Embed Size (px)
Citation preview

Current Protein and Peptide Science, 2006, 7, 255-280 255
1389-2037/06 $50.00+.00 © 2006 Bentham Science Publishers Ltd.
Cellobiose Dehydrogenase – A Flavocytochrome from Wood-Degrading,Phytopathogenic and Saprotropic Fungi
Marcel Zamocky1,2,*, R. Ludwig1,2, C. Peterbauer1,2, B.M. Hallberg3, C. Divne3, P. Nicholls4 andD. Haltrich1,2
1Department of Food Science and Technology, University of Natural Resources and Applied Life Sciences, Muthgasse18, A-1190 Vienna, Austria; 2Research Centre Applied Biocatalysis, Petersgasse 14, A-8010 Graz, Austria; 3Departmentof Biotechnology, Royal Institute of Technology, Albanova University Center, SCFAB, SE-106 91 Stockholm, Sweden;4Department of Biological Sciences, University of Essex, Wivenhoe Park, Colchester, Essex CO4 3SQ, UK
Abstract: Cellobiose dehydrogenase, the only currently known extracellular flavocytochrome, is formed not only by anumber of wood-degrading but also by various phytopathogenic fungi. This inducible enzyme participates in early eventsof lignocellulose degradation, as investigated in several basidiomycete fungi at the transcriptional and translational level.However, its role in the ascomycete fungi is not yet obvious. Comprehensive sequence analysis of CDH-encoding genesand their translational products reveals significant sequence similarities along the entire sequences and also a commondomain architecture. All known cellobiose dehydrogenases fall into two related subgroups. Class-I members are repre-sented by sequences from basidiomycetes whereas class-II comprises longer, more complex sequences from ascomycetefungi. Cellobiose dehydrogenase is typically a monomeric protein consisting of two domains joined by a protease-sensitive linker region. Each larger (dehydrogenase) domain is flavin-associated while the smaller (cytochrome) domainsare haem-binding. The latter shorter domains are unique sequence motifs for all currently known flavocytochromes. Eachcytochrome domain of CDH can bind a single haem b as prosthetic group. The larger dehydrogenase domain belongs tothe glucose-methanol-choline (GMC) oxidoreductase superfamily – a widespread flavoprotein evolutionary line. Thelarger domains can be further divided into a flavin-binding subdomain and a substrate-binding subdomain. In addition, theclass-II (but not class-I) proteins can possess a short cellulose-binding module of type 1 at their C-termini. All the cello-biose dehydrogenases oxidise cellobiose, cellodextrins, and lactose to the corresponding lactones using a wide spectrumof different electron acceptors. Their flexible specificity serves as a base for the development of possible biotechnologicalapplications.
Keywords: Cellobiose dehydrogenase, flavocytochrome, wood degrading fungi, GMC flavoenzyme superfamily.
1. INTRODUCTION (ENZYMES INVOLVED INLIGNOCELLULOSE BIODEGRADATION)
Cellulose, a long linear polymer consisting of glucosylunits connected by β-1,4-glycosidic linkages, is one of themost abundant polymers in our biosphere. This major renew-able carbon source frequently contains more than 10,000glucose units (in the secondary plant cell wall) and forms arigid, highly ordered structure built of elementary fibrils heldtogether mainly by hydrogen bonding and van der Waalsforces. Cellulose has a tendency to crystallise shortly afterbiosynthesis but less ordered amorphous regions can alsooccur in plant tissues [1]. In vascular plants the cellulose canbe associated with linear or branched polymers of hemicel-lulose, containing different pentose and hexose units. Inhardwood the major component of hemicellulose is xylan,which also constitutes an important renewable carbonsource. A heterogeneous network of branched phenylpro-panoid units embeds the cellulose in lignin. The polymerisa-
*Address correspondence to this author at the Department of Food Scienceand Technology, University of Natural Resources and Applied Life Sci-ences, Muthgasse 18, A-1190 Vienna, Austria and Research Centre AppliedBiocatalysis, Petersgasse 14, A-8010 Graz, Austria; Fax: +43 1 36006 6251;E-mail: [email protected]
tion pattern and assembly of lignin is guided by the orienta-tion of cellulose and the structure of hemicellulose and canoccur in variations known as softwood and hardwood lignin[2].
It is hence obvious that this chemically stable, robust andcomplex material of woody plants can be degraded only by aseries of enzymes with different specificities, which act to-gether in a strict synergism [3]. In the course of evolutionseveral lines of lignocellulolytic microorganisms appearedcapable of the enzymatic degradation of cellulose, hemicel-lulose, and lignin to form simple units that could be used ascarbon and energy sources. Fungi are currently the most im-portant and widespread group of organisms contributingmassively to wood decay. Concerted evolution of woodyplant-interacting fungi and their plant hosts can be tracedback to the Silurian period around 400 million years ago,when the first vascular plants emerged [4,5]. As the first vas-cular plants probably lacked highly developed root systems,symbiotic associations with fungal endophytes might havebeen crucial for their survival. During their further uniquephylogenetic radiation the fungi took advantage of the in-creasing availability of plant materials. Most representativesof this kingdom were adapted to a parasitic or (at least) aheterotrophic way of life so that they became dependent

256 Current Protein and Peptide Science, 2006, Vol. 7, No. 3 Zamocky et al.
upon extracellular nutritional sources, especially of carbonand nitrogen. They consequently evolved a secretion ma-chinery for the production of extracellular enzymes degrad-ing the abundant complex carbon and nitrogen sources fromwoody plants [6].
The array of enzymes involved in the degradation oflignocellulose-containing tissues of plants is representedmainly by hydrolases and oxidoreductases. Synchronisedaction of these enzymes significantly contributes to the recy-cling of plant biomass in the environment. Endo-cellulases[1,4-β-D-glucan-4-glucanohydrolases, E.C. 3.2.1.4] hydro-lyse the cellulose into small soluble sugars (mono-, di-, andtrisaccharides). Endocellulases cleave cellulose at varioussites along the polysaccharide chains. Exo-cellobio-hydrolases [1,4-β-D-glucan-4-cellobiohydrolases, E.C.3.2.1.91] release cellobiose units only from the chain ends ofintact crystalline cellulose. As a rule, both endo- andexocellulases occur in cellulolytic organisms as isozymemixtures, which exhibit synergism in their activity [7,8].Hence, through the action of several enzymes cellobiose isproduced as an important metabolic intermediate. Cellobiosein turn is hydrolysed by β-glucosidases [β-D-glucoside-O-glucohydrolases, E.C. 3.2.1.21] yielding two molecules ofglucose. In addition, cellobiose dehydrogenase [cello-biose:(acceptor) 1-oxidoreductase, E.C. 1.1.99.18] evolvedin fungi to oxidize cellobiose to cellobionic acid while con-currently a yet unspecified electron acceptor is reduced invivo.
While the polysaccharides in lignocellulose are mainlydegraded by various hydrolases, the oxidoreductases manga-nese peroxidase, lignin peroxidase, and laccase are involvedin lignin degradation by various pathways. Pathways thatinvolve peroxidases depend upon hydrogen peroxide gen-eration [9] by various flavin-containing oxidases. Such lignindepolymerising reactions are performed mainly by white-rotbasidiomycete fungi that also produce endo-xylanases [1,4-β-D-xylan-4-xylanohydrolases E.C. 3.2.1.8] specialised forthe degradation of hemicellulose [10]. All these enzymes areclosely interconnected with respect to biosynthesis andregulation. In the case of the oxidoreductases involved inlignocellulose cleavage an efficient cofactor regenerationprocess also takes place (discussed in a later section).
In this review we focus upon cellobiose dehydrogenase(CDH), its occurrence, regulation, sequence and structureanalysis, and its reaction specificity and kinetic mechanism.CDH was first described in 1974 by Westermark and Eriks-son [11] (originally termed cellobiose:quinone oxidoreduc-tase), the first cDNA sequence coding for a CDH was re-vealed in 1995 [12], and the structures of the separate do-mains of one representative were solved in 2000-2002[13,14]. Three decades of intensive investigation on theseunique cellobionolactone-forming oxidoreductases have elu-cidated much of their properties, and we can also begin tounderstand their physiological occurrence and role. Severalgenes coding for CDH have been successfully used for mo-lecular cloning and mutagenesis. Numerous biotechnologicalapplications of cellobiose dehydrogenases can now be fore-seen. In the following sections we will summarise the avail-able information concerning these unique extracellular fla-vocytochromes. Our review is a comprehensive update of
two previous reviews that appeared on the same topic in2000-2001 [15, 16]. Whereas those sections on the produc-tion of CDH in various fungi, catalytic mechanism and bio-technological applications are updated to the state of knowl-edge in 2005, the sections on the analysis of genes (includingthe phylogenetic relationship) and the description of 3Dstructures are completely new ones and were not addressedin the previous reviews.
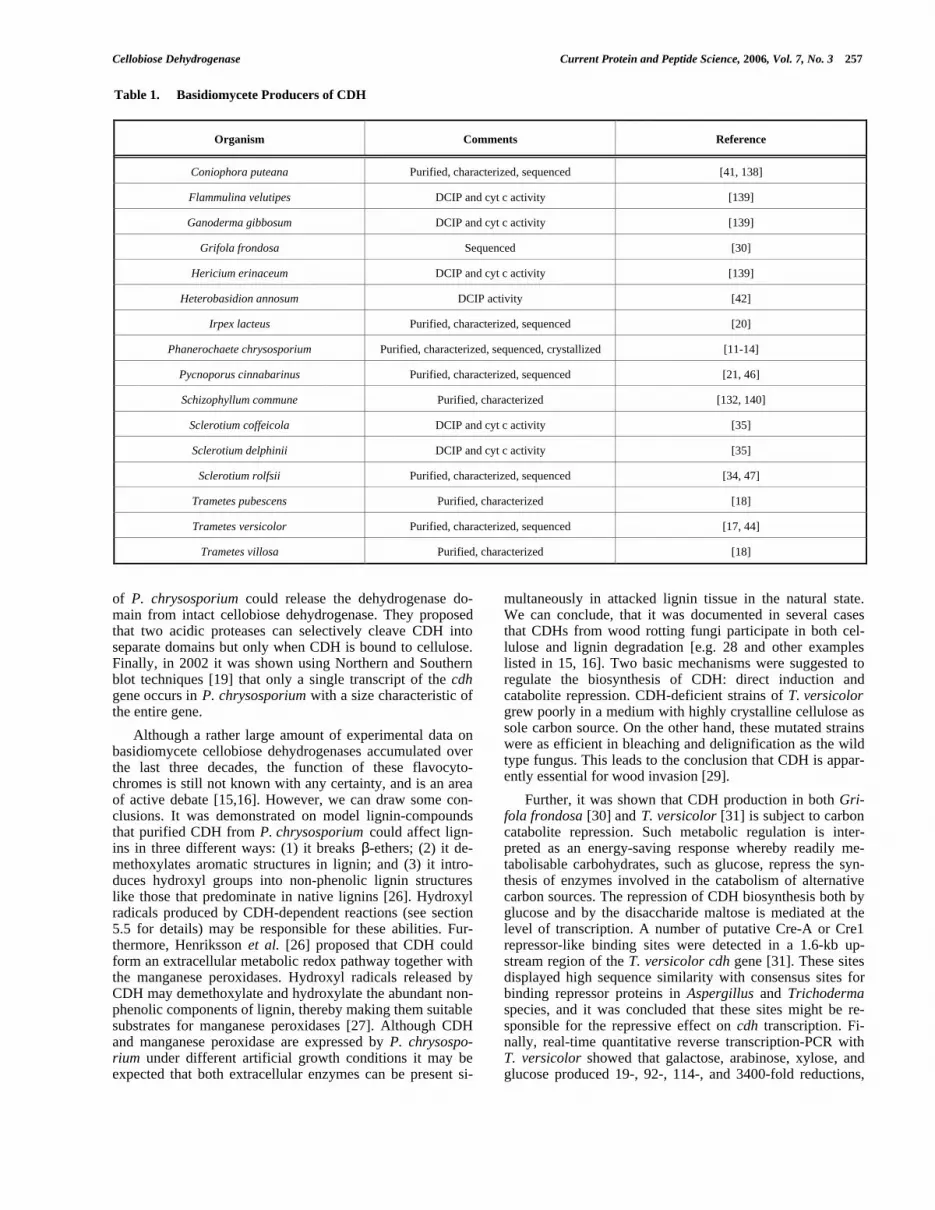
2. OCCURRENCE AND MICROBIAL PRODUCTIONOF CELLOBIOSE DEHYDROGENASES
2.1. Production of CDH in Basidiomycetes
Cellobiose dehydrogenase was first isolated from thebasidiomycetes Phanerochaete chrysosporium and Trametesversicolor [11,17] and identified as an oxidoreductase, re-ducing quinones in the presence of cellobiose but not of glu-cose. Later, it was observed that this lactone-releasing en-zyme is produced by numerous other wood-degrading fungi.Basidiomycete producers of CDH (both those well describedand others not yet characterized) are listed in Table 1. Typi-cally, CDH is secreted in the extracellular space during theexponential phase of growth when cellulose is the main car-bon source used for growth. The majority of investigatedCDH producers are white-rot fungi in which CDH secretionseems to be very common. For example all eleven screenedTrametes species were shown to exhibit significant CDHactivity when cultivated on a cellulose-containing medium[18]. The best investigated example is the production ofCDH by P. chrysosporium , in which under cellulolytic con-ditions this oxidoreductase represents about 0.5% of the ex-tracellular protein [19]. Further examples of white-rot CDHproducers are represented by Irpex lacteus [20] andPycnoporus cinnabarinus [21] both of which possess twodifferent CDHs when grown on cellulose as a carbon source.Notably, CDH from Py. cinnabarinus suppresses the forma-tion of the typical red pigment (cinnabarinic acid, a dimer of3-hydroxyantranilic acid 3-HAA), whereas laccase, in turn,forms this pigment by oxidizing the 3-HAA. A brown rotfungus, Coniophora puteana, with a CDH apparently in-volved in a Fenton-type reaction was also discovered [22].
Two types of flavoprotein, both of them capable of oxi-dizing cellobiose, were separately investigated for severalyears in numerous CDH-producing organisms. They wereclassified as (a) the flavohaemoprotein cellobiose dehydro-genase (CDH, E.C. 1.1.99.18; formerly cellobiose oxidase)and (b) the flavoprotein cellobiose:quinone oxidoreductase(CBQ, i.e., the separate dehydrogenase domain E.C. 1.1.5.1,this E.C. number deleted in 2002). The latter was for sometime believed to be a separate enzyme. In 1993 it was pro-posed that the name cellobiose oxidase is replaced by themore appropriate term cellobiose dehydrogenase because ofthe catalytic preference for electron acceptors other thanmolecular oxygen [23]. It was also demonstrated that cello-biose: quinone oxidoreductase from P. chrysosporium is abreakdown product of cellobiose dehydrogenase and thatboth proteins exhibited similar patterns after proteolytic di-gestion and cross reactivities with specific antibodies [24].Comprehensive evidence for native proteolytic processing ofcellobiose dehydrogenase was presented by Habu et al., [25].The authors showed that proteases from cellulolytic cultures
Cellobiose Dehydrogenase Current Protein and Peptide Science, 2006, Vol. 7, No. 3 257
of P. chrysosporium could release the dehydrogenase do-main from intact cellobiose dehydrogenase. They proposedthat two acidic proteases can selectively cleave CDH intoseparate domains but only when CDH is bound to cellulose.Finally, in 2002 it was shown using Northern and Southernblot techniques [19] that only a single transcript of the cdhgene occurs in P. chrysosporium with a size characteristic ofthe entire gene.
Although a rather large amount of experimental data onbasidiomycete cellobiose dehydrogenases accumulated overthe last three decades, the function of these flavocyto-chromes is still not known with any certainty, and is an areaof active debate [15,16]. However, we can draw some con-clusions. It was demonstrated on model lignin-compoundsthat purified CDH from P. chrysosporium could affect lign-ins in three different ways: (1) it breaks β-ethers; (2) it de-methoxylates aromatic structures in lignin; and (3) it intro-duces hydroxyl groups into non-phenolic lignin structureslike those that predominate in native lignins [26]. Hydroxylradicals produced by CDH-dependent reactions (see section5.5 for details) may be responsible for these abilities. Fur-thermore, Henriksson et al. [26] proposed that CDH couldform an extracellular metabolic redox pathway together withthe manganese peroxidases. Hydroxyl radicals released byCDH may demethoxylate and hydroxylate the abundant non-phenolic components of lignin, thereby making them suitablesubstrates for manganese peroxidases [27]. Although CDHand manganese peroxidase are expressed by P. chrysospo-rium under different artificial growth conditions it may beexpected that both extracellular enzymes can be present si-
multaneously in attacked lignin tissue in the natural state.We can conclude, that it was documented in several casesthat CDHs from wood rotting fungi participate in both cel-lulose and lignin degradation [e.g. 28 and other exampleslisted in 15, 16]. Two basic mechanisms were suggested toregulate the biosynthesis of CDH: direct induction andcatabolite repression. CDH-deficient strains of T. versicolorgrew poorly in a medium with highly crystalline cellulose assole carbon source. On the other hand, these mutated strainswere as efficient in bleaching and delignification as the wildtype fungus. This leads to the conclusion that CDH is appar-ently essential for wood invasion [29].
Further, it was shown that CDH production in both Gri-fola frondosa [30] and T. versicolor [31] is subject to carboncatabolite repression. Such metabolic regulation is inter-preted as an energy-saving response whereby readily me-tabolisable carbohydrates, such as glucose, repress the syn-thesis of enzymes involved in the catabolism of alternativecarbon sources. The repression of CDH biosynthesis both byglucose and by the disaccharide maltose is mediated at thelevel of transcription. A number of putative Cre-A or Cre1repressor-like binding sites were detected in a 1.6-kb up-stream region of the T. versicolor cdh gene [31]. These sitesdisplayed high sequence similarity with consensus sites forbinding repressor proteins in Aspergillus and Trichodermaspecies, and it was concluded that these sites might be re-sponsible for the repressive effect on cdh transcription. Fi-nally, real-time quantitative reverse transcription-PCR withT. versicolor showed that galactose, arabinose, xylose, andglucose produced 19-, 92-, 114-, and 3400-fold reductions,
Table 1. Basidiomycete Producers of CDH
Organism Comments Reference
Coniophora puteana Purified, characterized, sequenced [41, 138]
Flammulina velutipes DCIP and cyt c activity [139]
Ganoderma gibbosum DCIP and cyt c activity [139]
Grifola frondosa Sequenced [30]
Hericium erinaceum DCIP and cyt c activity [139]
Heterobasidion annosum DCIP activity [42]
Irpex lacteus Purified, characterized, sequenced [20]
Phanerochaete chrysosporium Purified, characterized, sequenced, crystallized [11-14]
Pycnoporus cinnabarinus Purified, characterized, sequenced [21, 46]
Schizophyllum commune Purified, characterized [132, 140]
Sclerotium coffeicola DCIP and cyt c activity [35]
Sclerotium delphinii DCIP and cyt c activity [35]
Sclerotium rolfsii Purified, characterized, sequenced [34, 47]
Trametes pubescens Purified, characterized [18]
Trametes versicolor Purified, characterized, sequenced [17, 44]
Trametes villosa Purified, characterized [18]

