентрации в бинарной с м е си с бензолом и ц и к л о гексан о м (p¡ (Е ) не за в и си т от N ).
Д ля в ы х о д а м е ч е н о г о б е н зо л а с л е д у е т п о л ь з о в а т ь с я у р а в н е н и е м ( 1), ко то р о е тр уд н о у п р о сти ть в в ед е н и е м к а к и х либо допущ ений. Р а сс м о т р и м у р авн ен и е ( 1) б о л ее п о д р о б н о . О бозначи м f = а х / [ а х + ( 1-х )Ь ] а = ст1п 1 7 1 , Ь = а 2п2 72 , x = N . Е сл и a = b , то f = x = N и вы хо д м е ч ен о го б ензол а должен линейно з а в и с е т ь о т концентраци и . Э то п р ед п о л а га ет р а в е н с т в о n j c r ^ i = п2ст2 72 , ч т о , ви ди м о , м о ж ет о с у щ е с т в и т ь с я для с т р о г о п о д о б н ы х с и с т е м , например ци клопентан —ц и к л о ге к с а н , г д е д е й ст ви т е л ь н о бы ла найдена а д - ди ти чц ость вы хо д о в м е ч е н ы х п р о д у к то в . В и ссл ед уем о м с л у ч а е , о чеви дн о ,
* Микросечение соударения, возможно, связано с вероятностью перекрывания атомной орбиты трития и молекулярной орбиты электронов связи.
Р Е А К Ц И И Г О Р Я Ч И Х А Т О М О В Т Р И Т И Я 297
Таблица 10
ОПРЕДЕЛЕНИЕ Т В ПОДВИЖНОЙ И НЕПОДВИЖНОЙ ГРУП П ЕС 3 Н7 ОН.
Образцы L¿2 IO 3 0,02г + (С 3 Н7ОН + С6 Н6 и л и С6 Н12) 0,02 мл. Поток нейтронов 2,5 • 101 1 н/см *сек. Время облучения 6,5 часа .
Температура при облучении 40°С. Разделение продуктов на колонках с 25%ТКФ (при 70°С) и ватой (при 120°С).
Объемная доля бензола
(циклогексана)
Выход от общей зарегистрированной активности, %
С 3 Н6ТОН С 3 Н 7О Т
а . Система СзН 7О Н + С бН6
0 , 24,9 24,9
0,117 21,3 18,4
0,338 18,4 1 1 , 6
0,544 14,3 3,7
0,735 7,6 2,4
0,915 1,51 1,39
б. Система С 3Н7ОН + C 6H i2 •
0 24,9 24,9
0,138 23,4 14,5
0,382 15,3 5,7
0,591 9,6 3,9
0,771 5,5 2,5
(Активность бензольной фракции не менялась при дополнительном пропускании через колонку с ватой. Полнота обмена трития в подвижной группе водорода контролировалась повторным пропусканием фракции спирта, вымороженной в ампуле, поставленной на выходе из колонки) .
ni a i Ti ¥= п2 72 i т .е . а #Ь . Не трудно показать, что при a>b ó2f/óx2>0, т.е . зависимость f от N имеет вогнутость к оси N. Аналогично, радиохимический выход С6Н5 T, R/N, или удельная активность зависят от N нелинейно. Кроме этого, так как Ri (С 6Н5Т) = F [р ! (Е )-р 2 (Е )] , то кривая R/N от N будет несимметрично расположена относительно оси N. Таким образом, ход зависимости R/N от N качественно согласуется с рис.З и 4.
В связи с этим концепция о влиянии процесса межмолекулярной передачи энергии [ 1 2 ] не является единственным возможным вариантом объяснения отклонений от аддитивнобти в выходах меченых продуктов, поскольку простые различия в микросечениях соударения или стерических параметрах приводят к таким же эффектам.
298 Э . С . Ф И Л А Т О В и д р .
СРАВНЕНИЕ РАСЧЕТНОГО И ЭКСПЕРИМЕНТАЛЬНОГО ОТНОШЕНИЯ ВЫХОДОВ HT/RTQH В РЕАКЦИЯХ ГОРЯЧИХ
АТОМОВ ТРИТИЯ С АЛИФАТИЧЕСКИМИ СПИРТАМИ
Таблица 11
(Исходное соотношение рассчитывалось по выходам продуктов из Н-бутанола)
СпиртыВ ыход HT/RTOH
HT RTOH расчетное экспериментальное
CH3OH 34 27 1,3 1,3
С2Н5ОН 41 25 1,3 1,6
С3Н7ОН 40 24 1,4 1,7
С4Н3ОН 38 25 1,5 1,5
i - C 4H9OH 32 30 1,4 1,1
втор-С 4НдОН 34 32 1,4 1,1
трет~С4НдОН 30 31 1,3 1,1
ЛИТЕРАТУРА
[1 ] Ф И Л А Т О В Э . С . , Н Е С М Е Я Н О В А н . Н . , Ч Е П Ы Ж Е В Ю .Б . , Радиохим ия, 6 , 5 (1964)[2 ] M A L L IN S O N J . , M IL L E R G . , SH A W P . , Rad. A c ta , 3 (1963) 136f 3 J K N IG H T В . , M I L L E R G . , S H A W P . , IN O R G J . N u c l . C h e m . , 23, 15 (1 96 1 )[4 ] S H A W P . , R a d .A c t a , 1 ,4 (1963 ) 177[5 ] E S T R U P R . , W O L F G A N G R . , J .A m . C h em . S o c . , 82, 11 (1960 ) 266516] H E N C H M A N М . , U R C H D . , W O L F G A N G R . , C an . J . C h e m ., 38, (1 9 6 0 ) 1722[7 ] R O W LA N D F., L E E J., M U SG R A V E H , W H IT E R „ Chem. E ff. Nucl. Trans..V ienna. 2, 67(1961)[ 8J LE E E., R O W LA N D F . , J. Am . Chem. S oc ., 85, 10 (1963) 2907 ~19) JU R Q E LE IT H . , W O L F G A N G R . . J. Am . Chem. S o c ., 85, (1963) 1057
[1 0 ) ROOT J ., R O W L A N D F . , J .A m . Chem. Soc., .85, (1963) 1021 111] ЯКУШИН Ф .С .,У с п ех и химии, 31, вы п.2, (1962) 241[12 ] АВДОНИНА Е . Н ., Радиохимия, 4, 5 (1962) 617[1 3 ] А В Д О Н И Н А Е . Н . , Н Е С М Е Я Н О В А н . Н . , Ун Х ао -м и н , Радиохим ия , 6 , 3 (1964) 323[1 4 ] Ф И Л А Т О В Е . С . , Н Е С М Е Я Н О В А н . Н ., Ч Е П Ы Ж Е В Ю .Б . , В е с т н и к 'м Г У , серия 6 ,
45 (1963)[1 5 ] ПОЗДЕЕВ В . В . , Н Е С М Е Я Н О В А н . Н ., Д ЗА Н ТИ ЕВ Б . Г ., Радиохимия, 4, 4 (1962) 404[1 6 ] П О З Д Е Е В В . В . , Н Е С М Е Я Н О В Ан . Н . , Д З А Н Т И Е В Б . Г . , Кинетика и катализ, 4, 2
(1963) 318[1 7 ] Ф И Л А Т О В Е . С ., Н Е С М Е Я Н О В А н .Н . , Вестник М Г У , сер .хим . , 4, 13 (1964)[1 8 ] SO K O LO W SK A A . , H ASK IN L . , R O W L A N D F . S . , J. A m . Chem . S o c ., 84, (1962) 2469 119] H O F F W . , R O W L A N D F .S . , J. A m . Chem . S o c ., 79. 18 (1957) 4867[2 0 ] ГУСЫ Н И Н В .И ., Т А Л Ь Р О З Е В . Л . , Труды 11 Всесою зного совещания no радиационной
химии, АН С С С Р , 1962, с т р .79[2 1 ] O D U M R . , W O L F G A N G R . , J. A m . Chem . Soc. 85, (1963) 1050[22 ] E L A T R A S H A . , JO N SE N R . , W O L F G A N G R . , J. Phys. C h e m ., 64, (1960) 785[23 ] Н ЕСМ ЕЯ Н О В А н .Н ., Ф И ЛАТОВ Э . С ., Г У С А К О В С К А Я И ., Радиохимия, 4, 4 (1962) 462[24 ] MOSER Н ., SHORES R . , J. P h y s .C h em ., 66, N 11, (1962) 2272

Р Е А К Ц И И Г О Р Я Ч И Х А Т О М О В Т Р И Т И Я 299
D IS C U S S IO N
A. G. MADDOCK: It seems to me that the results presented in the paper are strongly reminiscent of the radiolysis of the alcohols. CH2OH is a very prominent mass in the mass spectra of all alcohols. The Gch4 values for methyl and ethyl alcohols are approximately in the ratio of your yields of CH3T. In fact, it is known that about half of both these methane yields arise from hot processes or, as the radiation chemist would say, are molecular products. I should like to ask if all the studies you mentioned relate to unscavenged experiments,
A .N . NESMEYANOV: We believe that methyl and ethyl alcohols are obtained mainly in hot reactions. The products of the attack were studied on the basis of the tritium activity in the alcohols in question but, in experimental conditions, their radiolysis amounted to only a few per cent. No scavengers were added, since experience had shown that they did not affect the results.
F .S . ROWLAND: With regard to the intramolecular distribution of tritium in labelled ethyl alcohol after recoil tritium reactions, I might mention that the distribution of activity in ethanol has been found to be about 74±4% in the CH2T position for recoil tritium reactions in liquid ethanol*.I do not know of any comparable experiments in mixtures.
^ HOFF, W.J. Jr. and ROWLAND, F. S . , Studies of the Tritium Labelling Reaction. III. Alcohols
and Acetone, J. Amer. chem. Soc. 79 (1957) 4867.


REACTIONS OF HOT C138 ATOMS IN MIXTURES OF CARBON TETRACHLORIDE
WITH ALIPHATIC ALCOHOLS
L. VASÁROSCENTRAL RESEARCH IN STITU TE FOR PH YSICS,
BUDAPEST, HUNGARY
Abstract — Résumé — Аннотация — Resumen
REACTIONS OF HOT C l” ATOMS IN MIXTURES OF CARBON TETRACHLORIDE W ITH ALIPHATIC” ALCOHOLS. Investigations of the chemical effects of nuclear reactions in binary systems are expected to
yield much useful information. Study of the recoil processes of the halogen derivatives when the second
component is suitably chosen and its concentration varied in a wide range might permit inferences to be made
on the role and mechanism of the various stabilizing processes. Considering the results obtained with CCl^-Cli, CCl4-SiC l4, ССЦ-СеНб and CClj-c-hexane mixtures as well as the energy scavenger property of alcohol, it seemed of interest to study the contribution of the alcohols to the stabilization of hot C l38.
Chemical processes induced by hot C l38 from the nuclear reaction С1эт(п, y )C l38 were investigated in
mixtures of CC l4-ROH (where R = CH3- , C2H5~, CsH-j- and(CHs)£H -). The irradiations were performed
in the thermal column of the 2 MW VVRS reactor using rather short exposure times to keep the radiation chemical effects at negligible level. The organic fractions were separated from the inorganic ones by extraction
and the former were analysed by gas chromatographic method. Total retention and the yield of the complete
set of organic chlorine compounds were determined in terms of alcohol concentration.Some interesting results are that the yield of reaction products in which the OH radical of aliphatic
alcohol has been replaced by C l38 increases with increasing alcohol concentration with a simultaneous decrease
in the labelled CCI4 yield and that, in addition to the monochlorine derivates with less carbon atoms than
the alcohol molecule, a considerable amount of chloroform is formed with maximum yield at a given alcohol concentration.
The relative contributions of the hot and the epithermal stabilization processes of energetic C l38 and
the mechanism of the various reactions are discussed.
RÉACTIONS DES ATOMES seel CHAUDS DANS DES MELANGES DE TÉTRACHLORURE DE CARBONE
ET D 'U N ALCOOL ALIPHATIQUE, Les recherches sur les effets chimiques des réactions nucléaires dans les
systèmes binaires semblent devoir donner beaucoup de renseignements utiles. L’ étude des phénomènes de
recul dans les dérivés halogénés, lorsque le deuxième composant du mélange est choisi convenablement et que Ton fait varier sa concentration dans une gamme assez étendue, pourrait permettre d'élucider le rôle
et le mécanisme de divers processus de stabilisation. En considérant les résultats obtenus avec les mélanges
CCI4-C I2, CCI4-S ÍC I4, CC l4-C 6H6 et CC l4-cyclohexane, et tenant compte de la propriété de l'a lcoo l de
« b a la y e r » l'énergie, il a semblé intéressant d'étudier le rôle joué par les alcools dans la stabilisation de
98C1 chaud.L'auteur a étudié les réactions chimiques de 38C1 obtenu par le processus этС1(п,у)38С1 dans des mélanges
CCl4-ROH, o ù R représentait successivement les radicaux CH8-, C$HS- , C3H7- et (C H j^CH -. Il a procédé
aux irradiations dans la colonne thermique du réacteur VVRS de 2 MW avec des temps d'exposition assez
courts pour que les effets radiochimiques restent négligeables. Après avoir séparé les fractions inorganiques
des fractions organiques par extraction, il a analysé ces dernières par chromatographie gazeuse. Il a déterminé
la rétention totale et le rendement pour toute la série de composés organiques chlorés en fonction de la concentration de l'alcool.
Parmi les résultats intéressants, il cite les suivants: tout d'abord, le rendement en produits de la réaction
lorsque le radical OH de l'alcool aliphatique se trouve remplacé par 38C1 augmente lorsque la concentration
en alcool augmente, tandis que le rendement en CC l4 marqué diminue; d'autre part, en plus des dérivés
monochlorés contenant moins d'atomes de carbone que la molécule d'alcool, il se forme une quantité appréciable de chloroforme, le rendement étant maximum pour une certaine concentration de l'a lcoo l.
301

302 L. VASÁROS
L’auteur étudie dans quelle mesure interviennent les phénomènes de stabilisation d e 3*Cl, d'une part au niveau des hautes énergies, d'autre part au niveau épithermique. Il décrit, enfin, le mécanisme des
diverses réactions.
Х И М И Ч Е С К И Е РЕАК Ц И И Г О Р Я Ч И Х А Т О М О В Х Л О Р А -3 8 В С М Е С Я Х Ч Е Т Ы Р Е Х Х Л О Р И С Т О Г О У Г Л Е Р О Д А С А Л И Ф А Т И Ч Е С К И М И С П И Р Т А М И . Исследование реакций горячих атомов в бинарных системах представляет значительный интерес. При изучении атомов отдачи галогенов в их органических соединениях правильный подбор второго компонента и изменение состава смеси в широком интервале концентраций дают возможность для определения относительной роли различных процессов и механизмов стабилизации.
На основании опытных данных, полученных в см есях C C I4 — C I 2 , C C I4 —S ÍC I4 , C C I4 — C gH 6 и С С 14 цикло-СбН 12 , а также исходя из того, что алифатические спирты являются ак цепторами энергии возбуждения, считали целесообразным изучение роли алифатических спиртов в процессах стабилизации атомов отдачи хлора-38 .
Были изучены химические реакции горячих атомов хлора-38 , образующихся в результате ядерной реакции 37С1(п, у) 38С1 в системе СС14 — ROH (где: R = CH 3 , C 2 H5 , C 3 H7 И/СН3 /2С Н ) Облучение проводилось в термическом канале реактора В В Р —С с мощностью 2 М эв. Время облучения было достаточно коротким, чтобы свести к минимуму радиационно-химические эффекты .
Для разделения органической и неорганической фракции был использован экстракционный м етод, а органическая фракция бы ла анализирована путем газовой хром атограф ии .
Были определены общее удержание и выход целого ряда органических производных хлора. Установлено, что выход продуктов замещения ОН на хлор-38 растет с увеличением концентрации спирта в то же время выход меченого СС14 уменьшается. Наряду с монохлорпроизвод- ными с меньшим числом атомов углерода, чем в молекуле спирта, в значительном количестве образуется хлороформ, выход которого имеет максимум.
Рассмотрены относительная роль горячих и надтепловых процессов стабилизации горячих атомов хлора и механизм этих реакций.
REACCIONES DE ATOMOS заС1 CALIENTES EN MEZCLAS DE TETRACLORURO DE CARBONO Y ALCOHOLES ALIFATICOS. Cabe esperar que las investigaciones de los efectos químicos de las reacciones nucleares
en sistemas binarios proporcionen informaciones abundantes y útiles. El estudio de los procesos de retroceso
en los derivados halogenados, cuando se elige adecuadamente el segundo componente y se varía su concentración en un amplio intervalo, podría servir para sacar conclusiones sobre la función y e l mecanismo de los
diversos procesos de estabilización. Considerando los resultados obtenidos con las mezclas CC14-C12, CCl4-SiC^, CC^-CgHgy CCl4-ciclo-hexano, así como la propiedad del alcohol de actuar como absorbedor de la energía, parecía interesante estudiar la contribución de los alcoholes a la estabilización d e l38C l caliente.
Se investigaron los procesos químicos inducidos por átomos 3®C1 calientes, originados mediante la reacción
37C l(n ,y )38Cl, en mezclas de CC l4-ROH (siendo R = CH8-, C 2H5- , CjHy- у (СНз)2С Н -). Las irradiaciones
se efectuaron en la columna térmica del reactor VVRS, de 2 MW, empleando tiempos de exposición más
bien cortos, para mantener los efectos químicos de la radiación a un nivel despreciable. Las fracciones orgánicas se separaron de las inorgánicas, por extracción, y se analizaron las primeras por cromatografía en
fase gaseosa. Se determinó la retención total y el rendimiento de la serie completa de compuestos orgánicos
del cloro en función de la concentración del alcohol.Entre los resultados obtenidos, merece señalarse que el rendimiento de productos de reacción en los
cuales el radical OH del alcohol alifático ha sido sustituido por 3*C1 crece al aumentar la concentración de
alcohol, con una disminución simultánea del rendimiento de CC14 marcado; y que, además de los derivados
monoclorados con menos átomos de carbono que la molécula de alcohol, se forma una cantidad considerable
de cloroformo, alcanzándose un rendimiento máximo para una concentración de alcohol determinada.Se discuten las contribuciones relativas de los procesos de estabilización calientes y epitérmicos del
3BC1 de alta energía y e l mecanismo de las diversas reacciones.
INTRODUCTION
O n e o í the methods for studying the mechanism of chemical processes induced by recoiling atoms is to perform experiments with hot atoms in

REACTIONS OF HOT C l38 ATOMS 303
binary systems. In such mixtures it is easier to follow the fate of recoil atoms and to obtain information on the mechanism of hot atom reactions.
It is apparent from experimental results obtained in liquid organic binary systems with hot halogen atoms that the second component may act in various ways 11J . Some diluents such as SÍCI4 in the CCI4-SÍCI4 system(2] or CCI4 in the С2Н5ВГ-СС 14 system 13J reduce the organic yield, as is to be expected from the diluting effect, in terms of the physical impact theory. At the same time the addition of any radical scavenger, e .g . elementary halogens, even in very low concentration, hinders the diffusion controlled recombination reactions. Primary and secondary amines also reduce the organic yield, probably by their reactions with the excited molecules (Menshutkin reaction) 14, 5J. In С 2Н5В Г -С 6Н6 and СНгВгг-СбНб systems the energies of the excited recoil molecules seem to be transferred to benzene [3] and this results in the stabilization of the radioactive compounds.
It has been assumed from luminescence experiments on alcohols that it is the hydrocarbon chain of the alcohol molecule that serves as a "sponge transmitter" in the excitation energy transfer to the acting OH group,, and is actually capable of emitting the excitation energy [6 J . Intramolecular energy transfer of similar type can be held responsible for the high tritiated hydrocarbon yield in the irradiated L i2 CO3- aliphatic alcohol system [7] .
To obtain further information on the energy-accepting character of aliphatic alcohols as well as on the mechanism involveid in the reaction between Cl38 and the alcohol molecule, the Cl31 (n, 7 )Cl38 reaction inCC l4-ROH systems (where R = CH3-, C 2H 5-, CH3-C H 2-C H 2- , and (СНз)гСН-) has been investigated.
EXPERIM ENTAL
Carbon tetrachloride was photobrominated to eliminate possible unsaturations, then extracted with Na2S03 solution, washed by N a 2C 0 3 solution and distilled water, dried with CaCl2> twice fractionated and the medium fractions used. A ll the reagents were of G .R . purity.
The samples were irradiated in the thermal column of the 2 MW VVRS- type reactor with a neutron flux of about 1010n/cm2. s . To eliminate possible impurity effects, small amounts of Cl2 were added to each sample. The time of exposure was 2 h in each case, since it had been already established that in the interval from 1 h to 12 h irradiation time the total retention and the individual yields of the radioactive products did not vary with the time of exposure.
To determine the total organic retention the samples were extracted with 0.1N aqueous solution of NaCl containing Na2S03 . The activities of both the organic and inorganic fractions were measured by crystal scintillator counter. The relative yields of the various organic radioactive compounds were measured by gas chromatography. ,
The conditions of the chromatographic separation were:Column: 4 m, packed copper columnSolid support: 0. 2-0.3 mm "Thermolit" fire brick

304 L. VASÁROS
Stationary phase: 15wt.% tricresyl phosphate Temperature: 90°CCarrier-gas: H2, flow rate: 20 ml/min Sample introduction: bulb crusherThe inorganic activity was adsorbed on potassium ferrocyanide pre
column [8] . The assembly comprised two detectors. The catharometer was used for determining the chlorinated hydrocarbon retention time. Geiger counter method and automatic recorder served for measuring the activities in the different compounds. In Pig. 1 the radiochromatogram of irradiated 30 mole % CC l4-n -C 3H7OH mixture is given as an example.
Fig. 1
Radiochromatogram of irradiated CC14 -n -C 3H?OH mixture { N n- C 3H 7O H = 0 .3 m ole fraction)
RESULTS
Measured total retentions and the individual yields of the Cl38 active products in irradiated CCI4-ROH systems are plotted as a function of alcohol concentration in Figs. 2-5.
After pure carbon tetrachloride was irradiated, the total retention and the parent material yield were found to be 43% and 36% [9 ], respectively, which is in good agreement with Aten's results 110]. The total organic yield as well as that of the parent material first rapidly decreases with increasing alcohol concentration; above NROH =0.5 mole fraction, however, the yields attain afairly stable value: R totai = 14-18%, Reel, = 3-4%. In the CCI4- i- C3H7OH system the total retention is similarly 18% at 0. 9 mole fraction of alcohol but the radioactive carbon tetrachloride yield is lower,and changes in a different way from that observed in the case of other alcohols.
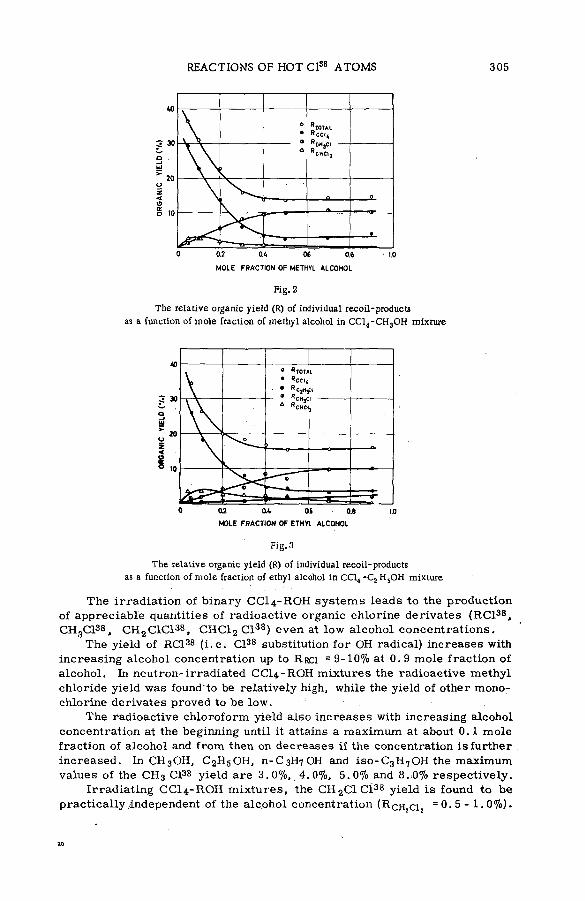
REACTIONS OF HOT C l38 ATOMS 305
0 0,2 0Л 06 0.6 ■ 1.0
MOLE FRACTION OF METHYL ALCOHOL
Fig. 2
The relative organic yield (R) o f individual recoil-products
as a function of m ole fraction of methyl alcohol in C C l4-C H 3OH mixture
0 02 0.4 as o.e 1.0
MOLE FRACTION OF ETHYL ALCOHOL
Fig.3
The relative organic yield (R) of individual recoil-products
■ as a function o f m ole fraction of ethyl alcohol in CC14 -C 2H 50H mixture
The irradiation of binary CCI4-ROH systems leads to the production of appreciable quantities of radioactive organic chlorine derivates (RC138, CH3C138, CH2C1C138, CHC12 Cl38) even at low alcohol concentrations.
The yield of RC138 (i.e . Cl38 substitution for OH radical) increases with increasing alcohol concentration up to Rrci = 9-10% at 0. 9 mole fraction of alcohol. In neutron-irradiated CCI4- ROH mixtures the radioactive methyl chloride yield was found'to be relatively high, while the yield of other monochlorine derivates proved to be low.
The radioactive chloroform yield also increases with increasing alcohol concentration at the beginning until it attains a maximum at about 0 .1 mole fraction of alcohol and from then on decreases if the concentration is further increased. In CH3OH, C2H5OH, П -С3Н7ОН and iso-C3H7OH the maximum values of the CH3 Cl38 yield are 3.0%, 4.0%, 5.0% and 8.;0% respectively.
Irradiating CCI4-ROH mixtures, the CH 2C lC l38 yield is found to be practically/independent of the alcohol concentration (Rch2ci2 = 0 .5 - 1 . 0%).
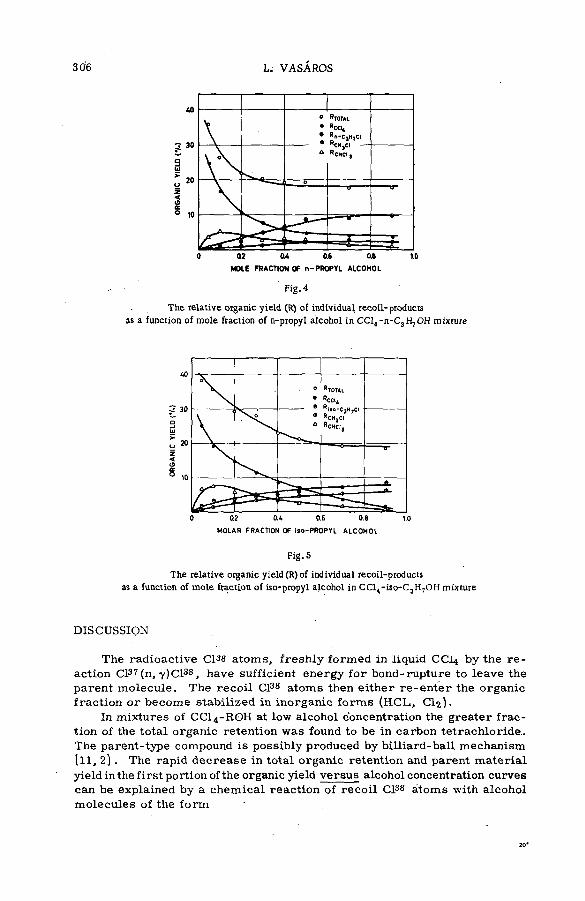
306 L. VASA ROS
М 0Л OS 0.8
MOLE FRACTION OF n-PROPYL ALCOHOL
10
Fig. 4 '
. The relative organic yield (R) of individual recoil- products
as a function of mole fraction of n-propyl alcohol in CCl4-n-C3H ,O H mixture
0 0.2 0.4 0.6 0.8 1.0
MOLAR FRACTION OF iso-PROPYL ALCOHOL
Fig. 5
The relative organic yield (R) of individual recoil-products
i a function of mole fraction of iso-propyl alcohol in CCl4-iso-C3H ,O H mixture
DISCUSSION
The radioactive Cl38 atoms, freshly formed in liquid CCJL4 by the re action Cl37 (n, y)Cl38, have sufficient energy for bond-rupture to leave the parent molecule. The recoil Cl38 atoms then either re-enter the organic fraction or become stabilized in inorganic forms (HCL, CI2 ).
In mixtures of CCI4-ROH at low alcohol concentration the greater fraction of the total organic retention was found to be in carbon tetrachloride. The parent-type compound is possibly produced by billiard-ball mechanism ( l l , 2] . The rapid decrease in total organic retention and parent material yield in the first portion of the organic yield versus alcohol concentration curves can be explained by a chemical reaction of recoil Cl38 atoms with alcohol molecules of the form

REACTIONS OF HOT Cl38 ATOMS 307
Cl38+R '-C -O H -» Cl38' + R '-C t y - (1)
Some of the newly formed radioactive carbon-tetrachloride molecules are possibly excited. The fate of these excited molecules depends on the way their excitation energy is lost. In systems like ССЦ-ROH energy transfer from some of the excited CCI3CI38 to alcohol molecules seems possible. Taking into consideration the experimental results, however, energy transfer appears to be negligible up to alcohol concentration Nrqh = 0.5 mole fraction. Thus it is assumed that some of the excited CCI3CI38 molecules, probably those with sufficient excitation energy for quasi-mono-molecular dissociation, undergo another type of reaction with the alcohol molecules that leads to the formation of radioactive chloroform. With increasing alcohol concentration the probability of energy transfer between newly formed radioactive carbon tetrachloride and alcohol molecules increases until at concentrations above 0.5 mole fraction the energy transfer may become dominant and actually lead to the stabilization of the excited parent molecules.
The excited carbon tetrachloride molecules may thus stabilize by the following reactions:
The CC12C138 radical in the reaction (2) may remove hydrogen atoms from any carbon atom of the alcohol molecule. This assumption is supported by the following experimental results. A considerable amount of CHCI3 is formed in CCI4- cyclohexane mixture exposed to ionizing radiation [12] , A similar mixture was irradiated in present experiments by slow neutrons and again the formation of CHC12C138 could be observed.
The radioactive chloroform yield increases with increasing alcohol concentration at the beginning because of the higher reaction probability between excited CCI3CI38 and alcohol molecules. On the other hand, the dilution of carbon-tetrachloride with alcohol, while decreasing the formation probability of strongly excited CCI3CI38 molecules, increases the probability of energy transfer. As a result of these competitive reactions the radioactive chloroform yield versus ROH concentration curve exhibits a maximum at about 0 .1 mole fraction of alcohol.
Some of the recoiling Cl38 atoms react with the alcohol molecules in the range of epithermal energies. This results in the production of various radioactive alkyl chlorides. The epithermal reaction probability is proportional to the alcohol concentration. A simplified scheme of these re actions can be written as , .
C C l3Cl38 + ROH -> C C 1 3C138 +ROH (2)
CCI3CI38 CC12C138+R0H ->CHCl2 Cl38+ROH (3)
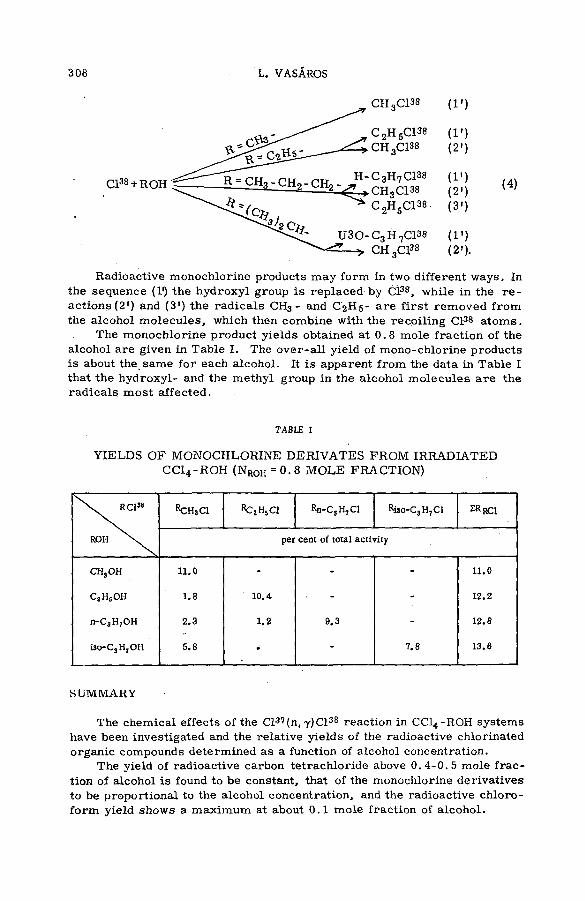
308 L. VASÁROS
d ’)
( ! ' )(2 -)
(1,) (4)( 2 - ) ™(3*)
(1 ')( 2 - ) -
Radioactive monochlorine products may form in two different ways. In the sequence (I1) the hydroxyl group is replaced by Cl38, while in the re actions (2 ') and (31) the radicals CH3 - and C 2H 5- are first removed from the alcohol molecules, which then combine with the recoiling Cl38 atoms.. The monochlorine product yields obtained at 0.8 mole fraction of the
alcohol are given in Table I. The over-all yield of mono-chlorine products is about the same for each alcohol. It is apparent from the data in Table I that the hydroxyl- and the methyl group in the alcohol molecules are the radicals most affected.
Cl38 + ROH
CH 3C138
C 2H 5C138CH 3C138
- C 3H7C138CH3C138C 2H5C138
изо- С3Н,С138 > CH 3C13S
TABLE I
YIELDS OF MONOCHLORINE DERIVATES FROM IRRADIATED CCI4-ROH (N roh =0.8 MOLE FRACTION)
SUMMARY
The chemical effects of the Cl37(n, y)Cl38 reaction in CCl4-ROH systems have been investigated and the relative yields of the radioactive chlorinated organic compounds determined as a function of alcohol concentration.
The yield of radioactive carbon tetrachloride above 0.4-0. 5 mole fraction of alcohol is found to be constant, that of the monochlorine derivatives to be proportional to the alcohol concentration, and the radioactive chloroform yield shows a maximum at about 0 .1 mole fraction of alcohol.

REACTIONS OF HOT C l38 ATOMS 309
It can be established from the experimental evidence that aliphatic alcohols may enter into chemical reaction with excited CC14 molecules and recoil Cl38 atoms, and at the same time may act as an energy acceptor particularly at concentrations above 0.5 mole fraction alcohol.
A C K N O W L E D G E M E N T S
Thanks are due to I. Kiss for assistance in preparing this paper, to R. Schiller for useful advice in developing the gas chromatographic technique, and to E.S. Filatov for helpful discussions.
R E F E R E N C E S
[1 ] Ф И Л А Т О В Э .С . , Исследование реакций горячих атомов брома в органических веществах. Автореферат на соискание ученой степени кандидата химических наук, И зд . М Г У , 1963.
[2 ] MILLER, J.M. and DODSON. R. W . . J. chem. Phys. Jjj (1950) 865.
[3 ] Ф И Л А Т О В Э . С . , Н Е С М Е Я Н О В А н .Н . , Ч Е П Ы Ж Е В Ю . Б . , Радиохимия 6 5 (1964) 595.[4 ] STOCKLIN, G . , SCHMIDT-BLEEK, F. and HERR, W . , in Chemical Effects of Nuclear Transformations J
IAEA, Vienna (1961) 245.
И Н Е С М Е Я Н О В А н .Н . , Ф И Л А Т О В Э . С . , Радиохимия 5 3 (1963) 378. .[6 ] ГУ С Ы Н И Н В . И . , Т А Л Ь Р О З Е В . Д . , "Т р . II. Всесою зного совещания по радиационной
химии", И зд . АН С С С Р , М . (1962) 79.
[7 ] Н Е С М Е Я Н О В А н . Н . , ЦЗЯН Т А Й -В А Н и Ф И Л А Т О В Э . С . , Радиохимия 5, 4 (1963) 515.[8 ] HARRIS, W .E ., McFADDEN, W. Н. and McINTOSH, R .G ., J. phys. Chem. 63 (1959) 1784.
[9 ] В А Ш А РО Ш Л . , Ф И Л А Т О В Э . C Н Е С М Е Я Н О В А н . Н . , Радиохим ия 6, 4 (1964 ) 484.[10] ATEN, A .H .W .,J r . and V A N RAAPHORST, J .G ., in Chemical Effects o f Nuclear Transformations J
IAEA, Vienna (1961) 203.[11] LIBBY, W .F . . J. Amer. chem. Soc. 69(1947) 2523.[12] STONE, J. A . and DYNE, P .J ., Cañad. J. Chem. 42 (1964) 669.


CHEMICAL EFFECTS OF NUCLEAR RECOIL IN ORGANIC HALIDE SYSTEMS:
A NEW THEORETICAL TREATMENT AND EXPERIMENTAL VERIFICATION OF THE THEORY
, S. S. KO N T IS* P. SA N ITW O N G S+ A N D M . WESTON
LONDONDERRY LABORATORY FOR R AD IO CH E M ISTRY,
U N IVERSITY OF DURHAM , DURHAM , UNITED K ING D O M
Abstract — Résumé — Аннотация — Resumen
CHEMICAL EFFECTS OF NUCLEAR RECOIL IN ORGANIC HALIDE SYSTEMS: A NEW THEORETICAL
TREATMENT AND EXPERIMENTAL VERIFICATION OF THE THEORY. Methods have been developed for calculating the organic retention of radiohalogen to be expected following neutron activation of mixtures of two organic halides and the corresponding free halogen. The methods are based on the concepts of the Libby
"billiard-ball" process of hot-atom labelling and retentions are expressed in terms of two types of parameters: ( 1 ) the fractional chance that collision of a hot radiohalogen atom with a particular molecule w ill lead to
retention of the hot atom in that molecule; (2 ) the upper and lower energy limits for what is termed a hot bromine atom. It is shown how the former type of parameter may be derived from experiments with binary
mixtures of an organic halide and the corresponding free halogen: the second type of parameter must be de
duced from nuclear and chemical data.
These methods have been tested by studies on the systems C 2H5Br/CCl4/Br2 and C 2H5Br/C6HsBr/Brf . The calculated retentions (using parameters derived from studies of the systems C 2H5Br/Br2, СС14/Вг2 and
CgH5Br/Br2) are in good agreement with those found experimentally in mixtures with a bromine mole-fraction
greater than about 0.1. It is therefore considered that the Libby mechanism is adequate to explain the observed organic retentions in such mixtures. As the bromine mole-fraction is reduced, below 0.1 the observed
retentions become progressively higher than calculated values. This is attributed to there being additional modes of hot-atom labelling which become operative when the bromine concentration is not high enough to
give adequate radical scavenging.
EFFETS CHIMIQUES DU RECUL NUCLÉAIRE DANS DES SYSTEMES ORGANIQUES HALOGENES : NOUVEAU TRAITEMENT THÉORIQUE ET VÉRIFICATION EXPÉRIMENTALE DE LA THÉORIE. Lesauteuts
ont mis au point des méthodes pour calculer la rétention organique du radiohalogène à laquelle on peut s'attendre
après activation par les neutrons de mélanges de deux halogénures organiques et de l ’halogène libre correspondant. Ces méthodes sont basées sur les principes du mécanisme de la «bou le de b illard» pour le marquage
par l'atome chaud, auquel Libby a donné son nom, et les rétentions sont exprimées en fonction de deux types
de paramètres, savoir: 1 . la probabilité que le choc entre un atome chaud de radiohalogène et une molécule
donnée détermine la rétention de l'atome chaud dans cette molécule; 2 . les limites supérieure et inférieure
des énergies pour lesquelles un atome de brome est dit chaud. Les auteurs montrent comment le premier type
de paramètre peut être dérivé d'expériences sur des mélanges binaires d'un halogénure organique et de l'halogène libre correspondant; le second type de paramètre doit être déduit des constantes nucléaires et chimiques.
Des études sur les systèmes СгЩВг/СС^/Вгг et CgHgBr/CeHgBr/Br^ ont permis de faire l'essai de ces
méthodes. Les valeurs de rétention calculées (en Utilisant des paramètres dérivés d'études des systèmes
С 2Н5ВГ/ВГ2, СС 14/ВГ2 et СбН5Вг/Вг2) concordent avec celles qui sont déterminées expérimentalement dans
des mélanges où la fraction molaire de brome est supérieure à 0,1 environ. Par conséquent, les auteurs con
sidèrent que le mécanisme Libby permet d'expliquer les rétentions organiques observées dans ces mélanges. Lorsque la fraction molaire de brome devient inférieure à 0 ,1 , les rétentions observées augmentent progressive
ment par rapport aux valeurs calculées, ce qui est sans doute dû au fait que d'autres modes de marquage par l ’atome chaud interviennent lorsque la concentration en brome n’est pas assez élevée pour assurer un bon
« b a la y a g e » des radicaux. .
* IAEA Fellow+ Former IAEA Fellow
311

312 S .S . KONTIS et al.
Х И М И Ч Е С К И Е В О З Д Е Й С Т В И Я Я Д Е РН О Й О Т Д А Ч И В О Р Г А Н И Ч Е С К И Х ГА Л О И Д Н Ы Х С И С Т Е М А Х : Н О В О Е Т Е О Р Е Т И Ч Е С К О Е Т О Л К О В А Н И Е И Э К С П Е Р И М Е Н Т А Л Ь Н О Е П О Д ТВ ЕРЖ Д Е Н И Е Т Е О Р И И . Были разработаны методы расчета органического удержания радиогалогенов, которые можно ожидать вследствие нейтронной активации смесей двух органических галоидов и соответствующих свободных гало ген о в . Эти методы основаны на концепциях процесса "бильярдного шара" Либби по мечению горячих атомов, и удержания выражаются в виде двух типов параметров, а именно: 1 ) фракционная возможность того, что столкновение го рячего атома радиоактивного галогена с какой-то определенной молекулой приведет к удержанию горячего атома в той м олекуле; 2 ) верхние и нижние пределы энергий для так называемого горячего атома брома. Показывается, как первый тип параметра может быть выведен из экспериментов с бинарными смесями органического галоидного соединения и соответствующего свободного галогена: второй тип параметра должен выводиться из ядерных и химических данных.
Эти методы проверялись с помощью исследований на систем ах H ^B r / C C I 4 / Б г 2 и С г Н5В г / С 6Н 5В г / В г 2 . Вычисленные удержания (с использованием параметров, выведенных из исследований систем С - Н-,В г / В C C W B r j и С е Н 5В г / В г г ) хорошо согласую тся с удержаниями, найденными экспериментально в смесях с фракцией граммолекулы брома больше 0 , 1 . Таким образом, полагают, что механизм Либби является подходящим для объяснения наблюдаемых органических удержаний в таких смесях. Если фракция граммолекулы брома опускается ниже О Д , наблюдаемые удержания становятся в прогрессивной степени выше, чем р ас считанные величины. Это объясняется дополнительными видами мечени* горячих атом ов, которые становятся действующими, если концентрация брома не является достаточно высокой, чтобы дать соответствующую очистку радикала.
EFECTOS QUIMICOS DEL RETROCESO NUCLEAR EN SISTEMAS DE HALUROS ORGANICOS: NUEVO
TRATAMIENTO TEORICO Y VERIFICACION EXPERIMENTAL DE LA TEORIA. Los autores elaboraron métodos
para calcular la probable retención orgánica de los halógenos radiactivos después de activar con neutrones
mezclas de dos haluros orgánicos y el correspondiente halógeno libre; Los métodos se basan en ¡a teoría del proceso de « bolas de billar » de Hbby para la marcación con átomos calientes ; la retención se expresa en
función de dos tipos de parámetros, a saber: 1 ) la probabilidad relativa de que ei choque de un átomo caliente
de halógeno radiactivo con una molécula determinada produzca la retención del átomo caliente en dicha
molécula; 2) los limites energéticos superior e inferior dentro de los cuales el átomo de bromo puede considerarse caliente. Los autores demuestran cómo el primer tipo de parámetro puede deducirse experimentalmente
de mezclas binarias de un haluro orgánico y el correspondiente halógeno libre; el segundo tipo de parámetro debe deducirse a partir de datos nucleares químicos.
Dichos métodos han sido ensayados en ¡os sistemas С гН5Вг/СС1,,/Вгг у С гН 5Br/C6H sBr/8r?. Losvalores
calculados para las retenciones (empleando parámetros derivados del estudio de los sistemas Сг H5 Br/Br 2 . СОа/Вгг у С6Н5Вг/Вгг> eoncuerdan satisfactoriamente con los dalos hallados experimentalmeme en mezclas
cuya fracción molar de bromo es superior a 0 , 1 . D e esta manera se considera que el mecanismo propuesto
por Líbby permite explicar ¡as retenciones orgánicas observadas en dichas mezclas. Cuando la fracción molar
de bromo se va reduciendo a valores inferiores a 0, 1 , las retenciones observadas se hacen progresivamente
mayores que los valores calculados. Este fenómeno se atribuye a la existencia de otros modos de marcación
con átomos calientes, que entran en juego cuando la concentración de bromo no es bastante elevada para
eliminar adecuadamente los radicales en virtud de mecanismos de depuración.
INTRODUCTION
Studies of the organic retention of the activity produced during neutron irradiation of liquid organic halides have been mainly concerned with either pure organic halides or binary mixtures of the organic halide with the corresponding halogen. The results of such studies depend., among other factors, on the mole fractions of the two components, and since these are not independently variable in a binary mixture, it appeared to us worthwhile to extend work in this field to ternary mixtures. (This has already been done for special purposes as, for example, in studies by MILMAN on the effect of a moderator on the ethyl bromide/bromine system [1 j,. ) It then becomes necessary to be able to relate results obtained from binary mixtures to those obtained from ternary mixtures. The treatment here reporte

NUCLEAR RECOIL IN ORGANIC HALIDE SYSTEMS 313
had therefore been developed with this aim and has now been experimentally tested for two ternary systems.
It had always been recognized that the success (or otherwise) of the treatment could be regarded as confirming (or otherwise) such features of the mechanism of organic retention as formed an essential part of the treatment. It was recently realized that the logical structure of this treatment was closely related to the treatment of ESTRUP and WOLFGANG [2] originally stated to explain the organic retentions observed when tritium atoms recoil in gaseous methane, but since adapted to other systems [3] . The adaption of the Estrup-Wolfgang (EW) treatment to liquid systems has been begun by MILMAN (4J and it may be that future interest in ternary systems would be most profitably devoted to using these systems to extend this development. Nevertheless, the original formulation of our treatment is reproduced here, since this determines the method of manipulating results and since this formulation appears to lead to agreement between results from binary and ternary mixtures, but the connection between this formulation and the Estrup-Wolfgang-Milman (EWM) treatment is developed in a series of notes.
THEORETICAL TREATMENT OF THREE COMPONENT SYSTEMS
For convenience this treatment is stated in terms of the С2Н5ВГ/ВГ2/CeH5 Br system: the applicability to two organic halide/halogen or halide/hydrocarbon/ halogen systems is obvious. Implicit or explicit assumptions in the treatment are discussed later in the notes.
If x, y and z are the mole fractions of ethyl bromide, bromine and phenyl bromide respectively, these quantities are equal to the fractional chance that a recoiling bromine atom will collide with part of a molecule of these same three materials (see note (i)). The parameters a, |3 and y are defined as being, respectively, the fractional chance that the recoiling bromine atom shall, in a single collision, become organically retained on colliding with an ethyl bromide molecule, inorganically retained on colliding with a bromine molecule or organically retained on colliding with a phenyl bromide molecule (see note (ii)). Thus in the first collisions of several recoil bromine atoms a fraction (ax + y z ) of these atoms becomes organically retained, a fraction /Зу becomes inorganically retained and a fraction p is elastically scattered where p is given by
p= [(l -a )x + (l-| 3) y + ( l - 7 )z ]
(1)= ( l -a x -0y-yz)
The elastically scattered recoil atoms will be only fractionally reduced in energy and will therefore suffer further collisions to each of which the same argument will apply except that the fraction of recoil atoms entering subsequent collisions will be p, p2, рз . . . ,pi . . . . etc. (see note (iii)). Now let E 0 be the initial energy of the recoil atom, E j be the energy below which

314 S. S. KONTIS et al.
the recoiling atom has insufficient energy to achieve organic retention in a collision with phenyl bromide and E 2 the corresponding energy for a collision with ethyl bromide (see note (iv)). Also let m be the average number of collisions occurring between E 0 and E j and n the average number of collisions occurring between E0 and E2. The commonly made assumption that recoil atoms (loosely described as thermalized) whose energy has been reduced below E 2 without capture are eventually scavenged by the reaction
* B r+ B r 2 -> * B r 2 + B r ' (a)1
is equivalent to imposing no lower limit on the retention process occurring when the recoil atom collides with a bromine molecule. The pattern of events
S ch e m a tic representation of retention m echanism
is now seen diagrammatically in Fig. 1, in which q (the equivalent, between the energy limits Ej and E 2, of p) is given by
q = [ l-orx-Py] (2 )
(see note (v )).The total organic retention, R, resulting from the scheme of Fig. 1 is
thus
R = (£*x+Yz) + p(<*x+-yz) + p2 (i*x+yz)........................ p m"1(ax+7 z)
. (3)+ pm.ax+pm. q.ax+p™. q 2.ax ................................ p“ . qn_m"1 .orx,

NUCLEAR RECOIL IN ORGANIC HALIDE SYSTEMS 315
and since both p and q are necessarily positive fractions these geometric series are convergent and can be summed yielding
1 - D m l - a n _ mR = (ax+yz) +ax. pm. ----- . (4)1 -p r 1 -q ' '
Substituting for p and q in the denominators yields finally
a x + y z , , i t k . a x m n - m 4
R = ^xT p y ^ ^ - P ) + - (1- q >■ (5)
Before this equation can be used it is necessary to calculate or measure the parameters a, (3 and y (when the parameters p and q become defined) and the parameters m and n. In calculations so far completed and reported here the parameters such as m have been calculated by the following procedure. The fractional energy retention per collision by the projectile sphere in the elastic collision of two spheres of masses mj and m 2 is given by
F m- л - m i + m iE rt(a v ) (m 1+m 2)2- (6 >
Defining the mean atomic weight of a three-component mixture, ma, by the equation
= x. M1 + y . M2+ z . M3 a x . N i + y . N a + z . N g * ' '
where the M¡ are the molecular weights and the N¡ are the numbers of atoms in the various molecules, then using ma and m b (the mass of a bromine atom) in place of mj and m2 in equation ( 6 ), the parameter m may then be calculated from the equation
m i +m i( m a + m b ) 2
(8)
and a similar equation gives n (see note (vi)).A method by which the parameters a, ¡3 and y maybe derived from studies
of the retention in binary mixtures is as follows. The equation corresponding to equation (5) for a binary mixture can be obtained from equation (5) by omitting the second term and setting z = 0 giving
axR = (i -p ).ax+py ^ (9)
in which p= (l-»x-|3y) and (x+y) = l . Hence, for solutions very dilutein

316 s. S. KONTIS et al.
bromine, у -> 0 therefore x -» 1 and p -» (1 - o r ) . Reciprocating equation (9) and inserting the approximation p = (1 -a ) yields ,
1 fl v m -1i - d + Ë . j H i - d - , ) } . (10)
A plot of 1/R versus y/x may therefore be expected to approximate to a straight lineas y-»0 . Moreover, the intercept at y/x = 0 would be [ l - ( l - « ) m] ' i , from which a may be obtained since m is calculable and, a having been obtained, ji can then be found from the equation Slope = (Intercept) /3¡a . A sim ilar treatment of the binary system implied by setting x = 0 instead of z = 0 . yields a value of y and a duplicate value of /3.
However, a complication here becomes important. It is generally recognized that reactions such as
C 2H 5‘ + * B r - » C 2H6*B r (b)
are responsible for producing additional retention when the bromine concentration becomes too low to scavenge thermalized bromine atoms by reaction (a). Such additional retention must be discounted in drawing the limiting slope required by this treatment on graphs of 1/R versus y/x. It is also to be expected that the retentions observed in ternary mixtures will exceed those calculated at low bromine concentrations due to this same cause.
Note (i) •
Setting the fractional chance that a recoil bromine atom w ill collide with an ethyl bromide, bromine or phenyl bromide molecule equal to x, y and z respectively appears to assume that these molecules have equal collisional cross-sectional areas and is in apparent conflict with Milman's definition (Ref .14J, Eq. 8 ) of the corresponding quantity, fj, in the EWM treatment. It will be noticed however that wherever x, y or z appear in the equations they are always accompanied by a, P or 7 respectively. We have therefore chosen for the moment to regai-d the cross-sectional area correction as being comprised within a, ¡3 and 7 and hence determined when these quantities are determined experimentally.
Note (ii)
The subsequent use of ( l -а ), ( l -P ) and ( I - 7 ) as the chances that the colliding recoil bromine atom will not be captured implies that collision with an organic molecule cannot lead to inorganic retention by a mechanism such ás
C2H 5B r+ *B r ' ->IC2H 5' + * B r ‘] + B r ’
I
C 2H4 + H * B r ,
(c)

NUCLEAR RECOIL IN ORGANIC HALIDE SYSTEMS 317
where square brackets are used to denote collision fragments held in a solvent cage. Apart from this restriction, the definition and use of a , fi and y make no assumption as to the detailed mechanism by which retention occurs.
Note ( i i i )
The use of the same values of a, fi and y for successive collisions implicitly assumes that a, fi and y are not functions of the energy of the recoil atom. Apart from the fact that they contain a cross-sectional area ternij a, (3 and y are by definition equivalent to the p¡ (E) of the EW treatment. But the use of the same a, fi and -/throughout the whole range of energies with the implied assumption that a, fi and y are independent of the recoil atom energy (which is the same assumption made by ROWLAND and COULTER
1 r°[5] ) makes our a, fi and y equivalent to integrals of the type ——- /p.(E). dE.L 0-& iJ 1
E‘Note (iv )
We shall follow the practice of identifying E j and E 2 respectively with the activation energies of the reactions ■
* B r ‘ +C 6H5Br -> C 6H5*B r + B r ‘ (d)
* B r ’ +C 2H5Br -» C 2H5* B r + B r ', (e)
and shall identify E 0 with the average recoil energy of bromine atoms calculated taking the y decay scheme of the (n, y) process into account so far as this is known. But .see also notes (v) and (vii).
N ote (v )
The scheme of Fig. 1 implies that retention reactions can occur even when the energy is as high as E0 and in this respect it differs from the model adopted by EW and EWM. The assumption of a range of energies below Eo during which retention cannot occur is essential to Estrup and Wolfgang in order to integrate equation (6 ) of Ref. [2b] and is anyway intuitively likely for atoms recoiling with an initial energy of 0.2 MeV, but such an assumption is not necessary for the method of calculation adopted here. Also, bearing in mind that some retention by LIBBY "billiard-ball" labelling [6 J is not definitely excluded, and that bromine recoil atoms probably have an average recoil energy of about 100 eV, there seems to be no good reason for supposing such a retention-free energy zone in liquid systems. It should also be noted that E Q is only an average initial energy: recoil bromine atomswith various energies about this value will be produced by the different yemission processes comprising the (n, 7 ) capture. (See also note (v ii).)