258 Current Protein and Peptide Science, 2006, Vol. 7, No. 3 Zamocky et al.
respectively, in cdh transcript levels [32]. In P. chrysospo-rium it was also shown by RT-PCR that the cdh gene is dif-ferentially regulated compared to the bgl (β-glucosidase)gene. Whereas glucose significantly decreased the transcrip-tion of both genes, cellobiose repressed only bgl but had noeffect on transcription of the cdh gene [33]. The expressionof CDH in various Trametes species was monitored viaWestern blot analysis of extracellular proteins under diversegrowth conditions [18]. Intact CDHs are predominant in richcellulose-containing media with complex organic nitrogensources while truncated (proteolytically processed) forms arefound only in minor amounts.
In addition to its role in white rot and brown rot fungiCDH is also produced by several plant pathogenic basidio-mycetes that are not implicated in wood decay. The bestknown, recently studied example is cellobiose dehydro-genase from Sclerotium rolfsii [34, teleomorph: Athelia rolf-sii]. S. rolfsii is a soil borne phytopathogenic fungus primar-ily attacking annuals and herbaceous perennials but alsosome younger woody plants. After addition of cellulose tothe cultivation medium S. rolfsii becomes an outstandingCDH producer forming 10- to 50-fold more extracellularCDH than reported from P. chrysosporium. CDH is alsointensively produced by related Sclerotium coffeicola andSclerotium delphinii species [35].
2.2. Production of CDH in Ascomycetes
The production of CDH by Ascomycetes has been lessintensively investigated and is only poorly understood com-pared to basidiomycete CDH (ascomycete producers of CDHare listed in Table 2). This flavocytochrome has, however,already been detected in the extracellular culture fluid of thesoft rot fungus Monilia sp. in the early 1980´s [36]. It wasdemonstrated to be an inducible enzyme with a slightly dif-ferent inducer profile from that for basidiomycete CDH.Schou et al. later purified and investigated the CDH from thethermophilic soft-rot fungus Humicola insolens [37] andsuggested a role in producing the Fenton´s reagent by re-ducing Fe(III) under alkaline conditions. Unlike basidiomy-cete CDHs the Humicola enzyme has an alkaline optimumfor certain electron acceptors. The flavin cofactor of H. in-solens CDH was found to be a mixture of 60% 6-hydroxy-
substituted flavin adenine dinucleotide (6-OH FAD) and40% FAD. Dehydrogenase domains carrying 6-OH FADexhibited lower rates of cellobiose oxidation [38]. A trulythermophilic CDH from Sporotrichum thermophile [39,synonym Thielavia heterothallica] exhibited a temperatureoptimum of 60°C. This fungus can degrade cellulose but notlignin and its thermostable CDH was produced in both cel-lulose- and cellobiose-supplemented cultures. Two differentforms of this thermostable CDH detected under variousgrowth conditions are probably due to posttranslationalmodifications of a single transcript. CDH was purified andcharacterised also from the asporogenic filamentous fungusINBI 2-26(-) [40] belonging to the genus of Chaetomium.This enzyme with a molecular weight of 95 kDa possessed arather high pH optimum between 5.5 – 7.0, and a character-istic absorption spectrum. This CDH exhibited affinity toamorphous cellulose, and interestingly the enzyme reducedby cellobiose was more stable than the nonreduced form.
3. PHYSICAL AND CHEMICAL PROPERTIES
Cellobiose dehydrogenase as isolated from the sourcesmentioned above is a rather complex protein that containstwo prosthetic groups: the haem group, and the flavin, FAD.There are significant differences between other types of fla-vocytochrome with respect to these two cofactors. The fla-vocytochrome from Shewanella frigidimarina contains haemc and FAD, while flavocytochrome b2 from Saccharomycescerevisiae contains haem b and flavin mononucleotide(FMN). In all known cellobiose dehydrogenases the haemprosthetic group is protoporphyrin IX (protohaem IX; haemb) with one haem per CDH molecule [34] as determined bythe classical pyridine haemochromogen assay. Similarly,FAD was shown spectrophotometrically to occur as onedinucleotide per CDH molecule and this was confirmed bythin-layer chromatography. CDH samples from H. insolens,S. rolfsii, P. chrysosporium and T. versicolor also contain 6-hydroxy-FAD [17, 38]. This modified cofactor has a morenegative midpoint redox potential than non-substituted FAD,which might enable CDH molecules to transfer electrons toelectron acceptors with lower redox potentials (e.g. Fe(III)-oxalate). Although the H. insolens enzyme with the modifiedcofactor had a decreased specific activity, this was not ob-served in S. rolfsii CDH [R. Ludwig & D. Haltrich unpub-
Table 2. Ascomycete Producers of CDH
Organism Comments Reference
Chaetomium sp. DCIP and Cyt c activity [36, 40, 66]
Corynascus thermophilus Purified, characterized, and partially sequenced [R. Ludwig et al. manuscript in preparation]
Humicola insolens Purified, characterized, sequenced [37]
Monilia sp. DCIP and Cyt c activity [36]
Myriococcum thermophilum Purified, characterized, sequenced [R. Ludwig et al. manuscript in preparation]
Neurospora crassa DCIP and Cyt c activity [139]
Thielavia heterothallica (Sporotrichum thermophile)
Purified, characterized, sequenced [39]

Cellobiose Dehydrogenase Current Protein and Peptide Science, 2006, Vol. 7, No. 3 259
lished results]. Henriksson et al. [15] suggested that reactiveoxygen species, such as hydroxyl radicals formed duringreaction with molecular oxygen, might be responsible for theflavin derivatisation.
Purified CDH exhibits a typical absorption spectrum. Theoxidised and the reduced spectra of the CDH from S. rolfsiiare shown in (Fig. 1). The major peak of the oxidised spec-trum at 421 nm is due to the haem cofactor, whereas thebroad shoulder between 450 and 500 nm is mainly attributedto the FAD group. Absorption coefficients for the oxidisedenzyme at 421, 460, 531, and 563 nm are 105, 24, 12, and 9mM-1 cm-1, respectively. Upon reduction of CDH by its sub-strate cellobiose or by sodium dithionite, strong peaks appearat 429, 533, and 564 nm, representing the Soret (or γ), β andα peaks of a typical haem protein. Absorption coefficients atthese wavelengths are 140, 17, and 28 mM-1 cm-1, respec-tively. In this state the absorption between 450 and 500 nmdecreases drastically due to reduction of the FAD. Compari-sons reveal very similar results with other CDH spectra withonly small differences in the position of the bands [17, 20,37, 38]. In the T. versicolor enzyme, the flavin-containingCBQ was separated from intact CDH, and the CBQ spectrumrecorded with rather small maxima at 457 nm and 496 nm.Reduction of FAD by dithionite or cellobiose eliminated thepeaks [17]. The flavin group in CDH is weakly fluorescentwith an emission maximum at 480 nm and excitationmaxima at 397 and 443 nm [37]. The separated cytochromedomains of the P. chrysosporium and H. insolens enzymeshave similar Soret maxima at 421 nm, and when treated withdithionite or ascorbate, they display a reduced haem spec-trum similar to that observed for the cellobiose-reduced in-tact enzyme [38].
Fig. (1). Absorption spectra of purified CDH from Sclerotium rolf-sii in the oxidised (grey line) and reduced state (black line). Thereduction was performed with cellobiose.
The chain lengths, molecular weights, and isoelectricpoints (pI) of different CDHs are listed in Table 3. A typicalCDH is a monomeric glycoprotein with a molecular mass ofapproximately 80 – 115 kDa and an acidic pI. A proteolyti-cally processed flavin-containing fragment “CBQ”, such asthat from T. versicolor, exhibits an upshift of pI from 4.2 to6.4., and might be a less stable protein [17].
Although the majority of reported purified CDHs arefunctional monomers, the enzymes from Coniophora
puteana [41] and Thielavia heterothallica [39] were reportedas dimeric, and that from Heterobasidion annosum [42] astetrameric. Hitherto, the only known crystal structures for aCDH are those of the individual domains of P. chrysospo-rium CDH [13-14, see sections 6.1 and 6.2 for details].These structures alone do not provide any reliable informa-tion on the biologically relevant oligomerisation state for thefull-length P. chrysosporium enzyme or for other CDHs.Thus, in order to evaluate the degree of oligomerisation andthe conformation of the interdomain peptide linker, tech-niques other than crystal-structure analysis should be consid-ered.
Like other secreted eukaryotic proteins, cellobiose dehy-drogenases are subjected to post-translational modificationsthat affect the properties of the mature protein significantly,including disulfide-bond formation, O- and N-linked glyco-sylation, and the processing of signal sequences. Glycosyla-tion is a common post-translational modification for fungalextracellular proteins. Presumably, all native CDHs are gly-coproteins, although the degree of glycosylation varies sig-nificantly, from 2% [37] up to 15% of the total CDH proteinmass [34]. Glucosamine, mannose and galactose were de-tected after total hydrolysis [37]. It seems that basidiomyceteCDHs are more glycosylated than their ascomycete counter-parts. Because fungi add O-linked oligosaccharides to theserine and threonine hydroxyl groups of secreted proteins,CDHs should be O-glycosylated mainly in the linker regionswhich have a high content of these amino acids (Fig. 2). O-linked glycosylation mostly involves mannose residues,whereas N-linked glycosylation is more complex [43]. Abranched heptasaccharide is first formed in the endoplasmicreticulum. After translocation three possible types of N-linked glycans can be incorporated into the final glycanstructure. In secreted fungal glycoproteins, the outer oligo-saccharide chain consists of a protein-linked core of two N-acetyl glucosamine residues, followed by a glycan tree ofeight to nine mannose residues. The consensus amino-acidsequence pattern for N-glycosylation is Asn-Xaa-Ser/Thrwhere Xaa is not proline. Several potential N-glycosylationsites are present in the primary structure of CDH. In Sporo-trichum thermophile CDH [39], six N-glycosylation sites arefound. The role of the glycan part of CDH is unclear, butdeglycosylated S. rolfsii CDH was found to be unstable [R.Ludwig & D. Haltrich unpublished results].
Several ascomycete CDHs are thermostable. In Thielaviaheterothallica CDH, the observed half life time was 10 hoursat 60°C and three hours at 70°C, respectively. This contrastswith P. chrysosporium CDH which exhibits a half life timeof only 25 minutes at 60°C and is rapidly inactivated at70°C. The thermophilic Chaetomiaceae CDHs (now com-prising four investigated members) contain a higher numberof disulfide bonds, salt bridges, and hydrogen bonds com-pared with their mesophilic counterparts. Thielavia het-erothallica has almost 3 times as many cysteine residues,and a two-fold higher content of charged (acidic and basic)residues compared with the basidiomycete CDHs [39].Moreover, thermophilic CDHs exhibit increased hydropho-bic interactions, as well as decreased solvent-exposed areasof their molecular surfaces, which all seem to contribute tothe increased thermostability.

260 Current Protein and Peptide Science, 2006, Vol. 7, No. 3 Zamocky et al.
A)
B)

Cellobiose Dehydrogenase Current Protein and Peptide Science, 2006, Vol. 7, No. 3 261
C)
Fig. (2). Selected parts of a multiple sequence alignment of 15 cellobiose dehydrogenases.A) Cytochrome domain in the area of haem ligands. Both ligands as well as a conserved disulfide bridge are marked with an asterisk. B) Mo-lecular signature around the βαβ mononucleotide binding motif known as Rossmann fold. C) Part near the C-terminus responsible for sub-strate binding. Essential histidin is marked with an asterisk. Numbers indicate position of the amino acid residues in the corresponding proteinsequence. Abbreviations are listed in table 3.
Table 3. Chain Length, Molecular Weight and Isoelectric Point of Different CDHs
Source of CDH, [accession nr.] Abbreviation Chain length [numberof amino acids]
Molecular weight [Da] pI
Aspergillus fumigatus [contig:asm50] Afumigatus 799 83100 6.4*
Aspergillus nidulans [XM_411367] Anidulans 761 81500 4.4*
Coniophora puteana [BAD32781] Coniophora 756 115000 4.6
Grifola frondosa [BAC20641] Gfrondosa 753 79600 4.3
Gibberella zeae [XM_389621] Gibberella 797 85900 6.7*
Humicola insolens [AF257654] Hinsolens 762 92000 4.0
Irpex lacteus [AB187223] Irpexlact 759 97000 5.1*
Magnaporte grisea [XM_360402] Mgrisea 840 87000 5.7*
Neurospora crassa [XP_322292] Ncrassa 1 806 86200 6.7*
Neurospora crassa [XP_325778] Ncrassa 2 805 86200 7.9*
Phanerochaete chrysosporium [U46081] Pchrysospo 755 89000 4.2
Pycnoporus cinnabarinus [AF081574] Pcinnabari 750 92000 3.8
Sclerotium rolfsii [AY187232] Srolfsii 752 101000 4.2
Thielavia heterothallica [AF074951] Theterotha 807 91000 4.1
Trametes versicolor [AF029668] Tversicolo 749 97000 4.2
References for these enzymes are the same as used in tables 1,2; * calculated from the amino acid composition.