318 S. S. KONTIS et al.
The definition of the mean atomic mass of a ternary mixture (a) ignores the fact that constituent atoms o f the severa l m olecules may present d ifferen t collision cross-sections, and (b) im p lic itly adopts the model of r e garding all atoms as being no more than loosely coupled (vide re f. [4 ], p. 184) and a ll equally accessible to a recoiling atom. This extreme model is one tested by other workers 14, 3a] and the correctness or otherwise of this model is a m atter that w ill be determined in part by the aptness of fit obtained in these calculations. At this prelim inary stage in the testing of this treatment we think that the approximation likely to be concealed in adopting this model w ill probably outweigh our failure to correct point (a) above. (See also note (v i i ) . )
Note (vi)
Note (v ii)
■ E rro rs introduced in the calculation of m using equation ( 8 ) are not c r itica l since m must be calculated in deriving a, fi and y from results for binary mixtures and recalculated when using ot, fi and y to obtain the re tention in a ternary m ixture. Such e rro rs are therefore la rge ly s e lfcancelling. In particular, points discussed in note (v i) are not critica l but in consequence it w ill be difficult to decide whether the "free-a tom " model is inadequate or not. Likew ise, values assumed fo r E 0, E j and E 2 are not critica l.
EXPERIM ENTAL
M ateria ls '
British Drug Houses Ltd. "Analar" grade bromine was used throughout these experiments without further purification. Carbon tetrachloride (B.D.H. "Analar" grade) and ethyl bromide (M ersey Chemicals "Analar" grade) were used as supplied fo r the system C2H 5Br/Br2 /CC14, but the ethyl bromide and phenyl bromide (B .D .H . laboratory grade) used fo r the С 2Н 5ВГ/ВГ2 /C6 H 5Br system were purified as follows.
Ethyl brom ide, a fter one in itia l d istillation, was shaken with concentrated sulphuric acid until no colouration developed in the acid, washed severa l times with dilute sodium carbonate solution, washed severa l times with distilled water, dried over anhydrous magnesium sulphate, and fra c tiona lly d istilled four tim es (37 cm X 2 cm glass-he lices packed column) with intermediate dryings over anhydrous magnesium sulphate. The fraction boiling between 38.3 and 38.5°C was retained for use.
Phenyl bromide, after one initial distillation, was dried over fused calcium chloride and fractionally distilled four times (30 cmX 2 cm glass-helices packed column) with intermediate dryings over fused calcium chloride. The fraction boiling between 155.5 and 156. 0°C was retained for use.

NUCLEAR RECOIL IN ORGANIC HALIDE SYSTEMS 319
Irrad ia tion p rocedure ,
A l e Ra/Ве neutron source was used fo r work on the C2 H5 Br/Br2/CCl4
system and a 3 с Sb/Be neutron source fo r С 2Н 5ВГ/ВГ2 /С6 Н5 ВГ solutions. The source was fixed in a central glass thimble surrounded by eight other glass thim bles, the whole assem bly being suspended in a la rge concrete- shielded tank of water. 1 0 m l aliquots of each solution, in glass-stoppe red tubes, were lowered into the glass thimbles fo r irradiation. To m inim ize neutron losses a ll glassware was of soda glass.
Irradiations w ere perform ed at room tem perature and in the dark (to preclude photochemical reactions), and irrad ia tion tim es w ere always as close to 17 h as was practicable to ensure that a ll samples contained the same re la tive activ ities of the various bromine iso topes.
E x tra c tion p ro ce d u re
Solutions from the С 2Н 5 ВГ/ВГ2 /СС14 system w ere extracted with 2N sodium hydroxide; and solutions from the С 2Н 5 ВГ/ВГ2 /С6 Н5 ВГ system with 2N sodium sulphite. To m in im ize the loss of brom ine the f ir s t ex traction was carried out in the irradiation tubes and the aqueous portions fo r subsequent extractions w ere used to rinse out the irrad iation tubes. Residual activ ity on the w alls of these tubes was always n eg lig ib le . A ll portions of aqueous extractant solutions were withdrawn from a burette to determ ine the volume o f aqueous solution (which varied according to the bromine concentration of the irradiated solution) when these portions were finally recombined. The addition of ca rr ie r bromide is unnecessary when extracting solutions containing bromine, and it was shown that the addition of ca rr ier made no difference to the retentions observed for the pure organic halides.
Counting procedure
Five m l samples of each of the aqueous and organic layers were counted in polythene tubes in a well-type thallium-activated sodium iodide 7 scintillation counter.
A ll samples from the С 2Н5 Вг/Вг2 /СбН5Вг system w ere counted 3 h after the end of the irradiation since it had been established that the complex decay curve at this period was such as to make decay corrections unnecessary over the time interval needed for counting samples.
Samples derived from the С2Н5 ВГ/ВГ2/C C I4 system contained initially 37.3 min C l38 activity. Settings of the pulse height analyser used in conjunction with the scintillation counter w ere found that m axim ized the (bromine counts)/(chlorine counts) ratio and samples from this system were counted 6 h from the end of the irradiation by which time Cl38 activities are almost dead.
A ll counts w ere corrected fo r background and, fo r samples from the C 2 H 5 B r/B r2/C C I 4 system, fo r decay i f necessary. Dead time corrections were negligible.

3 2 0 S. S. KONTIS eral.
The total volume of the organic layer remaining after extraction of the brom ine was calculated, using the known brom ine concentration. F rom this figure and the corrected count-rate fo r a 5 m l sample o f this la yer , the count-rate of the total organic layer was obtained. The volume of aque-
.ous extractant withdrawn from the burette was known, and the total volume of the aqueous layer was taken as this figure plus the volume of bromine extracted from the organic layer. This correction, though probably approximate, was never m ore than 2-3% and usually much sm aller, and so, even i f slightly erroneous, could not produce an appreciable over-a ll e rro r. The count-rate of the total aqueous layer was then calculated from the observed count-rate of the 5 ml sample of the aqueous layer and the fractional organic retention obtained as the ratio of the count-rate of the whole organic layer to the sum of the count-rates of the whole organic and aqueous layers. The uncertainties reported from the organic retentions are those calculated solely from the statistical uncertainties in the observed count-rates; other possible sources o f experim ental e r ro r are not included.
RESULTS
Calculations so fa r completed have been based on the assumption that E 0 should be equated with the in itia l re co il energy o f the brom ine atom s. Following RACK and GORDUS 17aj we have therefore used E0 =100 eV, this figu re being u ltim ately based on the re co il energy calculated by HSIUNG, HSIUNG and GORDUS [7b] fo r the Cl35(n, y)C136 reaction.
E j and E 2 are to be taken as the activation energ ies of the exchange reactions * B r ‘ +RX -» R* B r+ X ' that do not appear to have been measured. These have been estim ated (the supporting arguments being set out in the Appendix)with the result that we have taken:
F o r C2H5 B r/Br 2/ CgHg Br mixtures Ej = E 2 = 20 kcal (i. e . 0.8676 eV)F o r C 2H5 B r/ B r 2/CCl4 m ixtures E;i = 50 kcal
E 2 = 20 kcal and the second term in equation (5) as there stated.
The choice of E 1 = E2 fo r Gj H 5B r/B r2 /C6H 5Br mixtures has the effect o f making the second term in equation (5) redundant. This term computes the retention occurring between the energy lim its E^ and E2 (where only one organic retention process is operating) whereas the first term computes the retention occurring between the energy lim its E 0 and E } (where two organic retention processes are operating). Clearly, as long as E 0 is taken as 100 eV, E 2 ~ E 2 ~ 20 kcal m ole " 1 (i. e. 0.8676 eV), and as long as ] E j - E2) does not exceed 10 kcal4m ole-i ( i .e . 0.4338 eV), then the second term cannot exceed0.5% of the f ir s t te rm . A p ractica l reason fo r ignoring the second term is that until it is known which of ethyl and phenyl bromides has the greater activation energy fo r exchange it is not known whether the second term takes the form
Calculation of results -

NUCLEAR RECOIL IN ORGANIC HALIDE SYSTEMS 321
pm. ax/ (ax+j3y) (1 -qn"m) with q = (1 -a x -jiy ), o r the form
p m - ' Y z / O y + ' v z ) U - q n ‘ m )with q = (l-0 y -y z ). Since E j^ E 2for the C2H5 Br/Br2 /CCl4 system the second term has been used in calculations for this system.
The results fo r the binary mixtures СгН 5 В г/Вг2 and СбН5 Вг/Вг2 are shown in Table I and in F ig . 2(a, b) plotted in the form 1/R versus y/x. Included in F ig . 2(a) are some of the results of M ILM AN and SHAW [8 ] and it can be seen that their results and ours are in complete agreement. Published results [9] fo r phenyl bromide/bromine mixtures are too fragmentary to warrant comparison.
It w ill be reca lled that in drawing the lim iting slope on graphs o f 1/R versus y/x it is necessary to discount the additional retention occu rring by diffusive recombination at low bromine concentrations. Both curves of F ig . 2 show a fa ir ly sharp change of slope, at about y/x = 0 -3 X 1 0 '2 in F ig . 2(a) and at about y/x = 2 X 10-? in F ig . 2(b), and it is tempting to regard this change as being in ternal evidence of the onset o f d iffusive recom b ination. The weight of the chemical evidence is, however, that bromine concentrations as high as 0 . 1 m ole fraction 1 1 0 J a re needed to scavenge such diffusive reactions. In itially, therefore, lim iting slopes were drawn tangential to these curves at about у = 0 . 1 (y/x = 0 . 1 1 1 ) but this produced discrepant values of (3- Adjusting the lim iting slope on F ig . 2(b) to produce a concordant value o f /3 le ft us with the two lines shown which lead to the values:
F o r the C2 H5 B r/B r2 system a = 0-01600 = 0-0575
F o r the C6H 5B r/ B r2 system = 0.0252 ./3 = 0-0579
The results fo r C2 H5Br/Br2 /C6 H5Br mixtures are shown in Fig.3(a, b) and in Table 2. The convergence of the experim ental results from the ternary system with the curve fo r the C6H5 B r/B r2 binary system at those points where the ternary mixture becomes binary demonstrates the internal consistency of the work. As foreseen, the observed retentions become progress iv e ly higher than those calculated as the mole fraction of bromine is reduced below about 0-07: this e ffect is again to be attributed to fa ilu re to scavenge diffusive recombination reactions. At mole fractions oí bromine grea ter than about 0 - 1 it can be seen that the calculated line is in excellent agreement with the experimental results fo r the series of solutions at 0-32 mole fraction of phenyl bromide, and in quite good agreement for the series of solutions at 0.55 mole fraction of phenyl bromide.
This treatment was developed in response to the resu lts from the C 2H5 B r/B r2 /CCl4 system, but since work had to cease before the method of manipulating the results was fu lly worked out, deficiencies have become apparent. Our ea rly resu lts fo r the C 2H5 B r/B r2 system w ere in a g ree ment with F ig . 2(a) but more scattered and not so extensive. We have therefo re adopted the values of a and p derived from F ig . 2(a). Results fo r the

322 S. S. KONTIS et al.
TABLE I
THE BINARY SYSTEMS C2H 5Br/Br2 AND C6H5Br/Br2
M ole fractionFractio n al
reten tion
M ole fractionFraction al
retentionBr2
(У)
C 6 H 5Br
(z )
Br,
(У)
C2 H5 Br
0 0
0 . 0 027 0 .9 9 7 3 0 . 6 2 1 * 0 . 0 0 6 0 . 0 0 0 2 0 . 9998 0 .2 9 6 ± 0 .0 0 8
0 .0 0 7 9 0 .9 9 2 1 0 .4 7 7 ± 0 .0 0 6 0 .0 0 0 3 0 . 9 997 0 .2 9 4 ± 0 .0 0 8
0 .0 0 9 9 0 . 9 901 0 .4 3 2 ± 0 .0 0 4 0 .0 0 0 5 0 .9 9 9 5 0 .2 8 3 ± 0 .0 0 5
0 .0 1 8 7 0 .9 8 1 3 0 .4 3 7 ± 0 .0 0 6 0. 0 009 0 . 9991 0 .2 8 0 ± 0 . 0 0 2
0 .0 2 1 8 0 .9 7 8 2 0 .3 8 9 ± 0 . 008 0 . 0 0 1 2 0 .9 9 8 8 0 .2 7 1 ± 0 . 0 0 5
0 .0 2 6 7 0 .9 7 3 3 0 .3 9 7 ± 0 .0 0 9 0 .0 0 1 4 0 . 9986 0 .2 6 8 ± 0 .0 0 6
0 .0 3 4 5 0 .9 6 5 5 0 .3 7 3 ± 0 .0 0 5 0 .0 0 2 5 0 . 9 975 0 .2 6 6 ± 0 .0 0 8
0 .0 4 6 7 0 .9 5 3 3 0 .3 7 2 ± 0 .0 0 5 0 .0 0 3 1 0 .9 9 6 9 0 .2 5 7 ± 0 .0 0 5
0 .0 5 0 1 0 .9 4 9 9 0 .3 6 3 ± 0 .0 0 6 0 .0 0 4 3 0 .9 9 5 7 0 .2 5 2 ± 0 . 007
0 .0 7 2 2 0 . 9278 0 .3 3 1 ± 0 .0 0 8 0 .0 0 8 3 0 .9 9 1 7 0 .2 4 9 ± 0 .0 0 8
0 .0 9 9 8 0 . 9 0 0 2 0 .3 0 9 ± 0 .0 0 6 0 .0 1 2 4 0 .9 8 7 6 0 .2 4 2 ± 0 .0 0 6
0 .1 1 9 8 0 . 8 8 0 2 0 .2 8 3 ± 0 .0 0 5 0 .0 1 9 3 0 .9 8 0 7 0 .2 2 8 ± 0 .0 0 6
0 . Í2 4 0 0 .8 7 6 0 0 .2 8 9 ± 0 . 010 0 .0 2 3 3 0 .9 7 6 7 0 .2 3 1 ± 0 .0 0 6
0 .1 4 0 9 0 .8 5 9 1 0 .2 6 5 ± 0 .0 0 8 0 .0 2 6 9 0 . 9731 0 .2 2 4 ± 0 .0 0 7
0 .1 4 6 7 0 .8 5 3 3 0 .2 4 8 ± 0 .0 0 7 0 .0 2 9 1 0 .9 7 0 9 0 .2 2 3 .± 0 .0 0 9
0 .1 8 0 1 0 .8 1 9 9 0 .2 3 6 ± 0 .0 0 9 0 .0 4 0 7 0 .9 5 9 3 0 .2 1 7 ± 0 .0 0 9
0 .2 1 9 1 0 .7 8 0 9 0 .2 1 2 ± 0 .0 0 5 0 .0 5 0 2 0 .9 4 9 8 0 .2 0 8 ± 0 .0 0 4
0 .2 6 2 9 0 .7 3 7 1 0 .2 0 7 ± 0 . 006 0 .0 5 7 5 0 .9 4 2 5 0 .1 9 4 ± 0 .0 0 5
0 .2 9 4 2 0 . 705 8 0 .1 9 6 ± 0 .0 0 6 0 .0 6 1 2 0 . 9 388 0 .2 1 2 ± 0 . 0 0 5
0 .3 4 0 6 0. 6 5 9 4 0 .1 8 6 ± 0 .0 0 8 0 .0 7 9 9 0 .9 2 0 1 0 .1 7 9 ± 0 .0 0 6
0 .1 0 0 5 0 .8 9 9 5 0 .1 6 8 ± 0 .0 0 5
0 . 1 0 2 2 0 .8 9 7 8 0 .1 6 8 ± 0 . 0 0 5
0 . 1 2 0 1 0 . 8799 0 .1 5 7 ± 0 .0 0 8
0 .1 5 0 2 0 . 849 8 0 .1 4 4 ± 0 .0 0 4
0 .1 6 7 2 0 . 8328 0 .1 4 4 ± 0 .0 0 7
0 .1 8 0 1 0 .8 1 9 9 0 .1 3 5 ± 0 .0 0 7
0 .2 1 7 0 0 .7 8 3 0 0 .1 2 7 ± 0 .0 0 7
0 .3 1 2 7 0 .6 8 7 3 0 . 1 1 2 ± 0 . 006
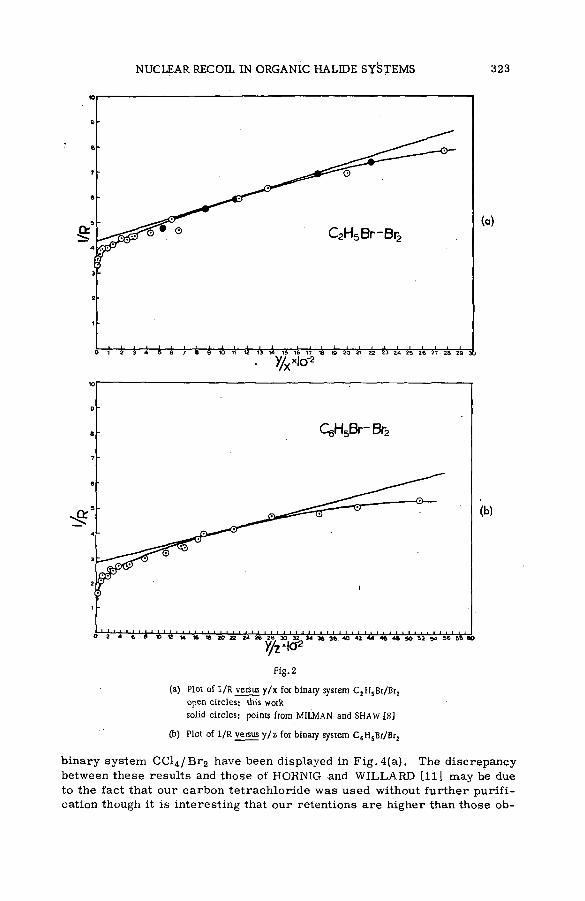
NUCLEAR RECOIL IN ORGANIC HALIDE SYSTEMS 323
(a) Plot of 1 /R versus y / x for binary system C 2 H5 B r/B r2
open c ir c le s : this work
solid c ir c le s : points from MILMAN and SHAW [ 8 ]
(b) Plot of 1 /R veisus y / z for binary system C 6 H 5 Br/B r2
binary system СС1 4/ВГ2 have been displayed in F ig . 4(a). The discrepancy between these results and those of HORNIG and W ILLARD [111 may be due to the fact that our carbon tetrach loride was used without fu rther p u rification though it is interesting that our retentions are higher than those ob-
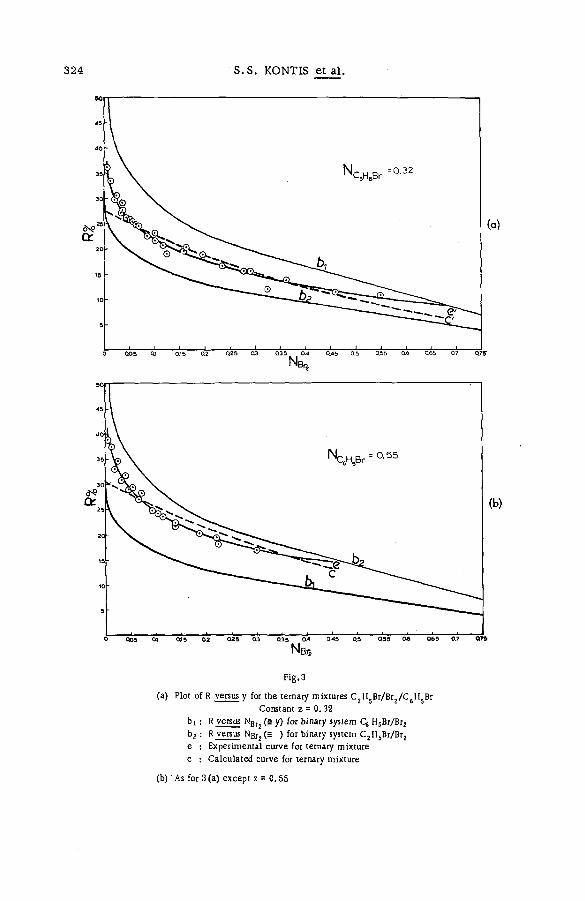
324 S.S. KONTIS et al.
F ig .3
(a ) Plot of R versus y for th e tern ary m ixtures C zH5 B r/B r2 / C 6 HsBr
Constant z = 0 .3 2
bi : R versus NB l2 ( = y ) for binary system Q H5 B r/B r2
b 2 : R versus Ng, 2 (= ) for binary system C 2 H5 B r/B r2
e : E xp erim ental cu rv e for ternary m ixtu re
с : C a lcu lated curve for tern ary m ixture
(b) ' As for 3 (a ) e x c e p t z = 0 .5 5
NUCLEAR RECOIL IN ORGANIC HALIDE SYSTEMS 325
TABLE II
THE TER NARY SYSTEM C2 H5 B r/B r2 /C6H 5Br
z = 0 .3 2 z = 0 . 55
M ole fraction
Fractio n al
reten tion
M ole fraction
Fraction al
retentionB r2
(У)
C 2 H 5Br
(x)
B r2
(У)
C 2 H 5Br
(X)
0 . 004 2 0 .6 7 5 8 0 .3 6 2 ± 0 .0 0 4 0 .0 0 4 3 0 .4 4 6 1 0 .3 8 9 ± 0 .0 0 6
0 .0 1 1 5 0 .6 6 8 7 0 .3 3 5 ± 0 . 003 0 .0 1 1 9 0 .4 3 85 0 . 3 7 5 ± 0 . 0 0 4
0 .0 1 8 2 0 .6 6 2 0 0 .2 9 8 ± 0 . 006 0 .0 1 8 6 0 .4 3 1 8 0 . 3 3 0 ± 0 . 0 0 4
0 .0 2 0 7 0 .6 5 9 5 0 .2 9 7 ± 0 . 0 0 6 0 .0 2 4 7 0 .4 2 5 7 0 .3 4 6 ± 0 .0 0 4
0 .0 2 4 3 0 .6 5 5 9 0 .3 0 5 ± 0 . 0 0 5 0 .0 3 2 4 0 .4 1 8 0 0 .3 0 8 ± 0 .0 0 9
0 .0 3 3 5 0 .6 4 6 7 0 .2 7 1 ± 0 . 0 0 5 0 .0 3 9 8 0 .4 1 0 6 0 .3 1 8 ± 0 . 005
0 .0 3 5 5 0 .6 4 4 7 0 .2 8 1 ± 0 .0 0 5 0 .0 4 6 9 0 .4 0 3 5 0 . 2 8 9 * 0 . 0 0 5
0 .0 3 6 4 0 .6 4 3 8 0 .2 7 7 ± 0 . 0 0 9 0 .0 5 4 9 0 .3 9 5 5 0 .2 9 3 ± 0 .0 0 5
0 . 043 8 0 .6 3 6 4 0 .2 6 2 ± 0 .0 0 6 0 .0 6 6 2 0 .3 8 4 2 0 .2 7 0 ± 0 . 006
0 .0 5 1 1 0 .6 2 9 1 0 .2 5 8 ± 0 .0 0 6 0 .0 7 1 2 0 .3 7 9 2 0 .2 8 3 ± 0 .0 0 5
0 .0 5 5 9 0 .6 2 4 3 0 .2 5 4 ± 0 . 00 5 0 .0 9 2 7 0 .3 5 7 7 0 .2 4 8 ± 0 .0 0 5
0 .0 6 1 9 0 .6 1 8 3 0 .2 4 8 ± 0 .0 0 5 0 .1 0 3 3 0 .3 4 7 1 0 .2 4 2 ± 0 .0 0 3
0 .0 6 7 0 0 .6 1 3 1 0 .2 4 5 ± 0 .0 0 5 0 .1 1 3 0 0 .3 3 7 4 0 .2 3 6 ± 0 .0 0 5
0 .0 8 3 7 0 .5 9 6 5 0 .2 2 6 ± 0 . 0 0 5 0 .1 3 7 8 0 .3 1 2 6 0 .2 2 4 ± 0 .0 0 6
0 .0 9 8 9 0 . 5813 0 .2 3 1 ± 0 .0 0 6 0 .1 3 8 1 0 .3 1 2 3 0 . 2 1 6 ± 0 . 006
' 0 .0 9 9 7 0 .5 8 0 5 0 .2 1 6 ± 0 . 0 0 4 0 .2 1 9 2 0 .2 3 1 2 0 .1 9 3 ± 0 .0 0 6
0 .1 1 6 7 0 . 5 6 3 5 0 .2 0 7 ± 0 . 0 0 5 0 .2 2 4 0 0 .2 2 6 4 0 .1 8 1 ± 0 . 0 0 4
0 .1 2 3 4 0 . 5 5 6 8 0 .1 9 0 ± 0 .0 0 5 0 .2 9 8 8 0 .1 5 1 6 0 .1 6 9 ± 0 . 007
0 .1 5 7 4 0 . 5228 0 .1 9 6 ± 0 .0 0 7
0 .1 6 2 2 0 .5 1 8 0 0 .2 0 3 ± 0 .0 0 5
0 .1 9 5 0 0 .4 8 5 2 0 .1 8 9 ± 0 . O il
0 .2 3 3 0 0 .4 4 7 2 0 .1 6 6 ± 0 .0 0 8
0 .2 7 6 6 0 .4 0 3 6 0 .1 5 6 ± 0 . 00 5
0 .2 9 0 0 0 .3 9 0 2 0 .1 5 7 ± 0 . 00 6
0 .3 2 2 2 0 .3 5 8 0 0 .1 2 1 ± 0 .0 0 7
0 .3 6 2 4 0 .3 1 7 8 0 .1 3 9 ± 0 . 00 5
0 .4 5 9 7 0 .2 2 0 5 0 .1 1 4 ± 0 . 00 5
0 . 5483 0 .1 3 1 9 0 .1 0 9 ± 0 . 00 6
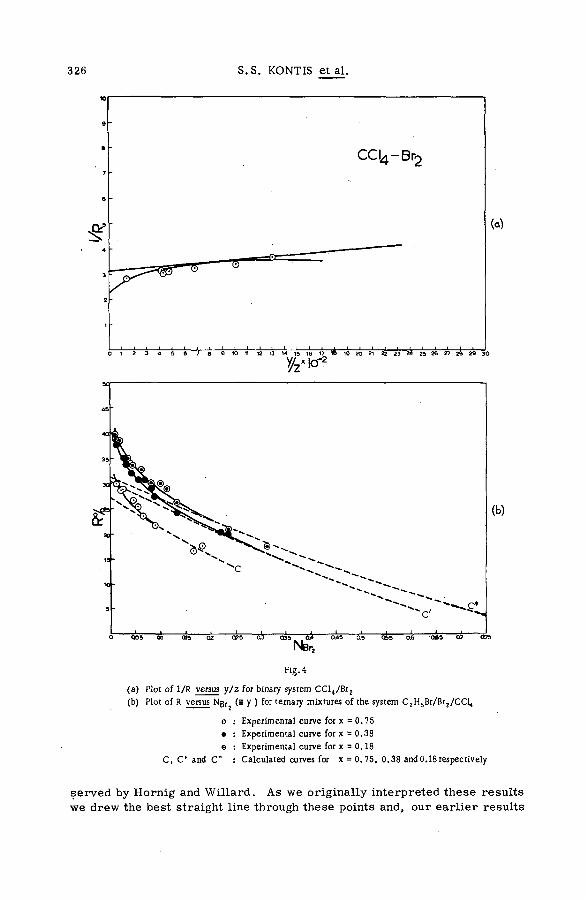
326 S. S. KONTIS et al.
(a ) Plot o f 1 /R versus y / z for binary system СС14 /В г г
(b) Plot o f R versus N gr2 (= у ) for tern ary m ixtures o f the system C ^ B r / B r j / C Q ,
о : E xperim ental cu rve for x = 0 . 7 5
• : Experim ental cu rv e for x = 0 .3 8
e : Experim ental curve for x = 0 .1 8
С , С ' and C " : C alcu lated curves for x = 0 . 7 5 , 0 .3 8 a n d 0 . 1 8 respectively
served by Hornig and W illard . As we orig ina lly interpreted these results we drew the best straight line through these points and, our ea rlie r results

NUCLEAR RECOIL IN ORGANIC HALIDE SYSTEMS 327
fo r the C 2H5 Br/Br2 system being then less precise, it was possible to obtain values of a, /3 and y that produced calculáted lines 1-2% below the co rresponding experim ental points. However, to avoid confusion on F ig . 4(b) where the upper two curves are close together, we have here chosen to select a value of у (=0-0504) that, with our established values of a and |3, produces lines in good agreem ent (as shown in F ig . 4(b)) with the resu lts fo r the ternary mixtures from the system C2 H5 Br/Br 2/CC14. Using the in tercept implied by this chosen value of y, we have drawn on F ig . 4(a) the slope shown. This produces a value of /3 (= 0-0695), which is within 20% of the value adopted from the C2H 5Br/Br2 system. Further, if it be assumed that the points o f F ig . 4(a) represent scatter about a curve s im ila r to those of F ig . 2 rather than a straight line, it is seen that this line is tangential to this curve at about у = 0-1 . It can be seen from F ig . 4(a) what degree of disregard of experimental points is needed to achieve the good fit on Fig.4(b).
DISCUSSION
It is an inelegance o f this treatment that there is no a p r io r i method o f determ ining on graphs such as F ig . 2 the position at which the lim iting slope should be drawn. We can expect that this weakness w ill be eliminated as m ore becomes known about the yields of individual products of organic retention reactions, particu larly those products arising from diffusive r e combination reactions. We did not consider that such studies as have been made along these lines [ 1 2 j were sufficiently conclusive to set lim its within which the lim itin g slope must be drawn. We have th ere fo re adopted our present procedure of testing whether values of a, 0 and у giving good agreement between calculation and experiment ior ternary mixtures are compatible with lim iting slopes drawn at reasonably low brom ine concentrations on graphs such as F i g . 2.
In assessing the value o f such comparisons it is important to rea lize that the freedom of manoeuvre is much more limited than might at first sight appear. Th is can be seen as fo llow s. In Table III are shown some of the partia l resu lts obtained in the calculation of the retention in a particu lar ternary mixture (Curve С of F ig . 4(b)). It can be seen that the fa ll in R as у increases depends almost entirely on the factors (ах+ут.)/(oix.+&y+yz) and a:x/(ax+£Sy), and acceptable behaviour of the calculated line w ill only be obtained if the ratio of (3 to the sum of a and у is approximately correct. The le v e l at which this trend in R operates is however determ ined almost ent ir e ly by the factors ( l - p m) and ( l - q n*m), and since p = (l-ax-fiy-yz) and q = ( l -ах-Эу) co rrec t magnitudes fo r pp and q n"m w ill only be obtained i f the sum of the values of a, f3 and у is approximately correct. When fitting to several curves for ternary mixtures (as in F igs.3 (a, b) or 4(b)) the correct rise from one series of m ixtures to another at a fixed mole fraction of one component w ill only be obtained i f the param eter a o r у associated with this component has been established with the co rrec t magnitude re la tive to the other two param eters. F inally, since a (o r y) and j3 a re linked by the equation (Slope) = (Intercept) /3/a and since a is an inverse function of the intercept, a and |3 cannot be adjusted independently. Taken together these lim itations place su ffic iently strict restric tions on the perm iss ib le
328 S. S. KONTIS et al.
TABLE III
P A R T IA L RESULTS OBTAINED IN THE CALCULATION OF THE RETENTION IN A PARTICU LAR TERNARY MIXTURE
УS
facto rl - p m
T
factorR
0 . 0 1
0 .0 9
0 .1 7
0 .2 9
0 .9 8 4 6
0 .8 6 3 2
0 .7 4 5 4
0 . 575 2
0 .2 9 4 3
0 .2 9 0 7
0 .2 8 7 0
0 .2 8 1 1
0 .9 1 4 0
0 .5 4 1 4
0 .3 8 4 6
0 .2 6 8 1
0 .0 1 4 6 3
0 .0 2 3 9 3
0 .0 3 2 5 5
0 .0 4 4 4 4
0 .2 9 9 1
0 .2 6 0 1
0 .2 2 2 8
0 .1 7 0 2
„ , a x + y z S fa cto r = -------- ------
_ . axa x + B y + y z a x + B y
values of a, fi and y that it may easily become impossible to achieve compatib ility between binary and ternary m ixtures. A potential use of this feature is mentioned la ter.
The additional condition that we have imposed in dealing with the C2H 5Br/Br2 / C6H5 B r system, namely, that concordant values of fi should be obtained from the two binary system s, stems from the defin ition of fi as a property o f the bromine m olecule. A t least three situations can be envisaged in which this criterion might be mistaken. F irs tly , i f brom ine is loose ly complexed to one organic halide and not to the other, d ifferen t values o f fi might be expected. Secondly, i f one organic halide o f a pa ir is much more or much less prone to produce inorganic retention by reactions such as reaction (c), different values of fi might again be expected. Thirdly, d ifferen t values of fi would a rise from the presence in one organic halide of an impurity capable of both trapping radiobromine atoms much more e ffec tiv e ly than m olecu lar brom ine and regeneration in, fo r exam ple, r e actions such as
* B r ‘ +A->X, X + B r 2 - » A + (inorganic * B r ) .
I f methods of determ ining where the lim iting slope should be drawn can be developed this uncertainty w ill be resolved , and measured d ifferences in fi would then be indicative of such special e ffects . In the meantime, however, it seems best to accept this criterion until it leads to contradiction.
Two possible complications associated with the organic halides we used with ethyl bromide should be mentioned. As a test of this treatment carbon tetrach loride has the advantage that the mean atomic weight shifts appreciably as the ethyl bromide/carbon tetrachloride ratio alters in a series of ternary m ixtures. Its disadvantage is that its chlorine atoms undergo (n, y) reactions. The effects of this are elim inated by the counting p rocedure, except in so fa r as the products of these reactions contribute to the

NUCLEAR RECOIL IN ORGANIC HALIDE SYSTEMS 329
retention of radiobrom ine. The method of determ ining y experim entally w ill, it is hoped, allow fo r this, but there may be unforeseen consequences in ternary solutions. Phenyl brom ide has the advantage that retentions in phenyl brom ide/brom ine solutions are genera lly h igher than in ethyl bromide/bromine mixtures, resulting in retentions in ternary mixtures considerably d ifferent from those observed in either binary system . But the v e ry fact that this la rge difference exists may be indicative o f special features occurring in phenyl bromide/bromine mixtures that may be modified or elim inated in ternary m ixtures.
Apart from a restriction already noted, the definitions of a, |3 and y do not otherwise place any lim itation on the detailed mechanism responsible fo r organic labelling. In particular, the mechanism is not confined to the concept, introduced by L IB B Y 16], of head-on co llis ions between atoms of equal mass. In other respects, however, this treatment follows the ideas developed by Libby in regarding a mixture as an assem bly of atoms and in the use ofEquation ( 6 ) to calculate the fractional energy retention per co llision. Excepting at low mole fractions of bromine we have shown that good agreement can be obtained between experimental retentions in ternary m ixtures and retentions calculated from parameters measured in binary systems. A t m ole fractions of brom ine g rea te r than are covered by our d irect ex perim ental results there are indications that a d iscrepancy is appearing, but in v iew of the approximations involved, the lik e ly experim ental uncertainties and the fact that experimental curves in this region are extrapolations, the discrepancy is not of sufficient magnitude to be significant. With the obviously wide lim its set by this treatment we there fo re conclude that the general ideas of the Libby model are compatible with experimental results.
There are severa l respects in which this treatm ent can be m odified: by using the logarithm ic energy decrement per collision, by assuming co llisions of the hot atom with molecules rather than with atoms, by making allowance fo r the d ifferen t cross-sectiona l areas of m olecules o r atoms, etc. These variations are currently being examined and w ill be tested against the present results. The discrim inatory power of the treatment is not high, but it is hoped that, by measuring the retentions in several series o f m ixtures at constant x and constant z the acceptable range of values of a, fi and y can be so severe ly restricted, that a satisfactory fit can be obtained on some assumptions but not on others. Some current suggestions as to mechanisms leading to organic retention may thus be elim inated.
It is interesting finally to compare our values of a, /3 and y with related parameters in the Estrup-Wolfgang kinetic treatment of hot atom reactions. It has been noted [2b] that mean values of the probability function p¡(E ) are given by
I.P ¡(E ) = ln ( E j / E 2) '
where I¡ is the reactiv ity in tegra l fo r a single species. Measured values of these in tegra ls have been listed by CROSS and W O LFG ANG [3b ], and fo r substitution reactions of halogen atoms with a hydrogen or halogen atom

3 3 0 S. S. KO.NTIS et al.
in halogeno-methane deriva tives these in tegra ls have values in the range 0- 06 to 0- 04. The EW treatment does not require values to be assigned to E j and E 2, only that these lim its shall ex ist. F o r com parison there fo re E j and E 2 may be taken as equal to our Eo and E 2 since this is the range over which oux d, P and y are assumed to be mean values and therefore equivalent to P j ( E ) over this range. The values of p¡(E ) derived from the listed values of I¡ therefore fa ll in the range 0-012 to 0- 008 (ln(100/0- 8676) = 4* 749 ~ 5). These values of p ¡(E ) re fe r however to retention in an individual species in the gas phase whereas our a, /3 and y re fe r to retention in a ll species present in the liquid phase. It is thus seen that our a, ¡3 and y are comparable with results derived from the EW treatment.
A C K N O W L E D G E M E N T S
P .S . and S .S .K . wish to express th e ir gratitude to the IA E A fo r the award o f Research Fellowships during the tenure o f which this work was carried out.
APPE ND IX
We requ ire the activation energies of the three reactions
-*B r' + С 2Н 5ВГ -» C2 H5 * B r+ B r ’ (A l )* B r ' + C 6H 5Br -> C6 H5* Br + Br* (A2)* B r ' + CC14 -*CC13* B r+C 1 ' . (A3)
LIBERATORE and WIIG [13] studied the exchange between radioactive bromine and gaseous ethyl brom ide and from a negative result concluded that the exchange had a re la t iv e ly high activation energy. Calculation by the sem i-em p irica l method yielded 25 kcal m ole -1 fo r the activation energy of reaction (A l ) . The other two reactions do not appear to have been studied with a view to determining the activation energy.
Many ionic exchange reactions sim ilar to (A l) and (A2) have been studied [14] and, fo r severa l aliphatic bromides, have been found to have activation energies about or just below 20 kcal m ole"1. The charge distribution occurring in the transition complex, ¿-B r- • • CRs* • • B r5- w ill help to stabilize the complex, but the eas ie r attraction of electrons to an entering bromine atom in the absence of a negative charge might well produce a compensatory lowering of energy fo r the corresponding radical complex. This argument suggests activation energies of about 20 kcal mole * 1 fo r reactions (A l) and (A2).
A probable low er lim it m aybe set as fo llow s: The activation energy of the reaction
B r ' +C C l3Br -» CC I3' + B r 2 (A4)
has been established [15] as 10.2 kcal m ole " 1 . As ОСС1^вг = 49 kcal and

NUCLEAR RECOIL IN ORGANIC HALIDE SYSTEMS 331
Овг-Вг =45*5 kcal, this reaction is 3'5 kcal m ole*1 endothermie leaving 6-7 kcal mole"1 as the "activation increm ent". The transition complex fo r this reaction is R-зС- • -B r • • -B r, and as electrons are not so fre e ly ava ilable on a carbon atom as on a bromine atom, it is likely that the transition com plex fo r reactions ( Al ) - (A3), B r - . . R 3C- • -B r, is o f h igher energy. A llow ing fo r this suggests that the "activation increm ent" o f reaction (A4) should be increased to perhaps 10-20 kcal and as reactions ( Al ) and (A2) are thermoneutral, this would be the activation energy. The absence of any suggestion of exchange reactions in the extensive literatu re of brom i- nation reactions may be taken as suggesting that the activation energies of the exchange reactions are higher than those typical of hydrogen abstractions that occur free ly with activation energies in the range 10-20 kcal m ole '1 U6J.
Reaction (A3) is ~ 30 kcal m ole-1 'endotherm ie (DCCi _C1 ~ 80 kcal D Cci,-Br = 49 kcai) and cannot have an activation energy less than th is. If the ''activation increm ent" is 10-20 kcal as suggested above, this leads to an activation energy of 40-50 kcal m o le-1 fo r reaction (A3).
R E F E R E N C E S
[1 ] M ILM AN, М . . R ad io ch im . A c ta 1 ( 1 9 6 2 ) 1 5 .
[2 ] ( a ) ESTRUP. P .J . and WOLFGANG, R. . J . A m e r. c h e m . S o c . 8 2 ( 1 9 6 0 ) 2 6 6 1 ;
(b ) ESTR U P, P . J . and WOLFGANG. R. , J . A m e r. c h e m . S o c . 8 2 ( 1 9 6 0 ) 2 6 6 5 .
[3 ] (a ) JURG ELEIT, H .C . and W OLFGANG. R . . J . A m er. c h e m . S o c . £ 5 ( 1 9 6 3 ) 1 0 5 7 ;
(b ) CROSS, R .J . and WOLFGANG, R . , R ad io ch im . A cta 2 (1 9 6 4 ) 1 1 2 .
[ 4 ] M ILM AN. М . , R ad io ch im . A cta 2 (1 9 6 4 ) 1 8 0 .
[ 5 ] ROWLAND, F .S . and CO U LTER , P . , R a d io ch im . A cta 2 (1 9 6 4 ) 1 6 3 .
[6 ] LIBBY, W . F . , J . A m e r. c h e m . S o c . 6 9 ( 1 9 4 7 ) 2 5 2 3 .
[ 7 ] ( a ) RA C K. E . P. and GORDUS, A . A . , J . phys. C h em . j ¡5 ( 1 9 6 1 ) 9 4 4 ;
(b ) HSIUNG, C . , HSIUNG, H. and GORDUS, A . A . . J . c h e m . Phys. 3 4 (1 9 6 1 ) 5 3 5 .
[ 8 ] M ILM AN , M . and SH AW , P . F . D . , J . c h e m . S o c . (1 9 5 7 ) 1 3 0 3 .
[9 ] WILLARD, J . E . , in C h e m ic a l E ffects o f N u clear Transform ations ¿ IA EA . V ienn a (1 9 6 1 ) 2 1 5 .
[1 0 ] M ILM AN , M. , R ad io ch im . A cta J. (1 9 6 2 ) 1 9 .
[1 1 ] HORNIG, J .F . and WILLARD, J . E. , J . A m e r. ch e m . S o c . 7 5 ( 1 9 5 3 ) 4 6 1 .
[1 2 ] HARRIS, W . E . , in C h e m ic a l E ffects of N u clear Transform ations ¿ IAEA , V ien n a (1 9 6 1 ) 2 2 9 .
[1 3 ] LIBERATORE. L .C . and W IIG, E .O . . J . c h e m . Phys. 8 ( 1 9 4 0 ) 3 4 9 .
[1 4 ] W AHL, A .C . and BONNER, N .A . , R ad io activ ity Applied to C h em istry , W iley , New Y o rk (1 9 5 1 ) 3 1 .
[1 5 ] SULLIVAN, J . H. and DAVIDSON, N. , J . c h e m . Phys. ¿ 9 (1 9 5 1 ) 1 4 3 .
[1 6 ] TRO TM AN -DICKENSON , A .F . . Gas K in e tic s , Butterw orth, London (1 9 5 5 ) 1 9 1 .