262 Current Protein and Peptide Science, 2006, Vol. 7, No. 3 Zamocky et al.
4. ANALYSIS AND CLASSIFICATION OF THEGENES CODING FOR CELLOBIOSE DEHYDRO-GENASE
Genes coding for CDHs were already identified in 21organisms. For most CDH genes, successful cloning wasachieved by reverse-transcription PCR and a rapid amplifi-cation of cDNA ends [12, 30, 44]. For this purpose, the totalmRNA was isolated from the mycelium grown on celluloseor cellobiose, but not on glucose. The average length of thecomplete cDNAs is 2480bp comprising, from the N termi-nus, the sequences coding for a signal peptide, the cyto-chrome domain, and the flavodehydrogenase domain. SomecDNAs contain an additional sequence, following the flavindomain region, that codes for a small cellulose-binding motifclassified as a carbohydrate-binding module of type 1 [seehttp://afmb.cnrs-mrs.fr/CAZY/]. In addition, the identifica-tion of a new CDH-coding gene in a complete genome isbased on sequence similarity along the whole coding se-quence, including the patterns of the cytochrome domain thatdistinguishes CDH from other members of the GMC-(glucose-methanol-choline)-superfamily of flavoproteins. Bythis procedure, new CDH genes were identified in recentlysequenced fungal genomes, although some open readingframes (ORFs) identified may be regarded as putative.
The CDH gene sequences have a relatively high GCcontent (average value of 57.14%, see Table 4) that corre-lates well with that expected for the respective organism.The lowest GC content is observed for the S. rolfsii sequence(47.93%), which is hitherto the only known example with a
significantly different GC content compared with other CDHsequences. Since only a few gene sequences have been ana-lysed from this fungus, it is uncertain whether the low GCcontent is representative for that of the full genome. In sev-eral cases, the corresponding gene was also monitored in thegenome. Interestingly, the majority of analysed fungal ge-nomes exhibit the presence of multiple CDH-encodingORFs. The two ORFs coding for P. chrysosporium CDH(accession # U50409 and U65888) share 98% sequenceidentity. Li et al. [45] identified these sequences as beingallelic variants of a heterokaryon with at least two distinctnuclei. A third variant of P. chrysosporium CDH (accession# X88897) contains one codon deletion, but has 97% identitywith the aforementioned sequences. In contrast to the above,two different genes presumably coding for CDH in the ge-nome of Neurospora crassa (accession # XM_322291 andXM32577) share only 57.5% sequence identity, and appearas phylogenetically different genes (Fig. 3). Other fungalspecies, both ascomycetes and basidiomycetes, possess sev-eral genes of different evolutionary history coding for thisessential degradative enzyme as well [M. Zamocky et al.,unpublished results].
For most of the investigated species, analysis of the ge-nomic DNA revealed the presence of introns in the CDH-coding sequences. For two allelic gene variants of P. chryso-sporium CDH, 14 exons were identified and introns werefound at the same positions [45]. All intron splice junctionsconform to the GT-AG rule. The exons 2 through 5 code forthe cytochrome domain, the fifth exon also for the linker
Table 4. GC content of Basidiomycete and Ascomycete DNA Sequences Coding for CDH
DNA coding for CDH from Genomic DNA or cDNA % GC
A. fumigatus Genomic 60.21
A. nidulans Genomic 58.60
C. puteana cDNA 57.35
G. frondosa cDNA 52.37
G. zeae Genomic 55.03
H. insolens cDNA 61.32
I. lacteus cDNA 53.29
M. grisea Genomic 58.30
N. crassa 1 Genomic 59.76
N. crassa 2 Genomic 54.72
P. cinnabarinus Genomic 55.28
P. chrysosporium Genomic 59.20
S. rolfsii (A. rolfsii) cDNA 47.93
T. heterothallica cDNA 62.67
T. versicolor Genomic 58.84
Average value 57.14

Cellobiose Dehydrogenase Current Protein and Peptide Science, 2006, Vol. 7, No. 3 263
Fig. (3). Reconstructed unrooted phylogenetic tree of 15 cellobiosedehydrogenases. The phylogeny was analysed using the Puzzleprogram, version 5.2 applying the WAG model of amino-acid sub-stitution and 500,000 puzzling steps [136]. The numbers in eachnode represent the likelihood output (according to Puzzle manual).Abbreviations of CDHs are the same as used in Figure 2 and Table3. The scale bar indicates the branch length corresponding to 0.1amino acid substitutions per site.
region. Exons 6 through 14 code for the flavin dehydro-genase domain. The genomic DNA with the ORF for T. ver-sicolor CDH [44] is interrupted by 14 introns. The averageintron length is 56 nt and the intron structure correspondswell to the consensus intron structure for Basidiomycetes.With the exception of an extra C-terminal intron, the intronpositions correspond to those of P. chrysosporium. TheCDH-coding region of the Pycnoporus cinnabarinus ge-nomic DNA revealed the presence of 16 exons interrupted by15 introns with an average length of 52 nt [46]. Since thesequence of neighbouring regions of the genome were identi-fied, it was possible to localise a putative TATA box-likeelement -53 bp relative to the start codon of CDH. Interest-ingly, no CAAT-like element was found in this relativelylong promoter region. Moreover, no polyadenylation se-quence consistent with the consensus eukaryotic sequencewas found in the 3´UTR (untranslated region) but a con-
served fungal and mammalian sequence motif(TGTGTTCG) was found in this region. The distribution ofintrons in genes coding for ascomycete CDH sequences isnot well documented but from the genome analysis it is clearthat some of these ORFs are also interrupted by several in-trons that are comparable in length with introns in basidio-mycete CDH sequences.
Multiple sequence alignment of the translation productsof 15 CDH sequences was performed with ClustalX. Thiscomplete alignment included both cytochrome and flavindehydrogenase domains and also the linker region connect-ing these. Selected parts are presented in (Fig. 2). Here wecan identify typical molecular signatures of these uniqueflavocytochromes [39]. In part A there is a conserved motifof the cytochrome domain comprising both ligands of thehaem group and also a region with a disulfide bridge be-tween two conserved cysteine residues. In part B there is thehighly conserved βαβ mononucleotide-binding Rossmannfold involved in binding of the flavin prosthetic group. Inpart C, high sequence conservation corresponds to a sub-strate-binding pattern. As a rule, the level of pairwise se-quence identity and similarity is much higher within the rep-resentative basidiomycete or ascomycete groups (65 to 83%)than between groups when ascomycete and basidiomyceterepresentatives are cross-compared (30 to 35%). This led tothe classification of all known CDH sequences into twoclasses [47]. Class-I is represented by basidiomycete CDHs.Their sequences are shorter, lacking a C-terminal carbohy-drate-binding module (CBM), but contain a highly conservedlinker region connecting the cytochrome and flavin domain.In contrast, class-II comprises the more complex ascomyceteCDHs that may use a distinct C-terminal CBM, and a lesswell conserved linker sequence. Hitherto, there is no knownsequence of a basidiomycete CDH that fits the class-II crite-ria or vice versa.
4.1. Phylogenetic Relationship
The phylogenetic relationships of cellobiose dehydro-genase and related sequences have been reported previously[47]. Whereas the cytochrome domains constitute a uniquephylogenetic entity, the flavodehydrogenase domains repre-sent an autonomous clade within the GMC flavoenzyme su-perfamily. All genes coding for CDH comprise a phyloge-netic cluster together with a separate clade for cholesteroloxidases and a further clade of bacterial dehydrogenases[47]. The two classes of cellobiose dehydrogenase genes thatare apparent from multiple sequence alignment (Fig. 2) arewell conserved, both in the cytochrome and in the flavode-hydrogenase domains. Here we present a new maximumlikelihood-based phylogenetic reconstruction based on theanalysis of the entire CDH-coding sequences (850 aminoacid positions used for the phylogeny, (Fig. 3). The existenceof two classes of cellobiose dehydrogenase genes is sup-ported statistically; the ORF for a putative CDH from Asper-gillus fumigatus is presented as an outgroup. We observelonger branches in the ascomycete class compared with thoseof the basidiomycete clade. The two different genes of Neu-rospora crassa are phylogenetically not close neighbours soin this fungus two independent gene lines for CDH may ex-ist. The putative sequence of A. nidulans CDH is separatedfrom other class-II representatives, and in class-I, the se-

264 Current Protein and Peptide Science, 2006, Vol. 7, No. 3 Zamocky et al.
quence of S. rolfsii CDH represents a similar case. The re-sulting unrooted phylogenetic tree is in good agreement withthe fungal phylogeny based on 18S-rDNA. Moreover, it sup-ports the outlined hypothesis of the evolutionary history forthis clade of the GMC superfamily [47]. The CDH-geneclade apparently diversified and adapted to a specific degra-dative function early in the evolution of the fungal ancestor.One of the earliest evolutionary events was probably theacquisition of a cytochrome domain. Separate cytochromedomains associated with a CBM exist in some fungal ge-nomes. One such representative from P. chrysosporium wasalready cloned and heterologously expressed [48] but itsphysiological function and mainly its phylogenetic originremain unclear. Although conserved in catalytically impor-tant areas the sequence similarity of these cytochrome genesto cytochrome domains of CDH is only moderate. Notably, avery high degree of sequence identity exists between thesecytochromes and cytochrome domains of putative iron re-ductases (data not shown). From the previous phylogeneticanalysis [47] it is, however, clear that the haem b-bindingdomains of all complete CDH genes have very probably acommon ancestor of fungal origin. If the fused cytochromeand flavodehydrogenase domains, containing different pros-thetic groups, can act synergistically, the complete fusedCDH gene may represent an evolutionary advantage for thehost. Further investigation is needed to study the phyloge-netic relationship between cytochrome domains of CDH,cytochromes associated with CBM and iron reductases (inpreparation).
5. CATALYTIC MECHANISM AND REACTIONKINETICS
5.1. Substrate Binding
The overall reaction of cellobiose dehydrogenase can bedivided into a reductive half-reaction and an oxidative half-reaction. The complete catalytic cycle of CDH is shown in(Fig. 4). During the reductive half-reaction, β-D-cellobiose is
Fig. (4). The proposed reaction mechanism of CDH. Cellobiose isoxidised to the corresponding lactone. Abbreviations used: F =CDH flavin group, b = CDH haem b group, Z = 2-electron acceptordyes, e.g. DCPIP, X = 1-electron acceptors, e.g. ferricyanide andmolecular oxygen, interacting with FAD, c = cytochrome c or 1-electron acceptors, interacting with haem b.
oxidised at the anomeric C1 carbon to yield cellobionolac-tone [23], which is further hydrolysed in bulk water to thecorresponding carboxylic acid [49]. Alternative substrates invivo are higher cello-oligosaccharides exhibiting slightlydecreased catalytic efficiencies (kcat/Km) [20, 23, 34, 50].Several reports also claim, albeit debated, that CDH oxidisesthe reducing ends of polysaccharide chains of differentpolymorphous modifications of cellulose [51, 52].
Henriksson et al. [50] suggested two binding sites in theCDH active site: one that binds the reducing glucosyl moietyof cellobiose where oxidation takes place (the C site; C forcatalytic), and another site that accounts for binding (the Bsite) of the glucosyl unit at the non-reducing end. The crys-tal-structure determination of the P. chrysosporium CDHflavoprotein domain with a bound substrate analogue, cello-bionolactam, has elucidated the necessary structural determi-nants for substrate binding in the two subsites, B and C, ofthe CDH active site [53]. These determinants include theexact position of the anomeric C1 atom in subsite C, thelinking oxygen in a β-1,4-linkage between subsites B and C,and the C2 and C3 hydroxyl groups in the glucosyl unit ofsubsite C. The key protein residues for hydrogen bonding insubsite C are His689, Asn732 and Asn688, which are allfully conserved among CDHs. In addition, two strategicallyplaced water molecules coordinated by Tyr609 provide ad-ditional hydrogen-bonding interactions with O3 and O6 inthis subsite. The importance of His689 and Asn732 for ca-talysis was confirmed kinetically by Rotsaert et al. [54] forP. chrysosporium CDH mutants. The His689 variants exhibitmore than 1000-fold lower kcat values for cellobiose andlactose, while the Km values for the same substrates aresimilar to that of the wild-type enzyme. This supports theproposed role of His689 as a general base in catalysis. TheAsn732 variants exhibit 5 to 4000-fold lower kcat values foroxidation of cellobiose and lactose, while the Km valuesrange from unperturbed to 60-fold higher than comparedwith the wild-type enzyme. This indicates that Asn732 par-ticipates in substrate binding as well.
In subsite B, the orientation of the C2, C3 and C6 hy-droxyl groups appear to be important. Hydrogen-bondingresidues are Glu279, Arg586 and Asn688, of which Arg586and Asn688 are fully conserved in CDHs. The structural dataprovide a rational explanation for the observed kinetic be-haviour of wild-type P. chrysosporium CDH on various sug-ars [34, 50, 55].
5.2. Catalytic Mechanism of the Reductive Half-Reaction
In line with the requirements listed above, kinetic data[50] showed that the β-1,4 linked disaccharides cellobiose,lactose and mannobiose are good or acceptable substrates forCDH, whereas the α-1,4 linked disaccharide maltose as wellas the monosaccharides mannose, galactose and allose arepoor or no substrates. The higher Km values for mannobioseand galactomannoside compared with cellobiose and lactoseare in agreement with the structural data, stressing the im-portance of the C2 hydroxyl groups in subsites C and B forsubstrate binding. The discrimination against glucose oxida-tion by P. chrysosporium CDH is very strong as shown by a87,000-fold larger catalytic efficiency for cellobiose com-pared with glucose [50]. The fact that monosaccharides are

Cellobiose Dehydrogenase Current Protein and Peptide Science, 2006, Vol. 7, No. 3 265
poor or no substrates for CDH is attributed to the presence ofvarious modes of non-productive binding in the two bindingsites. However, in this case one should expect strong inhibi-tion of CDH by monosaccharides, which is not confirmed[40, 50, 66].
In the case of P. chrysosporium CDH, thiocellobiose,which has a thioglucosidic instead of a glycosidic bond, wasoxidised with a kcat value that is 1.5 times higher comparedwith that for cellobiose, 25.6 s-1 and 15.7s-1, respectively.This was achieved despite a 10-fold increase in the Km value[50]. For the S. rolfsii enzyme, Baminger and co-workers[34] reported that the Km value increased 10-fold but that thekcat value remained unchanged. The disaccharide β-xylobioselacks both exocyclic C6-O6 groups. Based on kinetic data,this sugar was reported not to be a substrate for P. chryso-sporium CDH, thus suggesting, indirectly, that the C6 hy-droxyl groups are indeed important for substrate or transitionstate binding [50]. Other groups have reported that xylobiosecan act as an electron donor [34, 37] but the possibility ofcontamination with residual cellobiose must be considered[55]. Based on the highly resolved structural details of disac-charide binding in the cellobionolactam complex, xylobiosecould be a possible, but not necessarily good, substrate for
CDH since the C6 hydroxyl groups in subsites B and C ap-pear to be involved but not critical: the C6 hydroxyl in site Cinteracts with a water molecule, and the subsite B C6 hy-droxyl interacts with Asn688 [53]. Sugars that performpoorly as substrates violate one or several of the structuralcriteria stated above, resulting in low substrate affinityand/or specificity or lack of stabilisation of the transitionstate. Kinetic parameters for most CDHs and some substratesare shown in Table 5. The difference in substrate specificitybetween basidiomycete and ascomycete CDHs becomes ob-vious from this table. Ascomycete CDHs have lower Km val-ues for all examined electron donors compared with thebasidiomycete counterparts. The relative conversion rate formaltose and glucose, as compared with cellobiose, for theascomycete CDH from INBI 2-26(-), is 31% and 42%, re-spectively. The rather high relative velocity and affinity,which is not observed with basidiomycete CDHs, indicatesaltered substrate specificity. As expected, the data indicatethat ascomycete enzymes from thermophilic/thermotolerantorganisms have higher temperature optima.
5.3. Interdomain Electron TransferThe presence of two redox centres in CDH has fueled
speculations concerning the function of the haem domain. At
Table 5. Comparison of CDH from Various Sources and their Steady-State Kinetic Parameters for Some Electron Donors
Cellobiose Lactose Maltose Glucose e.a. pH T
Km kcat Km kcat Km kcat Km kcat
Organism
mM s-1 mM s-1 mM s-1 mM s-1 °C
Ref.
Basidiomycetes
0.042 14.8 0.68 11.4 3.65 1 380 1.2 DCIP 6.0 25 [72]
0.025 24 0.63 28.8 n.d. 0 n.d. 0 Cyt c 4.5 RT [23]
0.11 15.7 1.1 13.4 240 1.14 1600 2.64 DCIP 5.0 35 [50]
0.04 25.7 DCIP 4.5 RT [141]
P. chrysosporium
0.016 15.5 0.27 14.3 Cyt c 4.5 RT [54]
S. rolfsii 0.12 27 2.4 26 240 0.8 1250 1.5 DCIP 4.0 25 [34]
T. versicolor 0.12 6.1 DCIP 4.5 23 [17]
T. villosa 0.21 23.6 3.7 26.9 350 2.08 1300 1.92 DCIP 4.5 30 [18]
T. pubescens 0.21 21.9 2.4 25.8 390 1.92 890 1.31 DCIP 4.5 30 [18]
I. lacteus 0.034 14.6 1.85 17.7 0 0 Cyt c 4.5 30 [20]
C. puteana 0.046 45.6 0 Cyt c 4.0 30 [138]
S. commune 0.03 13 0.108 6 0 0 Cyt c 4.5 30 [140]
Ascomycetes
0.011 14 0.051 14 11 1.2 n.d. 0 DCIP 7.5 40 [37]H. insolens
0.034 1.53 DCIP 7.0 n.g. [73]
S. thermophile 0.0058 16.1 0.050 16.8 Cyt c 4.5 45 [39]
Chaetomium sp. 0.0047 8.9 0.056 14 n.d. 2.8a n.d. 3.8a DCIP 6.0 20 [40,66]
arecalculated from relative activity values; n.d…not determined; n.g...not given; e.a...electron acceptor used for activity assay; DCIP...2,6-dichloroindophenol; Cyt c...cytochrome c.