THE STEREOCHEMISTRY OF THE REACTIONS OF (n, y ) HALOGEN ATOMS WITH ALKYL HALIDES
IN THE LIQUID PHASE*
F .S . ROWLAND+, C .M . WAI, С . T . TING AND G. MILLER DEPARTMENTS OF CHEM ISTRY, UNIVERSITY OF KANSAS AND
UNIVERSITY OF CALIFORNIA IRVINE, UNITED STA TES OF AMERICA
Abstract — Résumé — Аннотация — Resumen
THE STEREOCHEMISTRY OF THE REACTIONS OF (n, y ) HALOGEN ATOMS W ITH ALKYL HALIDES
IN THE LIQUID PHASE. Th e reactio n s o f (n, y ) C l 38 atom s w ith m eso- and d l - 2 , 3 -d ich lo ro b u ta n e , as in
equation ( 1 ), have been investigated under a variety o f experim ental conditions, using radio gas chromatography
for product analysis.
C13 B + C H 3 C H -C H C H 3 --------- » C H 3 -C H -C H -C H 3 + C I . (1 )
I l I ICl Cl Cl C l38
In th e liquid phase a t 2 0 eC , th e ra tio of th e tw o ra d io a c tiv e d iastereo m ers form ed in this re a c tio n is 5 : 2
favouring th e irrad iated m o le cu le , for irradiations o f eith er the m eso or the dl form s. In the gas phase, the am ounts o f e a c h a re reduced by a fa c to r o f > 10 for both produ cts. T h ese results in d ica te th a t the
СНзСНС1СНС138СНз m o lecu les found in th e liquid phase are formed prim arily by com bin ation reaction s on
a tim e s c a le co m p a ra b le to th e ra c e m iz a tio n of CH 3 CH CHCICH 3 ra d ica ls , i . e . in ” c a g e " - l ik e processes.
Irradiations in th e liquid phase a t low er tem p eratu res or in the solid phase show th a t th e two diastereom ers
a re form ed in m o re n early equal am ounts than in the liquid a t room tem p e ra tu re .
T h e reactions o f (n, y )C l38 with either c is - or tran s- 1, 2 - dichloroethylene in the liquid phase produce
rad ioactiv e forms of each in both cases, with the yield of the labelled parent m o lecu le strongly predominating
over the opposite isom er in e a ch c a s e . These reactions again illustrate a substitution reactio n occurring on a
tim e s c a le co m p arab le to th at required for th e racem izatio n of radicals.
T h e re p la ce m e n t o f C l by B r 80m in an alo g y w ith equation (1 ) is m u ch m o re ste re o sp e cific than th e
corresponding C l 38 reactio n s, but ag ain ra d io a ctiv e form s o f both o f th e dl pairs o f C H 3 CHB1C H C IC H 3 a re
observed.
T h e substitution o f C l38 for H has been studied in d etail with C H 3 C1, and with both 1-ch lorob utan e and
2-ch lorob u tan e. R adioactive 1, 1 - , 1, 2 - , 1, 3* and 1 ,4-d ich lorob u tan e and 1, 3-dichloropropane are observed
from 1-ch lo ro b u tan e ; ra d io a ctiv e 1, 3 - , 2 , 2 - , 2 , 3 - and 2 , 4-d ich lo ro b u tan e , and 1 , 1 - and 1, 2 -d ic h lo ro -
propane a re found in 2 -ch lo ro b u tan e; the yields o f th e rem ain ing dichloropropanes and dichlorobutanes a re
negligib le in each case . These reactions are therefore highly specific and result from the replacem ent without
rearran gem ent o f the H atom s or C H 3 groups of the original m o le cu le . Th e yields of all products are roughly com p arab le w ith the number o f groups whose d isplacem ent leads to their form ation, but show som e c h e m ica l
selectiv ity in addition, e. g. the yields of m eso and d l-2 , 3-dichlorobu tane are not equal from 2-chlorobutan e.
STÉRÉOCHIMIE DES RÉACTIONS D'ATOM ES D'HALOGÈNES PRODUITS PAR UN PROCESSUS (n, y)
SUR DES HALOGÉNURES D’ALKYLES EN PHASE LIQUIDE. Les auteurs ont étudié la réaction de 38C l produit
par un processus (n, y) a v e c les m éso - e t d l- d ich lo ro -2 , 3-butanes, exp rim ée par l ’équation (1 ) dans diverses
conditions exp érim en tales e t ont fait l ’analyse des produits par radiochrom atograp hie gazeuse.
38C 1 + C H 3C H -C H C H 3 --------------------------------------------------------> C H 3 - C H - C H - C H 3 + CI . (1)
C l C l C l 38C1
* The experimental work was performed at the University of Kansas, and was supported by Atomic
Energy Commission Contract No. AT-(ll-l)-407.
+ Present address : Department of Chemistry, University of California Irvine, Irvine, Calif.
333

334 F. S. ROWLAND et al.
En phase liquide à 20*C , les deux diastéréom ères radioactifs se form ent dans la proportion de 5 /2 au profit de
la m o lé cu le irrad iée, qu’ il s 'agisse d e la form e rr.éso ou d e la form e d l. En phase g azeuse les quantités de
ch acu n de c e s produits sont réduites d'un fa c te u r supérieur à 10 . C es résultats indiquent que les m o lécu les
de СНзСНС1СН3 8 С1СНэ trouvées dans la phase liquide sont form ées principalem ent par des réactions de c o m
binaison d’une durée com parable à c e lle de la form ation de radicaux racém iques de CH3 CHCHCICH3 , c 'e s t - à -
d ire par un processus an alo gu e à « l ’effe t de c a g e » . Les irradiations en phase liqu ide à des tem p ératures
plus basses ou en phase solide m ontrent que les quantités de deux diastéréom ères produits se rapprochent davan
tag e de l 'é g a lité que lorsqu'il s 'ag it de liquides à la tem pérature am biante. .
Les réaction s d e 38C1 a v e c le d ic h lo ro -1 , 2 -é th y lè n e en phase liqu id e, q u 'il s 'agisse d e la form e c is
ou de la form e trans, produisent des formes radioactives de chacune de ces formes dans les deux cas, le rende
m ent en m o lécu les m ères m arquées prédom inan t toujours n e tte m e n t sur l ’iso m ère an tip ode. C es résultats
indiquent là en co re qu’une ré a c tio n d e substitution se produit, dont la d urée est co m p arab le à c e lle qui est
n écessaire pour la form ation d e rad icau x racém iqu es.
Le rem p lacem en t du ch lo re par 80rnBr dans-une ré a c tio n ca lq u é e sur la r é a c tio n ( l ) donne un résu ltat
plus stéréospécifique, m ais là encore, on observe des formes radioactives com portant l ’une et l 'a u tre paire dl
du CH 3 CHBrCHClCH3 .
La substitution du radiochlore à l'hydrogène a é té étudiée en détail av ec C H 3CI e ta v e c le c h lo ro -l-b u ta n e
e t l e c h lo ro -2 -b u ta n e . A p artir du c h lo r o -l -b u ta n e , on obtient les ra d io d ic h lo r o -1 ,1 - , - 1 , 2 - , - 1 , 3 - et
- l ,4 -b u ta n e s e t l e d i c h lo r o - l ,3 - p r o p a n e ; on obtient les ra d io d ich lo ro -1 ,3 - , - 2 , 2 - , - 2 , 3 - et -2 ,4 -b u ta n e s
e t les d ich lo ro -1 , 1 - e t - 1 , 2 -propanes dans le ch lo ro -2 -b utane; les rendem ents en autres dichloropropanes et
dichlorobutanes sont négligeables dans tous les cas. C es réaction s sont donc extrêm em en t spécifiques e t pro
viennent du rem p lacem en t sans réarran gem ent des atom es d'hydrogène ou des groupes m éth yle dans la m o lé
c u le p rim itiv e . Les rendem ents sont sensiblem ent co m p arab les au nom bre de groupes dont le d é p la ce m e n t
a perm is la form ation des produits, m ais ils accusent en outre une certa in e sé le ctiv ité chim ique (par exem p le,
le s re n d e m e n ts en m é s o - e t d l -d ic h lo r o -2 , 3 -b u ta n e s à p a rtir du c h lo r o -2 -b u ta n e s n e sont pas é g a u x ) .
С Т Е Р Е О Х И М И Я Р Е А К Ц И Й (n ,y ) Г А Л О Г Е Н Н Ы Х А Т О М О В С ГА Л О И Д Н Ы М И А Л К И Л А М И В Ж ИДКОЙ Ф А З Е . Р еак ц и и ( п ,у ) а т о м о в С 1 3 8 с м е з о - n d l - 2 ,3 -д и х л о р б у т а н о м , к а к э т о з а п исано в уравнени и 1 ) , и с с л е д о в а л и с ь в р а зн ы х эк сп е р и м ен т а л ь н ы х у сл о в и я х при и с п о л ь зо в а нии р а д и о га зо х р о м а т о гр а ф и и для а н а л и за п р о ду к та.
С 1 3 8 + С Н 3 С Н - С Н С Н 3 ------------ * С Н 3 - С Н - С Н - С Н з + С 1 (1 )
I l I Ic i c i С 1 С 1 3 8
В ж идкой ф а зе при т е м п е р а т у р е 20СС соотнош ен и е д в у х р а д и о ак ти в н ы х д и а с т е р е о и зо м е р о в , о б р а зо в а н н ы х в р е ак ц и и , р а в н я е т ся 5 : 2 , т я го т е я к облученной м о л е к у л е для облучения либо м е зо ф о р м ы , ли бо d l ф о р м ы . В г а з о в о й ф а зе к о л и ч ест в о к а ж д о го д и а с т е р е о и зо м е р а с о к р а щ а е т с я н а ф а к т о р > 1 0 . Э т и р е з у л ь т а т ы у к а з ы в а ю т , ч то м о л е к у л ы С Н з С Н С 1 С Н С 1 3 8 С Н 3 , н ай ден н ы е в жидкой ф а з е , о б р а зу ю т с я гл а в н ы м о б р а зо м ком бинацией реакций в п р ом еж утке в р е м е н и , ср а в н и м о м с р е ц е м и зац и е й р а д и к а л о в C H 3 C H C H C IC H 3 , т . е . в п р о ц е с с е , схо дн ом с " к л е т к о й " . О б лу ч ен и я в ж идкой ф а з е при б о л е е н и зк и х т е м п е р а т у р а х или в т в е р д о й ф а зе п о к а зы в а ю т , что эти д в а д и а с т е р е о и зо м е р а о б р азу ю т ся в б о лее оди н аковы х к о л и ч е ст в а х , чем в жидкой ф а зе при ком натной т е м п е р а т у р е .
Р е а к ц и и ( п ,у ) С 1 3 8 к а к с ц и с - , т а к и т р а н с - 1 ,2 -д и х л о р э т и л е н о м в ж идкой ф а з е даю т р а д и о ак ти вн ы е ф ормы к а ж д о го в обоих с л у ч а я х с в ы х о д о м м ечен ой р о д и тель ско й м о л ек у л ы , су щ е ст в е н н о п рео бладаю щ ей н ад противополож ны м и зо м ер о м в каж дом сл у ч а е . Э ти реакции ещ е р а з и ллю стрирую т реакцию за м е щ е н и я , происходящ ую во вр е м я , сравн и м ое с т е м , которое необходи м о для р ац ем и заци и р а д и к а л о в .
З ам е щ е н и е C l B r 80m по ан ал о ги и с у р авн ен и ем 1 ) я в л я е т с я б о лее стер ео сп ец и ф и ч н ы м , ч ем с о о т в е т с т в у ю щ и е р еакц и и С 1 3 8 , но с н о в а н а б л ю д а ю т с я р а д и о а к т и в н ы е ф орм ы о б еи х dl п ар С Н 3 С Н В г С Н С 1 С Н 3 .
З а м е щ е н и е в о д о р о д а С 1 3 8 подробно и зу ч а л о с ь с C H 3 C I , а т а к ж е с 1 - х л о р б у т а н о м и 2 - х л о р б у т а н о м . Р а д и о а к т и в н ы й 1 , 1 - , 1 , 2 - , 1 , 3 - и 1 ,4 -д и х л о р б у т а н и 1 ,3-д и хлор п р оп ан н а б л ю д аю тся в 1 -х л о р б у т а н е ; р ади о акти вн ы й 1 , 3 - , 2 , 2 - , 2 , 3 - и 2 ,4 -д и х л о р б у т а н и 1 , 1 - и 1 , 2 - ди хлорп роп ан н а х о д я т с я в 2 -х л о р б у т а н е ; в ы х о д ы о ст а ю щ и х ся ди хлор п р оп ан ов и д и х л о р о - бутано в н езн ач и тельн ы в каж дом с л у ч а е . П оэтом у эти реакции вы со ко специфичны и я вляю тс я р е з у л ь т а т о м за м е щ е н и я б е з п е р е гр у п п и р о в к и а т о м о в Н или гр у п п С Н 3 п ер во н а ч ал ь н о й

REACTIONS OF (n ,y ) HALOGEN ATOMS 335
м о л е к у л ы . В ы х о д ы в с е х п р одуктов гр у б о сравн и м ы с ч ислом гр у п п , зам ещ ен и е которы х приводи т к их о б р азо ва н и ю , но проявляю т некоторую дополнительную хи м и ческую , с е л е к т и в н о ст ь , т . е . вы хо д ы м е зо и d l - 2 , 3 - ди хлор бутан а неравны с 2 -х л о р б у т а н о м ,
ESTEREOQUIMICA DE LAS REACCIONES (n, y) DE ATOMOS DE HALOGENO CON HALUROS DE
ALQUILO EN FASE LIQUIDA. Se han investigado en distintas cond icio nes experim en tales las re a ccio n e s de
los átom os de 38C1 (n, y) con m e so - y d l - 2 , 3 - diclorobutano, ta le s c o m o la in d icad a en 1); los productos
se han an alizad o por rad iocro m ato g rafía de gases. En fase líquida
38 C1 + СНз С Н -С Н С Н з --------► С Н з -С Н -С Н -С Н з + C 1 (1)
' I l I IC l C l C l 38C1
a 20®C, la razón d e los dos d iastereóm eros rad iactiv o s form ados en esta re a cció n es de 5 : 2 , favorab le a la
m o lé cu la irrad iad a, tan to si se tra ta d e la form a m eso co m o de la dl. En fase gaseosa, la s can tid ad es de
c a d a una disminuyen m ás de 10 v e ce s para am bos productos. Estos resultados indican que las m o lécu las de
CH 3 CH C1CH 38C1CH 3 que se encuentran en la fase líquida se form an prin cipalm en te por re a ccio n e s d e c o m
binación con arreglo a una escala de tiem pos com parable a la de racem ización de los radicales CH 3CHCHClCHg,
esto es, en procesos d el tip o "e n ja u la ” . Las irrad iacio n es efectu ad as en fase líquida» a tem p eratu ras m ás
b ajas, o en fase sólida re v e la n que los dos d iastereó m eros se form an en can tid ad es que tien d en a ser m ás
ig u ales que en fase líqu ida a l a te m p eratu ra a m b ie n te .
En am bos casos, las re a ccio n e s del 38C1 (n, y) con c is - o trans- 1 , 2 -d ic lo ro e tile n o en fase líquida dan
origen a form as ra d ia ctiv a s d e c a d a com pu esto , predom inando fu ertem en te e l ren d im ien to d e la m o lé cu la
precursora m arcada sobre el del isóm ero opuesto. Estos procesos ponen una vez más de m anifiesto'una reacción
d e sustitución que se d esarrolla en una e s c a la d e tiem p os co m p arab le a la n e ce sa ria p ara la ra c e m iz a c ió n
de los radicales»
La sustitución de C1 por 80mBr en una re a cció n análoga a la (1 ) es m ucho m ás estereoespecífica que las
correspondientes reaccion es del 38C1, pero tam bién en este caso se observan formas radiactivas de ambos pares
dl d e C H 3CHBrCHClCH3 .
S e ha estudiado en d e ta lle la sustitución d e H p o r 3 8 C1 u tilizan d o C H 3 C I, a s í c o m o 1 -clo ro b u tan o y
2 -clo ro b u ta n o . S e h a observado que e l 1 -clo ro b u ta n o origina 1 , 1 - , 1 , 2 - , 1 , 3 - y 1 , 4 - diclorobutan o, a s í
c o m o 1, 3 -d iclo rop ro p an o rad iactiv os ; en e l 2 -clo ro b u ta n o se encu entran 1, 3 - , 2 , 2 - , 2 , 3 - y 2 , 4 - d ic lo
robutano, a s í c o m o 1 , 1 - y l , 2 - d iclorop ropano rad iactiv o s; los rendim ientos d e los dicloropropanos y d i -
clorobutanos restantes son despreciables en todo ca so . Estas re a ccio n e s son, pues, sum am ente esp ecíficas y
resultan de la sustitución sin reordenam iento de los áto m o s H o de los grupos C H 3 d e la m o lé cu la o rigin al.
Los rendim ientos de todos los productos son co m p arab les, en líneas generales, al número de grupos cuyo des
p la z a m ie n to origin a su fo rm a ció n , p ero presentan a d e m á s c ie r ta se le ctiv id a d q u ím ica , por e je m p lo , los
rend im ientos d e m eso y d l -2 , 3 -d ic lo ro b u ta n o no son igu ales a p artir d el 2 -clo ro b u ta n o .
INTRODUCTION
The reactions of radioactive halogen atoms in organic system s have been an important part of hot-atom chemistry since the initial experiments of SZILARD and CHALMERS [1]. A fter their demonstration of bond-breaking during reco il from 7 -em ission, the next chemical reactions observed and investigated were the substitution of the active halogen atom for an inactive one in an alkyl halide, as in equation ( 1 ), and the replacement of a hydrogen atom by the active halogen species. Since reaction (1) occurs
X* + RX ---- *RX* + X (1)
fo r essentially all alkyl halides in re latively high yields, the understanding

336 F. S. ROWLAND et al.
of this reaction is of central importance in the study of the chemical reactions of re co il halogen atoms [2 , 3].
E arlier studies of alkyl halide reactions under neutron irradiation have provided the basis for the widely used "b illia rd -b a ll" [4] and "random fra g mentation" [2] theoretical explanations for these events. The introduction o f accurate distillation techniques and then radio-gas chromatography p ro vided evidence that a large number of radioactive products were regularly obtained from reactions with pure compounds in the presence of scavengers, and indicated the necessity for at least as many reaction mechanisms to be simultaneously operative in order to account fo r the experimental observations. The "random fragmentation" model, suggesting combination o f the radioactive halogen atom with one of a "brush heap" of radicals form ed by its dissipated energy, was able to account qualitatively for the wide spectra o f observed products. The ''b illia rd -b a ll" hypothesis represented an opposite extrem e in that the energetic atom was postulated to displace only one struck atom of nearly equal mass, followed by combination with the fragment radical remaining. The "epitherm al atom " hypothesis m odified the original b illiard-ball approach to permit inelastic atom-molecule collisions, m olecu lar dissociations, and then atom -radica l combinations. Although both theories w ere put forw ard many years ago with only p r im itive data compared to what is now available, these models have continued to be the fram ework o f explanation fo r the increasing abundance o f data.
We have sought recen tly to investigate some alkyl halide systems by methods that have proved to be very informative when applied to recoil t r i tium studies of hydrocarbons and halocarbons. The most in form ative of these methods has involved the investigation of the stereochem istry of the substitution processes. The original re co il tritium studies of the s tereo chemistry of substitution were carried out with glucose and galactose, but subsequent studies were conducted with molecules containing a single asymm etric carbon atom (crystalline L(+)-alanine [5], gaseous sec-butyl alcohol)[6 ], necessitating chemical resolution o f optical isom ers. The present stereochemical experiments have utilized target molecules with two optically - active centres, thus providing either two d l-pairs or m eso- and dl-form s, and permitting gas chromatographic resolution of these products. This gas chromatographic technique is essential because of the short half-life of the C138, but has the added advantage o f provid ing much grea ter accuracy in measurement. .
We have, in addition, investigated the replacement of H by Cl38 in the various C-H positions of 2-chlorobutane. Again, the increased resolution and accuracy of radio-gas chromatography has permitted much more quantitative measurement of relative yields of products.
EXPERIM ENTAL
The radioactive Cl38 and Br80™ were formed by irradiation of alkyl halides fo r a few minutes in a neutron flux o f 1. 0X1011 n/cm2. s. The radioactive products were separated by gas chromatography, and measured with a proportional counter in the standard way [2] (see F ig . 1). Flow-through
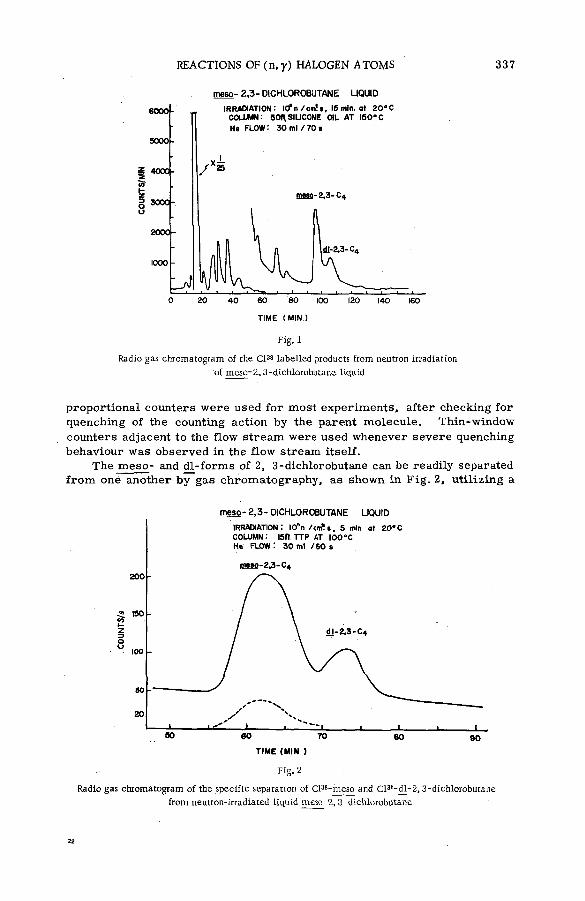
REACTIONS OF (n .y ) HALOGEN ATOMS 337
meso- 2,3- DICHLOROBUTANE LIQUID
TIME (M IN )
F ig . 1
Radio g as ch ro m a to g ra m of the C l38 la b e lle d products from neutron irrad iatio n
o f m e s o - 2 , 3 -d ic h lo to b u ta n e liquid
proportional counters were used for most experiments, after checking for quenching of the counting action by the parent molecule. Thin-window counters adjacent to the flow stream were used whenever severe quenching behaviour was observed in the flow stream itself.
The meso- and dl-forms of 2, 3-dichlorobutane can be readily separated from one another by gas chromatography, as shown in F ig . 2, u tiliz in g a
meso- 2 ,3- DICHLOROBUTANE LIQUID
TIME (MIN )
Fig-2R adio g a s c h ro m a to g ra m o f th e s p e c if ic se p a ra tio n o f C l38-m e s o and C l3 8 -d l -2 ,3 -d ic h lo r o b u ta n e
fro m n eu tro n -irra d ia te d liqu id m e s o -2 , 3 -d ic h lo ro b u ta n e ' -
22
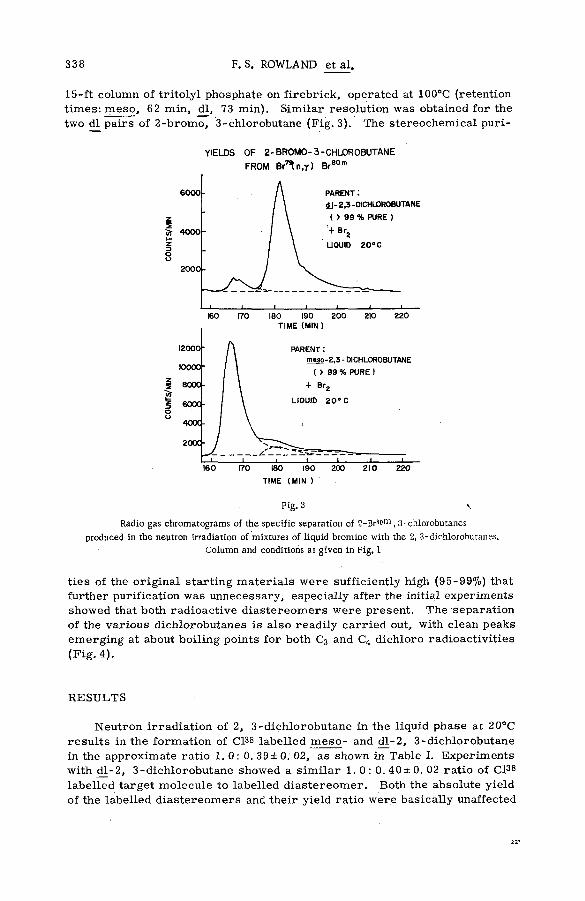
338 F.S . ROWLAND et al.
15-ft column of trito ly l phosphate on firebrick, operated at 100°C (retention times: meso, 62 min, dl, 73 min). Sim ilar resolution was obtained for the two dl pairs of 2-bromo, 3-chlorobutane (Fig. 3). The stereochemical puri-
YELDS OF 2- BROMO- 3 -CHUOROBUTANE
FROM В Л п /r ) Bre0m
6000
Jr 4000
2000
PARENT :Ü - 2,3 -DICHLOROBUTANE
( > 99 % PURE )
180 190 200TIME (MIN )
220
PARENT :meso-2.3- DICHLOROBUTANE
С > 99 % PURE )
+ Br9
ISO 190
TIME (MIN )
F ig . 3 V
Radio g as ch ro m a to g ra m s o f th e s p e c if ic se p a ra tio n o f 2 -B r 8“m , 3 -ch lo ro b u ta n e s
produced in th e neutron irra d ia tio n o f m ixtu res o f liquid b ro m in e w ith th e 2, 3 -d ich lo ro b u ta n e s .
C o lu m n and condition s as g iv e n in F ig . 1
ties of the original starting materials were sufficiently high (95-99%) that further purification was unnecessary, especially after the initial experiments showed that both radioactive diastereomers were present. The separation of the various dichlorobutanes is also readily carried out, with clean peaks emerging at about boiling points for both C3 and C4 dichloro radioactivities (F ig . 4).
RESULTS
Neutron irradiation of 2, З-diçhlorobutane in the liquid phase at 20°C results in the formation of Cl38 labelled m eso- and dl-2, 3-dichlorobutane in the approximate ratio 1. 0: 0. 39±0. 02, as shown in Table I. Experiments with dl-2, 3-dichlorobutane showed a sim ilar 1. 0: 0. 40±0. 02 ratio of Cl38
labelled target molecule to labelled diastereomer. Both the absolute yield of the labelled diastereomers and their yield ratio were basically unaffected
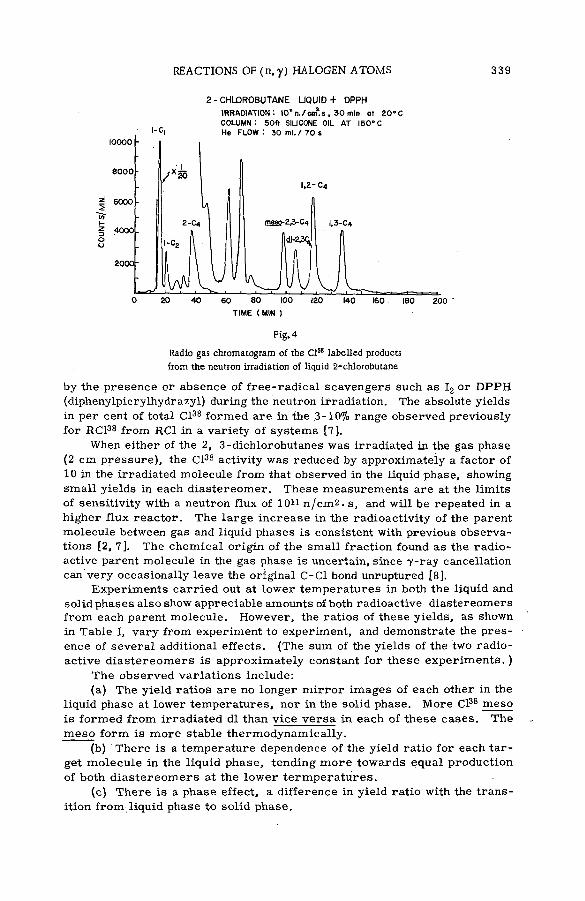
REACTIONS OF (n ,y ) HALOGEN ATOMS 3 3 9
2 - CHLOROBUTANE LIQUID + DPPHIRRADIATION '. IO” n ./cn f.s , 30 min al 20°C COLUMN : 50ft SIUCONE OIL AT 150" С
TIME ( MIN )
Fig. 4
Radio gas chromatogram of the Cl38 labelled products
from the neutron irradiation of liquid 2-chlorobutane
by the presence or absence of free-rad ica l scavengers such as I2 or DPPH (diphenylpicrylhydrazyl) during the neutron irradiation. The absolute yields in per cent of total Cl38 formed are in the 3-10% range observed previously for RC138 from RC1 in a variety of systems [7].
When either of the 2, 3-dichlorobutanes was irradiated in the gas phase (2 cm pressure), the Cl38 activity was reduced by approximately a factor of 10 in the irradiated molecule from that observed in the liquid phase, showing sm all yields in each diastereom er. These measurements are at the lim its of sensitivity with a neutron flux of 10*1 n/cm2. s, and w ill be repeated in a higher flux reactor. The la rge increase in the radioactivity o f the parent molecule between gas and liquid phases is consistent with previous observations [2, 7]. The chemical origin of the sm all fraction found as the rad ioactive parent molecule in the gas phase is uncertain, since 7 -ray cancellation can very occasionally leave the original C-Cl bond unruptured [8].
Experiments carried out at low er temperatures in both the liquid and solid phases also show appreciable amounts of both radioactive diastereomers from each parent molecule. However, the ratios of these yields, as shown in Table I, vary from experiment to experiment, and demonstrate the presence of several additional effects. (The sum of the yields of the two radioactive d iastereom ers is approxim ately constant fo r these experim ents. )
The observed variations include:(a) The yield ratios are no longer m irro r images of each other in the
liquid phase at lower temperatures, nor in the solid phase. More Cl38 meso is form ed from irradiated dl than v ice versa in each o f these cases. The meso form is m ore stable thermodynamically.
(b) There is a temperature dependence o f the yield ratio fo r each ta rget m olecule in the liquid phase, tending m ore towards equal production of both d iastereom ers at the low er term peratures.
(c ) There is a phase effect, a difference in yield ratio with the transition from liquid phase to solid phase.
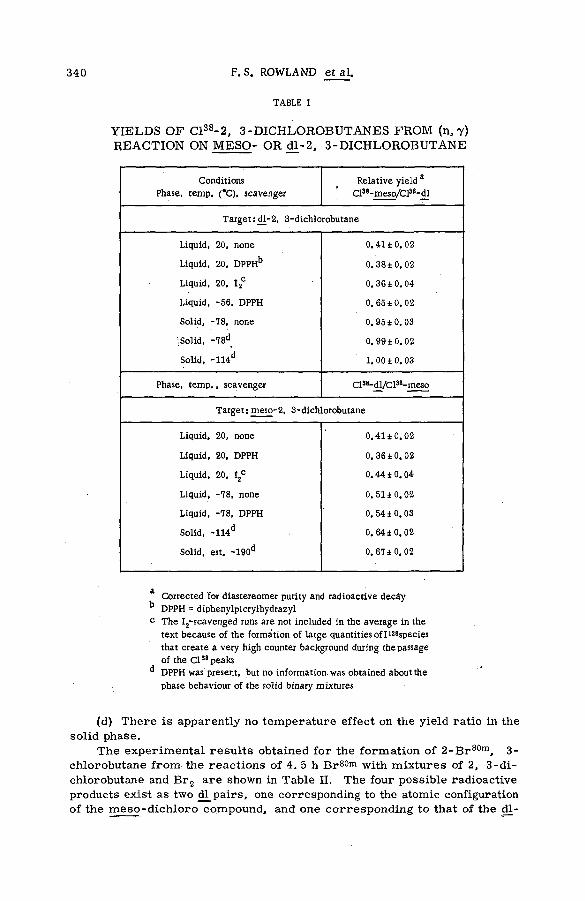
340 F. S. ROWLAND et al.
TABLE I
YIELDS OF Cl38- 2, 3-D ICHLOROBUTANES FROM (n, 7 ) REACTION ON MESO- OR dl-2, 3-DICHLOROBUTANE
Conditions
Phase, temp. (*C), scavenger
Relative yield a
’ Cl38-meso/Cl38-dl
Target: dl-2, 3-dichlorobutane
Liquid, 20, none 0.41 ±0 .02
Liquid, 20, DPPHb 0. 38 ± 0. 02
Liquid. 20. I2C 0.36±0. 04
Liquid, -56, DPPH 0. 65 ±0.02
Solid, -78, none 0. 95 ±0. 03
iSolid, -78d 0. 99 ±0.02
Solid, -114d 1.00±0.03
Phase, temp., scavenger Cl38-dl/Cl38-meso
Target: meso-2, 3-dichlorobutane
Liquid, 20, none ' 0 .41± 0 .02
Liquid, 20, DPPH 0.36 ±0 .0 2
Liquid, 20, I2C 0.44 ±0.04
Liquid, -78, none 0. 51 ±0. 02
Liquid, -78. DPPH ' 0 .54±0.03
Solid, -114d 0, 64± 0, 02
Solid, est. -190d 0. 67 ±0.02
Corrected for diastereomer purity and radioactive decay
DPPH = diphenylpicrylhydrazyl
The I2-scavenged runs are not included in the average in the
text because of the formation of large quantities of Iizsspecies
that create a very high counter background during the passage
of the Cl38peaks
DPPH was present, but no information was obtained about the
phase behaviour of the solid binary mixtures
(d) There is apparently no temperature effect on the yield ratio in the solid phase.
The experim ental resu lts obtained fo r the form ation o f 2 -B r80m, 3-chlorobutane from the reactions o f 4. 5 h B r80m with m ixtures of 2, 3-di- chlorobutane and B r2 are shown in Table II. The four possible radioactive products exist as two dl_ pairs, one corresponding to the atomic configuration o f the m eso-d ich loro compound, and one corresponding to that of the dl-
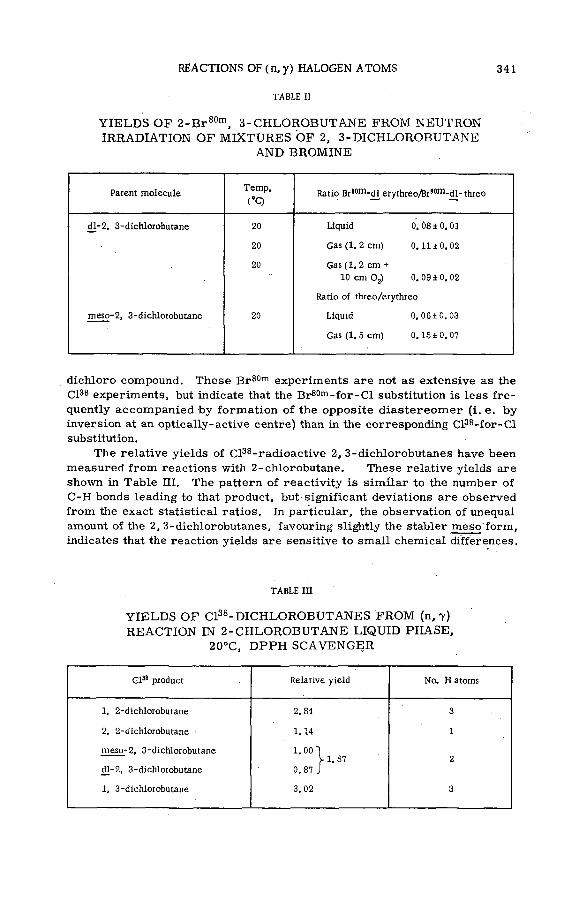
REACTIONS OF (n ,y ) HALOGEN ATOMS 341
TABLE II
Y IE LD S O F 2 -B r80m, 3-C H LO RO BU TANE FROM NEUTRON IR R AD IATIO N O F M IXTU RES OF 2, 3-D ICH LO RO BUTANE
AND BROM INE
Parent moleculeTemp.
C QRatio Br!om-dl erythreoÆr80m -dl- threo
dl-2, 3-dichlorobutane 20 Liquid 0. 08 ± 0.01
20 Gas (1. 2 cm) 0 .1 1 ±0.02
20 Gas (1, 2 cm +
10 cm Oj) 0.09 ±0.02
Ratio of threo/erythreo
meso-2, 3-dichlorobutane 20 . Liquid 0.06 ±0.03
Gas (1. 5 cm) 0.15 ±0.07
dichloro compound. These B r80m experiments are not as extensive as the Cl38 experiments, but indicate that the Br80111 -fo r-C l substitution is less fr e quently accompanied by form ation o f the opposite d iastereom er (i. e. by inversion at an optically-active centre) than in the corresponding Cl38-for-C l substitution.
The re lative yields of Cl38-radioactive 2, 3-dichlorobutanes have been measured from reactions with 2-chlorobutane. These re lative yields are shown in Table III. The pattern o f reactiv ity is s im ilar to the number of C-H bonds leading to that product, but significant deviations are observed from the exact statistical ratios. In particular, the observation of unequal amount of the 2, 3-dichlorobutanes, favouring slightly the stabler meso form, indicates that the reaction yields are sensitive to small chemical differences.
TABLE III
Y IE LD S OF Cl38-D ICH LO RO BU TANE S FROM (n, y) R E AC TIO N IN 2-C H LO RO BU TANE LIQU ID PHASE,
20°C, D PPH SCAVENGER
GI38 product Relative yield No. H atoms
1, 2-dichlorobutane 2.84 3 .
2, 2-dichlorobutane 1.14 1
meso-2, 3-dichlorobutane 1 .0 0 "I>-1. 87 2
cil-2, 3-dichlorobutane ' 0. 87 J1, 3-dichlorobutane 3.02 3

342 F. S. ROWLAND et a l.
Substitution o f C l38 fo r Cl at an asymmetric carbon '
Before the actual experiments were undertaken,three simple results seemed possible for experiments involving replacement of Cl by Cl38 at an asymmetric carbon position: (a) direct substitution with inversion, in closeanalogy with the well-known Walden inversion [9, 10]; (b) direct substitutionwith retention, as observed to be predominant in the recoil tritium reactions [5, 6]; and (c) combination of the Cl38 atom with a free radical to reform an equilibrium mixture of the two possible separable forms. The reaction of Cl38 with 2, 3-dichlorobutane as in equation (2)
Cl38+ C H 3- C H - C H - C H 3 CH3- C H - C H - C H 3+Cl , (2)i l i l
Cl Cl Cl38 Cl
has been the subject for our most extensive investigations of the stereochemistry of substitution. Since these experimental investigations have regularly shown appreciable, but unequal, amounts of the radioactive meso- and dl-form s, they do not conform to any of the three simple processes outlined above. N
The first conclusion to be drawn from the data of Table I is the much lower yield for reaction (2) in the gaseous phase than in any of the condensed phases. It is apparent that the large discrepancy indicates either a substantial decomposition of excited labelled molecules under the lower collision density conditions of the gas phase, or a substantially lower percentage of formation of a large fraction of the labelled molecules in the gas phase.
Separate experiments with CHeCi as the target have been performed, seeking the pressure dependence of yield that might characterize the secondary decompositions of the first possibility. No such pressure dependence was observed. Since excited CH3C138 should be much more susceptible to secondary decomposition than the Cl38- 2, 3-dichlorobutanes, we conclude that such decomposition reactions have a negligible effect in these halogen atom substitutions." The explanation for the enhanced yields in the condensed phase must then lie in the ability of the surrounding molecules to facilitate the combination of a Cl38 atom with a radical trapped nearby.
The most significant observations from Table I are that appreciable yields of both possible labelled diastereomers are found in each system, but that these yields are not necessarily "equilibrium" values. Thè mechanism of formation for these labelled products must involve the combination of the Cl38 atom with a chloro-sec-butyl radical under circumstances not permitting the complete racemization of this radical. Since the meso and dl forms are not thermodynamically equivalent (and are in fact readily separable by gas chromatography because of large differences in the dipole moments), the "equilibrium" concentration of the two forms for atom-plus- racemized-radical need not have equal amounts of each. However, the observation of the preference for the formation of the labelled parent molecule
DISCUSSION

REACTIONS OF (n,y) HALOGEN ATOMS 343
in both cases clearly indicates that the combination reaction is taking place on a time scale competitive with racemization of the radical itself.
The differences in the organic yields for gas and liquid phase experiments with alkyl halides have been discussed in term s of the effect of the solvent "cage " in holding two reac tive species in the near vic in ity of one another until combination can occur, and in terms of the presence of a "brush heap" of radicals form ed by the excited reco il atom [2, 3, 4]. The present experiments show that a racemization process is occurring in this system, but that it has not been completed p rior to the combination reaction in the liquid phase at 25°C. We interpret the observation of competitive racemization and combination reactions as strong support for the importance of "cage" effects in keeping the reacting species in close proximity.
The experiments carried out at lower temperatures show that both temperature and phase effects can be observed, leading to increased r a cemization of the radioactive product, but with no appreciable effect on the sum of the yield of the two diastereom ers. The results in the liquid phase are consistent with a slightly higher activation energy for combination (probably fo r diffusion together) than fo r racem ization. The low tem perature results demonstrate also that the physicochemical differences betwe'en diastereom ers also affect the degree of racemization observed fo r the rad ioactive products.
Substitution o f BrSOm f 0r Cl
The experiments with Br2 dissolved in 2, 3-dichlorobutane show the same qualitative pattern fo r B r 80m as fo r C l38 substitution, both possible dl pairs being observed. However, the quantitative measurements indicate less form ation o f the " in vers ion " product fo r the B r80111 case. A lo g ica l explanation o f the quantitative differences lies simply in the chemical d ifferences between Cl and Br atoms. However, the nuclear histories of (n, 7 ) reactions can be quite different, and the possibility must be' considered that other variations (charge state, excitation, etc. ) are as important as the atomic identity in determining the course of reaction. Similar experiments with the other (n, 7 ) bromine radioactivities, and with reco il products, from particle-em itting nuclear reactions, w ill be necessary to isolate the factors of importance.
Substitution o f Cl38 fo r H
The observation of a ll the dichlorobutanes possible by direct rep lacement of an H atom indicates that the radicals with which the Cl38 atom combines are formed approximately statistically from the parent molecule. However, no 1, 4 - dichlorobutane radioactivity was observed. S im ilarly, only those dichloropropanes are found that can be formed by substitution of Cl38
fo r a CH3 group with no rearrangem ent. In addition, the observed yields o f the dichlorobutanes do deviate from the exact 3 : 1 : 2 : 3 ra tio by sm all, but experim entally significant, amounts. These observations lead to the conclusion that these replacem ent reactions are quite sensitive to m inor differences in C-H bond environments, and do not produce radicals in an

344 F. S. ROWLAND et al.
indiscrim inate, highly excited manner. Further investigation into other target molecules w ill again be necessary to isolate the controlling factors o f these reactions. Nevertheless, the present results give great prom ise that these hot-atom chem istry experiments can be correla ted in a useful way with m ore usual organic chem ical reactions.
R E F E R E N C E S
[1] SZILARD, L. and CHALMERS, T .A . , Nature, Lond. 134(1934) 462,494.
[2] WILLARD, J. E ., in Chemical Effects of Nuclear Transformations I IAEA,Vienna (1961) 215.
£3] CAMPBELL, I .G . , in Advances in Inorganic and Radiochemistry 5, Academic Press, Inc. , New York
(1963) 135.
[4] LIBBY, W .F ., J. Amer. chem. Soc. 62(1940) 1930; 69(1947) 2523.
[5] KAY, J ,G , , MALSAN, R.P. and ROWLAND, F, S., J. Amer. chem. Soc. 81 (1959) 5050.
[6] HENCHMAN, M . and WOLFGANG, R., J. Amer, chem, Soc. 83 (1961) 2991.
[7] QUINLAN, J .E ., Ph. D. Thesis, University of Wisconsin (1958).
[8] GORDUS, A .A . and HSIUNG, C ., J. chem. Phys. 36(1962) 955. About 0 .l-o. 5% bond survival of
radioactive Br and I atoms after nuclear activation in gaseous systems.
[9] HUGHES, E. D . , JULIUSBERGER, F ., MASTERSON, S ., TOPLEY, B. and WEISS, J ., J. chem. Soc.,
(1935) 1525. '
[10] BUNTON, C. A . , "Nucleophilic Substitution at a Saturated Carbon Atom", Elsevier Publishing Co.,
New York, N .Y . , 1963.

REACTIONS OF (n.y) HALOGEN ATOMS 345
D IS C U S S IO N
(on the foregoing three papers)
M. M ILM AN: Using gas kinetics for liquid systems is a ve ry daring thing to do, and hence a cautious approach should be adopted to statements made on the subject and to the assumptions or im plications involved. I should like to make the follow ing three points regarding the kinetics of hot atoms in condensed media. F irst, since one only looks at hot organic yields and ignores or subtracts the diffusive thermal yield - although this part of the yield also comes from tagged radicals or atoms that were initially hot - param eters like a, ¡3 etc. are perhaps equivalent to, but not comparable with, the I 's as defined by the Estrup-Wolfgang treatment fo r gaseous systems. Secondly, a ll the recent work relating to the problem of the fractional energy loss per collision clearly shows that this parameter cannot be considered a p r io r i as being connected with elastic m olecular, atomic or inelastic collisions. A ll that can be done is to get this parameter from the experimental data and try to deduce from its order of magnitude something about the type of collisions involved. Thirdly, in liquids, the effect of im mediate recombination reactions of fragments formed by the direct hot r e actions should not be forgotten. The recombination reactions are typical of cage effects in liquids and can completely a lter the yie lds o f individual organic products, even more so when high concentrations of scavenger are present. The fact that the effects of such reactions on the total organic hot yield have been ignored should be clearly stated and justified.
F .S . ROWLAND: Many of the papers presented on liquid systems have dealt with experiments carried out in binary systems and the appropriate procedure fo r in terpreting those experiments. The kinetic theory o f hot reactions has often been used as the basis for the interpretations. Numerous experiments in binary recoil tritium systems have been carried out in recent years in our laboratory and in Professor Wolfgang's laboratory at Yale, and I w ill, i f I may, now present some of the recent conclusions based on the study of such systems.
In brief, the approach through the kinetic theory of hot reactions provides a functional relationship fo r the total yield of hot reactions in term s of two parameters, the reactiv ity integral, I, and the average logarithmic energy decrement, a. In one sim ple approximation, this yie ld takes the form, for apure molecule, of an exponential relationship, Рд = 1 -ехр(-1д /од ), and depends on the values of 1д and а д for this molecule itself. If this molecule is then studied in the presence of an excess of m olecule B, the yield from A also depends on the values of these param eters fo r molecule В - again, in one approximation for a non-reacting molecule В, =1-ехр(-^1д/о'дв ), in which ía is the fraction of hot collisions occurring with A, and адв simply approaches сев as the excess of В becomes larger and larger. The carrying out of experiments in binary systems can thus lead to measurements of the values of the param eters I A, од, а в , etc. The form of the kinetic theory suggests that experiments of this kind should be done and the results could lead at least to a series of relative values of these parameters for different molecules. The problem of the absolute evaluation of a fo r He4 - one value

346 F. S. ROWLAND et a l.
must be known as a standard i f absolute values are desired - has been d iscussed recently by Dr. Estrup*.
One question with regard to which some of these results have considerable interest has already been raised here, i .e . whether one should use the atomic mass of the struck atom or the molecular mass of the struck molecule in the estimation of a for a molecule through the formula (M -m)2 / (M+m)2 . Actual collisions are not lim ited to these two choices, and one should consider in each system the possib ility that the reco il atom collides with the molecule in a highly inelastic manner, losing much more energy than might be expected from the calculations if the mass of either the atom or the molecule in the elastic co llis ion form ula w ere used. The conclusion that the highly inelastic mechanism is operative fo r the reactions of reco il tritium atoms has been reached in the CH4 -D 2 system by Root and Rowland, in hydrocarbon-He4 systems by Rosenberg and Wolfgang, and in methyl fluoride-H e4 systems by Lee, M ille r and Rowland. Through experiments in binary mixtures, the values of a have been measured relative to о?не4 as a standard. Rosenberg and Wolfgang give relative a values of 2. 9 for propane, 5.0 for n-butane and 4.5 for butene-1; Lee, M iller and I give 2.8 for CH3F and 2. 3 fo r CD3 F. I do not wish to defend any of these numbers as being particularly accurate, or to compare relative values at this time, since the numbers involve various possible e r ro rs . The point I wish to make, however, is that a ll these molecules appear to be two to five times better moderators than He4. He4 is almost equal in mass to tritium and should therefore be a very efficient elastic moderator, with 98% maximum energy loss, and almost half, on the average, for billiard-ball collisions. Yet, the hydrocarbons and methyl fluorides are much better m oderators than He4 .
The correct value fo r а не4 is not certain, since the rig id sphere is not a ve ry good model fo r T -Н е 4 collisions in this energy range, and the choice of which interaction potential can best be used is not yet clear. N evertheless, these experiments indicate v e ry rapid energy loss in tritiu m - hydrocarbon mixtures, indicative of a large fraction of highly inelastic co llisions just above the hot chemical reaction range. The kinetic energy of the tritium atom is being converted into vibrational and rotational energy of the molecule with which it collides; at these tritium atom velocities, efficient translational-vibrational energy transfer is not surprising.
The general point should also be made that the behaviour observed in . these binary systems can be consistently explained through the kinetic theory of hot reactions.
A . GORDUS: It is reasonable to assume that T~C H 3F collisions are more effective than T -Не4 collisions in removing T kinetic energy, since I am confident everyone would agree that T + C H 3 F collisions could involve appreciable inelasticity. Those working in the atom-beam fie ld would, I am sure, pred ict that T -C H 3F collisions defin itely are not e lastic . M y concern is principally with the interpretations placed on experim entally determined a ratios. Bearing in mind our extensive computer evaluations of the mathematical formulations o f the neutron therm alization theory, I would be most reluctant to conclude, for example, that i f “ сн’ р/оне = 2 . 8
* ESTRUP, P. J . , Energy D eg rad ation o f E n e rg e tic A tôm s and H ot A tom R eaction s, I . c h e m . Phys.
41 (1964) 164.

REACTIONS OF (n ,y ) HALOGEN ATOMS 347
and ffcD,F /«He =2.3 this must imply that T -C H 3F collisions are more inelastic than T -C D 3F collisions.
A .G . MADDOCK: I think there is a connection between the comments by Pro fessor Gordus, the results Professor Rowland has just been describing and ea rlie r statements regarding the о -values. I fee l a ll the cases mentioned may be related owing to the effects produced by the rotational energy. In fact the temperature effect described in the paper presented by P ro fe sso r Rowland seems capable of only two explanations, i . e . that the "cage" becomes more rigid as the temperature falls, or that the rotational energy of the molecule involved in the replacement reaction is important. I wonder which explanation Pro fessor Rowland prefers and whether he has any information regarding temperature effects in the gaseous system.
F .S . ROW LAND: The question o f the tem perature effect in the C l38
work with 2, 3-dichlorobutanes is an interesting one. F irst, however, we need to recognize that the 3-ch loro-s ec . -butyl radical may not be typical. D r. A . P . Wolf has pointed out that this radical furnishes an excellent opportunity fo r neighbouring group effects of the Cl atom substituent on r e actions at the adjacent free radical position, and that these effects may be quite important in determining, for example, the meso/dl ratio in radioactive products.
I am inclined to explain the temperature effect as a competition between the racemization of a radical and the atom-radical combination process, with a tem perature coeffic ien t fo r this competition favouring racem ization at lower temperatures. The stiffening of the cage could be the source of such a temperature coefficient. However, at this stage, I do not regard this explanation as much m ore than a consistent hypothesis - we have not made any independent checks of its plausibility.
We have looked fo r temperature effects in reco il tritium systems over a temperature range of about 2 0 0 ° С without observing any differences other than those resulting from changes in density of the molecules in the system. That is, we have seen no temperature effect on the hot reactions themselves in re co il tritium systems, and therefore no gross effect of the changes in population o f rotational states.
In rep ly to D r. Gordus, I wish to re ite ra te what I said about binary system experiments during the discussion on paper SM-57/85. The systems can be accounted for in term s of the parameters a and I and the form alism of the kinetic theory of hot reactions. Whether the real significance of these parameters is precisely the same as their present definitions for the kinetic theory is something we want ve ry much to test. They certain ly describe the systems semi-quantitatively, and no inconsistencies in the relationships have yet been d iscovered. Whether acH,F is rea lly grea ter than ocDjF is not yet thoroughly established - our firs t measurement indicates that it is g rea ter. The re la tive values o f these two a 's to each other is m ore accu rately known than the m easurem ent o f e ith er value re la t iv e to o He4-
One last rem ark regarding inelastic collisions. The existence o f inelasticity in molecular collisions has, of course, been well-known to everybody fo r many years . However, the great importance o f such co llis ions from the point of view of the average energy loss in recoil systems has just

348 F. S. ROWLAND et a l.
been demonstrated. "H ot-atom " chemists have not been taking inelastic collisions into account, in many of the treatments of experimental data, and it is time for us to do so.

ORGANIC SYSTEMS (Session 4)


EFFECTS OF TEMPERATURE AND PRESSURE ON HOT-ATOM REACTIONS IN BROMOETHANE
A .J . COLE, M .D . M IA, G .E . MILLER* AND P. F . D. SHAW NUCLEAR PHYSICS LABORATORY,
OXFORD UNIVERSITY, OXFORD, UNITED KINGDOM
Abstract — Résumé — Аннотация — Resumen
EFFECTS OF TEMPERATURE AND PRESSURE ON H O T-A TO M REACTIONS IN BROMOETHANE. A
study has been m ad e o f th e yield s o f com pounds co n tain in g B r80*11 produced by irrad iatio n o f b ro m e th a n e -
brom ine m ixtu res w ith 1 4 MeV neutrons a t 18°C , -8 0 °C and -1 1 5 ° C , and a t atm o sp heric pressure, and also
at 18eC at pressures up to 1CM atm . In addition to compounds previously reported in this system, sm all quanti
ties of brom oethene, . 1 : 2 dibrom oethene, m o no-and dibromopropanes, and m on o-, d i-, tri* and te tra -b ro m o -
butanes have been found. There is also indirect eviden ce for the production of bromobutenes.
T h e diffusion-dependent reactions are co m p lex , and can be explained by assuming that pyrolysis of the
liquid occurs in the v icin ity o f the hot ato m to give brom oethyl rad icals and eth ylen e. Addition of radicals
to the la tte r then accou nts for the form ation of brom ides containing m ore than two carbon atom s and for their
diffu sion-depen dent yield s. Reduction in te m p eratu re or in crease in pressure g en erally cau ses an in cre a se
in yield a ttrib u tab le to a d ecreased ra te o f diffusion.
T h e e ffe ct is m ost m arked upon th e yield of 1: 2 dibrom oethane, which is larg ely produced by the d if
fusive reaction of brom oethyl radicals and w hich increases fourfold by the application of 1 0 4 atm because of
the suppression of the dissociation . .
CH2 -C H 2 Br £ CH2 = CH 2 +B r
under the influence of the "h o t-sp ik e ". By contrast, dissociation of the rad ical (CH3 -C H B r) into brom oethene
and a hydrogen atom is e n e rg e tica lly forbidden, and th e yield o f 1 : 1 dibrom oethane is roughly independent
of pressure. The variation with tem p erature and pressure of the yield of brom oethane a t large bromine con cen
trations is clo se to that predicted previously.
EFFETS DE LA TEMPÉRATURE ET DE LA PRESSION SUR LES RÉACTIONS DES ATOMES CHAUDS
DANS LE BROMOETHANE. Les auteurs ont étud ié les rendem ents en com posés contenan t 80mBr produits par
exposition de m élan g es d e b ro m o éth an e-b ro m e à des neutrons d e 14 MeV aux tem p ératures d e 1 8 e, -80® e t
-1 1 5 ° C e t à la pression atm osp h ériq u e, ainsi q u 'à la te m p ératu re d e 18®C e t à des pressions a lla n t ju squ 'à
l 0 4 a tm . En plus des com posés d éjà signalés, ils ont trouvé de faibles quantités de brom oéthylène, de dibrom o-
1 , 2 -é th y lèn e , d e m o n o -e t dibrom opropanes e t d e m on o -, d i-, t r i - e t tétrabrom obutanes; il existe en outre
des indices in directs d e la form ation d e brom obutènes.
Les réaction s qui dépendent de la diffusion sont com plexes-, on peut les expliquer en ad m ettan t qu'une
pyrolyse du liquide se produit dans le voisin age d e l 'a to m e chaud e t donne des rad icau x b rom oéth yle e t de
l'é th y lè n e . C 'e s t alors à l'ad d itio n d e rad icau x à l 'é th y lè n e que sont dus à la fois la form ation de bromures
conten ant plus de deux atom es d e carb o n e e t les rendem ents en ce s brom ures, qui dépendent de la diffusion.
D 'u n e m a n iè re g é n érale , une rédu ction d e la te m p é ra tu re ou un a ccro isse m e n t d e la pression en traîn e une
au g m en tatio n du ren dem ent im p u tab le à une d im inu tion de la v itesse d e diffusion.
L 'effe t est le plus m arqué sur le rendem ent en dibrom o-1, 2 -é th a n e f qui se produit surtout par la réaction
de diffusion des radicau x brom oéthyle et qui est quadruplé par l'app licatio n de Ю4 atm du fait de la suppression
d e la dissociation .
C H z -C H 2 B r ï* C H Z=C H Z+B r.
En revanch e, la dissociation du rad ical (C H 3 -C H B r) en brom oéthylène et hydrogène (CH 2 =CH Br + H) est im
* N ow a t th e D e p a rtm e n t o f C h e m is try , U n iv ersity o f K ansas.
351

352 A .J . COLE et a l.
possible, e t le rendem ent en d ib ro m o -1 ,1 -é th a n e est à peu près indépendant de la pression. La variation, en
fonction d e la tem p ératu re e t de la pression, du rendem ent en brom oéthane pour de fortes concen tration s de brom e est voisine d e c e l l e qui a v a it é té prévue précédem m ent par l'un des auteurs.
В Л И Я Н И Е Т Е М П Е Р А Т У Р Ы И Д А В Л Е Н И Я НА Р Е А К Ц И И Г О Р Я Ч И Х А Т О М О В В Б Р О М - Э Т А Н Е . И зу ч а л и сь вы ход ы соеди н ен и й , содерж ащ и х ВгЯОп^ которы й был получен облучением с м е с е й б р о м э т а н -б р о м и н а ней тр он ам и 14 М э в при т е м п е р а т у р а х 1 8 в С , - 8 0 ° С и - 1 1 5 ° С при а тм о сф ер н о м д а в л ен и и , а та к ж е при 1 8 ° С и д а вл ен и я х до 1 0 4 а т м о с ф е р . П ом им о соединений эт о й с и с т е м ы ,о ко то р ы х соо бщ а л ось р а н е е , бы ли най дены небольш и е к о л и ч е с т в а б р о м эт е н а , 1 : 2 д и б р о м эт е н а , м о н о - и ди бром пропанов и m o h o - , д и - , т р и - и т е т р а - б р о м б у т ан о в; е ст ь т а к ж е к о све н н о е д о к а з а т е л ь с т в о п р о и зв о д с т в а б р о м б у т е н о в .
Д и ф ф узи о н н о -зави си м ы е реакции являю тся ком плексны м и и м о гу т быть объяснены , предп оло ж и в, что пиролиз ж идкости происходит в о к р естн о ст и го р я ч е го а т о м а с получением б р о м - эт и л о вы х р ади к алов и эт и л е н а . Д о б авлен и е р ади к ало в к п о следн ем у о т в е ч а е т з а образовани е б р о м и до в, содер ж ащ и х б о лее ч ем 2 а т о м а у гл е р о д а и з а их д и ф ф у зи о н н о -за в и си м ы е вы х о д ы .
Понижение т ем п ер ату р ы или увели ч ен и е давл ен и я обычно в ы зы в а е т увели ч ен и е в ы х о д а, с в я з а н н о г о с ум еньш енной ск о р о ст ью ди ф ф узи и .
Э ф ф ект особен но зн ач и т елен для вы х о д а 1 : 2 ди б р о м о этан а, который п роизводится г л а в ным о б р азом реакц ией ди ф ф узии бр о м эти ловы х р ади к алов и которы й у ве л и ч и ва ет ся в четыре р а з а при применении 1 0 4 а т м о сф е р б л а го д а р я п о давлен и ю ди ссоц и ац и и
С Н 2 - С Н 2 В г - = С Н 2 = С Н 2 + В г
под д а в л е н и е м " г о р я ч е г о п и к а " . Н а п р о т и в , д и ссо ц и а ц и я р а д и к а л а (С Н 3 — С Н В г ) н а б р о м - э т е н и а т о м в о д о р о д а я в л я е т с я э н е р г е т и ч е с к и зап р ещ ен н о й , и в ы х о д 1 : 1 д и б р о м э т а н а гр у б о н е з а в и с и м от д а в л е н и я .
И зм е н е н и е в ы х о д а б р о м э т а н а в за в и с и м о с т и о т т е м п е р а т у р ы и д а в л е н и я при бо льш и х кон ц ен тр ац и ях бр о м и н а б л и зк о к т о м у , ч то п р е д с к а з ы в а л о с ь р а н е е .
EFECTOS DE LA TEMPERATURA Y DE LA PRESION EN LAS REACCIONES DE ATOMOS CALIENTES
EN EL BROMOETANO. S e ha estudiado el rendim iento d e los com puestos que c o n tie n e n 80rn Br, producidos
por irradiación d e m e z cla s de brom oetano y brom o con neutrones de 14 MéV a 18*C, - 8 0 eC y -115®C y a la
presión atm o sférica , así co m o a 18°C y presiones de hasta 1 0 4 a tm . Adem ás d e los com puestos y a conocidos
en este sistem a, se han h allad o pequeflas can tid ad es d e brom oetan o, 1 , 2 -d ib ro m o etan o , m ono y d ibrom o-
propanos y mono, di, tri y tetra bromobutanos; tam bién hay pruebas indirectas de producción debromobutenos.Las reaccio n es dependientes d e la difusión son co m p le ja s , y se pueden e x p lica r suponiendo que en las
proxim idades d el átom o c a lie n te se produce una pirólisis d el lfquido por la que se form an rad icales b rom o-
e tilo y etileno. La adición de radicales a este últim o determ ina la form ación de bromuros que contienen más
de dos átom os de carbono, a s í com o sus rendim ientos dependientes de la difusión. La disminución de la te m
peratura o el aum ento d e la presión se tra d u ce por lo g en eral en un in crem en to del rendim iento atribu ible a una m enor velocid ad d e difusión.
El. e fe c to es m ás acu sad o en e l ren d im ien to d e 1 , 2 -d ib ro m o e ta n o producido p rin cip a lm e n te por la re a cció n difusiva d e ra d ica le s brom oetilo , que se cu adrup lica a l a p lica r una presión d e 1 0 4 a tm debido a la
supresión de la d isociación
C H 2 - C H 2Br « ± C H 2 = C H 2 + Br
b ajo la influencia de la "punta té rm ic a " . En cam bio , la d isociación del rad ical (CH 3 -C H Br) en brom oetano
y un átom o de hidrógeno no es en erg éticam en te viab le , y el rendim iento de 1 , 1 -díbrom oetano es casi inde
pendiente de la presión. La v ariación del rendim iento de brom oetano para grandes concentraciones de bromo
en función de la tem peratura y de la presión se aproxim a al valor que se había previsto.