266 Current Protein and Peptide Science, 2006, Vol. 7, No. 3 Zamocky et al.
any given pH, the FAD of P. chrysosporium CDH is morereductive than the haem. Thus, during the reductive half-reaction, there will be a flow of electrons from the catalyticcentre to the haem domain. Such a flow is observed forclass-I CDHs only below pH 6, but for some class-II CDHsalso under neutral or slightly alkaline conditions [37,40,73].The experimentally determined redox potential [56] ofFox/Fred of the isolated flavin domain decreases with the in-crease in pH from 106 mV (pH 3.0) to -132 mV (pH 7.0).The redox potential of the haem domain is 190 mV (pH 3.0)and 130 mV (pH 7.0), respectively. The data for the haemdomain is in agreement with previous results of Kremer andWood [52] where the redox potential of the haem prostheticgroup at pH 4.0 was reported to be 165 mV.
For the catalytic role of the haem domain two differentmodels were suggested [15, 57]. The electron-chain modelstates that the electrons, one by one, are shuttled from FADto haem b before reduction of an external one-electron ac-ceptor occurs. The electron-sink model suggests that the roleof the haem b is to store electrons in order to ensure specificreaction conditions at the FAD for the reduction of electronacceptors. Both models may be valid for different electronacceptors. The preferred model for the electron transfer fromCDH to cytochrome c is the electron-chain model. Otherone- or two-electron acceptors can be reduced directly at theFAD centre, which would favour the electron-sink model.Unfortunately, the lack of data prevents more in-depth dis-cussion on this subject.
In order to shed light on the mechanism of electron trans-fer in CDH, the reduction of P. chrysosporium CDH by cel-lobiose has been investigated by rapid-kinetics studies usingstopped-flow spectrophotometry [56, 58-63]. The rate offlavin reduction depends on the cellobiose concentration butreaches a rate limit of approx. 20 s-1 at pH 6.0 [58]. Thisvalue has been confirmed (k lim ~ 29 s -1 at pH 6.0) by Igarashiet al. [56], but higher rates (klim ~ 68 s-1 at pH 4.0) are ob-tained at lower pH values with an optimum between pH 4and 5. Jones and Wilson [58] observed (under anaerobicconditions at pH 6.0) that the rapid reduction of the flavin isfollowed by a slower reduction of the haem, and that the rateof reduction decreases at higher cellobiose concentrations.This substrate-inhibition mechanism is pH dependent [56]being pronounced at higher, pH values. Stoica et al. [64]studied the substrate inhibition of P. chrysosporium CDHadsorbed on graphite electrodes through direct and mediatedelectron transfer between electrode and enzyme. Inhibitionoccurred only with cellobiose, but not with lactose. Theauthors suggest the formation of a [flavin radical-cellobiose]complex (a flavin semiquinone radical was detected with lowtemperature EPR [56, 62]) to be responsible for the reductionof the intramolecular electron transfer rate.
Jones and Wilson [58] noted that the rate constant forhaem reduction, unlike that for flavin reduction, increasedwith enzyme concentration, prompting the conclusion thatthe haem can be reduced also by flavin groups in differentmolecules. Samejima and Eriksson [65] observed bi-phasicbehaviour during the reduction of both FAD and haem.They, likewise, concluded that the electron-transfer mecha-nism for cellobiose oxidation could involve intermolecularelectron transfer between CDH molecules. The occurrence of
intermolecular electron transfer in vitro has been shown inanaerobic titration experiments of S. rolfsii CDH with cello-biose [66].
Samejima and Eriksson [65] observed a pH dependencyof the intramolecular electron-transfer in CDH. At pH 4.2,both FAD and haem are reduced rapidly by cellobiose,whereas at pH 5.9, only FAD reduction is fast, while thereduction of haem is extremely slow. This discrepancy isalso present in the pH dependency of cytochrome c reduc-tion. It acts as an effective electron acceptor at pH 4.2, but isreduced very slowly at pH 5.9. The authors conclude there-fore that the haem in CDH must be responsible for cyto-chrome c reduction.
A different theory was reported by another group [62]. Inthis study, it was confirmed that increasing the pH to 6.0slows down the rate of electron transfer from FAD to haemsignificantly. However, the authors suggested that the flavincofactor is responsible for the reduction of cytochrome c.The inhibition of the cytochrome c reductase activity of CDHby high cellobiose concentrations could, in the author’sopinion, indicate that the FAD semiquinone radical is re-sponsible for cytochrome c reduction, not the reduced haem,thus supporting the electron sink model. The role of thehaem would be to provide the FAD semiquinone radical.This hypothesis fueled the speculation that the site of carbo-hydrate reduction in P. chrysosporium CDH would lie at theinterface of the domains. However, this is not supported bythe crystal structure, since the catalytic centre is well buriedbelow the surface of the molecule.
Igarashi et al. [56] performed several experiments to elu-cidate the mechanism of intramolecular electron transfer inmore detail. It was observed that the reductive half-reactionsof flavin and haem were biphasic and monophasic, respec-tively. The behaviour of the second (intramolecular) phase offlavin conversion was almost similar to the rate of haem re-duction at all substrate concentrations tested, suggesting thatthe formation of flavin semiquinone and haem reductioninvolve the same electron-transfer reaction. The second (in-tramolecular) phase of flavin reduction and the haem reduc-tion were inhibited at higher pH values (almost zero at pH6.0), as well as by high cellobiose concentrations. The firstphase had a hyperbolic relation to substrate concentrationand was not slowed down (as was also observed by Jonesand Wilson [58]). The close correlation of the pH depend-ence of the dissociation constant of cellobiose observedduring FAD reduction with the substrate-inhibition constantof haem reduction is suggested to indicate that the binding ofcellobiose to the active site inhibits the electron transfer.
Additional proof for the intermolecular electron transferfrom haem to cytochrome c (electron chain theory) was pro-vided by the kinetic analysis of an F166Y mutant of P.chrysosporium CDH [67]. Phe166 is located in the cyto-chrome domain and approaches one of the propionates of thehaem. The mutation did not affect the pre-steady state re-duction of the flavin, but the rate of the subsequent electrontransfer from flavin to haem was reduced by a factor of 2.5.When the reduced wild-type and mutant protein are mixedwith cytochrome c, the haem oxidation and cytochrome creduction occurs synchronously, suggesting that the initialelectron is transferred from reduced haem to cytochrome c.

Cellobiose Dehydrogenase Current Protein and Peptide Science, 2006, Vol. 7, No. 3 267
In contrast, the steady-state phase of cytochrome c reductionwas dependent on the electron transfer from flavin to haem.The results of these aerobic experiments clearly indicate thatthe first and second electrons of two-electron reduced CDHare both transferred via haem, and that the redox reaction ofCDH involves an electron-transfer chain mechanism for cy-tochrome c reduction.
The conclusion that can be drawn from these reports onP. chrysosporium CDH is that the reduction of cytochrome cis carried out by the haem domain following flavin-to-haemelectron transfer, and that in this process, the relatively slowintramolecular electron transfer is rate limiting, especially ata pH above 5. Furthermore, the intramolecular electron-transfer rate is strongly pH dependent. Thus, at physiologicalpH (pH 3-4), the intramolecular electron-transfer ratereaches a maximum suggesting that electron transfer fromthe flavin domain to a one-electron acceptor via the haemdomain is the biologically relevant process.
The significance of the substrate inhibition by cellobiosein vivo is doubtful since cellobiose is likely to be present atlow concentrations only due to its efficient transport into thecell. Nevertheless, the substrate inhibition of intramolecularelectron transfer provides information about possible spatialand temporal relationships of substrate binding and electrontransport.
5.4. Catalytic Mechanism of the Oxidative Half-Reaction
The oxidative half-reaction proceeds by electron transferto either a two-electron acceptor or two equivalents of a one-electron acceptor [68]. The natural electron acceptors forCDH or the flavin fragment are uncertain and the multitudeof possibilities leaves space for speculation. A wide range ofelectron acceptors were reported to be reduced by theholoenzyme or the flavin fragment. Reduced CDH is reoxi-dised by electron acceptors such as 2,6-dichloro-indophenol(DCIP), 1,2- or 1,4-benzoquinone and their derivatives, theABTS cation radical, complexed metal ions such as Fe(III),Cu(II) and Mn(III), or oxygen [17]. The reduction of triio-dide was also reported [69]. The reduction of these electronacceptors usually takes place at the flavin [70], but asbasidiomycete CDHs can shuttle electrons efficiently to thehaem at pH values below 5.5 [59] and ascomycete CDHs canshuttle electrons efficiently to the haem even at neutral orincreased pH values [37, 40, 66], it is obvious that one-electron acceptors can also be reduced at the haem. It iscommon belief that two-electron acceptors are reduced di-rectly at the flavin domain, but opinions differ pertaining tothe reduction of one-electron acceptors. Cytochrome c cansolely interact with the haem domain of CDH and therebytransfer electrons from haem b to haem c. This special prop-erty can be used in an enzyme assay to distinguish betweenthe holoenzyme and the flavin fragment. CDH reacts alsowith cytochrome c-551 from Pseudomonas aeruginosa at thesame rate as with cytochrome c from horse heart [60]. Sev-eral other one-electron acceptors can be reduced by bothcentres but with varying catalytic efficiencies. Roy et al. [17]purified and characterised the T. versicolor holoenzyme andflavin domain. Both were similarly reduced by cellulose,cello-oligosaccharides and lactose, and oxidised by quinonesand organic radical species, but they differed in their ability
to reduce metal ion complexes. The flavin fragment devoidof the haem much less efficiently reduced Fe(III)-phenanthroline, Cu(II)-2,9-bathocuproinedisulfonate, orMn(III) malonate-complexes at pH 4.5 than the intact flavo-cytochrome.
The reactivity of different CDHs with several electronacceptors is shown in (Table 6) where some steady-statekinetic constants are listed. Again, the difference betweenbasidiomycete and ascomycete CDH is evident. In contrastto the electron donors, the Km values for electron acceptorsfor ascomycete CDHs are not lower than for the basidiomy-cete enzymes. For most electron acceptors, the most obviousdifference concerns the pH optima, which for ascomyceteCDHs are shifted to the less acidic or neutral region com-pared to the basidiomycete enzymes.
Studies on the nature of the flavin cofactor revealed thepresence of a fraction of 6-hydroxy-FAD in certain CDHpreparations [68]. Whether the more negative midpoint re-dox-potential (-255 mV at pH 7.0 for 6-hydroxy-FAD insolution compared to -219 mV for the unsubstituted FAD[71]) influences the catalytic activity with some co-substrateswas not yet proven unequivocally. Morpeth and Jones [72]found that purified CDH contained variable amounts of amodified flavin, 6-hydroxy-FAD. All of the isoenzymesfrom any one preparation displayed similar kinetic parame-ters. However, these varied between preparations. If the 6-hydroxy-FAD alters the catalytic properties, then this differ-ence could be a result of different amounts of the modifiedcofactor caused by variable culture or purification condi-tions. As mentioned in section 2.2 class-II CDH differ intheir higher 6-hydroxy-FAD content from class-I enzymes.Reconstitution experiments with the deflavo-proteins and 6-hydroxy-FAD resulted in flavin domains that were clearlyactive but their cellobiose oxidation rates were lower thanthose containing normal FAD [38]. Interestingly, the 6-hydroxy-FAD containing flavin domains showed a higheractivity (with ubiquinone) at pH 4.5 than at pH 7.0 whencompared to flavin domains reconstituted with normal FAD.
An astonishing observation for CDH from the ascomy-cete H. insolens was published by Xu et al. [73]. The authorsfound a peroxidase-like activity in the absence of cellobiose.When hydrogen peroxide was added, ABTS was slowly oxi-dised to the ABTS cation radical. This activity was approxi-mately 200-fold lower than ABTS radical reduction in thepresence of cellobiose under the given conditions. When noABTS was present, hydrogen peroxide led to a decrease inthe Soret band, indicating the oxidation of the haem domain.Whether this activity has any catalytic relevance in vivo, or isjust an artifact remains unclear. An alternative explanation ofthis activity might be the formation of highly reactive oxy-gen species that can directly oxidise ABTS.
5.5. Formation of Reactive Oxygen Species
Classically, two types of flavoenzyme are capable of re-ducing oxygen. First are those with flavin only as a pros-thetic group, such as glucose oxidase, D-amino acid oxidaseand related enzymes. These also include alternative glucoseoxidases from some CDH-secreting fungi [74]. When suchenzymes react with oxygen and a reducing substrate, thestoichiometric product is peroxide [75] although enzyme-

268 Current Protein and Peptide Science, 2006, Vol. 7, No. 3 Zamocky et al.
Table 6. Comparison of CDH from Various Sources Regarding their Steady State Kinetic Parameters for Electron Acceptors
DCIP Benzoquinone TBBQ e.d. T
Km kcat pH Km kcat pH Km kcat pH
Organism
µM s-1 µM s-1 µM s-1 °C
Ref.
Basidiomycetes
12 1.9 6.0 c 200 µM 25 [72]
15 15 6.0 130 21.4 6.0 c 200 µM 25 [23]
3.6 33 4.5 12 27 4.5 c 100 µM RT [141]
P. chrysosporium
6.4 27 4.5 c 200mM RT [54]
S. rolfsii 15 30 4.0 25 30 4.0 53 23 4.0 c 2 mM 25 [34]
T. versicolor 7.8 4.8 4.5 26 25 4.5 c 2 mM 23 [17]
T. villosa 9.9 39.1 5.5 11 20.8 4.5 41 16.9 4.5 l 30 mM 30 [18]
T. pubescens 4.9 25.6 19 27.5 4.5 18 24.2 4.5 l 30 mM 30 [18]
I. lacteus 12.9 15.2 4.5 54.8 6.98 4.5 c 100µM 30 [20]
C. puteana 13.5 17.4 4.0 c 500 µM 30 [138]
S. commune 10 63 4.5 c 1 mM 30 [140]
P. cinnabarinus 23 n.g. 4.5 45 n.g. 4.5 c 2 mM 25 [21]
Ascomycetes
26 17 7.5 132 21 7.5 c 225µM 40 [37]H. insolens
34 1.53 7.5 c n.g. n.g. [73]
S. thermophile 3.7 18.3 4.5 c 100 µM 45 [39]
INBI 2-26(-) 16 11.3 6.0 c 380 µM 20 [40]
Organism Cyt c Ferricyanide ABTS e.d. T
Km kcat pH Km kcat pH Km kcat pH °C
µM s-1 µM s-1 µM s-1
Ref.
Basidiomycetes
3100 0.3 6.0 c 200 µM 25 [72]
1.2 20.5 4.5 5.2 22 4.5 c 100 µM RT [23]
P. chrysosporium
0.7 14.2 4.5 c 200mM RT [141]
S. rolfsii 0.3 34 3.5 20 37 3.0 0.4 27 4.0 c 2 mM 25 [34]
T. versicolor 7.8 10.5 4.5 110 5.3 4.5 c 2 mM 23 [17]
T. villosa 1.8 32.5 3.5 5.5 36.9 4.0 0.20 16.1 4.0 l 30 mM 30 [18]
T. pubescens 0.69 29.4 3.5 8.6 27.8 4.0 0.28 18.1 4.0 l 30 mM 30 [18]
I. lacteus 3.98 17.6 4.5 34 9.33 4.5 30 [20]
C. puteana 126 8.1 4.0 c 500 µM 30 [138]
S. commune 2.8 25 4.5 71 13.5 4.5 c 1 mM 30 [140]
P. cinnabarinus 31 n.g. 4.5 210 n.g. 4.5 c 2 mM 25 [21]

Cellobiose Dehydrogenase Current Protein and Peptide Science, 2006, Vol. 7, No. 3 269
(Table 6) contd….
Organism Cyt c Ferricyanide ABTS e.d. T
Km kcat pH Km kcat pH Km kcat pH °C
µM s-1 µM s-1 µM s-1
Ref.
Ascomycetes
93 27 7.5 12 14 7.5 c 225µM 40 [37]H. insolens
36 1.53 7.5 48 7.5 n.g. 110 9.3 n.g. c n.g. n.g. [73]
S. thermophile 1.0 20.8 4.5 6.7 24.1 4.5 c 100 µM 45 [39]
INBI 2-26(-) 15.8 8.1 6.0 c 380 µM 20 [40]
n.d… not determined; n.g…not given; e.d...electron donor used for activity assay; l...lactose; c…cellobiose DCIP...2,6-dichloroindophenol, TBBQ...3,5-di-tert-butyl-1,2-benzoquinone, ABTS...2,2'-azino-bis(3-ethylbenzothiazoline-6-sulfonate) cation radical
bound superoxide may occur as a transient intermediate [76].The second flavoenzyme type comprises a number of differ-ent proteins all of which contain flavin plus a transitionmetal centre. These include the flavin/molybdenum/FeS en-zyme xanthine oxidase [77], the plasma membrane fla-vin/haem NADPH oxidase [78, 79], the mitochondrial mem-brane complex I NADH-Q reductase [80], and the mitochon-drial and prokaryotic complex II family, succinate-coenzymeQ reductase [81] and CoQ fumarate reductase [82]. Allmembers of this family can under some conditions react withoxygen in a one-electron process to generate superoxide.
As described above (section 2.1), the CDHs can exist inat least two forms, a holoenzyme haem-containing form(CDH) and a proteolytically derived flavin-containing frag-ment (CBQ). Both these forms can react with oxygen to pro-duce peroxide and superoxide as products. CDH can thusadopt alternative mechanisms resembling those of both thesimple flavin oxidases and the complex flavin oxidases.
CDH was originally described as cellobiose oxidoreduc-tase (CBO) because of its multiple redox capabilities. TheCBQ fragment is clearly an “aerobic dehydrogenase” to usethe classical terminology, producing hydrogen peroxide asthe product of oxygen reduction [65]. The holoenzyme(haem and flavin domain-containing) CDH also reacts withoxygen but in more complex ways. At an early stage in thestudy of these enzymes, Morpeth [68] identified superoxideas the product of the O2 reaction of P. chrysosporium CDHand cited the flavin semiquinone as reducing species. Even-tually superoxide decays to regenerate oxygen and peroxide,and therefore, under many conditions a simple stoichiometrycellobiose:O2:H2O2 of 1:1:1 can be obtained [23]. But in acomparative study of CDH and glucose oxidase, Nutt et al.[83] showed that only part of the oxygen taken up by CDHplus cellobiose became peroxide and the ratio was thereforesubstoichiometric. Cameron and Aust [84] considered that,although the initial O2:H2O2 stoichiometry might be 1.0,traces of heavy metals including iron can reduce the peroxideyield by secondary reactions giving and consuming radicalssuch as OH•.
Where does the oxygen react? In contrast to Morpeth’ssemiquinone suggestion [68], Wilson et al. [85] identifiedthe haem group as a candidate site. Later, the same group[63] redefined (for P. chrysosporium CDH) the relationships,
arguing for the flavin as the site of oxygen reduction to su-peroxide, and the haem as a site of superoxide reduction toperoxide. What is the evidence? Does the same mechanismapply to all types of CDH? What bearing does the oxygenreaction have on the ability of CDH to promote Fenton-likechemistry and consequent lignin degradation? Although es-sentially all CDH types investigated are rather specific forcellobiose as hydrogen donor (see section 6.3 following),several oxidants can act as acceptors and the biologicallysignificant acceptor remains uncertain. Ferricyanide and cy-tochrome c may be artificial analogues of naturally occurringferric complexes. Oxygen reacts solely with the flavin, cyto-chrome c with the haem group and ferricyanide with both, asdetermined using the P. chrysosporium enzyme [86]. Thesimplest reaction mechanism involving oxygen and one-electron acceptors may be written as in Eqs. 1-4:
CDHflavin + cellobiose ---> CDHflavinH2 + cellobionolactone (1)
CDHflavinH2 + CDHhaem b Fe3+ ---> CDHflavinH° + H+ +CDHhaem b Fe2+ (2)
CDHflavinH° + X ---> CDHflavin + XH (3)
CDHhaem b Fe2+ + Y + H+---> CDHhaem b Fe3+ + YH (4)
where X and Y are either one-electron acceptors(X=ferricyanide; Y=ferricyanide or cytochrome c) or oxygenand its intermediate reduction products, H2O2 and HO2.
Fig. 5 shows the time courses for reductions of flavin andhaem during aerobic cycles at pH 6 with limiting amounts ofcellobiose and excess of oxygen (which must therefore pro-vide both species X and Y) for the enzyme from S. rolfsii[35]. Flavin is reduced immediately and the haem reductionlags (cf. the corresponding reactions of P. chrysosporiumenzyme in ref. [86]). When cellobiose is exhausted bothgroups are reoxidised with the flavin reoxidation precedingthat of the haem. The cycle time corresponds to an oxygen-dependent cellobiose oxidation rate at least fourfold fasterthan either haem reduction or flavin reoxidation rates. Theinitial haem reduction and final reoxidation steps must there-fore be complemented by other faster processes during thecatalytic cycle.
Both catalase and superoxide dismutase (SOD) act toslow down the cycle (Fig. 6). The flavin redox cycles areshown in Fig. 6A, and the haem b cycles in Fig. 6B. The

270 Current Protein and Peptide Science, 2006, Vol. 7, No. 3 Zamocky et al.
Fig. (5). Time courses of Sclerotium rolfsii CDH flavin and haem b aerobic oxidoreduction by cellobiose (cf. the similar behaviour of Phan-erochaete chrysosporium described in ref. [62]). Data were obtained by successive scans of samples in semi-micro cuvettes (final volume0.95 mL) using a Cary 5E spectrophotometer. The experiment shown involved 0.46 µM CDH (haem b concentration based upon E(mM) of22.8 reduced minus oxidized at 564 nm) in pH 6.0, 50 mM potassium phosphate buffer at 30 °C and ambient oxygen levels of approx. 200µM with the reaction cycle started by addition of cellobiose (28 µM) as indicated. A t0.5off (the time to reach half-maximal reduction) of 450seconds gives a reaction rate of 0.06 µM cellobiose/second or 0.12 µelectrons/second and hence an enzyme turnover of 0.27 electron equiva-lents sec-1 haem b-1. [P. Nicholls unpublished results, cf. ref. 137].
A)
B)

Cellobiose Dehydrogenase Current Protein and Peptide Science, 2006, Vol. 7, No. 3 271
(Legend Fig. 6)
Fig. (6). Effects of SOD and catalase on aerobic Sclerotium rolfsii CDH cellobiose redox cycles (cf. the similar behaviour of Phanerochaetechrysosporium described in ref. [62]).A) Flavin cycles.Flavin reduction time courses were monitored at 510-483 nm (scale *3) using successive scans in a cary 5E spectrophotometer as in Fig. 5:the catalytic cycles show the effects of addition of SOD (40 µg/mL), catalase (40 µg/mL), and both enzymes together. 0.46 µM CDH (haemb) in 50 mM potassium phosphate buffer pH 6 at 30 °C was treated with 14 µM cellobiose; other conditions are as in Fig. 5.B) Haem b cycles.Haem reduction time courses were monitored at 564-550 nm. Experiments as in Fig. 6A. Conditions as in Fig. 5 and Fig. 6A. [P. Nicholls,unpublished results; cf. ref. 137].
duration of the flavin cycle, but not the final reoxidation rate,is SOD- and catalase-sensitive. The final post-steady-statereoxidation phase of the haem group depends upon both per-oxide and superoxide and is slowed markedly in the presenceof catalase plus SOD (Fig. 6B). SOD alone has its primaryaction upon cycle time, catalase slows the final oxidationrate, and the combination abolishes the fast haem reoxidationprocess. Either SOD, or catalase, diminishes the turnover byabout 50%. The two together give a rate only a little over25% of that in the uninhibited system. It is clear that the ini-tial reaction with oxygen must give superoxide (O2
•-); subse-quent reactions of both O2
•- and hydrogen peroxide must befaster than the oxygen reaction itself.
What is the oxygen-dependent step? It is almost certainlya reaction with the flavin semiquinone species generated inthe initial one-electron transfer to the haem b group.Prereduction of the latter with ascorbate diminishes the oxy-gen reaction rate [P. Nicholls, unpublished results]. The fla-vin semiquinone had already been identified as the candidateoxygen-reacting site by Morpeth twenty years ago [68].
Cytochrome c is a one-electron acceptor reduced by theenzyme [86]. The rate in electron equivalents/second at pH 6is about half that of oxygen [P. Nicholls, unpublished results;cf. data in Table 6]. CDH promotes oxidation of cellobioseby ferricyanide (cf. data in Table 6). With the S. rolfsii en-zyme, addition of SOD further decreases the cytochrome creduction rate by about 50%. This implies that the processoccurs partly via superoxide and partly by direct reactionwith the haem b [83]. The two rates are almost equal, as pre-dicted if the steps of Eqs. 1 & 2 are followed by those ofEqs. 3', 4’ and 5:
CDHflavinH• + O2 ---> CDHflavin + H+ + O2•- (3)'
CDHhaem b Fe2+ + cyt. c Fe3+---> CDHhaem b Fe3+ + cyt. c Fe2+ (4)'
O2•- + cyt. c Fe3+---> O2 + cyt. c2+ (5)
In Eq, 3', oxygen is the acceptor reoxidising the flavinsemiquinone after an electron has been transferred to haem b(Eq. 2 above). Two electrons then reach cytochrome c, oneby direct reaction with the haem b [83], the other through theintermediacy of superoxide anion. The overall reaction isthen that of an O2-mediated cellobiose-cytochrome c reduc-tase (for those CDHs active in the acidic range).
The CDH-promoted oxidation of cellobiose by ferricya-nide is much faster than by molecular O2, presumably be-cause ferricyanide (like two-electron acceptor dyes) can reactdirectly with the flavin in both fully reduced and semiqui-none states and therefore bypass haem b reduction. But the
steady state O2- reduction rate is faster than that of the flavin
reoxidation after substrate exhaustion, and much faster thanthe initial rate of haem b reduction seen in the cellobiose-O2
cycles (Fig. 6A and B). This demands an explanation.
The enzyme's oxygen cycle time is increased 2-fold byeither SOD or catalase and 4-fold by both. The intermediatesO2
•- and H2O2 must be produced during turnover and reactmore rapidly with reduced flavin and haem b than does oxy-gen itself. This is consistent with the steps of Eqs. 1 & 2 and3' (above) being followed in the absence of alternative one-electron acceptors by those in Eqs. 6 and 7:
CDHflavinH2 + O2•- + H+ ---> CDHflavinH• + H2O2 (6)
CDHflavinH• + H2O2 + H+ + CDHhaem b Fe2+ ---> CDHflavin +CDHhaem b Fe3+ + 2 H2O (7)
More broadly, the oxygen reactions provide evidence forthe possible occurrence of different oxidation mechanisms inthe steady state in the presence of excess cellobiose (involv-ing the three-electron reduced form of CDH) compared withthose involving one- or two-electron reduced enzyme formsas major intermediates during cellobiose depletion. The gen-eration of reactive oxygen species (H2O2 and O2
•- or HO2•)
may be an important component of the CDH-promoted oxi-dative attack on lignin (see section 6.6 following). Ecologi-cally available iron complexes (such as ferrous oxalate, cf.ref. [87]) may be reduced by the haem b group as are cyto-chrome c and ferricyanide. Peroxide-mediated reoxidation ina Fenton process can give rise to reactive hydroxyl radicalsas proposed by Wood and coworkers [52, 88] and discussedbelow.
It may here just be noted that the Wood mechanism is anindirect one. Formation of hydroxyl radicals takes place re-motely from the enzyme. But the enzyme alone can generatethe reactive oxygen species superoxide and peroxide. Andreoxidation of the haem b group itself by hydrogen peroxide[63] (Fig. 6B), indicated as a concerted two-electron processin Eq. 7, may also be part of a Fenton-like step producinghydroxyl radicals directly as in Eq. 8:
H2O2 + H+ + CDHhaem b Fe2+ -----> CDHhaem b Fe3+ + H2O + OH• (8)
The oxygen and transition metal redox cycles of CDHthus permit, at least in theory, both direct and indirect Fentonhydroxyl-radical generation. The indirect mechanism has theadvantage of keeping the destructive hydroxyl radical awayfrom the enzyme, as Hyde and Wood point out [22]. But thevarious options, both mechanistic and biological, remain tobe examined in detail.

272 Current Protein and Peptide Science, 2006, Vol. 7, No. 3 Zamocky et al.
6. STRUCTURE AND FUNCTION
6.1. Overall Structure of the CDH Cytochrome Domain
The inherent flexibility of the CDH molecule has effec-tively hindered crystal-structure analysis of the full-lengthhaemoflavoenzyme. Therefore, the crystal structures of theflavoprotein and the cytochrome domain of CDH were elu-cidated separately [13, 14]. The CDH cytochrome domain[13] comprises roughly 190 amino acids and folds into anellipsoidal anti-parallel β sandwich (Fig. 7A) with the sametopology as the heavy chain of the variable Fab domain. Thefold features a five-stranded and a six-stranded β sheet, re-ferred to as the inner and outer sheets, respectively. Besidesthe β structure, there is a short α-helical structure at the C-terminus. The haem group is bound in a pocket at one face ofthe β sandwich close to the surface of the molecule. Theconcave inner β sheet forms the framework of the pocket,and three loops protruding from this sheet wedge the pro-toporphyrin-IX ring, two from above (residues 62-68 and148-161) and one from below (residues 85-95). The packingof the pocket is tight with mainly hydrophobic residues,leaving little or no space for entry of additional molecules.The haem-iron atom lies almost perfectly in the least-squares-plane defined by the four pyrrole-nitrogen atoms,and the distances from the iron atom to the nitrogen atoms ofthe four haem-pyrrole rings are all close to 2.0 Å.
The haem iron is hexa-coordinated with Met65 andHis163 as axial ligands (Fig. 7A) and coordination distancesof approximately 2.0 Å and 2.3 Å are observed for His163Nε2 and Met65 Sγ, respectively. In addition, one watermolecule is within hydrogen-bonding distance (3.0 Å) ofHis163 Nδ1. The plane of the methionine side chain (CH3-S-CH2) forms an angle of ~30° with the plane of the pyrrolenitrogens of the protoporphyrin ring, whereas the histidineimidazole ring is close to perpendicular to the pyrrole-nitrogen plane. The planes defined by the CH3-S-CH2 atomsin Met65 and the imidazole ring of His163 are at an angle of~80°, which is relatively close to a perpendicular arrange-ment of the axial ligands. This arrangement is in agreementwith the unusual coordination geometry suggested by Coxand co-workers [89]. CDH represents the first haem b-containing enzyme with Met/His haem ligation, and theCDH cytochrome domain is only the second example of acytochrome with such a ligated b haem, the first having beensoluble cytochrome b562 from E. coli [90]. However, cyto-chrome b562 is an all-α four-helix bundle protein with nostructural relationship to the CDH cytochrome domain.
6.2. Overall Structure of the Flavin Dehydrogenase Do-main
As predicted from the sequence similarity [12, 91], theoverall structure of the 540-residue large flavoprotein do-main [14] resembles the p-hydroxybenzoate hydroxylase(PHBH)-like fold [92, 93] also described for other membersof the GMC superfamily. The fold typical for members ofthe GMC superfamily is represented by the structures ofcholesterol oxidase from Brevibacterium sterolicum andStreptomyces sp., glucose 1-oxidase from Aspergillus nigerand Penicillium amagasakiense, hydroxynitrile lyase fromPrunus dulcis, and pyranose 2-oxidase from Trametes multi-color and Phlebiopsis gigantea (see Protein Data Bank at
http://www.rcsb.org for details and references). The poly-peptide chain folds into two structurally distinct subdomains(Fig. 7B), one that binds the FAD cofactor noncovalently (Fsubdomain), and one that binds the substrate cellobiose (Ssubdomain). In the flavocytochrome, the N-terminal cyto-chrome domain ends at residue 190 and is followed by theinter-domain peptide linker (residues 191 to 215).
The F subdomain of CDH comprises residues 216 to 249,404 to 511 and 693 to 755. The structure is of type α/β andfeatures a six-stranded, mainly parallel, β-pleated sheetsandwiched between a β meander (a three-stranded antipar-allel β sheet) and three α helices. The binding of the adeno-sine diphosphate (ADP) moiety of the cofactor involves thecharacteristic βαβ mononucleotide-binding motif, or Ross-mann fold, frequently encountered in NAD and FAD de-pendent enzymes [94]. The F subdomain also contains threeadditional α helices that pack together to form a substructureon one side of this subdomain. The F subdomain forms aroof over the flavin-binding pocket.
The S subdomain consists of residues 250 to 403 and 512to 692, and the principal structural feature is a centraltwisted, seven-stranded β sheet with three α helices on oneface of the sheet, and the active site on the other. This βsheet provides a "floor" for the active site. A four-stranded,slightly irregular, antiparallel β sheet forms one side of theflavin-binding pocket close to the riboflavin moiety of theFAD cofactor, and also serves as a connection between the Fand S subdomains. At the beginning of the S subdomain,residues 250 to 299 form a loop-and-lid structure that deline-ates the active-site entrance (the loop, residues 250-288) andthe outer wall of the FAD-binding pocket (the lid, residues289-299).
In vitro, CDH adsorbs specifically to cellulose [95-97]through a binding site located in the FAD-binding dehydro-genase domain which does not coincide with the active site[96] Phylogenetic analysis [47] has shown that CDHs belongto one of two classes. Class-I comprises basidiomyceteCDHs that lack a characteristic cellulose-binding domain butnevertheless bind strongly to cellulose through an as yet uni-dentified mechanism; class-II consists of ascomycete CDHsthat carry a fungal type-1 carbohydrate-binding module(CBM1). P. chrysosporium CDH belongs to class-I, and se-quence [91] as well as crystal structure confirm the lack of adistinct CBM [14]. The structural basis for the strong ab-sorption of class-I CDHs to cellulose remains unclear.
6.3. Active-Site Structure and Substrate Binding
The isoalloxazine moiety of the flavin cofactor is locatedin the active site at the end of a 12 Å long tunnel leadingdown from the subdomain interface. The flavin in the crystalstructure is hydroxylated at C6 as predicted from its spectro-scopic features. Moreover, the isoalloxazine ring is bentalong the N5-N10 axis giving an angle of 22° between theplanes of the xylene and the pyrimidine rings (butterflybending). The amide moiety is positioned close to the N-terminal part of the C-terminal α helix, which may provide apositive dipole effect. Both the butterfly bending and thepositive dipole may modulate the redox potential of the fla-vin in its hydroquinone and semiquinone states. On the siside of the flavin, the Gly310 N atom forms a hydrogen bond