HOT-ATOM REACTIONS IN BROMOETHANE 353
Bromoethane-bromine mixtures have been irradiated with 14 M eV neutrons at different temperatures, and pressures up to 104 atm. A detailed study is made o f the variation o f the 1, 2 dibromoethane y ie ld ; enhanced caging o f reaction products, expected at sm alle r specific volum es o f the liquid, is not sufficient to-account fo r the results unless factors affecting the s ize and duration of the hot spike are also considered.
Y ie lds of 3 and 4 atom compounds are consistent with the intermediate form ation o f pyro lysis products from the hot spike. D iffusion-controlled reactions leading to bromoethane formation are more complex than thought previously, and hot-atom yie lds are less sensitive to change o f spec ific volume than predicted.
INTRODUCTION
Several authors [1] have observed that the total organic yield of halogen atoms activated by neutron capture in organic m edia increases when the irradiation temperature is decreased. This effect has been generally ascribed to enhanced caging of the products; no systematic study of the varia
tion o f the individual organic yie lds has been made, but A D IT Y A and W ILLARD [ le ] showed that the effect persisted in systems heavily scavenged with elem entary halogen, so that "hot" as w ell as diffusive yie lds are a ffected.
More recently [2] a marked variation of the parent yield has been found in bromoethane contamine sufficient bromine to suppress diffusive reactions. Both the magnitude and the variation of these yields can be estimated [3] by using a combination o f the "b illia rd -b a ll" theory o f L IB B Y [4 ] and the hot-zone theory, firs t applied to hot-atom systems by HARBOTTLE and SUTIN [5]. They can also be related, to a firs t approximation, to properties dependent upon the specific volume of the irradiated material.
The present work was undertaken in order to extend measurements to systems whose specific volume was altered by the application o f pressure rather than by thermal expansion. Since the steel pressure vesse l caused heavy attenuation o f therm al neutron fluxes it was necessary to use high energy neutrons, which are less easily absorbed, and to study bromine atoms activated predominantly by (n, 2n) reactions.
EXPERIMENTAL
Irradiations w ere made with 14 M eV neutrons from the H ^H2, n)He4 reaction fo r 1 h using a flux estimated at 108per s. Samples were out- gassed, sealed in pyrex ampoules and placed in cadmium containers near the target; to va ry temperature, the containers w ere held in a light metal box through which a ir, previously cooled in liquid nitrogen, was passed. Tem perature was controlled by vary ing the flow o f a ir, and monitored by thermocouples.
The pressure vesse l, which w ill be described m ore fu lly elsewhere, was made of steel and contained a cylindrical cavity closed with a Bridgman
23

354 A .J . COLE et a l.
tip and ram which could be held in position by means of a large nut threading on to the outside of the vesse l. To protect the vesse l from the corrosive action of bromine, samples were contained in a thin-walled Teflon capsule. This was placed in the cavity, which was filled with pure ethyl bromide to transm it the pressure produced when the tip and ram was pushed in by means of a hydraulic press. When the desired pressure had been obtained, the nut was tightened onto the ram and the assembly was removed from the press. P ressu res w ere calculated from the fo rce applied and the area of the tip; errors from friction between the tip and walls of the cavity were probably less than 5%.
A fter irradiation, an aliquot of the mixture was added to a. solution of brom ine in carbon tetrach loride. Half of this was extracted with sulphite and the activities of the extracted and unextracted portions determined the total organic yie ld . The rest of the sample was norm ally (procedure A ) extracted with sulphite; after addition of ca rr iers the mixture was separated by using a 3-m silicone-celite gas chromatographic column. The volume of the sample was normally about 1 to 1.5 ml. C arriers were detected at the exit of the column with a hot-w ire gauge and condensed in liquid air traps; their activity was subsequently determ ined with a G-M counter to give the re la tive yie lds of the various fractions. These were converted to absolute yields by assuming that the sum of the activities of the different fractions from the column was equivalent to the total organic yield.
When the irrad iated solution contained less than 10'1 m olar fraction Br2, an alternative procedure (B) was sometimes used; before extraction an excess o f elem entary bromine was added and the solution was allowed to stand fo r a few minutes in light in order to saturate olefinic compounds. C arriers added are shown in Table I; the fractions in brackets were co llected together.
Counting was done at least 3 h after the last chemical or physical operation in o rder to establish transient equilibrium between BrSOm and its daughter. B r78 (half life 6.4 min) from B r79(n, 2n)Br78, and directly formed B r80, had completely decayed before measurement; activity from Br82, produced by fast neutron capture, was negligible.
RESULTS
Altogether 130 analyses of this system have been made and the results presented here are therefore restricted to a selection chosen to illustrate some of the salient features.
Within experimental error,the results were independent of the duration of the irradiation, so that effects due to rad io lys is or to accumulation of decomposition products w ere not significant.
The distribution o f activity obtained in the presence of 10-5 molar frac tion of B r2 and at 18°C and 1 atm is given in Table I. There are significant differences in the yields obtained by the two analytical procedures; the addition o f bromine reduces the yields of a number o f ligh ter fractions, and increases those o f the heavier, indicating the presence o f unsaturated species containing active bromine; s im ilar evidence has been obtained with
23*
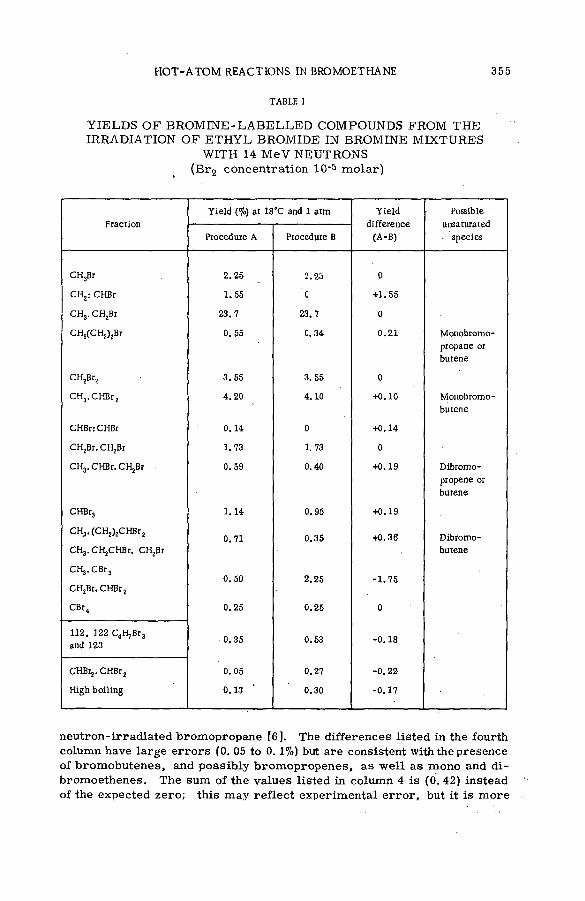
HOT-ATOM REACTIONS IN BROMOETHANE 355
TABLE I
Y IE LD S O F B R O M IN E -L A B E L L E D COMPOUNDS FROM THE IR R A D IA T IO N O F E T H Y L BROMIDE IN BROM INE M IXTU RES
W ITH 14 M eV NEUTRONS (B r2 concentration 10-5 m o lar)
Yield (%) at 18°C and 1 atm Yield Possible
difference unsaturatedггаспоп
Procedure A Procedure В (A-B) species
CHjBr 2.25 2.25 0
CH2: CHBr 1.55 0 +1.55
CH3. C H2Br 23.7 23.7 0
CH3(CH2)2Br 0.55 0.34 0 .2 1 Monobromo-
propane or
butene
CH 2Br2 ■ 3.55 3. 55 0
C H 3.CHBr2 4.20 4.10 +0 .1 0 Monobromo-
butene
CHBr: CHBr 0.14 0 +0.14
CH 2Br. CH2Br 1.73 1.73 0
CH3. CHBr. CH2Br . 0.59 0.40 +0.19 Dibromo-
propene or
butene
CHBr3 1.14 0.95 +0.19
CH3.(CH 2)2CHBr20.71 0.35 +0.36 Dibromo-
CH3.CR.CHBr. CH2Br butene
CH3.CBr3
CH2Br. CHBr20.50 2.25 -1.75
CBr4 0.25 0.25 0
112, 122 C4H,Br3 and 123
0.35 0.53 -0.18
CHBr2.CHBr2 0. 05 0.27 -0 . 2 2
High boiling 0.13 ' 0.30 -0.17
neutron-irradiated bromopropane [6]. The differences listed in the fourth column have large errors (0. 05 to 0. 1%) but are consistent with the presence o f bromobutenes, and possib ly brom opropenes, as w e ll as mono and di- bromoethenes. The sum of the values listed in column 4 is (0. 42) instead o f the expected zero; this may re flec t experimental e rro r, but it is more
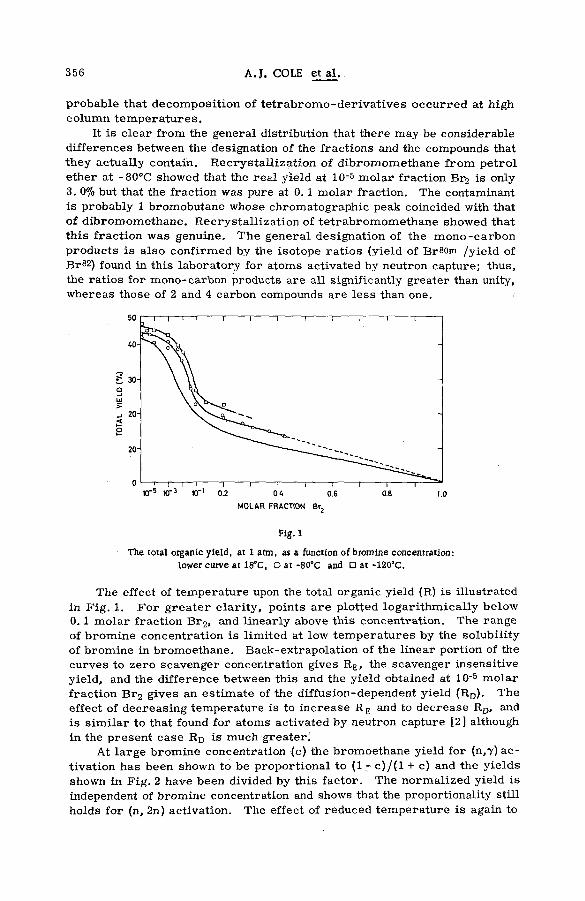
356 A .J . COLE et a l.
probable that decom position o f te trabrom o-deriva tives occurred at high column tem peratures.
It is clear from the general distribution that there may be considerable differences between the designation of the fractions and the compounds that they actually contain. R ecrysta lliza tion o f dibromomethane from petro l ether at -80°C showed that the rea l yie ld at 10-5 m olar fraction Br2 is only 3. 0% but that the fraction was pure at 0. 1 molar fraction. The contaminant is probably 1 bromobutane whose chromatographic peak coincided with that o f dibromomethane. R ecrysta llization o f tetrabromomethane showed that this fraction was genuine. The general designation of the mono-carbon products is also confirm ed by the isotope ratios (y ie ld o f Br80m /yield of Br82) found in this laboratory for atoms activated by neutron capture; thus, the ratios fo r mono-carbon products are all significantly greater than unity, whereas those of 2 and 4 carbon compounds are less than one. .
P i g - 1
The total organic yield, at 1 atm, as a function of bromine concentration:
lower curve at 18’C, О at -80°C and □ at -120°C.
The effect of temperature upon the total organic yield (R) is illustrated in P ig . 1. F o r g rea ter c la rity , points are plotted logarithm ica lly below 0. 1 m olar fraction B r2, and linearly above this concentration. The range of brom ine concentration is lim ited at low tem peratures by the solubility o f bromine in bromoethane. Back-extrapolation o f the linear portion of the curves to zero scavenger concentration gives RE, the scavenger insensitive yield , and the difference between this and the yie ld obtained at 10'5 m olar fraction B r2 gives an estim ate of the diffusion-dependent yie ld (R D). The effect of decreasing temperature is to increase R E and to decrease RD, and is s im ilar to that found for atoms activated by neutron capture [2] although in the present case RD is much greater:
At large bromine concentration (c ) the bromoethane yield fo r (n,y) activation has been shown to be proportional t o ( l - c ) / ( l + c ) and the yie lds shown in F ig . 2 have been divided by this factor. The norm alized yie ld is independent of bromine concentration and shows that the proportionality still holds fo r (n, 2n) activation. The effect of reduced temperature is again to
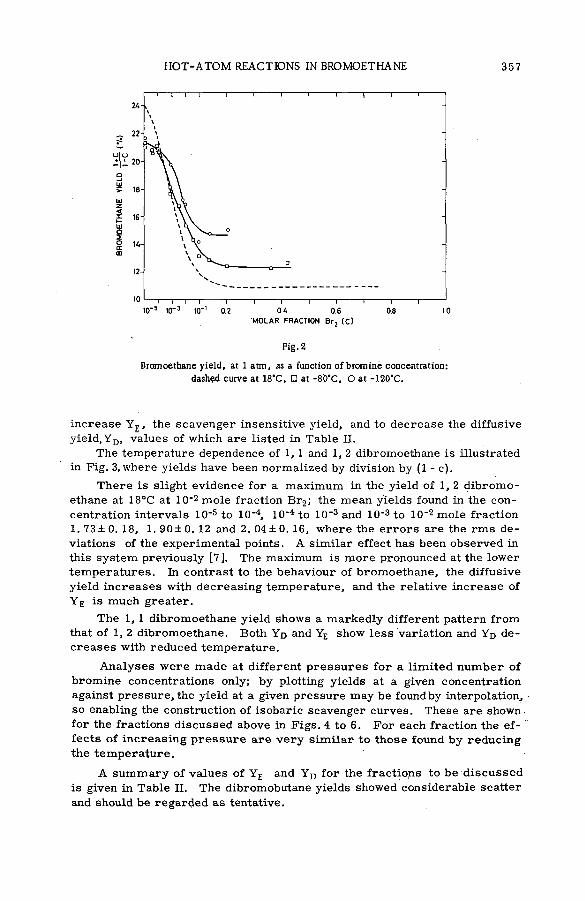
HOT-ATOM REACTIONS IN BROMOETHANE 357
F ig . 2
Bromoethane yield, at 1 atm, as a function of bromine concentration:
dashed curve at 18°C, □ at -80°C, О at -120°C.
increase Y g , the scavenger insensitive yield, and to decrease the diffusive yield, Y D, values of which are listed in Table II.
The temperature dependence of 1, 1 and 1, 2 dibromoethane is illustrated in Fig. 3, where yields have been normalized by division by (1 - c).
There is slight evidence for a maximum in the yie ld o f 1, 2 dibromoethane at 18°C at 10‘ 2 mole fraction Br2; the mean yields found in the concentration intervals 10"5 to 10‘4, 10'4 to 10"3 and 10"3 to 10"2mole fraction 1.73±0. 18, 1. 90± 0. 12 and 2. 04 ± 0. 16, where the e rro rs are the rms deviations of the experimental points. A sim ilar effect has been observed in this system previously [7 ]. The maximum is more pronounced at the lower tem peratures. In contrast to the behaviour o f bromoethane, the diffusive yie ld increases with decreasing temperature, and the re lative increase of Y E is much greater.
The 1, 1 dibromoethane yield shows a markedly different pattern from that o f 1, 2 dibromoethane. Both Y d and YE show less variation and Yd decreases with reduced temperature.
Analyses w ere made at d ifferen t pressures fo r a lim ited number o f bromine concentrations only; by plotting yields at a given concentration against pressure, the yield at a given pressure may be found by interpolation, so enabling the construction of isobaric scavenger curves. These are shown fo r the fractions discussed above in Figs. 4 to 6. For each fraction the e ffects o f increasing pressure are v e ry s im ilar to those found by reducing the tem perature.
A summary o f values o f Y E and Y D fo r the fractions to be discussed is given in Table II. The dibromobutane yields showed considerable scatter and should be regarded as tentative.
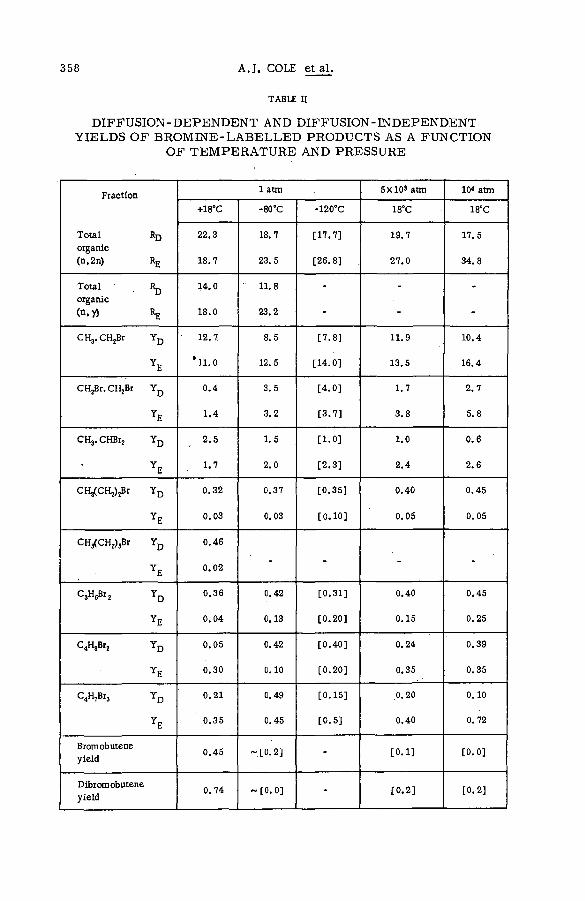
358 A .J . COLE et a l.
TABLE II
D IFFU S IO N -D E PE N D E N T AND D IFFU S IO N-IN D E PE N D E N T Y IE L D S O F B R O M IN E -L A B E L L E D PRO D UCTS AS A FU N CTIO N
O F T E M P E R A T U R E AND PRESSURE
Fraction1 atm 5 x 10s atm 104 atm
+18°C -80°C -120°C 18°C 18°C
Total rD 22.3 18.7 [17.7] 19.7 17.5
organic
(n ,2 n)re
18.7 23.5 [26.8] 27.0 34.8
Total«п
14.0 11. 8 - - -organic
(ft, y> re18.0 23.2 - - -
CHj. CH2Bry d
12.7 8.5 [7.8] 11.9 10.4
y e* 11.0 12.5 [14.0] 13.5 16.4
CHjBr. C H2Bry d
0 .4 3.5 [4.0] 1.7 2 .7
y e1.4 3.2 [3.7] 3.8 5.8
CH3. CHBrzy d
2 .5 1.5 [1 . 0] 1.0 0 .6
y e1.7 2 .0 [2.3] 2.4 2 .6
C H ^C H ^B ry d
0.32 0.37 [0.35] 0.40 0.45
y e0.03 0.03 [0. 10] 0.05 0. 05
C H / C H ^ ry d
0.46
y e0 .0 2
C 3H 6Br2y d
0.36 0.42 [0.31] 0.40 0.45
y e0.04 C. 13 [0 . 2 0] 0.15 0. 25
C4H8Br2y d
0.05 0.42 [0.40] 0.24 0.39
y e0.30 0.10 [0 . 2 0 ] 0.35 0.35
С4Н7ВГЗy d
0 .2 1 0.49 [0.15] 0.20 0 .1 0
y e0.35 0.45 [0.5] 0.40 0. 72
Bromobutene
yield0.45 ~ [ 0. 2 ] - [0 . 1] [0. 0]
Dibromobutene
yield0.74 ~ [ 0. 0] - [0. 2 ] [0 . 2]
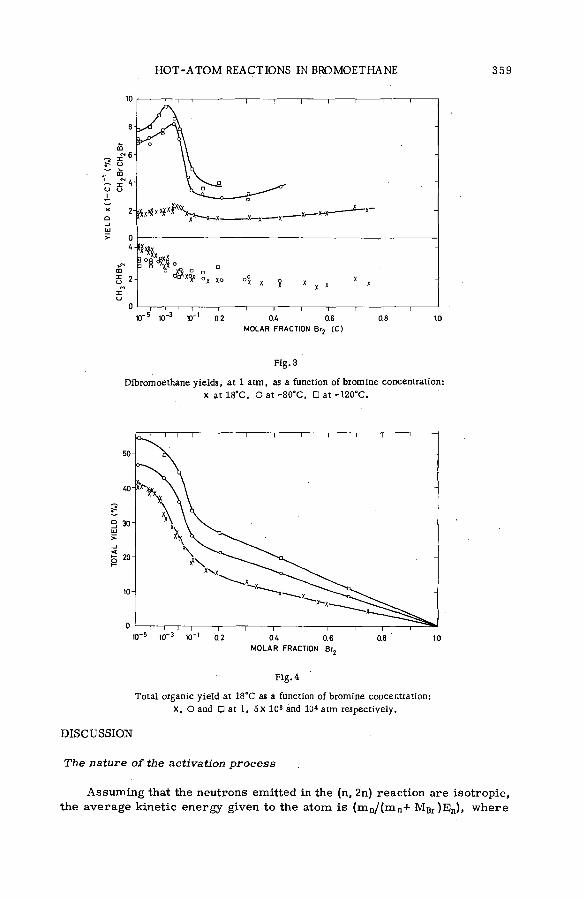
HOT-ATOM REACTIONS IN BROMOETHANE 359
Fig.3
Dibromoethane yields, at 1 atm, as a function of bromine concentration:
, X at 18”C , О at -80°C, □ at -120°C.
Fig. 4
Total organic yield at 18°C as a function of bromine concentration:
X, О and □ at 1, 5X 10s and 104 atm respectively.
DISCUSSION
The nature o f the activation process .
Assuming that the neutrons emitted in the (n, 2n) reaction are isotropic, the average kinetic energy given to the atom is (m n/(m n+ M Br)En), where
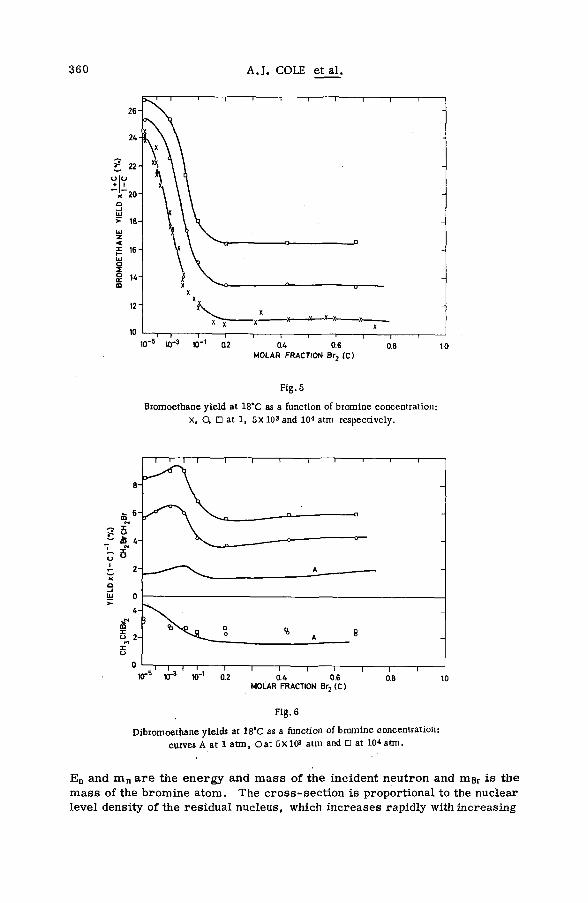
360 A . J . COLE et a l.
Fig. 5
Bromoethane yield at 18”C as a function of bromine concentration:
x, Q □ at 1 , 5X 103 and 104 atm respectively.
Fig. 6
Dibromoethane yields at 18°C as a function of bromine concentration:
curves A at 1 atm, Oat 5X103 atm and □ at 104 atm.
E„ and m nare the energy and mass o f the incident neutron and mar is the mass of the bromine atom. The cross-section is proportional to the nuclear leve l density o f the residual nucleus, which increases rapidly with increasing

HOT-ATOM REACTIONS IN BROMOETHANE 361
excitation energy, so that the BrSOm formed will, in general,de-excite finally by gam ma-ray emission. If the lifetim e of an excited state form ed during the gam m a-ray cascade is long (> 1 0 “19s) most of the damage produced in the liquid w ill have disappeared; alternatively, i f internal conversion o c curs, the atoms may become highly positive ly charged [8] by Auger processes, and the possibility that ions play a part in determining the final chemical combination o f the atom cannot be excluded.
Values o f Re found for (n,y) and (n, 2n) activation (Table II) are approximately equal and show sim ilar variation with temperature, so that there is no reason to suppose that the reactions contributing to RE d iffer in the two processes. By contrast, RDis considerably greater for the (n, 2n) reaction, indicating a more extensive diffusive zone, or a greater density of break-up products [9].
Before discussing the nature of the chemical reactions involved, factors affecting the size and duration of the thermal spike produced by the energetic atom w ill be examined. The range, r, of the reco il atom can be estimated from the formula [10];
n2TT2R47/2 + 2R
where n is the density o f atoms in the medium fo r s im ilar atoms; a and R are given by
a = ^ f§| and RE = 2Z2e2e ‘R/a;
E is here the kinetic energy and Z the atomic number o f the atom; e is the electronic charge and ав is the Bohr radius, a is an arbitary constant and has been taken as 2, since this value has been found em p irica lly to give better agreement with measured ranges [11]. Direct application of the fo r mula to the bromine atoms in bromoethane gives a range which is too large because energy loss to carbon and hydrogen atoms has been neglected. A correction fo r this can be made by estimating the rates of energy loss with a ll atoms using the same form of screened interaction potential [12]. In order to do this, the energy loss with carbon and bromine has been assumed to be through elastic co llis ions, and a correction o f the form applied previously [3] has been made to allow for the inelastic excitation of hydrogen atoms. Values of the ranges obtained are given in Table III, together with the mean temperature o f the therm al spike produced; this was calculated by assuming that (a) the spike is spherical, and of radius equal to half the range; (b) the heating is so rapid that there is no change in volume of the spike; and (c ) the specific heat at constant volume, calculated from data for the normal liquid, remains constant over the large changes of pressure and temperature within the spike. A more detailed analysis [3] shows that heating probably occurs within 5X10'13s, so that some expansion of the spike w ill have taken place before the heating is complete. This is neglected in the present model, in which the hot zone is regarded as a bubble, in the normal liquid, having an in itial radius of 25 Â and a uniform temperature

362 A .J . COLE et a l.
TA BLE III
C A L C U L A T E D RANGE AND M E AN T E M P E R A T U R E O F T H E R M A L SPIKE AS A FU N CTIO N O F .
K IN E T IC EN ERG Y O F BRO M INE ATO M
K in e tic en erg y o f Br a to m
(k e V )
R ange
(A )T e m p e ra tu re ch a n g e
(°C )
1 7 5 3 8 4 0 1 . 4
3 0 . 5 6 0 80
1 0 2 2 8 3 9 0
3 1 3 3 60 0
1 8 0 94 0
. 0 . 3 4 7 1 3 1 0
0 . 1 2 6 2 5 5 0
difference o f 2500°C. The term "tem perature" p roperly applies only to m acroscopic system s, and its use in the present case must be regarded tentatively.
Assuming that bromoethane behaves sim ilarly to a van der Waals gas, the pressure o f the spike is given by A T (9P/3T )v* (9P/3T)v should be a function of volume only and the available high pressure data [13] fo r it is represented to within 3% by the expression (373/(v-45. 5)) atm per deg, v being the molar volume in cm3. The growth, and consequently the cooling, o f such a bubble is determ ined by its adiabatic expansion [3] rather than by thermal conduction or radiation through its walls. The rate of expansionfor a gas obeying the law (p v ^ constant) has been given by LAMB [14] and is
Air Y 2Pn ггадз поэу -\d t у ~ 3 p ( Y - l ) L l R J I R J J '
where p is the density o f the surrounding medium, and P0 and R 0 are the in itia l pressure and radius of the bubble. Num erica l integration o f this equation gives R, and hence the volume, as a function of time, and the spike temperature can then be determined by the integration o f the adiabatic formula
c v6T + T (ap/9T )v av = o.
P V T data are too lim ited to enable the construction o f an equation of state, and hence to find a suitable value of the param eter y, which cannot be r e garded as a specific heat ratio in this case. The integrations were therefo re made with a rb itrarily chosen values of 1. 33 and 3. 0. The variation of temperature with tim e is shown in F ig . 7 fo r bromoethane under different in itial conditions, and it can be seen that the effect o f decreasing specific volume is to enhance quenching, owing to differences in the (9P/3T)v values

HOT-ATOM REACTIONS IN BROMOETHANE 363
F ig . 7
S p ike te m p e ra tu re as a fu nction o f t im e .
C u rv es 1 , 3 , 5 : y = 3 , P = 1 a t m , T = 1 8 °C , - 8 0 ° C , - 1 2 0 “C r e s p e c tiv e ly .
C u rv e 2 : y = 1 . 3 3 , P = 1 a t m , T = 1 8 °C .
C u rves 4 , 5 : 7 = 3 , T = 1 8 ° C , P = 5 X 1 0 3 and 1 0 4 a tm re s p e c tiv e ly .
and hence the initial pressures. The rate of quenching is not very sensitive to the value of y used (see Fig. 7).
The diffusive y ield o f the dibromoethanes
At low bromine concentrations, the yield of 1: 2 dibromoethane shows a maximum that can be explained on the assumption that active bromoethyl radicals are present. If these are unstable, or could react with other species, they might disappear before undergoing the reaction
CHaBr*. CH2. + B r2 -> CH2B r*. CH2Br + Br. (1)
Increasing brom ine concentration would then cause an enhanced y ie ld by competing m ore e ffective ly fo r radicals. Provid ing that the radicals were o f reasonably high stability they could survive fo r sufficient tim e fo r the rise to be apparent over a very low concentration range, and the effect would then be clearly distinguishable from the reduction in yield observed at higher concentrations. This decrease may be assumed to be due to the scavenging action of bromine upon inactive bromoethyl radicals which would otherwise combine diffusively with free active atoms. The combination of these effects would then produce the observed maximum. Reactions which could lead to the disappearance of the (CH2B r*. CH2. ) rad icals are
CH2B r*. CHj. + CH3. CH2Br -> CH3. CH2B r* + . CH2. CH2B r . , (2)
or
. CH2 - CH2B r* -* CH2 : CH2 + B r * . (3)

364 A . J . COLE et a l.
Another mechanism giving r is e to a maximum in the y ie ld cannot be excluded. A ctive brom oethyl radicals may be produced by diffusive r e action o f the active atom with ethylene produced by pyrolysis [15] in the hot spike
CH2: CH2 + B r* -» CH2Br*C H 2. , (4)
and then form 1: 2 dibromoethane as outlined above. On further increase o f brom ine concentration, exchange between the active atom and brom ine could occur and so decrease the y ie ld o f b rom oethyl rad ica ls from (4).
The sensitivity o f the over-a ll yield to change of both temperature and pressure strongly suggests that enhanced caging, caused by decreased specific volume of the liquid, is more important in determining the yie ld than the temperature dependence o f some reaction. Of the reactions considered, (2), and its counterpart involving inactive atoms, is most liab le to be a ffected by changes in caging: unless the bromine atom can diffuse from the ethene molecule recombination w ill be probable, so preserving the brom oethyl radicals and maintaining a large yield of 1: 2 dibromoethane.
This explanation would account a lso fo r the re la tive in sensitiv ity o f the 1: 1 dibromoethane y ie ld to changes o f temperature and pressure. In this case, CH3. CHBr. radicals must be responsible fo r the diffusive yield, but their dissociation into bromoethene molecules and hydrogen atoms is less probable because a large energy of activation is necessary; this must be sim ilar to that required for the dissociation of an ethyl radical into ethene and a hydrogen atom (between 38 and 43 kcal/mole [16]). By contrast, the activation energy required fo r (3) is probably [17] 13 kcal/mole.
It is now necessary to examine the conditions under which the brom oethyl rad ical yie ld is reduced by dissociation, and,in particular, whether it is in the hot spike, or the cooled diffusive zone. The rate of dissociation of the radicals may be written
dN/dt = - N[ к е '£/йт n е - ^ кт];
к is a rate constant, normally taken as 1013/s for unimolecular dissociation, E is the energy of activation and W that required to produce a "hole" in the liquid; (n e"W//RT) is then the probability that a "hole" exists in the cage consisting o f n (taken as 10) close neighbours so allowing the diffusion o f the bromine atom from the ethene molecule. Simple thermodynamic arguments show that the energy to produce a hole of volume ДУ against the "internal" and hydrostatic pressure is T(3P/9T)v AV. The mean life of a radical against dissociation is given by the reciprocal of the term in square brackets in the above expression; the ratio of the life tim es at 18°C and at 5X103 and 1 atm is 1. 7. If the over-a ll rate of reaction (1) is increased by a sim ila r factor at the lower pressure, fo r example, by increasing bromine concentration; sim ilar yields o f dibromoethane should be obtained from (1) in the concentration region 10"5 to 10’ 3 m olar fraction. In fact, yields d iffer by a factor of three, and the results therefore cannot be explained by a mechanism based upon dissociation of the radicals in the cold diffusive zone.
The probability of survival of bromoethyl radicals present in the initial hot spike can be found integrating the above expression over the range of
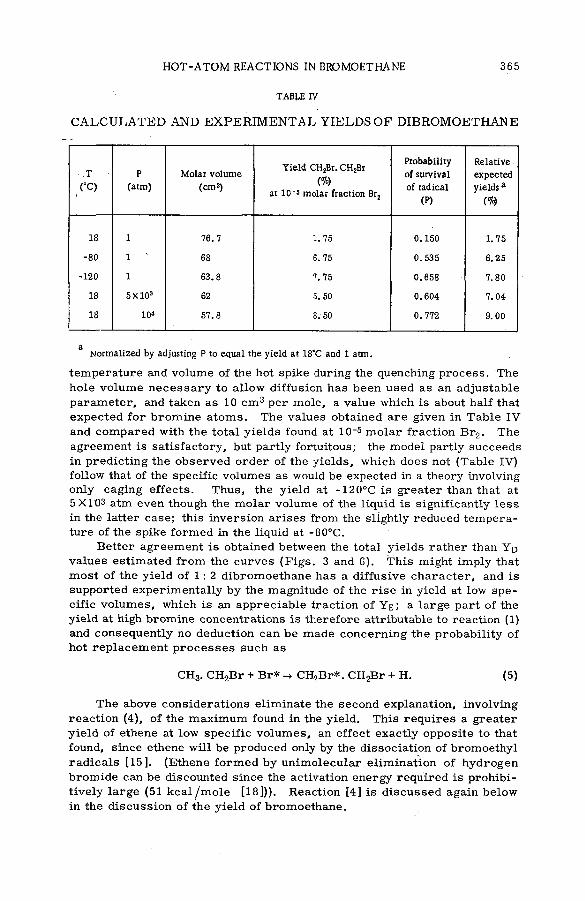
HOT-ATOM REACTIONS IN BROMOETHANE 365
TABLE IV
C A L C U L A T E D AND E X P E R IM E N T A L Y IE LD S O F D IBRO M O ETHANE
.T
CC)P
(atm)
Molar volume
(cm3)
Yield CH2Bi.CH2Br
(%)
at IO -5 molar fraction Br2
Probability
of survival
of radical
(P)
Relative
expected
yields a
№)
18 1 76.7 1.75 0.150 1.75
-80 1 ' 68 6.75 0.535 6.25
-120 1 63.8 7.75 0.658 7.80
18 5X103 62 5. 50 0.604 7. 04
18 104 57.8 8. 50 0.772 9.00
aNormalized by adjusting P to equal the yield at 18°C and 1 atm.
temperature and volume of the hot spike during the quenching process. The hole volume necessary to allow diffusion has been used as an adjustable parameter, and taken as 10 cm3 per mole, a value which is about half that expected fo r brom ine atoms. The values obtained are given in Table IV and compared with the total yie lds found at 10"5 m olar fraction B r2. The agreement is satisfactory, but partly fortuitous; the model partly succeeds in predicting the observed order o f the yields, which does not (Table IV ) follow that of the specific volumes as would be expected in a theory involving only caging effects. Thus, the y ie ld at -120°C is greater than that at 5X103 atm even though the m olar volume of the liquid is significantly less in the latter case; this inversion arises from the slightly reduced temperature of the spike formed in the liquid at -80°C.
Better agreement is obtained between the total yie lds rather than Y d values estimated from the curves (F igs . 3 and 6). This might im ply that most o f the y ie ld o f 1 : 2 dibromoethane has a diffusive character, and is supported experimentally by the magnitude o f the r is e in yield at low spec ific volumes, which is an appreciable fraction o f Y e ; a la rge part o f the yie ld at high bromine concentrations is therefore attributable to reaction (1) and consequently no deduction can be made concerning the probability of hot replacem ent processes such as
CH3. CH2Br + B r* -> CH2B r*. CH2Br + H. (5)
The above considerations elim inate the second explanation, involving reaction (4), o f the maximum found in the yield . This requ ires a greater y ie ld of ethene at low specific volumes, an e ffect exactly opposite to that found, since ethene w ill be produced only by the dissociation of bromoethyl rad icals [15]. (Ethene form ed by unimolecular elim ination o f hydrogen bromide can be discounted since the activation energy required is prohibit ive ly la rge (51 kcal/m ole [18])). Reaction [4] is discussed again below in the discussion o f the y ie ld o f bromoethane.

366 A.J. COLE et al.
Bromoethyl radicals may be produced initially by dissociation of hydrogen from bromoethane in a hot-atom collision, or by reactions of the type
X + CH3. CH2Br -» XH + CH2. CH2Br, (6)
where X may be Br, H, C2H5 etc. The heats of activation of these reactions have not been measured, but by analogy with sim ilar reactions are expected to be 10-15 kcal/m ole. The reaction rate constant w ill be high, since no diffusion is required, and the extent to which such reactions occur in the hot spike can be shown to be appreciable. The constancy o f the b rom oethyl radical yield, implicitly assumed above, is therefore open to question, and the yield may vary according to the initial state of the liquid.
Yields o f 3 and 4 carbon compounds
These are compatible with reactions involving pyrolysis products o f bromoethane (i. e. ethene and bromoethyl radicals) and of 1, 2 dibromoethane (i. e. bromoethene and dibromoethyl radicals whose formation has been suggested previously [19]). Under conditions prevailing in the spike, the latter could be formed by the reaction
CH2Br. CH2. + CH3. CH2Br -> CH2Br. CH2Br + CH3. CH2. ,
followed by hydrogen abstraction reactions sim ilar to (6).Monobromopropane and butane yie lds are completely suppressed by
brom ine, and are probably form ed by d iffusive reaction o f active atoms with rad ica ls form ed by the reaction
R. + CH2 = CH2 -> R. CH2. CH2. , (7)
where R is ethyl or methyl. Other reactions leading to the diffusive formation o f three and four carbon compounds are given below; these are sim ilar to those postulated by FOX and LIBBY to account for polymer formation in bromopropanes [la ]:
R CH2. CHBr. fa)/ B r
R. + CH2 = CHBr ------- -------> R. C2H3B r2 (8)1 \ (by diffusion) ¿ л 1
N[R . CHBr. CH2. ] (b)
1R. CH = CÜ2 + Br,
(R = CH3 or C2H5).
. CH2. CH2B r + CH¡ = CH2 -> CHjBr. (С Н ^С Н г (a) CHrjBr.fCH^CftjBr
(9)

HOT-ATOM REACTIONS IN BROMOETHANE 367
УCHsBr. CHg. CH2CHBr. (a)
. CH2. CH2B r + CH2: CHBr ----» С4Н7ВГ3Br
^ [CH2Br. CH2. CHBr. CH2. ] (b)I
CH2Br. CH2. CH = CH2
(10)
CH2Br. CHBr. CH2. CHBr. (a)
^ C4H6B r4
. ( 11)^ [С Н гВ г. CHBr. CHBr. CH2. ](b )I
CH2Br. CHBr. CH = CH2
In the reactions (7) to (11) the active bromine atom may be incorporated in the reacting radicals or bromoethene, or it may combine diffusively. The radicals (b) designated by square brackets are all labile, and should be sensitive to change o f specific volume o f the liquid, whereas the radicals (a) should show little change and behave sim ilarly to the CH3. CHBr. radicals involved in the formation o f 1 : 1 dibromoethene.
The general behaviour shown by the fractions (Table II) is as expected. A ll show changes with specific volume, the proportionate change being less than that found for 1: 2 dibromoethane. There is also evidence that the yield o f the bromobutenes is reduced when the m olar volume is decreased, in general agreement with the reduced dissociation expected fo r the (b) type radicals.
The y ie ld o f bromoethane
The reaction most often postulated [20] to account for the diffusive yield is
However, i f bromoethane is form ed at a ll by this route, it must be in re la tive ly low yield. Reasons fo r this are listed below.
There is considerable evidence [21] that the heats of activation and general probability of (13) is the same as that o f the corresponding reaction:
C2H5. + B r * - » C2H5B r*. ( 12)
(a) The suppression o f this reaction by brom ine scavenger has been ascribed [2 0 ] to the interception o f ethyl radicals by bromine:
C2H5. + B r , -* C2H5Br + Br. (13)
C2H5+ HBr -* GjH6 + Br, (14)
so that a sim ilar decrease in yield should also be caused by addition of hydrogen bromide. An earlie r experiment [2] using (n, 7 ) activation showed that there was no departure from linearity in the plot of bromoethane yield

3 6 8 A . J . COLE et a l.
against HBr concentration over the region IO-5 to 0. 6 mole fraction. This has been confirmed in the present work.
(b) Under hot-spike conditions the ethyl radical yield w ill be determined by reactions such as (6). At the highest spike tem peratures the reverse reaction w ill be equally probable, but as the spike cools less reactive radicals w ill assume a greater relative stability; since bromoethyl is less r e active than ethyl, requ iring m ore than 3 kcal/m ole activation energy to react with brom ine [17], the prim ary yie ld o f ethyl radicals may be considerably reduced. The reduction might be less at smaller specific volumes, owing to the decreased duration o f the spike, and an enhanced diffusive yield of bromoethane would be expected. The opposite e ffect is found, but this may re flec t a reduced number of bromine atoms available for diffusive r e action owing to non-dissociation of bromoethyl radicals, or the generally enhanced "hot" yields (Y E).
(c) If (7) occurs, the relative yield of bromopropane and butane (0. 67) should be proportional to that o f methyl and ethyl rad icals, and hence to the ratio of the diffusive yields of bromomethane and ethane. At 18°C and 1 atm y d (CH3Br) is approx. 1.3%, so that with these assumptions YD(C2H5Br) is expected to be only 1. 9%; that found is 12. 7%.
Other reactions that might contribute to YD{Cî,H5Br) are:(a) Reaction (2), which is required to explain the maximum in the d i
bromoethane yield . This w ill give a yie ld equal to the r ise in y ie ld o f d ibromoethane at low concentration, and maximally equal to Ун(СН2Вг. CHgBr).
(b) Exchange between bromine atoms and bromoethane. This has beenpostulated previously [20], but if its activation energy is as great as 25 kcal/mole [22], it is too slow to account fo r the present results, even under hot-spike conditions. '
A ll alternative explanations require either that (14) is improbable, or that exchange of atomic bromine is fast with bromine and slow with hydrogen bromide. Since the evidence for fast exchange [22] with hydrogen bromide is not convincing [23] a tentative explanation based on the second alternative is offered.
The sum of the Y D values for 1: 2 dibromoethane and bromoethane is nearly independent of the initial condition of the liquid. On the model proposed above, the diffusive yield of 1 : 2 dibromoethane depends upon the surv iva l of CH2. CH2Br radicals in the hot spike; it also requires that no significant yield of dibromoethane should be produced by the cold diffusive combination o f bromine atoms with ethene molecules followed by reaction with brom ine. The interdependence of the two yie lds th erefo re suggests that the probability o f bromoethane formation depends upon the availability of ethene and active bromine atoms in the diffusive zone. Reaction (4) is exotherm ic by 13-14 kcal/mole [15] and requires little activation energy. The heat o f reaction may therefore provide sufficient energy to enable the abstraction of hydrogen from a neighbouring bromoethane molecule, and hence the form ation of an active bromoethane molecule. Scavenging would then occur by exchange of the active atoms with elementary bromine; but, with the above assumption, there would be no marked scavenging by hydrogen brom ide.
On simple b illia rd -ba ll theory [4, 24] the scavenger-insensitive yield o f bromoethane should equal e/v, e and v being respective ly the energies
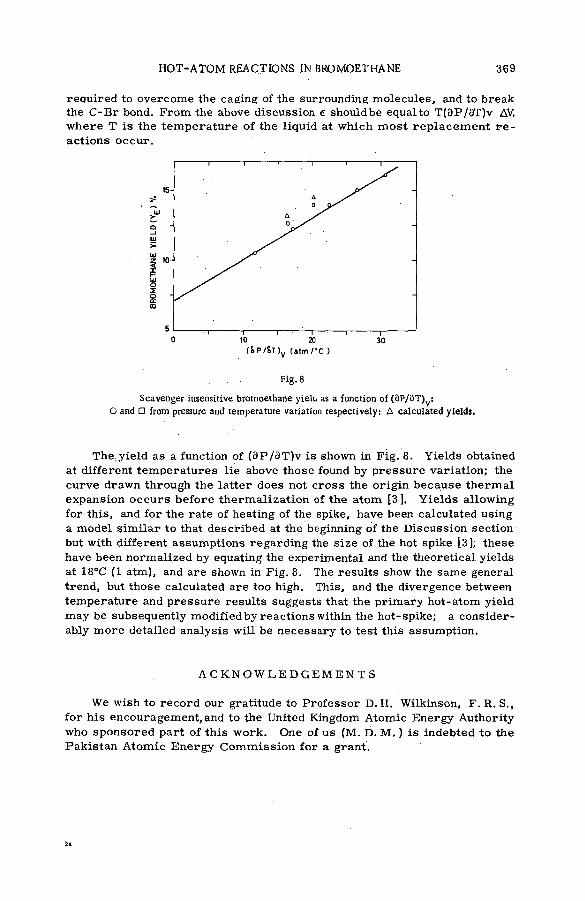
HOT-ATOM REACTIONS IN BROMOETHANE 3 6 9
required to overcome the caging of the surrounding molecules, and to break the C-Br bond. From the above discussion e shouldbe equal to T(3P/3T)v ДУ, where T is the tem perature o f the liquid at which m ost rep lacem ent r e actions occur.
. . Fig- 8
Scavenger Insensitive bromoethane yield as a function of (5P/3T)v:
О and □ from pressure and temperature variation respectively: Д calculated yields.
The yield as a function of (3P/3T)v is shown in Fig. 8. Yields obtained at different temperatures lie above those found by pressure variation; the curve drawn through the la tter does not cross the origin because therm al expansion occurs before therm alization of the atom [3]. Y ie ld s allow ing for this, and fo r the rate of heating of the spike, have been calculated using a model s im ilar to that described at the beginning of the Discussion section but with different assumptions regarding the size of the hot spike [3]; these have been normalized by equating the experimental and the theoretical yields at 18°C (1 atm), and are shown in Fig. 8. The results show the same general trend, but those calculated are too high. This, and the divergence between temperature and pressure results suggests that the primary hot-atom yield may be subsequently modified by reactions within the hot-spike; a considerably more detailed analysis w ill be necessary to test this assumption.
A C K N O W L E D G E M E N T S
We wish to record our gratitude to Professor D.H. Wilkinson, F. R. S., fo r his encouragement, and to the United Kingdom Atom ic Energy Authority who sponsored part o f this work. One of us (M. D. M. ) is indebted to the Pakistan Atom ic Energy Commission fo r a grant.

370 A.J. COLE et al.
R E F E R E N C E S
[1 ] (a ) FO X, M. and LIBBY, W . F . , J . c h e m . Phys. 2 0 (1 9 5 2 ) 4 8 7 . ■
(b) GOLDHABER, S . , CHIANG, R .S . H. and WILLARD, I .E . , J. Am er. chem. Soc. 73 (1 9 5 1 ) 2271 .
( c ) GOLDHABER, S. and WILLARD, J . E . . J . A m er. ch em . S o c . 7 4 (1 9 5 2 ) 3 18 .
(d) RICE, W. E. and WILLARD, J . E . , J . A m er. ch em . Soc. 7 5 (1 9 5 3 ) 6156 .
(e ) AD ITYA, S. and WILLARD, J . E . , J . A m er. ch em . Soc. (1 9 5 7 ) 3 3 6 7 .
(f ) ROY, J .C . , WILLIAMS, R. R. and HAMILL, W. H . , J . A m er. chem . Soc. 76 (1 9 5 7 ) 327Й.
[2 ] MALLINSON, J . H. , MILLER, G. E. and SHAW, P .F .D . , R a d io ch im . A cta 1_ (1 9 6 3 ) 136.
[ 3 ] SHAW, P .F .D . , R a d io ch im . A cta 1_( 1963) 177.
[4 ] LIBBY, W. F . , J . A m er. ch e m . S oc. 6 9 (1 9 4 7 ) 2523 .
[ 5 ] HARBOTTLE, G. and SUTIN , N. , J . phys. C h em .. 6 2 (1 9 5 8 ) 1 344 .
[ 6 ] CHIEN, I. C . W. and WILLARD, J. E . , J. A mer. ch em . Soc. 77 (1 9 5 5 ) 3 4 4 1 .
[ 7 ] HARRIS, W. E. , in C h e m ica l Effects of N uclear Transform ations I_ IAEA, Vienna (1 9 6 1 ) 229.
[ 8 ] WEXLER, S. and ANDERSON, G. R. , J . chem . Phys. 33 (1 9 6 0 ) 1325.
[ 9 ] MILMAN, M. and SHAW, P .F .D . , J . ch em . Soc. (1 9 5 7 ) 1325.
[1 0 ] HOLMES, D. K. and LE1BFRIED, G ., J . appl. Phys. 31 (1 9 6 0 ) 1046.
[ 1 1 ] Van LINT, V .A . J . , SC H M ITT, R A. and SUFFREDINI, C. S . , Phys. Rev. 121 (1 9 6 1 ) 1457.
[1 2 ] SEIT Z , F . , and KOEHLER, J . S . , in Solid S tate Physics 2 (SEIT Z , F. and TURNBULL, IX В. I. , E d s.),
A ca d e m ic Press I n c . , New York (1 9 5 6 ) 3 07 .
[1 3 ] BRIDGMAN, P. W . , Proc. A m er. A cad . Arts and S ci. 4 9 (1 9 1 3 ) 1.
[ 1 4 ] LAMB, H . , Phil. M ag. 4 5 (1 9 2 3 ) 2 5 8 .
[1 5 ] For review , see SEMENOV, N. N ., Som e problem s of C h em ical K inetics and R eactivity , Pergamon
Press, London (1 9 5 8 ) 2 2 8 . .
[ 1 6 ] BYW ATER, S. and STEA CIE, E. W. R . , I . ch em . Phys. 19 (1 9 5 1 ) 3 2 6 .
[ 1 7 ] SC H M ITZ, H . , SCHUMACHER, H .J . and JAGER, A . , Z . phys. C h em . , Lpz. 19 (1 9 5 1 ) 3 26 .
[ 1 8 ] BLADES, A .T . and MURPHY, G. W . , J . A m er. ch em . Soc. 74 (1 9 5 2 ) 6 2 1 9 .
[1 9 ] KNIGHT. B . , MILLER. G. E; and SHAW, P. F. D . . J . inorg. nucl. C h em . 23 (1 9 6 1 ) 15.
[ 2 0 ] (a ) LEVEY, G. and WILLARD, J . E . , J . A m er. ch em . S o c . 7 4 (1 9 5 2 ) 6161 .
(b) MILMAN, M . and SHAW, P .F .D . , ' J . c h e m . Soc. (1 9 5 7 ) 1 303 . See also [ l c ] and [ I f ] ,
[2 1 ] For sum m ary, see TROTM AN-DICKINSON, A. F ., Gas K in etics, Buttcrworths, London (1 9 5 5 ) 4 9 1 .
[ 2 2 ] LIBERATORE, L .C . and WIIG, E. O . , J . ch e m . Phys. 8 (1 9 4 0 ) 165 and 3 4 9 . .
[ 2 3 ] LIBBY. W. F . , J . ch e m . Phys. 8 (1 9 4 0 ) 3 48 .
[ 2 4 ] MILLER, J . M . , GRYDER, J .W . and DODSON, R .W . , I . ch e m . Phys. 18 (1 9 5 0 ) 5 7 9 .
24*
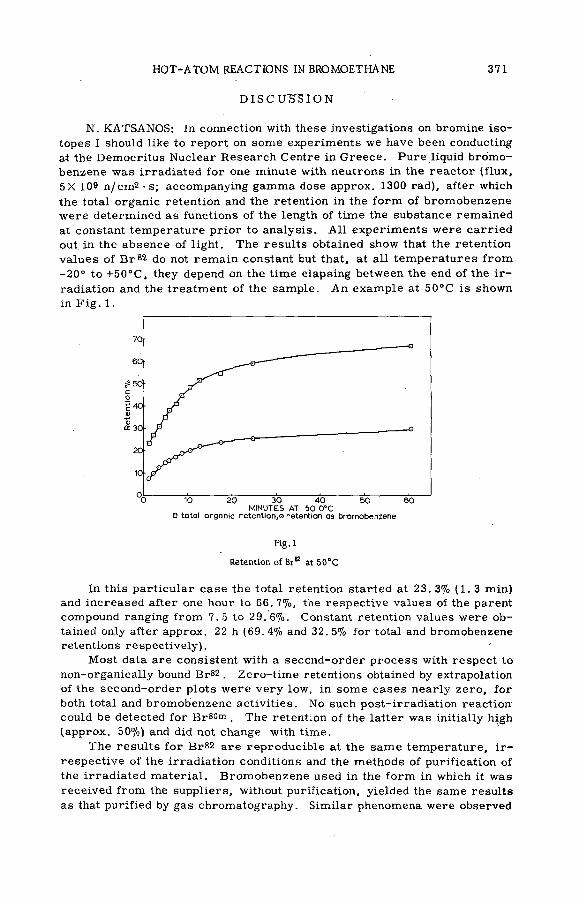
HOT-ATOM REACTIONS IN BROMOETHANE 371
D I S C U S I O N
N. KATSANOS: In connection with these investigations on bromine isotopes I should like to report on some experiments we have been conducting at the Democritus Nuclear Research Centre in Greece. Pure liquid bro'mo- benzene was irrad iated fo r one minute with neutrons in the reac tor (flux, 5X 109 n/cm2 • s; accompanying gamma dose approx. 1300 rad), after which the total organic retention and the retention in the form of bromobenzene w ere determined as functions of the length of tim e the substance remained at constant temperature p rior to analysis. A ll experim ents were carried out in the absence of light. The results obtained show that the retention values of B r® do not rem ain constant but that, at a ll tem peratures from -20° to +50°C, they depend on the tinje elapsing between the end of the i r radiation and the treatment of the sample. An example at 50°C is shown in F ig . 1.
MINUTES AT 50 0°C В total organic retention, о retention as bromobenzene
Fig. 1
Retention of Br82 at 50eC
In this particu lar case the total retention started at 23. 3% (1. 3 min) and increased after one hour to 66.1%, the respective values of the parent compound ranging from 7. 5 to .29. 6%. Constant retention values were obtained only after approx. 22 h (69. 4% and 32. 5% for total and bromobenzene retentions respectively). '
Most data are consistent with a second-order process with respect to non-organically bound B r82 . Zero-tim e retentions obtained by extrapolation of the second-order plots w ere ve ry low, in some cases nearly zero, fo r both total and bromobenzene activities. No such post-irradiation reaction could be detected fo r B r8°ni . The retention of the la tter was in itia lly high (approx. 50%) and did not change with time.
The resu lts fo r Br82 are reproducible at the same tem perature, i r respective of the irrad iation conditions and the methods of purification of the irrad iated m ateria l. Bromobenzene used in the form in which it was received from the suppliers, without purification, yielded the same results as that purified by gas chromatography. Sim ilar phenomena were observed

372 A .J . COLE et a l .
with m ixtures o f bromobenzene and n-heptane at 20, 0 and -28. 3°C, but the final retention values were lower than those in the pure system. This was almost en tirely due to the lower retention in the form of the parent compound.
A ll these phenomena are probably related to the newly discovered 6.2- min B r 82m # However, our experiments produced another unexpected result, namely that the post-irradiation reaction is temperature-dependent, being faster at low er tem peratures. Thus, quite apart from the effects which could be due to Br 82m I,T>B r82, it seems clear that some other phenomenon is also responsible fo r our results. We explain the negative temperature coefficient of the reaction by the hypothesis that radiobromine form s a charge-transfer com plex with inactive brom obenzene, analogous to that reported by Keefer and Andrews'1- for B r2 and bromobenzene. The experimentally observed rate constant, k, would then be a function of the equilibrium constant, K, fo r the complex formation and the true rate constant, k1, fo r the subsequent reaction of the complex. Since К w ill normally decrease as tem perature r is es , it is only necessary to assume that the increase in k' is of such a magnitude as to be outweighed by the decrease in K .
A .G . MADDOCK: I should like to make one rem ark in connection with the comparison of HBr and B r2 scavenging of C2 H 5 . Under the conditions relevant to your argument, it is surely important that the entropies as well as the enthalpies of activation are sim ilar. Have you any data on this point?
P . SHAW: No. I have assumed that the rates of reaction between ethyl radicals and HBr or B r2 in the liquid phase would be sim ilar to those found in the gas phase. It would be interesting to hear comments on this problem from P ro fessor W illard, who has used HBr as scavenger in the radiolysis of propyl brom ides. ,
J. W ILLARD: Our studies on the radiolysis of liquid П-С3Н7ВГ indicate that the rates at which rad icals react with dissolved H Br or B r 2 at room temperature are sim ilar. There is evidence that the activation energy for the reaction of radicals with HI is only 2 kcal/mole or so higher than fo r their reaction with I2 . HC1 is also known to be a good radical scavenger.
* J . A m er. ch em . S oc. 72 (1 9 5 0 ) 4 6 7 7 .

ВЗАИМОДЕЙСТВИЕ АТОМОВ ОТДАЧИ УГЛЕРОДА-14 В БИНАРНЫХ СИСТЕМАХ, СОДЕРЖАЩИХ
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
Л .П . ФИРСОВА, М .Ф . БАРАК АТ , М. Ф О РЫ С ЬиАн.Н . НЕСМЕЯНОВ МОСКОВСКИЙ ГОСУДАРСТВЕННЫЙ УНИВЕРСИТЕТ,М ОСКВА
СССР
Abstract — Résumé — Аннотация — Resumen
THE INTERACTION OF С 14 RECOIL ATOMS IN BINARY M IXTURES. A study was m ade of the inter
m olecu lar distribution of a c tiv ity in C14 -lab elled products, formed as a result o f the interaction of С 14 atom s
with com ponents of binary liquid m ixtures contain ing h e te ro cy clic com pounds. P articu lar attention was paid
to th e system s in d o le -a n ilin e , in d o le-p yrid in e , a -p ic o l in e - to lu e n e , a -p ic o le n e -a n i lin e , e t c . D ata w ere
obtained on the relation between the rad iochem ical yields of labelled products and the concentration of com po
nents in dilu te solutions and in com parable concentrations of substances in m ixtures.
Analysis of the experim en tal results for an additive system enabled us to d eterm in e the deviations from
th e ad d itiv ity p rin cip le . T h e deviation s observed w ere interpreted as th e result o f the p articip atio n o f the
second com ponent of the system in thé prim ary and secondary reactions accom panying nuclear transformations.
In considering the role of the second com ponent, we took into account the variations in its predominant reaction
m echanism s in various concentration ranges. Sp ecial attention was paid to the possibility of an interm olecular
tran sfer o f th e en e rg y o f a ra d ia tio n sh ield , and a lso o f th e m e c h a n ism o f th e corre sp o n d in g p ro ce sse s.
INTERACTION ENTRE ATOMES « C DE RECUL ET COMPOSANTS DE MÉLANGES BINAIRES. Les
auteurs étudient la distribution in term o lécu laire de la rad ioactiv ité dans les produits m arqués avec 14C qui se
form ent à la suite de l 'in te ra ctio n entre le c a rb o n e -1 4 et les com posants de m élan ges liquides binaires'
contenant des substances h étéro cy cliq iies. Ils e xam in en t notam m en t les systèm es in d o le -a n ilin e , in d o le -
pyxidine, a -p ic o l in e - to lu o l , a -p ic o l in e -a n il in e , e t c . Us ont obtenu des données sur la re la tio n en tre le
rendem ent radiochim ique en produits m arqués e t la concentration des com posants pour des solutions diluées et
pour des m élanges où les deux substances ont des concentrations com parables. .
En traitant les données expérim en tales selon un schém a additif, ils ont con staté des écarts par rapport à
la rè g le d ’a d d itiv ité . C e s é c a rts sont con sid érés c o m m e résultant du fa it que le d e u x iè m e co m p o san t du
systèm e p a rtic ip e au x réactio n s p rim aires e t secon d aires qui a cco m p a g n e n t le s tran sform ations n u clé a ire s .
En exam in an t le rô le du d eu xièm e com posant, les auteurs tiennent co m p te des p articu larités que présentent
les m écan ism es dom inants des réactio n s subies par c e com posant dans les divers in terv alles d e con cen tratio n .
Ils étudient n otam m ent la possibilité.d 'un transfert d ’én erg ie in term o lécu laire , ainsi que les m écanism es des
processus correspondants.
В З А И М О Д Е Й С Т В И Е А Т О М О В О Т Д А Ч И У Г Л Е Р О Д А - 1 4 В Б И Н А Р Н Ы Х С М Е С Я Х . И с с л е д о в а н о м еж м олекуляр н о е р асп р еделен и е акти вн ости в м ечен н ы хп о С 14 п родуктах, о б р азую щ ихся в р е з у л ь т а т е в за и м о д е й с т в и я а т о м о в у г л е р о д а - 1 4 с ко м п о н ен там и би нарны х ж идких с м е с е й , содер ж ащ и х ге те р о ц и к л и ч е ск и е соеди н ен и я. В ч а с т н о с т и , и зуч ены с и ст е м ы и н д о л - ани ли н , и н до л— пиридин, а -п и к о л и н —т о л у о л , а -п и к о л и н — анилин и т . д . П олучены дан н ы е по за в и с и м о с т и р ад и о х и м и ч е ск и х в ы х о д о в м е ч е н ы х п р одукто в от концентрации ко м п о н ен тов в о б л а с т и р а зб а в л е н н ы х р а с т в о р о в и в о б л а с т и с р а в н и м ы х ко н ц ен тр ац и й в е щ е с т в в с м е с я х .
При о б р аб о т к е эк сп е р и м е н т а л ь н ы х р е з у л ь т а т о в по адд и ти вн ой с х е м е б ш ш за ф и к си р о ван ы отклон ен и я о т п р ави ла а д д и т и в н о с т и . Н аб лю давш и еся отклон ен и я т р а к т у ю т ся к а к р е з у л ь т а т у ч а с т и я в т о р о го ко м п о н ен та с и с т е м ы в первичны х и втор и чн ы х р еак ц и я х со п р о во ж даю щ их ядер н ы е п р евр ащ ен и я. При обсуж ден и и роли в т о р о го ком понента у ч и ты ваю тся о т л и чия преобладаю щ и х м е х а н и зм о в е г о реакций в разли чн ы х концентрационных и н те р ва ла х . Р а с с м а т р и в а ю т с я в ч а с т н о с т и в о з м о ж н о с т ь м е ж м о л ек у л я р н о й п ер едач и эн е р ги и р адиационной за щ и т ы , а т а к ж е м е х а н и зм ы с о о т в е т с т в у ю щ и х п р о ц е с с о в .
INTERACCION DE LOS ATOMOS DE RETROCESO DEL *4С EN MEZCLAS BINARIAS. Los autores han
investigado la distribución in term olecu lar de la actividad en los productos m arcados con 14C , formados com o
373

374 Л .П. ФИРСОВА и др.
resultado de la in teracció n d e átom os d e 14C con los com ponen tes de m e z cla s binarias en estado líquido que
contienen compuestos h eterocíclico s. Prestaron particular atención a los sistemas indol-anílina, índol-piridina,
a -p ic o lin a -to lu e n o , a -p ic o lin a -a n ilin a , e t c . O btuvieron datos sobre los rendim ientos radioqufm icos de los
productos m arcados en función de la co n ce n tra ció n d e los com ponen tes en soluciones diluidas y en m ezclas
con co n cen tracio n es com p arab les de sustancias.
El análisis de los resultados exp erim en tales corresp ondientes a un sistem a de ad ició n , p erm itió a los
autores determ inar las desviaciones con respecto a la norm al. Estas desviaciones se consideraron com o resultado
de la p a rticip a ció n d e un segundo co m p o n en te d el sistem a en las re a c c io n e s p rim arias y secund arias que
acom pañan a las transform aciones nu cleares. Al exam in ar el papel desem peñado por e l segundo com ponente,
tuvieron en cuenta la variación de los m ecanism os de reacción predominantes en distintos intervalos de concen
tración. Examinaron en particular la posibilidad de una transferencia interm olecular de la energfa de resistencia
a la radiólisis, asf com o los m ecanismos de los procesos correspondientes.
При сравнении экспериментальных результатов по удельным активностям и выходам меченныхатомамиуглерода-14 продуктов облучения ряда бинарных смесей, содержащих гетероциклы, с величинами радиохимических выходов* и удельных активностей, рассчитанными по аддитивной схеме, наблюдались отклонения от линейности изменения с изменением концентрации растворов. В зависимости от характера добавки эти отклонения обнаруживались или только в области высоких концентраций или же были значительными уже в области разбавленных растворов.
Указанные отклонения'являются результатом участия молекул второго компонента как в первичных, так и во вторичных химических процессах, сопровождающих ядерные превращения. Одной из основных причин отклонений от аддитивности в системах с гетероциклическими соединениями так же, как и в других случаях, являются процессы межмолекулярной передачи энергии возбуждения или заряда и радиационной защиты.
Отличия концентрационных зависимостей р .х . выходов и удельных активностей в различных системах связаны с отличием механизмов преобладающих процессов с участием молекул добавок, т.к. именно этим определяются интервалы концентраций, в которых наиболее резко проявляется влияние второго компонента на величины р .х . выходов, удельных активностей и радиационной выживаемости. Действие веществ, принимающих участие в резонансных передачах энергии, например, как и действие акцепторов радикалов проявляется в полной мере даже при низких концентрациях, тогда как передача энергии при соударении становится заметной лишь при сравнимых концентрациях обоих компонентов.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Исследовались системы индол —анилин, индол —пиридин, индол — о'-пико- лин, а-пиколин — толуол, «-пиколин — анилин, р—пиколин — пиперидин, а-пико-' лин —3,5-диметилпиразол (ДМП) и й'-пиколин — 1,1-дифенил —2-пикрилгидра- зил (ДФПГ) в различных концентрационных интервалах. Образцы весом1 — 2 г запаивали после обезгаживания в кварцевые ампулы и облучали на реакторе при потоке нейтронов 0,87-Ю 13 н/см2сек. Время облучения составляло 200 часов. Сожжением навесок определялись суммарные актив
* Д а л е е принято со к р а щ е н и е: р . х . в ы х о д о в .
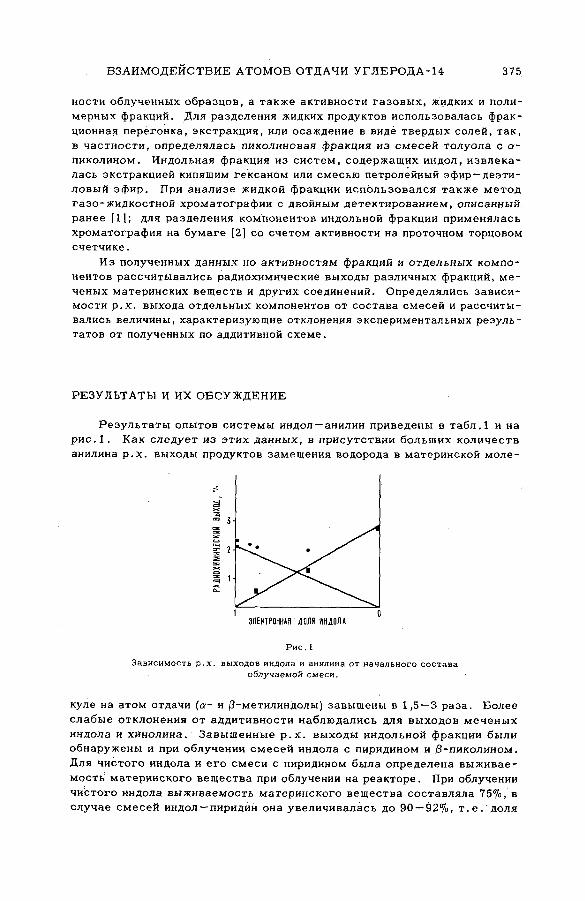
ВЗАИМОДЕЙСТВИЕ АТОМОВ ОТДАЧИ УГЛЕРОДА-14 375
ности облученных образцов, а также активности газовых, жидких и полимерных фракций. Для разделения жидких продуктов использовалась фракционная перегонка, экстракция, или осаждение в виде твердых солей, так, в частности, определялась пиколиновая фракция из смесей толуола с а - пиколином. Индольная фракция из систем, содержащих индол, извлекалась экстракцией кипящим гексаном или смесью петролейный эфир—деэти- ловый эфир. При анализе жидкой фракции использовался также метод газо-жидкостной хроматографии с двойным детектированием, описанный ранее f i l ; для разделения компонентов индольной фракции применялась хроматография на бумаге [2] со счетом активности на проточном торцовом счетчике.
Из полученных данных по активностям фракций и отдельных компонентов рассчитывались радиохимические выходы различных фракций, меченых материнских веществ и других соединений. Определялись зависимости р .х. выхода отдельных компонентов от состава смесей и рассчитывались величины, характеризующие отклонения экспериментальных результатов от полученных по аддитивной схеме.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Результаты опытов системы индол —анилин приведены в табл.1 и на рис.1. Как следует из этих данных, в присутствии больших количеств анилина р .х . выходы продуктов замещения водорода в материнской моле-
ЗЛЕКТРОННАЯ ДОЛЯ ИНДОЛА
Рис.1
Зависимость р .х . выходов индола и анилина от начального состава о б л у ч а е м о й смеси.
куле на атом отдачи (а - и (3-метилиндолы) завышены в 1,5 — 3 раза. Более слабые отклонения от аддитивности наблюдались для выходов меченых индола и хинолина. Завышенные р .х . выходы индольной фракции были обнаружены и при облучении смесей индола с пиридином и (8-пиколином. Для чистого индола и его смеси с пиридином была определена выживаемость материнского вещества при облучении на реакторе. При облучении чистого индола выживаемость материнского вещества составляла 75%, в случае смесей индол —пиридин она увеличивалась до 90 — 92%, т .е . доля
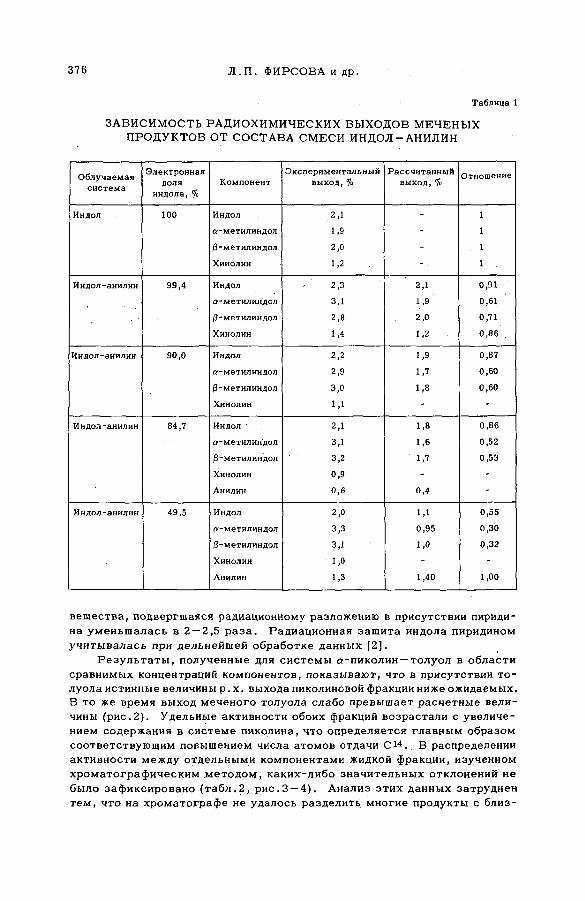
376 Л.П. ФИРСОВА и др.
ЗАВИСИМОСТЬ РАДИОХИМИЧЕСКИХ ВЫХОДОВ МЕЧЕНЫХ ПРОДУКТОВ ОТ СОСТАВА СМЕСИ ИНДОЛ-АНИЛИН
Таблица 1
Облучаемаясистема
Электронная доля
индола, %Компонент
Экспериментальный выход, %
Рассчитанный в ы хо д ,%
Отношение
Индол - 100 Индол . 2,1 - 1û—метилиндол 1,9 - 1/3-метилиндол 2,0 - 1Хинолин 1,2 . - ■ 1
Индол-анилин 99,4 Индол - 2,3 2,1 0,01¿у-метилиндол 3,1 1,9 0,61
/3-метилиндол 2,8 2,0 0,71
Хинолин 1,4 1,2 . 0,86 .
Индол-анилин 90,0 Индол 2,2 1,9 0,87
а-метилиндол 2,9 1 Д 0,60
/3-метилиндол 3,0 1,8 0,60
Хинолин 1,1 - '
Индол-анилин 84,7 Индол 2,1 1,8 0,86а-метилин'дол 3,1 1,6 0,52 .
jS-метилиндол ' 3,2 1,7 0,53
Хинолин 0,9 -
Анилин 0,6 0,4 '
Индол-анилин 49,5 Индол 2,0 1 Д 0,55
о'-метилиндол 3,3 0,95 0,30
|3-метилиндол 3,1 1,0 0,32
Хинолин ■ 1,0 - -
Анилин 1,3 1,40 1,00
вещества, подвергшаяся радиационному разложению в присутствии пиридина уменьшалась в 2 — 2,5 раза. Радиационная защита индола пиридином учитывалась при дельнейшей обработке данных [2].
Результаты , полученные для системы а-пиколин—толуол в области сравнимых концентраций компонентов, показывают, что в присутствии толуола истинные величины р .х . выхода пиколиновой фракции ниже ожидаемых. В то же время выход меченого толуола слабо превышает расчетные величины (рис.2). Удельные активности обоих фракций возрастали с увеличением содержания в системе пиколина, что определяется главным образом соответствующим повышением числа атомов отдачи C l4. В распределении активности между отдельными компонентами жидкой фракции, изученном хроматографическим методом, каких-либо значительных отклонений не было зафиксировано (т а б л .2, рис.3 — 4). Анализ этих данных затруднен тем, что на хроматографе не удалось разделить многие продукты с близ-

ВЗАИМОДЕЙСТВИЕ АТОМОВ ОТДАЧИ УГЛЕРОДА-14 377
' Рис . 2
Зависимость р .х . выходов фракций в системе толуол — а-пиколин от концентраций а-пиколина:
т толуол .о пиколиновая фракция
кими точками кипения, образующиеся из толуола и пиколина, например а-пиколин и п-, м-ксилолы .
Не нарушалась аддитивность выходов большинства меченых продуктов, полученных при облучении смесей о-пиколина с ДМП и пиперидином (табл.2, 3). В присутствии пиперидина, правда, падал р .х . выход бензола,по-видимому, за счет вторичного восстановления его в среде с большим содержанием водорода и значительно изменялся р .х . выход газовой фракции. Несколько увеличивался р .х . выход газов и в смесях с анилином. Для основных жидких меченых продуктов этих смесей каких-либо закономерных отклонений от линейности не отмечено (рис.5, 6).
Наиболее резкие отклонения от линейных зависимостей р .х . выходов от концентрации характерны для смесей о—пиколин —ДФПГ. В этом случае заметно меняется фракционное распределение активности, причем значительное увеличение выхбдов меченых высококипящих продуктов происходит за счет почти полного падения р .х . выходов материнских веществ и других соединений с точками кипения ниже 180°С. При концентрациях ДФПГ выше 0,005 мол % из низкокипящих жидких продуктов были обнаружены лишь следы толуола и бензол, выход которого в присутствии ДФПГ практически не изменялся (табл. 2). Действия добавок ДФПГ легко может быть интерпретировано как действие эффективного акцептора радикалов, что согласуется с ранее высказанным предположением, что большинство меченых продуктов при облучении пиколинов образуется за счет реакций в тепловой области [1]. Это относится прежде всего к продуктам замещения водорода в материнских молекулах на атомы отдачи С 14. Образование продуктов замещения азота и углерода кольца, по-видимому, более вероятно в области высоких энергий, чем и объясняется относительно слабое влияние добавок акцептора радикалов на р .х . выход этих вещ еств.
Результаты , полученные для смесей û'-пиколин с ДМП и пиперидином вполне укладываются в рамки представлений о конкурирующих добавках, не обладающих резко выраженным специфическим действием. В то же
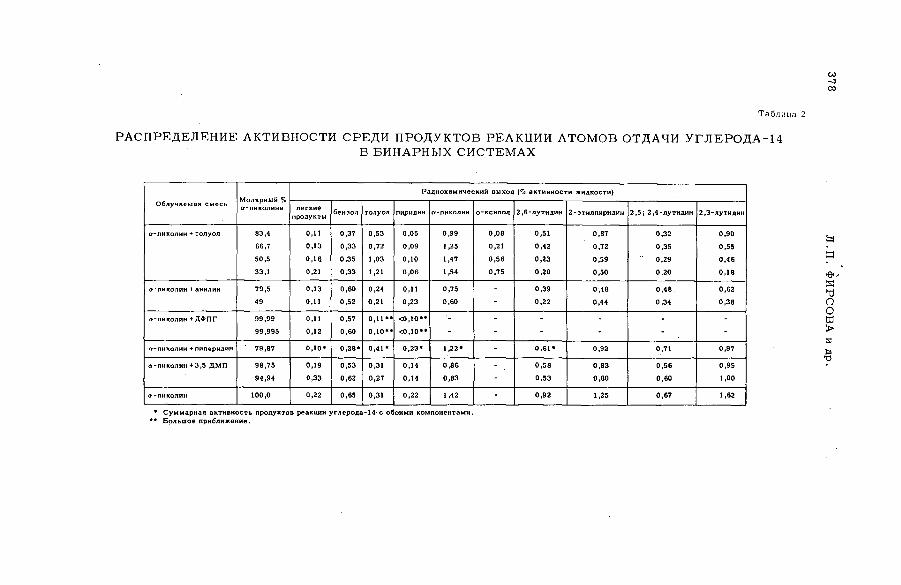
РАСП РЕ Д Е ЛЕ Н И Е АКТИВНОСТИ СРЕДИ П РО Д УК ТО В РЕАКЦИИ АТО М О В О ТДАЧИ У ГЛ Е Р О Д А -1 4 В БИНАРНЫ Х С И С ТЕ М АХ
Т а б л и ц а 2
О б л у ч а е м а я с м е с ьМ о л я р н ы й % а -г ш к о л и н а
Р а д и о х и м и ч е с к и й в ы х о д (Те а к т и в н о с т и ж и д к о ст и )
л е г к и еп р од ук ты
б е н з о л т о л у о л пиридин <у-пиколин о - к с и л о л 2 ,6 - л у т и д и н 2 -эт и лп и р н д и н 2 ,5 ; 2 ,4 - л у т и д и н 2 ,3 - л у т и д и н
а - п и к о л и н * т о л у о л 83,4 0,11 0 ,3 7 0 ,5 3 0 ,0 5 0 ,9 9 0 ,0 8 0 ,5 1 0 ,8 7 0 ,3 2 0 ,9 0
66,7 0 ,1 3 0 ,3 3 0 ,7 2 0 ,0 9 1 ,25 0 ,2 1 0 ,4 2 0 ,7 2 0 ,3 5 0 ,5 5
5 0 ,5 0 ,1 6 0 ,3 5 1 ,03 0 ,1 0 1 ,47 0 ,5 6 0 ,2 3 0 ,5 9 0 ,2 9 0 ,4 6
33,1 0 ,21 0 ,3 3 1,21 0 ,0 6 1 ,54 0 ,7 5 0 ,2 0 0 ,5 0 0 ,2 0 0 ,1 8
о -п и к о л и н + ан и ли н 7 9 ,5 0 ,1 3 0 ,6 0 0 ,2 4 0 ,11 0 ,7 5 - 0 ,3 9 0 ,4 8 0 ,4 8 0 ,6 2
49 0 ,11 0 ,5 2 0 ,2 1 0 ,2 3 0 ,6 0 - 0 ,2 2 0 ,4 4 0 ,3 4 0 ,3 8
о -п и к о л и н + Д Ф П Г 9 9 ,9 9 0 ,11 0 ,5 7 0 ,1 1 * * < 0 ,1 0 * * - - - - -
9 9 ,9 9 5 0 ,1 2 0 ,6 0 0 ,1 0 * * < 0 ,1 0 * * - - - - ' -
o’- п и к оли н + пипери ди н 7 9 ,8 7 0 ,1 0 * 0 ,2 8 * 0 ,4 1 * 0 ,2 3 * 1 ,2 2 * - 0 ,6 1 * 0 ,9 2 0,71 0 ,9 7
о - п и к о л и н + 3 ,5 Д М П 9 8 ,7 5 0 ,1 9 0 ,5 3 0 ,3 1 0 ,1 4 0 ,8 6 - . 0 ,5 8 0 ,8 3 0 ,5 6 0 ,9 5
9 4 ,9 4 0 ,3 3 0 ,6 2 0 ,2 7 0 ,1 4 0 ,8 3 - 0 ,5 3 0 ,8 0 0 ,6 0 1 ,00
о -п и к о л и н 1 00 ,0 0 ,2 2 0 ,6 5 0 ,3 1 0 ,2 2 1,42 0 ,9 2 1 ,25 0 ,6 7 1 ,62
* С у м м а р н а я а к т и в н о с т ь п р о д у к т о в р еа к ц и и у г л е р о д а - 1 4 - с о б о и м и к о м п о н е н т а м и . * * Б о л ь ш о е п р и б л и ж е н и е .
378 Л
.П.
ФИ
РСО
ВА
и
др.
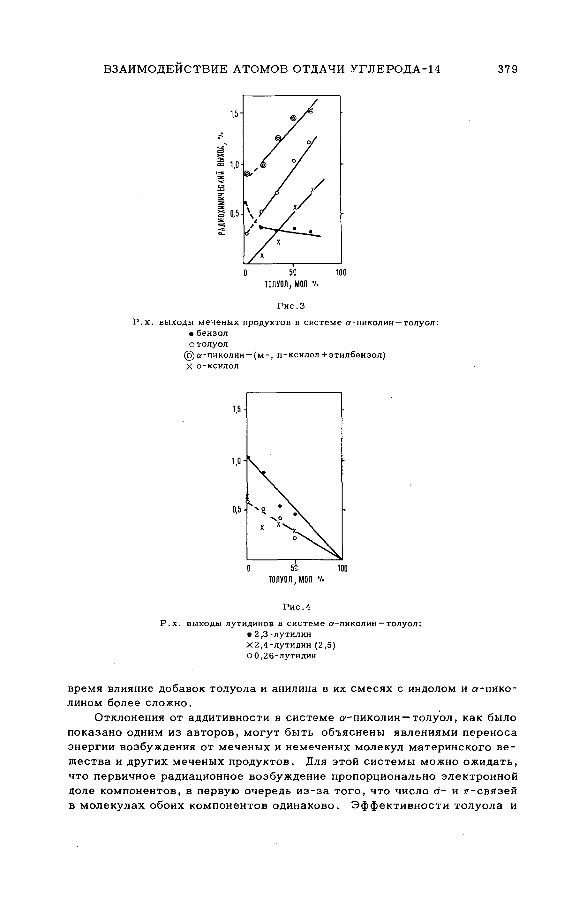
ВЗАИМОДЕЙСТВИЕ АТОМОВ ОТДАЧИ УГЛЕРОДА-14 379
ТОЛУОЛ, МОЛ V.
Рис.З
Р .х . выходы меченых продуктов в системе tf-пиколин —толуол: • бензол о толуол
(о)о ’-пиколин —(м - , п-ксилол + этилбензол)X о-ксилол
Рис . 4
Р .х . выходы лутидинов в системе û ’- п и к о л и н —толуол:• 2 ,3-лутилин Х2,4-лутидин (2,5)00,26-лутидин
время влияние добавок толуола и анилина в их смесях с индолом и а-пико- лином более сложно.
Отклонения от аддитивности в системе а-пиколин — толуол, как было показано одним из авторов, могут быть объяснены явлениями переноса энергии возбуждения от меченых и немеченых молекул материнского вещества и других меченых продуктов. Для этой системы можно ожидать, что первичное радиационное возбуждение пропорционально электронной доле компонентов, в первую очередь из-за того, что число ст- и ж-связей в молекулах обоих компонентов одинаково. Эффективности толуола и
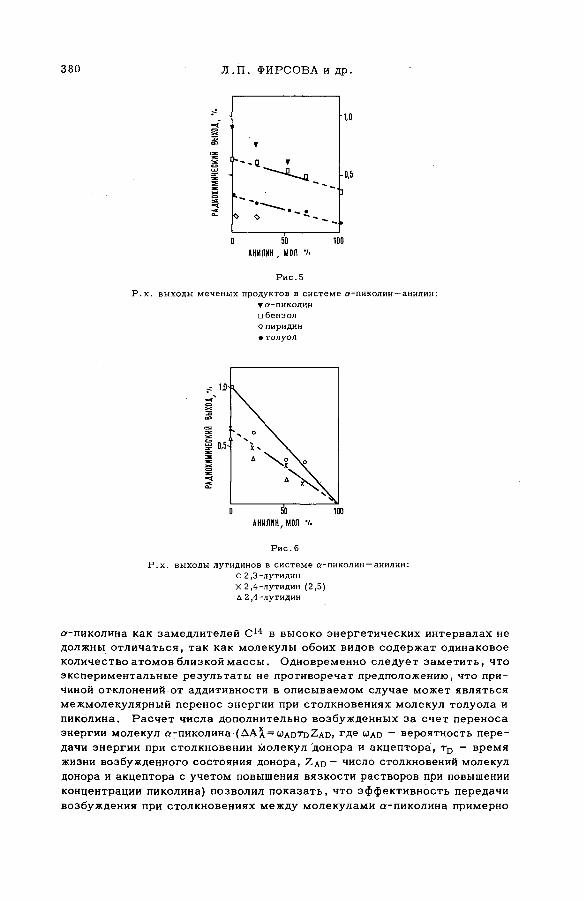
380 Л.П. ФИРСОВА и др.
Рис. 5
Р .х . выходы меченых продуктов в системе а-пиколин — анилин:▼ О '-П И К О Л И Н
□ бензол О пиридин • толуол
Р и с . 6Р .х . выходы лутидинов в системе а-пиколин—анилин:
о 2,3-лутидин Х2,4-лутидин (2,5) д 2,4-лутидин
се-пиколина как замедлителей С14 в высоко энергетических интервалах не должны отличаться, так как молекулы обоих видов содержат одинаковое количество атомов близкой массы. Одновременно следует заметить, что экспериментальные результаты не противоречат предположению, что причиной отклонений от аддитивности в описываемом случае может являться межмолекулярный перенос энергии при столкновениях молекул толуола и пиколина. Расчет числа дополнительно возбужденных за счет переноса энергии молекул «-пиколина (Д А а = uadtdZad, где идп - вероятность передачи энергии при столкновении молекул донора и акцептора, т0 - время жизни возбужденного состояния донора, Z ad — число столкновений молекул донора и акцептора с учетом повышения вязкости растворов при повышении концентрации пиколина) позволил показать, что эффективность передачи возбуждения при столкновениях между молекулами а-пиколина примерно

ВЗАИМОДЕЙСТВИЕ АТОМОВ ОТДАЧИ УГЛЕРОДА-14 381
на порядок ниже вероятности передачи при столкновениях молекул пиколи- на и толуола. Расчет был основан на предположении, что химические реакции между молекулами донора и акцептора в системе отсутствуют. В связи с чем, время жизни возбужденных состояний и вероятность переноса энергии в системе являются величинами независимыми от концентраций. Функцией концентраций а также вязкости растворов является лишь число межмолекулярных соударений.
Было установлено, что ход изменений истинных экспериментально наблюдаемых величин р .х . выходов с концентрацией укладывается в предложенную схему обработки, учитывающую передачу энергии возбуждения при столкновении между молекулами, если справедливо предположение, что ДАд является функцией числа соударений.
Изменение р .х . выхода донора может быть представлено в виде AHd= ДАр à + ДА£, где ДА| - величина учитывающая изменение радиационной выживаемости молекул, б - эффективность пометки передавших радиационное возбуждение молекул, ДА£ - величина, учитывающая изменение числа меченых молекул, возбужденных в реакциях пометки атомами отдачи С 14. Расчет удельных активностей (1= Б /N, где N - число молекул компонентов с учетом его радиационного разложения и концентрационной зависимости радиационной выживаемости) при изменении р .х . выходов за счет передачи энергии между молекулами одного вида или разных видов дает возможность предсказывать ход изменений удельных активностей в бинарных системах*.
Правда, для системы ог-пиколин — толуол это объяснение может быть не единственным. Если предположить возможность вторичных реакций между возбужденными мечеными молекулами одного компонента (А ) с мо- лёкулами другого компонента (В ), можно получить аналогичную картину изменения выходов в бинарных системах. Единственный критерий, позволяющий различить оба процесса (последовательные химические реакции и перенос энергии возбуждения) и связанный с симбатностью изменений р .х . выходов и удельных активностей, в исследованной системе применить сложно из-за указанных выше причин (при отсутствии переноса энергии ДАд = (JabtaZ ab, где идв — сечение реакции взаимодействия).
В результатах, полученных для систем, содержащих индол, наибольший интерес вызывает возможность сравнения данных по концентрационному изменению радиационной выживаемости материнских молекул и выходов меченых продуктов.
Отсутствие симбатности в изменениях этих величин указывает, в частности, на зависимость эффективности передачи избыточной.энергии при соударениях от уровня возбуждения [2,3 ] или на возможность передачи возбуждения другими путями. ,
В системах индол —анилин, индол —пиколин, индол —пиридин при интерпретации результатов необходимо учитывать также различия в массах молекул. Работами Вольфганга и других авторов [3] было'установлено, что в случае многокомпонентных систем возможно изменение энергетического спектра атомов отдачи за счет различной эффективности замедления их молекулами добавок. Это сопровождается также изменением суммарного
* Более подробно схема обработки данных по бинарным смесям описана в работе Л .П . Фирсовой, посланной в журнал "Радиохимия".

382 Л .П. ФИРСОВА и др.
РАСПРЕДЕЛЕНИЕ АКТИВНОСТИ ПО ФРАКЦИЯМ В ОБЛУЧЕННЫХ БИНАРНЫХ СМЕСЯХ
Таблица 3
СмесьСостав
<*-пиколина мол %
Радиохимический выход, %
газжидкаяфракция
полимер
Толуол — а-пиколин 83,4 3,0 96,8 0,266,7 4,6 95,4
50,5 3,2 96,5 0,3
33,1 3,8 96,2 0,2
Анилин —л-пиколин 79,5 0,82 99,0 0,249,0 3,3 99,5 0,231,1 7,5 92,3 0,2
ДФ ПГ — а-пиколин 99,990 1,3 98,7 0,199,995 1,2 98,7 0,1
Пиперидин—а-пиколин 79,9 6,1 93,7 0,2
ДМП — <*-пиколин . 98.8
94.9
2,22,0
97,6
97,8
0,20,2
выхода горячих реакций и выходов отдельных компонентов. При введении дополнительных упрощающих задачу условий, если принять, что сечение (и) не зависит от энергии и распределение атомов отдачи по энергиям описывается уравнением n(E) = -l/œE, на основании общего выражения для выхода реакций горячих атомов отдачи
Е1 .R = N Z J ' u j ( E ) Z j n ( E ) d E
можно показать, что в бинарной системе
R = N In E g / E i ,ai Z i + а г2 2
где N — число атомов отдачи; Z i ; Z 2 ~ число столкновения и a i ; — логарифмический декремент затухания для первого и второго компонентов соответственно. Если принять, что
п (Е ) = - — ехр
то концентрационная зависимость суммарного выхода в бинарной смеси имеет более сложный вид

ВЗАИМОДЕЙСТВИЕ АТОМОВ ОТДАЧИ УГЛЕРОДА-14 383
R = N “ lZ i j t « 2 Z 2aj Zj +Q-2Z2
р ц1г1*и1г гj e a¡ Zí +a2Z¡
E.
Приближенный расчет, проведенный без учета изменения п(Е) при реакциях горячего атома, показал, что экспериментальные данные в системах, содержащих индол не могут быть объяснены только замедляющим действием второго компонента или конкурирующими реакциями с его молекулами. По-видимому, и в этом случае для объяснения отклонения от правил аддитивности целесообразно использовать предположение о переносе энергии или последовательных реакциях в бинарных системах.
[1] Б А Р А К А Т М .Ф ., ФИРСОВА Л .П . , НЕСМЕЯНОВ А .Н . , Радиохимия, 6, вып. 5, 1964.[2] фИРСОВА JI. П ., ФОРЫ С Ь М ., Радиохимия, 6, вып. 5, 1964.[3] E S T R U P P .J . , W O LFG ANG R . , J. A m . Chem. Soc. 82, N 11, 2665, 1960.
Л И Т Е Р А Т У Р А


CHEMICAL BEHAVIOUR OF С11 IN LIQUID HYDROCARBONS*
A . F. V O IG T , D .E . CLARK A N D F .G . MESICH IN STITU TE OF A T O M IC RESEARCH AND DEPARTMENT OF CHEMISTRY,
IOW A STATE UNIVERSITY, AM ES, IO W A, UNITED STATES OF AMERICA
Abstract — Résumé — Аннотация — Resumen
CHEMICAL BEHAVIOUR OF С11 IN LIQUID HYDROCARBONS. Carbon-11 is produced by the C l2(y, n)Cu reaction in the bremsstrahlung beam o f a 70 MeV electron synchrotron. As target materials, liquid hydrocarbons with 5 and 6 carbons have been used, including normal, branched and alicyclic pentanes and hexanes as well as benzene. The behaviour o f C 11 has been studied by gas chromatographic separation
of the products, counting the C 11 in the gas stream in a cell placed in a w ell-type scintillation counter.In each experiment yields of different products were compared to the yield of acetylene as an internal
standard and either to a tantalum monitor or to the total C 11 produced as measured in the entire sample before separation. The flow counters were calibrated in terms of total C 11 produced in experiments in which the complete sample was burned toC 02 and passed through a flow counter.
Our earlier experiments were concerned only with the gaseous products that have now been well characterized for the various target molecules under different dosage conditions. Current experiments on product molecules similar in size to the target have proved very helpful in deciding on mechanisms for recombination of recoil atoms. Of particular interest is the yield of product with one carbon more than the target,the result of an addition reaction. The location of the additional atom on a target molecule having several types of addition sites gives information regarding the process itself. •
When the recoil atom is slowed to an energy at which it is possible for a bond to be formed, at least temporarily, the extra energy which the С 11 atom brings into the system may cause bond rupture elsewhere within the activated complex usually leading to a two-carbon product. I f the complex is able to hold together without rupturing, an additional product will result. Thus comparison of the yields of two-carbon compounds,
acetylene, ethylene and ethane, and the additional products provides valuable information regarding the energy at which stable bond formation can occur, and the nature o f the C u -containing group entering into the reaction.
COMPORTEMENT CHIMIQUE DE u c DANS LES HYDROCARBURES LIQUIDES. Le carbone-11 est produit par la réaction 12C(y, n)n C dans le rayonnement de freinage d'un synchrotron à électrons de 70 MeV. Comme cible, les auteurs ont utilisé des hydrocarbures liquides à 5 et 6 atomes de carbone, notamment les pentanes et hexanes normaux, ramifiés et alicycliques ainsi que le benzène. Ils ont étudié le comportement de UC en séparant les produits par chromatographie gazeuse et en comptant le 11C dans le courant de gaz à
l'intérieur d’une cellule placée dans un compteur à scintillations à puits.Lors de chaque expérience, ils ont comparé les rendements en divers produits au rendement en acétylène,
utilisé comme étalon interne, et, soit à un dispositif de contrôle au tantale, soit à la mesure de la quantité totale de 11C produite dans l'ensemble de l'échantillon avant séparation. Ils ont étalonné les compteurs à courant gazeux en tenant compte de la quantité totale de 11C qui est produite pendant les expériences au cours desquelles le carbone de l'échantillon est entièrement transformé en CQ par combustion, et qui passe
dans un compteur de ce type.Les expériences que les auteurs avaient faites antérieurement ne portaient que sur les produits gazeux
qui sont maintenant bien déterminés pour diverses molécules cibles dans différentes conditions de dosage. Les expériences en cours sur des molécules de produit d'une dimension semblable à celle de la cible se sont révélées très utiles pour déterminer les mécanismes de recombinaison des atomes de recul. Le rendement en produit comportant un atome de carbone de plus que la ciblé, qui est le résultat d’une réaction d'addition, revêt un
* Contribution No. 1622, Work was performed in the Ames Laboratory o f the United States Atomic Energy Commission,
38525