Cellobiose Dehydrogenase Current Protein and Peptide Science, 2006, Vol. 7, No. 3 273
with N5 of the flavin ring. The catalytically important aminoacids, Asn732 and His689 are both situated at the re side ofthe flavin ring (Fig. 8), a common feature in the GMC oxi-doreductase family.
The crystal structure of P. chrysosporium CDH flavo-protein domain with a bound competitive inhibitor, 5-amino-5-deoxy-cellobiono-1,5-lactam (Cblm; Ki ~0.25 mM at35°C), has also been reported [53]. The geometry of the in-hibitor is similar both to that of the product, and to that of apossible transition state. The flavin ring adopts a more planarconformation than in CDH without ligand. The glucosyl-binding sites and the substrate-binding residues are located atthe re face of the isoalloxazine ring (Fig. 8). The lactammoiety of Cblm, corresponding to the reducing end of cello-biose, is bound in site C. The endocyclic lactam nitrogen ispositioned near the flavin N5 (3.2 Å) and O4 (2.9 Å). The C1atom of the lactam moiety, which corresponds to the site ofoxidative attack in cellobiose, binds in a position 2.9 Å infront of and below the N5-C4a locus of the flavin ring. Ahydrogen bond is formed between the lactam O1 and the Nε2
atom of the catalytic residue His689. Considering the short-ness of this hydrogen bond (2.6 Å), the interaction is likelyto be strong. The glucosyl moiety of the ligand resides in siteB. From the number of possible interactions, site B is likelyto make a substantial contribution to cellobiose binding. Thisis in agreement with reported values of the kinetic constantsfor di- and monosaccharides [52].
With the exception of two important changes, the struc-tural features of the active site remain unperturbed uponligand binding. Firstly, the aromatic side chain of Tyr609flips from an energetically strained conformation in the un-liganded active site to a different conformation. In this newstate it interacts with a pair of water molecules bound to theO3 and O6 endocyclic atoms of the inhibitor in site C (Fig.8). These water molecules form the floor of the active site,and help position the reducing end of the inhibitor in front ofthe flavin ring. The interaction between the inhibitor C6 hy-droxyl and one of these water molecules may explain the lowactivity on xylobiose. In the absence of this interaction, thedegree of conformational freedom within the reducing end ofthe substrate would be high; this is probably the case withxylobiose. Secondly, the flipping of Tyr609 traps a watermolecule that has no obvious hydrogen-bonding partner ex-cept for a π-H2O interaction offered by the tyrosyl ring. Inthe unliganded active site, Tyr609 occupies the space takenup by this water molecule in the ligand complex. Tyr609 isconserved in all known CDH-sequences, further stressing itsprobable importance in catalysis.
6.4. Structural Implications for Catalysis
Similar specific relative geometry of the substrate andcofactor reactive groups is found in crystal structures ofligand complexes for nicotinamide-dependent [98], flavin-dependent [99] and quinone-dependent [100] oxidoreduc-tases, all of which have been assigned a hydride-transfer
Fig. (7). Presentation of the structure of the two (separated) domains of P. Chrysosporium CDH.A) Molecular fold of the CDH cytochrome b domain (PDB accession code 1D7C [13]). The cytochrome molecule has an immunoglobulin-like fold belonging to the Family-9 carbohydrate-binding module superfamily. The three loops in the cytochrome that corresponds to com-plementarity-determining regions in immunoglobulins are colored orange. The haem group and the haem-ligating residues His163 and Met65are shown as stick objects. Coordination of the protohaem Fe2+ atom by the protein ligands is indicated by dashed lines. Secondary structureis shown as follows: β strands, arrows; α helices, spirals. The picture was made with PyMOL (http://pymol.sourceforge.net).B) Ribbon drawing of the CDH flavodehydrogenase domain. The molecule consists of two subdomains: the Rossmann domain (purple) witha FAD/NAD(P)-binding domain fold; and a substrate-binding domain (blue) similar to the α+β sandwich fold of FAD-linked reductases(PDB accession code 1NAA [53]). The cellobionolactam inhibitor (green) is bound in the active site below the FAD cofactor (yellow). Theloop-and-lid segment that delineates the active-site entrance is highlighted in orange. Secondary structure is shown as follows: β strands,arrows; α helices, spirals. The picture was made with PyMOL (http://pymol.sourceforge.net).

274 Current Protein and Peptide Science, 2006, Vol. 7, No. 3 Zamocky et al.
mechanism. The close proximity between C1 and N5 inCDH, together with the relative geometry of the atoms, ap-pears to favour a general-base catalysed hydride-transfermechanism. General base-assisted deprotonation of the C1hydroxyl group by His689 and the associated expulsion of 1-H as hydride, via a planar or nearly planar TST, is consistentwith the experimentally observed binding of Cblm [53] (Fig.9). Based on crystal-structure analysis alone, it is howevernot possible to assess the relative timing of O-H and C-Hbond cleavages.
For choline oxidase, another representative of the GMCoxidoreductase superfamily, the relative timing of OH andCH bond cleavages has been investigated in detail by meansof primary deuterium and solvent kinetic isotope effects, incombination with steady-state and rapid kinetics approaches[101]. It was shown that the removal of the hydroxyl proton,and the transfer of the hydride from the substrate (choline)R-carbon to the flavin cofactor, occur in a stepwise fashionthat is consistent with the formation of a transient alkoxidespecies. Thus, in the case of choline oxidase, a chemicalmechanism is expected in which the choline hydroxyl protonis not in flight in the transition state for CH bond cleavage.The authors propose that flavin-dependent enzymes that oxi-dise unpolarised and polarised alcohols share a commoncatalytic mechanism where hydride transfer is facilitated bythe enzyme-catalysed formation of an alkoxide species.
Fig. (9). Superposition of the experimental cellobionolactam in-hibitor (Cblm) complex (orange carbon atoms; PDB accession code1NAA) with a theoretical model of cellobiose (green carbon atoms)bound in the active site of CDH [53]. Relevant hydrogen atoms arecoloured violet, oxygen atoms are red and nitrogen atoms blue. Thecatalytic subsite C is shown with the reducing-end lactam and glu-cosyl rings. In a nearly planar transition state resembling the in-hibitor, the 1-hydrogen would be suitably positioned for directtransfer as hydride to the flavin N5 atom (path indicated by dashedlines). The picture was made with PyMOL (http://pymol. source-forge.net).
Fig. (8). Details of the inhibitor-protein interactions of the CDH-cellobionolactam crystal complex. The lactam moiety of the inhibitor (cor-responding to the reducing-end glucosyl residue in cellobiose) is bound in the catalytic site C, and the non-reducing end glucosyl moiety insubsite B (PDB accession code 1NAA [53]). The catalytic residues His689 and Asn732, and the N5 atom of the flavin ring are all situated inclose proximity of the inhibitor C–1 carbon atom in site C. The FAD co-factor and the Cblm inhibitor are shown with yellow and green car-bon atoms, respectively. Protein carbon atoms are coloured beige. Oxygen atoms are red and nitrogen atoms are blue. Three water moleculesdiscussed in the text are shown as red spheres. All interactions between ligand and protein that are within hydrogen-bonding distance (< 3.2Å) are pictured as dashed lines. The picture was made with PyMOL (http://pymol.sourceforge.net).

Cellobiose Dehydrogenase Current Protein and Peptide Science, 2006, Vol. 7, No. 3 275
6.5 In Silico Docking of the CDH Domains
A possible mode of association between the domains inCDH was investigated by performing molecular dockingusing an exhaustive six-dimensional search procedure forsurface complementarity [M. Hallberg, unpublished results].In brief, the coordinates for the cytochrome domain of CDH(PDB accession code 1D7C [13]) and the flavodehydro-genase domain (PDB accession code 1KDG [14]) were usedto model the intact enzyme. The program HEX [102], whichuses spherical polar Fourier correlations, was used for theprotein-docking simulations with surface complementarity asthe main search criteria. The selected solution was furthersubjected to conjugate-gradient minimisation in CNS ex-cluding the X-ray term [103]. The solvent-excluded area wascalculated according to Lee & Richards [104] in CNS. Tocomplete the model of intact CDH, the linker peptide wasbuilt manually between the C-terminus of the cytochromedomain and the N-terminus of the dehydrogenase domainaccording to the amino-acid sequence [12; 45] using the pro-gram O [105].
Both haem-propionate groups in the crystal structure ofthe CDH cytochrome [13] are solvent exposed, and this faceof the molecule appears particularly suitable for interactingwith the flavoprotein domain to allow inter-domain electrontransfer between the prosthetic groups [13]. In another flavo-cytochrome, flavocytochrome b2 (FCB2), the cytochromeand flavodehydrogenase domains associate intimately toposition the haem and FMN cofactors with an edge-to-edgedistance of 9.7 Å to allow rapid inter-domain electron trans-fer [106]. In FCB2, a 15-residue long linker connects theflavoprotein and cytochrome domain, and domain mobilityhas been demonstrated by means of X-ray crystallography[106] and site-directed mutagenesis studies [107, 108]. Inone FCB2 mutant, a disulfide bond between the domains wasintroduced to function as a reversible lock [107]. For thisFCB2 variant, the locked form displayed a 10-fold lower rateconstant for haem reduction compared to that of the openform, whereas the rate constant for flavin reduction was onlyslightly affected. In addition, the length of the linker inFCB2 has been modified showing that the structural integrityof the linker is crucial for efficient flavin-to-haem electrontransfer [108]. CDH contains a 25-residue long linker be-tween the domains, comparable in length to that connectingthe catalytic and cellulose-binding domains in many cellu-lases. Although it remains to be proven, the presence of along spacer peptide makes it reasonable to assume a certaindegree of mobility of the domains in CDH with respect toeach other.
The interacting surfaces of the domains of flavocyto-chromes are highly complementary and their interaction hasa mainly non-ionic character. Thus, the strength of the inter-action is mainly attributed to the area of their shared surfaces[109]. The most favourable docking modes obtained posi-tioned the cytochrome domain close to active-site entranceof the flavin domain. The degree of surface complementarityis high between the surface around the active-site entranceon the flavoprotein domain and the haem-exposed face of thecytochrome. The surface area buried between the domains inthe selected CDH model is roughly 1440 Å
2, which is con-
siderably larger than the buried surface in FCB2 (~400 Å2),
but somewhat smaller than that of 1750 Å2 in flavocyto-
chrome c sulfide dehydrogenase from Chromatium vinosum[110]. In analogy with flavocytochrome b2 from Baker’syeast, surface-exposed tyrosine residues on the CDH cyto-chrome may play a role in communication between the flavinand haem redox centres in CDH. A potentially importantresidue in CDH is Tyr90, which forms a hydrogen bond toone of the haem-propionate groups [13]. Intrinsic backboneflexibility near Tyr90 may play a role in proper positioningof the tyrosine with respect to the flavoprotein domain, or indocking between the domains.
The docking simulation suggests that the cytochromealmost completely blocks the entrance to the active site inCDH (Fig. 10). In the docking model, at least one of thehaem propionates is pointing into the active site, resulting inan approximate distance of 8 Å between the haem propionateand the FAD isoalloxazine ring. In a protein medium, elec-trons normally do not tunnel over distances greater than 14 Å[111]. Although the edge-to-edge distance in the CDHdocking model is quite large (14 Å), this may be compen-sated for by the close proximity (8 Å) of one of the haempropionates to the flavin ring. If this model represents a validdocking mode, the haem propionates and the substrate wouldcompete for the active site, resulting in substrate-gated elec-tron transfer. This could explain the observation that exces-sive concentrations of cellobiose slow down or even inhibitelectron transfer [56, 62].
Fig. (10). Theoretical in silico model of full-length P. chrysospo-rium CDH with the N-terminal cytochrome b domain (right) andthe C-terminal flavodehydrogenase domain (left). The linker pep-tide connecting the two domains is shown. Secondary structure isshown as follows: β strands, arrows; α helices, spirals. All α helicesare coloured red. The β strands belonging to the cytochrome do-main are coloured light blue, and those belonging to the dehydro-genase domain dark blue. The co-factors are shown as stick models.Carbon atoms of the FAD and haem molecules are coloured yellowand light red, respectively. One haem propionyl group is occypyingthe entrance to the active site that leads to the FAD-binding pocketin the dehydrogenase domain. The picture was made with PyMOL(http://pymol.sourceforge.net).