386 A .F . VOIGT et al.
in t é r ê t p a r t ic u l ie r . L 'e m p l a c e m e n t d e l ’ a t o m e s u p p lé m e n t a i r e d a n s u n e m o l é c u l e c i b l e p o ss é d a n t p lu s ie u rs
e m p l a c e m e n t s p o s s ib le s p o u r l ’ a d d i t io n d e c e t a t o m e fo u r n it d e s r e n s e ig n e m e n t s su r l e p ro c e s su s l u i - m ê m e .
L o rsq u e l e r a le n t is s e m e n t d e l ’ a t o m e d e r e c u l e s t su ff isa n t p o u r q u 'u n e l ia is o n p u is se se fo rm e r , a u m o in s
p r o v is o ir e m e n t , l 'é n e r g i e s u p p lé m e n t a i r e q u e l ’ a t o m e d e n C c o m m u n iq u e a u s y s t è m e p e u t p ro v o q u e r a i l le u r s
d a n s l e c o m p le x e a c t i v é u n e ru p tu r e d e l ia i s o n d o n n a n t h a b it u e l le m e n t n a is s a n c e à un p ro d u it à d e u x a to m e s
d e c a r b o n e . S i l e c o m p l e x e p e u t s e m a in t e n ir sa n s r u p t u r e , i l e n r é s u lt e r a un p ro d u it d ’ a d d i t io n . A in s i , e n
c o m p a r a n t l e s re n d e m e n ts e n c o m p o s é s à d e u x a t o m e s d e c a r b o n e ( a c é t y lè n e , é t h y lè n e e t é th a n e ) à c e l u i en
p ro d u its d 'a d d it io n , o n o b tie n t d e s r e n s e ig n e m e n ts p r é c ie u x sur l ’ é n e r g ie q u i p e r m e t l a fo rm a tio n d 'u n e lia is o n
s t a b le e t l a n a tu r e d u g ro u p e c o n t e n a n t 1 1 C q u i e n tr e d a n s l a r é a c t io n ,
Х И М И Ч Е С К А Я Х А Р А К Т Е Р И С Т И К А У Г Л Е Р О Д А - 1 1 В Ж И Д К И Х У Г Л Е В О Д О Р О Д А Х . У г л е
р о д - 1 1 о б р а з у е т с я в р е з у л ь т а т е р е а к ц и и C i 2 («yf n ) C H в п у ч к е т о р м о з н о г о и з л у ч е н и я в с и н х р о
т р о н е с э н е р г и е й э л е к т р о н о в 7 0 М э в . В к а ч е с т в е в е щ е с т в а м и ш е н и п р и м е н я л и с ь ж и д к и е
у г л е в о д о р о д ы с п я т ь ю и ш е с т ь ю а т о м а м и у г л е р о д а , в т о м ч и с л е н о р м а л ь н ы е р а з в е т в л е н н ы е
и а л и ц и к л и ч е с к и е п е н т а н ы и г е к с а н ы , а т а к ж е б е н з о л . П о в е д е н и е С 1 1 и з у ч а л о с ь м е т о д о м
г а з о х р о м а т о г р а ф и ч е с к о г о р а з д е л е н и я п р о д у к т о в , с ч е т а а к т и в н о с т и С 1 1 в г а з о в о м п о т о к е
в к а м е р е , п о м е щ е н н о й в с ц и н т и л л я ц и о н н ы й с ч е т ч и к к а н а л ь н о г о т и п а .
В к а ж д о м э к с п е р и м е н т е в ы х о д ы р а з л и ч н ы х п р о д у к т о в с р а в н и в а л и с ь с в ы х о д о м а ц е т и л е н а
к а к в н у т р е н н е г о с т а н д а р т а , а т а к ж е л и б о с п о к а з а н и я м и р е г и с т р а т о р а т а н т а л а , л и б о с о б щ и м
к о л и ч е с т в о м о б р а з о в а в ш е г о с я С И , и з м е р е н н ы м в о в с е м о б р а з ц е п е р е д р а з д е л е н и е м . П р о
т о ч н ы е с ч е т ч и к и к а л и б р о в а л и с ь п р и у с л о в и и о б р а з о в а н и я в с е г о С и в х о д е э к с п е р и м е н т о в , в
п р о ц е с с е к о т о р ы х в е с ь о б р а з е ц с ж и г а л с я , п р е в р а щ а я с ь в С О 2 , и п р о х о д и л ч е р е з п р о т о ч н ы й
с ч е т ч и к .
Н а ш и б о л е е р а н н и е э к с п е р и м е н т ы б ы л и с в я з а н ы т о л ь к о с и с п о л ь з о в а н и е м г а з о о б р а з н ы х
п р о д у к т о в , к о т о р ы е в н а с т о я щ е е в р е м я х о р о ш о з а р е к о м е н д о в а л и с е б я в о т н о ш е н и и м о л е к у л
р а з л и ч н ы х в е щ е с т в м и ш е н и п р и р а з н ы х з н а ч е н и я х д о з ы . Т е к у щ и е э к с п е р и м е н т ы с п р о и з в о д
н ы м и м о л е к у л а м и , п о д о б н ы м и п о р а з м е р у м о л е к у л а м м а т е р и а л а м и ш е н и , о к а з а л и с ь о ч е н ь
п о л е з н ы м и п р и и з у ч е н и и в о п р о с а о м е х а н и з м а х р е к о м б и н а ц и и а т о м о в о т д а ч и . О с о б ы й и н т е
р е с п р е д с т а в л я е т в ы х о д п р о д у к т а с о д н и м а т о м о м у г л е р о д а б о л ь ш е п о с р а в н е н и ю с м и ш е н ь ю ,
ч т о я в л я е т с я р е з у л ь т а т о м н о в о й р е а к ц и и . Р а с п о л о ж е н и е д о п о л н и т е л ь н о г о а т о м а в м о л е к у л е
в е щ е с т в а м и ш е н и , к о т о р о е х а р а к т е р и з у е т с я н а л и ч и е м н е с к о л ь к и х д о п о л н и т е л ь н ы х т о ч е к , д а е т
и н ф о р м а ц и ю о т н о с и т е л ь н о с а м о г о п р о ц е с с а .
П р и з а м е д л е н и и а т о м а о т д а ч и д о в е л и ч и н ы э н е р г и и , п р и к о т о р о й в о з м о ж н о у с т а н о в л е н и е
с в я з и , п о к р а й н е й м е р е в р е м е н н о , и з л и ш н я я э н е р г и я , к о т о р у ю п р и н о с и т в с и с т е м у а т о м С И ,
м о ж е т п р и в е с т и к р а з р ы в у с в я з и г д е - л и б о в д р у г о м м е с т е в п р е д е л а х а к т и в и р у е м о г о к о м
п л е к с а , ч т о о б ы ч н о п р и в о д и т к о б р а з о в а н и ю д в у х у г л е р о д н о г о п р о д у к т а . Е с л и к о м п л е к с м о ж е т
б ы т ь с о х р а н е н б е з р а з р ы в а , т о о б р а з у е т с я д о п о л н и т е л ь н ы й п р о д у к т . Т а к и м о б р а з о м , с р а в
н е н и е в ы х о д о в д в у х у г л е р о д н ы х с о е д и н е н и й , а ц е т и л е н а , э т и л е н а и э т а н а и д о п о л н и т е л ь н ы х
п р о д у к т о в п р е д о с т а в л я е т ц е н н у ю и н ф о р м а ц и ю о т н о с и т е л ь н о э н е р г и и , п р и к о т о р о й в о з м о ж н о
о б р а з о в а н и е у с т о й ч и в о й с в я з и , и х а р а к т е р а с о д е р ж а щ е й С П г р у п п ы , в с т у п а ю щ е й в р е а к ц и ю .
C O M P O R T A M IE N T O Q U IM IC O D E L Н С EN H ID R O C A R B U R O S L IQ U ID O S . S e o b tu v o c a r b o n o - 1 1 p or
l a r e a c c ió n 1 г С ( у , п ) и С e n e l h a z d e r a d ia c ió n d e f r e n a d o d e un s in c ro tró n d e 7 0 M e V q u e a c e le r a e le c t r o n e s .
C o m o b la n c o s e u t i l iz a r o n h id r o c a r b u r o s l íq u id o s d e 5 y 6 á t o m o s d e c a r b o n o , e n tr e e l lo s p e n ta n o s y h e x a n o s
n o r m a le s , r a m i f i c a d o s y a l i c f c l i c o s , a s f c o m o b e n c e n o . S e e s t u d ió e l c o m p o r ta m ie n to d e l 1 1 С se p a r a n d o lo s
p ro d u c to s p o r c r o m a t o g r a f ía e n fa s e g a s e o s a y c o n t a n d o e l 1 1 С e n l a c o r r ie n t e g a se o s a d e u n a c e l d a c o lo c a d a
e n un c o n t a d o r d e c e n t e l l e o d e t ip o c a v i d a d . ,
En c a d a e x p e r im e n t o se c o m p a r a r o n lo s re n d im ie n to s d e lo s d ife r e n t e s p ro d u cto s a l re n d im ie n to d e a c e t i
le n o u t i l iz a d o c o m o p a tró n in te rn o , a s f c o m o o b ie n a l d e un m o n it o r d e t á n t a lo o b ie n a l 1 1 С t o t a l p ro d u c id o
y d e t e r m in a d o e n l a m u e s tr a in t e g r a a n t e s d e l a s e p a r a c ió n . L o s c o n t a d o r e s d e f lu jo se c a l ib r a r o n e n fu n c ió n
d e l n C t o t a l p r o d u c id o e n e x p e r im e n t o s e n q u e e l c a r b o n o d e l a m u e s t r a s e t r a n s fo r m ó t o t a lm e n t e e n C 0 2
p o r c o m b u s t ió n y s e h iz o p a s a r p o r un c o n t a d o r d e l . m e n c io n a d o t ip o .
L o s e x p e r im e n t o s a n t e r io r e s d e lo s a u t o r e s v e r s a r o n e x c l u s i v a m e n t e s o b r e lo s p r o d u c t o s g a s e o s o s q u e
a h o r a s e h a l l a n b ie n c a r á c t e r iz a d ó s p a r a d i fe r e n t e s m o lé c u la s b la n c o e n d is t in t a s c o n d ic io n e s d e d o s i f ic a c ió n .
L o s e x p e r im e n to s e n c u rso so b re m o lé c u la s a n á lo g a s e n ta m a ñ o a la s d e l b la n c o h a n c o n tr ib u id o a l a a c la r a c ió n
d e lo s m e c a n is m o s d e r e c o m b in a c i ó n d e lo s á t o m o s d e r e t r o c e s o . P a r t ic u la r interés r e v i s t e e l r e n d im ie n t o
d e p ro d u c to s c o n un á to m o d e c a r b o n o m á s q u e e l b la n c o , q u e r e s u lta n d e una r e a c c ió n d e a d ic ió n . L a l o c a l i

BEHAVIOUR OF C“ IN LIQUID HYDROCARBONS 387
z a c ió n d e l á to m o a d ic io n a l e n u n a m o lé c u la b la n c o q u e te n g a v a r io s lu g a r e s d e a d ic ió n p ro p o rc io n a in fo rm a c ió n
so b re l a n a t u r a le z a d e l p ro c e so .
C u a n d o e l á t o m o d e r e t r o c e s o e s m o d e r a d o y su e n e r g ía d is m in u y e h a s t a e l p u n to e n q u e e s p o s i b le l a
f o r m a c ió n d e un e n l a c e , a l m e n o s t e m p o r a l , e l e x c e s o d e e n e r g ía q u e e l á t o m o l l C c e d e a l s i s t e m a p u e d e
o r ig in a r l a ru p tu ra d e o tros e n la c e s d e l c o m p le jo a c t i v a d o , lo q u e s u e le d a r o r ig e n a un p ro d u c to d e do s á to m o s
d e c a r b o n o . S i e l c o m p le jo e s c a p a z d e c o n s e r v a r su in te g r id a d , s u e le r e s u lta r un p ro d u c to d e a d ic i ó n . P o r
ta n to , l a c o m p a r a c ió n d e l r e n d im ie n t o d e c o m p u e s to s d e dos á to m o s d e c a r b o n o ( a c e t i l e n o , e t i l e n o y e ta n o )
y d e p r o d u c to s d e a d ic i ó n p r o p o r c io n a u n a in fo r m a c ió n v a l i o s a s o b r e l a e n e r g í a a l a q u e p u e d e n fo r m a r s e
e n l a c e s e s t a b l e s , y s o b r e l a n a t u r a l e z a d e l g r u p o q u e c o n t i e n e e l U C y q u e p a r t i c i p a e n l a r e a c c i ó n .
The reco il chemistry o f carbon has received much attention in recent years. The development o f gas chromatographic methods by which com pounds containing the 20. 4 min C11 can be separated and the C11 determined in the short period of time available before it decays has made this an extrem ely interesting and active fie ld of hot atom research.
The behaviour o f carbon was the subject o f a session at the previous IAEA Symposium on the Chemical Effects of Nuclear Transform ations [1] and of several recent reviews [2, 3]. These references provide an excellent review of previous work and a complete bibliography.
This paper describes the recent work done in the Am es Laboratory using C11 as produced by the bremsstrahlung from an electron synchrotron operated at 47 or 70 M eV by the reaction C12(y , n)Cn . The irradiated compounds were liquid hydrocarbons with 5-7 carbon atoms, including straight and branched chain aliphatics/ alicyclics and benzene. E arlier papers from this Laboratory [4-6] presented results in which only the gaseous products were measured; the present paper reports- continued work of this type and also on products of s ize equal to and la rger than the irradiated compound.
E XPERIM ENTAL
M ateria ls and Apparatus
Research grade hydrocarbons obtained from the Phillips Petroleum Co. w ere used without additional purification. The samples w ere irrad iated in sm all P y rex bulbs containing about 0. 2 m l o f the liquid. These bulbs w ere filled on a vacuum rack using the freeze-thaw method of degassing. The 8. 1-h Taieom activity produced by the reaction Та181(т, n)Ta180m was used as a gam m a-ray monitor. For this purpose a tantalum monitor fo il was wrapped round the bulb. The bulb ,and monitor were placed in a holder which fitted into a re-entrant tube in the synchrotron's doughnut-shaped acceleration chamber [7]. The electron beam impinged on a lead target d irectly in front o f the sample. Thus the sample was subjected, in addition to the bremsstrahlung beam, to a somewhat degraded beam of electrons which penetrated this target.
In recent experiments, as an additional monitor, the total samples in the ir bulbs w ere m easured in a standard geom etry above a 2. 5X2. 5-cm N a l(T l) crystal. This is a better monitor than the tantalum fo il since the latter is not always located in exactly the same position with respect to the

388 A. F. VOIGT et al.
beam and the bulb. The total C11 activity also reflects changes in the beam intensity in the same way as the products, while the lon ger-lived T a l80m tends to smooth out any such changes. Decay curves of the total СП monitor showed only the C11 activity in the period from 14-90 min after the i r radiation. The reaction 0 16(т,огп)С11 is also observed in the synchrotron but was found to contribute <5% o f the C*1 a c tiv ity fro m a f i l le d bulb.
The chromatographic columns used varied according to the object of particular experiments. In a number of cases it was desired to separate ethane and ethylene, and fo r this separation s ilica ge l (14-20 mesh) was found to be the most e ffective column packing. In the experiments on the higher boiling components it was necessary to use other packings that did not give the best separations fo r the gases. Silicone o il (DC 703) and Apiezon greases L and M were used in these cases, 30 wt. % on 45-60 mesh firebrick in 10- to 20-ft columns.
Counting was done as in previous work using a v ia l through which the gas stream flowed that was placed in a well-type 5X5-cm Nal(T l) scintillation crystal. In order to reduce the background we have recently incorporated a discrim inator in the counting circu it that elim inates a ll signals c o rre s ponding to gam ma-ray energies less than 0. 4 MeV. This has proved quite effective in reducing background and hence improving the sensitivity, p ermitting the observation o f products of low yield.
The counting rates w ere displayed both on scalers and recording rate m eters, and results were obtained by allowing the sca ler to sum the counts over a peak, by determining the area on a recorder chart with a planimeter, or by using both procedures. Agreem ent between the two was very good.
D osim etry
Radiation o f severa l types contributes to the damage occurring in the samples. The accelerated electrons are not stopped by the lead target but pass through it and irrad ia te the sample. The brem sstrahlung produced in the target also irradiate the sample and while bremsstrahlung of all energies produce radiation effects, the production of C11 is lim ited to those with energies above the (y, n) threshold, 18. 7 M eV. Characteristic X -rays o f lead and other elements also contribute to the radiation field while not adding to the activity produced.
Another contributor to radiation damage is the recoiling C11 atom itself. The energy of these recoiling atoms can be estimated from published crosssection curves fo r the y, n reaction [8], which show peaks at energ ies o f 21. 6, 22, 23. 2 and 25. 4 M eV. Since the difference between the energy o f the exciting gamma-rays and the threshold w ill be divided between the neutrons and the C11 nuclei, the latter w ill have reco il energies of 0. 24, 0. 27,0. 37 and 0. 56 respective ly from gamma-rays of the above energies. Since a considerable contribution from gam m a-rays of h igher energies is also observed it is estimated that the average re co il energy o f the C11 nucleus is ~ 0 . 5 MeV. Prom the number of C11 nuclei produced, 109 to 1010 in most experiments, their contribution to the total radiation damage can be estim ated at 10'5 to 10"6 eV per molecule, which is very sm all compared to the total radiation. It is, however, localized in the regions where the C11 atom

BEHAVIOUR OF С11 IN LIQUID HYDROCARBONS 389
comes to rest and may contribute m ore to the effect of radiation on the r e actions than its m ere magnitude would suggest.
A characteristic of a synchrotron is that the radiation occurs in bursts. Pu lses o f electrons are produced with a duration o f about 4 X 1 O'8 s, and a frequency of 59 per s corresponding to a tim e interval between bursts of1. 7 XIQ-2 s. The secondary processes are essentially instantaneous so that the dose-rate during bursts is about 4X105 tim es the average dose-rate . The number of electrons per burst is of the order of 109, nearly all of which pass through the sample. Electrons of this energy w ill produce about 6200 ion pairs per mm in a liquid with the density of water. From these numbers the dose to the system can be estimated as 1.1 X1015 eV per burst or1.6X1024 eV per min during the burst. In the 0.2 g sample this is 8X1024 eV g"1 m in'1. The tim e-averaged dose-rate, which is the dose-rate usually considered, is thus about 2ХЮ 19 eV g '1m in”1.
In addition to this estim ate, severa l independent methods w ere used to measure the dose delivered to the samples. The Fricke dosimeter was firs t adapted to the sm all scale o f these samples with a 103 с Co60 source. The results using volumes of 0. 2 m l and m icroce lls fo r the spectrophotom eter w ere found to be reproducible to about 10%.
In the synchrotron beam the F ricke dosim eter was at the upper lim it of its usefulness. Very short irradiations, 5-10 s, were necessary in order to rem ain below 4X 104 rad, which is about the la rges t dose m easurable with this system. Because the instantaneous dose-rate is very high, much greater than the average ra te, it was necessary to use a low er value fo r G (Fe+3) than the usually áccepted one o f 15. 5 [9] . ANDERSON 1Ю] has studied the Fricke dosimeter under sim ilar conditions and obtained a value o f ~ 10 fo r G (Fe+3) at a dose-rate of 1025 eV g"1 min"1. Since the instantaneous dose-rate in this system approached this figure the value o f 10 was used for G(Fe+3). A series of measurements of the Fe+3 yield in irradiations lasting 5 to 10 s gave ~ 1. 2 X1019 eV g"1 min*1 as the over-a ll dose rate.
Samples o f a cobalt glass that is useful as a dosim eter at high dose- rates were obtained from the Bausch and Lomb Optical C o ., and calibrated at the cobalt-60 source against the Fricke dosim eter. A curve of changes in the optical density of the glass versus dose obtained with the Fricke dosim eter agreed with published resu lts fo r this glass [H ] . Samples o f the glass w ere irradiated at the sample position in the synchrotron and measured to obtain the dose-rate. The resu lt was approxim ately 1019 eV g"1 m in '1.
Observation o f the loss of iodine from a solution of iodine in 2, 2- dimethylbutane and the published value of 4. 0 for G(- I 2) in this system [6,12] led to another independent estimate o f the dose-rate as (l-2 )X 1 0 19e V g 'im in "1.
A conservative comparison o f these estim ates leads to a value o f 2X1019 eV g’ 1 min-1 fo r the average dose-rate or 3X10'3 eV per molecule per min. Since most irradiations lasted from 5 to 20 min, the total in tegrated dose was in the range 0. 01 to 0. 06 eV per m olecule fo r a beam of typ ical intensity.
At this leve l radiation damage certain ly contributes to the total yields o f labelled products. Experiments to determine the extent o f contribution o f radiation damage to the tota l y ie ld a re included in th is p rogram m e.

390 A . F. VOIGT et al.
In order to express the yield of the labelled compounds in terms of the total C11 produced, it is necessary to calibrate the flow-counter with a measurement o f total C11. This involves some procedure fo r converting the entire irradiated sample to a gas, and passing all this gas or an aliquot through the counting cell. In an earlier paper [4 ] experiments were reported in which carbon pellets were irradiated and burned to carbon dioxide, which was passed through the counter. A carbon disc monitor was used to relate the extent o f irradiation in these experiments with the usual irradiation of a compound followed by separation. In these experiments the yield of acetylene at room tem perature was found to be 15% from n-hexane and 14% from cyclohexane expressed as a percentage o f the tota l C11 produced.
Experim ents have since been conducted in which a sample o f cy c lo hexane was irradiated as in the usual separation experiment. The sample bulb was broken in a stream of helium and the cyclohexane was oxidized over hot copper oxide. A fte r rem ova l o f water vapour the tota l gas from the oxidation was collected, and aliquots were taken to be passed through the chromatograph and counter. In this way 10-12 check runs could be made fo r each irradiation. By comparing these experiments with irradiation of n-hexane by means o f tantalum monitors, the absolute y ie ld o f acetylene from n-hexane was found to be 13. 8± 1%, while that from cyclohexane itself was found to be 10. 8%. In the case of n-hexane this shows reasonable agreement with the ea rlie r result; fo r cyclohexane there is an appreciable difference.
Determination o f absolute yield
RESULTS
Gaseous products from various hydrocarbons
We have continued to examine the yie lds o f gaseous products, p a rticu larly methane and the C2 hydrocarbons from different target molecules: normal and branched hexanes, n-pentane; cyclohexane, cyclopentane and their methyl derivatives; and benzene. These yields have been determined in some cases in the presence o f iodine as a scavenger and as a function o f dose.
In a ll cases the yie ld of acetylene appears to be unaffected by the presence of scavenger and essentially independent of dose. There is some indication that the acetylene y ie ld decreases as the irrad ia tion tim e and dose increase. This is shown in Fig. 1 for the methylcyclopentane system. If this is real, it could be explained as due to gradual reduction o f the acetylene as its exposure to radiation and to the fre e hydrogen produced by radiation is increased. The magnitude of this effect is sm all and does not res tric t the use of acetylene as an internal monitor.
The e ffect o f radiation on the yie lds of methane, ethylene and ethane is noticeable; in the absence of iodine, all these yields increase with dose. Curves for the irradiation of methylcyclopentane, which are typical of other compounds as well, are shown in F igs. 2 and 3. The extent o f increase in
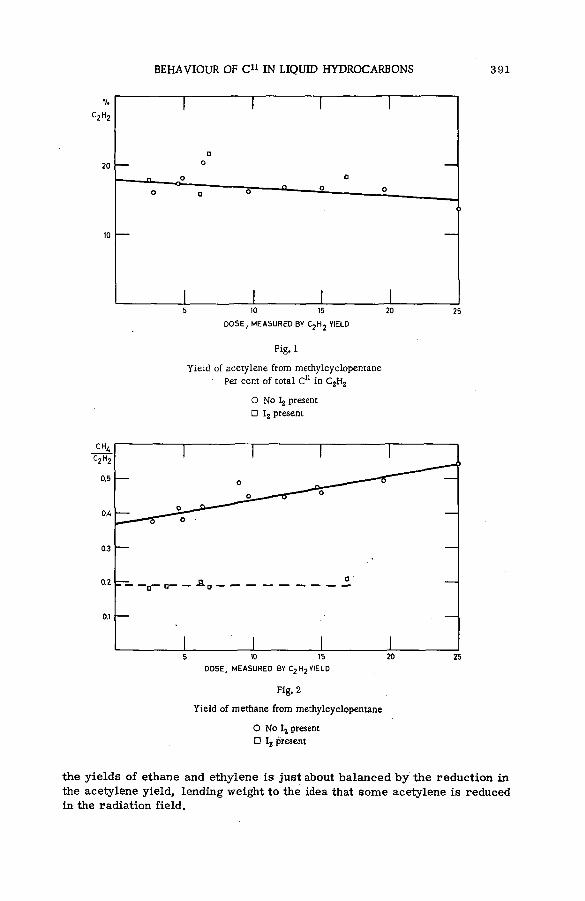
BEHAVIOUR OF C ^ IN LIQUID HYDROCARBONS 391
DOSE, MEASURED BY Cj Hj YIELD
Fig. 1
Yield of acetylene from methylcyclopentane
Per cent of total C11 in C2H2
О No I2 present
□ I2 present
DOSE, MEASURED BY C2 H2 YIELD
Fig. 2
Yield of methane from methylcyclopentane
О No Ij present
□ I2 present
the yields of ethane and ethylene is just about balanced by the reduction in the acetylene yield, lending weight to the idea that some acetylene is reduced in the radiation field.
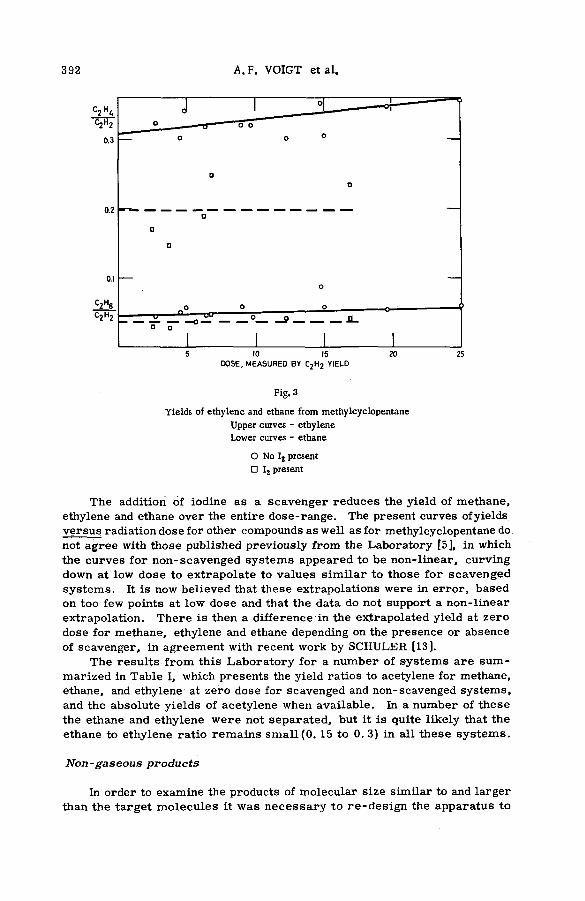
392 A. F. VOIGT et al.
Fig.3
Yields of ethylene and ethane from methylcyclopentane
Upper curves - ethylene
Lower curves - ethane
О No I2 present
□ I2 present
The addition of iodine as a scavenger reduces the yie ld of methane, ethylene and ethane over the entire dose-range. The present curves ofyields versus radiation dose for other compounds as well as for methylcyclopentane do not agree with those published previously from the Laboratory [5], in which the curves fo r non-scavenged systems appeared to be non-linear, curving down at low dose to extrapolate to values s im ila r to those fo r scavenged systems. It is now believed that these extrapolations were in error, based on too few points at low dose and that the data do not support a non-linear extrapolation. There is then a difference in the extrapolated yield at zero dose for methane, ethylene and ethane depending on the presence or absence of scavenger, in agreement with recent work by SCHULER [13].
The results from this Laboratory fo r a number o f systems are summarized in Table I, which presents the yield ratios to acetylene for methane, ethane, and ethylene at zero dose for scavenged and non-scavenged systems, and the absolute yields of acetylene when available. In a number of these the ethane and ethylene w ere not separated, but it is quite like ly that the ethane to ethylene ratio remains sm all (0. 15 to 0. 3) in a ll these systems.
Non-gaseous products
In order to examine the products of molecular size sim ilar to and larger than the target m olecules it was necessary to re-design the apparatus to
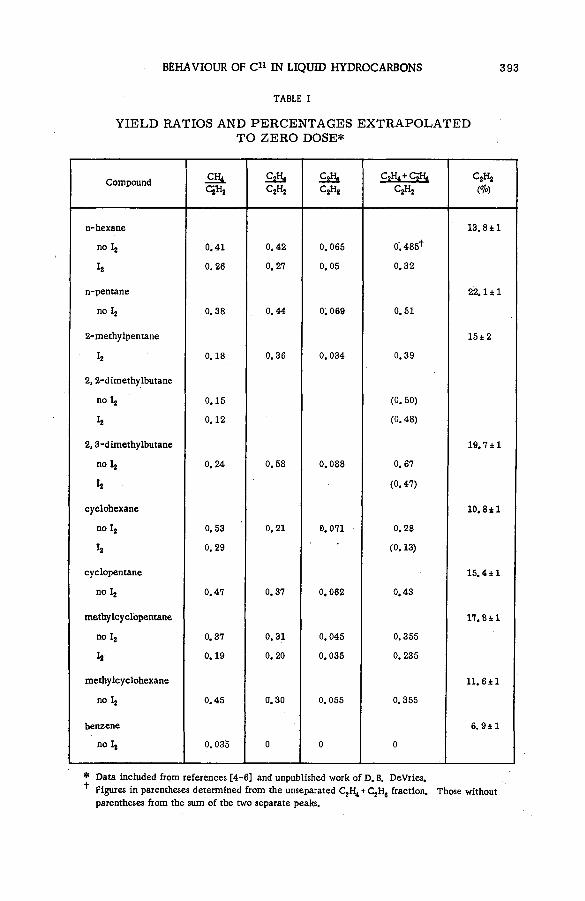
BEHAVIOUR OF С11 IN LIQUID HYDROCARBONS 393
TABLE I
Y IE L D RATIO S AND PE R C E N TAG E S E X T R A P O L A T E D TO ZE RO DOSE*
CompoundCHt
q p 2c a
C 2H2 C 2H2Q H i+ Q H fi
C 2H2C2H 2
<H°)
n-hexane 1 3 .8 *1
no 1¡ 0.41 0.42 0.065 0 .485+
к 0. 26 0.27 0. 05 0.32
n-pentane 22 .1 * 1
no 12 0.38 0.44 0.069 0.51
2-methylpentane 15* 2
h 0.18 0.36 0. 034 0.39
2, 2-dimethylbutane
no I2 0.15 (0. 50)
h 0 .1 2 (0.48)
2, 3-dimethylbutane 19. 7 i 1
no 0.24 0.58 0. 088 0. 67
h (0.47)
cyclohexane 10 .8 1 1
no I2 0.53 0 .21 0. 071 0. 28
h 0. 29 (0.13)
cyclopentane 1 5 . 4 i 1
no I2 0.47 0. 37 0. 062 0.43
methylcyclopentane 1 7 .8 *1
no I2 0.37 0.31 0 .0 4 5 0.355
h 0.19 0 .2 0 0. 035 0. 235
methylcyclohexane 1 1 . 6 * 1
no I2 0 .4 5 a . 30 0 .0 5 5 0.355
benzene 6 .9 * 1
no I2 0. 035 0 0 0
* D a t a i n c lu d e d f r o m r e f e r e n c e s [ 4 - 6 ] a n d u n p u b l is h e d w o r k o f D . B . D e V r i e s .
* F ig u r e s i n p a r e n t h e s e s d e t e r m i n e d f r o m t h e u n s e p a r a t e d C 2 H4 + C 2 H 6 f r a c t i o n . T h o s e w i t h o u t
p a r e n t h e s e s f r o m t h e s u m o f t h e t w o s e p a r a t e p e a k s .

394 A. F. VOIGT et al.
operate at elevated temperatures. The chromatographs were originally built with this in mind and were readily adapted. The flow-counting system presented some problems since condensation could occur in the flow counter. If a radioactive product condenses in the counter, its counting rate would be meaningless compared to that of the other non-condensable components. Since a scintillation counter cannot be operated effectively much above room temperature it was not possible to heat the counting system to prevent condensation. Steps taken to reduce the possibility of condensation, such as a smaller volume for the counting cell and a high flow rate for the helium carrier gas, proved to be effective.
The compounds irradiated so far in this part of the programme have been n-pentane, cyclopentane and benzene. In the cases of the two pentanes the columns employed were DC-703 silicone oil, Apiezon M and Apiezon L on firebrick.
Ten experiments on n-pentane gave the following average yields, as percentages of total C11: methane, 8.4; acetylene, 22.1; ethane plus ethylene,11.3; C3 compounds, 4. 7; C4 compounds, 7. 3; n-pentane, 3. 3; 3-methyl- pentane, 3.3; 2-methylpentane, 5. 9; n-hexane, 26. 2; hexenes, 2.2; total recovery, 94. 7.
In the case of cyclopentane, not all the higher boiling compounds have been identified and our results are somewhat preliminary. In five experiments the following product distribution was observed: methane, 7. 2; acetylene, 15..4; ethane plus ethylene, 6 . 7; C3 compounds, 3. 7; C4 compounds,3.3; C5 compounds, 1. 6 ; cyclopentane, 1. 3; methylcyclopentane, 7. 0; and four peaks of higher boiling components with activities of 23. 7, 10. 7, 11. 5 and 4. 9. The total recovery of C11 was 97%.
Nine experiments on benzene were run with separations on DC-703 and Apiezon L columns. Only four products were observed with yields as follows: methane, 0. 24; acetylene, 6 . 9; benzene, 11. 4; and toluene, 5. 7, representing a total recovery of 24. 2. .
DISCUSSION
The results of the experiments on gaseous products appear to be in general agreement with similar experiments from other laboratories and describable by the theories presented in recent review articles [2, 3]. The relatively large yield of acetylene in all the systems is a point of considerable interest. While it is possible that the methyne radical (ÔH11: ) may be the precursor of part of this yield, most of it probably results from reactions of a bare carbon atom in the epithermal region. Since in both gaseous and liquid systems the yield of acetylene is apparently insensitive to scavengers it is quite unlikely that radical mechanisms are important in its formation. The results appear to offer further evidence for the mechanism previously proposed based on insertion in the C-H bond [14, 15].
Present results on the ratio of ethane plus ethylene to acetylene in the presence of iodine scavenger agree with the hypothesis presented by RACK, LANG and VOIGT [6 ]. This was that the principal determining factor for this ratio is the nature of the group attacked by the С11(“СНЯ, -СНЙ or -CH).

BEHAVIOUR OF С11 IN LIQUID HYDROCARBONS 395
The ratio can be expressed as
(C2H4 +C 2H6)/C2H2 = aX + bY+cZ
in which X, Y and Z are the fractions of carbon atoms in the molecules that are present as CH3, CH2 and CH groups respectively. The coefficients a, b and с represent the distribution ratio (C9H4 + СгНб^С^Нг for each of the three types of carbon groups when present alone. From results on ethane [15], cyclohexane and benzene [6 ] values of 0. 6 6 , 0. 13 and 0 were taken for a, b and с leading to the predictions of Table II, in which they are compared with the most recent data for those systems studied in this Laboratory.
TABLE II
COMPARISON OF CALCULATED AND OBSERVED YIELD RATIOS, (C2H4+ С2Нб)/С2Н2
M olecule
Y ield ratioСгНг
Calculated Observed
n-hexane 0.31 0.32
• 2-methylpentane ' 0.37 0.39
2, 3-dimethylbutane 0.44 0.47
2, 2-dimethylbutane . 0.56 ■ 0.48
methylcyclopentane 0.20 0.23
The differences between these yields and those found for the same systems in the absence of scavenger are about what one would expect if the products with radical precursors are included with the hot-atom products.
The yields of addition products, molecules larger by one carbon than the irradiated material, are of considerable interest, and the comparison of their yield with that of acetylene should lead to a greater understanding of the hot-atom process. Acetylene is the most important product among the gases, apparently formed principally by hot-atom processes, and these addition products are the main non-gaseous ones. In examining the latter it has not been feasible thus far to add iodine scavenger since the separation of these products and the iodine derivatives of the gaseous products has not been established, but such studies are in progress.
Preliminary results on the hexanes produced from n-pentane do not agree with the statistical expectation pf 3:2:lfor the ratio n-hexane/2 methyl- pentane/3 methylpentane or the results of WOLF et al. on С14 products [16]. Our results give 8:1. 8:1 for this ratio. Some possible explanations are that radiation-induced reactions cause a change in this ratio or that the gas chromatographic separation of these isomers and related products is not

396 A. F. VOIGT et al.
complete, distorting the ratio. It is also possible that the process is not as simple as the statistical picture would predict.
Olefinic addition products should be of some importance if the radioactive and thermally energetic carbon is in the form of C11 or CH11 when bond insertion takes place. The low yield of hexenes from n-pentane, 2. 2% compared with 35.4% for hexanes, would lead to the belief thatthese additions do not largely result from attack by bare carbon but by methylene or to some extent by methyne groups. The results on the cyclopentane systems are still too preliminary to use for such hypotheses.
In the case of benzene the observed ratio of toluene to benzene, 0. 5, agrees with earlier results of SURYANARAYANA and WOLF [17], but the reported fraction of total carbon found in the two is much greater in these experiments (17 compared to 7%). Since the least accurate part of this type of experiment is the correlation with total С11 activity this disagreement is probably not important.
The process of formation of the addition products now appears to be describable in the following way. If, when the attack that results in a stable product occurs, the C11 is in the form of a bare carbon atom, it will have sufficient energy so that the intermediate is most likely to break down to give acetylene. If the Cu has lost enough energy to be in the form of a CH9 radical the intermediate will probably remain as an addition product. For the case between these, the methyne radical, both courses are open; the intermediate may break up leading to ethylene or it may stabilize giving olefinic addition products. In the case of n-pentane the ethylene/hexene ratio is ~ 5 indicating a greater probability for the first of these paths. Further work is in progress to describe these mechanisms more completely.
The very low total of C11 in the benzene system (~ 24%) presents a challenge to locate the rest of the C11. It appears that it must be in larger molecules, those with two benzene rings being fairly likely, particularly a structure like diphenylmethane.
A C K N O W L E D G E M E N T S
The continued assistance of the synchrotron crew in operating and maintaining the synchrotron is gratefully acknowledged. R. G. Clark and W. A. Stensland designed the gas chromatographs and assisted in many other ways in the instrumentation of these experiments.
R E F E R E N C E S
Cl] Special aspects o f hot-atom chemistry. I* Carbon, in Chemical Effects of Nuclear Transformations II IAEA, Vienna (1961) 3-63.
t2] WOLF, A .P . T h e reactions of energetic tritium and carbon atoms with organic compounds", Advances
in Physical Organic Chemistry H ( 1964) 202-2*73.[3] WOLFGANG, R., The hot-atom chemistry of gas phase systems, AEC Rpt NYO-1957-50 (1964).[4] LANG, C.E. and VOIGT, A .F ., J. phys. Chem. 65(1961) 1542.[5] RACK. E.P. and VOIGT. A .F ., J. phys. Chem. 67 (1963) 198.[ 6] RACK, E ,P ., LANG, C.E. and VOIGT, A .F ., J. chem. Phys. 38(1963) 1211.

BEHAVIOUR OF С11 IN LIQUID HYDROCARBONS 397
[7] BUREAU, A. J. and HAMMER, C. L ., Rev. sci. Instrum. 32 (1961) 93.[ 8] HAYWARD, E ., Rev. mod. Phys.. 35 (1963) 324.[9] SCHULER, R.H. and ALLEN. A .O ., J. chem. Phys. 24 (1956) 56.
[10] ANDERSON. A. R., J. phys. Chem. 66(1962) 180.
[11] BLAIR, G.E., I. Amer, ceram. Soc. 43 (1960) 426.[12] WEBER, E. H ., FORSYTH. P.F. and SCHULER, R. a , Rad. Res. 3 (1955) 68.[13] SCHULER, R.H.,J. phys. Chem. 68 (1964) 1618.
[14] MacKAY, C ., PANDOW, М ., POLAK, P. and WOLFGANG, R., Special aspects of hot-atom chemistry.I* Carbon, in Chemical Effects of Nuclear Transformations II IAEA, Vienna (1961) 23.
[15] MacKAY. C ., WOLFGANG. R ., J. Amer. chem. Soc. 83 (1961) 2399.[16] WOLF, A. P., "The reactions o f energetic tritium and carbon atoms with organic compounds". Advances
in Physical Organic Chemistry П ( 1964) 250.
[17] SURYANARAYANA, B. and WOLF, A .P .. J. phys. Chem. 62(1958) 1369.
D IS C U S S IO N
A. WOLF: Does Professor Voigt consider the value of 26.2% as a correct yield for n-hexane-CH, from n-pentane?
A .F . VOIGT: This is the result I have from the laboratory. It is, of course, possible that the peak is complex and that there is a second component.
A. WOLF: I would like to mention that Schrodt and Libby also noted a high yield of n-hexane in carbon-14 work on n-pentane. When we repeated this workj it became obvious that the n-hexane peak contained impurities that were difficult to remove. We finally obtained radiochemically pure material and were able to show that its yield was what we would predict from statistical insertion, i.e. about 6.5%.


ОБРАЗОВАНИЕ РАДИОАКТИВНЫХ ПОЛИМЕРНЫХ ПРОДУКТОВ ПРИ РЕАКЦИЯХ ПОЛИВАЛЕНТНЫХ
АТОМОВ ОТДАЧИ
Б .Г . ДЗАНТИЕВ, Р .А . СТУКАН, А .П . ШВЕДЧИКОВ и А .В . ШИШКОВ ИНСТИТУТ ХИМИЧЕСКОЙ ФИЗИКИ АН СССР, МОСКВА
СССР
Abstract — Résumé — Аннотация ■— Resumen
THE FORMATION OF POLYMERIC PRODUCTS IN REACTIONS OF POLYVALENT RECOIL ATOMS. One of the features o f the hot-atom reactions obtained as a result o f nuclear transformations is that labelled polymeric products can be formed. This tendency is very marked in the case o f polyvalent recoil atoms,
where the polymer yield can, in certain cases, reach an amount o f about 90% o f the total activ ity.The aim of the present research is a study of the behaviour of recoil atoms of sulphur-35 and carbon-14,
obtained in the nuclear reactions C l35(n, p)S35 and N14(nt p)C14 in gas and liquid phases. It can be assumed that in the stabilization process hot carbon atoms form methylene biradicals, whose behaviour, by reason of their reaction capacity, greatly resembles that o f atomic sulphur. The investigations were conducted like those for paraffins (CH4, C2 H6 ), and for cyclic hydrocarbons (cyclohexane, cyclohexene, benzene). The binary systems comprising hydrocarbons on the one hand and S3S and C14 hot-atom donors on the other were
subjected to irradiation. Compounds o f CCI 4, HC1 and ammonia were used as the donors. Irradiation was carried out on a reactor o f type IRT-1000 with a thermal neutron flux o f 1011 - 1012 n/cm2. s.
It is shown that for various compounds in the liquid phase, up to 60-90% o f the sulphur-35 becomes
stabilized in the form of a polymer, the yield of which is highly dependent on the composition, passing through the maximum at a nearly equimolecular ratio of components. In the gas phase the polymer yield amounts to 30-40% of the total activity. By means of paper radiochromatography it was established that labelled polymer products have a complex structure and are, at the least, a mixture o f compounds of two qualitatively different types whose yield changes in various ways depending upon the ratio of the components. An increase in irradiation time leads to an increase in the labelled polymer yield. In the case of the liquid phase system C 6H12- ССЦ, the molecular weight of the polymer was determined by capillary diffusion and found to be 5000 for the polymer o f one type and 500-1000 for the other. The formation of a labelled product with a high boiling point was also observed when pure CC I4 was irradiated.
Analogous experiments were carried out in an ethylene-ammonia system in the gas phase, at high pressure. It is shown that in this case, when puje anhydrous ammonia is irradiated the basic labelled product is C l4H4. However, when even small additions o f ethylene are made, a considerable proportion (up to 80%) o f C 14 becomes stabilized in the form o f a polymer, whose yield also varies extremely with the growth in the proportion o f ethylene in the system, attaining a maximum when there is 10-15% of C 2H4. It is shown that if С 14 polymer yield is plotted against composition, the shape o f the curve does not change when dose rate and flux are increased.
FORMATION DE POLYMÈRES LORS DE RÉACTIONS PROVOQUÉES PAR DES ATOMES DE RECUL POLYVALENTS. Les réactions que provoquent des atomes chauds obtenus à la suite de transformations nucléaires présentent la particularité de donner lieu à la formation de polymères marqués. Cette tendance est particulièrement nette dans le cas des atomes de recul polyvalents où le rendement en polymère peut parfois atteindre un ordre de grandeur approchant 90% de l'activité globale.
Le mémoire a pour objet d’ étudier la formation de polymères marqués lors de la stabilisation chimique des atomes 35S et 14C de recul obtenus au cours des processus 35Cl(n, p)35S et 14N(n, p)l4C en phase gazeuse et en phase liquide. On peut admettre que pendant la stabilisation les atomes chauds de carbone provoquent
la formation de radicaux doubles de méthylène dont la réactivité rappelle beaucoup le comportement du soufre
atomique. Les études ont porté tant sur les hydrocarbures saturés (CH4, C2H6) que sur les hydrocarbures cycliques (cyclohexane, cyclohexène, benzol). On a irradié des systèmes binaires hydrocarbure-substance génératrice d'atomes 35S ou 14C chauds. Comme substance génératrice d’ atomes chauds, on a utilisé CC14,
399

400 Б .Г . ДЗАНТИЕВ и др.
НС1 et NH3. L’ irradiation a été faite à l ’ intérieur d'un réacteur du type IRT-1000, dans un flux de neutrons thermiques de 1011 à 1012 n/cm2 • s.
Les auteurs montrent que pour différents hydrocarbures en phase liquide 60 à 90<#> de 35 S se stabilisent sous la forme d’un polymère dont le rendement dépend, dans les cas limites, de la composition du système et passe par un maximum lorsque le rapport moléculaire des composants du système approche de l ’unité. En phase gazeuse, le rendement en polymère représente 30 à 40% de l ’activité globale. Par radiochromatographie sur papier, on a établi que les polymères marqués sont d'une constitution complexe; il s’agit d'un mélange de deux composés de types qualitativement distincts et le rendement en ces deux composés varie différemment et dépend du rapport des composants du système. En augmentant la durée d'irradiation, on active la formation du polymère marqué. Pour le système СбН12-СС14> on a fait appel à la méthode de diffusion par tube capillaire pour déterminer le poids moléculaire du polymère contenant 35S, qui est de 5000 pour l ’un des polymères et de 500 à 1000 pour l ’autre. On a constaté qu'un produit marqué à point d’ ébullition élevé se formait également lorsqu’on irradiait du CC14 à l ’état pur.
Des expériences analogues ont été faites sur le système éthylène-ammoniac en phase gazeuse sous haute pression. Les auteurs montrent que lors de l'irradiation d'ammoniac seul, le principal produit marqué est 14CH4. Cependant, il suffit d'ajouter de petites quantités d’ éthylène pour qu’une fraction importante (jusqu'à 80%) de 14C se stabilise sous la forme d’ un polymère dont le rendement varie également, dans les cas limites, en fonction de la teneur du système en éthylène, en atteignant un maximum lorsque cette teneur est de 10 à 15%. On constate que la forme de la courbe qui exprime le rendement en polymère marqué par 14C en fonction de la composition du système ne subit aucune modification lorsqu’on a u g m e n te l'intensité de la dose et du flux.
О Б РА ЗО В АН И Е П О ЛИ М Е РН Ы Х П Р О Д У К Т О В ПРИ РЕ А К Ц И Я Х П О Л И В А Л Е Н ТН Ы Х А ТО М О В О Т Д А Ч И . Одной из особенностей реакций горячих атомов, получающихся в р е зультате ядерных превращений, является способность к образованию меченых полимерных продуктов. Наиболее ярко эта тенденция проявляется в случае поливалентных атомов отдачи, причем выход полимера может достигать в отдельных случаях величины порядка 90% от об щей активности.
Целью настоящей работы является исследование образования меченых полимерных продуктов в процессе химической стабилизации атомов отдачи серы-35 и углерода-14, получающихся по ядерным реакциям С135 /п, p/s3 и N ^ / n , р/С14 в газовой и жидкой фазах. Можно предположить, что в процессе стабилизации горячие атомы у г л е р о д а о б р а з у ю т метиленовые бирадикалы, которые по своей способности вступать в реакцию во многом напоминают поведение атомарной серы . Исследования проводились как для парафиновых (С Н 4, С 2Нб), так и для циклических (циклогексан, циклогексен, бензол ) углеводород . Облучению подвергались бинарные системы углеводород-датчик горячих атомов S35 и С 14. В качестве последнего использовались соединения C C I4, НС1 и аммиак. Облучение проводилось на реакторе типа ИРТ-1000 при потоке тепловы х нейтронов 10П —1012 неЙтрон/см2сек.
Показано, что для различных соединений в жидкой фазе до 60—90% серы-35 стабилизуется в форме полимера, выход которого экстремально зависит от состава, проходя через максимум при соотношении компонентов, близком к эквимолекулярному. В газовой фазе выход полимера составляет 30 — 40% от общей активности. Методом радиохроматографии на бумаге установлено, что меченые полимерные продукты имеют сложный состав и представляют собой смесь двух качественно отличных типов соединений, выход которых по-разному меняется в зависимости от соотношения компонентов. Увеличение времени облучения приводит к р о с т у
выхода меченого полимера. В случае жидкофазной системы C gH ^ —СС14 молекулярный вес S35-содержащего полимера, определялся методом диффузии из капилляра и оказался равным 5000 для полимера одного типа и 500 — 1000 для другого. Образование высококипящего меченого продукта наблюдалось также при облучении чистого C C I4.
Аналогичные опыты проводились в системе этилен —аммиак в газовой фазе при высоком давлении. Показано, что в этом случае при облучении чистого безводного аммиака основным меченым продуктом является С14Н 4. Однако уже малые добавки этилена приводят к тому, что значительная доля (до 80%) С *4 стабилизируется в форме полимера, выход которого также меняется экстремально с ростом доли этилена в системе, достигая максимума при 10 — 15% С2Н4 . Показано, что вид кривой выхода полимера С 14 не меняется с увеличением мощности дозы и потока.
FORMACION DE POLIMEROS EN LAS REACCIONES DE ATOMOS DE RETROCESO POLIVALENTES. Unadelascaracterfsticasdelasreaccionesdeátomoscalientes, obtenidoscomo resultado de las transformaciones

РАДИОАКТИВНЫЕ ПОЛИМЕРНЫЕ ПРОДУКТЫ 401
nucleares, es su capacidad de formar polímeros marcados. Esta tendencia es especialmente acusada en el caso de los átomos de retroceso polivalentes, en el que el rendimiento del polímero suele alcanzar un valor del orden del 90 °¡o de la actividad total.
Los autores investigaron la formación de productos polimerizados marcados durante el proceso de estabili
zación química de los átomos de retroceso azufre-35 y carbono-14, obtenidos en las reacciones nucleares respectivas 35Cl(n, p)3sS y l4N(n, p)14C en fase gaseosa y líquida. Puede suponerse que en el procesodeestabili- zación los átomos calientes del carbono originan la formación de birradicales metilénicos que, por su capacidad de reacción, tienen un comportamiento muy parecido al del azufre atómico. Las investigaciones se efectuaron con hidrocarburos parafínicos (CH4, C2 H 6) y cíclicos (ciclohexano, ciclohexeno, benceno). Irradiaron sistemas binarios compuestos por el hidrocarburo, por una parte, y por dadores de átomos calientes d e l35 F y del 14C,
por la otra. En calidad de dadores se utilizaron compuestos de CC1<, HCl y amoniaco. La irradiación se efectuó en un reactor de tipo IRT-1000, con un flujo de 1011 a 1012 neutrones térmicos/cm2. s.
Los autores demuestran que para diversos compuestos en fase líquida, del 60 al 90% del azufre-35 se estabiliza en forma de polímero, cuyo rendimiento depende de la composición, pasando por un máximo cuando la relación entre los componentes tiende a ser equimolecular. En fase gaseosa el rendimiento del polímero representa del 30 al 40% de la actividad total. Por radiocromatograffa sobre papel establecieron que los polímeros marcados poseéh una composición compleja y constituyen una mezcla de por lo menos dos tipos de compuestos cualitativamente diferentes, cuyo rendimiento varía de distinta manera en función de la relación entre los componentes. Un aumento del tiempo de irradiación se traduce en un mayor rendimiento del polímero marcado. Para el sistema C6H12 -C C I4 en fase líquida, el peso molecular del polímero se determinó por difusión capilar, hallándose valores de 5 000 para el polímero de un tipo y de 500 a 1000 para el otro. Se observó también la formación de un producto marcado de elevado punto de ebullición al irradiar el CC14 puro.
Se efectuaron experimentos análogos en un sistema etileno-amoniaco en fase gaseosa a elevada presión,
demostrándose que en este caso al irradiar el amoniaco anhidro puro, el principal producto marcado es el 14CH4. Sin embargo, la adición de pequeñas cantidades de etileno hace que una gran parte -hasta el 80% -
del 14C se estabilice en forma de polímero, cuyo rendimiento también varía considerablemente al aumentar la proporción de etileno en el sistema, alcanzando un máximo cuando esta proporción es del 10 al 15% de C2H4. Los autores demuestran que cuando el rendimiento del polímero marcado con 14C se representa en función de la composición, la forma de la curva obtenida no se modifica al aumentar la intensidad de dosis y el flujo neuttónico.
Целью настоящей работы является исследование меченых полимерных продуктов, образующихся при химической стабилизации атомов отдачи серы-35 и углерода-14. Факт образования высокомолекулярных продуктов при реакциях горячих атомов отмечался рядом авторов [1 — 8 ], однадсо нет никаких сведений ни о характере полимера, ни о возможных путях его образования. В то время как вопросам, связанным с такими обычными реакциями атомов отдачи, как отрыв и замещение атомов и атомных групп посвящено довольно большое число работ, механизм образования полимера практически нигде не рассматривается, хотя выход его часто оказывается весьма значительным. Ранними исследованиями, проводимыми в нашей лаборатории, было показано, что в случае атомов отдачи серы-35 выход полимерного продукта оказывается доминирующим и достигает величины порядка 60 — 90% [6 ],. Следует отметить, что высокомолекулярный продукт получался в очень малых количествах, обладая высокой удельной активностью.
Нами исследовалось взаимодействие горячих атомов серы и углерода, получающихся по ядерным реакциям Cl3 5 (п, p)S35 и N14(n ,p)G 14 с различными углеродами как алифатического так и циклического и ароматического ряда (метан, этан, этилен, циклогексан, циклогексен, бензол). Датчиком горячих атомов S35 служил CCI4 или НС1; в слчае С 14 использовался аммиак.
26