276 Current Protein and Peptide Science, 2006, Vol. 7, No. 3 Zamocky et al.
It has been noted that inter-domain electron transfer inCDH is strictly pH dependent such that electron transfer isinhibited at higher pH values. In order to explain this obser-vation, the crystallographic models of the individual P.chrysosporium CDH domains were used to render electro-static potential surfaces, coinciding with the modeled domaininterface, at two different pH values (pH 3.5 and pH 7.0). AtpH 3.5, the surfaces are essentially neutral whereas at pH7.0, the interacting surfaces of both domains are predomi-nantly negatively charged. At higher pH, severe repulsiveelectrostatic forces may effectively inhibit intimate domainassociation and accompanying inter-domain electron trans-fer. Thus, this explains, at least partly, the strong pH depend-ence of electron transfer.
6.6. Biological Function
Over the years, there have been probably as many func-tions proposed for CDH as there have been workers in-volved, perhaps even more because some investigators havesuggested several different functions. Here, we focus on oneleading hypothesis for the biological function of CDH,namely the hydroxyl radical hypothesis: Electrons from cel-lobiose oxidation are transferred one-by-one to the haemdomain and subsequently to a one-electron acceptor. In thehydroxyl-radical hypothesis [88], the electron acceptor isproposed to be a ferric complex, probably Fe(III)-oxalate,since oxalic acid is excreted by the fungus in large amountsduring cellulose degradation. The resulting ferrous ions canthen participate in a Fenton reaction in the presence of H2O2,produced by for instance pyranose oxidases (or from CDHitself - see above). The Fenton reaction has the form:
Fe(II) + H2O2 → Fe(III) + OH– + OH•
The hydroxyl radicals then react non-specifically withlignin or cellulose, since CDH binds specifically to the latter.Evidence for generation of hydroxyl radicals by CDH hasbeen provided, both directly [84] and indirectly [112]. Whensupplemented with ferric ions and hydrogen peroxide, CDHwas able to decrease the degree of polymerisation of pulp[113] and to reduce the viscosity of carboxymethyl cellulose(CMC). The attack on lignin is more debated. AlthoughCDH appears to depolymerise synthetic lignin in the testtube [112], a CDH-knockout mutant of T. versicolor showedno difference in its ability to degrade Kraft pulp lignin com-pared with the wild-type strain [29].
In theory, the generated hydroxyl radicals would attackboth lignin and cellulose. However, due to the strong bindingof CDH to cellulose and the expected short-range effect ofthe hydroxyl radicals, the radical-mediated attack may be-come physically restricted to the cellulosic regions in themore realistic experimental setup with Kraft pulp. But wouldnot the radicals destroy CDH as well as other enzymes pre-sent in the immediate vicinity of the attack? Indeed, Bao andRenganathan [28] showed that microcrystalline cellulose wasefficiently degraded using a combination of cellulase andCDH, but that degradation slowed down in the presence ofexcess CDH. Addition of catalase relieved inhibition byCDH significantly, although not completely. However, self-degradation or degradation of other enzymes present causedby the generated hydroxyl radicals is unlikely to be a prob-lem in vivo since only about 0.5 % of the protein mass se-
creted by the fungus constitutes CDH, and since the wood-based substrate will always be present in large excess overenzyme. A more detailed discussion about possible otherbiological functions of CDH are found in previous reviews[15, 16].
7. APPLICATIONS
Several interesting applications have been proposed util-ising the unique catalytic properties of CDH. Suggestionsconcern mainly two areas: analytical usage in biosensors andenzymatic assays for the detection and quantification of sub-strates and analogous molecules, as well as biotechnologicalapplications in pulp and paper processing, bioremedia-tion/biodegradation, and biocatalysis.
7.1. Analytical Usage
Due to its narrow specificity towards electron donors andits high sensitivity towards a multitude of electron acceptors,the enzyme has been used in several analytic assays and sen-sors.
Biosensors for the Detection of Electron Donors
A biosensor for the detection of disaccharides has beenprepared by crosslinking P. chrysosporium CDH in a poly-vinyl pyridine polymer functionalised with osmium com-plexes and polyamine groups (Os-pVP-PA) on a rotatingdisc electrode. The electrode was able to detect cellobioseand cellooligosaccharides up to a polymerisation degree of 6,lactose, and maltose [114]. Mounted in a flow cell togetherwith a glucose oxidase electrode, a CDH electrode preparedby the same group was used for post-column quantificationof glucose (50 µM – 50 mM), cellobiose (5 µM – 80 mM),and soluble cellodextrins [115]. In a different approach P.chrysosporium CDH was immobilised in an osmium-basedhydrogel on a graphite electrode and used together with asecond electrode containing oligosaccharide dehydrogenasein a dual-enzyme electrode cell for the detection of certainmono-, di-, and oligosaccharides. The enzyme electrodes haddifferent selectivities and the CDH-modified electrode re-sponded mainly to cellobiose and lactose, and to a far lesserdegree to maltose and glucose. A linear calibration rangefrom 25 µM to 3 mM for the analytes was obtained with thetwo working electrodes [116]. Another redox polymer-basedbiosensor was used for the detection of cellulase activity. P.chrysosporium CDH was immobilised by cross-linking in apolyvinyl pyridine polymer functionalised with osmiumcomplexes and ethylamine groups (Os-pVP-EA). The preci-sion is reported to be similar to the commonly used Somo-gyi-Nelson method but the range of detection is limited tothe area of linear response (100-300 µM cellobiose) and, dueto the kinetic properties of P. chrysosporium CDH, which asa basidiomycete CDH belongs to class-I, the sensitivity forglucose is nearly five orders of magnitude less. This dis-crimination of reducing monosaccharides is an interestingfeature of the biosensor when compared to the colorimetricmethod [117].
Biosensors for the Detection of Electron Acceptors
Detection of ortho- and para-diphenolic compounds wasaccomplished with P. chrysosporium CDH directly absorbedto graphite electrodes. The sensor efficiently discriminated

Cellobiose Dehydrogenase Current Protein and Peptide Science, 2006, Vol. 7, No. 3 277
between diphenols and monophenols, and was sensitiveenough to detect these in concentrations of as low as 5 nM[118]. The same electrode was used in an enzyme flow im-munoassay as the label detector. The substrate 4-aminophenol was oxidised at the electrode surface forming4-imino quinone, which was reduced back by CDH in thepresence of cellobiose. This cycling of the analyte was usedto amplify the signal [119].
The detection of phenolic compounds in waste water wasthe issue of another study. An amperometric biosensor basedon P. chrysosporium CDH was evaluated for its usefulnessas fast screening system for in-field detection. The authorsconclude that the biosensor results were overestimated whencompared to LC-MS results, but might be used for pre-screening waste water samples for positives [120].
The detection of catecholamines was reinvestigated andthe performance of P. chrysosporium and S. rolfsii CDHcompared [121]. The enzymes were immobilised on modi-fied graphite electrodes and used for the amperometric de-tection in the flow injection mode. The sensitivity, linearrange, and amplification factor was modulated by the cello-biose concentration in the buffer, which was used to recyclethe catecholamines between the graphite electrode and thereduced CDH. The result of this is an amplified responsesignal (the response current for 500 nM hydroquinone was62 fold increased in the presence of 200 µM cellobiose com-pared to the absence of cellobiose) enabling detection limitsbelow 1 nM. The electrodes prepared with enzymes fromdifferent sources show different characteristics, such as ahigher sensitivity with the enzyme from P. chrysosporium,which is the more suitable one for this application.
Other CDHs from basidiomycete and ascomycete sourceswere recently compared for their ability to directly exchangeelectrons (direct electron transfer DET) with different alka-nethiol-modified Au-electrodes. The basidiomycete CDHsfrom T. villosa and P. sordida exhibit excellent biocatalyticand redox currents, whereas for the ascomycete CDH fromM. thermophilum the DET communication is less efficient[64]. The promising results from this new study might pro-vide new input for the application of CDH in biosensors.
Enzymatic Assays
An assay for measuring the cellobiose produced in thecellulase saccharification process was developed to measurecellulase activity. It is based on the oxidation of cellobiosewith ferricyanide and cellobiose dehydrogenase fromSporotrichum thermophile. To eliminate the interference ofβ-glucosidase, inhibition with glucono-δ-lactone was sug-gested. The procedure gives a linear response between activ-ity and enzyme concentration, and between activity and timeof incubation. The sensitivity of the assay was improved byusing the formation of Prussian Blue, forming from the pro-duced ferrocyanide and ferric ions, which has a very highabsorption at 660 nm [122]. Another group used an electrodeto detect the oxidation of cellobiose. An oxidation-reductionpotential electrode was used in combination with the P.chrysosporium CDH-ferricyanide redox system for quantifi-cation [123].
A colorimetric cellobiose assay, albeit with DCIP or 3,5di-tert-butylbenzoquinone instead of ferricyanide, was used
for monitoring cellobiohydrolase purification from T. reeseiculture supernatant. The enzymes in the fractions weremonitored using carboxymethyl cellulose and the formedcellobiose was determined with CDH. An almost linear re-sponse in the range from 5 – 400 µM cellobiose was ob-tained, even in the presence of high amounts of glucose[124].
CDH was also successfully used to quantify lactose inmilk [125]. The assay is based on Sporotrichum thermophileCDH and on the experimental procedure suggested for cello-biose as described above [123].
7.2. Biotechnology
The still high price of CDH not yet encouraged largescale use of CDH in industrial processes. Although the het-erologous expression of several CDHs in Pichia pastorismight change this situation, for most applications the use offungal cultures producing the enzyme has been suggested.
Waste Removal / Bioremediation
Biodegradation of two superabsorbent polymers, acrosslinked, insoluble polyacrylate and an insoluble poly-acrylate/polyacrylamide copolymer were observed in P.chrysosporium cultures grown under CDH-inducing condi-tions [126]. A non-specific, free radical mechanism for de-polymerisation by hydroxyl radicals, produced in a Fentontype reaction, was proposed. The addition of iron increasedthe rate of solubilisation of both polymers. Solubilisation ofthe polymers was also observed in a system of purified CDH,cellobiose, ferric iron, hydrogen peroxide, and polyacrylate.For the practical application in the biodegradation of thesechemically very stable fibers used in hygiene products andfood packaging, the authors did not suggest the enzymaticapproach, but the use of the fungus grown under CDH in-ducing conditions [127]. A second mechanism for the bio-degradation of aliphatic halocarbons (the model substancebromotrichloromethane was dehalogenated) was also sug-gested. When oxalate, a secondary metabolite of white-rotfungi, was used as iron chelator in the above-mentioned re-action, it was oxidised by the hydroxyl radical to form thecarboxylate anion radical, a strong reductant [86]. Anotherremediation scenario might be the usage of CDH-producingfungi to degrade hexahydro-1,3,5-trinitro-1,3,5-triazine, amilitary high explosive compound that contaminates armyinstallations due to improper disposal of munition wastes[128]. A series of further compounds that can be degraded byP. chrysosporium cultures secreting wood-degrading en-zymes is given in [129]. The effects described are a result ofthe action of several enzymes including CDH. The applica-tion of CDH in this high-volume low-cost application is cer-tainly limited to the whole cell approach.
Finally, the CDH producing, nonsporulating mycelialfungus INBI 2-26(-) which was isolated from Vietnamesedioxin-contaminated soil was able to degrade atrazine, one ofthe most widespread triazine herbicides [130]. The degrada-tion of this compound (in the absence of laccase, which isnot produced) is attributed to CDH formed by the fungus.Again, the radical degradation mechanism via the Fentonreaction is suggested. Additionally, the authors could dem-

278 Current Protein and Peptide Science, 2006, Vol. 7, No. 3 Zamocky et al.
onstrate that CDH synthesis is stimulated when the xenobi-otic is present, even with glucose as the sole carbon source.
Enzymatic Treatment of Paper Pulp
Several authors suggested that CDH could be used as partof an enzyme complex to modify or break down celluloseand lignin in the pulp and paper industry [131]. The sugges-tion was to use it similar to other ligninolytic enzymes in thepresence of complexed iron [112]. The synergistic interac-tion of P. chrysosporium CDH with another ligninolytic en-zyme, manganese peroxidase, has been reported [27]. Otherreports of the successful use of CDH in kraft pulp delignifi-cation/bleaching include the Schizophyllum commune en-zyme [132] and the Humicola insolens enzyme [133].
CDH probably depolymerises lignin at the aromatic etherlinkage or by introducing hydroxyl groups, thereby transfer-ring non-phenolic lignin to phenolic lignin, that is suscepti-ble to an attack by the manganese peroxidase/laccase system[26]. Novo Nordisk A/S, Denmark, holds a patent for the useof a CDH with alkaline pH optimum (pH 5-9) for a pulpbleaching process, preferably in combination with xylanaseor other hemicellulases and cellulases, and with peroxidases[133]. The degree of bleaching has been moderate in allthese experiments.
Unfortunately, the unspecific attack by generating hy-droxyl radicals also affects cellulose fibres. The supplemen-tation of Douglas fir kraft pulp with CDH, cellobiose, andiron resulted in a substantial reduction in the degree of po-lymerisation of the pulp cellulose [113]. A cellulose-degrading effect was also reported for Irpex lacteus CDH[20] and for P. chrysosporium CDH [28]. The use of thepurified enzyme for bleaching is for the given reasons andthe high enzyme costs unlikely, but the application of wholeorganisms is a promising strategy. Nevertheless, the exactrole of CDH in these processes is still unclear.
Biocatalysis
The application of CDH in processes for the enzymaticconversion of carbohydrates in food industry requires thepurified enzyme. The high enzyme costs have to be justifiedby efficient usage or highly priced products. The productionof lactobionic acid with CDH, a redox mediator, and laccaseas regenerating enzyme was suggested [34]. This processprovides a high total turnover number for both enzymes,mainly depending on the chosen redox mediator. The highestspecific productivity of a typical conversion was reported tobe 32 g·L-1·h-1, resulting in a very high space-time yieldwhen compared with other enzymatic processes [134].
The use of this enzyme system for the production of alactose-free galacto-oligosaccharide mixture was also pro-posed. After transgalactosylation the residual lactose is con-verted to lactobionic acid by the above described enzymaticreaction, which can be easily removed by ion-exchangechromatography. The obtained product was of a considerablyhigher purity than other available products [135].
ACKNOWLEDGEMENTS
The authors are indebted to reviewer B for thoroughlyreading the manuscript and his critical remarks and valuablesuggestions.
REFERENCES
[1] Atalla, R.H. (1993) Trichoderma cellulases and other hydrolyases8, 25-39.
[2] Brunow, G., Kilpeläinen, I., Sipilä, J., Syrjäen, K., Karhunen, P.,Setälä, H., and Rummakko, P. (1998) ACS Symp. Series 697, 131-147.
[3] Beguin, P., and Aubert, J.P. (1994) FEMS Microbiol. Rev. 13, 25-58.
[4] Simon, L., Bousquet, J., Lévesque, R.C., and Lalonde, M. (1993)Nature 363, 67-69.
[5] Remy, W., Taylor, T.N., Hass, H., and Kerp, H. (1994) Proc. Natl.Acad. Sci. U.S.A. 91, 11841-11843.
[6] Carlile, M.J., and Watkinson, S.C. (1994) “The Fungi“ AcademicPress, New York.
[7] Barr, B.K., Hsieh, Y.-L., Ganem, B., and Wilson, D.B. (1996)Biochemistry 35, 586-592.
[8] Srisodsuk, M., Kleman-Leyer, K., Keränen, S., Kirk, T.K., andTeeri, T.T. (1998) Eur. J. Biochem. 251, 885-892.
[9] Périé, F.H., and Gold, M.H. (1991) Appl. Environ. Microbiol. 57,2240-2245.
[10] Harris, G.W., Pickersgill, R.W., Connerton, I., Debeire, P., Touzel,J.P., Breton, C., and Pérez, S. (1997) Proteins: Struct., Funct., &Genetics 29, 77-86.
[11] Westermark, U., and Eriksson, K.-E. (1974) Acta Chem. Scand. B28, 209-214.
[12] Raices, M., Paifer, E., Cremata, J., Montesino, R., Stahlberg, J.,Divne, C., Szabó, I.J., Henriksson, G., Johansson, G., and Petters-son, G. (1995) FEBS Lett. 369, 233-238.
[13] Hallberg, B.M., Henriksson, G., Pettersson, G., and Divne, C.(2000) Structure 9, 79-88.
[14] Hallberg, B.M., Henriksson, G., Pettersson, G., and Divne, C.(2002) J. Mol. Biol. 315, 421-434.
[15] Henriksson, G., Johansson, G., and Pettersson, G. (2000) J. Bio-technol. 78, 93-113.
[16] Cameron, M.D., and Aust, S.D. (2001) Enzyme Microb. Technol.28, 129-138.
[17] Roy, B.P., Dumonceaux, T., Koukoulas, A.A., and Archibald, F.S.(1996) Appl. Environ. Microbiol. 62, 4417-4427.
[18] Ludwig, R., Salamon, A., Varga, J., Zámocky, M., Peterbauer,C.K., Kulbe, K.D., and Haltrich, D. (2004) Appl. Microbiol. Bio-technol. 64, 213-222.
[19] Raices, M., Montesino, R., Cremata, J., Garcia, B., Perdomo, W.,Szabo, I., Henriksson, G., Hallberg, B.M., Pettersson, G., and Jo-hansson, G. (2002) Biochim. Biophys. Acta 1576, 15-22.
[20] Hai, P.Q., Nozaki, K., Amano, Y., and Kanda, T. (2000) J. Appl.Glycosci. 47, 311-318.
[21] Temp, E., and Eggert, C. (1999) Appl. Environ. Microbiol. 65, 389-395.
[22] Hyde, S.M., and Wood, P.M. (1997) Microbiology 143, 259-266.[23] Bao, W., Usha, S.N., and Renganathan, V. (1993) Arch. Biochem.
Biophys. 300, 705-713.[24] Wood, J.D., and Wood, P.M. (1992) Biochim. Biophys. Acta 1119,
90-96.[25] Habu, N., Samejima, M., Dean, J.F.D., and Eriksson K-E. (1993)
FEBS Lett. 327, 161-164.[26] Henriksson, G., Zhang, L., Li, J., Ljungquist, P., Reitberger, T.,
Pettersson, G., and Johansson, G. (2000) Biochim. Biophys. Acta1480, 83-91.
[27] Hildén, L., Johansson, G., Pettersson, G., Li, J., Ljungquist, P., andHenriksson, G. (2000) FEBS Lett. 477, 79-83.
[28] Bao, W., and Renganathan, V. (1992) FEBS Lett. 302, 77-80.[29] Dumonceaux, T., Bartholomew, K.A., Valeanu, L., Charles, T.C.,
and Archibald, F. (2001) Enzyme Microb. Technol. 29, 478-489.[30] Yoshida, M., Ohira, T., Igarashi, K., Nagasawa, H., and Samejima,
M. (2002) FEMS Microbiol. Lett. 217, 225-230.[31] Stapleton, P.C., and Dobson A.D. (2003) FEMS Microbiol. Lett.
221, 167-172.[32] Stapleton, P.C., O´Mahony, J., and Dobson, A.D. (2004) Can. J.
Microbiol. 50, 113-119.[33] Yoshida, M., Igarashi, K., Kawai, R., Aida, K., and Samejima, M.
(2004) FEMS Microbiol. Lett. 235, 177-182.[34] Baminger, U., Subramaniam, S.S., Renganathan, V., and Haltrich,
D. (2001) Appl. Environ. Microbiol. 67, 1766–1774.[35] Ludwig, R., and Haltrich, D. (2002) Lett. Appl. Microbiol. 35, 261-
266.