402 Б .Г . ДЗАНТИЕВ и др.
Облучению подвергались бинарные смеси вышеперечисленных у г л е водородов с соответствующими веществами-датчиками. Смеси помещались в кварцевые ампулы, замораживались жидким азотом и эвакуировались до давления 1 0 ~а мм рт .ст .
Облучение проводилось на реакторе при потоке тепловых нейтронов 1011 — 101а н/см2 сек. Время облучения варьировалось от 10 до 100 час в зависимости от характера системы. Опыты проводились с системами в газовой и жидкой фазах. После облучения образцы вскрывались, и путем вакуумной разгонки с инертными носителями отделялась легколетучая фракция. Полимерный остаток растворялся при нагревании в бензоле или толуоле и анализировался методом радиохроматографии на бумаге. Хроматографирование проводилось в восходящем токе растворителя, в качестве которого использовались смеси бутанол —вода —уксусная кислота или этанол —вода —уксусная кислота (1 :1 :1 ). Доля активности S35 , приходящаяся на полимерный продукт, определялась путем сжигания последнего по методике Кариуса. В этом случае измерялась активность бесконечно толстого слоя сульфата бария. В случае углерод-14 измерялась активность тонкой пленки полимера, полученной медленным выпариванием бензольного раствора. Общие активности определялись расчетным путем и по мониторам. Для измерения активности серы-35 и углерода-14 использовались торцовые счетчики со слюдяным окном толщиной 1 — 1,5 мг/см . Была проведена методом диффузии из капилляра в бесконечный объем [9,10] оценка молекулярных весов полимерных продуктов. Из уравнения Фика для случая диффузии из капилляра в бесконечный объем следует:
С/С0 = 8/тг2 - е '4 « 2 ' (1 )
где Со — начальная концентрация вещества в капилляре;С — концентрация вещества в момент времени t, в сутках;t — длина капилляра в см;D — коэффициент диффузии, см 2 /сутки.
При D t/ í2> 0,2 формула справедлива с достаточной точностью. Переходя от концентраций к активностям, формулу ( 1 ) можно представить в виде
lg jzy^ 0 = -0, 064 D t+a (2)
При постоянном коэффициенте диффузии имеем уравнение прямой, и Dможет быть определен по тангенсу угла наклона. Пользуясь соотношением Dn/m = const можно определить средний молекулярный вес неизвестного вещества по величине D для соединения с известным молекулярным весом. В качестве последних использовались меченные по сере-35 нонил- меркаптан и элементарная сера.
Были исследованы реакции горячей серы-35 в системах циклогексан — ССЦ, циклогексен —CCI4 и бензол —CCI4 в жидкой фазе. Полученные данные по зависимости выхода меченого полимера от состава системы представлены в та б л . 1 , из которой видно, что выход полимерного продукта меняется экстремально с изменением молярной доли CC I4 , проходя через максимум вблизи точки, отвечающей эквимолекулярному соотношению ком-
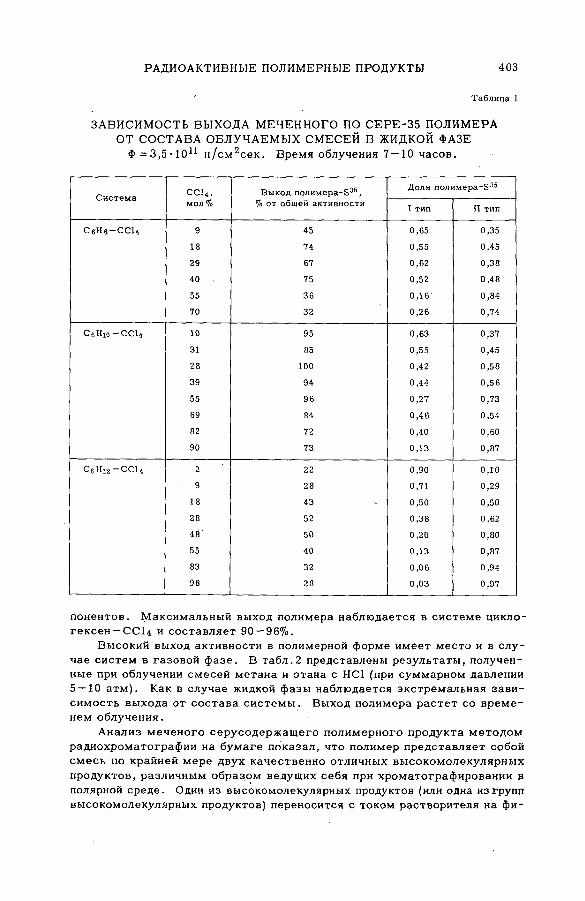
РАДИОАКТИВНЫЕ ПОЛИМЕРНЫЕ ПРОДУКТЫ 403
Таблица 1
ЗАВИСИМОСТЬ ВЫХОДА МЕЧЕННОГО ПО СЕРЕ-35 ПОЛИМЕРА ОТ СОСТАВА ОБЛУЧАЕМЫХ СМЕСЕЙ В ЖИДКОЙ ФАЗЕ
Ф =3 ,5 -10 11 н/см2 сек. Время облучения 7 —10 часов.
Система СС14, мол %
Выход полимера-S 35, % от общей активности
Доля полимера-S 35
I тип II тип
СбНе~СС14 9 45 0,65 0,35
18 74 0,55 0,45
29 67 0,62 0,38
40 - 75 0,52 0,48.
55 36 0,16 0,84
70 32 0,26 0,74
С 6Ню “ C C I4 10 95 0,63 0,37
31 85 0,55 0,45
28 100 0,42 0,58
39 94 0,44 0,56
55 96 0,27 0,73
69 84 0,46 0,54
82 72 0,40 0,60
90 73 0,13 0,87
Сб Hi2 —C C I4 2 22 0,90 0,10. 9 28 0,71 0,29
18 43 - 0,50 0,50
28 52 0,38 0,62
48 50 0,20 0,80
55 40 ОДЗ 0,87
83 32 0,06 0,94
98 28 0,03 0,97
понентов. Максимальный выход полимера наблюдается в системе цикло- гексен —CC I4 и составляет 90 — 96%.
Высокий выход активности в полимерной форме имеет место и в слу чае систем в газовой фазе. В табл. 2 представлены результаты, полученные при облучении смесей метана и этана с НС1 (при суммарном давлении 5 — 10 атм). Как в случае жидкой фазы наблюдается экстремальная зависимость выхода от состава системы . Выход полимера растет со временем облучения.
Анализ меченого серусодержащего полимерного продукта методом радиохроматографии на бумаге показал, что полимер представляет собой смесь по крайней мере двух качественно отличных высокомолекулярных продуктов, различным образом ведущих себя при хроматографировании в полярной среде. Один из высокомолекулярных продуктов (или одна из групп высокомолекулярных продуктов) переносится с током растворителя на фи-

404 Б .Г . ДЗАНТИЕВ и др.
ЗАВИСИМОСТЬ ВЫХОДА МЕЧЕННОГО ПО СЕРЕ-35 ПОЛИМЕРА ОТ СОСТАВА ОБЛУЧАЕМЫХ СМЕСЕЙ В ГАЗОВОЙ ФАЗЕ
Ф = 1,2-1011 н/см2сек
Таблица 2
СистемаВремя облучения,
часыНС1,
мол %Выход полимера-S35,
% от общей активности
И И
21 16,5
27 19
35 41,5
СН4-Н С 1 40 41 28
45 33,5
57 35
68 29
80 25
24,5 53
30,5 63,5CH4-H C I 8040,5 80
43 67
19 13,5
21 14
25 18,5
С2Н6~НС1 30 34 24
46 31
69 39
91 25
С 2Н6-Н С1 100 ' 40 62
ниш хроматограммы (R f~0 ,9 ), -полимер I типа-, а другой остается вблизи старта (Rf ~0,1 — 0,2), - полимер типа И. (рис.1).
Для одной из систем, а именно для системы циклогексан—CCI4 , методом препаративной хроматографии на колонке с набивкой из хроматографической бумаги Ватман-2 были выделены в чистом виде полимеры каждого типа и определены методом диффузии из капилляра их средние молекулярные веса. При этом оказалось, что оба типа полимеров не являются монодисперсными продуктами и имеют различные средние молекулярные веса (~500 для полимера типа II и ~5000 для полимера типа I).
Интересно отметить, что выход полимера II типа падает, а полимера типа I —растет с увеличением концентрации хлора в системе.
Реакции атомов отдачи С 14 исследовались при облучении безводного аммиака с добавками этилена. Полученные данные представлены на рис.2, из которого видно, что уже малые добавки этилена (2 — 5%) приводят к резкому увеличению выхода меченого полимерного продукта. Как и в случае серы-35, выход полимера экстремально зависит от состава системы, при-
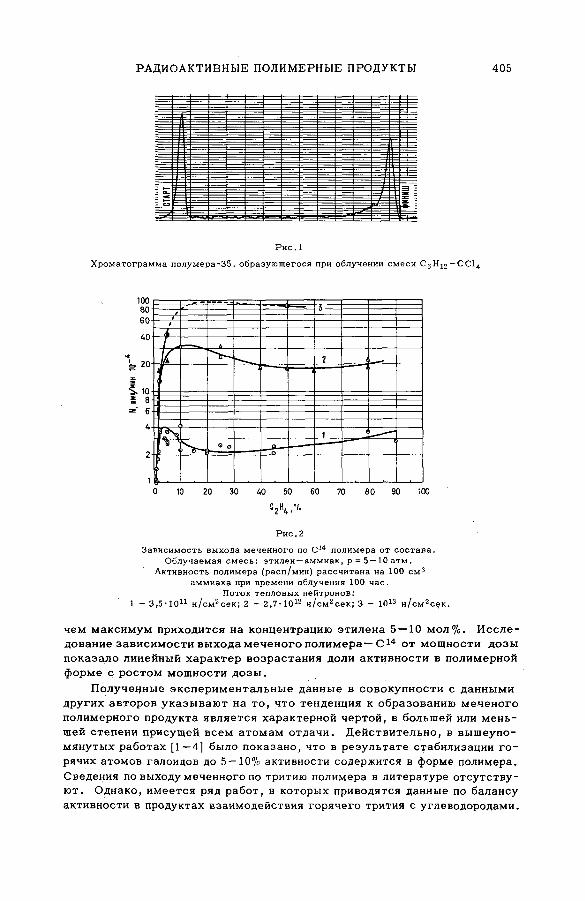
РАДИОАКТИВНЫЕ ПОЛИМЕРНЫЕ ПРОДУКТЫ 405
Рис. 1
Хроматограмма полумера-35, образующегося при облучении смеси С 6Н12 —СС14
Рис. 2
Зависимость выхода меченного по С14 полимера от состава.Облучаемая см есь : этилен —аммиак, р = 5 — 10 атм .
Активность полимера (расп/мии) рассчитана на 100 см 3 аммиака при времени облучения 100 час.
Поток тепловых нейтронов:1 — 3 ,5 -Ю 11 н/см2сек; 2 - 2,7*1012 н/см2сек; 3 — 1013 н/см2сек.
чем максимум приходится на концентрацию этилена 5 — 10 мол% . И сследование зависимости выхода меченого полимера—С 14 от мощности дозы показало линейный характер возрастания доли активности в полимерной форме с ростом мощности дозы.
Получецные экспериментальные данные в совокупности с данными других авторов указывают на то, что тенденция к образованию меченого полимерного продукта является характерной чертой, в большей или меньшей степени присущей всем атомам отдачи. Действительно, в вышеупомянутых работах [1—4] было показано, что в результате стабилизации го рячих атомов галоидов до 5 — 10% активности содержится в форме полимера. Сведения по выходу меченного по тритию полимера в литературе отсутствуют. Однако, имеется ряд работ, в которых приводятся данные по балансу активности в продуктах взаимодействия горячего трития с углеводородами.

406 Б .Г . ДЗАНТИЕВ и др.
Так, например, по данным Зауэра и Вилларда [11] выход меченых углеводородов > С 5 при облучении этилена и его смесей с алканами составляет 5 — 10% от общей активности трития.
Как показывает настоящее исследование, а также работы по горячим атомам углерода-14 [7,12] и мышьяка A s 76 [5], в случае поливалентных атомов отдачи выход меченого полимерного продукта значительно выше и в ряде случаев оказывается доминирующим.
Данные по анализу состава полимерного продукта и определению величин молекулярных весов позволяют сделать некоторые предположения0 характере меченых полимерных продуктов и о возможных путях их образования. Факт различного поведения серусодержащих полимеров типа1 и II в полярной среде при хроматографировании, а также различный характер зависимости величин их выходов от состава системы позволяет предположить, что полимерный продукт является полифункциональным соединением. Очевидно, полимерный продукт типа I представляет собой смесь углеводородных высокомолекулярных соединений, содержащих серу в виде сульфидных или сульфгидрильных групп, а полимер типа II является хлорсодержащим продуктом и образуется в основном за счет обрыва цепи полимеризации в треке горячего атома радикалами ССЬ? или С1’ .
Ранее было показано [6 ], что при реакциях горячих атомов серы в среде углеводородов образуется наряду с полимером целая гамма других меченых продуктов: алифатические и циклические меркаптаны и сульфиды, однако, активности их составляют незначительную долю от активности полимера.
Известно, что при радиолизе различных углеводородов в присутствии элементарной серы или серусодержащих соединений (сероводород, меркаптаны) основными продуктами наряду с водородом и углеводородами являются сероводород, меркаптаны, сульфиды и дисульфиды [13, 14]. Образование полимера, как правило, или вовсе не наблюдается или выход его незначителен. Аналогичная картина имеет место и в случае реакций серы в области тепловых энергий [15]. Однако, при реакциях горячих атомов серы сероводород отсутствует, а основным продуктом является меченый полимер. Интересно отметить, что при облучении быстрыми электронами (Е = 1,5 Мэв) смеси циклогексана с элементарной серой-35 образования меченого полимера нами не наблюдалось.
Таким образом, образование полимерного серусодержащего продукта является,по-видимому,специфической особенностью горячей серы и протекает, очевидно, в горячей или надтепловой области энергий в треке атома отдачи. Выход полимера в общем случае является функцией массы атома отдачи и его валентности. Действительно, в случае горячего трития выход полимера мал, в то время как энергия его велика. Несколько выше выход высокомолекулярного продукта для тяжелых атомов отдачи, как например атомов галоидов. Очевидно, для трития наиболее характерны соударения типа атом —атом, в то время как в случае горячих атомов с большой м ассой — атом—молекула.
При прохождении в углеводородной среде атом отдачи образует на своем пути цепь радикалов и возбужденных молекул, которые вступают в полимеризацию с захватом замедлившегося горячего атома. Очевидно, именно на последнем этапе определяющую роль начинает играть валент

РАДИОАКТИВНЫЕ ПОЛИМЕРНЫЕ ПРОДУКТЫ 407
ность атома отдачи. На образование полимерного продукта в треке горячего атома указывает и характер зависимости выхода полимера — С14 от мощности дозы. Действительно, если бы меченый полимерный продукт получался в результате захвата термализовавшегося атома отдачи радикалами, образованными за счет радиолиза облучаемой системы, то выход последнего должен был бы зависеть от \1у, где У — мощность дозы, а не быть пропорциональным У , как это имеет место. Данные по взаимодействию атомов отдачи С11, получаемого на ускорителе, с алканами показывают что выход "полимера" также составляет величину 30 — 50% [16,17], хотя в данном случае радиационные повреждения значительно меньше, чем в условиях облучения на реакторе. Таким образом, образование меченого полимера в первую очередь связано, по-видимому, не с побочными радиационнохимическими процессами, а обусловлено специфическими реакциями самого атома отдачи.
Процесс образования меченых по углероду и сере полимерных продуктов с высокой удельной активностью помимо теоретического интереса может иметь и ряд практических приложений, например, использоваться для концентрирования радиоактивных изотопов или для изготовления стандартных источников.
Л И Т Е Р А Т У Р А ’
[11 BOHEM AN E. G. , W IL L A R D J .E . J. Am . Chem Soc. 64, 6, 1342 (1942).[2] FO X M. S., L IB B Y W. F . , J. Chem. Phys. 20, 3, 487 (1952).[3] G O LD H ABER S. , CHIANG R. S . , W ILLA R D J .E ., JACS 73, 5, 2271 (1951).[4] НЕСМ ЕЯНОВ, А н .Н . , Ф ИЛАТОВ Э .С . , БОРИСОВ E . A . , Щ УКЛА Б .М . ,"C h em . E ffec ts
Nucl. T ran sform ation s". IA E A , v . 1, 259, Vienna, 1961.[5] C IFK A J . , M E L IC H A R Z ., C oll. Chech. Ch. Comm. 26, 5, 1403 (1961).[6] ДЗАНТИЕВ Б . Г . , Б А Р К А Л О В И .М ., Труды международной конференции по применению
изотопов. Копенгаген, 1960 (Radio isotopes in the P h ys ica l Sciences and Industry, IA E A , Vienna, v .3 , 27, 1962).
[7] SCHRODT A . G. , L IB B Y W . P . , J. A m . Chem. Soc. 78. 7, 1267 (1956).[8] АВДОНИНА E .H . , М У Д РА К . , НЕСМ ЕЯНОВ А н .Н . , "Радиохим ия", 5, 5, 633 (1963).[9 ]W A N G J .H . , ROBINSON С. V . , E D E LM A N J. S ., JACS 75, 2, 466 (1953).
[10] СПИЦЫН В .И . , СПИРИДОНОВ Ф .М . , КОЛЛИ И. Д . , ЖФХ, 32, 5, 1143 (1958)[11] SAUER М, С ., W ILLA R D J .E . , J. Phys. Chem. J54, 359 (1960).[12] JANG J ., W O LF A . P . , J. A m . Chem. Soc. 82, 17, 4488 (1960).]13] W A TA R U A. et a l. . Bull. Chem. Soc. Japan 37, 3, 353 (1964).[14] H E LM U T B . , D IE T E R H . , Z . Phys. Chem. (BRD) 33, 5 -6 , 352 (1962).[15] F A R M E R E. H ., S H IP L E Y F .W ., J. P o l. Sci. J_, 4, 293 (1946).[16] STÔ CK LIN G . , S T A N G L H ., CH RISTM AN D . , GUMMING J. B . , W O LF A . P . , J, Am .
Chem. Soc. 67, ', 1735 (1963).[17] STÓ CK LIN G ., W O LF A . P . , J. A m . Chem. Soc. 85, 2, 229 (1964).
D IS C U S S IO N
F . CACACE: I have a question on Dr. D zantiev's paper. A couple of years ago Dr. W olf and m yself studied the re co il chem istry of C11 in ammonia. We found that the yield of the two major recoil products, Cl l H4 and С ^Н з-К Н г, depended strongly on the radiation dose absorbed by the system.I wonder whether Dr. Dzantiev has taken these results into account. I am thinking particularly of the fact that the dose absorbed by this system ought to be re lative ly high.

408 Б .Г . ДЗАНТИЕВ и др.
В. D ZAN TIEV : When samples are subjected to in -p ile irradiation, radiochem ical p rocesses — and particu larly the production o f radicals through radiation — certainly play a role in the formation of the final forms of hot-atom chemical stabilization. The point we make in our paper, however, is that the formation pattern of high-molecular labelled products cannot be explained in term s of radiation polym erization alone. The labelled- polym er yie ld depends on the type of the hot atom involved (its mass and valency) and essentia lly the recom bination-polym erization process takes place in the track of a polyvalent heavy atom.
A . W OLF: With regard to the question of radiation polym erization versus hot-atom-induced polymerization we are in agreement with Dr .Dzantiev. The work of Yang and Wolf published in the Journal of the American Chemical Society in 1960* also indicated that the polymer observed from the NH3- CH4 system was probably formed in part by the hot-atom process since the specific activity was too high in the polymer to be accounted for by radiolytic processes alone.
In the paper re ferred to by Dr. Cacace we also delineated the effect of radiation damage in gaseous ammonia. The methane is form ed la rge ly by rad io lytic reduction of methylamine or the p recu rsor of methylam ine.
The determination of the molecular weight of the C14-containing polymer by the infinite cap illary method is a most interesting approach, and I very much hope that D r. Dzantiev and his group w ill now be able to determ ine the structure of this po lym er.
K. PA NEK: We have done quite a lot of work on irradiated alkyl chlorides but our results d iffer a little from those reported by Dr. Dzantiev. We have found, for instance, that the percentage of total sulphur activity incorporated in so-called "polym eric compounds" ranges from roughly 15 to 25% in ethyl and propyl chlorides. A m ore interesting feature is the occurrence of various chlorosulphides. On irradiation of ethyl chloride, fo r instance, we have found 10% of the total S35 activity in the form of dichlorodiethyl sulphide, i .e . as mustard gas (BP 215-7°C). It is known that all sim ilar compounds boil at even higher temperatures, but this does not necessarily mean, however, that they are polymeric in character.
In general, I fe e l that the resu lts o f investigations into the chem ical effects of polyvalent atoms in liquid media need to be interpreted most carefully. These systems seem to be of such complexity that under certain c ir cumstances our investigations are liable to lead to a dead end. To illustrate what I mean, I should like to present some other resu lts we have obtained.
In the low-boiling fractions, i. e. up to 200°C, we have found by means of radio-gas chromatography that at least 17 labelled compounds are present in irradiated propyl chloride. Most of these we have been unable to identify.
As to the mechanism responsible for the formation of these compounds, various hypotheses are possible at the present time and all of them are probably of equal valid ity. Unfortunately we are not able, on the basis of the available resu lts, to accept any of the possib le mechanisms considered.
B. DZANTIEV: I should like to draw attention to one of the main results of our work, namely that we have been able to determ ine the m olecu lar weight of in fin itesim ally sm all amounts of labelled polym eric products.
* J. Amer. chem. Soc. 82 (1960) 4488.

РАДИОАКТИВНЫЕ ПОЛИМЕРНЫЕ ПРОДУКТЫ 409
Th eir m olecular weight va ries between 500 and 5000, which, assuming 100 fo r the weight of an individual molecule, would correspond to between 5 and 50 molecules in the final product. This would be consistent with Dr. W olf's view that high-boiling products are products made up of groups of molecules. M y im pression is that the po lym er is form ed from a group of m olecu les in the ta il of the track (about a 1 0 0 th part of the tra il of a hot atom of sulphur or carbon) that contains 5 to 50 molecules.
F .S . ROWLAND: The separation of the actual hot-atom reactions from radiation chemical reactions in the same system is something that needs to be considered in every system investigated, and I should like to make a few remarks about polym er formation in recoil-tritium reactions in response to what P ro fessor Dzantiev has said. Almost the only system in which we have observed any considerable form ation of rad ioactive po lym er in recoil-tritium reactions themselves is the liquid benzene system, as was discussed ea r lie r . In this system, the initial hot radioactive radicals attack the benzene itself, perhaps a fter being firs t therm alized by collis ions, leading to polym er formation. Some formation of m oderately high-boiling radioactive molecules is observed with molecules such as ethylene, cyclo- hexene, e tc ., but in molecules such as ethyl alcohol or alkane hydrocarbons and other sim ilar molecules, almost all the tritium is found in very simple compounds. In most of these system s, however, radioactive species are form ed that may be very much m ore sensitive to radiation effects than the parent m olecule its e lf. With such a radioactive product that is sensitive to certain radiation-produced species — fo r example, H atoms attacking C2H 3T in an alkane — subsequent radiation-induced reactions can lead to the production of m aterials of higher molecular weight. The recoil tritium cyclopropane system is one system that is ve ry sensitive in this manner. We have observed radiation alteration of the product spectrum (rem oval of C2H3T ) even in irradiations of two hours' duration at 109n/cm2 -s. In most systems, however, the parent molecule is not so inert to attack by the radiation-produced species, and appreciable radiation effects are not observed until periods of.hours at 10П n/cm2 • s. At higher fluxes, or longer times of irradiation, radiation-damage alteration of the radioactive-product spectrum is quite common. Experim ents ca rr ied out in the presence of scavenger molecules usually suppress the rem oval of the sensitive radioactive species.


РЕАКЦИИ ГОРЯЧИХ АТОМОВ ТРИТИЯ С АМИНОКИСЛОТАМИ
Е.Ф . СИМОНОВ и Ан.Н. НЕСМЕЯНОВ МОСКОВСКИЙ ГОСУДАРСТВЕННЫЙ УНИВЕРСИТЕТ, МОСКВА
СССР
Abstract — Résumé — Аннотация — Resumen
REACTIONS OF HOT TRITIÚM ATOMS WITH AMINO ACIDS. In the existing literature there is a lack o f systematic data on the interaction of tritium recoil atoms with amino acids, yet such data, in conjunction with results already obtained for organic acids and amines, could help in determining the mechanism of hot reactions in relation to the structure o f compounds (chain length, functional substitutes).
A study was made o f the yields from the reaction o f hot tritium atoms: (1) with amino acids having
lengthened chains, and (2) with amino acids having a carbon chain o f constant length, but with various functional substitutes.
For this purpose mixtures of lithium carbonate and the acids under study were irradiated for 15 min with a slow neutron flux o f 0.87 x 1013 cm2/s. Analysis was carried out on a gas chromatography unit with interchangeable columns (molecular sieves, and liquid petrolatum on kieselguhr) and with paper chromatography.
Although the data obtained for the radiation survival capacity o f amino acids as a function of carbon
chain length were at variance with a basic tenet o f radiation chemistry according to which the conservation of molecules increases in proportion to the length o f their chains, the data can be explained in terms o f an intramolecular transfer of energy along the carbon chain fiom the collision site of the hot atom to the hydroxyl group, and subsequent "de-excitation"; on the other hand, although the energy, o f tritium recoil atoms is greater than that of the chemical bond, the latter nevertheless exerts an influence on the radiation conservation of molecules with a carbon chain of constant length but with various substitutes.
RÉACTIONS ENTRE ATOMES CHAUDS DE TRITIUM ET ACIDES AMINÉS. Les études publiées jusqu’à présent ne contiennent guère de renseignements systématiques sur les interactions entre atomes de tritium de recul et acides aminés. Pourtant, ces données, complétées par celles dont on dispose sur les acides organiques et les amines, pourraient aider à définir le mécanisme des réactions chaudes en fonction de la structure des composés (longueur de la chaîne, groupes de substitution fonctionnels).
Les auteurs ont étudié le rendement des réactions entre atomes chauds de tritium et acides aminés: 1 . lorsque la chaîne des acides aminés s'allonge; 2 . lorsque les acides aminés ont une chaîne carbonée de longueur identique mais possèdent des groupes de substitution fonctionnels différents.
A cette fin, ils ont irradié des mélanges de carbonate de lithium et d'acides à étudier dans un flux de neutrons lents de 0,87 • 1013n/cm2 • s, pendant 15 min. L'analyse a été faite au moyen d'un appareil de chromatographie gazeuse sur colonne à supports interchangeables (tamis moléculaires, huile de vaseline sur terré d'infusoires) et par chromatographie sur papier.
Les données obtenues sur la survie à l ’ irradiation des acides aminés en fonction de la longueur de la chaîne carbonée semblent contredire une règle fondamentale de radiochimie, selon laquelle la durée de conservation des molécules augmente avec l'allongement de leur chaîne, mais elles peuvent s'expliquer par un transfert intramoléculaire de l'excédent d'énergie le long de la chaîne carbonée - du point d’impact de l'atome chaud au groupe hydroxyle - et la dissipation consécutive de cet excédent; en revanche, bien que l ’énergie des atomes de tritium de recul dépasse l'énergie de liaison chimique, cette dernière agit malgré tout sur la conservation des molécules qui ont une chaîne carbonée de même longueur, mais possèdent des groupes de substitution différents. ■
РЕАКЦИИ Г О РЯ Ч И Х АТО М О В Т Р И ТИ Я С АМ ИНОКИСЛОТАМ И. В литературе в настоящее время отсутствую т систематические данные о взаимодействии атомов отдачи трития с аминокислотами. В то же время, такие данные в сочетании с имеющимися р езульта тами для органических кислот и аминов могли бы помочь установить механизм горячих реакций, в зависимости от структуры соединений (длина цепи, функциональные зам естители ).
411

412 Е.Ф. СИМОНОВ и Ан.Н. НЕСМЕЯНОВ
Были исследованы выходы реакции горячих атомов трития: 1) с аминокислотами при увеличении длины цепи последних, 2) с аминокислотами одной длины углеродной цепи, но разными функциональными заместителями.
Для этой цели смеси карбоната лития с исследуемыми кислотами были облучены потоком медленных нейтронов 0,87*1013 с м 2/сек в течение 15 минут. Анализ проводился на г а зовом хроматографе со сменными колонками (молекулярные сита, вазелиновое масло на диатомите) и с помощью бумажной хроматографии.
Полученные данные для радиационной выживаемости аминокислот в зависимости от длины углеродной цепи, хотя и противоречат основному положению радиационной химии, согласно которому сохранение молекул возрастает с удлинением их цепи, могут быть объяснены внутримолекулярной передачей энергии по углеродной цепи от места удара горячего атома к гидроксильной группе и последующим ее "высвечиванием", с другой стороны, хотя энергия атомов отдачи трития превосходит энергию химической связи, последняя все-таки оказывает свое влияние на радиационное сохранение м олекул одной длины углеродной цепи, но с разными зам естителям и .
REACCIONES DE ATOMOS DE TRITIO CALIENTES CON AMINOACIDOS. Las obras publicadas hasta la fecha no contienen datos sistemáticos sobre la interacción de los átomos de retroceso de tritio con aminoácidos. Tales datos, en conjunción con los resultados obtenidos para los ácidos orgánicos y las aminas, podrían ayudar a establecer el mecanismo de las reacciones de átomos calientes referido a la estructura de los compuestos (longitud de la cadena, naturaleza de los grupos funcionales).
Los autores investigaron los rendimientos de las reacciones de los átomos calientes de tritio: a) con aminoácidos, de longitud de cadena c r e c ie n t e ; b ) c o n a m in o á c id o s de longitud de cadena constante, pero que
contenían distintos grupos funcionales.Con esta finalidad irradiaron durante 15 min, con un flujo de 0, 87 . 1013 neutrones lentos/cm2. s,
mezclas de carbonato de litio con los aminoácidos que se querían estudiar. Efectuaron los análisis cromato-
gráficos en fase gaseosa con ayuda de columnas intercambiables (tamices moleculares, parafina líquida en diatomeas) asf como por cromatografía sobre papel.
Los datos obtenidos acerca de la resistencia a la radiólisis de los aminoácidos en función de la longitud de la cadena carbonada, aunque estén en contradicción con una de las reglas fundamentales de la radioquímica, a saber, que la resistencia de las moléculas aumenta con el aumento de la longitud de su cadena, pueden explicarse por la transferencia intramolecular de energía a lo largo de la cadena carbonada, desde el lugar de impacto del átomo caliente al grupo oxidrilo y su subsiguiente "desexcitación". En cambio, si bien la energía de los átomos de retroceso del tritio es mayor que la energía de enlace químico, esta última influye en la resistencia a la radiólisis de las moléculas cuya longitud de cadena es la misma, pero que contienen distintos grupos funcionales.
В работе исследовалось действие горячих атомов трития, получающихся по реакции L i6 (n ,a )H 3 на ряд аминокислот. Ставилась задача изучения влияния длины углеродной цепи и характера функциональной группы на радиационную устойчивость соединений, выход материнского соединения и ряда других продуктов, содержащих тритий, с целью выяснения применимости существующих концепций о характере реакций горячих атомов [ 1 ,2 ] к поликристаллическим образцам.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Смеси исследуемых аминокислот с карбонатом лития (1 :1 ) в запаянных, эвакуированных кварцевых ампулах облучались потоком медленных нейтронов 0,87*1013 см2/сек в течение 15 минут в ядерном реакторе типа ИРТ-1000. Анализ газовой фазы проводился методом газо-жидкостной хроматографии с использованием сменных колонок (молекулярные сита,

РЕАКЦИИ ГОРЯЧИХ АТОМОВ ТРИТИЯ 413
вазелиновое масло на диатомите). Облученные образцы перед анализом прогревались при 100° в течение нескольких часов. Конденсированная фаза исследовалась методом бумажной хроматографии в системах н- бутанол—уксусная кислота —вода (4: 1:5) и фенол, насыщенный водой (точность анализа 2,5—4%).
Для определения удельной активности продуктов разложения и радиохимического выхода материнских молекул проводилось препаративное разделений исследуемых соединений на бумаге с последующей их экстракцией и выделением. Так как в процессе хроматографирования подвижные атомы водорода "отмываются", идентифицированные соединения содержат атомы трития лишь в неподвижных положениях.
Измерение активности осуществлялось с помощью проточного 2 - 7гсчетчика в пропорциональном режиме.
РЕЗУЛЬТАТЫ РАБОТЫ И ИХ ОБСУЖДЕНИЕ
Результаты работы представлены в табл.1 и 2 и на рис. 1 — 9.Из табл.1 видно, что переход от простых аминокислот к более слож
ным не сопровождается существенными изменениями выхода активных газообразных продуктов. Более того, выход С3Н7Т остается постоянным, а выход С2Н5Т - меняется мало. Если сопоставить этот факт с данными для продуктов разложения различных материнских молекул, образующихся при отщеплении С2 Н5Т или С3 Н7Т (та бл .2), то оказывается, что удельная активность этих продуктов будет одинакова. Это означает, что независимо от длины углеродной цепи материнской молекулы отрыв С2 Н5Т и С3Н7Г - процесс равновероятностный. В то же время, чем длиннее цепь, тем выше удельная активность продуктов разложения с числом углеродных атомов на единицу меньше, чем в исходной молекуле, т .е . выживаемость максимальна при отрыве СН3 Т .С другой стороны, из таблицы 1 и рисунка 1 следует, что с увеличением длины углеродной цепи аминокислот радиационное сохранение материнской молекулы линейно падает. Объяснение этому факту можно было бы искать в возможности внутримолекулярной передачи энергии от места удара горячего атома по цепи к функциональной группе с последующим "высвечиванием". Однако со вершенно не ясна причина чрезвычайно низкого радиохимического выхода материнских соединений (т а б л .1 ). Можно лишь говорить о незначительном сохранении молекул после замещения атомов водорода на тритий. Однако зависимость радиохимического выхода и удельной активности от длины цепи углеродных атомов аминокислоты (рис . 2 и 3) характеризуется кривой с минимумом для аминомасляной кислоты . Это говорит о том, что наряду с уменьшением радиационной стойкости аминокислот, благодаря чему имеет место падение удельной активности от глицина до аминомасляной кислоты, наблюдается и какой-то другой процесс, приводящий к стабилизации молекул с 5 — 6 углеродными атомами.
Поскольку основное действие ионизирующих излучений на аминокислоты состоит в дезаминировании и декарбоксилировании [3,4J, целесообразно рассмотреть возможность протекания этих конкурирующих процессов.
Так как радиационное сохранение аминокислот изменяется линейно с увеличением длины углеродной цепи, а выход С 0 2 не подчиняется этой
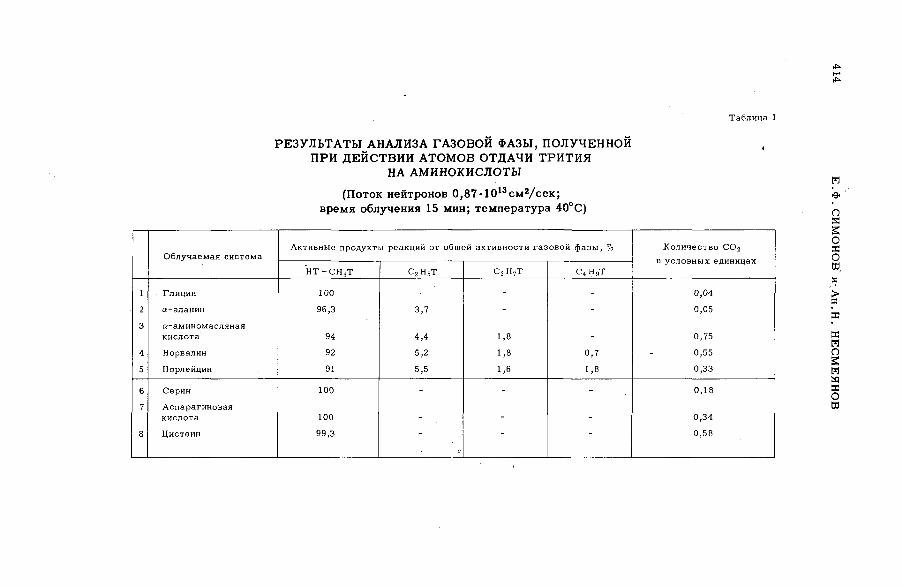
РЕЗУЛЬТАТЫ АНАЛИЗА ГАЗОВОЙ ФАЗЫ, ПОЛУЧЕННОЙ ,ПРИ ДЕЙСТВИИ АТОМОВ ОТДАЧИ ТРИТИЯ
НА АМИНОКИСЛОТЫ
(Поток нейтронов 0,87* 1013см2/сек; время облучения 15 мин; температура 40°С)
Таблица 1
Облучаемая системаАктивные продукты реакций от обшей активности газовой фазы, % Количество СОг
в условных единицахHT -t- СН3Т с 2 н 5т с 3 н 7т C4HgT
I Глицин 10 0 - * - - 0 ,0 4
2 а-аланин 9 6 ,3 3 ,7 - - 0 ,0 5
3 а-аминомаслянаякислота 94 4 ,4 1 ,8 - 0 ,7 5
4 Норвалин 92 5 ,2 1 ,8 0 ,7 - 0 ,5 5
5 Норлейцин . 91 5 ,5 1 ,6 1 ,8 0 ,3 3
6 Серии 100 - . - - . 0 , 1 8
7 Аспарагиноваякислота 10 0 - - - 0 , 3 4
8 Цистеин 9 9 ,3 ■ ■ 0 ,5 8
414 Е
.Ф.
СИ
МО
НО
В'и
Ан
.Н.
НЕ
СМ
ЕЯ
НО
В
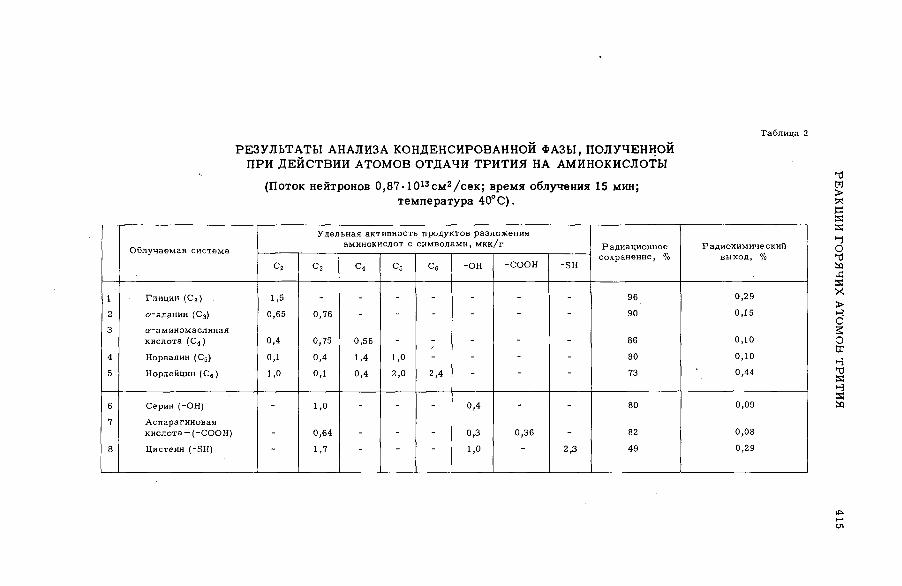
Таблица 2
РЕЗУЛЬТАТЫ АНАЛИЗА КОНДЕНСИРОВАННОЙ ФАЗЫ, ПОЛУЧЕННОЙ ПРИ ДЕЙСТВИИ АТОМ ОВ ОТДАЧИ ТРИТИЯ НА АМИНОКИСЛОТЫ
(Поток нейтронов 0,87-1013см2/сек; время облучения 15 мин; температура 40°С).
О б л у ч а е м а я с и с т е м а
У д е л ь н а я а к т и в н о с т ь п р о д у к т о в р а з л о ж е н и я а м и н о к и с л о т с с и м в о л а м и , м к к / г Р а д и а ц и о н н о е Р а д и о х и м и ч е с к и й
С2 с 3 С4 с 5 С6 -О Н - С О О Н -SHс о х р а н е н и е , % в ы х о д , %
1 Г л и ц и н ( С 2 ) . 1,5 - - - - - - - 96 0,29
2 с т - а л а н и н ( С 3) 0,65 0,76 - - - - - - 90 0,15 .
3 о ' - а м и н о м а с л я н а я
к и с л о т а { С 4 ) 0,4 0,75 0,56 - - - - - 86 0,104 Н о р в а л и н ( С 5) 0,1 0,4 1-4 1,0 - - - - - 80 0,105 Н о р л е й ц и н ( С 6 ) 1,0 0,1 0,4 2,0 2,4 - - - 73 ' 0,44
6 С е р и и ( - О Н ) - 1,0 - - - 0,4 - - 80 0,09
7 А с п а р а г и н о в а я
к и с л о т а — ( - С О О Н ) - 0,64 - - - 0 ,3 0,36 - 82 0 ,0 8
8 Цистеин (-SH ) 1,7 ■ ” “ 1,0■
2 ,3 49 0 ,2 9
РЕА
КЦ
ИИ
ГО
РЯЧ
ИХ
АТО
МО
В
ТРИТИ
Я
415

416 Е.Ф. СИМОНОВ и Ан.Н. НЕСМЕЯНОВ
Рис .1
Радиационное сохранение аминокислот с различной длиной углеродной цепи.
Р и с . 2
Радиохимический выход материнских молекул.
Рис .3
Удельная активность материнских молекул различной длины углеродной цепи.
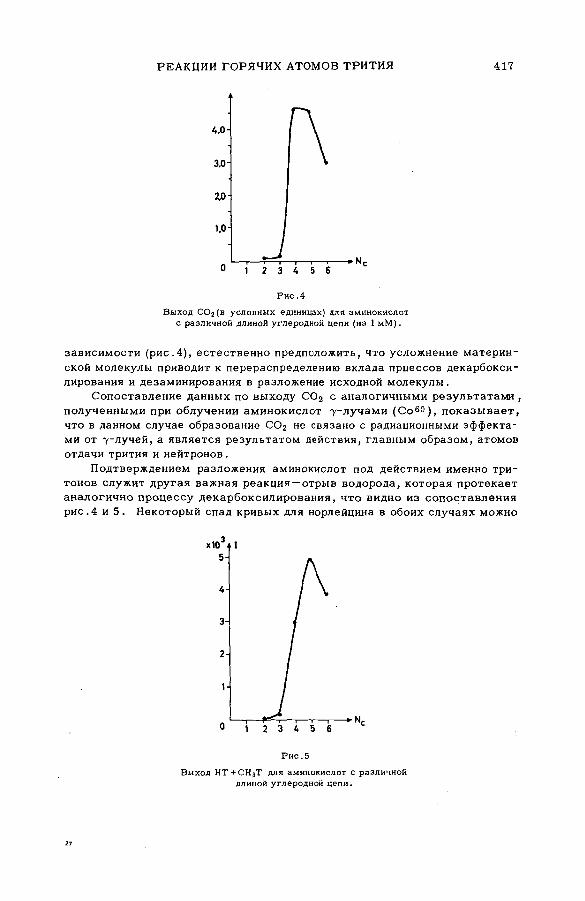
РЕАКЦИИ ГОРЯЧИХ АТОМОВ ТРИТИЯ 417
Рис .4
Выход СОа(в условных единицах) для аминокислот с различной длиной углеродной цепи (на 1 м М ).
зависимости (рис.4), естественно предположить, что усложнение материнской молекулы приводит к перераспределению вклада прцессов декарбокси- лирования и дезаминирования в разложение исходной молекулы.
Сопоставление данных по выходу С02 с аналогичными результатами , полученными при облучении аминокислот 7 - лучами (С о 60), показывает, что в данном случае образование С02 не связано с радиационными эффектами от 7 -лучей, а является результатом действия, главным образом, атомов отдачи трития и нейтронов.
Подтверждением разложения аминокислот под действием именно тритонов служит другая важная реакция —отрыв водорода, которая протекает аналогично процессу декарбоксилирования, что видно из сопоставления рис.4 и 5. Некоторый спад кривых для норлейцина в обоих случаях можно
Рис .5
Выход НТ + СН3Т для аминокислот с различной длиной углеродной цепи.
27

418 Е.Ф. СИМОНОВ и Ан.Н. НЕСМЕЯНОВ
отнести за счет разрыва связи углерод-углерод, происходящего в аминокислотах с длинной цепью.
При изучении ряда аминокислот (аланин, серин, аспарагиновая кислота и цистеин) одной длины углеродной цепи, но с различными заместителями было найдено, что наличие функциональных групп мало влияет на радиа-
Р и с .6Радиационное сохранение аминокислот с различными заместителями.
ционную устойчивость соединений (рис.6). Исключение составляет цистеин, для которого радиационная устойчивость много меньше, чем для остальных аминокислот. Заместитель изменяет соотношения конкурирующих процессов декарбоксилирования и дезаминирования из-за различной чувствительности к образующимся свободным радикалам. Особенно сильно эта чувствительность проявляется у тиольных групп, чем и объясняется то, что
Р и с .7
Выход СОа (в у с л о в н ы х е д и н и ц а х ) для различных заместителей (на 1 м М ).
цистеин —единственная кислота, которая не дезаминируется [5 ] . Следовательно, именно для этой аминокислоты должен в наибольшей степени происходить процесс декарбоксилирования. Оказалось, что цистеин действительно дает наибольшее количество СОг и что это количество резко падает с увеличением энергии связи заместителя с углеродом (уменьшается чувствительность к свободным радикалам).
27*
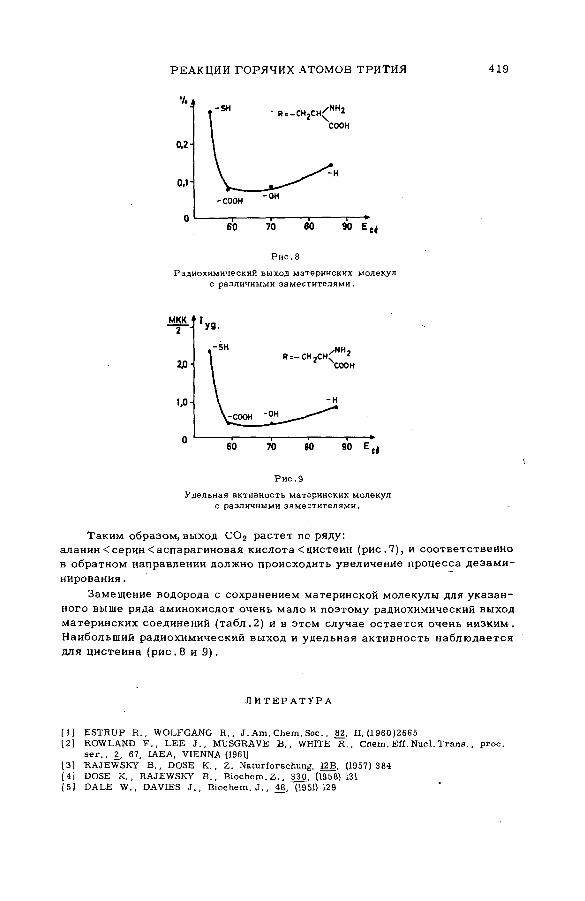
РЕАКЦИИ ГОРЯЧИХ АТОМОВ ТРИТИЯ 419
Р и с . 8Радиохимический выход материнских молекул
с различными заместителями.
Р и с .9
Удельная активность материнских молекулс различными заместителями. -
Таким образом, выход С 02 растет по ряду: аланин < серии < аспарагиновая кислота <цистеин (рис. 7), и соответственно в обратном направлении должно происходить увеличение процесса дезаминирования .
Замещение водорода с сохранением материнской молекулы для указанного выше ряда аминокислот очень мало и поэтому радиохимический выход материнских соединений (та бл .2) и в этом случае остается очень низким. Наибольший радиохимический выход и удельная активность наблюдается для цистеина (рис. 8 и 9).
Л И Т Е Р А Т У Р А
[1 ] E S TR U P R . , W O LFG ANG R . , J. Am . Chem. S oc ., 82. 11.(1960)2665[2 ] RO W LAND F . , LEE J ., M USGRAVE B . , W HITE R . , Chem. E ff. Nuel. T ra n s ., proc.
s e r . . 2, 67, IA E A , V IE N N A (1961)13] RAJEW SKY B ., DOSE K ., Z . Naturforschung, 12B, (1957) 384[4 ] DOSE K ., RAJEW SKY B . , B io ch em .Z ., 330, (1958) 131[5 ] D A LE W ., DAVIES J ., B iochem . J ., 48, (1951) 129 '


A TRIGGERING MECHANISM FOR THE PROMOTION OF THERMAL ANNEALING IN
CRYSTALLINE HEXABROMOETHANE BY RADIATION-PRODUCED DEFECTS
K .E . COLLINS DEPARTMENT OF CHEMISTRY,
STATE UNIVERSITY OF NEW YORK AT BUFFALO, BUFFALO, N .Y . ,
UNITED STATES OF AMERICA
Abstract — Resume — Аннотация — Resumen
A TRIGGERING MECHANISM FOR THE PROMOTION OF THERMAL ANNEALING IN CRYSTALLINE HEXABROMOETHANE BY RADIATION-PRODUCED DEFECTS. Thermal annealing of recoil-Br82 to the parent
form in hexabromoethane appears to be due to at least five different processes. Several o f these occur as independent processes in the absence of radiation-produced defects; the over-all kinetics of these processes are resolvable into first order components. However, a substantial fraction of the recoil-Br82 does not anneal to the parent hexabromoethane form, even at temperatures as high as 120°C, in the absence o f radiation- produced defects. When defects are produced in hexabromoethane crystals by ionizing radiation at -196®C prior to the introduction of the recoil-Br82 atoms, these defects tend to promote the subsequent thermal annealing of an additional fraction of the recoil-Br82 atoms. This fraction is a logarithmic function of the total dose.
The promotion of the "nonrannealing" Br82 species to species that anneal at temperatures in the 0° to 120°C range has recently been found to occur by a sequence o f steps, whereby increasing doses of ionizing radiation first promote one kind o f annealing process up to a point o f saturation, then proceed to promote other annealing processes. A model is introduced to account for the observed changes in the distribution of recoil-Br82 among the various annealing and non-annealing components as a function of the piior-inadiation dose. The model assumes a radiation-produced species that is stable in the crystalline hexabromoethane at -196°C. This species can diffuse at annealing temperatures to interact with recoil-Br82 sites and promote the thermal annealing of these sites. The kinetics of thermal annealing in hexabromoethane at several temperatures and at several dose levels are discussed in terms of this model. The possible extension of this model to radiation annealing in hexabromoethane and to other crystalline compounds is also discussed.
PRODUCTION DE DÉFAUTS RADIOINDUITS DANS DES CRISTAUX D'HEXABROMOÉTHANE POUR EN FAVORISER LE RECUIT. Le retour de 82Br de recul à la forme du générateur sous l'e ffe t d’un traitement thermique dans l ’hexabromoéthane semble être dû à cinq phénomènes différents pour le moins. Pour plusieurs,
il s’agit de processus indépendants en l'absence de défauts radioinduits; la cinétique globale de ces processus peut être résolue en des composants du premier ordre. Cependant, une partie importante de 82 Br de recul ne
reprend pas la forme du générateur dans l'hexabromoéthane, même à des températures qui atteignent 120eC, en l'absence de défauts radioinduits. Lorsque des rayonnements ionisants produisent des défauts dans des cristaux d’hexabromoéthane à -196°C avant introduction de 82 Br de recul, ces défauts ont tendance à favoriser le recuit thermique ultérieur d’ une fraction supplémentaire de 82 Br de recul. Cette fraction est une fonction logarithmique de la dose totale.
On a constaté récemment que la transition de l ’ espèce 82Br «n on susceptible de recu it» à l ’ espèce pouvant subir le recuit à une température comprise entre 0 et 120eC est une évolution en plusieurs étapes, dans
laquelle les doses croissantes de rayonnements ionisants favorisent d'abord un processus jusqu’à uncertain point de saturation, et ensuite d'autres processus de recuit. L'auteur présente un modèle qui permet d’ expliquer les changements observés dans la distribution de 82 Br de recul entre les divers composants susceptibles ou non de recuit en fonction de la dose d’ irradiation préalable. Le modèle suppose l ’existence d'une espèce radioinduite qui reste stable dans les cristaux d'hexabromoéthane à -196°C. Cette espèce peut diffuser aux températures
de recuit pour agir sur les positions 82Br de recul et favoriser ainsi le recuit thermique de ces positions. L'auteur étudie la cinétique du recuit thermique dans l ’hexabromoéthane, à plusieurs températures et pour des doses
421

422 К. Е. COLLINS
différentes, en se fondant sur ce modèle. Il envisage également la possibilité d'appliquer ce modèle au recuit par irradiation dans l ’hexabromoéthane et examine les résultats obtenus avec des cristaux d’autres composés;
г
МЕХАНИЗМ, ПРИВОДЯЩИЙ" К УСИЛЕНИЮ ТЕПЛОВОГО ОТЖ ИГА В КРИ СТАЛЛИ ЧЕСКОМ ГЕ К С А Б РО М Э ТАН Е ЗА С ЧЕТ ДЕФЕКТОВ, ВЫЗВАННЫХ ИЗЛУЧЕНИЕМ. Превращение атомов отдачи брома-82 в исходную форму гексабромэтана,по-видимому, достигается в р е зультате по крайней мере пяти различных процессов термического отж ига. Некоторые из них происходят в виде независимых процессов при отсутствии дефектов, вызванных излучением; общая кинетика этих процессов разлагается на компоненты первого порядка. Однако значительная часть атомов отдачи брома-82 не возвращается с помощью отжига в исходную
форму гексабромэтана даже при таких высоких температурах как 120® при отсутствии дефектов, вызванных излучением. Когда дефекты вызываются в кристаллах гексабромэтана за счет ионизирующей радиации при температуре -196е до введения атомов отдачи брома-82, эти дефекты имеют тенденцйю способствовать последующему тепловому отживу дополнительной части атомов отдачи брома-82. Эта часть является логарифмической функцией общей до зы .
Недавно было обнаружено, что преобразование "неотжигающихся" форм брома-82 в формы, которые отжигаются при температурах в интервале от 0 до 120° С, происходит постепенно, в результате чего увеличивающиеся дозы ионизирующего излучения сначала способствую т одному из видов процесса отжига вплоть до точки насыщения, а затем — другим процессам отж ига. Представляется модель для объяснения наблюдаемых изменений в распределении атомов отдачи брома-82 среди различных отжигающихся и неотжигающихся компонентов в зависимости от дозы предшествующего облучения. Модель предполагает наличие вызванной излучением формы, которая является устойчивой в кристаллическом гексабромэтане при тем пературе -19 6 °. Эта форма может диффундировать при температурах отжига и взаимодействовать с локациями атомов отдачи брома-82 и способствовать тепловому отжигу этих л о каций. Кинетика теплового отжига в гексабром этане при нескольких температурах и при нескольких дозах рассматривается с точки зрения этой модели. Рассм атривается также возможность распространения этой модели на радиационный отжиг в гексабром этане и на другие кристаллические соединения.
UN MECANISMO INICIADOR DE LA REGENERACION TERMICA EN HEXABROMOETANO CRISTALINO POR DEFECTOS PRODUCIDOS POR IRRADIACION. A l parecer, la regeneración térmica del 82 Br de retroceso a la forma original en el hexabromoetano es debida'por lo menos a cinco procesos diferentes. Varios de estos procesos se producen independientemente en ausencia de defectos producidos por irradiación y su cinética global puede reducirse a componentes de primer orden. Sin embargo, una fracción considerable del 82Br de retroceso no vuelve a la forma original de hexabromoetano, ni aun a temperaturas hasta de 120°C, en ausencia de defectos originados por irradiación. Cuando las radiaciones ionizantes producen defectos en cristales de hexabromoetano a -196°C antes de la introducción de átomos de 82 Br de retroceso, dichos defectos tienden a fomentar la regeneración térmica subsiguiente de una fracción adicional de los átomos de 82 Br de retroceso. Dicha fracción es función logarítmica de la dosis total.
Se ha encontrado recientemente que la transición de la especie de 82 Br "que no se regenera" a otra que se reeenera a temperaturas comprendidas entre 0* v 120eC, constituye una evolución en varias etapas, en la cual las dosis crecientes de radiaciones ionizantes favorecen en primer lugar una clase de proceso de regeneración hasta llegar a un punto de saturación para comenzar después a estimular otros procesos de regeneración. El autor propone un modelo para explicar las variaciones que se han observado en la distribución del 82Br de retroceso entre los diversos componentes, los que se regeneran y los que no se regeneran, en función de la dosis de irradiación previa. El modelo supone la existencia de una especie producida por irradiación que es estable en el hexabromoetano cristalino a. -196*C. Esta especie puede difundirse a las temperaturas de recocido para producir la interacción con los puntos de red del 82 Br de retroceso y fomentar la regeneración térmica de dichos puntos. Basándose en este modelo, se discute la cinética de la regeneración térmica en el hexabromoetano a diversas temperaturas y dosis. También se examina la posibilidad de aplicar este modelo a la "regeneración por irradiación” en el hexabromoetano, y a otros compuestos cristalinos. '

THERMAL ANNEALING IN CRYSTALLINE HEXABROMOETHANE 423
INTRODUCTION
Annealing processes that occur in the hot-atom chemistry of crystalline solids may be either loca lized reconstructions involving only a hot-atom - labelled species reacting with neighbouring fragments produced during the same nuclear event, or they may be processes involving other species that are produced external to the re co il site and diffuse to it. The annealing reactions that occur within isolated sites in the crysta l m atrix and that do not involve interaction with other, diffusing species have been term ed "in trinsic annealing reactions'1 LI ) . Two or m ore intrinsic annealing processes are thought to occur in hexabromoethane [1] . In addition, other annealing reactions in hexabromoethane involve the participation of species other than those produced in the nuclear activation event. The participation of species produced by ionizing radiation, whether introduced before, during o r a fter the introduction of the reco il atoms, has been found to greatly a ffect the course of the subsequent thermal annealing 12]. Qualitatively, this effect in hexabromoethane is one of promotion of the therm al annealing r e action. This promotion by radiation-produced defects has been regarded as the interaction of a crysta l defect, perhaps an exciton, electron, hole o r vacancy, with the re co il s ite. Thus the interaction can be pictured as a tr ig ger in g process in which either the diffusing defect deposits energy into the reco il site to activate annealing reactions, or the defect interacts with the reco il site to produce a strain relaxation whereby the activation energy required fo r subsequent reorganization (annealing) in the reco il site is lowered.
A s im ila r picture involving m igrating defects can be used to account fo r the radiation annealing reactions of hexabromoethane that occur when hexabromoethane is exposed to ionizing radiation at a suitably high temperature (e .g . 0°C) a fter the introduction of the reco il atoms into the crysta ls [2,3].
In addition to in trinsic therm al annealing, defect-prom oted therm al annealing and radiation annealing, the annealing of radiolytic bromine and the "annealing out" of radiation-produced defects have also been demonstrated in hexabromoethane [2] . The possib ility that the same o r s im ila r underlying processes contribute to both the hot-atom annealing and the annealing of radiolytic bromine was recognized severa l years ago (.2J. However, the recent demonstrations [4 ,5 ] of doped-crysta l annealing (which in potassium chromate bears even a quantitative s im ilarity to the hot-atom annealing case 15]) have re-em phasized that some hot-atom annealing r e actions may not require "hot" atoms. In other words, the kinetics ofthermal "hot-atom " annealing in crysta lline solids may be determ ined la rg e ly by bulk defect and im purity properties of the crystalline system', and the detailed structure of the hot atom reco il sites may be o f little importance to thè kinetics of the annealing reaction.
In the follow ing paragraphs a m odel fo r radiation-promoted annealing is suggested that incorporates this idea that species from the bulk o f the crysta ls can, in some cases, act as rate-determ in ing factors in the hot- atom annealing of hexabromoethane. Experimental facts about the severa l different types of annealing in hexabromoethane w ill be described in terms of the proposed model.