Cellobiose Dehydrogenase Current Protein and Peptide Science, 2006, Vol. 7, No. 3 279
[36] Dekker, R.F.H. (1988) Methods Enzymol. 160, 454-463.[37] Schou, C., Christensen, M.H., and Schülein, M. (1998) Biochem. J.
330, 565-571.[38] Igarashi, K., Verhagen, M.F.J.M., Samejima, M., Schülein, M.,
Eriksson, K.L., and Nishino, T. (1999) J. Biol. Chem. 274, 3338-3344.
[39] Subramaniam, S.S., Nagalla, S.R., and Renganathan, V. (1999)Arch. Biochem. Biophys. 365, 223-230.
[40] Vasil'chenko, L.G., Khromonygina, V.V., Karapetyan, K.N., Va-sil'chenko , O.V., and Rabinovich, M.L. (2005) J. Biotechnol. 119 ,44-59.
[41] Schmidhalter, D.R., and Canevascini, G. (1993) Arch. Biochem.Biophys. 300, 559-563.
[42] Hüttermann, A., and Noelle, A. (1982) Holzforschung 36, 283.[43] Macaluey-Patrick, S., Fazenda, M.L., McNeil, B., and Harvey,
L.M. (2005) Yeast 22, 249-270.[44] Dumonceaux, T.J., Bartholomew, K.A., Charles, T.C., Moukha,
S.M., and Archibald, F.S. (1998) Gene 210, 211-219.[45] Li, B., Nagalla, S.R., and Reganathan, V. (1997) Appl. Environ.
Microbiol. 63, 796-799.[46] Moukha, S.M., Dumonceaux, T.J., Record, E., and Archibald, F.S.
(1999) Gene 234, 23-33.[47] Zámocky, M., Hallberg, M., Ludwig, R., Divne, C., and Haltrich,
D. (2004) Gene 338, 1-14.[48] Yoshida, M., Igarashi, K., Wada, M., Kaneko, S., Suzuki, N.,
Matsumura, H., Nakamura, N., Ohno, H., and Samejima, M. (2005)Appl. Environ. Microbiol. 71, 4548-4555.
[49] Higham, C.W., Gordon-Smith, D., Dempsey, C.E., and Wood,P.M. (1994) FEBS Lett. 351, 128-132.
[50] Henriksson, G., Sild, V., Szabo, I.J., Pettersson, G., and Johansson,G. (1998) Biochim. Biophys. Acta 1383, 48-54.
[51] Canevascini, G., Borer, P., Dreyer, J.L. (1991) Eur. J. Biochem.198, 43-52.
[52] Kremer, S.M., and Wood, P.M. (1992) Eur. J. Biochem. 205, 133-138.
[53] Hallberg, B.M., Henriksson G., Pettersson G., Vasella A., andDivne C. (2003) J. Biol. Chem. 278, 7160-7166.
[54] Rotsaert, F.A.J., Renganathan, V. and Gold, M.H. (2003) Bio-chemistry 42, 4049-4056.
[55] Coudray, M.R., Canevascini, G., and Meier, H. (1982) Biochem. J.203, 277-284.
[56] Igarashi, K., Momohara, I., Nishino, T., and Samejima, M. (2002)Biochem. J. 365, 521-526.
[57] Henriksson, G., Johansson, G., Ruiz, A., and Uzcategui, E. (1993)Biochem. Biophys. Acta 1144, 184-190.
[58] Jones, G.D., and Wilson, M.T. (1988) Biochem. J. 256, 713-718.[59] Samejima, M., Phillips, R.S., and Eriksson, K.-E. (1992) FEBS
Lett. 306, 165-168.[60] Rogers, M.S., Jones, G.D., Antonini, G.,M., Wilson, M.T., and
Brunori, M. (1994) Biochem. J. 298, 329-334.[61] Wilson, M.T., and Liu, B.-L. (1994) Trans. Biochem. Soc. 22, 725-
728.[62] Cameron, M.D., and Aust, S.D. (2000) Biochemistry 39, 13595-
13601.[63] Mason, M.G., Wilson, M.T., Ball, A., and Nicholls, P. (2002)
FEBS Lett. 518, 29-32.[64] Stoica, L., ”Functional aspects of cellobiose dehydrogenase”
(2005) PhD Thesis University Lund, Sweden.[65] Samejima, M., and Eriksson, K.-E.L. (1992) Eur. J. Biochem. 207,
103-107.[66] Karapetyan, K.N., Fedorova,T.V., Vasil'chenko, L.G., Ludwig, R.,
Haltrich, D., and Rabinovich, M.L. (2005) J. Biotechnol.Doi:10.1016/j.jbiotec.2005.06.024.
[67] Igarashi, K., Yoshida, M., Matsumura, H., Nakamura, N., Ohno,H., Samejima, M., and Nishino, T. (2005) FEBS J. 272, 2869-2877.
[68] Morpeth, F.F. (1985) Biochem. J. 228, 557-564.[69] Bao, W., and Renganathan, V. (1991) FEBS Lett. 279, 30-32.[70] Cohen, J.D., Bao, W., Renganathan, V., Subramaniam, S.S., and
Loehr, T.M. (1997) Arch. Biochem. Biophys. 341, 321-328.[71] Massey, V., Schopfer, L.M., Nishino, T., and Nishino, T. (1988) J.
Biol. Chem. 264, 10567-10573.[72] Morpeth, F.F., and Jones, G.D. (1986) Biochem. J. 236, 221-226.[73] Xu, F., Golightly, E.J., Duke, K.R., Lassen, S.F., Knusen, B.,
Christensen, S., Brown, K.M., Brown, S.H., and Schülein, M.(2001) Enzyme Microb. Technol. 28, 744-753.
[74] Leitner, C., Volc, J., and Haltrich, D. (2001) Appl. Environ. Micro-biol. 67, 3636-3644.
[75] Roth, J.P., and Klinman, J. P. (2003) Proc. Natl. Acad. Sci. U.S.A.100, 62-67.
[76] Klinman, J.P. (2001) J. Biol. Inorg. Chem. 6, 1-13.[77] Hille, R., and Nishino, T. (1995) FASEB J. 9, 995-1003.[78] Hashida, S., Yuzawa, S., Suzuki, N.N., Fujioka, Y., Takikawa, T.,
Sumimoto, H., Inagaki, F., and Fujii, H. (2004) J. Biol. Chem. 279,26378-26386.
[79] Vignais, P.V. (2002) Cell Mol. Life Sci. 59, 1428-1459.[80] St-Pierre, J., Buckingham, J. A., Roebuck, S. J., and Brand, M. D.
(2002) J. Biol. Chem. 277, 44784-44790.[81] Ishii, N., Fujii, M., Hartman, P. S., Tsuda, M., Yasuda, K., Senoo-
Matsuda, N., Yanase, S., Ayusawa, D., and Suzuki, K. (1998) Na-ture 394, 694-697.
[82] Imlay, J.A. (1995) J. Biol. Chem. 270, 19767-19777.[83] Nutt, A., Salumets, A., Henriksson, G., Sild, V., and Johansson, G.
(1997) Biotechnol. Lett. 19, 379-383.[84] Cameron, M.D., and Aust, S. D. (1999) Arch. Biochem. Biophys.
367, 115-121.[85] Wilson, M.T., Hogg, N., and Jones, G. D. (1990) Biochem. J. 270,
265-267.[86] Mason, M.G., Nicholls, P., Divne, C., Hallberg, B. M., Henriksson,
G., and Wilson, M.T. (2003) Biochim. Biophys. Acta 1604, 47 - 54.[87] Park, J.S., Wood, P. M., Davies, M. J., Gilbert, B.C., and Whit-
wood, A.C. (1997) Free Radical Res. 27, 447-458.[88] Kremer, S.M., Wood, P. M. (1992) Eur. J. Biochem. 208, 807-814.[89] Cox, M.C., Rogers M.S., Cheesman, M., Jones, G.D., Thomson,
A.J., Wilson, M.T., and Moore, G.R (1992) FEBS Lett . 307, 233-236.
[90] Mathews, F.S., Bethge, P.H., and Czerwinski, E.W. (1979) J. Biol.Chem. 254, 1699-1706.
[91] Henriksson, G., Salumets, A., Divne, C., and Pettersson G. (1997)Biochem. J. 324, 833-838.
[92] Wierenga, R.K., DeJong, R.J, Kalk, K.H., and Hol, W.G.J. (1979)J. Mol. Biol. 131, 55-73.
[93] Mattevi, A. (1998) Biophys. Chem. 70, 217-222.[94] Rossmann, M.G., Liljas, A., Brändén, C.I., and Banaszak, L.J.
(1975) In "The Enzymes", Ed. P. D. Boyer, 11, 61-102.[95] Renganathan, V., Usha, S.N., and Lindenburg, F. (1990) Appl.
Microbiol. Biotechnol. 32, 609-613.[96] Henriksson, G., Petterson, G., Johansson, G., Ruiz, A., and Uz-
categui, E. (1991) Eur. J. Biochem. 196, 101-106.[97] Samejima, M., Ohkubo, T., Igarashi, K., Isogai, A., Kuga, S., Sugi-
yama, J., and Eriksson, K.-E. L. (1997) Biotechnol. Appl. Biochem.25, 135-141.
[98] Mattevi, A., Vanoni, M.A., Todone, F., Rizzi, M., Teplyakov, A.,Coda, A., Bolognesi, M., and Curti, B. (1996) Proc. Natl. Acad.Sci. U.S.A. 93, 7496-7501.
[99] Karplus, P.A., and Schulz, G.E. (1989) J. Mol. Biol. 210, 163-180.[100] Oubrie, A., Rozeboom, H.J., Kalk, K.H., Olsthoorn, A.J.J., Duine,
J.A., and Dijkstra, B.W. (1999) EMBO J 18, 5187-5194.[101] Fan, F., and Gadda, G. (2005) J. Am. Chem. Soc. 127, 2067-2074.[102] Ritchie, D.W., and Kemp, G.J.L. (2000) PROTEINS: Struct. Funct.
Genet. 39, 178-194.[103] Brunger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P.,
Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M.,Pannu, N.S., Read, R.J., Rice, L.M., Simonson, T. and Warren,G.L. (1998) Acta Crystallogr. D 54, 905-921.
[104] Lee, B., and Richards, F.M. (1971). J. Mol. Biol. 44, 379-400.[105] Jones, T.A., Zou, J.-Y., Cowan, S.W., and Kjeldgaard, M. (1991).
Acta Crystallogr. A 47, 110-119.[106] Xia, Z.-X., and Mathews, F.S (1990) J. Mol. Biol. 212, 837-863.[107] Chapman, SK., Reid, G.A., Bell, C., Short, D., and Daff, S. (1996).
Biochem. Soc. Trans. 24, 73-77.[108] Sharp, R.E., Chapman, S.K., and Reid, G.A. (1996) Biochemistry,
35, 891-899[109] Cunane, L.M., Chen, Z.W., Durley, R.C., Barton, J.D., and
Mathews, F.S. (1999) Biochem. Soc. Trans. 27, 179-184.[110] Chen, Z.-W., Koh, M., van Driessche, G., van Beeumen, J.J.,
Bartsch, R.G., Meyer, T.E., Cusanovich, M.A., and Mathews, F.S.(1994). Science 266, 430-432.
[111] Page, C.C., Moser, C.C., Chen, X., and Dutton, P.L. (1999) Nature402, 47-52.
[112] Henriksson, G., Ander, P., Pettersson, B., and Pettersson, G. (1995)Appl. Microbiol. Biotechnol. 42, 790-796.

280 Current Protein and Peptide Science, 2006, Vol. 7, No. 3 Zamocky et al.
[113] Mansfield, S.D., de Jong, E., and Saddler, J.N. (1997) Appl. Envi-ron. Microbiol. 63, 3804-3809.
[114] Elmgren, M., Lindquist, S.-E. and Henriksson, G. (1992) J. Elec-troanal. Chem. 341, 257-273.
[115] Nordling, M., Elmgren, M., Stahlberg, J., Pettersson, G. andLindquist, S.E. (1993) Anal. Biochem. 214, 389-396.
[116] Tessema, M., Larsson, T., Buttler, T., Csöregi, E., Ruzgas, T.,Nordling, M., Lindquist, S.-E., Pettersson, G. and Gorton, L.(1997) Anal. Chim. Acta 349, 179-188.
[117] Hilden, L., Eng, L., Johansson, G., Lindquist, S.-E. and Pettersson,G. (2001) Anal. Biochem. 290, 245-250.
[118] Lindgren, A., Stoica, L., Ruzgas, T., Ciucu, A. and Gorton, L.(1999) Analyst, 124, 527-532.
[119] Burestedt, E., Nistor, C., Schagerlöf, U. and Emneus, J. (2000)Anal. Chem. 72, 4171-4177.
[120] Nistor, C., Rose, A., Farre, M., Stoica, L., Wollenberger, U., Ruz-gas, T., Pfeiffer, D., Barcelo, D., Gorton, L. and Emneus, J. (2002)Anal. Chim. Acta 456, 3-17.
[121] Stoica, L., Lindgren-Sjölander, A., Ruzgas, T. and Gorton, L.(2004) Anal. Chem. 76, 4690-4696.
[122] Canevascini, G. (1985) Anal. Biochem. 147, 419-427.[123] Samejima, M., Sugiyama, J., Igarashi, K. and Eriksson, K. E.
(1998) Carbohydr. Res. 305, 281-288.[124] Kelleher, T.J., Montenecourt, B.S. & Eveleigh, D.E. (1987) Appl.
Microbiol. Biotechnol. 27, 299-305.[125] Canevascini, G., Etienne, K., and Meyer, H. (1982) Z. Lebensm.
Unters. Forsch. 175, 125-129.[126] Stahl, J.D., Cameron, M. D., Haselbach, J. and Aust, S. D. (2000)
Environ. Sci. Pollut. Res. 7, 83-88.[127] Cameron, M.D., Post, Z. D., Stahl, J. D., Haselbach, J. and Aust, S.
D. (2000) Environ. Sci. Pollut. Res. 7, 130-134.
[128] Stahl, J.D., Van Aken, B., Cameron, M. D. and Aust, S. D. (2001)Biorem. J. 5, 13-25.
[129] Cameron, M.D., Timofeevski, S. and Aust, S. D. (2000) Appl.Microbiol. Biotechnol. 54, 751-758.
[130] Khromonygina, V.V., Saltykova, A.I., Vasil'chenko, L.G., Kozlov,Yu. P., and Rabinovich, M.L. (2004) App. Biochem. Microbiol., 40,285-290.
[131] Ander, P., Daniel, G., Pettersson, B. and Westermark, U. (1996)ACS Symp. Ser. 655, 297-307.
[132] Fang, J., Huang, F. and Gao, P. (1999) Process Biochem. 34, 957-961.
[133] Schou, C., Schülein, M. and Vollmond, T. (1994) Patent WO94/01538.
[134] Ludwig, R., Ozga, M., Zamocky, M., Peterbauer, C., Kulbe, K. D.and Haltrich, D. (2004) Biocatal. Biotransf. 22, 97-104.
[135] Splechtna, B., Petzelbauer, I., Baminger, U., Haltrich, D., Kulbe,K. D. and Nidetzky, B. (2001) Enz. Microb. Technol. 29, 434-440.
[136] Schmidt, H.A., Strimmer, K., Vingron, M., and von Haeseler, A.(2002) Bioinformatics 18, 502-504.
[137] Nicholls, P., Zamocky, M., and Haltrich, D. (2003) “The roles ofperoxide and superoxide in the aerobic redox cycles of Sclerotiumrolfsii cellobiose dehydrogenase.” 679th. Biochem. Soc. Meeting,Essex, UK; abstracts.
[138] Kajisa, T., Yoshida, M., Igarashi, K., Katayama, A., Nishino, T.,and Samejima, M. (2004) J. Biosci. Bioeng. 98, 57-63.
[139] Fang, J., Qu, Y., and Gao, P. (1997) Biotechnol. Tech. 11, 195-197.[140] Fang, J., Qu, Y., and Gao, P. (1998) Arch. Biochem. Biophys. 353,
37-46.[141] Rotsaert, F.A., Li, B., Renganathan, V., and Gold, M.H. (2001)
Arch. Biochem. Biophys. 390, 206-214.
Received: October 10, 2005 Revised: December 22, 2005 Accepted: January 05, 2006





