424 К. Е. COLLINS
The hot-atom therm al annealing reaction in hexabromoethane appears to be a thermal conversion of radiobromine from the form Br| to the parent C2Bri? form 12]. S im ilarities in the general appearance o f the isotherms fo r annealing o f rad io ly tica lly produced brom ine and fo r annealing in the hot-atom case suggest that a corresponding incorporation of B r 2 -bromine into parent hexabromoethane also takes place in the annealing of radiolytic brom ine (.2 j . Thus we shall assume, fo r s im plicity, that both rad io lysis anealing and hot-atom annealing in hexabromoethane have the same anneal- able state o f brom ine. To distinguish the hot-atom and the rad io ly tica lly produced states, we shall re fe r to them as S (B r2) and S (B r2) (o r S* and S) respective ly. In addition, two or m ore other annealing states are present in the hot-atom case only; namely, the states involved in intrinsic annealing, I 3(B r f), 1г(Вг|), etc. .
Ionizing radiation produces not only the S (B r2) states but also species capable of promoting the annealing of states S (Br*) and S(Br2). We assume, fo r sim plicity, that there is one such in itia lly produced species, 6m, a mobile defect capable of triggering the annealing of S (Br2j or S (Br2) attempera- tures appreciably grea ter than -196°C but not at -196°C. The diffusing species, 6m, can have severa l possible fates at -196°C. They can interact with one another to form self-trapped pairs (such as a divacancy 16 J ) that can be dissociated at higher tem peratures. They can diffuse to the S (B rf) and S (Br2) sites at -196°C, as at higher tem peratures, but cannot tr ig g e r annealing at such low temperatures. A lternatively, they can be immobilized by trap-states present in the crystal matrix: 6ra +trap I -» 6 j. The trap states are pictured schem atically in F i g . l . These traps are filled in the o rder of their depth; the deepest traps, type I, are filled firs t, then the traps of type II, etc. Upon warming, mobile defects are released from the shallow traps at low er temperatures than are required to release defects from the
MODEL FOR RADIATION-PROMOTED ANNEALING
F ig .l
Trap states for hexabromoethane
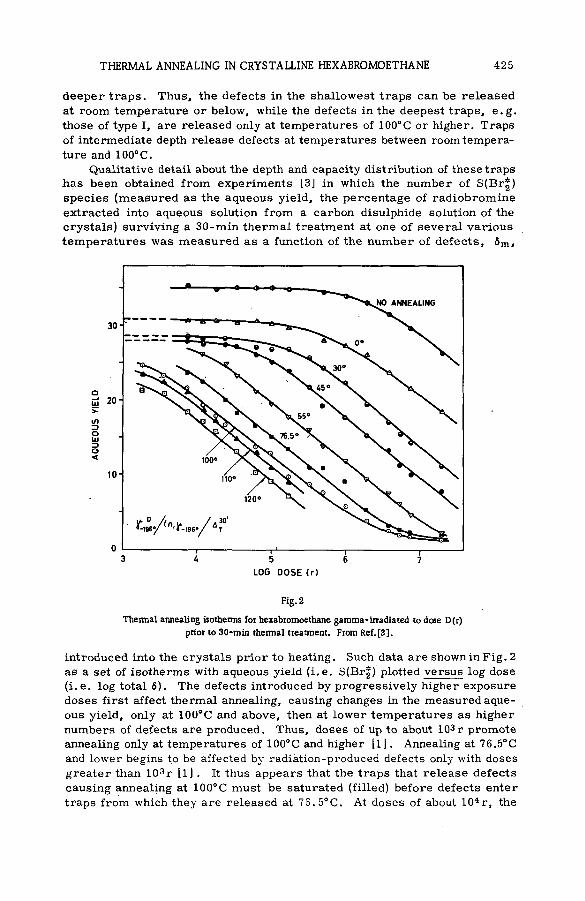
THERMAL ANNEALING IN CRYSTALLINE HEXABROMOETHANE 425
deeper traps. Thus, the defects in the shallowest traps can be re leased at room temperature or below, while the defects in the deepest traps, e .g . those of type I, are released only at temperatures of 100°C or higher. Traps of intermediate depth release defects at temperatures between room temperature and 100°C.
Qualitative detail about the depth and capacity distribution of these traps has been obtained from experiments 13J in which the number of S(Br£) species (measured as the aqueous yie ld , the percentage o f radiobrom ine extracted into aqueous solution from a carbon disulphide solution of the crysta ls ) surviving a 30-min therm al treatment at one o f severa l various tem peratures was measured as a function o f the number o f defects, 6m,
Fig. 2
Thermal annealing isotherms fo i hexabromoethane gamma-irradiated to dose D (r) prior to 30-min thermal treatment. From Ref.[3],
introduced into the crystals p r io r to heating. Such data are shown in F ig . 2 as a set of isotherms with aqueous yield ( i .e . S(Br|) plotted versus log dose ( i .e . log total 6). The defects introduced by progress ively higher exposure doses firs t affect thermal annealing, causing changes in the measured aqueous yie ld , only at 100°C and above, then at low er temperatures as higher numbers of defects are produced. Thus, doses of up to about 103r promote annealing only at temperatures of 100°C and higher [ l j . Annealing at 76.5°C and lower begins to be affected by radiation-produced defects only with doses grea ter than 103r [1] . It thus appears that the traps that re lease defects causing annealing at 100°C must be saturated (filled ) before defects enter traps from which they are released at 76.5°C. At doses of about 104r, the

4 2 6 К. Е. COLLINS
effect of dose on the 76.5°C annealing becomes negligible; i .e . those traps have become filled . Additional defects enter the shallower traps that affect annealing at low er tem peratures. Annealing at 0°C and low er is observed only at doses of substantially greater than 105r .
Thus, the defects from the firs t 103r enter and f i l l trap state I from which they are liberated at 100°C and higher. The additional defects introduced by irradiations of from 103 r to 105 r then f i l l trap states II and III, to be released at temperatures of 40°-80°C. Trap states IV and V are then filled , in turn, by doses o f 10br and above. The m obile defects are r e leased from these traps at temperatures low er than 40°C. The mobile defects, when released, can then interact with S ^ r^ ) and S (B r2) sites to tr ig g e r the reincorporation of bromine into hexabromoethane.
DESCRIPTION OF EXPERIM ENTAL RESULTS USING PROPOSED MODEL
Therm a l annealing
Three general types o f therm al annealing have been studied in hexabromoethane: conventional hot-atom annealing, annealing of radiation- produced defects (with hot-atom annealing as the measure of defects) and annealing of radiolytic brom ine. In conventional hot-atom annealing, the "in trinsic" annealing processes take place rapidly at room temperature and above, and are essentially complete in several minutes at 30-40°C or above [1] . Added to the intrinsic annealing are the radiation-defect promoted processes 12, 3] . Irradiation p r io r to the introduction of the hot atoms produces radiolytic S(Br2) states and trapped defects. At high (about 10br) predoses of ionizing radiation, most of the traps I-V are filled . A fter the introduction of the hot atoms, S(Br|), therm al treatment at a temperature of about 0°C results in some intrinsic annealing and some radiation-promoted annealing due to the reaction of mobile defects with S (B r2) sites follow ing the thermal liberation o f the trapped defects from the shallower traps, e .g . IV and V . At temperatures of 30°-80°C, more defect-promoted thermal annealing results from reaction of defects liberated from intermediate traps,II and III. At 100°C and higher, additional annealing results from defects liberated from the deep, type I, traps. Annealing isotherms are shown in F ig . 3a fo r hexabromoethane that received 2X 106r of gamma irradiation p r io r to the neutron activation. We see the intrinsic and shallow-trap annealing at 0°C, the intermediate trapped-defect-promoted annealing at 30°; 47°, 62° and 76.5°C, and the annealing caused by the defects from the type I traps at 100°C. The kinetics o f the observed therm al annealing might be expected to be quite complex since the thermal liberation of trapped defects, the diffusion of these defects through the crysta l and the actual annealing of the S(Br|) site after the triggering by the defect might a ll enter im portantly into determining the o ve r-a ll k inetics. The observed kinetics have been interpreted in term s of several firs t-o rd er processes and one second- order process [2] .
If the crysta ls of hexabromoethane are firs t irradiated to a high dose (about 106r ), to introducé S (B r2) sites and to f i l l most of the traps 1-V with defects, and the crysta ls a re then heated at a tem perature sufficient only
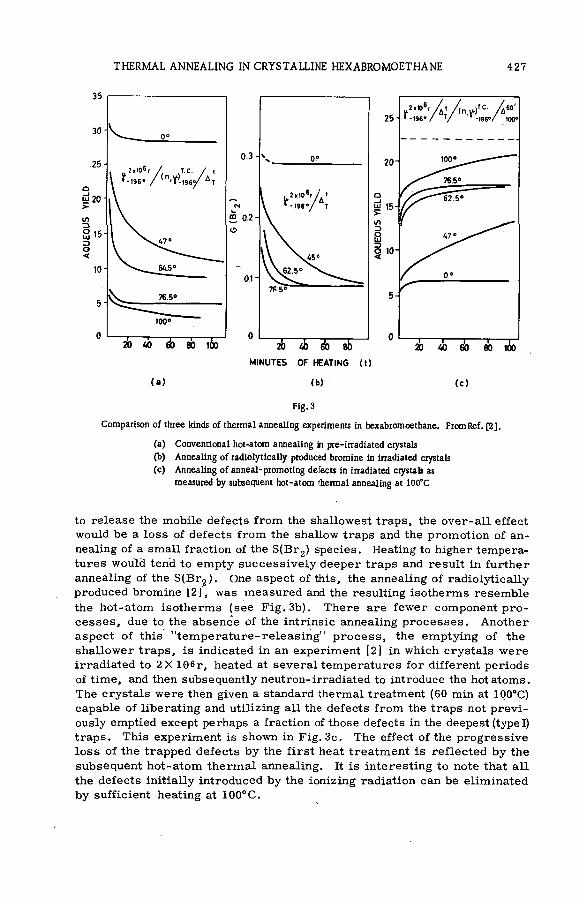
THERMAL ANNEALING IN CRYSTALLINE HEXABROMOETHANE 427
( a ) <b) ( e )
Fig.3
Comparison of three kinds of thermal annealing experiments in hexabromoethane. From Ref. [2].
(a) Conventional hot-atom annealing in pre-irradiated crystals
(b) Annealing of radiolytically produced bromine in irradiated crystals
(c) Annealing of anneal-promoting defects in irradiated crystals as
measured by subsequent hot-atom thermal annealing at 100°C
to release the mobile defects from the shallowest traps, the o ve r-a ll effect would be a loss of defects from the shallow traps and the promotion of annealing of a small fraction of the S (B r2) species. Heating to higher temperatures would tend to empty successively deeper traps and result in further annealing of the S (Br2). One aspect o f this, the annealing o f rad io lytica lly produced bromine 12], was measured and the resulting isotherms resemble the hot-atom isotherms (see F ig . 3b). There are few er component p rocesses, due to the absence of the intrinsic annealing processes. Another aspect o f this "tem pera tu re-re leas ing " process, the em ptying o f the shallower traps, is indicated in an experiment [2] in which crysta ls w ere irradiated to 2X 106r, heated at severa l temperatures fo r different periods of time, and then subsequently neutron-irradiated to introduce the hot atoms. The crystals were then given a standard thermal treatment (60 min at 100°C) capable o f liberating and utilizing a ll the defects from the traps not p rev iously emptied except perhaps a fraction of those defects in the deepest (type I) traps. This experiment is shown in Fig. 3c. The effect of the progressive loss o f the trapped defects by the f ir s t heat treatm ent is re flec ted by the subsequent hot-atom therm al annealing. It is in teresting to note that a ll the defects in itia lly introduced by the ionizing radiation can be elim inated by sufficient heating at 100°C.

428 К. Е. COLLINS
Ionizing radiation introduces mobile defects, 6m, into the crystals. A lthough the defects cannot cause annealing to occur at -196°C, they can trigger annealing at 0°C. Thus, crystals containing annealable S (B r2) are expected to show annealing when irradiated at o r above 0°C due to the-direct in teraction between 6m and S(Br|). The kinetics of radiation annealing are thus assumed to be described by an equation such as
= k [S (B r* )][6 m].
If the term [6m] were constant, radiation annealing would be a firs t-o rd er process. However, the defects, 5m, have severa l fates other than the anneal- promoting reaction with S(Br|). They can react with S(Br2), causing annealing o f these species, o r they can be trapped. Since the concentration of the S (Br2) states increases during an irradiation [7] the steady state value o f [6m] is expected to decrease. Th is resu lts in a deviation from fir s t - order annealing, as is shown in F ig . 4, which is a " firs t-o rd e r" plot of hot- atom radiation annealing data fo r hexabromoethane irrad iated at 0°C.
Radiation annealing
TIM E (m in )
Fig. 4
Fiist order plot of 0°C radiation annealing data for hexabromoethane. From Ref. [3].
Intensity e ffect
The intensity (dose-rate) employed in pre-irrad iation ( i . e . irradiation given at -196°C prior to neutron activation) o r in radiation annealing ( i . e . irradiation given at an annealing temperature a fter introduction of the hot atoms) has an important influence on the observed degree of annealing [3] . F igure 5a shows that higher intensities during pre-irrad iation give rise to
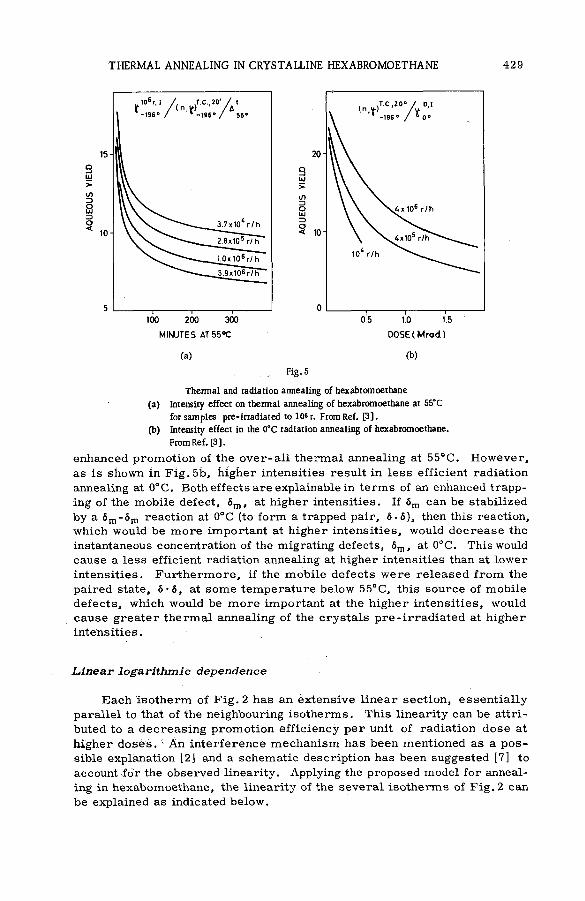
THERMAL ANNEALING IN CRYSTALLINE HEXABROMOETHANE 429
MINUTES AT 55°C
(a) (b)
Fig. 5
Thermal and radiation annealing of hexabromoethane
(a) Intensity effect on thermal annealing of hexabromoethane at 55“C
for samples pre-irradiated to 106r. From Ref. Р ].
(b) Intensity effect in the 0°C radiation annealing of hexabromoethane.
From Ref. [3].
enhanced prom otion of the o v e r -a ll therm al annealing at 55°C. H owever, as is shown in F ig . 5b, higher intensities result in less efficien t radiation annealing at 0°C. Both effects are explainable in term s of an enhanced trapping of the mobile defect, 6m, at higher intensities. I f 5m can be stabilized by a 6m-Sm reaction at 0°C (to form a trapped pair, 6-6), then this reaction, which would be m ore important at higher in tensities, would decrease the instantaneous concentration of the migrating defects, 6m, at 0°C. This would cause a less efficient radiation annealing at higher intensities than at lower in tensities. Furtherm ore, if the m obile defects w ere re leased from the paired state, S-б, at some temperature below 55°C, this source of mobile defects, which would be m ore important at the h igher in tensities, would cause g rea ter therm al annealing of the crysta ls p re-irrad ia ted at higher intensities.
L in e a r lo g a r ith m ic dependence
Each isotherm of F ig . 2 has an extensive lin ea r section, essen tia lly para lle l to that of the neighbouring isotherms. This linearity can be a ttr ibuted to a decreasing prom otion e ffic ien cy p er unit o f radiation dose at h igher dósés. ; Ah in terference mechanism has been mentioned as a possible explanation 12J and a schematic description has been suggested [7] to account fo r the observed linearity. Applying the proposed model for annealing in hexabomoethane, the lin earity of the severa l isotherm s of F ig . 2 can be explained as indicated below.

430 К. Е. COLLINS
If d 6 m signifies the number of mobile defects resulting from ал increment of irradiation dose, dD, and dS* is the additional number of S (Br2) species whose annealing is promoted by the defects, dô^ we can express the amount o f additional annealing, dS*, as the product o f the number of additional promoting defects, dôm, and the probability of interaction of an annealable S (B rf) site and a diffusing defect, 6ro. As reaction with the relat ive ly large numbers of S (B r2) species (produced by the ionizing radiation) is the principal fate of the defects, 6m, the probability of aôm-S (B r2 ) r e action is determined by the relative dilution factor, 1/S, where S is the number of S (B r2) species present. Therefore
dS* = k dôm (1/S),
where к is a proportionality constant. But with the increased number of defects, d6m, proportional to the dose increment, dD, and the number of S (B r2) species, S, proportional to the dose, D, we have
dS* = k' = k 'd (In D)
or
dS* _ k ,
d (In D) ’
which expresses the observed linear logarithm ic dependence.
SUM M ARY
The model suggested in this paper is an attempt to explain the various annealing data of hexabromoethane with a minimum number of assumptions. It is possible that this model, o r one s im ila r to it, can also be applied to hot-atom annealing in other crystalline systems. This model assumes that therm al annealing in hot-atom chem istry takes place by two d ifferent p rocesses; intrinsic annealing, i . e . loca lized reconstructions involving only the labelled species and its neighbouring fragments, and defect (radiation)- prom oted annealing. In this la tte r case, the annealing is assumed to be la rge ly due to the properties of the bulk crystals independent of the specific character of the "hot atom". That is, the detailed structure of the hot-atom site is assumed not to be a controlling factor in the annealing kinetics of the hot atoms. The annealing is thought to be promoted by mobile defects that can be introduced into the crystals and trapped in different trap states during irrad iation , to be subsequently re leased at elevated tem peratures to t r ig g e r annealing. The same m obile defects are assumed to prom ote radiation annealing.

THERMAL ANNEALING IN CRYSTALLINE HEXABROMOETHANE
A C K N O W L E D G E M E N T S
4 3 1
The author gratefu lly acknowledges the substantial contributions of P ro fe sso r J .E . W illard , D r. G. Harbottle and D r. C .H . Collins to thé development of the work reported in this paper.
R E F E R E N C E S
[1] COLLINS, K.E. and HARBOTTLE. G. , Parti, Radiochim. Acta (in press).[2] COLLINS, K.E. and WILLARD, J .E ., J. chem. Phys. 37 (1962) 1908.[3] COLLINS, K.E. and HARBOTTLE, G. , Part II, Radiochim. Acta (in press).[4] (a ) KAUCIC, S. and VLATKOVIC, M . , Croatica chemica Acta 35(1963) 305.
(b) APERS, D.J. , COLLINS, К. E ., COLLINS, C. H ., GHOOS, Y. F. and С APRON, P. C . , Radiochim. Acta (in press).
[5 ] COLLINS, C .H ., COLLINS, K .E ., GHOOS, Y. F ., APERS, D.J. and CAPRON, P. C. , submitted to Radiochim. Acta.
[6] VAN BUEREN, H. G. , Imperfections in Crystals, North-Holland Publishing Co. , Amsterdam (1961)610.[7] COLLINS, K .E ., Ph.D. Thesis, University o f Wisconsin (1961).


CHEMICAL EFFECTS OF THE NUCLEAR ISOMERIC - - TRANSITION OF Br.eom щ GLASSY AND POLYCRYSTALLINE ALKYL BROMIDES *
R. M . A . HAHNE A N D J. E. WILLARD DEPARTMENT OF, CH EM ISTRY, UNIVERSITY OF W ISCON SIN,
M A D ISO N , W IS. , UNITED STATES OF AM ERICA,
A N D KERNFORSCHUNGSANLAGE JÜLICH,. FEDERAL REPUBLIC OF GERMANY
Abstract — Résumé — Аннотация — Resumen
CHEMICAL EFFECTS OF THE NUCLEAR ISOMERIC TRANSITION OF Br80111 IN GLASSY AND POLYCRYSTALLINE ALKYL BROMIDES. The organic yields from BrBr8om undergoing the Br80m -> Br80 isomeric transition in polycrystalline n-C3H7Br are essentially independent o f Br2 concentration over a wide range, supporting other evidence that the bromine is present as a homogeneous solution and indicating that the fate o f the Br80 is determined very close to the site of birth. Organic yields from BrBr8om in n-C4H9Br are higher . in the glassy state than the polycrystalline state. In both glassy and polycrystalline П-С4Н9ВГ the organic yields when the Br80m is in the form of П-С4Н9 Br8om are much higher than when it is in the form o f BrBr80m.
In polycrystalline samples they are not appreciably changed by the presence o f 5x 10"3 mole fraction o f Br2 but are significantly reduced by this concentration o f bromine in glassy samples. Electron spin resonance observations show that the nature and annealing characteristics of the trapped radicals produced in solid n-C4HgBr by y-irradiation differ for the glassy and polycrystalline forms. There is also a substantial difference in the ratios of individual stable products formed by the y-irradiation of glassy n-C3H7Br compared with the crystalline form. Additional details are given in J. Amer. chem.Soc. 68 (1964) 2582.
EFFETS CHIMIQUES DE LA TRANSITION ISOMÉRIQUE NUCLÉAIRE DE 80mBr DANS LES BROMURES D’ALKYLE VITREUX ET POLYCRISTALLINS. Les rendements organiques de 8ümBr -> 80Br dans n-C3H 7Br polycristallin sont essentiellement indépendants de la concentration de ВГ2 dans une gamme étendue, ce qui fournit une nouvelle preuve de la présence du brome sous forme de solution homogène et montre que le sort de 808r est déterminé très près de l'endroit où il prend naissance. Les rendements organiques de 80rnBrBr dans n-C4H9Br sont plus élevés à l'état vitreux qu'à l'état polycristallin. Dans n-C4H9Br tant à l'état vitreux qu'à l'état polycristallin, les rendements organiques sont beaucoup plus élevés lorsque 8omBr est sous la forme de n*C4H9 80mBr que lorsqu’ il est sous la forme de 80mBrBr. Dans des échantillons polycristallins, ces rendements ne sont pas sensiblement modifiés par la présence d'une fraction de Br2 de 5* 10“3 mole, mais ils sont réduits de manière significative par cette concentration de brome dans des échantillons vitreux. D’après des observations sur la résonance de spin des électrons, la nature et les caractéristiques de recuit des radicaux piégés produits par irradiation gamma dans n-C4H9Br solide sont différentes pour les formes vitreuses et les formes polycristallines. Il existe également entre П-С3Н7ВГ vitreux et la forme cristalline une différence importante dans les rapports des différents produits stables formés par irradiation gamma. On trouvera des précisions supplémentaires dans J. Amer. chem. Soc. 68 (1964) 2582.
ХИМ ИЧЕСКИЕ ЭФФЕКТЫ Я Д Е РН О ГО И ЗО М ЕРИ Ч ЕС КО ГО П РЕ ВРАЩ ЕН И Я B r 80m В СТЕКЛОВИДНЫ Х И П О ЛИ К РИ СТАЛЛИ ЧЕ СКИ Х БРО М И СТЫ Х А Л К И Л А Х . Органические выходы из ВгВгвОт, претерпевающего изомерический переход B r 80m -* B r 80 в поликристал- лическом П-С3Н7ВГ, по существу не зависит от концентрации В гг в широком диапазоне, что является подтверждением того , что бром присутствует в виде гом огенного раствора, и показывает, что судьба В г 80 очень сильно зависит от места зарождения. Органические выходы из ВгВг80гпв п-С4НдВг являются более высокими при стекловидном состоянии, чем при поли- кристаллическом. Как при стекловидном, так и при поликристаллическом П-С4Н9ВГ органические выходы, когда Вг^ош находится в форме n-C^gBrSOm , являются значительно более
!,i Abstract onlyt paper has been published in J. phys. Chem. 68 (1964) 2582.
433

434 R. M. A. HAHNE and J. E. WILLARD
высокими, чем тогда, когда он находится в форме B rB r8°m. Присутствие В г 2 в количестве, составляющем 5<10~3 моля, не приводит к заметному изменению органических выходов в поли- кристаллических образцах, но приводит к значительному снижению их в стекловидных образцах. Наблюдения за электронным спиновым резонансом показывают, что характер и характеристики отжига захваченных радикалов, образовавшихся в твердом П-С4Н 9ВГ в результате облучения, не одинаковы для стекловидной и поликристаллической форм. Существует также значительная разница в пропорциях отдельных стабильных продуктов, образовавшихся в ре
зультате облучения стекловидного п - С з ^ В г , по сравнению с кристаллической формой. О стальные подробности содержатся в журнале Американского химического общества (journal o f the A m erican Chem ical Society) 68 (1964) 2582.
EFECTOS QUIMICOS DE LA TRANSICION ISOMERICA DEL 80mBr EN BROMUROS DE ALQUILO VITREOS Y POLICRISTALINOS. Los rendimientos orgánicos del Br8orn Br que experimenta la transición isomérica 8omBr-* 80Br en П-С3Н7ВГ policristalino son prácticamente independientes de la concentración de ВГ2 en una amplia gama, lo que constituye una prueba más de que el bromo se encuentra presente como solución homogénea e indica que lo que ocurre con el 80 Br queda determinado muy cerca del punto de origen. Los rendimientos orgánicos del Br8omBr en П-С4Н9ВГ son más elevados en el estado vitreo que en el policristalino. En el П-С4Н9ВГ, tanto vftreo como policristalino, los rendimientos orgánicos obtenidos cuando el 80mBr se encuentra en forma de П-С4Н9 8omBr son mucho mayores que cuando se presenta bajo la forma de Br80mBr. En las muestras policristalinas, la presencia de una fracción molar de 5 X10-3 de ВГ2 no modifica apreciable-
mente esos rendimientos, pero en las muestras vitreas esa concentración de bromo los reduce en forma considerable. Los estudios de la resonancia del spin electrónico muestran que la naturaleza y las características de regeneración de los radicales capturados que se han producido en el n-C4H9Br sólido por irradiación gamma son distintos para las formas vitrea y policristalina. También existe considerable diferencia en las razones entre los diversos productos estables formados por irradiación gamma del n-C3H 7Br vftreo, comparada con la irradiación de la forma cristalina. En el J. Amer. chem.Soc. 68(1964)2582 sedan detalles complementarios.

D I S C U S S I O N
(on the foregoing two papers)
A.G. MADDOCK: In connection with Dr. Collins' contribution, I should like to make a prelim inary statement on a c lose ly related, but not exactly sim ilar, mechanism for radiation annealing. E arlier work we have carried out in Cambridge suggests that during radiation annealing the energy supplied •is distributed in the following ways:
Hot zone —* reconstituted target molecules (1)
_ —« — --------- * I 1 Degradation to heat at vacancies (2)
I I Production of vacancies (3)
Dr. Baumgartner, working in my laboratory, has now shown that process (1) occurs in two steps. The first cannot be detected directly by chemical means and the second is a therm al step. At the tem perature of liquid nitrogen, radiation annealing appears to be suppressed if analysis is carried out on m aterial that is dissolved directly and is only warmed up as solution takes place (point A , fo r example). If, however, the m aterial is allowed to warm to room tem perature fo r two days before analysis, retention increases to point B, the value it would have assumed i f the irradiation had been carried out at room temperature. This thermal step shows the characteristic kinetics of the thermal annealing process.
log 100- R
______________________ _ A -1 9 6 "
вROOM TEMPERATURE
DATA FOR K2 C r04
DOSE
. F ig .l
Radiation annealing
G. H ARBO TTLE: This work of Dr. Baumgartner is very rem iniscent of the pre-irradiation effect found by Dr. Collins and myself in QîBrg. The only difference seems to be that, whereas in К 2СГО4 the ability of the gamma-introduced defects to cause annealing seems completely preserved, in C2B r6 it was found that pre-irradiation is much less efficient in producing annealing than post-irradiation.
In discussing defects it is important to rem em ber that even the most perfect crystals available still contain approximately 1016 vacancies/cm3 . In potassium chromate used for these investigations, their number is propa-
4 3 5

4 3 6 DISCUSSION
bly at least an order of magnitude higher. There are, therefore, plenty of traps available for holding the damage produced by reactor irradiation.
A .G . MADDOCK: That is of course entirely correct. But the doping experiments show that changes from 1016-* 1017-* 1018 defects/cm3 have a profound effect on annealing. All this means is that we cannot start our experiments from a zero base line; but so long as we confine our attention to differences.this does not matter too much.
J .I. VARGAS: In this connection, I should like to point out that apparently solid substances for which recoil studies have been made can be classified as belonging to one of two categories. There are substances fo r which the annealing rates are increased and there are substances for which the annealing rates are diminished. Phthalocyanines and acetyl acetonates belong to the second category, while chromâtes and hexa-bromoethane belong to the first. It seems to me that electron-hole reactions with the recoils could perhaps explain these results.
A . N. NESMEYANOV: I have a comment which is, strictly speaking, concerned with Dr. Willard's paper* but which also possibly has a bearing on the present discussion. I have the impression that a number of speakers during the discussions at the present session have been anticipating some of the problems that will be dealt with at later sessions devoted to solid compounds. Because of this, some of the examples that have been quoted with respect to transformations in inorganic systems may not be strictly applicable to organic systems.
Later on in the Symposium I hope to present some of our ideas on the stabilization of recoil atoms in solid inorganic substances. The closest approximation to our ideas is, Ithink, to be found in the views expressed by Dr. Willard, although in inorganic systems the process is somewhat different because of liquid-cage formation, which may not occur in organic compounds. Nevertheless, in all transformations occurring in solids the trend is towards the formation of compounds of maximum thermodynamic stability either through direct action in the liquid cage or in subsequent processes that take place as a result of annealing. In his discussion of the stabilization of hot bromine atoms in the gaseous, liquid and solid phases, Dr. Willard' feels that the billiard-ball collision mechanism is active in the gaseous phase but is doubtful in the liquid and solid phases. It would seem to me that the experimental data now available on the halogens — with the possible ex - ception of isomeric transitions — indicate, in a first approximation, that halogen systems are subject to the theory of elastic and inelastic collisions also in the liquid phase. If there were time, I could show you various plots that have been obtained by applying the simplest correlations of this theory with respect to the С2Н5ВГ-СС14 system. They show that such an approach, based on the utilization of geometrical collision cross-sections and the probability of a particular atom being hit, produces good results. It can also be shown that there is good agreement between the theory of elastic collisions and experimental data in the system where recoil bromine atoms replace various substituent atoms in the halobenzenes.* *
* WILLARD, J.E., "Chemical effects o f nuclear transformations of halogens in organic m ed ia” , these Proceedings I .
* # For data later submitted in writing see end of Discussion.

DISCUSSION 4 3 7
J. WILLARD: The question of whether a billiard-ball mechanism is involved is one that continues to be asked. The data Professor Nesmeyanov refers to certainly seem consistent with such a mechanism but I would have thought that they were perhaps also consistent with a number of other,mechanisms, and that we should keep an open mind on this subject. One fact that always comes to mind in this connection is that there is a constant organic yield of about 21% in the alkyl chloride homologous series from methyl chloride onwards up through three, four, five carbon atoms as well as in alkyl chlorides like methyl chloride dissolved to high dilutions in hydrocarbons. If there had been time during the discussions on liquid systems,I would have been interested to hear whether the author who reported on recoil chlorine and who also observed similar total retentions* had an explanation for this phenomenon. To my mind this is one of the puzzling small facts of hot-atom chemistry and it certainly does not seem consistent with the billiard-ball mechanism.
P. SHAW: I have one brief comment on the subject of the billiard-ball theory in liquids. By making a collision-to-collision calculation and by taking, into account the simultaneous heating of the liquid, I have found that the cal^ culated hot yield of bromoethane agrees with the observed value. However, similar treatment for bromoethane-bromine mixtures has shown that this approach cannot easily account for the observed dependence of yield upon bromine concentration. This suggests that some sort of statistical partitioning occurs between the bromine atoms present initially as bromoethane or elementary bromine. Such depehdence might be.a consequence of the pyrolytic reactions, postulated in our paper, which would tend to "mix in" thé active atom by transforming it from radical to molecule, etc. However,I should like to emphasize that if these reactions did occur they would modify the distribution of any atom that had undergone a billiard-ball reaction, so that no deduction concerning the probability of such reactions could be made directly from the observed yields.
F .S . ROWLAND: I wish to comment on the general use of the term "billiard-ball mechanism", because I believë that we need to keep this terminology in proper perspective. The idea of thé possible importance of billard-ball collisions was first put forward about 25 years ago to explain energy losses by high-energy atoms. From the beginning it has obviously been a highly simplified approximation, and it has proved to be a very useful concept that has lead to new experiments and a better understanding of hot- atom chemistry. It is now clear, however, that even helium atoms colliding with other helium atoms do not obey the "billiard-ball" laws of collision, and that therefore collisions of more reactive atoms with molecules are even less likely to be accurately described by such a model. The importance of inelastic scattering, isotropy, realistic interaction potentials, etc., will have to be recognized to an increasing extent. The data of hot-atom chemistry are much more sophisticated than they were 25 years ago, and numerous 1 other advances have been made, particularly in the study of chemical kinetics through molecular beams. It is really time for us to adopt more ad-
* VASAROS, L . , "Reactions o f hot C l38 atoms in mixtures o f carbon tetrachloride with aliphatic alcohols” , these Proceedings I . ■ . , . . .

438 DISCUSSION
vanced models of atomic-molecular interactions to match these other improvements, rather than to continue estimating energy losses from the simple value of m for the recoil atom and M for the struck species, either atom or molecule.
DATA LATER SUBMITTED IN WRITING BY A .N . NESMEYANOV
For calculations of the hot-reaction yield in an approximation based on a transfer of energy in the case of an elastic collision, the solution of the reasonance integral of the theory of neutron thermalization gives the following expression:
D f e EoR =■=■• — In — , (1)? v v ' '
where f =the geometrical probability of a collision between a recoil and another atom, equal to 4rf niN /[4riniN+(l-N) (Г1 + Г2 )* П2 ] in calculations of the yield of the parent compound (ri is the covalent radius of the recoil atom, гг is the radius of an environmental atom upon collision with which substitution reactions are possible, nj and П2 are the number of atoms in the molecule [ni relating to atoms equal in mass to the recoil atom, П2 being a second atom yielding competing reaction collisions], N is the molar fraction of the parent compound);
f = the mean logarithmic energy decrement
riN + (r 1 + r2) 2 (1-N)n2f 2 ' r*N + (r j+ r 2)2 (l-N)n2 ’
e = energy of the nucleus:v - energy of the chemical bond corrected for the Süss effect — ,M-m
v 1 = bond energy, M=mass of the molecule, m=mass of the atom to which the hot atom transmits part of its energy upon collision); and
Eo = the initial recoil energy.A variable in formula (1) that determines the yield of the hot reaction
is the ratio f/? , i .e .
R = const. f/§ . (2)
For the yield of СгНйВг82 in hot-atom reactions of Br82 in the C2H5Br-OCI4 system, expression (2) is as follows:
4r?N r?NR = const. — 0-----------------------------------------------------------5-------— = —5---—n —— . (3)4rfN + (r 1 + r2 )2(l-N)4Ç2 rfN + lrj + r 2) 2 (1-N)Ç2
The dependence of the yield of CçHôBr82 on the ratio f /l (parameter P)
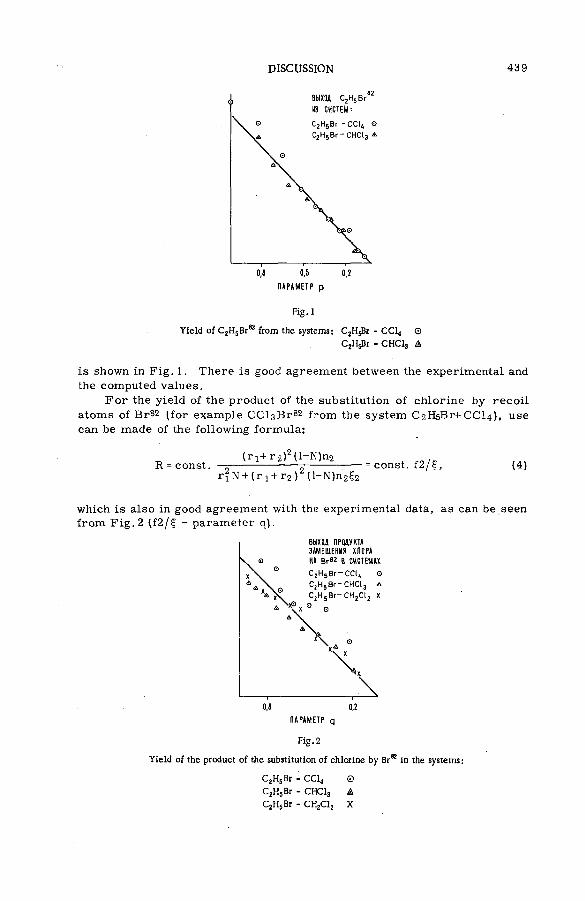
DISCUSSION 439
ПАРАМЕТР p
H g . l
Y ield o f C2H5Brœ from the systems: C^H^r - CC14 S , CjHsBr - CHCI3 A
is shown in Fig. 1. There is good agreement between the experimental and the computed values.
For the yield of the product of the substitution of chlorine by recoil atoms of Br®2 (for example СС1зВг82 from the system С2Н5ВГ+ ÇCI4 ), use can be made of the following formula:
(r i+ r 2)2 (l-N)n2 ,R = const, —z-------------------5----------:------- const. f2/|, (4)
rfN + lrj+rzrU-NJnsSs
which is also in good agreement with the experimental data, as can be seen from Fig. 2 (f2 /f - parameter q).
ПАРАМЕТР q
Fig. 2
Yield o f the product o f the substitution o f chlorine by Br82 in the systems:
C2H5Br - CC14 ©C2H5Br - CHCI3 AC2H5Br - CH2C12 X

440 DISCUSSION
It follows that the geometrical probability of a collision and the thermal- izing capacity of the environment determines the yields of products formed in elastic collisions between hot atoms and atoms of comparable mass.
The correctness of the theory of elastic collisions was also checked by studying the substitution of monoatomic derivatives of benzène by atoms- of Br82, ■
BrS2 + c 6H 5X -----»C6H5Br82 +X .
For the substituent series, the yield of bromobenzene amounted to 3, 20, 21 and 18%, which fits in well with the curve R = F(mx/mBr ), where m* = the mass of the substituent and твг = the mass of the bromine atom calculated by means of an approximation based on the existence of elastic collisions.

SYMPOSIUM ON CHEMICAL EFFECTS ASSOCIATED WITH NUCLEAR REACTIONS
; AND RADIOACTIVE TRANSFORMATIONS :
. HELD IN VIENNA, 7-11 DECEMBER 1964
Session 1
Session 2
Session 3
Session 4 R. HENRY
Session 5 J. I. VARGAS
Session 6 V. KAÍENA
Session 7 W. HERR
Session 8 G. HARBOTTLE
Session 9 A. N. NESMEYANOV
Osterreichische Studiengesell- schaft für Atomenergie GmbH
Atomic Energy of Canada Ltd.
Nuclear Physics Laboratory, Oxford University
Commisariat à l'Energie Atomique, Paris
Instituto de Química,Universidade de Minas Gerais, Belo Horizonte
Nuclear Research Institute of the Czechoslovak Academy of Sciences, ïteZ, Prague
Kernforschungsanlage Jülich and Institut für Kernchemie der Universitàt Koln, Cologne
Brookhaven National Laboratory
Moscow State University
CHAIRMEN OF SESSIONS
N. GETOFF
R. BETTS
P. SHAW
SECRETARIAT OF THE SYMPOSIUM
Scientific G. B. COOK Division of Research andSecretaries: Laboratories, IAEA
O. SUSCHNY Division of Research andLaboratories, IAEA
Administrative P. GHELARDONI Division of Scientific and TechnicalSecretary: Information, IAEA
441

442
Editor:
RecordsOfficers:
S. M. FREEMAN Division of Scientific and Technical Information, IAEA
T. J. JONES Division of Languages, IAEAT. B. DUNNE Division of Languages, IAEA

IAEA SALES AGENTS
Orders for Agency publications can be placed with your bookseller or any of our sa les agents listed below:
A U S T R A L IAHunter Publications,23 McKillop Street Melbourne, C .l
A U S T R IAGeorg Fromme & Co.Spengergasse 39 Vienna V .
B E L G IU MO ffice international de librairie 30, avenue Marnix B russels 5
B R A Z I LLivraria Kosmos Editora Rua do Rosario, 135 -137 Rio de JaneiroAgencia Expoente O scar M. Silva Rua Xavier de Toledo, 140- Ie Andar (Caixa P o sta l No. 5.614)S3o Paulo
BYELORUSSIAN SOVIET SOCIALIST REPUBLIC See under USSR
C A N A D AThe Queen*s Printer Ottawa, Ontario
C H I N A (Taiwan)Books and Scien tific Supplies Service, L td .,P .O . Box 83T aipei
DE N MA R KEjnar Munksgaard Ltd.6 Norregade Copenhagen К
F I N L A N DAkateeminen Kirjakauppa Keskuskatu 2 Helsinki
F R A N C EOffice international de documentation et librairie 48, rue G ay-Lussac Paris 5e
G E R M A N Y , Federal Republic of R . Oldenbourg Rosenheimer Strasse 145 8 Munich 8
I S R A E LHeiliger and Co.3 Nathan Strauss Street Jerusalem
I T A L YAgenzia Editoriale Intemazionale Organizzazioni Universali (A .E .I.O .U .) ViaM eravigli 16 Milan
J A P A NMaruzen Company Ltd.6 , Tori Nichome .Nihonbashi .(P.O . Box 605)Tokyo Central
M E X I C OLibraría Internacional Av. Sonora 206 Mexico I I , D .F .
N E T H E R L A N D SN.V. Martinus Nijhoff Lange Voorhout 9 The Hague
NEW Z E A L A N DWhitcombe & Tombs, Ltd.G .P.O . Box 1894 Wellington, C .l
N O R WA YJohan Grundt Tanum Karl Johans gate 43 Oslo
P A K I S T A NKarachi Education Society Haroon Chambers South Napier Road(P.O . Box No. 4866)Karachi, 2
P O L A N DOsrodek Rozpowszechniana ' Wydawnictw Naukowych Polsk a Akademia Nauk P afac Kultury i Nauki Warsaw
S O U T H A F R I C AVan Sch aik 's Bookstore (Pty) Ltd. L ibri Building Church Street (P .O . Box 724)Pretoria
S P A I NLibrería BoschRonda de la Universidad 11Barcelona

S W E D E NC .E . F ritzes Kungl. Hovbokhandel Fredsgatan 2 Stockholm 16
S W I T Z E R L A N D Librairie Payot Rue Grenus 6 1211 Geneva 11
T U R K E YLibrairie Hachette 469, Istik lâ l Caddesi Beyoglu, Istanbul
UKRAINIAN SOVIET SOCIALIST REPU BLIC See under USSR
UNION O F SOVIET SOCIALIST REPU BLICSMezhdunarodnaya Kniga Smolenskaya-Sennaya 32*34 Moscow G-200
UNITED KINGDOM OF GREAT BRITAINAND NORTHERN IRELAND
Her M ajesty 's Stationery Office P .O . Box 569 London, S .E .l
UNÍTED STA TES O F AMERICA National Agency for International Publications, Inc.317 E a s t 34th Street New York, N.Y. 10016 •
V E N E Z U E L ASr. Braulio Gabriel Chacaree Gobernador aC andilito 37 Santa R osalia (Apañado P o sta l 8092)Caracas D .F .
Y U G O S L A V I AJugoslovenska Knjiga T erazije 27 Belgrade
IAEA publications can also be purchased retail at the United Nations Bookshop at United Nations Headquarters, New York, at the news-stand at the Agency's Headquarters, Vienna, and at most conferences, symposia and seminars organized by the Agency. *
In order to facilita te the distribution of its publications, the Agency is prepared to accept payment in UNESCO coupons or in local currencies.
Orders and inquiries from countries where sa les agents have not yet been appointed may be sent to : .
Distribution and Sa les Unit, International Atomic Energy Agency, Kârntner Ring 11, Vienna I, Austria

I N T E R N A T I O N A L
A T O M I C ENERGY A G E N CY
V I E N N A , 1965
PRICE: USA a n d C a n a d a : USA $ 9 . 0 0
A u s t r i a a n d e j s e w h e r e : S 189.
( £ 2 . 14 . 0 ; F.Fr. 3 6 . - ; DM 31 , 50 )





























